Изобретение относится к нетоксичному полисилоксановому материалу (ПСН-материалу), при необходимости «созревшему» полисилоксановому материалу (сПСН-материалу), который предпочтительным образом образован как один из нескольких различных полисилоксановых материалов (ПСН-материалов). Такой сПСН (с означает созревший), согласно изобретению, может быть сформован, например, в биорассасывающиеся и/или биоактивные волокна в качестве одного из ПСН-материалов, и затем переработан далее в нетканый материал в качестве других ПСН-материалов. Представленное изобретение относится, кроме того, к способу получения при необходимости созревшего ПСН-материала, биорассасывающихся и/или биоактивных ПСН-материалов и применению этих материалов.
Существуют многочисленные попытки создать биорассасывающиеся материалы для различных применений в медицине и медицинской технике, а также в других технических областях, как, например, в технологии фильтров, биотехнологии или индустрии изоляционных материалов. В этих областях, к тому же, устанавливаются постоянно повышающиеся требования, в частности, к биологической активности и токсикологическим свойствам материалов.
Рассасывающиеся Si-полимеры - это известный уровень техники. В немецком патенте 196 09 551 С1 описаны биодеградирующие и биорассасывающиеся волокнистые структуры. Эти волокна могут быть получены в золь-гель-процессе таким образом, что из прядильной массы вытягивают волокна и их, в случае необходимости, сушат. Прядильная масса содержит одно или несколько частично или полностью гидролитически конденсированных соединений кремния, которые получаются за счет гидролитической конденсации мономеров общей формулы SiX4. Волокна имеют недостаток, состоящий в том, что они при деградации непосредственно после процесса прядения не показывают оптимальных результатов в тесте на цитотоксичность, и частично даже должны быть расценены как цитотоксичные. Такая токсичность непосредственно в использовании в медицине, в медицинской технике, в технологии фильтров, в биотехнологии или в индустрии изоляционных материалов, в частности в области заживления ран или фильтрации клеток из жидкостей тела, совсем нежелательна.
Способ получения волокон согласно немецкому патенту DE 196 09 551 С1 имеет, кроме того, недостаток, состоящий в том, что полученная смесь после удаления растворителя содержит твердое вещество и поэтому должна быть подвергнута фильтрации. Другие жидкие Si-полимеры, которые в некоторых случаях являются токсичными, не могут быть вообще удалены с помощью фильтрации. Кроме того, среди прочего, за счет образования твердой фазы и за счет необходимой стадии фильтрации большая часть Прядильного золя теряется. Согласно способу из немецкого патента DE 196 09 551 С1, во время выдерживания может происходить также образование незначительной доли гелеподобной фазы из высококонденсированных Si-соединений. Это также снижает долю прядильной золь-массы.
Задачей представленного изобретения является необходимости предоставить в распоряжение нетоксичный, биорассасывающийся и/или биоактивный материал, материалы, содержащие этот материал, и способ получения такого нетоксичного материала.
Биоактивность, согласно изобретению, означает положительное взаимодействие между материалом или материалами, с одной стороны, и тканью (например, тканью раны), с другой стороны, с последующей дифференциацией ткани и, как следствие из этого, связь или адгезию ткани вдоль граничной поверхности материал или материалы / ткань (реципиента).
Задача решается с помощью золя, соответственно, коллоидного раствора по пункту 1 формулы изобретения, который, согласно изобретению, обозначается также как ПСН-материал. Подобный коллоидный раствор получается за счет того, что (a) первая реакция гидролиза-конденсации (РГК) максимум одного остатка X одного или нескольких различных Si-соединений формулы (I):

в которой остатки X являются одинаковыми или различными и означают гидрокси, водород, галоген, амино, алкокси, ацилокси, алкилкарбонил и/или алкоксикарбонил и происходят от алкильных остатков, которые, при необходимости представляют собой замещенные линейные, разветвленные или циклические остатки с 1-20 атомами углерода, предпочтительно с 1-10 атомами углерода, и могут быть прерваны атомами кислорода или серы или аминогруппами, проводится при катализе кислыми агентами и начальном значении рН от 0 до ≤7, в присутствии водорастворимого растворителя, в течение промежутка времени от 1 до 192 часов при температуре от 0°°С до 80°С,
(b) вторая РГК материала, полученного в стадии (а), проводится при одновременном удалении растворителя за счет испарения в закрытом аппарате, в котором происходит перемешивание материала и испарение в вакууме между 1 и 1013 мбар и оптимально при непрерывной подаче химически инертного потока газа-носителя, при этом, по меньшей мере, один из параметров процесса - давление, поток газа-носителя и/или температура подгоняется в зависимости от времени, а температура испарения лежит между 30-90°С (30°С и 90°С) и эта стадия проводится до вязкости от 0,5 до 2 Па·с (при 4°С и 10 л/с).
c) указанный ПСН-материал в закрытом аппарате, в течение промежутка времени от нескольких минут до нескольких часов охлаждается, и
d) ПСН-материал, полученный из (с), посредством третьей РГК переводится в созревший полисилоксановый материал -(сПСН)-материал.
Остается отметить, что (не токсичный, биорассасывающийся и/или биоактивный) ПСН- или сПСН-материал согласно изобретению может быть получен, без необходимости включения в способ получения одной или нескольких стадий фильтрации. Это существенное отличие по отношению к способу, который известен из немецкого патента DE 1 96 09 551 С1. При необходимости к стадии (d) подключается четвертая РГК в виде одной из следующих стадий (е1) - (е4), с помощью которых из (сПСН)-материала, полученного в стадии (d), может быть произведен один из ПСН-материалов, таких как волокно-(е1), порошок (е2), монолит (е3) или материал с нанесенным покрытием (е4). В соответствии с этим указанные стадии включают следующие приемы:
(е1) формование ПСН-материала в биорассасывающиеся и/или биоактивные волокна;
(е2) переработка материала из стадии (d) в порошок, посредством того, что полученный сПСН-материал подвергается сушке, в частности лиофилизации, и высушенный сПСН-материал измельчается (размалывался) в порошок;
(е3) заливка сПСН-материала из стадии (d) в форму и его сушка;
(е4) нанесение сПСН-материала из стадии (d) на подлежащее нанесению покрытия тело или погружение его в сПСН-материал.
Особенно предпочтительно, когда сПСН-материал / сПСН-материалы при применении показывает, соответственно показывают, значение рН от 5 до 7, в частности ≥ 6, тем самым он/они обладает/ют приемлемой (физиологической) совместимостью. При рН 5 материал вследствие кислого характера уже не совместим. Так как в стадии (b) испарение происходит до небольшого содержания воды, сила кислоты в почти безводной системе не измеряется в виде определенного значения рН. Напротив, оптимальная буферизация (т.е. добавление подходящего буфера или щелочи) или снижение силы кислоты (например, в случае азотной кислоты за счет удаления/испарения NO или NO2) в (b) должно происходить таким образом, что сПСН-материал, полученный в конечном счете согласно (е), или ПСН-материалы, формованные из него, при разбавлении водой имеет/имеют значение рН от 5 до 7, в частности ≥6.
Для того, чтобы достигнуть этого, предпочтительно снизить силу кислоты в стадии (b) или буферизировать действие кислоты. Если это все же не происходит на стадии (b) или не достигается предпочтительный уровень, можно также осуществить это впоследствии в стадии (с) или (е) или непосредственно перед нанесением ПСН-материалов (например, на кожу или раны). Но регулировка необходимой силы или действия кислоты на стадии (b), согласно изобретению, является однозначно предпочтительным.
Уменьшение воздействия кислоты в одной из стадий (b), (с) или (е) или во время разбавления ПСН-материалов водой может происходить, в частности, с помощью трис(трис(гидроксиметил)-аминометана) в форме свободного основания или соли (например, трис-ацетата, трис-фосфата).
Ниже отдельные стадии вышеуказанных реакций обсуждаются более подробно.
Стадия (а)
На стадии (а) используется остаток X одного или нескольких различных Si-соединений формулы (I):
в которой остатки X являются одинаковыми или различными и означают гидрокси, водород, галоген, амино, алкокси, ацилокси, алкилкарбонил и/или алкоксикарбонил, и происходят от алкильных остатков, которые при необходимости представляют собой замещенные линейные, разветвленные или циклические остатки с 1-20 атомами углерода, предпочтительно с 1-10 атомами углерода, и могут быть прерваны атомами кислорода или серы или аминогруппами.
В предпочтительной форме исполнения согласно изобретению X в формуле (I) представляет собой при необходимости замещенный, линейный, разветвленный и/или циклический алкокси-остаток с 1-20 атомами углерода, предпочтительно с 1-10 атомами углерода. Особенно предпочтительно X в формуле (I) представляет собой при необходимости замещенный, линейный, разветвленный и/или циклический алкокси-остаток с 1-5 атомами углерода. Далее, особенно предпочтительными являются замещенные, но предпочтительно незамещенные линейные и/или разветвленные алкокси-остатки с 2-3 атомами углерода, как, например этокси, н-пропокси и/или изо-пропокси.
Согласно изобретению, в высшей степени предпочтительно применяется тетраэтоксисилан (ТЭОС) в качестве единственного Si-соединения формулы (I) в (первой) РКГ согласно изобретению.
Начальное значение рН устанавливают от 0 до ≤7, предпочтительно 2-3, 5, например, при помощи разбавленной азотной кислоты (например, 1 N, предпочтительно 0,01 N HNO3). Но подходящими являются в основном все кислые смеси и растворы, которые подходят для того, чтобы выработать локально NO или NO2. Это могут быть также кислые смеси и растворы, которые в физиологическом окружении с молекулярным кислородом ферментативно (с помощью нитроксид-синтазы, NOS) вырабатывают монооксид азота (NO), который снова в организме быстро превращается в NO2, или это могут быть также органические нитраты или нитратные эфиры (так называемые NO-донаторы), например, этилнитрат, которые образуют NO с помощью органической нитратредуктазы. Для такого ферментативного высвобождения NO необходимы тиольные группы (цистеин).
Наряду с разбавленной азотной кислотой, согласно изобретению, для установления рН до желаемого значения в области от слабо кислой до кислотности средней силы пригоден также водный или спиртовый (особенно предпочтительно водно-этанольный) раствор физиологически совместимой кислоты (например, лимонной, янтарной, винной, уксусной или аскорбиновой кислоты) и, по меньшей мере, одной незаменимой (например, L-аргинина, особенно предпочтительно: L-валина, L-лейцина, L-изолейцина, L-фенилаланина, L-тироксина, L-метионина, L-лицина или L-триптофана) или заменимой аминокислоты (например, L-глутамина, L-глутаминовой кислоты, L-аспарагина, L-аспарагиновой кислоты, L-цистеина, L-глицина, L-аланина, L-пролина, L-гистидина, L-тирозина) в качестве субстрата NOS.
Если для установления рН используется разбавленная азотная кислота (например, 0,01 N), то она предпочтительно используется в молярном соотношении [Si-соединение(я) формулы (I)]/[азотная кислота] от 110: 1 до 90: 1, особенно предпочтительным образом 100: 1. Выгодно использовать азотную кислоту так, чтобы молярное соотношение [Si-соединение формулы (I) (например, ТЭОС)]/HNO3 составляло примерно 100:1.
Водорастворимым растворителем, который, согласно изобретению, предпочтительно применяется на стадии (а), является этанол или смесь этанол-вода, который имеет задачу растворить Si-соединение(я) формулы (I) или, по меньшей мере, эмульгировать. Если Si-соединением формулы (I) является ТЭОС, то вода не растворяет/эмульгирует Si-соединение формулы (I), и поэтому предпочтительно смешивается с EtOH, выступающим в качестве связующего растворителя. Предпочтительное количество ETOH лежит в интервале от 1 до 1,5 моль/моль ТЭОС и составляет, согласно особенно предпочтительной форме исполнения, 1,26 моль/моль ТЭОС.
В высшей степени предпочтительная реакционная композиция приготавливается в соответствии с далее следующим массовым или мольным соотношениям. В реакционный сосуд помещается 1 моль ТЭОС, затем туда добавляется 1,26 моль EtOH. Эта смесь перемешивается, так что EtOH растворяет ТЭОС. Отдельно разбавляется 27,81 г 1 N HNO3 азотной кислоты (соответственно 1,75 г HNO3) при помощи 60,38 г H2O (воды) (общая масса разбавленной азотной кислоты составляет, таким образом, 88,19 г, из которых 86,44 г приходится на H2O, соответственно 4,8 моль, и 1,75 г на HNO3, соответственно 0,028 моль; мольное отношение H2O/HNO3 составляет 4,8/0,028=172). Затем к этанольному раствору ТЭОС добавляется 33,07 г разбавленной азотной кислоты (так что используется 1,8 моль H2O и 0,01 моль HNO3 на 1 моль ТЭОС).
Первая РГК протекает экзотермично. Первая РГК, согласно изобретению, раскрываясь на примере ТЭОС, означает, что в каждой молекуле ТЭОС гидролизуется по одной EtO-группе и полученная в результате ОН-группа конденсируется с димеризацией и отщеплением воды при постоянном перемешивании. Это означает, что оба раствора объединяют (например, ТЭОС в EtOH и разбавленную азотную кислоту) при комнатной температуре (КТ), при этом температура во время реакции 2 SiX4 (т.е., например, 2 ТЭОС) за счет гидролиза и конденсации каждого EtO-остатка в X3Si-O-SiX3 (например, (EtO)3Si-O-Si(EtO)3) при адиабатическом проведении реакции повышается примерно на 40°С. Начальную температуру при первой РГК в основном не оговаривают (так как реакция протекает и без того экзотермично). Она может быть комнатной, или быть ниже или выше соответствующей КТ, например 5, 10, 15, 20, 35, 45, 60 или 70°С. Она должна лишь лежать так высоко, чтобы могла протекать первая РГК.
Согласно изобретению особенно предпочтительно избегать гидролиза больше, чем одной EtO-группы на молекулу ТЭОС, КТ (примерно 20°С, при необходимости 18-25°С) является поэтому из экономических и практических предпосылок предпочтительной. Температуры, отклоняющиеся от комнатной, в области от 0°С до 80°С, предпочтительно в области от 10°С до 70°С или в области от 20°С до 60°С также подходят, при этом при превышении точки кипения при нормальном давлении требуется проведение реакции под давлением. Для температуры действует, естественно, обычное в химии соотношение, что более низкая температура требует более длительных времен реакции - и наоборот. В предпочтительной форме исполнения представленного изобретения указанная первая РГК проводится при комнатной температуре в течение 1-192 часов (ч), более предпочтительными являются временные области 8-24 часа. В качестве альтернативы предпочтительным является временная область от 8 до 17 ч.
Первая РГК проводится предпочтительно ступенчато в реакционном сосуде с перемешиванием. Si-соединение формулы (I) (например ТЭОС) и растворитель (например этанол) помещаются в реактор изначально. Затем происходит быстрая подача кислоты, предпочтительно в виде 0,01 N HNO3 (например, 0,01 моль HNO3 на моль ТЭОС). По причине кислотности реакционной смеси первая РГК протекает быстро, и содержимое сосуда нагревается примерно до температуры от 30°С до 40°С, прежде чем температура еще во время течения реакции (т.е. на стадии (а)) начнет снижаться (в результате охлаждения до температуры окружающего пространства или температуры нагревательного средства).
Стадия (b)
Вторая РГК материала, полученного в стадии (а), происходит в закрытом аппарате, в котором возможно смешение (например, ротационный испаритель, сосуд с перемешиванием), при одновременном удалении водорастворимого растворителя (например, воды, этанола) за счет испарения в вакууме между 1 и 1013 бар, предпочтительно при давлении <600 бар, и опционально при непрерывной подаче химически инертного газа-носителя (для частичного снижения давления испаряющихся компонентов), при этом, по меньшей мере, один из параметров процесса - давление, поток газа-носителя и/или температура подгоняется в зависимости от времени, и происходит испарение при реакционной температуре примерно от 30°С до 90°С, предпочтительно примерно между 60°С и 90°С, особенно предпочтительно примерно при 60-70°С и предпочтительным образом при умеренном перемешивании реакционной системы до вязкости смеси от 0,5 до 2 Па·с (при 4°С и 10 с-1), предпочтительно примерно до 1 Па·с (измерение при 4°С, скорость сдвига 10 с-1).
За счет поступательной реакции/полимеризации (определяемой по повышению вязкости) сдвигается фазовое равновесие, так что соответствующее равновесное давление растворителя в паровой фазе становится все ниже. Если равновесное давление снижается до общего давления в газовой фазе, то испарение прекращается.
Для того, чтобы далее испарить растворитель, должно снизиться давление, поток газа-носителя должен изменяться согласно условиям и/или должна быть повышена температура.
В предпочтительной форме исполнения изобретения испарение в стадии (b) происходит при постоянной температуре и изменяемом по времени давлении.
Стадия (b) должна, безусловно, протекать при отводе воды, чтобы не мог происходить дальнейший гидролиз. Температуры выше 60°С особенно предпочтительны, для того чтобы при концентрации HNO3, отчетливо повышающейся обычно в остаточном растворителе, благоприятствовать редуктивному превращению HNO3 в NO. Этот очень легко летучий газ (точка кипения при нормальном давлении примерно -150°С) после удаления из жидкой фазы при контакте с воздухом окисляется в легко кипящий NO2 (Т.кип. примерно 21°С), который удаляется из системы с отводимым потоком воздуха или газа. Таким путем концентрация кислоты в ПСН-материале ограничивается или снижается.
«Поток газа-носителя», согласно изобретению, означает газовый поток, который переносит газовый объем через жидкую фазу реакционной системы. Для соблюдения изобарных соотношений в реакционном сосуде тем самым должен отводиться газообразный объемный поток, который состоит как из газа-носителя, так и из компонента/компонентов, подлежащих испарению. Образующееся снижение парциального давления, т.е. уменьшение доли подлежащих испарению компонентов или смеси компонентов в газовом пространстве, повышает удаляющую силу для испарения растворителя на поверхности жидкости.
В особенно предпочтительной форме исполнения поток газа-носителя с помощью газового сепаратора, расположенного в газовом пространстве аппарата, разделяется, так что гарантируется достаточный обмен газа-носителя почти над самой поверхностью жидкости, однако все же без прямого конвективного обдува поверхности жидкости. Последнее в экстремальном случае может привести к локальному образованию геля, что нежелательно. Газовые сепараторы, с помощью которых может быть осуществлена эта форма исполнения, известны специалистам.
В предпочтительной форме исполнения изобретения в качестве химически инертного газа-носителя используется азот и/или воздух для снижения парциального давления.
В предпочтительной форме исполнения изобретения водорастворимый растворитель удаляется с помощью комбинации из вакуума и газа-носителя. Общее давление и поток газа-носителя при этой форме исполнения изобретения могут быть независимо друг от друга постоянными или подгоняться в зависимости от времени. Но в этой форме исполнения изобретения подгоняют в зависимости от времени также, по меньшей мере, один из параметров процесса - давление, поток газа-носителя и/или температуру. Тем самым возможно, например, достигнуть интегрально определенного времени реакции при желаемой степени испарения и/или подогнать скорость испарения к кинетике реакции.
В предпочтительной форме исполнения изобретения испарение происходит в стадии (b) при постоянной температуре и изменяющемся во времени давлении, при этом давление к концу второй РГК снижается, исходя из нормального давления или слегка пониженного давления, до <600 мбар, предпочтительно <500 мбар, особенно предпочтительно <100 мбар.
При комбинированном способе проведения процесса (вакуум с газом-носителем) предпочтительно постоянное или изменяющееся пониженное давление <600 мбар.
Особенно предпочтительно стадии (b), стадии так называемого реактивного испарения, позволить протекать так долго, пока вязкость не повысится примерно до 1 Па·с, и тем самым свойства геля, необходимые для проведения следующей за этим стадии (с), будут достигнуты. При слишком низкой вязкости (прерывание раньше времени) кинетика на стадии (с) будет слишком медленной. При более высокой вязкости образуются нежелательные гели, которые отрицательным образом влияют на дальнейшую переработку.
Более предпочтительным образом стадия (b) заканчивается посредством охлаждения до температур ниже 10°С и предпочтительно за счет установления нормального давления (примерно 1013 мбар).
Реактивное испарение происходит при соотношениях (парциального) давления и температуры, которые, с одной стороны, подходят для дальнейшего упаривания золя за счет испарения растворителя, с другой стороны, при использовании HNO3 благоприятствуют снижению силы кислоты, и, кроме того, из реакционно-кинетического слоя получаются (пред)-структуры, необходимые для последующих стадий процесса. Температуры около 70°С являются предпочтительными.
Если в стадии (а) в качестве кислоты используется азотная кислота, то в стадии (b) происходит возможное и предпочтительное снижение кислотности таким образом, что кислота во время реактивного испарения разлагается на NO и воду или в присутствии кислорода на NO2 и воду. Но N в виде NO или NO2 уносится затем только частично (наибольшая часть), часть (очень небольшая) остается в золе/коллоидном растворе. Но если используется система органическая кислота/аргинин вместо азотной кислоты, то происходит повышение рН или снижение кислотности, в случае, если это желаемо, например, с помощью трис-растворов (поскольку кислота, например уксусная кислота, не может быть унесена).
Поразительным образом было установлено, что при сохранении условий, как они были описаны выше для стадий (а) и (b), и после удаления растворителя в стадии (b) получается коллоидный раствор, который перед «созреванием» на стадии (d) не требует фильтрации, т.е. не содержит мешающих твердых веществ.
Стадия (с)
Эта стадия, которая является процессом охлаждения, отличается по смыслу тем, что коллоидный раствор, полученный на стадии (b), быстро, т.е. в течение времени от нескольких минут (предпочтительно в течение от 2 до 5 минут) до нескольких часов, предпочтительно от 0,2 до 5 часов, особенно предпочтительно в течение получаса, переводится в закрытый, предпочтительно в газодиффузионно-герметичный сосуд, и охлаждается до той температуры, при которой проводится стадия (d).
В соответствии с этим температуры, до которых идет охлаждение, составляют около -20°С - 10°С, предпочтительно 2°С - 4°С, особенно предпочтительно около 4°С. Попадание влаги, например в виде влаги из воздуха или влаги, присущей сосуду, безусловно следует устранить. При необходимости на этой стадии происходит также подгонка материала таким образом, чтобы значение рН дальнейших материалов, подлежащих использованию на теле, составляло от 5 до 7, предпочтительно >6.
Стадия (d)
Кинетически контролируемое созревание является составной частью способа согласно изобретению, и вообще дает возможность первой обработки, например, в виде формования или также покрытия материала реакционной смеси (ПСН-материала), полученной по стадии (с). На этой стадии (d) происходит третья РГК, при этом вязкость реакционной смеси на основе вышеописанной полимеризации повышается дальше.
Стадия (d), согласно изобретению, происходит в закрытых, предпочтительно газодиффузионно-герметичных сосудах, например, в так называемых стаканах для созревания, предпочтительно в сосудах, которые уже использовались для стадии (с). Попадание влаги или других газов, здесь следует упомянуть также CO2, безусловно следует устранить. Предпочтительное проведение стадии (d) происходит, согласно изобретению, при температуре от (выше) -20°С до 10°С в течение промежутка времени от 1 дня до 8 недель, предпочтительно при температуре от 2°С до 4°С и в течение промежутка времени от 3 до 18 дней. Особенно предпочтительно созревание проводится в течение промежутка времени от 10 до 14 дней, при 4°С, в частности посредством выдерживания реакционной смеси без тряски в закрытых, предпочтительно газодиффузионно-герметичных сосудах. Но созревание может производиться точно также при любой температуре в области от (выше) -20°С до 10°С.
Специалисту известно, что температура и время реакции являются двумя зависимыми друг от друга величинами, которые согласовываются друг с другом предпочтительно так, что сПСН-материал, полученный в стадии (d), принимает динамическую вязкость, которая делает его подготовленным и пригодным для проведения одной из стадий от (е1) до (е4). Если материал в стадии (е1) должен быть формован в волокно, то динамическая вязкость в конце (d) должна составлять примерно от 30 до 100 Па·с, предпочтительно от 45 до 60 Па·с (скорость сдвига 10 с-1 при 4°С), с фактором потерь (при 4°С, 10 1/с, 1%-ной деформации) от 2 до 5, предпочтительно от 2,5 до 3,5 (фактор потерь есть частное от деления вязкой доли к эластичной доле динамической вязкости). Если материал на стадии (е2) должен наоборот быть переработан в порошок, динамическая вязкость в конце (d) составляет примерно 60 Па·с (скорость сдвига 10 с-1 при 4°С). В случае переработки материала в монолит (на стадии (е3)) динамическая вязкость в конце (d) составляет больше 70 Па·с (скорость сдвига 10 с-1 при 4°С). И если материал на стадии (е4) должен быть использован для нанесения покрытия на корпус или поверхность, динамическая вязкость составляет, в зависимости от желаемой толщины слоя, величину меньше или равную 10 Па·с (скорость сдвига 10 с-1 при 4°С).
Слишком высокий фактор потерь означает слишком высокую эластичность материала, которая, например, противодействует образованию стабильной нити при прядении (образование геля, разрыв нити). При слишком низком факторе потерь материал настолько текуч, что стабильное образование нити невозможно (капли).
Конечным продуктом созревания в специальном стакане для созревания является золь (сПСН-материал) с определенными реологическими свойствами, среди прочего, структурной вязкостью. Структурная вязкость является свойством жидкости показывать при больших силах сдвига более низкую вязкость; чем сильнее сдвиг, который действует на жидкость, тем менее она вязкая (вязкотекучая). Снижение вязкости возникает за счет действия силы на полимеры в золе, которая ответственна за то, чтобы они выравнивались, и поэтому могли лучше скользить друг по другу; к этому же, в частности, для величины и формы структур, которые обнаруживают способность к прядению, см. Sakka в Sol-Gel Technology for Thin Films, Fibers, Preforms, Electronics and Specially Shapes, ed. L.C. Klein, Neyes, Park Ridge, N.Y., 1988, Seite 140 undAbbildung 2.7.).
Более предпочтительно согласно изобретению (значительно) подавлять конкурирующее образование трехмерной полимерной гель-сетки, конечный продукт способа по изобретению, согласно стадии (d), предпочтительным образом является гидрофобным, содержащим этокси-группы золем, без содержания геля, который (в значительной степени) свободен от воды.
Так как кинетически контролируемое созревание, а именно стадия (d), ниже -20°С протекает только очень медленно, ПСН-материал после стадии (с) может быть «заморожен» при температурах ниже -60°С. Поскольку это абсолютно предпочтительный вариант, когда ПСН-материал перед стадией (d) может точно также храниться и транспортироваться, как сПСН-материал после стадии (d).
Стадия (е1)
Процесс формования для переработки золя в волокно проводится при обычных условиях, как, например, описывается в немецких патентах DE 196 09 551 С1 и DE 10 2004 063 599 А1. При этом ПСН, например, через сосуд под давлением, выдувается через плато с соплами, содержащим до 250 отдельных сопел (давление в сосуде 1-100 бар, более предпочтительно 20 бар).
Золь, выходящий из (холодного) сопла при прохождении через (теплую) прядильную шахту, претерпевает следующую (четвертую) РГК, которая ответственна за то, чтобы материал, выходящий из сопла, через (молекулярное) поперечное сшивание, реагировал с образованием (стабильного) волокна. Прядильная шахта имеет обычно длину 1-5 м, более предпочтительно 2 м. Климат в прядильной шахте контролируемо регулируется относительно температуры и влажности (предпочтительными являются температуры между 20°С и 30°С и от -5 до 10°С точка росы), при необходимости здесь также происходит регулировка атмосферы при помощи других реагентов (например, этилнитрата).
Волокна после прохождения через прядильную шахту остаются, например, на раскладочном устройстве. Ширина ячейки образующейся таким образом копны волокна, среди прочего, устанавливается через скорости раскладочного механизма. Она составляет несколько см/с. За счет двухосевого движения образуется таким образом копна волокон (нетканый материал), в котором, в расчете на ТЭОС как исходный материал, содержащий кремний (Si), находятся еще свыше 30% этокси-групп.
Волокна, полученные согласно изобретению в стадии (е1), имеют на основе еще имеющихся этокси-групп известную гидрофобность. Они, кроме того, (в значительной степени) свободны от растворителя (вода, этанол).
В действительности предпочтительная форма исполнения состоит в том, чтобы получить волокна или нетканый материал по стадии (е1) или соответственно порошок, монолит и корпуса/поверхности с нанесенным покрытием по стадии: (е2), (е3) и (е4) и эти формы исполнения хранить, транспортировать и применять.
Если на стадии (а) в качестве кислоты используется разбавленная азотная кислота, происходит возможное предпочтительное снижение кислотности на стадии (е1), (е2), (е3) и (е4) таким образом, что оставшаяся, заключенная в материале часть HNO3 в виде NO или NO2, за счет продувки воздуха предпочтительно при 30°С улетучивается. Но если используется система (органическая кислота) / аргинин вместо азотной кислоты, происходит повышение рН или снижение кислотности, если это желательно, например, с помощью трис-раствора (поскольку кислота, например уксусная, не может быть удалена) непосредственно перед нанесением с помощью промывания в водном трис-растворе.
Стадия (е2)
Перед или также во время сушки к сПСН-материалу из стадии (d) (который на основе своей биоактивности может быть рассмотрен в качестве активного вещества) любые (другие) активные вещества, например фармацевтически активные вещества, могут быть добавлены или связаны ковалентно при помощи следующей, четвертой РГК (далее при использовании понятия «активное вещество» все-таки обычно имеется в виду не сПСН-материал из стадии (d), а другое активное вещество). Предпочтительно это должно происходить при производстве гомогенной смеси. В частности, в случае примешивания чувствительных к температуре активных веществ смесь из ПСН-материала и активного(ых) вещества(в) после четвертой РГК подвергается мягкой сушке, например, сушке распылением или вымораживанием. Если добавлено активное вещество, не чувствительное к температуре, или таковое вообще не добавляется, сушка может быть произведена при (заметно) повышенных температурах. При этом предпочтительно образуется биорассасывающийся и/или биоактивный матрикс. Этот матрикс подходит, в частности, также для капсулирования жидких активных веществ. Жидкости могут быть включены при длительной стабильности в матрикс и могут быть снова высвобождены контролируемым образом. Капсулирование способствует механической и химической стабилизации активных веществ, более удобному обращению с такими жидкими активными веществами и лекарственными средствами и помогает устранить неуправляемое увлажнение активных веществ. Понятно, что могут находиться другие вещества и/или активные вещества, приспособленные к соответствующему использованию в окончательном составе (порошок). Употребления без дополнительных активных веществ представляют собой, например, добавки для кожного крема, как они описаны, например, в http://http.V/www.photolagen.com.
Порошок может быть микропорошком или нанопорошком. Частицы микропорошка согласно изобретению имеют предпочтительно размер (средний диаметр) от 0,01 мкм до 100 мкм, в частности от 0,1 мкм до 20 мкм. Наночастицы порошка имеют, как правило, размер (средний диаметр) ≤ 100 нм.
Стадия (е3)
В другом исполнении к сПСН-материалу из стадии (d) (снова перед или во время сушки) может быть добавлено (дальнейшее) активное вещество, например фармацевтически активное вещество, или с помощью четвертой РГК может быть связано ковалентно. Затем, независимо от присутствия (дальнейшего) активного вещества, происходит литье сПСН-материала в форму. После сушки таким образом может быть получен монолит. Такие монолиты могут быть использованы в форме массивных имплантатов в качестве поставляющей активное вещество системы (Drug Delivery System), например, подкожно. Они могут быть использованы, например, как хранилище контрацептивов, и в течение длительного промежутка времени высвобождать активное вещество. Такие имплантаты согласно изобретению имеют хорошую совместимость. Монолиты предпочтительно могут иметь диаметр ≥0,5 мм. Альтернативным образом монолиты могут быть измельчены в порошок или размолоты.
Стадия (е4)
Созревший материал из стадии (d) может быть переработан также в покрытие. Для этого тело, на которое подлежит наносить покрытие, за счет погружения в сПСН-материал, за счет обливания сПСН-материалом или за счет центрифугирования или распыления сПСН-материала покрывается слоем материала. Предпочтительно покрытия наносятся на драже или капсулы, для чего спрессованные порошкообразные смеси лекарственных средств обволакивается биорассасывающимся и/или биоактивным покрытием из сПСН-материала. За счет этого может контролироваться и/или регулироваться высвобождение (дальнейших) активных веществ (например, через толщину слоя и/или последовательность слоев) внутри состава. Но подобное покрытие может быть нанесено также на часть тела-имплантата, за счет чего (биологическая) совместимость имплантата улучшается, например, смягчаются или предотвращаются реакции отторжения.
Согласно другому исполнению изобретения, высоковязкие золи, в частности гидрогели, могут быть дополнены или заменены сПСН-материалом согласно изобретению. Высоковязкие золи и гидрогели используются в медицине и в косметике в качестве активного вещества или носителя лекарственного средства. Вообще гидрогели используются часто в обеззараживании ран с большими поверхностями (обработка ран и заживление ран). Предпочтительно за счет добавки сПСН-материала может быть улучшена биологическая совместимость и тем самым заживление ран. Гидрогели согласно изобретению могут быть использованы предпочтительно как биорассасывающиеся и/или биоактивные продукты в медицине, в частности медицине человека и медицинской технике.
Дальнейшая переработка или применение волокна
Волокна как конечные продукты одного из предпочтительных способов согласно изобретению, включающих стадии от (а) до (d) и (е1), могут применяться как в качестве волокон, так и в качестве нетканого материала. Эти ПСН-материалы, такие как ПСН- и сПСН-материал, имеют чрезвычайно высокую способность к биорассасыванию и/или биоактивность.
Перед применением ПСН-материалов, предпочтительно непосредственно перед их применением, например, в качестве биорассасывающийся и/или биоактивных материалов, в медицине или медицинской технике (например, для обработки ран, заживления ран, в качестве сшивного хирургического материала или в качестве усиливающих волокон; см. также следующий ниже абзац) ПСН-материалы (волокно, порошок, монолит, раствор для нанесения покрытия) предпочтительно увлажняют водой, особенно предпочтительно при небольшом внешнем давлении. За счет увлажнения оставшиеся, еще имеющиеся в наличии этоксигруппы полностью гидролизуются, материалы тем самым становятся более гидрофильными. Как уже было упомянуто выше, указанное увлажнение может происходить в условиях повышения рН (например, в фосфатном буфере Н2РО4 -/НРО4 2-), в частности, если повышение значения рН не происходило уже на предшествующей стадии. При этом протекает шестая и последняя РГК, во время которой еще оставшиеся, не гидролизованные этоксигруппы удаляются из ПСН-материалов.
Следующее преимущество заключается в том, что ПСН- и сПСН-материалы, полученные согласно изобретению, и материалы, состоящие из них, по отношению к волокнам и волоконным материалам, которые были получены по способу из немецкого патента DE 196 09 551 С1, в тесте на цитотоксичность отчетливо показывают улучшенные значения. Эти улучшения были доказаны в тесте в присутствии мышиных фибробластов L929 (L929-Mausfibroplasten). Материалы, которые, согласно изобретению, получаются из стадий от (е1) до (е4), отличаются поэтому особенно хорошей биологической совместимостью.
Волокна или нетканые материалы, изготовленные согласно изобретению, могут быть использованы предпочтительно как биорассасывающийся и/или биоактивные материалы в медицине, медицинской технике, биотехнологии или индустрии изоляционных материалов. В частности, полученные согласно изобретению материалы преимущественно могут быть использованы в области обработки ран и заживления ран. Волокна могут быть использованы, например, как сшивной материал в хирургии или как усиливающие волокна. Нетканые материалы могут быть использованы предпочтительно при обеззараживании поверхностных ран, при фильтрации содержащихся в организме жидкостей (например, крови) или в области биореакторов в качестве основы для выращивания.
ПСН-материалы из (е1), (е2), (е3) и (е4) согласно изобретению, в которые может быть добавлено биологически активное вещество, то есть наряду с биоактивным Si-полимером содержат другое активное вещество, могут транспортировать их на собственное место действия или влиять на высвобождение активного вещества на месте действия. Эти материалы в дальнейшем обозначаются как носители активных веществ (Drug Delivery System). Применение созревших ПСН-материалов и ПСН-материалов согласно изобретению имеет преимущество, состоящее в том, что оба могут быть переработаны разносторонним образом, использованы и скомбинированы с различными (дальнейшими) активными веществами. Особенно предпочтительным является, если сПСН-материал согласно изобретению не образует при этом реакционных продуктов с другим активным веществом. ПСН-материалы согласно изобретению являются биорассасывающимися и/или биоактивными и показывают улучшенную цитотоксичность, что способствует улучшенной биологической совместимости материалов, которая непосредственно необходима в области медицины и медицинской техники.
Изобретение должно быть раскрыто на следующем примере, без возникновения в результате ограничений.
Все указанные вязкости были измерены на вискозиметре MCR 300 фирмы Physika при скорости сдвига 10 с-1 при 4°С.
Примеры
Пример 1: Биорассасывающийся и/или биоактивный сПСН-материал (золь) и его переработка в волокна и нетканое полотно.
В качестве эдукта для реакции гидролиза-конденсации 2,7 моль ТЭОС (тетраэтоксисилана) (562,4 г) помещали в реакционный сосуд. Прибавляли 3,4 моль (2,7×1,26) ETOH (156,8 г) в качестве растворителя. Смесь перемешивали. Отдельно разбавляли 1 N HNO3 (27,81 г) Н2О, и прибавляли к смеси ТЭОС-этанол. Полученная таким образом реакционная смесь содержала 1,8 моль H2O и 0,01 моль HNO3 на моль ТЭОС. Смесь перемешивали 18 часов.
Смесь, полученную согласно стадии а), после этого за счет испарения в ротационном испарителе (стадия b) при 70°С, при ступенчатом наложении вакуума 500-200 мбар, при медленном перемешивании (20 об/мин) почти полностью освобождали от воды и этанола. За счет высокой температуры HNO3 в значительной степени переходила в восстановленную форму NO2.
Золь имел вязкость примерно 1 Па·с (скорость сдвига 10 с-1 при 4°С), кислотность сильно снижалась.
Раствор на стадии (с) охлаждали в закрытом полипропиленовом стакане [сосуд для созревания] в течение 30 минут до 4°С, и при этой температуре на стадии (d) в сосуде для созревания подвергали созреванию 8 дней. Получали гомогенную однофазную золь-массу с вязкостью примерно 40 Па·с (скорость среза 10 с-1 при 4°С). Золь получался без видимой составной части твердой фазы.
Золь мог быть формован на стадии (е1) в волокна. Поэтому этот золь обозначается также как прядильная масса или сПСН-материал. Получение волокон проводили в обычной прядильной установке. Для этого прядильную массу помещали в охлажденной до -15°С цилиндр под давлением, в котором создавали давление 20 бар. Образовавшаяся в результате сила продавливала прядильную массу через сопла. Выходящие спряденные нити, в зависимости от локальной температуры и тем самым вязкости прядильной массы, имели диаметр от 30 до 70 мкм. Текучий, подобный меду материал падал под своим собственным весом в прядильную шахту длиной 2 м, находящуюся под цилиндром с давлением, и реагировал там с влагой воздуха, при этом текучесть нитей снижалась. В прядильной шахте температуру и влажность контролируемо регулировали. Температура составляла около 25°С и влажность воздуха около 35%. При выходе нитей на раскладочное устройство они сохраняли приблизительно свою цилиндрическую форму, но все-таки были еще так текучи, что они склеивались своими соприкасающимся поверхностями друг с другом в копну (нетканый материал).
Нетканый материал затем продували в сушильной камере примерно при 30°С и тем самым содержание кислоты еще дальше снижалось. Кислотность при этом снижалась до физиологически допустимой степени.
Полученное в примере 1 нетканое волокно подвергали тесту на циототоксичность согласно ISO 10993-5 (1999); EN 30993-5 (1994). Измеренная цитотоксичность в сравнении со значениями, полученными для контроля, показала, что нетканое волокно, полученное согласно изобретению, не показывает цитотоксичных свойств.
Пример сравнения
Эдукты ТЭОС (тетраэтоксисилан), EtOH, H2O и HNO3 смешивали в молярном соотношении 1:1,26; X; 0,01 (с X-1,6. 1,7. 1,8. 1,9 и 2,0) и сильно перемешивали 5 часов при комнатной температуре. Полученный в результате раствор помещали в один из открытых сосудов на водяную баню, нагретую до 70°С, в которой он (раствор) находился до определенной потери веса. Затем раствор охлаждали, и фильтровали через сетку из нержавеющей стали с шириной ячейки 1 мм × 1 мм. Фильтрат оставляли в закрытом сосуде при температуре 3°С для достижения созревшего состояния от 6 часов до 6 месяцев, в зависимости от потери веса. Полученная в результате прядильная масса была довольно гомогенной и некоторое время стабильной и способной к прядению. Изготовление волокон происходило на сушильной прядильной установке. Для этого прядильную массу помещали в прядильную головку, охлажденную до - 15°С, при давлении от 10 до 15 бар, сначала продавливали через сетку из нержавеющей стали с шириной отверстия сита 80×80 мкм и затем через сопло с диаметром 100 мкм. Полученную бесконечную нить после участка сушки в 1 м наматывали на вращающийся цилиндр. Полученные в результате волокна, в зависимости от образца, т.е. добавленного количества воды, имели круглую, овальную форму или форму кости в поперечном сечении с диаметрами между 5 мкм и 30 мкм. Поверхности поперечных сечений лежали между 100 мкм2 и 400 мкм2.
Поверхность волокна является гладкой и ни в коем случае не показывает волнового профиля.
Измерения прочности на разрыв волокон дали значения от 100 МПа до 800 МПа. В ИК-спектрах готового материала обнаруживаются: Si-OH - полосы при 950 см-1 и С-Н-сигналы при 3000 см-1. Имеются в наличии также частично гидролизованные и частично конденсированные волокна этоксисиланола. Примерно после 2 месяцев хранения при комнатной температуре в ИК-спектрах не обнаруживаются больше полос СН-колебаний. Волокна превращаются в частично конденсированные силаноловые волокна, которые стабильны в течение нескольких месяцев.
С полученными таким образом волокнами были проведены цитотоксичные измерения. Для волоконных материалов, полученных таким образом, в тесте на цитотоксичность согласно ISO 10993-5 (1999); EN 30993-5 (1994) были установлены цитотоксичные воздействия.
Также при этом могло быть вытянуто только 50% общего реакционного образца.
Изобретение относится к нетоксичному полисилоксановому материалу. Предложен биорассасывающийся полисилоксановый (ПСН)-материал для получения волокон, получаемый (а) первой реакцией гидролиза-конденсации (РГК) максимум одного остатка одного или нескольких различных Si-соединений формулы SiX4, в которой остатки Х являются одинаковыми или различными, проводимой при катализе кислыми агентами и при начальном значении рН от 0 до ≤7 в присутствии водорастворимого растворителя 1-192 ч при температуре 0-80°С, (b) второй РГК материала, полученного на стадии (а), проводимой при одновременном удалении растворителя за счет испарения в закрытом перемешивающем аппарате при непрерывной подаче химически инертного потока газа-носителя в вакууме 1-1013 мбар при температуре 30-90°С до вязкости 0,5-2 Па·с. На стадии (с) ПСН-материал охлаждают в закрытом аппарате до температуры -20 - +10°С, в течение 2 мин - 5 ч, и затем (d) ПСН-материал, полученный на стадии (с), путем третьей РГК переводят в созревший полисилоксановый материал (сПСН)-материал при температуре -20 - +10°С в течение от 1 дня до 8 недель, при этом стадию (d) осуществляют непосредственно после стадии (с). Технический результат - получаемый сПСН-материал пригоден для изготовления нетканого материала с улучшенной биологической переносимостью. 13 з.п. ф-лы, 2 пр.
1. Биорассасывающийся полисилоксановый (ПСН)-материал для получения волокон, получаемый за счет того, что
(a) первую реакцию гидролиза-конденсации (РГК) максимум одного остатка одного или нескольких различных Si-соединений формулы (I):
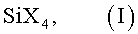
в которой остатки Х являются одинаковыми или различными и означают гидрокси, водород, галоген, амино, алкокси, ацилокси, алкилкарбонил и/или алкоксикарбонил и происходят от алкильных остатков, которые представляют собой при необходимости замещенные линейные, разветвленные или циклические остатки с 1-20 атомами углерода, предпочтительно с 1-10 атомами углерода, и могут быть прерваны атомами кислорода или серы или аминогруппами, проводят при катализе кислыми агентами и при начальном значении рН от 0 до ≤ 7, в присутствии водорастворимого растворителя, в течение промежутка времени от 1 до 192 ч при температуре от 0°С до 80°С,
(b) вторую РГК материала, полученного на стадии (а), проводят при одновременном удалении растворителя за счет испарения в закрытом аппарате, в котором осуществляют перемешивание материала, и опционально при непрерывной подаче химически инертного потока газа-носителя, при этом испарение проводят в вакууме между 1 и 1013 мбар при температуре между 30-90°С до вязкости от 0,5 до 2 Па·с (скорость сдвига 10 с-1 при 4°С) и, по меньшей мере, один из параметров процесса - давление, поток газа-носителя и/или температуру подгоняют в зависимости от времени,
с) указанный ПСП-материал охлаждают в закрытом аппарате до температуры от -20°С до +10°С, в течение от 2 мин до 5 ч, и
d) ПСН-материал, полученный на стадии (с), путем третьей РГК переводят в созревший полисилоксановый материал (сПСН)-материал при температуре от -20°С до +10°С в течение от 1 дня до 8 недель,
при этом стадию (d) осуществляют непосредственно после стадии (с).
2. Материал по п.1, отличающийся тем, что испарение на стадии (b) проводят при постоянной температуре и изменяющемся во времени давлении.
3. Материал по п.2, отличающийся тем, что испарение на стадии (b) проводят при изменяющемся пониженном давлении между 200 и 500 мбар.
4. Материал по п.1, отличающийся тем, что испарение на стадии (b) проводят с помощью потока химически инертного газа-носителя.
5. Материал по п.1, отличающийся тем, что водорастворимым растворителем является этанол или смесь этанол-вода.
6. Материал по п.1, отличающийся тем, что па стадии (а) рН устанавливают на значении от 0 до ≤7 при помощи разбавленной азотной кислоты или кислой смеси или раствора из (i) физиологически совместимой кислоты, такой как лимонная, янтарная, винная, уксусная или аскорбиновая кислота, и (ii) субстрата нитроксид-синтазы, такого, как аргинин.
7. Материал по п.6, отличающийся тем, что разбавленную азотную кислоту применяют в молярном соотношении Si-соединение(я) формулы (I) к азотной кислоте, равном от 90:1 до 110:1, предпочтительно 100:1.
8. Материал no п.1, отличающийся тем, что стадию (а) проводят при температуре от 20°С до 60°С в течение 8-24 ч.
9. Материал по п.1, отличающийся тем, что стадию (b) проводят при температуре между примерно 60 и 90°С.
10. Материал по п.1, отличающийся тем, что стадию (b) проводят до вязкости 1 Па·с (скорость сдвига 10 с-1 при 4°С).
11. Материал по п.1, отличающийся тем, что золь на стадии (с) охлаждают до температуры 4°С в течение 30 мин.
12. Материал по п.1, отличающийся тем, что созревание на стадии (d) проводят при температуре 4°С в течение 8 дней.
13. Материал по одному из пп.1-12, отличающийся тем, что стадию (d) проводят до вязкости материала от 30 до 100 Па·с (скорость сдвига 10 с-1 при 4°С), предпочтительно от 45 до 60 Па·с (скорость сдвига 10 с-1 при 4°С), с фактором потерь (скорость сдвига 10 с-1 при 4°С и при 1%-ной деформации) от 2 до 5, предпочтительно от 2,5 до 3,5.
14. Материал по п.1, отличающийся тем, что Si-соединением, применяемым на стадии (а), является тетраэтоксисилан.
| WO 00/50349 А2, 31.08.2000 | |||
| DE 19609551 C1, 17.07.1997 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОРГАНОСИЛОКСАНОВ ПУТЕМ ГИДРОЛИЗА ОРГАНОГАЛОГЕНСИЛАНОВ | 1997 |
|
RU2186794C2 |
| Способ получения полиэтоксисилоксанов | 1987 |
|
SU1581721A1 |

