По настоящей заявке испрашивается приоритет на основании заявки на патент США с порядковым номером 12/244968, озаглавленной «Соединения, композиции и способы, предназначенные для лечения β-амилоидных заболеваний и синуклеинопатий» и поданной 3 октября 2008 г. Esposito и соавторами.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к бис-дигидроксиарильным соединениям и их фармацевтически приемлемым солям, их синтезу, содержащим их фармацевтическим композициям и их применению для лечения Aβ-амилоидных заболеваний, например, болезни Альцгеймера, и синуклеинопатий, например, болезни Паркинсона, а также в производстве лекарственных средств, предназначенных для лечения упомянутых заболеваний.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Болезнь Альцгеймера характеризуется накоплением пептида, состоящего из 39-43 аминокислот и именуемого β-амилоидным белком или Aβ, в фибриллярной форме, существующего в виде агрегатов во внеклеточных амилоидных бляшках и в виде амилоида в стенках кровеносных сосудов мозга. Считается, что отложение агрегатов фибриллярного Aβ-амилоида при болезни Альцгеймера приносит вред пациенту и постепенно приводит к токсическому поражению и смерти нервных клеток, т.е. характерным признакам болезни Альцгеймера. Накапливающиеся данные позволяют считать амилоид, и более конкретно, образование, отложение, накопление и/или устойчивое существование Aβ-агрегатов, основным причинным фактором патогенеза болезни Альцгеймера. Кроме того, помимо болезни Альцгеймера, образование, отложение, накопление и устойчивое существование Aβ-агрегатов характерно для ряда других амилоидных заболеваний, в т.ч. синдрома Дауна, расстройств, включающих конгофильную ангиопатию, например, но не ограничиваясь этим, наследственную церебральную геморрагию голландского типа, а также церебральную β-амилоидную ангиопатию.
Болезнь Паркинсона является другим заболеванием человека, характеризующимся образованием, отложением, накоплением, агрегацией и/или устойчивым существованием аномальных отложений фибриллярного белка, который похож на амилоид по многим характеристикам. Считается, что при болезни Паркинсона накопление цитоплазмических телец Леви, состоящих из агрегатов или нитей α-синуклеина, играет важную роль в патогенезе и является мишенью для терапевтического воздействия. Новые средства или соединения, которые способны препятствовать образованию, отложению, накоплению, агрегации и/или устойчивому существованию α-синуклеина или разрушать ранее образовавшиеся фибриллы или агрегаты (или их части), рассматриваются в качестве потенциальных терапевтических средств для лечении болезни Паркинсона и родственных синуклеинопатий. Фрагмент α-синуклеина, состоящий из 35 аминокислот и обладающий способностью образовывать амилоид-подобные фибриллы или агрегаты, был обнаружен in vitro или в тканях мозга пациентов с болезнью Паркинсона. Этот фрагмент α-синуклеина является довольно важной терапевтической мишенью, поскольку считается, что он отвечает за образование телец Леви, которые наблюдаются у всех пациентов с болезнью Паркинсона, синуклеинопатиями и родственными расстройствами. Кроме того, белок α-синуклеин, который образует фибриллы или агрегаты и является Конго красный- и Тиофлавин S-положительным (особые красители, применяемые для выявления агрегатов амилоидных фибрилл), обнаружен в качестве составной части телец Леви в ткани мозга пациентов с болезнью Паркинсона, болезнью с тельцами Леви (Lewy in Handbuch der Neurologie, M. Lewandowski, ed., Springer, Berlin pp. 920-933, 1912; Pollanen et al., J. Neuropath. Exp. Neurol. 52:183-191, 1993; Spillantini et al., Proc. Natl. Acad. Sci. USA 95:6469-6473, 1998; Arai et al., Neurosci. Lett. 259:83-86, 1999), множественной системной атрофией (Wakabayashi et al., Acta Neuropath. 96:445-452, 1998), деменцией с тельцами Леви и вариантом болезни Альцгеймера с тельцами Леви. При болезни Паркинсона в мозге пациента появляются агрегаты, которые являются Конго красный- и Тиофлавин S-положительными, и в основном включают бета-складчатые листовые вторичные структуры.
Амилоид, как терапевтическая мишень при болезни Альцгеймера
Болезнь Альцгеймера является тяжелой экономической нагрузкой на общество. По данным современных исследований стоимость ухода за одним пациентом с болезнью Альцгеймера с тяжелыми когнитивными нарушениями на дому или в доме престарелых составляет более 47000 долларов в год (A Guide to Understanding Alzheimer's Disease and Related Disorders). Для заболевания, которое может продолжаться от 2 до 20 лет, общая стоимость болезни Альцгеймера для семей и для общества является непомерно высокой. Ежегодные экономические потери от болезни Альцгеймера в Соединенных Штатах с точки зрения затрат на уход и потерь в заработке, как самого пациента, так и тех, кто за ним ухаживает, оцениваются в 80-100 млрд. долларов (2003, Progress Report on Alzheimer's Disease).
Такрина гидрохлорид («Cognex») - первое одобренное FDA лекарственное средство для болезни Альцгеймера - представляет собой ингибитор ацетилхолинэстеразы (Cutler and Sramek, N. Engl. J. Med. 328:808-810, 1993). Однако было показано, что применение этого лекарственного средства позволяет добиться лишь ограниченного улучшения когнитивной способности у пациентов с болезнью Альцгеймера и само средство обладает значительными побочными эффектами, например, токсическим действием на печень. Второй одобренный FDA препарат, а именно донепезил («Aricept»), который также является ингибитором ацетилхолинэстеразы, более эффективен, чем такрин, демонстрируя небольшое улучшение когнитивных способностей у пациентов с болезнью Альцгеймера (Barner and Gray, Ann. Pharmacotherapy 32:70-77, 1998; Rogers and Friedhoff, Eur. Neuropsych. 8:67-75, 1998), но и он, по-видимому, не является средством излечения. Поэтому очевидно, что существует потребность в более эффективных средствах лечения пациентов с болезнью Альцгеймера.
Для болезни Альцгеймера характерно отложение и накопление пептида, содержащего 39-43 аминокислотных остатков и именуемого бета-амилоидным белком, Aβ или β/A4 (Glenner and Wong, Biochem. Biophys. Res. Comm. 120:885-890, 1984; Masters et al., Proc. Natl. Acad. Sci. USA 82:4245-4249, 1985; Husby et al., Bull. WHO 71:105-108, 1993). Aβ образуется при расщеплении протеазой более крупных белков-предшественников, именуемых белками-предшественниками β-амилоида (APP), три из которых существуют в виде нескольких альтернативно сшитых вариантов. Наиболее часто встречающиеся формы APP включают белки, состоящие из 695, 751 и 770 аминокислот (Tanzi et al., Nature 31:528-530, 1988).
Малый пептид Aβ является основным компонентом, который превращает отложения амилоида в мозге пациентов с болезнью Альцгеймера в «бляшки». Кроме того, болезнь Альцгеймера характеризуется наличием многочисленных нейрофибриллярных «клубков», состоящих из парных спиральных нитей, которые аномально накапливаются в нейрональной цитоплазме (Grundke-Iqbal et al., Proc. Natl. Acad. Sci. USA 83:4913-4917, 1986; Kosik et al., Proc. Natl. Acad. Sci. USA 83:4044-4048, 1986; Lee et al., Science 251:675-678, 1991). Следовательно, патологический признак болезни Альцгеймера заключается в наличии «бляшек» и «клубков», причем в центральном ядре бляшек откладывается β-амилоид. Другими основными типами поражений, обнаруженных в мозге пациентов с болезнью Альцгеймера, является накопление β-амилоида в стенках кровеносных сосудов, как в паренхиме мозга, так и в стенках сосудов мозговой оболочки, находящейся за пределами мозга. Отложения β-амилоида, находящиеся в стенках кровеносных сосудов, именуют цереброваскулярным амилоидом или конгофильной ангиопатией (Mandybur, J. Neuropath. Exp. Neurol. 45:79-90, 1986; Pardridge et al., J. Neurochem. 49:1394-1401, 1987).
В течение многих лет происходили непрерывные споры ученых о значении «β-амилоида» в болезни Альцгеймера и о том, являются ли характерные «бляшки» и «клубки» причиной или только следствием заболевания. Исследования последних нескольких лет показали, что β-амилоид действительно является причинным фактором для болезни Альцгеймера и его не следует рассматривать в качестве безвредного свидетельства заболевания. Было показано, что Aβ-белок, обнаруживаемый при болезни Альцгеймера, в клеточной культуре способен вызывать дегенерацию нервных клеток за короткий промежуток времени (Pike et al., Br.Res. 563:311-314, 1991; J. Neurochem. 64:253-265, 1995). Исследования дают основание предположить, что именно фибриллярная структура (в основном состоящая из β-складчатых листовых вторичных структур) отвечает за нейротоксическое действие. Кроме того, было обнаружено, что Aβ проявляет нейротоксичность в срезах культур гиппокампуса (Harrigan et al., Neurobiol. Aging 16:779-789, 1995) и вызывает смерть нервных клеток у трансгенных мышей (Games et al., Nature 373:523-527, 1995; Hsiao et al., Science 274:99-102, 1996). Инъекция Aβ Альцгеймера в мозг крысы также вызывает ухудшение памяти и нарушение деятельности нейронов (Flood et al., Proc. Natl. Acad. Sci. USA 88:3363-3366, 1991; Br. Res. 663:271-276, 1994).
Возможно наиболее убедительные доказательства того, что Aβ-амилоид принимает непосредственное участие в патогенезе болезни Альцгеймера, были получены в результате генетических исследований. Было обнаружено, что выработка Aβ может являться следствием мутаций в генах, кодирующих его предшественника, т.е. белка-предшественника β-амилоида (Van Broeckhoven et al., Science 248:1120-1122, 1990; Murrell et al., Science 254:97-99, 1991; Haass et al., Nature Med. 1:1291-1296, 1995). Выявление мутаций в гене белка предшественника β-амилоида, которые вызывают раннее начало семейной болезни Альцгеймера, является сильнейшим аргументом в пользу того, что амилоид является основным участником патогенетического процесса, лежащего в основе этого заболевания. Были обнаружены и описаны четыре мутации, вызывающие заболевание, что демонстрирует важность Aβ в появлении семейной болезни Альцгеймера (смотрите обзор в Hardy, Nature Genet. 1:233-234, 1992). Все перечисленные данные дают основание предположить, что разработка лекарственного средства, способного уменьшить, устранить или предотвратить образование, агрегацию, отложение, накопление и/или устойчивое существование фибриллярного Aβ в мозге пациентов, позволит получить эффективное терапевтическое средство.
Болезнь Паркинсона и синуклеинопатии
Болезнь Паркинсона представляет собой нейродегенеративное расстройство, патология которого характеризуется наличием телец Леви в цитоплазме (Lewy in Handbuch der Neurologie, M. Lewandowski, ed., Springer, Berlin pp. 920-933, 1912; Pollanen et al., J. Neuropath. Exp. Neurol. 52:183-191, 1993), основным компонентом которых являются нити, состоящие из α-синуклеина (Spillantini et al., Proc. Natl. Acad. Sci. USA 95:6469-6473, 1998; Arai et al., Neurosci. Lett. 259:83-86, 1999), т.е. белка, состоящего из 140 аминокислот (Ueda et al., Proc. Natl. Acad. Sci. USA 90:11282-11286, 1993). Были описаны две преобладающие мутации в гене α-синуклеина, вызывающие раннее семейное начало болезни Паркинсона, и это дает основание предположить, что тельца Леви механически способствуют дегенерации нейронов при болезни Паркинсона и родственных расстройствах (Polymeropoulos et al., Science 276:2045-2047, 1997; Kruger et al., Nature Genet. 18:106-108, 1998). Не так давно в результате исследований in vitro было продемонстрировано, что рекомбинантный α-синуклеин действительно может образовывать фибриллы или агрегаты, подобные тельцам Леви (Conway et al., Nature Med. 4:1318-1320, 1998; Hashimoto et al., Brain Res. 799:301-306, 1998; Nahri et al., J.Biol.Chem. 274:9843-9846, 1999). Важнее всего то, что обе мутации α-синуклеина, связанные с болезнью Паркинсона, ускоряют этот процесс агрегации, показывая, что эти исследования in vitro могут иметь отношение к патогенезу болезни Паркинсона. Агрегация альфа-синуклеина и образование фибрилл соответствуют критериям процесса полимеризации, зависимого от образования активных центров (Wood et al., J. Biol. Chem. 274:19509-19512, 1999). В этом отношении образование фибрилл или агрегация α-синуклеина сходны с соответствующими процессами для волокон β-амилоидного белка (Aβ) при болезни Альцгеймера. Как рекомбинантный белок альфа-синуклеин, так и не-Aβ компонент (известный как NAC), который представляет собой пептидный фрагмент α-синуклеина, состоящий из 35 аминокислот, обладают способностью образовывать фибриллы или агрегаты в случае инкубирования при 37°C и демонстрируют положительную реакцию на амилоидные красители, например, Конго красный (демонстрируя двойное лучепреломление в красных/зеленых лучах при наблюдении в поляризованном свете) и Тиофлавин S (демонстрируя положительную флуоресценцию) (Hashimoto et al., Brain Res. 799:301-306, 1998; Ueda et al., Proc. Natl. Acad. Sci. USA 90:11282-11286, 1993).
Синуклеины представляют собой семейство небольших предсинаптических нейрональных белков, состоящее из α-, β- и γ-синуклеинов, из которых только агрегаты α-синуклеина связаны с несколькими неврологическими заболеваниями (Ian et al., Clinical Neurosc. Res. 1:445-455, 2001; Trojanowski and Lee, Neurotoxicology 23:457-460, 2002). Роль синуклеинов (и в частности, альфа-синуклеина) в этиологии ряда нейродегенеративных заболеваний была установлена на основе ряда наблюдений. С точки зрения патологии синуклеин был идентифицирован как основной компонент телец Леви, т.е. включений, характерных для болезни Паркинсона, и его фрагмент был выделен из амилоидных бляшек, характерных для другого неврологического заболевания, а именно болезни Альцгеймера. С биохимической точки зрения было показано, что рекомбинантный α-синуклеин образует фибриллы или агрегаты, которые повторяют особенности ультраструктуры альфа-синуклеина, выделенного из тканей пациентов с деменцией с тельцами Леви, болезнью Паркинсона и множественной системной атрофией. Кроме того, идентификация мутаций в гене синуклеина, которые, хотя и в редких случаях, возникают при семейной болезни Паркинсона, продемонстрировала однозначную связь между синуклеиновой патологией и нейродегенеративными заболеваниями. Общее участие α-синуклеина в целом ряде заболеваний, таких как болезнь Паркинсона, деменция с тельцами Леви, множественная системная атрофия и вариант болезни Альцгеймера с тельцами Леви, привело к классификации этих заболеваний под обобщающим термином «синуклеинопатии».
Фибриллы или агрегаты α-синуклеина, возникающие при болезни Паркинсона, и волокна Aβ, возникающие при болезни Альцгеймера, преимущественно состоят из β-складчатых листовых структур. Для соединений, которые, как было найдено, ингибируют образование фибрилл Aβ-амилоида при болезни Альцгеймера, была также показана эффективность при ингибировании образования или агрегации фибрилл α-синуклеина, что показано в примерах настоящего изобретения. Следовательно, помимо эффективности в качестве средства лечения болезни Альцгеймера, эти соединения могли бы также служить в качестве средств лечения болезни Паркинсона и других синуклеинопатий.
Болезнь Паркинсона и болезнь Альцгеймера характеризуются неадекватным накоплением нерастворимых агрегатов, состоящих в основном из неправильно уложенных белков, которые обогащены β-складчатыми листовыми вторичными структурами (см. обзор в Cohen et al., Nature 426:905-909, 2003; Chiti et al., Annu. Rev. Biochem., 75:333-366, 2006). При болезни Паркинсона основной компонент этих агрегатов, являющихся частями телец Леви, представляет собой α-синуклеин, и мутации α-синуклеина, которые увеличивают его склонность к неправильной укладке и агрегации, наблюдаются при семейной болезни Паркинсона (Polymeropoulos et al., Science 276:2045-2047, 1997; Papadimitriou et al., Neurology 52:651-654, 1999).
Митохондриальная дисфункция, в частности, в результате нарушения комплекса I цепи переноса электронов, также является обычной особенностью болезни Паркинсона (Schapira et al., J. Neurochem., 54:823-827, 1990; обзор в Greenamyre et al., IUBMB Life, 52:135-141, 2001). Непосредственное доказательство митохондриальной недостаточности в этиологии болезни Паркинсона было впервые получено на основании наблюдения, что MPP+ (1-метил-4-фенил-2,3-дигидропиридиний), т.е. активный метаболит токсина паркинсонизма N-метил-4-фенил-1,2,3,6-тетрагидропиридина (MPTP), ингибирует упомянутый комплекс I (Nicklas et al., Life Sci., 36:2503-2508, 1985). Затем было показано, что ротенон, т.е. другой ингибитор комплекса I, является улучшенной моделью агрегации α-синуклеина, поскольку помимо изменений в поведении и потери дофаминергических нейронов, что наблюдается в модели MPTP, он воспроизводит упомянутые выше α-синуклеин-положительные цитоплазматические агрегаты. Проявляемую ротеноном токсичность этого типа можно наблюдать в целом ряде модельных систем, включая крыс (Betarbet et al., Nat. Neurosci., 3:1301-1306, 2000; Panov et al., J. Biol. Chem., 280:42026-42035, 2005), срезы крысиного мозга (Sherer et al., J. Neurosci., 23:10756-10764, 2003; Testa et al., Mol. Brain Res., 134:109-118, 2005), C. elegance (Ved et al., J. Biol. Chem., 280:42655-42668, 2005) и культивированные клетки (Sherer et al., J. Neurosci., 22:7006-7015, 2002), и было показано, что эта токсичность является следствием повышенного окислительного поражения митохондрий, которое, в свою очередь, является результатом ингибирования комплекса I.
Для лучшего понимания взаимосвязи окислительного поражения с патогенезом мутировавшего α-синуклеина исследователями была получена клеточная линия нейробластомы (с использованием клеток BE-M17), в которой имеет место избыточная экспрессия α-синуклеина A53T. В этих клетках, в ответ на действие различных средств, вызывающих окислительный стресс, происходит агрегация α-синуклеина A53T и усиливается митохондриальная дисфункция и смерть клеток (Ostrerova-Golts et al., J. Neurosci., 20:6048-6054, 2000). Эти клетки подвержены окислительному стрессу при обработке ротеноном и, следовательно, особенно применимы для тестирования средств, которые могли бы ингибировать агрегацию/образование фибрилл α-синуклеина.
Крайне необходимо обнаружение и идентификация новых соединений или средств, которые являются потенциальными терапевтическими средствами для прекращения образования, отложения, накопления и/или устойчивого существования амилоида, что имеет место при болезни Альцгеймера и болезни Паркинсона.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к бис-дигидроксиарильным соединениям и их фармацевтически приемлемым солям. Эти соединения применимы в лечении β-амилоидных заболеваний и синуклеинопатий.
Указанные соединения представляют собой соединения формулы:
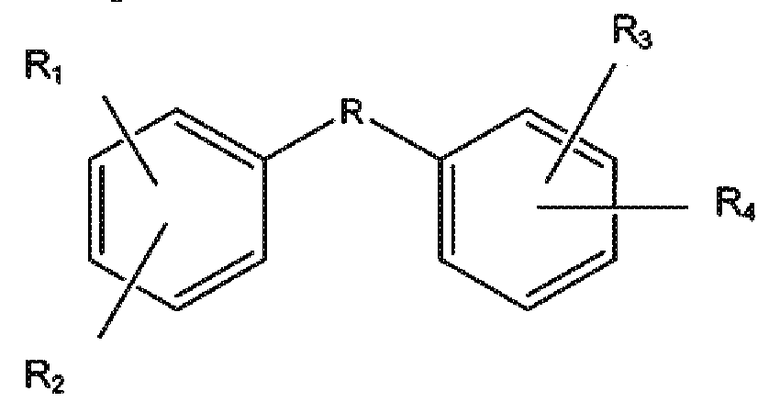 ,
,
где
R1, R2, R3 и R4 представляют собой гидроксильные группы, независимо находящиеся в одном из положений, выбранном из группы, состоящей из 2,3; 2,4; 2,5; 2,6; 3,5; 3,6; 4,5; 4,6 и 5,6, и R выбран из сульфонамида, гетероарила, трициклоалкила и -C(O)NR', где R' выбран из H или CH3, или их фармацевтически приемлемые эфиры или соли.
Кроме того, изобретение относится к любым фармацевтически приемлемым производным этих соединений, включая соли, сложные эфиры, простые или сложные эфиры енолов, ацетали, кетали, ортоэфиры, полуацетали, полукетали, сольваты, гидраты или пролекарства соединений. Фармацевтически приемлемые соли включают, не ограничиваясь этим, соли аминов, как, например, не ограничиваясь перечисленными, N,N'-дибензилэтилендиамина, хлорпрокаина, холина, аммиака, диэтаноламина и других гидроксиламинов, этилендиамина, N-метилглюкамина, прокаина, N-бензилфенэтиламина, 1-парахлорбензил-2-пирролидин-1'-илметилбензимидазола, диэтиламина и других алкиламинов, пиперазина, трис(гидроксиметил)аминометана, соли щелочных металлов, как, например, не ограничиваясь перечисленными, лития, калия и натрия, щелочноземельных металлов, как, например, не ограничиваясь перечисленными, бария, кальция и магния, соли переходных металлов, как, например, но не ограничиваясь этим, цинка, и другие соли металлов, как, например, но не ограничиваясь этим, остатков гидрофосфата натрия и динатрий фосфата, и, кроме того, но не ограничиваясь этим, соли минеральных кислот, как, например, не ограничиваясь указанным, гидрохлориды и сульфаты, соли органических кислот, как, например, не ограничиваясь этим, ацетаты, лактаты, малаты, тартраты, цитраты, аскорбаты, сукцинаты, бутираты, валераты и фумараты.
Кроме того, изобретение относится к фармацевтическим составам, предназначенным для введения любым подходящим путем и любыми подходящими средствами, содержащим эффективные концентрации одного или нескольких соединений по настоящему изобретению или их фармацевтически приемлемых производных, таких как соли, сложные эфиры, простые или сложные эфиры енолов, ацетали, кетали, ортоэфиры, полуацетали, полукетали, сольваты, гидраты или пролекарства соединений, которые доставляют действующее начало в количестве, эффективном для лечения амилоидных заболеваний.
Упомянутые составы представляют собой композиции, подходящие для введения любым желаемым путем, и в их число входят растворы, суспензии, эмульсии, таблетки, диспергируемые таблетки, пилюли, капсулы, порошки, сухие порошки для ингаляции, составы с замедленным высвобождением, аэрозоли для доставки в нос и дыхательные пути, пластыри для трансдермальной доставки, а также композиции для доставки любым другим подходящим путем. Композиции должны подходить для перорального введения, парентерального введения с помощью инъекций, включая подкожные, внутримышечные или внутривенные инъекции в форме водных или масляных растворов или эмульсий, трансдермального введения и других выбранных путей введения.
В настоящем изобретении разработаны способы с применением указанных соединений и композиций, предназначенные для разрушения, измельчения, удаления, уменьшения или выведения фибрилл или агрегатов β-амилоида и α-синуклеина, и, за счет этого, обеспечивающие новые пути лечения β-амилоидных заболеваний и синуклеинопатий.
Кроме того, в изобретении разработаны способы лечения, предупреждения или облегчения одного или нескольких симптомов амилоидных заболеваний или амилоидозов, включая, но не ограничиваясь этим, заболевания, связанные с образованием, отложением, накоплением или устойчивым существованием β-амилоидных фибрилл.
Способы лечения амилоидных заболеваний включают, не ограничиваясь этим, способы лечения болезни Альцгеймера, синдрома Дауна, наследственной церебральной геморрагии с амилоидизом голландского типа и церебральной β-амилоидной ангиопатии.
Кроме того, в изобретении разработаны способы лечения, предупреждения или облегчения одного или нескольких симптомов синуклеиновых заболеваний или синуклеинопатий. В одном из вариантов осуществления эти способы позволяют ингибировать или предупредить образование фибрилл α-синуклеина, ингибировать или предупредить рост фибрилл α-синуклеина и/или вызвать измельчение, разрушение и/или дезагрегацию ранее сформировавшихся агрегатов α-синуклеина и отложений белка, связанных с α-синуклеином. Синуклеиновые заболевания включают, не ограничиваясь указанными, болезнь Паркинсона, семейную болезнь Паркинсона, болезнь с тельцами Леви, вариант болезни Альцгеймера с тельцами Леви, деменцию с тельцами Леви, множественную системную атрофию и комплекс Гуама паркинсонизм-деменция.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Настоящая заявка на патент содержит по меньшей мере один рисунок, выполненный в цвете. Копии настоящей патентной публикации с цветным рисунком (рисунками) будут предоставлены в патентное бюро по запросу и после внесения необходимой оплаты.
На фиг.1A приведено несколько спектров кругового дихроизма, показывающих разрушение Aβ-фибрилл, характерных для болезни Альцгеймера, под действием тестируемых соединений в разбавлении 1:1 масс./масс. На фиг.1B графически показано ингибирование в %.
На фиг.2 приведены сравнительные спектры кругового дихроизма, показывающие, что α-синуклеин образует структуру, обогащенную β-слоями после 4 дней встряхивания при 37°C.
На фиг.3 приведено несколько спектров кругового дихроизма, показывающих, что тестируемые соединения ингибируют агрегацию α-синуклеина при разбавлении 1:1 масс./масс. На фиг.3B графически показано ингибирование в %.
На фиг.4 приведено несколько спектров кругового дихроизма, показывающих, что тестируемые соединения ингибируют агрегацию α-синуклеина при разбавлении 1:0,1 масс./масс. На фиг.4B графически показано ингибирование в %.
На фиг.5 графически суммированы результаты, полученные с использованием Тиофлавина T, относящиеся к ингибированию образования или агрегации фибрилл Aβ при действии тестируемых соединений.
На фиг.6 графически суммированы результаты, полученные с использованием Конго красного, относящиеся к ингибированию образования или агрегации фибрилл Aβ при действии тестируемых соединений.
На фиг.7 графически суммированы результаты, полученные с использованием Тиофлавина T, относящиеся к ингибированию образования или агрегации волокон α-синуклеина при действии тестируемых соединений.
На фиг.8 графически суммированы результаты, полученные с использованием Конго красного, относящиеся к ингибированию образования или агрегации фибрилл α-синуклеина при действии тестируемых соединений.
На фиг.9A-D приведены примеры флуоресцентных микрофотографий, демонстрирующих влияние ротенона на количество тиофлавин S-положительных агрегатов. На фиг.9A показан результат для чистого носителя, фиг.9B соответствует 1 мкМ ротенону, на фиг.9C показан результат для 5 мкМ при низком увеличении, и на фиг.9D - результат для 5 мкМ при высоком увеличении. На фиг.9E суммированы результаты исследования количественного изменения тиофлавин S-положительных агрегатов в ответ на обработку ротеноном.
На фиг.10A-C приведены примеры флуоресцентных микрофотографий, демонстрирующих уменьшение тиофлавин S-положительных агрегатов (зеленая флуоресценция) при действии положительного контрольного соединения. На фиг.10A показаны необработанные образцы, на фиг.10B - образцы, обработанные 500 нг/мл положительного контроля, и на фиг.10C - 1 мкг/мл положительного контрольного соединения. На фиг.10D суммированы результаты количественного анализа дозозависимого уменьшения агрегации.
На фиг.11A-D приведены примеры флуоресцентных микрофотографий, демонстрирующих дозозависимое влияние соединения 1 на образование в клетках тиофлавин S-положительных агрегатов (зеленого цвета) под действием ротенона. На фиг.11A показаны необработанные образцы (только ротенон), и на фиг.11B-D показаны образцы, обработанные, соответственно, 500 нг/мл, 1 мкг/мл и 2 мкг/мл соединения 1. На фиг.11E суммированы результаты количественного анализа действия соединения 1.
На фиг.12A-D приведены примеры флуоресцентных микрофотографий, демонстрирующих, что соединение 2 значительно уменьшает количество тиофлавин S-положительных агрегатов (зеленого цвета) в клетках, образовавшихся под действием ротенона. На фиг.12A показаны необработанные образцы (только ротенон), и на фиг.12B-D показаны образцы, обработанные, соответственно, 500 нг/мл, 1 мкг/мл и 2 мкг/мл соединения 2. На фиг.12E суммированы результаты количественного анализа анти-агрегирующего действия соединения 2.
На фиг.13A-D приведены примеры флуоресцентных микрофотографий, демонстрирующих, что соединение 3 дозозависимым образом уменьшает количество тиофлавин S-положительных агрегатов (зеленого цвета) в клетках, образовавшихся под действием ротенона. На фиг.13A показаны необработанные образцы (только ротенон), и на фиг.13B-D показаны образцы, обработанные, соответственно, 500 нг/мл, 1 мкг/мл и 2 мкг/мл соединения 3. На фиг.13E суммированы результаты количественного анализа анти-агрегирующего действия соединения 3.
На фиг.14A-D приведены примеры флуоресцентных микрофотографий, демонстрирующих, что соединение 4 дозозависимым образом незначительно уменьшает количество тиофлавин S-положительных агрегатов (зеленого цвета) в клетках, образовавшихся под действием ротенона. На фиг.14A показаны необработанные образцы (только ротенон), и на фиг.14B-D показаны образцы, обработанные, соответственно, 500 нг/мл, 1 мкг/мл и 2 мкг/мл соединения 4. На фиг.14E суммированы результаты количественного анализа действия соединения 4.
На фиг.15A-D приведены примеры флуоресцентных микрофотографий, демонстрирующих, что соединение 5 дозозависимым образом умеренно понижает количество тиофлавин S-положительных агрегатов (зеленого цвета) в клетках, образовавшихся под действием ротенона. На фиг.15A показаны необработанные образцы (только ротенон), и на фиг.15B-D показаны образцы, обработанные, соответственно, 500 нг/мл, 1 мкг/мл и 2 мкг/мл соединения 5. На фиг.15E суммированы результаты количественного анализа анти-агрегирующего действия соединения 5.
На фиг.16A-D приведены примеры флуоресцентных микрофотографий, демонстрирующих, что соединение 6 оказывает минимальное дозозависимое влияние на количество тиофлавин S-положительных агрегатов (зеленого цвета) в клетках, образовавшихся под действием ротенона. На фиг.16A показаны необработанные образцы (только ротенон), и на фиг.16B-D показаны образцы, обработанные, соответственно, 500 нг/мл, 1 мкг/мл и 2 мкг/мл соединения 6. На фиг.16E суммированы результаты количественного анализа действия соединения 6.
На фиг.17A-C приведены примеры флуоресцентных микрофотографий, демонстрирующих, что соединение 7 дозозависимым образом умеренно понижает количество тиофлавин S-положительных агрегатов (зеленого цвета) в клетках, образовавшихся под действием ротенона. На фиг.17A показаны необработанные образцы (только ротенон), и на фиг.17B-C показаны образцы, обработанные, соответственно, 500 нг/мл и 2 мкг/мл соединения 7. На фиг.17D суммированы результаты количественного анализа анти-агрегирующего действия соединения 7.
На фиг.18A-D приведены примеры флуоресцентных микрофотографий, демонстрирующих, что соединение 8 дозозависимым образом умеренно понижает количество тиофлавин S-положительных агрегатов (зеленого цвета) в клетках, образовавшихся под действием ротенона. На фиг.18A показаны необработанные образцы (только ротенон), и на фиг.18B-D показаны образцы, обработанные, соответственно, 500 нг/мл, 1 мкг/мл и 2 мкг/мл соединения 8. На фиг.18E суммированы результаты количественного анализа анти-агрегирующего действия соединения 8.
На фиг.19A-D приведены примеры флуоресцентных микрофотографий, демонстрирующих, что соединение 9 дозозависимым образом понижает количество тиофлавин S-положительных агрегатов (зеленого цвета) в клетках, образовавшихся под действием ротенона. На фиг.19A показаны необработанные образцы (только ротенон), и на фиг.19B-D показаны образцы, обработанные, соответственно, 500 нг/мл, 1 мкг/мл и 2 мкг/мл соединения 9. На фиг.19E суммированы результаты количественного анализа анти-агрегирующего действия соединения 9, где *p<0,05 соответствует только 1 мкМ ротенону.
Фиг.20 представляет собой график, демонстрирующий 35-45% уменьшение выживаемости клеток после двух дней обработки ротеноном по данным пробы на цитотоксичность XTT.
Фиг.21 представляет собой график, демонстрирующий способность положительного контрольного соединения ингибировать индуцированную ротеноном токсичность по данным пробы на цитотоксичность XTT.
Фиг.22A представляет собой график, демонстрирующий, что соединение 1 не является токсичным до концентрации 10 мкг/мл. Фиг.22B представляет собой график, демонстрирующий способность соединения 1 защищать от индуцированной ротеноном токсичности по данным пробы на цитотоксичность XTT.
Фиг.23A представляет собой график, демонстрирующий, что соединение 2 не является токсичным до концентрации 25 мкг/мл. Фиг.23B представляет собой график, демонстрирующий способность соединения 2 защищать от индуцированной ротеноном токсичности по данным пробы на цитотоксичность XTT.
Фиг.24A представляет собой график, демонстрирующий, что соединение 3 не является токсичным до концентрации 50 мкг/мл. Фиг.24B представляет собой график, демонстрирующий способность соединения 3 защищать от индуцированной ротеноном токсичности по данным пробы на цитотоксичность XTT.
Фиг.25A представляет собой график, демонстрирующий, что соединение 4 не является токсичным до концентрации 25 мкг/мл. Фиг.25B представляет собой график, демонстрирующий способность соединения 4 защищать от индуцированной ротеноном токсичности по данным пробы на цитотоксичность XTT.
Фиг.26A представляет собой график, демонстрирующий, что соединение 5 не является токсичным до концентрации 25 мкг/мл. Фиг.26B представляет собой график, демонстрирующий способность соединения 5 защищать от индуцированной ротеноном токсичности по данным пробы на цитотоксичность XTT.
Фиг.27A представляет собой график, демонстрирующий, что соединение 6 не является токсичным до концентрации 50 мкг/мл. Фиг.27B представляет собой график, демонстрирующий способность соединения 6 защищать от индуцированной ротеноном токсичности по данным пробы на цитотоксичность XTT.
Фиг.28A представляет собой график, демонстрирующий, что соединение 7 не является токсичным до концентрации 50 мкг/мл. Фиг.28B представляет собой график, демонстрирующий способность соединения 7 защищать от индуцированной ротеноном токсичности по данным пробы на цитотоксичность XTT.
Фиг.29A представляет собой график, демонстрирующий, что соединение 8 не является токсичным до концентрации 25 мкг/мл. Фиг.29B представляет собой график, демонстрирующий способность соединения 8 защищать от индуцированной ротеноном токсичности по данным пробы на цитотоксичность XTT.
Фиг.30A представляет собой график, демонстрирующий, что соединение 9 не является токсичным до концентрации 25 мкг/мл. Фиг.30B представляет собой график, демонстрирующий неспособность соединения 9 защищать от индуцированной ротеноном токсичности по данным пробы на цитотоксичность XTT.
Фиг.31 представляет собой график, демонстрирующий время прохождения по перекладине и эффект от введения соединений. Введение соединений 2 и 7 улучшает двигательную активность в тесте на прохождение по перекладине. После трех месяцев введения соединение 2 статистически значимо (p<0,05) улучшает двигательную активность (измеренную по уменьшению времени прохождения) в тесте на прохождение по перекладине на 49% по сравнению с мышами того же возраста, которым вводили носитель. После шести месяцев введения соединение 7 статистически значимо (p<0,05) улучшает двигательную активность в тесте на прохождение по перекладине на 35% по сравнению с мышью того же возраста, которой вводили носитель. Кроме того, соединение 7 демонстрирует общую тенденцию по улучшению двигательной активности на 39% в течение трех месяцев введения по сравнению с мышами того же возраста, которым вводили носитель.
Фиг.32 представляет собой график, на котором отображено время переворота в тесте с вертикальной стойкой и результат от введения соединений. Введение соединения 7 улучшает двигательную активность в тесте с вертикальной стойкой. Через 3 месяца введения соединение 7 способствует улучшению двигательной активности (измеренное по уменьшению времени переворота) в тесте с вертикальной стойкой, и через 6 месяцев введения соединение 7 статистически значимо (p<0,01) улучшает активность на 41% по отношению к активности перед началом введения. Через 6 месяцев введения соединения 7 активность примерно соответствует таковой у нетрансгенных мышей в возрасте 16 месяцев. Мыши, которым вводили носитель, демонстрировали примерно одинаковую активность до введения и через 3 и 6 месяцев введения.
Фиг.33 представляет собой график, на котором показано время в тесте на прохождение по перекладине и эффект от введения соединения 7. Через 6 недель введения соединение 7 статистически значимо улучшает двигательную активность в тесте на прохождение по перекладине (что определяли по 36% уменьшению времени прохождения) по сравнению с мышами того же возраста (**p<0,01), которым вводили носитель. Столбцы отображают среднее значение + SEM, n=8 на группу.
Фиг.34 (врезки A-F) представляют собой микрофотографии, которые демонстрируют, что обработка соединением 7 вызывает уменьшение уровней α-синуклеина в мозге трансгенных мышей в возрасте 18 месяцев, что показывают данные иммуногистохимического исследования. Мыши, которым вводили соединение 7 (врезки C-D), продемонстрировали значительно более низкие внутринейрональные уровни α-синуклеина в лобной доле коры, по сравнению с мышами, которым вводили носитель (врезки A-B). Ткани мозга нетрансгенных мышей дикого типа не подвергаются окрашиванию, характерному для человеческого α-синуклеина, и показаны в качестве контроля специфичности антитела для трансгенного человеческого α-синуклеина (врезки E и F). Анализ и количественная обработка изображений позволяет установить, что введение соединения 7 вызывает статистически значимое уменьшение количества α-синуклеин-положительных объектов на 81%. Данные выражены в виде площади, занятой α-синуклеин-положительными объектами, в процентах. Столбцы гистограммы отображают среднее значение + SEM, n=5 для группы, которой вводили носитель, n=11 для группы, которой вводили соединение 7, и n=4 для группы нетрансгенных (Non-Tg) мышей. ***p<0,001 при сравнении с группой, которой вводили носитель, по данным однопараметрического анализа ANOVA и post hoc критерия Тьюки-Крамера.
Фиг.35, врезка A представляет собой фотографию вестерн-блота, демонстрирующую уровни α-синуклеина в дисперсной фракции из передней части мозга α-синуклеин трансгенных мышей, которым в течение 6 месяцев вводили соединение 7 или носитель в качестве контроля. На врезке B показана гистограмма, отображающая средние интенсивности полос двух независимых вестерн-блотов в количественном выражении, которая демонстрируют статистически значимое уменьшение на 69% в среднем и на 58% у женских особей содержания мономерного α-синуклеина после введения соединения 7. Интенсивности полос мономерного α-синуклеина были нормализованы по интенсивности полосы 25 кДа (дополнительно введенный контроль). *p<0,05, **p<0,01 по отношению к контрольным группам, получавшим носитель. Бары отображают среднее значение + SEM.
Фиг.36, врезка A представляет собой фотографию вестерн-блота, демонстрирующую уровни α-синуклеина в цитозольной фракции из передней части мозга α-синуклеин трансгенных мышей, которым в течение 6 месяцев вводили соединение 7 или носитель в качестве контроля. На врезке B показана гистограмма, отображающая средние интенсивности полос вестерн-блотов, приведенных на врезке A, в количественном выражении, которая демонстрирует статистически значимое уменьшение на 73% в среднем и на 48% у женских особей содержания мономерного α-синуклеина после введения соединения 7. Интенсивности полос мономерного α-синуклеина были нормализованы по интенсивности полосы β-актина (дополнительно введенный контроль). *p<0,05 по отношению к контрольным группам, получавшим носитель. Бары отображают среднее значение + SEM.
Фиг.37, врезка A представляет собой фотографию вестерн-блота, демонстрирующую уровни α-синуклеина в дисперсной фракции из передней части мозга α-синуклеин трансгенных мышей в возрасте 4-5 месяцев, которым в течение 6 недель вводили соединение 7 или носитель в качестве контроля. На врезке B показана гистограмма, отображающая средние интенсивности полос четырех независимых вестерн-блотов в количественном выражении, которая показывает, что введение соединения 7 приводит к статистически значимому уменьшению на 45% содержания мономерного α-синуклеина по сравнению с контрольными животными, получавшими носитель. Интенсивности полос мономерного α-синуклеина были нормализованы по интенсивности полосы 25 кДа (дополнительно введенный контроль). *p<0,05 по отношению к контрольным группам, получавшим носитель. Бары отображают среднее значение + SEM.
Фиг.38, врезка A представляет собой фотографию вестерн-блота, демонстрирующую уровни α-синуклеина в цитозольной фракции из передней части мозга α-синуклеин трансгенных мышей в возрасте 4-5 месяцев, которым в течение 6 недель вводили соединение 7 или носитель в качестве контроля. На врезке B показана гистограмма, отображающая средние интенсивности полос четырех независимых вестерн-блотов в количественном выражении, которая показывает, что введение соединения 7 приводит к статистически значимому уменьшению на 71% содержания мономерного α-синуклеина по сравнению с контрольными животными, получавшими носитель. Интенсивности полос мономерного α-синуклеина были нормализованы по интенсивности полосы α-тубулина (дополнительно введенный контроль). *p<0,01 по отношению к контрольным группам, получавшим носитель. Бары отображают среднее значение + SEM.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения
В данной заявке приведенные ниже термины будут иметь следующие значения, безотносительно к тому, используются ли эти термины в других значениях где-либо в литературе или в технике известного уровня.
В настоящей заявке термин «амилоидные заболевания» или «амилоидозы» означает заболевания, связанные с образованием, отложением, накоплением или устойчивым существованием фибрилл Aβ-амилоида. Такие заболевания включают, не ограничиваясь этим, болезнь Альцгеймера, синдром Дауна, наследственную церебральную геморрагию с амилоидозом голландского типа и церебральную β-амилоидную ангиопатию.
В настоящей заявке термин «синуклеиновые заболевания» или «синуклеинопатии» относится к заболеваниям, связанным с образованием, отложением, накоплением, устойчивым существованием или агрегацией α-синуклеина. Эти заболевания включают, не ограничиваясь перечисленными, болезнь Паркинсона, семейную болезнь Паркинсона, болезнь с тельцами Леви, вариант болезни Альцгеймера с тельцами Леви, деменцию с тельцами Леви, множественную системную атрофию и комплекс Гуама паркинсонизм-деменция.
Термин «фибриллогенез» относится к образованию, отложению, накоплению, агрегации и/или устойчивому существованию фибрилл, нитей, включений, отложений β-амилоида, а также фибрилл, нитей, включений, отложений агрегатов и т.п. α-синуклеина.
Термин «ингибирование фибриллогенеза» относится к ингибированию образования, отложения, накопления, агрегации и/или устойчивого существования упомянутых фибрилл β-амилоида или фибриллоподобных отложений или агрегатов α-синуклеина.
Термины «разрушение фибрилл» или «нарушение фибриллогенеза» относятся к разрушению ранее образовавшихся агрегатов β-амилоида или α-синуклеина, которые обычно существуют в виде преобладающих β-складчатых листовых вторичных структур. Это разрушение под действием соединений, описанных в настоящем изобретении, может включать заметное уменьшение или разрушение агрегатов амилоида или синуклеина, которое можно оценить с помощью различных методик, как, например, флюорометрии с использованием Тиофлавина T, связывания Конго красного, спектров кругового дихроизма, тиофлавина S и клеточных анализов, например, агрегации α-синуклеина и пробы на цитотоксичность XTT, которые продемонстрированы в примерах, описанных в настоящем изобретении.
Термины «нейропротективное действие» или «нейропротективный» относятся к способности соединения защищать, уменьшать, облегчать, улучшать и/или ослаблять поражение нервных клеток (нейродегенерацию).
Термин «млекопитающие» включает как людей, так и млекопитающих, не являющихся людьми (кошек, собак и т.п.), лабораторных животных (например, мышей, крыс, морских свинок и т.п.) и сельскохозяйственных животных (крупный рогатый скот, лошадей, овец, коз, свиней и т.п.).
Термин «фармацевтически приемлемый эксципиент» означает любой эксципиент, который традиционно применяется при получении фармацевтических композиций и является в основном безопасным, нетоксичным и целесообразным, и этот термин включает эксципиенты, которые приемлемы для ветеринарного применения или фармацевтического применения в лечении человека. Эти эксципиенты могут быть твердыми, жидкими, полутвердыми или, в случае аэрозольных композиций, газообразными.
Термин «терапевтически эффективное количество» означает количество, которого, при введении субъекту или животному с целью лечения заболевания, достаточно для того, чтобы вызвать желаемую степень лечения, предупреждения или облегчения симптомов заболевания. «Терапевтически эффективное количество» или «терапевтически эффективная дозировка» в некоторых вариантах осуществления подавляет, уменьшает, прекращает или нарушает образование, отложение, накопление и/или устойчивое существование агрегатов β-амилоида или α-синуклеина, или же лечит, предупреждает или облегчает один или несколько симптомов заболевания, связанного с такими состояниями, как, например, амилоидные заболевания или синуклеопатии, причем все указанные эффекты имеют измеримую величину, как, например, в одном из вариантов осуществления не менее 20%, в другом варианте осуществления не менее 40%, в следующем варианте осуществления не менее 60% и в еще одном варианте осуществления не менее 80%, по отношению к субъекту, не получавшему препараты. Эффективные количества соединений по настоящему изобретению или их композиций, для лечения субъектов из числа млекопитающих, составляют от примерно 0,1 до примерно 1000 мг/кг массы тела субъекта в день, например, от примерно 1 до примерно 100 мг/кг/день, в другом варианте осуществления от примерно 10 до примерно 500 мг/кг/день. Считается, что композиции, раскрытые в настоящем изобретении, будут одновременно безопасными и эффективными в широком диапазоне дозировок.
Термин «компонент, обеспечивающий замедленное высвобождение» определяется в настоящей заявке как соединение или соединения, включая, но не ограничиваясь этим, полимеры, полимерные матрицы, гели, проницаемые мембраны, липосомы, микросферы и т.п. или их комбинации, которые способствуют замедленному высвобождению действующего ингредиента.
Если комплекс является водорастворимым, можно получить его раствор в подходящем буфере, например, фосфатно-солевом буфере или других физиологически совместимых растворах. С другой стороны, если полученный комплекс плохо растворим в водных растворителях, можно получить его препарат с помощью неионных ПАВ, таких как Tween или полиэтиленгликоль. Таким образом могут быть получены препараты соединений и их физиологически приемлемых растворителей для введения с помощью ингаляции или инсуффляции (через рот или через нос), или, например, перорального, буккального, парентерального или ректального введения.
В настоящем описании фармацевтически приемлемые производные соединения включают его соли, сложные эфиры, простые эфиры енолов, сложные эфиры енолов, ацетали, кетали, ортоэфиры, полуацетали, полукетали, сольваты, гидраты или пролекарства. Такие производные могут быть легко получены специалистом в данной области техники с использованием стандартных методик получения этих производных. Полученные соединения можно вводить животным или людям без заметного токсического воздействия, и они либо сами проявляют фармацевтическую активность, либо являются пролекарствами. Фармацевтически приемлемые соли включают, не ограничиваясь этим, соли аминов, как, например, не ограничиваясь перечисленными, N,N'-дибензилэтилендиамина, хлорпрокаина, холина, аммиака, диэтаноламина и других гидроксиламинов, этилендиамина, N-метилглюкамина, прокаина, N-бензилфенэтиламина, 1-парахлорбензил-2-пирролидин-1'-илметилбензимидазола, диэтиламина и других алкиламинов, пиперазина и трис(гидроксиметил)аминометана; соли щелочных металлов, как, например, не ограничиваясь перечисленными, лития, калия и натрия; щелочноземельных металлов, как, например, не ограничиваясь перечисленными, бария, кальция и магния; соли переходных металлов, как, например, но не ограничиваясь этим, цинка; и другие соли металлов, как, например, но не ограничиваясь этим, содержащие остатки гидрофосфата натрия и динатрий фосфата; и, кроме того, включают, не ограничиваясь этим, соли минеральных кислот, как, например, не ограничиваясь указанным, гидрохлориды и сульфаты; и соли органических кислот, как, например, не ограничиваясь этим, ацетаты, лактаты, малаты, тартраты, цитраты, аскорбаты, сукцинаты, бутираты, валераты и фумараты. Фармацевтически приемлемые сложные эфиры включают, не ограничиваясь этим, алкил, алкенил, алкинил, арил, гетероарил, аралкил, гетероаралкил, циклоалкил и гетероциклил содержащие сложные эфиры кислотных групп, в число которых входят, не ограничиваясь этим, карбоновые кислоты, фосфорные кислоты, фосфиновые кислоты, сульфоновые кислоты, сульфиновые кислоты и бороновые кислоты. Фармацевтически приемлемые простые эфиры енолов включают, не ограничиваясь этим, производные формулы C=C(OR), где R представляет собой водород, алкил, алкенил, алкинил, арил, гетероарил, аралкил, гетероаралкил, циклоалкил или гетероциклил. Фармацевтически приемлемые сложные эфиры енолов включают, не ограничиваясь этим, производные формулы C=C(OC(O)R), где R представляет собой водород, алкил, алкенил, алкинил, арил, гетероарил, аралкил, гетероаралкил, циклоалкил или гетероциклил. Фармацевтически приемлемые сольваты и гидраты представляют собой комплексы соединения с одной или несколькими молекулами растворителя или воды, например, от 1 до примерно 100, или от 1 до примерно 10, или от 1 до примерно 2, 3 или 4 молекул растворителя или воды.
В настоящем описании «лечение» означает любое действие, с помощью которого облегчаются или изменяются в благоприятную сторону один или несколько симптомов заболевания или расстройства. Лечение заболевания включает предупреждение появления заболевания у субъекта, который может быть предрасположен к этому заболеванию, но оно еще не началось или не проявились его симптомы (профилактическое лечение), подавление развития заболевания (замедление или остановку его развития), облегчение симптомов или побочных действий заболевания (включая паллиативное лечение) и смягчение заболевания (т.е. регрессию заболевания), например, путем разрушения ранее образовавшихся агрегатов β-амилоида или α-синуклеина. В настоящем описании облегчение симптомов конкретного расстройства путем введения конкретного соединения или его фармацевтической композиции относится к любому их уменьшению, независимо от того, является ли оно постоянным или временным, продолжительным или скоротечным, которое может быть отнесено к введению композиции или связано с ним.
В настоящем описании считается, что ингибирование образования, отложения, накопления, агрегации и/или устойчивого существования фибрилл α-синуклеина является эффективным лечением ряда заболеваний, связанных с α-синуклеином, например, болезни Паркинсона, болезни с тельцами Леви и множественной системной атрофии.
В настоящем описании «пролекарство» означает соединение, которое при введении in vivo претерпевает процесс метаболизма, состоящий из одной или нескольких стадий, или иным образом превращается в биологически, фармацевтически или терапевтически активную форму соединения. Для получения пролекарства фармацевтически активное соединение модифицируют таким образом, чтобы оно регенерировалось в результате метаболических процессов. Получение пролекарства может предназначаться для изменения метаболической стабильности или характеристик переноса лекарственного вещества в организме, для маскирования побочных действий или токсичности, для улучшения вкуса или запаха лекарственного средства или для изменения других характеристик и свойств лекарственного средства. Основываясь на знании фармакодинамических процессов и метаболизма лекарственных соединений in vivo, специалист в данной области техники, если ему известно фармацевтически активное соединение, может разработать соответствующие ему пролекарства (см., например, Nogrady (1985) Medicinal Chemistry a Biochemical Approach, Oxford University Press, New York, pages 388-392).
В заявке показаны структуры некоторых из соединений по настоящему изобретению. Названия соединений в различных случаях представляют собой наименования IUPAC [наименования, полученные согласно установленной IUPAC (International Union of Pure and Applied Chemistry - международный союз теоретической и прикладной химии) системе, совместно разработанной комиссией по номенклатуре в органической химии и комиссией по физической органической химии, и которую можно найти на сайте http://www.chem.qmul.ac.uk/iupac], наименования, полученные из наименований IUPAC с помощью добавлений или замен (например, при использовании названия «3,4-метилендиоксифенил», образованного от «фенила» вместо «бензо[1,3]диоксол-5-ил»), и наименования, образованные от наименований реагентов (например, при использовании названия «3,4-дигидроксианилид 3,4-дигидроксибензойной кислоты» вместо «N-(3,4-дигидроксифенил)-3,4-дигидроксибензамид»). Однако использованные наименования однозначно соответствуют химическим структурам, и считается, что их может легко понять рядовой специалист в данной области.
Термины «фармацевтическое средство», «фармакологическое средство» или «фармацевтическая композиция» относятся к соединению или комбинации соединений, применяемой для лечения, предпочтительно в чистой или почти чистой форме. В настоящем описании фармацевтические или фармакологические средства включают соединения по настоящему изобретению. Желательно очищать эти соединения до 80% чистоты и предпочтительно до 90% чистоты. Считается, что предпочтительно очищать соединения до чистоты 99,9%. В качестве теста или подтверждения чистоты можно указать на то, что соединение надлежащей степени чистоты могло бы дать хроматограмму ВЭЖХ, на которой специалист в данной области смог бы обнаружить единственную полосу в форме узкого пика.
Следует понимать, что соединения по настоящему изобретению могут содержать хиральные центры. Такие хиральные центры могут иметь (R) или (S) конфигурацию, или же соединение может включать хиральные центры в обеих указанных конфигурациях. Таким образом, соединения по настоящему изобретению могут являться эанантиомерно чистыми или представлять собой смеси стереоизомеров или диастереомеров. Что касается аминокислотных остатков, эти остатки могут быть либо в L-, либо в D-форме. Природные аминокислотные остатки в основном имеют L-конфигурацию. Если конфигурация не указана конкретно, остаток имеет L-конфигурацию. В настоящем описании термин «аминокислота» относится к α-аминокислотам, которые либо являются рацемическими, либо имеют D- или L-конфигурацию. Обозначение “d”, предшествующее обозначению аминокислоты (например, dAla, dSer, dVal и т.д.), относится к D-изомеру аминокислоты. Обозначение “dl”, предшествующее обозначению аминокислоты (например, dlPip), относится к смеси L- и D-изомеров аминокислоты. Необходимо понимать, что хиральные центры соединений, разработанных в настоящем изобретении, могут претерпевать эпимеризацию in vivo. В этом случае специалист в данной области техники поймет, что для соединений, которые подвергаются эпимеризации in vivo, введение соединения в его (R) форме эквивалентно введению соединения в его (S) форме.
В настоящем описании термин «практически чистый» означает достаточно чистый, для того чтобы показать отсутствие легко обнаруживаемых примесей по данным стандартных способов анализа, например, тонкослойной хроматографии (ТСХ), гель-электрофореза, высокоэффективной жидкостной хроматографии (ВЭЖХ) и масс-спектрометрии (МС), применяемых специалистами в данной области техники для оценки такой чистоты, или достаточно чистый для того, чтобы дальнейшая очистка не смогла бы заметно изменить физические и химические свойства вещества, например, ферментную или биологическую активность. Способы очистки соединений для получения практически чистых химических соединений известны специалисту в данной области. Однако практически чистые химические соединения могут быть смесью стереоизомеров. В этом случае дополнительная очистка могла бы увеличить удельную активность соединения.
В настоящем описании углеродные цепи алкилов, алкенилов и алкинилов, если не указано конкретно, содержат от 1 до 20 атомов углерода или от 1 или 2 до 16 атомов углерода и являются линейными или разветвленными. Углеродные цепи алкенилов, включающие от 2 до 20 атомов углерода, в некоторых вариантах осуществления содержат от 1 до 8 двойных связей, и углеродные цепи алкенилов, включающие от 2 до 16 атомов углерода, в некоторых вариантах осуществления содержат от 1 до 5 двойных связей. Углеродные цепи алкинилов, включающие от 2 до 20 атомов углерода, в некоторых вариантах осуществления содержат от 1 до 8 тройных связей, и углеродные цепи алкинилов, включающие от 2 до 16 атомов углерода, в некоторых вариантах осуществления содержат от 1 до 5 тройных связей. Типовые примеры алкильных, алкенильных и алкинильных групп по настоящему изобретению включают, не ограничиваясь перечисленными, метил, этил, пропил, изопропил, изобутил, н-бутил, втор-бутил, трет-бутил, изопентил, неопентил, трет-пентил, изогексил, аллил (пропенил) и пропаргил (пропинил). В настоящей заявке термины «низший алкил», «низший алкенил» и «низший алкинил» относятся к углеродным цепям, содержащим от примерно 1 или примерно 2 атомов углерода до примерно 6 атомов углерода. В настоящей заявке термин «алк(ен)(ин)ил» относится к алкильной группе, содержащей по меньшей мере одну двойную связь и по меньшей мере одну тройную связь.
В настоящем описании термин «циклоалкил» относится к насыщенной моно- или полициклической системе, содержащей в некоторых вариантах осуществления от 3 до 10 атомов углерода, в других вариантах осуществления от 3 до 6 атомов углерода; термины «циклоалкенил» или «циклоалкинил» относятся к моно- или полициклическим системам, которые, соответственно, включают по меньшей мере одну двойную связь и по меньшей мере одну тройную связь. Циклоалкенильные и циклоалкинильные группы в некоторых вариантах осуществления могут содержать от 3 до 10 атомов углерода, причем циклоалкенильные группы в других вариантах осуществления содержат от 4 до 7 атомов углерода, и циклоалкинильные группы в других вариантах осуществления содержат от 8 до 10 атомов углерода. Циклические системы циклоалкильных, циклоалкенильных и циклоалкинильных групп могут состоять из одного или двух или более циклов и могут быть соединены друг с другом с образованием конденсированных, мостиковых или спиро-структур. Термин «циклоалк(ен)(ин)ил» относится к циклоалкильной группе содержащей по меньшей мере одну двойную связь и по меньшей мере одну тройную связь.
В настоящем описании термин «арил» относится к ароматическим моноциклическим или полициклическим группам, содержащим от 6 до 19 атомов углерода. Арильные группы включают, не ограничиваясь этим, такие группы, как незамещенный или замещенный флуоренил, незамещенный или замещенный фенил и незамещенный или замещенный нафтил.
В настоящем описании термин «гетероарил» относится к моноциклической или полициклической ароматической системе, которая в некоторых вариантах осуществления состоит из примерно 5-15 атомов, где один или несколько атомов, в одном из вариантов осуществления от 1 до 3 атомов, циклической системы представляют собой гетероатомы, т.е. атомы элемента, отличающегося от углерода, в т.ч., но не ограничиваясь этим, азота, кислорода или серы. Гетероарильная группа может быть необязательно конденсирована с бензольным кольцом. Гетероарильные группы включают, не ограничиваясь перечисленными, фурил, имидазолил, пиримидинил, тетразолил, тиенил, пиридинил, пирролил, тиазолил, изотиазолил, оксазолил, изоксазолил, триазолил, хинолинил и изохинолинил, имидазол, тетразол и пиразол.
В настоящей заявке термин «гетероциклил» относится к моноциклической или полициклической неароматической системе, которая в одном из вариантов осуществления содержит от 3 до 10 атомов, в другом варианте осуществления от 4 до 7 атомов, в еще одном варианте осуществления от 5 до 6 атомов, где один или несколько из этих атомов в циклической системе, в некоторых из вариантов осуществления от 1 до 3, являются гетероатомами, т.е. атомами элементов, отличных от углерода, включая, но не ограничиваясь этим, атомы азота, кислорода или серы. В тех вариантах осуществления, где гетероатом(мы) является (являются) азотом, азот необязательно замещен алкилом, алкенилом, алкинилом, арилом, гетероарилом, аралкилом, гетероаралкилом, циклоалкилом, гетроциклилом, циклоалкилалкилом, гетероциклилалкилом, ацилом, гуанидино, или же атом азота может быть кватернизован с образованием аммонийной группы, заместитель в которой может быть выбран из перечисленных выше.
В настоящем описании термин «аралкил» относится к алкильной группе, в которой один из атомов водорода замещен арильной группой.
В настоящем описании термин «гетероаралкил» относится к алкильной группе, в которой один из атомов водорода замещен гетероарильной группой.
В настоящем описании термины «гало», «галоген» или «галогенид» относятся к F, Cl, Br или I.
В настоящем описании «псевдогалогениды» или «псевдогалогено» группы представляют собой группы, которые проявляют химические свойства, в основном похожие на свойства галогенов. Такие соединения можно применять теми же способами, что и галогениды, и обрабатывать таким же образом. Псевдогалогениды включают, не ограничиваясь этим, цианид, цианат, тиоцианат, селеноцианат, трифторметокси и азид.
В настоящем описании термин «галогеналкил» относится к алкильной группе, в которой один или несколько атомов водорода замещены атомами галогенов. Эти группы включают, не ограничиваясь этим, хлорметил, трифторметил и 1-хлор-2-фторэтил.
В настоящем описании термин «галогеналкокси» относится к группе RO-, в которой R представляет собой галогеналкильную группу.
В настоящем описании термины «сульфинил» или «тионил» относятся к группе -S(O)-. В настоящем описании термины «сульфонил» или «сульфурил» относятся к группе -S(O)2-. В настоящем описании термин «сульфо» относится к группе -S(O)2O-.
В настоящем описании термин «карбокси» относится к двухвалентному радикалу -C(O)O-.
В настоящем описании термин «аминокарбонил» относится к фрагменту -C(O)NH2.
В настоящем описании термин «алкиламинокарбонил» относится к фрагменту -C(O)NHR, в котором R означает алкил, в т.ч. низший алкил.
В настоящем описании термин «диалкиламинокарбонил» относится к группе -C(O)NR'R, где каждый из R и R' независимо представляет собой алкил, в т.ч. низший алкил; термин «карбоксамид» относится к группам формулы -NR'COR, где каждый из R' и R независимо представляет собой алкил, в т.ч. низший алкил.
В настоящем описании термин «арилалкиламинокарбонил» относится к группе -C(O)NRR', где один из R и R' является арилом, в т.ч. низшим арилом, например, фенилом, и другой из R или R' является алкилом, в т.ч. низшим алкилом.
В настоящем описании термин «ариламинокарбонил» относится к фрагменту -C(O)NHR, в котором R означает арил, в т.ч. низший арил, например, фенил.
В настоящем описании термин «гидроксикарбонил» относится к группе -COOH.
В настоящем описании термин «алкоксикарбонил» относится к группе -C(O)OR, где R означает алкил, в т.ч. низший алкил.
В настоящем описании термин «арилоксикарбонил» относится к группе -C(O)OR, где R означает арил, в т.ч. низший арил, например, фенил.
В настоящем описании термины «алкокси» и «алкилтио» относятся к группам RO- и RS-, где R является алкилом, в т.ч. низшим алкилом.
В настоящем описании термины «арилокси» и «арилтио» относятся к группам RO- и RS-, где R является арилом, в т.ч. низшим арилом, например, фенилом.
В настоящем описании термин «алкилен» относится к линейной, разветвленной или циклической, в некоторых вариантах осуществления линейной или разветвленной, двухвалентной алифатической углеводородной группе, в одном из вариантов осуществления включающей от 1 до примерно 20 атомов углерода, в другом варианте осуществления включающей от 1 до 12 атомов углерода. В другом варианте осуществления в число алкиленов входят низшие алкилены. В алкиленовую группу могут быть необязательно включены один или несколько атомов кислорода, серы, в т.ч. группы S(=O) и S(=O)2, или замещенные или незамещенные атомы азота, включая группы -NR- и -N+RR-, где заместитель(и) у атома азота представляет (представляют) собой алкил, арил, аралкил, гетероарил, гетероаралкил или COR', где R' означает алкил, арил, аралкил, гетероарил, гетероаралкил, -OY или -NYY, где Y означает водород, алкил, арил, гетероарил, циклоалкил или гетероциклил. Алкиленовые группы включают, не ограничиваясь этим, метилен (-CH2-), этилен (-CH2CH2-), пропилен (-(CH2)3-), метилендиокси (-O-CH2-O-) и этилендиокси (-O-(CH2)2-O-). Термин «низший алкилен» относится к алкиленовой группе, включающей от 1 до 6 атомов углерода. В некоторых вариантах осуществления алкиленовые группы представляют собой низший алкилен, включая алкилен, содержащий 1-3 атома углерода.
В настоящем описании термин «азаалкилен» относится к фрагменту -(CRR)n-NR-(CRR)m-, где каждый из коэффициентов n и m независимо является целым числом от 0 до 4. В настоящем описании термин «оксиалкилен» относится к фрагменту -(CRR)n-O-(CRR)m-, где каждый из коэффициентов n и m независимо является целым числом от 0 до 4. В настоящем описании термин «тиаалкилен» относится к -(CRR)n-S-(CRR)m-, -(CRR)n-S(=O)-(CRR)m- и -(CRR)n-S(=O)2-(CRR)m-, где каждый из коэффициентов n и m независимо является целым числом от 0 до 4.
В настоящем описании термин «алкенилен» относится к линейной, разветвленной или циклической, в одном из вариантов осуществления линейной или разветвленной, двухвалентной алифатической углеводородной группе, в некоторых вариантах осуществления включающей от 2 до примерно 20 атомов углерода и по меньшей мере одну двойную связь, в других вариантах осуществления включающей от 1 до 12 атомов углерода. В других вариантах осуществления в число алкениленовых групп входят низшие алкенилены. В алкениленовые группы могут быть необязательно включены один или несколько атомов кислорода, серы или замещенные или незамещенные атомы азота, где заместителем атома азота является алкил. Алкениленовые группы включают, не ограничиваясь этим, -CH=CH-CH=CH- и -CH=CH-CH2-. Термин «низший алкенилен» относится к алкениленовым группам, содержащим от 2 до 6 атомов углерода. В некоторых вариантах осуществления алкениленовые группы представляют собой низший алкенилен, в т.ч. алкенилен с 3-4 атомами углерода.
В настоящем описании термин «алкинилен» относится к линейной, разветвленной или циклической, в некоторых вариантах осуществления линейной или разветвленной, двухвалентной алифатической углеводородной группе, в одном из вариантов осуществления включающей от 2 до примерно 20 атомов углерода и по меньшей мере одну тройную связь, в других вариантах осуществления включающей от 1 до 12 атомов углерода. В других вариантах осуществления в число алкиниленовых групп входят низшие алкинилены. В алкиниленовые группы могут быть необязательно включены один или несколько атомов кислорода, серы или замещенные или незамещенные атомы азота, где заместителем атома азота является алкил. Алкиниленовые группы включают, не ограничиваясь этим, -C≡C-C≡C-, -C≡C- и -C≡C-CH2-. Термин «низший алкинилен» относится к алкиниленовым группам, содержащим от 2 до 6 атомов углерода. В некоторых вариантах осуществления алкиниленовая группа представляет собой низший алкинилен, в т.ч. алкинилен с 3-4 атомами углерода.
В настоящем описании термин «алк(ен)(ин)илен» относится к линейной, разветвленной или циклической, в некоторых вариантах осуществления линейной или разветвленной, двухвалентной алифатической углеводородной группе, в одном из вариантов осуществления включающей от 2 до примерно 20 атомов углерода и по меньшей мере одну тройную связь и по меньшей мере одну двойную связь; в других вариантах осуществления включающей от 1 до 12 атомов углерода. В других вариантах осуществления в число алк(ен)(ин)иленовых групп входят низшие алк(ен)(ин)илены. В алк(ен)(ин)иленовые группы могут быть необязательно включены один или несколько атомов кислорода, серы или замещенные или незамещенные атомы азота, где заместителем атома азота является алкил. Алк(ен)(ин)иленовые группы включают, не ограничиваясь этим, -C=C-(CH2)n-C≡C-, где n равно 1 или 2. Термин «низший алк(ен)(ин)илен» относится к алк(ен)(ин)иленовым группам, содержащим до 6 атомов углерода. В некоторых вариантах осуществления алк(ен)(ин)иленовая группа включает примерно 4 атома углерода.
В настоящем описании термин «циклоалкилен» относится к двухвалентной насыщенной моно- или полициклической системе, содержащей в некоторых вариантах осуществления от 3 до 10 атомов углерода, в других вариантах осуществления от 3 до 6 атомов углерода; термины «циклоалкенилен» и «циклоалкинилен» относятся к двухвалентным моно- или полициклическим системам, которые, соответственно, включают по меньшей мере одну двойную связь и по меньшей мере одну тройную связь. Циклоалкениленовые и циклоалкиниленовые группы в некоторых вариантах осуществления могут содержать от 3 до 10 атомов углерода, причем циклоалкениленовая группа в некоторых вариантах осуществления содержит от 4 до 7 атомов углерода, и циклоалкиниленовая группа в некоторых вариантах осуществления содержит от 8 до 10 атомов углерода. Циклические системы циклоалкиленовых, циклоалкениленовых и циклоалкиниленовых групп могут состоять из из одного цикла или двух или более циклов, которые могут образовывать конденсированную, мостиковую или спиро-структуру. Термин «циклоалк(ен)(ин)илен» относится к циклоалкиленовой группе, содержащей по меньшей мере одну двойную связь и по меньшей мере одну тройную связь.
В настоящем описании термин «арилен» относится к моноциклической или полициклической, в некоторых вариантах осуществления моноциклической, двухвалентной ароматической группе, в одном из вариантов осуществления включающей от 5 до 20 атомов углерода и по меньшей мере один ароматический цикл, в других вариантах осуществления включающей от 5 до 12 атомов углерода. В других вариантах осуществления в число ариленовых групп входят низшие арилены. Ариленовые группы включают, не ограничиваясь этим, 1,2-, 1,3- и 1,4-фенилены. Термин «низший арилен» относится к ариленовым группам, включающим 6 атомов углерода.
В настоящем описании термин «гетероарилен» относится к двухвалентной моноциклической или полициклической ароматической системе, содержащий в одном из вариантов осуществления от примерно 5 до примерно 15 атомов в цикле (циклах), где один или несколько из этих атомов циклической системы, в некоторых вариантах осуществления от 1 до 3, представляют собой гетероатомы, т.е. атомы элемента, отличного от углерода, в т.ч., но не ограничиваясь этим, атомы азота, кислорода или серы. Термин «низший гетероарилен» относится к гетероариленовым группам, содержащим в цикле 5 или 6 атомов.
Термин «гетероциклилен» относится к двухвалентной моноциклической или полициклической неароматической системе, включающей в некоторых вариантах осуществления от 3 до 10 атомов, в одном из вариантов осуществления от 4 до 7 атомов, в другом варианте осуществления от 5 до 6 атомов, где один или несколько циклических атомов, в т.ч. от 1 до 3, представляют собой гетероатомы, т.е. атомы элемента, отличного от углерода, в т.ч., но не ограничиваясь этим, атомы азота, кислорода или серы.
В настоящем описании термины «замещенный алкил», «замещенный алкенил», «замещенный алкинил», «замещенный циклоалкил», «замещенный циклоалкенил», «замещенный циклоалкинил», «замещенный арил», «замещенный гетероарил», «замещенный гетероциклил», «замещенный алкилен», «замещенный алкенилен», «замещенный алкинилен», «замещенный циклоалкилен», «замещенный циклоалкенилен», «замещенный циклоалкинилен», «замещенный арилен», «замещенный гетероарилен» и «замещенный гетероциклилен» относятся к алкилу, алкенилу, алкинилу, циклоалкилу, циклоалкенилу, циклоалкинилу, арилу, гетероарилу, гетероциклилу, алкилену, алкенилену, алкинилену, циклоалкилену, циклоалкенилену, циклоалкинилену, арилену, гетероарилену и гетероциклилену, соответственно, которые замещены одним или несколькими заместителями, в некоторых вариантах осуществления одним, двумя, тремя или четырьмя заместителями, где указанные заместители соответствуют определениям, данным в настоящем описании, в одном из вариантов осуществления выбраны из Q1.
В настоящем описании термин «алкилиден» относится к двухвалентной группе, например, =CR'R”, которая присоединена к одному атому другой группы с образованием двойной связи. Алкилиденовые группы включают, не ограничиваясь этим, метилиден (=CH2) и этилиден (=CHCH3). В настоящем описании термин «арилалкилиден» относится к алкилиденовой группе, в которой либо R', либо R” является арильной группой. «Циклоалкилиденовые группы» представляют собой такие группы, в которых R' и R” связаны друг с другом с образованием карбоцикла. «Гетероциклилиденовые» группы представляют собой такие группы, в которых по меньшей мере один из R' и R” содержит в своей цепи гетероатом, и R' и R” связаны друг с другом, образуя гетероцикл.
В настоящем описании термин «амидо» относится к двухвалентной группе -C(O)NH-. Термин «тиоамидо» относится к двухвалентной группе -C(S)NH-. Термин «оксиамидо» относится к двухвалентной группе -OC(O)NH-. Термин «тиаамидо» относится к двухвалентной группе -SC(O)NH-. Термин «дитиоамидо» относится к двухвалентной группе -SC(S)NH-. Термин «уреидо» относится к двухвалентной группе -HNC(O)NH-. Термин «тиоуреидо» относится к двухвалентной группе -NC(S)NH-.
В настоящем описании термин «семикарбазид» относится к группе -NHC(O)NHNH-. Термин «карбазат» относится к двухвалентной группе -OC(O)NHNH-. Термин «изотиокарбазат» относится к двухвалентной группе -SC(O)NHNH-. Термин «тиокарбазат» относится к двухвалентной группе -OC(S)NHNH-. Термин «сульфонилгидразид» относится к двухвалентной группе -SO2NHNH-. Термин «гидразид» относится к двухвалентной группе -C(O)NHNH-. Термин «азо» относится к двухвалентной группе -N=N-. Термин «гидразинил» относится к двухвалентной группе -NH-NH-.
В настоящем описании термин «сульфонамид» относится к фрагменту -RSO2NH2-, т.е. сульфоновой группе, соединенной с аминогруппой.
В настоящем описании термин «имидазол» относится к гетероциклическому ароматическому органическому соединению, имеющему общую формулу C3H4N2.
В настоящем описании термин «триазол» относится к любому из двух изомерных химических соединений, имеющих молекулярную формулу C2H3N3.
В настоящем описании термин «пиразол» относится к 5-членному гетероциклу, состоящему из трех атомов углерода и двух атомов азота в соседних положениях.
В настоящем описании термин «адамантан» относится к трициклоалкилу, имеющему общую формулу C10H16.
Если число единиц какой-либо замещающей группы не указано (например, галогеналкил), в молекуле может иметься один или несколько заместителей. Например, термин «галогеналкил» может включать один или несколько одинаковых или различных галогенов. В другом примере «C1-3 алкоксифенил» может включать одну или несколько одинаковых или различных алкоксигрупп, содержащих один, два или три атома углерода.
В настоящем описании аббревиатуры, применяемые для обозначения любых защитных групп, аминокислот и других соединений, если не указано иное, соответствуют их обычному применению, общепринятым обозначениям или рекомендациям комиссии по биохимической номенклатуре IUPAC-IUB (см. (1972) Biochem. 11:942-944).
Соединения по настоящему изобретению
Соединениями по настоящему изобретению являются:
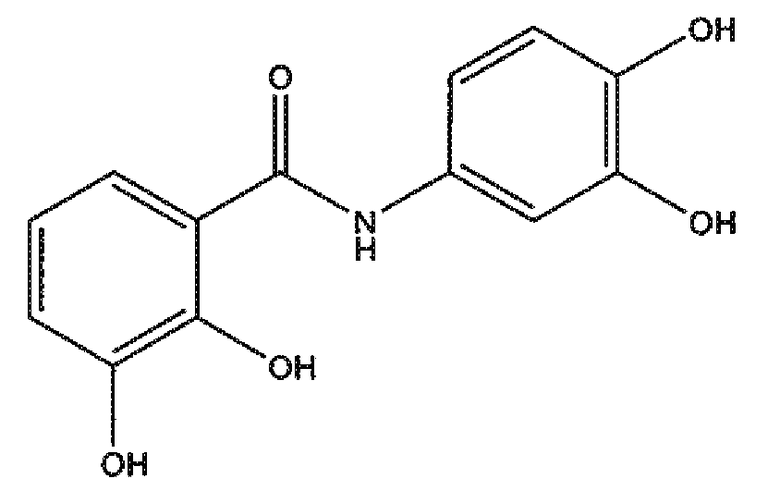
3,4-дигидроксианилид 2,3-дигидроксибензойной кислоты (соединение 1),
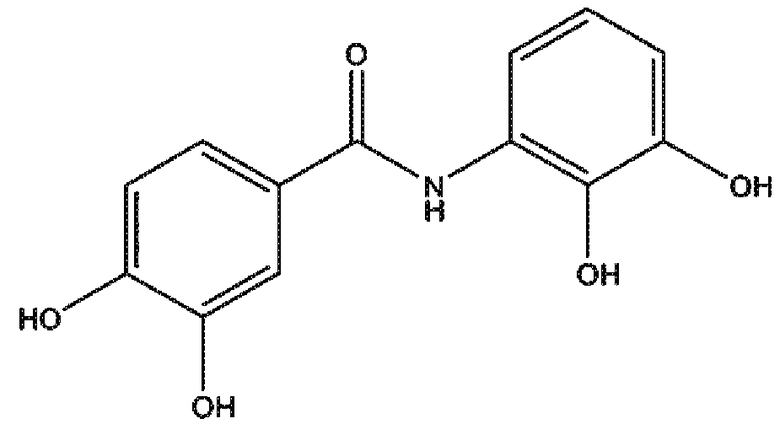
2,3-дигидроксианилид 3,4-дигидроксибензойной кислоты (соединение 2),
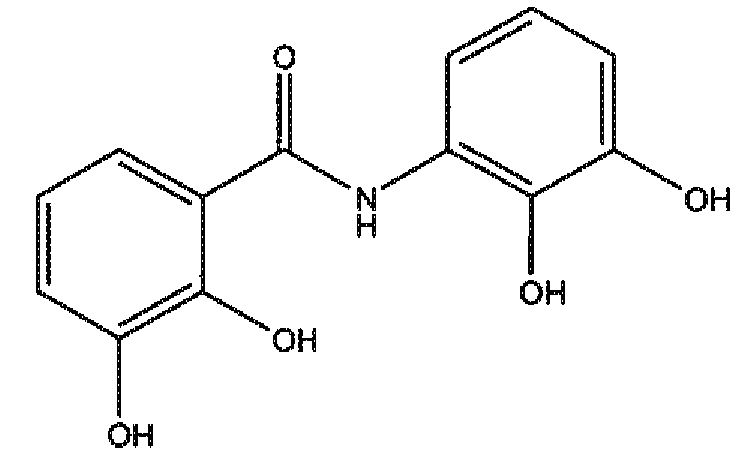
2,3-дигидроксианилид 2,3-дигидроксибензойной кислоты (соединение 3),
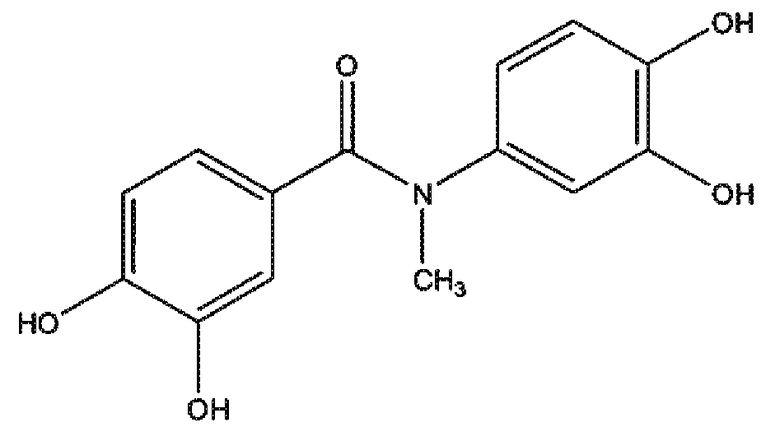
3,4-дигидрокси N-метиланилид 3,4-дигидроксибензойной кислоты (соединение 4),
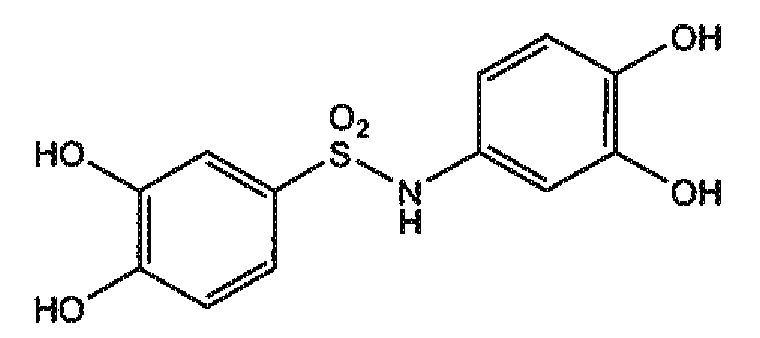
3,4-дигидроксифенилсульфонамид 3,4-дигидроксибензолсульфоновой кислоты (соединение 5),
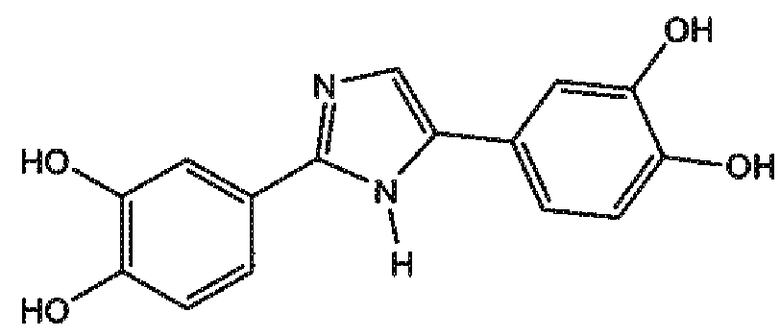
2,4-бис-(3,4-дигидроксифенил)имидазол (соединение 6),
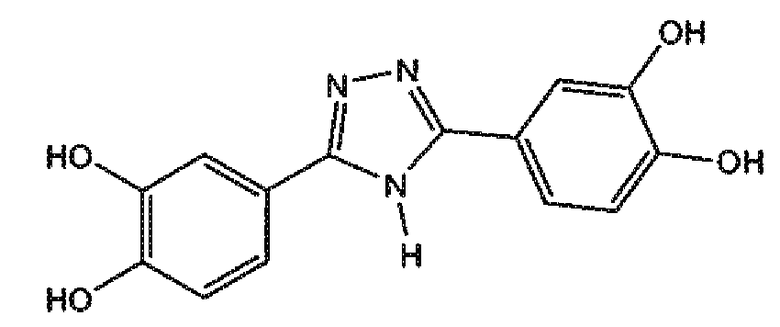
3,5-бис-(3,4-дигидроксифенил)-1,2,4-триазол (соединение 7),
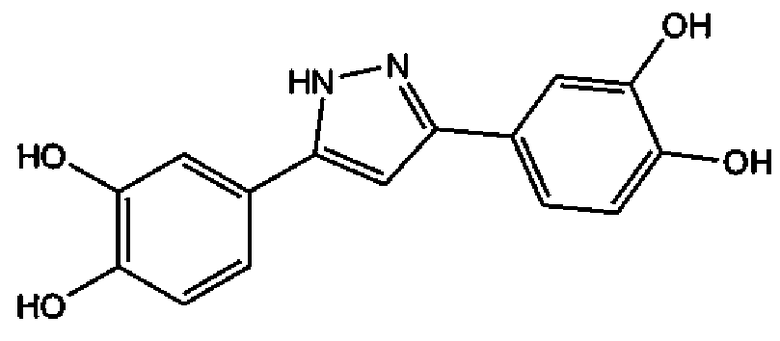
3,5-бис-(3,4-дигидроксифенил)пиразол (соединение 8),
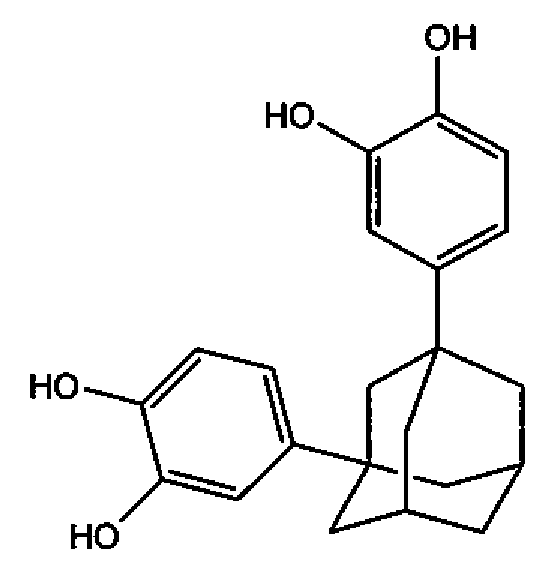
1,3-бис-(3,4-дигидроксифенил)адамантан (соединение 9).
Синтез соединений по настоящему изобретению
Соединения по настоящему изобретению могут быть получены способами, которые в основном известны рядовому специалисту в данной области техники, с учетом его знаний и содержания настоящего описания, в т.ч. примеров 1-5.
Исходные материалы и реагенты, применяемые при получении этих соединений, либо могут быть приобретены у коммерческих поставщиков, как, например, Aldrich Chemical Company (Milwaukee, WI), Bachem (Torrance, CA), Sigma (St.Louis, MO) или Lancaster Synthesis Inc. (Windham, NH), либо получены способами, хорошо известными рядовому специалисту в данной области, по методикам, описанным в следующих источниках: Fieser and Fieser's Reagents for Organic Synthesis, vols. 1-17, John Wiley and Sons, New York, NY, 1991; Rodd's Chemistry of Carbon Compounds, vols. 1-5 и supps., Elsevier Science Publishers, 1989; Organic Reactions, vols. 1-40, John Wiley and Sons, New York, NY, 1991; March J.: Advanced Organic Chemistry, 4th ed., John Wiley and Sons, New York, NY; и Larock: Comprehensive Organic Transformations, VCH Publishers, New York, 1989.
В большинстве случаев осуществляется введение защитных групп для гидроксильных групп соединения, которые удаляются на завершающих этапах синтеза. Подходящие защитные группы описаны в Greene et al., Protective Groups in Organic Synthesis, Second Edition, John Wiley and Sons, New York, 1991. Другие исходные вещества или промежуточные вещества начальных этапов синтеза могут быть получены по методикам, которые являются переработкой приведенных выше источников, например, по методикам, хорошо известным рядовому специалисту в данной области техники.
Исходные вещества, промежуточные вещества и соединения по настоящему изобретению могут быть выделены и очищены с использованием обычных методик, включая осаждение, фильтрование, перегонку, кристаллизацию, хроматографию и т.п. Соединения можно охарактеризовать с применением стандартных способов, включая определение физических констант и спектроскопические методики.
Фармакология и применимость
Соединения, разработанные в настоящем изобретении, могут применяться в чистом виде, вводиться в форме фармацевтически приемлемых солей, полученных из неорганических или органических кислот, или их можно применять в комбинации с одним или несколькими фармацевтически приемлемыми эксципиентами. Фраза «фармацевтически приемлемая соль» означает такие соли, которые, в рамках обоснованного суждения медицины, подходят для применения в контакте с тканями без неприемлемой токсичности, раздражения, аллергической реакции и т.п. и соответствуют разумному соотношению польза/риск. Фармацевтически приемлемые соли хорошо известны в технике. Соли можно получать либо in situ во время окончательного выделения и очистки соединений, предложенных в настоящем изобретении, либо отдельно при взаимодействии кислотного или основного лекарственного соединения с подходящим основанием или кислотой, соответственно. Типовые соли, полученные из органических или неорганических кислот, включают, не ограничиваясь этим, гидрохлориды, гидробромиды, гидройодиды, ацетаты, адипаты, альгинаты, цитраты, аспартаты, бензоаты, бисульфаты, глюконаты, фумараты, гидройодиды, лактаты, малеаты, оксалаты, пальмитаты, пектинаты, сукцинаты, тартраты, фосфаты, глутаматы и бикарбонаты. Типовые соли, полученные из органических или неорганических оснований, включают, не ограничиваясь этим, соли лития, натрия, калия, кальция, магния, аммония, моноалкиламмония, например, меглумина, диалкиламмония, триалкиламмония и тетраалкиламмония.
Можно менять фактические уровни дозировки действующих ингредиентов и способы введения фармацевтических композиций, разработанных в настоящем изобретении, для достижения эффективной реакции на терапию у конкретного пациента. Фраза «терапевтически эффективное количество» соединения по настоящему изобретению означает количество соединения, достаточное для лечения расстройства, при разумном соотношении польза/риск, применимом для любого медицинского лечения. Однако следует понимать, что решение об общем количестве соединения или композиции по настоящему изобретению, применяемом в течение суток, будет принимать лечащий врач в рамках тщательного медицинского обследования. Общая дневная доза соединений по настоящему изобретению может находиться в диапазоне от примерно 0,1 до примерно 1000 мг/кг/день. При пероральном введении дозировка может находиться в диапазоне от примерно 1 до примерно 500 мг/кг/день. Если это желательно, эффективную дневную дозу можно разделить на несколько отдельно вводимых доз; следовательно, одна доза композиции может включать соответствующие количества или их части, которые вместе составляют дневную дозу. Конкретный уровень терапевтически эффективной дозировки для любого конкретного пациента будет зависеть от ряда факторов, в т.ч. подвергаемого лечению расстройства и тяжести этого расстройства; истории заболевания пациента, активности конкретного применяемого соединения; конкретной применяемой композиции, возраста, массы тела, общего состояния здоровья, пола и рациона питания пациента, времени введения, пути введения, продолжительности лечения, скорости выведения конкретного применяемого соединения, других лекарственных средств, применяемых в комбинации или одновременно с конкретным применяемым соединением и т.п.
Соединения по настоящему изобретению можно включать в фармацевтические составы совместно с одним или несколькими нетоксичными фармацевтически приемлемыми разбавителями, носителями, вспомогательными добавками и антибактериальными и противогрибковыми средствами, например, парабенами, хлорбутанолом, фенолом, сорбиновой кислотой и т.п. В случае дисперсий надлежащую текучесть можно поддерживать, например, применением покрывающих материалов, таких как лецитин, поддержанием требуемого размера частиц и применением ПАВ. В некоторых случаях для продления действия лекарственного средства желательно понизить скорость абсорбции препарата из подкожной или внутримышечной инъекции. Этого можно добиться суспендированием кристаллического или аморфного лекарственного вещества в носителе, имеющем плохую растворимость в воде, например, в маслах. В этом случае скорость абсорбции лекарственного соединения будет зависеть от скорости растворения, которая, в свою очередь, может зависеть от размера и формы кристаллов. Пролонгированной абсорбции инъецированных фармацевтических форм можно добиться с применением агентов, вызывающих задержку абсорбции, например, моностерата алюминия или желатина.
Соединения по настоящему изобретению можно вводить энтерально или парентерально в твердых или жидких формах. Композиции, подходящие для парентеральной инъекции, могут включать физиологически приемлемые изотонические стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки, предназначенные для восстановления в стерильные растворы или дисперсии для инъекций. Примеры подходящих водных или неводных носителей, разбавителей, растворителей или наполнителей включают воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и т.п.), растительные масла (например, оливковое масло), подходящие для инъекций органические эфиры, например, этилолеат, а также их подходящие смеси. Эти композиции могут также содержать вспомогательные компоненты, например, консерванты, смачивающие, эмульгирующие и диспергирующие агенты. Суспензии помимо действующих соединений могут содержать суспендирующие агенты, например, этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтилен сорбита и сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант, или смеси этих соединений.
Соединения, разработанные в настоящем изобретении, можно также вводить с помощью инъекции или инфузии, подкожно, внутривенно, внутримышечно, интрастернально или интраназально, или с помощью инфузионных методик в форме стерильных инъецируемых или масляных суспензий. Соединение может применяться в форме стерильных водных или масляных суспензий для инъекции. В состав суспензий могут быть включены подходящие дисперсии смачивающих агентов и суспендирующих агентов известного уровня техники, которые были описаны выше. Стерильные препараты для инъекций могут также представлять собой стерильные растворы или суспензии для инъекции в нетоксичном разбавителе или растворителе, подходящем для парентеральных инъекций, например, раствор в 1,3-бутандиоле. В число подходящих носителей или растворителей, которые можно применять в данном случае, входят вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспензионной среды традиционно применяются стерильные нелетучие масла. С этой целью обычно можно применять любое нераздражающее нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, при получении составов для инъекции находят применение жирные кислоты, такие как олеиновая кислота. Можно подобрать схему дозировки для обеспечения оптимальной терапевтической реакции. Например, в течение дня можно вводить несколько отдельных доз, или доза может быть пропорционально уменьшена, в соответствии с требованиями терапевтической ситуации.
Формы для инъекции пролонгированного действия получают формированием матриц микрокапсул, в которой лекарственное средство заключено в биоразрушаемые полимеры, например, полилактид-полигликолид. В зависимости от соотношения лекарственного средства и полимера, и природы конкретного примененного полимера, можно регулировать скорость высвобождения действующего средства. Примеры других биоразрушаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Составы для инъекций пролонгированного действия получают также, заключая лекарственное средство в липосомы или микроэмульсии, которые совместимы с тканями организма. Составы для инъекций могут быть стерилизованы, например, фильтрованием через фильтр, удерживающий бактерии, или введением стерилизующих агентов в стерильные твердые композиции, которые можно растворить или диспергировать в стерильной воде или других подходящих для инъекций стерильных средах, непосредственно перед применением.
Твердые дозированные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В этих твердых дозированных формах действующее соединение может быть смешано по меньшей мере с одним инертным фрамацевтически приемлемым эксципиентом или носителем, например, цитратом натрия или дикальцийфосфатом и/или (a) наполнителями или средствами для увеличения объема, например, крахмалом, лактозой, сахарозой, глюкозой, маннитом и кремниевой кислотой; (b) связующими веществами, например, карбоксиметилцеллюлозой, альгинатами, желатином, поливинилпирролидоном, сахарозой и гуммиарабиком; (c) увлажнителями, например, глицерином; (d) дезинтегрирующими средствами, например, агар-агаром, карбонатом кальция, крахмалом картофеля или маниоки, альгиновой кислотой, некоторыми силикатами или карбонатом натрия; (e) средствами, замедляющими растворение, например, парафином; (f) ускорителями абсорбции, например, четвертичными аммониевыми соединениями; (g) смачивающими агентами, например, цетиловым спиртом или моностеаратом глицерина; (h) абсорбентами, например, каолином и бентонитовой глиной, и (i) смазывающими веществами, например, тальком, стеаратом кальция, стеаратом магния, твердыми полиэтиленгликолями, лаурилсульфатом натрия и их смесями. В случае капсул, таблеток или пилюль дозированная форма может включать также буферирующие средства. Твердые композиции подобного типа могут также применяться в качестве заполнителей в твердых и мягких желатиновых капсулах, при применении таких эксципиентов, как лактоза или молочный сахар, а также полиэтиленгликолей высокой молекулярной массы и т.п.
Твердые дозированные формы в виде таблеток, драже, капсул, пилюль и гранул можно получать с покрытиями и оболочками, например, кишечными покрытиями и другими покрытиями, хорошо известными в технике приготовления фармацевтических форм. Они необязательно могут содержать средства, придающие непрозрачность, и, кроме того, могут представлять собой такие композиции, которые высвобождают действующий ингредиент (ингредиенты) только или преимущественно в определенных участках кишечного тракта, необязательно с задержкой. Примерами композиций, которые можно применять для включения действующих агентов, являются полимерные вещества и воски. Таблетки содержат действующее соединение в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые подходят для производства таблеток. Такими наполнителями могут являться, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие средства, например, кукурузный крахмал или альгиновая кислота; связующие средства, например, кукурузный крахмал, желатин или гуммиарабик, и смазывающие средства, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут не иметь покрытия или они могут быть покрыты известными способами, чтобы замедлить разрушение и абсорбцию в желудочно-кишечном тракте и, тем самым, обеспечить непрерывное действие в течение более продолжительного периода. Например, можно применить материал, обеспечивающий задержку, такой как моностеарат глицерина или дистеарат глицерина. Составы, предназначенные для перорального применения, могут быть также помещены в твердые желатиновые капсулы, причем действующее соединение смешивают с твердым инертным разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в мягкие желатиновые капсулы, и в этом случае действующий ингредиент смешивают с водой или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Помимо действующих соединений, жидкие дозированные формы могут содержать инертные разбавители, обычно применяемые в технике, например, воду или другие растворители, солюбилизирующие агенты и эмульгаторы, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, масло зародышей, оливковое, касторовое и кунжутное), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли, эфиры жирных кислот и сорбитана, а также их смеси. Кроме инертных разбавителей пероральные композиции могут также включать адъюванты, например, смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, вкусовые добавки и ароматизаторы.
Водные суспензии содержат действующее соединение в смеси с эксципиентами, подходящими для получения водных суспензий. Такими эксципиентами являются суспендирующие средства, например, натрий карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, смола трагаканта и гуммиарабик; диспергирующие или смачивающие средства могут представлять собой природные фосфатиды, например, лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например, полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например, гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот, такими как гекситол, например, моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с частичными эфирами, полученными из жирных кислот и ангидридов гекситола, например, моноолеат полиэтиленсорбитана. Водные суспензии могут содержать также один или несколько консервантов, например, этил или н-пропил п-гидроксибензоат, один или несколько красителей, одну или несколько вкусоароматических добавок или один или несколько подсластителей, например, сахарозу или сахарин.
Масляные суспензии можно получать путем суспендирования действующего соединения в растительном масле, например, арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или минеральном масле, например, жидком парафине. Масляные суспензии могут содержать загуститель, например, пчелиный воск, твердый парафин или цетиловый спирт. Для получения пероральных препаратов с хорошими вкусовыми свойствами можно добавить подсластители, например, перечисленные ниже и вкусоароматические агенты. Эти композиции можно защищать от окисления добавлением антиоксиданта, например, аскорбиновой кислоты. Диспергируемые порошки и гранулы, подходящие для получения водной суспензии при добавлении воды, включают активный ингредиент в смеси с диспергирующим или смачивающим средством, суспендирующим средством и одним или несколькими консервантами. Примерами подходящих диспергирующих или смачивающих средств, а также суспендирующих средств, являются соединения уже описанные выше по тексту заявки. Кроме того, в состав могут входить дополнительные эксципиенты, например, подсластители и вкусоароматические агенты.
Фармацевтические формы соединений по настоящему изобретению могут также включать эмульсии масло-в-воде. Масляная фаза может представлять собой растительное масло, например, оливковое масло или масло арахиса, или минеральное масло, например, жидкий парафин, или их смесь. Подходящие эмульгаторы могут являться природными смолами, например, гуммиарабиком или смолой трагаканта, природными фосфатидами, например, соевым лецитином, и сложными эфирами или частичными сложными эфирами, полученными из жирных кислот и ангидридов гекситолов, например, моноолеатом сорбитана, а также продуктами конденсации указанных частичных эфиров с этиленоксидом, например, полиоксиэтиленсорбитан моноолеатом. Эмульсия может содержать также подсластители и вкусоароматические добавки. Сиропы и эликсиры могут включать подсластители, например, глицерин, сорбит или сахарозу. Такие составы могут содержать также мягчительное средство, консервант и вкусоароматическое или красящее средство.
В одном из вариантов осуществления, для легкости введения и однородности дозировок, соединения по настоящему изобретению включают в состав дозированных лекарственных форм. Термин «дозированная лекарственная форма» в настоящем описании относится к физически дискретным единицам, которые подходят в качестве разовых доз для субъектов, подвергаемых лечению; причем каждая из этих единиц содержит терапевтически эффективное количество соединения и по меньшей мере один фармацевтический эксципиент. Готовый лекарственный продукт должен включать дозированную лекарственную форму в контейнере, который имеет этикетку или снабжен описанием, в котором указан предполагаемый способ лечения, например, лечения β-амилоидного заболевания, например, амилоидоза, такого как болезнь Альцгеймера, или заболевания, связанного с образованием фибрилл α-синуклеина, например, болезни Паркинсона. Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые можно изготовить путем смешивания соединения по настоящему изобретению с подходящими нераздражающими эксципиентами или носителями, например, маслом какао, полиэтиленгликолем или воском для суппозиториев, которые являются твердыми при комнатной температуре, но жидкими при температуре тела, и, следовательно, плавятся в прямой кишке или вагинальной полости и высвобождают активное соединение.
Помимо этого, соединения по настоящему изобретению можно вводить в форме липосом. Способы получения липосом известны в технике (Prescott, Ed., Methods in Cell Biology 1976, Volume XIV, Academic Press, New York, N.Y.). Как известно в технике, липосомы в основном получают из фосфолипидов или других липидных веществ. Липосомы образованы одно- или многослойными гидратированными жидкими кристаллами, которые диспергированы в водной среде. Может применяться любой нетоксичный, физиологически приемлемый и метаболизируемый липид, способный к образованию липосом. Композиции по настоящему изобретению в форме липосом могут содержать, помимо самих соединений по настоящему изобретению, стабилизаторы, консерванты, эксципиенты и т.п. Предпочтительными липидами являются природные и синтетические фосфолипиды и фосфатидилхолины (лецитины).
Соединения по настоящему изобретению можно также вводить в форме «пролекарств», и в этом случае действующие фармацевтические ингредиенты высвобождаются in vivo в организме при контакте с гидролитическими ферментами, например, эстеразами и фосфатазами. Термин «фармацевтически приемлемые пролекарства» в настоящем описании относится к таким пролекарствам соединений по настоящему изобретению, которые согласно обоснованному суждению медицины подходят для применения в контакте с тканями, не проявляя неприемлемой токсичности, раздражения, аллергической реакции и т.п., и соответствуют разумному соотношению польза/риск, а также эффективны для намеченного применения. Всестороннее обсуждение приведено в работе T. Higuchi и V. Stella (Higuchi, T. and Stella, V. Pro-drugs as Novel Delivery Systems, V.14 of the A.C.S. Symposium Series; Edward B. Roche, Ed., Bioreversible Carriers in Drug Design 1987, American Pharmaceutical Association and Pergamon Press), которая включена в настоящую заявку с помощью ссылки.
Кроме того, соединения по настоящему изобретению или их фармацевтически приемлемые производные можно включать в состав препаратов, которые нацелены на конкретную ткань, рецептор или другую область организма субъекта, подвергаемого лечению. Многие из таких способов нацеливания хорошо известны специалистам в данной области техники. Все подобные способы нацеливания рассматриваются в настоящем изобретении для применения в композициях по настоящему изобретению. В качестве неограничивающих примеров способов нацеливания см., например, патенты США №№ 6316652, 6274552, 6271359, 6253872, 6139865, 6131570, 6120751, 6071495, 6060082, 6048736, 6039975, 6004534, 5985307, 5972366, 5900252, 5840674, 5759542 и 5709874.
В одном из вариантов осуществления липосомальные суспензии, включая липосомы, нацеленные на определенную ткань, например, липосомы, нацеленные на опухоль, также могут подходить в качестве фармацевтически приемлемых носителей. Такие липосомы можно получать способами, известными специалистам в данной области техники. Например, липосомальные составы можно получать, как описано в патенте США № 4522811. Вкратце, липосомы, например, многослойные везикулы (MLV), можно получать при высушивании яичного фосфатидилхолина и фосфатидилсерина мозга (в молярном соотношении 7:3) на внутренней поверхности колбы. Добавляют раствор соединения по настоящему изобретению в фосфатном буферном солевом растворе, в котором отсутствуют двухвалентные катионы (PBS), и встряхивают колбу до тех пор, пока липидная пленка не диспергируется. Полученные везикулы промывают для удаления неинкапсулированного соединения, осаждают центрифугированием и затем вновь суспендируют в PBS.
Составы с замедленным высвобождением
Настоящее изобретение включает также применение составов с замедленным высвобождением для доставки соединений по настоящему изобретению к желаемой мишени (т.е. мозгу или системным органам) при высоких циркулирующих уровнях (от 10-9 до 10-4 М). В предпочтительном варианте осуществления, для лечения болезни Альцгеймера или болезни Паркинсона, поддерживаются циркулирующие уровни соединений по настоящему изобретению до 10-7 М. Эти уровни соответствуют либо системной циркуляции в организме пациента, либо, в предпочтительном варианте осуществления, присутствуют в тканях мозга, и в наиболее предпочтительных вариантах осуществления локализованы в местах отложения фибрилл β-амилоида и α-синуклеина в мозге или других тканях.
Имеется в виду, что в течение определенного периода времени содержание соединений поддерживается на уровне, который является желательным, и может быть легко определен специалистом в данной области с использованием настоящего описания и соединений по настоящему изобретению. В предпочтительном варианте осуществления характерная особенность изобретения заключается во введении соединений с применением составов с замедленным высвобождением, так чтобы поддерживался постоянный уровень соединения в сыворотке от 10-8 до 10-6 М в течение 48-96 часов.
Такие составы с замедленным высвобождением и/или высвобождением в течение определенного периода времени могут быть изготовлены с применением средств доставки, обеспечивающих замедленное высвобождение, которые хорошо известны рядовым специалистам в данной области, например, описанных в патентах США №№ 3845770, 3916899, 3536809, 3598123, 4008719, 4710384, 5674533, 5059595, 5591767, 5120548, 5073543, 5639476, 5354556 и 5733566, содержание которых включено в настоящую заявку с помощью ссылки. Эти фармацевтические композиции могут применяться для обеспечения медленного или замедленного высвобождения одного или нескольких действующих соединений, и в них используются, например, гидроксипропилметилцеллюлоза, другие полимерные матрицы, гели, проницаемые мембраны, осмотические системы, многослойные покрытия, микрочастицы, липосомы, микросферы и т.п. Для применения с фармацевтическими композициями по настоящему изобретению можно без труда подобрать подходящие составы с замедленным высвобождением, известные специалисту в данной области, включая описанные в настоящей заявке. Так, например, настоящим изобретением охвачены дозированные лекарственные формы, подходящие для перорального введения, например, но не ограничиваясь этим, таблетки, капсулы, желатиновые капсулы, каплеты, порошки и т.д., которые адаптированы для обеспечения замедленного высвобождения.
В предпочтительном варианте осуществления составы с замедленным высвобождением содержат действующее соединение, а также, но не ограничиваясь перечисленным, микрокристаллическую целлюлозу, мальтодекстрин, этилцеллюлозу и стеарат магния. Как указано выше, все известные способы инкапсулирования, которые совместимы со свойствами раскрываемых соединений, входят в объем настоящего изобретения. В составах с замедленным высвобождением инкапсулирование осуществляется путем покрытия частиц или гранул фармацевтической композиции по настоящему изобретению слоем медленно растворимого полимера различной толщины или же путем микроинкапсулирования. В предпочтительном варианте осуществления, в составах с замедленным высвобождением инкапсулирование происходит с применением покрывающего материала различной толщины (например, от примерно 1 микрона до 200 микронов), что делает возможным растворение фармацевтической композиции за время от примерно 48 часов до примерно 72 часов после введения млекопитающему. В другом варианте осуществления материал покрытия представляет собой добавку, одобренную для употребления в пищу.
В другом варианте осуществления состав с замедленным высвобождением основан на растворении матрицы, которую получают прессованием в таблетку лекарственного средства с медленно растворимым полимерным носителем. В одном из предпочтительных вариантов осуществления покрытые частицы имеют размер в диапазоне от примерно 0,1 до примерно 300 микронов, как указано в патентах США №№ 4710384 и 5354556, которые включены в настоящую заявку посредством ссылки во всей полноте. Каждая из этих частиц имеет строение микроматрицы, где действующий ингредиент равномерно распределен в объеме полимера.
В настоящем изобретении раскрыты составы с замедленным высвобождением, подобные описанным в патенте США № 4710384, который включен в настоящую заявку с помощью ссылки во всей полноте, имеющие относительно высокое процентное содержание пластификатора в покрытии, для достижения значительной гибкости, с целью предотвращения заметного разрушения при прессовании. Удельное количество пластификатора меняется в зависимости от природы покрытия и конкретного примененного пластификатора. Это количество можно легко определить эмпирически путем тестирования характеристик высвобождения полученной таблетки. Если лекарственное средство высвобождается слишком быстро, необходимо применить большее количество пластификатора. Характеристики высвобождения являются также функцией толщины покрытия. Если применено значительное количество пластификатора, способность покрытия к обеспечению замедленного высвобождения падает. Следовательно, можно немного увеличить толщину покрытия, чтобы компенсировать повышенное содержание пластификатора. Как правило, в этом варианте осуществления пластификатор должен присутствовать в покрытии в количестве от примерно 15 до 30% от массы материала, обеспечивающего замедленное высвобождение, предпочтительно от 20 до 25%, и количество покрытия должно составлять от 10 до 25% от массы активного материала, предпочтительно от 15 до 20%. В покрытие можно вводить любой традиционный фармацевтически приемлемый пластификатор.
Соединения по настоящему изобретению можно включать в составы с замедленным высвобождением и/или высвобождением в течение определенного периода времени. Все фармацевтические продукты с замедленным высвобождением преследуют общую цель улучшения лекарственной терапии по сравнению с уровнем, которого можно добиться при применении их аналогов с обычным высвобождением. В идеальном случае применение оптимального препарата с замедленным высвобождением характеризуется минимумом лекарственного вещества, необходимого для лечения или контроля над тем или иным состоянием. Преимущества составов с замедленным высвобождением могут включать: 1) увеличенный период активности композиции, 2) уменьшенную частоту введения препарата и 3) более высокую податливость лечению со стороны пациента. Помимо этого составы с замедленным высвобождением можно применять для влияния на время начала действия или другие характеристики, например, содержание композиции в крови, и за счет этого воздействовать на проявление побочных эффектов.
Составы с замедленным высвобождением по настоящему изобретению предназначены для первоначального высвобождения определенного количества терапевтической композиции, которое сразу вызывает желаемый терапевтический эффект, и постепенного и непрерывного высвобождения остального количества композиции для поддержания достигнутого уровня терапевтического эффекта в течение продолжительного периода времени. Для достижения этого постоянного уровня в организме, терапевтическая композиция должна выделяться из дозированной формы с такой скоростью, которая позволит заместить композицию, подвергшуюся метаболизму и выведенную из организма.
Замедленное высвобождение действующего ингредиента можно стимулировать действием различных факторов, например, pH, температуры, ферментов, воды или других физиологических условий или соединений. Термин «компонент, обеспечивающий замедленное высвобождение» в контексте настоящего изобретения определяется как соединение или соединения, включая, но не ограничиваясь этим, полимеры, полимерные матрицы, гели, проницаемые мембраны, липосомы, микросферы и т.п. или их комбинации, которые способствуют замедленному высвобождению действующего ингредиента.
Если комплекс является водорастворимым, его можно получать в подходящем буфере, например, фосфатном буферном солевом растворе или других физиологически совместимых растворах. В качестве альтернативы, если конечный комплекс плохо растворим в водных растворителях, в его состав можно включать неионные ПАВ, например, Tween или полиэтиленгликоль. Так, соединения и их физиологически приемлемые растворители можно включать в составы, предназначенные для введения с помощью ингаляции или инсуффляции (через рот или через нос) или, например, для перорального, буккального, парентерального или ректального введения.
Можно получать препараты для перорального введения такого состава, который обеспечивает регулируемое высвобождение действующего соединения. В предпочтительном варианте осуществления соединения по настоящему изобретению включают в состав порошков с регулируемым высвобождением, состоящих из отдельных микрочастиц, которые можно легко включать в состав жидких лекарственных форм. Порошки с замедленным высвобождением включают частицы, содержащие действующий ингредиент и, необязательно, эксципиент с по меньшей мере одним нетоксичным полимером.
Эти порошки можно диспергировать или суспендировать в жидком носителе и они сохранят свои характеристики замедленного высвобождения в течение достаточного периода времени. Эти дисперсии или суспензии обладают как химической стабильностью, так и устойчивостью с точки зрения скорости растворения. Порошок может содержать эксципиент, включающий полимер, который может быть растворимым, нерастворимым, проницаемым, непроницаемым или биоразрушаемым. Эти полимеры могут являться полимерами или сополимерами. Полимер может быть природным или синтетическим полимером. Природные полимеры включают полипептиды (например, зеин), полисахариды (например, целлюлозу) и альгиновую кислоту. Типовые примеры синтетических полимеров включают, не ограничиваясь этим, полимеры, описанные в строках 33-45 стр. 3 патента США № 5354556, который во всей полноте включен в настоящую заявку посредством ссылки. Особенно подходящие полимеры включают, не ограничиваясь этим, полимеры, описанные в строке 46 стр.3 - строке 8 стр.4 патента США № 5354556, который во всей полноте включен в заявку посредством ссылки.
Составы с замедленным высвобождением соединений по настоящему изобретению могут быть предназначены для парентерального введения, например, путем внутримышечной инъекции или в виде имплантов для введения под кожу или в различные полости организма, а также представлять собой трансдермальные устройства. В одном из вариантов осуществления состав для внутримышечной инъекции получают в форме водных или масляных суспензий. В водных суспензиях эффект замедленного высвобождения частично достигается за счет уменьшения растворимости действующего соединения при образовании комплекса или уменьшения скорости растворения. Аналогичный подход применяется к масляным суспензиям и растворам, где скорость высвобождения действующего соединения определяется его фракционным распределением между маслом и окружающей водной средой. Подходят только те действующие соединения, которые растворимы в масле и обладают желаемыми характеристиками распределения. Масла, которые могут применяться для внутримышечной инъекции, включают, не ограничиваясь перечисленными, кунжутное, оливковое, арахисовое, кукурузное, миндальное, соевое, хлопковое и касторовое масла.
Высокоэффективной формой доставки лекарственных средств, которая обеспечивает замедленное высвобождение в течение периода от нескольких дней до нескольких лет, является введение имплантата, состоящего из полимерной матрицы, включающей лекарственное вещество, под кожу или в различные полости организма. Применяемый в имплантате полимерный материал, который должен быть биосовместимым и нетоксичным, включает, не ограничиваясь этим, гидрогели, силиконы, полиэтилены, сополимеры этилен-винилацетат или биоразрушаемые полимеры.
Оценка активности соединений
Биологическую активность соединений по настоящему изобретению в качестве средств, разрушающих/ингибирующих фибриллы β-амилоидного белка (Aβ), характерные для болезни Альцгеймера, и агрегаты α-синуклеина, характерные для болезни Паркинсона, оценивали путем определения эффективности соединений для уменьшения/разрушения ранее образованных амилоидных фибрилл, соответствующих болезни Альцгеймера (т.е. фибрилл, состоящих из Aβ 1-42), и агрегатов α-синуклеина, соответствующих болезни Паркинсона. В одном из исследований применяли флуориметрию с тиофлавином T для определения действия соединений и ЭДТУ (в качестве отрицательного контроля). В этом анализе тиофлавин T специфично связывается с фибриллярным амилоидом, и это связывание вызывает усиление флуоресценции при 485 нм, которое прямо пропорционально количеству присутствующих фибрилл. Чем выше флуоресценция, тем больше количество присутствующих фибрилл или агрегатов (Naki et al., Lab. Invest. 65:104-110, 1991; Levine III, Protein Sci. 2:404-410, 1993; Amyloid: Int. J. Exp. Clin. Invest. 2:1-6, 1995).
В анализе связывания Конго красного количественно определяется способность тестируемого соединения изменять связывание фибрилл амилоида (Aβ 1-42) или агрегатов α-синуклеина с Конго красным. В этом анализе фибриллы амилоида (Aβ 1-42) или агрегаты α-синуклеина и тестируемое соединение инкубируют в течение 3 дней и затем осуществляют вакуумное фильтрование через фильтр с размером пор 0,2 мкм. Затем определяют количество фибрилл Aβ 1-42 или агрегатов α-синуклеина, оставшихся на фильтре, по окрашиванию фильтра Конго красным. После соответствующего промывания фильтра любое ослабление интенсивности цвета Конго красного на фильтре в присутствии тестируемого соединения (по сравнению с окрашиванием амилоидного белка Конго красным в отсутствии тестируемого соединения) является показателем способности тестируемого соединения уменьшать/изменять количество агрегированных и конгофильных фибрилл Aβ 1-42 или агрегатов α-синуклеина.
Комбинированная терапия
В другом варианте осуществления соединения по настоящему изобретению можно вводить в комбинации или последовательно с другим терапевтическим средством. Эти другие терапевтические средства включают известные средства для лечения, предупреждения или облегчения одного или нескольких симптомов амилоидоза и синуклеиновых заболеваний. Такие терапевтические средства включают, не ограничиваясь этим, донепезила гидрохлорид (Aricept), ривастигмина тартрат (Exelon), такрина гидрохлорид (Cognex) и галантамина гидробромид (Reminyl).
Способы применения соединений и композиций
Соединения и композиции по настоящему изобретению применимы в способах лечения, предупреждения или облегчения одного или нескольких симптомов β-амилоидных заболеваний или расстройств, включая, но не ограничиваясь этим, заболевания, связанные с образованием, отложением, накоплением или устойчивым существованием фибрилл β-амилоида. В некоторых вариантах осуществления соединения и композиции по настоящему изобретению применяются для лечения, предупреждения или облегчения одного или нескольких симптомов заболеваний, включая, но не ограничиваясь этим, болезнь Альцгеймера, синдром Дауна, наследственную церебральную геморрагию с амилоидозом голландского типа и церебральную β-амилоидную ангиопатию.
Кроме того, в изобретении разработаны способы ингибирования или предупреждения образования фибрилл α-синуклеина, способы ингибирования или предупреждения роста фибрилл α-синуклеина и способы уменьшения, разрушения и/или дезагрегации ранее образовавшихся агрегатов α-синуклеина и белковых отложений, связанных с α-синуклеином.
В некоторых вариантах осуществления синуклеиновые заболевания или синуклеинопатии, которые лечат, предупреждают или симптомы которых облегчают с применением соединений и композиций по настоящему изобретению, включают, не ограничиваясь этим, заболевания, связанные с образованием, отложением, накоплением или устойчивым существованием агрегатов синуклеина, в т.ч. фибрилл α-синуклеина. В некоторых вариантах осуществления эти заболевания включают болезнь Паркинсона, семейную болезнь Паркинсона, болезнь с тельцами Леви, вариант болезни Альцгеймера с тельцами Леви, деменцию с тельцами Леви, множественную системную атрофию и комплекс Гуама паркинсонизм-деменция.
В одном из вариантов осуществления изобретение относится к соединению, выбранному из группы, состоящей из
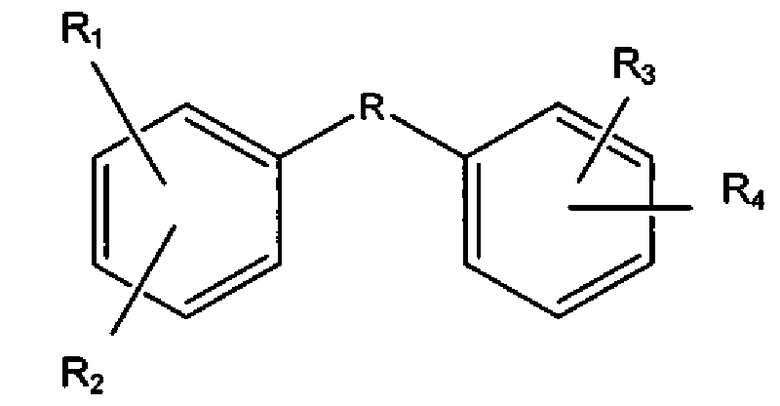 ,
,
где R1, R2, R3 и R4 представляют собой независимо расположенные гидроксильные группы и R выбран из гетероарила, -C(O)NR', сульфонамида, трициклоалкила или их фармацевтически приемлемых эфиров или солей, где R' выбран из H или CH3, и таких, что если R представляет собой -C(O)NR' и любая из пар гидроксильных групп R1 и R2 или R3 и R4 находится в положении 3,4, то две других гидроксильных группы находятся в одном из положений, выбранных из группы, состоящей из 2,3; 2,4; 2,5; 2,6; 3,5; 3,6; 4,5; 4,6 и 5,6.
В другом варианте осуществления изобретение относится к соединению, выбранному из группы, состоящей из
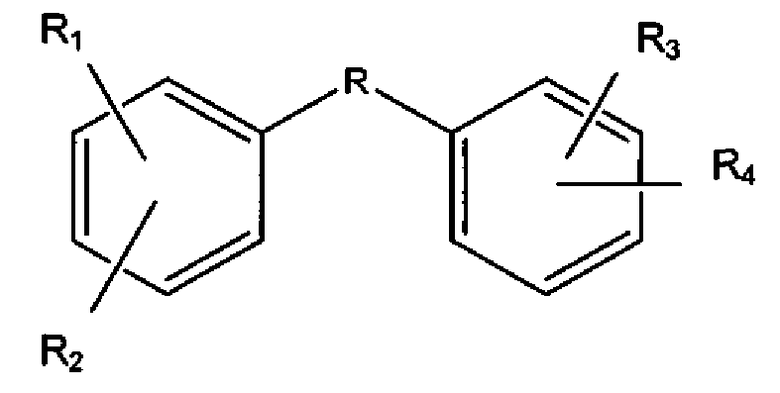 ,
,
где R1, R2, R3 и R4 представляют собой независимо расположенные гидроксильные группы, R означает -C(O)NR' и R' выбран из H или CH3, и если любая из пар гидроксильных групп R1 и R2 или R3 и R4 находится в положении 3,4, то две других гидроксильных группы находятся в одном из положений, выбранных из группы, состоящей из 2,3; 2,4; 2,5; 2,6; 3,5; 3,6; 4,5; 4,6 и 5,6, а также их фармацевтически приемлемых солей.
В другом варианте осуществления в изобретении разработана фармацевтическая композиция, включающая соединения по настоящему изобретению и фармацевтически приемлемый наполнитель.
В другом варианте осуществления в изобретении разработан способ лечения состояния, при котором происходит образование, отложение, накопление или устойчивое существование агрегатов Aβ-амилоида или α-синуклеина, включающий обработку агрегатов эффективным количеством соединений по настоящему изобретению.
В следующем варианте осуществления в изобретении разработан способ лечения β-амилоидных заболеваний или синуклеинопатий у страдающего ими млекопитающего, включающий введение терапевтически эффективного количества соединений по настоящему изобретению.
В другом варианте осуществления разработан способ улучшения двигательной активности млекопитающего, страдающего синуклеинопатией, включающий введение терапевтически эффективного количества соединения по настоящему изобретению.
В еще одном варианте осуществления разработан способ прекращения прогрессирования дефицита двигательной активности у млекопитающих, страдающих болезнью Паркинсона, включающий введение терапевтически эффективного количества соединения по настоящему изобретению.
Следующие далее по тексту неограничивающие примеры приведены только в качестве иллюстрации, и они не рассматриваются как ограничение объема настоящего изобретения, поскольку возможно существование большого числа их вариантов, которые соответствуют сути изобретения и не выходят за пределы его объема.
ПРИМЕРЫ
Общие методики проведения экспериментов
Все растворители перегоняли перед использованием и удаляли на роторном испарителе при температуре до 35°C. Для флэш-хроматографии на силикагеле использовали силикагель 60 фирмы Merck, 200-400 меш, 40-63 мкм. Для проведения ТСХ использовали пластинки Merck DC-plastikfolien Kieselgel 60 F254, причем визуализацию хроматограммы вначале осуществляли с помощью УФ-лампы и затем погружением в раствор ванилина (1% ванилин, 1% H2SO4 в EtOH) и нагреванием. Масс-спектры регистрировали на приборе Kratos MS-80. Спектры ЯМР регистрировали при 25°C при рабочей частоте 500 или 300 МГц для 1H и 125 или 75 МГц для 13C, на спектрометрах Varian INOVA-500 или VXR-300. Химические сдвиги приведены в м.д. по δ шкале и отнесены к сигналам растворителей: CHCl3 при 7,25 и CDCl3 при 77,0 м.д., или (CH3)2CO при 2,15 и (CD3)2CO при 30,5 м.д., или CH3OD при 3,30 и CD3OD при 39,0 м.д.
Условия проведения ВЭЖХ
Анализ образцов проводили на приборе Agilent HP1100, который функционировал под управлением программы EzChromElite и был снабжен колонкой C18 (Phenomenex Prodigy 5 мкм 100A, 250x4,6 мм) с предколонкой (Phenomenex ODS 4×3 мм, 5 мкм) при температуре 30°C. Пики регистрировались при 280 нм. Подвижная фаза представляла собой ацетонитрил в воде (с 0,1% ТФУ): t0=11%, t20=11%, t30=100%, t31=11%, t40=11%. Скорость потока составляла 1 мл/мин и объем вводимого образца равнялся 5 мкл.
Пример 1: Синтез сульфонамида 2
3,4-дигидроксифенилсульфонамид 3,4-дигидроксибензолсульфоновой кислоты (соединение 5)
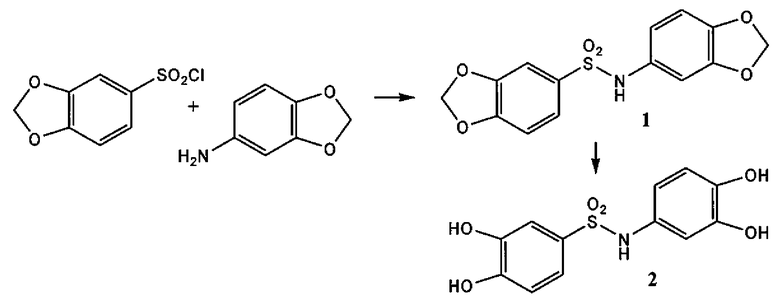
Синтез сульфонамида 2 осуществляли с помощью реакции 3,4-метилендиоксибензолсульфонилхлорида (полученного из 1,2-метилендиоксибензола (Tao, E.V.P.; Miller, W.D. патент США № 5387681, 1995)) с 3,4-метиендиоксианилином, которая приводила к образованию сульфонамида 1 с хорошим выходом. Снятие защиты трибромидом бора в стандартных условиях позволяло получить свободный фенолсульфонамид с приемлемым выходом.
К перемешиваемому раствору 1,3-бензодиоксол-5-сульфонилхлорида (Tao, E.V.P.; Miller, W.D. патент США № 5387681, 1995)) (1 г) в дихлорметане (DCM) (10 мл) добавляли раствор 3,4-метилендиоксианилина (0,62 г) в дихлорметане (10 мл) и затем пиридин (1 мл). Смесь кипятили с обратным холодильником в течение 2 ч, охлаждали, разбавляли дихлорметаном (150 мл), промывали водным раствором HCl (1М, 2×100 мл), высушивали и затем упаривали в вакууме, получая неочищенный продукт в виде коричневой смолы. Очистка колоночной хроматографией на силикагеле при элюировании 5-10% этилацетатом в дихлорметане приводила к получению чистого сульфонамида 1 в виде светло-коричневой смолы (1,34 г, 92%). Кристаллизация из 95% этанола позволяла получить продукт в виде бледно-коричневых кристаллов.
ВЭЖХ 29,6 минут.
1H ЯМР ((CD3)2CO) 8,75 (1H, с), 7,39 (2H, дд, J=2,9 Гц), 7,24 (1H, д, J=2 Гц), 7,02 (1H, д, J=9 Гц), 6,86 (1H, д, J=2 Гц), 6,81 (1H, д, J=9 Гц), 6,72 (2H, дд, J=2, 9 Гц), 6,23 (2H, с) и 6,06 (2H, с).
HREIMS (Масс-спектроскопия с ионизацией электронным ударом): найдено, 344,0201; MNa+, C14H11NNaO6S, вычислено 344,0199.
К раствору сульфонамида 1 (0,7 г) в сухом DCM (50 мл) добавляли трибромид бора (0,5 мл) и выдерживали смесь при комнатной температуре в течение 3 часов. Затем осторожно по каплям добавляли метанол (5 мл) и оставляли реакционную смесь при комнатной температуре на 24 часа. Полученную смесь упаривали в вакууме до объема 1 мл, затем добавляли дополнительное количество метанола (20 мл), и повторяли этот цикл четыре раза, после чего удаляли весь растворитель выпариванием в вакууме.
Очистка колоночной хроматографией на силикагеле при элюировании 0-20% метанолом в хлороформе приводила к получению продукта в виде бледно-коричневой смолы. Дополнительная очистка хроматографией на обращенной фазе при использовании колонки C-18 с оксидом кремния и при элюировании 0-50% ацетонитрилом в воде, с последующей лиофильной сушкой, позволяла получить чистый продукт 2 в виде светло-коричневого порошка (295 мг, 45%).
ВЭЖХ 12,9 минут 95%
1H ЯМР (CD3OD) 7,05 (1H, д, J=2 Гц), 7,03 (2H, дд, J=2, 9 Гц), 6,76 (1H, д} J=9 Гц), 6,57 (1H, д, J=2 Гц), 6,56 (1H, д, J=9 Гц) и 6,31 (2H, дд, J=2, 9 Гц).
HREIMS: найдено, 296,0241; M-, C12H10NO6S, вычислено 296,0234.
Пример 2: Синтез имидазола 4
2,4-бис-(3,4-дигидроксифенил)имидазол (соединение 6)
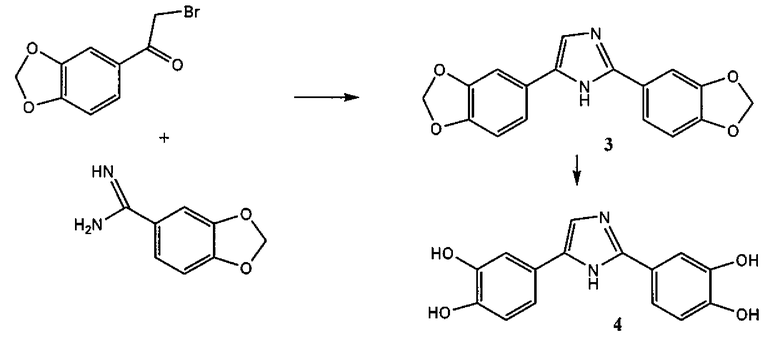
Имидазольный цикл был сформирован по методике, описанной Li и соавторами (Li et al., Organic Process Research and Development 2002, 6, 682-3), из амидинобензола, полученного из пиперонилонитрила (Thurkauf et al., J. Med Chem. 1995, 38(12), 2251-2255) и бромкетона (Castedo et al., Tetrahedron 1982, 38(11), 1569-70), синтезированного из 3,4-метилендиоксиацетофенона по методике, описанной Lee и соавторами (Korean Chem Soc. 2003, 24(4), 407-408). Снятие защиты с помощью трибромида бора в стандартных условиях позволило получить свободный фенолзамещенный имидазол с хорошим выходом.
Согласно способу, описанному Li, смесь 3-аминодибензола (Thurkauf et al., J. Med. Chem. 1995, 38(12), 2251-2255) (0,5 г, 3 ммоль) и бикарбоната калия (1,20 г, 12 ммоль) в тетрагидрофуране (ТГФ) (16 мл) и воде (4 мл) энергично нагревали до кипения с обратным холодильником. В течение 30 минут добавляли бромкетон (Castedo et al., Tetrahedron 1982, 38(11), 1569-70; и Lee et al., Korean Chem Soc. 2003, 24(4), 407-408) (0,729 г, 3 ммоль) в ТГФ (4 мл) и продолжали кипячение с обратным холодильником в течение еще 2 часов. Затем удаляли ТГФ выпариванием в вакууме, остаток экстрагировали этилацетатом, высушивали и упаривали в вакууме, получая неочищенный продукт в виде твердого вещества коричневого цвета. Кристаллизация из 95% этанола позволяла получить чистый имидазол 3 в виде бледно-желтого твердого кристаллического вещества (0,54 г, 58%).
ВЭЖХ 27,9 минут
1H ЯМР ((CD3)2CO) 7,45-7,70 (5H, м), 7,02 (1H, д, J=9 Гц, 6,95 (1H, д, J=9 Гц), 6,15 (2H, с) и 6,09 (2Н, с).
HREIMS: найдено, 309,0875; MH+, C17H12N2O4, вычислено 309,0870.
К раствору имидазола 3 (0,5 г) в сухом DCM (50 мл) добавляли трибромид бора (1,0 мл) и смесь оставляли стоять при комнатной температуре в течение 3 часов. Осторожно по каплям добавляли метанол (5 мл), и затем оставляли стоять реакционную смесь при комнатной температуре в течение 24 часов. Полученную смесь упаривали в вакууме до объема 1 мл, затем добавляли дополнительное количество метанола (30 мл), и повторяли этот цикл четыре раза, после чего удаляли весь растворитель выпариванием в вакууме.
Очистка колоночной хроматографией на силикагеле при элюировании 0-20% метанолом в хлороформе позволяла получить продукт 4 в виде бледно-коричневого твердого вещества (0,27 г, 58%).
ВЭЖХ 16,3 минуты 99%
1H ЯМР (CD3OD) 7,59 (1H, с), 7,36 (1H, д, J=2 Гц), 7,31 (2H, дд, J=2, 9 Гц), 7,16 (1H, д, J=2 Гц), 7,10 (2H, дд, J=2, 9 Гц), 6,98 (1H, д, J=9 Гц) и 6,88 (1H, д, J=9 Гц).
HREIMS: найдено, 285,0873; MH+, C15H13N2O4, вычислено 285,0870.
Пример 3: Синтез триазола 7
3,5-бис-(3,4-дигидроксифенил)-1,2,4-триазол (соединение 7)
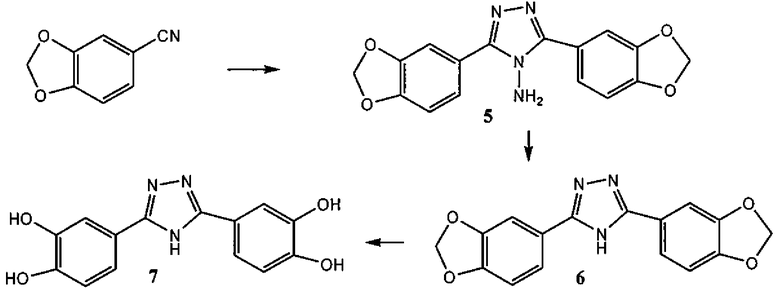
4-Аминотриазольный цикл получали димеризацией пиперонилонитрила способом, описанном Bentiss (Bentiss et al., J. Heterocyclic Chem. 1999, 36, 149-152), после чего проводили дезаминирование по методике, описанной Bentiss (Bentiss et al., J. Heterocyclic Chem. 2002, 39, 93-96), получая триазол 6 с хорошим выходом. Снятие защиты с применением трибромида бора в стандартных условиях позволяло получить свободный фенолзамещенный триазол 7 с хорошим выходом.
Согласно методике, описанной Bentiss (Bentiss et al., J. Heterocyclic Chem. 1999, 36, 149-152), смесь ароматического нитрила (1 г), гидразингидрата (1 г) и гидрохлорида гидразина (0,5 г), растворенную в этиленгликоле (5 мл), нагревали до 130°C в течение 5 часов. Раствор охлаждали, разбавляли водой (7 мл), отделяли твердый продукт фильтрованием, промывали DCM и высушивали, получая неочищенный продукт. Перекристаллизация из метанола позволяла получить чистый 4-аминотриазол 5 в виде бледно-желтого твердого вещества (0,65 г, 66%).
ВЭЖХ 27,0 минут
1H ЯМР ((CD3)2CO) 7,62 (2H, дд, J=2, 9 Гц), 7,42 (2H, д, J=2 Гц), 6,94 (2H, д, J=9 Гц), 6,15 (2H, с) и 5,93 (4H, с).
HREIMS: найдено, 325,0937; MH+, C16H13N4O4, вычислено 325,0931.
Согласно методике, описанной Bentiss (Bentiss et al., J. Heterocyclic Chem. 2002, 39, 93-96), к перемешиваемому раствору аминотриазола 5 (0,5 г) в водном растворе гипофосфорной кислоты (50%, 5 мл) медленно добавляли раствор нитрита натрия (0,6 г) в воде (1,5 мл). Полученную смесь перемешивали при комнатной температуре еще четыре часа, после чего собирали бледно-оранжевый осадок, промывали его водой и высушивали, получая триазол 6 (0,38 г, 80%).
ВЭЖХ 29,48 минут
1H ЯМР ((CD3)2CO) 7,81 (2H, дд, J=2, 9 Гц), 7,70 (2H, д, J=2 Гц), 7,10 (2H, д, J=9 Гц) и 6,20 (4H, с).
HREIMS: найдено, 310,0818; C16H12N3O4, вычислено 310,0822.
К раствору триазола 6 (0,5 г) в сухом DCM (50 мл) добавляли трибромид бора (1,0 мл) и смесь оставляли стоять при комнатной температуре в течение 3 часов. Осторожно по каплям добавляли метанол (5 мл), и затем оставляли стоять реакционную смесь при комнатной температуре в течение 24 часов. Полученную смесь упаривали в вакууме до объема 1 мл, затем добавляли дополнительное количество метанола (30 мл), и повторяли этот цикл четыре раза, после чего удаляли весь растворитель выпариванием в вакууме.
Очистка колоночной хроматографией на силикагеле при элюировании 0-20% метанолом в хлороформе позволяла получить продукт 7 в виде бледно-коричневого твердого вещества (0,24 г, 52%).
ВЭЖХ 16,1 минуты 97%
1H ЯМР (CD3OD) 7,46 (2H, д, J=2 Гц), 7,41 (2H, дд, J=2, 9 Гц), 7,15 (1H, с) и 6,96 (2H, д, J=9 Гц).
HREIMS: найдено, 286,0815; MH+, C14H12N3O4, вычислено 286,0822.
Пример 4: Синтез пиразола 9
3,5-бис-(3,4-дигидроксифенил)пиразол (соединение 8)
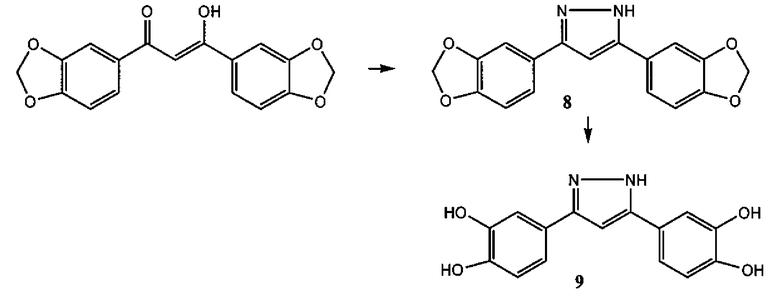
Взаимодействие 1,3-дикетона (Lopez et al., Planta Med. 1998, 64(1), 76-77) (полученного по методике, описанной Choshi et al., (Chem. Pharm. Bull. 1992, 40(4), 1047-1049)) с гидразингидратом по методике, описанной Fink et al. (Chemistry and Biology 1999, 6, 205-219) позволяло получить пиразол 8 с хорошим выходом. Снятие защиты с помощью трибромида бора в стандартных условиях позволяло получить свободный фенолзамещенный пиразол 9 с хорошим выходом.
Согласно методике, описанной Fink et al. (Chemistry and Biology 1999, 6, 205-219), суспензию дикетона (Choshi et al., Chem. Pharm. Bull. 1992, 40(4), 1047-1049; и Lopez et al., Planta Med. 1998, 64(1), 76-77) (1 г) и гидрохлорида гидразина (1 г, 5 экв.) в ДМФА/ТГФ (3:1, 12 мл) нагревали до кипения с обратным холодильником в течение 24 часов. Добавляли воду и экстрагировали смесь дихлорметаном, высушивали и упаривали в вакууме, получая неочищенный продукт 8 в виде твердого вещества желтого цвета. Очистка колоночной хроматографией на силикагеле при элюировании 0-20% этилацетатом в дихлорметане позволяла получить пиразол 8 в виде бледно-желтого твердого вещества (0,49 г, 50%).
ВЭЖХ 30,3 минуты
1H ЯМР ((CD3)2CO) 7,47 (2H, дд, J=2, 9 Гц), 7,46 (2H, д, J=2 Гц), 7,04 (1H, с), 7,02 (2H, д, J=9 Гц) и 6,14 (4H, с).
HREIMS: найдено, 309,0859; MH+, C17H13N2O4, вычислено 309,0870.
К раствору пиразола 8 (0,46 г) в сухом DCM (50 мл) добавляли трибромид бора (0,4 мл) и смесь оставляли стоять при комнатной температуре в течение 3 часов. Осторожно по каплям добавляли метанол (5 мл) и затем оставляли стоять реакционную смесь при комнатной температуре в течение 24 часов. Полученную смесь упаривали в вакууме до объема 1 мл, затем добавляли дополнительное количество метанола (30 мл), и повторяли этот цикл четыре раза, после чего удаляли весь растворитель выпариванием в вакууме.
Очистка колоночной хроматографией на силикагеле при элюировании 0-20% метанолом в хлороформе позволяла получить пиразол 9 в виде бледно-желтого твердого вещества (0,285 г, 67%).
ВЭЖХ 25,9 минуты 98%
1H ЯМР (CD3OD) 7,26 (2H, д, J=2 Гц), 7,22 (2H, дд, J=2, 9 Гц), 7,15 (1H, с) и 6,93 (2H, д, J=9 Гц).
HREIMS: найдено, 285,0879; C15H13N2O4, вычислено 285,0870.
Пример 5: Синтез адамантана 10
1,3-бис-(3,4-дигидроксифенил)адамантан (соединение 9)
Взаимодействие катехина с 1,3-адамантандиолом по методике, описанной Lu и соавторами (Lu et al., J. Med. Chem. 2005, 48(14), 4576-4585), позволяло получить аддукт 10 с приемлемым выходом.
Согласно методике, описанной Lu, раствор катехина (1,0 г) и адамантандиола (0,5 г) в метансульфоновой кислоте (2 мл) нагревали до 80°C в течение 3 часов, затем оставляли стоять при комнатной температуре в течение ночи. Добавляли воду и экстрагировали смесь 10% раствором метанола в хлороформе, после чего экстракт высушивали и упаривали в вакууме, получая твердое вещество белого цвета. Очистка колоночной хроматографией на силикагеле при элюировании 0-20% метанолом в хлороформе позволяла получить продукт в виде белого твердого вещества. Кристаллизация из смеси диэтиловый эфир/40% петролейный эфир привела к получению чистого продукта 10 в виде кристаллического твердого вещества белого цвета (210 мг, 20%).
ВЭЖХ 29,8 минуты 98%
1H ЯМР (CD3OD) 6,82 (2H, т, J=1,5 Гц), 6,68 (4H, д, J=1,5 Гц), 2,22 (2H, ушир.с), 1,87 (8H, м) и 1,77 (2H, ушир.с).
HREIMS: найдено, 387,1369; MCl-, C22H24ClO4, вычислено 387,1369.
Пример 6: Соединения по настоящему изобретению являются мощными разрушителями фибрилл или агрегатов Aβ 1-42, характерных для болезни Альцгеймера
Было обнаружено, что соединения, полученные в предыдущих примерах, являются мощными разрушителями/ингибиторами фибрилл или агрегатов β-амилоидного белка (Aβ), характерных для болезни Альцгеймера. В серии исследований было проанализировано, насколько эффективно соединения по настоящему изобретению вызывают распад/разрушение ранее образовавшихся амилоидных фибрилл (т.е. фибрилл, состоящих из Aβ 1-42), характерных для болезни Альцгеймера.
Часть A: флуорометрия с использованием тиофлавина T
В одном из исследований для определения действия соединений по настоящему изобретению и EDTA (в качестве отрицательного контроля) применяли флуорометрию с тиофлавином T. В этом исследовании тиофлавин Т специфично связывался с фибриллярным амилоидом, и это связывание вызывало усиление флуоресценции при 485 нм, которое было прямо пропорционально количеству образовавшихся фибрилл амилоида. Чем выше флуоресценция, тем больше количество образовавшихся фибрилл амилоида (Naki et al., Lab. Invest. 65:104-110, 1991; Levine III, Protein Sci. 2:404-410; Amyloid: Int. J. Exp. Clin. Invest. 2:1-6, 1995).
В данном исследовании 30 мкл 1 мг/мл раствора (в дистиллированной воде) предварительно фибриллизованного Aβ 1-42 (rPeptide Inc.) инкубировали при 37°C в течение 3 дней либо без добавок, либо в присутствии одного из тестируемых соединений или EDTA (массовые соотношения Aβ:тестируемое соединение 1:1, 1:0,1, 1:0,01 или 1:0,001). После трех дней инкубирования 50 мкл каждой из инкубируемых смесей переносили в лунку 96-луночного микротитровального планшета, содержащую 150 мкл дистиллированной воды и 50 мкл раствора тиофлавина Т (т.е. 500 мМ тиофлавин Т в 250 мМ фосфатном буфере, pH 6,8). Флуоресцентное излучение регистрировали при 485 нм (возбуждение на длине волны 444 нм), используя планшетный флуориметр для ELISA, после вычитания данных для холостого опыта, т.е. для буфера или соединения без добавок.
Результаты 3-дневного инкубирования графически показаны на фиг.5. Например, в то время как EDTA ('-C' на фиг.5) не вызывала заметного ингибирования фибрилл Aβ 1-42 при всех исследованных концентрациях, все тестируемые соединения вызывали дозозависимый распад/разрушение ранее образовавшихся фибрилл Aβ 1-42. Все испытанные соединения оказались эффективными при разрушении ранее образовавшихся фибрилл Aβ 1-42, аналогично результатам, полученным для положительного контрольного соединения ('+C' на фиг.5). Например, все соединения вызывали по меньшей мере 96% ингибирование при использовании массового соотношения Aβ:тестируемое соединение 1:1, по сравнению с 99% для контроля. При использовании массового соотношения Aβ:тестируемое соединение 1:0,1 уровни ингибирования находились в диапазоне от 86 до 95% по сравнению с 92% для контрольного соединения. Проведенное исследование показало, что соединения по настоящему изобретению являются мощными разрушителями/ингибиторами фибрилл Aβ, характерного для болезни Альцгеймера типа, и их действие обычно проявляется дозозависимым образом.
Часть B: Конго красный
В анализе связывания Конго красного количественно определяли способность тестируемого соединения изменять связывание β-амилоида с Конго красным. Для проведения анализа Aβ 1-42 (приготовленный как для анализа с тиофлавином Т) и тестируемые соединения инкубировали в течение 3 дней и затем осуществляли вакуумное фильтрование через 0,2-мкм фильтр. Затем определяли количество Aβ 1-42, удержанного фильтром, путем окрашивания фильтра Конго красным. После соответствующего промывания фильтра, любое уменьшение интенсивности цвета Конго красного на фильтре в присутствии тестируемого соединения (по сравнению с окрашиванием амилоидного белка Конго красным в отсутствии тестируемого соединения) являлось показателем способности тестируемого соединения уменьшать/изменять количество агрегированного и конгофильного Aβ.
В одном из исследований определяли способность фибрилл Aβ к связыванию Конго красного в отсутствии или присутствии возрастающих количеств тестируемых соединений или EDTA (массовые соотношения Aβ:тестируемое соединение 1:1, 1:0,1, 1:0,01 или 1:0,001). Результаты 3-дневного инкубирования графически показаны на фиг.6. В то время как EDTA ('-C' на фиг.6) не вызывала заметного ингибирования связывания фибрилл Aβ 1-42 с Конго красным при всех исследованных концентрациях, все тестируемые соединения вызывали дозозависимое ингибирование связывания фибрилл Aβ 1-42 с Конго красным, в некоторых случаях, превосходя результаты для положительного контрольного соединения ('+C' на фиг.6). Например, положительное контрольное соединение вызывало статистически значимое (p<0,01) 73,5% ингибирование связывания Конго красного с фибриллами Aβ 1-42 при использовании массового соотношения Aβ:тестируемое соединение 1:1, и статистически значимое (p<0,01) 10,4% ингибирование связывания Конго красного с фибриллами Aβ 1-42 при использовании массового соотношения Aβ:тестируемое соединение 1:0,1. Результаты для соединений 6, 8 и 9 превысили результаты положительного контроля для обоих указанных выше соотношений. Результаты этого исследования, как и результаты анализа с тиофлавином Т, показали, что соединения по настоящему изобретению являются мощными ингибиторами связывания фибрилл Aβ с Конго красным, и их действие обычно проявляется дозозависиммым образом.
Часть D: данные спектроскопии кругового дихроизма
Спектроскопия кругового дихроизма (CD) является методикой, которую можно использовать для определения влияния тестируемых соединений на разрушение вторичной структурной конформации фибрилл амилоида. В одном из исследований, которое описано в этом примере, спектроскопию кругового дихроизма применяли для определения влияния различных соединений по настоящему изобретению на β-листовую конформацию фибрилл Aβ 1-42. Для проведения исследования Aβ 1-42 (rPeptide Inc., Bogart, GA) сначала лиофилизовали из 50 мМ раствора NaOH, причем перед заморозкой и лиофилизацией поддерживали pH выше 10. Затем пептид разбавляли 20 мМ ацетатным буфером, pH 4,0, до концентрации 1 мг/мл. Разбавление и добавление тестируемых соединений или носителя осуществляли таким образом, чтобы конечная концентрация пептида равнялась 0,5 мг/мл и массовые соотношения Aβ1-42:тестируемые соединения составляли 1:1 и 1:0,1. Если тестируемые соединения не добавлялись, количество носителя, добавленного к реакционной смеси, было равно количеству, использованному для введения тестируемых соединений. Через 5 дней инкубирования при 37°C в присутствии соединений или носителя регистрировали CD-спектры на спектрополяриметре Jasco 810 (Easton, MD). Все CD-спектры регистрировали в 0,05- или 0,1-см кварцевых кюветах. Пропускание при различных длинах волн определяли в диапазоне 190-270 нм с шагом 0,1 нм при ширине полосы 2 нм и скорости сканирования 50 нм в минуту, времени отклика 1 сек и шаге данных 0,1 нм. Всю систему приводили в равновесие и непрерывно продували азотом при скорости 10 л/мин. Для обработки данных, перед инкубированием 10 раз регистрировали спектр Aβ 1-42 с добавлением носителя, усредняли и вычитали из результата усреднения 10 спектров «Aβ 1-42 + тестируемое соединение» или «Aβ 1-42 + носитель», после периода инкубирования. В усредненных спектрах эллиптичность в градусах преобразовывали в удельную эллиптичность, используя формулу [Ψ]=(Ψ°/d)×c, где Ψº означает эллиптичность в градусах, d означает длину пути света в мм, и c означает концентрацию в мг/мл. Таким образом, можно было оценить возникшие изменения структуры пептида, т.е. структуры, полученной после инкубирования, по сравнению со структурой, существовавшей в момент растворения.
На фиг.1A показаны некоторые CD-спектры, полученные в этом исследовании. Aβ 1-42 без добавления тестируемых соединений (на фиг.1A обозначен как «носитель») в 20 мМ ацетатном буфере после инкубирования, как правило, демонстрировал типичный CD-спектр амилоидного белка с большим количеством β-листовых структур, которым соответствует минимум, наблюдаемый при 218 нм. Однако, в присутствии некоторых тестируемых соединений, наблюдалось хорошо заметное разрушение β-листовых структур в фибриллах Aβ 1-42 (при значительном увеличении количества нерегулярных спиралей или α-спирали), что продемонстрировано уменьшением глубины минимума, наблюдаемого при 218 нм (по сравнению с Aβ 1-42 без тестируемых соединений).
На фиг.1B показано влияние соединений 1 и 2 на образование β-листовой структуры фибрилл Aβ 1-42 по сравнению с положительным контрольным соединением. Исследования кругового дихроизма показали, что соединения по настоящему изобретению обладают способностью вызывать распад/разрушение β-листовых структур, характерных для Aβ фибрилл болезни Альцгеймера. Кроме того, результаты этих исследований подтвердили результаты предыдущих примеров, в которых выполняли флуорометрию с использованием тиофлавина Т и анализ связывания Конго красного.
Пример 7: Соединения по настоящему изобретению являются мощными разрушителями фибрилл или агрегатов α-синуклеина, характерных для болезни Паркинсона
Было найдено, что тестируемые соединения по настоящему изобретению являются также мощными разрушителями/ингибиторами фибрилл или агрегатов α-синуклеина, характерных для болезни Паркинсона. Было показано, что α-синуклеин образует фибриллы или агрегаты в случае инкубирования при 37°C в течение нескольких дней. Постулируется, что α-синуклеин играет важную роль в патогенезе болезни Паркинсона и других синуклеинопатий. В этой серии исследований было проанализировано, насколько эффективно соединения по настоящему изобретению вызывают распад/разрушение ранее образовавшихся фибрилл или агрегатов α-синуклеина, характерных для болезни Паркинсона.
Часть A: Флуорометрия с использованием тиофлавина T
В одном из исследований для определения действия соединений по настоящему изобретению и EDTA (в качестве отрицательного контроля, (-C)) применяли флуорометрию с тиофлавином T. В этом исследовании тиофлавин Т специфично связывался с фибриллами или агрегатами α-синуклеина, и это связывание вызывало усиление флуоресценции при 485 нм, которое было прямо пропорционально количеству образовавшихся фибрилл или агрегатов α-синуклеина. Чем выше была флуоресценция, тем больше количество образовавшихся фибрилл или агрегатов α-синуклеина (Naki et al., Lab. Invest. 65:104-110, 1991; Levine III, Protein Sci. 2:404-410; Amyloid: Int. J. Exp. Clin. Invest. 2:1-6, 1995).
В данном исследовании 30 мкл 1 мг/мл раствора α-синуклеина (rPeptide Inc.) предварительно превращали в фибриллы при 37°C, перемешивая при 1400 об./мин в течение 4 дней, и затем инкубировали при 37°C в течение 3 дней либо без добавок, либо в присутствии одного из тестируемых соединений или EDTA (массовые соотношения α-синуклеин:тестируемое соединение 1:1, 1:0,1, 1:0,01 или 1:0,001). После трех дней инкубирования 50 мкл каждой из инкубированных смесей переносили в лунку 96-луночного микротитровального планшета, содержащую 150 мкл дистиллированной воды и 50 мкл раствора тиофлавина Т (т.е. 500 мМ тиофлавин Т в 250 мМ фосфатном буфере, pH 6,8). Флуоресцентное излучение регистрировали при 485 нм (возбуждение на длине волны 444 нм), используя планшетный флуориметр для ELISA, после вычитания данных для холостого опыта, т.е. для буфера или соединения без добавок.
Результаты 3-дневного инкубирования графически показаны на фиг.7. Например, в то время как EDTA не вызывала заметного ингибирования фибрилл или агрегатов α-синуклеина при всех исследованных концентрациях, все тестируемые соединения в той или иной степени вызывали дозозависимый распад/разрушение ранее образовавшихся фибрилл или агрегатов α-синуклеина. Например, при массовом соотношении α-синуклеин:соединение 1:0,01 положительное контрольное соединение (+C на фиг.7) вызывало статистически значимое (p<0,01) 77,4% ингибирование, тогда как другие исследованные соединения продемонстрировали ингибирование в диапазоне от 45 до 83%. Соединения 1, 4, 5 и 6 продемонстрировали результаты, очень близкие к результатам положительного контрольного соединения. Проведенное исследование показало, что соединения по настоящему изобретению являются мощными разрушителями/ингибиторами фибрилл или агрегатов α-синуклеина, характерных для болезни Паркинсона, и их действие обычно проявляется дозозависиммым образом.
Часть B: Конго красный
В анализе связывания Конго красного количественно определяли способность того или иного тестируемого соединения изменять связывание α-синуклеина с Конго красным. В этом анализе α-синуклеин (предварительно фибрилизованный, как в анализе с тиофлавином Т) и тестируемые соединения инкубировали в течение 3 дней и затем осуществляли вакуумное фильтрование через 0,2-мкм фильтр. После этого определяли количество α-синуклеина, удержанного фильтром, путем окрашивания фильтра Конго красным. После соответствующего промывания фильтра, любое уменьшение интенсивности цвета Конго красного на фильтре в присутствии тестируемого соединения (по сравнению с окрашиванием α-синуклеина Конго красным в отсутствии тестируемого соединения) являлось показателем способности тестируемого соединения уменьшать/изменять количество агрегированного и конгофильного α-синуклеина.
В одном из исследований определяли способность фибрилл или агрегатов α-синуклеина к связыванию Конго красного в отсутствии или присутствии возрастающих количеств тестируемых соединений или EDTA (при массовых соотношениях α-синуклеин:тестируемое соединение 1:1, 1:0,1, 1:0,01 или 1:0,001). Результаты 3-дневного инкубирования графически показаны на фиг.8. В то время как EDTA (-C) не вызывала заметного ингибирования связывания фибрилл α-синуклеина с Конго красным при всех исследованных концентрациях, тестируемые соединения вызывали дозозависимое ингибирование связывания α-синуклеина с Конго красным. Например, положительное контрольное соединение (+C) вызывало статистически значимое (p<0,01) 78,5% ингибирование связывания Конго красного с фибриллами или агрегатами α-синуклеина при массовом соотношении 1:1. При том же соотношении ингибирование для всех тестируемых соединений находилось в диапазоне от 60 до 100%. Результаты этого исследования показали, что соединения по настоящему изобретению являются также мощными ингибиторами связывания фибрилл α-синуклеина, характерных для болезни Паркинсона, с Конго красным, и их действие обычно проявляется дозозависиммым образом.
Часть C: Круговой дихроизм
Спектроскопия кругового дихроизма (CD) является методикой, которую можно использовать для определения влияния тестируемых соединений на разрушение вторичной структурной конформации α-синуклеина. Поскольку самопроизвольное объединение α-синуклеиновых мономеров в агрегаты или фибриллы невозможно без образования вторичной структуры, а именно бета-листов, можно использовать спектроскопию CD для определения способности тестируемых соединений ингибировать процессы фибриллизации или агрегации.
В одном из исследований, которое описано в этом примере, спектроскопию кругового дихроизма применяли для определения влияния различных соединений по настоящему изобретению на β-листовую конформацию α-синуклеина. Для проведения исследования α-синуклеин (rPeptide Inc., Bogart, GA) растворяли в 9,5 мМ фосфатном буфере (PBS) в концентрации 1 мг/мл. Полученный концентрированный раствор разбавляли тем же буфером и добавляли или тестируемое соединение, или носитель, так, чтобы конечная концентрация пептида составляла 0,25 мг/мл и массовые соотношения α-синуклеин:соединение составляли 1:1 и 1:0,1. Регистрировали CD-спектр образца, обработанного носителем, после чего инкубировали все образцы в течение 4 дней, и затем регистрировали спектры всех смесей α-синуклеин/соединение или α-синуклеин/носитель. CD-спектры регистрировали на спектрополяриметре Jasco 810 (Easton, MD). Все CD-спектры регистрировали в 0,10-см кварцевых кюветах. Пропускание при различных длинах волн определяли в диапазоне 190-270 нм с шагом 0,1 нм при ширине полосы 2 нм и скорости сканирования 50 нм/минута, времени отклика 32 сек и шаге данных 0,5 нм. Всю систему приводили в равновесие и непрерывно продували азотом при скорости 10 л/мин. Для обработки данных, перед инкубированием 10 раз регистрировали спектр буфера с добавлением носителя, усредняли и вычитали из результата усреднения 10 спектров «α-синуклеин + тестируемое соединение» или «α-синуклеин + носитель», после периода инкубирования. В усредненных спектрах эллиптичность в градусах преобразовывали в удельную эллиптичность, используя формулу [Ψ]=(Ψ°/d)×c, где Ψº означает эллиптичность в градусах, d означает длину пути света в мм, и c означает концентрацию в мг/мл. Таким образом, можно оценить возникшие изменения структуры пептида, т.е. структуры, полученной после инкубирования, по сравнению со структурой, существовавшей в момент растворения.
На фиг.2 показаны спектры CD, полученные для α-синуклеина в начале эксперимента и через 4 дня инкубирования при 37°C. α-Синуклеин в буфере PBS с добавкой носителя в начале эксперимента продемонстрировал неупорядоченную спиральную структуру, а через 4 дня инкубирования продемонстрировал типичный CD-спектр белка со значительной долей β-листовой структуры, на что указывает минимум при 218 нм. Однако, в присутствии некоторых из соединений, имело место заметное ингибирование формирования β-листовой структуры α-синуклеина, на что указывает уменьшение глубины минимума при 218 нм (по сравнению с образцом без тестируемых соединений).
На фиг.3A показаны некоторые из CD-спектров, полученных в данном исследовании. В начале эксперимента α-синуклеин обладал спектром, характерным для пептида с неупорядоченной спиралью, и этот спектр использовали в качестве контрольного, соответствующего 100% ингибированию образования β-листовой структуры. После инкубирования спектр α-синуклеина соответствовал тому, которого можно было ожидать для β-листовой структуры, он указывал на образование агрегатов высокого порядка и использовался в качестве контрольного, соответствующего 0% ингибирования. Образцы, использованные в качестве контрольных, обрабатывали носителем для гарантии количественного соответствия этих образцов и образцов, обработанных тестируемыми соединениями. Два полученных контрольных спектра давали возможность точного количественного определения ингибирования образования фибрилл в процентах для образцов, обработанных тестируемыми соединениями, поскольку их использовали в качестве положительного и отрицательного контроля, приняв образование фибрилл в этих образцах за 100% и 0%, соответственно. Несмотря на то, что процентные значения для контрольных образцов являются лишь оценочными, неточность этих значений не столь значительна, чтобы ставить их под сомнение, т.е. нижняя граница может соответствовать 0-5% ингибированию, тогда как верхняя граница может соответствовать 95-100% ингибированию. Контрольные образцы получали в каждой партии экспериментальных образцов, используя один и тот же концентрированный раствор α-синуклеина, который разделяли на аликвоты равного объема, добавляли к ним индивидуальное тестируемое соединение или носитель и параллельно запускали эксперимент, для гарантии точности количественного анализа. Спектры, приведенные на фиг.3A, были получены для массового соотношения α-синуклеин/тестируемое соединение 1:1. Эти CD-спектры продемонстрировали, что соединения по настоящему изобретению обладают способностью ингибировать образование β-листовых структур, характерных для фибрилл и агрегатов α-синуклеина болезни Паркинсона.
На фиг.3B показано влияние соединений на ингибирование β-листовых структур α-синуклеина по сравнению с положительным контрольным соединением (+C). Как уже было указано выше, положительный контроль (+С1) представляет собой спектр образца с носителем в начале эксперимента, тогда как отрицательный контроль (-С) представляет собой спектр образца с носителем после 4-дневного инкубирования.
На фиг.4A показаны некоторые CD-спектры, полученные в этом исследовании. Эти спектры получали и обрабатывали таким же образом, как спектры, приведенные на фиг.3A. На этих спектрах отсутствуют характерные признаки β-листовой структуры, обнаруженные в спектре образца с носителем.
На фиг.4B показано влияние соединений на ингибирование β-листовых структур α-синуклеина по сравнению с положительным контрольным соединением (+C). Как уже было указано выше, положительный контроль (+С1) представляет собой спектр образца с носителем в начале эксперимента, тогда как отрицательный контроль (-С) представляет собой спектр образца с носителем после 4-дневного инкубирования.
Результаты описанных исследований также подтверждают результаты предыдущих примеров, в которых использовалась флуорометрия с тиофлавином Т и анализ связывания Конго красного, что соединения по настоящему изобретению являются мощными агентами, препятствующими фибриллизации α-синуклеина.
Пример 8: Соединения по настоящему изобретению являются мощными разрушителями/ингибиторами агрегатов α-синуклеина, связанных с болезнью Паркинсона
Болезнь Паркинсона характеризуется накоплением нерастворимых внутринейронных агрегатов, именуемых тельцами Леви, основным компонентом которых является α-синуклеин (обзор в Dauer et al., Neuron, 39:889-909, 2003). Поскольку аутосомно-доминантные мутации в гене α-синуклеина вызывают подгруппу заболеваний, именуемых семейной болезнью Паркинсона, и поскольку эти мутации увеличивают вероятность агрегации α-синуклеина и образования телец Леви, предполагается, что агрегированный α-синуклеин непосредственно вовлечен в этиологию и прогрессирование заболевания (Polymeropoulos et al., Science 276:1197-1199, 1997; Papadimitriou et al., Neurology 52:651-654, 1999). Структурные исследования показали, что внутриклеточные тельца Леви содержат значительную долю неправильно уложенных белков с преобладанием β-складчатой листовой вторичной структуры. Описанные в данном примере исследования проводили с целью определения эффективности тестируемых соединений для ингибирования/разрушения агрегатов α-синуклеина, связанных с болезнью Паркинсона.
По этой причине, для исследования терапевтического потенциала соединений использовали два вида клеточных анализов. В обоих анализах для стимулирования митохондриального окислительного стресса и агрегации α-синуклеина применяли ротенон. В первом анализе использовали связывание флуоресцентного красителя тиофлавина S со структурами с высоким содержанием β-листов, в т.ч. фибриллами и агрегатами α-синуклеина. Поэтому для определения способности соединений по настоящему изобретению уменьшать количество агрегатов α-синуклеина проводили количественное определение степени тиофлавин S-положительного окрашивания фиксированных препаратов клеток. Во втором анализе определяли выживаемость клеток, используя пробу на цитотоксичность XTT, где выживаемость зависит от наличия неповрежденных, функционирующих митохондрий в живых клетках. Таким образом, пробу XTT на цитотоксичность использовали для выявления способности соединений по настоящему изобретению ослаблять токсическое действие на митохондрии и уменьшение выживаемости, связанное с накоплением агрегатов α-синуклеина. Говоря другими словами, проба на цитотоксичность XTT применялась для оценки нейрозащитной эффективности соединений. Эти исследования представлены в следующих примерах.
Для проведения описанных исследований использовали модельную клеточную культуру в которой вызывали экспериментальную агрегацию α-синуклеина. Получали клетки человеческой нейробластомы BE-M17, устойчиво трансфицированные A53T-мутантным человеческим α-синуклеином. Реагенты для культивирования клеток получали у Gibco/Invitrogen, и выращивали клетки в среде OPTIMEM, с добавками 10% FBS, пенициллина (1000 ед/мл), стрептомицина (100 мкг/мл) и 500 мкг/мл G418, которая была описана ранее (Osterova-Golts et al., J. Neurosci., 20:6048-6054, 2000).
Тиофлавин S широко применяется для обнаружения амилоид-содержащих структур in situ, включая ткани мозга (Vallet et al., Acta Neuropathol., 83:170-178, 1992) и клеточные культуры (Osterova-Golts et al., J. Neurosci., 20:6048-6054, 2000), в то время как тиофлавин T часто применяется in vitro, как реагент для анализа агрегации растворимых амилоидных белков в фибриллы, обогащенные β-складчатыми листовыми структурами (LeVine III, Prot. Sci., 2:404-410, 1993). Поэтому клеточные культуры исследовали с помощью гистохимии с тиофлавином S, для обнаружения агрегатов, содержащих значительные количества β-складчатых структур, которые сформировались в ответ на введение агентов, вызывающих окислительный стресс (в данном случае ротенона), следуя описанной ранее методике с незначительными изменениями (Osterova-Golts et al., J. Neurosci., 20:6048-6054, 2000). Вкратце, в этом исследовании клеточные культуры выращивали на стеклянных кюветах для микроскопии, покрытых поли-D-L-лизином в количестве примерно 3×104 клеток/см2. Через 24 часа клетки обрабатывали 500 нМ, 1 мкМ или 5 мкМ ротеноном (Sigma) или носителем (0,05% ДМСО), как показано на графике. Сразу же после добавления ротенона (или носителя) добавляли соединения в указанной концентрации или только среду для культивирования клеток (без соединения), в присутствии ротенона. Такую же обработку повторяли через 48 часов. Еще через 48 часов клетки фиксировали в течение 25 минут в 3% параформальдегиде. После промывания PBS клетки инкубировали с 0,015% тиофлавином S в 50% этаноле в течение 25 минут, два раза промывали 50% этанолом в течение четырех минут и два раза в течение 5 минут деионизированной водой, и затем готовили препараты, применяя среду на водной основе, разработанную для защиты от действия света. Агрегаты, которые связывали тиофлавин S, обнаруживали с помощью флуоресцентного микроскопа, используя набор фильтров High Q FITC (полоса пропускания 480-535 нм) и 20х объектив, если не указано иное. Для каждого препарата выбирали от 8 до 16 характерных участков, производили фотографирование и отдавали полученные изображения для оценки специалисту, который не был осведомлен о том, в каких условиях были получены изображения. Для оценки количества тиофлавин S-положительных агрегатов определяли общую площадь тиофлавин S-положительных включений в поле зрения. Для этой цели, используя программу Q-capture, убирали фоновую флуоресценцию, которая не превосходила заранее заданную величину или пороговый параметр интенсивности пикселя. Паразитную флуоресценцию, не связанную с клетками, удаляли в ручном режиме. Если не указано иное, приведенные данные представляют собой группу средних значений ± SEM. Статистический анализ производили с применением программы GraphPad Prism (GraphPad Inc). Различие между средними значениями (для двух образцов) оценивали с применением t-критерия Стьюдента. Различие между большим количеством средних значений оценивали с помощью однофакторного дисперсионного анализа ANOVA и затем критерия Тьюки для множественных сравнений.
Для подтверждения способности данного анализа количественно определять агрегаты, связывающие тиофлавин S, проводили окрашивание клеток BE-M17, избыточно экспрессирующих α-синуклеин A53T, и результаты этого окрашивания показали вызванное ротеноном дозозависимое увеличение тиофлавин S-положительных агрегатов по сравнению с обработанными носителем контрольными клетками (фиг.9A-D). Изображения с большим увеличением, полученные с помощью объектива 40х, показали, что тиофлавин S-положительные агрегаты являлись внутриклеточными и цитоплазменными (фиг.9D), по аналогии с аккумулированием внутрицитоплазменных телец Леви, которые являются патологическим признаком, связанным с болезнью Паркинсона. Количественное определение площади, занятой тиофлавин S-положительными агрегатами, позволило установить, что 5 мкМ ротенона достаточно для того, чтобы вызвать обильную агрегацию (фиг.9E), и, таким образом, данная доза является эффективной для тестирования способности соединений ослаблять образование этих агрегатов.
Используя описанный выше протокол, протестировали несколько соединений на их способность уменьшать, предупреждать образование или устранять тиофлавин S-положительные агрегаты в клетках BE-M17, обработанных ротеноном, которые избыточно экспрессируют A53T α-синуклеин. Ниже описаны примеры результатов, полученных в ходе экспериментов с этими соединениями.
В клетках, обработанных только 1 мкМ ротеноном, наблюдалось устойчивое наличие тиофлавин S-положительных агрегатов (фиг.10А). Добавление 500 нг/мл (фиг.10B) или 1 мкг/мл (фиг.10С) положительного контрольного соединения заметно уменьшило количество этих агрегатов, образовавшихся под действием ротенона, на 87% и 91%, соответственно (как показано на фиг.10D), по сравнению с клетками, обработанными только ротеноном. Следовательно, положительное контрольное соединение проявляет высокую эффективность в уменьшении, предупреждении образования и/или устранении тиофлавин S-положительных агрегатов в клетках, которые экспрессируют человеческий α-синуклеин A53T.
Добавление соединения 1 в количестве от 500 нг/мл до 2 мкг/мл (фиг.11B-D) не уменьшило количество индуцированных ротеноном агрегатов по отношению к клеткам, обработанным только ротеноном (фиг.11D и E).
Как показано на фиг.12, в клетках, обработанных 1 мкМ ротеноном, добавление 500 нг/мл и 2 мкг/мл соединения 2 уменьшило количество индуцированных ротеноном агрегатов на 39-44%, и в клетках, обработанных 5 мкМ ротеноном, добавление 1 мкг/мл соединения 2 заметно уменьшило количество индуцированных ротеноном агрегатов на 67% (фиг.12E).
На фиг.13A-E показаны результаты для соединения 3. В клетках, обработанных 1 мкМ ротеноном, добавление от 500 нг/мл до 2 мкг/мл соединения 3 уменьшило количество индуцированных ротеноном агрегатов на 41-63%, по сравнению с клетками, обработанными только ротеноном.
Добавление от 500 нг/мл до 2 мкг/мл соединения 4 не уменьшило количество индуцированных ротеноном агрегатов по сравнению с клетками, обработанными только ротеноном (фиг.14A-E).
На фиг.15A-E показаны результаты для соединения 5. В клетках, обработанных 1 мкМ ротеноном, добавление 1-2 мкг/мл соединения 5 уменьшило количество индуцированных ротеноном агрегатов на 25-49%, по сравнению с клетками, обработанными только ротеноном.
Добавление соединения 6 не оказало значительного влияния на количество индуцированных ротеноном агрегатов по сравнению с клетками, обработанными только ротеноном (фиг.16A-E).
В клетках, обработанных 1 мкМ ротеноном, добавление 500 нг/мл и 2 мкг/мл соединения 7 заметно уменьшило количество индуцированных ротеноном агрегатов на 60 и 74% (соответственно), по сравнению с клетками, обработанными только ротеноном. В клетках, обработанных 5 мкМ ротеноном, добавление 500 нг/мл и 2 мкг/мл соединения 7 уменьшило количество индуцированных ротеноном агрегатов на 31% и 67% (соответственно) (фиг.17A-E).
На фиг.18A-E показаны результаты для соединения 8. В клетках, обработанных 1 мкМ ротеноном, добавление 1 мкг/мл соединения 8 уменьшило количество индуцированных ротеноном агрегатов на 56% по сравнению с клетками, обработанными только ротеноном. В клетках, обработанных 5 мкМ ротеноном, добавление 1 или 2 мкг/мл соединения 8 уменьшило количество индуцированных ротеноном агрегатов на 48% и 38% (соответственно).
В клетках, обработанных 1 мкМ ротеноном, добавление от 500 нг/мл до 2 мкг/мл соединения 9 уменьшило количество индуцированных ротеноном агрегатов от 19 и 60% по сравнению с клетками, обработанными только ротеноном (фиг.19A-E). В клетках, обработанных 5 мкМ ротеноном, добавление соединения 9 не уменьшило количество индуцированных ротеноном агрегатов, хотя исходный уровень окрашивания был ниже, чем ожидалось при данной дозировке ротенона.
В результате, многие из протестированных соединений, в частности соединения 2, 3, 5, 7, 8 и 9, эффективно и значительно уменьшали, предупреждали образование, ингибировали и/или ликвидировали образование, отложение и/или накопление агрегатов α-синуклеина в клетках BE-M17, экспрессирующих A53T α-синуклеин.
Пример 9: Соединения по настоящему изобретению защищают от индуцированной ротеноном цитотоксичности
Пробу на цитотоксичность XTT (Roche Diagnostics, Mannheim, Germany) использовали в предыдущих экспериментах для демонстрации того, что A53T α-синуклеин потенцирует смерть клеток BE-M17 через механизм, зависящий от окислительного стресса (Osterova-Golts et al., J. Neurosci., 20:6048-6054, 2000). Исследования показали, что накопление агрегатов α-синуклеина в тельцах Леви механически способствует деградации нейронов при болезни Паркинсона и родственных расстройствах (Polymeropoulos et al., Science 276:2045-2047, 1997; Kruger et al., Nature Genet. 18:106-108, 1998). В настоящем примере пробу на цитотоксичность XTT (далее по тексту именуемая пробой XTT) использовали для измерения способности тестируемых соединений защищать от вызванной ротеноном цитотоксичности (нейрозащитная способность). Проба основана на том, что превращение желтой соли тетразолия XTT в оранжевый формазановый краситель (который поглощает свет в районе 490 нм) происходит только в метаболически активных жизнеспособных клетках. Следовательно, поглощение света при 490 нм пропорционально выживанию клеток. Для проведения этого анализа клетки помещали в 96-луночные чашки для культур тканей в количестве 104 клеток на лунку. Через 24 часа клетки обрабатывали 500 нМ или 2 мкМ ротеноном или носителем (0,05% ДМСО), как показано на фигуре. Сразу после добавления ротенона, добавляли соединения в указанных концентрациях. Для контроля добавляли соединения без ротенона (только носитель 0,05% ДМСО), что показало отсутствие токсичности протестированных дозировок соединений. К необработанным клеткам добавляли только среду для культивирования клеток (без соединения, с ротеноном или без него). После 40-44-часовой обработки удаляли культуральную среду и заменяли ее 100 мкл свежей среды и 50 мкл реакционной смеси для мечения XTT согласно рекомендациям производителя. Через пять-шесть часов измеряли поглощение при 490 нм и вносили поправку на поглощение при эталонной длине волны 700 нм. Обработка 500 нМ и 2 мкМ ротеноном, как правило, уменьшала количество живых клеток на 35-45% по сравнению с необработанным образцом без ротенона (фиг.20). Ингибирование гибели клеток в процентах рассчитывали как долю уменьшения поглощения, вызванного ротеноном (выживаемости), которую устраняло действие тестируемого соединения.
Обработка положительным контрольным соединением в концентрации 10-25 мкг/мл ингибировала индуцированное ротеноном уменьшение выживаемости на 25-33% при обеих дозировках ротенона (фиг.21).
Описанный выше эксперимент проводили для каждого из соединений, и результаты показаны на фиг.22-30. На врезке A фиг.22-30 графически показана токсичность соединения, в то время как на врезке B фиг.22-30 показано ингибирование соединением ротенон-индуцированного уменьшения выживаемости, измеренное при обеих дозировках ротенона.
Обработка соединением 1 в концентрации 10 мкг/мл показала, что данная дозировка не является токсичной, в то время как более высокие дозировки продемонстрировали определенную токсичность (фиг.22A). Обработка соединением 1 в концентрации 10 мкг/мл привела к ингибированию ротенон-индуцированного уменьшения выживаемости примерно на 18-27% при обеих дозировках ротенона (фиг.22B).
Обработка соединением 2 в концентрации 10-25 мкг/мл показала, что данная дозировка не является токсичной, в то время как дозировка 50 мкг/мл продемонстрировала определенную токсичность (фиг.23A). Обработка соединением 2 в концентрации 25 мкг/мл привела к ингибированию ротенон-индуцированного уменьшения выживаемости примерно на 20-28% при обеих дозировках ротенона (фиг.23B).
Обработка соединением 3 в концентрации 10-50 мкг/мл показала, что данное соединение является относительно нетоксичным при всех исследованных концентрациях (фиг.24A). Обработка соединением 3 в концентрации 10-50 мкг/мл привела к ингибированию ротенон-индуцированного уменьшения выживаемости примерно на 17-28% при обеих дозировках ротенона (фиг.24B).
Обработка соединением 4 в концентрации 10 и 25 мкг/мл показала, что данная дозировка не является токсичной, в то время как дозировка 50 мкг/мл продемонстрировала минимальную токсичность (фиг.25A). Обработка соединением 4 в концентрации 25 мкг/мл была особенно эффективной и привела к ингибированию ротенон-индуцированного уменьшения выживаемости примерно на 50% при дозировке ротенона 500 нМ, тогда как при дозировке 2 мкМ наблюдаемое ингибирование составляло примерно 26% (фиг.25B).
Обработка соединением 5 в концентрации 10 или 25 мкг/мл показала, что данная дозировка не является токсичной, в то время как дозировка 50 мкг/мл продемонстрировала минимальную токсичность (фиг.26A). Обработка соединением 5 не привела к какому-либо ингибированию ротенон-индуцированного уменьшения выживаемости при любых дозировках ротенона (фиг.26B).
Обработка соединением 6 в концентрации 10-50 мкг/мл показала, что данное соединение является нетоксичным при всех исследованных концентрациях (фиг.27A). Обработка соединением 6 в концентрации 50 мкг/мл при дозировке ротенона 500 нМ привела к ингибированию ротенон-индуцированного уменьшения выживаемости примерно на 10%, в то время как при дозировке ротенона 2 мкМ наблюдаемое ингибирование составляло примерно 12-16% для всех дозировок тестируемого соединения (фиг.27B).
Обработка соединением 7 в концентрации 1-50 мкг/мл показала, что данное соединение является нетоксичным, и даже при более высоких концентрациях (100-150 мкг/мл) была продемонстрирована лишь очень незначительная токсичность (фиг.28A). Обработка соединением 7 в концентрации 50 мкг/мл продемонстрировала максимальное ингибирование ротенон-индуцированного уменьшения выживаемости примерно на 25% при дозировке ротенона 500 нМ (фиг.28B).
Обработка соединением 8 в концентрации 10-25 мкг/мл показала, что данное соединение является относительно нетоксичным (фиг.29A). Несмотря на незначительную токсичность при 50 мкг/мл, соединение явно обеспечило определенную степень защиты от сильной токсичности ротенона. Этот вывод подтверждается морфологическим анализом (результаты не показаны). Обработка этим соединением в концентрации 50 мкг/мл образцов с концентрацией ротенона 1 и 2 мкМ продемонстрировала ингибирование ротенон-индуцированного уменьшения выживаемости примерно на 18% (фиг.29B).
Обработка соединением 9 в концентрации 10-25 мкг/мл показала, что это соединение является относительно нетоксичным, в то время при более высоких дозировках была продемонстрирована определенная токсичность (фиг.30A). Обработка соединением 9 не привела к какому-либо ингибированию ротенон-индуцированного уменьшения выживаемости при любых дозировках ротенона (фиг.30B).
В результате, многие из протестированных соединений оказались эффективными при ингибировании ротенон-индуцированной цитотоксичности, демонстрируя нейрозащитную активность против токсичности α-синуклеина.
Пример 10: Улучшение двигательной активности у α-синуклеин трансгенных мышей, которым вводили соединения по настоящему изобретению
Для оценки потенциальной эффективности соединений в мышиной модели, имеющей отношение к болезни Паркинсона, использовали трансгенных мышей, у которых наблюдается избыточная экспрессия человеческого α-синуклеина дикого типа под управлением мышиного промотора Thy-1 (Rockenstein E., et al., 2002. J. Neurosci Res 68:568-578). Было доказано, что трансгенные мыши, продуцирующие человеческий α-синуклеин, являются подходящей моделью болезни Паркинсона и, следовательно, удобной системой для тестирования потенциальных терапевтических агентов по ряду причин, включая следующие. (1) Наличие агрегатов α-синуклеина, которые можно обнаружить как иммуногистохимическими (окрашивание), так и биохимическими (вестерн-блоттинг) способами. Эти агрегаты подобны тельцам Леви (внутриклеточным включениям, в основном состоящим из α-синуклеина), которые являются патологическими признаками болезни Паркинсона (Rockenstein E., et al., 2002. J. Neurosci Res 68:568-578; и Hashimoto M., et al., 2003 Ann N Y Acad Sci 991:171-188). (2) Эти мыши испытывают дофаминергический дефицит при передаче сигнала между черным веществом и полосатым телом, что демонстрируется утратой тирозингидроксилаза-иммунореактивных нейронных проекций в полосатое тело (Hashimoto M., et al., 2003 Ann N Y Acad Sci 991:171-188). Этот дефицит также наблюдается у людей с болезнью Паркинсона. (3) У этих мышей проявляются нарушения в поведенческих тестах, зависящих от двигательной функции, например, в тесте на прохождение по перекладине, в т.ч. замедленность движения, потеря равновесия и координации, а также мышечная слабость (Fleming S.M., et al., 2004 J. Neurosci 24:9434-9440; и Fleming S.M., et al., 2006 Neuroscience 142:1245-1253).
Аналогичное нарушение двигательной функции наблюдается у людей с болезнью Паркинсона. С целью оценки потенциальной эффективности соединений для улучшения двигательной активности или минимизации нарушений, для животных, получавших соединения по настоящему изобретению или носитель, проводили требующий значительных усилий тест на прохождение по перекладине, и оценивали результаты до начала введения, в момент времени 0 месяцев, и затем после 3 и 6 месяцев введения. Если бы соединения проявляли эффективность, можно было бы ожидать, что мыши, получавшие эти соединения, справятся с тестом лучше, чем мыши того же возраста, получавшие носитель, и/или что введение тестируемых соединений могло бы облегчить возрастное снижение результатов в данной группе. Например, если тестируемые соединения эффективны, можно было бы ожидать, что получавшая их мышь пройдет по перекладине быстрее, по сравнению с мышью, получавшей носитель. Или же можно было бы ожидать, что возрастные нарушения в группе будут ослаблены (например, результаты после введения соединений могли бы быть аналогичны результатам до введения или даже превышать их, тогда как результаты мышей, получавших носитель, будут прогрессивно ухудшаться в течение того же периода времени).
Тест на прохождение по перекладине
В тесте на прохождение по перекладине, который является одним из показателей двигательной активности, мышей тренировали в течение двух дней, по пять попыток в день, проходить по сужающейся перекладине (разделенной на четыре сегмента), снабженной удерживающими бортиками, прикрепленными вдоль каждой стороны, и ведущей к клетке, в которой обитало животное. На третий день тест усложняли, помещая сетку с мелкими отверстиями над поверхностью перекладины, так чтобы между сеткой и поверхностью перекладины оставалось небольшое пространство около 1 см. Затем осуществляли видеозапись движения животных во время пяти попыток, и исследователь, не осведомленный о вводившихся животным препаратах, регистрировал время преодоления перекладины, число совершенных шагов и количество ошибок (Fleming S.M., et al., 2006 Neuroscience 142:1245-1253).
Трансгенные мыши, получавшие соединение 2 в течение трех месяцев, продемонстрировали заметное статистически значимое 49% улучшение (времени преодоления перекладины) по сравнению с контрольными мышами того же возраста (15 месяцев), получавшими носитель (фиг.31). Однако после шести месяцев введения результаты были аналогичны результатам мышей того же возраста, получавших носитель. В совокупности эти данные показывают, что соединение 2 вызывает задержку нарушения поведения в тесте на прохождение по перекладине.
Трансгенные мыши, получавшие соединение 7 в течение 6 месяцев, продемонстрировали заметное, статистически значимое 35% улучшение (времени преодоления перекладины) по сравнению с контрольными мышами того же возраста (15 месяцев), получавшими носитель (фиг.31). Кроме того, после лишь трех месяцев введения соединения 7, результаты в тесте были лучше на 39% по сравнению с контрольными животными, получавшими носитель (фиг.31). В совокупности эти данные показывают, что введение соединения 7 предупреждает возрастное прогрессирование нарушений в тесте на прохождение по перекладине.
Пример 11: Улучшение двигательной активности α-синуклеин трансгенных мышей, получавших соединения по настоящему изобретению
Для оценки потенциальной эффективности соединений по настоящему изобретению в тесте с вертикальной стойкой, определяли результаты мышей, получавших соединения и получавших носитель, до начала введения (0 месяцев) и повторно через 3 и 6 месяцев введения. Использовали таких же мышей, что и в описанном выше примере 10, и препараты вводили им ежедневно с помощью i.p. инъекций в количестве 50 мг/кг/день. Для оценки потенциальной эффективности в тесте на прохождение по перекладине, определяли результаты теста для мышей, получавших соединение и получавших носитель через 5-6 недель введения. Если бы соединения проявляли эффективность, можно было бы ожидать, что мыши, получавшие эти соединения, справятся с тестом лучше, чем мыши того же возраста, получавшие носитель, и/или что введение тестируемых соединений могло бы облегчить нарушения (улучшить результаты) с течением времени в данной группе. Например, если тестируемые соединения эффективны, можно было бы ожидать, что получавшая их мышь преодолеет перекладину быстрее, чем мышь, получавшая носитель. Или же можно было бы ожидать ослабления нарушений в группе с течением времени (например, результаты после введения соединений могли бы быть лучше, чем до введения соединений, тогда как результаты мышей, получавших носитель, будут такими же или прогрессивно ухудшаться в течение того же периода времени).
A) Тест с вертикальной стойкой
В тесте с вертикальной стойкой, который является одним из показателей двигательной активности, мышей тренировали в первый день (2 попытки) спускаться по деревянной стойке после того, как их помещали головой вверх на вершину стойки. На второй день мышей тестировали в пяти попытках, и исследователь, не осведомленный о вводимых мышам препаратах, регистрировал время задержки до переворота головой вниз (время переворота), время спуска (время движения) и общее время, проведенное на стойке.
Трансгенные мыши, получавшие соединение 7 в течение 3 месяцев (n=11) (в возрасте 15 месяцев), продемонстрировали тенденцию к улучшению результатов в тесте с вертикальной стойкой (уменьшение времени переворота) по отношению к их результатам до введения препаратов. Через 6 месяцев введения (n=11) соединения 7 мыши (в возрасте 18 месяцев) продемонстрировали статистически значимое 41% улучшение результатов в тесте с вертикальной стойкой по сравнению с собственными результатами до введения соединений, и их результаты были похожи на результаты нетрансгенных мышей в возрасте 16 месяцев (фиг.32). В противоположность этому, результаты мышей, получавших носитель, через 3 и 6 месяцев введения были аналогичны их результатам до начала введения. Приведенные данные показывают, что соединение 7 улучшает результаты животных в тесте с вертикальной стойкой.
B) Тест на прохождение по перекладине молодых трансгенных мышей
Тест на прохождение по перекладине с незначительными изменениями (2-дневный эксперимент вместо 3-дневного) использовали для измерения двигательной активности у молодых α-синуклеин трансгенных мышей (в возрасте 3 месяцев), которым в течение 6 недель вводили соединение 7 (или носитель контрольным животным) (n=8 на группу). Мышей тренировали в течение одного дня в шести попытках проходить по сужающейся перекладине (разделенной на четыре сегмента), снабженной удерживающими бортиками, прикрепленными вдоль обеих сторон, и ведущей к клетке, в которой обитало животное. На второй день тест усложняли, помещая сетку с мелкими отверстиями над поверхностью перекладины, так чтобы между сеткой и поверхностью перекладины оставалось небольшое пространство около 1 см. Затем осуществляли видеозапись движения мышей во время пяти попыток, и исследователь, не осведомленный о вводившихся животным препаратах, регистрировал время преодоления перекладины, число совершенных шагов и количество ошибок (Fleming S.M., et al., 2006 Neuroscience 142:1245-1253).
Трансгенные мыши, получавшие соединение 7 в течение 6 недель, продемонстрировали статистически значимое 36% улучшение (времени преодоления перекладины) по отношению к контрольным мышам того же возраста (4-5 месяцев), получавшим носитель (фиг.33). Не было зафиксировано статистически значимого влияния на количество шагов или количество поскальзываний на шаг (долю ошибок). В совокупности эти результаты показывают, что соединение 7 улучшает результаты теста на прохождение по планке.
Пример 12: Уменьшение α-синуклеин-положительного внутринейронного иммуноокрашивания в тканях мозга α-синуклеин трансгенных мышей
Иммуноокрашивание α-синуклеина и анализ полученных изображений проводили для того, чтобы установить, уменьшает ли введение соединения 7 уровни α-синуклеина у пожилых α-синуклеин трансгенных мышей (описанных выше). После блокирования связывания неспецифических антител, срезы мозга мышей инкубировали в течение ночи при 4°C с кроличьими поликлональными антителами против человеческого α-синуклеина (Chemicon AB5038P; разбавление 1:500). На второй день после тщательного промывания в солевом растворе с фосфатным буфером (PBS) срезы инкубировали с биотинилированным вторичным антителом (козье антикроличье антитело, VectorLabs) в разбавлении 1:200 в течение 1 часа при комнатной температуре. После тщательного промывания в PBS срезы инкубировали с комплексом авидин-биотин (ABC) производства Vector Labs в течение 30 минут при комнатной температуре. Затем срезы промывали PBS три раза и в течение 4 минут проводили реакцию с Vector DAB (согласно рекомендациям производителя). Реакцию DAB гасили промыванием буфером Tris. Готовили препараты срезов на предметных стеклах и высушивали их в течение ночи. Препараты покрывали покровными стеклами и регистрировали изображения.
Для анализа и количественной оценки изображений, три изображения коры головного мозга, типичных для группы мышей с анатомической точки зрения, оцифровывали (при увеличении 100х) при помощи цифровой камеры Q-Image и программы Q-Capture. Каждое изображение обрабатывали с использованием программы Image-Pro. К каждому изображению применяли заранее определенные пороговые значения сегментации цвета пиксела и минимальной площади объекта. Удаляли артефакты изображения, данные экспортировали и обрабатывали для определения площади (в %), занятой α-синуклеин-положительными иммуномеченными объектами.
Мышам в течение 6 месяцев ежедневно осуществляли i.p. инъекции соединений в количестве 50 мг/кг/день, начиная с возраста 12 месяцев и до возраста 18 месяцев. На фиг.34 видно, что у мышей, получавших соединение 7 (врезки C-D), в нейронах лобной коры головного мозга наблюдается значительно меньшее количество человеческого α-синуклеина, по сравнению с мышами, получавшими носитель (врезки A-B). Ткани мозга нетрансгенных мышей дикого типа не показали окрашивания, характерного для наличия человеческого α-синуклеина, и приведены в качестве контроля специфичности антитела для трансгенного человеческого α-синуклеина (E и F). Анализ и количественная оценка изображений показали, что введение соединения 7 вызывает статистически значимое уменьшение количества α-синуклеин-положительных объектов на 81%.
Введение соединения 7 в течение 6 месяцев резко снижает уровни α-синуклеина в лобной коре α-синуклеин-трансгенных мышей по сравнению с контрольными животными, получавшими носитель. Эти данные хорошо согласуются с данными предыдущих примеров, которые показывают улучшение двигательной активности у трансгенных мышей, получавших в течение 6 месяцев соединения по настоящему изобретению.
Пример 13: Уменьшение уровней α-синуклеина в мозге α-синуклеин трансгенных мышей
Электрофорез в полиакриламидном геле в присутствии додецилсульфата натрия (SDS-PAGE) и Вестерн-блоттинг использовали для определения концентрации α-синуклеина в дисперсной (клеточные мембраны) и цитозольной фракциях переднего отдела мозга у 18-месячных и 4-5 месячных трансгенных (описаны выше) и нетрансгенных мышей. Обе фракции содержали потенциально патогенные ресурсы агрегированного α-синуклеина, который в основном восстанавливался до мономера в восстановительных условиях SDS-PAGE.
После умерщвления α-синуклеин трансгенных мышей, последовавшего за 6-месячным введением соединений (для животных в возрасте 18 месяцев) или 6-недельным введением соединений (для животных в возрасте 4-5 месяцев), удаляли мозг и рассекали его пополам вдоль средней линии (схемы введения препаратов описаны выше). Те же операции осуществляли для контрольных животных обоих возрастов. Правые полушария рассекали по венечному шву, для получения передней и задней частей. Затем рассеченные полушария подвергали быстрой заморозке на бане сухой лед/этанол и хранили при -80°C. Затем ткани мозга подвергали биохимическому фракционированию для отделения растворимой цитозольной фракции от нерастворимой дисперсной (клеточные мембраны). Вкратце, ткани мозга гомогенизировали в 9 объемах буфера HEPES (свободного от ПАВ) с добавками ингибиторов протеаз и фосфатаз (Calbiochem). Осторожную гомогенизацию осуществляли тефлоновым пестиком в подходящем стеклянном гомогенизаторе (Kontes #19), для минимизации разрушения взаимодействия α-синуклеин/мембраны во время лизиса. Затем всю массу клеточных лизатов центрифугировали на низкой скорости для осаждения (P1) неразрушенных клеток и органелл (например, ядер). Затем супернатант (S1) центрифугировали при 225000 × g в течение 1 ч при 4°C для осаждения мембран (P2) (эта фракция может включать органеллы, например, митохондрии). Супернатант (S2) представлял собой «цитозольную» фракцию. Осадок (P2) ресуспендировали с помощью пипетки в 2,5 объемах гомогенизационного буфера, подвергали действию ультразвука на льду 5×1 сек и полученный препарат именовали «дисперсной» или «мембранной» фракцией. Общую концентрацию белка определяли с помощью анализа MicroBCA (Pierce). При проведении SDS-PAGE вводили равные количества белка (25 мкг) на каждую дорожку (Bio-Rad Criterion 4-12% Bis-Tris гели, 26 лунок на гель) и переносили на нитроцеллюлозу (BioRad). Одинаковую эффективность переноса пятен подтверждали с помощью обратимого окрашивания красителем Ponceau S и регистрации изображения на планшетном сканере. Пятна, из которых был удален краситель, блокировали и исследовали с помощью очищенного кроличьего поликлонального антитела AB5038P (Chemicon) и затем вторичного анти-кроличьего антитела IgG/HRP (Abcam) и хемолюминисцентного субстрата SuperSignal West Pico (Pierce) для обнаружения α-синуклеина. Специфичность антитела AB5038P в отношении α-синуклеина на вестерн-блотах подтверждали предварительной абсорбцией антитела 50х мольным избытком очищенного человеческого α-синуклеина, что устраняло все сигналы, кроме полосы, мигрирующей при 25 кДа (отмеченной на фигурах, как неспецифическая полоса 25 кДа).
Животным вводили соединения в течение 6 месяцев путем ежедневных i.p. инъекций 50 мг/кг/день, начиная с возраста 12 месяцев и до возраста 18 месяцев. Соединение 7 статистически значимо снижало уровни α-синуклеина на 69% в дисперсной фракции (фиг.35) и статистически значимо снижало уровни α-синуклеина на 73% в цитозольной фракции (фиг.36) по сравнению с мышами, получавшими носитель. Поскольку исследуемые группы включали животных обоих полов (обычно в группе имелось 2 самца), и поскольку в некоторых случаях наблюдалось изменение уровней α-синуклеина в зависимости от пола животного, авторы осуществили отдельное исследование самок. Важно отметить, что при отдельном исследовании женских особей соединение 7 статистически значимо снижало уровни α-синуклеина на 58% в дисперсной фракции (фиг.35) и на 48% в цитозольной фракции (фиг.36).
Для выяснения того, приведут ли соединения по настоящему изобретению к аналогичному снижению уровней α-синуклеина у молодых животных, получавших соединения в течение более короткого времени, соединения (или носитель) в течение 6 недель ежедневно вводили с помощью i.p. инъекций только самкам мышей в количестве 50 мг/кг/день, начиная примерно с 3-месячного возраста и до возраста примерно 4-5 месяцев. Введение соединения 7 статистически значимо уменьшало уровни α-синуклеина на 45% в дискретной фракции (фиг.37) и статистически значимо уменьшало уровни α-синуклеина на 71% в цитозольной фракции (фиг.38) по сравнению с мышами, получавшими носитель. Аналогичное уменьшение уровней α-синуклеина при введении соединения 7 (по отношению к контрольным животным, получавшим носитель) наблюдалось при анализе задней части мозга (без мозжечка) (данные не показаны).
В совокупности эти результаты дают основание предположить, что разрушение или уменьшение агрегатов α-синуклеина при обработке соединениями по настоящему изобретению может приводить к более растворимым формам α-синуклеина (включая мономеры), которые лучше поддаются выведению из организма.
Пример 14: Композиции соединений по настоящему изобретению
Как было указано ранее, соединения по настоящему изобретению желательно вводить в форме фармацевтических композиций. Подходящие фармацевтические композиции и способы их получения хорошо известны рядовым специалистам в данной области техники и описаны в таком руководстве, как Remington: The Science and Practice of Pharmacy, A. Gennaro, ed., 20th edition, Lippincott, Williams & Wilkins, Philadelphia, PA.
Ниже приведены типовые примеры композиций:
Состав для пероральных таблеток
Состав для получения пероральных таблеток соединений по настоящему изобретению готовят следующим образом:
Ингредиенты смешивают до достижения однородности, затем гранулируют с помощью воды и высушивают гранулят. Затем высушенный гранулят прессуют в таблетки такого размера, чтобы они включали подходящую дозу соединения. Таблетки необязательно снабжают покрытием, нанося суспензию пленкообразующего агента (например, гидроксипропилметилцеллюлозы), пигмента (например, диоксида титана) и пластификатора (например, диэтилфталата), и высушивая пленку выпариванием растворителя. Пленочное покрытие может включать, например, 2-6% массы таблетки.
Состав для пероральных капсул
Гранулятом, описанным в предыдущем разделе данного примера, заполняют твердые желатиновые капсулы такого размера, который соответствует предполагаемой дозе. Если это желательно, капсулу оборачивают полосой из подходящего материала для герметичности.
Состав для мягких желатиновых капсул
Состав для мягких желатиновых капсул готовят следующим образом
Соединение растворяют или диспергируют в полиэтиленгликоле и при необходимости добавляют загуститель. Затем мягкие желатиновые капсулы заполняют таким количеством состава, которого достаточно для обеспечения желаемой дозы соединения.
Парентеральный состав
Готовят следующий парентеральный состав:
Соединение растворяют в физиологическом растворе, затем полученный раствор стерилизуют и заполняют им флаконы, ампулы и шприцы в зависимости от необходимости.
Пероральный состав с регулируемым высвобождением
Состав с замедленным высвобождением можно приготовить по способу патента США № 4710348 следующим образом:
Один килограмм соединения по настоящему изобретению покрывают этилцеллюлозой Dow Type 10 в модифицированном устройстве для покрытия порошков Uni-Glatt. Распыляемый раствор представляет собой 8% раствор этилцеллюлозы в смеси 90% ацетона и 10% этанола. В качестве пластификатора добавляют касторовое масло в количестве 20% от массы этилцеллюлозы. Распыление состава осуществляют при следующих условиях: 1) скорость 1 литр/час; 2) заслонка 10-15%; 3) температура на входе 50°C; 4) температура на выходе 30°C; 5) процент покрытия 17%. Покрытое соединение просеивают, чтобы получить частицы размером от 74 до 210 микронов. Следят за тем, чтобы обеспечить хорошее перемешивание частиц разных размеров в указанном диапазоне. 400 мг покрытых частиц смешивают со 100 мг крахмала и эту смесь прессуют на ручном прессе под давлением 1,5 тн, получая 500-мг таблетку с контролируемым высвобождением.
Объем настоящего изобретения не ограничен конкретными вариантами осуществления, описанными в настоящей заявке. Действительно, различные модификации настоящего изобретения, помимо описанных в заявке, должны быть ясны специалисту в данной области техники из предыдущего описания. Предполагается, что эти модификации попадают в объем приложенной формулы изобретения. В описании упоминаются различные публикации, причем их содержание включено с помощью ссылки во всей их полноте.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ N-АРИЛБЕНЗАМИДЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ АМИЛОИДНЫХ ЗАБОЛЕВАНИЙ И СИНУКЛЕИНОПАТИИ | 2005 |
|
RU2381213C2 |
| СПОСОБЫ ПРОФИЛАКТИКИ И ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2009 |
|
RU2496502C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ НЕВРОЛОГИЧЕСКИХ НАРУШЕНИЙ | 2005 |
|
RU2387453C2 |
| СПЕЦИФИЧЕСКИЕ В ОТНОШЕНИИ АМИЛОИДА БЕТА (А БЕТА) 1-42 МОНОКЛОНАЛЬНЫЕ АНТИТЕЛА, ОБЛАДАЮЩИЕ ТЕРАПЕВТИЧЕСКИМИ СВОЙСТВАМИ | 2006 |
|
RU2551782C2 |
| МОНОКЛОНАЛЬНОЕ ТЕЛО ПРОТИВ АМИЛОИДА БЕТА | 2008 |
|
RU2571856C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ НЕЙРОДЕГЕНЕРАТИВНОГО ЗАБОЛЕВАНИЯ | 2012 |
|
RU2618412C2 |
| ГУМАНИЗИРОВАННЫЕ АНТИТЕЛА К β-АМИЛОИДНОМУ ПЕПТИДУ | 2008 |
|
RU2475500C2 |
| ТЕРАПЕВТИЧЕСКАЯ ВАКЦИНА | 2006 |
|
RU2440824C2 |
| 4-(3-((2-Адамантил)амино)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамид - новое средство для лечения болезни Альцгеймера | 2022 |
|
RU2783995C1 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ АМИЛОИДОЗОВ | 2009 |
|
RU2491953C2 |
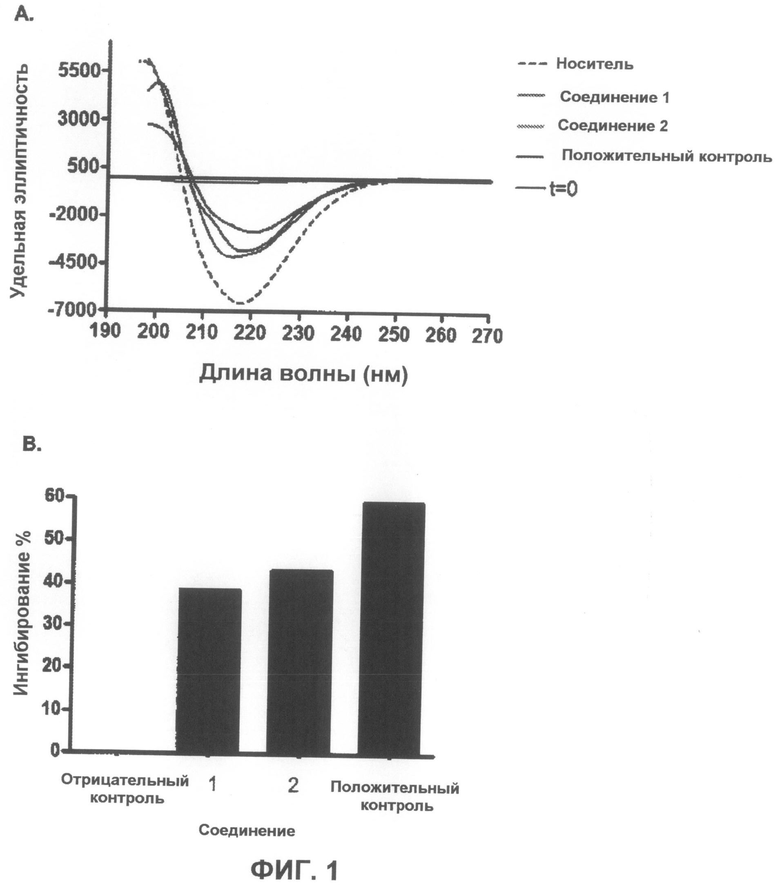
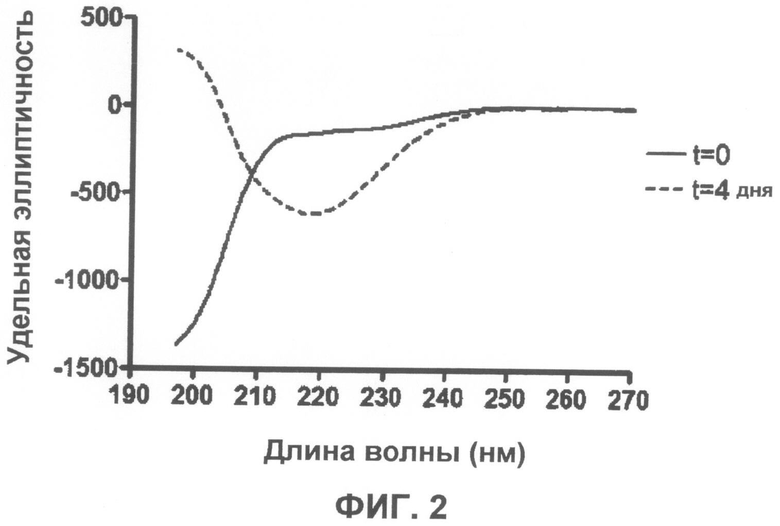
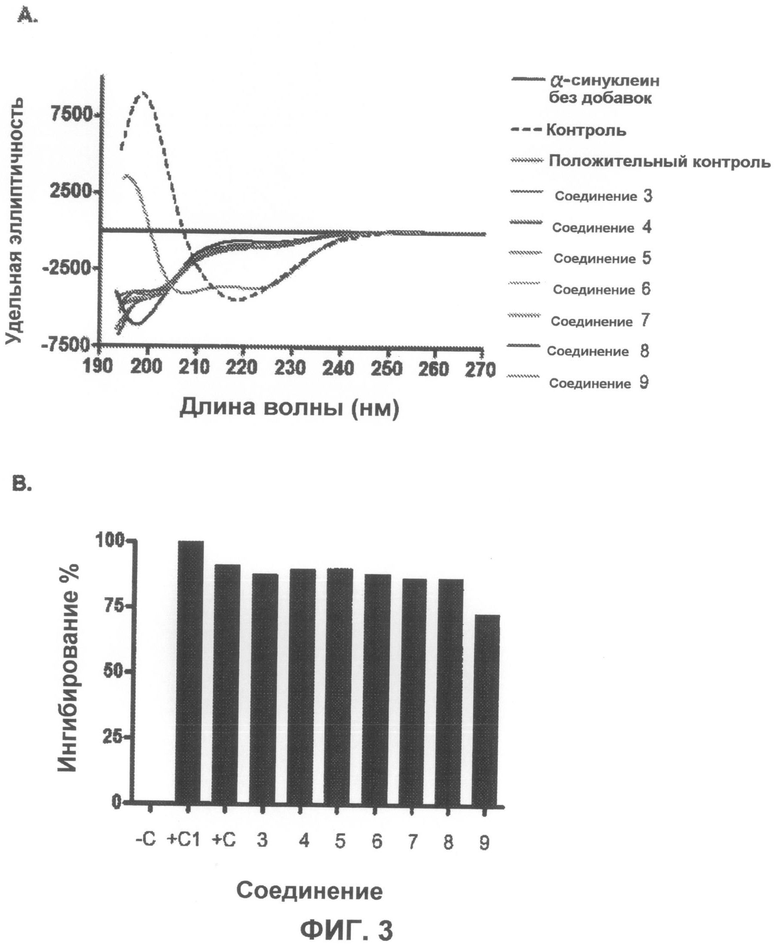
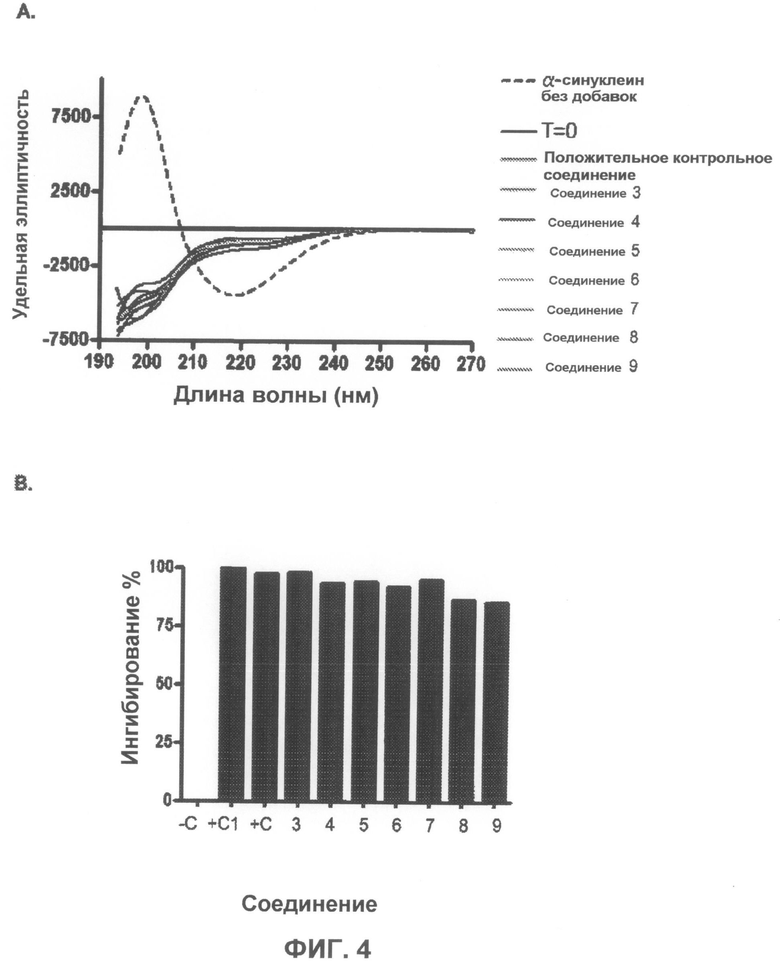
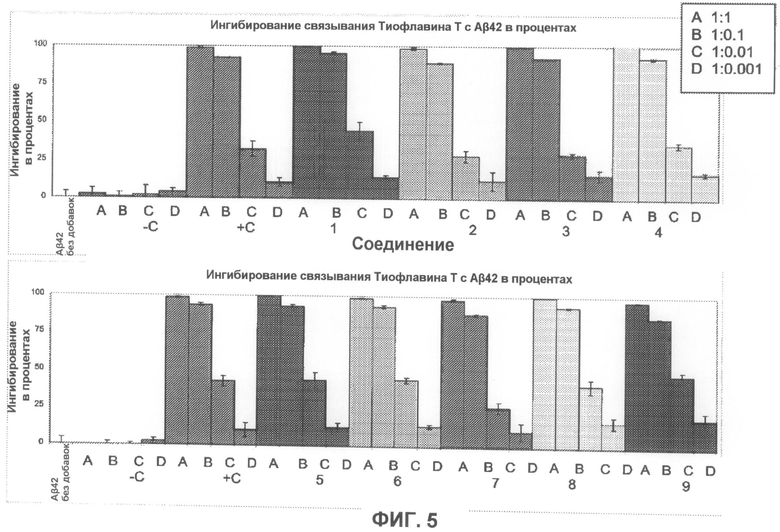
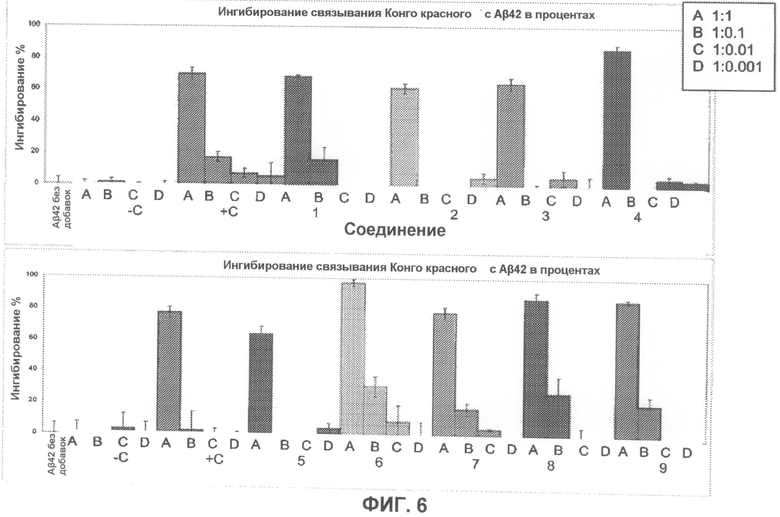
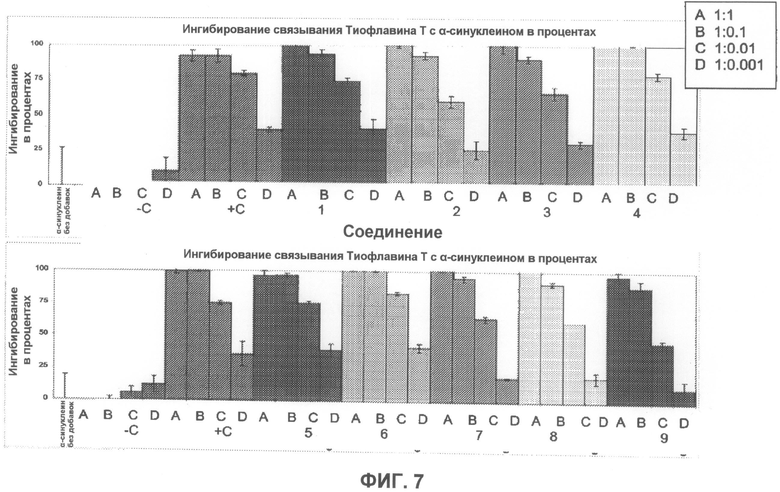
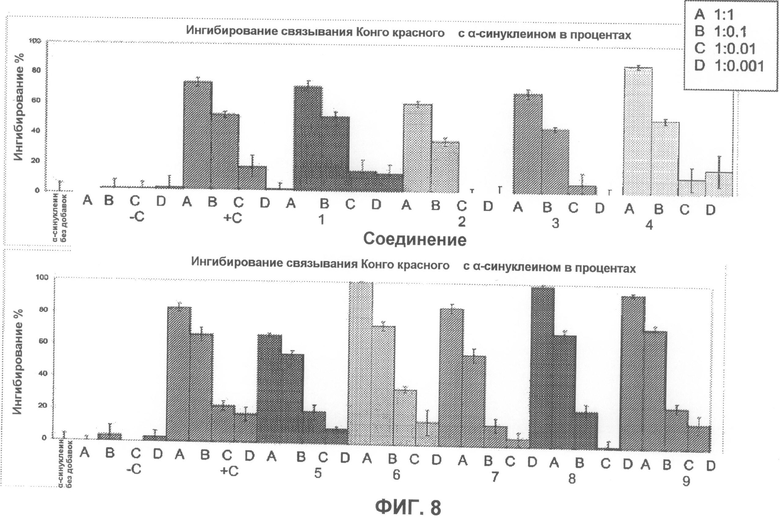
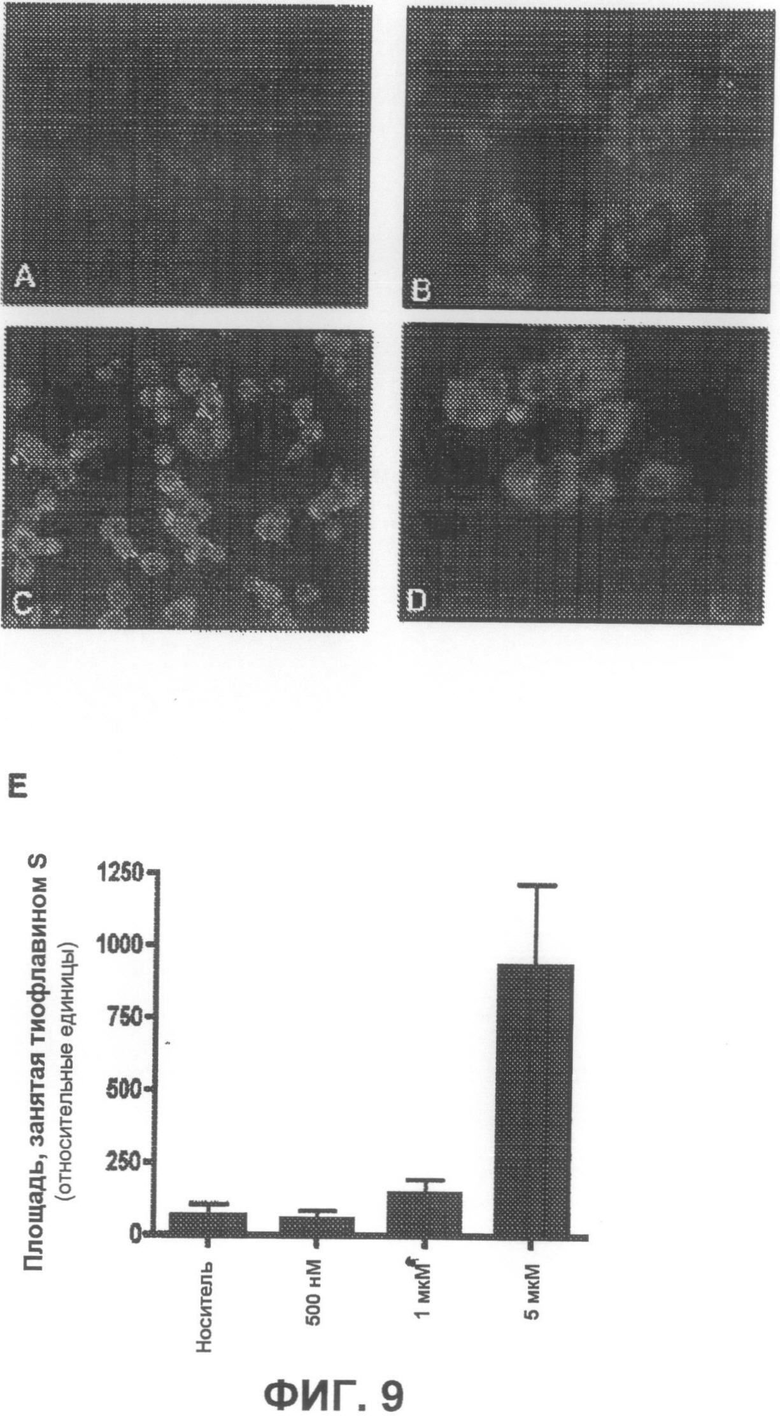
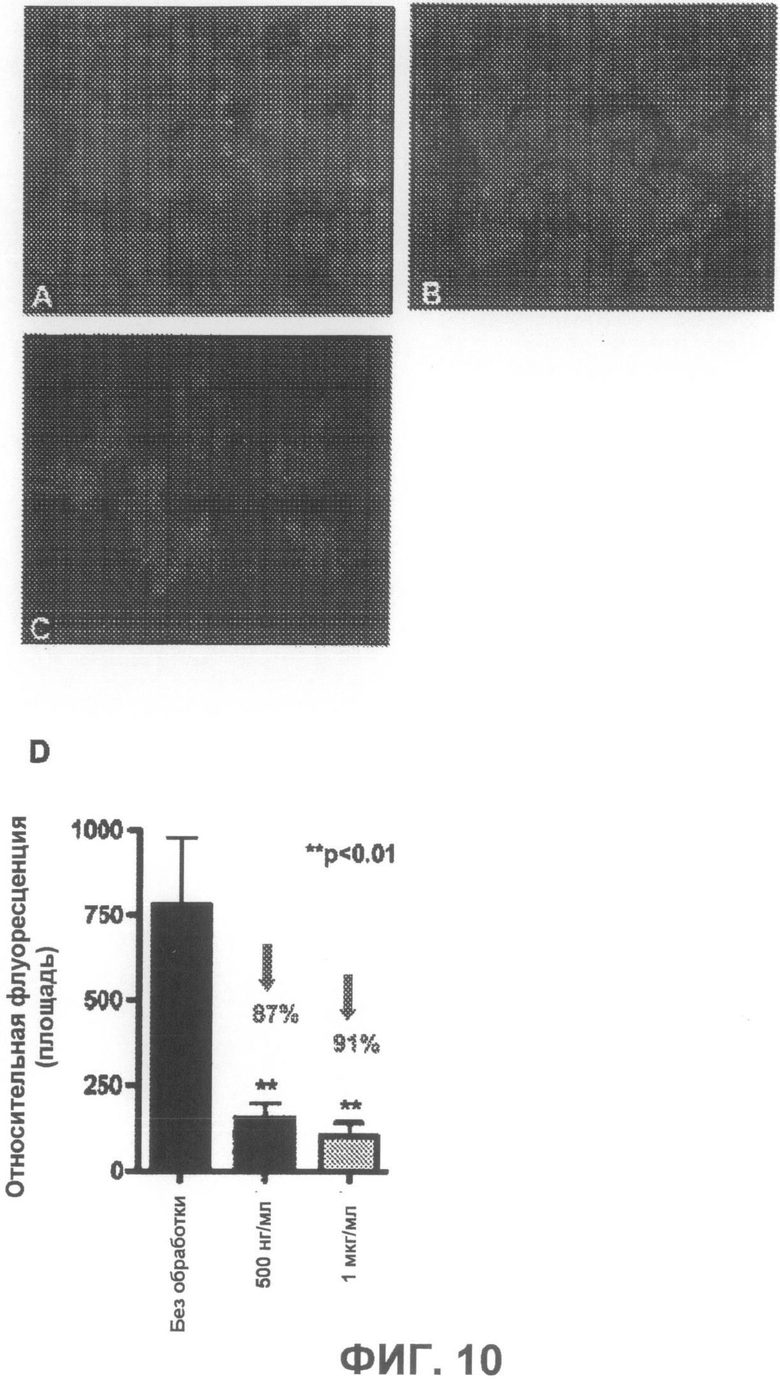
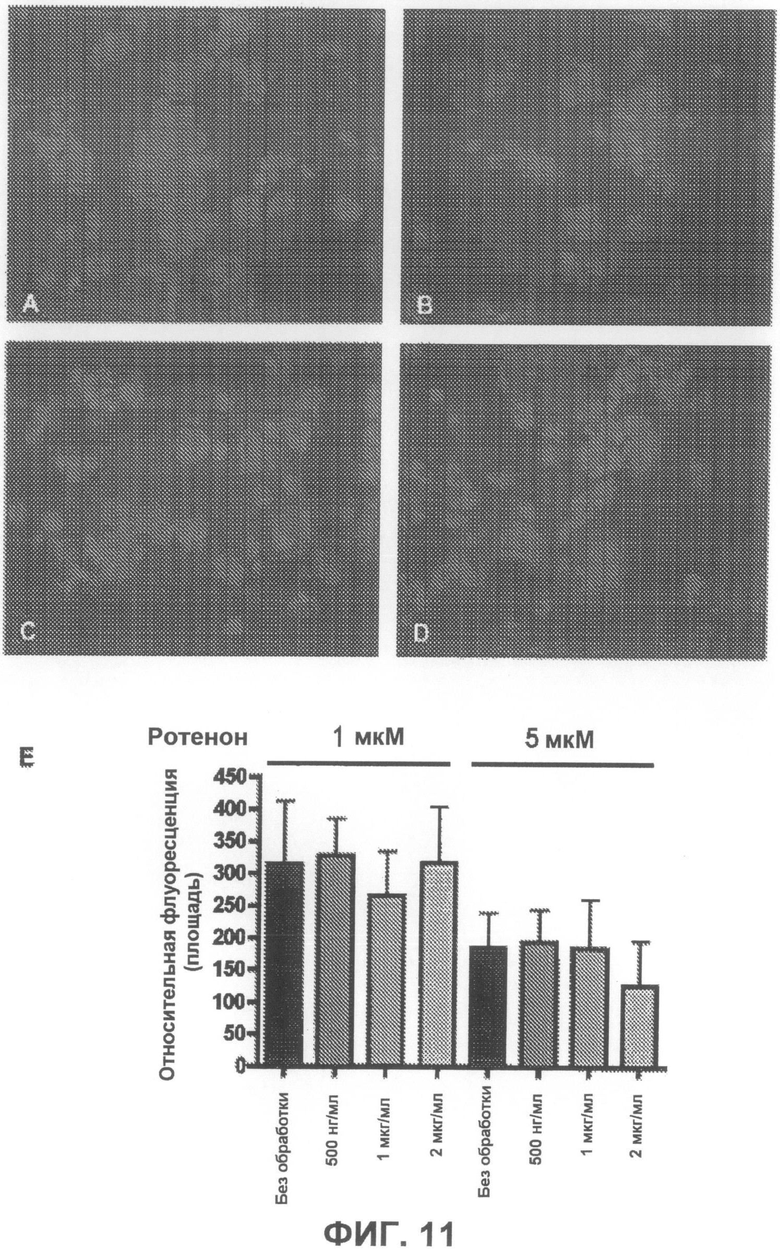
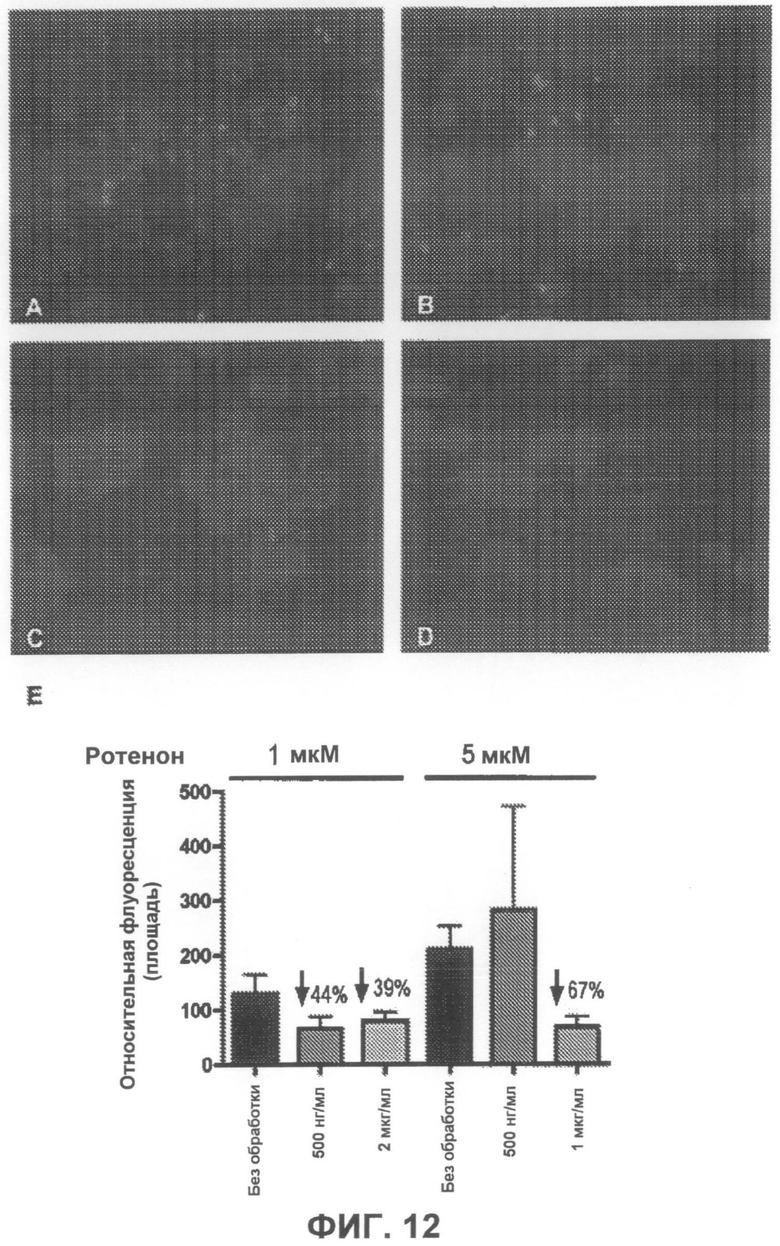
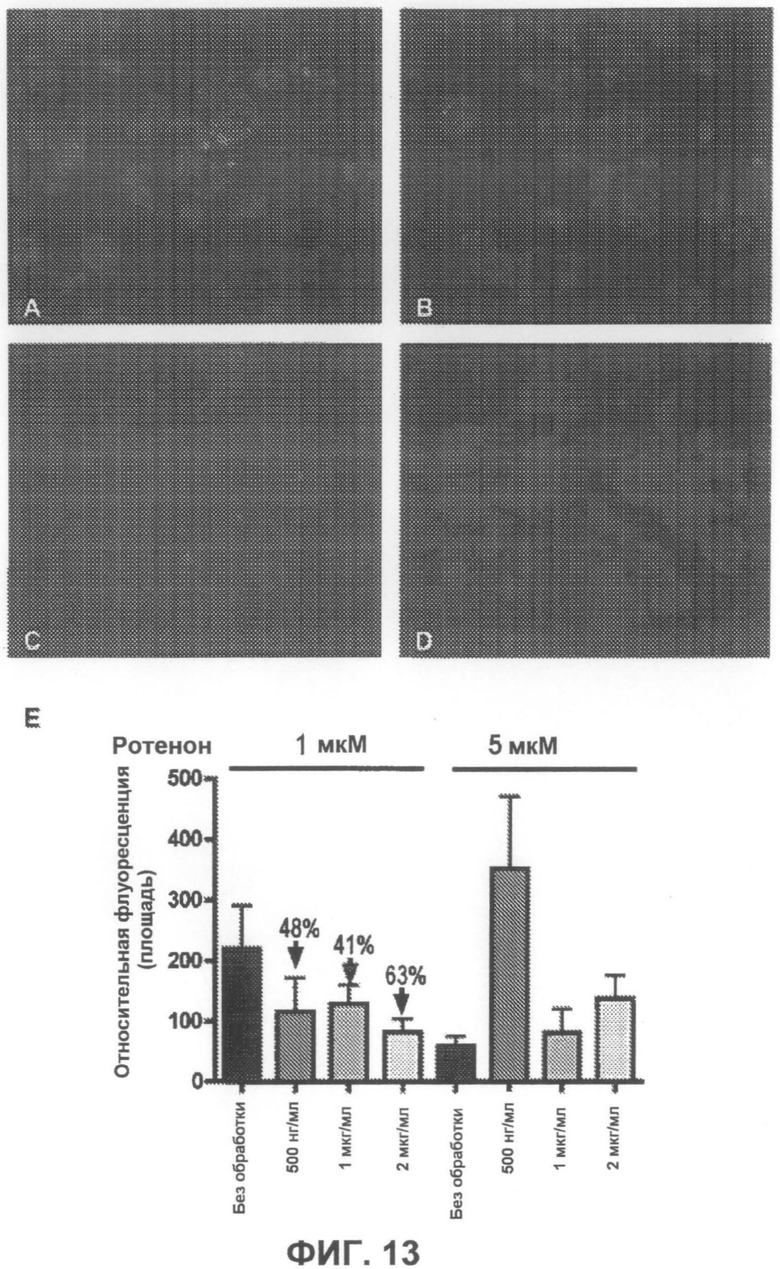
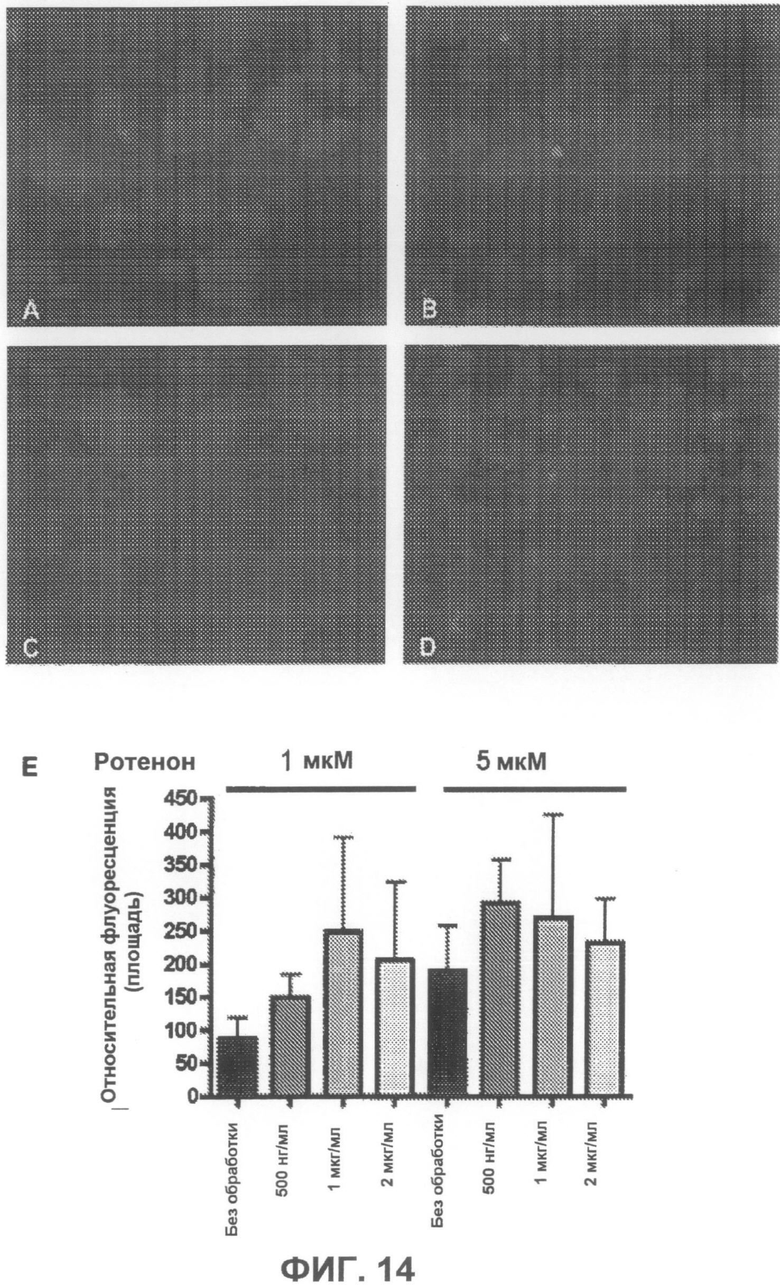
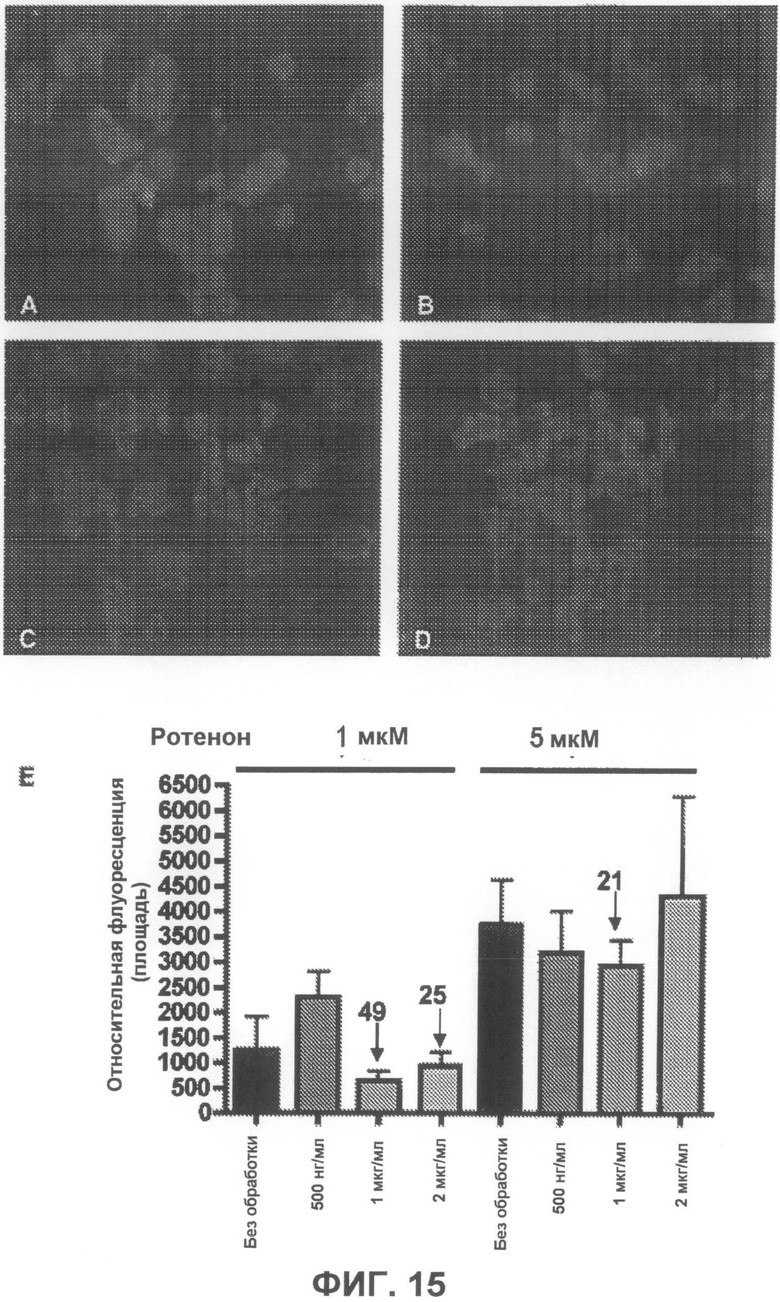
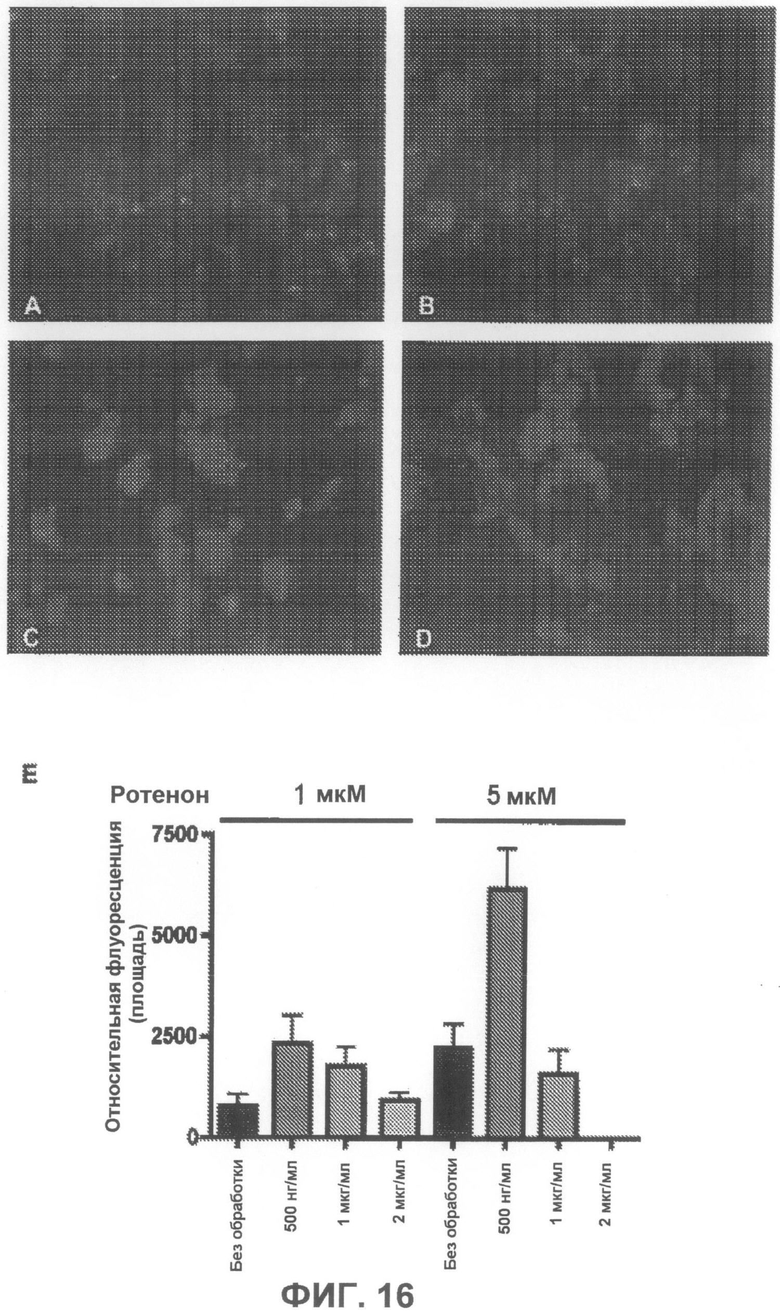
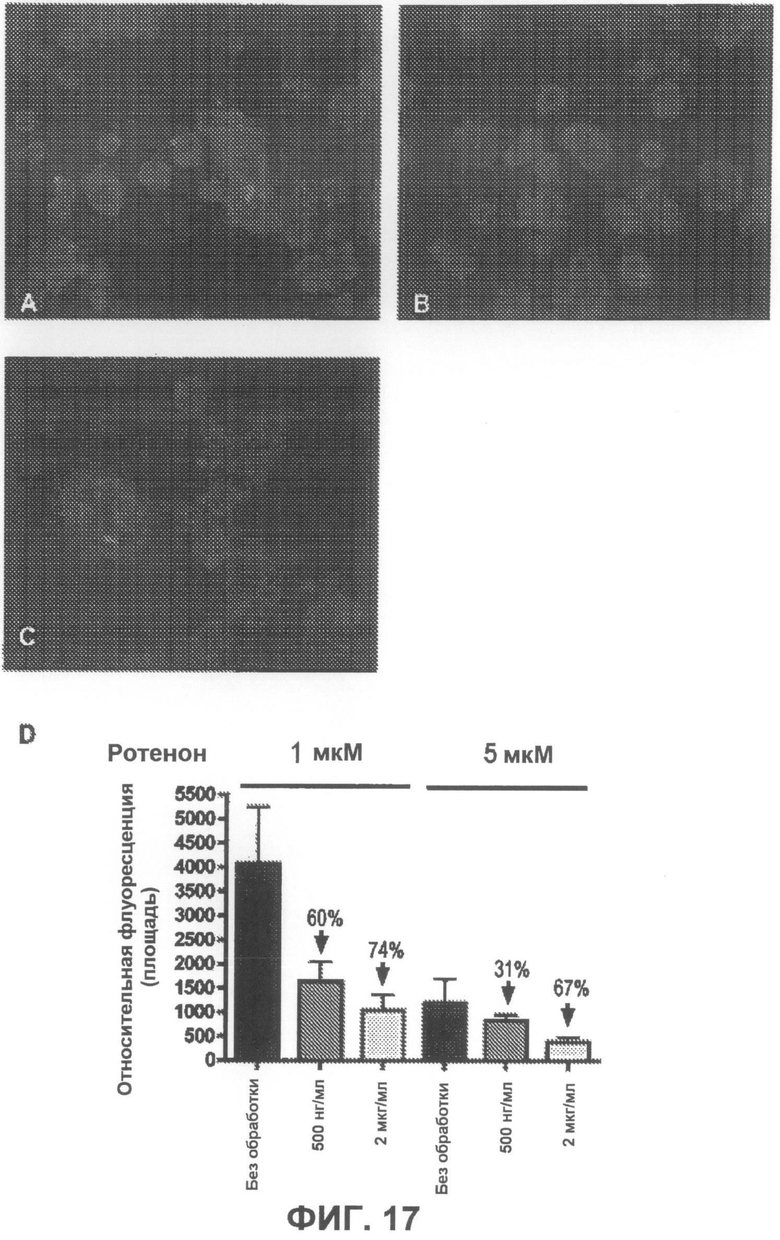
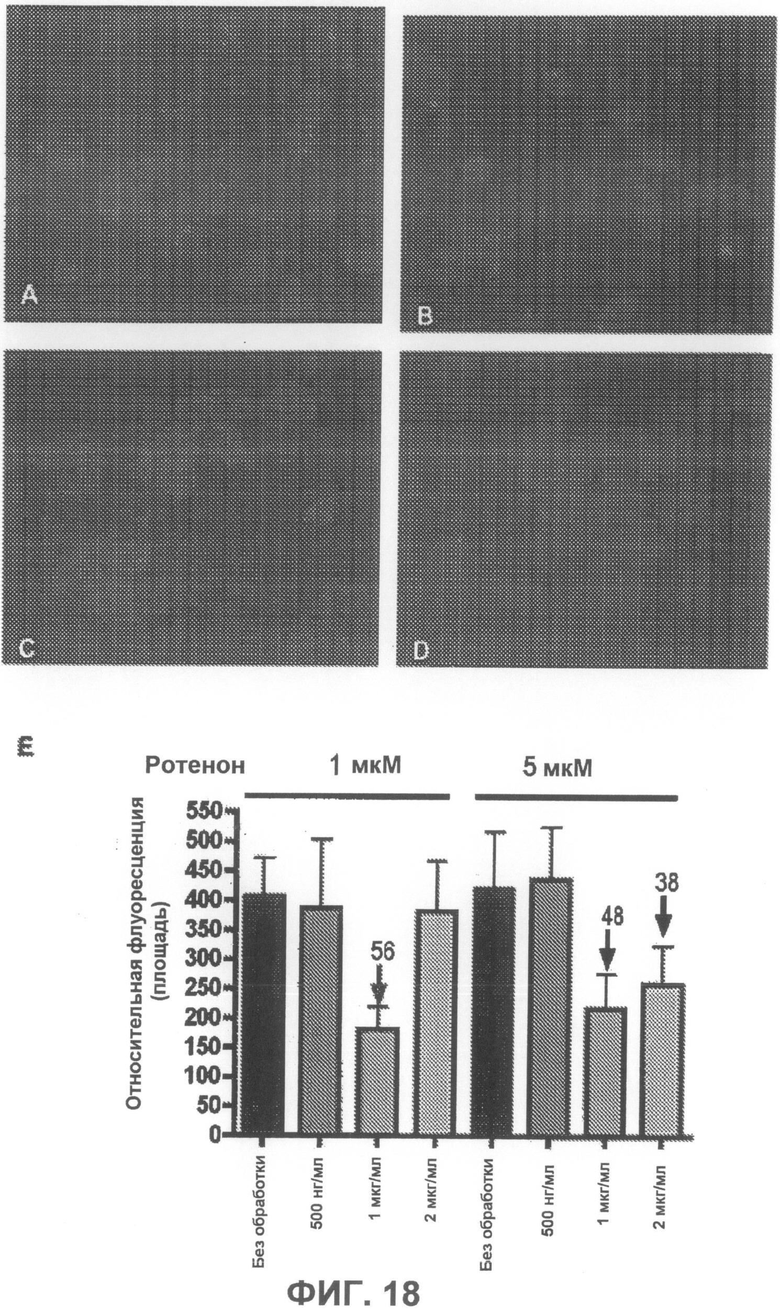
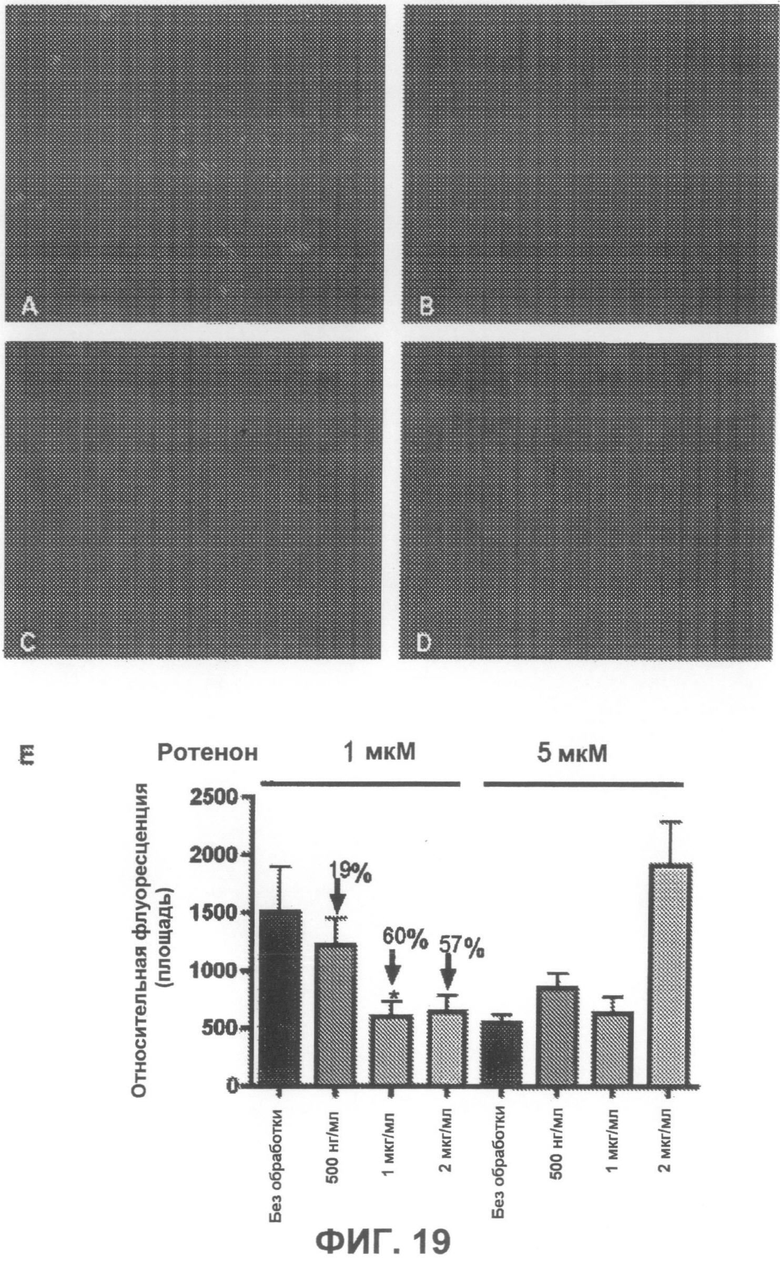
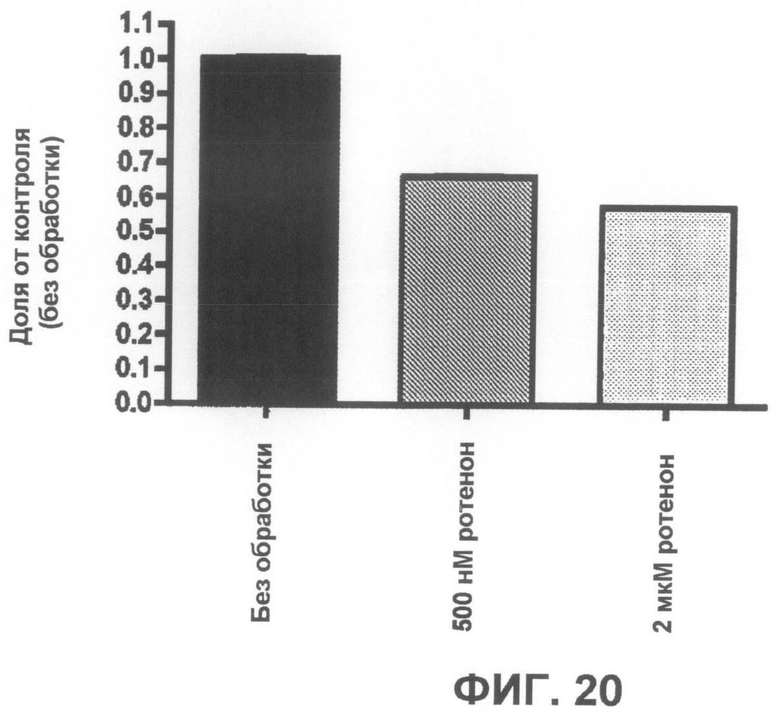
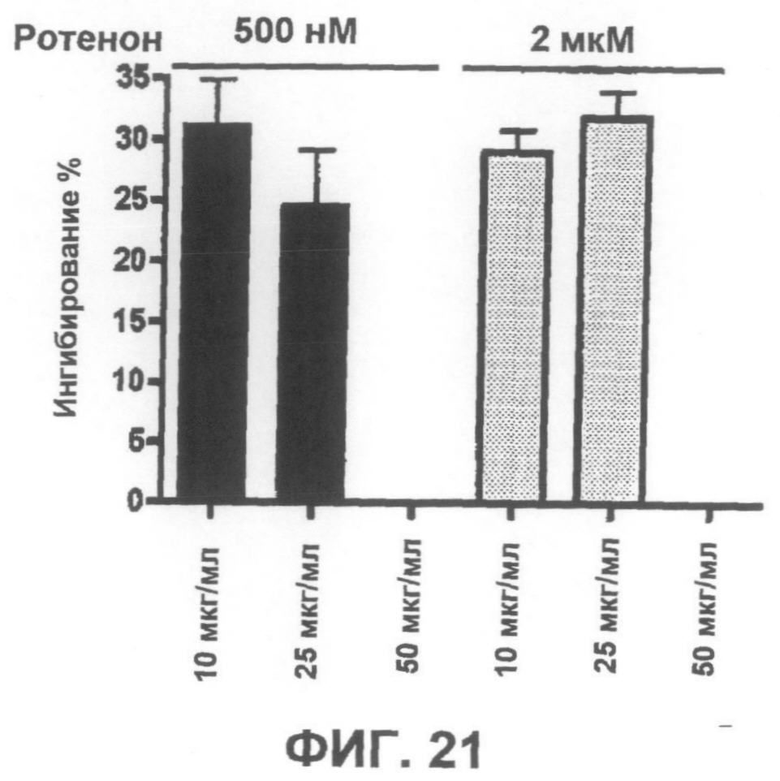
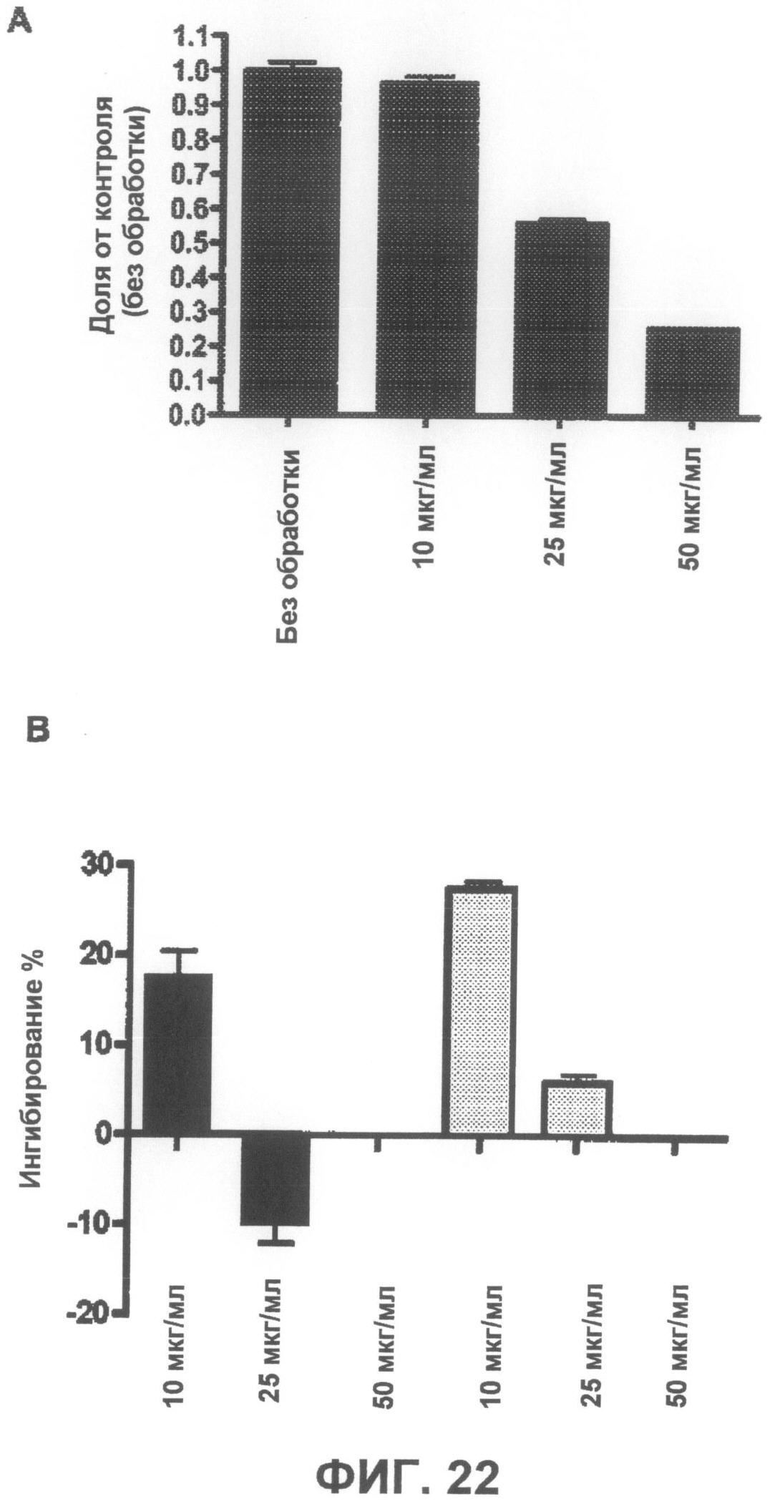
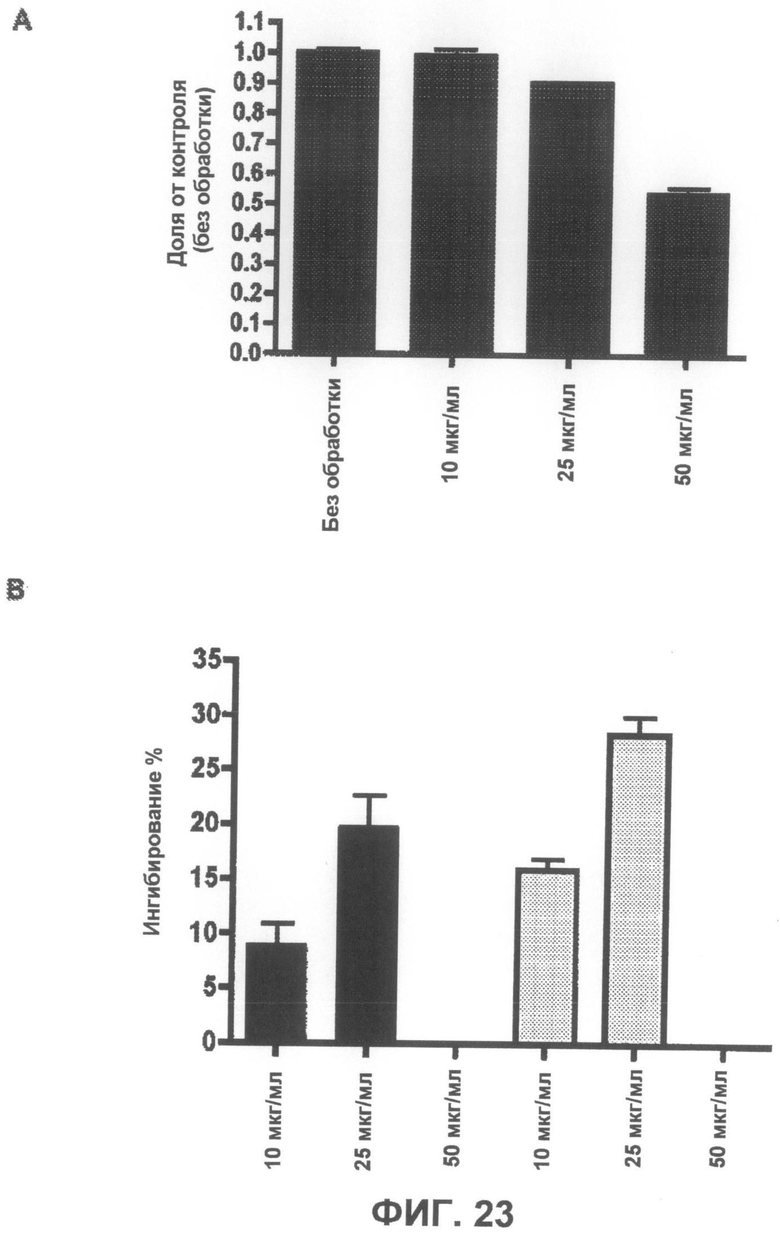
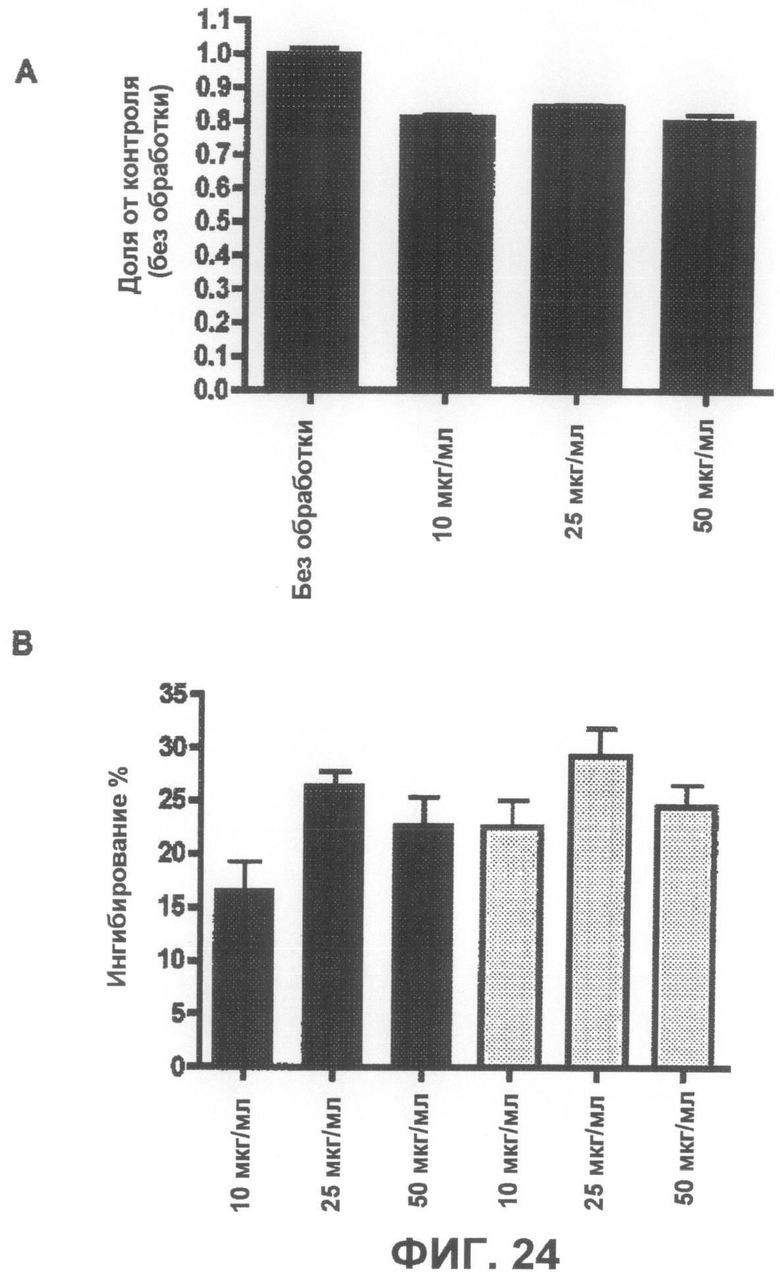

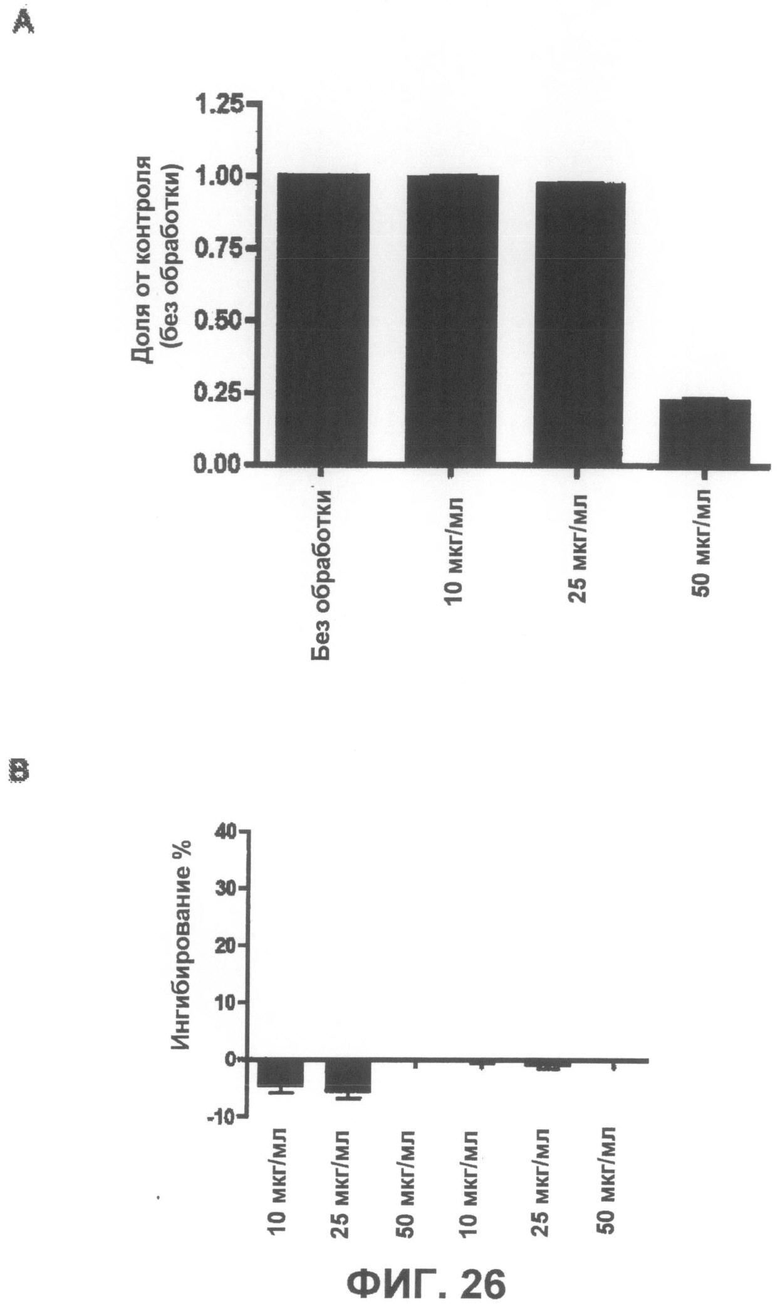
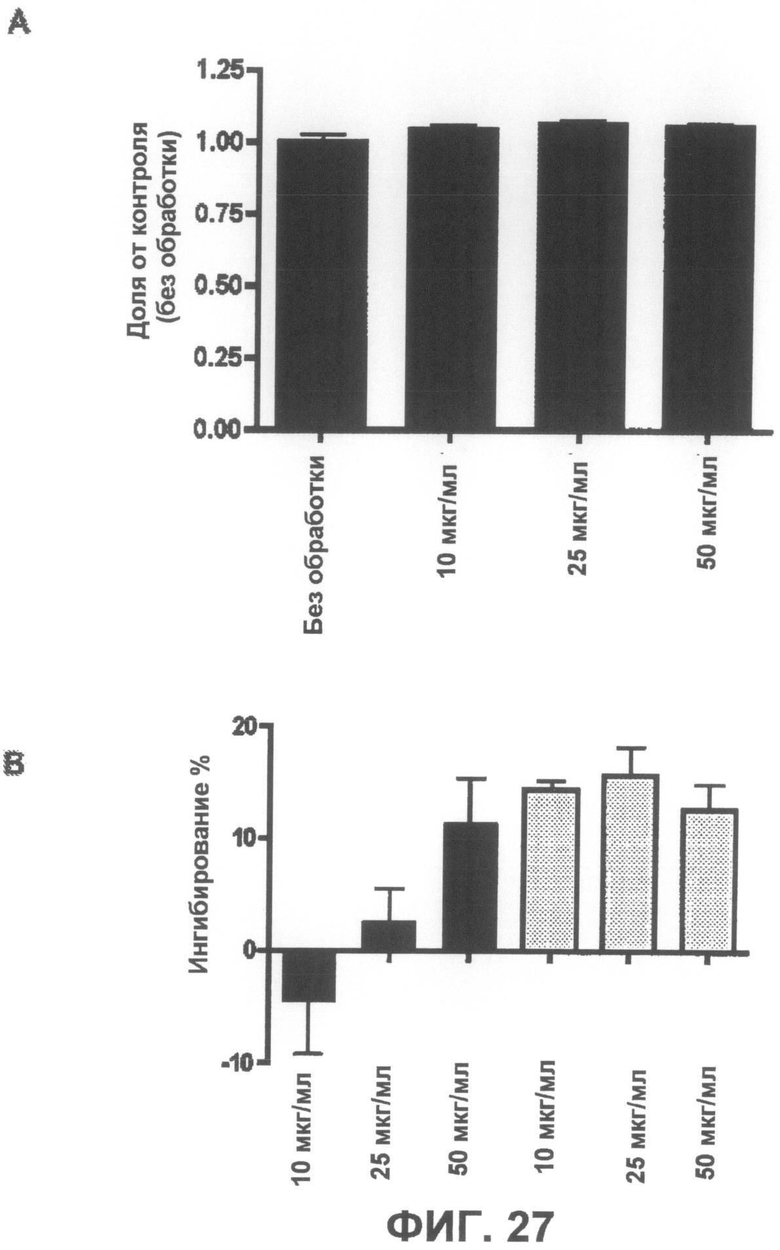
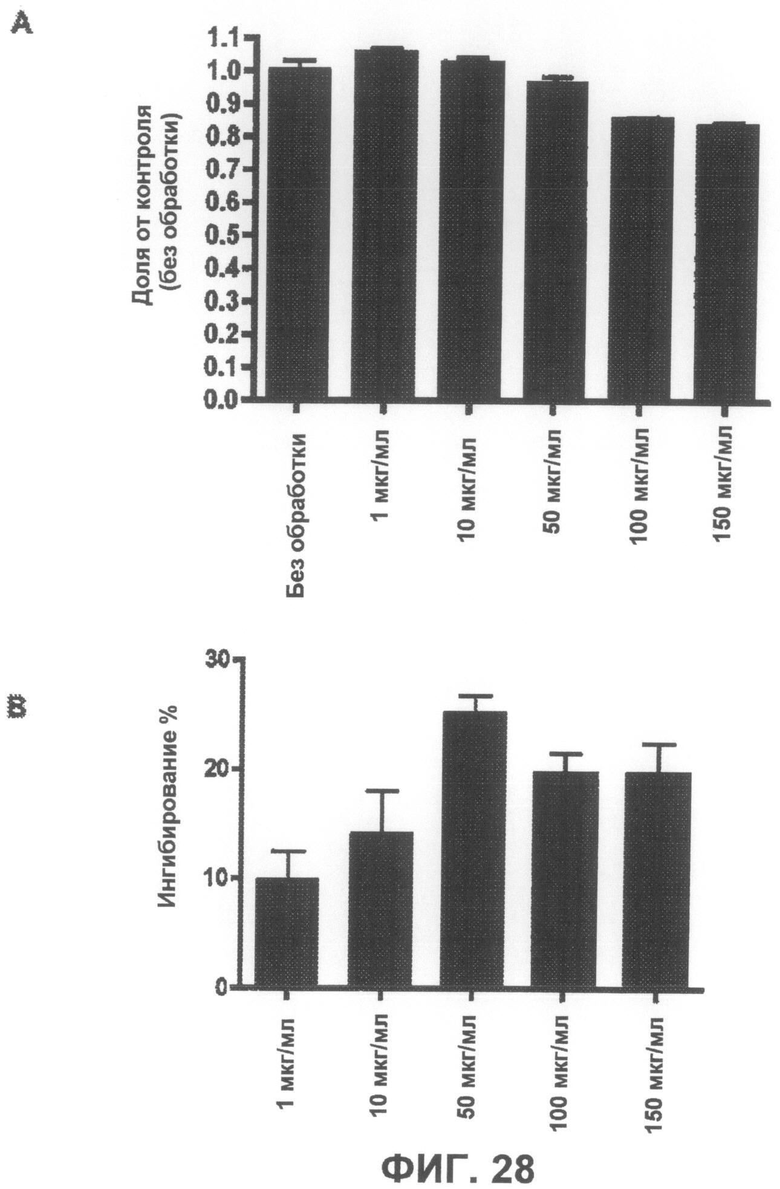
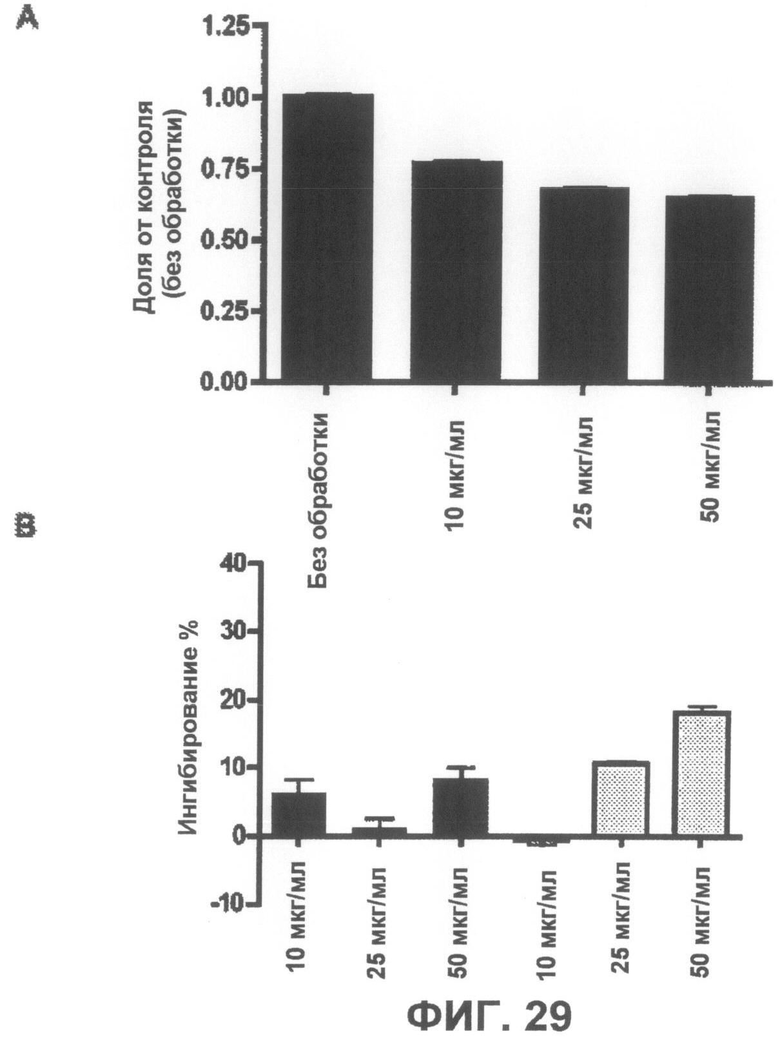
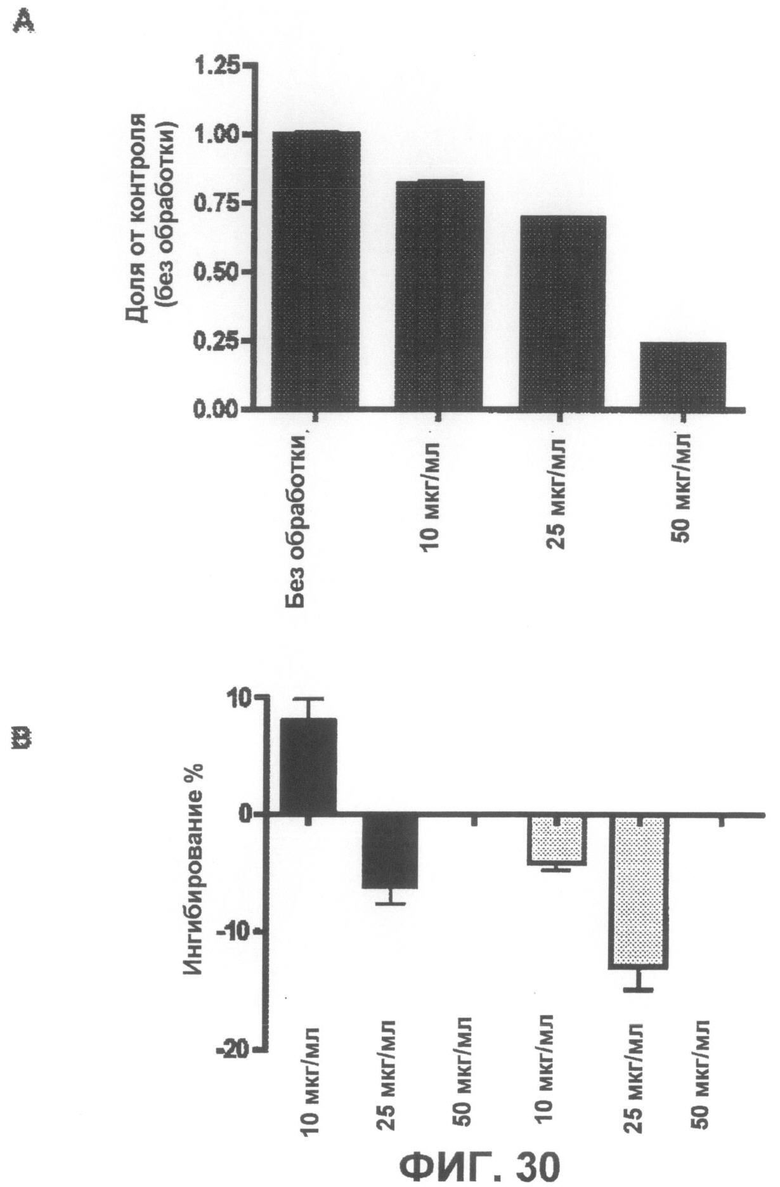
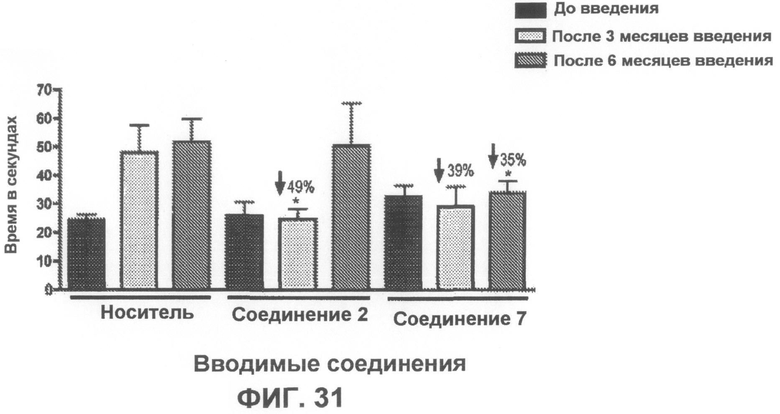

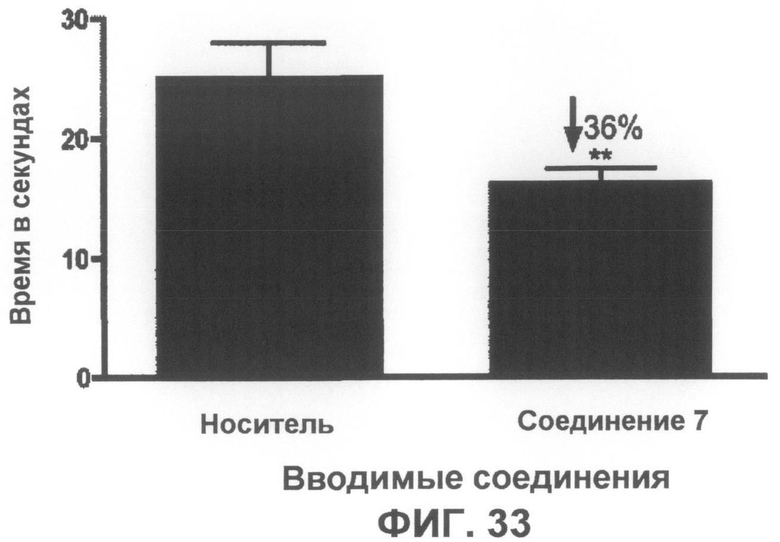
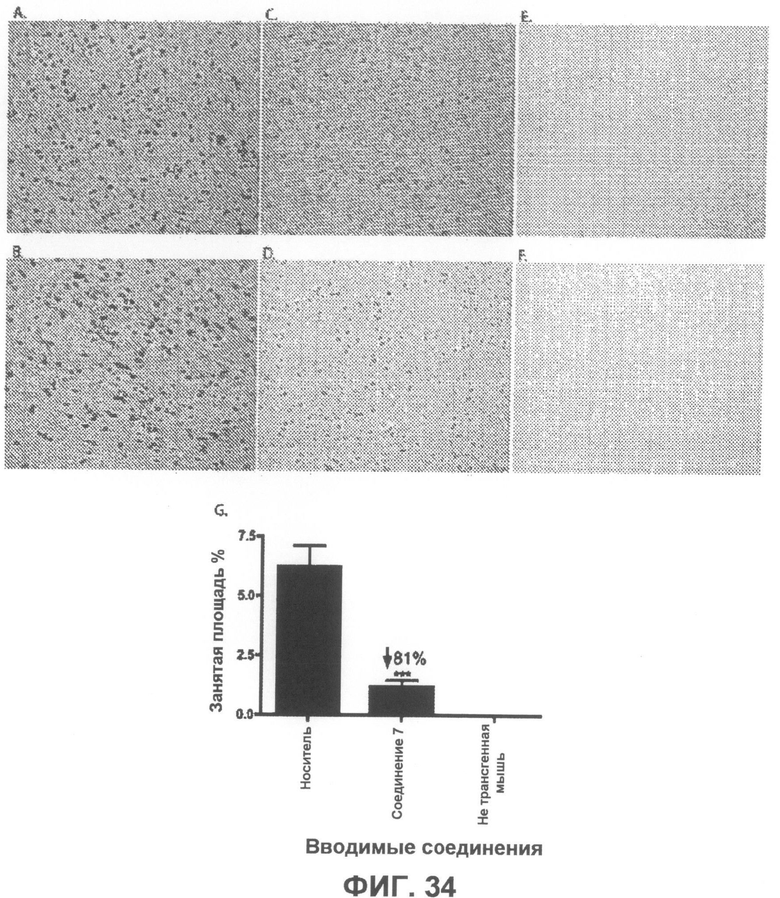
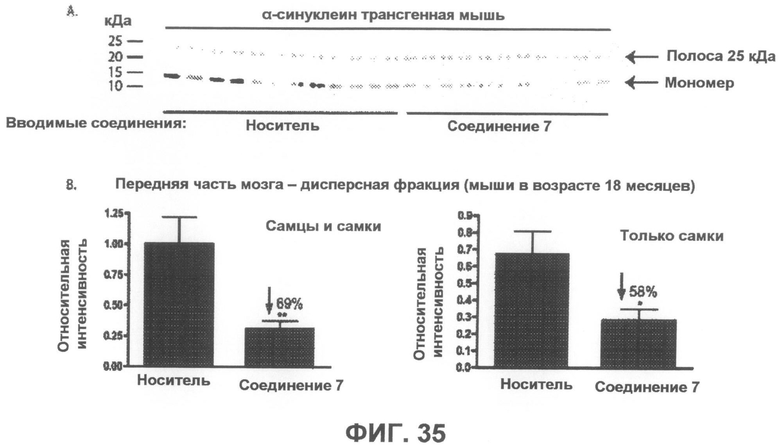
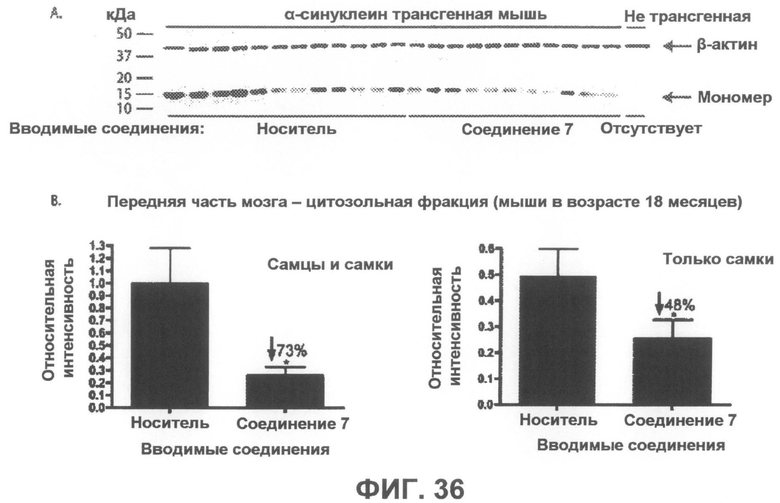
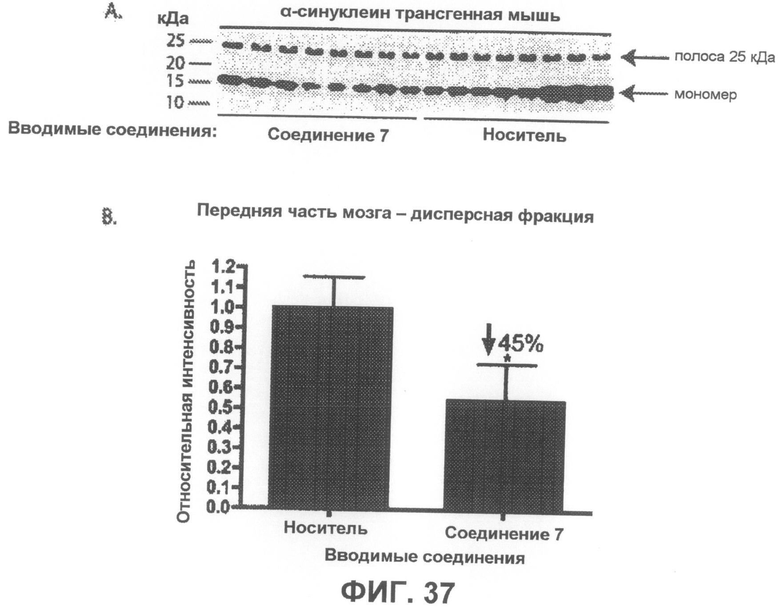
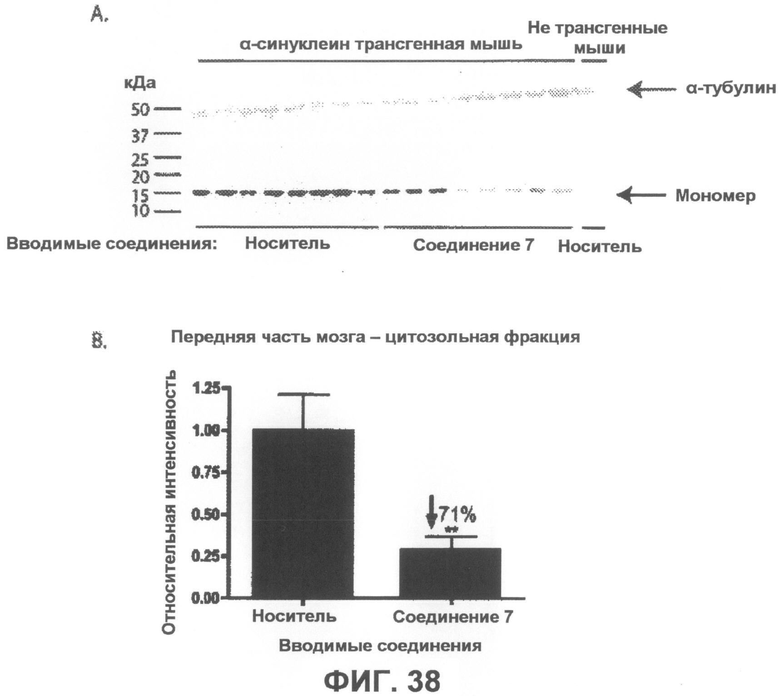
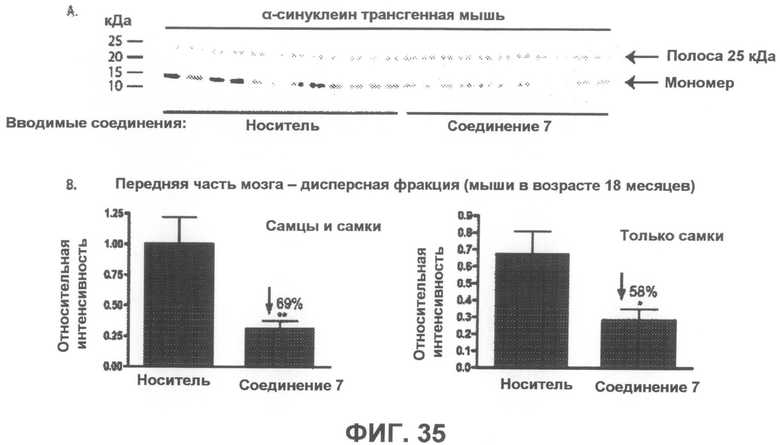
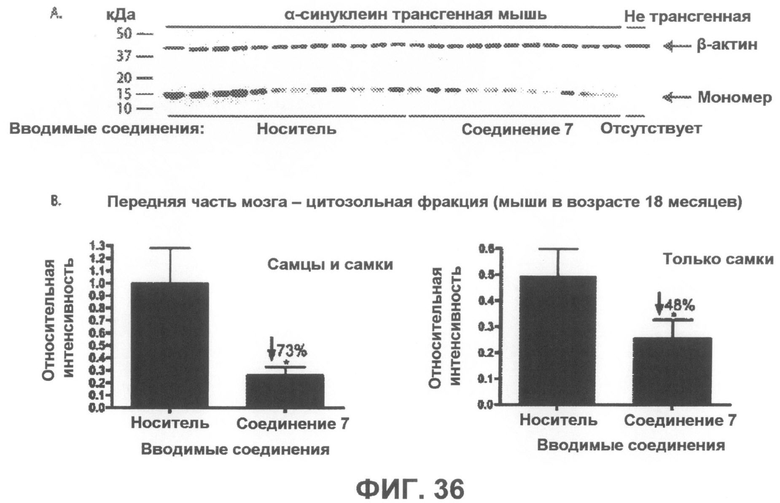
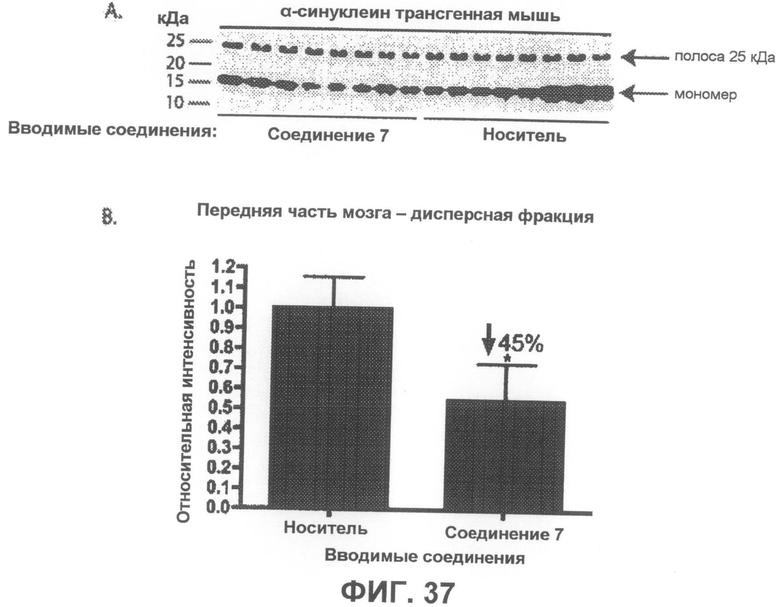
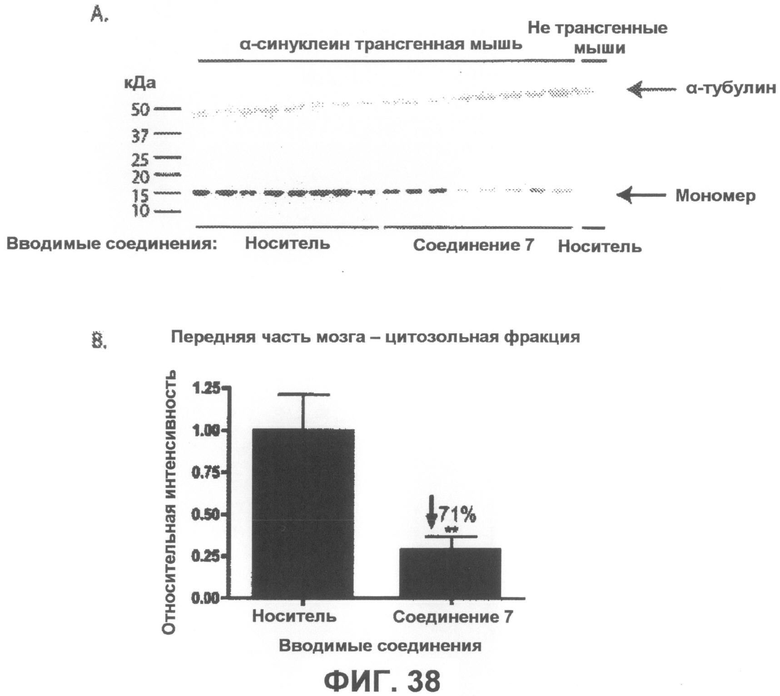
Изобретение относится к соединению формулы
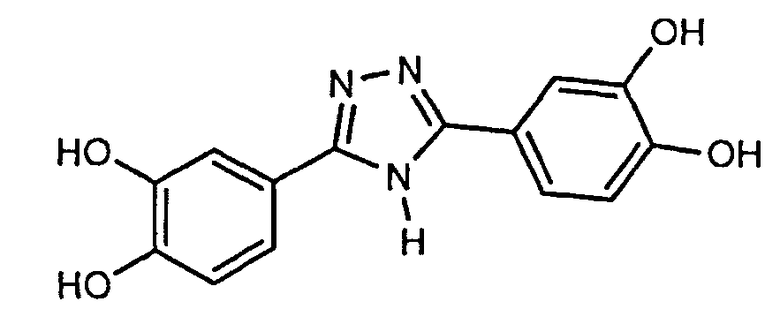
и к его фармацевтически приемлемой соли. Изобретение также касается фармацевтической композиции, предназначенной для лечения β-амилоидных заболеваний и синуклеинопатий, на основе указанного соединения. Технический результат - получено новое соединение и фармацевтическая композиция на его основе, которые могут найти применение в медицине для лечения таких заболеваний, как болезнь Альцгеймера и болезнь Паркинсона. 5 н. и 12 з.п. ф-лы, 38 ил., 14 пр.
1. Соединение формулы
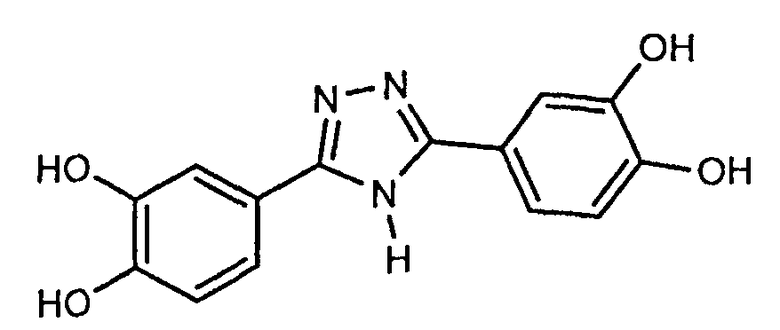
и его фармацевтически приемлемые соли.
2. Соединение по п.1, предназначенное для ингибирования образования, отложения, накопления или устойчивого существования агрегатов Аβ-амилоида или α-синуклеина.
3. Соединение по п.1, предназначенное для лечения β-амилоидных заболеваний и синуклеинопатий у млекопитающих, страдающих этими заболеваниями.
4. Соединение по п.3, где β-амилоидное заболевание выбрано из группы заболеваний, состоящей из болезни Альцгеймера и церебральной β-амилоидной ангиопатии.
5. Соединение по п.3, где β-амилоидное заболевание является болезнью Альцгеймера.
6. Соединение по п.3, где синуклеинопатия выбрана из группы, состоящей из болезни Паркинсона, семейной болезни Паркинсона, болезни с тельцами Леви и множественной системной атрофии.
7. Соединение по п.3, где синуклеинопатия представляет собой болезнь Паркинсона.
8. Соединение по п.1, предназначенное для улучшения двигательной активности у млекопитающих, страдающих от синуклеинопатии.
9. Соединение по п.1, предназначенное для прекращения прогрессирования двигательных нарушений у млекопитающих, страдающих от болезни Паркинсона.
10. Фармацевтическая композиция, предназначенная для лечения β-амилоидных заболеваний и синуклеинопатии у млекопитающих, страдающих этими заболеваниями, включающая соединение по п.1 и фармацевтически приемлемый эксципиент.
11. Применение соединения по п.1 для получения лекарственного средства для лечения β-амилоидных заболеваний и синуклеинопатии у млекопитающих, страдающих этими заболеваниями.
12. Применение по п.11, где β-амилоидное заболевание выбрано из группы заболеваний, состоящей из болезни Альцгеймера и церебральной β-амилоидной ангиопатии.
13. Применение по п.11, где β-амилоидное заболевание является болезнью Альцгеймера.
14. Применение по п.11, где синуклеинопатия выбрана из группы, состоящей из болезни Паркинсона, семейной болезни Паркинсона, болезни с тельцами Леви и множественной системной атрофии.
15. Применение по п.11, где синуклеинопатия представляет собой болезнь Паркинсона.
16. Применение соединения по п.1 для получения лекарственного средства для улучшения двигательной активности у млекопитающих, страдающих от синуклеинопатии.
17. Применение соединения по п.1 для получения лекарственного средства для прекращения прогрессирования двигательных нарушений у млекопитающих, страдающих от болезни Паркинсона.
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| US 6288061 B1, 11.09.2001 | |||
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), ПРОИЗВОДНЫЕ 3,5-ДИФЕНИЛ-1,2,4-ТРИАЗОЛОВ, В НЕЕ ВХОДЯЩИЕ | 1997 |
|
RU2208010C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ИНСЕКТИЦИДНОЕ И АКАРИЦИДНОЕ СРЕДСТВО | 1994 |
|
RU2131421C1 |

