Техническая область изобретения
Настоящее изобретение относится к визуализации in vivo, в частности к визуализации методом позитронно-эмиссионной томографии (PET) периферических бензодиазепиновых рецепторов (PBR). Предложен радиоиндикатор для PET на основе индола, который связывается с PBR с высокой аффинностью, имеет хорошее всасывание в головной мозг после введения, и который обладает способностью превосходного селективного связывания с PBR. Согласно настоящему изобретению предложено также соединение-предшественник, полезное в синтезе радиоиндикатора для PET по изобретению, а также способ синтеза указанного соединения-предшественника. В других аспектах данного изобретения предложен способ синтеза радиоиндикатора для PET по изобретению, включающий использование соединения-предшественника по изобретению, набор для осуществления указанного способа и кассету для проведения автоматизированного варианта указанного способа. Кроме того, согласно изобретению предложена радиофармацевтическая композиция, содержащая радиоиндикатор для PET по изобретению, а также способы применения указанного радиоиндикатора для PET.
Описание предшествующего уровня техники
Известно, что периферические бензодиазепиновые рецепторы (PBR) в основном локализованы в периферических тканях и глиальных клетках, но их физиологическая функция остается не до конца выясненной. PBR также называют белками-транслокаторами (TSPO). Известно, что внутриклеточно PBR локализуются на внешней митохондриальной мембране, что указывает на их потенциальную роль в модулировании митохондриальной функции и в иммунной системе. Считается также, что PBR вовлечены в клеточную пролиферацию, стероидогенез, транспорт кальция и клеточное дыхание.
Аномальная экспрессия PBR связана с воспалительными болезненными состояниями центральной нервной системы (ЦНС), включая рассеянный склероз (Banati et al., 2001 Neuroreport, 12(16): 3439-42; Debruyne et al., 2002 Acta Neurol Belg, 102(3):127-35), энцефалит Расмуссена (Banati et al., 1999 Neurology, 53(9):2199-203) церебральный васкулит (Goerres et al., 2001 Am J Roentgenol, 176(4):1016-8), герпетический энцефалит (Cagnin et al., 2001 Brain, 124(Pt 10): 2014-27) и СПИД-ассоциированную деменцию (Hammoud et al., 2005 J Neurovirol; 11(4):346-55).
Кроме того, в ЦНС связь с PBR была документально подтверждена при дегенеративных заболеваниях, таких как болезнь Паркинсона (Gerhard et al., 2006 Neurobiol Dis, 21(2):404-12; Ouchi et al., 2005 Ann Neurol, 57(2):161-2), кортикобазальная дегенерация (Gerhard et al., 2004 Mov Disord, 19(10): 1221-6), прогрессирующий надъядерный паралич (Gerhard et al., 2006 Neurobiol Dis, 21(2): 404-12), множественная системная атрофия (Gerhard et al., 2003 Neurology, 61(5):686-9), болезнь Гентингтона (Pavese et al., 2006 Neurology, 66(11):1638-43; Tai et al., 2007 Brain Res Bui,; 72(2-3):148-51), амиотрофический боковой склероз (Turner et al., 2004 Neurobiol Dis, 15(3):601-9) и болезнь Альцгеймера (Cagnin et al., 2001 Lancet, 358(9283):766; Yasuno et al., 2008 Biol Psychiatry, 64(10):835-41).
Было показано, что с аномальной экспрессией PBR связан целый ряд ишемических состояний ЦНС, в том числе ишемический инсульт (Gerhard et al., 2005 Neuroimage, 24(2): 591-5), повреждение периферических нервов (Banati et al., 2001 Neuroreport, 12(16):3439-42), эпилепсия (Sauvageau, 2002 Metab Brain Dis, 17(1):3-11; Kumar et al., 2008 Pediatr Neurol, 38(6)). Считают, что PBR является биомаркером для определения степени повреждения при травме головного мозга (Toyama et al., 2008 Ann Nucl Med, 22(5):417-24), и сообщают об увеличении экспрессии PBR в животной модели травматического повреждения головного мозга (Venneti et al., 2007 Exp Neurol, 207(1): 118-27). Интересно, что острый стресс коррелируется с увеличением экспрессии PBR в головном мозге, тогда как хронический стресс коррелируется с понижающей регуляцией PBR (Lehmann et al., 1999 Brain Res, 851(1-2):141-7). Было сообщение о возможности определения формы и размеров глиомы с использованием [11С]PK11195 для визуализации PBR (Junck et al., 1989 Ann Neurol, 26(6):752-8). PBR могут быть также ассоциированы с невропатической болью; Tsuda et al наблюдали активированную микроглию у субъектов с невропатической болью (2005 TINS 28(2) рр101-7).
На периферии экспрессия PBR связана с воспалением легких (Branley et al., 2008 Nucl. Med. Biol, 35(8):901-9), хроническим обструктивным заболеванием легких и астмой (Jones et al., 2003 Eur Respir J, 21(4):567-73), воспалительным заболеванием кишечника (Ostuni et al., 2010 Inflamm Bowel Dis, 16(9):1476-1487), ревматоидным артритом (van der Laken et al., 2008 Arthritis Rheum, 58(11): 3350-5), первичной фибромиалгией (Faggioli et al., 2004 Rheumatology, 43(10):1224-1225), повреждением нерва (Durrenberger et al., 2004 J Peripher Nerv Syst, 9(1):15-25), атеросклерозом (Fujimura et al., 2008 Atherosclerosis, 201(1):108-111), раком ободочной кишки, предстательной железы и молочной железы (Deane et al., 2007 Mol Cancer Res, 5(4):341-9; Miettinen et al., 1995 Cancer Res, 55(12):2691-5; Han et al., 2003 J Recept Signal Transduct Res, 23(2-3): 225-38), воспалением почек (Tarn et al., 1999 Nephrol Dial Transplant, 14(7): 1658-66; Cook et al., 1999 Kidney Int, 55(4):1319-26) и ишемическим реперфузионным повреждением (Zhang et al., 2006 J Am Coll Surg, 203(3):353-64).
Визуализация методом позитронно-эмиссионной томографии (PET) с использованием селективного лиганда PBR, (R)-[11C]PK11195, предусматривает использование наиболее широко распространенного индикатора воспаления центральной нервной системы (ЦНС). Однако известно, что (R)-[11C]PK11195 обладает высокой степенью связывания белка и низкой специфичностью к неспецифическому связыванию. К тому же, роль его метаболитов, меченных радиоизотопом, неизвестна, и количественное определение связывания требует сложного моделирования. Поэтому были предприняты усилия по разработке агента визуализации для PBR in vivo, который не имеет этих проблем. Одним таким агентом визуализации in vivo является производное трициклического индола, описанное в WO 2010/109007, которое обладает хорошей аффинностью к PBR, превосходным поглощением в головном мозге и специфичностью в отношении PBR, и высокая доля радиоактивности присутствует в головном мозге через 60 минут после инъекции родительского агента визуализации in vivo. В WO 2010/109007 раскрыто, что особенно предпочтительным агентом визуализации in vivo является следующее 18F-меченое соединение:
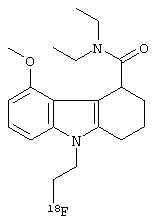
Существуют возможности для дополнительного улучшения визуализирующего агента для визуализации PBR in vivo.
Краткое изложение сущности изобретения
Согласно настоящему изобретению предложен радиоиндикатор для PET, который сохраняет преимущественные свойства известного радиоиндикатора для PET на основе трициклического индола, а также обладает целым рядом улучшенных свойств. Было продемонстрировано, что по сравнению с известным трициклическим радиоиндикатором для PET радиоиндикатор для PET по изобретению обладает улучшенной аффинностью связывания с PBR, слегка улучшенным профилем метаболизма с высокой долей активности через 60 минут после инъекции, представляющей собой активность в головном мозге, и значительно улучшенной специфичностью связывания с PBR-экспрессирующими тканями. Согласно настоящему изобретению также предложено соединение-предшественник, полезное в получении радиоиндикатора для PET по изобретению, а также способы получения указанного соединения-предшественника и указанного радиоиндикатора для PET. Согласно настоящему изобретению также предложена радиофармацевтическая композиция, содержащая радиоиндикатор для PET по изобретению. Способы применения радиоиндикатора для PET и радиофармацевтической композиции также предложены.
Подробное описание изобретения
Радиоиндикатор для PET
В одном аспекте настоящего изобретения предложен радиоиндикатор для позитронно-эмиссионной томографии (PET), имеющий следующую химическую структуру:
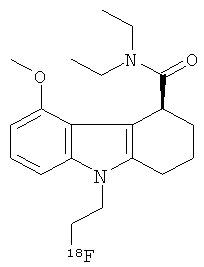 ,
,
где хиральный центр имеет (S) конфигурацию.
"Радиоиндикатор для PET" представляет собой химическое соединение, которое содержит позитрон-испускающий изотоп, причем данное химическое соединение предназначено для нацеливания на конкретную физиологию или патофизиологию в биологической системе. Присутствие позитрон-испускающего изотопа дает возможность детектировать радиоиндикатор для PET после введения в биологическую систему и тем самым способствовать обнаружению конкретной физиологии или патофизиологии.
Было показано, что радиоиндикатор для PET по изобретению обладает аффинностью, почти в 5 раз более высокой, чем аффинность его альтернативного энантиомера, и почти в два раза выше, чем аффинность рацемической смеси. Было также обнаружено, что радиоиндикатор для PET по изобретению лучше функционирует in vivo по сравнению с его альтернативным энантиомером. Радиоиндикатор для PET по изобретению также лучше функционирует in vivo по сравнению с рацемической смесью, содержащей указанный радиоиндикатор для PET и его альтернативный энантиомер.
Альтернативный энантиомер радиоиндикатора для PET по изобретению имеет следующую структуру:
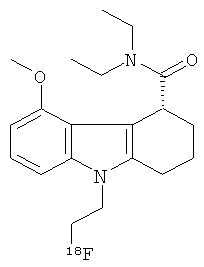 ,
,
где хиральный центр имеет (R) конфигурацию.
Используемый в настоящем изобретении термин "энантиомер" относится к энантиочистому соединению, т.е. к одной из двух зеркально противоположных форм оптически активной молекулы. Таким образом, энантиомер является соединением, имеющим только одну хиральность, где термин "хиральность" относится к такому свойству соединения, в силу которого он не имеет внутренней плоскости симметрии и не совмещается со своим зеркальным отражением. Признаком, который наиболее часто обусловливает хиральность в химических соединениях, является наличие асимметрического атома углерода. Эквимолярная смесь пары энантиомеров называется "рацематом" или "рацемической смесью".
В эксперименте по биораспределению, описанному в Примере 9, показано, что радиоиндикатор для PET по изобретению имеет улучшенное связывание с PBR-богатой тканью в головном мозге (т.е. в обонятельной луковице) по сравнению и с его альтернативным энантиомером и с рацемической смесью. Результаты эксперимента in vivo по блокированию, который описан в Примере 11, подтверждают эти данные. Результаты эксперимента, который описан в Примере 10, демонстрируют, что активность в головном мозге через 60 минут, обусловленная родительским соединением, улучшена в пользу радиоиндикатора для PET по изобретению по сравнению с рацемической смесью радиоиндикатора для PET и его альтернативного энантиомера. Более того, в эксперименте по авторадиографии, который описан в Примере 12, продемонстрировано, что радиоиндикатор для PET по изобретению в более высокой степени избирательно связывается с участками нейровоспаления по сравнению с рацемической смесью, содержащей указанный радиоиндикатор для PET и его альтернативный энантиомер. Было также установлено, что радиоиндикатор для PET по изобретению не подвергается рацемизации после инкубирования в плазме крови человека или во фракции S9 крысы в течение длительных периодов времени, как описано в Примере 8 ниже.
Соединение-предшественник
Радиоиндикатор для PET по изобретению может быть получен через подходящее соединение-предшественник. Поэтому в еще одном аспекте настоящего изобретения предложено соединение-предшественник для получения радиоиндикатора для PET по изобретению, где указанное соединение-предшественник представляет собой соединение формулы I:
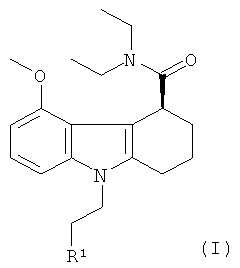 ,
,
где R1 представляет собой гидроксил или уходящую группу.
"Соединение-предшественник" содержит нерадиоактивное производное радиоиндикатора для PET по изобретению, рассчитанное на то, чтобы химическая реакция с удобной химической формой 18F происходила сайт-специфично, могла быть проведена в минимальное количество стадий (в идеале в одну стадию) и без необходимости в значительной очистке (в идеале без дополнительной очистки) с получением радиоиндикатора для PET по изобретению. Такие соединения-предшественники являются синтетическими и легко могут быть получены с хорошей химической чистотой.
"Уходящая группа" в контексте настоящего изобретения относится к атому или группе атомов, который(ая) удаляется как стабильный вид радикала в ходе реакции радиофторирования с замещением или заменой. Примерами подходящих уходящих групп являются галогеновые группы хлоро, бромо и йодо и сульфонатные эфирные группы мезилат, тозилат нозилат и трифлат. Предпочтительно указанная уходящая группа выбрана из мезилата, тозилата и трифлата и наиболее предпочтительно представляет собой мезилат. В тех случаях, когда уходящая группа представляет собой мезилат, соединение-предшественник в данном описании называется "соединением-предшественником 1".
Получение соединения-предшественника
Соединение-предшественник по изобретению может быть получено различными путями, каждый из которых составляет отдельный аспект настоящего изобретения.
Соответственно согласно настоящему изобретению предложен первый способ получения соединения-предшественника формулы I, как оно определено в данном описании, включающий:
(1) приготовление рацемической смеси указанного соединения-предшественника формулы I, как оно определено в данном описании, и соединения формулы II:
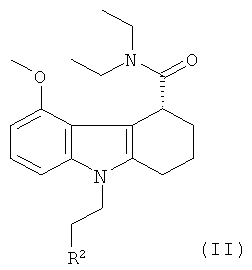 ,
,
где R2 такой, как определено выше для R1, и R1 и R2 одинаковые;
(2) отделение указанного соединения-предшественника формулы I от указанного соединения формулы II.
Стадию "отделения" указанного соединения-предшественника формулы I от указанного соединения формулы II проводят методом разделения энантиомеров. Подходящие методы разделения энантиомеров включают высокоэффективную жидкостную хроматографию (HPLC), сверхкритическую флюидную хроматографию (SFC), хроматографию в моделированном слое (SBC). Подробную оценку различных методов, которые могут быть использованы для разделения энантиомеров, можно найти в "Chiral Separation Techniques: a Practical Approach" (2007 Wiley; Subramanian, Ed.).
Схема 1
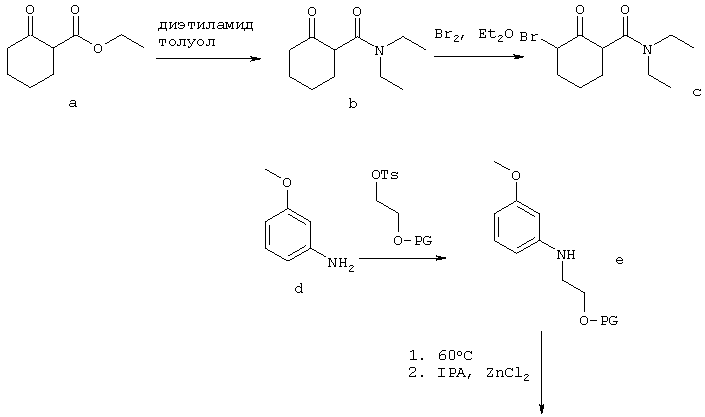
Схема 1, приведенная выше, иллюстрирует один способ получения рацемической смеси соединения-предшественника формулы I и соединения формулы II. На Схеме 1 PG означает защитную группу для гидроксильной группы; LG означает уходящую группу, как она определена в данном описании; OTs означает уходящую группу тозилат; и IPA означает изопропиловый спирт. Соединение g представляет собой соединение-предшественник по изобретению, где R1 представляет собой гидроксил. Подходящие защитные группы гидроксильной группы общеизвестны в данной области и включают ацетил, бензил, бензоил, силиловые эфиры, алкиловые эфиры и алкоксиметиловые эфиры. Защитные группы более подробно рассмотрены в Theorodora W. Greene and Peter G.M. Wuts, "Protective Groups in Organic Synthesis" (Fourth Edition, John Wiley & Sons, 2007). В контексте настоящего изобретения предпочтительной защитной группой для гидроксильной группы является бензил. Схема 1, приведенная выше, основана на способах получения сходных соединений, описанных в Napper et al., J Med Chem 2005, 48: 8045-54, и Davies et al., J Med Chem 1998, 41: 451-467.
Схема 2
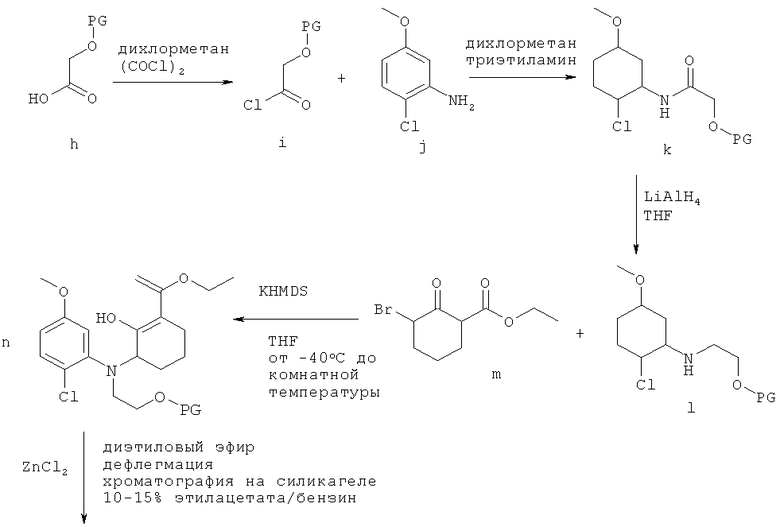
Схема 2 (продолжение)
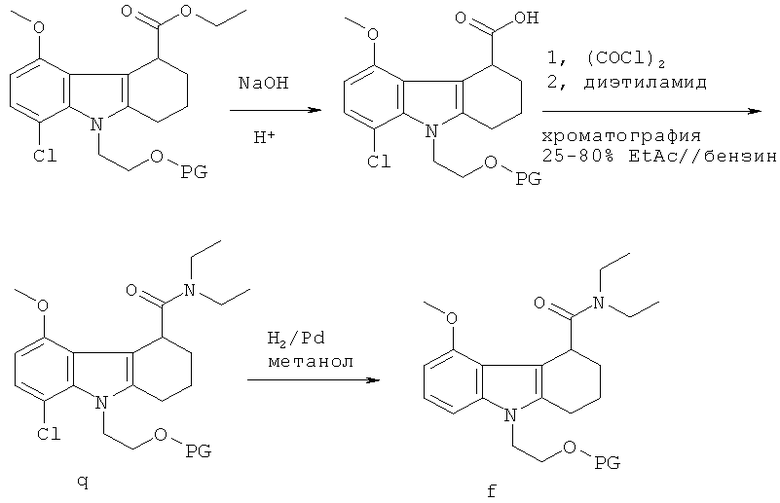
Альтернативный способ получения рацемической смеси соединения-предшественника формулы I и соединения формулы II иллюстрируется на Схеме 2, приведенной выше. На Схеме 2 PG означает защитную группу для гидроксильной группы, как она определена выше, THF означает тетрагидрофуран, KHMDS означает бис(триметилсилил)амид калия. Начиная с соединения f, Схема 2 продолжается, как показано на Схеме 1, от соединения f до получения в результате рацемической смеси. Схема 2 основана на способе, раскрытом в WO 2003/014082. Согласно этому пути синтеза хлор в нижнем положении с левой стороны кольца заставляет протекать циклизацию только одним способом. Однако когда авторы настоящего изобретения напрямую применили методы из WO 2003/014082 для получения рацемической смеси соединения-предшественника формулы I и соединения формулы II, выход оказался низким. Эта проблема была решена путем замены системы растворителей, используемой для стадии циклизации. В WO 2003/014082 стадию циклизации проводят в толуоле, тогда как авторы настоящего изобретения обнаружили, что оптимальные выходы достигаются при использовании диэтилового эфира вместо толуола. Продукт стадии циклизации растворяется в диэтиловом эфире, а не подвергшееся циклизации исходное соединение не растворяется. Не подвергшееся циклизации исходное соединение поэтому остается вместе с ZnCl2 на дне реакционного сосуда, а циклизованный продукт перемещается в диэтиловый эфир в верхней части реакционного сосуда.
Второй способ получения соединения-предшественника формулы I включает:
(1) приготовление соединения формулы III:
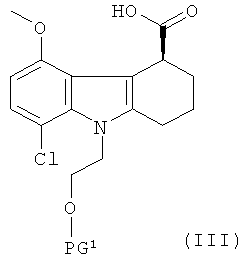 ,
,
где PG1 представляет собой защитную группу для гидроксильной группы;
(2) превращение указанного соединения формулы III в его соответствующий хлорангидрид;
(3) взаимодействие хлорангидрида, полученного на стадии (2), с диэтиламидом с получением соединения формулы IV:
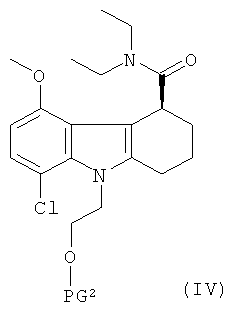 ,
,
где PG2 представляет собой защитную группу для гидроксильной группы и является такой же, как PG1;
(4) удаление защитной группы с соединения формулы IV, полученного на стадии (3), с получением гидроксильного производного;
(5) возможно присоединение уходящей группы, как она определена в данном описании.
Обе стадии (4) и (5), т.е. стадия (4), где R1 формулы I представляет собой гидроксил, и стадия (5), где R1 в формуле I представляет собой уходящую группу, приводят к образованию соединения-предшественника формулы I, как оно определено в данном описании.
Стадия (2) "превращения" указанного соединения формулы III в хлорангидрид может быть проведена с использованием реагента, выбранного из оксалилхлорида, тионилхлорида, трихлорида фосфора или пентахлорида фосфора. Оксалилхлорид является предпочтительным.
Стадия "удаления защитной группы" относится к удалению защитной группы для гидроксильной группы и может быть осуществлена способами, общеизвестными специалистам в данной области. Защитная группа PG1 для гидроксильной группы является такой, как определено выше для PG на Схеме 1. Используемый способ предназначен специально для конкретной защитной группы для гидроксильной группы. Типичные стратегии для удаления защитных групп для гидроксильной группы включают гидрогенолиз и обработку кислотой или основанием.
Стадия "присоединения" уходящей группы может быть осуществлена путем взаимодействия соединения g, указанного на приведенной выше Схеме 1, с галогенидным производным желаемой уходящей группы в подходящих реакционных условиях. Например, для присоединения мезилата соединение g, указанное на приведенной выше Схеме 1, может быть подвергнуто взаимодействию с метансульфонилхлоридом в присутствии основания, например аминного основания, такого кактриэтиламин.
На стадии (1) указанного второго способа получения соединения-предшественника формулы I соединение формулы III может быть приготовлено различными путями, например путем осуществления способа, включающего:
(а) приготовление рацемической смеси соединения формулы V и соединения формулы VI:
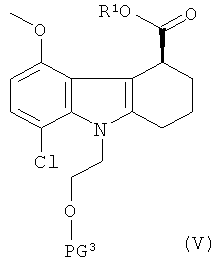
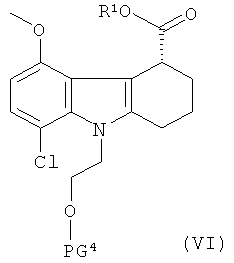
где:
R1 представляет собой хиральный спирт; и
PG3 и PG4 одинаковые, и каждая представляет собой защитную группу для гидроксильной группы;
(б) отделение соединения формулы V от соединения формулы VI;
(в) удаление R1 из отделенного соединения формулы V в кислотных условиях с получением указанного соединения формулы III.
Определение термина "рацемическая смесь" приведено в данном описании выше. Термин "хиральный спирт" относится к энантиомеру оптически активного спирта, и определение термина "энантиомер" приведено в данном описании выше. Термин "спирт" относится к органическому соединению, которое содержит гидроксильную группу, присоединенную к атому углерода. Предпочтительными хиральными спиртами для использования в описанном выше способе являются ментол и борнеол.
Хиральный спирт отщепляют от отделенного соединения формулы V кислотным гидролизом. Подходящие кислоты для использования на этой стадии включают соляную кислоту или серную кислоту, предпочтительно 2-молярную соляную кислоту или 1-молярную серную кислоту.
В альтернативном аспекте соединение формулы III может быть приготовлено с использованием способа, включающего:
(а) приготовление рацемической смеси указанного соединения формулы III и соединения формулы VIII:
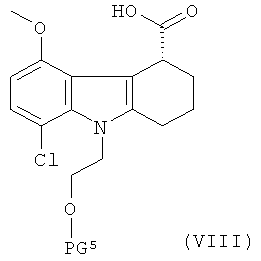 ,
,
где PG5 представляет собой защитную группу для гидроксильной группы и является такой же, как PG1, которая определена выше для соединения формулы III;
(б) взаимодействие смеси, которая определена в стадии (а), с оптически активным амином для отделения указанного соединения формулы III от указанного соединения формулы VIII.
Рацемическая смесь указанного соединения формулы III и указанного соединения формулы VIII может быть получена согласно способу, проиллюстрированному на Схеме 2 выше, где желаемая рацемическая смесь представляет собой соединение р, которое там проиллюстрировано.
Подходящий оптически активный амин для использования в описанном выше способе может быть выбран из S-альфа-метилбензиламина, R-(+)-N-(1- нафтилметил)-альфа-бензиламина, N-(2-гидрокси)этил-альфа-метилбензиламина и 1-(пара-толил)-этиламина. Другие оптически активные амины, подходящие для использования в описанном выше способе, легкодоступны из коммерческих источников, например от химической компании Aldrich.
В результате осуществления стадии (б) взаимодействия смеси со стадии (а) с оптически активным амином для отделения указанного соединения формулы III от указанного соединения формулы IV вначале образуются две диастереоизомерные соли. Эти диастереоизомерные соли разделяют путем кристаллизации из подходящего растворителя, такого как ацетон или этилацетат.Разделенные соли обрабатывают минеральной кислотой, такой как 2 н. соляная кислота или 1М серная кислота, для восстановления указанного соединения формулы III, отделенного от указанного энантиомера формулы VIII. Соединение формулы III затем выделяют экстракцией в этилацетат, отделяют от водного слоя и концентрируют в вакууме с получением энантиомера формулы III.
В еще одном варианте соединение формулы III может быть получено способом, включающим:
(а) приготовление рацемической смеси соединения формулы IX и соединения формулы X:
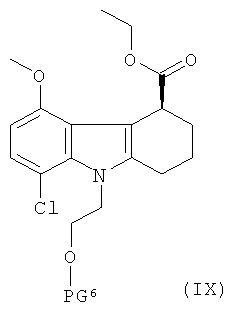
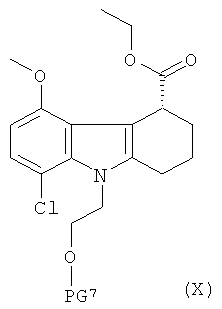 ,
,
где PG6 и PG7 одинаковые, и каждая представляет собой защитную группу для гидроксильной группы;
(б) взаимодействие смеси, которая определена в стадии (а), со стереоселективным ферментом с получением указанного соединения формулы III, где указанный стереоселективный фермент осуществляет гидролиз соединения формулы IX с расщеплением сложноэфирной связи.
Рацемическая смесь указанного соединения формулы IX и указанного соединения формулы X может быть получена способом, проиллюстрированным на Схеме 2, где желаемая рацемическая смесь представляет собой соединение о, которое там изображено.
Подходящий стереоселективный фермент для использования в описанном выше способе может быть выбран из липазы В Candida antarctica, эстеразы свиной печени, липазы свиной поджелудочной железы или других известных стереоселективных ферментов, которые действуют подобным образом.
Получение радиоиндикатора для PET
В следующем аспекте настоящего изобретения предложен способ получения радиоиндикатора для PET по изобретению, включающий взаимодействие соединения-предшественника формулы I с подходящим источником 18F. Реакция с 18F может быть осуществлена путем нуклеофильного замещения уходящей группы, присутствующей в положении R1 соединения-предшественника формулы I. Введение метки в соединение-предшественник может быть осуществлено в одну стадию в результате взаимодействия с подходящим источником [18F]-фторид-иона (18F-), который обычно получают в виде водного раствора, образующегося в результате ядерной реакции 18O(p,n)18F, и активируют путем добавления катионного противоиона и последующего удаления воды. Подходящие катионные противоионы должны иметь достаточную растворимость в безводном реакционном растворителе для поддержания растворимости 18F- . Поэтому противоионы, которые были использованы, включают большие, но мягкие ионы металлов, таких как рубидий и цезий, калия в виде комплекса с криптандом, таким как Kryptofix™, или тетраалкиламмониевые соли.
Предпочтительным противоионом является калий в виде комплекса с криптандом, таким как Kryptofix™, ввиду его хорошей растворимости в безводных растворителях и повышенной реакционной способности в отношении 18F-. 18F может быть также введен путем О-алкилирования гидроксильной группы в положении R1 в соединении-предшественнике соединением 18F(CH2)3-LG, где LG представляет собой уходящую группу, как она определена выше.
Более подробное обсуждение общеизвестных методов 18F-мечения можно найти в Главе 6 справочника "Handbook of Radiopharmaceuticals" (2003; John Wiley and Sons: M.J. Welch and C.S. Redvanly, Eds.).
В предпочтительном воплощении способ получения радиоиндикатора для PET по изобретению является автоматизированным. [18F]-радиоактивные индикаторы легко могут быть получены в автоматическом режиме при помощи автоматизированного аппарата для синтеза радиоактивных веществ. Есть несколько примеров таких аппаратов, которые коммерчески доступны, в том числе TRACERlab™ и FASTlab™ (оба коммерчески доступны от отделения GE Healthcare компании General Electric Company). Такой аппарат обычно содержит "кассету", часто одноразовую, в которой осуществляется радиохимический процесс, которая сопряжена с аппаратом для синтеза радиоактивных веществ. Кассета обычно включает в себя магистрали для текучей среды, реакционный сосуд и каналы для установки виал с реагентами, а также любые картриджи для твердофазной экстракции, используемые на стадиях очистки после синтеза радиоактивного вещества.
Таким образом, в другом аспекте настоящего изобретения предложена кассета для автоматизированного синтеза радиоиндикатора для PET, как он определен в данном описании, содержащая:
1) сосуд, содержащий соединение-предшественник формулы I, как он определен в данном описании; и
2) средства для элюирования сосуда стадии (1) подходящим источником 18F, как он определен в данном описании.
Для кассеты по изобретению подходящие и предпочтительные воплощения соединения-предшественника формулы I и подходящего источника 18F являются такими, как определено в данном описании выше.
Кассета дополнительно может содержать:
3) ионообменный картридж для удаления избытка 1SF.
Радиофармацевтическая композиция
В еще одном аспекте настоящего изобретения предложена радиофармацевтическая композиция, содержащая радиоиндикатор для PET, как он определен в данном описании, вместе с биосовместимым носителем, подходящим для введения млекопитающим.
"Биосовместимый носитель" представляет собой текучую среду, в частности жидкость, в которой радиоиндикатор для PET по изобретению суспендируют или растворяют, чтобы радиофармацевтическая композиция была физиологически переносимой, например чтобы ее можно было вводить в организм млекопитающего без токсического эффекта или чрезмерного дискомфорта. Биосовместимый носитель представляет собой соответственно инъецируемый носитель-жидкость, такой как стерильная, апирогенная вода для инъекций; водный раствор, такой как физиологический раствор (который предпочтительно может быть сбалансирован таким образом, чтобы конечный продукт для инъекций был либо изотоническим, либо негипотоническим); водный раствор одного или более регулирующих тоничность веществ (например, солей катионов плазмы с биосовместимыми противоионами), сахара (например, глюкозу или сахарозу), сахарные спирты (например, сорбит или маннит), гликоли (например, глицерин) или неионные полиолы (например, полиэтиленгликоли, пропиленгликоли и тому подобное). Биосовместимый носитель может также содержать биосовместимые органические растворители, такие как этанол. Такие органические растворители полезны для солюбилизации более липофильных соединений или композиций. Предпочтительно биосовместимый носитель представляет собой апирогенную воду для инъекций, изотонический физиологический раствор или водно-этанольный раствор. Подходящее значение рН биосовместимого носителя для внутривенных инъекций составляет от 4,0 до 10,5.
Радиофармацевтическую композицию можно вводить парентерально, т.е. инъекцией, и наиболее предпочтительно в виде водного раствора. Такая композиция возможно может содержать дополнительные ингредиенты, такие как буферные агенты; фармацевтически приемлемые солюбилизаторы (например, циклодекстрины или поверхностно-активные вещества, такие как Pluronic, Tween или фосфолипиды); фармацевтически приемлемые стабилизаторы или антиоксиданты (такие как этанол, аскорбиновая кислота, гентизиновая кислота или лара-аминобензойная кислота). В тех случаях, когда радиоиндикатор для PET по изобретению предоставляют в виде радиофармацевтической композиции, способ получения указанного радиоиндикатора для PET может дополнительно включать стадии, необходимые для получения радиофармацевтической композиции, например удаление органического растворителя, добавление биосовместимого буферного агента и любых возможных дополнительных ингредиентов. Для парентерального введения также необходимы стадии, обеспечивающие стерильность и апирогенность радиофармацевтической композиции. Такие стадии общеизвестны специалистам в данной области.
Способ визуализации методом PET
Радиоиндикатор для PET по изобретению полезен для детектирования in vivo экспрессии рецепторов PBR у субъекта. Поэтому в другом аспекте настоящего изобретения предложен способ визуализации методом PET для определения распределения и/или степени экспрессии PBR у субъекта, включающий:
1) введение указанному субъекту радиоиндикатора для PET, как он определен в данном описании;
2) предоставление возможности указанному радиоактивному индикатору для PET связаться с PBR у указанного субъекта;
3) детектирование сигналов, испускаемых 18F, который содержится в указанном связанном радиоиндикаторе для PET;
4) формирование изображения, отображающего локализацию и/или количество указанных сигналов; и
5) определение распределения и степени экспрессии PBR у указанного субъекта, где указанная экспрессия находится в прямой корреляции с указанными сигналами.
Стадию "введения" радиоиндикатора для PET предпочтительно проводят парентерально и наиболее предпочтительно внутривенно. Внутривенный путь введения является наиболее эффективным путем доставки радиоиндикатора для PET по всему организму субъекта и, следовательно, также через гематоэнцефалический барьер (ВВВ) и в контакт с PBR, экспрессированными в центральной нервной системе (ЦНС) указанного субъекта. Внутривенное введение не является существенным физическим вмешательством и не подвергает здоровье субъекта существенному риску. Радиоиндикатор для PET по изобретению предпочтительно вводят в виде радиофармацевтической композиции по изобретению, как она определена в данном документе. Стадия введения не является необходимой для полного определения способа визуализации методом PET по изобретению. Как таковой, способ визуализации методом PET по изобретению можно также рассматривать, как включающий определенные выше стадии (2)-(5), где указанный субъект стадии (2) является субъектом, которому предварительно был введен радиоиндикатор для PET по изобретению.
После стадии введения и перед стадией детектирования радиоиндикатору для PET предоставляют возможность связаться с PBR. Например, когда субъектом является интактное млекопитающее, радиоиндикатор для PET будет динамично продвигаться через организм млекопитающего, вступая в контакт с различными тканями в нем. Как только радиоиндикатор для PET вступает в контакт с PBR, происходит специфическое взаимодействие, так что клиренс радиоиндикатора для PET из ткани с PBR занимает больше времени, чем из ткани без PBR или с меньшим количеством PBR. Будет достигаться некоторая точка во времени, когда появляется возможность детектировать радиоиндикатор для PET, специфически связанный с PBR, как результат соотношения между радиоиндикатором для PET, связавшимся с PBR, и индикатором, который связался в ткани без PBR или в ткани с меньшим количеством PBR.
Стадия "детектирования" способа по изобретению включает детектирование сигналов, испускаемых 18F, который содержится в радиоиндикаторе для PET, с помощью детектора, чувствительного к указанным сигналам, т.е. РЕТ-камеры. Эту стадию детектирования можно также рассматривать как получение параметров сигналов.
Стадию "формирования" способа по изобретению проводят с помощью компьютера, который применяет алгоритм преобразования к полученным параметрам сигналов с получением набора данных. Этот набор данных затем обрабатывается для формирования изображений, показывающих локализацию и/или количество сигналов, испускаемых 18F. Испускаемые сигналы находятся в прямой корреляции с экспрессией PBR, так что стадия "определения" может быть выполнена путем оценки сформированного изображения.
"Субъектом" по изобретению может быть любой субъект-человек или субъект-животное. Предпочтительно субъектом по изобретению является млекопитающее. Наиболее предпочтительно указанный субъект представляет собой интактный организм млекопитающего in vivo. В особенно предпочтительном воплощении субъектом по изобретению является человек. Способ визуализации in vivo может быть использован для исследования PBR у здоровых субъектов или у субъектов, у которых, как известно или предполагается, имеется патологическое состояние, ассоциированное с аномальной экспрессией PBR (здесь далее "PBR-ассоциированное состояние"). Предпочтительно, указанный способ относится к визуализации in vivo субъекта, который, как известно или предполагается, имеет PBR-ассоциированное состояние и, следовательно, применим в способе диагностики указанного состояния.
Примеры таких PBR-ассоциированных состояний, когда может быть использована визуализация in vivo, включают рассеянный склероз, энцефалит Расмуссена, церебральный васкулит, герпетический энцефалит, СПИД-ассоциированную деменцию, болезнь Паркинсона, кортикобазальную дегенерацию, прогрессирующий надъядерный паралич, множественную системную атрофию, болезнь Гентингтона, амиотрофический боковой склероз, болезнь Альцгеймера, ишемический инсульт, повреждение периферического нерва, эпилепсию, травматическое повреждение головного мозга, острый стресс, хронический стресс, невропатическую боль, воспаление легких, хроническое обструктивное заболевание легких, астму, воспалительное заболевание кишечника, ревматоидный артрит, первичную фибромиалгию, повреждение нерва, атеросклероз, воспаление почек, ишемическое реперфузионное повреждение и рак, в частности рак ободочной кишки, предстательной железы или молочной железы. Радиоиндикатор для PET по изобретению благодаря его хорошему всасыванию в головном мозге пригоден, в частности, для визуализации ЦНС in vivo.
В альтернативном воплощении способ визуализации методом PET по изобретению можно проводить неоднократно во время осуществления схемы лечения указанного субъекта, причем указанная схема включает введение лекарственного средства для противодействия PBR-ассоциированному состоянию. Например, способ визуализации методом PET по изобретению можно проводить до, во время и после лечения лекарственным средством для противодействия PBR-ассоциированному состоянию. Таким способом эффект указанного лечения можно отслеживать во времени. PET особенно хорошо подходит для такого применения, поскольку этот метод имеет превосходные чувствительность и разрешение, так что даже относительно небольшие изменения в поражении можно наблюдать во времени, что особенно предпочтительно для мониторинга лечения.
В дополнительном аспекте настоящего изобретения предложен способ диагностики состояния, при котором имеет место повышающая регуляция PBR, включающий способ визуализации методом PET, как он определен выше, вместе с дополнительной стадией (6) соотнесения распределения и степени экспрессии PBR с конкретной клинической картиной.
В другом аспекте настоящего изобретения предложен радиоиндикатор для PET, как он определен в данном документе, для применения в определенном выше способе визуализации методом PET и определенном выше способе диагностики. Согласно настоящему изобретению также предложен радиоиндикатор для PET, как он определен в данном документе, для применения в изготовлении радиофармацевтической композиции, как она определена в данном документе, для применения в определенном выше способе визуализации методом PET и определенном выше способе диагностики.
Подходящие и предпочтительные аспекты любого признака, присутствующего во многих аспектах настоящего изобретения, являются такими, как определено для указанных признаков в первом аспекте, в котором они описаны в данном документе. Изобретение далее иллюстрируется примерами, не ограничивающими его объем.
Краткое описание Примеров
В Примере 1 описан синтез рацемической смеси, содержащей соединение-предшественник формулы I и энантиомер формулы II.
В Примере 2 описан синтез нерадиоактивной рацемической смеси, содержащей нерадиоактивный аналог радиоиндикатора для PET по изобретению вместе с его альтернативным энантиомером.
В Примере 3 описан синтез соединения-предшественника 1/активного энантиомера.
В Примере 4 описан синтез агента визуализации 1/активного энантиомера.
В Примере 5 описан синтез нерадиоактивного агента визуализации 1/активного энантиомера.
В Примере 6 описан способ, использованный для определения абсолютной стереохимии.
В Примере 7 описан анализ in vitro, использованный для оценки связывания нерадиоактивного рацемата 1 и его двух энантиомеров.
В Примере 8 описан способ, использованный для исследования хиральной стабильности радиоиндикатора для PET по изобретению in vitro.
В Примере 9 описан способ, использованный для оценки in vivo биораспределения радиоиндикатора для PET по изобретению, его альтернативного энантиомера и рацемической смеси двух энантиомеров.
В Примере 10 описан эксперимент для оценки метаболизма радиоиндикатора для PET по изобретению и рацемической смеси, содержащей указанный радиоиндикатор для PET и его альтернативный энантиомер.
В Примере 11 описан анализ на блокирование in vivo, использованный для оценки радиоиндикатора для PET по изобретению и рацемической смеси, содержащей указанный радиоиндикатор для PET и его альтернативный энантиомер.
В Примере 12 описана животная модель воспаления, использованная для оценки радиоиндикатора для PET по изобретению и рацемической смеси, содержащей указанный радиоиндикатор для PET и его альтернативный энантиомер.
Краткое описание графических материалов
Фиг.1 и Фиг.4 относятся к Примеру 4 и демонстрируют радиоактивные (вверху) и УФ (внизу) HPLC-хроматограммы, полученные с использованием полупрепаративного метода, для радиоиндикатора для PET по изобретению и его альтернативного энантиомера соответственно.
Фиг.2 и Фиг.5 относятся к Примеру 4 и демонстрируют HPLC-хроматограммы, полученные с использованием аналитического ахирального метода, для радиоиндикатора для PET по изобретению и его альтернативного энантиомера соответственно.
Фиг.3 и Фиг.6 относятся к Примеру 4 и демонстрируют HPLC-хроматограммы, полученные с использованием метода хиральной HPLC, для радиоиндикатора для PET по изобретению и его альтернативного энантиомера соответственно.
Фиг.7 относится к Примеру 8 и демонстрирует наложение хроматограмм радиоиндикатора для PET и альтернативного энантиомера, растворенных в ацетонитриле в концентрации 0,1 мг/мл.
Фиг.8а относится к Примеру 8 и демонстрирует хроматограмму радиоиндикатора для PET, растворенного в ацетонитриле в концентрации 0,1 мг/мл.
Фиг.8b относится к Примеру 8 и демонстрирует хроматограмму радиоиндикатора для PET (0,1 мг/мл), добавленного в плазму крови человека и экстрагированного перед инкубированием.
Фиг.8c относится к Примеру 8 и демонстрирует хроматограмму радиоиндикатора для PET (0,1 мг/мл), инкубированного с плазмой крови человека и экстрагированного.
Список сокращений, использованных в Примерах
Примеры
Пример 1: Синтез рацемической смеси мезилатного соединения-предшественника по изобретению ("соединение-предшественник 1") и его альтернативного энантиомера
Пример 1(а): Бензилоксиацетилхлорид (1)
К бензилоксиуксусной кислоте (10,0 г, 60,0 ммоль, 8,6 мл) в дихлорметане (50 мл) добавляли оксалилхлорид (9,1 г, 72,0 ммоль, 6,0 мл) и DMF (30,0 мг, 0,4 ммоль, 32,0 мкл), и эту смесь перемешивали при к.т.в течение 3 ч. Вначале происходило быстрое выделение газа при протекании реакции, но выделение прекращалось, когда реакция заканчивалась. Дихлорметановый раствор концентрировали в вакууме с получением смолы. Эту смолу обрабатывали дополнительным количеством оксалилхлорида (4,5 г, 35,7 ммоль, 3,0 мл), дихлорметаном (50 мл) и одной каплей DMF. Происходило быстрое выделение газа и реакционную смесь перемешивали в течение еще 2 ч. Реакционную смесь затем концентрировали в вакууме с получением 11,0 г (количественный выход) бензилоксиацетилхлорида (1) в виде смолы. Структура подтверждена 13С ЯМР (75 МГц, CDCl3) 5C 73.6, 74.8, 128.1, 128.4, 128.6, 130.0 и 171.9.
Пример 1(б): 2-Бензилокси-N-(2-хлор-5-метокси-фенил)ацетамид (2)
Бензилоксиацетилхлорид (1) (11,0 г, 60,0 ммоль) и гидрохлорид 2-хлор-5-метоксианилина (11,7 г, 60,2 ммоль) в дихлорметане (100 мл) перемешивали при 0°C и медленно добавляли триэтиламин (13,0 г, 126,0 ммоль, 18,0 мл) в течение 15 мин. Перемешиваемую реакционную смесь оставляли нагреваться до к.т.в течение 18 ч. Происходило осаждение гидрохлорида триэтиламина. Дихлорметановый раствор промывали 10%-ным водным раствором карбоната калия (50 мл), сушили над сульфатом магния и концентрировали в вакууме с получением 18,9 г (количественный выход) 2-бензилокси-N-(2-хлор-5-метокси-фенил)ацетамида (2) в виде смолы. Структура подтверждена 13С ЯМР (75 МГц, CDCl3): δC 55.6, 69.6, 73.6, 106.2, 111.1, 114.1, 127.7, 128.3, 128.6, 129.2, 134.6, 136.5, 158.9 и 167.7.
Пример 1(в): (2-Бензилокси-этил)-(2-хлор-5-метокси-фенил)амин (3)
2-Бензилокси-N-(2-хлор-5-метокси-фенил)ацетамид (2) (18,9 г, 62,0 ммоль) в THF (100 мл) перемешивали и медленно в течение 15 минут добавляли алюмогидрид лития (4,9 г, 130,0 ммоль). Сразу после добавления алюмогидрида лития происходило быстрое выделение газа водорода. Реакционную смесь затем нагревали до образования флегмы в течение 4 ч и оставляли стоять при к.т. в течение выходных дней. Реакционную смесь затем гасили добавлением к перемешиваемому раствору воды (50 мл) по каплям. Происходило интенсивное выделение водорода, вызывающее дефлегмацию реакционной смеси. Реакционную смесь затем концентрировали в вакууме до суспензии. Добавляли воду (200 мл) и этилацетат (200 мл) и смесь энергично встряхивали. Реакционную смесь затем фильтровали через целит для удаления выпавшего в осадок гидроксида алюминия, и этилацетатный раствор отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением 18,4 г (количественный выход) (2-бензилокси-этил)-(2-хлор-5-метоксифенил)амина (3) в виде смолы. Структура подтверждена 13С ЯМР (75 МГц, CDCl3) 6C 43.3, 55.3, 68.2, 73.0, 98.1, 101.8, 111.6, 127.6, 127.7, 128.4, 129.3, 137.9, 144.8 и 159.5.
Пример 1(г): Этиловый эфир 3-бром-2-гидрокси-циклогекс-1-ен карбоновой кислоты (4)
Этил-2-оксоциклогексанкарбоксилат (30 г, 176 ммоль, 28 мл) растворяли в диэтиловом эфире (30 мл) и охлаждали до 0°C в атмосфере азота. По каплям в течение 15 минут добавляли бром (28 г, 176 ммоль, 9,0 мл) и реакционную смесь оставляли нагреваться до к.т. в течение 90 мин. Смесь медленно вливали в ледяной насыщенный водный раствор карбоната калия (250 мл) и экстрагировали этилацетатом (3×200 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали, концентрировали в вакууме и сушили на вакуумной линии в течение 18 ч с получением 41,4 г (94%) этилового эфира 3-бром-2-гидрокси-1-ен карбоновой кислоты (4) в виде желтого масла. Структура подтверждена 13С ЯМР (75 МГц, CDCl3): 6C 14.1, 17.7, 21.8, 32.0, 60.0, 60.8, 99.7, 166.3, и 172.8.
Пример 1(д): Этиловый эфир 3-[(2-бензилокси-этил)-(2-хлор-5-метокси-фенил)-амино1-2-гидрокси-циклогекс-1-ен карбоновой кислоты (5)
(2-Бензилокси-этил)-(2-хлор-5-метоксифенил)амин (3) (10,0 г, 34,2 ммоль) перемешивали в сухом THF (100 мл) при -40°C в атмосфере азота и добавляли бис(триметилсилил)амид калия (143,0 мл 0,5 М раствора в толуоле, 72,0 ммоль) в течение 30 мин. Затем добавляли этиловый эфир 3-бром-2-гидроксициклогекс-1-ен карбоновой кислоты (4) (8,5 г, 34,2 ммоль) в сухом THF (10 мл) и смесь оставляли нагреваться до к.т. в течение периода времени 1,5 ч. Добавляли уксусную кислоту (10,0 г, 166 ммоль, 10,0 мл) и смесь концентрировали в вакууме для удаления THF. Добавляли этилацетат (200 мл) и 10%-ный водный раствор карбоната калия (100 мл) и смесь энергично встряхивали. Этилацетатный раствор отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением 16,5 г (количественный выход) этилового эфира 3-[(2-бензилокси-этил)-(2-хлор-5-метокси-фенил)-амино]-2-гидрокси-циклогекс-1-ен карбоновой кислоты (5) в виде смолы, которую использовали на следующей стадии неочищенной. HPLC (Gemini 150×4,6 мм, 50-95% метанола/вода за 20 мин) неочищенной реакционной смеси: 18,9 мин (38%), 19,2 мин (25%), 23,1 мин (28%).
Выделили один компонент реакционной смеси 13С ЯМР (75 МГц, CDCl3) 6С 14.3, 20.6, 21.8, 26.4, 38.6, 43.0, 55.8, 60.5, 68.7, 73.3, 93,4, 106.3, 108.2, 119.3, 121.5, 127.5, 127.6, 128.3, 135.7, 137.0, 137.9, 155.7 и 175.0.
Пример 1(е): Этиловый эфир 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (6)
Хлорид цинка (7,1 г, 52,0 ммоль) добавляли к этиловому эфиру 3-[(2-бензилокси-этил)-(2-хлор-5-метокси-фенил)-амино]-2-гидрокси-циклогекс-1-ен карбоновой кислоты (5) (8,0 г, 17,0 ммоль) в сухом диэтиловом эфире (150 мл) в атмосфере азота и нагревали при температуре дефлегмации в течение 5,5 ч. По мере дефлегмации реакционной смеси в результате реакции образовывалось густое коричневое плотное масло. Реакционную смесь затем охлаждали и надосадочный диэтиловый эфир декантировали, добавляли этилацетат (100 мл), промывали 2 н. HCl (50 мл) и 10%-ным водным раствором карбоната калия (50 мл). Диэтилэфирный слой отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением масла (2,0 г). Это неочищенное вещество очищали хроматографией на силикагеле, элюируя смесью бензин (А):этилацетат (В) (10-40% (В), 340 г, 22 CV (колоночных объемов), 150 мл/мин) с получением 1,8 г этилового эфира 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (6). Густой плотный коричневый слой обрабатывали этилацетатом (100 мл) и 2 н. HCI (50 мл). Этилацетатный раствор отделяли, промывали 10%-ным водным раствором карбоната калия (50 мл), сушили над сульфатом магния и концентрировали в вакууме с получением масла (5,2 г). Добавляли диэтиловый эфир (100 мл) и безводный хлорид цинка (7,0 г). Смесь нагревали при температуре дефлегмации в течение еще 5 суток. Эфирный слой декантировали с темной смолы, промывали 2 н. HCI (50 мл), сушили над сульфатом магния и концентрировали в вакууме с получением смолы (2,8 г). Эту смолу очищали хроматографией на силикагеле, элюируя смесью бензин (А): этил ацетат (В) (5-35% (В), 340 г, 150 мл/мин) с получением 2,1 г этилового эфира 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (6). В сумме получили 4,1 г (50%) этилового эфира 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (6). Структура подтверждена 13С ЯМР (75 МГц, CDCl3): 6C 14.4, 20.5, 22.3, 27.5, 40.2, 43.9, 55.0, 60.2, 70.7, 73.3, 100.2, 107.5, 108.4, 120.1, 122.8, 127.4, 127.5, 128.2, 132.0, 137.4, 138.1, 152.6 и 175.8.
Пример 1 (ж): 9-(2-Бензилокси-этил)-8-хлор-5-метокси-2,3.4,9-тетрагидро-1Н-карбазол-4-карбоновая кислота (7)
К этиловому эфиру 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (6) (2,0 г, 4,1 ммоль) в этаноле (50 мл) добавляли гидроксид натрия (1,1 г, 27,1 ммоль) и воду (5 мл) и смесь нагревали при 80°C в течение 18 ч. Этанол затем удаляли выпариванием в вакууме и остаток распределяли между диэтиловым эфиром (50 мл) и водой (50 мл). Диэтилэфирный слой отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением смолы (71,0 мг). Водный слой подкисляли до рН 1 добавлением 2 н. HCl (20 мл) и экстрагировали дихлорметаном (2×100 мл). Дихлорметановый слой сушили над сульфатом магния и концентрировали в вакууме с получением 1,6 г (87%) 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (7) в виде пены. Структура подтверждена 13С ЯМР (75 МГц; CDCl3): 6C 20.2, 22.2, 27.1, 39.7, 44.0, 55.1, 70.7, 73.3, 100.6, 106.3, 108.9, 123.0, 127.4, 127.5, 128.3, 132.0, 138.0 и 152.0.
Пример 1(з): 9-(2-Бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбонилхлорид (8)
9-(2-Бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновую кислоту (7) (1,5 г, 3,7 ммоль) растворяли в дихлорметане (50 мл) и добавляли оксалилхлорид (700 мг, 5,5 ммоль, 470 мкл) и DMF (1 каплю), и эту реакционную смесь перемешивали при 20°С в течение 2 ч. Происходило умеренное выделение газа в течение примерно 30 мин по мере протекания реакции. Реакционную смесь затем концентрировали в вакууме с получением 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбонилхлорида (8) в виде смолы, которую использовали на следующей стадии без очистки. Структура подтверждена 13С ЯМР (75 МГц; CDCl3): δС 20.8, 22.1, 26.4, 44.2, 51.8, 55.1, 70.7, 73.3, 100.7, 106.0, 108.6, 119.5, 123.4, 127.3, 127.7, 128.3, 131.9, 138.0, 138.2, 152.0 и 176.3.
Пример 1(и): Диэтиламид 9-(2-бензилокси-этил)-8-хлор-5-метокси-2.3.4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (9)
9-(2-Бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1 Н-карбазол-4-карбонилхлорид (8) (1,6 г, 3,7 ммоль) затем растворяли в дихлорметане (50 мл), охлаждали до 0°C, перемешивали и по каплям добавляли диэтиламин (810 мг, 11,0 ммоль, 1,1 мл). Реакционную смесь оставляли нагреваться до комнатной температуры в течение периода времени 18 ч. Реакционную смесь затем промывали 10%-ным водным раствором карбоната калия (50 мл), выделяли, сушили над сульфатом магния и концентрировали в вакууме до смолы. Это неочищенное вещество подвергали кристаллизации из диэтилового эфира с получением 1,2 г (71%) диэтиламида 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (9) в виде белого кристаллического твердого вещества. Структура подтверждена 13С ЯМР (75 МГц; CDCl3): δC 13.0, 14.5, 19.8, 22.2, 27.9, 36.4, 40.4, 41.9, 43.8, 55.0, 70.8, 73.3, 100.2, 108.5, 108.6, 119.9, 122.5, 127.4, 127.5, 128.3, 131.5, 137.8, 138.2, 152.4 и 174.5.
Пример 1(к): 9-(2-Бензилокси-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновая кислота-диэтиламин (10)
Диэтиламид 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (9) (1,0 г, 2,1 ммоль) в метаноле (100 мл) встряхивали с 10%-ным палладием на угле (1,0 г) и триэтиламином (2,9 мг, 2,9 ммоль, 4 мкл) в атмосфере газа водорода в течение 18 ч при 55°C. Реакционную смесь затем фильтровали через слой целита и фильтрат концентрировали в вакууме с получением смолы (908 мг). Смолу затем переносили в дихлорметан (100 мл) и промывали 5%-ным водным раствором карбоната калия (50 мл). Дихлорметановый раствор затем отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением смолы. Смолу затем подвергали кристаллизации из диэтилового эфира (50 мл) и кристаллы собирали фильтрованием с получением 523 мг (57%) 9-(2-бензилокси-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновая кислота-диэтиламина (10). Структура подтверждена 13С ЯМР (75 МГц; CDCl3): δС 13.1, 14.6, 20.1, 22.0, 28.1, 36.4, 40.5, 42.0, 43.0, 54.7, 68.8, 73.3, 99.4, 102.4, 107.8, 116.4, 121.2, 127.6, 127.6, 128.3, 135.6, 137.8, 138.0 153.6 и 175.0.
Пример 1 (л): 9-(2-Гидроксиэтил)-5-метокси-2,3,4,9-тетрагидро-1 Н-карбазол-4-карбоновая кислота-диэтиламин (11)
9-(2-Бензилокси-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновая кислота-диэтиламин (10) (1,0 г, 2,1 ммоль) в метаноле (50 мл) встряхивали с 10%-ным палладием на угле (300 мг) и избытком газообразного водорода в течение 18 ч при 55°C. Реакционную смесь затем фильтровали через слой целита и фильтрат концентрировали в вакууме с получением 578 мг (100%) 9-(2-гидроксиэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновая кислота-диэтиламина (11) в виде пены. Структура подтверждена 13С ЯМР (75 МГц; CDCl3): 6C 13.0, 14.4, 20.0, 22.0, 28.0, 36.4, 40.6, 42.0, 54.7, 60.6, 99.2, 102.6, 107.0, 116.7, 121.1, 136.1, 137.5, 138.0 153.5 и 175.7.
Пример 1(м): 2-(4-Диэтилкарбамил-5-метокси-1,2,3,4-тетрагидро-карбазол-9-ил)этиловый эфир метансульфоновой кислоты
9-(2-Гидроксиэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновая кислота-диэтиламин (11) (478 мг, 1,4 ммоль) в дихлорметане (30 мл) охлаждали до 0°C, добавляли метансульфонилхлорид (477 мг, 4,2 ммоль, 324 мкл) и триэтиламин (420 мг, 4.2 ммоль, 578 мкл), и смесь оставляли нагреваться до к.т.в течение ночи. Реакционную смесь промывали 5%-ным водным раствором карбоната калия. Слои разделяли. Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме с получением смолы (696 мг). Это неочищенное вещество очищали хроматографией на силикагеле, элюируя смесью бензин (А):этилацетат (В) (75-100% В, 22 CV, 120 г, 85 мл/мин) с получением 2-(4-диэтилкарбамил-5-метокси-1,2,3,4-тетрагидро-карбазол-9-ил)этилового эфира метансульфоновой кислоты в виде смолы, которую подвергали кристаллизации из диэтилового эфира с получением 346 мг (59%) бесцветного твердого вещества. Структура подтверждена 13С ЯМР (75 МГц; CDCl3): 6C 13.1, 14.5, 20.0, 21.9, 28.0, 36.3, 36.7, 40.3, 41.8, 41.9, 54.7, 68.1, 100.0, 102.0, 109.0, 116.4, 122.0, 135.1, 137.3, 153.8 и 174.6.
Пример 2: Синтез рацемической смеси нерадиоактивного аналога радиоиндикатора для PET по изобретению и его альтернативного энантиомера
Пример 2(а): Фторэтилтозилат (12)
2-Фторэтанол (640 мг, 10 ммоль, 0,6 мл) растворяли в пиридине (10 мл) в атмосфере азота. Раствор перемешивали при 0°C, и к этому раствору порциями добавляли тозилхлорид (4,2 г, 21,8 ммоль) за период времени 30 мин, поддерживая температуру ниже 5°C. Реакционную смесь перемешивали при 0°C в течение 3 ч. Медленно добавляли лед, затем добавляли воду (20 мл). Реакционную смесь экстрагировали в этилацетат и промывали водой. Избыток пиридина удаляли путем промывки 1 н. раствором HCl до тех пор, пока водный слой не становился кислотным. Избыток тозилхлорида удаляли путем промывки 1М водным раствором карбоната натрия. Органический слой промывали рассолом, сушили над сульфатом магния и концентрировали в вакууме с получением 2,1 г (98%) фторэтилтозилата (12) в виде бесцветного масла. Структура подтверждена 13С ЯМР (75 МГц, CDCl3): δC 21.6 (ССН3), 68.5 (d, JCF=173 Гц, OCH2CH2F), 80.6 (d, JCF=173 Гц, OCH2CH2F), 128.0, 129.9, 132.6 и 145.1.
Пример 2(б): 2-Хлор-5-метокси-фенил)(2-фторэтил)амин (13)
Гидрохлорид 2-хлор-5-метоксианилина (5,0 г, 26,0 ммоль) растворяли в DMF (50 мл) и добавляли гидрид натрия (2,3 г, 60% в масле, 57,0 ммоль). Реакционную смесь перемешивали в течение 30 минут при к.т. в атмосфере азота. По каплям добавляли фторэтилтозилат (12) (6,7 г, 31,0 ммоль) в DMF (5 мл) и реакционную смесь перемешивали при к.т. в течение 2 ч. Реакционную смесь затем нагревали при 100°C в течение 18 ч. Реакционной смеси давали возможность охладиться и растворитель удаляли при пониженном давлении. Остаток растворяли в этилацетате (100 мл) и промывали водой (2×100 мл). Органические фазы собирали, сушили над сульфатом магния и концентрировали в вакууме с получением коричневого масла, которое очищали хроматографией на силикагеле, элюируя смесью бензин (А):этилацетат (В) (5-30% (В), 330 г, 18,1 CV, 120 мл/мин) с получением 1,3 г (25%) 2-хлор-5-метокси-фенил)(2-фторэтил)амина (13) в виде желтого масла. Структура подтверждена 13С ЯМР (75 МГц; CDCl3): 6C 43.8 (d, JCF=23 Гц), 55.3, 82.0 (d, JCF=165 Гц), 98.1, 102.2, 111.6, 129.5, 144.1 и 159.5.
Пример 2(в): Этиловый эфир 3-[(2-хлор-5-метокси-Фенил)-(2-Фторэтил)амино1-2-гидрокси-циклогексил-1-ен карбоновой кислоты (14)
Раствор 2-хлор-5-метокси-фенил)(2-фторэтил)амина (13) (6,1 г, 30,0 ммоль) в THF (170 мл) охлаждали до -40°C. По каплям добавляли бис(триметилсилил)амид калия (126,0 мл 0,5 М раствора в толуоле, 63,0 ммоль) и реакционную смесь перемешивали в течение 30 мин при -40°C. По каплям при -40°C добавляли этиловый эфир 3-бром-2-гидрокси-циклогекс-1-ен карбоновой кислоты (4; получен согласно Примеру 1(г)) (7,4 г, 30,0 ммоль) в THF (30 мл). Охлаждающую баню удаляли и реакционную смесь перемешивали при к.т.в течение 4 ч.
Реакционную смесь гасили рассолом (300 мл) и экстрагировали в этилацетат (2×400 мл), сушили над сульфатом магния и концентрировали в вакууме с получением 12,0 г (количественный выход) этилового эфира 3-[(2-хлор-5-метокси-фенил)-(2-фторэтил)амино]-2-гидрокси-циклогексил-1-ен карбоновой кислоты (14) в виде коричневого масла, которое неочищенным использовали на следующей стадии. Структура в виде смеси изомеров подтверждена 1Н ЯМР (300 МГц, CDCl3): 6Н 1.08 (0.8Н, t, J=9 Гц, СО2СН2СН3), 1.22-1.33 (2.2 Н, m, СО2СН2СН3), 1-40-2.60 (7Н, m, 4-, 5-, и 6-СН2, CHN), 3.20-4.50 (10Н, m, NCH2CH2F, NCH2CH2F, ОСН3, CHCO2CH2CH3), 6.50-6.70 (1Н, m, СНС(ОСН3)СНСН), 6.95 (0.5Н, dd, J=3 и 6 Гц, СНС(ОСН3)СНСН), 7.08 (0.5Н, d, J=3 Гц, СНС(ОСН3)СНСН) и 7.20-7.30 (1Н, m, СНС(ОСН3)СНСН).
Пример 2(г) Этиловый эфир 8-хлор-9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (15)
Синтез этилового эфира 8-хлор-9-(2-фтор-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (15) вначале осуществляли с использованием условий, описанных в WO 2003/014082. Раствор 2-хлор-5-метокси-фенил)(2-фторэтил)амина (13; получен согласно Примеру 2(б)) (600 мг, 3,8 ммоль), в сухом THF (20 мл) охлаждали в ледяной бане и обрабатывали бис(триметилсилил)амидом калия (16 мл 0,5 М раствора в толуоле, 8,0 ммоль). Через 30 минут добавляли этиловый эфир 3-бром-2-гидрокси-циклогекс-1-ен карбоновой кислоты (4; получен согласно Примеру 1(г)) (1,04 г, 4,2 ммоль) в THF (4 мл), и реакционную смесь оставляли нагреваться до к.т. в течение 2 часов. Реакционную смесь гасили насыщенным раствором хлорида аммония и экстрагировали дважды диэтиловым эфиром. Экстракты промывали водой, рассолом, сушили и концентрировали в вакууме. Это неочищенное вещество очищали хроматографией на силикагеле, элюируя смесью бензина (А) и этилацетата (В) (2,5-50% В, 50 г, 25 CV, 40 мл/мин). Основная порция представляла собой смесь трех соединений. Эту смесь кипятили с обратным холодильником в толуоле (20 мл) с сухим хлоридом цинка (1,7 г, 12,6 ммоль) в течение ночи. Реакционную смесь концентрировали в вакууме и остаток распределяли между 1 н. HCl (25 мл) и этилацетатом (25 мл) и затем экстрагировали больше одного раза этилацетатом. Органические слои промывали водой и рассолом, сушили и концентрировали в вакууме с получением коричневого масла. 1Н ЯМР показал, что оно представляло собой смесь нескольких соединений. TLC на диоксиде кремния в диапазоне растворителей не смогла разделить эту смесь на отдельные порции. Сравнение 1Н ЯМР смеси с аутентичным образцом показало, что смесь содержит 25% этилового эфира 8-хлор-9-(2-фтор-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (15).
Затем осуществляли модифицированный способ. Этиловый эфир 3-[(2-хлор-5-метокси-фенил)-(2-фторэтил)амино]-2-гидрокси-циклогексил-1-ен карбоновой кислоты (14) (12,2 г, 30,0 ммоль) растворяли в диэтиловом эфире (250 мл) и добавляли хлорид цинка (16,4 г, 120,0 ммоль). Реакционную смесь нагревали при температуре дефлегмации в течение 16 ч. Добавляли этилацетат (500 мл) для полного растворения и промывали 2 н. раствором HCl (200 мл), водой (200 мл), 10%-ным водным раствором карбоната калия (200 мл), сушили над сульфатом магния и концентрировали в вакууме. Неочищенное вещество очищали хроматографией на силикагеле, элюируя смесью бензин (А).этилацетат (В) (5-20% В, 12 CV, 10 г, 100 мл/мин) с получением 5,3 г (50% за 2 стадии) этилового эфира 8-хлор-9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (15) в виде желтого твердого вещества. Структура подтверждена 13С ЯМР (75 МГц, CDCl3): δС 14.4, 20.4, 22.2, 27.4, 40.1, 44.2 (d, Jcf=23 Гц), 55.1, 60.2, 83.9 (of, JCF=173 Гц), 100.6, 107.9, 108.2, 119.8, 123.1, 131.9, 137.2, 152.7 и 175.7.
Пример 2(д): Этиловый эфир 9-(2-Фторэтил)-5-метокси-2,3,4.9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (16)
Этиловый эфир 8-хлор-9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (15) (5,3 г, 15,0 ммоль) растворяли в метаноле (180 мл) и добавляли триэтиламин (1,8 г, 18,0 ммоль, 2,5 мл) и 10% Pd/C (2 г в метаноле (20 мл)). Смесь помещали в гидрогенизатор Парра и встряхивали в течение 18 ч в атмосфере водорода. Реакционную смесь фильтровали через слой целита, промывали метанолом и растворитель удаляли в вакууме. Остаток растворяли в этилацетате (300 мл) и промывали 10%-ным водным раствором карбоната калия (200 мл), сушили над сульфатом магния и концентрировали в вакууме с получением 4,2 г (88%) этилового эфира 9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (16) в виде светло-коричневого твердого вещества. Структура подтверждена 13С ЯМР (75 МГц, CDCl3): δC 14.3, 20.6, 21.8, 27.6, 40.3, 43.3 (d, JCF=23 Гц), 54.9, 60.1, 82.0 (d, JCF=165 Гц), 99.8, 102.1, 107.3, 117.2, 121.8, 134.9, 137.6, 153.8 и 176.0.
HPLC (Gemini 150×4,6 мм, 50-95% метанол/вода за 20 мин) 13,6 мин (94%).
Пример 2(е): 9-(2-Фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновая кислота (17)
Этиловый эфир 8-хлор-9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (16) (380 мг, 1,2 ммоль) растворяли в этаноле (4 мл). Добавляли раствор гидроксида натрия (580 мг, 14,5 ммоль), растворенного в 6 мл воды. Реакционную смесь нагревали до образования флегмы в течение ночи. Растворитель удаляли в вакууме и неочищенную смесь разбавляли водой, подкисляли 2 н. HCl до кислой реакции и промывали дихлорметаном. Органические фазы объединяли и сушили над сульфатом магния и концентрировали в вакууме с получением 347 мг (количественный выход) 9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (17) в виде не совсем белого твердого вещества, которое неочищенным использовали на следующей стадии. Структура подтверждена 13С ЯМР (75 МГц; CDCl3): δC 20.4, 21.9, 27.2, 39.9, 43.3 (d, JCF=23 Гц), 55.1, 81.9 (d, JCF=173 Гц), 100.3, 102.8, 106.2, 117.1, 122.2, 135.6, 137.8, 153.3 и 180.8.
Пример 2(ж): 9-(2-Фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбонилхлорид (18)
Раствор 9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (17) (347 мг, 1,2 ммоль) в сухом дихлорметане (2 мл) перемешивали в атмосфере азота. Добавляли оксалилхлорид (453 мг, 3,6 ммоль, 300 мкл), затем добавляли каплю DMF. Реакционную смесь перемешивали при к.т. в атмосфере азота в течение 2 ч, затем упаривали в вакууме с получением 371 мг (количественный выход) 9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбонилхлорида в виде смолы, которую использовали на следующей стадии без очистки. Структура подтверждена 13С ЯМР (75 МГц, CDCl3): δC 20.2, 21.7, 26.4, 43.3 (d, JCF=23 Гц), 54.9, 80.5, 83.1, 100.2, 102.2, 105.8, 116.7, 122.4, 135.5, 137.4, 153.5 и 176.6.
Пример 2(з): Диэтиламид 9-(2-фторэтил)-5-метокси-2,3,4.9-тетрагидро-1Н-карбазол-4-карбоновой кислоты
9-(2-Фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбонилхлорид (18) (371 мг, 1,2 ммоль) растворяли в дихлорметане (2 мл) и охлаждали до 0°C. Затем добавляли диэтиламин (177 мг, 2,4 ммоль, 250 мкл) и реакционную смесь перемешивали в течение ночи при к.т. Реакционную смесь гасили 10%-ным водным раствором карбоната калия (2 мл). Дихлорметановый слой собирали через фазоразделитель, затем концентрировали в вакууме. Это неочищенное вещество очищали хроматографией на силикагеле, элюируя смесью бензин (А):этилацетат (В) (50-100% (В), 50 г, 35,2 CV, 40 мл/мин) с получением бледно-желтого твердого вещества. Это твердое вещество затем растирали с минимальным количеством диэтилового эфира с получением 240 мг (58%) диэтиламида 9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты. Структура подтверждена 13С ЯМР (75 МГц, CDCl3): δC 13.0, 14.6, 19.9, 21.9, 28.0, 36.3, 40.5, 41.9, 43.1 (d, JCF=23 Гц), 54.7, 82.0 (d, JCF=173 Гц), 99.7, 102.1, 108.3, 117.0, 121.5, 135.3, 137.4, 153.3 и 174.8.
Пример 3: Синтез соединения-предшественника 1 и его альтернативного энантиомера
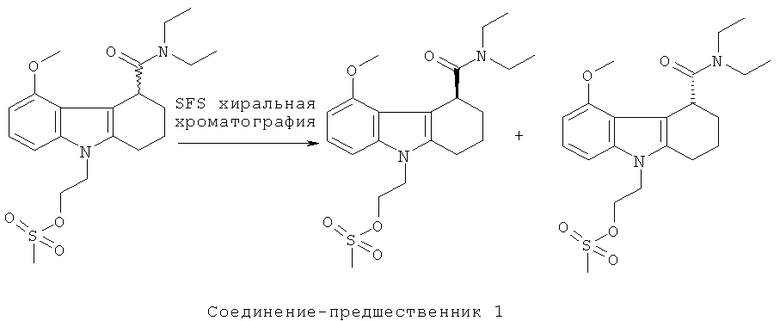
Рацемическую смесь соединения-предшественника 1 и его альтернативного энантиомера (полученную, как описано в Примере 1) разделяли на энантиомеры с использованием хиральной сверхкритической флюидной (СО2) хроматографии на колонке Kromasil Amycoat, 250×10 мм, 5 мкм, 100 Е, используя 30% IPA при 40°C при 13 мл в минуту с временем прогона 6 минут. 60 мг рацемата растворяли в 1,4-диоксане (2 мл) и до 200 мкл за раз впрыскивали для каждого прогона. Достигали исходного разделения двух энантиомеров. Аналитическое определение методом HPLC энантиомерной чистоты двух разделенных энантиомеров на 1С от Chiral Technologies, 250×4,6 мм, 5 мкм, изократический режим, 80:20 МеОН:IPA при 0,5 мл/мин и комнатной температуре показало энантиомерную чистоту 99,5% каждого из энантиомеров.
Пример 4: Синтез радиоиндикатора для PET по изобретению и его альтернативного энантиомера
Соединение-предшественник 1 и его энантиомер, полученные согласно Примеру 3, метили изотопом 18F, используя кассету FASTLab™ (GE Healthcare).
[18F]Фторид, доставленный из GE Healthcare с циклотрона GE PETrace, улавливали на QMA картридже. К222 (8 мг), KHCO3 (200 мкл, 0,1М водн.) и MeCN (1 мл) добавляли в виалу 1 с элюентом. 0,6 мл элюента из виалы 1 использовали для элюирования QMA картриджа. Сушку 18F элюата проводили при 100°C в течение 20 мин с последующим охлаждением до 86°C, после чего добавляли предшественник. 3 мг каждого соединения-предшественника растворяли в 1,6 мл CH3CN. 1 мл этого раствора добавляли в реакционный сосуд. Реакционный сосуд нагревали при 100°C в течение 15 минут. Реакционный сосуд затем ополаскивали 2 мл воды.
Полупрепаративную HPLC проводили следующим образом:
Аналитическую ахиральную HPLC проводили следующим образом:
Аналитическую хиральную HPLC проводили следующим образом:
EOS-выход радиоиндикатора для PET по изобретению составлял 32%, и выход его энантиомера составлял 19%.
Пример 5: Синтез нерадиоактивного аналога радиоиндикатора для PET по изобретению и его альтернативного энантиомера
Рацемическую смесь нерадиоактивного индикатора для PET по изобретению и его альтернативного энантиомера (полученную, как описано в Примере 2) разделяли на его энантиомеры хиральной сверхкритической флюидной (СО2) хроматографией (SFC) на колонке Kromasil Amycoat, 250×10 мм, 5 мкм, 100 Е, используя 20% IPA при 40°C при 14 мл в минуту с временем прогона 6 мин. 100 мг рацемической смеси растворяли в 1,4-диоксане (2,5 мл) и до 200 мкл за один раз впрыскивали для каждого прогона. Фракции отсекали по времени, чтобы не собирать смешанные фракции. Определение методом аналитической HPLC энантиомерной чистоты двух отдельных энантиомеров на 1С от Chiral Technologies, 250×4,6 мм, 5 мкм, изократический режим, 80:20 МеОН:IPA при 0,5 мл/мин и комнатной температуре показало энантиомерную чистоту 99,5% каждого энантиомера.
Пример 6: Определение абсолютной стереохимии методом спектроскопии колебательного кругового дихроизма
Тестировали нерадиоактивный аналог радиоиндикатора для PET по изобретению и его энантиомер, а также соединение-предшественник 1 и его энантиомер. Каждое тестируемое соединение растворяли в CDCl3 (5 мг/0,12 мл радиоиндикатора для PET и его энантиомера; 5 мг/0,15 мл для соединения-предшественника 1 и его энантиомера) и помещали в кювету с окнами из BaF2 и длиной оптического пути 100 мкм, IR- и VCD-спектры регистрировали на спектрометре Chiral/RTM VCD (BioTools, Inc.), оснащенном вспомогательным прибором DualPEM, с разрешением 4 см"1, 11 ч сбор данных для каждого образца и оптимизированным при 1400 см-1. IR-спектр растворителя регистрировали за 150 сканов. Регистрировали растворитель-субтрактивные IR-спектры и энантиомер-субтрактивные VCD-спектры. Оптическое вращение (OR) каждого тестируемого соединения измеряли с использованием поляриметра Jasco DIP-370 при 590 нм и 25°C.
(R)-конфигурация в каждом случае была построена с использованием Hyperchem (Hypercube, Inc., Gainesville, FL). Конформационный поиск проводили с использованием Hyperchem для всей структуры на уровне молекулярной механики. Вычисления геометрии, частоты и интенсивности IR и VCD выполняли на уровне DFT (функционал B3LYP/базисный комплект 6-31G(d)) с использованием Gaussian 09 (Gaussian Inc., Wallingford, CT). Вычисленные частоты приводили к масштабу 0,97 и интенсивности IR и VCD преобразовывали в лоренцевы полосы с полушириной 6 см-1 для сравнения с экспериментальными данными.
В отношении радиоиндикатора для PET и его энантиомера гауссовы вычисления 36 конформеров дали в результате десять конформеров с энергией в пределах 1 ккал/моль от самого низкоэнергетического конформера. Были вычислены оптимизированные геометрии четырех вычисленных конформеров с наименьшей энергией для (R)-конфигурации, и было сделано сравнение наблюдаемых спектров VCD и IR со спектрами десяти вычисленных конформеров с наименьшей энергией. На основе полной согласованности в паттерне VCD для наблюдаемой и больцмановской суммы вычисленных спектров десяти конформеров с наименьшей энергией нерадиоактивному аналогу радиоиндикатора для PET по изобретению была присвоена абсолютная конфигурация (S), и его энантиомеру была присвоена абсолютная конфигурация (R). Присвоение оценивали с помощью программы CompareVOA (Biotools), и доверительный уровень присваивания составил 100% на основе текущей базы данных, включающей 89 предшествующих точных присваиваний для разных хиральных структур.
Что касается соединения-предшественника 1 и его энантиомера, гауссовы вычисления 36 конформеров дали 9 конформеров с энергией в пределах 1 ккал/моль от конформера с наименьшей энергией. На основе полной согласованности в VCD паттерне для наблюдаемой и больцмановской суммы вычисленных спектров девяти конформеров с наименьшей энергией соединению-предшественнику 1 была присвоена абсолютная конфигурация (S), и его энантиомеру была присвоена абсолютная конфигурация (R). Присваивание оценивали с помощью программы CompareVOA, и доверительный уровень присваивания составил 96% на основе базы данных, включающей 89 предыдущих точных присваиваний для разных хиральных структур. Это присваивание согласуется с присваиванием конфигурации нерадиоактивному аналогу радиоиндикатора для PET по изобретению.
Пример 7: Анализ эффективности in vitro
Скрининг аффинности к PBR выполняли с использованием адаптированного метода, описанного в Le Fur et at., Life Sci. 1983; USA 33: 449-57. Тестировали нерадиоактивные аналоги радиоиндикатора для PET no изобретению и ассоциированный рацемат. Каждое тестируемое соединение (растворенное в 50 мМ Tris-HCl, pH 7,4, 10 мМ MgCl2, содержащем 1% DMSO) конкурировало за связывание либо с PBR сердца крысы Wistar, либо с PBR человека против 0,3 нм [3Н] PK-11195. Реакцию проводили в 50 мМ Tris-HCI, pH 7,4, 10 мМ MgCl2 в течение 15 минут при 25°C. Скрининг каждого тестируемого соединения выполняли при 6 разных концентрациях в 300-кратном диапазоне концентраций по оцениваемому значению Ki. Получены следующие данные:
Пример 8: Хиральный анализ стабильности in vitro
Нерадиоактивный аналог индикатора для PET по изобретению, полученный согласно Примеру 2, инкубировали (37°C) в плазме крови человека или во фракции S9 печени крысы в течение плоть до 4 часов. Энантиомеры экстрагировали из биологического материала путем преципитации белков. Твердый осадок отделяли от жидкой фазы, которую упаривали досуха. Сухой остаток растворяли в ацетонитриле.
В этом исследовании применяли систему HPLC Dionex Ultimate 3000, состоящую из двух насосов (микронасос LPG-3000 и насос Ultimate 3000), детектора, работающего в УФ/видимом диапазоне, автосамплера и двух переключающих клапанов. Один переключающий клапан был соединен с двумя насосами и автосамплером. Эта установка давала возможность использовать любой из насосов для впрыскивания в колонку. Насос, используемый для впрыскивания, был соединен только с SPE колонкой. После впрыскивания и элюирования SPE колонку промывали, используя насос для впрыскивания. Система была готова для нового впрыскивания, хотя хиральный анализ продолжался.
Другой переключающий клапан соединял аналитическую колонку и SPE колонку. После удерживания вещества на SPE колонке клапан переключался, и аналитический насос элюировал вещество из SPE колонки в аналитическую хиральную колонку. Направление потока элюирования менялось на обратное направлению удерживания. Аналитический насос был соединен только с аналитической системой и находился в режиме ожидания аналитов до тех пор, пока не начнется элюирование. И рабочее время удерживания на SPE колонке, и время элюирования из этой колонки варьировали с целью оптимизации этого двухстадийного процесса.
Аналитическая колонка: Chiralpak 1С 0,46×25 см с предварительной колонкой из того же материала 0,4×1 см.
SPE колонка: LiChrospher ADS RP-4 25×4 мм (RAM колонка), 25 мкм частицы. Отсечка молекулярной массы: 15 кДа (Merck).
Подвижная фаза: А: ацетат аммония 10 мМ, рН 7; В: 1:1 MeCN:MeOH.
Скорость потока: 300 мкл/мин.
Детектирование: УФ-детектирование при 230 нм.
Удерживание на SPE колонке: При удерживании аналита на SPE колонке применяли изократический режим с использованием 10% MeCN в 50 мМ ацетата аммония. Удерживание длилось 4 минуты, а затем клапан переключался.
Элюирование из SPE колонки и разделение: Элюирование начинали с использованием подвижной фазы, представляющей собой смесь 10% MeCN и 90% 10 мМ ацетата аммония. Через 5 минут клапан переключался назад на SPE колонку, которую промывали смесью 90% MeCN/MeOH в буфере. Градиент на аналитической колонке начинался при 65% органической фазы в буфере и изменялся до 85% органической фазы в буфере в течение 26,5 минут. Аналитическую колонку промывали в течение 3 мин смесью 70% MeCN/MeOH и затем стабилизировали при 10% MeCN/MeOH, чтобы подготовить систему разделения для следующего впрыскивания. Суммарное рабочее время составляло 40 минут.
Радиоиндикатор для PET в плазме крови не показал никаких хроматографических изменений после инкубирования в течение 4 часов. Хроматографические результаты были сравнимы с неинкубированным образцом в плазме крови и эталонным раствором радиоиндикатора для PET по изобретению. Рацемизация не наблюдалась.
Радиоиндикатор для PET по изобретению во фракции S9 печени крысы не подвергался рацемизации после инкубирования в течение 4 часов.
Пример 9: Биораспределение in vivo
Радиоиндикатор для PET по изобретению, его альтернативный энантиомер и рацемическую смесь их обоих тестировали в модели биораспределения in vivo.
Взрослым самцам крыс Wistar (200-300 г) инъецировали 1-3 МБк тестируемого соединения через латеральную хвостовую вену. Через 2, 10, 30 или 60 минут (n=3) после инъекции крыс подвергали эвтаназии и брали образцы тканей или жидкостей для измерения радиоактивности на гамма-счетчике.
Получили следующие важные данные:
Пример 10: Исследование метаболизма in vivo
Количество активности в головном мозге или плазме крови, имеющейся благодаря введению родительского тестируемого соединения, тестировали через вплоть до 1 часа после введения. Тестируемыми соединениями были радиоиндикатор для PET по изобретению и его ассоциированный рацемат.
Взрослым самцам крыс Wistar (150-200 г) инъецировали приблизительно 20 МБк тестируемого соединения. Образцы головного мозга и плазмы крови анализировали методом HPLC через 10, 30 и 60 минут после введения. Использовали следующие условия HPLC:
Получили следующие данные (где "пв" означает после впрыскивания):
Пример 11: Анализ на блокирование in vivo
Биораспределения in vivo радиоиндикатора для PET по изобретению по сравнению с его ассоциированным рацематом тестировали после предварительного введения их соответственных нерадиоактивных аналогов или предварительного введения известного PBR-специфичного лиганда, РК11195.
Взрослым самцам крыс Wistar (200-300 г) инъецировали приблизительно 3-4 МБк тестируемого соединения через латеральную хвостовую вену. РК11195 или нерадиоактивный аналог (оба по 3 мг/кг) вводили за 5 минут до введения тестируемого меченного радиоизотопом соединения. Через 30 минут после инъекции крыс подвергали эвтаназии и брали образцы тканей или жидкостей для измерения радиоактивности на гамма-счетчике.
Получили следующие важные данные:
Пример 12: Модель воспаления после аксотомии лицевого нерва
Связывание с сайтом очагового нейровоспаления тестировали методом ангиографии. Тестируемыми соединениями были радиоиндикатор для PET по изобретению и его ассоциированный рацемат.
Самцы крыс Wistar (200-300 г) либо были не подвергнуты никакому воздействию, либо были подвергнуты аксотомии лицевого нерва согласно методике, описанной в Graeber and Kreutzberg, J Neurocytol 1986; 15: 363-373. Различные ткани, включая ствол головного мозга и обонятельную луковицу, удаляли из животных и быстро замораживали в изопентане, затем хранили при -70°C до использования. Делали срезы тканей (12 мкм) и подтаявшими закрепляли их на слайдах Superfrost Plus. Слайды хранили при -70°C до использования.
Слайды сушили на воздухе, затем предварительно инкубировали в Tris-HCl буфере (170 мм, рН 7,4) в течение 5 мин при комнатной температуре. Добавляли 1000-кратный избыток нерадиоактивного РК11195 в концентрации 1 мкМ или нерадиоактивного аналога радиоиндикатора для PET по изобретению в концентрации 1 мкМ, затем инкубировали с Tris-HCl буфером (170 мМ, рН 7,4), содержащим 8 ГБк/мл тестируемого соединения в течение 60 минут. Реакцию затем останавливали промыванием срезов два раза в течение 5 минут каждый раз в ледяной буфере (Tris-HCI, 170 мМ, рН 7,4) и затем слайды окунали ненадолго в дистиллированную воду для промывки. Затем слайды сушили на воздухе и делали рентгеновский снимок. При экспозиции рентгеновского снимка использовали эталонный образец и для авторадиографии in vitro эталонный образец (20 мкм) отбирали из раствора и помещали на фильтровальную бумагу (зафиксированную на стекле) и экспонировали вместе со срезами. Время экспозиции составляло 24 часа. Данные анализировали путем очерчивания интересующих участков вокруг конкретных анатомических структур, а также вокруг блокированных образцов, эталонов и фона с использованием пакета программ MCID, используя шкалу градиента плотности в качестве калибровочной кривой, скорректированной в соответствии с эталонным образцом.
Были получены следующие важные данные:
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ ИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ЛИГАНДОВ PBR | 2010 |
|
RU2525196C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА, ПОДХОДЯЩИЕ ДЛЯ ВИЗУАЛИЗАЦИИ НЕЙРОВОСПАЛЕНИЯ | 2009 |
|
RU2512288C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2005 |
|
RU2382770C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2005 |
|
RU2430088C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ ФИЗИОЛОГИЧЕСКИХ И/ИЛИ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ, ОПОСРЕДОВАННЫХ LHRH РЕЦЕПТОРОМ, ПОСРЕДСТВОМ ВЫШЕНАЗВАННЫХ ПРОИЗВОДНЫХ | 2008 |
|
RU2497806C9 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2004 |
|
RU2318810C2 |
| ПРОИЗВОДНЫЕ 3-(ГЕТЕРОАРИЛАМИНО)-1,2,3,4-ТЕТРАГИДРО-9Н-КАРБАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ПРОСТАГЛАНДИНА D2 | 2011 |
|
RU2562255C2 |
| УПРОЩЕНИЕ СПОСОБА ПОЛУЧЕНИЯ СОЕДИНЕНИЯ-ПРЕДШЕСТВЕННИКА | 2011 |
|
RU2593372C2 |
| Новые производные имидазо[4,5-с]хинолина в качестве ингибиторов LRRK2 | 2018 |
|
RU2773516C2 |
| СПОСОБ СИНТЕЗА | 2011 |
|
RU2572554C2 |
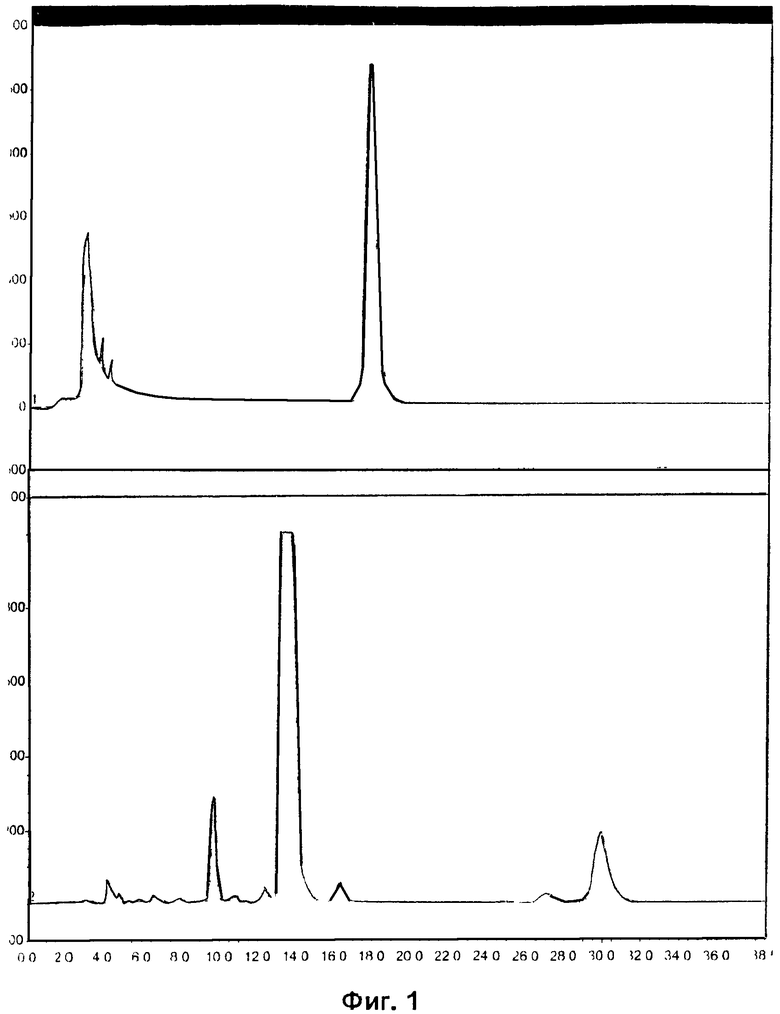
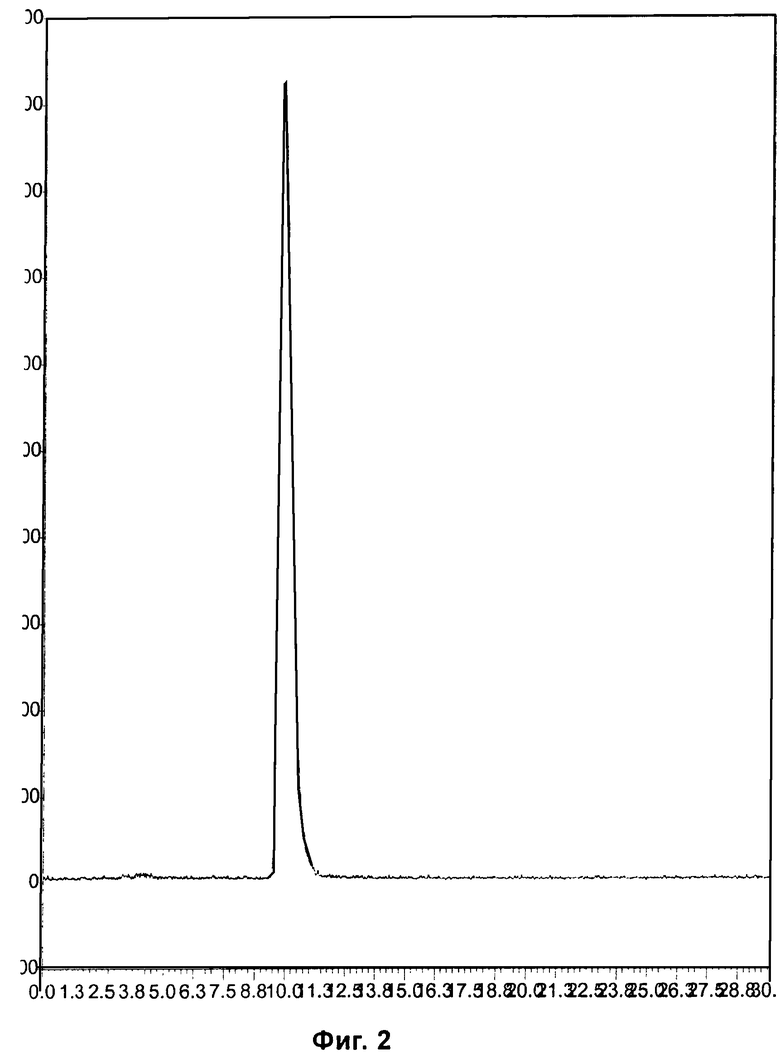
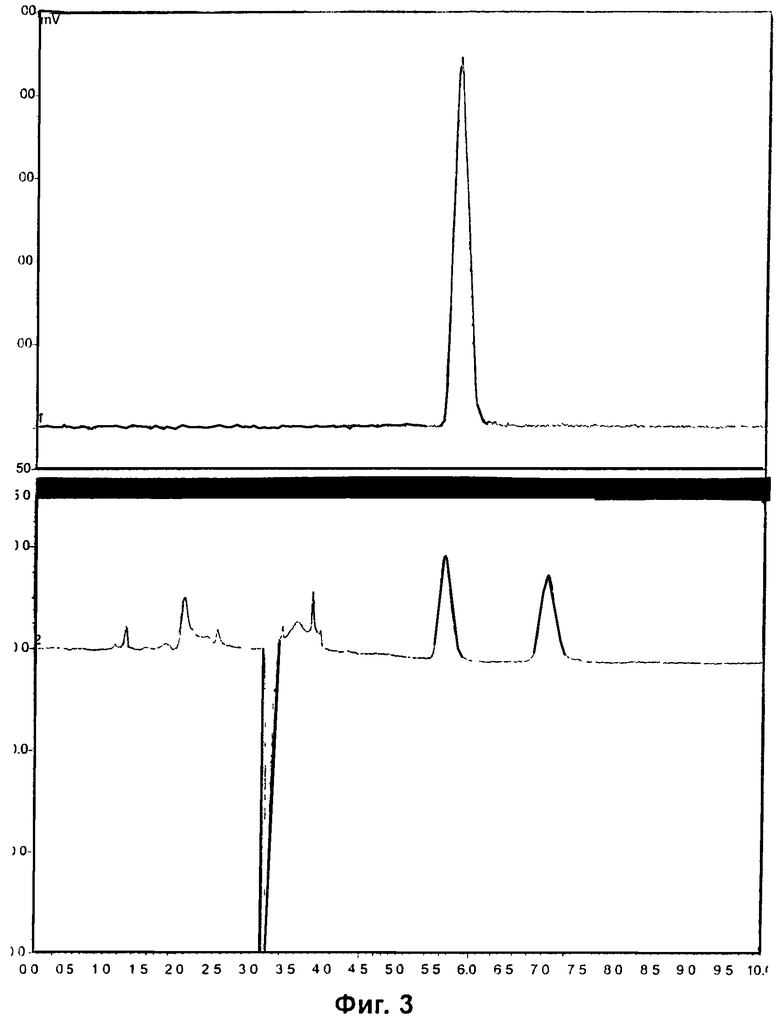
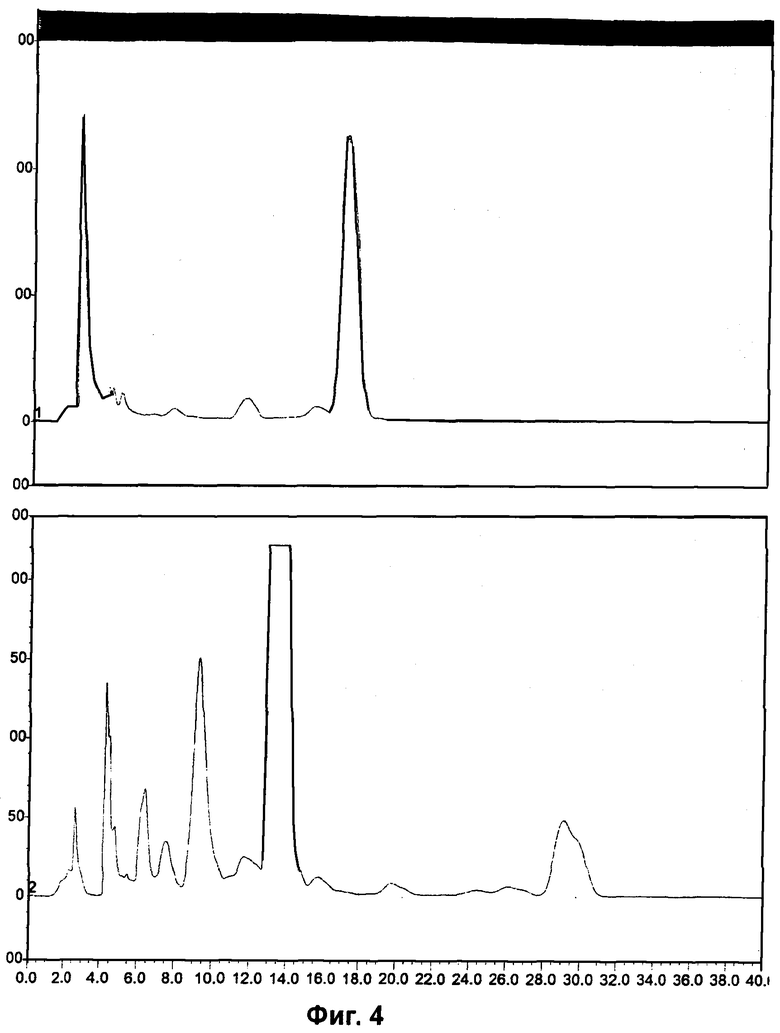
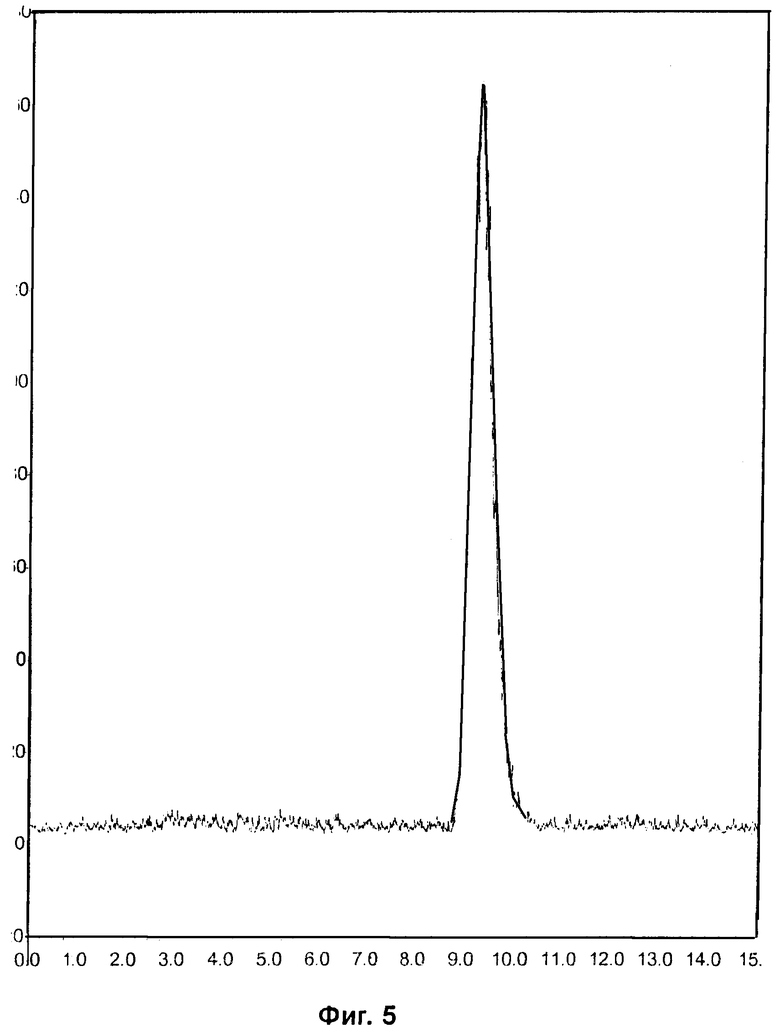
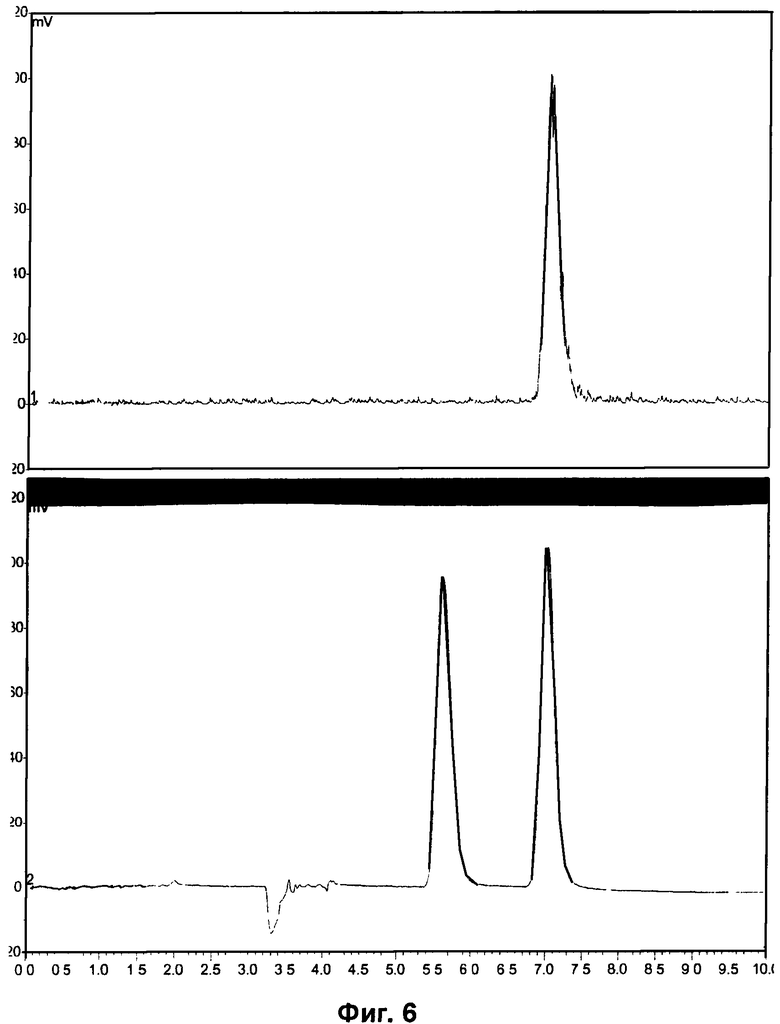
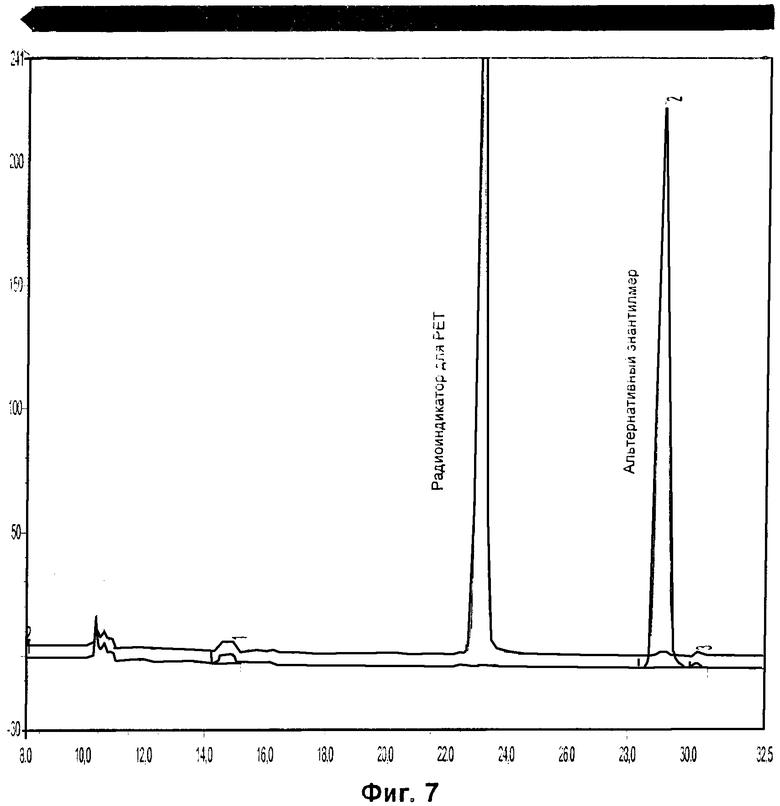
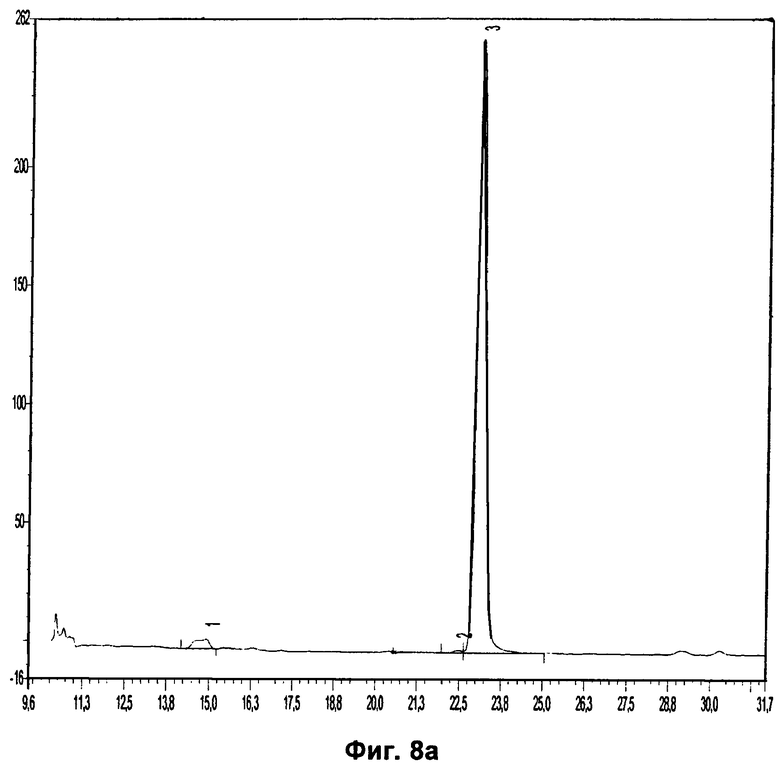
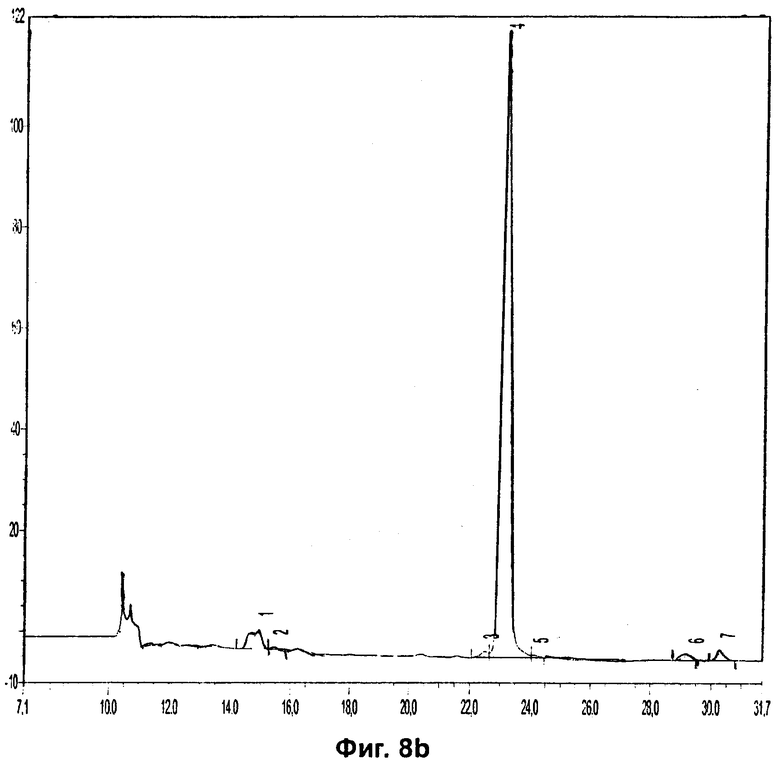
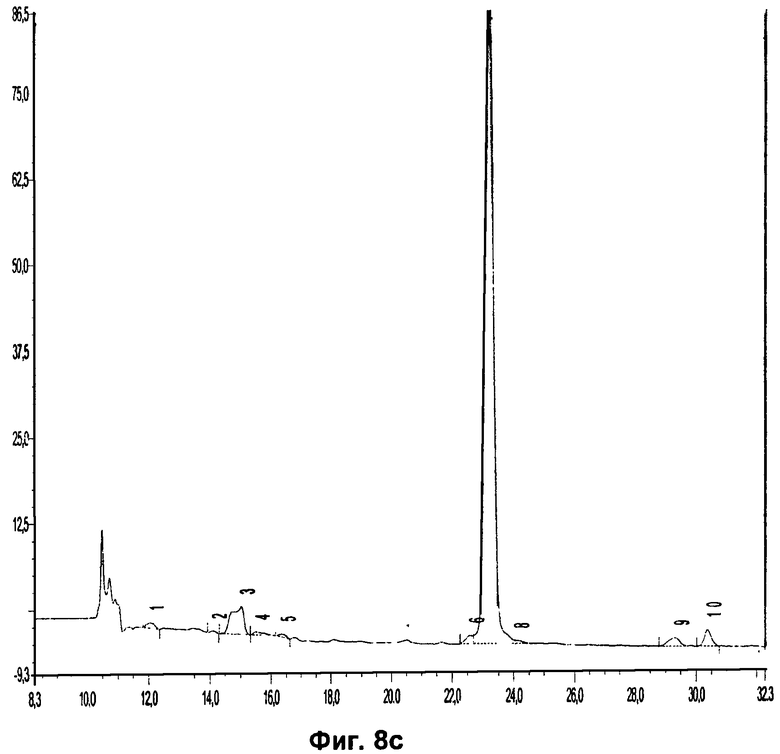
Изобретение относится к радиоиндикатору для PET, который обладает улучшенными свойствами в отношении визуализации периферических бензодиазепиновых рецепторов (PBR) по сравнению с известными радиоиндикаторами для PET. Согласно настоящему изобретению предложены также соединение-предшественник для получения радиоиндикатора для PET по изобретению и способы получения указанного соединения-предшественника и указанного радиоиндикатора для PET. Также предложена радиофармацевтическая композиция, содержащая радиоиндикатор для PET по изобретению и способы для применения радиоиндикатора для PET и радиофармацевтической композиции. 9 н. и 14 з.п. ф-лы, 8 ил., 12 пр.
1. Радиоиндикатор для позитронно-эмиссионной томографии (PET), имеющий следующую химическую структуру:
.
2. Соединение-предшественник для получения радиоиндикатора для PET по п.1, где указанное соединение-предшественник представляет собой соединение формулы I:
,
где R1 представляет собой гидроксил или уходящую группу.
3. Соединение-предшественник по п.2, где указанная уходящая группа выбрана из мезилата, тозилата и трифлата.
4. Соединение-предшественник по п.3, где указанная уходящая группа представляет собой мезилат.
5. Способ получения соединения-предшественника формулы I по любому из пп.2-4, включающий:
(1) приготовление рацемической смеси указанного соединения-предшественника формулы I и соединения формулы II:
,
где R2 такой, как определено для R1 в любом из пп.2-4, и R1 и R2 одинаковые;
отделение указанного соединения-предшественника формулы I от указанного соединения формулы II.
6. Способ по п.5, где указанная стадия отделения включает высокоэффективную жидкостную хроматографию, сверхкритическую флюидную хроматографию, хроматографию в моделированном слое.
7. Способ получения соединения-предшественника формулы I по любому из пп.2-4, включающий:
(1) приготовление соединения формулы III:
,
где PG1 представляет собой защитную группу для гидроксильной группы;
(2) превращение указанного соединения формулы III в его соответствующий хлорангидрид;
(3) взаимодействие хлорангидрида, полученного на стадии (2), с диэтиламидом с получением соединения формулы IV:
,
где PG2 представляет собой защитную группу для гидроксильной группы и является такой же, как PG1;
(4) удаление защитной группы с соединения формулы IV, полученного на стадии (3), с получением гидроксильного производного;
(5) возможно присоединение уходящей группы, как она определена в любом из пп.2-4 для соединения формулы I;
где соединение-предшественник формулы I получают либо на стадии (4), либо на стадии (5).
8. Способ по п.7, где указанная стадия (1) приготовления указанного соединения формулы III включает:
(а) приготовление рацемической смеси соединения формулы V и соединения формулы VI:
где:
R1 представляет собой хиральный спирт; и
PG3 и PG4 одинаковые, и каждая представляет собой защитную группу для гидроксильной группы;
(б) отделение соединения формулы V от соединения формулы VI;
(в) удаление R1 из отделенного соединения формулы V в кислотных условиях с получением в результате указанного соединения формулы III.
9. Способ по п.7, где указанная стадия (1) приготовления указанного соединения формулы III включает:
(а) приготовление рацемической смеси указанного соединения формулы III и соединения формулы VIII:
,
где PG5 представляет собой защитную группу для гидроксильной группы и является такой же, как PG1, которая определена в п.7;
(б) взаимодействие смеси, которая определена в стадии (а), с оптически активным амином для отделения указанного соединения формулы III от указанного соединения формулы VIII.
10. Способ по п.9, где указанный оптически активный амин выбран из S-альфа-метилбензиламина, R-(+)-N-(1-нафтилметил)-альфа-бензиламина, N-(2-гидрокси)этил-альфа-метилбензиламина и 1-(пара-толил)этиламина.
11. Способ по п.7, где указанная стадия (1) приготовления указанного соединения формулы III включает:
(а) приготовление рацемической смеси соединения формулы IX и соединения формулы X:
,
где PG6 и PG7 одинаковые, и каждая представляет собой защитную группу для гидроксильной группы;
(б) взаимодействие смеси, которая определена в стадии (а), со стереоселективным ферментом с получением указанного соединения формулы III, где указанный стереоселективный фермент осуществляет гидролиз соединения формулы IX с расщеплением сложноэфирной связи.
12. Способ по п.11, где указанный стереоселективный фермент выбран из липазы В Candida antarctica, эстеразы свиной печени или липазы свиной поджелудочной железы.
13. Способ получения радиоиндикатора для PET по п.1, включающий взаимодействие соединения-предшественника формулы I по любому из пп.2-4 с подходящим источником 18F.
14. Способ по п.13, где для указанного соединения-предшественника формулы I R1 представляет собой уходящую группу, и указанный подходящий источник 18F содержит 18F-фторид и катионный противоион.
15. Способ по п.14, где указанный катионный противоион выбран из рубидия, цезия, калия в виде комплекса с криптандом и тетраалкиламмониевой соли.
16. Способ по любому из пп.13-15, являющийся автоматизированным.
17. Кассета для проведения способа по п.16, содержащая:
(1) сосуд, содержащий соединение-предшественник по любому из пп.2-4; и
(2) средства для элюирования сосуда стадии (1) подходящим источником 18F, как он определен в любом из пп.13-15.
18. Кассета по п.17, которая дополнительно содержит (3) ионообменный картридж для удаления избытка 18F.
19. Радиофармацевтическая композиция, содержащая радиоиндикатор для PET по п.1 вместе с биосовместимым носителем, подходящим для введения млекопитающим.
20. Способ визуализации методом PET для определения распределения и/или степени экспрессии PBR (периферических бензодиазепиновых рецепторов) у субъекта, включающий:
1) введение указанному субъекту радиоиндикатора для PET по п.1;
2) предоставление возможности указанному радиоиндикатору для PET связаться с PBR у указанного субъекта;
3) детектирование сигналов, испускаемых 18F, который содержится в указанном связанном радиоиндикаторе для PET;
4) формирование изображения, отображающего локализацию и/или количество указанных сигналов; и
5) определение распределения и степени экспрессии PBR у указанного субъекта, где указанная экспрессия находится в прямой корреляции с указанными сигналами.
21. Способ визуализации методом PET по п.20, где указанный радиоиндикатор для PET вводят в виде радиофармацевтической композиции по п.19.
22. Способ визуализации методом PET по п.20 или 21, который проводят неоднократно во время осуществления схемы лечения указанного субъекта, причем указанная схема лечения включает введение лекарственного средства для противодействия PBR-ассоциированному состоянию.
23. Способ диагностики состояния, при котором происходит повышающая регуляция PBR, включающий способ визуализации методом PET по п.20 или 21 вместе с дополнительной стадией (6) соотнесения распределения и степени экспрессии PBR с конкретной клинической картиной.
| ТРИЦИКЛИЧЕСКИЕ ИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ЛИГАНДОВ PBR | 2010 |
|
RU2525196C2 |
| WO 2000044751 A1, 03.08.2000 | |||
| УСТАНОВКА ДЛЯ КОНДИЦИОНИРОВАНИЯ ВОЗДУХА В ЖЕЛЕЗНОДОРОЖНЫХ ВАГОНАХ | 0 |
|
SU248734A1 |
| KOZIKOWSKI A P ET AL: "Chemistry, binding affinities, and behavioral properties of a new class of 'antineophobic' mitochondrial DBI receptor complex (mDRC) ligands", JOURNAL OF MEDICINAL CHEMISTRY 1993 US, vol | |||
| Коридорная многокамерная вагонеточная углевыжигательная печь | 1921 |
|
SU36A1 |
| Прибор для промывания газов | 1922 |
|
SU20A1 |
| TAKETOSHI OKUBO ET | |||

