Область техники, к которой относится изобретение
Настоящее изобретение относится к новому способу синтеза этилового эфира (R,S)-3-[2-[(2-метоксифенокси)метил]-1,3-тиазолидин-3-ил]-3-оксипропионовой кислоты формулы (1)
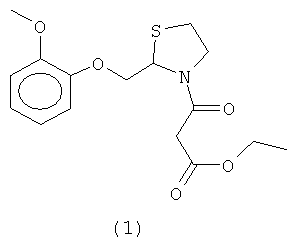
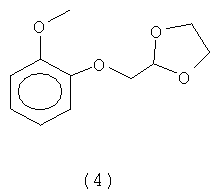
обычно известного под названием могуистеина. Изобретение также относится к новому интермедиату под названием 2-[(2-метоксифенокси)метил]-1,3-диоксолан (4).
Уровень техники
Могуистеин является противокашлевым средством периферического действия, проявившим сильный эффект при лечении сухого кашля, в основном связанного с респираторными нарушениями различной степени серьезности.
Соединения со структурой 3-ацил-2-замещеннго тиазолидина впервые описаны в EP 169581 B1 как средства, обладающие противокашлевой активностью.
Могуистеин был впервые получен и описан как предпочтительное соединение из семейства 3-ацил-2-замещенных тиазолидинов в EP 333080 B1.
Согласно описанию, приведенному в упомянутом патенте, получены N-ацил-замещенные тиазолидины, где ацил представляет собой β-карбонильный остаток, ответственный за их активность, которые являются более активными средствами, чем другие замещенные N-ацил-тиазолидины согласно патенту EP 169581 В1.
Согласно патенту ЕР 333080 В1 могуистеин получают, как описано в примере 1, по способу, включающему реакцию между (R,S)-2-(2-метокси-феноксиметил)-1,3-тиазолидином и этилмалонилхлоридом. Конкретно, вначале добавляют водный раствор KHCO3 к раствору (R,S)-2-(2-метокси-феноксиметил)-1,3-тиазолидина в этилацетате, затем после охлаждения по каплям добавляют раствор этилмалонилхлорида. Требуемое соединение получают путем кристаллизации из маслянистого остатка органической фазы.
Синтез могуистеина с применением этилмалонилхлорида в качестве реагента повторен и дополнительно детализирован теми же авторами EP 333080 B1 в статье под названием "N-Acyl-2-substituted-l,3-thiazolidines, a new class of non-narcotic antitussive agents: studies leading to the discovery of ethyl 2-[(2-methoxyphenoxy)methyl]-β-oxothiazolidine-3-propanoate" (J. Med. Chem 1995, 38, 508-525), в которой могуистеин отмечен как наиболее эффективный и безопасный противокашлевый агент семейства N-ацил-2-замещенных-1,3-тиазолидиновых соединений.
Согласно указанной статье могуистеин получают по способу, который первоначально включает получение (R,S)-2-(2-метокси-феноксиметил)-1,3-тиазолидина с последующей реакцией полученного соединения тиазолидина с этилмалонилхлоридом по способу, сходному с описанным в примере EP 333080 В1. Способ получения (R,S)-2-(2-метокси-феноксиметил)-1,3-тиазолидина, приведенный в статье, проводят в два этапа:
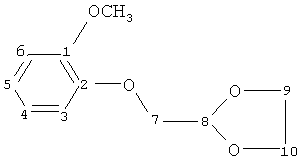
1) образование 2-(2-метоксифенокси)ацетальдегиддиэтилацеталя путем реакции при 150°C гваякол(2-метоксифенола) и 2-бромацетальдегиддиэтилацеталя формулы
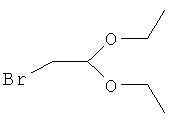
в N-метилпирролидоне и в присутствии K2CO3 с последующей перегонкой и
2) синтез (R,S)-2-(2-метокси-феноксиметил)-1,3-тиазолидина с помощью реакции 2-(2-метоксифенокси)ацетальдегидциэтилацеталя с цистеамином гидрохлоридом в смеси этанол/вода/HCl с последующим добавлением раствора NaOH и перекристаллизацией полученного осадка их смеси этанол/вода.
(R,S)-2-(2-метокси-феноксиметил)-1,3-тиазолидин, полученный по такому способу, далее вступает в реакцию с этилмалонилхлоридом в двухфазной системе из этил ацетата/воды в присутствии бикарбоната калия с получением продукта реакции, который промывают, сушат и обесцвечивают и концентрируют под вакуумом. Могуистеин получают из концентрата, разведенного гексаном, после фильтрации и промывки в виде продукта перекристаллизации из смеси этанол/вода.
Известные способы получения могуистеина включают, однако, реакцию замещенного тиазолидина с этилмалонилхлоридом.
Известно, что этилмалонилхлорид не очень стабильное и довольно дорогое соединение. Его получают путем довольно продолжительного синтеза, который включает получение моноэтилмалоновой кислоты (полученной при щелочном гидролизе диэтилмалонового эфира и последующей ацидификации путем вытеснения соли), которая должна вступать в реакцию с тионил хлоридом в присутствии дихлорметана с образованием этилмалонилхлорида. Дополнительно, полученный продукт нуждается в очистке путем перегонки под вакуумом, которую следует проводить с особой осторожностью, поскольку этилмалонилхлорид нестабилен при нагревании имеет тенденцию к разложению. Качество конечного продукта, т.е. этилмалонилхлорида, вследствие этого никогда не превышает 95%.
Таким образом есть потребность в способе, который не включает применения этилмалонилхлорида и приводит к образованию могуистеина высокой чистоты.
Согласно известному способу (R,S)-2-(2-метокси-феноксиметил)-1,3-тиазолидин получают путем реакции гваякола с бромацетальдегиддиацеталем. Воспроизводя все детали, описанные ранее в данной области техники, авторы настоящего изобретения повторили указанные реакции и обнаружили, что:
- может происходить нежелательный гидролиз с образованием свободного альдегида и этанола, который при реакции с гваяколом может дать в качестве побочного продукта 2-этоксианизол;
- интермедиат 2-(2-метоксифенокси)ацетальдегидциэтилацеталь, полученный в этой реакции, представляет собой черную жидкость, содержащую поликонденсированные соединения, обладающее при высвобождении такой же реакционной способностью, как 2-(2-метоксифенокси)ацетальдегиддиэтилацеталь.
Таким образом очевидно, что применение известного интермедиата, такого как на этапе 2) реакции, приводит к получению тиазолидина и впоследствии могуистеина с высоким содержанием примесей. Дополнительно, примеси высокой молекулярной массы трудно удалять с помощью обычных способов очистки и требуется перегонка под высоким вакуумом (114°C, 0,5 мм Hg).
Объектом изобретения таким образом является способ, приводящий к образованию могуистеина с высоким выходом и высокой чистотой, при этом недорогой и легко приспособляемый для промышленного масштаба производства.
Раскрытие изобретения
Цель изобретения достигнута с помощью способа синтеза могуистеина, включающего следующие стадии:
а) реакцию гваякола (2) с 2-X-метил-1,3-диоксоланом (3), где X представляет собой уходящую группу, для получения 2-[(2-метоксифенокси)метил]-1,3-диоксолана (4):
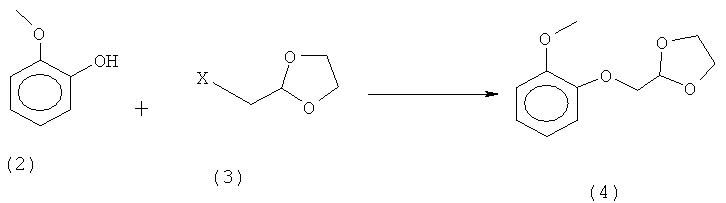
б) реакцию полученного 2-[(2-метоксифеыокси)метил]-1,3-диоксолана (4) с цистеамином (5) в присутствии кислоты с получением (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидина (6):
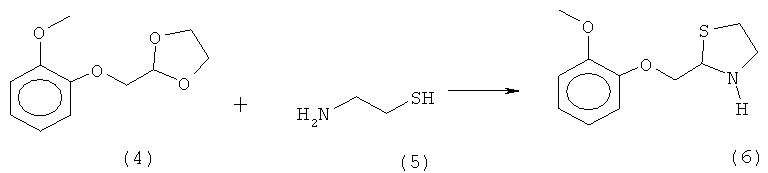
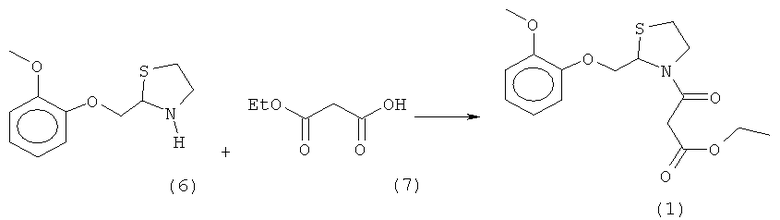
в) реакцию полученного (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидина (6) с моноэтилмалоновой кислотой (7) или ее солью в присутствии конденсирующего агента для получения могуистеина (1):
Способ согласно изобретению позволяет легко отделять с высокой чистотой в конце этапа а) соединение 2-[(2-метоксифенокси)метил]-1,3-даоксолан (4)? которое находится в виде твердого вещества с отличной стабильностью и способностью к кристаллизации. В соответствие с другим аспектом, изобретение также относится к интермедиату 2-[(2-метоксифенокси)метил]-1,3-диоксолану формулы (4):
Согласно настоящему изобретению молекула могуистеина формулы (1)
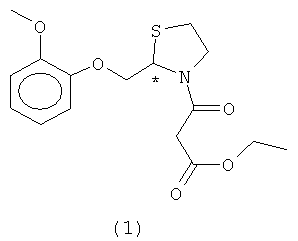
включает по определению любую стереохимическую конфигурацию относительно хирального центра в самой формуле, включая рацемическую смесь и энантиомеры, полученные по способам разделения, известным специалисту в данной области техники.
Краткое описание чертежей
Фигура 1 представляет собой схему для сравнения известного способа получения могуистеина, включающего применение соединений бромацетальдегиддиэтилацеталя и этилмалонилхлорида с предпочтительным способом его получения согласно изобретению.
Осуществление изобретения
Способ получения могуистеина согласно изобретению, по существу, включает три стадии:
а) реакцию гваякола (2) с 2-Х-метил-1,3-диоксоланом (3), где X представляет собой уходящую группу, для получения 2-[(2-метоксифенокси)метил]-1,3-диоксолана (4):
б) реакцию полученного 2-[(2-метоксифенокси)метил]-1,3-диоксолана (4) с цистеамином (5) в присутствии кислоты с получением (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидина (6):
в) реакцию полученного (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидина (6) с моноэтилмалоновой кислотой (7) или ее солью в присутствии конденсирующего агента для получения могуистеина (1).
Стадия а) способа согласно настоящему изобретению предпочтительно проходит в растворителях с высокой точкой кипения. Более предпочтительно, он проходит в растворителях, выбранных из группы, состоящей из 1-метокси-2-пропанола, N-метилпирролидона, диметилформамида, диметилацетамида, диглима (бис(2-метоксиэтил)эфира), этилцеллозольва, метилцеллозольва, этиленгликоля; еще более предпочтительно растворителем для проведения реакции является 1-метокси-2-пропанол.
На стадии а) применяют 2-Х-метил-1,3-диоксолан (3), где X представляет собой уходящую группу. Предпочтительно, уходящую группу выбирают из галогена, мезилата, тозилата, трифлата, где более предпочтителен галоген, который выбирают из группы, состоящей из хлора, брома и еще более предпочтительно - это бром.
Преимущественно, применением циклического диацеталя, 2-Х-метил-1,3-диоксолана (3) (более стабильного и труднее поддающегося гидролизу по сравнению с линейными диацеталями, такими как диметил и диэтил) сводит к минимуму присутствие конденсированных побочных продуктов, обусловленных образованием свободного альдегида.
Согласно изобретению в реакционной смеси преимущественно присутствует неорганическое основание в форме тонкого порошка, предпочтительно, K2CO3.
Молярное соотношение гваякола (2) и 2-Х-метил-1,3-диоксолана (3) предпочтительно находится в интервале от 1:1 до 1:1,5, более предпочтительно около 1:1,18. Стадию а) реакции, приводящую к формированию интермедиата (4), можно проводить при температуре между 100° и 140°C. Под конец стадии а) путем кристаллизации получают интермедиат (4) в виде кристаллического твердого вещества.
В одном осуществлении изобретения по техническим и экономическим соображениям благодаря превосходной стабильности и способности к кристаллизации получаемого интермедиата (4), предпочтительно проводить стадию а) в 1-метокси-2-пропаноле при температуре от 120°C до 129°C при молярном соотношении гваякола (2) к 2-Х-метил-1,3-диоксолану (3) примерно 1:1,18.
Стадия а) способа согласно изобретению таким образом позволяет получить новый интермедиат формулы 2-[(2-метоксифенокси)метил]-1,3-диоксолан (4), обладающий отличной стабильностью и способностью к кристаллизации. В этой связи получаемый циклический интермедиат появляется в конце реакции в виде практически белого твердого вещества с чистотой более 99%, который можно непосредственно применять на последующей стадии б). Преимущественно, для применения на последующей стадии для получения тиазолидина, этот интермедиат не требует длительной и дорогостоящей очистки, такой как перегонка под высоким вакуумом, которая необходима в случае интермедиата 2-(2-метоксифенокси)ацетальдегиддиэтилацеталя, получаемого на стадии 1) по способу, известному раннее в данной области техники. Дополнительно, этот интермедиат получается:
- с выходом более 80% относительно теоретического и в предпочтительном осуществлении с выходом более 85%;
- с чистотой более 99%.
Согласно изобретению стадия б) включает реакцию между интермедиатом 2-[(2-метоксифенокси)метил]-1,3-диоксоланом (4), полученным на стадии а), и цистеамином (5) в присутствии кислоты с получением получения (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидина (6).
Цистеамин (5) находится предпочтительно в виде соли, более предпочтительно в виде гидрохлорида или сульфата и еще более предпочтительно в виде гидрохлорида.
Молярное соотношение интермедиата (4) и цистеамина (5) предпочтительно находится в интервале от 1:1 до 1: 1,5, более предпочтительно оно составляет примерно 1:1,18.
Кислота в реакционной смеси предпочтительно представляет собой концентрированную соляную кислоту. Реакцию на стадии б) в предпочтительном осуществлении проводят преимущественно в присутствии изопропанола и деионизованной воды, но можно также применять другие растворители, такие как метанол и вода или этанол и вода.
Как только реакция на стадии б) завершается, тиазолидин получают путем кристаллизации. Неочищенный (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидин (6) на стадии б) получают с выходом более 80%, предпочтительно более 90% относительно теоретического значения и с чистотой выше 99%.
Стадия в) при получении могуистеина (1) включает реакцию между (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидином (6) и моноэтилмалоновой кислотой (7) или ее солью в присутствии конденсирующего агента.
Соль моноэтилмалоновой кислоты предпочтительно представляет собой соль щелочного металла, более предпочтительно это моноэтилмалонат калия.
Согласно изобретению моноэтилмалоновую кислоту можно добавлять как таковую или необходимым способом высвобождать из ее солей in situ.
Ряд солей моноэтилмалоновой кислоты высокой чистоты можно купить. Например, моноэтилмалонат калия доступен в продаже с чистотой выше 99% и с низким содержанием двухзамещенного моноэтилмалоната калия. Соли моноэтилмалоновой кислоты можно преимущественно получать путем гидролиза с применением необходимого диэтилмалонатного основания. Например, моноэтилмалонат калия можно преимущественно получать путем простого гидролиза диэтилмалоната с помощью раствора КОН в этаноле.
Реакция между моноэтилмалоновой кислотой (7) и тиазолидином (6) происходит в присутствии конденсирующего агента. Конденсирующий агент согласно изобретению может быть N,N′-дициклогексилкарбодиимидом или N,N′-диизопропилкарбодиимидом. Предпочтительно, это N,N′-дициклогексилкарбодиимид.
Для ускорения реакции преимущественно добавляют катализатор, такой как 1-гидроксибензотриазол гидрат или N-гидроксисукциниимид.
Молярное соотношение тиазолидина (6) и моноэтилмалоновой кислоты предпочтительно находится в интервале от 1:1 до 1:1,5 и более предпочтительно составляет примерно 1:1,10.
В предпочтительном осуществлении изобретения реакция на стадии в) происходит между солью моноэтилмалоновой кислоты (7) и тиазолидином (6) в присутствии кислоты, например, концентрированной соляной кислоты и N,N′-дициклогексилкарбодиимида в качестве конденсирующего агента.
Могуистеин (1) получают с выходом более 90% относительно теоретической величины и с чистотой выше 99%.
В наиболее предпочтительном осуществлении изобретения реакция на стадии в) происходит между тиазолидином (6) и моноэтилмалонатом калия (7) в присутствии концентрированной соляной кислоты, N,N′-дициклогексилкарбодиимида и этилацетата и приводит к образованию могуистеина, хлористого калия и 1,3-дициклогексилмочевины. После отделения нежелательных продуктов путем фильтрации получают могуистеин с высоким выходом и с чистотой выше 99,5%.
Таким образом, по способу согласно настоящему изобретению могуистеин получают с высоким выходом, высокой чистотой и с небольшими затратами, избегая недостатков ранее известного в данной области техники способа. Конкретно, как видно на приложенной фигуре 1, на которой слева показаны реагенты, применяемые по способу, известному ранее в данной области техники, и справа реагенты согласно предпочтительному осуществлению способа настоящего изобретения, получение нового циклического интермедиата (4) и применение моноэтилмалоновой кислоты (7) или одной из ее солей, например, калиевой соли, вместо этилмалонилхлорида, позволяет получать продукт могуистеин с чистотой выше 99%.
Дополнительно, обнаружено, что способ согласно изобретению является более экономически выгодным с одной стороны из-за более низкой стоимости сырья и с другой стороны, поскольку он не требует перегонки при критических условиях для удаления нежелательных побочных продуктов.
Изобретение далее описано в качестве отдельных осуществлений, приведенных в виде примеров, не ограничивающих область притязаний данного изобретения.
ПРИМЕРЫ
Пример 1
Стадия а) Получение интермедиата (4) формулы 2-[(2-метоксифенокси)метил]-1,3-диоксолан (4) в 1-метокси-2-пропаноле
Исходные материалы применяют в количествах, приведенных в нижеследующей таблице 1.
В атмосфере азота загружают тонкий порошок карбоната калия и 1-метокси-2-пропанол в совершенно сухой сосуд на 6 литров и перемешивают. Когда тонкой струей добавляют гваякол, температура спонтанно поднимается с 24°C до 42°C из-за экзотермической салификации и смесь значительно загустевает из-за выпадения в осадок соответствующей калийной соли. Массу затем нагревают до кипения (T=121-123°C) и по достижении этой температуры по каплям добавляют 2-бромметил-1,3-диоксолан в течение 1 часа и 30 мин в системе с обратных холодильником. Во время добавления присутствие реагента приводит к подъему температуры кипения смеси до T=127-129°C. Смесь держат с обратным холодильником в общей сложности в течение примерно 24 час от начала добавления бромдиоксолана путем прикапывания. После потребления реагента замечено, что температура кипения стремится упасть еще раз, возвращаясь к исходному значению T=121-123°C.
Смесь охлаждают до T=20±5°C и отбирают образец для контроля завершения реакции.
После подтверждения завершения реакции к смеси добавляют 50% раствор КОН. Поскольку температура смеси спонтанно поднимается примерно на 10°C, ее охлаждают до T=20±5°C и добавляют первую порцию деионизованой воды. Затем ожидают выпадения в осадок продукта, при необходимости добавляя затравку.
Для завершения осаждения далее добавляют вторую порцию деионизованной воды. Под конец добавления получается темная серо-зеленая текучая суспензия, которую выдерживают при T=20±5°C в течение по меньшей мере 2 час, затем фильтруют и тщательно промывают деионизованной водой до достижения pH промывной жидкости значения 7-8.
Получают 481,0 г сырого продукта цвета светлого лесного ореха, что соответствует 425,0 г сухого продукта.
Теоретический выход 504,54 г.
Выход в процентах относительно теоретического - 84,23%.
Далее проводят анализ полученного интермедиата (4) и получают следующие результаты:
Чистота согласно жидкостной хроматографии высокого разрешения (ВЭЖХ) 99,81%.
Чистота согласно газовой хроматографии (ГХ) 99,85%
Проводят также следующие анализы ля характеристики продукта.
Получены следующие результаты:
Точка плавления 48°C.
Элементарный анализ:
Обнаружено: C=62,83%; H=6,76%
Теоретическое значение: C=62,85%; H=6,71.
Масс-спектрометрический анализ:
CI+/MS 211 [MH+], 210 [(MH+)-H]+
ЯМР-спектроскопический анализ:
13C NMR-APT
56.3 (OMe), 65.5 (CH2), 70.5 (CH2), 102.4 (С8-CH), 112.4 (CH), 121.1 (СН), 122.2 (CH), 148.6 (C), 150.1 (C)
1H-NMR
3.84 (3H, s, OMe), 3.9-4.1 (4H, m, H-9/H-10), 4.07 (2Н, d, J=4Hz, H-7), 5.33 (1Н, t, J=4 Hz, H-8), 6.7-7.0 (4H, m).
Анализы подтверждают структуру интермедиата (4)
Пример 2
Стадия а) получения интермедиата (4) формулы 2-[(2-метоксифенокси)метил]-1,3-диоксолан (4) в N-метилпирролидоне
Следующие исходные материалы применяют в количествах, приведенных в таблице 2.
В атмосфере азота помещают тонкий порошок карбоната калия, гваякол и N-метилпирролидон в колбу на 3 литра при комнатной температуре. Смесь перемешивают и доводят температуру до 135±5°C; после достижения этой температуры по каплям добавляют 2-бромметил-1,3-диоксолан в течение по меньшей мере 2 час, поддерживая температуру в данном интервале. Под конец добавления реакционную смесь оставляют при температуре 135±5°C в течение еще на 8 час.(Общее время добавления 2-бромметил-1,3-диоксолана и время протекания реакции составляет примерно 10 час).
Смесь затем охлаждают до температуры примерно 25±5°C и отбирают образец для контроля завершения реакции.
После подтверждения завершения реакции добавляют 1400 мл воды для гашения и затем к смеси добавляют толуол для экстракции и нагревают до температуры Т=45±5°C. Перемешивание останавливают и оставляют смесь для разделения фаз примерно на 1 час.
Нижнюю водную фазу черного цвета отбрасывают и органическую фазу промывают 3 раза смесью вода/NaOH, поддерживая температуру на уровне 45±5°C и оставляя для разделения примерно на 30 мин. Органический экстракт бледно-янтарного цвета охлаждают до T=25±5°C и обрабатывают углем. Затем экстракт фильтруют на дикалитовой панели (фильтре) для получения абсолютно прозрачного фильтрата цвета желтой охры; панель промывают толуолом и объединяют промывной раствор с основным фильтратом.
Раствор в толуоле переносят в чистую колбу на 2 л, оборудованную системой для перегонки. Дистиллят концентрируют под вакуумом при температуре бани примерно 60±5°C до образования осадка.
Полученный интермедиат (4) образуется в виде плотной жидкости янтарного цвета, которая затвердевает при охлаждении.
Получают 252,8 г темно-желтого концентрата, из которого получается 153 г кристаллического продукта с чистотой 98%.
Теоретический выход 168,18 г.
Выход в процентах относительно теоретического значения 91,15%.
Примеры 3-31
Стадия а) Получение интермедиата (4) формулы 2-[(2-метоксифенокси)метил]-1,3-диоксолан (4)
По способу, описанному в примере 1, но с применением количеств, реакционных растворителей, продолжительности и температуры, указанных в таблице 3, получают интермедиат (4) с выходом и чистотой, указанными в нижеследующей таблице 4.
Таким образом, во всех примерах интермедиат (4) получают с высоким выходом и с высокой чистотой более 99%. Согласно настоящему изобретению, интермедиаты из примеров 1, 28, 29 и 31 обладают наилучшим видом и качеством.
Пример 32
Стадия б) Получение (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидина (6) Исходные материалы применяют в количествах, приведенных в таблице 5 ниже.
В атмосфере азота 2-[(2-метоксифенокси)метил]-1,3-диоксолан, как получен по примеру 2, цистеамина гидрохлорид, изопропанол, деионизованную воду и концентрированную соляную кислоту помещают в колбу на 1 л. Затем смесь перемешивают и нагревают и полученный раствор (обычно желтого цвета) доводят до температуры кипения (Ti=79°-80°C). После достижения температуры кипения раствор держат с обратным холодильником в течение по меньшей мере 8 час. Затем смесь охлаждают до температуры 25±5°C и проводят отбор образцов для определения полноты протекания реакции. Одновременно в колбу на 2 л помещают деионизованную воду для осаждения и 30% раствор NaOH и раствор охлаждают до температуры 0±5°C.
После подтверждения завершения реакции в колбу для осаждения в течение 10 мин переносят раствор гидрохлорида. По ходу добавления температура спонтанно поднимается до 13-15°C и смесь опять доводят до T=0±5°C.
Вначале продукт отделяется в виде клеевидной массы, которая в дальнейшем имеет тенденцию к хорошему кристаллообразованию. Для ускорения кристаллизации тиазолидина применяют затравку (тиазолидин характеризуется низкой точкой плавления, как и могуистеин).
Когда продукт хорошо закристаллизовался, смесь опять доводят до T=0±5°C и держат суспензию в таких условиях в течение по меньшей мере 8 час перед фильтрованием.
Полученный неочищенный продукт затем отфильтровывают и тщательно промывают деионизованной водой.
Получают 189,25 г продукта белого цвета с желтоватым оттенком, который высушивают в печи под вакуумом при T=35±5°C в течение примерно 24 час.
Выход составляет 139,0 г сухого продукта с едва заметным желтым оттенком.
Теоретический выход - 163,75 г.
Процент от теоретического выхода - 84,88%.
(R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидин (6) далее анализируют с получением следующих результатов.
Чистота по ВЭЖХ - 99,791%.
Титр HClO4 - 99,34%.
Примеры 33-50
Стадия б) Получение (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидина (6) По тому же способу и с применением тех же реагентов, как указано в примере 31, но с применением разных количеств интермедиата (4), полученного в примерах 1, 3-30, получают (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидин (6) с выходом и чистотой, как указано в таблице 6 ниже.
Применение на стадии б) согласно изобретению нового циклического интермедиата (4) позволяет получать тиазолидин (6) с выходом более 80% и с чистотой выше 99%.
Пример 51
Стадия в) Получение могуистеина
Исходные материалы применяют в количествах, приведенных в таблице 7 ниже.
В атмосфере азота тиазолидин из примера 33, моноэтилмалоновую кислоту (бесцветная жидкость) и первую порцию этилацетата помещают в однолитровую колбу при комнатной температуре. Смесь перемешивают и охлаждают до температуры 0±5°C. Одновременно во втором контейнере готовят раствор DCC (N,N′-дициклогексилкарбодиимида) в этилацетате, защищая его от попадания влаги. После достижения температуры 0±5°C по каплям добавляют раствор DCC (N,N′-дициклогексилкарбодиимида) в течение по меньшей мере 4 час. По окончании добавления смесь оставляют реагировать при Т=0±5°C в течение примерно 2 час. После проверки завершения реакции с помощью тонкослойной хроматографии добавляют 50% уксусную кислоту и раствор перемешивают в течение ночи при T=0±5°C. На следующее утро DCC (N,N′-дициклогексилкарбодиимид) отделяют фильтрованием, тщательно промывают этилацетатом и промывную жидкость объединяют с основным фильтратом.
Органическую фазу при перемешивании обрабатывают 20% раствором уксусной кислоты и нагревают до 30-35±5°C для ускорения разделения. После достижения этой температуры перемешивание прекращают и раствор оставляют для разделения фаз примерно на один час. Кислотную водную фазу (нижнюю) удаляют, органическую фазу обесцвечивают с помощью угля и фильтруют через декалит с получением совершенно прозрачного раствора соломенно-желтого цвета.
Фильтрат концентрируют под вакуумом на бане с температурой 50±5°C до получения остатка; остаток растворяют в 380 мл ацетона при комнатной температуре и переносят в колбу на 1 л.
Раствор могуистеина в ацетоне охлаждают до 0±5°C и как только эта температура достигнута, добавляют 380 мл воды для осаждения в течение одного часа; под конец добавления в раствор вносят затравку могуистеина. Через 13-30 мин продукт, который вначале отделяется в виде маслянистой клеевидной массы на стенках колбы, хорошо кристаллизуется. Как только продукт закристаллизовался, добавляют оставшиеся 80 мл воды.
Суспензию далее выдерживают при 0±5°C в течение по меньшей мере 8 час и оставляют в лаборатории на ночь при перемешивании.
Затем продукт отфильтровывают и тщательно промывают деионизованной водой.
Получают 81,30 г практически белого сырого продукта.
Продукт сушат в печи при 35±5°C до достижения величины KF<0,1%.
Получают 68,65 г практически белого сухого продукта.
Теоретический выход - 75,32 г.
Процент относительно теоретического выхода - 91,14%.
Чистота по ВЭЖХ - 99,76%.
Пример 52
Стадия в) Получение могуистеина
Исходные материалы применяют в количествах, приведенных в таблице 8 ниже.
В атмосфере азота (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидин из примера 33, однозамещенную калиевую соль, 1-гидроксибензоаттриазол гидрат (катализатор) и первую порцию растворителя ацетона помещают в однолитровую колбу при комнатной температуре. Суспензию охлаждают до температуры 0±5°C и по каплям добавляют концентрированную соляную кислоту в течение примерно 1 часа (на этом этапе наблюдается экзотермичность в течение всего добавления, температура спонтанно поднимается до 18-20°C). Одновременно во втором контейнере готовят раствор DCC (N,N′-дициклогексилкарбодиимида) в ацетоне, защищая его от попадания влаги. Когда белая суспензия достигает температуры 0±5°C, по каплям добавляют раствор DCC (N,N′-дициклогексилкарбодиимида) в течение по меньшей мере 3 час. По окончании добавления суспензию оставляют при T=0±5°C примерно на 2 часа. После проверки завершения реакции с помощью тонкослойной хроматографии (ТСХ) DCU (N,N′-дициклогексилмочевину) отделяют фильтрованием, тщательно промывают ацетоном и промывную жидкость объединяют с основным фильтратом.
Раствор концентрируют под вакуумом на бане с температурой 50±5°C до конечного объема примерно 300 мл и оставляют на ночь при комнатной температуре.
Полученный темно-желтый раствор обрабатывают 5,0 г угля в течение 10 мин. Затем раствор фильтруют через декалитовую панель с получением совершенно прозрачного желтого раствора, который переносят в колбу на 1 л. Фильтр промывают 80 мл ацетона и промывной раствор объединяют с основным фильтратом (300 мл + 80 мл = примерно 380 мл всего).
Раствор могуистеина в ацетоне охлаждают до 0±5°C и в течение часа добавляют 380 мл воды для осаждения; под конец добавления вносят затравку могуистеина. Через 15-30 мин продукт, который вначале отделяется в виде маслянистой клеевидной массы на стенках колбы, успешно кристаллизуется и гранулируется. Как только продукт закристаллизовался, добавляют оставшиеся 76 мл воды (380 мл = 76 мл = 456 мл; 380 мл ацетона/456 мл воды дают соотношение ацетон/вода = 1,0:1,2).
Суспензию далее выдерживают при 0±5°C в течение по меньшей мере 8 час и оставляют в лаборатории на ночь при перемешивании.
Затем продукт отфильтровывают и тщательно промывают деионизованной водой.
Получают 94,4 г практически белого продукта.
Полученный продукт сушат в печи при 35±5°C.
Получают 69,5 г белого сухого порошка с желтоватым оттенком.
Теоретический выход - 75,32 г.
Процент относительно теоретического выхода - 92,27%.
Чистота по ВЭЖХ - 99,81%.
Тест повторяют еще 4 раза с получением следующих значений чистоты, соответственно: 99,83%, 99,73%, 99,82%, 99,81%.
Чистота могуистеина, полученного по способу согласно изобретению, так высока, что дальнейшая стадия очистки не требуется.
Пример 53
Стадия в) Получение могуистеина
Исходные материалы применяют в количествах, приведенных в таблице 9 ниже.
В атмосфере азота тиазолидин из примера 33, однозамещенную калиевую соль и первую порцию этилацетата помещают в однолитровую колбу при комнатной температуре. Смесь перемешивают и охлаждают до температуры 0±5°C. По достижении указанной температуры по каплям добавляют концентрированную соляную кислоту в течение примерно 1 часа (на этом этапе наблюдается экзотермичность в течение всего добавления, температура спонтанно поднимается до 18-20°C). Одновременно во втором контейнере готовят раствор DCC (N,N′-дициклогексилкарбодиимида) в этилацетате, защищая его от попадания влаги. Когда белая суспензия достигает температуры 0±5°C, по каплям добавляют раствор DCC N,N′-дициклогексилкарбодиимида) в течение по меньшей мере 4 час. По окончании добавления смесь оставляют реагировать при Т=0±5°C в течение примерно 2 час. После проверки завершения реакции с помощью тонкослойной хроматографии добавляют 50% уксусную кислоту и смесь оставляют при перемешивании в течение ночи при T=0±5°C. На следующее утро DCU (N,N′-дициклогексилмочевину) отделяют фильтрованием, тщательно промывают этилацетатом и промывную жидкость объединяют с основным фильтратом.
Органическую фазу при перемешивании обрабатывают 20% раствором уксусной кислоты и нагревают до 30-35±5°C для ускорения разделения. После достижения этой температуры перемешивание прекращают и раствор оставляют для разделения фаз примерно на один час. Кислотную водную фазу (нижнюю) удаляют, органическую фазу обесцвечивают с помощью угля и фильтруют через декалит с получением совершенно прозрачного раствора соломенно-желтого цвета.
Фильтрат концентрируют под вакуумом на бане с температурой 50±5°C до получения остатка; остаток растворяют в 380 мл ацетона при комнатной температуре и переносят в колбу на 1 л.
Раствор могуистеина в ацетоне охлаждают до 0±5°C и как только эта температура достигнута, добавляют 380 мл воды для осаждения в течение одного часа; под конец добавления в раствор вносят затравку могуистеина. Через 13-30 мин продукт, который вначале отделяется в виде маслянистой клеевидной массы на стенках колбы, хорошо кристаллизуется и гранулируется. Как только продукт закристаллизовался, добавляют оставшиеся 80 мл воды.
Суспензию далее выдерживают при 0±5°C в течение по меньшей мере 8 час и оставляют в лаборатории на ночь при перемешивании.
Затем продукт отфильтровывают и тщательно промывают деионизованной водой.
Получают 79,49 г практически белого сырого продукта.
Продукт сушат в печи при 35±5°C до достижения величины KF<0,1%.
Получают 67,83 г практически белого сухого продукта.
Теоретический выход - 75,32 г.
Процент относительно теоретического выхода - 90,05%.
Чистота по ВЭЖХ - 99,88%.
Изобретение относится к способу синтеза могуистеина, представляющего собой этиловый эфир (R,S)-3-[2-[(2-метоксифенокси) метил]-1,3-тиазолидин-3-ил]-3-оксипропионовой кислоты, включающему а) реакцию гваякола с 2-Х-метил-1,3-диоксоланом с получением 2-[(2-метоксифенокси)метил]-1,3-диоксолана, б) взаимодействие с цистеамином в присутствии кислоты с получением (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидина, в) взаимодействие с моноэтилмалоновой кислотой или ее солью в присутствии конденсирующего агента с получением могуистеина. Могуистеин согласно изобретению получают с высоким выходом и чистотой. 17 з.п. ф-лы, 9 табл., 53 пр., и ил.
1. Способ синтеза могуистеина, который представляет собой этиловый эфир (R,S)-3-[2-[(2-метоксифенокси)метил]-1,3- тиазолидин-3-ил]-3-оксипропионовой кислоты, включающий следующие стадии:
а) реакцию гваякола (2) с 2-Х-метил-1,3-диоксоланом (3), где X представляет собой уходящую группу, для получения 2-[(2-метоксифенокси)метил]-1,3-диоксолана (4):
б) реакцию 2-[(2-метоксифенокси)метил]-1,3-диоксолана (4), полученного на стадии а), с цистеамином (5) в присутствии кислоты с получением (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидина (6):
в) реакцию (R,S)-2-[(2-метоксифенокси)метил]-1,3-тиазолидина (6) с моноэтилмалоновой кислотой (7) или ее солью в присутствии конденсирующего агента с получением могуистеина (1):
2. Способ по п.1, отличающийся тем, что уходящую группу X в формуле 2-Х-метил-1,3-диоксолана (3) на стадии а) выбирают из группы, состоящей из галогена, мезилата, тозилата и трифлата.
3. Способ по п.2, отличающийся тем, что галоген выбирают из группы, состоящей из брома и хлора, где бром предпочтителен.
4. Способ по п.1, отличающийся тем, что стадию а) проводят в растворителе, выбранном из группы, состоящей из 1-метокси-2-пропанола, N-метилпирролидона, диметилформамида, диметилацетамида, диглима (бис(2-метоксиэтил)эфира), этилцеллозольва, метилцеллозольва, этиленгликоля.
5. Способ по п.4, отличающийся тем, что растворитель представляет собой 1-метокси-2-пропанол.
6. Способ по п.1, отличающийся тем, что молярное соотношение гваякола (2) и 2-Х-метил-1,3-диоксолана (3) на стадии а) предпочтительно находится от 1:1 до 1:1,5, предпочтительно 1:1,18.
7. Способ по п.1, отличающийся тем, что реакция на стадии а) происходит при температуре от 100 до 140°C.
8. Способ по п.1, отличающийся тем, что на стадии а) K2СО3 в виде тонкого порошка также добавляют к реакционной смеси.
9. Способ по п.1, отличающийся тем, что стадию а) проводят в 1-метокси-2-пропаноле при температуре от 120 до 129°C при молярном соотношении гваякола (2) и 2-Х-метил-1,3-диоксолана (3) примерно 1:1,18.
10. Способ по п.1, отличающийся тем, что на стадии б) молярное соотношение интермедиата (4) и цистеамина (5) находится от 1:1 до 1:1,5, предпочтительно примерно 1:1,18.
11. Способ по п.1, отличающийся тем, что цистеамин (5) находится в виде соли.
12. Способ по п.11, отличающийся тем, что цистеамин присутствует в виде соли, представляющей собой гидрохлорид.
13. Способ по п.1, отличающийся тем, что кислота, присутствующая в реакционной смеси на стадии б), представляет собой концентрированную соляную кислоту.
14. Способ по п.1, отличающийся тем, что на стадии б) реакцию проводят в присутствии изопропанола и деионизованной воды.
15. Способ по п.1, отличающийся тем, что конденсирующий агент на стадии в) представляет собой N,N′-дициклогексилкарбодиимид.
16. Способ по п.1, отличающийся тем, что молярное соотношение тиазолидина (6) и моноэтилмалоновой кислоты (7) или ее соли на стадии в) находится от 1:1 до 1:1,5, предпочтительно 1:1,10.
17. Способ по п.1, отличающийся тем, что соль моноэтилмалоной кислоты (7) представляет собой моноэтилмалонат калия.
18. Способ по п.1, отличающийся тем, что реакция на стадии в) происходит между моноэтилмалонатом калия (7) и тиазолидином (6) в присутствии концентрированной соляной кислоты и N,N′-дициклогексилкарбодиимида в этилацетате.
| Corominos J | |||
| Pares; Bulto I.; Pfres R.; Villarroya Т.; Marty M.; Langer M | |||
| Structure of mephenoxalone// Chimie et Industrie | |||
| Приспособление к комнатным печам для постепенного сгорания топлива | 1925 |
|
SU1963A1 |
| ТОКОПРИЕМНИК ЭЛЕКТРОПОДВИЖНОГО СОСТАВА | 0 |
|
SU333080A1 |
| УСТРОЙСТВО для ИЗМЕРЕНИЯ ДОБРОТНОСТИ ПЬЕЗОЭЛЕКТРИЧЕСКИХ РЕЗОНАТОРОВ | 0 |
|
SU169581A1 |
| 1,3-ДИОКСОЛАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2276149C2 |

