Область техники, к которой относится изобретение
Настоящее изобретение относится к соединениям, предназначенным для лечения рака. Изобретение также относится к применению соединения для получения лекарственных средств для лечения рака. Также описан способ лечения рака у пациента, включая человека, где названный способ включает введение в организм пациента соединения, указанного выше. Изобретение дополнительно относится к фармацевтическим композициям, включающим вышеуказанное соединение и соединение с цитопротекторными свойствами. Изобретение также относится к применению фармацевтических композиций для получения лекарственных средств для лечения рака.
Уровень техники
EP 0910360, US 6147094, EP 0936915, US 6258828, EP 1054670, US 6310051, EP 1060174, US 6391895 раскрывают применение хелатирующих средств на основе дипиридоксила и их хелатов с металлами и применение определенных марганецсодержащих соединений, в особенности хелатов марганца, в медицине. Также раскрыто применение таких соединений, как цитопротекторные средства, в терапии рака. Процитированные выше документы раскрывают, что определенные хелатирующие средства, в особенности хелатирующие средства на основе дипиридоксила и аминополикарбоновой кислоты, и их хелаты металлов эффективны в лечении или предупреждении антрациклин-индуцированной кардиотоксичности, повреждений, вызванных ишемией-реперфузией, и атеросклероза. Хелатирующие средства на основе дипиридоксила и их хелаты с трехвалентными металлами были ранее описаны Taiferro (Inorg. Chem. 1984; 23: 1183-1192).
DPDP (N,N'-бис-(пиридоксал-5-фосфат)этилендиамин-N,N'-диуксусная кислота) и дефосфорилированный аналог PLED (N,N'-дипиридоксилэтилендиамин-N,N'-диуксусная кислота) являются дипиридоксильными соединениями, способными хелатировать металлы. Ранее было описано, что хелаты марганца этих соединений, MnDPDP и его дефосфорилированного аналога MnPLED, проявляют каталитическую антиоксидантную активность, т.е. активность, подобную супероксид-дисмутазе (SOD). Было показано, что эти соединения оказывают защитное действие на нормальные клетки, например, против цитостатических лекарственных средств и ишемии-реперфузии. Эта SOD-подобная активность представляет собой свойство, присущее окислительно-восстановительной активности марганца (Mn2+/Mn3+), связанного с DPDP/PLED (Brurok et al., Biochem. Biophys. Res. Commun. 1999; 254: 768-721), что объясняет защитный эффект. Таким образом, Brurok и сотрудники (1999) показали, что PLED металлический комплекс теряет его каталитическую активность после замещения обладающего окислительно-восстановительной активностью марганца на не обладающий окислительно-восстановительной активностью цинк (Zn2+).
Laurent et al. (Cancer Res. 2005; 6: 948-56) и Alexandre et al., J. Natl. Cancer Inst. 2006; 98: 236-44) недавно описали, что MnDPDP (эквивалентный готовому к использованию МРТ контрастному реагенту Тесласкан) не только повышает выживаемость нормальных клеток, но также увеличивает гибель раковых клеток в течение цитостатического лечения, например, оксаплатином. Цитостатические лекарственные препараты могут вызывать гибель раковых клеток путем повышения уровня внутриклеточного H2O2 и индуцируя апоптоз. Гипотеза Laurent et al. была такова, что MnDPDP из-за SOD-подобной активности увеличивает количество внутриклеточного H2O2 и, таким образом, действует синергетически с цитостатическими лекарственными препаратами. Поскольку базальный уровень H2O2 гораздо ниже в нормальных клетках по сравнению с раковыми клетками, авторы предположили, что подъем с низкого уровня H2O2 индуцирует клеточную выживаемость у нормальных клеток. Далее они предположили, что подъем с гораздо более высокого базального уровня H2O2 в раковых клетках в то же время приведет к апоптотическому сигналингу и, таким образом, клеточной смерти. Таким образом, авторы предположили, что оба этих эффекта, т.е. повышение гибели раковых клеток и выживаемости нормальных клеток, были вызваны SOD-подобной активностью MnDPDP, активностью, которая абсолютно зависима от окислительно-восстановительной активности марганца. Также было показано, что внутривенная инъекция как материнского соединения MnDPDP, так и его метаболита MnPLED мышам приводит к защите от определенных цитостатических лекарственных средств (EP0910360 и US6147094).
Когда MnDPDP внутривенно инъецировался людям, примерно 70% введенного марганца было высвобождено. Для использования в диагностической визуализации и для периодического терапевтического применения диссоциация марганца из MnDPDP не представляет большой проблемы. Однако, для более частого применения аккумулируемая марганцевая токсичность может представлять серьезную токсикологическую проблему, особенно когда дело касается нейротоксичности (Crossgrove & Zheng; NMR Biomed. 2004; 17: 544-53). Так, для частого терапевтического применения, такого как лечение рака, следует избегать соединений, которые должны распадаться с выделением марганца.
Несколько антираковых средств ассоциированы с вредным побочным действием. Паклитаксел, например, является одним таким цитостатическим лекарственным средством, которое проявляет антинеопластическую активность против множества злокачественных тканей, включая ткани молочной железы. Однако, при дозах, необходимых для достижения антинеопластического эффекта, паклитаксел обладает некоторыми вредными побочными эффектами, которые включают нарушения сердечного ритма, также как и гематологическую и гастроэнтерологическую токсичность. Оксалиплатин, в особенности в комбинации с 5-фторурацилом (5-FU), является другим примером цитостатического лекарственного средства, которое эффективно в лечении колоректального рака, но его применение ограничивается тяжелыми вредными побочными эффектами, в особенности гематологической токсичностью и нейротоксичностью. Тяжелые вредные побочные эффекты также ограничивают применение радиационной терапии рака.
Таким образом, существует неудовлетворенная медицинская необходимость в обнаружении новых хемотерапевтических лекарственных средств с меньшим количеством побочных эффектов в дополнение к обнаружению способов защиты нормальных клеток от повреждений, вызванных лечением рака.
Описание изобретения
Настоящее изобретение предоставляет соединение, обладающее способностью убивать раковые клетки, предназначенное для лечения рака. Изобретение также предоставляет применение соединения по изобретению для получения лекарственного средства для лечения рака. Изобретение также включает способ лечения рака у пациента включая человека, где названный способ включает введение в организм пациента соединения по изобретению. Также описывается фармацевтическая композиция, которая включает вышеуказанное соединение и второе соединение, имеющее цитопротекторное свойство, т.е. способность защищать нормальные клетки в течение лечения рака от побочных эффектов, вызванных хемотерапевтическими лекарственными средствами и радиацией. Также предоставлено применение фармацевтической композиции по изобретению для получения лекарственного средства для лечения рака. Изобретение также относится к способу лечения рака у пациента, включая человека, где названный способ включает введение в организм пациента соединения по изобретению.
В первом аспекте изобретение направлено на соединение формулы I

или его соль, предназначенное для лечения рака, где Х представляет CH или N,
каждый R1 независимо представляет водород или -CH2COR5;
R5 представляет гидрокси, необязательно гидроксилированный алкокси, амино или алкиламидо;
каждый R2 независимо представляет группу ZYR6; Z представляет связь или C1-3 алкиленовую или оксоалкиленовую группу, необязательно замещенную группой R7;
Y представляет связь, атом кислорода или группу NR6;
R6 является атомом водорода, группой COOR8, алкильной, алкенильной, циклоалкильной, арильной или аралкильной группой, необязательно замещенной одной или более группами из COOR8, CONR8 2, NR8 2, OR8, =NR8, =О, OP(O)(OR8)R7 и OSO3M;
R7 является гидрокси, необязательно гидроксилированной, необязательно алкоксилированной алкильной или аминоалкильной группой;
R8 является атомом водорода или необязательно гидроксилированной, необязательно алкоксилированной алкильной группой;
М является атомом водорода или одним эквивалентом физиологически приемлемого катиона; например, щелочным или щелочноземельным катионом, ионом аммония или катионом органического амина, таким как меглумин-ион;
R3 представляет C1-8 алкиленовую группу, предпочтительно C1-6, например C2-4 алкиленовую группу, 1,2-циклоалкиленовую группу, или 1,2-ариленовую группу, необязательно замещенную R7; и каждый R4 независимо представляет водород или C1-3 алкил, и где соединение необязательно является хелатом с одним или двумя Na+ или K+, где комбинация из одного Na+ и одного К+ также возможна.
В одном варианте осуществления изобретения R5 является гидрокси, C1-8 алкокси, этиленгликолем, глицерином, амино или C1-8 алкиламидо;
Z является связью или группой, выбранной из CH2, (CH2)2, CO, CH2CO, CH2CH2CO и CH2COCH2; Y является связью;
R6 является моно- или поли(гидрокси или алкоксилированной) алкильной группой или группой с формулой OP(O)(OR8)R7; и R7 является гидрокси или незамещенной алкильной или аминоалкильной группой.
В другом варианте осуществления изобретения R3 является этиленом и каждая группа R1 представляет -CH2COR5, в котором R5 является гидрокси.
В еще одном варианте осуществления изобретения соединение формулы I является N,N'-дипиридоксилэтилендиамин-N,N'-диуксусной кислотой (PLED).
В другом варианте осуществления изобретения соединение формулы I является N,N'-бис-(пиридоксал-5-фосфат)этилендиамин-N,N'-диуксусной кислотой (DPDP).
В другом варианте осуществления изобретения описано применение соединения формулы I, как определено выше, для получения лекарственного средства для лечения рака. Рак может быть любой разновидности рака, например, такой как лейкемия, рак молочной железы, колоректальный рак, рак печени и их метастазы. Лекарственное средство может дополнительно содержать фармацевтически приемлемые носители или наполнители.
Во втором аспекте изобретение направлено на способ лечения рака у пациента, включая человека, где указанный способ включает введение в организм пациента соединения формулы I в соответствии с изобретением.
В третьем аспекте изобретение направлено на фармацевтическую композицию, включающую первое соединение формулы I, как определено здесь ранее, и второе соединение, имеющее цитопротекторное свойство.
В одном из вариантов осуществления изобретения второе соединение, включенное в фармацевтическую композицию, является хелатом металла, представляющим соединение формулы I, как определено здесь ранее.
В еще одном варианте осуществления изобретения хелат металла, включенный в фармацевтическую композицию, имеет величину Ка предпочтительно в пределах от 108 до 1024, более предпочтительно в пределах от 1010 до 1022, и наиболее предпочтительно в пределах от 1012 до 1020.
В еще одном варианте осуществления изобретения хелат металла, включенный в фармацевтическую композицию, имеет более низкую величину Ка, чем величина Ка хелата железа (Fe3+), представляющего собой соединение формулы I, как определено здесь ранее, по крайней мере в 103 раза.
В еще другом варианте осуществления изобретения металл в хелате металла, включенном в фармацевтическую композицию, является марганцем (Mn2+ или Mn3+) или медью (Cu+ или Cu2+).
В другом варианте осуществления изобретения первое соединение в фармацевтической композиции является N,N'-дипиридоксилэтилендиамин-N,N'-диуксусной кислотой и второе соединение является хелатом металла, включающим N,N'-дипиридоксилэтилендиамин-N,N'-диуксусную кислоту. Металл в хелате металла предпочтительно является марганцем или медью.
В предпочтительном варианте осуществления изобретения первое соединение в фармацевтической композиции является N,N'-бис-(пиридоксал-5-фосфат)этилендиамин-N,N'-диуксусной кислотой и второе соединение является хелатом металла, включающим N,N'-дипиридоксилэтилендиамин-N,N'-диуксусную кислоту. Металл в хелате металла предпочтительно является марганцем или медью.
В еще одном варианте осуществления изобретения второе соединение в фармацевтической композиции по изобретению может предпочтительно составлять от 1/100 до 99/100 от первого соединения, по молярности.
В еще другом варианте осуществления изобретения предоставлена фармацевтическая композиция, предназначенная для лечения рака.
В четвертом аспекте изобретение предоставляет набор, включающий препарат первого активного ингредиента, который является соединением формулы I, как определено выше, препарат второго активного ингредиента, который является хелатом металла, включающим соединение формулы I, как определено выше, и необязательно инструкции для одновременного, последовательного или раздельного введения препаратов пациенту, нуждающемуся в этом.
В пятом аспекте изобретение предоставляет применение фармацевтической композиции по изобретению для получения лекарственного средства для лечения рака. Рак может быть любой разновидности рака, например, такой как лейкемия, рак молочной железы, колоректальный рак, рак печени и их метастазы.
В другом варианте осуществления изобретение предоставляет применение фармацевтической композиции по изобретению, где лекарственное средство дополнительно содержит фармацевтически приемлемые носители или наполнители.
В шестом аспекте изобретение предоставляет способ лечения рака у пациента, нуждающегося в таком лечении, включающий стадию введения указанному пациенту фармацевтической композиции, в соответствии с изобретением, в ингибирующем рак количестве, необязательно в комбинации с фармацевтически приемлемыми носителями или наполнителями. В одном из вариантов осуществления изобретения предоставлен способ, где фармацевтическая композиция вводится вместе с одним или более противораковым лекарственным(и) средством(ами). Противораковое лекарственное средство может быть любым лекарственным средством против рака, например, таким как доксорубицин, эпирубицин, оксиплатин, цисплатин, карбоплатин,паклитаксел, 5-фторурацил, циклофосфамид, гемцитабин, иринотекан и метотрексат. В другом варианте осуществления изобретения предоставляется способ, где описанная выше фармацевтическая композиция и одно или более другое противораковое лекарственное средство вводятся одновременно, раздельно или последовательно указанному пациенту, в дополнительном варианте осуществления изобретения предоставляется способ лечения как описано выше, где лечение комбинируется с радиационной терапией.
Должно также быть понятно, что изобретение включает применение фармацевтической композиции по изобретению для получения лекарственного средства для лечения рака.
Соединения формулы I, как определено выше, для применения по изобретению должны пониматься как терапевтически активные и физиологически предпочтительные соединения.
Как используется здесь, термины «алкил» и «алкилен» включают углеводороды с прямой и разветвленной цепью, насыщенные и ненасыщенные. Термин «1,2-циклоалкилен» включает как цис-, так и транс-циклоалкиленовые группы и алкилзамещенные циклоалкиленовые группы, имеющие 5-8 атомов углерода. Термин «1,2-арилен» включает фениловую и нафтиловую группы и их алкилзамещенные производные, имеющие 6-10 атомов углерода.
Если не указано обратного, любой алкильный, алкиленовый или алкениловый фрагмент может подходящим образом содержать 1-20, предпочтительно 1-8, более предпочтительно 1-6 и особенно предпочтительно 1-4 атома углерода.
Циклоалкильные, арильные или аралкильные фрагменты могут подходящим образом содержать 3-18, предпочтительно 5-12 и особенно предпочтительно 5-8 кольцевых атомов углерода. Предпочтительными являются арильные остатки, включающие фениловую или нафтиловую группы. В качестве аралкильных групп, фенил С1-8 арил, особенно бензил, являются предпочтительными.
Причем группы могут быть необязательно замещены гидроксильными группами, это может быть монозамещением или полизамещением, и, в случае полизамещения, алкокси- и/или гидроксизаместители могут замещать алкоксизаместители.
В формуле I R5 является предпочтительно гидроксилом, C1-8 алкокси, этиленгликолем, глицерином, амином или C1-8 алкиламидо. Предпочтительно каждая группа R1 представляет собой -CH2COR5, в котором R5 является гидрокси.
В соединении формулы I Z предпочтительно является связью или группой, выбранной из CH2, (CH2)2, CO, CH2CO, CH2CH2CO и CH2COCH2. Предпочтительно, Y представляет собой связь.
Соединение формулы I может иметь те же самые или различные R2 группы в двух пиридильных кольцах, и они могут быть присоединены в тех же самых или различных положениях кольца. Однако является особенно предпочтительным, чтобы замещение было в 5- и 6-положениях, наиболее предпочтительно в 6-положении, т.е. пара-положении к гидроксильной группе. Соединение, в котором R2 группы являются идентичными и идентично расположены, например, 6,6', являются наиболее предпочтительными.
Предпочтительными в качестве групп R6 являются моно- или поли(гидрокси или алкоксилированные) алкильные группы или группы формулы OP(O)(OR8)R7.
R7 предпочтительно является гидрокси или незамещенной алкильной или аминоалкильной группой.
Наиболее предпочтительные значения для группы R2 включают CHR7OCO(CH2)xPh и CHR7OCO(CH2CO)xPh (где значение х от 1 до 3), CHR7OCOBut, CH2N(H)R6', CH2N(H)R6', N(H)R6', N(R6')2, CH2OH, CH2OR6', COOR6', CON(H)R6', CON(R6')2 или OR6' (где R6' являются моно- или полигидроксилированными, предпочтительно C1-4, особенно предпочтительно C1-3, алкильной группой), (CH2)nCOOR7' (где значение n от 1 до 6), COOR7' (где R7' являются C1-4 алкильными, предпочтительно C1-3, особенно предпочтительно метильной группой) CH2OSO3 -М, CH2CH2COOH, CH2OP(O)(OH)(CH2)3NH2, CH2OP(O)(OH)CH3 или CH2OP(O)(OH)2 группами. Еще более предпочтительно, R2 представляет собой группу формулы CH2OP(O)(OH)2.
Соединения формулы I, в которых R3 является этиленом и R2 имеет одно из перечисленных выше значений, являются особенно предпочтительными.
Фармацевтические композиции по настоящему изобретению и препараты, включенные в набор по настоящему изобретению, могут быть составлены с использованием приемлемых фармацевтических или ветеринарных добавок, используемых для составления композиций, например, стабилизаторов, антиоксидантов, агентов, регулирующих осмолярность, буферов, агентов, регулирующих рН, и так далее, и могут быть в форме, подходящей для парентерального или энтерального введения, например, инъекцией или инфузией. Такие фармацевтические композиции по настоящему изобретению могут быть в приемлемой для введения фармацевтической форме, такой как таблетки, капсулы, порошки, растворы, суспензии, дисперсии, сиропы, суппозитории, и тому подобное.
Соединения формулы I и хелаты металлов, включающие соединения формулы I, могут формулироваться для введения с применением физиологически приемлемых носителей и/или вспомогательных средств способом, хорошо известным специалисту в данной области техники. Соединения формулы I и хелаты металлов, включающие соединения формулы I, могут, например, быть суспендированы или растворены в водной среде, необязательно с добавлением фармацевтически приемлемых вспомогательных средств. Лекарственное средство или формацевтическая композиция по настоящему изобретению могут быть введены различными путями, например, перорально, трансдермально, ректально, интратекально, местно или посредством ингаляции или инъекции, в особенности подкожной, внутримышечной, интраперитонеальной или внутривенной инъекции. Другие пути введения могут быть предусмотрены, если они увеличивают эффективность, биодоступность или переносимость продуктов. Наиболее приемлемый путь может быть выбран специалистом в данной области техники в соответствии с использованными составами.
Количество введенного пациенту лекарственного средства, ингибирующее рак, зависит от нескольких различных факторов таких как тип рака, возраст и вес пациента, и тому подобное, и лечащий врач будет вести лечение таким образом, чтобы подобрать дозировки в случае необходимости на основании лабораторных исследований.
Обычно дозы действующих веществ, т.е. первого и второго соединения, в фармацевтической композиции по настоящему изобретению будут содержать от 0,01 мкмоль соединения на килограмм веса пациента до 100 мкмоль соединения на килограмм веса пациента.
Фармацевтическая композиция по настоящему изобретению, таким образом, может содержать соединения формулы I, в особенности DPDP или его дефосфорилированные аналоги DPMP и PLED, обеспечивая способ лечения различных злокачественных заболеваний, как непосредственно, так и в комбинации с другими цитостатическими лекарственными средствами или радиационной терапией.
Если не все из лабильных водородов хелата по настоящему изобретению замещены образующим комплексы ионом металла, биопереносимость и/или растворимость хелатов может быть повышена путем замены оставшихся лабильных атомов водорода на физиологически биосовместимые катионы неорганических и/или органических оснований или аминокислот. Примеры подходящих неорганических катионов включают Li+, K+, Na+ и особенно Ca2+. Подходящие органические катионы включают аммоний, замещенный аммоний, этаноламин, диэтаноламин, морфолин, глутамин, N,N-диметилглюкамин, лизин, аргинин или орнитин.
Когда первое или второе соединение по изобретению несет общий заряд, его удобно использовать в форме соли с физиологически приемлемым противоионом, например, аммонием, замещенным аммонием, катионом щелочного металла или щелочноземельного металла (например, кальцием) или анионом, производным от неорганической или органической кислоты. В этом отношении меглуминовые соли являются особенно предпочтительными.
Терапевтические средства по настоящему изобретению могут быть составлены с использованием приемлемых фармацевтических или ветеринарных добавок, используемых для составления композиций, например, стабилизаторов, антиоксидантов, средств, доводящих осмолярность, подсластителей и так далее.
Как ранее описано, изобретение предоставляет соединение формулы I, как определено выше, предназначенное для лечения рака. Когда авторы настоящего изобретения сравнили MnDPDP и DPDP, они, к их удивлению, обнаружили, что DPDP был более эффективен, чем MnDPDP, в его способности убивать раковые клетки и пришли к выводу, что ранее описанная способность MnDPDP убивать злокачественные клетки является свойством, присущим DPDP. Таким образом, изобретение предоставляет новое соединение, предназначенное для лечения рака и одновременно позволяющее избежать проблемы, связанной с токсичностью, обусловленной высвобождением марганца.
Соединение можно, как ранее упоминалось, применять в комбинации со вторым соединением, имеющим цитопротекторные свойства. В варианте осуществления изобретения описано применение хелата металла, включающего соединение формулы I как соединения, имеющего цитопротекторные свойства. Неожиданно обнаружено, что этот хелат металла является гораздо более стабильным, чем MnDPDP, что позволяет избежать проблемы высвобождения металла. Таким образом, представлена подходящая комбинация лекарственных средств для лечения рака.
Стабильность MnDPDP после введения в организм человека, в соответствии с известным уровнем техники, является, главным образом, регулируемой константами стабильности между DPDP и Mn2+ другими конкурирующими металлами, в основном, не обладающими окислительно-восстановительной активностью Zn2+, который имеет большее сродство к DPDP , чем Mn2+ (Rocklage et al., Inorg. Chem. 1989; 28: 477-485 и Toft et al., Acta Radiol. 1997; 38: 677-689). После внутривенной инъекции человеку, в дополнение к диссоциации Mn2+, DPDP гидролизуется с отщеплением двух фосфатов, приводя к образованию PLED. Вскоре после внутривенной инъекции примерно 30% инъецированного MnDPDP трансформируется в MnPLED и, в соответствии с известным уровнем техники (Toft et al., 1997), PLED также диссоциирует с отщеплением Mn2+ фактически даже быстрее, чем DPDP. Такая динамика MnPLED в высшей степени подтверждается описанными в литературе константами стабильности (Rocklage et al., 1989).
Однако, новая интерпретация ранее опубликованных результатов фактически может позволить предположить, что MnPLED является гораздо более стабильным, чем MnDPDP (с точки зрения активности металла) в условиях in vivo. Если пересчитать данные концентраций в плазме, взятые из исследования Toft et al. 1997, то видно, что исчезновение MnDPDP и его 5 метаболитов из плазмы приблизительно соответствует таковому (между 30 и 60 минутами) для MnPLED (после фазы начального распределения). Все эти соединения удаляются из организма путем почечной экскреции, и если марганец отщепляется в результате диссоциации MnPLED, можно ожидать, что эти два процесса разнесены в течение этого периода времени. Эти сведения позволяют предположить, что MnPLED является стабильным в условиях in vivo.
Принимая во внимание описанные константы стабильности для Mn2+ и Fe3+, результаты в примере 3 дополнительно весьма очевидно подтверждают неожиданное предположение, что MnPLED является гораздо более стабильным, чем MnDPDP, в отношении диссоциации Mn2+. Более того, можно ожидать исходя из фармакокинетических данных, что целевые клетки и ткани не будут подвергаться воздействию концентраций, более высоких, чем 5 мкМ MnPLED, т.е. концентраций, при которых MnPLED, как ожидается, является стабильным.
Настоящее изобретение показывает, что MnPLED является гораздо более стабильным, чем MnDPDP, и, что более важно, возможно он может помочь избежать связанную с марганцем проблему токсичности при частом терапевтическом применении у человека.
Более того, следует подчеркнуть, что предварительное лечение мышей MnPLED показало, что он примерно в 100 раз более эффективен, чем MnDPDP (EP0910360 и US6147094). Это позволяет предположить, что доза MnPLED может быть значительно снижена по сравнению с MnDPDP, что дополнительно снизит токсикологический потенциал фармакологической композиции и, таким образом, дополнительно увеличит терапевтический индекс. К тому же, было показано, что более низкая доза MnPLED (3 мкмоль/кг), по сравнению с той, что используется в диагностической визуализации с помощью MnDPDP (5-10 мкмоль/кг), снижает размер инфаркта у свиней (Karlsson et al., Acta Radiol. 2001; 42: 540-547), и гораздо более низкие дозы были показаны как эффективные в той же животной модели (неопубликованные данные).
Примечательно, что MnDPDP не снижает размер инфаркта у свиней. Предположительно вследствие гораздо более быстрого замещения марганца на цинк в сравнении с человеком. Спустя десять минут после инъекции MnDPDP весь марганец замещается на цинк (Karlsson et al., 2001), что отличается от человека (и некоторых других исследованных видов), где примерно 30% инъецированного магния остается связанным с хелатирующим агентом в течение значительного количества времени. Как отмечено ранее, защита нормальных клеток, в этом случае клеток миокарда, зависит от окислительно-восстановительной активности марганца. В соответствии с известным уровнем техники (Rocklage et al., 1989), константа стабильности между Mn2+ и DPDP составляет 15,10 (logK), тогда как константа стабильности между Zn2+ и DPDP составляет 18,95, т.е. DPDP диссоциирует с образованием Mn2+ примерно в 100 раз быстрее, чем Zn2+. Соответствующие константы стабильности между Mn2+ и PLED, и Zn2+ и PLED составляют 12,56 и 16,68, соответственно, т.е. Mn2+ образуется в результате диссоциации примерно в 1000 раз быстрее, чем Zn2+. Исходя из этого и ранее опубликованной метаболической схемы (Toft et al., 1997) не ожидается какой-либо серьезной разницы в стабильности между MnDPDP и MnPLED в отношении замены марганца на цинк после введения свиньям. Выше отмеченное уменьшение инфаркта, наблюдаемое после введения MnPLED, но не MnDPDP, является, таким образом, неожиданным открытием. Однако, настоящее изобретение, как проиллюстрировано в примере 3 описания, приводит к разумному объяснению этого неожиданного явления, а именно MnPLED является более стабильным комплексом, чем MnDPDP, и, что более важно, позволяет избежать проблемы токсичности, обусловленной нестабильностью марганца.
Преимущество комбинирования DPDP-антираковой активности с MnPLED-цитопротекторной активностью по отношению к нормальным клеткам и тканям может быть продемонстрировано на примере проблемы, связанной с применением декстразоксана в качестве кардиопротекторного средства против антрациклин-индуцированной кардиотоксичности. Хотя это и не очевидно, декстразоксан не рекомендуется в начале терапии антрациклином у пациентов с метастазирующим раком молочной железы, из-за возможного снижения антиракового эффекта антрациклина (Yeh et al., Circulation 2004; 109: 3122-3132). Однако, как было продемонстрировано для MnDPDP авторами настоящего изобретения и другими исследователями, данные доклинического исследования весьма очевидно показывают, что это не является проблемой в случае подхода согласно настоящему изобретению. Одно возможное объяснение этого состоит в том, что MnDPDP имеет две различные и характерные активности, а именно его антираковая активность и его цитопротекторная активность, и которые в настоящем изобретении были дополнительно разделены на две химические структурные единицы, а именно DPDP, обладающую антираковой активностью, и MnPLED, обладающую цитопротекторной активностью в нормальных клетках и тканях.
Краткое описание чертежей
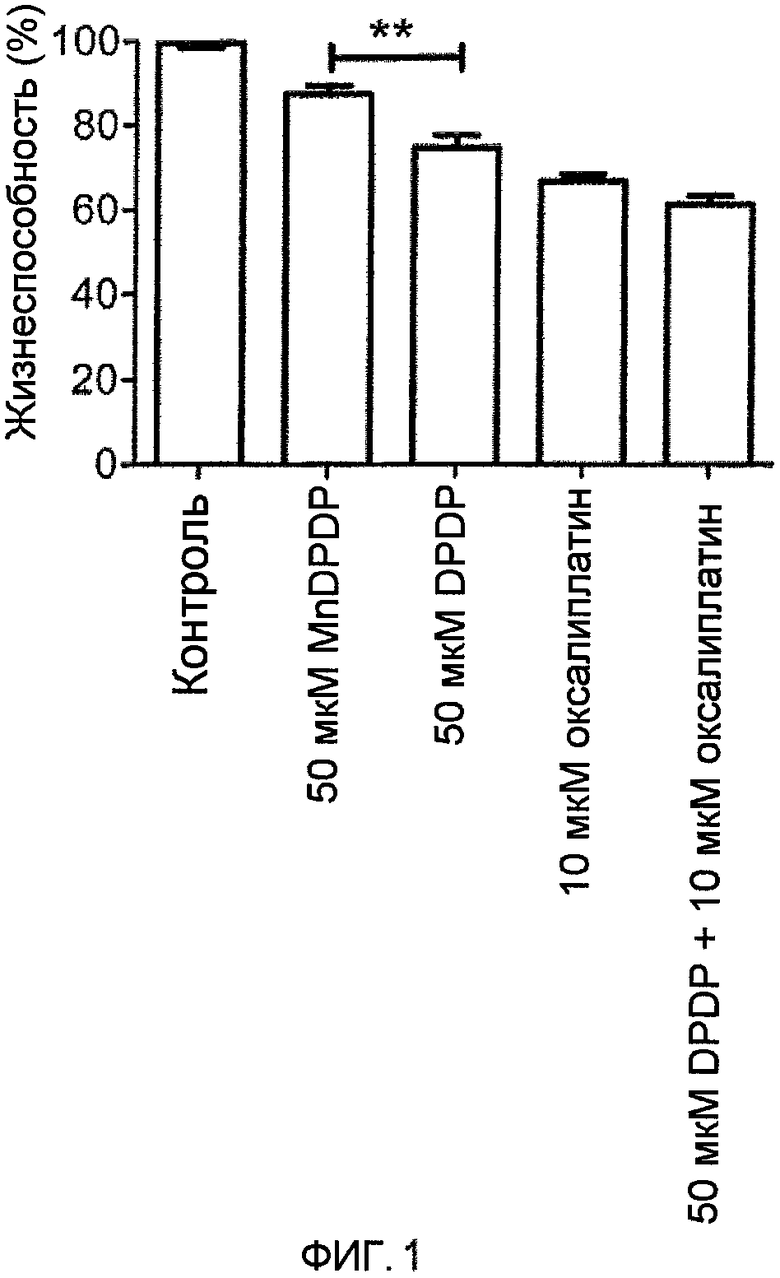
На фиг.1 представлены результаты МТТ анализа для SW480 клеток рака прямой кишки человека в отсутствие (контроль) или в присутствии MnDPDP, DPDP, оксалиплатина или DPDP + оксалиплатин (среднее ± SD; n=3).
На фиг.2 представлены результаты МТТ анализа для J774 клеток лимфомы мыши в отсутствие (контроль) или в присутствии MnDPDP, DPDP, оксалиплатина или DPDP + оксалиплатин (среднее ± SD; n=3).
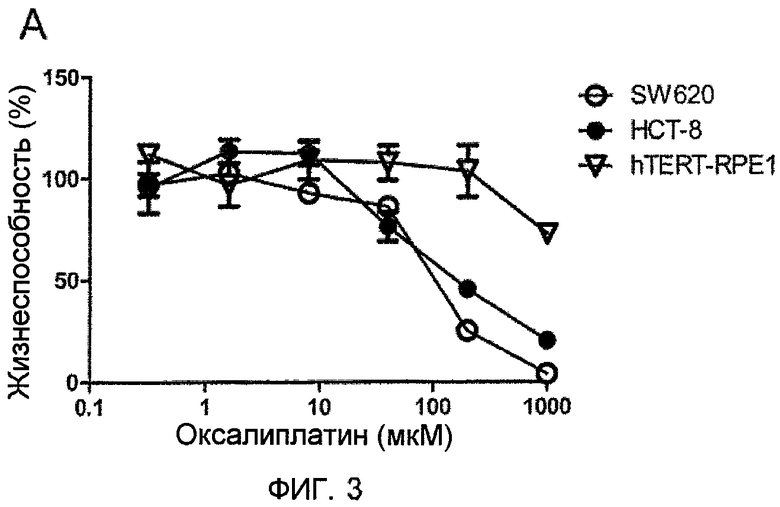
На фиг.3 показан цитостатический эффект возрастающих концентраций оксалиплатина для SW620, HCT-8 и hTERT-RPE1 клеток (А). Цитостатический эффект низких концентраций оксалиплатина в чистом виде или в комбинации с MnDPDP или DPDP для SW620 (B), HCT-8 (C), hTERT- RPE1 (D) клеток (среднее ± SD; n=3).
На фиг.4 представлены результаты анализа Фентона в присутствии различных концентраций DPDP, MnDPDP и MnPLED (среднее ± SD; n=3).
(А) контроли были поставлены в параллели и в конце экспериментов (среднее ± SD; n=3).
(В) контроли также включали отсутствие железа (-Fe), наличие хелатирующего железо средства десферриоксамина (15 мкМ DFO) или акцептора гидроксильных радикалов DMSO (10% DMSO) (среднее ± SD; n=3).
Примеры
Изобретение дополнительно продемонстрировано и описано в следующих не ограничивающих его объем примерах. Примеры должны пониматься как лишь иллюстрирующие изобретение, и изобретение не должно ими ограничиваться.
Пример 1
Цитостатическая активность DPDP и MnDPDP сравнивалась путем коинкубирования клеток рака прямой кишки человека (SW480) и клеток лимфомы мыши (J774) с MnDPDP, DPDP и/или оксалиплатином.
Способ
Жизнеспособность клеток была измерена с применением МТТ анализа. Коротко, 20000 SW480 или J774 клеток были высеяны в каждую лунку на 96-луночном планшете и выращивались в течение ночи в Dulbecco's Modified Eagle's Medium (DMEM), содержащей 10% фетальной бычьей сыворотки, 2 мМ L-глутамина, 100 Ul/мл пенициллина и 100 мкг/мл стрептомицина при 37°C в увлажненном воздухе с 5% CO2. Затем клетки были экспонированы на 24 часа в 50 мкМ MnDPDP, 50 мкМ DPDP или 10 мкМ оксалиплатином при 37°C. Эффект от комбинирования 50 мкМ DPDP и 10 мкМ оксалиплатина был также тестирован. Жизнеспособность клеток была затем определена путем добавления 5 мг/мл метилтиазолтетразолиума (МТТ) до конечной концентрации 0,5 мг/мл и инкубирования клеток еще 4 часа при 37°C. Голубой формазан, который формируется митохондриальными дегидрогеназами жизнеспособных клеток, был растворен в течение ночи при 37°C при добавлении 10% SDS и 10 мМ HCl до конечной концентрации 5% SDS и 5 мМ HCl. В заключение, абсорбция раствора была измерена при 570 нм с контролем сравнения 670 нм на считывающем устройстве для микропланшетов Spectramax 340 (Molecular Devices, Sunnyvale, CA, USA), подсоединенном к Apple Macintosh компьютеру с программой Softmax Pro V1.2.0 (Molecular Devices, Sunnyvale, CA, USA).
Результаты
Цитостатическая активность 50 мкМ DPDP была статистически достоверно более эффективной, чем цитостатическая активность 50 мкМ MnDPDP для SW480 клеток рака прямой кишки человека (unpaired t-test) (фиг.1). Была замечена тенденция, хотя и недостоверная статистически (p<0,07), что для клеток DPDP в комбинации с оксалиплатином был более эффективен, чем оксалиплатин в чистом виде. Соответствующая цитостатическая активность 50 мкМ DPDP и 50 мкМ MnDPDP для J774 клеток лимфомы мыши представлена на фиг.2, и, как очевидно из этого чертежа, DPDP был значительно более эффективен для уничтожения клеток лимфомы, чем MnDPDP. Соответственно, DPDP в комбинации с оксалиплатином был значительно более эффективен, чем оксалиплатин в чистом виде.
Выводы
При сравнении MnDPDP и DPDP было неожиданно обнаружено, что DPDP был более эффективен, чем MnDPDP в его способности убивать раковые клетки, и, следовательно, ранее описанная способность MnDPDP убивать раковые клетки является свойством, присущим DPDP.
Пример 2
Цитостатическая активность MnDPDP или DPDP в отсутствие или при наличии оксалиплатина была протестирована на клетках аденосаркомы человека (SW620), клетках илеоцекальной колоректальной аденосаркомы человека и нормальных клетках ретинального эпителия, иммортализированных теломеразой (hTERT-RPE1).
Способ
Клетки HCT-8 культивировались в RPMI 1640 среде с пируватом натрия (1 мМ) и 10% лошадиной сыворотки. SW620 выращивались в ATCC-сформулированной Leibovitz's L-15 среде с 10% фетальной бычьей сывороткой. Клетки инкубировались при 37°С во влажной атмосфере, содержащей 5% диоксида углерода. Клетки собирались в log-фазе для использования в эксперименте. Флуориметрический анализ цитотоксичности в культуре малого объема (FMCA) был применен для исследования цитостатической активности оксалиплатина, MnDPDP или DPDP и их комбинаций. FMCA основан на измерении флуоресценции, полученной в результате гидролиза диацетата флуоресцеина (FDA) до флуоресциина клетками с неповрежденными плазматическими мембранами. Коротко, 96-луночные микропланшеты были приготовлены перед использованием с раствором лекарственного средства в 3 параллелях с концентрацией, в 10 раз превышающей желаемую концентрацию лекарственного средства. Клеточная суспензия была высеяна в приготовленные планшеты с лекарственным средством в количестве 20000 клеток на лунку, и планшеты далее инкубировались в течение 72 часов. После инкубации планшеты были отмыты, добавлен FDA и после 50 минут инкубации индуцированная флуоресценция (возбуждение при 480 нм) была измерена при 538 нм на флуориметре (Fluorostar Optima, BMG Technologies). Флуоресценция пропорциональна количеству в лунке клеток с неповрежденными плазматическими мембранами.
Результаты
Цитостатическая активность увеличивающихся концентраций оксалиплатина для раковых клеток (SW620 и HCT-8) и нормальных клеток (hTERT-RPE1) показана на фиг.3А. Оксалиплатин продемонстрировал зависимый от концентрации цитостатический эффект для обеих раковых клеточных линий, и всего лишь легкий эффект при наибольших концентрациях для нормальных клеток. Ни MnDPDP, ни DPDP не продемонстрировали цитостатического эффекта для какой-либо из этих клеточных линий (не показано). Однако, 100 мкМ DPDP, но не MnDPDP, достоверно (unpaired t-test) усиливает цитостатический эффект низких концентраций (8 мкМ) оксалиплатина для обеих раковых клеточных линий, но не для нормальных клеток (фиг.3B-D).
Выводы
При сравнении MnDPDP и DPDP было неожиданно обнаружено, что DPDP был гораздо более эффективен, чем MnDPDP, в его способности увеличивать ингибирующую рак активность оксалиплатина, и, следовательно, описанная способность MnDPDP убивать раковые клетки является характеристикой, присущей DPDP.
Пример 3
Сравнивалась стабильность MnDPDP и MnPLED путем создания условий конкуренции с железом для хелатирующего агента DPDP, металлических комплексов MnDPDP и MnPLED в Fenton анализе.
Способ
Трехвалентное железо (10 мкМ) было частично восстановлено до его двухвалентной формы цистеином (100 мкМ) в 150 мМ ацетатном буфере. Н2О2 (100 мкМ) была добавлена для инициирования продукции гидроксильных радикалов (НО•). Последний окислял H2DCF (не флуоресцентный 2',7'-дихлордигидрофлуоресцеин; 5 мкМ) до флуоресцентного DCF (2',7'-дихлорфлуоресцеин). H2DCF был получен путем гидролиза его ацетатного эфира (H2DCF-DA). DMSO (10%) и DFO (10 мкМ) были использованы для демонстрации образования НО• и включения железа, соответственно. DPDP, MnDPDP и MnPLED при различных концентрациях были проанализированы на их железохелатирующую способность. Флуоресценция была измерена на FL600 Microplate Fluorescence reader (Bio-Tek.Winooski, VT, U.S.A.) при возбуждении 485 нм и эмиссии 530 нм.
Результаты
Фиг.4 показывает, что DPDP ингибировал Fenton реакцию в зависимости от дозировки; ингибирование начиналось при 0,1 мкМ и было полным при 10 мкМ. Паттерн ингибирования находился в соответствии с показанной высокой аффинностью DPDP к трехвалентному железу (logK=33,52). Однако, в случае MnPLED не наблюдалось какого-либо ингибирования вплоть до концентрации 5 мкМ, но при 10 мкМ ингибирование было полным. Железохелатирующая способность MnDPDP была значительно более высокой, чем таковая MnPLED.
Выводы
Настоящие результаты являются весьма неожиданными и противоречат тому, что можно ожидать от показанных железо- и марганец-хелатирующих потенциалов MnDPDP и MnPLED. Показанные константы стабильности между Fe3+ и DPDP и между Fe3+ и PLED составляют 33,52 и 36,88 (logK), соответственно, тогда как показанные константы стабильности между Mn2+ и DPDP и Mn2+ и PLED составляют 15,10 и 12,56, соответственно (Rocklage et al., 1989). Следовательно, можно было бы ожидать, что MnPLED будет гораздо лучшим ингибитором Fenton реакции, чем MnDPDP.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ТЕРАПЕВТИЧЕСКИЕ СПОСОБЫ, В КОТОРЫХ ПРИМЕНЯЕТСЯ КОМБИНАЦИЯ КОМПЛЕКСНОГО СОЕДИНЕНИЯ МАРГАНЦА И СОЕДИНЕНИЯ В ФОРМЕ, НЕ ЯВЛЯЮЩЕЙСЯ МАРГАНЦЕВЫМ КОМПЛЕКСОМ | 2010 |
|
RU2563825C2 |
| КАЛМАНГАФОДИПИР, НОВОЕ ХИМИЧЕСКОЕ СОЕДИНЕНИЕ И ДРУГИЕ СМЕШАННЫЕ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ С МЕТАЛЛАМИ, СПОСОБЫ ПОЛУЧЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ | 2012 |
|
RU2765805C2 |
| КАЛМАНГАФОДИПИР, НОВОЕ ХИМИЧЕСКОЕ СОЕДИНЕНИЕ И ДРУГИЕ СМЕШАННЫЕ КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ С МЕТАЛЛАМИ, СПОСОБЫ ПОЛУЧЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ | 2012 |
|
RU2622646C2 |
| СПОСОБ И КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКОВОГО ЗАБОЛЕВАНИЯ, ТОЗИЛАТ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ N-(4-ХЛОР-3-(ТРИФТОРМЕТИЛ)ФЕНИЛ)-N'-(4-(2-(N-МЕТИЛКАРБАМОИЛ)-4-ПИРИДИЛОКСИ)ФЕНИЛ)МОЧЕВИНЫ | 2002 |
|
RU2316326C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ С ИСПОЛЬЗОВАНИЕМ АЗАБИЦИКЛОСОЕДИНЕНИЯ ДЛЯ РАКОВОГО ЗАБОЛЕВАНИЯ | 2014 |
|
RU2657783C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИНГИБИТОР ГИСТОНДЕАЦЕТИЛАЗЫ | 2004 |
|
RU2322971C2 |
| ПРЕПАРАТ И КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ | 2017 |
|
RU2777595C2 |
| ПРОМЕЖУТОЧНЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 2013 |
|
RU2635359C2 |
| ЛИГАНДЫ СИГМА-РЕЦЕПТОРОВ ДЛЯ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ БОЛИ, ВЫЗВАННОЙ ХИМИОТЕРАПИЕЙ | 2010 |
|
RU2543382C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2017 |
|
RU2738837C2 |





Изобретение относится к способу лечения рака у пациента, где способ включает введение в организм пациента ингибирующего рак количества первого соединения формулы Способ лечения рака у пациента, включая человека, где указанный способ включает введение в организм пациента ингибирующего рак количества первого соединения формулы I
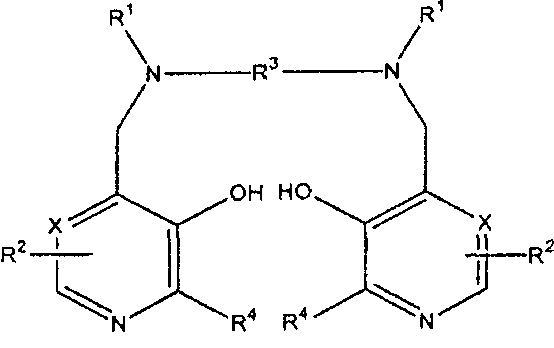
или его
физиологически приемлемой соли, где Х представляет СН или N, каждый R1 независимо представляет водород или -CH2COR5; R5 представляет гидрокси, необязательно гидроксилированный алкокси, амино или алкиламидо; каждый R2 независимо представляет группу ZYR6; Z представляет связь или C1-3 алкиленовую или оксоалкиленовую группу, необязательно замещенную группой R7; Y представляет связь, атом кислорода или группу NR6; R6 является атомом водорода, группой COOR8, алкильной, алкенильной, циклоалкильной, арильной или аралкильной группой, необязательно замещенной одной или более группами из COOR8, 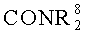
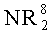
1. Способ лечения рака у пациента, включая человека, где указанный способ включает введение в организм пациента ингибирующего рак количества первого соединения формулы I
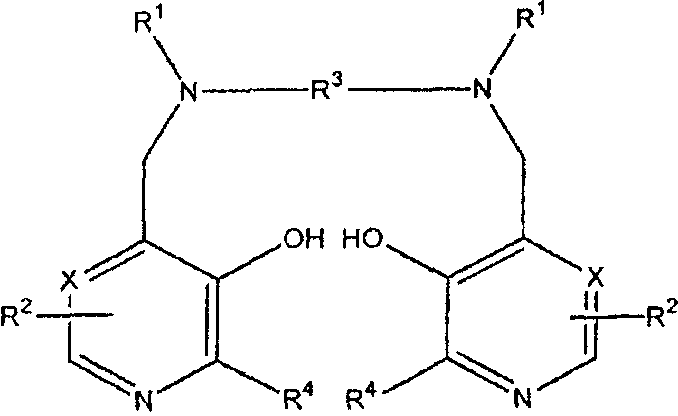
или его физиологически приемлемой соли,
где Х представляет СН или N,
каждый R1 независимо представляет водород или -CH2COR5;
R5 представляет гидрокси, необязательно гидроксилированный алкокси, амино или алкиламидо;
каждый R2 независимо представляет группу ZYR6;
Z представляет связь или С1-3 алкиленовую или оксоалкиленовую группу, необязательно замещенную группой R7;
Y представляет связь, атом кислорода или группу NR6;
R6 является атомом водорода, группой COOR8, алкильной, алкенильной, циклоалкильной, арильной или аралкильной группой, необязательно замещенной одной или более группами из COOR8,
R7 является гидрокси, необязательно гидроксилированной, необязательно алкоксилированной алкильной или аминоалкильной группой;
R8 является атомом водорода или необязательно гидроксилированной, необязательно алкоксилированной алкильной группой;
М является атомом водорода или одним эквивалентом физиологически приемлемого катиона;
R3 представляет C1-8 алкиленовую группу, 1,2-циклоалкиленовую группу или 1,2-ариленовую группу, необязательно замещенную R7; и каждый R4 независимо представляет водород или С1-3 алкил.
2. Способ по п.1, где
R5 является гидрокси, C1-8 алкокси, этиленгликоль, глицерин, амино или С1-8 алкиламидо;
Z является связью или группой, выбранной из СН2, (CH2)2, CO, СН2СО, СН2СН2СО и СН2СОСН2;
Y является связью;
R6 является моно- или поли(гидрокси или алкоксилированной) алкильной группой или группой формулы OP(O)(OR8)R7; и
R7 является гидрокси или незамещенной алкильной или аминоалкильной группой.
3. Способ по п.1, где R3 является этиленом и каждая группа R1 представляет собой -CH2COR5, в котором R5 является гидрокси.
4. Способ по п.1, где соединение является N,N'-дипиридоксилэтилендиамин-N,N'-диуксусной кислотой.
5. Способ по п.1, где соединение является N,N'-бис-(пиридоксал-5-фосфат)-этилендиамин-N,N'-диуксусной кислотой.
6. Способ по любому из пп.1-5, где рак выбран из лейкемии, рака молочной железы, рака прямой кишки, рака печени, лимфомы, колоректального рака и/или их метастаз.
7. Способ по любому из пп.1-5, где рак выбран из лимфомы, колоректального рака и/или их метастаз и рака молочной железы и/или его метастаз.
8. Способ по п.7, где рак выбран из рака прямой кишки и/или его метастаз.
9. Способ по п.7, где рак выбран из рака молочной железы и/или его метастаз.
10. Способ по п.7, где рак представляет собой лимфому.
11. Способ по любому из пп.1-5, где первое соединение вводят вместе с цитопротекторным количеством хелата металла соединения формулы I.
12. Способ по п.11, где указанный хелат металла имеет величину Ка в пределах от 108 до 1024.
13. Способ по п.11, где указанный хелат металла имеет более низкую величину Ка, чем величина Ка хелата железа (Fe3) соединения формулы I по крайней мере по фактору 103.
14. Способ по п.11, где металл является марганцем (Mn2+ или Mn3+) или медью (Cu+ или Cu2+).
15. Способ по п.11, где первое соединение является N,N'-дипиридоксилэтилендиамин- N,N'-диуксусной кислоты и хелат металла представляет собой хелат металла N,N'-дипиридоксилэтилендиамин- N,N'-диуксусной кислоты.
16. Способ по п.11, где первое соединение является N,N'-бис-(пиридоксал-5-фосфат)этилендиамин- N,N'-диуксусной кислотой и хелат металла представляет собой хелат металла N,N'-дипиридоксилэтилендиамин- N,N'-диуксусной кислоты.
17. Способ по любому из пп.1-5, где первое соединение вводят вместе с одним или более другим противораковыми лекарственными средствами, выбранными из группы, состоящей из доксорубицина, эпирубицина, оксиплатина, цисплатина, карбоплатина, паклитаксела, 5-фторурацила, циклофосфамида, гемцитабина, иринотекана и меторексата.
18. Способ по п.17, где первое соединение и одно или более других противораковых средств вводят указанному пациенту одновременно, раздельно или последовательно.
19. Способ по любому из пп.1-5, где первое соединение вводят в комбинации с радиационной терапией.
20. Фармацевтическая композиция для лечения рака у пациента, включая человека, содержащая первое соединение формулы I или его физиологически приемлемой соли и цитопротекторное количество хелата марганца N,N'-дипиридоксилэтилендиамин-N,N'-диуксусной кислоты (MnPLED):
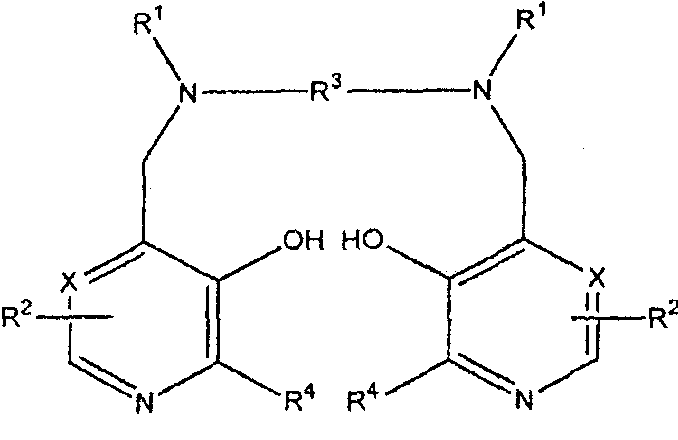
где Х представляет СН или N,
каждый R1 независимо представляет водород или -CH2COR5;
R5 представляет гидрокси, необязательно гидроксилированный алкокси, амино или алкиламидо;
каждый R2 независимо представляет группу ZYR6;
Z представляет связь или C1-3 алкиленовую или оксоалкиленовую группу, необязательно замещенную группой R7;
Y представляет связь, атом кислорода или группу NR6;
R6 является атомом водорода, группой COOR8, алкильной, алкенильной, циклоалкильной, арильной или аралкильной группой, необязательно замещенной одной или более группами из COOR8,
R7 является гидрокси, необязательно гидроксилированной, необязательно алкоксилированной алкильной или аминоалкильной группой;
R8 является атомом водорода или необязательно гидроксилированной, необязательно алкоксилированной алкильной группой;
М является атомом водорода или одним эквивалентом физиологически приемлемого катиона;
R3 представляет C1-8 алкиленовую группу, 1,2-циклоалкиленовую группу или 1,2-ариленовую группу, необязательно замещенную R7; и каждый R4 независимо представляет водород или C1-3 алкил.
21. Фармацевтическая композиция по п.20, где первое соединение представляет собой N,N'-бис-(пиридоксал-5-фосфат)этилендиамин-N,N'-диуксусную кислоту (DPDP).
22. Фармацевтическая композиция по п.20, где молярное отношение MnPLED первому соединению находится в диапазоне от 1/100 до 99/100.
23. Набор для лечения рака у пациента, включая человека, содержащий препарат первого активного ингредиента формулы I, препарат хелата металла N,N'-дипиридоксилэтилендиамин-N,N'-диуксусной кислоты (MnPLED) и, необязательно, инструкции для одновременного, последовательного или раздельного введения препаратов пациенту, нуждающемуся в этом:
Формула I

или его физиологически приемлемой соли,
где Х представляет СН или N,
каждый R1 независимо представляет водород или -CH2COR5;
R5 представляет гидрокси, необязательно гидроксилированный алкокси, амино или алкиламидо;
каждый R2 независимо представляет группу ZYR6;
Z представляет связь или C1-3 алкиленовую или оксоалкиленовую группу, необязательно замещенную группой R7;
Y представляет связь, атом кислорода или группу NR6;
R6 является атомом водорода, группой COOR8, алкильной, алкенильной, циклоалкильной, арильной или аралкильной группой, необязательно замещенной одной или более группами из COOR8,
R7 является гидрокси, необязательно гидроксилированной, необязательно алкоксилированной алкильной или аминоалкильной группой;
R8 является атомом водорода или необязательно гидроксилированной, необязательно алкоксилированной алкильной группой;
М является атомом водорода или одним эквивалентом физиологически приемлемого катиона;
R3 представляет C1-8 алкиленовую группу, 1,2-циклоалкиленовую группу или 1,2-ариленовую группу, необязательно замещенную R7; и каждый R4 независимо представляет водород или С1-3 алкил.
| Устройство для прессования порошков магнитно-твердых материалов | 1980 |
|
SU910360A2 |
| Способ остеосинтеза | 1982 |
|
SU1060174A1 |
| US 6391895 В1, 21.05.2002 | |||
| 0 |
|
SU282173A1 | |

