РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет предварительной заявки на патент США № 60/987958, поданной 14 ноября 2007 года, и предварительной заявки на патент США № 61/025458, поданной 1 февраля 2008 года. Содержание упомянутых выше заявок приводится здесь путем ссылки на них.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям-ингибиторам протеазы вируса гепатита С (HCV), обладающим противовирусной активностью в отношении HCV и применяющимся при лечении HCV инфекций. Более конкретно, изобретение относится к хиноксалин-содержащим соединениям, композициям, включающим такие соединения, и способам их применения, а также способам получения таких соединений.
УРОВЕНЬ ТЕХНИКИ
HCV является основной причиной не-A и не-B гепатита и становится все более и более серьезной проблемой для здравоохранения как развитых, так и развивающихся стран. По оценкам, во всем мире вирусом инфицировано свыше 200 миллионов человек, что больше примерно в пять раз, чем количество людей, инфицированных вирусом иммунодефицита человека (HIV). Вследствие того, что высокая доля людей поражена хроническими инфекциями, для пациентов, инфицированных HCV, существуют повышенный риск развития цирроза печени, последующей злокачественной гепатомы и смертельного заболевания печени. В западном мире HCV является самой распространенной причиной гепатоцеллюлярного рака и поводом для пересадки печени пациентам.
Существуют значительные препятствия для разработки лекарственных препаратов против HCV, которые включают, но этим не ограничиваются, персистенцию вируса, генетическое разнообразие вируса в процессе репликации в организме-хозяине, высокое число случаев развития мутантов вируса, резистентных к лекарственным средствам, и недостаток в воспроизводимых инфекционных культуральных системах и моделях мелких животных для репликации HCV и патогенеза. В большинстве случаев, при мягком течении инфекционной болезни и учитывая сложную биологию печени, следует тщательно выбирать антивирусные лекарственные средства, которые, вероятно, могут обладать значительными побочными эффектами.
В настоящее время в распоряжении имеются только два апробированных метода лечения инфекции HCV. Основная схема лечения обычно включает 3-12 месячный курс внутривенного введения интерферона-α (IFN-α), в то время как недавно опробованная терапия второго поколения включает совместный прием препаратов IFN-α и общих противовирусных нуклеозидных имитаторов, таких как рибавирин. Недостатком этих двух способов терапии является наличие связанных с интерфероном побочных эффектов, а также низкая эффективность по отношению к HCV инфекции. Поэтому, вследствие плохой переносимости и неудовлетворительной эффективности применяемых в настоящее время способов лечения, существует необходимость в разработке эффективных противовирусных средств для лечения HCV инфекции.
Для группы больных, в которой большинство людей являются хронически инфицированными и не обнаруживают симптомов заболевания, и прогнозы не известны, эффективное лекарственное средство должно, соответственно, обладать значительно меньшими побочными эффектами, чем доступные в настоящее время способы лечения. Неструктурный белок-3 вируса гепатита C (NS3) является протеолитическим ферментом, требующимся для процессирования вирусного полипротеина и, следовательно, вирусной репликации. Несмотря на громадное число вирусных вариантов, связанных с HCV инфекцией, активный центр NS3 протеазы остается практически неизменным, в силу чего его ингибирование может представлять собой перспективный метод воздействия. Недавнее достижение в терапии HIV путем применения ингибиторов протеазы подтверждает идею о том, что основной целью в борьбе против HCV является ингибирование NS3.
HCV является РНК-содержащим вирусом семейства Flaviridae. HCV геном заключен в оболочку и содержит молекулу одноцепочечной РНК, состоящую из примерно 9600 пар оснований. Он кодирует полипептид, состоящий из приблизительно 3010 аминокислот.
HCV полипротеин процессируется вирусной пептидазой и пептидазой организма-хозяина в 10 дискретных пептидов, которые выполняют целый ряд функций. Имеется три структурных белка, C, E1 и E2. Белок P7 выполняет неизвестную функцию и состоит из сильно изменяющейся последовательности. Имеется шесть неструктурных белков. NS2 является цинк-зависимой металлопротеиназой, которая функционирует совместно с частью NS3 белка. NS3 включает две каталитических функции (отдельно от его ассоциации с NS2): серинпротеазу на N-конце, которая требует NS4A в качестве кофактора, и функцию АТФаза-зависимой геликазы на карбоксильном конце. NS4A является прочно связанным, но не ковалентно, кофактором серинпротеазы.
NS3-NS4A протеаза является ответственной за расщепление четырех центров вирусного полипротеина. NS3-NS4A расщепление является автокаталитическим, происходящим в цис-положении. Оставшиеся три реакции гидролиза, NS4A-NS4B, NS4B-NS5A и NS5A-NS5B, все происходят в транс-положении. NS3 является серинпротеазой, которая структурно классифицируется как химотрипсин-подобная протеаза. В то время как NS серинпротеаза сама по себе обладает протеолитической активностью, фермент протеазы HCV не является эффективным ферментом с точки зрения катализа расщепления полипротеина. Было показано, что для этого усиления требуется центральная гидрофобная область NS4A белка. Для актов процессирования, по-видимому, необходимо образование комплекса NS3 белка с NS4A, усиливающее протеолитическую эффективность на всех центрах.
Общей стратегией при разработке противовирусных средств является инактивация ферментов, кодируемых вирусом, включая NS3, которые являются необходимыми для репликации вируса. Проводимые в настоящий момент исследования, направленные на поиск ингибиторов NS3 протеазы, были рассмотрены в обзоре S. Tan, A. Pause, Y. Shi, N. Sonenberg, Hepatitis C Therapeutics: Current Status and Emerging Strategies, Nature Rev. Drug Discov. 1, 867-881 (2002).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к хиноксалин-содержащим соединениям и их фармацевтически приемлемым солям, сложным эфирам или пролекарствам, и способам их применения для лечения инфекции гепатита C у субъекта, которому необходима такая терапия. Соединения по настоящему изобретению нарушают жизненный цикл вируса гепатита С, и их также применяют в качестве противовирусных средств. Кроме того, настоящее изобретение относится к фармацевтическим композициям, включающим упомянутые выше соединения, соли, сложные эфиры или пролекарства, для введения субъекту, страдающему от инфекции HCV. Настоящее изобретение дополнительно характеризует фармацевтические композиции, включающие соединение по настоящему изобретению (или его фармацевтически приемлемую соль, сложный эфир или лекарство) и другое анти-HCV средство, такое как интерферон (например, альфа-интерферон, бета-интерферон, консенсусный интерферон, пегилированный интерферон или альбумин или другой сопряженный интерферон), рибавирин, амантадин, другой ингибитор HCV протеазы или HCV полимеразы, геликазы или ингибитор участка внутренней посадки рибосомы. Изобретение также относится к способам лечения HCV инфекции у субъекта путем введения субъекту фармацевтической композиции по настоящему изобретению. Настоящее изобретение дополнительно относится к фармацевтическим композициям, включающим соединения по настоящему изобретению или их фармацевтически приемлемые соли, сложные эфиры или пролекарства в комбинации с фармацевтически приемлемым носителем или эксципиентом.
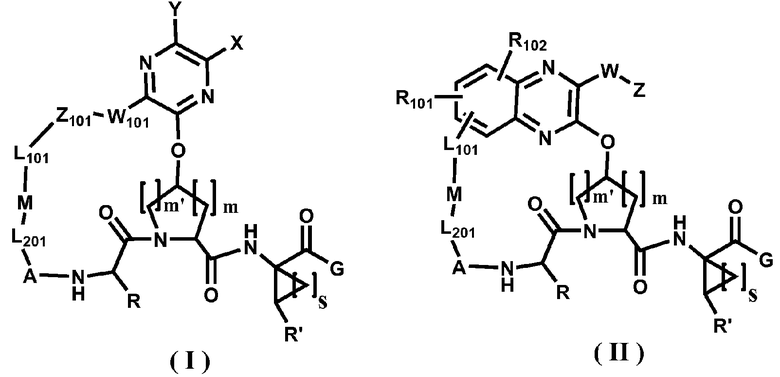
В одном варианте осуществления настоящего изобретения раскрываются соединения, представленные формулами I или II, или их фармацевтически приемлемые соли, сложные эфиры или пролекарства:
 ,
,
где
А отсутствует или его выбирают из -(C=O)-, -S(O)2, -C=N-OR1 или -C(=N-CN);
L201 отсутствует или его выбирают из -C1-C8 алкилена, -C2-C8 алкенилена или -C2-C8 алкинилена, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; замещенного -C1-C8 алкилена, замещенного -C2-C8 алкенилена или замещенного -C2-C8 алкинилена, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкилена или замещенного -C3-C12 циклоалкилена, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкенилена или замещенного -C3-C12 циклоалкенилена, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N;
M отсутствует или его выбирают из O, S, SO, SO2 или NR1; где R1 выбирают при каждом его появлении из группы, состоящей из:
(i) водорода;
(ii) арила; замещенного арила; гетероарила; замещенного гетероарила;
(iii) гетероциклоалкила или замещенного гетероциклоалкила;
(iv) -C1-C8 алкила, -C2-C8 алкенила или -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; замещенного -C1-C8 алкила, замещенного -C2-C8 алкенила или замещенного -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкила или замещенного -C3-C12 циклоалкила; -C3-C12 циклоалкенила или замещенного -C3-C12 циклоалкенила;
L101 отсутствует или его выбирают из -C1-C8 алкилена, -C2-C8 алкенилена или -C2-C8 алкинилена, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; замещенного -C1-C8 алкилена, замещенного -C2-C8 алкенилена или замещенного -C2-C8 алкинилена, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкилена или замещенного -C3-C12 циклоалкилена, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкенилена или замещенного -C3-C12 циклоалкенилена, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N;
Z101 отсутствует или его выбирают из арила, замещенного арила, гетероарила или замещенного гетероарила;
W101 отсутствует или его выбирают из -O-, -S-, -NR1-, -C(O)- или -C(O)NR1-;
X и Y, взятые вместе с атомами углерода, к которым они присоединены, образуют карбоциклический фрагмент или гетероциклический фрагмент. Карбоциклический фрагмент или гетероциклический фрагмент может быть выбран из арила, замещенного арила, гетероарила, замещенного гетероарила, циклоалкила, замещенного циклоалкила, циклоалкенила, замещенного циклоалкенила, гетероцикла или замещенного гетероцикла;
или X и Y вместе могут образовывать C2-C8-алкиленовую группу или C2-C8-гетероалкиленовую группу.
R101 и R102 независимо выбирают из группы, состоящей из:
(i) водорода, галогена, CN, CF3, N3, NO2, OR1, SR1, SO2R2, -NHS(O)2-R2, -NH(SO2)NR3R4, NR3R4, CO2R1, COR1, CONR1R2, N(R1)COR2;
(ii) арила; замещенного арила; гетероарила; замещенного гетероарила;
(iii) гетероциклоалкила или замещенного гетероциклоалкила;
(iv) -C1-C8 алкила, -C2-C8 алкенила или -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; замещенного -C1-C8 алкила, замещенного -C2-C8 алкенила или замещенного -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 и 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкила или замещенного -C3-C12 циклоалкила; -C3-C12 циклоалкенила или замещенного -C3-C12 циклоалкенила;
R и R' каждый независимо выбирают из группы, состоящей из:
(i) -C1-C8 алкила, -C2-C8 алкенила или -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; замещенного -C1-C8 алкила, замещенного -C2-C8 алкенила или замещенного -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкила или замещенного -C3-C12 циклоалкила; -C4-C12 алкилциклоалкила или замещенного -C4-C12 алкилциклоалкила; -C3-C12 циклоалкенила или замещенного -C3-C12 циклоалкенила; -C4-C12 алкилциклоалкенила или замещенного -C4-C12 алкилциклоалкенила;
(ii) арила; замещенного арила; гетероарила; замещенного гетероарила;
(iii) гетероциклоалкила или замещенного гетероциклоалкила;
(iv) водорода; дейтерия;
или R' является C1-C8-алкилом, замещенным одним или более атомами галогена, предпочтительно одним или более атомами фтора, хлора или брома. Предпочтительно, чтобы R' являлся -CHQ1Q2, где Q1 и Q2 независимо выбирают из галогена; предпочтительно F, Cl и Br;
G выбирают из -OH, -NHS(O)2-R2, -NH(SO2)NR3R4 и NR3R4;
R2 выбирают из:
(i) арила; замещенного арила; гетероарила; замещенного гетероарила;
(ii) гетероциклоалкила; замещенного гетероциклоалкила;
(iii) -C1-C8 алкила, -C2-C8 алкенила или -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N, замещенного -C1-C8 алкила, замещенного -C2-C8 алкенила или замещенного -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкила или замещенного -C3-C12 циклоалкила; -C3-C12 циклоалкенила или замещенного -C3-C12 циклоалкенила; гетероцикла; замещенного гетероцикла;
R3 и R4 независимо выбирают из:
(i) водорода;
(ii) арила; замещенного арила; гетероарила; замещенного гетероарила;
(iii) гетероциклоалкила или замещенного гетероциклоалкила;
(iv) -C1-C8 алкила, -C2-C8 алкенила или -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; замещенного -C1-C8 алкила, замещенного -C2-C8 алкенила или замещенного -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкила или замещенного -C3-C12 циклоалкила; -C3-C12 циклоалкенила или замещенного -C3-C12 циклоалкенила; гетероцикла или замещенного гетероцикла;
или альтернативно R3 и R4, взятые вместе с азотом, к которому они присоединены, образуют гетероцикл или замещенный гетероцикл;
Z выбирают из группы, состоящей из:
(i) водорода;
(ii) CN;
(iii) N3;
(iv) галогена;
(v) -NH-N=CHR1;
(vi) арила, замещенного арила;
(vii) гетероарила, замещенного гетероарила;
(viii) -C3-C12 циклоалкила, замещенного -C3-C12 циклоалкила, гетероциклоалкила, замещенного гетероциклоалкила;
(ix) -C1-C6 алкила, содержащего 0, 1, 2 или 3 гетероатома, выбранных из О, S или N, необязательно замещенного одним или более заместителями, выбранными из галогена, арила, замещенного арила, гетероарила или замещенного гетероарила;
(x) -C2-C6 алкенила, содержащего 0, 1, 2 или 3 гетероатома, выбранных из О, S или N, необязательно замещенного одним или более заместителями, выбранными из галогена, арила, замещенного арила, гетероарила или замещенного гетероарила;
(xi) -C2-C6 алкинила, содержащего 0, 1, 2 или 3 гетероатома, выбранных из О, S или N, необязательно замещенного одним или более заместителями, выбранными из галогена, арила, замещенного арила, гетероарила или замещенного гетероарила;
W отсутствует или его выбирают из алкилена, алкенилена, алкинилена, -O-, -S-, -NR1-, -C(O)NR1- или -C(O)-;
m является 0, 1, 2 или 3; предпочтительно 1;
m' является 0, 1, 2 или 3; предпочтительно 1; и
s является 1, 2, 3 или 4; предпочтительно 1.
В одной подгруппе соединений формул I и II:
M отсутствует или его выбирают из O или NR1;
Z101 является арилом, замещенным арилом, гетероарилом или замещенным гетероарилом;
W101 отсутствует или его выбирают из -O-, -S-, -NH-, -N(Me)-, -C(O)NH- или -C(O)N(Me)-;
X и Y, взятые вместе с атомами углерода, с которыми они соединены, образуют циклический фрагмент, который выбирают из арила, замещенного арила, гетероарила, замещенного гетероарила, гетероцикла или замещенного гетероцикла;
R101 и R102 независимо выбирают из группы, состоящей из:
(i) водорода, галогена, CN, CF3, NO2, OR1, SR1, -NHS(O)2-R2, -NH(SO2)NR3R4, NR3R4, CO2R1, COR1, CONR1R2, N(R1)COR2;
(ii) арила; замещенного арила; гетероарила; замещенного гетероарила;
(iii) гетероциклоалкила или замещенного гетероциклоалкила;
(iv) -C1-C8 алкила, -C2-C8 алкенила или -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; замещенного -C1-C8 алкила, замещенного -C2-C8 алкенила или замещенного -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкила или замещенного -C3-C12 циклоалкила; -C3-C12 циклоалкенила или замещенного -C3-C12 циклоалкенила;
R3 и R4 независимо выбирают из:
(i) водорода;
(ii) арила; замещенного арила; гетероарила; замещенного гетероарила;
(iii) гетероциклоалкила или замещенного гетероциклоалкила;
(iv) -C1-C8 алкила, -C2-C8 алкенила или -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; замещенного -C1-C8 алкила, замещенного -C2-C8 алкенила или замещенного -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкила или замещенного -C3-C12 циклоалкила; -C3-C12 циклоалкенила или замещенного -C3-C12 циклоалкенила; гетероцикла или замещенного гетероцикла;
W отсутствует или его выбирают из алкилена, алкенилена, алкинилена, -O-, -S-, -NH-, -N(Me)-, -C(O)NH- или -C(O)N(Me)-;
m = 0, 1 или 2;
m' = 1 или 2; и
s является 1.
В другом варианте осуществления настоящее изобретение характеризует фармацевтические композиции, включающие соединение по изобретению или его фармацевтически приемлемую соль, сложный эфир или пролекарство. В еще одном варианте осуществления настоящего изобретения раскрываются фармацевтические композиции, включающие терапевтически эффективное количество соединения по изобретению или его фармацевтически приемлемой соли, сложного эфира или пролекарства, в комбинации с фармацевтически приемлемым носителем или эксципиентом. В еще одном варианте осуществления изобретения раскрываются способы лечения инфекции гепатита C у пациента, который нуждается в таком лечении, при помощи указанных фармацевтических композиций.
Подробное описание изобретения
Первым вариантом осуществления изобретения является соединение, представленное описанными выше формулой I или формулой II, или его фармацевтически приемлемая соль, сложный эфир или пролекарство, само по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом.
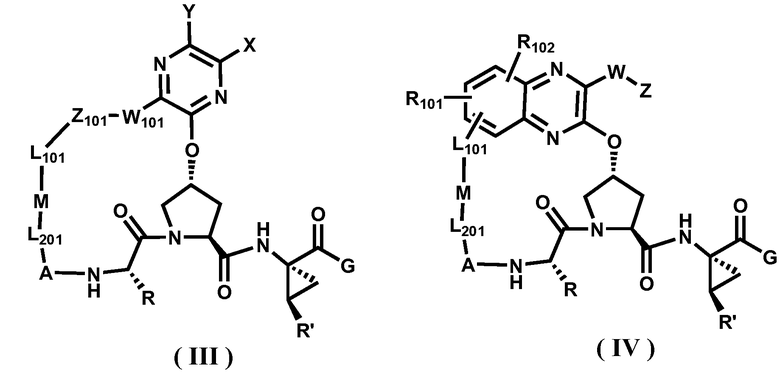
Другими вариантами осуществления изобретения являются соединения, представленные формулой III или IV:
 ,
,
или их фармацевтически приемлемая соль, сложный эфир или пролекарство, сами по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом, где R, R', A, L201, M, L101, Z101, W101, X, Y, R101, R102, W, Z и G определены выше.
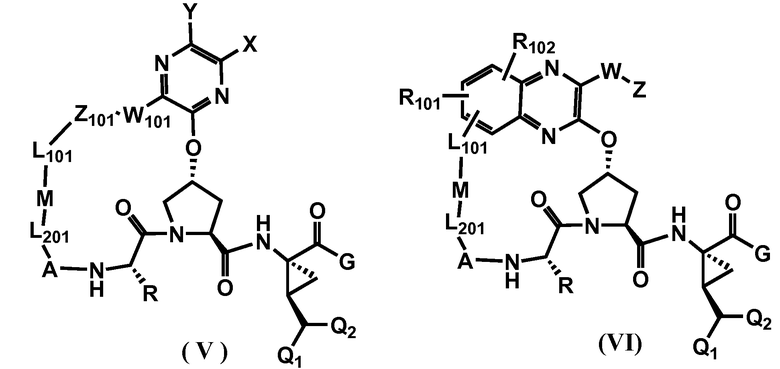
Другими вариантами осуществления изобретения являются соединения, представленные формулой V или VI:
 ,
,
или их фармацевтически приемлемая соль, сложный эфир или пролекарство, сами по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом, где Q1 и Q2 являются независимо фтором, хлором или бромом, и где R, A, L201, M, L101, Z101, W101, X, Y, R101, R102, W, Z и G определены выше. В особенно предпочтительных вариантах осуществления, Q1 и Q2 являются оба фтором.
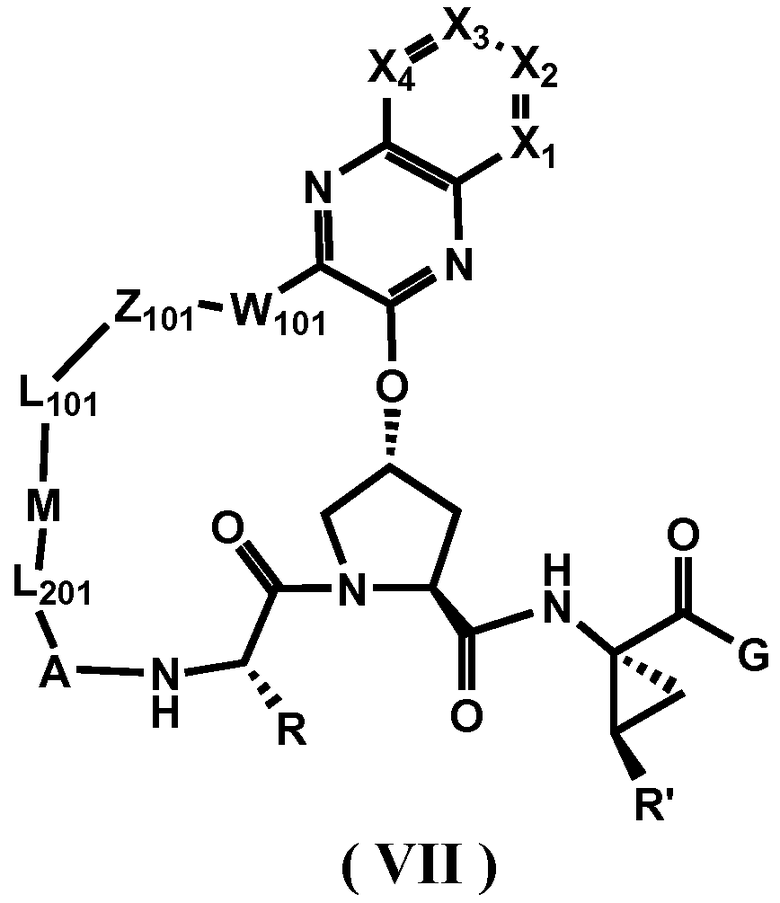
Другим вариантом осуществления изобретения является соединение, представленное формулой VII:
 ,
,
или его фармацевтически приемлемая соль, сложный эфир или пролекарство, само по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом, где X1-X4 независимо выбирают из -CR5 и N, где R5 независимо выбирают из:
(i) водорода; галогена; -NO2; -CN; N3; CF3;
(ii) -M-R4, M является O, S, NH;
(iii) NR3R4;
(iv) -C1-C8 алкила, -C2-C8 алкенила или -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; замещенного -C1-C8 алкила, замещенного -C2-C8 алкенила или замещенного -C2-C8 алкинила, каждый из которых содержит 0, 1, 2 или 3 гетероатома, выбранных из О, S или N; -C3-C12 циклоалкила или замещенного -C3-C12 циклоалкила; -C3-C12 циклоалкенила или замещенного -C3-C12 циклоалкенила;
(v) арила; замещенного арила; гетероарила; замещенного гетероарила;
(vi) гетероциклоалкила или замещенного гетероциклоалкила;
где R3, R4, R, R', A, L201, M, L101, Z101, W101 и G определены выше.
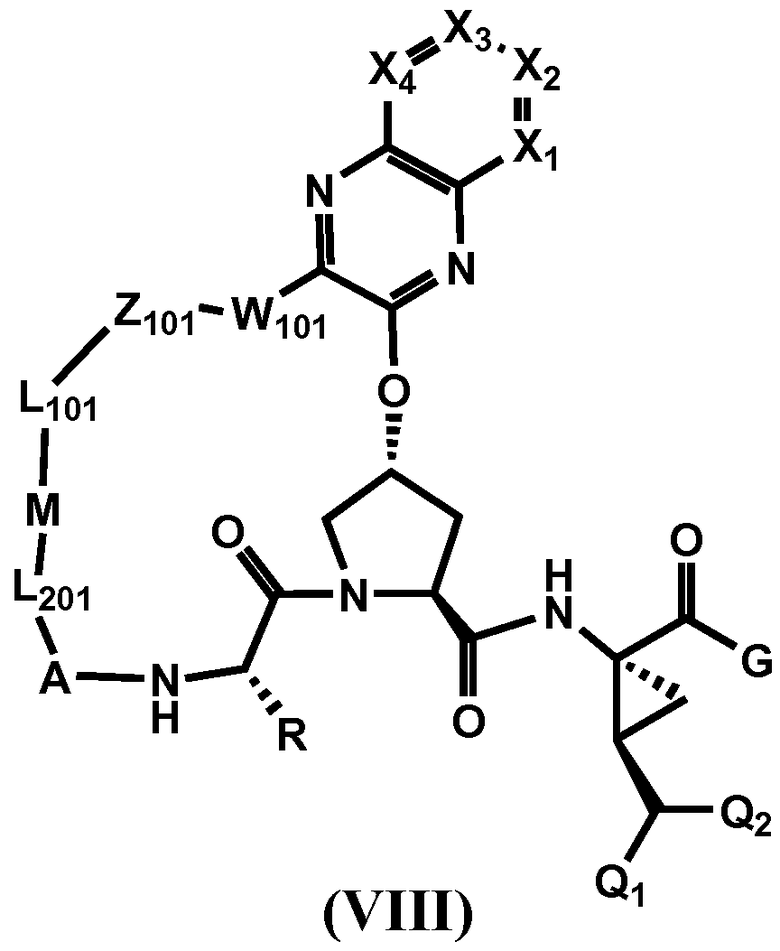
Другим вариантом осуществления изобретения является соединение, представленное формулой VIII:
 ,
,
или его фармацевтически приемлемая соль, сложный эфир или пролекарство, само по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом, где X1-X4 определены для формулы VII, и R, A, Q1, Q2, L201, M, L101, Z101, W101 и G определены выше.
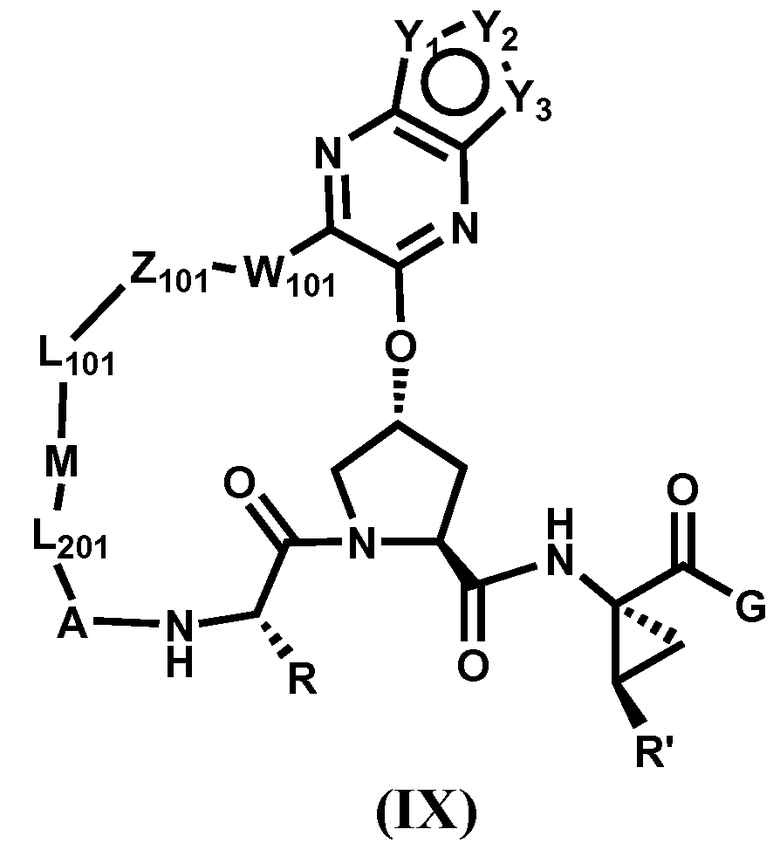
Другим вариантом осуществления изобретения является соединение, представленное формулой IX:
 ,
,
или его фармацевтически приемлемая соль, сложный эфир или пролекарство, само по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом, где Y1-Y3 независимо выбирают из CR5, N, NR5, S и O; где R5, R, R', A, L201, M, L101, Z101, W101 и G определены выше.
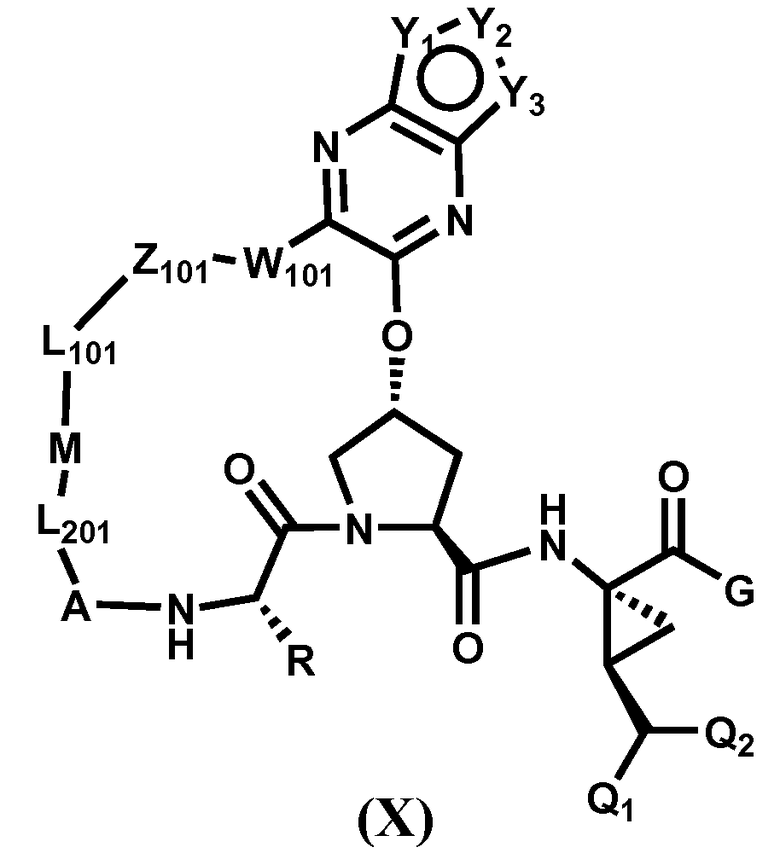
Другим вариантом осуществления изобретения является соединение, представленное формулой X:
 ,
,
его фармацевтически приемлемая соль, сложный эфир или пролекарство, само по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом, где Y1-Y3 определены для формулы IX, и R, Q1, Q2, A, L201, M, L101, Z101, W101 и G определены выше.
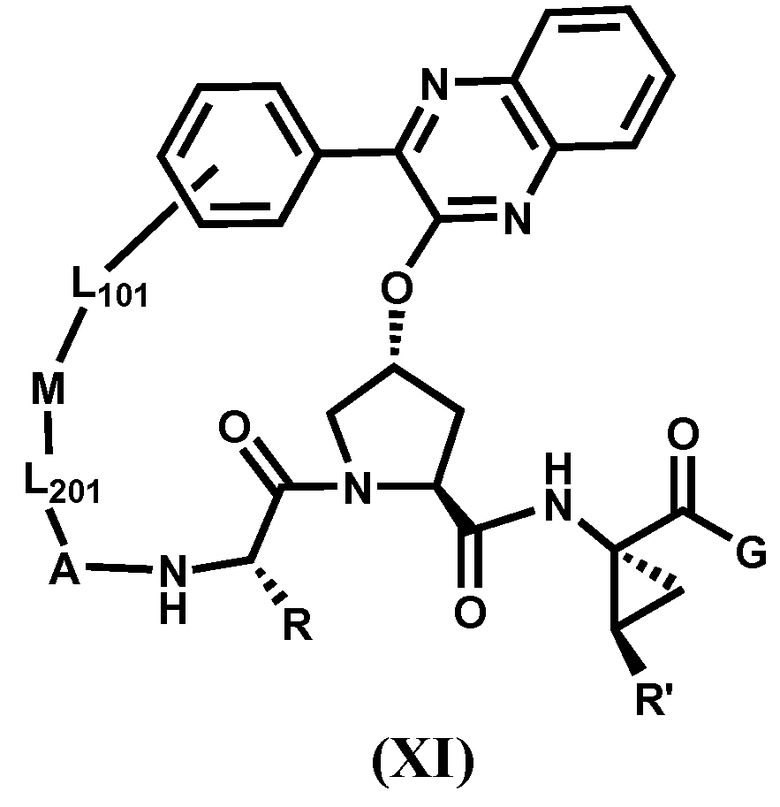
Другим вариантом осуществления изобретения является соединение, представленное формулой XI:
 ,
,
или его фармацевтически приемлемая соль, сложный эфир или пролекарство, само по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом, где R, R', A, L201, M, L101 и G определены выше.
Другим вариантом осуществления изобретения является соединение, представленное формулой XII:
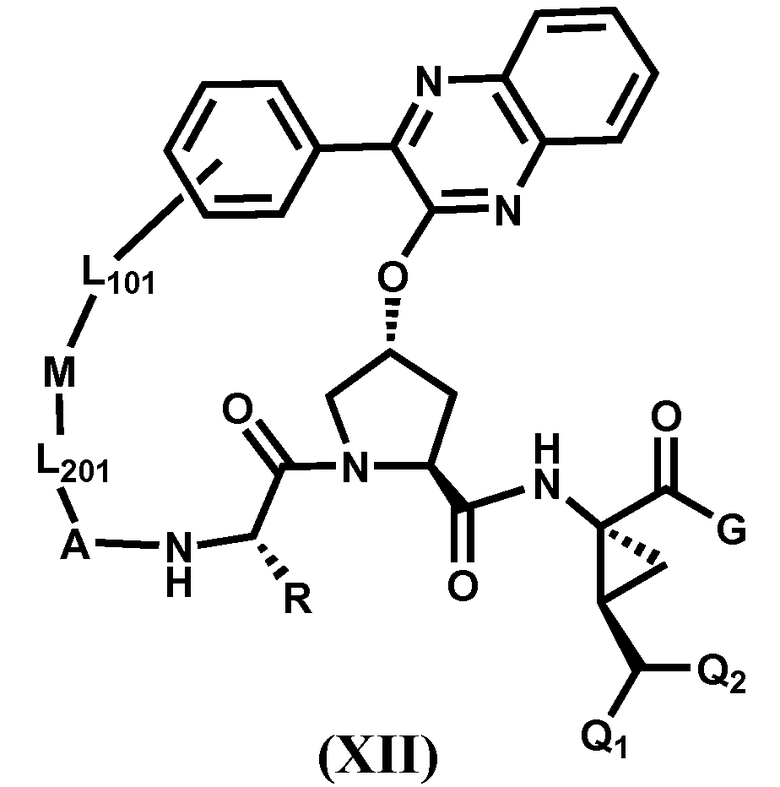 ,
,
его фармацевтически приемлемая соль, сложный эфир или пролекарство, само по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом, где R, Q1, Q2, A, L201, M, L101 и G определены выше.
Другим вариантом осуществления изобретения является соединение, представленное формулой XIII:
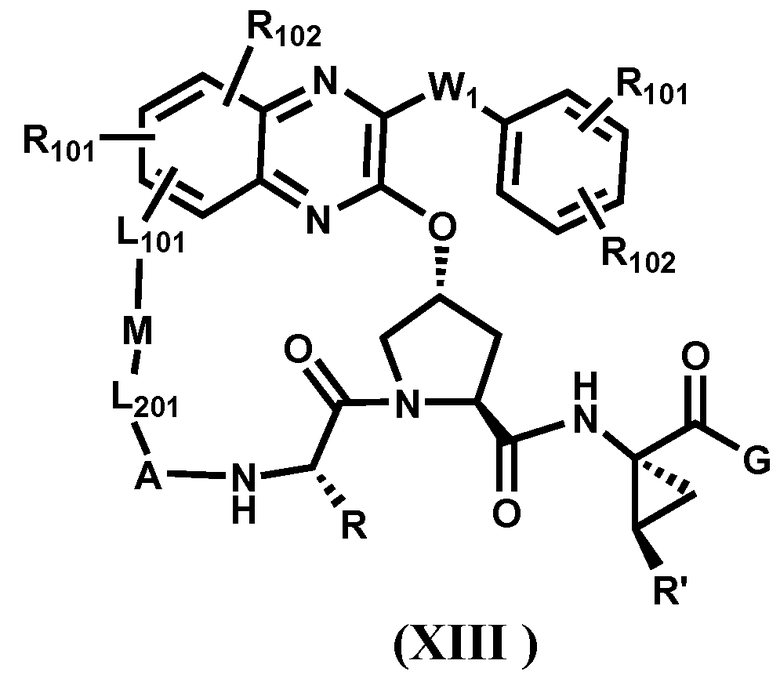 ,
,
или его фармацевтически приемлемая соль, сложный эфир или пролекарство, само по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом, где W1 отсутствует или его выбирают из C1-C4 алкилена, C2-C4 алкенилена, C2-C4 алкинилена; где R, R', A, L201, M, L101, R101, R102 и G определены выше.
Другим вариантом осуществления изобретения является соединение, представленное формулой XIV:
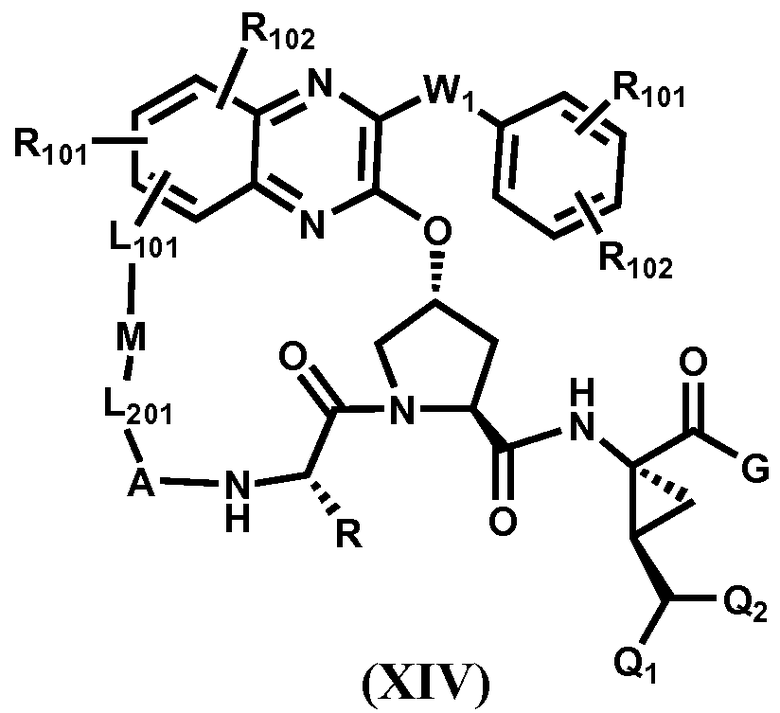 ,
,
его фармацевтически приемлемая соль, сложный эфир или пролекарство, само по себе или в комбинации с фармацевтически приемлемым носителем или эксципиентом, где W1 определено для формулы XIII, и R, Q1, Q2, A, L201, M, L101, R101, R102 и G определены выше.
Характерные соединения изобретения включают, но этим не ограничиваясь, следующие соединения (таблица 1) в соответствии с формулой XV, где R, M-L, Ar, R' и G определены для каждого примера в таблице 1.
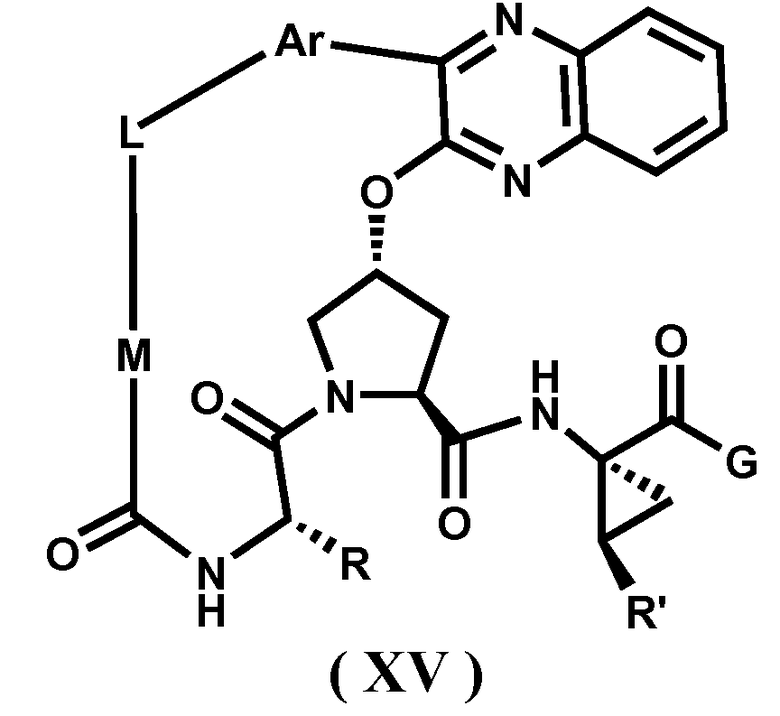
Характерные соединения изобретения также включают, но этим не ограничиваясь, следующие соединения (таблица 2) в соответствии с формулой XVI, где R, L-Ar, n, R' и G определены для каждого примера в таблице 2.
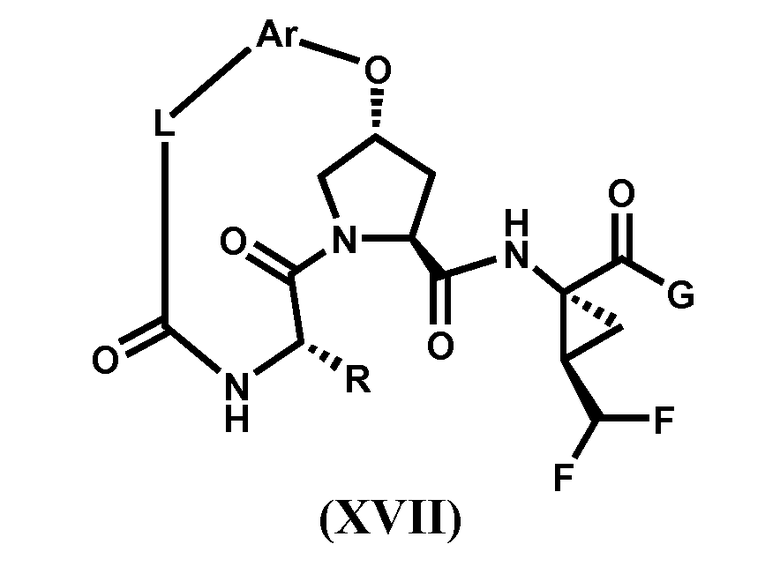
Характерные соединения изобретения также включают, но этим не ограничиваясь, следующие соединения (таблица 3) в соответствии с формулой XVII, где R, L-Ar, n и G определены для каждого примера в таблице 3.
Настоящее изобретение характеризует также фармацевтические композиции, включающие соединение по настоящему изобретению или его фармацевтически приемлемую соль, сложный эфир или пролекарство.
Соединения по настоящему изобретению могут быть введены в качестве единственного активного фармацевтического средства или использоваться в комбинации с одним или более средствами для лечения или предотвращения инфекций гепатита C или симптомов, связанных с HCV инфекцией. Другие средства, которые могут быть введены в комбинации с соединением или в комбинации с соединениями по изобретению, включают лекарственные средства для лечения заболевания, вызванного HCV инфекцией, которые подавляют репликацию HCV вируса на основе прямого или косвенного механизмов. Они включают такие средства, как иммуномодуляторы для организма-хозяина (например, интерферон-альфа, пегилированный интерферон-альфа, интерферон-бета, интерферон-гамма, CpG олигонуклеотиды и другие подобные иммуномодуляторы) или противовирусные соединения, которые ингибируют клеточные функции в организме-хозяине, такие как инозинмонофосфат дегидрогеназа (например, рибавирин и другие подобные соединения). Кроме того, они включают цитокины, которые модулируют иммунную функцию. Они также включают вакцины, содержащие HCV антигены или вспомогательные комбинации антигенов, направленные против HCV. Кроме того, они включают средства, которые взаимодействуют с клеточными компонентами организма-хозяина с блокированием синтеза белка вируса путем ингибирования стадии трансляции репликации вируса HCV, инициированной участком внутренней посадки рибосомы (IRES) или с блокированием созревания и высвобождения вирусной частицы при помощи средств, направленных на семейство виропорина мембранных белков, таких как, например, HCV P7, и другие подобные средства. Другие средства, которые могут быть введены в комбинации с соединением по настоящему изобретению, включают любое средство или комбинацию средств, которое ингибирует репликацию HCV путем направленного воздействия на белки вирусного генома, вовлеченного в вирусную репликацию. Эти средства включают, но этим не ограничиваясь, другие ингибиторы HCV РНК-зависимой РНК-полимеразы, такие как, например, ингибиторы полимеразы нуклеозидного типа, описанные в патентных документах WO0190121(A2) или патенте США № 6348587B1 или WO0160315 или WO0132153, или ненуклеозидные ингибиторы, такие как, например, бензимидазольные ингибиторы полимеразы, описанные в патентных документах EP 1162196Al или WO0204425, или ингибиторы HCV протеазы, такие как, например, ингибиторы типа пептидомиметика, такие как BILN2061 и другие подобные пептидомиметики, или ингибиторы HCV геликазы.
Другие средства, которые могут быть введены в комбинации с соединением по настоящему изобретению, включают любое средство или комбинацию средств, которое ингибирует репликацию других вирусов у людей, зараженных различными вирусами. Эти средства включают, но этим не ограничиваясь, медицинские препараты для заболевания, вызванного инфекцией гепатитом B (HBV), такие как, например, адефовир, ламивудин и тенофовир, или медицинские препараты для заболевания, вызванного инфекцией вирусом иммунодефицита человека (HIV), такие как, например, ингибиторы протеазы: ритонавир, лопинавир, индинавир, нелфинавир, саквинавир, ампренавир, атазанавир, типранавир, TMC-114, фосампренавир; ингибиторы обратной транскриптазы: зидовудин, ламивудин, диданозин, ставудин, тенофовир, залцитабин, абакавир, эфавиренз, невирапин, делавирдин, TMC-125; ингибиторы интенгразы: L- 870812, S-1360, или ингибиторы входа: энфувиртид (T-20), T-1249.
Соответственно, один аспект изобретения относится к способу лечения или предотвращения инфекции, вызванной РНК-содержащим вирусом, включающему совместное введение пациенту, при необходимости такого лечения, одного или более средств, выбранных из группы, состоящей из иммуномодулятора организма-хозяина и второго противовирусного средства или их комбинации, с терапевтически эффективным количеством соединения или комбинации соединений по изобретению или его фармацевтически приемлемой соли, стереоизомера, таутомера, пролекарства, соли пролекарства или их комбинации. Примерами иммуномодулятора организма-хозяина являются, но этим не ограничиваясь, интерферон-альфа, пегилированный-интерферон-альфа, интерферон-бета, интерферон-гамма, цитокин, вакцина, и вакцина, включающая антиген и адъювант, и указанное второе противовирусное средство ингибирует репликацию HCV или путем ингибирования клеточных функций организма-хозяина, связанных с вирусной репликацией, или путем направленного воздействия на белки вирусного генома.
Дополнительный аспект изобретения относится к способу лечения или предотвращения инфекции, вызванной РНК-содержащим вирусом, включающему совместное введение пациенту, при необходимости такого лечения, средства или комбинации средств, которые применяют для лечения или облегчения симптомов HCV инфекции, включая цирроз и воспаление печени, с терапевтически эффективным количеством соединения или комбинации соединений по изобретению или его фармацевтически приемлемой соли, стереоизомера, таутомера, пролекарства, соли пролекарства или их комбинации. Еще один аспект изобретения предлагает способ лечения или предотвращения инфекции, вызванной РНК-содержащим вирусом, включающему совместное введение пациенту, при необходимости такого лечения, одного или более средств, которые применяют для лечения пациентов от заболевания, вызванного инфекцией гепатита B (HBV), с терапевтически эффективным количеством соединения или комбинации соединений по изобретению или его фармацевтически приемлемой соли, стереоизомера, таутомера, пролекарства, соли пролекарства или их комбинации. Средством, которое применяют для лечения пациентов от заболевания, вызванного инфекцией гепатита B (HBV), может являться, например, но этим не ограничиваясь, L-дезокситимидин, адефовир, ламивудин или тенофовир или любая их комбинация. Пример РНК-содержащего вируса включает, но этим не ограничиваясь, вирус гепатита С (HCV).
Другой аспект изобретения предлагает способ лечения или предотвращения инфекции, вызванной РНК-содержащим вирусом, включающему совместное введение пациенту, при необходимости такого лечения, одного или более средств, которые применяют для лечения пациентов от заболевания, вызванного инфекцией вирусом иммунодефицита человека (HIV), с терапевтически эффективным количеством соединения или комбинации соединений по изобретению или его фармацевтически приемлемой соли, стереоизомера, таутомера, пролекарства, соли пролекарства или их комбинации. Средство, которое применяют для лечения пациентов от заболевания, вызванного инфекцией вирусом иммунодефицита человека (HIV), может включать, но этим не ограничиваясь, ритонавир, лопинавир, индинавир, нелфинавир, саквинавир, ампренавир, атазанавир, типранавир, TMC-114, фосампренавир, зидовудин, ламивудин, диданозин, ставудин, тенофовир, залцитабин, абакавир, эфавиренз, невирапин, делавирдин, TMC-125, L-870812, S-1360, энфувиртид (T-20) или T-1249 или любую их комбинацию. Пример РНК-содержащего вируса включает, но этим не ограничиваясь, вирус гепатита С (HCV). Кроме того, настоящее изобретение предлагает применение соединения или комбинации соединений по изобретению или его терапевтически приемлемой солевой формы, стереоизомера или таутомера, пролекарства, соли пролекарства или их комбинации, и одного или более средств, выбранных из группы, состоящей из иммуномодулятора организма-хозяина и второго противовирусного средства или их комбинации, для получения лекарственного препарата для лечения инфекции, вызванной РНК-содержащим вирусом, у пациента, в частности, вирусом гепатита С. Примерами иммуномодулятора организма-хозяина являются, но этим не ограничиваясь, интерферон-альфа, пегилированный-интерферон-альфа, интерферон-бета, интерферон-гамма, цитокин, вакцина, и вакцина, включающая антиген и адъювант, и указанное второе противовирусное средство ингибирует репликацию HCV или путем ингибирования клеточных функций организма-хозяина, связанных с вирусной репликацией, либо путем направленного воздействия на белки вирусного генома.
При применении для приведенных выше или других терапиий, комбинация соединения или соединений по изобретению вместе с определенными здесь выше одним или более средствами может быть использована в чистой форме или, когда такие формы существуют, в фармацевтически приемлемой солевой форме, форме пролекарства, соли пролекарства или их комбинации. Альтернативно такая комбинация терапевтических средств может быть введена в виде фармацевтической композиции, содержащей терапевтически эффективное количество представляющего интерес соединения или комбинации соединений или их фармацевтически приемлемой солевой формы, пролекарств или солей пролекарств, в комбинации с определенным выше одним или более средствами и фармацевтически приемлемым носителем. Такие фармацевтические композиции могут быть использованы для ингибирования репликации РНК-содержащего вируса, в частности, вируса гепатита С (HCV), путем контактирования указанного вируса с указанной фармацевтической композицией. Кроме того, такие композиции могут применяться для лечения или предотвращения инфекции, вызванной РНК-содержащим вирусом, в частности, вирусом гепатита С (HCV).
Поэтому дополнительный аспект изобретения относится к способу лечения или предотвращения инфекции, вызванной РНК-содержащим вирусом, в частности, вирусом гепатита С (HCV), включающему введение пациенту, при необходимости такого лечения, фармацевтической композиции, включающей соединение или комбинацию соединений по изобретению или его фармацевтически приемлемую соль, стереоизомер или таутомер, пролекарство, соль пролекарства или их комбинацию, одно или более определенных выше средств, и фармацевтически приемлемый носитель.
При введении в виде комбинации, терапевтические средства могут быть приготовлены в виде отдельных композиций, которые вводят одновременно или в течение заранее установленного периода времени или терапевтические средства могут вводиться в виде одной единичной дозированной формы.
Противовирусные средства, предлагаемые для использования в такой комбинированной терапии, включают средства (соединения или биологические препараты), которые являются эффективными при ингибировании образования и/или репликации вируса у млекопитающего, включая, но этим не ограничиваясь, средства, которые препятствуют либо протеканию механизмов в организме-хозяине, либо протеканию вирусных механизмов, необходимых для образования и/или репликации вируса у млекопитающего. Такие средства могут быть выбраны из еще одного анти-HCV средства, ингибитора HIV, ингибитора HAV и ингибитора HBV.
Другие анти-HCV средства включают те средства, которые являются эффективными для уменьшения или предотвращения развития симптомов или заболевания, связанных с гепатитом C. Такие средства включают, но этим не ограничиваясь, иммуномодулирующие вещества, ингибиторы HCV NS3 протеазы, другие ингибиторы HCV полимеразы, ингибиторы другой мишени в жизненном цикле HCV и другие анти-HCV средства, включая, но этим не ограничиваясь, рибавирин, амантадин, левовирин и вирамидин.
Иммуномодулирующие вещества включают те средства (соединения или биологические препараты), которые являются эффективными для усиления или потенцирования реакции иммунной системы у млекопитающего. Иммуномодулирующие вещества включают, но этим не ограничиваясь, ингибиторы инозинмонофосфат дегидрогеназы, такие как VX-497 (меримеподиб, Vertex Pharmaceuticals), интерфероны класса I, интерфероны класса II, консенсусные интерфероны, асиало-интерфероны, пегилированные интерфероны и конъюгированные интерфероны, включая, но этим не ограничиваясь, интерфероны, конъюгированные с другими белками, включая, но этим не ограничиваясь, человеческий альбумин. Интерфероны класса I являются группой интерферонов, которые все связаны с рецептором типа I, включая как природные, так и синтетически полученные интерфероны класса I, в то время как интерфероны класса II все связаны с рецептором типа II. Примеры интерферонов класса I включают, но этим не ограничиваясь, [альфа]-, [бета]-, [дельта]-, [омега]- и [тау]-интерфероны, в то время как примеры интерферонов класса II включают, но этим не ограничиваясь, [гамма]-интерфероны.
Ингибиторы HCV NS3 протеазы включают средства (соединения или биологические препараты), которые являются эффективными для ингибирования функции HCV NS3 протеазы у млекопитающего. Ингибиторы HCV NS3 протеазы включают, но этим не ограничиваясь, те соединения, которые описаны в патентных документах WO 99/07733, WO 99/07734, WO 00/09558, WO 00/09543, WO 00/59929, WO 03/064416, WO 03/064455, WO 03/064456, WO 2004/030670, WO 2004/037855, WO 2004/039833, WO 2004/101602, WO 2004/101605, WO 2004/103996, WO 2005/028501, WO 2005/070955, WO 2006/000085, WO 2006/007700 и WO 2006/007708 (все от Boehringer Ingelheim), WO 02/060926, WO 03/053349, WO03/099274, WO 03/099316, WO 2004/032827, WO 2004/043339, WO 2004/094452, WO 2005/046712, WO 2005/051410, WO 2005/054430 (все от BMS), WO 2004/072243, WO 2004/093798, WO 2004/113365, WO 2005/010029 (все от Enanta), WO 2005/037214 (Intermune) и WO 2005/051980 (Schering), и кандидаты, обозначенные как VX-950, ITMN-191 и SCH 503034.
Ингибиторы HCV полимеразы включают средства (соединения или биологические препараты), которые являются эффективными для ингибирования функции HCV полимеразы. Такие ингибиторы включают, но этим не ограничиваясь, ненуклеозидные и нуклеозидные ингибиторы HCV NS5B полимеразы. Примеры ингибиторов HCV полимеразы включают, но этим не ограничиваясь, те соединения, которые описаны в патентных документах: WO 02/04425, WO 03/007945, WO 03/010140, WO 03/010141, WO 2004/064925, WO 2004/065367, WO 2005/080388 и WO 2006/007693 (все от Boehringer Ingelheim), WO 2005/049622 (Japan Tobacco), WO 2005/014543 (Japan Tobacco), WO 2005/012288 (Genelabs), WO 2004/087714 (IRBM), WO 03/101993 (Neogenesis), WO 03/026587 (BMS), WO 03/000254 (Japan Tobacco) и WO 01/47883 (Japan Tobacco), и клинические кандидаты XTL-2125, HCV 796, R-1626 и NM 283.
Ингибиторы другого объекта в HCV жизненном цикле включают средства (соединения или биологические препараты), которые являются эффективными для ингибирования образования и/или репликации HCV, а не путем ингибирования функции HCV NS3 протеазы. Такие средства могут препятствовать механизмам либо организма-хозяина, либо HCV вирусным механизмам, необходимым для образования и/или репликации HCV. Ингибиторы другого объекта в HCV жизненном цикле включают, но этим не ограничиваясь, ингибиторы входа, средства, которые ингибируют объект, выбранный из геликазы, NS2/3 протеазы и участок внутренней посадки рибосомы (IRES), и средства, которые препятствовуют функции других вирусных объектов, включая, но этим не ограничиваясь, NS5A белок и NS4B белок.
Может так случиться, что пациент заражен вирусом гепатита С и одним или несколькими другими вирусами, включая, но этим не ограничиваясь, вирус иммунодефицита человека (HIV), вирус гепатита A (HAV) и вирус гепатита B (HBV). Поэтому предполагается также комбинированная терапия для лечения таких заражений разными вирусами путем совместного введения соединения согласно настоящему изобретению, по меньшей мере, с одним из ингибиторов HIV, ингибиторов HAV и ингибиторов HBV.
Согласно еще одному варианту осуществления, фармацевтические композиции по настоящему изобретению могут дополнительно включать ингибитор (ингибиторы) других мишеней жизненного цикла HCV, включая, но этим не ограничиваясь, геликазу, полимеразу, металлопротеазу и участок внутренней посадки рибосомы (IRES).
Согласно другому варианту осуществления, фармацевтические композиции по настоящему изобретению могут дополнительно включать другое противовирусное, антибактериальное, противогрибковое или противораковое средство, или иммуномодулятор. или другое терапевтическое средство.
Согласно еще одному варианту осуществления, настоящее изобретение включает способы лечения вирусной инфекции, такой как, но этим не ограничиваясь, гепатит C, у субъекта, при необходимости такого лечения, путем введения указанному субъекту эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли, сложного эфира или пролекарства.
Согласно другому варианту осуществления, настоящее изобретение включает способы лечения инфекций гепатита C у субъекта, при необходимости такого лечения, путем введения указанному субъекту эффективного по отношению к вирусу количества анти-HCV или эффективного с точки зрения ингибирования количества фармацевтической композиции по настоящему изобретению.
Дополнительный вариант осуществления настоящего изобретения включает способы обработки биологических образцов путем контактирования биологических образцов с соединениями по настоящему изобретению.
Еще одним аспектом настоящего изобретения является способ получения любого из описанных здесь соединений, используя любой из описанных здесь методов синтеза.
Предполагается, что используемый в этом изобретении ингибитор цитохром Р-450 монооксигеназы ингибирует метаболизм соединений по изобретению. Поэтому, ингибитор цитохром Р-450 монооксигеназы должен присутствовать в количестве, эффективном для ингибирования метаболизма ингибитора протеазы. Соответственно, ингибитор CYP вводят в таком количестве, чтобы повышалась биодоступность ингибитора протеазы по сравнению с биодоступностью в отсутствии ингибитора CYP.
В одном варианте осуществления, изобретение предлагает способы улучшения фармакокинетики соединений изобретения. Преимущества улучшения фармакокинетики лекарственных средств известны в техники (US 2004/0091527; US 2004/0152625; US 2004/0091527). Соответственно, один вариант осуществления этого изобретения предлагает способ введения ингибитора CYP3A4 и соединения по изобретению. Другой вариант осуществления этого изобретения предлагает способ введения соединения по изобретению и ингибитора изофермента 3A4 ("CYP3A4"), изофермента 2C19 ("CYP2C19"), изофермента 2D6 ("CYP2D6"), изофермента 1A2 ("CYP1A2"), изофермента 2C9 ("CYP2C9") или изофермента 2E1 ("CYP2E1"). В предпочтительном варианте осуществления, предпочтительно, чтобы ингибитор CYP ингибировал CYP3A4. Любой ингибитор CYP, который улучшает фармакокинетику соответствующей NS3/4A протеазы, может быть использован в способе по этому изобретению. Эти ингибиторы CYP включают, но этим не ограничиваясь, ритонавир (WO 94/14436), кетоконазол, тролеандомицин, 4-метилпиразол, циклоспорин, клометиазол, циметидин, итраконазол, флуконазол, миконазол, флувоксамин, флуоксетин, нефазодон, сертралин, индинавир, нелфинавир, ампренавир, фосампренавир, саквинавир, лопинавир, делавирдин, эритромицин, VX-944 и VX-497. Предпочтительные ингибиторы CYP включают ритонавир, кетоконазол, тролеандомицин, 4-метилпиразол, циклоспорин и клометиазол.
Следует иметь в виду, что желательной дополнительной характерной чертой этого изобретения является введение описываемой в изобретении комбинации пациенту в виде одной упаковки или упаковок с каждым препаратом, внутри которых вложен информационный вкладыш для пациента по правильному применению изобретения.
Согласно дополнительному аспекту изобретения, упаковка включает, по меньшей мере, соединение по изобретению и ингибитор CYP по изобретению и информационный вкладыш, содержащий инструкции по использованию комбинации по изобретению. В альтернативном варианте осуществления этого изобретения, фармацевтическая упаковка дополнительно включает одно или более из описанных здесь дополнительных средств. Дополнительное средство или средства могут предлагаться в одной и той же упаковке или в раздельных упаковках.
Другим аспектом этого изобретения является скомплектованный для пациента набор для применения при лечении HCV инфекции или для предотвращения HCV инфекции, включающий: один или множество фармацевтических препаратов каждого фармацевтического компонента; контейнер, вмещающий фармацевтический препарат (препараты) на время хранения и перед введением; и инструкции по введению лекарственного средства способом, являющимся эффективным для лечения или предотвращения HCV инфекции.
Соответственно, это изобретение предлагает наборы для одновременного или последовательного введения ингибитора NS3/4A протеазы по изобретению и CYP ингибитора (и, необязательно, дополнительного средства) или их производных, которые получают традиционным способом. Обычно такой набор может включать, например, композицию каждого ингибитора и, необязательно, дополнительное средство (средства) в фармацевтически приемлемом носителе (и в одном или во множестве фармацевтических препаратов) и письменные инструкции для одновременного или последовательного введения.
В другом варианте осуществления предлагается скомплектованный набор, который содержит одну или более дозированных форм для самостоятельного введение; контейнер, предпочтительно герметичный, для содержания дозированных форм во время хранения и до использования; и инструкции для пациента по введению лекарственного средства. Инструкциями могут обычно являться письменные инструкции на листе-вкладыше в упаковке, на наклейке и/или на других компонентах набора, а дозированной формой или формами являются описанные здесь дозированные формы. Каждая дозированная форма может быть индивидуально заключена, как, например, в лист ламината из металлической фольги и пластика, при этом каждая дозированная форма изолирована от других форм в индивидуальных ячейках или полых микросферах, или дозированные формы могут быть заключены в одном контейнере, как например, в пластмассовой бутылке. Настоящие наборы могут также обычно включать упаковочные средства для индивидуальных компонентов набора, то есть дозированных форм, контейнеров, и письменных инструкций по использованию. Такие упаковочные средства могут иметь форму картонной или бумажной коробки, мешочка из пластмассы или фольги, и так далее.
ОПРЕДЕЛЕНИЯ
Ниже приводится список определений для различных терминов, используемых при описании этого изобретения. Эти определения применимы к терминам, по мере того как они используются на протяжении всего этого описания и в пунктах формулы изобретения, если иное не ограничивает их в конкретных случаях, либо индивидуально, либо как часть более крупной группы.
Термин "вирусная инфекция" относится к внедрению вируса в клетки или ткани, например, вируса гепатита С (HCV). Обычно внедрение вируса также связано с репликацией. Вирусная инфекция может быть определена путем измерения титра вирусного антитела в образцах биологической жидкости, такой как кровь, с помощью, например, иммуноферментного анализа. Другие подходящие диагностические методы включают методики на молекулярном уровне, такие как RT-PCR (полимеразная цепная реакция с обратной транскрипцией), прямой метод улавливания гибридов, амплификация, основанная на последовательности нуклеиновых кислот, и другие подобные методы. Вирус может инфицировать орган, например, печень, и вызывать заболевание, например, гепатит, цирроз, хроническое заболевание печени и злокачественную гепатому.
Термин "противораковое средство" относится к соединению или лекарственному средству, способному предотвращать или ингибировать распространение злокачественного новообразования. Примеры таких средств включают цисплатин, актиномицин D, доксорубицин, винкристин, винбластин, этопозид, амсакрин, митоксантрон, тенипазид, таксол, колхицин, циклоспорин A, фенотиазины или тиоксантеры.
Термин "противогрибковое средство" следует использовать для описания соединения, которое может быть использовано для лечения грибковой инфекции, но не являющегося соединениями 3-AP, 3-AMP, пролекарствами 3-AP и 3-AMP согласно настоящему изобретению. Противогрибковые средства согласно настоящему изобретению включают, например, тербинафин, флуконазол, итраконазол, посаконазол, клотримазол, гризеофулвин, нистатин, толнафтат, каспофунгин, амфотерицин B, липосомальный амфотерицин B и липидный комплекс амфотерицина B.
Термин "антибактериальное средство" относится как к природным антибиотикам, продуцируемым микроорганизмами для подавления роста других микроорганизмов, так и к средствам, синтезируемым или модифицируемым в лаборатории, которые обладают либо бактерицидной, либо бактериостатической активностью, например, β-лактамные антибактериальные средства, гликопептиды, макролиды, хинолоны, тетрациклины и аминогликозиды. Обычно, если антибактериальное средство является бактериостатическим, это означает, что средство главным образом останавливает рост бактериальной клетки (но не убивает бактерии); если средство является бактерицидным, это означает, что средство убивает бактериальные клетки (и может останавливать их рост перед уничтожением бактерий).
Термин "иммуномодулятор" относится к любому веществу, предназначенному для изменения функционирования гуморальной или клеточной иммунной системы субъекта. Такие иммуномодуляторы включают ингибиторы воспаления, опосредованного тучными клетками, интерфероны, интерлейкины, простагландины, стероиды, кортикостероиды, колониестимулирующие факторы, хемотаксические факторы и так далее.
Используемый здесь термин "C1-C6 алкил" или "C1-C8 алкил" относится к насыщенным, линейным или разветвленным углеводородным радикалам, содержащим от одного до шести или от одного до восьми углеродных атомов, соответственно. Примеры C1-C6 алкильных радикалов включают, но этим не ограничиваясь, метильный, этильный, пропильный, изопропильный, н-бутильный, трет-бутильный, неопентильный, н-гексильный радикалы; и примеры C1-C8 алкильных радикалов включают, но этим не ограничиваясь, метильный, этильный, пропильный, изопропильный, н-бутильный, трет-бутильный, неопентильный, н-гексильный, гептильный, октильный радикалы.
Используемый здесь термин "C2-C6 алкенил" или "C2-C8 алкенил" обозначает группу, полученную из углеводородного фрагмента, где углеводородный фрагмент имеет, по меньшей мере, одну двойную связь углерод-углерод и содержит от двух до шести или от двух до восьми углеродных атомов, соответственно. Алкенильные группы включают, но этим не ограничиваясь, например, этенил, пропенил, бутенил, 1-метил-2-бутен-1-ил, гептенил, октенил и другие подобные группы.
Используемый здесь термин "C2-C6 алкинил" или "C2-C8 алкинил" обозначает группу, полученную из углеводородного фрагмента, где углеводородный фрагмент имеет, по меньшей мере, одну тройную связь углерод-углерод и содержит от двух до шести или от двух до восьми углеродных атомов, соответственно. Характерные алкинильные группы включают, но этим не ограничиваясь, например, этинил, 1-пропинил, 1-бутинил, гептинил, октинил и другие подобные группы.
Используемый здесь термин "C8-C8-циклоалкил" или "C3-C12-циклоалкил" обозначает группу, полученную из моноциклического или полициклического насыщенного карбоциклического кольца, где соединение из насыщенного карбоциклического кольца имеет от 3 до 8 или от 3 до 12 атомов в кольце, соответственно. Примеры C3-C8-циклоалкила включают, но этим не ограничиваясь, циклопропил, циклобутил, циклопентил, циклогексил, циклопентил и циклооктил; и примеры C3-C12-циклоалкила включают, но этим не ограничиваясь, циклопропил, циклобутил, циклопентил, циклогексил, бицикло[2.2.1]гептил и бицикло[2.2.2]октил.
Используемый здесь термин "C3-C8-циклоалкенил" или "C3-C12-циклоалкенил" обозначает группу, полученную из соединения с моноциклическим или полициклическим карбоциклическим кольцом, имеющим, по меньшей мере, одну двойную связь углерод-углерод, где соединение с карбоциклическим кольцом имеет от 3 до 8 или от 3 до 12 атомов в кольце, соответственно. Примеры C3-C8-циклоалкенила включают, но этим не ограничиваясь, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклооктенил, и другие подобные соединения; и примеры C3-C12-циклоалкенила включают, но этим не ограничиваясь, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклооктенил и другие подобные соединения.
Используемый здесь термин "арил" относится к моно- или бициклической карбоциклической кольцевой системе, имеющей одно или два ароматических кольца, включающих, но этим не ограничиваясь, фенил, нафтил, тетрагидронафтил, инданил, иденил и другие подобные кольца.
Используемый здесь термин "арилалкил" относится к C1-C3 алкильному или C1-C6 алкильному остатку, присоединенному к арильному кольцу. Примеры включают, но этим не ограничиваясь, бензил, фенетил и другие подобные группы.
Используемый здесь термин "гетероарил" относится к моно-, би- или трициклическому ароматическому радикалу или кольцу, имеющему от пяти до десяти атомов в кольце, из которых, по меньшей мере, один атом в кольце выбирают из S, O и N; где любой N или S, содержащийся в кольце, может являться необязательно окисленным. Гетероарил включает, но этим не ограничиваясь, пиридинил, пиразинил, пиримидинил, пирролил, пиразолил, имидазолил, тиазолил, оксазолил, изооксазолил, тиадиазолил, оксадиазолил, тиофенил, фуранил, хинолинил, изохинолинил, бензимидазолил, бензооксазолил, хиноксалинил и другие подобные гетероарилы.
Используемый здесь термин "гетероарилалкил" относится к C1-C3 алкильному или C1-C6 алкильному остатку, присоединенному к гетероарильному кольцу. Примеры включают, но этим не ограничиваясь, пиридинилметил, пиримидинилэтил и другие подобные гетероарилалкилы.
Используемый здесь термин "замещенный" относится к независимой замене одного, двух или трех или более атомов водорода в соединении на заместители, включающие, но этим не ограничиваясь этим, -F, -Cl, -Br, -I, -OH, замещенный гидрокси, -NO2, -CN, -NH2, N3, замещенный амино, алкокси, тиоалкокси, оксо, -галоген-C1-C12-алкил, -галоген-C2-C12-алкенил, -галоген-C2-C12-алкинил, -галоген-C3-C12-циклоалкил, -NH-C1-C12-алкил, -NH-C2-C12-алкенил, -NH-C2-C12-алкинил, -NH-C3-C12-циклоалкил, -NH-арил, -NH-гетероарил, -NH-гетероциклоалкил, -диалкиламино, -диариламино, -дигетероариламино, -O-C1-C12-алкил, -O-C2-C12-алкенил, -O-C2-C12-алкинил, -О-C3-C12-циклоалкил, -O-арил, -O-гетероарил, -O-гетероциклоалкил, -C(O)-C1-C12-алкил, -C(O)-C2-C12-алкенил, -C(O)- C2-C12-алкинил, -C(O)-C3-C12-циклоалкил, -C(O)-арил, -C(O)-гетероарил, -C(O)-гетероциклоалкил, -CONH2, -CONH-C1-C12-алкил, -CONH-C2-C12-алкенил, -CONH-C2-C12-алкинил, -CONH-C3-C12-циклоалкил, -CONH-арил, -CONH-гетероарил, -CONH-гетероциклоалкил, -OCO2-C1-C12-алкил, -OCO2-C2-C12-алкенил, -OCO2-C2-C12-алкинил, -OCO2-C3-C12-циклоалкил, -OCO2-арил, -OCO2-гетероарил, -OCO2-гетероциклоалкил, -OCONH2, -OCONH-C1-C12-алкил, -OCONH-C2-C12-алкенил, -OCONH-C2-C12-алкинил, -OCONH-C3-C12-циклоалкил, -OCONH-арил, -OCONH-гетероарил, -OCONH-гетероциклоалкил, -NHC(O)-C1-C12-алкил, -NHC(O)-C2-C12-алкенил, -NHC(O)-C2-C12-алкинил, -NHC(O)-C3-C12-циклоалкил, -NHC(O)-арил, -NHC(O)-гетероарил, -NHC(O)-гетероциклоалкил, -NHCO2-C1-C12-алкил, -NHCO2-C2-C12-алкенил, -NHCO2-C2-C12-алкинил, -NHCO2-C3-C12-циклоалкил, -NHCO2-арил, -NHCO2-гетероарил, -NHCO2-гетероциклоалкил, -NHC(O)NH2, -NHC(O)NH-C1-C12-алкил, -NHC(O)NH-C2-C12-алкенил, -NHC(O)NH-C2-C12-алкинил, -NHC(O)NH-C3-C12-циклоалкил, -NHC(O)NH-арил, -NHC(O)NH-гетероарил, -NHC(O)NH-гетероциклоалкил, NHC(S)NH2, -NHC(S)NH-C1-C12-алкил, -NHC(S)NH-C2-C12-алкенил, -NHC(S)NH-C2-C12-алкинил, -NHC(S)NH-C3-C12-циклоалкил, -NHC(S)NH-арил, -NHC(S)NH-гетероарил, -NHC(S)NH-гетероциклоалкил, -NHC(NH)NH2, -NHC(NH)NH-C1-C12-алкил, -NHC(NH)NH-C2-C12-алкенил, -NHC(NH)NH-C2-C12-алкинил, -NHC(NH)NH-C3-C12-циклоалкил, -NHC(NH)NH-арил, -NHC(NH)NH-гетероарил, -NHC(NH)NH-гетероциклоалкил, -NHC(NH)-C1-C12-алкил, -NHC(NH)-C2-C12-алкенил, -NHC(NH)-C2-C12-алкинил, -NHC(NH)-C3-C12-циклоалкил, -NHC(NH)-арил, -NHC(NH)-гетероарил, -NHC(NH)-гетероциклоалкил, -C(NH)NH-C1-C12-алкил, -C(NH)NH-C2-C12-алкенил, -C(NH)NH-C2-C12-алкинил, -C(NH)NH-C3-C12-циклоалкил, -C(NH)NH-арил, -C(NH)NH-гетероарил, -C(NH)NH-гетероциклоалкил, -S(O)-C1-C12-алкил, -S(O)-C2-C12-алкенил, -S(O)-C2-C12-алкинил, -S(O)-C3-C12-циклоалкил, -S(O)-арил, -S(O)-гетероарил, -S(O)-гетероциклоалкил, -SO2NH2, -SO2NH-C1-C12-алкил, -SO2NH-C2-C12-алкенил, -SO2NH-C2-C12-алкинил, -SO2NH-C3-C12-циклоалкил, -SO2NH-арил, -SO2NH-гетероарил, -SO2NH- гетероциклоалкил, -NHSO2-C1-C12-алкил, -NHSO2-C2-C12-алкенил, -NHSO2-C2-C12-алкинил, -NHSO2-C3-C12-циклоалкил, -NHSO2-арил, -NHSO2-гетероарил, -NHSO2-гетероциклоалкил, -CH2NH2, -CH2SO2CH3, -арил, -арилалкил, -гетероарил, -гетероарилалкил, -гетероциклоалкил, -C3-C12-циклоалкил, полиалкоксиалкил, полиалкокси, -метоксиметокси, -метоксиэтокси, -SH, -S-C1-C12-алкил, -S-C2-C12-алкенил, -S-C2-C12-алкинил, -S-C3-C12-циклоалкил, -S-арил, -S-гетероарил, -S-гетероциклоалкил, метилтиометил или -L'-R', где L' является C1-C6алкиленом, C2-C6алкениленом или C2-C6алкиниленом, и R' является арилом, гетероарилом, гетероциклом, C3-C12циклоалкилом или C3-C12циклоалкенилом. Следует иметь в виду, что арилы, гетероарилы, алкилы и другие подобные группы могут быть дополнительно замещены. В некоторых случаях каждый заместитель в замещенном фрагменте дополнительно необязательно замещен одной или более группами, при этом каждую группу независимо выбирают из -F, -Cl, -Br, -I, -OH, -NO2, -CN или -NH2. Когда имеет место, по меньшей мере, две замены атомов водорода заместителями, два заместителя могут быть взяты вместе с образованием циклоалкильного, циклоалкенильного или гетероциклического кольца.
В соответствии с изобретением любой из описанных здесь арилов, замещенных арилов, гетероарилов и замещенных гетероарилов может являться любой ароматической группой. Ароматические группы могут быть замещенными или незамещенными.
Следует иметь в виду, что любой описанный здесь алкильный, алкенильный, алкинильный, циклоалкильный и циклоалкенильный фрагмент может также являться алифатической группой, алициклической группой или гетероциклической группой. "Алифатическая группа" является неароматическим фрагментом, который может содержать любую комбинацию углеродных атомов, водородных атомов, атомов галогена, кислорода, азота или других атомов, и необязательно содержать одну или более единиц ненасыщенности, например, двойные и/или тройные связи. Алифатическая группа может быть линейной, разветвленной или циклической и предпочтительно содержать от примерно 1 до примерно 24 углеродных атомов, более типично, от примерно 1 до примерно 12 углеродных атомов. В дополнение к алифатическим углеводородным группам, алифатические группы включают, например, полиалкоксиалкилы, такие как полиалкиленгликоли, полиамины, и полиимины, например. Такие алифатические группы могут быть дополнительно замещены. Следует иметь в виду, что алифатические группы могут быть использованы вместо описанных здесь алкильных, алкенильных, алкинильных, алкиленовых, алкениленовых и алкиниленовых групп.
Используемый здесь термин "алициклическая" обозначает группу, полученную из моноциклического или полициклического насыщенного карбоциклического кольцевого соединения. Примеры включают, но этим не ограничиваясь, циклопропил, циклобутил, циклопентил, циклогексил, бицикло[2.2.1]гептил и бицикло[2.2.2]октил. Такие алициклические группы могут быть дополнительно замещены.
Термины "гетероциклоалкил" и "гетероцикл" являются взаимозаменяемыми и относятся к неароматическому 3-, 4-, 5-, 6- или 7-членному кольцу или конденсированной системе с би- или трициклической группой, где (i) каждое кольцо содержит от одного до трех гетероатома, независимо выбранных из кислорода, серы и азота, (ii) каждое 5-членное кольцо имеет от 0 до 1 двойной связи и каждое 6-шестичленное кольцо имеет от 0 до 2 двойных связей, (iii) гетероатомы азота и серы могут необязательно быть окисленными, (iv) гетероатом азота может необязательно быть четвертичным, (v) любое из приведенных выше колец может быть сконденсировано с бензольным кольцом, и (vi) остальные атомы в кольце являются углеродными атомами, которые могут быть необязательно оксо-замещенными. Характерные гетероциклоалкильные группы включают, но этим не ограничиваясь, [1,3]диоксолан, пирролидинил, пиразолинил, пиразолидинил, имидазолинил, имидазолидинил, пиперидинил, пиперазинил, оксазолидинил, изоксазолидинил, морфолинил, тиазолидинил, изотиазолидинил, хиноксалинил, пиридазинонил и тетрагидрофурил. Такие гетероциклические группы могут быть дополнительно замещены с получением замещенного гетероцикла.
Очевидно, что в различных вариантах осуществления изобретения предполагается, что замещенный или незамещенный алкил, алкенил, алкинил, циклоалкил, циклоалкенил, циклоалкинил, арилалкил, гетероарилалкил и гетероциклоалкил являются одновалентными или двухвалентными. Поэтому алкиленовая, алкениленовая и алкиниленовая, циклоалкиленовая, циклоалкениленовая, циклоалкиниленовая, арилалкиленовая, гетероарилалкиленовая и гетероциклоалкиленовая группы должны быть включены в приведенные выше определения, и их используют здесь для получения формул с соответствующей валентностью.
Используемый здесь термин "группа, активирующая гидроксигруппу" относится к лабильному химическому фрагменту, который известен в технике как активирующий гидроксигруппу, для того чтобы она уходила в процессе синтезов, таких как реакции замещения или реакции элиминирования. Примеры группы, активирующей гидроксигруппу, включают, но этим не ограничиваясь, мезилат, тозилат, трифлат, п-нитробензоат, фосфонат и другие подобные группы.
Используемый здесь термин "активированная гидроксигруппа" относится к гидроксигруппе, активированной определенной выше группой, активирующей гидроксигруппу, включающей, например, мезилатную, тозилатную, трифлатную, п-нитробензоатную, фосфонатную группы.
Используемый здесь термин "защищенная гидроксигруппа" относится к гидроксильной группе, защищенной с помощью определенной выше защитной группы для гидроксильной группы, включающей бензоильную, ацетильную, триметилсилильную, триэтилсилильную, метоксиметильную группы.
Используемые здесь термины "гало" и "галоген" относятся к атому, выбранному из фтора, хлора, брома и йода.
Описанные здесь соединения содержат один или более центров асимметрии и поэтому могут образовывать энантиомеры, диастереомеры и другие стереоизомерные формы, которые могут быть определены в терминах абсолютной стереохимии как (R)- или (S)- или как (D)- или (L)- для аминокислот. Предполагается, что настоящее изобретение включает все такие возможные изомеры, а также их рацемические и оптически чистые формы. Оптические изомеры могут быть получены из их соответствующих оптически активных предшественников при помощи описанных выше методов или путем разделения рацемических смесей. Разделение может быть проведено в присутствии агента для оптического расщепления, путем хроматографии или путем многократной кристаллизации или какой-либо комбинации этих методов, которые являются известными для специалистов в этой области. Дополнительные подробности относительно методов разделения можно найти в монографии Jacques, et al., Enantiomers, Racemates, and Resolutions (John Wiley & Sons, 1981). Когда описанные здесь соединения содержат олефиновые двойные связи или другие центры геометрической асимметрии, и если не указано иначе, то предполагается, что соединения включают как E, так и Z геометрические изомеры. Или же, также предполагается, что включаются все таутомерные формы. Конфигурацию появляющейся здесь любой двойной связи углерод-углерод выбирают только с точки зрения удобства, и предполагается, что она не обозначает конкретную конфигурацию, если в тексте не заявлено иначе; поэтому двойная связь углерод-углерод, изображенная здесь произвольно как транс, может являться цис, транс или смесью этих двух конфигураций в любом соотношении.
Используемый здесь термин "субъект" относится к млекопитающему. Поэтому к субъектам относятся, например, собаки, кошки, лошади, коровы, свиньи, морские свинки и другие подобные животные. Субъектом предпочтительно является человек. Когда субъектом является человек, субъекта здесь могут называть пациентом.
Используемый здесь термин "фармацевтически приемлемая соль" относится к тем солям соединений, получаемых с помощью способа по настоящему изобретению, которые, на основании обоснованного медицинского заключения, являются подходящими для использования при контакте с тканями людей и низших животных без чрезмерной токсичности, раздражения, аллергической реакции и других подобных явлений, и которые удовлетворяют разумному соотношению между выгодой и риском. Фармацевтически приемлемые соли хорошо известны в технике.
Используемый здесь термин "защитная группа для гидроксигруппы" относится к лабильному химическому фрагменту, который известен в технике в качестве фрагмента, защищающего гидроксигруппу от нежелательных реакций в процессе синтезов. После указанного синтеза (синтезов) описанная здесь защитная группа может быть селективно удалена. Известные в технике защитные группы для гидроксигруппы описаны в целом в монографии T.H. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999). Примеры защитных групп для гидроксигруппы включают бензилоксикарбонил, 4-нитро-бензилоксикарбонил, 4-бромбензил-оксикарбонил, 4-метокси-бензилоксикарбонил, метоксикарбонил, трет-бутоксикарбонил, изопропоксикарбонил, дифенил-метоксикарбонил, 2,2,2-трихлор-этоксикарбонил, 2-(триметилсилил)-этоксикарбонил, 2-фурфурилоксикарбонил, аллилоксикарбонил, ацетил, формил, хлорацетил, трифторацетил, метоксиацетил, феноксиацетил, бензоил, метил, трет-бутил, 2,2,2-трихлорэтил, 2-триметилсилилэтил, 1,1-диметил-2-пропенил, 3-метил-3-бутенил, аллил, бензил, пара-метоксибензилдифенилметил, трифенил-метил(тритил), тетрагидрофурил, метоксиметил, метилтиометил, бензилоксиметил, 2,2,2-трихлорэтоксиметил, 2-(триметилсилил)-этоксиметил, метансульфонил, пара-толуолсульфонил, триметилсилил, триэтилсилил, триизопропилсилил и другие подобные защитные группы. Предпочтительными защитными группами для гидроксигруппы для настоящего изобретения являются ацетил (Ac или -C(O)CH3), бензоил (Bz или -C(O)C6H5) и триметилсилил (TMS или -Si(CH3)3). Фармацевтически приемлемые соли подробно описаны в публикации Berge, et al. в J. Pharmaceutical Sciences, 66:1-19 (1977). Соли могут быть получены in situ во время окончательного выделения и очистки соединений по изобретению или отдельно путем взаимодействия функции свободного основания с подходящей органической кислотой. Примеры фармацевтически приемлемых солей включают, но этим не ограничиваясь, нетоксические соли присоединения кислоты, например, соли аминогруппы, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и хлорная кислота, или с органическими кислотами, такими как уксусная кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или путем использования других применяемых в технике методов, таких как ионный обмен. Другие фармацевтически приемлемые соли включают, но этим не ограничиваясь, адипатную, альгинатную, аскорбатную, аспартатную, бензолсульфонатную, бензоатную, бисульфатную, боратную, бутиратную, камфоратную, камфорсульфонатную, цитратную, циклопентанпропионатную, диглюконатную, додецилсульфатную, этансульфонатную, формиатную, фумаратную, глюкогептонатную, глицерофосфатную, глюконатную, гемисульфатную, гептаноатную, гексаноатную, гидройодидную, 2-гидроксиэтансульфонатную, лактобионатную, лактатную, лауратную, лаурилсульфатную, малатную, малеатную, малонатную, метансульфонатную, 2-нафталинсульфонатную, никотинатную, нитратную, олеатную, оксалатную, пальмитатную, памоатную, пектинатную, персульфатную, 3-фенилпропионатную, фосфатную, пикратную, пивалатную, пропионатную, стеаратную, сукцинатную, сульфатную, тартратную, тиоцианатную, пара-толуолсульфонатную, ундеканоатную, валератную соли и другие подобные соли. Характерные соли щелочных или щелочноземельных металлов включают натриевую, литиевую, калиевую, кальциевую, магниевую соли и другие подобные соли. Кроме того, фармацевтически приемлемые соли включают, когда это целесообразно, нетоксичные соли катионов аммония, четвертичного аммония и амина, образованные при использовании противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, алкил, имеющий от 1 до 6 углеродных атомов, сульфонат и арилсульфонат.
Используемый здесь термин "защитная группа для аминогруппы" относится к лабильному химическому фрагменту, который известен в технике, для защиты аминогруппы от нежелательных реакций в процессе синтезов. После указанного синтеза (синтезов) описанная здесь защитная группа для аминогруппы может быть селективно удалена. Известные в технике защитные группы для аминогруппы описаны в целом в монографии T.H. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999). Примеры защитных групп для аминогруппы включают, но этим не ограничиваясь, третбутоксикарбонил, 9-флуоренилметоксикарбонил, бензилоксикарбонил и другие подобные группы.
Используемый здесь термин "фармацевтически приемлемый сложный эфир" относится к сложным эфирам соединений, образованных с помощью способа по настоящему изобретению, которые гидролизуются in vivo и включают те эфиры, которые легко распадаются в человеческом организме с высвобождением исходного соединения или его соли. Подходящие эфирные группы включают, например, те эфирные группы, которые получают из фармацевтически приемлемых алифатических карбоновых кислот, в частности, алкановых, алкеновых, циклоалкановых и алкандикарбоновых кислот, в которых каждый алкильный или алкенильный фрагмент преимущественно имеет не более чем 6 углеродных атомов. Примеры конкретных сложных эфиров включают, но этим не ограничиваясь, формиаты, ацетаты, пропионаты, бутираты, акрилаты и этилсукцинаты.
Используемый здесь термин "фармацевтически приемлемые пролекарства" относится к тем пролекарствам соединений, получаемых с помощью способа по настоящему изобретению, которые, на основании обоснованного медицинского заключения, являются подходящими для использования при контакте с тканями людей и низших животных без чрезмерной токсичности, раздражения, аллергической реакции и других подобных явлений, и которые удовлетворяют разумному соотношению между выгодой и риском и эффективны при их предполагаемом использовании, а также, при наличии возможности, к цвиттерионным формам соединений по настоящему изобретению. Используемый здесь термин "пролекарство" означает соединение, которое способно к превращению in vivo в результате метаболизма (например, в результате гидролиза) с образованием любого соединения, определяемого формулами по настоящему изобретению. В технике известны различные формы пролекарств, например, описанные в следующих монографиях и публикациях Bundgaard, (ed.), Design of Prodrugs, Elsevier (1985); Widder, et al. (ed.), Methods in Enzymology, vol. 4, Academic Press (1985); Krogsgaard-Larsen, et al., (ed). "Design and Application of Prodrugs, Textbook of Drug Design and Development, Chapter 5, 113-191 (1991); Bundgaard, et al., Journal of Drug Deliver Reviews, 8:1-38(1992); Bundgaard, J. of Pharmaceutical Sciences, 77:285 et seq. (1988); Higuchi and Stella (eds.) Prodrugs as Novel Drug Delivery Systems, American Chemical Society (1975); и Bernard Testa & Joachim Mayer, "Hydrolysis In Drug And Prodrug Metabolism: Chemistry, Biochemistry And Enzymology", John Wiley and Sons, Ltd. (2002).
Термин "ацил" включает остатки, полученные из кислот, включающих, но этим не ограничиваясь, карбоновые кислоты, карбаминовые кислоты, угольные кислоты, сульфоновые кислоты и фосфористые кислоты. Примеры включают алифатические карбонилы, ароматические карбонилы, алифатические сульфонилы, ароматические сульфинилы, алифатические сульфинилы, ароматические фосфаты и алифатические фосфаты. Примеры алифатических карбонилов включают, но этим не ограничиваясь, ацетил, пропионил, 2-фторацетил, бутирил, 2-гидроксиацетил и другие подобные ацилы.
Используемый здесь термин "апротонный растворитель" относится к растворителю, который является относительно инертным к протонной активности, то есть, не действуя в качестве донора протона. Примеры включают, но этим не ограничиваясь, углеводороды, например, такие как гексан и толуол, галогенированные углеводороды, такие как, например, метиленхлорид, этиленхлорид, хлороформ и другие подобные углеводороды, гетероциклические соединения, такие как, например, тетрагидрофуран и N-метил-пирролидинон, и эфиры, такие как диэтиловый эфир, бис-метоксиметиловый эфир. Такие растворители хорошо известны специалистам в этой области, и индивидуальные растворители или их смеси могут являться предпочтительными для конкретных соединений и реакционных условий, в зависимости от таких факторов, например, как растворимость реагентов, реакционная способность реагентов и предпочтительные интервалы температур. Дополнительное обсуждение апротонных растворителей можно найти в учебниках по органической химии или специальных монографиях, например: Organic Solvents Physical Properties and Methods of Purification, 4th ed., edited by John A. Riddick et al., Vol. II, in the Techniques of Chemistry Series, John Wiley & Sons, NY, 1986.
Используемые здесь термины "протогенный органический растворитель" или "протонный растворитель" относятся к растворителю, который проявляет тенденцию к выделению протонов, такому как спирт, например, метанол, этанол, пропанол, изопропанол, бутанол, трет-бутанол и другие подобные растворители. Такие растворители являются хорошо известными для специалистов в этой области, и индивидуальные растворители или их смеси могут являться предпочтительными для конкретных соединений и реакционных условий, в зависимости от таких факторов, например, как растворимость реагентов, реакционная способность реагентов и предпочтительные интервалы температур. Дополнительное обсуждение протогенных растворителей можно найти в учебниках по органической химии или специальных монографиях, например: Organic Solvents Physical Properties and Methods of Purification, 4th ed., edited by John A. Riddick et al., Vol. II, in the Techniques of Chemistry Series. John Wiley & Sons, NY, 1986.
Комбинации заместителей и переменных, предусматриваемые этим изобретением, являются только теми комбинациями, которые приводят к образованию стабильных соединений. Используемый здесь термин "стабильное" относится к соединениям, которые обладают стабильностью, достаточной для того, чтобы их можно было производить, и которые сохраняют целостность соединения в течение достаточного периода времени, в течение которого его применяют для описанных здесь целей (например, для терапевтического или профилактического введения субъекту).
Синтезированные соединения могут быть выделены из реакционной смеси и дополнительно очищены при помощи такого метода, как колонная хроматография, высокоэффективная жидкостная хроматография или перекристаллизация. Кроме того, различные стадии синтеза могут быть осуществлены в иной последовательности или в ином порядке для получения требуемых соединений. Кроме того, описываемые здесь растворители, температуры, длительность реакций и так далее приводятся только в целях иллюстрации, и изменение реакционных условий могут приводить к образованию требуемых мостиковых макроциклических продуктов по настоящему изобретению. Синтетические химические трансформации и методики использования защитных групп (защита и снятие защиты), применяемые при синтезе описанных здесь соединений, включают, например, те, которые описаны в монографиях R. Larock, Comprehensive Organic Transformations. VCH Publishers (1989); T. W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2d. Ed., John Wiley and Sons (1991); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); и L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995).
Соединения по изобретению могут быть модифицированы для усиления селективных биологических свойств путем присоединения различных функциональностей с помощью описанных здесь синтетических способов. Такие модификации включают те модификации, которые повышают биологическое проникновение в данную биологическую систему (например, кровь, лимфатическую систему, центральную нервную систему), повышают пероральную усвояемость, увеличивают растворимость, для того чтобы сделать возможным введение путем инъекции, изменяют метаболизм и изменяют скорость выведения.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Фармацевтические композиции по настоящему изобретению включают терапевтически эффективное количество соединения по настоящему изобретению, приготовленное вместе с одним или более фармацевтически приемлемыми носителями. Используемый здесь термин "фармацевтически приемлемый носитель" обозначает нетоксичный, инертный, твердый, полутвердый или жидкий наполнитель, разбавитель, инкапсулирующий материал или вспомогательное вещество, способствующее процессу получению дозированной формы любого типа. Несколькими примерами материалов, которые могут служить в качестве фармацевтически приемлемых носителей, являются сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетатцеллюлоза; порошкообразная трагакантовая камедь; солод; желатин; тальк; эксципиенты, такие как масло какао и воски для суппозиториев; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор; раствор Рингера; этиловый спирт и растворы фосфатного буфера, а также другие нетоксичные совместимые лубриканты, такие как лаурилсульфат натрия и стеарат магния, а также, по усмотрению разработчика рецептур, в композиции могут присутствовать окрашивающие вещества, смазки для форм, вещества для нанесения покрытий, подсластители, ароматизирующие вещества и отдушки, консерваторы и антиоксиданты. Фармацевтические композиции по изобретению могут быть введены людям и другим животным перорально, ректально, парентерально, интрацистернально, интравагинально, интраперитонеально, местно (в виде порошков, мазей или капель), буккально или в виде орального или назального спрея.
Фармацевтические композиции по этому изобретению могут быть введены перорально, парентерально, путем ингаляции с помощью спрея, местно, ректально, назально, буккально, вагинально или при помощи имплантированного резервуара, предпочтительно путем перорального введения или введения путем инъекции. Фармацевтические композиции по изобретению могут содержать любые традиционные нетоксичные фармацевтически приемлемые носители, адьюванты или среды. В некоторых случаях pH препарата может быть скорректирован при помощи фармацевтически приемлемым кислот, оснований или буферов для повышения стабильности приготовленного соединения или его формы, в которой он доставляется в организм. Используемый здесь термин "парантерально" включает подкожные, внутрикожные, внутривенные, внутримышечные, внутрисуставные, внутриартериальные, интрасиновиальные, интрастернальные, интратекальные, интралезиональные и интракраниальные способы инъекции или инфузии.
Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Кроме активных соединений, жидкие дозированные формы могут содержать обычно используемые в технике инертные разбавители, такие как, например, вода или другие растворители, солюбилизаторы и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутилeнгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, из семян проросшей пшеницы, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и эфиры жирных кислот с сорбитаном, и их смеси. Кроме инертных разбавителей, композиции для перорального введения могут также включать адьюванты, такие как увлажняющие средства, эмульгаторы и суспендирующие средства, подсластители, ароматизирующие вещества и отдушки.
Инъецируемые препараты, например, стерильные инъецируемые водные или масляные суспензии, могут быть приготовлены в соответствии с известными способами с использованием подходящих диспергирующих или увлажняющих средств и суспендирующих средств. Стерильным инъецируемым препаратом может также быть стерильный инъецируемый раствор, суспензия или эмульсия в нетоксичном приемлемом для парентерального введения разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. К приемлемым средам и растворителям, которые могут быть использованы, относится вода, раствор Рингера, U.S.P. и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя и суспендирующей среды обычно используют стерильные нелетучие масла. По этой причине, может быть использовано любое легкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, при приготовлении инъецируемых препаратов используют жирные кислоты, такие как олеиновая кислота.
Инъецируемые препараты могут быть стерилизованы, например, путем фильтрации через задерживающий бактерии фильтр или путем введения стерилизующих веществ в форме стерильных твердых композиций, которые могут быть растворены или диспергированы в стерильной воде или другой пригодной для инъецирования стерильной среде перед использованием.
Для того чтобы пролонгировать действие лекарственного средства, часто является желательным замедление абсорбции лекарственного средства при подкожной или внутримышечной инъекции. Это может быть достигнуто за счет использования жидкой суспензии кристаллического или аморфного материала, который плохо растворим в воде. Тогда скорость абсорбции лекарственного средства зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. В качестве варианта, замедленная абсорбция парентерально вводимой дозированной формы достигается путем растворения или суспендирования лекарственного средства в масляной среде. Инъецируемые формы депо получают путем формирования микрокапсулированных матриц лекарственного средства в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от отношения лекарственного средства к полимеру и природы конкретного используемого полимера, можно регулировать скорость высвобождения лекарственного средства. Примеры других биоразлагаемых полимеров включают полиортоэфиры и полиангидриды. Инъецируемые депо препараты также получают путем включения лекарственного средства в липосомы или микроэмульсии, которые являются совместимыми с тканями организма.
Предпочтительными композициями для ректального или вагинального введения являются суппозитории, которые могут быть приготовлены путем смешения соединений по изобретению с подходящими нераздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но жидкими при температуре тела, и поэтому расплавляются в прямой кишке или вагинальной полости и высвобождают активное соединение.
Твердые дозированные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозированных формах активное соединение смешивают, по меньшей мере, с одним инертным, фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или дикальцийфосфат, и/или: a) наполнителями или расширителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремневая кислота, b) связующими, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и гуммиарабик, c) увлажнителями, такими как глицерин, d) дезинтегрантами, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, e) замедлителями растворения, такими как парафин, f) ускорителями абсорбции, такими как соединения четвертичного аммония, g) увлажняющими средствами, такими как, например, цетиловый спирт и глицеролмоностеарат, h) абсорбентами, такими как каолин и бентонитовая глина, и i) лубрикантами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль дозированная форма может также включать буферные вещества.
Твердые композиции аналогичного типа могут также быть использованы в качестве наполнителей в мягких и твердых наполняемых желатиновых капсулах, используя такие эксципиенты, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и другие подобные вещества.
Активные соединения могут также находиться в микроинкапсулированной форме с одним или более указанными выше эксципиентами. Твердые дозированные формы таблеток, драже, капсул, пилюль и гранул могут быть приготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, покрытия с регулируемым высвобождением и другие покрытия, хорошо известные в области фармацевтики. В таких твердых дозированных формах активное соединение может быть смешано, по меньшей мере, с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие дозированные формы в условиях обычной практики помимо инертных разбавителей могут также включать дополнительные вещества, например, лубриканты для таблетирования и другие вспомогательные вещества для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль дозированные формы могут также включать буферные вещества. Они могут необязательно содержать опалесцирующие компоненты и могут также представлять собой композицию, из которой они высвобождают только активный ингредиент (ингредиенты), или, предпочтительно, в конкретной части кишечника, необязательно, отсроченным образом. Примеры композиций для покрытия, которые могут быть использованы, включают полимерные вещества и воски.
Дозированные формы для местного или трансдермального введения соединения по изобретению включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, формы для ингаляции или пластыри. Активный компонент, в зависимости от обстоятельств, смешивают в стерильных условиях с фармацевтически приемлемым носителем и какими-либо необходимыми консервантами или буферами. Предполагается, что офтальмический препарат, глазные капли, глазные мази, порошки и растворы также входят в объем этого изобретения.
Мази, пасты, кремы и гели могут содержать, помимо активного соединения по изобретению, эксципиенты, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакантовую камедь, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремневую кислоту, тальк и оксид цинка или их смеси.
Порошки и спреи могут содержать помимо соединений по изобретению эксципиенты, такие как лактоза, тальк, кремневая кислота, гидроксид алюминия, силикаты кальция и порошок полиамида или смеси этих веществ. Спреи могут дополнительно содержать традиционные пропелленты, такие как хлорфторуглеводороды.
Трансдермальные пластыри имеют дополнительное преимущество с точки зрения обеспечения регулируемой доставки соединения в организм. Такие дозированные формы могут быть получены путем растворения или распределения соединения в соответствующей среде. Для интенсификации прохождения соединения через кожу могут также быть использованы усилители абсорбции. Скорость может регулироваться либо путем установки регулирующей скорость мембраны, либо путем диспергирования соединения в полимерной матрице или геле.
Противовирусная активность
Ингибирующее количество или доза соединений по настоящему изобретению может находиться в интервале от примерно 0,01 мг/кг до примерно 500 мг/кг, в качестве варианта, от примерно 1 до примерно 50 мг/кг. Ингибирующие количества или дозы будут также зависеть от способа введения, а также от возможности совместного применения с другими средствами.
Согласно способам лечения по настоящему изобретению, вирусные инфекции у субъекта, такого как человек или низшее млекопитающее, подвергают лечению или предотвращают, путем введения субъекту эффективного количества против вируса гепатита C соединения по настоящему изобретению или ингибирующего количества соединения по настоящему изобретению, в таких количествах и в течение такого времени, которые необходимы для достижения требуемого результата. Дополнительным способом по настоящему изобретению является обработка биологических образцов ингибирующим количеством соединения композиции по настоящему изобретению в таких количествах и в течение такого времени, которые необходимы для достижения требуемого результата.
Используемый здесь термин "эффективное количество против вируса гепатита C" соединения по изобретению обозначает достаточное количество соединения для снижения вирусной нагрузки в биологическом образце или у субъекта (например, приводящее, по меньшей мере, к 10%, предпочтительно, по меньшей мере, к 50%, более предпочтительно, по меньшей мере, к 80%, и наиболее предпочтительно, по меньшей мере, к 90% или 95% снижению вирусной нагрузки). Как это хорошо известно в медицине, эффективное количество соединения по изобретению против вируса гепатита C будет являться величиной, удовлетворяющей разумному отношению выгоды и риска, применяемому при любом медицинском лечении.
Термин "ингибирующее количество" соединения по настоящему изобретению означает достаточное количество для снижения нагрузки вируса гепатита C в биологическом образце или у субъекта (например, приводящее, по меньшей мере, к 10%, предпочтительно, по меньшей мере, к 50%, более предпочтительно, по меньшей мере, к 80%, и наиболее предпочтительно, по меньшей мере, к 90% или 95% снижению вирусной нагрузки). Следует иметь в виду, что когда указанное ингибирующее количество соединения по настоящему изобретению вводят субъекту, оно будет являться определяемой врачом величиной, удовлетворяющей разумному отношению выгоды и риска, применяемому при любом медицинском лечении. Используемый здесь термин "биологический образец (образцы)" означает вещество биологического происхождения у субъекта, в которое предполагается осуществить введение лекарственного средства. Примеры биологических образцов включают, но этим не ограничиваясь, кровь и ее компоненты, такие как плазма, тромбоциты, субпопуляции клеток крови и другие подобные компоненты; органы, такие как почка, печень, сердце, легкое и другие подобные органы; сперма и яйцеклетка; костный мозг и его компоненты; или стволовые клетки. Поэтому другим вариантом осуществления настоящего изобретения является способ обработки биологического образца путем контактирования указанного биологического образца с ингибирующим количеством соединения или фармацевтической композиции по настоящему изобретению.
При улучшении состояния субъекта, в случае необходимости, может быть введена поддерживающая доза соединения, композиции или комбинации по этому изобретению. В дальнейшем, доза или частота введения или и то, и другое могут быть уменьшены, в зависимости от симптомов, до уровня, при котором сохраняется улучшенное состояние, а когда облегчение симптомов достигает требуемого уровня, лечение может быть прекращено. Однако, при любом возобновлении симптомов заболевания, субъекту может потребоваться периодическое лечение на долгосрочной основе.
Следует иметь в виду, однако, что суммарное количество ежедневно применяемых соединений и композиций по настоящему изобретению будет определяться лечащим врачом в рамках обоснованного медицинского заключения. Конкретная ингибирующая доза для любого конкретного пациента будет зависеть от ряда факторов, включающих подвергаемое лечению заболевание и тяжесть заболевания; активность используемого конкретного соединения; используемую конкретную композицию; возраст, массу тела, общее состояние здоровья, пол и рацион пациента; время введения, способ введения и скорость выведения конкретного используемого соединения; продолжительность лечения; лекарственные средства, используемые в комбинации или которые совпадают по действию с конкретными используемыми соединениями; и другие подобные факторы, хорошо известные в медицине.
Суммарная дневная ингибирующая доза соединений по изобретению, введенная субъекту в виде разовой дозы или разделенных доз, может составлять, например, от 0,01 до 50 мг/кг массы тела или чаще всего от 0,1 до 25 мг/кг массы тела. Композиции разовых доз могут содержать такие количества или их доли, которые могут составлять дневную дозу. Обычно схемы лечение согласно настоящему изобретению включают введение пациенту, при необходимости такого лечения, от примерно 10 мг до примерно 1000 мг соединения (соединений) по изобретению в день в виде разовой дозы или разделенных доз.
Если не определено иначе, то все используемые здесь технические и научные термины соответствуют значению, которое является общеизвестным для любого специалиста в этой области. Содержание всех упомянутых здесь публикаций, патентов, опубликованных патентных заявок и других литературных источников приводится путем ссылки на них.
Сокращения
Сокращения, которые использованы при описании схем и в примерах, обозначают следующее:
ACN для ацетонитрила;
BME для 2-меркаптоэтанола;
BOP для гексафторфосфата бензотриазол-1-илокси-трис(диметиламино)фосфония;
COD для циклооктадиена;
DAST для диэтиламиносератрифторида;
DABCYL для 6-(N-4'-карбокси-4-(диметиламино)азобензол)-аминогексил-1-O-(2-цианоэтил)-(N,N-диизопропил)-фосфорамидита;
DCM для дихлорметана;
DIAD для диизопропилазодикарбоксилата;
DIBAL-H для диизобутилалюминийгидрида;
DIEA для диизопропилэтиламина;
DMAP для N,N-диметиламинопиридина;
DME для диметилового эфира этиленгликоля;
DMEM для среды Игла, модифицированной Дульбекко;
DMF для N,N-диметилформамида;
DMSO для диметилсульфоксида;
DUPHOS для  ;
;
EDANS для 5-(2-аминоэтиламино)нафталин-1-сульфоновой кислоты;
EDCI или EDC для гидрохлорида 1-(3-диэтиламинопропил)-3-этилкарбодиимида;
EtOAc для этилацетата;
HATU для гексафторфосфата O(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония;
Hoveyda's Cat. для дихлор(o-изопропоксифенилметилен) (трициклогексилфосфин)рутения(II);
KHMDS является бис(триметилсилил)амидом калия;
Ms для мезила;
NMM для N-4-метилморфолина;
PyBrOP для гексафторфосфата бром-три-пиролидино-фосфония;
Ph для фенила;
RCM для реакции метатезиса с закрытием кольца;
RT для обратной транскрипции;
RT-PCR для полимеразной цепной реакции с обратной транскриптазой;
TEA для триэтиламина;
TFA для трифторуксусной кислоты;
THF для тетрагидрофурана;
TLC для тонкослойной хроматографии;
TPP или PPh3 для трифенилфосфина;
tBOC или Boc для трет-бутилоксикарбонила; и
Xantphos для 4,5-бис-дифенилфосфанил-9,9-диметил-9H- ксантена.
Способы синтеза
Соединения и способы по настоящему изобретению будут лучше поняты при помощи следующих схем синтезов, иллюстрирующих способы, с помощью которых соединения по изобретению могут быть получены, и которые, как предполагается, являются только иллюстрацией и не ограничивают объем изобретения. Различные изменения и модификации раскрытых вариантов осуществления могут быть очевидными для специалистов в этой области, и такие изменения и модификации, включающие без ограничения те, которые относятся к химическим структурам, заместителям, производным и/или способам по изобретению, могут быть сделаны без отклонения от сущности изобретения и объема прилагаемых пунктов формулы изобретения.
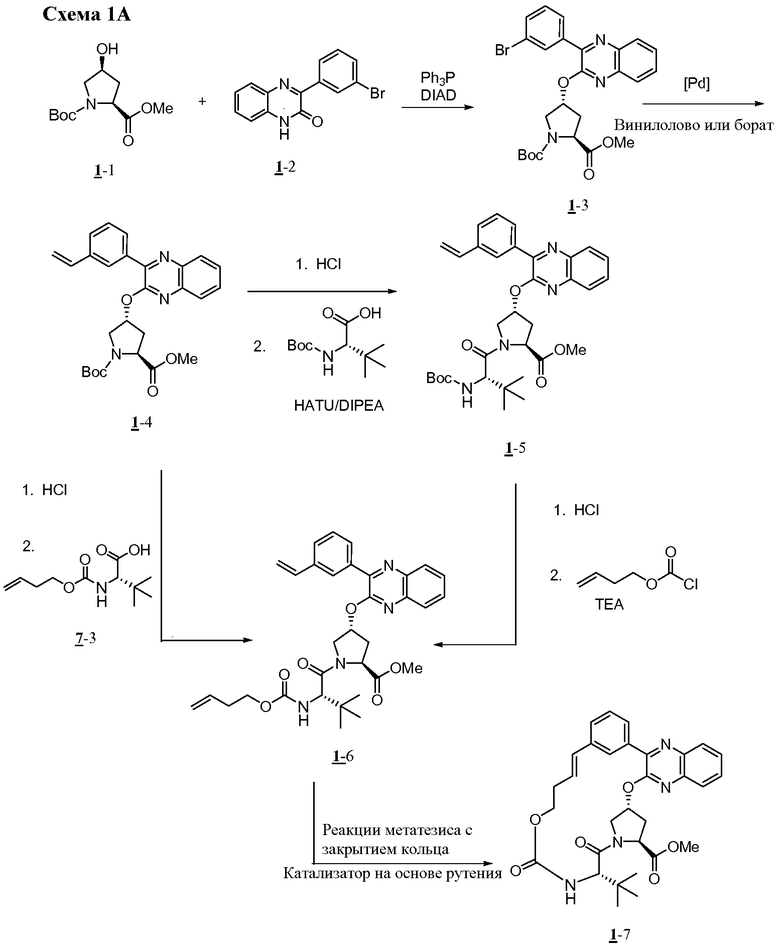
Получение соединений с хиноксалиниловым макроциклическим ядром иллюстрируется на схеме 1A. Производимый в промышленности Boc-гидроксипролин 1-1 приводили во взаимодействие с производным хиноксалина 1-2 (по поводу получении аналогов хиноксалина смотрите схемы 3-6) при условиях реакции Митсунобу с образованием соединения 1-3. Было обнаружено, что присоединение по карбонильному кислороду приводит к образованию требуемого соединения. Подробное обсуждение идентификации и исследования неожидаемых оксопродуктов реакции присоединения Митсунобу приведено в примерах. По поводу дополнительных подробностей о реакции Митсунобу смотрите публикации O. Mitsunobu, Synthesis 1981, 1-28; D. L. Hughes, Org. React. 29, 1-162 (1983); D. L. Hughes, Organic Preparations and Procedures Int. 28, 127-164 (1996); и J. A. Dodge, S. A. Jones, Recent Res. Dev. Org. Chem. 1, 273-283 (1997). Введение винильной группы осуществляли при помощи реакции Сузуки (винилборат/палладий) или реакции Стилла (винилолово/палладий) с получением соединения 1-4. Снятие защиты с соединения 1-4 с помощью HCl с последующей реакцией сочетания (HATU/DMF) с Boc-L-трет-лейцином давало соединение 1-5. Снятие защиты с соединения 1-5 с помощью HCl с последующей реакцией с 3-бутенил-хлорформиатом приводило к образованию макроциклического соединения предшественника 1-6. В качестве варианта, соединение 1-6 может быть получено путем непосредственного сочетания промежуточного соединения 1-4 (после снятия защиты с Boc группы) с соответствующим карбаматом аминокислоты 7-3 (по поводу получения карбаматных производных аминокислоты смотрите схему 7). Реакция метатезиса с закрытием кольца соединения 1-6 с катализатором на основе рутения дает требуемое макроциклическое промежуточное соединение 1-7 (по поводу дополнительных подробностей относительно реакции метатезиса с закрытием кольца смотрите недавние обзоры: Grubbs et al., Ace. Chem. Res., 1995, 28, 446; Shrock et al., Tetrahedron 1999, 55, 8141; Furstner, A. Angew. Chem. Int. Ed. 2000, 39, 3012; Trnka et al., Ace. Chem. Res. 2001, 34, 18, и Hoveyda et al., Chem. Eur. J. 2001, 7, 945).
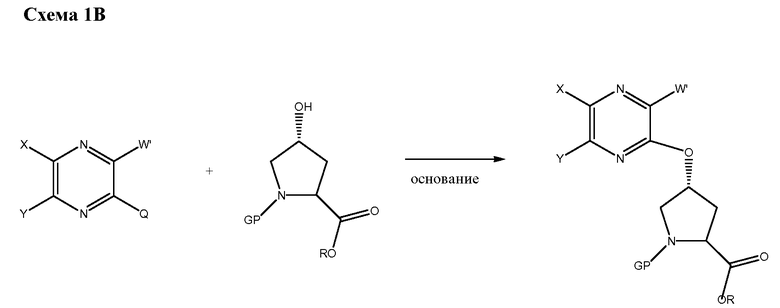
Альтернативный способ синтеза структур с хиноксалиновым ядром показан на схеме 1B, которая иллюстрирует реакцию замещения замещенного хиноксалингалогенида с помощью амино-защищенного гидроксипролина в присутствии основания, такого как NaH или NaOtBu. На этой схеме PG является защитной группой для амина, Q является галогеном, предпочтительно бромом, хлором или йодом, X и Y имеют значения, данные для этих переменных в формуле I, W' имеет значения, данные для L101-W101- в формуле I и Z-W- в формуле II, и R является водородом или алкилом.
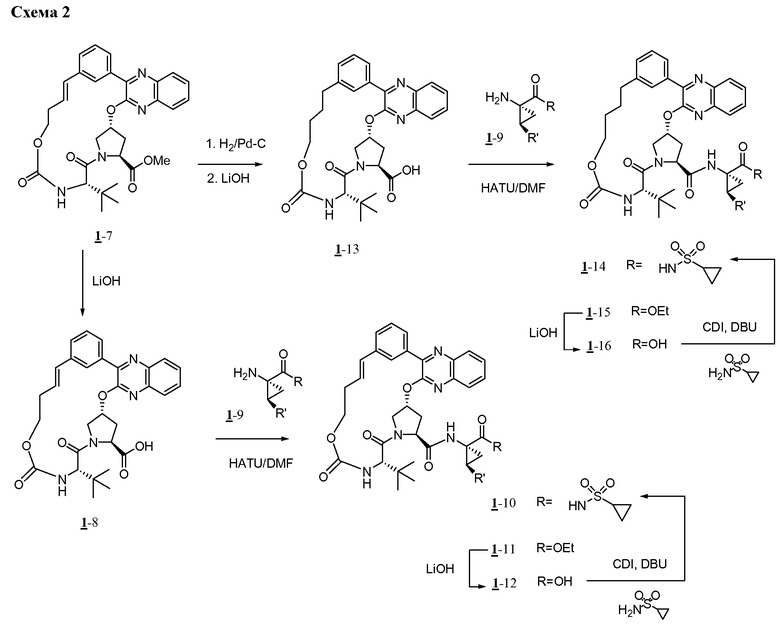
Синтез хиноксалиниловых макроциклических соединений в качестве ингибиторов HCV протеазы приведен на схеме 2. Гидролиз соединения 1-7 дает соответствующую карбоновую кислоту 1-8, которая в сочетании с аминосульфинимидом или эфиром аминокислоты 1-9 (по поводу получения смотрите схемы 8 и 9) дает соединение 1-10 или эфир 1-11. Гидролиз соединения 1-11 приводит к образованию соединения 1-12. Соединение 1-10 может также быть получено из кислоты 1-12, как показано на схеме 2. Гидрирование соединения 1-7 дает соединение 1-13, которое превращают в соединения 1-14 и 1-16 при помощи таких же химических реакций, как и в случае получения соединений 1-10 и 1-12.
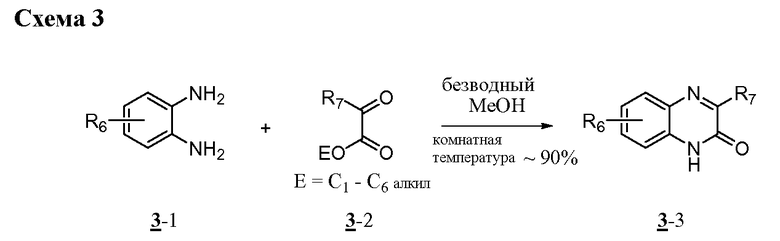
Различные хиноксалиновые производные соединения 1-2 (то есть соединение формулы 3-3) могут быть получены путем реакции конденсации фенилдиаминов формулы 3-1, где R6 определен ранее, с кетокислотами или эфирами формулы 3-2, где R7 является определенным выше W-Z, в безводном метаноле при комнатной температуре (по поводу дополнительных подробностей этой реакции смотрите Bekerman et al., J. Heterocycl. Chem. 1992, 29, 129-133). Примеры фенилдиаминов, подходящих для получения хиноксалиновых производных формулы 3-3, включают, но этим не ограничиваясь, 1,2-диамино-4-нитробензол, o-фенилендиамин, 3,4-диаминотолуол, 4-хлор-1,2-фенилендиамин, метил-3,4-диамино-бензоат, бензо[1,3]диоксол-5,6-диамин, 1,2-диамино-4,5-метилен- диоксибензол, 4-хлор-5-(трифторметил)-1,2-бензолдиамин и другие подобные фенилдиамины. Примеры кетокислот, подходящих для описанной на схеме 3 реакции включают, но этим не ограничиваясь, бензоилмуравьиную кислоту, фенилпировиноградную кислоту, индол-3-глиоксиловую кислоту, индол-3-пировиноградную кислоту, нитрофенилпировиноградную кислоту, (2-фурил)глиоксиловую кислоту и другие подобные кетокислоты. Примеры кетоэфиров, подходящих для описанной на схеме 3 реакции, включают, но этим не ограничиваясь, этил тиофен-2-глиоксилат, этил 2-оксо-4-фенилбутират, этил 2-(формиламино)-4-тиазолилглиоксилат, этил-2-амино-4-тиозолилглиоксилат, этил-2-оксо-4-фенилбутират, этил-(5- бромтиен-2-ил)глиоксилат, этил-3-индолилглиоксилат, этил-2-метилбензоилформиат, этил-3-этилбензоилформиат, этил-3- этилбензоилформиат, этил-4-циано-2-оксобутират, метил(1-метилиндолил)-3 -глиоксилат и другие подобные кетоэфиры.
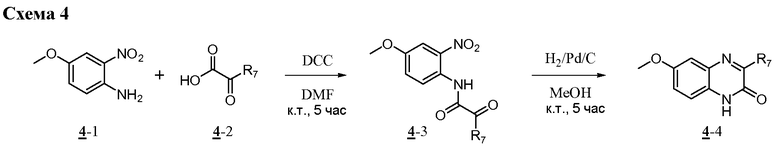
3,6-замещенные хиноксалин-2-оны формулы 4-4, где R7 является определенным ранее W-Z, могут быть получены региоселективно замещением в 6-положение, исходя из реакции амидного сочетания 4-метокси-2-нитроанилина 4-1 и замещенной глиоксиловой кислоты 4-2 с получением соединения 4-3. 3,6-Замещенный хиноксалин-2-он 4-4 получали путем каталитического восстановления нитросоединения 4-3 с последующей реакцией конденсации. Другие заместители могут быть введены в соединение 4-4 в результате использования других 2-нитроанилинов. Примеры кетокислот, подходящих для описанной в схеме 4 реакции, включают, но этим не ограничиваясь, бензоилмуравьиную кислоту, фенилпировиноградную кислоту, индол-3-глиоксиловую кислоту, индол-3-пировиноградную кислоту, нитрофенилпировиноградную кислоту, (2-фурил)- глиоксиловую кислоту и другие подобные кетокислоты. Примеры 2-нитроанилинов, подходящих для описанной в схеме 4 реакции, включают, но этим не ограничиваясь, 4-этокси-2-нитроанилин, 4-амино-3-нитробензотрифторид, 4,5-диметил-2-нитроанилин, 4-фтор-2-нитроанилин, 4-хлор-2-нитроанилин, 4-амино-3-нитрометилбензоат, 4-бензоил-2-нитроанилин, 3-бром-4-метокси-2-нитроанилин, 3'-амино-4'-метил-2-нитроацетофенон, 5-этокси-4-фтор-2-нитроанилин, 4-бром-2-нитроанилин, 4-(трифторметокси)-2-нитроанилин, этил-4-амино-3-нитробензоат, 4-бром-2-метил-6-нитроанилин, 4-пропокси-2-нитроанилин, 5-(пропилтио)-2-нитроанилин и другие подобные нитроанилины.
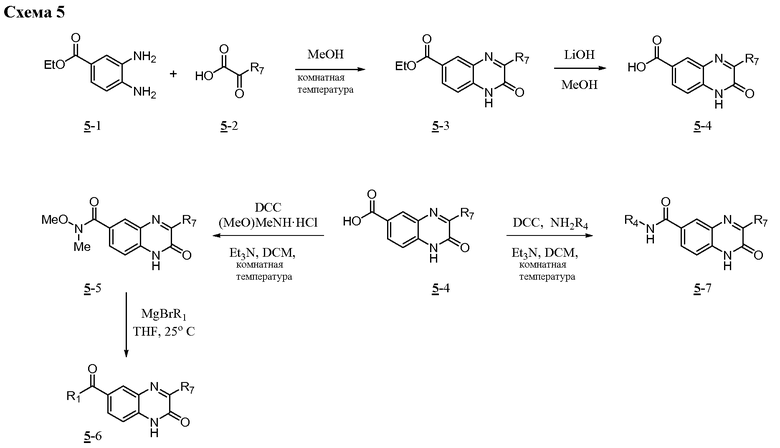
Промежуточное соединение 3-замещенной 2-оксо-1,2-дигидро-хиноксалин-6-карбоновоой кислоты 5-4 может быть получено путем реакции конденсации этил 3,4-диаминбензоата (5-1) с оксоуксусной кислотой формулы 5-2, где R7 = W-Z, определенные ранее, при помощи метода, описанного ранее в схеме 3 (по поводу дополнительных подробностей смотрите Bekerman et al., J. Heterocycl. Chem. 1992, 29, 129-133). Получаемый этиловый эфир 5-3 затем подвергали гидролизу при помощи LiOH в MeOH при комнатной температуре с получением промежуточного соединения карбоновой кислоты 5-4.
Карбоновая кислота 5-4 затем может быть превращена в замещенный кетон 5-6 (где R1 определен ранее) с помощью амида Вейнреба 5-5 и последующей обработки различными реактивами Гриньяра (по поводу подробностей получения и применения амида Вейнреба смотрите Weinreb et al. Tetrahedron Lett. 1977, 33, 4171; Weinreb et al., Synth. Commun. 1982, 12, 989; и смотрите B.S. Furniss, A.J. Hannaford, Р.W.G. Smith, A.R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, 5th ed., Longman, 1989). Реакцию присоединения проводили в инертном растворителе, обычно при низких температурах. Подходящие растворители включают, но этим не ограничиваясь, тетрагидрофуран, диэтиловый эфир, 1,4-диоксан, 1,2-диметоксиэтан и гексаны. Предпочтительным растворителем являлся тетрагидрофуран или диэтиловый эфир. Предпочтительно проводить реакцию при температурах от -78°C до 0°C.
В качестве варианта карбоновая кислота 5-4 может быть использована для получения амидов формулы 5-7, где R4 определен ранее, способом, описанном в общих чертах в схеме 5. Все многочисленные соединения хиноксалин-2-она, описанные в схеме 5, затем подвергают реакции сочетания с образованием макроциклического предшественника при использовании описанных выше реакционных условий Митсунобу.
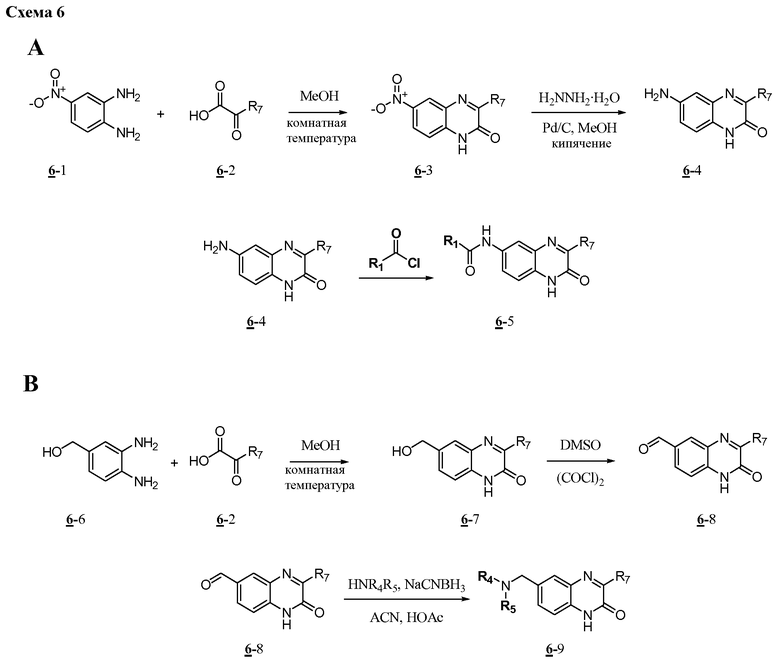
Затем могут быть получены соединения 6-замещенного хиноксалин-2-она с помощью общих методик, приведенных на схеме 6.
A . Восстановление 6-нитро и образование амида
6-нитро-1H-хиноксалин-2-он (6-3) может быть получен с помощью описанного ранее способа из 3,4-диаминонитробензола и оксоуксусной кислоты формулы 6-2, где R7 = W-Z описан ранее. Восстановление нитрогруппы в 6-положении может быть осуществлено с катализатором Pd/C с помощью H2NNH2·H2O при кипячении с обратным холодильником в MeOH. 6-Положение амина 6-4 при обработке его широким рядом хлорангидридов дает различные амиды формулы 6-5, где R1 определен ранее.
B . Окисление бензилового спирта и восстановительное аминирование
Хиноксалин-2-он формулы 6-7 может быть получен путем реакции конденсации 3,4-диаминобензилового спирта и различных оксоуксусных кислот формулы 6-2, где R7 = W-Z определен ранее. Получаемый бензиловый спирт 6-7 может затем быть окислен при реакционных условиях Сверна или любых других условиях для окисления, с получением альдегида формулы 6-8. По поводу дополнительных подробностей относительно реакции Сверна смотрите A. J. Mancuso, D. Swern, Synthesis 1981, 165-185 passim; T. T. Tidwell, Org. React. 1990, 39, 297-572 passim. По поводу других реакционных условий для окисления смотрите B. S. Furniss, A. J. Hannaford, Р.W.G. Smith, A. R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, 5th ed., Longman, 1989. Последующие реакции восстановительного аминирования с первичными или вторичными аминами в присутствии NaCNBH3 и уксусной кислоты могут давать соединения формулы 6-9, где R4 и R5 определены выше.
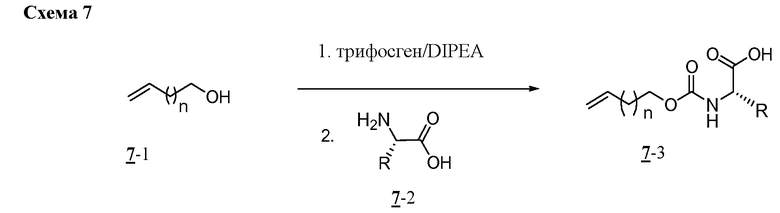
Получение карбаматов аминокислот 7-3 показано на схеме 7; n является целым числом от 0 до 5. Соответствующий олефиновый спирт взаимодействует с трифосгеном в присутствии DIPEA, с последующим добавлением соответствующих аминокислот 7-2 с получением требуемых карбаматов аминокислот 7-3.
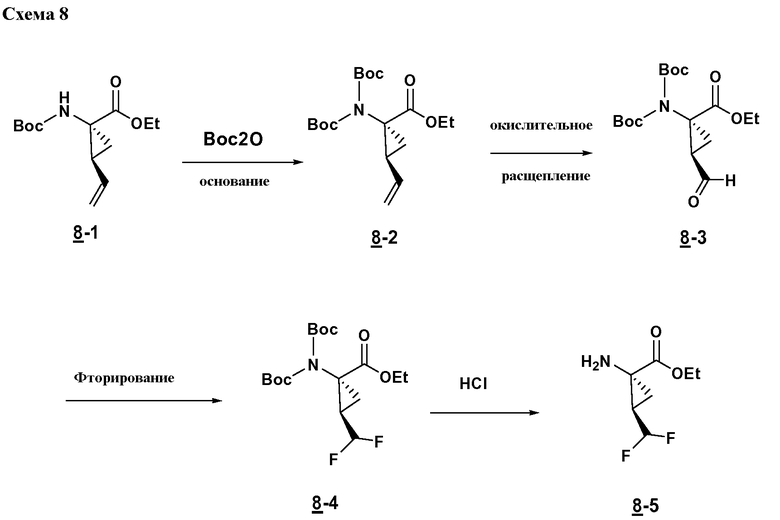
Синтез производных дифторметил P1 аминокислоты 8-5 (то есть 1-9 при R=OEt и R'=CF2H) показан на схеме 8. Моно-Boc эфир аминокислоты был затем защищен как бис-Boc эфир аминокислоты 8-2. Окислительное расщепление соединения 8-2 приводило к образованию альдегида 8-3, который затем превращали в дифторметильное соединение 8-4 при помощи производных аминосератрифторида, таких как диэтиламиносератрифторид (DAST). Снятие защиты с соединения 8-4 с помощью HCl давало требуемое соединение дифторметил P1 8-5.
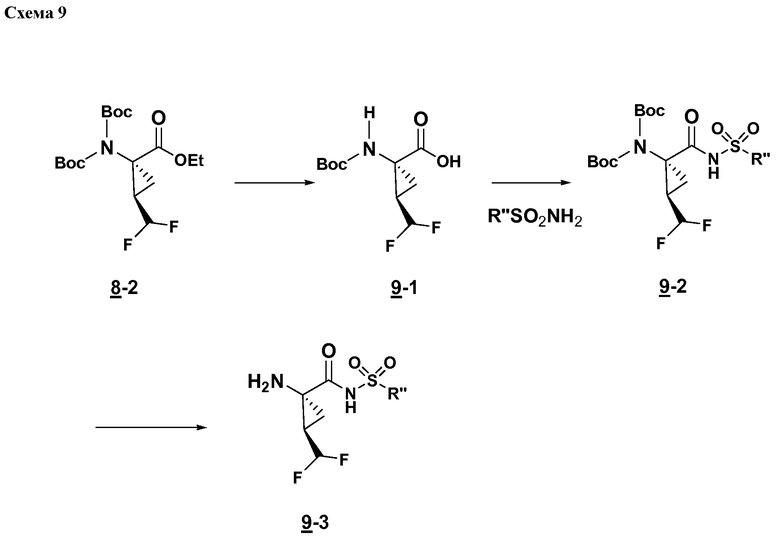
Производное дифторметил P1 сульфонамида 9-3 (то есть 1-9 с R=NHS(O)2R" и R=CF2H) получали, как показано на схеме 9. Гидролиз соединения 8-2 давал кислоту 9-1, которую превращали в соединение 9-2 при помощи CDI/R"SO2NH2/DBU или EDC/DMAP/R"SO2NH2. Снятие защиты с соединения 9-2 давало требуемое промежуточное соединение 9-3.
Содержание всех цитируемых здесь источников в любой печатной форме, электронной форме, в виде считываемой компьютером информации с носителя или в другой форме, включая, но этим не ограничиваясь, рефераты, статьи, журналы, публикации, тексты, учебники, интернет-ресурсы, базы данных, патенты и патентные публикации, приводится здесь путем ссылки на них.
ПРИМЕРЫ
Соединения и способы по настоящему изобретению будут более понятны при помощи приводимых далее примеров, которые, как предполагается, являются только иллюстрацией и не ограничивают объем изобретения. Различные изменения и модификации раскрытых вариантов осуществления будут очевидными для специалистов в этой области, и такие изменения и модификации, включающие без ограничения изменения и модификации, относящиеся к химическим структурам, заместителям, производным, составам и/или способам по изобретению, могут быть сделаны без отклонения от сущности изобретения и объема прилагаемых пунктов формулы изобретения.
Пример 1. Соединение формулы XV, где R= , M-L=
, M-L= , Ar=
, Ar= , R' = CF2H, GF = OH
, R' = CF2H, GF = OH
Стадия 1A
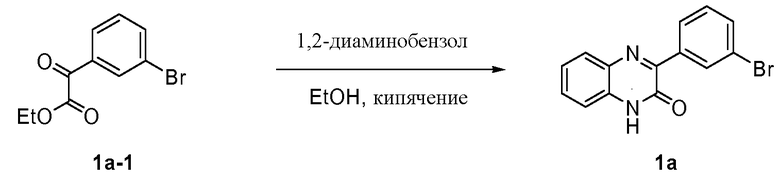
Смесь соединения 1a-1 (0,84 г, 7,77 ммоль), 1,2-диаминобензола (2 г, 7,77 ммоль) и этанола (10 мл) кипятили с обратным холодильником в течение 2 часов, охлаждали до комнатной температуры, фильтровали, промывали холодным этанолом и сушили при пониженном давлении с получением соединения 1a (2,12 г), которое непосредственно использовали на следующей стадии.
Стадия 1B
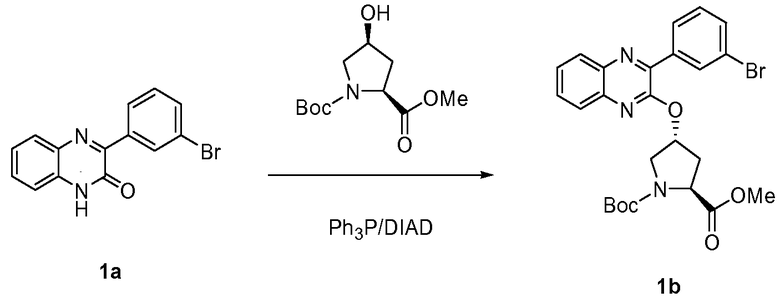
К смеси приведенного выше соединения 1a (1,02 г, 3,39 ммоль), метилового эфира Boc-L-цис-гидроксипролина (0,831 г, 3,38 ммоль) и трифенилфосфина (1,8 г, 6,86 ммоль) в THF при 0°C добавляли по каплям DIAD (1,33 мл, 6,76 ммоль). Полученную смесь выдерживали при 0°C в течение 15 минут, перед тем как подогреть до комнатной температуры. Через 18 часов смесь концентрировали под вакуумом, и остаток очищали с помощью хроматографии (гексан/EtOAC = от 1:0 до 4:1) с получением соединения 1b (1,74 г). MS(ESI): m/e 528,02 (M+H).
В качестве варианта промежуточное соединение 1b получали из соединения 1-2, как показано ниже:
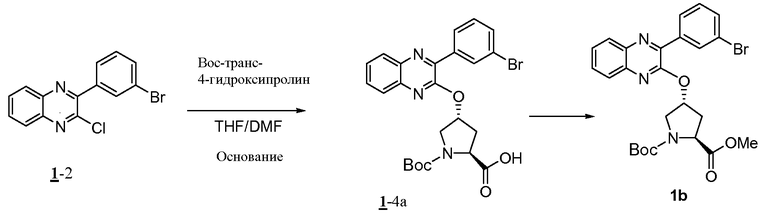
В круглодонную колбу емкостью 12 л загружали Boc-транс-4-гидроксипролин (141 г, 0,61 моль), THF (3,3 л). Смесь охлаждали до 0°C, медленно добавляли через капельную воронку раствор NaOtBu (175,9 г, 1,83 моль) в DMF (0,8 л), при этом температура внутри колбы не превышала 10°C. Ледяную баню удаляли, и смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную колбу помещали опять при 0°C, и осторожно добавляли порциями соединение 1-2 (195 г, 0,61 моль). Тонкослойная хроматография показывала отсутствие исходного материала после 2 часов. Реакцию останавливали с помощью воды (2 л) и концентрировали с удалением большей части THF. Добавляли еще 2 л воды, и смесь дважды экстрагировали трет-бутилметиловым эфиром (4 л + 3 л). К водному слою добавляли 4 л EtOAc, медленно добавляли 10% лимонную кислоту для корректировки pH до 4-5. Разделяли две фазы, водный слой экстрагировали с помощью EtOAc (2×3 л). Объединенные органические слои промывали рассолом, сушили над MgSO4 и концентрировали с получением коричневого масла (356 г). Этот неочищенный материал растворяли в MeOH/CH2Cl2 (1,5 л/1,6 л), охлаждали до 0°C, и добавляли по каплям Me3SiCH2N2 (2M в гексане, 640 мл, 1,28 моль). После 3,5 часов добавление заканчивали, и тонкослойная хроматография показывала оставшийся исходный материал. Реакционную смесь концентрировали, и остаток кристаллизовали из MeOH (~500 мл) с получением 275 г соединения 1b. Маточный раствор очищали на колонке с силикагелем с получением еще 22 г продукта, (суммарный выход: 88% из соединения 1-2).
Стадия 1C
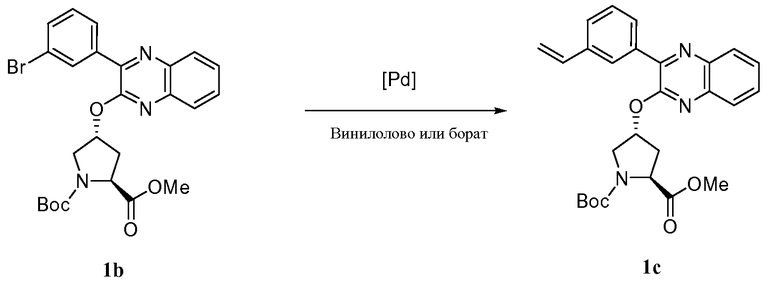
К смеси соединения 1b (0,6 г, 1 ммоль), винилтрифторбората калия (0,52 г, 2 ммоль), триэтиламина (0,55 мл) и этанола (20 мл) добавляли комплекс хлорида 1,1'-бис(дифенилфосфино)-ферроценпалладия(II) с CH2Cl2 (40 мг, 0,048 ммоль). Полученную смесь перемешивали при 75°C в течение 15 часов, охлаждали до комнатной температуры, останавливали реакцию с помощью 10% водного раствора KHSO4, экстрагировали этилацетатом (3x). Объединенные органические слои сушили (MgSO4), концентрировали под вакуумом, и остаток очищали хроматографией (гексан/EtOAC = от 1:0 до 4:1) с получением 1c (0,49 г). MS(ESI): m/z 476,21 (M+H).
Стадия 1D
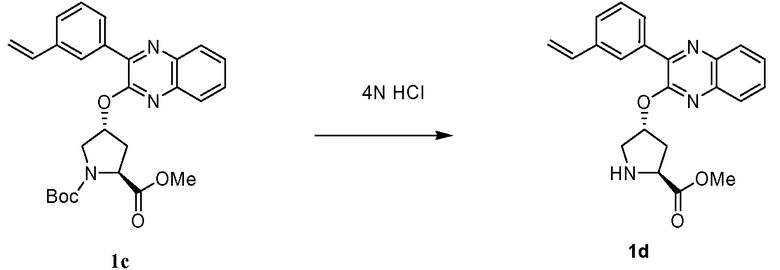
Раствор соединения 1c (1 ммоль) в дихлорметане (2 мл) обрабатывали смесью 4M HCl/диоксан (4 мл, 16 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали в вакууме досуха с получением HCl соли соединения 1d (100%). MS (ESI): m/e 376,15 (M+H).
Стадия 1E
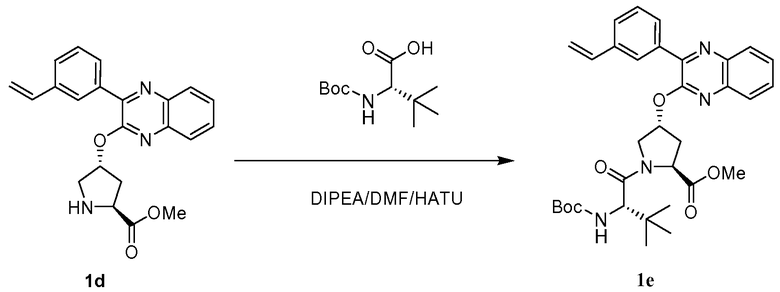
К раствору соединения 1d (HCl соль, 100 мг, 0,223 ммоль), Boc-L-трет-лейцина (67 мг, 0,29 ммоль) и DIPEA (0,24 мл, 1,37 ммоль) в DMF (3 мл) при 0°C добавляли HATU (110 мг, 0,29 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, разбавляли EtOAc и промывали полунасыщенным водным раствором NaCl. Органическую фазу сушили над безводным MgSO4, фильтровали и затем концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (гексан/EtOAC = от 9:1 до 4:1) с получением соединения 1e (128 мг). MS (ESI): m/e 589,44 (M+H), 489,36 (M-Boc).
Стадия 1F
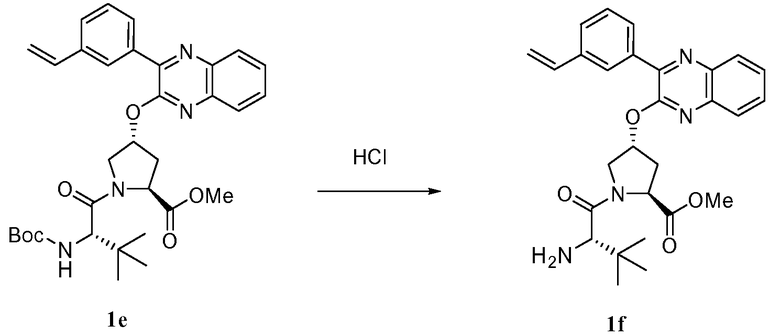
Раствор соединения 1e (117 мг, 0,2 ммоль) в дихлорметане (1 мл) обрабатывали смесью 4M HCl/диоксан (3 мл, 12 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали под вакуумом досуха с получением HCl соли соединения 1f (100%). MS (ESI): m/z 489,45 (M+H).
Стадия 1G
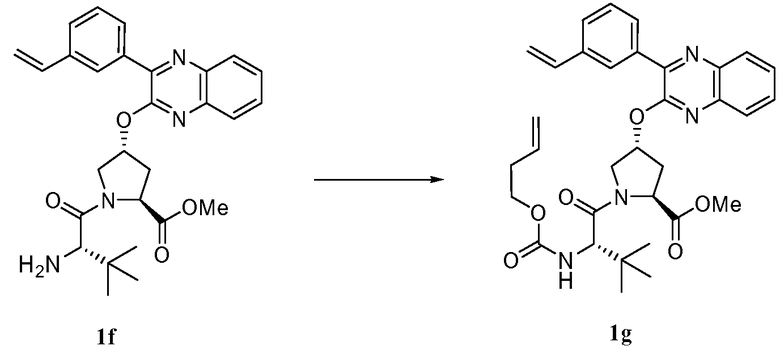
Соединение 1f (0,198 ммоль) растворяли в дихлорметане (3 мл), охлаждали до 0°C, обрабатывали триэтиламином (120 мкл, 4 экв.), затем 3-бутенилхлорформиатом (0,041 мл, 0,33 ммоль). Смесь перемешивали при комнатной температуре в течение от 0,5 до 1 часа, разбавляли этилацетатом, дважды промывали полунасыщенным водным раствором NaCl, сушили (MgSO4) и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (гексаны/EtOAc = от 6:1 до 4:1) с получением соединения 1g (95 мг). MS(ESI): m/z 587,47 (M+H).
В качестве варианта соединение 1g получали путем реакции сочетания соединения 1d и 1g-1, как показано ниже:
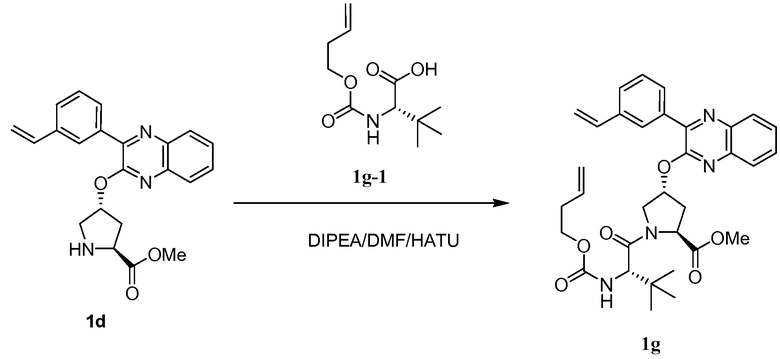
К раствору соединения 1d (HCl соль, 10,78 ммоль), соединения 1g-1 (2,97 г, 13,12 ммоль) и DIPEA (5,6 мл, 32,3 ммоль) в DMF (30 мл) при 0°C добавляли порциями HATU (5,12 г, 13,5 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, разбавляли EtOAc и промывали четыре раза полунасыщенным водным раствором NaCl. Органическую фазу сушили над безводным MgSO4, фильтровали и затем концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (гексан/EtOAC = от 9:1 до 4:1) с получением соединения 1g (6,1 г).
Стадия 1H
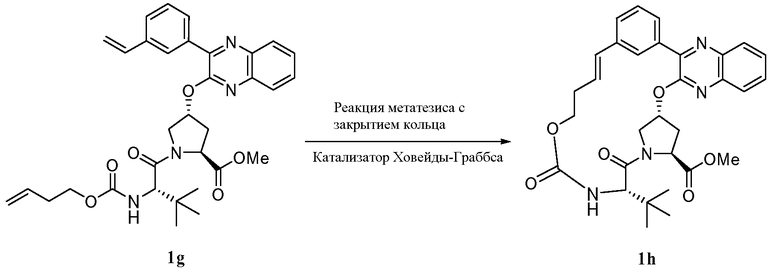
К раствору соединения 1g (95 мг, 0,16 ммоль) в дихлорметане (20 мл) добавляли катализатор Ховейды-Граббса первого поколения или аналогичный катализатор (5 моль.% экв.). Реакционную смесь перемешивали при 40°C в течение 20 часов. Растворитель затем испаряли, и остаток очищали флэш-хроматографией на силикагеле с использованием градиента элюирования (гексан/EtOAC = от 9:1 до 7:3) с получением макроциклического соединения 1h (58 мг). MS (ESI) m/z 559,30 (M+H).
Стадия 1I
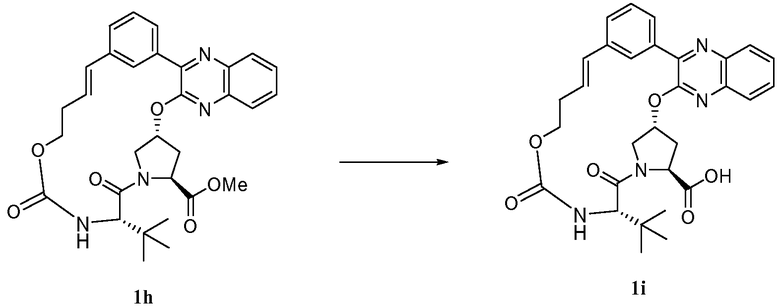
К раствору соединения 1h (58 мг, 0,103 ммоль) в THF/MeOH (3 мл/1,5 мл) добавляли 1н раствор гидроксида лития (1,5 мл, 1,5 ммоль). Смесь перемешивали при комнатной температуре в течение 20 часов. Большую часть органических растворителей испаряли под вакуумом, и полученный остаток разбавляли водой и подкисляли до pH от 5 до 6. Смесь три раза экстрагировали EtOAc. Объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали под вакуумом с получением 1i (100%). MS(ESI): m/z 545,24 (M+H), 551,25 (M+Li).
Стадия 1J
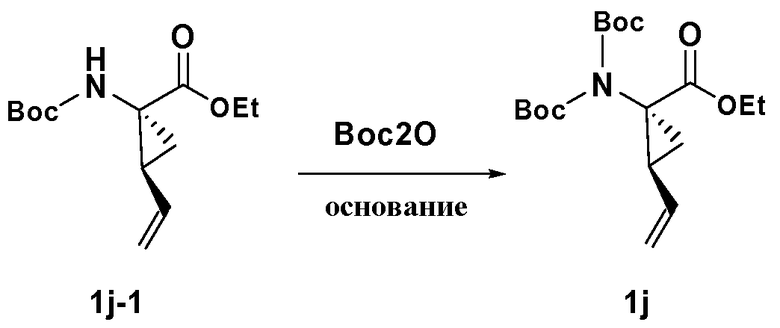
К раствору соединения 1j-1 (6,6 г, 25,85 ммоль) в THF (115 мл) при -78°C медленно добавляли NaHMDS (1,0M в THF, 28,5 мл, 28,5 ммоль). Затем смесь перемешивали при -78°C в течение часа, добавляли Boc2O (6,8 г, 1,2 экв.) в THF (15 мл). Полученную смесь перемешивали, и температуру постепенно повышали до комнатной температуры в течение ночи. Реакционную смесь разбавляли EtOAc, промывали рассолом (2x), сушили (MgSO4) и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (гексан/EtOAc = от 1:0 до 85:15) с получением 1j (8,05 г).
Стадия 1K
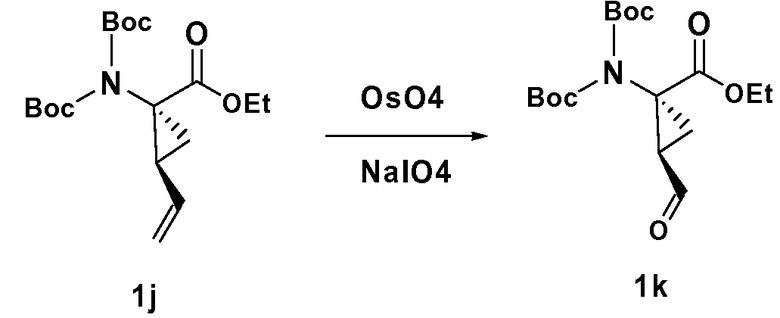
К раствору соединения 1j (0,5 г, 1,4 ммоль) в изопропаноле (5 мл) добавляли NaIO4 (0,9 г, 4,2 ммоль), затем воду (5 мл). К этой интенсивно перемешиваемой смеси добавляли OsO4 (0,4% водный раствор, 0,22 мл, 2,5% экв.). Полученную смесь перемешивали при комнатной температуре в течение 4 часов, разбавляли EtOAc, промывали водным раствором NaHCO3, водным раствором Na2S2O3, рассолом, сушили (MgSO4) и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (гексан/EtOAc = от 1:0 до 85:15) с получением 1k (0,37 г).
Стадия 1L
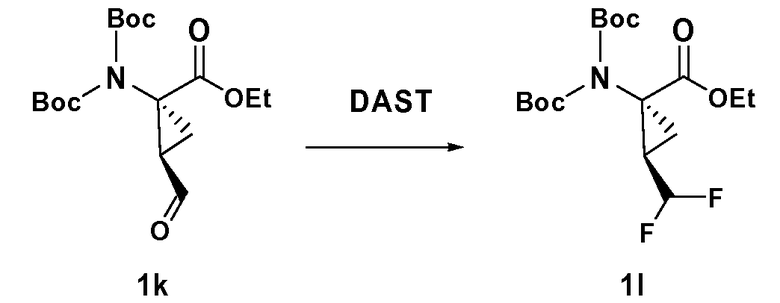
К раствору соединения 1k (2,9 г, 8,1 ммоль) в дихлорметане (25 мл) при -78°C добавляли диэтиламиносератрифторид (DAST) (2,7 мл, 20,25 ммоль). Полученную смесь перемешивали при -78°C в течение часа, затем температуру постепенно повышали до комнатной температуры в течение 6 часов, разбавляли EtOAc, промывали водным раствором NaHCO3 (2x), рассолом, сушили (MgSO4) и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (гексан/EtOAc = от 1:0 до 85:15) с получением 1l (1,49 г). Извлекали исходный материал 1k (1,2 г).
Стадия 1M
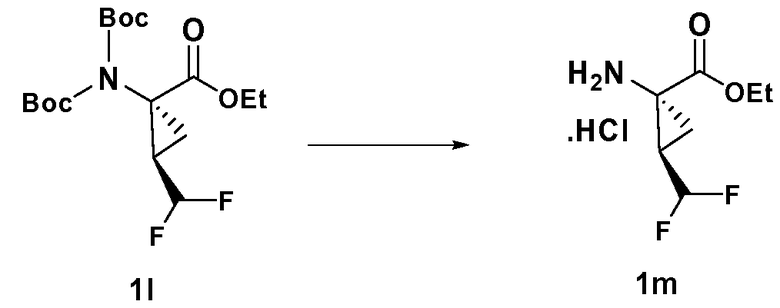
Раствор соединения 1l (491 мг, 1,29 ммоль) в дихлорметане (1 мл) обрабатывали 4н HCl в 1,4-диоксане (6 мл, 24 ммоль). Смесь перемешивали при комнатной температуре в течение часа, концентрировали досуха с получением 1m (~100%).
Стадия 1N
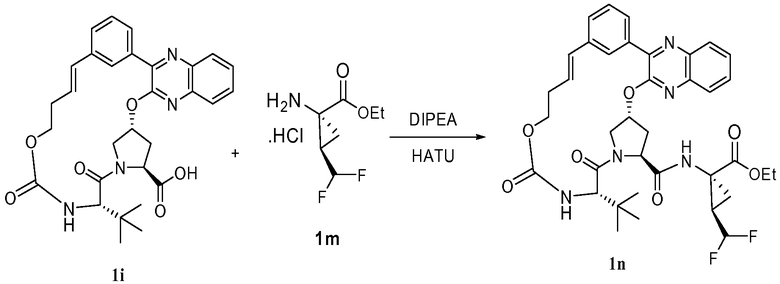
К раствору соединения 1i (5,28 ммоль), соединения 1m (5,808 ммоль) и DIPEA (3,6 мл, 3,9 экв.) в DMF (22 мл) при 0°C добавляли HATU (2,31 г, 6,07 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов, разбавляли EtOAc и промывали четыре раза полунасыщенным раствором NaCl. Органическую фазу сушили над безводным MgSO4, фильтровали и затем концентрировали под вакуумом. Остаток очищали на колонке с силикагелем (гексан/EtOAc = от 9:1 до 6:4) с получением соединения 1n (2,85 г). MS (ESI): m/z 706,53 (M+H).
Стадия 1O
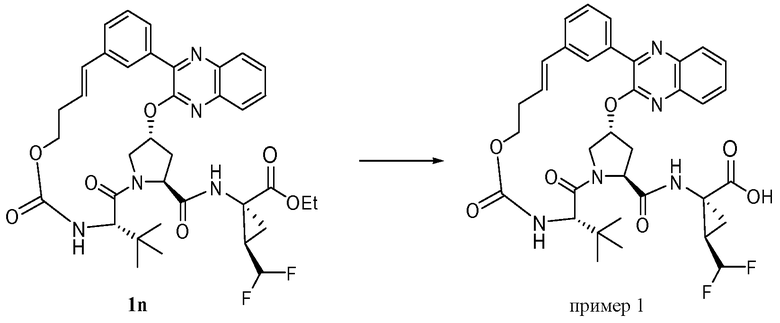
К раствору соединения 1n (2,85 г, 4,04 ммоль) в THF/MeOH (28 мл-12 мл) при 0°C добавляли моногидрат гидроксида лития (1,02 г, 24,2 ммоль), затем воду (12 мл). Смесь перемешивали при комнатной температуре в течение 2 часов, охлаждали до 0°C, подкисляли с помощью 1н HCl до pH от 5 до 6. Некоторую часть органических растворителей удаляли под вакуумом, и полученную смесь три раза экстрагировали EtOAc (3×150 мл). Объединенные органические экстракты промывали рассолом (30 мл), сушили (MgSO4), фильтровали и концентрировали под вакуумом с получением названного соединения (~100%). MS(ESI): m/z 678,41 (M+H).
Пример 2. Соединение формулы XV, где R=, M-L=, Ar=, R' = CF2H, G=
Метод 1
Стадия 2A
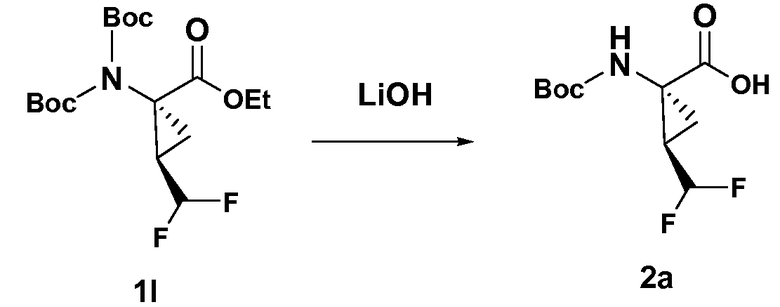
Этиловый эфир 1l (50 г, 132 ммоль) растворяли в THF (400 мл), затем дополнительно разбавляли с помощью MeOH (160 мл) и воды (160 мл). Добавляли LiOH∙H2O (27,6 г, 660 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор подкисляли до pH ~2 с помощью 1M HCl, затем экстрагировали с помощью DCM (3×500 мл). Объединенные органические порции сушили (Na2SO4), фильтровали и концентрировали. Неочищенную карбоновую кислоту 2a непосредственно использовали на следующей стадии без какой-либо дополнительной очистки.
Стадия 2B
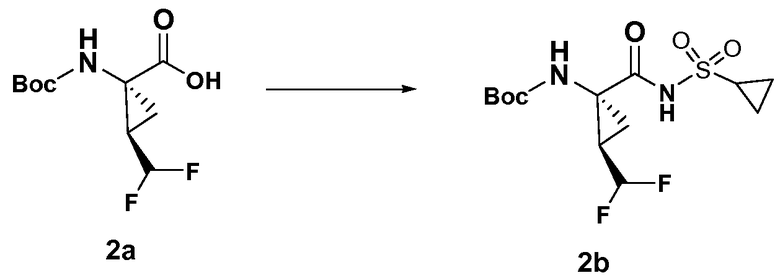
Карбоновую кислоту 2a (~132 ммоль) растворяли в DCM (400 мл), затем охлаждали до 0°C. Добавляли DMAP (40,3 г, 330 ммоль), сульфонамид (16,0 г, 132 ммоль) и EDC (63,3 г, 330 ммоль), и реакционную смесь перемешивали при 0°C в течение 1 часа, подогревали до комнатной температуры, затем перемешивали еще в течение 48 часов. Реакционную смесь разбавляли с помощью EtOAC (1,5 л) и экстрагировали при помощи 1н HCl (2×750 мл). Объединенные водные слои обратно экстрагировали с помощью EtOAc, и объединенные EtOAc слои промывали рассолом (1 л). Органические порции сушили (Na2SO4), фильтровали и концентрировали. Неочищенное масло очищали при помощи хроматографии на SiO2 с использованием 60% EtOAc/гексаны с получением требуемого сульфонимида 2b (40 г, 85%, за две стадии).
Стадия 2C
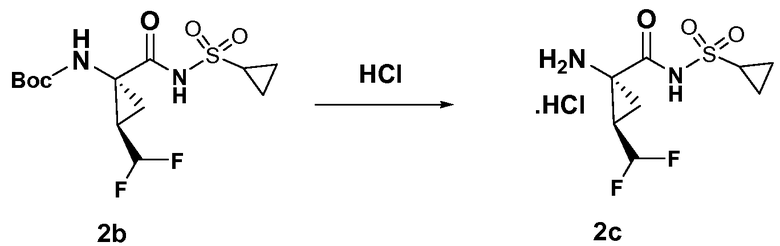
Сульфонимид 2b (40 г, 112 ммоль) непосредственно загружали с 4M HCl раствором в диоксане (282 мл, 1,13 моль), и полученный раствор перемешивали при комнатной температуре в течение 1,5 часа. Реакционную смесь концентрировали под вакуумом, и неочищенное масло растворяли в минимальном количестве DCM и растирали с гексаном (гексан/DCM = 3,5/1,5). Твердое вещество затем отфильтровывали и сушили с получением 32,4 г целевого соединения 2c.
Стадия 2D
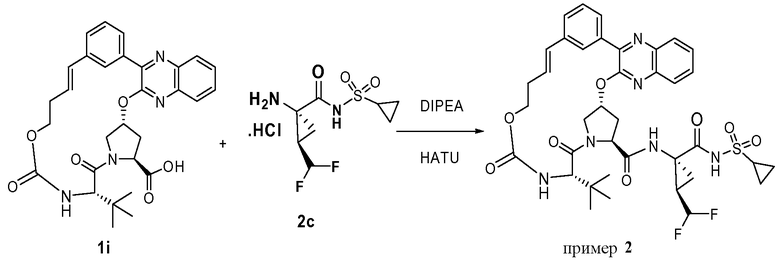
К раствору соединения 1i (11 мг, 0,02 ммоль), соединения 2c (1 экв.) и DIPEA (0,05 мл, 0,287 ммоль) в DMF (1 мл) при 0°C добавляли HATU (24 мг, 0,063 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, разбавляли с помощью EtOAc и промывали четыре раза полунасыщенным водным раствором NaCl. Органическую фазу сушили над безводным MgSO4, фильтровали и затем концентрировали под вакуумом. Остаток очищали с помощью препаративной ВЭЖХ с получением названного соединения (6 мг). MS(ESI); m/z 781,20 (M+H).
Метод II
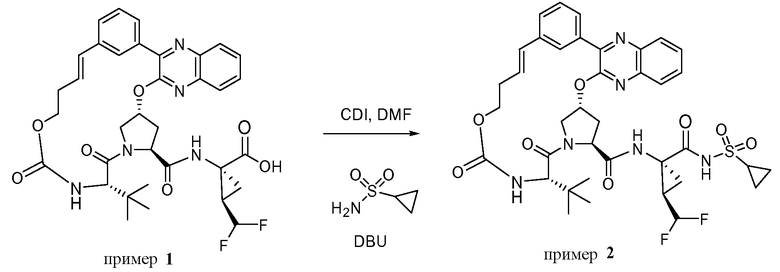
Соединение из примера 1 (2,78 г, 4,04 ммоль) и карбонилдиимидазол (1,11 г, 6,85 ммоль) растворяли в 22 мл безводного DMF, и полученный раствор перемешивали при 40°C в течение 1 часа. К реакционной смеси добавляли циклопропилсульфонамид (1,22 г, 10,06 ммоль), а затем DBU (0,96 мл, 6,4 ммоль). Реакционную смесь перемешивали при 40°C в течение 5 часов. Реакционную смесь разбавляли этилацетатом (400 мл), промывали водой (2×50 мл), 0,5M KH2PO4 (2×50 мл), насыщенным водным раствором NaCl (50 мл), сушили над безводным MgSO4 и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (гексаны/ EtOAc = от 1:1 до 0:1) с получением названного соединения (2,4 г). MS(ESI); m/z 781,20 (M+H).
Пример 3. Соединение формулы XV, где R=, M-L= , Ar=
, Ar= , R'=
, R'= , G=
, G=
Стадия 3A
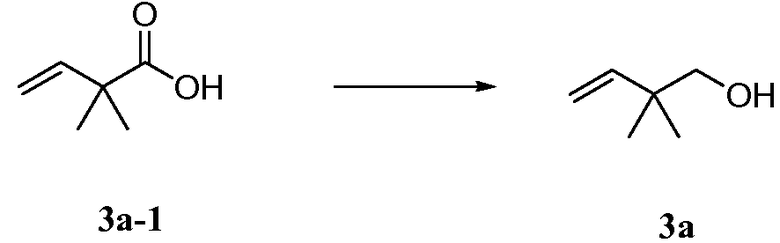
К раствору соединения 3a-1 (21,7 г, 0,19 моль) в диэтиловом эфире (100 мл) при 5-10°C добавляли по каплям LAH (1M в эфире, 200 мл, 0,2 моля). Смесь затем перемешивали при комнатной температуре в течение 1 часа, охлаждали до 0-5°C. Медленно добавляли EtOAc (5 мл), затем осторожно добавляли 6н HCl (200 мл), воду (50 мл) и эфир (50 мл). Отделенную водную фазу затем экстрагировали эфиром (2×250 мл), объединенные органические слои промывали рассолом (75 мл), сушили (сульфат натрия) и концентрировали при 0°C для удаления эфира. Отгонка при пониженном давлении давала соединение 3a (10,5 г).
Стадия 3B
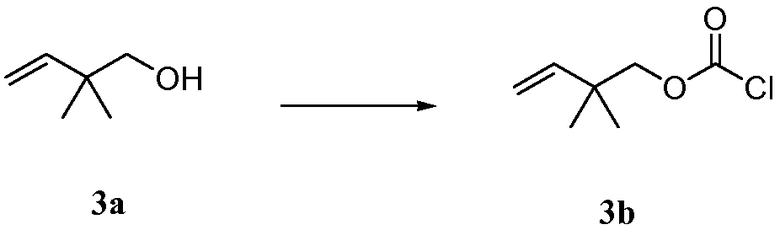
К раствору фосгена в толуоле (20 масс.%, 100 мл, 188 ммоль) при 10°C добавляли по каплям соединение 3a в течение 20 минут. Смесь перемешивали при комнатной температуре в течение 3 часов, испаряли совместно с метиленхлоридом (5×40 мл) с получением соединения 3b (15,6 г, 77% чистоты).
Стадия 3C
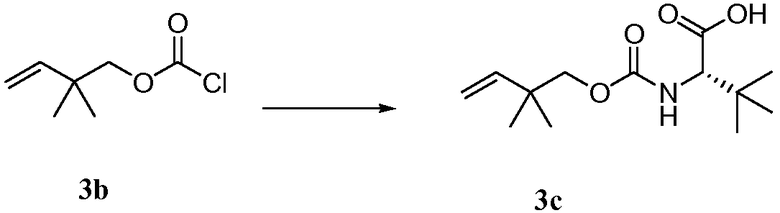
К смеси L-трет-лейцина (5,36 г, 40,8 ммоль) и 1,4-диоксана (22 мл) медленно добавляли 2н NaOH, при этом внутренняя температура не превышала 30°C. Смесь охлаждали до 10-15°C, и медленно добавляли соединение 3b (10,6 г, 77%, 49 ммоль). Смесь перемешивали при комнатной температуре в течение 15 часов, затем при 60°C в течение 3 часов, охлаждали до комнатной температуры, промывали метиленхлоридом (3×35 мл). Корректировали рН водной фазы до величины pH от 2 до 3 с помощью 6н HCl, экстрагировали при помощи EtOAc (3×70 мл). Объединенные EtOAc слои промывали рассолом (20 мл), сушили (сульфат натрия) и концентрировали досуха с получением соединения 3c (8,3 г).
Стадия 3D
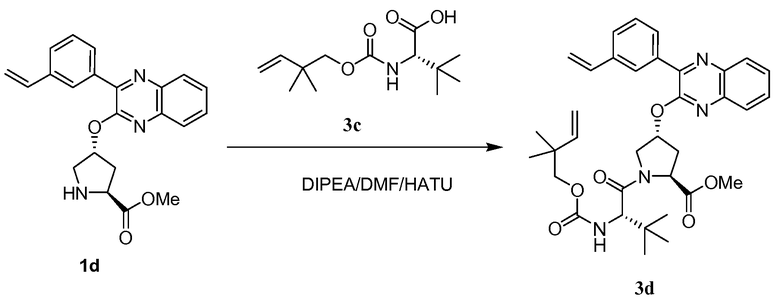
К раствору соединения 1d (HCl соль, 13,57 ммоль), соединения 3c (3,84 г, 14,9 ммоль) и DIPEA (7,1 мл, 40,7 ммоль) в DMF (45 мл) при 0°C добавляли порциями HATU (5,95 г, 15,6 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, разбавляли с помощью EtOAc (400 мл) и промывали водой, четыре раза полунасыщенным водным раствором NaCl. Органическую фазу сушили над безводным Na2SO4, фильтровали и затем концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (гексан/EtOAC = от 9:1 до 4:1) с получением соединения 3d (8,6 г).
Стадия 3E
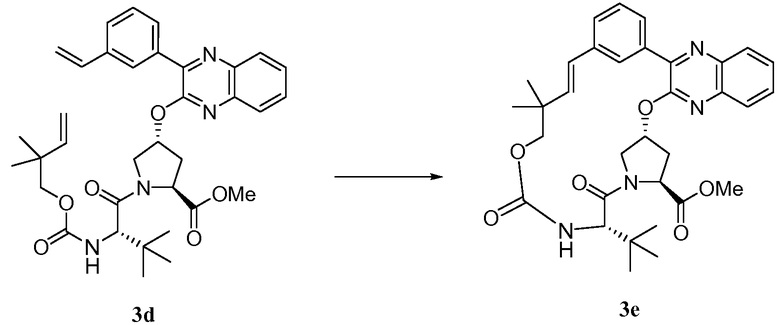
Раствор соединения 3d (4,04 г, 6,75 ммоль) в толуоле (1200 мл) продували азотом в течение 0,5 часа и добавляли катализатор на основе рутения Zhan 1B (0,5 г, 0,675 ммоль). Смесь перемешивали при 110°C в течение 8 часов, охлаждали до комнатной температуры, добавляли 2-меркаптоникотиновую кислоту (1,014 г, 6,7 ммоль) и DIPEA (1,2 мл, 6,75 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, фильтровали и концентрировали. Остаток растворяли в EtOAc, промывали водным раствором NaHCO3, рассолом, сушили (сульфат натрия). Затем испаряли растворитель, и остаток очищали флэш-хроматографией на силикагеле с использованием градиента элюирования (EtOAc/гексан от 0% до 25%) с получением соединения 3e (2,6 г). MS (ESI) m/z 587,30 (M+H).
Стадия 3F
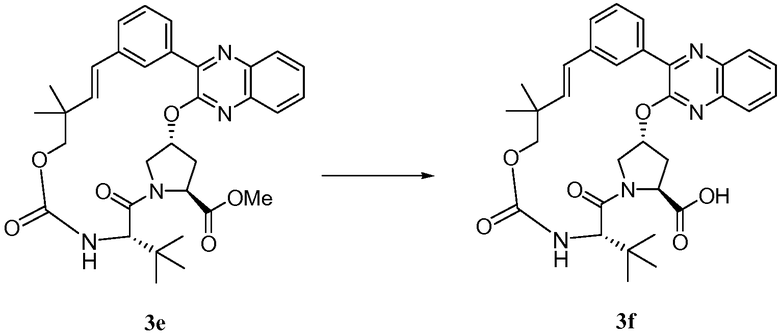
К раствору соединения 3e (3,1 г, 5,28 ммоль) в THF/MeOH (32 мл/14 мл) при 0°C добавляли гидрат гидроксида лития (1,11 г, 26,45 ммоль), затем воду (14 мл). Смесь перемешивали при комнатной температуре в течение часа, помещали при 0°C, добавляли по каплям 1н HCl (~29 мл) до тех пор, пока значение pH смеси не устанавливалось ~5. Смесь экстрагировали с помощью EtOAc (3×80 мл), объединенные органические слои промывали рассолом (30 мл), сушили (Na2SO4), фильтровали и концентрировали под вакуумом с получением 3f (~100%). MS(ESI): m/z 573,39 (M+H).
Стадия 3G
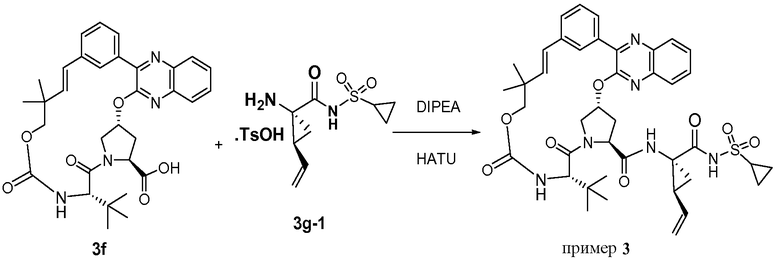
К раствору соединения 3f (16 мг, 0,028 ммоль), соединения 3g-1 (14 мг, 0,034 ммоль) и DIPEA (0,025 мл, 0,14 ммоль) в DMF (1,8 мл) при 0°C добавляли HATU (15 мг, 0,039 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов и очищали с помощью препаративной ВЭЖХ с получением названного соединения (16 мг). MS(ESI): m/z 785,26 (M+H).
Пример 4. Соединение формулы XV, где R=, M-L=, Ar=, R'= , G=
, G=
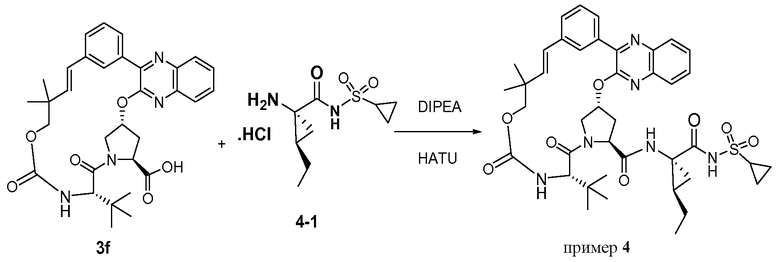
К раствору соединения 3f (15 мг, 0,026 ммоль), соединения 4-1 (8,4 мг, 0,031ммоль) и DIPEA (0,025 мл, 0,14 ммоль) в DMF (1,8 мл) при 0°C добавляли HATU (15 мг, 0,039 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов и очищали с помощью препаративной HPLC с получением названного соединения (15 мг). MS(ESI): m/z 787,39 (M+H).
Пример 5. Соединение формулы XV, где R=, M-L=, Ar=, R'=CF2H, G=
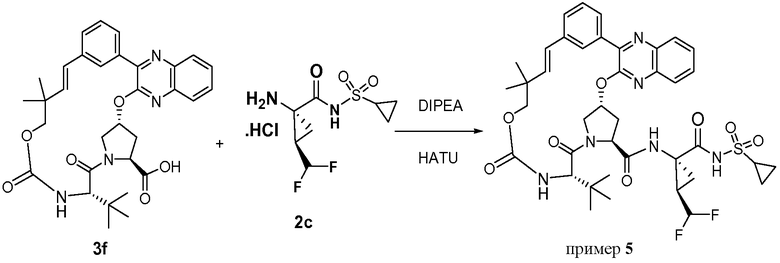
К раствору соединения 3f (1,284 г, 2,07 ммоль), соединения 2c (0,681 г, 2,34 ммоль) и DIPEA (1,1 мл, 3 экв.) в DMF (16 мл) при 0°C добавляли порциями HATU (0,914 г, 2,4 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, разбавляли с помощью EtOAc (180 мл) и промывали водой (20 мл), полунасыщенным водным раствором NaCl (20 мл), водным раствором 0,5M KH2PO4 (2×20 мл), рассолом (20 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (EtOAc/гексан от 25% до 50%) с получением названного соединения (1,3 г). MS(ESI); m/z 809,55 (M+H).
Пример 6. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/e 767,35 (M+H).
Пример 7. Соединение формулы XV, где R=, M-L=, Ar=, R'= , G=
, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/e 757,51 (M+H).
Пример 8. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 771,23 (M+H).
Пример 9. Соединение формулы XV, где R=, M-L=, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/z 795,34 (M+H).
Пример 10. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Стадия 10A
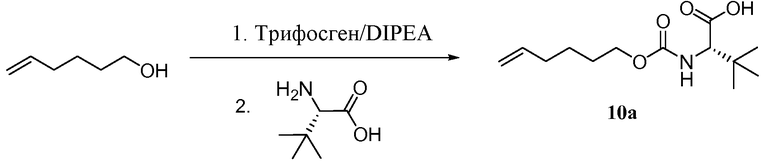
К раствору гекс-5-ен-1-ола (1,3 мл, 10,9 моль), трифосгена (1,46 г, 4,92 ммоль) в 1,4-диоксане (21 мл) при 0°C добавляли по каплям DIPEA (1,7 мл, 9,72 ммоль). Смесь перемешивали при комнатной температуре в течение часа, охлаждали до 0°C. К ней медленно добавляли L-трет-лейцин (1,28 г, 9,72 ммоль), растворенный в 1н NaOH (9,8 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи, концентрировали с удалением половины объема диоксана, обрабатывали с помощью 1н NaOH (25 мл), промывали эфиром (3×30 мл). Водную фазу подкисляли до pH 2~3 с помощью 6н HCl, затем экстрагировали дихлорметаном (3×30 мл). Объединенные органические слои сушили (MgSO4), концентрировали досуха с получением соединения 10a (2,5 г), которое непосредственно использовали на следующей стадии.
Стадия 10B
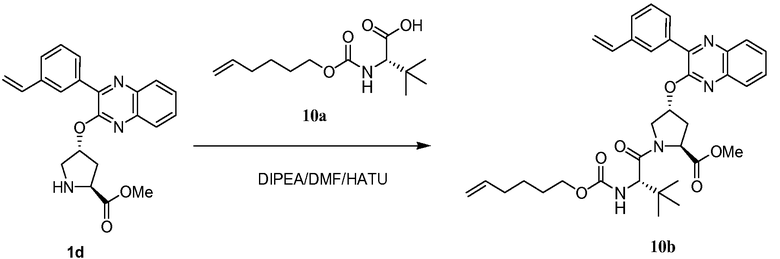
К раствору соединения 1d (HCl соль, 98 мг, 0,218 ммоль), соединения 10a (90 мг, 0,35 ммоль) и DIPEA (0,24 мл, 1,37 ммоль) в DMF (3 мл) при 0°C добавляли HATU (133 мг, 0,35 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, разбавляли с помощью EtOAc и четыре раза промывали полунасыщенным водным раствором NaCl. Органическую фазу сушили над безводным MgSO4, фильтровали и затем концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (гексан/EtOAC = от 6:1 до 4:1) с получением соединения 10b (119 мг). MS (ESI): m/e 615,45 (M+H).
Стадия 10C
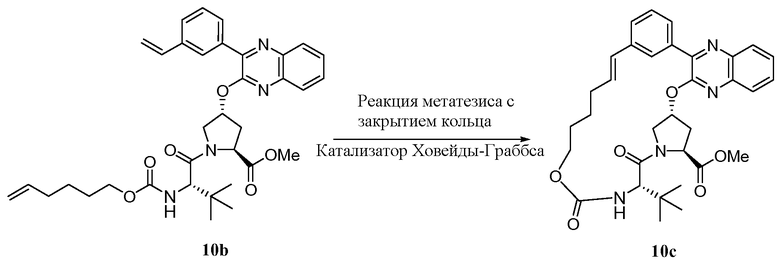
К раствору соединения 10b (119 мг, 0,19 ммоль) в дихлорметане (20 мл) добавляли катализатор Ховейды-Граббса первого поколения (5 моль.% экв.). Реакционную смесь перемешивали при 40°C в течение 20 часов. Растворитель затем испаряли, и остаток очищали с помощью флэш-хроматографии на силикагеле с использованием градиента элюирования (гексан/EtOAC = от 9:1 до 7:3) с получением макроциклического соединения 10c (58 мг). MS (ESI) m/z 587,44 (M+H).
Стадия 10D
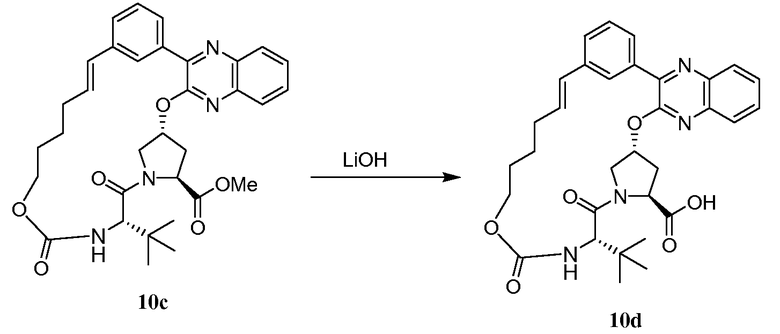
К раствору соединения 10c (58 мг, 0,099 ммоль) в THF/MeOH (3 мл/1,5 мл) добавляли 1н раствор гидроксида лития (1,5 мл, 1,5 ммоль). Смесь перемешивали при комнатной температуре в течение 20 часов. Большую часть органических растворителей испаряли под вакуумом, и полученный остаток разбавляли водой и подкисляли до pH от 5 до 6. Смесь три раза экстрагировали с помощью EtOAc. Объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали под вакуумом с получением соединения 10d (100%). MS(ESI): m/z 573,31 (M+H), 579,32 (M+Li).
Стадия 10E
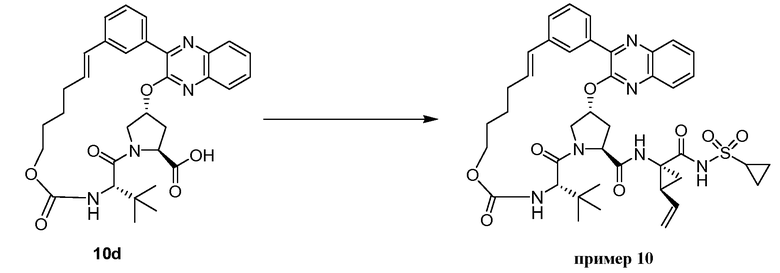
Это соединение получали из соединения 10d по методике, аналогичной методике, описанной в примере 3. MS (ESI): m/z 785,31 (M+H).
Пример 11. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/z 795,53 (M+H).
Пример 12. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/z 795,58 (M+H).
Пример 13. Соединение формулы XV, где R=, M-L= , Ar=, R'=
, Ar=, R'= , G=
, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 785,29 (M+H).
Пример 14. Соединение формулы XV, где R=, M-L= , Ar=, R'=
, Ar=, R'= , G=
, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 4. MS (ESI): m/z 787,39 (M+H).
Пример 15. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/e 809,28 (M+H).
Пример 16. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/e 771,11 (M+H).
Пример 17. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/z 795,21 (M+H).
Пример 18. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 799,25 (M+H).
Пример 19. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 4. MS (ESI): m/z 801,22 (M+H).
Пример 20. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/z 823,21 (M+H).
Пример 21. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 813,42 (M+H).
Пример 22. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 4. MS (ESI): m/z 815,43 (M+H).
Пример 23. Соединение формулы XV, где R=, M-L=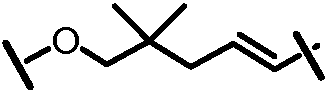 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/z 837,41 (M+H).
Пример 24. Соединение формулы XV, где R=, M-L=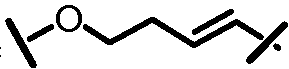 ,
, 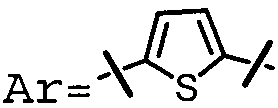 , R'=, G=
, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 763,25 (M+H).
Пример 25. Соединение формулы XV, где R=, M-L=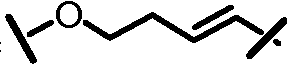 , , R'=CF2H, G=
, , R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/z 787,22 (M+H).
Пример 26. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Стадия 26A
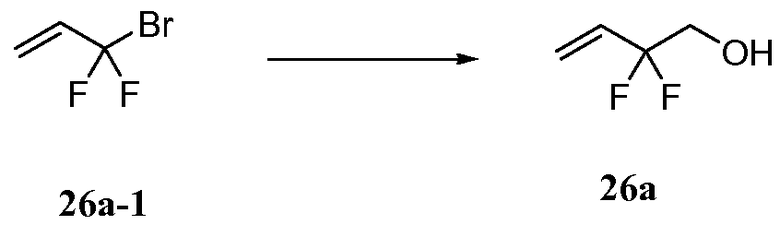
К раствору соединения 26a-1 (3,4 г, 21,6 ммоль), HCOOEt (3,5 мл, 43,2 ммоль) в смеси THF (100 мл)-эфир (30 мл)-пентан (30 мл) при -95°C (жидкий N2 и толуольная баня) добавляли по каплям н-BuLi (2,5 M в гексане, 10 мл, 25 ммоль). Смесь перемешивали при этой температуре в течение 1,5 часов, и температуру бани постепенно поднимали до 0°C в течение 2-3 часов, останавливали реакцию с помощью водного раствора NH4Cl (1,4 г в 20 мл воды). Смесь перемешивали в течение 15 минут. К отделенному органическому слою добавляли MeOH (50 мл), затем добавляли порциями NaBH4 (1 г) при 0°C. Смесь перемешивали при комнатной температуре в течение 0,5 часа, охлаждали до 0°C, осторожно останавливали реакцию с помощью 1н HCl до установления pH ~3, экстрагировали эфиром (3×). Объединенные органические слои промывали водным раствором NaHCO3, рассолом, сушили (Na2SO4) и концентрировали при 0°C. Остаток очищали ректификацией с получением соединения 26a (1,3 г).
Стадия 26B
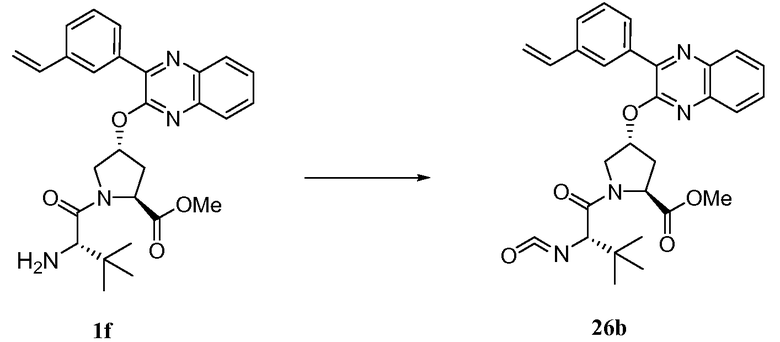
К раствору соединения 1f (6,23 г, 11,10 ммоль) в метиленхлориде (100 мл) при 0°C добавляли пиридин (5,4 мл, 67 ммоль), затем по каплям добавляли раствор фосгена (20 масс.% в толуоле, 17 ммоль) в течение 0,5 часа, смесь перемешивали при комнатной температуре в течение 0,5 часа, разбавляли с помощью EtOAc (500 мл), промывали водой (70 мл), рассолом (2×70 мл), сушили (сульфат натрия), фильтровали и концентрировали досуха с получением соединения 26b (5,3 г). MS(ESI): m/z 515,28 (M+H).
Стадия 26C
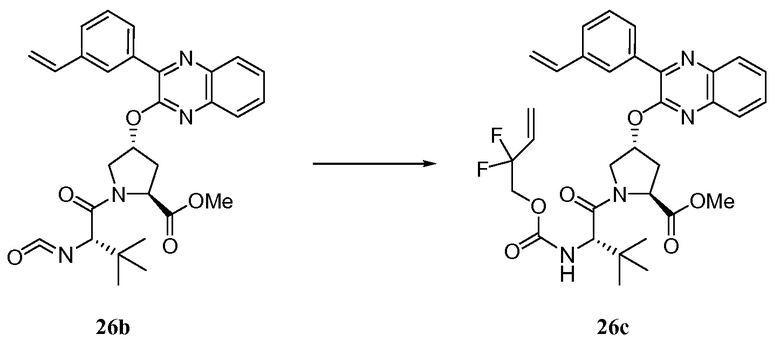
К раствору соединения 26b (4,89 г, 9,5 ммоль) и соединения 26a (~1,4 экв.) в метиленхлориде (65 мл) добавляли молекулярное сито (4Å, 5 г). Затем смесь перемешивали при комнатной температуре в течение 0,5 часа, добавляли DBU (2,1 мл, 16,4 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали. Остаток очищали при помощи флэш-хроматографии на силикагеле с использованием градиента элюирования (EtOAC/гексан от 0% до 25%) с получением соединения 26c (6,1 г). MS (ESI) m/z 623,34 (M+H).
Стадия 26D
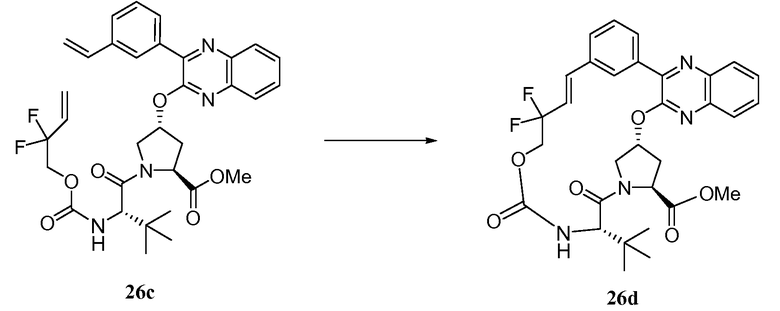
Соединение 26d получали из соединения 26c с помощью методик, аналогичных методикам, описанным на стадии 3E примера 3. MS (ESI) m/z 595,01 (M+H).
Стадия 26E
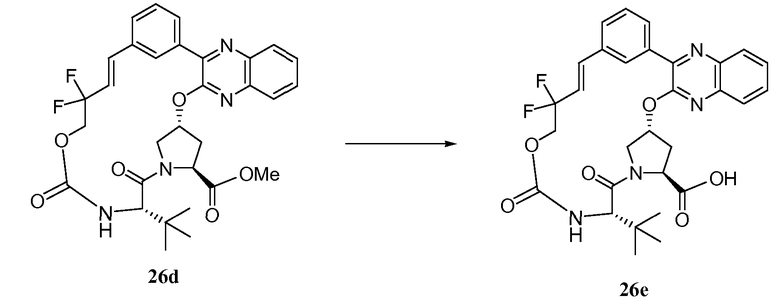
Соединение 26e получали из соединения 26d с помощью методик, аналогичных методикам, описанным на стадии 3F примера 3. MS (ESI) m/z 581,15 (M+H).
Стадия 26F
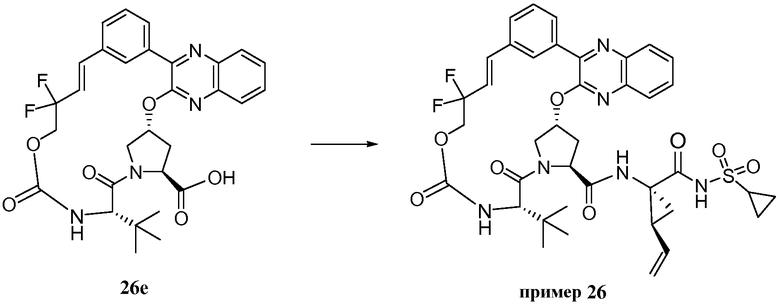
Соединение примера 26 получали из соединения 26e с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 793,20 (M+H).
Пример 27. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали из соединения 26e с помощью методик, аналогичных методикам, описанным в примере 4. MS (ESI): m/z 795,43 (M+H).
Пример 28. Соединение формулы XV, где R=, M-L=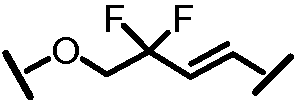 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали из соединения 26e с помощью методик, аналогичных методикам, описанным в примере 5. MS (ESI): m/z 817,27 (M+H).
Пример 29. Соединение формулы XV, где R=, M-L=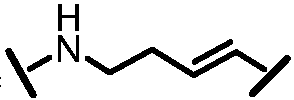 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 756,23 (M+H).
Пример 30. Соединение формулы XV, где R=, M-L=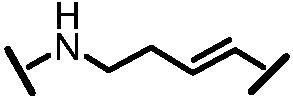 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/z 780,19 (M+H).
Пример 31. Соединение формулы XV, где R=, M-L=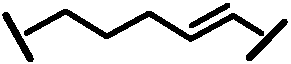 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 755,13 (M+H).
Пример 32. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 2 или примере 5. MS (ESI): m/z 779,26 (M+H).
Пример 33. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Стадия 33A
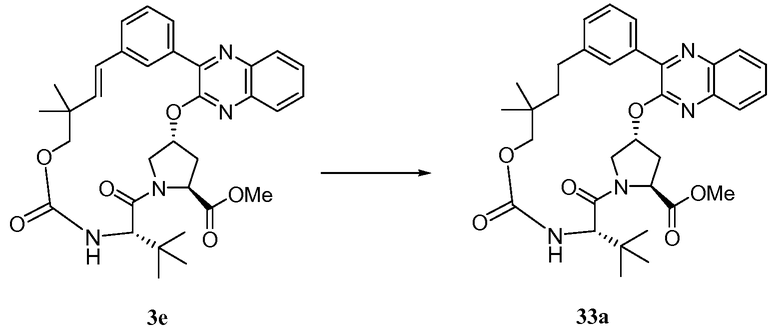
Смесь соединения 3e (1,52 г, 2,59 ммоль), Pd-C (10 масс.% 182 мг) и EtOAc (38 мл) гидрировали в течение 10 часов, фильтровали и концентрировали. Остаток очищали при помощи флэш-хроматографии на силикагеле с использованием градиента элюирования (EtOAC/гексан от 0% до 25%) с получением соединения 33a (1,3 г). MS (ESI): m/z 589,27 (M+H).
Стадия 33B
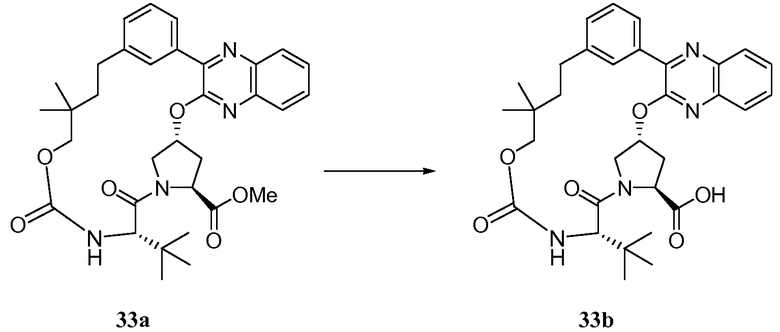
Соединение 33b получали из соединения 33a с помощью методик, аналогичных методикам, описанным на стадии 3F примера 3. MS(ESI): m/z 575,15 (M+H).
Стадия 33C
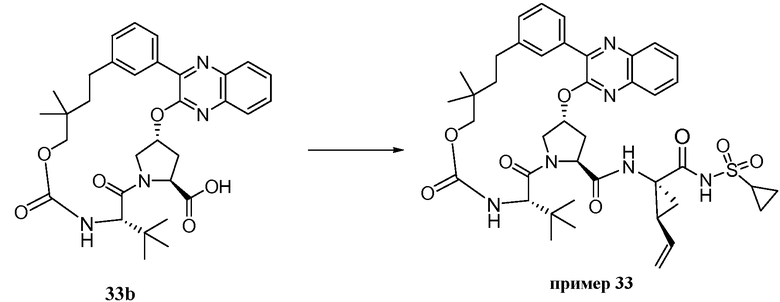
Соединение примера 33 получали из соединения 33b с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 778,39 (M+H).
Пример 34. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали из соединения 33b с помощью методик, аналогичных методикам, описанным в примере 4. MS (ESI): m/z 789,37 (M+H).
Пример 35. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали из соединения 33b с помощью методик, аналогичных методикам, описанным в примере 5. MS (ESI): m/z 811,26 (M+H).
Пример 36. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 35. MS (ESI): m/z 769,35 (M+H).
Пример 37. Соединение формулы XV, где R=, M-L=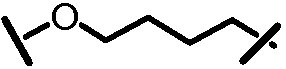 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 34. MS (ESI): m/z 761,62 (M+H).
Пример 38. Соединение формулы XV, где R=, M-L=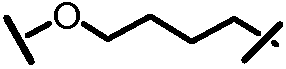 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 35. MS (ESI): m/z 783,38 (M+H).
Пример 39. Соединение формулы XV, где R=, M-L=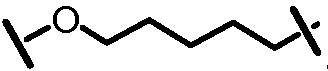 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 33. MS (ESI): m/z 773,49 (M+H).
Пример 40. Соединение формулы XV, где R=, M-L=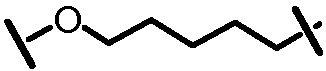 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 34. MS (ESI): m/z 775,36 (M+H).
Пример 41. Соединение формулы XV, где R=, M-L=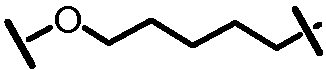 , Ar=, R'=CF2Н, G=
, Ar=, R'=CF2Н, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 35. MS (ESI): m/z 797,34 (M+H).
Пример 42. Соединение формулы XV, где R=, M-L=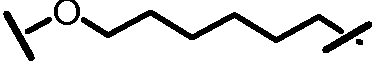 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 33. MS (ESI): m/z 787,41 (M+H).
Пример 43. Соединение формулы XV, где R=, M-L=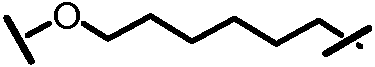 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 34. MS (ESI): m/z 789,26 (M+H).
Пример 44. Соединение формулы XV, где R=, M-L=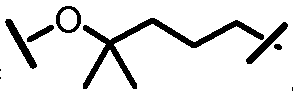 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 35. MS (ESI): m/z 811,28 (M+H).
Пример 45. Соединение формулы XV, где R=, M-L=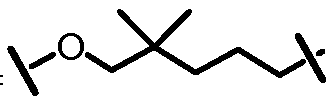 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 33. MS (ESI): m/z 801,57 (M+H).
Пример 46. Соединение формулы XV, где R=, M-L=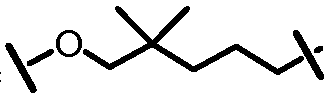 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 34. MS (ESI): m/z 803,57 (M+H).
Пример 47. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 35. MS (ESI): m/z 825,50 (M+H).
Пример 48. Соединение формулы XV, где R=, M-L=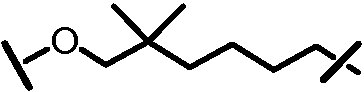 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 33. MS (ESI): m/z 815,46 (M+H).
Пример 49. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 34. MS (ESI): m/z 817,46 (M+H).
Пример 50. Соединение формулы XV, где R=, M-L=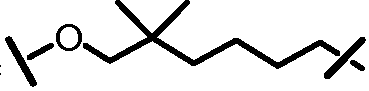 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 35. MS (ESI): m/z 839,46 (M+H).
Пример 51. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Стадия 51A
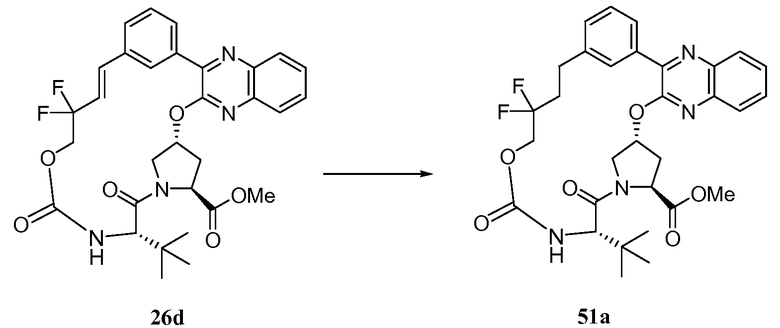
Смесь соединения 26d (2,55 г, 4,286 ммоль), Pd-C (10 масс.% 255 мг), DIPEA (1,5 мл) и EtOAc (65 мл) гидрировали в течение 4 часов, фильтровали и концентрировали. Остаток очищали при помощи флэш-хроматографии на силикагеле с использованием градиента элюирования (EtOAC/гексан от 0% до 25%) с получением соединения 51a (1,83 г). MS (ESI): m/z 597,05 (M+H).
Стадия 51B
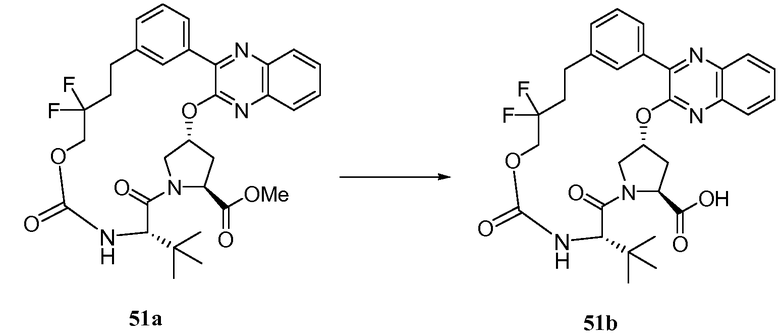
Соединение 51b получали из соединения 51a с помощью методик, аналогичных методикам, описанным на стадии 3F примера 3. MS (ESI) m/z 583,04 (M+H).
Стадия 51C
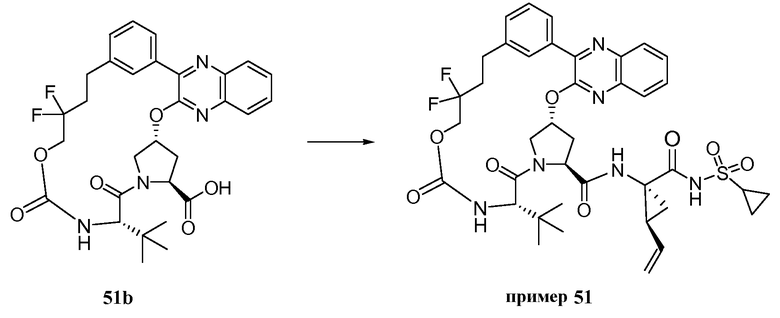
Соединение примера 51 получали из соединения 51b с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 795,27 (M+H).
Пример 52. Соединение формулы XV, где R=, M-L=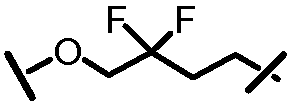 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали из соединения 51b с помощью методик, аналогичных методикам, описанным в примере 4. MS (ESI): m/z 797,43 (M+H).
Пример 53. Соединение формулы XV, где R=, M-L=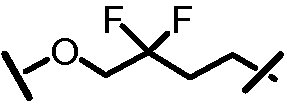 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали из соединения 51b с помощью методик, аналогичных методикам, описанным в примере 5. MS (ESI): m/z 819,43 (M+H).
Пример 54. Соединение формулы XV, где R=, M-L=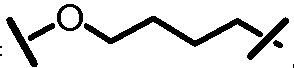 , Ar=, R'=CF2H, G=ОН
, Ar=, R'=CF2H, G=ОН
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 1. MS (ESI): m/z 680,46 (M+H).
Пример 55. Соединение формулы XV, где R=, M-L=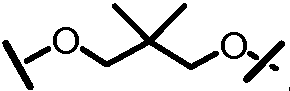 , Ar=, R'=, G=
, Ar=, R'=, G=
Стадия 55A
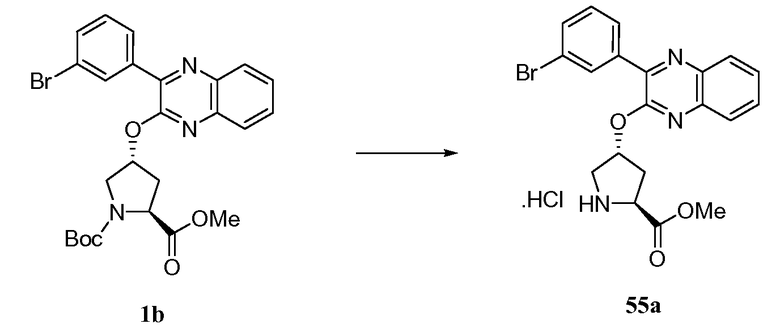
Раствор соединения 1b (1 г, 1,9 ммоль) в дихлорметане (1 мл) обрабатывали смесью 4M HCl/диоксан (6 мл, 24 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 часов и концентрировали под вакуумом досуха с получением HCl соли соединения 55a (100%).
Стадия 55B
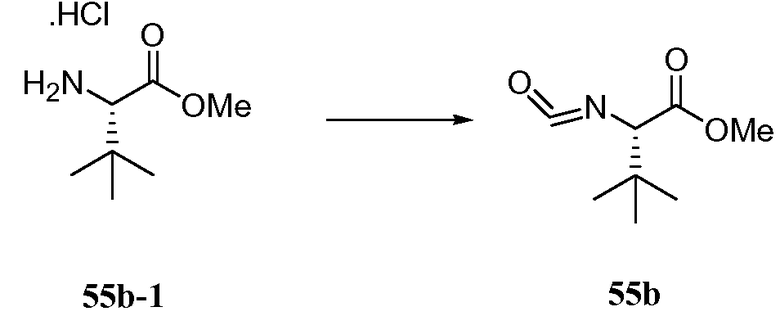
К раствору соединения 55b-1 (1,046 г, 5,5 ммоль) в дихлорметане (30 мл) при 0°C добавляли пиридин (2,23 мл), затем по каплям добавляли раствор фосгена (в толуоле, 20 масс.%, 4,4 мл, 8,3 ммоль). Смесь перемешивали при 0°C в течение 2 часов, разбавляли с помощью EtOAc, промывали 1н HCl, рассолом, сушили (сульфат натрия) и концентрировали досуха с получением соединения 55b (0,95 г).
Стадия 55C
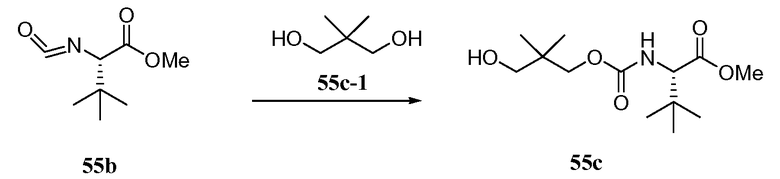
К раствору соединения 55b (0,228 г, 1,33 ммоль) и соединения 55c-1 (1,4 г, 13,3 ммоль) в смеси дихлорметан (10 мл)-DMF (2 мл) при комнатной температуре добавляли DBU (0,3 мл, 2,18 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, разбавляли с помощью EtOAc, промывали водой, дважды рассолом, 1н HCl, рассолом, сушили (сульфат натрия) и концентрировали. Остаток очищали при помощи флэш-хроматографии на силикагеле с использованием градиента элюирования (EtOAC/гексан от 0% до 30%) с получением соединения 55c (0,303 г).
Стадия 55D
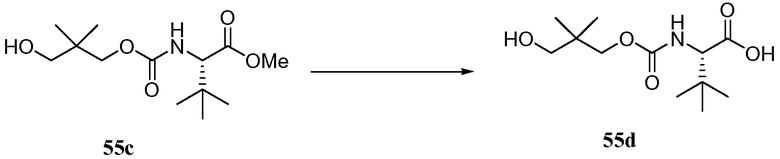
К раствору соединения 55c (0,255 г, 0,93 ммоль) в THF/MeOH (6 мл/2,6 мл) при 0°C добавляли гидрат гидроксида лития (0,195 г, 4,6 ммоль), затем воду (2,6 мл). Смесь перемешивали при комнатной температуре в течение 16 часов, помещали при 0°C, добавляли по каплям 1н HCl до тех пор, пока рН смеси не становилась равной ~3. Смесь три раза экстрагировали с помощью EtOAc, объединенные органические слои промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали под вакуумом с получением соединения 55d (0,227 г).
Стадия 55E
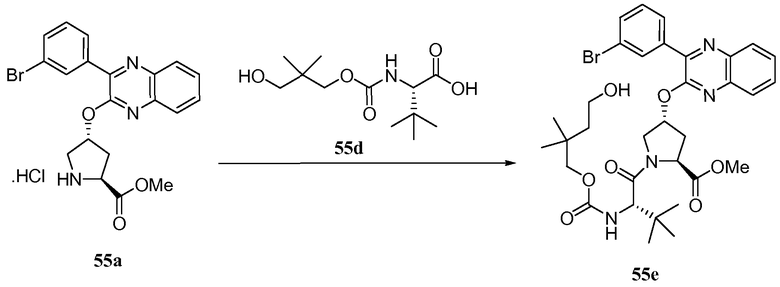
К раствору соединения 55a (0,52 г, 1,1 ммоль), соединения 55d (0,93 ммоль) и DIPEA (0,81 мл, 5 экв.) в DMF (8 мл) при 0°C добавляли порциями HATU (0,407 г, 1,07 ммоль). Смесь перемешивали при комнатной температуре в течение 16 часов, разбавляли с помощью EtOAc, промывали один раз водой, четыре раза рассолом. Органическую фазу сушили над безводным сульфатам натрия, фильтровали и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (EtOAc/гексан от 0% до 40%) с получением 55e (0,328 г). MS(ESI); m/z 671,32, 673,32 (M+H).
Стадия 55F
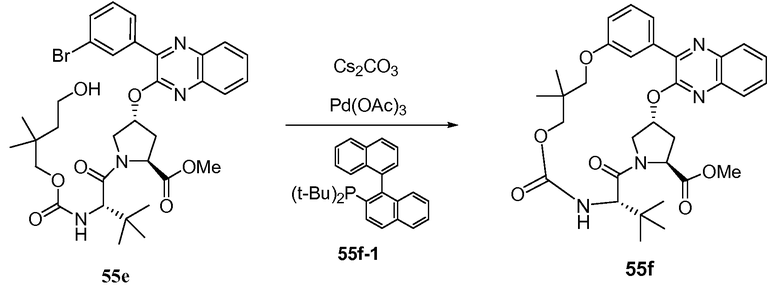
Смесь соединения 55e (0,161 г, 0,24 ммоль), карбоната цезия (0,156 г, 0,48 ммоль) и толуола (10 мл) продували азотом в течение 5 минут. Добавляли ацетат палладия (11 мг, 0,049 ммоль) и лиганд 55f-1 (24 мг, 0,06 ммоль). Смесь перемешивали при 80°C в течение 19 часов, охлаждали до комнатной температуры, фильтровали, промывали с помощью EtOAc и концентрировали. Остаток очищали хроматографией на силикагеле (EtOAc/гексан от 0% до 25%) с получением соединения 55f (0,035 г). MS(ESI); m/z 591,61(M+H).
Стадия 55G
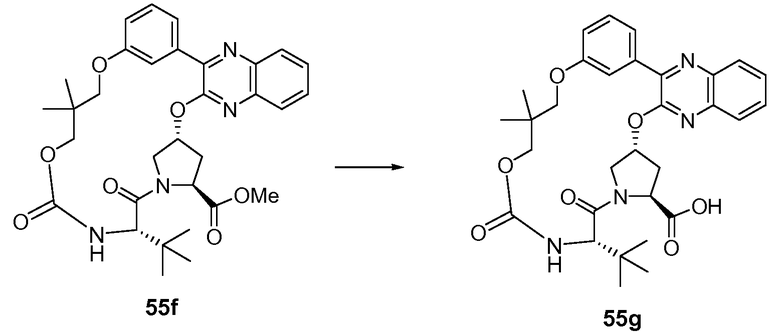
Соединение 55g получали из соединения 55f с помощью методик, аналогичных методикам, описанным на стадии 3F примера 3. MS(ESI): m/z 577,55 (M+H).
Стадия 55H
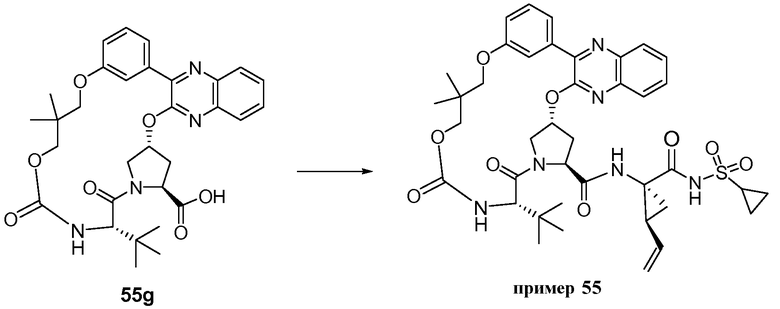
Соединение примера 55 получали из соединения 55g с помощью методик, аналогичных методикам, описанным в примере 3. MS (ESI): m/z 789,34 (M+H).
Пример 56. Соединение формулы XV, где R=, M-L=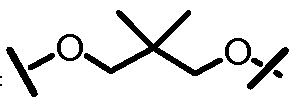 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали из соединения 55g с помощью методик, аналогичных методикам, описанным в примере 4. MS (ESI): m/z 790,92 (M+H).
Пример 57. Соединение формулы XV, где R=, M-L=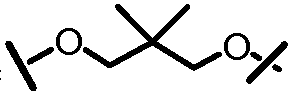 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали из соединения 55g с помощью методик, аналогичных методикам, описанным в примере 5. MS (ESI): m/z 813,74 (M+H).
Пример 58. Соединение формулы XV, где R=, M-L=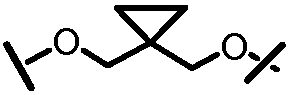 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 55. MS (ESI): m/z 787,20 (M+H).
Пример 59. Соединение формулы XV, где R=, M-L=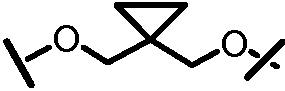 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 56. MS (ESI): m/z 789,22 (M+H).
Пример 60. Соединение формулы XV, где R=, M-L=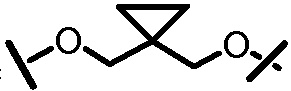 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 57. MS (ESI): m/z 811,59 (M+H).
Пример 61. Соединение формулы XV, где R=, M-L=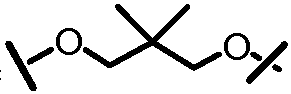 , Ar=
, Ar= , R'=, G=
, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 55. MS (ESI): m/z 807,17 (M+H).
Пример 62. Соединение формулы XV, где R=, M-L=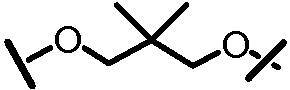 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 56. MS (ESI): m/z 809,10 (M+H).
Пример 63. Соединение формулы XV, где R=, M-L=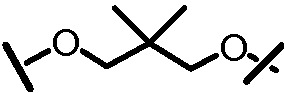 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 57. MS (ESI): m/z 831,24 (M+H).
Пример 64. Соединение формулы XV, где R=, M-L=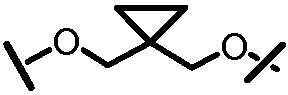 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 55. MS (ESI): m/z 805,14 (M+H).
Пример 65. Соединение формулы XV, где R=, M-L=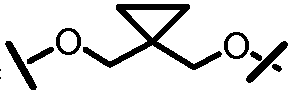 , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 56. MS (ESI): m/z 807,03 (M+H).
Пример 66. Соединение формулы XV, где R=, M-L=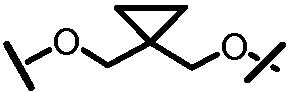 , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 57. MS (ESI): m/z 828,97 (M+H).
Пример 67. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 55. MS (ESI): m/z 797,11 (M+H).
Пример 68. Соединение формулы XV, где R=, M-L= , Ar=, R'=, G=
, Ar=, R'=, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 56. MS (ESI): m/z 799,06 (M+H).
Пример 69. Соединение формулы XV, где R=, M-L= , Ar=, R'=CF2H, G=
, Ar=, R'=CF2H, G=
Это соединение получали с помощью методик, аналогичных методикам, описанным в примере 57. MS (ESI): m/z 821,54 (M+H).
Примеры 70-1506, соединения формулы XV в таблице 1, получают, следуя методикам, описанным в примерах 1-69 и в разделе "Способы синтеза".
Примеры 1507-1554, соединения формулы XVI в таблице 2, получают, следуя методикам, описанным в примерах 1-69 и в разделе "Способы синтеза".
Примеры 1555-1586, соединения формулы XVII в таблице 3, получают, следуя методикам, описанным в примерах 1-69 и в разделе "Способы синтеза".
Соединения по настоящему изобретению проявляют эффективные ингибирующие свойства по отношению к HCV NS3 протеазе. Следующие примеры описывают исследования, в которых соединения по настоящему изобретению могли быть испытаны на анти-HCV действие.
Пример 1587. Исследование фермента протеазы NS3/NS4a
Исследуется активность и ингибирование HCV протеазы с использованием внутренне блокированного флуорогенного субстрата. DABCYL и EDANS группы прикреплены к противоположным концам короткого пептида. Блокирование EDANS флуоресценции с помощью DABCYL группы облегчается при протеолитическом расщеплении. Флуоресценцию измеряют с помощью прибора Molecular Devices Fluoromax (или аналогичного) при длине волны возбуждения 355 нм и длине волны излучения 485 нм.
Исследование проводят в 96-луночных наполовину белых планшетах фирмы Corning (VWR 29444-312 [Corning 3693]) с полноразмерной NS3 протеазой HCV генотипа 1b, связанной с NS4A кофактором (конечная концентрация фермента от 1 до 15 нM). Буфер для исследования дополняют с помощью 10 мкM NS4A кофактора Pep 4A (Anaspec 25336 или полученный собственными силами, молекулярная масса 1424,8). RET S1 (Ac-Asp-Glu-Asp(EDANS)-Glu-Glu-Abu-[COO]Ala-Ser-Lys-(DABCYL)-NH2, AnaSpec 22991, молекулярная масса 1548,6) используют в качестве флуорогенного пептидного субстрата. Буфер для исследования содержит 50 мM Hepes при pH 7,5, 30 мM NaCl и 10 мM BME. Реакция фермента происходит в течение 30 минут при комнатной температуре в отсутствии и присутствии ингибиторов.
Пептидные ингибиторы HCV Inh 1 (Anaspec 25345, молекулярная масса 796,8) Ac-Asp-Glu-Met-Glu-Glu-Cys-OH [-20°C] и HCV Inh 2 (Anaspec 25346, молекулярная масса 913,1) Ac-Asp-Glu-Dif-Cha-Cys-OH используются в качестве ссылочных соединений.
Значения IC50 вычисляют с использованием программного продукта XLFit в системе ActivityBase (IDBS) по уравнению 205: y=A+((B-A)/(1+((C/x)^D))).
Пример 1588. Исследование репликона на основе клеток
Количественное определение РНК репликона HCV (исследование на основе клеток HCV) осуществляют с помощью клеточной линии Huh 11-7 (Lohmann, et al. Science 285:110-113, 1999). Клетки высевают при плотности 4×103 клеток/лунка в 96-луночных планшетах в питательную среду, содержащую DMEM (с высокой концентрацией глюкозы), 10% фетальной телячьей сыворотки, пеницилин-стрептомицин и неосновные аминокислоты. Клетки инкубируют в инкубаторе с 7,5% CO2 при 37°C. В конце периода инкубирования суммарную РНК извлекают и очищают от клеток при помощи набора Ambion RNAqueous 96 Kit (Catalog No. AM1812). Для того чтобы увеличить HCV РНК, с тем чтобы можно было обнаружить достаточный материал с помощью HCV специфического зонда (ниже), праймеры, специфичные на HCV (ниже), содействуют как обратной транскрипции HCV РНК, так и амплификации cДНК путем полимеразной цепной реакции (PCR) при помощи набора TaqMan One-Stage RT-PCR Master Mix Kit (Applied Biosystems catalog no. 4309169). Нуклеотидные последовательности RT-PCR праймеров, которые расположены в NS5B области генома HCV, являются следующими:
HCV прямой праймер "RBNS5bfor"
5 'GCTGCGGCCTGTCGAGCT (SEQ ID NO: 1):
HCV обратный праймер "RBNS5Brev"
5 'CAAGGTCGTCTCCGCATAC (SEQ ID NO: 2).
Обнаружение RT-PCR продукта осуществляется при помощи системы обнаружения последовательности (SDS) Applied Biosystems (ABI) Prism 7500, которая обнаруживает флуоресценцию, которая излучается в случае, когда зонд, который помечен флуоресцентным репортерным красителем и гасящим флуоресценцию красителем, разрушается во время PCR реакции. Увеличение количества флуоресценции измеряют во время каждого цикла PCR, и оно отражает увеличение количества RT-PCR продукта. А именно, количественное определение основано на пороговом цикле, когда кривая амплификации пересекает определенный порог флуоресценции. Сравнение пороговых циклов образца с известным стандартом позволяет с высокой чувствительностью измерять относительную концентрацию темплата в различных образцах (ABI User Bulletin #2 December 11, 1997). Данные анализируют при помощи программного продукта ABI SDS версия 1.7. Относительная концентрация темплата может быть преобразована в число копий РНК путем использования калибровочной кривой для HCV РНК эталонов с известным числом копий (ABI User Bulletin #2 December 11, 1997).
RT-PCR продукт определяли при помощи следующего меченого зонда:
5' FAM-CGAAGCTCCAGGACTGCACGATGCT-TAMRA (SEQ ID NO: 3)
FAM = флуоресцентный репортерный краситель.
TAMRA = гасящий флуоресценцию краситель.
RT реакцию проводят при 48°C в течение 30 минут, затем проводят PCR. Параметры термоциклера, используемые для PCR реакции на системе обнаружения последовательности ABI Prism 7500, являются следующими: один цикл при 95°C, 10 минут, затем 40 циклов, каждый из которых включает одно инкубирование при 95°C в течение 15 секунд и второе инкубирование при 60°C в течение 1 минуты.
Чтобы нормализовать данные относительно молекулы внутреннего контроля внутриклеточной РНК, RT-PCR проводят на глицеральдегид-3-фосфат-дегидрогеназе (GAPDH) клеточной информационной РНК. Число копий GAPDH является стабильным в используемых клеточных линиях. GAPDH RT-PCR осуществляют на том же образце РНК, из которого определяют число копий HCV. GAPDH праймеры и зонды содержатся в наборе ABI Pre-Developed TaqMan Assay Kit (catalog no. 4310884E). Отношение HCV/GAPDH РНК используют для вычисления активности соединений, оцениваемых для ингибирования HCV РНК репликации.
Активность соединений в качестве ингибиторов HCV репликации (исследование на основе клеток) в репликоне, содержащем клеточные линии Huh-7.
Воздействие конкретного противовирусного соединения на уровни РНК репликона HCV в клетках Huh-11-7 определяют путем сравнения количества HCV РНК, нормализованного к GAPDH (например, отношение HCV/GAPDH), в клетках, подвергнутых воздействию соединения, по отношению к клеткам, подвергнутым действию среды DMSO (отрицательный контроль). Конкретно, клетки высевают с плотностью 4×103 клеток/лунка в 96-луночный планшет и инкубируют или: 1) со средой, содержащей 1% DMSO (0% ингибирование в качестве контроля), или 2) со средой (1% DMSO), содержащей определенную концентрацию соединения. Описанные выше 96-луночные планшеты затем инкубируют при 37°C в течение 4 дней (определение EC50). Процент ингибирования определяют как:
% ингибирования = 100-100*S/C1,
где
S = отношение число копий HCV РНК/число копий GAPDH РНК в образце;
C1 = отношение число копий HCV РНК/число копий GAPDH РНК при 0% ингибировании в качестве контроля (среда 1% DMSO).
Кривую зависимости "доза-эффект" для ингибитора получают путем последовательного добавления соединений, трехкратного разбавления в течение трех регистраций в лунках, начиная с самой высокой концентрации конкретного соединения при 1,5 мкM и заканчивая самой низкой концентрацией 0,23 нM. Дополнительные серии разведения (например, от 500 нM до 0,08 нM) проводят, если величина EC50 не ложится хорошо на кривую. EC50 определяют при помощи IDBS Activity Base program "XL Fit", используя для аппроксимации 4-параметрическую нелинейную регрессию (модель # 205 версия 4.2.1, форма 16).
Несмотря на то, что это изобретение было конкретно представлено и описано со ссылками на его предпочтительные варианты осуществления, для специалистов в этой области является очевидным, что в нем могут быть сделаны различные изменения по форме и в деталях без отклонения от объема изобретения, определяемого прилагаемыми пунктами формулы изобретения.



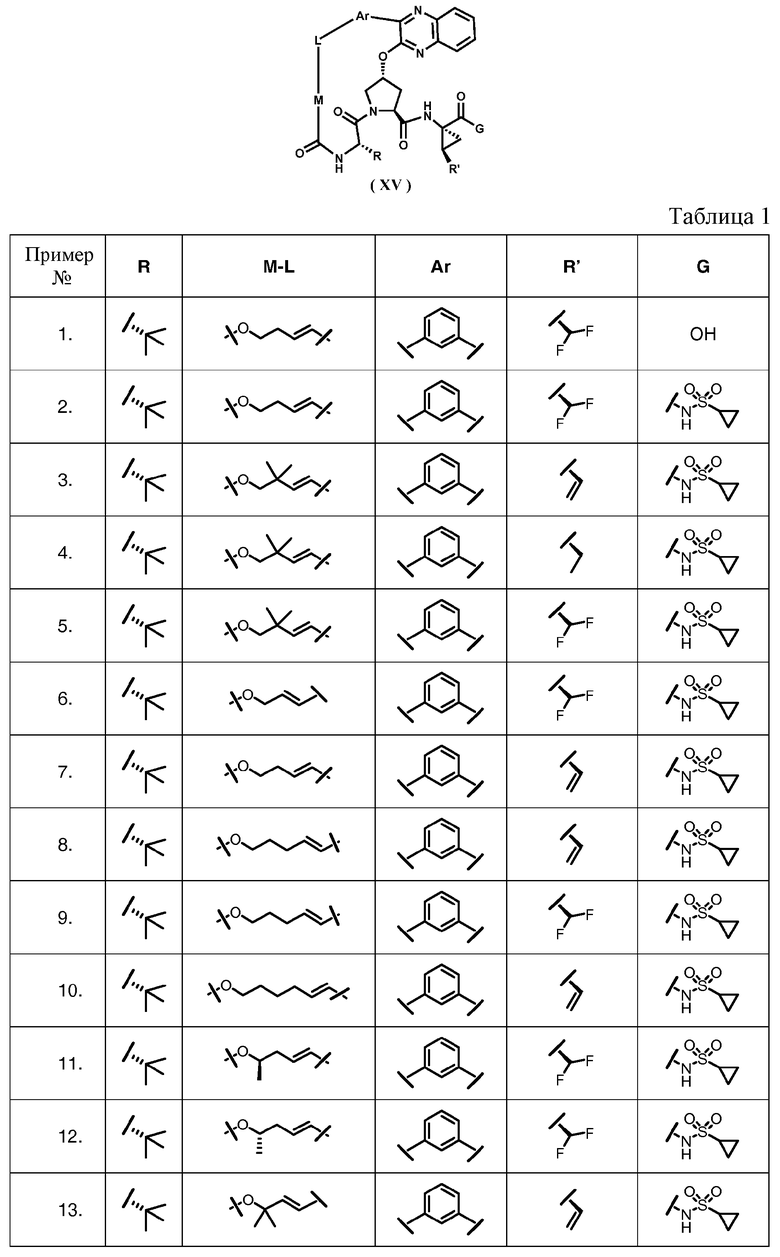
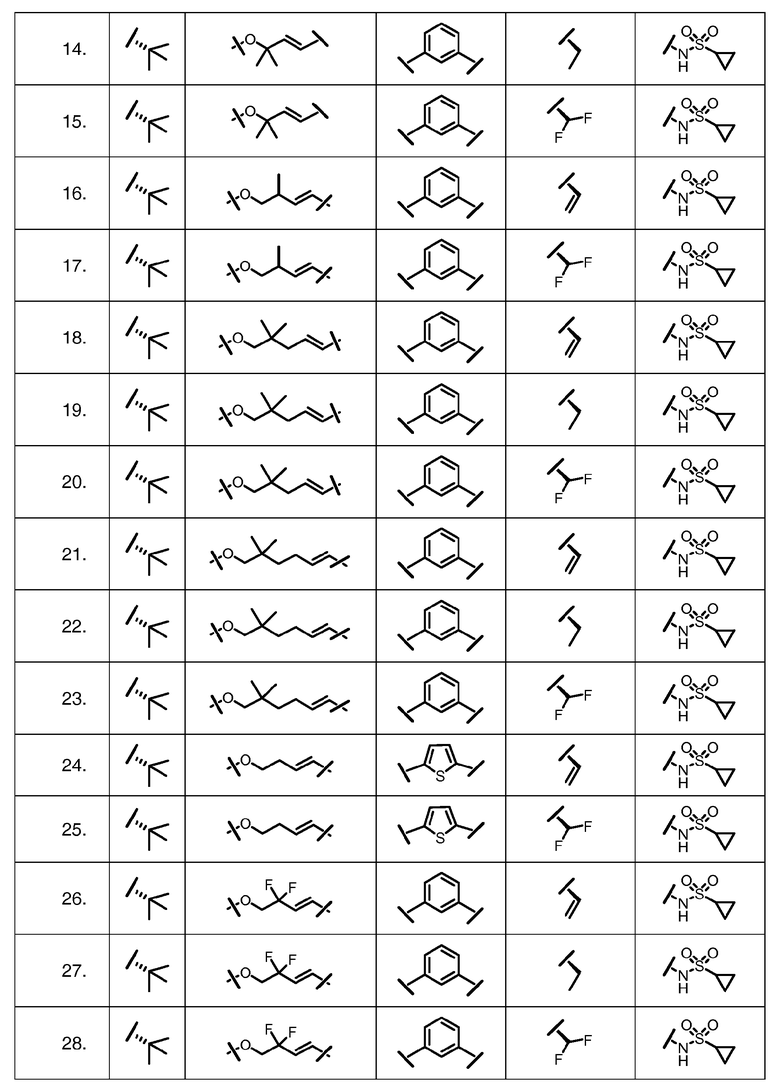
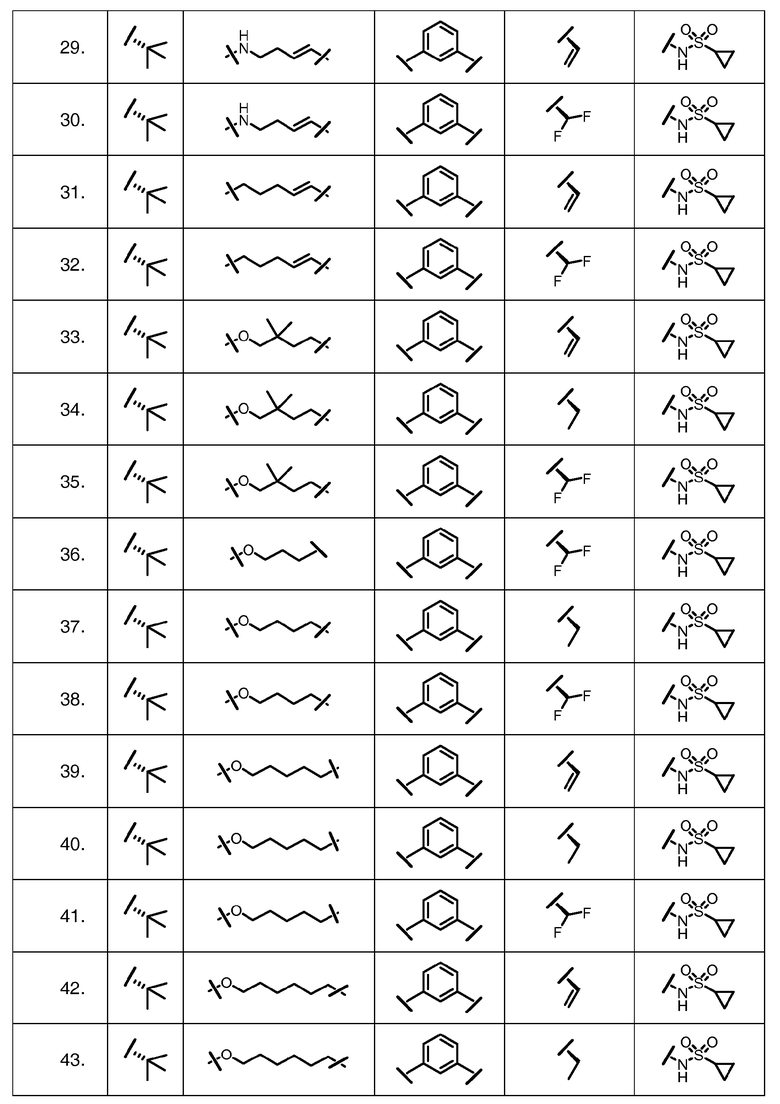
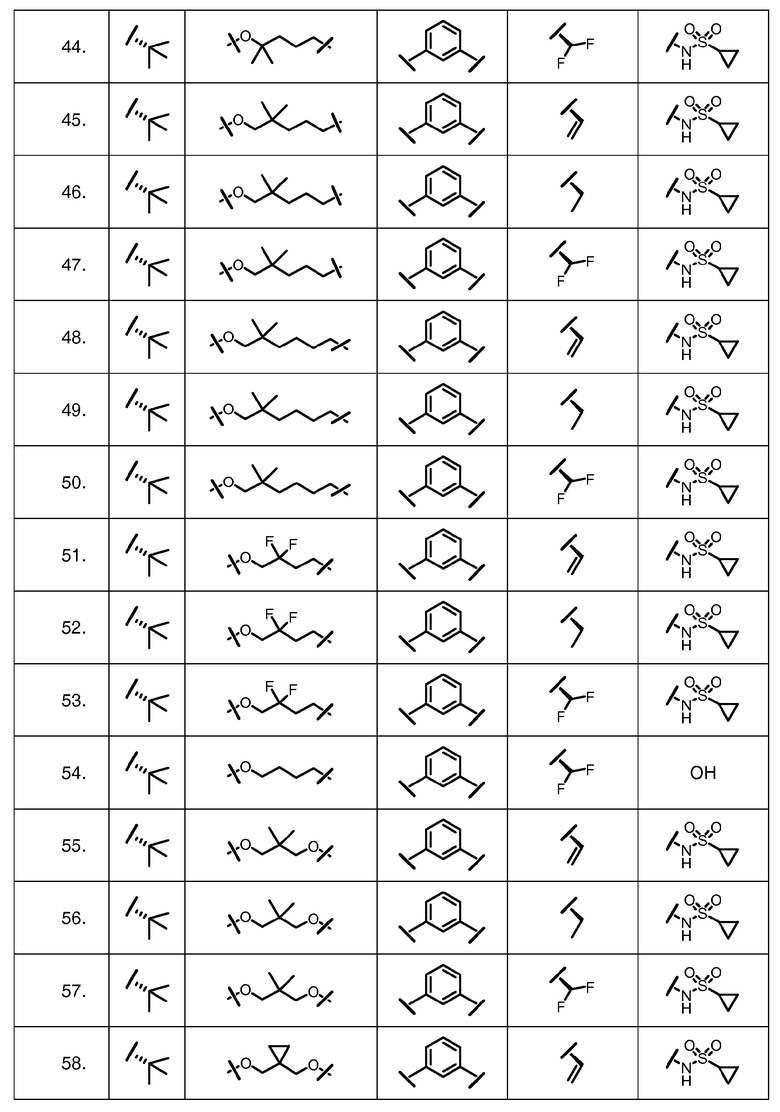
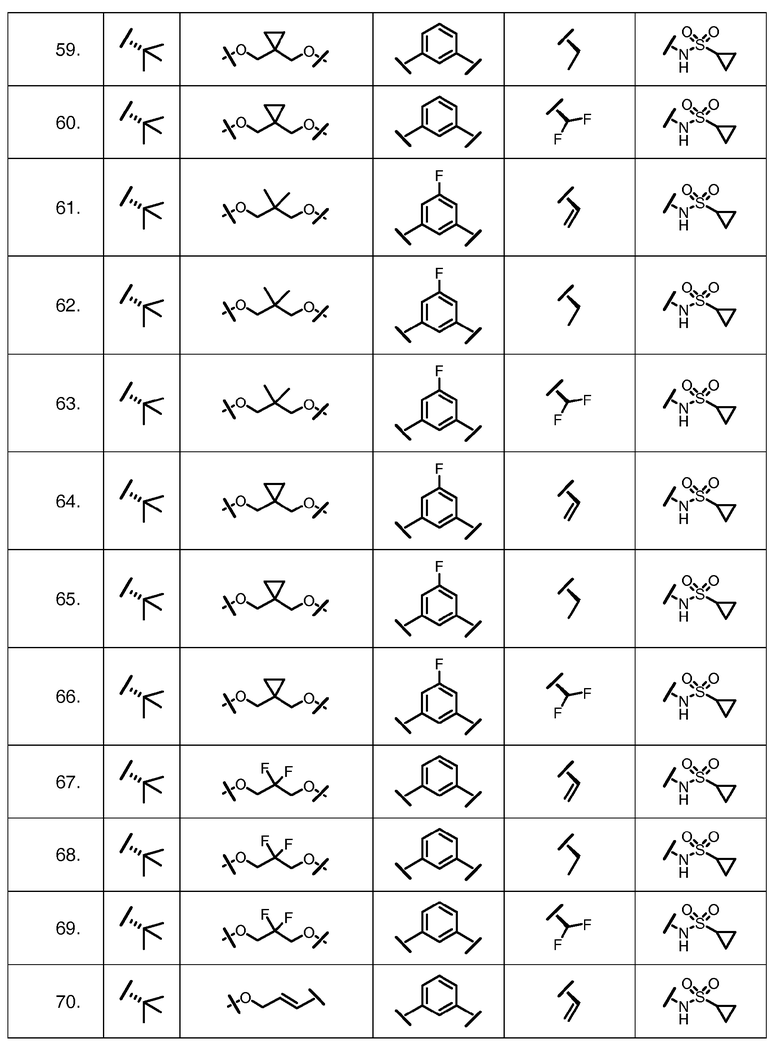
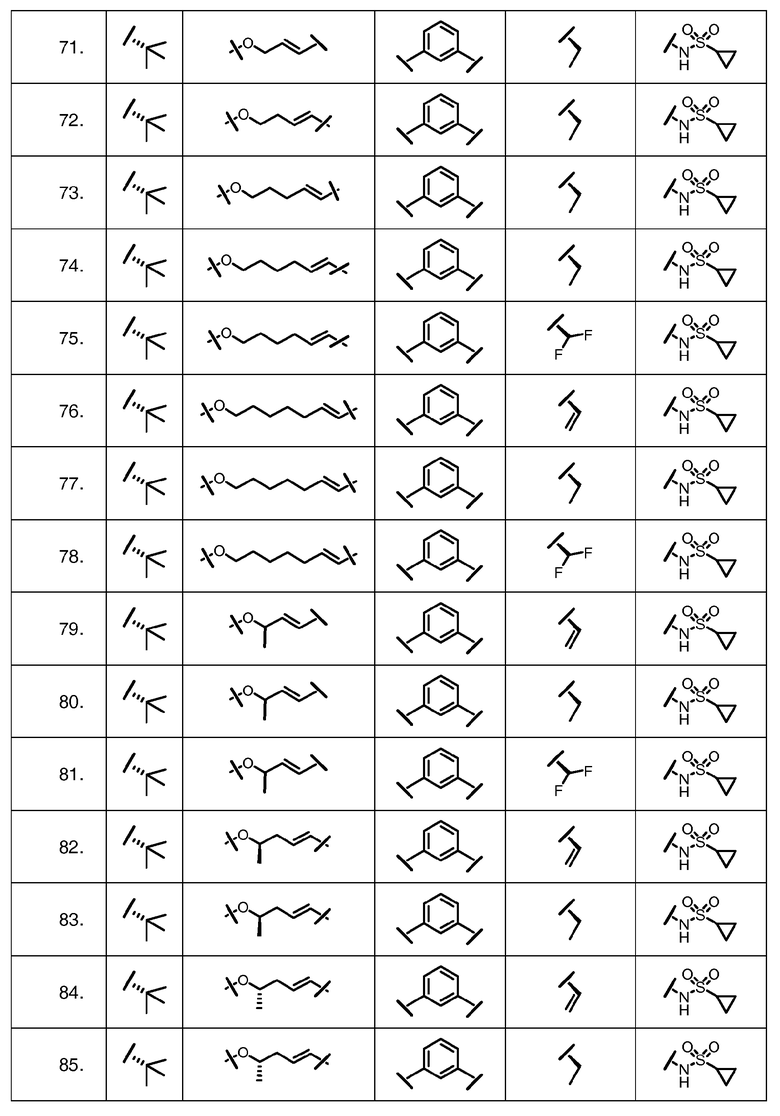
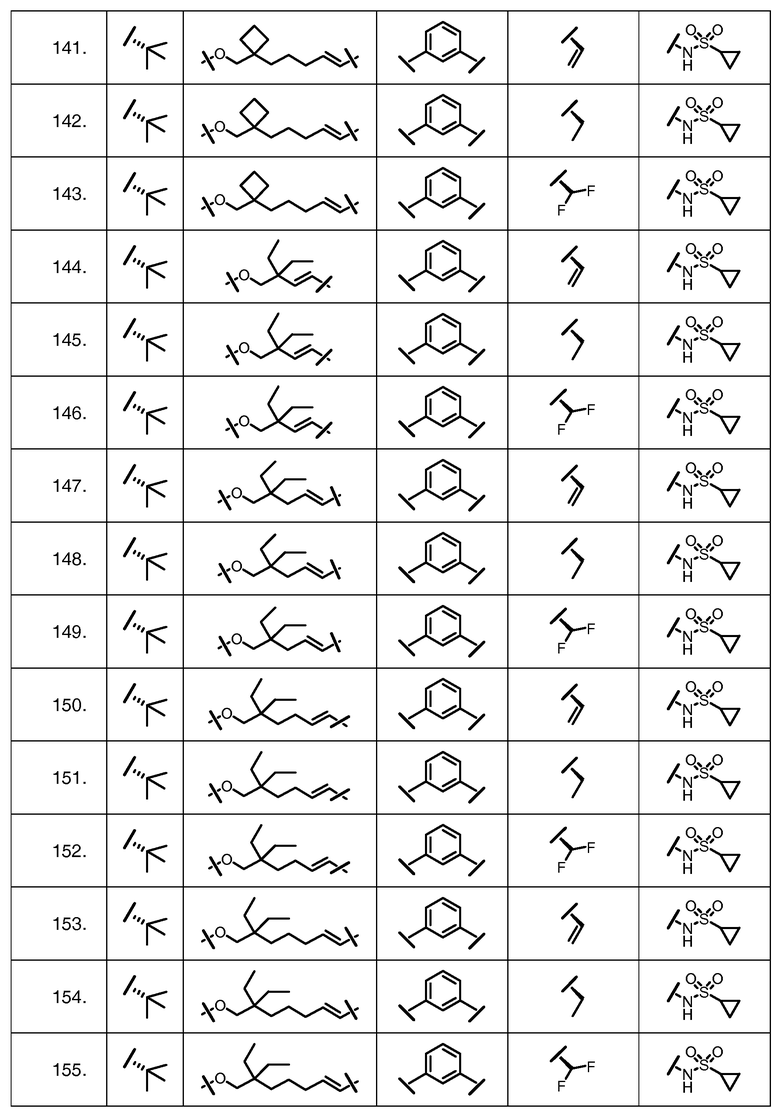
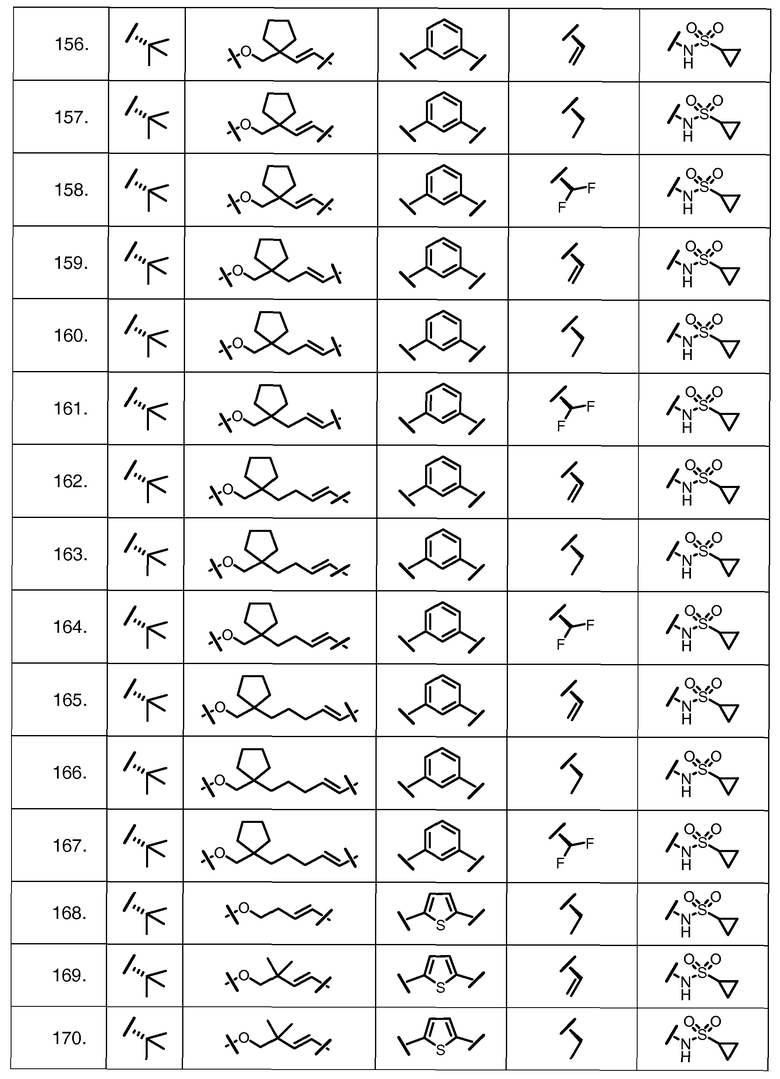
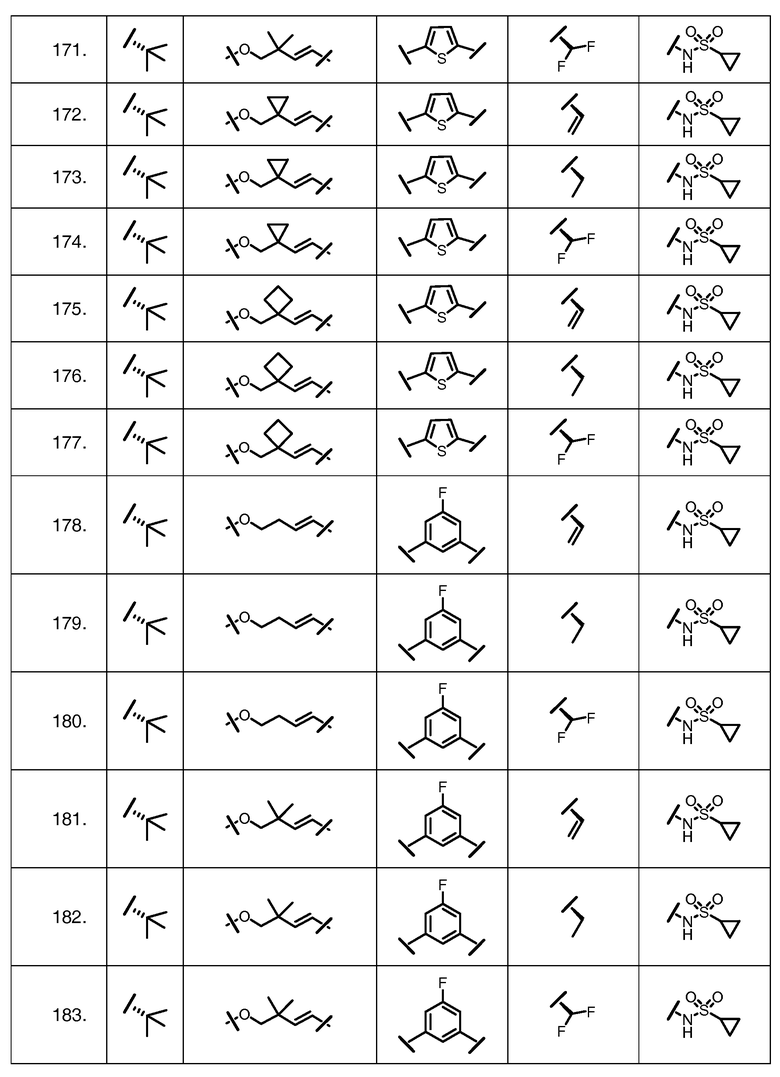
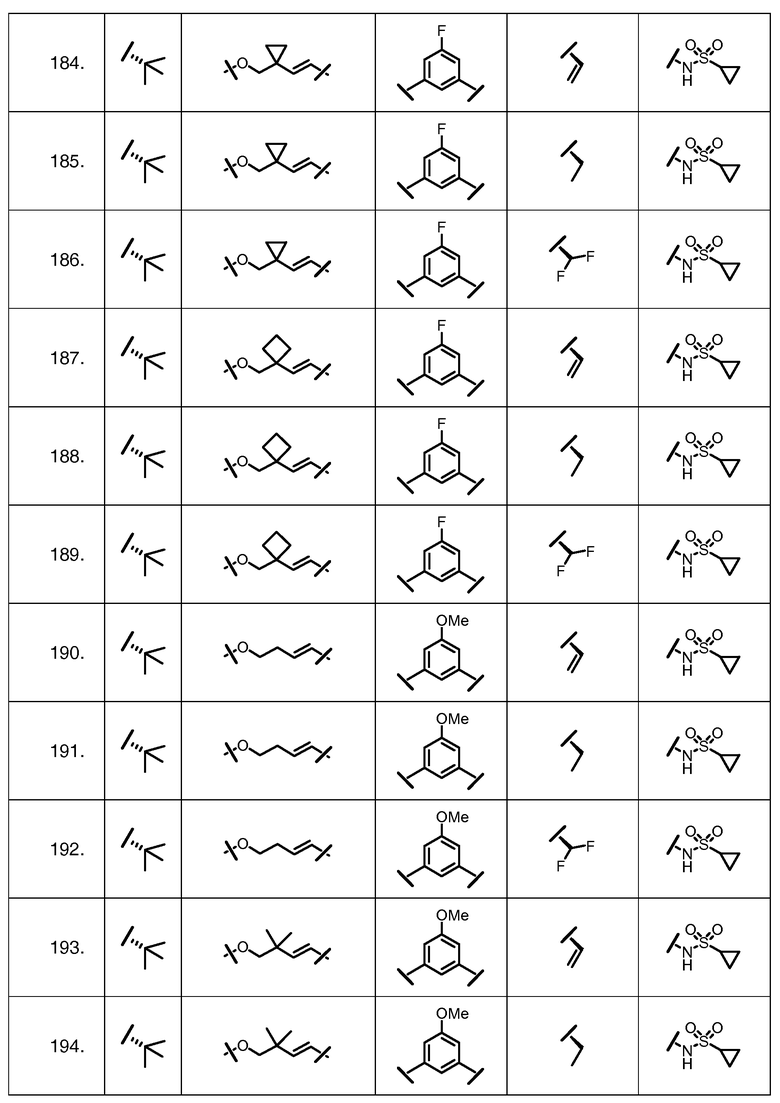
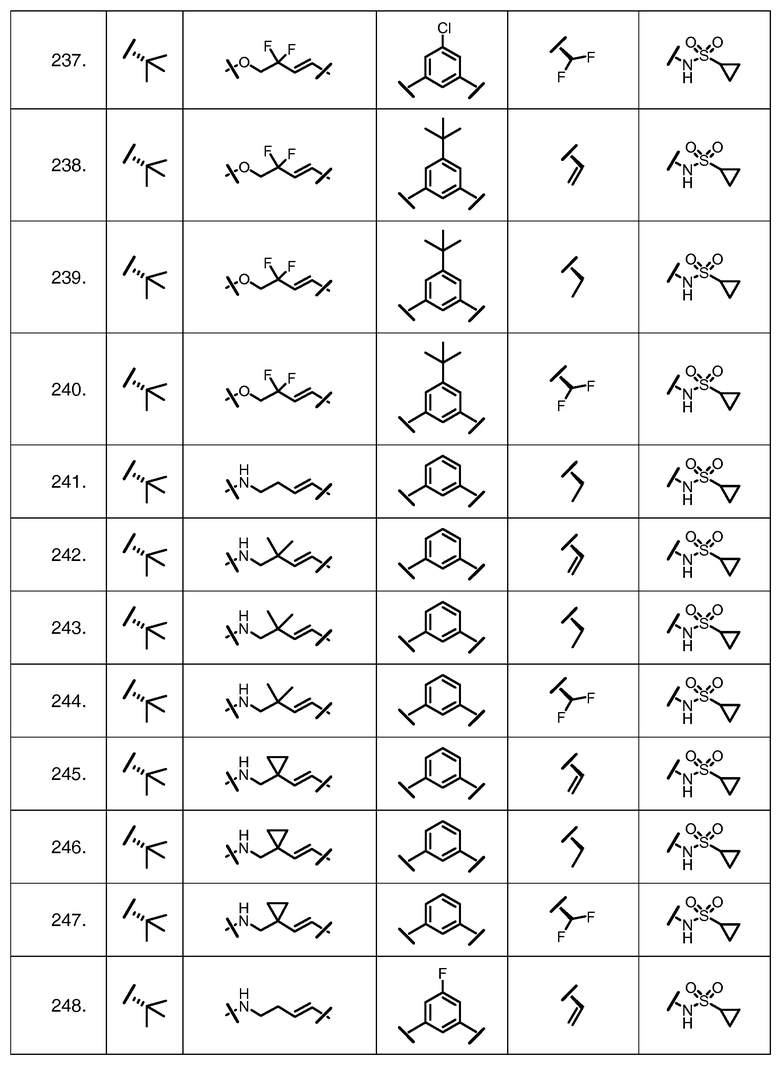
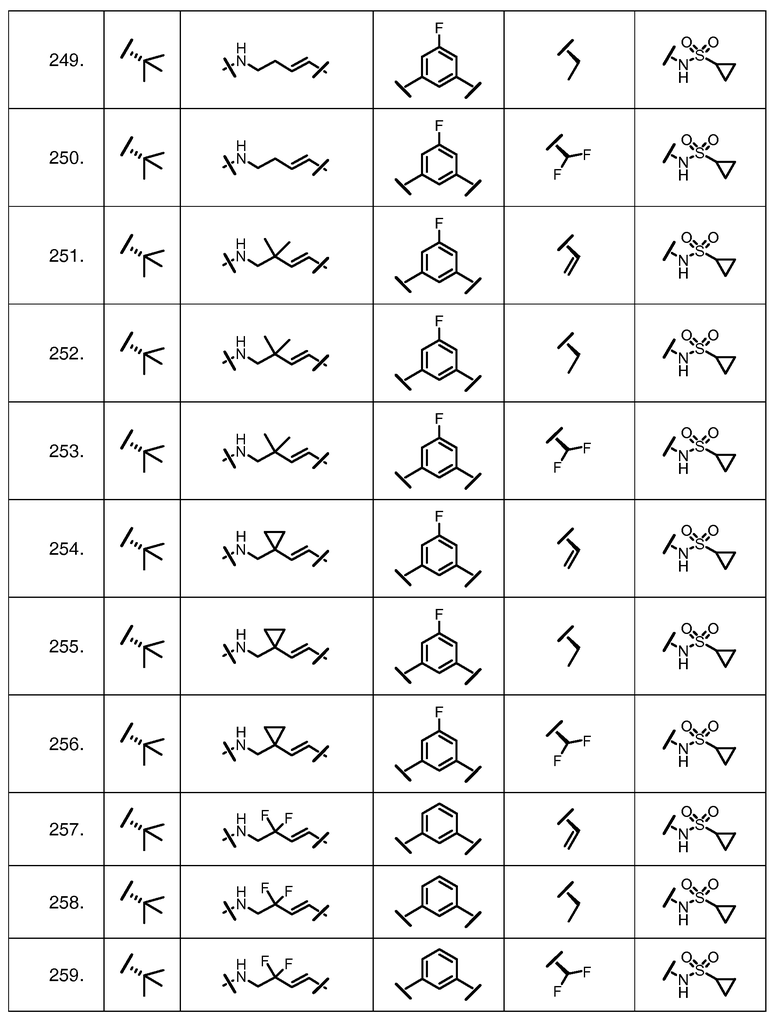
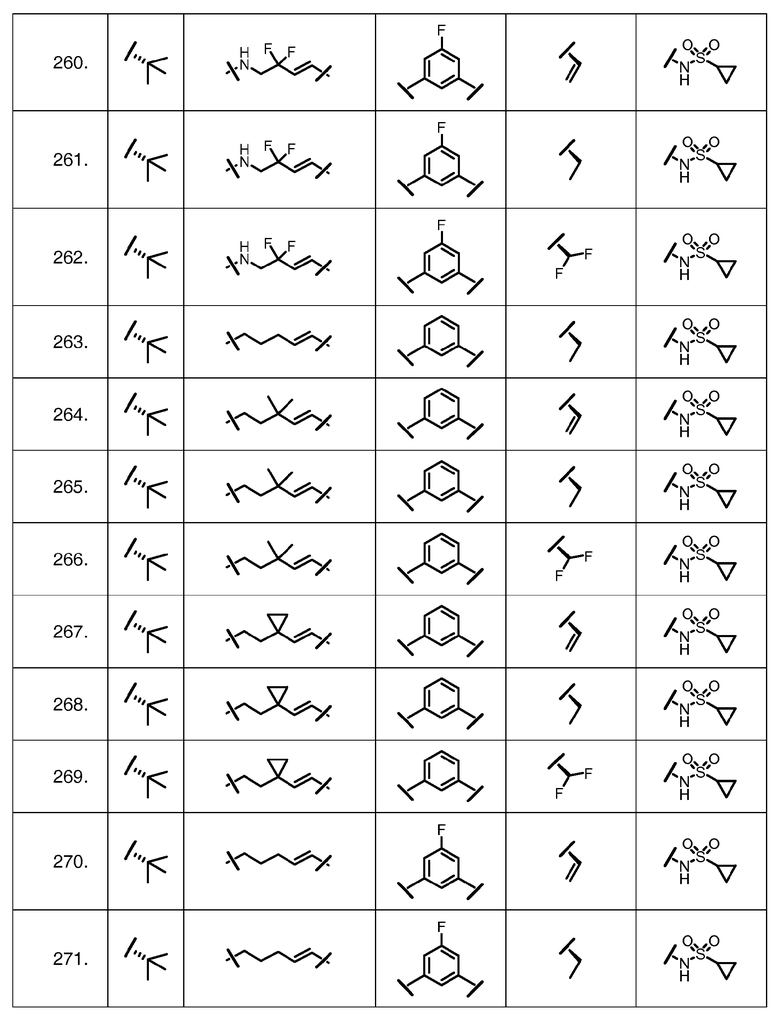
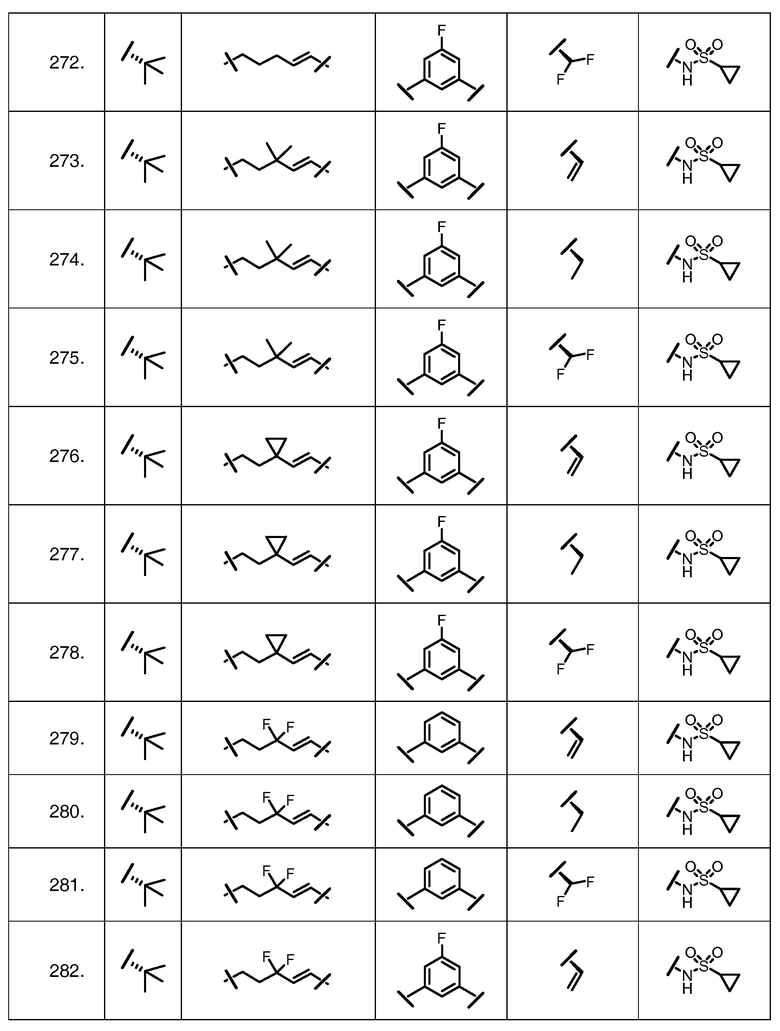
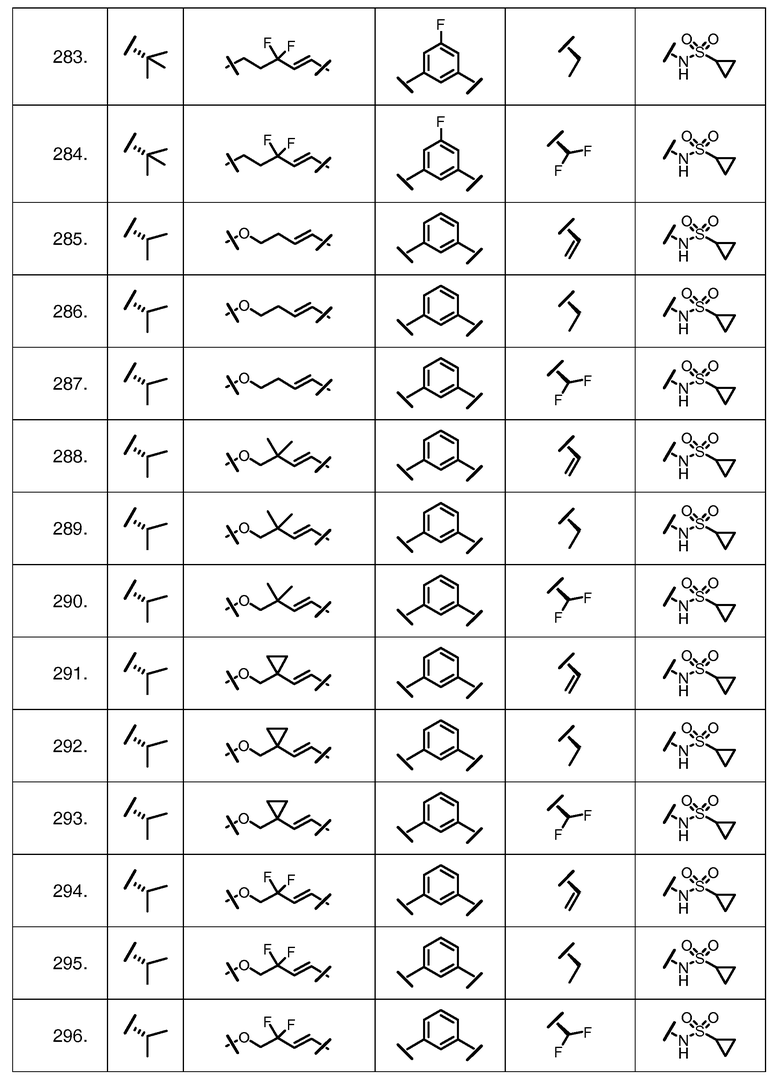
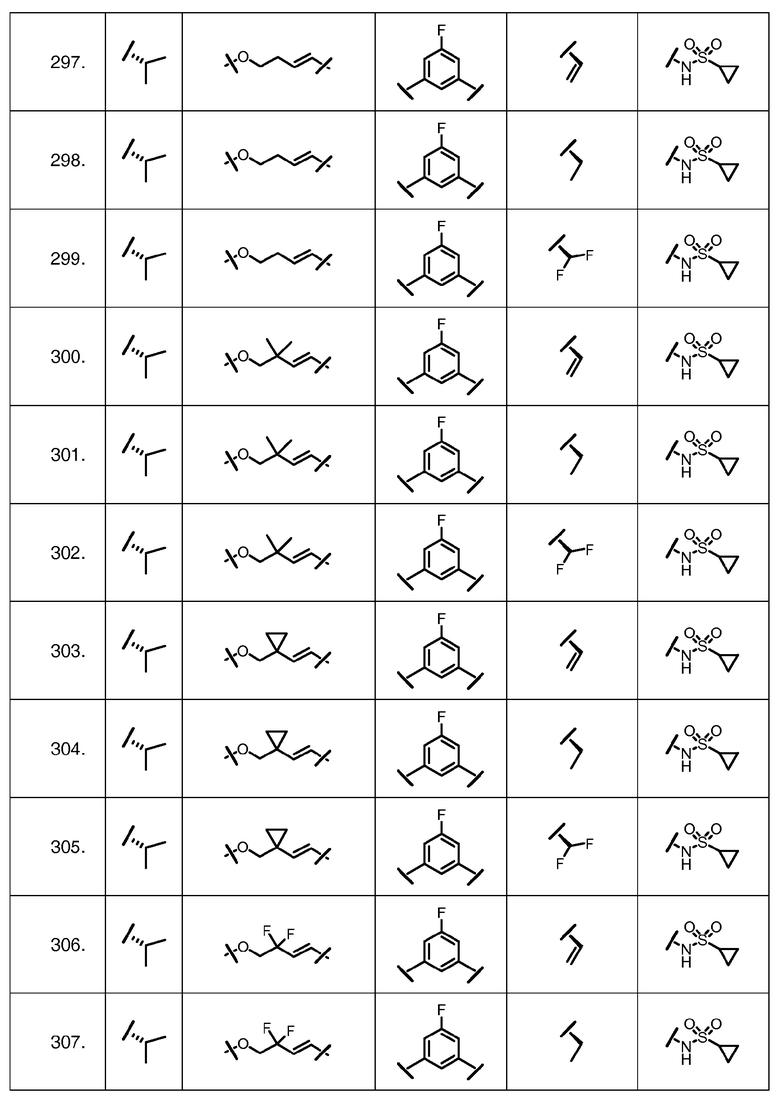
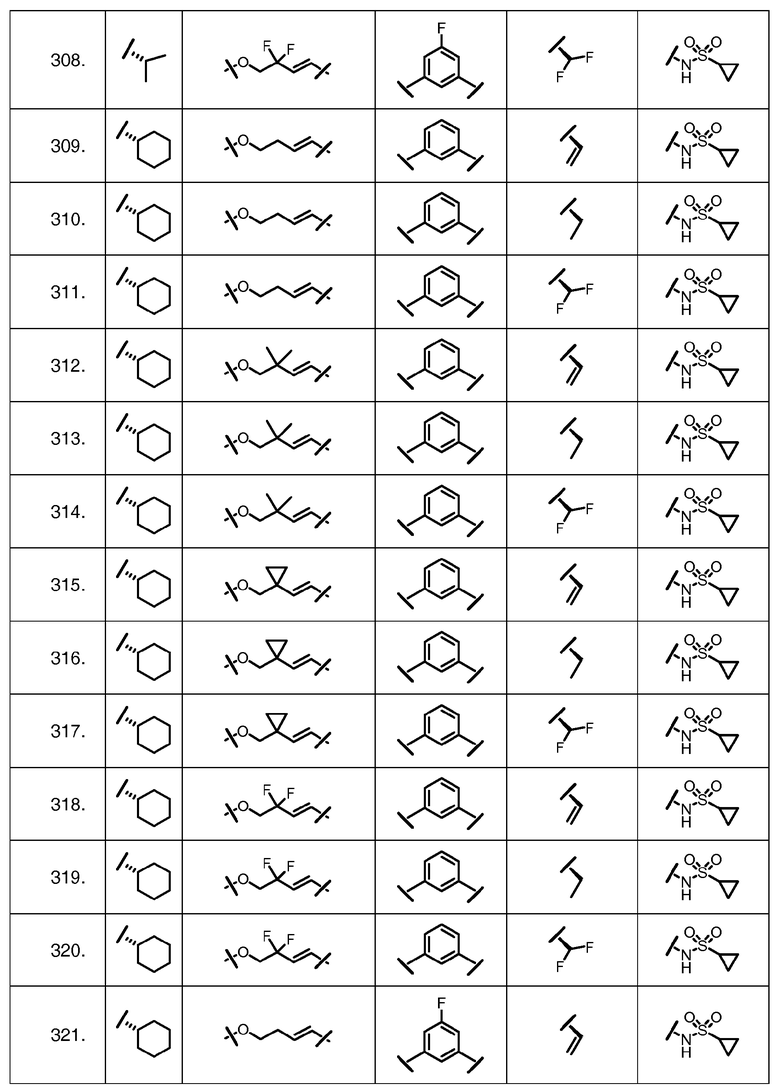
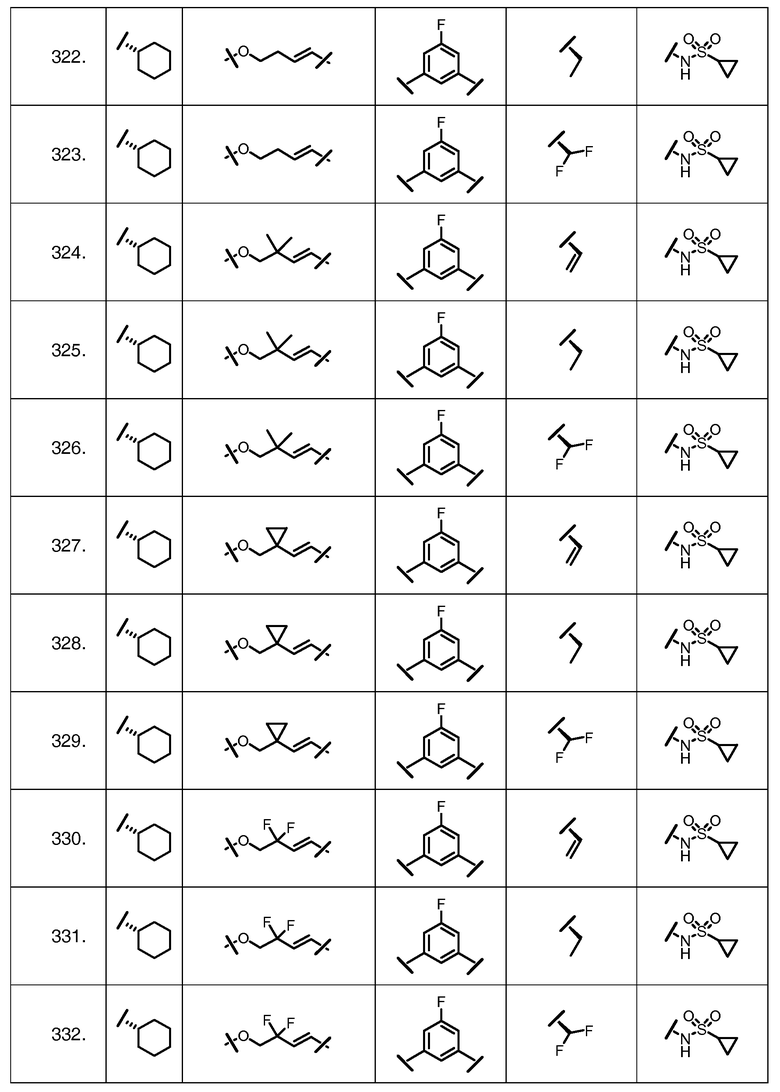
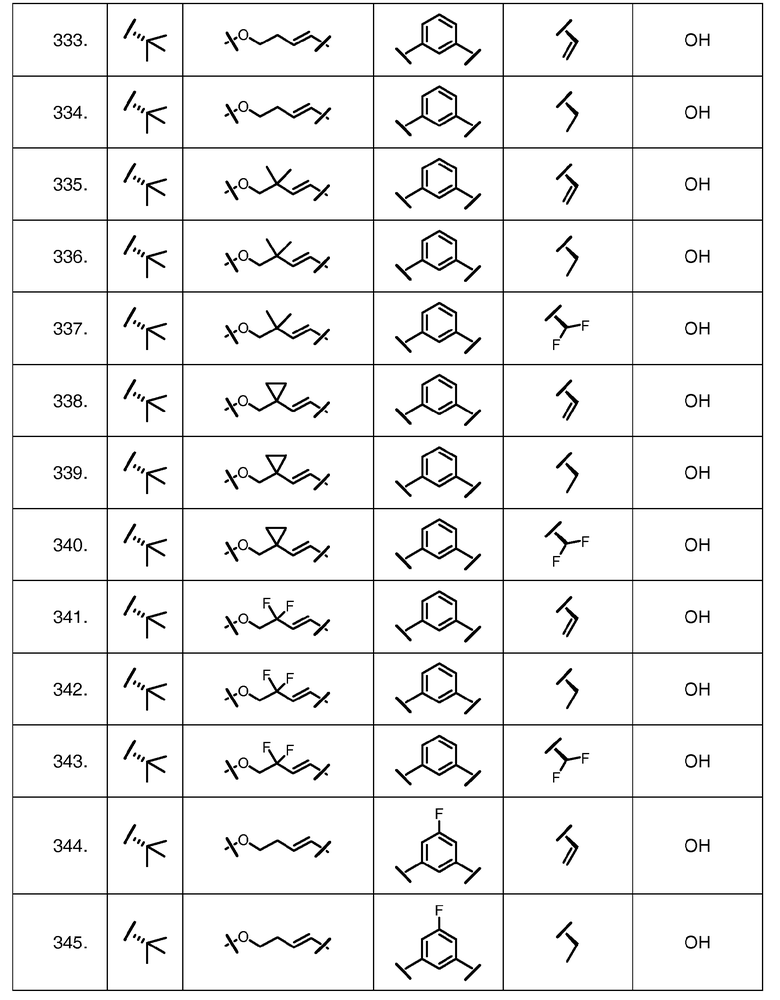
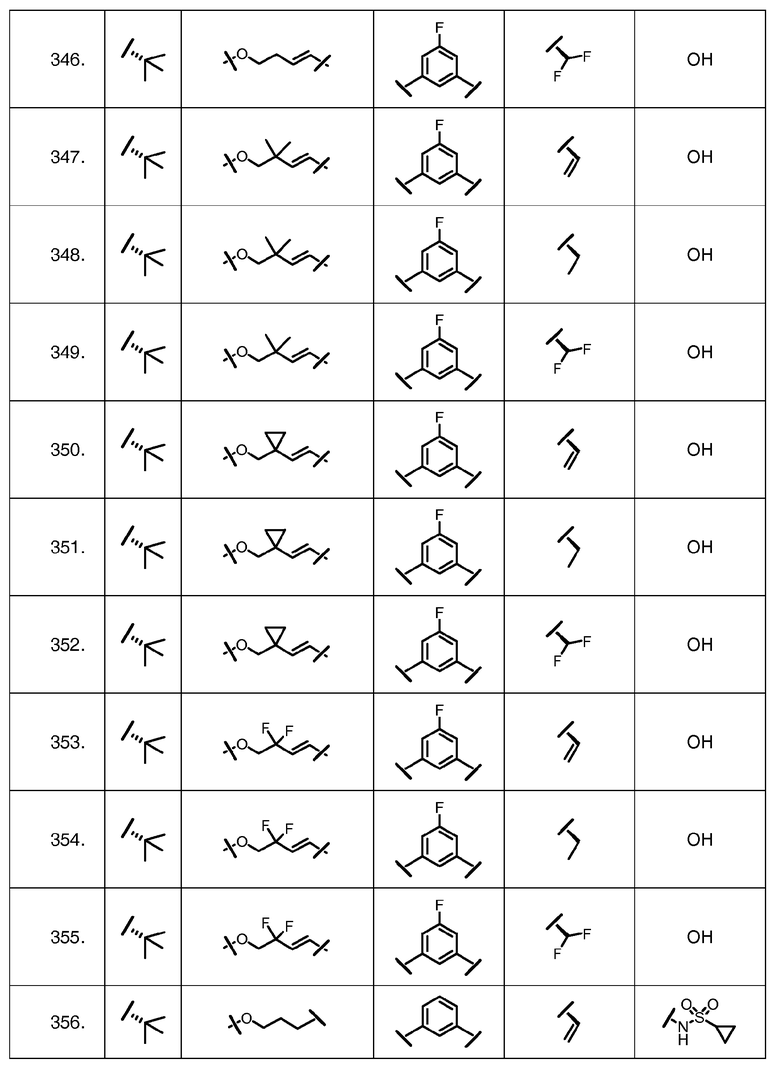
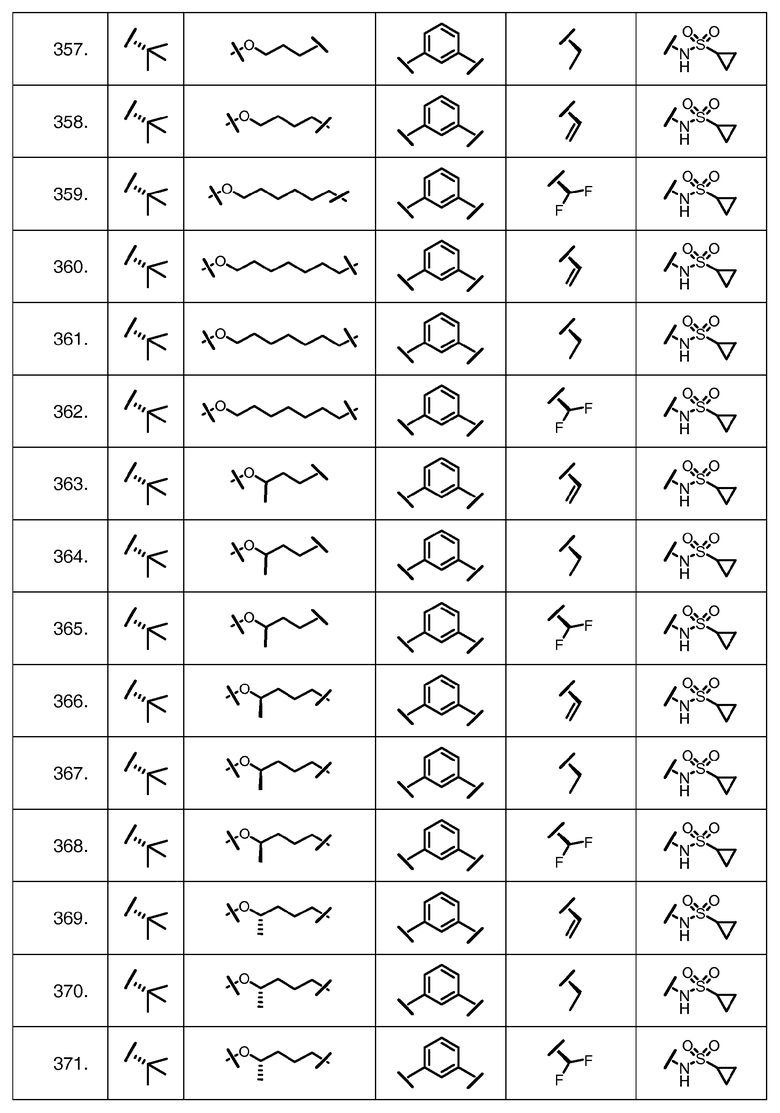

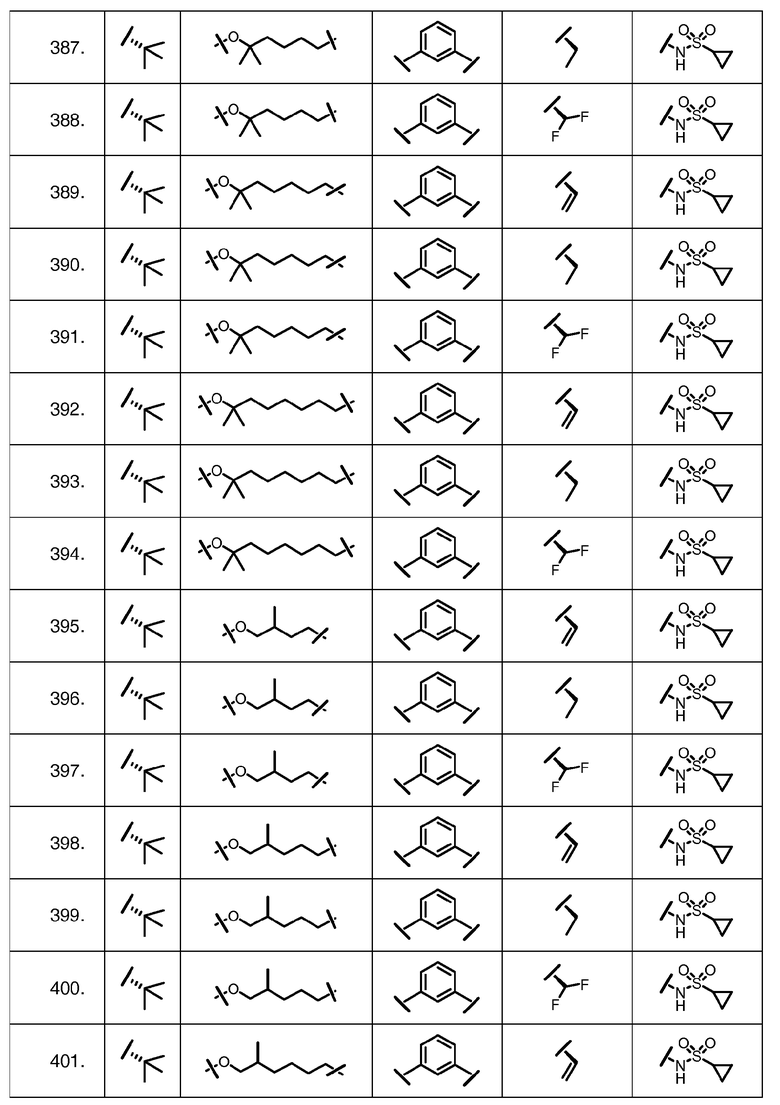
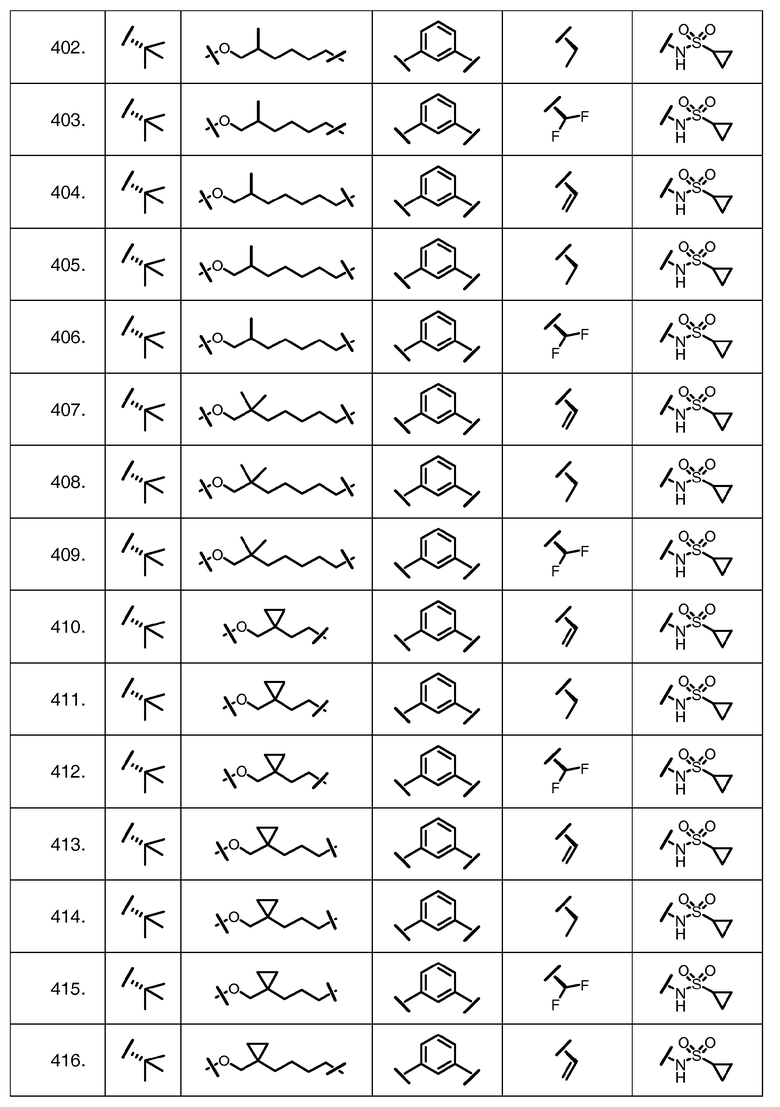
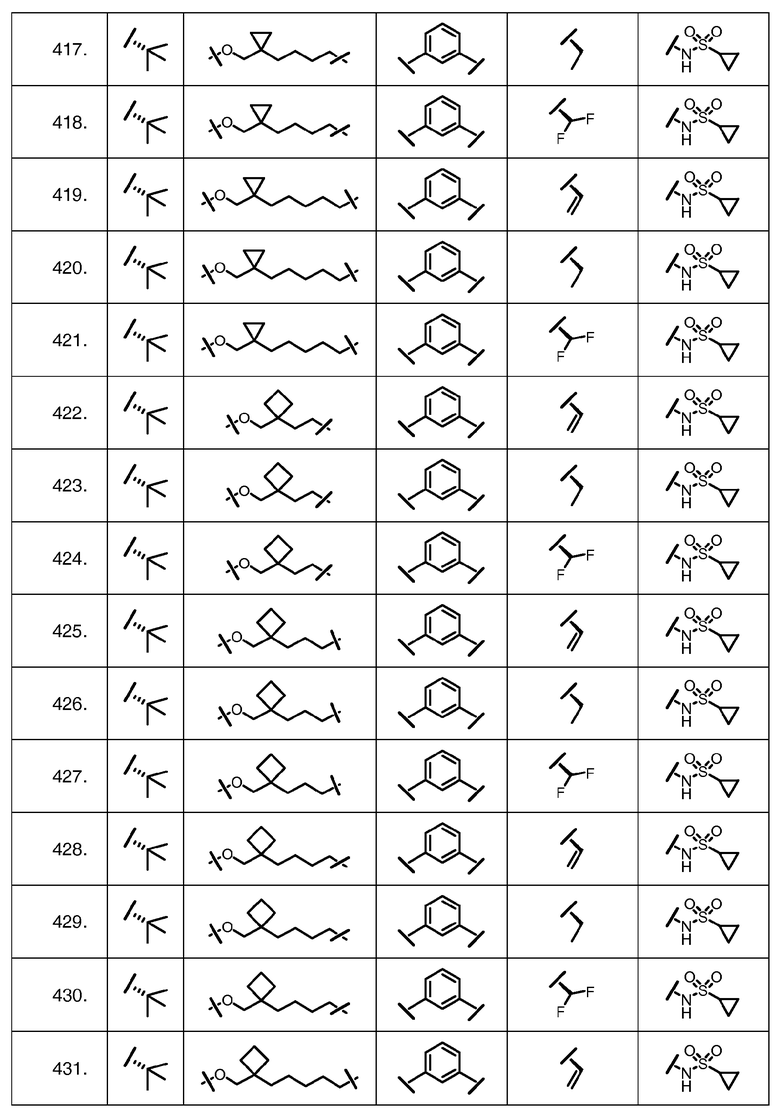
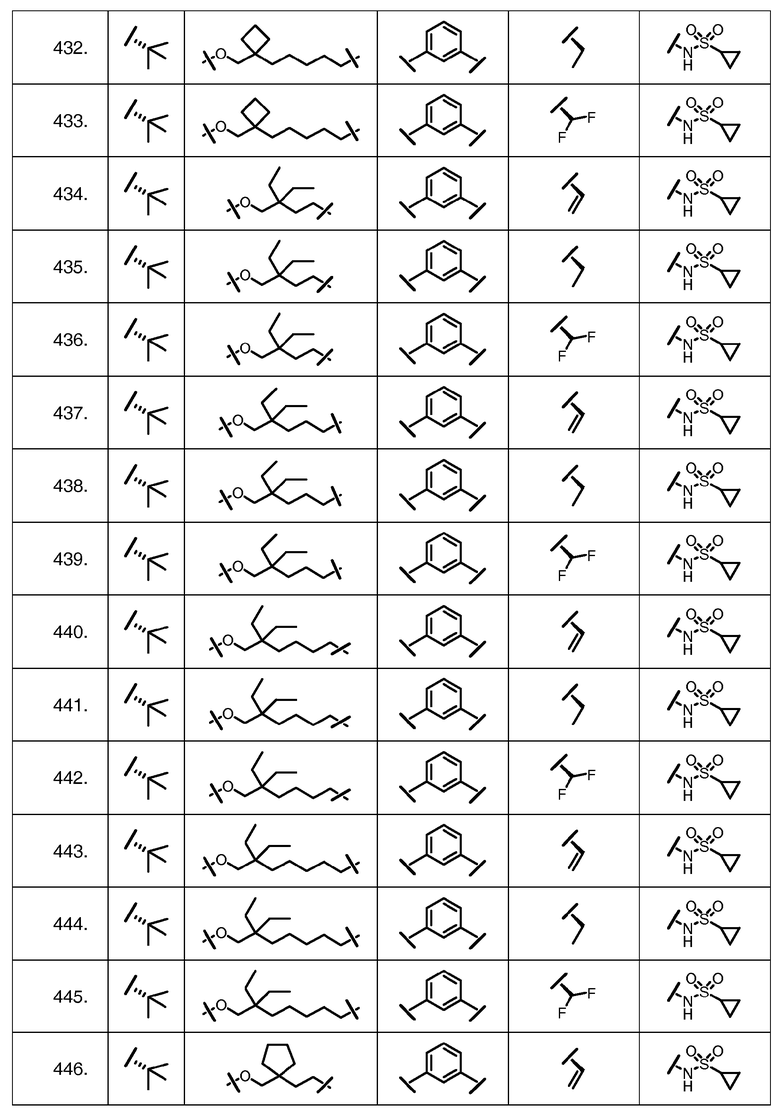
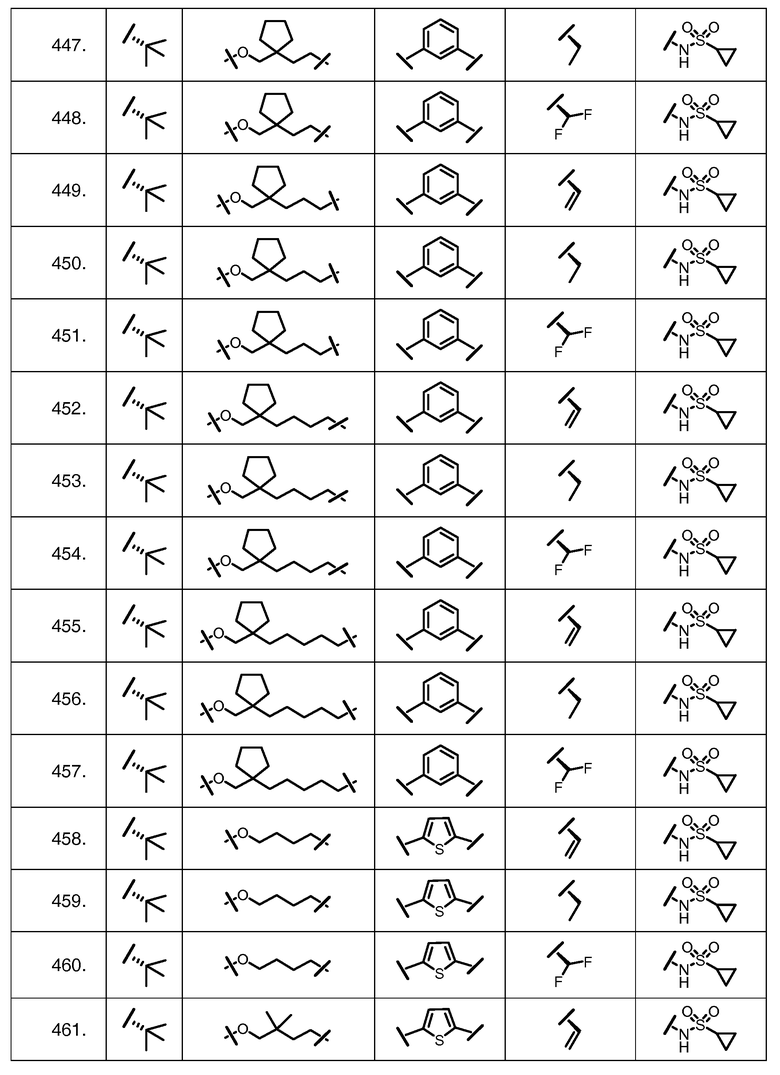
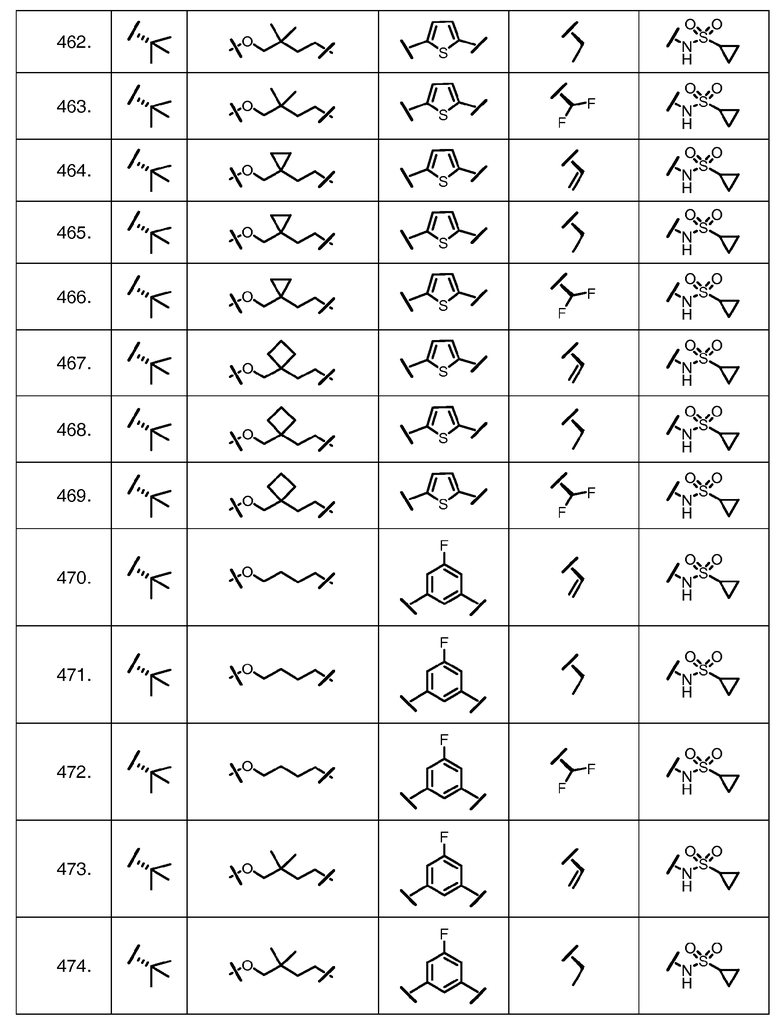
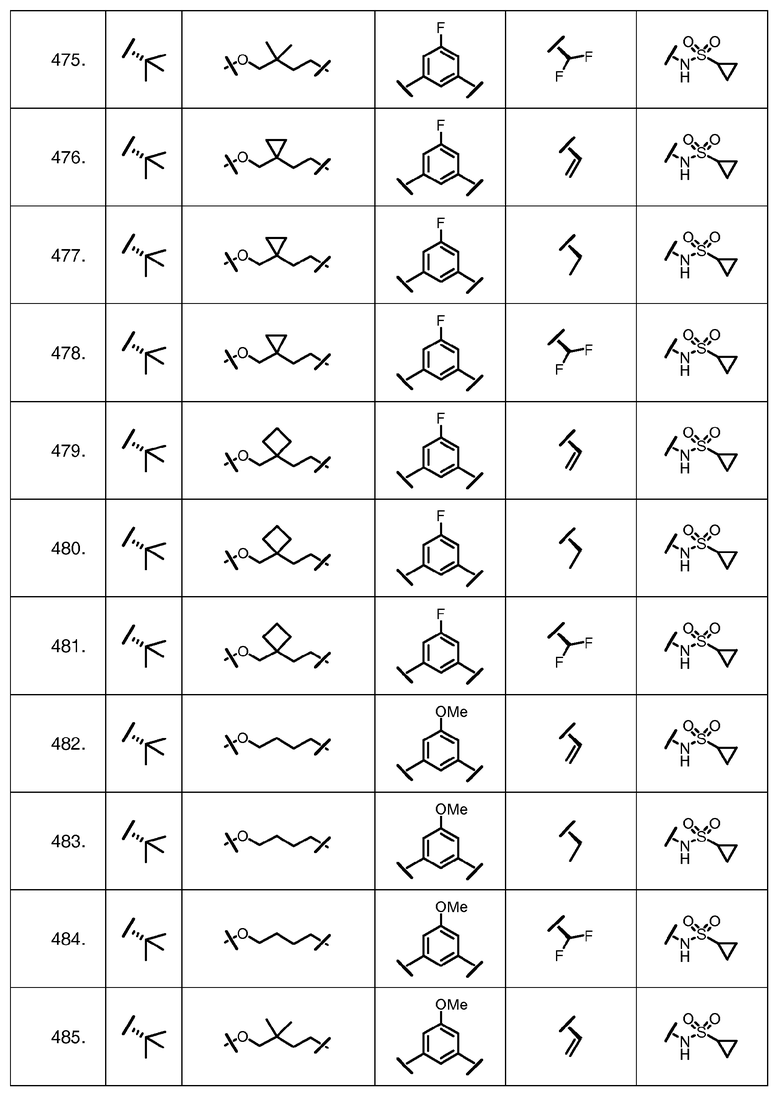
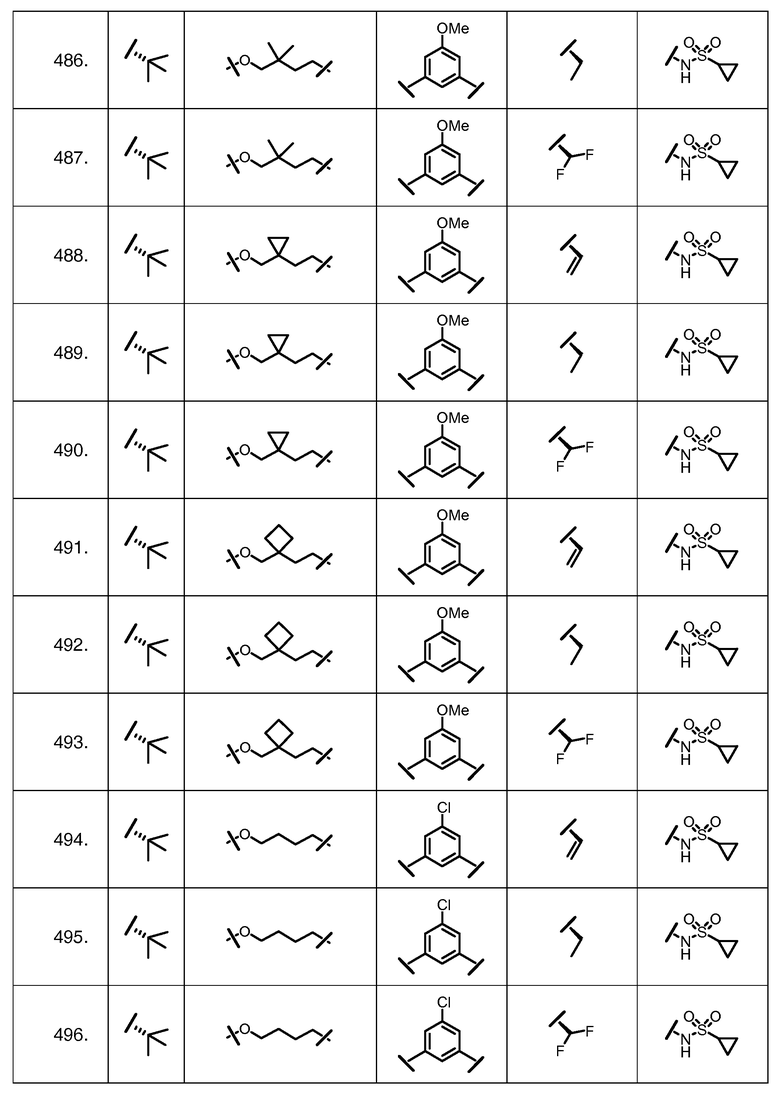
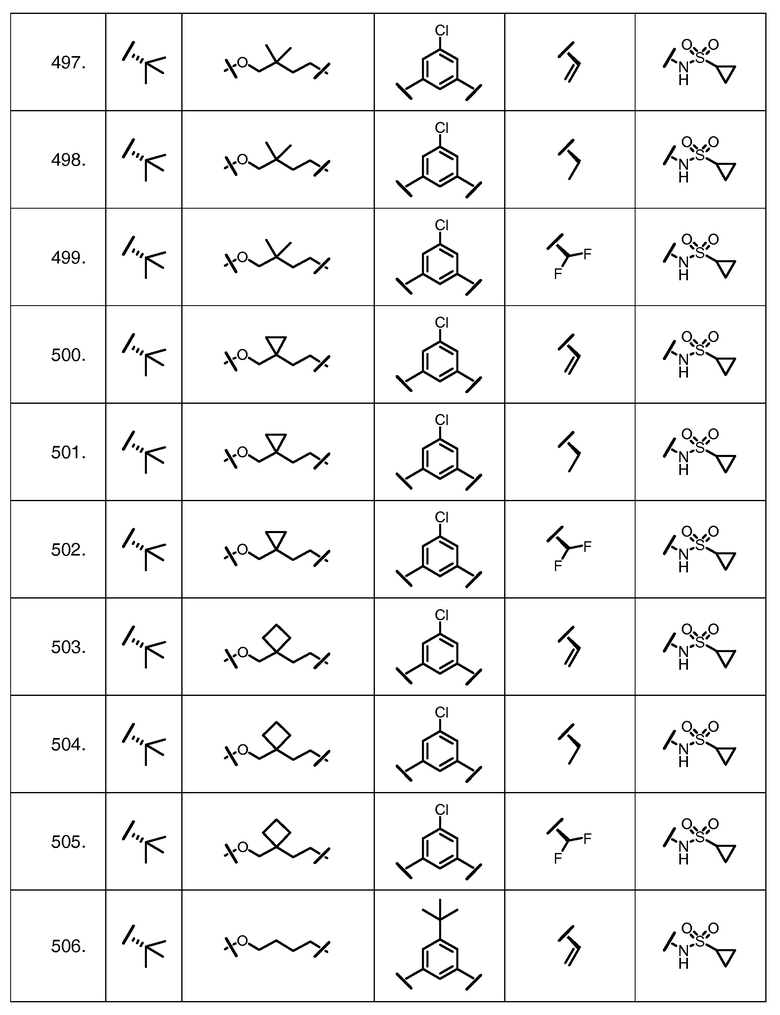
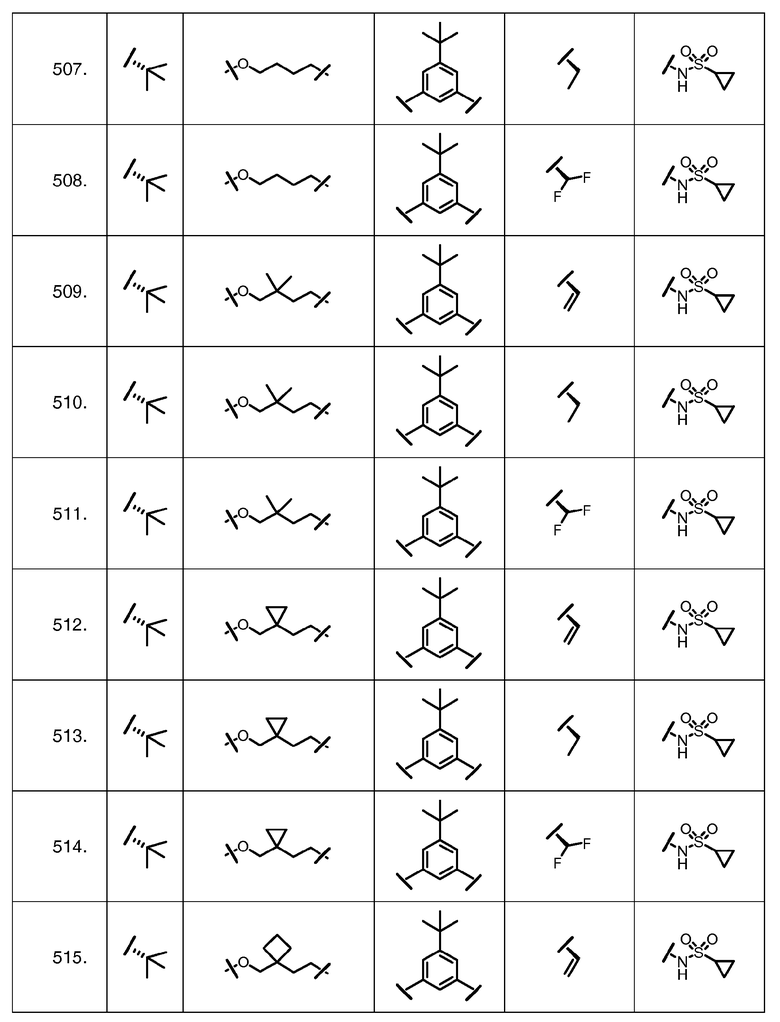
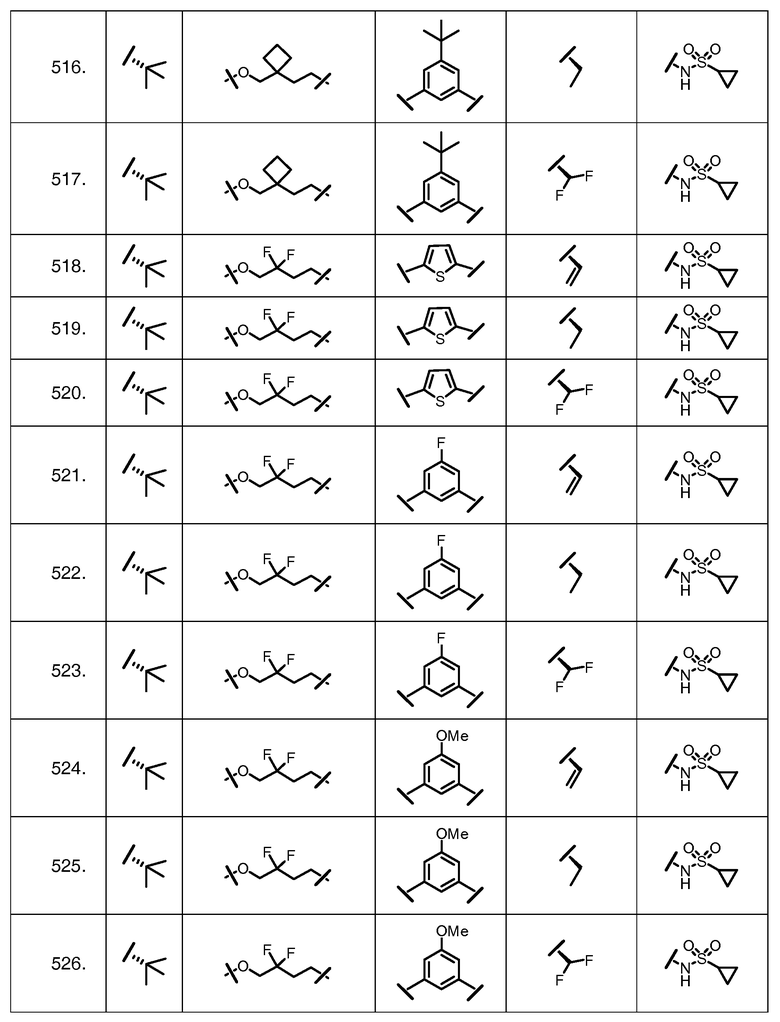
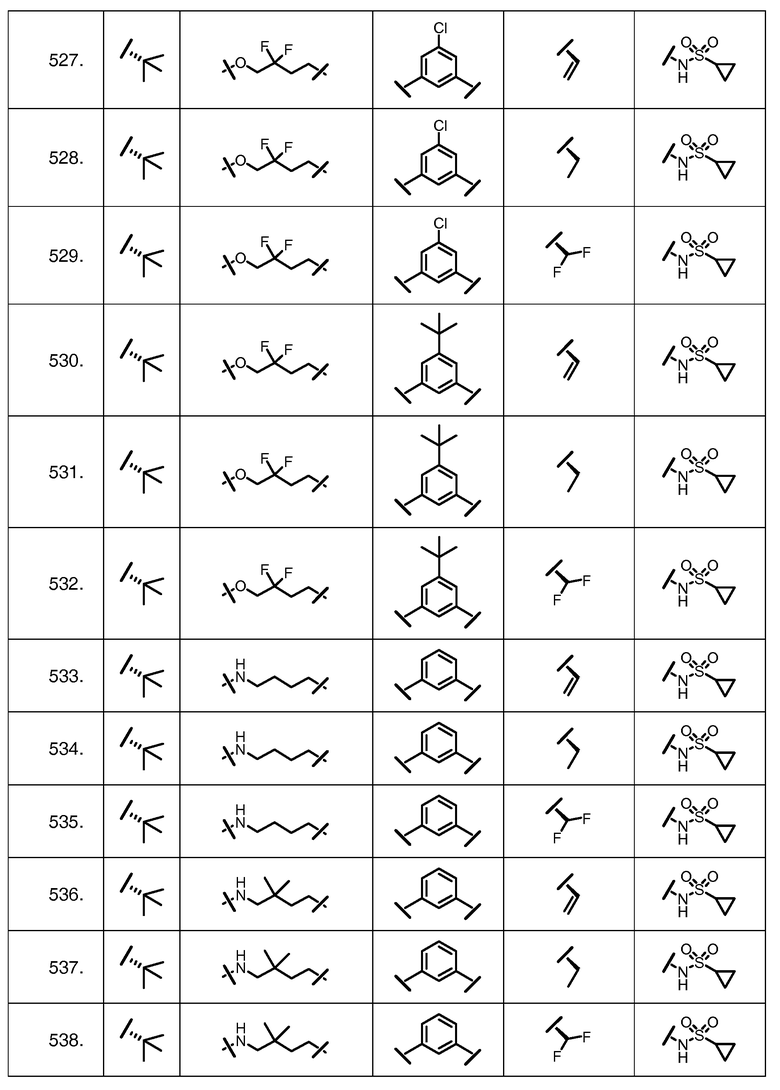
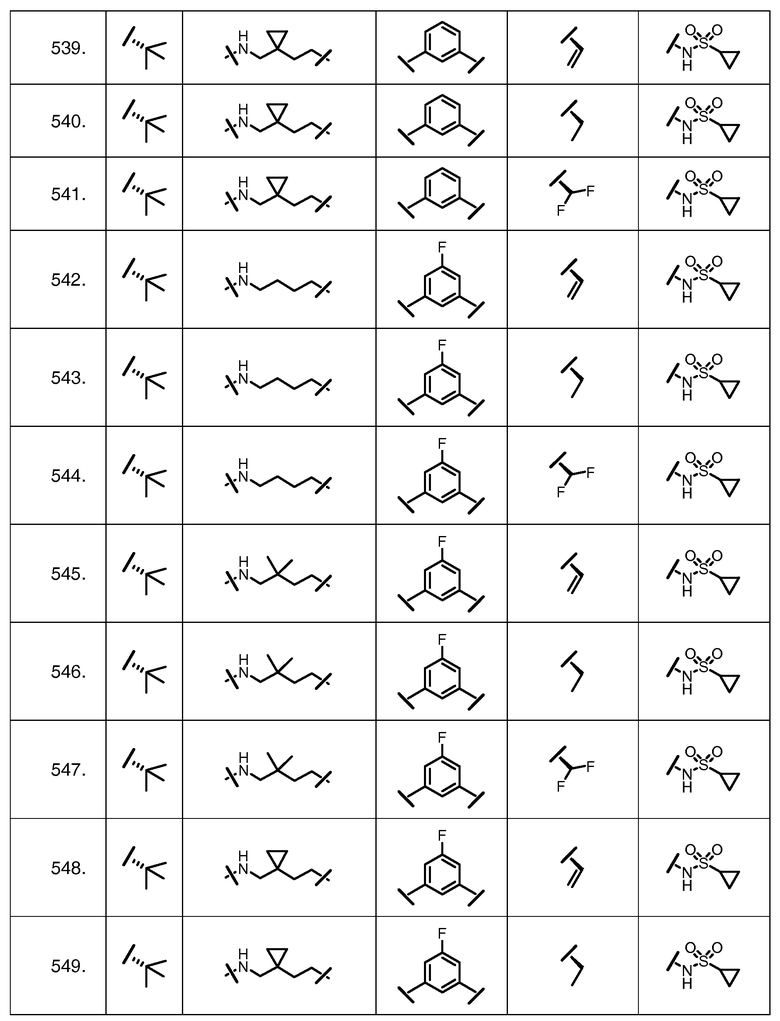
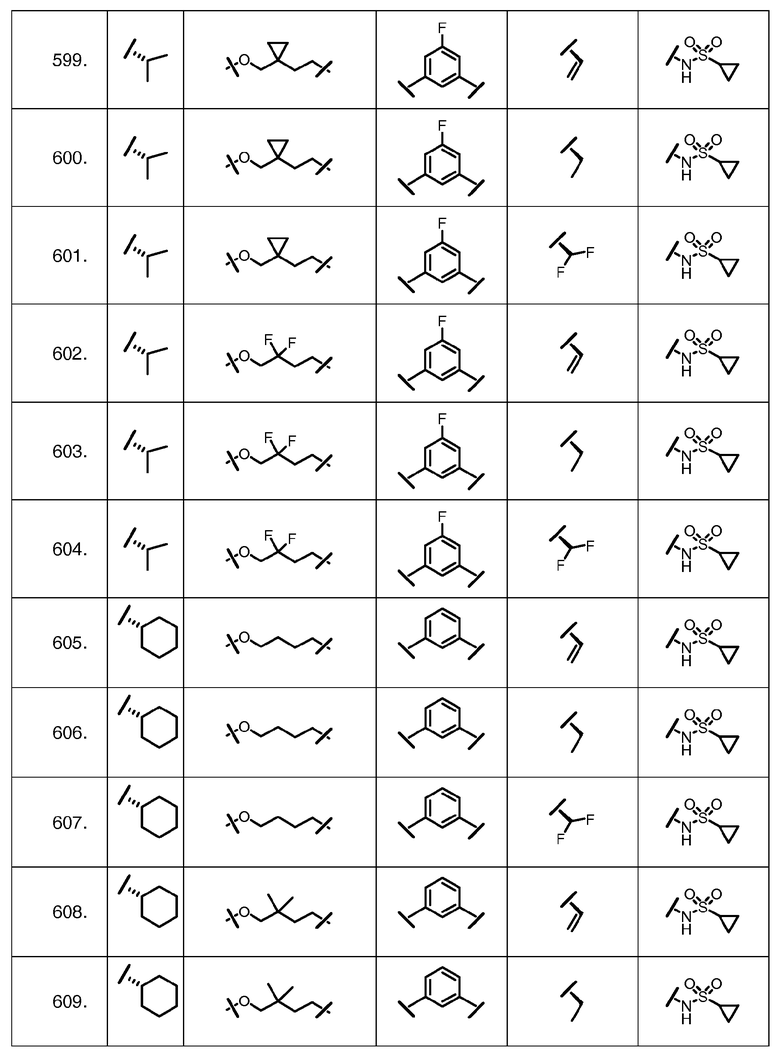
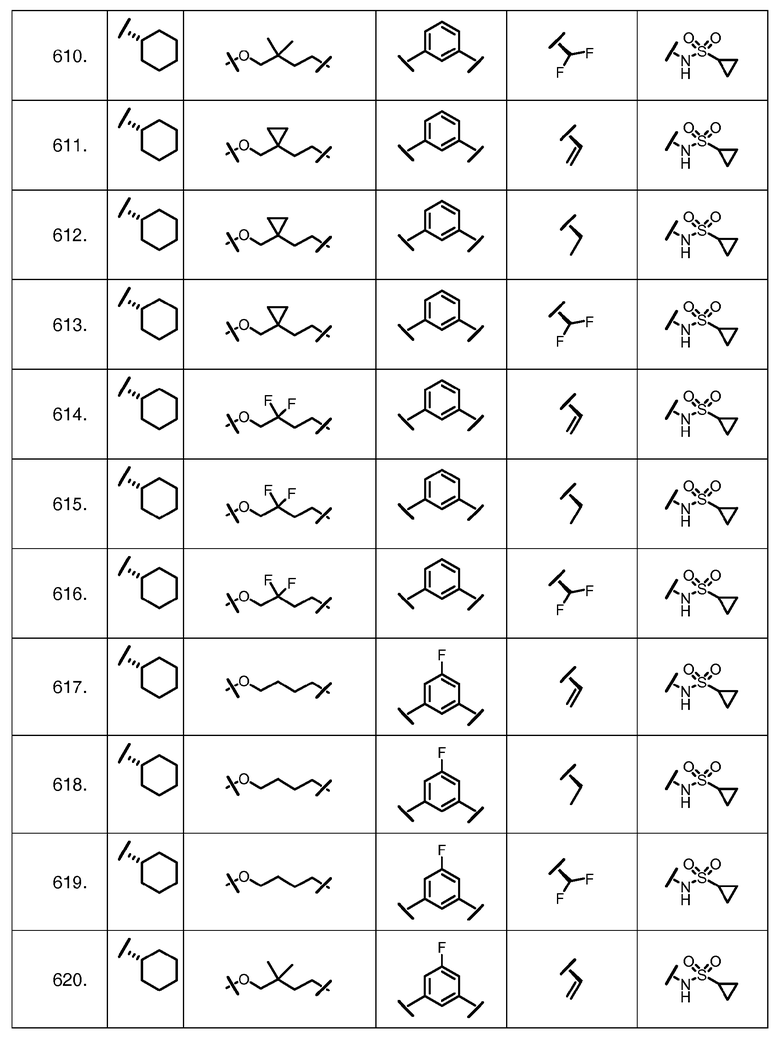
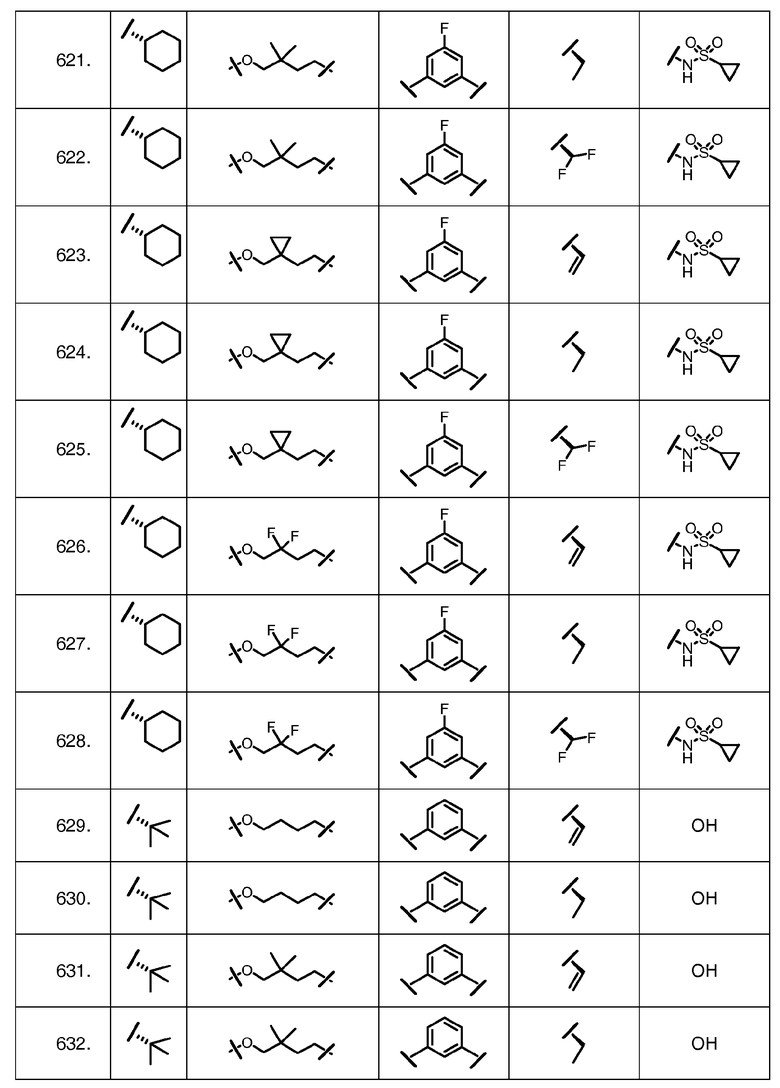
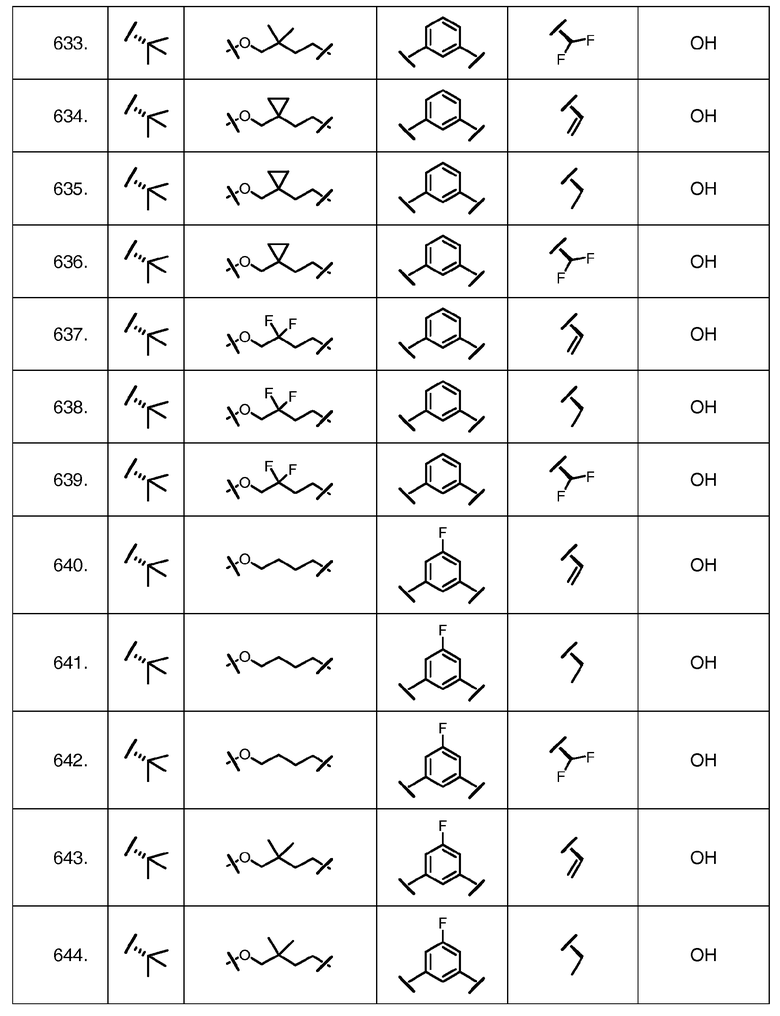
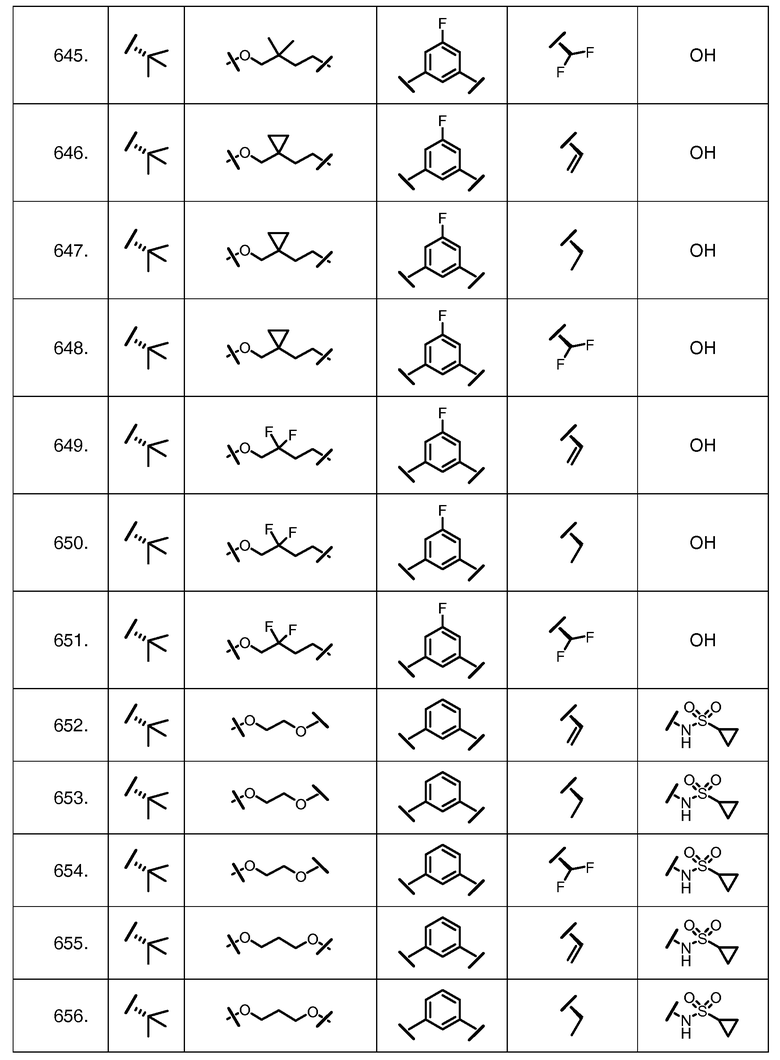
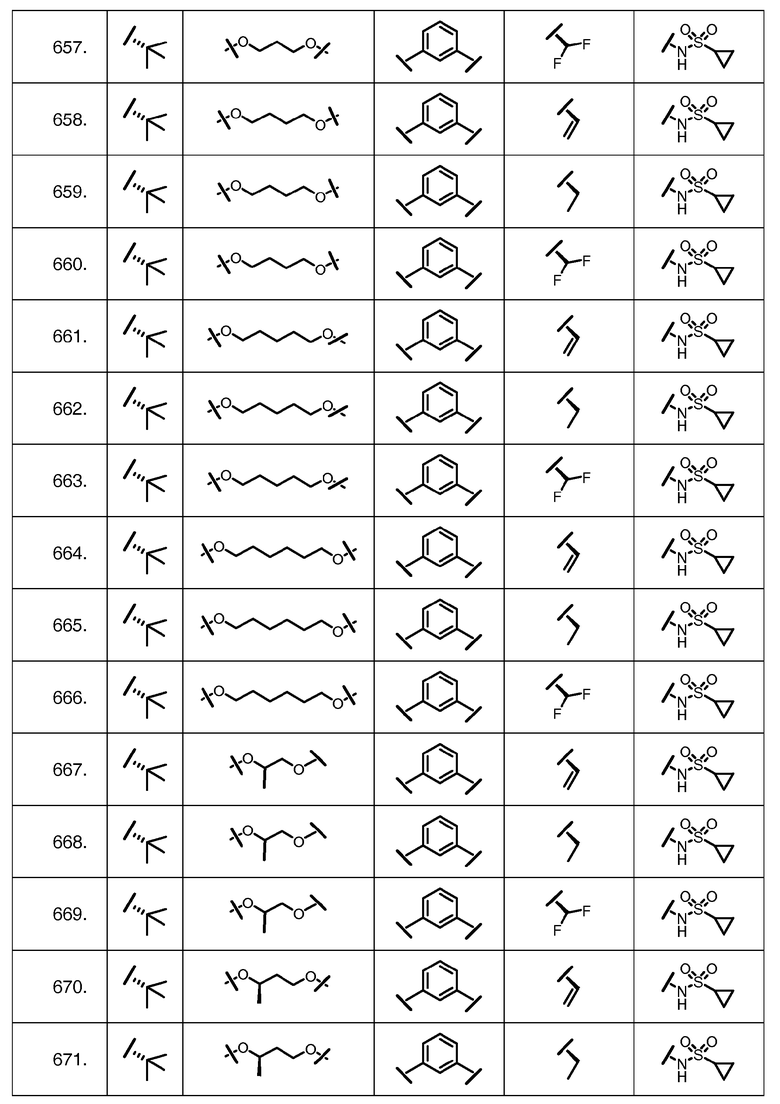
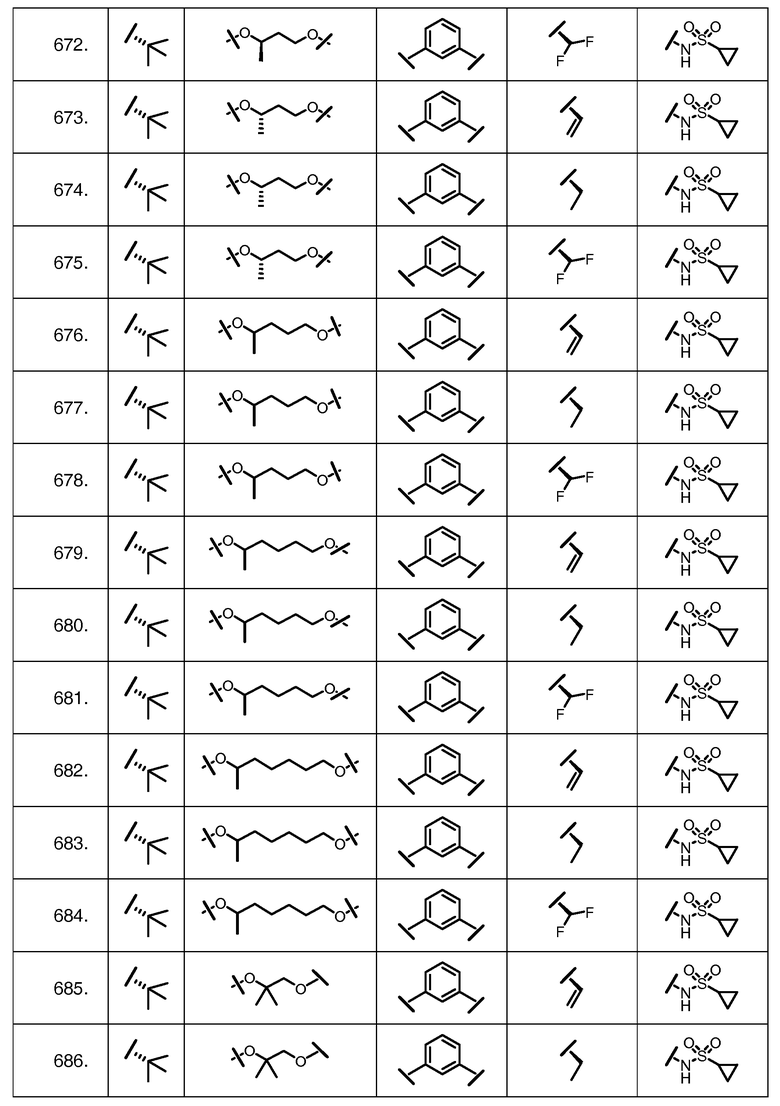
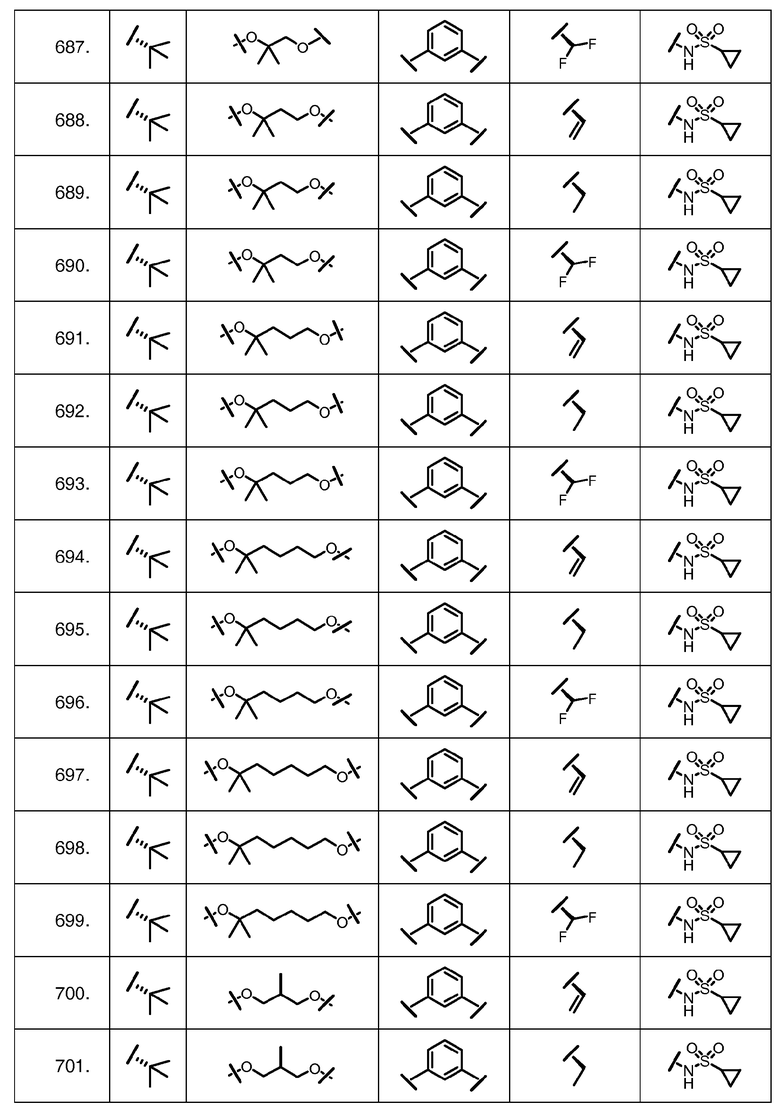
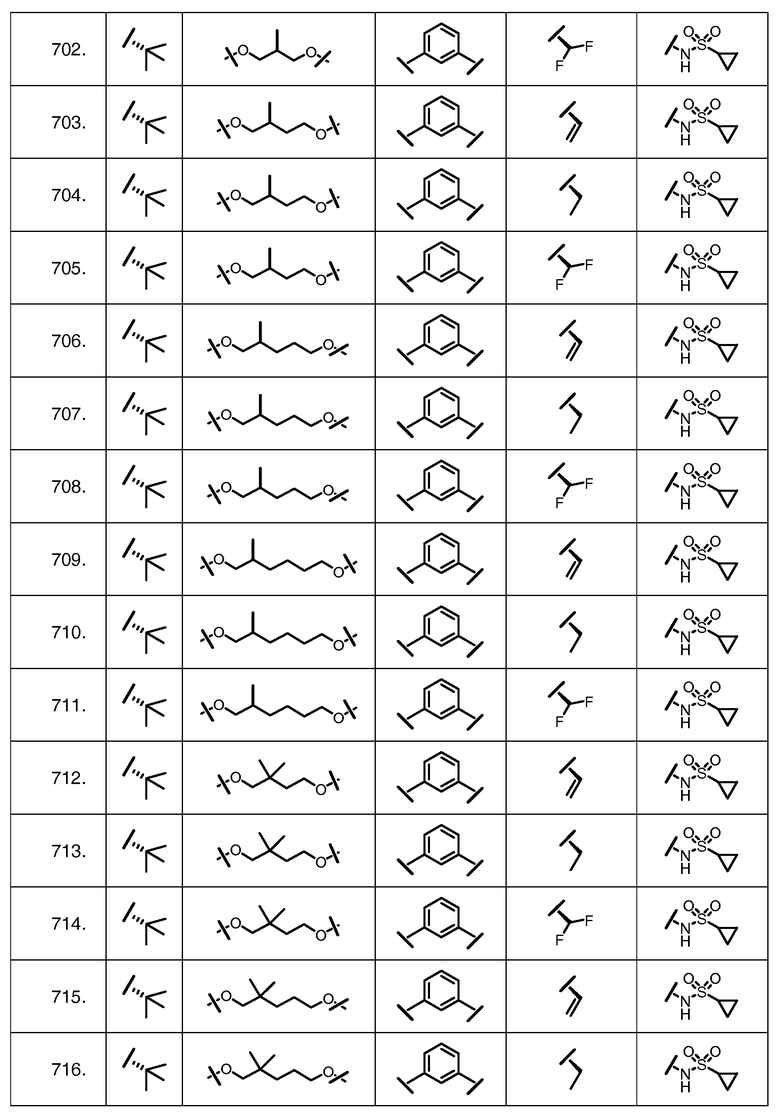
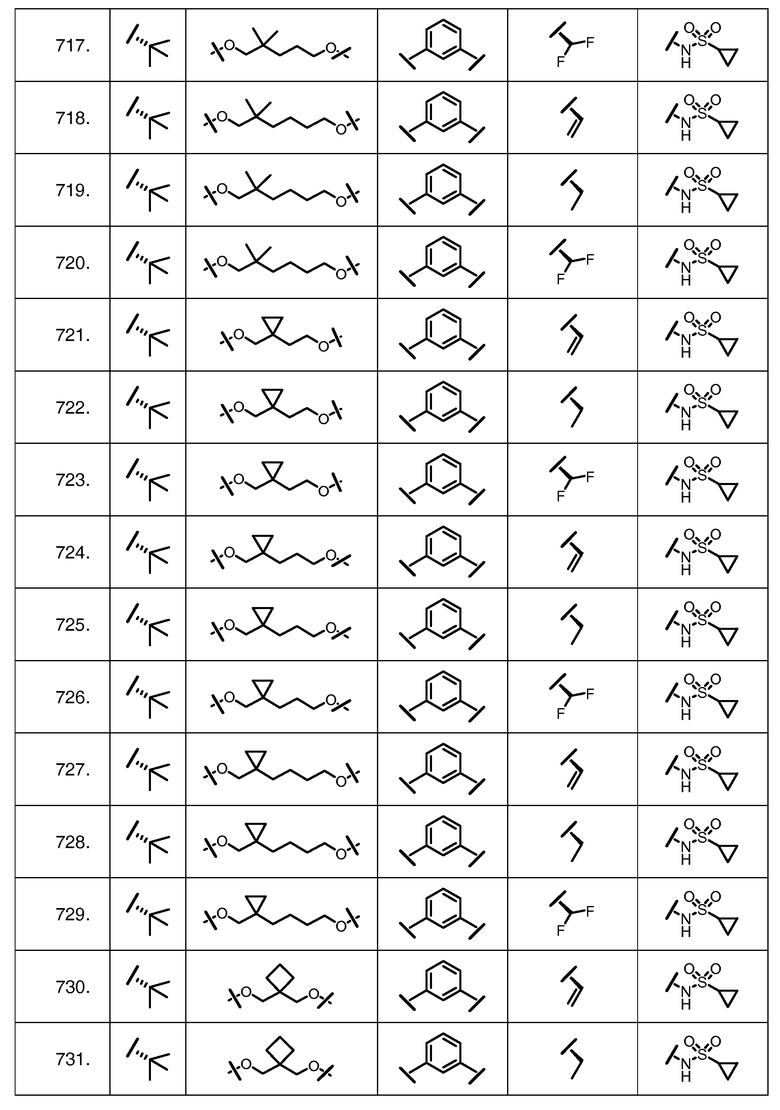
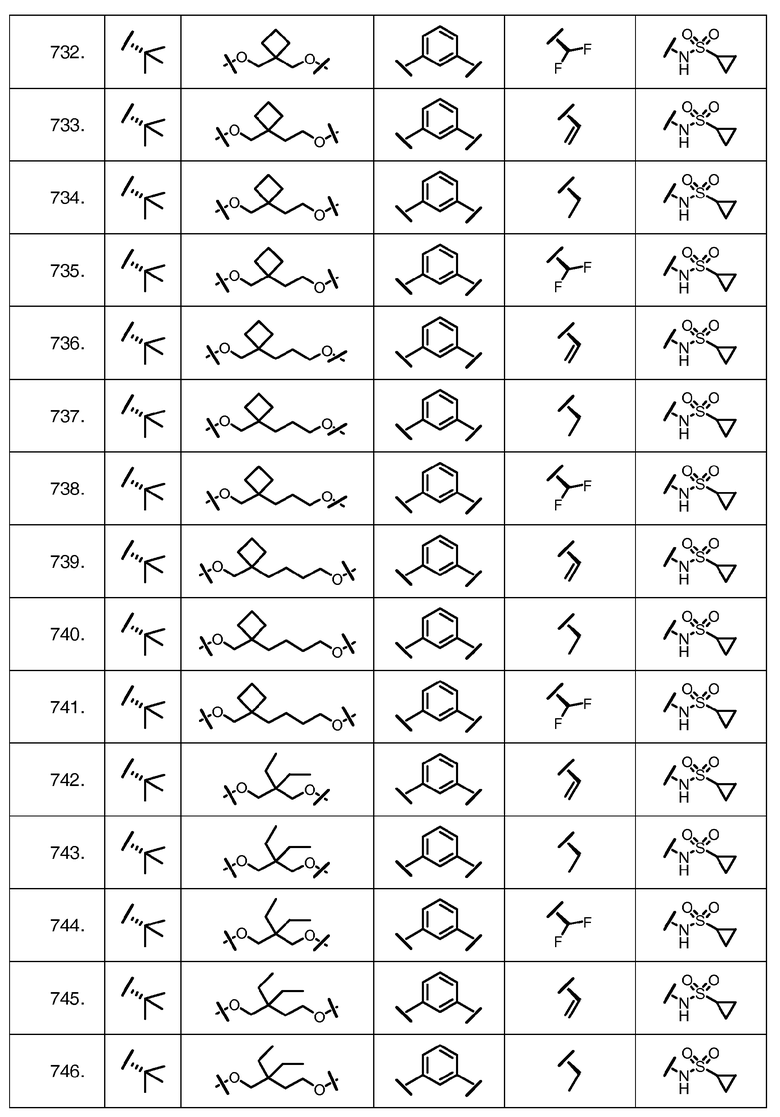
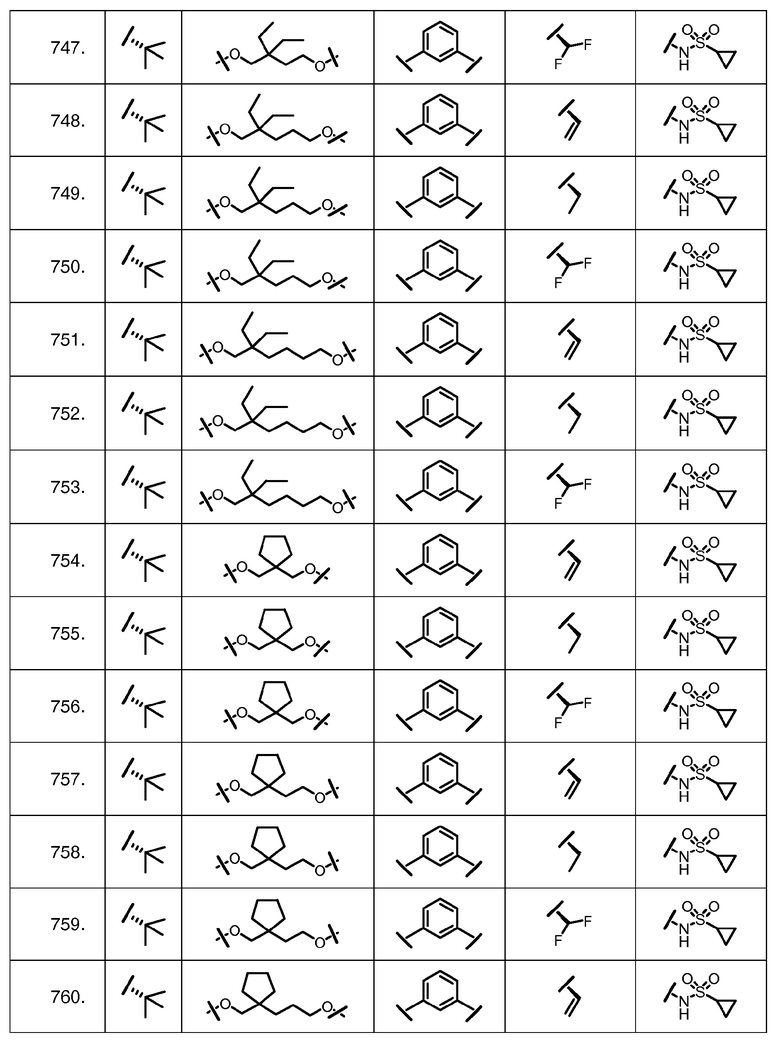
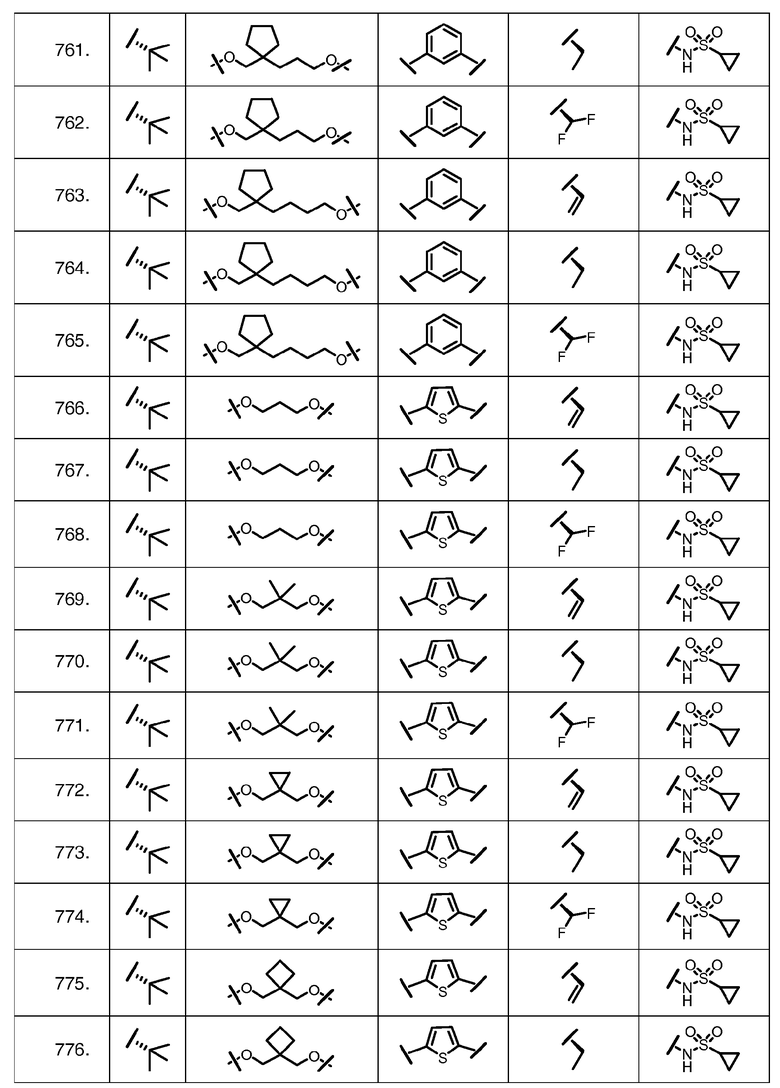
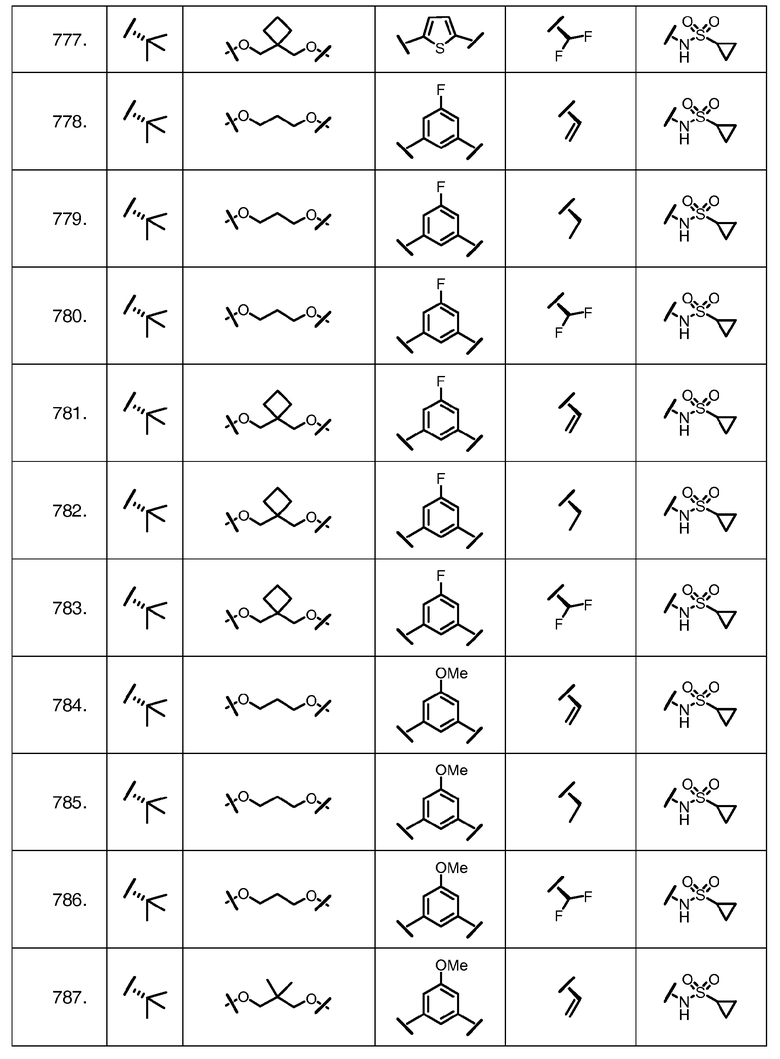
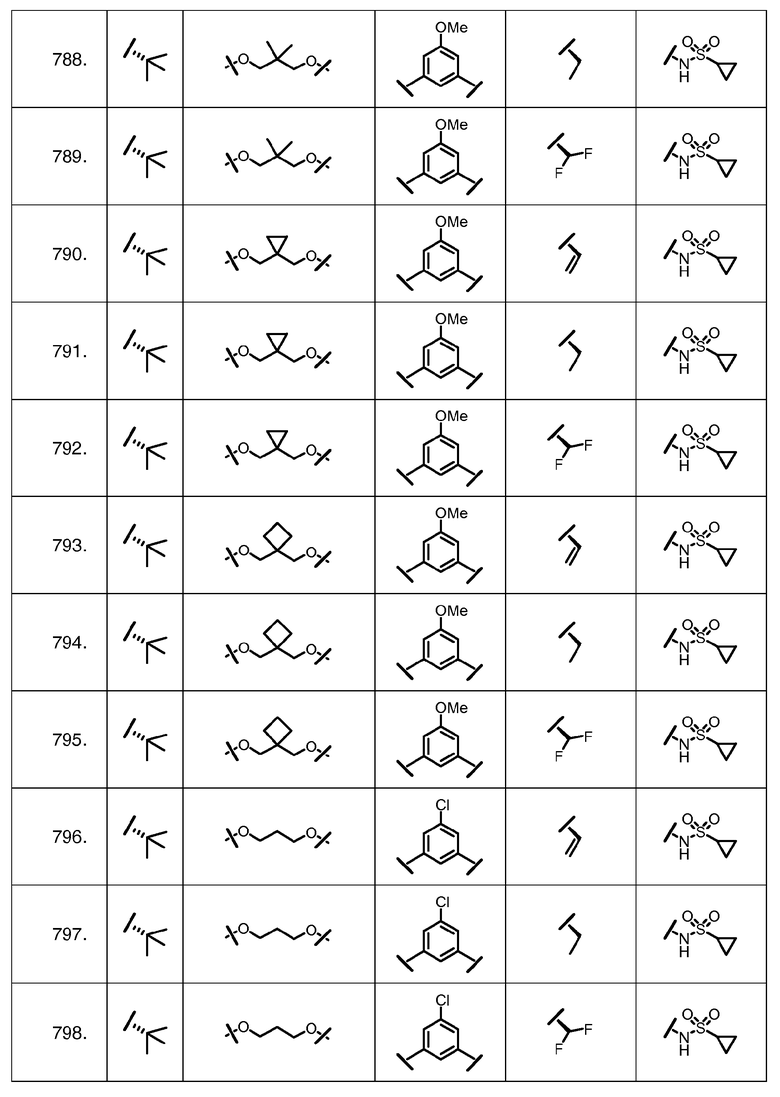
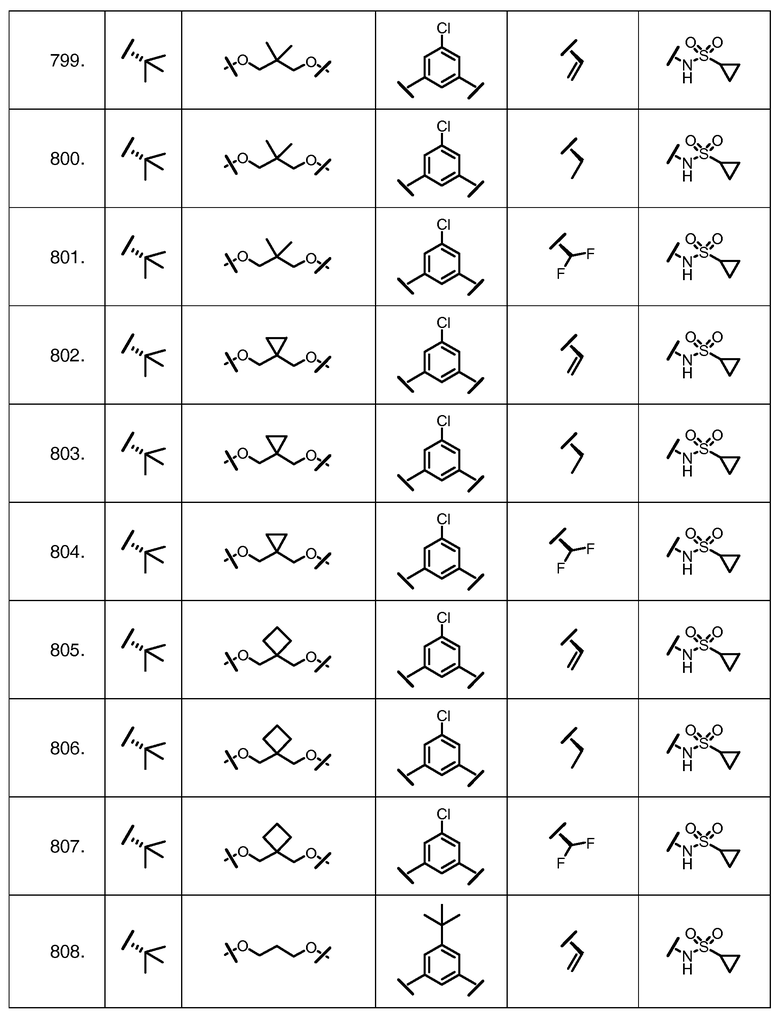
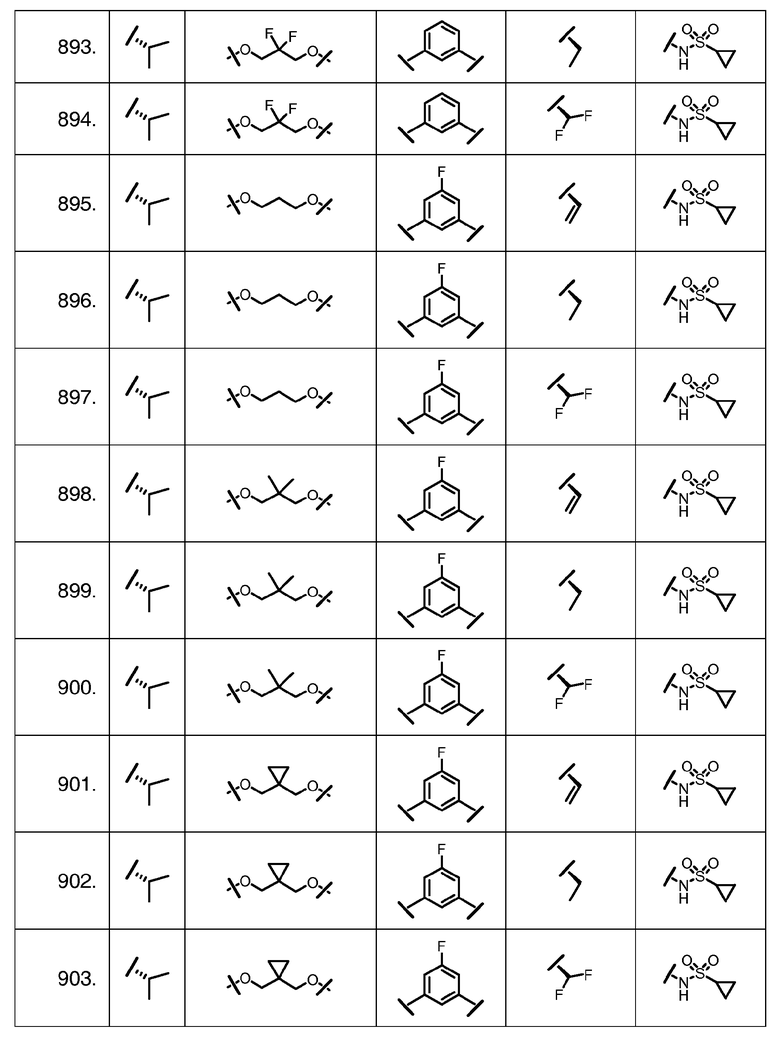
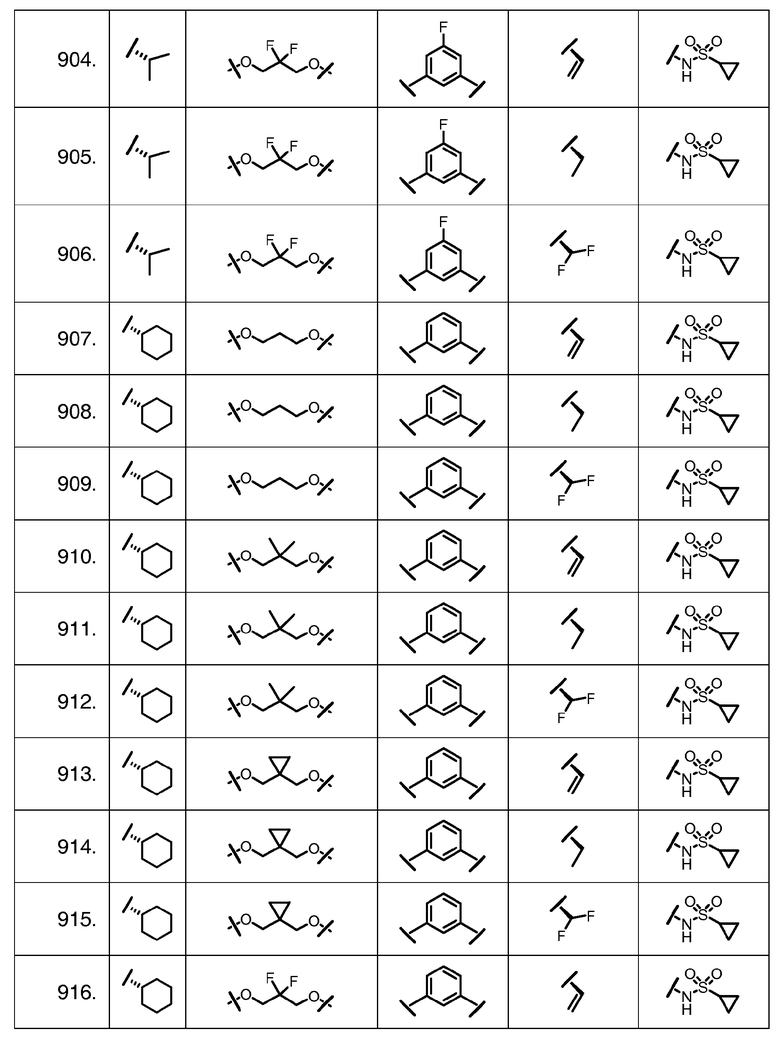
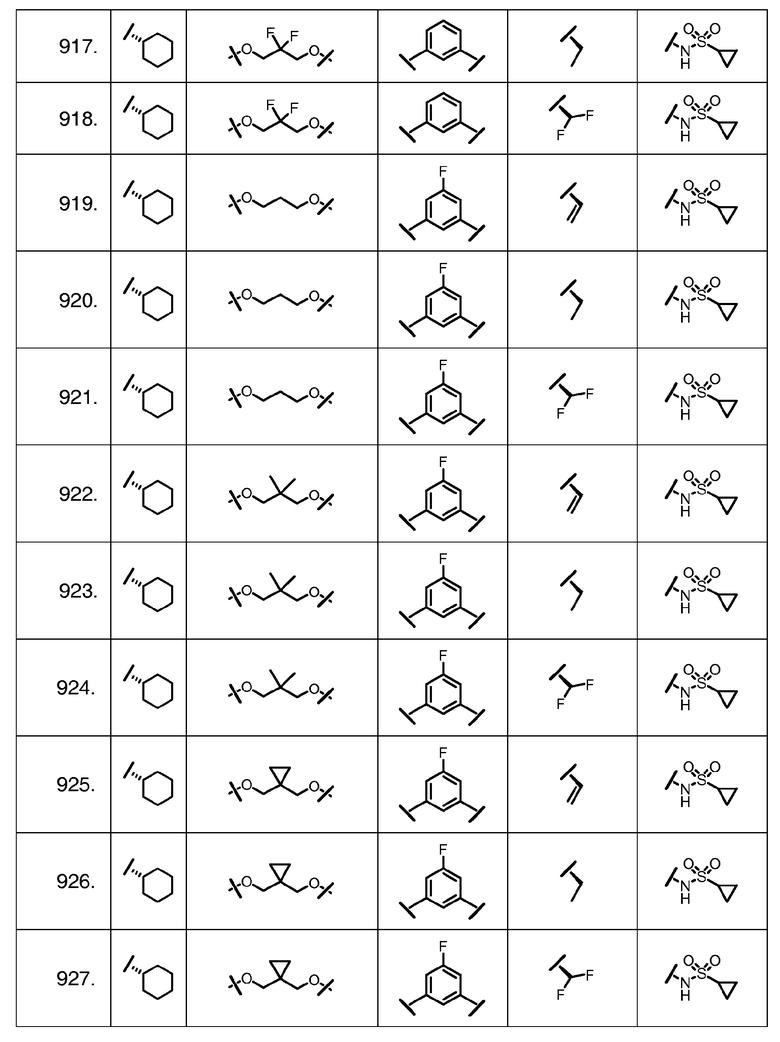
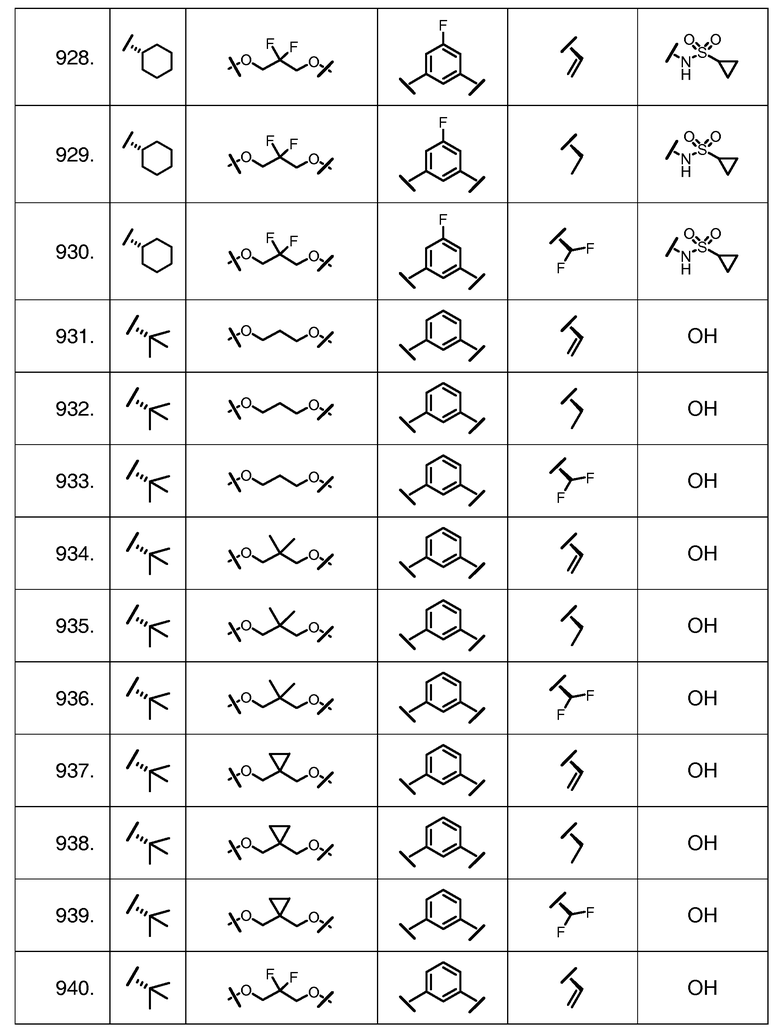
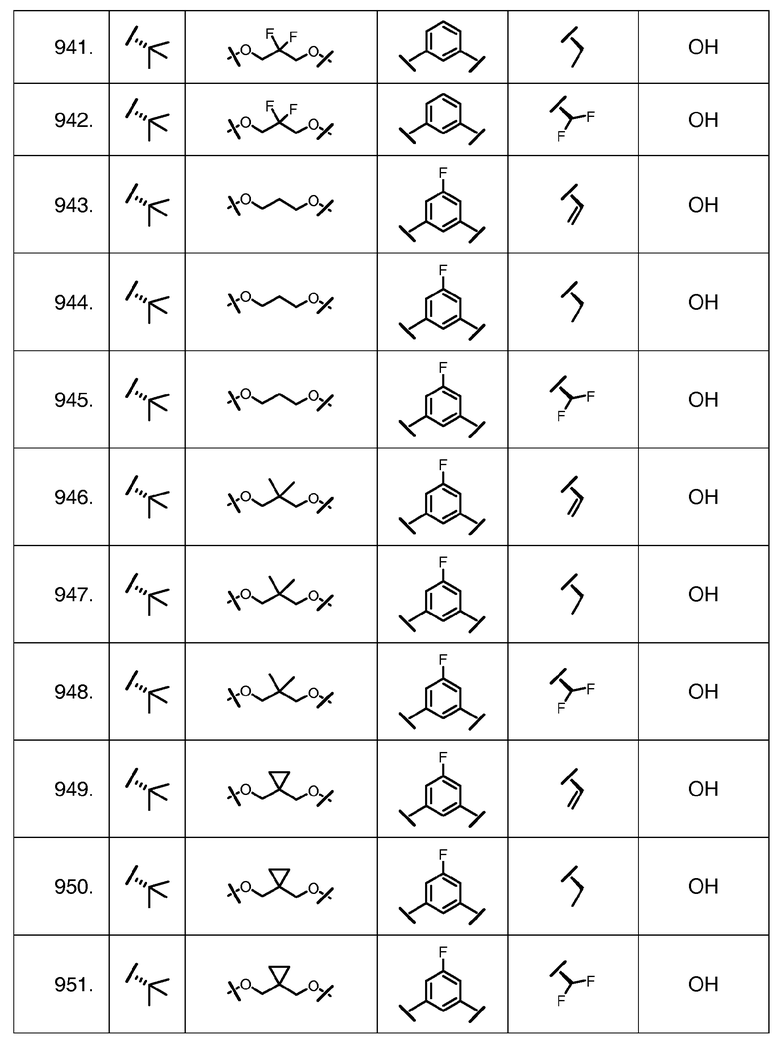
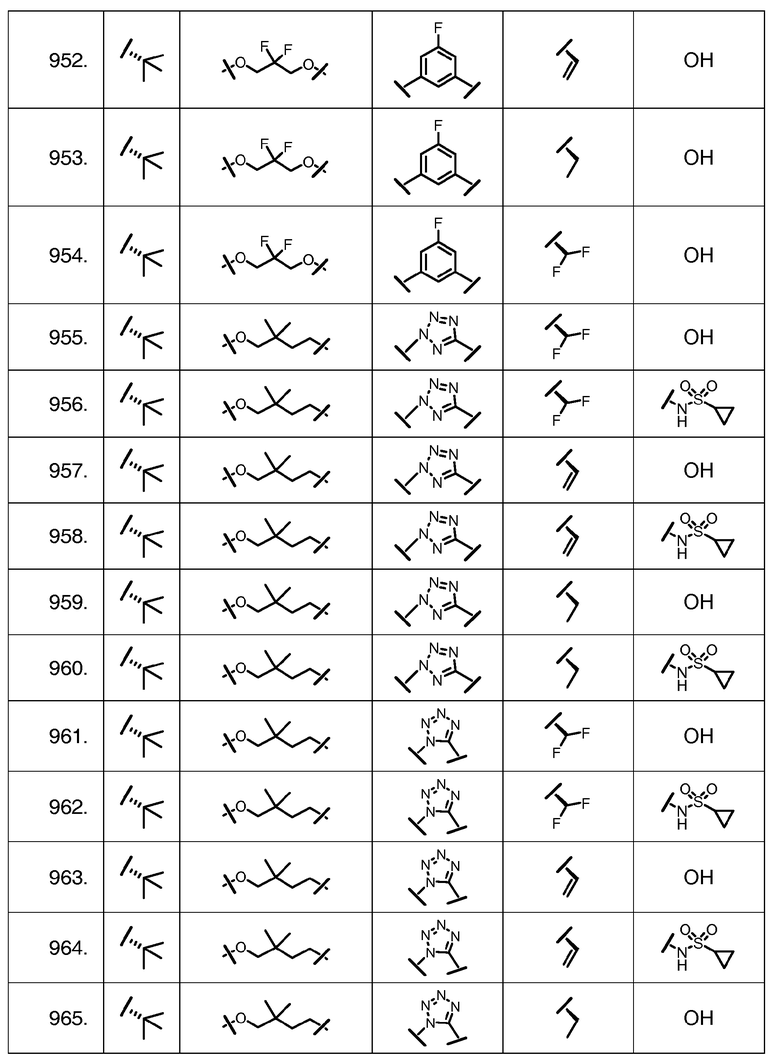
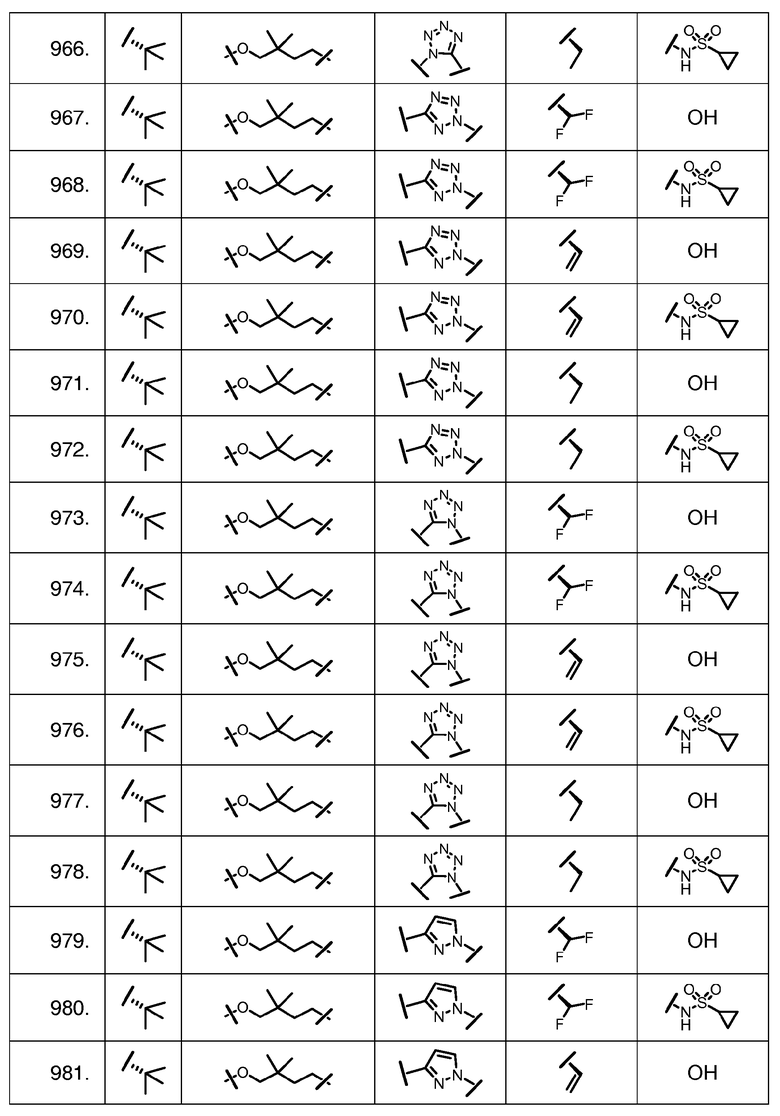
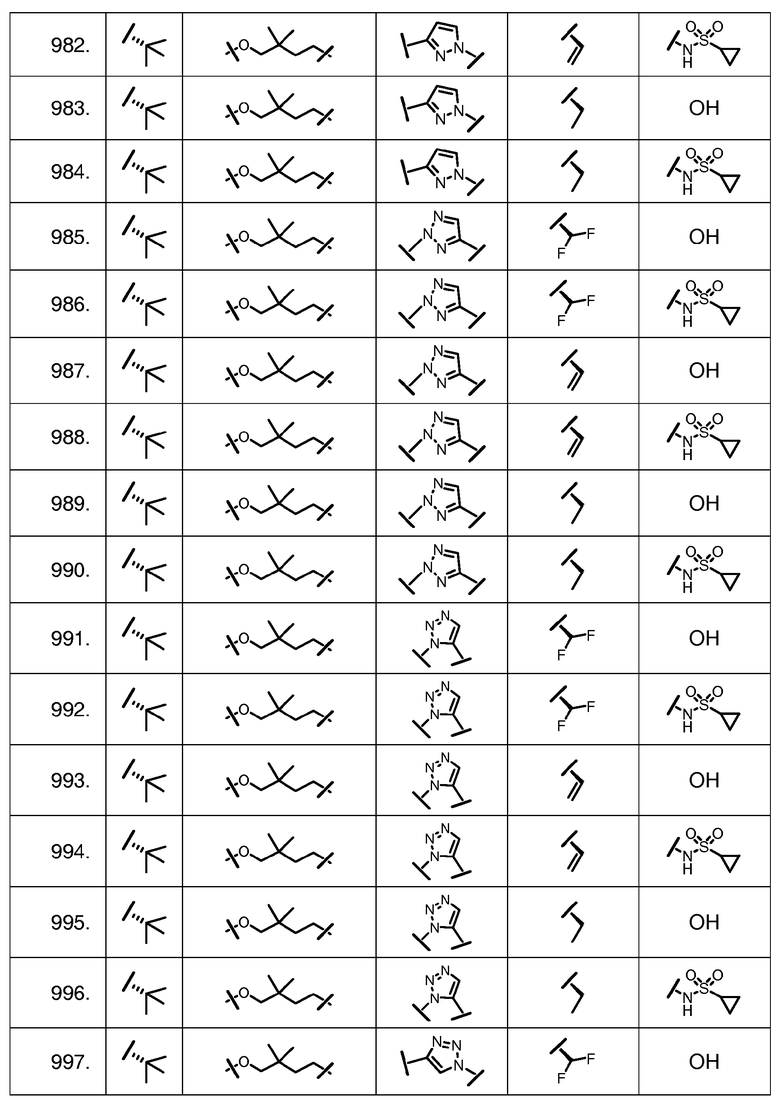
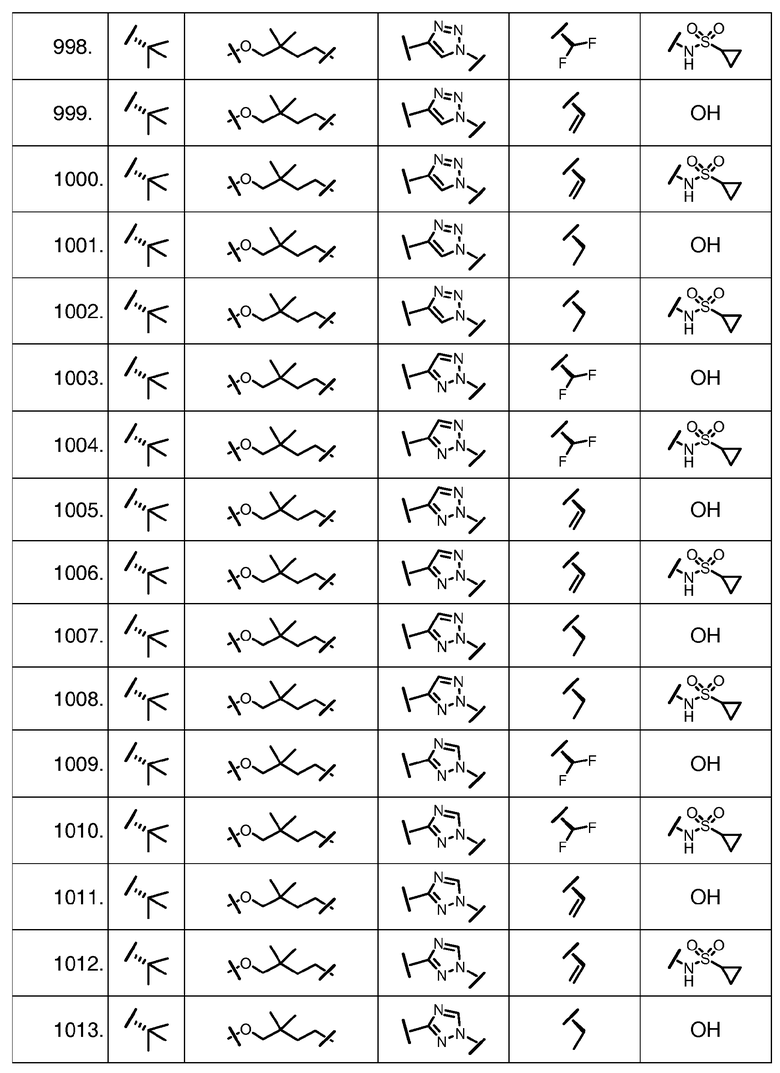
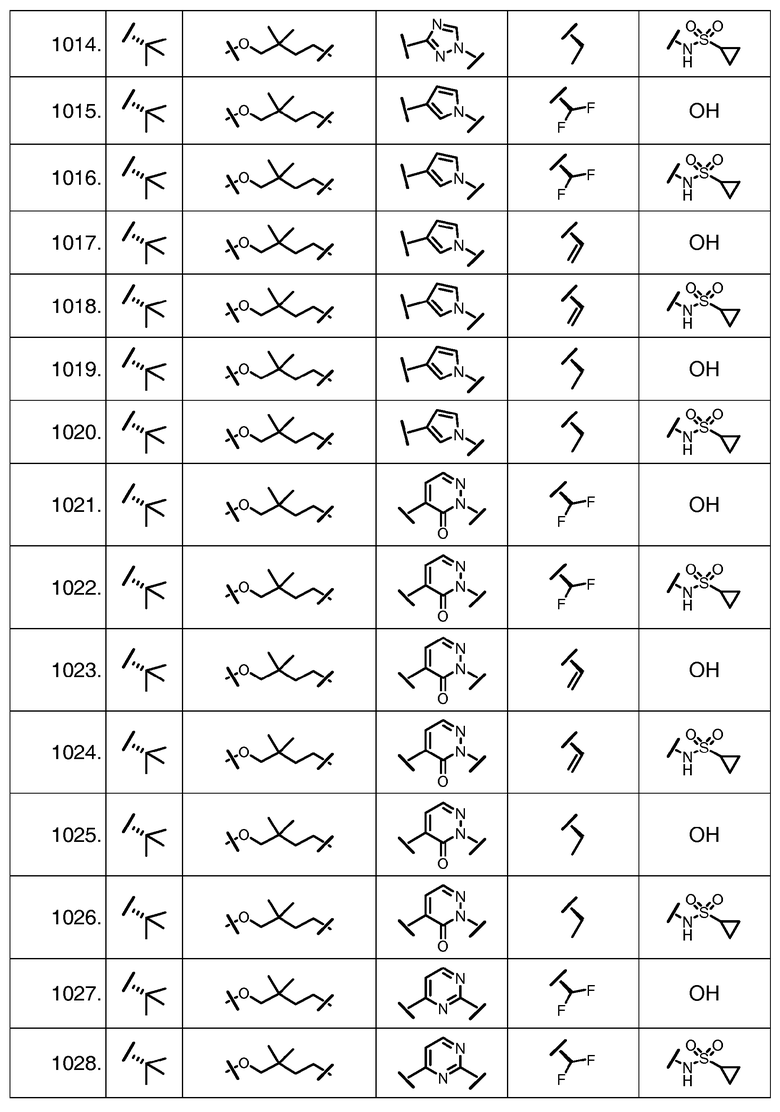
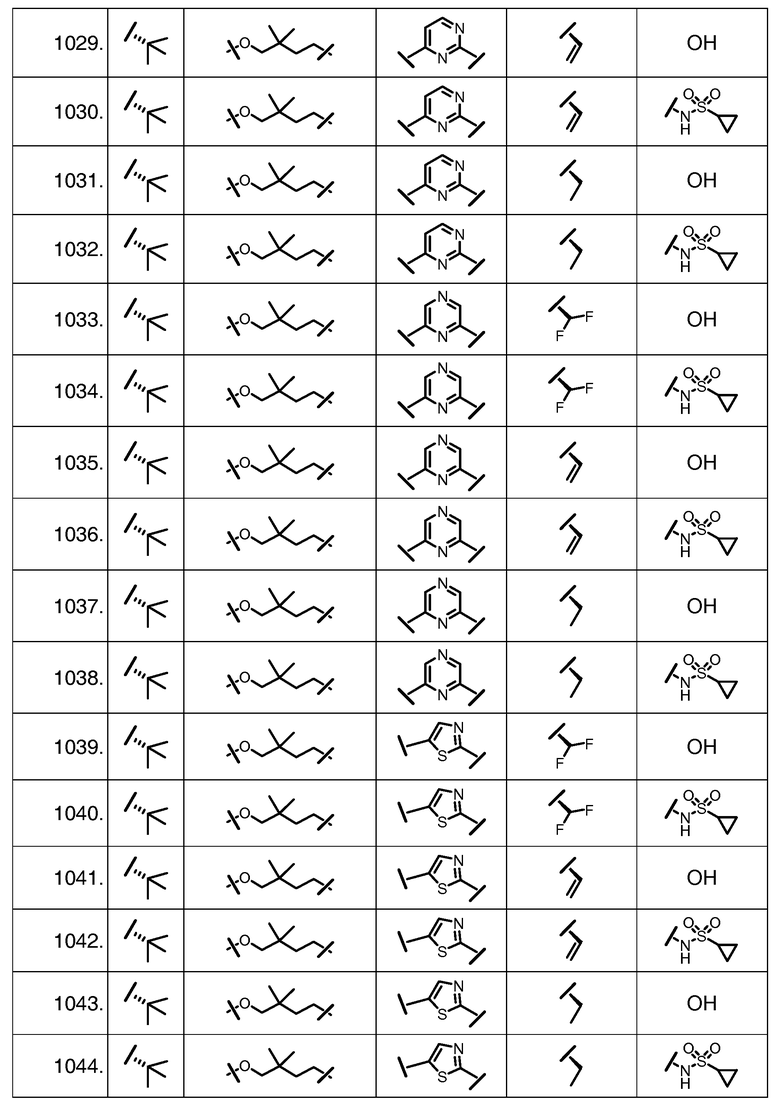
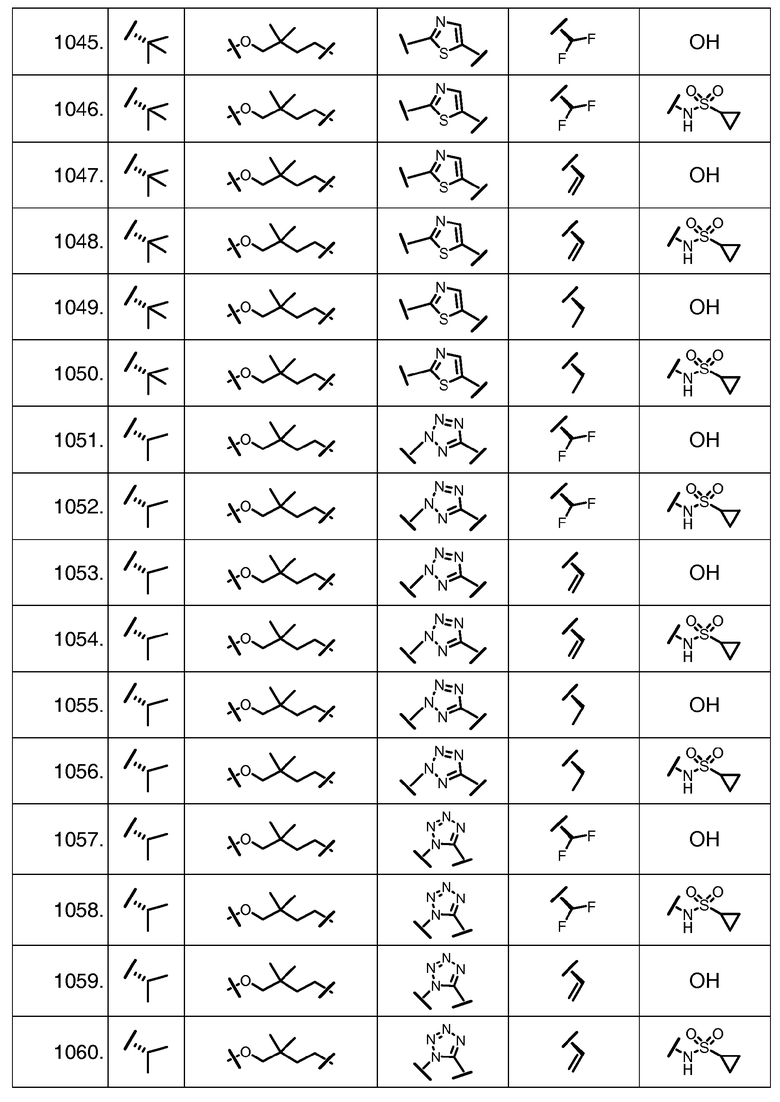
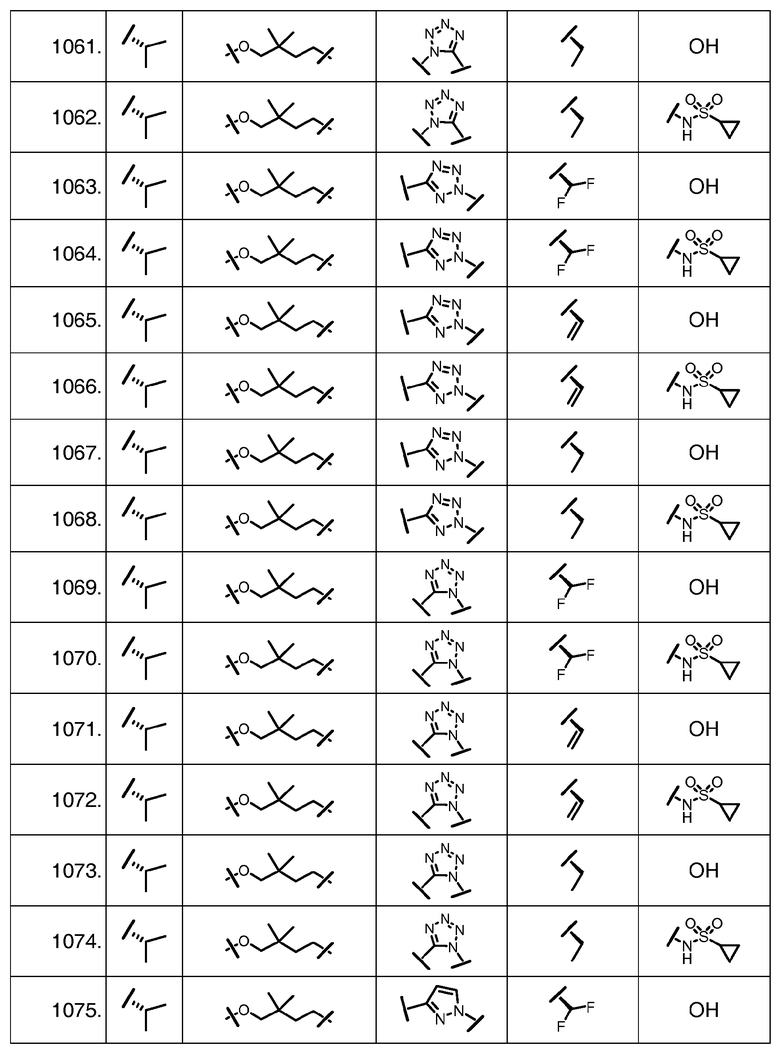
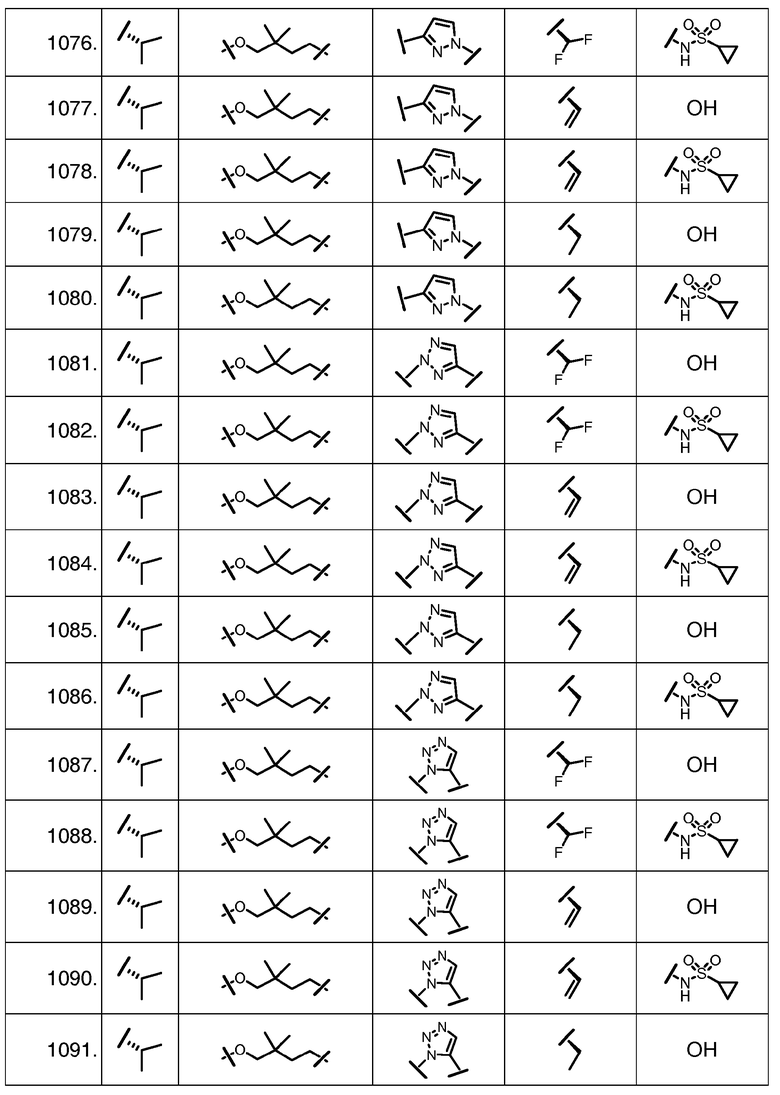
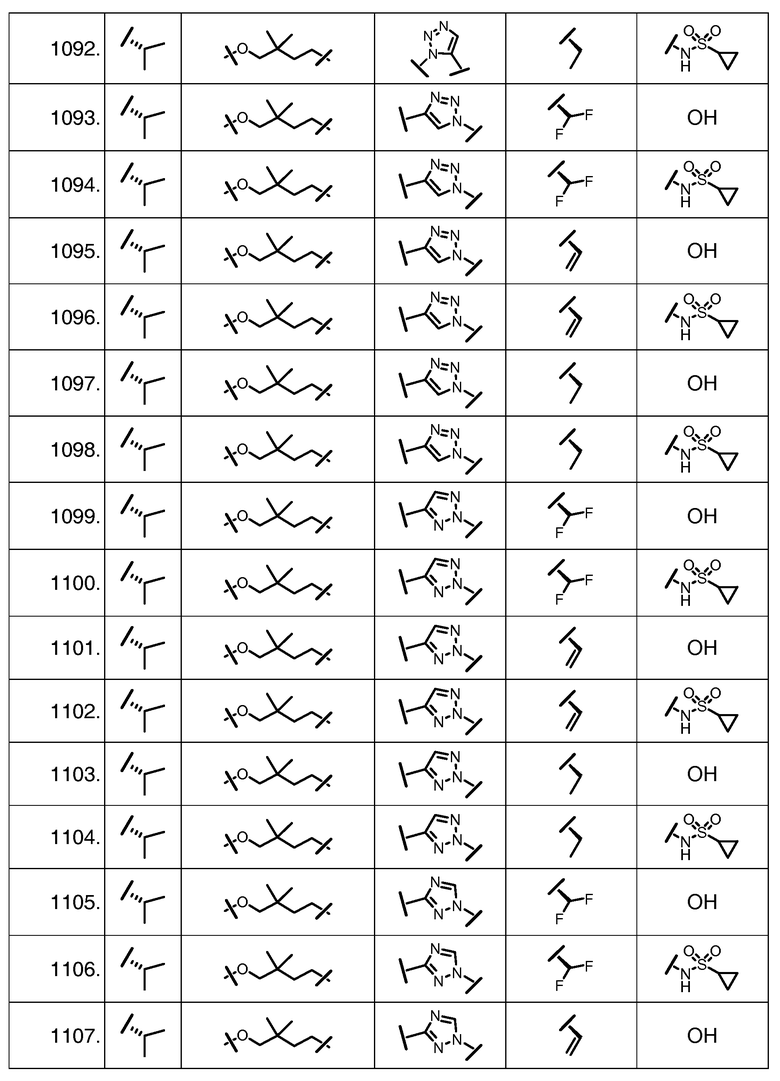
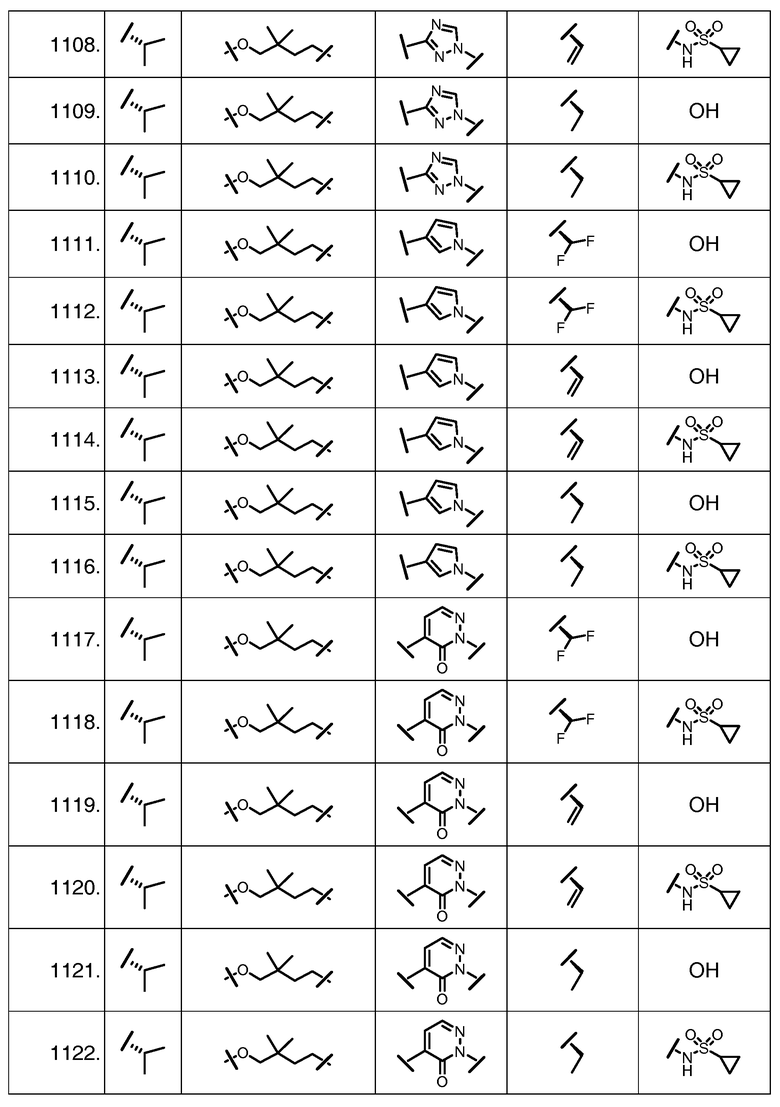
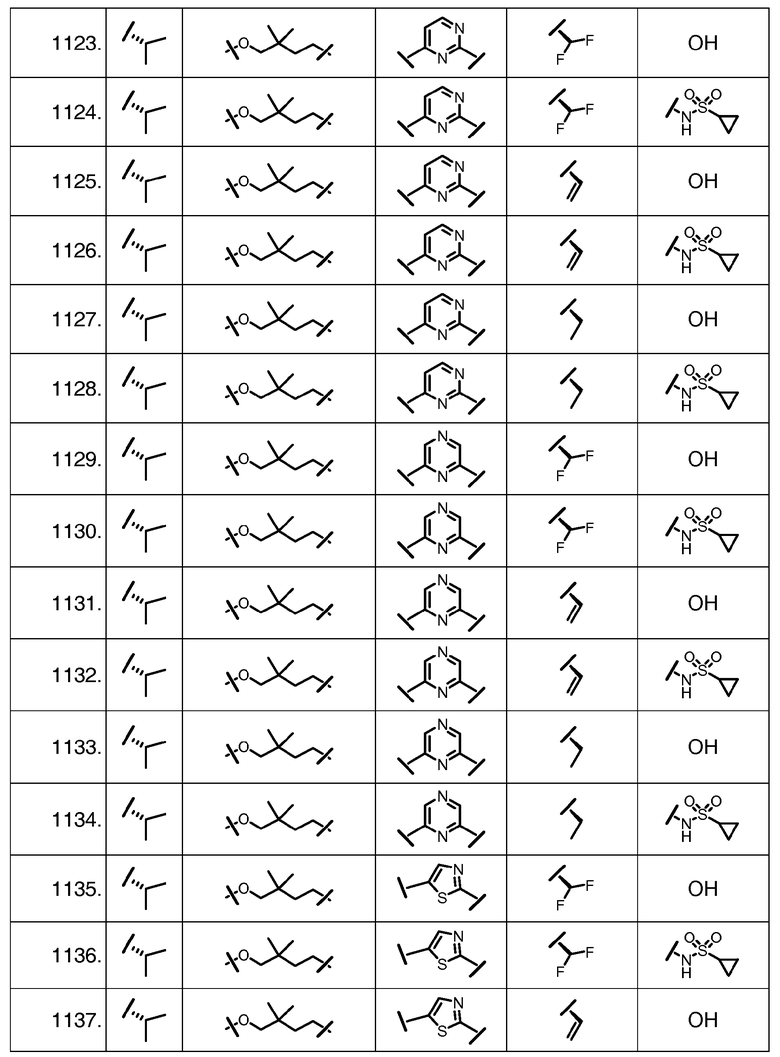
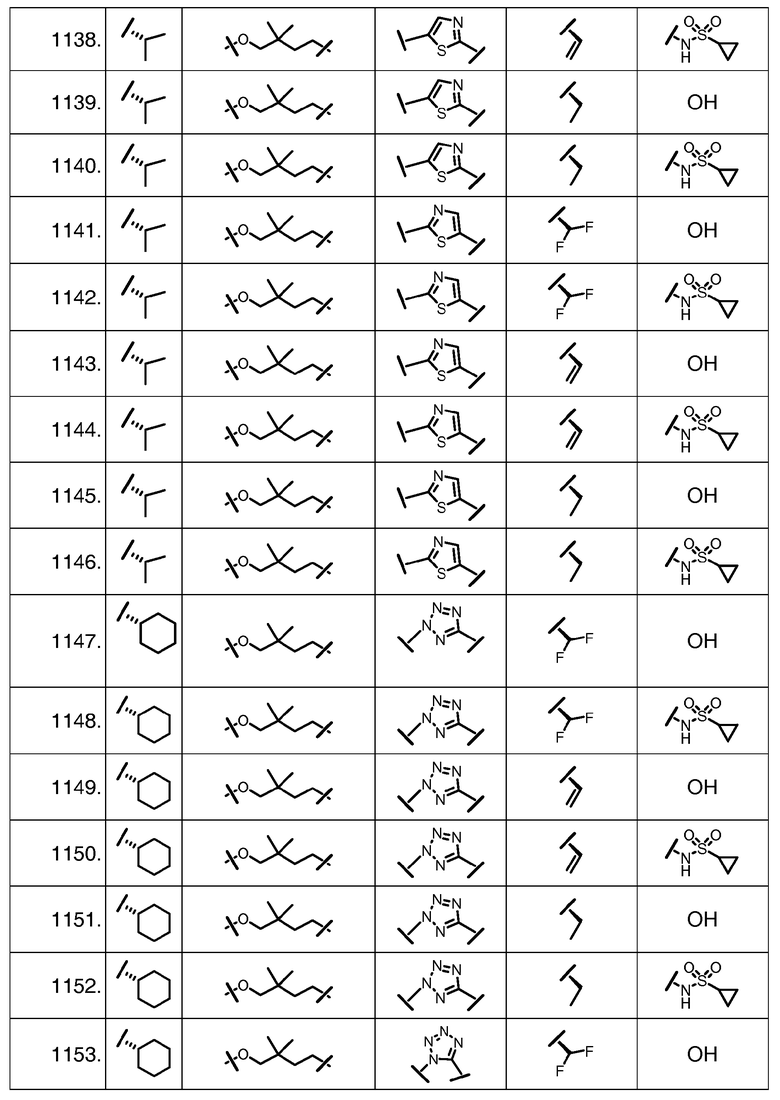
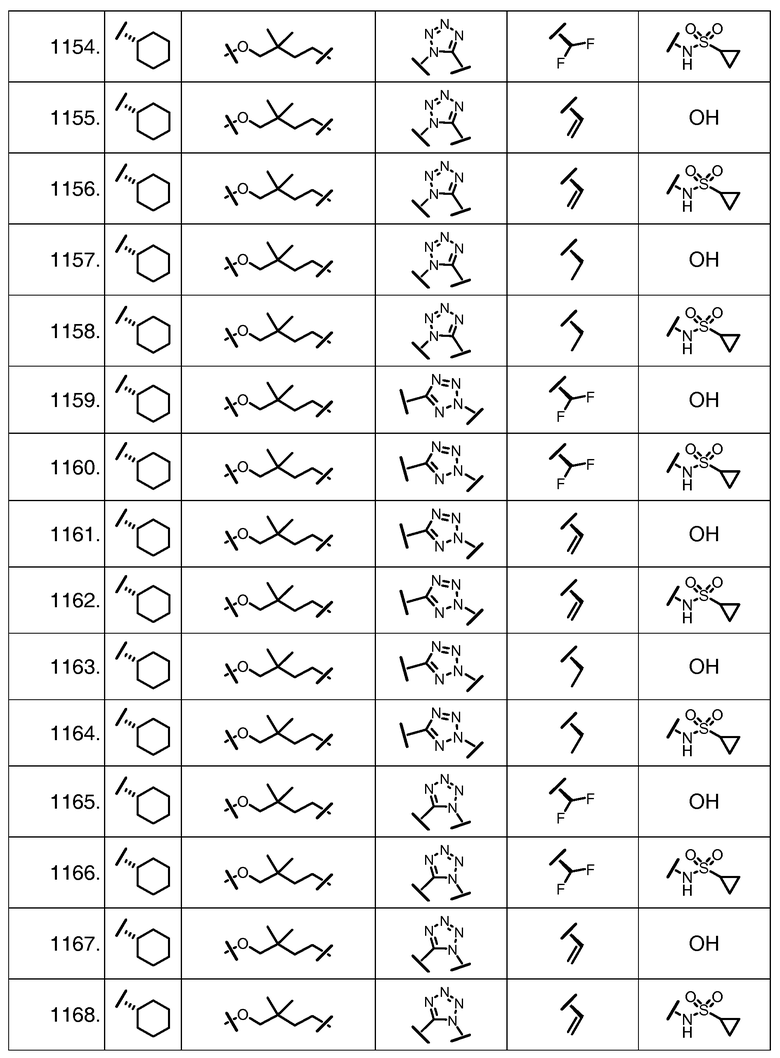
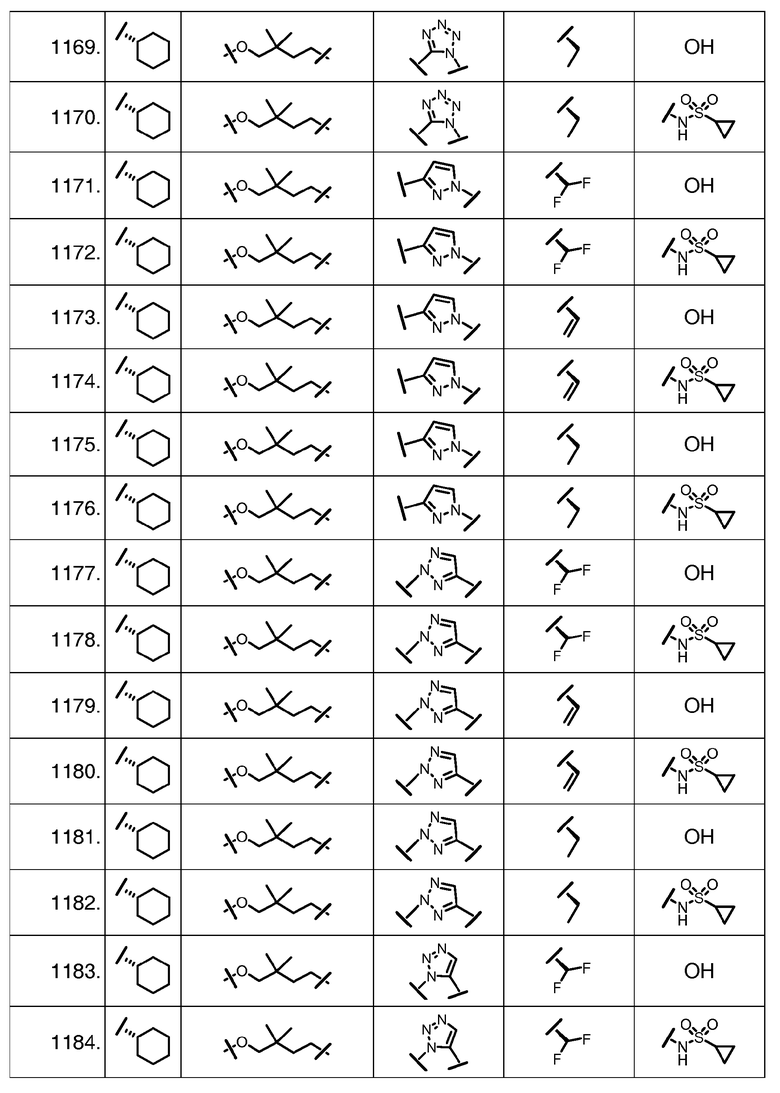
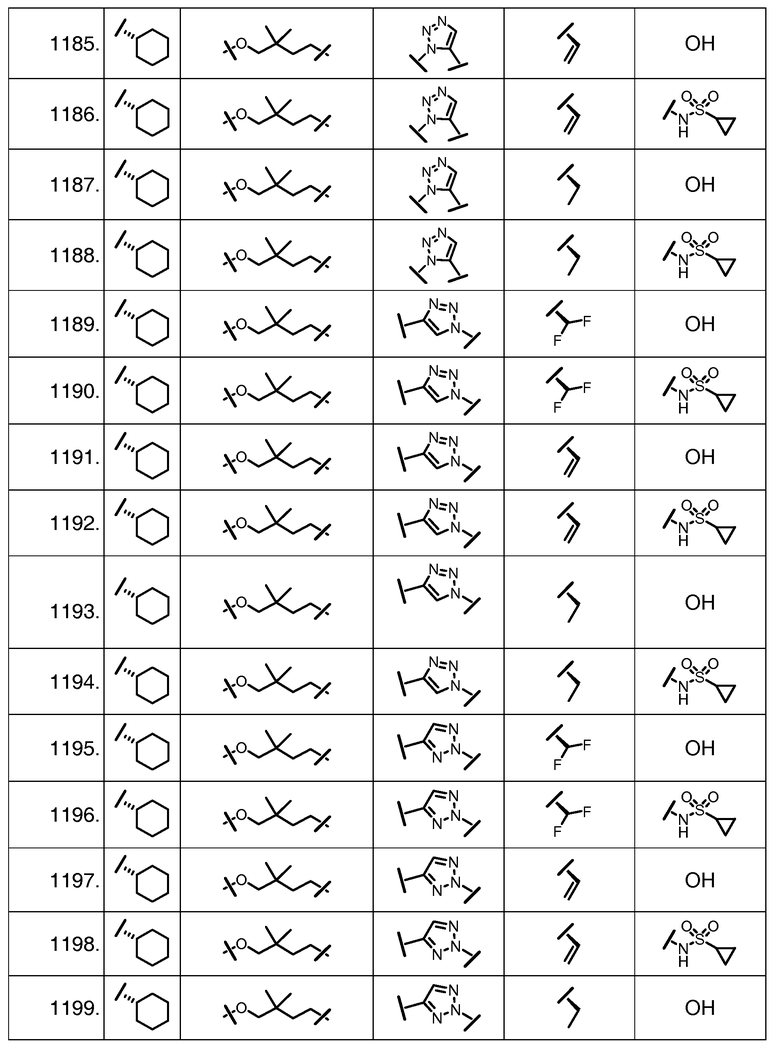
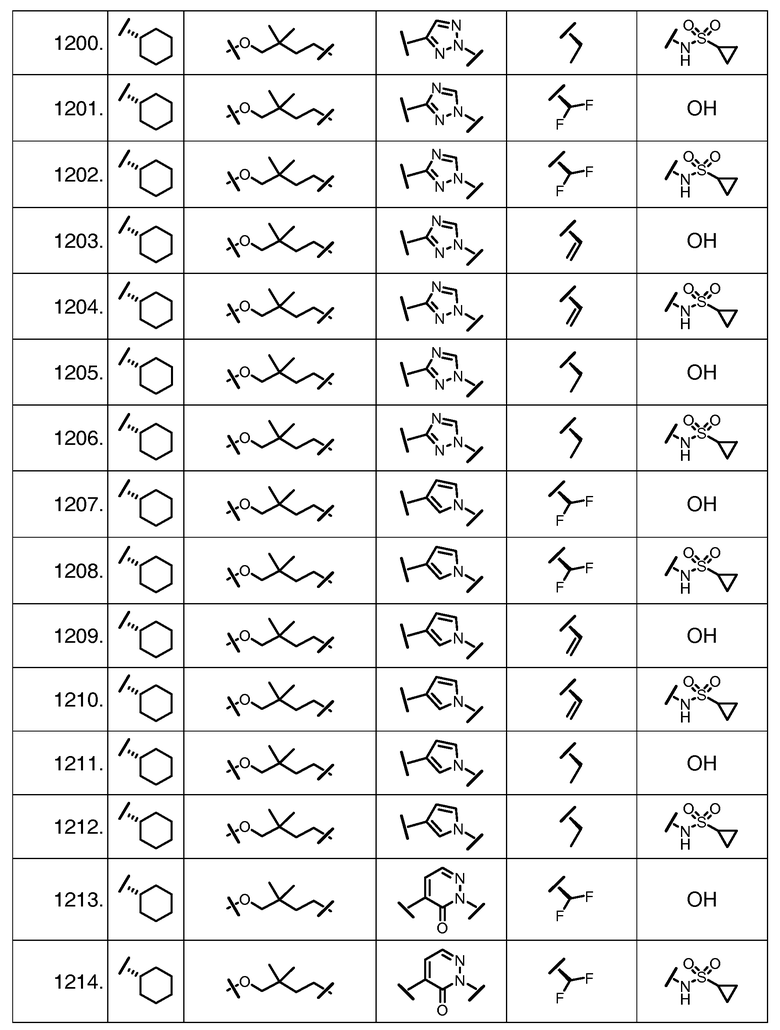
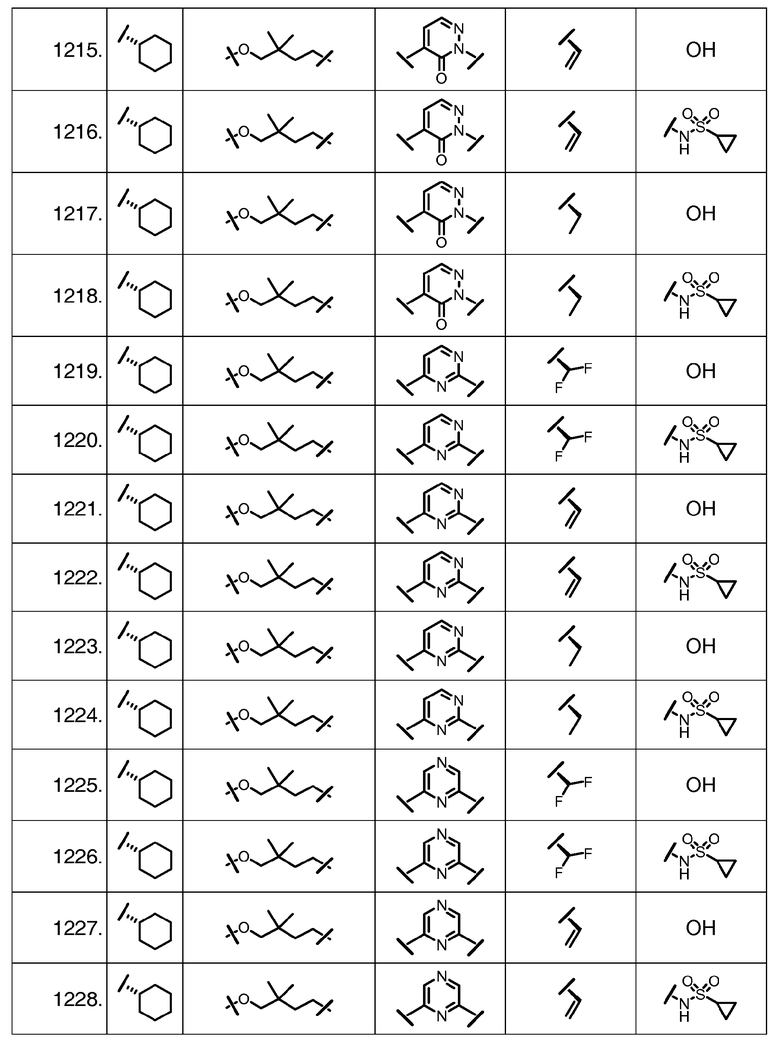
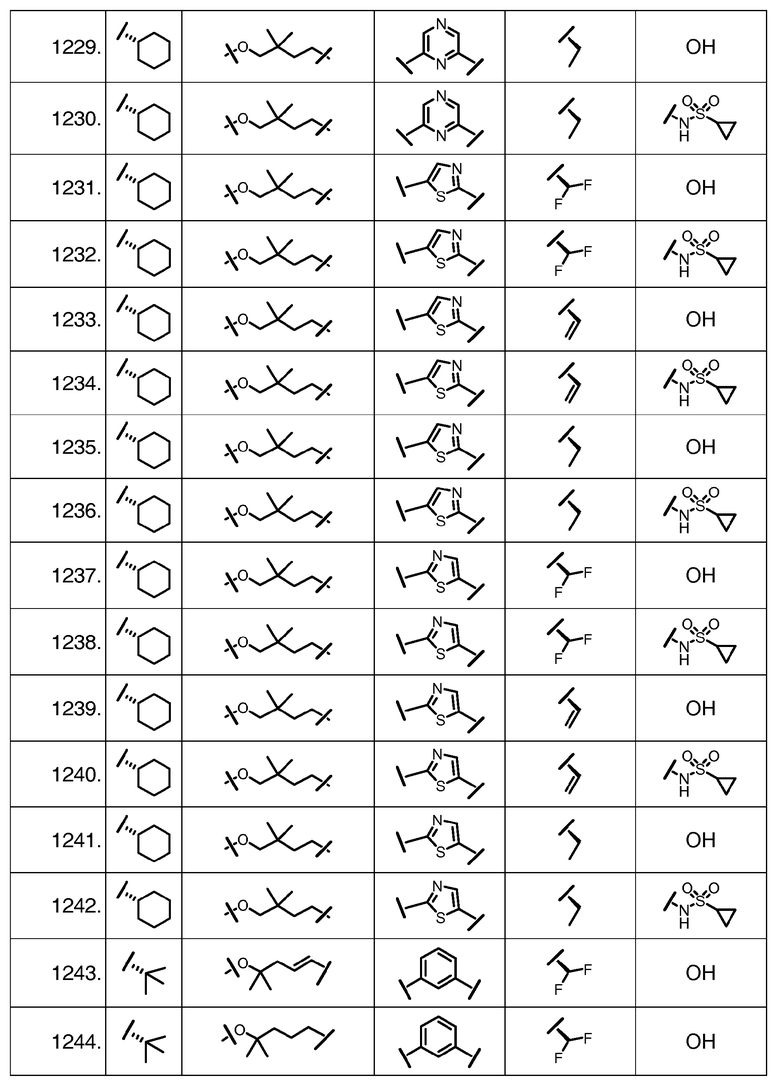
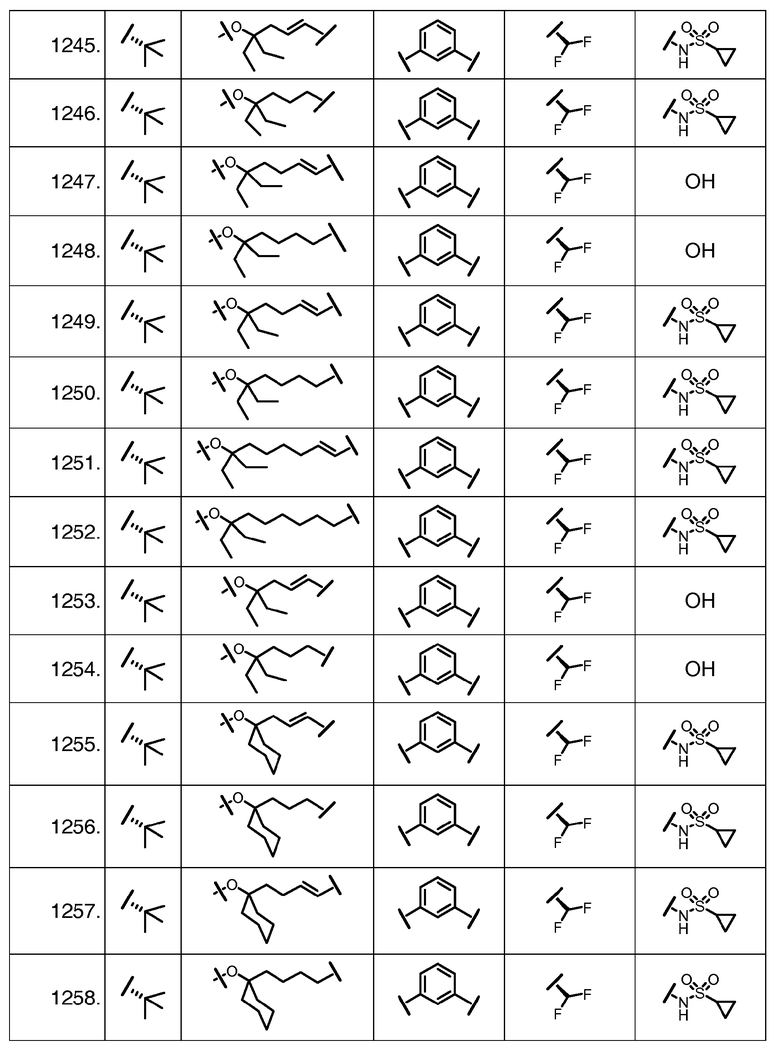
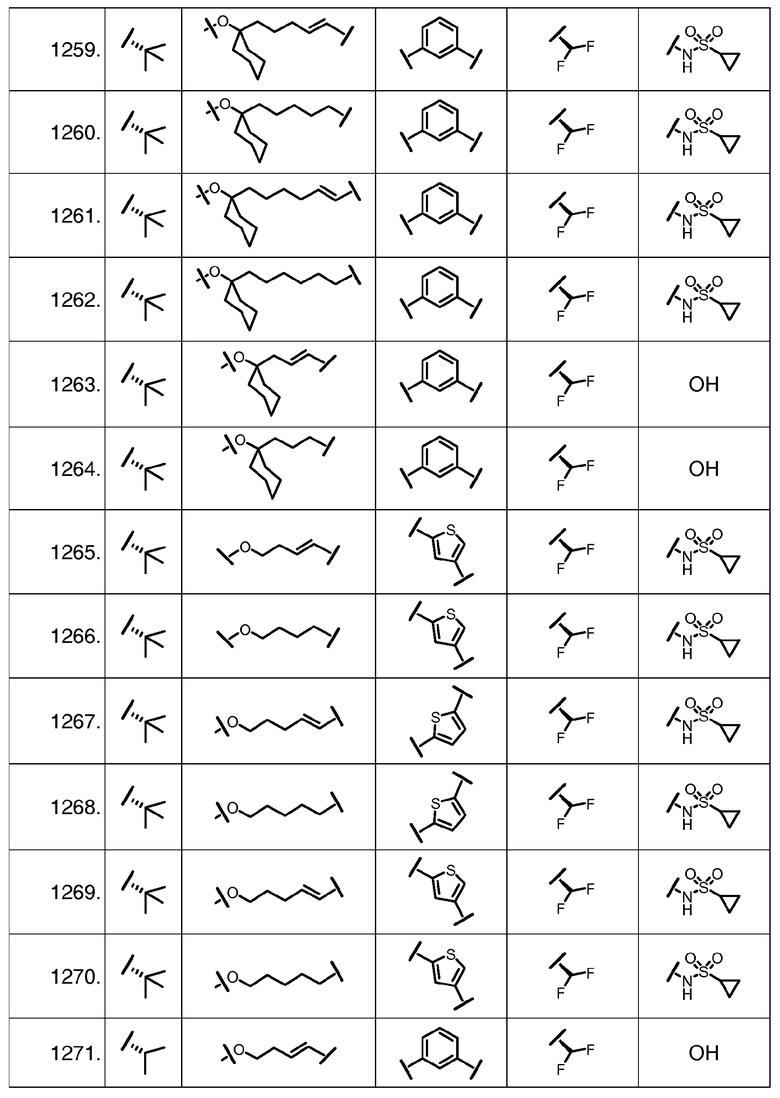
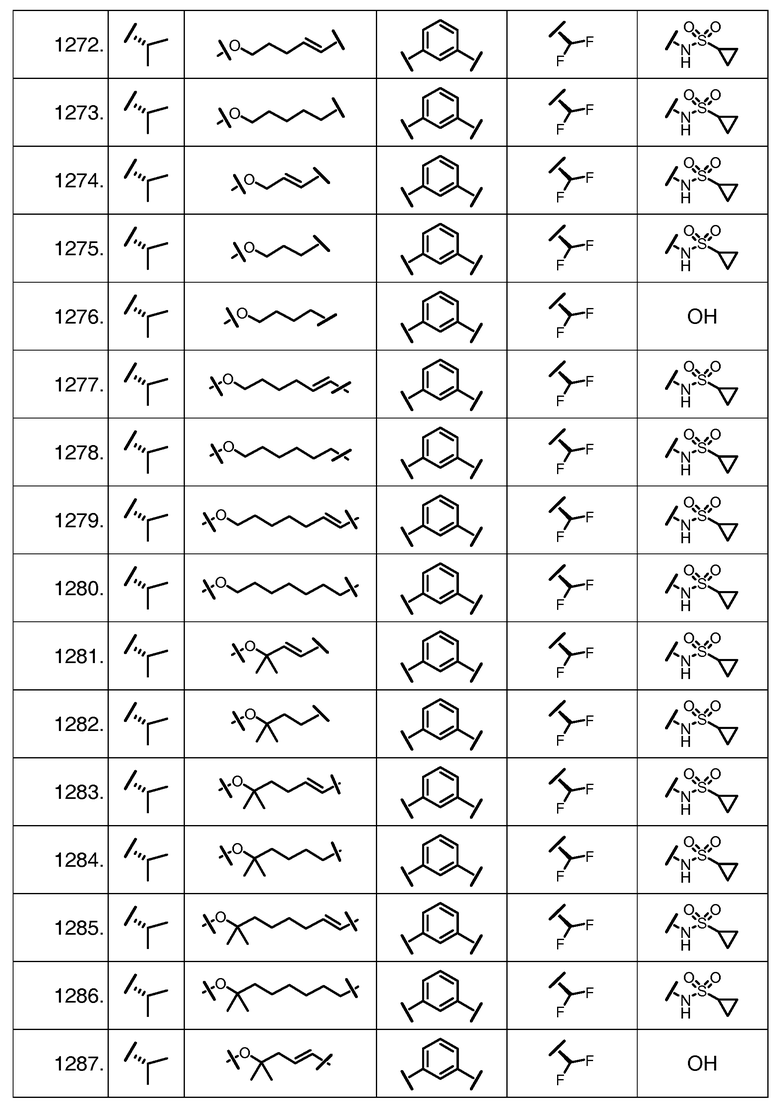
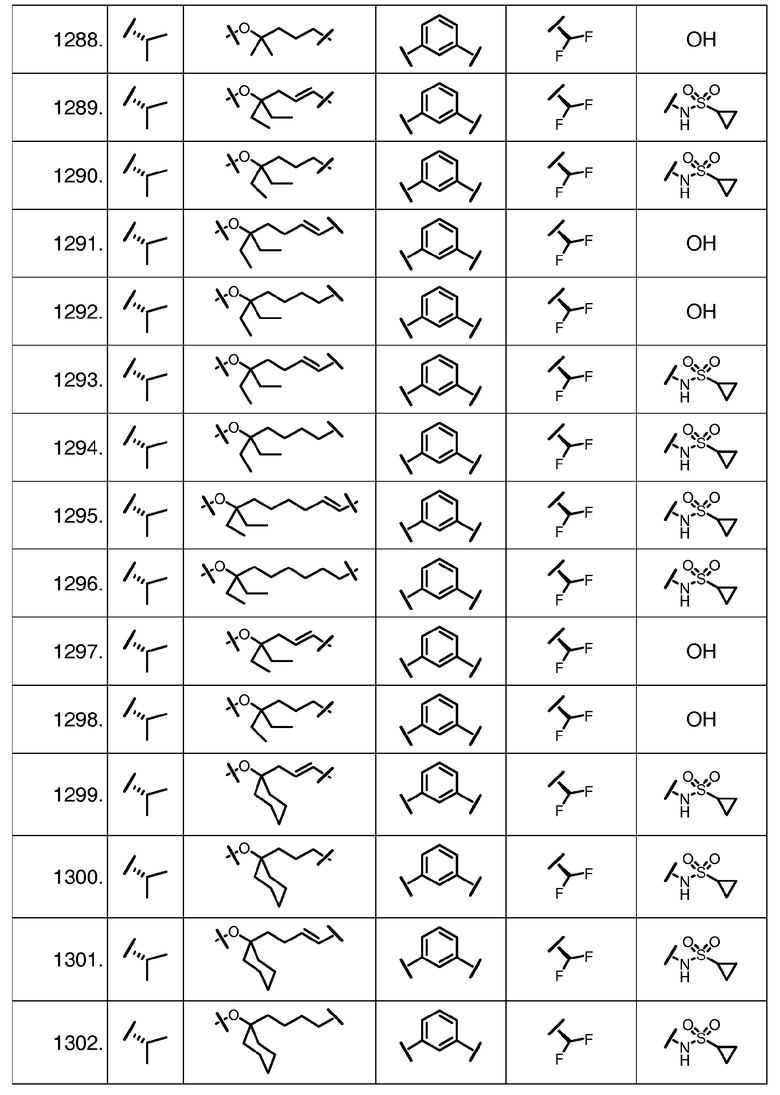
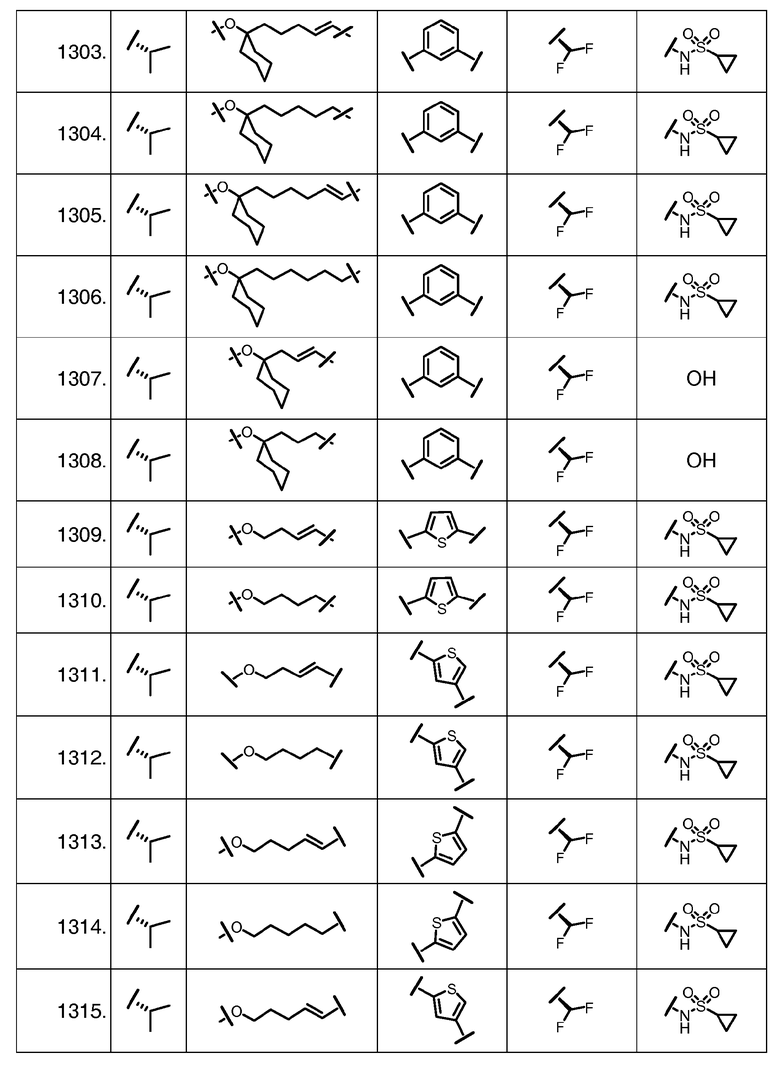

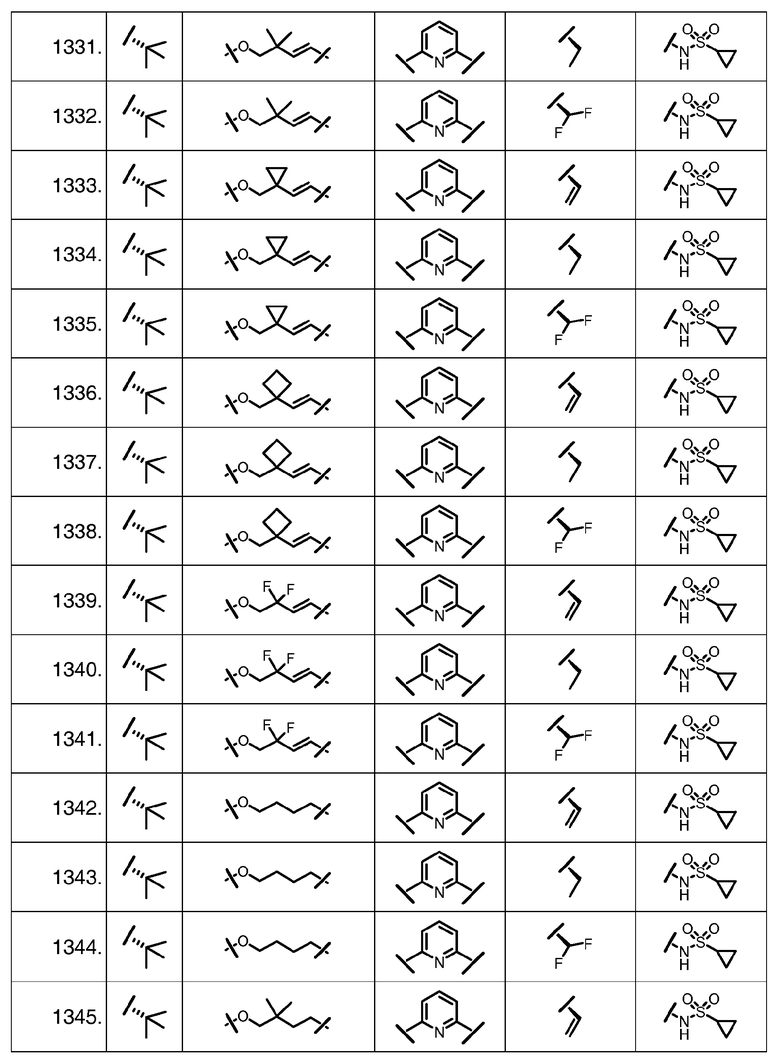
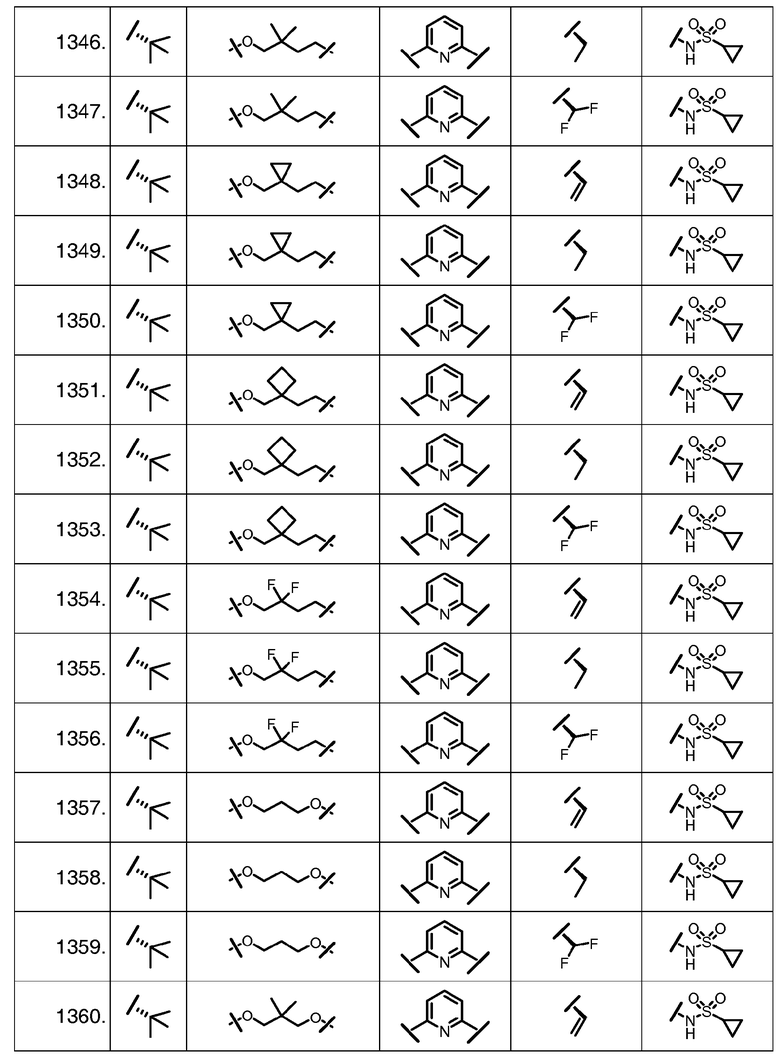
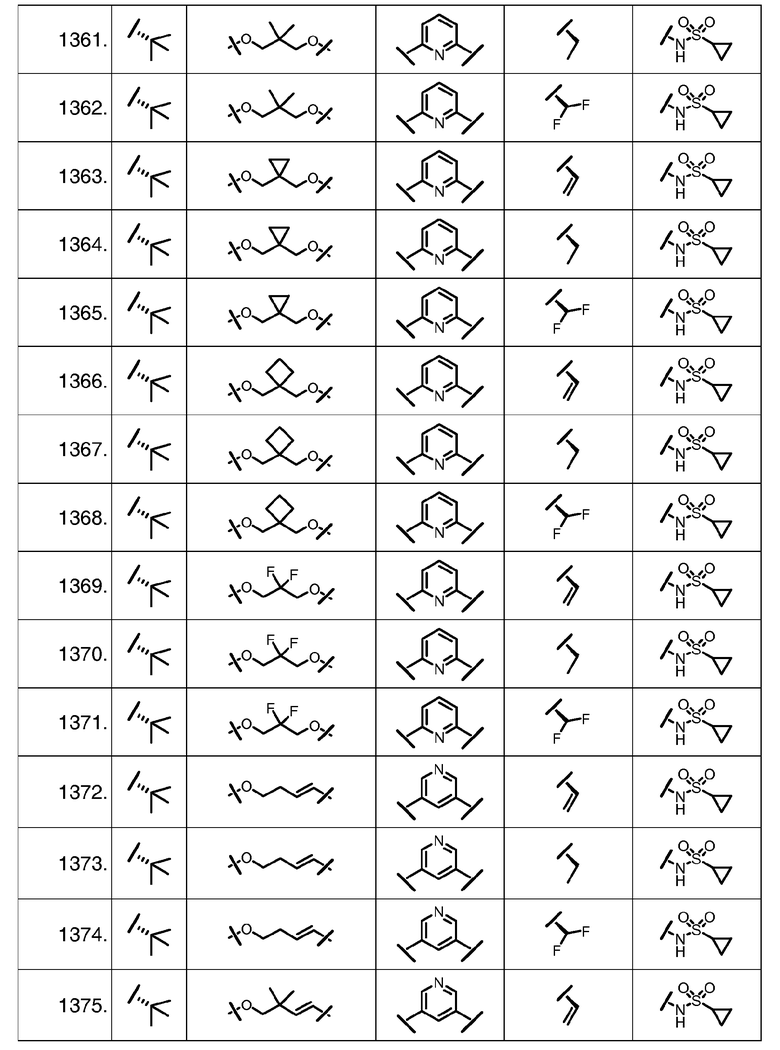
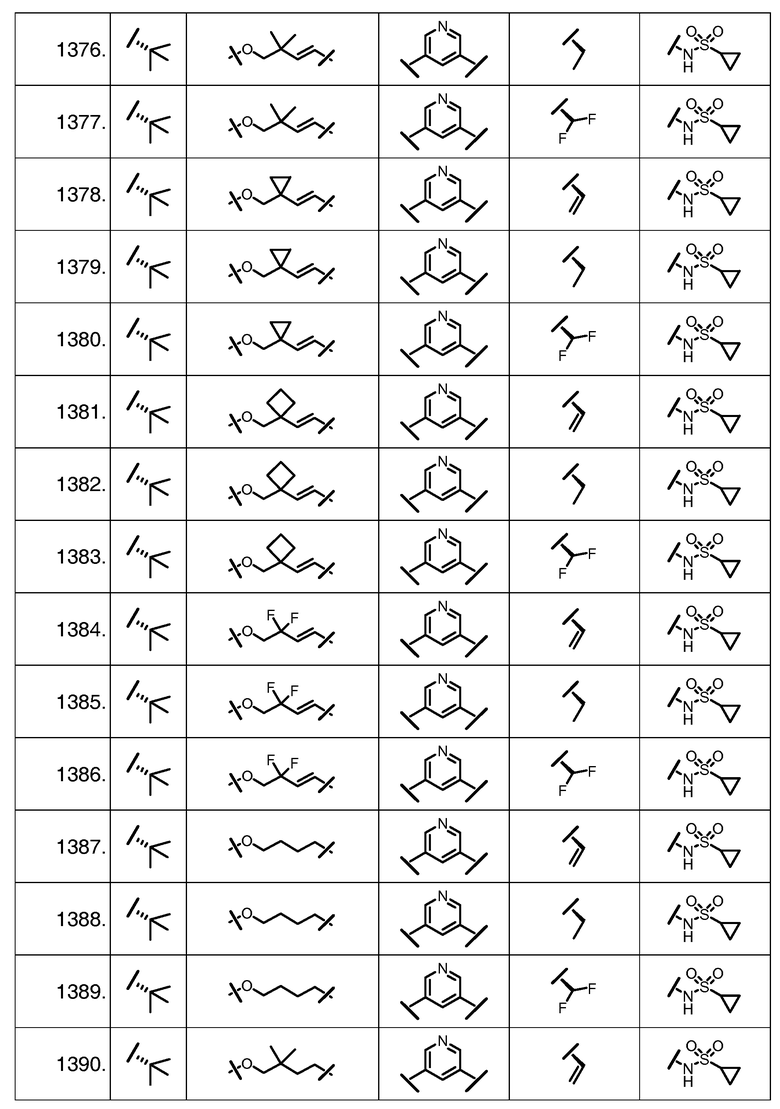
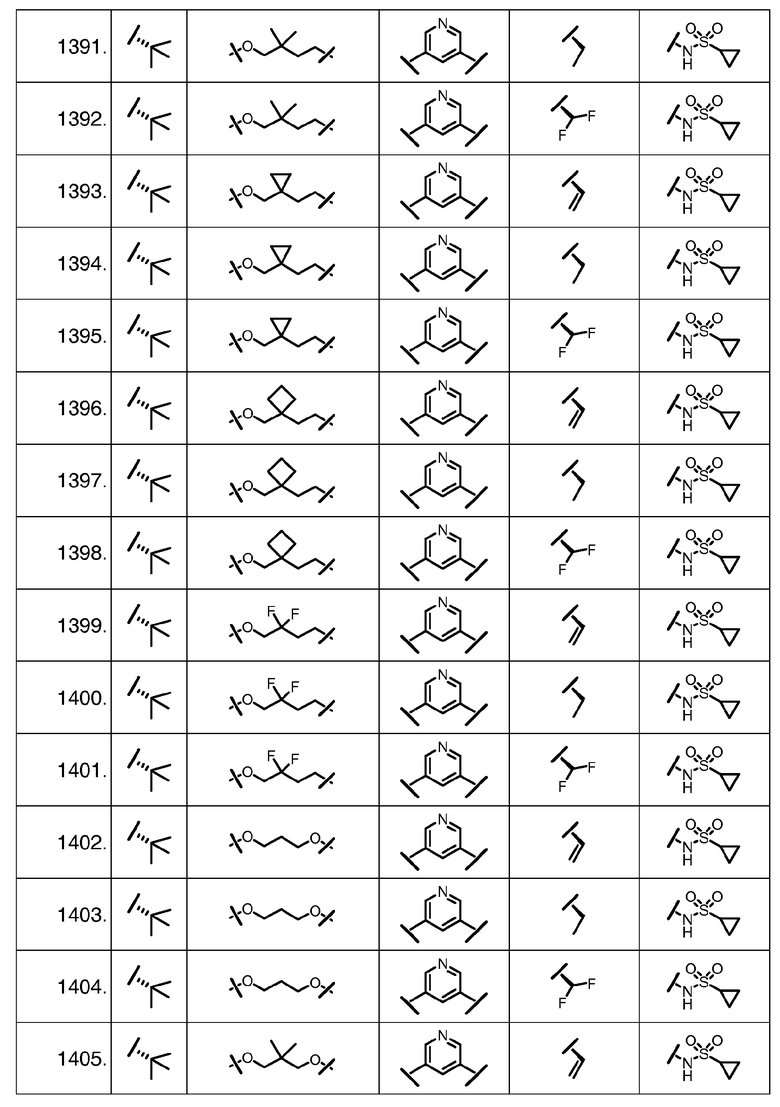
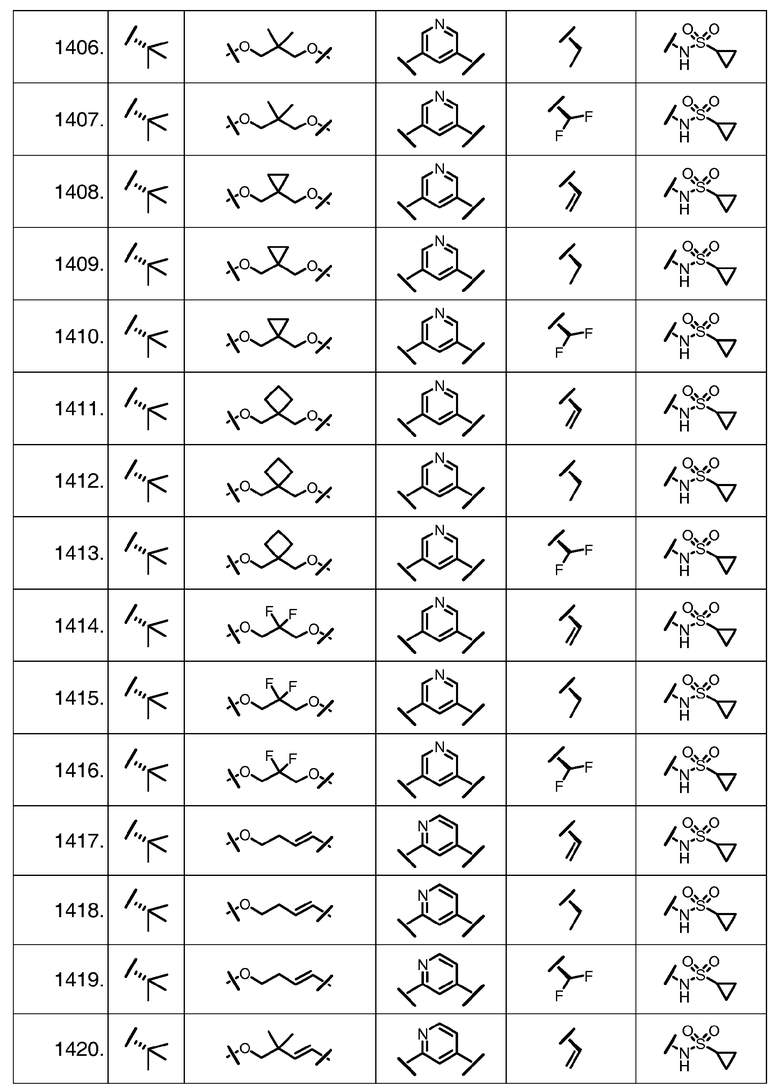
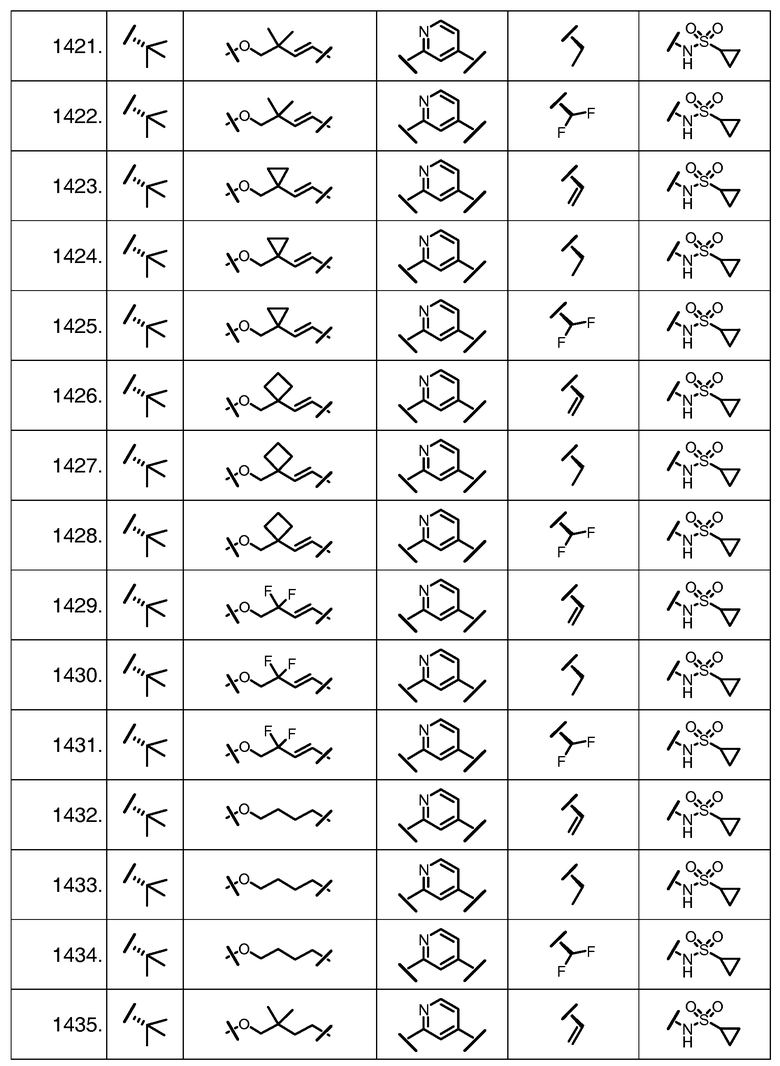
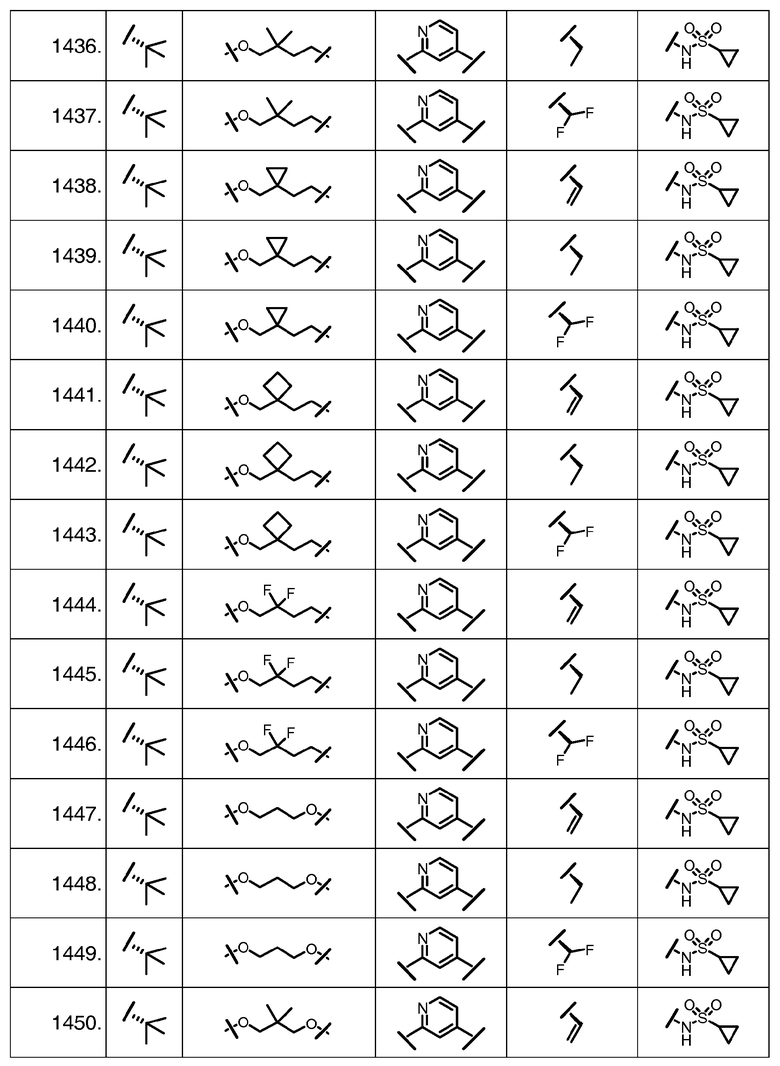
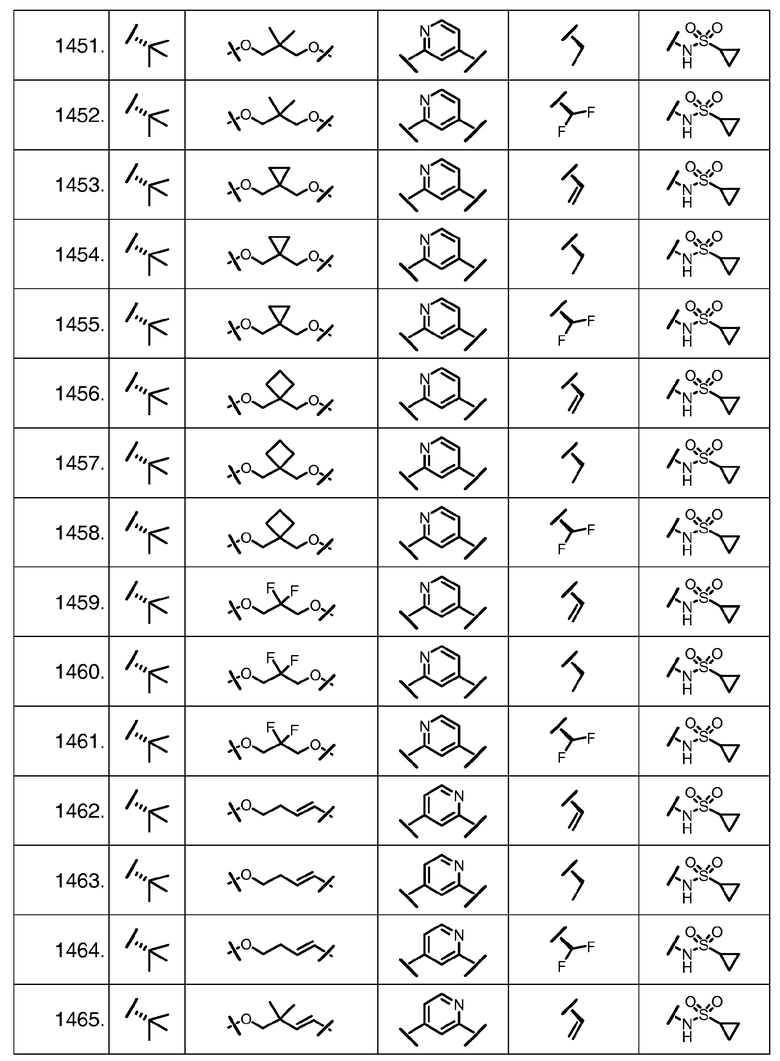
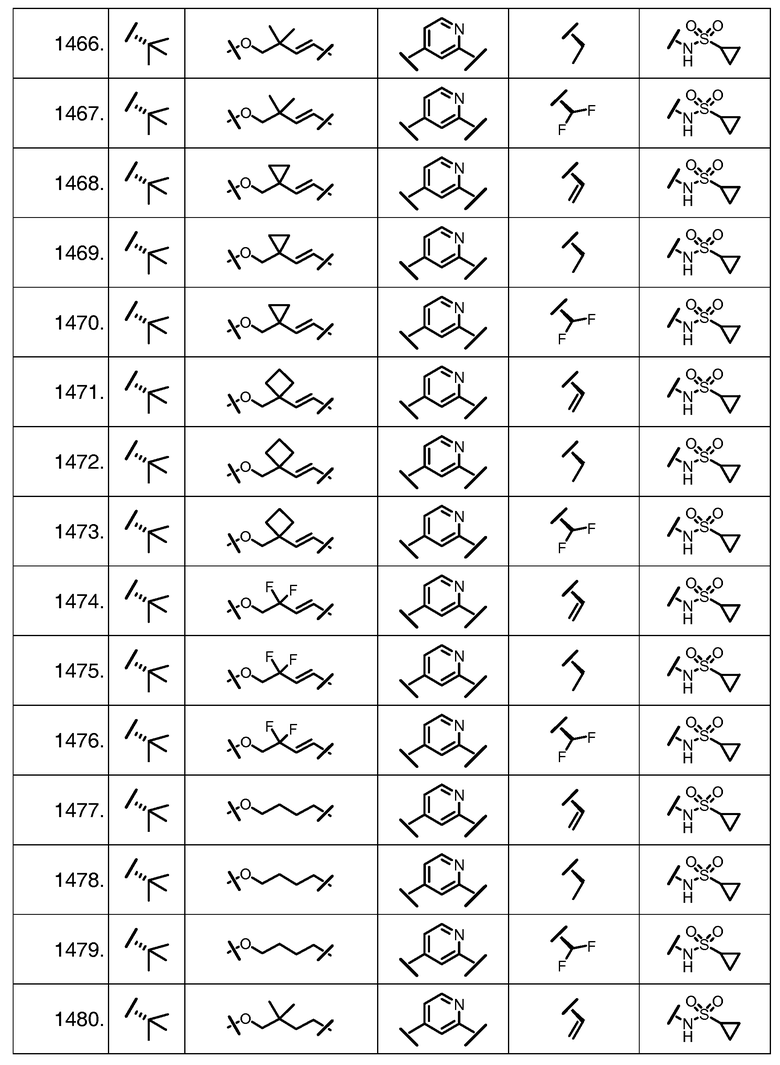
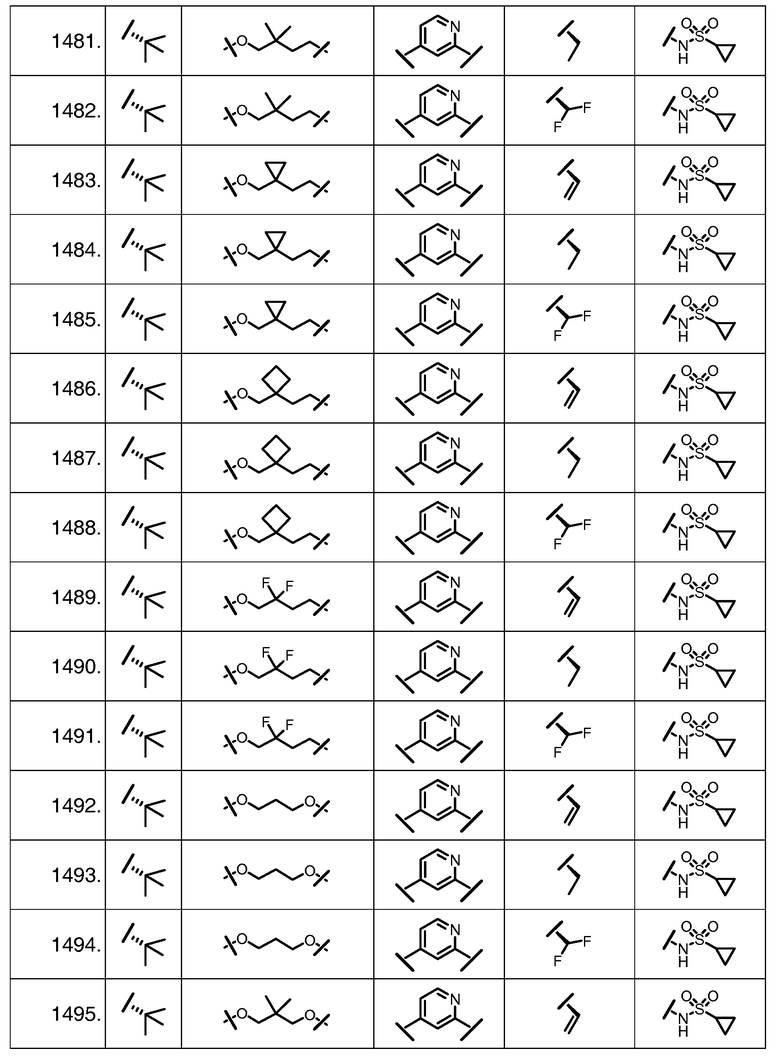
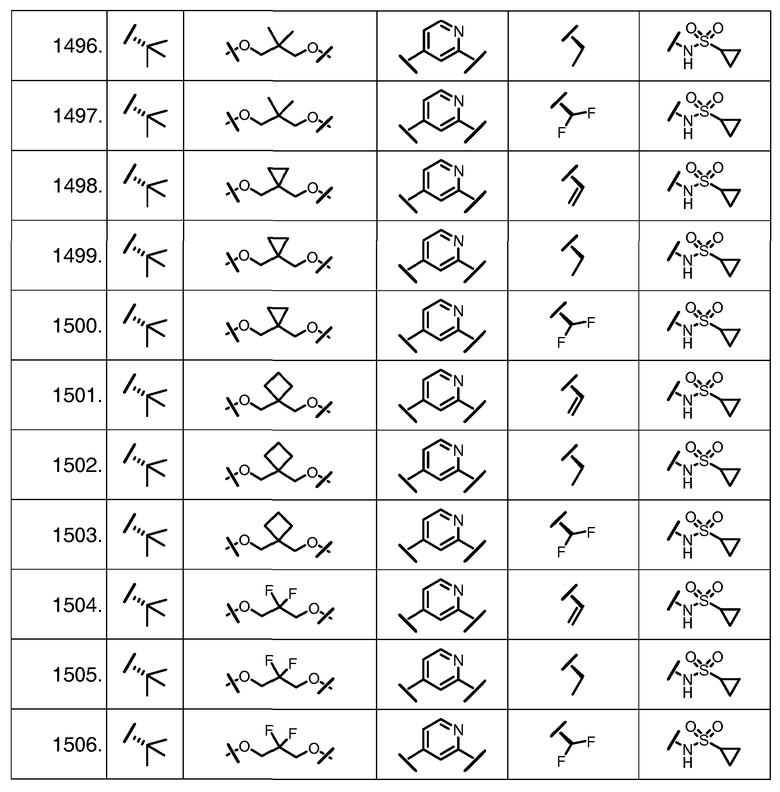
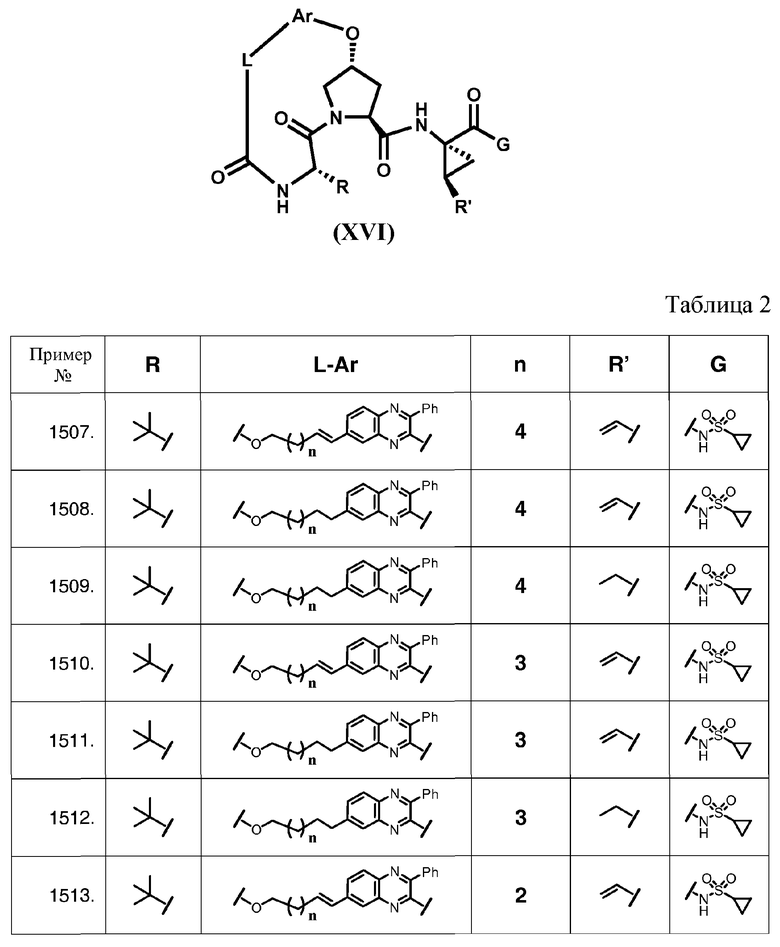
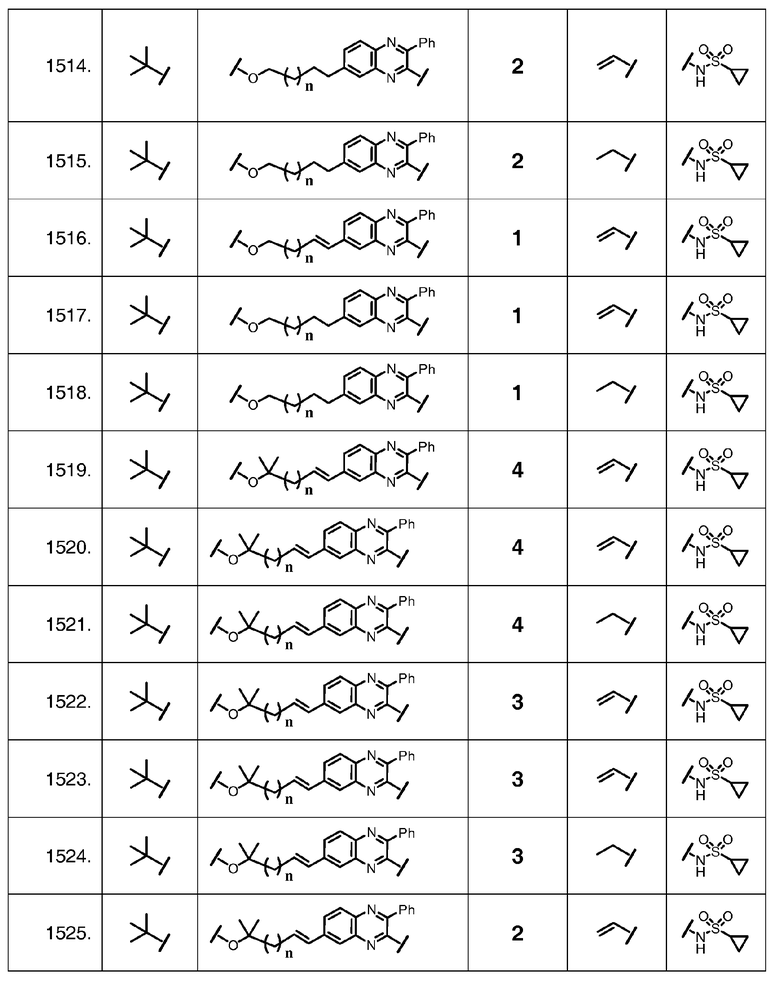
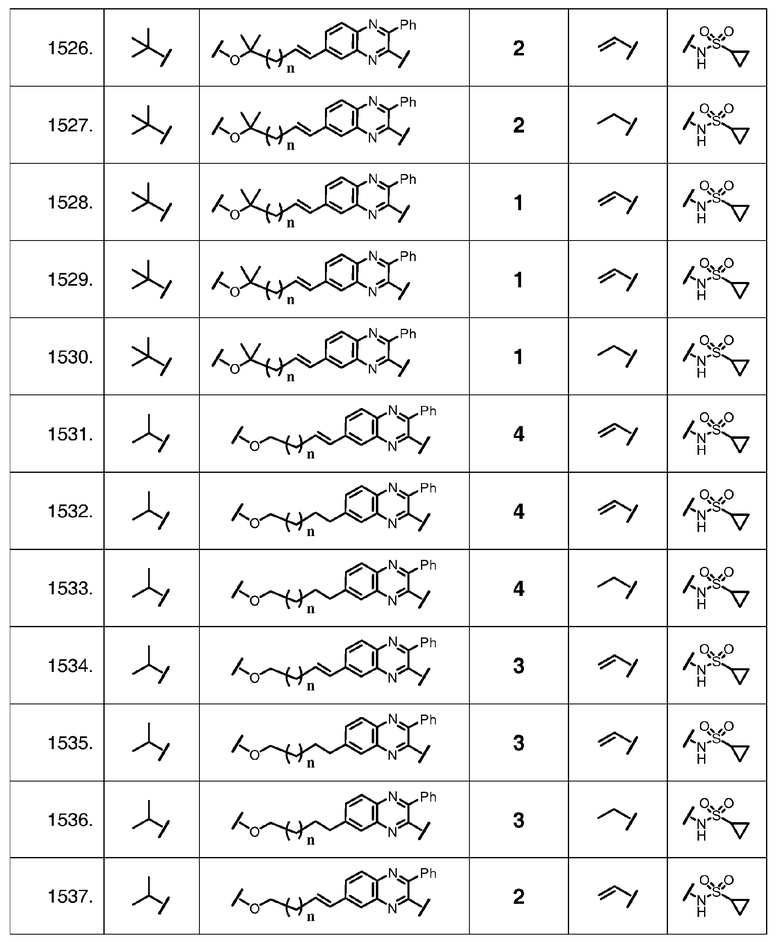
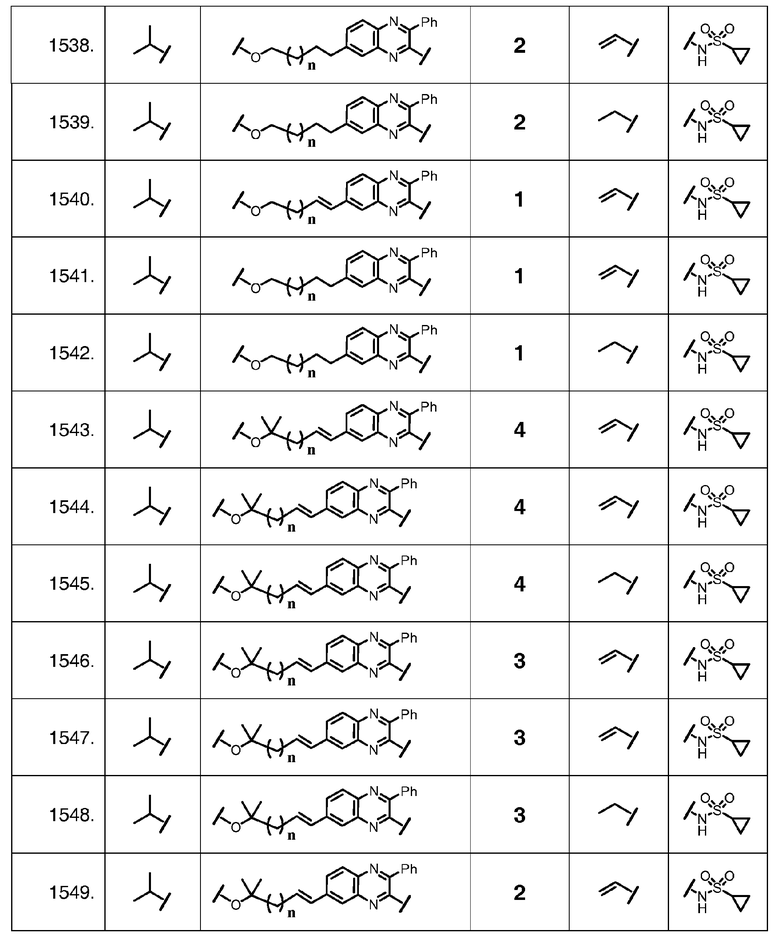
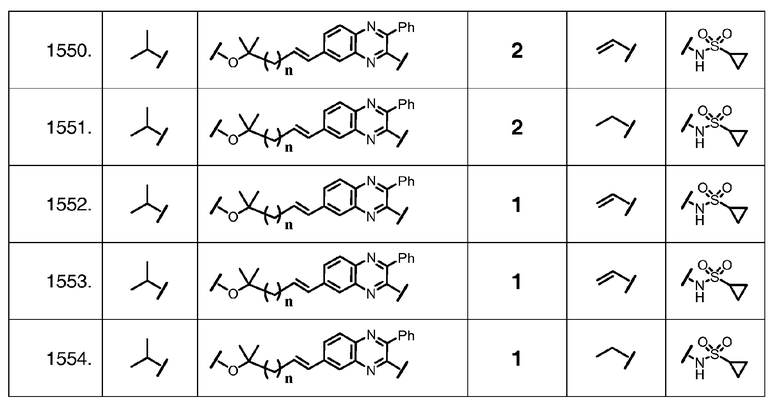
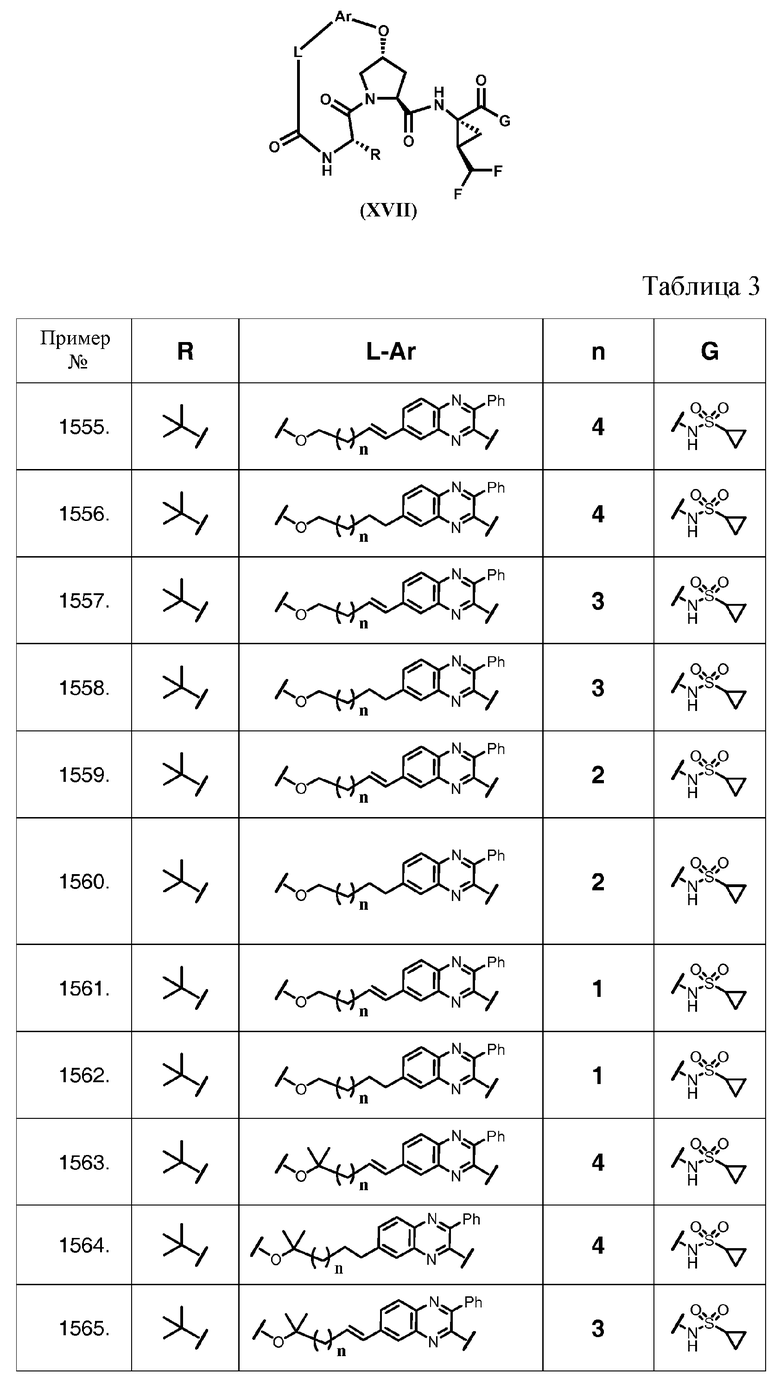
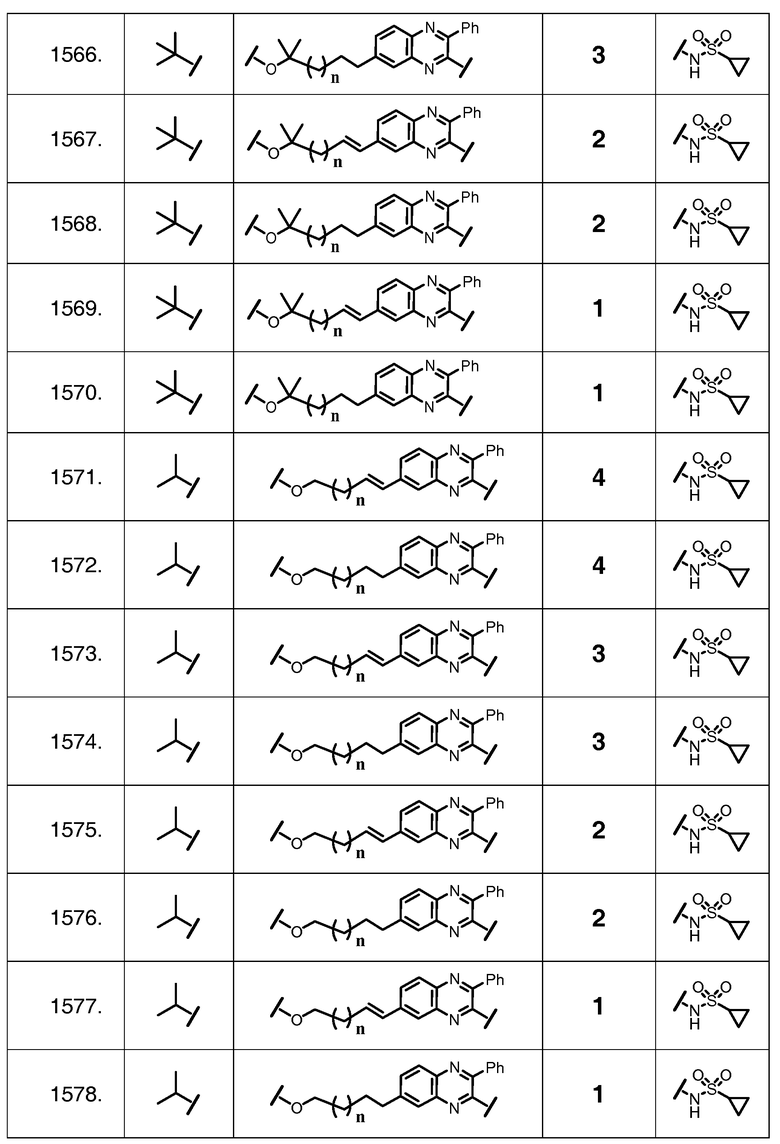
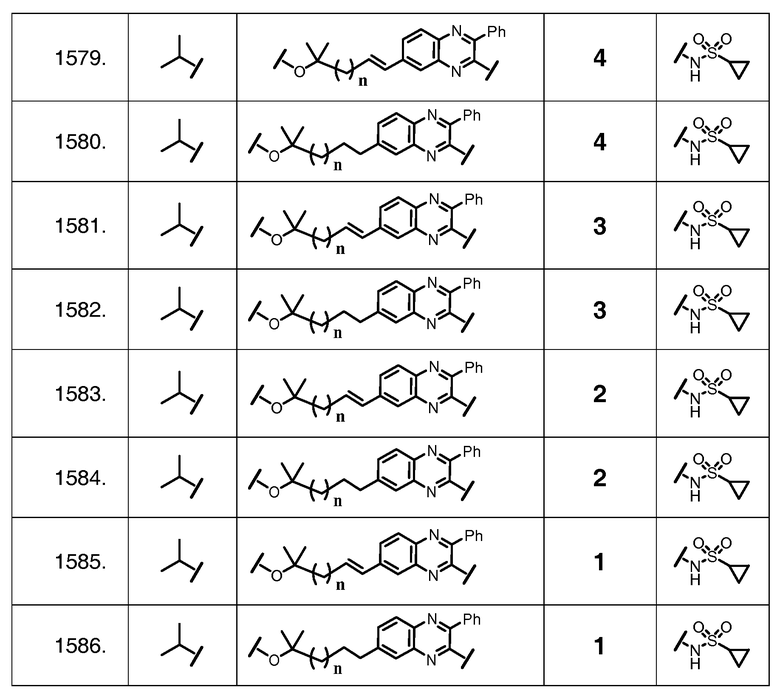
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИНОКСАЛИНИЛМАКРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ СЕРИНПРОТЕАЗЫ ВИРУСА ГЕПАТИТА С | 2007 |
|
RU2475494C2 |
| ИНГИБИРУЮЩИЕ HCV ХИМИЧЕСКИЕ СОЕДИНЕНИЯ, ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ПРИМЕНЕНИЯ | 2014 |
|
RU2671194C2 |
| ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2544010C2 |
| β-D-2'-ДЕЗОКСИ-2'-α-ФТОР-2'-β-С-ЗАМЕЩЕННЫЕ-2-МОДИФИЦИРОВАННЫЕ-N6-ЗАМЕЩЕННЫЕ ПУРИНОВЫЕ НУКЛЕОТИДЫ ДЛЯ ЛЕЧЕНИЯ ВЫЗВАННЫХ HCV ЗАБОЛЕВАНИЙ | 2016 |
|
RU2764767C2 |
| ПРОИЗВОДНЫЕ ЖЕЛЧНЫХ КИСЛОТ В КАЧЕСТВЕ АГОНИСТОВ FXR/TGR5 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2712099C2 |
| НЕЛИНЕЙНЫЕ САМОРАСЩЕПЛЯЮЩИЕСЯ ЛИНКЕРЫ И ИХ КОНЪЮГАТЫ | 2017 |
|
RU2755899C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗЫ | 2015 |
|
RU2686740C2 |
| СОЕДИНЕНИЯ ПУРИНОНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2013 |
|
RU2655388C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, ИХ КОМПОЗИЦИИ И ПРИМЕНЕНИЕ | 2014 |
|
RU2696585C2 |
| ПРОИЗВОДНЫЕ ЖЕЛЧНОЙ КИСЛОТЫ В КАЧЕСТВЕ АГОНИСТОВ FXR/TGR5 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2707280C2 |
Изобретение относится к соединениям формулы I

где A является -(C=O); L201 отсутствует; M отсутствует или его выбирают из O или NR1; где R1 выбирают при каждом его появлении из водорода; L101 выбирают из -C1-C8 алкилена, -C2-C8 алкенилена или C1-C8 алкилен-O; или -C1-C8 алкилена, -C2-C8 алкенилена или C1-C8 алкилен-O, каждый из которых замещен независимо 1, 2 или 3 заместителями, выбранными из C1-C8 алкила или галогена, или два атома водорода на атоме углерода -C1-C8 алкилена, -C2-C8 алкенилена или C1-C8 алкилен-O могут быть заменены заместителями, где два заместителя, взятые вместе, образуют C3-C12 циклоалкильное кольцо; Z101 является фенилом; фенилом, замещенным 1, 2 или 3 галогенами; или тиенилом; W101 отсутствует; X и Y, взятые вместе с атомами углерода, к которым они присоединены, образуют фенильное кольцо; R является -C1-C8 алкилом; R' является -C1-C8 алкилом, -C2-C8 алкенилом или CHQ1Q2; Q1 и Q2 каждый независимо является F, Cl или Br; G выбирают из -OH или -NHS(O)2-R2; R2 является -C3-C12 циклоалкилом; m является 1; m' является 1; и s является 1. Изобретение также относится к фармацевтической композиции, ингибирующей активность серинпротеазы, в частности активность протеазы NS3-NS4A вируса гепатита C (HCV), на основе указанных соединений. Технический результат: получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения гепатита C. 5 н. и 14 з.п. ф-лы, 3 табл., 1588 пр.
1. Соединение формулы I:
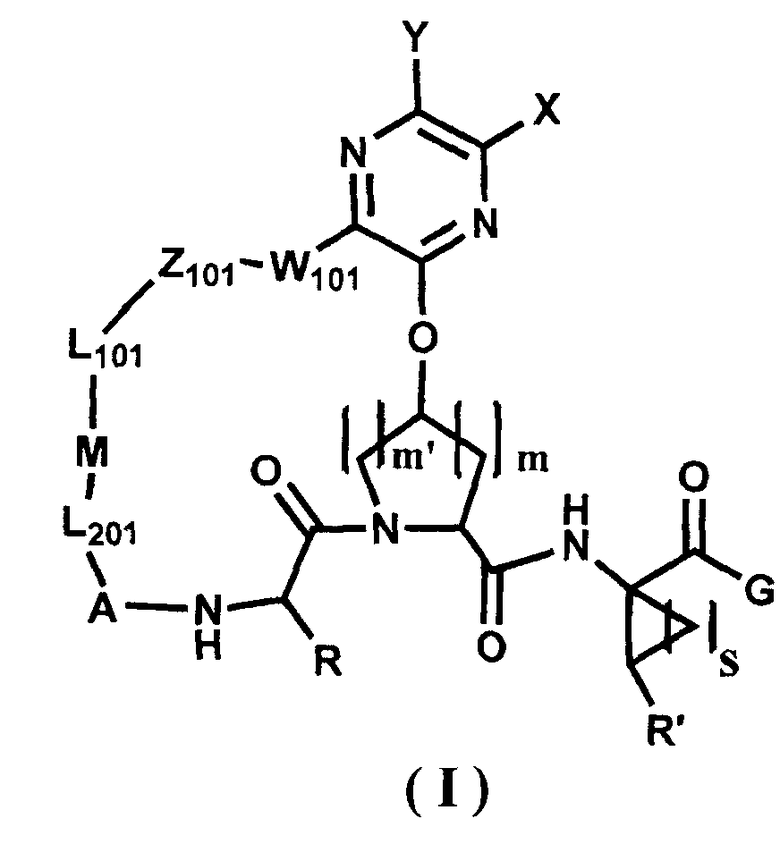
где A является -(C=O);
L201 отсутствует;
M отсутствует или его выбирают из O или NR1; где R1 выбирают при каждом его появлении из водорода;
L101 выбирают из -C1-C8 алкилена, -C2-C8 алкенилена или C1-C8 алкилен-O; или -C1-C8 алкилена, -C2-C8 алкенилена или C1-C8 алкилен-O, каждый из которых замещен независимо 1, 2 или 3 заместителями, выбранными из C1-C8 алкила или галогена, или два атома водорода на атоме углерода -C1-C8 алкилена, -C2-C8 алкенилена или C1-C8 алкилен-O могут быть заменены заместителями, где два заместителя, взятые вместе, образуют C3-C12 циклоалкильное кольцо;
Z101 является фенилом; фенилом, замещенным 1, 2 или 3 галогенами; или тиенилом;
W101 отсутствует;
X и Y, взятые вместе с атомами углерода, к которым они присоединены, образуют фенильное кольцо;
R является -C1-C8 алкилом;
R' является -C1-C8 алкилом, -C2-C8 алкенилом или CHQ1Q2;
Q1 и Q2 каждый независимо является F, Cl или Br;
G выбирают из -OH или -NHS(O)2-R2;
R2 является - C3-C12 циклоалкилом;
m является 1;
m' является 1; и
s является 1.
2. Соединение по п.1, где соединением является соединение формулы III:
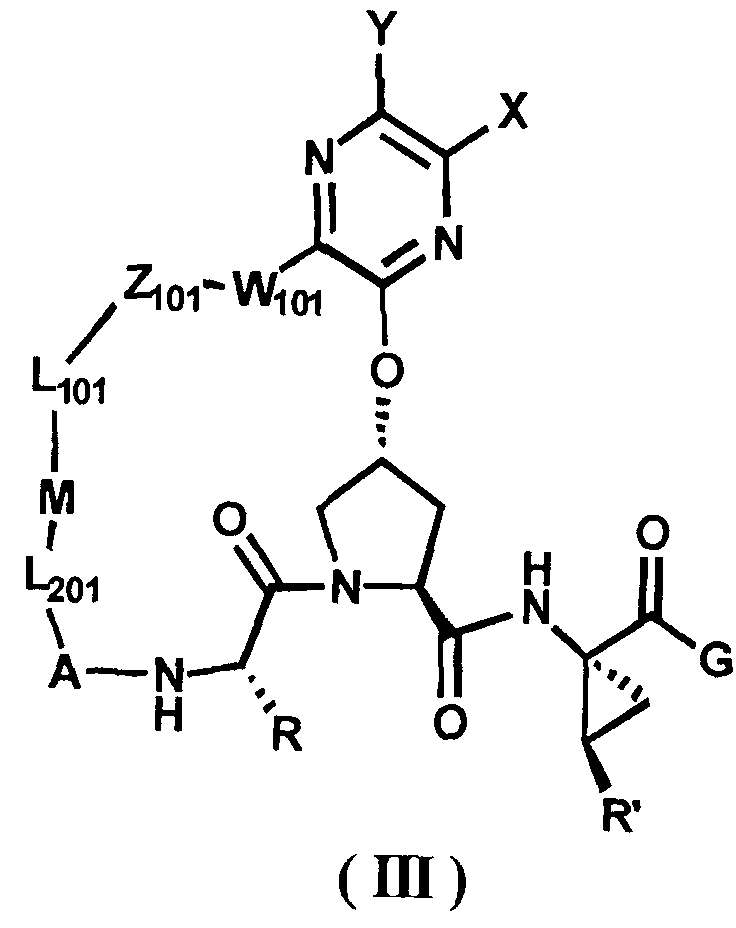
где R, R', A, L201, M, L101, Z101, W101, X, Y и G определены в п.1.
3. Соединение по п.1, где соединением является соединение формулы V:
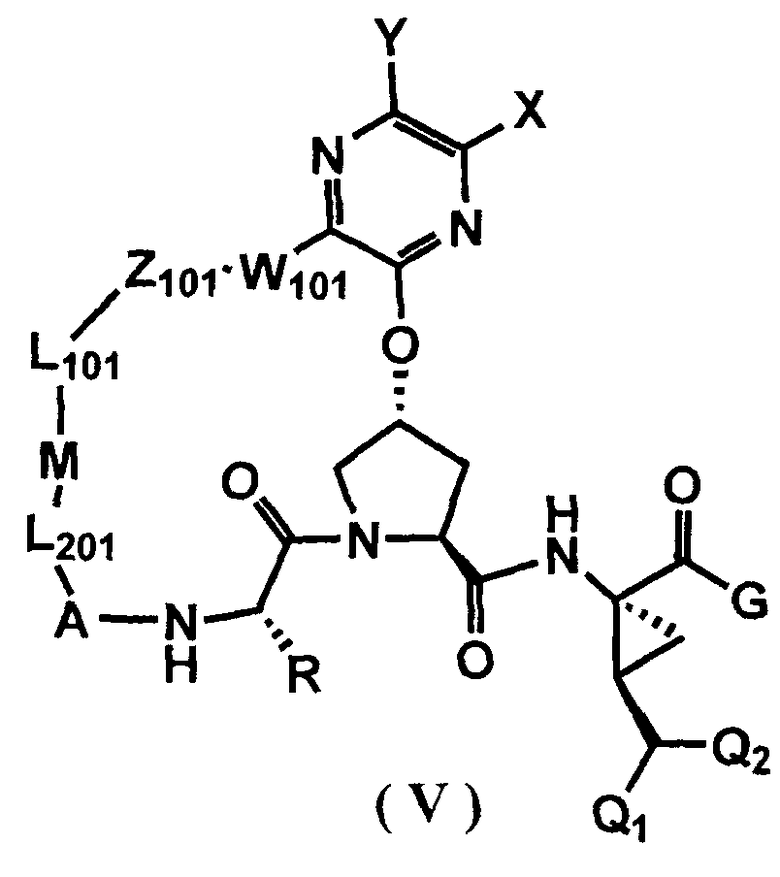
где Q1 и Q2 каждый независимо выбирают из F, Cl и Br, и R, A, L201, М, L101, Z101, W101, X, Y и G определены в п.1.
4. Соединение по п.1, где соединением является соединение формулы XI:
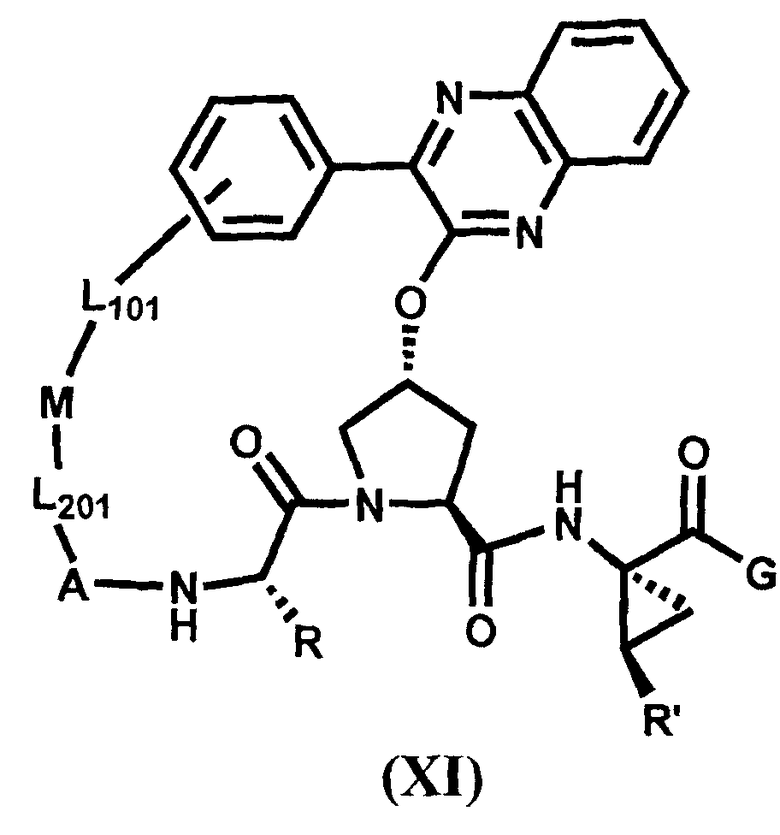
где R, R', A, L,201, M, L101 и G определены в п.1.
5. Соединение по п.1, где соединением является соединение формулы XII:
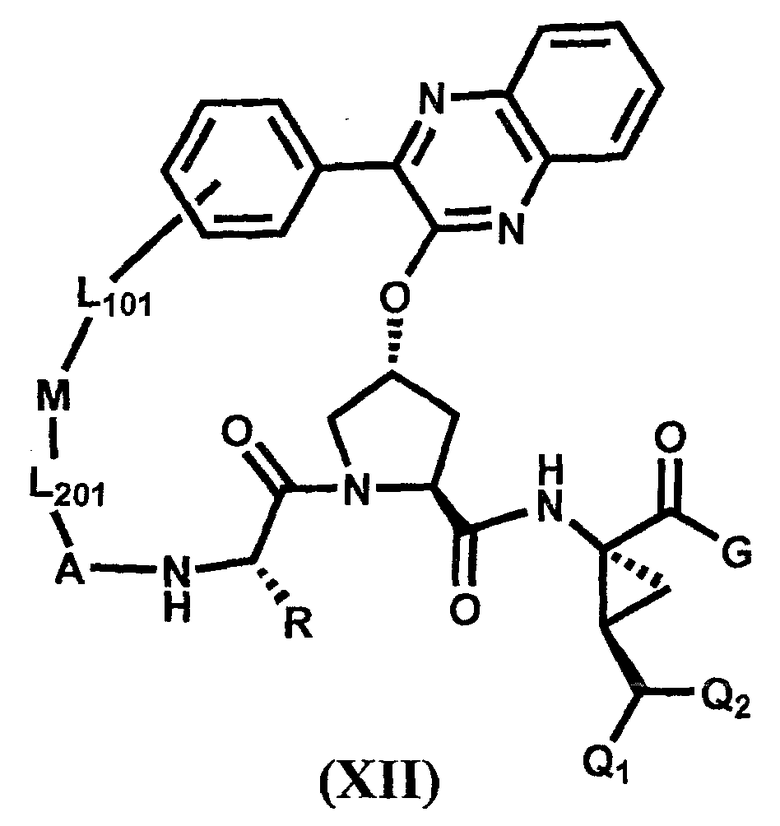
где Q1 и Q2 являются независимо фтором, хлором или бромом, и R, A, L201, M, L101 и G определены в п.1.
6. Соединение по п.1, такое как приведенное в таблице ниже:
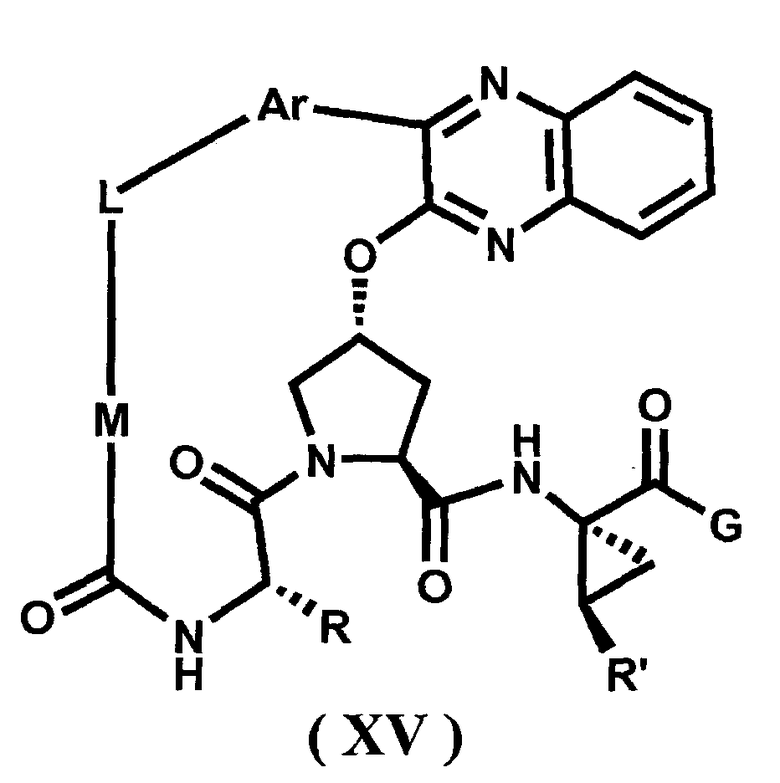
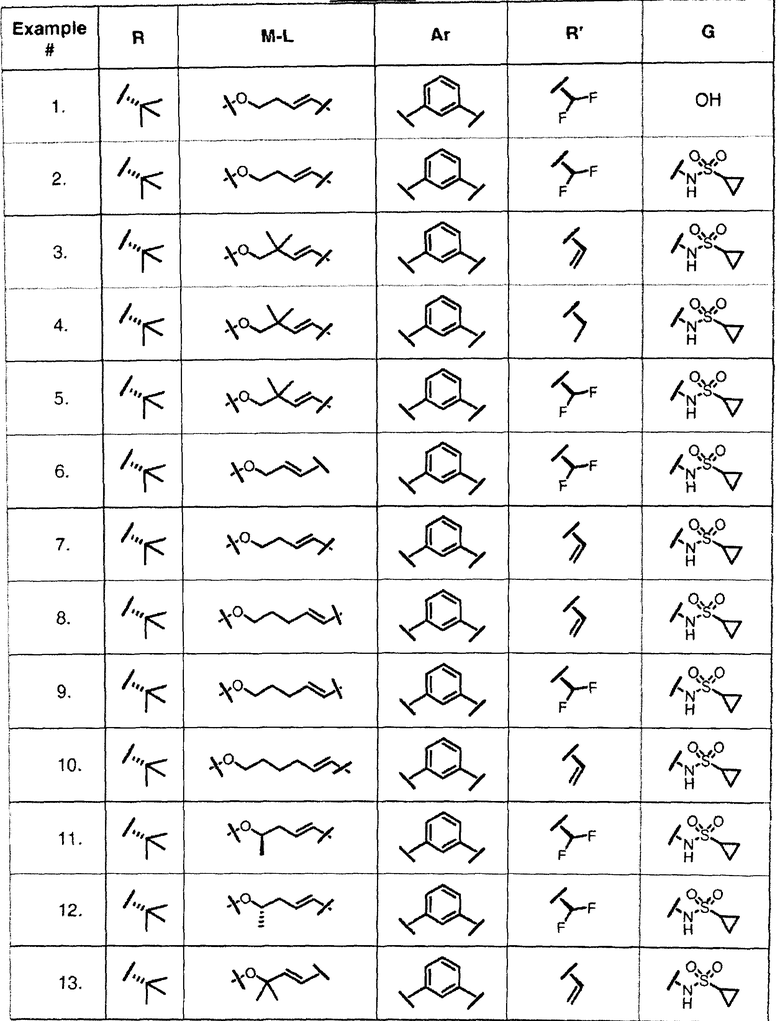
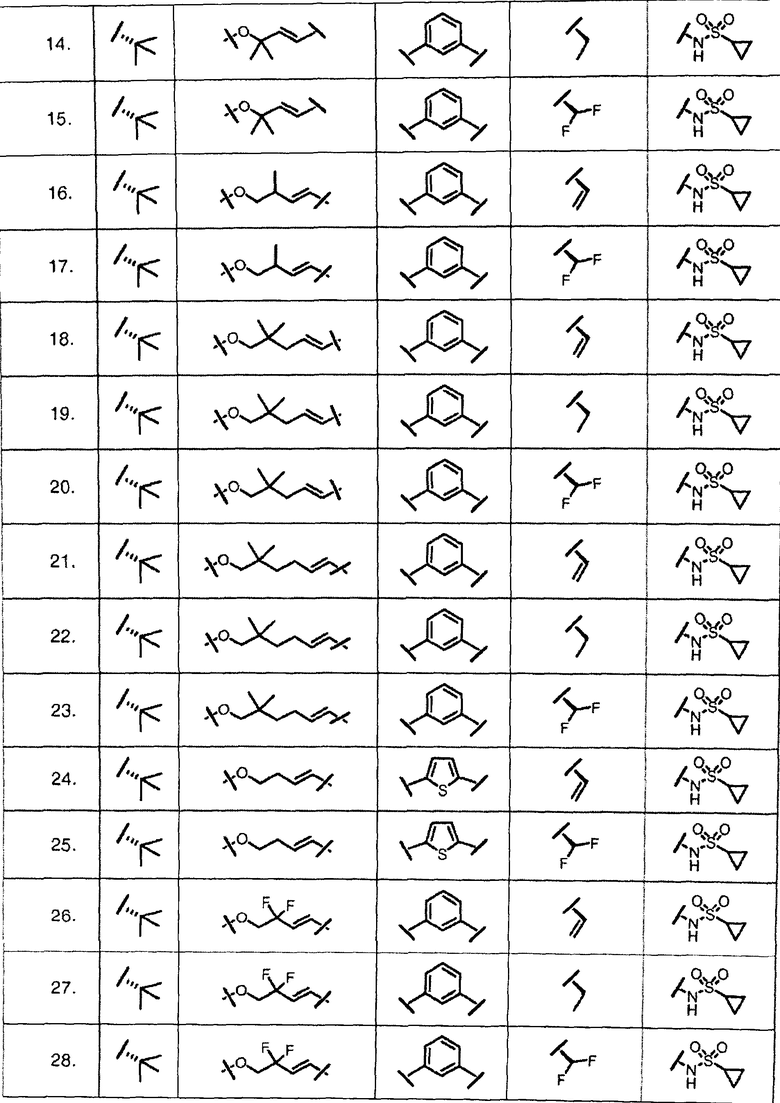
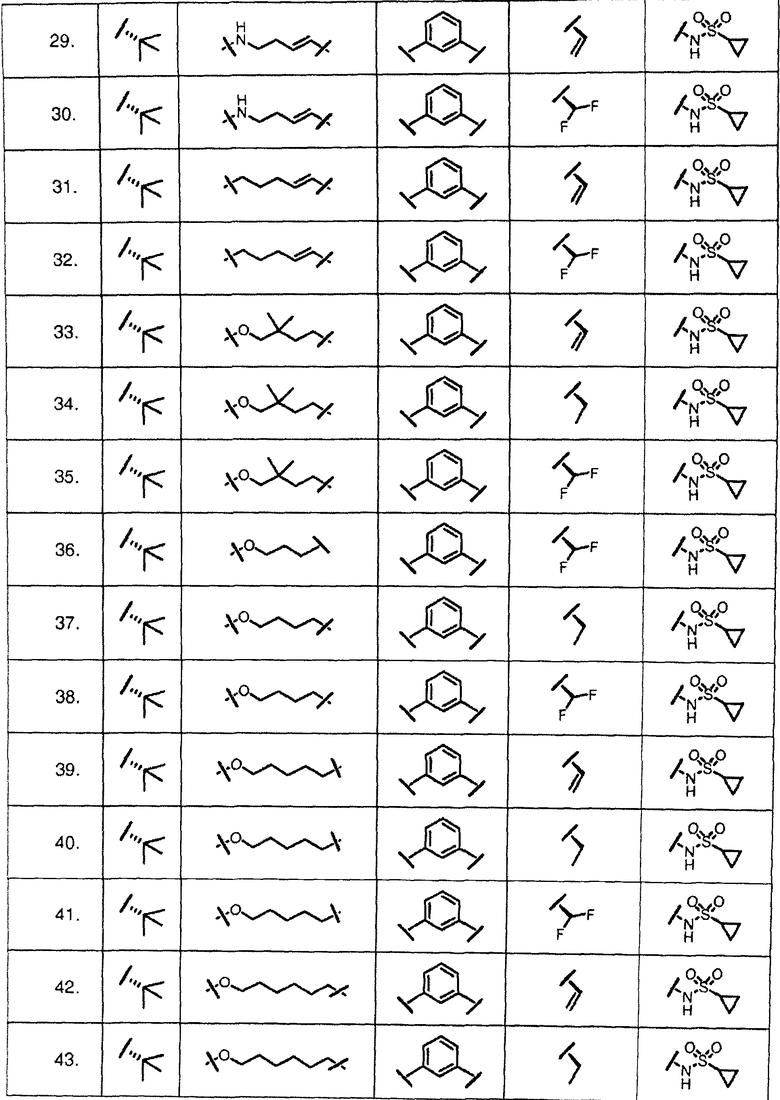
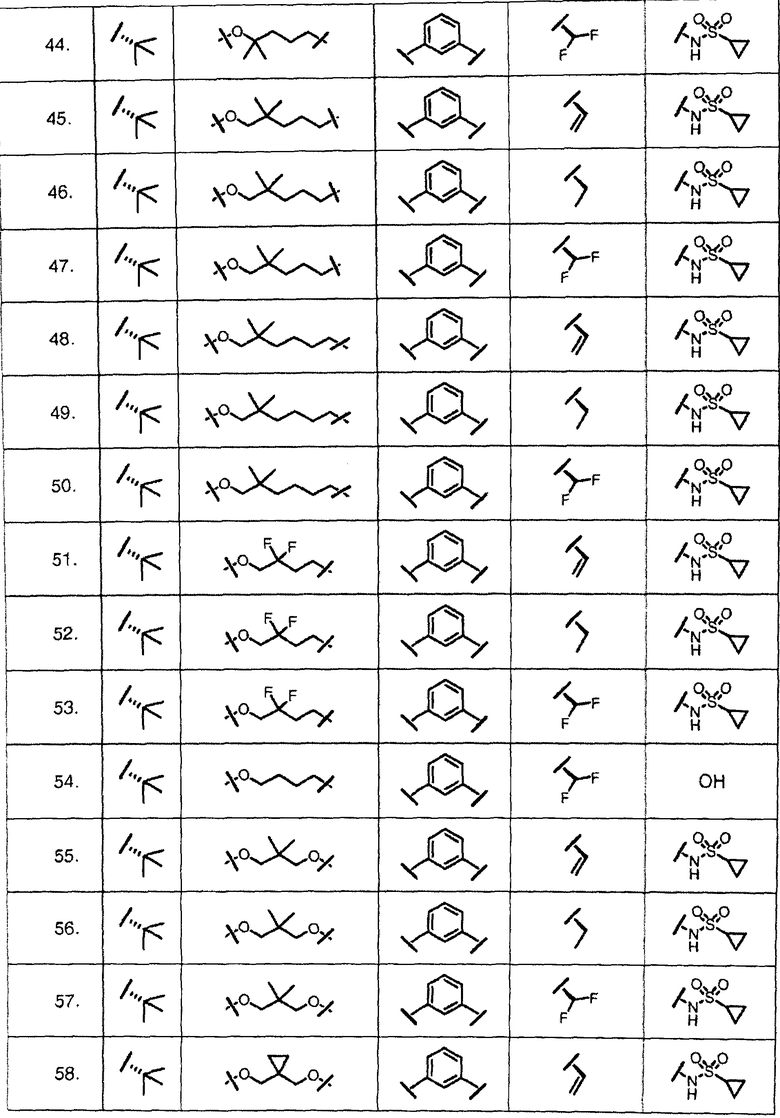
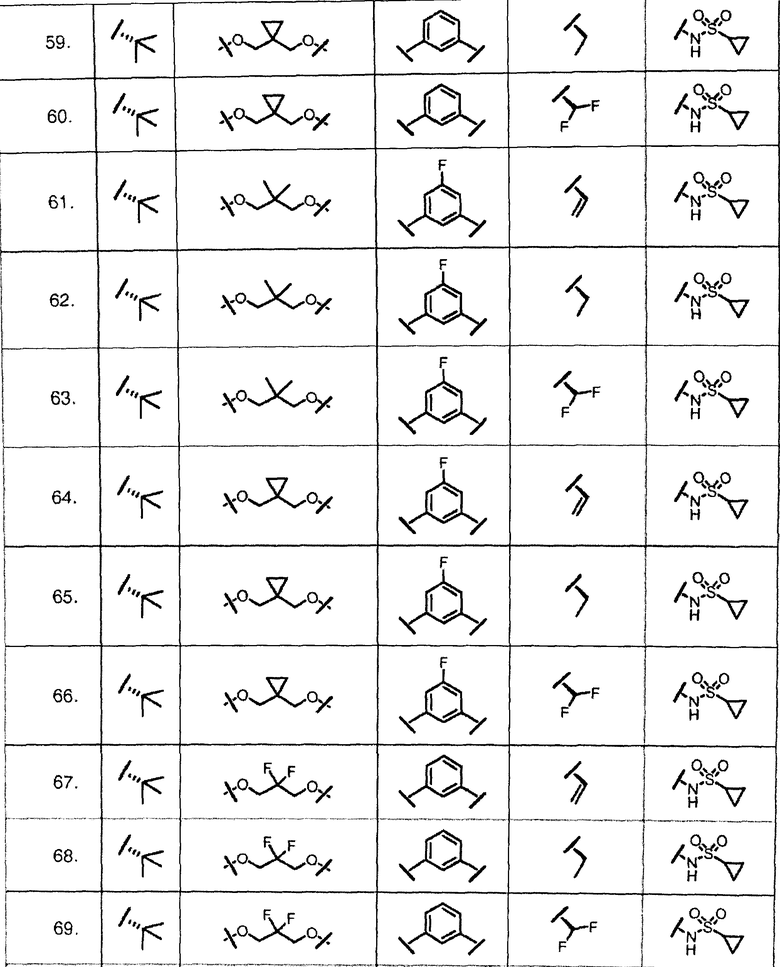
7. Фармацевтическая композиция, применяемая для лечения вирусной инфекции, представляющей собой гепатит C, у субъекта, включающая ингибирующее количество соединения по п.1, в комбинации с фармацевтически приемлемым носителем или эксципиентом.
8. Способ лечения вирусной инфекции, представляющей собой гепатит С, у субъекта, включающий введение субъекту ингибирующего количества фармацевтической композиции по п.7.
9. Способ ингибирования репликации вируса гепатита С, включающий введение количества фармацевтической композиции по п.7, которое ингибирует NS3 протеазу вируса гепатита С.
10. Способ по п.8, дополнительно включающий введение одновременно дополнительного средства против вируса гепатита С.
11. Способ по п.10, где указанное дополнительное средство против гепатита С выбирают из группы, состоящей из α-интерферона, β-интерферона, рибавирина и амантадина.
12. Способ по п.10, где указанное дополнительное средство против гепатита С является ингибитором геликазы, полимеразы, металлопротеазы или IRES вируса гепатита С.
13. Фармацевтическая композиция по п.1, дополнительно включающая еще одно анти-HCV средство.
14. Фармацевтическая композиция по п.7, дополнительно включающая средство, выбранное из интерферона, рибавирина, амантадина, другого ингибитора HCV протеазы, ингибитора HCV полимеразы, ингибитора HCV геликазы или ингибитора участка внутренней посадки рибосомы.
15. Фармацевтическая композиция по п.7, дополнительно включающая пегилированный интерферон.
16. Фармацевтическая композиция по п.7, дополнительно включающая еще одно противовирусное, антибактериальное, противогрибковое или противораковое средство или иммуномодулятор.
17. Композиция по п.7, дополнительно включающая ингибитор монооксигеназы цитохрома Р450 или его фармацевтически приемлемую соль.
18. Композиция по п.17, где ингибитором монооксигеназы цитохрома Р450 является ритонавир.
19. Способ совместного введения пациенту, нуждающемуся в лечении от вируса гепатита С, включающий введение ингибитора монооксигеназы цитохрома Р450 или его фармацевтически приемлемой соли и соединения по п.1.
| WO 2007016441 A1, 08.02.2007 | |||
| Гаечный ключ | 1927 |
|
SU9064A1 |
| US 20070054842 A1, 08.03.2007 | |||
| US 20070060510 A1, 15.03.2007 | |||
| US 20040106559 A1, 03.06.2004. | |||

