Область изобретения
Настоящее изобретение относится к визуализации in vivo и, в частности, визуализации in vivo периферического бензодиазепинового рецептора (PBR). Предложен индольный in vivo визуализирующий агент, который с высокой аффинностью связывается с PBR, обладает хорошим поглощением в головном мозге после введения и который обладает хорошим селективным связыванием с PBR. В изобретении также предложено соединение-предшественник, полезное в синтезе in vivo визуализирующего агента по изобретению, а также способ синтеза указанного соединения-предшественника. Другие аспекты изобретения включают способ синтеза in vivo визуализирующего агента по изобретению, включающий применение соединения-предшественника, набор для осуществления указанного способа и кассету для осуществления автоматического варианта указанного способа. Кроме того, в изобретении предложена радиофармацевтическая композиция, содержащая in vivo визуализирующий агент по изобретению, а также способы применения указанного in vivo визуализирующего агента.
Предшествующий уровень техники
Известно, что периферический бензодиазепиновый рецептор (PBR) в основном локализован в периферических тканях и глиальных клетках, но его физиологическую функцию еще предстоит прояснить. Известно, что внутриклеточно PBR локализован на внешней митохондриальной мембране, что свидетельствует о возможной роли в модулировании митохондриальной функции и в иммунной системе. Кроме того, предполагалось, что PBR вовлечен в клеточную пролиферацию, стероидогенез, поток кальция и клеточное дыхание.
Аномальная экспрессия PBR ассоциируется с воспалительными болезненными состояниями центральной нервной системы (ЦНС), включающими рассеянный склероз (Banati era/2001 Neuroreport; 12(16): 3439-42; Debruyne et al 2002 Acta Neurol Beig; 102(3): 127-35), энцефалит Расмуссена (Banati et al 1999 Neurology; 53(9): 2199-203), церебральный васкулит (Goerres era/2001 Am J Roentgenol; 176(4): 1016-8), герпетический энцефалит (Cagnin et al 2001 Brain; 124(Pt 10): 2014-27) и СПИД-ассоциированную деменцию (Hammoud et al 2005 J Neurovirol; 11(4): 346-55).
Кроме того, в ЦНС документально подтверждена связь с PBR при дегенеративных заболеваниях, таких как болезнь Паркинсона (Gerhard et al 2006 Neurobiol Dis; 21(2): 404-12; Ouchi et al 2005 Ann Neurol; 57(2): 161-2), кортикобазальная дегенерация (Gerhard et al 2004 Mov Disord; 19(10): 1221-6), прогрессирующий надъядерный паралич (Gerhard et al 2006 Neurobiol Dis; 21(2): 404-12), множественная системная атрофия (Gerhard era/2003 Neurology; 61(5): 686-9), болезнь Гентингтона (Pavese era/2006 Neurology; 66(11): 1638-43; Tai et al 2007 Brain Res Bull; 72(2-3): 148-51), боковой амиотрофический склероз (Turner et al 2004 Neurobiol Dis; 15(3): 601-9) и болезнь Альцгеймера (Cagnin et а/2001 Lancet; 358(9283): 766; Yasuno era/2008 Biol Psychiatry; 64(10): 835-41).
Показано, что с аномальной экспрессией PBR связан ряд ишемических состояний ЦНС, включающих: ишемический инсульт (Gerhard et al 2005 Neuroimage; 24(2): 591-5), поражение периферических нервов (Banati et al 2001 Neuroreport; 12(16):3439-42), эпилепсию (Sauvageau 2002 Metab Brain Dis; 17(1): 3-11; Kumar et al 2008 Pediatr Neurol; 38(6)). PBR был указан как биомаркер для определения степени поражения при травматическом повреждении головного мозга (Toyama et al 2008 Ann Nucl Med; 22(5): 417-24), с сообщением об увеличении экспрессии PBR в животной модели травматического повреждения головного мозга (Venneti et al 2007 Exp Neurol; 207(1): 118-27). Интересно то, что острый стресс коррелирует с увеличением экспрессии PBR в головном мозге, в то время как хронический стресс коррелирует с понижающей регуляцией PBR (Lehmann et al 1999 Brain Res; 851(1-2): 141-7). Сообщалось о возможности установления границ глиомы с использованием [11C]PK11195 для визуализации PBR (Junck er а/1989 Ann Neurol; 26(6): 752-8). PBR также может ассоциироваться с невропатической болью, Tsuda et al обнаружил активированную микроглию у субъектов с невропатической болью (2005 TINS 28(2) рр101-7).
На периферии экспрессия PBR связана с воспалением легких (Branley et а/2008 Nucl. Med. Biol; 35(8): 901-9), хроническим обструктивным заболеванием легких и астмой (Jones et al 2003 Eur Respir J; 21(4): 567-73), воспалительным заболеванием кишечника (Ostuni et al Inflamm Bowel Dis; 2010 онлайн публикация), ревматоидным артритом (van der Laken et al 2008 Arthritis Rheum; 58(11): 3350-5), первичной фибромиалгией (Faggioli et al 2004 Rheumatology; 43(10): 1224-1225), поражением нервов (Durrenberger et al 2004 J Peripher Nerv Syst; 9(1): 15-25), атеросклерозом (Fujimura et al 2008 Atherosclerosis; 201(1): 108-111), раком толстой кишки, предстательной железы и молочной железы (Deane et al 2007 Mol Cancer Res; 5(4): 341-9; Miettinen et al 1995 Cancer Res; 55(12): 2691-5; Han et al 2003 J Recept Signal Transduct Res; 23(2-3): 225-38), воспалением почек (Tarn et al 1999 Nephrol Dial Transplant; 14(7): 1658-66; Cook et al 1999 Kidney Int; 55(4): 1319-26) и ишемически-реперфузионным повреждением (Zhang et al 2006 J Am Coil Surg; 203(3): 353-64).
Визуализация при помощи позитронно-эмиссионной томографии (PET) с использованием PBR-селективного лиганда (R)-[11C]PK11195 обеспечивает обобщенный показатель воспаления центральной нервной системы (ЦНС). Однако известно, что (R)-[11C]PK11195 обладает высоким связыванием с белком и низким соотношением специфического связывания к неспецифическому связыванию. Кроме того, роль его меченных радиоизотопами метаболитов не известна, и количественная оценка связывания требует сложного моделирования.
В данной области техники известны трициклические индольные соединения. В Davies et al (J. Med. Chem. 1998; 41(4): 451-67) раскрыт класс трициклических индольных соединений и они охарактеризованы как агонисты и антагонисты мелатонина. В Napper et al (J. Med. Chem. 2005; 48: 8045-54) раскрыта и обсуждается взаимосвязь структура-активность для класса трициклических индольных соединений в контексте селективного ингибирования фермента SIRT1 (сиртуин-1), являющегося членом семейства ферментов, которые удаляют ацетильные группы из лизиновых остатков в гистонах и других белках. Другой класс трициклических индольных соединений раскрыт в US 6451795 и обсуждается как полезный в лечении связанных с PBR болезненных состояний. В US 6451795 раскрыты значения IC50 для наиболее активных соединений в диапазоне от 0,2 нМ до 5,0 нМ и указано, что эти соединения полезны для предупреждения или лечения периферических невропатий и для лечения центральных нейродегенеративных заболеваний.
В Okubu et al (Bioorganic & Medicinal Chemistry 2004 12 3569-80) описаны дизайн, синтез и структура группы тетрациклических индольных соединений, а также их аффинность к PBR (низкие значения IC50 порядка примерно 0,4 нМ). В WO 2007/057705, переданной заявителю настоящего изобретения, раскрыты тетрациклические индольные производные, меченные рядом in vivo визуализирующих группировок. Предпочтительными in vivo визуализирующими группировками, раскрытыми в WO 2007/057705, являются такие группировки, которые подходят для визуализации при помощи позитронно-эмиссионной томографии (PET) или однофотонной эмиссионной томографии (SPECT), наиболее предпочтительно PET.
Кроме того, в находящейся на одновременном рассмотрении патентной заявке РСТ/ЕР2009/062827 описаны тетрациклические in vivo визуализирующие агенты, представляющие собой индольные производные, аналогичные агентам из WO 2007/057705.
Тетрациклические индольные производные, описанные в WO 2007/057705 и в находящейся в одновременном рассмотрении патентной заявке РСТ/ЕР2009/062827, обладают хорошей аффинностью в отношении PBR рецептора, и высокая доля радиоактивности в головном мозге в течение 60 минут после инъекции представляет собой родительский in vivo визуализирующий агент. Хотя эти тетрациклические индольные производные также достигают приемлемой начальной концентрации в головном мозге крысы в исследованиях биораспределения, поглощение все еще остается относительно низким и может быть улучшено. Авторы настоящего изобретения также обнаружили, что относительное удерживание в обонятельной луковице (области головного мозга, имеющей наивысшую концентрацию рецептора PBR) этих тетрацикпических индольных производных не достаточно высоко, как это требуется для визуализации in vivo. Таким образом, имеется сфера применения PBR in vivo визуализирующего агента, который сохраняет полезные свойства тетрациклических индольных in vivo визуализирующих агентов из вышеописанного уровня техники, но обладает улучшенным поглощением в головном мозге и улучшенным специфическим связыванием с рецептором PBR.
Краткое изложение сущности изобретения
В настоящем изобретении предложено новое трициклическое индольное соединение, подходящее для применения в качестве in vivo визуализирующего агента. В настоящем изобретении также предложено соединение-предшественник, полезное в синтезе in vivo визуализирующего агента по изобретению, а также способ синтеза указанного соединения-предшественника. Также предложен способ получения in vivo визуализирующего агента, включающий применение соединения-предшественника по изобретению. Кроме того предложена фармацевтическая композиция, содержащая in vivo визуализирующий агент по изобретению, а также набор, подходящий для облегчения приготовления этой фармацевтической композиции. В еще одном аспекте предложено применение in vivo визуализирующего агента для in vivo визуализации состояния, ассоциированного с аномальной экспрессией PBR. In vivo визуализирующий агент по настоящему изобретению сохраняет полезные свойства известных тетрациклических in vivo визуализирующих агентов в сочетании с улучшенным поглощением в головном мозге и специфичностью в отношении периферического бензодиазепинового рецептора.
Подробное описание изобретения
Визуализируюший агент
В одном из аспектов настоящего изобретения предложен in vivo визуализирующий агент формулы I:
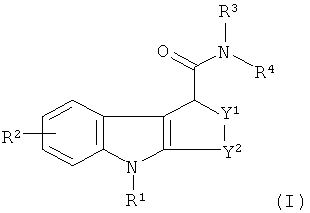
где:
R1 представляет собой C1-3алкил или C1-3фторалкил;
R2 представляет собой водород, гидроксил, галогено, циано, C1-3алкил,
C1-3алкокси, C1-3фторалкил или C1-3фторалкокси;
R3 и R4 независимо представляют собой C1-3алкил, C7-10аралкил, или R3 и R4 вместе с атомом азота, к которому они присоединены, образуют азотсодержащее C1-6алифатическое кольцо, возможно содержащее 1 дополнительный гетероатом, выбранный из азота, кислорода и серы;
Y1 представляет собой О, S, SO, SO2 или CH2; и
Y2 представляет собой СН2, СН2-СН2, СН(СН3)-СН2 или СН2-СН2-СН2;
и где формула I, как она определена, содержит атом, который является радиоизотопом, подходящим для визуализации in vivo.
"In vivo визуализирующий агент" в контексте настоящего изобретения представляет собой радиоактивно меченное соединение, подходящее для визуализации in vivo. Используемый здесь термин "визуализация in vivo" относится к таким методам, которые неинвазивно позволяют получать изображения всех или части внутренностей субъекта.
Если не указано иное, термин "алкил" самостоятельно или в комбинации означает прямой или разветвленный алкильный радикал, предпочтительно содержащий от 1 до 3 атомов углерода. Примеры таких радикалов включают метил, этил и пропил.
Если не указано иное, термин "алкокси" означает алкильный радикал, как он определен выше, содержащий простую эфирную связь, а термин "эфирная связь" относится к группе -С-O-С-. Примеры подходящих алкильных эфирных радикалов включают метокси, этокси и пропокси.
Термин "галоген" или "галогено-" означает заместитель, выбранный из фтора, хлора, брома или йода. "Галогеноалкил" и "галогеноалкокси" представляют собой алкильную и алкокси группу соответственно, как они определены выше, замещенную одним или более галогенами. В случае галогеноалкильных и галогеноалкокси заместителей галоген подходящим образом заменяет водород в конце радикала, т.е. получается -алкилен-галоген или -алкоксилен-галоген. Термин "алкилен" относится к двухвалентной группе -(СН2)n-, где n равен 1-3, и термин "алкоксилен" относится к алкиленовой группе, содержащей простую эфирную связь, где простая эфирная связь является такой, как определено выше.
Термин "циано" относится к группе -CN.
Термин "гидроксил" относится к группе -ОН.
Термин "аралкил" относится к группе - алкилен-фенил, где алкилен является таким, как определено выше.
"Азот-содержащее C4-6алифатическое кольцо" представляет собой насыщенное C4-6алкильное кольцо, содержащее гетероатом азот. Примеры включают пирролидинильные, пиперидинильные и морфолинильные кольца.
Термин "содержит атом, который представляет собой радиоизотоп, подходящий для визуализации in vivo", означает, что в формуле I, как она определена выше, изотопная форма одного из атомов представляет собой радиоизотоп, подходящий для визуализации in vivo. Для того чтобы быть подходящим для визуализации in vivo, радиоизотоп является обнаруживаемым извне после введения указанному субъекту.
Если хиральный центр или другая форма изомерного центра присутствует в in vivo визуализирующем агенте по настоящему изобретению, то все формы такого изомера, включая энантиомеры и диастереоизомеры, охвачены настоящим изобретением. In vivo визуализирующие агенты по изобретению, содержащие хиральный центр, могут быть использованы в виде рацемической смеси или в виде энантиомерно-обогащенной смеси, или рацемическая смесь может быть разделена с использованием хорошо известных способов, и отдельный энантиомер может быть использован самостоятельно.
Предпочтительные визуализирующие агенты
R1 предпочтительно представляет собой метил или C2-3фторалкил и наиболее предпочтительно -этилен-F (т.е. -CH2-CH2-F).
R2 предпочтительно представляет собой водород, галогено, C1-3алкокси или C1-3фторалкокси. R2 наиболее предпочтительно представляет собой водород, галогено или C1-3алкокси и особенно наиболее предпочтительно водород, фтор или метокси. Когда R2 представляет собой заместитель, тогда он предпочтительно находится в положении 5 или 6 и наиболее предпочтительно выбран из 5-метокси, 6-метокси, 5-фтор и 6-фтор.
R3 и R4 предпочтительно независимо представляют собой метил, этил или бензил и наиболее предпочтительно оба являются этилом.
Альтернативно предпочтительно R3 и R4 вместе с атомом азота, к которому они присоединены, образуют азот-содержащее C5-6алифатическое кольцо.
Y1 предпочтительно представляет собой CH2.
Для наиболее предпочтительных in vivo визуализирующих агентов по настоящему изобретению Y2 представляет собой CH2-CH2.
Предпочтительный in vivo визуализирующий агент по изобретению подходит для визуализации с использованием однофотонной эмиссионной компьютерной томографии (SPECT) или позитронно-эмиссионной томографии (PET). Для SPECT подходящий радиоизотоп представляет собой гамма-излучающий радиоактивный галоген. Примерами гамма-излучающих радиоактивных галогенов, подходящих для применения в настоящем изобретении, являются 123I, 131I и 77Br. Предпочтительный гамма-излучающий радиоактивный галоген представляет собой 123I. Когда радиоизотоп in vivo визуализирующего агента представляет собой 123I, тогда предпочтительно, чтобы R2 представлял собой 123I. Для PET подходящий радиоизотоп представляет собой позитронно-эмиссионный радиоактивный неметалл. Примерами позитронно-эмиссионных радиоактивных неметаллов, подходящих для применения в настоящем изобретении, являются 11С, 18F и 124I. Предпочтительные позитронно-эмиссионные радиоактивные неметаллы представляют собой 11С и 18F. В случае 11C предпочтительно, чтобы R1 представлял собой 11С-метил. Когда радиоизотоп представляет собой 18F, тогда предпочтительно, чтобы R1 представлял собой C2-3[18F]фторалкил, наиболее предпочтительно -этилен-18F.
Предпочтительно, чтобы in vivo визуализирующий агент по изобретению подходил для PET визуализации, и 18F является предпочтительным радиоизотопом, подходящим для PET-визуализации. Предпочтение PET в способе по изобретению является следствием ее исключительной чувствительности и разрешающей способности, так что даже относительно небольшие изменения в поражении со временем могут быть обнаружены. PET-сканеры обычно измеряют радиоактивные концентрации в пикомолярном диапазоне. Микро-PET сканеры в настоящее время приближаются к пространственному разрешению приблизительно 1 мм, а клинические сканеры -приблизительно 4-5 мм.
Предпочтительный in vivo визуализирующий агент формулы 1 имеет формулу 1а:
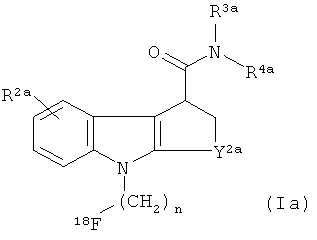
где:
R2a представляет собой водород, галогено или C1-3алкокси;
R3a и R4a независимо представляют собой метил, этил или бензил, или вместе с атомом азота, к которому они присоединены, образуют пирролидинильное, пиперидинильное, азепанильное или морфолинильное кольцо;
Y2а представляет собой CH2, CH2-CH2, СН(CH3)-CH2 или CH2-CH2-CH2; и
n равен 1, 2 или 3.
В формуле Ia R3a и R4a предпочтительно оба представляют собой этил, или R3a представляет собой метил и R4a представляет собой бензил, или вместе с атомом азота, к которому они присоединены, образуют азепанильное кольцо.
R2а предпочтительно представляет собой водород, метокси или фтор.
Y2a предпочтительно представляет собой CH2-CH2 или СН(CH3)-CH2.
n предпочтительно равен 2.
В предпочтительном in vivo визуализирующем агенте формулы Ia:
R3a и R4a оба представляют собой этил, или R3a представляет собой метил и R4a представляет собой бензил, или вместе с атомом азота, к которому они присоединены, они образуют азепанил;
R2a представляет собой водород, метокси или фтор;
Y2a представляет собой CH2-CH2 или СН(CH3)-CH2; и
n равен 2.
Неограничивающими примерами in vivo визуализирующих агентов формулы Ia являются следующие:
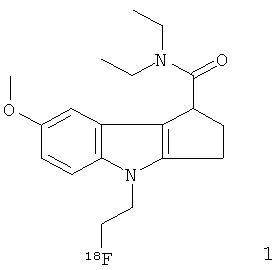
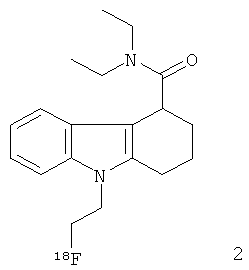
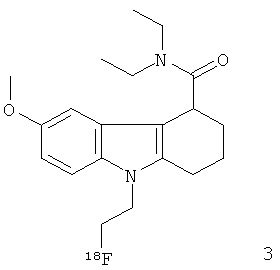
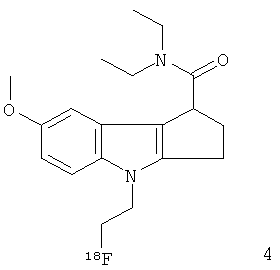

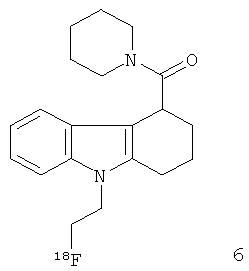
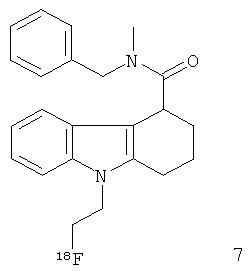
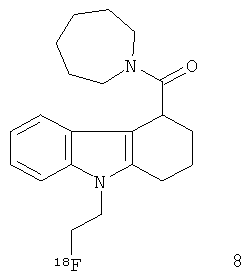

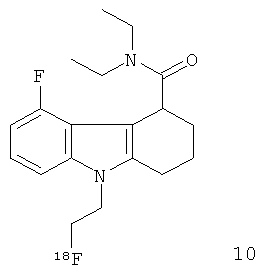
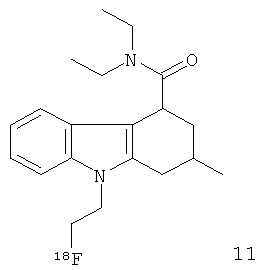
Из приведенных выше in vivo визуализирующих агентов 1-11, in vivo визуализирующие агенты 5, 6, 7, 9, 10 и 11 являются предпочтительными, in vivo визуализирующие агенты 5 и 10 являются наиболее предпочтительными и in vivo визуализирующий агент 5 является особенно предпочтительным. Для любого in vivo визуализирующего агента по настоящему изобретению энантиомерно чистая форма является особенно предпочтительной.
Соединение-предшественник
В еще одном аспекте настоящего изобретения предложено соединение-предшественник для получения in vivo визуализирующего агента по изобретению, где указанное соединение-предшественник имеет формулу II:
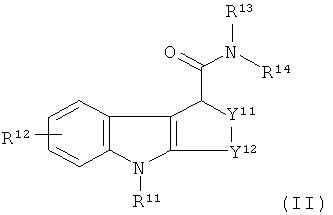
где один из R11 и R12 содержит химическую группу, которая вступает во взаимодействие с подходящим источником радиоизотопа, как определено выше для in vivo визуализирующего агента по изобретению, так что in vivo визуализирующий агент по изобретению образуется при взаимодействии указанного соединения-предшественника с указанным подходящим источником указанного радиоизотопа, а другой из R11 и R12 является таким, как определено для R1 и R2, соответственно, и возможно содержит защитную группу; и
R13-14 и Y11-12 являются такими, как определено здесь для R3-4 и Y1-2, соответственно, и возможно каждый дополнительно содержит защитную группу.
"Соединение-предшественник" содержит нерадиоактивное производное радиоактивно меченного соединения, сконструированное так, что химическая реакция с удобной химической формой детектируемой метки происходит сайт-специфически; может быть проведена за минимальное количество стадий (в идеале за одну стадию); и без необходимости в значительной очистке (в идеале без дополнительной очистки), с получением желаемого in vivo визуализирующего агента. Такие соединения-предшественники являются синтетическими и легко могут быть получены с хорошей химической чистотой.
Под термином "защитная группа" подразумевают группу, которая ингибирует или подавляет нежелательные химические реакции, но которая сконструирована таким образом, чтобы быть достаточно реакционноспособной, чтобы она могла быть отщеплена от интересующей функциональной группы с получением желательного продукта в достаточно мягких условиях, которые не модифицируют остальную часть молекулы. Защитные группы хорошо известны специалистам в данной области техники и описаны в 'Protective Groups in Organic Synthesis', Theorodora W. Greene and Peter G. M. Wuts, (Third Edition, John Wiley & Sons, 1999).
Термин "подходящий источник радиоизотопа" означает радиоизотоп в химической форме, которая реакционноспособна с заместителем соединения-предшественника, так что радиоизотоп ковалентно присоединяется к соединению-предшественнику. Для каждого конкретного радиоизотопа, представленного в следующем разделе, обсуждается один или более чем один подходящий источник радиоизотопа. Специалисту в области in vivo визуализирующих агентов знакомы эти и другие источники радиоизотопов, подходящие для применения в настоящем изобретении.
Схема 1 ниже представляет собой общую реакционную схему, которая показывает, как получить соединения, которые сами могут быть использованы в качестве соединений-предшественников или могут быть превращены в соединения-предшественники за небольшое количество дополнительных стадий. R11-14 и Y11-12 на схеме 1 являются такими, как определено выше для формулы II.
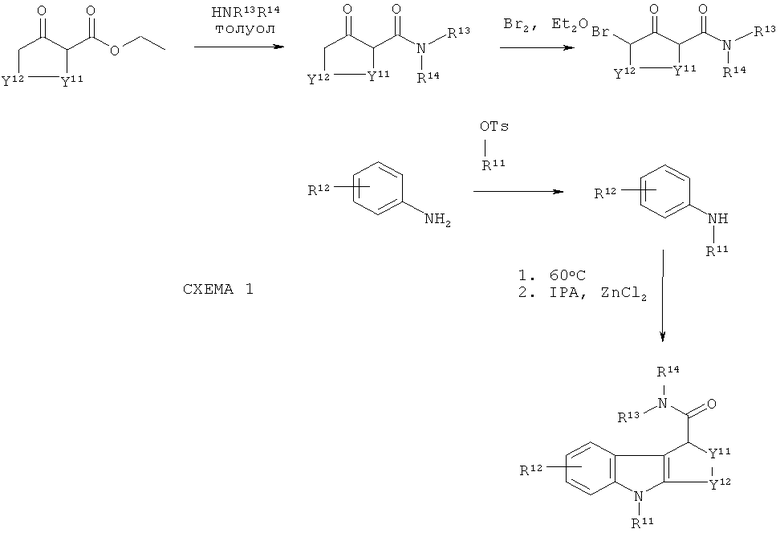
Альтернативно, когда R12 соединения-предшественника находится в верхнем положении кольца, тогда может быть использован общий путь синтеза, проиллюстрированный на схеме Ia ниже:
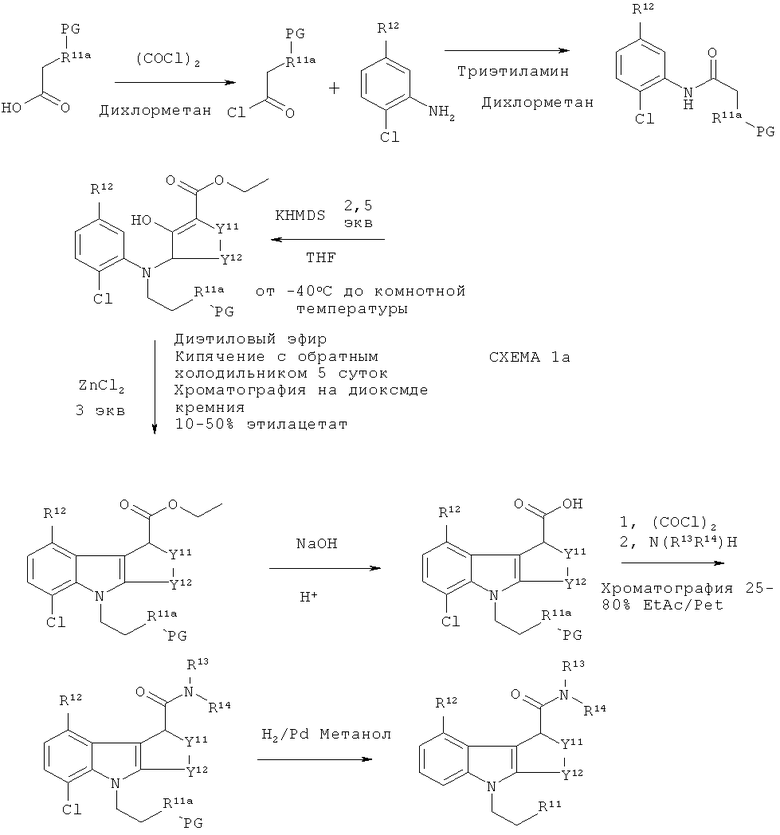
На схеме Ia выше -R11a-PG представляет защищенную группу R11, где R11 является таким, как подходящим образом и предпочтительно определено здесь. Когда R11 представляет собой гидрокси, тогда -R11a-PG может представлять собой, например, -O-бензил. R12-14 и Y11-12 являются такими, как подходящим образом и предпочтительно предложено для формулы II выше, при условии, что R12 не является хлоро. В этом пути синтеза хлор в нижнем положении кольца способствует прохождению циклизации только одним путем, так что образуется только один изомер. Аналогичный способ раскрыт в WO 2003/014082. Однако, когда авторы настоящего изобретения применили руководство из WO 2003/014082 для получения соединений-предшественников по настоящему изобретению, выход был низким (смотри пример 2(d)). Эта проблема была преодолена путем замены системы растворителей, используемых на стадии циклизации. В WO 2003/014082 стадию циклизации осуществляют в толуоле, в то время как авторы настоящего изобретения обнаружили, что оптимальные выходы получаются, когда вместо толуола используют диэтиловый эфир. Продукт стадии циклизации растворяется в диэтиловом эфире, в то время как не подвергшееся циклизации исходное соединение не растворяется. Таким образом, не подвергшееся циклизации исходное соединение остается с ZnCl2 в нижней части реакционного сосуда, а подвергшийся циклизации продукт переходит в диэтиловый эфир в верхней части реакционного сосуда.
Таким образом, в отдельном аспекте настоящего изобретения предложен способ получения соединения-предшественника формулы IIb:
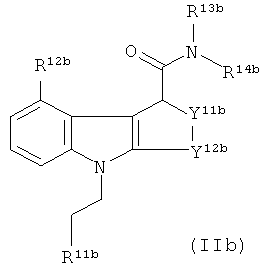
где:
R11b является таким, как определено на схеме 1а для R11a;
R12b-14b являются такими, как определено для R12-14 формулы II, при условии, что R12b не является хлоро; и
Y11b-12b являются такими, как определено для Y11-12 формулы II;
где указанный способ включает взаимодействие соединения формулы IIc с ZnCl2:
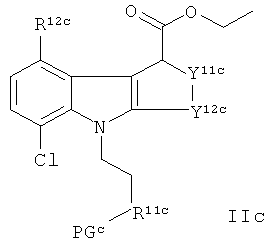
где R12c, Y11c и Y12c являются такими, как подходящим образом и предпочтительно определено здесь для R12, Y11 и Y12 соответственно, и PGc представляет собой защитную группу;
с образованием соединения формулы IId:
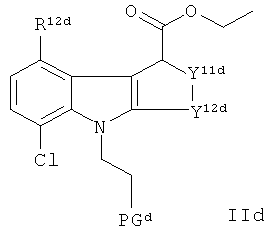
где R12d, Y11d, Y12d и PGd являются такими, как определено для R12c, Y11c, Y12c и PGc соответственно;
где указанное взаимодействие осуществляют в системе растворителей, содержащей диэтиловый эфир.
Предпочтительно, указанная защитная группа PGc, PGd представляет собой - бензил. Соединение-предшественник формулы IIb представляет собой предпочтительное соединение-предшественник формулы II.
Когда радиоизотоп in vivo визуализирующего агента представляет собой 18F, тогда внесение метки 18F может быть достигнуто посредством нуклеофильного замещения уходящей группы из соединения-предшественника. Подходящие уходящие группы включают Cl, Br, I, тозилат (OTs), мезилат (OMs) и трифлат (OTf). Еще одна стратегия может заключаться в том, чтобы иметь подходящую уходящую группу вместо алкиламидной группы, представленной на соединении-предшественнике. В обоих случаях соединение-предшественник можно пометить за одну стадию путем взаимодействия с подходящим источником [18F]-фторид-иона (18F-), который обычно получают в виде водного раствора в результате ядерной реакции 18O(p,n)18F, и приводят в реакционноспособное состояние путем добавления катионного противоиона и последующего удаления воды. 18F также может быть введен путем O-алкилирования гидроксильных групп в соединении-предшественнике при помощи 18F(CH2)3-LG, где LG представляет собой уходящую группу, как определено выше. Альтернативно, радиоактивный атом фтора может быть присоединен при помощи прямой ковалентной связи к ароматическому кольцу, такому как бензольное кольцо. Для арильных систем 18F-фторидное нуклеофильное замещение из арилдиазониевой соли, арилнитросоединения или арилчетвертичной аммониевой соли являются подходящими путями получения арил-18F-производных.
Любая из вышеприведенных схем 1 или 1а может быть продолжена с получением соединений-предшественников, подходящих для получения 18F in vivo визуализирующих агентов по изобретению, как показано на схеме 2 ниже:
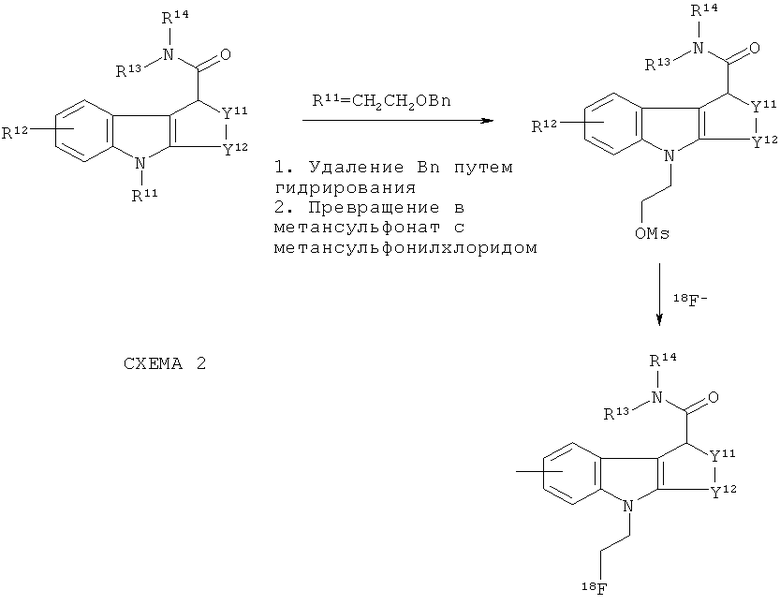
Исходные соединения и промежуточные соединения имеются в продаже или известны из опубликованных научных статей, например Napper et al J Med Chem 2005; 48: 8045-54; Davies et al J Med Chem 1998; 41: 451-467.
В предпочтительном соединении-предшественнике формулы II для
получения in vivo визуализирующего агента, содержащего 18F, R11 представляет собой C1-3алкилен-LG, где LG представляет собой уходящую группу. Наиболее предпочтительное такое соединение-предшественник имеет формулу IIа:
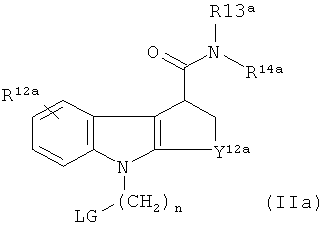
где:
LG выбрана из мезилата, тозилата и трифлата; и R12a-14а, Y12a и m являются такими, как подходящим образом и предпочтительно определено выше для R2a-4a, Y2a и n соответственно, в формуле Ia.
Неограничивающими примерами предпочтительных соединений-предшественников формулы IIа являются следующие:
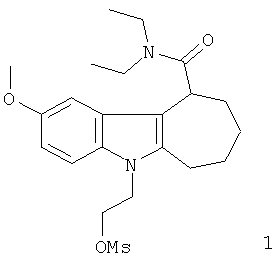
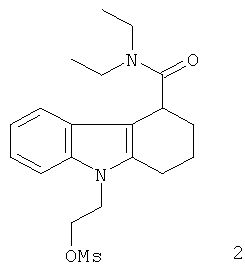
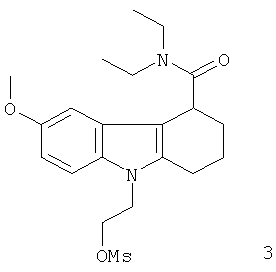

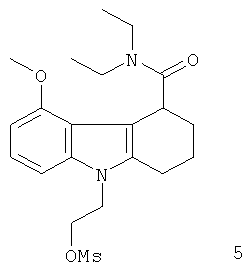
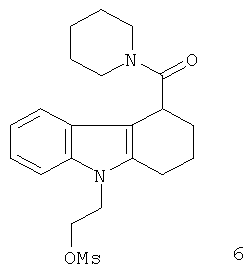
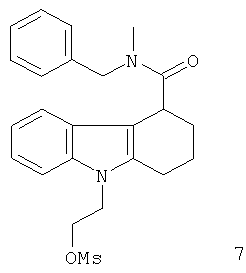
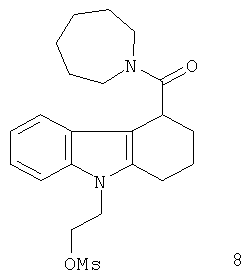
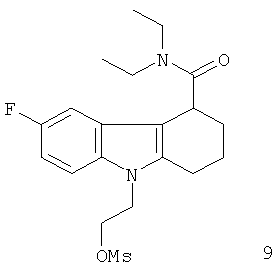
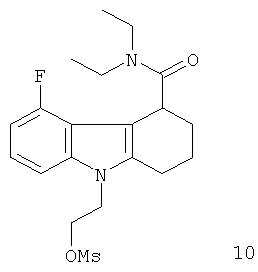
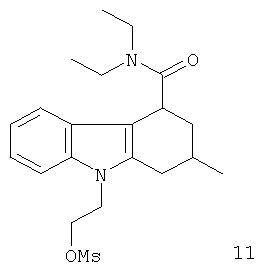
Из вышеприведенных соединений-предшественников 1-11 соединения-предшественники 5, 6, 7, 9, 10 и 11 являются предпочтительными, соединения-предшественники 5 и 10 являются наиболее предпочтительными и соединение-предшественник 5 является особенно предпочтительным.
11C-меченые PET соединения, содержащие изотопный индикатор, могут быть синтезированы путем взаимодействия соединения-предшественника с 11С-метилйодидом. Поскольку период полураспада 11С составляет только 20,4 минут, важно, чтобы промежуточный 11С-метилйодид обладал высокой специфической активностью и, следовательно, чтобы его получали с использованием реакционного процесса, который был бы настолько быстрым, насколько это возможно. Подробный обзор таких методов введения 11С-меток можно найти в Antoni et al "Aspects on the Synthesis of 11C-Labelled Compounds" in Handbook of Radiopharmaceuticals, Ed. M.J.Welch и C.S.Redvanly (2003, John Wiley and Sons).
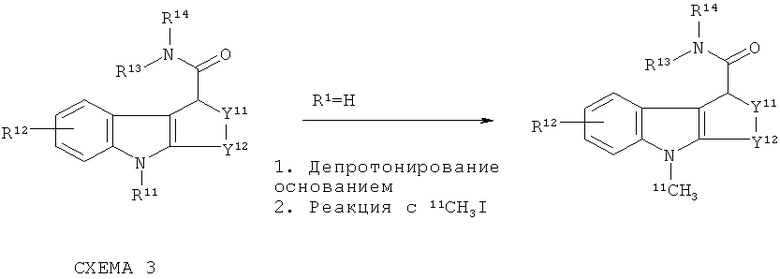
11C-меченые in vivo визуализирующие агенты по изобретению могут быть получены путем продолжения вышеприведенной схемы 1, как показано на схеме 3 ниже:
Когда визуализирующая группировка представляет собой радиоактивный йод, тогда предпочтительные соединения-предшественники представляют собой соединения, содержащие производное, которое претерпевает электрофильное йодирование. Примерами являются металлоорганические производные, такие как триалкилстаннан (например, триметилстаннил или трибутилстаннил), или триалкилсилан (например, триметилсилил), или борорганическое соединение (например, боронатные эфиры или органотрифторбораты).
Для электрофильного радиоактивного йодирования соединение-предшественник предпочтительно содержит: активированное металлоорганическое соединение-предшественник (например, триалкилолово, триалкилсилил или борорганическое соединение). Соединения-предшественники и способы введения радиоактивного йода в органические молекулы описаны в Bolton (J. Lab. Сотр. Radiopharm. 2002; 45: 485-528). Подходящие борорганические соединения на основе боронатного эфира и их получение описаны в Kabalaka et al (Nucl. Med. Biol., 2002; 29: 841-843 и 2003; 30: 369-373). Подходящие органотрифторбораты и их получение описаны в Kabalaka et al (Nucl. Med. Biol., 2004; 31: 935-938). Предпочтительные соединения-предшественники для радиоактивного йодирования содержат металлоорганическое соединение-предшественник, наиболее предпочтительно триалкилолово.
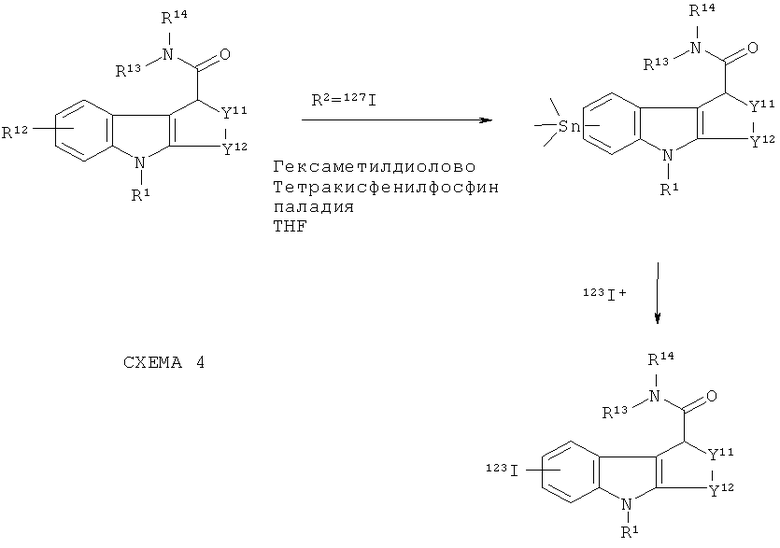
Меченные радиоактивный йодом in vivo визуализирующие агенты по изобретению могут быть получены путем продолжения вышеприведенной схемы 1, как проиллюстрировано на приведенной ниже схеме 4:
Радиоактивное бромирование может быть осуществлено при помощи способов, аналогичных способам, описанным выше для радиоактивного йодирования. Kabalka и Varma привели обзор различных способов синтеза радиоактивных галогенированных соединений, включающих радиобромированные соединения (Tetrahedron 1989; 45(21): 6601-21).
Соединение-предшественник по изобретению в идеале предложено в стерильной апирогенной форме. Соответственно, соединение-предшественник можно использовать для изготовления фармацевтической композиции, содержащей in vivo визуализирующий агент вместе с биосовместимым носителем, подходящим для введения млекопитающему. Соединение-предшественник также подходит для включения в качестве компонента в набор или кассету для изготовления такой фармацевтической композиции. Эти аспекты более подробно обсуждаются ниже.
В еще одном предпочтительном воплощении соединение-предшественник связан с твердой фазой. Соединение-предшественник предпочтительно поставляется ковалентно присоединенным к твердой несущей матрице. Таким образом, желательный продукт образуется в растворе, в то время как исходные вещества и примеси остаются связанными с твердой фазой. В качестве примера такой системы соединения-предшественники для твердофазного электрофильного фторирования при помощи 18F-фторида описаны в WO 03/002489, и соединения-предшественники для твердофазного нуклеофильного фторирования при помощи 18F-фторида описаны в WO 03/002157.
Способ получения
В еще одном аспекте настоящего изобретения предложен способ получения in vivo визуализирующего агента по изобретению, при котором:
(1) берут соединение-предшественник по изобретению;
(2) берут подходящий источник указанного радиоизотопа, как определено здесь;
(3) приводят соединение-предшественник со стадии (1) во взаимодействие с радиоизотопом со стадии (2) с получением in vivo визуализирующего агента по изобретению.
На стадии (1) соединение-предшественник может быть получено в растворе в наборе или в кассете, подходящей для применения с аппаратом для автоматического синтеза, или альтернативно присоединено к твердому носителю, как описано выше в описании соединения-предшественника. Набор и кассета образуют дополнительные аспекты изобретения и более подробно будут обсуждаться ниже.
Стадия "взаимодействия" соединения-предшественника с радиоизотопом включает приведение двух реагирующих веществ вместе в условиях реакции, подходящих для образования желаемого in vivo визуализирующего агента с настолько высоким радиохимическим выходом (RCY), насколько это возможно. Некоторые конкретные синтетические пути получения in vivo визуализирующих агентов по настоящему изобретению представлены в нижеприведенном экспериментальном разделе.
Для способа получения по изобретению подходящие и предпочтительные воплощения in vivo визуализирующего агента, соединения-предшественника и радиоизотопа являются такими, как уже здесь предложено.
Набор и кассета
В еще одном аспекте настоящего изобретения предложен набор для получения in vivo визуализирующего агента по изобретению, где указанный набор содержит соединение-предшественник по изобретению, так, что взаимодействие со стерильным источником радиоизотопа позволяет получить желаемый in vivo визуализирующий агент с минимальным количеством манипуляций. Данные соображения особенно важны, когда радиоизотоп обладает относительно коротким периодом полураспада, и для облегчения обработки и, следовательно, уменьшенной дозы радиации для радиофармацевта. Соединение-предшественник предпочтительно представлено в наборе в лиофилизированной форме, и реакционная среда для восстановления таких наборов предпочтительно представляет собой биосовместимый носитель.
"Биосовместимый носитель" представляет собой жидкость, в частности жидкость, в которой in vivo визуализирующий агент суспендирован или растворен, так что композиция является физиологически приемлемой, т.е. может быть введена в организм млекопитающего без токсичности или чрезмерного дискомфорта. Биосовместимый носитель подходящим образом представляет собой инъецируемую жидкость-носитель, такую как стерильная апирогенная вода для инъекции; водный раствор, такой как физиологический раствор (который предпочтительно может быть сбалансирован таким образом, что конечный продукт для инъекции является или изотоническим или не гипотоническим); водный раствор одного или более веществ, регулирующих ионную силу (например, солей катионов плазмы крови с биосовместимыми противоионами), сахаров (например, глюкозы или сахарозы), сахарных спиртов (например, сорбита или маннита), гликоли (например, глицерин) или другие неионные полиольные вещества (например, полиэтиленгликоли, пропиленгликоли и т.п.). Биосовместимый носитель также может содержать биосовместимые органические растворители, такие как этанол. Такие органические растворители полезны для солюбилизации более липофильных соединений или композиций. Предпочтительно биосовместимый носитель представляет собой апирогенную воду для инъекции, изотонический физиологический раствор или водный этанольный раствор. pH биосовместимого носителя для внутривенной инъекции подходящим образом находится в диапазоне от 4,0 до 10,5.
В наборе по изобретению соединение-предшественник предпочтительно представлен в закрытом контейнере, который обеспечивает поддержание стерильности и/или радиоактивной безопасности, с возможным присутствием инертного газа в свободном пространстве (например, азота или аргона), в то же время позволяет добавлять и отбирать растворы при помощи шприца. Предпочтительный закрытый контейнер представляет собой флакон, закрытый перегородкой, где газонепроницаемая крышка фиксируется при помощи дополнительного укупорочного средства (обычно из алюминия). Дополнительным преимуществом таких закрытых контейнеров является то, что крышка, при необходимости, может выдерживать вакуум, например, для смены газа в свободном пространстве или дегазирования растворов.
Предпочтительные воплощения соединения-предшественника при использовании в наборе являются такими, как описано ранее.
Соединение-предшественник для применения в наборе можно использовать в асептических условиях изготовления с получением желаемого стерильного апирогенного вещества. Соединение-предшественник альтернативно можно использовать в нестерильных условиях, а затем окончательно стерилизовать с использованием, например, гамма-излучения, автоклавирования, сухого жара или химической обработки (например, этиленоксидом). Предпочтительно, соединение-предшественник предложено в стерильной апирогенной форме. Наиболее предпочтительно стерильное апирогенное соединение-предшественник предложено в герметично закрытом контейнере, как описано ранее.
Предпочтительно, все компоненты набора являются одноразовыми, чтобы минимизировать возможности загрязнения между использованиями и для обеспечения стерильности и гарантии качества.
[18F]-Радиоактивные индикаторы, в частности, в настоящее время часто для удобства готовят на автоматическом аппарате для радиохимического синтеза. Имеется несколько имеющихся в продаже примеров такого аппарата, включая Tracerlab™ и Fastlab™ (GE Healthcare Ltd). Такой аппарат обычно содержит "кассету", часто одноразовую, в которой проводится радиохимическая реакция, которая прикрепляется к аппарату для обеспечения радиохимического синтеза. Кассета обычно включает жидкостные магистрали, реакционный сосуд и порты для присоединения флаконов с реагентами, а также любые картриджи для твердофазной экстракции, используемые на стадиях очистки после радиохимического синтеза.
Таким образом, в еще одном аспекте настоящего изобретения предложена кассета для автоматического синтеза in vivo визуализирующего агента, как определено здесь, содержащая:
(1) сосуд, содержащий соединение-предшественник, как оно определено здесь; и
(2) средства для элюирования из сосуда с подходящим источником указанного радиоизотопа, подходящего для in vivo визуализации, как она определена здесь.
Для кассеты по изобретению подходящие и предпочтительные воплощения соединения-предшественника и подходящего источника радиоактивного изотопа являются такими, как определено здесь выше.
Кассета дополнительно может включать:
(3) ионообменный картридж для удаления избытка радиоизотопа; и, возможно,
(4) когда соединение-предшественник содержит одну или более чем одну защитную группу, картридж для удаления защиты с образующегося в результате радиоактивно меченного продукта с образованием in vivo визуализирующего агента, как определено здесь.
Радиофармацевтическая композиция
В еще одном дополнительном аспекте настоящего изобретения предложена "радиофармацевтическая композиция", которая представляет собой композицию, содержащую in vivo визуализирующий агент по изобретению вместе с биосовместимым носителем в форме, подходящей для введения млекопитающему. Биосовместимый носитель является таким, как определено выше в отношении набора по изобретению. Для радиофармацевтической композиции по изобретению подходящие и предпочтительные воплощения in vivo визуализирующего агента являются такими, как ранее определено в описании.
Радиофармацевтическая композиция может быть введена парентерально, т.е. путем инъекции, и наиболее предпочтительно представляет собой водный раствор. Такая композиция возможно может содержать дополнительные ингредиенты, такие как буферы; фармацевтически приемлемые солюбилизаторы (например, циклодекстрины или поверхностно-активные вещества, такие как Pluronic, Tween или фосфолипиды); фармацевтически приемлемые стабилизаторы или антиоксиданты (такие как аскорбиновая кислота, гентизиновая кислота или пара-аминобензойная кислота). Когда in vivo визуализирующий агент по изобретению предложен в виде радиофармацевтической композиции, способ получения указанного in vivo визуализирующего агента может дополнительно включать стадии, требующиеся для изготовления радиофармацевтической композиции, например удаление органического растворителя, добавление биосовместимого буфера и любые дополнительные возможные ингредиенты. Для парентерального введения также необходимо учитывать стадии, гарантирующие, что радиофармацевтическая композиция является стерильной и апирогенной.
Способы применения
В еще одном аспекте настоящего изобретения предложен способ визуализации in vivo для определения распределения и/или степени экспрессии PBR у субъекта, при котором:
(1) указанному субъекту вводят in vivo визуализирующий агент по изобретению;
(2) обеспечивают возможность указанному in vivo визуализирующему агенту связываться с PBR у указанного субъекта;
(3) детектируют способом in vivo визуализации сигналы, испускаемые радиоизотопом указанного in vivo визуализирующего агента;
(4) создают изображение, типичное для расположения и/или количества указанных сигналов; и
(5) определяют распределение и степень экспрессии PBR у указанного субъекта, где указанная экспрессия находится в прямой корреляции с указанными сигналами, испускаемыми указанным in vivo визуализирующим агентом.
Для способа визуализации in vivo по изобретению подходящие и предпочтительные воплощения in vivo визуализирующего агента являются такими, как описано ранее в описании изобретения.
"Введение" in vivo визуализирующего агента предпочтительно осуществляют парентерально и наиболее предпочтительно внутривенно. Внутривенный путь представляет собой наиболее эффективный путь для доставки in vivo визуализирующего агента в организм субъекта и таким образом также через гематоэнцефалический барьер (ВВВ) и в контакт с PBR, экспрессирующимися в центральной нервной системе (CNS) указанного субъекта. Кроме того, внутривенное введение не является существенным физическим вмешательством или значительным риском для здоровья. In vivo визуализирующий агент по изобретению предпочтительно вводят в виде фармацевтической композиции по изобретению, как определено здесь. Способ визуализации in vivo по изобретению также можно понять как содержащий определенные выше стадии (ii)-(v), осуществляемые в отношении субъекта, которому предварительно вводят in vivo визуализирующий агент по изобретению.
После стадии введения и до стадии детектирования in vivo визуализирующему агенту дают возможность связываться с PBR. Например, когда субъект представляет собой интактное млекопитающее, тогда in vivo визуализирующий агент динамически перемещается в организме млекопитающего, вступая в нем в контакт с различными тканями. Как только in vivo визуализирующий агент вступает в контакт с PBR, происходит специфическое взаимодействие, так что клиренс in vivo визуализирующего агента из ткани с PBR происходит медленнее, чем из ткани без PBR или с меньшим количеством PBR. Будет достигнут определенный момент времени, когда обнаружение in vivo визуализирующего агента, специфически связанного с PBR, становится возможным как результата соотношения in vivo визуализирующего агента, связанного с тканью, содержащей PBR, и связанного с тканью без PBR или с меньшим количеством PBR. В идеале такое отношение составляет примерно 2:1.
Стадия "обнаружения" способа по изобретению включает обнаружение сигналов, испускаемых радиоизотопом, при помощи детектора, чувствительного к указанным сигналам. Эта стадия обнаружения также может быть понята как получение данных о сигнале. Однофотонная эмиссионная томография (SPECT) и позитронно-эмиссионная томография (PET) являются наиболее подходящими методами in vivo визуализации для применения в способе по изобретению. PET представляет собой предпочтительный способ in vivo визуализации для применения в способе по изобретению.
Стадию "генерирования" способа по изобретению осуществляют при помощи компьютера, который применяет алгоритм реконструкции к полученным данным о сигнале с получением набора данных. Этим набором данных затем манипулируют для генерирования изображений, показывающих расположение и/или количество сигналов, испускаемых указанным радиоизотопом. Испускаемые сигналы находятся в прямой корреляции с экспрессией PBR, так что стадия "определения" может быть осуществлена путем оценки полученного изображения.
"Субъектом" по изобретению может являться любой человек или животное. Предпочтительно субъект по изобретению представляет собой млекопитающее. Наиболее предпочтительно, указанный субъект представляет собой организм интактного млекопитающего in vivo. В особенно предпочтительном воплощении субъект по изобретению представляет собой человека. In vivo способ визуализации может быть использован для исследования PBR у здоровых субъектов или субъектов, которые, как известно или предполагается, имеют патологическое состояние, ассоциированное с аномальной экспрессией PBR (здесь "PBR-состояние"). Предпочтительно, указанный способ относится к визуализации in vivo субъекта, который, как известно или предполагается, имеет PBR-состояние и, следовательно, полезен для диагностики указанного состояния.
Примеры таких состояний PBR, при которых визуализация in vivo может быть полезна, включают рассеянный склероз, энцефалит Расмуссена, церебральный васкулит, герпетический энцефалит, СПИД-ассоциированную деменцию, болезнь Паркинсона, кортико-базальную дегенерацию, прогрессирующий надъядерный паралич, множественную системную атрофию, болезнь Гентингтона, боковой амиотрофический склероз, болезнь Альцгеймера, ишемический инсульт, повреждение периферических нервов, эпилепсию, травматическое поражение головного мозга, острый стресс, хронический стресс, невропатическую боль, воспаление легких, хроническое обструктивное заболевание легких, астму, воспалительное заболевание кишечника, ревматоидный артрит, первичную фибромиалгию, поражение нервов, атеросклероз, воспаление почек, ишемически-реперфузионное поражение и рак, в частности рак толстого кишечника, предстательной железы или молочной железы. In vivo визуализирующие агенты по изобретению особенно подходят для визуализации in vivo ЦНС благодаря их хорошему поглощению в головном мозге.
В альтернативном воплощении способ визуализации in vivo по изобретению может быть осуществлен повторно в схеме лечения указанного субъекта, где указанная схема включает введение лекарственного средства для борьбы с PBR-состоянием. Например, способ визуализации in vivo по изобретению может быть осуществлен до, во время и после лечения лекарственным средством для борьбы с PBR-состоянием. Таким образом, эффект указанного лечения можно наблюдать по времени. Предпочтительно для этого воплощения метод визуализации in vivo представляет собой PET (позитронно-эмиссионная томография). PET обладает превосходной чувствительностью и разрешением, так что даже относительно небольшие изменения поражения могут быть обнаружены по времени, что особенно благоприятно для наблюдения за лечением.
В еще одном аспекте настоящего изобретения предложен способ диагностики PBR-состояния. Способ диагностики по изобретению включает способ визуализации in vivo, как он определенный выше, вместе с дополнительной стадией (6), на которой относят распределение и степень экспрессии PBR к конкретной клинической картине, т.е. дедуктивной фазе принятия медицинского решения.
В еще одном аспекте настоящего изобретения предложен in vivo визуализирующий агент, как он определено здесь, для применения в способе диагностики, как он определен здесь.
В еще одном аспекте настоящего изобретения предложен in vivo визуализирующий агент, как он определен здесь, для применения в изготовлении радиофармацевтической композиции, как она определена здесь, для применения способе диагностики, как он определен здесь.
Изобретение проиллюстрировано при помощи ряда неограничивающих объем изобретения примеров.
Краткое описание примеров
Пример 1 описывает синтез соединения-предшественника 5 и визуализирующего агента 5.
Пример 2 описывает синтез нерадиоактивного аналога визуализирующего агента 5.
Пример 3 описывает синтез соединения-предшественника 6 и визуализирующего агента 6.
Пример 4 описывает синтез нерадиоактивного аналога визуализирующего агента 6.
Пример 5 описывает синтез соединения-предшественника 7 и визуализирующего агента 7.
Пример 6 описывает синтез нерадиоактивного аналога визуализирующего агента 7.
Пример 7 описывает синтез соединения-предшественника 9 и визуализирующего агента 9.
Пример 8 описывает синтез нерадиоактивного аналога визуализирующего агента 9.
Пример 9 описывает синтез соединения-предшественника 10 и визуализирующего агента 10.
Пример 10 описывает синтез нерадиоактивного аналога визуализирующего агента 10.
Пример 11 описывает синтез соединения-предшественника 11 и визуализирующего агента 11.
Пример 12 описывает синтез нерадиоактивного аналога визуализирующего агента 11.
Пример 13 описывает энантиомерное разделение соединения-предшественника 5.
Пример 14 описывает энантиомерное разделение нерадиоактивного визуализирующего агента 5.
Пример 15 описывает in vitro анализ активности, который использовали для тестирования аффинности в отношении PBR.
Пример 16 описывает способ биораспределения, который использовали для проверки эффективности визуализирующих агентов по изобретению in vivo.
Пример 17 описывает синтез нерадиоактивного аналога предшествующего тетрациклического индольного визуализирующего агента.
Пример 18 описывает синтез предшествующего тетрациклического индольного визуализирующего агента.
Перечень сокращений, используемых в примерах
Примеры
Пример 1: Синтез метансульфоновой кислоты 2-(4-диэтилкарбамил-5-метокси-1,2,3,4-тетрагидро-карбазол-9-ил)этилового эфира (соединение-предшественник 5) и 9-(2-[18F]фтор-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (визуализирующий агент 5)
Пример 1(а): Бензилокси-ацетилхлорид (1)
К бензилоксиуксусной кислоте (10,0 г, 60,0 ммоль, 8,6 мл) в дихлорметане (50 мл) добавляли оксалилхлорид (9,1 г, 72,0 ммоль, 6,0 мл) и DMF (30,0 мг, 0,4 ммоль, 32,0 мкл) и перемешивали при к.т. в течение 3 ч. Сначала происходило быстрое выделение газа по мере прохождения реакции, но выделение прекращалось, когда реакция завершалась. Дихлорметановый раствор концентрировали в вакууме с получением смолы. Эту смолу обрабатывали дополнительным количеством оксалилхлорида (4,5 г, 35,7 ммоль, 3,0 мл), дихлорметана (50 мл) и одной каплей DMF. Происходило быстрое выделение газа, и реакционную смесь перемешивали в течение еще 2 ч. Реакционную смесь затем концентрировали в вакууме с получением 11,0 г (количественный выход) бензилоксиацетилхлорида (1) в виде смолы. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 73.6, 74.8, 128.1, 128.4, 128.6, 130.0 и 171.9.
Пример 1(b): 2-Бензилокси-N-(2-хлор-5-метокси-фенил)ацетамид (2)
Бензилокси-ацетилхлорид (1) (11,0 г, 60,0 ммоль) и 2-хлор-5-метоксианилина гидрохлорид (11,7 г, 60,2 ммоль) в дихлорметане (100 мл) при 0°C перемешивали и триэтиламин (13,0 г 126,0 ммоль, 18,0 мл) медленно добавляли в течение 15 мин. Перемешиваемую реакционную смесь оставляли нагреваться до к.т. в течение 18 ч. Происходит сильное осаждение триэтиламина гидрохлорида. Дихлорметановый раствор промывали 10% водным карбонатом калия (50 мл), сушили над сульфатом магния и концентрировали в вакууме с получением 18,9 г (количественный выход) 2-бензилокси-Т-(2-хлор-5-метокси-фенил)ацетамида (2) в виде смолы. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 55.6, 69.6, 73.6, 106.2, 111.1, 114.1, 127.7, 128.3, 128.6, 129.2, 134.6, 136.5, 158.9 и 167.7.
Пример 1(с): (2-Бензилокси-этил)-(2-хлор-5-метоксифенил)амин (3)
2-Бензилокси-N-(2-хлор-5-метокси-фенил)ацетамид (2) (18,9 г, 62,0 ммоль) в THF (100 мл) перемешивали и медленно добавляли алюмогидрид лития (4,9 г, 130,0 ммоль) в течение 15 мин. Происходило быстрое выделение газообразного водорода после добавления первой порции алюмогидрида лития. Реакционную смесь затем нагревали до температуры дефлегмации в течение 4 ч и оставляли при к.т. на выходные дни. Реакцию затем гасили путем добавления по каплям воды (50 мл) к перемешиваемому раствору. Происходило сильное выделение водорода, вызывающее дефлегмацию реакционной смеси. Реакционную смесь затем концентрировали в вакууме до суспензии. Добавляли воду (200 мл) и этилацетат (200 мл) и смесь интенсивно встряхивали. Реакционную смесь затем фильтровали через целит для удаления осажденного гидроксида алюминия и этилацетатный раствор отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением 18,4 г (количественный выход) (2-бензилокси-этил)-(2-хлор-5-метоксифенил)амина (3) в виде смолы. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 43.3, 55.3, 68.2, 73.0, 98.1, 101.8, 111.6, 127.6,127.7, 128.4, 129.3, 137.9, 144.8 и 159.5.
Пример 1(d): 3-Бром-2-гидрокси-циклогекс-1-енкарбоновой кислоты этиловый эфир (4]
Этил-2-оксоциклогексанкарбоксилат (30 г, 176 ммоль, 28 мл) растворяли в диэтиловом эфире (30 мл) и охлаждали до 0°C в атмосфере азота. Бром (28 г, 176 ммоль, 9,0 мл) добавляли по каплям в течение 15 мин и реакционную смесь оставляли нагреваться до к.т. в течение 90 мин. Смесь медленно выливали в ледяной насыщенный водный карбонат калия (250 мл) и экстрагировали этилацетатом (3×200 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали, концентрировали в вакууме и сушили в вакуумной линии в течение 18 ч с получением 41,4 г (94%) 3-бром-2-гидрокси-1-енкарбоновой кислоты этилового эфира (4) в виде желтого масла. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 14.1, 17.7, 21.8, 32.0, 60.0, 60.8, 99.7, 166.3 и 172.8.
Пример 1(e): 3[(2-Бензилокси-этил)-(2-хлор-5-метокси-фенил)-амино]-2-гидрокси-циклогекс-1-ен карбоновой кислоты этиловый эфир (5)
(2-Бензилокси-этил)-(2-хлор-5-метоксифенил)амин (3) (10,0 г, 34,2 ммоль) перемешивали в обезвоженном THF (100 мл) при -40°C в атмосфере азота и бис(триметилсилил)амид калия (143,0 мл 0,5 М раствора в толуоле, 72,0 ммоль) добавляли в течение 30 мин. Затем добавляли 3-бром-2-гидроксициклогекс-1-енкарбоновой кислоты этиловый эфир (4) (8,5 г, 34,2 ммоль) в обезвоженном THF (10 мл) и оставляли нагреваться до к.т. в течение 1,5 ч. Добавляли уксусную кислоту (10,0 г, 166 ммоль, 10,0 мл) и концентрировали в вакууме для удаления THF. Добавляли этилацетат (200 мл) и 10% водный карбонат калия (100 мл) и смесь интенсивно встряхивали. Этилацетатный раствор отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением 16,5 г (количественный выход) 3[(2-бензилокси-этил)-(2-хлор-5-метокси-фенил)-амино]-2-гидрокси-циклогекс-1-ен-карбоновой кислоты этилового эфира (5) в виде смолы, которую использовали неочищенной на следующей стадии. HPLC (высокоэффективная жидкостная хроматография) (Gemini 150×4,6 мм, 50-95% метанол/вода в течение 20 мин) неочищенной реакционной смеси, 18,9 мин (38%), 19,2 мин (25%), 23,1 мин (28%).
Выделяли один компонент реакционной смеси 13С NMR (75 МГц, CDCl3): δC 14.3, 20.6, 21.8, 26.4, 38.6, 43.0, 55.8, 60.5, 68.7, 73.3, 93,4, 106.3, 108.2, 119.3, 121.5, 127.5, 127.6, 128.3, 135.7, 137.0, 137.9, 155.7 и 175.0.
Пример 1(f): 9-(2-Бензилокси-этил)-8-хлор-5-метокси-2,3,4,9, -тетрагидро-1Н-карбазол-4-карбоновой кислоты этиловый эфир (6)
Хлорид цинка (7,1 г, 52,0 ммоль) добавляли к 3[(2-бензилокси-этил)-(2-хлор-5-метокси-фенил)-амино]-2-гидрокси-циклогекс-1-енкарбоновой кислоты этиловому эфиру (5) (8,0 г, 17,0 ммоль) в обезвоженном диэтиловом эфире (150 мл) в атмосфере азота и нагревали с обратным холодильником в течение 5,5 ч. По мере дефлегмации реакционной смеси в ней образовывалось густое коричневое масло. Реакционную смесь затем охлаждали и надосадочный диэтиловый эфир декантировали, добавляли этилацетат (100 мл), промывали 2 н. HCl (50 мл) и 10% водным карбонатом калия (50 мл). Слой диэтилового эфира отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением масла (2,0 г). Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя смесью бензин (А):этилацетат (В) (10-40% (В), 340 г, 22 CV, 150 мл/мин) с получением 1,8 г 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9,-тетрагидро-1Н-карбазол-4-карбоновой кислоты этилового эфира (6). Густой плотный коричневый слой обрабатывали этилацетатом (100 мл) и 2 н. HCl (50 мл). Этилацетатный раствор отделяли, промывали 10%-ным водным карбонатом калия (50 мл), сушили над сульфатом магния и концентрировали в вакууме с получением масла (5,2 г). Добавляли диэтиловый эфир (100 мл) и сухой хлорид цинка (7,0 г). Смесь нагревали с обратным холодильником в течение еще 5 суток. Эфирный слой декантировали с темной смолы, промывали 2 н. HCl (50 мл), сушили над сульфатом магния и концентрировали в вакууме с получением смолы (2,8 г). Эту смолу очищали посредством хроматографии на силикагеле, элюируя смесью бензин (А):этилацетат (В) (5-35% (В), 340 г, 150 мл/мин) с получением 2,1 г 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9,-тетрагидро-1Н-карбазол-4-карбоновой кислоты этилового эфира (6). Все полученное вещество представляло собой 4,1 г (50%) 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9,-тетрагидро-1Н-карбазол-4-карбоновой кислоты этилового эфира (6). Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 14.4, 20.5, 22.3, 27.5, 40.2, 43.9, 55.0, 60.2, 70.7, 73.3, 100.2, 107.5, 108.4, 120.1, 122.8, 127.4, 127.5, 128.2, 132.0, 137.4, 138.1, 152.6 и 175.8.
Пример 1(g): 9-(2-Бензилокси-этил]-8-хлор-5-метокси-2,3,4,9,-тетрагидро-1Н-карбазол-4-карбоновая кислота (7)
К 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9,-тетрагидро-1Н-карбазол-4-карбоновой кислоты этиловому эфиру (6) (2,0 г, 4,1 ммоль) в этаноле (50 мл) добавляли гидроксид натрия (1,1 г, 27,1 ммоль) и воду (5 мл) и нагревали при 80°C в течение 18 ч. Этанол затем удаляли путем упаривания в вакууме и остаток распределяли между диэтиловым эфиром (50 мл) и водой (50 мл). Слой диэтилового эфира отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением смолы (71,0 мг). Водный слой подкисляли до pH 1 при помощи 2 н. HCl (20 мл) и экстрагировали дихлорметаном (2×100 мл). Дихлорметановый слой сушили над сульфатом магния и концентрировали в вакууме с получением 1,6 г (87%) 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9,-тетрагидро-1Н-карбазол-4-карбоновой кислоты (7) в виде пены. Структуру подтверждали при помощи 13С NMR (75 МГц; CDCl3): δC 20.2, 22.2, 27.1, 39.7, 44.0, 55.1, 70.7, 73.3, 100.6, 106.3, 108.9, 123.0, 127.4, 127.5, 128.3, 132.0, 138.0 и 152.0.
Пример 1(h): 9-(2-Бензилокси-этил)-8-хлор-5-метокси-2,3,4,9,-тетрагидро-1H-карбазол-4-карбонилхлорид (8)
9-(2-Бензилокси-этил)-8-хлор-5-метокси-2,3,4,9,-тетрагидро-1Н-карбазол-4-карбоновую кислоту (7) (1,5 г, 3,7 ммоль) растворяли в дихлорметане (50 мл) и добавляли оксалилхлорид (700 мг, 5,5 ммоль, 470 мкл) и DMF (1 каплю) и реакционную смесь перемешивали при 20°C в течение 2 ч. Происходило умеренное выделение газа в течение приблизительно 30 мин по мере прохождения реакции. Реакционную смесь затем концентрировали в вакууме с получением 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9,-тетрагидро-1Н-карбазол-4-карбонилхлорида (8) в виде смолы, которую использовали на следующей стадии без очистки. Структуру подтверждали при помощи 13С NMR (75 МГц; CDCl3): δC 20.8, 22.1, 26.4, 44.2, 51.8, 55.1, 70.7, 73.3, 100.7, 106.0, 108.6, 119.5, 123.4, 127.3, 127.7, 128.3, 131.9, 138.0, 138.2, 152.0 и 176.3.
Пример 1(i): 9-(2-Бензилокси-этил)-8-хлор-5-метокси-2,3,4.9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (9)
9-(2-Бензилокси-этил)-8-хлор-5-метокси-2,3,4,9,-тетрагидро-1Н-карбазол-4-карбонилхлорид (8) (1,6 г, 3,7 ммоль) затем растворяли в дихлорметане (50 мл), охлаждали до 0°C, перемешивали и по каплям добавляли диэтиламин (810 мг, 11,0 ммоль, 1,1 мл). Реакционную смесь оставляли нагреваться до комнатной температуры в течение 18 ч. Реакционную смесь затем промывали 10%-ным водным карбонатом калия (50 мл), разделяли, сушили над сульфатом магния и концентрировали в вакууме до смолы. Неочищенное вещество кристаллизовали из диэтилового эфира с получением 1,2 г (71%) 9-(2-бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (9) в виде белого кристаллического твердого вещества. Структуру подтверждали при помощи 13С NMR (75 МГц; CDCl3): δC 13.0, 14.5, 19.8, 22.2, 27.9, 36.4, 40.4, 41.9, 43.8, 55.0, 70.8, 73.3, 100.2, 108.5, 108.6, 119.9, 122.5, 127.4, 127.5, 128.3, 131.5, 137.8, 138.2, 152.4 и 174.5.
Пример 1(j): 9-(2-Бензилокси-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламин (10)
9-(2-Бензилокси-этил)-8-хлор-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (9) (1,0 г, 2,1 ммоль) в метаноле (100 мл) встряхивали с 10% палладием на углероде (1,0 г), триэтиламином (2,9 мг, 2,9 ммоль, 4 мкл) в атмосфере газообразного водорода в течение 18 ч при 55°C. Реакционную смесь затем фильтровали через слой целита и фильтрат концентрировали в вакууме с получением смолы (908 мг). Эту смолу затем переносили в дихлорметан (100 мл) и промывали 5% водным раствором карбоната калия (50 мл). Дихлорметановый раствор затем отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением смолы. Смолу затем кристаллизовали из диэтилового эфира (50 мл) и кристаллы собирали посредством фильтрации с получением 523 мг (57%) 9-(2-бензилокси-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламина (10). Структуру подтверждали при помощи 13С NMR (75 МГц; CDCl3): δC 13.1, 14.6, 20.1, 22.0, 28.1, 36.4, 40.5, 42.0, 43.0, 54.7, 68.8, 73.3, 99.4, 102.4, 107.8, 116.4, 121.2, 127.6,127.6, 128.3, 135.6, 137.8, 138.0 153.6 и 175.0.
Пример 1(k): 9-(2-Гидроксиэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламин (11)
9-(2-Бензилокси-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламин (10) (1,0 г, 2,1 ммоль) в метаноле (50 мл) встряхивали с 10% палладием на углероде (300 мг) и избытком газообразного водорода в течение 18 ч при 55°C. Реакционную смесь затем фильтровали через слой целита и фильтрат концентрировали в вакууме с получением 578 мг (100%) 9-(2-гидроксиэтил)-5-метокси-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты диэтиламина (11) в виде пены. Структуру подтверждали при помощи 13С NMR (75 МГц; CDCl3): δC 13.0, 14.4, 20.0, 22.0, 28.0, 36.4, 40.6, 42.0, 54.7, 60.6, 99.2, 102.6, 107.0, 116.7, 121.1, 136.1, 137.5, 138.0 153.5 и 175.7.
Пример 1(l): Метансульфоновой кислоты 2-(4-диэтилкарбамил-5-метокси-1,2,3.4-тетрагидро-карбазол-9-ил)этиловый эфир (соединение-предшественник 5)
9-(2-Гидроксиэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламин (11) (478 мг, 1,4 ммоль) в дихлорметане (30 мл) охлаждали до 0°C, добавляли метансульфонилхлорид (477 мг, 4,2 ммоль, 324 мкл) и триэтиламин (420 мг, 4,2 ммоль, 578 мкл) и оставляли нагреваться до к.т. в течение ночи. Реакционную смесь промывали 5% водным раствором карбоната калия. Слои отделяли. Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме с получением смолы (696 мг). Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя смесью бензин (А):этилацетат (В) (75-100% В, 22 CV, 120 г, 85 мл/мин) с получением метансульфоновой кислоты 2-(4-диэтилкарбамил-5-метокси-1,2,3,4-тетрагидро-карбазол-9-ил)этилового эфира (соединение-предшественник 5) в виде смолы, которая кристаллизовалась из диэтилового эфира с получением 346 мг (59%) бесцветного твердого вещества. Структуру подтверждали при помощи 13С NMR (75 МГц; CDCl3): δC 13.1, 14.5, 20.0, 21.9, 28.0, 36.3, 36.7, 40.3, 41.8, 41.9, 54.7, 68.1, 100.0, 102.0, 109.0, 116.4, 122.0 135.1, 137.3, 153.8 и 174.6.
Пример 1(m): 9-(2-[18F]Фтор-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (визуализируюший агент 5)
[18F] Фторид получен из GE Healthcare на циклотроне GE РЕТгасе. Kryptofix 2,2,2 (2 мг, 5 мкмоль), бикарбонат калия (0,1 моль дм-3, 0,1 мл, 5 мг, 5 мкмоль) и ацетонитрил (0,5 мл) добавляли к [18F]F-/H2O (примерно 400 МБк, 0,1-0,3 мл) в реакционном сосуде COC (циклоолефиновый сополимер). Смесь сушили путем нагревания при 100°C в потоке азота в течение 20-25 мин. После сушки и без охлаждения соединение-предшественник 5 (0,5-1 мг, 1,2-2,4 мкмоль) в ацетонитриле (1 мл) добавляли в реакционный сосуд COC и нагревали при 100°C в течение 10 мин. После охлаждения реакционную смесь удаляли и реакционный сосуд COC промывали водой (1,5 мл) и добавляли к основной неочищенной реакционной смеси.
После этого неочищенный продукт наносили на полупрепаративную HPLC: колонка HICHROM ACE 5 С18 (100×10 мм вн.д. (внутренний диаметр)), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 3 мл/мин; 0-1 мин 40% В; 1-20 мин 40-95% В; длина волны 254 нм; tR визуализирующего агента 5 16 мин. Очищенный на HPLC пик визуализирующего агента 5 разбавляли до объема 10 мл водой и адсорбировали на картридже tC18 Sep-Pak (lite). Картридж промывали водой (2 мл) и элюировали безводным этанолом (0,5 мл), а затем физиологическим раствором Дульбекко, забуференным фосфатом (4,5 мл). Радиохимический выход 30±7% (n=4), не скорректированный на распад, время 90-120 мин, радиохимическая чистота ≥99%.
Аналитическая-HPLC: колонка Phenomenex Luna C18 (150×4,6 мм вн.д.), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 1 мл/мин; 0-1 мин 40% В; 1-20 мин 40-95%В; длина волны 230 нм; tR визуализирующего агента 5 16 мин. На Фиг.1 продемонстрировано совместное элюирование визуализирующего агента 5 и нерадиоактивного визуализирующего агента 5.
Пример 2: Синтез 9-(2-фтор-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (нерадиоактивный визуализирующий агент 5)
Пример 2(а): Фторэтилтозилат (12)
2-Фторэтанол (640 мг, 10 ммоль, 0,6 мл) растворяли в пиридине (10 мл) в атмосфере азота. Раствор перемешивали при 0°С и тозилхлорид (4,2 г, 21,8 ммоль) добавляли порциями к раствору в течение 30 мин, поддерживая температуру ниже 5°C. Реакционную смесь перемешивали при 0°C в течение 3 ч. Медленно добавляли лед, затем воду (20 мл). Реакционную смесь экстрагировали в этилацетат и промывали водой. Избыток пиридина удаляли путем промывания 1 н. раствором HCl до тех пор, пока водный слой не станет кислым. Избыток тозилхлорида удаляли путем промывания 1 М водным карбонатом натрия. Органический слой промывали рассолом, сушили над сульфатом магния и концентрировали в вакууме с получением 2,1 г (98%) фторэтилтозилата (12) в виде бесцветного масла. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δH 21.6 (CCH3), 68.5 (d, JCF=173 Гц, OCH2CH2F), 80.6 (d, JCF=173 Гц, OCH2 CH2F), 128.0, 129.9, 132.6 и 145.1.
Пример 2(b): (2-хлор-5-метокси-фенил)(2-фторэтил)амин (13)
2-Хлор-5-метоксианилина гидрохлорид (5,0 г, 26,0 ммоль) растворяли в DMF (50 мл) и добавляли гидрид натрия (2,3 г, 60% в масле, 57,0 ммоль). Реакционную смесь перемешивали в течение 30 минут при к.т. в атмосфере азота. Фторэтилтозилат (12) (6,7 г, 31,0 ммоль) в DMF (5 мл) добавляли по каплям и реакционную смесь перемешивали при к.т. в течение 2 ч. Реакционную смесь затем нагревали при 100°C в течение 18 ч. Реакционную смесь оставляли нагреваться и растворитель удаляли при пониженном давлении. Остаток растворяли в этилацетате (100 мл) и промывали водой (2×100 мл). Органические вещества собирали, сушили над сульфатом магния и концентрировали в вакууме с получением коричневого масла, которое очищали посредством хроматографии на силикагеле, элюируя смесью петролейный эфир (А):этилацетат (В) (5-30% (В), 330 г, 18,1 CV, 120 мл/мин) с получением 1,3 г (25%) (2-хлор-5-метокси-фенил)(2-фторэтил)амина (13) в виде желтого масла. Структуру подтверждали при помощи 13С NMR (75 МГц; CDCl3): δc 43.8 (d, JCF=23 Гц), 55.3, 82.0 (в, JCF=165 Гц), 98.1, 102.2, 111.6, 129.5, 144.1 и 159.5.
Пример 2(c): 3-[(2-Хлор-5-метокси-фенил)-(2-фторэтил)амино]-2-гидрокси-циклогексил-1-енкарбоновой кислоты этиловый эфир (14)
Раствор 2-хлор-5-метокси-фенил(2-фторэтил)амина (13) (6,1 г, 30,0 ммоль) в THF (170 мл) охлаждали до -40°C. По каплям добавляли бис(триметилсилил)амид калия (126,0 мл 0,5 М раствора в толуоле, 63,0 ммоль) и реакционную смесь перемешивали в течение 30 мин при -40°C.) По каплям добавляли 3-бром-2-гидрокси-циклогекс-1-енкарбоновой кислоты этиловый эфир (4; полученный в соответствии с примером 1(d)) (7,4 г, 30,0 ммоль) в THF (30 мл) при -40°C. Охлаждающую баню удаляли и реакционную смесь перемешивали при к.т. в течение 4 ч. Реакционную смесь гасили рассолом (300 мл) и экстрагировали в этилацетат (2×400 мл), сушили над сульфатом магния и концентрировали в вакууме с получением 12,0 г (количественный выход) 3-[(2-хлор-5-метокси-фенил)-(2-фторэтил)амино]-2-гидрокси-циклогексил-1-енкарбоновой кислоты этилового эфира (14) в виде коричневого масла, которое использовали неочищенным на следующей стадии. Структуру в виде смеси изомеров подтверждали при помощи 1H NMR (300 МГц, CDCl3):δH 1.08 (0.8Н, t, J=9 Гц, CO2CH2CH 3), 1.22-1.33 (2.2H, m, CO2CH2CH 3), 1.40-2.60 (7H, m, 4-, 5- и 6-CH2, CHN), 3.20-4.50 (10H, m, NCH 2CH2F, NCH2CH 2F, OCH 3, CHCO2CH 2CH3), 6.50-6.70 (1H, m, CHC(OCH3)CHCH), 6.95 (0.5Н, dd, J=3 и 6 Гц, CHC(OCH3)CHCH), 7.08 (0.5Н, d, J=3 Гц, СНС(ОСН3)СНСН) и 7.20-7.30 (1Н, m, СНС(ОСН3)СНСН).
Пример 2(d): 8-хлор-9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-каобазол-4-карбоновой кислоты этиловый эфир (15)
Синтез 8-хлор-9-(2-фтор-этил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этилового эфира (15) сначала пытались осуществить с использованием условий, описанных в WO 2003/014082. Раствор (2-хлор-5-метокси-фенил)(2-фторэтил)амина (13; полученного в соответствии с примером 2(b)) (600 мг, 3,8 ммоль) в безводном THF (20 мл) охлаждали в ледяной бане и обрабатывали бис(триметилсилил)амидом калия (16 мл 0,5 М раствора в толуоле, 8,0 ммоль). Через 30 минут добавляли 3-бром-2-гидрокси-циклогекс-1-енкарбоновой кислоты этиловый эфир (4; полученный в соответствии с примером 1(d)) (1,04 г, 4,2 ммоль) в THF (4 мл) и реакционную смесь оставляли нагреваться до к.т. в течение 2 часов. Реакционную смесь гасили насыщенным раствором хлорида аммония и дважды экстрагировали эфиром. Экстракты промывали водой, рассолом, сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (2,5-50% В, 50 г, 25 CV, 40 мл/мин). Основное пятно представляло собой смесь трех соединений. Эту смесь кипятили с обратным холодильником в толуоле (20 мл) с безводным хлоридом цинка (1,7 г, 12,6 ммоль) в течение ночи. Реакционную смесь концентрировали в вакууме и остаток распределяли между 1 н. HCl (25 мл) и этилацетатом (25 мл) и затем еще раз экстрагировали этилацетатом. Органические слои промывали водой и рассолом, сушили и концентрировали в вакууме с получением коричневого масла. 1H NMR указывала на то, что оно представляло собой смесь нескольких соединений. ТСХ на диоксиде кремния в ряде растворителей не смогла разделить эту смесь на отдельные пятна. Сравнение 1H NMR смеси с аутентичным образцом указывало на то, что эта смесь содержала примерно 25% 8-хлор-9-(2-фтор-этил)-5-метокси-2,3,4,9-тетрагидро-1 Н-карбазол-4-карбоновой кислоты этилового эфира (15).
Затем осуществляли модифицированный способ. 3-[(2-Хлор-5-метокси-фенил)-(2-фторэтил)амино]-2-гидрокси-циклогексил-1-енкарбоновой кислоты этиловый эфир (14) (12,2 г, 30,0 ммоль) растворяли в диэтиловом эфире (250 мл) и добавляли хлорид цинка (16,4 г, 120,0 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 16 ч. Для полного растворения добавляли этилацетат (500 мл) и промывали 2 н. HCl (200 мл), водой (200 мл), 10% водным карбонатом калия (200 мл), сушили над сульфатом магния и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя смесью бензин (А): этилацетат (В) (5-20% В, 12 CV, 10 г, 100 мл/мин) с получением 5,3 г (50% за 2 стадии) 8-хлор-9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этилового эфира (15) в виде желтого твердого вещества. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 14.4, 20.4, 22.2, 27.4, 40.1, 44.2 (d, JCF=23 Гц), 55.1, 60.2, 83.9 (d, JCF=173 Гц), 100.6, 107.9, 108.2, 119.8, 123.1, 131.9, 137.2, 152.7 и 175.7.
Пример 2(е): 9-(2-Фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этиловый эфир (16)
8-Хлор-9-(2-Фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этиловый эфир (15) (5,3 г, 15,0 ммоль) растворяли в метаноле (180 мл) и добавляли триэтиламин (1,8 г, 18,0 ммоль, 2,5 мл) и 10% Pd/C (2 г в метаноле (20 мл)). Смесь помещали в гидрогенизатор Парра и встряхивали в течение 18 ч в атмосфере водорода. Реакционную смесь фильтровали через слой целита, промывали метанолом и растворитель удаляли в вакууме. Остаток растворяли в этилацетате (300 мл) и промывали 10% водным карбонатом калия (200 мл), сушили над сульфатом магния и концентрировали в вакууме с получением 4,2 г (88%) 9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этилового эфира (16) в виде светло-коричневого твердого вещества. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 14.3, 20.6, 21.8, 27.6, 40.3, 43.3 (d, JCF=23 Гц), 54.9, 60.1, 82.0 (d, JCF=165 Гц), 99.8, 102.1, 107.3, 117.2, 121.8, 134.9, 137.6, 153.8 и 176.0.
HPLC (Gemini 150×4,6 мм, 50-95% метанол/вода в течение 20 мин) 13,6 мин (94%).
Пример 2(f): 9-(2-Фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-кар6оновая кислота (17)
8-Хлор-9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этиловый эфир (16) (380 мг, 1,2 ммоль) растворяли в этаноле (4 мл). Добавляли раствор гидроксида натрия (580 мг, 14,5 ммоль), растворенного в 6 мл воды. Реакционную смесь нагревали до температуры дефлегмации в течение ночи. Растворитель удаляли в вакууме и неочищенную смесь разбавляли водой, подкисляли 2 н. HCl до кислого состояния и промывали дихлорметаном. Органические вещества объединяли и сушили над сульфатом магния и концентрировали в вакууме с получением 347 мг (количественный выход) 9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (17) в виде не совсем белого твердого вещества, которое использовали неочищенным на следующей стадии. Структуру подтверждали при помощи 13С NMR (75 МГц; CDCl3): δC 20.4, 21.9, 27.2, 39.9, 43.3 (d, JCF=23 Гц), 55.1, 81.9 (d, JCF=173 Гц), 100.3, 102.8, 106.2, 117.1, 122.2, 135.6, 137.8, 153.3 и 180.8.
Пример 2(g): 9-(2-Фторэтил)-5-метокси-2,3.4,9-тетрагидро-1Н-карбазол-4-карбонилхлорид (18)
Раствор 9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (17) (347 мг, 1,2 ммоль) в обезвоженном дихлорметане (2 мл) перемешивали в атмосфере азота. Добавляли оксалилхлорид (453 мг, 3,6 ммоль, 300 мкл), а затем каплю DMF. Реакционную смесь перемешивали при к.т. в атмосфере азота в течение 2 ч, затем упаривали в вакууме с получением 371 мг (количественный выход) 9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбонилхлорида в виде смолы, которую использовали на следующей стадии без очистки. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 20.2, 21.7, 26.4, 43.3 (d, JCF=23 Гц), 54.9, 80.5, 83.1, 100.2, 102.2, 105.8, 116.7, 122.4, 135.5, 137.4, 153.5 и 176.6.
Пример 2(h): 9-(2-Фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (нерадиоактивный визуализируюший агент 5)
9-(2-Фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбонилхлорид (18) (371 мг, 1,2 ммоль) растворяли в дихлорметане (2 мл) и охлаждали до 0°C. Затем добавляли диэтиламин (177 мг, 2,4 ммоль, 250 мкл) и реакционную смесь перемешивали в течение ночи при к.т. Реакционную смесь гасили 10% водным карбонатом калия (2 мл). Дихлорметановый слой собирали через фазоразделитель, затем концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя смесью бензин (А):этилацетат (В) (50-100% (В), 50 г, 35,2 CV, 40 мл/мин) с получением бледно-желтого твердого вещества. Это твердое вещество затем растирали с минимальным количеством диэтилового эфира с получением 240 мг (58%) 9-(2-фторэтил)-5-метокси-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (нерадиоактивного визуализирующего агента 5). Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 13.0, 14.6, 19.9, 21.9, 28.0, 36.3, 40.5, 41.9, 43.1 (d, JCF=23 Гц), 54.7, 82.0 (d, JCF=173 Гц), 99.7, 102.1, 108.3, 117.0, 121.5, 135.3, 137.4, 153.3 и 174.8.
Пример 3: Синтез метансульфоновой кислоты 2-[4-(пиперидин-1-карбонил)-1,2,3,4-тетрагидро-карбазол-9-ил]-этилового эфира (соединение-предшественник 6) и [9-(2-[18F]фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-ил]-пиперидин- 1-ил-метанона (визуализирующий агент 6)
Пример 3(а): 2-(пиперидин-1-карбонил)-циклогексанон (19)
Этил-2-оксоциклогексан-карбоксилат (5,3 г, 31 ммоль, 5,0 мл) DMAP (1,05 г, 9,4 ммоль) и пиперидин (5,3 г, 63 ммоль, 6,2 мл) в толуоле (100 мл) кипятили с обратным холодильником течение 4 суток. Реакционную смесь оставляли охлаждаться и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (20 - 80% (В), 100 г, 8 CV, 85 мл/мин) с получением 6,26 г (96%) 2-(пиперидин-1-карбонил)-циклогексанона (19) в виде белого твердого вещества. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 23.5, 24.5, 25.5, 26.2, 27.1, 30.4, 41.9, 42.9, 46.8, 54.2, 167.6, 207.6.
Пример 3(b): 2-Бром-6-(пиперидин-1-карбонил)-циклогексанон (20)
2-(Пиперидин-1-карбонил)-циклогексанон (19) (4,0 г, 19 ммоль) растворяли в диэтиловом эфире (5 мл) и охлаждали до 0°C в атмосфере Ns. Бром (5,9 г, 19 ммоль, 1,0 мл) добавляли по каплям в течение 15 мин и реакционную смесь оставляли нагреваться до комнатной температуры в течение 90 мин. Твердое вещество собирали посредством фильтрации с получением 5,86 г (количественный выход) 2-бром-6-(пиперидин-1-карбонил)-циклогексанона (20) в виде белого твердого вещества, которое использовали на следующей стадии без очистки. Структуру подтверждали при помощи 13С NMR (75 МГц, DMSO (диметилсульфоксид)-d6): δC 17.3, 24.2, 25.3, 25.8, 32.5, 44.0,51.6, 108.3, 145.5, 167.8.
Пример 3(с): (2-Бензилокси-этил}-фенил-амин (21)
В круглодонной колбе анилин (2,0 г, 21,5 ммоль, 2,0 мл), 2,6-лутидин (2,30 г, 21,5 ммоль) и бензил-2-бромэтиловый эфир (4,6 г, 21,5 ммоль, 3,4 мл) объединяли в DMF (10 мл) и перемешивали при 100°C в течение ночи. Реакционную смесь оставляли охлаждаться и затем разбавляли этилацетатом (50 мл). Ее промывали водой (3×20 мл) и органические вещества сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (0-50% В, 100 г, 19,5 CV, 85 мл/мин) с получением 2,22 г (37%) (2-бензилокси-этил)-фенил-амина (21) в виде желтого масла. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3) δC 43.6, 68.6, 73.2, 113.1, 117.5, 127.5, 127.7, 128.4, 129.1, 138.2, 148.1.
Пример 3(d): [9-(2-Бензилокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-ил]-пиперидин-1-ил-метанон (22)
Смесь 2-бром-6-(пиперидин-1-карбонил)-циклогексанона (20) (1,5 г, 5,2 ммоль) и (2-бензилокси-этил)-фениламина (21) (3,2 г, 10,4 ммоль) перемешивали в атмосфере N2 при 50°C в течение 3 ч, и реакционная смесь становилась коричневой. Полученную смесь растворяли в пропан-2-оле (5 мл) и добавляли сухой хлорид цинка (2,13 г, 15,6 ммоль). Смесь нагревали до температуры дефлегмации в атмосфере N2 в течение 16 ч и затем концентрировали в вакууме. Остаток растворяли в этилацетате (100 мл) и промывали 2 н. HCl (30 мл), водой (2×30 мл) и водным раствором карбоната калия (2×30 мл), затем сушили и концентрировали в вакууме. Неочищенное вещество очищали при помощи картриджа SCX и затем при помощи хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (30-100% В, 12 г, 41 CV, 30 мл/мин) с получением 600 мг (27%) [9-(2-бензилокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-ил]-пиперидин-1-ил-метанона (22) в виде масла. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 21.5, 21.7, 24.5, 25.7, 26.3, 273, 37.7, 42.8, 43.1, 46.7, 60.2, 68.7, 73.1, 108.2, 108.7, 117.8, 118.9, 120.5, 126.4, 127.3, 127.4, 128.1, 136.2, 137.8, 172.9.
Пример 3(е): [9-(2-Гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-ил]-пиперидин-1-ил-метанон (23)
К раствору [9-(2-бензилокси-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-ил]-пиперидин-1-ил-метанона (22) (600 мг, 1,4 ммоль) в метаноле (15 мл) добавляли суспензию Pd/C (200 мг) в метаноле (10 мл). Смесь помещали в гидрогенизатор Парра и встряхивали в течение 24 ч в атмосфере азота. Реакционную смесь фильтровали через слой целита, промывали метанолом и концентрировали в вакууме. Неочищенное вещество растирали с получением 332 мг (71%) [9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-ил]-пиперидин-1-ил-метанона (23) в виде белого твердого вещества. Структуру подтверждали при помощи 13C NMR (75 МГц, CDCl3): δc 21.2, 21.9, 24.7, 27.4, 36.4, 43.4, 45.0, 47.0, 60.9, 107.8, 109.0, 117.7, 119.0, 120.7, 126.6, 136.2, 137.2, 173.5.
Пример 3(f): Метансульфоновой кислоты 2-[4-(пиперидин-1-карбонил)-1,2,3,4-тетрагидро-карбазол-9-ил]-этиловый эфир (соединение-предшественник 6)
К раствору [9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-ил]-пиперидин-1-ил-метанона (23) (260 мг, 0,8 ммоль) в дихлорметане (15 мл) добавляли пиридин (633 мг, 8,0 ммоль, 0,65 мл). Реакционную смесь охлаждали до 0°C и добавляли метансульфонилхлорид (458 мг, 4,0 ммоль, 0,31 мл). Реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. Смесь промывали 2 н. HCI (2×50 мл) и водой (2×50 мл), сушили и концентрировали в вакууме. Неочищенное вещество растирали с диэтиловым эфиром с получением 263 мг (82%) метансульфоновой кислоты 2-[4-(пиперидин-1-карбонил)-1,2,3,4-тетрагидро-карбазол-9-ил]-этилового эфира (соединение-предшественник 6) в виде белого твердого вещества. Структуру подтверждали при помощи 13C NMR (75 МГц, CDCl3): δc 21.4, 21.8, 24.7, 25.9, 26.9, 27.4, 36.6, 36.8, 41.7, 43.3, 47.0, 67.9, 108.5, 109.5, 118.4, 119.7, 121.3, 126.9, 136.2, 172.7.
Пример 3(g): [9-(2-[18F]Фтор-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-ил]-пиперидин-1-ил-метанон (визуализирующий агент 6)
Введение в соединение-предшественник 6 метки 18F осуществляли, как описано в примере 1(m).
Полупрепаративная HPLC: колонка HICHROM АСЕ 5 С18 (100×10 мм вн.д.), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 3 мл/мин; 0-1 мин 50% В; 1-20 мин 50-95%В; длина волны 254 нм; tR визуализирующего агента 617 мин.
Аналитическая HPLC: колонка Phenomenex Luna C18 (150×4,6 мм вн.д.), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 1 мл/мин; 0-1 мин 50%В; 1-20 мин 50-95%В; длина волны 230 нм; tR визуализирующего агента 6 16 мин.
Радиохимический выход 23±2% (n=3) не скорректированный на распад, время 90-120 мин, радиохимическая чистота ≥99%. На Фиг.2 показано совместное элюирование визуализирующего агента 6 и нерадиоактивного визуализирующего агента 6.
Пример 4: Синтез (9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-ил]-пиперидин-1-ил-метанона (нерадиоактивного аналога визуализирующего агента 6)
Пример 4(а): (2-Фтор-этил)-фенил-амин (24)
В круглодонной колбе анилин (0,5 г, 5,4 ммоль), 2,6-лутидин (0,58 г, 5,4 ммоль) и 2-фторэтилтозилат (12; полученный в соответствии с примером 2(а)) (1,17 г, 5,4 ммоль) объединяли в DMF (2,5 мл) и перемешивали при 100°C в течение ночи. Реакционную смесь оставляли охлаждаться и затем разбавляли этилацетатом (50 мл). Ее промывали водой (3×20 мл), и органические вещества сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (100 г, 0-100% В, 18 CV, 85 мл/мин) с получением 435 мг (60%) (2-фтор-этил)-фенил-амина (24) в виде желтого масла. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 3.41 (1Н, t, J=3 Гц, NCH 2CH2F), 3.50 (1Н, t, J=3 Гц, NCH 2CH2F), 3.93 (1Н, s, br), 4.54 (1Н, t, J=3 Гц, NCH2CH 2F), 4.71 (1Н, t, J=3 Гц, NCH2CH 2F), 6.65-6.82 (3H, m, 2×NCCH, NCCHCHCH), 7.14-7.28 (2Н, m, 2×NCCHCHCH).
Пример 4(b): [9-(2-Фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-ил]-пиперидин-1-ил-метанон (нерадиоактивный визуализиоующий агент 6)
Смесь 2-бром-6-(пиперидин-1-карбонил)-циклогексанона (20; полученного в соответствии с примером 3(b)) (500 мг, 1,7 ммоль) и (2-фтор-этил)-фенил-амина (24) (890 мг, 3,5 ммоль) перемешивали в атмосфере N2 при 50°C в течение 3 ч, и реакционная смесь становилась коричневой. Полученную смесь растворяли в пропан-2-оле (2 мл) и добавляли сухой хлорид цинка (682 мг, 5 ммоль). Смесь нагревали до температуры дефлегмации в атмосфере N2 в течение 16 ч и затем концентрировали в вакууме. Остаток растворяли в этилацетате (50 мл) и промывали 2 н. HCl (20 мл), водой (2×20 мл) и водным раствором карбоната калия (2×20 мл), затем сушили и концентрировали в вакууме. Неочищенное вещество растирали с диэтиловым эфиром с получением 151 мг(27%) [9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-ил]-пиперидин-1-ил-метанона (нерадиоактивный визуализирующий агент 6) в виде белого твердого вещества. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 21.6, 21.8, 24.7, 26.5, 26.9, 27.4, 37.3, 43.1 (d, JCF=45 Гц), 47.0, 82.1 (d, JCF=173 Гц), 108.5, 108.9, 118.6, 119.4, 121.0, 126.8, 136.2, 172.7.
Пример 5: Синтез метансульфоновой кислоты 2-[4-(бензил-метил-карбамоил)-1,2,3,4-тетрагидро-карбазол-9-ил]-этилового эфира (соединение-предшественник 7) и 9-(2-[18F](фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты бензил-метил-амида (визуализирующий агент 7)
Пример 5(а): 9-(2-Бензилокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этиловый эфир (25)
Смесь (2-бензилокси-этил)-фенил-амина (21; полученного в соответствии с примером 3(с)) (8,0 г, 26 ммоль) и 3-бром-2-гидрокси-циклогекс-1-енкарбоновой кислоты этилового эфира (4; полученного в соответствии с примером 1(d)) (3,2 г, 13 ммоль) перемешивали в атмосфере N2 при 50°C в течение 3 ч, и реакционная смесь становилась коричневой. Полученную смесь растворяли в пропан-2-оле (30 мл) и добавляли сухой хлорид цинка (10,6 г, 78 ммоль). Смесь нагревали до температуры дефлегмации в атмосфере N2 в течение 16 ч и затем концентрировали в вакууме. Остаток растворяли в этилацетате (300 мл) и промывали 2 н. HCl (100 мл), водой (2×100 мл) и водным раствором карбоната калия (2×100 мл), затем сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (2,5-40% В, 17 CV, 330 г, 100 мл/мин) с получением 3,49 г (72%) 9-(2-бензилокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этилового эфира (25) в виде масла. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 14.2, 20.5, 21.8, 26.5, 38.6, 42.9, 60.4, 68.7, 73.2, 106.4, 108.8, 118.7, 120.7, 127.4, 127.5, 128.3, 136.2, 136.9, 137.8, 175.0.
Пример 5(b): 9-(2-Бензилокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (26)
9-(2-Бензилокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этиловый эфир (25) (35 г, 9,3 ммоль) растворяли в этаноле (9 мл) и затем добавляли NaOH (1,56 г) в воде (15 мл). Реакционную смесь кипятили с обратным холодильником в течение 2 ч. Реакционную смесь концентрировали в вакууме и остаток разбавляли водой и промывали дихлорметаном (2×150 мл). Водный слой добавляли по каплям к 2 н. HCl (150 мл) и затем экстрагировали в дихлорметане (3 х 150 мл). Органические вещества сушили и концентрировали в вакууме с получением 2,48 г (92%) 9-(2-бензилокси-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты (26) в виде желтого твердого вещества, которое использовали на следующей стадии без очистки. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 20.4, 21.8, 26.4, 38.3, 42.9, 68.7, 73.3, 105.7, 108.8, 118.7, 119.3, 102.9, 127.4, 127.6, 128.3, 136.2, 137.1, 137.8, 108.9.
Пример 5(с): 9-(2-Бензилокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты бензил-метил-амид (27)
9-(2-Бензилокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновую кислоту (26) (600 мг, 1,7 ммоль) растворяли в обезвоженном DCM (8 мл) в атмосфере азота и добавляли оксалилхлорид (393 мг, 3,1 ммоль, 0,26 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, и происходило интенсивное выделение газа. Реакционную смесь концентрировали в вакууме, затем перерастворяли в дихлорметане (8 мл), охлаждали до 0°C и добавляли N-бензилметиламин (412 мг, 3,4 ммоль, 0,44 мл). Реакционную смесь нагревали до комнатной температуры в течение ночи. Реакционную смесь промывали 5% водным раствором карбоната калия, сушили и концентрировали в вакууме с получением коричневого масла. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (30% В, 10 г) с получением 246 мг (64%) 9-(2-бензилокси-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты бензил-метил-амида (27) в виде желтого масла. Структуру подтверждали при помощи 1H NMR (CDCl3): δH 1.60-2.30 (4H, m, CHCH 2CH 2CH2), 2.70-2.90 (2Н, m, CHCH2CH2CH 2), 3.10 (1.5Н, s, N(CH 3)CH2Ph), 3.13 (1.5Н, s, N(CH 3)CH2Ph), 3.73 (2Н, t, J=6 Гц, NCH 2CH2O), 4.10-4.30 (3H, m, NCH2CH 2O, СНСН2СН2СН2), 4.42 (1H, s, OCH 2Ph), 4.44 (1H, s, OCH 2Ph), 4.80 (1H, s, N(СН3)CH2Ph), 4.81 (1H, s, N(CH3)CH 2Ph), 6.90-7.50 (14H, m).
Пример 5(d): 9-(2-Гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты бензил-метил-амид (28)
К раствору 9-(2-бензилокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты бензил-метил-амида (27) (246 мг, 0,5 ммоль) в метаноле (15 мл) добавляли суспензию Pd/C (200 мг) в метаноле (10 мл). Смесь помещали в гидрогенизатор Парра и встряхивали в течение 24 ч в атмосфере азота. Реакционную смесь фильтровали через слой целита, промывали метанолом и концентрировали в вакууме с получением 36 мг (20%) 9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты бензил-метил-амида (28) в виде зеленого масла, которое использовали на следующей стадии без очистки. Структуру подтверждали при помощи 1H NMR (CDCl3): δH 1.80-2.20 (4Н, m), 2.70-3.00 (2Н, m), 3.20-4.30 (10Н, т), 6.90-7.50 (9Н, m).
Пример 5(е) Метансульфоновой кислоты 2-[4-(бензил-метил-карбамоил)-1,2,3,4-тетрагидро-карбазол-9-ил]-этиловый эфир (соединение-предшественник 7)
К раствору 9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1 Н-карбазол-4-карбоновой кислоты бензил-метил-амида (28) (36 мг, 0,1 ммоль) в дихлорметане (2 мл) добавляли пиридин (7,91 г, 1,0 ммоль, 8,1 мл). Реакционную смесь охлаждали до 0°C и добавляли метансульфонилхлорид (57 мг, 0,5 ммоль, 0,04 мл). Реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. Смесь промывали 2 н. HCl (2×10 мл) и водой (2×10 мл), сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (20-80% В, 4 г, 45 CV, 18 мл/мин) с получением 14 мг (32%) метансульфоновой кислоты 2-[4-(бензил-метил-карбамоил)-1,2,3,4-тетрагидро-карбазол-9-ил]-этилового эфира (соединение-предшественник 7) в виде желтого масла. Структуру подтверждали при помощи 1H NMR (CDCl3): δH 1.10-2.40 (5Н, m), 2.51 (1.5Н, s, OSO2CH 3), 2.54 (1.5Н, s, OSO2CH 3), 2.70-2.90 (2H, m), 3.08 (1.5H, s, NCH 3), 3.15 (1.5H, s, NCH 3), 3.40-3.70 (1H, m), 4.10-4.80 (4H, m), 7.00-7.50 (9H, m).
Пример 5(f): 9-(2-[18F]Фтор-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты бензил-метил-амид (визуализирующий агент 7)
Введение в соединение-предшественник 7 метки 18F осуществляли, как описано в примере 1(m). Полупрепаративная HPLC: колонка HICHROM АСЕ 5 С18 (100×10 мм вн.д.), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 3 мл/мин; 0-1 мин 50% В; 1-20 мин 50-95% B; длина волны 254 нм; tR визуализирующего агента 7, 17 мин.
Аналитическая-HPLC: колонка Phenomenex Luna С18 (150×4,6 мм вн.д.), размер частиц 5 мкм; подвижная фаза A: вода, подвижная фаза В: метанол; градиент потока: 1 мл/мин; 0-1 мин 50% В; 1-20 мин 50-95% В; длина волны 230 нм; tR визуализирующего агента 7 16 мин. Радиохимический выход 23±2% (n=3), нескорректированный на распад, время 90-120 мин, радиохимическая чистота ≥99%. Фиг.3 демонстрирует совместное элюирование визуализирующего агента 7 и нерадиоактивного визуализирующего агента 7.
Пример 6: 9-(2-Фтор-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты бензил-метил-амид (нерадиоактивный визуализирующий агент 7)
Пример 6(a): 3-Бром-2-гидрокси-циклогекс-1-енкарбоновой кислоты этиловый эфир (29)
Этил-2-оксоциклогексанкарбоксилат (5,0 г, 29 ммоль, 4,7 мл) растворяли в диэтиловом эфире (5 мл) и охлаждали до 0°C в атмосфере N2. Бром (4,6 г, 29 ммоль, 4,2 мл) добавляли по каплям в течение 15 мин и реакционную смесь оставляли нагреваться до комнатной температуры в течение 90 мин. Смесь медленно выливали в ледяной насыщенный водный раствор карбоната натрия (40 мл) и экстрагировали этилацетатом (3×40 мл). Объединенные органические слои сушили и концентрировали в вакууме с получением 5,96 г (81%) 3-бром-2-гидрокси-циклогекс-1-енкарбоновой кислоты этилового эфира (29) в виде бледно-желтого масла, которое использовали на следующей стадии без очистки. Структуру подтверждали при помощи 13C NMR (75 МГц, DMSO-d6): δc 14.14, 17.65, 21.77, 32.02, 59.95, 60.83, 99.70, 166.33, 172.81.
Пример 6(b) 9-(2-Фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этиловый эфир (30)
Смесь (2-фтор-этил)-фенил-амина (24; полученного в соответствии с примером 4(а)) (560 мг, 4,0 ммоль) и 3-бром-2-гидрокси-циклогекс-1-енкарбоновой кислоты этилового эфира (29) (500 мг, 2,0 ммоль) перемешивали в атмосфере N2 при 50°C в течение 3 ч, и реакционная смесь становилась коричневой. Полученную смесь растворяли в пропан-2-оле (4 мл) и добавляли сухой хлорид цинка (820 мг, 6 ммоль). Смесь нагревали до температуры дефлегмации в атмосфере N2 в течение 16 ч и затем концентрировали в вакууме. Продукт растворяли в смеси этилацетат/эфир (30 мл/150 мл) и промывали 2 н. HCl (40 мл), водой (2×100 мл) и водным раствором карбоната калия (2×100 мл), затем сушили и концентрировали с получением 447 мг (91%) 9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этилового эфира (30) в виде желтого масла, которое использовали на следующей стадии без очистки. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 14.3, 20.4, 21.7, 26.4, 38.5, 43.1 (d, JCF=15 Гц), 60.6, 76.6, 77.0, 77.4, 82.1 (d, JCF=173 Гц), 106.9, 108.5, 118.9, 119.4, 121.1, 127.1, 136.2, 136.7, 174.9.
Пример 6(с): 9-(2-Фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновая кислота (31)
9-(2-Фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты этиловый эфир (30) (380 мг, 1,3 ммоль) растворяли в этаноле (3 мл) и затем добавляли NaOH (520 мг) в воде (5 мл). Реакционную смесь кипятили с обратным холодильником в течение 2 ч. Реакционную смесь концентрировали в вакууме и остаток разбавляли водой и промывали дихлорметаном (2×50 мл). Водный слой добавляли по каплям к 2 н. HCl (50 мл) и затем экстрагировали в дихлорметане (3×50 мл). Органические вещества сушили и концентрировали в вакууме с получением 130 мг (37%) 9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты (31) в виде желтого твердого вещества, которое использовали на следующей стадии без очистки. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δ 1.90-2.42 (4Н, m, 2- и 3-СН 2), 2.60-2.91 (2H, m, 1-СН 2), 3.94 (1H, t, J=6 Гц, 4-СН), 4.30 (1Н, t, J=6 Гц, NCH 2CH2F), 4.37 (1H, t, J=6 Гц, NCH 2CH2F), 4.59 (1H, t, J=6 Гц, NCCH2CH 2F), 4.74 (1Н, t, J=6 Гц, NCH 2CH2F), 7.05-7.26 (3Н, m, ArH), 7.59 (1Н, d, J=9 Гц, ArH).
Пример 6(d): 9-(2-Фтор-этил)-2.3,4.9-тетрагидро-1 Н-карбазол-4-карбонилхлорид (32)
9-(2-Фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновую кислоту (31) (0,5 г, 1,91 ммоль) в обезвоженном дихлорметане (6 мл) перемешивали в атмосфере азота при комнатной температуре с оксалилхлоридом (490 мг, 3,8 ммоль, 0,34 мл) и каплей DMF. Реакционную смесь концентрировали в вакууме с получением 545 мг (количественный выход) 9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбонилхлорида (32), который использовали на следующей стадии без очистки. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 20.2, 21.6, 26.7, 43.1, 43.4, 50.6, 80.9, 83.1, 105.3, 108.8, 118.3, 120.0, 121.6, 126.5, 136.2, 137.5, 176.1.
Пример 6(е) 9-(2-Фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты бензил-метил-амид (нерадиоактивный визуализируюший агент 7)
9-(2-Фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбонилхлорид (32) (110 мг, 0,4 ммоль) растворяли в дихлорметане (1 мл) и охлаждали до 0°C. Затем добавляли N-бензилметиламин (92 мг, 0,8 ммоль, 98 мкл) и реакционную смесь перемешивали в течение ночи при к.т. Реакционную смесь гасили 10% водным раствором карбоната калия (2 мл). Дихлорметановый слой собирали через фазоразделитель, затем концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (20-100% В, 12 г, 30 CV, 30 мл/мин) с получением 39 мг (28%) 9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты бензил-метил-амида (нерадиоактивный визуализирующий агент 7). Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.75-2.32, (4Н, m, 2- и 3-СН 2), 2.68-2.86 (2Н, m, 1-СН 2), 3.10 (1Н, s, NCH 3), 3.14 (2Н, s, NCH 3), 4.17-4.39 (3Н, m, NCH 2CH2F и 4-СН 2), 4.52-4.87 (4Н, m, NCH 2Ph и NCH 2CH2F), 6.96-7.42 (9Н, m, ArH).
Пример 7: Синтез метансульфоновой кислоты 2-(4-диэтилкарбамоил-6-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этилового эфира (соединение-предшественник 9) и 6-фтор-9-(2-[18F]фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (визуализируюший агент 9)
Пример 7(а): 2-Бензилокси-N-(4-фтор-фенил]-ацетамид (33)
К раствору бензилоксиуксусной кислоты (4,6 г, 28,0 ммоль, 4,0 мл) в DCM (52 мл) добавляли оксалилхлорид (7,7 г, 61 ммоль, 5,3 мл) и каплю DMF. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Избыток оксалилхлорида удаляли в вакууме с получением бензилокси-ацетилхлорида. Неочищенный хлорангидрид разбавляли в DCM (100 мл) и добавляли триэтиламин (5,3 мл, 41,6 ммоль, 4,2 г), затем 4-фторанилин (3,5 г, 32 ммоль, 3,0 мл). Реакционную смесь перемешивали при к.т. в течение ночи. Реакцию затем гасили 1 М водной HCI (100 мл), сушили и концентрировали в вакууме с получением 7,1 г (95%) 2-бензилокси-N-(4-фтор-фенил)-ацетамида (33) в виде желтого масла, которое использовали на следующей стадии без очистки. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 69.2, 73.5, 115.4 (d, JCF=22 Гц), 121.4 (d, JCF=7 Гц), 127.9, 128.2, 128.5, 132.5 (d, JCF=3 Гц), 136.3, 157.6, 160.8 и 167.5.
Пример 7(b): (2-Бензилокси-этил)-(4-фтор-фенил)-амин (34)
К суспензии LAH (алюмогидрида лития) (1,25 г, 27 ммоль) в обезвоженном диэтиловом эфире (100 мл) добавляли по каплям раствор 2-бензилокси-N-(4-фтор-фенил)-ацетамида (33) (6,9 г, 27 ммоль) в обезвоженном диэтиловом эфире (100 мл). Добавление происходило так, чтобы поддерживать кипячение с обратным холодильником. После завершения добавления реакционную смесь нагревали до температуры дефлегмации в течение 4 ч, затем выливали в смесь лед-воду и добавляли DCM. Для разрушения соли алюминия добавляли 2 М водный раствор гидроксида натрия до получения сильнощелочного значения pH. Слои разделяли и водный слой промывали DCM, сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (5-50% В, 100 г, 12 CV, 60 мл/мин) с получением 5,5 г (84%) (2-бензилокси-этил)-(4-фтор-фенил)-амина (34) в виде желтого масла. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 44.0, 68.3, 72.8, 113.7 (d, JCF=7 Гц), 115.3 (d, JCF=22 Гц), 127.5, 127.6 (d, JCF=3 Гц), 128.3, 137.8, 144.5, 154.1 и 157.2.
Пример 7(с): 3-Бром-2-оксо-циклогексанкарбоновой кислоты диэтиламид (35)
Этил 2-циклогексан-карбоксилат (7,50 мл, 47,0 ммоль), DMAP (1,72 г, 14,1 ммоль) и диэтиламин (9,77 мл, 94,0 ммоль) кипятили с обратным холодильником в течение 72 часов в толуоле (100 мл). Реакционную смесь оставляли охлаждаться и толуол удаляли при пониженном давлении. Неочищенное масло очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (1:1, 100 г, SiО2) с получением 6,8 г (73%) 2-оксо-циклогексанкарбоновой кислоты диэтиламина в виде оранжевого масла. Структуру подтверждали при помощи 13С NMR (CDCl3) δ 11.1, 12.7, 21.3, 24.9, 28.5, 39.4, 39.6, 51.7, 166.5, 205.9.
2-Оксо-циклогексанкарбоновой кислоты диэтиламин (3,56 мл, 19,3 ммоль) растворяли в диэтиловом эфире (5 мл) и охлаждали при перемешивании до 0°C в атмосфере N2. Бром (0,99 мл, 19,3 ммоль) добавляли по каплям в течение 15 минут и реакционную смесь оставляли нагреваться до комнатной температуры в течение 3 часов. Твердое вещество осаждалось из реакционной смеси. Его собирали посредством фильтрации и промывали эфиром с получением 5,85 г (109%) 3-бром-2-оксо-циклогексанкарбоновой кислоты диэтиламида (35) в виде бледно-желтого твердого вещества. Структуру подтверждали при помощи 13С NMR (CDCl3): δ 11.2, 12.8, 22.7, 28.8, 37.6, 37.9, 39.4,51.0,55.7, 165.5, 197.2.
Пример 7(d): 9-(2-Бензилокси-этил)-6-фтор-2,3,4,9-тетрагидро- 1Н-карбазол-4-карбоновой кислоты диэтиламид (36)
Смесь 2-бензилокси-N-(4-фтор-фенил)-ацетамида (33) (5,3 г, 22 ммоль) и 3-бром-2-оксо-циклогексанкарбоновой кислоты диэтиламида (35) (3,0 г, 13 ммоль) перемешивали в атмосфере N2 при 50°C в течение 3 ч, и реакционная смесь становилась коричневой. Полученную смесь растворяли в пропан-2-оле (30 мл) и добавляли сухой хлорид цинка (9,0 г, 66 ммоль). Смесь нагревали до температуры дефлегмации в атмосфере N2 в течение 16 ч и затем концентрировали в вакууме. Остаток растворяли в этилацетате (300 мл) и промывали 2 н. HCl (100 мл), водой (2×100 мл) и водным раствором карбоната калия (2×100 мл), затем сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (10-50% В, 100 г) с получением 196 мг (11%) 9-(2-бензилокси-этил)-6-фтор-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (36) в виде белого твердого вещества. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.14 (3Н, t, J=7 Гц, N(СН2СН 3)2), 1.30 (3Н, t, J=7 Гц, N(СН2СН 3)2), 1.60-2.60 (4Н, m, 2- и 3-СН 2), 2.70-2.85 (2Н, m, 1-СН 2), 3.10-3.65 (4Н, m, N(СН 2СН3)2 и NCH 2CH2OBn), 3.66-3.75 (1Н, m, 4-СН), 4.00-4.25 (2Н, m, NCH 2CH2OBn), 4.41 (2Н, s, OCH 2Ph), 6.75-6.95 (2Н, m, NCCHCHCFCH), 7.05-7.15 (1Н, m, NCCHCHCFCH) и 7.16-7.25 (5Н, m, Ph).
Пример 7(d): 6-Фтор-9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (37)
К раствору 9-(2-бензилокси-этил)-6-фтор-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (36) (600 мг, 1,4 ммоль) в метаноле (40 мл) добавляли суспензию Pd/C (100 мг) в метаноле (5 мл). Смесь помещали в гидрогенизатор Парра и встряхивали в течение 24 ч в атмосфере азота. Реакционную смесь фильтровали через слой целита, промывали метанолом и концентрировали в вакууме с получением 460 мг (80%) 6-фтор-9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (37) в виде желтого масла, которое использовали на следующей стадии без очистки. Структуру подтверждали при помощи 1H NMR (300 МГц, MeOD-d6) δH 1.18 (3Н, t, J=9 Гц, N(СН2СН 3)2), 1.35 (3Н, t, J=9 Гц, N(СН2СН 3)2), 1.80-2.20 (4Н, m, 2- и 3-СН 2), 2.69-3.88 (2Н, m, 1-СН 2), 3.40-3.86 (6Н, m, N(СН 2СН3)2 и NCH 2CH2OH), 4.03-4.22 (3Н, m, NCH 2CH2OH и 4-СН), 6.75-6.95 (2Н, m, NCCHCHCFCH) и 7.05-7.15 (1H, m, NCCHCHCFCH).
Пример 7(е): Метансульфоновой кислоты 2-(4-диэтилкарбамоил-6-фтор-1,2,3.4-тетрагидро-карбазол-9-ил)-этиловый эфир (соединение-предшественник 9)
К раствору 6-фтор-9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (37) (460 мг, 1,4 ммоль) в дихлорметане (20 мл) добавляли пиридин (1,11 г, 14,0 ммоль, 1,1 мл). Реакционную смесь охлаждали до 0°C и добавляли метансульфонилхлорид (722 мг, 6,3 ммоль, 0,5 мл). Реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. Смесь промывали 2 н. HCl (2×30 мл) и водой (2×30 мл), сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (A) и этилацетатом (B) (0-100% (В), 10 г, 45 CV, 30 мл/мин), затем растирали с диэтиловым эфиром с получением 166 мг (30%) метансульфоновой кислоты 2-(4-диэтилкарбамоил-6-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этилового эфира (соединение-предшественник 9) в виде белого твердого вещества. Структуру подтверждали при помощи 13C NMR (75 МГц, CDCl3): δc 12.9, 15.0, 21.1, 27.7, 36.1, 36.7, 40.6, 41.7, 67.8, 103.3 (d, JCF=23 Гц), 108.7, 109.0, 109.1, 109.4 (d, JCF=5 Гц), 126.9 (d, JCF=10 Гц), 132.4, 138.4, 156.1, 159.2 и 173.3.
Пример 7(f): 6-Фтор-9-(2-[18F]фтор-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты диэтиламид (визуализирующий агент 9)
Введение в соединение-предшественник 9 метки 18F осуществляли, как описано в примере 1(m).
Полупрепаративная HPLC: колонка HICHROM АСЕ 5 С18 (100×10 мм вн.д.), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 3 мл/мин; 0-1 мин 40% B; 1-20 мин 40-95% В; длина волны 254 нм; tR визуализирующего агента 9 15 мин.
Аналитическая-HPLC: колонка Phenomenex Luna С18 (150×4,6 мм вн.д.), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 1 мл/мин; 0-1 мин 50% B; 1-20 мин 50-95% В; длина волны 230 нм; tR визуализирующего агента 9 14 мин. Радиохимический выход 26±8% (n=4), нескорректированный на распад, время 90-120 мин, радиохимическая чистота ≥99%. На Фиг.4 показано совместное элюирование визуализирующего агента 9 и нерадиоактивного визуализирующего агента 9.
Пример 8: Синтез 6-фтор-9-(2-Фтор-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты диэтиламида (нерадиоактивного визуализирующего агента 9)
Пример 8(a): (2-Фтор-этил)-(4-фтор-фенил)-амин (38)
В круглодонной колбе 4-фторанилин (1,3 г, 11,6 ммоль, 1,6 мл), 2,6-лутидин (1,24 г, 11,6 ммоль) и 2-фторэтил-тозилат (12; полученный в соответствии с примером 2(a)) (2,5 г, 11,6 ммоль) объединяли в DMF (5 мл) и перемешивали при 100°C в течение ночи. Реакционную смесь оставляли охлаждаться и затем разбавляли этилацетатом (100 мл). Ее промывали водой (3×40 мл) и органические вещества сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (10% В, 100 г, 12 CV, 60 мл/мин) с получением 383 мг (20%) (2-фтор-этил)-(4-фтор-фенил)-амина (38) в виде желтого масла. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 3.30-3.35 (1Н, m, NCH 2CH2F), 3.40-3.45 (1Н, m, NCH 2CH2F), 3.90 (1Н, s, br, NH), 4.53 (1Н, t, J=3 Гц, NCH 2CH2F), 4.69 (1Н, t, J=3 Гц, NCH 2CH2F), 6.51-6.72 (2H, m, 2×NCCH), 6.85-7.05 (2H, m, 2×NCCHCH).
Пример 8(b): 6-Фтоо-9-(2-фтор-этил)-2,3,4,9-тетрагидро- 1Н-карбазол-4-карбоновой кислоты диэтиламид (нерадиоактивный визуализирующий агент 9)
Смесь 3-бром-2-оксо-циклогексанкарбоновой кислоты диэтиламида (35; полученного в соответствии с примером 7(с)) (336 мг, 1,2 ммоль) и (2-фтор-этил)-(4-фтор-фенил)-амин (38) (383 мг, 2,4 ммоль) перемешивали в атмосфере N2 при 50°C в течение 3 ч, и реакционная смесь становилась коричневой. Полученную смесь растворяли в пропан-2-оле (2 мл) и добавляли сухой хлорид цинка (491 мг, 3,6 ммоль). Смесь нагревали до температуры дефлегмации в атмосфере N2 в течение 16 ч и затем концентрировали в вакууме. Остаток растворяли в этилацетате (20 мл) и промывали 2 н. HCl (10 мл), водой (2×10 мл) и водным раствором карбоната калия (2×5 мл), затем сушили и концентрировали в вакууме. Неочищенное вещество растирали с диэтиловым эфиром с получением 40 мг (10%) 6-фтор-9-(2-фтор-этил)-2,3,4,9-тетрагидро-1 Н-карбазол-4-карбоновой кислоты диэтиламида (нерадиоактивный визуализирующий агент 9) в виде белого твердого вещества. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.13 (3Н, t, J=9 Гц, N(СН2СН 3)2), 1.30 (3Н, t, J=9 Гц, N(СН2СН 3)2), 1.55-2.14 (4Н, m, 2- и 3-СН 2), 2.78-2.86 (2H, т, 1-СН2), 3.36-3.67 (4Н, m, N(СН 2СН3)2), 4.00-4.10 (1Н, m, 4-СН), 4.30 (2H, dm, J=21 Гц, NCH 2CH2F), 4.60 (2H, dm, J=41 Гц, NCH 2CH2F), 6.75-6.95 (2H, m, NCCHCHCFCH) и 7.05-7.15 (1Н, т, NCCHCHCFCH).
Пример 9: Синтез метансульфоновой кислоты 2-(4-диэтилкарбамоил-5-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этилового эфира (соединение-предшественник 10) и 5-фтор-9-(2-[18F]фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (визуализирующий агент 10)
Пример 9(а): 2-Бензилокси-N-(3-фтор-фенил)-аиетамид (39)
К раствору бензилоксиуксусной кислоты (4,65 г, 28 ммоль, 4,0 мл) в DCM (52 мл) добавляли оксалилхлорид (7,7 г, 61 ммоль, 5,3 мл) и каплю DMF. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Избыток оксалилхлорида удаляли в вакууме и неочищенный хлорангидрид карбоновой кислоты разбавляли DCM (100 мл), и добавляли триэтиламин (5,3 мл, 41,6 ммоль, 4,2 г), а затем 3-фторанилин (3,5 г, 32 ммоль, 3,0 мл). Реакционную смесь перемешивали при к.т. в течение ночи. Реакцию затем гасили 1 М водной HCl (100 мл), сушили и концентрировали в вакууме с получением 7,10 г (95%) 2-бензилокси-N-(3-сртор-фенил)-ацетамида (39) в виде желтого масла, которое использовали на следующей стадии без очистки. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 69.2, 73.5, 106.9, 107.2, 111.0 (d, JCF=24 Гц), 114.9 (d, JCF=3 Гц), 127.8, 128.2, 128.5, 129.7 (d, JCF=9 Гц), 136.2 и 167.6.
Пример 9(b): (2-Бензилокси-этил)-(3-фтор-фенил)-амин (40)
К суспензии LAH (1,25 г, 27 ммоль) в обезвоженном диэтиловом эфире (100 мл) добавляли по каплям раствор 2-бензилокси-N-(3-фтор-фенил)-ацетамида (39) (7,0 г, 27 ммоль) в обезвоженном диэтиловом эфире (100 мл). Добавление было таким, что поддерживалось кипячение с обратным холодильником. После завершения добавления реакционную смесь нагревали до температуры дефлегмации в течение 4 ч, затем выливали в смесь лед-вода и добавляли DCM. Для разрушения соли алюминия добавляли 2 М водный раствор гидроксида натрия до достижения сильного щелочного pH. Слои разделяли и водный слой промывали DCM, сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (5-50% В, 100 г, 12 CV, 60 мл/мин) с получением 4,1 г (84%) (2-бензилокси-этил)-(3-фтор-фенил)-амина (40) в виде желтого масла. Структуру подтверждали при помощи 13С NMR (75 МГц, CDCl3): δC 43.3, 68.2, 73.0, 99.4 (d, JCF=24 Гц), 103.5, 103.8, 108.8, 127.4 (d, JCF=3 Гц), 127.6, 128.4, 130.0 (d, JCF=9 Гц) и 138.8.
Пример 9(с): 9-(2-Бензилокси-этил)-5-фтор-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (41)
Смесь 3-бром-2-оксо-циклогексанкарбоновой кислоты диэтиламида (35; полученного в соответствии с примером 7(с)) (2,3 г, 10 ммоль) и (2-бензилокси-этил)-(3-фтор-фенил)-амина (40) (4,1 г, 17 ммоль) перемешивали в атмосфере N2 при 50°C в течение 3 ч, и реакционная смесь становилась коричневой. Полученную смесь растворяли в пропан-2-оле (10 мл), и добавляли сухой хлорид цинка (4,09 г, 30 ммоль). Смесь нагревали до температуры дефлегмации в атмосфере N2 в течение 16 ч и затем концентрировали в вакууме. Остаток растворяли в этилацетате (200 мл) и промывали 2 н. HCl (50 мл), водой (2×50 мл) и водным раствором карбоната калия (2×50 мл), затем сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (5-100% В, 100 г, 28 CV, 60 мл/мин) с получением 1,3 г (30%) 9-(2-бензилокси-этил)-5-фтор-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (41) вместе с изомером 9-(2-бензилокси-этил)-7-фтор-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида в виде смеси, которую использовали на следующей стадии без очистки. Структуру 9-(2-бензилокси-этил)-5-фтор-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (41) подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.10-1.40 (6Н, m, N(СН2СН 3)2), 1.60-2.60 (4Н, m, 2- и 3-СН 2), 2.70-2.85 (2Н, m, 1-СН 2), 3.10-3.65 (4Н, m, N(СН 2СН3)2 и CH 2CH2OBn), 4.00-4.30 (3Н, m, CH 2CH2OBn и 4-СН), 4.43 (2Н, s, OCH 2Ph), 6.55-6.65 (1H, m, NCCHCHCHCF), 6.90-7.05 (1H, m, NCCHCHCHCF), 7.05-7.15 (1H, m, NCCHCHCHCF) и 7.16-7.25 (5Н, m, Ph).
Структуру 9-(2-бензилокси-этил)-7-фтор-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.10-1.40 (6Н, m, N(СН2СН 3)2), 1.60-2.60 (4Н, m, 2- и 3-СН 2), 2.70-2.85 (2Н, m, 1-СН 2), 3.10-3.65 (4Н, m, N(СН 2СН3)2 и NCH 2CH2OBn), 4.00-4.30 (3Н, m, NCH2CH 2Obn и 4-СН), 4.55 (2Н, s, OCH 2Ph), 6.70-6.80 (1H, m, NCCHCFCHCH) и 7.00-7.40 (7Н, m, NCCHCFCHCH и Ph).
Пример 9(d): 5-Фтор-9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (42)
К раствору смеси 9-(2-бензилокси-этил)-5-фтор-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (41) и 9-(2-бензилокси-этил)-7-фтор-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (1,3 г, 3,0 ммоль) в метаноле (75 мл) добавляли суспензию Pd/C (200 мг) в метаноле (10 мл). Смесь помещали в гидрогенизатор Парра и встряхивали в течение 24 ч в атмосфере азота. Реакционную смесь фильтровали через слой целита, промывали метанолом и концентрировали в вакууме с получением 743 мг (80%) смеси 5-фтор-9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (42) и 7-фтор-9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида в виде желтого масла, которое использовали на следующей стадии без очистки. Структуру 5-фтор-9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (55) подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.10-1.40 (6Н, m, N(СН2СН 3)2), 1.60-2.60 (4Н, m, 2- и 3-СН 2), 2.70-2.85 (2Н, m, 1-СН 2), 3.10-3.65 (4Н, m, N(СН 2СН3)2 и CH 2CH2OH), 4.00-4.30 (3Н, m, CH2CH 2OH, 4-СН), 6.55-6.65 (1Н, m, NCCHCHCHCF), 6.90-7.05 (1Н, m, NCCHCHCHCF) и 7.05-7.15 (1H, m, NCCHCHCHCF).
Структуру 7-фтор-9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.10-1.40 (6Н, m, N(СН2СН 3)2), 1.60-2.60 (4Н, m, 2- и 3-СН 2), 2.70-2.85 (2Н, m, 1-СН 2), 3.10-3.65 (4Н, m, N(СН2СН 3)2 и CH 2CH2OH), 4.00-4.30 (3Н, m, NCH 2CH2OH, 4-СН), 6.70-6.80 (1Н, m, NCCHCFCHCH) и 7.00-7.40 (2Н, m, NCCHCFCHCH).
Пример 9(е): Метансульфоновой кислоты 2-(4-диэтилкарбамоил-5-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этиловый эфир (соединение-предшественник 10)
К раствору смеси 5-фтор-9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (42) и 7-фтор-9-(2-гидрокси-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (743 мг, 2,2 ммоль) в дихлорметане (30 мл) добавляли пиридин (1,74 г, 22,0 ммоль, 1,8 мл). Реакционную смесь охлаждали до 0°C и добавляли метансульфонилхлорид (1,01 г, 8,8 ммоль, 0,7 мл). Реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. Смесь промывали 2 н. HCl (2×50 мл) и водой (2×50 мл), сушили и концентрировали в вакууме. Неочищенное вещество очищали при помощи полупрепаративной HPLC, элюируя водой (А) и метанолом (B) (Gemini 5u, С18, 110A, 150×21 мм, 50-95% B в течение 20 мин, 21 мл/мин), с получением 10 мг (1%) метансульфоновой кислоты 2-(4-диэтилкарбамоил-7-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этилового эфира в виде белого твердого вещества и 30 мг (9%) смеси метансульфоновой кислоты 2-(4-диэтилкарбамоил-7-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этилового эфира и метансульфоновой кислоты 2-(4-диэтилкарбамоил-5-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этилового эфира (соединение-предшественник 10) в виде белого твердого вещества. С использованием этих условий очистки- -метансульфоновой кислоты 2-(4-диэтилкарбамоил-5-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этиловый эфир (соединение-предшественник 10) не может быть выделен в виде отдельного компонента. Структуру метансульфоновой кислоты 2-(4-диэтилкарбамоил-7-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этилового эфира подтверждали при помощи 1H NMR (300 МГц, CDCl3): δн 1.18 (3H, t, J=7 Гц, N(CH2CH 3)2), 1.39 (3H, t, J=7 Гц, N(CH2CH 3)2) 1.70-2.30 (4H, m, 2-и 3-СН 2), 2.58 (3H, s, OSO2CH 3), 2.60-2.80 (2H, m, 1-CH 2), 3.40-3.65 (4H, m, N(CH 2CH3)2), 4.02 (1H, t, J=6 Гц, 4-CH), 4.20 (2H, t, J=7 Гц, NCH 2CH2OMs), 4.35 (2H, t, J=7 Гц, NCH2CH 2OMs), 6.70-6.85 (1H, m, NCCHCFCHCH), 6.90-7.00 (1H, m, NCCHCFCHCH) и 7.05-7.15 (2Н, m, NCCHCFCHCH).
Структуру метансульфоновой кислоты 2-(4-диэтилкарбамоил-5-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этилового эфира (соединение-предшественник 10) подтверждали при помощи 1H NMR (300 МГц, CDCl3): δн 1.18 (3H, t, J=7 Гц, N(CH2CH 3)2), 1.39 (3H, t,J=7 Гц, N(CH2CH 3)2) 1.70-2.30 (4H, m, 2- и 3-CH 2), 2.58 (3H, s, OSO2CH 3), 2.60-2.80 (2H, m, 1-СН 2), 3.40-3.65 (4Н, m, N(CH 2CH3)2), 4.15 (1H, m, 4-CH), 4.20 (2Н, t,J=7 Гц, NCH 2CH2OMs), 4.35 (2H, t, J=7 Гц, NCH2CH 2OMS), 6.55-6.65 (1H, m, NCCHCHCHCF), 6.90-7.05 (1H, m, NCCHCHCHCF) и 7.05-7.15 (1H, m, NCCHCHCHCF).
Пример 9(f): 5-Фтор-9-(2-[18F]фтор-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты диэтиламид (визуализирующий агент 10)
Смесь соединения-предшественника 10 и метансульфоновой кислоты 2-(4-диэтилкарбамоил-7-фтор-1,2,3,4-тетрагидро-карбазол-9-ил)-этилового эфира использовали в реакции введения радиоизотопной метки. Введение метки 18F осуществляли, как описано в примере 1(m). Получали 7-фтор-9-(2-[18F]фтор-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты диэтиламид - визуализирующий агент 10.
Полупрепаративная HPLC: колонка HICHROM АСЕ 5 С18 (100 ×10 мм вн.д.), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 3 мл/мин; 0-1 мин 50% В; 1-20 мин 50-95% В; длина волны 254 нм; tR визуализирующего агента 10 15 мин; tR 7-фтор-9-(2-[18F]фтор-этил-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты диэтиламид 14 мин.
Аналитическая-HPLC: колонка Phenomenex Luna C18 (150×4,6 мм вн.д.), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 1 мл/мин; 0-1 мин 50% В; 1-20 мин 50-95%В; длина волны 230 нм; tR визуализирующего агента 10 16 мин; tR 7-фтор-9-2-[18F]фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида 14 мин. Радиохимический выход визуализирующего агента 10 8,7±1% (n=3), нескорректированный на распад, время 90-120 мин, радиохимическая чистота ≥99%. На Фиг.5 показан визуализирующий агент 10 (верх) и 7-фтор-9-(2-[18F]фтор-этил)-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты диэтиламид (середина) и 7-фтор-9-(2-[18F]фтор-этил-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты диэтиламид (низ).
Пример 10: Синтез 5-Фтор-9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (нерадиоактивный визуализирующий агент 10)
Пример 10(a): (2-Фтор-этил}-(3-фтор-фенил)-амин (43)
3-Фторанилин (1,4 г, 11,6 ммоль, 1,2 мл) и 2-фторэтилтозилат (12; полученный в соответствии с примером 2(а)) (2,5 г, 11,6 ммоль) и лутидин (1,24 г, 11,6 ммоль) перемешивали и нагревали в DMF (5 мл) при 100°C в течение ночи. Реакционную смесь оставляли охлаждаться и затем разбавляли этилацетатом (100 мл). Ее промывали водой (3×40 мл) и органические вещества сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (10% В, 100 г, 12 CV, 60 мл/мин) с получением 184 мг (10%) (2-фтор-этил)-(3-фтор-фенил)-амина (43) в виде желтого масла. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 3.37 (1Н, q, J=6 Гц, NCH 2CH2F), 3.46 (1Н, q, J=6 Гц, NCH 2CH2F), 4.12 (1Н, s, br, NH), 4.54 (1Н, t, J=3 Гц, NCH2CH 2F), 4.69 (1H, t, J=3 Гц, NCH 2CH2F), 6.31-6.50 (3Н, m, NCCHCHCH), 7.10-7.25 (1H, m, NCCHCF).
Пример 10(b): 5-Фтор-9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (нерадиоактивный визуализирующий агент 10)
Смесь 3-бром-2-оксо-циклогексанкарбоновой кислоты диэтиламида (35; полученного в соответствии с примером 7(с)) (161 мг, 0,6 ммоль) и (2-фтор-этил)-(3-фтор-фенил)-амина (43) (184 мг, 1,2 ммоль) перемешивали в атмосфере N2 при 50°C в течение 3 ч, и реакционная смесь становилась коричневой. Полученную смесь растворяли в пропан-2-оле (1 мл) и добавляли сухой хлорид цинка (245 мг, 1,8 ммоль). Смесь нагревали до температуры дефлегмации в атмосфере N2 в течение 16 ч и затем концентрировали в вакууме. Остаток растворяли в этилацетате (10 мл) и промывали 2 н. HCl (5 мл), водой (2х5 мл) и водным раствором карбоната калия (2×5 мл), затем сушили и концентрировали в вакууме. Неочищенное вещество очищали при помощи полупрепаративной HPLC, элюируя водой (А) и метанолом (В) (Gemini 5u, C18, 110А, 150×21 мм, 50-95% B в течение 20 мин, 21 мл/мин) с получением 20 мг (6%) 7-фтор-9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида в виде белого твердого вещества и 10 мг (3%) 5-фтор-9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (нерадиоактивный визуализирующий агент 10) в виде белого твердого вещества. Структуру 7-фтор-9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.14 (ЗН, t, J=7 Гц, N(СН2СН 3)2), 1.33 (3Н, t, J=7 Гц, N(СН2СН 3)2), 1.80-2.15 (4Н, m, 2- и 3-СН 2), 2.70-2.80 (2Н, m, 1-СН 2), 3.50-3.80 (4Н, m, N(СН 2СН3)2), 4.20-4.35 (1H, m, 4-СН), 4.40 (2Н, dm, J=21 Гц, NCH 2CH2F), 4.60 (2Н, dm, J=41 Гц, NCH 2CH2F), 6.70-6.80 (1H, m, NCCHCFCHCH) и 7.00-7.10 (2Н, m, NCCHCFCHCH).
Структуру 5-фтор-9-(2-фтор-этил)-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (нерадиоактивный визуализирующий агент 10) подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.14 (3Н, t, J=7 Гц, N(СН2СН 3)2), 1.33 (3Н, t, J=7 Гц, N(СН2СН 3)2), 1.80-2.15 (4Н, m, 2- и 3-СН 2), 2.70-2.80 (2Н, m, 1-СН 2), 3.50-3.80 (4Н, m, N(СН 2СН3)2), 4.20-4.35 (1H, m, 4-СН), 4.40 (2Н, dm, J=21 Гц, NCH 2CH2F), 4.60 (2H, dm, J=41 Гц, NCH 2CH2F), 6.55-6.65 (1H, m, NCCHCHCHCF), 6.90-7.05 (1H, m, NCCHCHCHCF) и 7.05-7.15 (1H,m, NCCHCHCHCF).
Пример 11: Метансульфоновой кислоты 2-(4-диэтилкарбамоил-2-метил-1.2.3.4-тетрагидро-карбазол-9-ил)-этиловый эфир (соединение-предшественник 11) и 9-(2-[18F]фтор-этил)-2-метил-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (визуализируюший агент 11)
Пример 11(а): 4-(4-Метил-циклогекс-1-енил)-морфолин (44]
В колбе, снабженной ловушкой Дина-Старка, раствор 4-метилциклогексанона (20,1 г, 179,3 ммоль, 22 мл) и морфолина (31,3 г, 359,0 ммоль, 31,4 мл) кипятили с обратным холодильником в бензоле (55 мл) в течение 26 часов. Бензол удаляли в вакууме и неочищенный продукт очищали путем перегонки при пониженном давлении с получением 23 г (70%) 4-(4-метил-циклогекс-1-енил)-морфолина (44) в виде масла (точка кипения 120°C при 10 мм рт.ст.(1,33 кПа)). Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 0.94 (3Н, d, J=6,0 Гц, СН 3), 1.15-1.35 (1H, m, CH 2CH=CN), 1.50-1.80 (3Н, m, СН2СН 2СНСН3), 2.00-2.25 (4Н, m, CH 2CH=CN и СН2СН 2СНСН3), 2.65-2.95 (4Н, m, OCH2NCH 2), 3.73 (4Н, t, J=6,0 Гц, OCH 2NCH2) и 4.60-4.65 (1H, m, CH2CH=CN).
Пример 11(b): 5-Метил-2-оксо-ииклогексанкарбоновой кислоты этиловый эфир (45)
К раствору 4-(4-метил-циклогекс-1-енил)-морфолина (44) (23 г, 127,0 ммоль) в бензоле (55 мл) добавляли этилхлорформиат (7,5 г, 69,0 ммоль, 6,6 мл) в атмосфере азота, в то время как раствор энамина быстро перемешивали. После кипячения с обратным холодильником в течение 18 ч раствор охлаждали и фильтровали. Осадок энамина гидрохлорида промывали с обезвоженным эфиром. Фильтрат и промывки возвращали в реакционную колбу и добавляли 10% водную HCl (40 мл). Смесь интенсивно перемешивали в течение 15-30 мин. Слои разделяли, водный слой экстрагировали этилацетатом (2×100 мл) и объединенные органические слои концентрировали в вакууме. Неочищенное вещество очищали путем перегонки при пониженном давлении с получением 12,5 г (53%) 5-метил-2-оксо-циклогексанкарбоновой кислоты этилового эфира (45) в виде масла (т.к. 85°C-90°C при 10 мм рт.ст.(1,33 кПа)). Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 0.85-0.95 (3Н, m, СН 3), 1.17 (ЗН, t, J=7 Гц, ОСН2СН 3), 1.25-2.00 (5Н, m, 5-СН, 4- и 6-СН 3), 2.15-2.40 (3Н, m, 1-CH и 3-СН 2) и 4.00-4.20 (2Н, т, CH 2CH3).
Пример 11 (с): 5-Метил-2-оксо-циклогексанкарбоновой кислоты диэтиламид (46)
5-Метил-2-оксо-циклогексанкарбоновой кислоты этиловый эфир (45) (5,9 г, 32 ммоль), DMAP (1,12 г, 10 ммоль) и диэтиламин (4,7 г, 65 ммоль, 6,7 мл) в толуоле (90 мл) кипятили с обратным холодильником течение 4 суток. Реакционную смесь оставляли охлаждаться, и толуол удаляли при пониженном давлении с получением желтого масла. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (20-50% В, 80 г) с получением 4,4 г (65%) 5-метил-2-оксо-циклогексанкарбоновой кислоты диэтиламида (46) в виде желтого масла. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 0.8-1.05 (9Н, m, СН 3 и N(СН2СН 3)2), 1.05-2.10 (5Н, m, 5-CH и 4- и 6-СН 2), 2.15-2.80 (2Н, m, 3- СН 2), 2.95-3.55 (5Н, m, 1-CH и N(CH 2CH3)2).
Пример 11(d): 3-Бром-2-гидрокси-5-метил-и,иклогекс-1-енкарбоновой кислоты диэтиламид (47)
5-Метил-2-оксо-циклогексанкарбоновой кислоты диэтиламид (46) (4,4 г, 21 ммоль) растворяли в диэтиловом эфире (5 мл) и охлаждали до 0°C в атмосфере N2. Бром (3,32 г, 21 ммоль, 1,1 мл) добавляли по каплям в течение 15 мин и реакционную смесь оставляли нагреваться до комнатной температуры в течение 90 мин. Смесь медленно вливали в ледяной насыщенный водный раствор карбоната натрия (40 мл) и экстрагировали этилацетатом (3×40 мл). Объединенные органические слои сушили и концентрировали в вакууме с получением 6,1 г (количественный выход) 3-бром-2-гидрокси-5-метил-циклогекс-1-енкарбоновой кислоты диэтиламида (47) в виде не совсем белого твердого вещества. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 0.8-1.20 (9Н, m, СН 3 и N(СН2СН 3)2), 1.80-2.40 (5Н, m, СН 2СН(СН3)СН2), 3.15-3.55 (4Н, т, N(СН 2СН3)2), 4.65-4.74 (1Н, т, CHBr) и 12.04 (1H, s, OH).
Пример 11(е): 9-(2-Бензилокси-этил)-2-метил-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (48)
Смесь 3-бром-2-гидрокси-5-метил-циклогекс-1-енкарбоновой кислоты диэтиламида (47) (4,0 г, 14 ммоль) и (2-бензилокси-этил)-фенил-амина (21; полученного в соответствии с примером 3(с)) (6,3 г, 28 ммоль) перемешивали в атмосфере N2 при 50°C в течение 3 ч, и реакционная смесь становилась коричневой. Полученную смесь растворяли в пропан-2-оле (14 мл) и добавляли сухой хлорид цинка (5,72 г, 42 ммоль). Смесь нагревали до температуры дефлегмации в атмосфере N2 в течение 16 ч и затем концентрировали в вакууме. Остаток растворяли в этилацетате (200 мл) и промывали 2 н. HCl (50 мл), водой (2×50 мл) и водным раствором карбоната калия (2×50 мл), затем сушили и концентрировали в вакууме. Неочищенную смесь очищали при помощи картриджа SCX (40 мл) и затем при помощи хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (10-50% В, 100 г, 12 CV, 85 мл/мин), с получением 467 мг (8%) 9-(2-бензилокси-этил)-2-метил-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (48) в виде белого твердого вещества. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.20-1.40 (9Н, m, СН3 и N(СН2СН3)2), 1.90-2.20 (3Н, m, 2-СН и 3-CH2), 2.35-2.45 (1Н, m, 1-СН2), 2.85-2.95 (1Н, m, 1-СН2), 3.40-3.70 (4Н, m, N(СН2СН3)2), 3.70-3.80 (1Н, m, 4-CH), 4.10-4.30 (4Н, m, NCH2CH2OBn), 4.43 (2Н, s, OCH2Ph) и 7.00-7.30 (9Н, m, CHCHCHCH и Ph).
Пример 11(f): 9-(2-Гидрокси-этил)-2-метил-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламид (49)
К раствору 9-(2-бензилокси-этил)-2-метил-2,3,4,9-тетрагидро-1 Н-карбазол-4-карбоновой кислоты диэтиламида (48) (460 мг, 1,1 ммоль) в метаноле (25 мл) добавляли суспензию Pd/C (100 мг) в метаноле (5 мл). Смесь помещали в гидрогенизатор Парра и встряхивали в течение 24 ч в атмосфере азота. Реакционную смесь фильтровали через слой целита, промывали метанолом и концентрировали в вакууме с получением 250 мг (79%) 9-(2-гидрокси-этил)-2-метил-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (49) в виде желтого масла, которое использовали на следующей стадии без очистки. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.20-1.40 (9Н, m, CH 3 и N(СН2СН 3)2), 1.90-2.20 (3Н, m, 2-CH и 3-CH 3), 2.35-2.45 (1Н, m, 1-CH 2), 2.85-2.95 (1Н, m, 1-CH 2), 3.40-3.70 (4Н, m, N(CH 2CH3)2), 3.70-3.80 (1H, m, 4-CH), 4.10-4.30 (4H, m, NCH 2CH 2OH), 6.91 (1H, t, J=7 Гц, NCCHCHCHCH), 7.00 (1H, t, J=7 Гц, NCCHCHCHCH), 7.12 (1H, d, J=7 Гц, NCCHCHCHCH) и 7.15 (1H, d, J=7 Гц, NCCHCHCHCH).
Пример 11(g): Метансульфоновой кислоты 2-(4-диэтилкарбамоил-2-метил-1,2,3,4-тетрагидро-карбазол-9-ил)-этиловый эфир (соединение-предшественник 11)
К раствору 9-(2-гидрокси-этил)-2-метил-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты диэтиламида (49) (250 мг, 0,8 ммоль) в дихлорметане (10 мл) добавляли пиридин (633 мг, 8,0 ммоль, 0,6 мл). Реакционную смесь охлаждали до 0°C и добавляли метансульфонилхлорид (367 мг, 3,2 ммоль, 0,2 мл). Реакционную смесь оставляли нагреваться до комнатной температуры в течение ночи. Смесь промывали 2 н. HCl (2×20 мл) и водой (2×20 мл), сушили и концентрировали в вакууме. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя бензином (A) и этилацетатом (B) (0-100% В, 10 г, 34 CV, 30 мл/мин), затем растирали с диэтиловым эфиром с получением 250 мг (80%) метансульфоновой кислоты 2-(4-диэтилкарбамоил-2-метил-1,2,3,4-тетрагидро-карбазол-9-ил)-этилового эфира (соединение-предшественник 11) в виде белого твердого вещества. Структуру подтверждали при помощи 13C NMR (75 МГц, CDCl3)^ δс 12.9, 13.0, 15.2, 22.0, 29.7, 30.2, 36.7, 36.8, 40.8, 41.6, 42.0, 67.8, 108.6, 109.5, 118.6, 119.6, 121.2, 126.4, 136.2, 136.4, 173.7.
Пример 11(h): 9-(2-[18F]Фтор-этил)-2-метил-2,3,4,9-тетрагидро-1H-карбазол-4-карбоновой кислоты диэтиламид (визуализирующий агент 11)
Введение в соединение-предшественник 11 метки 18F осуществляли, как описано в примере 1(m).
Полупрепаративная HPLC: колонка HICHROM АСЕ 5 С18 (100×10 мм вн.д.), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 3 мл/мин; 0-26 мин 50% В; длина волны 254 нм; tR визуализирующего агента 11 15 мин.
Аналитическая-HPLC: колонка Phenomenex Luna С18 (150×4,6 мм вн.д.), размер частиц 5 мкм; подвижная фаза А: вода, подвижная фаза В: метанол; градиент потока: 1 мл/мин; 0-1 мин 40% В; 1-20 мин 40-95% В; длина волны 230 нм; tR визуализирующего агента 11 17 мин. Радиохимический выход 14±13% (n=3), нескорректированный на распад, время 90-120 мин, радиохимическая чистота ≥99%. На Фиг.6 показано совместное элюирование визуализирующего агента 11 и нерадиоактивного визуализирующего агента 11.
Пример 12: синтез 9-(2-фтор-этил)-2-метил-2,3,4,9-тетрагидро-1Н-карбазол-4-карбоновой кислоты диэтиламида (нерадиоактивный визуализирующий агент 11)
Смесь 3-бром-2-гидрокси-5-метил-циклогекс-1-енкарбоновой кислоты диэтиламида (47; полученного в соответствии с примером 11(d)) (2,0 г, 7 ммоль) и (2-фтор-этил)-сренил-амина (24; полученного в соответствии с примером 4(а)) (1,9 г, 14 ммоль) перемешивали в атмосфере N2 при 50°C в течение 3 ч, и реакционная смесь становилась коричневой. Полученную смесь растворяли в пропан-2-оле (7 мл) и добавляли сухой хлорид цинка (2,86 г, 21 ммоль). Смесь нагревали до температуры дефлегмации в атмосфере N2 в течение 16 ч и затем концентрировали в вакууме. Остаток растворяли в этилацетате (100 мл) и промывали 2 н. HCl (30 мл), водой (2×30 мл) и водным раствором карбоната калия (2×30 мл), затем сушили и концентрировали в вакууме. Неочищенную смесь очищали при помощи картриджа SCX (40 мл) и затем при помощи хроматографии на силикагеле, элюируя бензином (А) и этилацетатом (В) (0-100% В, 100 г, 12 CV, 85 мл/мин) с получением 400 мг (17%) 9-(2-фтор-этил)-2-метил-2,3,4,9-тетрагидро-1 Н-карбазол-4-карбоновой кислоты диэтиламида (нерадиоактивный визуализирующий агент 11) в виде белого твердого вещества. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δH 1.10-1.35 (9Н, m, СН 3 и N(СН2СН 3)2), 1.95-2.10 (2Н, m, 3- СН 2), 2.30-2.50 (1Н, m, 2-CH), 2.70-2.80 (2Н, m, 1-СН 2), 3.40-3.70 (4Н, m, N(СН 2СН3)2), 4.05-4.15 (1Н, m, 4-СН), 4.30 (2Н, dm, J=21 Гц, NCH2CH 2F), 4.65 (2Н, dm, J=41 Гц, NCH2CH 2F) и 7.00-7.30 (4Н, т, NCCHCHCHCH.
Пример 13: Энантиомерное разделение соединения-предшественника 5

Соединение-предшественник 5 (полученный, как описано в примере 1) разделяли на его энантиомеры, используя хиральную суперкритическую флюидную (CO2) хроматографию на колонке Kromasil Amycoat, 250×10 мм, 5 мкм, 100 А с использованием 30% IPA (изопропилового спирта) при 40°C со скоростью 13 мл/мин и временем прогона 6 мин. Соединение-предшественник 5 (60 мг) растворяли в 1,4-диоксане (2 мл) и до 200 мкл единовременно инжектировали для каждого прогона. Достигали основного разделения двух энантиомеров. Определение при помощи аналитической HPLC энантиомерной чистоты двух разделенных энантиомеров на 1C от Chiral Technologies, 250×4,6 мм, 5 мкм, изократический прогон, при 0,5 мл/мин 80:20-МеОН: IPA и комнатной температуре указывало на энантиомерную чистоту 99,5% каждого из энантиомеров.
Пример 14: Энантиомерное разделение нерадиоактивного визуализируюшего агента 5
Нерадиоактивный визуализирующий агент 5 (полученный, как описано в примере 2) разделяли на его энантиомеры, используя хиральную суперкритическую флюидную (CO2) хроматографию на колонке Kromasil Amycoat, 250×10 мм, 5 мкм, 100 Ǻ с использованием 20% IPA при 40°C со скоростью 14 мл/мин и времени прогона 6 мин. Соединение 5 (100 мг) растворяли в 1,4-диоксане (2,5 мл) и для каждого прогона инжектировали вплоть до 200 мкл единовременно. Фракции отсекали по времени, чтобы гарантировать, что смешанные фракции не были собраны. Определение при помощи аналитической HPLC энантиомерной чистоты двух разделенных энантиомеров на IC от Chiral Technologies, 250×4,6 мм, 5 мкм, изократический прогон, 80:20-МеОН: 1РА при 0,5 мл/мин и комнатной температуре указывало на энантиомерную чистоту 99,5% каждого из энантиомеров.
Пример 15: Анализ активности in vitro
Аффинность в отношении PBR исследовал, используя способ, адаптированный из Le Fur et al (Life Sci. 1983; USA 33: 449-57). Нерадиоактивные аналоги in vivo визуализирующих агентов по изобретению тестировали вместе с нерадиоактивным аналогом предшествующего тетрациклического индольного визуализирующего агента (из находящейся в одновременном рассмотрении патентной заявки РСТ/ЕР2009/062827; синтез описан в примере 17 ниже):
Каждое тестируемое соединение (растворенное в 50 мМ Tris-HCI, pH 7,4, 10 мМ MgCl2, содержащем 1% DMSO) конкурировало за связывание с сердечными PBR крысы Вистар с 0,3 нм [3Н] PK-11195. Реакцию осуществляли в 50 мМ Tris-HCl, pH 7,4 10 мМ MgCl2 в течение 15 минут при 25°C. Каждое тестируемое соединение исследовали при 6 отличающихся концентрациях в 300-кратном диапазоне концентраций вокруг ожидаемой К, (константа ингибирования). Были получены следующие данные:
Пример 16: Метод биораспределения in vivo
Визуализирующие агенты по изобретению тестировали в модели биораспределения in vivo вместе с предшествующим тетрациклическим индольным визуализирующим агентом (из находящейся в одновременном рассмотрении патентной заявки РСТ/ЕР2009/062827; синтез описан в примере 18 ниже).
Взрослым самцам крыс Вистар (200-300 г) инъецировали 1-3 МБк тестируемого соединения через латеральную хвостовую вену. Через 2, 10, 30 или 60 мин (n=3) после инъекции крыс умерщвляли и ткани или жидкости отбирали для радиоактивного измерения на гамма-счетчике.
Были получены следующие данные:
На Фиг.7-13 показан профиль биораспределения в головном мозге тетрациклического визуализирующего агента и визуализирующих агентов 5-7 и 9-11 соответственно. Можно видеть, что in vivo визуализирующие агенты по настоящему изобретению обладают хорошим поглощением в головном мозге и улучшенным специфическим поглощением в тканях, экспрессирующих PBR, по сравнению с тетрациклическим визуализирующим агентом.
Пример 17: Получение (+-)-11-(2-фторэтил}-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты диэтиламида (нерадиоактивный тетраииклический индольный визуализирующий агент)
17(а): (+-)-4-Оксо-тиохроман-2-карбоновой кислоты диэтиламид (50)
(+-)-4-Оксо-тиохроман-2-карбоновую кислоту (10,4 г, 50 ммоль), полученную, как описано в Т. Okubo et al (Bioorg. Med. Chem. 2004; 12: 3569-3580), в обезвоженном DCM (100 мл) перемешивали в атмосфере азота при комнатной температуре с оксалилхлоридом (12,6 г, 100 ммоль) и одной каплей DMF в течение 18 ч. Реакционную смесь затем упаривали в вакууме до смолы и затем перерастворяли в DCM (100 мл), охлаждали до 0°C в ледяной бане, перемешивали и обрабатывали по каплям диэтиламином (8,03 г, 110 ммоль) в DCM (20 мл) в течение 1 ч. Реакционную смесь оставляли нагреваться до комнатной температуры в течение 1 ч и добавляли 10% водный раствор карбоната калия (100 мл) и реакционную смесь интенсивно перемешивали. DCM раствор отделяли. Водный раствор экстрагировали двумя дополнительными партиями DCM (100 мл), и объединенные экстракты сушили над сульфатом магния. DCM раствор концентрировали в вакууме с получением темно-зеленого масла, которое кристаллизовалось при стоянии. Кристаллическое твердое вещество растирали с диэтиловым эфиром (50 мл) и фильтровали с получением указанного в заголовке соединения (50) (8,57 г, 65%) в виде бледно-зеленого твердого вещества. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δ 1.06 (t, J=7,1 Гц, 3H), 1.23 (t, J=7,1 Гц, 3H), 3.0-3.5 (m, 6H), 4.25 (m, 1H), 7.15-7.21 (m, 2H), 7.32-7.39 (m, 1H), 8.10-8.14 (m, 1H).
17(b): (+-)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]-флуорен-6-карбоновой кислоты диэтиламид (51)
К раствору (+-)-4-оксо-тиохроман-2-карбоновой кислоты диэтиламида (50) (1,32 г, 5,0 ммоль) и 4-метоксифенилгидразина гидрохлорида (0,87 г, 5,0 ммоль) в этаноле (10 мл) добавляли концентрированную серную кислоту (0,73 мл, 1,35 г, 13,8 ммоль) в атмосфере азота. Реакционную смесь кипятили с обратным холодильником в течение 24 ч. После охлаждения реакционную смесь фильтровали, твердое вещество промывали этанолом, сушили в вакууме (45°C) с получением указанного в заголовке соединения (51) (1,05 г, 57%) в виде бледно-желтого твердого вещества. Структуру подтверждали при помощи 13С NMR (75 МГц, DMSO-d6): δ 10.5, 12.7, 32.7, 37.9, 39.5, 53.0, 97.6, 103.3, 109.87, 109.92, 120.3, 123.5, 123.8, 124.3, 124.7, 124.9, 127.8, 129.4, 131.8, 151.3, 166.2.
17(c): (+-)-11-(2-Фторэтил)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты диэтиламид (нерадиоактивный аналог предшествующего тетрациклического индольного визуализирующего агента)
К раствору (+-)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[а]флуорен-6-карбоновой кислоты диэтиламида (51) (150 мг, 0,41 ммоль; полученного в соответствии с примером 17(b)) в обезвоженном DMF (4 мл), добавляли 2-фторэтил-тозилат (166 мг, 0,82 ммоль), полученный, как описано в L. Cronin et al (J. Org. Chem. 2004; 69: 5934-5946), а затем 60% дисперсию гидрида натрия в минеральном масле (34 мг, 0,82 ммоль) в атмосфере азота. Реакционную смесь нагревали при 80°C в течение 1 ч. После охлаждения растворители удаляли в вакууме, остаток гасили водой (30 мл), экстрагировали DCM (2×30 мл), сушили (MgSO4) и растворители удаляли в вакууме. Остаток очищали при помощи колоночной хроматографии на диоксиде кремния, элюируя 5-10% смесью EtOAc/CH2Cl2. Неочищенное твердое вещество гасили смесью эфир/петролейный спирт, фильтровали, сушили в вакууме (45°C) с получением указанного в заголовке соединения (нерадиоактивный визуализирующий агент) (77 мг, 46%) в виде бледно-коричневого твердого вещества. Структуру подтверждали при помощи 1H NMR (300 МГц, CDCl3): δ 1.12 (t, J=7,0 Гц, 3Р), 1.36 (t,J=7,0 Гц,3H), 3.25-3.70 (m, 4Н), 3.83 (s, 3H), 4.45-4.70 (m, 2Н), 4.80 (t, J=5,2 Гц, 1H), 4.96 (t, J=5,2 Гц, 1H), 5.09 (s, 1H), 6.84-6.93 (m, 2Н), 7.13-7.32 (m, 3Н), 7.46 (m, 1H), 7.58 (d, J=8,0 Гц, 1Н).
Пример 18: Синтез (+-)-11-(2-[18F]фторэтил)-8-метокси-6,11-дигидро-5-тио-11-аза-бензо[а]флуорен-6-карбоновой кислоты диэтиламида (тетрациклический индольный визуализирующий агент)
18F/воду добавляли к K222 (4 мг), водному K2CO3 (50 мкл 0,1 молярного раствора) и ацетонитрилу (500 мкл) в реакционном сосуде и сушили в течение 20-30 мин при 100°C в потоке азота. Добавляли этил-1,2-дитозилат (4 мг) в ацетонитриле (1000 мкл) и нагревали при 100°C в течение 10 мин. Реакционную смесь охлаждали и очищали при помощи полупрепаративной HPLC и собирали фракцию, содержащую 18F-фторэтилтозилат. Эту фракцию разбавляли H2O до объема приблизительно 20 мл, наносили на кондиционированный light t-C18 sep pak и промывали H2O (1×2 мл). Sep pak сушили на линии N2 в сильном потоке в течение 20 мин. 18F фторэтилтозилат затем элюировали с DMF (500 мкл).
Соединение-предшественник (+-)-8-метокси-6,11-дигидро-5-тиа-11-аза-бензо[a]флуорен-6-карбоновой кислоты диэтиламид (51; полученный в соответствии с примером 17(b)) (13 мг) в DMF (250 мкл) добавляли во второй реакционный сосуд и продували N2 в течение 5 мин. Затем добавляли в атмосфере азота NaH (1,3 мг) в DMF (2×250 мкл) и реакционный сосуд нагревали при 45°C в течение 0,5-1 ч. В него затем добавляли полученный выше 18F фторэтилтозилат в DMF и нагревали при 100°C в течение 10 мин в продуваемом N2 реакционном сосуде. Реакционную смесь охлаждали и вымывали из реакционного сосуда водой (1 мл). Раствор фильтровали через шприцевой фильтр и очищали на препаративной HPLC. Собирали фракцию, содержащую основной радиоактивный пик. Ее разбавляли H2O до объема примерно 10 мл и наносили на кондиционированный light C18 sep pak, промывали H2O (1×2 мл) и элюировали EtOH (0,5 мл) во флакон Р6 и добавляли физиологический раствор, забуференный фосфатом (5 мл).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТРИЦИКЛИЧЕСКОГО ИНДОЛА В КАЧЕСТВЕ ЛИГАНДОВ PBR | 2011 |
|
RU2551423C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА, ПОДХОДЯЩИЕ ДЛЯ ВИЗУАЛИЗАЦИИ НЕЙРОВОСПАЛЕНИЯ | 2009 |
|
RU2512288C2 |
| МОДУЛЯТОРЫ ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ | 2017 |
|
RU2738646C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2005 |
|
RU2430088C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2005 |
|
RU2382770C2 |
| ПРОИЗВОДНЫЕ 3-(ГЕТЕРОАРИЛАМИНО)-1,2,3,4-ТЕТРАГИДРО-9Н-КАРБАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ПРОСТАГЛАНДИНА D2 | 2011 |
|
RU2562255C2 |
| Новые производные имидазо[4,5-с]хинолина в качестве ингибиторов LRRK2 | 2018 |
|
RU2773516C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА ВАЗОПРЕССИНА V | 2005 |
|
RU2370497C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ ФИЗИОЛОГИЧЕСКИХ И/ИЛИ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ, ОПОСРЕДОВАННЫХ LHRH РЕЦЕПТОРОМ, ПОСРЕДСТВОМ ВЫШЕНАЗВАННЫХ ПРОИЗВОДНЫХ | 2008 |
|
RU2497806C9 |
| НОВЫЕ 2-ГЕТЕРОАРИЛ-ЗАМЕЩЕННЫЕ БЕНЗОТИОФЕНЫ И БЕНЗОФУРАНЫ 709 | 2008 |
|
RU2472789C2 |
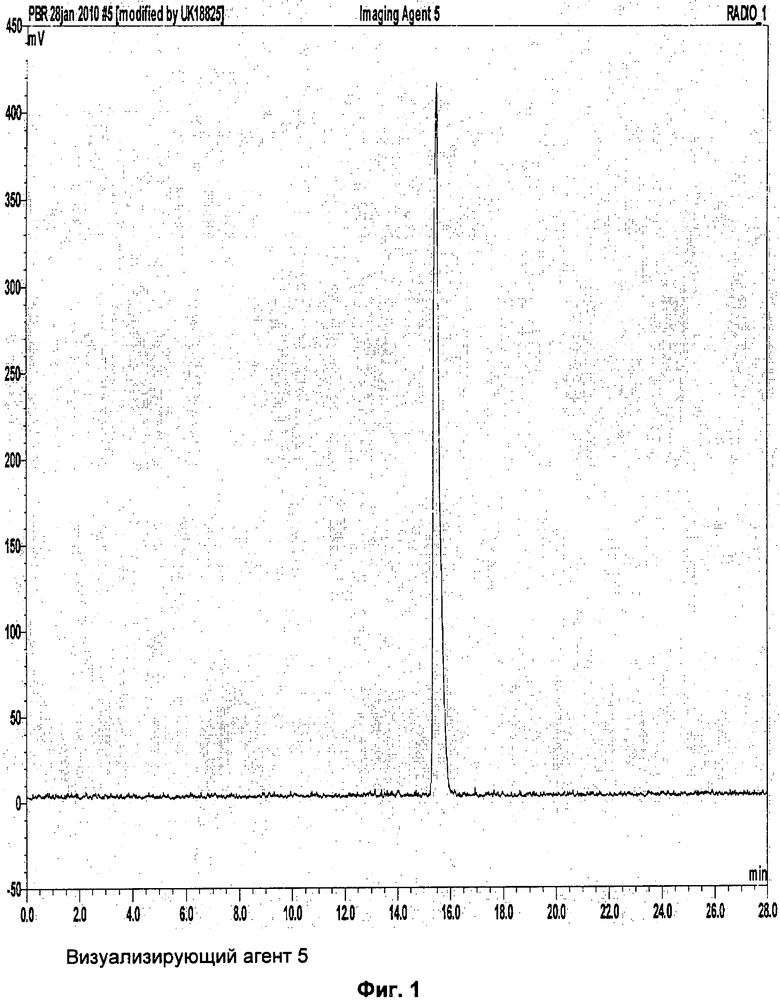
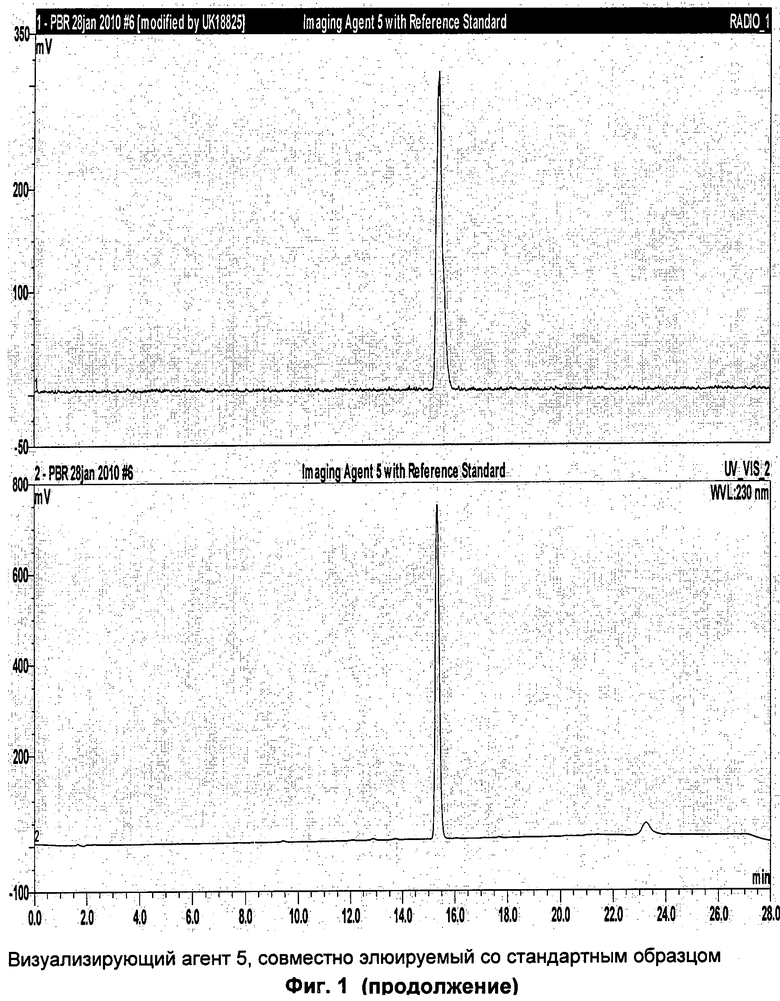
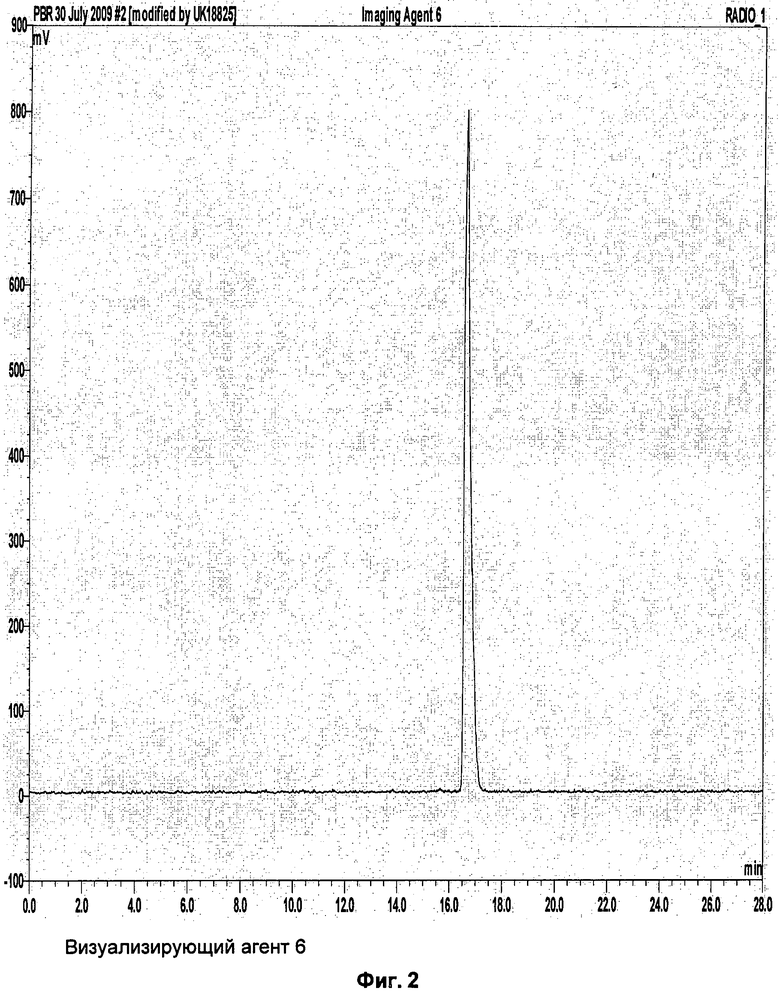
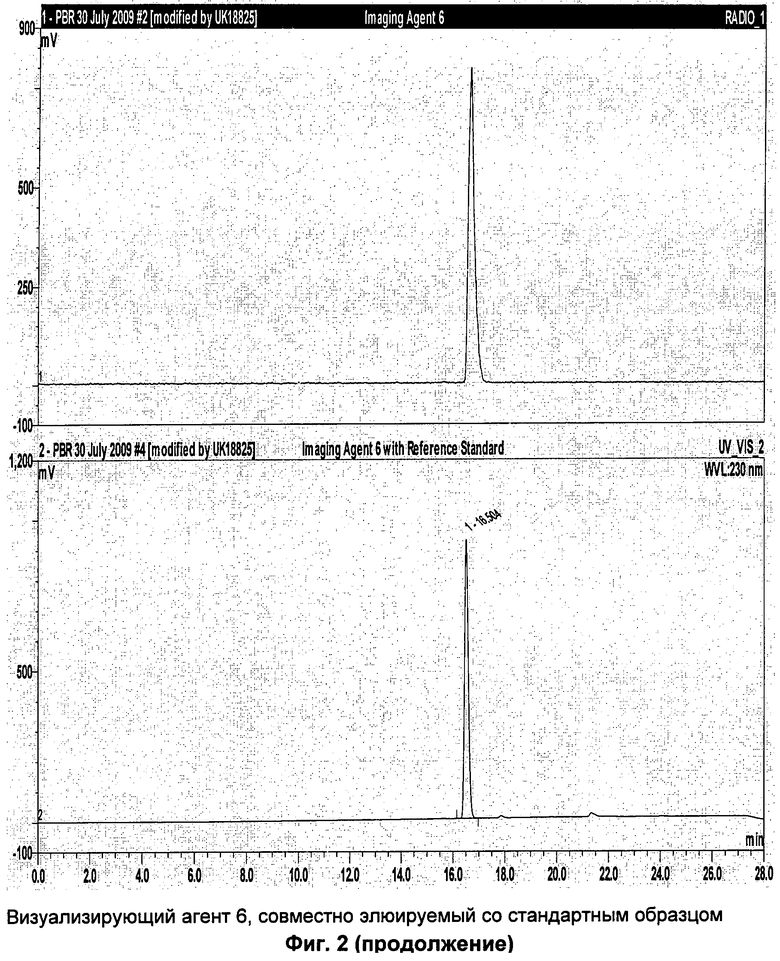
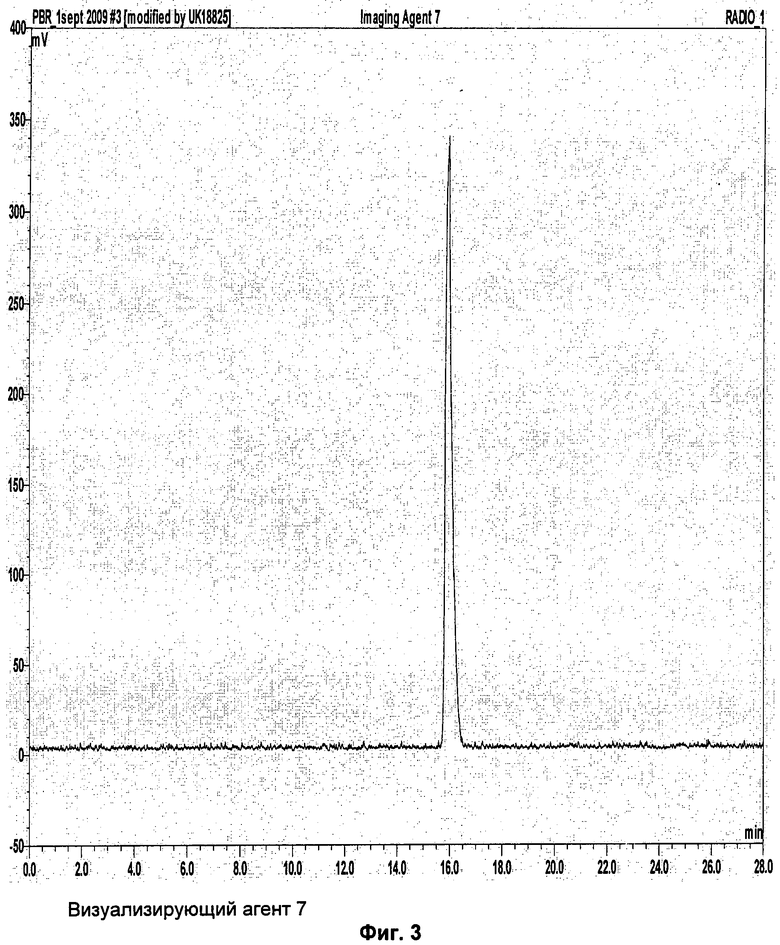
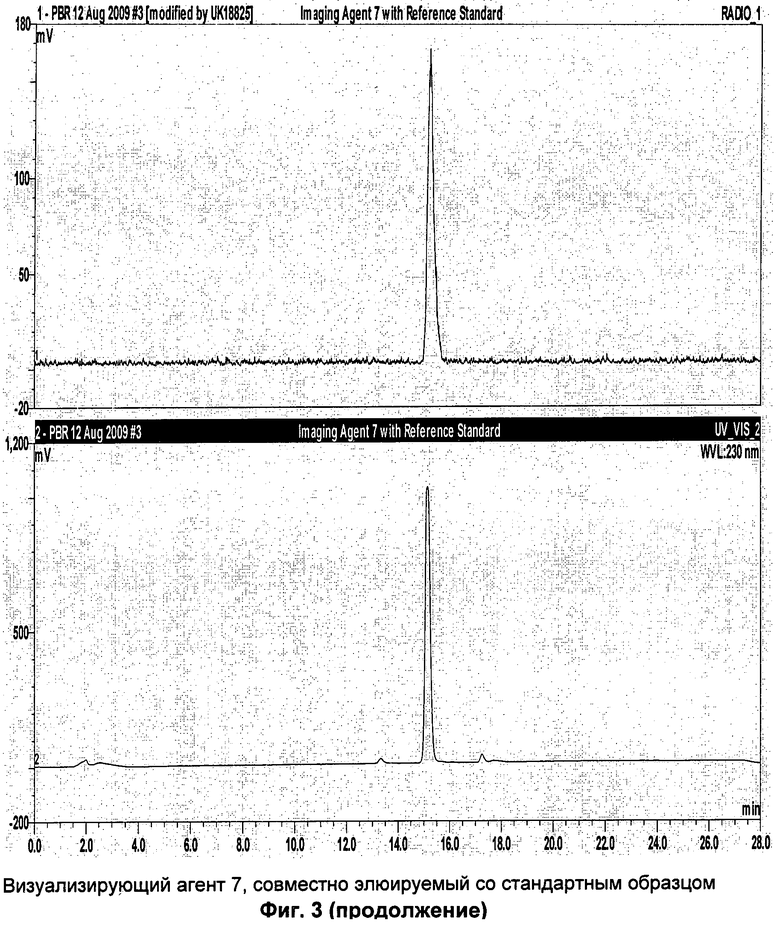
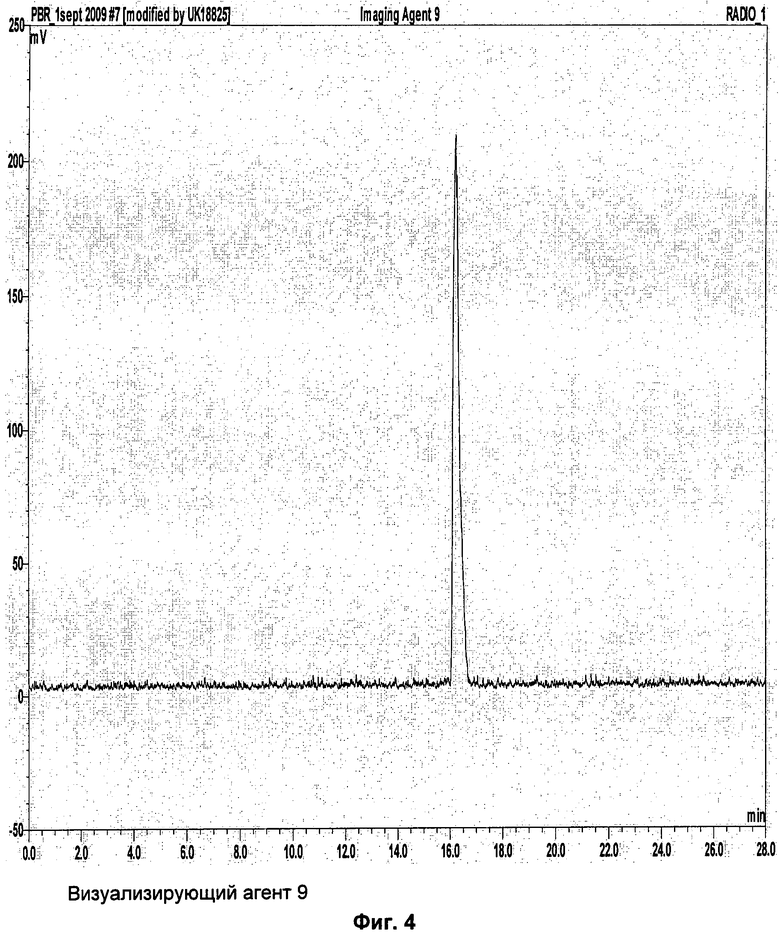
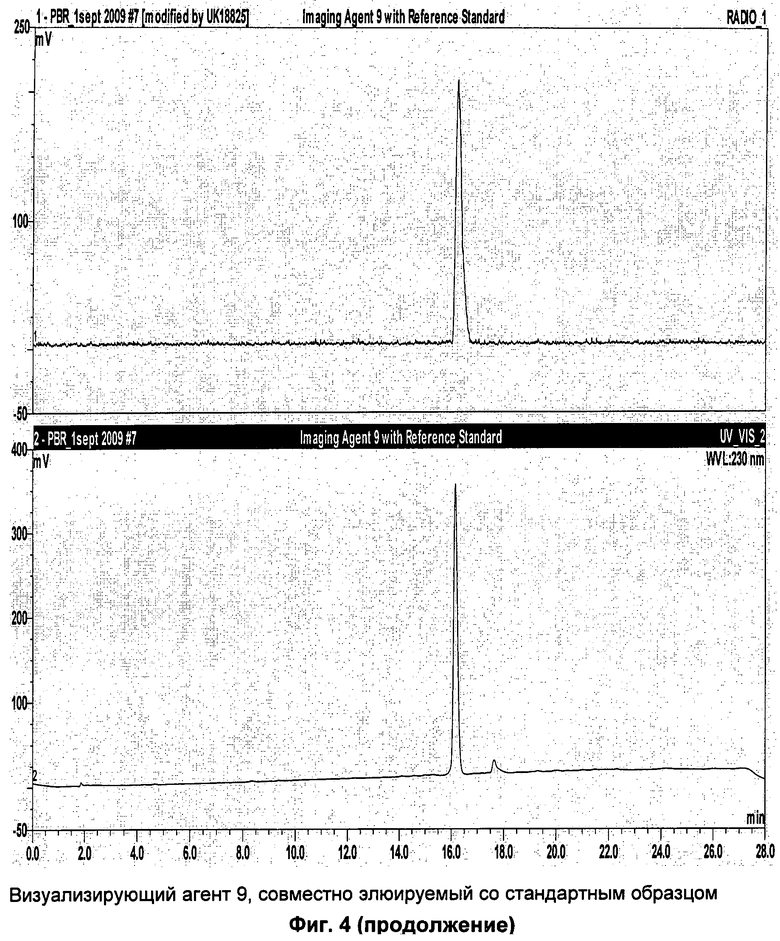
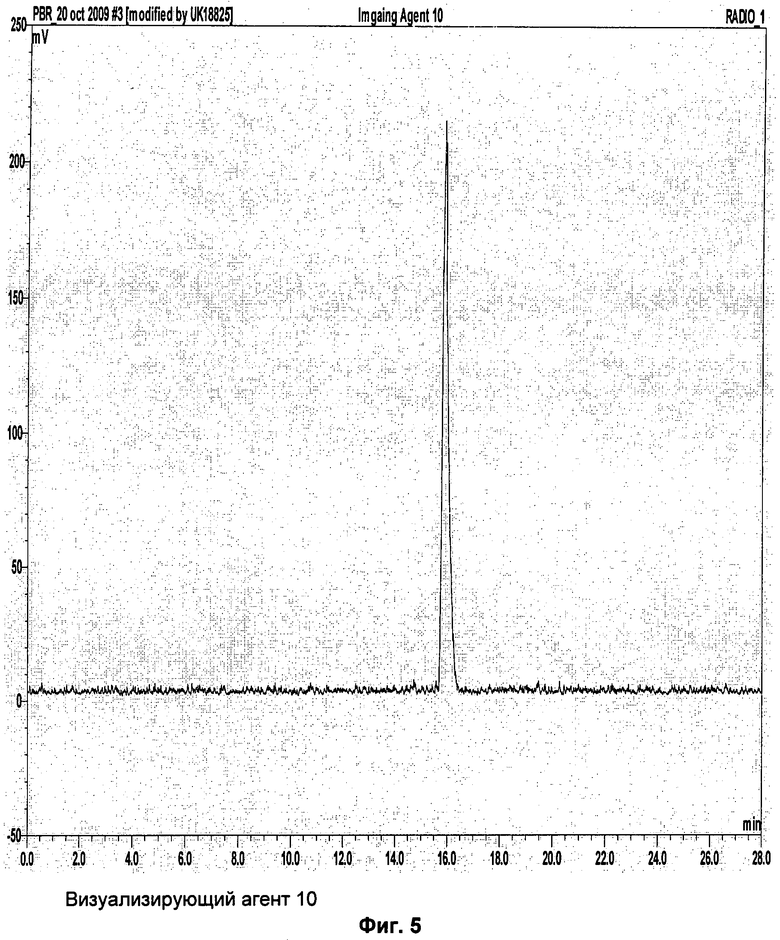

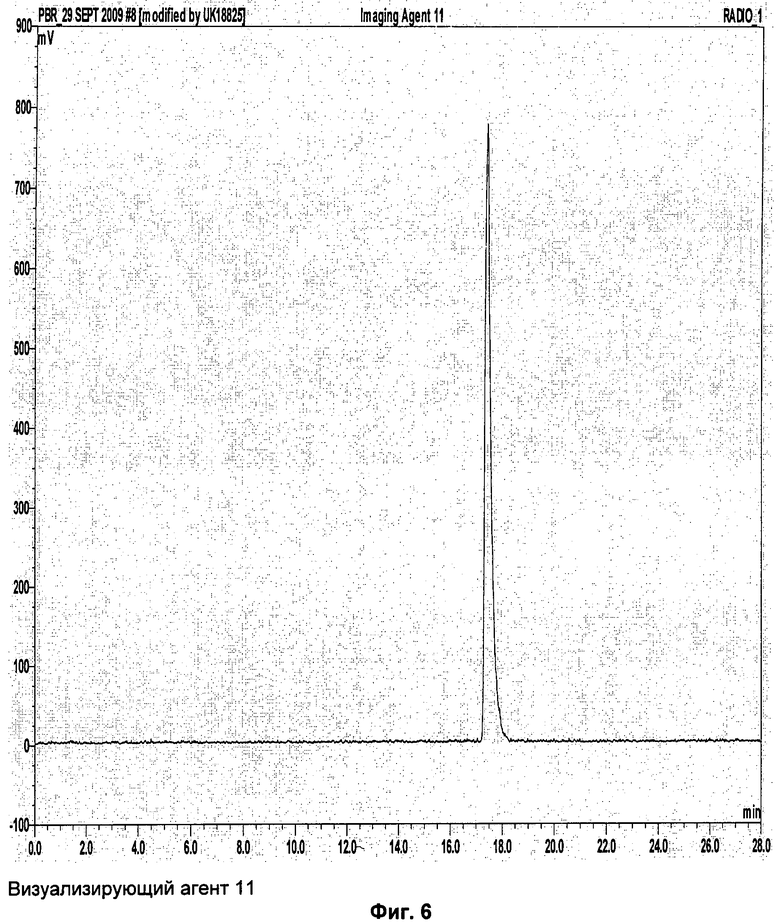
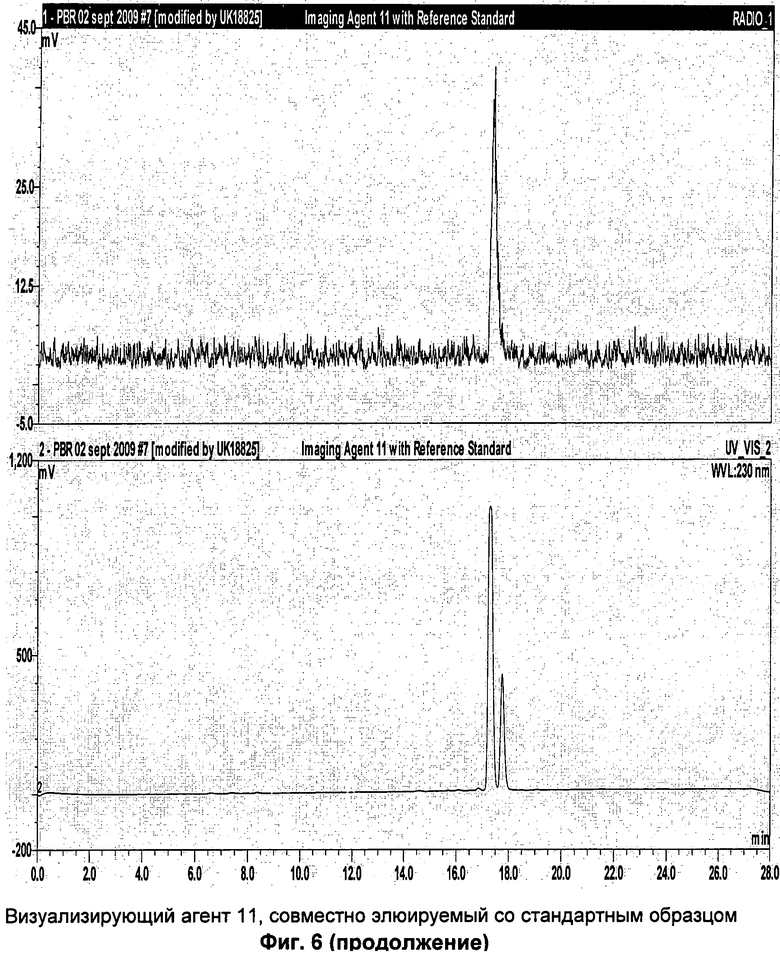
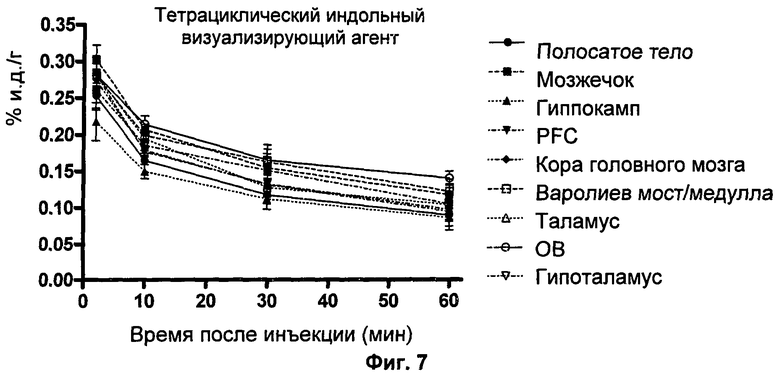

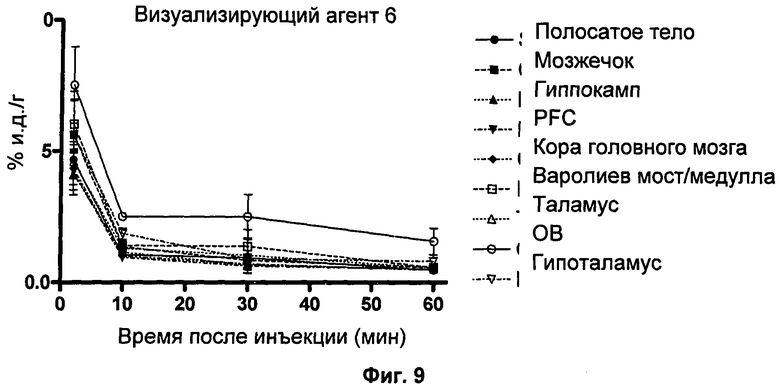

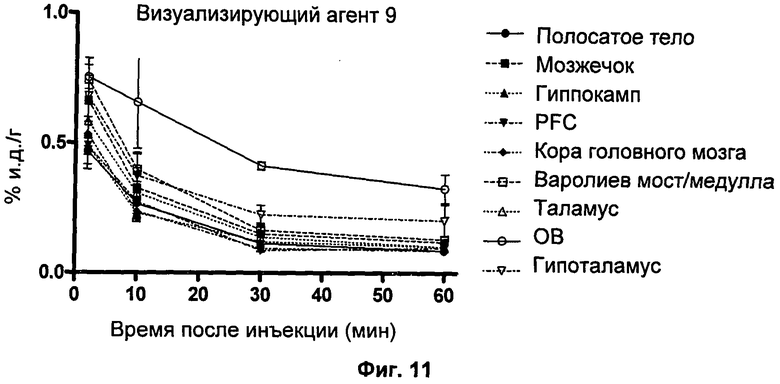
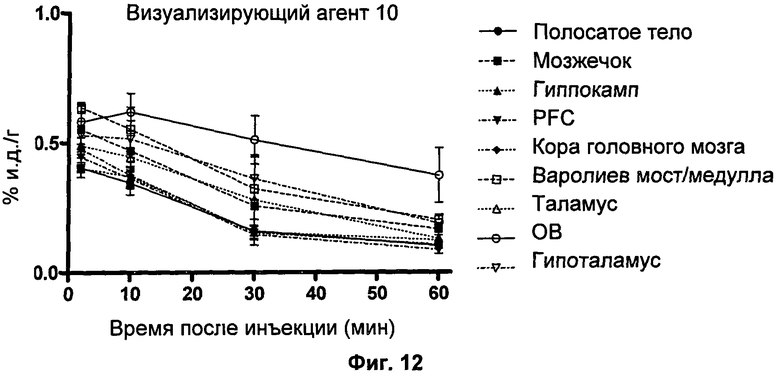
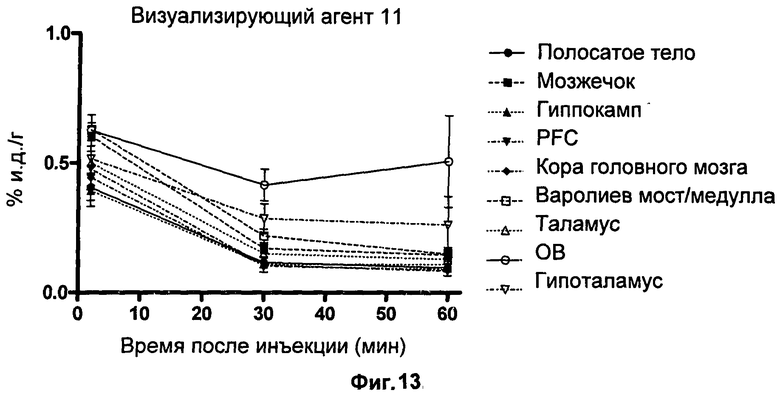
Изобретение относится к новым in vivo визуализирующим агентам формулы I
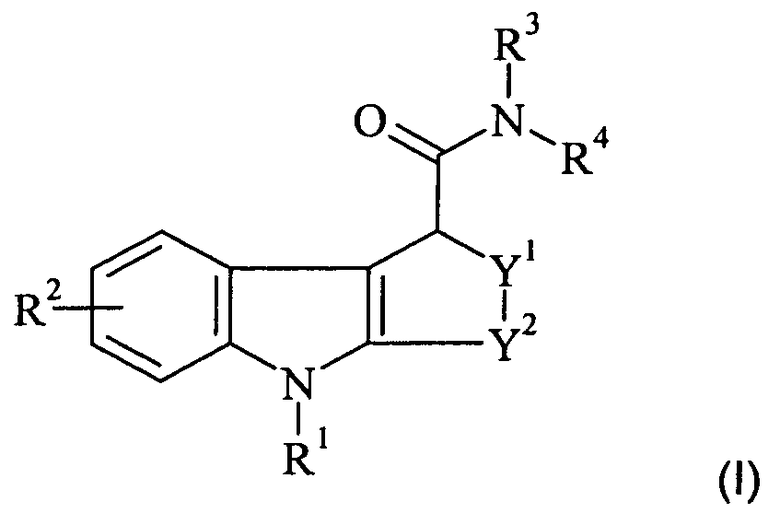
где R1 означает C1-3алкил, C1-3фторалкил; R2 означает Н, ОН, галогено, C1-3алкил, C1-3алкокси, C1-3фторалкил, C1-3фторалкокси; R3 и R4 независимо означают С1-3алкил, C7-C10аралкил или R3 и R4 вместе с атомом азота, к которому они присоединены, образуют азотсодержащее C4-6алифатическое кольцо; Y1 означает CH2; Y2 означает CH2, CH2-CH2, CH(CH3)-CH2 или CH2-CH2-CH2; и где формула I, как она определена, содержит атом, представляющий собой радиоизотоп, подходящий для визуализации in vivo, выбранный из 11C, 18F, 77Br, 123I, 124I и 131I. Указанные соединения с высокой аффинностью связываются с PBR, обладают хорошим поглощением в головном мозге после введения. Описан способ получения in vivo визуализирующего агента и набор для осуществления указанного способа. Описана радиофармацевтическая композиция, содержащая in vivo визуализирующий агент. 8 н. и 13 з.п. ф-лы, 13 ил., 18 пр.
1. In vivo визуализирующий агент формулы I
где R1 представляет собой C1-3алкил или C1-3фторалкил;
R2 представляет собой водород, гидроксил, галогено, C1-3алкил, C1-3алкокси, C1-3фторалкил или C1-3фторалкокси;
R3 и R4 независимо представляют собой C1-3алкил, C7-C10аралкил или R3 и R4 вместе с атомом азота, к которому они присоединены, образуют азотсодержащее C4-6алифатическое кольцо;
Y1 представляет собой CH2 и
Y2 представляет собой CH2, CH2-CH2, CH(CH3)-CH2 или CH2-CH2-CH2;
и где формула I, как она определена, содержит атом, представляющий собой радиоизотоп, подходящий для визуализации in vivo, выбранный из 11C, 18F, 77Br, 123I, 124I и 131I.
2. In vivo визуализирующий агент по п.1, где R1 представляет собой метил или C2-3фторалкил.
3. In vivo визуализирующий агент по п.1, где R2 представляет собой водород, галогено, C1-3алкокси или C1-3фторалкокси.
4. In vivo визуализирующий агент по п.1, где R3 и R4 независимо представляют собой метил, этил или бензил.
5. In vivo визуализирующий агент по п.1, где R3 и R4 вместе с атомом азота, к которому они присоединены, образуют азот-содержащее C5-6алифатическое кольцо.
6. In vivo визуализирующий агент по любому из пп.1-5, где Y2
представляет собой CH2-CH2.
7. In vivo визуализирующий агент по п.1 формулы Ia
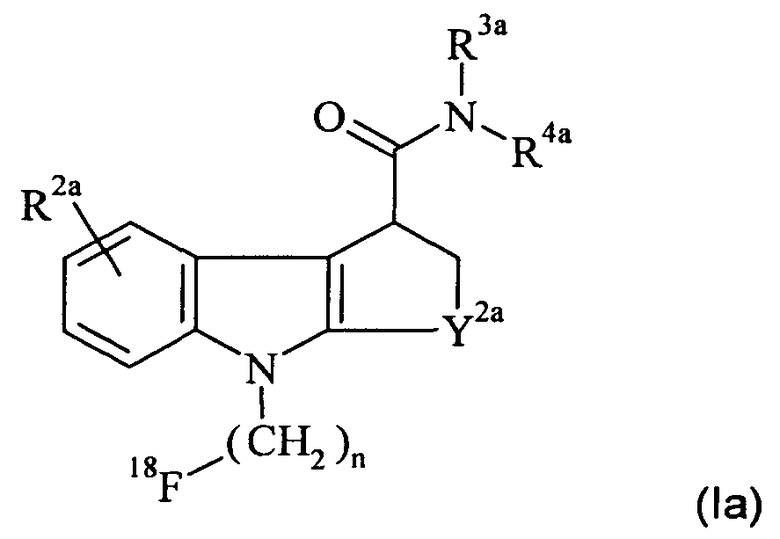
где R2a представляет собой водород, галогено или C1-3алкокси;
R3a и R4a независимо представляют собой метил, этил или бензил или вместе с атомом азота, к которому они присоединены, образуют пирролидинильное, пиперидинильное или азепанильное кольцо;
Y2a представляет собой CH2, CH2-CH2, CH(CH3)-CH2 или CH2-CH2-CH2 и
n равен 1, 2 или 3.
8. In vivo визуализирующий агент по п.7, где
R3a и R4a оба представляют собой этил, или R3a представляет собой метил и R4a представляет собой бензил, или вместе с атомом азота, к которому они присоединены, образуют азепанильное кольцо;
R2a представляет собой водород, метокси или фторо;
Y2a представляет собой CH2-CH2 или CH(CH3)-CH2 и
n равен 2.
9. In vivo визуализирующий агент по п.8, имеющий следующую химическую структуру:
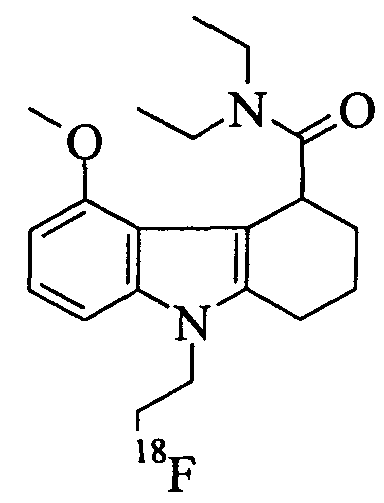
10. Соединение-предшественник для получения in vivo
визуализирующего агента по любому из пп.1-9, имеющее формулу II
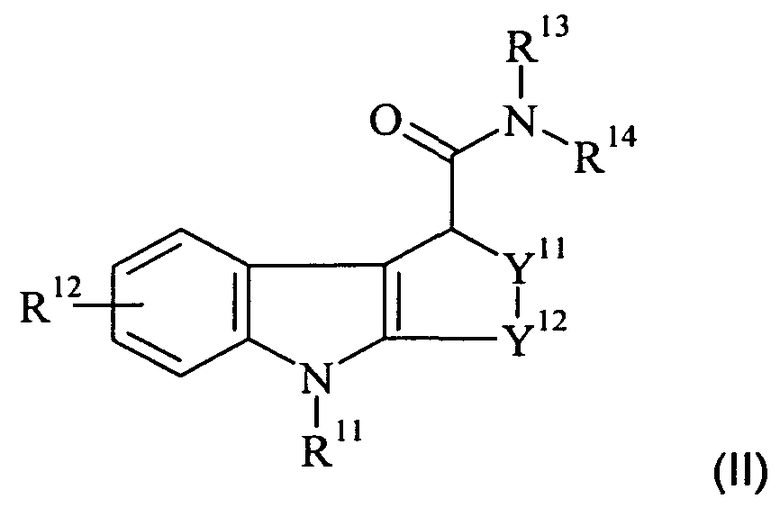
где R11 представляет собой C1-3-алкилен-LG, где LG представляет собой уходящую группу, выбранную из мезилата, тозилата и трифлата;
R12 представляет собой водород, гидроксил, галогено, C1-3алкил, C1-3алкокси, C1-3фторалкил или C1-3фторалкокси;
R13-14 независимо представляют собой C1-3алкил, C7-10аралкил или R13-14 вместе с атомом азота, к которому они присоединены, образуют азотсодержащее C4-6алифатическое кольцо;
Y11 представляет собой CH2 и
Y12 представляет собой CH2, CH2-CH2, CH(CH3)-CH2 или CH2-CH2-CH2.
11. Соединение-предшественник по п.10, представляющее собой соединение формулы IIа
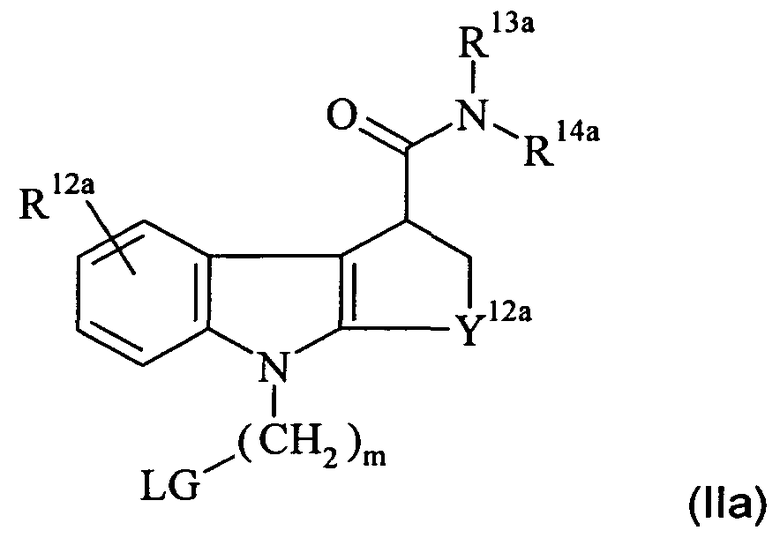
где LG является такой, как определено в п.10; и
R12a-14a, Y12a и m являются такими, как определено в любом из пп.7 и 8 для R2a-4a, Y2a и n соответственно.
12. Соединение-предшественник по п.10, представляющее собой соединение формулы IIb
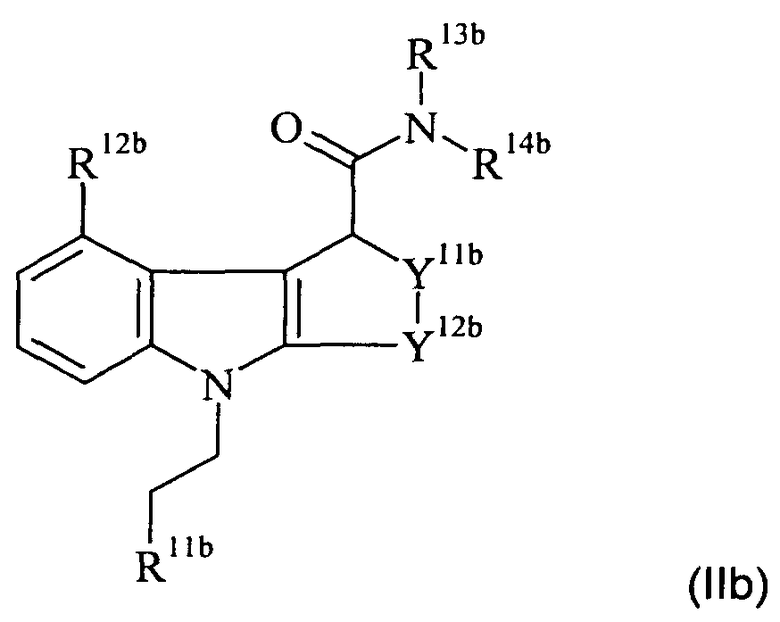
где R11b представляет собой мезилат, тозилат или трифлат;
R12b-14b являются такими, как определено для R12-14 в п.10, при условии, что R12b не является хлоро; и
Y11b-12b являются такими, как определено для Y11-12 в п.10.
13. Способ получения in vivo визуализирующего агента, содержащего атом, представляющий собой 18F радиоизотоп, по любому из пп.1-9, при котором:
(1) берут соединение-предшественник по любому из пп.10-12;
(2) берут источник, содержащий 18F радиоизотоп;
(3) приводят соединение-предшественник со стадии (1) во взаимодействие с радиоизотопом со стадии (2) с получением указанного in vivo визуализирующего агента.
14. Способ по п.13, который является автоматизированным.
15. Набор для осуществления способа по п.13, содержащий предшественник по любому из пп.10-12.
16. Кассета для осуществления способа по п.14, включающая:
(1) сосуд, содержащий соединение-предшественник по любому из пп.10-12; и
(2) средство для элюирования из сосуда со стадии (1) с источником радиоактивного изотопа 18F.
17. Кассета по п.16, дополнительно содержащая:
(3) ионообменный картридж для удаления избытка радиоизотопа.
18. Радиофармацевтическая композиция, обладающая аффинностью к PBR (периферическому бензодиазепиновому рецептору), содержащая in vivo визуализирующий агент по любому из пп.1-9 вместе с биосовместимым носителем в форме, подходящей для введения млекопитающему.
19. Способ визуализации in vivo для определения распределения и/или степени экспрессии PBR у субъекта, при котором:
(1) указанному субъекту вводят in vivo визуализирующий агент по любому из пп.1-9;
(2) позволяют указанному in vivo визуализирующему агенту связаться с PBR указанного субъекта;
(3) обнаруживают посредством метода in vivo визуализации сигналы, испускаемые радиоизотопом указанного in vivo визуализирующего агента;
(4) создают изображение, типичное для расположения и/или количества указанных сигналов; и
(5) определяют распределение и степень экспрессии PBR у указанного субъекта, где указанная экспрессия находится в прямой корреляции с указанными сигналами, испускаемыми указанным in vivo визуализирующим агентом.
20. Способ визуализации in vivo по п.19, который осуществляют неоднократно в схеме лечения указанного субъекта, где указанная схема включает введение лекарственного средства для борьбы с PBR-состоянием.
21. Способ диагностики состояния, при котором PBR активирован, включающий способ визуализации in vivo по п.19 вместе с дополнительной стадией (6) отнесения распределения и степени экспрессии PBR к конкретной клинической картине.
| WO 00/44751 A1 (SANOFI SYNTHELABO), 03.08.2000 | |||
| J.Med | |||
| Chem | |||
| Способ изготовления фанеры-переклейки | 1921 |
|
SU1993A1 |
| Bioorganic & Medicinal Chemistry, 12 (2004) 3569-3580 | |||
| Eur | |||
| J | |||
| Med.Chem | |||
| Предохранительное устройство для паровых котлов, работающих на нефти | 1922 |
|
SU1996A1 |
| FARMACO, 50, (5), 311-320, (1995) | |||
| Bioorganic & Medicinal Chemistry, 14, (2006), 3938-3946 | |||
| Bioorganic & Medicinal Chemistry, 14, (2006), 7582-7591 | |||
| УСТАНОВКА ДЛЯ КОНДИЦИОНИРОВАНИЯ ВОЗДУХА В ЖЕЛЕЗНОДОРОЖНЫХ ВАГОНАХ | 0 |
|
SU248734A1 |

