ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Данное изобретение относится к способу получения слитка сплава, и более конкретно, к способу получения слитка сплава, такого как высокосортная нержавеющая сталь и суперсплав, для которого требуется ультравысокая чистота (ультранизкое содержание примесей). А именно, данное изобретение относится к способу получения слитка сплава ультравысокой чистоты для практических целей, который имеет массу 10 кг или более.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Примесные элементы, такие как углерод (C), азот (N), кислород (O), фосфор (P) и сера (S), известны как оказывающие неблагоприятное воздействие на коррозионную стойкость в сплавах. Как также известно, снижение содержания таких примесных элементов до максимально возможной степени приводит к значительному повышению коррозионной стойкости сплава.
При массовом производстве нержавеющей стали в обычных установках для комбинированной плавки в электропечи с аргонокислородным обезуглероживанием (или установках с обезуглероживанием кислородом в вакууме) общее содержание ([C]+[N]+[O]+[P]+[S]) этих примесных элементов находится при примерно 250 млн-1, даже когда выполняется обработка по удалению примесных элементов посредством рафинирования в ковше.
В противоположность этому, при вакуумной индукционной плавке получают слиток сплава из высокочистого исходного материала сплава, такого как, например, электролитическое железо, электролитический никель или металлический хром, с применением вакуумной индукционной плавильной печи. Поэтому содержание примесных элементов может быть уменьшено до примерно от 10 до 20 млн-1 для [P] и [S], примерно от 20 до 30 млн-1 для [N] и [O] и примерно от 30 до 50 млн-1 для [C]. Однако высокочистые исходные материалы сплава дороги, и, следовательно, вакуумная индукционная плавка не может быть использована для массового производства.
Для вакуумной индукционной плавки обычно используется огнеупорный тигель. Поэтому, как известно, трудно уменьшить содержание примесных элементов, таких как P и N, в расплаве при получении высокохромистой нержавеющей стали. Это является следствием фундаментальных проблем, рассмотренных ниже. Рафинирование с удалением P в расплавленной стали обычно выполняется в виде окислительного рафинирования. При окислительном рафинировании P в расплавленной стали преобразуется в шлакоподобный оксид фосфора (P2O5) и удаляется, будучи абсорбированным в шлак. Однако, когда применяется окислительное рафинирование для получения высокохромистой нержавеющей стали, окисляется не только P в расплавленной стали, но также и хром (Cr), в качестве одного из компонентов сплава. Содержание Cr в стали становится тем самым недостаточным.
В соответствии с этим в 1970-х годах был разработан метод восстановительного рафинирования, раскрытый в Непатентном документе 1, в качестве метода удаления примесных элементов, таких как фосфор (P), при производстве высокохромистой нержавеющей стали. А именно, рафинирование расплава нержавеющей стали (SUS304) в качестве расходуемого электродного материала выполняется в водоохлаждаемом медном тигле, имеющем внутренний диаметр ⌀70 мм, который использован в установке для электрошлакового переплава (ESR), посредством использования CaF2 в качестве плавильного шлака и образования шлаковой ванны вследствие плавления металлического кальция в CaF2. В результате удаляются, например, фосфор (P), олово (Sn), свинец (Pb), мышьяк (As), сурьма (Sb), висмут (Bi), кислород (O), сера (S), селен (Se), теллур (Te), азот (N) и подобное в качестве примесных элементов в нержавеющей расплавленной стали. Непатентный документ 1 является первым сообщением о восстановительном рафинировании с применением металлического кальция, и является сообщением о возможности удаления, в принципе, примесных элементов, таких как фосфор (P) или подобного, присутствующих в Cr-содержащем сплаве, посредством восстановительного рафинирования. Однако процессы ESR, используемые в этом документе, требуют протекания переменного электрического тока через саму шлаковую ванну, так что шлаковая ванна образуется за счет результирующего теплового сопротивления. Поэтому увеличение добавляемого количества металлического кальция в качестве пути улучшения результатов рафинирования приводит к существенному снижению электрического сопротивления самой шлаковой ванны, так что не достигается достаточное количество выделяемого тепла, и образование самой шлаковой ванны затрудняется. Соответственно, вышеуказанный процесс не являлся процессом для практических целей.
С тех пор был разработан метод восстановительного рафинирования с применением индукционной плавильной печи магнитнолевитационного типа (индукционной плавильной печи с холодным тиглем), снабженный водоохлаждаемым медным тиглем, как раскрыто в Патентных документах с 1 по 3 и Непатентном документе 2. В этом методе рафинирования ванна расплава образуется посредством плавления нержавеющей стали индукционным нагревом и посредством добавления рафинирующего агента в форме металлического кальция и фторида кальция (CaF2) к ванне расплава, чтобы удалить, тем самым, примесные элементы, такие как фосфор (P).
А именно, первоначально формируется слой расплавленного фторида кальция, при применении фторида кальция (CaF2) в качестве флюса, и затем металлический кальций плавится в слое расплавленного фторида кальция. Металлический кальций может реагировать с фосфором (P) в ванне расплава, чтобы образовать фосфид кальция (Ca3P2). Фосфид кальция абсорбируется в ванне фторида кальция. Тем самым выполняется дефосфоризация. В этой реакции рафинирования обязательно использование расплавленного флюса, такого как CaF2 или подобного, в котором можно плавить металлический кальций. Соответственно, в качестве реактора должен быть использован водоохлаждаемый медный тигель, который не реагирует с расплавленным CaF2 или Ca. То есть этот метод восстановительного рафинирования не может быть использован при обычной вакуумной индукционной плавке, для которой используются огнеупорные тигли.
В этом методе восстановительного рафинирования очистка с удалением фосфора (P) и подобного выполняется посредством загрузки от 0,8 до 2 кг нержавеющей стали (SUS316L), в качестве исходного материала сплава, в водоохлаждаемый медный тигель, имеющий внутренний диаметр ⌀60 мм или внутренний диаметр ⌀84 мм, и посредством образования небольшой ванны расплава. Поэтому методы рафинирования, раскрытые в Патентных документах с 1 по 3 и Непатентном документе 2, подобно методу рафинирования, раскрытому в Непатентном документе 1, являются контрольно-проверочными испытаниями в небольших ваннах расплава, в которых полученные слитки представляют собой лишь слитки для исследования, имеющие самое большее массу менее 2 кг. Для того, чтобы получить слитки для практических целей массой 10 кг или более, необходимо, поэтому, создать новый метод восстановительного рафинирования для большой индукционной плавильной печи с холодным тиглем.
В качестве метода крупномасштабного индукционного плавления с холодным тиглем авторы изобретения создали метод крупномасштабного индукционного плавления с холодным тиглем с применением водоохлаждаемого медного тигля, имеющего внутренний диаметр ⌀400 мм или более, раскрытый в Патентном документе 4. При разработке этого метода индукционного плавления, однако, было найдено, что поведение расплава и шлака в водоохлаждаемом медном тигле, имеющем внутренний диаметр ⌀200 мм или более, проявляет значительные флуктуации по сравнению с водоохлаждаемым медным тиглем, имеющим внутренний диаметр менее чем ⌀100 мм, и было найдено, что регулируемое рафинирование в большой индукционной плавильной печи с холодным тиглем становится тем более трудным, чем в большей степени повышается чистота расплава (т.е. чем в большей степени снижается содержание примесных элементов). В результате неясно, может ли метод восстановительного рафинирования, как раскрыто в Патентных документах с 1 по 3, быть применим в производстве слитков сплава ультравысокой чистоты в ванне расплава массой 10 кг или более, которая считается ванной расплава для практических целей. Даже при условии, что такой метод восстановительного рафинирования может быть применен, невозможно спрогнозировать, в свете вышеуказанных данных исследований, проведенных авторами изобретения, конкретные условия, которые необходимы для стабильного функционирования в практическом масштабе, на основании Патентных документов с 1 по 3, за исключением случая, в котором условия восстановительного рафинирования, раскрытого в Патентных документах с 1 по 3, могут быть использованы без модификации при восстановительном рафинировании в большой индукционной плавильной печи с холодным тиглем, или случая, в котором метод восстановительного рафинирования, раскрытый в Патентных документах с 1 по 3, может быть оптимизирован до условий функционирования большой индукционной плавильной печи с холодным тиглем. Поэтому, становится необходимым создание отдельного метода рафинирования расплава в практическом масштабе.
Восстановительное рафинирование, как раскрыто в Патентных документах с 1 по 3, основывается на металлическом кальции, так что содержание Ca в слитках сплава нержавеющей стали или подобного после восстановительного рафинирования достигает нескольких сотен млн-1 (ppm). Слитки сплава, подвергнутые такому восстановительному рафинированию, могут обладать ухудшенной коррозионной стойкостью вследствие высокой концентрации Ca. Предпочтительно, поэтому, Ca дополнительно удаляется из расплава после восстановительного рафинирования.
Патентный документ 5 раскрывает способ получения слитка сплава ультравысокой чистоты посредством использования, в качестве первичного слитка, слитка сплава, полученного выполнением восстановительного рафинирования, раскрытого в Патентных документах с 1 по 3, с применением индукционной плавильной печи с холодным тиглем, и последующего удаления кальция, содержащегося в первичном слитке при давлении атмосферы ниже чем 0,5 Па, с применением электроннолучевой плавильной печи. В результате получают слиток сплава ультравысокой чистоты, удовлетворяющий условию [C]+[N]+[O]+[P]+[S]≤100 млн-1 и [Ca]≤10 млн-1.
Даже после выполнения восстановительного рафинирования в соответствии с методом получения по Патентному документу 5, однако, дефосфоризация, обезуглероживание и/или раскисление были недостаточными в некоторых случаях, в которых [C]+[N]+[O]+[P]+[S]>100 млн-1, в зависимости от количества металлического кальция и флюса и в зависимости от условий функционирования. То есть способ получения слитков сплава в практическом масштабе не мог быть предоставлен. Кроме того, электроннолучевая плавка должна быть выполнена в условиях ультраглубокого вакуума, т.е. при давлении атмосферы ниже чем 0,5 Па, и, следовательно, производственные расходы возрастают по мере того, как увеличивается продолжительность производства. Электроннолучевая плавка при давлении атмосферы выше чем 0,5 Па являлась, таким образом, предпочтительной.
Когда недорогие исходные материалы, такие как скрап нержавеющей стали, углеродистая сталь, феррохромный материал и подобное, используются в качестве исходных материалов для плавки (исходного материала сплава) при индукционном плавлении с холодным тиглем (CCIM), тогда углерод (C), кремний (Si), марганец (Mn), алюминий (Al) и подобное примешиваются к расплаву из исходного материала для плавки на стадии плавления. Когда используется скрап сплава ультравысокой чистоты, такого как нержавеющая сталь ультравысокой чистоты, в качестве исходного материала для плавки, напротив, примесные элементы, такие как фосфор (P), сера (S), олово (Sn), свинец (Pb) и подобное, практически не примешиваются к расплаву. Однако кремний (Si), алюминий (Al), титан (Ti), цирконий (Zr), гафний (Hf), бор (B) и подобное примешиваются к расплаву из исходного материала для расплава. Поэтому необходимо выполнять рафинирование с удалением элементов, таких как C, Si, Mn, Al, Ti, Zr и B, поступающих из исходного материала для плавки, в соответствии с целевым составом сплава.
Патентный документ 6 раскрывает способ удаления алюминия как примесного элемента, который плавится в расплаве при индукционном плавлении с холодным тиглем. А именно, первоначально образуется ванна расплава посредством плавления 2 кг высокохромной ферритной жаропрочной стали (Fe-10Cr) в качестве исходного материала для плавки в водоохлаждаемом медном тигле, имеющем внутренний диаметр ⌀84 мм, который предоставлен в плавильной печи для левитационной плавки с холодным тиглем. Затем 10 г оксида железа добавляют к ванне расплава, чтобы окислить Al, который не расплавлен в расплаве, и образуют тем самым оксид алюминия (неметаллическое включение), такой как глинозем или т.п., который не плавится в расплаве. После этого добавляют 75 г фторида кальция (CaF2) в качестве флюса, чтобы обеспечить удаление оксида алюминия посредством абсорбции во флюсе на базе CaF2.
В Патентном документе 6 применение оксида железа в качестве окислителя алюминия эффективно для рафинирования с удалением алюминия, поскольку оксид железа выбран в качестве оксида элемента, обладающего меньшим сродством к кислороду по сравнению с алюминием. Однако, как в примерах Патентного документа 6, практически не могут быть удалены элементы, такие как углерод (C), кремний (Si), бор (B) и подобные, обладающие более высоким сродством к кислороду по сравнению с алюминием. Предположительно, только алюминий удаляется оксидом железа в соответствии с механизмом реакции, который отличается от механизма реакции, представленного в Патентном документе 6. Таким образом, неясно, будет ли элемент, подлежащий удалению, удаляться даже посредством применения окислителя в форме оксида элемента, обладающего меньшим сродством к кислороду по сравнению с удаляемым элементом, и таким же образом неясно, будет ли удален элемент, обладающий более высоким сродством к кислороду по сравнению с элементом, подлежащим удалению, в соответствии с признаками, раскрытыми в Патентном документе 6.
Поэтому неясно, могут ли быть удалены Si, Mn и B до целевой величины, даже посредством применения метода окислительного рафинирования Патентного документа 6, в случае, когда требуется содержание [Si]<0,01 масс.%, [Mn]<0,01 масс.% и [B]<1 млн-1 в нержавеющей стали ультравысокой чистоты, от которой требуется чрезвычайно высокая коррозионная стойкость. Метод окислительного рафинирования в Патентном документе 6 является контрольно-проверочным испытанием в небольшой ванне расплава, которая образована в водоохлаждаемом медном тигле, имеющем внутренний диаметр ⌀84 мм, и неясно, применим ли данный метод к ванне расплава практического масштаба массой 10 кг или более. Даже если данный метод применим к такой ванне расплава, остаются неясными конкретные условия окислительного рафинирования, требующиеся для стабильного функционирования.
Патентный документ 7 раскрывает индукционную плавильную печь с холодным тиглем, в которой используется тигель (тигель на базе галогенида), в котором слой галогенида, содержащий галогенид кальция, такой как фторид кальция, сформирован на внутренней стороне тигля, при индукционном плавлении с холодным тиглем. Хотя повреждение тигля подавляется в этой индукционной плавильной печи с холодным тиглем, все время протекает реакция вследствие контакта между галогенидом, таким как фторид кальция, и расплавом, на части внутренней стенки тигля на базе галогенида. Соответственно, оперативное управление является более трудным, чем в случае применения обычного водоохлаждаемого медного тигля.
В качестве метода получения слитков сплава на базе Ni или нержавеющей стали повышенной чистоты, имеется метод производства слитков электроннолучевой плавкой, раскрытый в Патентном документе 8, который отличается от вышеописанных методов индукционного плавления с холодным тиглем. Однако электроннолучевая плавка обычно используется для плавления металлов с высокой температурой плавления, таких как Ti, Nb и Ta, и, тем не менее, методы рафинирования с удалением примесных элементов, таких как углерод (C) и кислород (O), в нержавеющей стали посредством электроннолучевой плавки остаются неясными. В частности, полностью неясны конкретные условия для стабильного обезуглероживания и раскислительного рафинирования до [C]≤10 млн-1 и [O]≤10 млн-1.
СПИСОК ДОКУМЕНТОВ, ОТНОСЯЩИХСЯ К ПРЕДШЕСТВУЮЩЕМУ УРОВНЮ ТЕХНИКИ
[ПАТЕНТНЫЕ ДОКУМЕНТЫ]
Патентный документ 1: JP H11-246910 A
Патентный документ 2: JP 2002-69589 A
Патентный документ 3: JP 2003-55744 A
Патентный документ 4: JP H11-310833 A
Патентный документ 5: JP 2007-154214 A
Патентный документ 6: JP 2003-342629 A
Патентный документ 7: JP 2007-155141 A
Патентный документ 8: JP 2008-274340 A
[НЕПАТЕНТНЫЕ ДОКУМЕНТЫ]
Непатентный документ 1: Y. Nakamura et al.: Refining of 18%Cr-8%Ni Steel with Ca-CaF2 Solution, Transaction ISIJ, Vol. 16 (1976), p.623.
Непатентный документ 2: Iwasaki, Sakuraya, Fukuzawa: Production of Super-low Phosphorus Stainless Steel by Cold Crucible Levitation Melting: Iron and Steel Vol. 88 (2002), No. 7, p. 413.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение создано, принимая во внимание вышеуказанные обстоятельства, и его целью является предоставление способа получения, в практическом масштабе, слитка сплава, имеющего чрезвычайно низкое содержание по меньшей мере отдельных элементов (фосфора (P), углерода (C), кальция (Ca) или кислорода (O)) из числа примесных элементов.
Одним аспектом изобретения является способ получения слитка сплава, включающий: стадию загрузки исходного материала сплава в холодный тигель в индукционной плавильной печи с холодным тиглем и образования ванны расплава исходного материала сплава индукционным нагревом в атмосфере инертного газа; стадию продолжения индукционного нагрева и добавления первого рафинирующего агента к ванне расплава и затем уменьшения содержания по меньшей мере фосфора из числа примесных элементов, присутствующих в ванне расплава; и стадию формирования слитка сплава посредством затвердевания расплава, содержание фосфора в котором было уменьшено, при этом первый рафинирующий агент представляет собой смесь металлического кальция и флюса, содержащего галогенид кальция; флюс содержит фторид кальция и по меньшей мере один компонент из оксида кальция и хлорида кальция при доле общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция в интервале от 5 до 30 масс.%; и доля массы металлического кальция в первом рафинирующем агенте по отношению к массе ванны расплава перед добавлением первого рафинирующего агента составляет 0,4 масс.% или более.
Другим аспектом изобретения является способ получения слитка сплава, включающий: стадию загрузки исходного материала сплава в холодный тигель в индукционной плавильной печи с холодным тиглем и образования ванны расплава исходного материала сплава индукционным нагревом в атмосфере инертного газа; стадию продолжения индукционного нагрева и добавления второго рафинирующего агента к ванне расплава и после этого продолжения индукционного нагрева в течение 15 минут или более в вакуумированном состоянии, образованном откачиванием инертного газа во внешнюю атмосферу, и затем уменьшения содержания по меньшей мере углерода и кальция из числа примесных элементов, присутствующих в ванне расплава; и стадию формирования слитка сплава посредством затвердевания расплава, содержание углерода и кальция в котором было уменьшено, при этом второй рафинирующий агент представляет собой смесь флюса и первого оксида, содержащего один, два или более видов из числа оксидов основного составного элемента в исходном материале сплава; флюс содержит фторид кальция и по меньшей мере один компонент из оксида кальция и хлорида кальция при доле общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция в интервале от 5 до 30 масс.%; и масса первого оксида во втором рафинирующем агенте находится в интервале от 0,2 до 4 по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить по меньшей мере углерод и кальций из числа примесных элементов, присутствующих в ванне расплава; и доля массы флюса во втором рафинирующем агенте по отношению к массе ванны расплава перед добавлением второго рафинирующего агента находится в интервале от 0,5 до 5 масс.%.
Еще одним аспектом изобретения является способ получения слитка сплава, включающий: стадию подачи электрода из исходного материала в электроннолучевую плавильную печь с холодным подом и облучения электронным пучком электрода из исходного материала при давлении атмосферы ниже чем 5×10-4 мбар и последующего образования ванны расплава на холодном поде в электроннолучевой плавильной печи с холодным подом; стадию добавления третьего рафинирующего агента к ванне расплава и затем уменьшения содержания углерода, в качестве примесного элемента, присутствующего в ванне расплава; и стадию формирования слитка сплава посредством затвердевания расплава, содержание углерода в котором было уменьшено, при этом третий рафинирующий агент представляет собой второй оксид, содержащий один, два или более видов из числа оксидов основного составного элемента в исходном материале для электрода; и масса третьего рафинирующего агента находится в интервале от 1 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава.
Цели, признаки, особенности и преимущества данного изобретения станут более очевидными из представленного ниже подробного описания и чертежей.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 представляет собой схематическое изображение, иллюстрирующее индукционную плавильную печь с холодным тиглем.
Фиг.2 представляет собой графики, иллюстрирующие соотношение между долей добавляемого металлического кальция и степенью дефосфоризации, и соотношение между долей добавляемого металлического кальция и степенью деазотирования.
Фиг.3 представляет собой графики, схематически иллюстрирующие изменение степени дефосфоризации и степени деазотирования со временем выдержки ванны расплава.
Фиг.4 представляет собой графики, включающие график (Фиг.4(a)), иллюстрирующий модель реакции восстановительного рафинирования посредством добавления первого рафинирующего агента, график (Фиг.4(b)), иллюстрирующий соотношение между параметром в этой модели реакции (константой скорости плавления Kmelt(Ca+Flx)) первого рафинирующего агента и временем, необходимым для плавления, и график (Фиг.4(c)), иллюстрирующий соотношение между другим параметром в модели реакции (константой скорости испарения Ca Kev(Ca)) и степенью дефосфоризации.
Фиг.5 представляет собой график, иллюстрирующий соотношение между внутренним диаметром водоохлаждаемого медного тигля и соответствующим временем выдержки после добавления рафинирующего агента.
Фиг.6 представляет собой графики, иллюстрирующие взаимосвязь между степенью обезуглероживания и долей добавляемого оксида железа (WFe3O4/MFeO) и долей добавляемого флюса {Flx}M при рафинировании с добавлением оксида железа/окислительном вакуумном рафинировании.
Фиг.7 представляет собой графики, иллюстрирующие взаимосвязь между степенью обескремнивания и долей добавляемого оксида железа (WFe3O4/MFeO) и долей добавляемого флюса {Flx}M при рафинировании добавлением оксида железа/окислительном вакуумном рафинировании.
Фиг.8 представляет собой схематическое изображение, иллюстрирующее электроннолучевую плавильную печь с холодным подом.
Фиг.9 представляет собой график, иллюстрирующий взаимосвязь между степенью обезуглероживания и долей добавляемого оксида железа (WFe2O3/MFeO) при окислительном рафинировании электроннолучевой плавкой в печи с холодным подом.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
(Первый вариант осуществления)
Для того, чтобы оценить, применимы ли методы восстановительного рафинирования, изложенные в Патентных документах с 1 по 3, также к ванне расплава (также называемой ванной плавления сплава) размера от 10 до 50 кг, авторы изобретения выполнили различные испытания, которые включали образование 20 кг ванны расплава нержавеющей стали (состава SUS310) с применением индукционной плавильной печи с холодным тиглем, снабженной вакуумной камерой и водоохлаждаемым медным тиглем, имеющим внутренний диаметр ⌀220 мм, с последующим добавлением фторида кальция и металлического кальция, чтобы образовать ванну расплава. А именно, исходные материалы сплава в форме коммерчески доступного феррохромного материала, низкоуглеродистой стали, электролитического никеля и подобного загружали в водоохлаждаемый медный тигель. Затем атмосферу в вакуумной камере удаляли и создавали атмосферу инертного газа посредством обеспечения от 600 до 800 гПа (мбар) газообразного Ar для того, чтобы подавить окислительные потери Ca. Образовывали 20 кг ванну расплава нержавеющей стали (состава SUS310). После этого к ванне расплава добавляли рафинирующий агент, образованный смешиванием 400 г порошкового фторида кальция (CaF2) и 100 г металлического кальция в виде частиц. Металлический кальций начинал интенсивно испаряться сразу же после добавления рафинирующего агента, и образовывался материал в виде черного дыма (пыли), так что в течение нескольких секунд свет, излучаемый поверхностью ванны расплава, больше не мог наблюдаться.
При обычных плавках поддерживается состояние ванны расплава наряду с тем, что визуально контролируется расплавленное состояние поверхности ванны расплава. В вышеуказанном испытании, напротив, поверхность ванны расплава невозможно было наблюдать сразу после добавления рафинирующего агента. Поэтому авторы изобретения выполняли операцию затвердевания ванны расплава в водоохлаждаемом медном тигле посредством отключения источника питания для индукционного нагрева через 1 минуту после добавления рафинирующего агента. А именно, операция затвердевания включала отключение вышеуказанного источника питания и выдерживание водоохлаждаемого медного тигля в течение ночи.
Вакуумную камеру открывали на следующий день. Большинство черной пыли было осаждено падением или прилипанием на дно и боковые стенки вакуумной камеры. Блок из затвердевшей нержавеющей стали, так же как и затвердевший шлак (CaF2-Ca), находился в водоохлаждаемом медном тигле. Диаметр вышеуказанного блока был меньше, вследствие усадки при затвердевании, чем в момент времени, когда был отключен источник питания, и, следовательно, он мог быть извлечен из водоохлаждаемого медного тигля. Соответственно, были извлечены блок из затвердевшей нержавеющей стали и шлак CaF2-Ca.
Первоначально визуально обследовали водоохлаждаемый медный тигель на наличие повреждений. Следы эрозии под действием шлака CaF2-Ca отсутствовали, и было установлено, что водоохлаждаемый медный тигель сам по себе является неповрежденным. Весь металлический кальций, добавленный в качестве рафинирующего агента, был расплавлен, однако часть фторида кальция осталась в форме порошка и прилипла к верху затвердевшего шлака (CaF2-Ca). То есть фторид кальция не смог расплавиться полностью.
Затем из извлеченного затвердевшего слитка (блок из отвержденной нержавеющей стали) отбирали образцы для анализа и анализировали их и исследовали. Результаты указывали, что содержание [P], которое составляло примерно 0,018 масс.% в ванне расплава, теперь составляло примерно 0,014 масс.%. Таким образом, было найдено, что некоторая часть, хотя и небольшая, фосфора могла быть удалена рафинированием. А именно, было найдено, что рафинирование с удалением было возможно, в принципе, также в ванне расплава массой 10 кг или более, в практическом масштабе. Однако также было найдено, что отдельное исследование для соответствующих условий рафинирования требовалось для того, чтобы достигнуть достаточного эффекта рафинирования с удалением.
Поэтому авторы изобретения провели тщательное исследование, сосредоточенное на составе рафинирующего агента, добавляемого к ванне расплава, и доле рафинирующего агента по отношению к ванне расплава, с целью удаления в достаточной степени по меньшей мере фосфора (P), из числа примесных элементов, из ванны расплава практического масштаба, такой, что масса конечного слитка составляет 10 кг или более. В результате, авторы изобретения нашли, что возможно получить, в практическом масштабе, слиток сплава, имеющий чрезвычайно низкое содержание по меньшей мере фосфора (P) из числа примесных элементов, посредством способа получения, который включает образование ванны расплава в холодном тигле, который предоставлен в индукционной плавильной печи с холодным тиглем, добавление рафинирующего агента к ванне расплава, чтобы удалить тем самым примесные элементы, и затвердевание расплава, из которого были удалены примесные элементы, чтобы образовать слиток сплава, при условии, что в данном способе:
(1) В качестве рафинирующего агента используется первый рафинирующий агент, который представляет собой смесь металлического кальция и флюса, содержащего галогенид кальция;
(2) В качестве флюса здесь используется флюс на базе галогенида кальция, содержащий фторид кальция и по меньшей мере один компонент из числа оксида кальция и хлорида кальция, при условии, что доля общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция находится в интервале от 5 до 30 масс.%;
(3) Доля массы металлического кальция в первом рафинирующем агенте по отношению к массе ванны расплава перед добавлением первого рафинирующего агента устанавливается равной 0,4 масс.% или более.
Авторы изобретения выполнили первый вариант осуществления данного изобретения на основе результатов вышеуказанного исследования.
Целью первого варианта осуществления в соответствии с данным изобретением является предоставление способа получения, в практическом масштабе, слитка сплава, имеющего чрезвычайно низкое содержание по меньшей мере фосфора (P) из числа примесных элементов.
Первый вариант осуществления данного изобретения будет разъяснен далее при ссылках на сопроводительные чертежи.
Первый вариант осуществления в соответствии с данным изобретением представляет собой способ получения слитка сплава, данный способ включает: стадию загрузки исходного материала сплава в холодный тигель в индукционной плавильной печи с холодным тиглем и образование ванны расплава исходного материала сплава индукционным нагревом в атмосфере инертного газа; стадию продолжения индукционного нагрева и добавления первого рафинирующего агента к ванне расплава, и затем уменьшения содержания по меньшей мере фосфора из числа примесных элементов, присутствующих в ванне расплава; и стадию формирования слитка сплава посредством отверждения расплава, содержание фосфора в котором было уменьшено.
(Индукционная плавильная печь с холодным тиглем)
Фиг.1 представляет собой схематическое изображение, иллюстрирующее индукционную плавильную печь с холодным тиглем. В качестве индукционной плавильной печи с холодным тиглем в способе получения слитков сплава в данном варианте осуществления может быть использована, например, индукционная плавильная печь 1 с холодным тиглем, проиллюстрированная схематически на Фиг.1. Индукционная плавильная печь 1 с холодным тиглем, которая является примером плавильного оборудования, относящегося к индукционной плавильной печи магнитнолевитационного типа с холодным тиглем (CCIM), содержит узел 2 подачи исходного материала, вакуумную камеру 4, холодный тигель 3, размещенный в вакуумной камере 4, и катушку 5, расположенную таким образом, что она окружает внешнюю периферию боковой стороны холодного тигля 3 в вакуумной камере 4.
Узел 2 подачи исходного материала обеспечивает подачу исходных материалов сплава различных видов в холодный тигель 3. Катушка 5 расположена таким образом, что намотана по спирали вокруг боковой стороны холодного тигля 3, оставляя открытыми его верхний и нижний концы, и таким образом, что отделена на небольшое расстояние от поверхности холодного тигля 3. Катушка 5 плавит, посредством индукционного нагрева, исходный материал сплава в области (области индукционного нагрева) холодного тигля 3, вокруг которой намотана катушка 5, чтобы тем самым образовать ванну расплава 6. Затвердевший слой (настыль) 8 образуется, вследствие охлаждения ванны расплава 6, в области без индукционного нагрева, которая расположена ниже области индукционного нагрева холодного тигля 3. Узел 2 подачи исходного материала обеспечивает подачу рафинирующего агента для удаления рафинированием примесных элементов в ванну расплава 6.
Атмосфера в вакуумной камере 4 регулируется. Для того, чтобы предотвратить потери на испарение компонентов сплава из готового сплава, атмосфера может поддерживаться в виде атмосферы инертного газа, образуемой введением газообразного аргона (Ar) или газообразного гелия (He) в вакуумную камеру 4. Для того, чтобы образовать атмосферу инертного газа, предпочтительно вакуумную камеру 4 вакуумируют с применением вакуумного насоса, и после этого инертный газ, такой как газообразный Ar или подобное, вводят в вакуумную камеру 4. Это обусловлено тем, что расплавленный металлический кальций, используемый в качестве рафинирующего агента, чрезвычайно активен, так что присутствие газообразного кислорода или подобного в вакуумной камере 4 вызывает уменьшение количества Ca, вследствие окисления, перед реакцией рафинирования. Утечки из вакуумной камеры 4 предпочтительно уменьшаются до предельно возможной степени при восстановительном рафинировании с применением металлического кальция. Атмосфера в вакуумной камере 4 может поддерживаться в виде разреженной атмосферы посредством непрерывной откачки газа из внутреннего пространства вакуумной камеры 4.
Холодный тигель 3 должен лишь являться таким тиглем, что ванна расплава 6 не реагирует со слоем 7 расплавленного шлака, который образуется вокруг ванны расплава 6. Примеры холодного тигля 3 включают, например, металлические тигли, поверхность которых охлаждается охлаждающим агентом, например, водоохлаждаемые медные тигли. Обычный огнеупорный тигель, однако, не может быть использован вместо холодного тигля 3. Это обусловлено тем, что в данном варианте осуществления необходимо использовать флюс, содержащий галогенид кальция, такой как фторид кальция (CaF2) или подобное, в качестве флюса при рафинировании. Если используется обычный огнеупорный тигель, то такой огнеупорный тигель существенно повреждается расплавленной массой галогенида кальция, например расплавленного фторида кальция, и риск серьезного повреждения возникает вследствие, например, парового взрыва или эрозии водоохлаждаемой медной катушки для нагревания в индукционной плавильной печи.
Для того, чтобы обеспечить возможность какой-либо горячей обработки в производстве изделий в практическом масштабе, слитки сплава должны весить по меньшей мере примерно 10 кг. Поэтому для того, чтобы образовать ванну расплава 6 массой 10 кг или более внутренний диаметр D холодного тигля 3 является предпочтительно диаметром 0,2 м или более. Это обусловлено тем, что масса ванны расплава 6, которая может быть образована, меньше, и может оказаться невозможным стабильное образование ванны расплава массой 10 кг или более, если внутренний диаметр холодного тигля 3 составляет не более чем 0,2 м.
В данном варианте осуществления, первоначально, исходный материал сплава загружается, посредством узла 2 подачи исходного материала, в холодный тигель 3 индукционной плавильной печи 1 с холодным тиглем. В атмосфере инертного газа исходный материал сплава плавится посредством индукционного нагрева катушкой 5, чтобы образовать, тем самым, ванну 6 расплава для регулирования содержания компонентов до заданного состава сплава (стадия образования ванны расплава). Затем первый рафинирующий агент добавляют к ванне 6 расплава при продолжающемся индукционном нагреве катушкой 5 (т.е. в состоянии, в котором поддерживается ванна 6 расплава), чтобы удалить, тем самым, по меньшей мере фосфор из числа примесных элементов в ванне расплава 6 (стадия рафинирования). Расплав после рафинирования (из которого удален фосфор) оставляют затвердевать, чтобы образовать слиток сплава (стадия формирования слитка). Слиток сплава приготавливают, таким образом, в данном варианте осуществления в результате этих стадий.
На стадии формирования слитка в данном варианте осуществления индукционный нагрев может быть прерван, например, после процесса перевода примесей в шлак, и расплав в холодном тигле 3 может быть отвержден, чтобы образовать, тем самым, слиток сплава. В качестве холодного тигля 3 здесь может быть использован холодный тигель с подъемным дном, в котором дно может перемещаться вверх и вниз, так что слиток сплава может быть сформирован последовательным затвердеванием расплава от дна, посредством опускания дна вместе с ванной расплава вниз к области без индукционного нагрева, в то время как ванна расплава поддерживается в области индукционного нагрева холодного тигля; в качестве варианта, расплав после рафинирования может быть введен из холодного тигля 3 в форму для затвердевания в ней, чтобы образовать, тем самым, слиток сплава.
В данном варианте осуществления гранулированные, в виде пластин или в виде дисков металлы, сплавы и подобное могут быть использованы в качестве исходных материалов сплава. Форма, чистота и состав исходных материалов сплава могут быть выбраны в соответствии с целевым составом слитка сплава. В случае, когда, например, должна быть получена высоконикелевая высокохромистая нержавеющая сталь, могут быть использованы феррохромный материал, низкоуглеродистая сталь (конвертерная сталь) и электролитический никель. Для того, чтобы дополнительно увеличить чистоту слитка сплава (дополнительно уменьшить содержание примесных элементов), слиток сплава, полученный в данном варианте осуществления, может быть также использован в качестве исходного материала сплава.
Данный вариант осуществления предоставляет возможность получения слитков сплава разных компонентных составов. Например, может быть получен сплав, имеющий Fe в качестве основного компонента (слиток сплава на базе Fe), сплав, имеющий Ni в качестве основного компонента (слиток сплава на базе Ni), сплав, имеющий Fe и Ni в качестве основных компонентов (слиток сплава на базе Fe-Ni), или сплав, имеющий Co в качестве основного компонента (слиток сплава на базе Co). Способ получения слитков сплава в данном варианте осуществления особенно подходит для получения слитка сплава на базе Fe, слитка сплава на базе Ni и слитка сплава на базе Fe-Ni.
(Восстановительное рафинирование с применением индукционной плавильной печи с холодным тиглем)
В данном варианте осуществления ванна 6 расплава образуется в холодном тигле 3 индукционной плавильной печи 1 с холодным тиглем. После этого выполняется рафинирование посредством удаления примесных элементов, таких как фосфор (P), сера (S), азот (N), олово (Sn), свинец (Pb) и бор (B), с помощью добавления рафинирующего агента, который удовлетворяет условиям с (1) по (3) ниже, для массы M (г) ванны 6 расплава. Затвердевший слой (настыль) 8 образуется ниже ванны 6 расплава. Фиг.1 схематически изображает состояние рафинирования в данном варианте осуществления.
Условия рафинирования в данном варианте осуществления были выяснены в результате нескольких опытов и исследований с применением индукционной плавильной печи 1 с холодным тиглем, имеющей водоохлаждаемый медный тигель (холодный тигель 3), внутренний диаметр которого составлял ⌀220 мм.
(1) В качестве рафинирующего агента используется первый рафинирующий агент, который представляет собой смесь металлического кальция и флюса, содержащего галогенид кальция.
При восстановительном рафинировании с применением металлического кальция (также называемом восстановительным рафинированием посредством Ca) необходимо надежным образом подавать металлический кальций к ванне 6 расплава. Температура кипения чистого металлического кальция составляет 1484°C. Температура ванны расплава, образованной CCIM, составляет примерно 1520°C для сплава на базе Fe, и примерно 1450°C для сплава на базе Ni. При получении сплава на базе Fe, сплава на базе Ni или сплава на базе Fe-Ni добавление металлического кальция как чистого вещества к ванне 6 расплава приводит к испарению большинства металлического кальция, так что в ванне практически не остается металлический кальций для реакции рафинирования. Как известно, металлический кальций плавится в расплавленном галогениде кальция (далее в данном документе, галогениде Ca), таком как расплавленный фторид кальция (CaF2) или подобное. Температура плавления фторида кальция составляет примерно 1410°C, что ниже температуры ванны 6 расплава. Поэтому может быть вызвано образование слоя 7 расплавленного шлака, с помощью передачи тепла от ванны расплава 6, посредством добавления галогенида кальция, такого как фторид кальция, в ванну 6 расплава. Поэтому, металлический кальций может присутствовать в ванне 6 расплава посредством применения, в качестве рафинирующего агента, первого рафинирующего агента, который представляет собой смесь металлического кальция и флюса, содержащего галогенид кальция.
(2) Флюс содержит фторид кальция и по меньшей мере один компонент из числа оксида кальция и хлорида кальция, при условии, что доля общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция (т.е. общая масса оксида кальция плюс хлорида кальция/масса фторида кальция) находится в интервале от 5 до 30 масс.%.
В Патентных документах с 1 по 3 и Непатентных документах 1 и 2 используется лишь один фторид кальция (CaF2) в качестве флюса в рафинирующем агенте. Однако температура образованной ванны расплава ниже при получении сплава на базе Ni или нержавеющей стали, имеющей значительное содержание Ni, посредством CCIM, чем при получении сплава на базе Fe. Поэтому применение одного лишь фторида кальция (CaF2) в качестве флюса часто приводит к завершению реакции рафинирования в состоянии, в котором фторид кальция остается в порошковой форме без плавления. Вследствие этого было трудно поддерживать эффективным образом металлический кальций в ванне флюса (т.е. обеспечивать присутствие металлического кальция, не связанного с другими элементами, в расплавленном флюсе), и достижение эффекта рафинирования посредством удаления примеси будет менее вероятным. В результате множества опытов авторы изобретения нашли, что плавление флюса может быть вызвано простым образом в ванне расплава посредством применения флюса в форме смеси (называемым также композиционным флюсом на базе галогенида кальция или композиционным флюсом на базе галогенида Ca), содержащего фторид кальция (CaF2) в качестве основного компонента и соединение, которое понижает температуру плавления самого флюса. Вышеуказанное соединение является по меньшей мере одним из числа оксида кальция (CaO) и хлорида кальция (CaCl2). Это обусловлено тем, что оксид кальция и хлорид кальция являются соединениями, которые уменьшают температуру плавления флюса, содержащего фторид кальция в качестве основного компонента, при том, что оказывают лишь небольшое влияние на реакцию рафинирования.
Флюс (т.е. флюс на базе галогенида кальция) в первом рафинирующем агенте представляет собой смесь, содержащую фторид кальция в качестве основного компонента и содержащую по меньшей мере один компонент из числа оксида кальция и хлорида кальция, при условии, что доля общей массы “оксида кальция и хлорида кальция” по отношению к массе фторида кальция находится в интервале от 5 до 30 масс.%. Смешивание небольшого количества CaO и/или CaCl2 с CaF2 приводит к снижению температуры плавления флюса и обеспечению простым образом формирования слоя 7 расплавленного шлака посредством передачи тепла от ванны 6 расплава. Если доля массы оксида кальция по отношению к массе фторида кальция превышает 30 масс.%, то флюс не плавится легко, и текучесть шлаковой ванны уменьшается. Это может быть обусловлено остаточным CaO, помимо прочих факторов. Поэтому верхний предел для доли массы оксида кальция по отношению к массе фторида кальция устанавливается равным 30 масс.%. Хлорид кальция оказывает значительное влияние, заключающееся в снижении температуры плавления флюса, и эффективен при получении слитков высоконикелевого сплава, имеющего низкую температуру плавления. Однако имеют место значительные потери на испарение хлорида кальция, и операция рафинирования становилась нестабильной при использовании флюса, в котором доля массы хлорида кальция по отношению к массе фторида кальция превышает 30 масс.%. Соответственно, верхний предел для доли массы хлорида кальция по отношению к массе фторида кальция устанавливается равным 30 масс.%. В таком случае, верхний предел доли общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция аналогичным образом устанавливается равным 30 масс.%.
Если доля общей массы “оксида кальция и хлорида кальция” по отношению к массе фторида кальция была меньше чем 5 масс.%, то температура плавления флюса снижалась лишь незначительно в отдельных случаях, и фторид кальция не был в состоянии расплавиться ко времени окончания реакции рафинирования, в случае, когда флюс содержал фторид кальция и оксид кальция, в случае, когда флюс содержал фторид кальция и хлорид кальция, и также в случае, когда флюс содержал фторид кальция, оксид кальция и хлорид кальция. Поэтому нижний предел доли общей массы “оксида кальция и хлорида кальция” по отношению к массе фторида кальция устанавливается равным 5 масс.%.
Флюс в первом рафинирующем агенте может содержать другие соединения (например, галогенид кальция, иной, чем фторид кальция) в таком количестве, что соединение не оказывает влияния на повышение температуры плавления флюса, и при условии, что соединение оказывает лишь малое влияние на реакцию рафинирования.
Примеры флюса в первом рафинирующем агенте включают, например, CaF2-CaO (от 5 до 30 масс.%), CaF2-CaCl2 (от 5 до 30 масс.%), CaF2-(CaO+CaCl2) (от 5 до 30 масс.%). При этом, CaF2-CaO (от 5 до 30 масс.%) представляет собой флюс, полученный смешиванием от 5 до 30 масс.% оксида кальция с фторидом кальция (доля массы W(CaO) оксида кальция в смеси по отношению к массе W(CaF2) фторида кальция в смеси, а именно W(CaO)/W(CaF2)). Кроме того, CaF2-CaCl2 (от 5 до 30 масс.%) представляет собой флюс, полученный смешиванием от 5 до 30 масс.% хлорида кальция с фторидом кальция (доля массы W(CaCl2) хлорида кальция в смеси по отношению к массе W(CaF2) фторида кальция в смеси, а именно W(CaCl2)/W(CaF2)). Также, CaF2-(CaO+CaCl2) (от 5 до 30 масс.%) представляет собой флюс, полученный смешиванием суммарно от 5 до 30 масс.% оксида кальция и хлорида кальция с фторидом кальция (доля суммы массы W(CaO) оксида кальция и массы W(CaCl2) хлорида кальция в смеси по отношению к массе W(CaF2) фторида кальция в смеси, т.е. (W(CaO)+W(CaCl2))/W(CaF2)).
Для того, чтобы получить сплав на базе Fe-Ni, имеющий низкое содержание Ni, эффективно использование смеси CaF2-CaO (20 масс.%), имеющей сравнительно высокую температуру плавления, (далее в данном документе также называемой “CaF2-20CaO”) в качестве флюса в первом рафинирующем агенте.
(3) Доля массы металлического кальция в первом рафинирующем агенте по отношению к массе ванны расплава перед добавлением первого рафинирующего агента составляет 0,4 масс.% или более.
Авторы изобретения нашли, что степень дефосфоризации при одной операции рафинирования может быть значительно улучшена, когда доля {Ca}M (масс.%) массы WCa (кг) металлического кальция в первом рафинирующем агенте по отношению к массе M (кг) ванны 6 расплава перед добавлением первого рафинирующего агента составляет 0,4 масс.% или более, т.е. когда выполняется условие 0,4≤{Ca}M.
При этом {Ca}M определяется в соответствии с выражением, приведенным ниже.
{Ca}M=WCa/M×100
В этом выражении, WCa обозначает массу (кг) металлического кальция в первом рафинирующем агенте, и M обозначает массу (кг) ванны 6 расплава перед добавлением первого рафинирующего агента.
Описанная ниже доля {Flx}M (масс.%) массы WFlx (кг) флюса в первом рафинирующем агенте по отношению к массе M (кг) ванны 6 расплава перед добавлением первого рафинирующего агента определяется в соответствии с выражением, приведенным ниже.
{Flx}M=WFlx/M×100
В этом выражении, WFlx обозначает массу (кг) флюса в первом рафинирующем агенте, и M обозначает массу (кг) ванны 6 расплава перед добавлением первого рафинирующего агента.
В данном описании, доля массы {Ca}M металлического кальция в первом рафинирующем агенте по отношению к массе ванны расплава перед добавлением первого рафинирующего агента также называется как “доля добавляемого металлического кальция по отношению к ванне расплава”, “доля добавляемого металлического кальция” или “доля добавляемого металлического Ca”. Доля массы {Flx}M флюса в первом рафинирующем агенте по отношению к массе ванны расплава перед добавлением первого рафинирующего агента также называется как “доля добавляемого флюса по отношению к ванне расплава” или “доля добавляемого флюса”.
При обычных операциях количество металлического кальция и количество флюса при рафинировании регулируются на основании концентрации металлического кальция в рафинирующем агенте (сумма общего металлического кальция плюс флюс) и концентрации флюса в рафинирующем агенте. Однако желательное количество металлического кальция и/или количество флюса может быть легко определено непосредственным образом, если регулирование выполняется на основании доли массы WCa (кг) металлического кальция по отношению к массе M (кг) ванны 6 расплава и доли массы WFlx (кг) флюса по отношению к массе M (кг) ванны 6 расплава. Форма отображения величин, использованная в данном описании, поэтому, представляет собой массовую долю металлического кальция по отношению к ванне расплава и массовую долю флюса по отношению к ванне расплава, как в определении {Ca}M и {Flx}M. Масса M ванны 6 расплава была установлена равной массе исходного материала сплава перед загрузкой в водоохлаждаемый медный тигель (холодный тигель 3).
Подробное разъяснение приведено далее в отношении подходящей величины (0,4≤{Ca}M) доли добавляемого металлического кальция.
Металлический кальций начинал испаряться сразу же, вызывая образование черного дыма, при добавлении смеси (т.е. первого рафинирующего агента) металлического кальция и флюса на базе галогенида кальция к ванне 6 расплава. Добавленный металлический кальций был в значительной степени потерян, вследствие потерь на испарение металлического кальция, когда доля добавляемого металлического кальция была небольшой ({Ca}M<0,1). Поэтому, практически не был достигнут эффект рафинирования с удалением. Когда доля добавляемого металлического кальция была большой (0,4≤{Ca}M), напротив, черный дым, образуемый в течение времени от нескольких секунд до нескольких десятков секунд после добавления металлического кальция, поднимался в вакуумной камере 4, так что черный дым блокировал свет, испускаемый поверхностью ванны 6 расплава, и становилось трудно наблюдать состояние поверхности ванны 6 расплава.
На первоначальной стадии исследований в отношении подходящей величины доли добавляемого металлического кальция испытания были проведены при условиях, когда доля добавляемого металлического кальция была небольшой, принимая во внимание то, что состояние поверхности ванны 6 расплава невозможно было визуально наблюдать после добавления первого рафинирующего агента. Например, было выполнено испытание, при котором ванну расплава 20 кг (Fe-20Ni-25Cr) образовывали в водоохлаждаемом медном тигле (холодном тигле 3), имеющем диаметр ⌀220 мм, и 30 г металлического кальция и 270 г CaF2-CaO (25 масс.%) добавляли в качестве первого рафинирующего агента к ванне расплава ({Ca}M=0,15%, {Flx}M=1,35%). При таких условиях, видимость была плохой вследствие пыли из-за испарения Ca, однако поверхность ванны 6 расплава можно было более или менее наблюдать.
При таких условиях, однако, степень дефосфоризации составляла примерно от 15 до 30%. Эффект рафинирования посредством удаления примесей, таких как фосфор (P) и подобного, был совершенно неудовлетворительным. Степень дефосфоризации (на которую в данном документе также делается ссылка как на «степень de-[P]») ηp (%) и степень деазотирования (на которую в данном документе также делается ссылка как на «степень de-[N]») ηN (%) определяются в соответствии с выражениями, приведенными ниже.
ηp=([P]0-[P])/[P]0×100
В этом выражении, [P]0 обозначает концентрацию фосфора (масс.%) в расплаве перед рафинированием, и [P] обозначает концентрацию фосфора (масс.%) в расплаве после рафинирования.
ηN=([N]0-[N])/[N]0×100
В этом выражении, [N]0 обозначает концентрацию азота (масс.%) в расплаве перед рафинированием, и [N] обозначает концентрацию азота (масс.%) в расплаве после рафинирования.
Было найдено, что не происходит повреждения водоохлаждаемого медного тигля (холодного тигля 3) вследствие многочисленных опытов рафинирования при условии небольшой доли добавляемого металлического кальция {Ca}M, даже в случае многочисленных опытов рафинирования, которые включают добавление металлического кальция и флюса на базе галогенида кальция (т.е. добавление первого рафинирующего агента). Было найдено, что рафинирование могло быть продолжено, даже если ванна 6 расплава не наблюдалась визуально непосредственным образом. Соответственно, затем были проведены различные опыты рафинирования при условии большой доли добавляемого металлического кальция {Ca}M и доли добавляемого флюса {Flx}M.
При операции рафинирования при условии большой доли добавляемого металлического кальция (0,4≤{Ca}M) черный дым, образуемый в течение времени от нескольких секунд до нескольких десятков секунд после добавления металлического кальция, поднимался в вакуумной камере 4. Свет, испускаемый от поверхности ванны 6 расплава, блокировался, и становилось невозможным визуальное наблюдение состояния поверхности ванны 6 расплава. Однако было найдено, что добавление металлического кальция в таком количестве, что даже не мог быть виден свет, испускаемый от ванны 6 расплава, приводило к улучшению эффекта рафинирования дефосфоризацией.
Многочисленные опыты рафинирования выполняли при условиях, при которых доля добавляемого металлического кальция {Ca}M и доля добавляемого флюса {Flx}M были модифицированы различным образом, в пределах интервала массы M ванны 6 расплава от 20 кг до 50 кг. Результаты испытаний выявили взаимосвязь между степенью дефосфоризации ηP и долей добавляемого металлического кальция {Ca}M и взаимосвязь между степенью деазотирования ηN и долей добавляемого металлического кальция {Ca}M. Взаимосвязь между долей добавляемого металлического кальция {Ca}M и степенью дефосфоризации (степенью de-[P]) отображена на графике Фиг.2(a). Взаимосвязь между долей добавляемого металлического кальция {Ca}M и степенью деазотирования (степенью de-[N]) отображена на графике Фиг.2(b).
Фиг.2(a) показывает, что степень дефосфоризации ηP увеличивается с увеличением доли добавляемого металлического кальция {Ca}M по отношению к ванне расплава. Более конкретно, при ориентации на верхнюю предельную величину степени дефосфоризации, последняя резко возрастает в интервале {Ca}M от 0 до примерно 0,4 и становится равной примерно 90%, когда {Ca}M составляет примерно 0,4. После этого верхняя предельная величина степени дефосфоризации возрастает умеренно для {Ca}M вплоть до примерно 1,0 и достигает максимальной величины 100% для {Ca}M примерно 1,0 или более. При ориентации на нижний предел степени дефосфоризации, последняя умеренно возрастает в интервале {Ca}M от 0 до примерно 0,5 и становится равной примерно 9% для {Ca}M примерно 0,5. После этого, нижний предел величины степени дефосфоризации возрастает резко до {Ca}M примерно 1,0, до величины, составляющей примерно 41% при {Ca}M примерно 1,0. Нижний предел степени дефосфоризации затем возрастает несколько сильнее для интервала {Ca}M от примерно 1,0 до примерно 1,7 и достигает максимальной величины примерно 71% для {Ca}M примерно 1,7 или более. Такое изменение в степени дефосфоризации (изменение в ηP/{Ca}M) показывает, что реакция дефосфоризации протекает резким образом, когда {Ca}M составляет 0,5 или более. Поэтому, доля добавляемого металлического кальция {Ca}M устанавливается равной 0,4 или более. Предпочтительно, доля добавляемого металлического кальция {Ca}M составляет 0,5 или более, более предпочтительно 1,0 или более. В результате может быть достигнут высокий эффект дефосфоризации в одной операции рафинирования.
Несколько операций рафинирования (восстановительного рафинирования посредством Ca) могут быть повторены в случае, когда сплав ультравысокой чистоты (например [P]<2 млн-1]), имеющий низкое содержание примесного элемента, приготавливается с применением недорогого исходного материала (например, феррохромного материала, имеющего величину [P]0= от 200 до 300 млн-1), имеющего значительное содержание примесного элемента. А именно, слиток сплава (первичный слиток), полученный на первой стадии получения, используется в качестве исходного материала сплава на второй стадии получения, и после этого n-й первичный слиток используется в качестве исходного материала сплава на n-й стадии получения (n представляет собой натуральное число, равное 3 или более).
Фиг.2(b) показывает, что степень деазотирования ηN увеличивается с увеличением доли добавляемого металлического кальция {Ca}M по отношению к ванне расплава. Более конкретно, при ориентации на верхнюю предельную величину степени деазотирования, последняя резко возрастает в интервале {Ca}M от 0 до примерно 0,4 и становится равной примерно 76%, когда {Ca}M составляет примерно 0,4. После этого верхняя предельная величина степени деазотирования возрастает умеренно для {Ca}M вплоть до примерно 1,1 и достигает максимальной величины примерно 92% для {Ca}M примерно 1,1 или более. При ориентации на нижнюю предельную величину степени деазотирования, последняя монотонно возрастает в интервале {Ca}M от 0 до примерно 0,35, достигая величины примерно 30% для {Ca}M примерно 0,35, и после этого резко возрастает до {Ca}M примерно 0,5 и достигает величины примерно 50% для {Ca}M примерно 0,5 или более. Такое изменение в степени деазотирования (изменение в ηN/{Ca}M) показывает, что реакция деазотирования протекает резким образом, когда {Ca}M составляет 0,35 или более. Для целей деазотирования, поэтому, доля добавляемого металлического кальция {Ca}M устанавливается равной 0,35 или более, предпочтительно 0,5 или более и более предпочтительно 1,1 или более.
Степень деазотирования проявляет тенденцию к несколько меньшей величине по сравнению со степенью дефосфоризации. Это может быть объяснено загрязнением азотом, поскольку газообразный азот в воздухе может легко протекать в вакуумную камеру 4 вследствие небольшой негерметичности во время операции рафинирования.
В отношении доли добавляемого металлического кальция, предполагается, что, в принципе, более высокая величина {Ca}M влечет за собой больший эффект в отношении надежного удаления примеси. При фактической операции рафинирования, однако, доля {Ca}M больше чем 1,5, при предоставлении возможности проведения самой операции восстановительного рафинирования посредством Ca, тем не менее, приводит к прилипанию {Ca}M к слитку после операции рафинирования и увеличенному количеству абсорбированного металлического кальция. В результате, становится вероятным возникновение, например, проблема воспламенения пыли испаренного Ca во время последующей обработки, например, при извлечении слитков. На m-й стадии рафинирования расплава, на которой (m-1)-й слиток (m является натуральным числом, равным 2 или более) представляет собой исходный материал сплава, имеют место существенные эксплуатационные препятствия вследствие, например, образования пыли из-за испарения кальция, расплавленного в слитке. В свете вышеуказанных проблем, предпочтительно поддержание {Ca}M≤1,5, с точки зрения выполнения стабильной операции рафинирования.
В данном варианте осуществления предпочтительно выполняется приведенное ниже условие (4), также с точки зрения дополнительного улучшения эффекта дефосфоризации. Авторы изобретения пришли к этому открытию в результате вышеуказанных многократных опытов рафинирования.
(4) Доля массы флюса в первом рафинирующем агенте по отношению к массе ванны расплава перед добавлением первого рафинирующего агента (доля добавляемого флюса {Flx}M по отношению к ванне расплава) равна или больше чем доля массы металлического кальция в первом рафинирующем агенте по отношению к массе ванны расплава (доля добавляемого металлического кальция {Ca}M по отношению к ванне расплава), т.е. соответствует выражению, приведенному ниже.
{Ca}M≤{Flx}M
Очевидной причиной является то, что способность металлического кальция к захватыванию фосфора дополнительно увеличивается, при практически полном отсутствии потерь на испарение добавленного металлического кальция, когда выполняется вышеуказанное условие (4).
Для того, чтобы металлический кальций был стабильно расплавлен во флюсе (т.е. для того, чтобы обеспечить присутствие металлического кальция в расплавленном флюсе без связывания с другими элементами), более предпочтительно, {Flx}M в 1,5 раза или более превышает {Ca}M, т.е. выполняется условие {Ca}M×1,5≤{Flx}M.
Предпочтительно, один лишь флюс добавляется к ванне расплава предварительно, перед добавлением первого рафинирующего агента (смеси металлического кальция и флюса (флюса на базе галогенида кальция)). В результате перед добавлением первого рафинирующего агента образуется слой расплавленного шлака на базе галогенида кальция, что позволяет сдержать потери на испарение добавляемого металлического кальция. Эффективным являлось установление количества предварительно добавляемого флюса (также называемого заранее добавляемым флюсом), которое удовлетворяет условию {Ca}M≤{Flx}M0. При этом, {Flx}M0 обозначает долю (масс.%) массы WFlx0 (кг) заранее добавляемого флюса по отношению к массе M (кг) ванны 6 расплава перед добавлением первого рафинирующего агента. {Flx}M0 также называют долей предварительного добавления или долей добавления заранее добавляемого флюса по отношению к массе M (кг) ванны 6 расплава.
При восстановительном рафинировании посредством Ca (восстановительном рафинировании металлическим кальцием - галогенидом Ca) в данном варианте осуществления слой 7 расплавленного шлака должен быть сформирован в результате передачи тепла от ванны 6 расплава. Слой 7 расплавленного шлака было трудно сформировать, если общее количество {Ca}M, {Flx}M плюс {Flx}M0 было чрезмерно большим. Результаты множества опытов показывают, что, предпочтительно, общее количество {Ca}M, {Flx}M плюс {Flx}M0 находится в пределах 5% массы M ванны 6 расплава.
При этом S легко удалялась посредством восстановительного рафинирования в данном варианте осуществления, в числе примесных элементов, иных, чем P и N. Эффект удаления, в основном равный его величине для P и N, был получен для следовых и случайных элементов, таких как Sn, Pb, Sb и подобное. Степень удаления B при {Ca}M=1,0 составляла примерно 20%, и, следовательно, было возможно рафинирование посредством отделения и удаления B.
Таким образом, высокая степень дефосфоризации может быть обеспечена установлением доли добавляемого {Ca}M металлического кальция по отношению к ванне 6 расплава, составляющей 0,4 масс.% или более, т.е. посредством выполнения условий с (1) по (3) выше. Однако эффект рафинирования с удалением изменяется в зависимости от времени выдержки ванны 6 расплава после добавления рафинирующего агента к ванне 6 расплава. Это является одной из причин, почему верхняя предельная величина и нижняя предельная величина степени дефосфоризации, так же как и верхняя предельная величина и нижняя предельная величина степени деазотирования, принимают разные значения для одной и той же доли добавляемого металлического кальция, как проиллюстрировано на Фиг.2(a) и Фиг.2(b). Рафинирование должно быть выполнено при подходящем времени выдержки в ванне расплава 6 для того, чтобы достигнуть более высокого эффекта рафинирования с удалением.
Соответственно, авторами изобретения выполнены описанные ниже опыты.
Материал нержавеющей стали (Fe-20Ni-25Cr, Fe-35Ni-25Cr) или подобное загружали в водоохлаждаемый медный тигель 3, в индукционную плавильную печь 1 с холодным тиглем, имеющую водоохлаждаемый медный тигель (холодный тигель 3) с внутренним диаметром ⌀220 мм, и формировали ванны 6 расплава массой M (20 кг, 40 кг, 50 кг) посредством индукционного нагрева. После этого добавляли флюсы (80 масс.% CaF2-20 масс.% CaO, 80 масс.% CaF2-10 масс.% CaCl2-10 масс.% CaO или подобное) при условии {Flx}M0=1,5%, чтобы образовать, тем самым, слои 7 предварительно расплавленного шлака перед рафинированием. Затем добавляли первый рафинирующий агент (смесь металлического кальция и флюса) при условиях {Ca}M=1,0% и {Flx}M=1,5%, и операции индукционного нагрева продолжали в течение заданного промежутка времени, чтобы поддерживать ванны 6 расплава и расплавленные слои 7 шлака (от 2 минут до 60 минут). Источник питания для индукционного нагрева отключали сразу же после этого (т.е. прерывали индукционный нагрев), и ванны 6 расплава отверждали быстрым охлаждением в водоохлаждаемом медном тигле 3. После этого анализировали содержание примесных элементов, таких как фосфор (P), азот (N) и подобное в быстро охлажденном и отвержденном слитке.
Фиг.3 иллюстрирует результаты опытов, выполненных, как указано выше. Фиг.3(a) представляет собой график, схематически иллюстрирующий изменение степени дефосфоризации в зависимости от времени выдержки ванны 6 расплава. Фиг.3(b) представляет собой график, схематически иллюстрирующий изменение степени деазотирования в зависимости от времени выдержки ванны 6 расплава.
Условия испытания для результатов испытания, схематически проиллюстрированных на Фиг.3(a), являются следующими. Масса ванны 6 расплава была установлена равной 20 кг. Масса флюса (заранее добавляемого флюса) добавляемого предварительно перед рафинированием была установлена равной 300 г ({Flx}M0=1,5). Масса металлического кальция, добавляемого в качестве первого рафинирующего агента, была установлена равной 200 г ({Ca}M=1,0), и масса флюса была установлена равной 300 г ({Flx}M=1,5).
Условия испытания для результатов испытания, схематически проиллюстрированных на Фиг.3(b), являются следующими. Масса ванны 6 расплава составляла 50 кг. Масса заранее добавляемого флюса была установлена равной 750 г ({Flx}M0=1,5). Масса металлического кальция, добавляемого в качестве первого рафинирующего агента, была установлена равной 500 г ({Ca}M=1,0), и масса флюса была установлена равной 750 г ({Flx}M=1,5).
Как иллюстрируют Фиг.3(a) и 3(b), как степень дефосфоризации, так и степень деазотирования проявляют более низкие величины сразу после добавления первого рафинирующего агента (смеси металлического кальция и флюса). После этого, как степень дефосфоризации, так и степень деазотирования увеличиваются и достигают максимальных величин при времени выдержки от 4 до 7 минут, в случае 20 кг ванны 6 расплава (Фиг.3(a)), и при времени выдержки от 10 до 17 минут в случае 50 кг ванна 6 расплава (Фиг.3(b)). Однако было найдено, что как степень дефосфоризации, так и степень деазотирования имеют тенденцию к уменьшению при продолжающемся далее поддержании состояния расплава (расплавленного состояния) в течение длительных периодов времени. Такая же тенденция наблюдалась для ванны 6 расплава, имеющей массу 40 кг. Степень дефосфоризации и степень деазотирования становились максимальными при времени от 8 до 11 минут в случае 40 кг ванны 6 расплава. При этом 600 г флюса (заранее добавляемого флюса) добавляли предварительно, перед рафинированием, к 40 кг ванны 6 расплава, для выполнения условия {Flx}M0=1,5. После этого добавляли 400 г металлического кальция и 600 г флюса, в качестве условия, в соответствии с которым первый рафинирующий агент удовлетворяет выражениям {Ca}M=1,0 и {Flx}M=1,5.
Результаты опытов показали, что время, проходящее до достижения максимального эффекта рафинирования, имеет тенденцию к уменьшению для случаев, в которых количество добавленного рафинирующего агента было невелико (т.е. случаев, в которых масса ванны 6 расплава была небольшой).
Фиг.3 показывает, что высокая степень дефосфоризации и высокая степень деазотирования достигаются, когда время выдержки ванны 6 расплава после добавления первого рафинирующего агента (смеси металлического кальция и флюса) находится в пределах соответствующего интервала времени. Поэтому метод рафинирования может включать регулирование вышеуказанного времени выдержки расплава, с тем, чтобы оно находилось в пределах соответствующего интервала времени, в соответствии с условиями, такими как внутренний диаметр D водоохлаждаемого медного тигля 3, масса M ванны 6 расплава и добавляемое количество первого рафинирующего агента (масса металлического кальция плюс масса флюса).
На основании результатов вышеуказанных опытов, показывающих, что время выдержки ванны 6 расплава, при котором степень дефосфоризации становится максимальной, изменяется, когда изменяется размер ванны 6 расплава, и количество добавляемого первого рафинирующего агента (количество металлического кальция плюс количество флюса) изменяется соответственно, предполагается, что на стадии рафинирования данного варианта осуществления, реакция дефосфоризации и реакция деазотирования протекают в соответствии с механизмом реакции рафинирования, представленным ниже.
Металлический кальций и флюс в первом рафинирующем агенте, который добавлен к ванне 6 расплава, начинают плавиться вследствие передачи тепла от ванны 6 расплава. Слой 7 расплавленного шлака (слой Ca + флюс) образуется непрерывным образом в результате этого плавления. Представленные ниже реакция дефосфоризации и реакция деазотирования ускоряются в соответствии с увеличением количества металлического кальция, расплавленного в шлаке.
2[P]+3(Ca)→(Ca3P2)
2[N]+3(Ca)→(Ca3N2)
Вместе с этим, Ca в слое 7 расплавленного шлака продолжает испаряться в виде газообразного кальция. Вследствие этих потерь на испарение, как полагают, концентрация Ca в слое 7 расплавленного шлака постепенно снижается после полного растворения Ca в слое 7 расплавленного шлака. Давление пара элементарного металлического кальция очень высокое. Поэтому скорость испарения Ca из слоя 7 расплавленного шлака довольно велика, и снижение концентрации Ca в слое 7 расплавленного шлака также довольно быстрое. Когда концентрация Ca в слое 7 расплавленного шлака уменьшается, Ca, который присутствует в форме соединения кальция (Ca3P2, Ca3N2 и подобного), абсорбированного слоем 7 расплавленного шлака, возвращается к первоначальному металлическому состоянию вследствие разложения соединений кальция. В результате, P и N, образованные при этом разложении, переходят снова в ванну расплава 6. А именно, так называемая реакция рефосфоризации и подобного в соответствии с выражением, приведенным ниже, имеет место в слое 7 расплавленного шлака.
(Ca)→Ca (г)↑
(Ca3P2)→2[P]+3(Ca)
(Ca3N2)→2[N]+3(Ca)
Поэтому, если время выдержки расплава при рафинировании больше необходимого, то P и N, абсорбированные в слой 7 расплавленного шлака, переходят назад в ванну 6 расплава, и не достигается достаточный эффект рафинирования (эффект дефосфоризации, эффект деазотирования). Соответственно, регулирование времени, прошедшего после добавления рафинирующего агента является ключевым при восстановительном рафинировании с применением металлического кальция. Масса добавляемого первого рафинирующего агента (масса металлического кальция и масса флюса в первом рафинирующем агенте) изменяется в зависимости от массы ванны 6 расплава. При регулировании времени выдержки расплава, поэтому, интервал подходящего времени выдержки расплава изменяется в зависимости от массы металлического кальция и массы флюса, которые добавлены. Величина образуемого слоя 7 расплавленного шлака изменяется также в зависимости от внутреннего диаметра водоохлаждаемого медного тигля 3, и, следовательно, последний фактор должен также приниматься во внимание.
Авторы изобретения, как проиллюстрировано на Фиг.4(a), разработали модель состояния при рафинировании расплава, схематически проиллюстрированного на Фиг.1. Фиг.4(a) представляет собой схему, иллюстрирующую модель реакций восстановительного рафинирования, возникающих при добавлении первого рафинирующего агента (смеси металлического кальция и флюса). На Фиг.4(a), цифры от 0 до 6 обозначают временные отрезки, [P]0 до [P]6 обозначают концентрацию фосфора в расплаве на каждом временном отрезке, и [Ca]0 до [Ca]6 обозначают концентрацию кальция в слое 7 расплавленного шлака на каждом временном отрезке. Участки штриховки с наклоном влево и вниз обозначают ванну расплава, а участки штриховки с наклоном вправо и вниз обозначают слой первого рафинирующего агента. Участки горизонтальной штриховки обозначают слой 7 расплавленного шлака.
Авторы изобретения разработали описанную ниже модель реакции. Металлический кальций и флюс в первом рафинирующем агенте, которые добавлены поверх ванны 6 расплава, плавятся последовательно вследствие передачи тепла от ванны 6 расплава, и тем самым образуется слой 7 расплавленного шлака (стадии от временного отрезка 0 до 4 на Фиг.4(a)). В этой модели реакции реакция дефосфоризации и реакция деазотирования протекают, как только образуется слой 7 расплавленного шлака, при концентрации P и концентрации N в ванне 6 расплава, и концентрация Ca3P2 и концентрация Ca3N2 в слое 7 расплавленного шлака, помимо других факторов, определяется локальным равновесием с Ca в слое 7 расплавленного шлака. После формирования цельного слоя 7 расплавленного шлака (стадия от временного этапа 4 на Фиг.4(a)), однако, концентрация Ca в слое 7 расплавленного шлака уменьшается с течением времени вследствие потерь на испарение Ca из слоя 7 расплавленного шлака. Реакции при локальном равновесии являются причиной повторного увеличения концентрации P и концентрации N в ванне 6 расплава, сопровождающего это понижение концентрации Ca.
В этой модели реакции, величины двух параметров (константы скорости плавления Kmelt(Ca+Flx) (кг/с/м2), при которой металлический кальций и флюс, добавленные в качестве первого рафинирующего агента, плавятся вследствие передачи тепла от ванны расплава 6 в расчете на единицу площади поверхности и единицу времени, и константы скорости испарения Ca Kev(Ca) (кг/с/м2), при которой металлический кальций испаряется из слоя 7 расплавленного шлака в расчете на единицу времени и единицу площади поверхности) были определены таким образом, чтобы они соответствовали результатам опытов для соответствующих примеров массы ванны 6 расплава в 20 кг, 40 кг и 50 кг.
Слой 7 расплавленного шлака образовывался в течение примерно от 1 минуты до 2 минут при добавлении 200 г 80 масс.% CaF2-20 масс.% CaO в водоохлаждаемый медный тигель 3 ⌀220 мм. В таком случае, согласно оценке, константа скорости плавления (Kmelt(Ca+Flx)) первого рафинирующего агента находится в интервале от примерно 0,017 до 0,088 (кг/с/м2). Температура плавления шлака снижается, если присутствует металлический кальций, и, следовательно, скорость плавления ожидается несколько выше ее величины для 80 масс.% CaF2-20 масс.% CaO. Рассчитанная константа скорости испарения (Kev(Ca)) пара Ca из слоя 7 расплавленного шлака (например, 25 масс.% Ca-60 масс.% CaF2-15 масс.% CaO или т.п.) составляет примерно от 1 до 2 (кг/с/м2) в соответствии с выражением для определения константы скорости испарения в вакууме, на основании давления пара чистого Ca. В данном варианте осуществления, однако, восстановительное рафинирование посредством Ca выполняется, например, с применением газообразного Ar при давлении примерно 1 атмосфера. Поэтому ожидается, что скорость испарения будет ниже вышеуказанной величины. В связи с этим R. G. Ward (JISI, Vol. 201 (1963), p.11) сообщает, что скорость испарения Mn в ванне расплавленной стали в атмосфере газообразного Ar при давлении 1 атмосфера составляет примерно 1/100 от скорости испарения Mn в ванне расплавленной стали в вакууме. В предположении аналогичного поведения для Ca, величину Kev(Ca) можно считать составляющей примерно от 0,01 до 0,02 (кг/с/м2). Поэтому 750 г флюса на базе галогенида кальция добавляли к ванне 6 расплава массой 50 кг, в качестве условия соответствия выражению {Flx}M0=1,5%, чтобы сформировать слой 7 расплавленного шлака. После этого добавляли 500 г металлического кальция и 750 г флюса, в качестве условия соответствия выражениям {Ca}M=1,0% и {Flx}M=1,5%, и при этом рассчитывали время, необходимое для плавления добавленного рафинирующего агента в случае, когда константа скорости плавления рафинирующего агента была модифицирована в соответствии с моделью реакции, для условий испытания, при которых опыты восстановительного рафинирования посредством Ca выполнились посредством варьирования времени рафинирования. Результаты представлены на Фиг.4(b). В этом опыте, время реакции, за которое была достигнута наибольшая степень дефосфоризации, найдено находящимся в интервале от примерно 10 до 17 минут (от 600 до 1020 секунд). Соответственно, может быть установлено, что первый рафинирующий агент плавится полностью, приводя к образованию слоя 7 расплавленного шлака, в пределах этого промежутка времени (от 600 до 1020 секунд). Поэтому, Kmelt(Ca+Flx) находится в интервале от примерно 0,03 до 0,06 (кг/с/м2). Эта величина по существу совпадает с константой скорости, оцененной на основании скорости плавления флюса.
Затем величина Kev(Ca) была модифицирована, с помощью Kmelt(Ca+Flx)=0,045, чтобы определить получаемую степень дефосфоризации. Результаты представлены на Фиг.4(c). Степень дефосфоризации, полученная при фактическом испытании рафинирования, находится в интервале от примерно 88% до 94%. Поэтому, соответствующая ей величина Kev(Ca) находится в интервале от примерно 0,002 до 0,012 (кг/с/м2). Эта величина по существу совпадает с величиной константы скорости испарения Ca, оцененной на основании давления пара чистого Ca. Таким образом, получена действительная величина.
При определении величин для результатов опытов для ванн расплава 40 кг и 20 кг, Kmelt(Ca+Flx) принималась величиной от 0,04 до 0,07 и от 0,03 до 0,06 (кг/с/м2), соответственно, и Kev(Ca) принималась величиной от 0,002 до 0,012 и от 0,002 до 0,012 (кг/с/м2), соответственно, на основании вышеуказанной модели реакции. Патентный документ 2 и Непатентный документ 2 сообщают результаты опытов при изменении рафинирующего агента в ванне расплава 1,6 кг, которая образована в водоохлаждаемом медном тигле ⌀84 мм, и к которой добавляется Ca+CaF2 в качестве рафинирующего агента. Патентный документ 2 и Непатентный документ 2 указывают, что наибольшая степень дефосфоризации (например, от 85 до 95%) достигается через 1-2 минуты после добавления. При анализе этих данных на основании той же самой модели реакции, Kmelt(Ca+Flx) для испытания в водоохлаждаемом медном тигле ⌀84 мм составляла примерно от 0,04 до 0,09 (кг/с/м2), и Kev(Ca) составляла примерно 0,008 до 0,0027 (кг/с/м2). По сравнению с испытанием в водоохлаждаемом медном тигле 3 ⌀220 мм, скорости приобретали несколько большие величины в обоих случаях, однако являлись в основном идентичными. Было найдено, что вышеуказанная модель реакции может быть использована для восстановительного рафинирования посредством Ca с применением водоохлаждаемого медного тигля 3 практически любого диаметра.
В таком случае, было найдено, что интервал для вышеуказанной Kmelt(Ca+Flx) при полном расплавлении первого рафинирующего агента (смеси металлического кальция и флюса) по отношению к массе ванны 6 расплава от 20 до 50 кг составлял примерно 0,03≤Kmelt(Ca+Flx)≤0,1, и для Kev(Ca) составлял примерно 0,002≤Kev(Ca)≤0,012, принимая во внимание изменчивость данных, наряду с другими факторами.
С применением вышеуказанных величин, становится возможным установление подходящего время выдержки расплава в случае, когда первый рафинирующий агент (смесь металлического кальция и флюса) добавляется после образования ванны 6 расплава в водоохлаждаемом медном тигле 3 произвольного размера. В количественном отношении, более высокая степень дефосфоризации и степень деазотирования могут быть получены, как проиллюстрировано на Фиг.3, установлением времени выдержки T (минуты) ванны 6 расплава, после добавления первого рафинирующего агента (смеси металлического кальция и флюса) к ванне 6 расплава, в интервале от T1 (минуты) до T2 (минуты).
Времена T1 и T2 могут быть установлены указанным ниже образом.
При этом T1 (минуты) составляет 1/2 от времени, требуемого для полного плавления рафинирующего агента после добавления первого рафинирующего агента (смеси металлического кальция и флюса).
T1=WCa+Flx/S/Kmelt(Ca+Flx)/60/2
В этом выражении, WCa+Flx представляет собой общую массу (кг) первого рафинирующего агента, выраженную как M×({Ca}M+{Flx}M)/100 на основании массы M (кг) ванны 6 расплава, доли добавляемого металлического кальция {Ca}M (масс.%) и доли добавляемого флюса {Flx}M (масс.%); и S представляет собой площадь горизонтального поперечного сечения (м2) водоохлаждаемого медного тигля 3, выраженную как πD2/4 на основании внутреннего диаметра D (м) водоохлаждаемого медного тигля 3.
При этом время, которое равно двукратному времени T1, рассчитанному в предположении, что Kmelt(Ca+Flx) (кг/с/м2)=0,1, представляет собой время, требующееся для формирования слоя 7 расплавленного шлака посредством полного плавления добавленного рафинирующего агента. Это время (T1×2), которое представляет собой время, при котором эффект рафинирования наибольший, проявляет, однако, значительную изменчивость на практике. Поэтому, время T1, являющееся 1/2 от этого времени, было установлено в качестве минимального времени выдержки ванны 6 расплава. Соответственно, более высокая степень дефосфоризации и степень деазотирования может быть получена выдерживанием расплава в течение времени T1 или более.
При этом T2 (минуты) представляет собой время, в течение которого 1/2 металлического кальция в добавленном первом рафинирующем агенте удалено из слоя 7 расплавленного шлака вследствие потерь на испарение.
T2=WCa/2/S/Kev(Ca)/60
В этом выражении WCa обозначает массу (кг) металлического кальция в первом рафинирующем агенте, выраженную как WCa=M×{Ca}M/100 на основании массы M (кг) ванны 6 расплава и доли добавляемого металлического кальция {Ca}M (масс.%).
Вышеуказанное время T2, рассчитанное при условии, что Kev(Ca) (кг/с/м2)=0,002, представляет собой время, при котором 1/2 добавленного металлического кальция теряется вследствие испарения. Соответственно, увеличенная степенью дефосфоризации и деазотирования может быть достигнута посредством завершения выдержки расплава в момент времени T2 или ранее.
При фактической операции рафинирования имеет место несущественная потребность в выдержке до тех пор, пока не будет потеряно 1/2 добавленного металлического кальция. В качественном отношении, эффект рафинирования является максимальным в то время (время, примерно равное двукратному T1), когда слой 7 расплавленного шлака образуется посредством плавления общего количества добавленного первого рафинирующего агента (смеси металлического кальция и флюса); соответственно, полагают, что охлаждение и затвердевание в этот момент времени ванны 6 расплава так быстро, насколько это возможно, эффективно в подавлении, например, реакции рефосфоризации, которая сопутствует потерям Ca на испарение. При фактической операции рафинирования, однако, визуальное наблюдение самой поверхности ванны 6 расплава невозможно вследствие пыли, которая возникает от испарения Ca за короткий промежуток времени, от нескольких секунд до нескольких десятков секунд, после добавления первого рафинирующего агента. Поэтому, невозможно проверить, посредством визуального наблюдения, что наступил момент времени, при котором первый рафинирующий агент полностью расплавлен, и управление процессом должно осуществляться на основании одного лишь времени.
Что касается эффекта рафинирования с удалением примеси, то в этом случае градиент изменения со временем степени дефосфоризации и степени деазотирования является крутым на стадии, на которой рафинирующий агент плавится, как проиллюстрировано на Фиг.3. Напротив, изменение со временем степени дефосфоризации и степени деазотирования становится менее крутым на стадии, на которой, как только слой 7 расплавленного шлака сформирован полностью, концентрация Ca в слое 7 расплавленного шлака снижается, поскольку происходит испарение Ca. Поэтому, более надежно устанавливать до некоторой степени продолжительное время выдержки, для того, чтобы надежным образом обеспечить степень дефосфоризации. Соответственно, верхний предел устанавливается как время (T2), при котором половина Ca в шлаке потеряна вследствие испарения.
При этом T1 и T2 задаются выражениями, приведенными ниже, в которых t0 обозначает момент времени, в который первый рафинирующий агент добавляется к ванне расплава, t1 обозначает момент времени, к которому прошла половина времени до плавления всего первого рафинирующего агента от момента времени t0, и t2 обозначает момент времени, к которому прошло время, которое требуется для испарения половины металлического кальция, от момента времени t0.
T1=t1-t0
T2=t2-t0
На основании этих соотношений, T1 и T2, в качестве соответствующих времен выдержки расплава для внутреннего диаметра D (м) водоохлаждаемого медного тигля 3, вычисляли для одного из примеров массы ванны 6 расплава (L/D=0,75: масса цилиндрической ванны 6 расплава, имеющей высоту (L) в водоохлаждаемом медном тигле высотой, эквивалентной 0,75 от внутреннего диаметра водоохлаждаемого медного тигля 3), массы заранее добавляемого флюса ({Flx}M0=1,5) и массы первого рафинирующего агента ({Ca}M=1,0,{Flx}M=1,5). Результаты представлены на Фиг.5.
Фиг.5 показывает, что соответствующее время выдержки изменяется в зависимости от внутреннего диаметра водоохлаждаемого медного тигля 3. Несмотря на изменчивость в данных опытов, более подходящий интервал времени выдержки расплава проходит от времени, рассчитанного для Kmelt(Ca+Flx) (кг/с/м2)=0,06, в качестве T1, до времени, рассчитанного для Kev(Ca) (кг/с/м2)=0,006, в качестве T2, т.е. находится внутри интервала, обозначенного пунктирной линией с одиночными точками и пунктирной линией с двойными точками на Фиг.5.
В таком случае, даже более высокая степень дефосфоризации и степень деазотирования (предпочтительно, например, 50% или более при одной операции рафинирования во время массового производства), может быть получена посредством выдерживания посредством выдерживания ванны расплава в течение времени, которое удовлетворяет вышеуказанному соотношению (T1≤T≤T2), при операции рафинирования, в которой первый рафинирующий агент (смесь металлического кальция и флюса) добавляется к ванне 6 расплава, образованной в водоохлаждаемом медном тигле 3 диаметром ⌀200 мм или более.
Соответственно, содержание примесных элементов, таких как фосфор (P), сера (S), олово (Sn), свинец (Pb) и подобное, может быть уменьшено до 2 млн-1 или менее установлением доли {Ca}M добавляемого металлического кальция по отношению к ванне расплава, как 0,5 масс.% или более, установлением доли {Flx}M добавляемого флюса по отношению к ванне расплава, равной доле {Ca}M добавляемого металлического кальция по отношению к ванне расплава или более, и установлением время выдержки T ванны расплава при T1≤T≤T2.
Например, исходные материалы сплава в виде феррохромного материала и низкоуглеродистой стали (конвертерной стали), имеющих значительное содержание примесных элементов, плюс высокочистый электролитический Ni в качестве исходного материала, загружали в водоохлаждаемый медный тигель 3 ⌀220 мм для применения в индукционной плавильной печи 1 с холодным тиглем 3, имеющей этот водоохлаждаемый медный тигель 3. Затем ванну 6 расплава массой 50 кг из нержавеющей стали (Fe-20Ni-25Cr) образовывали в водоохлаждаемом медном тигле 3 индукционным нагревом. После этого, к ванне 6 расплава добавляли первый рафинирующий агент (смесь металлического кальция и флюса), с применением CaF2-20CaO в качестве флюса, при выполнении следующих условий: {Ca}M=1,0 и {Flx}M=1,5. При этом выполняется операция рафинирования, в которой ванна 6 расплава поддерживается в течение от 11 до 15 минут, посредством непрерывного индукционного нагрева. В этом случае, концентрация примесных элементов в ванне 6 расплава на первоначальной стадии смешивания исходных материалов сплава составляет примерно [P]0=250 млн-1 и [N]0=250 млн-1. На протяжении восстановительного рафинирования посредством Ca реакция дефосфоризации и реакция деазотирования протекали до степени дефосфоризации примерно 90% и степени деазотирования примерно 85%, так что концентрация примесных элементов после окончания рафинирования составляет примерно [P]1=25 млн-1 и [N]1=40 млн-1. Расплав после завершения рафинирования затвердевает с образованием первичного слитка. При применении этого первичного слитка в качестве исходного материала сплава выполняется второе восстановительное рафинирование посредством Ca при тех же самых условиях, что и при первом рафинировании. Концентрация примесных элементов после завершения второго рафинирования составляет примерно [P]2=3 млн-1 и [N]2=6 млн-1. Расплав после завершения второго рафинирования затвердевает с образованием вторичного слитка. При применении этого вторичного слитка в качестве исходного материала сплава выполняется третье восстановительное рафинирование посредством Ca при тех же самых условиях, что и при первом рафинировании. Концентрация примесных элементов после завершения третьего рафинирования может составлять [P]3<2 млн-1, [N]3<2 млн-1. Таким образом, концентрация примесных элементов может достигать величины 2 млн-1 или менее, которая является пределом аналитического обнаружения в современных методах химического анализа. При этом, [P]0 и [N]0 обозначают концентрацию фосфора и концентрацию азота в ванне 6 расплава перед восстановительным рафинированием посредством Ca; [P]1, [P]2, [P]3 обозначают концентрацию фосфора в ванне 6 расплава после завершения процессов первого, второго и третьего восстановительного рафинирования посредством Ca, соответственно; и [N]1, [N]2, [N]3 обозначают концентрацию азота в ванне 6 расплава после завершения процессов первого, второго и третьего восстановительного рафинирования посредством Ca, соответственно.
В индукционной плавильной печи 1 с холодным тиглем, имеющей водоохлаждаемый медный тигель 3 ⌀220 мм, получали слиток сплава восстановительным рафинированием посредством добавления рафинирующего агента, который удовлетворял условиям с (1) по (4), изложенным в первом варианте осуществления, и с применением в качестве исходных материалов сплава, например, электролитического железа, электролитического Ni и металлического Cr, коммерчески доступных высокочистых исходных материалов, которые могут быть сравнительно легко приобретены. Содержание примесных элементов в ванне 6 расплава перед рафинированием (т.е. содержание примесных элементов на первоначальной стадии смешивания исходных материалов сплава) и содержание примесных элементов в слитке сплава измеряли методами химического анализа. Было найдено, что [P]1<1 млн-1, [S]1<1 млн-1 для [P]0=10 млн-1, [S]0=10 млн-1. Было найдено, что содержание следовых и случайных элементов, таких как Sn, Pb и Sb в слитке сплава, посредством анализа масс-спектрометрией с тлеющим разрядом (GD-MS), составляло 1 млн-1 или менее для каждого элемента. Таким образом, было найдено, что содержание примесных элементов может быть уменьшено до 2 млн-1 или менее в результате всего лишь одного восстановительного рафинирования посредством Ca в первом варианте осуществления. При этом, [P]0 и [S]0 обозначают концентрацию фосфора и концентрацию серы в ванне 6 расплава перед восстановительным рафинированием посредством Ca; и [P]1 и [S]1 обозначают концентрацию фосфора и концентрацию серы в ванне 6 расплава после завершения восстановительного рафинирования посредством Ca.
Таким образом, могут быть произведены в практическом масштабе слитки сплава, в которых содержание примесных элементов, таких как фосфор (P) или подобное, составляет 2 млн-1 или менее, посредством выполнения восстановительного рафинирования, в котором к ванне расплава добавляется рафинирующий агент, удовлетворяющий условиям с (1) по (4) данного варианта осуществления.
(Второй вариант осуществления)
Значительное количество металлического кальция остается в слитках сплава, полученных рафинированием с применением металлического кальция, в частности, в слитках сплава, который содержит Ni в качестве компонента сплава. Например, концентрация остаточного кальция в сплаве, имеющем 0 (нулевое) содержание Ni составляет примерно 0,02 масс.%. В противоположность этому, концентрация остаточного кальция составляет примерно 0,05 масс.% в сплаве, имеющем содержание Ni примерно 20 масс.%; концентрация остаточного кальция составляет примерно 0,09 масс.% в сплаве, имеющем содержание Ni примерно 35 масс.%; концентрация остаточного кальция составляет примерно 0,12 масс.% в сплаве, имеющем содержание Ni примерно 45 масс.%; и концентрация остаточного кальция составляет примерно 0,5 масс.% в сплаве, имеющем содержание Ni примерно 60 масс.%. Как известно, коррозионная стойкость ухудшается щелочноземельными металлами и щелочными металлами, такими как Ca, которые присутствуют в слитке сплава. Поэтому в качестве способа получения коррозионностойких материалов требуется способ, в котором получают слиток сплава, имеющий содержание Ca не более чем 0,001 масс.%, предпочтительно не более чем 1 млн-1.
Восстановительное рафинирование, выполненное с применением металлического кальция в индукционных плавильных печах с холодным тиглем, часто влечет за собой увеличение содержания таких элементов, как углерод, алюминий и кремний, в качестве примесей в расплаве, которые восстанавливаются легче, чем Ca. Например, при этом может происходить увеличение концентрации до примерно 30 млн-1 для C и примерно до 50 млн-1 для Al и Si. Считается, что это может быть обусловлено восстановлением, посредством металлического кальция, органических материалов и керамики, которые прилипли к вакуумной камере индукционной плавильной печи, и последующей абсорбцией ванной 6 расплава. Эти примесные элементы также должны быть удалены в соответствии с целевым составом сплава.
В зависимости от вида применения, может также требоваться сплав ультравысокой чистоты, в котором [C]<10 млн-1, [Si]<0,01 масс.% и [Ca]<10 млн-1. В таком случае, становится необходимым удаление примесных элементов, таких как C, Si и подобного, вместе с Ca на стадии рафинирования. Кроме того, бор может существенно ухудшать коррозионную стойкость, и, следовательно, требуются также некоторые сплавы ультравысокой чистоты, в которых [B]<1 млн-1.
В свете вышеуказанного, авторы изобретения выполнили тщательное исследование, сосредоточенное на составе рафинирующего агента, добавляемого к ванне расплава, и доле рафинирующего агента по отношению к ванне расплава, для того, чтобы удалить в достаточной степени по меньшей мере углерод и кальций, из числа примесных элементов, из ванны расплава практического масштаба, такой, что масса конечного слитка составляла 10 кг или более. В результате, авторы изобретения нашли, что возможно получить, в практическом масштабе, слиток сплава, имеющий чрезвычайно низкое содержание по меньшей мере углерода (C) и кальция (Ca), из числа примесных элементов, посредством способа получения, который включает образование ванны расплава в холодном тигле, который предоставлен в индукционной плавильной печи с холодным тиглем, добавление рафинирующего агента к ванне расплава, чтобы удалить тем самым примесные элементы, и затвердевание расплава, из которого были удалены примесные элементы, чтобы образовать слиток сплава, при условии, что в данном способе:
(5) В качестве рафинирующего агента используется второй рафинирующий агент, который представляет собой смесь флюса и первого оксида, содержащего один, два или более видов оксидов основного составного элемента в исходном материале сплава;
(6) Флюс на базе галогенида кальция используется в качестве флюса;
(7) Масса первого оксида во втором рафинирующем агенте устанавливается в интервале от 0,2 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить по меньшей мере углерод и кальций из числа примесных элементов, присутствующих в ванне расплава; и
(8) Доля массы флюса во втором рафинирующем агенте по отношению к массе ванны расплава перед добавлением второго рафинирующего агента устанавливается в интервале от 0,5 до 5 масс.%.
Авторы изобретения выполнили второй вариант осуществления данного изобретения на основе результатов вышеуказанного исследования.
Целью второго варианта осуществления в соответствии с данным изобретением является предоставление способа получения, в практическом масштабе, слитка сплава, имеющего чрезвычайно низкое содержание по меньшей мере углерода (C) и кальция (Ca) из числа примесных элементов.
Второй вариант осуществления данного изобретения будет разъяснен далее при ссылках на сопроводительные чертежи.
Второй вариант осуществления в соответствии с данным изобретением представляет собой способ получения слитка сплава, данный способ включает: стадию загрузки исходного материала сплава в холодный тигель в индукционной плавильной печи с холодным тиглем и образование ванны расплава исходного материала сплава индукционным нагревом в атмосфере инертного газа; стадию продолжения индукционного нагрева и добавления второго рафинирующего агента к ванне расплава, и затем уменьшения содержания по меньшей мере углерода и кальция из числа примесных элементов, присутствующих в ванне расплава; и стадию формирования слитка сплава посредством отверждения расплава, содержание углерода и кальция в котором было уменьшено.
В качестве индукционной плавильной печи с холодным тиглем в способе получения слитков сплава в данном варианте осуществления может быть использована, например, индукционная плавильная печь 1 с холодным тиглем, проиллюстрированная схематически на Фиг.1.
В данном варианте осуществления, первоначально, исходный материал сплава загружается, посредством узла 2 подачи исходного материала, в холодный тигель 3 индукционной плавильной печи 1 с холодным тиглем. Исходный материал сплава плавится посредством индукционного нагрева катушкой 5, чтобы образовать, тем самым, ванну 6 расплава для регулирования содержания компонентов до заданного состава сплава (стадия образования ванны расплава). Затем второй рафинирующий агент добавляют к ванне 6 расплава при продолжающемся индукционном нагреве катушкой 5 (т.е. в состоянии, в котором поддерживается ванна 6 расплава), чтобы удалить, тем самым, по меньшей мере углерод и кальция из числа примесных элементов в ванне 6 расплава (стадия рафинирования). Расплав после рафинирования (удаления углерода и кальция) оставляют затвердевать, чтобы образовать слиток сплава (стадия формирования слитка). Слиток сплава приготавливают, таким образом, в данном варианте осуществления в результате этих стадий.
На стадии формирования слитка данного варианта осуществления, аналогично первому варианту осуществления, индукционный нагрев может быть прерван, например, после процесса перевода примесей в шлак, и расплав в холодном тигле 3 может быть отвержден, чтобы образовать, тем самым, слиток сплава. В качестве холодного тигля 3 здесь может быть использован холодный тигель с подъемным дном, в котором дно может перемещаться вверх и вниз, так что слиток сплава может быть сформирован последовательным затвердеванием расплава от дна, посредством опускания дна вместе с ванной расплава вниз к области без индукционного нагрева, в то время как ванна расплава поддерживается в области индукционного нагрева холодного тигля; или, в качестве варианта, расплав после рафинирования может быть введен из холодного тигля 3 в форму для затвердевания в ней, чтобы образовать, тем самым, слиток сплава.
В данном варианте осуществления гранулированные, в виде пластин или в виде дисков металлы, сплавы и подобное могут быть использованы в качестве исходного материала сплава. Форма, чистота и состав исходного материала сплава могут быть выбраны в соответствии с целевым составом слитка сплава. Например, может быть использован легированный черный металл, такой как коммерчески доступные скрап нержавеющей стали, низкоуглеродистая сталь или феррохром, которые являются недорогими исходными материалами. Это обусловлено тем, что, в отличие от восстановительного рафинирования, в данном варианте осуществления окислительное рафинирование предоставляет возможность удаления C, Si, Mn, Al и Ca, присутствующих в больших количествах, при уровне в интервале от нескольких сотен до нескольких тысяч млн-1, до уровня в несколько млн-1 или менее.
Например, здесь может быть использован скрап сплава ультравысокой чистоты, который, хотя имеет достаточно низкое содержание таких примесных элементов, как P, S, Sn или Pb, содержит, тем не менее, добавленные к нему Al, Ti, Zr или Si, или высокочистый металл, такой как электролитическое железо, электролитический никель, металлический хром или подобное, которые содержат Si, Al или B в качестве примесных элементов. Это обусловлено тем, что окислительное рафинирование в данном варианте осуществления предоставляет возможность уменьшения содержания Ca, Al, Ti, Zr, Si и B.
Для того, чтобы дополнительно увеличить чистоту слитка сплава (дополнительно уменьшить содержание примесных элементов), слиток сплава, полученный в данном варианте осуществления, может быть также использован в качестве исходного материала сплава.
Данный вариант осуществления предоставляет возможность получения слитков сплава разных компонентных составов. Например, в нем может быть получен слиток сплава на базе Fe, слиток сплава на базе Ni, слиток сплава на базе Fe-Ni или слиток сплава на базе Co. Способ получения слитков сплава в данном варианте осуществления особенно подходит для получения слитка сплава на базе Fe, слитка сплава на базе Ni и слитка сплава на базе Fe-Ni.
(Окислительное рафинирование с применением индукционной плавильной печи с холодным тиглем)
В данном варианте осуществления рафинирование посредством удаления активных элементов, присутствующих в расплаве, таких как углерод (C), кремний (Si), алюминий (Al), титан (Ti), цирконий (Zr), гафний (Hf) и бор (B), может быть выполнено посредством загрузки исходных материалов сплава в холодный тигель 3 в индукционной плавильной печи 1 с холодным тиглем, образования ванны 6 расплава в атмосфере инертного газа, и добавления после этого рафинирующего агента, который удовлетворяет условиям с (5) по (8) ниже. Затвердевший слой (настыль) 8 образуется ниже ванны 6 расплава. Фиг.1 схематически изображает состояние рафинирования в данном варианте осуществления. В качестве инертного газа при этом используется газообразный Ar, газообразный He или подобное. Для того, чтобы образовать атмосферу инертного газа в вакуумной камере 4, внутреннее пространство вакуумной камеры 4 вначале вакуумируют с применением вакуумного насоса и затем инертный газ, такой как газообразный Ar, вводят в вакуумную камеру 4.
Условия рафинирования в данном варианте осуществления были выяснены в результате нескольких опытов и исследований с применением индукционной плавильной печи 1 с холодным тиглем, имеющей водоохлаждаемый медный тигель (холодный тигель 3), внутренний диаметр которого составлял ⌀220 мм.
(5) В качестве рафинирующего агента используется второй рафинирующий агент, который представляет собой смесь флюса и первого оксида, содержащего один, два или более видов из оксидов основного составного элемента в исходном материале сплава.
Во время получения слитка сплава с применением индукционной плавильной печи 1 с холодным тиглем должна быть ускорена реакция обезуглероживания в соответствии с выражением, приведенным ниже, для того, чтобы удалить углерод в качестве примесного элемента в расплаве.
[C]+[O]→CO (г)↑
Для того, чтобы ускорить эту реакцию, к ванне 6 расплава должен подаваться кислород и образованный газообразный монооксид углерода (CO (г)) должен удаляться, чтобы уменьшать, тем самым, парциальное давление CO. Способ подачи кислорода к ванне 6 расплава может включать, например, вдувание газообразного кислорода в ванну расплава. Эффективный способ удаления CO (г), однако, включает вакуумирование и непрерывное удаление образованного газообразного монооксида углерода из вакуумной камеры 4. Соответственно, добавление оксида основного составного элемента исходного материала сплава в твердом состоянии является более эффективным способом подачи кислорода в ванну 6 расплава, чем вдувание газообразного кислородсодержащего вещества. В частности, концентрация остаточного кислорода в слитке сплава составляет часто 5 млн-1 или менее, так что в результате ванна расплава находится в состоянии, свободном от источников кислорода, в случае, когда ванна расплава образуется с применением исходного материала сплава в виде слитка сплава, подвергнутого восстановительному рафинированию посредством металлического кальция. Соответственно, необходима подача источника кислорода для того, чтобы ускорить вышеуказанную реакцию обезуглероживания.
При этом, “оксид основного составного элемента исходного материала сплава” представляет собой соединение, включенное в исходный материал сплава и являющееся соединением атомов кислорода и основного составного элемента в слитке сплава. В случае, когда, например, слиток сплава представляет собой сплав на базе Fe, основным составным элементом в слитке сплава является Fe, и оксид основного составного элемента исходного материала сплава представляет собой оксид железа, такой как Fe3O4, Fe2O3 или т.п. В случае, когда, например, слиток сплава представляет собой сплав на базе Fe-Ni, основными составными элементами в слитке сплава являются Fe и Ni, и оксид основного составного элемента исходного материала сплава представляет собой оксид железа или оксид никеля. В случае, когда, например, слиток сплава представляет собой сплав на базе Ni, основным составным элементом в слитке сплава является Ni, и оксид основного составного элемента исходного материала сплава представляет собой оксид никеля. В случае, когда, например, слиток сплава представляет собой сплав на базе Co, основным составным элементом в слитке сплава является Co, и оксид основного составного элемента исходного материала сплава представляет собой оксид кобальта. Эти оксиды находятся в твердой форме в момент времени, в который они добавляются к ванне расплава. Оксид основного составного элемента исходного материала сплава действует в качестве окислителя в реакции рафинирования (вышеуказанной реакции обезуглероживания) в данном варианте осуществления, и является поэтому окислителем, содержащим оксид металла. Первый оксид, действующий в качестве окислителя во втором рафинирующем агенте, может быть одного вида или комбинацией двух или более видов, из числа оксидов основного составного элемента.
Элементы сплава, такие как Si, Al, Ti, Zr, Hf, B, Ca и т.п., являются активными металлическими элементами, оксиды которых термодинамически более стабильны, чем оксиды железа и никеля. Поэтому, чтобы удалить эти элементы сплава, должны иметь место описанные ниже реакции окислительного рафинирования, и соответствующие результирующие оксиды должны быть удалены посредством выделения в шлак.
[Si]+2[O]→(SiO2)
2[Al]+3[O]→(Al2O3)
[Ti]+2[O]→(TiO2)
[Zr]+2[O]→(ZrO2)
2[B]+3[O]→(B2O3)
[Ca]+[O]→(CaO)
Хотя Ca в расплаве удаляется посредством испарения, даже когда он находится в расплавленном состоянии, некоторое количество Ca остается в слитке сплава. Выполнение окислительного рафинирования таким же образом важно для того, чтобы удалить Ca в расплаве. При окислении и абсорбции в шлак эти элементы реагируют с CaO в слое 7 расплавленного шлака и становятся более стабильными соединениями, посредством чего активность таких компонентов падает в слое 7 расплавленного шлака. Эта более низкая активность эффективна для реакции рафинирования с удалением. Например, SiO2, сформированный посредством окисления Si, реагирует с CaO с образованием стабильного соединения, такого как Ca2SiO4 или т.п. Это эффективно, в результате, для снижения активности SiO2 в слое 7 расплавленного шлака и содействия протеканию реакции окисления Si. Аналогичным образом, Al2O3, TiO2, B2O3 и т.п. реагируют с CaO с образованием соединений, посредством чего имеет место падение их активности в слое 7 расплавленного шлака. Это эффективно для рафинирования посредством окислительного удаления этих элементов. Соответственно, эффективно установление количества CaO в слое 7 расплавленного шлака в соответствии с количеством этих различных оксидов, которые образуются в ходе реакций окисления.
Второй рафинирующий агент на стадии рафинирования данного варианта осуществления должен ускорить вышеуказанную реакцию обезуглероживания и вышеуказанную реакцию окисления металлических активных элементов, в качестве реакций рафинирования с удалением активных элементов. Поэтому, второй рафинирующий агент содержит первый оксид, который действует в качестве окислителя вышеуказанной реакции обезуглероживания, и вышеуказанный флюс (флюс на базе галогенида кальция), который вызывает стабильную абсорбцию оксидов, образованных в результате реакции окисления вышеуказанных металлических активных элементов, в слое шлака.
Например, Fe2O3 или Fe3O4 используются в качестве оксидов железа, которые образуют типичный источник кислорода. Оксиды, образованные в результате реакции окисления, должны стабильно абсорбироваться в слое 7 расплавленного шлака. Поэтому флюс, получаемый от добавления CaO к флюсу на базе галогенида Ca, такого как фторид кальция (CaF2) и хлорид кальция (CaCl2), используется в качестве флюса, обладающего высокой способностью к абсорбции кислорода.
(6) Флюс во втором рафинирующем агенте представляет собой флюс на базе галогенида кальция.
Компонент этого флюса имеет такой же состав, что и флюс в первом рафинирующем агенте. Это обусловлено тем, что снижение температуры плавления флюса, чтобы способствовать плавлению добавленного флюса, посредством передачи тепла от ванны 6 расплава, и образованию, тем самым, слоя 7 расплавленного шлака, является эффективным для содействия протеканию реакций. Флюс, который содержит CaO, более эффективен для абсорбции оксидов, образованных в ходе реакции окисления. Еще более эффективной является компонентная система, имеющая включенное в нее предварительно необходимое количество CaO для перевода образованных оксидов в стабильные соединения.
(7) Масса первого оксида во втором рафинирующем агенте составляет от 0,2 до 4 кратное превышение по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить по меньшей мере углерод и кальций из числа примесных элементов, присутствующих в ванне расплава.
В случае, когда, например, оксид железа (FexOy) используется в качестве оксида основного составного элемента исходного материала сплава во втором рафинирующем агенте, тогда масса WFexOy (кг) добавляемого оксида железа (FexOy) и рассчитанная масса MFeO (кг) оксида железа (FexOy) удовлетворяют выражению, приведенному ниже, на основании вышеуказанного соотношения.
0,2×MFeO≤WFexOy≤4,0×MFeO
При этом, MFeO получена посредством выражения, приведенного ниже, с применением массы ванны расплава и концентрации активных элементов в ванне расплава.
MFeO=M/100×([C]/12,01+2×[Si]/28,09+1,5×[Al]/26,98+2×[Ti]/47,9+2×[Zr]/91,22+2×[Hf]/178,49+1,5×[B]/10,811+[Ca]/40,08-[O]/15,9994)/y×(55,85×x+16,0×y)
M: масса (кг) ванны 6 расплава
[C]: концентрация (масс.%) C в ванне 6 расплава
[Si]: концентрация (масс.%) Si в ванне 6 расплава
[Al]: концентрация (масс.%) Al в ванне 6 расплава
[Ti]: концентрация (масс.%) Ti в ванне 6 расплава
[Zr]: концентрация (масс.%) Zr в ванне 6 расплава
[Hf]: концентрация (масс.%) Hf в ванне 6 расплава
[B]: концентрация (масс.%) B в ванне 6 расплава
[Ca]: концентрация (масс.%) Ca в ванне 6 расплава
[O]: концентрация (масс.%) O в ванне 6 расплава.
Во время восстановительного рафинирования посредством Ca происходит некоторое загрязнение C, Si, Al и т.п. из огнеупорного листа для приема брызг расплава, который размещен на внешней периферии холодного тигля 3 (водоохлаждаемого медного тигля). Соответственно, расчет количества добавляемого кислорода должен принимать во внимание захватываемое количество во время восстановительного рафинирования посредством Ca. А именно, результаты опытов выявили возможность загрязнения вследствие захвата на величину примерно 30 млн-1 для [C] и примерно 50 млн-1 для каждого из [Si] и [Al]. Концентрации различных элементов в вышеуказанной расчетной формуле должны быть использованы с применением смешанных расчетных величин, которые используют, например, величины, полученные анализом загруженных исходных материалов. Вышеуказанная расчетная формула представляет собой формулу для вычисления, в случае, когда кислород подается в форме оксида железа FexOy, требуемого числа молей кислорода для преобразования [C] в газообразный CO, [Si] в шлак SiO2, [Al] в шлак Al2O3, [Ti], [Zr], [Hf] в шлак TiO2, шлак ZrO2 и шлак HfO2, соответственно, [B] в шлак B2O3 и [Ca] в шлак CaO, с помощью деления на атомную массу соответствующего элемента, и для вычисления, на основании этого, необходимой массы оксида железа FexOy. Величины концентрации [C], [Si], [Al], [Ti], [Zr], [Hf], [B], [Ca] и т.п. рассчитываются на основании величин, полученных анализом смешанного расплавленного исходного материала.
Хотя это не включено в вышеуказанную расчетную формулу, она должна быть дополнена необходимым количеством кислорода для преобразования, если они присутствуют в качестве активных металлов, например, щелочных металлов (EI), щелочноземельных металлов (EII), Y, лантаноидов и актиноидов (ER) и т.п., в шлаки EI2O, EIIO, Y2O3 и ER2O3, соответственно. Марганец (Mn) не является таким активным как Fe и сравнительно менее легко удаляется окислительным рафинированием. Кроме того, марганец типично легко удаляется испарением, когда плавится при высоком вакууме. Поэтому, марганец не является целью для удаления окислительным рафинированием. Когда оксид никеля или оксид кобальта используется в качестве окислителя, Fe (атомная масса: 55,85) в вышеуказанном выражении необходимо лишь преобразовать в Ni (атомная масса: 58,71) или Co (атомная масса: 58,93).
В результате нескольких приготовлений и опытов рафинирования было найдено, что масса оксида железа WFexOy (кг), которая должна быть добавлена, когда главной целью является обезуглероживание, составляет 0,2 или более от рассчитанного количества MFeO (кг), и доля добавляемого флюса {Flx}M должна составлять 0,5 масс.% или более, как проиллюстрировано на Фиг.6(a). Степень обезуглероживания существенно снижается, и эффекта рафинирования труднее достигнуть, когда WFexOy является WFexOy<0,2×MFeO и {Flx}M является {Flx}M<0,5. Что касается рафинирования посредством окисления и удаления Si и B, то их оксиды обладают меньшей термодинамической стабильностью, чем оксиды таких элементов, как Al и Ti, и было найдено, что задание массы добавляемого оксида железа, чтобы удовлетворять условию 1,0×MFeO<WFexOy, и задание {Flx}M, чтобы удовлетворять 3,0<{Flx}M, было эффективным для достижения степени обескремнивания 50% или более, как проиллюстрировано посредством соотношения между WFe3O4/MFeO и {Flx}M и степенью обескремнивания на Фиг.7. Для того, чтобы улучшить степень обескремнивания более надежным образом, WFexOy должна быть установлена в интервале от примерно 1,5 до 2 кратного превышения по отношению к MFeO. Это обусловлено тем, что не весь добавляемый оксид железа используется в реакциях. С другой стороны, добавление массы оксида железа, превышающей в 4 раза или более требуемое количество, приводит к чрезмерному увеличению окислительных потерь компонентов сплава, таких как Cr или т.п., что нежелательно.
Рафинирование с удалением элементов, таких как C, Al, Ti, Zr, Hf, Ca и т.п., оксиды которых являются термодинамически стабильными, протекает, когда выполняется вакуумное окислительное рафинирование с заданием массы добавляемого оксида железа в интервале от 0,2 до примерно 1,0 по отношению к рассчитанному количеству MFeO; однако рафинирование с удалением Si, B и т.п. часто не может протекать заметным образом в этом случае. Тем не менее, Si, B и т.п. могут быть удалены установлением количества добавляемого оксида железа, составляющего 1,0 или более по отношению к количеству MFeO, и установлением доли добавляемого флюса {Flx}M, составляющей 3 масс.% или более. Например, [B] составляет 50 млн-1 перед рафинированием и становится 1 млн-1 после рафинирования. Также [Si] составляет 0,22 масс.% перед рафинированием и меньше чем 0,01 масс.% после рафинирования. Рафинированию с удалением Si и B может, кроме того, способствовать установление количества добавляемого оксида железа при 2,0 или более по отношению к количеству MFeO, при применении флюса на базе CaF2-CaO или флюса на базе CaF2-(CaO+CaCl2), и задании доли добавляемого флюса {Flx}M, составляющей 3,0 масс.% или более по отношению к массе ванны 6 расплава.
Если масса добавляемого оксида железа больше, чем в 4 раза превышает массу MFeO, реакция окисления является чрезвычайно бурной, и окислительные потери Cr и т.п., как элементов сплава, становятся значительными. Поэтому масса, которая больше чем в 4 раза превышает массу MFeO, не является целесообразной.
В конкретном примере вышеупомянутых величин, например, ванну расплава массой 50 кг образуют с применением нержавеющей стали, имеющей состав Fe-20Ni-25Cr, феррохромного материала и низкоуглеродистой стали или т.п. в качестве коммерчески доступных недорогих исходных материалов, плюс исходный материал, являющийся электролитическим Ni, в качестве высокочистого исходного материала. При этом, концентрации примесных элементов в ванне расплава составляли примерно [C]=0,02, [Si]=0,21, [Al]=0,015, [Ti]=0,0, [B]=0,005, [Ca]=0,001 и [O]=0,02 масс.%. Рассчитанное молярное количество кислорода, требующееся для окисления вышеуказанных элементов, составляет 8,9 молей. В случае подачи в форме оксида железа Fe3O4, кислород в форме оксида железа Fe3O4 соответствует MFeO=515 г. Поэтому добавление примерно 800 г (превышение примерно в 1,5 раза) Fe3O4 вызывает снижение [C] до примерно 0,005 масс.% и снижение [Al] до примерно 0,003 масс.%. Однако имело место лишь незначительное снижение для [Si], до примерно 0,19 масс.%, и [B], до примерно 0,004 масс.%. Тем не менее, [Si] снижается до 0,01 масс.% или менее, и [B] снижается до примерно 0,0001 масс.%, при добавлении 1300 г (превышение примерно в 2,5 раза) Fe3O4. Используемым при этом флюсом являлся CaF2-CaO (25 масс.%) и доля его добавления {Flx}M=4,0% (2000 г).
Найдено, что такое рафинирование делает возможным выполнение рафинирования с удалением до концентрации примеси ([Si]<0,01 масс.%), сравнимой с величиной, достигаемой при применении высокочистого исходного материала, такого как электролитическое железо, электролитический никель, металлический хром или подобное, даже когда феррохромный материал, низкоуглеродистая сталь или т.п., которые являются коммерчески доступными недорогими исходными материалами, используются в качестве исходных материалов для плавки.
В случае, когда удаление Si или B не является необходимым, может быть выполнено рафинирование, в котором удаление C сделано предварительно, при подавлении снижения содержания Si или B, посредством добавления оксида железа в количестве примерно 1,5 по отношению к количеству MFeO, посредством применения композиционного флюса на базе галогенида Ca с небольшим содержанием CaO и установления доли добавляемого {Flx}M флюса в интервале от примерно 0,5 до 2%.
(8) Доля массы металлического кальция во втором рафинирующем агенте по отношению к массе ванны расплава перед добавлением второго рафинирующего агента находится в интервале от 0,5 до 5 масс.%.
Если доля добавляемого флюса слишком мала, то не достигается эффект абсорбции образованного оксида. Поэтому количество флюса должно составлять по меньшей мере 0,5 масс.% от массы ванны 6 расплава. С другой стороны, когда {Flx}M составляет 5,0 масс.% или более, как в случае восстановительного рафинирования с применением металлического кальция, передача тепла от ванны 6 расплава является недостаточной, и затруднено образование слоя 7 расплавленного шлака. Поэтому верхний предел устанавливается равным 5,0 масс.%.
В данном варианте осуществления предпочтительно добавляется рафинирующий агент (второй рафинирующий агент), являющийся смесью флюса и оксида железа; после этого откачивается инертный газ из камеры (вакуумной камеры 4) (выполняется вакуумирование), и это состояние (вакуумное состояние (состояние откачки)) поддерживается в течение 15 минут или более.
Например, вакуумирование выполняется в камере с применением центробежного насоса с масляным уплотнением, если слой 7 расплавленного шлака образуется посредством добавления второго рафинирующего агента, являющегося смесью флюса и оксида железа, к ванне 6 расплава. В зависимости от обстоятельств, вакуумирование выполняется с применением механического насоса, диффузионного насоса или т.п., и окислительное рафинирование выполняется в разреженной атмосфере.
Тонкие капли (брызги) рассеиваются от ванны 6 расплава, когда степень вакуума составляет 10 гПа или ниже. Это явление, как полагают, обусловлено газообразным CO (г), высвобождающимся вследствие реакции C и O, который вызывает разлетание окружающего расплавленного металла. При этом явлении, реакция заканчивается, когда концентрация углерода снижается в ванне 6 расплава. Например, проявление сильного разбрызгивания заканчивается примерно через 10 минут, и [C] снижается до величины примерно от 20 до 60 млн-1, также в случае первоначальной концентрации [C]0=250 млн-1. Также наблюдается то, что выдерживание ванны 6 расплава под вакуумом вызывает увеличение температуры поверхности ванны 6 расплава и облегчает плавление добавленного флюса. Посредством этого, слой 7 расплавленного шлака образуется стабильным образом.
После такой реакции обезуглероживания фрагменты оплавленного шлака, которые, как считается, являются оксидами, наблюдаются как плавающие на поверхности ванны 6 расплава. Эти фрагменты исчезают посредством постепенной абсорбции в слое 7 расплавленного шлака флюса. Через примерно 15 минут, практически отсутствует всплывание оксидного шлака, и наблюдается, что количество оксида, плавающего на поверхности ванны 6 расплава, становится меньше. Соответственно, расплав должен выдерживаться в течение 15 минут или более.
Операция добавления оксида железа к ванне расплава раскрыта в Патентном документе 5. А именно, оксид железа и CaF2 добавляются к ванне расплава массой 2 кг в водоохлаждаемом медном тигле ⌀84 мм, в атмосфере инертного газа. Условия являются такими же, что и в отношении признаков применения оксида железа. В данном изобретении, однако, образуется ванна 6 расплава массой 10 кг или более в водоохлаждаемом медном тигле 3, имеющем диаметр ⌀200 мм или более, и разъясняются условия добавления оксида железа и легкоплавкого композиционного флюса на базе галогенида Ca в практическом масштабе. Этот признак отличается от обычного способа. В данном изобретении, сверх того, реакция обезуглероживания ускоряется посредством операции вакуумирования. В результате, это ускоряет образование слоя 7 расплавленного шлака и абсорбцию образованных оксидов. Этот признак отличен от обычного способа. А именно, основным отличительным признаком данного изобретения является точное определение условий для получения конкретного эффекта рафинирования.
(Третий вариант осуществления)
В способе получения слитков сплава второго варианта осуществления, как описано выше, углерод и кальций, в качестве примесных элементов, удаляются из ванны расплава практического масштаба посредством окислительного рафинирования в индукционной плавильной печи с холодным тиглем. В индукционной плавильной печи с холодным тиглем, однако, ванна расплава образуется в холодном тигле. Поэтому, соотношение площадь поверхности/объем ванны расплава сравнительно невелико, и удаление образованного газообразного CO является недостаточным в отдельных случаях, даже при выполнении окислительного рафинирования при высоком вакууме. То есть в отдельных случаях обезуглероживание не происходит, и затрудняется удаление углерода до [C]<10 млн-1. При этом концентрация [C]<10 млн-1 может быть достигнута окислительным рафинированием посредством загрузки большого количества окислителя, чтобы принудительно удалить углерод, однако тогда [O] в ванне расплава существенно повышается, до величины, превышающей 100 млн-1.
Поэтому авторы изобретения провели тщательное исследование, нацеленное на способ плавления, состав рафинирующего агента, добавляемого к ванне расплава, количество добавляемого рафинирующего агента и интервал подходящего давления атмосферы во время плавления исходных материалов, для того, чтобы удалить в достаточной степени кислород и углерод, в качестве примесных элементов, из ванны расплава практического масштаба, такой, что масса конечного слитка составляет 10 кг или более. В результате, авторы изобретения нашли, что возможно получение, в практическом масштабе, слитка сплава, имеющего чрезвычайно низкое содержание углерода (C) и кислорода (O), из числа примесных элементов, посредством способа получения, который включает облучение электронным пучком электрода из исходного материала, подаваемого в электроннолучевую плавильную печь с холодным подом, чтобы образовать, тем самым, ванну расплава на холодном поде, предусмотренном в плавильной печи; удаление примесных элементов посредством добавления рафинирующего агента к ванне расплава; и затвердевание расплава, из которого были удалены примесные элементы, чтобы образовать, тем самым, слиток сплава, при условии, что в данном способе:
(9) Внутреннее пространство электроннолучевой плавильной печи с холодным подом во время облучения электронным пучком устанавливается при давлении атмосферы ниже чем 5×10-4 мбар;
(10) В качестве рафинирующего агента используется третий рафинирующий агент, который представляет собой второй оксид, содержащий один, два или более видов из оксидов основного составного элемента в исходном материале для электрода;
(11) Масса третьего рафинирующего агента устанавливается равной от 1 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава.
Авторы изобретения выполнили третий вариант осуществления данного изобретения на основе результатов вышеуказанного исследования.
Целью третьего варианта осуществления в соответствии с данным изобретением является предоставление способа получения, в практическом масштабе, слитка сплава, имеющего чрезвычайно низкое содержание углерода (C) и кислорода (O) из числа примесных элементов.
Третий вариант осуществления данного изобретения будет разъяснен далее со ссылкой на сопроводительные чертежи.
Третий вариант осуществления в соответствии с данным изобретением представляет собой способ получения слитка сплава, данный способ включает: стадию подачи электрода из исходного материала в электроннолучевую плавильную печь с холодным подом и облучения электронным пучком электрода из исходного материала при давлении атмосферы ниже чем 5×10-4 мбар и последующего образования ванны расплава на холодном поде в электроннолучевой плавильной печи с холодным подом; стадию добавления третьего рафинирующего агента к ванне расплава и затем уменьшения содержания углерода, в качестве примесного элемента, присутствующего в ванне расплава; и стадию формирования слитка сплава посредством затвердевания расплава, содержание углерода в котором было уменьшено.
(Электроннолучевая плавильная печь с холодным подом)
Фиг.8 представляет собой схематическое изображение, иллюстрирующее электроннолучевую плавильную печь 11 с холодным подом. Например, электроннолучевая плавильная печь 11 с холодным подом, схематически проиллюстрированная на Фиг.8, может быть использована в качестве электроннолучевой плавильной печи с холодным подом в способе получения слитков сплава данного варианта осуществления. Электроннолучевая плавильная печь 11 с холодным подом содержит вакуумную камеру 4; механизм для подачи исходного материала, не показан, который подает электрод 12 из исходного материала в вакуумную камеру 4; электронные пушки 14, которые вызывают плавление переднего конца электрода 12 из исходного материала, посредством облучения его электронным пучком, обозначенным пунктирной линией на фигуре; холодный под 9, размещенный внутри вакуумной камеры 4, который удерживает ванну расплава, образуемую посредством плавления; и холодный тигель 10, размещенный внутри вакуумной камеры 4, который образует слиток посредством приема расплава, который выпускается из холодного пода 9.
После образования ванны 13 расплава на Фиг.8, механизм для подачи исходного материала подает электрод 12 из исходного материала вместе с оксидом железа, в качестве третьего рафинирующего агента, к одному из концов холодного пода 9. В качестве альтернативы, может быть предусмотрен дополнительный узел подачи исходного материала для подачи третьего рафинирующего агента в электроннолучевой плавильной печи 11 с холодным подом. Предоставление, таким образом, узла подачи исходного материала делает возможным добавление третьего рафинирующего агента (окислителя (FexOy или т.п.)) к ванне расплава 13 на холодном поде 9, в соответствии с состоянием плавления электрода 12 из исходного материала.
Холодный под 9 может быть резервуаром, который не реагирует с ванной 13 расплава или со слоем расплавленного шлака, который образуется вокруг ванны 13 расплава. Примеры холодного пода 9 включают, например, водоохлаждаемый блюдцеобразный резервуар, изготовленный из меди таким образом, что внутренние размеры резервуара для плавления составляют 0,2×0,2 м или более. Это обусловлено тем, что реакция рафинирования, которая сопровождается испарением или газификацией примесных элементов, ускоряется посредством образования ванны расплава, имеющей большое соотношение площадь поверхности/объем (т.е. мелкой ванны расплава с большой площадью поверхности), посредством применения водоохлаждаемого блюдцеобразного резервуара, изготовленного из меди и имеющего большую площадь поверхности. Холодный тигель 10 может быть тиглем, который не реагирует с расплавом. Примеры холодного тигля 10 включают, например, водоохлаждаемый медный кристаллизатор с подъемным дном.
Плавление и рафинирование в вакууме выполняются в электроннолучевой плавильной печи 11 с холодным подом с применением электронных пучков в качестве источника тепла, в условиях глубокого вакуума. Поэтому становится возможным выполнение рафинирования в вакууме с удалением азота (N), марганца (Mn) и т.д., которые не удалось удалить рафинированием с удалением углерода, в качестве примесного элемента, до максимальной степени или восстановительным рафинированием с применением первого рафинирующего агента в индукционной плавильной печи с холодным тиглем или окислительным рафинированием с применением второго рафинирующего агента.
Электроннолучевая плавильная печь 11 с холодным подом снабжена холодным подом 9 (водоохлаждаемым блюдцеобразным резервуаром, изготовленным из меди), имеющим большое соотношение площадь поверхности/объем по сравнению с холодным тиглем, предоставленным в индукционной плавильной печи с холодным тиглем. Поэтому примесные элементы могут быть удалены более надежным образом, чем в индукционной плавильной печи с холодным тиглем, в ходе реакции рафинирования, которая сопровождается газификацией или испарением.
В данном варианте осуществления, первоначально, электрод 12 в виде стержня или в виде чушки из исходного материала (исходного материала сплава) подается от боковой стороны, противоположной выпускному отверстию холодного пода 9. Электрод 12 из исходного материала плавится на холодном поде 9 посредством облучения электронным пучком, чтобы образовать, тем самым, ванну 13 расплава (стадия образования ванны расплава). Затем третий рафинирующий агент добавляется к ванне 13 расплава, чтобы удалить углерод, в качестве примесного элемента, присутствующего в ванне 13 расплава (стадия рафинирования). Затем избыточный расплав после рафинирования инжектируется, через выпускное отверстие холодного пода 9, в холодный тигель 10, который расположен рядом с холодным подом 9. После этого расплав в холодном тигле 10 оставляют затвердевать, чтобы образовать слиток сплава (стадия формирования слитка). Затем посредством последовательного вытягивания вниз слитка сплава приготавливают удлиненные слитки практического масштаба, имеющие конечную массу 10 кг или более.
В данном варианте осуществления в качестве исходного материала электрода может быть использован металл, сплав или подобное в соответствии с целевым составом слитка сплава в виде стержня или в виде чушки. Для того, чтобы дополнительно увеличить чистоту слитка сплава (дополнительно уменьшить содержание примесных элементов), слиток сплава, полученный в данном варианте осуществления, может быть также использован в качестве исходного материала сплава.
Данный вариант осуществления предоставляет возможность получения слитков сплава разных компонентных составов. Например, в нем может быть получен слиток сплава на базе Fe, слиток сплава на базе Ni, слиток сплава на базе Fe-Ni или слиток сплава на базе Co.
(Окислительное рафинирование с применением электроннолучевой плавильной печи с холодным подом)
В данном варианте осуществления рафинирование посредством удаления углерода (C) и кислорода (O), в качестве примесных элементов, выполняется посредством образования ванны 13 расплава в холодном поде 9 электроннолучевой плавильной печи 11 с холодным подом, с последующей операцией рафинирования, которая удовлетворяет условиям с (9) по (11) ниже. Фиг.8 схематически изображает состояние рафинирования в данном варианте осуществления.
Условия рафинирования в данном варианте осуществления были выяснены в результате нескольких опытов и исследований с применением электроннолучевой плавильной печи 11 с холодным подом, при условии, что конечный вакуум в вакуумной камере 4 составлял порядка 10-6 мбар.
(9) Получение выполняется в условиях глубокого вакуума (<5×10-4 мбар).
Это обусловлено тем, что чем выше степень вакуума (т.е. чем ниже давление атмосферы), тем легче протекает реакция обезуглероживания. Предпочтительно, глубина вакуума является такой высокой, насколько это возможно, чтобы уменьшить содержание углерода до максимально возможной степени. Основной причиной задания степени вакуума (давления атмосферы) меньше чем 5×10-4 мбар является то, что небольшое количество газообразного Ar вводится в отдельных случаях в вакуумную камеру 4. Если инертный газ, такой как газообразный Ar или т.п., не вводится в вакуумную камеру 4, то получение предпочтительно выполняется при давлении атмосферы ниже чем 1×10-4 мбар.
(10) В качестве рафинирующего агента используется третий рафинирующий агент, который представляет собой второй оксид, содержащий один, два или более видов из оксидов основного составного элемента в исходном материале для электрода;
Во время получения слитка сплава с применением индукционной плавильной печи 11 с холодным тиглем должна быть ускорена реакция обезуглероживания в соответствии с выражением, приведенным ниже, для того, чтобы удалить углерод в качестве примесного элемента, из расплава.
[C]+[O]→CO (г)↑
Однако, если концентрация кислорода в ванне 13 расплава недостаточна, то обезуглероживание не происходит, даже в условиях глубокого вакуума, когда глубина вакуума составляет <5×10-4 мбар. Соответственно, становится необходимой подача кислорода, требующегося для окисления углерода. Электроннолучевая плавка выполняется в условиях глубокого вакуума, и, следовательно, подача газообразного кислорода затруднена. Поэтому эффективной является подача, вместе с электродом из исходного материала, источника кислорода в форме твердого оксида основного составного элемента исходного материала электрода (например, высокочистого оксида железа или т.п.). А именно, эффективно использование оксида основного составного элемента исходного материала электрода в качестве рафинирующего агента (третьего рафинирующего агента) при обезуглероживании рафинированием с удалением. В этом случае, твердый оксид в форме микропорошка, например, микропорошок оксида железа, становится рассеянным посредством вовлечения в газовый поток на первоначальной стадии вакуумирования при электроннолучевой плавке. Этот микропорошок достигает вакуумного насоса и повреждает последний. Поэтому предпочтительно предварительное приведение твердого оксида в виде микропорошка в объемную форму. Когда, например, используется микропорошок оксида железа в качестве оксида основного составного элемента исходного материала для электрода, предпочтительно предварительно выполнять спекание или подобное и добавлять оксид железа в гранулированной форме.
При этом, “оксид основного составного элемента исходного материала для электрода” представляет собой соединение, включенное в исходный материал электрода и являющееся соединением атомов кислорода и основного составного элемента в слитке сплава. В случае, когда, например, слиток сплава представляет собой сплав на базе Fe, основным составным элементом в слитке сплава является Fe, и оксид основного составного элемента исходного материала для электрода представляет собой оксид железа, такой как Fe3O4, Fe2O3 или подобное. В случае, когда, например, слиток сплава представляет собой сплав на базе Fe-Ni, основными составными элементами в слитке сплава являются Fe и Ni, и оксид основного составного элемента исходного материала для электрода представляет собой оксид железа или оксид никеля. В случае, когда, например, слиток сплава представляет собой сплав на базе Ni, основным составным элементом в слитке сплава является Ni, и оксид основного составного элемента исходного материала для электрода представляет собой оксид никеля. В случае, когда, например, слиток сплава представляет собой сплав на базе Co, основным составным элементом в слитке сплава является Co, и оксид основного составного элемента исходного материала для электрода представляет собой оксид кобальта. Эти оксиды находятся в твердой форме в момент времени, когда они добавляются к ванне расплава. Оксид основного составного элемента исходного материала для электрода действует в качестве окислителя в реакции рафинирования (вышеуказанной реакции обезуглероживания) в данном варианте осуществления, и является поэтому окислителем, содержащим оксид металла. Третий рафинирующий агент, являющийся вторым оксидом, который действует в качестве окислителя, может быть оксидом одного вида из числа оксидов основного составного элемента в исходном материале для электрода или комбинацией двух или более видов.
(11) Масса третьего рафинирующего агента устанавливается равной от 1 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава.
Авторы изобретения экспериментально исследовали изменение в содержании примесного элемента в соответствии с количеством добавляемого третьего рафинирующего агента и нашли, что реакция обезуглероживания активируется, когда количество добавляемого третьего рафинирующего агента постепенно увеличивается от 0 (нуля), при этом реакция обезуглероживания прогрессирует в достаточной степени, когда масса добавляемого третьего рафинирующего агента представляет собой массу, по существу равную “расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава”. Авторы изобретения нашли, что содержание кислорода в слитке имеет тенденцию к повышению, когда, в результате увеличения количества добавляемого третьего рафинирующего агента, масса добавляемого третьего рафинирующего агента превышает по существу в четыре раза “рассчитанную массу, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава”.
Соответственно, масса третьего рафинирующего агента устанавливалась равной от 1 до 4 кратному превышению по отношению к “расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава”. Авторы изобретения нашли, что содержание углерода становится по существу наименьшей величиной (пределом аналитического обнаружения), и содержание кислорода становится по существу наименьшей величиной (пределом аналитического обнаружения), когда масса третьего рафинирующего агента составляет от 2 до 3 кратное превышение рассчитанной массы. Предпочтительно, масса третьего рафинирующего агента от 2 до 3 раз выше рассчитанной массы.
В случае, когда оксид железа (FexOy) используется в качестве оксида основного составного элемента в исходном материале для электрода, масса WFexOy (кг) добавляемого оксида железа (FexOy) и рассчитанная масса MFeO (кг) оксида железа (FexOy) удовлетворяют выражению, приведенному ниже, на основании вышеуказанного соотношения.
1,0×MFeO≤WFexOy≤4,0×MFeO
При этом, MFeO задается выражением, приведенным ниже, на основании массы WM (кг) исходного материала для электрода и содержания углерода [C] (масс.%) в исходном материале для электрода.
MFeO=WM/100×([C]/12,01-[O]/16,0)/y×(55,85×x+16,0×y)
Оксид никеля (NixOy) или т.п. может быть использован вместо оксида железа в качестве оксида основного составного элемента в исходном материале для электрода. В этом случае достаточно использовать атомную массу (58,71) Ni вместо атомной массы (55,85) Fe в вышеприведенном выражении для расчета MFeO.
При этом была исследована взаимосвязь между долей WFe2O3 по отношению к MFeO (т.е. WFe2O3/MFeO, также называемой долей добавляемого оксида железа) и степенью обезуглероживания η[C]=([C]0-[C]EB)/[C]0×100(%) в случае, когда нержавеющая сталь сравнительно высокой чистоты была использована в качестве исходного материала для электрода. Результаты представлены на Фиг.9. Нижняя предельная величина степени обезуглероживания составляет примерно 40% или более, когда доля добавляемого оксида железа (WFe2O3/MFeO) составляет 1 или более, и центральное значение степени обезуглероживания составляет примерно 50% или более. Это указывает на то, что реакция обезуглероживания ускоряется.
Авторы изобретения нашли, что электроннолучевая плавка с холодным подом была высокоэффективна для отделения, посредством всплывания, неметаллических включений в слитке и также высокоэффективна в качестве процедуры удаления кислорода. Было найдено, что способ получения слитков сплава в данном варианте осуществления предоставляет возможность формирования слитков сплава, в которых [C]≤10 млн-1 и [O]≤10 млн-1. Также было найдено, что слитки сплава, имеющие [C]<5 млн-1 и [O]<5 млн-1, на уровне предела аналитического обнаружения или ниже, могут быть получены посредством оптимизации массы третьего рафинирующего агента. Во многих случаях, содержание марганца (Mn), в качестве компонента сплава, снижается до [Mn]≤0,01 масс.% после рафинирования, вследствие удаления испарением во время процесса электроннолучевой плавки.
(Комбинация процессов рафинирования с первого по третий вариантов осуществления)
Предпочтительно, комбинируются любые два или более процесса рафинирования с первого по третий вариантов осуществления.
А именно, при этом предпочтительно выполняется процесс рафинирования, который является подходящей комбинацией процесса восстановительного рафинирования в первом варианте осуществления (также называемого процессом восстановительного рафинирования посредством Ca) и процесса окислительного рафинирования во втором варианте осуществления (также называемого процессом окислительного рафинирования). Посредством восстановительного рафинирования в первом варианте осуществления примесные элементы, такие как фосфор (P), сера (S), азот (N), следовые и случайные элементы (Sn, Pb, As, Sb, Bi, Se и т.п.), бор (B) и т.п., удаляются из ванны 6 расплава, компоненты которой регулируются до заданного состава сплава. Посредством вакуумного окислительного рафинирования во втором варианте осуществления удаляются бор (B), углерод (C), кремний (Si), алюминий (Al), титан (Ti), цирконий (Zr), кальций (Ca) и щелочные металлы. В результате, содержание примесей, таких как фосфор (P), сера (S), олово (Sn) и свинец (Pb), может быть уменьшено до 2 млн-1 или менее. Также содержание азота (N) может быть уменьшено до 5 млн-1 или менее, и содержание кремния (Si), алюминия (Al), титана (Ti), циркония (Zr) и т.п. может быть доведено до 100 млн-1 или менее. Кроме того, содержание углерода (C) может быть уменьшено до 50 млн-1 или менее, и содержание кремния (Si), кальция (Ca) и т.п. может быть уменьшено до 1 млн-1 или менее.
Вследствие процесса восстановительного рафинирования в первом варианте осуществления происходит захватывание элементов, таких как C, Si, Al, Ca и т.п. Предпочтительно, поэтому, процесс окислительного рафинирования (например, процесс вакуумного окислительного рафинирования) во втором варианте осуществления выполняется после восстановительного рафинирования посредством Ca. В случае, когда содержание C, Si, Al, Ti и т.п., которые являются примесными элементами в исходном материале расплава, очень высокое, эффективным является снижение содержания C, Si, Al, Ti и т.п. посредством выполнения окислительного рафинирования перед восстановительным рафинированием посредством Ca.
В случае, когда, например, скрап или т.п. нержавеющей стали используется в качестве исходного материала сплава (исходного материала сплава), содержание углерода может быть высоким, и содержание компонентов сплава, таких как Si, Al, Ti и т.п. может быть примерно 1 масс.%, в зависимости от состава сплава. Также, например, сплав может иметь добавленные к нему примерно несколько десятков млн-1 Zr, B и т.п. В таких случаях, необходимо значительно увеличить количество оксида железа, который добавляется во время окислительного рафинирования. Окислительное рафинирование, выполненное таким образом при увеличенном количестве добавленного оксида железа, будет, вероятно, проявлять значительно более высокое содержание [O] в ванне 6 расплава. Когда активный металлический элемент сплава добавляется к ванне 6 расплава, имеющей высокое содержание кислорода, для того, чтобы отрегулировать содержание компонентов, входящих в конечный состав сплава, могут иметь место отклонения от желаемых содержаний компонентов вследствие существенного снижения усвоения, так же как и значительное увеличение количества неметаллических включений. В таких случаях окислительное рафинирование предпочтительно выполняется разделенным на два этапа. Первоначально, C, Si, Al, Ti, B и т.п. удаляются посредством выполнения окислительного рафинирования (например, вакуумного окислительного рафинирования). После этого, P, S, Sn, Pb, N и т.п. удаляются первоначально восстановительным рафинированием посредством Ca. Соответственно, C, Si, Al, Ca и т.п., содержащиеся в интервале от нескольких десятков до нескольких сотен млн-1, захваченные во время процесса восстановительного рафинирования посредством Ca, удаляются после этого в ходе повторного (второго) вакуумного окислительного рафинирования, которое выполняется снова, чтобы предотвратить, тем самым, избыточное содержание остаточного кислорода O в ванне 6 расплава.
Таким образом, рафинирование расплава с получением слитков сплава на базе Fe, слитков сплава на базе Fe-Ni и слитков сплава на базе Ni ультравысокой чистоты (с ультранизким содержанием примесей) становится возможным посредством выполнения рафинирования при использовании соответствующей комбинации процесса восстановительного рафинирования посредством Ca и процесса окислительного рафинирования (например, процесса вакуумного окислительного рафинирования). Соответственно, могут быть получены слитки сплава, очищенного до ультравысокой чистоты.
Предпочтительно, выполняются также обезуглероживание и раскисление, в соответствии с процессом окислительного рафинирования в третьем варианте осуществления, с применением вторичного слитка (исходного материала сплава) в виде слитка, полученного комбинированием процесса восстановительного рафинирования в первом варианте осуществления и процесса окислительного рафинирования во втором варианте осуществления, или слитка, полученного в результате образования сплава посредством добавления к вышеуказанному слитку, описанного ниже компонента сплава на базе раскисляющего элемента.
Предпочтительно, выполняются также обезуглероживание и раскисление, в соответствии с процессом окислительного рафинирования в третьем варианте осуществления, с применением первичного слитка (исходного материала сплава) в виде слитка, полученного в соответствии с процессом окислительного рафинирования во втором варианте осуществления, или слитка, полученного в результате образования сплава посредством добавления к вышеуказанному слитку, описанного ниже компонента сплава на базе раскисляющего элемента. А именно, слиток сплава получают посредством рафинирования с удалением примесных элементов, таких как C, Al, Ti, Zr, Ca, Si, B и т.п. посредством выполнения окислительного рафинирования (например, вакуумного окислительного рафинирования), в котором добавляется второй рафинирующий агент, с применением индукционной плавильной печи 1 с холодным тиглем, имеющей холодный тигель 3 с внутренним диаметром ⌀200 мм или более, при массе ванны расплава 10 кг или более. В качестве альтернативы, получают слиток, в котором, в зависимости от обстоятельств, к вышеуказанному слитку сплава дополнительно добавляется компонент сплава на базе раскисляющего элемента. Предпочтительно рафинирование с удалением выполняется после этого, посредством обезуглероживания и раскисления, в соответствии с процессом окислительного рафинирования в третьем варианте осуществления, со снижением до [C]<10 млн-1 и [O]<10 млн-1. При этом становится возможным получение высокочистого (с ультранизким содержанием примесей) слитка сплава на базе Fe, слитка сплава на базе Fe-Ni и слитка сплава на базе Ni.
Процесс окислительного рафинирования в способе получения слитков сплава во втором варианте осуществления может следовать за процессом восстановительного рафинирования в способе получения слитков сплава в первом варианте осуществления. Более конкретно, рафинирование с удалением P, S, Sn, Pb, N, B и т.п. может быть выполнено проведением восстановительного рафинирования с применением металлического кальция, в котором добавляется первый рафинирующий агент, при массе ванны расплава 10 кг или более, с применением индукционной плавильной печи 1 с холодным тиглем, имеющей холодный тигель 3 с внутренним диаметром ⌀200 мм или более; после этого рафинирование с удалением примесных элементов, таких как C, Al, Ti, Zr, Ca, Si, B и т.п. может быть выполнено посредством окислительного рафинирования (например, вакуумного окислительного рафинирования), в котором добавляется второй рафинирующий агент; и расплав после рафинирования оставляется для затвердевания, чтобы получить, тем самым, слиток сплава. В качестве альтернативы, может быть получен слиток, в котором, в зависимости от обстоятельств, к вышеуказанному слитку сплава дополнительно добавляется компонент сплава на базе раскисляющего элемента. Рафинирование с удалением может быть выполнено после этого, посредством обезуглероживания и раскисления, в соответствии с процессом окислительного рафинирования в третьем варианте осуществления, со снижением до [C]<10 млн-1 и [O]<10 млн-1. В результате вышеуказанного способа становится возможным получение ультрачистого (с ультранизким содержанием примесей) слитка сплава на базе Fe, слитка сплава на базе Fe-Ni и слитка сплава на базе Ni.
(Добавление компонента сплава на базе раскисляющего элемента)
В случае использования исходного материала сплава в форме слитка, полученного в результате процесса окислительного рафинирования во втором варианте осуществления, или слитка, полученного в результате соответствующей комбинации процесса восстановительного рафинирования в первом варианте осуществления и процесса окислительного рафинирования во втором варианте осуществления, предпочтительно выполнять легирование (образование сплава) посредством добавления компонента сплава на базе раскисляющего элемента к ванне расплава, которая образуется из вышеуказанного слитка. Примеры компонентов сплава на базе раскисляющего элемента включают, например, такие элементы, как Si, Al, Ti, Zr и B. В результате при этом может быть получен слиток сплава на базе Fe, слиток сплава на базе Fe-Ni или слиток сплава на базе Ni ультравысокой чистоты (с ультранизким содержанием примесей) при заданном составе сплава. Предпочтительно, восстановительное рафинирование посредством Ca и т.д. выполняется в случае, когда содержание P, S, N, Sn, Pb и т.п. в слитке высокое.
При получении слитков сплава ультравысокой чистоты возникает состояние с высоким содержанием кислорода, присутствующего в ванне 6 расплава, когда вакуумное окислительное рафинирование или окислительное рафинирование выполняется в качестве последнего процесса. При добавлении компонента сплава на базе активного элемента (на базе раскисляющего элемента) к ванне 6 расплава, содержащей большое количество кислорода, образуется его оксид в качестве продукта раскисления. Для того, чтобы отделить и удалить такие оксидные продукты раскисления из ванны 6 расплава, эффективным является добавление флюса на базе галогенида кальция вместе с компонентом сплава. Подходящее количество добавляемого флюса на базе галогенида кальция {Flx}M в этом случае находится в интервале от примерно 0,5 до 2 масс.%. При этом, восстановительное рафинирование посредством Ca и т.д. должно быть выполнено в случае, когда содержание P, S, N, Sn, Pb и т.п. в слитке высокое.
В данном описании, [X] обозначает содержание (концентрацию) элемента X, содержащегося в сплаве (или расплаве), и “удаление” примесных элементов из сплава (или расплава) охватывает не только случаи, когда примесные элементы, присутствующие в сплаве (или расплаве), полностью удаляются, но также и случаи, когда содержание примесных элементов уменьшается. В данном описании различные элементы также обозначаются символами элементов. При этом, ультравысокая чистота означает, что общее суммарное содержание элементов, присутствующих в слитке сплава, из которого элементы должны быть удалены, меньше чем 100 млн-1. В первом варианте осуществления, например, элементом, подлежащим удалению, является, по меньшей мере, фосфор. Поэтому, ультравысокая чистота означает выполнение условия [P]<100 млн-1. Во втором варианте осуществления элементами, подлежащими удалению, являются по меньшей мере углерод и кальций. Поэтому, ультравысокая чистота означает выполнение условия [C]+[Ca]<100 млн-1. В третьем варианте осуществления элементами, подлежащими удалению, являются по меньшей мере углерод и кислород. Поэтому, ультравысокая чистота означает выполнение условия [C]+[O]<40 млн-1.
ПРИМЕРЫ
Данное изобретение поясняется более подробно далее на основании примеров, однако изобретение не ограничивается каким-либо образом данными примерами. Примеры могут быть выполнены посредством подходящей модификации при том, что они соответствуют цели предмета изобретения, изложенного ранее или позднее в данном описании, и все такие модификации включены в технический объем данного изобретения.
В примерах эффект рафинирования по данному изобретению был оценен с применением плавильной печи, схематически проиллюстрированной на Фиг.1 и Фиг.8. Основные технические характеристики плавильной печи представлены ниже.
частота: 3000 Гц
число сегментов: 24,
водоохлаждаемый,
изготовленный из меди
максимальная выходная мощность: 300 кВт
диффузионный насос
(Пример A: опыт восстановительного рафинирования с применением индукционной плавильной печи с холодным тиглем)
В данном примере выполняли опыт восстановительного рафинирования, в котором используется первый рафинирующий агент, являющийся смесью металлического кальция и флюса. А именно, первоначально исходный материал сплава в форме феррохромного материала и низкоуглеродистой стали, в качестве недорогих исходных материалов, имеющих значительное содержание примесных элементов, загружали в водоохлаждаемый медный тигель (холодный тигель 3), имеющий внутренний диаметр ⌀220 мм, который был предоставлен в индукционной плавильной печи 1 с холодным тиглем. Ванны 6 расплава, содержащие типы сплавов (Fe-20Ni-25Cr, Fe-35Ni-25Cr), представленные в Таблице 1 в качестве целевых составов, образовывали в атмосфере газообразного аргона. Затем к ваннам 6 расплава добавляли металлический кальций и флюс, представленные в Таблице 1, в качестве первого рафинирующего агента, и расплав выдерживали в течение от 2,0 до 60,0 минут, чтобы выполнить, тем самым, восстановительное рафинирование. Содержание фосфора [P]0 перед рафинированием и содержание фосфора [P]1 после рафинирования определяли масс-спектрометрией с тлеющим разрядом (GD-MS) и вычисляли степень дефосфоризации. Результаты измерений представлены в Таблице 1 вместе с условиями испытания. В Таблице 1, CaF2-20CaO обозначает CaF2-CaO (20 масс.%), CaF2-10CaO обозначает CaF2-CaO (10 масс.%) и CaF2-10CaO-10CaCl2 обозначает CaF2-(CaO+CaCl2)(20 масс.%).
Как показывает Таблица 1, ванна расплава могла визуально наблюдаться также после добавления первого рафинирующего агента в Сравнительном примере A1, в котором доля добавляемого металлического кальция {Ca}M=0,2, однако лишь небольшой эффект дефосфоризации мог быть достигнут. В Примере A1, в котором доля добавляемого металлического кальция {Ca}M=0,4, в противоположность этому, ванна расплава не могла визуально наблюдаться после добавления первого рафинирующего агента, однако был достигнут значительный эффект дефосфоризации. Более высокий эффект дефосфоризации был достигнут в Примере A2 с долей добавляемого металлического кальция {Ca}M=1,0, т.е. увеличенным количеством добавляемого металлического кальция. Очень хороший эффект дефосфоризации был достигнут в Примерах с A3 по A6, в которых количество добавляемого металлического кальция было значительным (сравнительно высокая доля добавляемого металлического кальция ({Ca}M≥0,5)) и было большим количество добавляемого флюса по отношению к металлическому кальцию (доля добавляемого флюса {Flx}M≥{Ca}M). Очень хороший эффект дефосфоризации был достигнут также в случае (Пример A13) большой ванны расплава (50,0 кг) с 2,5-кратным увеличением массы.
При ориентации далее на время выдержки расплава, превосходный эффект дефосфоризации был достигнут посредством регулирования подходящим образом времени выдержки расплава для каждого типа сплава в Примерах с A7 по A11 (сплавы Fe-20Ni-25Cr) и Примерах с A12 по A16 (сплавы Fe-35Ni-25Cr), в которых изменялось лишь время выдержки расплава.
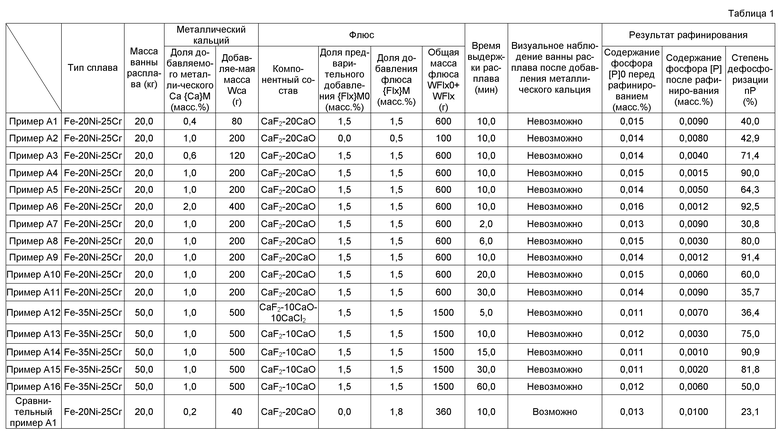
(Пример B: испытание 1 окислительного рафинирования с применением индукционной плавильной печи с холодным тиглем)
Испытание окислительного рафинирования выполняли с применением смеси (т.е. второго рафинирующего агента) флюса и оксида железа в качестве первого оксида. А именно, первоначально исходный материал сплава в виде низкоуглеродистой стали с ультранизким содержанием углерода и феррохромного материала (FeCr), в качестве недорогих исходных материалов, имеющих значительное содержание примесных элементов, например углерода (C), кремния (Si) и т.п., плюс электролитический никель, являющийся высокочистым исходным материалом, загружали в водоохлаждаемый медный тигель, предоставленный в индукционной плавильной печи 1 с холодным тиглем. Ванны расплава 6, содержащие типы сплавов (Fe-35Ni-25Cr, Fe-20Ni-25Cr), представленные в Таблице 2 в качестве целевых составов, образовывали в атмосфере газообразного аргона. Затем, к ваннам расплава 6 добавляли флюсы и оксиды железа, представленные в Таблице 2, в качестве второго рафинирующего агента. После этого расплав выдерживали в течение от 10,0 до 30,0 минут в состоянии вакуумированния, при котором газообразный аргон откачивали во внешнюю атмосферу, чтобы выполнить, тем самым, окислительное рафинирование. Содержание углерода [C], содержание кремния [Si], содержание кальция [Ca] и содержание бора [B] после рафинирования определяли масс-спектрометрией с тлеющим разрядом (GD-MS). Результаты измерений представлены в Таблице 2 вместе с условиями испытания. Таблица 2 показывает, что вышеуказанные исходные материалы сплава содержат значительное количество углерода и кремния в качестве примесных элементов и содержат также примерно 50 млн-1 бора (B) в качестве примесного элемента.
В Таблице 2 CaF2-15CaO-5CaCl2 обозначает CaF2-(CaO+CaCl2) (20 масс.%), CaF2-20CaO обозначает CaF2-CaO (20 масс.%), CaF2-21CaO обозначает CaF2-CaO (21 масс.%) и CaF2-22CaO обозначает CaF2-CaO (22 масс.%), соответственно.
Как показывает Таблица 2, количество добавляемого оксида железа в Сравнительном примере B1, в котором WFe3O4/MFeO=0,15, было слишком мало (WFe3O4/MFeO<0,2), и, соответственно, не был достигнут высокий эффект обезуглероживания. В противоположность этому, Примеры B1 и B2, которые удовлетворяли условиям WFe3O4/MFeO≥0,2 и {Flx}M≥0,5 с целью обезуглероживания, проявили достаточный эффект обезуглероживания. Примеры с B3 по B5, которые удовлетворяли условиям WFe3O4/MFeO≥1,0 и {Flx}M≥3,0, с целью обескремнивания (Si) и удаления бора (B), проявили достаточный эффект обезуглероживания и также удовлетворительные эффекты обескремнивания и удаления бора. Количество добавляемого оксида железа в Сравнительном примере B2, в котором WFe3O4/MFeO=4,5, было чрезмерно большим (WFe3O4/MFeO>4), и, соответственно, происходило значительное рассеивание (разбрызгивания) капель в ходе реакции обезуглероживания во время рафинирования в вакууме, что делало невозможным стабильное выполнение операции рафинирования.
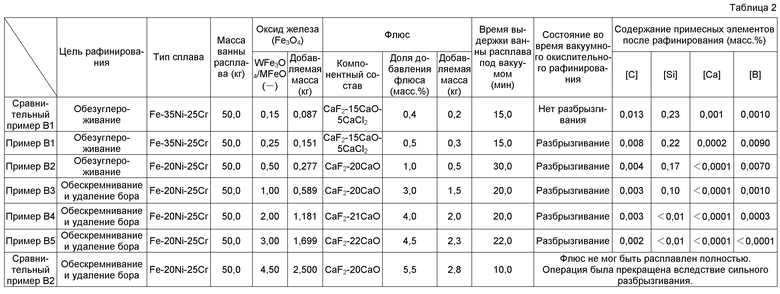
(Пример C: испытание 2 окислительного рафинирования с применением индукционной плавильной печи с холодным тиглем)
Испытание восстановительного рафинирования с применением той же самой индукционной плавильной печи с холодным тиглем, что и в Примере A, выполняли с использованием феррохромного материала, стали с ультранизким содержанием углерода и электролитического никеля в качестве исходных материалов сплава и приготавливали слиток сплава (первичный слиток). При использовании первичного слитка в качестве исходного материала сплава выполняли испытание окислительного рафинирования таким же образом, что и в Примере B при условиях испытания Таблицы 3, вместо условий испытания Таблицы 2. Величины содержания углерода, кремния, кальция и бора после рафинирования определяли масс-спектрометрией с тлеющим разрядом (GD-MS). Результаты измерений представлены в Таблице 3 вместе с условиями испытания.
Как показывает Таблица 3, количество добавляемого оксида железа в Сравнительном примере C1, в котором WFe3O4/MFeO=0,1, было слишком мало (WFe3O4/MFeO<0,2), и, соответственно, не был достигнут высокий эффект обезуглероживания. В противоположность этому, Примеры C1 и C2, которые удовлетворяли условиям WFe3O4/MFeO≥0,2 и {Flx}M≥0,5 с целью обезуглероживания, проявили достаточный эффект обезуглероживания. Примеры с C3 по C5, которые удовлетворяли условиям WFe3O4/MFeO≥1,0 и {Flx}M≥3,0, с целью обескремнивания (Si) и удаления бора (B), проявили достаточный эффект обезуглероживания и также удовлетворительные эффекты обескремнивания и удаления бора. Количество добавляемого оксида железа в Сравнительном примере C2, в котором WFe3O4/MFeO=5,0, было чрезмерно большим (WFe3O4/MFeO>4), и, соответственно, происходило значительное рассеивание (разбрызгивания) капель в ходе реакции обезуглероживания во время рафинирования в вакууме, что делало невозможным стабильное выполнение операции рафинирования.
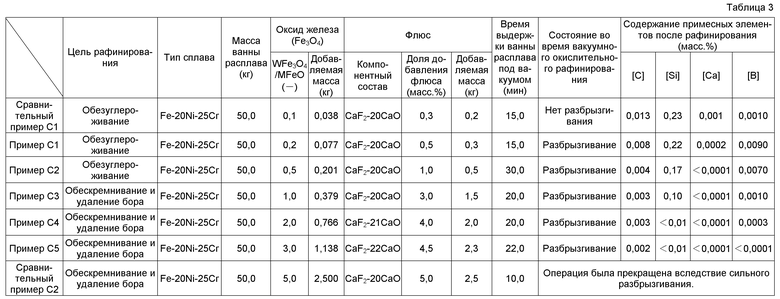
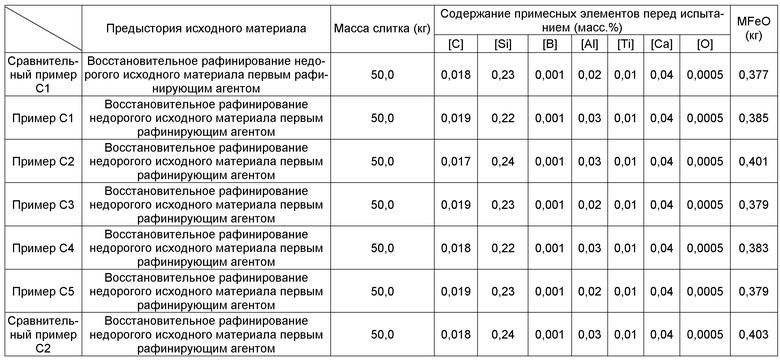
(Пример D: испытание 1 с использованием электроннолучевой плавки с холодным подом)
Испытание окислительного рафинирования выполняли с применением второго оксида, в качестве третьего рафинирующего агента. А именно, первоначально электрод 12 из исходного материала подавали до боковой стороны, противоположной выпускному отверстию холодного пода 9 посредством механизма для подачи исходного материала электроннолучевой плавильной печи 11 с холодным подом, и образовывали ванну 13 расплава, содержащую сплав типа (Fe-20Ni-25Cr), представленный в Таблице 4 в качестве целевого состава, на холодном поде 9 в разреженной атмосфере при условии, что степень вакуума была меньше чем 5×10-4 мбар. Затем к ванне расплава 13 на холодном поде 9 добавляли Fe2O3 (второй оксид) в гранулированной форме, полученный спеканием микрочастиц порошкового Fe2O3 при 1250°C, в качестве третьего рафинирующего агента, чтобы выполнить, тем самым окислительное рафинирование. Содержание углерода [C] и содержание кислорода [O] после рафинирования определяли масс-спектрометрией с тлеющим разрядом (GD-MS). Результаты измерений представлены в Таблице 4 вместе с условиями испытания.
Содержание (масс.%) примесных элементов в электроде 12 из исходного материала перед испытанием составляло [C]=0,005, [O]=0,003. Величина MFeO, как вычислено из содержания примесных элементов, составляла 0,011 кг.
Как показывает Таблица 4, Сравнительный пример D1, в котором WFe2O3/MFeO=0,5, не смог проявить высокий эффект обезуглероживания. Это может быть обусловлено тем, что количество добавляемого оксида железа было слишком мало, и, соответственно, количество Fe2O3 было меньше (WFe2O3/MFeO<1), чем количество кислорода, требуемое для ожидаемой реакции обезуглероживания, так что, в результате, протекание реакции обезуглероживания было незначительным.
В противоположность этому, Примеры с D1 по D4, которые удовлетворяли условию 1≤WFe2O3/MFeO≤4, проявляли достаточные эффекты обезуглероживания и раскисления. В частности, Примеры D2 и D3, которые удовлетворяли условию 2≤WFe2O3/MFeO≤3, проявляли очень высокие эффекты обезуглероживания и раскисления, в результате которых общее суммарное содержание углерода и содержание кислорода было меньше чем 15 млн-1.
Сравнительный пример D2, в котором WFe2O3/MFeO=5,0, проявлял достаточный эффект обезуглероживания, однако эффект раскисления отсутствовал полностью, так что содержание кислорода было увеличено по сравнению с его величиной перед рафинированием. Это может быть обусловлено чрезмерно большим количеством добавляемого оксида железа, так что количество Fe2O3 (WFe2O3/MFeO>4) было больше, чем количество кислорода, требуемое для ожидаемой реакции обезуглероживания, т.е. кислород подавался сверх его количества, расходуемого в реакции обезуглероживания.
(-)
(Пример E: испытание 2 с использованием электроннолучевой плавки с холодным подом)
Испытание окислительного рафинирования с применением той же самой индукционной плавильной печи с холодным тиглем, что и в Примере B, выполняли с использованием феррохромного материала, стали с ультранизким содержанием углерода и электролитического никеля в качестве исходных материалов сплава и приготавливали слиток сплава (первичный слиток). Затем выполняли испытание окислительного рафинирования посредством электроннолучевой плавки с холодным подом таким же образом, что и в Примере D, с применением этого первичного слитка в качестве исходного материала для электрода, однако теперь при условиях испытания Таблицы 5 вместо условий испытания Таблицы 4. Содержание углерода и кислорода после рафинирования определяли масс-спектрометрией с тлеющим разрядом (GD-MS). Результаты измерений представлены в Таблице 5 вместе с условиями испытания.
Содержание (масс.%) примесных элементов в электроде 12 из исходного материала перед испытанием составляло [C]=0,0045, [O]=0,0025. Величина MFeO, как вычислено из содержания примесных элементов, составляла 0,011 кг.
Как показывает Таблица 5, Сравнительный пример E1, в котором WFe2O3/MFeO=0,80, не смог проявить высокий эффект обезуглероживания. Это может быть обусловлено тем, что количество добавляемого оксида железа было слишком мало, и, соответственно, количество Fe2O3 было меньше (WFe2O3/MFeO<1), чем количество кислорода, требуемое для ожидаемой реакции обезуглероживания, так что, в результате, протекание реакции обезуглероживания было незначительным.
В противоположность этому, Примеры с E1 по E4, которые удовлетворяли условию 1≤WFe2O3/MFeO≤4, проявляли значительные эффекты обезуглероживания и раскисления. В частности, Примеры E2 и E3, которые удовлетворяли условию 2≤WFe2O3/MFeO≤3, проявляли очень высокие эффекты обезуглероживания и раскисления, в результате которых общее суммарное содержание углерода и содержание кислорода составляло 15 млн-1 или менее.
Сравнительный пример E2, в котором WFe2O3/MFeO=5,00, проявлял достаточный эффект обезуглероживания, однако эффект раскисления отсутствовал полностью, так что содержание кислорода было увеличено по сравнению с его величиной перед рафинированием. Это может быть обусловлено чрезмерно большим количеством добавляемого оксида железа, так что количество Fe2O3 (WFe2O3/MFeO>4) было больше, чем количество кислорода, требуемое для ожидаемой реакции обезуглероживания, т.е. кислород подавался сверх его количества, расходуемого в реакции обезуглероживания.
(-)
(Пример F: испытание 3 с использованием электроннолучевой плавки с холодным подом)
Восстановительное рафинирование, аналогичное Примеру A, и окислительное рафинирование, аналогичное Примеру B, выполняли в индукционной плавильной печи с холодным тиглем, с использованием феррохромного материала, стали с ультранизким содержанием углерода и электролитического никеля в качестве исходных материалов сплава, чтобы приготовить слиток сплава. Затем выполняли испытание окислительного рафинирования посредством электроннолучевой плавки с холодным подом таким же образом, что и в Примере D, с применением этого слитка в качестве исходного материала для электрода, однако теперь при условиях испытания Таблицы 6 вместо условий испытания Таблицы 4. Содержание углерода и кислорода после рафинирования определяли масс-спектрометрией с тлеющим разрядом (GD-MS). Результаты измерений представлены в Таблице 6 вместе с условиями испытания.
Содержание (масс.%) примесных элементов в электроде 12 из исходного материала перед испытанием составляло [C]=0,005, [O]=0,003. Величина MFeO, как вычислено из содержания примесных элементов, составляла 0,011 кг.
Как показывает Таблица 6, Сравнительный пример F1, в котором WFe2O3/MFeO=0,5, не смог проявить высокий эффект обезуглероживания. Это может быть обусловлено тем, что количество добавляемого оксида железа было слишком мало, и, соответственно, количество Fe2O3 было меньше (WFe2O3/MFeO<1), чем количество кислорода, требуемое для ожидаемой реакции обезуглероживания, так что, в результате, протекание реакции обезуглероживания было незначительным.
В противоположность этому, Примеры с F1 по F4, которые удовлетворяли условию 1≤WFe2O3/MFeO≤4, проявляли значительные эффекты обезуглероживания и раскисления. В частности, Примеры F2 и F3, которые удовлетворяли условию 2≤WFe2O3/MFeO≤3, проявляли очень высокие эффекты обезуглероживания и раскисления, в результате которых общее суммарное содержание углерода и содержание кислорода составляло 15 млн-1 или менее.
Сравнительный пример F2, в котором WFe2O3/MFeO=5,0, проявлял достаточный эффект обезуглероживания, однако эффект раскисления отсутствовал полностью, так что содержание кислорода было увеличено по сравнению с его величиной перед рафинированием. Это может быть обусловлено чрезмерно большим количеством добавляемого оксида железа, так что количество Fe2O3 (WFe2O3/MFeO>4) было больше, чем количество кислорода, требуемое для ожидаемой реакции обезуглероживания, т.е. кислород подавался сверх его количества, расходуемого в реакции обезуглероживания.
(-)
(Пример G: сравнение величин содержания примесных элементов)
Как подробно разъясняется ниже, содержание примесных элементов в слитках нержавеющей стали ультравысокой чистоты двух видов (Fe-20Ni-25Cr-0.2Ti и Fe-35Ni-25Cr-0.2Ti), приготовленных комбинированием способов получения слитков сплава в соответствии с первого по третий вариантами осуществления, сравнивали с содержанием примесных элементов в обычных высокочистых слитках. Детали будут пояснены ниже.
Первоначально были получены первичные слитки Fe-20Ni-25Cr-0.2Ti и Fe-35Ni-25Cr-0.2Ti, соответственно, посредством выполнения восстановительного рафинирования (CCIM восстановительного рафинирования) с добавлением рафинирующего агента, которое удовлетворяло условиям с (1) по (4), изложенным в первом варианте осуществления, при использовании исходных материалов сплава в форме низкоуглеродистой стали (конвертерной стали) и феррохромного материала, имеющих высокое содержание примесных элементов, и применении индукционной плавильной печи 1 с холодным тиглем, имеющей водоохлаждаемый медный тигель 3 ⌀220 мм. Затем, при использовании этих первичных слитков в качестве исходных материалов сплава получали вторичные слитки посредством выполнения вакуумного окислительного рафинирования (окислительного рафинирования CCIM), в котором добавляли рафинирующий агент, который удовлетворял условиям с (5) по (8), изложенным во втором варианте осуществления, при применении индукционной плавильной печи 1 с холодным тиглем. Затем, при использовании этих вторичных слитков в качестве исходных материалов сплава получали конечные слитки посредством вакуумного окислительного рафинирования (окислительного рафинирования EB), которое удовлетворяло условиям с (9) по (11), изложенным в третьем варианте осуществления, в электроннолучевой плавильной печи 11 с холодным подом, имеющей водоохлаждаемый блюдцеобразный резервуар 9, изготовленный из меди. Величины содержания примесных элементов в конечных слитках определяли в соответствии с обычными методами химического анализа для сталей и в соответствии с масс-спектрометрией с тлеющим разрядом (GD-MS), подходящей для микроанализа. При применении методов химического анализа все слитки обнаружили [P]<2 млн-1 (равно или ниже предельной величины аналитического обнаружения), и [P]=1,54 млн-1 и [P]=0,65 млн-1 анализом масс-спектрометрией с тлеющим разрядом (GD-MS). Величины содержания других примесных элементов (величин, полученных анализом), определенные масс-спектрометрией с тлеющим разрядом (GD-MS), представлены в Таблице 1. Для сравнения в Таблице 1 представлены также результаты измерений содержания примесных элементов в коммерчески доступной нержавеющей стали (SUS304ULC) и обычном высокочистом слитке (полученном вакуумной индукционной плавкой - с электронным пучком (VIM-EB) с применением электролитического железа, электролитического никеля и металлического хрома в качестве исходных материалов для плавки). Таблица 1 показывает, что слиток повышенной чистоты (слиток, свободный от примесей) может быть получен комбинированием способов получения слитков сплава в соответствии с первого по третий вариантами осуществления. А именно, рафинирование в первом варианте осуществления очень эффективно для удаления P и S, а также для удаления следовых и случайных элементов, таких как Sn, Sb, Pb и т.п. Из Таблицы 1 очевидна повышенная чистота по сравнению с обычным высокочистым слитком. Хотя содержание азота не могло быть выявлено масс-спектрометрией с тлеющим разрядом (GD-MS), методы химического анализа показали, что азот был удален до величины содержания менее чем 5 млн-1, т.е. [N]<5 млн-1, что является пределом аналитического обнаружения, во всех вышеуказанных конечных слитках.
При этом, GD-MS, относящаяся к методу анализа, известному как масс-спектрометрия с тлеющим разрядом, является методом анализа, который предоставляет возможность микроанализа, например, металлических элементов, полупроводниковых элементов и диэлектрических элементов вплоть до уровня примерно 0,01 млн-1. Например, анализ масс-спектрометрией с тлеющим разрядом (GD-MS) используется для микроанализа полупроводниковых материалов и т.п.
Как подробно описано выше, особенностью данного изобретения является способ получения слитка сплава, включающий: стадию загрузки исходного материала сплава в холодный тигель в индукционной плавильной печи с холодным тиглем и образования ванны расплава исходного материала сплава индукционным нагревом в атмосфере инертного газа; стадию продолжения индукционного нагрева и добавления первого рафинирующего агента к ванне расплава и затем уменьшения содержания по меньшей мере фосфора из числа примесных элементов, присутствующих в ванне расплава; и стадию формирования слитка сплава посредством затвердевания расплава, содержание фосфора в котором было уменьшено, при этом первый рафинирующий агент представляет собой смесь металлического кальция и флюса, содержащего галогенид кальция; флюс содержит фторид кальция и по меньшей мере один компонент из оксида кальция и хлорида кальция при доле общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция в интервале от 5 до 30 масс.%; и доля массы металлического кальция в первом рафинирующем агенте по отношению к массе ванны расплава перед добавлением первого рафинирующего агента составляет 0,4 масс.% или более.
Вышеуказанный способ предоставляет возможность получения в практическом масштабе слитков сплава, имеющих массу, например, 10 кг или более, и имеющих чрезвычайно низкое содержание фосфора (P).
В вышеуказанном способе получения, предпочтительно, доля массы флюса в первом рафинирующем агенте по отношению к массе ванны расплава перед добавлением первого рафинирующего агента равна или более доле массы металлического кальция в первом рафинирующем агенте по отношению к массе ванны расплава.
Потери на испарение металлического кальция существенно уменьшены благодаря вышеуказанному способу. В результате, это обеспечивает получение слитка сплава с еще более низким содержанием фосфора (P).
В вышеуказанном способе получения, предпочтительно, ванна расплава выдерживается при продолжающемся индукционном нагреве от момента времени, при котором первый рафинирующий агент добавляется к ванне расплава, вплоть до заданного момента времени между t1 и t2, на стадии уменьшения содержания фосфора. t1 обозначает момент времени, к которому прошла половина времени до плавления всего первого рафинирующего агента от момента времени, при котором первый рафинирующий агент добавляется к ванне расплава, и t2 обозначает момент времени, к которому прошло время, которое требуется для испарения половины металлического кальция в первом рафинирующем агенте, от момента времени, при котором первый рафинирующий агент добавляется к ванне расплава.
Вышеуказанный способ предоставляет возможность установления количественным образом подходящего времени выдержки расплава. В результате могут быть достигнуты еще более высокая степень дефосфоризации и более высокая степень деазотирования.
В вышеуказанном способе получения, предпочтительно, процесс от стадии образования ванны расплава вплоть до стадии формирования слитка сплава выполняется по меньшей мере один раз после стадии формирования слитка сплава, посредством применения слитка сплава в качестве исходного материала сплава.
Способ предоставляет возможность получения, в практическом масштабе, слитков сплава, имеющих еще более низкие величины содержания фосфора (P) и азота (N).
Вышеуказанный способ получения, предпочтительно, также включает: стадию загрузки в холодный тигель слитка, образованного затвердеванием, в качестве первичного слитка и образование ванны расплава первичного слитка из первичного слитка посредством индукционного нагрева в атмосфере инертного газа; стадию продолжения индукционного нагрева и добавления второго рафинирующего агента к ванне расплава первичного слитка, и последующего уменьшения содержания по меньшей мере углерода и кальция из числа примесных элементов, присутствующих в ванне расплава; и стадию формирования слитка сплава посредством отверждения расплава, содержание углерода и кальция в котором было уменьшено, при этом второй рафинирующий агент представляет собой смесь флюса и первого оксида, содержащего один, два или более видов оксидов основного составного элемента в первичном слитке; масса первого оксида во втором рафинирующем агенте находится в интервале от 0,2 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить по меньшей мере углерод и кальций из числа примесных элементов, присутствующих в ванне расплава первичного слитка; и доля массы флюса во втором рафинирующем агенте по отношению к массе ванны расплава первичного слитка перед добавлением второго рафинирующего агента находится в интервале от 0,5 до 5 масс.%.
Способ предоставляет возможность выполнения надежным образом рафинирования с удалением углерода (C) и кальция (Ca) в качестве примесных элементов и обеспечивает поэтому получение в практическом масштабе слитков, имеющих чрезвычайно низкое содержание углерода, кальция и фосфора.
В этом случае, предпочтительно, второй рафинирующий агент добавляется к ванне расплава первичного слитка, и после этого ванна расплава первичного слитка выдерживается при продолжающемся индукционном нагреве в течение 15 минут или более в вакуумированном состоянии, образованном откачиванием инертного газа во внешнюю атмосферу, на стадии уменьшения содержания углерода и кальция. Это позволяет также уменьшить содержание оксида в расплаве.
Вышеуказанный способ получения, предпочтительно, также включает: стадию загрузки в холодный тигель, в качестве вторичного слитка, слитка сплава, образованного затвердеванием расплава, содержание углерода и кальция в котором было уменьшено, и образования ванны расплава вторичного слитка из вторичного слитка индукционным нагревом в атмосфере инертного газа; стадию продолжения индукционного нагрева и добавления компонента сплава на базе раскисляющего элемента в ванну расплава вторичного слитка, чтобы, тем самым, образовать сплав; и стадию формирования слитка сплава посредством отверждения расплава, который образует сплав.
Вышеуказанный способ получения, предпочтительно, также включает: стадию подачи в электроннолучевую плавильную печь с холодным подом слитка сплава, образованного затвердеванием, в качестве электрода из исходного материала, и облучения электрода из исходного материала электронным пучком при давлении атмосферы ниже чем 5×10-4 мбар, и последующего образования ванны расплава из исходного материала для электрода на холодном поде в электроннолучевой плавильной печи с холодным подом; стадию добавления третьего рафинирующего агента к ванне расплава исходного материала для электрода и последующего уменьшения содержания углерода в качестве примесного элемента, присутствующего в ванне расплава; и стадию формирования слитка сплава посредством отверждения расплава, содержание углерода в котором было уменьшено, при этом третий рафинирующий агент представляет собой второй оксид, содержащий один, два или более видов из числа оксидов основного составного элемента в исходном материале для электрода; и масса третьего рафинирующего агента составляет от 1 до 4 кратное превышение по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава исходного материала для электрода.
Способ предоставляет возможность выполнения надежным образом рафинирования с удалением углерода (C) и кислорода (O) в качестве примесных элементов и обеспечивает поэтому получение в практическом масштабе слитков, имеющих чрезвычайно низкое содержание углерода, кислорода и фосфора.
Другой особенностью данного изобретения является способ получения слитка сплава, включающий: стадию загрузки исходного материала сплава в холодный тигель в индукционной плавильной печи с холодным тиглем и образования ванны расплава исходного материала сплава индукционным нагревом в атмосфере инертного газа; стадию продолжения индукционного нагрева и добавления второго рафинирующего агента к ванне расплава и после этого продолжения индукционного нагрева в течение 15 минут или более в вакуумированном состоянии, образованном откачиванием инертного газа во внешнюю атмосферу, и затем уменьшения содержания по меньшей мере углерода и кальция из числа примесных элементов, присутствующих в ванне расплава; и стадию формирования слитка сплава посредством затвердевания расплава, содержание углерода и кальция в котором было уменьшено, при этом второй рафинирующий агент представляет собой смесь флюса и первого оксида, содержащего один, два или более видов из числа оксидов основного составного элемента в исходном материале сплава; флюс содержит фторид кальция и по меньшей мере один компонент из оксида кальция и хлорида кальция при доле общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция в интервале от 5 до 30 масс.%; и масса первого оксида во втором рафинирующем агенте находится в интервале от 0,2 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить по меньшей мере углерод и кальций из числа примесных элементов, присутствующих в ванне расплава; и доля массы флюса во втором рафинирующем агенте по отношению к массе ванны расплава перед добавлением второго рафинирующего агента находится в интервале от 0,5 до 5 масс.%.
Вышеуказанный способ предоставляет возможность получения в практическом масштабе слитков сплава, имеющих массу, например, 10 кг или более, и имеющих чрезвычайно низкое содержание углерода (C) и кальция (Ca).
В вышеуказанном способе получения, предпочтительно, флюс представляет собой смесь фторида кальция и оксида кальция, при доле массы оксида кальция по отношению к массе фторида кальция в интервале от 5 до 30 масс.%, или смесь фторида кальция, оксида кальция и хлорида кальция, при доле общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция в интервале от 5 до 30 масс.%; масса первого оксида во втором рафинирующем агенте находится в интервале от 2 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить по меньшей мере углерод, кальций, алюминий и кремний из числа примесных элементов, присутствующих в ванне расплава; и доля массы флюса во втором рафинирующем агенте по отношению к массе ванны расплава перед добавлением второго рафинирующего агента находится в интервале от 3 до 5 масс.%.
Вышеуказанный способ предоставляет возможность выполнения, более надежным образом, рафинирования с удалением углерода (C) и кальция (Ca), в качестве примесных элементов, и обеспечивает также удаление кремния (Si) в качестве примесного элемента. Поэтому, способ обеспечивает получение в практическом масштабе слитков, имеющих еще более низкое содержание углерода, кальция и фосфора и имеющих низкое содержание кремния.
Вышеуказанный способ получения, предпочтительно, также включает: стадию загрузки в холодный тигель, в качестве первичного слитка, слитка сплава, образованного затвердеванием расплава, содержание углерода и кальция в котором было уменьшено, и образования ванны расплава первичного слитка из первичного слитка индукционным нагревом в атмосфере инертного газа; стадию продолжения индукционного нагрева и добавления компонента сплава на базе раскисляющего элемента в ванну расплава первичного слитка, чтобы, тем самым, образовать сплав; и стадию формирования слитка сплава посредством отверждения расплава, который образует сплав.
Вышеуказанный способ получения, предпочтительно, также включает: стадию подачи в электроннолучевую плавильную печь с холодным подом слитка сплава, образованного затвердеванием, в качестве электрода из исходного материала, и облучения электрода из исходного материала электронным пучком при давлении атмосферы ниже чем 5×10-4 мбар, и последующего образования ванны расплава из исходного материала для электрода на холодном поде в электроннолучевой плавильной печи с холодным подом; стадию добавления третьего рафинирующего агента к ванне расплава исходного материала для электрода и последующего уменьшения содержания углерода в качестве примесного элемента, присутствующего в ванне расплава; и стадию формирования слитка сплава посредством отверждения расплава, содержание углерода в котором было уменьшено, при этом третий рафинирующий агент представляет собой второй оксид, содержащий один, два или более видов из числа оксидов основного составного элемента в исходном материале для электрода; и масса третьего рафинирующего агента составляет от 1 до 4 кратное превышение по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава исходного материала для электрода.
Способ предоставляет возможность выполнения надежным образом рафинирования с удалением углерода (C) и кислорода (O) в качестве примесных элементов и обеспечивает поэтому получение в практическом масштабе слитков, имеющих чрезвычайно низкое содержание углерода, кислорода и кальция.
Еще одной особенностью данного изобретения является способ получения слитка сплава, включающий: стадию подачи электрода из исходного материала в электроннолучевую плавильную печь с холодным подом и облучения электронным пучком электрода из исходного материала при давлении атмосферы ниже чем 5×10-4 мбар и последующего образования ванны расплава на холодном поде в электроннолучевой плавильной печи с холодным подом; стадию добавления третьего рафинирующего агента к ванне расплава и затем уменьшения содержания углерода, в качестве примесного элемента, присутствующего в ванне расплава; и стадию формирования слитка сплава посредством затвердевания расплава, содержание углерода в котором было уменьшено, при этом третий рафинирующий агент представляет собой второй оксид, содержащий один, два или более видов из числа оксидов основного составного элемента в исходном материале для электрода; и масса третьего рафинирующего агента находится в интервале от 1 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава.
Способ предоставляет возможность выполнения надежным образом рафинирования с удалением углерода (C) и кислорода (O) в качестве примесных элементов. Поэтому способ обеспечивает получение в практическом масштабе слитков, имеющих ультранизкое содержание углерода (например, [C]<10 млн-1) и ультранизкое содержание кислорода (например, [O]<10 млн-1), которые трудно получить, когда применяется индукционная плавильная печь с холодным тиглем.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Способ получения слитка сплава по данному изобретению, при его применении, делает возможным получение, в практическом масштабе, слитков сплава ультравысокой чистоты, в которых масса конечного слитка составляет, например, 10 кг или более. Поэтому, данное изобретение расширяет область сплавов ультравысокой чистоты.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПЕРЕРАБОТКИ РАДИОКТИВНЫХ ОТХОДОВ, ОБРАЗУЮЩИХСЯ В ПРОЦЕССЕ РАЗРУШЕНИЯ ОБЛУЧЕННЫХ ТЕПЛОВЫДЕЛЯЮЩИХ СБОРОК РЕАКТОРОВ НА БЫСТРЫХ НЕЙТРОНАХ, МЕТОДОМ ИНДУКЦИОННОГО ШЛАКОВОГО ПЕРЕПЛАВА В ХОЛОДНОМ ТИГЛЕ | 2018 |
|
RU2765028C1 |
| СЛИТОК ИЗ РАДИОАКТИВНЫХ МЕТАЛЛИЧЕСКИХ ОТХОДОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1998 |
|
RU2145126C1 |
| Способ получения электродов из сплавов на основе алюминида титана | 2016 |
|
RU2630157C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУПЕРСПЛАВОВ НА ОСНОВЕ НИКЕЛЯ, ЛЕГИРОВАННЫХ РЕДКОЗЕМЕЛЬНЫМИ МЕТАЛЛАМИ | 2014 |
|
RU2572117C1 |
| МЕТАЛЛИЧЕСКИЕ КОМПОЗИЦИОННЫЕ МАТЕРИАЛЫ НА ОСНОВЕ АЛЮМИНИЕВЫХ СПЛАВОВ, АРМИРОВАННЫХ КЕРАМИЧЕСКИМИ ЧАСТИЦАМИ TIB | 1996 |
|
RU2159823C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЛИГАТУРЫ НИКЕЛЬ-РЕДКОЗЕМЕЛЬНЫЙ МЕТАЛЛ | 2014 |
|
RU2556176C1 |
| СПОСОБ ПЕРЕРАБОТКИ МЕТАЛЛИЧЕСКИХ ОТХОДОВ ПРОИЗВОДСТВА ЛИТЫХ ПОСТОЯННЫХ МАГНИТОВ | 2005 |
|
RU2323268C2 |
| ФЛЮС ДЛЯ ПЛАВКИ МЕДНЫХ СПЛАВОВ | 1990 |
|
SU1795662A1 |
| ПЛАВИЛЬНАЯ ПЕЧЬ С ХОЛОДНЫМ ПОДОМ | 2009 |
|
RU2413017C2 |
| СПОСОБ РАФИНИРОВАНИЯ МЕДИ И СПЛАВОВ НА МЕДНОЙ ОСНОВЕ | 2000 |
|
RU2185455C1 |
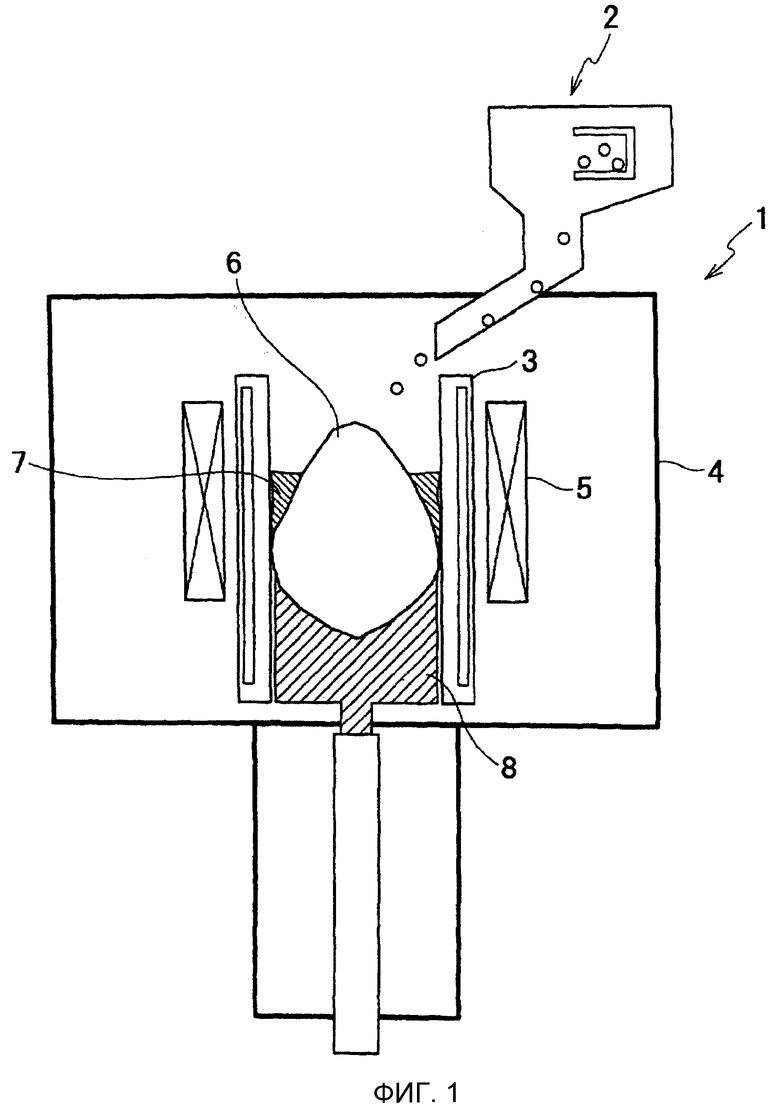
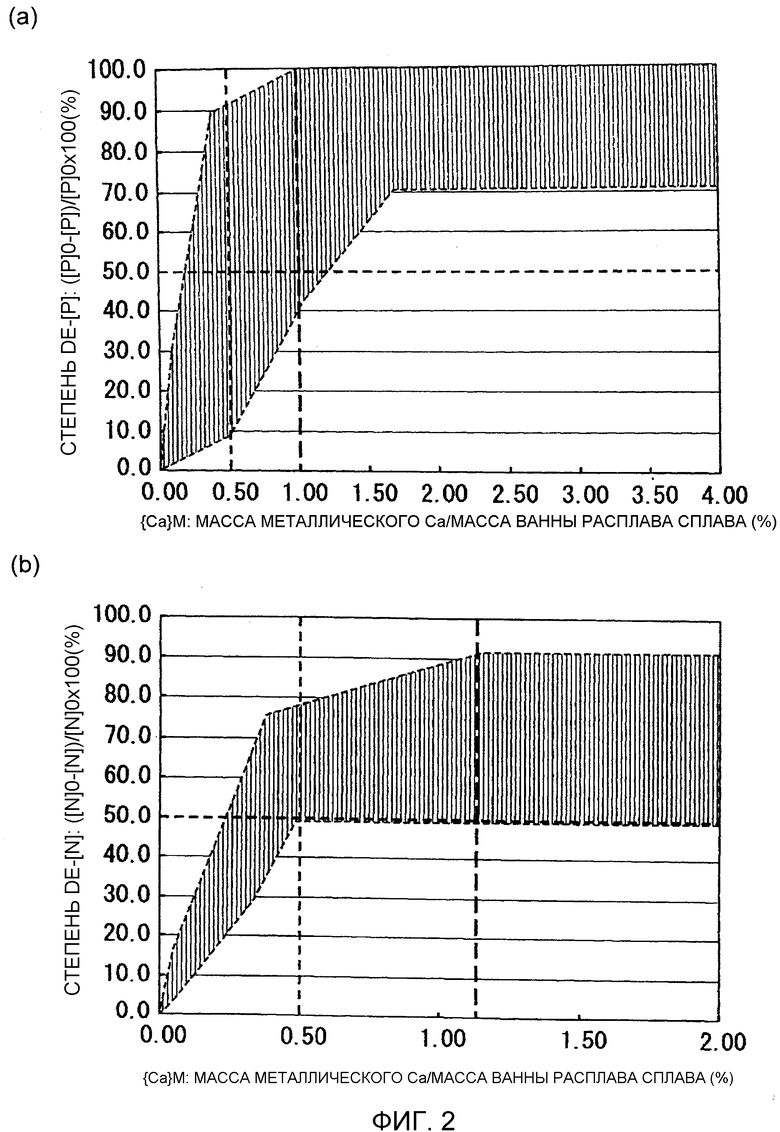
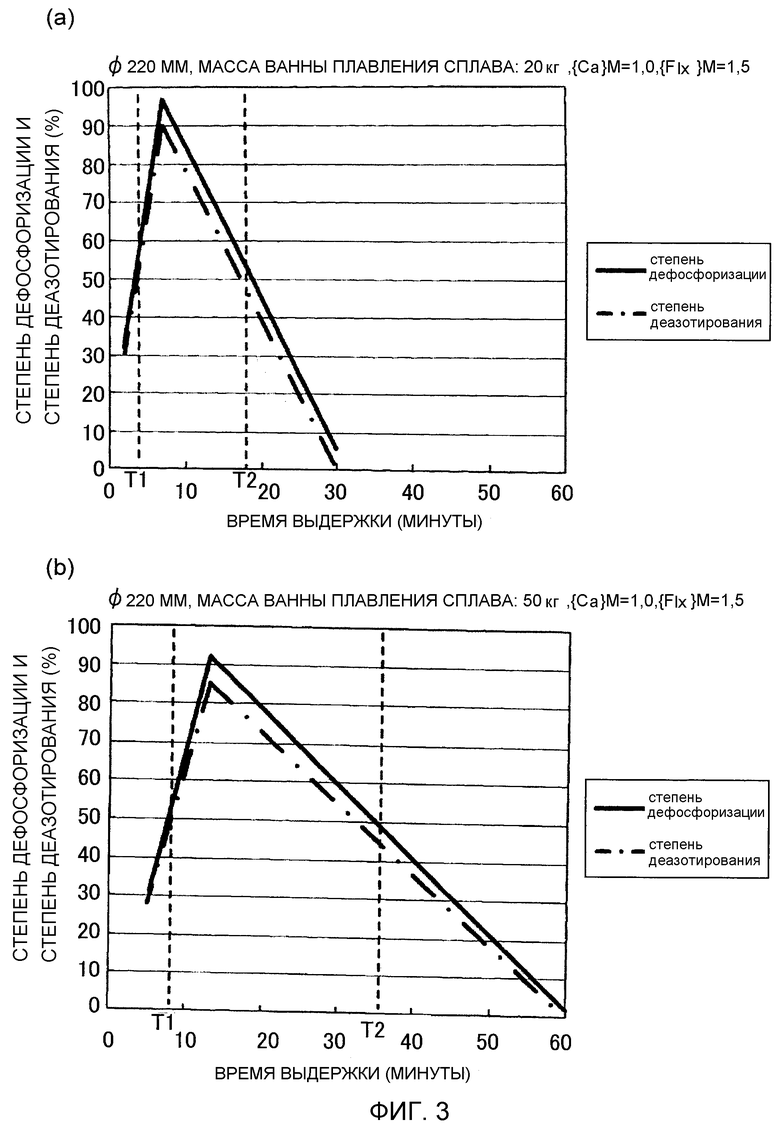
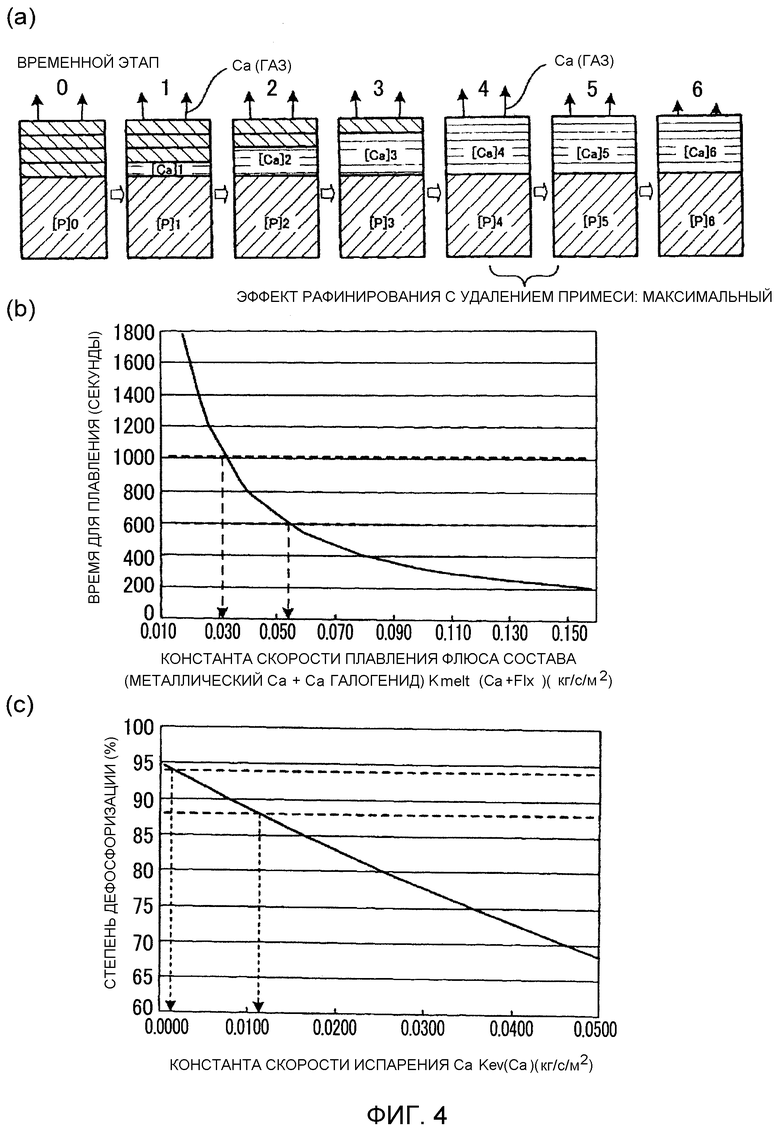
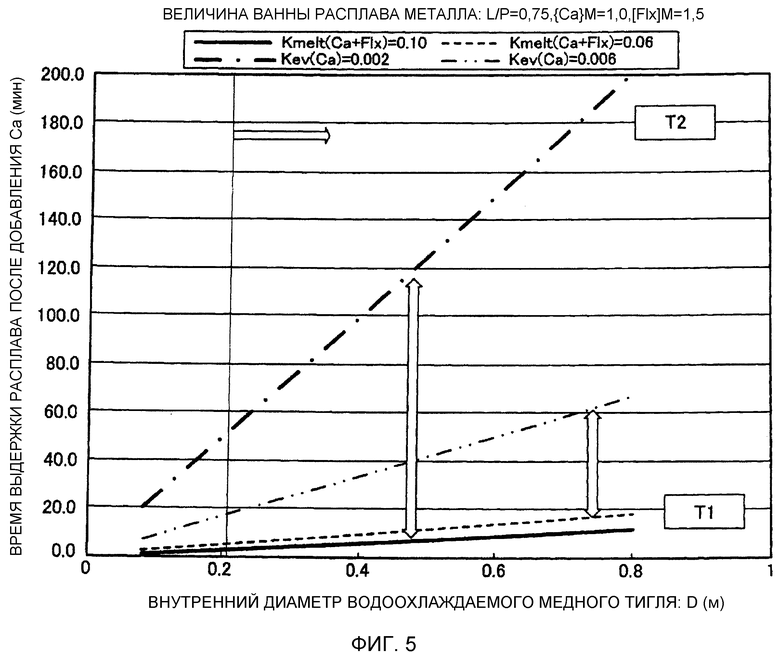
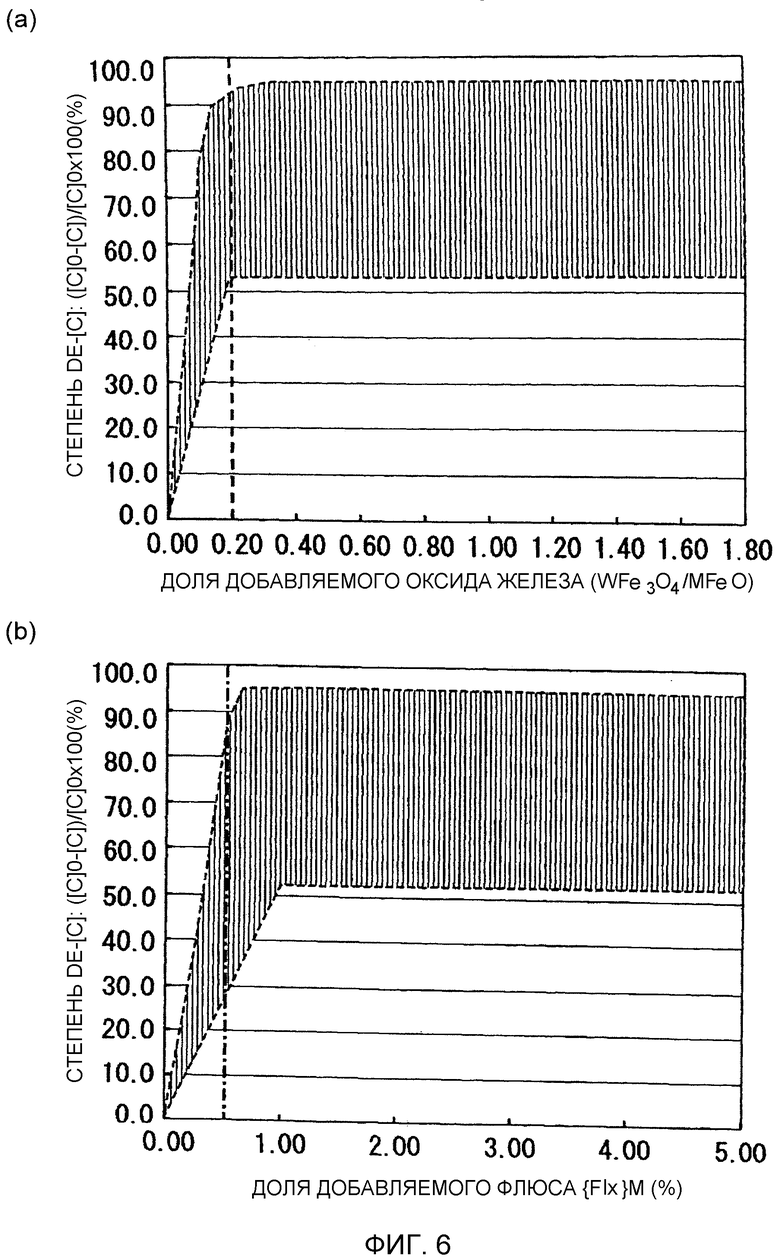
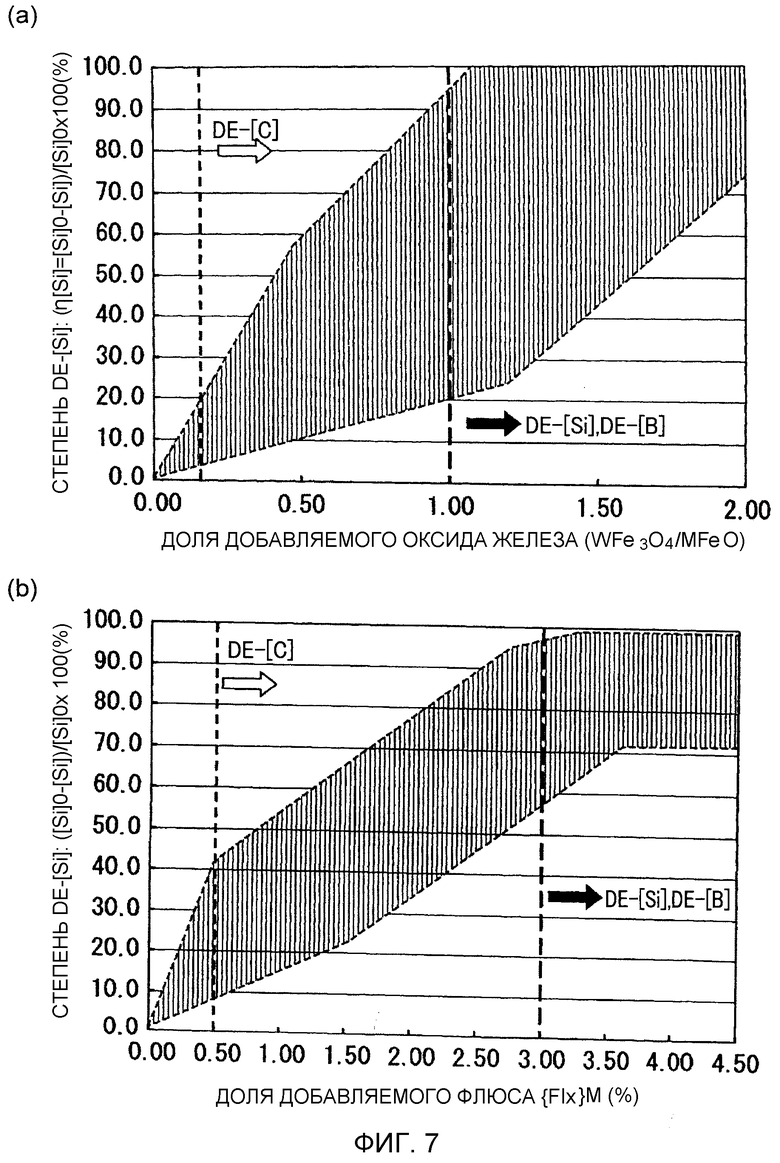
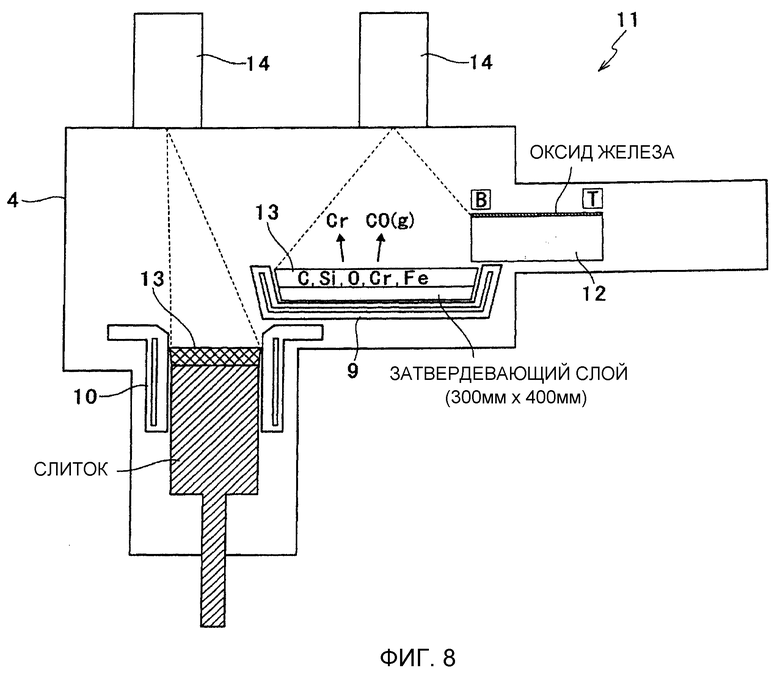
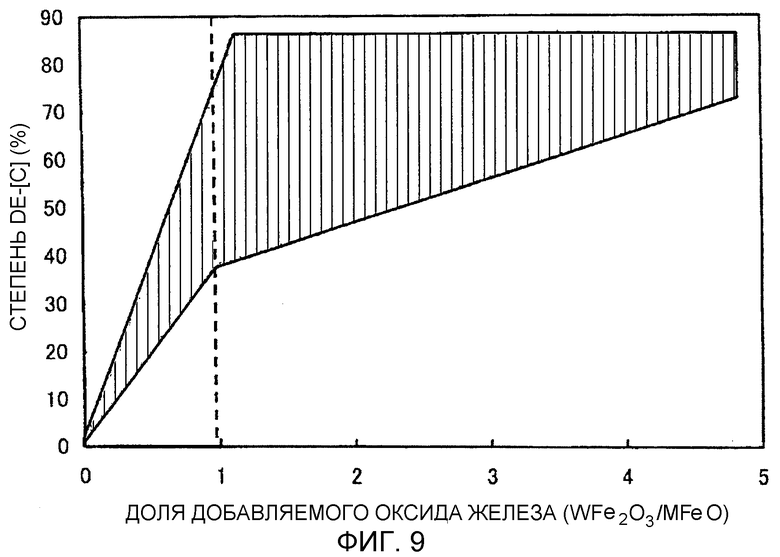
Изобретение относится к специальной электрометаллургии и может быть использовано для получения слитка сплава высокой чистоты. Способ включает: стадию загрузки исходного материала сплава в холодный тигель в индукционной плавильной печи с холодным тиглем и образование ванны расплава исходного материала сплава индукционным нагревом в атмосфере инертного газа, стадию продолжения индукционного нагрева и добавления первого рафинирующего агента к ванне расплава, и затем уменьшения содержания по меньшей мере фосфора из числа примесных элементов, присутствующих в ванне расплава, и стадию формирования слитка сплава посредством отверждения расплава, содержание фосфора в котором было уменьшено. Первый рафинирующий агент представляет собой смесь металлического кальция и флюса, при этом флюс содержит фторид кальция и по меньшей мере один компонент из оксида кальция и хлорида кальция. Массовая доля суммарного содержания оксида кальция и хлорида кальция по отношению к фториду кальция находится в интервале от 5 до 30 масс.%, и массовая доля металлического кальция по отношению к ванне расплава составляет 0,4 масс.% или более. Слиток сплава также может быть получен в электроннолучевой печи с холодным подом. Изобретение позволяет получить слиток сплава высокой чистоты с минимальным содержанием фосфора за счет использования рафинирующего агента специального состава. 3 н. и 10 з.п. ф-лы, 7 табл., 9 ил.
1. Способ получения слитка сплава, включающий:
стадию загрузки исходного материала сплава в холодный тигель в индукционной плавильной печи с холодным тиглем и образования ванны расплава исходного материала сплава индукционным нагревом в атмосфере инертного газа;
стадию продолжения индукционного нагрева и добавления первого рафинирующего агента к ванне расплава и затем уменьшения содержания по меньшей мере фосфора из числа примесных элементов, присутствующих в ванне расплава; и
стадию формирования слитка сплава посредством затвердевания расплава, содержание фосфора в котором было уменьшено, в котором первый рафинирующий агент представляет собой смесь металлического кальция и флюса, содержащего галогенид кальция;
флюс содержит галогенид кальция в виде фторида кальция и по меньшей мере один компонент из оксида кальция и хлорида кальция при доле общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция в интервале от 5 до 30 мас.%; и
доля массы металлического кальция в первом рафинирующем агенте по отношению к массе ванны расплава перед добавлением первого рафинирующего агента составляет 0,4 мас.% или более.
2. Способ по п.1, в котором
доля массы флюса в первом рафинирующем агенте по отношению к массе ванны расплава перед добавлением первого рафинирующего агента равна или более чем доля массы металлического кальция в первом рафинирующем агенте по отношению к массе ванны расплава.
3. Способ по п.1, в котором
ванна расплава выдерживается при продолжающемся индукционном нагреве от момента времени, при котором первый рафинирующий агент добавляется к ванне расплава, вплоть до заданного момента времени между t1 и t2, на стадии уменьшения содержания фосфора,
где t1 обозначает момент времени, к которому прошла половина времени до плавления всего первого рафинирующего агента от момента времени, при котором первый рафинирующий агент добавляется к ванне расплава, и t2 обозначает момент времени, к которому прошло время, которое требуется для испарения половины металлического кальция в первом рафинирующем агенте, от момента времени, при котором первый рафинирующий агент добавляется к ванне расплава.
4. Способ по п.1, в котором
процесс от стадии образования ванны расплава вплоть до стадии формирования слитка сплава выполняется по меньшей мере один раз после стадии формирования слитка сплава, посредством использования слитка сплава в качестве исходного материала сплава.
5. Способ по п.1, дополнительно включающий:
стадию загрузки в холодный тигель слитка, сформированного посредством затвердевания расплава, в качестве первичного слитка и образование ванны расплава из первичного слитка посредством индукционного нагрева в атмосфере инертного газа;
стадию продолжения индукционного нагрева и добавления второго рафинирующего агента к ванне расплава из первичного слитка, и последующего уменьшения содержания по меньшей мере углерода и кальция из числа примесных элементов, присутствующих в ванне расплава; и
стадию формирования слитка сплава посредством отверждения расплава, содержание углерода и кальция в котором было уменьшено, в котором
второй рафинирующий агент представляет собой смесь флюса и первого оксида, содержащего один, два или более видов оксидов основного составного элемента в первичном слитке;
масса первого оксида во втором рафинирующем агенте находится в интервале от 0,2 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить по меньшей мере углерод и кальций из числа примесных элементов, присутствующих в ванне расплава первичного слитка; и
доля массы флюса во втором рафинирующем агенте по отношению к массе ванны расплава первичного слитка перед добавлением второго рафинирующего агента находится в интервале от 0,5 до 5 мас.%.
6. Способ по п.5, в котором
второй рафинирующий агент добавляют к ванне расплава из первичного слитка, и после этого ванну расплава из первичного слитка выдерживают при продолжающемся индукционном нагреве в течение 15 мин или более в вакуумированном состоянии, образованном откачиванием инертного газа во внешнюю атмосферу, на стадии уменьшения содержания углерода и кальция.
7. Способ по п.6, дополнительно включающий:
стадию загрузки в холодный тигель, в качестве вторичного слитка, слитка сплава, сформированного посредством затвердевания расплава, содержание углерода и кальция в котором было уменьшено, и образования ванны расплава вторичного слитка из вторичного слитка индукционным нагревом в атмосфере инертного газа;
стадию продолжения индукционного нагрева и добавления компонента сплава на базе раскисляющего элемента в ванну расплава из вторичного слитка, чтобы образовать сплав; и
стадию формирования слитка сплава посредством отверждения расплава, который образует сплав.
8. Способ по п.1, дополнительно включающий:
стадию подачи в электронно-лучевую плавильную печь с холодным подом упомянутого слитка сплава, сформированного посредством затвердевания расплава, в качестве электрода из исходного материала, и облучения электрода из исходного материала электронным пучком при давлении атмосферы ниже, чем 5·10-4 мбар, и последующего образования ванны расплава из исходного материала для электрода на холодном поде в электронно-лучевой плавильной печи с холодным подом;
стадию добавления третьего рафинирующего агента к ванне расплава исходного материала для электрода и последующего уменьшения содержания углерода в качестве примесного элемента, присутствующего в ванне расплава; и
стадию формирования слитка сплава посредством отверждения расплава, содержание углерода в котором было уменьшено, в котором
третий рафинирующий агент представляет собой второй оксид, содержащий один, два или более видов из числа оксидов основного составного элемента в исходном материале для электрода; и
масса третьего рафинирующего агента составляет от 1 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава исходного материала для электрода.
9. Способ получения слитка сплава, включающий:
стадию загрузки исходного материала сплава в холодный тигель в индукционной плавильной печи с холодным тиглем и образования ванны расплава исходного материала сплава индукционным нагревом в атмосфере инертного газа;
стадию продолжения индукционного нагрева и добавления второго рафинирующего агента к ванне расплава и после этого продолжения индукционного нагрева в течение 15 мин или более в вакуумированном состоянии, образованном откачиванием инертного газа во внешнюю атмосферу, и затем уменьшения содержания по меньшей мере углерода и кальция из числа примесных элементов, присутствующих в ванне расплава; и
стадию формирования слитка сплава посредством отверждения расплава, содержание углерода и кальция в котором было уменьшено, в котором второй рафинирующий агент представляет собой смесь флюса и первого оксида, содержащего один, два или более видов из числа оксидов основного составного элемента в исходном материале сплава;
флюс содержит фторид кальция и по меньшей мере один компонент из оксида кальция и хлорида кальция при доле общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция в интервале от 5 до 30 мас.%; и
масса первого оксида во втором рафинирующем агенте находится в интервале от 0,2 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить по меньшей мере углерод и кальций из числа примесных элементов, присутствующих в ванне расплава; и
доля массы флюса во втором рафинирующем агенте по отношению к массе ванны расплава перед добавлением второго рафинирующего агента находится в интервале от 0,5 до 5 мас.%.
10. Способ по п.9, в котором
флюс представляет собой смесь фторида кальция и оксида кальция, при доле массы оксида кальция по отношению к массе фторида кальция в интервале от 5 до 30 мас.%, или смесь фторида кальция, оксида кальция и хлорида кальция, при доле общей массы оксида кальция и хлорида кальция по отношению к массе фторида кальция в интервале от 5 до 30 мас.%;
масса первого оксида во втором рафинирующем агенте находится в интервале от 2 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить по меньшей мере углерод, кальций, алюминий и кремний из числа примесных элементов, присутствующих в ванне расплава; и
доля массы флюса во втором рафинирующем агенте по отношению к массе ванны расплава перед добавлением второго рафинирующего агента находится в интервале от 3 до 5 мас.%.
11. Способ по п.9, дополнительно включающий:
стадию загрузки в холодный тигель, в качестве первичного слитка, слитка сплава, сформированного посредством затвердевания расплава, содержание углерода и кальция в котором было уменьшено, и образования ванны расплава из первичного слитка индукционным нагревом в атмосфере инертного газа;
стадию продолжения индукционного нагрева и добавления компонента сплава на базе раскисляющего элемента в ванну расплава из первичного слитка, чтобы, тем самым, образовать сплав; и
стадию формирования слитка сплава посредством отверждения расплава, который образует сплав.
12. Способ по п.9, дополнительно включающий:
стадию подачи в электроннолучевую плавильную печь с холодным подом упомянутого слитка сплава, сформированного посредством затвердевания расплава, в качестве электрода из исходного материала, и облучения электрода из исходного материала электронным пучком при давлении атмосферы ниже чем 5×10-4 мбар, и последующего образования ванны расплава из исходного материала для электрода на холодном поде в электроннолучевой плавильной печи с холодным подом;
стадию добавления третьего рафинирующего агента к ванне расплава исходного материала для электрода и последующего уменьшения содержания углерода в качестве примесного элемента, присутствующего в ванне расплава; и
стадию формирования слитка сплава посредством отверждения расплава, содержание углерода в котором было уменьшено, в котором
третий рафинирующий агент представляет собой второй оксид, содержащий один, два или более видов из числа оксидов основного составного элемента в исходном материале для электрода; и
масса третьего рафинирующего агента составляет от 1 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава исходного материала для электрода.
13. Способ получения слитка сплава, включающий:
стадию подачи исходного материала в виде электрода в электроннолучевую плавильную печь с холодным подом и облучения электронным пучком электрода из исходного материала при давлении атмосферы ниже чем 5·10-4 мбар и последующего образования ванны расплава на холодном поде в электроннолучевой плавильной печи с холодным подом;
стадию добавления третьего рафинирующего агента к ванне расплава и затем уменьшения содержания углерода в качестве примесного элемента, присутствующего в ванне расплава; и
стадию формирования слитка сплава посредством отверждения расплава, содержание углерода в котором было уменьшено, в котором
третий рафинирующий агент представляет собой второй оксид, содержащий один, два или более видов из числа оксидов основного составного элемента в исходном материале для электрода; и
масса третьего рафинирующего агента составляет от 1 до 4 кратного превышения по отношению к расчетной массе, которая рассчитана для того, чтобы полностью окислить углерод из числа примесных элементов, присутствующих в ванне расплава.
| Кипятильник для воды | 1921 |
|
SU5A1 |
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| СПОСОБ ИНДУКЦИОННОЙ ПЛАВКИ ЛИТЬЯ МЕТАЛЛОВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2003 |
|
RU2319752C2 |
| СПОСОБ ПЛАВКИ И ЛИТЬЯ МЕТАЛЛА | 1999 |
|
RU2209842C2 |

