ПРИОРИТЕТ
Настоящая заявка испрашивает приоритет на основании IN2544/MUM/2012, поданной 31 августа 2012 г., и IN678/MUM/2013, поданной 06 марта 2013 г., содержание которых полностью включено в настоящую заявку посредством ссылки.
Область техники
Настоящее изобретение относится к способу получения тенелиглиптина, 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1H-пиразол-5-ил)-1-пиперазинил]-2-пирролидинилкарбонил}-1,3-тиазолидина и его фармацевтически приемлемых солей. Новый способ направлен на улучшение в получении тенелиглиптина, которое подходит для применения в промышленности и обеспечивает простое и экономически целесообразное получение тенелиглиптина и его солей, которые имеют более высокие чистоту и выход.
Уровень техники
Тенелиглиптин, химическое название 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1H-пиразол-5-ил)-1-пиперазинил]-2-пирролидинилкарбонил}-1,3-тиазолидин, структурно представляет собой соединение формулы (I):
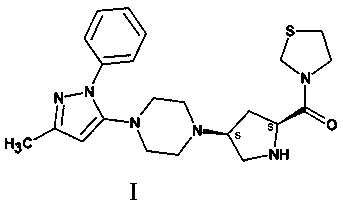
Патент США №7074794 (US'794) описывает способ получения тригидрохлоридной соли соединения I. В способе, описанном в (US'794), для синтеза трет-бутил-4-ацетоацетилпиперазин-1-карбоксилата, который является одним из ключевых промежуточных соединений в синтезе соединения I из 1-трет-бутилоксикарбонилпиперазина, применяют дикетен. Существует несколько известных недостатков указанного способа, которые среди прочего включают нестабильность дикетена, что затрудняет его реализацию. К сожалению, для дикетеновой реакции известно немного альтернатив.
Таким образом, задача настоящего изобретения заключается в создании способа для преодоления вышеуказанных проблем, а также в создании простых, экономически целесообразных и подходящих для применения в промышленности способов получения тенелиглиптина и его фармацевтически приемлемой соли или сольвата. Тенелиглиптин и его фармацевтически приемлемая соль или сольват, полученный способом согласно настоящему изобретению, имеет более высокие выход и чистоту.
Краткое описание изобретения
В одном варианте реализации согласно настоящему изобретению предложен способ получения тенелиглиптина, соединения формулы I, или его соли или гидрата, включающий:

а) осуществление взаимодействия соединения формулы 11 с бис-(2-хлорэтил)амином или его N-защищенным производным или солью,

или
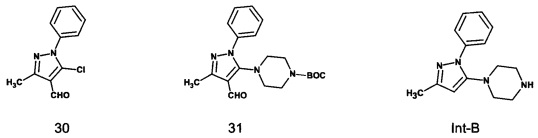
а) осуществление взаимодействия 5-хлор-3-метил--фенил-1H-пиразол-4-карбальдегида, соединения формулы 30, с пиперазином или его N-защищенным производным,

с получением соединения формулы Int-B или его N-защищенного производного или соли;
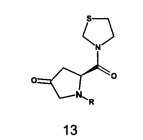
b) осуществление взаимодействия соединения формулы Int-B или его N-защищенного производного или соли с соединением формулы 13 с получением соединения формулы 14,

где R представляет собой аминозащитную группу, выбранную из группы, состоящей из аралкила, ацила, низшего алкоксикарбонила, аралкилоксикарбонила, низшего алкансульфонила, арилсульфонила, три-(низший алкил)силила, трифосгена; и
c) снятие защиты с соединения формулы 14 с получением тенелиглиптина, соединения формулы I, или его соли или гидрата.
В одном варианте реализации согласно настоящему изобретению предложен аморфный тенелиглиптин.
В одном варианте реализации согласно настоящему изобретению предложен способ получения тенелиглиптина 2,5 гидробромида или его гидрата, включающий кристаллизацию тенелиглиптина 2,5 гидробромида или его гидрата из растворителя, выбранного из метанола, н-бутанола, трет-бутанола, N,N-диметилацетамида, N,N-диметилформамида, тетрагидрофурана, тетрагидропирана, 1,4-диоксана, пропилацетата, изопропилацетата и метилэтилкетона, метилизобутилкетона и их смесей.
В одном варианте реализации согласно настоящему изобретению предложен по существу чистый тенелиглиптина 2,5 гидробромида гидрат, имеющий чистоту по меньшей мере 99% по данным высокоэффективной жидкостной хроматографии.
В одном варианте реализации согласно настоящему изобретению предложено соединение, выбранное из следующих;


В одном варианте реализации согласно настоящему изобретению предложен тенелиглиптина 2,5 гидробромида гидрат, который содержит менее чем 0,1% любого из нижеприведенных соединений по данным высокоэффективной жидкостной хроматографии.

В одном варианте реализации согласно настоящему изобретению предложено применение бис-(2-хлорэтил)-амина или N-boc-бис-(2-хлорэтил)-амина или его соли для получения соединения формулы Int-B или тенелиглиптина.
В одном варианте реализации согласно настоящему изобретению предложено применение 5-хлор-3-метил-1-фенил-1H-пиразол-4-карбальдегида или трет-бутил-4-(4-формил-3-метил-1-фенил-1H-пиразол-5-ил)-пиперазин-1-карбоксилата для получения соединения формулы Int-B или тенелиглиптина.
Краткое описание чертежей
Фиг. 1: Рентгенограмма порошковой рентгеновской дифракции (PXRD) тенелиглиптина 2,5 гидробромида гидрата, который по существу охарактеризован в Примере 6.
Фиг. 2: ИК-спектр тенелиглиптина 2,5 гидробромида гидрата, который по существу охарактеризован в Примере 6.
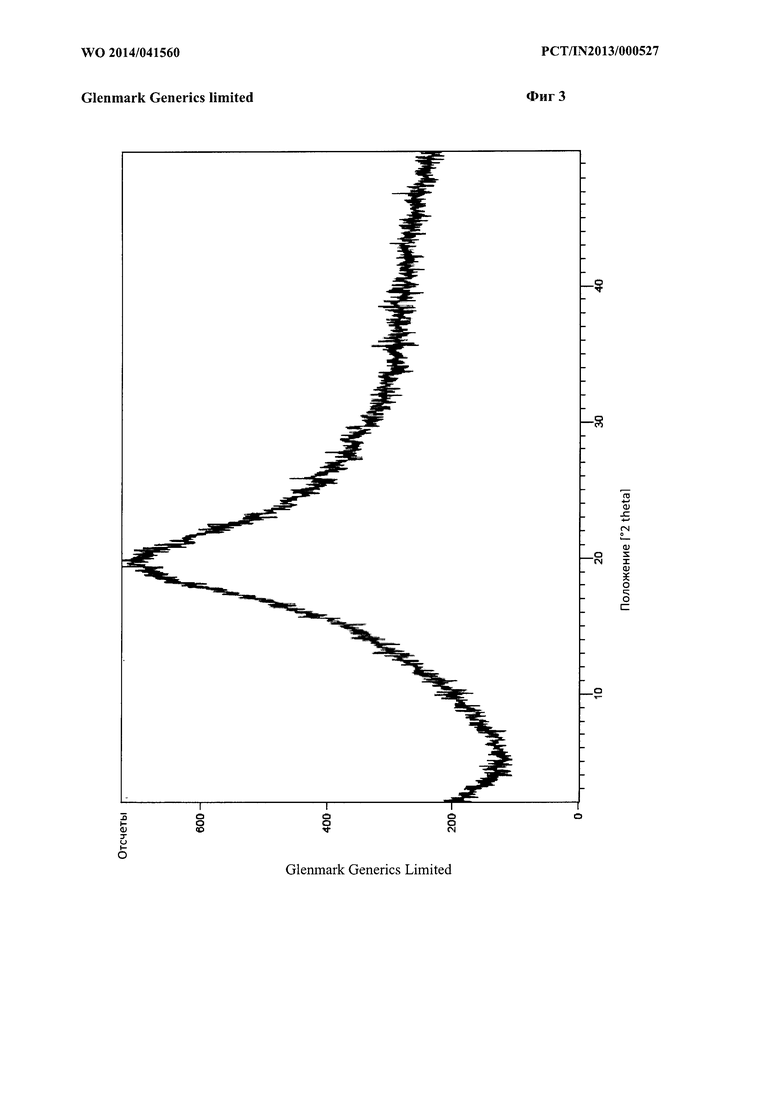
Фиг. 3: Рентгенограмма порошковой рентгеновской дифракции (PXRD) тенелиглиптина, который по существу охарактеризован в примере 14 способе А.
Фиг. 4: ИК-спектр тенелиглиптина, который по существу охарактеризован в примере 14 способа А.
Фиг. 5: ВЭЖХ-хроматограмма тенелиглиптина 2,5 гидробромида гидрата, который по существу охарактеризован в Примере 6.
Подробное описание изобретения
В одном варианте реализации согласно настоящему изобретению предложен способ получения тенелиглиптина, соединения формулы I, или его соли или гидрата, включающий:

а) осуществление взаимодействия соединения формулы 11 с бис-(2-хлорэтил)-амином или его N-защищенным производным или солью,

или
а) осуществление взаимодействия 5-хлор-3-метил-1-фенил-1H-пиразол-4-карбальдегида, соединения формулы 30, с пиперазином или его N-защищенным производным

с получением соединения формулы Int-B или его N-защищенного производного или соли;
b) осуществление взаимодействия соединения формулы Int-B или его N-защищенного производного или соли с соединением формулы 13 с получением соединения формулы 14,

где R представляет собой аминозащитную группу, выбранную из группы, состоящей из аралкила, ацила, низшего алкоксикарбонила, аралкилоксикарбонила, низшего алкансульфонила, арилсульфонила, три-(низший алкил)силила, трифосгена; и
c) снятие защиты с соединения формулы 14 с получением тенелиглиптина, соединения формулы I, или его соли или гидрата.
В одном варианте реализации настоящего изобретения на стадии а) вышеуказанного способа осуществляют взаимодействие соединения формулы 11 с бис-(2-хлорэтил)амином или его N-защищенным производным или солью в подходящем растворителе и в присутствии подходящего основания с получением соединения формулы Int-B или его производного или соли.
Термин «N-защищенное производное» обозначает аминозащитную группу, выбранную из группы, состоящей из аралкила, ацила, низшего алкоксикарбонила, аралкилоксикарбонила, низшего алкансульфонила, арилсульфонила и три-(низшийалкил)силила. Предпочтительная аминозащитная группа представляет собой низший алкоксикарбонил, такой как трет-бутоксикарбонил.
Подходящее основание может быть выбрано из органических или неорганических оснований. Неорганическое основание может быть выбрано, не ограничиваясь ими, из гидроксидов, таких как гидроксид натрия, гидроксид калия; карбонатов, таких как карбонат натрия, карбонат калия; бикарбонатов, таких как бикарбонат натрия, бикарбонат калия, гидридов, таких как гидрид натрия, алкоксидов, таких как метоксид натрия, метоксид калия, трет-бутоксид калия; при этом органическое основание может быть выбрано, не ограничиваясь ими, из (R)-(+)-2,2'-бис-(дифенилфосфин)-1,1'-бинафтила, триэтиламина, триметиламина, пиридина, диизопропиламина и диметиламинопиридина. Предпочтительно, основание представляет собой гидрид натрия.
Подходящий растворитель может быть выбран, не ограничиваясь ими, из галогенированных углеводородов, таких как метиленхлорид, этиленхлорид, хлороформ и четыреххлористый углерод; спиртов, таких как метанол, этанол, 1-пропиловый спирт, 2-пропанол, трет-бутанол; сложных эфиров, таких как этилацетат, изопропилацетат и бутилацетат; амидов, таких как формамид, N,N-диметилформамид, N,N-диметилацетамид; диметилсульфоксид; нитрилов, таких как ацетонитрил, пропионитрил; эфиров, таких как диэтиловый эфир, диизопропиловый эфир, трет-бутилметиловый эфир, 1,4-диоксан, тетрагидрофуран; углеводородов, таких как бензол, толуол, циклогексан, метилциклогексан и толуол; или их смесей. Предпочтительно, растворитель представляет собой N,N-диметилформамид.
В одном варианте реализации настоящее изобретение относится к способу получения соли Int-B, включающему осуществление взаимодействия соединения формулы Int-B с подходящей кислотой.
Подходящая кислота может быть выбрана из группы, состоящей из уксусной кислоты, трифторуксусной кислоты, хлористоводородной кислоты, бромистоводородной кислоты, азотной кислоты, щавелевой кислоты, малоновой кислоты, янтарной кислоты, фосфорной кислоты, малеиновой кислоты, нафтален-1-сульфоновой кислоты, нафтален-2-сульфоновой кислоты, галловой кислоты, (+)камфорсульфоновой кислоты, (-)камфорсульфоновой кислоты, фумаровой кислоты, L-винной кислоты, этандисульфоновой кислоты, лимонной кислоты, щавелевой кислоты, малоновой кислоты, яблочной кислоты, серной кислоты, хлористоводородной кислоты, п-толуолсульфоновой кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты и им подобных. Предпочтительно получают ацетатную соль соединения формулы Int-B.
В одном варианте реализации настоящего изобретения соединение формулы 11 в N,N-диметилформамиде реагирует с бис-(2-хлорэтил)-амином или его N-защищенным производным или солью в присутствии гидрида натрия. Реакцию проводят при температуре от примерно 25 до примерно 35°C. Предпочтительно реакцию проводят при температуре от примерно 20 до примерно 30°C. Таким образом, полученное соединение формулы Int-B далее взаимодействует с уксусной кислотой в подходящем растворителе. Ацетатную соль соединения формулы Int-B отделяют посредством способов, известных в данной области, например, фильтрацией или центрифугированием.
Подходящий растворитель для получения соли соединения формулы Int-B может быть выбран, не ограничиваясь ими, из галогенированных углеводородов, таких как метиленхлорид, этиленхлорид, хлороформ и четыреххлористый углерод; спиртов, таких как метанол, этанол, 1-пропиловый спирт, 2-пропанол, трет-бутанол; сложных эфиров, таких как этилацетат, изопропилацетат и бутилацетат; амидов, таких как формамид, N,N-диметилформамид, N,N-диметилацетамид; нитрилов, таких как ацетонитрил, пропионитрил; эфиров, таких как диэтиловый эфир, диизопропиловый эфир, трет-бутилметиловый эфир, 1,4-диоксан, тетрагидрофуран; углеводородов, таких как бензол, толуол, циклогексан, метилциклогексан; или их смесей. Предпочтительно, растворитель представляет собой толуол.
В одном варианте реализации настоящего изобретения на стадии а) вышеуказанного способа 5-хлор-3-метил-1-фенил-1H-пиразол-4-карбальдегид, соединение формулы 30, взаимодействует с пиперазином или его N-защищенным производным с получением соединения формулы Int-B или его N-защищенного производного или соли.
Реакцию можно проводить в присутствии подходящего растворителя и основания.
Растворитель может быть выбран, не ограничиваясь ими, из галогенированных углеводородов, таких как метиленхлорид, этиленхлорид, хлороформ и четыреххлористый углерод; спиртов, таких как метанол, этанол, 1-пропиловый спирт, 2-пропанол, трет-бутанол; сложных эфиров, таких как этилацетат, изопропилацетат и бутилацетат; амидов, таких как формамид, N,N-диметилформамид, N-N-диметилацетамид; нитрилов, таких как ацетонитрил, пропионитрил; эфиров, таких как диэтиловый эфир, диизопропиловый эфир, трет-бутилметиловый эфир, 1,4-диоксан, тетрагидрофуран; углеводородов, таких как бензол, толуол, циклогексан, метилциклогексан; или их смесей. Предпочтительно, растворитель представляет собой N,N-диметилформамид.
Подходящее основание может быть выбрано из органических или неорганических оснований. Неорганическое основание может быть выбрано, не ограничиваясь ими, из гидроксидов, таких как гидроксид натрия, гидроксид калия; карбонатов, таких как карбонат натрия, карбонат калия; бикарбонатов, таких как бикарбонат натрия, бикарбонат калия, гидридов, таких как гидрид натрия, алкоксидов, таких как метоксид натрия, метоксид калия, трет-бутоксид калия; при этом органическое основание может быть выбрано, не ограничиваясь ими, из (R)-(+)-2,2'-бис-(дифенилфосфин)-1,1'-бинафтила, триэтиламина, триметиламина, пиридина, диизопропиламина и диметиламинопиридина. Предпочтительно, основание представляет собой карбонат калия.
В одном варианте реализации настоящего изобретения 5-хлор-3-метил-1-фенил-1H-пиразол-4-карбальдегид, соединение формулы 30, взаимодействует с трет-бутиловым сложным эфиром пиперазин-1-карбоновой кислоты с получением трет-бутил-4-(4-формил-3-метил-1-фенил-1H-пиразол-5-ил)пиперазин-1-карбоксилата, соединения формулы 31,

которое далее преобразуют в соединение формулы Int-B.
Реакцию проводят в диапазоне температуры от примерно 25 до 200°C. Предпочтительно, реакцию проводят примерно при 120-140°C.
В одном варианте реализации настоящего изобретения 5-хлор-3-метил-1-фенил-1H-пиразол-4-карбальдегид, соединение формулы 30, взаимодействует с трет-бутиловым сложным эфиром пиперазин-1-карбоновой кислоты в присутствии диметилформамида и карбоната калия с получением трет-бутил-4-(4-формил-3-метил-1-фенил-1H-пиразол-5-ил)-пиперазин-1-карбоксилата, соединения формулы 31.
Трет-бутил-4-(4-формил-3-метил-1-фенил-1H-пиразол-5-ил)пиперазин-1-карбоксилат, соединение формулы 31, превращают в 1-(3-метил-1-фенил-1H-5-ил)-пиперазин.
Превращение можно проводить посредством взаимодействия с кислотой, выбранной из группы, состоящей из хлористоводородной кислоты, трифторуксусной кислоты, серной кислоты, бромистоводородной кислоты, п-толуолсульфоновой кислоты, трехбромистого бора, муравьиной кислоты. Предпочтительно, кислота представляет собой пара-толуолсульфоновую кислоту.
Превращение можно проводить в инертном растворителе или без растворителя. Инертный растворитель может быть выбран из воды, спиртов, таких как метанол, этанол, 1-пропанол, 2-пропанол, трет-бутанол; кетонов, таких как ацетон, метилэтилкетон; нитрилов, таких как ацетонитрил, пропионитрил; амидов, таких как формамид, N,N-диметилформамид, N,N-диметилацетамид, 1,3-диметил-2-имидазолидинон; эфиров, таких как диэтиловый эфир, диизопропиловый эфир, трет-бутилметиловый эфир, 1,4-диоксан, тетрагидропиран, тетрагидрофуран; сложных эфиров, таких как этилформиат, этилацетат, пропилацетат; галогенированных углеводородов, таких как метилендихлорид, хлороформ, 1,2-дихлорэтан; углеводородов, таких как н-гексан, циклогексан, бензол, толуол и метилциклогексан; сульфоксидов, таких как диметилсульфоксид; полярных растворителей, таких как сульфолан, гексаметилфосфориламид; или их смесей. Предпочтительно, растворитель представляет собой метанол.
В одном варианте реализации настоящего изобретения трет-бутил-4-(4-формил-3-метил-1-фенил-1H-пиразол-5-ил)пиперазин-1-карбоксилат, соединение формулы 31, обрабатывают пара-толуолсульфоновой кислотой в метаноле с получением соединения формулы Int-B.
Превращение можно проводить при температуре от примерно 0 до примерно 100°C. Предпочтительно, реакцию проводят при температуре от примерно 75 до примерно 80°C.
В одном варианте реализации настоящего изобретения на стадии b) вышеуказанного способа соединение формулы Int-B или его N-защищенное производное или соль взаимодействует с соединением формулы 13 с получением соединения формулы 14.

В одном варианте реализации настоящего изобретения вышеуказанный способ осуществляют в присутствии подходящего растворителя и восстанавливающего агента с получением соединения формулы 14.
Подходящий растворитель может быть выбран, не ограничиваясь ими, из галогенированных углеводородов, таких как метиленхлорид, этиленхлорид, хлороформ и четыреххлористый углерод; спиртов, таких как метанол, этанол, 1-пропиловый спирт, 2-пропанол, трет-бутанол; сложных эфиров, таких как этилацетат, изопропилацетат и бутилацетат; амидов, таких как формамид, N,N-диметилформамид, N,N-диметилацетамид; нитрилов, таких как ацетонитрил, пропионитрил; эфиров, таких как диэтиловый эфир, диизопропиловый эфир, трет-бутилметиловый эфир, 1,4-диоксан, тетрагидрофуран, тетрагидропиран; углеводородов, таких как бензол, толуол, циклогексан, метилциклогексан; или их смесей. Предпочтительно, растворитель представляет собой метилендихлорид.
Подходящий восстанавливающий агент может быть выбран из борогидридов, таких как борогидрид натрия, борогидрид калия, цианоборогидрид натрия, триацетоксиборогидрид натрия; гидридов, таких как гидрид лития и алюмогидрид лития. Предпочтительно, восстанавливающий агент представляет собой триацетоксиборогидрид натрия.
Аминозащитная группа, обозначенная как R в соединениях формулы 13 и 14, может быть выбрана из группы, состоящей из аралкила, такого как бензил, п-нитробензил, бензгидрил, тритил; ацила, такого как формил, ацетил, пропионил, метоксиацетил, метоксипропионил, бензоил, тиенилацетил, тиазолилацетил, тетразолилацетил, тиазолилглиоксилоил, тиенилглиоксилоил; низшего алкоксикарбонила, такого как метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил; аралкилоксикарбонила, такого как бензилоксикарбонил, п-нитробензилоксикарбонил, 9-флуоренилметилоксикарбонил; низшего алкансульфонила, такого как метансульфонил, этансульфонил; арилсульфонила, такого как толуолсульфонил; три-(низший алкил)силила, такого как триметилсилил; и трифосгена.
В одном варианте реализации настоящего изобретения на стадии b), когда R представляет собой ацетил, способ включает обеспечение взаимодействия (5S)-1-ацетил-5-(1,3-тиазолидин-3-карбонил)пирролидин-3-она, соединения формулы 29,

с соединением формулы Int-B с получением 1-[(2S,4S)-4-[4-(3-метил-1-фенил-1H-пиразол-5-ил)пиперазин-1-ил]-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-ил]этанона, соединения формулы 32.
В одном варианте реализации настоящего изобретения на стадии b), когда R представляет собой 9-флуоренилметилоксикарбонил (Fmoc), способ включает обеспечение взаимодействия 9Н-флуорен-9-илметил (2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)-пирролидин-1-карбоксилата, соединения формулы 19,
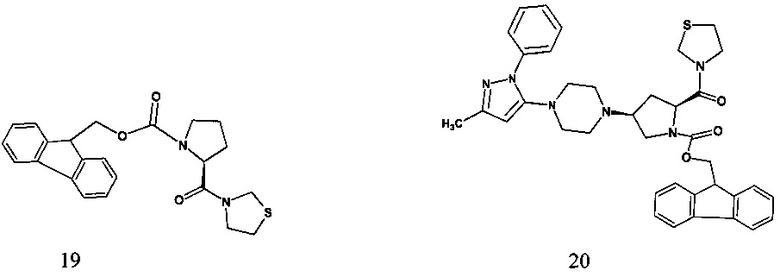
с соединением формулы Int-B с получением 3-{(2S,4S)-1-9Н-флуорен-9-илметоксикарбонил-4-[4-(3-метил-1-фенил-5-пиразолил)-1-пиперазинил]-2-пирролидинилкарбонил}-1,3-тиазолидина, соединения формулы 20.
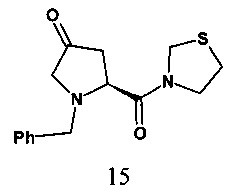
В одном варианте реализации настоящего изобретения на стадии b) R представляет собой бензил, способ включает обеспечение взаимодействия 1-бензил-(2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)пирролидина, соединения формулы 15, с соединением формулы Int-B

с получением 3-{(2S,4S)-1-бензил-4-[4-(3-метил-1-фенил-5-пиразолил)-1-пиперазинил]-2-пирролидинилкарбонил}-1-,3-тиазолидина, соединения формулы 16.
В одном варианте реализации настоящего изобретения на стадии b) R представляет собой бензилоксикарбонил, способ включает обеспечение взаимодействия бензилоксикарбонил-(2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-карбоксилата, соединения формулы 21, с соединением формулы Int-B

с получением 3-{(2S,4S)-1-бензилоксикарбонил-4-[4-(3-метил-1-фенил-5-пиразолил)-1-пиперазинил]-2-пирролидинилкарбонил}-1,3-тиазолидина, соединения формулы 22.
В одном варианте реализации настоящего изобретения на стадии b) R представляет собой трифенилметил, способ включает обеспечение взаимодействия 1-тритил-(2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)пирролидина, соединения формулы 17, с соединением формулы Int-B

с получением 3-{(2S,4S)-1-тритил-4-[4-(3-метил-1-фенил-5-пиразолил)-1-пиперазинил]-2-пирролидинилкарбонил}-1,3-тиазолидина, соединения формулы 18.
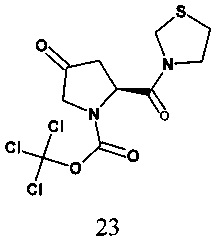
В одном варианте реализации настоящего изобретения на стадии b) R представляет собой трихлорметоксикарбонил, способ включает обеспечение взаимодействия трихлорметоксикарбонил-(2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-карбоксилата, соединения формулы 23, с соединением формулы Int-B

с получением 3-{(2S,4S)-1-трихлорметоксикарбонил-4-[4-(3-метил-1-фенил-5-пиразолил)-1-пиперазинил]-2-пирролидинилкарбонил}-1-,3-тиазолидина, соединения формулы 24.
В одном варианте реализации настоящего изобретения на стадии с) вышеуказанного способа с соединения формулы 14 снимают защиту с применением подходящих реагентов с получением тенелиглиптина, соединения формулы I, или его соли или гидрата.
Подходящие реагенты в зависимости от типа защитной группы могут быть выбраны из группы, состоящей из кислоты, такой как хлористоводородная кислота, трифторуксусная кислота, бромистоводородная кислота, серная кислота, бромистоводородная кислота, п-толуолсульфоновая кислота, трехбромистый бор, муравьиная кислота; восстановителя, такого как палладий/углерод, ацетат палладия или гидроксид палладия, основания, такого как пиперидин, аммиак, метиламин и циклогексиламин.
Реакцию снятия защиты можно проводить в инертном растворителе или без растворителя. Инертный растворитель может быть выбран из воды, спиртов, таких как метанол, этанол, 1-пропанол, 2-пропанол, трет-бутанол; кетонов, таких как ацетон, метилэтилкетон; нитрилов, таких как ацетонитрил, пропионитрил; амидов, таких как формамид, N,N-диметилформамид, N,N-диметилацетамид, 1,3-диметил-2-имидазолидинон; эфиров, таких как диэтиловый эфир, диизопропиловый эфир, трет-бутилметиловый эфир, 1,4-диоксан, тетрагидрофуран, тетрагидропиран; сложных эфиров, таких как этилформиат, этилацетат, пропилацетат; галогенированных углеводородов, таких как дихлорметан, хлороформ, метилендихлорид; углеводородов, таких как н-гексан, циклогексан, бензол, толуол и метилциклогексан; сульфоксидов, таких как диметилсульфоксид; полярных растворителей, таких как сульфолан, гексаметилфосфориламид; или их смесей.
В одном варианте реализации настоящего изобретения на стадии с), когда R представляет собой ацетил, способ включает снятие защиты с 1-[(2S,4S)-4-[4-(3-метил-1-фенил-1H-пиразол-5-ил)пиперазин-1-ил]-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-ил]этанона, соединения формулы 32, посредством неорганической кислоты, выбранной из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты; неорганического основания, выбранного из группы, состоящей из гидроксида натрия, гидроксида калия, карбоната натрия, карбоната калия и им подобных, с получением тенелиглиптина, соединения формулы I, или его соли или гидрата.
В одном варианте реализации настоящего изобретения с 1-[(2S,4S)-4-[4-(3-метил-1-фенил-1H-пиразол-5-ил)-пиперазин-1-ил]-2-(1,3-тиазолидин-3-илкарбонил)-пирролидин-1-ил]-этанона, соединения формулы 32, снимают защиту посредством применения водного раствора бромистоводородной кислоты в 2-пропаноле.
Реакцию снятия защиты проводят при температуре от примерно 0 до 120°C. Предпочтительно, при температуре примерно 80-85°C.
В одном варианте реализации настоящего изобретения на стадии с), когда R представляет собой 9-флуоренилметилоксикарбонил (Fmoc), с 3-{(2S,4S)-1-9Н-флуорен-9-илметоксикарбонил-4-[4-(3-метил-1-фенил-5-пиразолил)-1-пиперазинил]-2-пирролидинилкарбонил}-1-,3-тиазолидина, соединения формулы 20, снимают защиту посредством применения подходящего основания, выбранного из группы, состоящей из неорганического или органического основания. Органическое основание может быть выбрано из диметиламинопиридина, диизопропиламина, пиридина, пиперидина, триэтиламина, триметиламина и им подобных. Предпочтительно, основание представляет собой пиперидин.
Реакцию снятия защиты проводят при температуре от примерно 0 до 120°C. Предпочтительно, при температуре от примерно 80 до примерно 85°C.
В одном варианте реализации согласно настоящему изобретению предложен способ получения тенелиглиптина, соединения формулы I, или его соли, включающий: а) обеспечение взаимодействия соединения формулы Int-B или его N-защищенного производного или соли с соединением формулы 29 с получением соединения формулы 32; и b) снятие защиты с соединения формулы 32 с получением тенелиглиптина, соединения формулы I, или его соли.
В одном варианте реализации настоящего изобретения на вышеуказанной стадии а) осуществляют взаимодействие соединения формулы 29 с соединением формулы Int-B с получением соединения формулы 32.
Реакцию можно проводить в присутствии подходящего восстанавливающего агента, как описано выше.
Реакцию проводят в течение периода от примерно 3 до 15 часов. Предпочтительно, реакцию проводят в течение периода от примерно 4 до 5 часов. Соединение формулы 31 отделяют посредством способов, известных в данной области, таких как, фильтрация, концентрирование и им подобных.
В одном варианте реализации настоящего изобретения на стадии b) вышеуказанного способа тенелиглиптин получают посредством снятия защиты с соединения формулы 32, как описано выше.
В одном варианте реализации согласно настоящему изобретению предложен аморфный тенелиглиптин.
В одном варианте реализации согласно настоящему изобретению предложен аморфный тенелиглиптин, характеризующийся 1H ЯМР (300 МГц, ДМСО-D6), имеющий пики при 1,55, 2,14, 2,2-2,22, 2,43-2,76, 2,92-3,07, 3,63-3,84, 4,42-4,66, 5,78, 7,24-7,29, 7,43-7,47, 7,72-7,75.
В одном варианте реализации согласно настоящему изобретению предложен аморфный тенелиглиптин, характеризующийся пиками ИК-спектра при примерно 3443,97, 2952,9, 2826,5, 1639,5, 1556,4, 1504,5, 1016,23, 915,9, 764,18 см-1.
В одном варианте реализации согласно настоящему изобретению предложен тенелиглиптин, который по существу охарактеризован на Фиг. 3.
В одном варианте реализации согласно настоящему изобретению предложен тенелиглиптин, который по существу охарактеризован на Фиг. 4.
В одном варианте реализации согласно настоящему изобретению предложен кристаллический тенелиглиптин.
В одном варианте реализации согласно настоящему изобретению предложен способ очистки тенелиглиптина без применения колоночной хроматографии, включающий обработку тенелиглиптина растворителем, выбранным из группы, состоящей из спиртов, эфиров, сложных эфиров, амидов, нитрилов, сульфоксидов, кетонов, углеводородов, ацетатов, галогенированных углеводородов, воды или их смесей.
В одном варианте реализации согласно настоящему изобретению предложен способ очистки тенелиглиптина, включающий обработку тенелиглиптина смесью сложного эфира и спирта.
В одном варианте реализации согласно настоящему изобретению предложен способ очистки тенелиглиптина, включающий обработку тенелиглиптина 1-пропанолом.
В одном варианте реализации согласно настоящему изобретению предложен тенелиглиптин, имеющий чистоту примерно 99% и выше по данным ВЭЖХ (высокоэффективной жидкостной хроматографии).
Соль соединения формулы I может быть получена посредством способов, известных в данной области. Например, соль согласно настоящему изобретению может быть получена посредством взаимодействия соединения формулы I с органической или неорганической кислотой, выбранной из группы, состоящей из хлористоводородной кислоты, серной кислоты, уксусной кислоты, трифторуксусной кислоты, бромистоводородной кислоты, азотной кислоты, метансульфокислоты, малеиновой кислоты, толуолсульфокислоты, бензолсульфокислоты, нафтален-1-сульфоновой кислоты, нафтален-2-сульфоновой кислоты, галловой кислоты, (+)-камфорсульфоновой кислоты, (-)-камфорсульфоновой кислоты, фумаровой кислоты, серной кислоты, янтарной кислоты, L-винной кислоты, этандисульфоновой кислоты, лимонной кислоты, щавелевой кислоты, яблочной кислоты, малеиновой кислоты, малоновой кислоты и фосфорной кислоты. Предпочтительно, получают бромистоводородную соль соединения формулы I.
В одном варианте реализации настоящего изобретения соль соединения формулы I может быть получена в присутствии подходящего растворителя. Подходящий растворитель может быть выбран из группы, состоящей из метанола, н-бутанола, трет-бутанола, пропилацетата, бутилацетата, эфиров, таких как тетрагидрофуран, тетрагидропиран, диэтиловый эфир, диизопропиловый эфир, метил-трет-бутиловый эфир; кетонов, таких как ацетон, метилэтилкетон, амидов, таких как N,N-диметилформамид, N,N-диметилацетамид; нитрилов, таких как ацетонитрил, углеводородов, таких как толуол, ксилол, циклогексан, метилциклогексан; галогенированных углеводородов, таких как метилендихлорид, хлороформ, этилендихлорид. Предпочтительно, растворитель представляет собой смесь метанола и трет-бутанола.
В одном варианте реализации согласно настоящему изобретению предложен способ получения тенелиглиптина 2,5 гидробромида или его гидрата, который включает обработку тенелиглиптина бромистоводородной кислотой или бромистоводородной кислотой в уксусной кислоте в растворителе, выбранном из метанола и трет-бутанола.
В одном варианте реализации настоящего изобретения тенелиглиптин растворяют в смеси метанола и трет-бутанола. Раствор нагревают до температуры от примерно 30°C до примерно температуры кипения смеси растворителей. Предпочтительно раствор нагревают до примерно 70-75°C. При данной температуре к раствору добавляют водный раствор бромистоводородной кислоты. Реакционную смесь охлаждают до температуры от примерно -5 до примерно 25°C. Тенелиглиптина 2,5 гидробромида гидрат отделяют посредством способов, известных в данной области, таких как, фильтрация, центрифугирование и им подобных.
В одном варианте реализации согласно настоящему изобретению предложен способ получения тенелиглиптина 2,5 гидробромида гидрата, включающий кристаллизацию тенелиглиптина 2,5 гидробромида гидрата из растворителя, выбранного из группы, состоящей из метанола, н-бутанола, трет-бутанола, пропилацетата, изопропилацетата, бутилацетата, эфиров, таких как диэтиловый эфир, тетрагидрофуран, тетрагидропиран, диизопропиловый эфир, метил-трет-бутиловый эфир; кетонов, таких как ацетон, метилэтилкетон, амидов, таких как N,N-диметилформамид, N,N-диметилацетамид; нитрилов, таких как ацетонитрил, углеводородов, таких как толуол, ксилол, циклогексан, метилциклогексан; галогенированных углеводородов, таких как метилендихлорид, хлороформ, этилендихлорид и их смесей. Предпочтительно, растворитель представляет собой метанол или смесь метанола и трет-бутанола.
В одном варианте реализации согласно настоящему изобретению предложен способ получения тенелиглиптина 2,5 гидробромида гидрата, включающий кристаллизацию тенелиглиптина 2,5 гидробромида гидрата из метанола и н-бутанола.
В одном варианте реализации согласно настоящему изобретению предложен способ получения тенелиглиптина 2,5 гидробромида гидрата, включающий кристаллизацию тенелиглиптина 2,5 гидробромида гидрата из N,N-диметилацетамида и тетрагидрофурана.
В одном варианте реализации согласно настоящему изобретению предложен способ получения тенелиглиптина 2,5 гидробромида гидрата, включающий кристаллизацию тенелиглиптина 2,5 гидробромида гидрата из N,N-диметилформамида и тетрагидрофурана.
В одном варианте реализации согласно настоящему изобретению предложен способ получения тенелиглиптина 2,5 гидробромида гидрата, включающий кристаллизацию тенелиглиптина 2,5 гидробромида из метанола.
В одном варианте реализации настоящего изобретения тенелиглиптина 2,5 гидробромида гидрат контактирует с метанолом и реакционную смесь нагревают до кипения с получением раствора. Раствор может быть обработан углем и профильтрован в горячем виде через кизельгуровый фильтр. Реакционную смесь охлаждают до температуры примерно 15-20°C и отделяют чистый тенелиглиптина 2,5 гидробромида гидрат.
В одном варианте реализации согласно настоящему изобретению предложен способ очистки тенелиглиптина 2,5 гидробромида гидрата способом с применением растворителя/антирастворителя.
В одном варианте реализации согласно настоящему изобретению предложен по существу чистый тенелиглиптина 2,5 гидробромид, имеющий чистоту по меньшей мере 99%, что измерено посредством высокоэффективной жидкостной хроматографии.
В одном варианте реализации согласно настоящему изобретению предложен по существу чистый тенелиглиптина 2,5 гидробромида гидрат, имеющий чистоту по меньшей мере 99,7% что измерено посредством высокоэффективной жидкостной хроматографии.
В одном варианте реализации согласно настоящему изобретению предложен по существу чистый тенелиглиптина 2,5 гидробромида гидрат, имеющий чистоту по меньшей мере 99,9%, что измерено посредством высокоэффективной жидкостной хроматографии.
В одном варианте реализации согласно настоящему изобретению предложен по существу чистый тенелиглиптина 2,5 гидробромида гидрат, который по существу соответствует фиг. 1.
В одном варианте реализации согласно настоящему изобретению предложен по существу чистый тенелиглиптина 2,5 гидробромида гидрат, который по существу охарактеризован на Фиг. 2
В одном варианте реализации согласно настоящему изобретению предложен тенелиглиптин или его соль или сольват, имеющий менее чем примерно 0,2% (2S,4R)-изомера тенелиглиптина или его соли или сольвата, имеющий менее чем примерно 0,15% (2S,4R)-изомера тенелиглиптина или его соли или сольвата, имеющий менее чем примерно 0,1% (2S,4R)-изомера тенелиглиптина или его соли или сольватапо данным хиральной хроматографии. Предпочтительно, (2S,4R)-изомер тенелиглиптина или его соли или сольвата не обнаруживают в тенелиглиптине или его соли или сольвате.
В одном варианте реализации согласно настоящему изобретению предложен тенелиглиптин или его соль или сольват, имеющий менее чем примерно 0,2% (2R,4S)-изомера тенелиглиптина или его соли или сольвата, имеющий менее чем примерно 0,15% (2R,4S)-изомера тенелиглиптина или его соли или сольвата, имеющий менее чем примерно 0,1% (2R,4S)-изомера тенелиглиптина или его соли или сольватапо данным хиральной хроматографии. Предпочтительно, (2R,4S) изомер тенелиглиптина или его соли или сольвата не обнаруживают в тенелиглиптине или его соли или сольвате.
В одном варианте реализации согласно настоящему изобретению предложен тенелиглиптин или его соль или сольват, имеющий менее чем примерно 0,2% (2R,4R)-изомера тенелиглиптина или его соли или сольвата, имеющий менее чем примерно 0,15% (2R,4R)-изомера тенелиглиптина или его соли или сольвата, имеющий менее чем примерно 0,1% (2R,4R)-изомера тенелиглиптина или его соли или сольватапо данным хиральной хроматографии. Предпочтительно, (2R,4R)-изомер тенелиглиптина или его соли или сольвата не обнаруживают в тенелиглиптине или его соли или сольвате.
В одном варианте реализации согласно настоящему изобретению предложен тенелиглиптин или его соль или сольват, имеющий химическую чистоту не менее чем примерно 99,5% по данным ВЭЖХ (высокоэффективной жидкостной хроматографии) и хиральную чистоту примерно 100% по данным хиральной хроматографии.
В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 19.
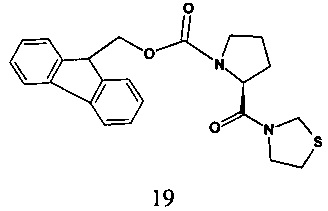
В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 19, характеризующееся по данным 1H ЯМР (300 МГц, ДМСО-d6) пиками при 2,43, 2,96-3,12, 3,60-3,95, 4,19-4,36, 4,41-4,63, 4,75-4,83, 5,02-5,11, 7,31-7,44, 7,58-7,67, 7,89-7,91.
В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 20.
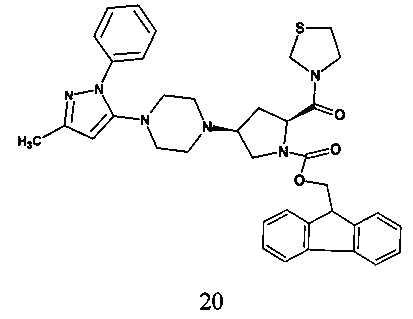
В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 20, характеризующееся по данным 1H ЯМР (300 МГц, ДМСО-d6) пиками при 2,14, 2,40, 2,73-3,08, 3,24-3,34, 3,47-3,82, 4,15-4,69, 5,78, 7,27-7,60, 7,62-7,67, 7,72-7,69, 7,87-7,89.
В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 28.

В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 28, характеризующееся по данным 1H ЯМР (300 МГц, ДМСО-d6) пиками при 2,07, 2,19-2,30, 3,31, 3,50-3,53, 3,79-3,84, 4,18-4,21, 4,49-4,77.
В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 29.

В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 29, характеризующееся по данным 1H ЯМР (300 МГц, CDCl3) пиками при 1,98, 2,91-3,00, 3,09-3,12, 3,59-3,63, 3,92-3,98, 4,10-4,16, 4,31-4,35, 4,40-4,50, 4,62-4,65, 4,91-4,94, 5,10-5,18.
В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 31.

В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 31, характеризующееся по данным 1H ЯМР (300 МГц, CDCl3) пиками при 1,4, 2,4, 3,07, 3,4, 7,29-7,46, 9,94.
В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 32.

В одном варианте реализации согласно настоящему изобретению предложено соединение формулы 32, характеризующееся по данным 1H ЯМР (300 МГц, ДМСО-D6) пиками при 1,48-1,52, 1,94, 2,14, 2,5-2,78, 2,88-3,2, 3,6-4,79, 5,79, 7,27, 7,45, 7,77.
В одном варианте реализации согласно настоящему изобретению предложен тенелиглиптина 2,5 гидробромида гидрат, имеющий менее чем 0,1% любого из нижеследующих соединений:

В одном варианте реализации согласно настоящему изобретению предложено применение бис-(2-хлорэтил)амина или N-boc-бис-(2-хлорэтил)амина или его соли для получения соединения формулы Int-B или тенелиглиптина.
В одном варианте реализации согласно настоящему изобретению предложено применение 5-хлор-3-метил-1-фенил-1H-пиразол-4-карбальдегида или трет-бутил-4-(4-формил-3-метил-1-фенил-1H-пиразол-5-ил)-пиперазин-1-карбоксилата для получения соединения формулы Int-B или тенелиглиптина.
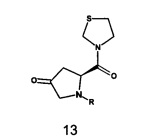
В одном варианте реализации согласно настоящему изобретению предложен способ получения соединения формулы 13, включающий:
a) осуществление взаимодействия транс-4-гидрокси-L-пролина с аминозащитной группой с образованием N-защищенного L-транс-4-гидроксипролина;
b) осуществление взаимодействия N-защищенного L-транс-4-гидроксипролина с 1,3-тиазолидином в присутствии гидроксибензотриазола (HOBt) и 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (EDC.HCl) с образованием N-защищенного (2S)-4-гидрокси-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-карбоксилата;
c) осуществление взаимодействия N-защищенного (2S)-4-гидрокси-2-(1,3-тиазолидин-3-илкарбонил)-пирролидин-1-карбоксилата с комплексом триоксида серы и пиридина с образованием N-защищенного (2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-карбоксилата.
В одном варианте реализации согласно настоящему изобретению предложен способ получения соединения формулы 15, включающий превращение N-бензил-транс-4-гидрокси-2-пирролидинилкарбонил-1,3-тиазолидина в 1-бензил-(2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)пирролидин, соединение формулы 15.
В одном варианте реализации согласно настоящему изобретению предложен способ получения соединения формулы 17, включающий превращение N-тритил-транс-4-гидрокси-2-пирролидинилкарбонил-1,3-тиазолидина в N-тритил-4-оксо-2-пирролидинилкарбонил-1,3-тиазолидин, соединение формулы 17.

В одном варианте реализации согласно настоящему изобретению предложен способ получения соединения формулы 19, включающий превращение N-9Н-флуорен-9-илметилоксикарбонил-транс-4-гидрокси-2-пирролидинилкарбонил-1,3-тиазолидина в 9Н-флуорен-9-илметил-(2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-карбоксилат, соединение формулы 19.

В одном варианте реализации согласно настоящему изобретению предложен способ получения соединения формулы 21, включающий превращение N-бензилоксикарбонил-транс-4-гидрокси-2-пирролидинилкарбонил-1,3-тиазолидина в бензил-(2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-карбоксилат, соединение формулы 21.

В одном варианте реализации согласно настоящему изобретению предложен способ получения соединения формулы 23, включающий превращение N-трихлорметил-транс-4-гидрокси-2-пирролидинилкарбонил-1,3-тиазолидина в трихлорметил-(2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-карбоксилат, соединение формулы 23.
В одном варианте реализации согласно настоящему изобретению предложен способ получения соединения формулы 29, включающий превращение 1-[(2S,4R)-4-гидрокси-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-ил]этанона в (5S)-1-ацетил-5-(1,3-тиазолидин-3-илкарбонил)-пирролидин-3-он, соединение формулы 29.

В одном варианте реализации согласно настоящему изобретению предложен способ

получения соединения формулы 14, как отображено на нижеприведенной схеме:
В одном варианте реализации согласно настоящему изобретению предложен способ получения соединения формулы 14, включающий:
a) превращение транс-4-гидрокси-1-пролина в соединение формулы 25, где R представляет собой тот же заместитель, который указан выше,
b) превращение соединения формулы 25 в соединение формулы 26,
c) осуществление взаимодействия соединения формулы 26 с 1-(3-метил-1-фенил-1H-пиразол-5-ил)-пиперазином или его солью с получением соединения формулы 27; и
d) осуществление взаимодействия соединения формулы 27 с 1,3-тиазолидином с получением соединения формулы 14.
Хотя настоящее изобретение было описано с точки зрения конкретных вариантов его реализации, некоторые модификации и эквиваленты очевидны для специалистов в данной области и предполагаются в объеме настоящего изобретения.
Следующие примеры приведены, чтобы дать возможность специалисту в данной области осуществить изобретение, и лишь иллюстрируют изобретение. Примеры не следует рассматривать как ограничивающие объем изобретения, как определено в формуле изобретения.
Инструментальные параметры порошковой рентгеновской дифрактометрии (PXRD):
Измерения проводят на рентгеновском дифрактометре Philips модели XPERT-PRO (PANalytical), детектор: X'celerator [1] с применением Cu лампы с типом и длиной волны рентгеновского излучения: K-Alphal [А] и 1,54060 при следующих условиях: Настройки генератора: 40 мА/45 кВ, Время шага: 50, Размер шага: 0,0170, Ширина пика 2,00 и начальный угол (°) 2,0 и конечный угол: 50,0, Тип сканирования: непрерывный; измерение проводят при 25°C. Прибор XRPD калибруют с применением стандартов NIST SRM 6-40С кремния и NIST SRM 1976 Оксид алюминия.
Подготовка образца: берут примерно 20 мг образца и применяют для наполнения канавки держателя стандартного образца кремния с применением техники верхней загрузки. Держатель образца далее помещают на оптическом пути рентгеновских лучей и сканируют с применением нижеописанных параметров. Полученные данные рентгеновской порошковой дифракции объединяют с применением программного обеспечения High Score Plus Software.
Инструментальные параметры ядерного магнитного резонанса (метод ЯМР) для тенелиглиптина 2,5 гидробромида гидрата:
Спектр протонного ЯМР записывают в CDCl3 и ДМСО-d6 с применением ЯМР-спектрометра Varian 300 МГц. Величины химических сдвигов (δ) в ppm соотносят с тетраметилсиланом (TMS), используемым в качестве внутреннего стандарта.
Инструментальные параметры ИК:
ИК-спектры с Фурье преобразованием (FTIR spectra) записывают на FTIR-спектрофотометре Spectrum One Perkin-Elmer, оснащенном ДТГС-детектором. Спектр записывают с применением таблеток KBr в диапазоне от 4000 см-1 до 450 см-1, применяя три скана на образец, принимая воздух в качестве стандарта. Примерно 200 мг KBr, предварительно высушенного при 200°C и охлажденного, взвешивают и растирают в ступке до мелкого порошка. Добавляют примерно 2,0 мг тестируемого образца и хорошо перемешивают и растирают до мелкого порошка. Небольшое количество порошка берут для получения тонкой полупрозрачной пеллеты. Эту тонкую пеллету затем помещают в держатель образцов, который затем помещают в Фурье-спектрофотометри сканируют в диапазоне 4000-450 см-1. Данные обрабатывают с применением программного обеспечения Spectrum One Software.
Инструментальные параметры ВЭЖХ:
Расходные материалы для ВЭЖХ:
Реагенты и растворители: вода (milli Q эквивалент), метанол (градиентной чистоты), натриевая соль октансульфоновой кислоты (ЧДА), ортофосфорная кислота (ЧДА).
Условия хроматографирования:
Прибор: высокоэффективный жидкостной хроматограф, оснащенный четырехканальными градиентными насосами, УФ-детектором с переменной длиной волны, который соединен с регистратором данных и программным обеспечением для интегрирования или равноценным ему,
Колонка: Inertsil ODS 3V, 250×4,6 мм, 5 мкм или эквивалентная
Подвижная фаза: A = буфер, B = метанол (градиентная программа)
Буфер: 0,01 М натриевой соли октансульфоновой кислоты в воде, pH доводят до 3,5 раствором разбавленной ортофосфорной кислоты, фильтруют через 0,45 мкм фильтровальную бумагу и дегазируют.
Разбавитель: буфер : метанол (10:90 об./об.); Скорость потока: 1,0 мл/минута
Рабочая длина волны детектора: УФ 210 нм, Температура колонки: 25°C, Объем вводимой пробы: 10 мкл; Время анализа: 60 минут
Градиентная программа:

Следует отметить, что тенелиглиптин элюируется при времени удерживания примерно 25 минут, а пик гидробромида (HBr) при времени удерживания примерно 1,9 минут.
Приготовление испытуемого раствора: (Готовят две пробы)
Точно взвешивают около 25 мг испытуемого образца и помещают его в мерную колбу объемом 25 мл. Добавляют 15 мл разбавителя и обрабатывают ультразвуком для растворения. Доводят разбавителем до метки и перемешивают.
Методика:
Вводят холостую пробу, а затем вводят каждый испытуемый раствор. Записывают хроматограммы для всех проб. Не обращают внимания на пики, обусловленные холостой пробой и гидробромидом. Соотношение веществ рассчитывают методом нормализации площадей.
ПРИМЕРЫ
Пример-1. Синтез 1,3-тиазолидина
К насыщенному раствору карбоната натрия (2,5 кг) в воде (17,5 л), добавляли цистеамина гидрохлорид (500 г, 4,40 моль). Реакционную смесь затем охлаждали до 18-20°C и добавляли 35-37% раствор формальдегида (350 г) и перемешивали при 18-20°C в течение 2 ч. Реакционную смесь экстрагировали дихлорметаном и сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 375 г титульного соединения в виде масла. Его применяли на следующей стадии без очистки.
1H ЯМР (300 МГц, CDCl3): δ 2,82-2,887 (t, 2Н, J=6,0 Гц), 3,147-3,180 (t, 2Н, J=5,8 Гц), 4,16 (s, 2Н) MS (m/z): 90,16 [М+Н]+
Пример-2. Синтез (2S,4R)-1-[(9H-флуорен-9-илметокси)карбонил]-4-гидроксипирролидин-2-карбоновой кислоты
К раствору (2S,4R)-4-гидрокси-L-пролина (100 г) в тетрагидрофуране (200 мл) добавляли бикарбонат натрия (80 г), воду (400 мл) и раствор 9-флуоренилметилоксикарбонил (Fmoc) хлорида (226 г) (в 200 мл ТГФ) при 25-30°C. Реакционную смесь перемешивали при примерно 25-30°C в течение 10-12 ч. После окончания реакции добавляли воду. Водную реакционную массу промывали диизопропиловым эфиром (DIPE) и подкисляли 1н. хлористоводородной кислотой. Реакционную смесь перемешивали в течение 2-3 часов. Твердое вещество собирали посредством фильтрации с получением 150 г титульного соединения в виде белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6): δ 1,89-2,24 (m, 2Н), 3,34-3,43 (m, 2Н), 3,43-3,54 (m, 0,5Н), 4,12-4,21 (m, 3Н), 4,25 (s, 2Н), 4,28-4,42 (m, 0,5Н), 5,16 (brs, 1H), 7,29-7,34 (m, 2Н), 7,38-7,65 (m, 2Н), 7,63-7,65 (m, 2Н), 7,87-7,89 (m, 2Н)
Температура плавления: 188-190°C
Масс-спектр (М+Н): 354,33
Пример-3. Синтез 3-[(2S,4R)-1-флуоренилметоксикарбонил-4-гидрокси-2-пирролидинилкарбонил]-1,3-тиазолидина.
К раствору соединения примера 2 (100 г) в метилендихлориде (MDC) (500 мл) добавляли НОВТ (гидроксибензотриазол) (19,11 г), N-метилморфолин (28,63 г) и 1,3-тиазолидин (соединение примера 1) (25,23 г) при 0-5°C. После перемешивания в течение 30 мин добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (65,2 г). Вышеуказанную реакционную массу затем перемешивали в течение 24 ч при 25-30°C. Реакционную смесь контролировали посредством ТСХ (тонкослойной хроматографии). Затем реакционную массу упаривали при пониженном давлении. К полученному маслянистому остатку добавляли раствор гидроксида натрия (22,64 г NaOH в 500 мл воды). Вышеуказанный водный раствор промывали DIPE (диизопропиловым эфиром). Затем pH водного раствора доводили до 8-9 50% водным раствором HCl. Вышеуказанный раствор экстрагировали этилацетатом и сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученное маслянистое вещество кристаллизовали из изопропилового спирта (IPA) с получением 55,0 г титульного соединения в виде почти белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-d6): - δ 1,85-1,89 (m, 1H), 2,17-2,20 (m, 1H), 2,91-3,10 (m, 2Н), 3,35-3,63 (m, 5Н), 4,17-4,38 (m, 5Н), 4,45-4,74 (m, 2Н), 5,1 (d, 1H), 7,31-7,42 (m, 4Н), 7,56-7,65 (m, 2Н), 7,87-7,91 (m, 2Н).
Масс-спектр (М+Н): - 425,18.
Пример-4 Синтез 9Н-флуорен-9-илметил-(2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)-пирролидин-1-карбоксилата (соединение 13, где R=Fmoc)
К раствору соединения примера 3 (25 г) и триэтиламина (50 мл) в MDC (50 мл) и диметилсульфоксиде (250 мл), добавляли комплекс триоксида серы и пиридина (28,15 г) при 0°C. Перемешивание продолжали в течение 2 ч при 0-5°C. После окончания реакции вышеуказанную реакционную массу гасили в водном растворе HCl и экстрагировали MDC. Затем слой MDC промывали 10% водным раствором карбоната натрия, после чего солевым раствором и сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный маслянистый остаток коричневого цвета затем очищали посредством колоночной флэш-хроматографии (30-40% ацетон в гексане) с получением титульного соединения в виде твердого вещества кремового цвета (15,0 г).
1H ЯМР (300 МГц, ДМСО-d6): - δ 2,43 (m, 1H), 2,96-3,12 (m, 2Н), 3,60-3,95 (m, 4Н), 4,19-4,36 (m, 4Н), 4,41-4,63 (m, 2Н), 4,75-4,83 (m, 1H), 5,02-5,11 (m, 1H), 7,31-7,44 (m, 4Н), 7,58-7,67 (m, 2Н), 7,89-7,91 (m, 2Н).
Пример 5а. Синтез 1-(3-метил-1-фенил-1H-пиразол-5-ил)пиперазина ацетата (Int-B):
Стадия 1: К перемешанному раствору 3-аминокротононитрила (60 г) и 1н. HCl (600 мл) добавляли фенилгидразин (72 мл). Реакционную массу перемешивали в течение 4 ч при 110-115°C. Реакционную смесь охлаждали до 25-30°C и гасили в ледяной воде (3,0 л). Затем ее нейтрализовали раствором бикарбоната натрия. Осажденное твердое вещество затем перемешивали, фильтровали и сушили с получением 110 г соединения стадии 1.
1H ЯМР (300 МГц, ДМСО-D6) δ 2,05 (s, 3Н), 5,25 (s, 2Н), 5,30 (s, 1H), 7,25 (t, 1H, J=6,9 Гц), 7,43 (t, 2Н, J=7,8 Гц), 7,56 (d, 2Н, J=7,8 Гц); APCI-MS (m/z) 174,25 (М+Н)+.
Стадия 2: К перемешиваемому раствору гидрида натрия (60% дисперсия в минеральном масле, 69 г, 1,734 моль) и N,N-диметилформамида (ДМФ 400 мл) медленно добавляли раствор соединения стадии 1 (50 г, 0,289 моль в 50 мл ДМФ) при 0-5°C и перемешивали в течение 0,5 ч. Через 30 минут к реакционной массе добавляли раствор бис-(2-хлорэтил)-амина гидрохлорида (55 г) в 50 мл ДМФ при 0-5°C и перемешивали в течение ночи при 25-30°C. Реакционную массу медленно гасили в ледяной воде и затем фильтровали. Водный слой подщелачивали раствором KOH и экстрагировали MDC. Слой MDC сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного соединения (60 г). К вышеуказанному неочищенному соединению добавляли этилацетат, активированный уголь и перемешивали в течение 15 минут при 50°C. Реакционную массу фильтровали, и фильтр промывали этилацетатом, и фильтрат концентрировали с получением неочищенного соединения (60 г). К вышеуказанному неочищенному соединению в течение 15 минут добавляли толуол и уксусную кислоту. Реакционную массу перемешивали в течение 2 ч при 0-5°C и осажденное твердое вещество фильтровали и промывали охлажденным толуолом с получением 17 г ацетатной соли в виде белого твердого вещества.
1H ЯМР (300 МГц, ДМСО-D6) δ 1,87 (s, 3Н), 2,14 (s, 3Н), 2,72-2,74 (m, 8Н), 5,77 (s, 1H), 7,26 (t, 1H, J=7,5 Гц), 7,44 (t, 2Н, J=7,8 Гц), 7,74 (d, 2Н, J=7,8 Гц).
Пример 5b. Синтез 1-(3-метил-1-фенил-1H-пиразол-5-ил)пиперазина ацетата (Int-B):
Стадия 2: N-boc-бис-(2-хлорэтил)-амин: К перемешиваемой смеси бис-(2-хлорэтил)-амина гидрохлорида (20 г) и метилендихлорида (MDC) (100 мл) добавляли триэтиламин (12,45 г) в один прием, после чего в течение 30 минут добавляли трет-бутоксикарбонильный (boc) ангидрид при 25-30°C. Полученную реакционную массу перемешивали в течение периода от примерно 12 до примерно 18 часов при 25-30°C. К вышеуказанной реакционной массе добавляли воду и отделяли слой MDC, который сушили над безводным сульфатом натрия и концентрировали с получением 25 г светло-желтого масла.
1H ЯМР (300 МГц, CDCl3) δ 1,47 (s, 9Н), 3,65 (m, 8Н). APCI-MS (m/z) 243,43(М+Н)+.
Стадия 3: К перемешиваемой смеси соединения примера 5а стадии 1 (10 г) и N,N-диметилформамида (70 мл) медленно добавляли гидрид натрия (6,9 г), (60% дисперсия в минеральном масле) при 0-5°C и перемешивали в течение 1,0 ч. К данной смеси через 30 минут добавляли вышеуказанное масло из стадии 2 (14 г) в 30 мл ДМФ при 0-5°C и перемешивали в течение ночи при примерно 25-30°C. Реакционную массу медленно гасили в ледяной воде и фильтровали через кизельгуровый фильтр. Водную фазу экстрагировали этилацетатом. Этилацетатную фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного вещества (25 г). Неочищенное вещество очищали посредством элюирования н-гексаном и этилацетатом (80:20) с получением 6 г продукта в виде почти белого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 1,44 (s, 9Н), 2,27 (s, 3Н), 2,80 (brs, 4Н), 3,42 (brs, 4Н), 5,67 (s, 1H), 7,26 (t, 1H, J=7,5 Гц), 7,40 (t, 2H, J=7,8 Гц), 7,74 (d, 2H, J=7,8 Гц).
INT-B:
К перемешиваемому раствору соединения стадии 3 (6 г) и метилендихлорида (60 мл) добавляли трифторуксусную кислоту (30 мл) при 25-30°C и перемешивали в течение 1,5 ч. Растворитель упаривали при пониженном давлении и к данному остатку добавляли воду (3,0 л). Смесь промывали диэтиловым эфиром. Водный слой подщелачивали раствором бикарбоната натрия и смесь экстрагировали хлороформом. Слой хлороформа сушили над сульфатом натрия и упаривали при пониженном давлении с получением остатка (4,0 г). В течение 15 минут добавляли толуол и уксусную кислоту (1 мл) и перемешивали в течение 1,5-2,0 ч при 0-5°C и осажденное твердое вещество отфильтровывали с получением 3,2 г ацетатной соли в виде белого твердого вещества. 1H ЯМР (300 МГц, ДМСО-D6) δ 1,87 (s, 3Н), 2,14 (s, 3Н), 2,72-2,74 (m, 8Н), 5,77 (s, 1H), 7,26 (t, 1H, J=7,5 Гц), 7,44 (t, 2Н, J=7,8 Гц), 7,74 (d, 2Н, J=7,8 Гц).
Пример 6. Синтез тенелиглиптина гемипентагидробромида:
Стадия 1: К раствору 9Н-флуорен-9-илметил-(2S)-4-оксо-2-(1,3-тиазолидин-3-илкарбонил)-пирролидин-1-карбоксилата (12,0 г), 1-(3-метил-1-фенил-1H-пиразол-5-ил)-пиперазина ацетата (10,3 г) в дихлорметане (120 мл), добавляли триацетоксиборогидрид натрия (9,02 г) и перемешивали смесь в течение 4-5 ч при 25-30°C. После окончания реакции к реакционной смеси добавляли воду и перемешивали в течение 15 мин. Органический слой отделяли и промывали водой и солевым раствором. Растворитель упаривали при пониженном давлении с получением промежуточного соединения (18 г).
Стадия 2: К раствору стадии 1 (18,0 г) в дихлорметане (190,0 мл), добавляли пиперидин (2,90 г) добавляли и реакционную смесь перемешивали в течение 5-6 ч при 25-30°C. После окончания реакции к реакционной смеси добавляли воду (60,0 мл) и подкисляли водный слой с помощью хлористоводородной кислоты. Реакционную смесь перемешивали в течение 15 мин и разделяли слои. К дихлорметановому слою добавляли водный раствор карбоната натрия и перемешивали. Слои разделяли и органический слой промывали солевым раствором. Продукт отделяли посредством концентрирования органического слоя при пониженном давлении (10,5 г).
К продукту стадии 2 (10,5 г) добавляли метанол (12,0 мл) и трет-бутиловый спирт (96,0 мл) и нагревали до 70-75°C. К реакционной смеси с температурой 70-75°C медленно добавляли водный раствор HBr (9,0 мл) и перемешивали в течение 1 ч. Реакционнную смесь охлаждали до 25-30°C и далее перемешивали в течение 2-3 ч при 25-30°C. Осадок собирали посредством фильтрации и промывали трет-бутиловым спиртом (24,0 мл). Полученное твердое вещество сушили при пониженном давлении при 50-55°C с получением неочищенного соединения тенелиглиптина 2,5 гидробромида (11,0 г). Затем продукт кристаллизовали с применением метанола или смеси метанола и трет-бутилового спирта с получением чистого продукта.
Кристаллизация: Неочищенное соединение тенелиглиптина 2,5 гидробромид (11,0 г) растворяли в метаноле (22,0 мл) посредством нагревания до температуры кипения. Данный раствор фильтровали в горячем виде. Реакционную смесь оставляли остывать и перемешивали при 25°C в течение 1 ч и затем при 15-20°C в течение 1 ч. Осадок собирали посредством фильтрации, промывали метанолом и сушили при пониженном давлении при 55-60°C с получением чистого тенелиглиптина 2,5 гидробромида (8,0 г). Чистота по методу ВЭЖХ: 99,7%, хиральная чистота: 100%, содержание воды: 5,3%, содержание HBr: 33,8%. Дифракция рентгеновских лучей: таблица для тенелиглиптина 2,5 гидробромида:

Результаты пиков хроматограммы ВЭЖХ.
Результаты пиков

Пример 7. Ацетилирование транс-4-гидрокси-L-пролина:
К перемешивамой суспензии 4-гидрокси-L-пролина (131 г) в дистиллированной воде (300 мл) по каплям добавляли уксусный ангидрид (110 мл) в течение 1 ч, температуру реакционной массы медленно увеличивали до 50°C. После добавления реакционную смесь выдерживали при 40-50°C в течение 2 ч. После окончания реакции уксусную кислоту и воду упаривали под вакуумом. Продукт получали в виде вязкого сиропа, который растирали с ацетонитрилом с получением продукта в виде белого твердого вещества (116 г, выход 70%).
1H ЯМР (300 МГц, ДМСО): δ 1,943 (m, 3Н), 2,05-2,08 (m, 1H), 3,33-3,37 (m, 2Н), 3,57-3,62 (m, 1H), 4,16-4,21 (m, 1H), 4,31-4,50 (m, 1H), 5,14 (brs, 1H) D2O обменивающийся, 12,37 (brs, 1H) D2O обменивающийся, MS (m/z): 174,54 [М-Н]+, SOR:  (c=4, вода),
(c=4, вода),
Температура плавления: 127-132°C.
Пример 8.1-[(2S,4R)-4-гидрокси-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-ил]-этанон
К раствору N-ацетил-4-гидрокси-L-пролина (1,5 г) в метилендихлориде (15 мл), добавляли HOBt (0,61 г), N-метилморфолин (1,04 г) и 1,3-тиазолидин (0,76 г) при 0-5°C. После перемешивания в течение 30 мин добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (2,61 г). Вышеуказанную реакционную массу перемешивали в течение 24 ч при 25-30°C. Реакционную смесь упаривали под вакуумом с получением остатка, который растворяли в растворе гидроксида натрия (2,50 г NaOH в 20 мл воды). Вышеуказанный водный раствор промывали диизопропиловым эфиром (DIPE) и разделяли. Полученный водный слой подкисляли 50% водным раствором HCl до pH 8-9. Раствор экстрагировали этилацетатом, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта. Очищали колоночной флэш-хроматографией (на силикагеле, элюируя 35% этилацетатом в гексане) с получением титульного соединения (0,7 г, выход 33%).
1H ЯМР (300 МГц, ДМСО-d6): - δ 2,07 (s, 3Н), 2,19-2,30 (m, 4Н), 3,31 (m, 1H), 3,50-3,53 (m, 1H), 3,79-3,84 (m, 2Н), 4,18-4,21 (m, 2Н), 4,49-4,77 (m, 4H)MS (m/z): 245,38
Пример 9. (5S)-1-ацетил-5-(1,3-тиазолидин-3-илкарбонил)пирролидин-3-он:
К раствору вышеуказанного чистого соединения (0,6 г), триэтиламина (1,71 мл) в MDC (1,2 мл) и диметилсульфоксиде (3 мл) добавляли комплекс триоксида серы и пиридина (1,5 г, 0,009 моль) при 0°C.Перемешивали в течение последующих 2 ч при 0-5°C. После окончания реакции вышеуказанную реакционную массу гасили в водном растворе HCl и экстрагировали MDC. Объединенный слой MDC промывали водным раствором 10% карбоната натрия, после чего солевым раствором и далее сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток коричневого цвета очищали посредством колоночной флэш-хроматографии (35% этилацетата в гексане) с получением чистого продукта в виде вязкого масла (200 мг, выход 35%).
1H ЯМР (300 МГц, CDCl3): δ 1,98 (s, 3Н), 2,91-3,00 (m, 2Н), 3,09-3,12 (m, 2Н), 3,59-3,63 (m, 2Н), 3,92-3,98 (m, 1H), 4,10-4,16 (m, 2Н), 4,431-4,35 (m, 1H), 4,40-4,50 (m, 1H), 4,62-4,65 (m, 1H), 4,91-4,94 (m, 1H), 5,10-5,18 (m, 1H). MS (m/z): 243,69 [М+Н]+
Пример 10. Синтез 5-хлор-3-метил-1-фенил-1H-пиразол -4-карбальдегида
К перемешиваемому раствору 210 г оксихлорида фосфора (125 мл) по каплям добавляли 47,5 г (50 мл) N,N-диметилформамида при 10-15°C и выдерживали реакционную массу в течение 40 мин при 25-30°C. К вышеуказанному раствору добавляли 3-метил-1-фенил-пиразол-5-он (25 г) при 25-30°C. Реакционную смесь нагревали на масляной бане при 110-115°C в течение 1 ч. После окончания реакции ее охлаждали до 10-15°C. Реакционную смесь гасили в 1500 мл ледяной воды и перемешивали в течение 2-3 ч при 25-30°C. Продукт осаждали и фильтровали. Его промывали 250 мл воды. Полученное влажное твердое вещество сушили при 60°C в течение 12 ч с получением 5-хлор-3-метил-1-фенил-1H-пиразол-4-карбальдегида в виде светло-желтого твердого вещества (22 г, 70%).
1H ЯМР (300 МГц, CDCl3): δ 2,4 (s, 3Н), 7,54-7,61 (m, 5Н), 9,90 (s, 1H),
Температура плавления: 133-134°C.
Пример 11. Получение трет-бутил4-(4-формил-3-метил-1-фенил-1H-пиразол-5-ил)-пиперазин-1-карбоксилата
Проводили реакцию раствора трет-бутилового сложного эфира пиперазин-1-карбоновой кислоты (2,4 г) в ДМФ (10 мл) с 5-хлор-3-метил-1-фенил-1H-пиразол-4-карбальдегидом (1 г) в присутствии карбоната калия (2,17 г) при 25-30°C. Реакционную смесь нагревали при 120-130°C в течение 15 ч. Реакцию останавливали в воде и подкисляли концентрированной HCl при 25°C, затем реакционную массу трижды экстрагировали этилацетатом. Объединенный слой этилацетата упаривали под вакуумом с получением 1,9 г в качестве остатка, который применяли на следующей стадии. Чистое соединение получали посредством колоночной хроматографии.
1H ЯМР (300 МГц, CDCl3): δ 1,4 (s, 9Н), 2,4 (s, 3Н), 3,07 (bs, 4Н), 3,4 (bs, 4Н), 7,29-7,46 (m, 5Н), 9,94 (s, Н). MS m/z: 371,61 (М+Н)+
Пример 12. Получение 1-(3-метил-1-фенил-1H-пиразол-5-ил)пиперазина
Раствор трет-бутил-4-(4-формил-3-метил-1-фенил-1H-пиразол-5-ил)пиперазин-1-карбоксилата (1,9 г неочищенного вещества) в метаноле (19 мл) обрабатывали PTSA (п-толуолсульфоновая кислота 2,9 г) при 80°C в течение 2-3 ч. После окончания реакции метанольную смесь упаривали и гасили в воде и дважды промывали этилацетатом. Водный слой подщелачивали с применением карбоната натрия и экстрагировали MDC. Объединенный слой MDC упаривали с получением продукта в виде твердого вещества (0,4 г).
1H ЯМР (300 МГц, CDCl3): δ 2,27 (s, 3Н), 2,94-2,86 (m, 8Н), 5,6 (s, Н), 7,26-7,22(t, Н), 7,4-7,38(t, 2Н), 7,77-7,74 (d, 2Н); MS m/z: 243,77 (М+Н)+
Пример 13. 1-[(2S,4S)-4-[4-(3-метил-1-фенил-1H-пиразол-5-ил)пиперазин-1-ил]-2-(1,3-тиазолидин-3-илкарбонил)пирролидин-1-ил]этанон:
Способ А:
К перемешиваемому раствору (5S)-1-ацетил-5-(1,3-тиазолидин-3-илкарбонил)-пирролидин-3-она (5,5 г) и 1-(3-метил-1-фенил-1H-пиразол-5-ил)пиперазина (5,8 г) в дихлорметане (55 мл) добавляли уксусную кислоту (1,15 мл), после чего добавляли триацетоксиборогидрид натрия (7,3 г). Реакционную смесь перемешивали в течение 3-4 ч при комнатной температуре. После окончания реакции добавляли воду и перемешивали в течение 15 минут. Затем водный слой экстрагировали дихлорметаном. Объединенный органический слой промывали водой, после чего промывали солевым раствором и концентрировали при пониженном давлении с получением 8,6 г остатка в виде коричневого масла. Его очищали посредством колоночной хроматографии на силикагеле (5% метанол в этилацетате) с получением титульного соединения (6,6 г).
1H ЯМР (300 МГц, ДМСО-D6): δ 1,48-1,52 (m, 1H), 1,94 (s, 3Н), 2,14 (s, 3Н), 2,5-2,78 (m, 8Н), 2,88-3,2 (m, 3Н), 3,6-4,79 (m, 8Н), 5,79 (s, 1H), 7,27 (t, 1H, J=7,8 Гц), 7,45 (t, 2Н, J=7,8 Гц), 7,77 (d, 2Н, J=7,8 Гц); ESI-MS (m/z): 469,18 [М+Н]+.
Способ В
К перемешиваемому раствору (5S)-1-ацетил-5-(1,3-тиазолидин-3-илкарбонил)-пирролидин-3-она (10 г) и 1-(3-метил-1-фенил-1H-пиразол-5-ил)пиперазина (10 г) в дихлорметане (100 мл) добавляли уксусную кислоту (2,1 мл), после чего добавляли триацетоксиборогидрид натрия (13,2 г). Реакционную смесь перемешивали в течение 3-4 ч при 25-30°C. После окончания реакции добавляли воду и перемешивали в течение 15 минут. Затем водный слой экстрагировали дихлорметаном. Объединенный органический слой промывали водой, после чего промывали солевым раствором и концентрировали при пониженном давлении получением продукта (18 г).
Пример 14: {(2S,4S)-4-[4-(3-метил-1-фенил-1H-пиразол-5-ил)-пиперазин-1-ил]-пирролидин-2-ил}(1,3-тиазолидин-3-ил)метанон (тенелиглиптин):
Способ А
К перемешиваемому раствору 3-((2S,4S)-1-(ацетил)-4-[4-(3-метил-1-фенил-1H-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил)тиазолидина (1,0 г) в IPA (15,0 мл) медленно добавляли водный раствор бромистоводородной кислоты (2,2 мл, 48% в воде) при 80-85°C. Полученную смесь перемешивали в течение 16 ч при 80-85°C. После окончания реакции реакционную смесь полностью выпаривали при пониженном давлении при температуре ниже 50°C. К полученной неочищенной маслянистой массе добавляли воду (10,0 мл) и этилацетат (10,0 мл) и перемешивали. Слои разделяли и к водному слою добавляли твердый бикарбонат натрия для достижения pH 8. Водный слой экстрагировали метилендихлоридом, сушили над сульфатом натрия, и упаривали при пониженном давлении с получением неочищенного маслянистого остатка. Остаток очищали посредством колоночной хроматографии на силикагеле (25% метанол в этилацетате) с получением титульного соединения в виде твердого вещества (360 мг).
1H ЯМР (300 МГц, ДМСО-D6): δ 1,55 (m, 1H), 2,14 (s, 3Н), 2,2-2,22 (m, 1H), 2,43-2,76(m, 9Н), 2,92-3,07 (m, 3Н), 3,63-3,84 (m, 4Н), 4,42-4,66 (m, 2Н), 5,78 (s, 1H), 7,24-7,29 (t, 1H, J=7,5 Гц), 7,43-7,47 (t, 2Н, J=7,8 Гц), 7,72-7,75 (d, 2Н, 8,1 Гц); ESI-MS (m/z): 427,34 [М+Н]+.
{(2S,4S)-4-[4-(3-метил-1-фенил-1H-пиразол-5-ил)-пиперазин-1-ил]пирролидин-2-ил}(1,3-тиазолидин-3-ил)метанон 2,5 гидробромида гидрат.
Вышеуказанный полученный тенелиглиптин (100,0 мг) растворяли в этаноле (1,0 мл) при 80°C и добавляли водный раствор бромистоводородной кислоты (105 мг, 48%) при 80°C. Смесь перемешивали в течение 30 мин и постепенно охлаждали смесь при 25-30°C. Осажденное твердое вещество собирали посредством фильтрации с получением титульного соединения (80 мг) в виде твердого вещества кремового цвета.
1H ЯМР (300 МГц, ДМСО-D6): δ 2,17 (s, 3Н), 3,06-3,14 (m, 6Н), 3,44 (brs, 4Н), 3,7-3,77(m, 2Н), 3,86-4,09 (m, 8Н), 4,44-4,75 (m, 3Н), 5,95 (s, 1H), 7,32 (t, 1H, J=7,8 Гц), 7,48 (t, 2Н, J=7,8 Гц), 7,79 (d, 2Н, 7,8 Гц), 9,19 (brs, 1H), 9,93 (brs, 1H).
Способ В:
К перемешиваемому раствору 3-((2S,4S)-1-(ацетил)-4-[4-(3-метил-1-фенил-1H-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил)тиазолидина (1,0 г) в IPA (10 мл) медленно добавляли водный раствор бромистоводородной кислоты (2,5 мл, 48% в воде) при 80-85°C. Полученную смесь перемешивали в течение 4 ч при 80-85°C. После окончания реакции реакционную смесь охлаждали до 25-30°C и добавляли воду (30 мл), затем перемешивали в течение 15 мин, после чего добавляли этилацетат (15 мл) и перемешивали. Разделяли слои. Водный слой подщелачивали 10% раствором карбоната натрия (8-9 pH). Его экстрагировали метилендихлоридом. Объединенный слой метилендихлорида промывали 15 мл воды и сушили над сульфатом натрия и упаривали при пониженном давлении с получением титульного продукта (0,5 г). Для получения гидробромидной соли его применяли без очистки. Его растворяли в IPA и фильтровали. Фильтрат обрабатывали водным раствором бромистоводородной кислоты (0,5 мл) при 25-30°C в течение 12 ч с получением гидробромидной соли (0,3 г).
Пример 15. Кристаллизация тенелиглиптина 2,5 гидробромида гидрата с применением смеси метанола и трет-бутанола:
Тенелиглиптина 2,5 гидробромида гидрат (23 г) растворяли в трет-бутаноле (92 мл) и метаноле (235 мл) при температуре кипения. Раствор охлаждали до 25-30°C и перемешивали в течение 3-4 часов. Твердое вещество фильтровали и промывали метанолом и сушили при пониженном давлении при 50°C в течение 12 с получением тенелиглиптина 2,5 гидробромида гидрата (19 г). ВЭЖХ чистота 99,43%.
Пример 16. Кристаллизация тенелиглиптина 2,5 гидробромида гидрата с применением смеси н-бутанола и метанола:
Тенелиглиптина 2,5 гидробромида гидрат (27 г) растворяли в н-бутаноле (108 мл) и метаноле (220 мл) при температуре кипения. Раствор охлаждали до 25-30°C и перемешивали в течение 3-4 часов. Твердое вещество фильтровали и промывали метанолом и сушили при пониженном давлении при 50°C в течение 12 ч с получением тенелиглиптина 2,5 гидробромида гидрата (20,5 г). ВЭЖХ чистота 99,6%.
Пример 17. Кристаллизация тенелиглиптина 2,5 гидробромида гидрата с применением смеси DMA и ТГФ:
Тенелиглиптина 2,5 гидробромида гидрат (10,0 г) растворяли в DMA (диметилацетамид 100 мл). К данному раствору добавляли ТГФ (тетрагидрофуран, 350 мл) при 25-30°C. Раствор перемешивали в течение 3-4 часов. Твердое вещество фильтровали и промывали ТГФ и сушили при пониженном давлении при 50°C в течение 12 ч с получением тенелиглиптина 2,5 гидробромида гидрата (9,0 г). ВЭЖХ чистота 99,25%.
Пример 18. Кристаллизация тенелиглиптина 2,5 гидробромида гидрата с применением смеси ДМФ и ТГФ:
Тенелиглиптина 2,5 гидробромида гидрат (10,0 г) растворяли в ДМФ (диметилформамид 100 мл). К данному раствору добавляли ТГФ (тетрагидрофуран, 250 мл) при 25-30°C. Раствор охлаждали до 25-30°C и перемешивали в течение 12-20 часов. Твердое вещество фильтровали и промывали ТГФ и сушили при пониженном давлении при 50°C в течение 12 ч с получением тенелиглиптина 2,5 гидробромида гидрата (8,25 г). ВЭЖХ чистота 99,3%.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ БЕНЗОПИПЕРАЗИН, В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНОВ ВЕТ | 2014 |
|
RU2720237C2 |
| АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 2000 |
|
RU2269525C2 |
| АЗОТСОДЕРЖАЩИЕ БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2835672C1 |
| ИНГИБИТОРЫ MAGL НА ОСНОВЕ ПИРАЗОЛА | 2018 |
|
RU2789157C2 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693629C2 |
| СРЕДСТВА, ИНДУЦИРУЮЩИЕ АПОПТОЗ, ДЛЯ ЛЕЧЕНИЯ РАКА, ИММУННЫХ И АУТОИММУННЫХ ЗАБОЛЕВАНИЙ | 2011 |
|
RU2568611C2 |
| ЛЕКАРСТВЕННЫЕ ФОРМЫ, СОДЕРЖАЩИЕ ОКСАЛАТНЫЕ СОЛИ ТЕНЕЛИГЛИПТИНА И ИХ СОЛЬВАТЫ | 2018 |
|
RU2742418C1 |
| ПРОИЗВОДНЫЕ АЗАСПИРОАЛКАНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕАЗ | 2004 |
|
RU2379303C2 |
| ХИНОЛИНКАРБОКСАМИДНЫЕ И ХИНОЛИНКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ mGLuR2-НЕГАТИВНЫХ АЛЛОСТЕРИЧЕСКИХ МОДУЛЯТОРОВ, КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2610262C2 |
| КРИСТАЛЛИЧЕСКИЕ ПРОИЗВОДНЫЕ 1-МЕТИЛКАРБАПЕНЕМА | 2000 |
|
RU2214411C2 |




Изобретение относится к способу получения тенелиглиптина, соединения формулы I, или его соли, или гидрата. Способ включает а) осуществление взаимодействия соединения формулы 11 с бис-(2-хлорэтил)амином, или его N-защищенным производным, или его солью с получением соединения формулы Int-B, или его N-защищенного производного, или их соли или а) осуществление взаимодействия 5-хлор-3-метил-1-фенил-1Н-пиразол-4-карбальдегида, соединения формулы 30, с трет-бутиловым сложным эфиром пиперазин-1-карбоновой кислоты с получением трет-бутил-4-(4-формил-3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-карбоксилата, соединения формулы 31, с которого далее снимают защиту с получением соединения формулы Int-B; b) осуществление взаимодействия соединения формулы Int-B, или его N-защищенного производного, или их соли с соединением формулы 13 с получением соединения формулы 14, где R представляет собой аминозащитную группу, выбранную из группы, состоящей из аралкила, ацила, низшего алкоксикарбонила, аралкилоксикарбонила, низшего алкансульфонила, арилсульфонила, три-(низший алкил)силила, трифосгена; и c) снятие защиты с соединения формулы 14 с получением тенелиглиптина, соединения формулы I, или его соли, или гидрата. Также изобретение относится к способу получения тенелиглиптина 2,5 гидробромида гидрата, включающему кристаллизацию тенелиглиптина 2,5 гидробромида или его гидрата из растворителя, выбранного из группы, состоящей из метанола, н-бутанола, трет-бутанола, диметилацетамида, диметилформамида, тетрагидрофурана и их смесей. Способы по изобретению позволяют получить тенелиглиптин или его соли с высокими выходами и чистотой. 2 н. и 6 з.п. ф-лы, 5 ил., 18 пр.
 ,
,  ,
, ,
, ,
,  ,
,
1. Способ получения тенелиглиптина, соединения формулы I, или его соли, или гидрата, включающий:

а) осуществление взаимодействия соединения формулы 11 с бис-(2-хлорэтил)амином, или его N-защищенным производным, или его солью с получением соединения формулы Int-B, или его N-защищенного производного, или их соли
 ,
,
или
а) осуществление взаимодействия 5-хлор-3-метил-1-фенил-1Н-пиразол-4-карбальдегида, соединения формулы 30, с трет-бутиловым сложным эфиром пиперазин-1-карбоновой кислоты с получением трет-бутил-4-(4-формил-3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-карбоксилата, соединения формулы 31, с которого далее снимают защиту с получением соединения формулы Int-B
b) осуществление взаимодействия соединения формулы Int-B, или его N-защищенного производного, или их соли с соединением формулы 13 с получением соединения формулы 14

где R представляет собой аминозащитную группу, выбранную из группы, состоящей из аралкила, ацила, низшего алкоксикарбонила, аралкилоксикарбонила, низшего алкансульфонила, арилсульфонила, три-(низший алкил)силила, трифосгена; и
c) снятие защиты с соединения формулы 14 с получением тенелиглиптина, соединения формулы I, или его соли, или гидрата.
2. Способ по п. 1, отличающийся тем, что на стадии b) R представляет собой ацетил или R представляет собой 9-флуоренилметилоксикарбонил, при этом способ включает
осуществление взаимодействия соединения формулы 29 с соединением формулы Int-В с получением соединения формулы 32 или


осуществление взаимодействия соединения формулы 19 с соединением формулы Int-В с получением соединения формулы 20


3. Способ по п. 1, дополнительно включающий обработку тенелиглиптина бромистоводородной кислотой с получением тенелиглиптина 2,5 гидробромида гидрата.
4. Способ по п. 1, отличающийся тем, что снятие защиты проводят реагентом, выбранным из группы, состоящей из кислоты, восстанавливающего агента и основания.
5. Способ получения тенелиглиптина 2,5 гидробромида гидрата, включающий кристаллизацию тенелиглиптина 2,5 гидробромида или его гидрата из растворителя, выбранного из группы, состоящей из метанола, н-бутанола, трет-бутанола, диметилацетамида, диметилформамида, тетрагидрофурана и их смесей.
6. Способ по п. 5, отличающийся тем, что тенелиглиптина 2,5 гидробромида гидрат кристаллизуют из метанола.
7. Способ по п. 1, отличающийся тем, что соединение выбрано из следующих:

8. Способ по п. 1, отличающийся тем, что бис-(2-хлорэтил)амин, или N-boc-бис-(2-хлорэтил)амин, или их соль, или 5-хлор-3-метил-1-фенил-1Н-пиразол-4-карбальдегид, или трет-бутил-4-(4-формил-3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-карбоксилат применяют для получения соединения формулы Int-B, которое применяют для получения тенелиглиптина.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| ЕР 1854795 А1, 14.11.2007 | |||
| Паровая форсунка | 1927 |
|
SU13119A1 |
| ЕР 1894567 А1, 05.03.2008. | |||

