Область изобретения
Настоящее изобретение относится к визуализации in vivo и, в частности, к визуализации in vivo периферического бензодиазепинового рецептора (PBR). Предлагается тетрациклический индольный агент визуализации in vivo, который связывается с PBR с высокой аффинностью, хорошо поглощается в головном мозге после введения и который предпочтительно связывается с тканями, экспрессирующими более высокие уровни PBR. В настоящем изобретении также предлагается соединение-предшественник, полезное в синтезе агента визуализации in vivo no изобретению, а также способ синтеза указанного агента визуализации in vivo, включающий применение указанного соединения-предшественника, и набор для выполнения указанного способа. Также предлагается кассета для автоматизированного синтеза агента визуализации in vivo. Кроме того, в изобретении предлагается радиофармацевтическая композиция, содержащая агент визуализации in vivo по изобретению, а также способы применения указанного агента визуализации in vivo.
Описание уровня техники
Периферический бензодиазепиновый рецептор (PBR), который также известен как транслокаторный белок (TSPO), как известно, главным образом локализован в периферических тканях и глиальных клетках, но его физиологическая функция ясно не истолкована. Известно, что внутриклеточно PBR локализуется на внешней митохондриальной мембране, что указывает на потенциальную роль в модулировании митохондриальной функции и в иммунной системе. Кроме того, предполагалось, что PBR вовлечен в клеточную пролиферацию, стероидогенез, ток кальция и клеточное дыхание. PBR ассоциируется с множеством состояний, включая острый и хронический стресс, тревогу, депрессию, болезнь Паркинсона, болезнь Альцгеймера, повреждение головного мозга, рак (Gavish et al. Pharm. Rev. 1999; 51:629), болезнь Хантингтона (Meβmer and Reynolds Neurosci. Lett. 1998; 241:53-6), астму (Pelaia et al. Gen. Pharmacol. 1997; 28(4):495-8), ревматоидный артрит (Bribes et al. Eur. J.Pharmacol. 2002; 452(1):111-22), атеросклероз (Davies et al. J.Nucl. Med. 2004; 45:1898-1907) и рассеянный склероз (Banati et al. 2000 Brain; 123:2321). PBR также может быть связан с невропатической болью. Tsuda et al. наблюдали активированную микроглию у субъектов с невропатической болью (2005 TINS 28(2) р.101-7).
Лиганды, имеющие высокую аффинность к PBR, известны в данной области техники. Класс индольных соединений, обладающих аффинностью к PBR (значения IC50 для наиболее активных соединений от 0,2 нМ и до 5,0 нМ), раскрыт в US 6451795. Утверждается, что соединения, раскрытые там, являются полезными для предупреждения или лечения периферических невропатий и для лечения нейродегенеративных заболеваний центральной нервной системы. Okubu et al. (Bioorganic & Medicinal Chemistry 2004; 12:3569-80) описывают разработку, синтез и структуру группы тетрациклических индольных соединений, обладающих аффинностью к PBR (низкие значения IC50 составляют примерно 0,4 нМ). В данной публикации Okubu et al. отсутствует обсуждение конкретных применений данных соединений.
In vivo визуализация PBR также известна в данной области техники. Визуализация посредством позитрон-эмиссионной томографии (PET) с использованием селективного лиганда PBR, (R)-[11C]PK11195, обеспечивает общий индикатор воспаления центральной нервной системы (ЦНС). Несмотря на успешное применение (R)-[11C]PK11195, он имеет ограничения. Известно, что он обладает высоким связыванием с белком и связыванием от низкоспецифичного до неспецифичного (Lockhart et al. Nucl Med Biol. 30(2):199-206). Роль его меченных радиоизотопами метаболитов не известна, и для количественного определения связывания требуется сложное моделирование. Были попытки предложить соединения, обладающие высокой аффинностью и селективностью к PBR, чтобы обеспечить возможность улучшенного измерения PBR в ЦНС. [11C]DAA1106 и [18F]FЕОАА1106 представляют собой радиолиганды для PET на основе арилоксианилиновых соединений и были исследованы на людях (Ikomo et al. J.Cereb. Blood Flow Metab. 2007; 27:173-84 и Fujimura et al. J.Nuc. Med. 2006; 47:43-50). Однако кинетические свойства таких соединений не идеальны и могут ограничить их применение в количественных исследованиях. В попытке дополнительно улучшить такие радиолиганды Briard et al. (J.Med. Chem. 2008; 51:17-30) сообщили о другом арилоксианилиновом производном, PBR28. 11C-меченый вариант PBR28 инъецировали обезьянам для оценки его кинетики в головном мозге с использованием PET. [11C]PBR28 показал высокое поглощение головным мозгом, хорошее специфическое связывание с PBR-экспрессирующими тканями и кинетические свойства, более подходящие для визуализации in vivo. PBR-связывающие пиразолопиримидиновые соединения также оценивали в качестве РЕТ-радиолигандов для нацеливания на PBR. В James et al. (J. Nuc. Med. 2008; 49(5):814-22) сообщалось, что радиолиганд для PET, [18F]-DPA-714, обладает высокой аффинностью к PBR и селективным поглощением посредством PBR в головном мозге павиана после внутривенного введения. Сообщалось, что кинетика поглощения головным мозгом [18F]-DPA-714 медленнее чем, но подобна по характеру [11С]DAA1106 и [18F]FEDAA1106. В WO 2007/057705 раскрыты тетрациклические индольные соединения, меченные визуализирующей группировкой, которые являются подходящими для визуализации in vivo. Показано, что агенты визуализации in vivo, приведенные в примерах в WO 2007/057705, обладают хорошей аффинностью к PBR, со значениями K1 в конкурентном анализе с [3H]-PK-11195 от 1,0 нМ до 0,1 нМ. Однако в настоящее время авторы данного изобретения обнаружили, что селективность таких соединений в отношении PBR-экспрессирующих тканей не является идеальной для визуализации in vivo экспрессии PBR в центральной нервной системе.
Существуют пределы улучшения известных тетрациклических индольных соединений с целью получения альтернативных агентов визуализации in vivo для оценки экспрессии PBR в центральной нервной системе.
Краткое изложение сущности изобретения
В настоящем изобретении предлагаются агенты визуализации in vivo на основе тетрациклических индольных соединений. По сравнению с известными агентами визуализации in vivo на основе тетрациклических индольных соединений агенты визуализации in vivo по настоящему изобретению обладают лучшими свойствами для визуализации in vivo. Агенты визуализации in vivo по настоящему изобретению обладают подходящими свойствами связывания с периферическим бензодиазепиновым рецептором, а также хорошим поглощением в головном мозге и кинетикой in vivo после введения субъекту.
Краткое описание графических материалов
Фиг 1: Типичные ауторадиограммы, показывающие связывание агента 1 визуализации in vivo в поврежденном (справа) ядре лицевого нерва крысы с FNA (аксотомия лицевого нерва).
Фиг.2. Относительная эффективность связывания агента визуализации in vivo в ядре лицевого нерва крысы через семь дней после FNA. Данные представлены как среднее ±50 (стандартное отклонение) от 24 отдельных срезов у 1 животного.
Подробное описание изобретения
Агент визуализации in vivo
В одном аспекте настоящего изобретения предлагается агент визуализации in vivo Формулы I:
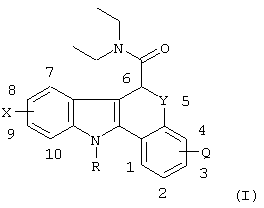
или его соль или сольват, где:
Q представляет собой водород или фтор;
Х представляет собой водород, фтор, бром, йод, гидрокси, C1-6алкил, C1-6галогеноалкил, C1-6алкокси или C1-6алкиламид;
Y представляет собой S, SO или SO2; и,
R представляет собой водород, C1-6алкил или C1-6фторалкил;
и где по меньшей мере один атом указанного агента визуализации in vivo Формулы I представляет собой радиоизотоп, подходящий для визуализации in vivo; где если указанный радиоизотоп представляет собой радиоизотоп углерода, то он является карбонильным углеродом;
при условии, что если Y представляет собой S, то Q или Х не являются оба водородом.
Термин "визуализация in vivo" при использовании в данном описании изобретения относится к тем способам, которые неинвазивно создают изображения всего или части внутреннего аспекта субъекта изобретения. Предпочтительные способы визуализации in vivo для использования в настоящем изобретении представляют собой однофотонную эмиссионную компьютерную томографию (SPECT) и позитрон-эмиссионную томографию (PET), причем PET является особенно предпочтительной. Преимущество для PET в способе по изобретению обусловлено ее превосходной чувствительностью и разрешением, так что даже относительно небольшие изменения в повреждении можно наблюдать в динамике по времени. Сканеры PET в плановом порядке измеряют концентрации радиоактивности в пикомолярном диапазоне. Сканеры микро-РЕТ в настоящее время приближаются к пространственному разрешению примерно 1 мм, а клинические сканеры - примерно 4-5 мм.
"Агент визуализации in vivo" Формулы I содержит радиоизотоп, подходящий для визуализации in vivo. Такой "радиоизотоп, подходящий для визуализации in vivo" представляет собой радиоизотопную форму одного из атомов, определенных выше, для агента визуализации in vivo Формулы I. Чтобы быть подходящим для визуализации in vivo, как определено в данном описании изобретения, радиоизотоп предпочтительно представляет собой гамма- или позитронный излучатель, тем самым делая возможным обнаружение агента визуализации in vivo снаружи от субъекта после введения.
Подходящие соли согласно изобретению включают (1) физиологически приемлемые соли присоединения кислоты, например полученные из неорганических кислот, например соляной, бромистоводородной, фосфорной, метафосфорной, азотной и серной кислот, и произведенные из органических кислот, например винной, трифторуксусной, лимонной, яблочной, молочной, фумаровой, бензойной, гликолевой, глюконовой, янтарной, метансульфоновой и пара-толуолсульфоновой кислот; и (2) физиологически приемлемые соли оснований, такие как соли аммония, соли щелочных металлов (например, соли натрия и калия), соли щелочноземельных металлов (например, соли кальция и магния), соли с органическими основаниями, такими как триэтаноламин, N-метил-D-глюкамин, пиперидин, пиридин, пиперазин и морфолин, и соли с аминокислотами, такими как аргинин и лизин.
Подходящие сольваты согласно изобретению включают сольваты, образованные с этанолом, водой, солевым раствором, физиологического буфера и гликоля.
Если не указано иное, термин "алкил" сам по себе или в комбинации означает алкильный радикал с прямой или разветвленной цепью, предпочтительно содержащий от 1 до 6 атомов углерода, наиболее предпочтительно от 1 до 4 атомов углерода. Примеры таких радикалов включают, без ограничения ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил.
Если не указано иное, термин "алкокси" сам по себе или в комбинации означает алкилэфирный радикал, где термин алкил такой, как определено выше. Примеры подходящих алкилэфирных радикалов включают, без ограничения ими, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изо-бутокси, втор-бутокси, трет-бутокси.
"Алкиламид" представляет собой алкильную группу, как определено выше, связанную с амидом, где амид представляет собой группу -C(=O)-NR'R” где R' и R” представляют собой независимо водород или углеводородный радикал.
Термин "галоген" или "галогено-" означает заместитель, выбранный из фтора, хлора, брома или йода. "Галогеноалкил" представляет собой алкильную группу, как определено выше, замещенную одним или более галогеном.
Термин "гидрокси" относится к радикалу -ОН.
В предпочтительном воплощении Q представляет собой водород.
Х предпочтительно представляет собой водород, фтор, бром, йод, гидрокси, С1-4алкил, C1-4галогеноалкил, C1-4алкокси или C1-4алкиламид. Х наиболее предпочтительно представляет собой водород или С1-4алкокси.
Y предпочтительно представляет собой S или SO2. Y наиболее предпочтительно представляет собой S.
R предпочтительно представляет собой водород, С1-4алкил или C1-4фторалкил. R наиболее предпочтительно представляет собой С1-4фторалкил.
В предпочтительном воплощении Формулы I:
Х представляет собой водород, фтор, бром, йод, гидрокси, С1-4алкил, C1-4галогеноалкил, C1-4алкокси или C1-4алкиламид;
Y представляет собой S или SO2; и
R представляет собой водород, С1-4алкил или С1-4фторалкил.
В наиболее предпочтительном воплощении Формулы I:
Q представляет собой водород;
Х представляет собой С1-4алкокси;
Y представляет собой S; и
R представляет собой С1-4фторалкил.
В альтернативном предпочтительном воплощении Формулы I:
Q представляет собой фтор;
Х представляет собой водород;
Y представляет собой S; и
R представляет собой С1-4фторалкил.
Предпочтительные радиоизотопы, подходящие для визуализации in vivo по настоящему изобретению, представляют собой гамма-излучающие радиоактивные галогены и позитрон-излучающие радиоактивные неметаллы.
Примерами гамма-излучающих радиоактивных галогенов, подходящих для применения в настоящем изобретении, являются 123I, 131I и 77Br. Предпочтительным гамма-излучающим радиоактивным галогеном является 123I.
Примерами позитрон-излучающего радиоактивного неметалла, подходящего для применения в настоящем изобретении, являются 11С, 13N, 18F и 124I. Предпочтительным позитрон-излучающим радиоактивным неметаллом является 18F. 18F является наиболее предпочтительным радиоизотопом, подходящим для визуализации in vivo по настоящему изобретению.
В предпочтительном воплощении для агента визуализации in vivo Формулы I Х представляет собой 123I, 124I или 131I, 18F или C1-4[18F]-фторалкил.
В альтернативном предпочтительном воплощении для агента визуализации in vivo Формулы I R представляет собой C1-4[18F]-фторалкил.
В еще одном альтернативном предпочтительном воплощении для агента визуализации in vivo Формулы I карбонильный углерод представляет собой 11С.
Неограничивающими примерами некоторых предпочтительных агентов визуализации in vivo по настоящему изобретению являются следующие:

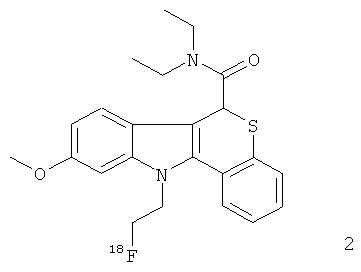
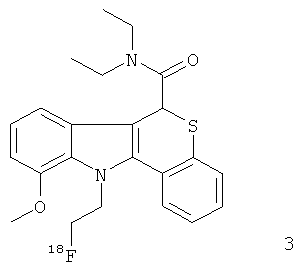
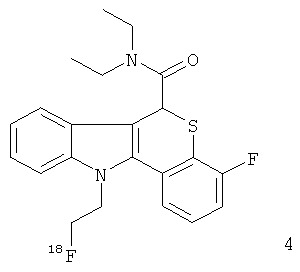
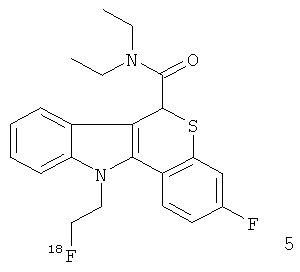
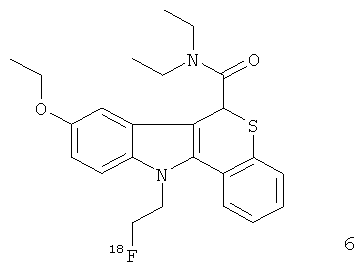

Из рассмотренных выше агентов визуализации in vivo агент визуализации 2 является наиболее предпочтительным.
Способы синтеза, используемые для получения этих агентов визуализации in vivo, описаны в экспериментальном разделе ниже. Эффективность этих нерадиоактивных вариантов агентов визуализации in vivo по настоящему изобретению измеряли в анализе in vitro, как описано в Примере 10.
В Примерах 7-9 описано, как получить радиофторированные агенты визуализации in vivo 1-7. Специалисту известно, что при обращении с 18F масштаб и используемые условия являются различными исходя из безопасности и практических соображений. Обзор получения 18F PET индикаторов см. в главах 1 и 2 of "Principles and Practice of Positron Emission Tomography" (2002 Lippincott Williams & Wilkins; Wahl and Buchanan, Eds.). Агенты визуализации in vivo испытывали на животной модели биораспределения (Пример 11) и их биораспределение сравнивали с биораспределением соединения [18F]FE-PBR из уровня техники (полученного согласно Примеру 14 в WO 2007/057705):

В Таблице 1 ниже представлены данные, полученные в анализе аффинности in vitro, а также в изучении биораспределения in vivo. Нерадиоактивные аналоги испытывали в анализе аффинности in vitro, и меченные радиоизотопом варианты оценивали в анализе биораспределения.
Данные иллюстрируют, что эффективность нерадиоактивных вариантов агентов визуализации in vivo 1-7 выгодно отличается от соединения из уровня техники, [18F]FE-PBR. Кроме того, данные показывают, что агенты визуализации in vivo 1-7 по изобретению удерживаются значительно дольше в 0 В по сравнению с полосатым телом через 30 минут после инъекции по сравнению с [18F]FE-PBR]. Известно, что PBR в значительных количествах экспрессируется в OB по сравнению с другими областями головного мозга крыс (см. "Handbook of Substance Abuse", Tarter, Ammerman and Ott; Springer 1998:398-99), эти данные неожиданно демонстрируют, что агенты визуализации in vivo 1-7 обладают улучшенной селективностью в отношении PBR, чем ранее приведенный в качестве примера агента визуализации in vivo [18F]FE-PBR.
Агент визуализации in vivo 1 дополнительно анализировали в ауторадиографической модели, как описано в Примере 12 ниже. Значительно более высокие уровни радиоактивности были обнаружены на поврежденной области ядра лицевого нерва (см. Фиг.1 и 2). Средняя интенсивность в поврежденной области составляла 7,75±0,95 по сравнению с 3,73±0,36 в неповрежденной области. Соотношение между двумя областями составляло 8,23±2,36. Так как повреждение имело более высокую экспрессию PBR по сравнению с нормой, эти данные подтверждают вывод, сделанный из данных по биораспределению, что агент визуализации in vivo 1 обладает хорошей селективностью в отношении PBR.
Следовательно, агенты визуализации in vivo по настоящему изобретению обладают неожиданно лучшими свойствами в отношении визуализации in vivo PBR по сравнению с известными PBR-связывающими агентами визуализации in vivo на основе тетрациклического индола.
Соединение-предшественник
В другом аспекте настоящего изобретения предлагается соединение-предшественник Формулы II:
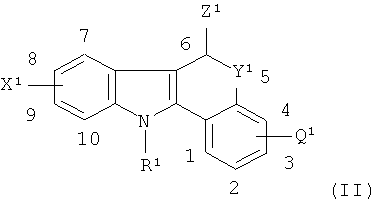
где один из R1, X1 или Z1 содержит химическую группу, которая взаимодействует с подходящим источником радиоизотопа, где указанный радиоизотоп является таким, как это целесообразно и предпочтительно определено в данном описании изобретения, так что агент визуализации in vivo, как целесообразно и предпочтительно определено в данном описании изобретения, образуется при взаимодействии указанного соединения-предшественника с указанным подходящим источником указанного радиоизотопа;
и где:
если R1 не содержит указанную химическую группу, то он является таким, как целесообразно и предпочтительно определено в данном описании изобретения для R в Формуле I, и, возможно, дополнительно содержит защитную группу;
если X1 не содержит указанную химическую группу, то он является таким, как целесообразно и предпочтительно определено в данном описании изобретения для Х Формулы I, и, возможно, дополнительно содержит защитную группу;
если Z1 не содержит указанную химическую группу, то он представляет собой -С(=O)-N-(СН2-СН3)2 и, возможно, дополнительно содержит защитную группу;
Q1 является таким, как целесообразно и предпочтительно определено в данном описании изобретения для Q в Формуле I; и
Y1 является таким, как целесообразно и предпочтительно определено в данном описании изобретения для Y в Формуле I, и, возможно, дополнительно содержит защитную группу.
"Соединение-предшественник" содержит производное меченного радиоизотопом соединения, сконструированное так, что химическое взаимодействие с подходящей химической формой детектируемой метки происходит сайт-специфически; может быть проведено с минимальным количеством стадий (идеально в одну стадию); и без необходимости в значительной очистке (идеально без дополнительной очистки), с получением целевого агента визуализации in vivo. Такие соединения-предшественники являются синтетическими и могут быть легко получены с хорошей химической чистотой. Соединение-предшественник, возможно, может содержать защитную группу для некоторых функциональных групп соединения-предшественника.
Термин "защитная группа" означает группу, которая ингибирует или сдерживает нежелательные химические взаимодействия, но которую конструируют достаточно реакционноспособной, чтобы ее можно было отщеплять от функциональной группы, о которой идет речь, при достаточно мягких условиях, чтобы не модифицировать остальную часть молекулы. После снятия защиты получают целевой продукт. Защитные группы хорошо известны специалисту в данной области техники и подходящим образом выбираются из: для аминогрупп - Вос (где Вос представляет собой трет-бутилоксикарбонил), Fmoc (где Fmoc представляет собой флуоренилметоксикарбонил), трифторацетил, аллилоксикарбонил, Dde [то есть 1-(4,4-диметил-2,6-диоксоциклогексилиден)этил] или Npys (то есть 3-нитро-2-пиридинсульфенил); и для карбоксильных групп - метиловый эфир, трет-бутиловый эфир или бензиловый эфир. Для гидроксильных групп подходящие защитные группы представляют собой: метил, этил или трет-бутил; алкоксиметил или алкоксиэтил; бензил; ацетил; бензоил; тритил (Trt) или триалкилсилил, такой как тетрабутилдиметилсилил. Применение дополнительных защитных групп описано в «Protective Groups in Organic Synthesis», Theorodora W. Greene and Peter G.M. Wuts (Third Edition, John Wiley & Sons, 1999).
Термин "подходящий источник радиоизотопа" означает радиоизотоп в химической форме, которая вступает в реакцию с заместителем соединения-предшественника, так что радиоизотоп ковалентно присоединяется к соединению-предшественнику.
Для каждого конкретного радиоизотопа, присутствующего в следующем разделе, обсуждается один или более подходящих источников этого радиоизотопа. Специалист в области агентов визуализации in vivo знаком с этими и другими источниками радиоизотопов, которые подходят для применения в настоящем изобретении.
Когда радиоизотоп агента визуализации in vivo представляет собой 18F, атом радиофтора может образовывать часть фторалкильной или фторалкоксигруппы, так как алкилфториды устойчивы к метаболизму in vivo. Альтернативно, атом радиофтора может присоединяться с помощью прямой ковалентной связи с ароматическим кольцом, таким как бензольное кольцо.
Радиофторирование можно выполнять с помощью прямого введения метки, используя взаимодействие 18F-фторида с подходящей химической группой в соединении-предшественнике, имеющем подходящую уходящую группу, такую как алкилбромид, алкилмезилат или алкилтозилат.
18F также может быть введен посредством О-алкилирования гидроксильных групп с помощью 18F(CH2)3OMs или 18F(CH2)3Br.
Для арильных систем 18F-фторидное нуклеофильное замещение из соли арилдиазония, арильного нитросоединения и/или арильной четвертичной аммониевой соли являются подходящими путями к арил-18F производным. Такая стратегия является подходящей, например, для введения 18F в положения 1-4 или 7-10 Формулы I.
Альтернативно, введение метки 18F может быть достигнуто посредством нуклеофильного замещения уходящей группы из производного Формулы I. Подходящие уходящие группы включают Cl, Br, I, тозилат (OTs), мезилат (OMs) и трифлат (OTf). Такие производные являются соединениями-предшественниками для получения соединений для визуализации in vivo no изобретению.
Другой стратегией будет иметь подходящую уходящую группу, как определено выше, на месте алкиламидной группы, присутствующей в соединении-предшественнике. В данном способе можно вводить метку в соединение-предшественник в одну стадию посредством взаимодействия с подходящим источником [18F]фторид-иона (18F-), который обычно получают в виде водного раствора из ядерной реакции 18O(p,n)18F и делают реакционно-способным путем добавления катионного противоиона и последующего удаления воды. Для данного способа соединения-предшественники обычно избирательно защищают химически так, чтобы радиофторирование имело место в конкретном месте соединения. Подходящие защитные группы уже упоминались ранее.
Если радиоизотоп представляет собой 18F, то предпочтительно, чтобы или X1, или R1 содержал или:
(1) алкилгалогенид или алкилсульфонат (такой, как алкилбромид, алкилмезилат или алкилтозилат) для нуклеофильного замещения; или
(2) гидроксил (для введения 18F посредством O-алкилирования гидроксильных групп с помощью, например, 18F(CH2)3OMs или 18F(CH2)3Br).
Типичная схема взаимодействия для получения некоторых 18F-агентов визуализации in vivo по изобретению проиллюстрирована ниже:

где Х такой, как определено для Формулы I, и n равен от 0 и до 5. К.т. означает комнатную температуру, и OMs означает мезилат.
Альтернативная общая схема взаимодействия для получения некоторых 18F-агентов визуализации in vivo по изобретению проиллюстрирована ниже:

где X такой, как определено для Формулы I, и n равен от 0 и до 5, и OTs означает тозилат.
11C-меченые индикаторные соединения для PET можно синтезировать путем взаимодействия соединения-предшественника с 11C-метилйодидом. Так как период полураспада 11С составляет только 20,4 минут, важно, чтобы промежуточный 11C-метилйодид имел высокую удельную активность и, следовательно, чтобы его получали, используя способ взаимодействия, который является настолько быстрым, насколько это возможно. Детальное рассмотрение таких способов введения метки 11С можно найти в Antoni et al. "Aspects on the Synthesis of 11C-Labelled Compounds" в Handbook of Radiopharmaceuticals, Ed.M.J.Welch and C.S.Redvanly (2003, John Wiley and Sons).
Если агент визуализации in vivo по настоящему изобретению помечен при помощи 11С, то 11С представляет собой карбонильный углерод. Следовательно, это означает, что 11С может присутствовать на месте карбонильного углерода в Формуле I или альтернативно в X, когда Х представляет собой C1-6алкиламид.
11C-меченый агент визуализации in vivo Формулы I может быть получен с использованием следующей схемы взаимодействия:

где R3, X3 и Y3 в Формуле IIb и Формуле Id такие, как описано для R, Х и Y из Формулы I; и
Z3 представляет собой субстрат, подходящий для катализаторов - переходных металлов, например водород, галогенид, бороновую кислоту, OTf, оловоорганическое соединение.
Способы синтеза 13N-меченых соединений описаны в Clark and Aigbirhio ("Chemistry of Nitrogen-13 and Oxygen-15" в "Handbook of Radiopharmaceuticals"; 2003 Wiley: Welch and Redvanly, Eds.). Например, агент визуализации in vivo Формулы I может быть получен посредством нуклеофильного замещения галогена в подходящем соединении-предшественнике с 13N-меченым диэтиловым амином с получением целевого амида.
Если визуализирующая группировка представляет собой радиойод, предпочтительными соединениями-предшественниками являются такие, которые содержат производное, которое либо подвергается электрофильному или нуклеофильному йодированию, либо подвергается конденсации с меченым альдегидом или кетоном. Примерами первой категории являются:
а) металлорганические производные, такие как триалкилстаннан (например, триметилстаннил или трибутилстаннил), или триалкилсилан (например, триметилсилил), или борорганическое соединение (например, боронатные эфиры или органотрифторбораты);
б) ароматические кольца, активированные в отношении электрофильного йодирования (например, фенолы) и ароматические кольца, активированные в отношении нуклеофильного йодирования (например, соль арил-йодония, арил-диазония, соли арил-триалкиламмония или нитроарильные производные).
Для радиойодирования соединение-предшественник предпочтительно содержит: арилйодид или -бромид (для разрешения обмена радиойода); арильное кольцо активированного соединения-предшественника (например, фенольную группу); металлорганическое соединение-предшественник (например, триалкилолово, триалкилсилил или борорганическое соединение); или органическое соединение-предшественник, такое как триазены, или подходящую уходящую группу для нуклеофильного замещения, такую как соль йодония. Соединения-предшественники и способы введения радиойода в органические молекулы описаны в Bolton (J.Lab.Comp. Radiopharm. 2002; 45:485-528). Соединения-предшественники и способы введения радиойода в белки описаны в Wilbur (Bioconj. Chem. 1992; 3(6):433-470). Подходящие борорганические соединения боронатных эфиров и их получение описаны в Kabalaka et al. (Nucl. Med. Biol., 2002; 29:841-843 и 2003; 30:369-373). Подходящие органотрифторбораты и их получение описаны в Kabalaka et al. (Nucl. Med. Biol., 2004; 31:935-938). Предпочтительные соединения-предшественники для радиойодирования содержат металлорганическое соединение-предшественник, наиболее предпочтительно триалкилолово.
Примеры арильных групп, к которым может быть присоединен радиоактивный йод, представлены ниже:
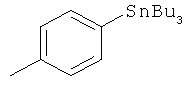
Обе группы содержат заместители, которые обеспечивают возможность легкого замещения радиойодом на ароматическом кольце. Альтернативные заместители, содержащие радиоактивный йод, можно синтезировать путем непосредственного йодирования с помощью обмена радиогалогена, например

Если радиоизотоп представляет собой радиойод, то X1 в Формуле II вместе с ароматической группой, к которой он присоединен, образует:
1) ароматическое кольцо, замещенное или металлорганическим производным, или борорганическим соединением;
2) ароматическое кольцо, активированное в отношении электрофильного радиойодирования (например, фенолы); или
3) ароматическое кольцо, активированное в отношении нуклеофильного радиойодирования (например, соль арилйодония, арилдиазоний, соли арилтриалкиламмония или нитроарильные производные).
Такие соединения-предшественники легко превращаются в радиойодированные агенты визуализации in vivo по изобретению посредством замещения радиойодом.
Радиобромирование может быть достигнуто способами, аналогичными описанным выше для радиойодирования. Kabalka и Varma сделали обзор различных способов синтеза радиогалогенированных соединений, включая радиобромированные соединения (Tetrahedron 1989; 45(21): 6601-21).
Соединение-предшественник по изобретению в идеале предлагается в стерильной, апирогенной форме. Соединение-предшественник, таким образом, можно использовать для получения фармацевтической композиции, содержащей агент визуализации in vivo вместе с биосовместимым носителем, подходящим для введения млекопитающему. Соединение-предшественник также является подходящим для включения в качестве компонента в набор для получения такой фармацевтической композиции.
В предпочтительном воплощении соединение-предшественник предлагается в растворе и в виде части набора или кассеты, сконструированной для применения в приборе для автоматизированного синтеза. Эти аспекты обсуждаются более подробно ниже по отношению к дополнительным аспектам изобретения.
В другом предпочтительном воплощении соединение-предшественник связано с твердой фазой. Соединение-предшественник предпочтительно поставляют ковалентно соединенным с твердой матрицей-подложкой. Таким способом целевой продукт образуется в растворе, в то время как исходные вещества и примеси остаются связанными с твердой фазой. В качестве примера такой системы в WO 03/002489 описаны соединения-предшественники для твердофазного электрофильного фторирования 18F-фтором, и в WO 03/002157 описаны соединения-предшественники для твердофазного нуклеофильного фторирования 18F-фтором.
Способ получения агента визуализации in vivo
В еще одном аспекте настоящего изобретения предлагается способ получения агента визуализации in vivo по изобретению, где указанный способ включает:
1) получение соединения-предшественника Формулы II, как определено выше;
2) получение подходящего источника указанного радиоизотопа, как определено выше;
3) взаимодействие соединения-предшественника со стадии (1) с радиоизотопом со стадии (2) с получением агента визуализации in vivo по изобретению.
На стадии (1) соединение-предшественник может быть представлено в растворе в наборе или в кассете, подходящих для использования с прибором для автоматизированного синтеза, или, альтернативно, присоединенным к твердой подложке, как описано выше в описании соединения-предшественника. Набор и кассета образуют дополнительные аспекты изобретения и подробно обсуждаются ниже.
Подходящими источниками радиоизотопа являются такие, как описано выше в отношении соединения-предшественника по изобретению.
Стадия "взаимодействия" соединения-предшественника с радиоизотопом охватывает объединение вместе двух реагентов в условиях взаимодействия, подходящих для образования целевого агента визуализации in vivo с насколько возможно высоким радиохимическим выходом (RCY). Конкретные пути синтеза для получения агентов визуализации in vivo по настоящему изобретению представлены в экспериментальном разделе ниже.
Набор для получения агента визуализации in vivo
В еще одном аспекте настоящего изобретения предлагается набор для получения агента визуализации in vivo по изобретению, где указанный набор содержит соединение-предшественник Формулы II, как описано выше, такой, что взаимодействие со стерильным источником радиоизотопа дает целевой агент визуализации in vivo с минимальным количеством манипуляций. Такие соображения особенно важны, когда радиоизотоп имеет относительно короткий период полураспада и для облегчения обработки и, следовательно, пониженной дозы радиации для радиофармацевта. Соединение-предшественник предпочтительно присутствует в наборе в лиофилизированной форме, и реакционная среда для восстановления в таких наборах предпочтительно представляет собой биосовместимый носитель.
"Биосовместимый носитель" представляет собой жидкую среду, в частности жидкость, в которой суспендируют или растворяют агент визуализации in vivo, так что композиция является физиологически переносимой, то есть может быть введена в организм млекопитающего без токсичности или чрезмерного дискомфорта. Биосовместимый носитель является подходящей инъецируемой жидкостью-носителем, такой как стерильная апирогенная вода для инъекций; водный раствор, такой как солевой раствор (который можно успешно уравновешивать так, чтобы конечный продукт для инъекции являлся или изотоническим, или негипотоническим); водный раствор одного или более регулирующих тоничность веществ (например, солей катионов плазмы с биосовместимыми противоионами), сахара (например, глюкоза или сахароза), сахарные спирты (например, сорбитол или маннитол), гликоли (например, глицерин) или другие неионные полиоловые вещества (например, полиэтиленгликоли, пропиленгликоли и тому подобное). Биосовместимый носитель может также содержать биосовместимые органические растворители, такие как этанол. Такие органические растворители являются полезными для солюбилизации более липофильных соединений или композиций. Предпочтительно биосовместимый носитель представляет собой апирогенную воду для инъекций, изотонический солевой раствор или водный раствор этанола. рН биосовместимого носителя для внутривенной инъекции подходящим образом находится в диапазоне от 4,0 до 10,5.
В наборе по изобретению соединение-предшественник предпочтительно присутствует в герметично закрытом контейнере, что позволяет сохранять стерильность и/или радиоактивную безопасность плюс, возможно, инертный газ (например, азот или аргон) в свободном пространстве над продуктом, в то же время обеспечивая возможность добавления и извлечения растворов с помощью шприца. Предпочтительный герметично закрытый контейнер представляет собой запечатанный мембраной флакон, где газонепроницаемую крышку обжимают дополнительным укупорочным средством (обычно из алюминия). Дополнительным преимуществом таких герметично закрытых контейнеров состоит в том, что крышка может выдерживать вакуум, если это желательно, например, для замены газа в свободном пространстве над продуктом или дегазирования растворов.
Предпочтительные воплощения соединения-предшественника при использовании в наборе такие, как описано в данном описании изобретения.
Соединение-предшественник для применения в наборе можно использовать в асептических условиях изготовления с получением целевого стерильного непирогенного вещества. Соединение-предшественник можно альтернативно использовать в нестерильных условиях с последующей термической стерилизацией с использованием, например, гамма-излучения, автоклавирования, сухого тепла или химической обработки (например, этиленоксидом). Предпочтительно соединение-предшественник представлено в стерильной апирогенной форме. Наиболее предпочтительно стерильное апирогенное соединение-предшественник представлено в герметично закрытом контейнере, как описано выше.
Предпочтительно все компоненты набора являются одноразовыми, чтобы минимизировать возможности загрязнения между прогонами и гарантировать стерильность и обеспечение качества.
В предпочтительном воплощении набор может содержать кассету, которая может быть вставлена в подходящим образом адаптированный автоматизированный синтезатор, описанный более подробно ниже. Такой набор обычно включает средства для фторирования фторид-ионом и также может содержать колонку для удаления нежелательных фторид-ионов. Реагенты, растворители и другие расходные материалы, требующиеся для синтеза, можно также включать вместе с носителем данных, таким как компакт-диск с записанным программным обеспечением, что позволяет управлять автоматизированным синтезаторам так, чтобы отвечать требованиям конечного пользователя в отношении концентрации, объема, времени доставки и так далее.
[18F]-Радиометки для РЕТ-визуализации в настоящее время часто легко получают на приборе для автоматизированного радиосинтеза. В продаже имеется несколько примеров такого прибора, включая Tracerlab и Fastlab (GE Heathcare). Такой прибор обычно содержит "кассету", часто одноразовую, в которой осуществляют радиохимию, которую устанавливают в прибор для осуществления радиосинтеза. Кассета обычно включает каналы для текучих веществ, реакционный сосуд и отверстия для принимающих реагент флаконов, а также любые картриджи для твердофазной экстракции, используемые на стадиях очистки после радиосинтеза.
Следовательно, в другом аспекте настоящего изобретения предлагается кассета для прибора автоматизированного синтеза, содержащая соединение-предшественник в герметично закрытом контейнере, как описано выше в данном описании изобретения. В настоящем изобретении также предлагается кассета для автоматизированного синтеза агента визуализации in vivo, как определено в данном описании изобретения, содержащая:
1) сосуд, содержащий соединение-предшественник, как определено в данном описании изобретения; и
2) средства для элюирования сосуда подходящим источником радиоизотопа, где указанный радиоизотоп такой, как определено в данном описании изобретения.
Кассета дополнительно может содержать:
3) ионообменный картридж для удаления избытка радиометки; и, возможно,
4) картридж для удаления защиты с полученного меченного радиоизотопом продукта с образованием агента визуализации in vivo, как определено в данном описании изобретения.
Радиофармацевтическая композиция
В еще одном аспекте настоящего изобретения предлагается "радиофармацевтическая композиция", которая представляет собой композицию, содержащую агент визуализации in vivo по изобретению вместе с биосовместимым носителем в форме, подходящей для введения млекопитающему. Биосовместимый носитель является таким, как определено выше в отношении набора по изобретению.
Радиофармацевтическую композицию можно вводить парентерально, то есть посредством инъекции, и она наиболее предпочтительно представляет собой водный раствор. Такая композиция, возможно, может содержать дополнительные ингредиенты, такие как буферы; фармацевтически приемлемые солюбилизаторы (например, циклодекстрины или поверхностно-активные вещества, такие как Pluronic, Твин или фосфолипиды); фармацевтически приемлемые стабилизаторы или антиоксиданты (такие, как аскорбиновая кислота, гентизиновая кислота или пара-аминобензойная кислота). Когда агент визуализации in vivo по изобретению представлен в виде радиофармацевтической композиции, способ получения указанного агента визуализации in vivo может дополнительно содержать стадии, требующиеся для получения радиофармацевтической композиции, например удаление органического растворителя, добавление биосовместимого буфера и любых возможных дополнительных ингредиентов. Для парентерального введения также следует осуществлять стадии для обеспечения стерильности и апирогенности радиофармацевтической композиции.
Способ визуализации in vivo
В еще одном аспекте настоящего изобретения предлагается способ визуализации in vivo для определения распределения и/или величины экспрессии PBR у субъекта, включающий:
1) введение указанному субъекту агента визуализации in vivo, как определено в данном описании изобретения;
2) обеспечение возможности связывания указанного агента визуализации in vivo с PBR указанного субъекта;
3) обнаружение посредством способа визуализации in vivo сигналов, испускаемых радиоизотопом указанного агента визуализации in vivo;
4) формирование визуального представления расположения и/или величины указанных сигналов; и
5) определение распределения и величины экспрессии PBR у указанного субъекта, где указанная экспрессия прямо коррелирует с указанными сигналами, испускаемыми указанным агентом визуализации in vivo.
Для способа визуализации in vivo по изобретению агент визуализации in vivo является таким, как определено ранее в описании.
"Введение" агента визуализации in vivo предпочтительно осуществляют парентерально и наиболее предпочтительно внутривенно. Внутривенный путь представляет собой наиболее эффективный способ доставки агента визуализации in vivo по всему организму субъекта и, следовательно, также через гематоэнцефалический барьер (ВВВ) и в контакт с PBR, экспрессируемыми в указанном субъекте. Агент визуализации in vivo по изобретению предпочтительно вводят в виде фармацевтической композиции по изобретению, как определено в данном описании изобретения.
После стадии введения и до стадии обнаружения агента визуализации in vivo оставляют для связывания с PBR. Например, когда субъект представляет собой необработанное млекопитающее, агент визуализации in vivo динамически перемещается по организму млекопитающего, вступая в контакт с различными тканями в нем. Как только агент визуализации in vivo вступает в контакт с PBR, имеет место специфическое взаимодействие такое, что клиренс агента визуализации in vivo из ткани с PBR осуществляется дольше, чем из ткани без PBR или с небольшим количеством PBR. Будет достигнут определенный момент времени, когда обнаружение агента визуализации in vivo, специфически связанного с PBR, становится возможным в виде результата соотношения между агентом визуализации in vivo, связанным с тканью с PBR, против связанного в ткани без PBR или с небольшим количеством PBR. Такое идеальное соотношение составляет около 2:1.
Стадия "обнаружения" способа по изобретению включает обнаружение сигналов, испускаемых радиоизотопом, посредством детектора, чувствительного к указанным сигналам. Данную стадию обнаружения можно также понимать как получение данных сигнала. Однофотонная эмиссионная томография (SPECT) и позитрон-эмиссионная томография (PET) являются наиболее подходящими методами визуализации in vivo для применения в способе по изобретению. PET является предпочтительным методом визуализации in vivo для применения в способе по изобретению.
Стадию "формирования" способа по изобретению выполняют с помощью компьютера, который применяет алгоритм реконструкции к полученным данным сигнала с получением базы данных. Затем этой базой данных манипулируют для формирования изображений, показывающих расположение и/или количество сигналов, испускаемых указанным радиоизотопом. Испускаемые сигналы непосредственно коррелируют с экспрессией PBR, так что стадию "определение" можно выполнять, оценивая сформированное изображение.
"Субъект" по изобретению может являться любым человеком или животным. Предпочтительно субъект по изобретению представляет собой млекопитающее. Наиболее предпочтительно указанный субъект представляет собой организм необработанного млекопитающего in vivo. В особенно предпочтительном воплощении субъект по изобретению является человеком. Способ визуализации in vivo можно использовать для изучения PBR у здоровых субъектов или у субъектов, про которых известно или предполагается, что они имеют патологическое состояние, ассоциированное с аномальной экспрессии PBR ("PBR-состояние"). Предпочтительно указанный способ относится к визуализации in vivo субъекта, про которого известно или предполагается, что он имеет PBR-состояние и, следовательно, для него полезен способ диагностики указанного состояния. Примеры таких PBR-состояний, при которых может быть использована визуализация in vivo, включают невропатологии, такие как болезнь Паркинсона, рассеянный склероз, болезнь Альцгеймера и болезнь Хантингтона, где присутствует нейровоспаление. Другие PBR-состояния, которые можно с пользой визуализировать с помощью соединений по изобретению, включают невропатическую боль, артрит, астму, атеросклероз, а также злокачественные заболевания, такие как колоректальный рак и рак молочной железы. Агенты визуализации in vivo по изобретению особенно пригодны для визуализации in vivo центральной нервной системы (ЦНС) вследствие их хорошего поглощения головным мозгом.
В альтернативном воплощении способ визуализации in vivo по изобретению можно выполнять неоднократно во время прохождения схемы лечения указанного субъекта, где указанная схема включает введение лекарственного средства для борьбы с PBR-состоянием. Например, способ визуализации in vivo по изобретению может быть осуществлен до, во время и после лечения лекарственным средством для борьбы с PBR-состоянием. Таким образом, за эффектом указанного лечения можно наблюдать во времени. Предпочтительно для данного воплощения способом in vivo визуализации является PET. PET имеет исключительную чувствительность и разрешение, так что даже относительно небольшие изменения в повреждении могут наблюдаться во времени, что является полезным для мониторинга лечения. Сканеры PET обычно измеряют концентрации радиоактивности в пикомолярном диапазоне. В настоящее время сканеры микро-РЕТ приближаются к пространственному разрешению примерно 1 мм, а клинические сканеры - примерно 4-5 мм.
В еще одном аспекте настоящего изобретения предлагается способ диагностики PBR-состояния. Способ диагностики по изобретению включает способ визуализации in vivo, как определено выше, вместе с дополнительной стадией (6) отнесения распределения и величины экспрессии PBR к конкретной клинической картине, то есть дедуктивной фазой принятия медицинского решения.
В еще одном аспекте настоящего изобретения предлагается агент визуализации in vivo, как он определен в данном описании изобретения, для применения в способе диагностики, как определено в данном описании изобретения.
В еще одном аспекте настоящего изобретения предлагается агент визуализации in vivo, как он определен в данном описании изобретения, для применения в изготовлении радиофармацевтической композиции, как она определена в данном описании изобретения, для применения в способе диагностики, как он определен в данном описании изобретения.
Краткое описание Примеров
Все реагенты получали от Sigma Aldrich.
В Примерах 1-6 описан синтез нерадиоактивных вариантов различных агентов визуализации in vivo по изобретению.
В Примерах 7-9 описано, как получать 18F-меченые агенты визуализации in vivo по изобретению.
В Примере 10 описан анализ эффективности in vitro, используемый для измерения PBR-аффинности агентов визуализации по изобретению.
В Примере 11 описано, как выполняли исследования биораспределения на животных.
В Примере 12 описана животная модель аксотомии лицевого нерва и ее использование в изучении ауторадиографии.
Перечень сокращений, используемых в Примерах
Примеры
Пример 1: Получение диэтиламида (+-)-11-(2-фторэтил)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]-флуорен-6-карбоновой кислоты (нерадиоактивный агент визуализации 1)
Пример 1(1): диэтиламид (+-)-4-оксо-тиохроман-2-карбоновой кислоты

(+-)-4-Оксо-тиохроман-2-карбоновую кислоту (10,4 г, 50 ммоль), полученную как описано в Т.Okubo et al. (Bioorg. Med. Chem. 2004; 12:3569-3580), в безводном DCM (100 мл), перемешивали атмосфере азота при комнатной температуре с оксалилхлоридом (12,6 г, 100 ммоль) и одной каплей DMF в течение 18 ч. Затем реакционную смесь упаривали в вакууме с получением смолы и затем снова растворяли в DCM (100 мл), охлаждали до 0°С на ледяной бане, перемешивали и обрабатывали по каплям диэтиламином (8,03 г, 110 ммоль) в DCM (20 мл) в течение 1 ч. Реакционную смесь оставляли нагреваться до комнатной температуры в течение 1 ч, добавляли 10%-ный водный раствор карбоната калия (100 мл) и реакционную смесь интенсивно перемешивали. Раствор DCM отделяли. Водный раствор экстрагировали еще двумя порциями DCM (100 мл) и объединенные экстракты сушили над сульфатом магния. Раствор DCM концентрировали в вакууме с получением темно-зеленого масла, которое кристаллизовалось при стоянии. Кристаллическое твердое вещество растирали с диэтиловым эфиром (50 мл) и фильтровали с получением указанного в заголовке соединения (8,57 г, 65%) в виде бледно-зеленого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 1.06 (t, J=7,1 Гц, 3Н), 1.23 (t, J=7,1 Гц, 3Н), 3.0-3.5 (m, 6H), 4.25 (m, 1H), 7.15-7.21 (m, 2H), 7.32-7.39 (m, 1H), 8.10-8.14 (m, 1H).
13С ЯМР (75 МГц, CDCl3) δ 12.9, 14.8, 40.1, 40.7, 42.3, 42.5, 125.8, 127.2, 128.7, 130.8, 133.4, 137.9, 167.9, 193.1
Пример 1(2): Диэтиламид (+-)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]-флуорен-6-карбоновой кислоты

К раствору диэтиламида (+-)-4-оксо-тиохроман-2-карбоновой кислоты (1,32 г, 5,0 ммоль; Пример 1(1)) и 4-метоксифенил-гидразина гидрохлорида (0,87 г, 5,0 ммоль) в этаноле (10 мл) добавляли концентрированную серную кислоту (0,73 мл, 1,35 г, 13,8 ммоль) в атмосфере азота. Реакционную смесь нагревали при температуре дефлегмации в течение 24 ч. После охлаждения реакционную смесь фильтровали, твердое вещество промывали этанолом, сушили в вакууме (45°С) с получением указанного в заголовке соединения (1,05 г, 57%) в виде бледно-желтого твердого вещества.
1H ЯМР (300 МГц, DMSO-d6) δ 0.97 (t, J=6,8 Гц, 3Н), 1.28 (t, J=6,8 Гц, 3Н), 3.25 (m, 2H), 3.60 (m, 2H), 3.74 (s, 3Н), 5.59 (s, 1H), 6.80 (m, 2H), 7.10-7.35 (m, 4H), 7.75 (d, J=7,3 Гц, 1H), 11.52 (s, 1H, NH).
13С ЯМР (75 МГц, DMSO-d6) δ 10.5, 12.7, 32.7, 37.9, 39.5, 53.0, 97.6, 103.3, 109.87, 109.92, 120.3, 123.5, 123.8, 124.3, 124.7, 124.9, 127.8, 129.4, 131.8, 151.3, 166.2
m/z (ES+) 367,1 (M+H).
Пример 1(3): Диэтиламид (+-)-11-(2-фторэтил)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты
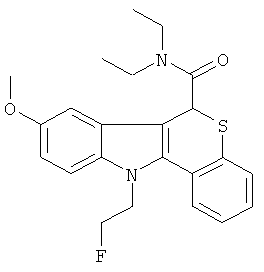
К раствору диэтиламида (+-)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]-флуорен-6-карбоновой кислоты (150 мг, 0,41 ммоль; Пример 1(2)) в безводном DMF (4 мл) добавляли 2-фторэтил-тозилат (166 мг, 0,82 ммоль), полученный, как описано в L.Cronin et al. (J.Org.Chem. 2004; 69:5934-5946), затем добавляли 60%-ные дисперсии гидрида натрия в минеральном масле (34 мг, 0,82 ммоль) в атмосфере азота. Реакционную смесь нагревали при 80°С в течение 1 ч. После охлаждения растворители удаляли в вакууме, остаток гасили водой (30 мл), экстрагировали DCM (2×30 мл), сушили (MgSO4) и растворители удаляли в вакууме. Остаток очищали колоночной хроматографией на диоксиде кремния, элюируя 5-10% смесью EtOAc/CH2Cl2. Неочищенное твердое вещество гасили смесью эфир/уайт-спирит, фильтровали, сушили в вакууме (45°С) с получением указанного в заголовке соединения (77 мг, 46%) в виде бледно-коричневого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 1.12 (t, J=7,0 Гц, 3Н), 1.36 (t, J=7,0 Гц, 3Н), 3.25-3.70 (m, 4H), 3.83 (s, 3Н), 4.45-4.70 (m, 2H), 4.80 (t, J=5,2 Гц, 1Н), 4.96 (t, J=5,2 Гц, 1Н), 5.09 (s, 1Н), 6.84-6.93 (m, 2H), 7.13-7.32 (m, 3Н), 7.46 (m, 1H), 7.58 (d, J=8,0 Гц, 1Н).
13С ЯМР (75 МГц, CDCl3) δ 12.9, 14.9, 37.3, 41.1, 42.5, 45.5, 45.8, 55.9, 81.2, 83.5, 100.4, 110.1, 111.09, 111.12, 112.8, 124.31, 124.35, 125.2, 126.5, 127.1, 127.6, 128.8, 132.2, 134.4, 137.0, 154.8, 168.0.
19F ЯМР (282 МГц, CDCl3) δ -219.4, -219.5, -219.6, -219.65, -219.73, -219.8, -219.9
m/z (ES+) 413,1 (M+H).
Пример 2: Получение диэтиламида (+-)-11-(2-фторэтил)-10-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]-флуорен-6-карбоновой кислоты (нерадиоактивный агент визуализации 3)
Пример 2(1): диэтиламид (+-)-10-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]-флуорен-6-карбоновой кислоты

Данное соединение получали, как описано в Примере 1(2), за исключением того, что 2-метоксифенил-гидразина гидрохлорид использовали вместо 4-метоксифенил-гидразина гидрохлорида. Соединение получали с выходом 40%.
m/z (ES+) 367,0 (M+H).
Пример 2(2): диэтиламид (+-)-11-(2-фторэтил)-10-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты
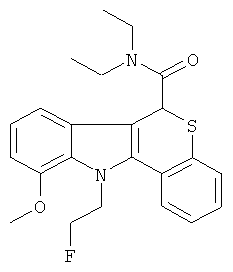
Данное соединение получали, как описано в Примере 1(3), за исключением того, что диэтиламид (+-)-10-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]-флуорен-6-карбоновой кислоты (Пример 2(1)) использовали вместо диэтиламида (+-)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]-флуорен-6-карбоновой кислоты. После перекристаллизации (эфир) получали с 10% выходом в виде белого твердого вещества.
1H ЯМР (300 МГц, CDCl3) δ 1.09 (t, J=7,0 Гц, 3Н), 1.35 (t, J=7,0 Гц, 3Н), 3.25-3.67 (m, 4H), 3.95 (s, 3Н), 4.70-4.96 (m, 4H), 5.04 (s, 1H), 6.67 (m, 1H), 7.04 (m, 2H), 7.16 (m, 1H), 7.29 (m, 1H), 7.45 (m, 1H), 7.77 (m, 1H).
m/z (ES+) 413,1 (M+H).
Пример 3: Получение диэтиламида (+-)-4-фтор-11-(2-фторэтил)-6,11-дигидро-5-тиа-11-аза-бензо[а]-(флуорен-6-карбоновой кислоты (нерадиоактивный агент визуализации 4)
Пример 3(i): (+-)-8-Фтор-4-оксо-тиохроман-2-карбоновая кислота

В круглодонной колбе 2-фтортиофенол (5,0 г, 39,0 ммоль, 4,16 мл) и фуран-2,5-дион (3,82 г, 39,0 ммоль) в толуоле (12 мл) перемешивали при 50°С в течение 40 минут. Затем добавляли триэтиламин (100 мкл) в толуоле (5 мл) в течение 10 минут, обеспечивая, чтобы температура взаимодействия не повышалась выше 60°С. Затем реакционную смесь нагревали при 70°С в течение 20 минут. Затем реакционную смесь концентрировали в глубоком вакууме с получением неочищенного продукта в виде масла. Данное вещество растворяли в DCM (75 мл), охлаждали в ледяной бане и обрабатывали трихлоридом алюминия (7,78 г, 58,5 ммоль) небольшими порциями так, чтобы поддерживать температуру ниже 10°С. Реакционную смесь нагревали до к.т. и наблюдали интенсивное выделение газообразного хлористого водорода, и реакционная смесь становилась очень вязкой и окрашивалась в красный цвет. После перемешивания при к.т. в течение 1,5 часов реакционную смесь разбавляли DCM (50 мл), чтобы сделать ее менее вязкой, и медленно вливали в интенсивно перемешиваемую концентрированную соляную кислоту (30 мл) и лед (30 г) в 2 л конической колбе. Реакционную смесь интенсивно перемешивали и разбавляли дополнительной порцией DCM (500 мл) и изопропилового спирта (50 мл) для растворения любого твердого вещества, которое кристаллизовалось. Слой DCM отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением коричневого твердого вещества. Неочищенное твердое вещество очищали растиранием с диэтиловым эфиром и кремовое твердое вещество собирали фильтрацией с получением 2,5 г (28%) 8-фтор-4-оксо-тиохроман-2-карбоновой кислоты.
1H ЯМР (300 МГц; DMSO-d3): δ 3.04-3.20 (2Н, m, 3-Н), 4.51 (1Н, dd, J=4 и 6 Гц, 2-Н), 7.26-7.34 (1Н, m, 6-H), 7.45-7.52 (1Н, m, 7-H), 7.82 (1Н, dd, J=1 и 8 Гц, 5Н). 13С ЯМР (75 МГц; DMSO-d3): δ 40.5, 40.7, 119.8, 120.1, 123.88, 123.92, 126.0, 126.1, 131.9, 156.1, 159.2, 171.5, 191.2, 191.3.
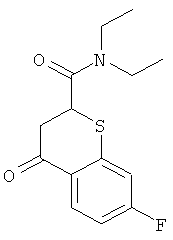
Пример 3(2): диэтиламид (+-)-8-фтор-4-оксо-тиохроман-2-карбоновой кислоты

8-Фтор-4-оксо-тиохроман-2-карбоновую кислоту (2,5 г, 11,1 ммоль) в безводном DCM (50 мл) перемешивали в атмосфере азота при комнатной температуре в течение 18 ч с оксалилхлоридом (2,81 г, 22, ммоль, 1,93 мл) и одной каплей DMF для катализа взаимодействия. Кислота была сначала нерастворимой, но растворялась по мере взаимодействия с получением оранжевого прозрачного раствора через 2 часа и затем окрашивалась в черный цвет через 24 ч. Затем реакционную смесь упаривали в вакууме до смолы для удаления избытка оксалилхлорида и выполняли 1H и 13С ЯМР в CDCl3 для подтверждения окончания реакции. Затем реакционную смесь снова растворяли в DCM (50 мл), охлаждали до 0°С на ледяной бане, перемешивали и обрабатывали по каплям диэтиламином (1,66 г, 22,7 ммоль, 2,05 мл) в DCM (20 мл) в течение 1 ч. Реакционную смесь оставляли нагреваться до комнатной температуры в течение периода 1 ч. Затем реакционную смесь гасили путем добавлением 5% раствора карбоната калия (100 мл) и реакционную смесь интенсивно перемешивали. Раствор DCM отделяли и сушили над сульфатом магния. Две дополнительные порции DCM (100 мл) встряхивали с водным раствором и затем отделяли и сушили над сульфатом магния. Объединенные растворы DCM концентрировали в вакууме с получением коричневого твердого вещества. Неочищенное твердое вещество очищали посредством горячей перекристаллизации из этилацетата и бензина с получением 1,73 г (56%) диэтиламида 8-фтор-4-оксо-тиохроман-2-карбоновой кислоты в виде желтых кристаллов.
1H ЯМР (300 МГц; CDCl3): δ 1.07 (3Н, t, J=7 Гц, N(СН2СН3)2), 1.26 (3Н, t, J=7 Гц, N(СН2СН3)2), 3.02-3.55 (6Н, m, 2-H и N(СН2СН3)2), 4.24-4.27 (1Н, m, 2-Н), 7.15-7.19 (2Н, m, 6-H и 7-Н), 7.93-7.97 (1Н, m, 5-H).
LC-MS: m/z рассчитано для C14H16FNO2S 281,1; получено 282,0 (М+Н)+
Пример 3(3): диэтиламид (+-)-4-фтор-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты
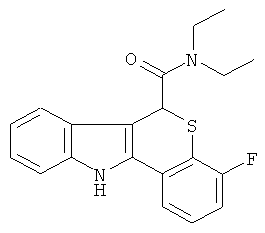
Диэтиламид 8-фтор-4-оксо-тиохроман-2-карбоновой кислоты (1,7 г, 6,0 ммоль) и фенилгидразин (0,65 г, 6,0 ммоль, 0,6 мл) в этаноле (10 мл) и серную кислоту (конц., 0,8 мл) перемешивали при температуре дефлегмации в течение ночи. После охлаждения реакционную смесь фильтровали и собирали белое твердое вещество с получением 1,4 г (80%) неочищенного вещества (чистота 90%). Неочищенное твердое вещество (500 мг) очищали посредством горячей перекристаллизации из этанола с получением 277 мг (13%) диэтиламида 4-фтор-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты в виде белых кристаллов. Структуру подтверждали с помощью 1H ЯМР (300 МГц; DMSO-d6): δ 0.96 (3Н, t, J=7 Гц, N(СН2СН3)2), 1.30 (3Н, t, J=9 Гц, N(СН2СН3)2), 3.19-3.25 (2Н, m, N(СН2СН3)2), 3.56-3.66 (2Н, m, N(СН2СН3)2), 5.76 (1Н, s, 6-H), 7.02-7.45 (6Н, m, ArH), 7.65 (1Н, dd, J=1 и 6 Гц, ArH), 11.8 (1H, s, NH).
LC-MS: m/z рассчитано для C20H19FN2OS 354,2; получено 355,0(М+Н)+
Пример 3(4): диэтиламид (+-)-4-фтор-11-(2-фторэтил)-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты
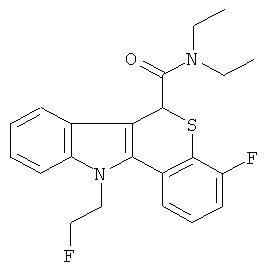
Диэтиламид (+-)-4-фтор-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (0,10 г, 0,28 ммоль) растворяли в безводном DMF (6 мл) при комнатной температуре в атмосфере азота. Добавляли фторэтил-тозилат (0,12 г, 0,12 ммоль) и затем NaH (0,02 г, 0,56 ммоль, 60% в масле). Реакционную смесь нагревали до 80°С в течение 1 часа. Растворитель удаляли при пониженном давлении и остаток растворяли в DCM и промывали водой. Органические вещества сушили над MgSO4, фильтровали и упаривали досуха. Неочищенное вещество кристаллизовалось из метанола с получением 34,4 мг (30%) диэтиламида 4-фтор-11-(2-фтор-этил)-6,11-дигидро-6-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты в виде белого твердого вещества. 1H ЯМР (300 МГц, DMSO-d6) δ 0.94 (3Н, t, J=7 Гц, N(СН2СН3)2), 1.29 (3Н, t, J=7 Гц, N(СН2СН3)2), 3.14-3.26 (2Н, m, N(СН2СН3)2), 3.55-3.65 (2Н, m, N(СН2СН3)2), 4.65-4.95 (4Н, m, NCH2CH2F), 5.62 (1Н, s, 6-Н), 7.12-7.37 (4Н, т, ArH), 7.48 (1Н, d, J=9 Гц, ArH), 7.61-7.68 (2Н, m, ArH).
LC-MS: m/z рассчитано для C22H22F2N2OS 401,1; получено 401,1 (М+Н)+.
Пример 4: Получение диэтиламида (+-)3-фтор-11-(2-фторэтил)-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (нерадиоактивный агент визуализации 5)
Пример 4(1): (+-)-7-Фтор-4-оксо-тиохроман-2-карбоновая кислота

В круглодонной колбе перемешивали 3-фтортиофенол (10,0 г, 71,3 ммоль, 8,85 мл) и фуран-2,5-дион (7,0 г, 71,3 ммоль) в толуоле (12 мл) при 50°С в течение 40 минут. Затем добавляли триэтиламин (26 мкл) в толуоле (1 мл) в течение 10 минут, обеспечивая, чтобы температура взаимодействия не повышалась выше 60°С. Затем реакционную смесь нагревали при 70°С в течение 20 минут. Затем реакционную смесь концентрировали в глубоком вакууме с получением неочищенного продукта в виде масла. Это вещество растворяли в DCM (75 мл), охлаждали на ледяной бане и обрабатывали трихлоридом алюминия (7,78 г, 58,5 ммоль) небольшими порциями так, чтобы поддерживать температуру ниже 10°С. Реакционную смесь нагревали до к.т., происходило интенсивное выделение газообразного хлористого водорода, и реакционная смесь становилась очень вязкой и окрашивалась в красный цвет. Затем после перемешивания при комнатной температуре в течение 1,5 часов реакционную смесь разбавляли DCM (50 мл), чтобы сделать ее менее вязкой, и медленно вливали в интенсивно перемешиваемую концентрированную соляную кислоту (30 мл) и лед (30 г) в 2 л коническую колбу. Реакционную смесь интенсивно перемешивали и разбавляли дополнительной порцией DCM (500 мл) и изопропилового спирта (50 мл) для растворения любого твердого вещества, которое кристаллизовалось. Слой DCM отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением коричневого твердого вещества. Твердое вещество растирали с диэтиловым эфиром и затем фильтровали с получением 4,2 г (48%) 7-фтор-4-оксо-тиохроман-2-карбоновой кислоты в виде кремового твердого вещества. 1H ЯМР (300 МГц; DMSO-d3): 6 3.00-3.16 (2Н, m, 3-Н), 4.44 (1Н, dd, J=5 и 10 Гц, 2-Н), 7.08 (1Н, td, J1=3 и 9 Гц, 6-Н), 7.30 (1Н, dd, J=5 и 10 Гц, ArH), 8.01 (1Н, dd, J1=5 и 10 Гц, ArH). 13С ЯМР (75 МГц; DMSO-d3): δ 38.0, 39.6, 111.1, 111.3, 111.5, 111.8, 125.0, 125.1, 129.0, 129.2, 139.6, 139.7, 160.9, 164.3, 169.5, 188.9.
Пример 4(2): диэтиламид (+-)-7-фтор-4-оксо-тиохроман-2-карбоновой кислоты
7-Фтор-4-оксо-тиохроман-2-карбоновую кислоту (4 г, 17,7 ммоль) в безводном DCM (50 мл) перемешивали в течение 18 ч в атмосфере азота при комнатной температуре с оксалилхлоридом (4,49 г, 35,4 ммоль, 3,1 мл) и одной каплей DMF для катализа реакции. Сначала кислота была нерастворимой, но растворялась при взаимодействии с получением оранжевого прозрачного раствора через 2 часа и затем окрашивалась в черный цвет через 18 ч. Затем реакционную смесь упаривали в вакууме до смолы для удаления избытка оксалилхлорида и выполняли 1H и 13С ЯМР в CDCl3 для подтверждения окончания взаимодействия. Затем реакционную смесь снова растворяли в DCM (50 мл), охлаждали до 0°С на ледяной бане, перемешивали и обрабатывали по каплям диэтиламином в DCM (10 мл) в течение 1 ч. Реакционную смесь оставляли нагреваться до комнатной температуры в течение периода 1 ч. Затем реакционную смесь гасили добавлением 5% раствора карбоната калия (50 мл) и реакционную смесь интенсивно перемешивали. Раствор DCM отделяли и сушили над сульфатом магния. Две дополнительные порции DCM (50 мл) встряхивали с водным раствором и затем отделяли и сушили над сульфатом магния. Объединенные растворы DCM концентрировали в вакууме с получением коричневого твердого вещества, которое кристаллизовалось при стоянии с получением 5,03 г (количественно) диэтиламида 7-фтор-4-оксо-тиохроман-2-карбоновой кислоты. Структуру подтверждали с помощью 1H ЯМР (300 МГц; CDCl3): 5 1.07 (3Н, t, J=7 Гц, N(СН2СН3)2), 1.24 (3Н, t, J=7 Гц, N(СН2СН3)2), 2.99-3.50 (6Н, m, 2-Н и N(СН2СН3)2), 4.24-4.27 (1Н, m, 2-H), 6.83-6.94 (2H, m 6-H и 8-H), 8.15 (1H, dd, J=6 и 9 Гц, 5-H).
LC-MS: m/z рассчитано для C14H16FNO3S 281,1; получено 282,0 (М+Н)+.
Пример 4(3): диэтиламид (+-)-3-фтор-6,11-дигидро-5-тиа-11-аза-бензо[a]флуорен-6-карбоновой кислоты

Диэтиламид 7-фтор-4-оксо-тиохроман-2-карбоновой кислоты (2,5 г, 8,9 ммоль) и фенилгидразин (0,96 г, 8,9 ммоль, 0,9 мл) в этаноле (10 мл) и серную кислоту (конц., 1,2 мл) перемешивали при температуре дефлегмации в течение ночи. Неочищенное твердое вещество очищали посредством горячей перекристаллизации из этанола с получением 1,49 г (47%) диэтиламида 3-фтор-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты в виде белых кристаллов. 1H ЯМР (300 МГц; DMSO-d6): δ 0.96 (3Н, t, J=6 Гц, N(СН2СН3)2), 1.29 (3Н, t, J=6 Гц, N(CH2CH3)2), 3.19-3.25 (2Н, m, N(СН2СН3)2), 3.55-3.61 (2Н, m, N(СН2СН3)2), 5.66 (1Н, s, 6-H), 7.03 (1Н, td, J=1 и 8 Гц, ArH), 7.09-7.18 (2Н, m, ArH), 7.25 (1Н, dd, J=3 и 9 Гц, ArH), 7.35 (1Н, d, J=8 Гц, ArH), 7.41 (1Н, d, J=8 Гц, ArH), 7.81 (1Н, dd, J=6 и 9 Гц, ArH), 11.68(1 Н, s, NH).
LC-MS: m/z рассчитано для C20H19FN2OS 352,1; получено 353,2 (М+Н)+.
Пример 4(4): диэтиламид (+-)3-фтор-11-(2-(фтор-этил)-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты

Диэтиламид 3-фтор-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (0,20 г, 0,56 ммоль) растворяли в безводном DMF (6 мл) при комнатной температуре в атмосфере азота. Добавляли фторэтилтозилат (0,25 г, 1,13 ммоль) и затем NaH (0,05 г, 1,13 ммоль, 60% в масле). Реакционную смесь нагревали до 80°С в течение 1 часа. Растворитель удаляли при пониженном давлении и остаток растворяли в DCM и промывали водой. Органические вещества сушили над MgSO4, фильтровали и упаривали досуха. Неочищенное вещество очищали при помощи полупрепаративной HPLC, элюируя водой (А) и ацетонитрилом (В) (Gemini 5 мкм, С18, 110 А, 150×21 мм, 5-95% В в течение 20 мин, 21 мл/мин), с получением 79,9 мг (35%) диэтиламида 3-фтор-11-(2-фтор-этил)-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты в виде белого твердого вещества. 1Н ЯМР (300 МГц, DMSO-d6) δ 0.95 (3Н, t, J=9 Гц, N(СН2СН3)2), 1.88 (3Н, t, J=9 Гц, N(СН2СН3)2), 3.14-3.26 (2Н, m, М(СН2СН3)2), 3.51-3.67 (2Н, m, N(СН2СН3)2), 4.58-4.97 (4Н, m, NCH2CH2F), 5.53 (1Н, s, 6-H), 7.12-7.27 (3Н, m, ArH), 7.38-4.47 (2Н, m, ArH), 7.61 (1Н, d, J=9 Гц, ArH), 7.80-7.86 (1Н, m, ArH).
LC-MS: m/z рассчитано для C22H22F2N2OS 401,1; получено 401,1 (М+Н)+.
Пример 5: Получение диэтиламида (+-)8-этокси-11-(2-фторэтил)-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (нерадиоактивный агент визуализации 6)
Пример 5(1): диэтиламид (+-)11-[2-(трет-бутил-диметил-силанилокси)]этил]-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты
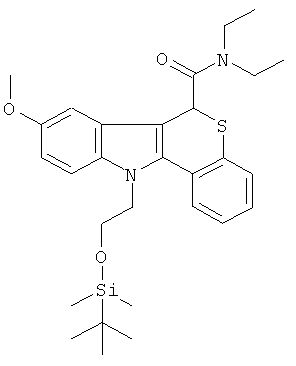
К раствору диэтиламида (+-)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты из Примера 1(2) (2,0 г, 5,40 ммоль) в безводном DMF (20 мл) добавляли 60% дисперсию гидрида натрия в минеральном масле (240 мг, 6,0 ммоль) и смесь перемешивали при комнатной температуре в течение 5 мин в атмосфере азота. Добавляли 2-(бромэтокси)-трет-бутил-диметилсилан (2,6 г, 10,8 ммоль) и смесь перемешивали в течение 4 ч. Растворители удаляли в вакууме, остаток гасили водой (30 мл), экстрагировали DCM (2×30 мл), сушили (MgSO4) и растворители удаляли в вакууме. Остаток очищали колоночной хроматографией на диоксиде кремния, элюируя смесью 3% EtOAc/CH2Cl2, с получением указанного в заголовке соединения (2,0 г, 70%) в виде желтого твердого вещества.
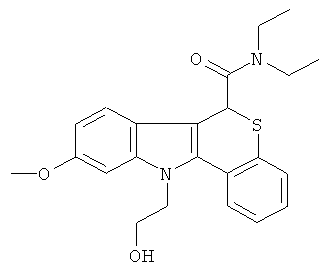
Пример 5(2): диэтиламид (+-)11-[2-гидроксиэтил]-8-гидрокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты

К раствору диэтиламида (+-)11-[2-(трет-бутил-диметил-силанилокси)]этил]-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (1,0 г, 1,91 ммоль) в безводном DCM (60 мл) при -78°С добавляли трибромид бора (11,5 мл, 1 М в DCM, 11,5 ммоль). Раствор оставляли нагреваться до к.т. и перемешивали в течение 24 ч. Растворители удаляли в вакууме, гасили метанолом (40 мл) и добавляли 1 н. HCl (10 мл), кипятили при температуре дефлегмации в течение 1 ч. Растворители удаляли в вакууме, смесь растворяли в метаноле (5 мл), гасили водой (100 мл), фильтровали, сушили в вакууме (45°С) с получением указанного в заголовке соединения (0,77 г, 100%) в виде светло-коричневого порошка.
Пример 5(3): диэтиламид (+-)11-[2-гидроксиэтил]-8-этокси-6,11-дигидро-5-тиа-11-аза-бензо[a]флуорен-6-карбоновой кислоты
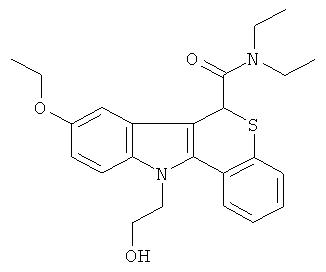
К раствору диэтиламида (+-)11-[2-гидроксиэтил]-8-гидрокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (400 мг, 1,01 ммоль) в безводном DMF (4 мл) при 0°С добавляли 60% дисперсию гидрида натрия в минеральном масле (40 мг, 1,01 ммоль). Смесь перемешивали при 0°С в течение 10 мин в атмосфере азота. Добавляли этилбромид (218 мг, 2,0 ммоль, 150 мкл) и смесь перемешивали в течение 24 ч. Растворители удаляли в вакууме, остаток гасили водой (30 мл), экстрагировали DCM (2×30 мл), сушили (MgSO4) и растворители удаляли в вакууме. Остаток очищали колоночной хроматографией на диоксиде кремния, элюируя смесью 40-60% EtOAc/CH2Cl2, с получением указанного в заголовке соединения (340 мг, 79%) в виде белого твердого вещества.
Пример 5(4): диэтиламид (+-)11-[2-метансульфоксиэтил]-8-этокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты
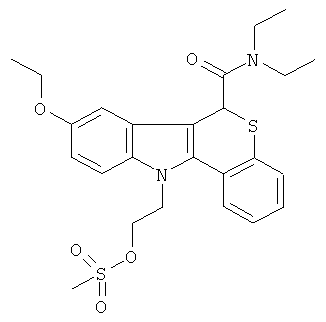
К суспензии диэтиламида (+-)11-[2-гидроксиэтил]-8-этокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (0,34 г, 0,80 ммоль) в безводном DCM (15 мл) добавляли пиридин (0,63 г, 8,0 ммоль, 0,65 мл). Реакционную смесь охлаждали до 0°С и добавляли метансульфонилхлорид (0,37 г, 3,2 ммоль, 0,25 мл). Реакционную смесь перемешивали при к.т. в течение 3 ч. Смесь промывали с помощью 0,5 М HCl (2×20 мл), затем водой 2×20 мл), сушили (MgSO4) и растворитель удаляли при пониженном давлении. Остаток очищали посредством колоночной хроматографии на диоксиде кремния, элюируя смесью 20% EtOAc/CH2Cl2. Остаток гасили смесью эфир/уайт-спирит, фильтровали, сушили в вакууме (45°С) с получением указанного в заголовке соединения (0.38 г, 95%) в виде светло-желтого твердого вещества.
Пример 5(5): диэтиламид (+-)11-[2-фторэтил]-8-этокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты
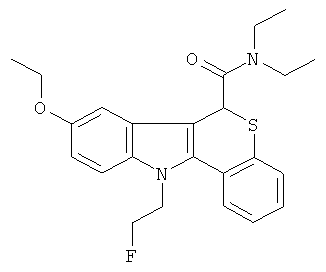
К раствору диэтиламида (+-)11-[2-метансульфоксиэтил]-8-этокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (100 мг, 0,20 ммоль) в безводном ацетонитриле (5 мл) в атмосфере азота добавляли 1,0 М TBAF в THF (тетрагидрофуран) (0,4 мл, 0,4 ммоль). Смесь нагревали до 80°С в течение 2 ч. Растворители удаляли в вакууме и остаток очищали колоночной хроматографией на диоксиде кремния, элюируя смесью 5-10% EtOAc/CH2Cl2, с получением указанного в заголовке соединения (26 мг, 31%) в виде желтого твердого вещества.
Пример 6: Получение диэтиламида (+-)7-метокси-11-(2-фторэтил)-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (нерадиоактивный агент визуализации 2) и диэтиламида (+-)9-метокси-11-(2-(фторэтил)-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (нерадиоактивный агент визуализации 7)
Пример 6(1): диэтиламид 7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты и диэтиламид 9-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты
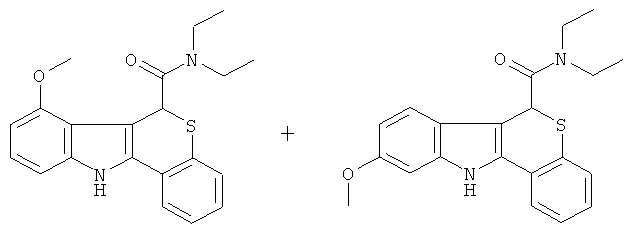
К раствору диэтиламида (+-)-4-оксо-тиохроман-2-карбоновой кислоты (3,33 г, 12,6 ммоль) (Пример 1(1)) и 3-метоксифенил-гидразина гидрохлорида (2,2 г, 12,6 ммоль) в этаноле (30 мл) добавляли концентрированную серную кислоту (1,83 мл, 3,40 г, 11,5 ммоль) в атмосфере азота. Реакционную смесь нагревали при температуре дефлегмации в течение 24 ч. После охлаждения реакционную смесь фильтровали, твердое вещество промывали этанолом, сушили в вакууме (45°С) с получением смеси диэтиламида 7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты и диэтиламида 9-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]-флуорен-6-карбоновой кислоты (3,2 г, 69%) в виде бледно-белого твердого вещества.
Пример 6(2): диэтиламид 11-(2-фторэтил)-7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты и диэтиламид 11-(2-фторэтил)-9-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты

К раствору смеси изомеров диэтиламида 7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты и диэтиламида 9-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]-флуорен-6-карбоновой кислоты (1,0 г, 2,73 ммоль) (полученных согласно Примеру 6(1)) в безводном DMF (10 мл) добавляли 2-фторэтил-тозилат (1,2 г, 5,46 ммоль), затем 60% дисперсию гидрида натрия в минеральном масле (131 мг, 5,46 ммоль) в атмосфере азота. Реакционную смесь нагревали при 80°С в течение 1 ч. После охлаждения растворители удаляли в вакууме, остаток гасили водой (30 мл), экстрагировали DCM (2×30 мл), сушили (MgSO4) и растворители удаляли в вакууме. Остаток очищали колоночной хроматографией на диоксиде кремния, элюируя смесью 5-10% EtOAc/CH2Cl2, с получением смеси изомеров (1,0 г, 89%). Затем вещество (400 мг) очищали с помощью HPLC, элюируя водой (А) и метанолом (В) (Gemini 5u, С18, 110 А, 150×21 мм, 70-95% В в течение 20 мин, 21 мл/мин), с получением 240 мг диэтиламида 9-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты в виде желтого твердого вещества и 100 мг диэтиламида 7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты в виде белого твердого вещества.
Пример 7: Получение 18F-меченых агентов визуализации 2 и 7
Пример 7(1): диэтиламид 11-[2-(трет-бутил-диметил-силанилокси)]этил]-7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты и диэтиламид 11-[2-(трет-бутил-диметил-силанилокси)]этил]-9-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты

К раствору смеси изомеров, полученной согласно Примеру 6(1), (2,0 г, 5,46 ммоль) в безводном DMF (20 мл) добавляли 60% дисперсию гидрида натрия в минеральном масле (240 мг, 6,0 ммоль) и смесь перемешивали при комнатной температуре в течение 5 мин в атмосфере азота. Добавляли 2-(бромэтокси)-трет-бутил-диметилсилан (2,6 г, 10,9 ммоль) и смесь перемешивали в течение 4 ч. Растворители удаляли в вакууме, остаток гасили водой (30 мл), экстрагировали DCM (2×30 мл), сушили (MgSO4) и растворители удаляли в вакууме. Остаток очищали колоночной хроматографией на диоксиде кремния, элюируя смесью 5% EtOAc/CH2Cl2, с получением смеси изомеров диэтиламида 11-[2-(трет-бутил-диметил-силанилокси)]этил]-7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты и диэтиламида 11-[2-(трет-бутил-диметил-силанилокси)]этил]-9-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (2,53 г, 88%) в виде желтого твердого вещества.
Пример 7(2): диэтиламид 11-[2-гидроксиэтил]-7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты и диэтиламид 11-[2-(2-гидроксиокси)]этил]-7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты

К раствору смеси изомеров, полученной согласно Примеру 7(1), (2,5 г, 4,76 ммоль) в безводном THF (40 мл) добавляли TBAF 1,0 М в THF (9,5 мл, 9,5 ммоль) в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 4 ч. Растворители удаляли в вакууме, остаток очищали колоночной хроматографией на диоксиде кремния, элюируя 40%-ной смесью EtOAc/CH2Cl2, с получением смеси изомеров диэтиламида 11-[2-гидроксиэтил]-7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты и диэтиламида 11-[2-(2-гидроксиокси)]этил]-7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (1,90 г, 97%) в виде бледно-желтого твердого вещества.
Пример 7(3): диэтиламид (+-)11-[2-метансульфоксиэтил]-7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты и диэтиламид (+-)11-2-(метансульфоксиэтил)-9-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты

К раствору смеси изомеров, полученной согласно Примеру 7(2), (1,0 г, 2,4 ммоль), растворенной в безводном DCM (30 мл), добавляли пиридин (1,9 г, 24,0 ммоль, 1,9 мл). Реакционную смесь охлаждали до 0°С и добавляли метансульфонилхлорид (1,1 г, 9,6 ммоль, 0,74 мл). Реакционную смесь перемешивали при к.т. в течение 4 ч. Смесь промывали 0,5 М HCl (2×20 мл), затем водой (2×20 мл), сушили (MgSO4) и растворитель удаляли при пониженном давлении. Остаток очищали колоночной хроматографией на диоксиде кремния, элюируя смесью 20% ETOАс/СН2Cl2, с получением смеси изомеров (1,0 г, 85%). Затем вещество (400 мг) очищали с помощью HPLC, элюируя водой (А) и метанолом (В) (Gemini 5 мкм, С18, 110 А, 150×21 мм, 5-95% В в течение 30 мин, 21 мл/мин), с получением 170 мг диэтиламида 11-[2-метансульфоксиэтил]-7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты в виде белого твердого вещества и 60 мг диэтиламида 11-2-(метансульфоксиэтил)-9-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты в виде белого твердого вещества.
Пример 7(4): Метод прямого введения метки
Соединения-предшественники диэтиламида 11-[2-метансульфоксиэтил]-7-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты и диэтиламида 11-2-(метансульфоксиэтил)-9-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты, полученные согласно Примеру 7(3), метили радиоизотопом методом прямого введения метки с получением агентов визуализации 2 и 7 соответственно. В реакционный сосуд добавляли 18F-воду, затем К222 (2 мг) в ацетонитриле (500 мкл) и KHCO3 (0,1 мол. дм-3, 50 мкл) и сушили при 100°С в течение 20-30 мин. Добавляли предшественник (0,5-1 мг) в ацетонитриле (1000 мкл). Реакционный сосуд герметично закрывали и нагревали при 100°С в течение 10 мин. Реакционную смесь охлаждали, вымывали из реакционного сосуда водой (1,5 мл) и очищали посредством полупрепаративной HPLC. Фракцию, содержащую основной радиоактивный продукт, собирали и разбавляли до объема 10 мл с помощью H2O. Ее загружали на кондиционированный light sep pak C18, промывали H2O (1×2 мл) и продукт элюировали с помощью EtOH (0,5 мл) во флакон Р6 и добавляли PBS (5 мл).
Пример 8: Получение 18F-меченых агентов визуализации 1, 3, 4 и 5
Пример 8(1): Получение соединений-предшественников
Соединения-предшественники:
(а) диэтиламид (+-)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (полученный согласно Примеру 1(2));
(б) диэтиламид (+-)-10-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (полученный согласно Примеру 2(1));
(в) диэтиламид (+-)-4-фтор-6,11-дигидро-5-тиа-11-аза-бензо(а]флуорен-6-карбоновой кислоты (полученный согласно Примеру 3(3)); и
(г) (+-)3-фтор-11-(2-фтор-этил)-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты диэтиламид (полученный согласно Примеру 4(4))
метили радиоизотопом, используя способ непрямого введения метки, описанный ниже, для получения 18F-меченых агентов визуализации 1, 3, 4 и 5 соответственно.
Пример 8(2): способ непрямого введения метки
18F/воду добавляли к К222 (4 мг), водному K2CO3 (50 мкл 0,1 молярного раствора) и ацетонитрилу (500 мкл) в реакционном сосуде и сушили в течение 20-30 мин при 100°С в потоке азота. Добавляли этил-1,2-дитозилат (4 мг) в ацетонитриле (1000 мкл) и нагревали при 100°С в течение 10 мин. Реакционную смесь охлаждали и очищали посредством полупрепаративной HPLC и собирали фракцию, содержащую 18F-фторэтилтозилат. Эту фракцию разбавляли H2O до объема примерно 20 мл, загружали на кондиционированный light sep pak t-C18 и промывали Н2О (1×2 мл). Sep pak сушили в линии N2 с высокой скоростью потока в течение 20 мин. Затем 18F фторэтилтозилат элюировали DMF (500 мкл).
Отдельно добавляли предшественник (13 мг) в DMF(250 мкл) во второй реакционный сосуд и продували с помощью N2 в течение 5 мин. Затем добавляли NaH (1,3 мг) в DMF (2×250 мкл) в атмосфере азота и реакционный сосуд нагревали при 45°С в течение 0,5-1 ч. Затем в него добавляли 18F-фторэтилтозилат в DMF, полученный выше, и нагревали при 100°С в течение 10 мин в реакционном сосуде, продуваемом N2. Реакционную смесь охлаждали и вымывали из реакционного сосуда водой (1 мл). Раствор фильтровали через шприцевой фильтр и очищали на препаративной HPLC. Собирали фракцию, содержащую основной радиоактивный пик. Ее разбавляли до объема приблизительно 10 мл с помощью H2O и загружали на кондиционированный light sep pak t-C18, промывали Н2O (1×2 мл) и элюировали ЕtОН (0,5 мл) во флакон Р6 и добавляли фосфатно-солевой буферный раствор (5 мл).
Пример 9: Получение 18F-меченого агента визуализации 6
Диэтиламид (+-)11-[2-метансульфоксиэтил]-8-этокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты (полученный согласно Примеру 5(4)) метили радиоизотопом, используя способ прямого введения метки, описанный в Примере 7(4) выше.
Пример 10: Анализ эффективности in vitro
Соединения сортировали по их аффинности к PBR, используя способ, основанный на Le Fur et al. (Life Sci. 1983; USA 33: 449-57).
Соединения, подлежащие тестированию (растворенные в 50 мМ Трис-HCl, рН 7,4, 10 мМ MgCl2, содержащем 1% DMSO), конкурировали за связывание с PBR сердца крыс Вистар (Wistar) против 0,3 нМ [3H]-PK-11195. Взаимодействие выполняли в 50 мМ Трис-HCl, рН 7,4 10 мМ MgCl2 в течение 15 минут при 25°С.
Соединения исследовали при 6 разных концентрациях в 300-кратном диапазоне концентраций вокруг оцениваемой Ki. Результаты представлены в Таблице 1 выше и демонстрируют, что эффективность соединений по изобретению лучше по сравнению с эффективностью соединений из уровня техники.
Пример 11: Метод биораспределения in vivo
Агенты визуализации in vivo 1-7, которые получены в Примерах 7, 8 и 9 выше, испытывали в модели биораспределения in vivo и их биораспределение сравнивали с биораспределением соединения из известного уровня техники [18F]FE-PBR (полученного согласно Примеру 14 из WO 2007/057705).
Взрослых самцов крыс Вистар (200-300 г) инъецировали с помощью 1-3 мБк каждого агента визуализации in vivo через боковую хвостовую вену. На 2, 10, 30 или 60 мин (n=3) после инъекции крыс умерщвляли и ткани или жидкости отбирали для радиоактивного измерения посредством подсчета с помощью жидкостной сцинтилляции.
По сравнению с [18F]FE-PBR соединения по изобретению демонстрировали более высокое отношение связывания обонятельная луковица: полосатое тело (см. Таблицу 1 выше).
Пример 12: Ауторадиография с использованием модели аксотомии лицевого нерва (FNA)
Для исследований in vivo использовали самцов крыс Вистар (180-200 г). Под анестезией изофлураном удаляли шерсть на правой стороне ушной области. Выполняли внутриушной надрез и идентифицировали главный ствол лицевого нерва. Лицевой нерв разрезали за ухом на выходе из шилососцевидного отверстия. Рану зашивали и животных оставляли восстанавливаться. Через семь суток после операции животным инъецировали ~5-10 мБк агента визуализации in vivo 1 через боковую хвостовую вену. Животных умерщвляли через 30 минут и ствол головного мозга удаляли и замораживали в изопентане. Криостатные срезы (12 мкм) ствола головного мозга, содержащие оба лицевых нервных центра, закрепляли на стеклянные пластинки и подвергали воздействию люминесцентного экрана в течение ночи.
Затем экраны сканировали на установке для визуализации люминофора Storm (GE Healthcare) и полученный снимок анализировали и количественно определяли, используя ImageQuant TL (GE Healthcare). Фиг.1 и 2 иллюстрируют данные, полученные в данном эксперименте.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ ИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ЛИГАНДОВ PBR | 2010 |
|
RU2525196C2 |
| ПРОИЗВОДНЫЕ ТРИЦИКЛИЧЕСКОГО ИНДОЛА В КАЧЕСТВЕ ЛИГАНДОВ PBR | 2011 |
|
RU2551423C2 |
| АГЕНТЫ, СВЯЗЫВАЮЩИЕСЯ С PSMA, И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2494096C2 |
| ТЕТРАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2010 |
|
RU2725140C2 |
| ТЕТРАЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2010 |
|
RU2585622C2 |
| НОВЫЕ 2-ГЕТЕРОАРИЛ-ЗАМЕЩЕННЫЕ БЕНЗОТИОФЕНЫ И БЕНЗОФУРАНЫ 709 | 2008 |
|
RU2472789C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| АЛКИЛСУЛЬФОНАМИДХИНОЛИНЫ С АФФИННОСТЬЮ К РЕЦЕПТОРАМ NK-3 | 2006 |
|
RU2421447C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ИНГИБИРОВАНИЯ ПУТИ JAK | 2011 |
|
RU2672100C2 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2013 |
|
RU2654692C2 |


Изобретение относится к соединению Формулы I, которое представляет собой агент визуализации in vivo периферического бензодиазепинового рецептора (PBR):

где Q представляет собой водород или фтор; Х представляет собой водород или С1-4алкокси; Y представляет собой S; и R представляет собой C1-6фторалкил; и где по меньшей мере один атом указанного агента визуализации in vivo Формулы I представляет собой радиоизотоп, подходящий для визуализации in vivo, представляющий собой либо гамма-излучающий радиоактивный галоген, либо позитрон-излучающий радиоактивный неметалл; и где если указанный радиоизотоп является радиоизотопом углерода, то он является карбонильным углеродом. 7 н. и 8 з.п. ф-лы, 2 ил., 1 табл., 12 пр.
1. Агент визуализации in vivo Формулы I:
или его соль или сольват, где:
Q представляет собой водород или фтор;
Х представляет собой водород или С1-4алкокси;
Y представляет собой S; и
R представляет собой C1-6фторалкил;
и где по меньшей мере один атом указанного агента визуализации in vivo Формулы I представляет собой радиоизотоп, подходящий для визуализации in vivo, представляющий собой либо гамма-излучающий радиоактивный галоген либо позитрон-излучающий радиоактивный неметалл; и где если указанный радиоизотоп является радиоизотопом углерода, то он является карбонильным углеродом; при условии, что Q или Х оба не являются водородом.
2. Агент визуализации in vivo по п.1, где:
Q представляет собой водород;
Х представляет собой С1-4алкокси;
Y представляет собой S; и
R представляет собой С1-4фторалкил.
3. Агент визуализации in vivo по п.1, где:
Q представляет собой фтор;
Х представляет собой водород;
Y представляет собой S; и
R представляет собой С1-4фторалкил.
4. Агент визуализации in vivo по п.1, где указанный радиоизотоп представляет собой позитрон-излучающий радиоактивный неметалл, выбранный из 11С, 13N, 18F и 124I.
5. Агент визуализации in vivo по п.4, где R представляет собой С1-4[18F]-фторалкил.
6. Агент визуализации in vivo по п.1, который выбран из:

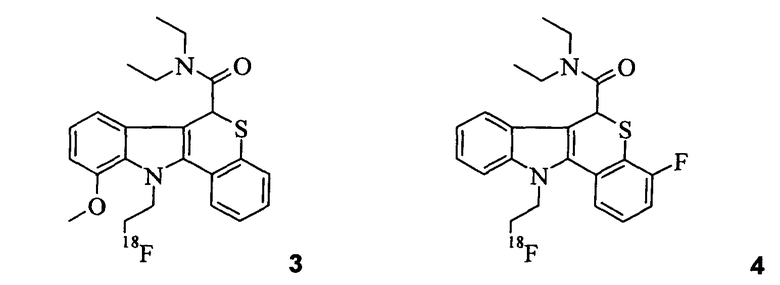
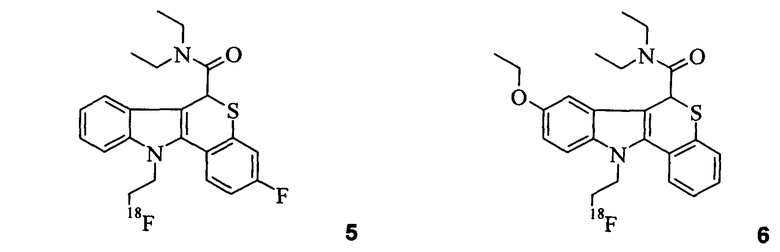

7. Соединение-предшественник, полезное в получении агента визуализации in vivo по любому из пп.1-6, имеющее Формулу II:
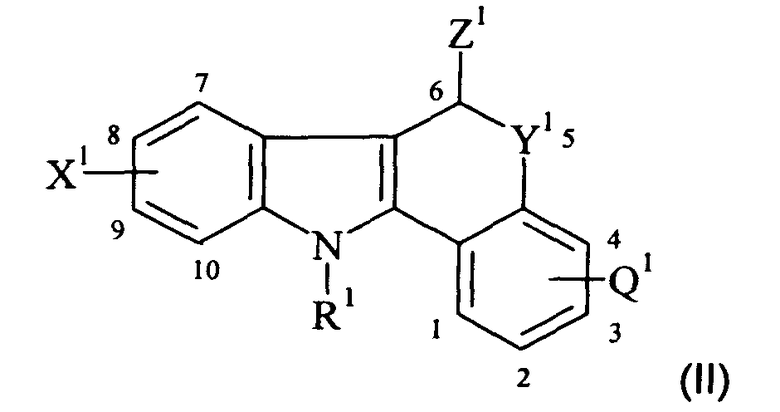
где один из R1, X1 или Z1 содержит химическую группу, которая взаимодействует с подходящим источником радиоизотопа, где указанный радиоизотоп является таким, как определено в п.1 или 4, так что указанный агент визуализации in vivo образуется при взаимодействии указанного соединения-предшественника с указанным подходящим источником указанного радиоизотопа;
и где:
когда R1 не содержит указанную химическую группу, тогда он является таким, как определено для R в любом из пп.1, 2 и 3, и, возможно, дополнительно содержит защитную группу;
когда X1 не содержит указанную химическую группу, тогда он является таким, как определено для Х в любом из пп.1, 2 и 3, и, возможно, дополнительно содержит защитную группу;
когда Z1 не содержит указанную химическую группу, тогда он представляет собой -С(=O)-N-(СН2-СН3)2 и, возможно, дополнительно содержит защитную группу;
Q1 является таким, как определено для Q в любом из пп.1, 2 и 3; и
Y1 является таким, как определено для Y в любом из пп.1, 2 и 3, и, возможно, дополнительно содержит защитную группу.
8. Способ получения агента визуализации in vivo по любому из пп.1-6, при котором:
(1) берут соединение-предшественник Формулы II по п.7;
(2) берут подходящий источник указанного радиоизотопа, где указанный радиоизотоп является таким, как определено в любом из пп.1 или 4;
(3) осуществляют взаимодействие соединения-предшественника со стадии (1) с источником радиоизотопа со стадии (2) с получением указанного агента визуализации in vivo.
9. Кассета для автоматизированного синтеза агента визуализации in vivo по любому из пп.1-6, где указанная кассета содержит:
(1) сосуд, содержащий соединение-предшественник по п.7; и
(2) средство для элюирования сосуда с подходящим источником радиоизотопа, где указанный радиоизотоп является таким, как определено в любом из пп.1 или 4.
10. Радиофармацевтическая композиция, содержащая агент визуализации in vivo по любому из пп.1-6 вместе с биосовместимым носителем в форме, подходящей для введения млекопитающему.
11. Способ визуализации in vivo для определения распределения и/или величины экспрессии PBR (периферического бензодиазепинового рецептора) у субъекта, включающий:
(1) введение указанному субъекту агента визуализации in vivo по любому из пп.1-6;
(2) обеспечение возможности связывания указанного агента визуализации in vivo с PBR у указанного субъекта;
(3) определение посредством визуализации in vivo сигналов, излучаемых радиоизотопом указанного агента визуализации in vivo;
(4) создание изображения, отображающего локализацию и/или количество указанных сигналов; и
(5) определение распределения и величины экспрессии PBR у указанного субъекта, где указанная экспрессия прямо коррелирует с указанными сигналами, излучаемыми указанным агентом визуализации in vivo.
12. Способ визуализации in vivo по п.11, который выполняют неоднократно во время курса лечения указанного субъекта, где указанный курс включает введение лекарственного средства для противодействия PBR-состоянию.
13. Способ диагностики состояния, при котором PBR активирован, включающий способ визуализации in vivo по п.11 или 12, вместе с дополнительной стадией (6) отнесения распределения и величины экспрессии PBR к конкретной клинической картине.
14. Агент визуализации in vivo по любому из пп.1-6 для применения в способе диагностики по п.13.
15. Агент визуализации in vivo по любому из пп.1-6 для применения в изготовлении радиофармацевтической композиции по п.10 для применения в способе диагностики по п.13.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ ПИРИДАЗИНО[4,5-B]ИНДОЛ-1-АЦЕТАМИДА ДЛЯ ПРИГОТОВЛЕНИЯ ЛЕКАРСТВ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С ДИСФУНКЦИЕЙ БЕНЗОДИАЗЕПИНОВЫХ РЕЦЕПТОРОВ ПЕРИФЕРИЧЕСКОГО ТИПА | 2000 |
|
RU2243764C2 |

