РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет по заявке США US 62/140,927, поданной 31 марта 2015 года, и заявке США US 62/287,267, поданной 26 января 2016 года. Описание вышеуказанных заявок полностью включено в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение в целом относится к соединениям, применимым в качестве модуляторов FXR/TGR5 и их фармацевтическим композициям. В частности, настоящее изобретение относится к производным желчных кислот и способам их получения и применения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Фарнезоидный Х-рецептор (FXR) представляет собой орфанный ядерный рецептор, первоначально идентифицированный из библиотеки кДНК печени крысы (ВМ. Forman, et al., Cell, 1995, 81(5), 687-693), который наиболее тесно связан с рецептором экдизона насекомых. FXR является членом семейства ядерных рецепторов лиганд-активируемых факторов транскрипции, которое включает рецепторы для стероидных, ретиноидных гормонов и гормонов щитовидной железы (DJ. Mangelsdorf, et al., Cell, 1995, 83(6), 841-850). Соответствующими физиологическими лигандами FXR являются желчные кислоты (D. Parks et al., Science, 1999, 284(5418), 1362-1365). Наиболее сильной является хенодезоксихолевая кислота (CDCA), которая регулирует экспрессию нескольких генов, участвующих в гомеостазе желчных кислот. Фарнезол и его производные, называемые вместе фарнезоидами, первоначально были описаны как активаторы ортолога крысы при высокой концентрации, но они не активируют рецептор человека или мыши. FXR экспрессируется в печени, по всему желудочно-кишечному тракту, включая пищевод, желудок, двенадцатиперстную кишку, тонкую кишку, толстую кишку, яичник, надпочечник и почку. Помимо контроля внутриклеточной экспрессии гена FXR, по-видимому, также участвует в паракринной и эндокринной сигнальной системе, активируя экспрессию цитокинового фактора роста фибробластов (J. Holt et al., Genes Dev., 2003, 17(13), 1581-1591; Т. Inagaki et al., Cell Metab., 2005, 2(4), 217-225).
Соединения, имеющие малые молекулы, которые действуют как модуляторы FXR, раскрыты в следующих публикациях: WO 2000/037077, WO 2003/015771, WO 2004/048349, WO 2007/076260, WO 2007/092751, WO 2007/140174, WO 2007/140183, WO 2008/051942, WO 2008/157270, WO 2009/005998, WO 2009/012125, WO 2008/025539 и WO 2008/025540. В последнее время были рассмотрены другие модуляторы FXR, имеющие малые молекулы (R.C. Buijsman et al., Curr. Med. Chem., 2005, 12, 1017-1075).
Рецептор TGR5 представляет собой рецептор, связанный с G-белком, определенный как рецептор клеточной поверхности, чувствительный к желчным кислотам (ЖК). Было обнаружено, что первичная структура TGR5 и его чувствительность к желчным кислотам является высококонсервативной среди TGR5 человека, быка, кролика, крысы и мыши, и таким образом указывает на то, что TGR5 имеет важные физиологические функции. Было обнаружено, что TGR5 широко распространен не только в лимфоидных тканях, но и в других тканях. Высокие уровни мРНК TGR5 были обнаружены в плаценте, селезенке и моноцитах/макрофагах. Показано, что желчные кислоты индуцируют интернализацию слитого белка TGR5 от клеточной мембраны к цитоплазме (Kawamata et al., J. Bio., Chem., 2003, 278, 9435). Было установлено, что TGR5 идентичен hGPCR19, представленному Takeda et al., FEBS Lett. 2002, 520, 97-101.
TGR5 связан с внутриклеточным накоплением цАМФ, который широко экспрессируется в различных типах клеток. В то время как активация данного мембранного рецептора в макрофагах снижает продуцирование провоспалительных цитокинов (Kawamata, Y., et al., J. Biol. Chem. 2003, 278, 9435-9440), стимуляция TGR5 жирными кислотами в адипоцитах и миоцитах увеличивает потребление энергии (Watanabe, M., et al., Nature, 2006, 439, 484-489). Последний эффект связан с цАМФ-зависимой индукцией йодотиронин-дейодиназы 2-го типа (D2), которая путем локального превращения Т4 в Т3 вызывает повышенную активность гормона щитовидной железы. В соответствии с ролью TGR5 в контроле энергетического метаболизма, у самок мышей с нокаутом TGR5 наблюдается значительное накопление жира с увеличением массы тела при постановке на диету с высоким содержанием жиров, что указывает на то, что отсутствие TGR5 снижает потребление энергии и вызывает ожирение (Maruyama, Т., et al., J. Endocrinol. 2006, 191, 197-205). Кроме того, в соответствии с вовлечением TGR5 в энергетический гомеостаз, сообщалось также, что активация мембранного рецептора желчными кислотами способствует продуцированию глюкагоноподобного пептида 1 (GLP-1) в мышиных энтероэндокринных клеточных линиях (Katsuma, S., Biochem. Biophys. Res. Commun., 2005, 329, 386-390). Исходя из всех вышеприведенных наблюдений TGR5 является привлекательной мишенью для лечения заболеваний, например, ожирения, диабета и метаболического синдрома.
В дополнение к применению агонистов TGR5 для лечения и профилактики метаболических заболеваний, соединения, которые модулируют модуляторы TGR5, также применимы для лечения других заболеваний, например, заболеваний центральной нервной системы, а также воспалительных заболеваний (WO 01/77325 и WO 02/84286). Модуляторы TGR5 также обеспечивают способы регулирования гомеостаза желчных кислот и холестерина, поглощения жирных кислот и расщепления белков и углеводов.
Существует потребность в разработке модуляторов FXR и/или TGR5 для лечения и профилактики заболеваний. Настоящее изобретение выявляет соединения, содержащие фрагменты аминокислот, мочевины, сульфомочевины или сульфонамида, которые модулируют FXR и/или TGR, а также способы применения этих соединений для лечения заболевания.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном аспекте изобретение относится к соединениям, представленным формулой I, или к фармацевтически приемлемым солям, стереоизомерам, сольватам, гидратам или их комбинации:
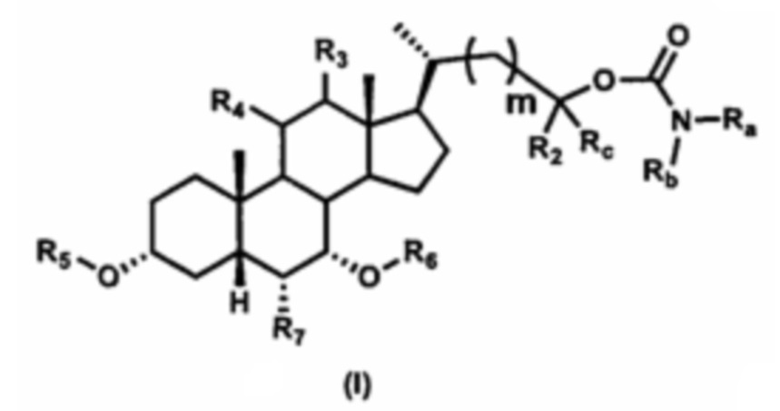
где:
Ra выбран из группы, состоящей из:
1) водорода;
2) замещенного или незамещенного -C1-C6 алкокси;
3) замещенного или незамещенного -C1-C8 алкила;
4) замещенного или незамещенного -C2-C8 алкенила;
5) замещенного или незамещенного -C2-C8 алкинила;
6) замещенного или незамещенного арилалкила;
7) замещенного или незамещенного арила.
Rb выбран из группы, состоящей из:
1) водорода;
2) замещенного или незамещенного -C1-C8 алкила;
3) замещенного или незамещенного -C2-C8 алкенила;
4) замещенного или незамещенного -C2-C8 алкинила;
5) замещенного или незамещенного арилалкила;
6) замещенного или незамещенного арила;
7) -C(O)NR10R11;
8) -C(O)NHSO2R1;
9) -SO2R1; и
10) -C(O)R1;
Альтернативно, Ra и Rb вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо.
R1 выбран из группы, состоящей из:
1) галогена;
2) гидроксила;
3) замещенного или незамещенного -C1-C8 алкила;
4) замещенного или незамещенного -C2-C8 алкенила;
5) замещенного или незамещенного -C2-C8 алкинила;
6) замещенного или незамещенного -C3-C8 циклоалкила;
7) замещенного или незамещенного арила;
8) замещенного или незамещенного арилалкила;
9) замещенного или незамещенного гетероциклоалкила;
10) замещенного или незамещенного гетероарила;
11) замещенного или незамещенного гетероарилалкила; и
12) -NR10R11.
R2 выбран из группы, состоящей из:
1) водорода;
2) замещенного или незамещенного -C1-C8 алкила;
3) замещенного или незамещенного -C2-C8 алкенила;
4) замещенного или незамещенного -C2-C8 алкинила;
5) замещенного или незамещенного арилалкила; и
6) замещенного или незамещенного арила.
Предпочтительно R2 представляет собой водород или метил.
Rc выбран из группы, состоящей из:
1) водорода;
2) замещенного или незамещенного -C1-C8 алкила;
3) замещенного или незамещенного -C2-C8 алкенила;
4) замещенного или незамещенного -C2-C8 алкинила;
5) замещенного или незамещенного арилалкила;
6) замещенного или незамещенного арила.
Альтернативно, R2 и Rc вместе с атомом углерода, к которому они присоединены, образуют циклическое кольцо, предпочтительно циклоалкил или циклоалкилен.
m выбран из 0, 1, 2 и 3, предпочтительно m составляет от 0 до 2.
R3 представляет собой водород, гидроксил, -OSO3H, -OSO3-, -ОАс, -ОРО3Н2 или -ОРО32-; предпочтительно R3 представляет собой водород.
R4 представляет собой водород, галоген, CN, N3, гидроксил, -OSO3H, -OSO3-, -ОАс, -ОРО3Н2, -ОРО32-, -SR2 или -NHR2, где R2 является таким, как определено ранее; предпочтительно R4 представляет собой водород.
Альтернативно, R3 и R4 вместе с атомами углерода, к которым они присоединены, образуют -СН=СН-, или циклоалкильное кольцо, или гетероциклоалкильное кольцо, такое как, но не ограничиваясь указанными, циклопропил или эпоксид.
R5 и R6 независимо выбраны из водорода или гидроксизащитной группы, такой как, но не ограничиваясь указанными, ацетил, триметилсилил или бензил; предпочтительно R5 и R6 представляют собой водород.
R7 выбран из группы, состоящей из:
1) водорода;
2) галогена;
3) замещенного или незамещенного -C1-C8 алкила;
4) замещенного или незамещенного -C2-C8 алкенила;
5) замещенного или незамещенного -C2-C8 алкинила; и
6) замещенного или незамещенного -C3-C8 циклоалкила; предпочтительно R7 представляет собой C1-C4 алкил, более предпочтительно R7 представляет собой этил.
R10 и R11 каждый независимо выбран из водорода, замещенного или незамещенного -C1-C8 алкила, замещенного или незамещенного -C2-C8 алкенила, замещенного или незамещенного -C2-C8 алкинила и замещенного или незамещенного -C3-C8 циклоалкила, или R10 и R11 вместе с атомом азота, к которому они присоединены, образуют гетероциклическое кольцо.
Каждая предпочтительная группа, указанная выше, может быть взята в сочетании с одной, любой или всеми другими предпочтительными группами.
В другом варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения, или комбинацию соединений по настоящему изобретению, или фармацевтически приемлемую соль, стереоизомер, сольват, гидрат или их комбинацию в сочетании с фармацевтически приемлемым носителем или эксципиентом.
В другом варианте осуществления настоящее изобретение относится к способу профилактики или лечения FXR-опосредованного заболевания или состояния. Способ включает введение терапевтически эффективного количества соединения формулы (I). Настоящее изобретение также относится к применению соединения формулы (I) для получения лекарственного средства для профилактики или лечения FXR-опосредованного заболевания или состояния.
В еще одном варианте осуществления настоящее изобретение относится к способу профилактики или лечения TGR5-опосредованного заболевания или состояния. Способ включает введение терапевтически эффективного количества соединения формулы (I). Настоящее изобретение также относится к применению соединения формулы (I) для получения лекарственного средства для профилактики или лечения TGR5-опосредованного заболевания или состояния.
В некоторых вариантах осуществления заболевание, которое связано с модулированием рецептора TGR5, выбрано из метаболического заболевания, воспалительного заболевания, заболевания печени, аутоиммунного заболевания, заболевания сердца, заболевания почек, ракового заболевания и заболевания желудочно-кишечного тракта.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
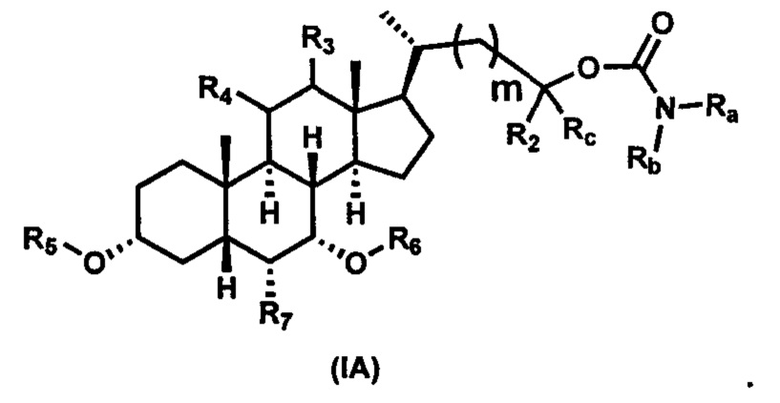
Первым вариантом осуществления изобретения является соединение, представленное формулой I, как описано выше, или его фармацевтически приемлемая соль, гидрат, сольват, сложный эфир или пролекарство. В предпочтительных соединениях формулы I R2, Rc, R3, R4, R5 и R6 каждый представляет собой водород и R7 представляет собой этил.
В предпочтительных вариантах осуществления соединения по изобретению имеют стереохимию, указанную в формуле IA:
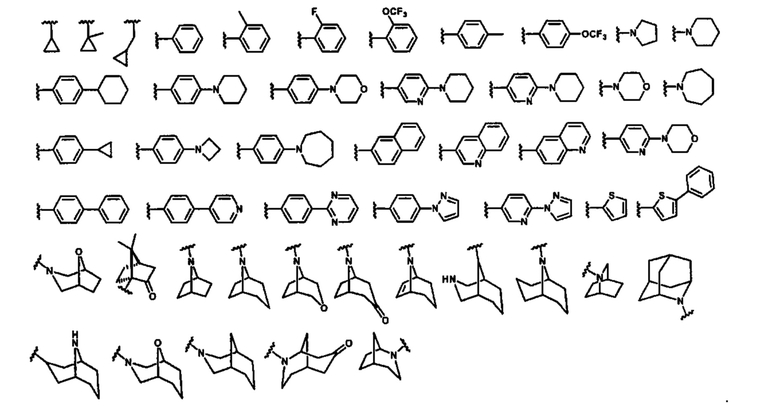
В некоторых вариантах осуществления соединений по изобретению Ra представляет собой C1-C4-алкил; галогенированный C1-C4-алкил; C1-C4-алкенил; фенил-C1-C4-алкил; замещенный или незамещенный C3-C6-циклоалкил; C1-C6-циклоалкил-C1-C4-алкил; гетероарил, такой как 5- или 6-членный гетероарил; или замещенный или незамещенный арил, такой как замещенный или незамещенный фенил или нафтил. В этом варианте осуществления Rb предпочтительно представляет собой водород или C1-C4-алкил, более предпочтительно водород или метил.
В некоторых вариантах осуществления соединений по изобретению Ra выбран из группы, состоящей из метила, этила, изопропила, бутила, трет-бутила, пропила, бензила, винила, аллила, CF3,  ,
,  ,
, 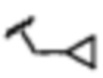 ,
, 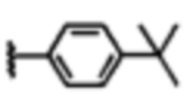 и
и 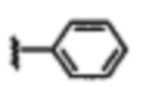 .
.
В других вариантах осуществления соединений по изобретению Ra, Rb и атом азота, к которому они присоединены, образуют гетероциклоалкильное или гетероциклоалкенильное кольцо, предпочтительно 3-8-членное гетероциклоалкильное или 3-8-членное гетероциклоалкенильное и более предпочтительно 3-6-членный гетероциклоалкил или 3-6-членный гетероциклоалкенил. В некоторых вариантах осуществления Ra, Rb и атом азота, к которому они присоединены, образуют C3-C8-гетероциклоалкильное или C3-C8-гетероциклоалкенильное кольцо, более предпочтительно C3-C6-гетероциклоалкильное или C3-C6-гетероциклоалкенильное кольцо. В некоторых вариантах осуществления Ra, Rb и атом азота, к которому они присоединены, образуют кольцо, выбранное из:
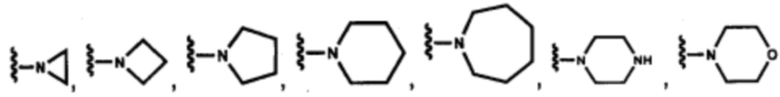 и
и 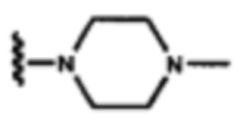 .
.
В некоторых вариантах осуществления соединений по изобретению Rb представляет собой -C(O)NHSO2R1, -SO2R1 или -C(O)R1. R1 предпочтительно представляет собой амино, алкиламино, диалкиламино, галоген, C1-C4-алкил; галогенированный C1-C4-алкил; C1-C4-алкенил; фенил-C1-C4-алкил; замещенный или незамещенный C3-C6-циклоалкил; C3-C6-циклоалкил-C1-C4-алкил; C3-C6-гетероциклоалкил; C3-C6-гетероциклоалкил-C1-C4-алкил; гетероарил, такой как 5- или 6-членный гетероарил; или замещенный или незамещенный арил, такой как замещенный или незамещенный фенил или нафтил, включая 4-трет-бутилфенил. Ra предпочтительно представляет собой водород или C1-C4-алкил, более предпочтительно водород или метил и наиболее предпочтительно водород.
В некоторых вариантах осуществления соединений по изобретению R1 выбран из группы, состоящей из фтора, амино, метила, этила, изопропила, бутила, трет-бутила, пропила, бензила, аллила, винила, CF3; циклогексила, циклопентила и групп, перечисленных ниже:
Второй вариант осуществления изобретения представляет собой соединение, представленное формулой II, или его фармацевтически приемлемую соль, гидрат, сольват, сложный эфир или пролекарство,
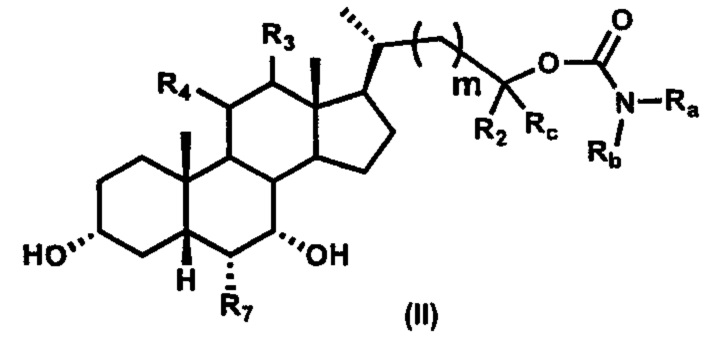
где Ra, Rb, Rc, R2, R3, R4, R7 и m являются такими, как определено ранее.
Третий вариант осуществления изобретения представляет собой соединение, представленное формулой III, или его фармацевтически приемлемую соль, гидрат, сольват, сложный эфир или пролекарство,
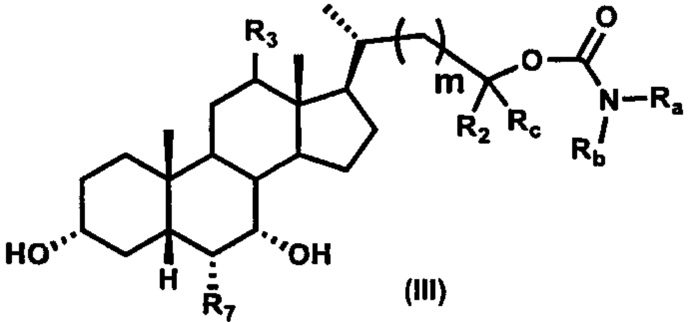
где Ra, Rb, Rc, R2, R3, R7 и m являются такими, как определено ранее.
Иллюстративные структуры формулы (III) включают, но не ограничиваются указанными, формулу (от III-1 до III-18), где Ra, Rb, Rc, R1, R2, R7 и m являются такими, как определено ранее:
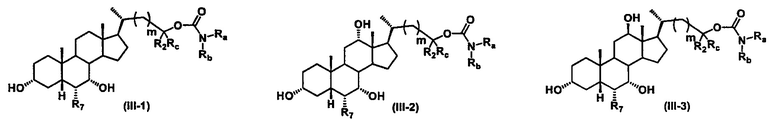
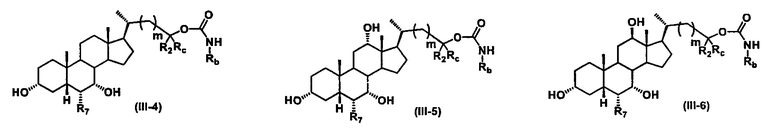
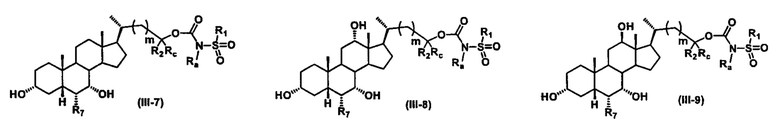
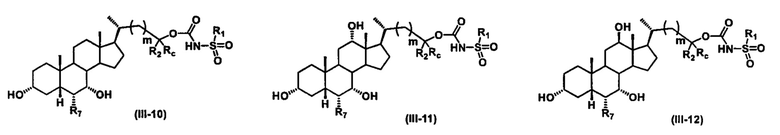
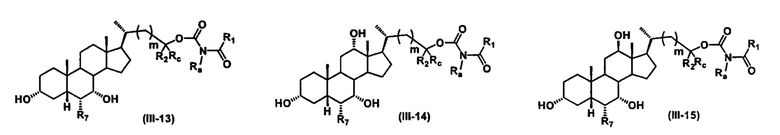
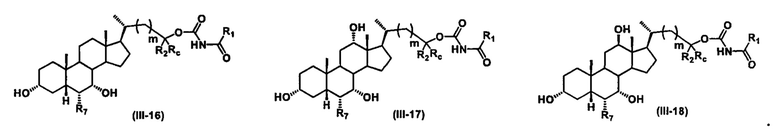
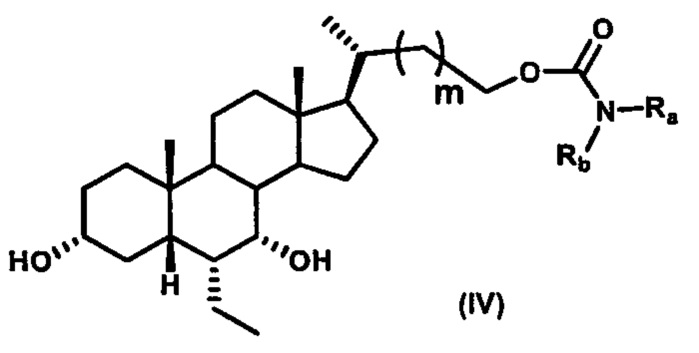
Четвертый вариант осуществления изобретения представляет собой соединение, представленное формулой IV, или его фармацевтически приемлемую соль, сольват, гидрат, сложный эфир или пролекарство,
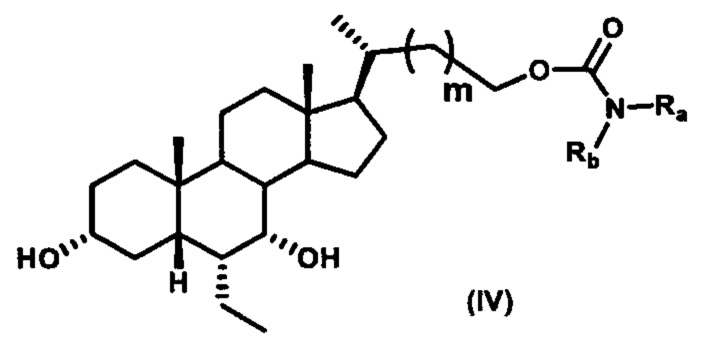
где Ra, Rb и m являются такими, как определено ранее.
Типичные соединения по изобретению включают, но не ограничиваются указанными, следующие соединения (от соединения 1 до соединения 99 в Таблице 1) согласно формуле IV, где Ra, Rb и m представлены для каждого соединения в Таблице 1.
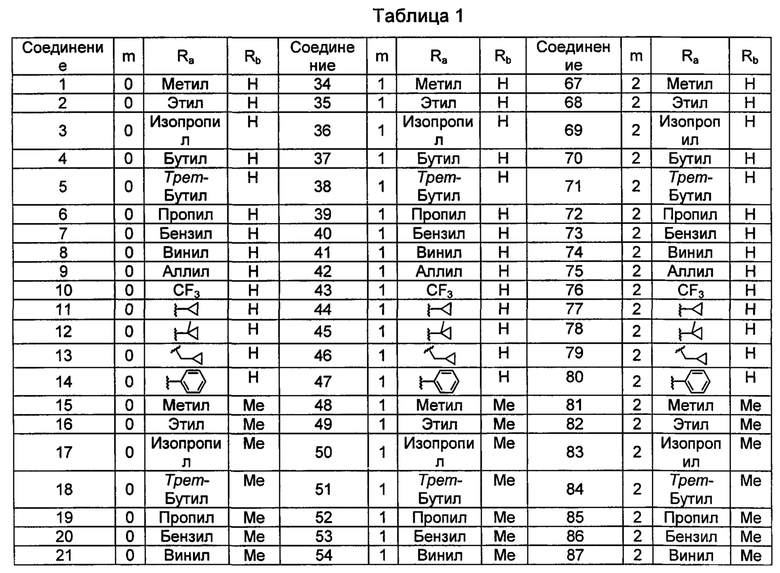
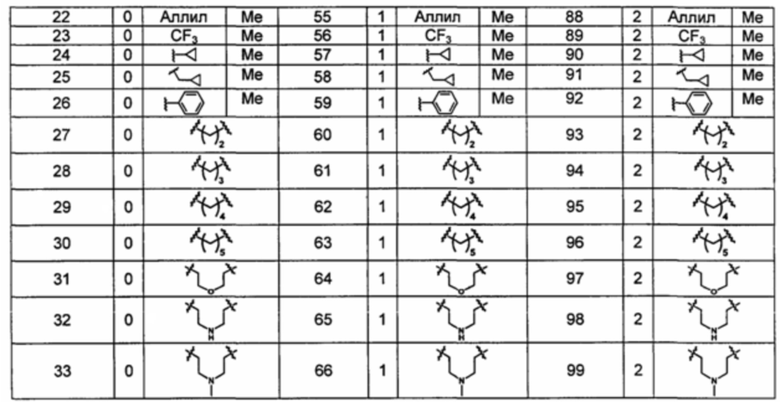
Пятый вариант осуществления изобретения представляет собой соединение, представленное формулой V, или его фармацевтически приемлемую соль, сольват, гидрат, сложный эфир или пролекарство,
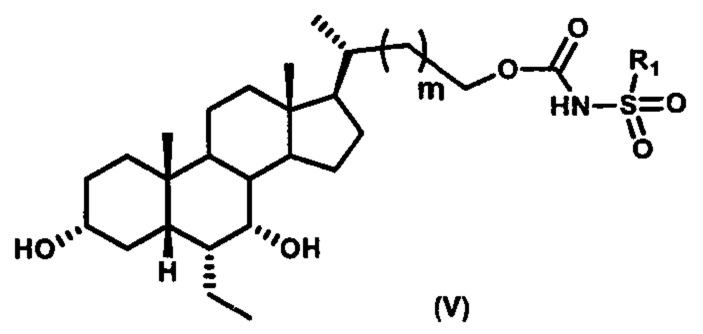
где R1 и m являются такими, как определено ранее.
Типичные соединения по изобретению включают, но не ограничиваются указанными, следующие соединения (от соединения 100 до соединения 180 в Таблице 2) согласно формуле V, где R1 и m представлены для каждого соединения в Таблице 2.
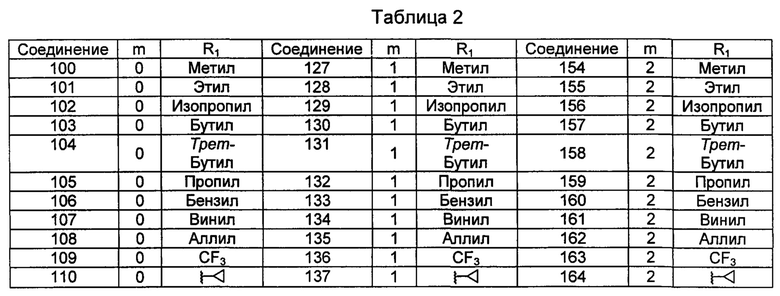
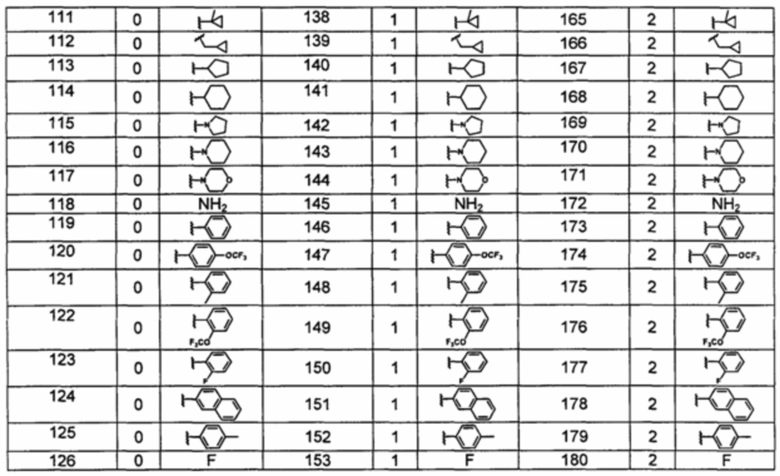
Шестой вариант осуществления изобретения представляет собой соединение, представленное формулой VI, или его фармацевтически приемлемую соль, сольват, гидрат, сложный эфир или пролекарство,
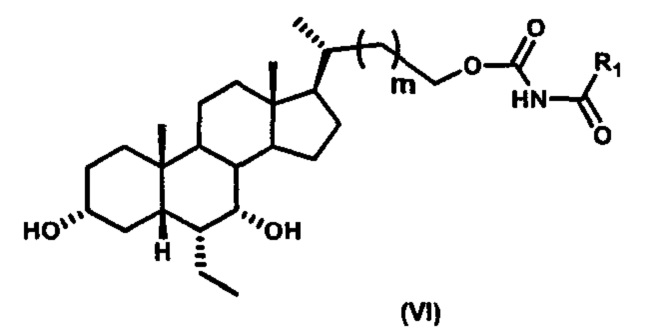
где R1 и m являются такими, как определено ранее.
Типичные соединения по изобретению включают, но не ограничиваются указанными, следующие соединения (от соединения 181 до соединения 261 в Таблице 3) согласно формуле VI, где R1 и m представлены для каждого соединения в Таблице 3.
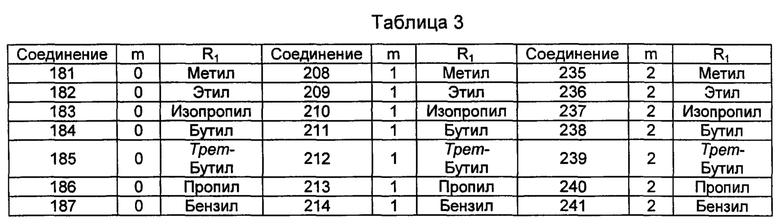
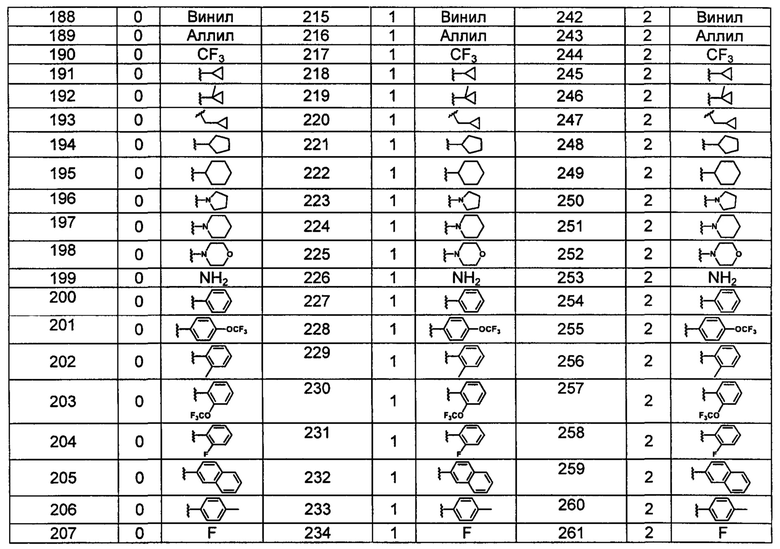
Понятно, что описание настоящего изобретения, представленное здесь, должно толковаться в соответствии с законами и принципами химической связи. В некоторых случаях может потребоваться удаление атома водорода для размещения заместителя в любом заданном положении.
Следует также понимать, что соединения по настоящему изобретению могут содержать один или более асимметричных атомов углерода и могут существовать в рацемических, диастереоизомерных и оптически активных формах. Вместе с тем следует понимать, что некоторые соединения по настоящему изобретению могут существовать в различных таутомерных формах. Предполагается, что все таутомеры входят в объем настоящего изобретения. В некоторых вариантах осуществления настоящее изобретение относится к способу профилактики или лечения FXR-опосредованного заболевания или состояния. Способ включает введение терапевтически эффективного количества соединения формулы (I). Настоящее изобретение также относится к применению соединения формулы (I) для получения лекарственного средства для профилактики или лечения FXR-опосредованного заболевания или состояния.
В некоторых вариантах осуществления FXR-опосредованное заболевание или состояние представляет собой сердечно-сосудистое заболевание, атеросклероз, артериосклероз, гиперхолестеринемию или гиперлипидемию, хроническое заболевание печени, заболевание желудочно-кишечного тракта, почечное заболевание, метаболическое заболевание, раковое заболевание (например, колоректальный рак) или неврологические показания, такие как инсульт.
В некоторых вариантах хроническое заболевание печени представляет собой первичный билиарный цирроз (ПБЦ), церебросухожильный ксантоматоз (ЦСК), первичный склерозирующий холангит (ПСХ), холестаз, обусловленный действием лекарственных средств, внутрипеченочный холестаз беременных, холестаз, связанный с парентеральным питанием (ХПП), холестаз, связанный с чрезмерным развитием микрофлоры или сепсисом, аутоиммунный гепатит, хронический вирусный гепатит, алкогольную болезнь печени, неалкогольную жировую болезнь печени (НЖБП), неалкогольный стеатогепатит (НАСГ), связанная с трансплантацией печени реакция «трансплантат против хозяина», регенерация печени живого донора, врожденный фиброз печени, желчнокаменная болезнь, гранулематозное заболевание печени, внутри- или внепеченочное злокачественное образование, синдром Шегрена, саркоидоз, болезнь Вильсона, болезнь Гоше, гемохроматоз или дефицит альфа-1-антитрипсина. В некоторых вариантах осуществления заболевание желудочно-кишечного тракта представляет собой воспалительное заболевание кишечника (ВЗК) (в том числе болезнь Крона и язвенный колит), синдром раздраженного кишечника (СРК), чрезмерное развитие микрофлоры, мальабсорбцию, постлучевой колит или микроскопический колит.
В некоторых вариантах осуществления почечное заболевание представляет собой диабетическую нефропатию, фокально-сегментарный гломерулоскпероз (ФСГ), гипертонический нефроскпероз, хронический гломерулонефрит, хроническую трансплантационную гломерулопатию, хронический интерстициальный нефрит или поликистозную болезнь почек.
В некоторых вариантах осуществления сердечно-сосудистое заболевание представляет собой атеросклероз, артериосклероз, дислипидемию, гиперхолестеринемию или гипертриглицеридемию.
В некоторых вариантах осуществления метаболическое заболевание представляет собой резистентность к инсулину, диабет типа I и типа II или ожирение.
В еще одном варианте осуществления настоящее изобретение относится к применению соединения или фармацевтической композиции по изобретению для получения лекарственного средства для лечения или профилактики заболевания у субъекта, который включает модуляцию рецептора TGR5. Изобретение относится к способу лечения или профилактики заболевания, включающему модуляцию рецептора TGR5 у субъекта путем введения соединения или фармацевтической композиции по изобретению.
В некоторых вариантах осуществления заболевание, которое включает модуляцию рецептора TGR5, выбрано из метаболического заболевания, воспалительного заболевания, заболевания печени, аутоиммунного заболевания, заболевания сердца, заболевания почек, ракового заболевания и заболевания желудочно-кишечного тракта.
В одном аспекте изобретение относится к применению, где заболевание представляет собой воспалительное заболевание, выбранное из аллергии, остеоартрита, аппендицита, бронхиальной астмы, панкреатита, аллергической сыпи и псориаза. Изобретение относится к способу лечения или профилактики воспалительного заболевания, выбранного из аллергии, остеоартрита, аппендицита, бронхиальной астмы, панкреатита, аллергической сыпи и псориаза.
В одном аспекте изобретение относится к применению, где заболевание представляет собой аутоиммунное заболевание, выбранное из ревматоидного артрита, рассеянного склероза и диабета типа I. Изобретение относится к способу лечения или профилактики аутоиммунного заболевания, выбранного из ревматоидного артрита, рассеянного склероза и диабета типа I.
В одном аспекте настоящее изобретение относится к применению, где заболевание представляет собой заболевание желудочно-кишечного тракта, выбранное из воспалительного заболевания кишечника (болезнь Крона, язвенный колит), синдрома короткого кишечника (постлучевой колит), микроскопического колита, синдрома раздраженного кишечника (мальабсорбция), и чрезмерного развития микрофлоры. Изобретение относится к способу лечения или профилактики заболевания желудочно-кишечного тракта, выбранного из воспалительного заболевания кишечника (болезнь Крона, язвенный колит), синдрома короткого кишечника (постлучевой колит), микроскопического колита, синдрома раздраженного кишечника (мальабсорбция) и чрезмерного развития микрофлоры.
В одном аспекте изобретение относится к применению, где заболевание представляет собой заболевание почек, выбранное из диабетической нефропатии, хронической почечной недостаточности, гипертонического нефроскпероза, хронического гломерулонефрита, хронической трансплантационной гломерулопатии, хронического интерстициального нефрита и поликистозной болезни почек. Изобретение относится к способу лечения или профилактики заболевания почек, выбранного из диабетической нефропатии, хронической почечной недостаточности, гипертонического нефросклероза, хронического гломерулонефрита, хронической трансплантационной гломерулопатии, хронического интерстициального нефрита и поликистозной болезни почек.
В одном аспекте изобретение относится к применению, где заболевание представляет собой раковое заболевание, выбранное из колоректального рака, рака печени, гепатоцеллюлярной карциномы, холангиокарциномы, рака почки, рака желудка, рака поджелудочной железы, рака предстательной железы и инсуланомы. Изобретение относится к способу лечения или профилактики рака, выбранного из колоректального рака, рака печени, гепатоцеллюлярной карциномы, холангиокарциномы, рака почки, рака желудка, рака поджелудочной железы, рака предстательной железы и инсуланомы.
В одном аспекте соединение представляет собой селективный агонист FXR больше чем активатор TGR5.
В одном аспекте соединение представляет собой селективный агонист TGR5 больше чем активатор FXR.
В одном аспекте соединение представляет собой двойной агонист как для FXR, так и для TGR5.
Еще одним аспектом настоящего изобретения является способ получения любого из соединений, представленных здесь, с применением любого из синтетических средств, представленных здесь.
ОПРЕДЕЛЕНИЯ
Ниже перечислены определения различных терминов, используемых для описания настоящего изобретения. Данные определения применимы к терминам, используемым в настоящем описании и формуле изобретения, если в конкретных случаях не ограничены иным образом, либо индивидуально, либо как часть большей группы.
Используемый в данном документе термин «алкил» относится к насыщенной, одновалентной углеводородной группе с линейной или разветвленной цепью. Предпочтительные алкильные радикалы включают C1-C6 алкильные и C1-C8 алкильные радикалы. Примеры C1-C6 алкильных групп включают, но не ограничиваются указанными, метил, этил, пропил, изопропил, н-бутил, трет-бутил, неопентил, н-гексил, а примеры C1-C8 алкильных групп включают, но не ограничиваются указанными, метил, этил, пропил, изопропил, н-бутил, трет-бутил, неопентил, н-гексил, гептил и октил.
Используемый в данном документе термин «алкенил» относится к одновалентной группе, полученной от углеводородного фрагмента, путем удаления одного атома водорода, где углеводородный фрагмент имеет по меньшей мере одну углерод-углеродную двойную связь. Предпочтительные алкенильные группы включают C2-C6 алкенильные и C2-C8 алкенильные группы. Алкенильные группы включают, но не ограничиваются указанными, например, этенил, пропенил, бутенил, 1-метил-2-бутен-1-ил, гептенил, октенил и тому подобное.
Используемый в данном документе термин «алкинил» относится к одновалентной группе, полученной от углеводородного фрагмента, путем удаления одного атома водорода, где углеводородный фрагмент имеет по меньшей мере одну углерод-углеродную тройную связь. Предпочтительные алкинильные группы включают C2-C6 алкинильные и C2-C8 алкинильные группы. Типичные алкинильные группы включают, но не ограничиваются указанными, например, этинил, 1-пропинил, 1-бутинил, гептинил, октинил и тому подобное.
Термин «карбоцикл» относится к насыщенной (например, «циклоалкильной»), частично насыщенной (например, «циклоалкенильной» или «циклоалкинильной») или полностью ненасыщенной (например, «арильной») кольцевой системе, не содержащей гетероатомный кольцевой атом. «Кольцевые атомы» или «члены кольца» представляют собой атомы, связанные вместе с образованием кольца или колец. Если карбоциклическая группа представляет собой двухвалентный фрагмент, связывающий два других элемента в изображенной химической структуре (такой как Z в формуле IA), карбоциклическая группа может быть присоединена к двум другим элементам через любые два замещаемых кольцевых атома. C4-C6 карбоцикл имеет от 4 до 6 кольцевых атома.
Используемый в данном документе термин «циклоалкил» относится к одновалентной группе, полученной от моноциклического или полициклического насыщенного карбоциклического циклического соединения путем удаления одного атома водорода. Предпочтительные циклоалкильные группы включают C3-C8 циклоалкильные и C3-C12 циклоалкильные группы. Примеры C3-C8 циклоалкила включают, но не ограничиваются указанными, циклопропил, циклобутил, циклопентил, циклогексил, циклопентил и циклооктил; и примеры C3-C12 циклоалкила включают, но не ограничиваются указанными, циклопропил, циклобутил, циклопентил, циклогексил, бицикло[2.2.1]гептил и бицикло[2.2.2]октил.
Используемый в данном документе термин «циклоалкенил» относится к одновалентной группе, полученной от моноциклического или полициклического карбоциклического кольцевого соединения, имеющего по меньшей мере одну углерод-углеродную двойную связь, путем удаления одного атома водорода. Предпочтительные циклоалкенильные группы включают C3-C8 циклоалкенильные и C3-C12 циклоалкенильные группы. Примеры C3-C8 циклоалкенила включают, но не ограничиваются указанными, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклооктенил и тому подобное; и примеры C3-C12 циклоалкенила включают, но не ограничиваются указанными, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклооктенил и тому подобное.
Используемый в данном документе термин «арил» относится к радикалу моно- или бициклической карбоциклической кольцевой системы, имеющему одно или два ароматических кольца, включая, но не ограничиваясь указанными, фенил, нафтил, тетрагидронафтил, инданил, инденил и тому подобное.
Используемый в данном документе термин «арилалкил» относится к C1-C3 алкильному или C1-C6 алкильному радикалу, присоединенному к арильному кольцу. Примеры включают, но не ограничиваются указанными, бензил, фенэтил и тому подобное. Термин «замещенный арилалкил» означает арилалкильную функциональную группу, в которой арильная группа является замещенной.
Используемый в данном документе термин «гетероарил» относится к моно-, би- или трициклическому ароматическому радикалу или кольцу, имеющему от пяти до десяти кольцевых атомов, из которых по меньшей мере один кольцевой атом выбран из S, О и N; где любые N или S, содержащиеся в кольце, могут быть необязательно окислены. Предпочтительные гетероарильные группы являются моноциклическими или бициклическими. Гетероарильные группы включают, но не ограничиваются указанными, пиридинил, пиразинил, пиримидинил, пирролил, пиразолил, имидазолил, тиазолил, оксазолил, изооксазолил, тиадиазолил, оксадиазолил, тиофенил, фуранил, хинолинил, изохинолинил, бензимидазолил, бензооксазолил, хиноксалинил и тому подобное.
Используемый в данном документе термин «гетероарилалкил» относится к C1-C3 алкильному или C1-C6 алкильному радикалу, присоединенному к гетероарильному кольцу. Примеры включают, но не ограничиваются указанными, пиридинилметил, пиримидинилэтил и тому подобное. Термин «замещенный гетероарилалкил» относится к гетероарилалкильной функциональной группе, в которой гетероарильная группа является замещенной.
Используемый в данном документе термин «алкокси», отдельно или в сочетании с другими терминами, относится, если не указано иное, к алкильной группе, имеющей указанное число атомов углерода, связанных с остальной частью молекулы с помощью атома кислорода, такой как, например, метокси, этокси, 1-пропокси, 2-пропокси (изопропокси) и высшим гомологам и изомерам. Предпочтительные алкокси представляют собой (C1-C3) алкокси.
Понятно, что любые алкильные, алкенильные, алкинильные, циклоалкильные, гетероциклические и циклоалкенильные фрагменты, описанные в данном документе, также могут представлять собой алифатическую группу или алициклическую группу.
Используемый в данном документе термин «замещенный» относится к независимому замещению одного, двух или трех или более атомов водорода заместителями, включая, но не ограничиваясь указанными, дейтерий, -F, -Cl, -Br, -I, -ОН, защищенный гидрокси, -NO2, -CN, -NH2, N3, защищенный амино, алкокси, тиоалкокси, оксо, C1-C12-алкил, C2-C12-алкенил, C2-C12-алкинил, C3-C12-циклоалкил-галоген-C1-C12-алкил, -галоген-C2-C12-алкенил, -галоген-C2-C12-алкинил, -галоген-C3-C12-циклоалкил, -NH-C1-C12-алкил, -NH-C2-C12-алкенил, -NH-C2-C12-алкинил, -NH-C3-C12-циклоалкил, -NH-арил, -NH-гетероарил, -NH-гетероциклоалкил, -диалкиламино, -диариламино, -дигетероариламино, -O-C1-C12-алкил, -O-C2-C12-алкенил, -O-C2-C12-алкинил, -O-C3-C12-циклоалкил, -O-арил, -O-гетероарил, -O-гетероциклоалкил, -С(O)-C1-C12-алкил, -С(O)-C2-C12-алкенил, -С(O)-C2-C12-алкинил, -С(O)-C3-C12-циклоалкил, -С(O)-арил, -С(O)-гетероарил, -С(O)-гетероциклоалкил, -CONH2, -CONH-C1-C12-алкил, -CONH-C2-C12-алкенил, -CONH-C2-C12-алкинил, -CONH-C3-C12-циклоалкил, -CONH-арил, -CONH-гетероарил, -CONH-гетероциклоалкил, -OCO2-C1-C12-алкил, -ОСО2-C2-C12-алкенил, -OCO2-C2-C12-алкинил, -OCO2-C3-C12-циклоалкил, -OCO2-арил, -OCO2-гетероарил, -OCO2-гетероциклоалкил, -OCONH2, -OCONH-C1-C12-алкил, -OCONH-C2-C12-алкенил, -OCONH-C2-C12-алкинил, -OCONH-C3-C12-циклоалкил, -OCONH-арил, -OCONH-гетероарил, -OCONH-гетероциклоалкил, -NHC(O)-C1-C12-алкил, -NHC(O)-C2-C12-алкенил, -NHC(O)-C2-C12-алкинил, -NHC(O)-C3-C12-циклоалкил, -NHC(O)-арил, -NHC(O)-гетероарил, -NHC(O)-гетероциклоалкил, -NHCO2-C1-C12-алкил, -NHCO2-C2-C12-алкенил, -NHCO2-C2-C12-алкинил, -NHCO2-C3-C12-циклоалкил, -NHCO2-арил, -NHCO2-гетероарил, -NHCO2-гетероциклоалкил, -NHC(O)NH2, -NHC(O)NH-C1-C12-алкил, NHC(O)NH-C2-C12-алкенил, -NHC(O)NH-C2-C12-алкинил, -NHC(O)NH-C3-C12-циклоалкил, -NHC(O)NH-арил, -NHC(O)NH-гетероарил, -NHC(O)NH-гетероциклоалкил, NHC(S)NH2, -NHC(S)NH-C1-C12-алкил, -NHC(S)NH-C2-C12-алкенил, -NHC(S)NH-C2-C12-алкинил, -NHC(S)NH-C3-C12-циклоалкил, -NHC(S)NH-арил, -NHC(S)NH-гетероарил, -NHC(S)NH-гетероциклоалкил, -NHC(NH)NH2, -NHC(NH)NH-C1-C12-алкил, -NHC(NH)NH-C2-C12-алкенил, -NHC(NH)NH-C2-C12-алкинил, -NHC(NH)NH-C3-C12-циклоалкил, -NHC(NH)NH-арил, -NHC(NH)NH-гетероарил, -NHC(NH)NH-гетероциклоалкил, -NHC(NH)-C1-C12-алкил, -NHC(NH)-C2-C12-алкенил, -NHC(NH)-C2-C12-алкинил, -NHC(NH)-C3-C12-циклоалкил, NHC(NH)-арил, -NHC(NH)-гетероарил, -NHC(NH)-гетероциклоалкил, -C(NH)NH-C1-C12-алкил, -C(NH)NH-C2-C12-алкенил, -С(NH)NH-C2-C12-алкинил, -C(NH)NH-C3-C12-циклоалкил, -С(NH)NH-арил, -С(NH)NH-гетероарил, -С(NH)NH-гетероциклоалкил, -S(O)-C1-C12-алкил, -S(O)-C2-C12-алкенил, -S(O)-C2-C12-алкинил, -S(O)-C3-C12-циклоалкил, -S(O)-арил, -S(O)-гетероарил, -S(O)-гетероциклоалкил-SO2NH2, -SO2NH-C1-C12-алкил, -SO2NH-C2-C12-алкенил, -SO2NH-C2-C12-алкинил, -SO2NH-C3-C12-циклоалкил, -SO2NH-арил, -SO2NH-гетероарил, -SO2NH-гетероциклоалкил, -NHSO2-C1-C12-алкил, -NHSO2-C2-C12-алкенил, -NHSO2-C2-C12-алкинил, -NHSO2-C3-C12-циклоалкил, -NHSO2-арил, -NHSO2-гетероарил, -NHSO2-гетероциклоалкил, -CH2NH2, -CH2SO2CH3, -арил, -арилалкил, -гетероарил, -гетероарилалкил, -гетероциклоалкил, -C3-C12-циклоалкил, полиалкоксиалкил, полиалкокси, -метоксиметокси, -метоксиэтокси, -SH, -S-C1-C12-алкил, -S-C2-C12-алкенил, -S-C2-C12-алкинил, -S-C3-C12-циклоалкил, -S-арил, -S-гетероарил, -S-гетероциклоалкил, метилтиометил, или -L'-R', где L' представляет собой C1-C6-алкилен, C2-C6-алкенилен или C2-C6-алкинилен, и R' представляет собой арил, гетероарил, гетероциклическое соединение, C3-C12-циклоалкил или C3-C12-циклоалкенил. Понятно, что арилы, гетероарилы, алкилы и тому подобное могут быть дополнительно замещены. В некоторых случаях каждый заместитель в замещенном фрагменте дополнительно необязательно замещен одной или несколькими группами, каждая из которых независимо выбрана из -F, -Cl, -Br, -I, -ОН, -NO2, -CN или -NH2.
В соответствии с изобретением любой из арилов, замещенных арилов, гетероарилов и замещенных гетероарилов, описанных в данном документе, может быть любой ароматической группой. Ароматические группы могут быть замещенными или незамещенными.
Понятно, что любой алкильный, алкенильный, алкинильный, циклоалкильный и циклоалкенильный фрагменты, описанные в данном документе, также могут быть алифатической группой, алициклической группой или гетероциклической группой. «Алифатическая группа» представляет собой неароматический фрагмент, который может содержать любую комбинацию атомов углерода, атомов водорода, атомов галогена, кислорода, азота или других атомов и необязательно содержать одну или более единиц ненасыщенности, например, двойные и/или тройные связи. Алифатическая группа может быть линейной, разветвленной или циклической и предпочтительно содержать от приблизительно 1 до приблизительно 24 атомов углерода, более типично от приблизительно 1 до приблизительно 12 атомов углерода. В дополнение к алифатическим углеводородным группам, алифатические группы включают, например, полиалкоксиалкилы, такие как полиалкиленгликоли, полиамины и полиимины. Такие алифатические группы могут быть дополнительно замещены. Понятно, что алифатические группы могут быть использованы вместо алкильных, алкенильных, алкинильных, алкиленовых, алкениленовых и алкиниленовых групп, описанных в данном документе.
Используемый в данном документе термин «алициклический» относится к одновалентной группе, полученной от моноциклического или полициклического насыщенного карбоциклического кольцевого соединения путем удаления одного атома водорода. Примеры включают, но не ограничиваются указанными, циклопропил, циклобутил, циклопентил, циклогексил, бицикло[2.2.1]гептил и бицикло[2.2.2]октил. Такие алициклические группы могут быть дополнительно замещены.
Термины «гетероциклический» и «гетероциклоалкил» могут быть взаимозаменяемы и относятся к неароматическому кольцу или би- или трициклической конденсированной, мостиковой или спиросистеме, где (i) каждая кольцевая система содержит по меньшей мере один гетероатом независимо выбранный из кислорода, серы и азота, (ii) каждая кольцевая система может быть насыщенной или ненасыщенной, (iii) гетероатомы азота и серы могут быть необязательно окислены, (iv) гетероатом азота может быть необязательно кватернизован, (v) любое из выше описанных колец может быть конденсировано с ароматическим кольцом и (vi) остальные кольцевые атомы представляют собой атомы углерода, которые могут быть необязательно оксозамещены. Типичные гетероциклоалкильные группы включают, но не ограничиваются указанными, 1,3-диоксолан, пирролидинил, пиразолинил, пиразолидинил, имидазолинил, имидазолидинил, пиперидинил, пиперазинил, оксазолидинил, изоксазолидинил, морфолинил, тиазолидинил, изотиазолидинил, хиноксалинил, пиридазинонил и тетрагидрофурил. Такие гетероциклические группы могут быть дополнительно замещены. Гетероарильные или гетероциклические группы могут быть С-присоединенными или N-присоединенными (если возможно). Примеры включают, но не ограничиваются указанными, 3-азабицикло[3.3.1]нонанил, 2-окса-7-азасприо[4.4]нонанил и тому подобное.
Очевидно, что в различных вариантах осуществления изобретения замещенные или незамещенные алкильные, алкенильные, алкинильные, циклоалкильные, циклоалкенильные, циклоалкинильные, арилалкильные, гетероарилалкильные и гетероциклоалкильные группы являются одновалентными или двухвалентными. Таким образом, алкиленовые, алкениленовые и алкиниленовые, циклоалкиленовые, циклоалкениленовые, циклоалкиниленовые, арилалкиленовые, гетероарилалкиленовые и гетероциклоалкиленовые группы должны быть включены в приведенные выше определения и применимы для получения соединений формул, приведенных в данном документе, с надлежащей валентностью.
Используемый в данном документе термин «гидроксиактивирующая группа» относится к лабильному химическому фрагменту, который известен в данной области техники для активации гидроксигруппы, таким образом, что он исчезает в ходе стадии синтеза, например в реакции замещения или элиминирования. Примеры гидроксиактивирующей группы включают, но не ограничиваются указанными, мезилат, тозилат, трифлат, пара-нитробензоат, фосфонат и тому подобное.
Используемый в данном документе термин «активированная гидрокси» относится к гидроксигруппе, активированной гидроксиактивирующей группой, как определено выше, включая, например, мезилат, тозилат, трифлат, пара-нитробензоат, фосфонат.
Используемый в данном документе термин «защищенная гидрокси» относится к гидроксигруппе, защищенной гидроксизащитной группой, как определено выше, включая бензоильную, ацетильную, триметилсилильную, триэтилсилильную и метоксиметильную группы.
Используемый в данном документе термин «гидроксизащитная группа» относится к лабильному химическому фрагменту, который известен в данной области техники для защиты гидрокси группы от нежелательных реакций в ходе стадий синтеза. После указанной стадии(ий) синтеза гидроксизащитная группа, как описано в данном документе, может быть селективно удалена. Гидроксизащитные группы, известные в области техники, раскрыты, как правило, в Т.Н. Greene and P.O., S.M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999). Примеры гидроксизащитных групп включают бензилоксикарбонил, 4-нитробензилоксикарбонил, 4-бромбензилоксикарбонил, 4-метоксибензилоксикарбонил, метоксикарбонил, трет-бутоксикарбонил, изопропоксикарбонил, дифенилметоксикарбонил, 2,2,2-трихлорэтоксикарбонил, 2-(триметилсилил)этоксикарбонил, 2-фурфурилоксикарбонил, аллилоксикарбонил, ацетил, формил, хлорацетил, трифторацетил, метоксиацетил, феноксиацетил, бензоил, метил, трет-бутил, 2,2,2-трихлорэтил, 2-триметилсилилэтил, 1,1-диметил-2-пропенил, 3-метил-3-бутенил, аллил, бензил, пара-метоксибензилдифенилметил, трифенилметил (тритил), тетрагидрофурил, метоксиметил, метилтиометил, бензилоксиметил, 2,2,2-трихлорэтоксиметил, 2-(триметилсилил)этоксиметил, метансульфонил, пара-толуолсульфонил, триметилсилил, триэтилсилил, триизопропилсилил, и тому подобное. Предпочтительными гидроксизащитными группами по настоящему изобретению являются ацетил (Ас или -С(О)СН3), бензоил (Bz или -С(O)С6Н5) и триметилсилил (TMS или -Si(СН3)3).
Используемые в данном документе термины «гало» и «галоген» относятся к атому, выбранному из фтора, хлора, брома и йода.
Термин «водород» включает водород и дейтерий. Кроме того, раскрытие атома элемента включает все изотопы этого элемента, если полученное соединение является фармацевтически приемлемым.
В некоторых вариантах осуществления соединения каждой формулы в данном описании включают изотопно-меченые соединения. «Изотопно-меченое соединение» представляет собой соединение, в котором по меньшей мере одно положение атома обогащено определенным изотопом обозначенного элемента до уровня, который значительно превышает распространенность в природе этого изотопа. Например, одно или более положений атомов водорода в соединении могут быть обогащены дейтерием до уровня, который значительно превышает распространенность в природе дейтерия, например, обогащение до уровня по меньшей мере 1%, предпочтительно по меньшей мере 20% или по меньшей мере 50%. Такое дейтерированное соединение может, например, метаболизироваться более медленно, чем его недейтерированный аналог, и, следовательно, иметь более длительный период полувыведения при введении субъекту. Такие соединения могут быть синтезированы с применением способов, известных в данной области, например, путем использования дейтерированных исходных веществ. Если не указано обратное, изотопно-меченые соединения являются фармацевтически приемлемыми.
Термин «уходящая группа» означает функциональную группу или атом, которые могут быть замещены другой функциональной группой или атомом в реакции замещения, такой как реакция нуклеофильного замещения. В качестве примера, типичные уходящие группы включают группы хлор, бром и йод; сульфоновые сложноэфирные группы, такие как мезилат, тозилат, брозилат, нозилат и тому подобное; и ацилоксигруппы, такие как ацетокси, трифторацетокси и тому подобное.
Соединения, описанные в данном документе, содержат один или более асимметричных центров и, таким образом, образуют энантиомеры, диастереомеры и другие стереоизомерные формы, которые могут быть определены с точки зрения абсолютной стереохимии как (R)- или (S)- или как (D)- или (L)- для аминокислот. Настоящее изобретение предусматривает включение всех таких возможных изомеров, а также их рацемических и оптически чистых форм. Оптические изомеры могут быть получены из их соответствующих оптически активных предшественников с помощью описанных выше методик или путем разделения рацемических смесей. Разделение может быть выполнено в присутствии разделяющего агента, путем хроматографии или повторной кристаллизации, или некоторой комбинацией этих техник, которые известны специалистам в данной области техники. Более подробную информацию о разделении можно найти в Jacques, et al., Enantiomers, Racemates and Resolutions (John Wiley & Sons, 1981). Когда описанные в данном документе соединения содержат олефиновые двойные связи или другие центры геометрической асимметрии, и, если не указано иное, предполагается, что соединения включают как Е, так и Z геометрические изомеры. Аналогично, подразумевается, что все таутомерные формы также включены. Конфигурация любой углерод-углеродной двойной связи, приведенной в данном документе, выбрана только для удобства и не предназначена для обозначения конкретной конфигурации, если в тексте не указано иного; таким образом, углерод-углеродная двойная связь, изображенная произвольно в данном документе, как транс, может быть цис, транс или смесью обоих в любой пропорции.
Используемый в данном документе термин «субъект» относится к млекопитающему. Таким образом, к субъекту относятся, например, собаки, кошки, лошади, коровы, свиньи, морские свинки и тому подобные. Предпочтительно, субъект представляет собой человека. Когда субъект представляет собой человека, субъект может упоминаться в данном документе как пациент.
Используемый в данном документе термин «фармацевтически приемлемая соль» относится к таким солям соединений, образованных способом по настоящему изобретению, которые с медицинской точки зрения подходят для применения в контакте с тканями людей и низших животных без нежелательной токсичности, раздражения, аллергической реакции и тому подобного, и соответствуют разумному соотношению польза/риск. Фармацевтически приемлемые соли хорошо известны в данной области техники.
Berge, et al. подробно описывает фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 66: 1-19 (1977). Соли могут быть получены in situ во время окончательного выделения и очистки соединений по изобретению или отдельно путем реакции свободного основания с подходящей органической кислотой. Примеры фармацевтически приемлемых солей включают, но не ограничиваются указанными, соли присоединения нетоксичных кислот, например соли аминогруппы, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота и перхлорная кислота, или с органическими кислотами, такими как уксусная кислота, малеиновая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота, или с применением других способов, используемых в данной области техники, таких как ионный обмен. Другие фармацевтически приемлемые соли включают, но не ограничиваются указанными, адипат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептаноат, гексаноат, гидроиодид, 2-гидроксиэтансульфонат, лактобионат, лактат, лаурат, лаурилсульфат, малат, малеат, малонат, метансульфонат, 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, пара-толуолсульфонат, ундеканоат, валерат и тому подобное. Типичные соли щелочных или щелочноземельных металлов включают соли натрия, лития, калия, кальция, магния и тому подобное. Другие фармацевтически приемлемые соли включают, при необходимости, нетоксичные аммониевые, четвертичные аммониевые и аминокатионы, образованные с использованием противоионов, таких как галогенид, гидроксид, карбоксилат, сульфат, фосфат, нитрат, алкил, содержащий от 1 до 6 атомов углерода, сульфонат и арилсульфонат.
Фармацевтически приемлемые соли также могут быть получены депротонированием исходного соединения подходящим основанием, в результате чего образуется анионное сопряженное основание исходного соединения. В таких солях противоион является катионом. Подходящие катионы включают катионы аммония и металлов, такие как катионы щелочных металлов, включая Li+, Na+, K+ и Cs+ и катионы щелочноземельных металлов, такие как Mg2+ и Са2+.
Используемый в данном документе термин «аминозащитная группа» относится к лабильному химическому фрагменту, который известен в данной области техники для защиты аминогруппы от нежелательных реакций в ходе стадии синтеза. После указанной стадии(ий) синтеза аминозащитная группа, как описано в данном документе, может быть селективно удалена. Аминозащитные группы, известные в области техники, раскрыты, как правило, в Т.Н. Greene and P.G. M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York (1999). Примеры аминозащитных групп включают, но не ограничиваются указанными, трет-бутоксикарбонил, 9-флуоренилметоксикарбонил, бензилоксикарбонил и тому подобное.
Используемый в данном документе термин «фармацевтически приемлемый сложный эфир» относится к сложным эфирам соединений, образованных способом по настоящему изобретению, которые гидролизуются in vivo и включают те, которые легко расщепляются в организме человека с высвобождением исходного соединения или его соли. Подходящие сложноэфирные группы включают, например, те, которые получены из фармацевтически приемлемых алифатических карбоновых кислот, особенно алкановых, алкеновых, циклоалкановых и алкандиовых кислот, в которых каждый алкильный или алкенильный фрагмент преимущественно имеет не более 6 атомов углерода. Примеры конкретных сложных эфиров включают, но не ограничиваются указанными, формиаты, ацетаты, пропионаты, бутираты, акрилаты и этилсукцинаты.
Используемый в данном документе термин «фармацевтически приемлемые пролекарства» относится к таким пролекарствам соединений, образованных способом по настоящему изобретению, которые с медицинской точки зрения подходят для применения в контакте с тканями людей и низших животных без нежелательной токсичности, раздражения, аллергической реакцией и тому подобным, соответствуют разумному соотношению польза/риск и эффективны для их предполагаемого применения, а также цвиттерионные формы, где это возможно, соединений по настоящему изобретению. Термин «пролекарство», используемый в данном документе, означает соединение, которое превращается in vivo метаболическими средствами (например, путем гидролиза) с получением любого соединения, подпадающего под формулы настоящего изобретения. Различные виды пролекарств известны в данной области техники, например, как раскрыто в Bundgaard, (ed.), Design of Prodrugs, Elsevier (1985); Widder, et al. (ed.), Methods in Enzymology, Vol. 4, Academic Press (1985); Krogsgaard-Larsen, et al., (ed). Design and Application of Prodrugs, Textbook of Drug Design and Development, Chapter 5, 113-191 (1991); Bundgaard, et al., Journal of Drug Deliver Reviews, 8:1-38(1992); Bundgaard, J. of Pharmaceutical Sciences, 77:285 et seq. (1988); Higuchi and Stella (eds.) Prodrugs as Novel Drug Delivery Systems, American Chemical Society (1975); и Bernard Testa & Joachim Mayer, Hydrolysis In Drug And Prodrug Metabolism: Chemistry, Biochemistry And Enzymology, John Wiley and Sons, Ltd. (2002).
Используемый в данном документе термин «лечение» означает ослабление, уменьшение, снижение, устранение, изменение или улучшение, то есть вызывание регресса стадии заболевания или состояния. Лечение может также включать в себя ингибирование, то есть остановку развития, существующей стадии заболевания или состояния, а также ослабление или облегчение, то есть вызывание регресса существующей стадии заболевания или состояния, например, когда стадия заболевания или состояние уже могут присутствовать.
Используемый в данном документе термин «профилактика» означает полное или почти полное прекращение течения заболевания или состояния от возникновения у пациента или субъекта, особенно когда пациент или субъект предрасположены к такому или подвержены риску возникновения стадии заболевания или состояния.
Кроме того, соединения по настоящему изобретению, например, соли соединений, могут существовать как в гидратированной, так и в негидратированной (безводной) форме или в виде сольватов с другими молекулами растворителя. Неограничивающие примеры гидратов включают моногидраты, дигидраты и т.д. Неограничивающие примеры сольватов включают сольваты этанола, сольваты ацетона и т.д.
«Сольваты» означают формы присоединения растворителей, которые содержат либо стехиометрические, либо не стехиометрические количества растворителя. Некоторые соединения имеют тенденцию захватывать фиксированное молярное отношение молекул растворителя в кристаллическом твердом состоянии, образуя таким образом сольват. Если растворителем является вода, образовавшийся сольват представляет собой гидрат, когда растворителем является спирт, образовавшийся сольват представляет собой алкоголят. Гидраты образуются путем сочетания одной или более молекул воды с одним из веществ, в которых вода сохраняет свое молекулярное состояние как H2O, причем такая комбинация способна образовывать один или более гидратов.
Используемый в данном документе термин «аналог» относится к химическому соединению, которое структурно похоже на другое, но немного отличается по составу (как при замене одного атома атомом другого элемента или в присутствии определенной функциональной группы, или замене одной функциональной группы другой функциональной группой). Таким образом, аналогом является соединение, которое аналогично или сопоставимо по функции и внешнему виду с эталонным соединением.
Используемый в данном документе термин «апротонный растворитель» относится к растворителю, который является относительно инертным по отношению к протонной активности, то есть не действует в качестве донора протонов. Примеры включают, но не ограничиваются указанными, углеводороды, такие как гексан и толуол, например, галогенированные углеводороды, такие как, например, метиленхлорид, этиленхлорид, хлороформ и тому подобное, гетероциклические соединения, такие как, например, тетрагидрофуран и N-метилпирролидинон, и простые эфиры, такие как диэтиловый эфир, бис-метоксиметиловый эфир. Такие растворители хорошо известны специалистам в данной области техники, и отдельные растворители или их смеси могут быть предпочтительными для конкретных соединений и условий реакции в зависимости от таких факторов, как, например, растворимость реагентов, реакционная способность реагентов и предпочтительные диапазоны температур. Дополнительные обсуждения апротонных растворителей можно найти в учебниках по органической химии или в специализированных монографиях, например: Organic Solvents Physical Properties and Methods of Purification, 4th ed., edited by John A. Riddick et al., Vol. II, in the Techniques of Chemistry Series, John Wiley& Sons, NY, 1986.
Используемые в данном документе термины «протогенный органический растворитель» или «протонный растворитель» относятся к растворителю, который способен обеспечивать протоны, такому как спирт, например метанол, этанол, пропанол, изопропанол, бутанол, трет-бутанол и тому подобное. Такие растворители хорошо известны специалистам в данной области, и отдельные растворители или их смеси могут быть предпочтительными для конкретных соединений и условий реакции в зависимости от таких факторов, как, например, растворимость реагентов, реакционная способность реагентов и предпочтительные диапазоны температур. Дополнительные обсуждения протогенных растворителей можно найти в учебниках по органической химии или в специализированных монографиях, например: Organic Solvents Physical Properties and Methods of Purification, 4th ed., edited by John A. Riddick et al., Vol. II, in the Techniques of Chemistry Series, John Wiley & Sons, NY, 1986.
Комбинации заместителей и переменных, предусмотренных настоящим изобретением, представляют собой только те, которые приводят к образованию стабильных соединений. Используемый в данном документе термин «стабильный» относится к соединениям, которые обладают достаточной стабильностью для производства, которая поддерживает целостность соединения в течение достаточного периода времени для применения по назначениям, подробно описанным в данном документе (например, терапевтическое или профилактическое введение субъекту).
Синтезированные соединения могут быть отделены от реакционной смеси и дополнительно очищены способом, таким как колоночная хроматография, высокоэффективная жидкофазная хроматография или перекристаллизация. Кроме того, различные стадии синтеза могут быть выполнены в альтернативной последовательности или порядке с получением желаемых соединений. Кроме того, растворители, температуры, длительности реакции и т.д., описанные в данном документе, предназначены только для иллюстрации, и изменение условий реакции может приводить к желаемым мостиковым макроциклическим продуктам по настоящему изобретению. Трансформации синтетической химии и методология защитных групп (защита и снятие защиты), применимые для синтеза соединений, описанных в данном документе, включают, например, раскрытые в R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 2d. Ed., John Wiley and Sons (1991); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); и L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995).
Соединения по настоящему изобретению могут быть модифицированы путем добавления различных функциональных возможностей с помощью синтетических средств, приведенных в данном документе, для повышения селективных биологических свойств. Такие модификации включают те, которые увеличивают биологическое проникновение в заданную биологическую систему (например, кровь, лимфатическую систему, центральную нервную систему), увеличивают пероральную доступность, повышают растворимость, делая возможным введение путем инъекции, изменяют метаболизм и изменяют скорость выделения.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Фармацевтические композиции по настоящему изобретению содержат терапевтически эффективное количество соединения по настоящему изобретению, получаемого вместе с одним или более фармацевтически приемлемыми носителями. Используемый в данном документе термин «фармацевтически приемлемый носитель» означает нетоксичный, инертный твердый, полутвердый или жидкий наполнитель, разбавитель, материал для инкапсулирования или вспомогательный состав любого типа. Некоторыми примерами материалов, которые могут служить в качестве фармацевтически приемлемых носителей, являются сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; эксципиенты, такие как масло какао и суппозиторные воски; масла, такие как арахисовое масло, хлопковое масло; сафлоровое масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли; такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический физиологический раствор; раствор Рингера; этиловый спирт и фосфатные буферные растворы, а также другие нетоксичные совместимые смазывающие вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, разделительные вещества, покрывающие вещества, подсластители, ароматизаторы и отдушки, консерванты и антиоксиданты также могут присутствовать в композиции согласно решению составителя рецептуры. Фармацевтические композиции по настоящему изобретению могут быть введены людям и другим животным перорально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местно (как в виде порошков, мазей или капель), буккально или в виде перорального или назального спрея.
Фармацевтические композиции по настоящему изобретению могут быть введены перорально, парентерально, путем ингаляционного спрея, местно, ректально, назально, буккально, вагинально или через имплантированный резервуар, предпочтительно путем перорального введения или введения путем инъекции. Фармацевтические композиции по настоящему изобретению могут содержать любые обычные нетоксичные фармацевтически приемлемые носители, адъюванты или разбавители. В некоторых случаях рН состава можно регулировать с помощью фармацевтически приемлемых кислот, оснований или буферных растворов для повышения стабильности полученного соединения или его формы доставки. Используемый в данном документе термин «парентеральный» включает подкожную, внутрикожную, внутривенную, внутримышечную, интраартикулярную, внутриартериальную, интрасиновиальную, внутригрудинную, интратекальную, внутриочаговую и внутричерепную инъекцию или способы инфузии.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, из зародышей, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот сорбитана и их смеси. Помимо инертных разбавителей, композиции для перорального введения могут также содержать адъюванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, ароматизаторы и отдушки.
Инъекционные формы, например, стерильные инъекционные водные или масляные суспензии, могут быть получены в соответствии с известным уровнем техники с применением подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильная инъекционная форма может также представлять собой стерильный инъекционный раствор, суспензию или эмульсию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых разбавителей и растворителей, которые могут быть применены, могут быть вода, раствор Рингера, U.S.Р. и изотонический раствор хлорида натрия. Кроме того, стерильные, нелетучие масла обычно применяют в качестве растворителя или суспендирующей среды. Для этого назначения можно использовать любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для получения инъекционных форм применяют жирные кислоты, такие как олеиновая кислота.
Инъекционные составы могут быть стерилизованы, например, путем фильтрации через бактериальный удерживающий фильтр или путем введения стерилизующих агентов в форме стерильных твердых композиций, которые можно растворить или диспергировать в стерильной воде или другой стерильной инъекционной среде перед применением.
Чтобы продлить действие лекарственного средства, часто желательно замедлять абсорбцию лекарственного средства от подкожной или внутримышечной инъекции. Это может быть достигнуто за счет применения жидкой суспензии кристаллического или аморфного материала с плохой водорастворимостью. Скорость поглощения лекарственного средства тогда зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристалла и кристаллической формы. Альтернативно, замедленное всасывание парентерально вводимой лекарственной формы осуществляют путем растворения или суспендирования лекарственного средства в масляном разбавителе. Инъекционные депо-формы получают путем формирования микрокапсульных матриц лекарственного средства в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от соотношения лекарственного средства и полимера, и природы конкретного используемого полимера скорость высвобождения лекарственного средства можно контролировать. Примеры других биодеградируемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъекционные депо-составы также получают путем захвата лекарственного средства в липосомы или микроэмульсии, которые совместимы с тканями организма.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые могут быть получены путем смешивания соединений по настоящему изобретению с подходящими не раздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или суппозиторный воск, которые являются твердыми при температуре окружающей среды, но становятся жидкими при температуре тела и, следовательно, плавятся в прямой кишке или влагалищной полости с высвобождением активного соединения.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешивают с по меньшей мере одним инертным, фармацевтически приемлемым эксципиентом или носителем, таким как цитрат натрия или дикальцийфосфат, и/или: а) наполнителем или заполнителем, таким как крахмалы, лактоза, сахароза, глюкоза, маннит, и кремниевая кислота, b) связующими веществами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и аравийская камедь, с) увлажнителями, такими как глицерин, d) дезинтегрирующими агентами, такими как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия, е) замедляющими растворение агентами, такими как парафин, f) ускорителями абсорбции, такими как соединения четвертичного аммония, g) смачивающими агентами, такими как, например, цетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолин и бентонитовая глина и i) смазывающими вещества, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль лекарственная форма может также содержать буферные агенты.
Твердые композиции аналогичного типа также могут быть использованы в качестве наполнителей в мягких и труднозаполняемых желатиновых капсулах с применением таких эксципиентов, как лактоза или молочный сахар, а также полиэтиленгликолей высокой молекулярной массы и тому подобное.
Активные соединения также могут быть в микроинкапсулированной форме с одним или более эксципиентами, как указано выше. Твердые лекарственные формы таблеток, драже, капсул, пилюль и гранул могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, контролирующие высвобождение покрытия и другие покрытия, хорошо известные в фармацевтической области техники. В таких твердых лекарственных формах активное соединение можно смешивать по меньшей мере с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы могут также включать, в качестве обычной практики, дополнительные вещества, отличные от инертных разбавителей, например, смазывающие вещества для таблетирования и другие средства для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль лекарственные формы могут также содержать буферные агенты. Они могут необязательно содержать замутняющие агенты и могут также быть такого состава, что они высвобождают активный ингредиент(ы) только, или предпочтительно, в определенной части кишечного тракта, необязательно с задержкой. Примеры капсулирующих композиций, которые могут быть применены, включают полимерные вещества и воски.
Лекарственные формы для местного или трансдермального введения соединения по настоящему изобретению включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, ингалянты или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферными растворами, которые могут потребоваться. Офтальмологический состав, ушные капли, глазные мази, порошки и растворы также рассматриваются как входящие в объем настоящего изобретения.
Мази, пасты, кремы и гели могут содержать в дополнение к активному соединению по настоящему изобретению эксципиенты, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремниевые кислоты, тальк и оксид цинка или их смеси.
Порошки и спреи могут содержать помимо соединений по настоящему изобретению эксципиенты, такие как лактоза, тальк, кремниевая кислота, гидроксид алюминия, силикаты кальция и порошок полиамида или смеси этих веществ. Спреи могут дополнительно содержать обычные пропелленты, такие как хлорфторуглеводороды.
У трансдермальных пластырей есть дополнительное преимущество обеспечения контролируемой доставки соединения в организм. Такие лекарственные формы могут быть получены путем растворения или распределения соединения в соответствующей среде. Усилители абсорбции также могут быть использованы для увеличения тока соединения через кожу. Скорость можно регулировать либо путем обеспечения мембраны, контролирующей скорость, либо путем диспергирования соединения в полимерной матрице или геле.
Если не указано иное, все технические и научные термины, используемые в данном документе, имеют значение, известное среднему специалисту в данной области техники. Все публикации, патенты, опубликованные заявки на патенты и другие ссылки, упомянутые в данном документе, настоящим включены в качестве ссылки во всей их полноте.
Сокращения
Сокращения, которые были использованы в описаниях схем и нижеследующих примеров:
ACN для ацетонитрил;
ВМЕ для 2-меркаптоэтанола;
ВОР для гексафторфосфата бензотриазол-1-илокси-трис(диметиламино)фосфония;
BzCl для бензоилхлорида;
CDI для карбонилдиимидазола;
COD для циклооктадиена;
DABCO для 1,4-диазабицикло[2.2.2]октана;
DAST для диэтиламиносульфата трифторида;
DABCYL для 6-(N-4'-карбокси-4-(диметиламино)азобензол)-аминогексил-1-O-(2-цианоэтил)-(N,N-диизопропил)-амидофосфита;
DBU для 1,8-диазабициклоундец-7-ена;
DCC для N,N'-дициклогексилкарбодиимида;
DCM для дихлорметана;
DIAD для диизопропилазодикарбоксилата;
DIBAL-H для диизобутилалюминийгидрида;
DIPEA для диизопропилэтиламина;
DMAP для N,N-диметиламинопиридина;
DME для диметилового эфира этиленгликоля;
DMEM для среды Игла, модифицированной по способу Дульбекко;
DMF для N,N-диметилформамида;
DMSO для диметилсульфоксида;
DSC для N,N'-дисукцинимидилкарбоната;
DPPA для дифенилфосфорилазида;
DUPHOS для 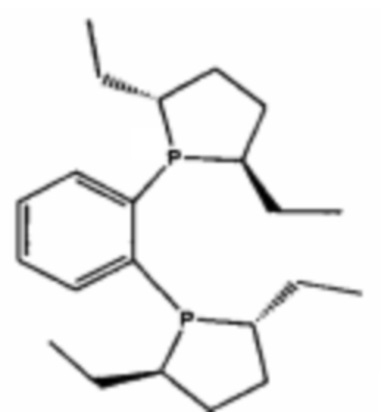 ;
;
EDANS для 5-(2-амино-этиламино)нафталин-1-сульфоновой кислоты;
EDCl или EDC для 1-(3-диэтиламинопропил)-3-этилкарбодиимида гидрохлорида;
EtOAc для этилацетата;
EtOH для этилового спирта;
HATU для O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата;
HCl для соляной кислоты;
Hoveyda's Cat. для дихлор(орто-изопропоксифенилметилен) (трициклогексилфосфин)рутения(II);
In для индия;
KHMDS представляет собой бис(триметилсилил)амид калия;
Ms для метилсульфонила;
NMM для N-4-метилморфолина;
NMI для N-метилимидазола;
NMO для N-4-метилморфолин-N-оксида;
PyBrOP для бромтрипиролидинфосфония гексафторфосфата;
Ph для фенила;
RCM для метатезисной реакции с закрытием цикла;
RT для обратной транскрипции;
RT-PCR для полимеразной цепной реакции с обратной транскрипцией;
ТВМЕ для трет-бутилметилового эфира;
TEA для триэтиламина;
Tf2O для ангидрида трифторметансульфокислоты;
TFA для трифторуксусной кислоты;
THF для тетрагидрофурана;
TLC для тонкослойной хроматографии;
(TMS)2NH для гексаметилдисилазана;
TMSOTf для триметилсилилтрифторметансульфоната;
TBS для трет-бутилдиметилсилила;
TMS для триметилсилила;
ТРАР тетрапропиламмония перрутената;
ТРР или PPh3 для трифенилфосфина;
TrCl для тритилхлорида;
DMTrCl для 4,4'-диметокситритилхлорида;
tBOC или Boc для трет-бутилоксикарбонила.
Способы синтеза
Соединения и способы по настоящему изобретению станут более поняты в сочетании со следующими схемами синтеза, которые иллюстрируют способы, с помощью которых могут быть получены соединения по изобретению, которые предназначены только для иллюстрации и не ограничивают объем изобретения. Различные изменения и модификации раскрытых вариантов осуществления будут очевидны для специалистов в данной области и такие изменения и модификации, включая, без ограничения, те, которые связаны с химическими структурами, заместителями, производными и/или способами по изобретению, могут быть осуществлены без отступления от сущности изобретения и объема прилагаемой формулы изобретения.
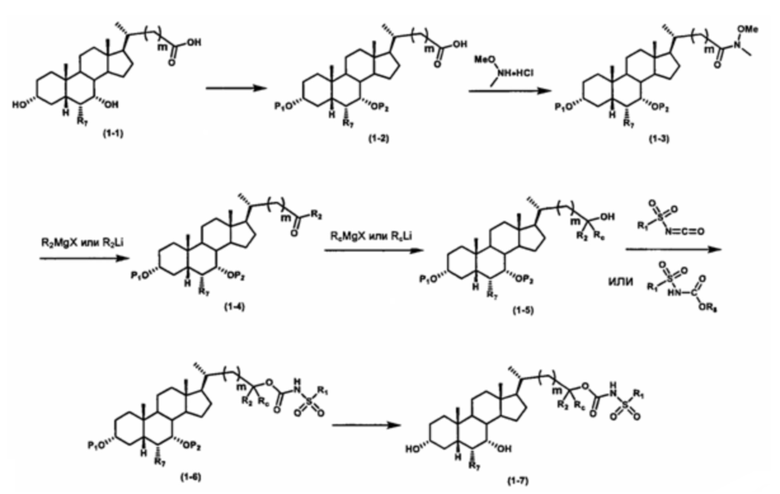
Как показано на схеме 1, новые аналоги желчной кислоты соединения формулы (1-7) получают из соединения формулы (1-1), где R1, R2, Rc, m и R7 являются такими, как определено ранее, R8 представляет собой замещенный или незамещенный -C1-C8 алкил; замещенный или незамещенный -C2-C8 алкенил; замещенный или незамещенный -C2-C8 алкинил; замещенный или незамещенный арилалкил; или замещенный или незамещенный арил; и Р1 и Р2 представляют собой гидроксизащитные группы. Таким образом, две гидроксильные группы соединения формулы (1-1) защищены группами P1 и P2 с получением соединения формулы (1-2). P1 и P2 могут быть одинаковыми или разными. P1 и P2 могут представлять собой любую гидроксизащитную группу, такую как, но не ограничиваясь указанными, Ac, Bz, хлорацетил, TES, TBS, MOM и Bn. Более подробное обсуждение методик, реагентов и условий для защиты гидроксильной группы раскрыто в литературе, например T.W. Greene and P.G.M. Wuts in "Protective Groups in Organic Synthesis" 3rd ed., John Wiley & Son, Inc., 1999. Затем соединение формулы (1-2) реагирует с N,O-диметилгидроксиамина гидрохлоридом с получением соединения формулы (1-3) в присутствии связующего реагента, такого как, но не ограничиваясь указанными, HATU, EDCl, DCC, HBTU и т.д. и основания, такого как, но не ограничиваясь указанными, TEA, DIPEA, DMAP и т.д. Соединение формулы (1-3) превращают в кетон формулы (1-4) путем взаимодействия с реактивом Гриньяра R2MgX или литиевым реактивом R2Li. Реакционный растворитель может представлять собой, но не ограничиваясь указанными, THF, эфир и толуол. Предпочтительным растворителем является THF. Температура реакции составляет от -78°С до 40°С. Соединение формулы (1-4) превращают в спирт формулы (1-5) путем взаимодействия с реактивом Гриньяра RcMgX или литиевым реактивом RcLi. Реакционный растворитель может представлять собой, но не ограничиваясь указанными, THF, эфир и толуол. Предпочтительным растворителем является THF. Температура реакции составляет от -78°С до 40°С. Реакция с сульфонилизоцианатами или сульфонилкарбаматами приводит к получению соединения формулы (1-6). Затем снятие защитных групп P1 и P2 приводит к получению соединения сульфонилмочевины формулы (1-7). Более подробное обсуждение методик, реагентов и условий для снятия гидроксизащитных групп описано в литературе, например T.W. Greene and P.G.M. Wuts in "Protective Groups in Organic Synthesis" 3rd ed., John Wiley & Son, Inc., 1999.
Альтернативно, если Rc=R2, промежуточное соединение формулы (1-5) может быть получено из сложного эфира соединения формулы (1-2) путем взаимодействия с избыточным количеством реактива Гриньяра RcMgX или литиевого реактива RcLi.
Схема 1
Как показано на схеме 2, альтернативно, соединение формулы (2-3) получают из соединения формулы (1-2). P1 и P2 могут быть одинаковыми или разными. P1 и P2 могут представлять собой любую гидроксизащитную группу, такую как, но не ограничиваясь указанными, Ac, Bz, хлорацетил, TES, TBS, MOM и Bn. Более подробное обсуждение методик, реагентов и условий для защиты гидроксильной группы описано в литературе, например T.W. Greene and P.G.M. Wuts in "Protective Groups in Organic Synthesis" 3rd ed., John Wiley & Son, Inc., 1999.
Затем соединение формулы (1-2) превращают в спирт формулы (2-1) с использованием подходящего восстанавливающего реагента, такого как, но не ограничиваясь указанными, LiAlH4, ВН3 и т.д. Растворителем реакции может быть, но не ограничиваясь указанными, THF, эфир и толуол. Предпочтительным растворителем является THF. Температура реакции составляет от -20°С до 40°С. Реакция с сульфонилизоцианатами или сульфонилкарбаматами приводит к получению соединения формулы (2-2). Затем снятие защитных групп P1 и P2 приводит к получению соединения сульфонилмочевины формулы (2-3). Более подробное обсуждение методик, реагентов и условий для снятия защиты гидроксизащитных групп описано в литературе, например T.W. Greene and P.G.M. Wuts in "Protective Groups in Organic Synthesis" 3rd ed., John Wiley & Son, Inc., 1999.
Схема 2
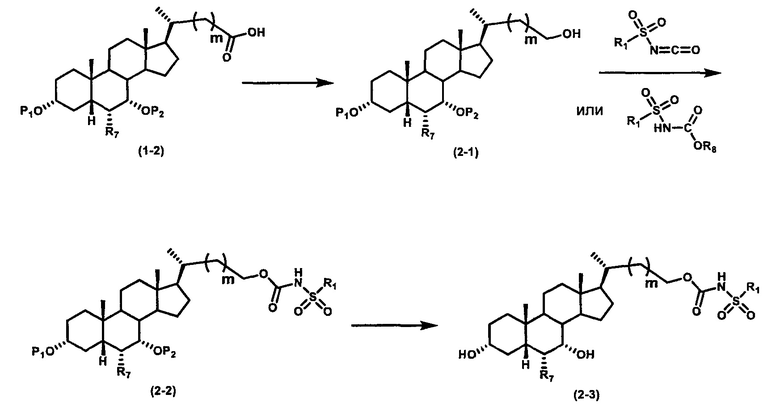
Схема 3 иллюстрирует получение карбаматного соединения формулы (3-2) из соединения формулы (1-5), где R2, Ra, Rc, m, R7 и R8 являются такими, как определено ранее, Р1 и Р2 представляют собой гидроксизащитные группы. Таким образом, соединение формулы (1-5) реагирует с изоцианатами или карбаматами с получением соединения формулы (3-1) в присутствии основания, такого как, но не ограничиваясь указанными, DBU, TEA, DIPEA, DMAP и т.д. Затем снятие защитных групп P1 и P2 приводит к получению соединения мочевины формулы (3-2). Более подробное обсуждение методик, реагентов и условий для снятия защиты гидроксизащитных групп описано в литературе, например T.W. Greene and P.G.M. Wuts in "Protective Groups in Organic Synthesis", 3rd ed., John Wiley & Son, Inc., 1999.
Схема 3
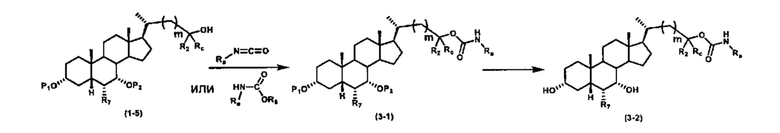
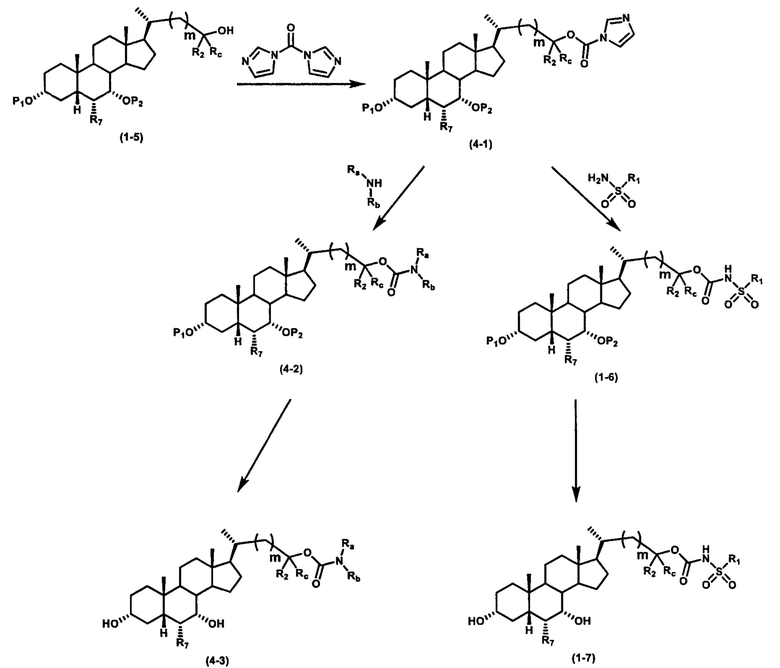
Схема 4 иллюстрирует альтернативный способ получения карбаматных соединений формулы (4-3) и (1-7) из соединения формулы (1-5), где R1, R2, Ra, Rb, Rc, m и R7 являются такими, как определено ранее, P1 и Р2 представляют собой гидроксизащитные группы. Таким образом, соединение формулы (1-5) превращают в соединение формулы (4-1) путем взаимодействия с CDI в присутствии основания, такого как, но не ограничиваясь указанными, DBU, TEA, DIPEA и DMAP. Затем соединение формулы (4-1) приводят во взаимодействие с амином RaNHRb или сульфонамидом R1SO2NH2 однореакторным способом с получением карбаматных соединений формулы (4-2) и (1-6). Дальнейшее снятие защиты гидроксизащитной группы P1 и Р2 приводит к получению соединения формул (4-3) и (1-7). Более подробное обсуждение методик, реагентов и условий для защиты и снятия гидроксизащитных групп и аминозащитной группы раскрыто в литературе, например T.W. Greene and P.G.M. Wuts in "Protective Groups in Organic Synthesis" 3rd ed., John Wiley & Son, Inc., 1999.
Схема 4
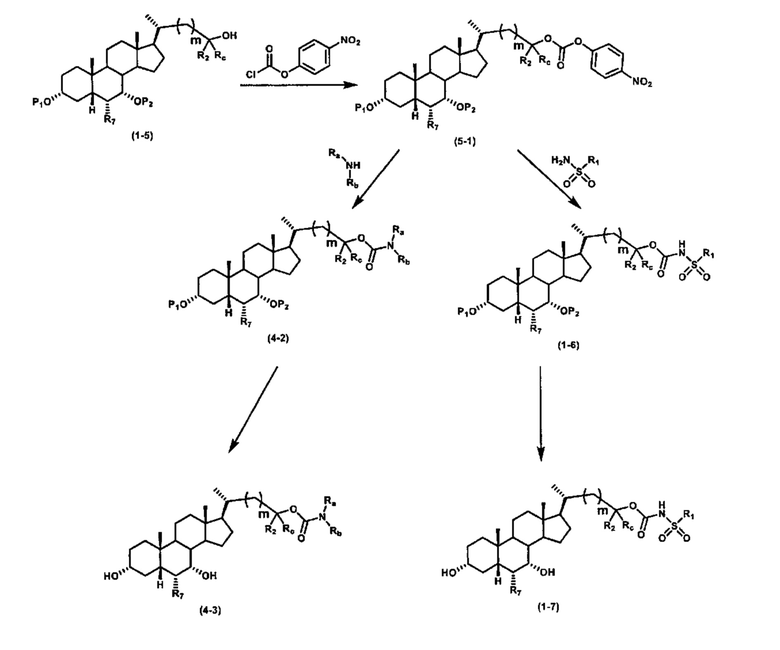
Альтернативный способ получения карбаматных соединений формулы (4-2) и (1-7) показан на схеме 5, где R1, R2, Ra, Rb, Rc, m и R7 являются такими, как определено ранее, Р1 и Р2 представляют собой гидроксизащитные группы. Соединение формулы (1-3) реагирует с пара-нитрофенилхлорформиатом с получением карбонатного соединения формулы (5-1) в присутствии основания. Подходящие основания включают, но не ограничиваются указанными, триэтиламин, диизопропилэтиламин, DBU, N-метилморфолин и DMAP. Реакцию проводят в апротонном растворителе, таком как, но не ограничиваясь указанными, CH2Cl2, DMF или THF. Температура реакции может изменяться от 0°С до приблизительно 50°С. Соединение формулы (5-1) приводят во взаимодействие с амином RaNHRb или сульфонамидом R1SO2NH2 однореакторным способом с получением карбаматных соединений формул (4-2) и (1-6). Дальнейшее снятие гидроксизащитной группы Р1 и P2 приводит к получению соединения формулы (4-3) и (1-7). Более подробное обсуждение методик, реагентов и условий для защиты и снятия гидроксизащитных групп и аминозащитной группы раскрыто в литературе, например T.W. Greene and P.G.M. Wuts in "Protective Groups in Organic Synthesis" 3rd ed., John Wiley & Son, Inc., 1999.
Схема 5
R1S(O)2NH2 на схеме 4 и схеме 5 могут быть синтезированы следующими способами, но не ограничиваясь указанными.
Способ 1
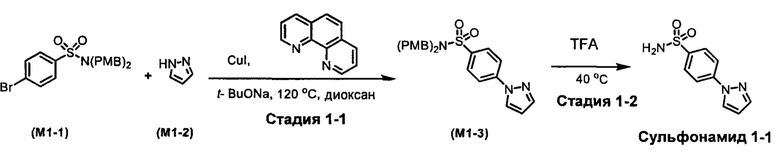
Стадия 1-1. Синтез М1-3
Смесь CuI (230 мг, 1,2 ммоль) и 1,10-фенантролина (220 мг, 1,2 ммоль) в 1,4-диоксане (15 мл) перемешивали при комнатной температуре в течение 10 мин в атмосфере N2. Затем к смеси добавляли 4-бром-N,N-бис(4-метоксибензил)бензолсульфонамид (соединение М1-1, 480 мг, 1 ммоль), 1Н-пиразол (210 мг, 3 ммоль) и t-BuONa (300 мг, 3 ммоль). После добавления эту смесь перемешивали при 120°С в течение 15 часов в атмосфере N2. Полученную смесь вливали в воду (200 мл) и экстрагировали EtOAc (100 мл × 2). Объединенный органический слой сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали флэш-хроматографией на силикагеле, градиент элюирования от 20 до 50% EtOAc в петролейном эфире. Чистые фракции выпаривали досуха с получением желаемого соединения М1-3 (150 мг, 32%) в виде желтого твердого вещества.
Стадия 1-2. Синтез сульфонамида 1-1
N,N-бис(4-метоксибензил)-4-(1Н-пиразол-1-ил)бензолсульфонамид (соединение М1-3; 150 мг, 0,32 ммоль) растворяли в TFA (3 мл) и перемешивали при 40°С в течение 3 ч. Реакционную смесь концентрировали, добавляли воду (10 мл), доводили рН до 8 насыщенным NaHCO3 и экстрагировали EtOAc (10 мл × 2). Объединенную органическую фазу сушили над безводным Na2SO4, фильтровали и выпаривали с получением желаемого соединения (63 мг, 82%) в виде белого твердого вещества.
Следующие сульфонамиды синтезировали с применением вышеуказанного способа 1.
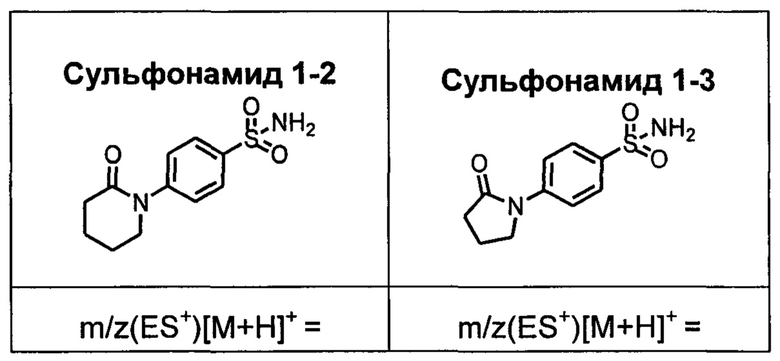
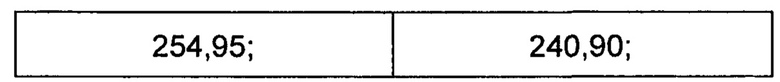
Способ 2
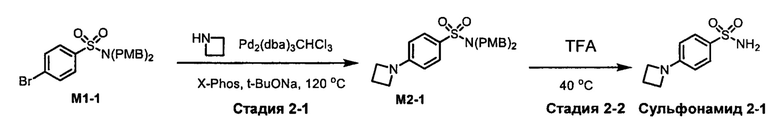
Стадия 2-1. Синтез М2-1
Смесь 4-бром-N,N-бис(4-метоксибензил)бензолсульфонамида (соединение М1-1; 192 мг, 0,4 ммоль), азетидина (96 мг, 1,6 ммоль), t-BuONa (80 мг, 0,8 ммоль), Pd2(dba)3CHCl3 (40 мг, 0,04 ммоль) и X-Phos (76 мг, 0,16 ммоль) в толуоле (10 мл) перемешивали при 120°С в течение 15 часов в атмосфере N2. Полученную смесь вливали в воду (50 мл) и экстрагировали EtOAc (30 мл × 2). Органический слой сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали флэш-хроматографией на силикагеле, градиент элюирования от 20 до 50% EtOAc в петролейном эфире. Чистые фракции выпаривали досуха с получением желаемого соединения (100 мг, 55%) в виде желтого твердого вещества.
Стадия 2-2. Синтез сульфонамида 2-1
4-(Азетидин-1-ил)-N,N-бис(4-метоксибензил)бензолсульфонамид (соединение М2-1; 90 мг, 0,2 ммоль) растворяли в TFA (5 мл). Полученный раствор перемешивали при 40°С в течение 3 часов. Реакционную смесь концентрировали, добавляли воду (100 мл), затем доводили рН до 8 насыщенным NaHCO3 и экстрагировали EtOAc (100 мл × 2). Органический слой сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали флэш-хроматографией на силикагеле, градиент элюирования от 40 до 60% EtOAc в петролейном эфире. Чистые фракции выпаривали досуха с получением желаемого соединения (35 мг, 8 3%) в виде желтого твердого вещества. m/z (ES+) [M*2+H]+=424,95.
Следующие сульфонамиды синтезировали с применением общей методики способа 2.
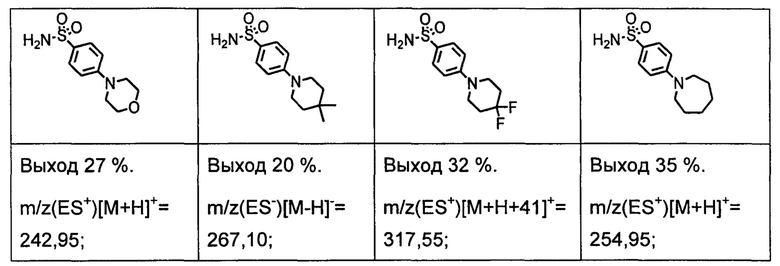
Способ 3
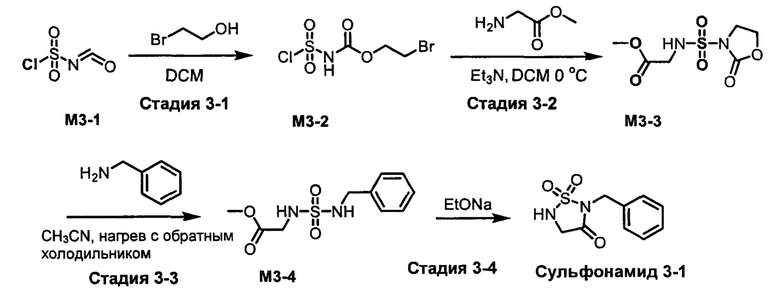
Стадия 3-1. Синтез М3-2
2-Бромэтанол (14,2 г, 100 ммоль) добавляли по каплям к хлорсульфонил изоцианату (соединение М3-1; 12,5 г, 100 ммоль) в DCM (50 мл) при 0°С в течение 10 минут в атмосфере азота. Полученную смесь перемешивали при 0°С в течение 0,5 часа. Реакционную смесь использовали непосредственно на следующей стадии.
Стадия 3-2. Синтез М3-3
К раствору метил-2-аминоацетата (8,9 г, 100 ммоль) и Et3N (20,2 г, 200 ммоль) в DMF (100 мл) добавляли 2-бромэтилхлорсульфонилкарбамат (соединение М3-2; 26,6 г, 100 ммоль) в течение 40 минут при охлаждении ледяной баней в атмосфере азота. Смесь перемешивали при 0°С в течение 5 часов, затем в течение 12 часов при комнатной температуре. Добавляли DCM (500 мл) и последовательно промывали 1Н HCl (200 мл × 2) и солевым раствором (200 мл × 2). Органический слой сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали флэш-хроматографией на силикагеле, градиент элюирования от 20 до 40% EtOAc в петролейном эфире. Чистые фракции выпаривали досуха с получением желаемого соединения (8,9 г, 37%) в виде желтого твердого вещества.
Стадия 3-3. Синтез М3-4
Метил-2-(2-оксооксазолидин-3-сульфонамидо)ацетат (соединение М3-3, 2,38 г, 10 ммоль) добавляли к фенилметанамину (1,28 г, 12 ммоль) и Et3N (3,1 г, 30 ммоль) в CH3CN (50 мл). Полученный раствор перемешивали при нагревании с обратным холодильником в течение 2 часов. Реакционную смесь концентрировали и очищали флэш-хроматографией на силикагеле, градиент элюирования от 20 до 50% EtOAc в петролейном эфире. Чистые фракции выпаривали досуха с получением желаемого соединения (1,5 г, 58%) в виде желтого твердого вещества.
Стадия 3-4. Синтез сульфонамида 3-1
EtONa (5,1 г, 75 ммоль) добавляли к метил-2-(N-бензилсульфамоиламино)ацетату (соединение М3-4, 3,9 г, 15 ммоль) в EtOH (50 мл) при комнатной температуре. Смесь перемешивали при 50°С в течение 5 часов. Затем смесь концентрировали и очищали флэш-хроматографией на силикагеле, градиент элюирования от 30 до 70% EtOAc в петролейном эфире. Чистые фракции выпаривали досуха с получением желаемого соединения (1,2 г, 35%) в виде желтого твердого вещества.
Способ 4
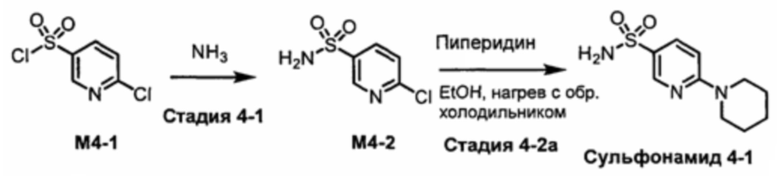
Стадия 4-1. Синтез М4-2
Аммиак (20 мл) добавляли к 6-хлорпиридин-3-сульфонилхлориду (соединение М4-1; 2,0 г, 9,48 ммоль) в MeCN (5 мл). Смесь перемешивали при комнатной температуре в течение 30 мин. Смесь разбавляли EtOAc (100 мл) и промывали солевым раствором. Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флэш-преп-ВЭЖХ ((IntelFlash-1): Колонка, С18; подвижная фаза MeCN/H2O=0/100, увеличивающаяся до MeCN/H2O=10/90 в течение 20 мин, УФ-детектор 254 нм) с получением РН-ЕТА-С-330-1 (1,45 г, 79,6%) в виде белого твердого вещества. m/z (ES+) [М+Н]+=192,70; ВЭЖХ tR (время удержания) = 0,403 мин.
Стадия 4-2а. Синтез сульфонамида 4-1
К пиперидину (4 мл) добавляли раствор 6-хлорпиридин-3-сульфонамида (соединение М4-2; 400 мг, 2,08 ммоль) в этаноле (5 мл). Смесь перемешивали при 90°С в течение ночи. Смесь разбавляли EtOAc (100 мл) и промывали солевым раствором. Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флэш-преп-ВЭЖХ ((IntelFlash-1): колонка, С18; подвижная фаза MeCN/H2O=0/100, увеличивающаяся до MeCN/H2O=20/80 в течение 20 мин, УФ-детектор 254 нм) с получением РН-ЕТА-С-330-2 (280 мг, 55,8%) в виде желтого твердого вещества. m/z (ES+) [М+Н]+=241,95; ВЭЖХ tR=0,787 мин.
Стадия 4-2b. Синтез сульфонамида 4-2
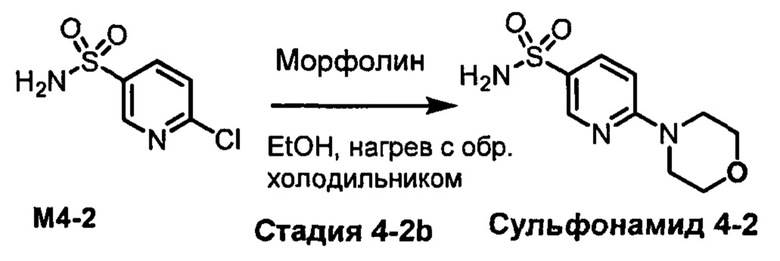
К морфолину (4 мл) добавляли раствор М4-2 (400 мг, 2,08 ммоль) в этаноле (5 мл). Смесь перемешивали при 90°С в течение ночи. Смесь разбавляли EtOAc (100 мл) и промывали насыщенным солевым раствором. Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флэш-преп-ВЭЖХ ((IntelFlash-1): колонка, С18, подвижная фаза MeCN/H2O=0/100, увеличивающаяся до MeCN/H2O=20/80 в течение 20 мин, УФ-детектор 254 нм) с получением РН-ЕТА-С-331-1 (290 мг, 57,4%) в виде желтого твердого вещества. m/z (ES-) [M-H]-=242,00; ВЭЖХ tR=0,496 мин.
Следующие сульфонамиды синтезировали с применением общей методики способа 4.
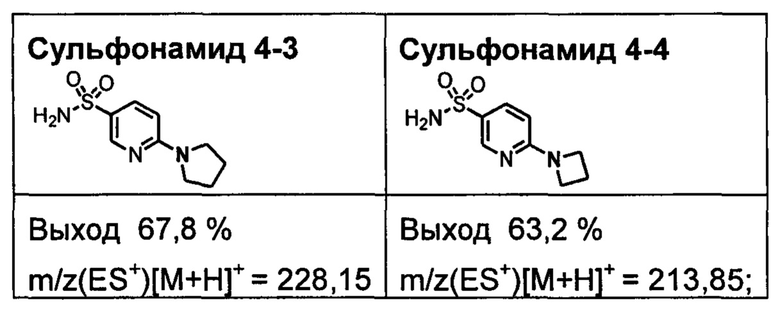
Способ 5
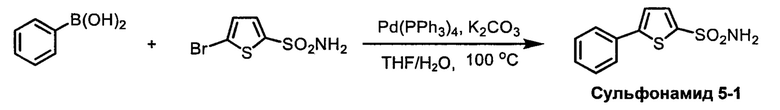
Pd(PPh3)4 (115 мг, 0,05 ммоль) добавляли в фенилборную кислоту (300 мг, 2,4 ммоль), 5-бромтиофен-2-сульфонамид (500 мг, 2 ммоль) и K2CO3 (1,2 г, 10 ммоль) в THF (10 мл) и H2O (2 мл). Смесь перемешивали при 90°С в течение 1 часа. Смесь гасили водой, экстрагировали этилацетатом (50 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией на силикагеле, градиент элюирования от 50 до 100% EtOAc в петролейном эфире. Чистые фракции выпаривали досуха с получением целевого соединения (310 мг, 62,8%) в виде желтого твердого вещества. m/z (ES-) [M-H]-=237,95, tR=0,872 мин.
Следующие сульфонамиды синтезировали с применением общей методики способа 5.
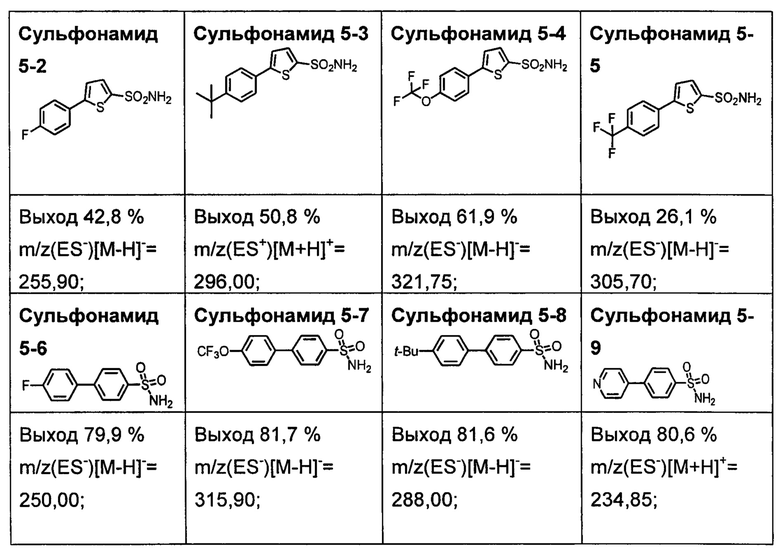
Способ 6
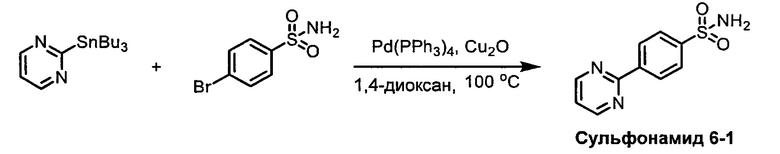
Pd(PPh3)4 (98 мг, 0,08 ммоль) добавляли к 2-(трибутилстаннил)пиримидину (375 мг, 1,02 ммоль), 4-бромбензолсульфонамиду (200 мг, 0,85 ммоль) и Cu2O (12 мг, 0,08 ммоль) в 1,4-диоксане (5 мл). Смесь перемешивали при 100°С в течение ночи в атмосфере N2. Реакционную смесь разбавляли керосином, добавляли насыщенный раствор KF и осажденный станнилфторид отфильтровывали и фильтрат экстрагировали EtOAc (20 мл), промывали водой (10 мл), сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали флэш-хроматографией на силикагеле, градиенто элюирования от 50 до 100% EtOAc в петролейном эфире. Чистые фракции выпаривали досуха с получением желаемого сульфонамида 6-1 (120 мг, 60%) в виде белого твердого вещества, m/z (ES+) [M*2+Н]+=470,85, tR=0,602 мин.
Способ 7
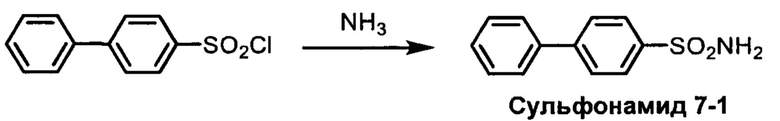
Раствор бифенил-4-сульфонилхлорида (200 мг, 0,79 ммоль) в NH3/МеОН (3 мл, 7 М) перемешивали в течение ночи при комнатной температуре. Метанол удаляли при пониженном давлении и добавляли EtOAc (10 мл). Затем промывали последовательно водой (10 мл) и насыщенным NaCl (10 мл). Органическую фазу сушили над Na2SO4, затем фильтровали и выпаривали с получением бифенил-4-сульфонамида 7-1 (170 мг, 93%) в виде белого твердого вещества. m/z (ES-) [M-H]-=232,10, ВЭЖХ tR=0,650 мин.
Следующие сульфонамиды синтезировали с применением общей методики способа 7.
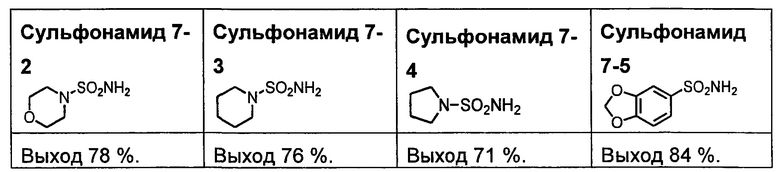
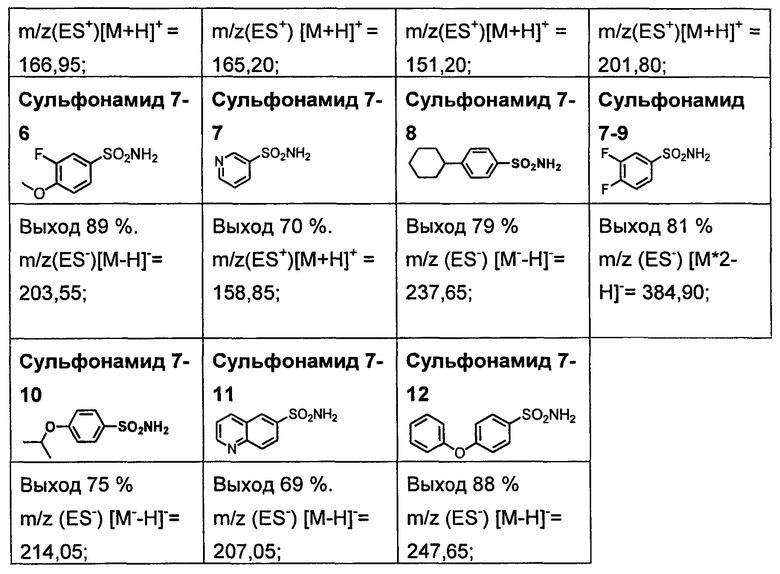
Способ 8
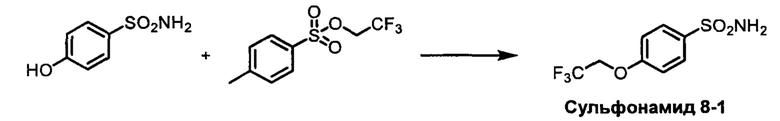
4-гидроксибензолсульфонамид (500 мг, 2,89 ммоль) растворяли в DMA (5 мл). Полученный раствор нагревали до 50°С, затем медленно добавляли 50% KOH (0,5 мл). Смесь выдерживали при 60°С в течение примерно 30 минут. Образовавшуюся воду выпаривали при пониженном давлении. Медленно добавляли 2,2,2-трифторэтил-4-метилбензолсульфонат, поддерживая температуру реакционной смеси между 120°С и 140°С. Затем реакционную смесь перемешивали при 130°С в течение 3 часов. Остаток растворяли в воде и экстрагировали EtOAc (20 мл × 3). Объединенный органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали флэш-преп-ВЭЖХ: колонка С18; подвижная фаза, MeCN/H2O=0/100, увеличивающаяся до MeCN/H2O=55/45 в течение 30 мин; УФ-детектор 254 нм, с получением желаемого сульфонамида 8-1, 4-(2,2,2-трифторэтокси)бензолсульфонамида (300 мг, 40%) в виде желтого твердого вещества. m/z (ES-) [М-Н]-=253,95; ВЭЖХ tR=1,172 мин.
Способ 9
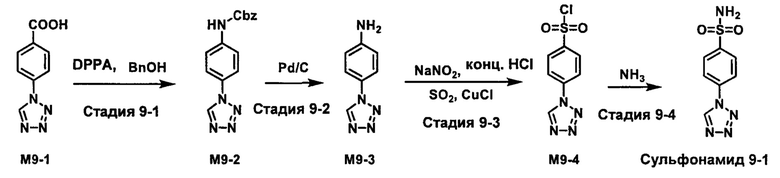
Стадия 9-1. Синтез М9-2
Смесь 4-(1Н-тетразол-1-ил)бензойной кислоты (соединение М9-1, 500 мг, 2,64 ммоль), DPPA (868 мг, 3,16 ммоль), Et3N (800 мг, 7,92 ммоль) и BnOH (5,7 г, 53 ммоль) в 1,4-диоксане/DMSO (15 мл / 10 мл) перемешивали при 100°С в течение 5 часов в атмосфере N2. Полученную смесь вливали в воду (200 мл) и экстрагировали EtOAc (100 мл × 2). Органический слой сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали флэш-хроматографией на силикагеле, градиент элюирования от 30 до 50% EtOAc в петролейном эфире. Чистые фракции выпаривали досуха с получением желаемого соединения (600 мг, 72%) в виде белого твердого вещества.
Стадия 9-2. Синтез М9-3
К раствору бензил-4-(1Н-тетразол-1-ил)фенилкарбамата (соединение М9-2, 295 мг, 1 ммоль) в МеОН (20 мл) добавляли Pd/C (100 мг, 10%) в атмосфере N2. Суспензию дегазировали под вакуумом и продували Н2 несколько раз. Смесь перемешивали в атмосфере водорода при комнатной температуре в течение 2 часов. Затем Pd/C отфильтровывали и фильтрат концентрировали, получая желаемое соединение (152 мг, 93%) в виде белого твердого вещества.
Стадия 9-3. Синтез М9-4
К раствору 4-(1Н-тетразол-1-ил)бензоламина (соединение М9-3, 113 мг, 0,7 ммоль) и концентрированной HCl (2,8 мл) в CH3CN/СН3СООН (8 мл/1 мл) добавляли NaNO2 (58 мг, 0,875 ммоль) в воде (1 мл) при -10°С. Смесь перемешивали при 0°С в течение 0,5 часа, затем по каплям добавляли насыщенный SO2 в АсОН (5 мл, 1,4 ммоль) со скоростью, поддерживающей температуру реакции ниже 5°С. По каплям добавляли раствор дигидрата хлорида меди(I) (148 мг, 0,875 ммоль) в воде (1 мл). Реакционную смесь перемешивали при 0°С в течение 30 минут и затем при комнатной температуре в течение 3 часов. Смесь вливали в ледяную воду (100 мл) и экстрагировали EtOAc (20 мл × 2). Органический слой концентрировали с получением неочищенного соединения в виде желтого твердого вещества, которое непосредственно использовали на следующей стадии.
Стадия 9-4. Синтез сульфонамида 9-1
К раствору 4-(1Н-тетразол-1-ил)бензол-1-сульфонилхлорида (соединение М9-4, 122 мг, 0,5 ммоль) в CH3CN (5 мл) добавляли гидроксид аммония (2 мл). Смесь перемешивали при комнатной температуре в течение 30 мин. Затем добавляли воду (20 мл) и экстрагировали EtOAc (20 мл × 2). Органический слой сушили над Na2SO4 и концентрировали с получением желаемого 4-(1Н-тетразол-1-ил)бензолсульфонамида, сульфонамида 9-1 (80 мг, 71%) в виде желтого твердого вещества.
Способ 10
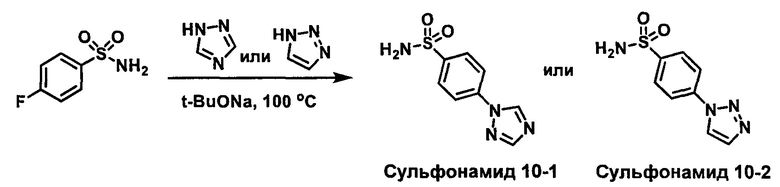
К раствору 4-фторбензолсульфонамида (175 мг, 1 ммоль) и 1H-1,2,4-триазола/1Н-1,2,3-триазола (140 мг, 2 ммоль) в DMSO (10 мл) добавляли t-BuONa (200 мг, 2 ммоль) в атмосфере азота. Смесь перемешивали при 100°С в течение 48 часов. Затем добавляли воду (100 мл) и экстрагировали EtOAc (20 мл × 2). Органический слой сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали флэш-хроматографией на силикагеле, градиент элюирования от 20 до 40% EtOAc в петролейном эфире. Чистые фракции выпаривали досуха, получая сульфонамид 10-1, 4-(1Н-1,2,4-триазол-1-ил)бензолсульфонамид (95 мг, выход 42%) в виде желтого твердого вещества. ВЭЖХ tR=0,482 мин, или сульфонамид 10-2, 4-(1Н-1,2,3-триазол-1-ил)бензолсульфонамид, выход 49%, ВЭЖХ tR=0,492 мин.
Способ 11
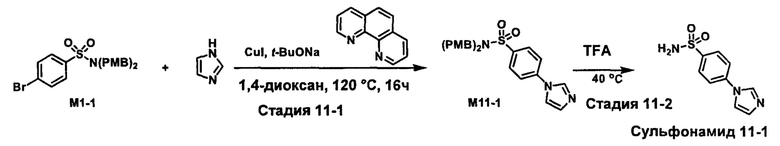
Стадия 11-1. Синтез М11-1
Смесь 4-бром-N,N-бис(4-метоксибензил)бензолсульфонамида (соединение М1-1, 200 мг, 0,42 ммоль), 1Н-имидазола (86 мг, 1,26 ммоль), CuI (96 мг, 0,51 ммоль), t-BuONa (121 мг, 1,26 ммоль) и 1,10-фенантролина (91 мг, 0,50 ммоль) в 1,4-диоксане (4 мл) перемешивали при 120°С в течение 16 часов в атмосфере N2. Реакционную смесь концентрировали и очищали флэш-хроматографией на силикагеле, градиент элюирования от 0 до 10% МеОН в DCM. Чистые фракции выпаривали досуха с получением желаемого сульфонамида 11-1 (120 мг, выход 61,5%) в виде желтого твердого вещества. m/z (ES+) [М+Н]+=464,10; ВЭЖХ tR=0,780 мин.
Стадия 11-2. Синтез сульфонамида 11-1
4-(1Н-имидазол-1-ил)-N,N-бис(4-метоксибензил)бензолсульфонамид (соединение М11-1; 120 мг, 0,26 ммоль) растворяли в TFA (2 мл). Полученный раствор перемешивали при 40°С в течение 2 часов. Реакционную смесь концентрировали, добавляли воду (100 мл), затем доводили рН до 8 насыщенным NaHCO3 и экстрагировали EtOAc (20 мл × 2). Органический слой сушили над Na2SO4, фильтровали и выпаривали с получением желаемого сульфонамида 11-1 (50 мг, выход 86%) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. m/z (ES+) [М+H]+=223,90; ВЭЖХ tR=0,187 мин.
Способ 12
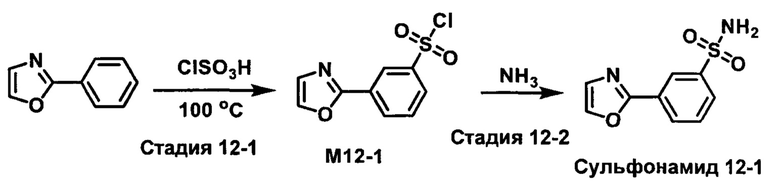
Стадия 12-1. Синтез М12-1
Раствор 2-фенилоксазола (200 мг, 1,4 ммоль) в сульфохлоридной кислоте (5 мл) перемешивали в течение 15 часов при 100°С в атмосфере N2. Полученную смесь вливали в ледяную воду (50 мл) и экстрагировали EtOAc (20 мл × 2). Органический слой сушили над Na2SO4 и концентрировали с получением неочищенного соединения М12-1 (180 мг, 52%) в виде желтого масла.
Стадия 12-2. Синтез сульфонамида 12-1
К раствору 4-(оксазол-2-ил)бензол-1-сульфонилхлорида (соединение М12-1, 121 мг, 0,5 ммоль) в CH3CN (5 мл) добавляли гидроксид аммония (2 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 30 мин. Затем добавляли воду (20 мл) и экстрагировали EtOAc (20 мл × 2). Органический слой сушили над Na2SO4 и концентрировали с получением желаемого сульфонамида 12-1, 3-(оксазол-2-ил)бензолсульфонамида (98 мг, 87%) в виде желтого твердого вещества.
Способ 13
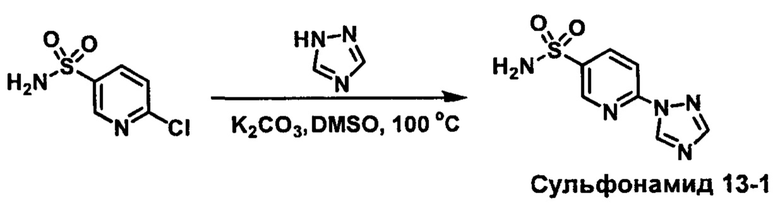
6-Хлорпиридин-3-сульфонамид (200 мг, 1,04 ммоль) добавляли к смеси 1Н-1,2,4-триазола (72 мг, 1,04 ммоль) и K2CO3 (287 мг, 2,08 ммоль) в DMSO (2 мл) при 100°С. Затем перемешивали в течение 16 часов при 100°С. Реакционную смесь разбавляли EtOAc (20 мл) после охлаждения до комнатной температуры, затем промывали насыщенным NaCl (10 мл × 2). Органическую фазу сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали флэш-хроматографией на силикагеле, градиент элюирования от 0 до 10% МеОН в DCM. Чистые фракции выпаривали досуха с получением желаемого сульфонамида 13-1, 6-(1Н-1,2,4-триазол-1-ил)пиридин-3-сульфонамида (70 мг, выход = 61,5%) в виде желтого твердого вещества, m/z (ES+) [M+H]+=225,85; ВЭЖХ tR=0,417 мин.
Следующие сульфонамиды синтезировали с применением общей методики способа 13.
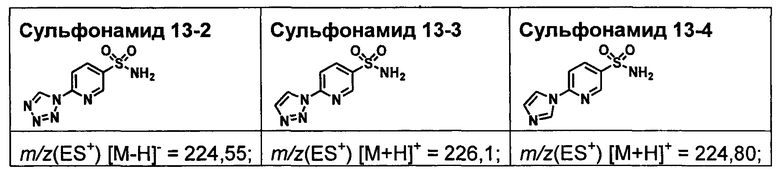
Способ 14
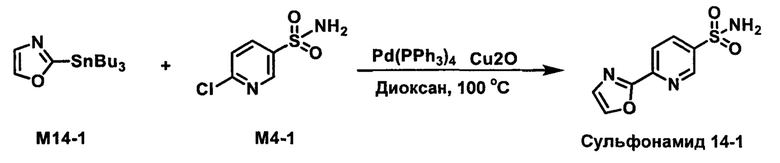
В 10-миллилитровую пробирку для проведения реакции под воздействием микроволнового излучения, содержащую азот, помещали 2-(трибутилстаннил)оксазол (соединение М14-1; 358 мг, 1 ммоль), 6-хлорпиридин-3-сульфонамид (соединение М4-1; 174 мг, 0,9 ммоль) Cu2O (143 мг, 1 ммоль), Pd(PPh3)4 (115,5 мг, 0,1 ммоль) и диоксан (5 мл). Полученную смесь перемешивали при микроволновом нагревании 100°С в течение 4 часов. После охлаждения до комнатной температуры раствор концентрировали и остаток растворяли в этилацетате (20 мл), затем промывали водой (10 мл × 2), насыщенным NaCl (10 мл × 2). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией на силикагеле, градиент элюирования от 0 до 10% МеОН в DCM с получением желаемого сульфонамида 14-1, 6-(оксазол-2-ил)пиридин-3-сульфонамида, в виде желтого твердого вещества (60 мг, 26,7%). m/z (ES+) [М+Н]+=225,80.
Способ 15
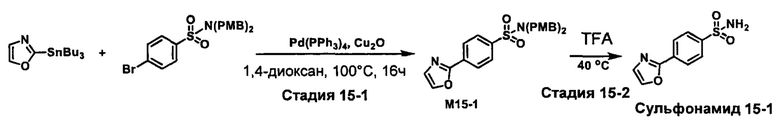
Стадия 15-1. Синтез М15-1
Смесь 2-(трибутилстаннил)оксазола (310 мг, 0,867 ммоль), 4-бром-N,N-бис(4-метоксибензил)бензолсульфонамида (282 мг, 0,788 ммоль), Cu2O (75 мг, 0,525 ммоль) и Pd(PPh3)4 (61 мг, 0,053 ммоль) в диоксане (10 мл) нагревали до 100°С в течение 1 часа в атмосфере N2. Смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток очищали колонкой с силикагелем с применением РЕ/ЕА=3/1 с получением 200 мг (выход 82%) N,N-бис(4-метоксибензил)-4-(оксазол-2-ил)бензолсульфонамида в виде белого твердого вещества. m/z (ES+) [M+H]+=465,20.
Стадия 15-2. Синтез сульфонамида 15-1
Смесь N,N-бис(4-метоксибензил)-4-(оксазол-2-ил)бензолсульфонамида (200 мг, 0,43 ммоль) в TFA (5 мл) перемешивали при 40°С в течение 3 ч и затем концентрировали. Остаток растворяли в EtOAc (10 мл), последовательно промывали насыщенным раствором NaHCO3 (10 мл × 1) и солевым раствором (10 мл × 1), органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением 90 мг (выход 93%) сульфонамида 15-1, 4-(оксазол-2-ил)бензолсульфонамида, в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. m/z (ES+) [M+H]+=224,90.
Способ 16
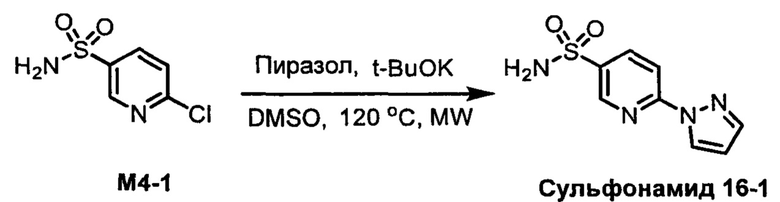
t-BuOK (176 мг, 1,57 ммоль) добавляли к М4-1 (200 мг, 1,04 ммоль) и пиразолу (142 мг, 2,08 ммоль) в DMSO (5 мл). Смесь нагревали в микроволновом аппарате при 120°С в течение 120 минут. Смесь разбавляли водой (20 мл) и доводили рН до 7 1Н HCl. Смесь экстрагировали EtOAc (30 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью флэш-преп-ВЭЖХ: колонка С18; подвижная фаза, MeCN/H2O=0/100, увеличивающаяся до MeCN/H2O=25/75 в течение 30 мин; УФ-детектор 254 нм, с получением РН-ЕТА-С-332-1 (150 мг, 42,7%) в виде желтого твердого вещества, сульфонамида 16-1. m/z (ES+) [M+H]+=224,80; ВЭЖХ tR=0,395 мин.
ПРИМЕРЫ
Соединения и способы по настоящему изобретению станут более поняты в сочетании со следующими примерами, которые предназначены только для иллюстрации и не ограничивают объем изобретения. Различные изменения и модификации раскрытых вариантов осуществления будут очевидны для специалистов в данной области и такие изменения и модификации, включая, без ограничения, те, которые связаны с химическими структурами, заместителями, производными, рецептурами и/или способами по изобретению, могут быть осуществлены без отступления от сущности изобретения и объема прилагаемой формулы изобретения.
Пример 1
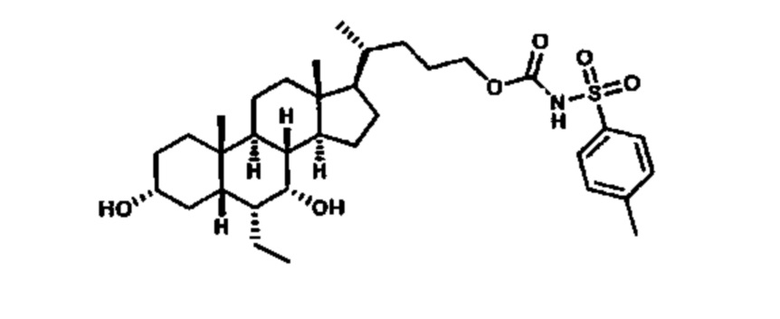
Стадия 1-1
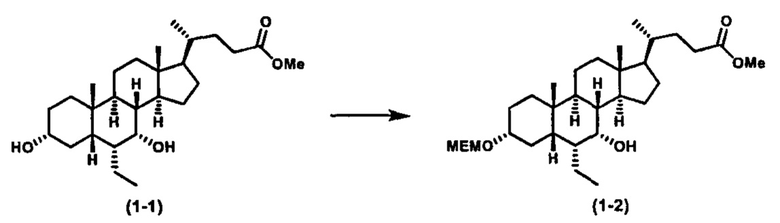
MEMCl добавляли к перемешиваемому раствору (1-1) (4,35 г, 10,0 ммоль) и DIPEA (10,3 мл, 30 ммоль) в DCM (100 мл) при 0°С в атмосфере N2. Полученной реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение ночи, затем гасили водой (50 мл) и 1Н HCl (50 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением 6,5 г неочищенного продукта (1-2), который использовали непосредственно для следующей стадии.
ЖХ-МС измерено 2М+NH4 1062,83 (вычислено 1062,81).
Стадия 1-2
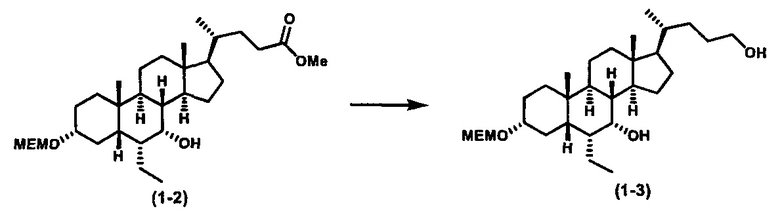
Вышеуказанный неочищенный продукт (1-2) (4,18 г, 8,0 ммоль) сначала растворяли в THF (30 мл) при 0°С в атмосфере N2, добавляли сухой МеОН (1,28 мл, 32 ммоль) с последующим медленным добавлением LiBH4 (697 мг, 32 ммоль). Смесь перемешивали при 0°С в течение 6 ч, анализ ТСХ и ЖХ-МС показал частичную конверсию исходного материала, затем добавляли больше LiBH4 (348 мг, 16 ммоль). Смеси давали нагреться до комнатной температуры и перемешивали в течение ночи, гасили 1 М водным раствором NaOH (20 мл) и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором и концентрировали. Остаток очищали хроматографией на силикагеле с применением смеси гексан/ацетон (от 100/0 до 60/40, 10 мин) с получением спиртового продукта (1-3) (3,2 г, выход 94% на основании 86% конверсии) в виде белой пены.
ЖХ-МС измерено 2М+NH4 1006,83 (вычислено 1006,83).
Стадия 1-3
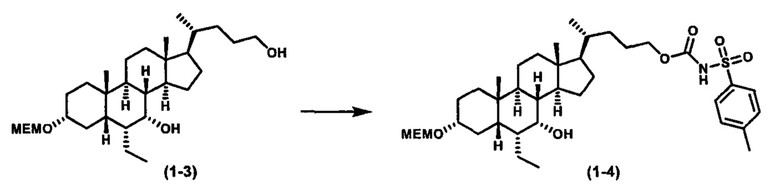
К перемешиваемому раствору спирта (1-3) (99 мг, 0,2 ммоль) и DIPEA (35 мкл, 0,2 ммоль) в THF (3 мл) при комнатной температуре добавляли по каплям пара-толуолсульфонилизоцианат (31 мкл, 0,2 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 30 мин, затем гасили солевым раствором (10 мл) и экстрагировали этилацетатом (40 мл). Органический слой промывали солевым раствором и концентрировали. Остаток очищали хроматографией на силикагеле с использованием смеси гексан/ацетон (от 100/0 до 60/40, 10 мин) с получением сульфонилкарбамата (1-4) в виде бесцветного масла (69 мг, 50%).
ЖХ-МС измерено М-1 690.33 (вычислено 690,41)
Стадия 1-4
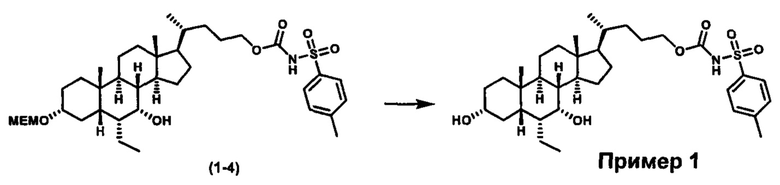
Карбамат (1-4) (69 мг, 0,1 ммоль) сначала растворяли в THF (2 мл) при комнатной температуре. Затем добавляли 37% HCl (0,1 мл, 1,2 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч, гасили насыщенным раствором NaHCO3 и экстрагировали этилацетатом. Органический слой промывали солевым раствором и концентрировали. Остаток очищали хроматографией на силикагеле с использованием смеси гексан/ацетон (от 100/0 до 60/40, 10 мин) с получением желаемого карбамата Примера 1 (30 мг, 50%) в виде белого твердого вещества после лиофилизации в течение ночи из МеСМ/H2O (1/1,2 мл).
ЖХ-МС измерено М-1 602,29 (вычислено 602,33).
Пример 2
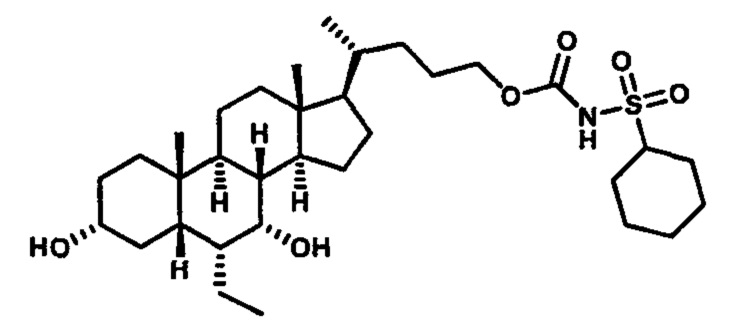
Стадия 2-1
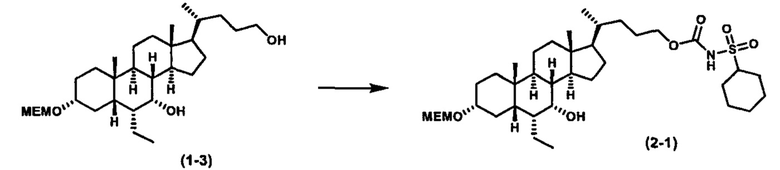
Раствор спирта (1-3) (99 мг, 0,2 ммоль) и CDI (65 мг, 0,4 ммоль) в MeCN/THF (1/1, 1 мл) перемешивали при комнатной температуре в течение 1,5 ч, затем добавляли циклогексансульфонамид (98 мг, 0,6 ммоль) и DBU (89 мкл, 0,6 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем гасили насыщенным раствором соли и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором и концентрировали. Остаток очищали хроматографией на силикагеле с использованием смеси гексан/ацетон (от 100/0 до 60/40, 10 мин) с получением сульфониларбамата (2-1) (100 мг, 73%) в виде бесцветного масла.
ЖХ-МС измерено М-1 682,36 (вычислено 682,44).
Стадия 2-2.
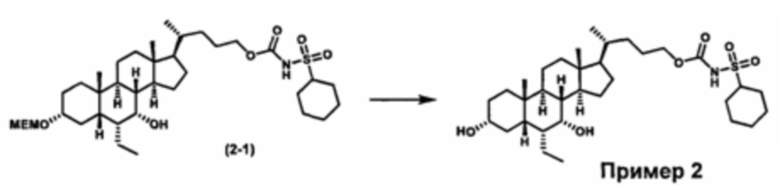
Карбамат (2-1) (100 мг, 0,15 ммоль) сначала растворяли в THF (3 мл) при комнатной температуре. Затем добавляли 37% HCl (0,1 мл, 1,2 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, гасили насыщенным NaHCO3 и экстрагировали этилацетатом. Органический слой промывали солевым раствором и концентрировали. Остаток очищали хроматографией на силикагеле с использованием смеси гексан/ацетон (от 100/0 до 60/40, 10 мин) с получением желаемого карбамата Примера 2 (45 мг, 45%) в виде белого твердого вещества после лиофилизации в течение ночи из MeCN/H2O (1/1, 2 мл).
ЖХ-МС измерено М-1 594,31 (вычислено 594,39).
Пример 3:
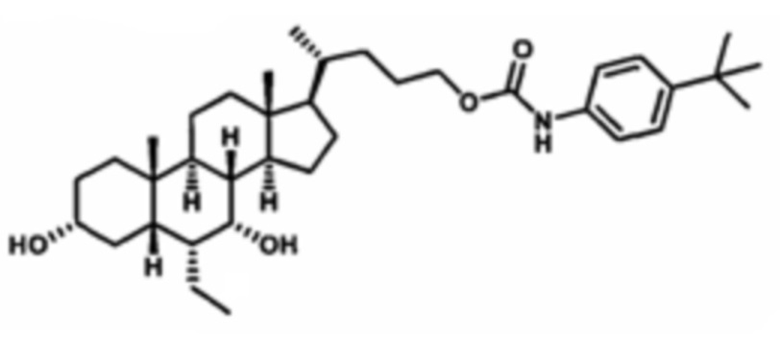
Стадия 3-1
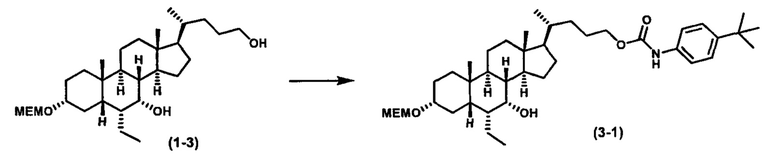
Раствор спирта (1-3) (99 мг, 0,2 ммоль) и CDI (65 мг, 0,4 ммоль) в MeCN/THF (1/1, 1 мл) перемешивали при комнатной температуре в течение 1,5 ч, добавляли 4-трет-бутиланилин (90 мг, 0,6 ммоль) и DBU (89 мкл, 0,6 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 18 ч, затем гасили насыщенным солевым раствором и экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором и концентрировали. Остаток очищали хроматографией на силикагеле с использованием смеси гексан/ацетон (от 100/0 до 60/20, 10 мин) с получением карбамата (3-1) (87 мг, 65%) в виде бесцветного масла.
ЖХ-МС измерено М+NH4 687,64 (вычислено 687,53).
Стадия 3-2.
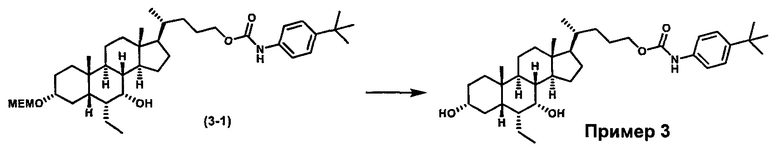
Карбамат (3-1) (87 мг, 0,13 ммоль) сначала растворяли в THF (3 мл) при комнатной температуре. Затем добавляли 37% HCl (0,1 мл, 1,2 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, гасили насыщенным NaHCO3 и экстрагировали этилацетатом. Органический слой промывали солевым раствором и концентрировали. Остаток очищали хроматографией на силикагеле с использованием смеси гексан/ацетон (от 100/0 до 60/30, 10 мин) с получением желаемого карбамата Примера 3 (65 мг, 87%) в виде белого твердого вещества после лиофилизации в течение ночи из MeCN/H2O (1/1, 2 мл).
ЖХ-МС измерено M+NH4 599,48 (вычислено 599,47).
Пример 4:
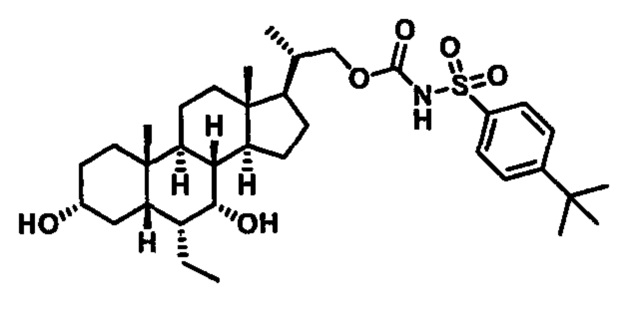
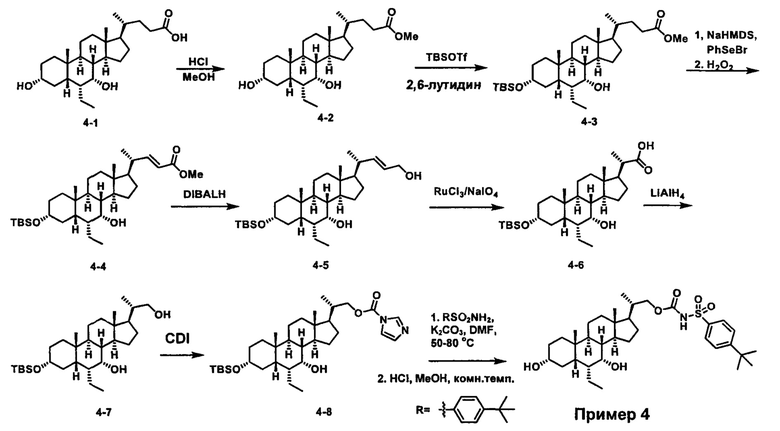
Стадия 4-1:
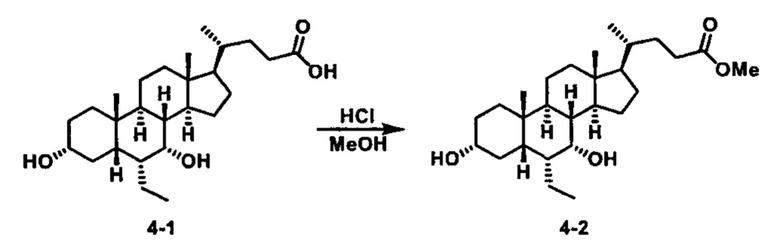
HCl (10 мл, 37%) добавляли к соединению 4-1 (15,0 г) в МеОН (100 мл) при 0°С. Раствор перемешивали при комнатной температуре в течение 1 часа. Раствор выпаривали досуха, затем остаток разбавляли EtOAc (200 мл), последовательно промывали насыщенным раствором NaHCO3 (30 мл × 2) и насыщенным NaCl (30 мл × 1). Органический слой сушили над Na2SO4, фильтровали и выпаривали с получением соединения 4-2 (15,6 г, 95,7%) в виде белого твердого вещества, которое использовали непосредственно на следующей стадии.
Стадия 4-2:
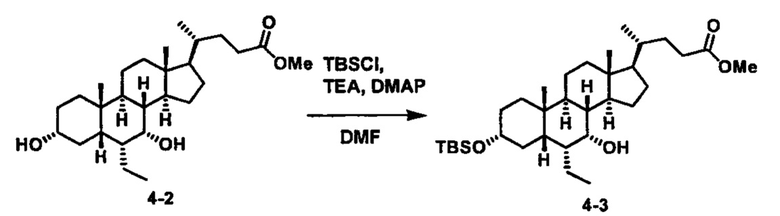
TBSCl (10,9 г, 72 ммоль) добавляли к соединению 4-2 (15,6 г, 36 ммоль), ТЕА (10,9 г, 108 ммоль) и DMAP (0,22 г, 1,8 ммоль) в DMF (50 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 3 часов. К смеси добавляли 200 мл воды и экстрагировали EtOAc (100 мл × 3). Объединенную органическую фазу промывали насыщенным NaCl (100 мл × 3), затем сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали на колонке с силикагелем (от 20 до 40% EtOAc в петролейном эфире) с получением соединения 4-3 (15,8 г, 82%) в виде желтого твердого вещества.
Стадия 4-3:
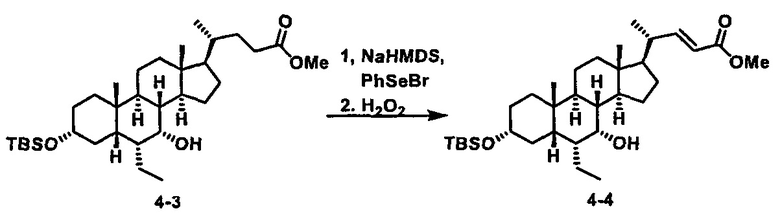
NaHMDS (2,0 М в THF, 30 мл, 60,54 ммоль) добавляли по каплям к соединению 4-3 (15,8 г, 28,83 ммоль) в THF (100 мл) при -78°С в течение 1 часа. Смесь перемешивали при -78°С в течение 1,5 ч, затем к реакционной смеси добавляли по каплям PhSeBr (8,16 г, 34,6 ммоль) в THF (40 мл). Смесь перемешивали при -78°С в течение 2,5 часов и при комнатной температуре 30 мин. Добавляли насыщенный NH4Cl (30 мл) при 0°С, экстрагировали EtOAc (100 мл × 2), объединенную органическую фазу промывали насыщенным NaCl (30 мл × 1), сушили над Na2SO4, фильтровали. Раствор в EtOAc обрабатывали 30% Н2О2 (10 мл) при 0°С, затем раствор перемешивали при комнатной температуре в течение 40 минут. Раствор последовательно промывали насыщенным NaHCO3 (30 мл) и насыщенным NaCl (30 мл). Органическую фазу сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали на колонке с силикагелем (от 10 до 20% EtOAc в петролейном эфире) с получением соединения 4-4 (10,6 г, 67,9 %) в виде желтого масла.
Стадия 4-4:
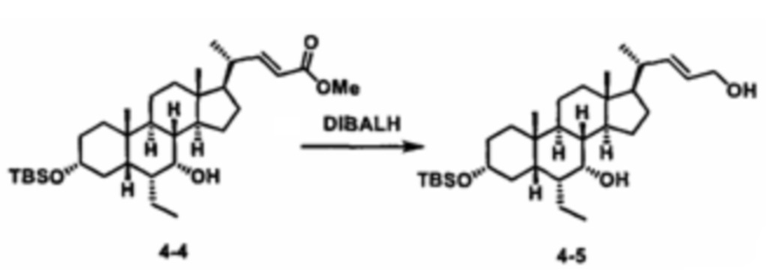
DIBALH (1,0 М в PhMe, 85 мл, 85 ммоль) добавляли по каплям к соединению 4-4 (10,6 г, 19,4 ммоль) в DCM (100 мл) при -78°С в течение 1 часа. Раствор нагревали до 0°С в течение 30 мин, затем добавляли МеОН (100 мл) для гашения реакции. К замутненной смеси добавляли виннокислый калий-натрий (водный раствор, 100 мл) и реакционную смесь интенсивно перемешивали в течение 12 часов. Смесь экстрагировали EtOAc (100 мл × 3), последовательно промывали водой и солевым раствором (100 мл × 3), сушили, фильтровали. Остаток очищали на колонке с силикагелем (от 20 до 40% EtOAc в петролейном эфире) с получением соединения 4-5 (7,5 г, 74,6%) в виде белого твердого вещества.
Стадия 4-5:
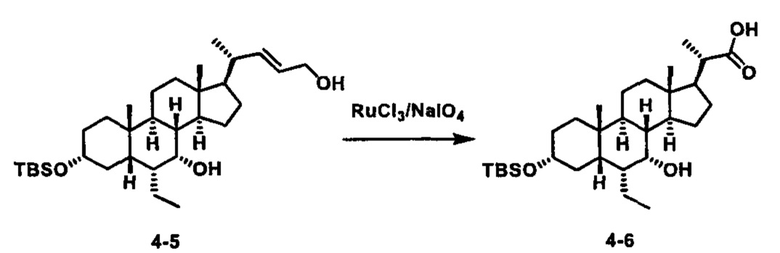
NaIO4 (13,5 г, 59,37 ммоль) добавляли к соединению 4-5 (7,5 г, 14,48 ммоль) и RuCl3 (0,155 г, 0,72 ммоль) в CCl4 (30 мл), MeCN (30 мл) и H2O (50 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 3 часов. Смесь гасили водой, экстрагировали EtOAc (100 мл), промывали водой и солевым раствором (100 мл × 3), сушили, фильтровали, получая соединение 4-6 (6,6 г, неочищенное) в виде твердого вещества черного цвета и использовали непосредственно в следующей стадии.
Стадия 4-6:
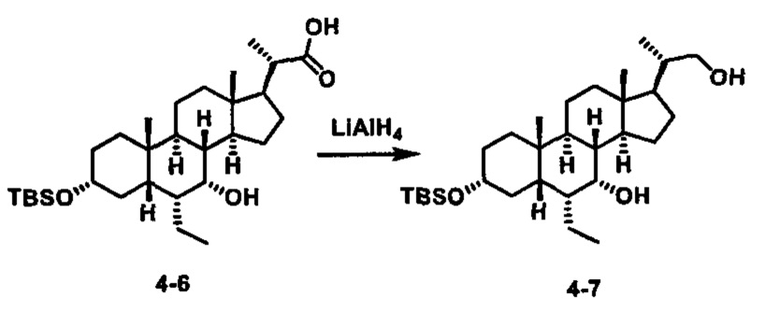
LiAlH4 (1,48 г, 39 ммоль) добавляли к соединению 4-6 (6,6 г, 13 ммоль) в THF (60 мл) при 0°С. Полученную смесь перемешивали при комнатной температуре в течение 3 часов. Смесь гасили водой при 0°С, экстрагировали ЕА (100 мл × 3), объединенную органическую фазу промывали солевым раствором (100 мл × 3), сушили, фильтровали. Остаток очищали хроматографией на силикагеле (от 50 до 100% EtOAc в петролейном эфире) с получением соединения 4-7 (3,5 г, 49,3% в 2 стадиях) в виде белого твердого вещества.
Стадия 4-7:
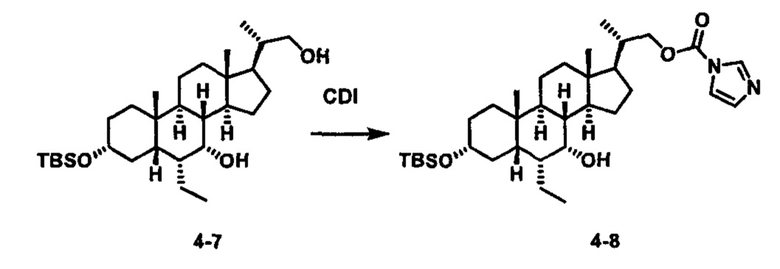
CDI (0,95 г, 5,9 ммоль) добавляли к спирту 4-7 (2,40 г, 4,9 ммоль) и DIEA (0,95 г, 7,3 ммоль) в DCM (30 мл), смесь перемешивали при комнатной температуре в течение 2 часов. Остаток выпаривали и разбавляли EtOAc (50 мл), последовательно промывали насыщенным NaHCO3 (20 мл × 2) и водой (20 мл × 1). Затем органический слой сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали на колонке с силикагелем (от 0 до 50% EtOAc в петролейном эфире) с получением соединения 4-8 (2,40 г, 84,0%) в виде белого твердого вещества.
Стадия 4-8:
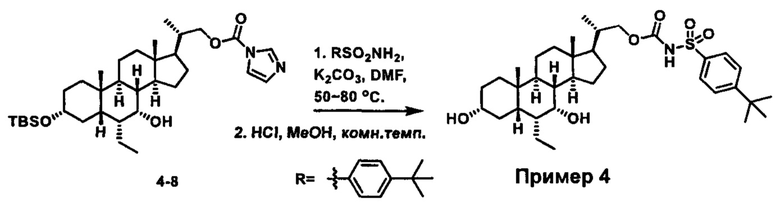
Соединение 4-8 (100 мг, 0,17 ммоль) добавляли к p-tBuPhSO2NH2 (0,26 ммоль) и K2CO3 (70,5 мг, 0,51 ммоль) в DMF (2 мл) при комнатной температуре, затем смесь перемешивали при температуре от 50 до 80°С в течение 4 часов. После охлаждения до комнатной температуры добавляли воду (10 мл), экстрагировали EtOAc (10 мл × 2), сушили над Na2SO4, фильтровали и выпаривали. Остаток растворяли в МеОН (2 мл), затем добавляли 1 каплю 37% HCl. Смесь перемешивали при комнатной температуре в течение 10 минут, затем разбавляли EtOAc (50 мл) и последовательно промывали насыщенным NaHCO3 (10 мл × 2) и солевым раствором (10 мл × 1). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флэш-преп-ВЭЖХ ((IntelFlash-1): колонка, С18, подвижная фаза, MeCN/H2O, УФ-детектор 254), с получением соединения Примера 4, ИЭР-МС m/z=616,40 [М+H]+.
Приведенные ниже соединения Примеров 5-44 и Примеров 44а-44р в Таблице 4 были получены способами, аналогичными описанным в Примере 4.
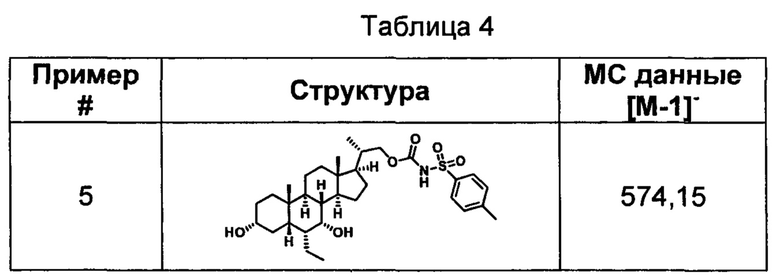
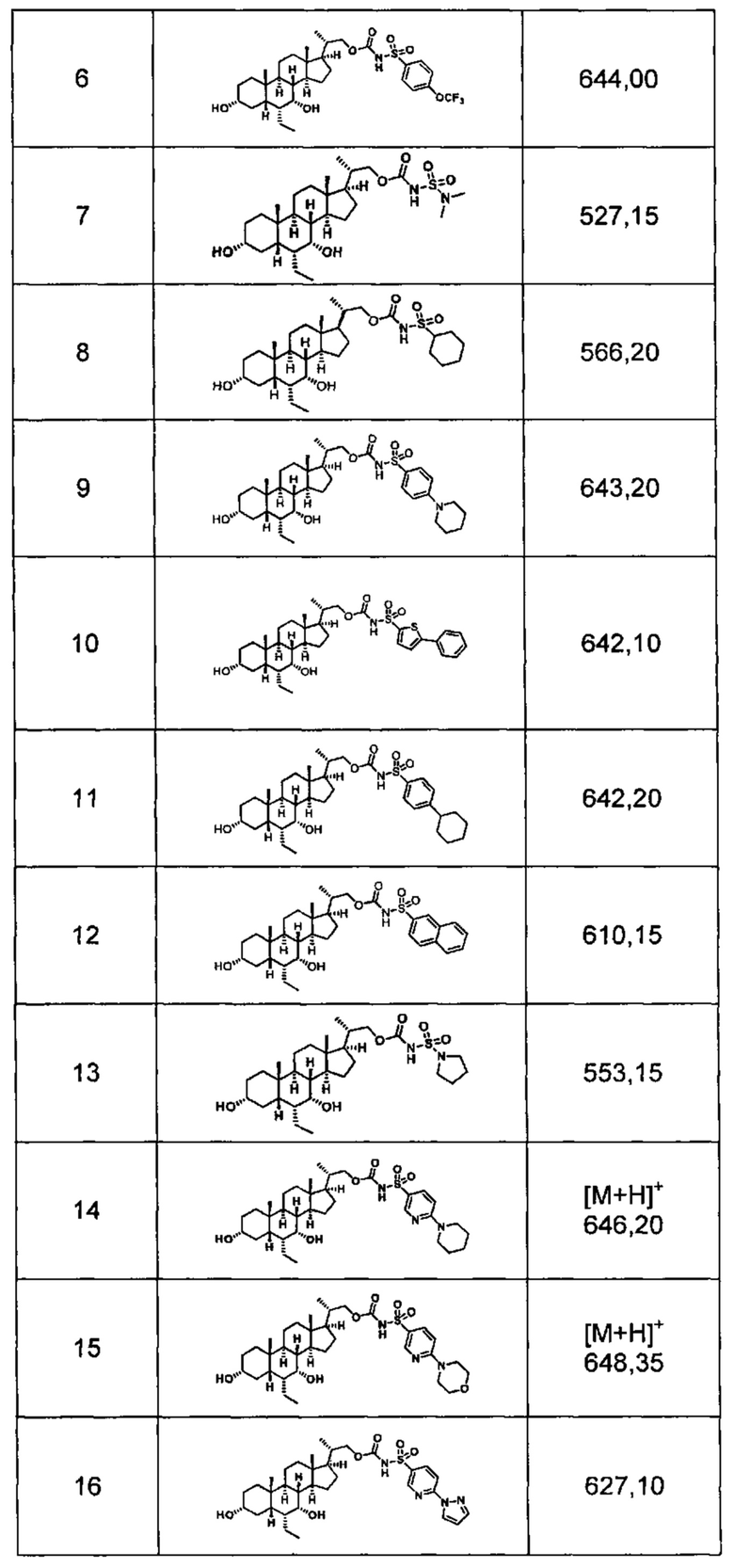
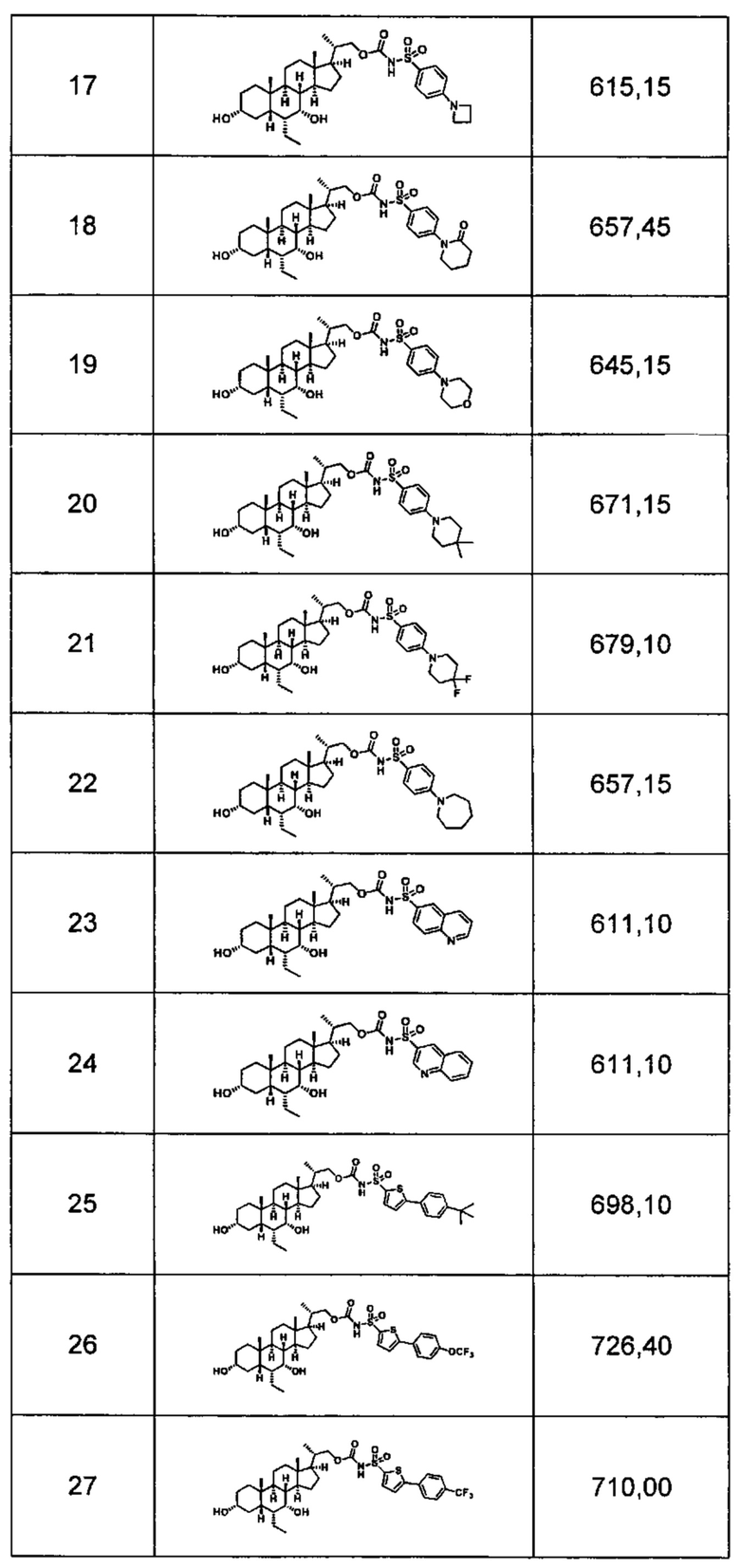
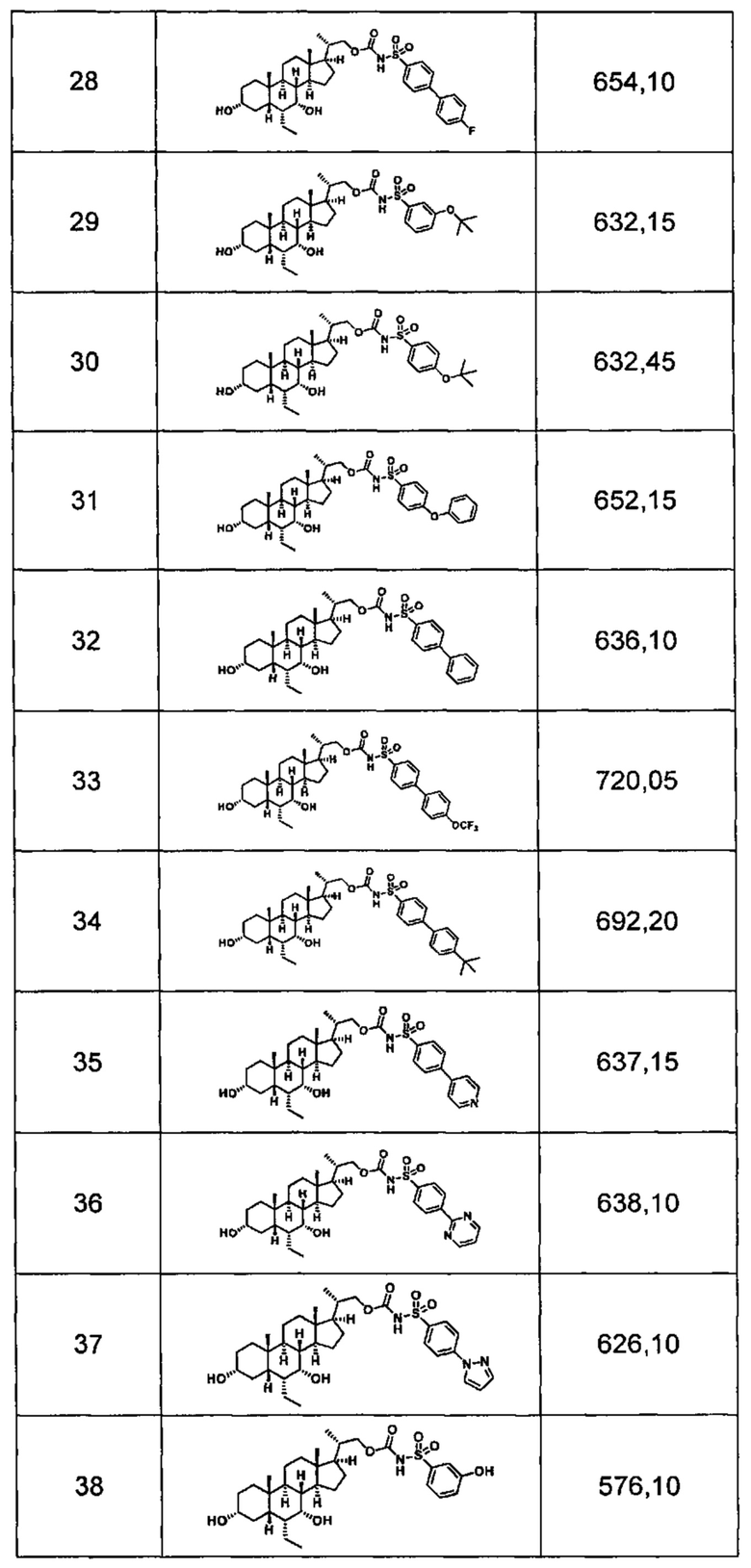
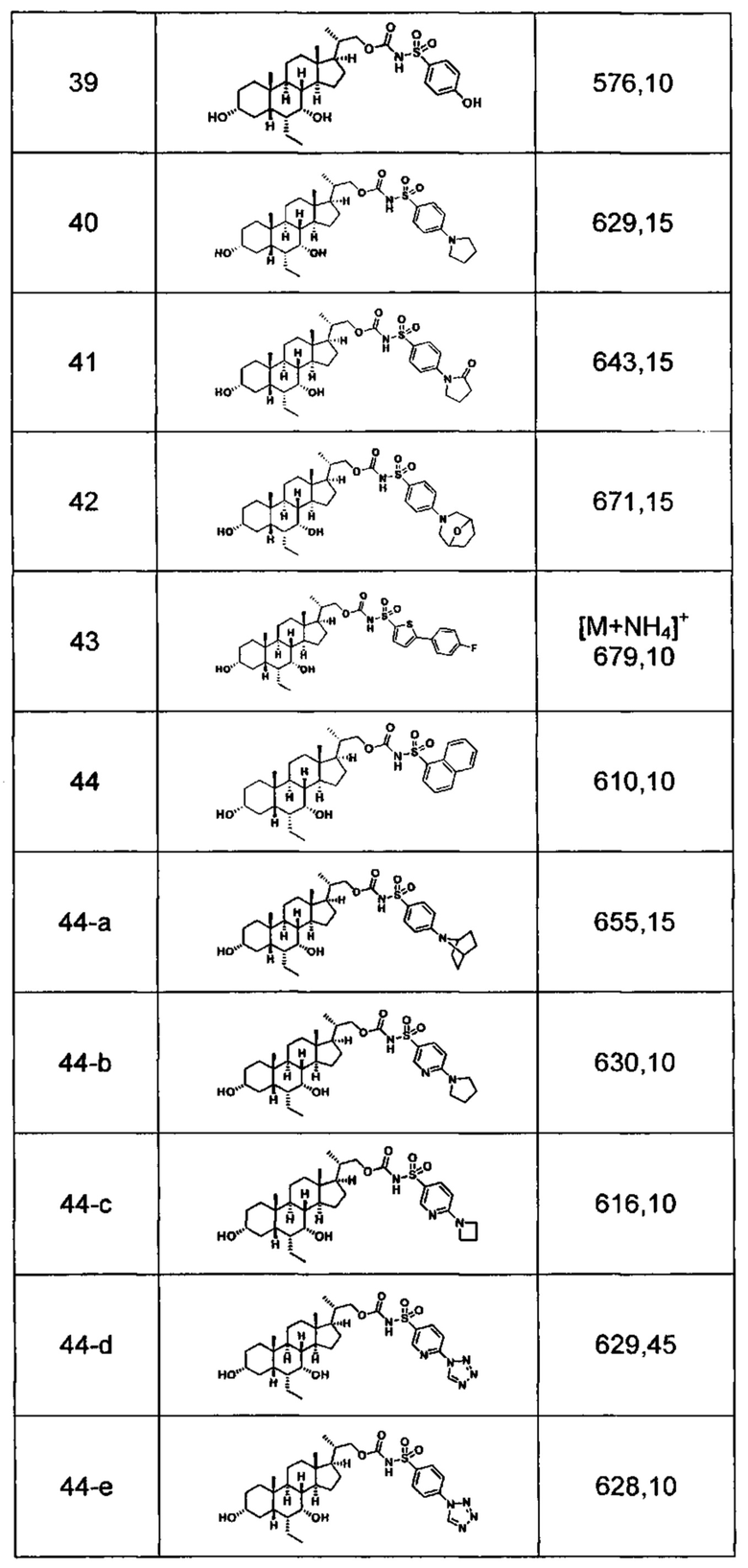
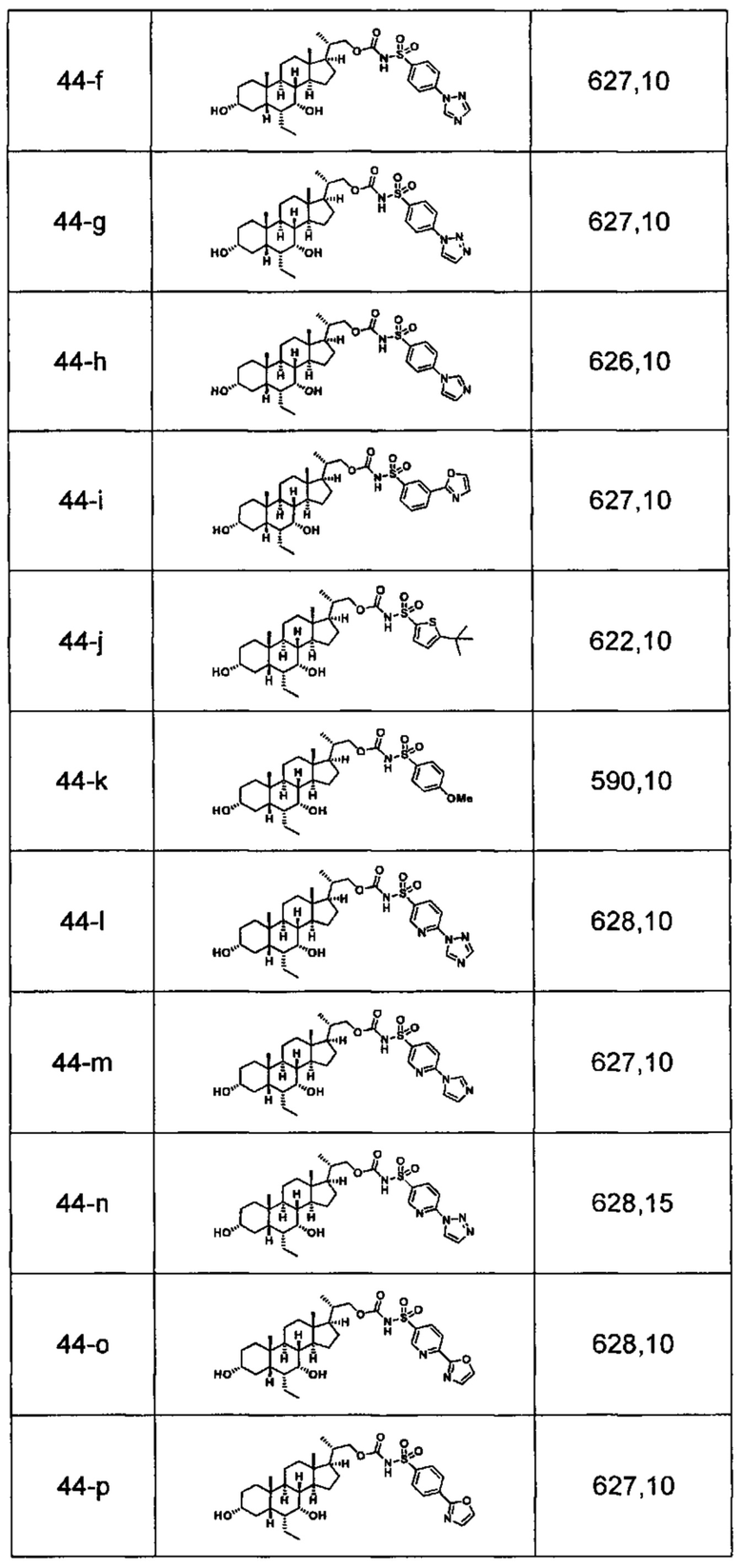
Пример 45
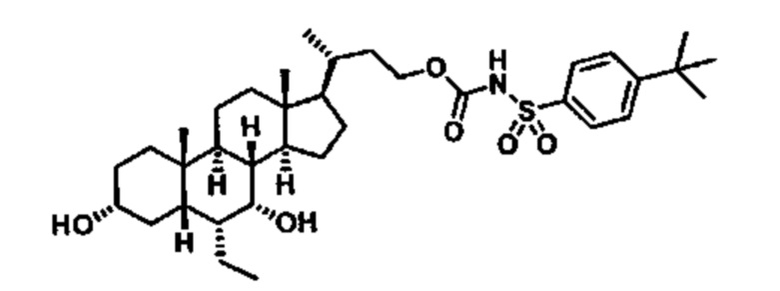
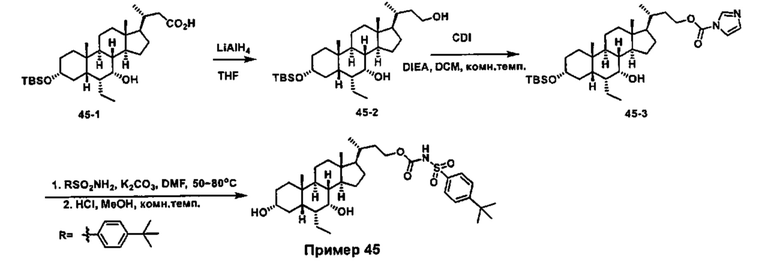
Стадия 45-1
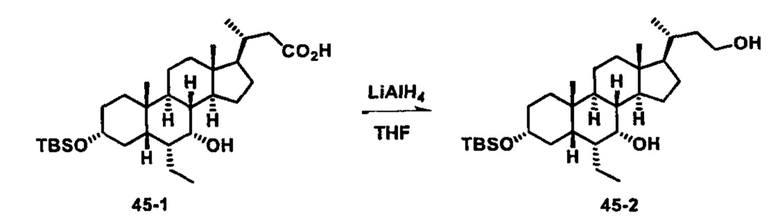
LiAlH4 (2,6 г, 0,067 моль) добавляли к соединению 45-1 (7 г, 0,014 моль) в THF (150 мл) при 0°С в атмосфере N2, полученную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли 300 мл воды при 0°С, экстрагировали EtOAc (100 мл × 3), объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали, остаток очищали на колонке с силикагелем (от 20 до 40% EtOAc в петролейном эфире) с получением целевого соединения 45-2 (5,4 г, 79,3%) в виде белого твердого вещества.
Стадия 45-2:
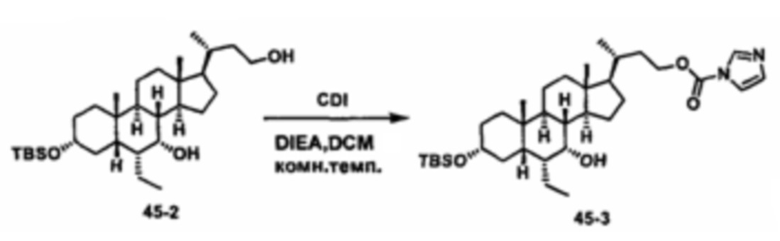
CDI (2,03 г, 12,2 ммоль) добавляли к соединению 45-2 (5,3 г, 10,5 ммоль) и DIEA (2,03 г, 15,7 ммоль) в DCM (30 мл), смесь перемешивали при комнатной температуре в течение 2 часов. Остаток выпаривали и разбавляли EtOAc (50 мл), последовательно промывали насыщенным NaHCO3 (20 мл × 2) и водой (20 мл × 1). Затем органический слой сушили над Na2SO4, фильтровали и выпаривали. Остаток очищали на колонке с силикагелем (от 0 до 50% EtOAc в петролейном эфире) с получением соединения 45-3 (4,6 г, 73,2%) в виде белого твердого вещества.
Стадия 45-3:
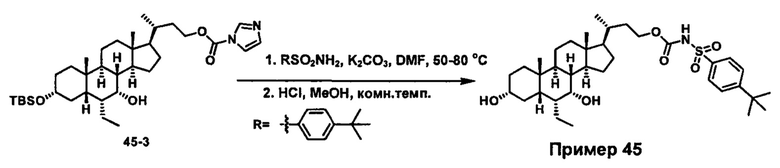
Соединение 45-3 (100 мг, 0,17 ммоль) добавляли к р-tBuPhSO2NH2 (0,25 ммоль) и K2CO3 (70,5 мг, 0,51 ммоль) в DMF (2 мл) при комнатной температуре, затем смесь перемешивали при температуре от 50 до 80°С в течение 4 часов. После остывания до комнатной температуры добавляли воду (10 мл), экстрагировали EtOAc (10 мл × 2), сушили над Na2SO4, фильтровали и выпаривали. Остаток растворяли в МеОН (2 мл), затем добавляли 1 каплю 37% HCl. Смесь перемешивали при комнатной температуре в течение 10 минут, затем разбавляли EtOAc (50 мл) и последовательно промывали насыщенным NaHCO3 (10 мл × 2) и солевым раствором (10 мл × 1). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флэш-преп-ВЭЖХ ((IntelFlash-1): колонка, С18, подвижная фаза, MeCN/H2O, УФ-детектор 254 нм) с получением целевого соединения Примера 45, ИЭР-МС m/z=596,60 [М+1-2H2O]+.
Приведенные ниже соединения. Примеров 46-104 и Примеров 104а-104k в Таблице 5 были получены способами, аналогичными описанным в Примере 45.
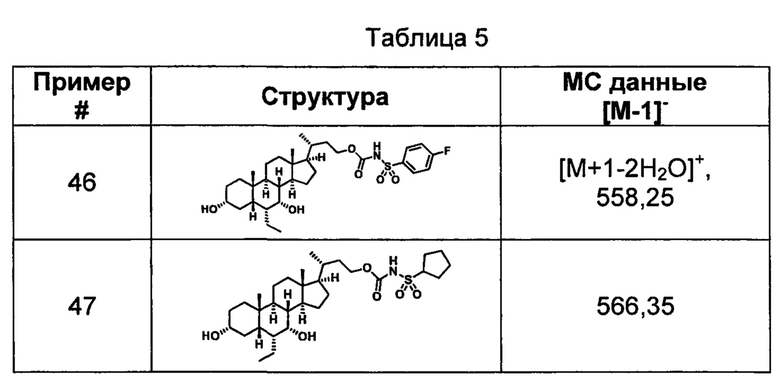
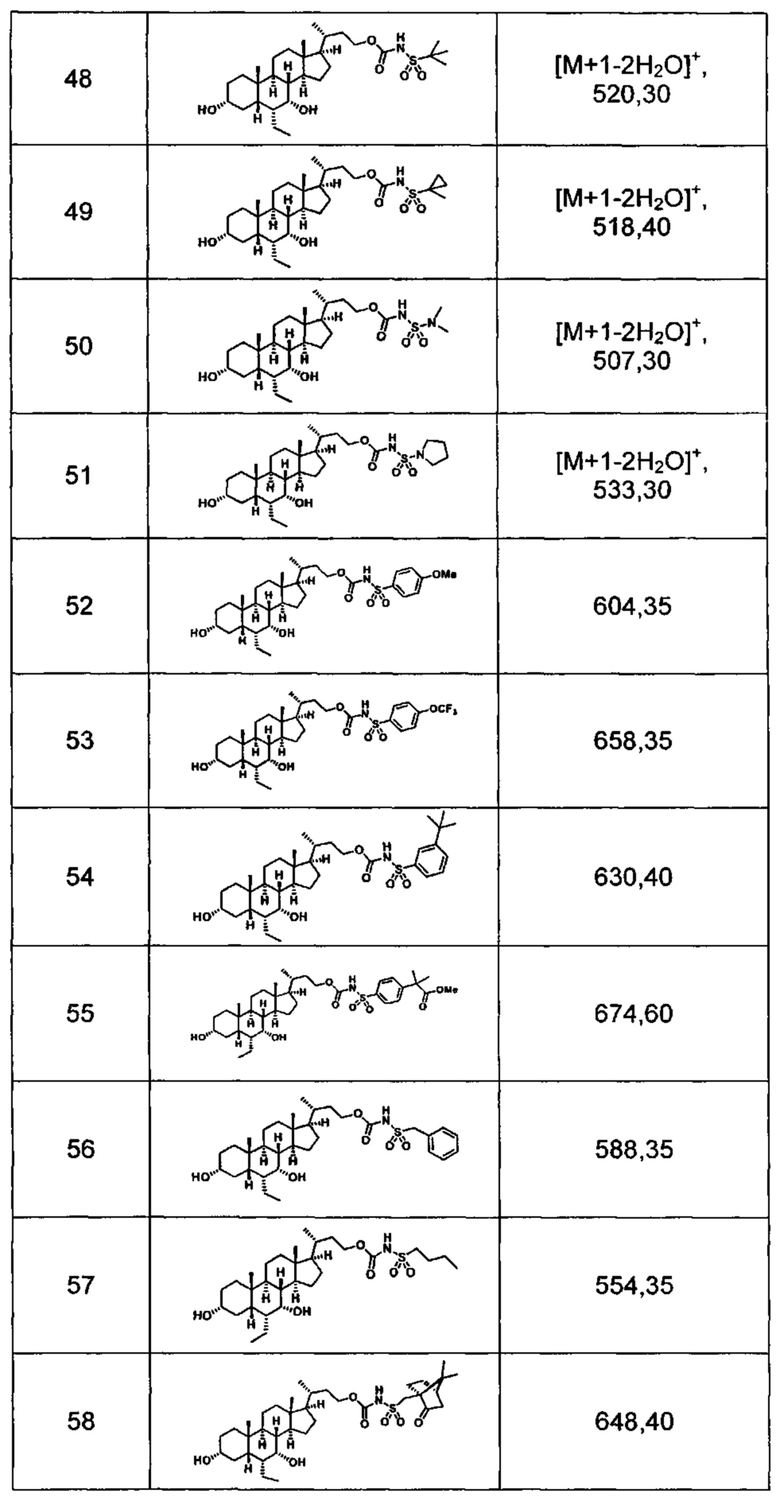
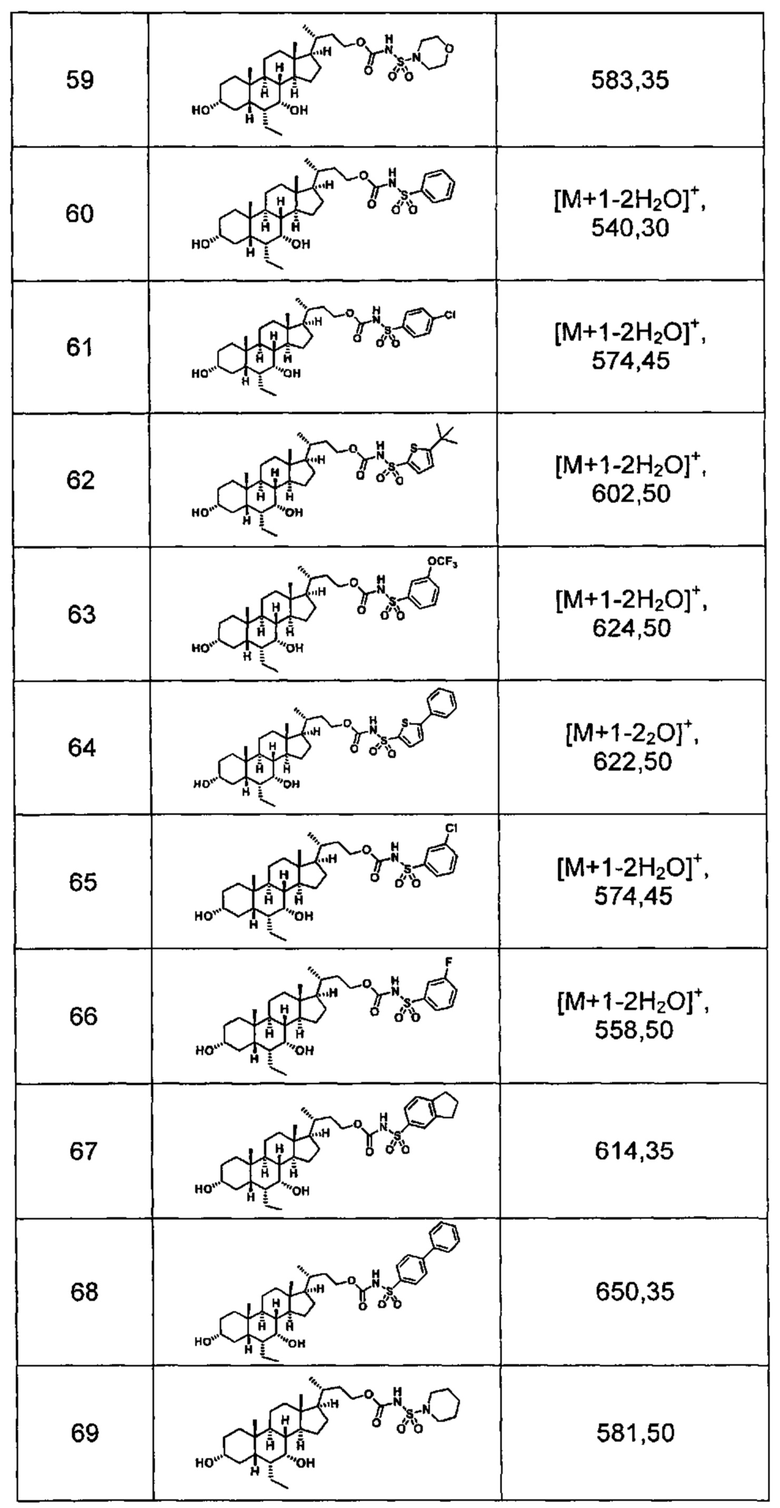
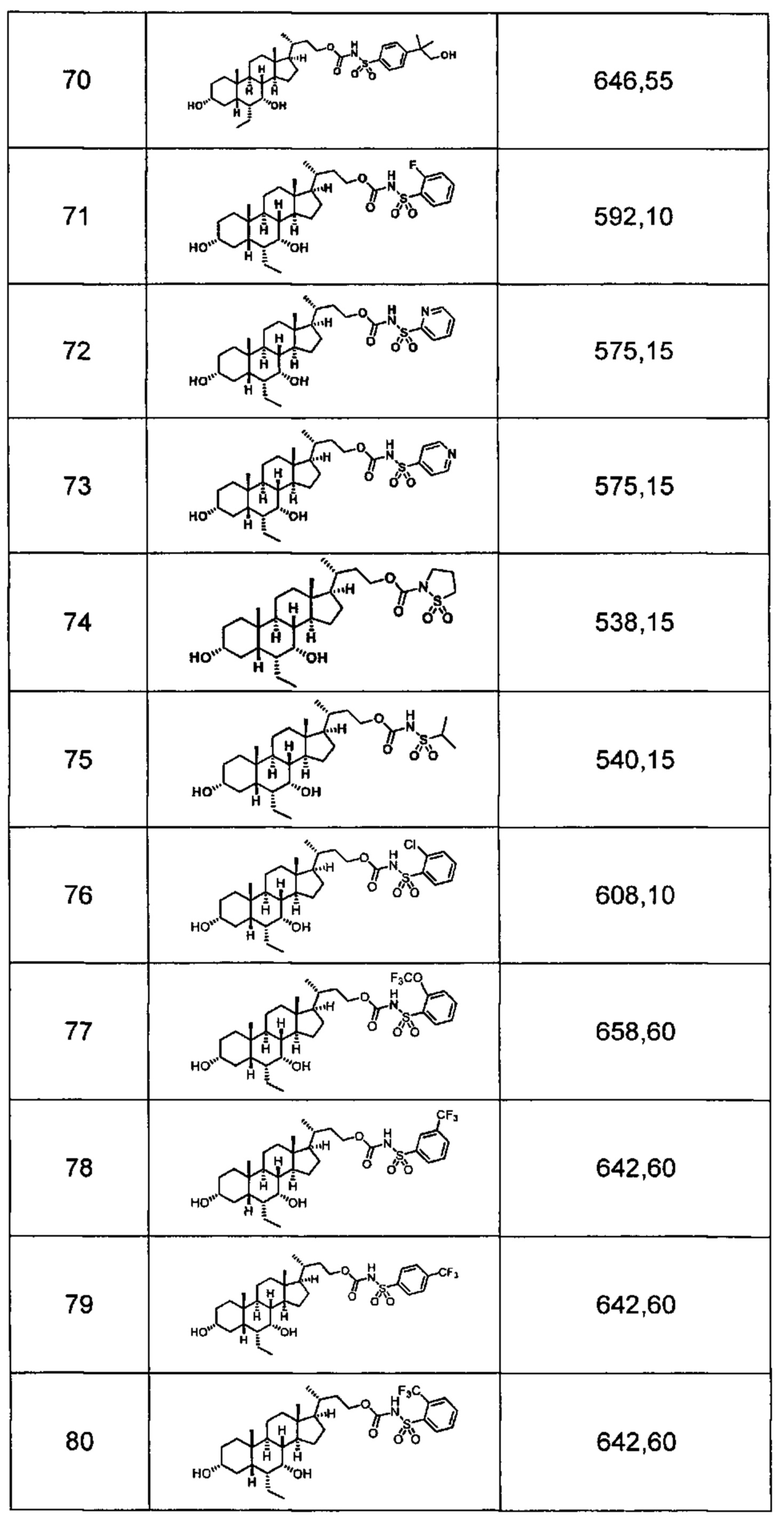
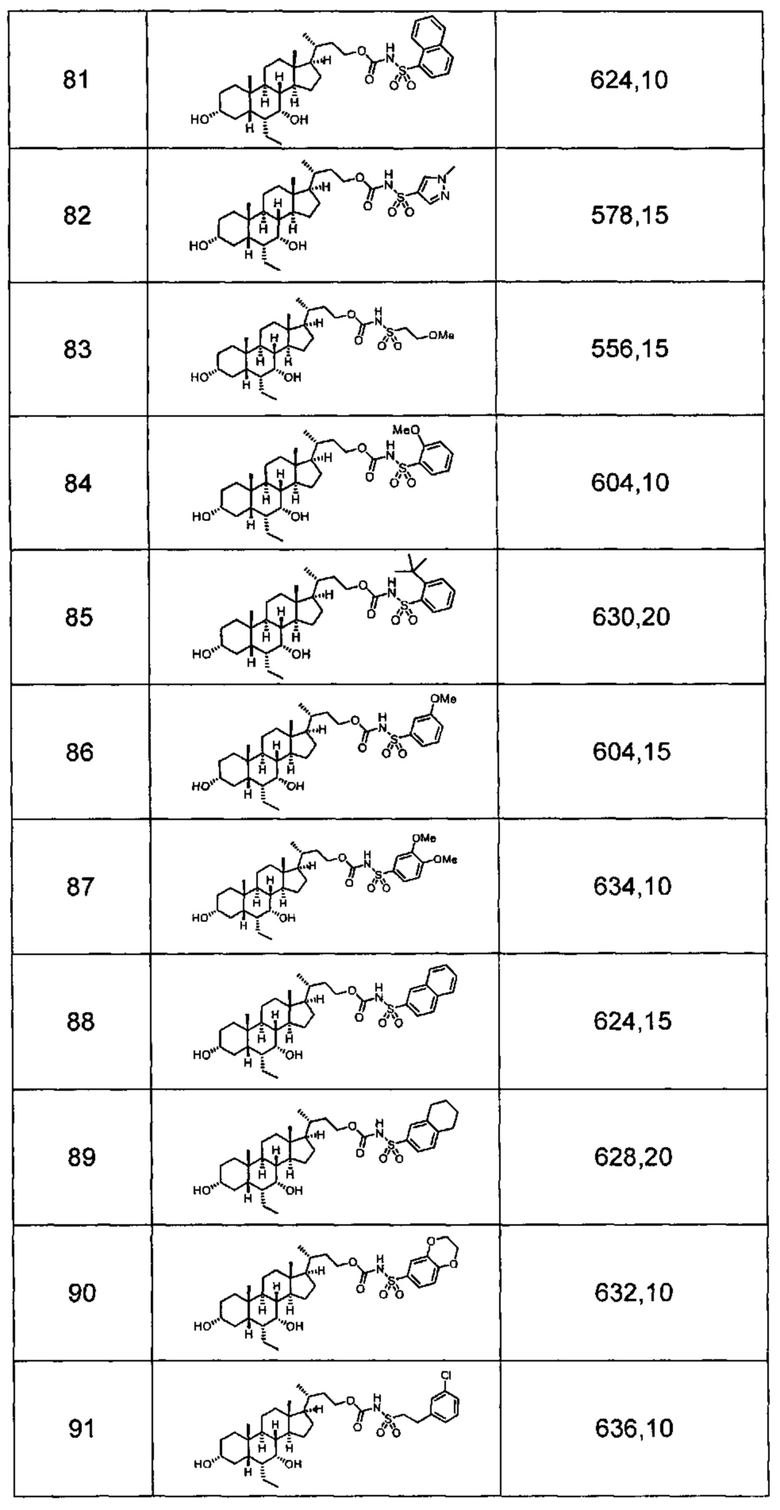
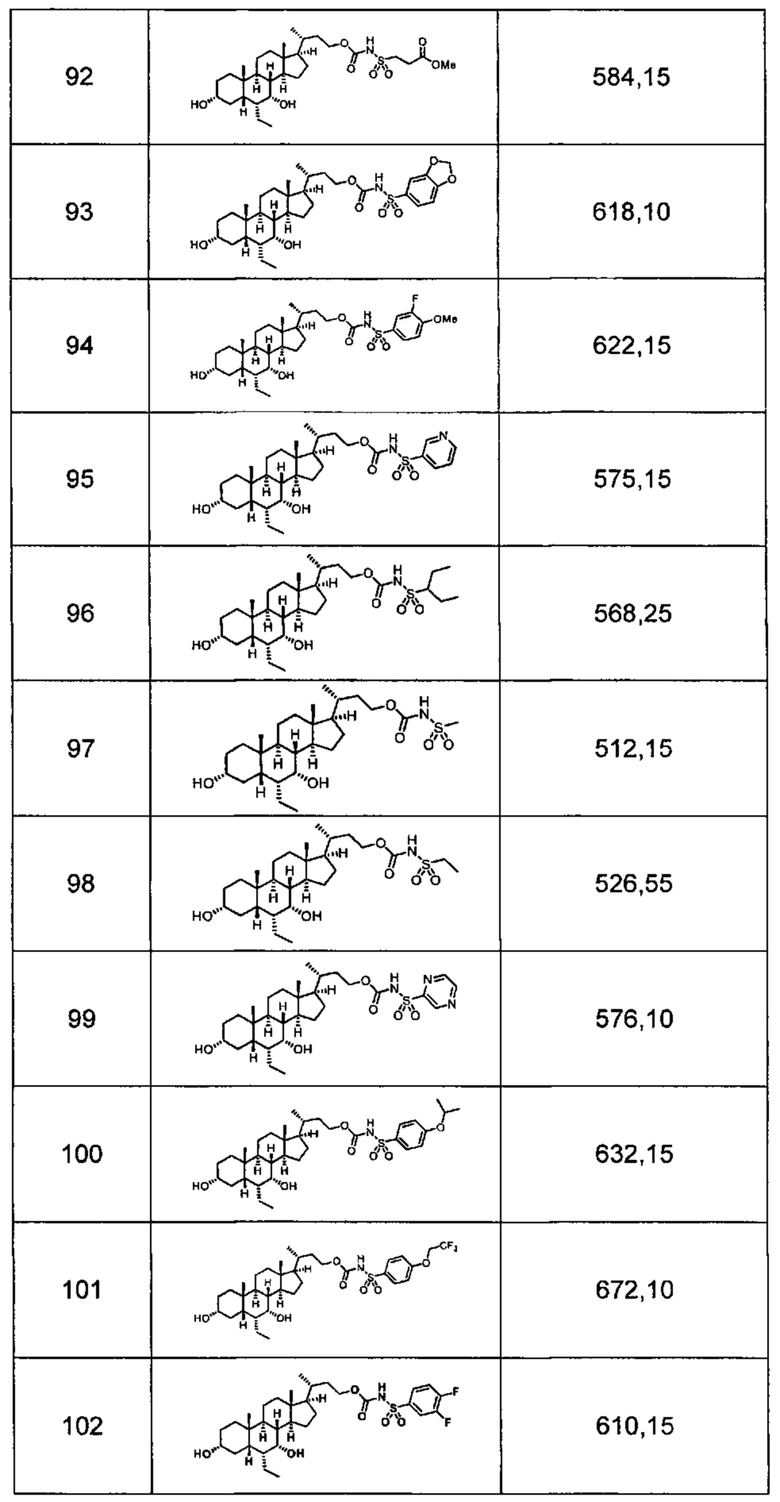
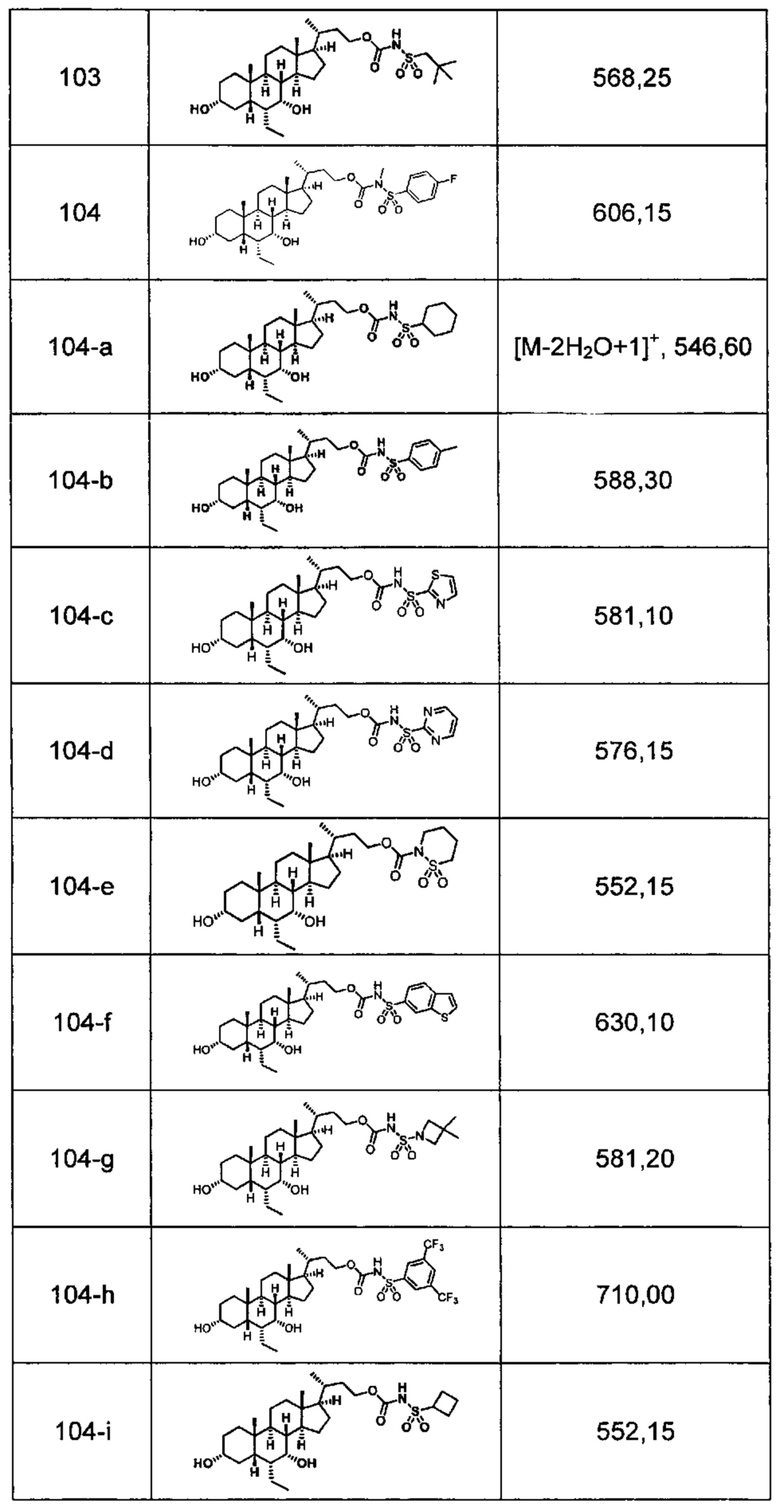
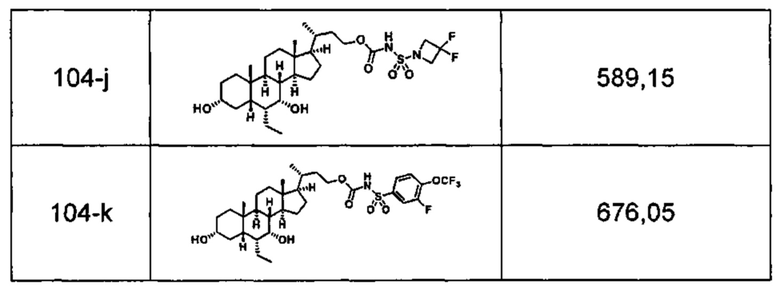
Пример 105
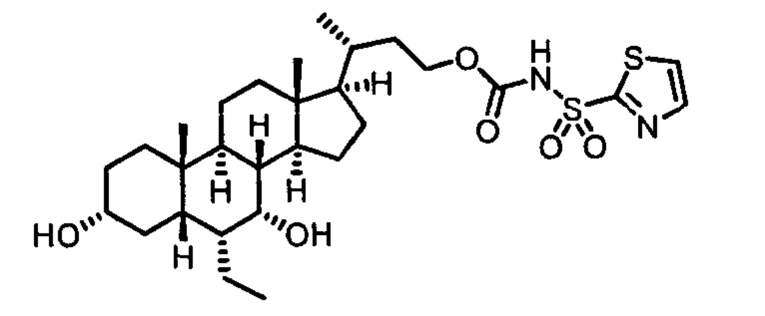
Стадия 105-1:
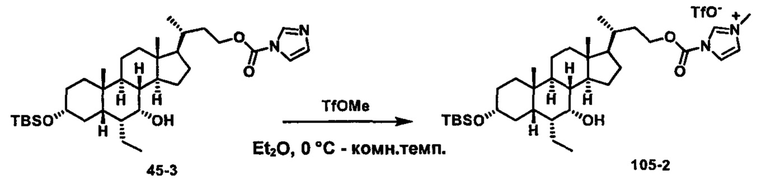
TfOMe (164 мг, 1,00 ммоль) добавляли к промежуточному соединению 45-3 (400 мг, 0,67 ммоль) в Et2O (2 мл) при 0°С, затем смесь перемешивали при комнатной температуре в течение 1 часа. Смесь фильтровали и соединение 105-2 получали в виде белого твердого вещества (430 мг, 84,4%).
Стадия 105-2:
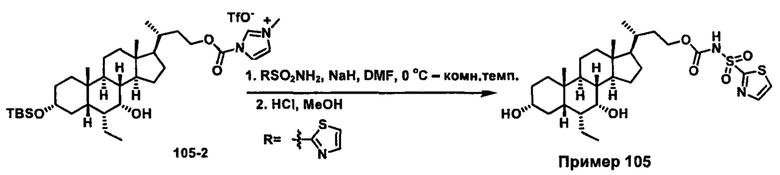
NaH (30 мг, 0,5 ммоль, 60%) добавляли к тиазол-2-сульфонамиду (0,32 ммоль) в THF при 0°С и перемешивали при 0°С в течение 1 часа. Затем к вышеуказанной смеси при 0°С добавляли соединение 105-3 (190 мг, 0,25 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Смесь гасили насыщенным NH4Cl (10 мл), экстрагировали EtOAc (20 мл × 2), сушили над Na2SO4, фильтровали и выпаривали. Остаток растворяли в МеОН (2 мл), затем добавляли 1 каплю 37% HCl. Смесь перемешивали при комнатной температуре в течение 10 минут, затем разбавляли EtOAc (50 мл) и последовательно промывали насыщенным NaHCO3 (10 мл × 2) и солевым раствором (10 мл × 1). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флэш-преп-ВЭЖХ ((IntelFlash-1): колонка, С18, подвижная фаза, MeCN/H2O, УФ-детектор 254 нм) с получением целевого соединения Примера 105, ИЭР-МС m/z=581,10 [М-1]-.
Приведенные ниже соединения Примеров 106-109 в Таблице 6 были получены способами, аналогичными описанным в Примере 105.
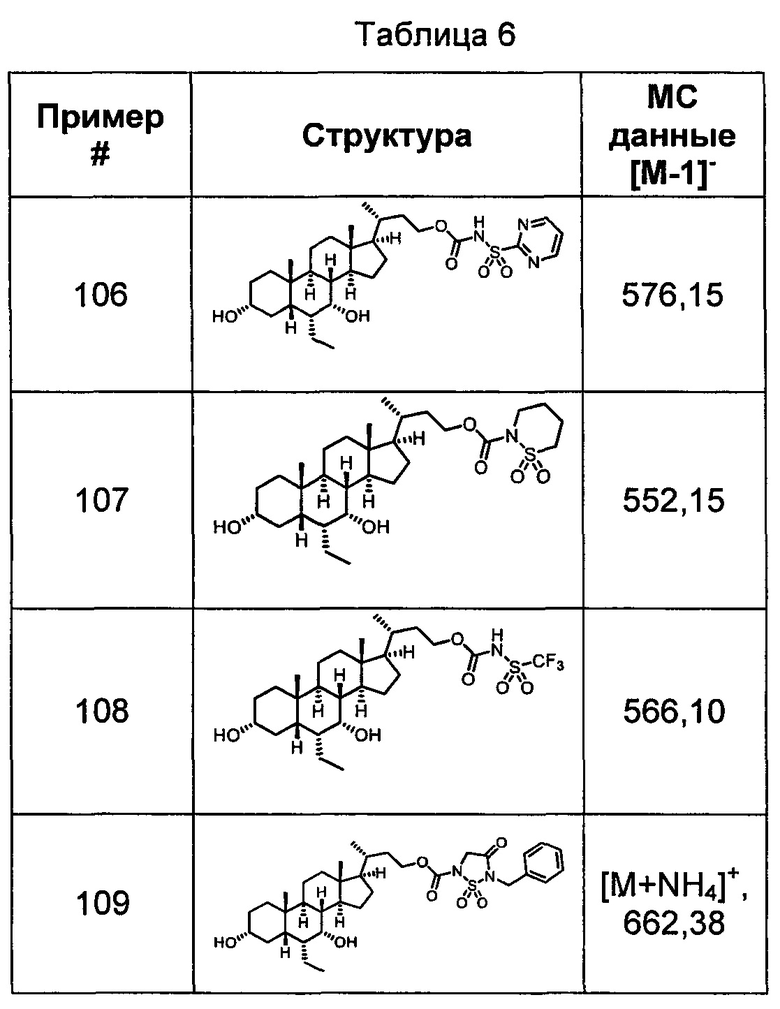
Пример 110:
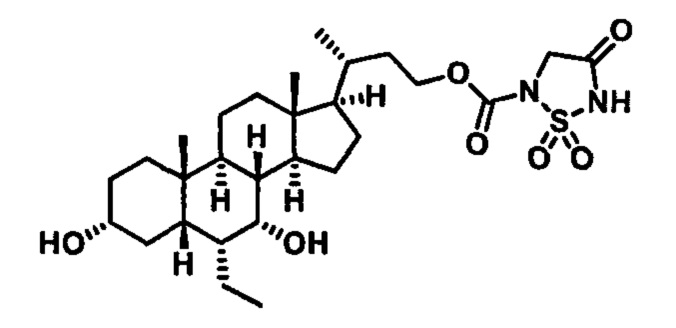
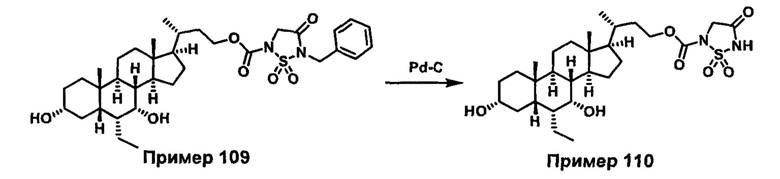
К раствору соединения 109 (30 мг) в МеОН (2 мл) добавляли Pd-C (3 мг, 10%) в атмосфере N2. Суспензию дегазировали под вакуумом и продували H2 несколько раз. Смесь перемешивали в атмосфере H2 при комнатной температуре в течение 1 часа. Затем фильтровали, фильтрат выпаривали досуха, получая 110 (23 мг, 91,9%) в виде белого твердого вещества, ИЭР-МС m/z=553,10 [М-1]-.
Пример 111:
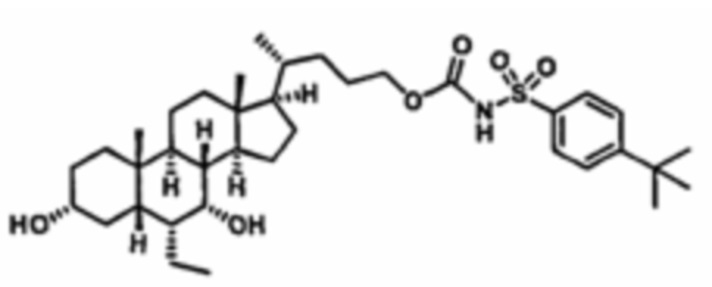
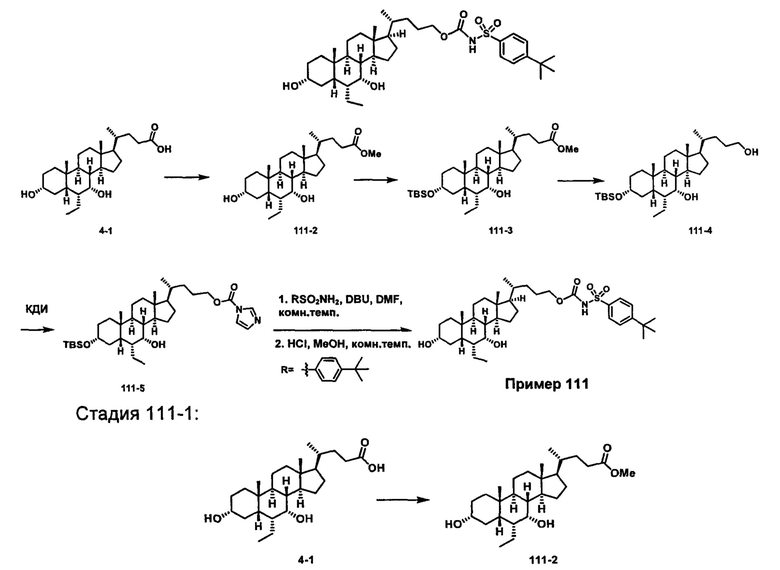
К раствору соединения 4-1 (6,5 г, 15,5 ммоль) в МеОН (130 мл) добавляли серную кислоту (98%, 0,13 мл). Раствор перемешивали при 23°С в течение 24 ч и концентрировали в вакууме. Очистка остатка на колонке с силикагеле (105 г) с 0-30% ацетоном в гексане приводила к получению соединения 111-2 (6,6 г, выход 98%).
Стадия 111-2:
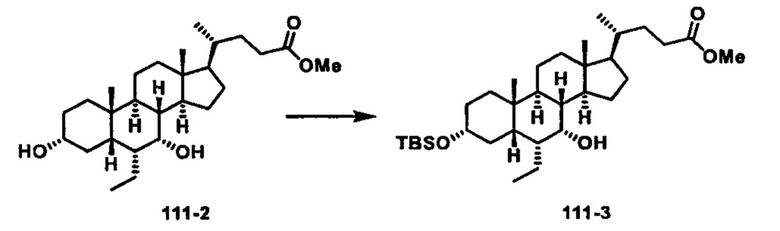
К раствору соединения 111-2 (6,39 г, 14,7 ммоль) в DMF (30 мл) добавляли имидазол (2,20 г, 32,3 ммоль) и TBSCl (2,33 г, 15,5 ммоль). Смесь перемешивали при 23°С в течение 20 ч, гасили буфером с рН 7 и экстрагировали МТВЕ. Органический слой промывали насыщенным NaCl, сушили над Na2SO4 и концентрировали в вакууме. Очистка остатка на колонке с силикагелем (105 г) с 0-50% EtOAc/гексаном приводила к получению соединения 111-3 (7,59 г, выход 94%).
Стадия 111-3:
К раствору 111-3 (7,59 г, 13,85 ммоль) в THF добавляли по каплям LiBH4 (2,0 М в THF, 41,6 ммоль, 20,8 мл) и безводный МеОН (1,7 мл, 41,6 ммоль). Смесь перемешивали при 23°С в течение 15 ч, медленно гасили водой (100 мл) и экстрагировали EtOAc. Водный слой подкисляли 1 М HCl до рН 5 и экстрагировали EtOAc (2 раза). Органические слои объединяли, промывали 1 М раствором HCl, насыщенным NaCl, сушили над Na2SO4 и концентрировали в вакууме. Очистка остатка на колонке с силикагелем (105 г) с 0-30% EtOAc/гексаном приводила к получению соединения 111-4 (6,5 г) с выходом 90%.
Стадия 111-4:
CDI (0,37 г, 2,3 ммоль) добавляли к спирту 111-4 (1,00 г, 1,9 ммоль) и DIEA (0,37 г, 2,9 ммоль) в DCM (20 мл), смесь перемешивали при комнатной температуре в течение 2 часов. Получали раствор промежуточного соединения 111-5, который использовали непосредственно на следующей стадии.
Стадия 111-5:
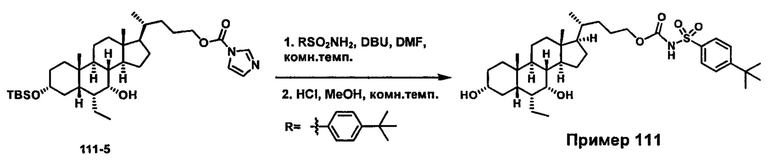
Смесь p-tBuPhSO2NH2 (0,3 ммоль), DBU (92,6 мг, 0,61 ммоль) в DMF (2 мл) добавляли к одному образцу промежуточного соединения 111-5, указанного выше, смесь перемешивали при комнатной температуре в течение ночи. Воду (10 мл) добавляли после остывания до комнатной температуры, экстрагировали EtOAc (10 мл × 2), сушили над Na2SO4, фильтровали и выпаривали. Остаток растворяли в МеОН (2 мл), затем добавляли 1 каплю 37% HCl. Смесь перемешивали при комнатной температуре в течение 10 минут, затем разбавляли EtOAc (50 мл) и последовательно промывали насыщенным NaHCO3 (10 мл × 2) и солевым раствором (10 мл × 1). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флэш-преп-ВЭЖХ ((IntelFlash-1): колонка, С18, подвижная фаза, MeCN/H2O, УФ-детектор 254 нм) с получением целевого соединения Примера 111 ИЭР-МС m/z=644,40 [М-1]-.
Нижеприведенные соединения Примеров 112-120 в Таблице 7 были получены способами, аналогичными описанным в Примерах 111.
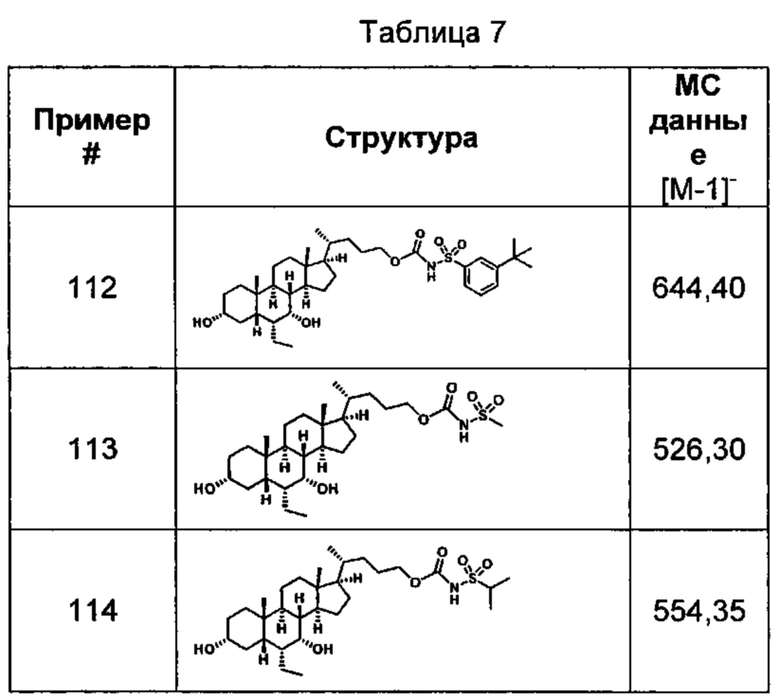
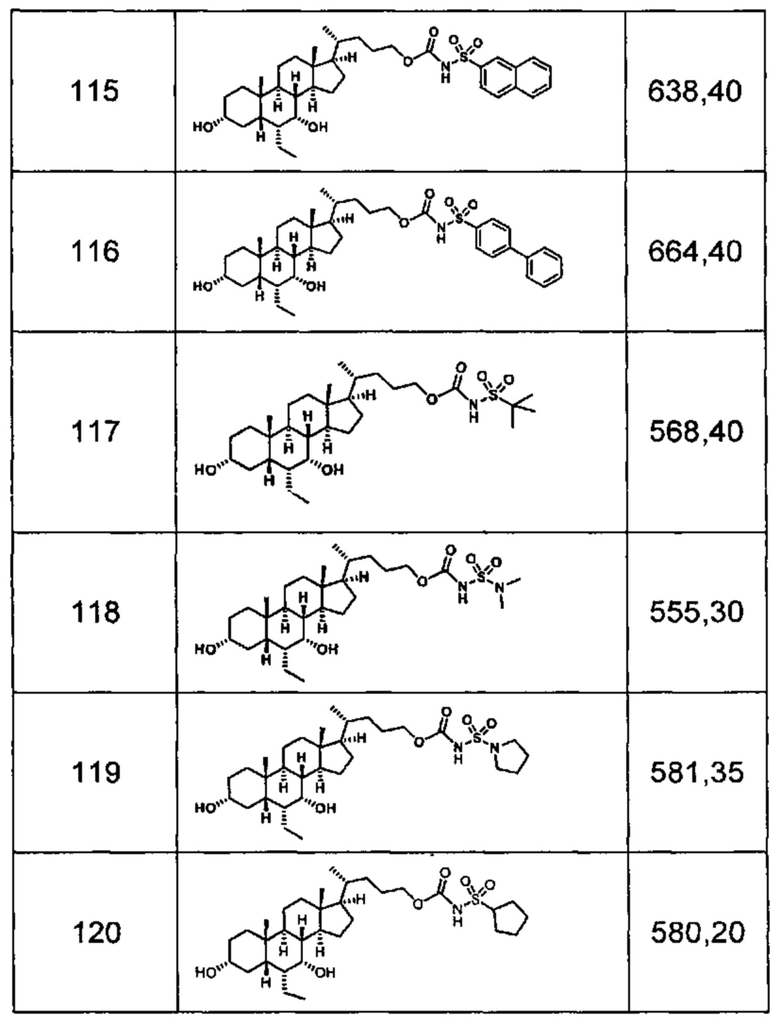
Пример 209
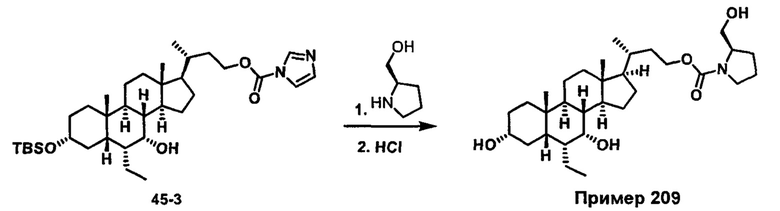
К раствору (R)-пирролидин-2-илметанола (51 мг, 0,501 ммоль) и DBU (127 мг, 0,833 ммоль) в DCM (5 мл) добавляли соединение 45-3 (100 мг, 0,167 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили водой (10 мл) и экстрагировали EtOAc (10 мл × 3). Объединенную органическую фазу сушили над Na2SO4, фильтровали и концентрировали.
Остаток растворяли в МеОН (2 мл). К раствору добавляли 5 мкл 37% HCl и смесь перемешивали при комнатной температуре в течение 10 минут, разбавляли EtOAc (20 мл) и последовательно промывали насыщенным бикарбонатом натрия (10 мл) и солевым раствором (10 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали с помощью флэш-препр-ВЭЖХ ((IntelFlash-1): колонка, С18, подвижный слой, MeCN/H2O, УФ-детектор 254 нм) с получением 16,6 мг соединения Примера 209 в виде белого твердого вещества. [М-1]-, 518,20.
Нижеприведенные соединения Примеров 121-208 и Примеров 210-218 в Таблице 8 были получены способами, аналогичными описанным в Примерах 209, начиная с соединений 4-8, 111-5 или 45-3.
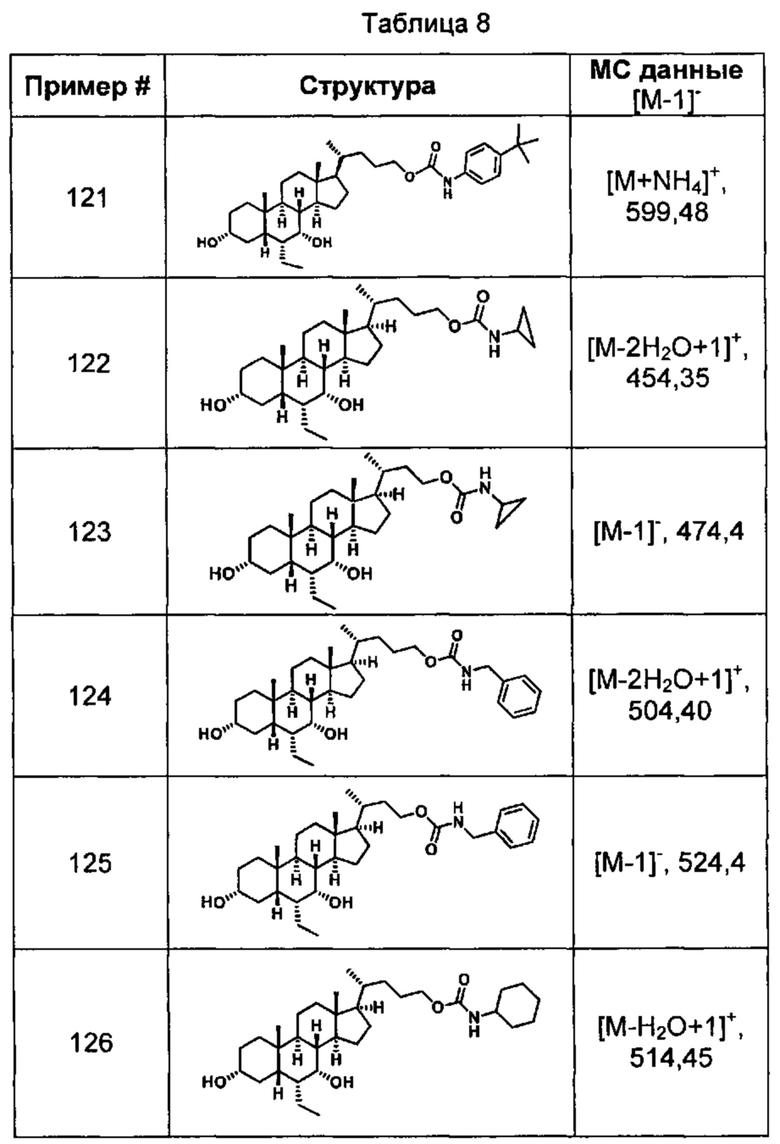
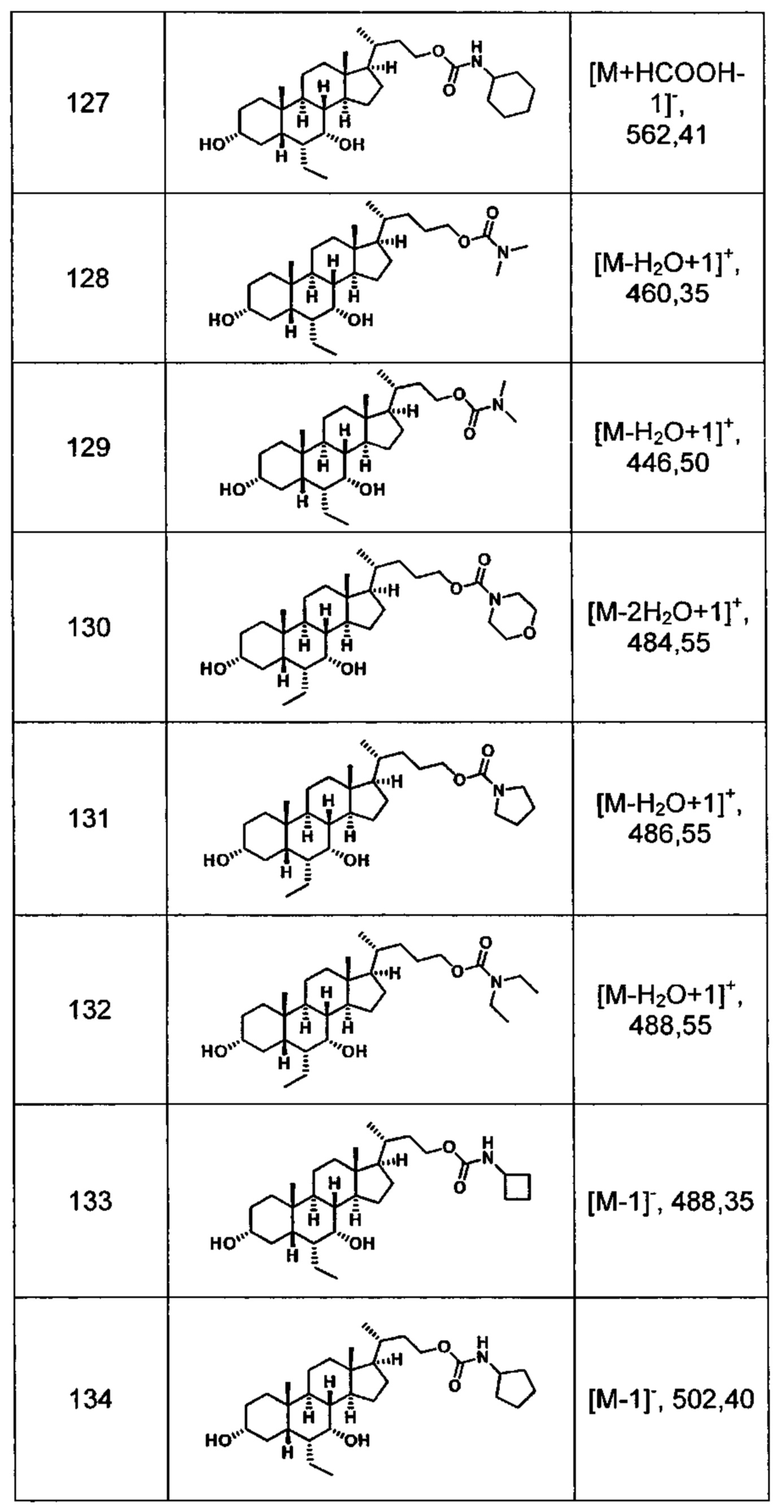
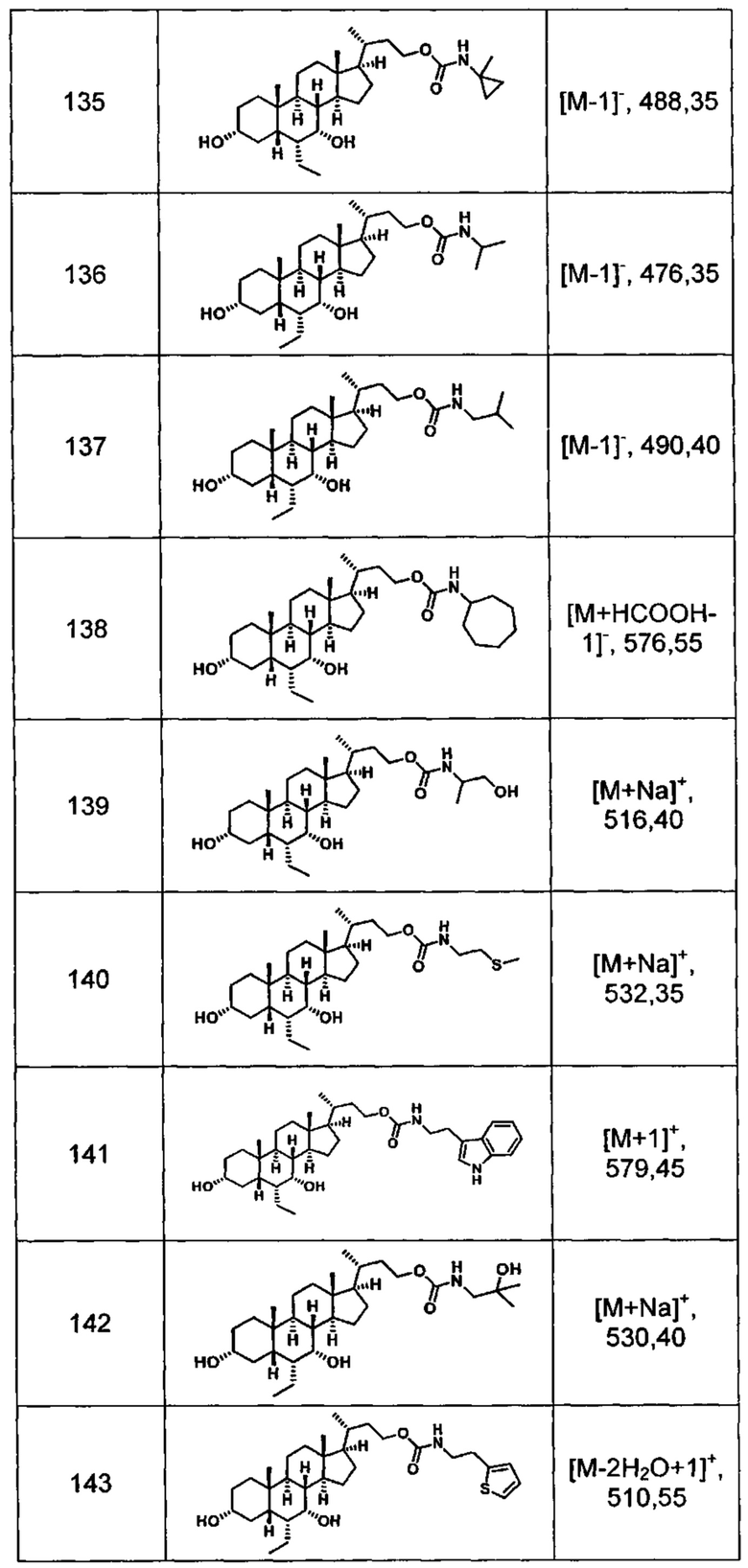
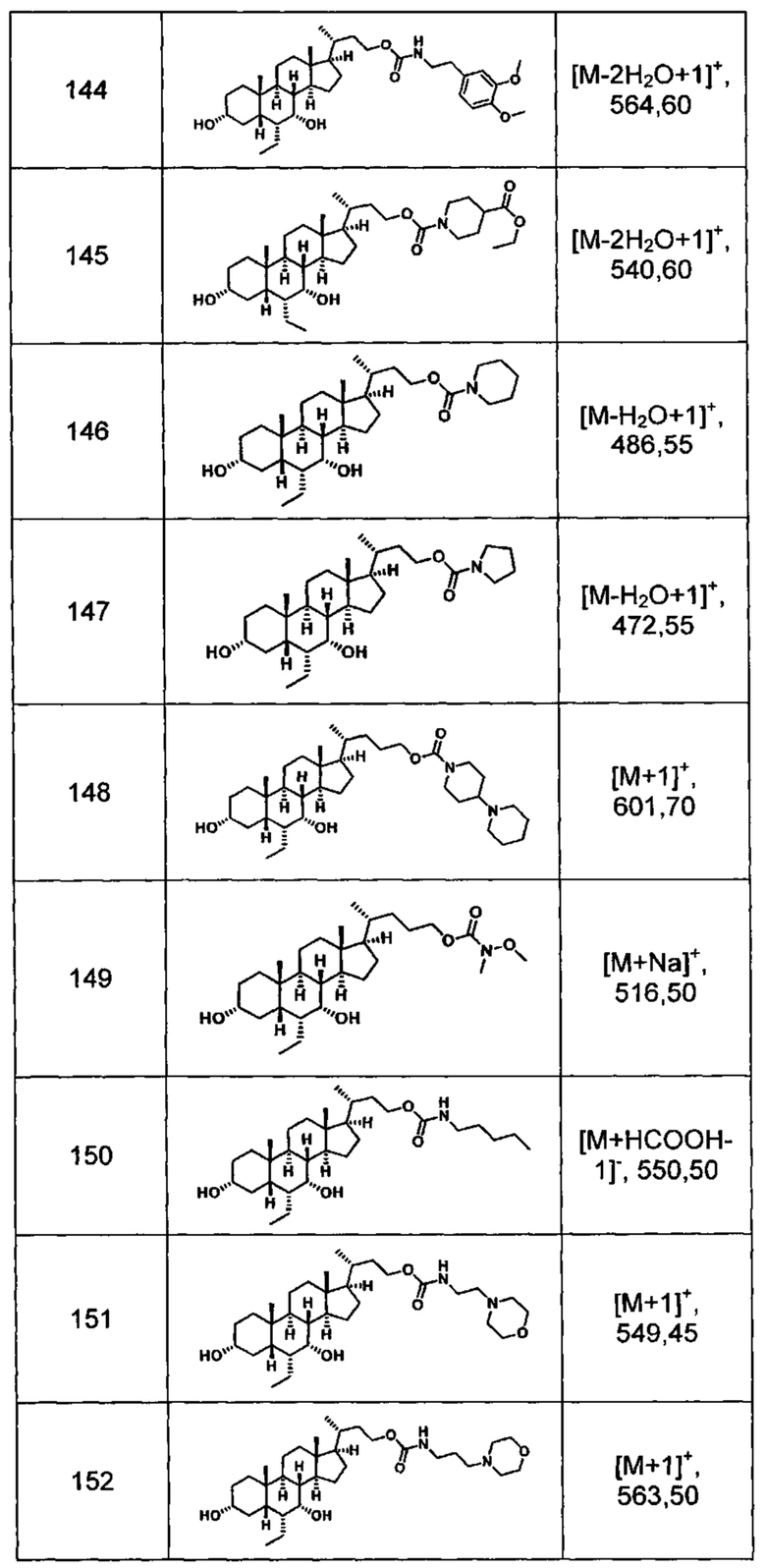
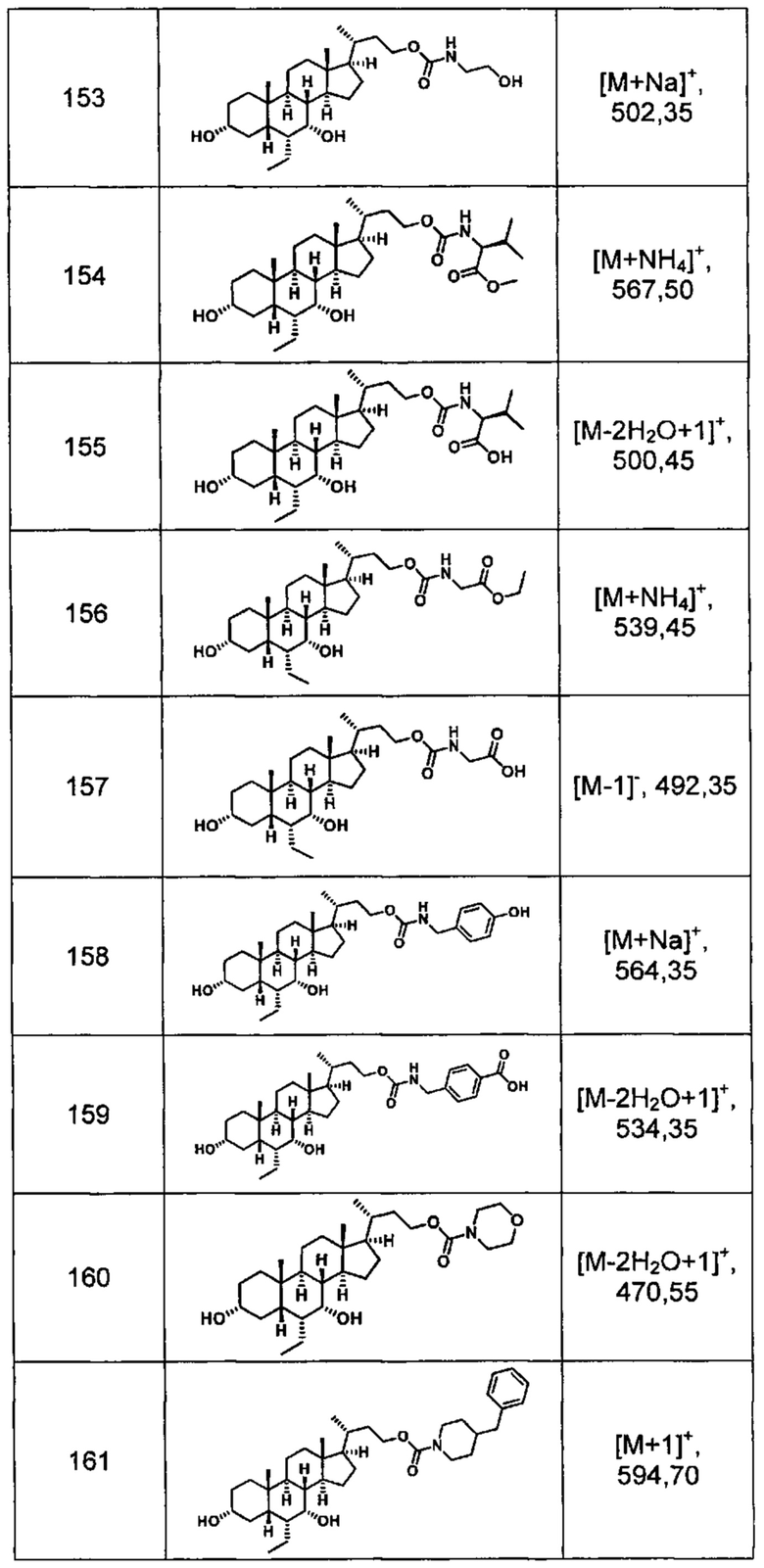
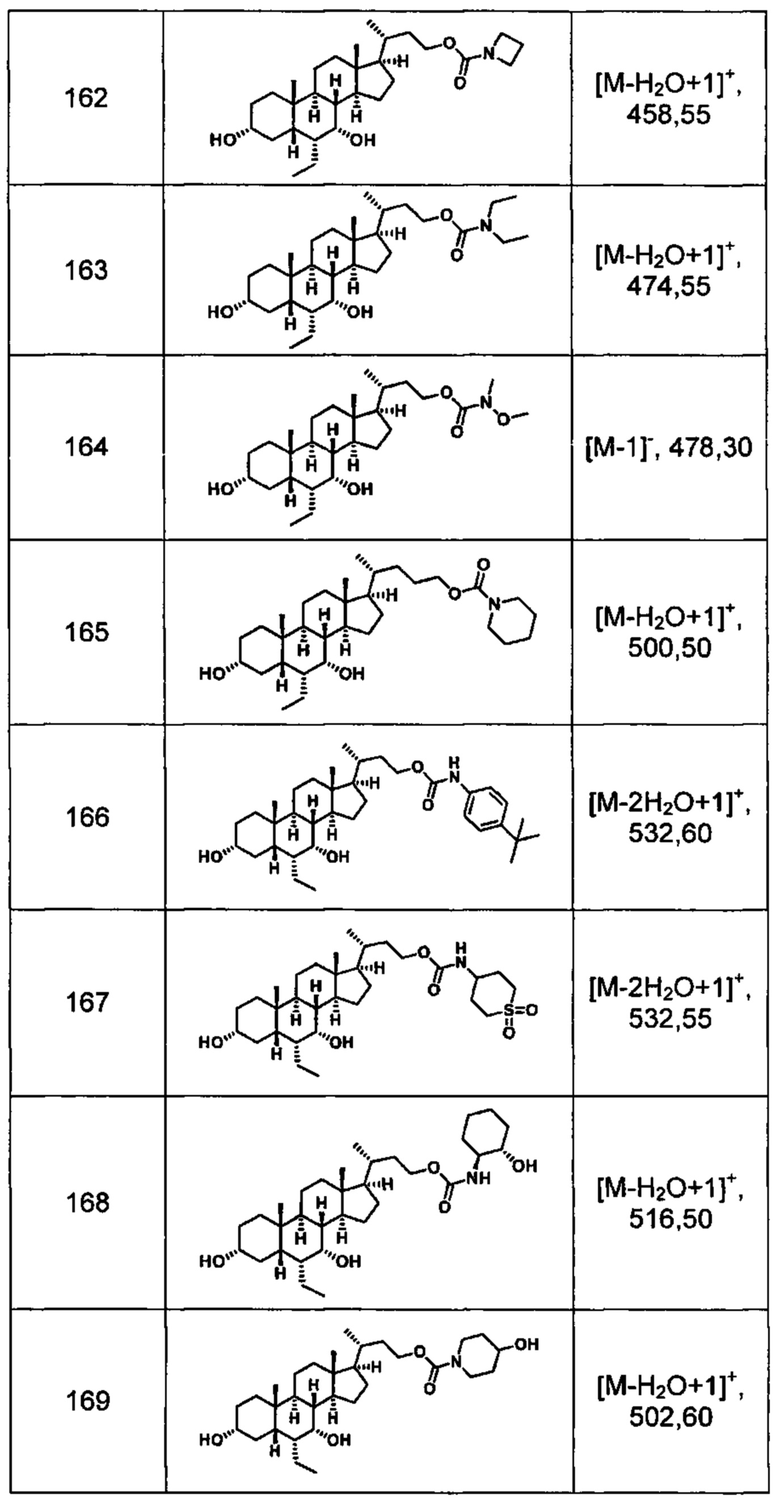
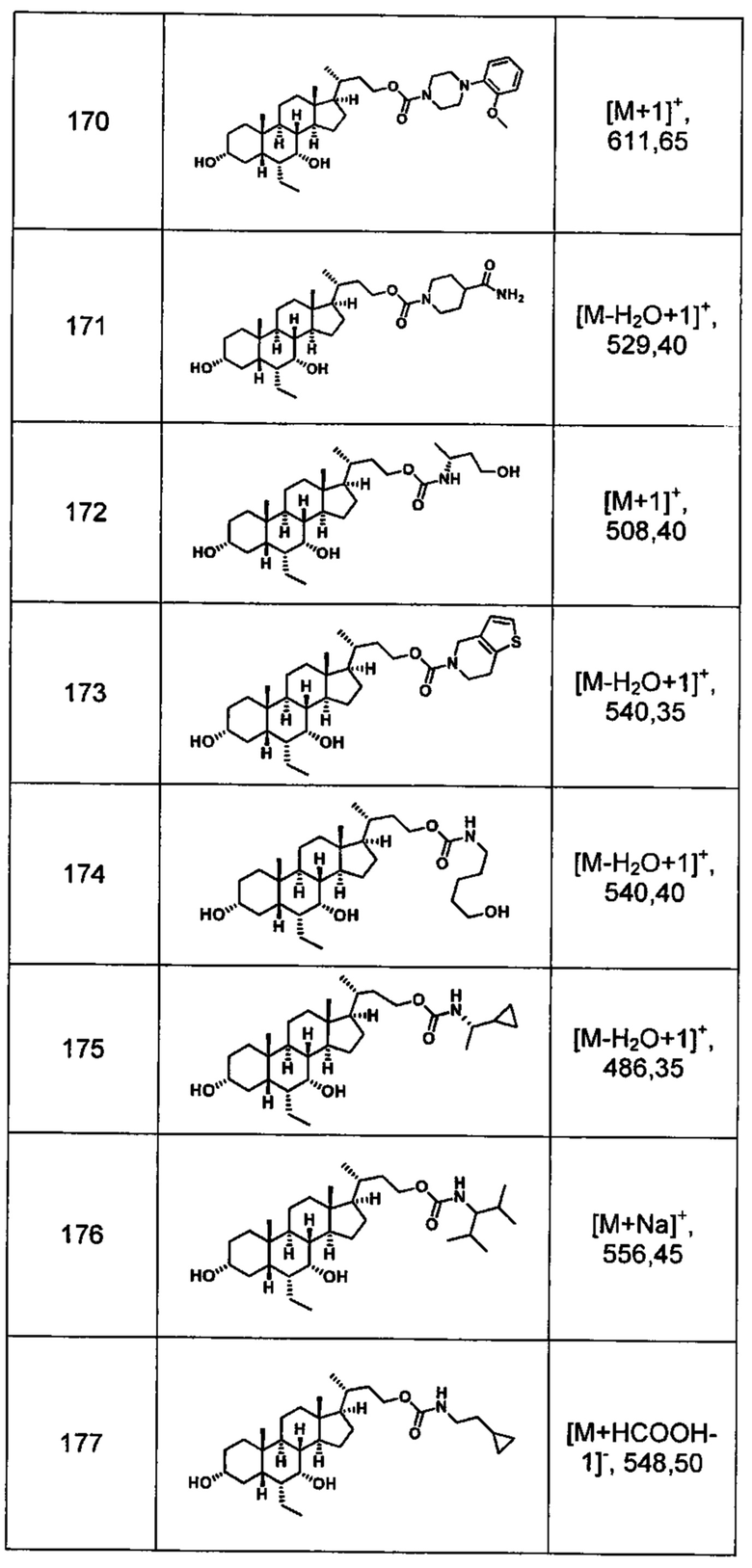
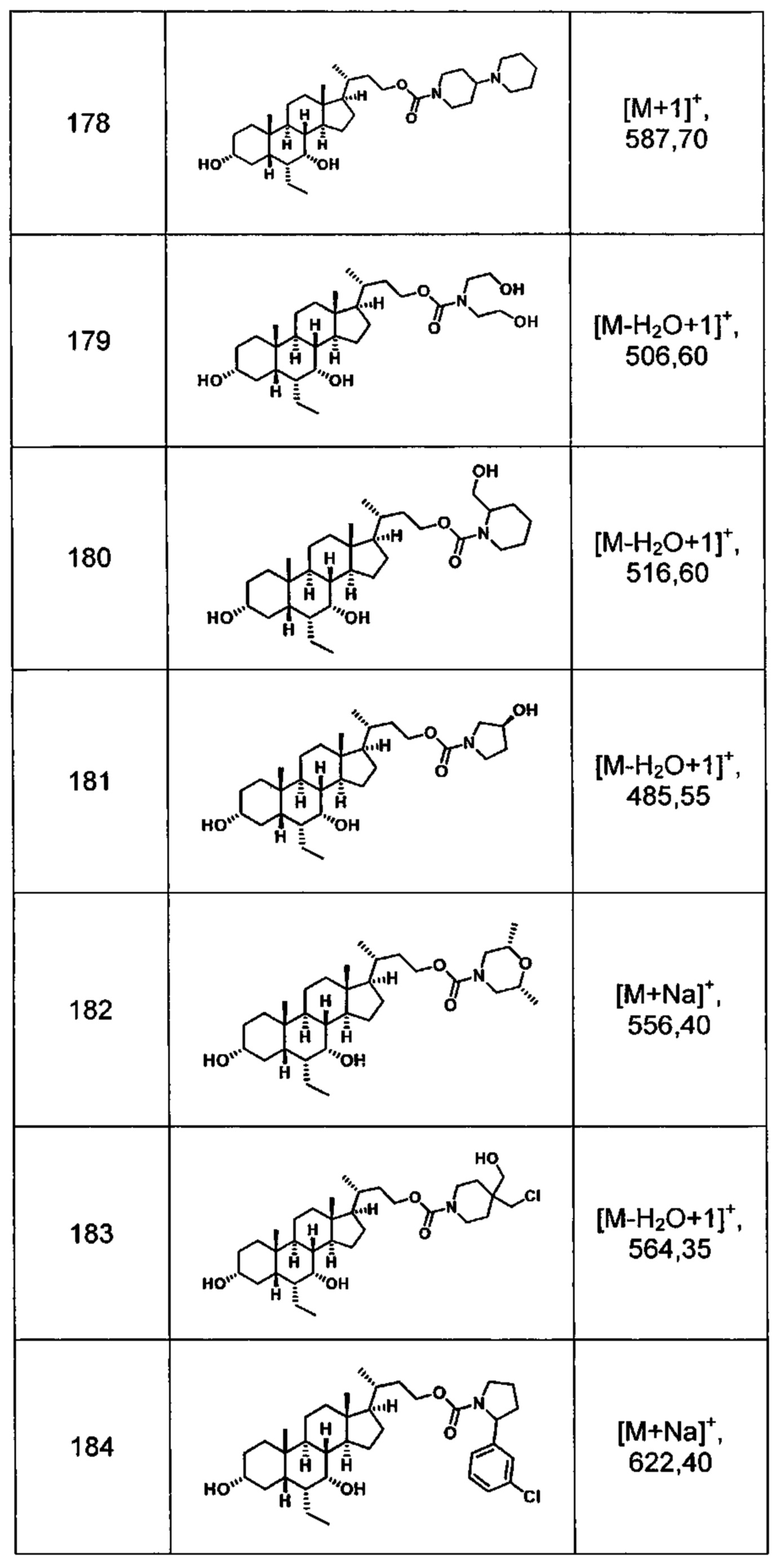
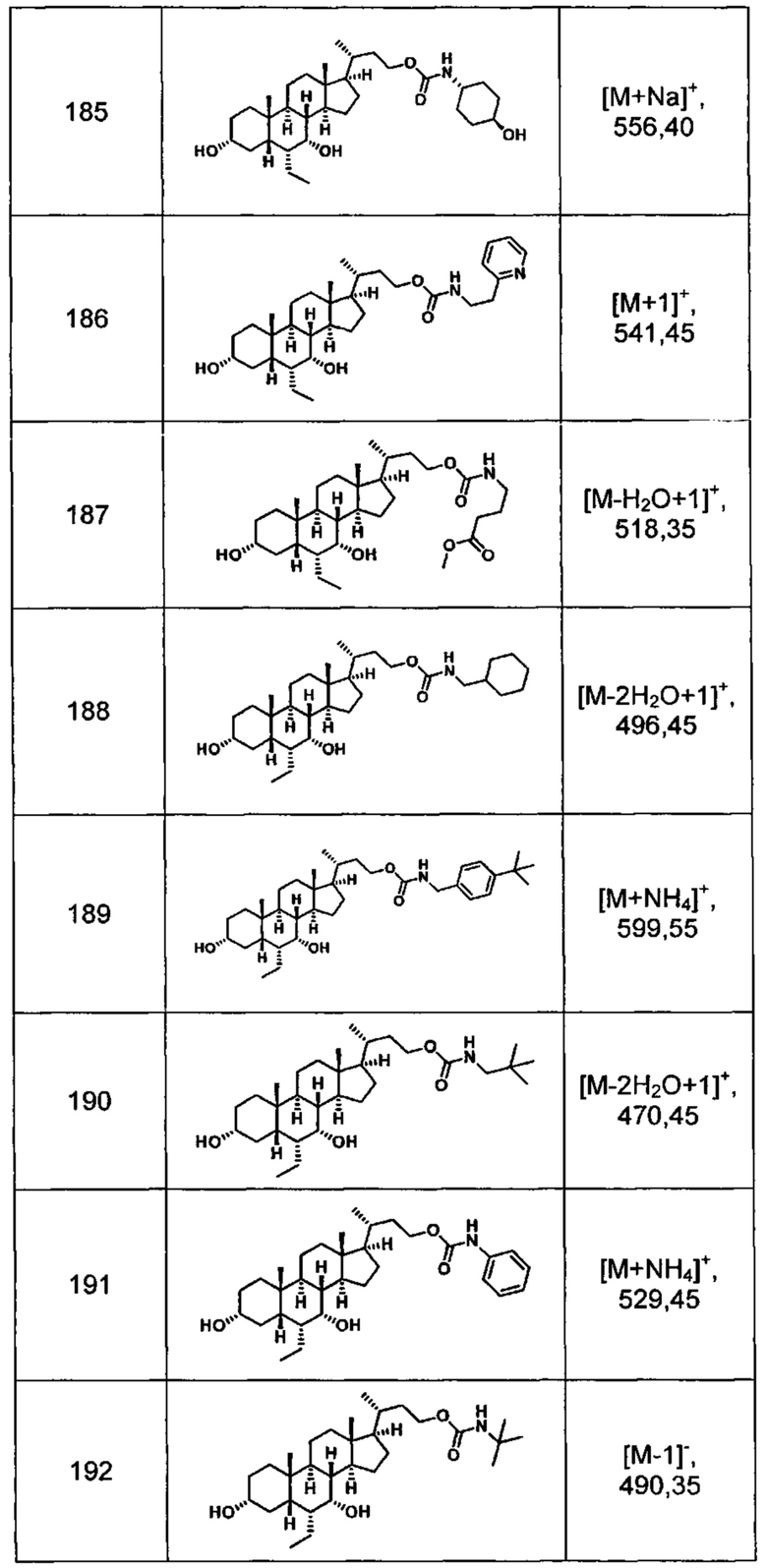
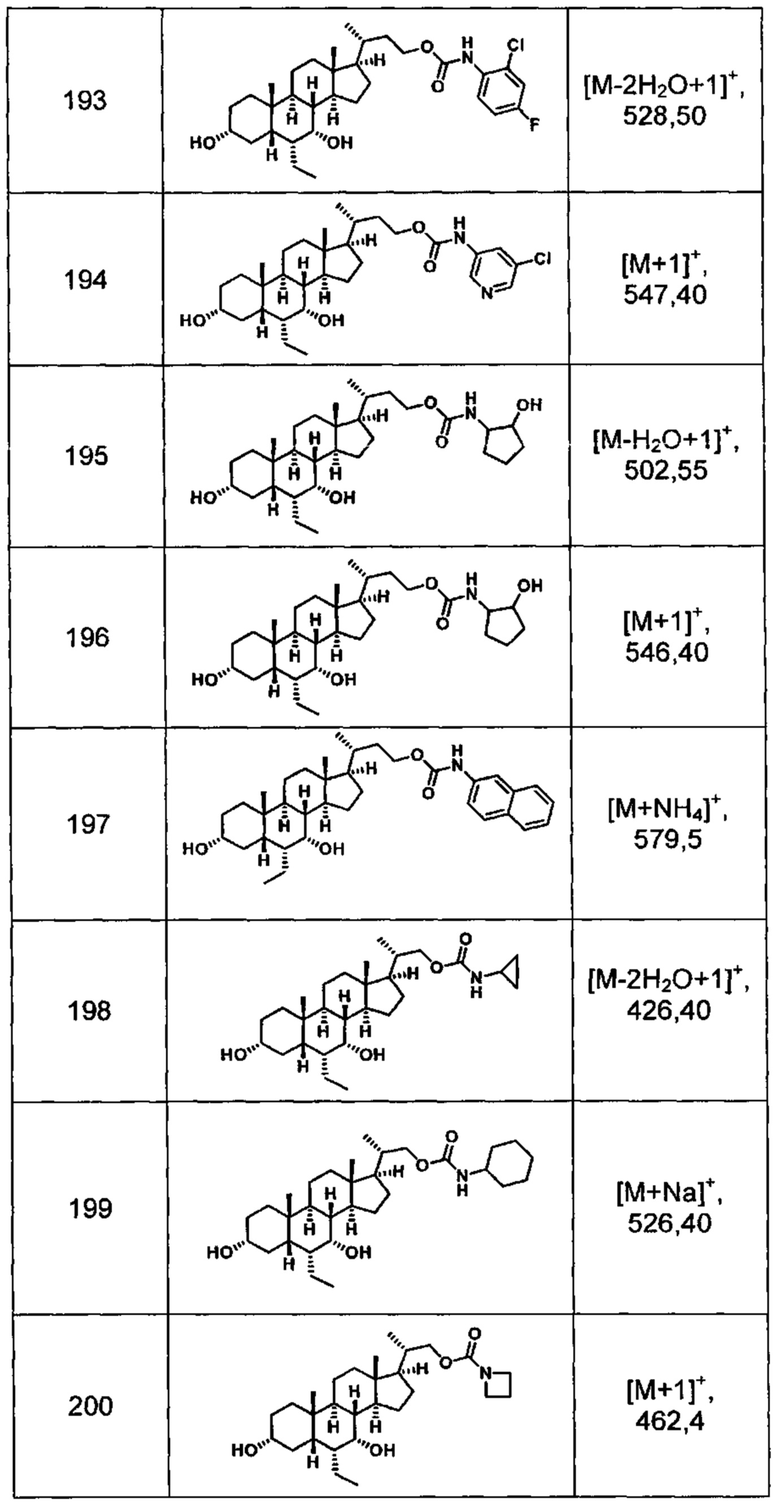
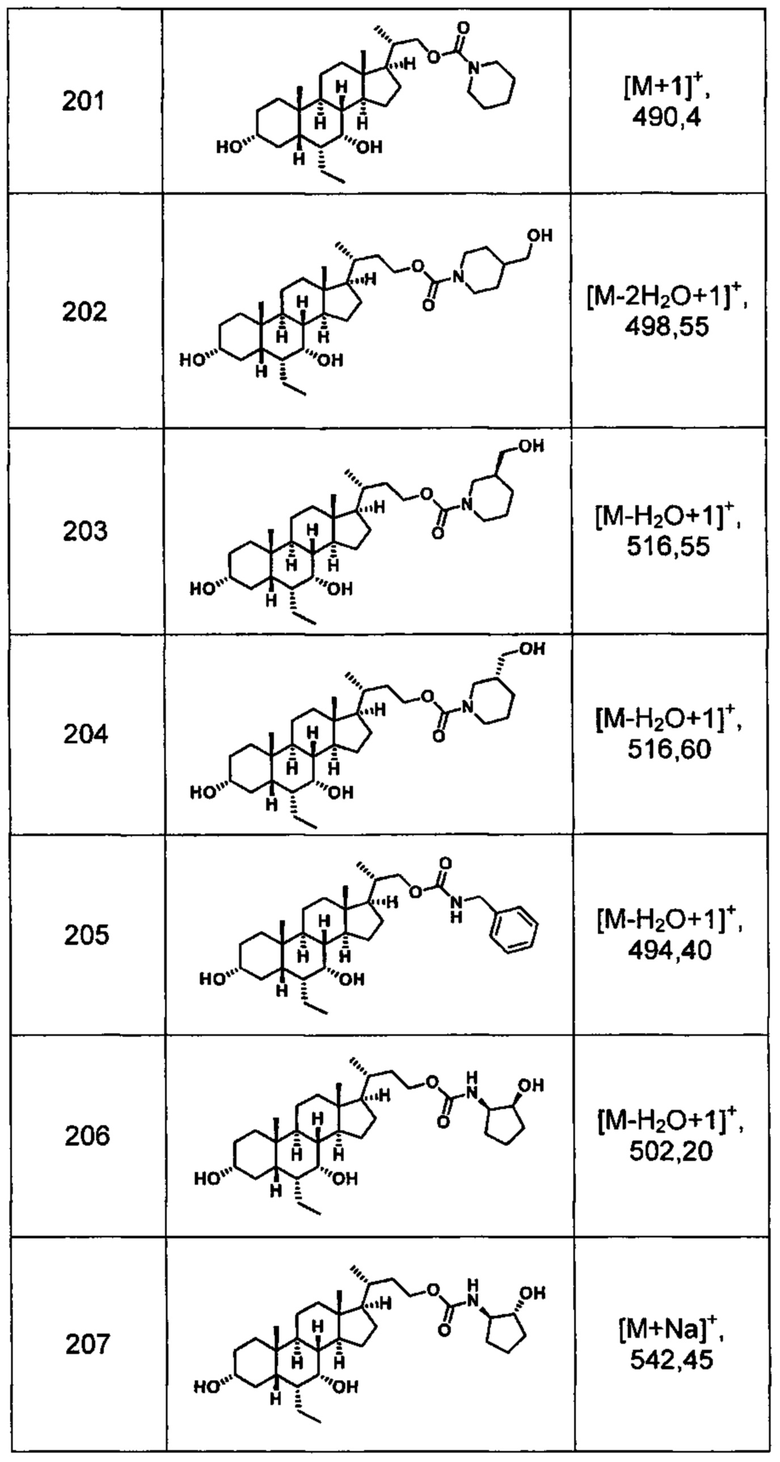
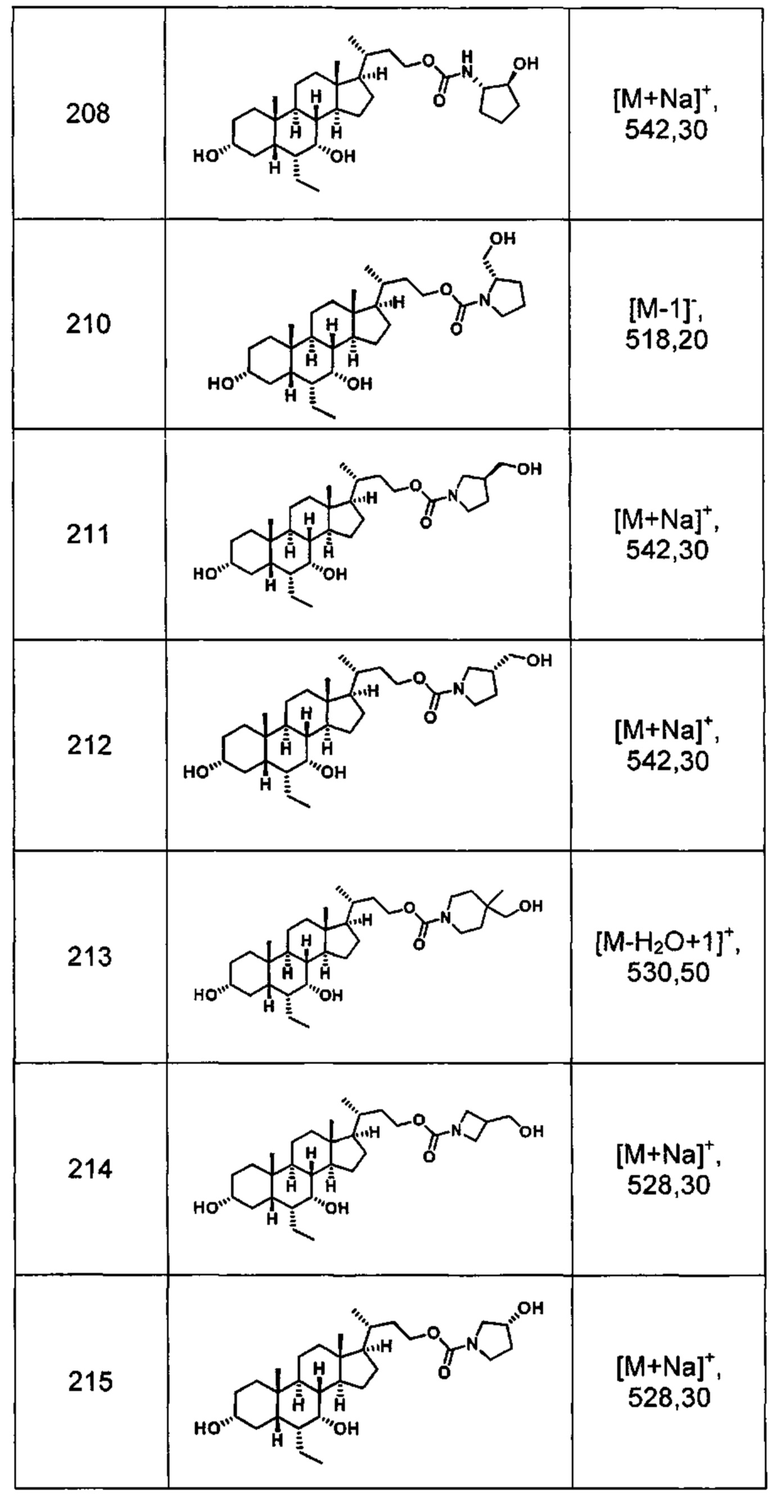
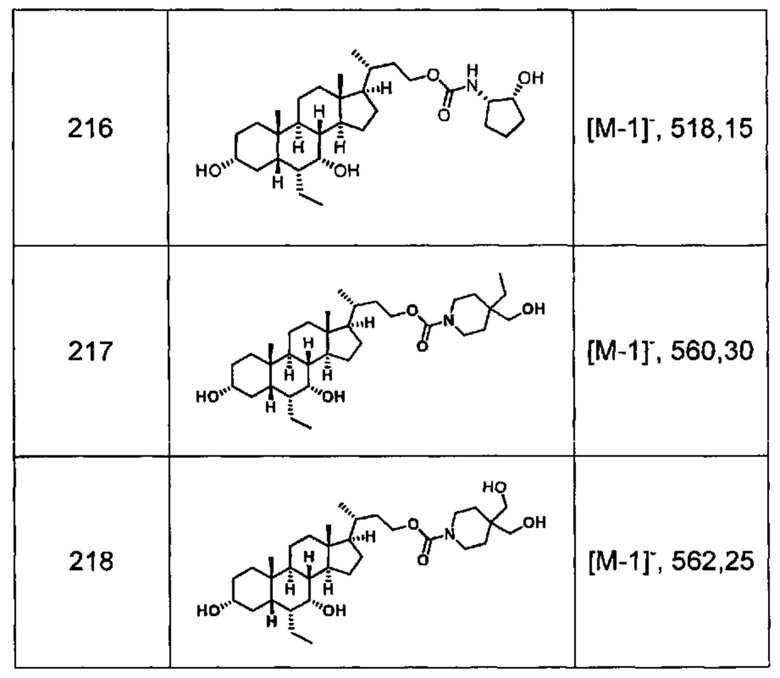
АНАЛИЗЫ
Анализ FXR человека (NR1H4)
Определение лиганд-опосредованной трансактивации, управляемой промотором Gal4, для количественного определения опосредованной связыванием лиганда активации FXR. Набор для репортерного анализа FXR, приобретенный у Indigo Bioscience (номер по каталогу: IB00601) для определения активности и эффективности соединения, разработанного Enanta, которое может вызвать активацию FXR. Принципиальное применение этой репортерной системы анализа заключается в количественной оценке функциональной активности FXR человека. В анализе используют клетки млекопитающего, не относящегося к человеку, клетки СНО (яичника китайского хомячка), сконструированные для экспрессии белка NR1H4 человека (называемого FXR). Репортерные клетки также включают кДНК, кодирующую люциферазу жука, которая катализирует субстраты и дает излучение фотонов. Интенсивность люминесценции реакции количественно определяют с помощью люминометра, считывающего планшет, Envision. Репортерные клетки включают репортерный ген люциферазы, функционально связанный с чувствительным к FXR промотором. Таким образом, количественные изменения экспрессии люциферазы в обработанных репортерных клетках обеспечивают чувствительное суррогатное измерение изменений активности FXR. EC50 и эффективность (нормализованная к CDCA, установленной как 100%) определяют с помощью XLFit. Анализ проводят в соответствии с инструкциями производителя. Кратко, анализ проводили на белых 96-луночных планшетах с использованием конечного объема 100 мкл, содержащего клетки с различными дозами соединений. Репортерные клетки извлекали из хранилища, температура в котором - 80°С. Замороженные клетки размораживали путем переноса в пробирку с замороженными клетками 10 мл среды для восстановления клеток, нагретой до температуры 37°С. Пробирку с репортерными клетками закрывали и немедленно помещали ее на водяную баню с температурой 37°С на 5-10 минут. Пробирку с суспензией репортерных клеток извлекали из водяной бани. Наружную поверхность пробирки санировали с помощью 70% спиртового тампона, а затем переносили ее в культуральный вытяжной шкаф. В каждую лунку 96-луночного планшета для анализа распределяли 90 мкл клеточной суспензии. Планшет переносили в инкубатор при 37°С, для того, чтобы клетки прикрепились ко дну лунки. Соединения разбавляли в планшете для разведения (ПР) и вводили клетки в аналитический планшет (АП). Содержание DMSO в образцах поддерживали на уровне 0,2%. Клетки инкубировали в течение дополнительных 22 часов перед измерением активности люциферазы. За тридцать минут до количественной оценки активности FXR проявляющий субстрат и проявляющий буфер доставали из холодильника и помещали их в область с низким освещением, чтобы привести их в равновесие с комнатной температурой. Крышку планшета снимали и удаляли всю содержащуюся среду, поместив ее в соответствующий контейнер для отходов. По перевернутому планшету аккуратно стучали над чистой абсорбирующей бумажной салфеткой, чтобы удалить остаточные капли. Клетки оставались плотно прикрепленными ко дну лунок. Добавляли 100 мкл реагента для выявления люциферазы в каждую лунку аналитического планшета. Аналитический планшет остывал при комнатной температуре в течение не менее 5 минут после добавления реагента для выявления люциферазы. Устанавливали инструмент (Envision) для выполнения одного 5-секундного встряхивания планшета перед считыванием первой аналитической лунки. Время считывания может составлять 0,5 секунды (500 мсек) на лунку. EC50 и эффективность (нормализованная к CDCA, установленной как 100%) определяют с помощью XLFit.
In vitro анализ активности TGR5 человека (GPBAR1)
Активность и эффективность соединений по изобретению в отношении рецептора TGR5 оценивали с применением анализов in vitro, которые проводили с применением экспресс-набора от DiscoverX (CAMP HUNTER™ eXpress GPBAR1 CHO-K1 GPCR Assay; номер по каталогу: 95-0049E2CP2S) GPBAR1 (рецептор желчной кислоты 1, связанный с G-белком) кодирует член суперсемейства рецептора, связанного с G-белком (GPCR). Активация GPBAR1 после связывания лиганда инициирует серию каскадов передачи сигнала, которые приводят к клеточному ответу. Обработка клеток СНО, экспрессирующих GPBAR1, желчными кислотами индуцирует продукцию внутриклеточного цАМФ и интернализацию рецептора. Активность и эффективность соединения для активации GPBAR1 определяли путем измерения уровней циклического аденозинмонофосфата (циклического АМФ или цАМФ) в живых клетках с применением конкурентного иммуноанализа на основе комплементации фрагментов белка (EFC).
Кратко, после посева клеток в белый 96-луночный микропланшет его помещали во влажный инкубатор при 37°С, 5% CO2 на от 18 до 24 часов перед тестированием. На второй день переходили к соответствующему протоколу цАМФ Hunter eXpress в соответствии с инструкциями производителя. Соединение агониста в DMSO растворяли в желаемой стоковой концентрации и готовили 3-кратные серийные разведения соединения агониста в буфере для клеточного анализа. Концентрацию каждого разведения готовили при 4 × конечной концентрации (то есть 15 мкл соединения + 45 мкл буфера для клеточного анализа/реактива цАМФ антитела). Для каждого разведения конечная концентрация растворителя должна оставаться постоянной. 15 мкл разведенного соединения переносили на аналитический планшет и инкубировали планшет в течение 30 минут при 37°С. После инкубации с агонистами добавляли 60 мкл рабочих реактивов для обнаружения цАМФ/смеси раствора цАМФ (буфер для лизиса цАМФ, субстратный реагент 1, раствор цАМФ D) в соответствующие лунки. Инкубацию осуществляли в течение 1 часа при комнатной температуре (23°С), защищая от света. Добавляли 60 мкл раствора цАМФ А в соответствующие лунки. Инкубацию осуществляли в течение 3 часов при комнатной температуре (23°С), защищая от света. Проводили считывание образцов стандартного люминесцентного планшетного ридера Envision. Осуществляли вычисление среднего значения EC50 после преобразования логарифма.
Для оценки агонистической активности FXR соединений, раскрытых в примерах, а также для эталонного соединения, диапазоны активности определяли в анализе FXR человека (NR1H4), как указано ниже в Таблице 9. Эффективность нормализовали к CDCA, установленной как 100%. (А = EC50 < 0,1 мкМ, В = 0,1 мкМ < ЕС50 < 1,0 мкМ; С = 1,0 мкМ < EC50 < 10 мкМ; D = EC50 > 10 мкМ).
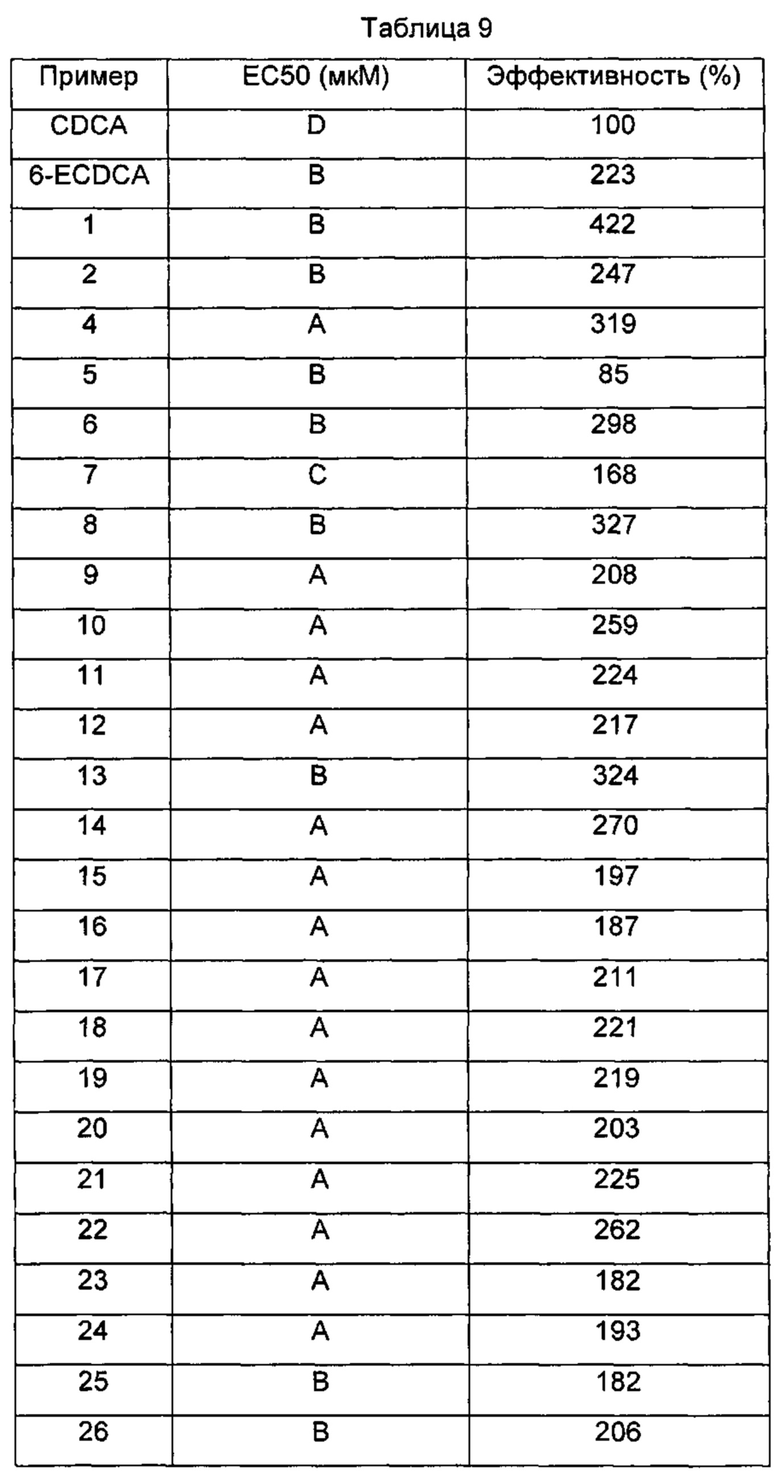
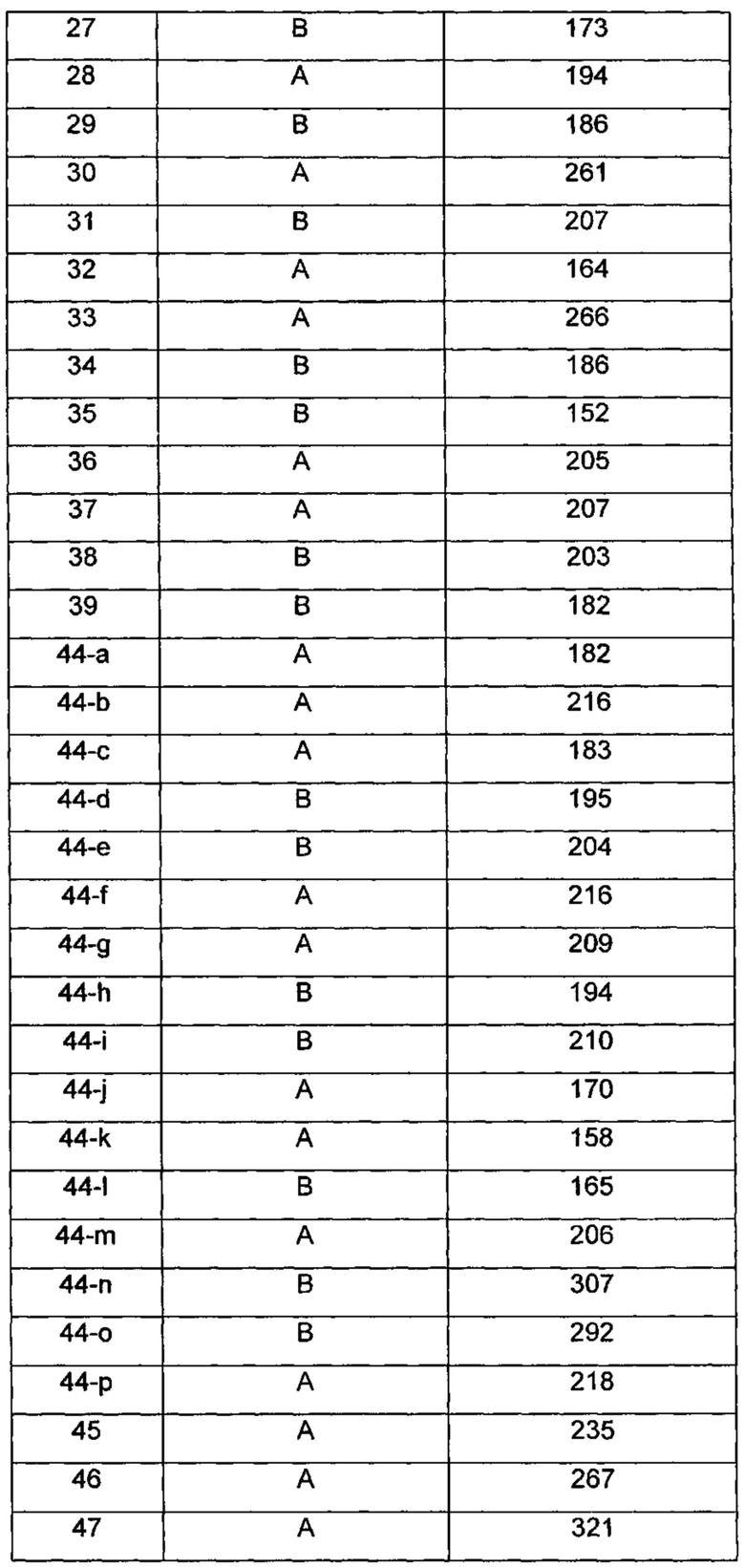
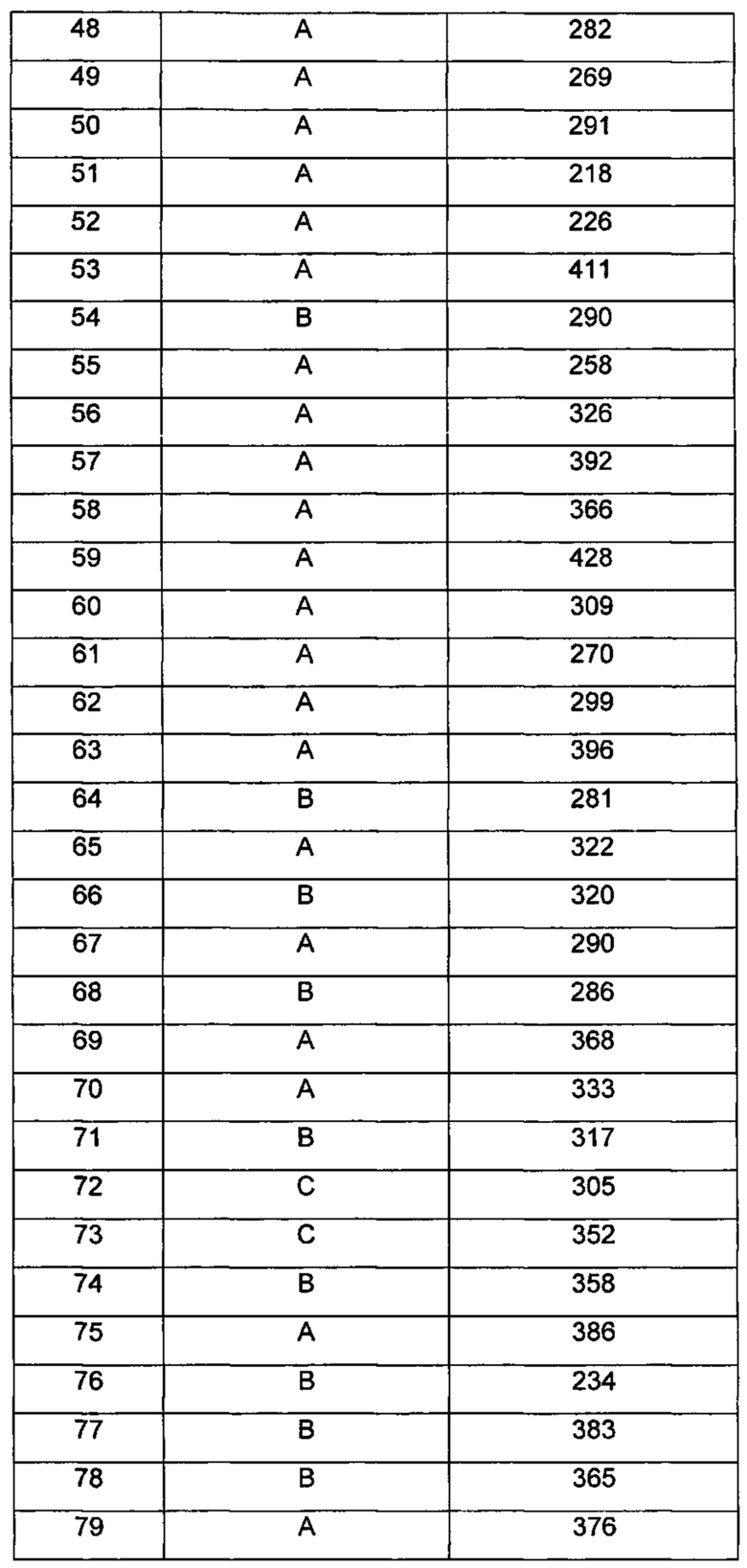
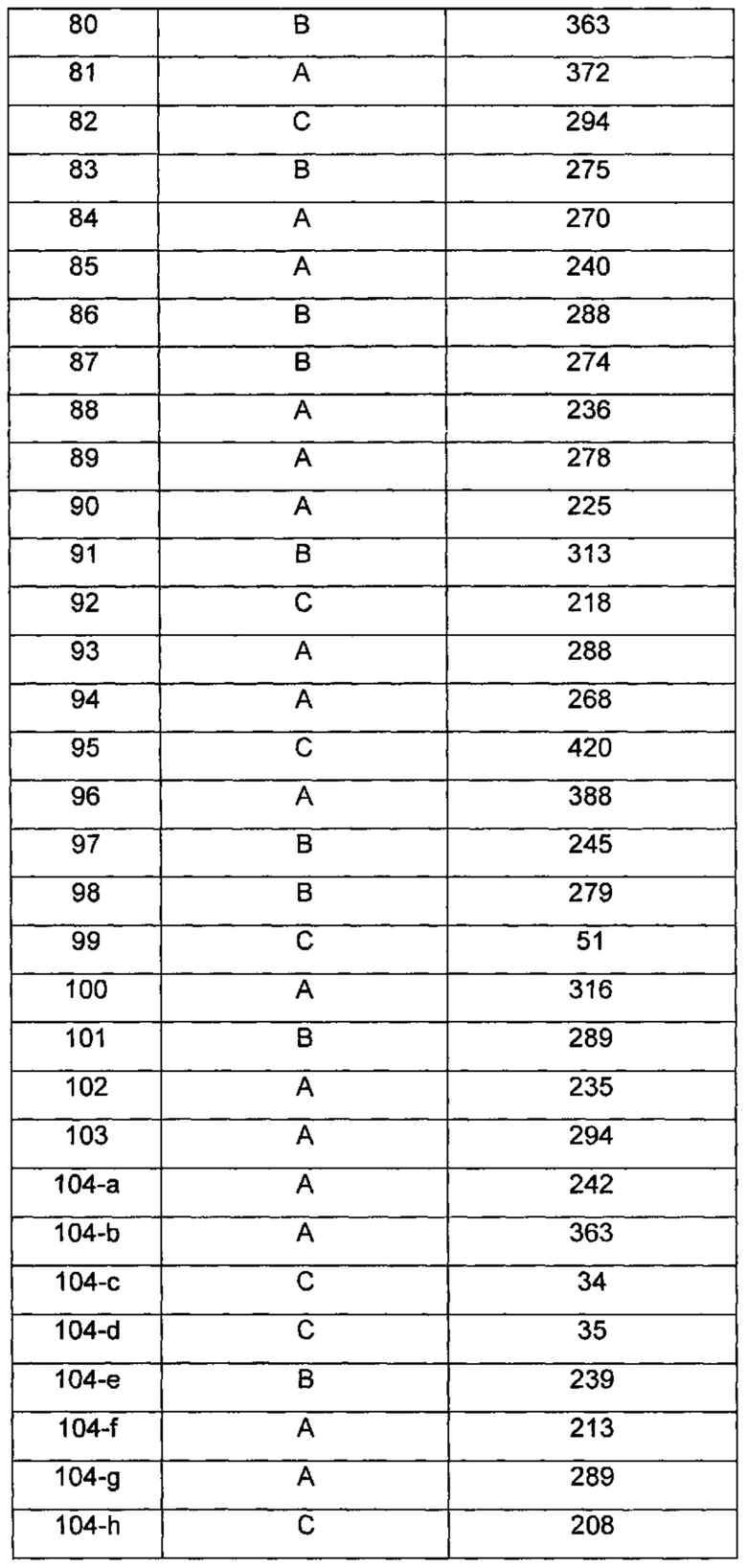
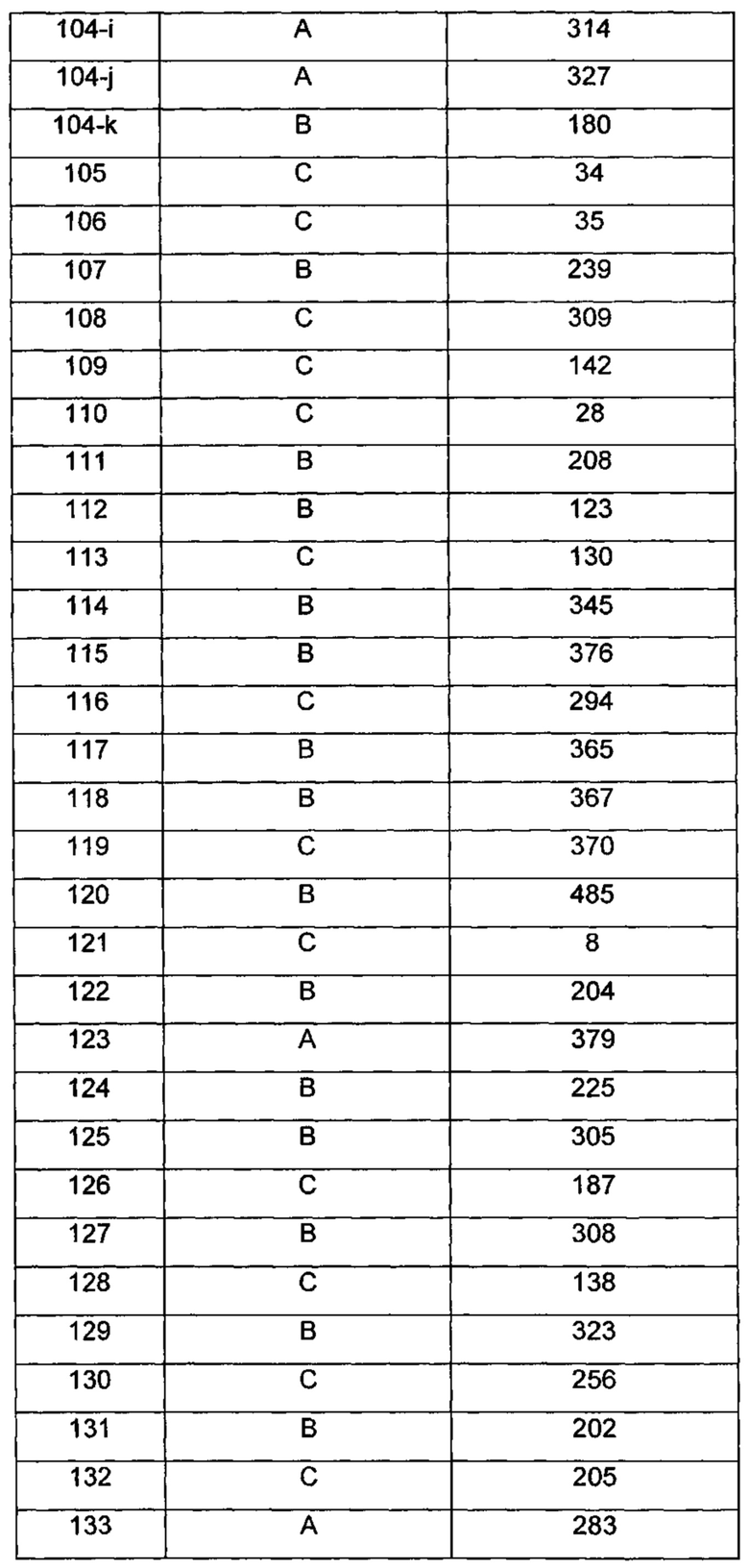
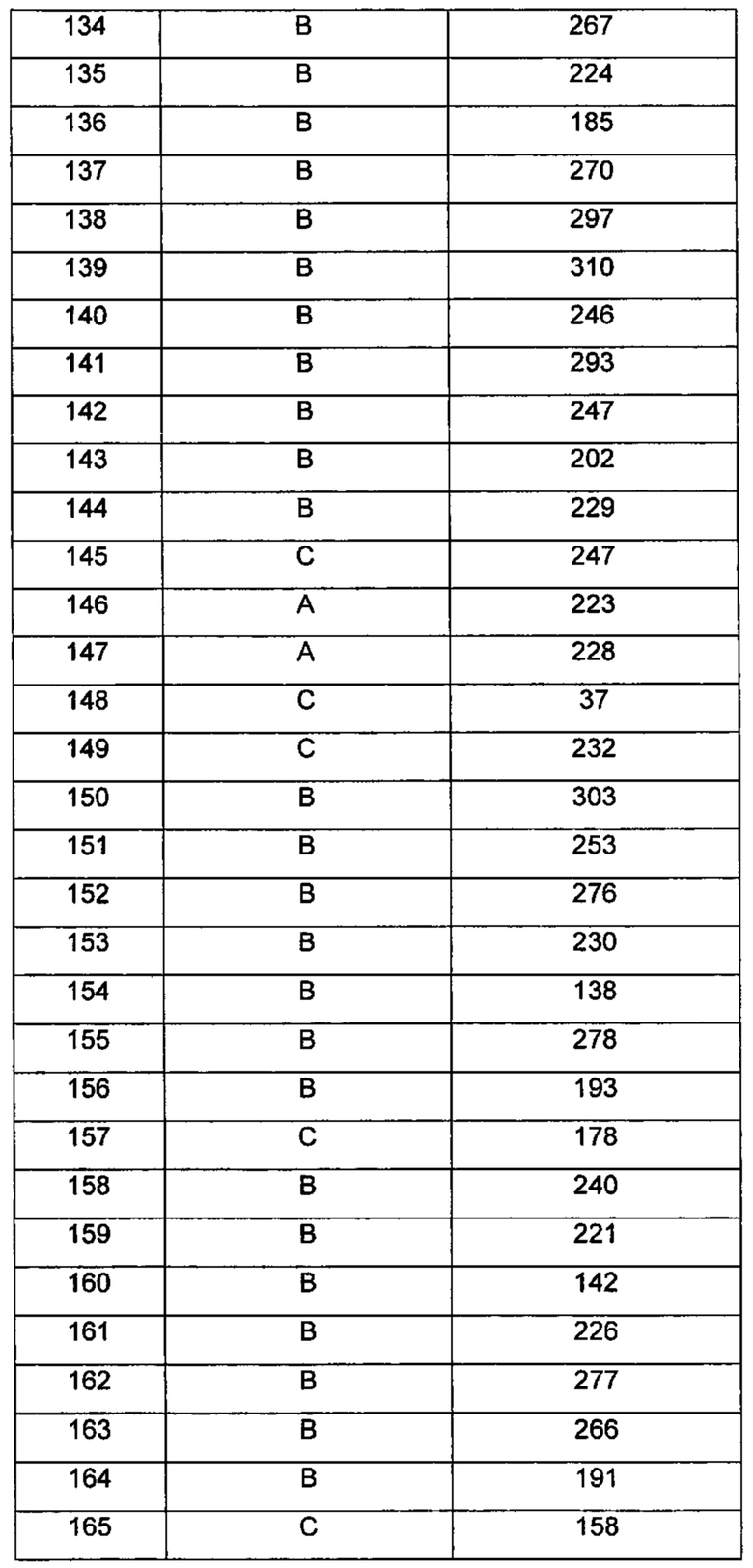
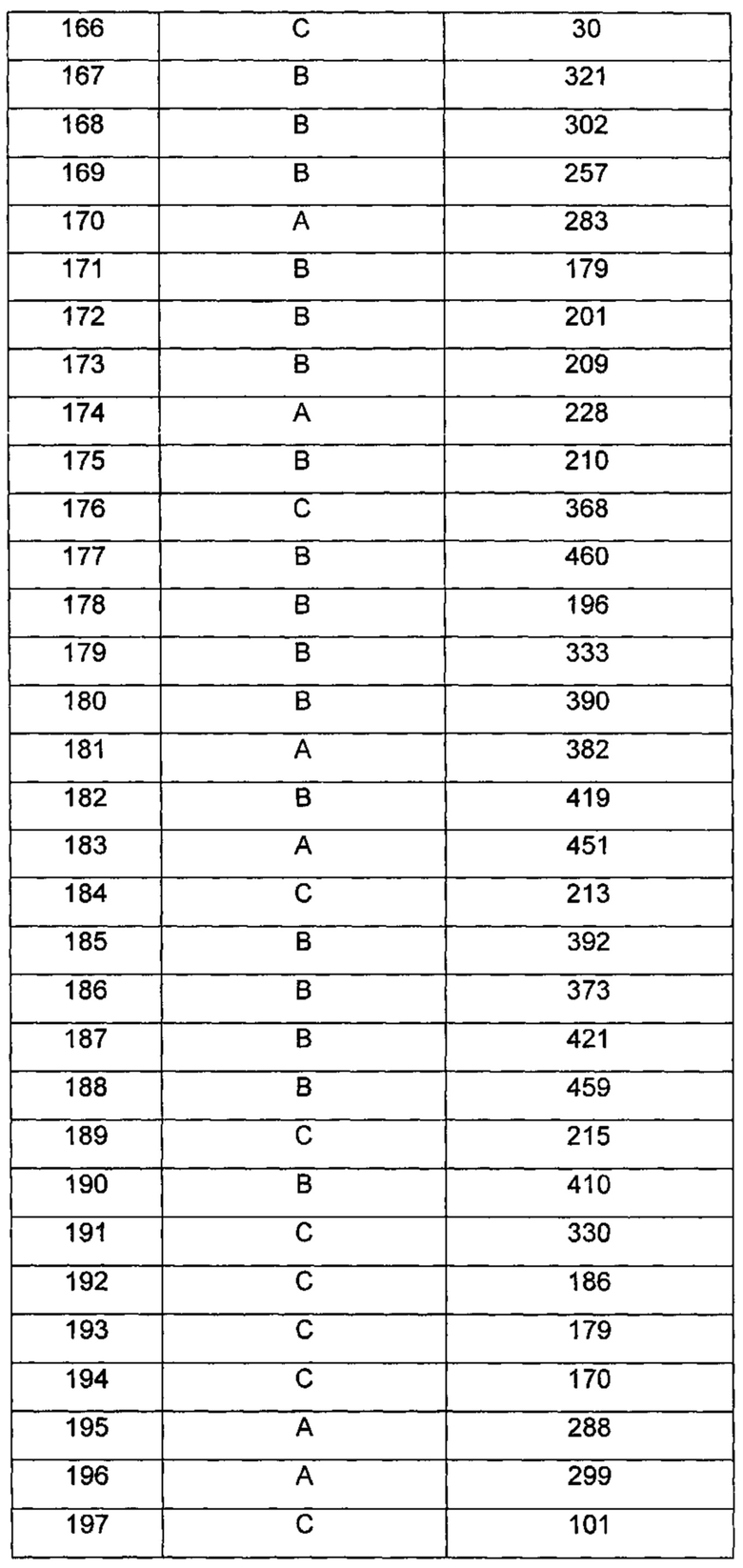
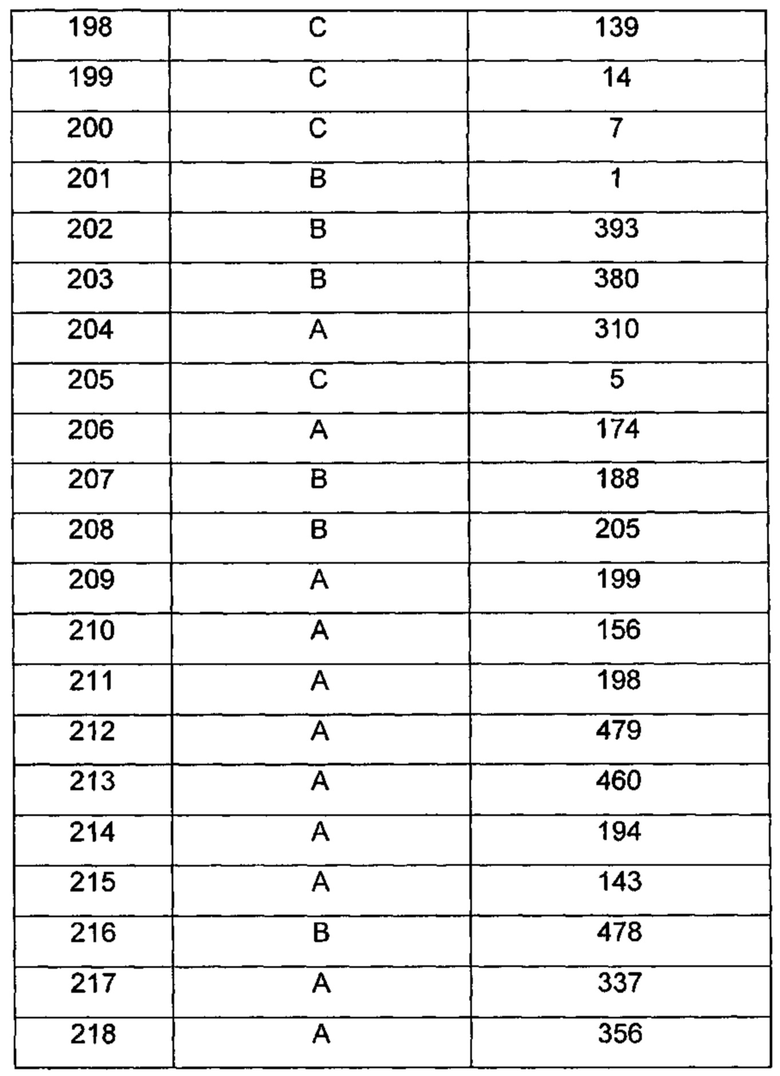
Хотя это изобретение было подробно продемонстрировано и описано со ссылками на предпочтительные варианты его осуществления, специалистам в данной области техники будет понятно, что в него могут быть внесены различные изменения в форме и деталях без отступления от объема изобретения, охватываемого прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЖЕЛЧНОЙ КИСЛОТЫ В КАЧЕСТВЕ АГОНИСТОВ FXR/TGR5 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2707280C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФОНИЛКАРБАМАТНЫХ ПРОИЗВОДНЫХ ЖЕЛЧНЫХ КИСЛОТ | 2018 |
|
RU2791682C2 |
| ХИНОКСАЛИНСОДЕРЖАЩИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ВИРУСА ГЕПАТИТА С | 2008 |
|
RU2493160C2 |
| ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С | 2010 |
|
RU2544010C2 |
| ТЕТРАЗОЛ-СОДЕРЖАЩИЕ ИНГИБИТОРЫ РЕГУЛИРУЮЩЕЙ АПОПТОТИЧЕСКИЕ СИГНАЛЫ КИНАЗЫ 1 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2807545C2 |
| АНАЛОГИ ДИАЗОНАМИДА | 2012 |
|
RU2608308C2 |
| ИНГИБИТОРЫ MEK И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2812929C1 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2808166C2 |
| 19-НОР C3, 3-ДИЗАМЕЩЕННЫЕ C21-N-ПИРАЗОЛИЛЬНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2675855C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2684103C2 |
Изобретение относится к соединению, представленному формулой I, или его фармацевтически приемлемой соли. В формуле I Ra представляет собой водород; Rb представляет собой -SO2R1; R2 представляет собой водород; Rc представляет собой водород; m выбран из 0, 1 и 2; R3 представляет собой водород; R4 представляет собой водород; R5 и R6 представляют собой водород; R7 представляет собой незамещенный -C1-C8 алкил; значения остальных радикалов указаны в формуле изобретения. Изобретение также относится к индивидуальным соединениям, к способу профилактики или лечения FXR-опосредованного заболевания или состояния у субъекта, нуждающегося в этом, к фармацевтической композиции. Технический результат: получены новые соединения формулы I, обладающие свойствами модулятора FXR. 4 н. и 20 з.п. ф-лы, 9 табл., 218 пр.
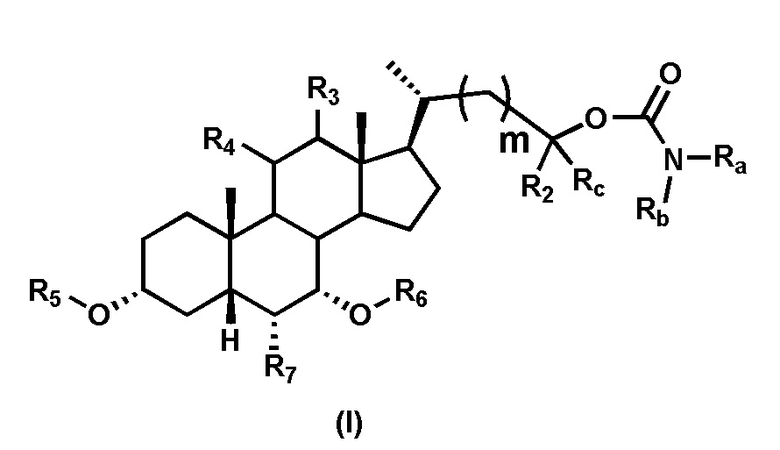
1. Соединение, представленное формулой I, или его фармацевтически приемлемая соль:
где:
Ra представляет собой водород;
Rb представляет собой -SO2R1;
R1 выбран из группы, состоящей из:
1) -C1-C8 алкила, необязательно замещенного галогеном, фенилом, необязательно замещенным галогеном, 7-членным бициклическим циклоалкилом, замещенным оксо и С1-С6-алкилом, С1-С6-алкокси, С1-С6-алкоксикарбонилом;
2) -C3-C8 циклоалкила, необязательно замещенного С1-С6-алкилом;
3) незамещенного нафтила; незамещенного 1,2,3,4-тетрагидронафтила; незамещенного инданила; или фенила, необязательно замещенного гидрокси, галогеном, С1-С6-алкилом, гидроксиС1-С6-алкилом, галогенС1-С6-алкилом, С1-С6-алкокси, галогенС1-С6-алкокси, С3-С8циклоалкилом, 4-7-членным гетероциклоалкилом, в котором один или два кольцевых атома выбраны из N и О, и который необязательно замещен оксо, С1-С6-алкилом, галогеном, 7-8-членным бициклическим гетероциклоалкилом, в котором один или два кольцевых атома выбраны из N и О; и фенилом, который необязательно замещен галогеном, галогенС1-С6-алкокси, С1-С6-алкилом, фенокси, 5-6-членным гетероарилом, в котором один-четыре кольцевых атома выбраны из N и О, С1-С6-алкоксикарбонилС1-С6-алкилом;
4) 5-6-членного гетероциклоалкила в котором два кольцевых атома представляют собой N, O или S, необязательно замещённого окси,
5) 5-6-членный гетероарил, в котором 1-2 кольцевых атома выбраны из S и N, необязательно замещенный С1-С6-алкилом, 4-6-членным гетероциклоалкилом, в котором 1-2 кольцевых атома выбраны из N и O, фенилом, который необязательно замещен галогеном, С1-С6-алкилом, галогенС1-С6-алкокси, галогенС1-С6-алкилом; 5-членный гетероарил, в котором 2-4 кольцевых атома выбраны из N и O; 9-10-членного бициклического гетероарила, в котором 1-2 кольцевых атома выбраны из N, O и S, и который может быть частично насыщен; и
6) -NR10R11;
R2 представляет собой водород;
Rc представляет собой водород;
m выбран из 0, 1 и 2;
R3 представляет собой водород;
R4 представляет собой водород;
R5 и R6 представляют собой водород;
R7 представляет собой незамещенный -C1-C8 алкил; и
R10 и R11 каждый независимо выбран из водорода, незамещенного -C1-C8 алкила, или R10 и R11 вместе с атомом азота, к которому они присоединены, образуют 4-6-членное гетероциклическое кольцо, необязательно замещенное C1-C6 алкилом или галогеном.
2. Соединение по п. 1, в котором R7 представляет собой этил.
3. Соединение по п. 1, выбранное из соединений формулы V, или его фармацевтически приемлемая соль:
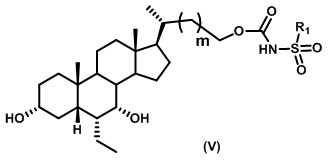
где R1 и m представлены для каждого соединения в Таблице 2:
Таблица 2
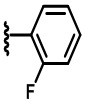
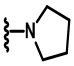
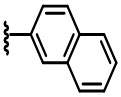
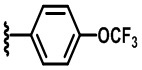
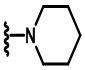
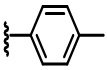
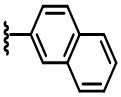
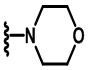
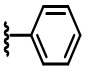
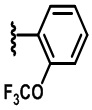
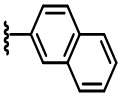
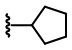
4. Соединение, выбранное из соединений, представленных ниже, или его фармацевтически приемлемая соль:
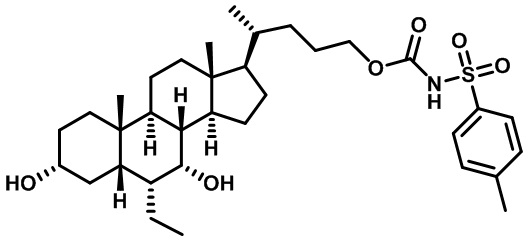
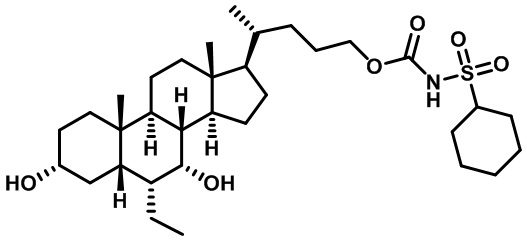
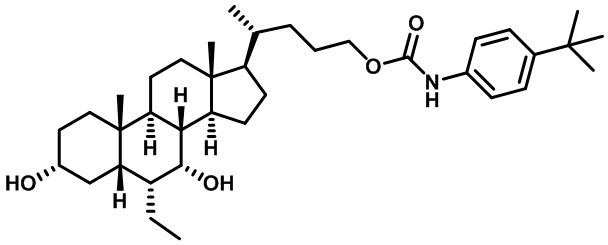
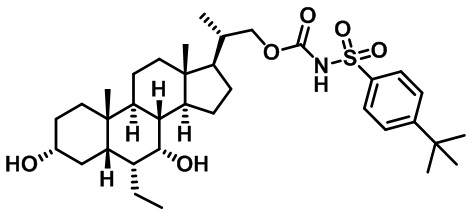
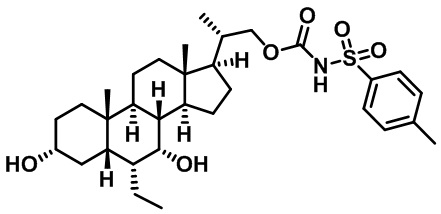
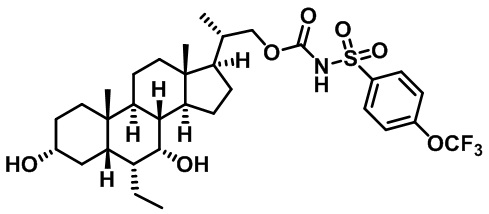
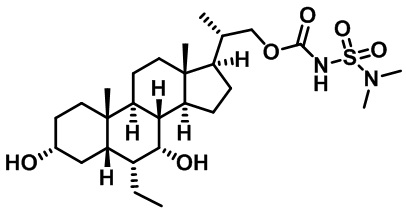
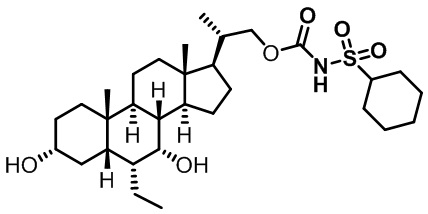
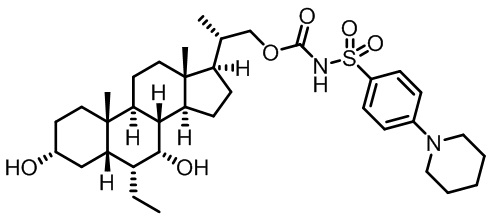
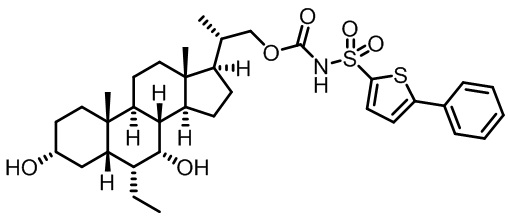
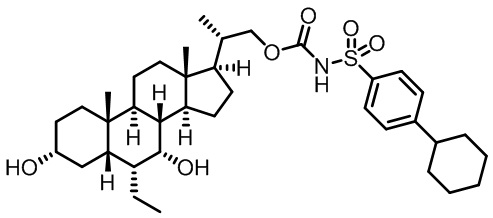
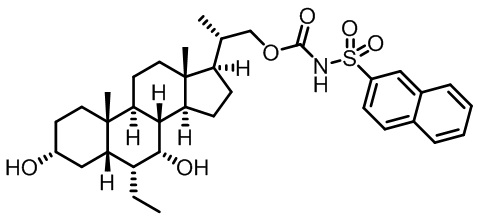
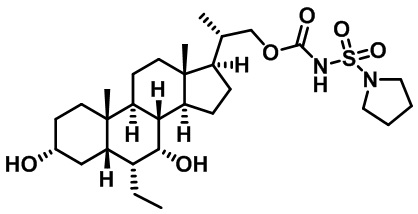
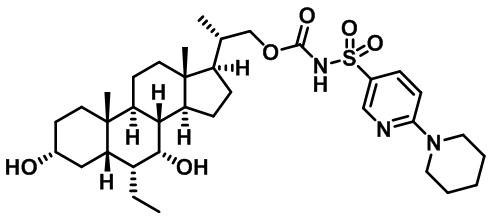
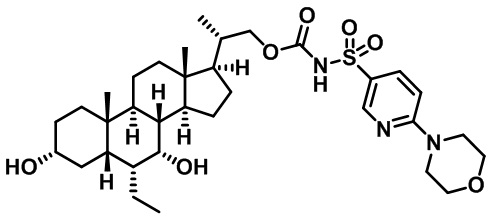
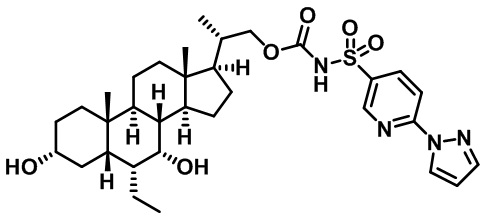
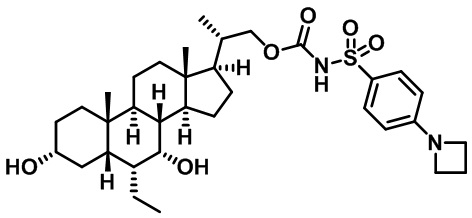
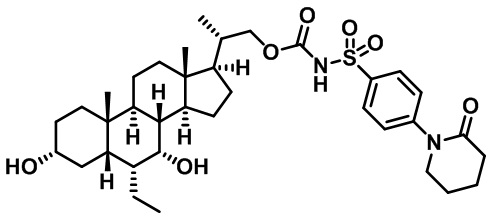
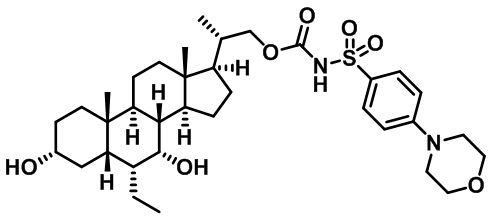
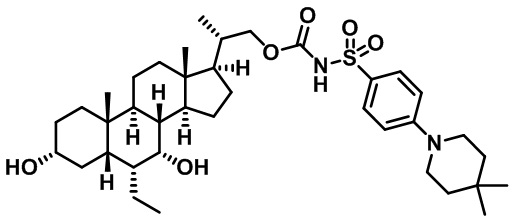
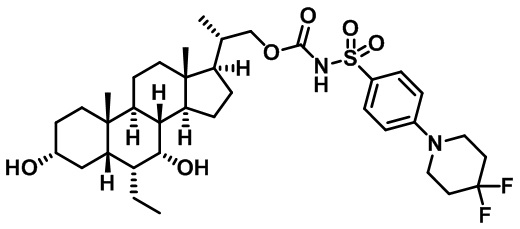
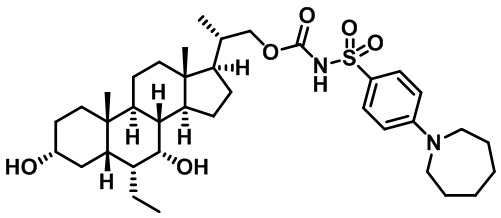
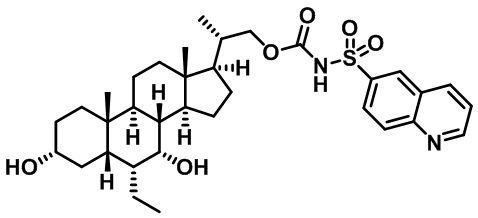
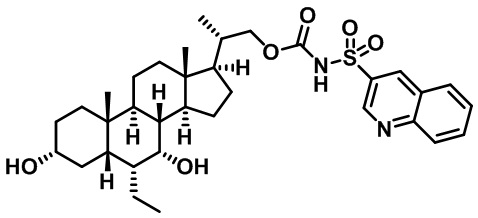
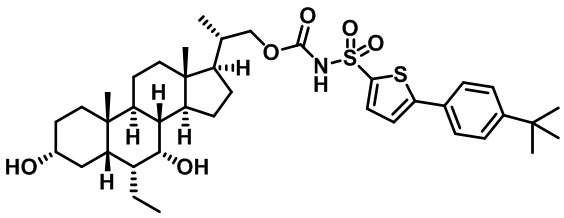
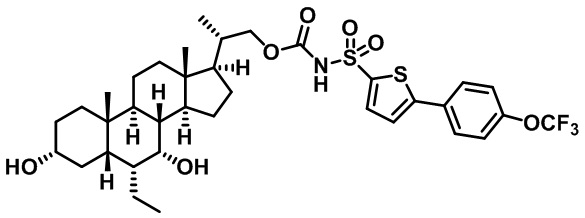
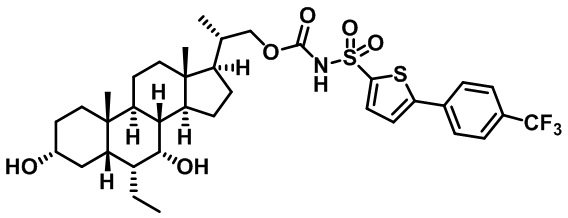
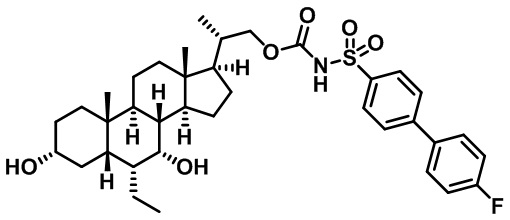
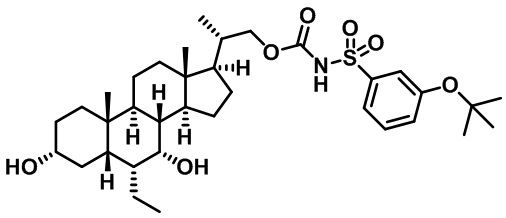
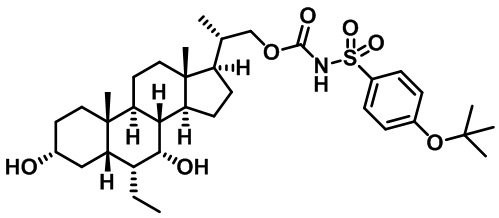
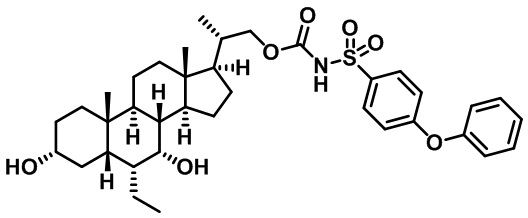
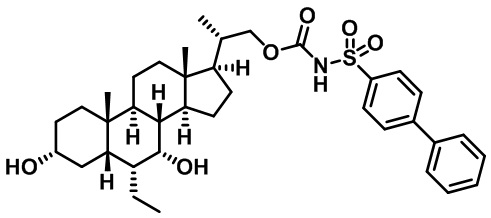
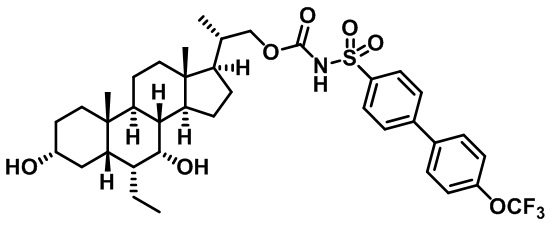
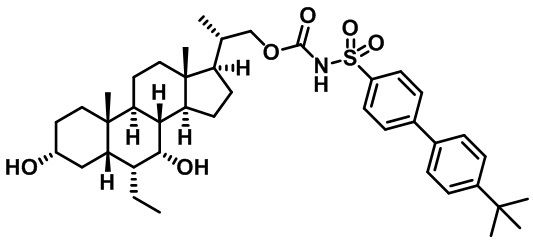
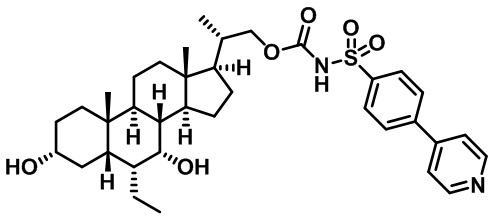
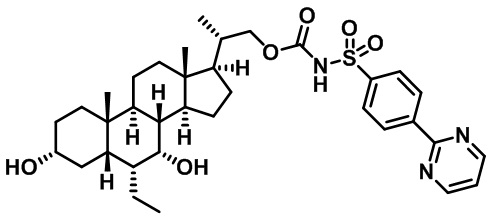
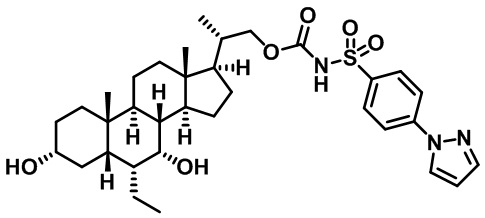
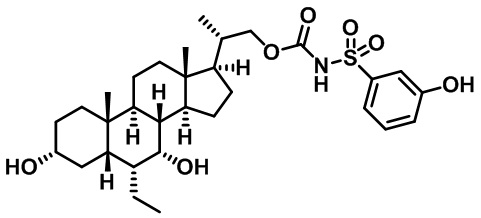
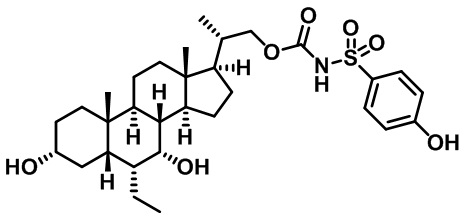
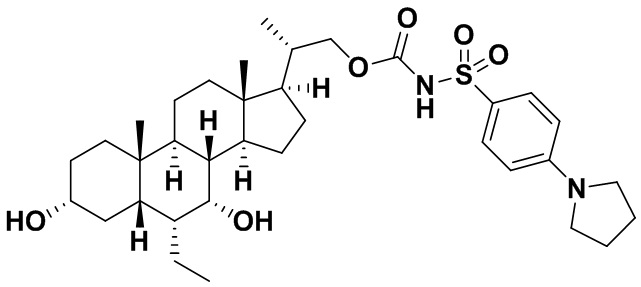
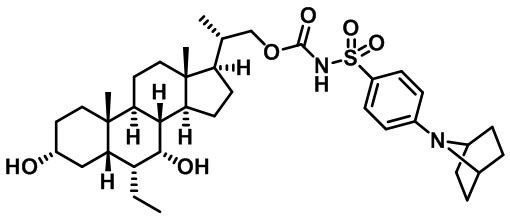
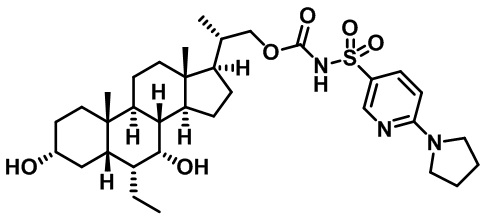
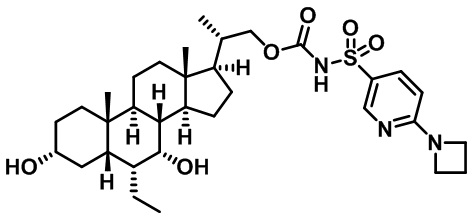
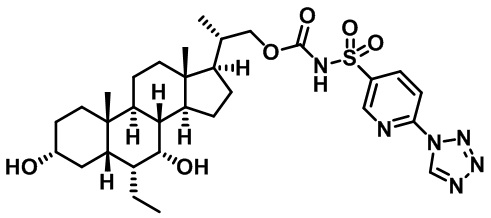
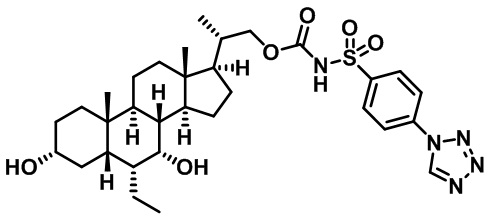
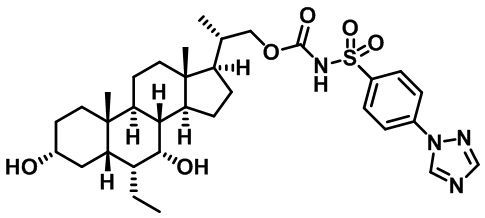
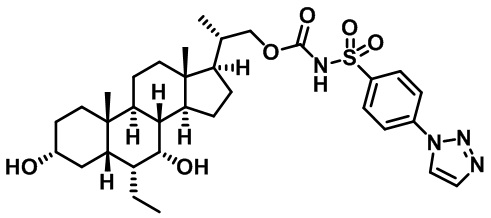
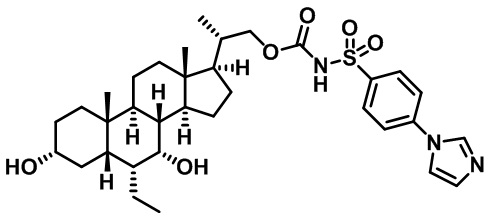
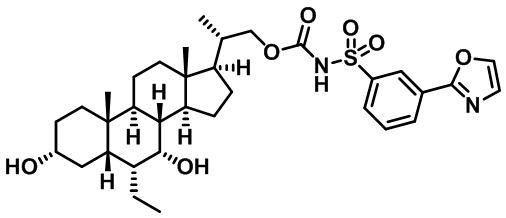
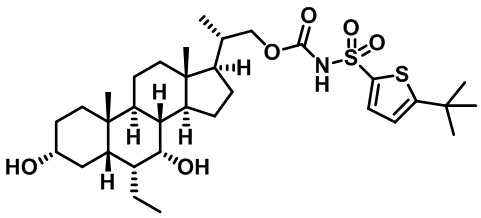
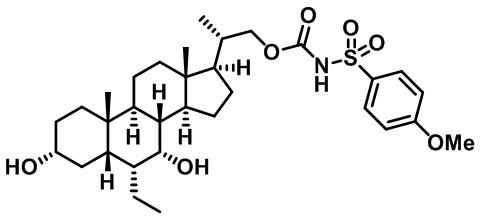
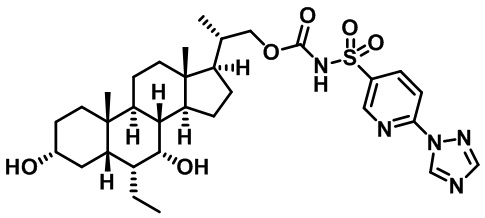
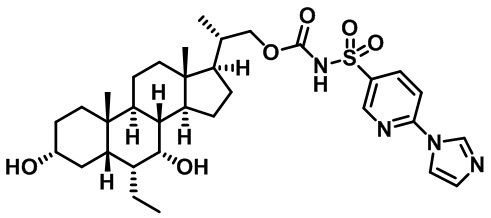
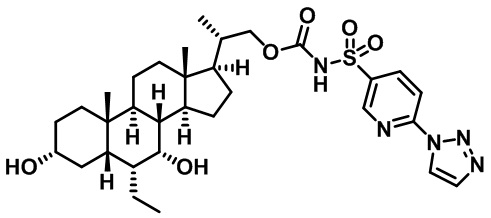
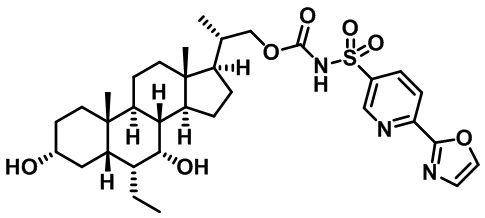
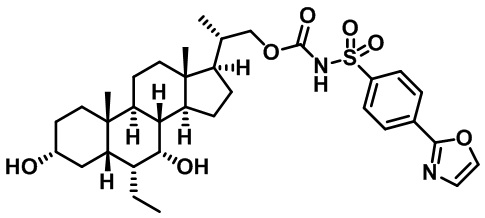
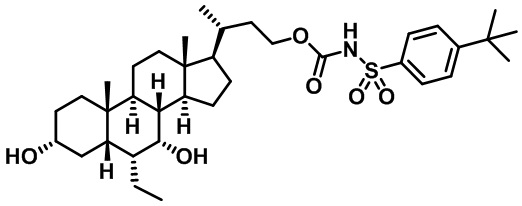
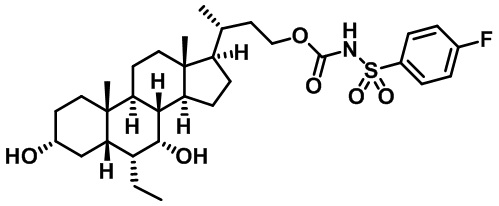
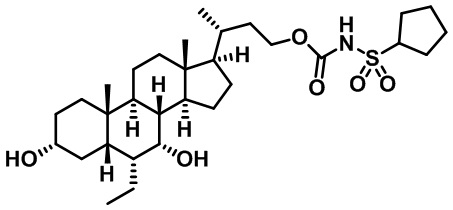
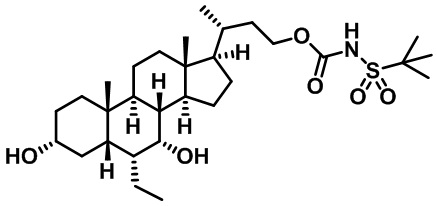
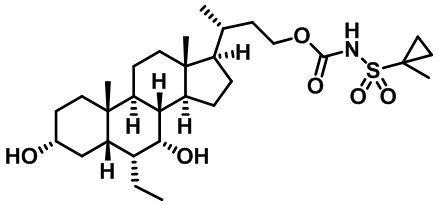
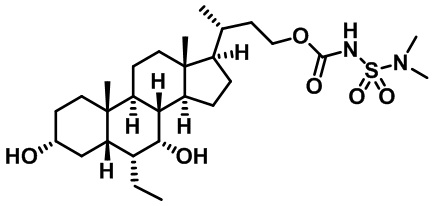
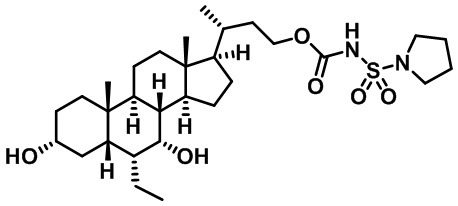
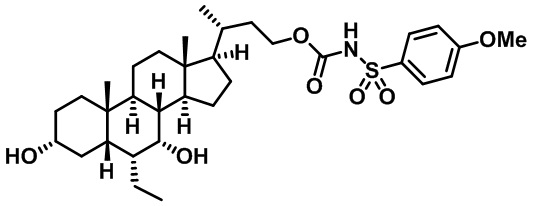
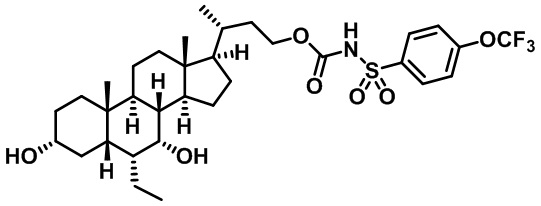
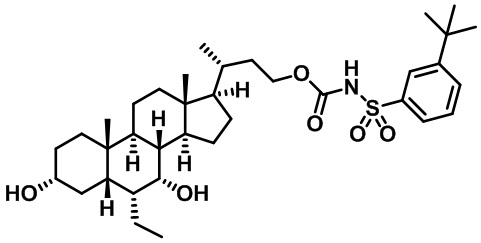
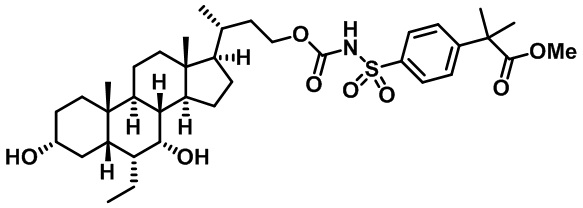
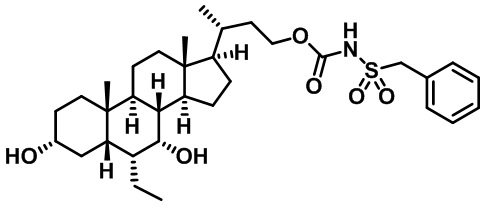
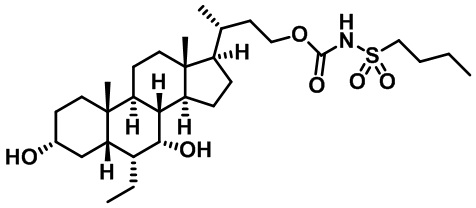
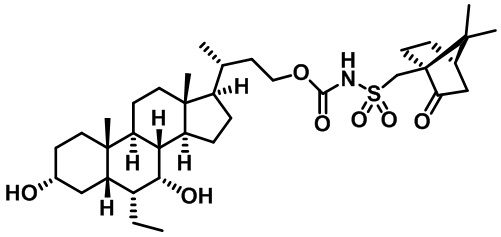
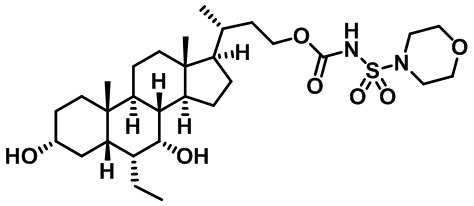
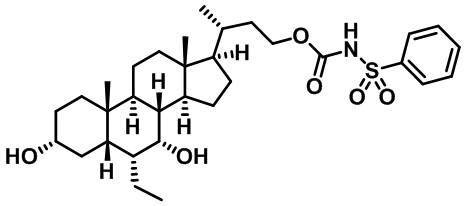
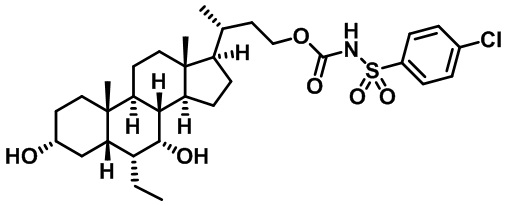
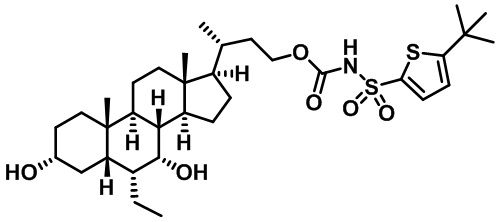
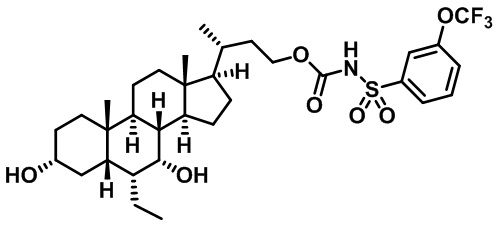
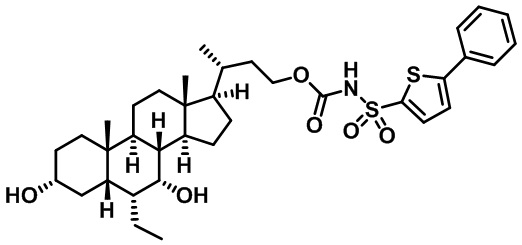
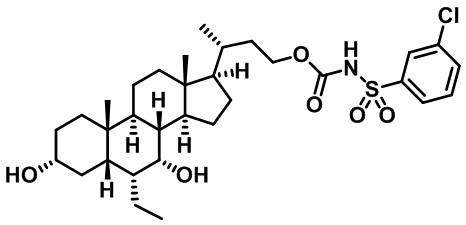
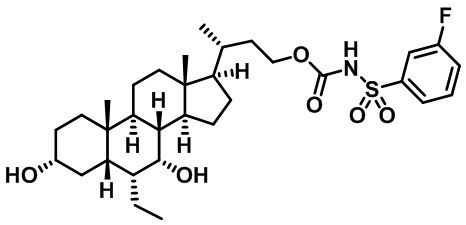
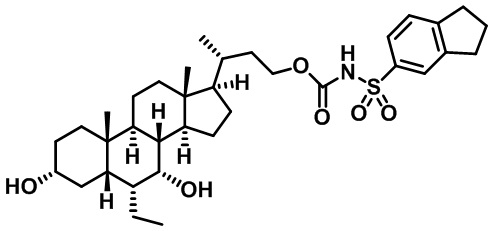
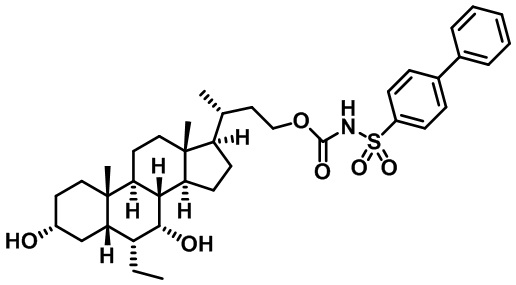
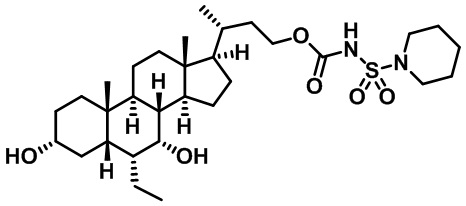
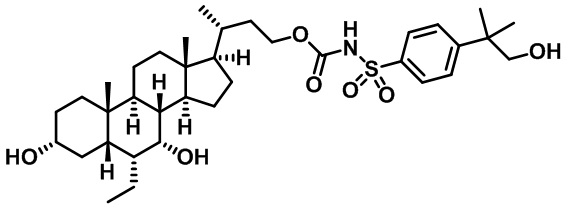
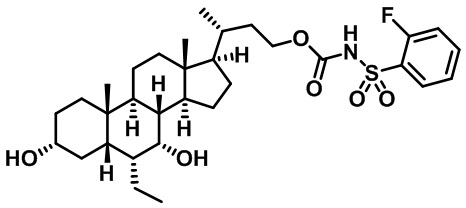
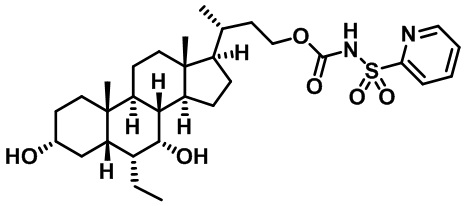
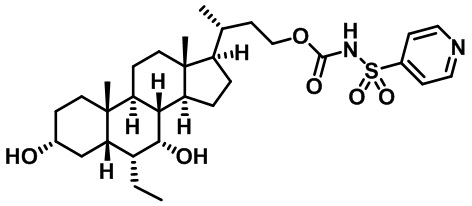
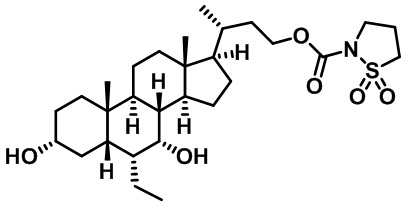
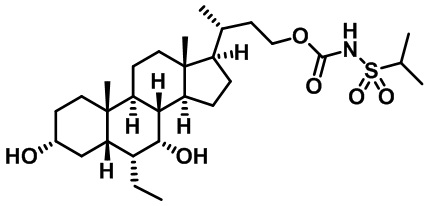
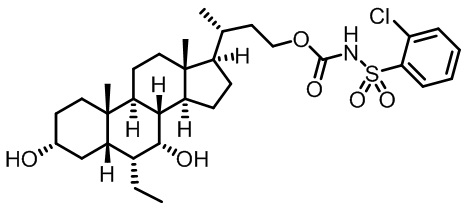
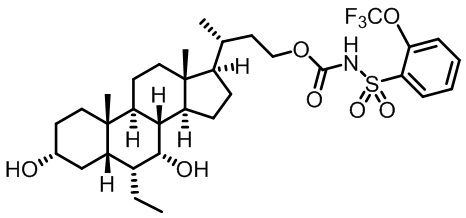
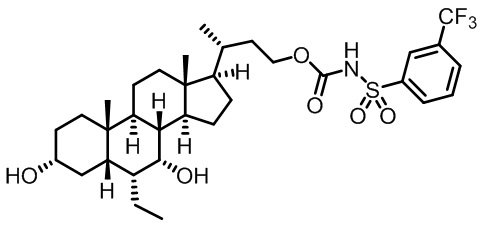
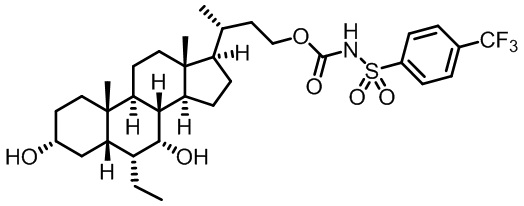
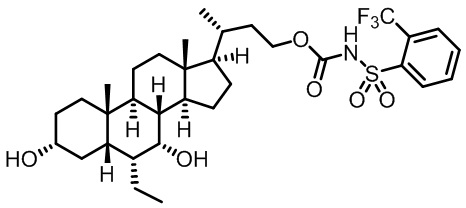
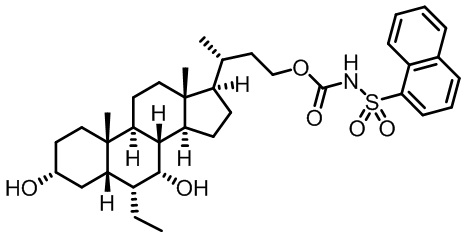
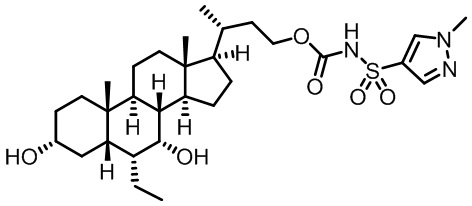
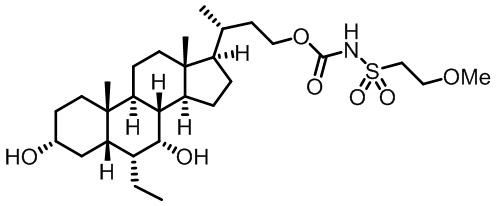
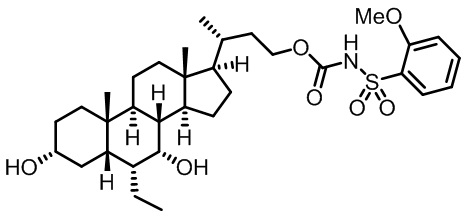
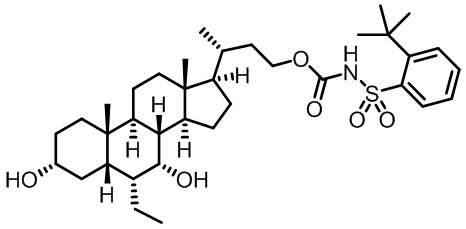
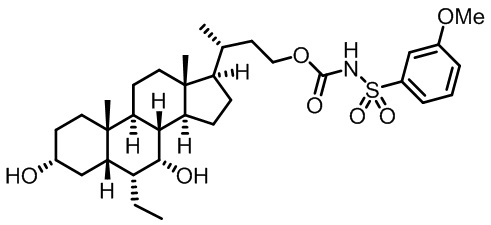
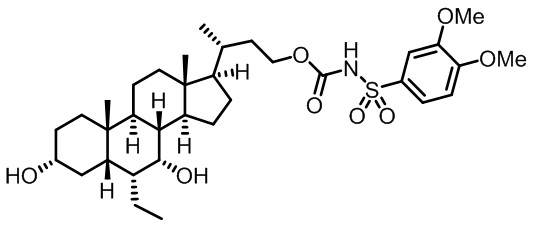
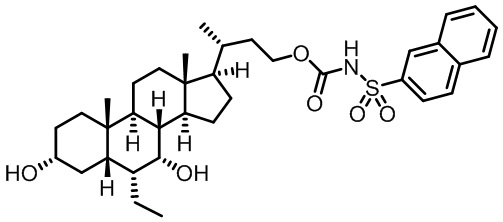
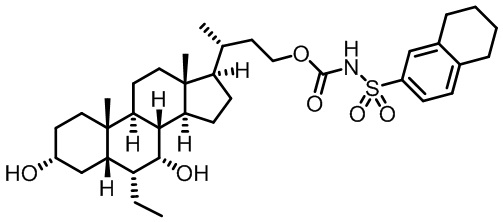
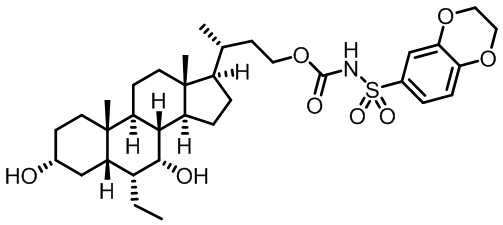
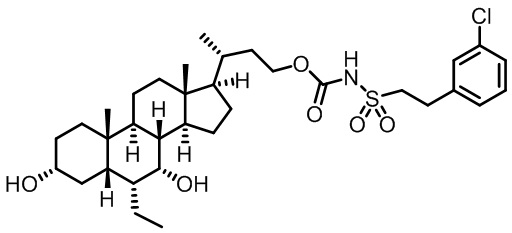
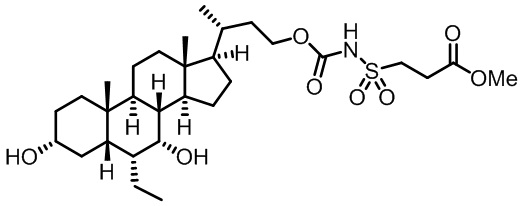
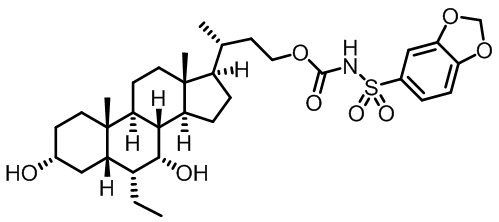
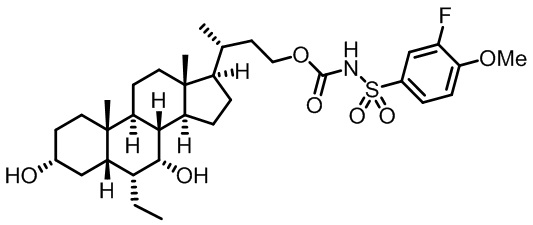
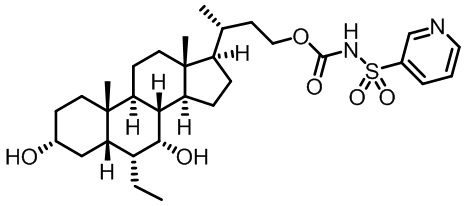
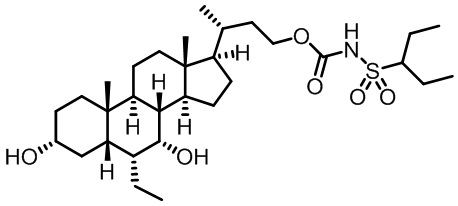
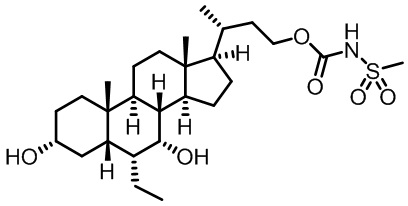
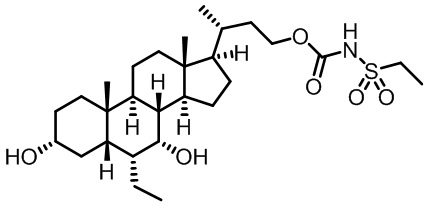
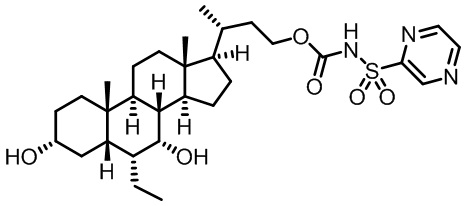
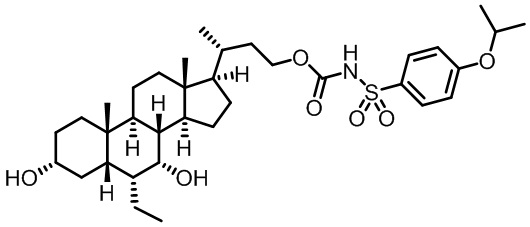
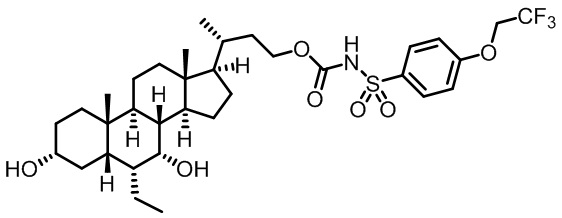
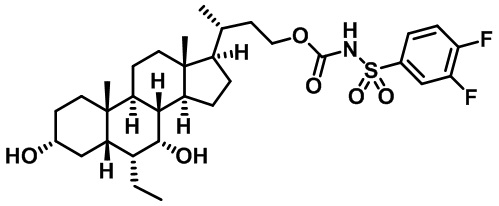
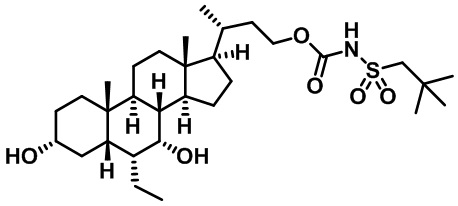
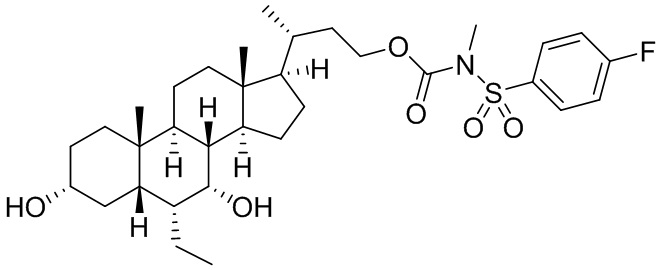
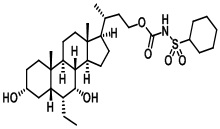
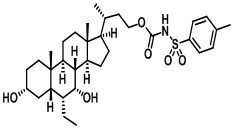
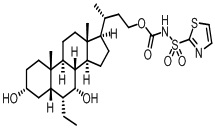
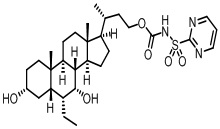
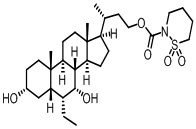
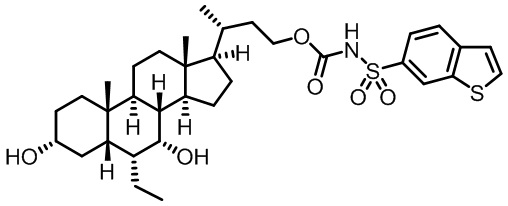
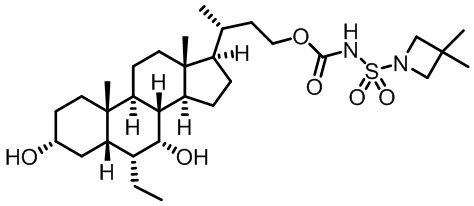
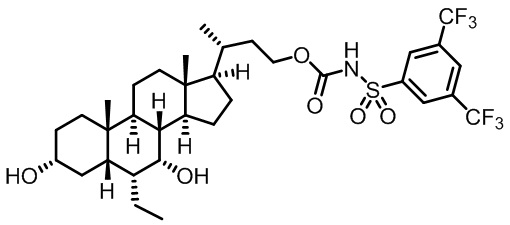
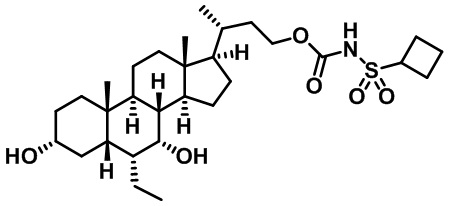
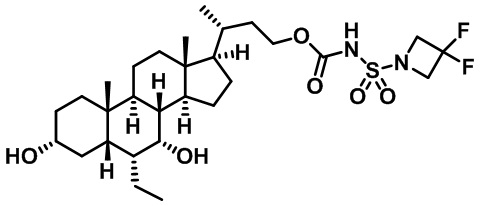
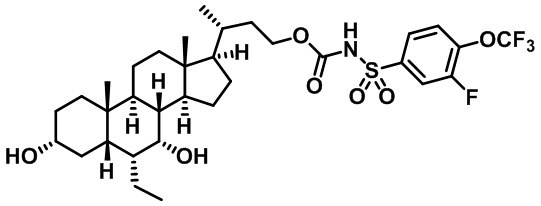
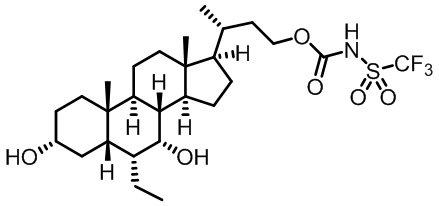
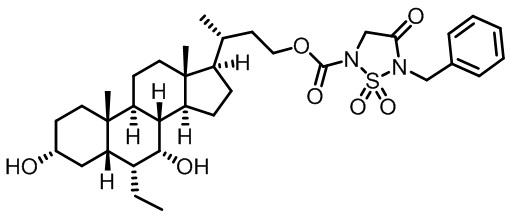
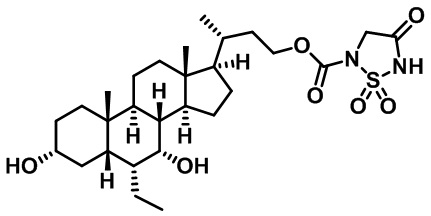
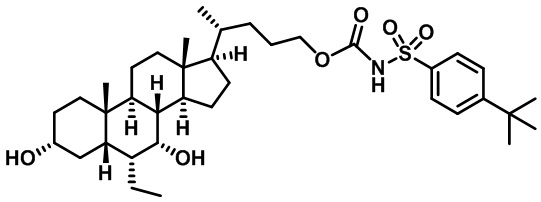
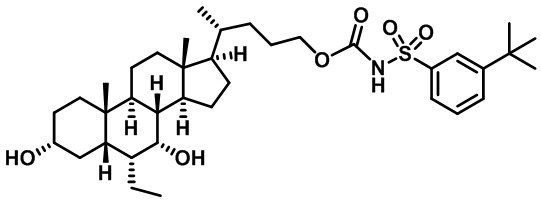
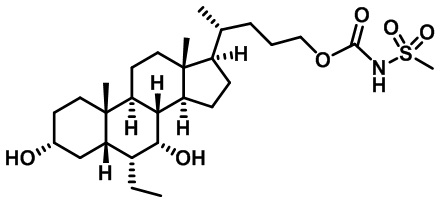
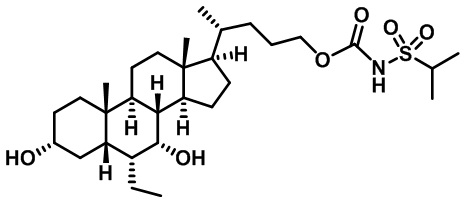
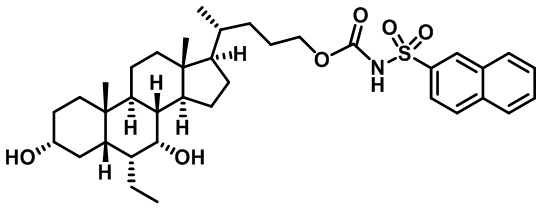
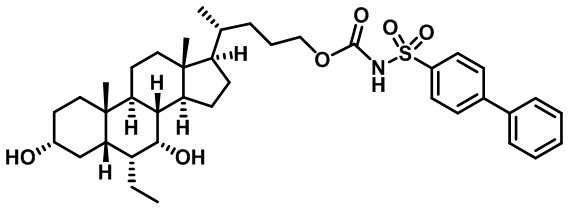
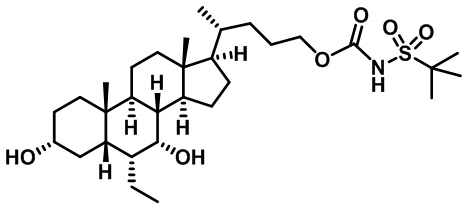
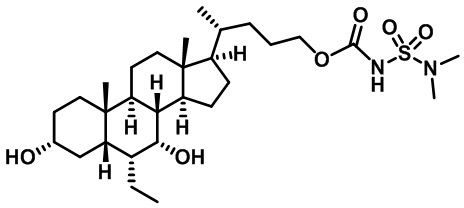
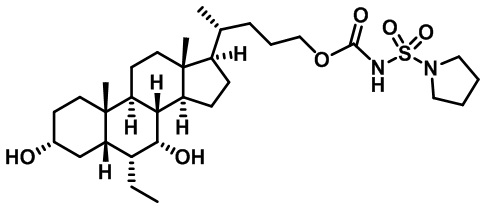
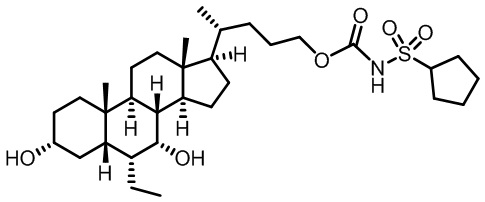
5. Соединение по п. 4, имеющее структуру
,
или его фармацевтически приемлемая соль.
6. Соединение по п. 4, имеющее структуру
,
или его фармацевтически приемлемая соль.
7. Соединение по п. 4, имеющее структуру
,
или его фармацевтически приемлемая соль.
8. Соединение по п. 4, имеющее структуру
,
или его фармацевтически приемлемая соль.
9. Соединение по п. 4, имеющее структуру
,
или его фармацевтически приемлемая соль.
10. Соединение по п. 4, имеющее структуру
,
или его фармацевтически приемлемая соль.
11. Соединение по п. 4, имеющее структуру
,
или его фармацевтически приемлемая соль.
12. Соединение по п. 4, имеющее структуру
,
или его фармацевтически приемлемая соль.
13. Способ профилактики или лечения FXR-опосредованного заболевания или состояния у субъекта, нуждающегося в этом, включающий введение этому субъекту терапевтически эффективного количества соединения по любому из пп. 1-12.
14. Способ по п. 13, где FXR-опосредованное заболевание или состояние выбрано из группы, состоящей из первичного билиарного цирроза, церебросухожильного ксантоматоза, первичного склерозирующего холангита, алкогольной болезни печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, атеросклероза, гиперхолестеринемии, гипертриглицеридемии, диабета типа II и гепатоцеллюлярной карциномы.
15. Способ по п. 13, где FXR-опосредованное заболевание или состояние выбрано из группы, состоящей из первичного билиарного цирроза, неалкогольного стеатогепатита и неалкогольной жировой болезни печени.
16. Способ по любому из пп. 13-15, в котором соединение представляет собой соединение структуры
или его фармацевтически приемлемую соль.
17. Способ по любому из пп. 13-15, в котором соединение представляет собой соединение структуры
или его фармацевтически приемлемую соль.
18. Способ по любому из пп. 13-15, в котором соединение представляет собой соединение структуры
или его фармацевтически приемлемую соль.
19. Способ по любому из пп. 13-15, в котором соединение представляет собой соединение структуры
или его фармацевтически приемлемую соль.
20. Способ по любому из пп. 13-15, в котором соединение представляет собой соединение структуры
или его фармацевтически приемлемую соль.
21. Способ по п. 13 или 14, в котором соединение представляет собой соединение структуры
или его фармацевтически приемлемую соль.
22. Способ по любому из пп. 13-15, в котором соединение представляет собой соединение структуры
или его фармацевтически приемлемую соль.
23. Способ по любому из пп. 13-15, в котором соединение представляет собой соединение структуры
или его фармацевтически приемлемую соль.
24. Фармацевтическая композиция для лечения заболевания или состояния, выбранного из группы, состоящей из первичного билиарного цирроза, церебросухожильного ксантоматоза, первичного склерозирующего холангита, алкогольной болезни печени, неалкогольной жировой болезни печени, неалкогольного стеатогепатита, атеросклероза, гиперхолестеринемии, гипертриглицеридемии, диабета типа II и гепатоцеллюлярной карциномы, содержащая терапевтически эффективное количество соединения по любому из пп. 1-12 и фармацевтически приемлемый носитель.
| WO 0228881 A1, 11.04.2002 | |||
| EA 200970698 А1, 26.02.2010 | |||
| US 2014206657 A1, 24.07.2014 | |||
| WO 2015017813 A2, 05.02.2015 | |||
| R | |||
| Pellicciari et al | |||
| "Bile Acid Derivatives as Ligands of the Farnesoid X Receptor | |||
| Synthesis, Evaluation, and Structure - Activity Relationship of a Series of Body and Side Chain Modified Analogues of Chenodeoxycholic Acid" |

