ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к противовирусному соединению, более конкретно, соединению, проявляющему высокую селективность и физиологическую активность против вируса иммунодефицита человека (ВИЧ), к способу его получения и к его применению.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Синдром приобретенного иммунодефицита (СПИД) вызывается вирусом иммунодефицита человека (ВИЧ). Существует два типа ВИЧ, ВИЧ-1 и ВИЧ-2, и наиболее распространенным в мире является ВИЧ-1. Для лечения СПИДа были разработаны ингибиторы ферментов в соответствии с механизмами действия ВИЧ. В зависимости от точки действия эти ингибиторы классифицируются как нуклеозидный ингибитор обратной транскриптазы (NRTI), ингибитор протеазы (PI), ингибитор слияния и ингибитор интегразы.
Ингибиторы интегразы подразделены на ингибиторы каталитических центров и ингибиторы некаталитических центров. На сегодняшний день активно ведутся исследования ингибиторов каталитического центра интегразы, и были разработаны три вида лекарственных средств, которые являются коммерчески доступными. Ралтегравир, разработанный в 2008 году, является репрезентативным лекарственным средством. Между тем механизм действия ингибиторов некаталитического центра интегразы был предложен Ziger Debyser, et al. (Frauke Christ, Zeger Debyser et al., Nature Chemical Biology, 2010, Vol. 6, 442), и разработка ингибиторов для этого механизма действия активно продолжалась.
Кроме того, проводились различные исследования для разработки лекарственных средств для эффективного лечения резистентных вирусов. Такие химиотерапевтические средства вводят в комбинации из двух или четырех лекарственных средств, которые ингибируют различные механизмы действия, которые называются высокоактивными антиретровирусными терапиями (HAART), что приводит к значительным эффектам продления жизни. Однако, несмотря на такие усилия, СПИД полностью не излечивается, и из-за токсичности лекарственного средства и проявления резистентности к существующим терапевтическим средствам постоянно требуется разработка новых лекарственных средств.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Техническая задача, которая должна быть решена
В попытке решить вышеуказанные проблемы авторами настоящего изобретения были осуществлены глубокие исследования для поиска новых лекарственных средств против СПИДа, и в результате было обнаружено, что соединения пирролопиридина, имеющие новый скелет, оказывают ингибирующее действие на пролиферацию ВИЧ. Настоящее изобретение было создано на основании таких открытий.
Следовательно, одна цель настоящего изобретения состоит в том, чтобы обеспечить новое пирролопиридиновое соединение и его фармацевтически приемлемую соль, которое проявляет ингибирующее действие на пролиферацию ВИЧ-1 путем ингибирования активности ферментов интегразы ВИЧ-1, а также демонстрирует превосходные результаты в испытаниях свойств и основной токсичности лекарственного средства.
Другой целью настоящего изобретения является обеспечение способа получения нового пирролопиридинового соединения, как описано выше, и его фармацевтически приемлемой соли.
Еще одной целью настоящего изобретения является обеспечение фармацевтической композиции, включающей указанное выше соединение в качестве активного ингредиента.
Техническое решение
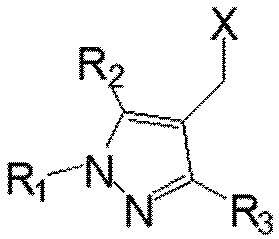
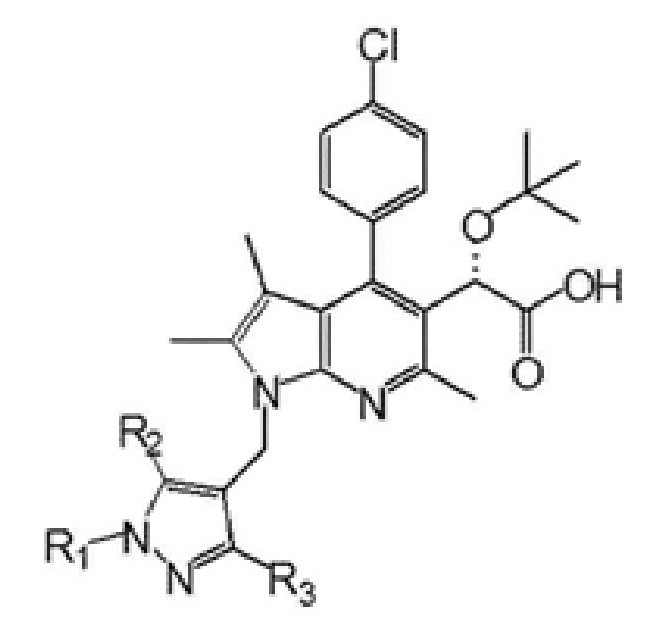
Первый аспект настоящего изобретения обеспечивает соединение, представленное следующей химической формулой I, его рацемат или стереоизомер или его фармацевтически приемлемую соль:
Химическая формула I
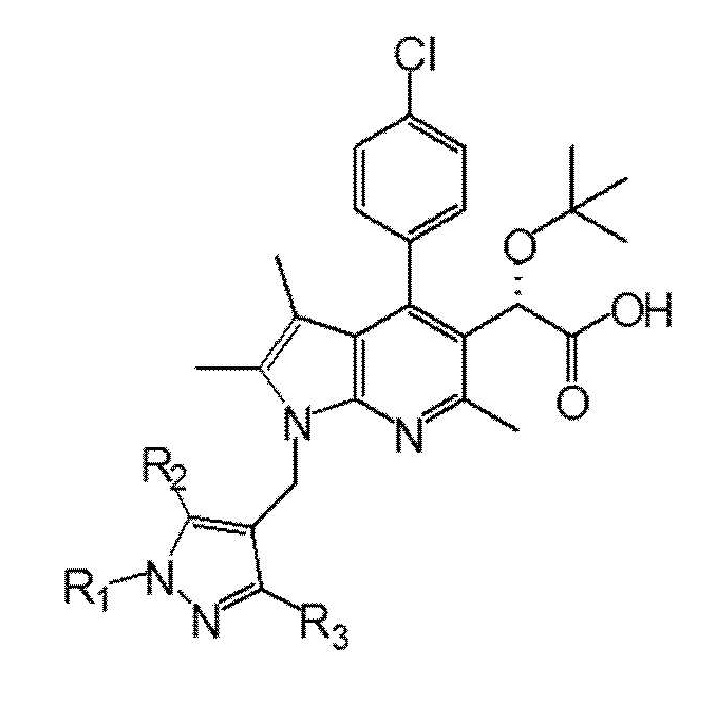
где,
R1 выбран из группы, состоящей из C1–6 алкила, незамещенного или замещенного атомом галогена, бензила, незамещенного или замещенного C1–3 алкилом или галогеном, C1–3 алкилоксиметила, C1–3 алкилкарбамата и сульфонила, незамещенного или замещенного C1–3 алкилом, и
R2 и R3 каждый независимо представляют собой водород, C1–6 алкил или атом галогена.
В одном варианте осуществления настоящее изобретение обеспечивает соединение, где R1 представляет собой C1–6 алкил, его рацемат или стереоизомер или его фармацевтически приемлемую соль.
В другом варианте осуществления настоящее изобретение обеспечивает соединение, где R1 представляет собой метил и R2 и R3 каждый независимо представляют собой водород, метил или хлор, его рацемат или стереоизомер или его фармацевтически приемлемую соль.
В еще одном варианте осуществления настоящее изобретение обеспечивает соединение, где R1 представляет собой метил и оба R2 и R3 представляют собой водород, его рацемат или стереоизомер или его фармацевтически приемлемую соль.
В частности, атом галогена относится к атому хлора, брома или фтора.
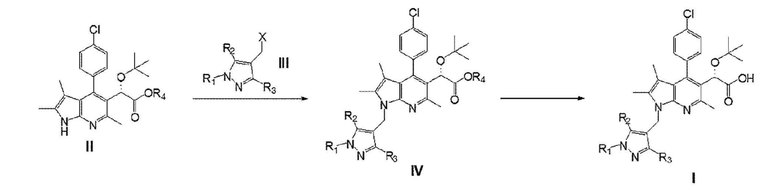
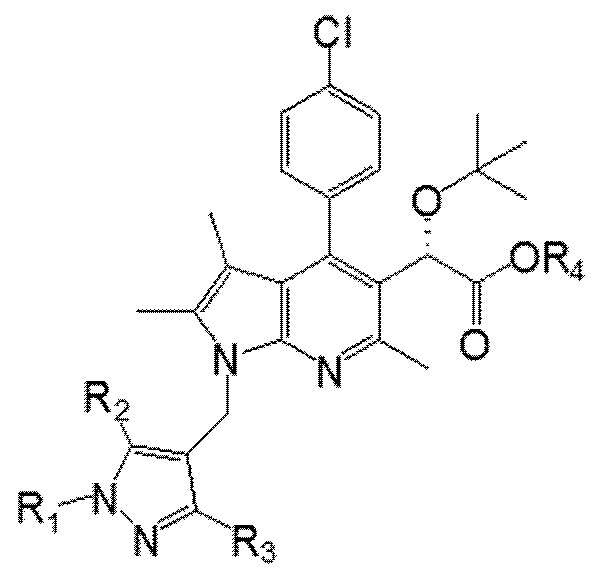
Второй аспект настоящего изобретения обеспечивает способ получения соединения Химической формулы I в соответствии со схемой реакции 1, представленной ниже:
Схема реакции 1
В частности, способ получения соединения химической формулы I,
Химическая формула I
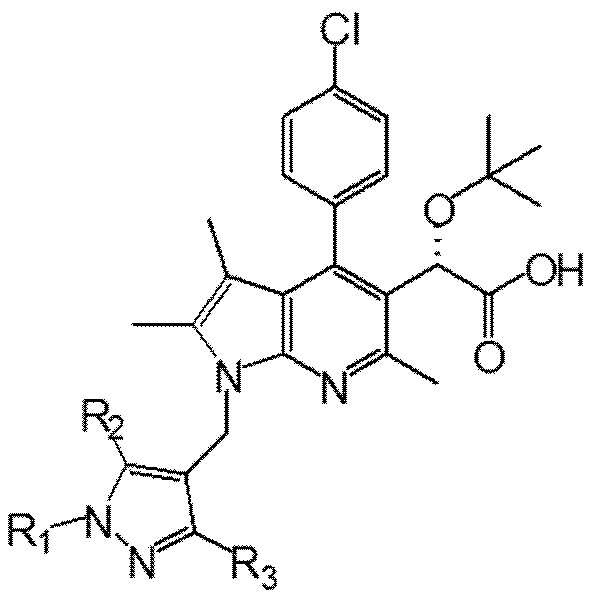 включает:
включает:
1) первую стадию взаимодействия соединения, представленного химической формулой II, с соединением, представленным химической формулой III, с получением соединения, представленного химической формулой IV,
Химическая формула II
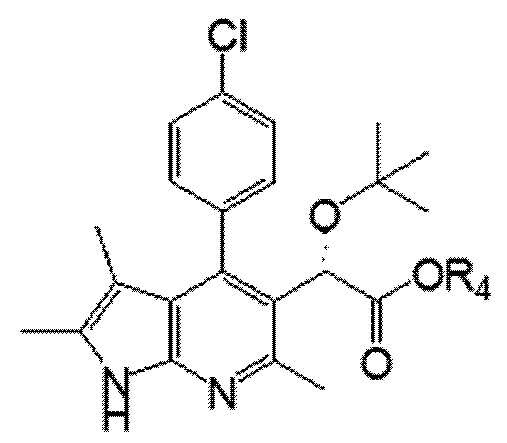
Химическая формула III
Химическая формула IV
 и
и
2) вторую стадию гидролиза соединения, представленного химической формулой IV,
где
R1 выбран из группы, состоящей из C1–6 алкила, незамещенного или замещенного атомом галогена, бензила, незамещенного или замещенного C1–3 алкилом или атомом галогена, C1–3 алкилоксиметила, C1–3 алкилкарбамата и сульфонила, незамещенного или замещенного C1–3 алкилом,
R2 и R3 каждый независимо представляют собой водород, C1–6 алкил или атом галогена,
R4 представляет собой C1–6 алкил и
X представляет собой галоген, метансульфонил, толуолсульфонил или трифторметансульфонил.
В частности, R4 может представлять собой метил или этил, и X может представлять собой хлор или пара-толуолсульфонил.
На первой стадии способа получения соединения химической формулы I молярное соотношение между соединением химической формулы II и соединением химической формулы III предпочтительно составляет от 1:2 до 1:5, но не ограничивается этим.
На первой стадии растворитель реакции может представлять собой дихлорметан, диметилформамид, тетрагидрофуран или любую их комбинацию, но не ограничивается этим.
Первую стадию можно осуществлять в течение от 2 часов до 18 часов, но не ограничиваясь этим.
Первую стадию можно осуществлять в присутствии карбоната цезия, и диметилформамид предпочтительно используют в качестве растворителя.
На первой стадии карбонат цезия используют в количестве предпочтительно от 2 до 5 эквивалентов по отношению к соединению химической формулы II.
На данном этапе, но не ограничиваясь этим, температура реакции предпочтительно составляет от 40°С до 100°С, и время реакции предпочтительно составляет от 4 часов до 18 часов.
Например, соединение, представленное химической формулой II, которое используют в качестве исходного вещества для получения соединения химической формулы I в соответствии с настоящим изобретением, можно получить в соответствии со способом, раскрытым в примере получения заявки WO 2013/073875A1.
На второй стадии гидролиз можно осуществлять с использованием гидроксида лития, гидроксида кальция, гидроксида бария или гидроксида калия, но не ограничиваясь ими. Предпочтительно, можно использовать гидроксид калия или гидроксид лития.
В гидролизе гидроксид калия или гидроксид лития можно использовать в количестве от 3 до 8 эквивалентов по отношению к соединению химической формулы IV, но не ограничиваясь этим.
Гидролиз на второй стадии можно осуществлять при комнатной температуре, или альтернативно при 35°С - 50°С.
В гидролизе в качестве растворителя можно использовать воду, метанол, тетрагидрофуран или любую их комбинацию, но не ограничиваясь этим.
В одном варианте осуществления гидролиз осуществляют с гидроксидом лития в смешанном растворителе, например, 4N гидроксид натрия/метанол или тетрагидрофуран/метанол/вода.
Гидролиз, в частности, можно осуществлять в течение 6 часов - 18 часов, но не ограничиваясь этим.
Третий аспект настоящего изобретения обеспечивает противовирусную композицию, включающую соединение, представленное химической формулой I, описанной выше, его рацемат или стереоизомер или его фармацевтически приемлемую соль.
В частности, вышеуказанная композиция представляет собой композицию против вируса иммунодефицита человека (ВИЧ).
В настоящем изобретении конкретным примером соединения химической формулы I может быть (S)-2-(трет-бутокси)-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1-метил-1H-пиразол-4-ил)метил)-1H-пирроло[2,3-b]пиридин-5-ил)уксусная кислота или
(S)-2-(трет-бутокси)-2-(1-((5-хлор-1,3-диметил-1H-пиразол-4-ил)метил)-4-(4-хлорфенил)-2,3,6-триметил-1H-пирроло[2,3-b]пиридин-5-ил)уксусная кислота.
Соединение химической формулы I, полученное, как указано выше, может образовывать соль, в частности, фармацевтически приемлемую соль. Подходящая фармацевтически приемлемая соль конкретно не ограничивается, при условии, что она является солью, обычно используемой в данной области, такой как кислотно-аддитивная соль (см., J. Pharm. Sci., 1977, 66, 1).
Предпочтительный пример кислоты для фармацевтически приемлемой кислотно-аддитивной соли включает неорганическую кислоту, такую как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, ортофосфорная кислота или серная кислота; или органическую кислоту, такую как метансульфоновая кислота, бензолсульфоновая кислота, толуолсульфоновая кислота, уксусная кислота, пропионовая кислота, молочная кислота, лимонная кислота, фумаровая кислота, яблочная кислота, янтарная кислота, салициловая кислота, малеиновая кислота, глицерофосфорная кислота или ацетилсалициловая кислота.
Фармацевтически приемлемую соль металла также можно получить в соответствии с обычным способом с использованием основания. Например, соединение химической формулы I можно растворить в избыточном количестве раствора гидроксида щелочного металла или гидроксида щелочноземельного металла, нерастворенную соль соединения можно отфильтровать, а затем фильтрат можно выпарить и высушить с получением фармацевтически приемлемой металлической соли соединения.
Фармацевтически неприемлемую соль или сольват соединения химической формулы I можно использовать в качестве промежуточного соединения при получении соединения химической формулы I или его фармацевтически приемлемой соли или сольвата.
Соединение химической формулы I в соответствии с настоящим изобретением включает не только его фармацевтически приемлемые соли, но также его сольваты и гидраты, которые могут быть получены из таких соединений. Стереоизомеры соединения, представленного химической формулой I, и его промежуточные соединения можно получить в соответствии с общепринятым способом.
Кроме того, соединение химической формулы I в соответствии с настоящим изобретением можно получить либо в кристаллической форме, либо в некристаллической форме. Когда соединение химической формулы I получают в кристаллической форме, оно может быть необязательно гидратировано или сольватировано.
Кроме того, настоящее изобретение обеспечивает противовирусную композицию, включающую, в качестве активного ингредиента, описанное выше соединение химической формулы I или его фармацевтически приемлемую соль, гидрат или сольват. В этом случае противовирусная представляет собой, в частности, композицию против вируса иммунодефицита человека (ВИЧ).
В экспериментальных примерах настоящего изобретения было обнаружено, что соединение, представленное химической формулой I, является превосходным веществом с низкой цитотоксичностью, превосходным эффектом ингибирования ВИЧ и высокой физиологической активностью, и которое при этом демонстрирует безопасность в испытании на базовую токсичность и обладает подходящей растворимостью для свойств лекарственного средства.
Фармацевтическая композиция в соответствии с настоящим изобретением может быть сформулирована в форму для перорального введения или для инъекций. Например, лекарственная форма для перорального введения включает таблетку, капсулу и т.п., и такая лекарственная форма содержит разбавитель (например, лактозу, декстрозу, сахарозу, маннит, сорбит, целлюлозу и/или глицин) и глидант (например, диоксид кремния, тальк, стеариновую кислоту или магниевую или кальциевую соль стеариновой кислоты или полиэтиленгликоль) в дополнение к активному ингредиенту. Таблетка также может содержать связующее, такое как алюмосиликат магния и крахмальная паста, желатин, трагакант, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы или поливинилпиколидин, и в зависимости от конкретного случая она может содержать разрыхлитель, такой как крахмал, агар, альгиновая кислота или ее натриевая соль, или кипящую смесь и/или абсорбент, краситель, ароматизатор и подсластитель. Композиция для инъекций предпочтительно представляет собой изотонический водный раствор или суспензию.
Вышеуказанная композиция может быть стерилизована и/или может содержать адъювант, такой как консервант, стабилизатор, смачиваемый порошок или ускоритель эмульгирования, соль для регулирования осмотического давления и/или буфер, и любое другое терапевтически полезное вещество.
Вышеуказанную лекарственную форму можно получить обычным способом смешивания, гранулирования или нанесения покрытия и она может содержать активный ингредиент в диапазоне приблизительно от 0,1 до 75 масс.%, и предпочтительно в диапазоне приблизительно от 1 до 50 масс.%. Стандартная лекарственная форма для млекопитающего с массой тела приблизительно от 50 до 70 кг содержит приблизительно от 10 до 200 мг активного ингредиента.
Предпочтительная дозировка соединения по изобретению варьируется в зависимости от состояния и массы тела пациентов, прогрессирования заболеваний, формы лекарственных средств, пути и времени введения, но ее могут правильно выбрать специалисты в данной области. Суточную дозу можно вводить пероральным или парентеральным путем в виде разовой или дробных доз.
Фармацевтическую композицию по настоящему изобретению можно вводить млекопитающему, включая крысу, мышь, домашнее животное, человека и т.п., различными путями. Можно рассматривать все пути введения, и композицию можно вводить, например, перорально, ректально или путем внутривенной, внутримышечной, подкожной, внутриматочной, дуральной или интрацеребровентрикулярной инъекции.
Полезные эффекты
Соединение, представленное химической формулой I в соответствии с настоящим изобретением, его рацемат или стереоизомер, или фармацевтически приемлемая соль, гидрат или сольват такого соединения демонстрируют высокую селективность и физиологическую активность против вируса иммунодефицита человека (ВИЧ), с низкой токсичностью и, таким образом, полезны для лечения вирусной инфекции, в частности, инфекции вируса иммунодефицита человека (ВИЧ).
Подробное описание вариантов осуществления
Далее настоящее изобретение будет описано более подробно со ссылкой на следующие примеры получения и примеры. Однако следующие примеры получения и примеры приведены только для иллюстративных целей, и объем настоящего изобретения не ограничивается ими.
Пример получения 1: Получение гидрохлоридной соли 4-(хлорметил)-1-метил-1H-пиразола
Дихлорметан (1,8мл) и триэтиламин (2 капли) добавляли к (1-метил-1H-пиразол-4-ил)метанолу (380 мг, 3,39 ммоль), полученному в соответствии с известным способом (Frey, R. R,; at al, J. Med. Chem., 2008, 51, 3777-3787), и затем полученную смесь охлаждали до 0°С. Раствор, в котором тионилхлорид (0,62 мл) был растворен в толуоле (1,8 мл), медленно добавляли к этой смеси и смесь перемешивали в течение 2 часов при 30°С. Растворитель и избыточное количество тионилхлорида удаляли из реакционного раствора при пониженном давлении с получением целевого соединения. Соединение использовали в следующей реакции без очистки.
Пример получения 2: Получение 4-(бромметил)-5-хлор-1,3-диметил-1H-пиразола
(5-Хлор-1,3-диметил)-1H-пиразол-4-ил)метанол (937 мг, 5,8 ммоль), полученный в соответствии с известным способом (Attardo, G.; Tripthy, S., PCT Int. Appl. 2010, WO 2010-132999 A1), растворяли в дихлорметане (40 мл) и затем охлаждали до 0°С. К этой смеси медленно добавляли раствор, в котором трибромид фосфора (0,54 мл, 5,8 ммоль) был разбавлен дихлорметаном (5 мл), и затем полученную смесь перемешивали в течение 1,5 часов при комнатной температуре. Растворитель удаляли из реакционного раствора при пониженном давлении с получением целевого соединения. Соединение использовали в следующей реакции без очистки.
Пример 1: (S)-2-(трет-бутокси)-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1-метил)-1H-пиразол-4-ил)метил)-1H-пирроло[2,3-b]пиридин-5-ил)уксусная кислота.
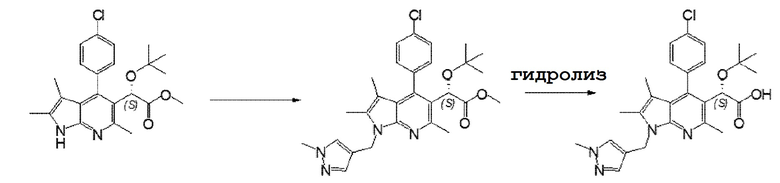
Стадия 1: Метил(S)-2-(трет-бутокси)-2-(4-(4-хлорфенил)-2,3,6-триметил-1H-пирроло[2,3-b]пиридин-5-ил)ацетат (700 мг, 1,69 ммоль) растворяли в диметилформамиде (14мл) и затем добавляли карбонат цезия (2,75 г, 8,45 ммоль) и 10 капель триэтиламина. После того, как температура достигала 40°С, соединение, полученное в Примере получения 1 (560 мг, 3,39 ммоль), добавляли порциями к этой смеси в течение 1 часа. Полученную смесь перемешивали в течение 18 часов при этой же температуре до завершения реакции. Полученную смесь перемешивали в течение 18 часов при этой же температуре для завершения реакции. Реакционный раствор охлаждали на ледяной бане и добавляли к нему воду (50 мл) и полученный раствор перемешивали в течение 10 минут. Полученные твердые вещества отфильтровывали и промывали водой. Полученное твердое вещество, без сушки, очищали колоночной хроматографией на силикагеле (элюент: этилацетат/н-гексан=1/2 и 1/1) с получением целевого соединения (430 мг, 50%).
1H-ЯМР(CDCl3, 500 МГц) δ 1,01(с, 9H), 1,49(с, 3H), 2,30(с, 3H), 2,75(с, 3H), 3,69(с, 3H), 3,83(с, 3H), 5,11(с, 1H), 5,32(с, 2H), 7,30(м, 2H), 7,44-7,47(м, 4H); MS(EI, m/e)= 509(M+).
Стадия 2: Соединение (369 мг, 0,724 ммоль), полученное на Стадии 1, растворяли в тетрагидрофуране (5,5 мл), затем добавляли 4N гидроксид натрия в метаноле (0,98 мл) и полученную смесь перемешивали в течение 18 часов при 35°С. Реакционный раствор охлаждали до 10°С и затем нейтрализовали путем добавления 4N хлористоводородной кислоты. Затем растворитель удаляли из реакционного раствора при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле (элюент: дихлорметан/метанол=95/5 и 90/10) с получением целевого соединения (260 мг, 73%) в виде белого твердого вещества.
1H-ЯМР(CD3OD, 500 МГц) δ 1,00(с, 9H), 1,52(с, 3H), 2,31(с, 3H), 2,72(с, 3H), 3,80(с, 3H), 5,14(с, 1H), 5,37(шир.с, 2H), 7,34-7,53(м, 6H);
MS(EI, m/e)= 495(M+).
Пример 2: (S)-2-(трет-бутокси)-2-(1-((5-хлор-1,3-диметил-1H-пиразол-4-ил)метил)-4-(4-хлорфенил)-2,3,6-триметил-1H-пирроло[2,3-b]пиридин-5-ил)уксусная кислота.
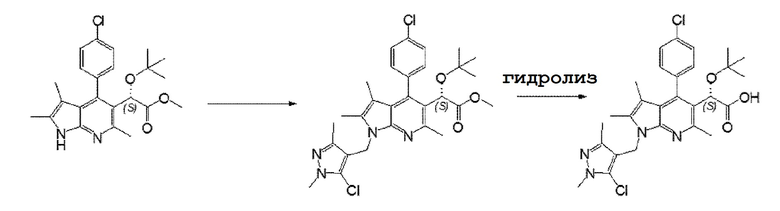
Целевое соединение (30 мг, 44%) получали путем взаимодействия метил(S)-2-(трет-бутокси)-2-(4-(4-хлорфенил)-2,3,6-триметил-1H-пирроло[2,3-b]пиридин-5-ил)ацетата (200 мг, 0,48 ммоль) и соединения, полученного в примере получения 2 (432 мг, 1,44 ммоль), таким же способом, как в Примере 1.
1H-ЯМР(CD3OD, 500 МГц) δ 1,00(с, 9H), 1,49(с, 3H), 1,90(с, 3H), 2,19(с, 3H), 2,68(с, 3H), 3,75(с, 3H), 5,20(с, 1H), 2,28(дд, J=40,7, 15,8 Гц, 2H), 7,24(д, J=7,4 Гц, 1H), 7,47-7,39(м, 2H), 7,63(д, J=7,4 Гц, 1H);
MS(EI, m/e)= 544(M+).
Экспериментальный Пример 1: Исследование ингибиторных эффектов в отношении ВИЧ-1 (дикого/мутантного типа) и испытание на цитотоксичность соединения по настоящему изобретению
Для изучения ингибиторных эффектов соединения по настоящему изобретению в отношении ВИЧ-1 (дикого/мутантного типа) осуществляли испытание для определения эффекта ингибирования ВИЧ-1 (дикого/мутантного типа) in vitro следующим образом в соответствии с известным способом (H. Tanaka et al., J. Med. Chem., 1991, 34, 349). Клетки МТ-4 использовали в качестве клеток-хозяев, и исследовали степень ингибирования и цитотоксичности соединения по настоящему изобретению для инфицированных вирусом клеток МТ-4.
Сначала клетки МТ-4 диспергировали в культуральной среде в концентрации 1 × 104 клеток/лунка, и ВИЧ-1 инокулировали так, чтобы концентрация составляла 500 TCI50 (концентрация, при которой инфицировано 50% клеток)/лунка. Сразу после инокуляции клеточную дисперсию переносили по 100 мкл на плоскую микротитровальную пластинку, содержащую образец соединения по настоящему изобретению. Образец инкубировали приблизительно в течение 4-5 дней при 17°С и эффект ингибирования вируса определяли методом МТТ. Кроме того, жизнеспособность экспериментально инфицированных клеток наблюдали с использованием МТТ метода для определения степени цитотоксичности. В качестве сравнительного соединения использовали азидотимидин (AZT), Ралтегравир, Долутегравир И Элвитегравир. Результаты показаны в таблицах 1 и 2 ниже.
** ИК50: полумаксимальная ингибирующая концентрация
Экспериментальный Пример 2: Фармакокинетическое испытание соединения по настоящему изобретению
Осуществляли эксперименты для определения изменений кинетики in vivo, включая абсорбцию, дистрибуцию, метаболизм и экскрецию in vivo, соединения примера 1 по настоящему изобретению. Вставляли трубку в яремную вену и бедренную вену крысы. Лекарственное средство вводили в бедренную вену в случае внутривенного введения, и лекарственное средство вводили в полость рта в случае перорального введения. Кровь собирали из яремной вены в заранее установленное время.
Концентрация дозы составляла 1 мг/кг для внутривенного введения и 2 мг/кг для перорального введения. После центрифугирования крови для отделения плазмы образцы плазмы и мочи предварительно обрабатывали подходящим органическим растворителем, а затем концентрацию лекарственного средства анализировали при помощи ЖХ-МС/МС. На основании данных концентрации лекарственного средства в крови в зависимости от времени, которые анализировали после перорального и внутривенного введения, некомпартментный фармакокинетический параметр рассчитывали при помощи WinNonlin (Pharsight, США).
Экспериментальный Пример 3: Испытание метаболической стабильности соединения по настоящему изобретению in vitro
Испытание на метаболическую стабильность in vitro осуществляли для соединения Примера 1 по настоящему изобретению. Для подтверждения метаболической стабильности in vitro наблюдали период полужизни соединения в микросомах печени. Осуществляли взаимодействие лекарственного соединения с NADPH с использованием видоспецифической (крысы, собаки, обезьяны и человека) микросомы печени, содержащей различные метаболизирующие ферменты, и затем определяли период полужизни лекарственного средства путем количественного определения методом ЖХ–МС/МС в минутах. Было обнаружено, что соединение Примера 1 является стабильным соединением с периодом полужизни 2 или 3 часа или более.
(T1/2, мин.)
(T1/2, мин.)
(T1/2, мин.)
(T1/2, мин.)
(Тестостерон)
Экспериментальный Пример 4: Испытание ингибирования CYP450 соединением по настоящему изобретению
Испытание ингибирования CYP450 осуществляли для соединения по настоящему изобретению. К микросомам печени человека (0,25 мг/мл), 0,1 M фосфатного буфера (pH 7,4) и коктейлям лекарственных средств из пяти ферментов, метаболизирующих лекарственные средства (CYP1A2, CYP2C9, CYP2D6, CYP3A4 и CYPC19) (Коктейль A: Фенацетин 50 мкМ, S–мефенитоин 100 мкМ, декстрометорфан 5 нМ, мидазолам 2,5 мкМ, Коктейль B: толбутамид 100 мкМ) добавляли соединение Примера 1 в концентрации 0 и 10 мкМ, соответственно, и затем культивировали при 37°С в течение 15 минут. Затем для остановки реакции к смеси добавляли ацетонитрильный раствор, содержащий внутренний стандарт (хлорпропамид), и полученную смесь центрифугировали (14000 об/мин, 4°С) в течение 5 минут. Супернатант затем вводили в систему ЖХ/МС/МС и метаболиты субстратных лекарственных средств одновременно анализировали, чтобы таким образом оценить активность испытываемого соединения, ингибирующего ферменты, метаболизирующие лекарственные средства. Было определено, что соединение Примера 1 не проявляет ингибирующей активности против таких пяти CYP ферментов.
Экспериментальный Пример 5: анализ hERG K+ канала для оценки соединения по настоящему изобретению
Анализ hERG K+ канала осуществляли для прогнозирования кардиотоксичности соединения по настоящему изобретению. hERG-активность соединения измеряли при помощи HERG-HEK293 с использованием автоматического планарного пэтч-кламп анализа [PatchXpress 7000A]. Этот метод является наиболее известным методом исследования ионного канала, в котором поток ионов через канал измеряют непосредственно при помощи фиксации потенциала. ИК50 значение в анализе hERG K+ канала для соединения Примера 1 составило 66,7 мкМ. Значение ИК50 ниже 10 мкМ является критерием, для которого определено, что возможно проявление кардиотоксичности. Поэтому соединение Примера 1, значение которого было больше такого критерия, идентифицировали как безопасное.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРРОЛОПИРИДИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2815649C1 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2833354C1 |
| НОВЫЕ ХИНАЗОЛИОНОВЫЕ ПРОИЗВОДНЫЕ, ИНГИБИРУЮЩИЕ PI3K, И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2017 |
|
RU2702904C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-A]ПИРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2017 |
|
RU2725147C1 |
| КОНДЕНСИРОВАННЫЕ КОЛЬЦЕВЫЕ ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ TRK | 2015 |
|
RU2708674C2 |
| ФТОРЗАМЕЩЕННЫЕ (3R,4R,5S)-5-ГУАНИДИНО-4-АЦИЛАМИНО-3-(ПЕНТАН-3-ИЛОКСИ)ЦИКЛОГЕКСЕН-1-КАРБОНОВЫЕ КИСЛОТЫ, ИХ ЭФИРЫ И СПОСОБ ПРИМЕНЕНИЯ | 2012 |
|
RU2489422C1 |
| НОВАЯ СОЛЬ ТЕНОФОВИРА ДИЗОПРОКСИЛА | 2015 |
|
RU2660438C1 |
| НОВОЕ ЗАМЕЩЕННОЕ ДЕЙТЕРИЕМ ПРОИЗВОДНОЕ ПИРИМИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2020 |
|
RU2811770C1 |
| НОВОЕ ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ, ОБЛАДАЮЩЕЕ ЭФФЕКТОМ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2744168C1 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ПИРИМИДИНА, ПРОЯВЛЯЮЩЕЕ ИНГИБИРУЮЩЕЕ ДЕЙСТВИЕ НА РОСТ РАКОВЫХ КЛЕТОК, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО | 2020 |
|
RU2834201C1 |
Изобретение относится к соединению химической формулы I, в которой R1 представляют собой C1–6 алкил и R2 и R3 каждый независимо представляют собой водород. Изобретение также относится к применению соединения. Технический результат: получено новое соединение химической формулы I, которое эффективно при лечении или ингибировании вируса иммунодефицита человека (ВИЧ). 2 н. и 1 з.п. ф-лы, 6 табл., 9 пр.
Химическая формула I
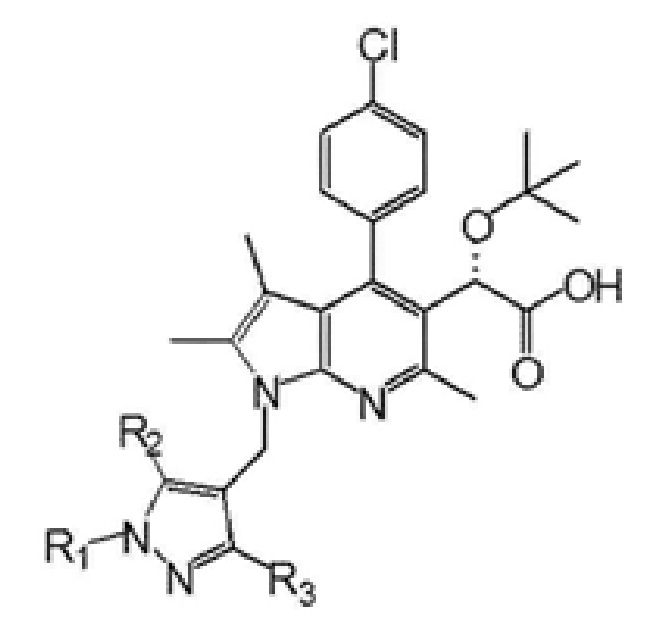
1. Соединение, представленное следующей химической формулой I:
Химическая формула I
 ,
,
где
R1 представляют собой C1–6 алкил и
R2 и R3 каждый независимо представляют собой водород.
2. Соединение по п. 1,
где R1 представляет собой метил.
3. Применение соединения по п. 1 или 2 для лечения или ингибирования вируса иммунодефицита человека (ВИЧ).
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| EA 201000777 A1, 30.12.2010 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

