Перекрестная ссылка на родственные заявки
[0001] Настоящая заявка испрашивает преимущество заявки США с серийным номером 61/734900, поданной 7 декабря 2012 года, и заявки США серийный номер 61/783238, поданной 14 марта 2013 года, каждая из которых включена в настоящее описание посредством отсылки в их полном объеме.
Область техники
[0002] Настоящее изобретение относится к борсодержащим соединениям, композициям, составам и их применению в качестве ингибиторов бета-лактамазных ферментов и в качестве антибактериальных агентов.
Уровень техники
[0003] Антибиотики являются наиболее эффективными лекарственными препаратами для клинического лечения вызванных бактериями инфекционных заболеваний. Для них существует широкий рынок, благодаря тому, что они обладают такими преимуществами как хорошее антибактериальное действие в сочетании с ограниченными побочными эффектами. Из антибиотиков широко используются бета-лактамные антибиотики (например пенициллины, цефалоспорины и карбапенемы), посколько они характеризуются сильным бактерицидным действием и низкой токсичностью.
[0004] Чтобы противостоять действию различных бета-лактамов, бактерии эволюционировали, что привело к возникновению у них способности продуцировать варианты дезактивирующих бета-лактамы ферментов, называемых бета-лактамазами, и обмениваться данным "инструментом" внутри вида и между видами. Данные бета-лактамазы классифицируются как основанные на "серине" или на "металле", соответственно, по наличию ключевого остатка серина или атома цинка в активном центре фермента. Быстрое распространение данного механизма резистентности бактерий может серьезно ограничивать возможности лечения бета-лактамами в клинике и в обществе.
Сущность изобретения
[0005] В настоящем изобретении описаны соединения, которые модулируют активность бета-лактамаз. В некоторых вариантах осуществления описанные здесь соединения ингибируют бета-лактамазы. В некоторых вариантах осуществления описанные здесь соединения применимы в лечении бактериальных инфекций.
[0006] В одном из аспектов настоящего изобретения предложены соединения формулы I или формулы Ia или их фармацевтически приемлемые соли, сольваты, полиморфы, стереоизомеры, таутомеры, пролекарства, метаболиты, N-оксиды или изомеры:
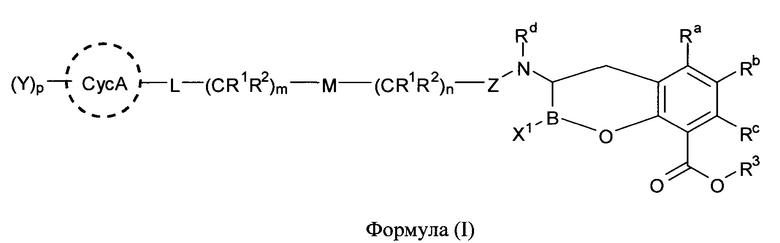
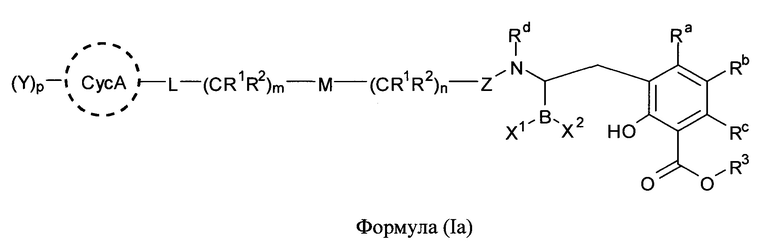
где:
L представляет собой связь, -CR1R2-, >C=O, или =CR1-;
М представляет собой связь, -O-, -S-, -S(O)-, SO2- или -N(R4)-;
m равно 0, 1 или 2;
n равно 0, 1, 2 или 3;
при условии, что,
когда n равно 0, то М представляет собой связь;
р равно 0, 1, 2, 3 или 4;
при условии, что
когда р равно 0, то L представляет собой -CR1R2- или =CR1-;
X1 и X2 независимо выбраны из -ОН, -OR8 или F;
Z представляет собой >С=O, >C=S, или >SO2;
СусА представляет собой необязательно замещенный 3-10-членный неароматический карбоцикл, в котором необязательная олефиновая функциональная группа неароматического карбоцикла не присоединена непосредственно к заместителю - атому кислорода, атому серы или атому азота;
Ra, Rb и Rc независимо выбраны из группы, состоящей из атома водорода, атома фтора, атома хлора, атома брома, необязательно замещенного C1-C6-алкила, необязательно замещенного C3-C6-циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного арила, необязательно замещенного гетероарила, -ОН, -OR10, -NR4R5 и -SR10;
каждый R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома фтора, атома хлора, атома брома, необязательно замещенного C1-C6-алкила, необязательно замещенного C3-C6-циклоалкила, -ОН, -OR10, -SR10 и -NR4R5,
или R1 и R2, взятые вместе, образуют оксогруппу, оксим, или необязательно замещенный карбоцикл, или необязательно замещенный гетероцикл с атомом углерода, к которому они присоединены;
R3 представляет собой атом водорода, необязательно замещенный C1-C6-алкил, или фармацевтически приемлемое пролекарство;
каждый Rd, R4 и R5 независимо выбран из группы, состоящей из атома водорода, -ОН, -CN, необязательно замещенного C1-C6-алкила, необязательно замещенного алкоксиалкила, необязательно замещенного гидроксиалкила, необязательно замещенного аминоалкила, необязательно замещенного циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного циклоалкилалкила, необязательно замещенного гетероциклилалкила, необязательно замещенного аралкила, необязательно замещенного гетероаралкила, (полиэтиленгликоль)этила и необязательно замещенного сахарида;
или R4 и R5, взятые вместе, образуют необязательно замещенный гетероцикл с атомом азота, к которому они присоединены;
R8 представляет собой необязательно замещенный C1-C6-алкил, необязательно замещенный C3-C6-циклоалкил или фармацевтически приемлемую боронатную сложноэфирную группу;
R10 представляет собой необязательно замещенный C1-C6-алкил или необязательно замещенный C3-C6-циклоалкил
и каждый Y независимо представляет собой группу, содержащую от 1 до 50 атомов, отличных от атома водорода, выбранных из группы, состоящей из С, N, О, S и Р.
[0007] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb и Rc независимо выбраны из группы, состоящей из атома водорода, атома фтора, атома хлора, необязательно замещенного C1-C6-алкила, необязательно замещенного C3-C6-циклоалкила, -ОН, -OR10, -NR4R5 и -SR10. В некоторых вариантах осуществления Ra, Rb и Rc независимо представляют собой атом водорода, атом фтора или атом хлора. В предпочтительных вариантах осуществления Ra, Rb и Rc представляют собой атом водорода.
[0008] В некоторых вариантах осуществления соединения формулы I или формулы Ia R3 представляет собой атом водорода, метил, этил, пропил, бутил или изопропил. В предпочтительных вариантах осуществления R3 представляет собой атом водорода.
[0009] В некоторых вариантах осуществления соединения формулы I или формулы Ia X1 и X2 представляют собой -ОН.
[0010] В некоторых вариантах осуществления соединения формулы I или формулы Ia Rd представляет собой атом водорода или С1-С4-алкил. В предпочтительных вариантах осуществления Rd представляет собой атом водорода.
[0011] В некоторых вариантах осуществления соединения формулы I или формулы Ia Z представляет собой >С=O или >SO2. В предпочтительных вариантах осуществления Z представляет собой >С=O.
[0012] В некоторых вариантах осуществления соединения формулы I или формулы Ia L представляет собой собой -CR1R2- или =CR1-; М представляет собой -O-, -S-, -SO2- или -N(R4)-; m равно 0 или 1; и n равно 1 или 2. В некоторых вариантах осуществления L представляет собой связь, -CR1R2- или =CR1-; М представляет собой связь или -O-; m равно 0; и n равно 1 или 2. В других вариантах осуществления L представляет собой связь или >С=O; М представляет собой связь или -N(R4)-; и m и n равны 0. В других вариантах осуществления L представляет собой связь; М представляет собой связь; и m или n равны 1. В некоторых вариантах осуществления L представляет собой -CR1R2- or =CR1-; M представляет собой связь; и m и n равны 0. В некоторых вариантах осуществления L представляет собой -CR1R2- или =CR1-; М представляет собой связь; и m или n равны 1.
[0013] В некоторых вариантах осуществления соединения формулы I или формулы Ia СусА выбран из группы, состоящей из циклопропана, циклобутана, циклопентана, циклогексана, циклогептана, циклооктана, циклопентена, циклогексена, циклогептена и циклооктена, где олефиновая функциональная группа циклопентена, циклогексена, циклогептена и циклооктена не присоединена непосредственно к заместителю - атому кислорода, атому серы или атому азота. В некоторых вариантах осуществления СусА представляет собой циклобутан, циклопентан, циклогексан или циклогексен, где олефиновая функциональная группа циклогексена не присоединена непосредственно к заместителю - атому кислорода, атому серы или атому азота. В других вариантах осуществления СусА выбран из группы, состоящей из бицикло[3.3.0]октана, бицикло[4.3.0]нонана, цис-декалина, транс-декалина, бицикло[2.1.1]гексана, бицикло[2.2.1]гептана, бицикло[2.2.2]октана, бицикло[3.2.2]нонана и бицикло[3.3.2]декана. В предпочтительных вариантах осуществления СусА представляет собой циклобутан, циклопентан и циклогексан. В некоторых вариантах осуществления соединения формулы I или формулы Ia по меньшей мере один Y выбран из группы, состоящей из атома фтора, атома хлора, атома брома, необязательно замещенного С1-С6-алкила, необязательно замещенного С3-С6-циклоалкила, необязательно замещенного гетероцикла, необязательно замещенного арила, необязательно замещенного гетероарила, =O, -ОН, -OR10, -SR10, -NR4R5, -(CR6R7)vNR4R5, -(CR6R7)vNR4R5(CR6R7)vNR4R5, -NR4R5(CR6R7)vR6, -NR4(CR6R7)vNR4R5, -NR4(CR6R7)vNR4R5(CR6R7)vNR4R5, -O(CR6R7)vNR4R5, -S(O)0,1,2(CR6R7)vNR4R5, -N(R4)C(O)(CR6R7)vNR4R5, -(CR6R7)vN(R4)C(O)(CR6R7)vNR4R5, -(CR6R7)vNR4(CR6R7)vNR4R5, -NR4(CR6R7)vOR10, -NR4(CR6R7)vS(O)0,1,2R10, -C(O)NR4(CR6R7)vNR4R5, -S(O)0,1,2NR4(CR6R7)vNR4R5, -NR5C(O)NR4(CR6R7)vNR4R5, -OC(O)NR4(CR6R7)vNR4R5, -NR5C(=NR7)NR4(CR6R7)vNR4R5, -N(R4)C(=NR5)R6, -(CR6R7)vN(R4)C(=NR5)R6, -NR4(CR6R7)vN(R4)C(=NR5)R6, -O(CR6R7)vN(R4)C(=NR5)R6, -S(O)0,1,2(CR6R7)vN(R4)C(=NR5)R6, -(CR6R7)vC(=NR5)NR4R5, -NR4(CR6R7)vC(=NR5)NR4R5, -O(CR6R7)vC(=NR5)NR4R5, -S(O)0,1,2(CR6R7)vC(=NR5)NR4R5, -(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4(CR6R7)vN(R4)C(=NR5)NR4R5, -O(CR6R7)vN(R4)C(=NR5)NR4R5, -S(O)0,1,2(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4C(=NR5)NR4C(=NR5)NR4R5, -(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4(CR6R7)vNR4C(=NR4)NR4R5, -O(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -S(O)0,1,2-(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4C(=NR5)NR4R5, -C(=NR4)NR4R5, -C(=NR4)NR4C(O)R6, -NR4SO2R6, -NR4C(O)R6, -NR4C(=O)OR6, -C(O)NR4R5, -(CR6R7)vC(O)NR4R5, -SO2NR4R5, -гетероарил-NR4R5, гетероциклил-NR4R5, -гетероарил-N(R4)C(=NR5)NR4R5, -гетероциклил-N(R4)C(=NR5)NR4R5, -N(R4)-гетероарил-NR4R5, -N(R4)-гетероциклил-NR4R5, -(CR6R7)vгетероарил-NR4R5, -(CR6R7)vгетероциклил-NR4R5, -(CR6R7)vгетероарил-N(R4)C(=NR5)NR4R5, -(CR6R7)vгетероциклил-N(R4)C(=NR5)NR4R5, -NR4R5(CR6R7)vгетероциклил-C(=NR5)NR4R5-(CR6R7)vгетероарила, -(CR6R7)vгетероциклила, -О-гетероарила, -О-гетероциклила, -NR4(CR6R7)vгетероарила, -NR4(CR6R7)vгетероциклила, -O(CR6R7)vгетероарила, -O(CR6R7)vгетероциклила, -NR4(CR6R7)vNR5-гетероарила, -NR4(CR6R7)vNR5-гетероциклила, -O(CR6R7)vNR5-гетероарила, -O(CR6R7)vNR5-гетероциклила, -O(CR6R7)vO-гетероциклила, -NR4R5R9+Q-, -(CR6R7)vNR4R5R9+Q-, -NR4(CR6R7)vNR4R5R9+Q-, -NR4R9+(CR6R7)vNR4R5R9+Q-2, -(CR6R7)v(T)+Q- и -O(CR6R7)vNR4R5R9+Q-;
где:
каждый Т независимо выбран из группы, состоящей из пиридин-1-ила, пиримидин-1-ила и тиазол-3-ила;
каждый Q независимо представляет собой фармацевтически приемлемый противоион; и каждый v независимо обозначает 1, 2, 3 или 4;
или Y, взятый вместе с атомом углерода, к которому он присоединен, образует необязательно замещенный спирокарбоцикл или необязательно замещенный спирогетероцикл;
или два Y, взятые вместе с атомами углерода, к которым они присоединены, образуют необязательно замещенный карбоцикл или необязательно замещенный гетероцикл;
каждый R6 и R7 независимо выбран из группы, состоящей из атома водорода, атома фтора, атома хлора, атома брома, необязательно замещенного C1-C6-алкила, необязательно замещенного алкоксиалкила, необязательно замещенного гидроксиалкила, необязательно замещенного С3-С6-циклоалкила, -ОН, -OR10, -SR10, -NR4R5, -NR4C(O)R5, -NR4C(O)OR5, -NR4C(O)NR5, -C(O)OR5, -C(O)NR4R5, -C(N=R5)NR4R5 -NR4SO2R5, необязательно замещенного гетероциклила, необязательно замещенного арила и необязательно замещенного гетероарила;
или R6 и R7, взятые вместе, образуют оксогруппу, оксим, или необязательно замещенный карбоцикл, или необязательно замещенный гетероцикл с атомом углерода, к которому они присоединены;
каждый R9 представляет собой независимо необязательно замещенный C1-С6-алкил. В некоторых вариантах осуществления по меньшей мере один Y содержит от 1 до 6 основных атома азота. В некоторых вариантах осуществления по меньшей мере один Y содержит 1, 2 или 3 основных атомов азота. В некоторых вариантах осуществления по меньшей мере один Y содержит 2 основных атома азота.
[0014] В некоторых вариантах осуществления соединения формулы I или формулы Ia по меньшей мере один Y выбран из группы, состоящей из атома фтора, атома хлора, необязательно замещенного C1-С6-алкила, =O, -ОН, -OR10, -NR4R5, -(CR6R7)vNR4R5, -NR4(CR6R7)vNR4R5, -(CR6R7)vNR4R5(CR6R7)vNR4R5, -NR4R5(CR6R7)vR6, -NR4R5(CR6R7)vгетероциклил-C(=NR5)NR4R5, -NR4(CR6R7)vNR4C(=NR4)NR4R5, -NR4(CR6R7)vNR4R5(CR6R7)vNR4R5, -O(CR6R7)vNR4R5, -N(R4)C(O)(CR6R7)vNR4R5, -(CR6R7)vN(R4)C(O)(CR6R7)vNR4R5, -C(O)NR4(CR6R7)vNR4R5, -S(O)0,1,2NR4(CR6R7)vNR4R5, -NR5C(O)NR4(CR6R7)vNR4R5, -OC(O)NR4(CR6R7)vNR4R5, -NR5C(=NR7)NR4(CR6R7)vNR4R5, -N(R4)C(=NR5)R6, -(CR6R7)vN(R4)C(=NR5)R6, -NR4(CR6R7)vN(R4)C(=NR5)R6, -O(CR6R7)vN(R4)C(=NR5)R6, -(CR6R7)vC(=NR5)NR4R5, -NR4(CR6R7)vC(=NR5)NR4R5, -O(CR6R7)vC(=NR5)NR4R5, -(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4(CR6R7)vN(R4)C(=NR5)NR4R5, -O(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4C(=NR5)NR4C(=NR5)NR4R5, -(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -O(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4C(=NR5)NR4R5, -C(=NR4)NR4R5, -C(=NR4)NR4C(O)R6, -NR4SO2R6, -NR4C(O)R6, -NR4C(=O)OR6, -C(O)NR4R5, -(CR6R7)vC(O)NR4R5, -гетероарил-NR4R5, -гетероциклил-NR4R5, -гетероарил-N(R4)C(=NR5)NR4R5, -гетероциклил-N(R4)C(=NR5)NR4R5, -N(R4)-гетероарил-NR4R5, -N(R4)-гетероциклил-NR4R5, -(CR6R7)vгетероарил-NR4R5, -(CR6R7)vгетероциклил-NR4R5, -(CR6R7)vгетероарил-N(R4)C(=NR5)NR4R5, -(CR6R7)vгетероциклил-N(R4)C(=NR5)NR4R5, -(CR6R7)vгетероарила, -(CR6R7)vгетероциклила, -О-гетероарила, -О-гетероциклила, -NR4(CR6R7)vгетероарила, -NR4(CR6R7)vгетероциклила, -O(CR6R7)vгетероарила, -O(CR6R7)vгетероциклила и -O(CR6R7)vО-гетероциклила. В некоторых вариантах осуществления по меньшей мере один Y выбран из группы, состоящей из атома фтора, необязательно замещенного C1-С6-алкила, -ОН, -NR4R5, -(CR6R7)vNR4R5, -(CR6R7)vNR4R5(CR6R7)vNR4R5, -NR4R5(CR6R7)vR6, -NR4R5(CR6R7)vгетероциклил-C(=NR5)NR4R5, -NR4(CR6R7)vNR4C(=NR4)NR4R5, -NR4(CR6R7)vNR4R5(CR6R7)vNR4R5, -NR4(CR6R7)vNR4R5, -O(CR6R7)vNR4R5, -C(O)NR4(CR6R7)vNR4R5, -NR5C(O)NR4(CR6R7)vNR4R5, -NR5C(=NR7)NR4(CR6R7)vNR4R5, -N(R4)C(=NR5)R6, -(CR6R7)vN(R4)C(=NR5)R6, -NR4(CR6R7)vN(R4)C(=NR5)R6, -(CR6R7)vC(=NR5)NR4R5, -NR4(CR6R7)vC(=NR5)NR4R5, -(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4C(=NR5)NR4C(=NR5)NR4R5, -(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4C(=NR5)NR4R5, -C(=NR4)NR4R5, -C(=NR4)NR4C(O)R6, -NR4C(O)R6, -(CR6R7)vC(O)NR4R5, -гетероциклил-NR4R5, -гетероциклил-N(R4)C(=NR5)NR4R5, -N(R4)-гетероциклил-NR4R5, -(CR6R7)vгетероциклил-NR4R5, -(CR6R7)vгетероциклил-N(R4)C(=NR5)NR4R5, -(CR6R7)vгетероциклила и -NR4(CR6R7)vгетероциклила. В других вариантах осуществления по меньшей мере один Y выбран из группы, состоящей из -гетероарил-NR4R5, -гетероциклил-NR4R5, -гетероарил-N(R4)C(=NR5)NR4R5, -гетероциклил-N(R4)C(=NR5)NR4R5, -N(R4)-гетероарил-NR4R5, -N(R4)-гетероциклил-NR4R5, -гетероарил-C(=NR5)NR4R5, -гетероциклил-C(=NR5)NR4R5, -(CR6R7)vгетероарил-NR4R5,-(CR6R7)vгетероциклил-NR4R5, -(CR6R7)vгетероарил-N(R4)C(=NR5)NR4R5 и -(CR6R7)vгетероциклил-N(R4)C(=NR5)NR4R5. В предпочтительных вариантах осуществления по меньшей мере один Y выбран из группы, состоящей из -NR4R5, -NR4C(=NR5)NR4R5, -C(=NR4)NR4R5, -N(R4)C(=NR5)R6,-(CR6R7)vNR4R5,-(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4(CR6R7)vNR4R5, -NR4(CR6R7)vOR10, -(CR6R7)vNR4(CR6R7)vNR4R5,NR5C(=NR5)NR4(CR6R7)vNR4R5, -NR4(CR6R7)vN(R4)C(=NR5)NR4R5, -NR5C(O)CR6(NR4R5)(CR6R7)vNR4R5, -(CR6R7)vC(=NR5)NR4R5, -(CR6R7)vN(R4)C(O)(CR6R7)vNR4R5,-C(=NR4)NR4C(O)R6, -NR4(CR6R7)v гетероарила и -O(CR6R7)vNR4R5.
[0015] В некоторых вариантах осуществления р равно 0, 1, 2, 3 или 4. В некоторых вариантах осуществления р равно 1 или 2. В некоторых вариантах осуществления р равно 1.
[0016] В некоторых вариантах осуществления соединения формулы I или формулы Ia R4 и R5 независимо выбраны из группы, состоящей из атома водорода, ОН, необязательно замещенного C1-C6-алкила, необязательно замещенного алкоксиалкила, необязательно замещенный гидроксиалкила, необязательно замещенного гетероциклила. В предпочтительных вариантах осуществления R4 и R5 независимо представляют собой атом водорода или необязательно замещенный C1-С6-алкил.
[0017] В некоторых вариантах осуществления соединения формулы I или формулы Ia R6 и R7 независимо выбраны из группы, состоящей из атома водорода, необязательно замещенного C1-С6-алкила, -ОН, -NR4R5 и необязательно замещенного гетероциклила, или R6 и R7, взятые вместе, образуют необязательно замещенный гетероцикл с атомом углерода, к которому они присоединены. В предпочтительных вариантах осуществления R6 и R7 независимо представляют собой атом водорода, атом фтора или необязательно замещенный C1-С6-алкил. В некоторых вариантах осуществления  представляет собой
представляет собой  В некоторых вариантах осуществления
В некоторых вариантах осуществления 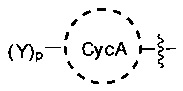 представляет собой
представляет собой  . В некоторых вариантах осуществления Y представляет собой -NR4(CR6R7)vNR4R5. В некоторых вариантах осуществления Y представляет собой -NR4(CR6R7)vNR4C(=NR4)NR4R5. В некоторых вариантах осуществления Y представляет собой -NR4R5. В других вариантах осуществления Y представляет собой -NR4C(=NR4)NR4R5. В некоторых вариантах осуществления Y представляет собой -(CR6R7)vNR4R5. В некоторых вариантах осуществления Y представляет собой -(CR6R7)vNR4C(=NR4)NR4R5. В некоторых вариантах осуществления v представляет собой 2. В некоторых вариантах осуществления v представляет собой 1. В некоторых вариантах осуществления каждый из R4 и R5 выбран из Н, необязательно замещенного C1-С6-алкила или необязательно замещенного С3-С6-циклоалкила. В некоторых вариантах осуществления каждый R4, R6 и R7 представляет собой Н.
. В некоторых вариантах осуществления Y представляет собой -NR4(CR6R7)vNR4R5. В некоторых вариантах осуществления Y представляет собой -NR4(CR6R7)vNR4C(=NR4)NR4R5. В некоторых вариантах осуществления Y представляет собой -NR4R5. В других вариантах осуществления Y представляет собой -NR4C(=NR4)NR4R5. В некоторых вариантах осуществления Y представляет собой -(CR6R7)vNR4R5. В некоторых вариантах осуществления Y представляет собой -(CR6R7)vNR4C(=NR4)NR4R5. В некоторых вариантах осуществления v представляет собой 2. В некоторых вариантах осуществления v представляет собой 1. В некоторых вариантах осуществления каждый из R4 и R5 выбран из Н, необязательно замещенного C1-С6-алкила или необязательно замещенного С3-С6-циклоалкила. В некоторых вариантах осуществления каждый R4, R6 и R7 представляет собой Н.
[0018] В некоторых вариантах осуществления соединения формулы I или формулы Ia данное соединение выбрано из группы, представленной следующими структурами:
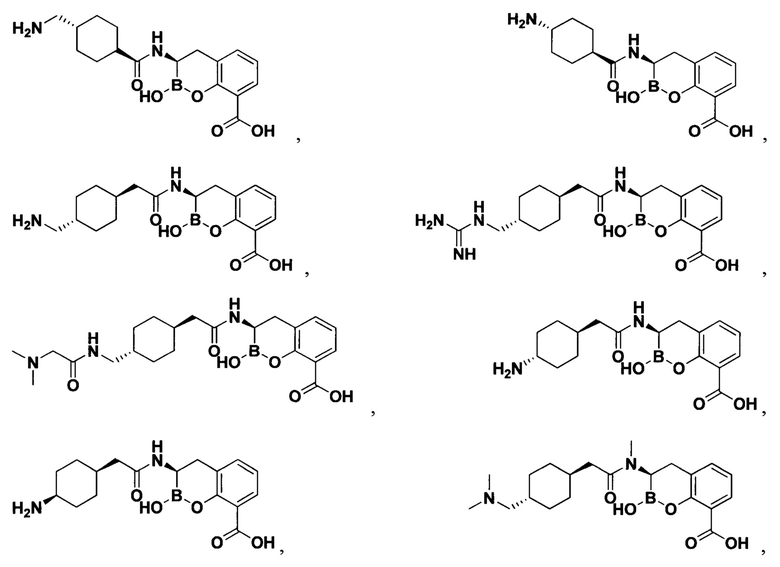
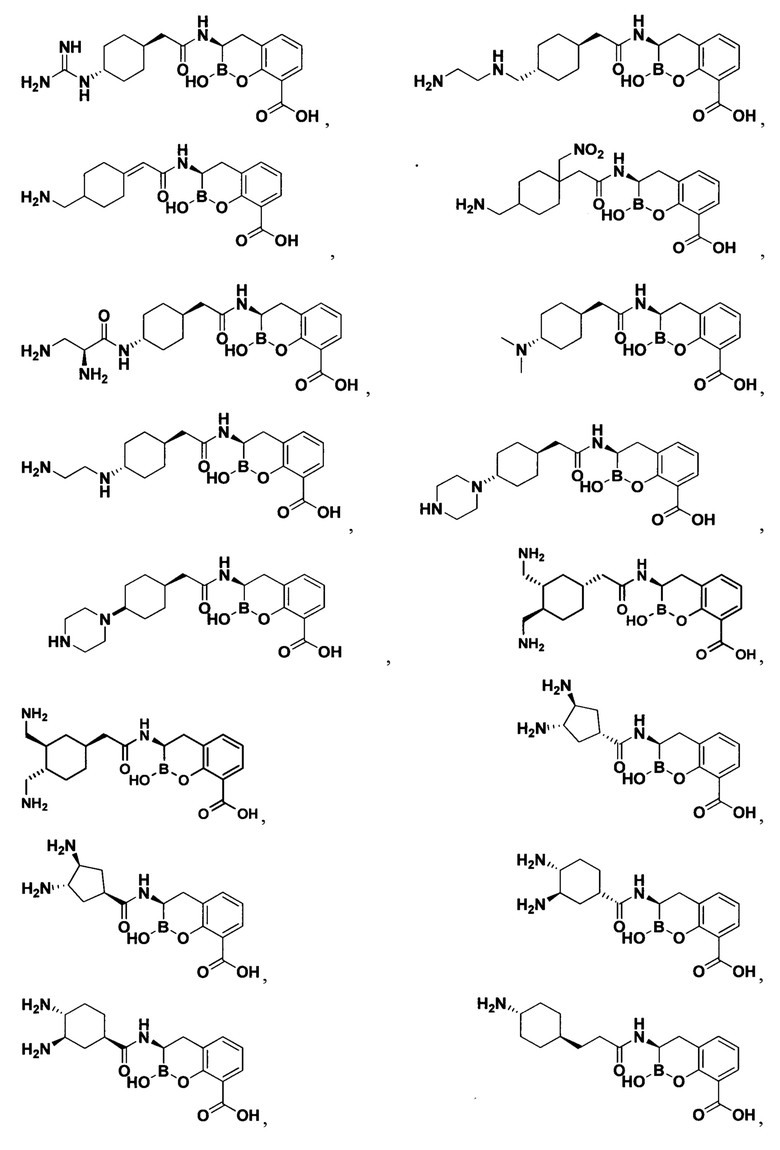
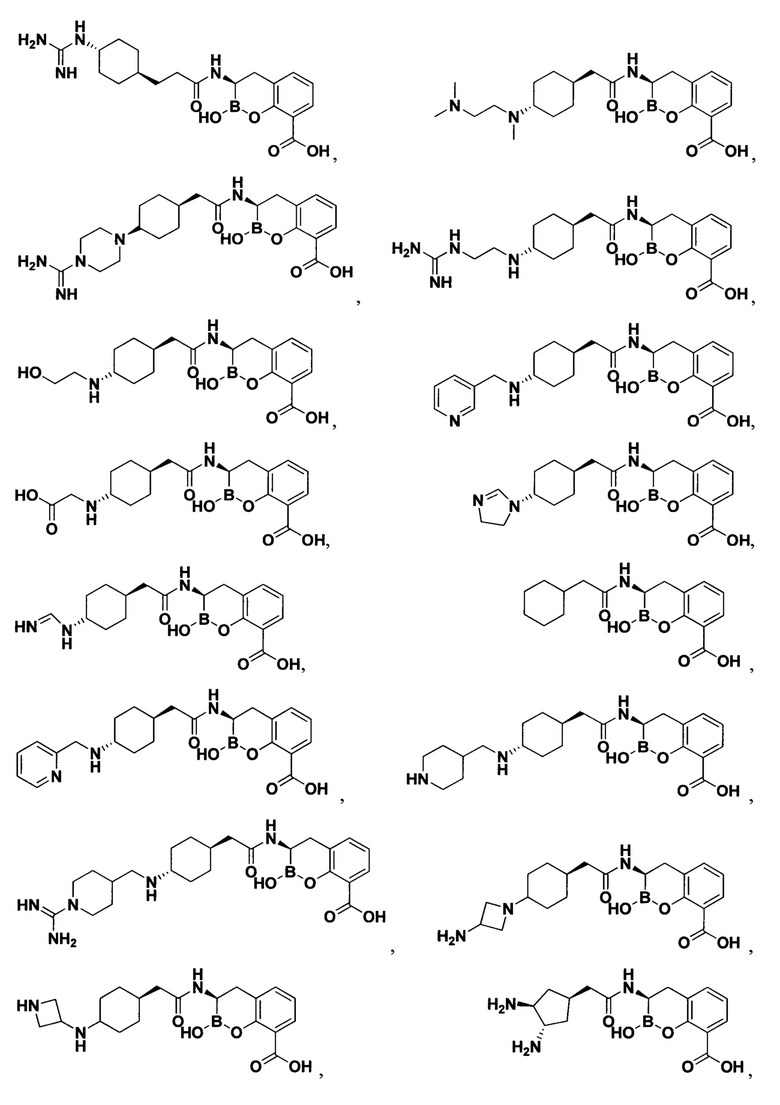
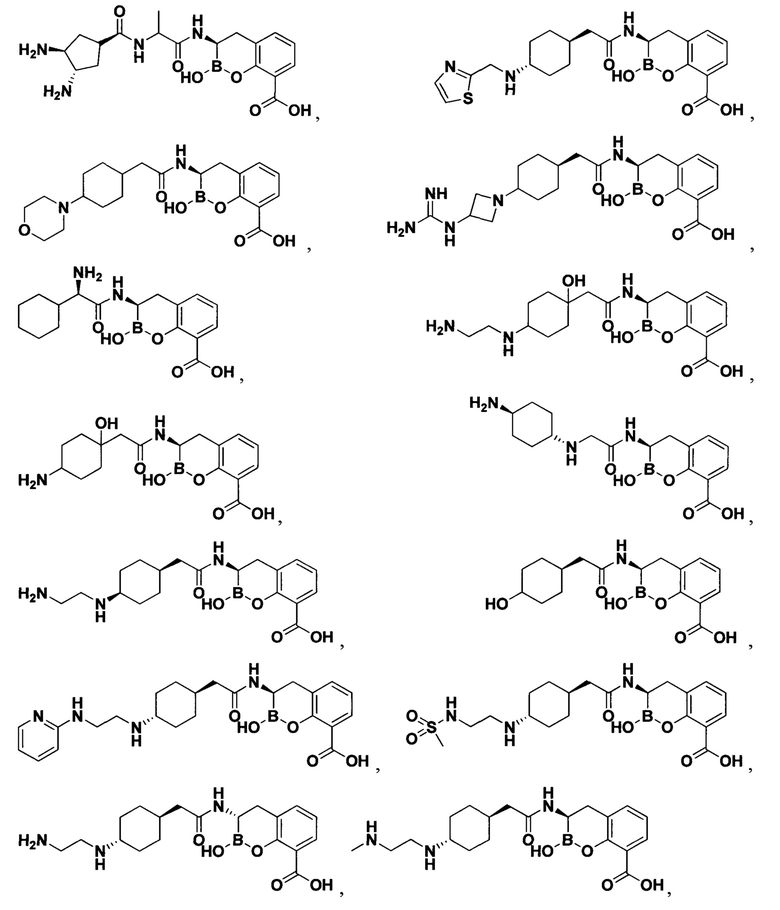
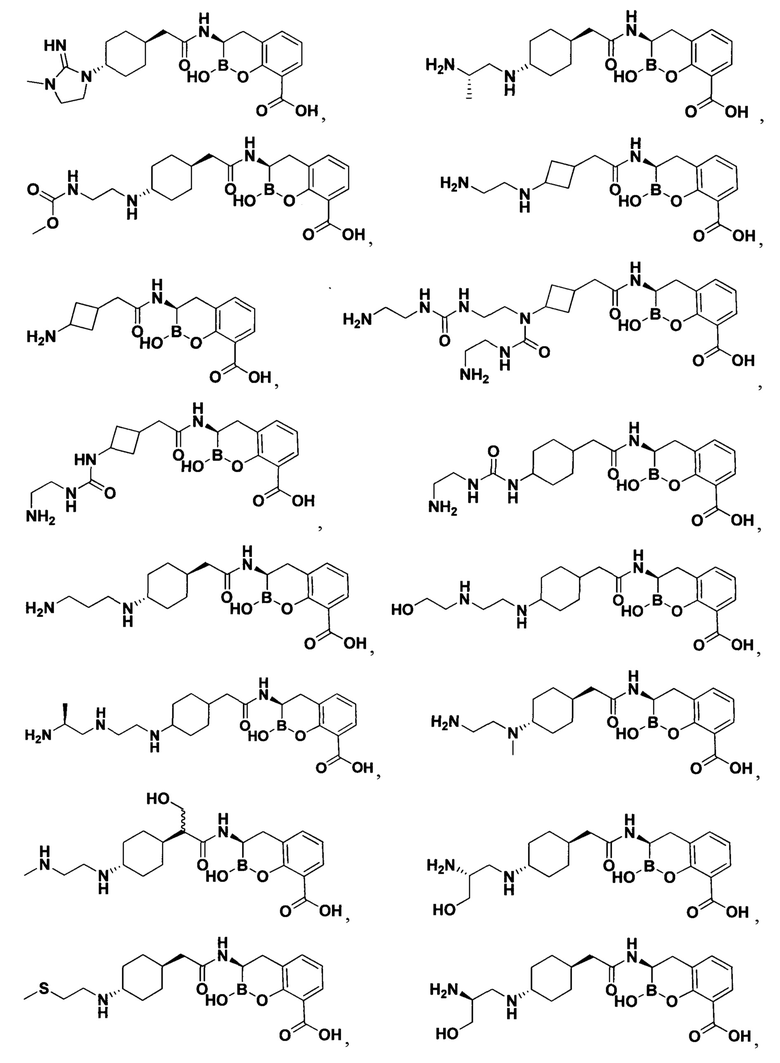

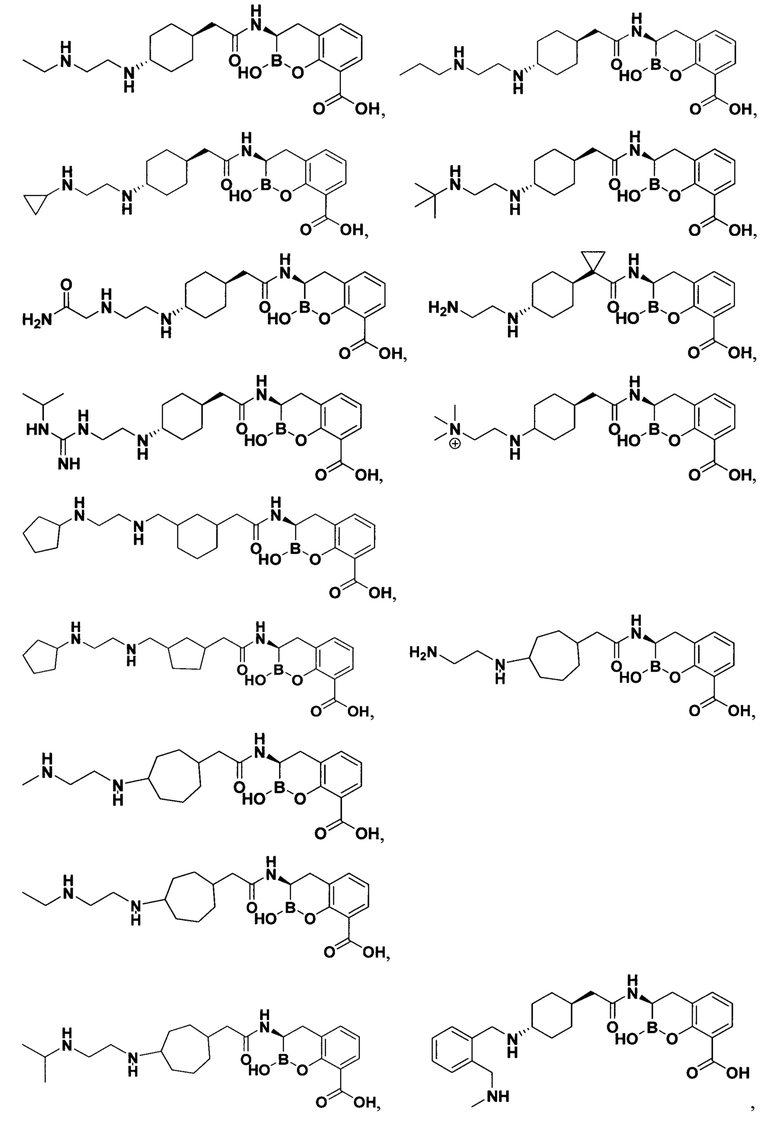
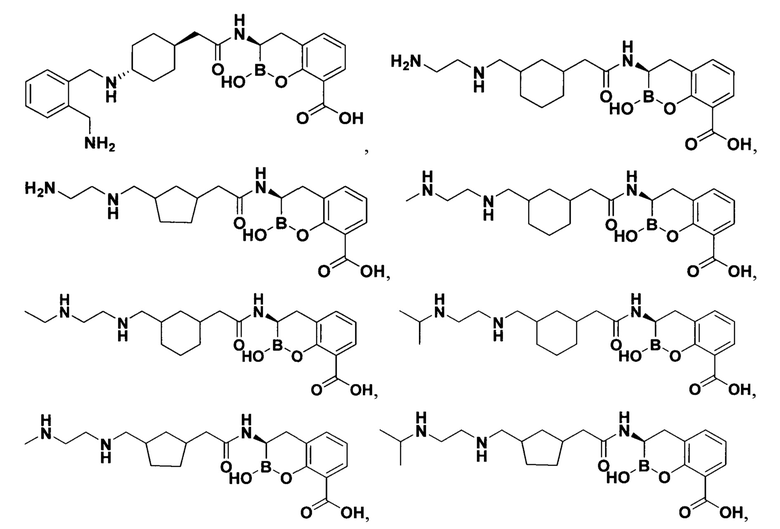
или его фармацевтически приемлемой соли, сольвата, полиморфа, стереоизомера, таутомера, пролекарства, метаболита, N-оксида или изомера, где соединение присутствует в закрытой, циклической форме в соответствии с формулой I и, как показано в структурах выше, раскрытой, ациклической форме в соответствии с формулой Ia, или их смеси. В некоторых вариантах осуществления соединение формулы I или формулы Ia представляет собой стереоизомер, представленный любой из упомянутых выше структур. В некоторых вариантах осуществления соединение формулы I или формулы Ia представляет собой энантиомер стереоизомера, представленного любой из упомянутых выше структур. В некоторых вариантах осуществления соединение формулы I или формулы Ia представляет собой диастереомер стереоизомера, представленного любой из упомянутых выше структур. В некоторых вариантах осуществления соединение формулы I или формулы Ia представляет собой смесь энантиомеров и/или диастереомеров стереоизомера, представленного любой из упомянутых выше структур. В некоторых вариантах осуществления соединение формулы I или формулы Ia представляет собой рацемат стереоизомера, представленного любой из упомянутых выше структур.
[0019] В другом аспекте настоящего изобретения представлены фармацевтические композиции, содержащие соединение формулы I или формулы Ia, как описано здесь, или его фармацевтически приемлемую соль, сольват, полиморф, стереоизомеры, таутомер, пролекарство, метаболит, N-оксид или изомер и фармацевтически приемлемый наполнитель. В некоторых вариантах осуществления фармацевтическая композиция дополнительно содержит бета-лактамный антибиотик. В некоторых вариантах осуществления бета-лактамный антибиотик представляет собой пенициллин, цефалоспорин, карбапенем, монобактам, мостиковый монобактам или их комбинации.
[0020] В дополнительном аспекте настоящего изобретения представлены способы лечения бактериальной инфекции у субъекта, включающие введение субъекту фармацевтической композиции, как здесь описано, необязательно в сочетании с бета-лактамным антибиотиком. В некоторых вариантах осуществления способы лечения бактериальной инфекции у субъекта включают введение субъекту фармацевтической композиции, как описано здесь, в сочетании с бета-лактамным антибиотиком.
ВКЛЮЧЕНИЕ ПОСРЕДСТВОМ ССЫЛКИ
[0021] Все публикации, патенты и патентные заявки, упомянутые в данном описании, включены посредством ссылки в такой же степени, как если бы каждая отдельная публикация, патент или заявка на патент была конкретно и индивидуально указана как включенная посредством ссылки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0022] Бета-лактамазы обычно подразделяются на 4 класса, а именно на классы А, В, С и D по классификации Ambler, исходя из их аминокислотных последовательностей. Ферменты, образующие классы А, С и D, представляют собой бета-лактамазы, содержащие остаток серина в активном центре, в то время как ферменты класса В являются Zn-зависимыми. Цефалоспорины и карбапенемы последних поколений разрабатывались с тем, чтобы придать им способность избегать дезактивирующего действия ранних вариантов бета-лактамаз, содержащих остаток серина в активном центре. Тем не менее, недавнее взрывоподобное увеличение количества новых версий имеющих остаток серина в активном центре бета-лактамаз, таких как, например, ферменты бета-лактамазы расширенного спектра (БЛРС, Extended-Spectrum Beta-Lactamase (ESBL)) класса А, карбапанемазы класса А (например, KPC-2), хромосомальные и плазмид-опосредованные цефалоспориназы класса С (AmpC, CMY и т.д.) и оксасиллиназы класса D, а также металло-бета-лактамазы класса В (например, VIM, NDM), имело следствием уменьшение применимости семьи бета-лактамных антибиотиков, в том числе, относящихся к более поздним поколениям бета-лактамных лекарственных препаратов, что представляет собой серьезную медицинскую проблему. Действительно, количество каталогизированных бета-лактамаз, содержащих остаток серина в активном центре, выросло от менее чем десяти в 1970 году до более чем 750 вариантов (смотри, например, "Jacoby & Bush, Амино Acid Sequences for ТЕМ, SHV и ОХА Extended-Spectrum и Inhibitor Resistant β-Lactamases", на сайте клиники Lahey).
[0023] Коммерчески доступные ингибиторы бета-лактамаз (клавулановая кислота, сульбактам, тазобактам) были разработаны для решения проблемы с бета-лактамазами, которые были клинически значимыми в 1970-х и 1980-х годах (например, пенициллиназы). Данные ингибиторы бета-лактамаз обладают слабой активностью в отношении разнообразия бета-лактамазных ферментов (как с остатком серина, так и с атомом металла в активном центре), приобретающих клиническую значимость в настоящее время. Кроме того, данные ингибиторы ферментов доступны только в фиксированных комбинациях с производными пенициллина. Комбинации с цефалоспоринами (или карбапенемами) не являются клинически доступными. Данный факт, в сочетании с увеличением использования цефалоспоринов и карбапенемов новейшего поколения, служит движущим фактором отбора и распространения новых вариантов бета-лактамаз (ESBL, карбапанемазы, хромосомальные и плазмид-опосредованные цефалоспориназы класса С и оксасиллиназы класса D и т.д.). Сохраняя хорошую ингибиторную активность в отношении ESBL, ингибиторы бета-лактамаз предыдущих поколений в значительной степени неэффективны против новых карбапанемаз класса А и класса В, против хромосомальных и плазмид-опосредованных цефалоспориназ класса С и против многих из оксасиллиназ класса D.
[0024] Для решения этой проблемы растущей терапевтический уязвимости, и с учетом того, что имеются три основных молекулярных класса бета-лактамаз с остатком серина в активном центре, и один крупный класс металло-бета-лактамаз, и каждый из данных классов содержит значительное число вариантов бета-лактамаз, авторы определили подход для разработки новых ингибиторов бета-лактамаз с широким функциональным спектром. В частности, авторы определили подход для разработки соединений, которые активны в отношении бета-лактамазных ферментов, как с остатком серина в активном центре, так и с атомом металла в активном центре. Соединения по настоящему изобретению демонстрируют высокую активность в отношении всех четырех основных классах бета-лактамаз.
[0025] Настоящее изобретение относится к некоторым соединениям на основе бора (бороновые кислоты и циклические сложные эфиры бороновых кислот), которые являются ингибиторами бета-лактамазы и антибактериальными соединениями. Соединения и их фармацевтически приемлемые соли могут быть использованы отдельно или в сочетании с бета-лактамными антибиотиками при лечении бактериальных инфекций, устойчивых к антибиотикам, особенно, бактериальных инфекций. Некоторые варианты осуществления включают в себя соединения, композиции, фармацевтические композиции, их применение и получение.
Определения
[0026] В последующем описании некоторые конкретные детали изложены для того, чтобы обеспечить полное понимание различных вариантов осуществления. Тем не менее специалисту в данной области техники будет понятно, что изобретение может быть реализовано на практике без таких подробностей. В других случаях хорошо известные структуры не были показаны или описаны подробно, чтобы избежать излишнего усложнения описаний вариантов осуществления. Если контекст не требует иного, во всем описании и в формуле изобретения, которая следует, слово "содержать" и его вариации, такие как "содержит" и "содержащий" должны толковаться в открытом включительном смысле, то есть как "включая, но не ограничиваясь ими". Дополнительно, заголовки, представленные здесь, предназначены только для удобства, а не для интерпретации объема и смысла заявленного изобретения.
[0027] Ссылка в данном описании на "один вариант осуществления" или "вариант осуществления" означает, что конкретный признак, структура или характеристика, описанные в связи с вариантом осуществления, включены по меньшей мере в один вариант осуществления. Таким образом, появления фраз "в одном варианте осуществления" или "в варианте осуществления" в различных местах по всему данному описанию не обязательно относятся к одному варианту осуществления изобретения. Дополнительно, конкретные признаки, структуры или характеристики могут быть объединены любым подходящим образом в одном или нескольких вариантах осуществления. Кроме того, как используется в данном описании и прилагаемой формуле изобретения, формы единственного числа "a," "an" и "the" включают ссылки на множественное число, если из содержания явно не следует иное. Следует также отметить, что термин "или" обычно используется в значении, включающем "и/или", если из содержания явно не следует иное.
[0028] Термин "антибиотик" относится к соединению или композиции, которое уменьшает жизнеспособность микроорганизма или которое ингибирует рост или пролиферацию микроорганизма. Фраза "ингибирует рост или пролиферацию" означает увеличение времени генерации (т.е. времени, необходимого для деления бактериальной клетки или для удвоения популяции) по меньшей мере приблизительно в 2 раза. Предпочтительными антибиотиками являются те, которые могут увеличить время генерации по меньшей мере приблизительно в 10 раз или более (например, по меньшей мере приблизительно в 100 раз или даже до бесконечности, как в случае гибели всех клеток). Как используется в данном описании, антибиотик дополнительно предназначен для того, чтобы включать противомикробное, бактериостатическое или бактерицидное средство. Примеры антибиотиков, подходящих для использования по отношению к настоящему изобретению, включают пенициллины, цефалоспорины и карбапенемы.
[0029] Термин "β-лактамный антибиотик" относится к соединению с антибиотическими свойствами, которое содержит β-лактамную функциональную группу. Неограничивающие примеры β-лактамных антибиотиков, которые можно применять по отношению к настоящему изобретению, включают пенициллины, цефалоспорины, пенемы, карбапенемы и монобактамы.
[0030] Термин "β-лактамаза" обозначает белок, способный к инактивации β-лактамного антибиотика. β-Лактамаза может быть ферментом, который катализирует гидролиз β-лактамного кольца β-лактамного антибиотика. Особый интерес в данном описании представляют микробные β-лактамазы. β-лактамаза может быть, например, β-лактамазой, содержащей в активном центре остаток серина, или металло-β-лактамазой. Интересующие β-лактамазы включают те, которые описаны на постоянном сайте, который отслеживает номенклатуру бета-лактамаз (www.lahey.org), и в публикации Bush, K. and G.A. Jacoby. 2010. An updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 54: 969-976. β-Лактамазы, представляющие особый интерес в данном описании, включают β-лактамазы, найденные в бактериях, такие как β-лактамазы класса А, включая подклассы SHV, СТХ-М и KPC, β-лактамазы класса В, такие как VIM, β-лактамазы класса С (как хромосомальные, так и плазмид-опосредованные) и β-лактамазы класса D. Термин "ингибитор β-лактамазы" относится к соединению, которое способно ингибировать β-лактамазную активность. Подавление β-лактамазной активности означает ингибирование активности β-лактамазы класса А, В, С или D. Для антимикробных приложений ингибирование на уровне 50% ингибирующей концентрации предпочтительно достигается на уровне, равном или ниже около 100 мкг/мл, или на уровне, равном или ниже около 50 мкг/мл, или на уровне, равном или ниже около 25 мкг/мл. Термины β-лактамазы "класса А", "класса В", "класса С" и "класса D" понятны специалистам в данной области техники и описаны в публикации Bush, K. and G.A. Jacoby. 2010. An updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 54: 969-976.
[0031] Термины ниже, как они использованы в настоящем описании, имеют следующие значения, если не указано иное:
[0032] Термин "амино" относится к радикальной группе -NH2.
[0033] "Циано" или "нитрил" относится к радикальной группе -CN.
[0034] "Гидрокси" или "гидроксил" относится к радикальной группе -ОН.
[0035] "Нитро" относится к радикальной группе -NO2.
[0036] "Оксо" относится к заместителю =O.
[0037] "Оксим" относится к заместителю =N-OH.
[0038] "Тиоксогруппа" относится к заместителю =S.
[0039] Термин "алкил" относится к насыщенной углеводородной монорадикальной группе с необязательно замещенной прямой цепью или необязательно замещенной разветвленной цепью, имеющей от одного до десяти атомов углерода, более предпочтительно от одного до шести атомов углерода, в которой sp3-гибридизированный углерод алкильного остатка присоединен к остальной части молекулы одинарной связью. Примеры включают, но не ограничиваются ими, метил, этил, n-пропил, изопропил, 2-метил-1-пропил, 2-метил-2-пропил, 2-метил-1-бутил, 3-метил-1-бутил, 2-метил-3-бутил, 2,2-диметил-1-пропил, 2-метил-1-пентил, 3-метил-1-пентил, 4-метил-1-пентил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 2,2-диметил-1-бутил, 3,3-диметил-1-бутил, 2-этил-1-бутил, n-бутил, изобутил, втор-бутил, трет-бутил, n-пентил, изопентил, неопентил, трет-амил и гексил, и алкильные группы большего размера, такие как гептил, октил и тому подобное. Всякий раз, когда он появляется в данном документе, числовой диапазон, такой как "С1-С6-алкил" или "C1-6-алкил", означает, что алкильная группа может состоять из 1-го атома углерода, 2-х атомов углерода, 3-х атомов углерода, 4-х атомов углерода, 5-ти атомов углерода или 6-ти атомов углерода, хотя настоящее определение также охватывает появление термина "алкил", где нет обозначения числового диапазона. Если не указано иное, специально в данном описании, алкильная группа может быть необязательно замещена, как описано ниже, например, оксогруппой, аминогруппой, нитрилом, нитрогруппой, гидроксильной группой, алкилом, алкиленом, алкинилом, алкокси-группой, арилом, циклоалкилом, гетероциклилом, гетероарилом и подобными.
[0040] Термин "алкенил" относится к углеводородной монорадикальной группе с необязательно замещенной прямой цепью или необязательно замещенной разветвленной цепью, имеющей одну или более углерод-углеродных двойных связей и имеющей от двух до десяти атомов углерода, более предпочтительно от двух до около шести атомов углерода, где sp2-гибридизированный углерод алкенильного остатка присоединен к остальной части молекулы одинарной связью. Группа может находиться либо в цис-, либо в транс-конформации у двойной связи(ей), и следует понимать, что она включает оба изомера. Примеры включают, но не ограничиваются ими, этенил (-CH=CH2), 1-пропенил (-CH2CH=CH2), изопропенил [-С(CH3)=CH2], бутенил, 1,3-бутадиенил и тому подобное. Всякий раз, когда он появляется в данном документе, числовой диапазон, такой как "С2-С6-алкенил" или "С2-6-алкенил" означает, что алкенильная группа может состоять из 2-х атомов углерода, 3-х атомов углерода, 4-х атомов углерода, 5-ти атомов углерода или 6-ти атомов углерода, хотя настоящее определение также охватывает появление термина "алкенил", где нет обозначения числового диапазона.
[0041] Термин "алкинил" относится к углеводородной монорадикальной группе с необязательно замещенной прямой цепью или необязательно замещенной разветвленной цепью, имеющей одну или более углерод-углеродных тройных связей и имеющей от двух до десяти атомов углерода, более предпочтительно от двух до около шести атомов углерода. Примеры включают, но не ограничиваются ими, этинил, 2-пропинил, 2-бутинил, 1,3-бутадинил и тому подобное. Всякий раз, когда он появляется в данном документе, числовой диапазон, такой как "С2-С6-алкинил" или "С2-6-алкинил", означает, что алкинильная группа может состоять из 2-х атомов углерода, 3-х атомов углерода, 4-х атомов углерода, 5-ти атомов углерода или 6-ти атомов углерода, хотя настоящее определение также охватывает появление термина "алкинил", где нет обозначения числового диапазона.
[0042] "Алкилен" или "алкиленовая цепь" относится к прямой или разветвленной двухвалентной углеводородной цепи. Если не указано иное, специально в данном описании, алкиленовая группа может быть необязательно замещена, как описано ниже.
[0043] Термин "алкокси" относится к радикальной группе формулы -ORa, где Ra представляет собой алкильный радикал, как определено. Если не указано иное, специально в данном описании, алкокси-группа может быть необязательно замещена, как описано ниже.
[0044] Термин "арил" относится к радикальной группе, полученной из углеводородной кольцевой системы, содержащей водород, от 6 до 30 атомов углерода и по меньшей мере одно ароматическое кольцо. Арильная радикальная группа может быть моноциклической, бициклической, трициклической или тетрациклической кольцевой системой, которая может включать конденсированные или мостиковые кольцевые системы. Арильные радикальные группы включают, но не ограничиваются ими, арильные радикалы, полученные из углеводородных кольцевых систем ацеантрилена, аценафтилена, ацефенантрилена, антрацена, азулена, бензола, хризена, фторантена, флуорена, as-индацена, s-индацена, индана, индена, нафталина, феналена, фенантрена, плейадена, пирена и трифенилена. Если не указано иное, специально в данном описании, термин "арил" или префикс "ар" (например, в термине "аралкил") предназначен для включения арильных радикалов, которые являются необязательно замещенными.
[0045] Термин "циклоалкил" или "карбоцикл" относится к стабильному, неароматическому, моноциклическому или полициклическому карбоциклическому кольцу, которое может включать конденсированные или мостиковые кольцевые системы, которое является насыщенным или ненасыщенным. Типичные циклоалкилы или карбоциклы включают, но не ограничиваются ими, циклоалкилы, имеющие от трех до пятнадцати атомов углерода, от трех до десяти атомов углерода, от трех до восьми атомов углерода, от трех до шести атомов углерода, от трех до пяти атомов углерода или от трех до четырех атомов углерода. Моноциклические карбоциклы или циклоалкилы включают, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил, циклогептил и циклооктил. Полициклические карбоциклы или циклоалкилы включают, например, адамантил, норборнил, декалинил, бицикло[3.3.0]октан, бицикло[4.3.0]нонан, цис-декалин, транс-декалин, бицикло[2.1.1]гексан, бицикло[2.2.1]гептан, бицикло[2.2.2]октан, бицикло[3.2.2]нонан, и бицикло[3.3.2]декан, и 7,7 диметил-бицикло[2.2.1]гептанил. Если иное специально не оговорено в описании, циклоалкильная или карбоциклическая группа может быть необязательно замещена. Иллюстративные примеры циклоалкильных групп включают, но не ограничиваются ими, следующие фрагменты:

 и тому подобное.
и тому подобное.
[0046] Термин "аралкил" означает радикальную группу формулы -(алкилен)-R, в которой R представляет собой арил, как определено выше.
[0047] "Циклоалкилалкил" означает радикальную группу формулы -(алкилен)-R, в которой R представляет собой циклоалкил, как определено выше; например, циклопропилметил, циклобутилметил, циклопентилэтил или циклогексилметил и тому подобное.
[0048] Термин "конденсированный" относится к любой кольцевой структуре, описанной в настоящем изобретении, которая присоединена к существующей кольцевой структуре. Когда конденсированное кольцо представляет собой гетероциклическое кольцо или гетероарильное кольцо, любой атом углерода в существующей кольцевой структуре, которая становится частью конденсированного гетероциклического кольца или конденсированного гетероарильного кольца, может быть замещен атомом азота.
[0049] "Гало" или "галоген" относится к брому, хлору, фтору или иоду.
[0050] "Галогеналкил" относится к алкильной радикальной группе, как определено выше, которая замещена одной или более галогеновыми радикальными группами, как определено выше, такой как трифторметил, дифторметил, фторметил, трихлорметил, 2,2,2-трифторэтил, 1,2-дифторэтил, 3-бром-2-фторпропил, 1,2-дибромэтил и тому подобное. Если не указано иное, специально в данном описании, галогеналкильная группа может быть необязательно замещена.
[0051] "Галогеналкокси" аналогично относится к радикальной группе формулы -ORa, в которой Ra представляет собой галогеналкильную радикальную группу, как определено. Если не указано иное, специально в данном описании, галогеналкокси-группы могут быть необязательно замещены, как описано ниже.
[0052] Термин "гетероциклоалкил" или "гетероциклил" или "гетероциклическое кольцо" или "гетероцикл" относится к стабильному, 3-24-членному неароматическому кольцевому радикалу, содержащему от 2 до 23 атомов углерода и от одного до 8 гетероатомов, выбранных из группы, состоящей из атома азота, атома кислорода, атома фосфора и атома серы. Если не указано иное, специально в данном описании, гетероциклический радикал может быть моноциклической, бициклической, трициклической или тетрациклической кольцевой системой, которая может включать конденсированные или мостиковые кольцевые системы; и атомы азота, углерода или серы в гетероциклильном радикале могут быть необязательно окислены; атом азота может быть необязательно кватернизирован; и гетероциклический радикал может быть частично или полностью насыщенным. Примеры таких гетероциклических радикалов включают, но не ограничиваются ими, азетидинил, диоксоланил, тиенил[1,3]дитианил, декагидроизохинолил, имидазолинил, имидазолидинил, изотиазолидинил, изоксазолидинил, морфолинил, октагидроиндолил, октагидроизоиндолил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, оксазолидинил, пиперидинил, пиперазинил, 4-пиперидонил, пирролидинил, пиразолидинил, хинуклидинил, тиазолидинил, тетрагидрофуранил, тритианил, тетрагидропиранил, тиоморфолинил, тиаморфолинил, 1-оксо-тиоморфолинил, 1,1-диоксо-тиоморфолинил, 12-краун-4, 15-краун-5, 18-краун-6, 21-краун-7, аза-18-краун-6, диаза-18-краун-6, аза-21-краун-7 и диаза-21-краун-7. Если не указано иное, специально в данном описании, гетероциклическая группа могут быть необязательно замещена. Иллюстративные примеры гетероциклоалкильных групп, называемых также неароматическими гетероциклами, включают в себя:
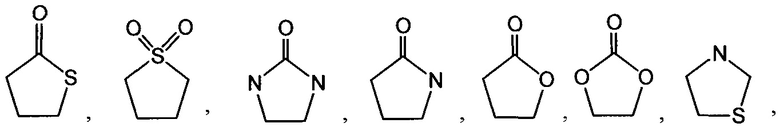
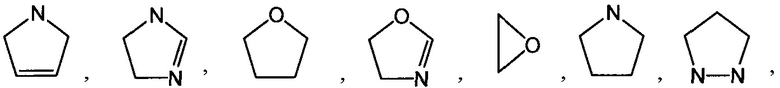


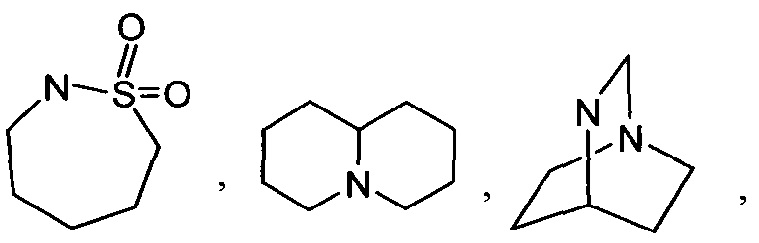
и тому подобное. Термин "гетероциклоалкил" также включает в себя все кольцевые формы углеводов, включая, но не ограничиваясь ими, моносахариды, дисахариды и олигосахариды. Если не указано иное, гетероциклоалкилы имеют в кольце от 2 до 10 атомов углерода. Понятно, что, при обращении к числу атомов углерода в гетероциклоалкиле, число атомов углерода в гетероциклоалкиле не совпадает с общим числом атомов (включая гетероатомы), которые составляют гетероциклоалкил (т.е. скелетных атомов гетероциклоалкильного кольца). Если не указано иное, специально в данном описании, гетероциклоалкильная группа может быть необязательно замещена.
[0053] "Гетероарил" относится к радикальной группе, представляющей собой от 5- до 14-членную кольцевую систему, содержащей атомы водорода, от одного до тринадцати атомов углерода, от одного до шести гетероатомов, выбранных из группы, состоящей из атома азота, атома кислорода, атома фосфора и атома серы, и по меньшей мере одно ароматическое кольцо. Для целей настоящего изобретения гетероарильный радикал может быть моноциклической, бициклической, трициклической или тетрациклической кольцевой системой, которая может включать конденсированные или мостиковые кольцевые системы; и атомы азота, углерода или серы в гетероарильном радикале могут быть необязательно окислены; атом азота может быть необязательно кватернизирован. Примеры включают, но не ограничиваются ими, азепинил, акридинил, бензимидазолил, бензотиазолил, бензиндолил, бензодиоксолил, бензофуранил, бензоксазолил, бензотиазолил, бензотиадиазолил, бензо[b][1,4]диоксипинил, 1,4-бензодиоксанил, бензонафтофуранил, бензоксазолил, бензодиоксолил, бензодиоксинил, бензопиранил, бензопиранонил, бензофуранил, бензофуранонил, бензотиенил (бензотиофенил), бензотриазолил, бензо[4,6]имидазо[1,2-а]пиридинил, карбазолил, циннолинил, дибензофуранил, дибензотиофенил, фуранил, фуранонил, изотиазолил, имидазолил, индазолил, индолил, индазолил, изоиндолил, индолинил, изоиндолинил, изохинолинил, индолизинил, изоксазолил, нафтиридинил, оксадиазолил, 2-оксоазепинил, оксазолил, оксиранил, 1-оксидопиридинил, 1-оксидопиримидинил, 1-оксидопиразинил, 1-оксидопиридазинил, 1-фенил-1H-пирролил, феназинил, фенотиазинил, феноксазинил, фталазинил, птеридинил, пуринил, пирролил, пиразолил, пиридинил, пиразинил, пиримидинил, пиридазинил, хиназолинил, хиноксалинил, хинолинил, хинуклидинил, изохинолинил, тетрагидрохинолинил, тиазолил, тиадиазолил, триазолил, тетразолил, триазинил и тиофенил (то есть тиенил). Если не указано иное, специально в данном описании, гетероарильная группа может быть необязательно замещена.
[0054] Все вышеперечисленные группы могут быть либо замещенными, либо незамещенными. Термин "замещенный", используемый в настоящем описании, означает, что любая из упомянутых выше групп (например, алкил, алкилен, алкокси, арил, циклоалкил, галогеналкил, гетероциклил и/или гетероарил) может быть дополнительно функционализирована, где по меньшей мере один атом водорода заменен связью с заместителем, являющимся атомом, отличным от водорода. Если не указано специально в данном описании, замещенная группа может содержать один или более заместителей, выбранных из: оксогруппы, аминогруппы, -CO2H, нитрила, нитрогруппы, гидроксила, тиоокси-группы, алкила, алкилена, алкокси-группы, арила, циклоалкила, гетероциклила, гетероарила, диалкиламинов, ариламинов, алкилариламинов, диариламинов, триалкиламмоний(-N+R3), N-оксидов, имидов и енаминов; атом кремния в таких группах, как триалкилсилильные группы, диалкиларилсилильные группы, алкилдиарилсилильные группы, триарилсилильные группы, перфторалкил или перфторалкокси-группа, например, трифторметил или трифторметокси-группа. "Замещенный" также означает любую из упомянутых выше групп, в которой один или несколько атомов водорода замещены связью более высокого порядка (например, двойной или тройной связью) с гетероатомом, таким, как кислород в оксо-, карбонильной, карбоксильной и эфирной группах; и азот в таких группах, как имины, оксимы, гидразоны и нитрилы. Например, "замещенный" включает любую из вышеупомянутых групп, в которой один или несколько атомов водорода замещены -NH2, -NRgC(=O)NRgRh, -NRgC(=O)ORh, -NRgSO2Rh, -OC(=O)NRgRh, -ORg, -SRg, -SORg, -SO2Rg, -OSO2Rg, -SO2ORg, =NSO2Rg и -SO2NRgRh. В изложенном выше Rg и Rh являются одинаковыми или различными и независимо представляют собой атом водорода, алкил, алкокси-группу, алкиламиногруппу, тиоалкил, арил, аралкил, циклоалкил, циклоалкилалкил, галогеналкил, гетероциклил, N-гетероциклил, гетероциклилалкил, гетероарил, N-гетероарил и/или гетероарилалкил. В дополнение, каждый из упомянутых выше заместителей может быть необязательно замещен одним или более из упомянутых выше заместителей. Кроме того, любая из вышеупомянутых групп может быть замещена, чтобы включать в себя один или более внутренних атомов кислорода, серы или азота. Например, алкильная группа может быть замещена одним или более внутренних атомов кислорода с образованием эфирной или полиэфирной группы. Подобным образом, алкильная группа может быть замещена одним или более внутренних атомов серы с образованием тиоэфира, дисульфида и т.д.
[0055] Термин "необязательный" или "необязательно" означает, что описанное далее событие или обстоятельство может произойти или может не произойти, и что описание включает случаи, когда упомянутое событие или обстоятельство имеет место, и случаи, в которых это не так. Например, "необязательно замещенный алкил" означает либо "алкил", либо "замещенный алкил", как определено выше. Кроме того, необязательно замещенная группа может быть незамещенной (например, -CH2CH3), полностью замещенной (например, -CF2CF3), монозамещенной (например, -CH2CH2F) или замещенной на каком-либо промежуточном уровне между полностью замещенной и монозамещенной (например, -CH2CHF2, -CH2CF3, -CF2CH3, -CFHCHF2 и т.д.). Специалистам в данной области техники будет понятно в отношении любой группы, содержащей один или несколько заместителей, что такие группы не предназначены для введения какой-либо замены или паттернов замен (например, замещенный алкил включает в себя необязательно замещенные циклоалкильные группы, которые, в свою очередь, определяются как включающие необязательно замещенные алкильные группы, потенциально до бесконечности), которые являются пространственно нереализуемыми и/или синтетически неосуществимыми. Таким образом, любые описанные заместители в целом следует понимать как имеющие максимальную молекулярную массу, составляющую около 1000 дальтон, а более типично, до около 500 дальтон.
[0056] Термин "эффективное количество" или "терапевтически эффективное количество" относится к количеству соединения, вводимого млекопитающему, либо в виде единичной дозы, либо в виде части серии доз, которое является эффективным для получения желаемого терапевтического эффекта,
[0057] Термин "лечение" индивидуума (например млекопитающего, такого как человек) или клетки представляет собой любой тип вмешательства, используемый в попытке изменить естественный ход событий у индивидуума или клетки. В некоторых вариантах осуществления лечение включает введение фармацевтической композиции после начала патологического события или контакта с возбудителем и включает стабилизацию состояния (например, состояние не ухудшается) или облегчения состояния. В других вариантах осуществления лечение также включает профилактическое лечение (например, введение композиции, описанной в настоящем описании, когда имеется подозрение, что индивидуум страдает от бактериальной инфекции).
[0058] "Таутомер" относится к протонному переходу от одного атома в молекуле к другому атому той же молекулы. Соединения, представленные в настоящем документе, могут существовать в виде таутомеров. Таутомеры представляют собой соединения, которые являются взаймопревращаемыми посредством переноса атома водорода, сопровождаемого "взаимным перебросом" (switch) одинарной связи и прилегающей двойной связи. В системах связей, где возможна таутомеризация, будет существовать химическое равновесие таутомеров. Рассматриваются все таутомерные формы соединений, описанных в данном документе. Точное соотношение таутомеров зависит от нескольких факторов, в том числе от температуры, растворителя и значения рН. Некоторые примеры таутомерных взаимопревращений включают:
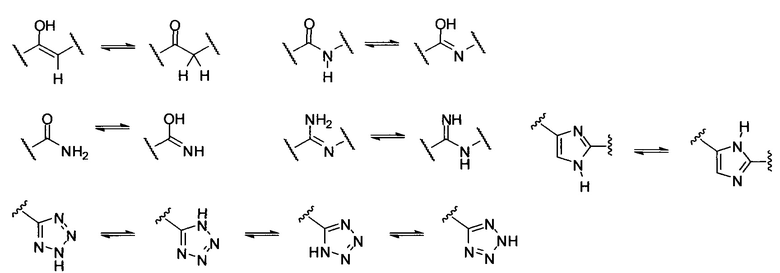
[0059] "Метаболит" соединения, раскрытого в настоящем описании, является производным данного соединения, которое образуется, когда соединение метаболизируется. Термин "активный метаболит" относится к биологически активному производному соединения, которое образуется, когда соединение метаболизируется. Термин "метаболизируется", как используется в настоящем описании, относится к сумме процессов (в том числе, но не ограничиваясь ими, реакциям гидролиза и реакциям, катализируемых ферментами, таким, как окислительные реакции), при которых конкретное вещество изменяется организмом. Таким образом, ферменты могут продуцировать специфические структурные изменения в соединении. Например, цитохром Р450 катализирует различные окислительные и восстановительные реакции, тогда как уридиндифосфатглюкуронилтрансферазы катализируют перенос активированной молекулы глюкуроновой кислоты на ароматические спирты, алифатические спирты, карбоновые кислоты, амины и свободные сульфгидрильные группы. Более подробную информацию о метаболизме можно получить из руководства The Pharmacological Basis of Therapeutics, 9th Edition, McGraw-Hill (1996). Метаболиты соединений, раскрытых в настоящем описании, могут быть определены либо введением соединений хозяину и анализом образцов ткани, полученных от хозяина, либо инкубацией соединений с печеночными клетками in vitro и анализом полученных соединений. Оба способа хорошо известны в данной области техники. В некоторых вариантах осуществления метаболиты соединения образуются посредством окислительных процессов и соответствуют соответствующему гидрокси-содержащему соединению. В некоторых вариантах осуществления соединение метаболизируется до фармакологически активных метаболитов.
Соединения
[0060] В настоящем изобретении описаны соединения, которые модулируют активность бета-лактамазы. В некоторых вариантах осуществления описанные в настоящем изобретении соединения ингибируют бета-лактамазы. В некоторых вариантах осуществления описанные в настоящем изобретении соединения являются применимыми в лечении бактериальных инфекций. В некоторых вариантах осуществления бактериальная инфекция представляет собой инфекцию верхних или нижних дыхательных путей, инфекцию мочевыводящих путей, внутрибрюшную инфекцию или инфекцию кожи.
[0061] В одном из аспектов настоящего изобретения предложены соединения формулы I или формулы Ia или их фармацевтически приемлемые соли, сольваты, полиморфы, стереоизомеры, таутомеры, пролекарства, метаболиты, N-оксиды или изомеры:

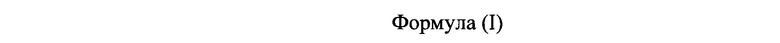
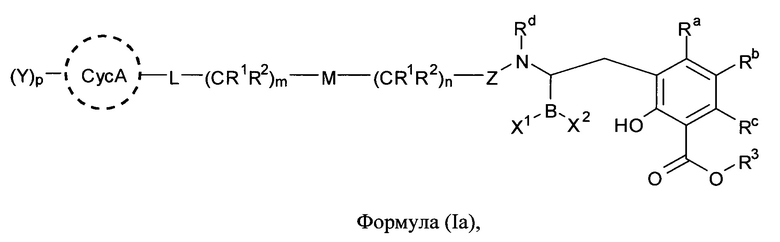
где:
L представляет собой связь, -CR1R2-, >С=O, или =CR1-;
М представляет собой связь, -O-, -S-, -S(O)-, SO2- или -N(R4)-;
m равно 0, 1 или 2;
n равно 0, 1, 2 или 3;
при условии, что,
когда n равно 0, то М представляет собой связь;
р равно 0, 1, 2, 3 или 4;
при условии, что
когда р равно 0, то L представляет собой -CR1R2- или =CR1-;
X1 и X2 независимо выбраны из -ОН, -OR8 или F;
Z представляет собой >С=O, >C=S, или >SO2;
СусА представляет собой необязательно замещенный 3-10-членный неароматический карбоцикл, в котором необязательная олефиновая функциональная группа неароматического карбоцикла не присоединена непосредственно к заместителю - атому кислорода, атому серы или атому азота;
Ra, Rb и Rc независимо выбраны из группы, состоящей из атома водорода, атома фтора, атома хлора, атома брома, необязательно замещенного C1-С6-алкила, необязательно замещенного С3-С6-циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного арила, необязательно замещенного гетероарила, -ОН, -OR10, -NR4R5 и -SR10;
каждый R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома фтора, атома хлора, атома брома, необязательно замещенного C1-С6-алкила, необязательно замещенного С3-С6-циклоалкила, -ОН, -OR10, -SR10 и -NR4R5,
или R1 и R2, взятые вместе, образуют оксогруппу, оксим, или необязательно замещенный карбоцикл, или необязательно замещенный гетероцикл с атомом углерода, к которому они присоединены;
R3 представляет собой атом водорода, необязательно замещенный C1-С6-алкил, или фармацевтически приемлемое пролекарство;
каждый Rd, R4 и R5 независимо выбран из группы, состоящей из атома водорода, -ОН, -CN, необязательно замещенного C1-С6-алкила, необязательно замещенного алкоксиалкила, необязательно замещенного гидроксиалкила, необязательно замещенного аминоалкила, необязательно замещенного циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного арила, необязательно замещенного гетероарила, необязательно замещенного циклоалкилалкила, необязательно замещенного гетероциклилалкила, необязательно замещенного аралкила, необязательно замещенного гетероаралкила, (полиэтиленгликоль)этила и необязательно замещенного сахарида;
или R4 и R5, взятые вместе, образуют необязательно замещенный гетероцикл с атомом азота, к которому они присоединены;
R8 представляет собой необязательно замещенный C1-С6-алкил, необязательно замещенный С3-С6-циклоалкил или фармацевтически приемлемую боронатную сложноэфирную группу;
R10 обозначает необязательно замещенный C1-С6-алкил или необязательно замещенный С3-С6-циклоалкил
и каждый Y независимо представляет собой группу, содержащую от 1 до 50 атомов, отличных от атома водорода, выбранных из группы, состоящей из С, N, О, S и Р.
[0062] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb и Rc независимо выбраны из группы, состоящей из атома водорода, атома фтора, атома хлора, необязательно замещенного C1-С6-алкила, необязательно замещенного С3-С6-циклоалкила, -ОН, -OR10, -NR4R5 и -SR10. В некоторых вариантах осуществления Ra, Rb и Rc независимо представляют собой атом водорода, атом фтора или атом хлора. В предпочтительных вариантах осуществления Ra, Rb и Rc представляют собой атом водорода.
[0063] В некоторых вариантах осуществления соединения формулы I или формулы Ia R3 представляет собой атом водорода, метил, этил, пропил, бутил или изопропил. В предпочтительных вариантах осуществления R3 представляет собой атом водорода.
[0064] В некоторых вариантах осуществления соединения формулы I или формулы Ia X1 и X2 представляют собой -ОН.
[0065] В некоторых вариантах осуществления соединения формулы I или формулы Ia Rd представляет собой атом водорода или С1-С4-алкил. В предпочтительных вариантах осуществления Rd представляет собой атом водорода.
[0066] В некоторых вариантах осуществления соединения формулы I или формулы Ia Z представляет собой >С=O или >SO2. В предпочтительных вариантах осуществления Z представляет собой >С=O.
[0067] В некоторых вариантах осуществления соединения формулы I или формулы Ia L представляет собой -CR1R2- или =CR1-. В некоторых вариантах осуществления L представляет собой связь. В некоторых вариантах осуществления соединения формулы I или формулы Ia M представляет собой -O-, -S-, -SO2- или -N(R4)-. В некоторых вариантах осуществления М представляет собой связь или -O-. В дальнейших вариантах осуществления М представляет собой связь. В некоторых вариантах осуществления соединения формулы I или формулы Ia m равно 0 или 1. В некоторых вариантах осуществления m равно 0. В других вариантах осуществления m равно 1. В некоторых вариантах осуществления соединения формулы I или формулы Ia n равно 1 или 2. В некоторых вариантах осуществления n равно 1. В дальнейших вариантах осуществления n равно 0. В других вариантах осуществления n равно 2. В некоторых вариантах осуществления m и n равны 0. В некоторых вариантах осуществления m и n равны 1.
[0068] В некоторых вариантах осуществления соединения формулы I или формулы Ia L представляет собой -CR1R2- или =CR1-; М представляет собой -O-, -S-, -SO2- или -N(R4)-; m равно 0 или 1; и n равно 1 или 2. В некоторых вариантах осуществления L представляет собой связь, -CR1R2- или =CR1-; М представляет собой связь или -O-; m равно 0; и n равно 1 или 2. В дальнейших вариантах осуществления L представляет собой связь или >С=O; М представляет собой связь или -N(R4)-; и m и n равны 0. В некоторых вариантах осуществления L представляет собой >С=O; М представляет собой -N(R4)-; и m и n равны 0. В некоторых вариантах осуществления L представляет собой связь; М представляет собой связь; и m и n равны 0. В других вариантах осуществления L представляет собой связь; М представляет собой связь; и m или n равны 1. В некоторых вариантах осуществления L представляет собой -CR1R2- или =CR1-; М представляет собой связь; и m и n равны 0. В некоторых вариантах осуществления L представляет собой -CR1R2- или =CR1-; М представляет собой связь; и m и n равны 1.
[0069] В некоторых вариантах осуществления соединения формулы I или формулы Ia СусА выбран из группы, состоящей из циклопропана, циклобутана, циклопентана, циклогексана, циклогептана, циклооктана, циклопентена, циклогексена, циклогептена и циклооктена, где олефиновая функциональная группа циклопентена, циклогексена, циклогептена и циклооктена не присоединена непосредственно к заместителю - атому кислорода, атому серы или атому азота. В некоторых вариантах осуществления СусА представляет собой циклобутан, циклопентан, циклогексан или циклогексен, где олефиновая функциональная группа циклогексена не присоединена непосредственно к заместителю - атому кислорода, атому серы или атому азота. В других вариантах осуществления СусА выбран из группы, состоящей из бицикло[3.3.0]октана, бицикло[4.3.0]нонана, цис-декалина, транс-декалина, бицикло[2.1.1]гексана, бицикло[2.2.1]гептана, бицикло[2.2.2]октана, бицикло[3.2.2]нонана и бицикло[3.3.2]декана. В некоторых вариантах осуществления СусА представляет собой циклопентан. В предпочтительных вариантах осуществления СусА представляет собой циклогексан. В некоторых вариантах осуществления СусА представляет собой циклогексан, ковалентно связанный с одним Y и L; упомянутые ковалентные связи в 1,4-транс-расположении.
[0070] В некоторых вариантах осуществления соединения формулы I или формулы Ia каждый Y выбирают из группы, состоящей из
атома фтора, атома хлора, атома брома, необязательно замещенного C1-С6-алкила, необязательно замещенного С3-С6-циклоалкила, необязательно замещенного гетероцикла, необязательно замещенного арила, необязательно замещенного гетероарила, =O, -ОН, -OR10, -SR10, -NR4R5, -(CR6R7)vNR4R5, -(CR6R7)vNR4R5(CR6R7)vNR4R5, -NR4R5(CR6R7)vR6, -NR4(CR6R7)vNR4R5, -NR4(CR6R7)vNR4R5(CR6R7)vNR4R5, -O(CR6R7)vNR4R5, -S(O)0,1,2(CR6R7)vNR4R5, -N(R4)C(O)(CR6R7)vNR4R5, -(CR6R7)vN(R4)C(O)(CR6R7)vNR4R5, -(CR6R7)vNR4(CR6R7)vNR4R5, -NR4(CR6R7)vOR10, -NR4(CR6R7)vS(O)0,1,2R10, -C(O)NR4(CR6R7)vNR4R5, -S(O)0,1,2NR4(CR6R7)vNR4R5, -NR5C(O)NR4(CR6R7)vNR4R5, -OC(O)NR4(CR6R7)vNR4R5, -NR5C(=NR7)NR4(CR6R7)vNR4R5, -N(R4)C(=NR5)R6, -(CR6R7)vN(R4)C(=NR5)R6, -NR4(CR6R7)vN(R4)C(=NR5)R6, -O(CR6R7)vN(R4)C(=NR5)R6, -S(O)0,1,2(CR6R7)vN(R4)C(=NR5)R6, -(CR6R7)vC(=NR5)NR4R5, -NR4(CR6R7)vC(=NR5)NR4R5, -O(CR6R7)vC(=NR5)NR4R5, -S(O)0,1,2(CR6R7)vC(=NR5)NR4R5, -(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4(CR6R7)vN(R4)C(=NR5)NR4R5, -O(CR6R7)vN(R4)C(=NR5)NR4R5, -S(O)0,1,2(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4C(=NR5)NR4C(=NR5)NR4R5, -(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4(CR6R7)vNR4C(=NR4)NR4R5, -O(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -S(O)0,1,2-(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4C(=NR5)NR4R5, -C(=NR4)NR4R5, -C(=NR4)NR4C(O)R6, -NR4SO2R6, -NR4C(O)R6, -NR4C(=O)OR6, -C(O)NR4R5, -(CR6R7)vC(O)NR4R5, -SO2NR4R5, -гетероарил-NR4R5, гетероциклил-NR4R5, -гетероарил-N(R4)C(=NR5)NR4R5, -гетероциклил-N(R4)C(=NR5)NR4R5, -N(R4)-гетероарил-NR4R5, -N(R4)-гетероциклил-NR4R5, -(CR6R7)vгетероарил-NR4R5, -(CR6R7)vгетероциклил-NR4R5, -(CR6R7)vгетероарил-N(R4)C(=NR5)NR4R5, -(CR6R7)vгетероциклил-N(R4)C(=NR5)NR4R5, -NR4R5(CR6R7)vгетероциклил-C(=NR5)NR4R5-(CR6R7)vгетероарила, -(CR6R7)vгетероциклила, -О-гетероарила, -О-гетероциклила, -NR4(CR6R7)vгетероарила, -NR4(CR6R7)vгетероциклила, -O(CR6R7)vгетероарила, -O(CR6R7)vгетероциклила, -NR4(CR6R7)vNR5-гетероарила, -NR4(CR6R7)vNR5-гетероциклила, -O(CR6R7)vNR5-гетероарила, -O(CR6R7)vNR5-гетероциклила, -O(CR6R7)vO-гетероциклила, -NR4R5R9+Q-, -(CR6R7)vNR4R5R9+Q-, -NR4(CR6R7)vNR4R5R9+Q-, -NR4R9+(CR6R7)vNR4R5R9+Q-2, -(CR6R7)v(T)+Q- и -O(CR6R7)vNR4R5R9+Q-;
где:
каждый Т независимо выбран из группы, состоящей из пиридин-1-ила, пиримидин-1-ила и тиазол-3-ила;
каждый Q независимо представляет собой фармацевтически приемлемый противоион; и
каждый v независимо обозначает 1, 2, 3 или 4;
или Y, взятый вместе с атомом углерода, к которому он присоединен, образует необязательно замещенный спирокарбоцикл или необязательно замещенный спирогетероцикл;
или два Y, взятые вместе с атомами углерода, к которым они присоединены, образуют необязательно замещенный карбоцикл или необязательно замещенный гетероцикл;
каждый R6 и R7 независимо выбран из группы, состоящей из атома водорода, атома фтора, атома хлора, атома брома, необязательно замещенного C1-С6-алкила, необязательно замещенного алкоксиалкила, необязательно замещенного гидроксиалкила, необязательно замещенного С3-С6-циклоалкила, -ОН, -OR10, -SR10, -NR4R5, -NR4C(O)R5, -NR4C(O)OR5, -NR4C(O)NR5, -C(O)OR5, -C(O)NR4R5, -C(N=R5)NR4R5 -NR4SO2R5, необязательно замещенного гетероциклила, необязательно замещенного арила и необязательно замещенного гетероарила;
или R6 и R7, взятые вместе, образуют оксогруппу, оксим, или необязательно замещенный карбоцикл или необязательно замещенный гетероцикл с атомом углерода, к которому они присоединены;
и каждый R9 представляет собой независимо необязательно замещенный C1-С6-алкил.
[0071] В некоторых вариантах осуществления соединения формулы I или формулы Ia по меньшей мере один Y выбран из группы, состоящей из атома фтора, атома хлора, необязательно замещенного C1-С6-алкила, =O, -ОН, -OR10, -NR4R5, -(CR6R7)vNR4R5, -NR4(CR6R7)vNR4R5, -(CR6R7)vNR4R5(CR6R7)vNR4R5, -NR4R5(CR6R7)vR6, -NR4R5(CR6R7)vгетероциклил-C(=NR5)NR4R5, -NR4(CR6R7)vNR4C(=NR4)NR4R5, -NR4(CR6R7)vNR4R5(CR6R7)vNR4R5, -O(CR6R7)vNR4R5, -N(R4)C(O)(CR6R7)vNR4R5, -(CR6R7)vN(R4)C(O)(CR6R7)vNR4R5, -C(O)NR4(CR6R7)vNR4R5, -S(O)0,1,2NR4(CR6R7)vNR4R5, -NR5C(O)NR4(CR6R7)vNR4R5, -OC(O)NR4(CR6R7)vNR4R5, -NR5C(=NR7)NR4(CR6R7)vNR4R5, -N(R4)C(=NR5)R6, -(CR6R7)vN(R4)C(=NR5)R6, -NR4(CR6R7)vN(R4)C(=NR5)R6, -O(CR6R7)vN(R4)C(=NR5)R6, -(CR6R7)vC(=NR5)NR4R5, -NR4(CR6R7)vC(=NR5)NR4R5, -O(CR6R7)vC(=NR5)NR4R5, -(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4(CR6R7)vN(R4)C(=NR5)NR4R5, -O(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4C(=NR5)NR4C(=NR5)NR4R5, -(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -O(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4C(=NR5)NR4R5, -C(=NR4)NR4R5, -C(=NR4)NR4C(O)R6, -NR4SO2R6, -NR4C(O)R6, -NR4C(=O)OR6, -C(O)NR4R5, -(CR6R7)vC(O)NR4R5, -гетероарил-NR4R5, -гетероциклил-NR4R5, -гетероарил-N(R4)C(=NR5)NR4R5, -гетероциклил-N(R4)C(=NR5)NR4R5, -N(R4)-гетероарил-NR4R5, -N(R4)-гетероциклил-NR4R5, -(CR6R7)vгетероарил-NR4R5, -(CR6R7)vгетероциклил-NR4R5, -(CR6R7)vгетероарил-N(R4)C(=NR5)NR4R5, -(CR6R7)vгетероциклил-N(R4)C(=NR5)NR4R5, -(CR6R7)vгетероарила, -(CR6R7)vгетероциклила, -О-гетероарила, -О-гетероциклила, -NR4(CR6R7)vгетероарила, -NR4(CR6R7)vгетероциклила, -O(CR6R7)vгетероарила, -O(CR6R7)vгетероциклила и -O(CR6R7)vО-гетероциклила. В некоторых вариантах осуществления по меньшей мере один Y выбран из группы, состоящей из атома фтора, необязательно замещенного C1-С6-алкила, -ОН, -NR4R5, -(CR6R7)vNR4R5, -(CR6R7)vNR4R5(CR6R7)vNR4R5, -NR4R5(CR6R7)vR6, -NR4R5(CR6R7)vгетероциклил-C(=NR5)NR4R5, -NR4(CR6R7)vNR4C(=NR4)NR4R5, -NR4(CR6R7)vNR4R5(CR6R7)vNR4R5, -NR4(CR6R7)vNR4R5, -O(CR6R7)vNR4R5, -C(O)NR4(CR6R7)vNR4R5, -NR5C(O)NR4(CR6R7)vNR4R5, -NR5C(=NR7)NR4(CR6R7)vNR4R5, -N(R4)C(=NR5)R6, -(CR6R7)vN(R4)C(=NR5)R6, -NR4(CR6R7)vN(R4)C(=NR5)R6, -(CR6R7)vC(=NR5)NR4R5, -NR4(CR6R7)vC(=NR5)NR4R5, -(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4C(=NR5)NR4C(=NR5)NR4R5, -(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4(CR6R7)vC(=NR4)NR5C(=NR4)NR4R5, -NR4C(=NR5)NR4R5, -C(=NR4)NR4R5, -C(=NR4)NR4C(O)R6, -NR4C(O)R6, -(CR6R7)vC(O)NR4R5, -гетероциклил-NR4R5, -гетероциклил-N(R4)C(=NR5)NR4R5, -N(R4)-гетероциклил-NR4R5, -(CR6R7)vгетероциклил-NR4R5, -(CR6R7)vгетероциклил-N(R4)C(=NR5)NR4R5, -(CR6R7)vгетероциклила и -NR4(CR6R7)vгетероциклила. В других вариантах осуществления по меньшей мере один Y выбран из группы, состоящей из -гетероарил-NR4R5, -гетероциклил-NR4R5, -гетероарил-N(R4)C(=NR5)NR4R5, -гетероциклил-N(R4)C(=NR5)NR4R5, -N(R4)-гетероарил-NR4R5, -N(R4)-гетероциклил-NR4R5, -гетероарил-C(=NR5)NR4R5, -гетероциклил-C(=NR5)NR4R5, -(CR6R7)vгетероарил-NR4R5, -(CR6R7)vгетероциклил-NR4R5, -(CR6R7)vгетероарил-N(R4)C(=NR5)NR4R5 и -(CR6R7)vгетероциклил-N(R4)C(=NR5)NR4R5. В предпочтительных вариантах осуществления по меньшей мере один Y выбран из группы, состоящей из -NR4R5, -NR4C(=NR5)NR4R5, -C(=NR4)NR4R5, -N(R4)C(=NR5)R6,-(CR6R7)vNR4R5,-(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4(CR6R7)vNR4R5, -NR4(CR6R7)vOR10, -(CR6R7)vNR4(CR6R7)vNR4R5, NR5C(=NR5)NR4(CR6R7)vNR4R5, -NR4(CR6R7)vN(R4)C(=NR5)NR4R5, -NR5C(O)CR6(NR4R5)(CR6R7)vNR4R5, -(CR6R7)vC(=NR5)NR4R5, -(CR6R7)vN(R4)C(O)(CR6R7)vNR4R5, -C(=NR4)NR4C(O)R6, -NR4(CR6R7)vгетероарила и -O(CR6R7)vNR4R5.
[0072] В некоторых вариантах осуществления р равно 0, 1, 2, 3 или 4. В некоторых вариантах осуществления р равно 1 или 2. В других вариантах осуществления р равно 1.
[0073] В некоторых вариантах осуществления соединения формулы I или формулы Ia каждый R4 и R5 независимо выбран из группы, состоящей из атома водорода, ОН, необязательно замещенного C1-С6-алкила, необязательно замещенного алкоксиалкила, необязательно замещенный гидроксиалкила, необязательно замещенного гетероциклила. В предпочтительных вариантах осуществления каждый R4 и R5 независимо представляет собой атом водорода или необязательно замещенный C1-С6-алкил.
[0074] В некоторых вариантах осуществления соединения формулы I или формулы Ia каждый R6 и R7 независимо выбран из группы, состоящей из атома водорода, необязательно замещенного C1-С6-алкила, -ОН, -NR4R5 и необязательно замещенного гетероциклила, или R6 и R7, взятые вместе, образуют необязательно замещенный гетероцикл с атомом углерода, к которому они присоединены. В предпочтительных вариантах осуществления каждый R6 и R7 независимо представляет собой атом водорода, атом фтора или необязательно замещенный C1-С6-алкил.
[0075] В некоторых вариантах осуществления соединения формулы I или формулы Ia R6 и R7 независимо выбраны из группы, состоящей из атома водорода, необязательно замещенного C1-С6-алкила, -ОН, -NR4R5 и необязательно замещенного гетероциклила, или R6 и R7, взятые вместе, образуют необязательно замещенный гетероцикл с атомом углерода, к которому они присоединены. В предпочтительных вариантах осуществления R6 и R7 независимо представляют собой атом водорода, фтора или необязательно замещенный C1-С6-алкил. В некоторых вариантах осуществления представляет собой В некоторых вариантах осуществления представляет собой . В некоторых вариантах осуществления Y представляет собой -NR4(CR6R7)vNR4R5. В некоторых вариантах осуществления Y представляет собой -NR4(CR6R7)vNR4C(=NR4)NR4R5. В некоторых вариантах осуществления Y представляет собой -NR4R5. В других вариантах осуществления Y представляет собой -NR4C(=NR4)NR4R5. В некоторых вариантах осуществления Y представляет собой -(CR6R7)vNR4R5. В некоторых вариантах осуществления Y представляет собой -(CR6R7)vNR4C(=NR4)NR4R5. В некоторых вариантах осуществления v равно 2. В некоторых вариантах осуществления v равно 1. В некоторых вариантах осуществления каждый из R4 и R5 выбран из Н, необязательно замещенного C1-С6-алкила или необязательно замещенного С3-С6-циклоалкила. В некоторых вариантах осуществления каждый R4, R6 и R7 представляет собой Н.
[0076] В некоторых вариантах осуществления соединения формулы I или формулы Ia каждый Y определяет включение отличных от водорода атомов. Например, в некоторых вариантах осуществления каждый Y содержит по меньшей мере 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, 16, 18, 20, 24, 28, 32, 36, 40, 50 или 60 отличных от водорода атомов. В некоторых вариантах осуществления каждый Y содержит менее чем 50, 40, 36, 32, 28, 24, 20, 18, 16, 14, 12, 10, 9, 8, 7, 6, 5, 4, 3 или 2 отличных от водорода атомов. В некоторых вариантах осуществления каждый Y независимо представляет собой группу, содержащую от 1 до 50 атомов, отличных от атома водорода. В некоторых вариантах осуществления отличные от водорода атомы представляют собой любой атом, который не является атомом водорода. В некоторых вариантах осуществления атомы, отличные от водорода, представляют собой атомы, которые обычно встречаются в органических молекулах. В некоторых вариантах осуществления атомы, отличные от водорода, представляют собой атомы, выбранные из группы, состоящей из С, N, О, S и Р. В некоторых вариантах осуществления каждый Y независимо представляет собой группу, содержащую от 1 до 50 атомов, отличных от атома водорода, выбранных из группы, состоящей из С, N, О, S и Р.
[0077] В некоторых вариантах осуществления соединения формулы I или формулы Ia каждый Y определяется его молекулярной формулой. Например, в некоторых вариантах осуществления каждый Y имеет формулу CwHxNyOz; где каждый w независимо равен от 0 до 30; каждый х независимо равен от 1 до 69; каждый y независимо равен от 1 до 8; и каждый z независимо равен от 0 до 10. В некоторых вариантах осуществления каждый Y имеет формулу CwHxNyOz; где каждый w независимо равен от 0 до 10; каждый х независимо равен от 1 до 25; каждый y независимо равен от 1 до 4; и каждый z независимо равен от 0 до 3. В некоторых вариантах осуществления каждый y равен 2.
[0078] В некоторых вариантах осуществления соединения формулы I или формулы Ia каждый Y определяется его молекулярной массой. В некоторых вариантах осуществления каждый Y имеет молекулярную массу, составляющую, например, менее 500, 450, 400, 350, 300, 250, 200, 190, 180, 170, 160, 150, 140, 130, 120, 110, 100, 90, 80, 75, 70 или 50 дальтон. В некоторых вариантах осуществления каждый Y имеет молекулярную массу, составляющую менее 200 дальтон. В некоторых вариантах осуществления каждый Y имеет молекулярную массу, составляющую менее 150 дальтон. В некоторых вариантах осуществления каждый Y имеет молекулярную массу, составляющую от 30 до 280 дальтон.
[0079] В некоторых вариантах осуществления соединения формулы I или формулы Ia каждый Y определяется количеством основных атомов азота, которое он содержит. Например, каждый Y может содержать 10, 9, 8, 7, 6, 5, 4, 3, 2, 1 или 0 основных атомов азота. В некоторых вариантах осуществления каждый Y содержит от 1 до 6 основных атома азота. В некоторых вариантах осуществления каждый Y содержит 1, 2 или 3 основных атомов азота. В некоторых вариантах осуществления каждый Y содержит 2 основных атомов азота. В некоторых вариантах осуществления по меньшей мере один Y содержит от 1 до 6 основных атома азота. В некоторых вариантах осуществления по меньшей мере один Y содержит 1, 2 или 3 основных атомов азота. В некоторых вариантах осуществления по меньшей мере один Y состоит из 2-х основных атома азота. Основной атом азота представляет собой атом азота, который может быть, по меньшей мере, частично протежированным в по существу нейтральном водном буфере. Например, основной атом азота может быть атомом азота аминогруппы или атомом азота в функциональной группе, такой как алкиламин, циклоалкильный амин, гетероциклоалкильная группа, содержащая атом азота гетероарильная группа, амидин или гуанидин.
[0080] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; X2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y представляет собой группу, содержащую 2 основных атома азота. В некоторых вариантах осуществления основные атомы азота каждый представляют собой атом в аминогруппе и амидиновой группе, группе гуанидина, гетероцикло-алкильной группе и гетероарильной группе или алкиламиногруппе. В некоторых вариантах осуществления Y содержит две аминогруппы. В некоторых вариантах осуществления Y содержит две группы гуанидина. В некоторых вариантах осуществления Y содержит аминогруппу и группу гуанидина.
[0081] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; X2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y имеет формулу CwHxNyOz; где каждый w равен от 0 до 10; каждый х равен от 1 до 25; каждый y равен от 1 до 4; и каждый z равен от 0 до 3. В некоторых вариантах осуществления y равен 2. В некоторых вариантах осуществления y равен 4.
[0082] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; X2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y имеет молекулярную массу от 30 до 280 дальтон и Y содержит по меньшей мере 1 основной атом азота. В некоторых вариантах осуществления Y имеет молекулярную массу от 30 до 280 дальтон и Y содержит по меньшей мере 2 основных атома азота. В некоторых вариантах осуществления Y представляет собой алкильную группу, содержащую 2 аминогруппы, и Y имеет молекулярную массу от 30 до 280 дальтон.
[0083] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; X2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y представляет собой -NR4(CR6R7)vNR4R5; и v равно 2. В некоторых вариантах осуществления каждый R6 и R7 независимо выбран из группы, состоящей из Н, метила или ОН. В некоторых вариантах осуществления каждый R6 и R7 независимо представляет собой Н или метил. В некоторых вариантах осуществления каждый R6 и R7 представляет собой Н. В некоторых вариантах осуществления каждый R4 представляет собой Н. В некоторых вариантах осуществления R5 выбран из Н, необязательно замещенного C1-С6-алкила, необязательно замещенного С3-С6-циклоалкила. В некоторых вариантах осуществления R5 выбран из Н, необязательно замещенного C1-С6-алкила, необязательно замещенного С3-С6-циклоалкила. В некоторых вариантах осуществления R5 выбран из группы, состоящей из метила, этила, пропила, изопропила и Н. В некоторых вариантах осуществления каждый R4, R6 и R7 представляет собой Н, и R5 выбран из Н, необязательно замещенного С1-С6-алкила и необязательно замещенного С3-С6-циклоалкила. В некоторых вариантах осуществления каждый R4 независимо представляет собой Н или необязательно замещенный C1-С3-алкил; каждый R6 и R7 представляют собой Н; и R5 выбран из Н, необязательно замещенного C1-С6-алкила, необязательно замещенного С3-С6-циклоалкила. В некоторых вариантах осуществления R5 представляет собой группу гуанидина. В некоторых вариантах осуществления R5 представляет собой амидиновую группу. В некоторых вариантах осуществления СусА представляет собой транс-1,4-циклогексил.
[0084] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; X2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y представляет собой -NR4R5. В некоторых вариантах осуществления R4 и R5 каждый выбран из группы, состоящей из Н, гуанидина, амидина, необязательно замещенного алкила и гетероциклоалкила. В некоторых вариантах осуществления R4 и R5 каждый представляют собой Н. В некоторых вариантах осуществления R5 представляет собой группу амидина. В некоторых вариантах осуществления R5 представляет собой группу гуанидина.
[0085] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; X2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y представляет собой -(CR6R7)vNR4R5; и v равно 1 или 2. В некоторых вариантах осуществления R4 и R5 каждый выбран из группы, состоящей из Н, гуанидина, амидина, необязательно замещенного алкила и гетероциклоалкила. В некоторых вариантах осуществления v равно 1; R6 и R7 каждый представляют собой Н или метил; и R4 и R5, каждый независимо, представляют собой Н, необязательно замещенный C1-С6-алкил или необязательно замещенный С3-С6-циклоалкил. В некоторых вариантах осуществления v равно 1; R6 и R7 каждый представляет собой Н или метил; и R4 и R5, каждый независимо, представляют собой Н, необязательно замещенный C1-С6-алкил или необязательно замещенный С3-С6-циклоалкил. В некоторых вариантах осуществления v равно 1; R6 и R7 каждый представляет собой Н или метил; и R4 и R5, каждый независимо, представляют собой Н, C1-С6-алкил или С3-С6-циклоалкил. В некоторых вариантах осуществления v равно 1; R6 и R7 каждый представляют собой Н или метил; и R4 и R5 каждый представляют собой Н или метил; и R4 и R5 каждый представляют собой Н.
[0086] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; Х2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y представляет собой -(CR6R7)vNR4C(=NR4)NR4R5; и v равно 1 или 2. В некоторых вариантах осуществления R4 и R5 каждый выбран из группы, состоящей из Н, необязательно замещенного C1-С6-алкила или необязательно замещенного С3-С6-циклоалкила. В некоторых вариантах v равно 1; R6 и R7 каждый представляют собой Н или метил; и R4 и R5, каждый независимо, представляют собой Н, необязательно замещенный C1-С6-алкил или необязательно замещенный С3-С6-циклоалкил. В некоторых вариантах осуществления v равно 1; R6 и R7 каждый представляют собой Н; и R4 и R5, каждый независимо, представляют собой Н, необязательно замещенный C1-С6-алкил или необязательно замещенный С3-С6-циклоалкил. В некоторых вариантах осуществления v равно 1; R6 и R7 каждый представляют собой Н; и R4 и R5 каждый представляют собой Н, C1-С6-алкил или С3-С6-циклоалкил. В некоторых вариантах осуществления v равно 1; R6 и R7 каждый представляют собой Н или метил; и R4 и R5 каждый представляют собой Н.
[0087] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; X2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y представляет собой -NR4(CR6R7)vNR4C(=NR4)NR4R5; и v равно 2. В некоторых вариантах осуществления R4 и R5 каждый выбран из группы, состоящей из Н, необязательно замещенного алкила и гетероциклоалкила. В некоторых вариантах осуществления R6 и R7 каждый представляют собой Н или метил; и R4 и R5, каждый независимо, представляют собой Н, необязательно замещенный C1-С6-алкил или необязательно замещенный С3-С6 циклоалкил. В некоторых вариантах осуществления; R6 и R7 каждый представляют собой Н; и R4 и R5, каждый независимо, представляют собой Н, необязательно замещенный C1-С6-алкил или необязательно замещенный С3-С6 циклоалкил. В некоторых вариантах осуществления R6 и R7 каждый представляют собой Н; и R4 и R5 каждый представляют собой Н, С1-С6-алкил или С3-С6-циклоалкил. В некоторых вариантах осуществления R6 и R7 каждый представляют собой Н; и R4 и R5 каждый представляют собой Н или метил; и R4 и R5 каждый представляют собой Н.
[0088] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; X2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y представляет собой -(CR6R7)v; v равно 1 или 2; каждый R7 представляет собой Н или метил; и по меньшей мере один R6 представляет собой -C(N=R5)NR4R5. В некоторых вариантах осуществления каждый из R4 и R5 выбран из Н, необязательно замещенного C1-С6-алкила и необязательно замещенного С3-С6-циклоалкила. В некоторых вариантах осуществления каждый из R4 и R5 выбран из Н, С1-С6-алкила и С3-С6-циклоалкила. В некоторых вариантах осуществления каждый из R4 и R5 выбран из Н и метила. В некоторых вариантах осуществления каждый из R4 и R5 представляет собой Н.
[0089] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; X2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y представляет собой -NR4(CR6R7)vNR4R5; v равно 2; каждый из R6 и R7 независимо выбран из группы, состоящей из Н, метила или ОН; каждый R4 представляет собой Н; R5 выбран из Н, необязательно замещенного С1-С6-алкила, необязательно замещенного С3-С6-циклоалкила; и СусА представляет собой необязательно замещенный 3-10-членный неароматический карбоцикл, в котором необязательная олефиновая функциональная группа неароматического карбоцикла не присоединена непосредственно к заместителю - атому кислорода, серы или азота. В некоторых вариантах осуществления СусА выбран из группы, состоящей из циклопропана, циклобутана, циклопентана, циклогексана, циклогептана, циклооктана, циклопентена, циклогексена, циклогептена, циклооктена, и где олефиновая функциональная группа циклопентена, циклогексена, циклогептена и циклооктена не присоединена непосредственно к заместителю - атому кислорода, серы или азота. В некоторых вариантах осуществления СусА представляет собой циклобутан, циклопентан, циклогексан или циклогексен, где олефиновая функциональная группа циклогексена не присоединена непосредственно к заместителю - атому кислорода, серы или азота. В других вариантах осуществления, СусА выбран из группы, состоящей из бицикло[3.3.0]октана, бицикло[4.3.0]нонана, цис-декалина, транс-декалина, бицикло[2.1.1]гексана, бицикло[2.2.1]гептана, бицикло[2.2.2]октана, бицикло[3.2.2]нонана и бицикло[3.3.2]декана. В некоторых вариантах осуществления СусА представляет собой циклопентан. В предпочтительных вариантах осуществления СусА представляет собой циклогексан. В некоторых вариантах осуществления СусА является циклогексаном, ковалентно связанным с одним Y и L; упомянутые ковалентных связей в 1,4-транс-расположении.
[0090] В некоторых вариантах осуществления соединения формулы I или формулы Ia Ra, Rb, Rc, Rd и R3 представляют собой Н; X1 представляет собой ОН; X2, если присутствует, представляет собой ОН, Z представляет собой >С=O; n равно 0; m равно 1; р равно 1; М и L каждый представляют собой связь; R1 and R2 каждый представляют собой Н; СусА представляет собой 1,4-циклогексил; и Y представляет собой -NR4(CR6R7)vNR4R5; и v равно 2; каждый R6 и R7 независимо выбран из группы, состоящей из Н, метила или ОН; каждый R4 представляет собой Н; R5 выбран из Н, необязательно замещенного С1-С6-алкила, необязательно замещенного С3-С6-циклоалкила; и Z выбран из группы, состоящей из >С=O, >C=S или >SO2.
Получение соединений
[0091] В настоящем изобретении описаны соединения формулы I или формулы Ia, которые ингибируют активность бета-лактамаз, и способы их получения. Также в настоящем изобретении описаны фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, фармацевтически активные метаболиты и фармацевтически приемлемые пролекарства таких соединений. Также предложены фармацевтические композиции, содержащие по меньшей мере одно такое соединение или фармацевтически приемлемую соль, фармацевтически приемлемый сольват, фармацевтически активный метаболит или фармацевтически приемлемое пролекарство такого соединения и фармацевтически приемлемый наполнитель.
[0092] Соединения формулы I или формулы Ia могут быть синтезированы с помощью стандартных синтетических реакций, известных специалистам в данной области техники, или с помощью способов, известных в данной области техники. Реакции могут быть использованы в линейной последовательности с получением соединений, или они могут быть использованы для синтеза фрагментов, которые впоследствии присоединяются способами, известными в данной области техники.
[0093] Исходный материал, используемый для синтеза соединений, описанных в данном изобретении, может быть синтезирован или может быть получен из коммерческих источников, таких как, но не ограничиваясь ими, Aldrich Chemical Co. (Milwaukee, Wisconsin), Bachem (Torrance, California) или Sigma Chemical Co. (St. Louis, Mo.). Описанные в настоящем изобретении соединения и другие родственные соединения, имеющие различные заместители, могут быть синтезированы с использованием способов и материалов, известных специалистам в данной области техники, как описано, например, у March, Advanced Organic Chemistry 4th Ed., (Wiley 1992); Carey and Sundberg, Advanced Organic Chemistry 4th Ed., Vols. A and В (Plenum 2000, 2001); Green and Wuts, Protective Groups in Organic Synthesis 3rd Ed., (Wiley 1999); Fieser and Fieser's Reagents for Organic Synthesis, Volumes 1-17 (John Wiley and Sons, 1991); Rodd's Chemistry of Carbon Compounds, Volumes 1-5 and Supplemental (Elsevier Science Publishers, 1989); Organic Reactions, Volumes 1-40 (John Wiley and Sons, 1991); и Larock's Comprehensive Organic Transformations (VCH Publishers Inc., 1989). (Все из которых включены посредством ссылки во всей их полноте). Другие способы синтеза соединений, описанных в настоящем изобретении, могут быть найдены в Международной патентной публикации WO 01/01982901, Arnold et al. Bioorganic & Medicinal Chemistry Letters 10 (2000) 2167-2170; Burchat et al. Bioorganic & Medicinal Chemistry Letters 12 (2002) 1687-1690. Общие способы получения соединения, как описано в настоящем изобретении, могут происходить из известных реакций в данной области техники, и реакции могут быть модифицированы с использованием соответствующих реагентов и условий, как должно быть очевидно специалисту в данной области техники, для введения различных групп, найденных в формулах, как представлено в настоящем описании.
[0094] Продукты реакций могут быть выделены и очищены, если желательно, с использованием обычных способов, включая, но не ограничиваясь ими, фильтрование, перегонку, кристаллизацию, хроматографию и тому подобное. Такие материалы могут быть охарактеризованы с использованием обычных способов, включая физические константы и спектральные данные.
[0095] Соединения, описанные в настоящем изобретении, могут быть получены в виде одного изомера или смеси изомеров.
Дополнительные формы описанных в настоящем изобретении соединений
Изомеры
[0096] В некоторых вариантах осуществления, в связи с оксофильной природой атома бора, соединения, описанные в настоящем изобретении, могут преобразоваться в альтернативные формы или существовать в равновесии с альтернативными формами, особенно в средах, которые содержат воду (водный раствор, плазма и т.д.). Соответственно, соединения, описанные в настоящем изобретении, могут существовать в равновесии между "закрытой" циклической формой, показанной в формуле I, и "открытой" ациклической формой, показанной на фигуре Ia. Кроме того, соединения, описанные в настоящем изобретении, могут объединяться во внутримолекулярные димеры, тримеры и связанные с ними комбинации.
[0097] Дополнительно, в некоторых вариантах осуществления описанные в настоящем изобретении соединения существуют в виде геометрических изомеров. В некоторых вариантах осуществления соединения, описанные в настоящем изобретении, имеют одну или более двойных связей. Соединения, представленные в настоящем описании, включают все цис-, транс-, син-, анти-, противо- (entgegen (Е)), и вместе- (zusammen (Z)) изомеры, а также их соответствующие смеси. В некоторых случаях соединения существовуют в виде таутомеров. Соединения, описанные в данном изобретении, включают все возможные таутомеры в пределах формул, описанных в настоящем изобретении. В некоторых случаях описанные в настоящем изобретении соединения могут иметь один или более хиральных центров, и каждый центр существует в конфигурации R или конфигурации S. Соединения, описанные в настоящем изобретении, включают все диастереомерные, энантиомерные и эпимерные формы, а также их соответствующие смеси. В дополнительных вариантах осуществления соединений и способов, представленных в настоящем описании, смеси энантиомеров и/или диастереомеров, полученных из одной препаративной стадии, комбинации или взаимопревращения могут быть использованы для приложений, описанных в данном изобретении. В некоторых вариантах осуществления соединения, описанные в настоящем изобретении, получены в виде их отдельных стереоизомеров взаимодействием рацемической смеси соединения с оптически активным разделяющим агентом с образованием пары диастереоизомерных соединений, разделением диастереомеров и выделением оптически чистых энантиомеров. В некоторых вариантах осуществления диссоциирующие комплексы являются предпочтительными (например, кристаллические диастереомерные соли). В некоторых вариантах осуществления диастереомеры имеют различные физические свойства (например, температуры плавления, кипения, растворимость, реактивность, и т.д.) и разделены, воспользовавшись такими различиями. В некоторых вариантах осуществления диастереомеры разделены хиральной хроматографией или предпочтительно посредством способов разделения/разрешения на основе различий в растворимости. В некоторых вариантах осуществления оптически чистый энантиомер затем извлекают вместе с разделяющим агентом любым практическим способом, который не приводит к рацемизации.
Меченые соединения
[0098] В некоторых вариантах осуществления описанные в настоящем изобретении соединения существуют в их меченных изотопами формах. В некоторых вариантах осуществления способы, описанные в настоящем изобретении, включают в себя способы лечения заболеваний введением таких меченных изотопами соединений. В некоторых вариантах осуществления способы, раскрытые в настоящем описании, включают способы лечения заболеваний введением таких меченных изотопами соединений в виде фармацевтических композиций. Таким образом, в некоторых вариантах осуществления соединения, раскрытые в настоящем описании, включают меченные изотопами соединения, которые идентичны тем, которые приведены в настоящем изобретении, но за исключением того, что один или более атомов заменены атомами, имеющими атомную массу или массовое число, отличные от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2H, 3H, 13С, 14С, 15N, 18O, 17О, 31Р, 32P, 35S, 18F и 36Cl, соответственно. Соединения, описанные в данном изобретении, и их метаболиты, фармацевтически приемлемые соли, сложные эфиры, пролекарства, сольваты, гидраты или их производные, которые содержат вышеупомянутые изотопы и/или другие изотопы других атомов, входят в объем настоящего изобретения. Некоторые меченные изотопами соединения, например, такие, в которые включены радиоактивные изотопы, такие как 3H и 14С, могут быть использованы в анализах распределения лекарственного средства и/или субстрата в тканях. Третированные, т.е. 3H, и углерод-14, т.е. 14C, изотопы являются особенно предпочтительными вследствие простоты их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, приводит к некоторым терапевтическим преимуществам в результате более высокой метаболической стабильности, например, увеличению in vivo периода полураспада или снижению требований дозировки. В некоторых вариантах осуществления меченные изотопами соединения, их фармацевтически приемлемую соль, сложный эфир, пролекарство, сольват, гидрат или производное получают любым подходящим способом.
[0099] В некоторых вариантах осуществления описанные в настоящем изобретении соединения помечены другим способом, включая, но не ограничиваясь ими, использование хромофоров или флуоресцентных групп, биолюминесцентные маркеры или хемилюминесцентные маркеры.
Фармацевтически приемлемые соли
[00100] В некоторых вариантах осуществления описанные в настоящем изобретении соединения существуют в виде их фармацевтически приемлемых солей. В некоторых вариантах осуществления способы, описанные в настоящем изобретении, включают в себя способы лечения заболеваний путем введения таких фармацевтически приемлемых солей. В некоторых вариантах осуществления способы, описанные в настоящем изобретении, включают в себя способы лечения заболеваний путем введения таких фармацевтически приемлемых солей в фармацевтических композициях.
[00101] В некоторых вариантах осуществления соединения, описанные в настоящем изобретении, обладают кислотными или основными группами и, следовательно, взаимодействуют с любым(ой) из ряда неорганических или органических оснований и неорганических и органических кислот с образованием фармацевтически приемлемой соли. В некоторых вариантах осуществления данные соли получают in situ во время конечного выделения и очистки соединений по изобретению или отдельным взаимодействием очищенного соединения в его свободной форме с подходящей кислотой или основанием, и, таким образом, с образованием выделенной соли.
[00102] Примеры фармацевтически приемлемых солей включают соли, полученные взаимодействием соединений, описанных в настоящем изобретении, с неорганической, органической кислотой или неорганическим основанием, причем такие соли включают ацетат, акрилат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бисульфит, бромид, бутират, бутин-1,4-диоат, камфорат, камфорсульфонат, капроат, каприлат, хлорбензоат, хлорид, цитрат, циклопентанпропионат, деканоат, диглюконат, дигидрофосфат, динитробензоат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептаноат, глицерофосфат, гликолат, гемисульфат, гептаноат, гексаноат, гексин-1,6-диоат, гидроксибензоат, γ-гидроксибутират, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, иодид, изобутират, лактат, малеат, малонат, метансульфонат, манделатметафосфат, метансульфонат, метоксибензоат, метилбензоат, моногидрофосфат, 1-нафталенсульфонат, 2-нафталенсульфонат, никотинат, нитрат, пальмоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, пиросульфат, пирофосфат, пропионат, фталат, фенилацетат, фенилбутират, пропансульфонат, салицилат, сукцинат, сульфат, сульфит, сукцинат, суберат, себацат, сульфонат, тартрат, тиоцианат, тозилат ундеканоат и ксилолсульфонат.
[00103] Кроме того, соединения, описанные в настоящем изобретении, могут быть получены в виде фармацевтически приемлемых солей, образованных взаимодействием соединения в форме свободного основания с фармацевтически приемлемой неорганической или органической кислотой, включая, но не ограничиваясь ими, неорганические кислоты, такие как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, метафосфорная кислота и тому подобное; и органические кислоты, такие как уксусная кислота, пропионовая кислота, капроновая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, п-толуолсульфоновая кислота, винная кислота, трифторуксусная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, арилсульфоновая кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-метилбицикло-[2.2.2]окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 4,4'-метиленебис-(3-гидрокси-2-ен-1-карбоновая кислота), 3-фенилпропионовая кислота, триметилуксусная кислота, третичная бутилуксусная кислота, лаурилсульфаткислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота и муконовая кислота. В некоторых вариантах осуществления другие кислоты, такие как щавелевая, хотя сами по себе не являются фармацевтически приемлемыми, используются в получении солей, подходящих в качестве промежуточных соединений при получении соединений по изобретению и их фармацевтически приемлемых кислотно-аддитивных солей.
[00104] В некоторых вариантах осуществления те соединения, описанные в данном изобретении, которые содержат свободную кислотную группу, взаимодействуют с подходящим основанием, таким как гидроксид, карбонат, бикарбонат, сульфат, фармацевтически приемлемый катион металла, с аммиаком или с фармацевтически приемлемым органическим первичным, вторичным, третичным или четвертичным амином. Типичные соли включают соли щелочных металлов или соли щелочноземельных металлов, таких как литий, натрий, калий, кальций и магний, и соли алюминия и тому подобное. Иллюстративные примеры оснований включают гидроксид натрия, гидроксид калия, гидроксид холина, карбонат натрия N+(C1-4-алкил)4 и тому подобное.
[00105] Типичные органические амины, подходящие для образования основно-аддитивных солей, включают этиламин, диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и тому подобные. Следует понимать, что описанные в настоящем изобретении соединения также включают в себя кватернизацию любых основных азотсодержащих групп, которые они содержат. Следует понимать, что описанные в настоящем изобретении соединения также включают в себя кватернизацию любых борсодержащих групп, которые они содержат. Такая кватернизация может возникнуть в результате обработки соединения бора, являющегося кислотой Льюиса, основанием Льюиса с образованием комплекса или соли. В некоторых вариантах осуществления такой кватернизацией получают водо- или маслорастворимые или диспергируемые продукты.
Сольваты
[00106] В некоторых вариантах осуществления описанные в настоящем изобретении соединения существуют в виде сольватов. Настоящее изобретение относится к способам лечения заболеваний введением таких сольватов. Настоящее изобретение также относится к способам лечения заболеваний введением таких сольватов в виде фармацевтических композиций.
[00107] Сольваты содержат либо стехиометрические, либо нестехиометрические количества растворителя и, в некоторых вариантах осуществления, образуются в процессе кристаллизации с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное. Гидраты образуются, когда растворителем является вода, или алкоголяты образуются, когда растворителем является спирт. Сольваты соединений, описанных в настоящем изобретении, могут быть получены или образованы в ходе процессов, описанных в данном изобретении. Только в качестве примера, гидраты соединений, описанных в настоящем изобретении, могут быть легко получены перекристаллизацией из водного/органического растворителя смеси, с использованием органических растворителей, включая, но не ограничиваясь ими, диоксан, тетрагидрофуран или метанол. Кроме того, представленные в настоящем описании соединения могут существовать в несольватированных, а также в сольватированных формах. В целом, сольватированные формы считаются эквивалентными несольватированным формам для целей соединений и способов, представленных в настоящем описании.
Полиморфы
[00108] В некоторых вариантах осуществления описанные в настоящем изобретении соединения существуют в виде полиморфов. Настоящее изобретение относится к способам лечения заболеваний путем введения таких полиморфных форм. Настоящее изобретение также относится к способам лечения заболеваний путем введения таких полиморфных форм в виде фармацевтических композиций.
[00109] Таким образом, соединения, описанные в настоящем изобретении, включают все их кристаллические формы, известные как полиморфы. Полиморфы включают различные механизмы кристаллической упаковки одного и того же химического состава соединения. В некоторых случаях полиморфы имеют различные рентгеновские дифракционные картины, инфракрасные спектры, точки плавления, плотность, твердость, форму кристаллов, оптические и электрические свойства, стабильность и растворимость. В некоторых случаях различные факторы, такие как растворитель при перекристаллизации, скорость кристаллизации и температура хранения приводят к доминированию одной кристаллической формы.
Пролекарства
[00110] В некоторых вариантах осуществления описанные в настоящем изобретении соединения существуют в форме Пролекарства. Настоящее изобретение относится к способам лечения заболеваний путем введения таких пролекарств. Настоящее изобретение также относится к способам лечения заболеваний путем введения таких пролекарств в фармацевтических композициях.
[00111] Пролекарства представляют собой, как правило, предшественники лекарств, которые после введения индивидууму и последующего поглощения превращаются в активный или более активный вид посредством какого-либо процесса, такого как преобразование в процессе метаболизма. Некоторые пролекарства имеют химическую группу, присутствующую в пролекарстве, которая делает его менее активным и/или придает растворимость или некоторую другую особенность препарату. После того, как химическая группа была отщеплена от пролекарства и/или модифицирована, генерируется активный лекарственный препарат. Пролекарства часто применяются, так как в некоторых ситуациях их легче вводить, чем исходный лекарственный препарат. Они, например, биодоступны при пероральном введении, тогда как исходный лекарственный препарат не является биодоступным. В некоторых случаях пролекарство имеет также улучшенную растворимость в фармацевтических композициях, по сравнению с исходным лекарственным препаратом. Например, без ограничения, пролекарством будет соединение, как описано в настоящем изобретении, которое вводят в виде сложного эфира ("пролекарство"), чтобы облегчить перенос через клеточную мембрану, где растворимость в воде является отрицательным качеством в отношении мобильности, но который затем метаболическим путем гидролизуется до карбоновой кислоты, активного соединения, после попадания внутрь клетки, где растворимость в воде является полезным качеством. Еще одним примером пролекарства может быть короткий пептид (полиаминокислота), соединенный с кислотной группой, где пептид метаболизируется с выявлением активного фрагмента. (Смотрите, например, публикацию Bundgaard, "Design and Application of Prodrugs" в руководстве A Textbook of Drug Design and Development, Krosgaard-Larsen and Bundgaard, Ed., 1991, Chapter 5, 113-191, которая включена в настоящее описание посредством ссылки).
[00112] В некоторых вариантах осуществления пролекарства выполнены в виде обратимых производных лекарств для использования в качестве модификаторов для улучшения транспорта лекарства в сайт-специфические ткани. Принцип конструкции пролекарств до настоящего времени заключался в том, чтобы увеличить эффективную растворимость в воде терапевтического соединения для нацеливания на области, где вода является основным растворителем.
[00113] Дополнительно, пролекарственные производные соединений, описанных в данном изобретении, могут быть получены способами, описанными в данном изобретении, которые иначе известны в данной области техники (более подробно см. Saulnier et al., Bioorganic and Medicinal Chemistry Letters, 1994, 4, 1985). В качестве лишь примера, соответствующие пролекарства могут быть получены реагированием недериватизированного соединения в подходящим карбамилирующим агентом, таким как, но не ограничиваясь ими, 1,1-асилоксиалкилкарбанохлоридат, p-нитрофенилкарбонат или тому подобное. Формы пролекарств описанных в настоящем изобретении соединений, где пролекарство метаболизируется in vivo с получением производного, как изложено в данном описании, включены в объем формулы изобретения. Действительно, некоторые из описанных в данном изобретении соединений являются пролекарствами для другого производного или активного соединения.
[00114] В некоторых вариантах осуществления пролекарства включают соединения, в которых аминокислотный остаток или полипептидная цепь из двух или более (например, два, три или четыре) аминокислотных остатков ковалентно присоединены посредством амидной или сложноэфирной связи к свободной амино-, гидрокси- или карбоксильной группе соединений по настоящему изобретению. Аминокислотные остатки включают, но не ограничиваются ими, 20 встречающихся в природе аминокислот, а также включает в себя 4-гидроксипролин, гидроксилизин, демозин, изодемозин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляную кислоту, циртуллин, гомоцистеин, гомосерин, орнитин и метионинсульфон. В других вариантах осуществления пролекарства включают соединения, в которых остаток нуклеиновой кислоты или олигонуклеотид из двух или более (например, два, три или четыре) остатков нуклеиновой кислоты ковалентно присоединен к соединению по настоящему изобретению.
[00115] Фармацевтически приемлемые пролекарства соединений, описанных в настоящем изобретении, также включают в себя, но не ограничиваются ими, сложные эфиры, карбонаты, тиокарбонаты, N-ацильные производные, N-ацилоксиалкильные производные, четвертичные производные третичных аминов, N-основания Манниха, основания Шиффа, конъюгаты аминокислот, сложные эфиры фосфорной кислоты, соли металлов и сульфонатные эфиры. Соединения, имеющие свободные амино-, амидо-, гидрокси- или карбоксильные группы, могут быть преобразованы в пролекарства. Например, из свободных карбоксильных групп могут быть получены производные в виде амидов или алкиловых сложных эфиров. В некоторых случаях все такие группы пролекарств включают группы, включая, но не ограничиваясь ими, функциональные группы простого эфира, амина и карбоновой кислоты.
[00116] Пролекарства с функциональными гидроксигруппами включают сложные эфиры, такие как, хотя и не ограничиваясь ими, ацилоксиалкиловые (например, ацилоксиметиловый, ацилоксиэтиловый) сложные эфиры, алкоксикарбонилоксиалкиловые сложные эфиры, алкиловые эфиры, сложные ариловые эфиры, сложные эфиры фосфорной кислоты, эфиры сульфокислот, сульфатные эфиры и дисульфид-содержащие сложные эфиры; простые эфиры, амиды, карбаматы, гемисукцинаты, диметиламиноацетаты и фосфорилоксиметилоксикарбонилы, как указано в Advanced Drug Delivery Reviews 1996, 19, 115.
[00117] Пролекарства, представляющие собой аминовые производные, включают, но не ограничиваются ими, следующие группы и комбинации групп:
а также сульфаниламиды и фосфонамиды.
[00118] В некоторых случаях сайты на каких-либо частях ароматических колец восприимчивы к различным метаболическим реакциям, поэтому включением соответствующих заместителей в ароматические кольцевые структуры можно уменьшить, свести к минимуму или устранить данный метаболический путь.
Метаболиты
[00119] В некоторых вариантах осуществления соединения формулы I или формулы Ia восприимчивы к различным метаболическим реакциям. Таким образом, в некоторых вариантах осуществления введение соответствующих заместителей в структуру снизит, сведет к минимуму или устранит метаболический путь. В конкретных вариантах осуществления соответствующий заместитель, который уменьшит или устранит восприимчивость ароматического кольца к метаболическим реакциям, только в качестве примера, представляет собой атом галогена или алкильную группу.
[00120] В дополнительных или других вариантах осуществления соединения формулы I или формулы Ia, описанные в настоящем изобретении, метаболизируются при введении в организм, чтобы произвести метаболит, который затем используется для получения желаемого эффекта, в том числе, желательного терапевтического эффекта.
Фармацевтические композиции/Препараты
[00121] В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы I или формулы Ia, как описано в настоящем изобретении, или его фармацевтически приемлемую соль, полиморф, сольват, пролекарство, N-оксид или изомер и фармацевтически приемлемый наполнитель. В некоторых вариантах осуществления фармацевтическая композиция дополнительно содержит бета-лактамный антибиотик. В некоторых вариантах осуществления бета-лактамным антибиотиком является пенициллин, цефалоспорин, карбапенем, монобактам, мостиковый монобактам или их комбинация.
[00122] В некоторых вариантах осуществления соединения, описанные в настоящем изобретении, входят в состав фармацевтических композиций. Фармацевтические композиции получают обычным способом с использованием одного или более фармацевтически приемлемых неактивных ингредиентов, которые облегчают переработку активных соединений в препараты, которые могут быть использованы фармацевтически. Правильный состав зависит от выбранного пути введения. Краткое обобщение сведений о фармацевтических композициях, описанных в настоящем изобретении, можно найти, например, в Remington: The Science and Practice of Pharmacy, Nineteenth Ed (Easton, Pa.: Mack Publishing Company, 1995); Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania 1975; Liberman, H.A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, New York, N.Y., 1980; и Pharmaceutical Dosage Forms and Drug Delivery Systems, Seventh Ed. (Lippincott Williams & Wilkins 1999), которые включены в настоящее описание посредством ссылки для такого раскрытия.
[00123] В настоящем изобретении представлены фармацевтические композиции, которые включают соединение формулы I или формулы Ia и по меньшей мере один фармацевтически приемлемый неактивный ингредиент. В некоторых вариантах осуществления описанные в настоящем изобретении соединения вводят в виде фармацевтических композиций, в которых соединение формулы I или формулы Ia смешано с другими активными ингредиентами, как в комбинированной терапии. В других вариантах осуществления фармацевтические композиции включают другие медицинские или фармацевтические агенты, носители, адъюванты, консерванты, стабилизаторы, смачивающие или эмульгирующие агенты, способствующие растворению вещества, соли для регулирования осмотического давления и/или буферы. В других вариантах осуществления фармацевтические композиции включают другие терапевтически ценные вещества.
[00124] Фармацевтическая композиция, в целях настоящего изобретения, относится к смеси соединения формулы I или формулы Ia с другими химическими компонентами (например, фармацевтически приемлемые вспомогательные вещества), такими как носители, наполнители, связующие вещества, наполнители, суспендирующие агенты, ароматизаторы, подсластители, дезинтегрирующие агенты, диспергирующие агенты, поверхностно-активные вещества, смазывающие вещества, красители, разбавители, растворители, увлажняющие агенты, пластификаторы, стабилизаторы, усилители проницаемости, смачивающие агенты, антипенные агенты, антиоксиданты, консерванты или комбинация их одного или более. Фармацевтическая композиция облегчает введение соединения в организм. При осуществлении способов лечения или применения, описанных в настоящем изобретении, терапевтически эффективные количества описанных в настоящем изобретении соединений вводят в виде фармацевтической композиции в организм млекопитающего, имеющего заболевание, расстройство или состояние, подлежащее лечению. В некоторых вариантах осуществления млекопитающее представляет собой человека. Терапевтически эффективное количество может широко варьировать в зависимости от тяжести заболевания, возраста и относительного состояния здоровья субъекта, активности используемого соединения и других факторов. Соединения могут быть использованы по отдельности или в сочетании с одним или более терапевтическими агентами в качестве компонентов смесей.
[00125] Фармацевтические составы, описанные в настоящем изобретении, вводят субъекту соответствующими путями введения, включая, но не ограничиваясь ими, пероральный, парентеральный (например, внутривенный, подкожный, внутримышечный), интраназальный, трансбуккальный, местный, ректальный или трансдермальный пути введения. Описанные фармацевтические композиции по данному изобретению включают, но не ограничиваются ими, водные жидкие дисперсии, жидкости, гели, сиропы, эликсиры, взвеси, суспензии, самоэмульгирующие дисперсии, твердые растворы, дисперсии липосом, аэрозоли, твердые лекарственные формы для перорального введения, порошки, составы с немедленным высвобождением, составы с контролируемым высвобождением, быстротающие составы, таблетки, капсулы, пилюли, порошки, драже, шипучие составы, лиофилизированные композиции, составы с замедленным высвобождением, составы с расширенным высвобождением, составы с пульсирующим высвобождением, составы в форме мультипартикулята и смешанные составы с немедленным и контролируемым высвобождением.
[00126] Фармацевтические композиции, включающие соединения формулы I или формулы Ia, изготовлены обычным способом, например, только в качестве примера, посредством обычного смешивания, растворения, гранулирования, дражирования, порошкования, эмульгирования, инкапсулирования, "внедрения" или прессования.
[00127] Фармацевтические композиции будут содержать по меньшей мере одно соединение формулы I или формулы Ia в качестве активного ингредиента в форме свободной кислоты или свободного основания или в форме фармацевтически приемлемой соли. Кроме того, способы и фармацевтические композиции, описанные в настоящем изобретении, включают в себя применение N-оксидов (при необходимости), кристаллических форм, аморфных фаз, а также активных метаболитов таких соединений, обладающих активностью того же типа. В некоторых вариантах осуществления описанные в настоящем изобретении соединения существуют в несольватированной форме или в сольватированной формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное. Считается, что сольватированные формы соединений, представленных в настоящем описании, также раскрыты в настоящем изобретении.
[00128] Фармацевтические препараты для перорального применения получают смешиванием одного или более твердого наполнителя с одним или более из описанных в настоящем изобретении соединений, необязательного измельчения полученной смеси и обработки смеси гранул после добавления подходящих вспомогательных веществ, при желании, для получения таблеток или ядер драже. Подходящие наполнители включают, например, такие наполнители, как сахара, в том числе, лактоза, сахароза, маннит или сорбит; целлюлозные препараты, такие как, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, камедь трагаканта, метилцеллюлоза, микрокристаллическая целлюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза натрия; или другие, такие как: поливинилпирролидон (ПВП или повидон) или фосфат кальция. При желании, добавляют дезинтегрирующие агенты, такие как сшитая кроскармеллоза натрия, поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия. В некоторых вариантах осуществления красители или пигменты добавляют к покрытиям таблеток или драже для идентификации или характеристики различных комбинаций доз активных соединений.
[00129] Фармацевтические препараты, которые вводят перорально, включают капсулы push-fit, изготовленные из желатина, а также мягкие герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбитол. Капсулы push-fit содержат активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими веществами, такими как крахмалы, и/или смазывающими веществами, такими как тальк или стеарат магния, и, необязательно, стабилизаторами. В мягких капсулах активные соединения растворяют или суспендируют в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. В некоторых вариантах осуществления добавлены стабилизаторы.
[00130] В некоторых вариантах осуществления могут быть использованы системы доставки фармацевтических соединений, такие как, например, липосомы и эмульсии. В некоторых вариантах осуществления композиции, представленные в настоящем описании, также могут включать в себя мукоадгезивный полимер, выбранный из, например, карбоксиметилцеллюлозы, карбомера (полимер акриловой кислоты), поли(метилметакрилата), полиакриламида, поликарбофила, сополимера акриловая кислота/ бутилакрилат, альгината натрия и декстрана.
Комбинированное лечение
[00131] Соединения согласно формуле I или формулы Ia могут быть использованы в сочетании с одним или более антибиотиком в лечении бактериальных инфекций. Такие антибиотики могут быть поэтому введены обычно используемым путем и в обычно используемом количестве, одновременно или последовательно, с соединением формулы I или Ia. Когда соединение формулы I или Ia используется одновременно с одним или более антибиотиком, предпочтительной является фармацевтическая композиция в единичной дозированной форме, содержащей такие другие лекарственные средства и соединение по настоящему изобретению. Однако комбинированная терапия может также включать терапии, при которых соединение формулы I или Ia и один или более антибиотиков вводят согласно различным перекрывающимся графикам введения. Также предполагается, что, при использовании в сочетании с одним или несколькими антибиотиками, антибиотики могут быть использованы в более низких дозах, чем когда каждый из антибиотиков используется порознь.
[00132] Соответственно, фармацевтические композиции по настоящему изобретению также включают те, которые содержат один или более антибиотиков, в дополнение к соединению в соответствии с формулой I или формулы Ia. В некоторых вариантах осуществления фармацевтическая композиция, содержащая соединение формулы I или Ia, дополнительно содержит бета-лактамный антибиотик. В некоторых вариантах осуществления бета-лактамным антибиотиком является пенициллин, цефалоспорин, карбапенем, монобактам, мостиковый монобактам или их комбинация.
[00133] Приведенные выше комбинации включают сочетания соединения формулы I или Ia не только с одним антибиотиком, но также с двумя или более антибиотиками. Кроме того, соединения формулы I или Ia, или в сочетании с антибиотиками или сами по себе, могут быть использованы в сочетании с другими препаратами, которые используются в профилактике, лечении, контроле, улучшении или снижении риска бактериальных инфекций или заболеваний, связанных с бактериальными инфекциями. Такие другие лекарственные средства могут быть поэтому введены обычно используемым путем и в обычно используемом количестве, одновременно или последовательно, с соединением формулы I или Ia. Когда соединение формулы I или Ia используется одновременно с одним или несколькими другими лекарственными средствами, предпочтительной является фармацевтическая композиция, содержащая такие другие лекарственные средства в дополнение к соединению по настоящему изобретению. Соответственно, фармацевтические композиции по настоящему изобретению также включают те фармацевтические композиции, которые также содержат один или более других активных ингредиентов, в дополнение к соединению формулы I или Ia. Массовое отношение соединения формулы I или Ia ко второму активному ингредиенту может варьировать и будет зависеть от эффективной дозы каждого ингредиента. Как правило, будет использоваться эффективная доза каждого ингредиента.
[00134] В некоторых вариантах осуществления соединения формулы I или формулы Ia используются в сочетании с одним или более антибиотиков в лечении бактериальных инфекций. В некоторых вариантах осуществления бактериальная инфекция представляет собой инфекцию верхних или нижних дыхательных путей, инфекцию мочевыводящих путей, внутрибрюшное инфицирование или инфицирование кожи. В некоторых вариантах осуществления один или более антибиотиков выбирают из β-лактамных антибиотиков. β-лактамные антибиотики включают, но не ограничиваются ими, пенициллины, пенемы, карбапенемы, цефалоспорины, цефамицины, монобактамы или их комбинации. Пенициллины, включают, но не ограничиваются ими, амоксициллин, ампициллин, ацидоциллин, азлоциллин, бакампициллин, бензатин бензилпенициллин, феноксиметилпенициллин бензатин, бензилпенициллин (G), карбенициллин, кариндациллин, клометоциллин, клоксациллин, диклоксациллин, епициллин, флуклоксациллин, гетациллин, мециллинам, метампициллин, метициллин, мезлоциллин, нафциллин, оксациллин, пенамециллин, фенетициллин, феноксиметилпенициллин (V), пиперациллин, пивампициллин, пивмециллинам, прокаин бензилпенициллин, пропициллин, сульбенициллин, талампициллин, темоциллин, тикарциллин. Пенемы включают, но не ограничиваются им, фаропенем. Карбапенемы включают, но не ограничиваются ими, биапенем, эртапенем, дорипенем, имипенем, меропенем, панипенем. Цефалоспорины/цефамицины включают, но не ограничиваются ими, цефацитрил, цефаклор, цефадроксил, цефалексин, цефалоглицин, цефалолониум, цефалоридин, цефалотин, цефамандол, цефапирин, цефатрицин, цефазафлур, цефацедон, цефазолин, цефбуперазон, цефкапен, цефдалоксим, цефдинир, цефдиторен, цефепим, цефетамет, цефиксим, цефменоксим, цефметазол, цефминокс, цефодицим, цефонисид, цефоперазон, цефоранид, цефотаксим, цефотетан, цефотиам, цефовесин, цефокситин, цефозопран, цефримизол, цефрирамид, цефпиром, цефподоксим, цефргозил, цефхином, цефхином, цефгадин, цефгохдин, цефсулодин, цефтаролин фозамил, цефтазидим, цефтерам, цефтезол, цефтибутен, цефтиофур, цефтиолен, цефтизоксим, цефтобипрол, цефтриаксон, цефуроксим, цефузонам, фломоксеф, латамоксеф, лоракарбеф. Монобактамы включают, но не ограничиваются ими, ацтреонам, карумонам, нокардицин А, тигемонам.
Введение фармацевтической композиции
[00135] Подходящие способы введения включают, но не ограничиваются ими, пероральное, внутривенное, ректальное, аэрозольное, парентеральное, глазное, легочное, трансмукозальное, трансдермальное, вагинальное, ушное, назальное и местное введение. Кроме того, только в качестве примера, парентеральная доставка включает внутримышечную, подкожную, внутривенную, интрамедуллярную инъекции, а также интратекальную, прямую внутрижелудочковую, внутрибрюшинную, внутрилимфатическую и интраназальную инъекции.
[00136] В некоторых вариантах осуществления соединения формулы I или формулы Ia и их композиции вводят любым подходящим способом. Способ введения может быть выбран на основе, например, того, желательно ли местное или системное лечение, и в зависимости от области, подлежащей обработке. Например, композиции могут быть введены перорально, парентерально (например, внутривенная, подкожная, внутрибрюшинная или внутримышечная инъекция), ингаляцией, экстракорпорально, местно (в том числе трансдермально, офтальмологически, вагинально, ректально, интраназально) или тому подобное.
[00137] Парентеральное введение композиции, если используется, обычно характеризуется инъекцией. Композиции для инъекции могут быть приготовлены в виде обычных форм, либо в виде жидких растворов, либо в виде суспензий, твердых форм, подходящих для растворения суспензии в жидкости перед инъекцией, либо в виде эмульсий. Сравнительно недавно пересмотренный подход для парентерального введения включает использование системы с медленным высвобождением или замедленным высвобождением, так что сохраняется постоянная доза.
Аналитические способы определения антибактериальной активности
[00138] Аналитические способы определения ингибирования бета-лактамазной активности хорошо известны в данной области. Например, можно использовать способность соединения ингибировать бета-лактамазную активность в стандартном аналитическом определении ингибирования фермента (см., например, Page, Biochem J, 295: 295-304 (1993)). Бета-лактамазы для использования в таких аналитических определениях могут быть очищены из бактериальных источников или предпочтительно получены способами, основанными на применении рекомбинантных ДНК, поскольку гены и кДНК-клоны, кодирующие многие бета-лактамазы, известны (см., например, Cartwright & Waley, Biochem J 221: 505-12 (1984)).
[00139] Альтернативно, можно определить чувствительность бактерий к ингибитору, о которых известно, что они продуцируют бета-лактамазу, или которые были модифицированы с целью продукции ими бета-лактамазы. Другие аналитические способы определения ингибирования бактерий включают диффузию в агаровом диске и разбавление в агаре (см., например, Traub & Leonhard, Chemotherapy 43 159-67 (1997)). Таким образом, бета-лактамазу можно заингибировать контактированием бета-лактамазного фермента с эффективным количеством соединения согласно изобретению или контактированием бактерий, которые продуцируют бета-лактамазные ферменты, с эффективным количеством такого соединения, так что бета-лактамаза в бактериях вступает в контакт с ингибитором. Контактирование может происходить in vitro или in vivo. "Контактирование" означает, что бета-лактамаза и ингибитор сводятся вместе таким образом, что ингибитор может связываться с бета-лактамазой. Количества соединения, эффективные для ингибирования бета-лактамазы, можно определить эмпирически, и выполнение таких определений находится в пределах квалификации в данной области техники. Ингибирование включает в себя как снижение, так и полное подавление бета-лактамазной активности.
Способы
[00140] Настоящее изобретение также относится к способам ингибирования роста бактерий, например, посредством снижения бактериальной устойчивости к β-лактамному антибиотику, причем такие способы включают контактирование бактериальной культуры клеток или бактериально инфицированной культуры клеток, тканей или организма с бета-лактамазным ингибитором, описанным в настоящем изобретении. Предпочтительно, бактерии, которые подлежат ингибированию введением бета-лактамазного ингибитора формулы I или Ia, представляют собой бактерии, устойчивые к бета-лактамным антибиотикам. Термин "устойчивый" хорошо понятен обычному специалисту в данной области техники (см., например, Payne et al., Antimicrobial Agents and Chemotherapy 38 767-772 (1994), Hanaki et al., Antimicrobial Agents and Chemotherapy 30 1120-1126 (1995)).
[00141] Данные способы могут применяться для ингибирования роста бактерий в различных контекстах. В некоторых вариантах осуществления соединение формулы I или Ia вводят экспериментальной культуре клеток in vitro, чтобы предотвратить рост бактерий, резистентных к бета-лактамам. В некоторых других вариантах осуществления соединение формулы I или Ia вводят млекопитающему, включая человека, чтобы предотвратить рост резистентных к бета-лактамам бактерий in vivo. Способ согласно данному варианту осуществления включает в себя введение терапевтически эффективного количества бета-лактамазного ингибитора в течение терапевтически эффективного периода времени млекопитающему, включая человека. Предпочтительно, бета-лактамазный ингибитор вводят в виде фармацевтической композиции, как описано выше. В некоторых вариантах осуществления бета-лактамный антибиотик вводят совместно с бета-лактамазным ингибитором, как описано выше.
[00142] В другом аспекте настоящего изобретения предложены способы лечения бактериальной инфекции, причем способ включает введение субъекту фармацевтической композиции, содержащей соединение формулы I или формулы Ia или его фармацевтически приемлемую соль и фармацевтически приемлемый наполнитель. В некоторых вариантах осуществления способ лечения бактериальной инфекции у субъекта включает введение субъекту фармацевтической композиции, как описано в настоящем изобретении, необязательно в сочетании с бета-лактамным антибиотиком. В некоторых вариантах осуществления бактериальная инфекция представляет собой инфекцию верхних или нижних дыхательных путей, инфекцию мочевыводящих путей, внутрибрюшную инфекцию или кожную инфекцию.
[00143] В некоторых вариантах осуществления инфекция, которая подлежит лечению или предотвращению, содержит бактерию, которая включает 


[00144] В некоторых вариантах осуществления инфекция, которая подлежит лечению или предотвращению, содержит бактерию, которая включает 

ПРИМЕРЫ
Список сокращений
[00145] Следует понимать, что в приведенном выше и во всем описании изобретения следующие аббревиатуры, если не указано иное, имеют следующие значения:
Общие примеры получения соединений по изобретению
[00146] Исходные материалы и промежуточные соединения для соединений по данному изобретению могут быть получены посредством применения или адаптации способов, описанных ниже, их очевидных химических эквивалентов или, например, как описано в литературе, такой как The Science of Synthesis, Volumes 1-8. Editors E.M. Carreira et al. Thieme publishers (2001-2008). Подробная информация о реагентах и вариантах реакций также доступна по поискам структур и реакций с использованием коммерческих компьютерных поисковых систем, таких как Scifinder (www.cas.org) или Reaxys (www.reaxys.com).
[00147] Некоторые соединения по настоящему изобретению (I) (СХЕМА 1) получают из соответствующих сложных эфиров бороновой кислоты (II) с защищенными функциональными группами обработкой кислотой Льюиса, такой как BCl3, в растворителе, таком как дихлорметан, при температуре от -78°С до 0°C с последующим гашением водой.

[00148] Альтернативно, (I) получают из соединения (II) обработкой соединения (II) водной соляной кислотой (в интервале от 3 до 5 M) в диоксане при температуре от комнатной температуры до 100°С.
[00149] Необходимые сложные эфиры бороновой кислоты (II) получают (СХЕМА 2) связыванием амина (III) с (карбоновой или сульфоновой) кислотой (IV). Данное преобразование осуществляется сначала активацией кислотной функциональной группы, такой как хлорангидрид, ангидрид или реакционноспособный сложный эфир (Va, Vв или Vc), с последующей обработкой активированного субстрата с соединением (III) в растворителе, таком как DMF, DMA, NMP, THF или дихлорметан (или их смесь), приблизительно при комнатной температуре, как правило, в присутствии ненуклеофильного основания, такого как 4-метил-морфолин, триэтиламин или диизопропилэтиламин.
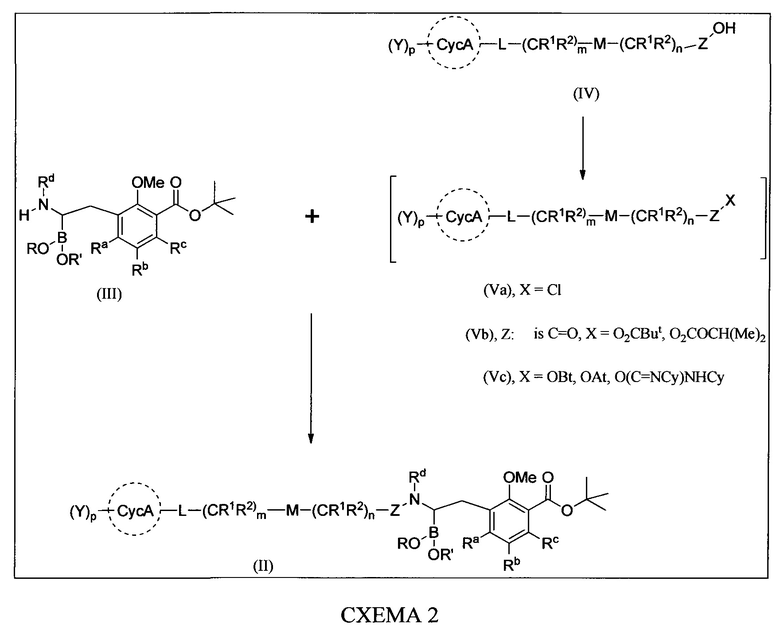
[00150] Образование хлорангидрида (Va) включает обработку соединения (IV) с хлорирующим агентом, таким как тионилхлорид, пентахлорид фосфора или оксалилхлорид, в растворителе, таком как дихлорметан, в присутствии катализатора, такого как DMF, при температуре, близкой к комнатной температуре. В некоторых случаях DMF также используется в качестве сорастворителя. Образование ангидрида (Vb) (Z представляет собой С=O) включает обработку соединения (IV) с пространственно затрудненным хлорангидридом или хлорформиатом, таким как триметилацетилхлорид или изопропилхлорформиат, в инертном растворителе, таком как дихлорметан, в присутствии ненуклеофильного основания, такого как триэтиламин или диизопропиламин, при комнатной температуре или ниже. Получение активированного эфира (Vc) включает обработку соединения (IV) с системой активирующего реагента, такого как EDCI, DCC/HOBt, HATU, реагенты ВОР или TBTU, в растворителе, таком как DMF, DMA, NMP или дихлорметан, при комнатной температуре или ниже (International Journal of Pharmaceutical Sciences Review and Research (2011), 8(1), 108-119).
[00151] Необходимые кислоты (IV) получают посредством ряда различных последовательностей реакций. Хотя среди иллюстративных примеров, приведенных ниже, существуют общие темы и стратегии, выбор соответствующей последовательности реакций (в том числе требований в отношении защитных групп) диктуется природой и расположением функциональной группы, присутствующей в молекуле-мишени, и, следовательно, может включать в себя очевидные адаптации проиллюстрированных способов для того, чтобы применить их в каждом конкретном случае.
[00152] В случае, когда Y1 связан с СусА через функциональную аминогруппу, требуемые кислоты (IV) (СХЕМА 3) удобно получать из соответствующего замещенного карбоциклического кетона (VI). Например, обработка соединения (VI) с подходящим амином (VII) в присутствии восстанавливающего агента, такого как три-ацетоксиборгидрид натрия, цианоборгидрид натрия или боргидрид натрия, в растворителе, таком как дихлорметан, 1,2-дихлор-этан, THF, метанол, уксусная кислота или их смесь, при температуре, близкой к комнатной температуре температуре, дает сложный эфир (VIII). В случае, когда требуется использование первичного амина (VII, R5=Н), соединение (VIII) можно также получить обработкой эквимолярной смеси (VI) и (VII, R5=Н) с кислотой Льюиса/осушителем, таким как Ti(OEt)4, в растворителе, таком как дихлорметан или 1,2-дихлорэтан, при комнатной температуре или выше, чтобы обеспечить промежуточный имин. Затем следует восстановление имина с боргидридом натрия, в растворителе, таком как метанол, при температуре от -78°С до комнатной температуры.
[00153] Кислоту (IV) получают из сложного эфира (VIII) формальным гидролизом сложноэфирной функциональной группы. Условия реакции применяются в зависимости от типа используемого сложного эфира. В случае метила, этила или другого простого алкила, гидролиз обычно достигается кратковременной обработкой водным основанием, таким как гидроксид натрия или гидроксид лития, в смеси растворителей THF, воды и метанола. Тем не менее, можно также использовать другие, используемые для защиты карбоксильной функциональной группы защитные группы, например, бензил, аллил, 2-триметилсилил-этил или 2,2,2-трихлорэтил. В данных случаях, преобразование сложного эфира в соответствующую кислоту достигается с помощью стандартных процедур снятия защиты, известных в литературе (Greene's Protective Groups in Organic Synthesis. Fourth Edition. John Wiley & Sons, Inc. 2006).
[00154] В некоторых случаях удобно выполнить последовательность реакций восстановительного аминирования с использованием кето-производного кислоты (VI, R=H). В данном случае обработка эквимолярной смеси кето-кислоты (VI, R=H) и амина (VII) с водородом в растворителе, таком как метанол, в присутствии катализатора, такого как палладий на угле, непосредственно дает кислоту (IV).
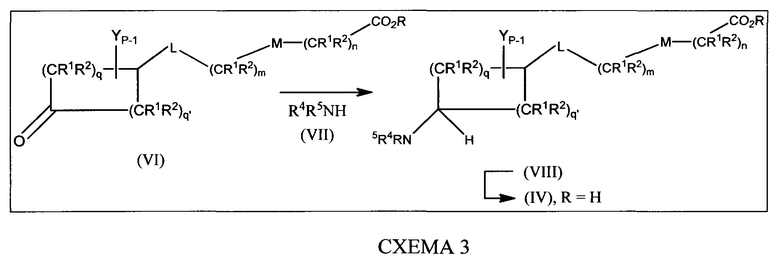
[00155] В другом подходе к аминосвязанным системам (СХЕМА 4), обработка кетона (VI) с восстанавливающим агентом, таким как боргидрид натрия, в метаноле при около 0°С или L-селектридом в THF при температуре от -78°С до комнатной температуры, дает спирт (IX). Обработка спирта (IX) с метансульфонилхлоридом или р-толуол-сульфонилхлоридом, в присутствии ненуклеофильного основания, такого как триэтиламин или DIEA, в растворителе, таком как дихлорметан или пиридин, при температуре около 0°С, дает соответствующий сульфонатный эфир (X). Замещение сульфонатной группы азидом обработкой соединения (X) с азидом натрия или азидом тетраалкиламмония в растворителе, таком как DMA, DMF, NMP, ацетонитрил или DMSO, при температуре от комнатной температуры до 120°С, дает азид (XI). Восстановление азида с трифенилфосфином и водой в THF при температуре, близкой к комнатной температуре температуре, (реакция Штаудингера) дает первичный амин (XII). Далее получение производных соединения (XII), где это уместно, может быть достигнуто восстановительным аминированием с соответствующим альдегидом или кетоном, с использованием условий, описанных для получения соединения (XV).
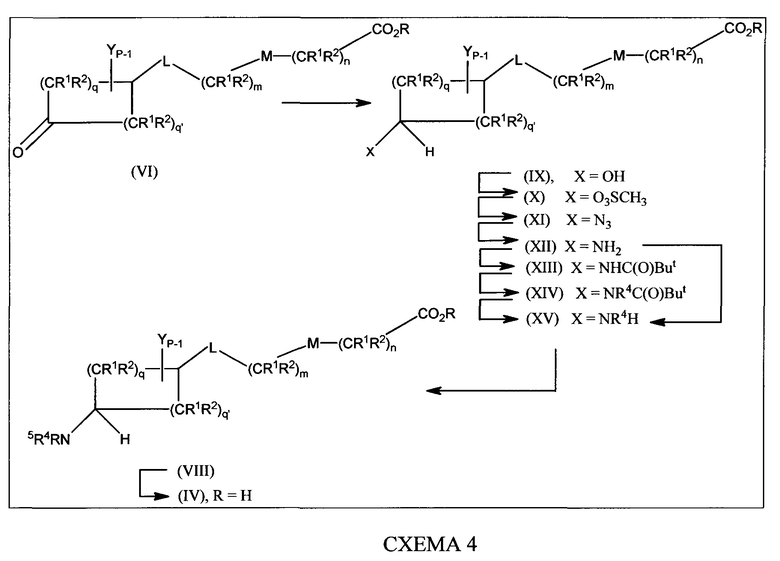
[00156] Альтернативно, образование N-ВОС производного (XII) обработкой BOC2O в присутствии ненуклеофильного основания, такого как триэтиламин или DIEA, в растворителе, таком как дихлорметан, при температуре, близкой к комнатной температуре, дает карбамат (XIII). Обработка соединения (XIII) с алкилгалогенидом или сульфонатом в присутствии основания, такого как гидрид натрия, карбонат калия или тетраметилгуанидин, в растворителе, таком как DMF, DMA, NMP, THF, DMPU или этанол (или их смеси), при комнатной температуре или ниже, обеспечивает соединение (XIV). Расщепление группы ВОС кислотой, такой как TFA, в дихлорметане или HCl в диоксане, этилацетате или эфире, при температуре, близкой к комнатной температуре, дает вторичный амин (XV). Далее получение производных (XV), где это уместно, может быть достигнуто восстановительным аминированием с соответствующим альдегидом или кетоном, с использованием условий, описанных для получения соединения (VIII). Гидролиз соединения (VIII), как уже отмечалось, дает соединение (IV).
[00157] В случае, когда Y1 представляет собой гуанидин, гуанидиновую группу получают из соответствующего карбоциклического первичного (XII) или вторичного амина (XV) (СХЕМА 5) обработкой реагентом, таким как 1,3-ди-трет-бутилоксикарбонил-S-метилизотиомочевина, в растворителе, таком как DMF (Synthesis, (2004), 37-42) или пиридин, при комнатной температуре или выше, или обработкой N,N'-бис-(ВОС)-1Н-пиразол-1-карбоксамидином в присутствии основания, такого как диизопропилэтиламин, в растворителе, таком как DMF или DMA, при температуре, близкой к комнатной температуре. Селективное расщепление функциональной сложноэфирной группы, как уже было описано, обеспечивает соответствующую кислоту (IV).
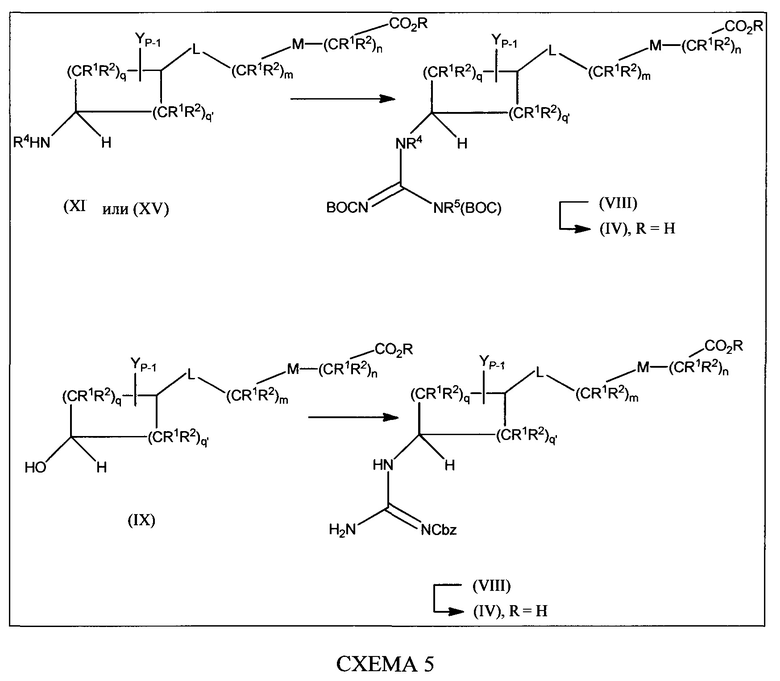
[00158] Альтернативно, гуанидинильная группа может быть введена обработкой соответствующего карбоциклического спирта (IX) с реагентом, таким как Cbz-гуанидин, в присутствии трифенилфосфина и диэтил-азо-дикарбоксилата, в растворителе, таком как THF (Mitsunobu conditions: Chemical Reviews, (2009), 109, 2551-2651), с получением непосредственно соединения (VIII).
[00159] В случае, когда Y1 представляет собой амидин, связанный с СусА через атом азота, требуемые кислоты (СХЕМА 6) получают из соответствующего первичного (XII) или вторичного амина (XV) обработкой соответствующим алкилтиоимидатом, таким как производное 2-нафтилметилтиоимидат (XVI), в растворителе, таком как этанол, при температуре от 0°С до комнатной температуры (Tetrahedron Letters, (1997), 38(2), 179-182), что дает соединение (XVII). Защита амидина (XVII) в качестве производного карбамата, такого как ВОС или Cbz, в стандартных условиях, с последующим селективным гидролизом сложного эфира дает кислоту (IV).
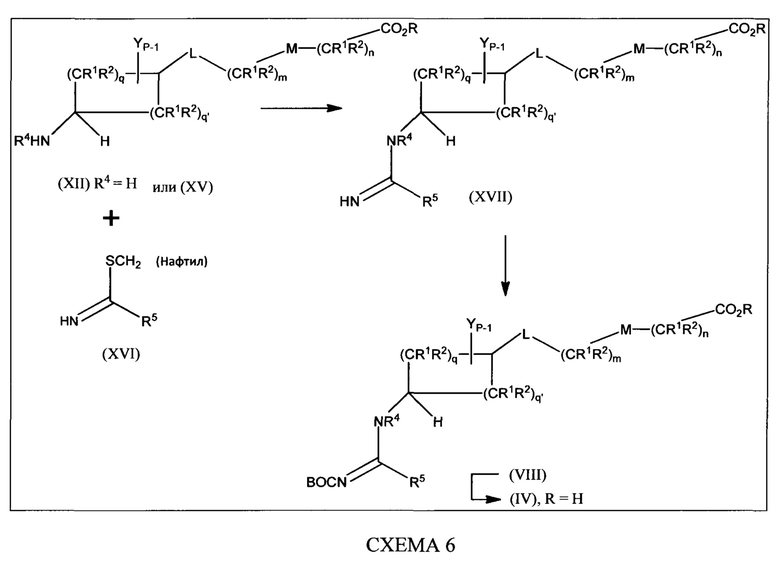
[00160] В случае, когда Y1 представляет собой амидин, связанный с СусА через атом углерода, функциональная группа амидина (СХЕМА 7) вводится превращением соответствующего карбоциклического кетона (XVIII) в соответствующий экзоциклический нитрил (XIX) обработкой соединения (XVIII) с толуолсульфонил-метилизоцианидом (Journal of Organic Chemistry (1977), 42(19), 3114-18), в присутствии основания, такого как KOBut, в растворителе, таком как DMSO или DME, содержащем около 2% трет-бутанола или этанола, при температуре от 0°С до 50°С. За обработкой соединения (XIX) с HCl в метаноле с образованием соответствующего имидата (XX) следует реакция взаимодействия данного промежуточного продукта с соответствующим амином (R4R5NH) в растворителе, таком как метанол или THF, при температуре, близкой к комнатной температуре, с получением амидина (XXI). В некоторых случаях удобно вызвать непосредственное образование амидина из нитрила (XIX) с использованием подходящего амида метил-хлоралюминия, в растворителе, таком как толуол, при приблизительно 80°С (Tetrahedron Letters, (1990), 31(14), 1969-1972). Кроме того, в случае, когда R5 представляет собой Н, функциональную группу амидина можно также ввести обработкой соответствующего карбоциклического нитрила (XIX) с гидроксиламином или O-алкил-гидроксиламином, чтобы дать N-гидроксил-(или алкокси)-амидин (XXII, R5 представляет собой ОН, OR). За этим следует удаление защитных групп каталитическим гидрогенолизом, чтобы обеспечить амидин (XXIII). Селективное ацилирование амидиновой функциональной группы в соединении (XXIII) обработкой ВОС-ангидридом или Cbz-хлоридом, как описано ранее, дает первичный спирт (XXIV). Преобразование первичного спирта (XXIV) в соответствующую кислоту (IV) осуществляется с использованием одного из нескольких протоколов, таких как окисление NaIO4 с каталитическим RuCl3 в смеси растворителей вода/CCl4/CH3CN в соотношении 3/2/2, при температуре, близкой к комнатной температуре (Journal of Organic Chemistry, (1981), 46(19), 3936-8) или спиридиндихроматом в DMF (Tempahedron Letters, (1979). 20 (52), 399).
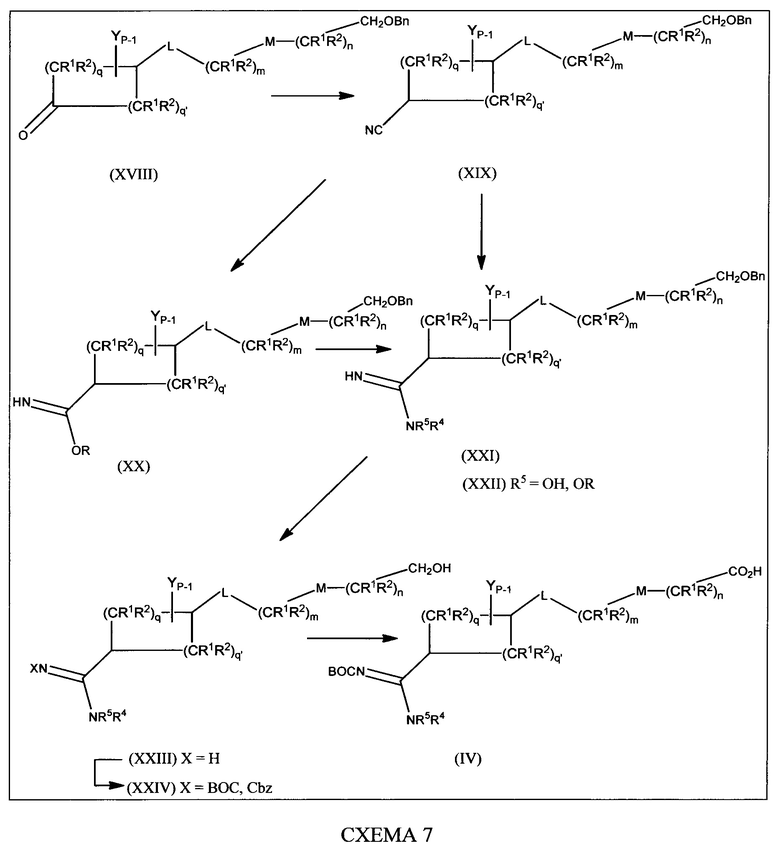
[00161] В некоторых случаях первичный спирт (XXIV) окисляют до соответствующей карбоновой кислоты с использованием двухстадийной процедуры, вовлекая начальное окисление до альдегида с использованием системы на основе окислителя DMSO, такое как окисление по Сверну (Swern) (Organic Reactions. (1990), 39, 297-572.), или обработкой избытком реагента Десс-Мартина в растворителе, таком как дихлорметан, при температуре, близкой к комнатной температуре. Последующее окисление альдегида осуществляют обработкой хлоритом натрия/дигидрофосфатом натрия в присутствии тетраметилэтилена, в растворителе, таком как трет-бутанол/вода, при температуре, близкой к комнатной температуре (Journal of Organic Chemistry, (1980), 45, 4825).
[00162] В случае, когда Y1 представляет собой азот-замещенную метиленовую группу, требуемые кислоты (СХЕМА 8) получают из соответствующих карбоциклических кетонов (XVIII) преобразованием функциональной группы кетона, во-первых, в соответствующее гидроксил-метильное производное обработкой с реагентом олефинирования, таким как метилтрифенилфосфонийбромид, в присутствии гексаметилдисилазида натрия, в растворителе, таком как THF, при около 0°С (реакция Виттига (Wittig)), или обработкой литий-триметилсилилметаном/CeCl3 при температуре от около 0°С до комнатной температуры, в растворителе, таком как THF или простой эфир, (реакция Петерсона (Peterson)) (Journal of Organic Chemistry, (1987) 52(2), 281-3), или в результате реакции с Petasis-модифицированным реагентом Tebbe (дициклопентадиенил-диметилтитан) в THF/толуоле при около 60°С (Journal of the American Chemical Society, (1990), 112 (17) 6392-6394), с получением соединения (XXV). Затем следует окисление гидроборированием экзоциклического алкена в соединении (XXV) с реагентом, таким как боран THF или алкильное производное, при около 0°С, в растворителе, таком как THF, с последующей окислительным обработкой перекисью водорода с водн. NaOH, чтобы обеспечить соединение (XXVI). Преобразование гидроксиметила (XXVI) в функционализованное амино-метил-производное (XXVII) осуществляется преобразованием в соответствующий тозилат, азид и первичный амин, как описано выше. Альтернативно, окисление соединения (XXVI) до альдегида (XXVIII) с последующим восстановительным аминированием соединения (XXVIII) с амином (R4R5NH), как уже было описано, также обеспечивает амин (XXVII). Преобразование соединения (XXVII) в требуемую кислоту (IV) осуществляется модификацией боковой цепи, как описано выше.
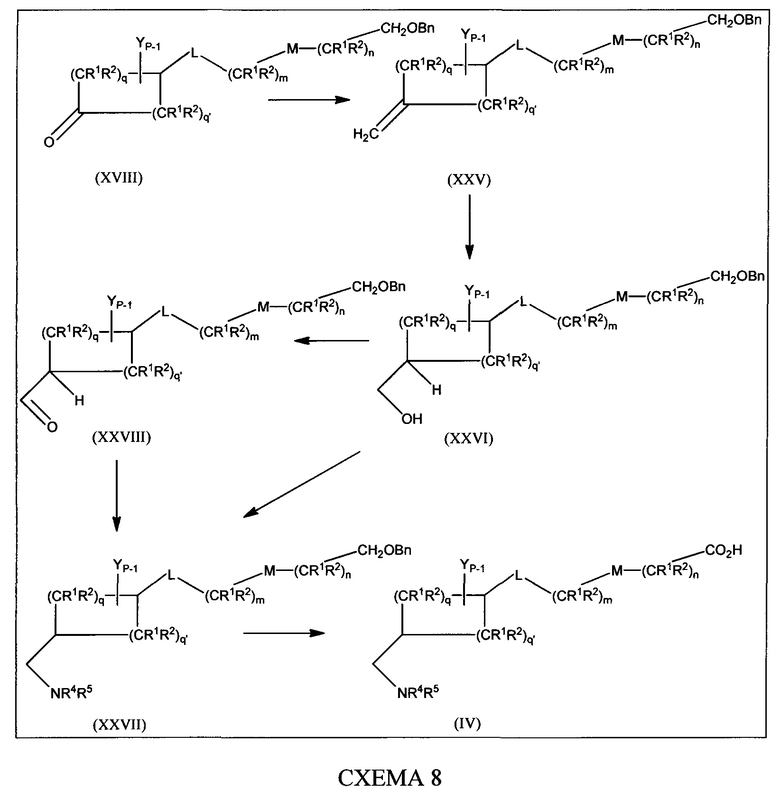
[00163] В альтернативном подходе к системам, где Y1 представляет собой азот-замещенную метиленовую группу, СусА представляет собой 5-7-членный карбоцикл и Z представляет собой карбонильную группу, требуемые карбоновые кислоты (IV) получают из производного акриламида (XXIX) (СХЕМА 9). Соединение (XXIX) конденсируется, в реакции Дильса-Альдера (Diels Alder), с соответствующим образом замещенным диеном, таким как силокси-диен (XXX), в инертном растворителе, таком как толуол, ксилол или DMA, при температуре от 70° до 190°С, с получением карбоциклического силил-енольного простого эфира (XXXI).
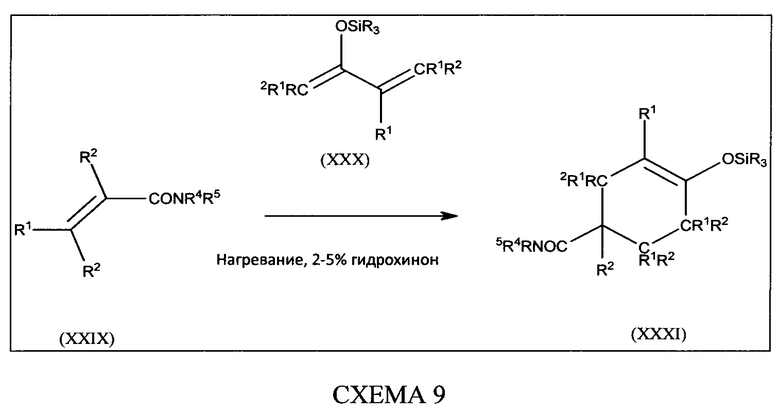
[00164] Карбоциклический силил-енольный простой эфир (XXXI) затем перерабатывают, чтобы обеспечить некоторые из требуемых карбоновых кислот (IV), с применением известных преобразований функциональных групп. Например, (СХЕМА 10), обработка соединения (XXXI) с N-фенил-трифлимидом и CsF в герметичной системе, используя растворитель, такой как DME, при температуре, близкой к комнатной температуре или ниже, дает соответствующий енолтрифлат (XXXII) {Journal of the American Chemical Society, (2002) 124, 11290-11291). Метокси-карбонилирование соединения (XXXII) с окисью углерода/метанолом в присутствии ненуклеофильного основания, такого как триэтиламин, и катализатора, такого как Pd(OAc)2, в сочетании с 1,3-бис-(дифенилфосфино-)пропаном, в растворителе, таком как DMSO, при температуре от 50 до 100°С дает ненасыщенный сложный эфир (XXXIII). Ненасыщенный сложный эфир (XXXIII) восстанавливается с использованием гетерогенного катализатора Pd, Rh или Pt, такого как 10% Pd на угле, в атмосфере водорода (от 1 до 4 атм), в растворителе, таком как этилацетат, метанол или THF (или их смесь), при температуре от комнатной температуры до 70°C с получением насыщенного сложного эфира (XXXIV). Кроме того, в некоторых случаях, ненасыщенный сложный эфир (XXXIII) может быть восстанавлен обработкой избытком магния, в растворителе, таком как метанол, при температуре, близкой к комнатной температуре, (Tetrahedron Letters, (1986); 27(21), 2409-2410) с получением соединения (XXXIV).

[00165] В подходящих случаях селективное восстановление амидной функциональной группы промежуточного продукта (XXXIV) с получением соответствующего амина (XXXV) (СХЕМА 11) осуществляется обработкой соединения (XXXIV) с силановым восстановителем, таким как дифенилсилан, и родиевым катализатором, таким как родийгидрокарбонилтрифенилфосфин, в растворителе, таком как THF, при близкой к комнатной температуре (Tetrahedron Letters, (1998); 39(9), 1017), или обработкой 1,1,3,3-тетраметилдисилоксаном и каталитическим количеством гидрата платинохлористоводородной кислоты, в растворителе, таком как толуол, при температуре от комнатной температуры до 90°С (Journal of the American Chemical Society, (2009); 131(41), 15032-15040).

[00166] Функциональную сложноэфирную группу в соединении (XXXV) подвергают гидролизу с получением требуемой кислоты (IV), с кратковременной обработкой основанием, таким как гидроксид натрия или гидроксид лития, в растворителе, таком как THF/метанол/вода, при температуре, близкой к комнатной температуре.
[00167] Альтернативно, восстановление функциональной амидной группы в соединении (XXXIV) с сопутствующим восстановлением сложноэфирной группы, с целью получения соединения (XXXVI), осуществляется обработкой соединения (XXXIV) с гидридным восстанавливающим агентом, таким как алюмогидрид лития, в эфирном растворителе, таком как THF, диэтиловый эфир, диметокси-этан или метил-отреот-бутиловый сложный эфир, при температуре от 0°С до 65°С. Преобразование полученного аминоспирта (XXXVI, R4, R5 представляет собой Н) в соответствующую кислоту (IV) осуществляют дериватизацией амина и окислением первичного спирта, как уже описано.
[00168] Соответствующую одноуглеродную родственную карбоновую кислоту (IV), где СусА представляет собой шестичленное кольцо, L, М представляют собой связь, m равно 0, n равно 1, Y1 представляет собой (R4R5NCH2) (СХЕМА 12), также получают из карбоциклического силил-енольного простого эфира (XXXI). Обработкой (XXXI) с карбонатом калия в метаноле при температуре, близкой к комнатной температуре, или гидратом фторида тетра-п-бутиламмония в растворителе, таком как THF (и, если применимо, забуференном с эквимолярным количеством уксусной кислоты), получают кетон (XXXVII). Обработкой соединения (XXXVII) с триалкилфосфоноацетатом, таким как триэтилфосфоноацетат, в растворителе, таком как THF, в присутствии основания, такого как гидрид натрия, при температуре от около -5°С до комнатной температуры получают соответствующий α,β-ненасыщенный сложный эфир (XXXVIII) (Liebigs Annalen/Recueil, (1997), 7, 1283-1301). Восстановление алкеновой и амидной функциональных групп в соединении (XXXVIII) и последующая обработка (гидролиз сложного эфира или окисление спирта), аналогично тому, как описано выше, дает необходимую кислоту (IV).
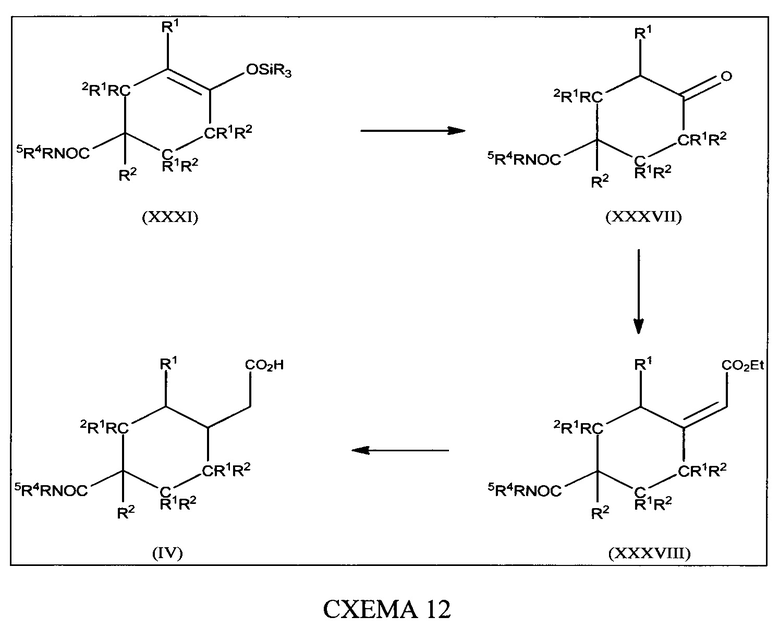
[00169] Карбоновую кислоту (IV), где СусА представляет собой шестичленное кольцо, L, М представляют собой связь, m равно 0, n равно 2, Y1 представляет собой (R4R5NCH2) (СХЕМА 13), получают из спирта (XXXVI) окислением в соответствующий альдегид (XXXIX) в стандартных условиях с последующей реакцией Хорнера-Вадсворта-Эммонса (Hoemer-Wadsworth-Emmons), как описано выше, чтобы обеспечить α,β-ненасыщенный сложный эфир (XL). Восстановление алкена в (XL) и гидролиз сложного эфира, аналогично описанному выше, дает необходимую кислоту (IV).
[00170] Карбоновую кислоту (IV), где СусА представляет собой шестичленное кольцо, L представляет собой О, М представляет собой связь, m равно 0, n равно 1, Y1 представляет собой (R4R5NCH2) (СХЕМА 14), получают из кетона (XXXVII) обработкой с реагентом, таким как боргидрид натрия, в метаноле при около 0°С, или L-селектрид в THF, при температуре от -78°С до комнатной температуры, с получением спирта (XLI). Конденсация соединения (XLI) с этилдиазоацетатом в присутствии катализатора, такого как Rh(асас)-димер, в растворителе, таком как дихлорметан, обеспечивает алкоксиацетатное производное (XLII). Селективное восстановление амида и гидролиз сложного эфира (XLII), как уже описано, дает (IV).
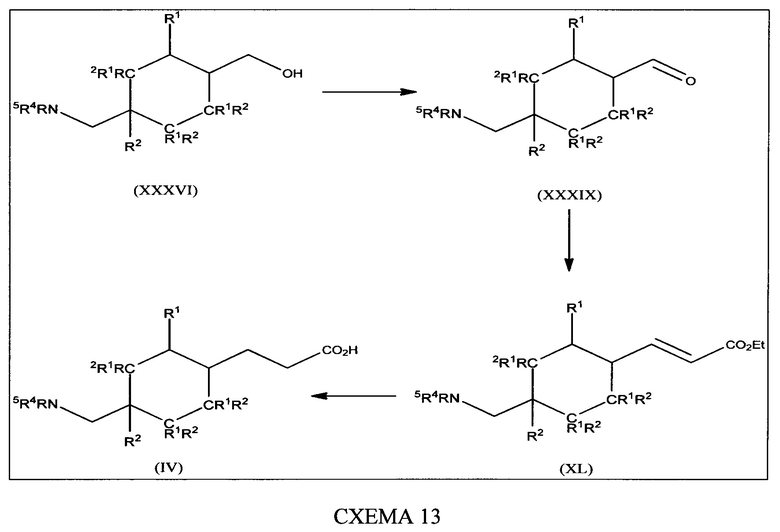
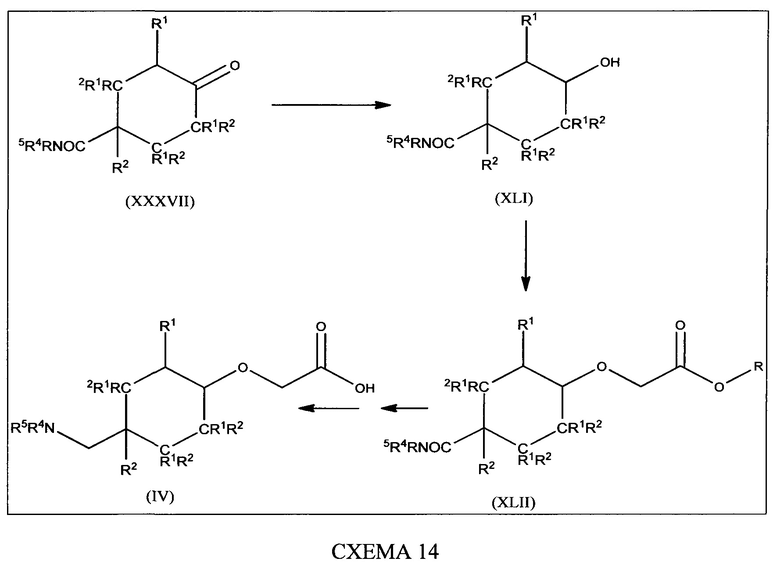
[00171] Карбоновую кислоту (IV), где СусА представляет собой шестичленное кольцо, L представляет собой NR4, М представляет собой связь, m равно 0, Y1 представляет собой (R4R5NCH2), (СХЕМА 15) получают из кетона (XXXVII) обработкой соответствующим аминоэфиром (XLIII) в условиях восстановительного аминирования, уже описанных для получения соединения (XLIV). Селективное восстановление амида и гидролиз сложного эфира (XLIV), как уже было описано, обеспечивает необходимую кислоту (IV).
[00172] Семичленные карбоциклические системы (IV) (СХЕМА 16) получают расширением кольца карбоциклического-силил-енольного простого эфира (XXXI).
[00173] Например, обработка (XXXI) с карбен-синтетическим эквивалентом, например, реагентом Симмонса-Смита (Simmons Smith), с последующим мягким окислением полученного циклопропана, с использованием такого реагента, как FeCl3 (Journal of Organic Chemistry, (1985), 50(4), 531-534) или CAN/NaOAc (Organic Letters, (2007), 9(7), 1323-1326), обеспечивает семичленный енон (XLV). Обработка (XLV) с силаном, например, PhMesSiH, в присутствии катализатора, такого как катализатор Уилкинсона (Wilkinson's catalyst) (Ph3P)3RhCl, в чистом виде или в растворителе, таком как бензол или толуол, обеспечивает силил-енольный простой эфир (XLVI) с расширенным кольцом. Данный семичленный карбоцикл обрабатывают непосредственно аналогичным способом с получением родственного ему соединения (XXXI) с шестичленным кольцом, описанного выше, чтобы предоставить необходимые кислоты (IV, СусА представляет собой гептан).
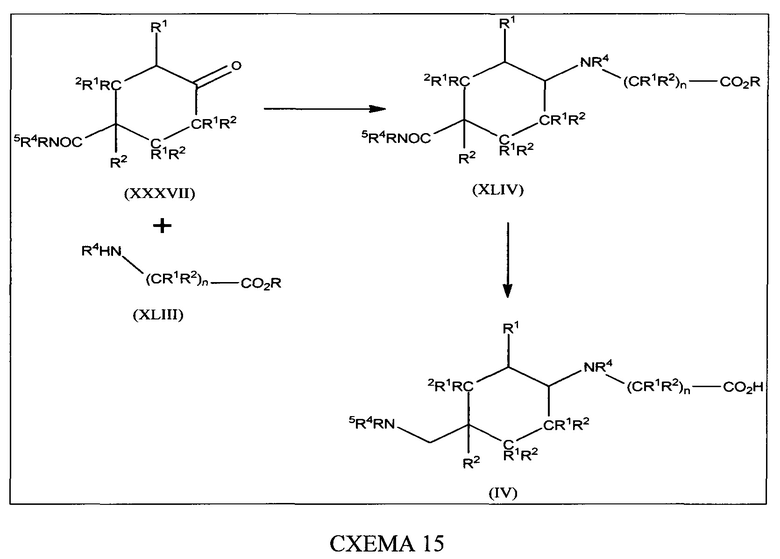
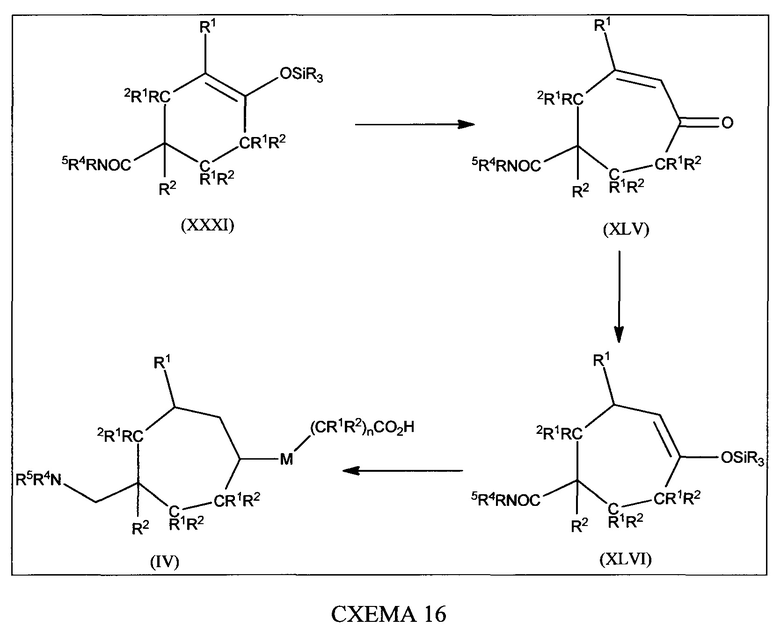
[00174] В некоторых случаях удобно получать карбоциклические системы (IV, СусА представляет собой циклопентан) (СХЕМА 17) из кетона (XXXI) реакцией перегруппировки Фаворского соответствующего α-гало-кетона (XLVII) (Current Organic Chemistry (2005), 9(17), 1713-1735). Например, обработка соединеня (XXXI) с галогенирующим реагентом, таким как бром, NCS, или пиридиний трибромид, в инертном растворителе, таком как дихлорметан или гексан, при температуре от -78°С до комнатной температуры обеспечивает α-гало-кетон (XLVII). Применение реакции перегруппировки Фаворского к условиям (XLVII) (медленное добавление к суспензии метоксида натрия в эфир, при температуре, близкой к комнатной температуре) затем дает замещенный сложным эфиром карбоцикл (XLVIII) с уменьшенным кольцом.
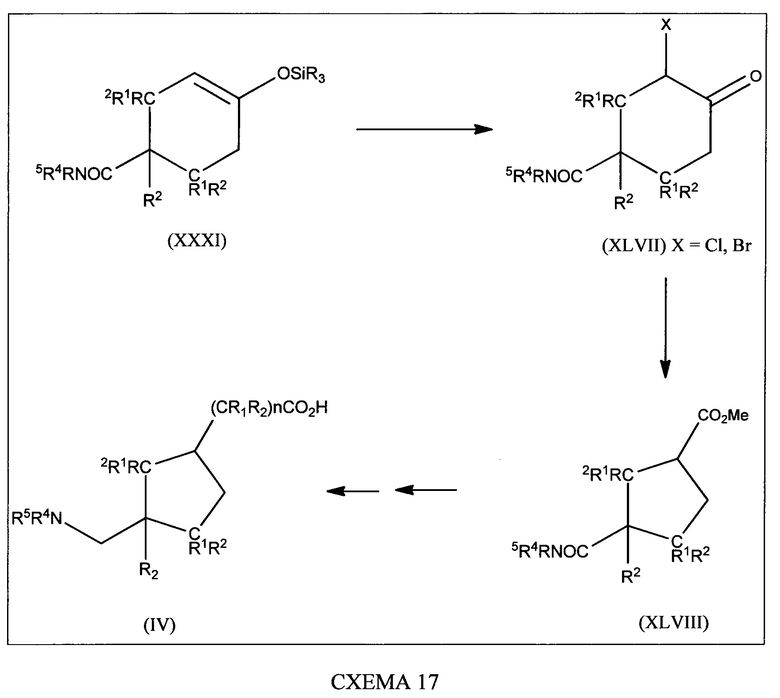
[00175] Данное промежуточное соединение обрабатывают непосредственно аналогичным способом с получением родственного ему соединения (XXXIV) с шестичленным кольцом, описанного выше, чтобы предоставить необходимые кислоты (IV).
[00176] В некоторых случаях карбоциклические системы (IV, СусА представляет собой циклопентан) получают реакцией циклоприсоединения между акриламидным производным (XXIX) и 2-((триметилсилил)метил)аллилацетатом (XLIX) в растворителе, таком как THF, толуол или ксилол, в присутствии катализатора, такого как бис(дифенилфосфинопропан)/ацетат палладия, при температуре от 70° до 160°С (Journal of the American Chemical Society. (1979), 101(21), 6429-6431) с получением соединения (L). Экзоциклический олефин в (L) подвергают окислительному расщеплению обработкой с каталитическими количествами осмия (Org. Synth. Oxid. Met. Compd. (1986), 633-93. Publisher: Plenum, New York) в присутствии совместного оксиданта, такого как N-метилморфолин-N-оксид, в растворителе, таком как трет-бутанол/вода с получением соответствующего ди-гидрокси-производного (LI). Данный диол подвергается затем окислительному расщеплению с использованием периодата натрия, в растворителе, таком как THF/вода, при температуре, близкой к комнатной температуре, с образованием (LII).
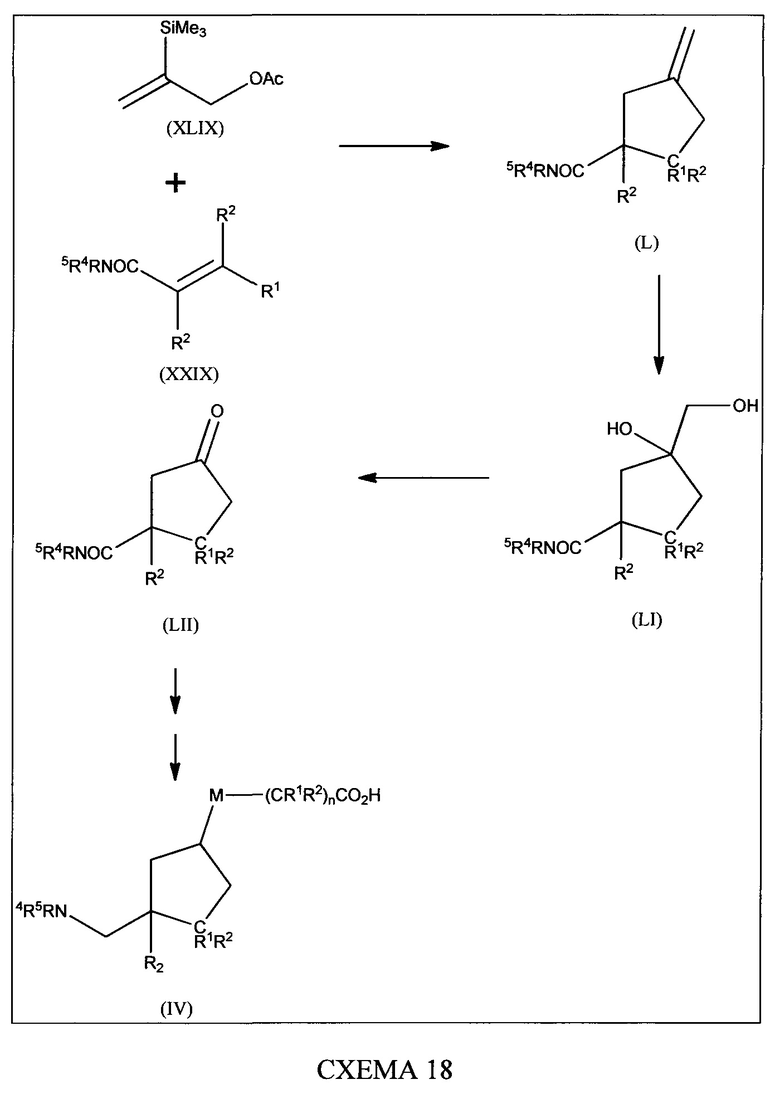
[00177] Карбоциклический кетон (LII) затем обрабатывают непосредственно аналогичным способом с получением родственного ему соединения (XXXVII) с шестичленным кольцом, чтобы обеспечить соответствующие кислоты (IV).
[00178] В случае, когда оба Y1 и Y2 представляют собой азот-замещенные метиленовые группы, так что Y1 и Y2 расположены вицинально в карбоцикле, требуемые кислоты (IV) получают, как показано выше (СХЕМЫ 9-18), за исключением того, что начинают с диамида фумаровой или малеиновой кислоты, вместо акриламида.
[00179] В случае, когда СусА представляет собой 5-, 6- или 7-членный карбоцикл, где Y3 и Y4, взятые вместе, образуют кольцо, требуемые карбоновые кислоты (IV) (СХЕМА 19) получают способами, описанными выше, за исключением того, что в реакции Дильса-Альдера используют циклический силокси-диен, такой как (LIII), чтобы обеспечить бициклический силил-енольный простой эфир (LIV). Исходные диеновые структуры получают из соответствующего α,β-ненасыщенного кетона (LV) обработкой соответствующего енона с триалкилсилилтрифлатом в присутствии основания, такого как триэтиламин, протонной губки или DBU, в растворителе, таком как простой эфир, при температуре 0°С или выше.
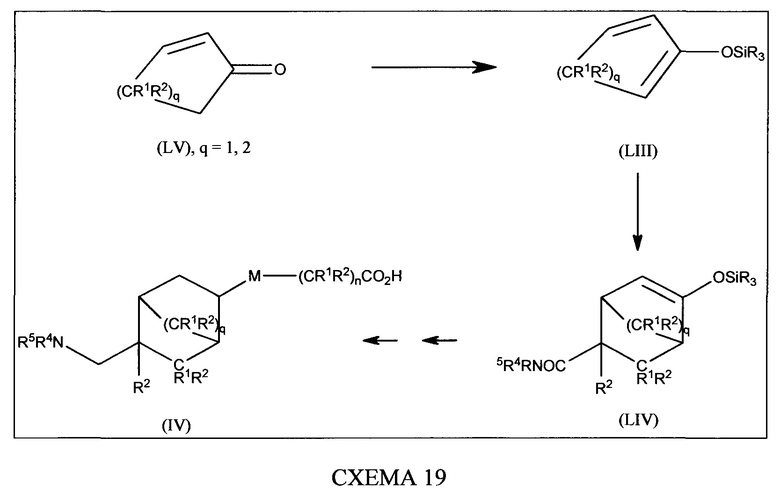
[00180] В случае, когда СусА представляет собой 5-, 6- или 7-членный карбоцикл, М представляет собой связь. О, NR4, Y1 и Y2 каждый связаны с СусА через атом азота и Y1 и Y2 расположены вицинально по отношению к другу другу в карбоцикле, требуемые кислоты (IV) (СХЕМА 20) получают из соответствующих циклических олефинов (LVI). Например, обработка (LVI) с азидом натрия в присутствии мягкого окислителя, такого как Mn(ОАс)3(H2O)2, и кислоты, такой как уксусная кислота или трифторуксусная кислота, в растворителе, таком как ацетонитрил, при температуре от -30°С до 0°С, обеспечивает диазид (LVII) преимущественно в конфигурации транс-изомера (Synthetic Communications, 28(10), 1913-1922; 1998). Последующее восстановление бис-азида обработкой восстановителем, таким как трифенилфосфин, в растворителе, таком как THF, с последующим in situ гидролизом промежуточного соединения аза-фосфорана добавлением избытка воды приводит к получению бис-амина (LVIII). В некоторых случаях восстановление бис-азида также достигается обработкой газообразным водородом в присутствии катализатора, такого как палладий на угле, в растворителе, таком как THF или метанол.
[00181] Данный бис-первичный амин (LVIII) защищен как ВОС, Cbz или другое подходящее N-защищенное производное (Greene's Protective Groups in Organic Synthesis; 4th Edition: John Wiley & Sons, Inc., 2006). Например, обработка соединения (LVIII) с соответствующим ангидридом или эфиром хлормуравьиной кислоты в присутствии основания, такого как триэтиламин, в растворителе, таком как THF или дихлорметан, при температуре, близкой к комнатной температуре, обеспечивает промежуточное карбаматное соединение (LIX). Где уместно, карбамат дополнительно подвергают дериватизации обработкой с соответствующим алкилирующим агентом в присутствии основания, такого как K2CO3, в растворителе, таком как DMF, DMA, или ацетонитрил, чтобы получить соединение (LX). Удаление защитной группы карбамата, с последующей обработкой полученного вторичного амина с альдегидом или кетоном в присутствии восстанавливающего агента, такого как боргидрид натрия, цианоборгидрид натрия или триацетоксиборгидрид натрия, в растворителе, таком как дихлорметан, 1,2-дихлорэтан, метанол или THF, при температуре, близкой к комнатной температуре, предоставляет соединение (LXI). Гидролиз сложного эфира из соединения (LXI), как описано выше, дает требуемую кислоту (IV).
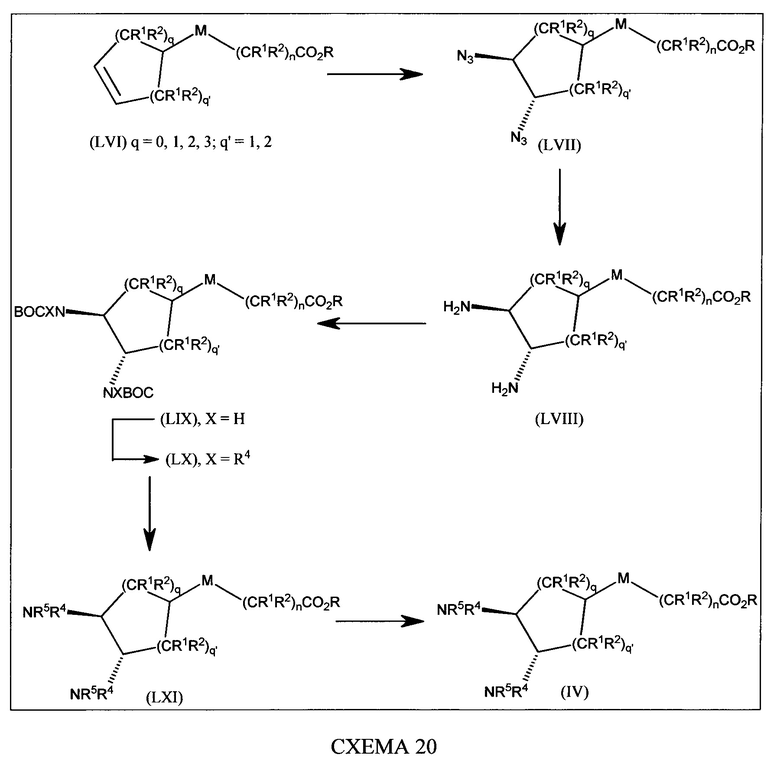
[00182] Альтернативно, обработка соответствующего циклического олефина (LVI) (СХЕМА 21) с окислителем, таким как мета-хлорпербензойная кислота, в растворителе, таком как дихлорметан, при около 0°С дает соответствующий циклический эпоксид (LXII). Раскрытие кольца эпоксида обработкой азидом натрия и хлоридом аммония в растворителе, таком как этанол, полиэтиленгликоль, или DMF/вода, при температуре от комнатной температуры до 80°С обеспечивает транс-гидрокси-азид (LXIII). Реакция взаимодействия соединения (LXIII) с метансульфонилхлоридом в пиридине при 0°С дает мезилат (LXIV). Обработка соединения (LXIV) с азидом тетрабутиламмония в растворителе, таком как толуол, обеспечивает цис-ориентированный бис-азид (LXV). Обработку промежуточного соединения (LXV) осуществляют, как описано выше, чтобы обеспечить кислоты (IV).
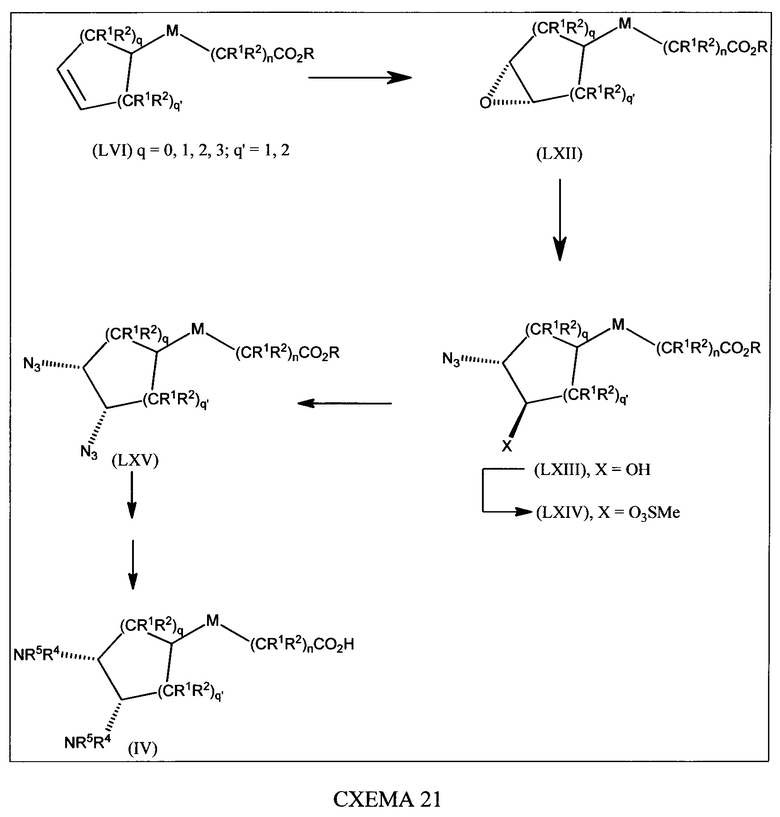
[00183] Где уместно, циклические олефины (LVI) (СХЕМА 22) получают из ациклических исходных материалов реакцией обмена олефинов. Например, обработка сложного эфира (LXVI) с сильным основанием, таким как LDA, в растворителе, таком как THF, THF/DMPU или DME, при температуре от -78°С до 0°С приводит к образованию соответствующего енолята лития. Данный енолят лития обрабатывают подходящим галоген-алкильным олефином с получением бис-олефина (LXVII). Обработка соединения (LXVII) с одним из целого ряда катализаторов метатезиса по Граббсу или Шроку (Grubbs или Schrock) (Tetrahedron, (2012), 68(2) 397-421: Organic Letters, (2007), 9(23), 4885-4888: Tetrahedron, (2004), 60, 7117-7139) в растворителе, таком как дихлорметан, при комнатной температуре или выше, или в водном растворе диметилового эфира PEG500 обеспечивает циклический олефин (LVI, М представляет собой связь, n равно 0).
[00184] Следует отметить, что циклические силил-енольные простые эфиры или алкил-енольные простые эфиры, соответствующие соединению (LVI), которые являются предшественниками циклических кетонов, также удобно получать таким подходом с использованием подходящего ациклического силильного или алкиленольного эфира в реакции метатезиса с циклизацией (Tetrahedron Letters, (2001), 42 (45), 8023).
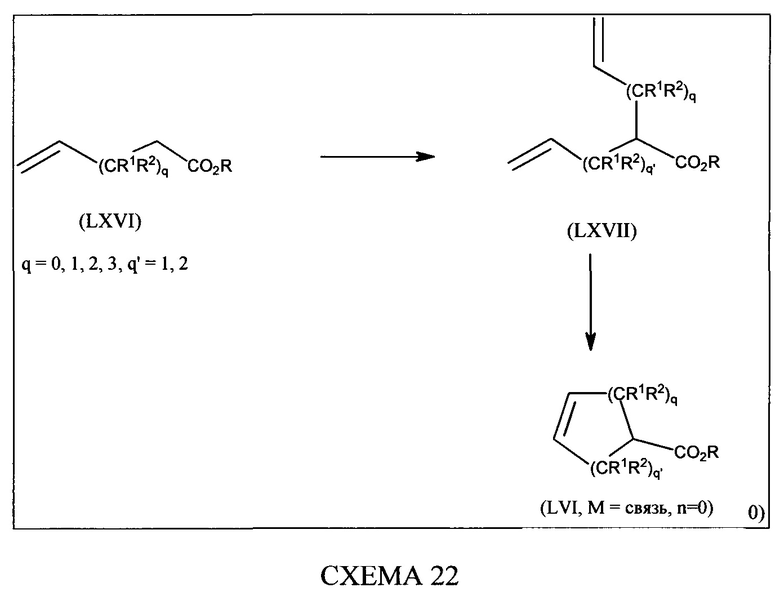
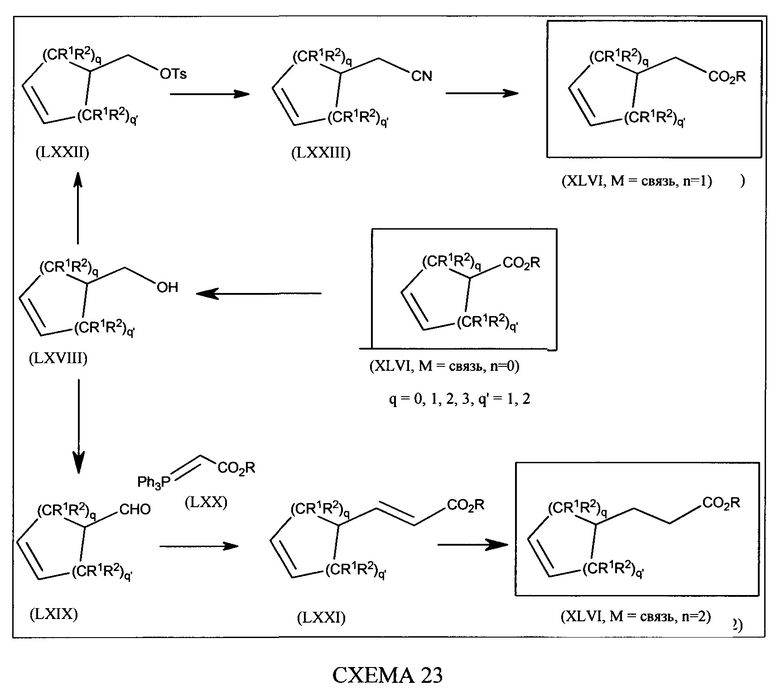
[00185] Последующие стандартные преобразования функциональной группы в экзоциклической алкоксикарбонильной группе (LVI, М представляет собой связь, n равно 0) предоставляют широкий спектр гомологичных алкоксикарбонил-алкилзамещенных циклических олефинов (СХЕМА 23). Например, восстановление функциональной сложноэфирной группы обработкой восстанавливающим агентом, таким как DIBALH, в растворителе, таком как толуол, THF или дихлорметан, при температуре от -78° до 0°С дает первичный спирт (LXVIII). Окисление соединения (LXVIII) с одним из целого ряда окислителей, описанных выше, (например, реагент Десс-Мартина), в растворителе, таком как дихлорметан, дает соответствующий альдегид (LXIX). Олефинирование по Виттигу (LXIX) с использованием трифенилфосфоранилидин-ацетатного сложного эфира (LXX) в растворителе, таком как толуол или THF, при температуре от комнатной температуры до 80°С обеспечивает ненасыщенный сложный эфир (LXXI). Селективное восстановление конъюгированной двойной связи в (LXXI) до соответствующего насыщенного эфира (LVI, n равно 2) осуществляют обработкой с лентой магния, в растворителе, таком как метанол, при температуре, близкой к комнатной температуре.
[00186] Альтернативно, обработка спирта (LXVIII) с n-толуолсульфонилхлоридом в растворителе, таком как пиридин или дихлорметан, в присутствии основания, такого как триэтиламин или DMAP, при температуре от -20°С до комнатной температуры обеспечивает тозилатное производное (LXXII). Удаление тозильной группы в (LXXII) обработкой цианидом натрия в DMFA, DMA или DMSO, при температуре от комнатной температуры до 160°С обеспечивает нитрил (LXXIII). Сольволиз данного нитрила обработкой HCl в соответствующем спирте (таком как метанол или этанол) дает (LVI, M представляет собой связь, n равно 1).
[00187] Реакция метатезиса с циклизацией может быть использована для обеспечения доступа к (LVI, M представляет собой О или NR4) с применением соответствующего алкокси- или аминозамещенного бис-алкенового субстрата (СХЕМА 24). Например, реакция присоединения подходящего реактива Гриньяра (Grignard) к соответствующим образом замещенному альдегиду или имину (LXXIV) обеспечивает вторичный спирт (сульфинамин) (LXXV). Алкилирование или восстановительное аминирование соединения (LXXV) в стандартных условиях обеспечивает соединение (LXXVI). Подвергание соединения (LXXVI) воздействию условиям реакции метатезиса с циклизацией, как описано выше, обеспечивает карбоциклический алкен (LVI, M представляет собой О, NR4).
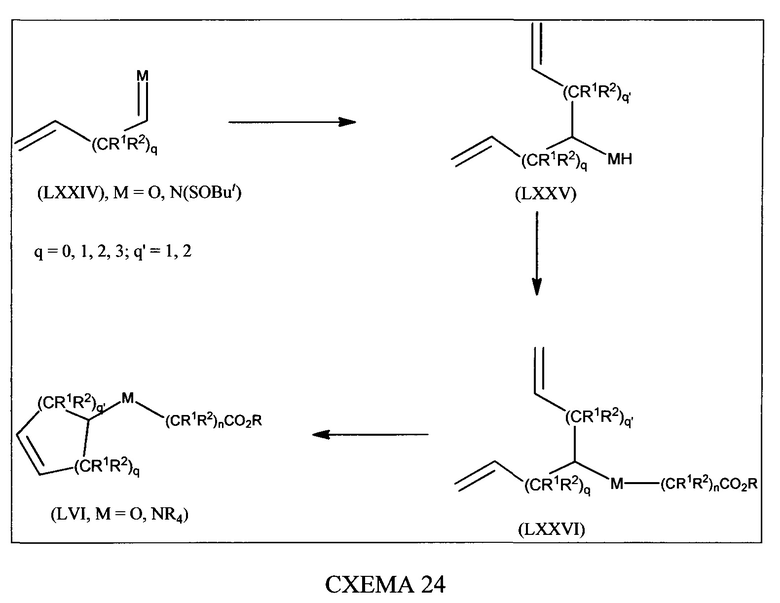
[00188] В случае, когда СусА представляет собой 4-7-членный карбоцикл, Y1 представляет собой 1-амино-трет-амино-алкильную группу (t равно 2, 3, 4), требуемые кислоты (IV) (СХЕМА 25) обычно получают последовательным включением функциональной аминогруппы в соответствующий гидроксил-замещенный или карбокси-замещенный углеродный каркас. Например, селективное восстановление функциональной сложноэфирной группы (LXXVIII) до соответствующего спирта (LXXIX) осуществляют обработкой восстанавливающим агентом, таким как боран: THF, в растворителе, таком как THF, при температуре, близкой к комнатной температуре. Окисление первичного спирта в соответствующий альдегид (LXXX) достигается применением одного из широкого спектра стандартных протоколов окисления, такого как окисление по Сверну (Swern) или окисления с реагентом Десс-Мартина. Альдегид затем обрабатывают замещенным олефином алкильным реактивом Гриньяра в растворителе, таком как THF, диэтиловый эфир или DME, при температуре от около -50°С до комнатной температуры с получением вторичного спирта (LXXXI). В некоторых случаях, когда требуемый замещенный олефином алкил является аллилом (t равно 1), конденсацию с альдегидом (LXXX) также можно осуществить с использованием аллилбороната или аллилсилана в присутствии кислоты Льюиса, такой как TiCl4 или эфират BF3, в растворителе, таком как дихлорметан, при температуре от -78°С до комнатной температуры. Замещенный олефином вторичный спирт (LXXXI) затем превращают в соответствующий амин обработкой p-толуолсульфонилхлорида в присутствии основания, такого как триэтиламин или DMAP, в растворителе, таком как дихлорметан или пиридин, при температуре от -20°С до комнатной температуры, получая соответствующий тозилат (LXXXII). Тозилат обрабатывают солью азида, такой как азид натрия, азид тетрабутиламмония, в растворителе, таком как DMF, DMA или DMSO, при температуре от комнатной температуры до 160°С, с получением соответствующего азида (LXXXIII). Данное промежуточное соединение восстанавливают до первичного амина (LXXXIV), а затем подвергают дериватизации, как описано выше, с получением соединения (LXXXV). Альтернативно, введение функциональной первичной аминогруппы осуществляют превращением вторичного спирта (LXXXI) до соответствующего фталимида (LXXXVI) в условиях реакции Мицунобу (Mitsunobu) (Chemical Reviews, (2009) 2551-2651) с последующим снятием защиты с фталимида обработкой избытком гидразина в растворителе, таком как этанол, при комнатной температуре или выше, чтобы обеспечить первичный амин (LXXXIV).
[00189] Введение второй функциональной аминогруппы в (LXXXV) осуществляется с помощью окислительного расщепления олефина, как описано выше, чтобы предоставить альдегид (LXXXVII). Данное промежуточное соединение превращают в требуемый амин (LXXXVIII) непосредственным восстановительным алкилированием или восстановлением до первичного спирта с использованием восстанавливающего агента, такого как боргидрид натрия, в растворителе, таком как THF/метанол, при температуре от -78°С до 0°С, с последующей дериватизацией полученного первичного спирта (через соответствующий тозилат, азид и первичный амин), как описано выше. Обработка боковой цепи соединения (LXXXVIII), как описано ранее, дает соединение (IV).
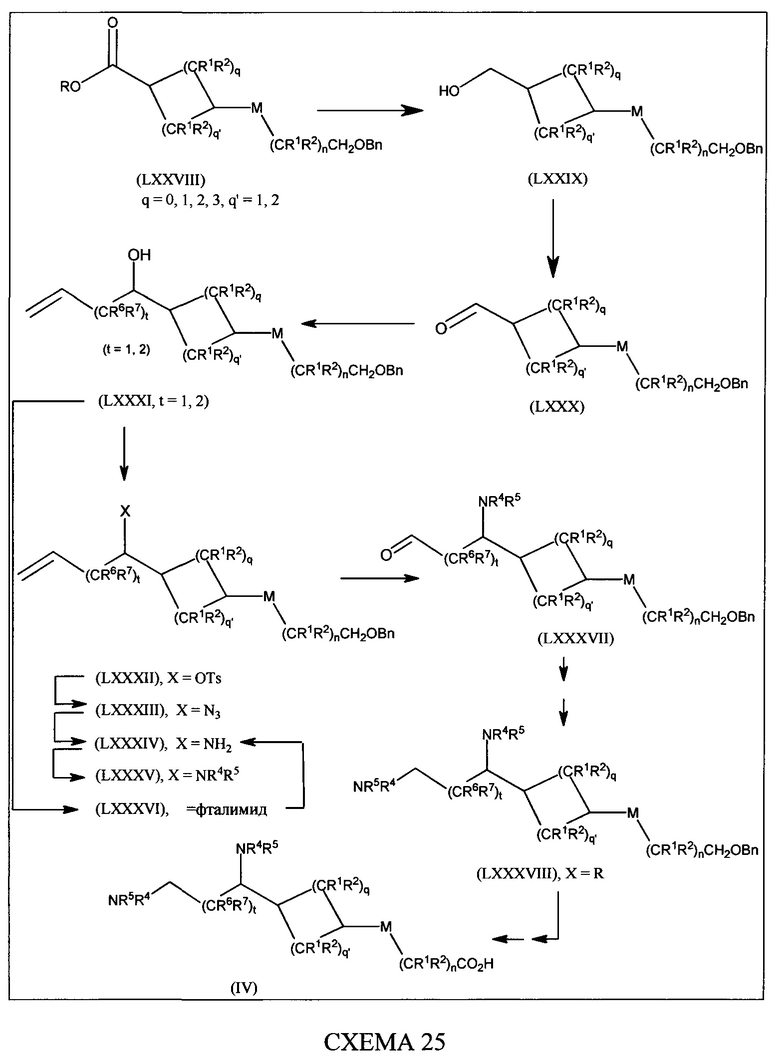
[00190] В частном случае, где Y1 представляет собой необязательно замещенную 1-амино-2-амино-этильную группу, введение диамино-функциональной группы может быть достигнуто прямым аминированием соответствующего экзоциклического алкена (Mn(OAc)3/NaN3/TFA, затем восстановление). Необходимый экзоциклический олефин получают олефинированием соединения (LXXX) по Виттигу, Петерсону или Tebbe, как уже описано.
[00191] В случае, когда СусА представляет собой необязательно замещенный 4-7-членный карбоцикл, Y1 представляет собой необязательно замещенную трет-амино-алкильную группу (t равно 1, 2, 3, 4), Y2 представляет собой необязательно замещенный амин и оба Y1 и Y2 присоединены к одному и тому же атому углерода кольца из СусА, требуемые кислоты (IV) (схема 26) получают из предшественника (LXXXIX) соответствующего карбоциклического кетона обработкой трет-бутилсульфинамином (Chemical Reviews, (2010), 110(6), 3600-3740), обычно в растворителе, таком как THF или метанол, в присутствии Ti(OEt)4, при температуре от комнатной температуры до 60°С. Полученный трет-бутилсульфинимин затем подвергают реакции конденсации с соответствующим металлоорганическим реагентом, например, реактивом Гриньяра с замещенной олефином алкильной группой (или, если применимо, CeCl3-модифицированным реактивом Гриньяра) в растворителе, таком как THF, эфир, дихлорметан или толуол, при температуре от -60 до 0°С, чтобы получить замещенный сульфинамином карбоцикл (ХС). Удаление тионильной группы осуществляют обработкой кислотой, такой как трифторуксусная кислота, в растворителе, таком как дихлорметан, при температуре, близкой к комнатной температуре, с получением соответствующего первичного амина (XCI, R4, R5 представляет собой Н). Группу первичного амина подвергают дериватизации или защищают, в соответствующих случаях, описанными выше способами. Обработку функциональной группы алкена для обеспечения соответствующего амина осуществляют окислительным расщеплением (OsO4/NMO, затем NaIO4), затем проводят восстановительное аминирование полученного альдегида с получением соединения (XCII). Альтернативно, гидридное восстановление альдегида и дериватизация спирта, как описано выше, также обеспечивает соединение (XCII). Преобразование соединения (XCII) до требуемой кислоты осуществляют обработкой кислотной группы предшественника боковой цепи Y, как уже описано.
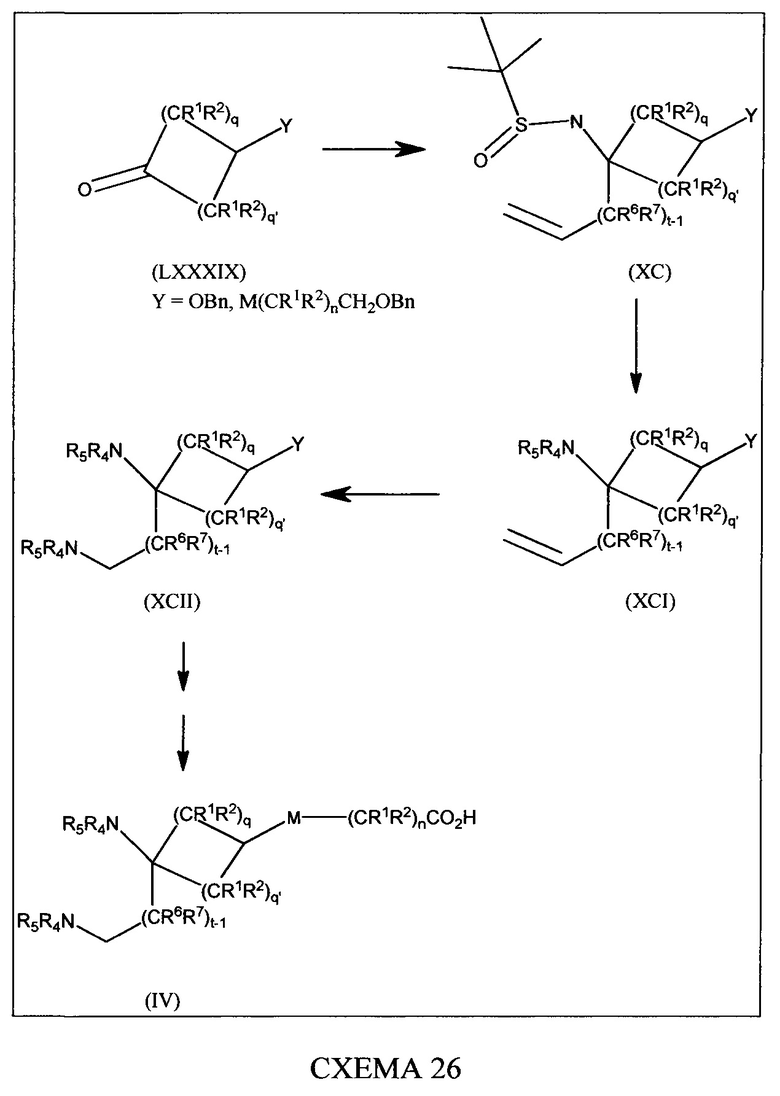
[00192] В конкретном случае, когда t равно 1, (IV) получают из кетона (LXXXIX) реакцией по Strecker (Synthesis, (2007), 1230-1234. Organic Letters, (2008), 10, 1509-1512) с последующим восстановлением полученной нитрильной группы. Последующую обработку данного промежуточного соединения с получением соединения (IV) проводят, как уже описано.
[00193] В одном варианте осуществления вышеприведенной системы, где СусА представляет собой необязательно замещенный 4-7-членный карбоцикл, Y1 представляет собой трет-амино-алкильную группу (t равно 2, 3, 4), Y2 представляет собой амин, оба Y1 и Y2 присоединены к тому же атому углерода кольца из СусА и Y1 и Y2 соединены друг с другом через их заместителей (группой СН2), требуемые кислоты (IV) (схема 27) получают из промежуточного продукта (XCI) обработкой с OsO4 в растворителе, таком как ацетон/вода, в присутствии окислителя, такого как N-метил-морфолин-N-оксид, с получением соответствующего диола. Данное промежуточное соединение обрабатывают периодатом натрия в THF/вода с образованием усеченного альдегида (XCIII). (XCIII) затем конденсируют с производным сложного эфира α-фосфоноглицина, таким как ВОС-защищенный α-фосфоно-глицин-триметиловый сложный эфир, в присутствии основания, такого как трет-бутоксид калия, в растворителе, таком как дихлорметан, при температуре от -78°С до -50°C с получением ненасыщенного эфира (XCIV) (Tetrahedron, (2001), 57, 6463). (XCIV) селективно гидрируют с использованием катионного катализатора фосфин-фосфита родия с получением соответствующего насыщенного-ВОС-ТСЕОС-защищенного сложного аминоэфира. С данного промежуточного соединения снимают защиту в стандартных условиях (цинк/уксусная кислота в THF) с получением Вос-защищенного аминоэфира (XCV).
[00194] Циклизация сложного аминоэфира в присутствии основания, такого как DBU, в растворителе, таком как толуол, при температуре от комнатной температуры до 110°С обеспечивает пактам (XCVI). Селективное восстановление третичного амида до соединения (XCVI) с использованием силанового восстанавливающего агента в присутствии родийгидрокарбонилтрифенилфосфина или платинохлористоводородной кислоты, как описано выше, с последующей дериватизацией амина обеспечивает (XCVII). Последующая обработка боковой цепи Y, как описано выше, обеспечивает необходимые кислоты (IV).
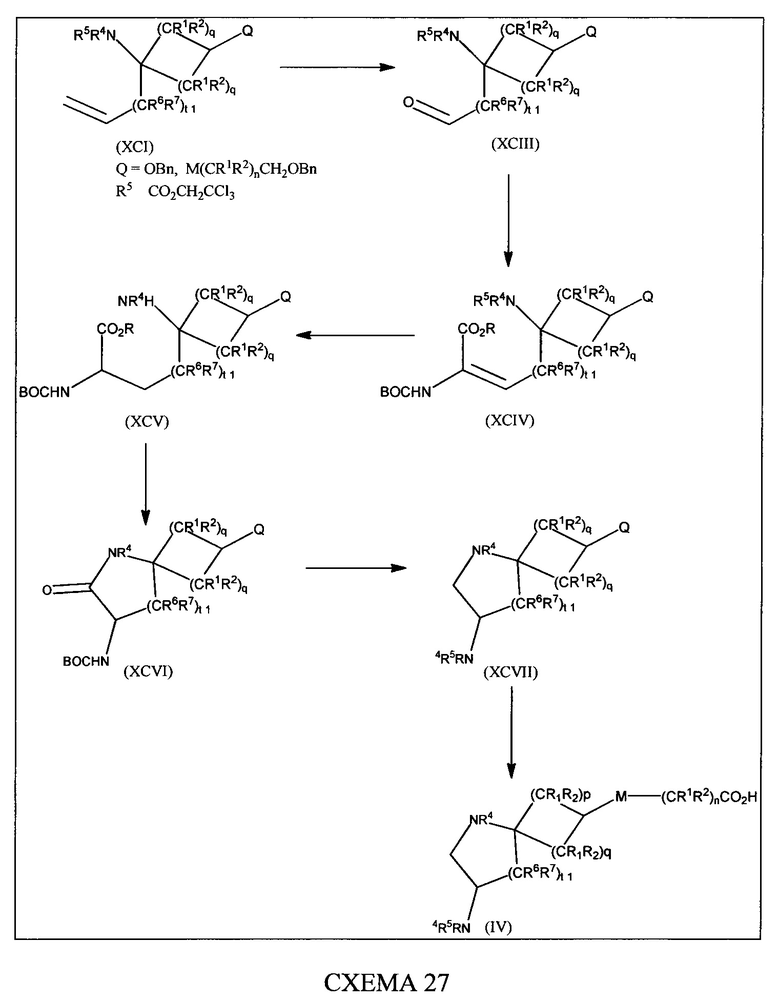
[00195] В случае, когда СусА представляет собой необязательно замещенный 4-7-членный карбоцикл, Y1 представляет собой необязательно замещенную трет-амино-алкильную группу (t равно 2, 3, 4), Y2 представляет собой необязательно замещенный амин, и кольцевой атом углерода из СусА, присоединенный к Y1, и кольцевой атом углерода из СусА, присоединенный к Y2, расположены вицинально относительно друг друга, требуемые кислоты (IV) (СХЕМА 28) могут быть получены из соответствующего циклического кетона (XCVIII). Например, в случае, когда t равно 2, обработка (XCVIII) с триалкилсилилтрифлатом в присутствии основания, такого как триэтиламин, в растворителе, таком как эфир, дает соответствующий силиленольный простой эфир (XCIX). Реакция взаимодействия данного промежуточного соединения с нитро-олефином (R6R7C=CR6NO2) в присутствии кислоты Льюиса, такой как TiCl4/(TiOiPr)4 или SnCl4, в растворителе, таком как дихлорметан, при температуре от -78°С до комнатной температуры обеспечивает замещенный нитроалкилом циклический кетон (С) (Journal of the American Chemical Society, 106(7), 2149-56; 1984; Helvetica Chimica Acta, 82(11), 1829-1842; 1999; Canadian Journal of Chemistry, 65(4), 836-50; 1987). Гидридное восстановление данного промежуточного соединения с использованием реагента, такого как литийалюминийгидрид, в растворителе, таком как THF, при температуре от -20°С до температуры флегмы дает аминоспирт (CI). Аминогруппу соединения (CI) подвергают селективной дериватизации, как описано выше, с получением (CII). Введение второй аминогруппы осуществляют дериватизацией карбоциклического спирта (CII) с использованием способов, описанных выше, чтобы обеспечить соединение (CIII). Удаление бензильной защитной группы эфира и окисление полученного первичного спирта осуществляют, как описано выше, чтобы получить необходимую карбоновую кислоту (IV).
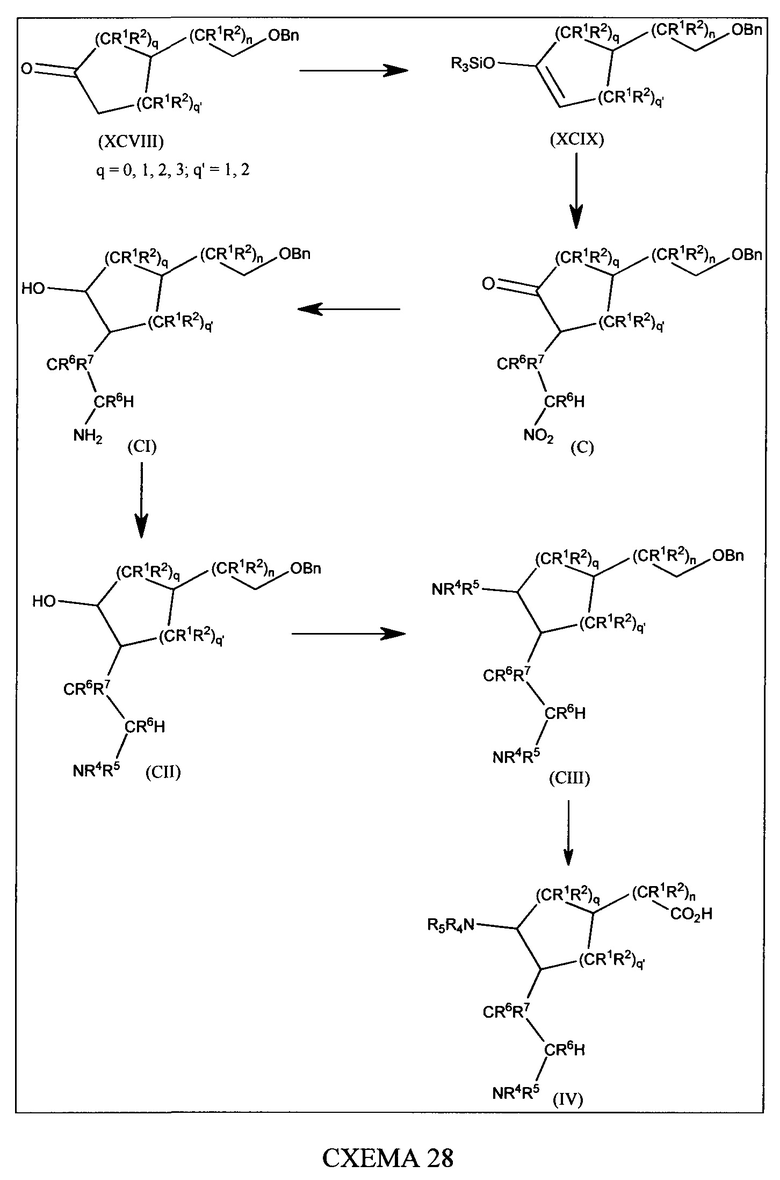
[00196] В случае, когда t равно 1 (СХЕМА 29), введение надлежащей функциональной группы боковой цепи достигается реакцией взаимодействия силиленольного простого (XCIX) с солью иминия (в соответствующих случаях, получаемой in situ из N,O-ацеталя и триметилсилилтрифлата) в растворителе, таком как дихлорметан, при температуре от -78°С до комнатной температуры, чтобы обеспечить аминокетон (CIV). Обработка кетона в (CIV), как описано выше, обеспечивает диамино-функционализированный каркас (CV). После этого удаление бензилового простого эфира и окисление полученного первичного спирта обычным способом дает требуемую кислоту (IV).
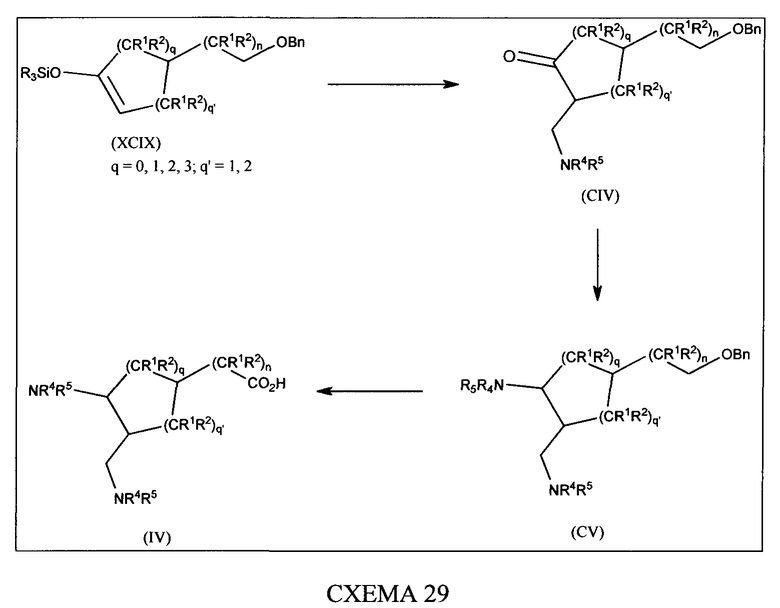
[00197] В случае, когда t равно 3 (СХЕМА 30), введение соответствующей функциональной группы боковой цепи достигается в результате реакции взаимодействия силиленольного простого эфира (XCIX) с метиллитием, в растворителе, таком как THF, при температуре от -20° до 0°С, получая региоселективно соответствующий енолят лития. Затем следует обработка енолята с электрофильным соединением, таким как акриламид (или акрилонитрил), с получением соединения (CIV). Обработка кетона, как уже описано, обеспечивает амин-функционализированный амид (CVII). Восстановление амида с восстановителем, таким как литийалюминийгидрид, в растворителе, таком как THF, при температуре от -10°С до температуры флегмы дает диамин (CVIII). Данное промежуточное соединение затем обрабатывают, как описано выше (дебензилирование, затем окисление), чтобы обеспечить необходимую кислоту (IV).
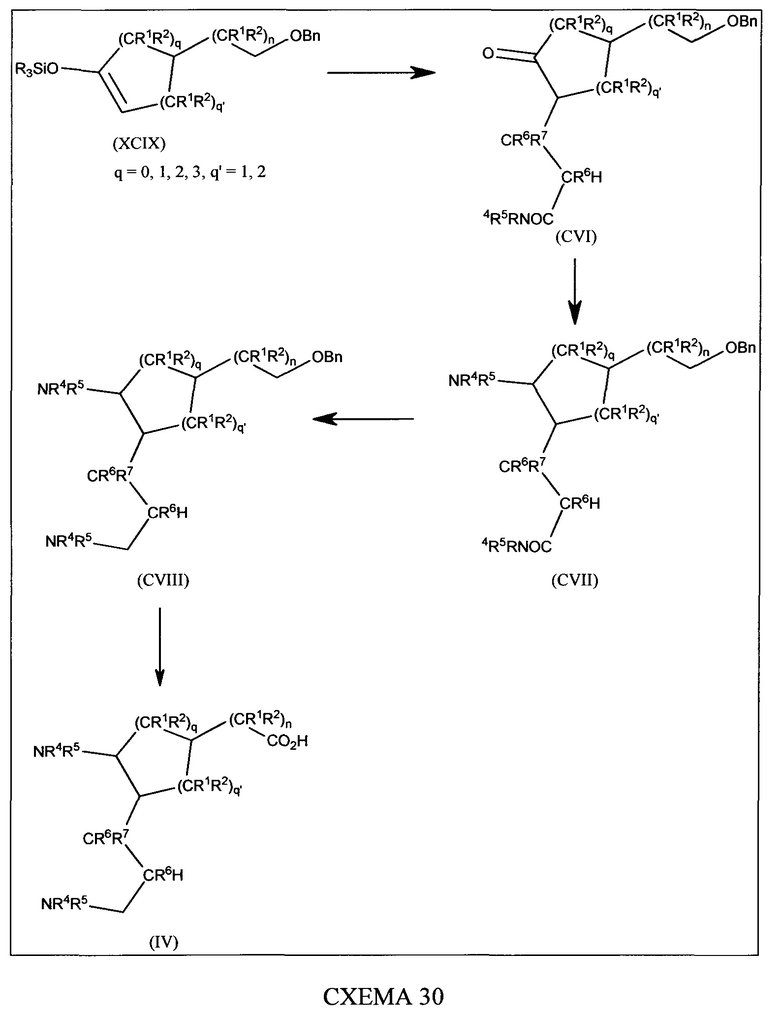
[00198] В случае, когда А представляет собой необязательно замещенный 4-7-членный карбоцикл, Y1 представляет собой необязательно замещенную r-аминоалкильную группу (t равно 2, 3, 4), Y2 представляет собой необязательно замещенный амино-метилен и оба Y1 и Y2 присоединены к одному и тому же атому углерода кольца А, требуемые кислоты (IV) (СХЕМА 31) получают из карбоциклического кетона (CIX). Например, преобразование (CIX) до соответствующего экзоциклического нитрила (СХ) осуществляют обработкой с TOSMIC и основанием, как описано ранее. Обработка соедиднения (СХ) с основанием, таким как LDA, LHMDS или NaHMDS в растворителе, таком как THF, и имином, таким как трет-бутилсульфинимин (Chemical Reviews, (2010), 110(6), 3600-3740), дает замещенный трет-бутилсульфинаминометилом нитрил (CXI). Удаление сульфинильной группы обработкой кислотой, такой как трифторуксусная кислота, в дихлорметане с последующей дериватизацией первичного амина, как описано выше, обеспечивает аминонитрил (CXII). Восстановление нитрила (CXII) алюмогидридом лития, как описано выше, дает первичный амин (CXIII). Данный амин дополнительно подвергают дериватизации, в случае необходимости, как описано выше (это может также включать в себя временную дериватизацию с использованием легко удаляемой защитной группы), чтобы получить (CXIV). Последующая обработка экзоциклического бензилового простого эфира в (CXIV), используя обычные процедуры, обеспечивает необходимую кислоту (IV).
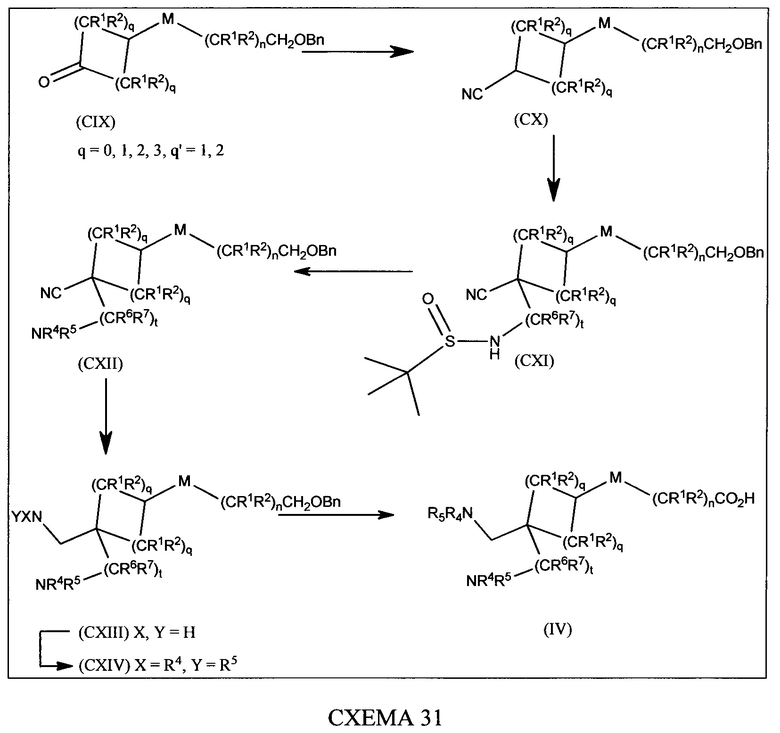
[00199] В случае, когда СусА является 6- или 7-членный карбоциклом, Y1 представляет собой необязательно замещенную r-аминоалкильную группу (t равно 1, 2, 3), Y2 представляет собой необязательно замещенный амин, и кольцевой атом углерода из СусА, присоединенный к Y1, и кольцевой атом углерода из СусА, присоединенный к Y2, разделены метиленом, требуемые кислоты (IV) (СХЕМА 32) получают из соответствующим образом замещенного карбоциклического енона, такого как (CXV). Например, в случае, когда t равно 1, соответственно функционализированная одноуглеродная группа вводится обработкой карбоциклического енона (CXV) с системой цианирующего реагента, такого как триметилсилилцианид или цианистый водород, в присутствии основания, такого как KF, Cs2CO3, Bu4NF или тетраметилгуанидин, в подходящем растворителе, таком как метанол, THF, DMA или ацетонитрил, чтобы получить соединение (CXVI). Введение нитрила может быть также осуществлено обработкой енона (CXV) с цианидом диэтилалюминия в растворителе, таком как бензол, толуол или дихлорметан, при температуре от около -20°С до комнатной температуры. Последующая дериватизация карбоциклического кетона (CXVI) (через восстановительное аминирование, превращение в азид, восстановление азида и дальнейшую дериватизацию результирующего первичного амина), как описано выше, дает замещенный 1-амино-3-циано-группой карбоцикл (CXVII). Данное промежуточное соединение преобразуют в искомое аминометильное производное восстановлением нитрила и функционализацией полученного первичного амина (CXVIII), как уже было описано, чтобы обеспечить (CXIX). Обработка боковой цепи обычным способом обеспечивает (IV).
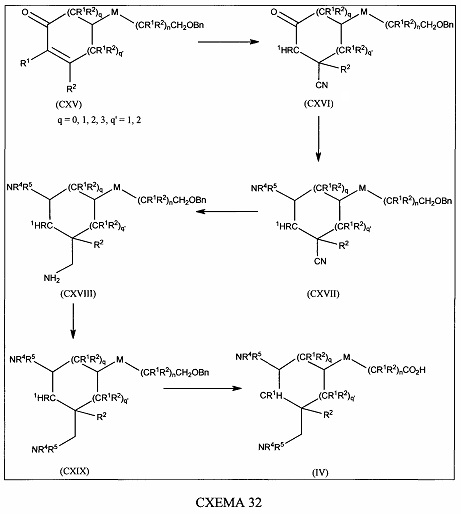
[00200] В случае, когда t равно 2 или 3 (схема 33), соответственно функционализированный углеродный каркас получают обработкой карбоциклического енона (CXV) с аллилсиланом в некоординирующем растворителе, таком как дихлорметан, в присутствии кислоты Льюиса, такой как TiCl4, SnCl4 или эфират BF3, при температуре от -78°С до комнатной температуры с получением функционализированного олефином карбоциклического кетона (СХХ). Обработка функциональной группы кетона в (СХХ), как уже было описано, дает (CXXI). Окислительное расщепление олефина в (CXXI) и обработка полученного альдегида до соответствующего амина, как уже описано, обеспечивает (CXXII). Данное промежуточное соединение затем преобразуют в требуемую кислоту (IV, t равно 2) обработкой боковой цепи обычным способом. Альтернативно, гидроборирование олефина (CXXI) обработкой с бораном, например, 9-BBN, в растворителе, таком как THF, при температуре около 0°C с последующей окислительной обработкой (NaOH/H2O2) обеспечивает (CXXIII). Последующая обработка соединения (CXXIII), как уже было описано, вводит функциональную аминогруппу с получением (CXXIV). Обработка боковой цепи данного промежуточного соединения дает кислоту (IV, t равно 3).
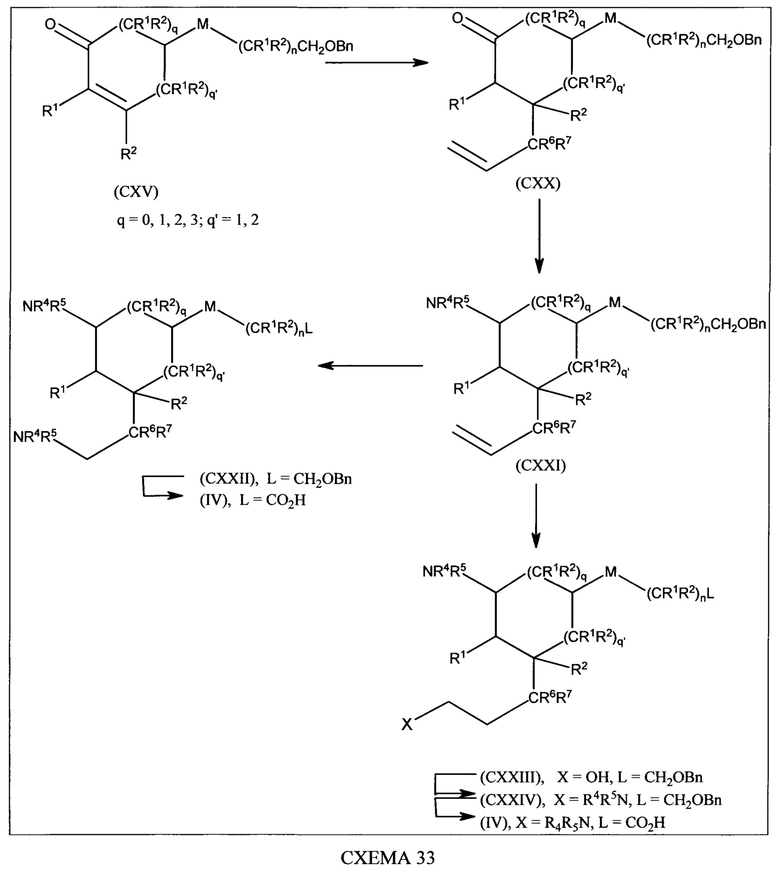
[00201] В случае, когда А представляет собой необязательно замещенный 6- или 7-членный карбоцикл, Y1 представляет собой необязательно замещенный амин, Y2 представляет собой необязательно замещенный амин и кольцевой атом углерода из СусА, присоединенный к Y1, и кольцевой атом углерода из СусА, присоединенный к Y2, разделены метиленом, требуемые кислоты (IV) (СХЕМА 34) получают из соответствующим образом замещенного карбоциклического енона, такого как (CII), обработкой соответствующего амина в присутствии катализатора, такого как RuCl3, Cu(асас)2, FeCl3, Ce(NH4)NO3 Pd(acac)2NH4PF6, в растворителе, таком как вода или полиэтиленгликоль (Green Chemistry, (2006), 8(4), 356-358; Synthesis, (2005), (13), 2129-2136; Helvetica Chimica Acta, (2004), 87(6), 1522-1526; Advanced Synthesis & Catalysis, (2005), 347(6), 763-766; Synthetic letters, (2006), (10), 1549-1553), при температуре от комнатной температуры до 80°C с получением упомянутого β-амино-кетона (CXXV). Последующая обработка функциональной группы кетона, как описано выше, дает диамин (CXXVI). Преобразование (CXXVI) в кислоту (IV) достигается обработкой боковой цепи, как описано ранее.
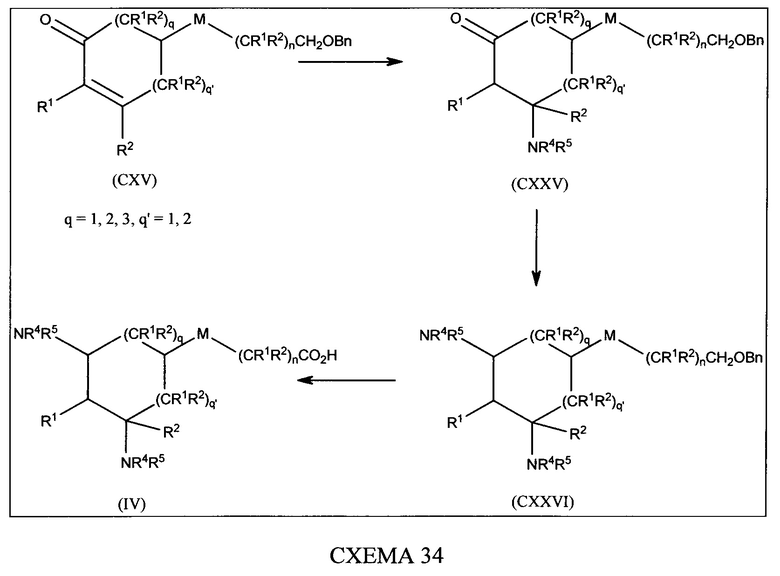
[00202] В случае, когда Y1 представляет собой необязательно замещенную амино-алкил-окси-группу, требуемые кислоты (IV) (СХЕМА 35) получают обработкой соответствующего карбоциклического спирта (CXXVII) с алкилгалогенидом, замещенным фталимидом или азидом, в присутствии основания, такого как гидрид натрия, в растворителе, таком как DMF, DMA, DMSO, при температуре от 5°С до 80°С (также могут быть использован катализатор, такой как иодид тетрабутиламмония) с получением соответствующих, замещенных скрытой аминогруппой алкокси-карбоциклов (CXXVIII и CXXIX), соответственно. Раскрытие скрытой аминогруппы обработкой соединения (CXXVIII) с гидразином в этаноле или обработка соединения (CXXIX) с трифенилфосфином и водой в THF дает соответствующий первичный амин (СХХХ). Дериватизация аминогруппы, как описано выше, дает соединение (CXXXI). Обработка соединения (CXXXI) по стандартной методике дает необходимую кислоту (IV).

[00203] Альтернативно (СХЕМА 36), обработка спирта (CXVII) с системой реагентов, такой как йод/имидазол/Ph3P или NBS/Ph3P, или с соответствующим сульфонилхлоридом (или ангидридом) в присутствии основания, такого как пиридин или триэтиламин, в растворителе, таком как THF или дихлорметан, дает соответствующий иодид, бромид или сульфонат (CXXXII). Реакция (CXXXII) с соответствующим спиртом в присутствии основания, такого как гидрид натрия, в растворителе, таком как THF, DMF, DMA или TBTU, или их смеси, обеспечивает алкокси-замещенные карбоциклы (CXXVIII) и (CXXIX). Преобразование данных промежуточных соединений до соединения (IV) осуществляется, как уже описано.
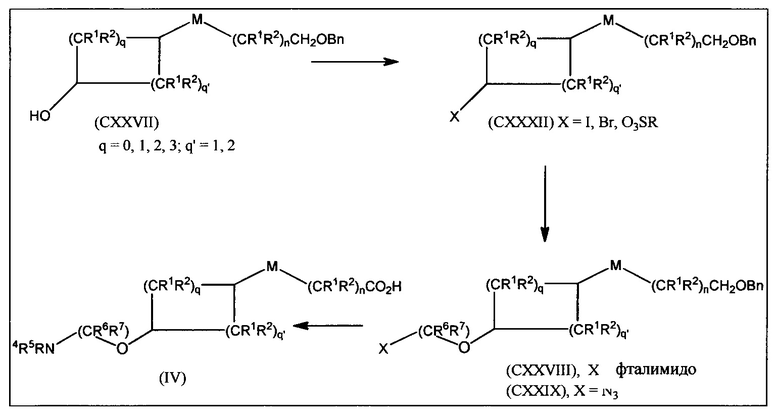
[00204] Формула (I) предложена в случае, когда Y1 и Y2, взятые вместе с атомом углерода или атомами углерода, к которым они присоединены, образуют необязательно замещенный карбоцикл или необязательно замещенный гетероцикл. Конкретный случай данного осуществления (СХЕМА 37) имеется, когда СусА представляет собой циклогексан, Y1 представляет собой (N-метил-гуанидинил)метильную группу, Y2 представляет собой метальную группу, Y1 и Y2 расположены вицинально относительно друг друга в СусА, и Y1 и Y2 связаны друг с другом формальным слиянием двух метальных групп с образованием замещенного пиперидина. В данном конкретном случае требуемые кислоты (IV) получают из соответствующего пиперидинона, такого как (CXXXIII). Например, обработка соединения (CXXXIII) с α-метилбензиламином (Bioorganic and medicinal Chemistry Letters, (2008), 18(4), 1312-1317) и соответствующим металвинилкетоновым производным в толуоле с последующей циклизацией с метоксидом натрия в метаноле дает бициклический кетон (CXXXIV). Гидрирование соединения (CXXXIV) с использованием катализатора, такого как палладий на угле, в растворителе, таком как метанол, и введение гуанидинильной группы, как уже было описано, дает кетон (CXXXV). Обработка кетона (CXXXV), как уже отмечалось, обеспечивает необходимые кислоты (IV, М представляет собой связь; n равно 1, 2). Альтернативно, восстановление соединения (CXXXV) с гидридным восстанавливающим агентом, таким как боргидрид натрия, в метаноле при температуре от -78°С до 0°С дает соответствующий спирт, который обрабатывают этилдиазоацетатом и каталитическим количеством Rh(acac)2-димера в растворителе, таком как дихлорметан, с получением эфира (CXXXVI). Сапонификация соединения (CXXXVI) кратковременной обработкой гидроксидом лития в смеси THF/метанол/вода дает кислоту (IV, М представляет собой связь; n равно 1).
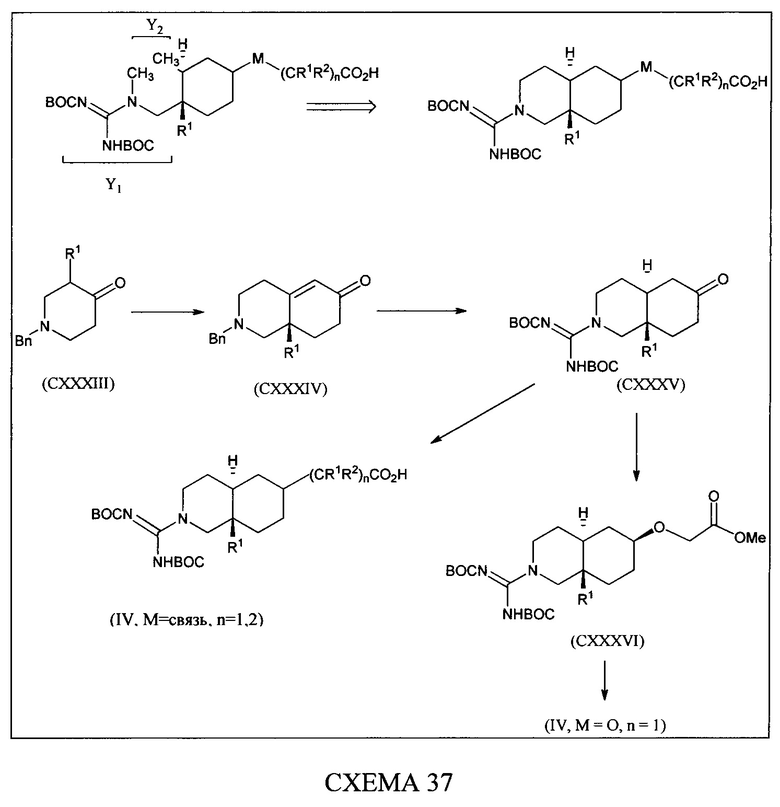
[00205] В случае, когда Z представляет собой сульфонильную группу (СХЕМА 38), необходимую сульфоновую кислоту получают из соответствующей активированной карбоновой кислоты (V) обработкой гидрокситиопиридоном натрия в растворителе, таком как дихлорметан, при температуре, близкой к комнатной температуре с получением промежуточного соединения эфира Бартона (CXXXVII). Соединение (CXXXVII) обрабатывают йодоформом в CCl4 под вольфрамовой УФ-лампой при температуре, близкой к температуре кипения, чтобы обеспечить продукт декарбоксилативного йодирования (CXXXVIII) (Journal of Organic Chemistry, 75(19), 6489-6501; 2010). Альтернативно, обработка кислоты (IV) с иодозо-бензол-диацетатом и иодом в CCl4, под вольфрамовой УФ-лампой, около температуры флегмы (Journal of Organic Chemistry, (1986), 51, 402) обеспечивает соединение CXXXVIII) непосредственно. Обработка соединения (CXXXVIII) с сульфитом натрия в водном этаноле, изопропаноле или ацетоне при температуре от 60 до 90°C с последующим подкислением дает сульфоновую кислоту (IV). Кроме того, обработка соединения (CXXXVIII) с тиомочевиной в ацетоне, при температуре около 60°С обеспечивает производное соль изотиоурония (CXXXIX) (Synthetic Letters, (2010), 7, 1037). Расщепление соединения (CXXXVIII) с водн. тиосульфатом натрия дает тиол (CXL). Обработка соединения (CXL) надмуравьиной кислотой (муравьиная кислота и водный раствор H2O2 при от близкой к 0°С до комнатной температуры) дает (IV).
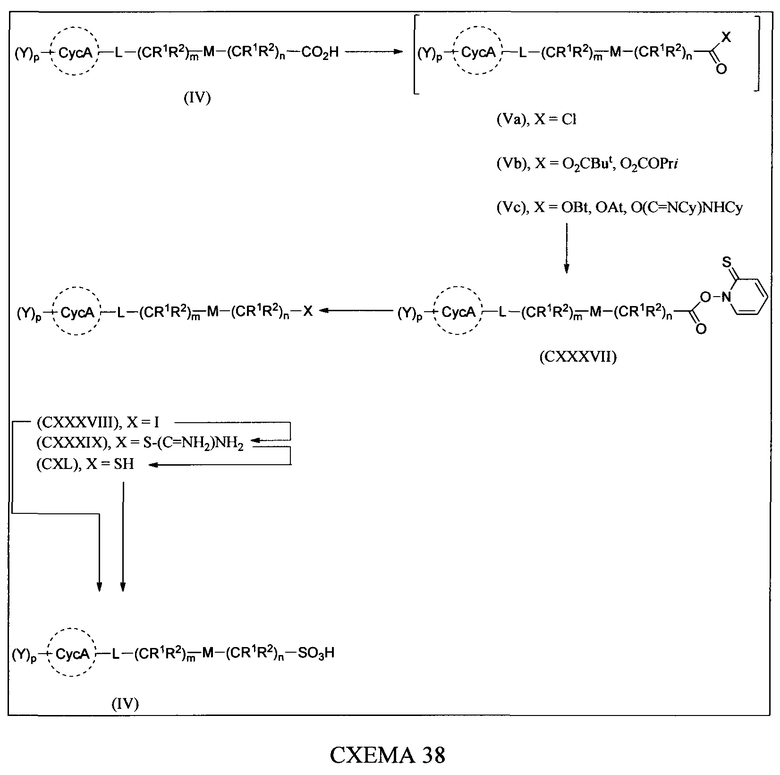
Примеры синтеза
[00206] Следующие способы получения соединений формулы I или формулы Ia и промежуточных продуктов приведены, чтобы дать возможность специалистам в данной области техники более четко понимать и осуществить на практике настоящее изобретение. Они не должны рассматриваться как ограничивающие объем изобретения, а только как иллюстративные и представляющие его.
Пример 1: (R)-3-(транс-4-(аминометил)циклогексанкарбоксамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
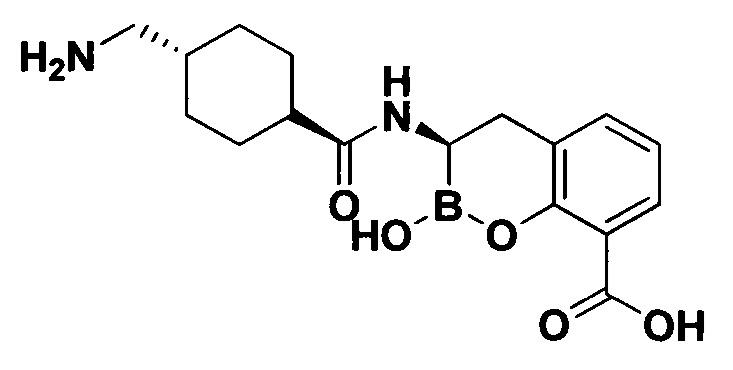
Стадия 1: Синтез трет-бутилового сложного эфира 3-((2R)-2-(транс-4-((трет-бутоксикарбониламино)метил)циклогексанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата.
[00207] К безводному CH2Cl2 (0,61 мл, 9,4 ммоль) в THF (20 мл) в атмосфере аргона при -100°С (МеОН/жидк. N2) по каплям добавляли n-BuLi (2,7 мл, 2,5 М в гексане) и реакционную смесь перемешивали при той же температуре в течение 30 мин. Раствор трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты (2.37 г, 5,92 ммоль) в THF (5 мл) добавляли в течение 10 мин. Через 20 мин охлаждающую баню удаляли и реакционную смесь медленно нагревали до 0°С и перемешивали при той же температуре в течение 1 часа. Затем реакционную смесь охлаждали до -78°С, медленно добавляли LHMDS (8,0 мл, 1М в THF) и полученную реакционную смесь перемешивали, давая нагреться до комнатной температуры постепенно в течение ночи. Безводный МеОН (0,29 мл, 7,1 ммоль) добавляли при -10°С, реакционную смесь перемешивали при той же температуре в течение 1 часа и затем при комнатной температуре в течение 1 часа.
[00208] В отдельной колбе, содержащей 0,386 г 4-((трет-бутоксикарбониламино)метил)циклогексанкарбоновой кислоты (1,5 ммоль), добавляли безводный CH2Cl2 (12 мл). К данной реакционной смеси добавляли NMM (0,22 мл, 2 ммоль), а затем HATU (0,570 г, 1,5 ммоль). Добавляли DMF (1 мл) и полученный раствор перемешивали при комнатной температуре (RT) в течение 1 часа, после чего часть раствора из описанной выше реакционной смеси (1,5 ммоль) добавляли в колбу и реакционную смесь перемешивали в течение 2 ч. Реакцию гасили добавлением воды (30 мл) и водную фазу экстрагировали этилацетатом (3×50 мл). Органическую фазу промывали насыщенным раствором соли, высушивали над Na2SO4 и концентрировали в вакууме с получением неочищенного продукта, который очищали флэш-хроматографией на силикагеле (гексан/EtOAc, от 2:1 до 1:2) с получением продукта (200 мг, выход 20%). ESI-MS m/z 669,1 (MH)+.
Стадия 2: Синтез (R)-3-(транс-4-(аминометил)циклогексанкарбоксамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00209] К раствору трет-бутил-3-((2R)-2-(транс-4-((трет-бутоксикарбониламино)метил)циклогексанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата со стадии 1 (200 мг, 0,30 ммоль) в безводном CH2Cl2 (5 мл) при -78°С добавляли BCl3 (2,1 мл, 1М в DCM, 2,1 ммоль), и реакционную смесь перемешивали при той же температуре в течение 1 часа, при чего реакционную смесь нагревали до 0°С и перемешивали при той же температуре в течение дополнительного 1 часа. Реакцию гасили добавлением воды (5 мл) при 0°С. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации, получая продукт (30 мг) в виде белого твердого вещества. ESI-MS m/z 347 (МН)+.
Пример 2: (R)-3-(транс-4-аминоциклогексанкарбоксамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
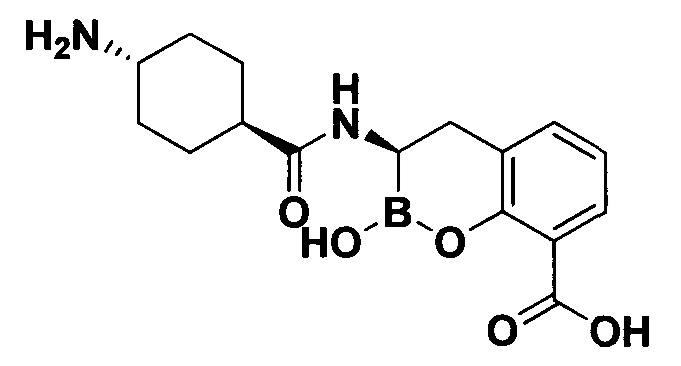
Стадия 1: Синтез трет-бутилового сложного эфира 3-((2R)-2-(транс-4-(трет-бутоксикарбониламино)циклогексанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата.
[00210] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и tran-4-(трет-бутоксикарбониламино)циклогексанкарбоновой кислоты в соответствии с процедурой, описанной на стадии 1 в примере 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (гексан/EtOAc, от 2:1 до 1:2). ESI-MS m/z 655,1 (МН)+.
Стадия 2: Синтез (R)-3-(транс-4-аминоциклогексанкарбоксамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00211] Соединение получали из трет-бутилового сложного эфира 3-((2R)-2-(транс-4-(трет-бутоксикарбониламино)циклогексанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 333 (МН)+.
Пример 3: (R)-3-(2-транс-4-(аминометил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
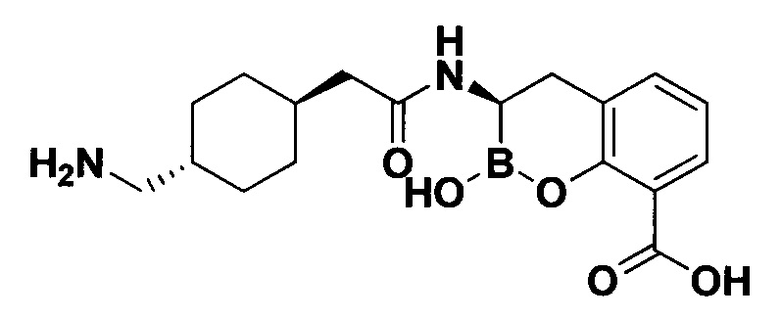
Стадия 1: Синтез трет-бутилового сложного эфира 3-((2R)-2-(2-транс-4-((трет-бутоксикарбониламино)метил)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата.
[00212] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и 2-(транс-4-((трет-бутоксикарбониламино)метил)циклогексил)уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (гексан/этилацетат, от 2:1 до 1:2). ESI-MS m/z 683.1 (МН)+.
Стадия 2: Синтез (R)-3-(2-транс-4-(аминометил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00213] Соединение получали из трет-бутил-3-((2R)-2-(2-транс-4-((трет-бутоксикарбониламино) метил)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 361 (МН)+.
Пример 4: (R)-3-(2-(транс-4-(гуанидинометил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
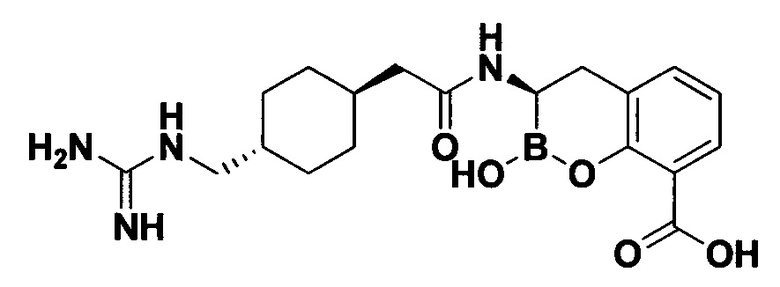
Синтез (R)-3-(2-(транс-4-(гуанидинометил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00214] К (R)-3-(2-транс-4-(аминометил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоте из примера 3 (12 мг) в МеОН (2 мл) добавляли трет-бутил(1Н-пиразол-1-ил)метандиилидендикарбамат (12 мг) и перемешивали в течение 4 ч. Растворитель удаляли в вакууме. Остаток растворяли в 4N HCl в диоксане (2 мл) и перемешивали в течение 2 ч. Растворитель удаляли в вакууме и неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 403 (МН)+.
Пример 5: (R)-3-(2-(транс-4-((2-(диметиламино)ацетамидо)метил) циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
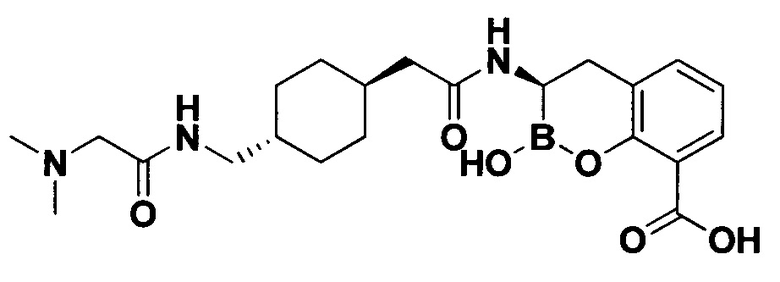
Стадия 1: Синтез 3-((2R)-2-(2-(транс-4-(аминометил)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты.
[00215] К трет-бутил-3-((2R)-2-(2-(транс-4-((трет-бутоксикарбониламино)метил)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоату (422 мг, 0,75 ммоль из примера 3, стадия 1) в колбу добавляли 4N HCl в диоксане (3 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После удаления растворителей получали продукт в виде желтой пены.
Стадия 2: Синтез 3-((2R)-2-(2-(транс-4-((2-(диметиламино)ацетамидо)метил)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты.
[00216] К 3-((2R)-2-(2-(транс-4-(аминометил)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоте со стадии 1, в THF (5 мл), добавляли TEA (0,35 мл), а затем 2-бромацетилбромид (0,07 мл, 0,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли воду, и водную фазу экстрагировали EtOAc. Органическую фазу высушивали и концентрировали с получением неочищенного продукта, который растворяли в THF (5 мл) и добавляли диметиламин (1 мл, 2N в THF). После перемешивания при комнатной температуре в течение 8 ч летучие компоненты удаляли в вакууме и остаток использовали на следующей стадии без дополнительной очистки. ESI-MS m/z 612,1 (МН)+.
Стадия 3: Синтез (R)-3-(2-(транс-4-((2-(диметиламино)ацетамидо)метил) циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00217] Соединение получали из 3-((2R)-2-(2-(транс-4-((2-(диметиламино)ацетамидо)метил)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 446 (МН)+.
Пример 6: (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
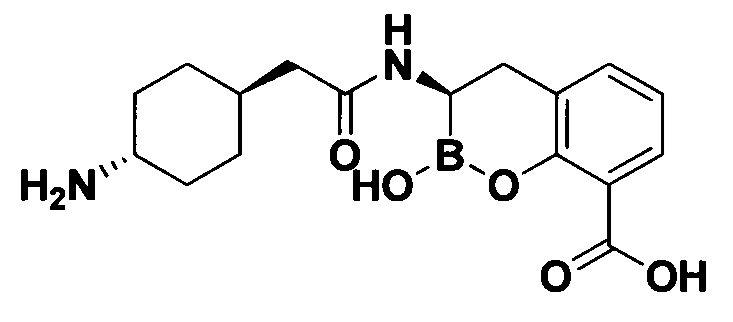
Стадия 1: Синтез трет-бутилового сложного эфира 3-((2R)-2-(2-(транс-4-(трет-бутоксикарбониламино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты.
[00218] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и 2-(транс-4-(трет-бутоксикарбониламино)циклогексил)уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (гексан/EtOAc, от 2:1 до 1:2). ESI-MS m/z 613,1 (МН)+.
Стадия 2: Синтез (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00219] Соединение получали из 3-((2R)-2-(2-(транс-4-(трет-бутоксикарбониламино)циклогексил) ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 347 (МН)+.
Пример 7: (R)-3-(2-(цис-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
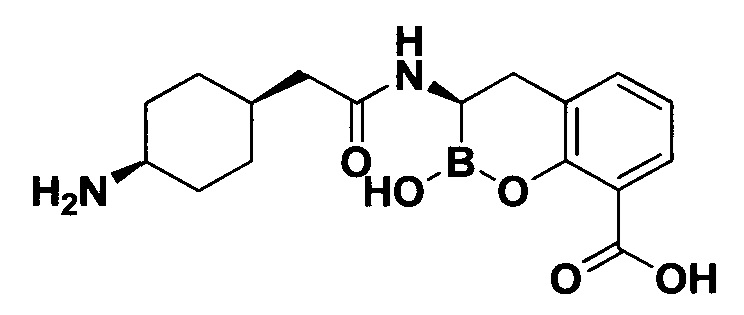
Стадия 1: Синтез трет-бутилового сложного эфира 3-((2R)-2-(2-(цис-4-(трет-бутоксикарбониламино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты.
[00220] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и 2-(цис-4-(трет-бутоксикарбониламино)циклогексил)уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (гексан/EtOAc, от 2:1 до 1:2). ESI-MS m/z 613,1 (МН)+.
Стадия 2: Синтез (R)-3-(2-(цис-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00221] Соединение получали из 3-((2R)-2-(2-(цис-4-(трет-бутоксикарбониламино)циклогексил) ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 347 (МН)+.
Пример 8: (R)-3-(2-(транс-4-((диметиламино)метил)циклогексил)-N-метилацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота

Стадия 1: [4-(бензилоксикарбониламино-метил)циклогексил]-уксусной кислоты.
[00222] Бензилхлорформиат (1,5 мл, 10,5 ммоль) и гидроксид натрия (12,5 мл, 1 н, 12,5 ммоль) добавляли к раствору транс-(4-аминометилциклогексил)уксусной кислоты (2,08 г, 10,0 ммоль) в THF (32 мл) и Н2О (16 мл). Реакционную смесь перемешивали при комнатной температуре в течение 17 ч. Реакцию гасили 1N HCl и экстрагировали с EtOAc (2×). Объединенные органические слои промывали солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали с получением 1,91 г (выход 62%) продукта, который переносили на следующую стадию без очистки. ESI-MS m/z 306 (МН)+.
Стадия 2: Трет-бутиловый сложный эфир 3-[2-{2-[4-(бензилоксикарбониламино-метил)-циклогексил]-ацетиламино}-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты.
[00223] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и [4-(бензилоксикарбониламино-метил)циклогексил]-уксусной кислоты, следуя процедуре, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (10-100% этилацетам/гексан) ESI-MS m/z 717 (МН)+.
Стадия 3: Трет-бутиловый сложный эфир 3-[2-[2-(4-аминометил-циклогексил)-ацетиламино]-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты.
[00224] Раствор трет-бутилового сложного эфира 3-[2-{2-[4-(бензилоксикарбониламино-метил)-циклогексил]-ацетиламино}-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты (1,56 г, 2,18 ммоль) в МеОН (22 мл) продували аргоном в течение 5 минут. Добавляли палладий на угле (10%, 0,153 г), колбу вакуумировали, и реакционную смесь перемешивали в атмосфере водорода в течение 6,5 часов. Реакционную смесь фильтровали через пористый фильтр с целитом (Celite-plugged filter frit), промывали МеОН и DCM, и концентрировали с получением 1,29 г неочищенного продукта, который переносили на следующую стадию без очистки. ESI-MS m/z 583 (МН)+.
Стадия 4: Трет-бутиловый сложный эфир 3-[2-{[2-(4-диметиламинометил-циклогексил)-ацетил]-метил-амино}-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты.
[00225] К раствору трет-бутилового сложного эфира 3-[2-[2-(4-аминометил-циклогексил)-ацетиламино]-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты (0,208 г, 0,357 ммоль) в DCM (3,8 мл) в атмосфере аргона добавляли DIEA (0,18 мл, 1,03 ммоль) и йодистый метил (0,068 мл, 1,09 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакцию гасили МеОН, концентрировали и переносили на следующую стадию без очистки. ESI-MS m/z 611 (МН)+.
Стадия 5: (R)-3-(2-(транс-4-((диметиламино)метил)циклогексил)-N-метилацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
[00226] Соединение получали из трет-бутилового сложного эфира 3-[2-{[2-(4-диметиламинометил-циклогексил)-ацетил]-метил-амино}-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 403 (МН)+.
Пример 9: (R)-3-(2-(транс-4-гуанидиноциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
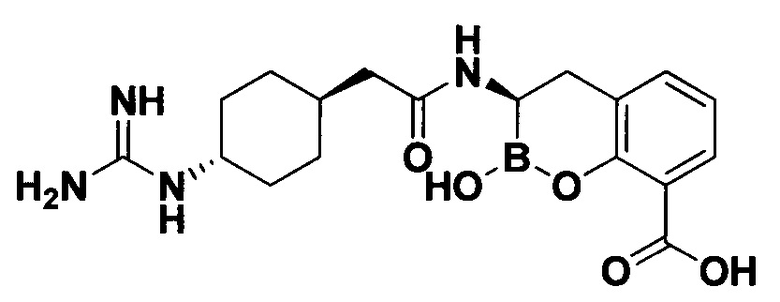
Синтез (R)-3-(2-(транс-4-гуанидиноциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00227] Соединение получали из 3-((R)-2-(2-(транс-4-аминоциклогексил)ацетамидо)-2-бороноэтил)-2-гидроксибензойной кислоты (пример 6) в соответствии с процедурой, описанной в примере 4. ESI-MS m/z 389 (МН)+.
Пример 10: (R)-3-(2-(транс-4-((2-аминоэтиламино)метил)циклогексил) ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
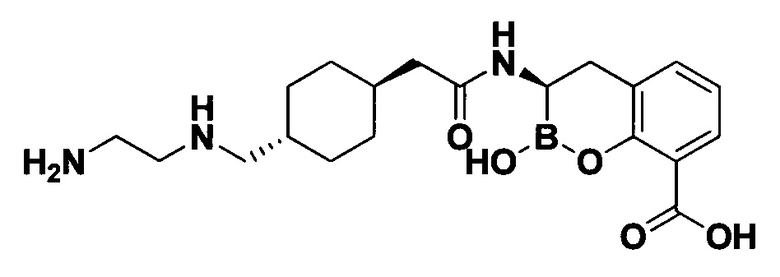
Стадия 1: Трет-бутиловый сложный эфир 3-[2-(2-{4-[(2-трет-бутоксикарбониламино-этиламино)-метил]-циклогексил}-ацетиламино)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты.
[00228] Закрываемую герметично реакционную пробирку заполняли трет-бутиловым эфиром 3-[2-[2-(4-аминометил-циклогексил)-ацетиламино]-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты (0,205 г, 0,352 ммоль), карбонатом калия (0,057 г, 0,412 ммоль), 2-(Вос-амино)этилбромидом (0,095 г, 0,424 ммоль) и DMF (3,0 мл). Пробирку герметично закрывали, и реакционную смесь нагревали при 65°С в течение 24 часов. Реакционную смесь охлаждали до комнатной температуры. Реакционную смесь разбавляли EtOAc и промывали 5%-ным водным раствором LiCl (2×) и насыщенным солевым раствором. Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением 0,120 г неочищенного продукта, который переносили на следующей стадии без очистки. ESI-MS m/z 726 (МН)+.
Стадия 2: (R)-3-(2-(транс-4-((2-аминоэтиламино)метил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
[00229] Соединение получали из трет-бутилового сложного эфира 3-[2-(2-{4-[(2-трет-бутоксикарбониламино-этиламино)-метил]-циклогексил}-ацетиламино)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 404 (МН)+.
Пример 11: (3R)-3-(2-(4-(аминометил)циклогексилиден)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
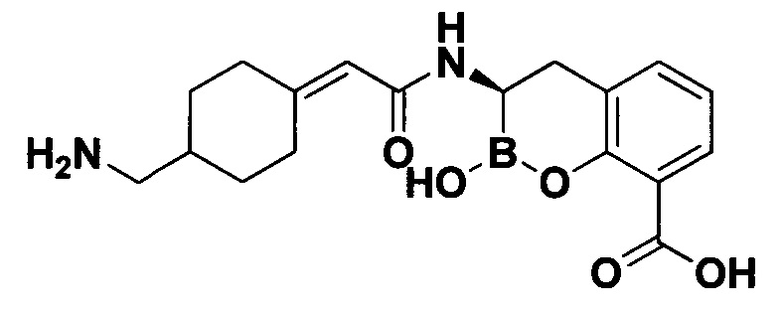
Стадия 1: Этиловый эфир [4-(трет-бутоксикарбониламино-метил)циклогексилиден]-уксусной кислоты.
[00230] Трет-бутоксид калия (0,479 г, 4,27 ммоль) добавляли к раствору триэтилфосфоноацетата (0,85 мл, 4,28 ммоль) в DMF (6,5 мл) в атмосфере аргона, и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Трет-бутиловый сложный эфир (4-оксо-циклогексилметил)-карбаминовой кислоты (0,643 г, 2,83 ммоль) в DMF (6,5 мл) добавляли по каплям в течение 8 мин. После 20 мин перемешивания наблюдали осадок и добавляли дополнительное количество DMF (6,5 мл), и реакционную смесь перемешивали в течение дополнительных 17 часов. Реакционную смесь выливали в ледяную H2O и экстрагировали Et2O (3×). Объединенные органические слои промывали Н2О и насыщенным солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали. Флэш-хроматография (0-30% EtOAc/гексан) давала 0,784 г (93%) продукта. ESI-MS m/z 298 (МН)+.
Стадия 2: [4-(трет-бутоксикарбониламино-метил)циклогексилиден]-уксусная кислота.
[00231] К раствору этилового эфира [4-(трет-бутоксикарбониламино-метил)циклогексилиден]-уксусной кислоты (0,518 г, 1,74 ммоль) в МеОН (16 мл) и THF (4 мл) добавляли гидроксид натрия (9,0 мл, 1N, 9,0 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 23 часов. Реакцию гасили Н2О и экстрагировали с помощью EtOAc (2×). Водный слой подкисляли до значения рН около 1 с помощью 1N HCl и экстрагировали этилацетатом (3×). Объединенные органические слои высушивали над Na3SO4, фильтровали и концентрировали в вакууме с получением 0,300 г (64%) продукта. ESI-MS m/z 270 (МН)+.
Стадия 3: Трет-бутиловый сложный эфир 3-[2-{2-[4-(трет-бутоксикарбониламино-метил)-циклогексилиден]-ацетиламино}-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты.
[00232] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-[2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-2-(триметилсиланил-амино)-этил]-бензойной кислоты и [4-(трет-бутоксикарбониламино-метил)циклогексилиден]-уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-100% EtOAc/гексан). ESI-MS m/z 681 (МН)+.
Стадия 4: (3R)-3-(2-(4-(аминометил)циклогексилиден)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
[00233] Соединение получали из трет-бутилового сложного эфира 3-[2-{2-[4-(трет-бутоксикарбониламино-метил)-циклогексилиден]-ацетиламино}-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 359 (МН)+.
Пример 12: (R)-3-(2-(4-(аминометил)-1-(нитрометил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
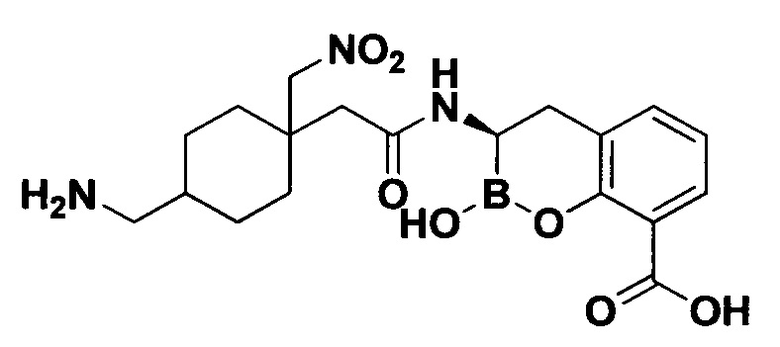
Стадия 1: Этиловый эфир [4-(трет-бутоксикарбониламино-метил)-1-нитрометил-циклогексил]-уксусной кислоты.
[00234] Закрываемую герметично реакционную пробирку заполняли 2,8,9-триизобутил-2,5,8,9-тетраазафенален-1-фосфабицикло[3.3.3]ундеканом (0,16 мл, 0,450 ммоль) и THF (1,0 мл) в атмосфере аргона. Добавляли нитрометан (0,23 мл, 4,24 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 5 мин, затем охлаждали до 0°С в течение 15 мин. Медленно добавляли этиловый эфир [4-(трет-бутоксикарбониламино-метил)циклогексилиден]-уксусной кислоты (0,261 г, 0,878 ммоль) в THF (1,5 мл), и реакционную смесь перемешивали при комнатной температуре в течение 15 мин. Пробирку герметично закрывали и реакционную смесь нагревали при 70°С в течение 17 часов. Реакционную смесь охлаждали до комнатной температуры, гасили 0,5 М HCl, и экстрагировали этилацетатом (2×). Объединенные органические слои промывали насыщенным раствором NaHCO3 и насыщенным солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали с получением 0,304 г неочищенного продукта, который переносили на следующую стадию без очистки. ESI-MS m/z 359 (МН)+.
Стадия 2: [4-(трет-бутоксикарбониламино-метил)-1-нитрометил-циклогексил]-уксусная кислота.
[00235] К раствору этилового эфира [4-(трет-бутоксикарбониламино-метил)-1-нитрометил-циклогексил]-уксусной кислоты (0,304 г, 0,848 ммоль) в МеОН (5,0 мл) и THF (1,5 мл) добавляли гидроксид натрия (4,2 мл, 1 н, 4,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь подкисляли до значения рН около 2 с помощью 1N HCl и экстрагировали этилацетатом (3×). Объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали с получением 0,308 г неочищенного продукта, который переносили на следующую стадию без очистки. ESI-MS m/z 331 (МН)+.
Стадия 3: Трет-бутиловый сложный эфир 3-[2-{2-[4-(трет-бутоксикарбониламино-метил)-1-нитрометил-циклогексил]-ацетиламино}-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты.
[00236] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-[2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-2-(триметилсиланил-амино)-этил]-бензойной кислоты и [4-(трет-бутоксикарбониламино-метил)-1-нитрометил-циклогексил]-уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-100% EtOAc/гексан). ESI-MS m/z 742 (MH)+.
Стадия 4: (R)-3-(2-(4-(аминометил)-1-(нитрометил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
[00237] Соединение получали из трет-бутилового сложного эфира 3-[2-{2-[4-(трет-бутоксикарбониламино-метил)-1-нитрометил-циклогексил]-ацетиламино}-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 420 (МН)+.
Пример 13: (R)-3-(2-(транс-4-((S)-2,3-диаминопропанамидо)циклогексил) ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
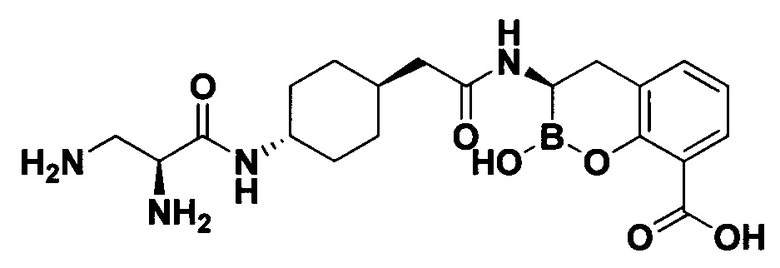
Синтез 3-((R)-2-бороно-2-(2-транс-4-((S)-2,3-диаминопропанамидо)циклогексил)ацетамидо)этил)-2-гидроксибензойной кислоты.
[00238] К (S)-2-(бензилоксикарбониламино)-3-(трет-бутоксикарбониламино) пропионовой кислоте (85 мг, 0,25 ммоль) в DCM/DMF (4 мл, 1/1) добавляли NMM (1,2 экв), а затем HATU (95 мг, 0,26 моль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. В отдельном флаконе трет-бутиловый сложный эфир 3-((2R)-2-(2-(транс-4-(трет-бутоксикарбониламино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты (133 мг, 0.2 ммоль, пример 6) обрабатывали 4N HCl в диоксане (2 мл), и полученный раствор перемешивали при комнатной температуре в течение 2 часов. Растворитель затем удаляли при пониженном давлении. К данному остатку добавляли активный сложный эфир, полученный выше, и реакционную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду, и водную фазу экстрагировали EtOAc. Органическую фазу промывали 1N HCl, насыщ. NaHCO3, насыщенным солевым раствором, высушивали и концентрировали в вакууме с получением продукта (0,100 г) в виде коричневого масла без дальнейшей очистки. ESI-MS m/z 833,1 (МН)+. Затем остаток растворяли в МеОН и добавляли кат. Pd на угле (20 мг) и перемешивали в атмосфере водорода в течение ночи. После фильтрования и удаления растворителя остаток обрабатывали BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1 с получением указанного в заголовке соединения. ESI-MS m/z 433 (МН)+.
Пример 14: (R)-3-(2-(транс-4-(диметиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
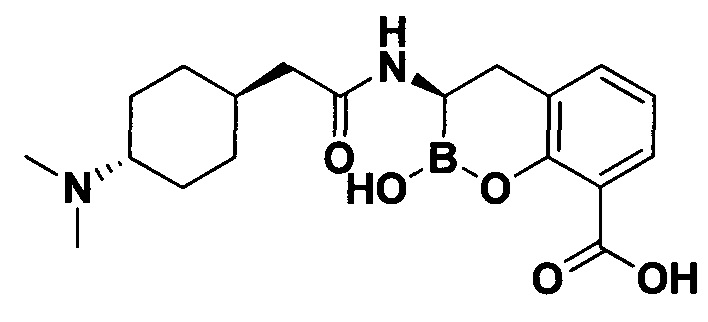
Синтез (R)-3-(2-(транс-4-(диметиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00239] К (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоте (10,0 мг из примера 6) в МеОН (5 мл) добавляли формальдегид (1,0 мл, 37% раствор), а затем 10% Pd/C (20 мг). Реакционную смесь гидрировали в создаваемой с помощью баллона атмосфере Н2 в течение 3 ч. Реакционную смесь фильтровали и растворитель удаляли в вакууме. Конечный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 433 (МН)+.
Пример 15: (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
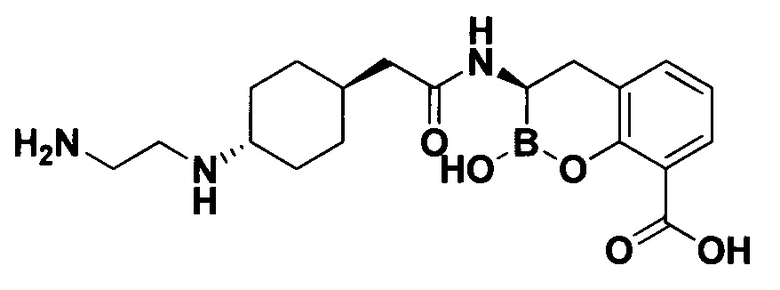
Стадия 1: Синтез (R)-3-(2-(транс-4-(2-(трет-бутоксикарбониламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00240] К (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоте (пример 6, 15 мг) в МеОН (2 мл) добавляли трет-бутил-2-охоэтилкарбамат (20 мг). Добавляли Pd/C (10% по весу, 10 мг) и реакционную смесь перемешивали в создаваемой с помощью баллона атмосфере Н2 в течение ночи. Реакционную смесь фильтровали и растворитель удаляли при пониженном давлении, и остаток использовали на следующей стадии без дополнительной очистки. ESI-MS m/z 490,1 (МН)+.
Стадия 2: Синтез (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00241] К (R)-3-(2-(транс-4-(2-(трет-бутоксикарбониламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоте (20 мг) в колбе добавляли 1 мл 4N HCl в диоксане. Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли в вакууме, и остаток очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 390 (МН)+.
Пример 16: (R)-2-гидрокси-3-(2-(транс-4-(пиперазин-1-ил)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
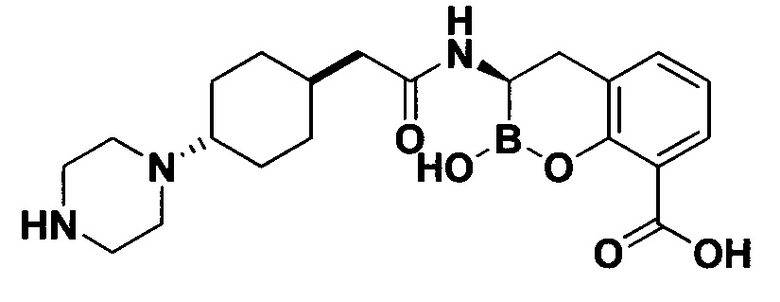
Стадия 1: Синтез 2-(4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)циклогексил)уксусной кислоты.
[00242] К 2-(4-оксоциклогексил)уксусной кислоте (0,576 г, 3,7 ммоль) и трет-бутил-пиперазин-1-карбоксилату (0,700 г, 3,7 ммоль) в МеОН добавляли Pd на угле (80 мг), и полученную реакционную смесь перемешивали в атмосфере водорода в течение ночи. Катализатор удаляли фильтрацией, и растворитель удаляли при пониженном давлении с получением кислоты в виде белого твердого вещества (1,2 г, 99%).
Стадия 2: Синтез трет-бутилового сложного эфира 4-(4-{3-трет-бутоксикарбонил-2-метокси-фенил)-1-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этилкарбамоил]-метил}-циклогексил)-пиперазине-карбоновой кислоты.
[00243] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и 2-(4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)циклогексил)-уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (гексан/EtOAc, от 2:1 до 1:2). ESI-MS m/z 638,1 (МН)+.
Стадия 3: Синтез (R)-2-гидрокси-3-(2-(транс-4-(пиперазин-1-ил)циклогексил) ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00244] Соединение получали из трет-бутилового сложного эфира 4-(4-{3-трет-бутоксикарбонил-2-метокси-фенил)-1-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этилкарбамоил]-метил}-циклогексил)-пиперазине-карбоновой кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ с получением транс-изомера в качестве основного продукта, который высушивали с помощью лиофилизации. ESI-MS m/z 416 (МН)+.
Пример 17: (R)-2-гидрокси-3-(2-(цис-4-(пиперазин-1-ил)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
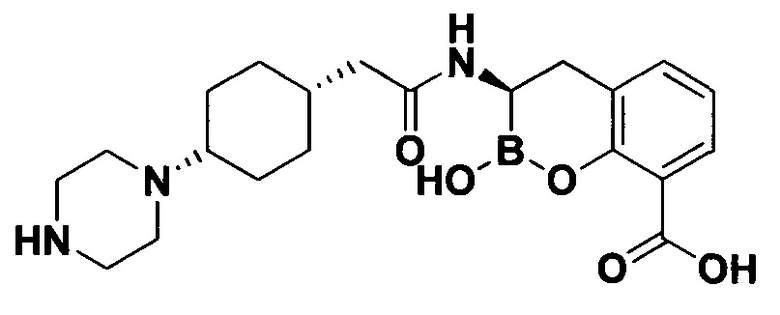
Синтез (R)-2-гидрокси-3-(2-(цис-4-(пиперазин-1-ил)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00245] Соединение получали из трет-бутилового сложного эфира 4-(4-{3-трет-бутоксикарбонил-2-метокси-фенил)-1-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этилкарбамоил]-метил}-циклогексил)-пиперазине-карбоновой кислоты (из примера 16, стадия 2) и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ с получением цис-изомера, как побочного продукта, который высушивали с помощью лиофилизации. ESI-MS m/z 416 (МН)+.
Пример 18: (3R)-3-[[2-[(1S,3S,4S)-3,4-бис(аминометил)циклогексил]ацетил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновая кислота и (3R)-3-[[2-[(1R,3R,4R)-3,4-бис(аминометил)циклогексил]ацетил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновая кислота
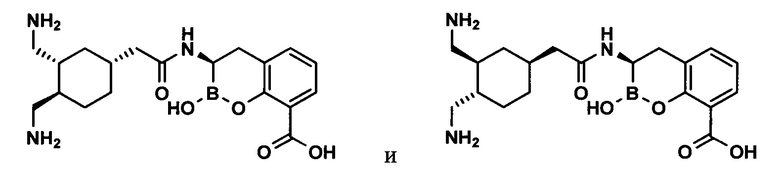 и
и
Стадия 1: Синтез (Е)-N,N,N',N'-тетрабензилбут-2-ендиамида.
[00246] К охлажденному (-5°С) раствору N,N-дибензиламина (19,3 мл, 100 ммоль) в DCM (200 мл) добавляли TEA (13,8 мл, 100 ммоль). К данному раствору добавляли по каплям в течение 5 мин фумароилхлорид (4,2 мл, 40 ммоль). По окончании добавления охлаждающую баню удаляли, и перемешивание продолжали в течение 1 часа. Данный раствор разбавляли DCM, промывали HCl (ок. 100 мл 1М водн.), затем водой (2×), высушивали над MgSO4 и концентрировали. Твердый остаток кристаллизовали из горячего (109°С) толуола с получением указанного в заголовке соединения (17,9 г) в виде белого твердого вещества. ESI-MC (m/z) 475 (МН)+.
Стадия 2: Синтез (транс)-N,N,N',N'-тетрабензил-4-оксо-циклогексан-1,2-дикарбоксамида.
[00247] К суспензии (Е)-N,N,N',N'-тетрабензилбут-2-ендиамида (10,6 г, 22 ммоль) в орто-ксилоле (106 мл) добавляли гидрохинон (180 мг, 1,63 ммоль), а затем 2-триметилсилилокси-1,3-бутадиен (9.48 г, 66 ммоль). Полученную смесь продували аргоном, затем герметизировали. Затем смесь нагревали до 135°С и перемешивали при данной температуре в течение 87 часов. Полученный раствор охлаждали, затем концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле (240 г двуокиси кремния, элюция 10%-ным этилацетатом в гексане) с получением 12.81 г продукта в виде масла. Данный неочищенный продукт растворяли в THF (10 мл) и МеОН (40 мл). К данному раствору добавляли карбонат калия (2.76 г, 20 ммоль). Полученную смесь перемешивали в течение 10 мин, затем разбавляли Et2O, промывали водой и насыщенным солевым раствором, высушивали над MgSO4 и концентрировали. Остаток очищали хроматографией на силикагеле (160 г двуокиси кремния, элюция 20% этилацетат/20% дихлорметан в гексане) с получением указанного в заголовке соединения (10.11 г) в виде масла. ESI-MC (m/z) 545 (МН)+.
Стадия 3: Синтез этилового эфира 2-[(транс)-3,4-бис(дибензилкарбамоил)циклогексилиден]ацетата.
[00248] К охлажденной (-5°С) суспензии гидрида натрия (846 мг, 60% дисперсии в минеральном масле, 21,2 ммоль) в THF (60 мл) добавляли по каплям триэтилфосфоноацетат (4,2 мл, 21,2 ммоль). По окончании добавления охлаждающую баню удаляли и перемешивание продолжали в течение 20 мин. Полученный раствор охлаждали до -5°С. К данному раствору добавляли раствор (транс)-N,N,N',N'-тетрабензил-4-оксо-циклогексан-1,2-дикарбоксамида (10.11 г, 18,4 ммоль) в THF (10 мл). По окончании добавления охлаждающую баню удаляли, и перемешивание продолжали в течение 30 мин. К данному раствору добавляли HCl (30 мл, 1М водный). Полученную смесь разбавляли Et2O, промывали насыщенным солевым раствором, высушивали над MgSO4 и концентрировали. Остаток очищали хроматографией на силикагеле (160 г двуокиси кремния, элюция 20% этилацетат/10% дихлорметана в гексане) с получением указанного в заголовке соединения (9.89 г) в виде масла (смесь 4:1 и Z-изомеры). ESI-MC (m/z) 615 (МН)+.
Стадия 4: Синтез (рацемического)-метил-2-[(1R,3R,4R)-3,4-бис(дибензилкарбамоил)циклогексил]ацетата.
[00249] К раствору этил-2-[(транс)-3,4-бис(дибензилкарбамоил)циклогексилиден]ацетата (9.27 г, 15 ммоль) в сухом метаноле (75 мл) добавляли ленту магния (1,08 г, 45 ммоль). Суспензию перемешивали в течение 5 часов. К данному гомогенному раствору добавляли дополнительную порцию ленты магния (200 мг, 8,2 ммоль). Данную смесь перемешивали в течение 17 часов. Полученный раствор экстрагировали EtOAc, промывали насыщенным солевым раствором, высушивали над MgSO4 и концентрировали. Остаток очищали хроматографией на силикагеле (элюент: 20% этилацетата/10% дихлорметана в гексане) с получением указанного в заголовке соединения (8.27 г) в виде пены. ESI-MC (m/z) 617 (МН)+.
Стадия 5: Синтез (рацемического)-2-[(1R,3R,4R)-3,4-бис[(дибензиламино)метил]циклогексил]этанола.
[00250] К охлажденному (-10°С) раствору (рацемического)-метил-2-[(1R,3R,4R)-3,4-бис(дибензилкарбамоил)циклогексил]ацетата (8.18 г, 13,5 ммоль) в THF (40 мл) добавляли по каплям литийалюминийгидрид (50 мл, 1М в THF). По окончании добавления охлаждающую баню удаляли и перемешивание продолжали в течение 15 мин. Полученный раствор нагревали до 55°С и перемешивали при данной температуре в течение 2,5 часов. Данный раствор охлаждали до 0°С. К полученному раствору добавляли по каплям воду (1,9 мл), затем NaOH (1,9 мл, 5М водный) затем воду (0,95 мл). Полученную смесь разбавляли Et2O (100 мл), фильтровали через целит, и фильтрат концентрировали в вакууме с получением указанного в заголовке соединения в виде масла (8,1 г). ESI-MC (m/z) 547 (МН)+.
Стадия 6: Синтез (рацемического)-трет-бутил-N-[[(1S,2S,4S)-2-[(трет-бутоксикарбониламино)метил]-4-(2-гидроксиэтил)циклогексил]метил]карбамата.
[00251] К смеси (рацемического)-2-[(1R,3R,4R)-3,4-бис[(дибензиламино)метил]циклогексил]этанола (8,1 г, 13 ммоль) и гидроксида палладия на углероде (1,0 г, 20%-ный палладий по весу) добавляли МеОН (70 мл). Полученную смесь продували газообразным водородом (1 атм) и перемешивали в течение 17 часов. Систему затем продували аргоном, разбавляли DCM и фильтровали через целит. Фильтрат концентрировали под вакуумом. Остаток растворяли в DCM (30 мл) и THF (20 мл). К данному раствору добавляли DIEA (4,6 мл, 26 ммоль), а затем ди-трет-бутилдикарбонат (5.66 г, 26 ммоль). Полученный раствор перемешивали в течение 2 ч, затем разбавляли с EtOAc, промывали водой и насыщенным солевым раствором, высушивали над MgSO4 и концентрировали. Остаток очищали хроматографией на силикагеле (160 г двуокиси кремния, элюция 80%-ным этилацетатом в гексане) с получением указанного в заголовке соединения (3,1 г) в виде вязкого масла. 1Н ЯМР (CDCl3) δ 0,7-1,0 (m, 1Н), 1.05-1.35 (m, 4H), 1.4-1.75 (m, 4H), 1,46 (s, 18H), 1.75-1.96 (m, 2H), 2,9 -3.05 (m, 2H), 3.05-3.75 (bm, 5H), 4,6-4,8 (bd, 1Н), 4,95 (bs, 1Н).
Стадия 7: Синтез (рацемической)-2-[(1S,3S,4S)-3,4-бис[(трет-бутоксикарбониламино)метил]циклогексил]-уксусной кислоты.
[00252] К раствору (рацемического)-трет-бутил-N-[[(1S,2S,4S)-2-[(трет-бутоксикарбониламино)метил]-4-(2-гидроксиэтил)циклогексил]метил]карбамата (772 мг, 2 ммоль) в ацетонитриле (3 мл), четыреххлористого углерода (3 мл) и воды (4,5 мл) добавляли периодат натрия (1,28 г, 6 ммоль) с последующим добавлением RuCl3⋅H2O (21 мг, 0,1 ммоль). Полученную смесь перемешивали в течение 2 ч, затем разбавляли с EtOAc, промывали насыщенным солевым раствором, высушивали над MgSO4 и концентрировали. Остаток переносили в насыщенный раствор карбоната натрия и экстрагировали этилацетатом. Водную фазу подкисляли HCl (2М водная) и экстрагировали EtOAc. Органический экстракт промывали насыщенным солевым раствором, высушивали над MgSO4 и концентрировали. Данный остаток очищали хроматографией на силикагеле (элюент: этилацетат) с получением указанного в заголовке соединения (394 мг) в виде вязкого масла. 1H ЯМР (DMSO-d6) δ 0,7-1,0 (m, 1H), 1,0-1,9 (м, 9Н), 1,39 (s, 18H), 2,0-2,3 (m, 2H), 2.6-3.18 (m, 4H), 6,6-6,8 (bm, 2H).
Стадия 8: Синтез трет-бутил-3-((2R)-2-(-[(1S,3S,4S)-3,4-бис[(трет-бутоксикарбониламино)метил]циклогексил]ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и трет-бутил-3-((2R)-2-(-[(1R,3R,4R)-3,4-бис[(трет-бутоксикарбониламино)метил]циклогексил]ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата.
[00253] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и (рацемической)-2-[(1S,3S,4S)-3,4-бис[(трет-бутоксикарбониламино)метил]циклогексил]-уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. За исключением того, что неочищенный продукт очищали хроматографией на силикагеле (элюент: 30%-ный этилацетат в гексане, 60%-ный этилацетат в гексане) с получением указанного в заголовке соединения E8I-MC (m/z) 834 (MNa)+.
Стадия 9: Синтез (3R)-3-[[2-[(1S,3S,4S)-3,4-бис(аминометил)циклогексил]ацетил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновой кислоты и (3R)-3-[[2-[(1R,3R,4R)-3,4-бис(аминометил)циклогексил]ацетил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновой кислоты.
[00254] Соединение получали из трет-бутил-3-((2R)-2-(-[(1S,3S,4S)-3,4-бис[(трет-бутоксикарбониламино)метил]циклогексил]ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и трет-бутил-3-((2R)-2-(-[(1R,3R,4R)-3,4-бис[(трет-бутоксикарбониламино)метил]циклогексил]ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата в соответствии с процедурой, описанной на стадии 2 примера 1. ESI-MS (m/z) 390 (МН)+.
Пример 19: (3R)-3-[[(3S,4S)-3,4-диаминоциклопентанкарбонил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновая кислота и (3R)-3-[[(3R,4R)-3,4-диаминоциклопентанкарбонил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновая кислота
 и
и
Стадия 1: Синтез (рацемического)-этил(3S,4S)-3,4-диазидоциклопентанкарбоксилата.
[00255] К охлажденной (-20°С) суспензии триацетата марганца (8.02 г, 30 ммоль) в ацетонитриле (120 мл) добавляли азид натрия (3,4 г, 50 ммоль). К данной смеси по каплям добавляли раствор этил-циклопент-3-ен-1-карбоксилата (1,4 г, 10 ммоль) в TFA (14 мл) в течение приблизительно 10 мин. Полученную смесь перемешивали при температуре от -25 до -19°С в течение 3 ч, затем давали нагреться до комнатной температуры и перемешивали в течение 17 часов. К данной смеси добавляли тиосульфат натрия (30 мл, 10% водный). Данную смесь перемешивали в течение 5 мин, затем экстрагировали гексаном. Гексановый экстракт промывали насыщенным раствором бикарбоната натрия (3 раза), затем насыщенным солевым раствором, высушивали над MgSO4 и концентрировали. Данный остаток очищали хроматографией на силикагеле (90 г двуокиси кремния, элюция 10%-ным этилацетатом в гексане) с получением указанного в заголовке соединения (1,278 г) в виде масла. 1Н ЯМР (DMSO-d6) δ 1,15 (t, J=7 Гц, 3Н), 1.68-1.91 (m, 2H), 2.13-2.35 (m, 2H), 3,01 (m, 1H), 3,97 (m, 2H), 4,05 (q, J=7 Гц, 2Н).
Стадия 2: Синтез (рацемического)этил(3S,4S)-3,4-бис(трет-бутоксикарбониламино) циклопентанкарбоксилата.
[00256] К раствору (рацемического)-этил(3S,4S)-3,4-диазидоциклопентанкарбоксилата (3,8 г, 16,9 ммоль) в THF (85 мл) добавляли трифенилфосфин (9,7 г, 37.18 ммоль). Полученный раствор перемешивали в течение 17 часов. К данному раствору добавляли воду (4,2 мл). Полученный раствор перемешивали в течение 6 ч. К данному раствору добавляли DIEA (8,9 мл, 51 ммоль), а затем ди-трет-бутилдикарбонат (11,05 г, 51 ммоль). Данную смесь перемешивали в течение 2 ч, затем концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле (230 г диоксида кремния, элюируя смесью 20%-ного этилацетата в гексане) с получением указанного в заголовке соединения (3,79 г) в виде белого твердого вещества. 1Н ЯМР (DMSO-d6) δ 1,15 (t, J=7 Гц, 3Н), 1,36 (s, 18Н), 1.5-1.66 (m, 2H), 1.91-2.11 (m, 2H), 2,81 (m, 1H), 3,61 (bm, 2H), 4,01 (q, J=7 Гц, 2), 6,8 (bs, 2H).
Стадия 3: Синтез (рацемического)-(3S,4S)-3,4-бис(трет-бутоксикарбониламино) циклопентанкарбоксилата.
[00257] К раствору (рацемического)этил(3S,4S)-3,4-бис(трет-бутоксикарбониламино)циклопентанкарбоксилата (790 мг, 2,12 ммоль) в МеОН (3 мл), THF (6 мл) добавляли гидроксид натрия (3 мл, 1 М водного раствора). Полученный раствор перемешивали в течение 20 мин, затем подкисляли HCl (1М водн.). Данную смесь разбавляли EtOAc, промывали насыщенным солевым раствором, высушивали над MgSO4 и концентрировали с получением указанного в заголовке соединения (671 мг) в виде твердого вещества. 1Н-ЯМР (DMSO-d6) δ 1,38 (s, 18H), 1.46-1.62 (m, 2H), 1.87-2.11 (m, 2H), 2,77 (m, 1H), 3,6 (bm, 2H), 6,75 (bs, 2H), 12,15 (bs, 1H).
Стадия 4: Синтез трет-бутил-3-((2R)-2-((3S,4S)-3,4-бис(трет-бутоксикарбониламино)циклопентанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и трет-бутил-3-((2R)-2-(3R,4R)-3,4-бис(трет-бутоксикарбониламино)циклопентанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата.
[00258] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и (рацемического)-(3S,4S)-3,4-бис(трет-бутоксикарбониламино)циклопентанкарбоксилата в соответствии с процедурой, описанной на стадии 1 примера 1. За исключением того, что неочищенный продукт очищали хроматографией на силикагеле (элюируя 30%-ным этилацетатом в гексане, затем 60%-ным этилацетатом в гексане) с получением указанного в заголовке соединения ESI-MC (m/z) 778 (MNa)+.
Стадия 5: Синтез (3R)-3-[[(3S,4S)-3,4-диаминоциклопентанкарбонил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновой кислоты и (3R)-3-[[(3R,4R)-3,4-диаминоциклопентанкарбонил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновой кислоты.
[00259] Соединение получали из трет-бутил-3-((2R)-2-((3S,4S)-3,4-бис(трет-бутоксикарбониламино)циклопентанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и трет-бутил-3-((2R)-2-(3R,4R)-3,4-бис(трет-бутоксикарбониламино)циклопентанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата в соответствии с процедурой, описанной на стадии 2 примера 1. ESI-MS (m/z) 334 (МН)+.
Пример 20: (3R)-3-[[(1S,3S,4S)-3,4-диаминоциклогексанкарбонил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновая кислота и (3R)-3-[[(1R,3R,4R)-3,4-диаминоциклогексанкарбонил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновая кислота
 и
и
Стадия 1: Синтез (рацемического)метил(1S,3R,4R)-3,4-диазидоциклогексанкарбоксилата.
[00260] Соединение получали из метилциклогексен-3-ен-1-карбоксилата с использованием по существу той же процедуры, описанной на стадии 1 примера 19. Данный материал использовали без дополнительной очистки
Стадия 2: Синтез (рацемического)-метил(1S,3R,4R)-3,4-бис(трет-бутоксикарбониламино)циклогексанкарбоксилата.
[00261] Соединение получали из (рацемического)-метил(3R,4R)-3,4-диазидоциклогексанкарбоксилата с использованием по существу той же процедуры, описанной на стадии 2 примера 19. 1Н ЯМР (CDCl3) δ 1.3-1.45 (m, 3H), 1,40 (s, 18H), 1,91 (m, 1H), 2,10 (m, 1H), 2,41 (m, 1H)m, 2,72 (m, 1H), 3,28 (m, 1H), 3,52 (m, 1H), 3,68 (s, 3H), 4,79 (bs, 1H), 4,93 (bs, 1H).
Стадия 3: Синтез (рацемической)-(1S,3R,4R)-3,4-бис(трет-бутоксикарбониламино)циклогексанкарбоновой кислоты.
[00262] Соединение получали из (рацемического)-метил(1S,3R,4R)-3,4-бис(трет-бутоксикарбониламино)циклогексанкарбоксилата с использованием по существу той же процедуры, описанной на стадии 3 примера 19. 1H ЯМР (CDCl3) δ 1.28-1,62 (m, 3H), 1,42 (s, 18H), 1,86 (m, 1H), 2,20 (m, 1H), 2,47 (m, 1H), 2,75 (m, 1H), 3,32 (m, 1H), 3,53 (m, 1H), 4.91 (bd, 1H), 5,55 (bd, 1H).
Стадия 4: Синтез трет-бутил-3-((2R)-2-((1S,3R,4R)-3,4-бис(трет-бутоксикарбониламино)циклогексанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и трет-бутил-3-((2R)-2-((1R,3S,4S)-3,4-бис(трет-бутоксикарбониламино)циклогексанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата.
[00263] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и (рацемической)-(1S,3R,4R)-3,4-бис(трет-бутоксикарбониламино)циклогексанкарбоновой кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. За исключением того, что неочищенный продукт очищали хроматографией на силикагеле (элюирование 30%-ным этилацетатом в гексане, затем 60%-ным этилацетатом в гексане) с получением указанного в заголовке соединения ESI-MC (m/z) 792 (MNa)+.
Стадия 5: Синтез (3R)-3-[[(1S,3S,4S)-3,4-диаминоциклогексанкарбонил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновой кислоты и (3R)-3-[[(1R,3R,4R)-3,4-диаминоциклогексанкарбонил]амино]-2-гидрокси-3,4-дигидро-1,2-бензохаборинин-8-карбоновой кислоты.
[00264] Соединение получали из трет-бутил-3-((2R)-2-((1S,3R,4R)-3,4-бис(трет-бутоксикарбониламино)циклогексанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и трет-бутил-3-((2R)-2-((1R,3S,4S)-3,4-бис(трет-бутоксикарбониламино)циклогексанкарбоксамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата в соответствии с процедурой, описанной на стадии 2 примера 1. ESI-MS (m/z) 348 (МН)+.
Пример 21: (R)-3-(3-(транс-4-аминоциклогексил)пропанамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
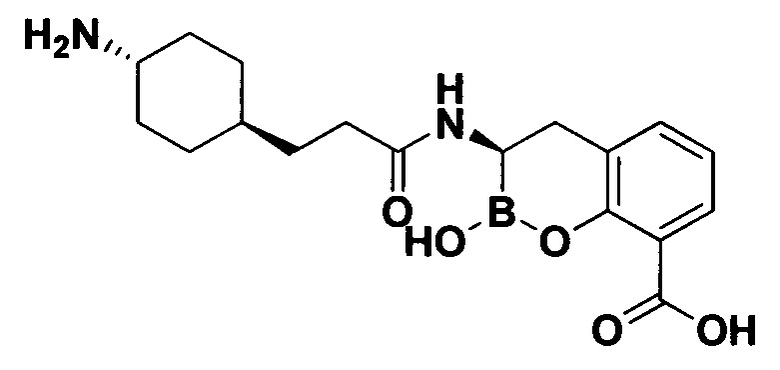
Стадия 1: Синтез трет-бутилового сложного эфира 3-((2R)-2-(3-(транс-4-(трет-бутоксикарбониламино)циклогексил)пропанамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты.
[00265] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и 3-(транс-4-(трет-бутоксикарбониламино)циклогексил)пропановой кислоты в соответствии с процедурой, описанной на стадии 1 в примере 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (гексан/EtOAc, от 2:1 до 1:2). ESI-MS m/z 683,1 (МН)+.
Стадия 2: Синтез (R)-3-(3-(транс-4-аминоциклогексил)пропанамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00266] Соединение получали из трет-бутилового сложного эфира 3-((2R)-2-(3-(транс-4-(трет-бутоксикарбониламино)циклогексил)пропанамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 361 (МН)+.
Пример 22: (R)-3-(3-(транс-4-гуанидиноциклогексил)пропанамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
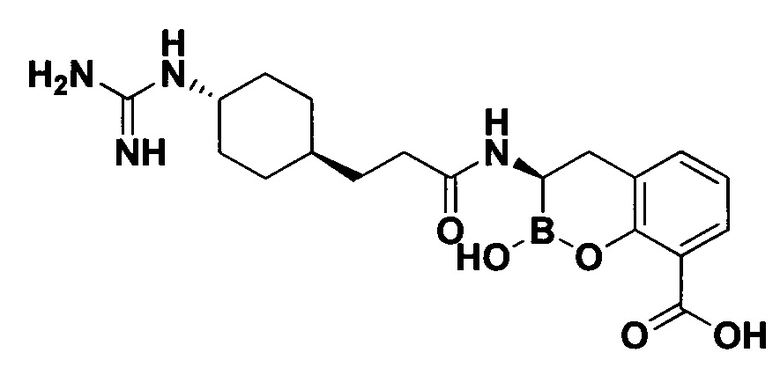
Синтез (R)-3-(3-(транс-4-гуанидиноциклогексил)пропанамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00267] Соединение получали из (R)-3-(3-(транс-4-аминоциклогексил)пропанамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 21), следуя способу, описанному в примере 4. ESI-MC m/z 403 (МН)+.
Пример 23: (R)-3-(2-(транс-4-((2-(диметиламино)этил)(метил)амино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
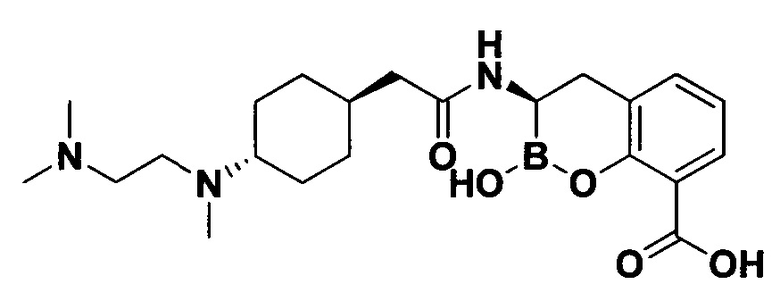
Синтез (R)-3-(2-(транс-4-((2-(диметиламино)этил)(метил)амино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00268] Соединение получали из (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 15) в соответствии с процедурой, описанной в примере 14. ESI-MS m/z 432 (МН)+.
Пример 24: (R)-3-(2-(транс-4-(4-карбамимидоилпиперазин-1-ил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
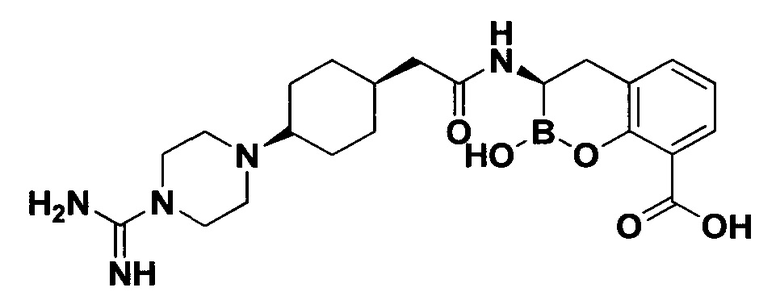
Синтез (R)-3-(2-(транс-4-(4-карбамимидоилпиперазин-1-ил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00269] Соединение получали из (R)-2-гидрокси-3-(2-(цис-4-(пиперазин-1-ил)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 17) в соответствии с процедурой, описанной в примере 4. ESI-MC m/z 458 (МН)+.
Пример 25: (R)-3-(2-(транс-4-(2-гуанидиноэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
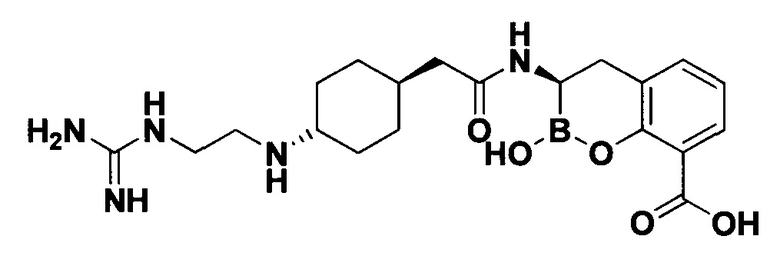
Синтез (R)-3-(2-(транс-4-(2-гуанидиноэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00270] Соединение получали из (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 15) в соответствии с процедурой, описанной в примере 4. ESI-MC m/z 432 (МН)+.
Пример 26: (R)-2-гидрокси-3-(2-(транс-4-(2-гидроксиэтиламино)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
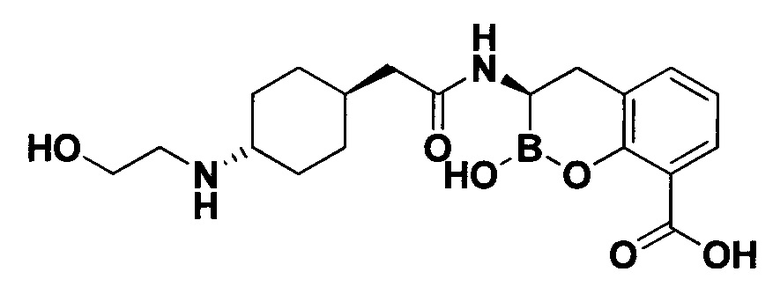
Стадия 1: Синтез (R)-3-(2-(транс-4-(2-(трет-бутилдиметилсилилокси)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00271] Соединение получали из (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 15) и 2-(трет-бутилдиметилсилилокси)ацетальдегида в соответствии с процедурой, описанной на стадии 1 примера 15. ESI-MS m/z 505 (МН)+.
Стадия 2: Синтез (R)-2-гидрокси-3-(2-(транс-4-(2-гидроксиэтиламино)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00272] К (R)-3-(2-(транс-4-(2-(трет-бутилдиметилсилилокси)этиламино)циклогексил) ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоте со стадии 1 (100 мг) в колбе добавляли 4 мл 4N HCl в диоксане. Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли в вакууме и остаток очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 391 (МН)+.
Пример 27: (R)-2-гидрокси-3-(2-(транс-4-(пиридин-3-илметиламино)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
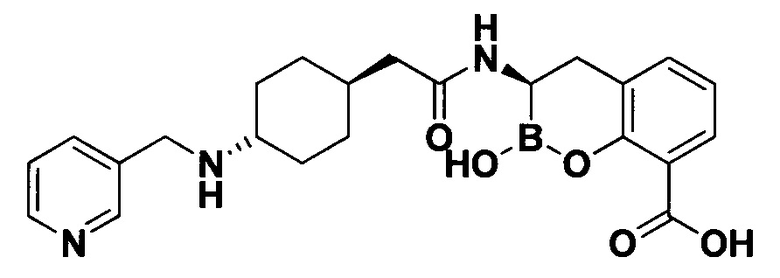
Синтез (R)-3-(2-(транс-4-(2-гуанидиноэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00273] К 40 мг (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 6) в МеОН (5 мл) добавляли TEA (0,03 мл), а затем никотинальдегид (20 мг), АсОН (0,01 мл) и триацетоксиборгидрид натрия (25 мг). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли в вакууме и остаток очищали с помощью ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение. ESI-MS m/z 438 (МН)+.
Пример 28: (R)-3-(2-(транс-4-(карбоксиметиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
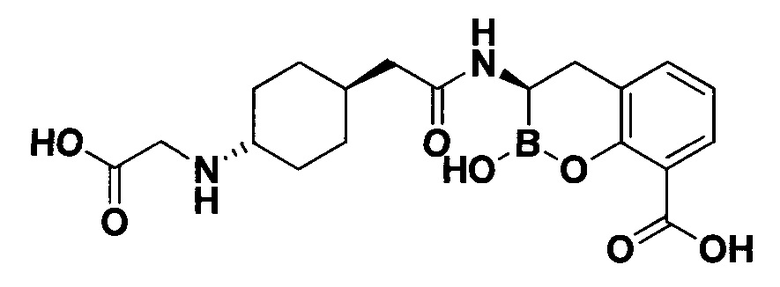
[00274] К (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоте (пример 6) в МеОН добавляли TEA (2,5 экв), а затем этиловый эфир бромуксусной кислоты (1,2 экв). Реакционную смесь перемешивали при комнатной температуре в течение ночи. К данной реакционной смеси затем добавляли 1N NaOH и перемешивали в течение 6 ч. После концентрирования в вакууме добавляли 1N HCl для доведения занчения рН до 1. Реакционную смесь очищали с помощью ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение. ESI-MS m/z 405 (МН)+.
Пример 29: (R)-3-(2-(транс-4-(4,5-дигидро-1Н-имидазол-1-ил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
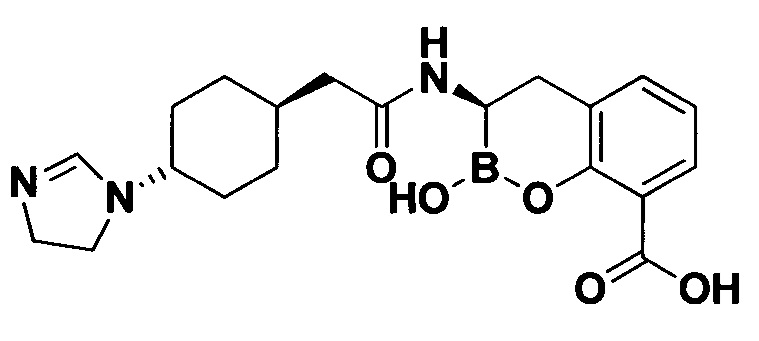
[00275] К (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоте (пример 15) в МеОН добавляли DIEA (2,5 экв), а затем изопропилформимидат-гидрохлорид (1,2 экв). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем смесь концентрировали в вакууме и неочищенный продукт очищали с помощью ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение. ESI-MS m/z 400 (МН)+.
Пример 30: (R)-3-(2-(транс-4-формимидамидоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
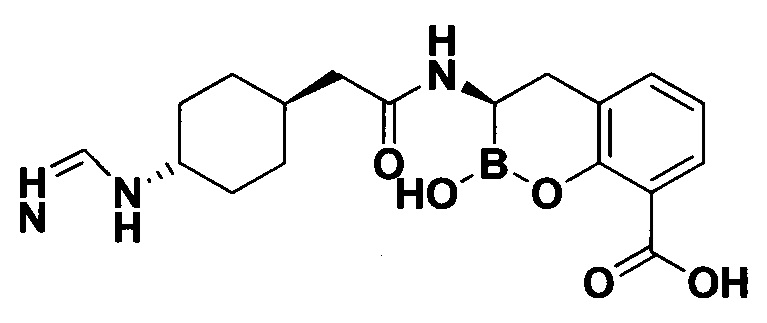
[00276] К (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоте (пример 6) в МеОН добавляли DIEA (2,5 экв), а затем изопропилформимидат-гидрохлорид (1,2 экв). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем смесь концентрировали в вакууме и неочищенный продукт очищали с помощью ВЭЖХ с обращенной фазой, получая указанное в заголовке соединение. ESI-MS m/z 374 (МН)+.
Пример 31: (R)-3-(2-циклогексилацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновая кислота
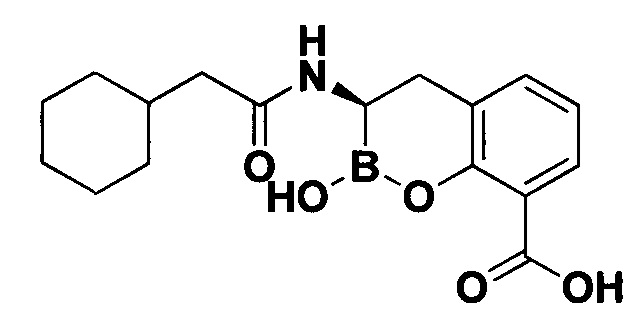
Стадия 1: Синтез трет-бутилового сложного эфира 3-((2R)-2-(2-циклогексилацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата.
[00277] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и 2-циклогексилуксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (Haxane/EtOAc).
Стадия 2: Синтез (R)-3-(3-(транс-4-аминоциклогексил)пропанамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00278] Соединение получали из трет-бутилового сложного эфира 3-((2R)-2-(2-циклогексилацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 332 (МН)+.
Пример 32: (R)-2-гидрокси-3-(2-(транс-4-(пиридин-2-илметиламино)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
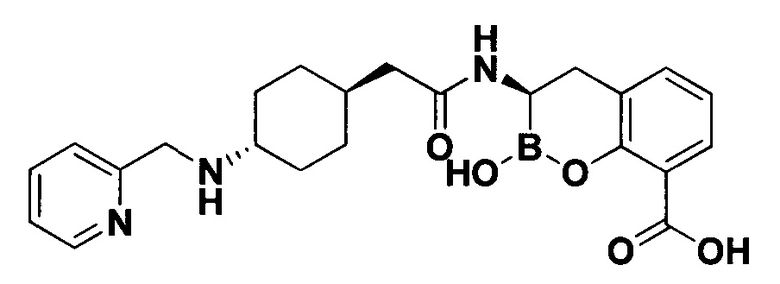
Синтез (R)-2-гидрокси-3-(2-(транс-4-(пиридин-2-илметиламино)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты
[00279] Соединение получали из (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 6) и пиколинальдегида в соответствии с процедурой, описанной в примере 27. Продукт очищали ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 438 (МН)+.
Пример 33: (R)-2-гидрокси-3-(2-(транс-4-(пиперидин-4-илметиламино)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
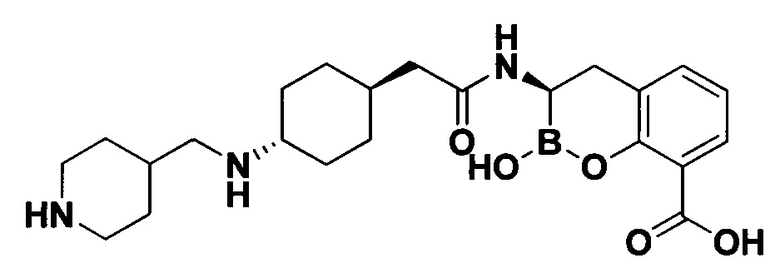
Синтез (R)-2-гидрокси-3-(2-(транс-4-(пиперидин-4-илметиламино)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00280] Соединение получали из (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 6) и трет-бутил-4-формилпиперидин-1-карбоновой кислоты в соответствии с процедурой, описанной в примере 27. Вос-группу удаляли обработкой 4N HCl в диоксане. Продукт очищали ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 444 (МН)+.
Пример 34: (R)-3-(2-(транс-4-((1-карбамимидоилпиперидин-4-ил)метиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота

Синтез (R)-3-(2-(транс-4-((1-карбамимидоилпиперидин-4-ил)метиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00281] Соединение получали из (R)-2-гидрокси-3-(2-(транс-4-(пиперидин-4-илметиламино)циклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 33) и трет-бутил-(1Н-пиразол-1-ил)метандиилидендикарбамата в соответствии с процедурой, описанной на примере 4. Полученный продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 486 (МН)+.
Пример 35: (3R)-3-(2-(4-(3-аминоазетидин-1-ил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
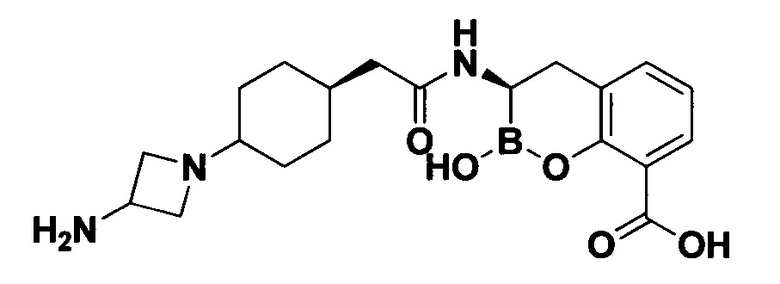
Синтез (3R)-3-(2-(4-(3-аминоазетидин-1-ил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00282] Соединение получали из трет-бутилового сложного эфира азетидин-3-илкарбамата в соответствии с процедурой, описанной в примере 16. Продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 402 (МН)+.
Пример 36: (3R)-3-(2-(4-(азетидин-3-иламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
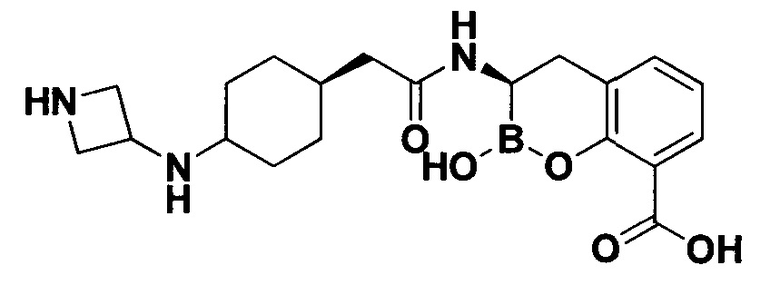
Синтез (3R)-3-(2-(4-(азетидин-3-иламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00283] Соединение получали из трет-бутил-3-амино-1-карбоксилата по способу, описанному в примере 16. Продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 402 (МН)+.
Пример 40: (R)-2-гидрокси-3-(2-(4-морфолиноциклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
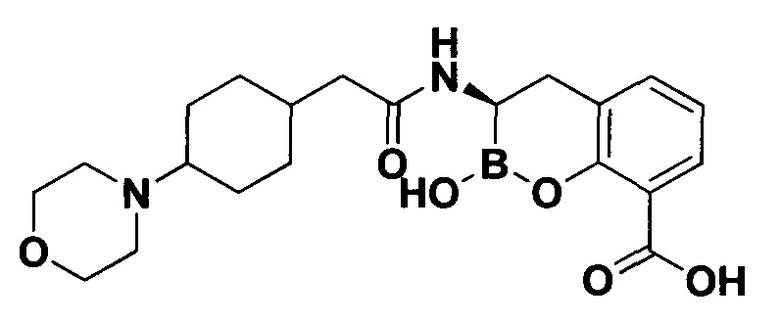
Синтез (R)-2-гидрокси-3-(2-(4-морфолиноциклогексил)ацетамидо)-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00284] Соединение получали из морфолина и 2-(4-оксоциклогексил)уксусной кислоты в соответствии с процедурой, описанной в примере 16. Продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 417 (MH)+.
Пример 41: (3R)-3-(2-(4-(3-гуанидиноазетидин-1-ил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота

Синтез (3R)-3-(2-(4-(3-гуанидиноазетидин-1-ил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00285] Соединение получали из (3R)-3-(2-(4-(3-аминоазетидин-1-ил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 35) и трет-бутил-(1Н-пиразол-1-ил)метандиилиденедикарбамата в соответствии с процедурой, описанной в примере 4. Полученный продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 444 (МН)+.
Пример 42: (R)-3-((R)-2-амино-2-циклогексилацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота

Синтез (R)-3-((R)-2-амино-2-циклогексилацетамидо)-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00286] Соединение получали из (R)-2-(трет-бутоксикарбониламино)-2-циклогексилуксусной кислоты с использованием процедуры, описанной на стадии 1 и стадии 2, пример 1. Указанный продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 347 (МН)+.
Пример 43: 3-{2-[4-(2-амино-этиламино)-1-гидрокси-циклогексил]-ацетиламино}-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
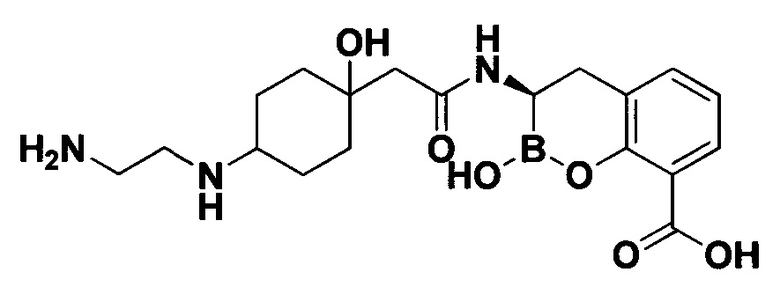
Стадия 1: Синтез (1-гидрокси-4-оксо-циклогексил)бензилового сложного эфира уксусной кислоты.
[00287] К суспензии цинковой пыли (2,06 г, 31,5 ммоль) в диэтиловом эфире (50 мл) в атмосфере аргона добавляли триметилсилил хлорид (3,0 мл, 23,6 ммоль), и реакционную смесь перемешивали в течение 15 мин при комнатной температуре. Затем реакционную смесь нагревали с обратным холодильником в течение 25 мин. Добавляли бензилбромацетат (3,9 мл, 24,6 ммоль) и 1,4-циклогександионмоноэтиленацеталь (3,05 г, 19,6 ммоль), и реакционную смесь выдерживали при температуре дефлегмации в течение 1,3 ч. Затем реакционную смесь охлаждали до комнатной температуры, гасили 1N HCl (125 мл) и перемешивали в течение ночи. Водный слой экстрагировали Et2O (3×). Объединенные органические слои промывали насыщ. NaHCO3, высушивали над MgSO4, фильтровали и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-60% EtOAc: гексан) с получением 2,79 г (54%) чистого продукта. ESI-MS m/z 285 (M+Na)+.
Стадия 2: Синтез бензилового сложного эфира [4-(2-трет-бутоксикарбониламино-этиламино)-1-гидрокси-циклогексил]-уксусной кислоты.
[00288] Этилат титана (0,47 мл, 2,24 ммоль) добавляли к раствору бензилового сложного эфира (1-гидрокси-4-оксо-циклогексил)-уксусной кислоты (1,22 г, 4,65 ммоль) и 2-(Вос-амино)этиламина (0,97 г, 6,05 ммоль) в DCM (5,0 мл) в атмосфере аргона. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь концентрировали в вакууме. Остаток разбавляли метанолом (23 мл) в атмосфере аргона и охлаждали до -78°С. Добавляли триацетоксиборгидрид натрия (1,49 г, 7,03 ммоль) одной порцией, и реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение ночи. Реакцию гасили 10% раствором Na2CO3 и экстрагировали этилацетатом (2×). Объединенные органические слои промывали насыщенным солевым раствором, высушивали над Na2SO4, фильтровали и концентрировали. Неочищенный продукт применяли на следующей стадии без очистки. ESI-MS m/z 407 (МН)+.
Стадия 3: Синтез бензилового сложного эфира {4-[трет-бутоксикарбонил(2-трет-бутоксикарбониламино-этил)амино]-1-гидрокси-циклогексил}уксусной кислоты.
[00289] Триэтиламин (1,1 мл, 7,89 ммоль) и ди-трет-бутилдикарбонат (1,22 г, 5,59 ммоль) добавляли к раствору бензилового сложного эфира [4-(2-трет-бутоксикарбониламино-этиламино)-1-гидрокси-циклогексил]-уксусной кислоты (1,89 г, 4,65 ммоль) в DCM (46 мл) в атмосфере аргона. Реакционную смесь перемешивали при комнатной температуре в течение 17 ч. Реакцию гасили насыщенным солевым раствором и экстрагировали DCM (2×). Объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-75% EtOAc: гексан). ESI-MS m/z 507 (МН)+.
Стадия 4: Синтез бензилового сложного эфира {4-[трет-бутоксикарбонил(2-трет-бутоксикарбониламино-этил)амино]-1-гидрокси-циклогексил}-уксусной кислоты.
[00290] Раствор {4-[трет-бутоксикарбонил(2-трет-бутоксикарбониламино-этил)амино]-1-гидрокси-циклогексил}-уксусной кислоты (0,540 г, 1,07 ммоль) в метаноле (15 мл) продували аргоном в течение 5 мин. Добавляли палладий на угле (10%, 0,127 г), из колбы откачивали воздух, и реакционную смесь перемешивали в атмосфере Н2 в течение 19 ч. Реакционную смесь фильтровали через пористый фильтр с целитом (Celite-plugged filter frit), промывали метанолом и DCM и концентрировали. Неочищенный продукт применяли на следующей стадии без очистки. ESI-MS m/z 439 (M+Na)+.
Стадия 5: Синтез трет-бутилового сложного эфира 3-[2-(2-{4-[трет-бутоксикарбонил-(2-трет-бутоксикарбониламино-этил)-амино]-1-гидрокси-циклогексил}-ацетиламино)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты.
[00291] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и {4-[трет-бутоксикарбонил(2-трет-бутоксикарбониламино-этил)амино]-1-гидрокси-циклогексил}-уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (5-100% EtOAc: гексан). ESI-MS m/z 828 (МН)+.
Стадия 6: Синтез 3-{2-[4-(2-амино-этиламино)-1-гидрокси-циклогексил]-ацетиламино}-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00292] Соединение получали из трет-бутилового сложного эфира 3-[2-(2-{4-[трет-бутоксикарбонил-(2-трет-бутоксикарбониламино-этил)-амино]-1-гидрокси-циклогексил}-ацетиламино)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 406 (МН)+.
Пример 44: 3-[2-(4-Амино-1-гидрокси-циклогексил)-ацетиламино]-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновая кислота
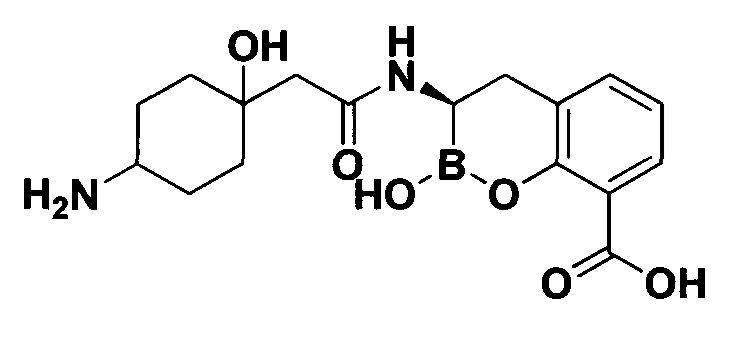
Стадия 1: Синтез бензилового сложного эфира (4-бензиламино-1-гидрокси-циклогексил)-уксусной кислоты.
[00293] Соединение получали из бензилового сложного эфира (1-гидрокси-4-оксо-циклогексил)-уксусной кислоты и бензиламина в соответствии с процедурой, описанной на стадии 2 примера 43. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-10% СН3ОН : CH2Cl2). ESI-MS m/z 354 (МН)+.
Стадия 2: Синтез (4-трет-бутоксикарбониламино-1-гидрокси-циклогексил)-уксусной кислоты.
[00294] Раствор бензилового сложного эфира (4-бензиламино-1-гидрокси-циклогексил)-уксусной кислоты (1,41 г, 3,99 ммоль) и ди-трет-бутилдикарбоната (0,952 г, 4,36 ммоль) в этаноле (35 мл) продували аргоном в течение 5 мин. Добавляли гидроксид палладия (20%, 0,608 г), из колбы откачивали воздух, и реакционную смесь перемешивали в атмосфере Н2 при 65°С в течение 43 ч. Реакционную смесь охлаждали до комнатной температуры, фильтровали через пористый фильтр с целитом, промывали метанолом и DCM и концентрировали. Неочищенный продукт применяли на следующей стадии без очистки. ESI-MS m/z 296 (M+Na)+.
Стадия 3: Синтез трет-бутилового сложного эфира 3-[2-[2-(4-трет-Бутоксикарбониламино-1-гидрокси-циклогексил)-ацетиламино]-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты.
[00295] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)-бензойной кислоты и (4-трет-бутоксикарбониламино-1-гидрокси-циклогексил)-уксусной кислоты по способу, описанному на стадии 1 в примере 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (5-100% EtOAc : гексан). ESI-MS m/z 685 (МН)+.
Стадия 4: Синтез 3-[2-(4-амино-1-гидрокси-циклогексил)-ацетиламино]-2-гидрокси-3,4-дигидро-2Н-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00296] Соединение получали из трет-бутилового сложного эфира 3-[2-[2-(4-трет-бутоксикарбониламино-1-гидрокси-циклогексил)-ацетиламино]-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 363 (МН)+.
Пример 45: 3-[2-(4-Амино-циклогексиламино)-ацетиламино]-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
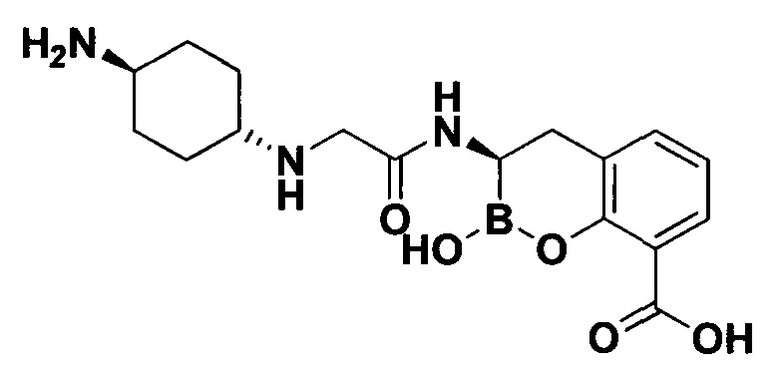
Стадия 1: Синтез бензилового сложного эфира (4-трет-бутоксикарбониламино-циклогексиламино)-уксусной кислоты.
[00297] К суспензии транс-N-Вос-1,4-диаминоциклогексана (0,256 г, 1,19 ммоль) и карбоната калия (0,663 г, 4,80 ммоль) в ацетонитриле (15 мл) и DMF (5 мл) добавляли бензилбромацетат (0,21 мл, 1,33 м моль) в атмосфере аргона, и реакционную смесь перемешивали при комнатной температуре в течение 19 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщ. NaHCO3 и насыщенным солевым раствором. Органический слой высушивали над Na2SO4, фильтровали и концентрировали. Неочищенный продукт применяли на следующей стадии без очистки. ESI-MS m/z 363 (МН)+.
Стадия 2: Синтез бензилового сложного эфира [трет-бутоксикарбонил (4-трет-бутоксикарбониламино-циклогексил)амино]-уксусной кислоты.
[00298] Соединение получали из бензилового сложного эфира (4-трет-бутоксикарбониламино-циклогексиламино)-уксусной кислоты и ди-трет-бутилдикарбоната в соответствии с процедурой, описанной на стадии 3 примера 43. Неочищенный продукт очищали флэш-хроматографией на силикагеле (от 0 до 50% EtOAc : гексан). ESI-MS m/z 463 (МН)+.
Стадия 3: Синтез [трет-бутоксикарбонил-(4-трет-бутоксикарбониламино-циклогексил)амино]-уксусной кислоты.
[00299] Раствор бензилового сложного эфира [трет-бутоксикарбонил-(4-трет-бутоксикарбониламино-циклогексил)амино]-уксусной кислоты (0,277 г, 0,599 ммоль) в метаноле (6 мл) продували аргоном в течение 5 мин. Добавляли гидроксид палладия (20%, 0,053 г), откачивали воздух из колбы, и реакционную смесь перемешивали в атмосфере Н2 в течение 19 ч. Реакционную смесь фильтровали через пористый фильтр с целитом, промывали метанолом и DCM и концентрировали. Неочищенный продукт применяли на следующей стадии без очистки. ESI-MS m/z 395 (M+Na)+.
Стадия 4: Синтез трет-бутилового сложного эфира 3-[2-{2-[трет-бутоксикарбонил-(4-трет-бутоксикарбониламино-циклогексил)-амино]-ацетиламино}-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты.
[00300] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и [трет-бутоксикарбонил-(4-трет-бутоксикарбониламино-циклогексил)амино]-уксусной кислоты в соответствии с процедурой, описанной в стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (от 5 до 100% EtOAc : гексан). ESI-MS m/z 784 (МН)+.
Стадия 5: Синтез 3-[2-(4-амино-циклогексиламино)-ацетиламино]-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00301] Соединение получали из трет-бутилового сложного эфира 3-[2-{2-[трет-бутоксикарбонил-(4-трет-бутоксикарбониламино-циклогексил)-амино]-ацетиламино}-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)-этил]-2-метокси-бензойной кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 362 (МН)+.
Пример 46: (R)-3-(2-(цис-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
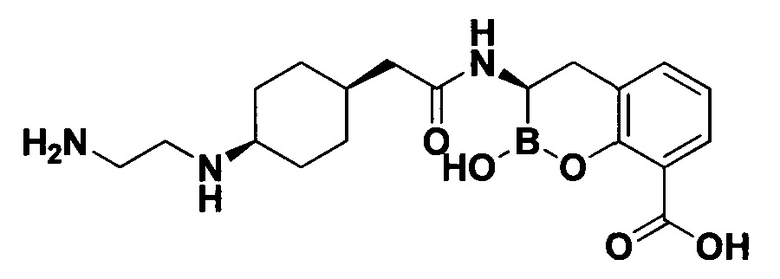
Синтез (R)-3-(2-(цис-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00302] Соединение получали из (R)-3-(2-(цис-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 7) в соответствии с процедурой, описанной в примере 10. Продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 390 (МН)+.
Пример 47: (3R)-2-гидрокси-3-(2-(4-гидроксициклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
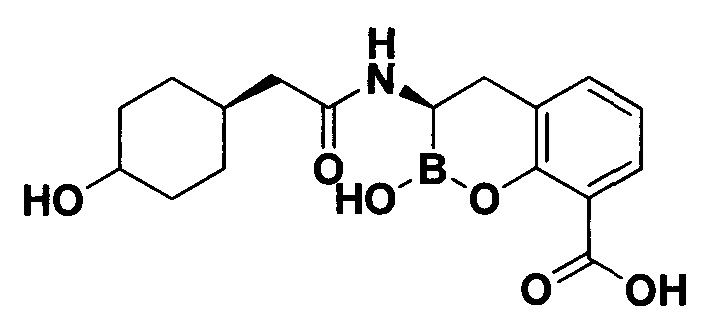
Стадия 1: Синтез трет-бутилового сложного эфира 3-((2R)-2-(гексагидробензо[d][1,3,2]диоксаборол-2-ил)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата
[00303] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)-бензойной кислоты и 2-(4-оксоциклогексил)-уксусной кислоты соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (Haxane/EtOAc). ESI-MS m/z 568,1 (МН)+.
Стадия 2: Синтез (3R)-2-гидрокси-3-(2-(4-гидроксициклогексил)ацетамидо)-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00304] (R)-2-гидрокси-3-(2-(4-оксоциклогексил)ацетамидо)-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновую кислоту получали из трет-бутил-3-((2R)-2-(гексагидробензо[d][1,3,2]диоксаборол-2-ил)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата, как описано на стадии 2, пример 1. К неочищенному продукту в МеОН и H2O добавляли NaBH4. Полученную реакционную смесь перемешивали при комнатной температуре в течение 4 ч. После удаления метанола продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 348 (МН)+.
Пример 48: (R)-2-гидрокси-3-(2-(транс-4-(2-(пиридин-2-иламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
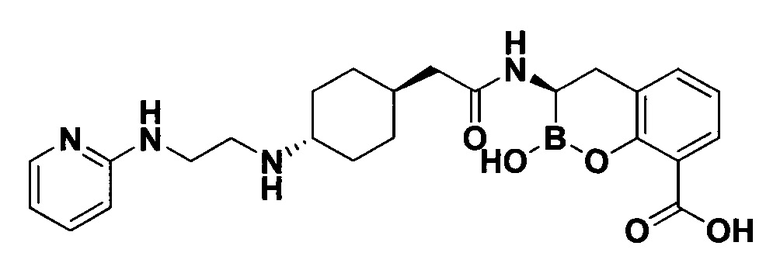
Стадия 1: Синтез (+) пинандиолового сложного эфира [(1S)-2-(3-трет-бутоксикарбонил-2-метокси-фенил)-1-хлорэтил]бороновой кислоты.
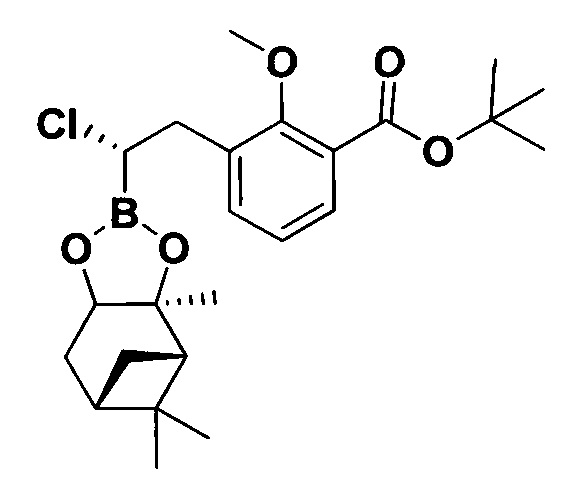
Стадия 1а: Синтез (+) пинандиолового сложного эфира 3-карбокси-2-метокси-фенилбороновой кислоты.
[00305] К смеси 3-карбокси-2-метокси-фенилбороновой кислоты (35.0 г, 178,5 ммоль) и (+) пинандиола (30.35 г, 178,5 ммоль) добавляли толуол (400 мл). Полученную смесь перемешивали в течение 3 ч, затем концентрировали в вакууме (28 мм рт.ст., температура бани 40°С). Полученное твердое вещество высушивали азеотропной смесью толуола (2 раза, приблизительно 100 мл). Данный остаток высушивали при высоком вакууме (прибл. 1 мм рт.ст.) при комнатной температуре в течение 17,5 часов с получением неочищенного указанного в заголовке соединения, которое можно использовать без дополнительной очистки. 48.06 г данного неочищенного продукта перекристаллизовывали из 150 мл смеси хлороформ/гексан (1:5 об./об.) с получением очищенного указанного в заголовке соединения. Маточный раствор, полученный после кристаллизации, концентрировали и очищали хроматографией на силикагеле (120 г двуокиси кремния, элюируя от 40 до 100% этилацетата в гексане), чтобы дать дополнительную партию указанного в заголовке соединения.
Стадия 1b: Синтез (+) пинандиолового сложного эфира 3-трет-бутилоксикарбонил-2-метокси-фенил бороновой кислоты.
[00306] К перекристаллизованному (+) пинандиоловому сложному эфиру 3-карбокси-2-метокси-фенилбороновой кислоты (9.90 г, 30 ммоль) добавляли тионилхлорид (20 мл, класс) с выходом из реакционной колбы через CaCl2-ловушку. Полученный раствор нагревали на масляной бане при 95°С и перемешивали в течение 1 часа. Данный раствор охлаждали до комнатной температуры в течение около 10 мин, затем концентрировали при пониженном давлении (от 20 до 30 мм рт.ст., 35°С), пока не была достигнута постоянная масса в 10,57 г.
В отдельной колбе к охлажденному (-5°С) раствору трет-BuOH (3.7 мл, 39 ммоль) в THF (100 мл) по капле добавляли BuLi (14,4 мл, 2,5 М в гексане, 36 ммоль) в течение около 5 мин. После завершения добавления полученный раствор перемешивали в течение 20 мин. К данному раствору добавляли неочищенный хлорид кислоты (выше) (10.57 г, 30 ммоль) в THF (15 мл) в течение около 30 сек. По окончании добавления охлаждающую баню удаляли и перемешивание продолжали в течение 4 ч. К данному раствору добавляли HCl (50 мл, 0,2 М водный). Смесь (рН=3) экстрагировали эфиром, и эфирный экстракт промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (120 г двуокиси кремния, элюируя с от 2 до 20% этилацетата в гексане) с получением указанного в заголовке соединения в виде твердого вещества кремового цвета.
Стадия 1 с: Синтез (+)-пинандиолового сложного эфира (3-трет-бутоксикарбонил-2-метокси-фенил)метилбороновой кислоты.
[00307] К охлажденному (внешняя температура -100°С) раствору (+) пинандиолового сложного эфира 3-трет-бутилоксикарбонил-2-метокси-фенилбороновой кислоты (34,6 г, 89,6 ммоль) и хлор-иодометана(10,3 мл, 140 ммоль) в THF (250 мл) добавляли по каплям, по стенке колбы, BuLi (54 мл, 2,5 М в гексане, 135 ммоль) в течение 80 мин. После завершения добавления реакционную смесь перемешивали 15 мин. К полученному раствору добавляли ZnCl2 (90 мл, 1М в эфире), по каплям, вниз по стороне колбы, в течение около 40 мин. По окончании добавления охлаждающую баню удаляли и перемешивание продолжали в течение 16,5 ч. Реакционную смесь разбавляли NH4Cl (300 мл, насыщенным водным раствором) и этилацетатом (700 мл). Отделенный органический экстракт промывали дополнительной порцией водного насыщенного NH4Cl (100 мл) и насыщенным солевым раствором (100 мл), высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (224 г диоксида кремния, элюируя гексаном (1 л), затем 10% этилацетата в гексане (2 л) с получением указанного в заголовке соединения в виде бесцветного масла. Данный материал медленно кристаллизовался при -10°С.
Стадия 1d: Синтез (+) пинандиолового сложного эфира [(1S)-2-(3-трет-бутоксикарбонил-2-метокси-фенил)-1-хлорэтил]бороновой кислоты
[00308] Способ 1: К охлажденному (наружная температура -100°С) раствору дихлорметана (2,27 мл, 35 ммоль) в THF (44 мл) добавляли по каплям, вниз по стороне колбы, BuLi (8,88 мл, 2,5 М в гексане, 22 ммоль) в течение 45 мин. После добавления приблизит. 80% BuLi образовывался белый осадок. После завершения добавления реакционную смесь перемешивали в течение 30 мин. К данной смеси добавляли по каплям, вниз по стороне колбы, (+)-пинандиоловый сложный эфир (3-трет-бутоксикарбонил-2-метокси-фенил)метилбороновой кислоты (8,0 г, 20 ммоль) в THF (20 мл) в течение приблизительно 30 мин. После завершения добавления полученный раствор перемешивали в течение 5 минут. К данному раствору по каплям добавляли, по стенке колбы, ZnCl2 (22 мл, 1М в эфире), в течение около 12 мин. По окончании добавления охлаждающую баню удаляли и заменяли на баню с температурой -10°С. Реакционную смесь перемешивали в течение 1,25 ч. К данному раствору добавили ледяной эфир (300 мл) и ледяной насыщенный водный NH4Cl (125 мл). Органическую фазу промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле (120 г диоксида кремния, элюируя с от 2 до 20% этилацетата в гексане) с получением указанного в заголовке соединения в виде бесцветного масла. Данный материал медленно кристаллизовался при -10°.
[00309] Способ 2: (+)-пинандиоловый сложный эфир (3-трет-бутоксикарбонил-2-метокси-фенил)метилбороновой кислоты (2,0 г, 5 ммоль) и дихлорметан (1,6 мл, 25 ммоль) в THF (20 мл) перемешивали при -60°С в течение 30 мин. К данному раствору добавляли LDA (6,5 ммоль, 2 М раствор от Aldrich) в течение 10 мин. Полученную реакционную смесь перемешивали при -60°С в течение 20 мин. ZnCl2 (8,75 ммоль, 1М раствор в эфире) медленно добавляли при -60°С. Реакционную смесь перемешивали при температуре от -50 до -60°С в течение 30 мин. Полученную смесь нагревали до 0°С в течение 1 часа, после чего добавляли 10% раствор H2SO4 (10 мл) и реакционную смесь перемешивали в течение 10 мин. После разделения фаз органическую фазу промывали водой и насыщенным солевым раствором. Органическую фазу затем высушивали и концентрировали в вакууме. Затем остаток очищали флэш-хроматографией на силикагеле (EtOAc/гексан : 4/1) с получением указанного в заголовке соединения.
Стадия 2: Синтез этил-2-[4-[2-(2-пиридиламино)этиламино]циклогексил]ацетата.
[00310] К смеси 2-(R-[2-(амино)-этил]-амино)-пиридина (685 мг, 5 ммоль) и этил-2-(4-оксоциклогексил)ацетата (786 мг, 4 ммоль) добавляли дихлорметан (4 мл) с последующим добавлением этилата титана (420 μL, 2 ммоль, технического сорта). Полученную смесь перемешивали в течение 4 ч, затем концентрировали при пониженном давлении. Остаток растворяли в метаноле (10 мл) и охлаждали до -78°С. К данному раствору добавляли боргидрид натрия (228 мг, 6 ммоль). По окончании добавления охлаждающую баню удаляли и перемешивание продолжали в течение 1,25 ч. Смесь разбавляли дихлорметаном и выливали в насыщенный раствор карбоната натрия (15 мл). Органическую фазу отделяли, высушивали над сульфатом магния и концентрировали с получением неочищенного указанного в заголовке соединения в виде смеси транс : цис-изомеров с соотношением 6:1. Данную смесь использовали без дополнительной очистки.
Стадия 3: Синтез этил-2-[4-[трет-бутоксикарбонил-[2-[трет-бутоксикарбонил(2-пиридил)амино]этил]амино]циклогексил]ацетата.
[00311] К раствору этил-2-[4-[2-(2-пиридиламино)этиламино]циклогексил]ацетата (1,31 г, 4 ммоль) в дихлорметане (12 мл) добавляли ди-трет-бутилдикарбонат (2,18 г, 10 ммоль) с последующим добавлением диизопропилэтиламина (1,76 мл, 10 ммоль). Полученный раствор перемешивали в течение 4 ч, разбавляли этилацетатом, промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (30 г диоксида кремния, элюировали смесью 20% этилацетата/10% дихлорметана в гексане) с получением указанного в заголовке соединения в виде смеси транс : цис-изомеров с соотношением 6:1.
Стадия 4: Синтез 2-[4-[трет-бутоксикарбонил[2-[трет-бутоксикарбонил-(2-пиридил)амино]этил]амино]циклогексил]-уксусной кислоты.
[00312] К раствору этил-2-[4-[трет-бутоксикарбонил[2-[трет-бутоксикарбонил-(2-пиридил)амино]этил]амино]циклогексил]ацетата (968 мг, 1,91 ммоль) в THF (3 мл); метанола (3 мл) и воды (6 мл) добавляли моногидрат гидроксида лития (397 мг, 9,7 ммоль). Полученный раствор перемешивали в течение 2,25 ч, затем подкисляли до значения рН 3 с помощью 1 н HCl. Полученную смесь экстрагировали дихлорметаном (4 раза). Объединенный органический экстракт высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (10 г диоксида кремния; элюировали от 40 до 100% этилацетата в гексане) с получением указанного в заголовке соединения в виде смеси транс : цис-изомеров с соотношением 6:1.
Стадия 5: Синтез трет-бутил-2-метокси-3-((2R)-2-(2-(транс-4-(2-(пиридин-2-иламино)этиламино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)бензоата.
[00313] К охлажденному (-78°С) раствору (+) пинандиолового сложного эфира [(1S)-2-(3-трет-бутоксикарбонил-2-метокси-фенил)-1-хлорэтил]бороновой кислоты (со стадии 1) (1,35 г, 3 ммоль) в THF (9 мл) добавляли по каплям раствор бистриметилсилиламида лития (3,0 мл, 1М в THF, 3 ммоль). По окончании добавления охлаждающую баню удаляли и перемешивание продолжали в течение 16,75 часов. Полученный раствор, который представлял собой около 0,25 М бензойной кислоты, 3-[(2R)-2-[бис(триметилсилил)амино]-2-[(3aS,4S,6S,7aR)-гексагидро-3а,5,5-триметил-4,6-метано-1,3,2-бензодиоксаборол-2-ил]этил]-2-метокси,1,1-диметилэтиловый эфир в THF, использовали без дальнейшей очистки.
[00314] В отдельной колбе к смеси 2-[4-[трет-бутоксикарбонил[2-[трет-бутоксикарбонил(2-пиридил)амино]этил]амино]циклогексил]-уксусной кислоты (477 мг, 1 ммоль) и HATU (418 мг, 1,1 ммоль) добавляли DMA (3 мл) с последующим добавлением N-метил-морфолина (120 μL, 1,1 ммоль). Полученный раствор перемешивали в течение 90 минут. К данному раствору добавляли раствор бензойной кислоты, 3-[(2R)-2-[бис(триметилсилил)амино]-2-[(3aS,4S,6S,7aR)-гексагидро-3а,5,5-триметил-4,6-метано-1,3,2-бензодиоксаборол-2-ил]этил]-2-метокси,1,1-диметилэтиловый эфир (4 мл, 0,25 М в THF 1 ммоль). Полученную смесь перемешивали в течение 2,5 ч, разбавляли этилацетатом, промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (10 г диоксида кремния; элюировали 20-100%-ным раствором этилацетата в гексане) с получением указанного в заголовке соединения.
Стадия 6: Синтез (R)-2-гидрокси-3-(2-(транс-4-(2-(пиридин-2-иламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00315] К раствору трет-бутил-2-метокси-3-((2R)-2-(2-(транс-4-(2-(пиридин-2-иламино)этиламино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)бензоата (396 мг, 0,45 ммоль) в 1,4-диоксане (6 мл) добавляли раствор HCl (6 мл, 3М раствор в воде). Полученный раствор нагревали с обратным холодильником и перемешивали при данной температуре в течение 3,5 часов. Полученную смесь охлаждали до комнатной температуры и экстрагировали эфиром (2×). Оставшийся водный раствор очищали обращенно-фазовой ВЭЖХ Phenomenex Luna С 18 колонке 35×75 мм; скорость потока 40 мл/мин; элюировали от 5 до 70% CH3CN в H2O/0,1% TFA в течение 8 минут. Указанное в заголовке соединение выделяли в виде соли TFA лиофилизацией.
Пример 49: (R)-2-гидрокси-3-(2-(транс-4-(2-(метилсульфонамидо)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
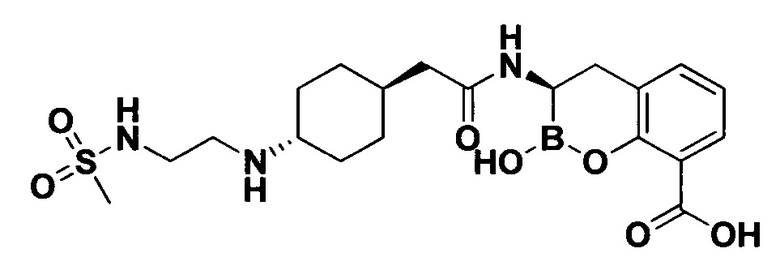
Стадия 1: Синтез трет-бутил-N-[2-(метансульфонамидо)этил]карбамата.
[00316] К охлажденному (-10°С) раствору 2-трет-бутил-N-(2-аминоэтил)карбамата (1,60 г, 10 ммоль) в дихлорметане (25 мл) добавляли триэтиламин (1,38 мл, 10 ммоль) с последующим добавлением метансульфонилхлорида (770 μл, 10 ммоль). Полученный раствор перемешивали в течение 5 минут, затем охлаждающую баню удаляли и перемешивание продолжали в течение 1 часа. Затем реакционную смесь разбавляли этилацетатом, промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением указанного в заголовке соединения в виде твердого вещества. Данный материал использовали без дальнейшей очистки.
Стадия 2: Синтез N-(2-аминоэтил)метансульфонамида; 2,2,2-соли трифторуксусной кислоты.
[00317] К раствору трет-бутил-N-[2-(метансульфонамидо)этил]карбамата (2,23 г, 9,4 ммоль) в дихлорметане (30 мл) добавляли трифторуксусную кислоту (7,5 мл). Полученный раствор перемешивали в течение 1,5 часов, затем концентрировали под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества. Данный материал использовали без дальнейшей очистки.
Стадия 3: Синтез этил-2-[4-[2-(метансульфонамидо)этиламино]циклогексил]ацетата.
[00318] К смеси этил-2-(4-оксоциклогексил)ацетата (736 мг, 4 ммоль) и N-(2-аминоэтил)метансульфонамида; 2,2,2-соли трифторуксусной кислоты (1,26 г, 5 ммоль) в дихлорметане (6 мл) добавляли триэтиламин (690 μл, 5 ммоль) с последующим добавлением титана (IV) этоксида (420 μл, 2 ммоль, технического сорта). Полученную непрозрачную смесь перемешивали в течение 4 часов, затем концентрировали при пониженном давлении. Остаток растворяли в метаноле (6 мл). Данную смесь охлаждали до -78°С. К полученной смеси добавляли боргидрид натрия (187 мг, 4,8 ммоль). По окончании добавления охлаждающую баню убирали и перемешивание продолжали в течение 15,5 часов. Полученную смесь концентрировали при пониженном давлении с получением густой пасты. Данный остаток растворяли в дихлорметане (40 мл). К данной смеси добавляли Na2CO3 (5,5 мл, насыщенный водный раствор). Полученную смесь перемешивали в течение 5 минут. К данной смеси добавляли целит (1,8 г). Полученную смесь перемешивали в течение 5 минут, затем фильтровали через слой целита. Фильтрат промывали насыщенным водным раствором Na2CO3, высушивали над сульфатом магния и концентрировали с получением указанного в заголовке соединения в виде смеси транс : цис-изомеров с соотношением 6:1. Данный материал использовали без дальнейшей очистки.
Стадия 4: Синтез этил-2-[4-[трет-бутоксикарбонил[2-(метансульфонамидо)этил]амино]циклогексил]ацетата.
[00319] К раствору этил-2-[4-[2-(метансульфонамидо)этиламино]циклогексил]ацетата (1,1 г, 4 ммоль) в дихлорметане (12 мл) добавляли ди-трет-бутилдикарбонат (1,74 г, 8 ммоль) с последующим добавлением диизопропилэтиламина (1,4 мл, 8 ммоль). Полученный раствор перемешивали в течение 4 ч, затем разбавляли дихлорметаном, промывали водой, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (25 г диоксида кремния; элюировали от 5 до 50% этилацетата в гексане) с получением указанного в заголовке соединения в виде смеси 6:1 транс : цис-изомеров в виде масла.
Стадия 5: Синтез транс-2-[4-[трет-бутоксикарбонил[2-(метансульфонамидо)этил]амино]циклогексил]-уксусной кислоты.
[00320] К раствору этил-2-[4-[трет-бутоксикарбонил[2-(метансульфонамидо)этил]амино]циклогексил]ацетата (0.842 г, 2,06 ммоль) в метаноле (4 мл); THF (4 мл) и Waterl (4 мл) добавляли моногидрат гидроксида лития (252 мг, 6 ммоль). Полученный раствор перемешивали в течение 2 часов, затем подкисляли HCl (7 мл, 1М водн.), Разбавляли этилацетатом, промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток растирали с эфиром с получением указанного в заголовке соединения в виде твердого вещества.
Стадия 6: Синтез (+) пинандиолового сложного эфира [(1R)-1-[[2-[4-[трет-бутоксикарбонил-[2-(метансульфонамидо)этил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
[00321] Указанное в заголовке соединение получали, используя ту же самую процедуру, как описано в примере 48; стадия 4, за исключением использования транс-2-[4-[трет-бутоксикарбонил[2-(метансульфонамидо)этил]амино]циклогексил]уксусной кислоты вместо 2-[4-[трет-бутоксикарбонил-[2-[трет-бутоксикарбонил(2-пиридил)амино]этил]амино]циклогексил]уксусной кислоты.
Стадия 7: Синтез (R)-2-гидрокси-3-(2-(транс-4-(2-(метилсульфонамидо)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00322] К раствору (+) пинандиолового сложного эфира [(1R)-1-[[2-[4-[трет-бутоксикарбонил-[2-(метансульфонамидо)этил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты (460 мг, 0,55 ммоль) в 1,4-диоксане (4 мл) добавляли HCl (4 мл, 3М раствор в воде). Полученный раствор нагревали с обратным холодильником и перемешивали при данной температуре в течение 3,5 часов. Полученную смесь охлаждали до комнатной температуры и экстрагировали эфиром (2 раза). Оставшийся водный раствор концентрировали до объема в 25%, а остаток очищали с помощью обращенно-фазовой ВЭЖХ-колонки Phenomenex Luna С18 35×75 мм; скорость потока 40 мл/мин; элюировали 5-70% CH3CN в H2O/0,1% TFA в течение 8 минут. Указанное в заголовке соединение выделяли в виде соли TFA лиофилизацией.
Пример 50: (S)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
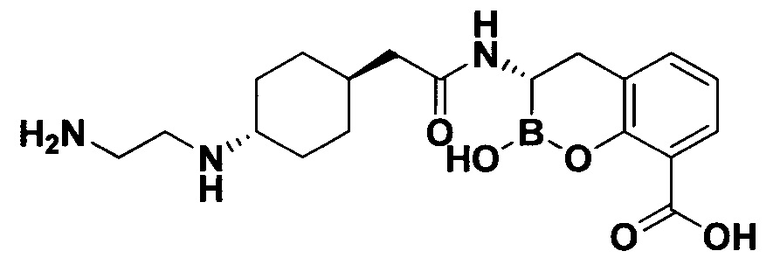
Стадия 1: Синтез 3-йодо-2-метокси-бензальдегида.
[00323] К раствору 3-йодо-2-гидрокси-бензальдегида (4,0 г, 16.06 ммоль) в DMA (32 мл) добавляли карбонат цезия (5,85 г, 18 ммоль) с последующим добавлением метилиодида (1.12 мл, 18 ммоль). Полученную смесь перемешивали в течение 4,75 ч, затем разбавляли эфиром, промывали водой, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (50 г диоксида кремния; элюировали от 0 до 20% этилацетата в гексане) с получением указанного в заголовке соединения в виде масла.
Стадия 2: Синтез 3-йодо-2-метокси-бензойной кислоты.
[00324] К раствору 3-йодо-2-метокси-бензальдегида (3.68 г, 14 ммоль) в трет-бутаноле (70 мл) добавляли 2,3-диметил-бут-2-ен (7 мл) с последующим добавлением раствора, содержащего динатрийгидрофосфат моногидрат (7,56 г 56 ммоль) и хлорит натрия (7,56 г, прибл. 66 ммоль, технический сорт) в воде (70 мл). Полученную смесь перемешивали в течение 20 минут, затем разбавляли этилацетатом, промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Твердый остаток перекристаллизовывали из циклогексана с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 3: Синтез 3-йодо-2-метокси-бензоилхлорида.
[00325] К 3-йодо-2-метокси-бензойной кислоте (8.29 г, 29,8 ммоль) добавляли тионилхлорид (15 мл). Полученный раствор перемешивали в течение 2 минут, затем нагревали до 82°С и перемешивали при данной температуре в течение 30 минут. Раствор затем концентрировали при пониженном давлении с получением указанного в заголовке соединения. Данный материал использовали без дальнейшей очистки.
Стадия 4: Синтез трет-бутил-3-йодо-2-метокси-бензоата.
[00326] К охлажденному (-5°С) раствору трет-бутанола (12,87 мл, 30 ммоль) в THF (30 мл) добавляли по каплям BuLi (12,0 мл, 2,5 М в гексане, 30 ммоль). По окончании добавления раствор перемешивали в течение 20 минут. К данному раствору добавляли раствор 3-йодо-2-метокси-бензоилхлорида (8,8 г, 29,8 ммоль) в THF (12 мл). По окончании добавления охлаждающую баню удаляли и перемешивание продолжали в течение 1,5 часа. Данный раствор разбавляли этилацетатом, промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (50 г диоксида кремния; элюировали от 0 до 20% этилацетата в гексане) с получением указанного в заголовке соединения в виде масла.
Стадия 5: Синтез (-)-пинандиолового сложного эфира (3-трет-бутоксикарбонил-2-метокси-фенил)метилбороновой кислоты.
[00327] К охлажденному (-40°С) раствору трет-бутил-3-йодо-2-метокси-бензоата (3.34 г, 10 ммоль) в THF (25 мл) добавляли по каплям комплекс изопропилмагний хлорид: хлорид лития (7,69 мл, 1,3 М в THF, 10 ммоль). По окончании добавления раствор перемешивали в течение 20 мин, затем охлаждали до -78°С. К данному раствору добавили по каплям, вниз, по внутренней стороне колбы (-)-пинандиоловый сложный эфир хлорметилбороновой кислоты (2,28 г, 10 ммоль) в THF (2 мл), (-)-пинандиоловый сложный эфир хлорметилбороновой кислоты готовили в соответствии с публикацией Strynadka et al. Biochemistry 2000, 39, 5312; за исключением использования (-) пинандиола вместо (+) пинандиола). Полученный раствор перемешивали в течение 45 минут. К данному раствору добавили по каплям ZnCl2 (10 мл, 1М в эфире, 10 ммоль). Полученную смесь перемешивали в течение 5 минут, затем охлаждающую баню удаляли и перемешивание продолжали в течение 2 часов. Данную смесь разбавляли эфиром, промывали 0,1 М HCl и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (50 г диоксида кремния; элюировали от 0 до 20% этилацетата в гексане) с получением указанного в заголовке соединения в виде масла. Данный материал кристаллизуется при стоянии при -10°С.
Стадия 6: Синтез (-) пинандиолового сложного эфира (1R)-2-(3-трет-бутоксикарбонил-2-метокси-фенил)-1-хлорэтил]бороновой кислоты.
[00328] К охлажденному (-100°С) раствору дихлорметана (518 μл, 8 ммоль) в THF (10 мл) добавляли по каплям, вниз, по внутренней стороне колбы, BuLi (2,0 мл, 2,5 М в гексане, 5 ммоль) в течение около 20 минут. Образуется осадок после добавления около 75% от BuLi. После окончания добавления полученный мутный раствор перемешивали в течение 40 минут. К данной смеси добавляли по каплям, вниз, по внутренней стороне колбы, раствор (-)-пинандиолового сложного эфира (3-трет-бутоксикарбонил-2-метокси-фенил)метилбороновой кислоты (1,8 г, 4,5 ммоль) в THF (4 мл). По окончании добавления добавляли раствор ZnCl2 (5 мл, 1 М в эфире, 5 ммоль), по капле, в течение около 8 минут. Баню с охлаждением до -100°С заменяли на баню с охлаждением до -10°С и полученную смесь перемешивали в течение 1 часа. К данной смеси добавляли холодный насыщенный раствор NH4Cl с последующим добавлением холодного эфира (5°С). Органическую фазу отделяли, промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (25 г диоксида кремния; элюировали от 0 до 20% этилацетата в гексане) с получением указанного в заголовке соединения в виде масла. Данный материал кристаллизуется при стоянии при -10°С.
Стадия 7: Синтез этил-2-[4-[трет-бутоксикарбонил[3-(трет-бутоксикарбониламино)этил]амино]циклогексил]ацетата.
[00329] Указанное в заголовке соединение (выделено в виде смеси транс : цис-изомеров в соотношении 6,8:1) получали с использованием по существу той же процедуры, которая описана в примере 48, стадия 2, за исключением использования трет-бутил-этил-2-[4-[3-(трет-бутоксикарбониламино)этиламино]циклогексил]ацетата вместо этил-2-[4- [2-(2-пиридиламино)этиламино]циклогексил]ацетата.
Стадия 8: Синтез транс-2-[4-[трет-бутоксикарбонил[3-(трет-бутоксикарбониламино)этил]амино]циклогексил]-уксусной кислоты.
Указанное в заголовке соединение получали с использованием по существу той же процедуры, которая описана в примере 63, стадия 3, за исключением использования этил-2-[4-[трет-бутоксикарбонил[3-(трет-бутоксикарбониламино)этил]амино]циклогексил]ацетата вместо этил-2-[4-[трет-бутоксикарбонил[3-(трет-бутоксикарбониламино)пропил]амино]циклогексил]ацетата.
Стадия 9: Синтез (-) пинандиолового сложного эфира [(1S)-1-[[2-[транс-4-[трет-бутоксикарбонил-[2-(трет-бутоксикарбониламино)этил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
[00330] К охлажденному (-20°С) раствору (-) пинандиолового сложного эфира [(1R)-2-(3-трет-бутоксикарбонил-2-метокси-фенил)-1-хлорэтил]бороновой кислоты (430 мг, 1 ммоль) в THF (2 мл) добавляли по каплям раствор бистриметилсилиламида лития (1,0 мл, 1М в THF, 1 ммоль). По окончании добавления охлаждающую баню удаляли и перемешивание продолжали в течение 1 часа. Полученный раствор приблизительно 0.29 М в (-) пинандиоловом сложном эфире [(1S)-1-[бис(триметилсилил)амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты использовали без дополнительного воздействия.
[00331] В отдельной колбе к смеси 2-[4-транс-[трет-бутоксикарбонил[2-(трет-бутоксикарбониламино)этил]амино]циклогексил]уксусной кислоты (400 мг, 1 ммоль) и HATU (418 мг, 1,1 ммоль) добавляли DMA (2 мл) с последующим добавлением N-метил-морфолина (120 μл, 1,1 ммоль). Полученный раствор перемешивали в течение 90 минут. К данному раствору добавили раствор приблизительно 0.29М (-) пинандиолового сложного эфира [(1S)-1-[бис(триметилсилил)амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты, полученный выше. Полученную смесь перемешивали в течение 4 часов, разбавляли этилацетатом, промывали водой и насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (10 г диоксида кремния; элюировали 20-100%-ным раствором этилацетата в гексане) с получением указанного в заголовке соединения в виде пены.
Стадия 10: Синтез (S)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00332] К охлажденному (-78°С) раствору (-) пинандиолового сложного эфира [(1S)-1-[бис(триметилсилил)амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты (468 мг, 0,59 ммоль) в дихлорметане (2 мл) добавляли по каплям раствор BCl3 (3 мл, 1 М в CH2Cl2, 3 ммоль). После завершения добавления полученную в результате смесь перемешивали в течение 30 минут, затем нагревали до 0°С в течение 30 минут. К данной смеси добавляли воду (6 мл). Полученную смесь оставляли нагреваться до комнатной температуры в течение 20 мин. Данную смесь экстрагировали эфиром и оставшуюся водную фазу очищали ВЭЖХ с обращенной фазой (Phenomenex Luna С18 колонка 35×75 мм; скорость потока 40 мл/мин; элюент: от 5 до 45% CH3CN в H2O/0,1% TFA в течение 8 минут). Указанное в заголовке соединение выделяли в виде соли TFA лиофилизацией.
[00333] ESI-MS m/z 390 (МН)+.
Пример 51: (R)-2-гидрокси-3-(2-(транс-4-(2-(метиламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
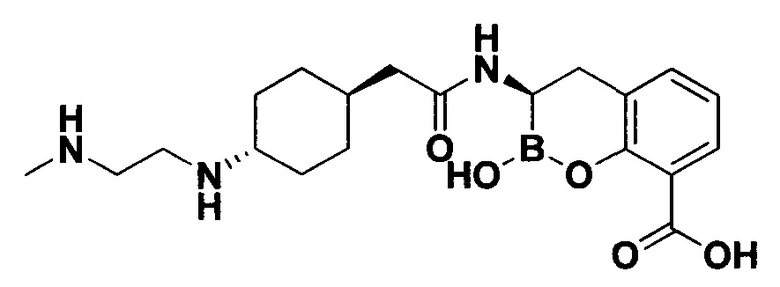
[00334] Соединение получали из 2-(транс-4-(трет-бутоксикарбонил(2-(трет-бутоксикарбонил(метил)амино)этил)амино)циклогексил)уксусной кислоты в соответствии с процедурой, описанной на стадии 1 и стадии 2 примера 1. Продукт очищали ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 404 (МН)+.
Пример 52: (R)-2-гидрокси-3-(2-(транс-4-(2-имино-3-метилимидазолидин-1-ил)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо [е][1,2]оксаборинин-8-карбоновая кислота
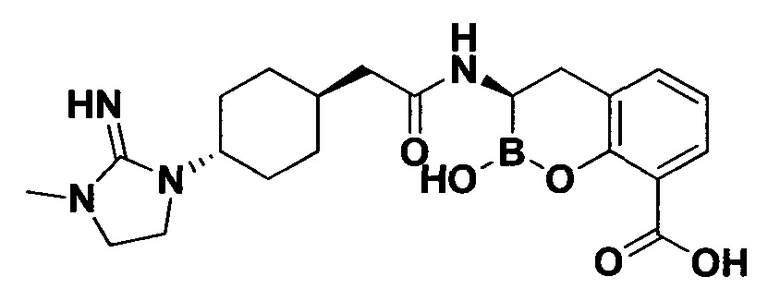
Синтез (R)-2-гидрокси-3-(2-(транс-4-(2-имино-3-метилимидазолидин-1-ил)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00335] К (R)-2-гидрокси-3-(2-(транс-4-(2-(метиламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоте из примера 51 (10 мг) в МеОН (1 мл) добавляли трет-бутил-(1Н-пиразол-1-ил)метандиилиденедикарбамат (12 мг) и TEA (0,1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Растворитель удаляли в вакууме. Остаток обрабатывали смесью TFA (3 мл) и DCM (2 мл) и перемешивали в течение 1 часа. Затем растворитель удаляли в вакууме и неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 429 (МН)+.
Пример 53: (R)-3-(2-(транс-4-((S)-2-аминопропиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
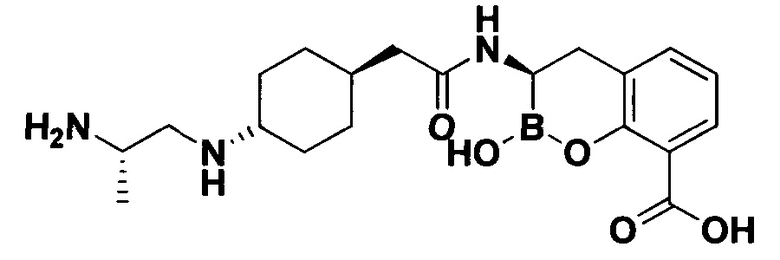
Стадия 1: Синтез 3-((2R)-2-(2-(транс-4-аминоциклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)этил)-2-метоксибензойной кислоты.
[00336] К трет-бутил-3-((2R)-2-(2-(tran-4-(трет-бутоксикарбониламино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоату со стадии 1 примера 6 (640 мг) добавляли 4 N HCl в диоксане (4 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли диэтиловый эфир для осаждения продукта в виде твердого вещества (500 мг), которое использовали непосредственно на следующей стадии.
Стадия 2: Синтез 3-((2R)-2-(2-(транс-4-((S)-2-(трет-бутоксикарбониламино) пропиламино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты.
[00337] Соединение получали из 3-((2R)-2-(2-(транс-4-амино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты и (S)-трет-бутил-1-оксопропан-2-илкарбамата в соответствии с процедурой, описанной в примере 27.
Стадия 3: Синтез (R)-3-(2-(транс-4-((S)-2-аминопропиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00338] К 3-((2R)-2-(2-(транс-4-((S)-2-(трет-бутоксикарбониламино)пропиламино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоте со стадии 2, в колбе, добавляли 3N водн. HCl, и реакционную смесь перемешивали при кипячении с обратным холодильником в течение 1 часа. Полученный продукт очищали с помощью ВЭЖХ с обращенной фазой и высушивали с помощью лиофилизации. ESI-MC m/z 409 (МН)+.
Пример 54: (R)-2-гидрокси-3-(2-(транс-4-(2-(метоксикарбониламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновая кислота
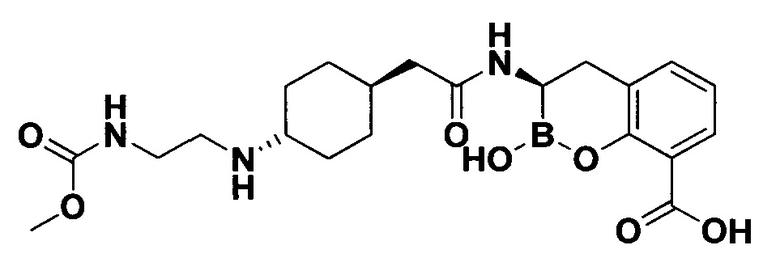
Синтез (R)-2-гидрокси-3-(2-(транс-4-(2-(метоксикарбониламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00339] К (R)-2-гидрокси-3-(2-(транс-4-(2-(метиламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоте из примера 51 (39 мг) в смеси H2O (1 мл), THF (1 мл) и МеОН (1 мл) добавляли NaHCO3 (200 мг), с последующим добавлением метилхлорформиата (1,2 экв). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем растворитель удаляли в вакууме и неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 448 (МН)+.
Пример 55: (R)-3-(2-(3-(2-аминоэтиламино)циклобутил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
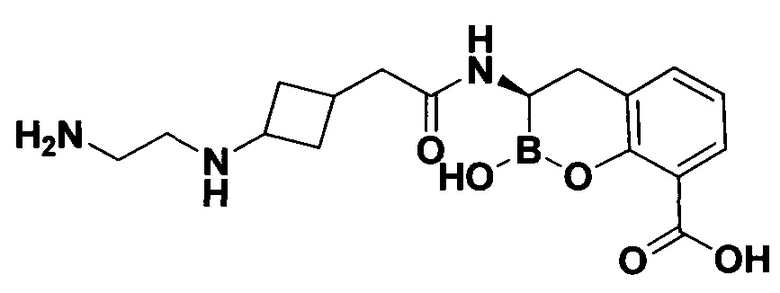
Стадия 1: Синтез метил-2-(3-(2-(трет-бутоксикарбониламино)этиламино)циклобутил)ацетата.
[00340] К метил-2-(3-охоциклобутил)ацетату (284 мг) и трет-бутил-2-аминоэтилкарбамату (336 мг) в колбе добавляли МеОН (10 мл) и Pd/C (10%, 50 мг). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. В конце реакции катализатор отфильтровывали через целит и растворитель удаляли при пониженном давлении. Неочищенный продукт (577 мг) использовали на следующей стадии без дополнительной очистки.
Стадия 2: Синтез метил-2-(3-(трет-бутоксикарбонил(2-(трет-бутоксикарбониламино)этил)амино)циклобутил)ацетата.
[00341] К продукту со стадии 1 в DCM (15 мл) добавляли TEA (0,35 мл) и ди-трет-бутилдикарбонат (480 мг). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Органическую фазу промывали 1N HCl, водой и насыщенным солевым раствором, высушивали над сульфатом натрия. Удалением растворителей при пониженном давлении получали продукт (1,0 г) без дополнительной характеристики.
Стадия 3: Синтез 2-(3-(трет-бутоксикарбонил(2-(трет-бутоксикарбониламино)этил)амино)циклобутил)уксусной кислоты.
[00342] К метил-2-(3-(трет-бутоксикарбонил (2-(трет-бутоксикарбониламино)этил)амино)циклобутил)ацетату, полученному на стадии 2 в смеси МеОН и Н2О добавляли 1N NaOH (8 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. Половину растворителей удаляли при пониженном давлении и добавляли 1N HCl для доведения значения рН раствора до 4. Водную фазу экстрагировали этилацетатом три раза. Объединенную органическую фазу высушивали и концентрировали в вакууме с получением кислоты (0,9 г).
Стадия 4: Синтез трет-бутил-3-((R)-2-(2-(3-(трет-бутоксикарбонил(2-(трет-бутоксикарбониламино)этил)амино)циклобутил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата.
[00343] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и 2-(3-(трет-бутоксикарбонил(2-(трет-бутоксикарбониламино)этил)амино)циклобутил)уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (гексан/этилацетат, от 2:1 до 1:2). ESI-MS m/z 784,1 (MH)+.
Стадия 5: Синтез (R)-3-(2-(3-(2-аминоэтиламино)циклобутил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00344] Соединение получали из трет-бутил-3-((R)-2-(2-(3-(трет-бутоксикарбонил(2-(трет-бутоксикарбониламино)этил)амино)циклобутил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 362 (МН)+.
Пример 56: (R)-3-(2-(3-аминоциклобутил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
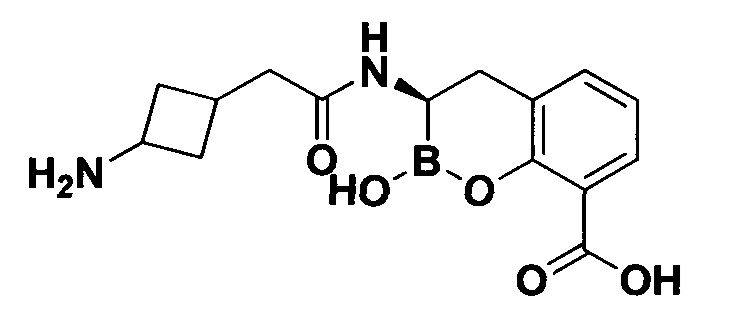
Стадия 1: Синтез метил-2-(3-(2,4-диметоксибензиламино)циклобутил)ацетата.
[00345] К метил-2-(3-охоциклобутил)ацетату (426 мг) и (2,4-диметоксифенил)метанамину (501 мг) в колбе добавляли МеОН (20 мл) и Pd/C (10%, 100 мг). Реакционную смесь перемешивали в атмосфере водорода в течение ночи. Катализатор удаляли фильтрованием через слой целита. После удаления растворителя получали неочищенный продукт (850 мг), который использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2: Синтез метил-2-(3-(трет-бутоксикарбонил (2,4-диметоксибензил)амино)циклобутил)ацетата.
[00346] К продукту со стадии 1 в DCM (20 мл) добавляли TEA (0,56 мл) и ди-трет-бутилдикарбонат (900 мг). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Органическую фазу промывали 1N HCl, водой и насыщенным солевым раствором, высушивали над безводным сульфатом натрия. Удаление растворителей при пониженном давлении давало продукт, который очищали флэш-хроматографией (0,75 г).
Стадия 3: Синтез 2-(3-(трет-бутоксикарбонил(2,4-диметоксибензил)амино)циклобутил)уксусной кислоты.
[00347] К метил-2-(3-(трет-бутоксикарбонил(2,4-диметоксибензил)амино)циклобутил)ацетату, полученному на стадии 2, в смеси МеОН, THF и Н2О добавляли 1N NaOH (10 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Половину растворителей удаляли при пониженном давлении и добавляли 1N HCl для доведения значения рН раствора до 4. Водную фазу экстрагировали этилацетатом три раза. Объединенную органическую фазу высушивали и концентрировали в вакууме с получением кислоты (0,67 г).
Стадия 4: Синтез трет-бутил-3-((2R)-2-(2-(3-(трет-бутоксикарбониламино)циклобутил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата.
[00348] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6] дец-4-илметил)бензойной кислоты и 2-(3-(трет-бутоксикарбонил(2,4-диметоксибензил)амино)циклобутил)уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (гексан/EtOAc, от 2:1 до 1:2). ESI-MS m/z 641,1 (МН)+.
Стадия 5. Синтез (R)-3-(2-(3-(2-аминоэтиламино)циклобутил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00349] Соединение получали из трет-бутил-3-((2R)-2-(2-(3-(трет-бутоксикарбониламино)циклобутил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 319 (МН)+.
Пример 57: (R)-3-(2-(3-(3-(2-аминоэтил)-1-(2-(3-(2-аминоэтил)уреидо)этил)уреидо)циклобутил)ацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновая кислота
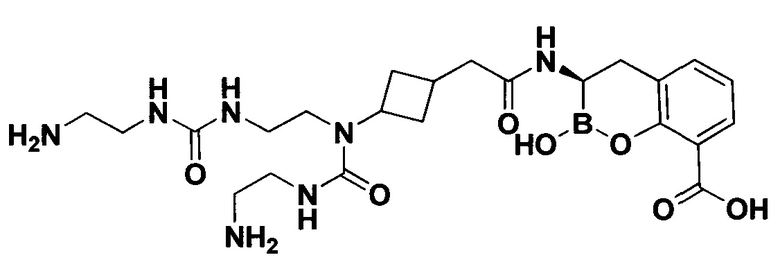
Стадия 1: Синтез 3-((2R)-2-(2-(3-(2-аминоэтиламино)циклобутил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты.
[00350] Соединение получали из трет-бутил-3-((R)-2-(2-(3-(трет-бутоксикарбонил(2-(трет-бутоксикарбониламино)этил)амино)циклобутил)ацетамидо)-2(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата (Стадия 4 примера 55) и 4N HCl в соответствии с процедурой, описанной на стадии 1 примера 53.
Стадия 2: Синтез (R)-3-(2-(3-(3-(2-аминоэтил)-1-(2-(3-(2-аминоэтил)уреидо)этил) уреидо)циклобутил)ацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00351] К 1,1'-карбонилдиимидазолу (180 мг) в DCM (5 мл) добавляли трет-бутил-2-аминоэтилкарбамат (160 мг). Полученную реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Часть данного раствора (1 мл) затем добавляли к 3-((2R)-2-(2-(3-(2-аминоэтиламино)циклобутил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоте со стадии 1 (20 мг) в DMF и TEA (2 экв) в другой колбе. Реакционную смесь перемешивали в течение ночи. Добавляли воду и экстрагировали этилацетатом. Объединенные органические фазы высушивали и концентрировали в вакууме с получением продукта, который использовали непосредственно на следующей стадии.
Стадия 3: Синтез (R)-3-(2-(3-(3-(2-аминоэтил)-1-(2-(3-(2-аминоэтил)уреидо)этил) уреидо)циклобутил)ацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00352] Соединение получали из (R)-3-(2-(3-(3-(2-аминоэтил)-1-(2-(3-(2-аминоэтил)уреидо)этил)уреидо)циклобутил)ацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 534 (МН)+.
Пример 58: (R)-3-(2-(3-(3-(2-аминоэтил)уреидо)циклобутил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
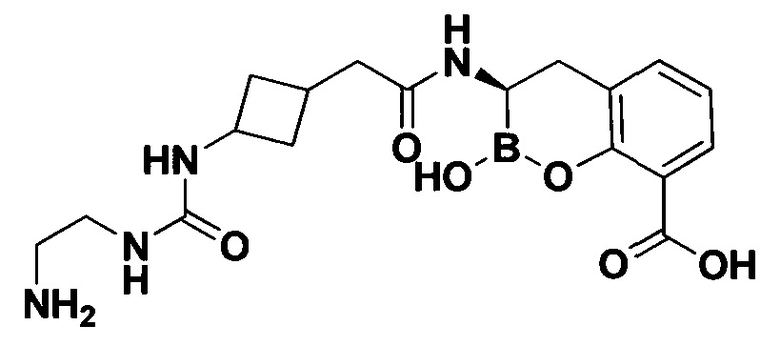
Синтез (R)-3-(2-(3-(3-(2-аминоэтил)уреидо)циклобутил)ацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00353] Соединение получали из трет-бутил-3-((2R)-2-(2-(3-(трет-бутоксикарбониламино)циклобутил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата (Стадия 4 примера 56) в соответствии с процедурой, описанной в примере 57. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 405 (МН)+.
Пример 59: (R)-3-(2-(транс-4-(3-(2-аминоэтил)уреидо)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
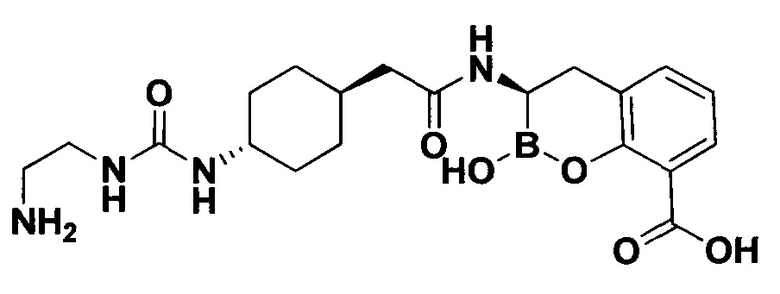
Синтез (R)-3-(2-(транс-4-(3-(2-аминоэтил)уреидо)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00354] Соединение получали из 3-((2R)-2-(2-(транс-4-аминоциклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)этил)-2-метоксибензойной кислоты (Стадия 1 примера 53) в соответствии с процедурой, описанной в примере 57. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 433 (МН)+.
Пример 60: (R)-3-(2-(транс-4-(3-аминопропиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
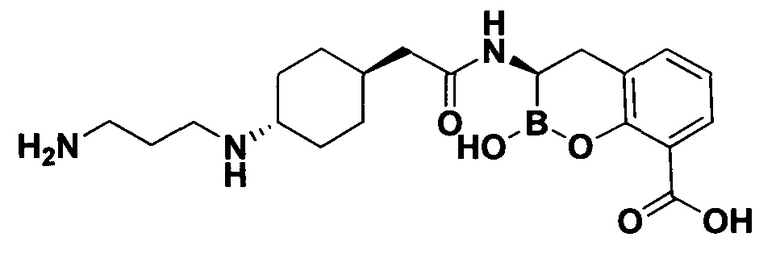
Стадия 1: Синтез этил-2-[4-[3-(трет-бутоксикарбониламино)пропиламино]циклогексил]ацетата.
[00355] Указанное в заголовке соединение (выделено в виде смеси транс : цис-изомеров с соотношением 4:1) получали с использованием по существу той же процедуры, которая описана в примере 48, стадия 1, за исключением использования трет-бутил-N-(3-аминопропил)карбамата вместо 2-(N-[2-(амино)этил]амино)пиридина.
Стадия 2: Синтез этил-2-[4-[трет-бутоксикарбонил[3-(трет-бутоксикарбониламино) пропил]амино]циклогексил]ацетата.
[00356] Указанное в заголовке соединение (выделено в виде смеси транс : цис-изомеров с соотношением 4:1) получали с использованием по существу той же процедуры, которая описана в примере 48, стадия 2, за исключением использования трет-бутил-этил-2-[4-[3-(трет-бутоксикарбониламино)пропиламино]циклогексил]ацетата вместо этил-2-[4-[2-(2-пиридиламино)этиламино]циклогексил]ацетата.
Стадия 3. Синтез транс-2-[4-[трет-бутоксикарбонил[3-(трет-бутоксикарбониламино)пропил]амино]циклогексил]-уксусной кислоты.
[00357] К раствору этил-2-[4-[трет-бутоксикарбонил[3-(трет-бутоксикарбониламино) пропил]амино]циклогексил]ацетата (1,53 г, 3,47 ммоль) в метаноле (3 мл) и THF (3 мл) добавляли раствор NaOH (7,5 мл, 1М водный). Полученный раствор перемешивали в течение 2,75 часа подкисленным с HCl (8 мл, 1 М водным раствором). Данную смесь экстрагировали этилацетатом, промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток растворяли в эфире (5 мл). К данному раствору добавляли (-) α-метил-бензиламин (428 μл, 347 ммоль) и полученный раствор выдерживали в течение ночи. Кристаллическую массу отфильтровывали, промывали эфиром, и собранное твердое вещество перекристаллизовывали из смеси изопропанол/эфира с получением 1,1 г твердого продукта. Данное вещество суспендировали в этилацетате, промывали 1 М HCl водной, затем насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали с получением указанного в заголовке соединения.
Стадия 4: Синтез (+) пинандиолового сложного эфира [1-[[2-[4-[трет-бутоксикарбонил-[3-(трет-бутоксикарбониламино)пропил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
[00358] Указанное в заголовке соединение получали с использованием по существу той же процедуры, описанной в примере 50; стадия 9, за исключением использования транс-2-[4-[трет-бутоксикарбонил[3-(трет-бутоксикарбониламино)пропил]амино]циклогексил]-уксусной кислоты вместо 2-[4-транс-[трет-бутоксикарбонил-[2-(трет-бутоксикарбониламино)этил]амино]циклогексил]-уксусной кислоты.
Стадия 5: Синтез (R)-3-(2-(транс-4-(3-аминопропиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00359] Указанное в заголовке соединение получали с использованием по существу той же процедуры, описанной в примере 49; стадия 7, за исключением использования (+) пинандиолового сложного эфира [1-[[2-[4-[трет-бутоксикарбонил-[3-(трет-бутоксикарбониламино)пропил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты вместо (+) пинандиолового сложного эфира [(1R)-1-[[2-[4-[трет-бутоксикарбонил-[2-(метансульфонамидо)этил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
Пример 61: (R)-2-гидрокси-3-(2-(транс-4-(2-(2-гидроксиэтиламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновая кислота
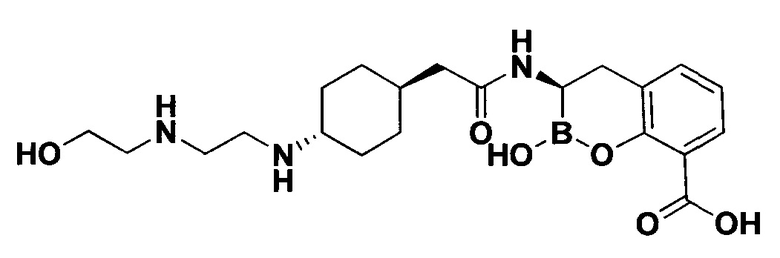
Стадия 1: Синтез (R)-3-(2-(транс-4-(2-(2-(трет-бутилдиметилсилилокси)этиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00360] К (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоте из примера 15 (92 мг) в МеОН (2 мл) добавляли TEA (70 μл), уксусную кислоту (30 μл), 2-(трет-бутилдиметилсилилокси)ацетальдегид (35 мг) и триацетоксиборгидрид натрия (212 мг). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли при пониженном давлении, и продукт использовали на следующей стадии без дополнительной очистки.
Стадия 2: Синтез (R)-2-гидрокси-3-(2-(транс-4-(2-(2-гидроксиэтиламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00361] К соединению со стадии 1 добавляли смесь TFA (2 мл) и Н2О (0,2 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем растворители удаляли в вакууме и остаток очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 434 (МН)+.
Пример 62: (R)-3-(2-(транс-4-(2-((S)-2-аминопропиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
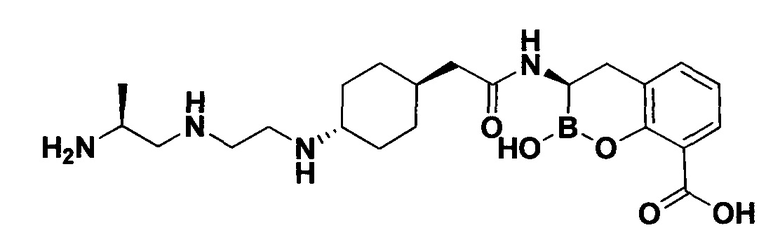
Стадия 1: Синтез (R)-3-(2-(транс-4-(2-((S)-2-(трет-бутоксикарбониламино)пропиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00362] К (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновой кислоте из примера 15 (92 мг) в МеОН (2 мл) добавляли TEA (70 μл), уксусную кислоту (30 μл), (S)-трет-бутил-1-оксопропан-2-илкарбамат (86 мг) и триацетоксиборгидрид натрия (212 мг). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли, и продукт использовали на следующей стадии без дополнительной очистки.
Стадия 2: Синтез (R)-3-(2-(транс-4-(2-((S)-2-аминопропиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00363] К соединению со стадии 1 добавляли 3N HCl (2 мл) и полученную реакционную смесь нагревали с обратным холодильником в течение 1 часа. Затем растворители удаляли в вакууме, и остаток очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 447 (МН)+.
Пример 63: (R)-3-(2-(транс-4-((2-аминоэтил)(метил)амино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
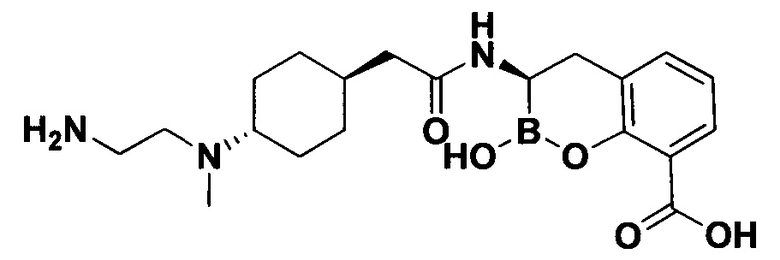
Стадия 1: Синтез этилового эфира 2-[4-[2-(трет-бутоксикарбониламино)этиламино]циклогексил]ацетата.
[00364] Указанное в заголовке соединение (выделено в виде смеси транс : цис-изомеров с соотношением 6,5:1) получали с использованием по существу той же процедуры, которая описана в примере 48, стадия 1, за исключением использования трет-бутил-N-(3-аминоэтил)карбамата вместо 2-(N-[2-(амино)этил]амино)-пиридина.
Стадия 2: Синтез этил-2-[4-[2-(трет-бутоксикарбониламино)этил-метил-амино]циклогексил]ацетата.
[00365] К раствору этил-2-[4-[2-(трет-бутоксикарбониламино)этиламино]циклогексил]ацетата (326 мг, 1 ммоль) в дихлорметане (4 мл) добавляли формалин (97 μл, 1,2 ммоль) затем уксусную кислоту (60 μл, 1 ммоль) и триацетоксиборгидрид натрия (255 мг, 1,2 ммоль). Полученный непрозрачный раствор перемешивали в течение 19 часов. К данной смеси добавляли карбонат натрия (2 мл, насыщенный водный раствор). Смесь разбавляли этилацетатом и разделяли. Органический экстракт промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (10 г диоксида кремния, элюировали смесью от 2 до 20% метанола в дихлорметане) с получением указанного в заголовке соединения.
Стадия 3: Синтез 2-[4-[2-(трет-бутоксикарбониламино)этил-метил-амино]циклогексил]-уксусной кислоты.
[00366] К раствору этил-2-[4-[2-(трет-бутоксикарбониламино)этил-метил-амино]циклогексил]ацетата (231 мг, 0,67 ммоль) в метаноле (2 мл) и THF (2 мл) добавляли NaOH (1 мл, 2М водный раствор). Полученный раствор перемешивали в течение 2,75 часа, затем концентрировали при пониженном давлении до около ¼ от первоначального объема. Остаток подкисляли HCl (3 мл, 1 М водного раствора). Данный раствор очищали с помощью обращенно-фазовой хроматографии на силикагеле (30 г диоксида кремния C18, элюировали смесью 5-100% ацетонитрил/вода/0,1% TFA), получая указанное в заголовке соединение.
Стадия 4: Синтез (+) пинандиолового сложного эфира [1-[[2-[4-[2-(трет-бутоксикарбониламино)этил-метил-амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
[00367] Указанное в заголовке соединение получали с использованием по существу той же процедуры, описанной в примере 50; стадия 9, за исключением использования 2-[4-[2-(трет-бутоксикарбониламино)этил-метил-амино]циклогексил] -уксусной кислоты вместо 2-[4-транс-[трет-бутоксикарбонил-2-[(трет-бутоксикарбониламино)этил]амино]циклогексил]-уксусной кислоты.
Стадия 6: Синтез (R)-3-(2-(транс-4-((2-аминоэтил)(метил)амино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00368] Указанное в заголовке соединение получали с использованием по существу той же процедуры, описанной в примере 49; стадия 7, за исключением использования (+) пинандиолового сложного эфира [1-[[2-[4-[2-(трет-бутоксикарбониламино)этил-метил-амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты вместо (+) пинандиолового сложного эфира [(1R)-1-[[2-[4-[трет-бутоксикарбонил-[2-(метансульфонамидо)этил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
Пример 64: (R)-2-гидрокси-3-(3-гидрокси-2-(транс-4-(2-(метиламино)этиламино)циклогексил)пропанамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
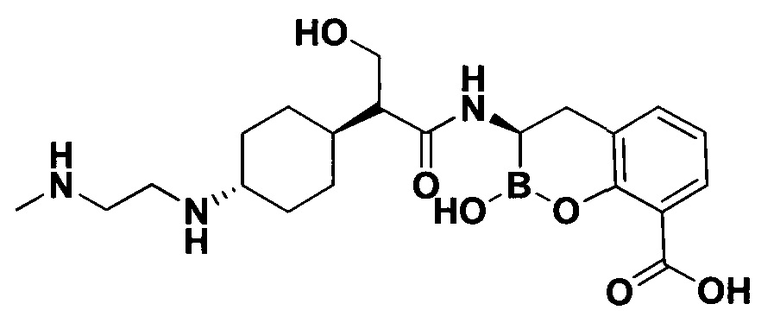
Стадия 1: Синтез трет-бутил-3-((2R)-2-(2-(транс-4-(трет-бутоксикарбонил(2-(трет-бутоксикарбонил(метил)амино)этил)амино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-метоксибензоата.
[00369] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и 2-(транс-4-(трет-бутоксикарбонил(2-(трет-бутоксикарбонил(метил)амино)этил)амино)циклогексил)уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1. Неочищенный продукт очищали флэш-хроматографией на силикагеле (гексан/EtOAc, от 2:1 до 1:2).
Стадия 2. Синтез трет-бутил-3-((2R)-2-(2-(транс-4-(трет-бутоксикарбонил(2-(трет-бутоксикарбонил(метил)амино)этил)амино)циклогексил)-3-гидроксипропанамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата.
[00370] К трет-бутил-3-((2R)-2-(2-(транс-4-(трет-бутоксикарбонил(2-(трет-бутоксикарбонил(метил)амино)этил)амино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоату (110 мг) в THF (5 мл) добавляли LDA (2 М в бензоле, 160 μл) при -76°С. Реакционную смесь перемешивали при той же температуре в течение 30 мин, после чего добавляли формальдегид (20 мг). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 4 ч. Добавляли навыщенный солевой раствор и экстрагировали этилацетатом. Органическую фазу высушивали и концентрировали с получением неочищенного продукта, который использовали на следующей стадии без дополнительной очистки.
Стадия 3: Синтез (R)-2-гидрокси-3-(3-гидрокси-2-(транс-4-(2-(метиламино)этиламино)циклогексил)пропанамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00371] Соединение получали из трет-бутил-3-((2R)-2-(2-(транс-4-(трет-бутоксикарбонил(2-(трет-бутоксикарбонил(метил)амино)этил)амино)циклогексил)-3-гидроксипропанамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата и BCl3 в соответствии с процедурой, описанной на стадии 2 примера 1. Неочищенный продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 434 (МН)+.
Пример 65: (R)-3-(2-(транс-4-((R)-2-амино-3-гидроксипропиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
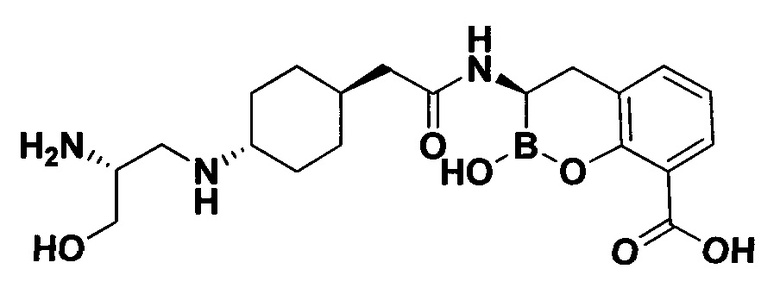
Стадия 1: Синтез бензил 2-[транс-4-(амино)циклогексил]ацетата.
[00372] К раствору 2-[4-(трет-бутоксикарбониламино)циклогексил]-уксусной кислоты (1,0 г, 4 ммоль) в DMF (10 мл) добавляли K2CO3 (600 мг, 4 ммоль) с последующим добавлением бензилбромида (0,5 мл, 4,2 ммоль). Полученную смесь перемешивали в течение 16,75 часов, разбавляли эфиром, промывали водой (2×), затем насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток растворяли в дихлорметане (12 мл). К данному раствору добавляли TFA (3 мл). Полученный раствор перемешивали в течение 2,5 часов, затем концентрировали при пониженном давлении. Остаток растворяли в этилацетате, промывали насыщенным раствором бикарбоната натрия, затем насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 2: Синтез трет-бутил-(3R-)-4-[[[4-(2-бензилокси-2-оксо-этил)циклогексил]-трет-бутоксикарбонил-амино]метил]-2,2-диметил-оксазолидин-3-карбоксилата.
[00373] К раствору трет-бутил-(3R)-4-формил-2,2-диметил-оксазолидин-3-карбоксилата (460 мг, 2 ммоль) в дихлорметане (6 мл) добавляли бензил-2-[транс-4-(амино)циклогексил]ацетата (494 мг, 2 ммоль) с последующим добавлением триацетоксиборгидрида натрия (636 мг, 3 ммоль) и уксусной кислоты (120 μл, 2 ммоль). Полученный раствор перемешивали в течение 16,75 часов. К раствору добавляли карбонат натрия (насыщенный водный раствор). Смесь разбавляли этилацетатом, промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток растворяли в дихлорметане (6 мл). К данному раствору добавили ди-трет-бутилдикарбонат (654 мг, 3 ммоль) с последующим добавлением триэтиламина (460 μL, 3,3 ммоль). Данный раствор перемешивали в течение 2 часов, затем разбавляли эфиром, промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (25 г диоксида кремния; элюировали с от 10 до 40% этилацетата в гексане) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 3: Синтез 2-[4-[трет-бутоксикарбонил-[[(3R)-3-трет-бутоксикарбонил-2,2-диметил-оксазолидин-4-ил]метил]амино]циклогексил]уксусной кислоты.
[00374] К раствору трет-бутил-(3R)-4-[[[4-(2-бензилокси-2-охо-этил)циклогексил]-трет-бутоксикарбонил-амино]метил]-2,2-диметил-оксазолидин-3-карбоксилата (520 мг, 0,928 ммоль) в этилацетате (4 мл) добавляли палладий на угле (56 мг, 10%-ный палладий на сухом порошке углерода). Смесь дегазировали, продували газообразным водородом и перемешивали при данной атмосфере в течение 20 минут. Систему снова дегазировали, затем продували аргоном. Данную смесь разбавляли дихлорметаном, фильтровали через целит и концентрировали при пониженном давлении. Остаток растирали в порошок с гексаном с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 4: Синтез (+) пинандиолового сложного эфира [1-[[2-[4-[трет-бутоксикарбонил-[[(3R)-3-трет-бутоксикарбонил-2,2-диметил-оксазолидин-4-ил]метил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
[00375] Указанное в заголовке соединение получали с использованием по существу той же процедуры, описанной в примере 50; стадия 9, за исключением использования 2-[4-[трет-бутоксикарбонил-[[(3R)-3-трет-бутоксикарбонил-2,2-диметил-оксазолидин-4-ил]метил]амино]циклогексил]уксусной кислоты вместо 2-[4-транс-[трет-бутоксикарбонил-[2-(трет-бутоксикарбониламино)этил]амино]циклогексил]уксусной кислоты.
Стадия 5: Синтез (R)-3-(2-(транс-4-((R)-2-амино-3-гидроксипропиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00376] Указанное в заголовке соединение получали с использованием по существу той же процедуры, описанной в примере 49; стадия 7, за исключением использования (+) пинандиолового сложного эфира [1-[[2-[4-[трет-бутоксикарбонил-[[(3R)-3-трет-бутоксикарбонил-2,2-диметил-оксазолидин-4-ил]метил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты вместо (+) пинандиолового сложного эфира [(1R)-1-[[2-[4-[трет-бутоксикарбонил-[2-(метансульфонамидо)этил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
Пример 66: (R)-2-гидрокси-3-(2-(транс-4-(2-(метилтио)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
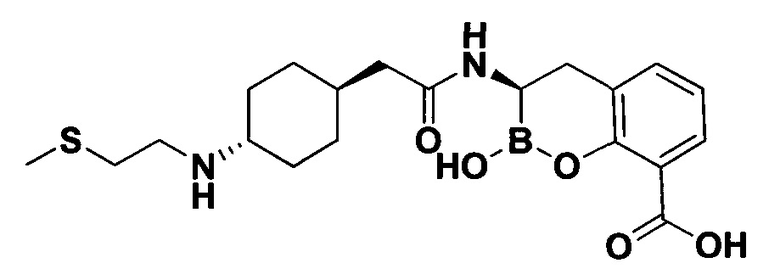
Стадия 1: Синтез этилового эфира 2-[4-(2-метилсульфанилэтиламино)циклогексил]ацетата.
[00377] Указанное в заголовке соединение получали с использованием по существу той же процедуры, которая описана в примере 48, стадия 1, за исключением использования 2-(метилтио)этиламина вместо 2-(N-[2-(амино)этил]амино)-пиридина.
Стадия 2: Синтез этилового эфира 2-[4-[трет-бутоксикарбонил(2-метилсульфанилэтил)амино]циклогексил]ацетата.
[00378] Указанное в заголовке соединение (выделено в виде смеси транс : цис-изомеров с соотношением 6,8:1) получали с использованием по существу той же процедуры, которая описана в примере 48, стадия 2, за исключением использования этил-2-[4-(2-метилсульфанилэтиламино)циклогексил]ацетата вместо этил-2-[4-[2-(2-пиридиламино)этиламино]циклогексил]ацетата.
Стадия 3: Синтез 2-[4-[трет-бутоксикарбонил(2-метилсульфанилэтил)амино]циклогексил]-уксусной кислоты.
[00379] К раствору этил-2-[4-[трет-бутоксикарбонил(2-метилсульфанилэтил)амино]циклогексил]ацетата (2.11 г, 5,89 ммоль) в метаноле (10 мл) и THF (10 мл) добавили NaOH (10 мл, 1 М водный). Полученный раствор перемешивали в течение 3,75 ч, а затем подкисляли HCl (2М, водн., до значения рН 2). Данную смесь экстрагировали этилацетатом, промывали насыщенным солевым раствором, высушивали над сульфатом магния и концентрировали. Остаток очищали хроматографией на силикагеле (25 г диоксида кремния, элюировали смесью от 20 до 60% этилацетата в гексане) с получением указанного в заголовке соединения в виде смеси транс : цис-изомеров с соотношением 6,9:1.
Стадия 4: Синтез (+) пинандиолового сложного эфира [2-(3-трет-бутоксикарбонил-2-метокси-фенил)-1-[[2-[4-[трет-бутоксикарбонил(метилсульфанилметил)амино]циклогексил]ацетил]амино]этил]бороновой кислоты.
[00380] Указанное в заголовке соединение получали с использованием по существу той же процедуры, описанной в примере 50; стадия 9, за исключением использования 2-[4-[трет-бутоксикарбонил(2-метилсульфанилэтил)амино]циклогексил]уксусной кислоты вместо 2-[4-транс-[трет-бутоксикарбонил-[2-(трет-бутоксикарбониламино)этил]амино]циклогексил]уксусной кислоты.
Стадия 5: Синтез (R)-2-гидрокси-3-(2-(транс-4-(2-(метилтио)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00381] Указанное в заголовке соединение получали с использованием по существу той же процедуры, описанной в примере 49; стадия 7, за исключением использования (+) пинандиолового сложного эфира [2-(3-трет-бутоксикарбонил-2-метокси-фенил)-1-[[2-[4-[трет-бутоксикарбонил(метилсульфанилметил)амино]циклогексил]ацетил]амино]этил]бороновой кислоты вместо (+) пинандиолового сложного эфира [(1R)-1-[[2-[4-[трет-бутоксикарбонил-[2-(метансульфонамидо)этил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
Пример 67: (R)-3-(2-(транс-4-((S)-2-амино-3-гидроксипропиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
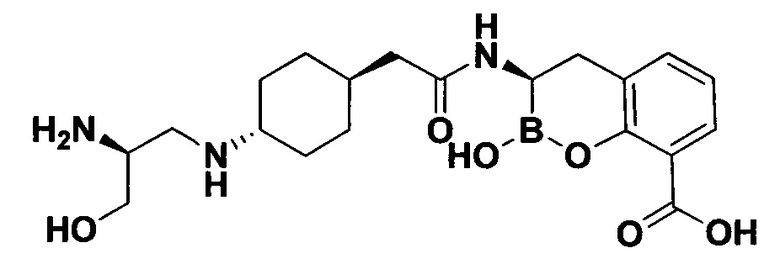
Стадия 1: Синтез трет-бутил-(3S)-4-[[[4-(2-бензилокси-2-охо-этил)циклогексил]-трет-бутоксикарбонил-амино]метил]-2,2-диметил-оксазолидин-3-карбоксилата.
[00382] Указанное в заголовке соединение получали с использованием по существу той же процедуры, которая описана в примере 65; стадия 2, за исключением использования трет-бутил-(3S)-4-формил-2,2-диметил-оксазолидин-3-карбоксилата вместо трет-бутил-(3R)-4-формил-2,2-диметил-оксазолидин-3-карбоксилата.
Стадия 2: Синтез 2-[4-[трет-бутоксикарбонил-[[(3S)-3-трет-бутоксикарбонил-2,2-диметил-оксазолидин-4-ил]метил]амино]циклогексил]уксусной кислоты.
[00383] Указанное в заголовке соединение получали с использованием по существу той же процедуры, которая описана в примере 65; стадия 3, за исключением использования трет-бутил-(3S)-4-[[[4-(2-бензилокси-2-охо-этил)циклогексил]-трет-бутоксикарбонил-амино]метил]-2,2-диметил-оксазолидин-3-карбоксилата вместо трет-бутил-(3R)-4-[[[4-(2-бензилокси-2-охо-этил)циклогексил]-трет-бутоксикарбонил-амино]метил]-2,2-диметил-оксазолидин-3-карбоксилата.
Стадия 3: Синтез (+) пинандиолового сложного эфира [1-[[2-[4-[трет-бутоксикарбонил-[[(3S)-3-трет-бутоксикарбонил-2,2-диметил-оксазолидин-4-ил]метил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
[00384] Указанное в заголовке соединение получали с использованием по существу той же процедуры, описанной в примере 50; стадия 9, за исключением использования 2-[4-[трет-бутоксикарбонил-[[(3S)-3-трет-бутоксикарбонил-2,2-диметил-оксазолидин-4-ил]метил]амино]циклогексил]уксусной кислоты вместо 2-[4-транс-[трет-бутоксикарбонил-[2-(трет-бутоксикарбониламино)этил]амино]циклогексил]уксусной кислоты.
Стадия 4: Синтез (R)-3-(2-(транс-4-((S)-2-амино-3-гидроксипропиламино)циклогексил) ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00385] Указанное в заголовке соединение получали с использованием по существу той же процедуры, описанной в примере 49; стадия 7, за исключением использования (+) пинандиолового сложного эфира [1-[[2-[4-[трет-бутоксикарбонил-[[(3S)-3-трет-бутоксикарбонил-2,2-диметил-оксазолидин-4-ил]метил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты вместо (+) пинандиолового сложного эфира [(1R)-1-[[2-[4-[трет-бутоксикарбонил-[2-(метансульфонамидо)этил]амино]циклогексил]ацетил]амино]-2-(3-трет-бутоксикарбонил-2-метокси-фенил)этил]бороновой кислоты.
Пример 68: (R)-3-(2-(транс-4-(2-аминоацетамидо)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота
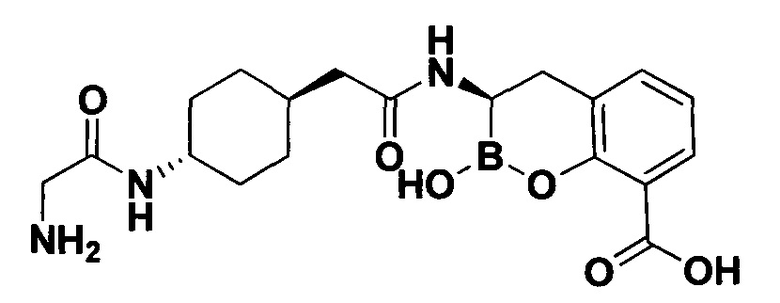
Стадия 1: Синтез 3-((2R)-2-(2-(транс-4-(2-(трет-бутоксикарбониламино)ацетамидо)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты.
[00386] К 2-(трет-бутоксикарбониламино)уксусной кислоте (175 мг) в колбе добавляли HATU (380 мг) и N-метилморфолин (0,56 мл). После перемешивания при комнатной температуре в течение 1 часа, полученный раствор добавляли к 3-((2R)-2-(2-(транс-4-аминоциклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-ил)этил)-2-метоксибензойной кислоте со стадии 1 примера 53 (110 мг) в DMF (3 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. Воду добавляли к реакционной смеси и экстрагировали этилацетатом. Органическую фазу высушивали и концентрировали в вакууме с получением неочищенного продукта, который использовали на следующей стадии без дополнительной очистки.
Стадия 2: Синтез (R)-3-(2-(транс-4-(2-аминоацетамидо)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00387] Соединение получали из 3-((2R)-2-(2-(транс-4-(2-(трет-бутоксикарбониламино) ацетамидо)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензойной кислоты в соответствии с процедурой, описанной на стадии 2 примера 62. Продукт очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 404 (МН)+.
Пример 69: (R)-3-(2-(транс-4-(бис((1Н-имидазол-2-ил)метил)амино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
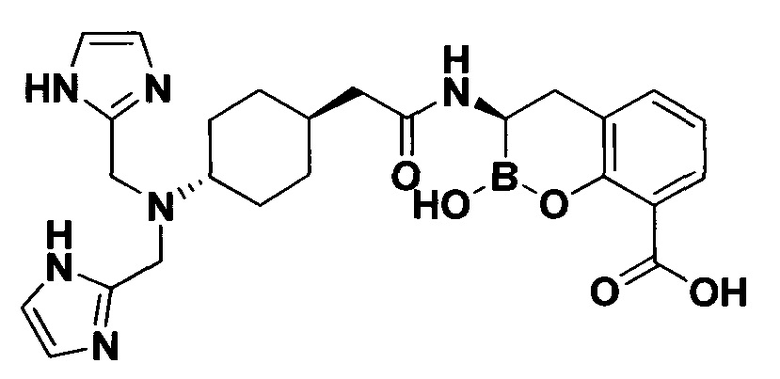
Синтез (R)-3-(2-(транс-4-(бис((1Н-имидазол-2-ил)метил)амино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00388] Соединение получали из (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 6) и 1N-имидазол-2-карбальдегида в соответствии с процедурой, описанной в примере 27. Продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 507 (МН)+.
Пример 70: (R)-3-(2-(транс-4-((1Н-имидазол-5-ил)метиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
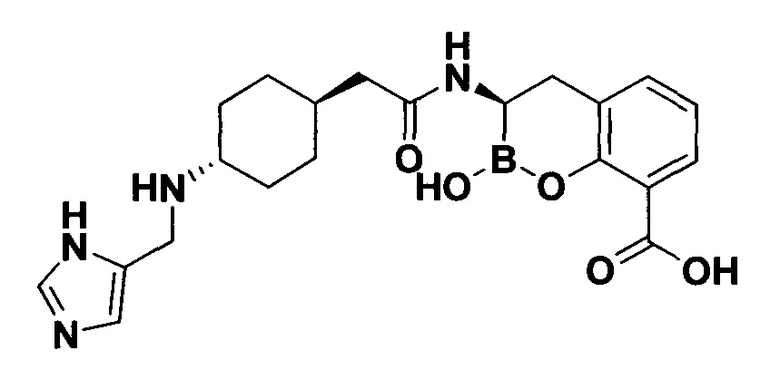
Синтез (R)-3-(2-(транс-4-((1Н-имидазол-5-ил)метиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00389] Соединение получали из (R)-3-(2-(транс-4-аминоциклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты (пример 6) и 1N-имидазол-5-карбальдегида в соответствии с процедурой, описанной в примере 27. Продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 427 (МН)+.
Пример 71: (R)-2-гидрокси-3-(2-(транс-4-(2-(изопропиламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
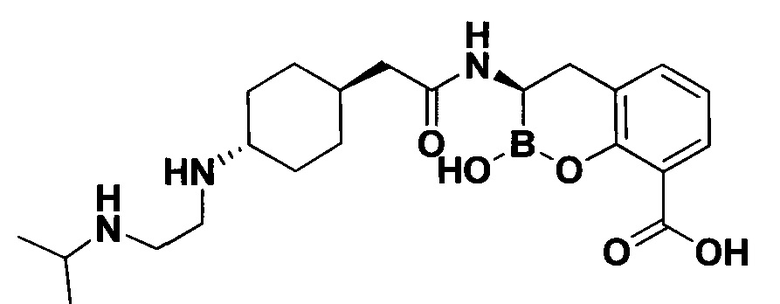
Синтез (R)-2-гидрокси-3-(2-(транс-4-(2-(изопропиламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00390] К (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоте из примера 15 (92 мг) в МеОН (2 мл) добавляли TEA (70 μL), уксусную кислоту (30 μл), ацетон (0,1 мл) и триацетоксиборгидрид натрия (212 мг). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли, и продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 432 (МН)+.
Пример 72: (R)-2-гидрокси-3-(2-транс-4-(2-(пиримидин-2-иламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
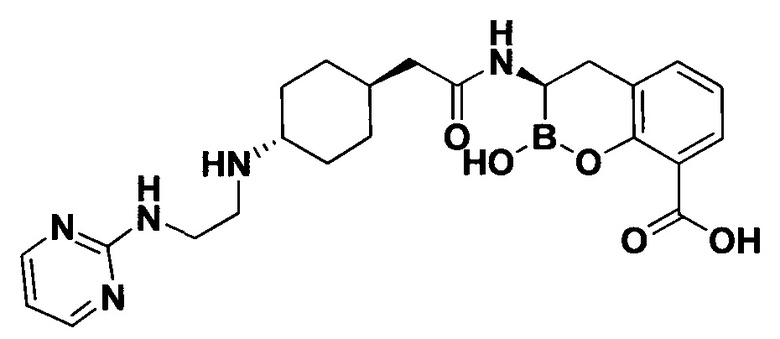
Синтез (R)-2-гидрокси-3-(2-транс-4-(2-(пиримидин-2-иламино)этиламино)циклогексил)ацетамидо)-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00391] К (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоте из примера 15 (46 мг) в МеОН (2 мл) добавляли TEA (70 μл) и 2-хлорпиримидин (25 мг). Реакционную смесь перемешивали при 70°С в течение ночи. Растворитель удаляли, и продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 468 (МН)+.
Пример 73: (R)-3-(2-(транс-4-(2-(циклопентиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
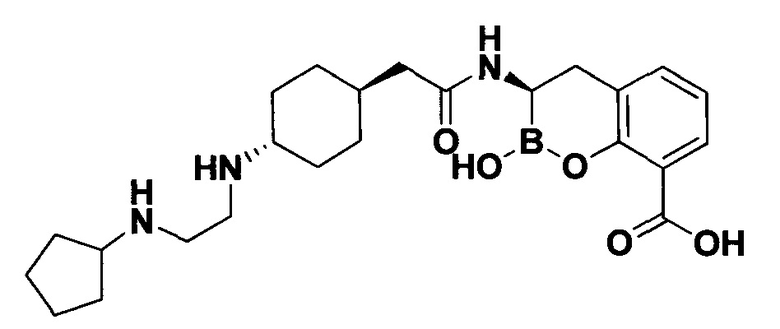
Синтез (R)-3-(2-(транс-4-(2-(циклопентиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00392] Соединение получали из (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты и циклопентанона в соответствии с процедурой, описанной в примере 71. Продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 458 (МН)+.
Пример 74: (R)-3-(2-(транс-4-(2-(циклопропилметиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
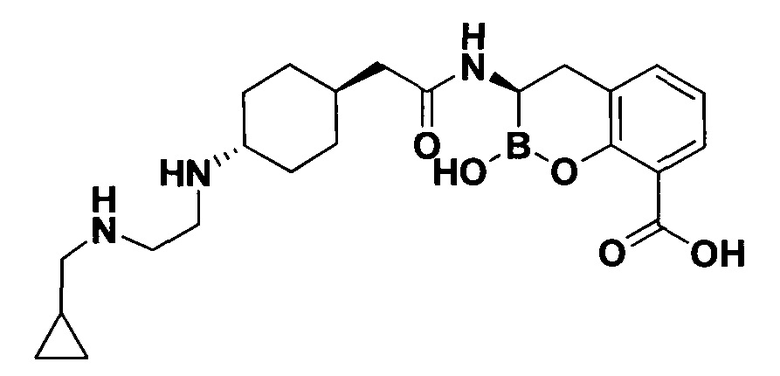
Синтез (R)-3-(2-(транс-4-(2-(циклопропилметиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00393] Соединение получали из (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты и циклопропанкарбальдегида в соответствии с процедурой, описанной в примере 71. Продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 444 (МН)+.
Пример 75: (R)-3-(2-(транс-4-(2-(бис(циклопропилметил)амино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2N-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
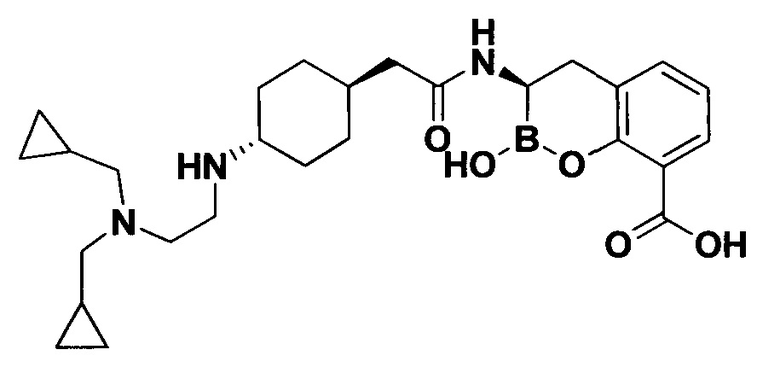
Синтез (R)-3-(2-(транс-4-(2-(бис(циклопропилметил)амино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00394] Соединение получали из (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты и циклопропанкарбальдегида в соответствии с процедурой, описанной в примере 71. Продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 498 (МН)+.
Пример 76: (R)-3-(2-(транс-4-(1,3-диаминопропил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.

Синтез 2-(транс-4-(2,2,12,12-тетраметил-4,10-диоксо-3,11-диокса-5,9-диазатридекан-6-ил)циклогексил)уксусной кислоты.
Стадия 1: Синтез этил-2-(1,4-диоксаспиро[4,5]декан-8-ил)ацетата.
[00395] К охлажденной (0°С) суспензии NaH (60%, 4,4 г, 110 ммоль) в THF (200 мл) добавляли триэтилфосфоноацетат (25,16 г, 110 ммоль) с такой скоростью, чтобы получить слабое выделение газа. После завершения добавления гомогенный раствор перемешивали в течение 30 мин. К данному раствору добавляли 1,4-циклогександионмоноэтиленкеталь (15,62 г, 100 ммоль) в THF (40 мл) в течение 10 мин. После полного добавления ледяную баню удаляли и продолжали перемешивание в течение 3 ч. Реакцию гасили добавлением насыщенного водного раствора NH4Cl, экстрагировали EtOAc, промывали насыщенным солевым раствором, высушивали над Na2SO4. Фильтрованием и выпариванием досуха получали неочищенный продукт, который использовали на следующей стадии без дополнительной очистки.
[00396] Упомянутый выше неочищенный продукт (25,3 г, 100 ммоль) растворяли в МеОН (80 мл) и добавляли 10% Pd/C (1 г). Полученную смесь гидрировали при 35 пси в течение 3 ч. После фильтрации и упаривания остаток очищали колоночной флэш-хроматографией (элюент: от 20% EtOAc в гексане до 30%) и получали продукт в виде бесцветного масла (20 г, 87%).
Стадия 2: Синтез 2-(1,4-диоксаспиро[4,5]декан-8-ил)этанола.
[00397] К раствору этил-2-(1,4-диоксаспиро[4,5]декан-8-ил)ацетата, полученного на стадии 1, (3,31 г, 14,5 ммоль) в Et2O (80 мл) при 0°С в атмосфере N2 добавляли LiAlH4 (1M в THF, 13,66 мл, 13,66 ммоль) в течение 15 мин. Полученную смесь перемешивали при 0°С в течение еще 20 мин и гасили добавлением насыщенного водного раствора NH4Cl, экстрагировали EtOAc, высушивали над Na2SO4. Фильтрование и выпаривание досуха давало спирт в виде белого твердого вещества (2,76 г, 100%). LC/MC: 187,1 (МН)+.
Стадия 3: Синтез 8-(2-(бензилокси)этил)-1,4-диоксаспиро[4,5]декана.
[00398] К предварительно охлажденной (0°С) суспензии NaH (60% в минеральн., 0,44 г, 11 ммоль) в THF (20 мл) добавляли 2-(1,4-диоксаспиро[4,5]декан-8-ил)этанол (1,86 г, 10 ммоль) в THF (10 мл). Полученный раствор перемешивали при 0°С в течение 10 мин и оставляли нагреваться до комнатной температуры и перемешивали в течение 1 часа. К упомянутой выше смеси добавляли бензилбромид (1,78 г, 15 ммоль) и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили добавлением насыщенного водного раствора NH4Cl, экстрагировали EtOAc, высушивали над Na2SO4. Очисткой колоночной флэш-хроматографией (элюент: от 20% EtOAc в гексане до 30%) получали продукт (2,3 г, 83%). LC/MC: 277,1 (МН)+.
Стадия 4: Синтез 4-(2-(бензилокси)этил)циклогексанона.
[00399] К раствору 8-(2-(бензилокси)этил)-1,4-диоксаспиро[4,5]декана (2,30 г, 8,32 ммоль) в ацетонитриле (18 мл) добавляли раствор 6N HCl и полученный раствор перемешивали при комнатной температуре в течение 2 ч. После удаления ацетонитрила выпариванием остаток нейтрализовали твердым NaHCO3, экстрагировали EtOAc, высушивали над Na2SO4. Фильтрование и выпаривание досуха дает кетон в виде белого твердого вещества (1,84 г, 95,5%).
Стадия 5: Синтез ((2-(4-метиленециклогексил)этокси)метил)бензола.
[00400] К охлажденной (-78°С) смеси Ph3PCH2Br (5,42 г, 14,88 ммоль) в THF (25 мл) добавляли KOtBu (1M в THF, 17,05 мл, 17,05 ммоль) в атмосфере N2. Полученную смесь перемешивали при 0°С в течение 1 ч и нагревали до комнатной температуры в течение еще 1,5 ч. Затем реакционную смесь охлаждали до -40°С, добавляли по каплям раствор 4-(2-(бензилокси)этил)циклогексанона (1,8 г, 7,75 ммоль) в THF (15 мл). Затем смесь перемешивали при комнатной температуре в течение ночи и гасили насыщенным солевым раствором, экстрагировали EtOAc, высушивали над Na2SO4. Очистка колоночной флэш-хроматографией (элюент: от 20% EtOAc в гексане до 30%) дает указанный в заголовке продукт (1,77 г, 99%). LC/MC: 231,1 (МН)+.
Стадия 6: Синтез (транс-4-(2-(бензилокси)этил)циклогексил)метанола
[00401] К охлажденному (0°С) раствору ((2-(4-метиленециклогексил)этокси)метил)бензола (9,68 г, 41,66 ммоль) в THF (200 мл) добавляли комплекс B2H6Me2S (2 М в THF, 41,66 мл, 83.32 ммоль) в атмосфере N2. После перемешивания при 0°С в течение 2 ч и при комнатной температуре в течение еще 2 ч, раствор охлаждали до 0°С и добавляли по каплям смесь 3М водного раствора NaOH (34 мл) и 30%-ного раствора перекиси водорода (34 мл). Полученную смесь перемешивали при 0°С в течение 1 ч и при комнатной температуре в течение 1,5 ч. Водная обработка и очистка колоночной флэш-хроматографией (элюент: 40% EtOAc в гексане) дает продукт в виде желтого масла (7,04 г, 68%). LC/MC: 249,1 (МН)+.
Стадия 7: Синтез (транс-4-(2-(бензилокси)этил)циклогексанкарбальдегида.
[00402] Раствор DMSO (2,20 мл, 31 ммоль) в DCM (45 мл) по каплям добавляли к предварительно охлажденному (-78°С) раствору оксалилхлорида (2,9 мл, 33,8 ммоль) в DCM (45 мл) в атмосфере N2. После перемешивания при -78°С в течение 10 мин добавляли по каплям раствор (транс-4-(2-(бензилокси)этил)циклогексил)метанола (7,0 г, 28,18 ммоль) в DCM (45 мл). Полученный раствор перемешивали при -78°С в течение 15 мин, и добавляли TEA (23,6 мл, 169,1 ммоль). Раствор перемешивали при -78°С в течение 15 мин и при комнатной температуре в течение 20 мин. Реакционную смесь разбавляли DCM, промывали раствором насыщенного солевого раствора и 1M HCl, высушивали над Na2SO4. Очисткой колоночной флэш-хроматографией (элюент: 20% EtOAc в гексане) получали целевой продукт альдегид в виде желтого масла (6,86 г, 98,8%).
Стадия 8: Синтез (R,Е)-N-((транс-4-(2-(бензилокси)этил)циклогексил)метилен)-2-метилпропан-2-сульфинамида.
[00403] К раствору (транс-4-(2-(бензилокси)этил)циклогексанкарбальдегида (6,45 г, 26,18 ммоль) и (R)-(+)-трет-бутилсульфинамида (3,49 г, 28,8 ммоль) в THF (66 мл) добавляли этоксид титана (IV) (8,78 мл, 41,89 ммоль) в атмосфере N2. Полученный раствор перемешивали при комнатной температуре в течение 20 ч и гасили добавлением по каплям насыщенного раствора NaHCO3. Смесь энергично перемешивали в течение 30 мин и фильтровали через слой целита, концентрировали. Очисткой колоночной флэш-хроматографией (элюент: от 20% EtOAc в гексане до 30%). Получали упомянутый в заголовке продукт (8,38 г, 92%) LC/MC: 350,1 (МН)+.
Стадия 9: Синтез (R)-N-(1-(транс-4-(2-(бензилокси)этил)циклогексил)бут-3-енил)-2-метилпропан-2-сульфинамида.
[00404] К охлажденному (0°С) раствору (R,Е)-N-((транс-4-(2-(бензилокси)этил)циклогексил)метилен)-2-метилпропан-2-сульфинамида в DCM (65 мл) добавляли хлорид аллилмагния (2М в THF, 5,87 мл, 11,74 ммоль) в атмосфере N2. Полученную смесь перемешивали при 0°С в течение 1 ч и гасили добавлением насыщенного раствора NH4Cl, фракции разделяли. Водную фазу экстрагировали этилацетатом и объединенные органические фазы промывали насыщенным солевым раствором, высушивали над Na2SO4. Очисткой колоночной флэш-хроматографией (элюент: 20% EtOAc в гексане) получали продукт (2,57 г, 100%). LC/MC: 392,1 (МН)+.
Стадия 10: Синтез трет-бутил-1-((транс-4-(2-(бензилокси)этил)циклогексил)бут-3-енилкарбамата
[00405] К перемешиваемому раствору (R)-N-(1-(транс-4-(2-(бензилокси)этил)циклогексил)бут-3-енил)-2-метилпропан-2-сульфинамида (2,56 г, 6,5 ммоль) в МеОН (3,5 мл) добавляли раствор 4М HCl в 1,4-диоксане (3,25 мл, 13 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 30 мин и затем концентрировали. Остаток растворяли в EtOAc, промывали насыщенным солевым раствором, высушивали над Na2SO4. Фильтрованием и выпариванием досуха получали неочищенный продукт.
[00406] К охлажденному (0°С) раствору вышеупомянутого неочищенного продукта в DCM (40 мл) добавляли ди-трет-бутилдикарбонат (1,75 г, 7,8 ммоль) и TEA (3,62 мл, 26 ммоль). Смесь медленно нагревали до RT и перемешивали при комнатной температуре в течение 4 ч. Реакцию гасили добавлением насыщенного раствора NH4Cl, фракции разделяли. Водную фазу экстрагировали DCM и объединенные DCM-экстракты промывали 20%-ной лимонной кислотой, насыщенным солевым раствором, высушивали над Na2SO4. Очистка колоночной флэш-хроматографией (элюент: 20% EtOAc в гексане) дает продукт в виде бесцветного геля (2,78 г, 40%). LC/MC: 388,1 (МН)+.
Стадия 11: Синтез трет-бутил-1-(транс-4-(2-(бензилокси)этил)циклогексил)-3-охопропилкарбамата.
[00407] К раствору трет-бутил-1-((транс-4-(2-(бензилокси)этил)циклогексил)бут-3-енилкарбамата (6,5 ммоль) в 1,4-диоксане (118 мл) и воде (38 мл) добавляли N-метилморфолин-N-оксид (1,52 г, 13 ммоль) и OsO4 (4% масс. в воде, 1,4 мл, 0,23 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 18 ч и добавляли NaIO4 (4,87 г, 22,75 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 ч и гасили добавлением насыщенного водного раствора Na2S2O3. После водной обработки остаток очищали колоночной флэш-хроматографией (элюент : 10% EtOAc в гексане до 30%) с получением альдегида в виде бесцветного масла (2,36 г, 89,7%) LC/MC: 412,1 (MNa)+.
Стадия 12: Синтез трет-бутил-1-(транс-4-(2-гидроксиэтил)циклогексил)-3-охопропилкарбамата.
[00408] К раствору альдегида со стадии выше (2,36 г, 6,06 ммоль) в МеОН (20 мл) добавляли 10% Pd/C (0,2 г). Полученную смесь гидрировали с помощью баллона с Н2 в течение ночи при комнатной температуре. Фильтрование и выпаривание досуха дает продукт в виде бесцветного геля (1,87 г, 100%). LC/MC: 322,1 (MNa)+.
Стадия 13: Синтез 2-(транс-4-(1-(трет-бутоксикарбониламино)-3-оксопропил)циклогексил)этилацетата.
[00409] К раствору трет-бутил-1-(транс-4-(2-гидроксиэтил)циклогексил)-3-охопропилкарбамата (1,88 г, 6,28 ммоль) в DCM (60 мл) добавляли DMAP (кат. количество), с последующим добавлением TEA (2,63 мл, 18,84 ммоль) и уксусного ангидрида (0,89 мл, 9,42 ммоль) при 0°С в атмосфере N2. Смесь перемешивали при комнатной температуре в течение 3 ч и разбавляли DCM, гасили добавлением водного раствора NaHCO3. Органическую фазу отделяли и высушивали над Na2SO4. Очистка колоночной флэш-хроматографией (элюент: 40% EtOAc в гексане) дает продукт в виде бесцветного геля (1,90 г, 88,6%).
Стадия 14: Синтез 2-(транс-4-(1-(трет-бутоксикарбониламино)-3-гидроксипропил)циклогексил)этилацетата.
[00410] К охлажденному (этиленгликоль + сухой лед) раствору 2-(транс-4-(1-(трет-бутоксикарбониламино)-3-оксопропил)циклогексил)этилацетата (1,88 г, 5,5 ммоль) в этаноле (90 мл) добавляли NaBH4 (0,212 г, 5,5 ммоль). Смесь перемешивали при -10 ~ 15°С в течение 10 мин и при 0°С в течение 10 мин, а затем гасили добавлением насыщенного водного раствора NH4Cl (30 мл) и насыщенного раствора соли (30 мл). После удаления летучих компонентов испарением, водный остаток экстрагировали этилацетатом, высушивали над Na2SO4. Очисткой колоночной флэш-хроматографией (элюент: 30% EtOAc в гексане до 40%) получали спирт продукт (1,0 г, 53%). LC/MC: 366,1 (MNa)+.
Стадия 15: Синтез 2-(транс-4-(3-азидо-1-(трет-бутоксикарбониламино)пропил)циклогексил)этилацетата
[00411] К охлажденному (0°С) раствору 2-(транс-4-(1-(трет-бутоксикарбониламино)-3-гидроксипропил)циклогексил)этилацетата (1,0 г, 2,9 ммоль) в DCM (15 мл) добавляли TEA (0,81 мл, 5,82 ммоль) и метансульфонилхлорид (0,34 мл, 4,35 ммоль) в атмосфере N2. Смесь перемешивали при комнатной температуре в течение 3 ч и разбавляли DCM, промывали водным раствором NH4Cl, высушивали над Na2SO4. Очисткой колоночной флэш-хроматографией (элюент: 40% EtOAc в гексане) получали мезилат в виде желтого масла (0,96 г, 79%). LC/MC: 444,1 (MNa)+. Смесь мезилата (0,95 г, 2,25 ммоль) и NaN3 (1,17 г, 18 ммоль) в DMF (25 мл) нагревали при 80°С в течение ночи. После водной обработки неочищенный продукт очищали колоночной флэш-хроматографией (элюент: 30% EtOAc в гексане) с получением азида в виде желтого масла (0,74 г, 89%). LC/MC: 391,1 (MNa)+.
Стадия 16: Синтез 2-(транс-4-(3-амино-1-(трет-бутоксикарбониламино)пропил)циклогексил)этилацетата.
[00412] Смесь азида со стадии 15 (0,72 г, 1,95 ммоль) и 10% Pd/C (0,1 г) в МеОН (20 мл) гидрировали с помощью баллона с Н2 при комнатной температуре в течение 18 ч. Фильтрование и выпаривание досуха дает амин с количественным выходом. LC/MC: 343,1 (МН)+.
Стадия 17: Синтез 2-(транс-4-(2,2,12,12-тетраметил-4,10-диоксо-3,11-диокса-5,9-диазатридекан-6-ил)циклогексил)этилацетата.
[00413] К охлажденному (0°С) раствору 2-(транс-4-(3-амино-1-(трет-бутоксикарбониламино)пропил)циклогексил)этилацетата (1,95 ммоль) в DCM (15 мл) добавляли ди-трет-бутилдикарбонат (0,53 г, 2,34 ммоль) и TEA (1,09 мл, 7,8 ммоль). Смесь медленно нагревали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. После гашения водным раствором NH4Cl реакционную смесь экстрагировали DCM, промывали с помощью 0,5 н водного раствора HCl, насыщенным солевым раствором и высушивали над Na2SO4. Фильтрованием и выпариванием досуха получали неочищенный продукт. LC/MC: 465,1 (MNa)+.
Стадия 18: Синтез трет-бутил-1-(4-(2-гидроксиэтил)циклогексил)пропан-1,3-диилдикарбамата.
[00414] К раствору 2-(транс-4-(2,2,12,12-тетраметил-4,10-диоксо-3,11-диокса-5,9-диазатридекан-6-ил)циклогексил)этилацетата (1,95 ммоль) в МеОН (5 мл) добавляли K2CO3 (0,135 г, 0,975 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 3 ч. Реакцию гасили добавлением водного раствора NH4Cl. После удаления метанола выпариванием остаток экстрагировали с помощью EtOAc, высушивали над Na2SO4. Фильтрование и выпаривание досуха давало продукт в виде белой пены (0,71 г, 91%). LC/MC: 423,1 (MNa)+.
Стадия 19: Синтез 2-(транс-4-(2,2,12,12-тетраметил-4,10-диоксо-3,11-диокса-5,9-диазатридекан-6-ил)циклогексил)уксусной кислоты.
[00415] Смесь трет-бутил-1-(4-(2-гидроксиэтил)циклогексил)пропан-1,3-диилдикарбамата (1,95 ммоль), гидрата хлорида рутения (III) (0,008 г, 0,039 ммоль), периодата натрия (1,67 г, 2,8 ммоль) в четыреххлористом углероде (10 мл), ацетонитриле (10 мл) и воде (10 мл) перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь охлаждали до 0°С и добавляли 0,5 HCl (10 мл), экстрагировали DCM, высушивали над Na2SO4. Очистка колоночной флэш-хроматографией (элюент: 40% EtOAc в гексане) давала указанную в заглавии кислоту в виде белой пены (0,67 г, 83%). LC/MC: 437,1 (MNa)+.
Синтез (R)-3-(2-(транс-4-(1,3-диаминопропил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00416] К охлажденному (-25°С) раствору (+) пинандиолового сложного эфира [(1S)-2-(3-трет-бутоксикарбонил-2-метокси-фенил)-1-хлорэтил]бороновой кислоты (пример 48, стадия 1, 0,41 г, 0,92 ммоль) в THF (2 мл) добавляли LHMDS (1 мл, 1М в THF) в атмосфере N2. После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч. Между тем, в отдельную колбу, к смеси 2-(транс-4-(2,2,12,12-тетраметил-4,10-диоксо-3,11-диокса-5,9-диазатридекан-6-ил)циклогексил)уксусной кислоты (0,38 г, 0,92 ммоль) и HATU (0,38 г, 1 ммоль) добавляли DMA (2 мл) и 4-метилморфолин (0,11 мл) и полученную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 1,5 ч. Через 1,5 ч два раствора смешивали и перемешивали при комнатной температуре в течение ночи. После водной обработки остаток очищали FC-хроматографией (элюент: 30% EtOAc в гексане до 40%, до 50%) с получением продукта (0,25 г, 33%). LC/MC: 848,2 (MNa)+.
[00417] К раствору указанного выше продукта (0,22 г, 0,266 ммоль) в 1,4-диоксане (0,6 мл) добавляли 3N водного раствора HCl (3 мл). Полученную смесь нагревали при 100°С в течение 3 ч. После охлаждения до комнатной температуры остаток экстрагировали эфиром и водный остаток концентрировали. ВЭЖХ с обращенной фазой и лиофилизацией собранной фракции получали указанное в заголовке соединение в виде белого твердого вещества. LC/MC: 404,1 (МН)+.
Пример 77: (R)-3-(2-(транс-4-(1,2-диаминоэтил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
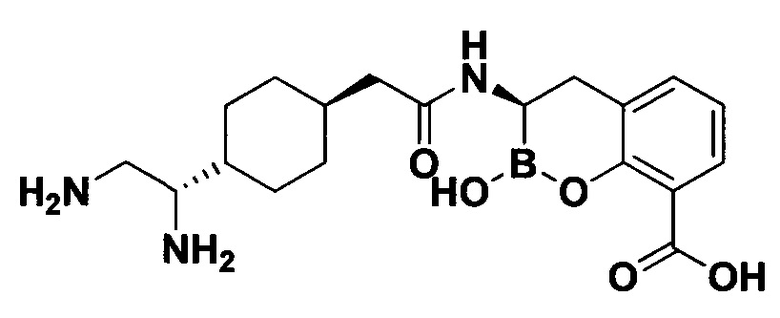
Синтез 2-(транс-4-(2,2,11,11-тетраметил-4,9-диоксо-3,10-диокса-5,8-диазадодекан-6-ил)циклогексил)уксусной кислоты.
Стадия 1: Синтез этил-2-(4-оксоциклогексил)ацетата.
[00418] К раствору этил-2-(1,4-диоксаспиро[4,5]декан-8-ил)ацетата (полученного на стадии 1, пример 77, 4,71 г, 19,44 ммоль) в ацетонитриле (45 мл) добавляли водный раствор 6N HCl (45 мл). Полученные смеси перемешивали при комнатной температуре в течение 2 ч и нейтрализовали твердым NaHCO3 до значения рН 8, экстрагировали этилацетатом. Органическую фазу промывали насыщенным солевым раствором, высушивали над Na2SO4. Фильтрование и выпаривание досуха дает кетона в виде бесцветного масла (2,97 г, 78,1%).
Стадия 2: Синтез этил-2-(4-метиленециклогексил)ацетата.
[00419] К охлажденной (0°С) суспензии метилтрифенилфосфонийбромида (8,58 г, 23,5 ммоль) в THF (60 мл) добавляли KOtBu (3,17 г, 28,3 ммоль) порциями в атмосфере N2. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 1 ч. Полученную смесь охлаждали до 0°С и добавляли раствор этил-2-(4-оксоциклогексил)ацетата (2,9 г, 15,7 ммоль) в THF (15 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 ч и при 50°С в течение ночи. После охлаждения до комнатной температуры реакционную смесь гасили добавлением насыщенного NH4Cl, экстрагировали этилацетатом, промывали насыщенным солевым раствором, высушивали над Na2SO4. Очистка колоночной флэш-хроматографией (элюент: 20% EtOAc в гексане) дает продукт в виде бесцветного масла (2,11 г, 73,7%).
Стадия 3: Синтез этил-2-(транс-4-(гидроксиметил)циклогексил)ацетата.
[00420] 9-BBN (0,5 N в тетрагидрофуране, 57,5 мл, 28,75 ммоль) добавляли к раствору этил-2-(4-метиленциклогексил)ацетата (2,10 г, 11,5 ммоль) в THF (20 мл) при 0°С в атмосфере N2. Смесь нагревали до комнатной температуры и перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь охлаждали до 0°С и добавляли по каплям смесь 20%-ного раствора NaOAc (40 мл) и 30% H2O2 (30 мл). Полученную смесь нагревали до комнатной температуры и перемешивали в течение 40 мин, гасили насыщенным раствором NH4Cl, разбавляли EtOAc и разделяли. Органическую фазу промывали насыщенным раствором Na2S2O3, насыщенным солевым раствором и высушивали над Na2SO4. Очисткой колоночной флэш-хроматографией (элюент: 30% EtOAc в гексане до 40%) получали спиртовый продукт в виде бесцветного масла (1,66 г, 72,1%). LC/MC: 201,1 (МН)+.
Стадия 4: Синтез этил-2-(транс-4-формилциклогексил)ацетата.
[00421] Раствор DMSO (0,64 мл, 9,06 ммоль) в DCM (2 мл) по каплям добавляли к предварительно охлажденному (-78°С) раствору оксалилхлорида (0,85 мл, 9,9 ммоль) в DCM (2 мл) в атмосфере N2. После перемешивания при -78°С в течение 10 мин добавляли по каплям раствор этилового эфира 2-(транс-4-(гидроксиметил)циклогексил)ацетата (1,65 г, 8,23 ммоль) в DCM (12 мл). Полученный раствор перемешивали при -78°С в течение 15 мин и добавляли TEA (6,89 мл). Раствор перемешивали при -78°С в течение 15 мин и при комнатной температуре в течение 20 мин. Реакционную смесь разбавляли DCM, промывали 1М HCl раствором и насыщенным солевым раствором, высушивали над Na2SO4. Очистка колоночной флэш-хроматографией (элюент: от 20% EtOAc в гексане до 30%) дает указанное продукт кетон в виде масла желтого цвета (0,76 г, 47%). LC/MC: 199,1 (МН)+.
Стадия 5: Синтез этилового эфира 2-(транс-4-(амино(циано)метил)циклогексил) ацетата.
[00422] Смесь этил-2-(транс-4-формилциклогексил)ацетата (0,75 г, 3,78 ммоль), NaCN (0,21 г, 4,28 ммоль), насыщенного водного раствора NH4OH (0,53 мл) и NH4Cl (0,24 г) в этаноле (22 мл) и воде (11 мл) нагревали при 70°С в течение ночи. После удаления органического растворителя остаток экстрагировали EtOAc, промывали насыщенным раствором NaHCO3 и насыщенным солевым раствором, высушивали над Na2SO4. Фильтрованием и выпариванием досуха получали неочищенный продукт. LC/MC: 225,1 (MH)+.
Стадия 6: Синтез этил-2-(транс-4-(трет-бутоксикарбониламино)(циано)метил)циклогексил)ацетата.
[00423] К раствору этил-2-(транс-4-(амино(циано)метил)циклогексил)ацетата (3,70 ммоль) в THF (25 мл) добавляли ди-трет-бутилдикарбонат (1,25 г, 5,55 ммоль) и NaHCO3 (0,62 г, 7,4 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Водная обработка и очистка колоночной флэш-хроматографией (элюент: 30% EtOAc в гексане) давала продукт (0,54 г, 45%). LC/MC: 347,1 (MNa)+.
Стадия 7: Синтез этил-2-(транс-4-(2-амино-1-(трет-бутоксикарбониламино)этил)циклогексил)ацетата
[00424] К раствору этил-2-(транс-4-(трет-бутоксикарбониламино)(циано)метил)циклогексил)ацетата (1,63 ммоль) в уксусной кислоте (15 мл) добавляли Pd(OH)2 (0,2 г). Полученную смесь гидрировали при комнатной температуре при атмосферном давлении в 55 пси, в течение 3 дней. Фильтрованием и выпариванием досуха получали неочищенный продукт. LC/MC: 329,1 (МН)+.
Стадия 8: Синтез этил-2-(транс-4-(2,2,11,11-тетраметил-4,9-диоксо-3,10-диокса-5,8-диазадодекан-6-ил)циклогексил)ацетата.
[00425] Смесь этил-2-(транс-4-(2-амино-1-(трет-бутоксикарбониламино)этил)циклогексил)ацетата (1,63 ммоль), ди-трет-бутилдикарбоната (0,44 г, 1,96 ммоль) и TEA (2,27 мл, 16,3 ммоль) в DCM (15 мл) перемешивали при комнатной температуре в течение ночи. Реакцию гасили добавлением насыщенного раствора NH4Cl и разделяли. Органическую фазу промывали 0,5 н HCl, насыщенным солевым раствором и высушивали над Na2SO4. Очистка колоночной флэш-хроматографией (элюент: от 20% EtOAc в гексане до 30%) давала продукт в виде бесцветного масла (0,58 г, 83%). LC/MC: 451,1 (MNa)+.
Стадия 9: Синтез 2-(транс-4-(2,2,11,11-тетраметил-4,9-диоксо-3,10-диокса-5,8-диазадодекан-6-ил)циклогексил)уксусной кислоты.
[00426] К раствору этил-2-(транс-4-(2,2,11,11-тетраметил-4,9-диоксо-3,10-диокса-5,8-диазадодекан-6-ил)циклогексил)ацетата (0,58 г, 1,35 ммоль) в THF (5 мл) и МеОН (5 мл) добавляли 3N NaOH (2,25 мл, 6,75 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 ч. После удаления летучих компонентов выпариванием остаток подкисляли с помощью 0,5N HCl раствором до значения рН около 4,5 и экстрагировали с помощью EtOAc, высушивали над Na2SO4. Фильтрование и выпаривание досуха дает кислоту с количественным выходом. LC/MC: 423,1 (MNa)+.
Синтез (R)-3-(2-(транс-4-(1,2-диаминоэтил)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00427] Соединение получали из 2-(транс-4-(2,2,11,11-тетраметил-4,9-диоксо-3,10-диокса-5,8-диазадодекан-6-ил)циклогексил)уксусной кислоты согласно процедуре связывания и удаления защитной группы, описанной в примере 76. Получали продукт в виде белого твердого вещества. LC/MC: 390,1 (МН)+.
Пример 78: (R)-3-(2-(транс-4-(2-(этиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
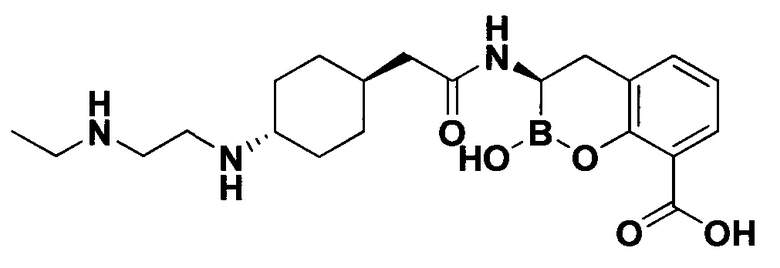
Синтез (R)-3-(2-(транс-4-(2-(этиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00428] Получали из (R)-3-(2-(транс-4-(2-аминоэтиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты и уксусного альдегида в соответствии с процедурой, описанной в примере 71. Продукт очищали с помощью ВЭЖХ с обращенной фазой с получением указанного в заголовке соединения. ESI-MS m/z 418 (MH)+.
Пример 79: (R)-3-(2-(транс-4-(2-(диметиламино)этиламино)циклогексил)ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновая кислота.
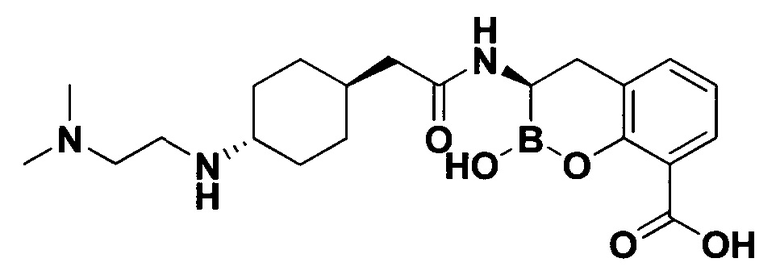
Стадия 1: Синтез 2-(транс-4-(трет-бутоксикарбонил (2-(диметиламино)этил)амино)циклогексил)уксусной кислоты.
[00429] К 2-(транс-4-(трет-бутоксикарбониламино)циклогексил)уксусной кислоте (1,47 г) в DMF (10 мл) добавляли Na2CO3 (0.907 г) и бензилбромид (0,75 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли воду и экстрагировали этилацетатом. Органическую фазу высушивали и концентрировали с получением желаемого продукта в виде белого твердого вещества (1,60 г). К данному твердому веществу добавили 4N HCl (10 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Диэтиловый эфир затем добавляли к реакционной смеси для осаждения гидрохлорида бензил-2-(транс-4-аминоциклогексил)ацетата в виде белого твердого вещества.
[00430] К гидрохлориду бензил-2-(транс-4-аминоциклогексил)ацетата (860 мг) в DMF добавляли K2CO3 (414 мг) и 2-бром-N,N-диметилетанамин соль HBr (700 мг). Полученную реакционную смесь перемешивали при 60°С в течение ночи. Затем добавляли воду и экстрагировали этилацетатом. Органическую фазу высушивали и концентрировали с получением неочищенного продукта, который использовали непосредственно на следующей стадии.
[00431] К указанному выше продукту в DCM (20 мл) добавляли TEA (1 мл) и ди-трет-бутилдикарбонат (1,5 г). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Органическую фазу промывали насыщенным солевым раствором, высушивали и концентрировали. Остаток очищали с помощью ВЭЖХ. К данному продукту в МеОН (10 мл) добавляли Pd/C (10%, 50 мг) и реакционную смесь перемешивали в атмосфере водорода в течение ночи. Катализатор отфильтровывали через слой целита и растворитель удаляли при пониженном давлении с получением 2-(транс-4-(трет-бутоксикарбонил(2-(диметиламино)этил)амино)циклогексил)уксусной кислоты в виде желтой пены (250 мг).
Стадия 2: Синтез трет-бутил-3-((2R)-2-(2-(транс-4-(трет-бутоксикарбонил(2-(диметиламино)этил)амино)циклогексил)ацетамидо)-2-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дек-4-ил)этил)-2-метоксибензоата
[00432] Соединение получали из трет-бутилового сложного эфира 2-метокси-3-(2,9,9-триметил-3,5-диокса-4-бора-трицикло[6.1.1.02,6]дец-4-илметил)бензойной кислоты и 2-(транс-4-(трет-бутоксикарбонил(2-(диметиламино)этил)амино)циклогексил)уксусной кислоты в соответствии с процедурой, описанной на стадии 1 примера 1.
Стадия 3: Синтез (R)-3-(2-(транс-4-(2-(диметиламино)этиламино)циклогексил) ацетамидо)-2-гидрокси-3,4-дигидро-2H-бензо[е][1,2]оксаборинин-8-карбоновой кислоты.
[00433] К соединению со стадии 2 (40 мг) добавляли 3N HCl (2 мл), и полученную реакционную смесь нагревали с обратным холодильником в течение 1 часа. Затем растворители удаляли в вакууме, и остаток очищали обращенно-фазовой препаративной ВЭЖХ и высушивали с помощью лиофилизации. ESI-MS m/z 418 (МН)+.
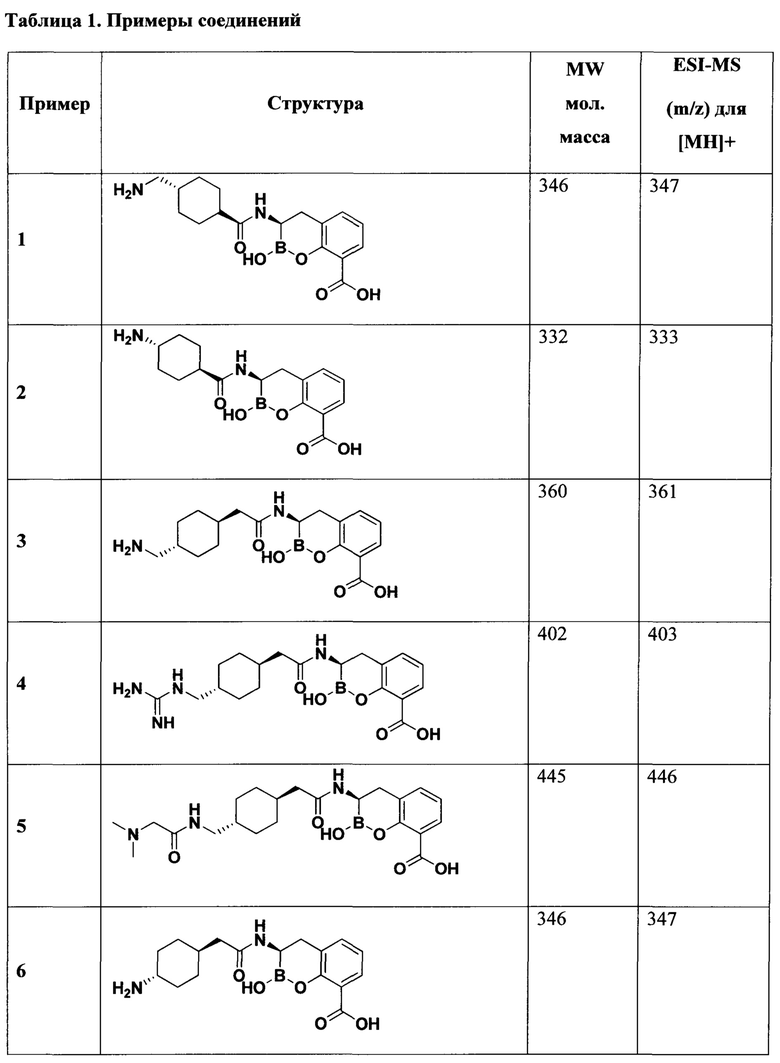
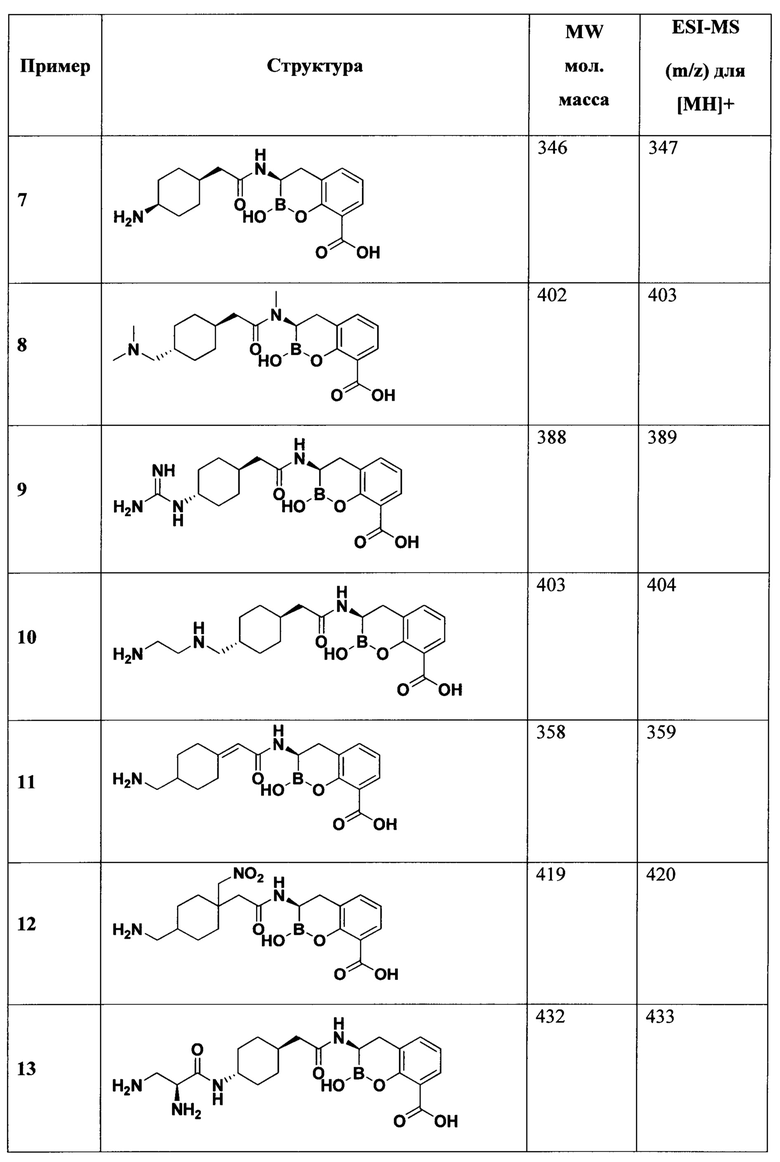
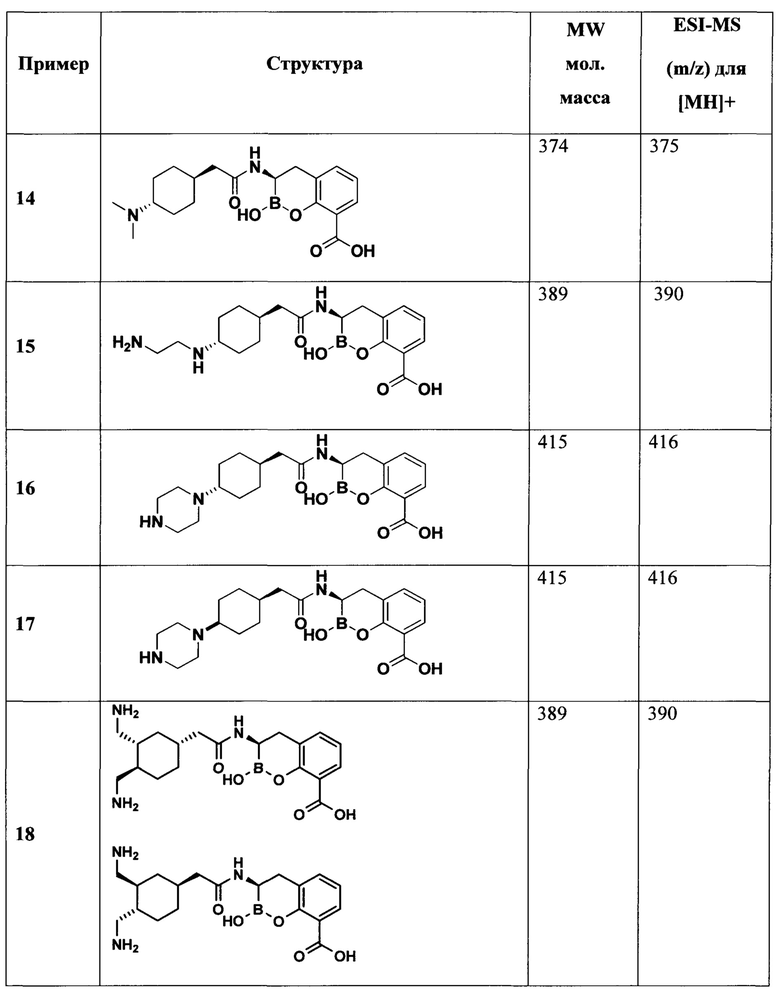
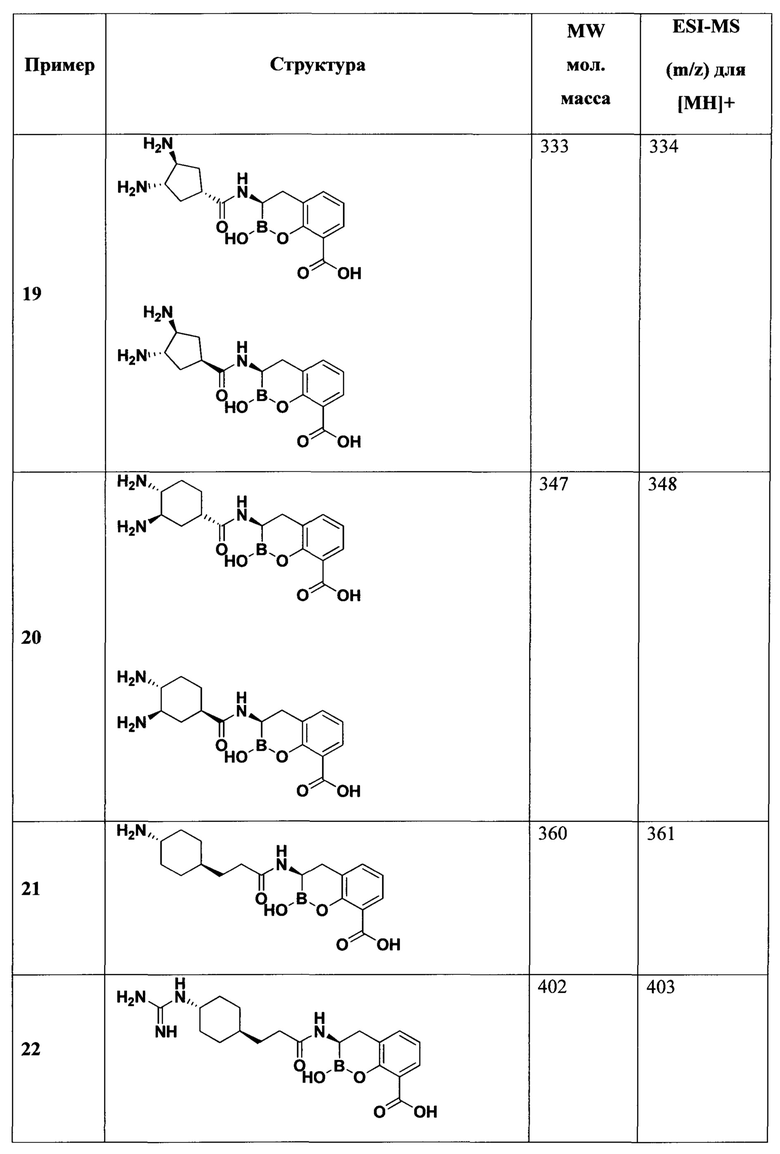
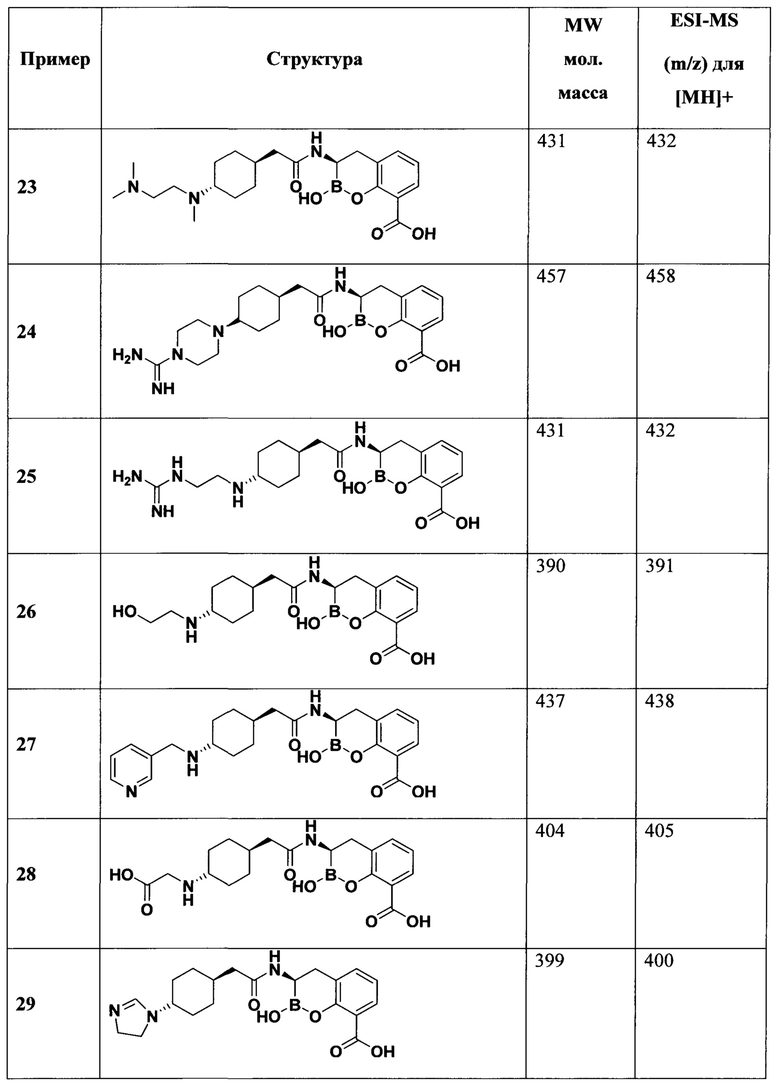
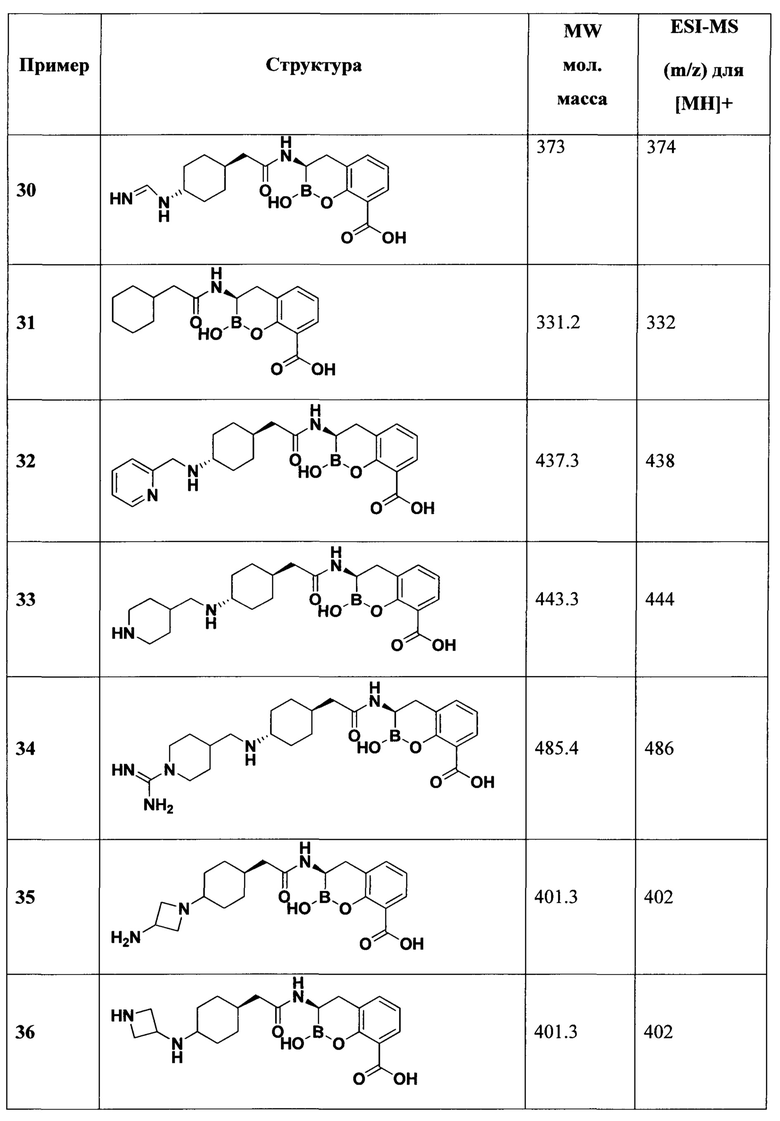
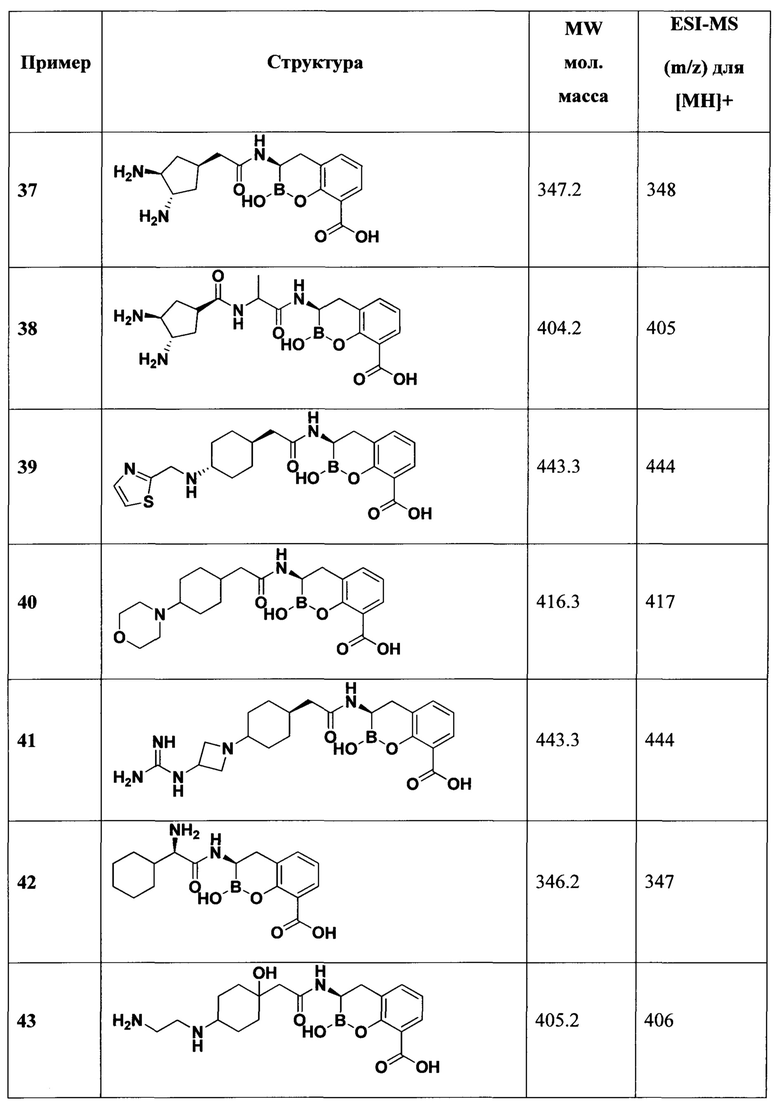
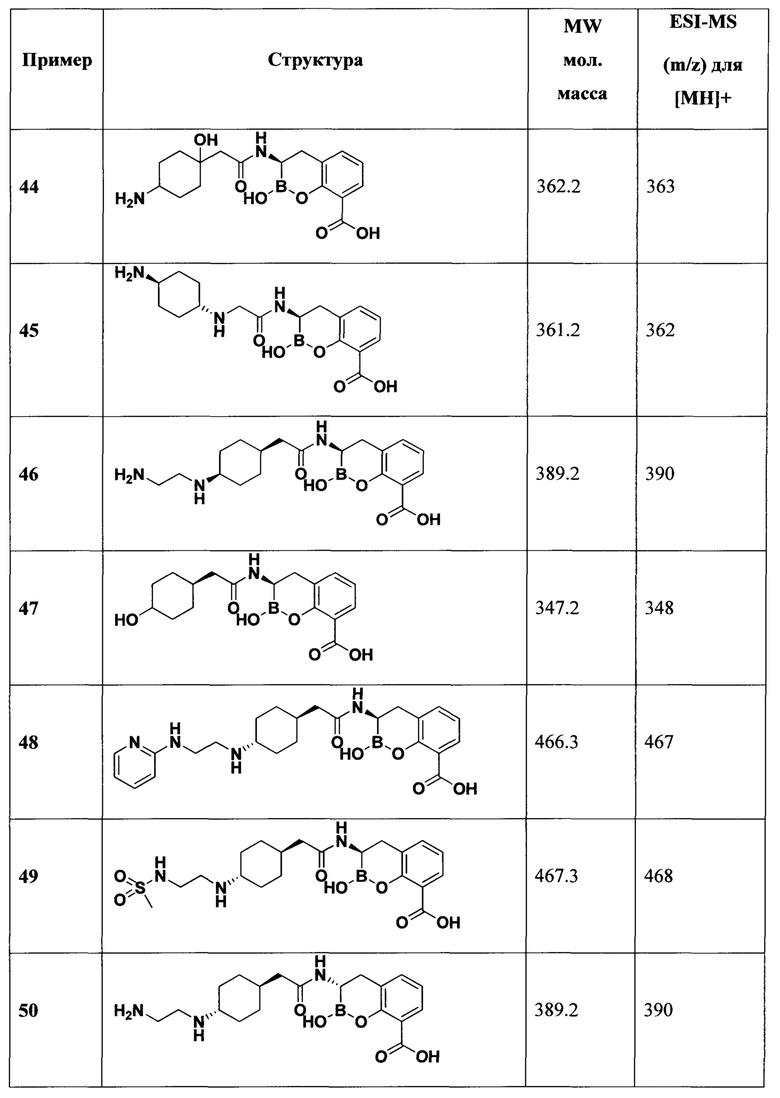
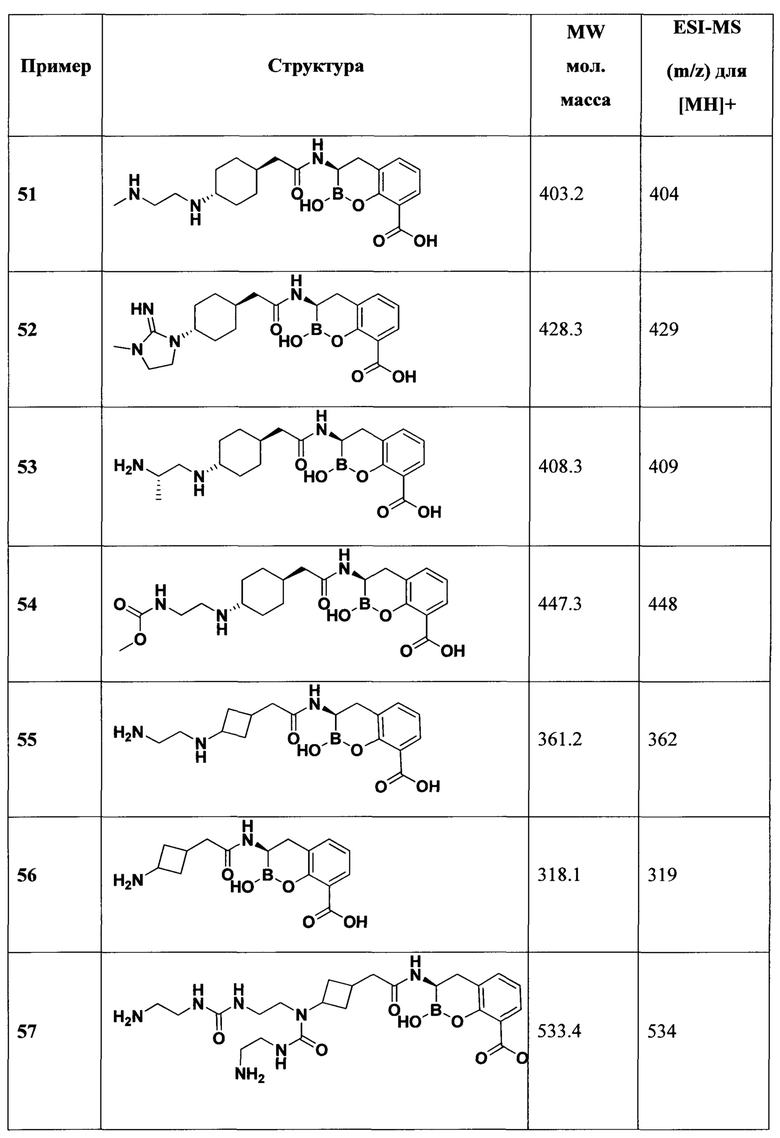
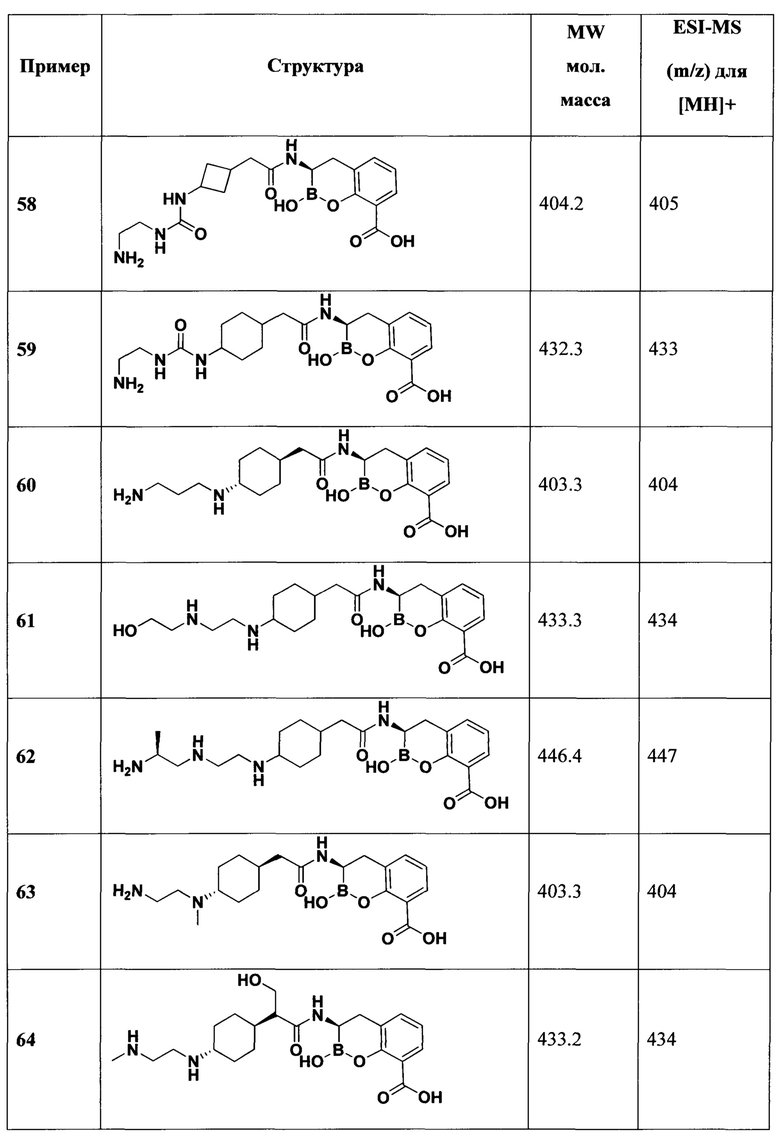
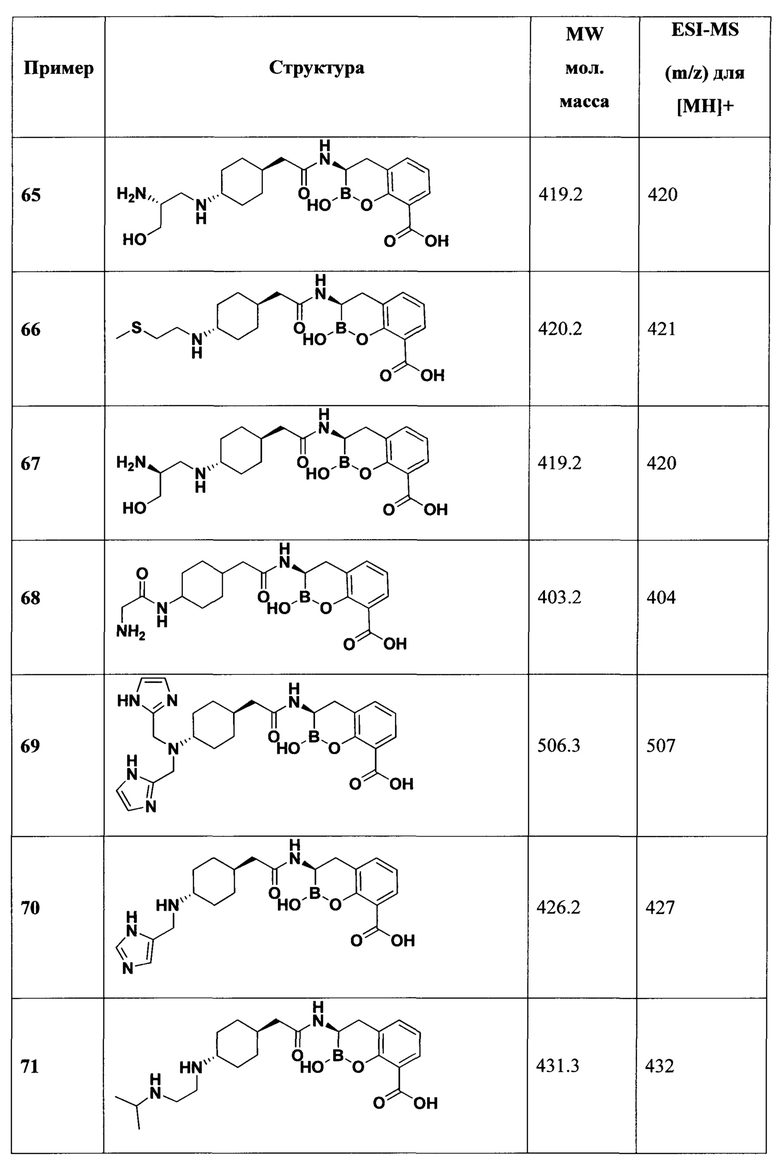
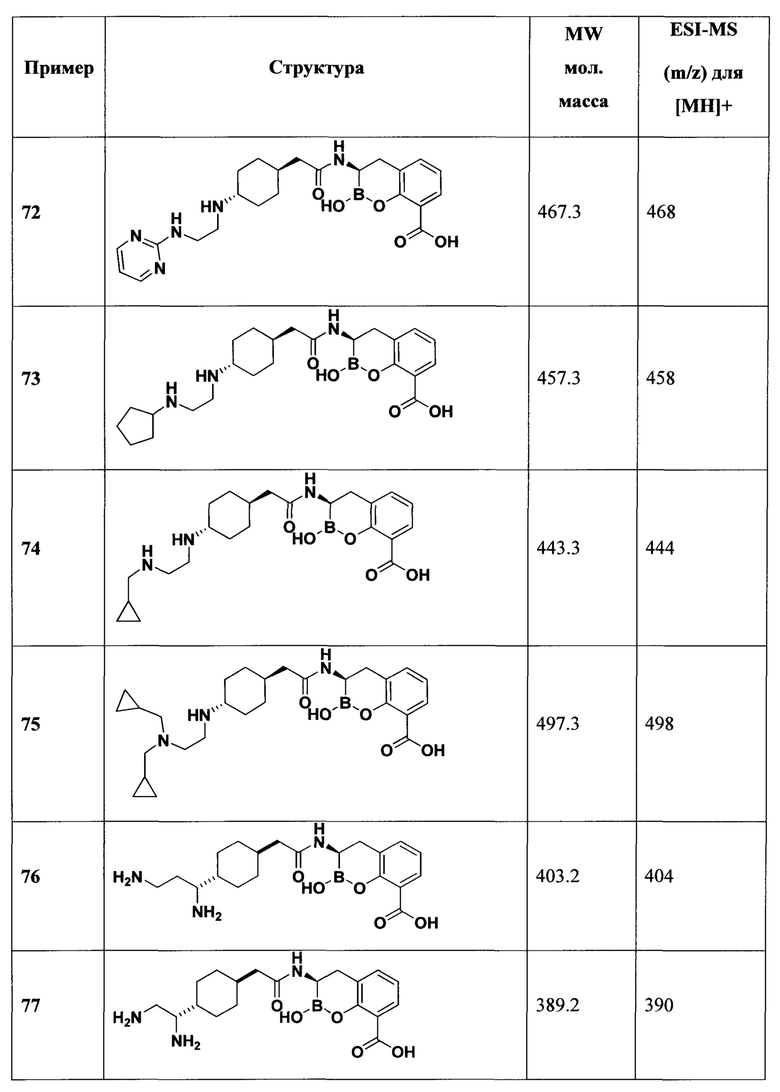
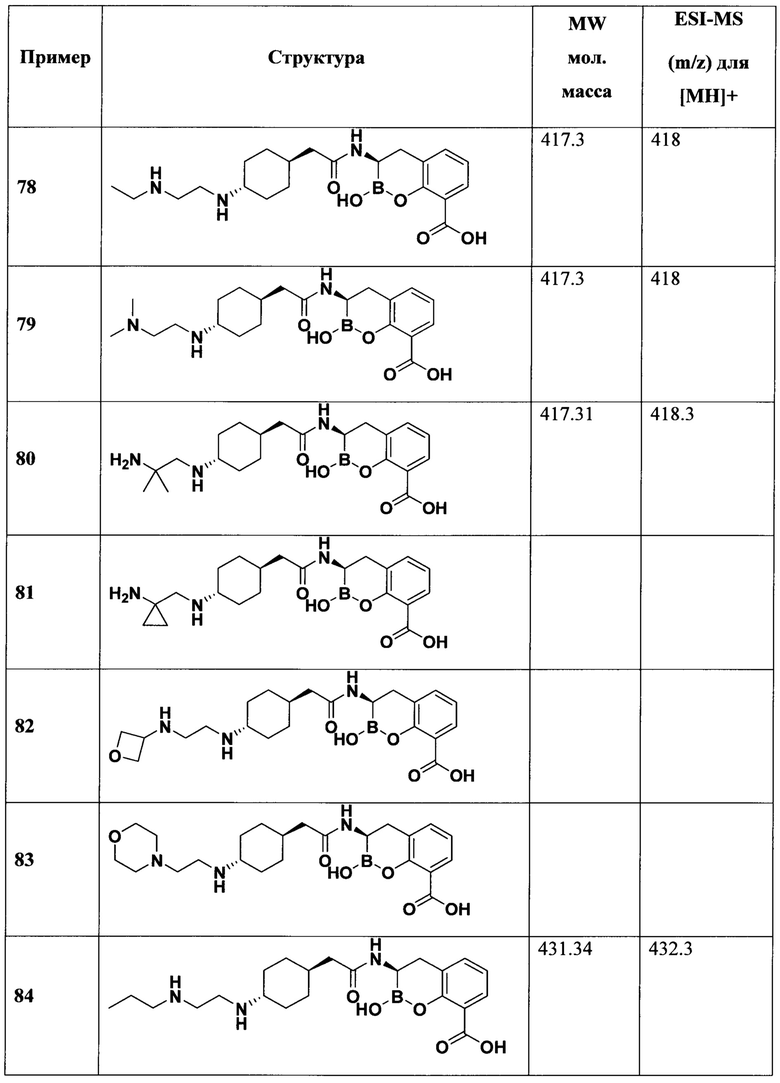
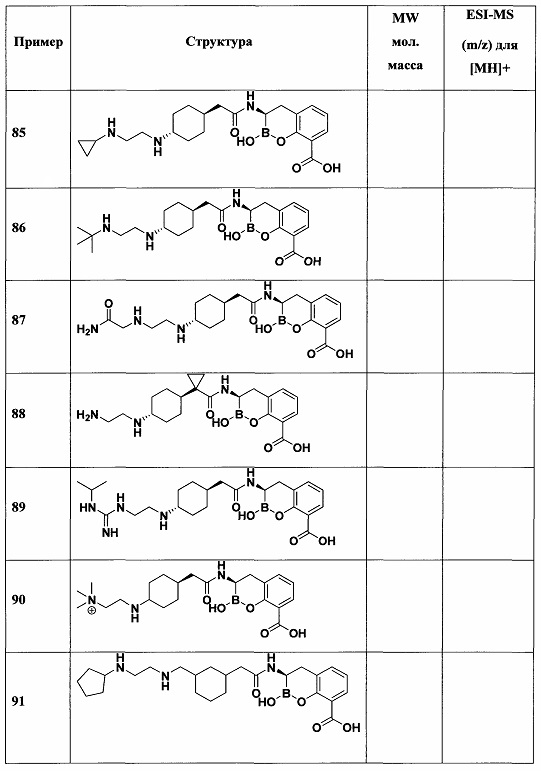
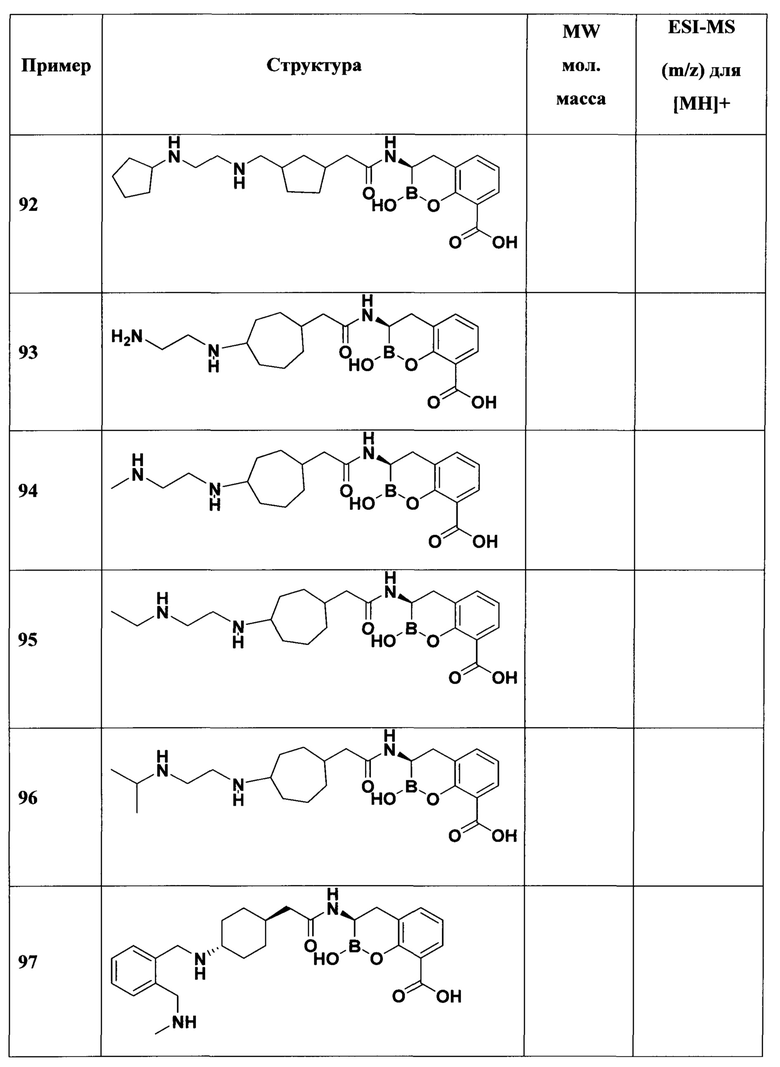
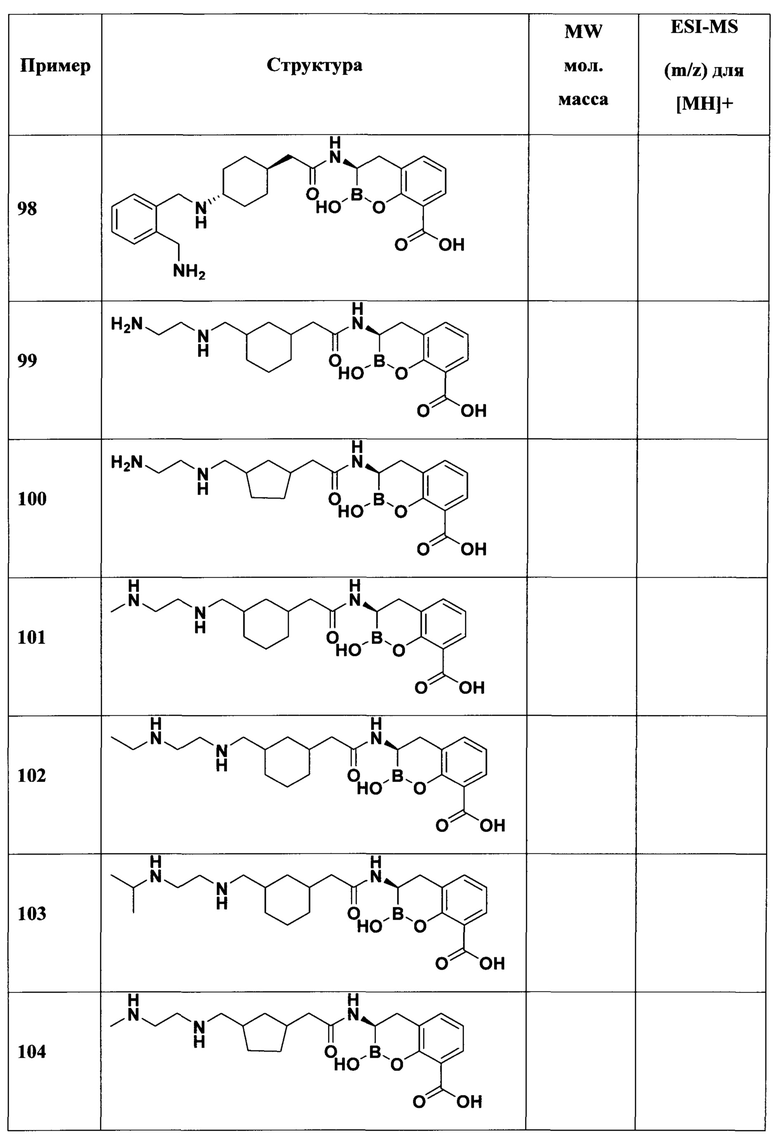
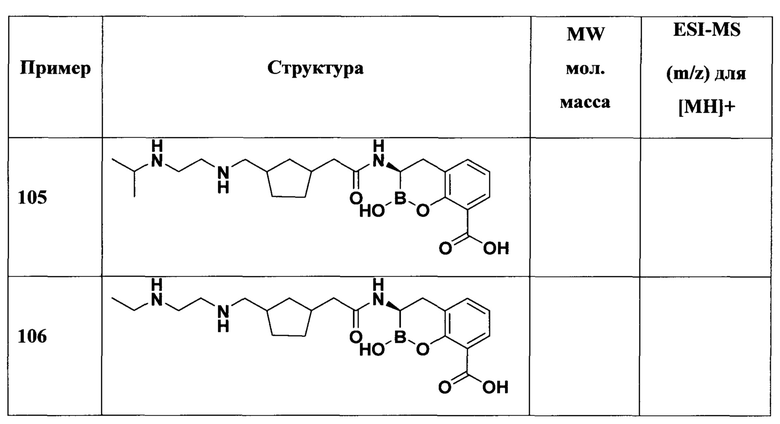
ПРИМЕР 107: Композиция соединения формулы I или формулы Ia для парентерального введения
[00434] Для приготовления фармацевтической композиции для парентерального введения, подходящей для введения инъекцией, 100 мг соединения формулы I или формулы Ia или его водорастворимой фармацевтически приемлемой соли растворяли в DMSO и затем смешивали с 10 мл стерильного 0,9%-ного солевого раствора. Смесь помещают в единицу дозирования, подходящую для введения инъекцией.
ПРИМЕР 108: Композиция соединения формулы I или формулы Ia для перорального введения
[00435] Для приготовления фармацевтической композиции для перорального введения 400 мг соединения формулы I или формулы Ia, а также следующие ингредиенты тщательно перемешивают и прессуют в таблетки с одной насечкой.


[00436] Следующие ингредиенты тщательно смешивали и загружали в твердые желатиновые капсулы.

Биологические примеры
Пример I: Экспериментальный способ аналитического определения β-лактамазной ферментативной активности
Выделение β-лактамаз.
[00437] В случае β-лактамаз SHV-5, KPC-2, p99AmpC и ОХА-1, бактериальные клетки E.coli BL21 (DE3), несущие плазмиды экспрессии (экспрессирующих белки в виде нативных непомеченных белков) для отдельных β-лактамаз, выращивали в 1 л культуральной среды Superbroth (Teknova Inc. Hollister, CA) с добавлением 100 мкг/мл канамицина и среды отбора 1×5052 (0,5% глицерина, 0,05% глюкозы и 0,2% α-лактозы) при 35°С в течение от 18 до 20 часов. Клетки собирали центрифугированием (4000 × g, 4°С, 20 мин), ресуспендировали в 50 мл 10 мМ HEPES, рН 7,5 (1/20 от первоначального объема). Клетки лизировали обработкой ультразвуком (5 импульсов продолжительностью 45 секунд) при 45 Вт на льду. Лизаты осветляли центрифугированием при 10000 × g в течение 40 минут при 4°С. Пробы разводили в 5 раз в 50 мМ ацетате натрия, рН 5,0, хранили в течение ночи при 4°С, после чего их центрифугировали при 10000×g в течение 30 минут для осветления и фильтровали через фильтры с диаметром пор в 0,45 мкм. Образцы наносили на катионообменную колонку с САРТО S-сефарозой объемом в 5 мл (GE Healthcare), предварительно уравновешенную 50 мМ ацетатом натрия, рН 5.0. Колонку промывали 5 объемами колонки 50 мМ ацетата натрия, рН 5,0, чтобы смыть несвязавшийся белок, и использовали линейный градиент NaCl (от 0 до 500 мм) для элюирования белка (более 16 объемов колонки) из колонки. Фракции анализировали на наличие β-лактамазной активности с помощью Centa (Calbiochem, Gibbstown, NJ) или Nitrocefin (EMD Millipore chemicals, Darmstadt, Germany) в качестве репортерного β-лактамазного субстрата для анализа активности в отдельных фракциях. Фракции, обладающие активностью, объединяли, концентрировали и дополнительно очищали гель-фильтрационной хроматографией на гель-фильтрационной колонке с Superdex 75 prep grade (GE Healthcare, Piscataway, NJ), предварительно уравновешенной в 50 мМ Hepes, рН 7,5, 150 мМ NaCl. Фракции, обладающие активностью, собирали, концентрировали, измеряли количественно содержание белка способом определения белка с ВСА (Thermo Scientific, Rockford, IL), диализовали в PBS и замораживали при -80°С в 20%-ном глицерине до использования.
[00438] В случае металло-β-лактамазы VIM-2, процедура была идентична со следующими исключениями, во-первых, значение рН во фракции с белком не доводили до рН 5 с 50 мМ ацетата натрия, во-вторых, стадию хроматографии проводили на анионообменной колонке с 5 мл Q-Sepharose, предварительно уравновешенной 50 мМ Hepes, рН 7,5, и элюирование белка достигалось линейным градиентом NaCl (от 0 до-600 мМ). В заключение, для очистки VIM-2 требуется вторичная хроматография (3-я стадия) на анионообменной колонке с Q-Sepharose для достижения приемлемой чистоты (более 90%).
Ингибирование β-лактамазы.
[00439] Для определения уровня ингибирования β-лактамазных ферментов соединения разводили в PBS при рН 7,4 с получением концентрации в интервале от 100 до 0,00005 мкМ в 96-луночных микротитровальных планшетах. Добавляли равный объем разбавленного исходного раствора фермента, и планшеты инкубировали при 37°С в течение 15 мин. В качестве субстрата для р99 АМРС, VIM-2 и окса-1 использовали Nitrocefin, который добавляли в каждую лунку в конечной концентрации, равной 100 мкМ. Оптическое поглощение при 486 нм контролировали немедленно в течение 10 мин с использованием спектрофотометра для измерения поглощения в микропланшетах (Biotek Powerwave XS2 microplate spectrophotometer), используя пакет программного обеспечения GEN5 (Biotek Instruments, Winooski VT). Аналогичным образом, имипенем использовали в качестве субстрата для KPC-2 и цефотаксим использовали для SHV-5, в то время как изменения оптического поглощения при гидролизе β-лактамного кольца контролировали при длинах волн, равных 300 нм и 260 нм, соответственно, в УФ-прозрачных 96-луночных микропланшетных аналитических планшетах. Максимальную скорость гидролиза сравнивали со скоростями гидролиза в контрольных лунках (без ингибиторов), и процент ингибирования ферментов рассчитывали для каждой концентрации ингибитора. Концентрацию ингибитора, необходимую для того, чтобы уменьшить начальную скорость гидролиза субстрата на 50% (IC50), рассчитывали как остаточную активность β-лактамазы при длине волны, равной 486 нм, с использованием пакета программного обеспечения GraFit version 7 kinetics (Erithacus Software, Surrey, UK).
Пример II: Ингибирование различных β-лактамаз соединениями, служащими примерами
[00440] Используя методику, описанную выше, примеры по настоящему изобретению оценивали по их способности ингибировать β-лактамазные ферменты из всех четырех классов по классификации Ambler (от А до D). Результаты данных анализов приведены в таблице 3 для ферментов, представляющих различные подтипы (обратите внимание, что SHV-5 является представителем β-лактамаз расширенного спектра класса А, согласно классификации Ambler, KPC-2 служит примером карбапенемазы класса А, Р99 представляет хромосомальную AmpC класса С, ОХА-1 представляет оксацилиназу класса D и VIM-2 представляет цинк-зависимую металло-β-лактамазу класса В, также обладающую карбапенемазной активностью), где А представляет величину IC50 в интервале от 10 до 100 мкМ, В представляет величину IC50 в интервале от 1 до 10 мкм, С представляет величину IC50 в интервале от 0,1 до 1 мкМ, и D представляет величину IC50 менее 0,1 мкМ. NT = не тестировали.
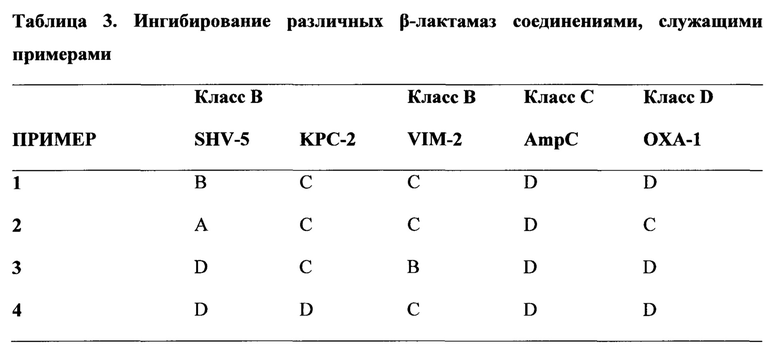
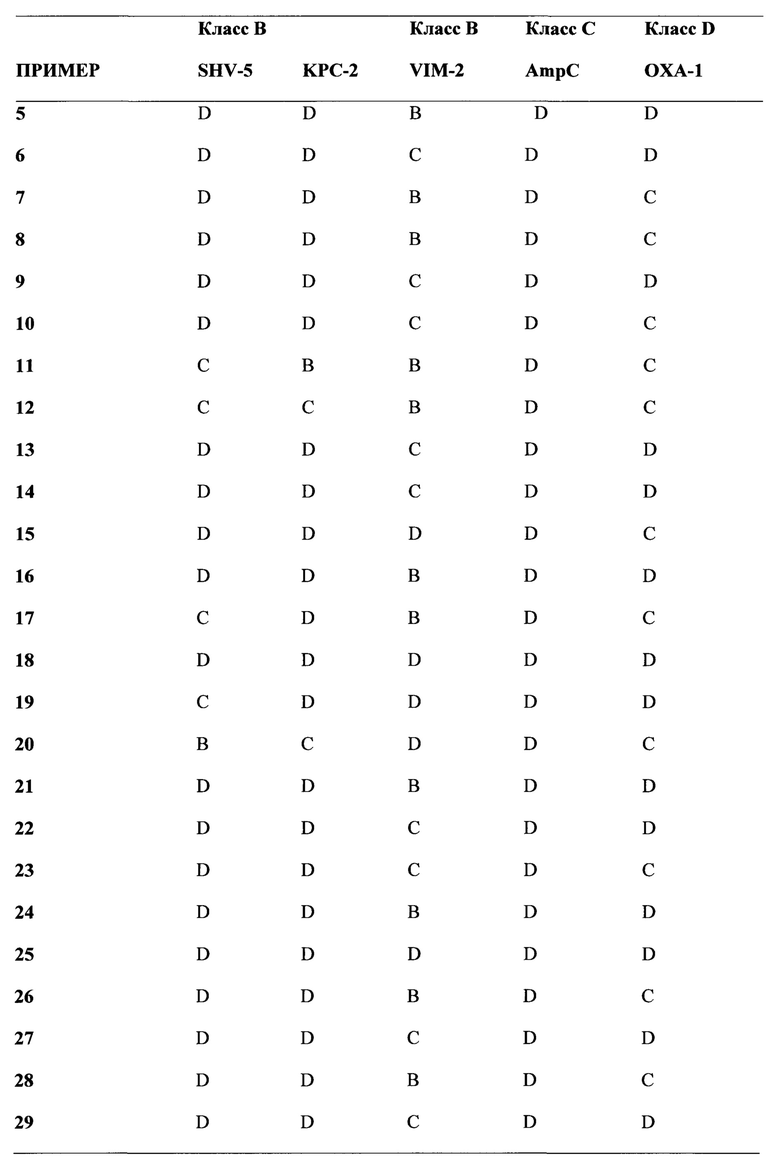

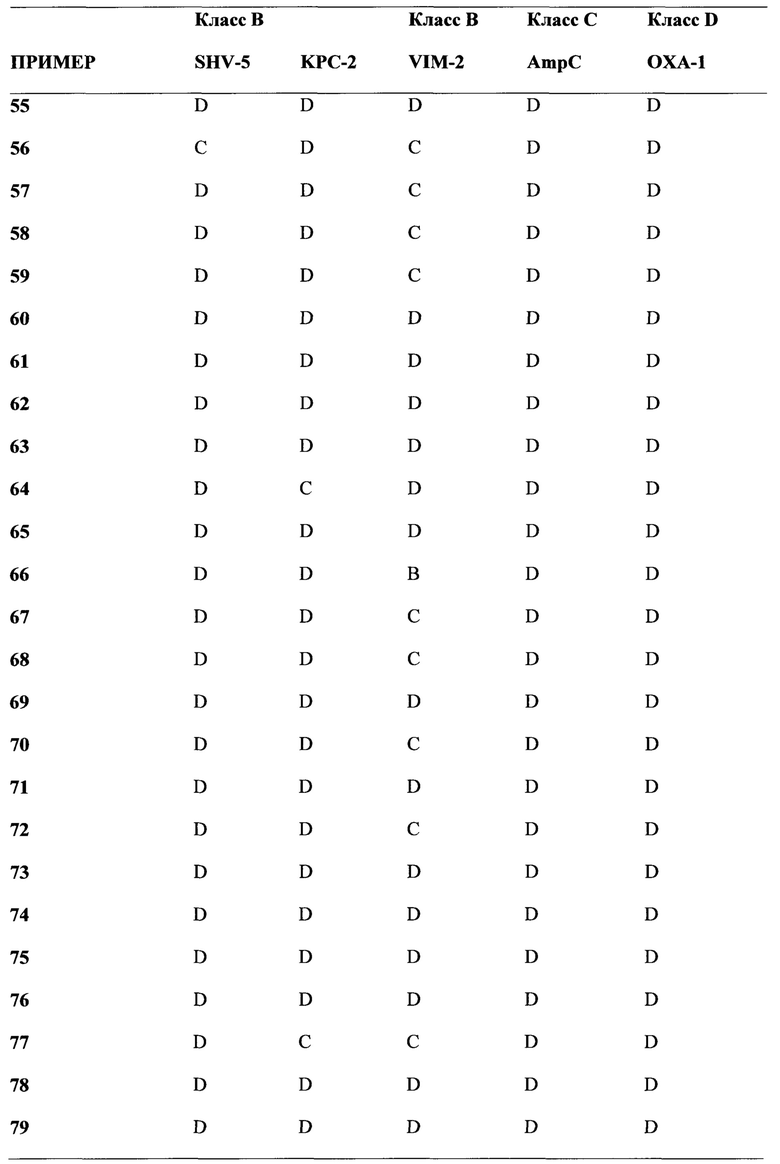
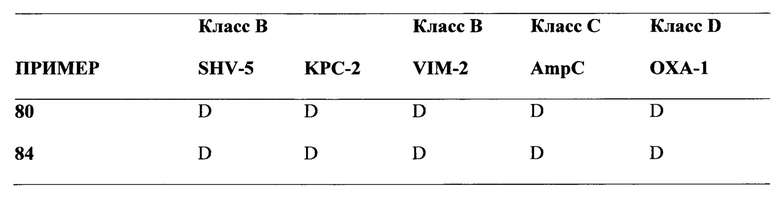
Пример III: In vitro антибактериальные аналитические способы определения ингибирования β-лактамаз
[00441] Для определения способности тестируемых соединений усиливать ингибирование роста бактериальных штаммов, которые продуцируют бета-лактамазные ферменты, использовали классические аналитические способы определения MIC, основанные на микроразведениях клеток в бульоне. Использовали шесть штаммов бактерий, производящих бета-лактамазные ферменты: E. coli, экспрессирующий бета-лактамазу расширенного спектра (БЛРС, Extended Spectrum Beta-Lactamase (ESBL)) CTX-M-15 класса А, E. cloacae, экспрессирующий фермент Р99 класса С, K. pneumoniae, экспрессирующий карбапенемазу KPC-2 класса A, P. aeruginosa, экспрессирующий карбапенемазу VIM-2 класса В, K. pneumoniae, экспрессирующий карбапенемазу класса А KPC-2 и карбапенемазу класса В VIM-24, и S. aureus, продуцирующий пенициллиназу РС-1 класса А. Аналитическое определение проводили в культуральной среде Cation Adjusted Mueller Hinton Broth (CAMHB, BD # 212322, BD Diagnostic Systems, Sparks, MD). Штаммы бактерий выращивали в течение от 3 до 5 часов в бульоне САМВН. Тестируемые соединения добавляли в планшет для микротитрования в 2-кратных серийных разведениях в САМНВ в конечной концентрации в интервале от 32 мкг/мл до 0,25 мкг/мл. Наложение САМНВ, содержащей бета-лактам, добавляли к соединениям с конечной статической концентрацией, равной 4 мкг/мл. Цефтазидим (CAZ, Sigma# С3809-1 Г, Sigma-Aldrich, St. Louis, МО) использовали в качестве партнера-антибиотика для E. coli, экспрессирующей бета-лактамазу CTX-M-15 ESBL класса А по классификации Ambler (собственная MIC при добавлении без партнера >128 мкг/мл), и E. cloacae, экспрессирующей бета-лактамазу Р99 класса С (собственная MIC при добавлении без партнера равна 128 мкг/мл). Меропенем (Меро, USP # 1392454, Pharmacopeia, Rockville, MD) использовали в качестве партнера-антибиотика для K. pneumoniae, экспрессирующей карбапенемазу KPC-3 класса А по классификации Ambler (собственная MIC при добавлении без партнера >128 мкг/мл), P. aeruginosa, экспрессирующей карбапенемазу VIM-2 класса А (собственная MIC при добавлении без партнера равна 16 мкг/мл), и K. pneumoniae, экспрессирующей карбапенемазу KPC-2 класса А и карбапенемазу VIM-4 класса В (собственная MIC при добавлении без партнера 64 мкг/мл). Пиперациллин (Pip, Fisher # ICN15626801, MP Biomidiсаls, Solon, ОН) использовали в качестве партнера-антибиотика для S. aureus, продуцирующего пенициллиназу РС-1 класса А (собственная MIC при добавлении без партнера равна 64 мкг/мл). Титрование тестируемых соединений с считыванием MIC показывает концентрацию исследуемого вещества, необходимую для того, чтобы достаточно ингибировать активность бета-лактамазного фермента и защитить внутреннюю антибактериальную активность бета-лактама. В дополнение к титрованию испытываемых соединений, также протестировали значения MIC из панели контрольных бета-лактамов, чтобы гарантировать, что штаммы ведут себя последовательно от теста к тесту. После добавления испытуемого соединения и антибиотиков, планшеты могут быть инокулированы в соответствии со способом CLSI микроразведений бульона. После инокуляции планшеты инкубировали в течение от 16 до 20 часов при температуре 37°C, затем определяли визуально минимальную ингибирующую концентрацию (МИК, Minimal Inhibitory Concentration (MIC)) испытуемого соединения.
[00442] Используя методику, описанную выше, примеры по настоящему изобретению оценивали на их способность ингибировать рост бактерий, продуцирующих β-лактамазы, в присутствии β-лактамного антибиотика.
[00443] Типичные результаты приведены в таблице 3, где А представляет величину MIC более 16 мкг/мл, В представляет величину MIC в интервале от 1 до 16 мкг/мл включительно, и С представляет величину MIC менее 1 мкг/мл. NT = не тестировали.
Пример IV: In vitro антибактериальная активность соединений, служащих примерами
[00444] Используя методику, описанную выше в ПРИМЕРЕ III, служащие примерами соединения формулы I или формулы Ia оценивали на их способность ингибировать рост бактерий, продуцирующих β-лактамазы, в присутствии β-лактамного антибиотика.
[00445] Типичные результаты приведены в таблице 4, где А представляет MIC фиксированного β-лактамного антибиотика в присутствии более 32 мкг/мл β-лактамазного ингибитора из служащих примерами соединений, В представляет MIC в присутствии от 8 до 32 мкг/мл β-лактамазного ингибитора из служащих примерами соединений, и С представляет MIC в присутствии ≤4 мкг/мл β-лактамазного ингибитора из служащих примерами соединений. NT = не тестировали.
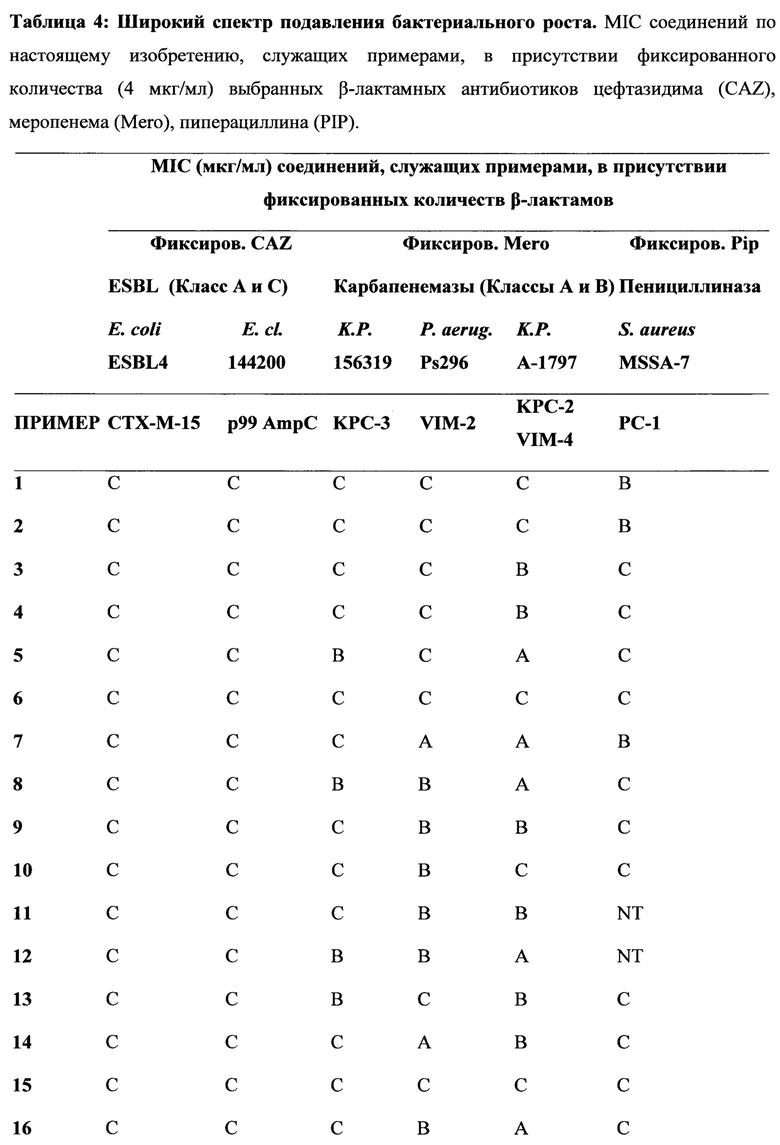
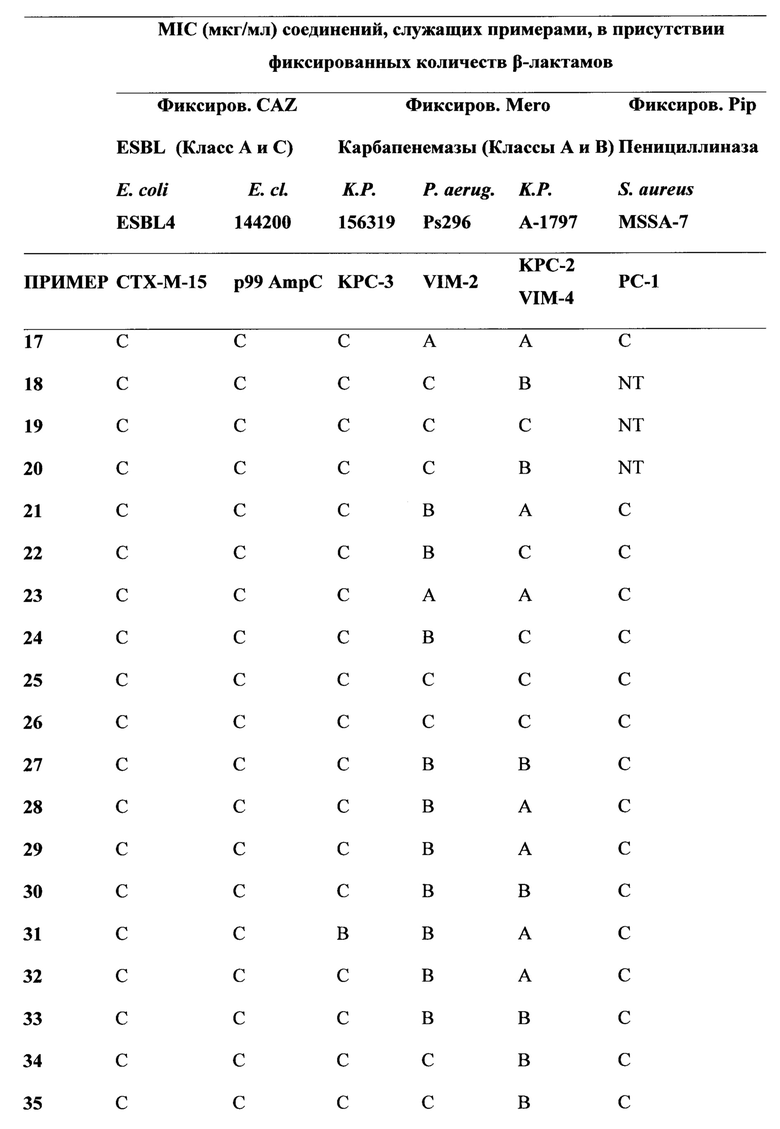
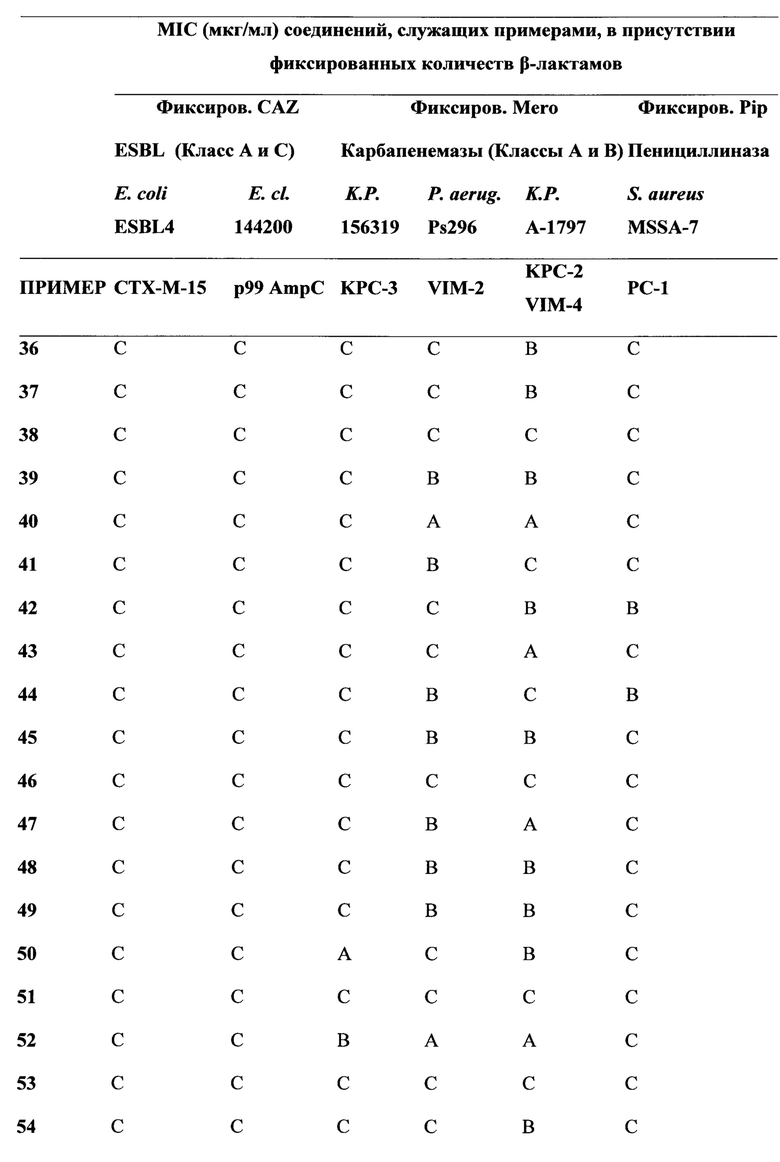
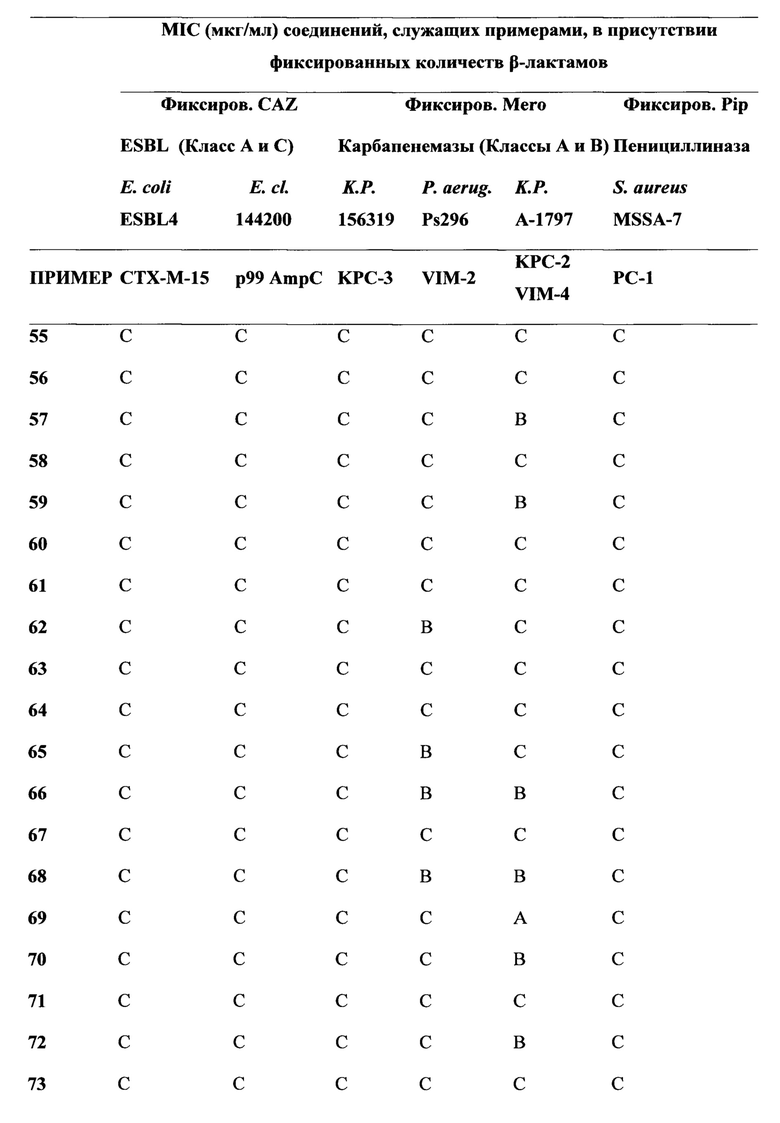

[00446] В то время как предпочтительные варианты осуществления настоящего изобретения были показаны и описаны в настоящем описании, специалистам в данной области техники будет очевидно, что такие варианты осуществления приведены в качестве лишь примера. Многочисленные вариации, изменения и замены теперь будут очевидны специалистам в данной области техники без отступления от сущности изобретения. Следует понимать, что различные альтернативы вариантам осуществления описанного в настоящем описании изобретения могут быть использованы в практической реализации изобретения. Предполагается, что нижеследующая формула изобретения определяет объем настоящего изобретения, и что способы и структуры в пределах объема данной формулы изобретения и ее эквивалентов покрыты ими.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗЫ | 2015 |
|
RU2686740C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2018 |
|
RU2793315C2 |
| ИНГИБИТОРЫ MAGL | 2017 |
|
RU2754536C1 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ИНДУКЦИИ АПОПТОЗА РАКОВЫХ КЛЕТОК | 2003 |
|
RU2379056C2 |
| АЗАБИЦИКЛО-ХИНОЛОН-КАРБОНОВЫЕ КИСЛОТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1989 |
|
RU2049777C1 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| ДВОЙНЫЕ МОДУЛЯТОРЫ 5-HT И D-РЕЦЕПТОРОВ | 2008 |
|
RU2480466C2 |
| 3-ОКСО-3,9-ДИГИДРО-1Н-ХРОМЕНО[2,3-c]ПИРРОЛЫ В КАЧЕСТВЕ АКТИВАТОРОВ ГЛЮКОКИНАЗЫ | 2011 |
|
RU2603191C2 |
| СКОНДЕНСИРОВАННЫЕ С ГЕТЕРОЦИКЛИЧЕСКИМ КОЛЬЦОМ ПРОИЗВОДНЫЕ ПИРИМИДИНА | 1996 |
|
RU2136683C1 |
| ПРОИЗВОДНЫЕ НИТРИЛОВ β-АМИНОКИСЛОТ | 2001 |
|
RU2245871C2 |
Изобретение относится к борсодержащему соединению формулы (I) или его фармацевтически приемлемой соли или стереоизомеру:
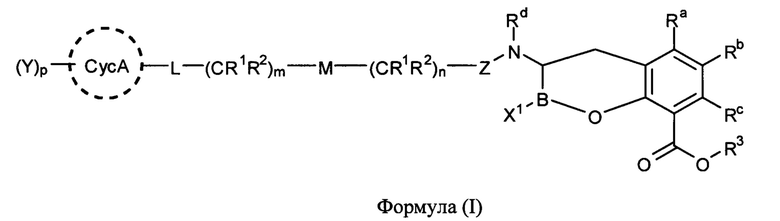
В формуле (I) L представляет собой связь, -CR1R2-, >С=O, или =CR1-; М представляет собой связь или -N(R4)-; m равно 0 или 1; n равно 0 или 1; при условии, что, когда n равно 0, то М представляет собой связь; p равно 1 или 2; X1 представляет собой -OH; Z представляет собой >С=O; СусА представляет собой циклобутан, циклопентан или циклогексан; Ra, Rb и Rc представляют собой атом водорода; каждый R1 и R2 независимо выбраны из группы, состоящей из атома водорода, C1-С6-алкила и -NR4R5, R3 представляет собой атом водорода; Rd выбран из группы, состоящей из атома водорода и C1-C6 алкила; каждый R4 и R5 независимо выбран из группы, состоящей из атома водорода, C1-С6-алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклила, 5-6-членного гетероарила, C1-С6 алкил(3-6-членного циклоалкила) и C1-С6 алкил(5-6-членного гетероарила); или R4 и R5, взятые вместе, образуют 3-6-членный гетероцикл с атомом азота, к которому они присоединены; каждый R6 и R7 независимо выбраны из группы, состоящей из атома водорода, C1-С6 алкила, -ОН, -SR10, -NR4R5, -NR4C(O)OR5, -C(O)OR5 и -NR4SO2R5; каждый R10 независимо представляет собой C1-С6-алкил; и каждый Y независимо выбран из группы, состоящей из -ОН, -NR4R5, -(CR6R7)vNR4R5, -NR4(CR6R7)vNR4R5, -N(R4)C(O)(CR6R7)vNR4R5, -(CR6R7)vN(R4)C(O)(CR6R7)vNR4R5, -NR5C(O)NR4(CR6R7)vNR4R5, -N(R4)C(=NR5)R6, -(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4C(=NR5)NR4R5, -(CR6R7)vNR4(CR6R7)vNR4R5, -NR4(CR6R7)vR6, -NR4(CR6R7)vNR4(CR6R7)vNR4R5, --(3-6-членный гетероциклил)NR4R5, -(3-6-членный гетероциклил)N(R4)C(=NR5)NR4R5, -NR4(CR6R7)v(5-6-членный гетероарил), -NR4(CR6R7)v-(3-6-членный гетероциклил), -NR4(CR6R7)vNR5-(5-6-членный гетероарил) и -NR4(CR6R7)v-(3-6-членный гетероциклил)-C(=NR5)NR4R5; и v равно 1, 2 или 3. Также предложены фармацевтическая композиция и способ лечения бактериальной инфекции. Соединение формулы (I) применяется для лечения бактериальной инфекции. 3 н. и 22 з.п. ф-лы, 4 табл., 108 пр., 38 схем.
1. Соединение формулы (I) или его фармацевтически приемлемая соль или стереоизомер:
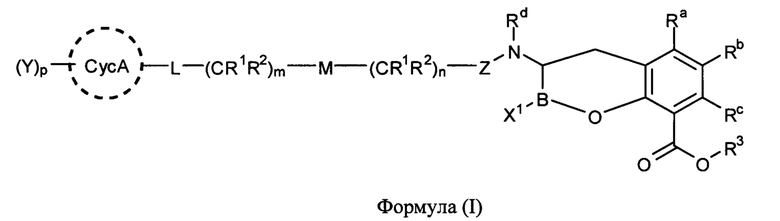
где:
L представляет собой связь, -CR1R2-, >С=O, или =CR1-;
М представляет собой связь или -N(R4)-;
m равно 0 или 1;
n равно 0 или 1;
при условии, что,
когда n равно 0, то М представляет собой связь;
p равно 1 или 2;
X1 представляет собой -ОН;
Z представляет собой >С=O;
СусА представляет собой циклобутан, циклопентан или циклогексан;
Ra, Rb и Rc представляют собой атом водорода;
каждый R1 и R2 независимо выбраны из группы, состоящей из атома водорода, C1-С6-алкила и -NR4R5,
R3 представляет собой атом водорода;
Rd выбран из группы, состоящей из атома водорода и C1-C6 алкила;
каждый R4 и R5 независимо выбран из группы, состоящей из атома водорода, C1-С6-алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклила, 5-6-членного гетероарила, C1-С6 алкил(3-6-членного циклоалкила) и C1-С6 алкил(5-6-членного гетероарила);
или R4 и R5, взятые вместе, образуют 3-6-членный гетероцикл с атомом азота, к которому они присоединены;
каждый R6 и R7 независимо выбраны из группы, состоящей из атома водорода, C1-С6 алкила, -ОН, -SR10, -NR4R5, -NR4C(O)OR5, -C(O)OR5 и -NR4SO2R5;
каждый R10 независимо представляет собой C1-С6-алкил;
и каждый Y независимо выбран из группы, состоящей из -ОН, -NR4R5, -(CR6R7)vNR4R5, -NR4(CR6R7)vNR4R5, -N(R4)C(O)(CR6R7)vNR4R5, -(CR6R7)vN(R4)C(O)(CR6R7)vNR4R5, -NR5C(O)NR4(CR6R7)vNR4R5, -N(R4)C(=NR5)R6, -(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4(CR6R7)vN(R4)C(=NR5)NR4R5, -NR4C(=NR5)NR4R5, -(CR6R7)vNR4(CR6R7)vNR4R5, -NR4(CR6R7)vR6, -NR4(CR6R7)vNR4(CR6R7)vNR4R5, --(3-6-членный гетероциклил)NR4R5, -(3-6-членный гетероциклил)N(R4)C(=NR5)NR4R5, -NR4(CR6R7)v(5-6-членный гетероарил), -NR4(CR6R7)v-(3-6-членный гетероциклил), -NR4(CR6R7)vNR5-(5-6-членный гетероарил) и -NR4(CR6R7)v-(3-6-членный гетероциклил)-C(=NR5)NR4R5; и
v равно 1, 2 или 3.
2. Соединение по п. 1, в котором Rd представляет собой атом водорода.
3. Соединение по п. 1, в котором:
L представляет собой связь или >С=O;
М представляет собой связь или -N(R4)-; и
m и n равны 0.
4. Соединение по п. 1, в котором:
L представляет собой связь;
М представляет собой связь; и
m или n равны 1.
5. Соединение по п. 1, в котором:
L представляет собой -CR1R2- или =CR1-;
М представляет собой связь; и
m и n равны 0.
6. Соединение по п. 1, в котором:
L представляет собой -CR1R2 - или =CR1-;
М представляет собой связь; и
m или n равны 1.
7. Соединение по п. 1, в котором СусА представляет собой циклогексан.
8. Соединение по п. 1, в котором R4 и R5 независимо представляют собой атом водорода или C1-С6-алкил.
9. Соединение по п. 1, в котором R6 и R7 независимо представляют собой атом водорода или C1-С6-алкил.
10. Соединение по п. 1, в котором  представляет собой
представляет собой 
11. Соединение по п. 10, в котором представляет собой 
12. Соединение по п. 10, в котором Y представляет собой -NR4(CR6R7)vNR4R5.
13. Соединение по п. 10, в котором Y представляет собой -NR4(CR6R7)vNR4C(=NR4)NR4R5.
14. Соединение по п. 10, в котором Y представляет собой -NR4R5.
15. Соединение по п. 10, в котором Y представляет собой -NR4C(=NR4)NR4R5.
16. Соединение по п. 10, в котором Y представляет собой -(CR6R7)vNR4R5.
17. Соединение по п. 10, в котором Y представляет собой -(CR6R7)vNR4C(=NR4)NR4R5.
18. Соединение по п. 16, в котором v равно 2.
19. Соединение по п. 17, в котором v равно 1.
20. Соединение по п. 1, в котором каждый из R1 и R2 представляют собой атом водорода.
21. Соединение по п. 1, в котором соединение выбрано из группы, представленной следующими структурами:

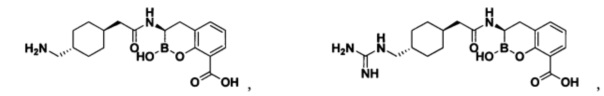
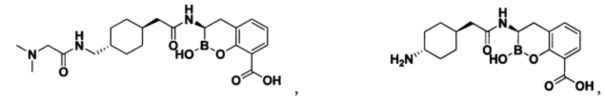


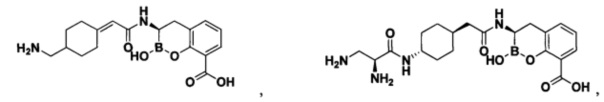
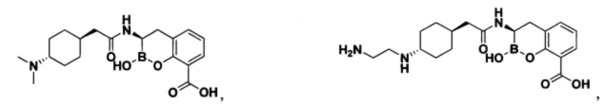
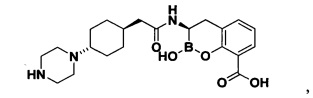
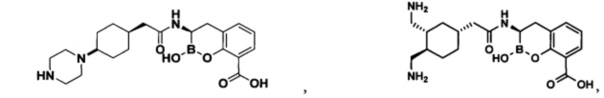



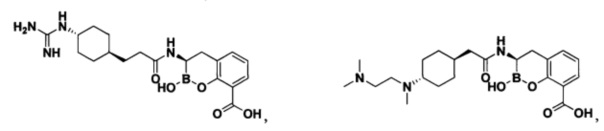
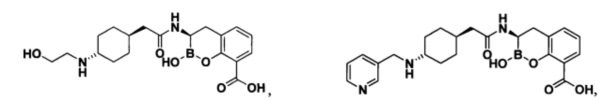
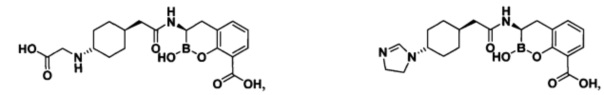
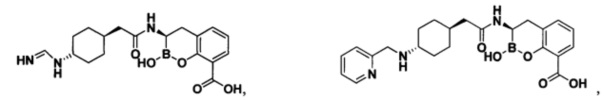
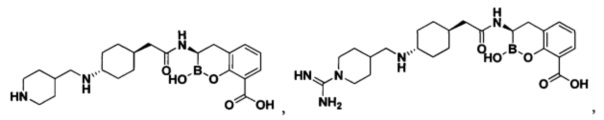
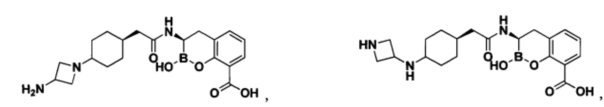


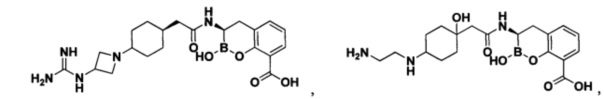


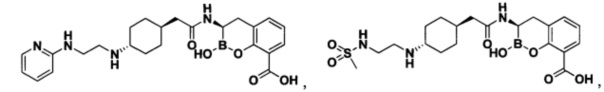
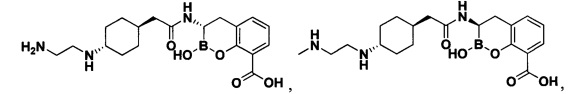
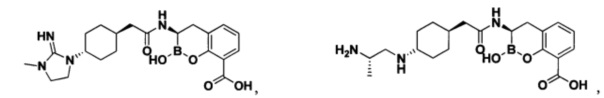
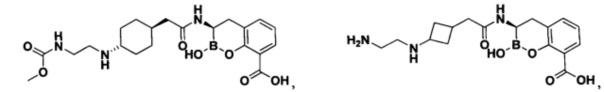

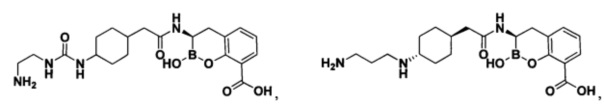
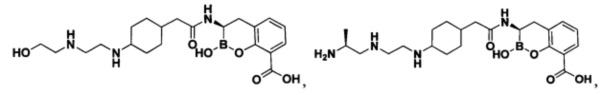
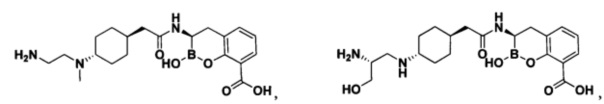
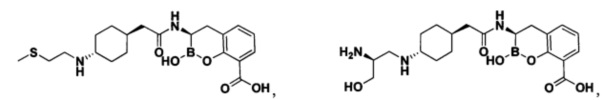

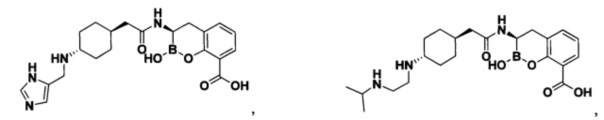
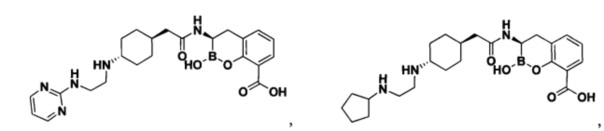


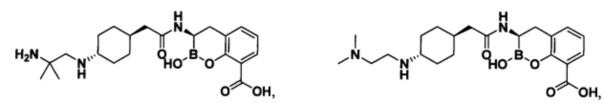
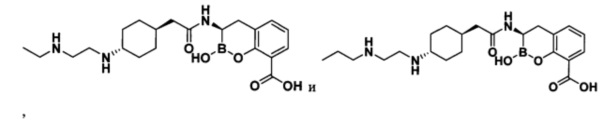
или его фармацевтически приемлемой соли или стереоизомера.
22. Фармацевтическая композиция для применения в лечении бактериальной инфекции, содержащая соединение по п. 1, или его фармацевтически приемлемую соль или стереоизомер, и фармацевтически приемлемый наполнитель.
23. Фармацевтическая композиция по п. 22, дополнительно содержащая бета-лактамный антибиотик.
24. Фармацевтическая композиция по п. 23, в которой бета-лактамным антибиотиком является пенициллин, цефалоспорин, карбапенем, монобактам, мостиковый монобактам, или их комбинация.
25. Способ лечения бактериальной инфекции у субъекта, включающий введение субъекту фармацевтической композиции по п. 22, в комбинации с бета-лактамным антибиотиком.
| WO 2010056827 A1, 20.05.2010 | |||
| US 20100292185 A1, 18.11.2010 | |||
| US 7271186 B1, 18.09.2007 | |||
| OLIV E | |||
| et al., Design, synthesis, crystal structures and antimicrobial activity of sulfonamide boronic acids as beta-lactamase inhibitors, J | |||
| Med | |||
| Chem., 2010, v | |||
| Веникодробильный станок | 1921 |
|
SU53A1 |
| Санный велосипед | 1925 |
|
SU7852A1 |
| ИНГИБИТОРЫ БЕТА-ЛАКТАМАЗ | 2009 |
|
RU2445314C9 |

