Изобретение относится к химии силиконов, а именно - к гидролизу галосиланов, преимущественно органогалогенсиланов, с целью получения поли(органо)силоксанов. Предлагаемый способ основан на очень высокой реакционной способности связей Si-Cl к воде и в меньшей степени связей SiOR (R = алкил).
Органогалогенсиланы в результате гидролиза и поликонденсации (гомо- и гетероконденсации) превращаются в промежуточные полиорганосилоксаны (олигоорганосилоксаны) с линейной или циклической структурой, которые полимеризуются и образовывают поперечные связи в среде силиковых масел или силиконовых эластомеров с более высоким молекулярным весом.
Диметилдихлоросилан является органогалогенсиланом, который хорошо известен как исходный продукт для реакции гидролиза/конденсации.
Технологические процессы, использующие указанную реакцию гидролиза/конденсации Me2SiCl2, являются непрерывными процессами и превосходно освоены промышленностью. Это описано, например, Х.К. Лихтенвальнером и М.М. Спрунгом в Энциклопедии науки о полимерах, т.12, Вилей и сыновья, г. Нью-Йорк, 1970 г. (Encyclopedia of Polymer science Vol. 12 Wiley and Sons, New-York, 1970).
В результате гидролиза Me2SiCl2 образуется сложная смесь из циклических и линейных олигоорганосилоксанов. Классически источником реакционной воды для гидролиза служит раствор хлористо-водородной кислоты. С целью максимальной рентабельности способа образующуюся при гидролизе хлористо-водородную кислоту улавливают и используют, например, в реакции с метанолом для получения хлорметана, являющегося основным продуктом при синтезе диметилдихлорсилана, посредством прямого синтеза.
В известном уровне техники используются разные варианты гидролиза органогалогенсиланов, например Me2SiCl2.
Из патентной заявки Франции 2512028 известен способ гидролиза органогалогенсиланов, в частности Me2SiCl2, при котором источником воды при гидролизе органогалогенсилана выступает водный раствор хлористо-водородной кислоты с содержанием не менее 35 вес.% хлористого водорода, при этом данный раствор используют в количестве, при котором молярное соотношение между Н2О и органохлорсиланом колеблется от 10 до 30. Гидролиз протекает за одну операцию. Целями, достигаемыми при этом, являются, во-первых, существенное уменьшение весового процента галогена (хлорида) в гидролизате на базе полученного полиорганосилоксана и, во-вторых, обеспечение максимального выхода циклополиорганосилоксанов. При температурах реакции в диапазоне от 20 до 30oС концентрация водного раствора НСl составляет 37 вес.% или меньше. В любом случае, какой бы ни была температура, указанный раствор НСl никогда не оказывается насыщенным в примерах упомянутой патентной заявки 2512028.
Недостатком такого гидролиза в присутствии избыточной воды являются значительные количества водной хлористо-водородной кислоты. Однако ввиду того, что наиболее легко утилизируемой НСl является газообразная НСl, то необходима дистилляция водной НСl, которая является весьма дорогостоящей.
Кроме того, гидролиз в присутствии избыточного количества воды крайне изотермичен, что неизбежно ведет к технологическим трудностям.
Изобретение, описанное в патентной заявке Франции 2512099, безуспешно ставит своей целью устранение указанных недостатков, применяя практически стехиометрическое количество воды при одностадийном гидролизе диметилдихлоросилана. Это позволяет получать безводную (газообразную) хлористо-водородную кислоту, насыщенный водный раствор хлористо-водородной кислоты и гидролизат полиорганосилоксана. Если водная НСl в конце гидролиза оказывается насыщенной, то согласно примерам это ни в коем случае не относится к исходному раствору НСl. См. пример 3:37 вес.% при 60oС. Следовательно, отсюда можно заключить, что полученная при реакции НСl не выделилась в полном объеме в газообразной форме.
Изобретение по FR-A-2518099 ставит своей целью устранить избыточные количества воды, не создавая в то же время условий для дефицита воды, которые известны при получении линейных полиорганосилоксанов, не прошедших полностью гидролиз и содержащих галогены (хлориды) в концевых положениях. Это совершенно негативное явление, поскольку приходится завершать гидролиз при больших количествах воды, что ведет к образованию водной хлористо-водородной кислоты, создающей трудности и осложнения в той мере, в какой сложны ее рециклирование и/или повторная обработка по низкой стоимости.
Не представляется возможным проводить такой дополнительный гидролиз и с применением незначительных количеств воды, поскольку в таких условиях начинается поликонденсация олигоорганосилоксанов, что имеет своим прямым следствием неблагоприятное возрастание вязкости.
Патент США 5169970 описывает способ гидролиза органохлорсиланов, при котором гидролиз протекает в два этапа:
- первый этап гидролиза состоит в гидролизе органохлорсилана с использованием преимущественно стехиометрического количества воды и с получением в результате этого гидролизата из полиорганосилоксанов,
- на втором этапе гидролиза полученный на первом его этапе гидролизат подвергают той же операции, но на этот раз с использованием избыточного стехиометрического количества воды, при этом в качестве источника воды используется раствор НСl определенной концентрации.
Предполагается, что данный способ позволит получить гидролизат полиорганосилоксана без недостатков, присущих известным способам, на стадии гидролиза, использующих либо избыток воды, либо ее недостаток, либо ее стехиометрическое количество по отношению к органогалогенсиланы. Данный способ позволяет якобы регулировать вязкость полученного полиорганосилоксана.
Однако указанный способ несовершенен в той части, что он прозводит газообразную НСl при низком давлении, вследствие чего усложняется улавливание и утилизация последней.
Принцип гидролиза в два этапа также известен в способе получения полидиметилсилоксана и описан в заявке на европейский патент 0658588. Согласно этому способу на первом этапе осуществляют реакцию между диметилдихлорсиланом и водой с использованием водного раствора хлористо-водородной кислоты концентрацией 25 вес.% с целью получения сырого гидролизата, состоящего из циклических и линейных α, ω -дихлорполидиметилсилоксанов и газообразного хлористого водорода.
На втором этапе сырой гидролизат обрабатывают водяным паром с целью снижения в нем содержания хлора при дополнительном гидролизе, в ходе которого также образуется водная хлористо-водородная кислота. Последняя рециклируется для использования на первом этапе процесса.
Давление хлористо-водородного газа на первом этапе составляет от 0,15 до 0,5 МПа, преимущественно от 0,25 до 0,35 МПа. Температура на первом этапе комнатная, в то время как на втором этапе температура составляет от 110 до 160oС. При завершении первого этапа гидролиза в полученном полидиметилсилоксане содержится 50% циклических олигомеров и 50% линейных α, ω -хлористых олигомеров.
Применение НСl такой концентрации на первом этапе гидролиза и высокой температуры на втором этапе затрудняет стабилизацию и регулирование вязкости целевых продуктов.
В заявке на патент DD-227145 описан способ получения гидролизата нейтрального диметилдихлоросилана, характеризующегося небольшой вязкостью и стойкостью при хранении, а также остаточным содержанием связей SiCl, составляющим менее 10 ppm. Данный способ предусматривает обработку с целью удаления остатков групп SiCl и водной НСl, содержащихся в гидролизате диметилдихлорсилана, путем пропускания последнего после его промывки и/или нейтрализации через пористый материал в форме листа. Данный материал представляет собой волокнистый материал типа бумаги или ткани (шерсть, целлюлоза, полиэфир, стекловолокно). Указанная фильтрация прозводится при температуре окружающей среды и позволяет получить гидролизат с остаточным содержанием SiCl 2 ppm и с необнаруживаемой концентрацией НСl.
Такой способ обработки гидролизата дихлордиметилсилана позволяет несомненно уменьшить примеси в готовом продукте, однако эффективность данного способа может быть обеспечена только в том случае, когда на предшествующих этапах получают продукт, способный обрабатываться коалесценцией на пористой основе.
Из данного обзора уровня техники следует, что желательно располагать способом получения полиорганосилоксанов путем гидролиза органогалогенсиланов, который бы
- позволял получить продукт гидролиза типа галогенид водорода в газообразной форме и находящийся под давлением,
- возволял устранить по-возможности полностью связи 
- обеспечивал получение конечного гидролизата в виде полиорганосилоксана, свободного или почти свободного от наличия кислотно-водных капелек,
- позволял устранить образование больших количеств кислотно-водных потоков, вызывающих проблему их переработки,
- позволял обеспечить контроль за вязкостью полиорганосилоксана,
- позволял производить полиорганосилоксан высокого качества и, в особенности, высокой чистоты для обеспечения его максимальной стабильности.
Одной из основных задач настоящего изобретения является устранение указанного недостатка путем разработки способа, удовлетворяющего перечисленным выше требованиям.
Другой задачей настоящего изобретения является разработка способа получения полиорганосилоксана, при котором гидролиз протекает при дефиците воды по отношению к стехиометрическим количествам на начальном его этапе и при котором отпадает необходимость в последующем применении больших количеств воды для удаления остатков концевых связей галоген-Si, которые присутствуют в слишком большой концентрации после первого гидролиза. В результате этого исключаются галогенсодержащие водные потоки, трудные для рециклирования и/или повторной обработки ввиду постоянно присутствующих в них силоксановых примесей.
Еще одной задачей настоящего изобретения является разработка способа получения полиорганосилоксанов путем гидролиза органогалогенсиланов, обеспечивающего возможность проведения постоянного контроля за вязкостью реакционной среды.
Следующей задачей настоящего изобретения служит разработка способа гидролиза органогалогенсилана для получения полиорганосилоксанов, свободных или практически свободных от остаточных галогенов, а также от водных капелек с растворенным веществом, образованным водородным галогенидом, образующегося в процессе гидролиза.
Другой задачей настоящего изобретения является разработка способа получения полиорганосилоксанов путем гидролиза органогалогенсиланов, который легко и экономично осуществляем (высокая производительность и рентабельность).
Еще одной задачей настоящего изобретения является разработка способа гидролиза органогалогенсиланов для получения полиорганосилоксанов, который может быть осуществлен в непрерывном или периодическом режиме, удовлетворяя в том или ином случае указанные выше требования.
Названные задачи достигаются настоящим изобретением, которое касается способа получения полиорганосилоксанов путем гидролиза органогалогенсиланов формулы (I) RaR1 bSiXc, где
- R, R1 - радикалы, одинаковые или разные, означают водовод или линейные или разветвленные алкилы С1-C6, арилы, алкил-арилы или аралкилы;
- Х означает галоген;
- а+b+с=4 и 0 < с < 4;
отличающегося тем, что осуществляют:
а) по меньшей мере три последовательных этапа гидролиза (1), (2), (3) органогалогенсиланов формулы (I), реакционная среда на которых имеет возрастающую гидролизующую силу,
б) и при необходимости по меньшей мере один этап коалесценции (4), причем этап (1) гидролиза осуществляют под давлением в присутствии водного насыщенного раствора S1 галогенида водорода и при температуре и давлении реакционной среды.
Заявитель неожиданно установил, что
- сочетание, с одной стороны, по меньшей мере трех последовательных этапов гидролиза с возрастающей гидролизующей силой и, с другой стороны, первым этапом гидролиза, проводимым под давлением в присутствии водной фазы, насыщенной галогенидом водорода, с получением соответствующих продуктов гидролиза галогенсилановых связей 
- позволяет получить чистые полиорганосилоксаны, не содержащие остаточных групп SiХ и следов водной фазы.
Такой результат тем хорош, что может быть достигнут относительно просто и экономично, при высокой производительности и эффективности в непрерывном или периодическом режиме.
Кроме того, способ согласно изобретению не образует больших количеств загрязненных галогенидами эффлюентов. Следовательно, способ безвреден в отношении его воздействия на окружающую среду.
Необходимо также отметить, что проблема регулирования вязкости реакционной среды, которая непосредственно зависит от содержания галогенида водорода, решается согласно изобретению, благодаря использованию возрастающего гидролизующего действия и насыщению исходной водной фазы галогенидом водорода.
Не желая быть связанным теорией, можно полагать, что способ согласно изобретению позволяет оптимизировать гидролиз, сводя до минимума явления связывания гидролизной воды, обусловленного гидратацией галогенида водорода.
Под возрастающим гидролизующим действием в данном изобретении понимается нуклеофильный градиент реакционной среды, распространяющийся на три этапа гидролиза (1), (2), (3). Более точно, это означает, что реактивность водной фазы по отношению к связям =SiX возрастает по мере осуществления этапов 1, 2, 3.
Что касается понятия насыщения галогенидом водорода водной фазы реакционной среды на этапе (1), то следует понимать максимальную концентрацию растворенного в воде галогенида водорода, т.е. реакционная среда находится в газовой атмосфере, создаваемой газообразным галогенидом водорода.
Очевидно, что степень насыщения зависит от природы растворенного вещества, а также от температуры и давления среды. Средний специалист превосходно умеет управлять насыщением раствора S1 на этапе (1).
Согласно предпочтительному варианту осуществления способа согласно изобретению применяется вариант непрерывного действия.
Галогеном Х преимущественно является хлор, тогда получаемым галогенидом является НСl. В целях упрощения используют галоген X или хлор (Сl), галогенид водорода (НХ) и хлорид водорода (НСl).
Предпочтительно, чтобы этап (1) гидролиза под давлением состоял из следующих операций:
по меньшей мере один гидролиз (11) органогалогенсиланов формулы (I) для получения сырого по меньшей мере трехфазного гидролиза, содержащего:
- газовую фазу, образованную газообразным галогенидом водорода под давлением,
- силоксановую жидкость F1, состоящую преимущественно из линейных α, ω -дигалогенированных олигомеров и, возможно, в меньшей мере из цикличных олигомеров,
- водную фазу, содержащую раствор S1, насыщенный галогенидом водорода;
по меньшей мере одно разделение (12) типа газ/жидкость для получения газообразного галогенида водорода под давлением, с одной стороны, и смеси F1/S1, с другой стороны;
по меньшей мере одно разделение (13) типа жидкость/жидкость с получением потоков F1 и S1, причем последний целесообразно рециклировать на этап (11);
при необходимости по меньшей мере одну очистку (14) газа, полученного на этапе (12), предпочтительно путем конденсации;
при необходимости по меньшей мере одно дополнительное дегазирование (15) жидкости F1, получаемой на (13), предпочтительно путем декомпрессии.
Предпочтительно, чтобы данная реакция (11) протекала непрерывно при интенсивном перемешивании в закрытом сосуде, например, при температуре от 10 до 50oС и давлении, создаваемом газообразным галогенидом водорода, образовавшимся при гидролизе. На практике давление газообразного галогенида водорода устанавливается равным или более 0,10 МПа, предпочтительно от 0,15 до 1 МПа.
Подача реактивов на вход реактора на этапе (11) регулируется таким образом, чтобы массовое соотношение подаваемых фаз RM1 = водная фаза/органогалогенсилан формулы (I) составляло ≥ 2, предпочтительно от 3 до 15.
Показатель RM является одной из важных характеристик способа согласно изобретению, поскольку он вместе с концентрацией галогенида водорода в водной фазе и режимом перемешивания (в числе прочего) определяет концентрацию воды на поверхности раздела "водная фаза/органогалогенсилан формулы (I)". Гидролизующая сила среды зависит от концентрации воды на поверхности раздела фаз.
Предпочтительно, чтобы получаемый в конце этапа (11) сырой гидролизат был трехфазным.
На практике продукт F1 образован смесью линейных α, ω -дигалогенированных олигоорганосилоксанов и цикличных олигоорганосилоксанов, в которой
- первые составляют по меньшей мере 50, предпочтительно по меньшей мере 60 мол.% от смеси,
- а вторые - самое большее 50, предпочтительно самое большее 40 мол.% от смеси.
Указанная силоксановая жидкость F1, именуемая также "хлорсилоксом", содержит, например, около 8-13 вес.% остаточного галогена типа НХ и 
Предпочтительно, чтобы продолжительность реакции на этапе (11) составляла от 30 с до 5 мин, преимущественно от 1 до 2 минут.
Газообразный галогенид водорода, получаемый под давлением в конце этапа (12) разделения "газ/жидкость" может быть утилизирован, например, если речь идет о НСl, то его можно использовать в промышленном способе (при большом расходе) получения хлорида метила путем реакции с метанолом. Хлорид метила является основным сырьем в химии органосиланов.
При желании можно производить очистку газа, получаемого на этапе (12) под давлением, путем обработки (14), которой является, например, конденсация при температуре от 0o до -10oС, что позволяет удалить летучие легкие полисилоксаны.
Как уже уточнялось выше, предпочтительно рециклировать хлористо-водородный раствор S1, образующийся при разделении "жидкость/жидкость" этапа (13). Такое рециклирование на этап (11) влияет на качество конечного силоксанового масла или жидкости. Согласно предпочтительному варианту изобретения рециклирование является единственным или главным источником питания реакции хлористо-водородным раствором S1 на этапе (11). Таким образом, соотношение по массе рециклируемого S1 к органогалогенсилану формулы (I) согласно данной гипотезе будет эквивалентно RM1, которое определено выше, т.е. более или равно 2, предпочтительно от 3 до 15 или очень близко к RM1.
Возможное дегазирование на этапе (15) жидкости F1 осуществляется посредством, например, мгновенной декомпрессии при ΔP порядка 0,2 МПа. Таким образом получают газообразную хлористо-водородную кислоту, которая может быть непосредственно использована при промышленном синтезе. Такое дегазирование (15) позволяет повысить выход газообразной хлористо-водородной кислоты по меньшей мере на 3%.
Предпочтительно, чтобы этап (2) включал в себя по меньшей мере один влажный гидролиз (21), осуществляемый при перемешивании и при температуре от 10 до 50oС, при котором силоксановую жидкость F1, образовавшуюся на этапе (1), подвергают взаимодействию с концентрированным водным раствором S2 галогенида водорода. Концентрация растворенного в S2 вещества составляет часть эталонной концентрации насыщения (ЭКН) того же вещества, растворенного в водном растворе, при тех же значениях температуры и давления, причем указанная часть составляет 45-75%, предпочтительно 50-72% от ЭКН. Величина массового соотношения подаваемых фаз RM2= водная фаза/жидкость F1 устанавливается на величине, превышающей или равной 1,5, предпочтительно 2-10, с тем, чтобы обеспечить гидролиз связей 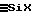 силоксановой жидкости F1 на уровне не менее 95%, предпочтительно не менее 98%, и получить таким образом силоксановую жидкость F2, состоящую главным образом из линейных α, ω -дигидроксилированных или α, ω -дигалогенированных олигомеров, и возможно, в меньшей степени, из цикличных олигомеров.
силоксановой жидкости F1 на уровне не менее 95%, предпочтительно не менее 98%, и получить таким образом силоксановую жидкость F2, состоящую главным образом из линейных α, ω -дигидроксилированных или α, ω -дигалогенированных олигомеров, и возможно, в меньшей степени, из цикличных олигомеров.
На данном этапе (2) влажного гидролиза используют гидролизующую силу, превышающую эту же силу на этапе (1), причем такое увеличение достигается благодаря увеличению относительной доли воды в гидролизной среде и, следовательно, благодаря снижению концентрации водной НСl. Согласно изобретению такой влажный гидролиз (2) имеет своей целью доведение гидролиза связей SiX до величины, достаточно близкой к 100%, т.е., например, ≥ 95%, предпочтительно ≥ 98%.
Регулирование гидролизующей силы гидролизной среды на этапе (2) может быть прервано, в частности:
- с помощью концентрации НСl, которая преимущественно составляет 45-75% от ЭКН,
- с помощью температуры.
Одной из характеристик способа согласно изобретению является тот факт, что при заданных рабочих условиях можно манипулировать концентрацией НСl для регулирования протекания реакции.
В любом случае такая концентрация представляет собой особенно критический фактор, поскольку хлоросилоксаны, гидролизованные на этапе (2), содержат почти столько же связей = SiX, что и связей =SiOH. Это означает, что риск поликонденсации велик, при этом известно, что главным следствием такой реакции является неблагоприятное повышение вязкости полиорганосилоксанов и, следовательно, всей реакционной среды.
Другим важным параметром является продолжительность реакции, протекающей между F1 и водной фазой S2 на этапе (21). Предпочтительно, чтобы такая продолжительность составляла от 4 до 20 мин, преимущественно от 7 до 12 минут.
Предпочтительно, чтобы на этапе (21) смесь жидкость/жидкость поддерживалась однородной, что достигается интенсивным перемешиванием реакционной среды (например, использование реактора-смесителя или замкнутого контура).
На практике силоксановая жидкость F2, называемая также "кислотным силоксом", содержит, например, около 0,6-3 вес.% остаточного галогена типа НХ и 
В качестве примера, концентрация галогенида водорода в S2 составляет порядка 30% в экспериментальных условиях, указываемых ниже: температура - около 25oС, время протекания реакции на этапе (21) - от 7 до 12 минут.
В соответствии с частным вариантом осуществления изобретения на этапе (2) дополнительно предусмотрена по меньшей мере одна операция разделения (22), предпочтительно отстаиванием силоксановой жидкости F2, выходящей после этапа (21), от концентрированного водного раствора S2, причем последний предпочтительно по крайней мере частично рециклируют на влажный гидролиз (21) этапа (2) и/или на гидролиз под давлением (11) этапа (1) для подачи в качестве добавочной гидролизной воды.
Разделение (22) производят, например, гравитационным отстаиванием.
Возможности рециклирования остатка S2 на этап (11) и/или этап (21) повышают экономичность способа согласно изобретению.
Силоксановая жидкость F2, выходящая после этапа (2), содержит еще небольшое количество гидролизуемых связей 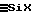 и капельки галогенсодержащего (хлористо-водородного) раствора S2. Поэтому для завершения гидролиза и очистки осуществляют затем этап (3) глубокого гидролиза, включающий по меньшей мере одну обработку (31) силоксановой жидкости F2, полученной на этапе (2) с помощью водной фазы разбавленного водного раствора S3, содержание воды в которой больше или равно 90 вес.%, предпочтительно составляет от 92 до 97%, причем количества используемых на этапе (31) F2 и S3 должны быть такими, чтобы массовое соотношение подаваемых фаз RМ3 = водная фаза/жидкость F2 (кислотный силокс) превышало или было равно 1,5, предпочтительно 2-10.
и капельки галогенсодержащего (хлористо-водородного) раствора S2. Поэтому для завершения гидролиза и очистки осуществляют затем этап (3) глубокого гидролиза, включающий по меньшей мере одну обработку (31) силоксановой жидкости F2, полученной на этапе (2) с помощью водной фазы разбавленного водного раствора S3, содержание воды в которой больше или равно 90 вес.%, предпочтительно составляет от 92 до 97%, причем количества используемых на этапе (31) F2 и S3 должны быть такими, чтобы массовое соотношение подаваемых фаз RМ3 = водная фаза/жидкость F2 (кислотный силокс) превышало или было равно 1,5, предпочтительно 2-10.
В том случае, когда содержание воды менее 100%, остальное до 100% составляет растворенное вещество. Растворенное вещество, содержащееся из расчета не более 10 вес. % в водной фазе предпочтительно 3-8%, содержит в большем количестве по меньшей мере одну соль и, возможно, в меньшем количестве по меньшей мере один кислый или основный агент.
Указанная обработка (3 л) позволяет завершить гидролиз остаточных связей 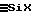 в среде F2, которая преобразуется в этом случае в силоксановую жидкость F3 и состоит главным образом из линейных α, ω -дигидроксилированных олигомеров и в меньшей степени из цикличных олигомеров.
в среде F2, которая преобразуется в этом случае в силоксановую жидкость F3 и состоит главным образом из линейных α, ω -дигидроксилированных олигомеров и в меньшей степени из цикличных олигомеров.
Предпочтительно, чтобы время протекания реакции при глубоком гидролизе (31) превышало или было равно 5 мин, преимущественно 10 мин. Однако, разумеется, в любом случае окончание такой реакции (31) определяется моментом, когда будут гидролизованы все группы  Специалист сможет определить такой момент с тем, чтобы свести к минимуму продолжительность осуществления промышленного способа.
Специалист сможет определить такой момент с тем, чтобы свести к минимуму продолжительность осуществления промышленного способа.
Согласно наиболее предпочтительному варианту осуществления изобретения, водная фаза S3 содержит растворенное вещество, количество которого составляет от 3 до 8 вес.% в водной фазе и которая содержит в наибольшем количестве по меньшей мере одну соль и в наименьшем - НСl.
Согласно одной характеристики изобретения соль или соли, применяемые на этапе (31), выбраны из группы: NaCl, LiCl, NaHCO3, Na2CO3.
Такая соль (соли) может быть введена непосредственно в водную фазу Sз и/или получена на месте путем нейтрализации всего количества или только части присутствующего галогенида водорода посредством по меньшей мере одного основного агента, предпочтительно выбранного из следующих оснований: NaOH, КОН, LiOH, причем NaOH особенно предпочтителен.
Возможное присутствие соли в водной фазе, которая может быть получена путем нейтрализации HCl посредством основания, позволяет оптимизировать этап разделения (32) (отстаивание).
Согласно предпочтительной характеристике изобретения этап (31) протекает при температуре свыше или равной 60oС, предпочтительно при температуре от 70 до 100oС и еще более предпочтительно при температуре от 80 до 90oС.
Предпочтительно, чтобы в F3 содержание остаточного галогена типа НХ и = SiX составляло менее или равно 60 pmm, предпочтительно менее или равно 30 pmm; необходимо отметить, что содержание остаточного галогена типа =SiX составляет, как правило, менее 1 ppm. Реакция протекает полностью.
На практике предпочтительно, чтобы была предусмотрена гомогенизация реакционной среды на этапе (31) (например, использование реактора-смесителя или замкнутого контура).
Этап (3) содержит, кроме того, по меньшей мере одну операцию разделения (32), предпочтительно отстаиванием силоксановой жидкости F3, выходящей после (31), от водной фазы, причем последняя после рекуперации рециклируется по меньшей мере частично по меньшей мере на этап (31) и при необходимости на этап (этапы) (11) и/или (21), поскольку упомянутая водная фаза не содержит соли. Такое отстаивание позволяет получить масло или силоксановую жидкость F3, в которой все или почти все связи =SiX подвергнуты гидролизу.
Силоксановая жидкость f3, рекуперируемая в конце этапа разделения (32), полностью гидролизована, однако еще содержит несколько капелек водной фазы.
Поэтому согласно изобретению силоксановую жидкость F3 дополнительно направляют на этап (4) коалесценции, заключающейся главным образом в пропускании жидкости F3 через пористый материал, с тем чтобы извлечь капельки водной кислотной фазы, содержащиеся в F3, и получить силоксановую жидкость F4, состоящую главным образом из линейных α, ω -дигидроксилированных олигомеров с содержанием остаточного галогена менее или равным 10 ppm, как правило 2 ppm, и в меньшей степени из цикличных олигомеров (30%).
Предпочтительно, пористый материал, применяемый на этапе (4), выбирают из следующей группы материалов:
- осажденная двуокись кремния,
- тканые или нетканые волокнистые подложки, например из стекловолокна и/или полимерных волокон (целлюлоза, полиэфир, полипропилен), стекловолокно,
- цеолиты
- и их смеси.
Согласно предпочтительной и оригинальной характеристике изобретения силоксановые жидкости F3 и силоксановая жидкость F4, в которой отсутствуют связи =SiX, практически не содержат больше капелек водной фазы и имеют вязкость при 25oС менее или равную 50 мПа•с, преимущественно 40 мПа•с и более предпочтительно 25-35 мПа•с.
Такая низкая вязкость наиболее желательна, поскольку она существенно облегчает переработку масс, получаемых в процессе способа. Помимо этого очень важна последовательность этапов способа согласно изобретению, обеспечивающих в конечном итоге получение полиорганосилоксана высокого качества и высокой чистоты и делающих способ легко осуществимым и воспроизводимым.
Более конкретно, обработка жидкости F3 фильтрацией через пористую подложку, позволяющая обеспечить коалеcценцию остаточных капелек водной фазы, происходит предпочтительно в форме потока (расход на единицу площади) при скорости от 1 до 10 см/мин и при температуре порядка, например, 50oС.
Предпочтительно, чтобы конечное содержание линейных α, ω -дигалогенированных олигомеров в жидкости F4 составляло от 50 до 70 вес.%, остальное приходится на циклические олигоорганосилоксаны.
В связи с тем что, как было сказано выше, предпочтительным галогеном Х - в рамках изобретения - является хлор, то в качестве неограничивающего примера предпoчитают использовать в качестве исходного органогалосилана Me2SiCl2, MeSiCl3 или Ме3SiСl.
Каким бы ни был режим осуществления способа согласно изобретению: непрерывный или периодический, средний специалист полностью в состоянии подобрать необходимую аппаратуру (закрытые или незакрытые реакторы, отстойники, детендеры, устройства коалесценции. . . ), позволяющую осуществить рассматриваемый способ.
Изобретение будет лучше понято благодаря приводимым ниже примерам на получение полидиметилсилоксанового масла F4 путем гидролиза диметилдихлорсилана (Me2SiCl2). Эти примеры наглядно показывают все преимущества и возможные варианты осуществления способа согласно изобретению.
Прилагаемый чертеж изображает блок-схему способа, приведенную в качестве неограниченного примера для иллюстрации изобретения. Чертеж иллюстрирует способ, осуществляемый в непрерывном режиме, при котором поток 1 продукта Me2SiCl2 направляется на этап (11) гидролиза, осуществляемого под давлением. Потоки 14 и 15 хлористо-водородного раствора S1, образованные соответственно рециклируемыми растворами хлористо-водородной кислоты S3 и S2 с этапов 3 и 2, непосредственно направляются на гидролиз (11).
При этом гидролизе образуется сырой гидролизат, содержащий силоксановую жидкость F1, газообразную НСl и водную фазу, образованную насыщенным раствором S1 хлористо-водородной кислоты. Данный гидролизат представлен потоком 2, который направляют на этап (12) разделения "газ/жидкость", в результате чего образуется газовый поток 3, направляемый на очистку (14), проводимую путем конденсации при температуре -5oС, причем данная очистка (14) позволяет получить газообразную НСl при высоком давлении в виде потока 6. На этапе разделения (12) образуется также поток 4, состоящий из смеси "жидкость/жидкость", которую подвергают разделению на этапе (13), в процессе которого образуется поток 5 раствора S1, рециклируемого на стадию (11), и поток 7, состоящий из силоксановой жидкости (хлорсилокс F1).
Хлорсилокс F1 подвергают дегазации на этапе (15) декомпрессией, в результате чего образуется газовый поток 8, состоящий из НСl низкого давления, и поток 9, образованный дегазированной силоксановой жидкостью F1.
Последняя подвергается влажному гидролизу на этапе (21), для осуществления которого подают поток 10 для подпитки водой и поток 17 для подпитки рециклируемым водным раствором S2 хлористо-водородной кислоты. Влажный гидролиз (21) позволяет получить поток 11, состоящий из жидкой смеси силоксановой жидкости F2 с раствором S2 хлористо-водородной кислоты. Данный поток 11 подвергается разделению жидкость/жидкость на этапе (22) отстаиванием. В результате получают, с одной стороны, поток 13, состоящий из водного раствора S2 HCl и разделяемый на поток 17, рециклируемый на этап (21), и на поток 14, рециклируемый на этап (11) в виде раствора S2. Избыток S2, рециклируемого потоком 14, удаляется в виде потока 15. При разделении (22) образуется также поток 12, состоящий из силоксановой жидкости F2 и направляемый на глубокий гидролиз (31), для чего подают поток 16 едкого натра и поток 22 декантированного водного раствора. Поток 18, образующийся на этапе (31), представляет собой жидкую смесь полиорганосилоксана с хлористо-водородным раствором S3. После разделения на этапе (32) потока 18 образуется поток 20 хлористо-водородного раствора S3 и поток 19 силоксановой жидкости F3.
Полученная силоксановая жидкость F3 затем подвергается коалесценции (4), в результате чего образуется поток 21, состоящий из силоксановой жидкости F4 или полидиметилсилоксанового масла, из которого почти полностью удалены капельки водной HCl.
ПРИМЕР 1
1-й этап:
В блок, состоящий из реактора, дегазатора, отстойника и работающий при рабочем давлении b=0,3 МПа (абс.) и температуре Т=30oС, подают:
- Me2SiCl2, 1,98 кг/ч,
- раствор HCl с концентрацией 26%, 0,34 кг/ч,
- водный раствор HCl концентрацией 44%, из расчета 23,6 кг/ч,
таким образом, чтобы массовое соотношение RM1 подаваемых фаз, водной и силоксановой, составляло 12.
Хлорсилокс или силоксановая жидкость F1, получаемая после этапа (15), производилась в количестве 1,2 мг/ч.
Весовой состав F1 следующий:
- 9,7% остаточного хлора (типа НСl и =SiCl),
- 33% циклических олигоорганосилоксанов,
- 67% линейных, α, ω -хлорированных олигоорганосилоксанов.
Хлорсилокс F1 имеет вязкость, равную 4,7 мПа•с.
2-й этап:
Силоксановую жидкость F1 вводят в другой блок, состоящий из реактора и отстойника, из расчета 1,2 кг/ч. Одновременно подают в указанный блок "реактор/отстойник" воду из расчета 0,31 кг/ч и водный раствор НСl с концентрацией 28,7% из расчета 5 кг/ч таким образом, чтобы RM2=4,4.
Давление внутри реактора на этапе (2) составляет 0,1 МПа, температура - порядка 30oС.
силоксановую жидкость F2, полученную на этапе (22) (обозначаемую также кислотным силоксом), производят в количестве 1,17 кг/ч. Указанный кислотный силокс F2 содержит 1,3 вес.% остаточного хлора (типа НСl и =SiCl).
Вязкость кислотного силокса F2 составляет 28 мПа•с при 25oС.
F2 содержит 37 вес. % циклического полидиметилсилоксана и 63 вес.% α, ω -дихлорированного и α, ω -дигидроксилированного полидиметилсилоксана.
3-й этап:
Кислотный силокс F2 подают в блок, состоящий из реактора и отстойника, из расчета 1,17 кг/ч одновременно с подачей водного раствора едкого натра с концентрацией 3 вес.% NaOH при расходе 0,46 кг/ч и с подачей рециклированной водной фазы, полученной на этапе разделения (32) с расходом 4,5 мг/ч.
RМ3=4,2.
Концентрация NaCl (соль при нейтрализации) в водной фазе составляет 4,2 вес.%.
Давление внутри блока "реактор/отстойник" составляет 0,1 МПа, температура - 85oС.
Силоксановую жидкость F3, полученную в конце этапа (32), производят в количестве 1,14 кг/ч.
Данная силоксановая жидкость F3, называемая также нейтральным силоксом, содержит 50 ppm остаточного хлора (типа НСl и =SiCl; при этом содержание хлора только типа =SiCl менее 1 ppm), 37 вес.% циклического полидиметилсилоксана и 63 вес.% линейного практически α, ω -дигидроксилированного полидиметилсилоксана.
Вязкость F3 составила 37 мПа•с при 25oС.
4-й этап:
Нейтральный силоксан F3 фильтровали через пористую основу, выполненную из осажденной двуокиси кремния, с порами среднего диаметра 350 мкм. Расход подаваемой F3 составляет 1,14 кг/ч, скорость прохождения через пористую основу - 1,5 см/мин, давление - 0,1 МПа, температура 20oС.
Полидиметилсилоксан F4 получают из расчета 1,14 кг/ч. В нем содержалось 1 ppm общего остаточного хлора, 63 вес.% линейного α, ω -дигидроксилированного полидиметилсилоксана и 37 вес.% циклического полидиметилсилоксана.
Вязкость F4 составляет 37 мПа•с при 25oС.
Описывается способ получения полиорганосилоксанов, заключающийся в гидролизе органогалогенсиланов формулы I RаR1 bSiXc, в которой R, R1 - радикалы, одинаковые или разные, означают водород или линейные или разветвленные алкилы C1-C6, арилы, алкиларилы или аралкилы; Х означает галоген; а+b+с=4 и 0 < с < 4; заключающемся в том, что осуществляют а) по меньшей мере три последовательных этапа гидролиза (1), (2), (3), на которых реакционная среда имеет возрастающую гидролизующую силу на органогалосиланы (1), б) и при необходимости по меньшей мере один этап коалесценции (4), причем этап (1) представляет собой гидролиз под давлением в присутствии водного раствора S1 насыщенного галогенида водорода, проводимый при давлении и температуре реакционной среды. Техническим результатом является практически полное устранение связей ≡Si-X, получение полиорганосилоксана высокого качества, высокой чистоты для обеспечения его максимальной стабильности. 17 з.п.ф-лы, 1 ил.
RаRb 1SiXc,
в которой R, R1 - радикалы, одинаковые или разные, означают водород или линейные или разветвленные алкилы C1-C6, арилы, алкиларилы или аралкилы;
Х означает галоген;
а+b+с=4 и 0<с<4,
отличающийся тем, что осуществляют по меньшей мере три последовательных этапа гидролиза (1), (2), (3) органогалогенсиланов формулы 1, реакционная среда на которых имеет возрастающую гидролизующую силу, и при необходимости осуществляют по меньшей мере один этап коалесценции, при этом этап (1) представляет собой гидролиз под давлением в присутствии насыщенного водного раствора S1 галогенида водорода, проводимый при давлении и температуре реакционной среды.
а) по меньшей мере один этап гидролиза (l1) органогалогенсиланов формулы I с получением сырого, по меньшей мере трехфазного гидролизата, содержащего газовую фазу, образованную газообразным галогенидом водорода под давлением, силоксановую жидкость F1, состоящую преимущественно из линейных α, ω -дигалогенированных олигомеров и, возможно, в меньшей мере из цикличных олигомеров, водную фазу, содержащую насыщенный галогенидом водорода раствор S1;
б) по меньшей мере одно разделение (12) газ/жидкость с получением газообразного галогенида водорода под давлением, с одной стороны, и смеси F1/S1, с другой стороны;
в) по меньшей мере одно разделение (13) жидкость/жидкость с получением раствора F1 и S1, причем водную фазу преимущественно рециклируют на первый этап гидролиза (11);
г) при необходимости по меньшей мере одну очистку (14) газа, получаемого на этапе (12), предпочтительно путем конденсации;
д) при необходимости, по меньшей мере одно дополнительное дегазирование (15) жидкости F1, полученной на этапе разделения (13), предпочтительно путем декомпрессии.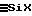 силоксановой жидкости F1 на уровне не менее 95%, предпочтительно не менее 98%, с получением таким образом силоксановой жидкости F2, состоящей главным образом из линейных α, ω-дигидроксилированных или α, ω- дигалогенированных олигомеров, и возможно в меньшей мере из цикличных олигомеров.
силоксановой жидкости F1 на уровне не менее 95%, предпочтительно не менее 98%, с получением таким образом силоксановой жидкости F2, состоящей главным образом из линейных α, ω-дигидроксилированных или α, ω- дигалогенированных олигомеров, и возможно в меньшей мере из цикличных олигомеров. в среде F2, с получением силоксановой жидкости F3, состоящей главным образом из линейных α, ω - гидроксилированных олигомеров и в меньшей мере из цикличных олигомеров.
в среде F2, с получением силоксановой жидкости F3, состоящей главным образом из линейных α, ω - гидроксилированных олигомеров и в меньшей мере из цикличных олигомеров.
| US 5169970 А, 08.12.1992 | |||
| Устройство для приема команд телеуправления локомотивом | 1976 |
|
SU658588A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОРГАНОСИЛОКСАНОВЫХ СМОЛ | 0 |
|
SU407931A1 |

