Область техники
Изобретение относится к новым соединениям со спирохиральной углеродной основой, способам их получения и фармацевтическим композициям, содержащим такие соединения.
Уровень техники
Быстрый экономический рост и разработка лекарственных средств в последнее время привели к избыточному питанию и к увеличению количества населения преклонного возраста, результатом чего является ожирение и резкое увеличение числа пациентов с жировой дистрофией печени из-за ожирения и увеличение числа страдающих от остеопороза из-за старения.
В течение длительного времени полагали, что жировая ткань защищает ткани организма и сохраняет тепло тела и является хранилищем энергии для физической активности. Однако результаты многих последних исследований показывают, что жировая ткань играет важную роль в физиологии и генезисе человеческого организма. В частности, обнаружены факты, что материалы, способные регулировать различные виды физиологической активности, такие как энергетический баланс, регулирование сахара в крови, регуляция чувствительности к инсулину, генерация кровеносных сосудов и т.п., например адипсин, TNFa, лептин и т.д., секретируются в адипоцитах один за другим, и, таким образом, адипоциты стали центром внимания в отношении регуляции метаболизма в человеческом организме.
С другой стороны, так как ожирение вызывает тяжелые социальные болезни, активно осуществляется разработка лекарственных средств для ингибирования образования адипоцитов. Однако даже хотя быстрое возрастание числа пациентов с неалкогольной жировой дистрофией печени из-за ожирения отражает серьезную угрозу здоровью современного населения, лекарственное средство для эффективного лечения в таком случае до сих пор не разработано.
Остеопороз является результатом нарушения остеогенного баланса между способностью остеобластов к костеобразованию и костеабсорбирующей способностью остеокластов. Известно, что генерация остеобластов и остеокластов регулируется с учетом гормонов, поступающих извне питательных веществ и генов, но многие гены, которые являются непосредственно причинами костных болезней, пока не обнаружены.
Большинство лекарственных средств, используемых в настоящее время в способах лечения, ингибируют костеабсорбирующую способность костных клеток для уравновешивания с образованием костных клеток. Однако такие лекарственные средства имеют серьезное побочное действие и недостаточный клинический эффект, и поэтому необходима разработка новой концепции лекарственных средств. Хотя многие исследователи пытаются разрабатывать лекарственные средства, способные промотировать образование костных клеток, иными словами, активацию остеобластов, новые лекарственные средства с благоприятным действием пока еще не разработаны.
Описание
Техническая задача
Целью настоящего изобретения является новое соединение с в высшей степени превосходной способностью дифференцировки остеобластов.
Другой целью настоящего изобретения является новое соединение с превосходной способностью ингибировать дифференцировку адипоцитов.
Еще одной целью настоящего изобретения является новое соединение с селективной и превосходной антагонистической активностью против Х-рецептора печени (LXR).
Еще одной целью настоящего изобретения является новое соединение, ингибирующее биосинтез и абсорбцию жира в печени.
Еще одной целью настоящего изобретения является фармацевтическая композиция для лечения остеопороза, жировой дистрофии печени или ожирения, содержащая такое новое соединение в качестве активного компонента.
Техническое решение
В одном общем аспекте изобретение относится к соединению формулы 1, приведенной ниже, его стереоизомеру, его энантиомеру, его предшественнику, способному к гидролизу in vivo, или его фармацевтически приемлемой соли.
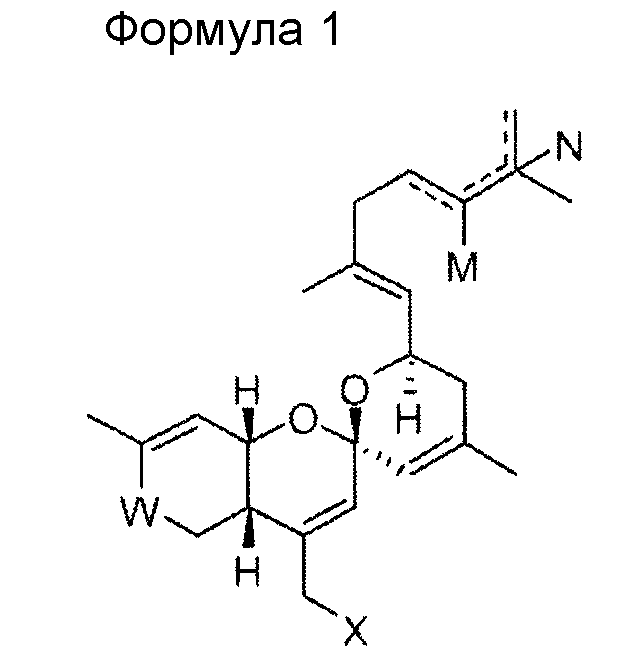
В формуле 1
W представляет собой СО или CHOR1;
Х представляет собой N3, NHR2, OR2, SR2, SeR2 или TeR2;
R1 и R2 выбирают, независимо, из водорода, линейного или разветвленного алкила С1~C8, алкенила С2~C8, алкинила С2~C8, циклоалкила С3~C8, арила С6~С20, гетероарила С4~С20 или 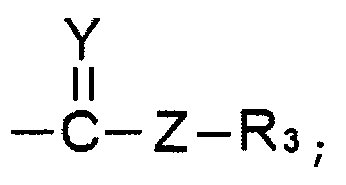
Y представляет собой О, S или NR4;
Z представляет собой простую связь, NH, O, S, Se или Те;
R3 и R4 выбирают, каждый независимо, из водорода, линейного или разветвленного алкила С1~C8, алкенила С2~C8, алкинила С2~C8, циклоалкила С3~C8, арила С6~С20, гетероарила С4~С20; и
М и N представляют собой, каждый независимо, водород, ОН или отсутствуют; при этом атом углерода, связанный с М или N, образует простую связь или двойную связь с другими атомами углерода, и число двойных связей составляет одну или менее для каждого из атомов углерода.
В другом общем аспекте изобретение относится к способу получения соединения формулы 1, включающему
(а) нарезание и сушку губки Phorbas sp. с последующей экстракцией с использованием спирта С1~C4;
(b) обработку экстракта, полученного на стадии (а), с использованием воды и метиленхлорида и затем удаление растворителя из органического слоя с последующей обработкой с использованием н-гексана и смешанного растворителя из метанола и воды; и
(с) удаление растворителя из слоя метанольной аликвоты, полученной на стадии (b), и последующее получение аликвоты хроматографией с использованием диоксида кремния в качестве неподвижной фазы и с использованием метанольного раствора в качестве элюента, причем метанольный раствор содержит или не содержит 20 мас.% или менее воды относительно его общей массы.
В еще одном общем аспекте изобретение относится к фармацевтической композиции для лечения остеопороза, включающей соединение формулы 1, его стереоизомер, его энантиомер, его предшественник, способный к гидролизу in vivo, или его фармацевтически приемлемую соль в качестве фармацевтически приемлемого носителя и активного агента.
В еще одном общем аспекте изобретение относится к фармацевтической композиции для лечения жировой дистрофии печени, включающей соединение формулы 1, его стереоизомер, его энантиомер, его предшественник, способный к гидролизу in vivo, или его фармацевтически приемлемую соль в качестве фармацевтически приемлемого носителя и активного агента.
В еще одном общем аспекте изобретение относится к фармацевтической композиции для лечения ожирения, включающей соединение формулы 1, его стереоизомер, его энантиомер, его предшественник, способный к гидролизу in vivo, или его фармацевтически приемлемую соль в качестве фармацевтически приемлемого носителя и активного агента.
В еще одном общем аспекте изобретение относится к фармацевтической композиции для антагонизации (как антагонисту) Х-рецептора печени (LXR), включающей соединение формулы 1, его стереоизомер, его энантиомер, его предшественник, способный к гидролизу in vivo, или его фармацевтически приемлемую соль в качестве фармацевтически приемлемого носителя и активного агента.
Благоприятные действия
Соединение формулы 1 по настоящему изобретению имеет в высшей степени превосходную способность дифференцировки остеобластов, и поэтому ожидается, что соединение по настоящему изобретению может играть весьма передовую роль в лечении остеопороза. Кроме того, соединение формулы 1 по настоящему изобретению обладает сильной антагонистической эффективностью против Х-рецепторов печени для ингибирования синтеза жира и абсорбции жира в печени, и поэтому ожидается, что соединение по настоящему изобретению может являться весьма эффективным при лечении жировой дистрофии печени.
Более того, соединение формулы 1 по настоящему изобретению обладает превосходной способностью ингибировать дифференцировку адипоцитов, и поэтому ожидается, что соединение по настоящему изобретению можно использовать при лечении ожирения.
Описание чертежей
Указанные выше и другие цели, особенности и преимущества настоящего изобретения станут очевидны из последующего описания предпочтительных воплощений изобретения, приведенных в сочетании с прилагаемыми чертежами, в которых
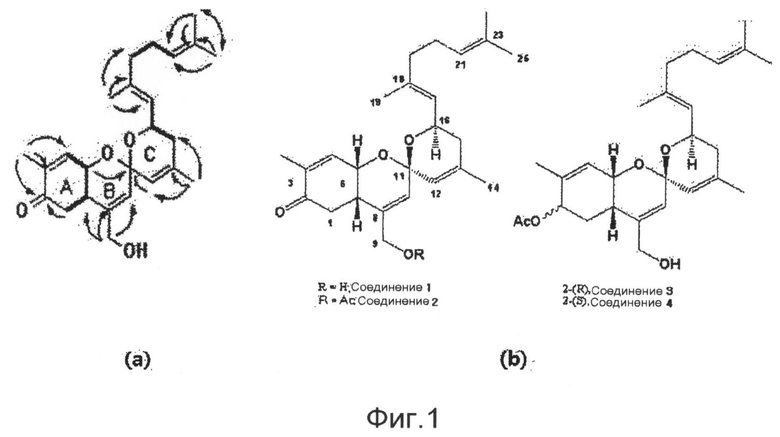
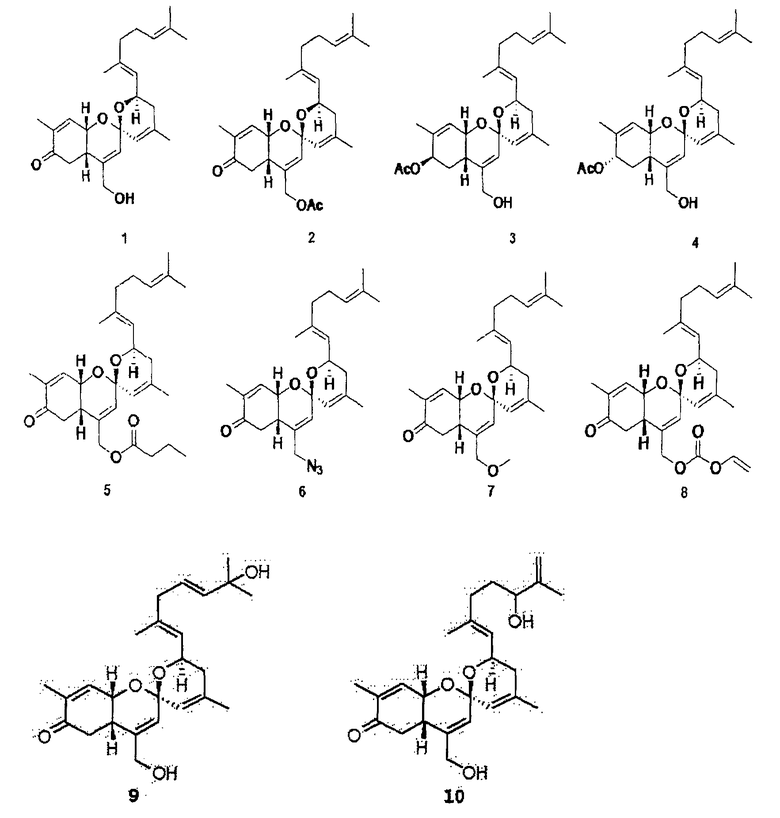
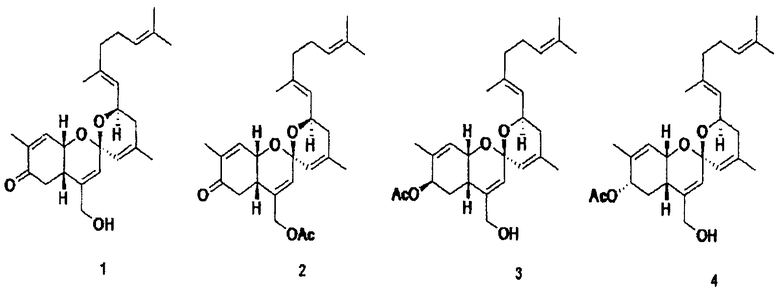
фиг.1 показывает корреляцию водорода (сплошные линии) и корреляцию НМВС (стрелки, показывающие корреляцию связывания от ядра водорода к ядру углерода), полученные экспериментом COSY (а), и показывает структуры соединений 1-4 по настоящему изобретению (b);
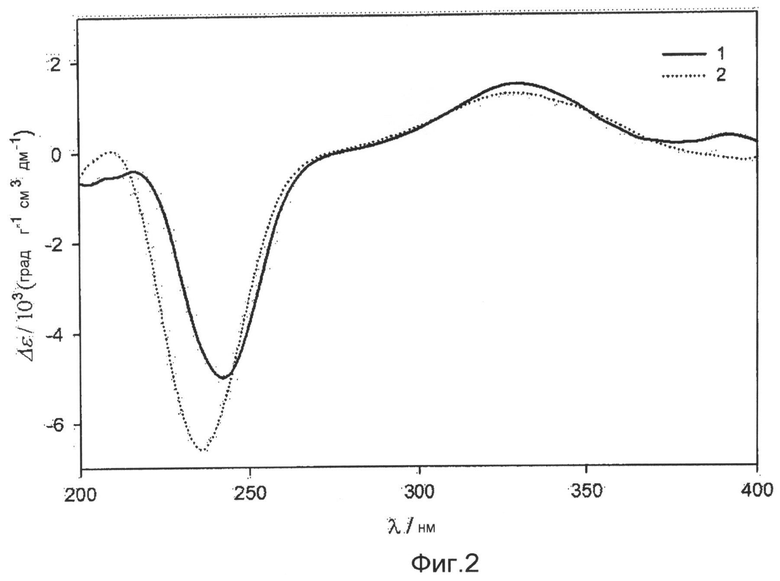
фиг.2 показывает спектры кругового дихроизма, которые дают соединения 1 и 2 по настоящему изобретению;
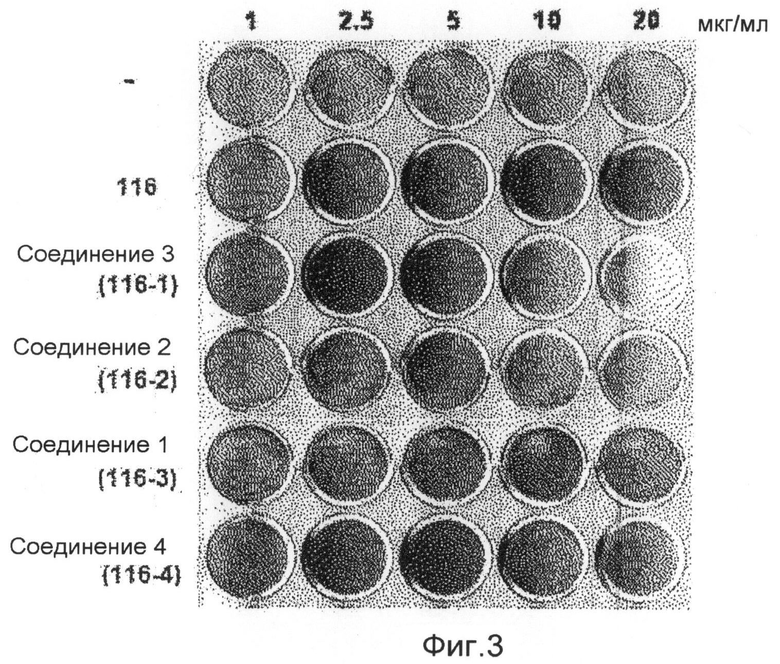
фиг.3 показывает картину, представляющую результаты измерения способности дифференцировки остеобластов аликвоты экстракта 116V и соединений 1-4 по настоящему изобретению (экспериментальный пример 1);
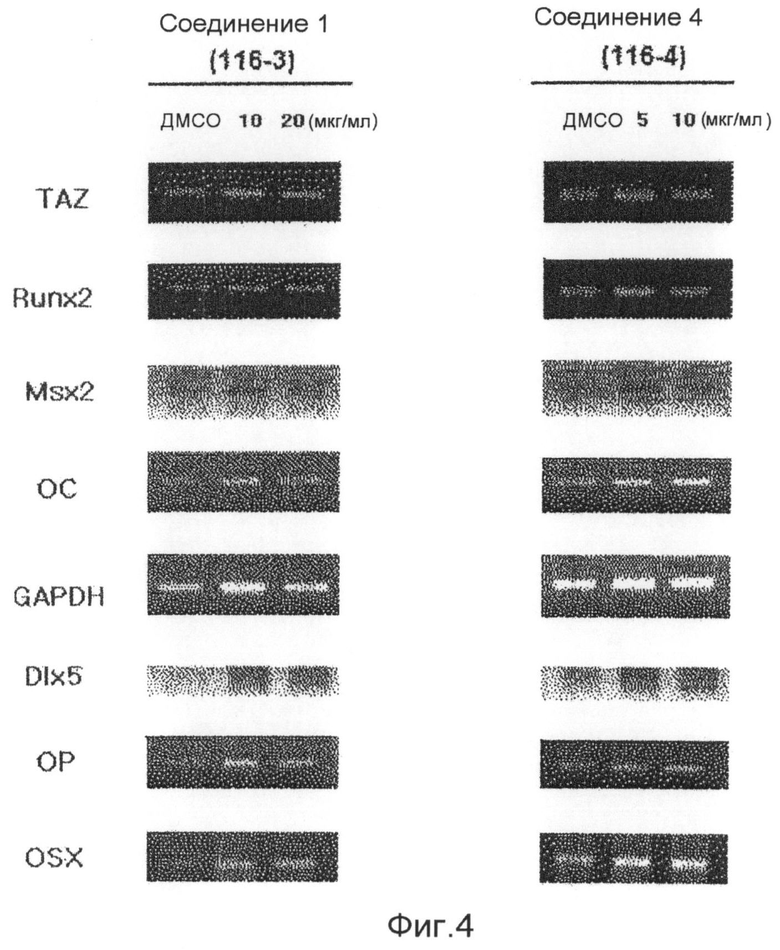
фиг.4 показывает данные ОТ-ПЦР (RTPCR), которые подтверждают степени транскрипции факторов признаков дифференцировки остеобластов (Runx2, остеокальцина, Msx2 и т.д.) в реальном времени ПЦР (ОТ-ПЦР) после обработки клеточных линий С3Н/10Т1/2 соединениями 1-4 по настоящему изобретению в течение 6 суток (экспериментальный пример 1);
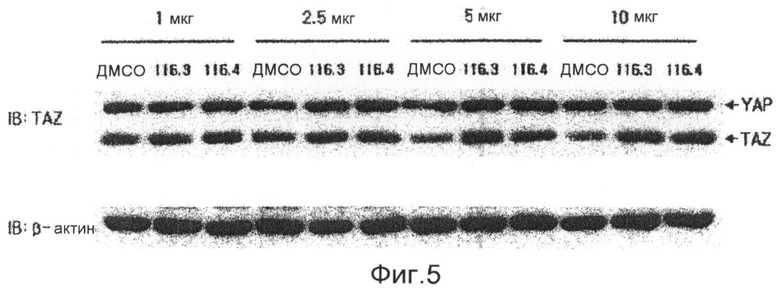
фиг.5 показывает данные вестерн-блоттинга, которые подтверждают экспрессию белка фактора признаков дифференцировки остеобластов TAZ, с использованием вестерн-блоттинга после обработки клеточных линий С3Н/10Т1/2 соединениями 1-4 по настоящему изобретению в течение 6 суток (экспериментальный пример 1);
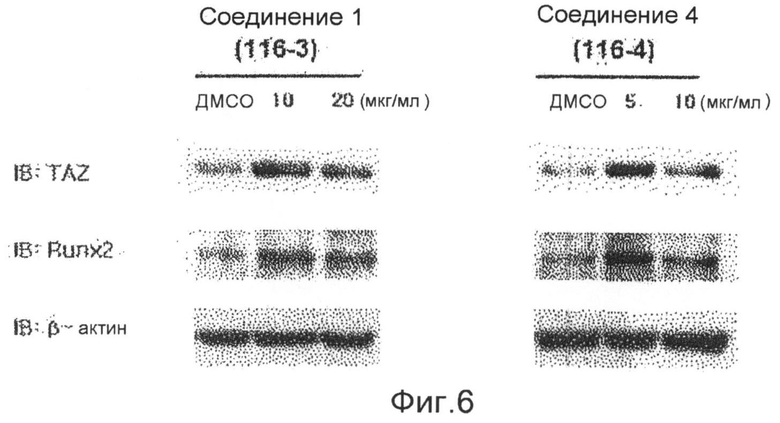
фиг.6 показывает данные вестерн-блоттинга, которые подтверждают экспрессию белков факторов признаков дифференцировки остеобластов TAZ и Runx2, с использованием вестерн-блоттинга после обработки клеточных линий С3Н/10Т1/2 соединениями 1-4 по настоящему изобретению в течение 6 суток (экспериментальный пример 1);
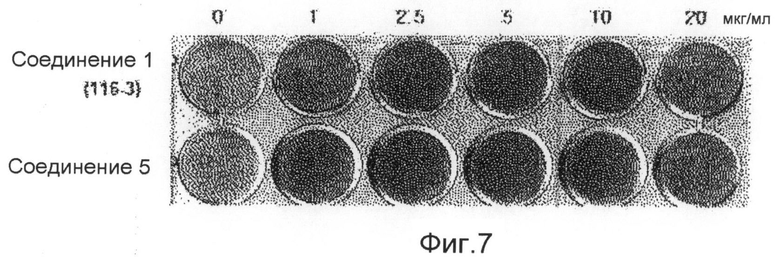
фиг.7 показывает картину, представляющую результат измерения способности дифференцировки остеобластов по соединению 5 по настоящему изобретению в экспериментальном примере 1;
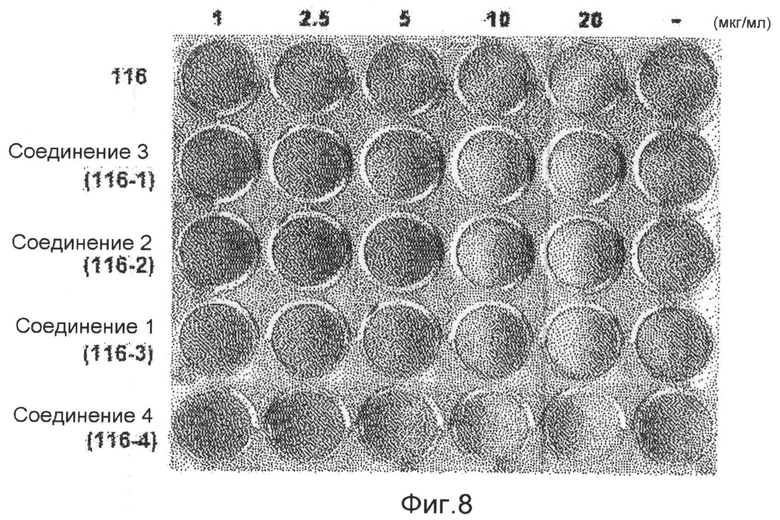
фиг.8 показывает картину, представляющую результаты измерения способности дифференцировки адипоцитов (С3Н/10Т1/2) аликвоты экстракта 116V и соединений 1-4 по настоящему изобретению в экспериментальном примере 2;
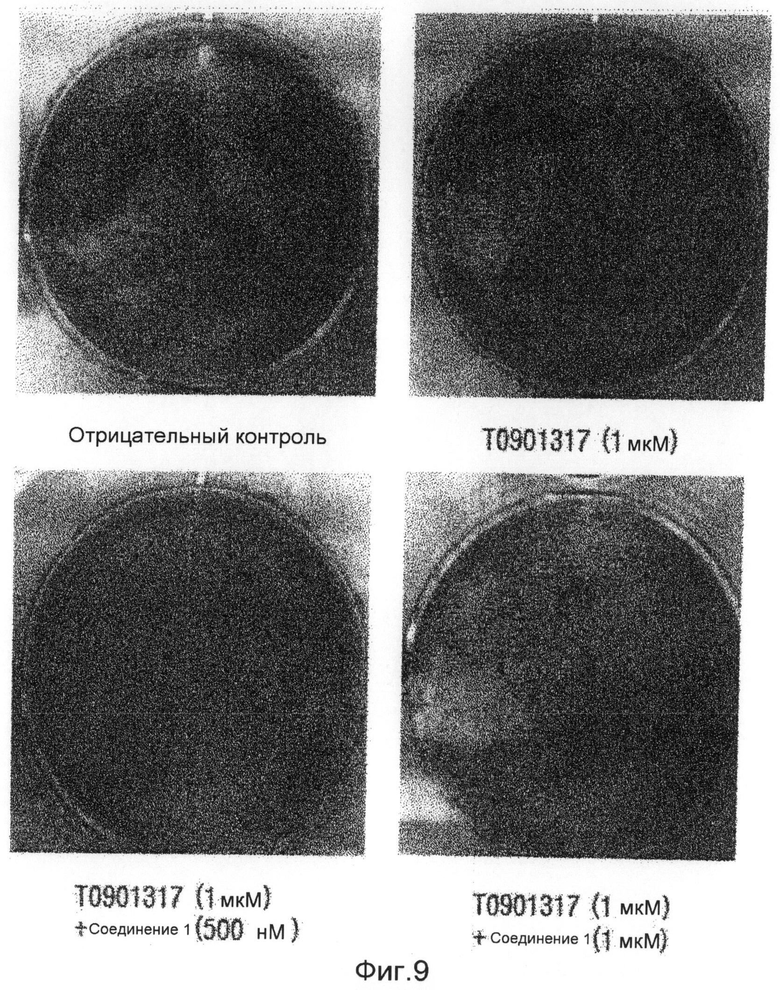
фиг.9 показывает картину, представляющую результаты измерения способности ингибировать дифференцировку адипоцитов (3Т3-L1) соединения 1 по настоящему изобретению в экспериментальном примере 2;
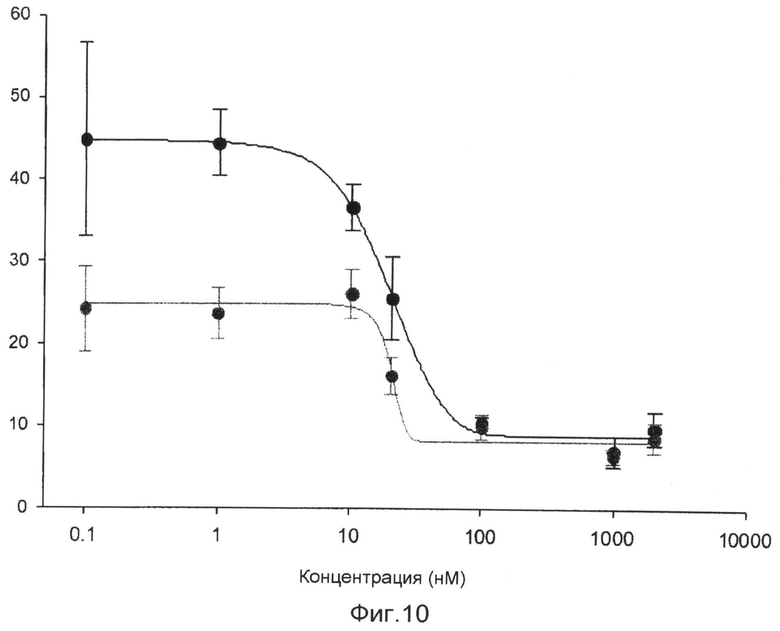
фиг.10 показывает график, представляющий результат измерения антагонистической активности соединения 1 по настоящему изобретению против ядерного рецептора LXR в экспериментальном примере 3;
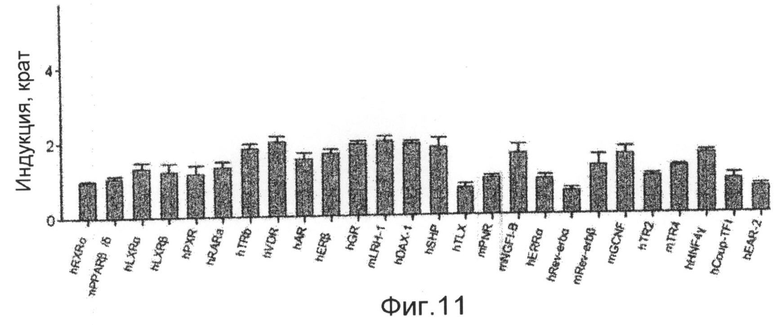
фиг.11 показывает график, представляющий результаты измерения селективной активности соединения 1 по настоящему изобретению в отношении различных ядерных рецепторов в экспериментальном примере 3;
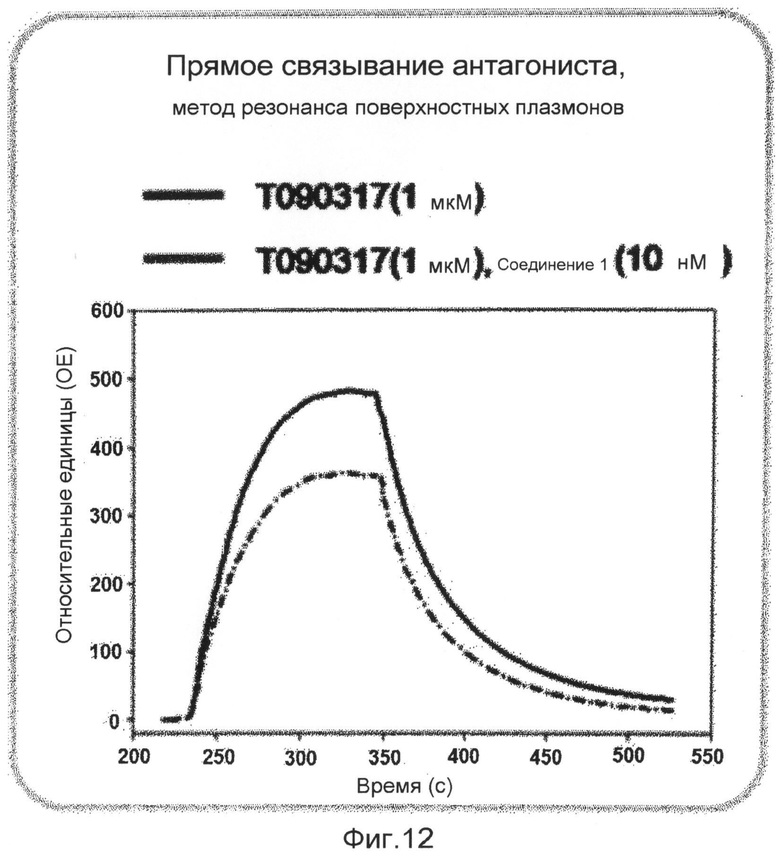
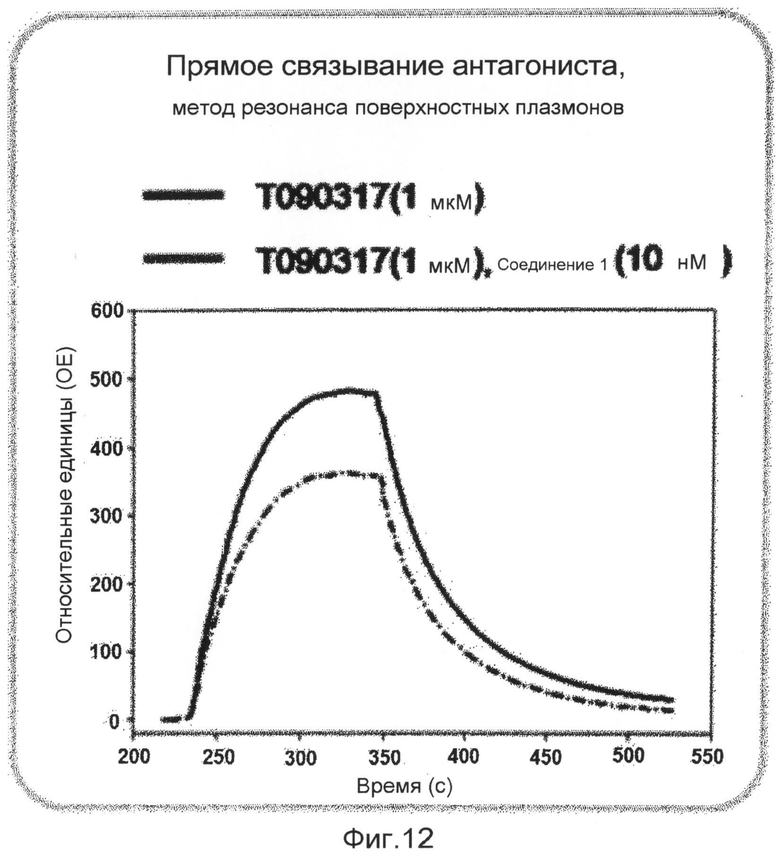
фиг.12 показывает график, представляющий результаты измерения непосредственного связывания соединения 1 по настоящему изобретению на белке ядерного рецептора LXR в экспериментальном примере 3;
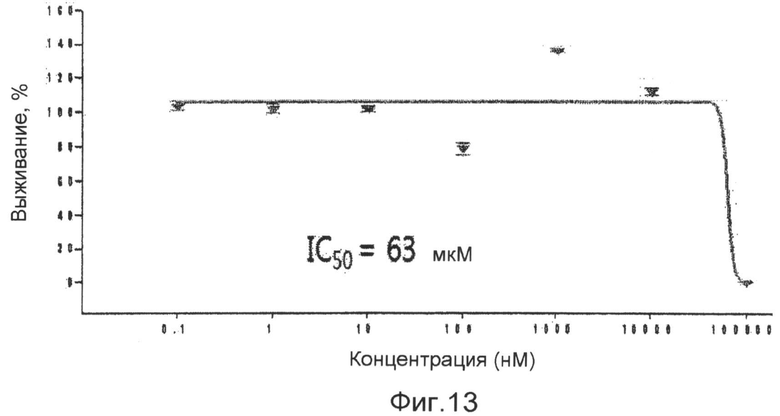
фиг.13 показывает график, представляющий результат измерения цитотоксичности соединения 1 по настоящему изобретению на клетках селезенки мыши в экспериментальном примере 4;
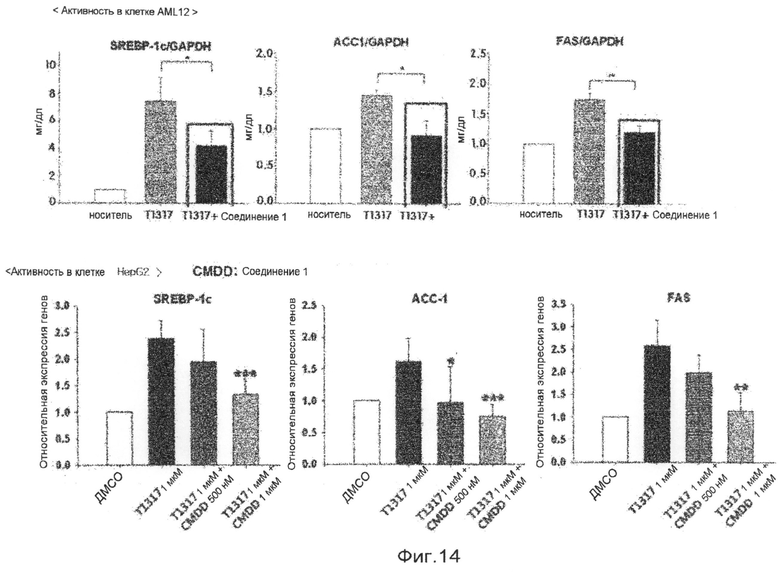
фиг.14 показывает диаграммы, представляющие результаты измерения регуляции экспрессии генов соединением 1 по настоящему изобретению в клетках печени (клетки AML12 и HepG2) в экспериментальном примере 5;
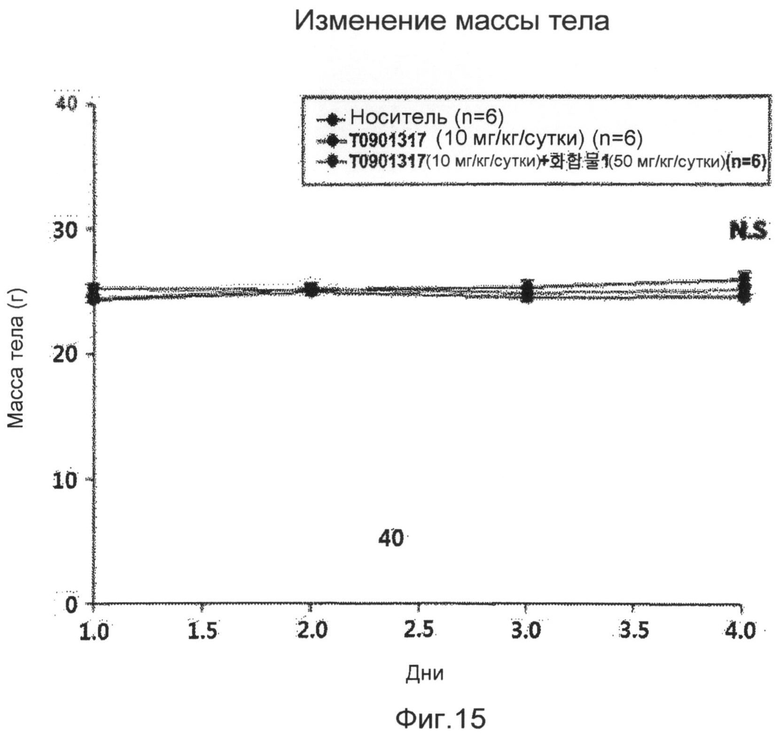
фиг.15 показывает график, представляющий изменения массы тела в периоды введения для групп обработки и контроля, когда мышам в экспериментальном примере 6 вводят соединение 1 по настоящему изобретению;
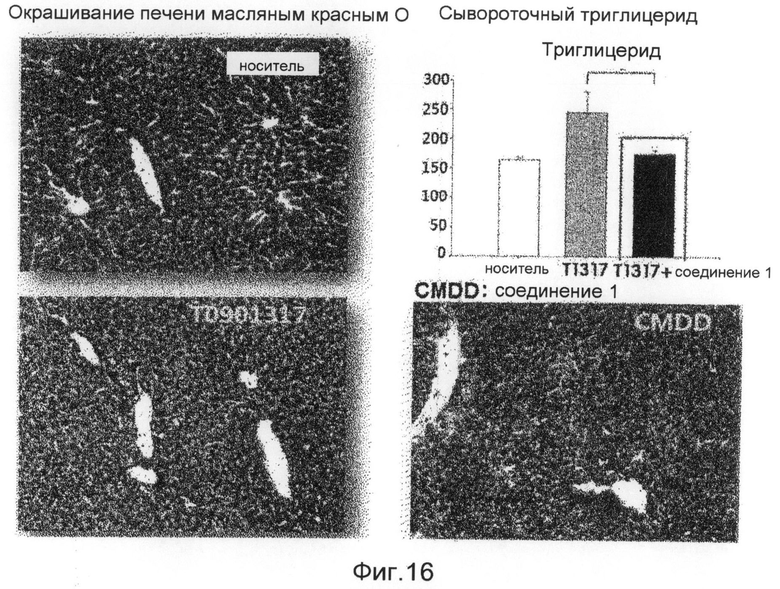
фиг.16 показывает диаграмму и картины, представляющие эффективность ингибирования жировой дистрофии печени соединением 1 по настоящему изобретению на животной модели заболевания в экспериментальном примере 6; и
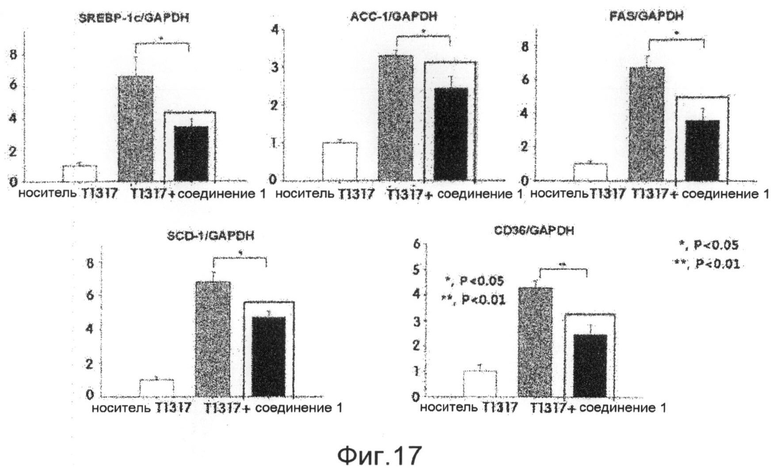
фиг.17 показывает диаграммы, представляющие результаты измерения эффективности регуляции экспрессии генов, проявляемой соединением 1 по настоящему изобретению, на животной модели заболевания после того, как указанная эффективность на таких животных подтверждена в ходе экспериментального примера 6.
Наилучший способ осуществления изобретения
Настоящее изобретение относится к соединению приведенной ниже формулы 1, его стереоизомеру, его энантиомеру, его предшественнику, способному к гидролизу in vivo, или его фармацевтически приемлемой соли.
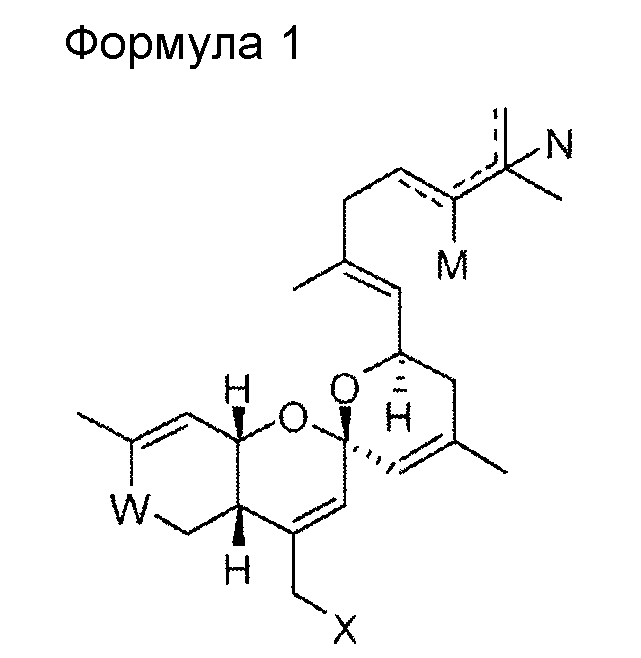
В формуле 1
W представляет собой СО или CHOR1;
Х представляет собой N3, NHR2, OR2, SR2, SeR2 или TeR2;
R1 и R2 выбирают, независимо, из водорода, линейного или разветвленного алкила С1~C8, алкенила С2~C8, алкинила С2~C8, циклоалкила С3~C8, арила С6~С20, гетероарила С4~С20 или 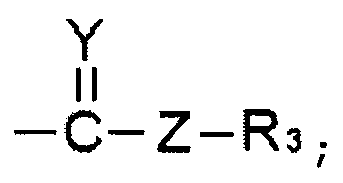
Y представляет собой О, S или NR4;
Z представляет собой простую связь, NH, O, S, Se или Те;
R3 и R4 выбирают, каждый независимо, из водорода, линейного или разветвленного алкила С1~C8, алкенила С2~C8, алкинила С2~C8, циклоалкила С3~C8, арила С6~С20 или гетероарила С4~С20; и
М и N представляют собой, каждый независимо, водород, ОН или отсутствуют; при этом атом углерода, связанный с М или N, образует простую связь или двойную связь с другими атомами углерода, и число двойных связей составляет одну или менее для каждого из атомов углерода.
Соединение формулы 1 выделяют из материала (KNUE116), экстрагированного из Phorbas sp., обитающей в стране, или синтезируют с использованием выделенного соединения в качестве исходного материала, и нового соединения со спирохиральной углеродной основой. Соединение формулы 1 по новому промотирует дифференцировку остеобластов, прекрасно ингибирует способность адипоцитов к дифференцировке и подавляет синтез жира и абсорбцию жира в печени. Поэтому ожидается, что соединение формулы 1 может играть передовую роль при лечении остеопороза, лечении жировой дистрофии печени и лечении ожирения.
Конкретные примеры соединений формулы 1 приводятся далее.
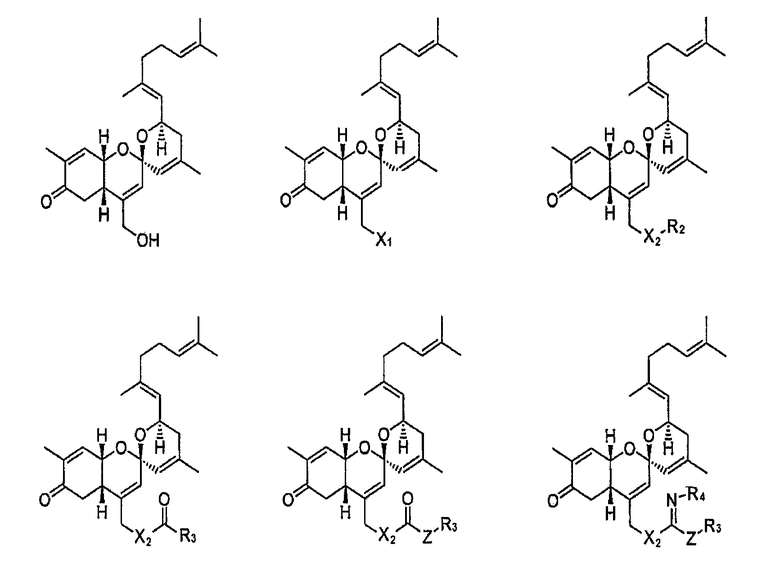
В указанных выше формулах
Х1 представляет собой N3, NH2, OH, SH, SeH или TeH;
Х2 представляет собой NH, O, S, Se или Te;
Z представляет собой простую связь, NH, O, S, Se или Те;
R1 представляет собой водород, линейный или разветвленный алкил С1~C8, алкенил С2~C8, алкинил С2~C8, циклоалкил С3~C8, арил С6~С20, гетероарил С4~С20 или 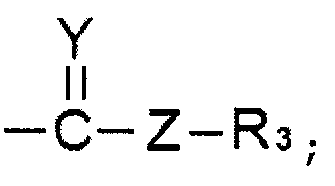 и
и
каждый из R2, R3 и R4 представляет собой водород, линейный или разветвленный алкил С1~C8, алкенил С2~C8, алкинил С2~C8, циклоалкил С3~C8, арил С6~С20 или гетероарил С4~С20.
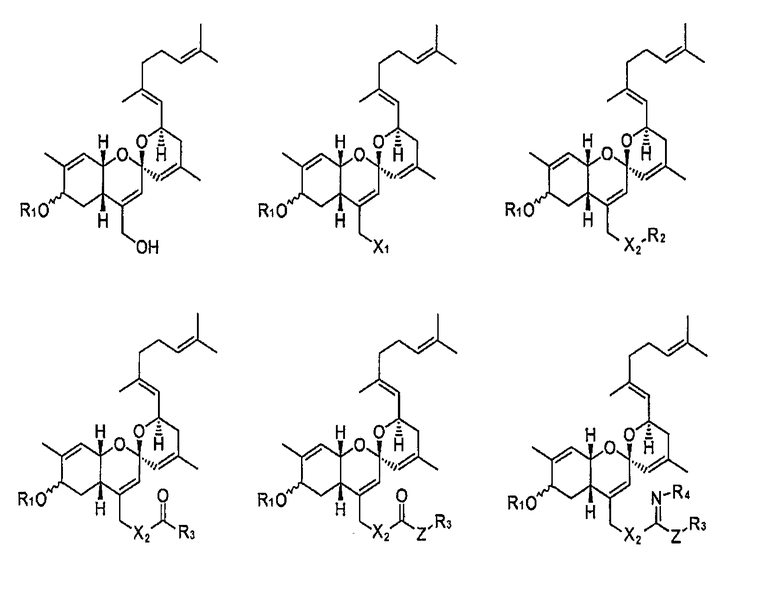
В другом предпочтительном соединении из числа соединений указанной выше формулы 1 W представляет собой СО или CHOR1; Х представляет собой N3, NHR2, OR2, SR2, SeR2 или TeR2; R1 и R2 выбирают, независимо, из водорода, линейного или разветвленного алкила С1~C8, алкенила С2~C8, алкинила С2~C8 или Y представляет собой О, S или NR4; Z представляет собой простую связь, NH, O или S; R3 и R4 выбирают, каждый независимо, из водорода, линейного или разветвленного алкила С1~C8, алкенила С2~C8 или алкинила С2~C8; и М и N представляют собой, каждый независимо, водород, ОН или отсутствуют, при этом атом углерода, связанный с М или N, образует простую связь или двойную связь с другими атомами углерода, и число двойных связей составляет одну или менее для каждого из атомов углерода.
В другом предпочтительном соединении из числа соединений указанной выше формулы 1, где W представляет собой СО или CHOR1, Х представляет собой N3, OR2 или SR2; R1 и R2 выбирают, каждый независимо, из водорода, линейного или разветвленного алкила С1~C8, алкенила С2~C8, алкинила С2~C8 или Y представляет собой О или S; Z представляет собой простую связь; R3 выбирают из водорода, линейного или разветвленного алкила С1~C8, алкенила С2~C8 или алкинила С2~C8; и М и N представляют собой, каждый независимо, водород, ОН или отсутствуют; при этом атом углерода, связанный с М или N, образует простую связь или двойную связь с другими атомами углерода, и число двойных связей составляет одну или менее для каждого из атомов углерода.
Конкретные примеры соединений формулы 1 приводятся далее.
Далее настоящее изобретение относится к способу получения соединения формулы 1.
Способ получения по настоящему изобретению включает:
(а) нарезание и сушку губки Phorbas sp. с последующей экстракцией с использованием спирта С1~C4;
(b) обработку экстракта, полученного на стадии (а), с использованием воды и метиленхлорида и затем удаление растворителя из органического слоя снова с последующей обработкой с использованием н-гексана и смешанного растворителя из метанола и воды; и
(с) удаление растворителя из слоя метанольной аликвоты, полученной на стадии (b), и последующее получение аликвоты хроматографией с использованием диоксида кремния в качестве неподвижной фазы и с использованием метанольного раствора в качестве элюента, причем метанольный раствор содержит или не содержит 20 мас.% или менее воды относительно его общей массы.
Также способ получения может дополнительно включать после стадии (с) стадию (d) очистку аликвоты, полученной на стадии (с).
На стадии (а) для сушки можно использовать сушку вымораживанием, и в качестве спирта С1~С4 можно использовать метанол. Экстракцию можно выполнять при комнатной температуре, и предпочтительно, в течение 2 часов или более.
На стадии (b) смешанный растворитель из метанола и воды может содержать 60~90 мас.% метанола и 10~40 мас.% воды относительно общей массы растворителя.
На стадии (с) можно выполнять флэш-хроматографию с обращенной фазой. Хроматографию можно выполнять один раз или более в порядке от элюента, имеющего наивысшую полярность, до элюента, имеющего наименьшую полярность, путем использования в качестве элюента смешанного растворителя из воды и метанола с наивысшей полярностью перед использованием в качестве элюента метанольного раствора, содержащего или не содержащего 20 мас.% или менее воды от общей массы элюента. В частности, в качестве элюента можно использовать смесь воды и метанола.
На стадии (d) очистку можно выполнять высокоэффективной жидкостной хроматографией (ВЭЖХ), и в качестве элюента можно использовать смесь 50~80 мас.% ацетонитрила (ACN) и 20~50 мас.% воды относительно общей массы элюента.
Между тем, соединения формулы 1 по настоящему изобретению можно синтезировать с использованием в качестве исходного материала соединений, выделенных вышеуказанными способами, и такого способа как реакция этерификации (получения сложного эфира), реакция азидного замещения, реакция этерификации (получения простого эфира) или подобная реакция.
Кроме того, настоящее изобретение относится к фармацевтической композиции для лечения остеопороза, жировой дистрофии печени и ожирения, включающей соединение формулы 1, его стереоизомер, его энантиомер, его предшественник, способный к гидролизу in vivo, или его фармацевтически приемлемую соль в качестве фармацевтически приемлемого носителя и активного агента.
Кроме того, настоящее изобретение относится к фармацевтической композиции для антагонизации Х-рецептора печени (LXR), включающей соединение формулы 1, его стереоизомер, его энантиомер, его предшественник, способный к гидролизу in vivo, или его фармацевтически приемлемую соль в качестве фармацевтически приемлемого носителя и активного агента.
В фармацевтической композиции фармацевтически приемлемая соль может представлять собой носитель или среду, применимые при введении лекарственных средств, и можно использовать без ограничения любой материал, обычно используемый в технике. Например, можно использовать растворитель, диспергатор, наполнители, сухие разбавители, связующие, смачивающие вещества, вещества, способствующие рассыпанию, поверхностно-активные вещества или подобное.
Фармацевтическую композицию по настоящему изобретению можно получить в формате перорального препарата, такого как порошок, гранула, таблетка, капсула, суспензия, эмульсия, сироп, аэрозоль, или подобного, препарата для наружного применения, суппозитория, стерильного раствора для инъекций или подобном формате.
Дозировка соединения формулы 1, его стереоизомера, его энантиомера, его предшественника, способного к гидролизу in vivo, или его фармацевтически приемлемой соли по настоящему изобретению может изменяться в зависимости от состояния, массы тела и степени заболевания пациентов, типов препаратов лекарственных средств, способов введения и периодов введения, но может быть правильно установлена специалистами в данной области техники. Например, может назначаться дозировка от 0,01 мг/кг до 200 мг/кг в сутки. Введение может осуществляться один раз в сутки или несколько раз в сутки. Соответственно, дозировка не ограничивает объем настоящего изобретения в каком-либо аспекте.
Фармацевтическую композицию по настоящему изобретению можно вводить млекопитающим, таким как крысы, мыши, домашний скот, люди и т.п., различными способами. Все типы введения, известные ранее, могут быть использованы, например, для введения можно использовать ректальный, внутривенный, внутримышечный, подкожный, интраутериндуральный путь или интрацеребровентрикулярную инъекцию.
Предпочтительный вариант осуществления изобретения
Далее в данном описании будут подробно описываться воплощения настоящего изобретения с обращением к прилагаемым чертежам. Однако воплощения используются для пояснения настоящего изобретения на примерах, и настоящее изобретение может различным образом модифицироваться и изменяться, не будучи ограничено указанными воплощениями.
Пример 1. Выделение и очистка новых соединений
Губку Phorbas sp., обитающую в стране, собирают с использованием кожаных скуб, режут на куски размером примерно 10 см или меньше и сушат вымораживанием в течение 3 суток, и получают сухие материалы с массой сухого вещества примерно 1 кг. К высушенным материалам добавляют 3,0 л метанола и затем выполняют экстракцию при комнатной температуре в целом дважды в течение 2 суток. Экстракт обрабатывают с использованием воды и метиленхлорида и затем из органического слоя удаляют растворитель вакуумным испарением с последующей обработкой с использованием н-гексана и смешанного растворителя с 85 мас.% метанола и 15 мас.% воды. Из слоя аликвоты с 85 мас.% метанола удаляют растворитель и получают аликвоту примерно в 5 г. С полученной аликвотой выполняют флэш-хроматографию с обращенной фазой на диоксиде кремния. В данном случае для обращенной фазы в качестве неподвижной фазы используют диоксид кремния С18 и используют элюент в порядке от высокой полярности до низкой полярности, иными словами, в порядке 50% воды/50% метанола, 40% воды/60% метанола, 30% воды/70% метанола, 20% воды/80% метанола, 10% воды/90% метанола, 100% метанола и 100% ацетона. Измеряют способность дифференцировки остеобластов материала, соответствующего каждому слою. Результаты показывают, что способность дифференцировки остеобластов обнаруживается в аликвоте 10% воды/90% метанола (116V) и аликвоте 100% метанола (116VI), каждую из двух аликвот получают в количестве примерно 1 г.
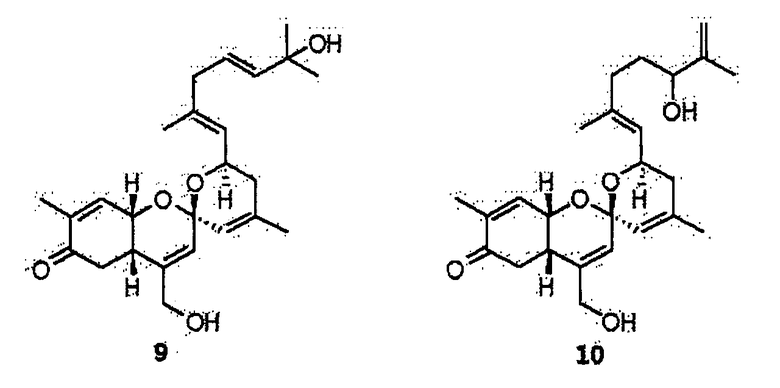
Для того чтобы очистить соединения из двух аликвот с активностью, выполняют полупрепаративную ВЭЖХ с обращенной фазой. Сначала выполняют хроматографию аликвоты 116V в условиях, указанных далее, и получают соединения 1, 9 и 10.
[Колонка YMC ODSC18, диаметр частиц 5 мкм, размер колонки 250×10 мм (длина × диаметр), скорость элюции 2,0 мл/мин, детектор с измерением показателя преломления, элюент - смесь 65 мас.% ацетонитрила (ACN) и 35 мас.% воды.]
Когда впрыскивают 50 мг такой аликвоты, выделяют компоненты в форме масла оранжевого цвета при времени удерживания примерно 33 минуты (соединение 1), 15 минут (соединение 9) и 40 минут (соединение 10) в количествах 25 мг, 1,5 мг и 1,0 мг, соответственно. Такую же ВЭЖХ также используют для аликвоты 116VI, но для разделения аддитивных компонентов используют другие условия в отношении проявляющего растворителя. В данном случае используют смесь 70 мас.% ацетонитрила и 30 мас.% воды. Общее время проявления при каждой обработке составляет примерно полтора часа. Когда за один раз впрыскивают 5 мг аликвоты жидкости 116VI, соединение 2 в форме масла оранжевого цвета, соединение 3 и, в заключение, соединение 4 выделяют, очищают и получают при времени удержания примерно 1 час 10 минут, 1 час 40 минут и, в заключение, 1 час 57 минут в количествах 0,5 мг, 0,08 мг и, в заключение, 0,004 мг.
Пример 2. Анализ химических структур новых соединений
Сначала измеряют спектры водородного ядерного магнитного резонанса полученных из аликвот 116V и 116VI соединений 1-4, 9 и 10 для проверки их чистоты, и затем получают спектроскопические данные с использованием приборов, указанных далее. Масс-спектрометр (спектрометр JMS700 от Jeol Inc.) используют для измерения молекулярной массы соответствующих соединений, и затем используют спектрометр ядерного магнитного резонанса (спектрометр VNMRS 500 от Varian Inc.) для анализа их точных химических структур. Кроме того, используют спектрометр Cary50 (от Varian Inc.) и спектрометр FT_IR 4100 (от JACSO Inc.) для измерения ультрафиолетовых полос поглощения и инфракрасных полос поглощения молекул соединений, соответственно, и используют поляризатор Р1010 (JACSO Inc.) для измерения их углов поляризации.
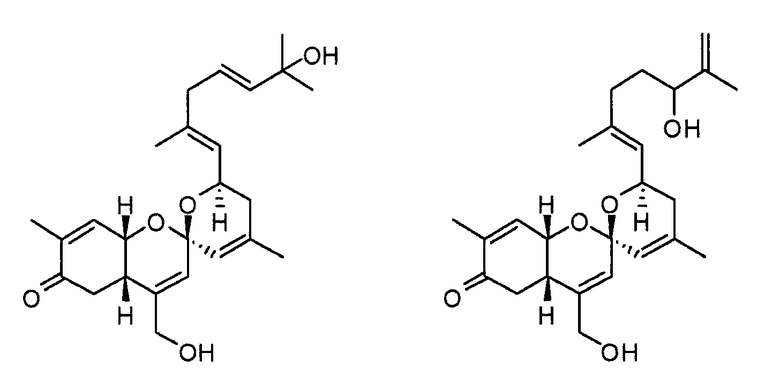
Соединение 1 выделяют в форме масла бледно-оранжевого цвета, и данные высокоэффективной FAB-масс-спектроскопии ([M+H]+ m/z 399,2533) показывают, что соединение 1 имеет молекулярную формулу С25Н34О4. Из характеристических полос поглощения анализом инфракрасного спектра при 3433 см-1 и 1680 см-1 полагают, что соединение 1 содержит гидроксильную группу и карбонильную функциональную группу. Для определения структуры соединения используют С13 ЯМР и Н ЯМР.
Величины химических сдвигов для соединения 1 суммированы ниже в таблице 1.

Выполняют эксперимент ROESY для определения относительной стереоструктуры данного соединения. Из NOE между водородом (4,49 ч/млн) и водородом (2,59 ч/млн) определяют, что циклы А и В связаны в цис-конфигурации. Стереоконфигурацию цикла С можно определить согласно информации NOE между водородом (5,28 ч/млн) и водородом (5,54 ч/млн) и между водородом (5,28 м.д.) и водородом (2,43 ч/млн). Наконец, пространственную конфигурацию водорода у С-16 можно оценить из констант взаимодействия (11,3, 7,8, 3,4 Гц) между близкими атомами водорода, которая косвенно доказывается фактом, что водород Н-19 метила имеет соотношения NOE с Н-5 и Н-6.
Абсолютную стереохимическую структуру соединения 1 определяют анализом спектра кругового дихроизма (CD). Абсолютную стереоконфигурацию хиральных центров в циклогексиноне определяют согласно правилу секторов Snatzke. Когда хиральный центр С-7 в циклогексиноне цикла А имеет абсолютную (S) стереоконфигурацию, соединение 1 обнаруживает в спектре кругового дихроизма положительное поглощение в переходной области nπ* (330 нм-1~350 нм-1). Так как соединение 1 обнаруживает положительное поглощение при 330 нм-1~350 нм-1, абсолютную конфигурацию С-7 в соединении 1 определяют как (S)-стереоконфигурацию (фиг.2).
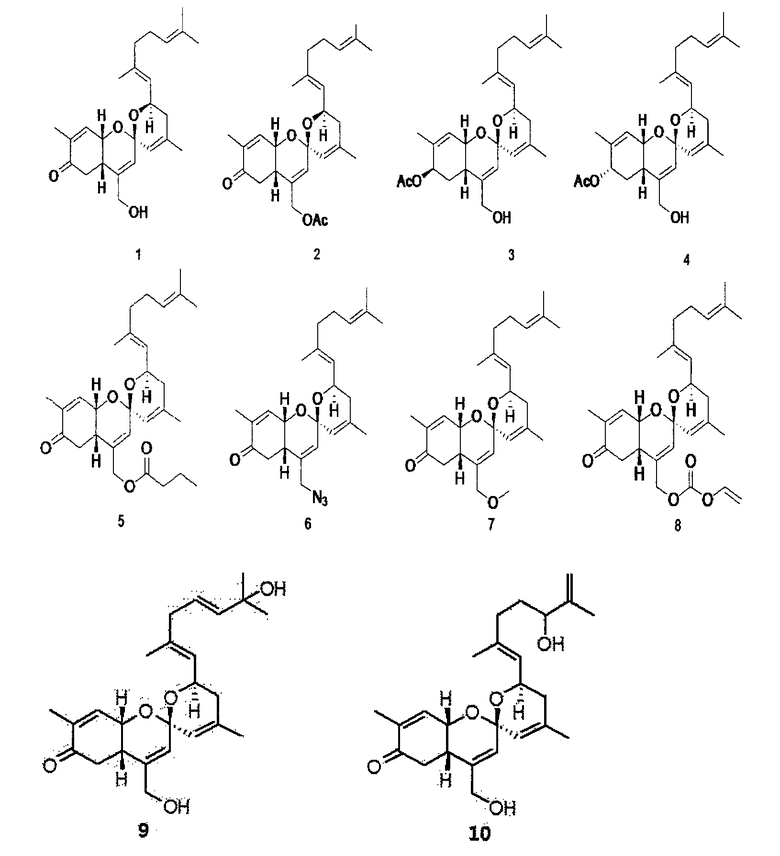
Химические структуры других пяти соединений определяют с использованием вышеуказанного способа. Данные углеродного ЯМР и данные водородного ЯМР соответствующих трех соединений приводятся ниже в таблицах 2-4, и физические и спектроскопические данные сведены в таблице 5.


желтый
поглощения
(см-1)
поглощения
(нм)
в МеОН
(с 0,15)
(с 0,15)
(с 0,10)
(с 0,10)
(с 0,15)
(с 0,15)
Пример 3. Синтез производного соединения 1 реакцией этерификации (получения сложного эфира)
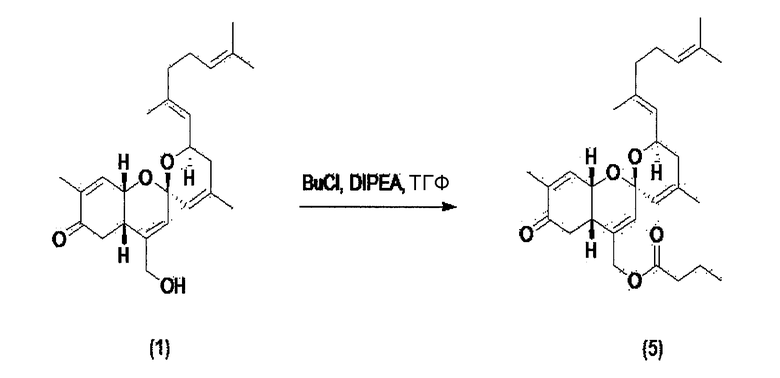
Соединение 1 по настоящему изобретению растворяют в тетрагидрофуране, и затем температуру понижают до 0~5°С. К раствору последовательно добавляют диизопропилэтиламин и бутирилхлорид. Полученный материал перемешивают при 0~5°С в течение 1 часа и экстрагируют, добавляя этилацетат и воду, и затем слой органического растворителя отделяют и перегоняют. Оставшийся материал очищают колоночной флэш-хроматографией, и получают соединение 5.
Соединение 5 (С29Н41О5): [M+H]+ = 469,29.
Пример 4. Синтез производного соединения 1 реакцией азидного замещения
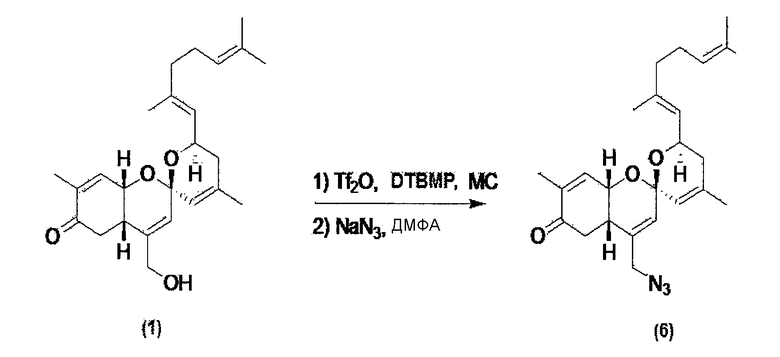
Соединение 1 по настоящему изобретению растворяют в метиленхлориде, и затем температуру понижают до 0~5°С. К раствору последовательно добавляют ди-трет-бутилметилпиридин и трифторметансульфоновый ангидрид. Полученный материал перемешивают в течение 30 минут и экстрагируют, добавляя метиленхлорид и воду. Слой органического растворителя отделяют и перегоняют, и весь растворитель выпаривают. Оставшийся материал снова растворяют в диметилформамиде, и к раствору добавляют азид натрия. Полученный материал перемешивают при комнатной температуре в течение 3 часов, разбавляют, добавляя метиленхлорид, и затем несколько раз промывают водой. Слой органического растворителя отделяют и перегоняют, и затем оставшийся материал очищают колоночной флэш-хроматографией, и получают соединение 6.
Соединение 6 (C25H34N3O3): [M+H]+ = 424,26.
Пример 5. Синтез простого эфирного производного соединения 1
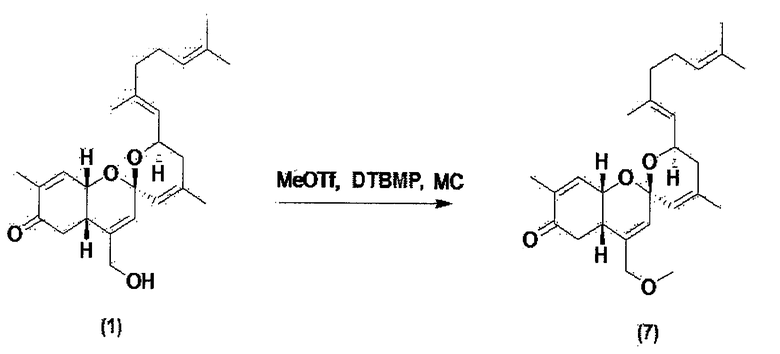
Соединение 1 по настоящему изобретению и ди-трет-бутилметилпиридин растворяют в метиленхлориде, и затем к раствору добавляют метантрифторсульфонат. Полученный материал перемешивают при комнатной температуре в течение 3 часов, и затем растворитель выпаривают. Оставшийся материал очищают колоночной флэш-хроматографией и получают соединение 7.
Соединение 7 (С26Н37О4): [M+H]+ = 413,27.
Пример 6. Синтез карбонатного производного соединения 1
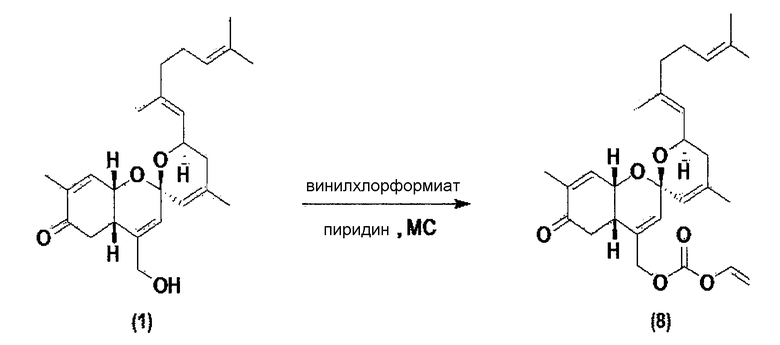
Соединение 1 по настоящему изобретению растворяют в метиленхлориде и затем температуру понижают до 0~5°С. К раствору последовательно добавляют пиридин и винилхлорформиат. Полученный материал перемешивают при комнатной температуре в течение 1 часа, разбавляют метиленхлоридом и затем промывают водой. Слой органического растворителя отделяют и перегоняют, и затем оставшийся материал очищают колоночной флэш-хроматографией, и получают соединение 8.
Соединение 8 (С28Н37О6): [M+H]+ = 469,26.
Экспериментальный пример 1. Измерение активности новых соединений в отношении образования остеобластов (анализ с осаждением кальция)
Клетки C3H/10Т1/2, которые представляют собой мышиные мезенхимные клетки-предшественники, закупленные у АТСС, разводят в среде DMEM (модифицированная по способу Дульбекко среда Игла), содержащей 5,958 г/л HEPES, 3,7 г/л бикарбоната натрия и 10% FBS (сыворотка плода коровы), и культивируют в 24-луночных культуральных планшетах при плотности 4×104 клеток/лунку в присутствии 5% СО2 при 37°С в течение 2 суток. Культивируемые клетки выращивают в культуральных планшетах до 90~100% слияния, клетки культивируют в среде DMEM, содержащей 10% FBS, к которой добавлены 10 мМ β-глицерофосфата и 50 мкг/мл аскорбиновой кислоты, в присутствии 5% СО2 при 37°С в течение 6 суток, для того чтобы вызывать дифференцировку в остеобласт. Во время дифференцировки среду заменяют через день. Клеточную линию C3H/10Т1/2, в которой индуцируют дифференцировку в остеобласт, промывают один раз PBS (забуференный фосфатом физиологический раствор) и фиксируют 70% этанолом при 20°С в течение 1 часа. После фиксации клетки промывают холодным PBS три раза и окрашивают 40 мМ раствором красителя ализаринового красного S при комнатной температуре в течение 20 минут. Раствор красителя удаляют, и клетки три раза промывают дистиллированной водой для того, чтобы селективно наблюдать только клетки, дифференцированные в остеобласт.
Аликвоту 116V и аликвоту 116VI, экстрагированные из губок и обработанные, растворяют в растворителе ДМСО и используют для обработки клеточной линии C3H/10Т1/2 мезенхимных клеток-предшественников в концентрациях 1, 2,5, 5, 10 и 20 мкг/мл. В результате показывают, что способность дифференцировки остеобластов заметно повышается в зависимости от концентрации, и отмечают слабую цитотоксичность при концентрации 20 мкг/мл. После этого такой же эксперимент выполняют с соединением 1 (1163), соединением 2 (1162), соединением 3 (1161) и соединением 4 (1164), которые выделяют в чистом виде из аликвот 116V и 116VI, и в результате можно утверждать, что способность дифференцировки остеобластов в соединениях 1, 3 и 4 возрастает. Концентрации, при которых проявляется максимальная активность, несколько различаются. Соединение 3 (116-1) показывает максимальную активность при 2,5 мкг/мл, и его цитотоксичность можно наблюдать при последующих концентрациях. Соединение 1 (116-3) показывает активность, значительно возрастающую при концентрациях до 10 мкг/мл в зависимости от концентрации, и цитотоксичность при концентрации 20 мкг/мл, подобно активности выделенной аликвоты 116V без очистки. Соединение 4 показывает максимальную активность при концентрации 5 мкг/мл, и его цитотоксичность наблюдают при последующих концентрациях (фиг.3). Для того чтобы исследовать механизм, касающийся способности дифференцировки остеобластов, клеточные линии C3H/10Т1/2 обрабатывают соединением 1 и соединением 4, соответственно, в течение 6 суток. Обнаружено, с использованием ПЦР (ОТ-ПЦР) в реальном времени, что степень транскрипции факторов признаков дифференцировки (Runx2, остеокальцина, Msx2 и т.д.) остеобласта заметно возрастает (фиг.4). Также клеточные линии C3H/10Т1/2 обрабатывают соединением 1 и соединением 4, соответственно, в течение 6 суток. Обнаружено, с использованием вестерн-блоттинга, что экспрессия белков Runx2 и TAZ возрастает (фиг. 5 и 6). Следовательно, можно принять за основу, что соединения по настоящему изобретению приводят к возрастанию количеств белков Runx2 и TAZ через регуляцию после транскрипции, и, таким образом, можно промотировать дифференцировку остеобласта. Более того, можно убедиться, что сумма белков Runx2 и TAZ возрастает за счет обработки соединениями, и, таким образом, активность Runx2-опосредуемой транскрипции возрастает. Соединение 5, которое представляет собой сложноэфирное производное соединения 1 (1163), синтезируют для того, чтобы получить материал с еще более превосходной биоактивностью, и определяют структуру соединения 5, полученного таким образом. Кроме того, в анализе с осаждением кальция на физиологическую активность (способность дифференцировки остеобластов) полученного соединения найдено, что соединение 5, которое является производным соединения 1, также промотирует дифференцировку остеобласта на уровне, схожем с уровнем соединения 1 (фиг.7). Поэтому ожидается, что соединения по настоящему изобретению и их производные промотируют дифференцировку остеобласта и, таким образом, играют передовую роль в лечении остеопороза.
Экспериментальный пример 2. Определение способности новых соединений ингибировать дифференцировку адипоцитов
Клетки C3H/10Т1/2, которые представляют собой мышиные мезенхимные клетки-предшественники, закупленные у АТСС, разводят в среде DMEM (модифицированная по способу Дульбекко среда Игла), содержащей 5,958 г/л HEPES, 3,7 г/л бикарбоната натрия и 10% FBS (сыворотка плода коровы), и культивируют в 24-луночных культуральных планшетах при плотности 4×104 клеток/лунку в присутствии 5% СО2 при 37°С в течение 2 суток. Когда культивируемые клетки в культуральных планшетах вырастают до 90~100% слияния, клетки культивируют в среде DMEM, содержащей 10% FBS, к которой добавлены 5 мкг/мл инсулина, 1 мкМ дексаметазона и 5 мкМ троглитазона, в присутствии 5% СО2 при 37°С в течение 6 суток, для того, чтобы вызывать дифференцировку в адипоцит. Во время дифференцировки среду заменяют через день. Клеточную линию C3H/10Т1/2, в которой индуцируют дифференцировку в адипоцит, фиксируют 3,7% формальдегидом при комнатной температуре в течение 30 минут. Масляный раствор красного О, растворенного в изопропаноле в концентрации 0,5%, разбавляют в дистиллированной воде в соотношении 6:4, фильтруют через фильтр 0,2 мкм и добавляют к фиксированной клеточной линии, которую окрашивают в течение 1 часа. Для того чтобы наблюдать только клетки, дифференцированные в адипоцит, раствор красителя удаляют, и клетки два раза промывают дистиллированной водой.
Образец 116V, экстрагированный из губки, растворяют в растворителе ДМСО, и обрабатывают клеточные линии C3H/10Т1/2, которые представляют собой мышиные мезенхимные клетки-предшественники, в концентрациях 1, 2,5, 5, 10 и 20 мкг/мл. Результаты показывают, что способность дифференцировки адипоцитов заметно снижается в зависимости от концентрации. Затем получают образцы соединения 1 (1163), соединения 2 (1162), соединения 3 (1161) и соединения 4 (1164), которые выделяют в чистом виде из аликвот 116V и 116VI, и выполняют такой же эксперимент. Экспериментальные результаты показывают, что способность дифференцировки адипоцитов в значительной степени снижается при концентрации 10 мкг/мл. В частности, образец соединения 3 (116-1) показывает способность ингибировать дифференцировку адипоцитов даже при низкой концентрации (1 мкг/мл) при сравнении с другими видами. Заметной цитотоксичности во время дифференцировки адипоцитов не наблюдают ни в одном из образцов (фиг.8). В результате проверки эффективности соединения 1 на клетках 3Т3-L1 оказывается, что соединение 1 показывает великолепную ингибирующую способность в отношении дифференцировки адипоцитов даже на таких клетках (фиг.9).
Экспериментальный пример 3. Измерение антагонистической эффективности и селективности новых соединений в отношении Х-рецептора печени (LXR)
В исследовании трансфекции используют линию животных клеток CV-1. Клетки культивируют в среде DMEM в устройстве для культивирования клеток, содержащем 5% диоксида углерода, при 37°С. Среда содержит 10% FBS (сыворотка плода коровы), 100 Е/мл пенициллина и 100 мкг/мл стрептомицина. В день 1 эксперимента клетки CV-1 высевают в 96-луночные планшеты при плотности 5000 клеток/лунку. В день 2 высеянные клетки трансфицируют плазмидой, экспрессирующей GAL-hLXR, плазмидой, экспрессирующей ген люциферазы, и плазмидой, экспрессирующей β-галактозидазу, с использованием трансфицирующего реагента Superfect (QIAGEN). Через 16 часов трансфицированные клетки обрабатывают соединением 1, растворенным в диметилсульфоксиде (ДМСО), в различных концентрациях, вместе с агонистом ТО901317 (2,5 мкМ). Клетки, обработанные диметилсульфоксидом в конечной концентрации 1%, используют в качестве группы отрицательного контроля, и клетки, обработанные ТО901317 в конечной концентрации 500 нМ, используют в качестве группы положительного контроля. Клетки культивируют в течение 24 часов и лизируют с использованием буфера для лизиса. К клеткам добавляют люциферин для измерения люциферазной активности с использованием люминометра. После добавления реагента ONPG измеряют β-галактозидазную активность с использованием аппарата для прочтения планшетов ELISA. Измеренную величину люциферазной активности исправляют на величину активности β-галактозидазы. Результаты показывают, что соединение 1 имеет величины IC50 18,7 и 2,4 нМ в отношении LXRα и LXRβ, соответственно (фиг.10). Кроме того, с использованием такого же метода измеряют активности на различных ядерных рецепторах для того, чтобы измерить селективность в отношении различных ядерных рецепторов. Однако соединение 1 никогда не показывает активности в отношении других ядерных рецепторов (фиг.11). Также эксперимент Biacore доказывает, что соединение 1 непосредственно связывается с белком LXR (фиг.12).
Экспериментальный пример 4. Измерение цитотоксичности новых соединений
Используют клетки селезенки мыши в измерении цитотоксичности в анормальных клетках. Клетки селезенки мыши получают следующим образом. Селезенку 5~6-недельной мыши тонко крошат, и только флотирующие клетки селезенки фильтруют через ячеистый материал с порами размером 100 мкм. Эритроциты, смешанные с клетками селезенки, лизируют с использованием буфера для лизиса эритроцитов, и затем удаляют центрифугированием, осаждением и промывкой клеток. Полученные клетки селезенки высевают в 96-луночные планшеты в концентрации 5×105 клеток/лунку. В планшетах клетки селезенки обрабатывают соединением формулы 1 для измерения токсичности в соответствии с концентрациями. На следующий день клетки, культивированные в течение 16~18 часов, обрабатывают в анализе Titer-Glo Luminescent Cell Viability Assay (Promega), и через 10 минут жизнеспособность клеток измеряют с использованием люминометра. Путем определения на клетках селезенки мыши величины IC50 63 мкМ показано, что соединение 1 имеет небольшую цитотоксичность в отношении здоровых клеток, которая больше в 1000 раз или более в сравнении с антагонистической концентрацией против LXR (фиг.13).
Экспериментальный пример 5. Проверка функции экспрессии генов новыми соединениями в клетках печени
Для того чтобы проверить эффективность разработанного антагониста Х-рецептора печени, идентифицируют функцию регуляции экспрессии генов в клетках печени мыши и клетках печени человека. В данном эксперименте используют клетки печени мыши AML 12 и клетки печени человека HepG2. Клетки AML12 культивируют в среде DMEM в инкубаторе для клеток с 5% диоксида углерода при 37°С. Среда содержит 10% сыворотки плода коровы (FBS), 100 Е/мл пенициллина и 100 мкг/мл стрептомицина. В день 1 эксперимента клетки AML12 высевают в 6-луночные планшеты. В день 2, когда клетки вырастают до 80% слияния, среду заменяют на среду DMEM, не содержащую сыворотку, и затем три лунки на группу обработки обрабатывают ТО901317 и разработанным антагонистом Х-рецептора печени. Клетки, обработанные диметилсульфоксидом в конечной концентрации 0,2%, используют в качестве группы отрицательного контроля, и клетки, обработанные ТО901317 в конечной концентрации 500 нМ, используют в качестве группы положительного контроля. Соединение 1, разработанное для раскрытия эффективности антагониста, используют одно в концентрации 1 мкМ или используют вместе с 500 нМ ТО901317. После инкубации в течение 18 часов все РНК клеток печени экстрагируют с использованием набора для экстракции РНК RNeasy (QIAGEN). Экстрагированные РНК определяют количественно, и 1 мкг экстрагированных РНК на каждый образец используют в синтезе кДНК. В синтезе кДНК используют набор для синтеза кДНК Transcriptor First Strand cDNA Synthesis (Roche). Осуществляют генетический анализ на синтезированных кДНК клеток печени с использованием полимеразной цепной реакции в реальном времени. Синтезированные для полимеразной цепной реакции в реальном времени кДНК смешивают с праймером, селективным для АСС1 или гена актина, и QuantiTech Master Mix (QIAGEN). Полимеразную цепную реакцию выполняют в 45 циклах 90°С в течение 10 секунд, 60°С в течение 15 секунд и 72°С в течение 20 секунд. Полимеразную цепную реакцию выполняют трижды для каждого образца кДНК. Для того чтобы сравнить экспрессированное количество каждого гена на группу обработки друг с другом, для каждого образца получают величины Ct с использованием программы анализа полимеразной цепной реакции в реальном времени. Величины Ct каждой группы обработки сравнивают с величинами Ct группы отрицательного контроля, и вычисляют различия в экспрессированном количестве генов. Различие в экспрессированном количестве гена, представляющего интерес, на каждую группу обработки корректируют различием в экспрессированном количестве гена GADPH. Экспериментальные результаты показывают, что соединение 1 весьма эффективно ингибирует экспрессию вызывающих жировую дистрофию печени генов биосинтеза жирных кислот SREBP1c, ACC и FAS (фиг.14).
Экспериментальный пример 6. Измерение эффективности ингибирования жировой дистрофии печени новых соединений на экспериментальных животных моделях
Для того чтобы проверить эффективность ингибирования жировой дистрофии печени соединения 1, разработанного в настоящем изобретении, в данном эксперименте используют мышей C57BL/6. Модель жировой дистрофии печени создают введением ТО901317, который вызывает жировую дистрофию печени, мышам C57BL/6 в возрасте 10 недель, причем в это время мышей кормят общей пищевой добавкой. Эффективность ингибирования жировой дистрофии печени соединения 1 наблюдают при пероральном введении указанного соединения. Мышиный корм с 0,75% только карбоксиметилцеллюлозы как средства для доставки лекарственного средства используют для группы отрицательного контроля, и мышиный корм только с ТО901317 используют для группы положительного контроля. Кроме того, для того чтобы проанализировать экспрессию генов печени мыши C57BL/6, печени мышей группы отрицательного контроля, группы положительного контроля и группы обработки экстрагируют и обрабатывают тризолом для получения РНК. Полученные РНК определяют количественно с использованием абсорбционного спектрометра (Nanodrop), и из РНК методом ОТ-ПЦР с использованием олиго-dT и обратной транскриптазы получают кДНК с тем же количеством соответствующих групп. Полимеразную цепную реакцию в реальном времени выполняют с использованием в качестве матриц кДНК, которые получают для анализа изменения мРНК в группах, и использованием праймеров генов-транспортеров, связанных с синтезом жира и поступлением жира в печень. Экспериментальные результаты показывают, что не имеется различий в массе тела в группе обработки и контрольных группах, когда соединение 1 вводят мышам с вызванной жировой дистрофией печени, и соединение 1 также проявляет превосходную эффективность ингибирования жировой дистрофии печени на экспериментальной животной модели (фиг. 15 и 16). Кроме того, результаты анализа экспрессии генов показывают, что соединение 1 весьма эффективно ингибирует экспрессию генов синтеза жирных кислот, которые вызывают жировую дистрофию печени, и генов-транспортеров, переносящих жиры в печень (фиг.17). Соответственно, ожидается, что соединения по настоящему изобретению и их производные играют весьма передовую роль при лечении алкогольной жировой дистрофии печени, неалкогольной жировой дистрофии печени и дистрофии печени вследствие вирусной инфекции.
Изобретение относится к новому соединению со спирохиральной углеродной основой, или его фармацевтически приемлемой соли общей формулы 1
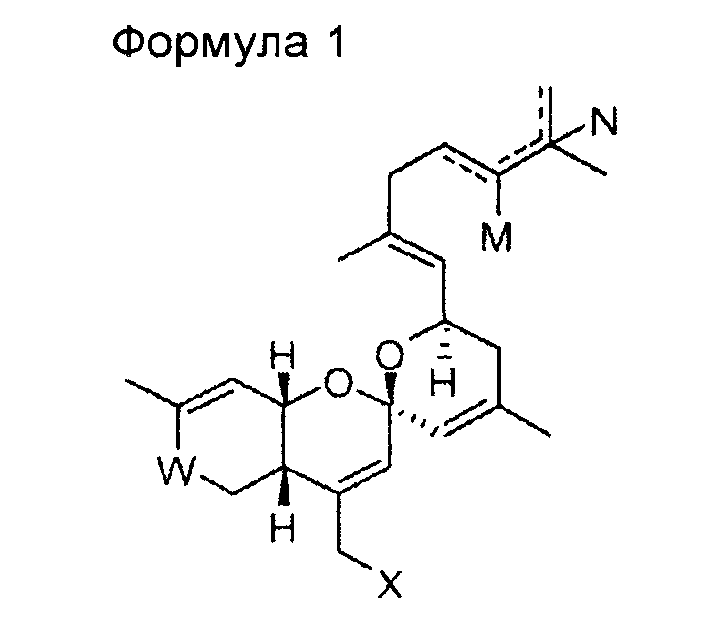
где W представляет собой СО или СНО(С=O)СН3; Х представляет собой N3 или OR2; R2 представляет собой водород, линейный или разветвленный алкил С1~С8 или 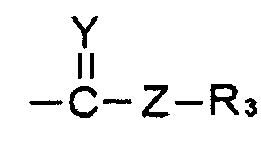 ; Y представляет собой О; Z представляет собой простую связь или O; R3 представляет собой линейный или разветвленный алкил С1~С8 или алкенил С2~С8, и М и N представляют собой, каждый независимо, водород, ОН или отсутствуют; при этом атом углерода, связанный с М или N, образует простую связь или двойную связь с другими атомами углерода, и число двойных связей составляет одну или менее для каждого из атомов углерода. Изобретение относится к способу получения и фармацевтическим композициям. Соединение со спирохиральной углеродной основой обладает превосходной активностью дифференцировки остеобластов, активностью ингибирования тучных клеток и активностью ингибирования синтеза жирных кислот в печени. Поэтому соединение будет играть передовую роль при лечении остеопороза, жировой дистрофии печени и ожирения. 6 н. и 7 з.п. ф-лы, 6 пр., 5 табл., 17 ил.
; Y представляет собой О; Z представляет собой простую связь или O; R3 представляет собой линейный или разветвленный алкил С1~С8 или алкенил С2~С8, и М и N представляют собой, каждый независимо, водород, ОН или отсутствуют; при этом атом углерода, связанный с М или N, образует простую связь или двойную связь с другими атомами углерода, и число двойных связей составляет одну или менее для каждого из атомов углерода. Изобретение относится к способу получения и фармацевтическим композициям. Соединение со спирохиральной углеродной основой обладает превосходной активностью дифференцировки остеобластов, активностью ингибирования тучных клеток и активностью ингибирования синтеза жирных кислот в печени. Поэтому соединение будет играть передовую роль при лечении остеопороза, жировой дистрофии печени и ожирения. 6 н. и 7 з.п. ф-лы, 6 пр., 5 табл., 17 ил.
1. Соединение приведенной ниже формулы 1, или его фармацевтически приемлемая соль,
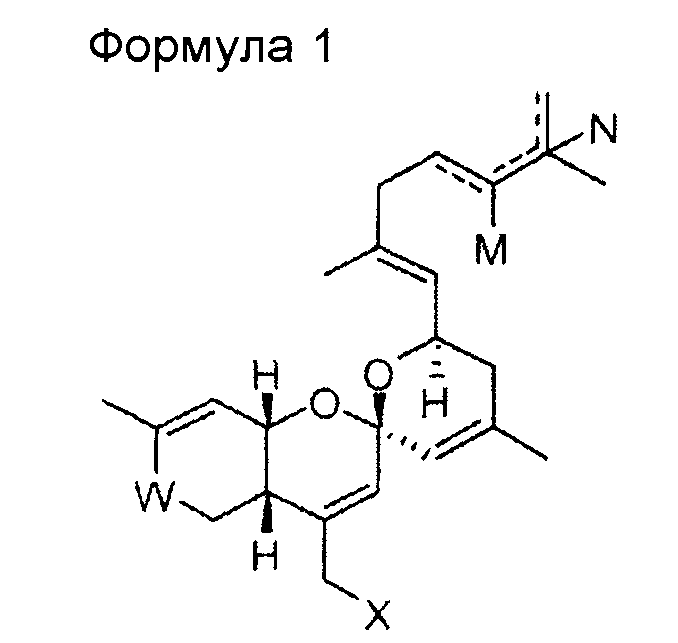
где W представляет собой СО или СНО(С=O)СН3;
Х представляет собой N3 или OR2;
R2 представляет собой водород, линейный или разветвленный алкил С1~С8 или 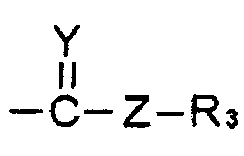 ;
;
Y представляет собой О;
Z представляет собой простую связь или О;
R3 представляет собой линейный или разветвленный алкил С1~С8 или алкенил С2~С8; и
М и N представляют собой, каждый независимо, водород, ОН или отсутствуют; при этом атом углерода, связанный с М или N, образует простую связь или двойную связь с другими атомами углерода, и число двойных связей составляет одну или менее для каждого из атомов углерода.
2. Соединение формулы 1 или его фармацевтически приемлемая соль по п.1,
при этом W представляет собой СО или СНО(С=O)СН3;
Х представляет собой OR2;
R2 представляет собой водород.
3. Соединение или его фармацевтически приемлемая соль по п.1,
при этом W представляет собой СО;
Х представляет собой N3 или OR2;
R2 выбирают из линейного или разветвленного алкила С1~С8 или 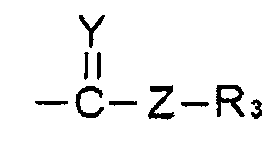 ;
;
Y представляет собой О;
Z представляет собой О или простую связь;
R3 представляет собой линейный или разветвленный алкил С1~С8 или алкенил С2~С8; и
М и N представляют собой, каждый независимо, водород, ОН или отсутствуют; при этом атом углерода, связанный с М или N, образует простую связь или двойную связь с другими атомами углерода, и число двойных связей составляет одну или менее для каждого из атомов углерода.
4. Соединение, его стереоизомер, или его фармацевтически приемлемая соль по п.1, при этом соединение формулы 1 выбирают из группы, состоящей из
5. Способ получения соединения формулы 1, включающий
(a) нарезание и сушку губки Phorbas sp. с последующей экстракцией с использованием спирта С1~С4;
(b) обработку экстракта, полученного на стадии (а), с использованием воды и метиленхлорида и затем удаление растворителя из органического слоя с последующей обработкой с использованием н-гексана и смешанного растворителя из метанола и воды; и
(c) удаление растворителя из слоя метанольной аликвоты, полученной на стадии (b), и последующее получение аликвоты хроматографией с использованием диоксида кремния в качестве неподвижной фазы и с использованием метанольного раствора в качестве элюента, причем метанольный раствор содержит или не содержит 20 мас.% или менее воды относительно его общей массы, где соединения представляют собой
6. Способ по п.5, при этом на стадии (а) для сушки используют сушку вымораживанием и в качестве спирта С1~С4 используют метанол.
7. Способ по п.5, при этом на стадии (b) смешанный растворитель из метанола и воды содержит 60~90 мас.% метанола и 10~40 мас.% воды относительно общей массы растворителя.
8. Способ по п.5, при этом на стадии (с) выполняют хроматографию один раз или более в порядке от элюента, имеющего наивысшую полярность, до элюента, имеющего наименьшую полярность, путем использования в качестве элюента смешанного растворителя из воды и метанола с наивысшей полярностью перед использованием в качестве элюента метанольного раствора, содержащего или не содержащего 20 мас.% или менее воды от общей массы элюента.
9. Способ по п.5, также включающий (d) очистку аликвоты, полученной на стадии (с), при этом очистку выполняют высокоэффективной жидкостной хроматографией (ВЭЖХ), и в качестве элюента используют смесь 50~80 мас.% ацетонитрила (ACN) и 20~50 мас.% воды относительно общей массы элюента.
10. Фармацевтическая композиция для лечения остеопороза, включающая соединение формулы 1, или его фармацевтически приемлемую соль по п.1 в качестве фармацевтически приемлемого носителя и активного агента.
11. Фармацевтическая композиция для лечения жировой дистрофии печени, включающая соединение формулы 1, или его фармацевтически приемлемую соль по п.1 в качестве фармацевтически приемлемого носителя и активного агента.
12. Фармацевтическая композиция для лечения ожирения, включающая соединение формулы 1, или его фармацевтически приемлемую соль по п.1 в качестве фармацевтически приемлемого носителя и активного агента.
13. Фармацевтическая композиция для антагонизации Х-рецептора печени (LXR), включающая соединение формулы 1, или его фармацевтически приемлемую соль по п.1 в качестве фармацевтически приемлемого носителя и активного агента.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Алдошин С.М | |||
| // Успехи химии, т.59, №7, (1990), с.1144-1178. | |||

