ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Известно несколько способов, применимых для синтеза пептидов. Классическим способом является жидкофазный пептидный синтез (LPPS), предпочтительный для получения больших количеств пептидов. Другим современным и широко используемым способом синтеза пептидов является твердофазный пептидный синтез (SPPS), где растущую пептидную цепь ковалентно присоединяют к смоле на твердой подложке, до отщепления пептидной цепи от подложки по достижении желаемой длины и последовательности. В этих способах реакционноспособные боковые цепи включенных аминокислот необходимо защищать во избежание других взаимодействий, кроме желаемого образования новых пептидных связей в растущем пептиде. В дополнение, во избежание побочных взаимодействий между добавляемыми аминокислотами, а также включения нескольких аминокислот на каждой стадии, в добавляемых аминокислотах обычно защищают α-аминогруппу. Таким образом, синтез становится одним из повторяющихся циклов снятия защиты с α-аминогруппы пептида, присоединенного к твердой фазе, с последующим сочетанием с одной аминокислотной единицей с защищенной α-аминогруппой.
Дегареликс представляет собой антагонист гонадотропин-рилизинг гормона (GnRH) для применения в лечении рака предстательной железы. Дегареликс обладает незамедлительным началом действия и подавляет гонадотропины, тестостерон и простатоспецифический антиген (PSA). Дегареликс представляет собой синтетический декапептид формулы Ac-D-2Nal-D-Phe(4Cl)-D-3Pal-Ser-4Aph(Hor)-D-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-NH2.
Пятая аминокислотная группировка с N-конца дегареликса соответствует искусственной аминокислоте Aph(L-hor). Aph(L-Hor) означает (L-гидрооротил)-4-аминофенилаланин. В данной области техники известно (Koedjikov, А. Н. et al., J. Chem. Soc. Perkin, Trans. 2, 1984, pages 1077-1081; Kaneti, J. et. al., Org. Biomol. Chem., 2004, pages 1098-1103), что в основных условиях соединения, содержащие дигидроурациловую группировку, проходят перегруппировку с образованием соединений, содержащих гидантоиновую группировку. Соответствующая перегруппировка Aph(L-Hor) показана ниже (слева сверху: дигидроурациловая группировка I, N-4-(L-гидрооротиламино)-фенилаланин, R=-CH2CHNH2COOH; справа внизу: гидантоиновая группировка II, N-4-[2-(5-гидантоил)-ацетил)-фенилаланин).
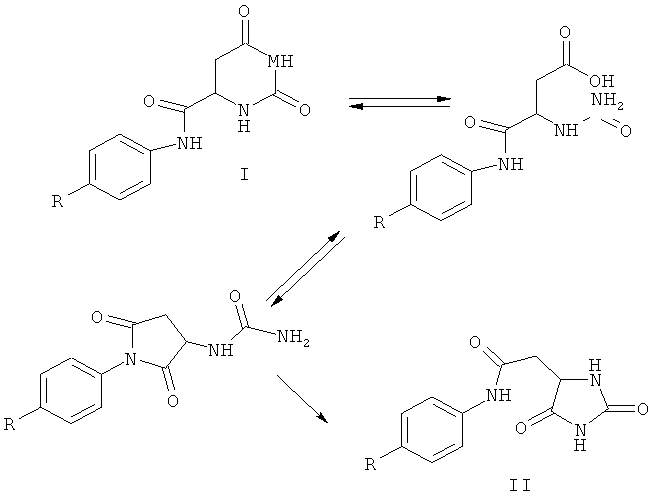
При перегруппировке происходит превращение дигидроурациловой группировки I в гидантоиновую группировку II. Ожидают, что группировка L-Hor в 4Aph(L-Hor), являясь группировкой дигидроурацилового типа, будет проходить такую перегруппировку в процессе получения дегареликса, при котором используют основные условия. Авторы изобретения подтвердили это путем приведения промежуточных продуктов пептидного синтеза, содержащих α-аминогруппу Fmoc-защищенной концевой 4Арh(Ноr), в контакт с NaOH или с органическим основанием дициклогексиламином (DCHA). Было обнаружено, что полученный при снятии защиты продукт содержал примесь, до нескольких % по массе, соответствующего гидантоинового продукта перегруппировки. Таким образом, можно ожидать, что в синтезе дегареликса промежуточный продукт Fmoc-4Aph(Hor)-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-NH-смола будет частично проходить перегруппировку с образованием Fmoc-X-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-МН-смола, где X представляет собой 4-([2-(5-гидантоил)]ацетиламино)-фенилаланин, при снятии защиты в основных условиях. Следовательно, можно, таким образом, ожидать, что продукт дегареликс, полученный с помощью Fmoc-4Aph(Hor)-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-NH-смолы, будет содержать примесь соответствующего количества Ac-D-2Nal-D-Phe(4Cl)-D-3Pal-Ser-X-D-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-NH2. Дегареликс является активным ингредиентом лекарственного средства для введения людям. По этой причине он не должен быть загрязнен какими-либо примесями, превышающими 0,3% по массе продукта. Таким образом, в дегареликсе, подходящем для применения у людей, количество гидантоинового побочного продукта более 0,3% по массе недопустимо. Поскольку побочный продукт, содержащий гидантоиновую группировку, структурно очень сходен с дегареликсом, их разделение затруднительно. Ожидают, что попытки разделения приведут к значительной потере продукта. Поэтому при получении дегареликса фармацевтической марки с использованием защитной группы Fmoc следует избегать основных условий.
Синтез дегареликса раскрыт в US 5925730 А. Предпочтительная защитная группа для α-аминогруппы в этом синтезе, которая была использована во всех Примерах, представляет собой трет-бутилоксикарбонильную группу (Вос). В дополнение, для этой цели раскрыт широкий спектр других хорошо известных защитных групп, таких как флуоренилметилоксикарбонильная группа (Fmoc).
Преимущество Вос-группы состоит в том, что защищенные ей α-аминогруппы можно деблокировать в кислых условиях стандартной обработкой трифторуксусной кислотой (TFA).
Недостатком TFA является ее высокая токсичность для человека, что создает риск для производственного персонала. Другим недостатком TFA является ее токсичность для окружающей среды, что либо делает ее утилизацию дорогостоящей, либо, при ее неправильной утилизации, приводит к загрязнению окружающей среды.
ЗАДАЧА ИЗОБРЕТЕНИЯ
Задачей изобретения является обеспечение способа получения дегареликса, не представляющего риска для здоровья человека, в частности менее опасного для здоровья человека, чем способ, раскрытый в US 5925730 А.
Также в задачу изобретения входит обеспечение способа получения дегареликса, не представляющего риска для окружающей среды, в частности менее опасного для окружающей среды, чем способ, раскрытый в US 5925730 А.
Кроме того, в задачу изобретения входит обеспечение способа получения дегареликса, менее дорогостоящего, чем способы, известные в данной области техники.
Другие аспекты задачи изобретения будут очевидны из следующего краткого изложения сущности изобретения, нескольких предпочтительных воплощений, раскрытых в форме примеров, и приложенной формулы изобретения.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Авторы изобретения неожиданно обнаружили, что фармацевтически чистый дегареликс может быть получен твердофазным синтезом с использованием Fmoc в качестве защитной группы для α-аминогруппы. «Фармацевтически чистый» означает продукт, не содержащий более 0,3% по массе любой отдельной примеси. Неожиданно, группировка Aph(L-Hor) не подвергается перегруппировке во время твердофазного синтеза, несмотря на проведение нескольких циклов Fmoc-защиты и снятия Fmoc-защиты в основных условиях.
Аминокислоты с Fmoc-защищенными α-аминогруппами присоединяют к смоле и затем друг к другу постадийным, циклическим и последовательность-зависимым образом. После каждой стадии присоединения аминокислоты проводят стадию снятия защиты для удаления защитной группы Fmoc и обеспечения возможности присоединения следующей аминокислоты. Снятие защиты проводят с использованием основания. Предпочтительно, для снятия защиты используют основание, представляющее собой пиперидин или пиперидин, замещенный алкилом, в органической среде.
Предпочтительно, проводят защиту боковых цепей для защиты боковых цепей особенно реакционноспособных или неустойчивых аминокислот во избежание побочных реакций и/или разветвления растущей молекулы.
Защитные группы боковых цепей удаляют по достижении полной длины растущего пептида.
Таким образом, согласно настоящему изобретению раскрыт способ получения дегареликса, Ac-D-2Nal-D-Phe(4Cl)-D-3Pal-Ser-4Aph(Hor)-D-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-NH2, содержащего 0,3% по массе или менее, в частности 0,1% по массе или менее, особенно 0,01% по массе или менее Ac-D-2Nal-D-Phe(4Cl)-D-3Pal-Ser-X-D-4Aph(Cbm)-Leu-lLys-Pro-D-Ala-NH2, где Х представляет собой 4-([2-(5-гидантоил)]ацетиламино)-фенилаланин, включающий постадийный синтез на твердой подложке, содержащей аминогруппу, связанную с подложкой, где стадия включает: обеспечение раствора аминокислоты или пептида, где α-аминогруппа защищена Fmoc; приведение подложки в контакт с этим раствором в присутствии реагента для образования пептидной связи между карбоксильной группой растворенной аминокислоты или пептида и аминогруппой, связанной с подложкой, на время, достаточное для образования указанной пептидной связи; удаление Fmoc путем приведения подложки в контакт с органическим основанием в органическом растворителе. Предпочтительное органическое основание представляет собой пиперидин. Другие предпочтительные органические основания представляют собой пиперидины, замещенные С-алкилом, в частности 2-алкилпиперидин, 3-алкилпиперидин, 2,4-диалкилпиперидин, 2,5-диалкилпиперидин, 2,6-диалкилпиперидин, где алкил представляет собой разветвленную или неразветвленную цепь из 1-6 атомов углерода, в частности метил или этил, особенно метил. Предпочтительный растворитель представляет собой диметилформамид. Другой предпочтительный растворитель представляет собой диэтилформамид. Другие предпочтительные растворители представляют собой N-метилпирролидон (NMP) или N,N-диметилацетамид (DMA). Предпочтительный реагент для образования пептидной связи включает N,N'-диизопропилкарбодиимид. Предпочтительно, аминогруппа, связанная с подложкой, представляет собой α-аминогруппу фрагмента дегареликса, связанного с подложкой. Также предпочтительно, пептид, защищенный Fmoc, представляет собой фрагмент дегареликса. Предпочтительная подложка представляет собой подложку, выбранную из смолы Rink amide AM и смолы Rink amide MBHA. Предпочтительный способ отделения дегареликса от подложки представляет собой обработку кислотой.
Согласно предпочтительному аспекту изобретения раскрыт дегареликс, полученный способом по изобретению, содержащий 0,3% по массе или менее Ac-D-2Nal-D-Phe(4Cl)-D-3Pal-Ser-X-D-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-NH2, где Х представляет собой 4-([2-(5-гидантоил)]ацетиламино)-фенилаланин, в частности 0,1% по массе или менее, особенно 0,01% по массе или менее.
Согласно другому предпочтительному аспекту изобретения раскрыто применение Fmoc в твердофазном синтезе для получения дегареликса, содержащего 0,3% по массе или менее, более предпочтительно 0,1% по массе или менее, наиболее предпочтительно 0,01% по массе или менее Ac-D-2Nal-D-Phe(4Cl)-D-3Pal-Ser-X-D-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-NH2, где Х представляет собой 4-([2-(5-гидантоил)]ацетиламино)-фенилаланин.
Изобретение будет теперь описано более подробно со ссылкой на графический материал и несколько предпочтительных воплощений, описанных в примерах.
ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВОПЛОЩЕНИЙ
Аббревиатуры
ПРИМЕР 1
Образование гидантоина в синтезе дегареликса. Перегруппировку гидрооротовой группы в гидантоинацетильную группу при получении дегареликса наблюдали на двух стадиях и при двух сериях основных условий.
Первая перегруппировка происходила при основных экстракциях сегмента Z-Ser(tBu)-4Aph(Hor)-D-4Aph(tBu-Cbm)-Leu-ILys(Boc)-Pro-D-Ala-NH2. pH корректировали до 9,1 в органической/водной двухфазной системе с использованием концентрированного раствора NaOH, что приводило к образованию 4,5% по массе гидантоинового аналога. По-видимому, механизм включал две стадии: (а) гидролиз 6-членной гидрооротовой группировки в основных условиях с последующим замыканием кольца с образованием 5-членного гидантоинового аналога в кислых условиях.
Вторую перегруппировку наблюдали при упаривании сегмента Z-Ser(tBu)-4Aph(Hor)-D-4Aph(tBu-Cbm)-Leu-OH-DCHA. После предшествующих экстракций Z-Ser(tBu)-4Aph(Hor)-D-4Aph(tBu-Cbm)-Leu-OH растворяли в смеси этилацетата и бутанола-2. Добавляли DCHA (2,5 экв.), поскольку сегмент выделяли в форме соли DCHA после выпаривания растворителя, за которым следовала стадия осаждения. В определенной партии были идентифицированы как гидантоиновый аналог, так и гидролизованная форма (упомянутая выше). Количественное определение гидантоина было невозможным ввиду недостаточного отделения от других продуктов посредством высокоэффективной жидкостной хроматографии (HPLC); образование гидролизованной формы происходило в количестве 1,34% по массе объединенных продуктов. Экспериментальные данные показали, что степень перегруппировки/гидролиза была связана с количеством DCHA, использованным в способе.
Следующий эксперимент обеспечил дополнительное доказательство нестабильности гидрооротовой группировки в основных условиях. Z-Ser(tBu)-4Aph(Hor)-D-4Aph(tBu-Cbm)-Leu-OH-DCHA (67 мМ) растворяли во влажном 2-BuOH с 167 мМ (2,5 экв.) DCHA при 31°С. Через 25 часов происходило образование 1,3% гидантоинового аналога и 0,3% гидролизованного промежуточного продукта.
ПРИМЕР 2
Стабильность дегареликса в DBU/DMF и пиперидине/DMF. Стабильность дегареликса исследовали в условиях, соответствующих условиям, используемым для удаления Fmoc-группы при SPPS. Известно, что гидрооротовая группа в боковой цепи 4Арh(Ноr), аминокислотного остатка №5 в последовательности дегареликса, чувствительна к основаниям и подвержена перегруппировке в гидантоинацетильную группу. Все способы SPPS, известные авторам изобретения, основаны на Вос-химии.
Образцы дегареликса растворяли в 20% пиперидине/DMF; 2% DBU в DMF и 2% DBU+5% воды в DMF соответственно. Через 20 часов образцы анализировали посредством HPLC и определяли количество гидантоинового аналога.
Использование 2% DBU/DMF приводило к образованию 1,8% гидантоина. Сходным образом, в присутствии 5% воды (имитирующей влажный DMF) количество было увеличено до 7%. Неожиданно, использование 20% пиперидина в DMF не приводило к образованию гидантоинового аналога, указывая на то, что эта смесь может быть полезной для SPPS дегареликса с использованием Fmoc.
ПРИМЕР 3
Синтез и очистка дегареликса с использованием Fmос-смолы Rink amide AM
Стадия 1. Fmoc-смолу Rink amide AM (64 г; замещение 0,67 ммоль/г) помещали в реакционный аппарат и промывали 1,9 л DMF. К набухшей смоле добавляют 250 мл 20% пиперидина в DMF и перемешивают 20 минут. Реакционный аппарат опорожняют через фильтр в дне аппарата, воздействуя на реакционный аппарат вакуумом, и проводят вторую обработку 250 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат снова опорожняют, воздействуя на него вакуумом, с последующей промывкой пептидной смолы с использованием 2 л DMF. Затем реакционный аппарат опорожняют, используя вакуум. Теперь пептидная смола готова к стадии 2.
Стадия 2. Раствор 27,0 г Fmoc-D-Ala-OH (2 экв.), 14,3 г HOBt и 13,2 мл DIC растворяют в 250 мл DMF и оставляют активироваться в течение 15 минут, после чего выливают в реакционный аппарат, содержащий пептидную смолу.
По истечении времени реакции 1 ч к раствору добавляют 2,2 мл NMM и реакции позволяют протекать еще час. Затем к смеси добавляют 30 мл уксусного ангидрида и 2 мл NMM и оставляют перемешиваться в течение 15 минут. Затем реакционный аппарат опорожняют, используя вакуум. Пептидную смолу промывают 2 л DMF. После воздействия на реакционный аппарат вакуумом с удалением DMF пептидную смолу обрабатывают 250 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат опорожняют, используя вакуум, и проводят вторую обработку 250 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат снова опорожняют, используя вакуум, и пептидную смолу промывают 2 л DMF. Теперь она готова к стадии 3.
Стадия 3. Раствор 29 г Fmoc-L-Pro-OH (2 экв.), 14,3 г HOBt и 13,2 мл DIC растворяют в 250 мл DMF и оставляют активироваться в течение 25 минут, после чего выливают в реакционный аппарат, содержащий пептидную смолу. После 75 минут реакции к раствору добавляют 2,2 мл NMM и реакции позволяют протекать еще час. Затем к смеси добавляют 30 мл уксусного ангидрида и 2 мл NMM и оставляют перемешиваться в течение 15 минут. Затем реакционный аппарат опорожняют, используя вакуум. Для промывки пептидной смолы используют DMF (2,6 л). После воздействия на реакционный аппарат вакуумом с удалением DMF пептидную смолу обрабатывают 250 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат опорожняют, используя вакуум, и проводят вторую обработку 250 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат снова опорожняют, используя вакуум, и пептидную смолу промывают 2 л DMF. Теперь она готова к стадии 4.
Стадия 4. Раствор 33 г Fmoc-L-ILys(Boc)-OH (1,5 экв.), 10,7 г HOBt и 10,1 мл DIC растворяют в 250 мл DMF и оставляют активироваться в течение 0,5 часа, после чего выливают в реакционный аппарат, содержащий пептидную смолу. После 2 ч реакции к раствору добавляют 2,2 мл NMM и реакции позволяют протекать еще час. Затем к смеси добавляют 30 мл уксусного ангидрида и 2,2 мл NMM и оставляют перемешиваться в течение 15 минут, после чего реакционный аппарат опорожняют, используя вакуум. Пептидную смолу промывают DMF (3 л). После воздействия на реакционный аппарат вакуумом с удалением DMF пептидную смолу обрабатывают 250 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат опорожняют, используя вакуум, и проводят вторую обработку 250 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат снова опорожняют, используя вакуум, и пептидную смолу промывают 3,5 л DMF. Теперь она готова к стадии 5.
Стадия 5. Раствор 38 г Fmoc-L-Leu-OH (2,5 экв.), 18 г HOBt и 16,8 мл DIC растворяют в 250 мл DMF и оставляют активироваться в течение 0,5 часа, после чего выливают в реакционный аппарат, содержащий пептидную смолу. После 2 ч реакции к раствору добавляют 2,2 мл NMM и реакции позволяют протекать еще 50 минут. Затем к смеси добавляют 30 мл уксусного ангидрида и 2 мл NMM и оставляют перемешиваться в течение 15 минут. Затем реакционный аппарат опорожняют, используя вакуум. Для промывки пептидной смолы используют DMF (2,6 л). После воздействия на реакционный аппарат вакуумом с удалением DMF пептидную смолу обрабатывают 250 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат опорожняют, используя вакуум, и проводят вторую обработку 250 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат снова опорожняют, используя вакуум, и пептидную смолу промывают 2,5 л DMF. Теперь она готова к стадии 6.
Стадия 6. Раствор 32 г Fmoc-D-4Aph(tBu-Cbm)-OH (1,5 экв.), 10,7 г HOBt и 10,1 мл DIC растворяют в 250 мл DMF и оставляют активироваться в течение 1 часа, после чего выливают в реакционный аппарат, содержащий пептидную смолу. После 20 минут реакции к раствору добавляют 22 мл NMM и реакции позволяют протекать еще 20 ч. Затем к смеси добавляют 30 мл уксусного ангидрида и 2 мл NMM и оставляют перемешиваться в течение 15 минут. Затем реакционный аппарат опорожняют, используя вакуум. Пептидную смолу отмывают 4 л DMF. После воздействия на реакционный аппарат вакуумом с удалением DMF пептидную смолу обрабатывают 250 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат опорожняют, используя вакуум, и проводят вторую обработку 250 мл 20% пиперидина в DMF продолжительностью 20 минут. Реакционный аппарат снова опорожняют, используя вакуум, и пептидную смолу промывают 3,4 л DMF. Теперь она готова к стадии 7.
Стадия 7. Раствор 35 г Fmoc-L-4Aph(L-Hor)-OH (1,5 экв.), 11 г HOBt и 10,1 мл DIC растворяют в 350 мл DMF и оставляют активироваться в течение 1 часа, после чего выливают в реакционный аппарат, содержащий пептидную смолу. После 50 минут реакции к раствору добавляют 2,2 мл NMM и реакции позволяют протекать еще 21,5 ч. Реакционный аппарат опорожняют, используя вакуум. Пептидную смолу промывают 4,4 л DMF. После воздействия на реакционный аппарат вакуумом с удалением DMF пептидную смолу обрабатывают 350 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат опорожняют, используя вакуум, и проводят вторую обработку 350 мл 20% пиперидина в DMF продолжительностью 20 минут. Реакционный аппарат снова опорожняют, используя вакуум, и пептидную смолу промывают 4,4 л DMF. Теперь она готова к стадии 8.
Стадия 8. Fmoc-L-Ser(tBu)-OH (2,5 экв.) (41 г), 17,9 г HOBt, 16,8 мл DIC и 4.9 мл NMM растворяют в 500 мл DMF и выливают в реакционный аппарат, содержащий пептидную смолу. Реакции позволяют протекать 3,5 часа. Затем реакционный аппарат опорожняют, используя вакуум. Пептидную смолу промывают 4,2 л DMF. После воздействия на реакционный аппарат вакуумом с удалением DMF пептидную смолу обрабатывают 375 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат опорожняют, используя вакуум, и проводят вторую обработку 375 мл 20% пиперидина в DMF продолжительностью 20 минут. Реакционный аппарат снова опорожняют, используя вакуум, и пептидную смолу промывают 4,2 л DMF. Теперь она готова к стадии 9.
Стадия 9. Раствор 25 г Fmoc-D-3Pal-OH (1,5 экв.), 10,7 г HOBt, 10,1 мл DIC и 4,9 мл NMM растворяют в 400 мл DMF и выливают в реакционный аппарат, содержащий пептидную смолу. Реакции позволяют протекать 4,5 часа. Затем реакционный аппарат опорожняют, используя вакуум. Пептидную смолу промывают 4,2 л DMF. После воздействия на реакционный аппарат вакуумом с удалением DMF пептидную смолу обрабатывают 375 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат опорожняют, используя вакуум, и проводят вторую обработку 375 мл 20% пиперидина в DMF продолжительностью 20 минут. Реакционный аппарат снова опорожняют, используя вакуум, и пептидную смолу промывают 4,2 л DMF. Теперь она готова к стадии 10.
Стадия 10. Раствор 27 г Fmoc-D-Phe(4CI)-OH (1,5 экв.), 10,7 г HOBt, 10,1 мл DIC и 4,9 мл NMM растворяют в 400 мл DMF и выливают в реакционный аппарат, содержащий пептидную смолу. Реакции позволяют протекать 10 часов. Реакционный аппарат опорожняют, используя вакуум. Смолу промывают 5,5 л DMF. После воздействия на реакционный аппарат вакуумом и удаления DMF пептидную смолу обрабатывают 375 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат опорожняют, используя вакуум, и проводят вторую обработку 375 мл 20% пиперидина в DMF продолжительностью 20 минут. Реакционный аппарат снова опорожняют, используя вакуум, и пептидную смолу промывают 5 л DMF. Теперь она готова к стадии 11.
Стадия 11. Раствор 28 г Fmoc-D-2Nal-OH (1,5 экв.), 10,7 г HOBt, 10,1 мл DIG и 4,9 мл NMM растворяют в 400 мл DMF и выливают в реакционный аппарат, содержащий пептидную смолу. Реакции позволяют протекать 2,5 часа. Реакционный аппарат опорожняют, используя вакуум. Пептидную смолу промывают 5,2 л DMF. После воздействия на реакционный аппарат вакуумом и удаления DMF пептидную смолу обрабатывают 375 мл 20% пиперидина в DMF в течение 20 минут. Реакционный аппарат опорожняют, используя вакуум, и проводят вторую обработку 375 мл 20% пиперидина в DMF продолжительностью 20 минут. Реакционный аппарат снова опорожняют, используя вакуум, и пептидную смолу промывают 5 л DMF. Теперь она готова к стадии 12.
Стадия 12. Ацетилимидазол (3 экв.) (14,5 г) и 4,9 мл NMM растворяют в 400 мл DMF и выливают в реакционный аппарат. Через 1,5 часа реакционный аппарат опорожняют, воздействуя на него вакуумом. Пептидную смолу промывают 5 л DMF и реакционный аппарат опорожняют, используя вакуум.
Стадия 13. Пептидную смолу промывают IPA и сушат в вакууме. Пептидную смолу отделяли (129,8 г; выход 96%).
Стадия 14. Сухую пептидную смолу (60 г) суспендируют в 600 мл TFA в течение 25 ч при комнатной температуре. Затем вливают в смесь 2,4 л воды, 620 г ацетата аммония, 600 мл этанола и 600 мл уксусной кислоты. рН смеси корректируют до 3-4 с использованием TFA и фильтруют.
Стадия 15. Проводят очистку продукта с использованием двухстадийного протокола очистки. На первой стадии используют колонку (2,5 см х 34 см), заполненную материалом С-18, с обращенной фазой и системой буферов, состоящей из буфера А (0,12% водная TFA) и буфера В (99,9% этанол). В колонку вносят объем фильтрованного раствора со стадии 14, соответствующий 1,6 г продукта. Очистку проводят с использованием ступенчатого градиента, начиная с 2-3 объемов колонки с 10% В, 5-7 объемов колонки с 29% В и градиента от 29% В до 50% В за 3 объема колонки при скорости потока 70 мл/мин. Эту процедуру повторяют до тех пор, пока не обработают весь фильтрованный раствор со стадии 14. Все собранные фракции анализируют посредством аналитической HPLC. Фракции, содержащие продукт с чистотой более 94%, объединяют. Вторую стадию очистки проводят с использованием колонки (2,5 см × 34 см), заполненной материалом С-18, с обращенной фазой и системой буферов, состоящей из буфера А (1% водная уксусная кислота), буфера В (99,9% этанол) и буфера С (0,5 М водный ацетат аммония). Из объединенных фракций, содержащих продукт, в колонку вносят количество, эквивалентное 1,3 г продукта, и очистку проводят с использованием ступенчатого градиента, начиная с 2-3 объемов колонки с 10% В+90% С с последующими 2-3 объемами колонки с 90% А+10% В. Продукт элюируют с использованием 24% В+76% А. Фракции, содержащие продукт с приемлемой степенью чистоты, объединяют и обессоливают с использованием той же колонки. Обессоливание проводят с использованием буфера А (1% водная уксусная кислота) и буфера В (99,9% этанол). В колонку вносят объем объединенной очищенной фракции, соответствующий 1,6 г продукта, и для отмывки продукта от ацетата аммония используют 2-3 объема колонки буфера А. Затем продукт элюируют с использованием 50% буфера А+50% буфера В. Раствор очищенного продукта, содержащий 50% этанола, концентрируют на роторном испарителе. После удаления всего этанола оставшийся раствор, содержащий продукт, лиофилизируют. Получают в общей сложности 11,8 г (общий выход 37%) дегареликса в форме пушистого твердого вещества. 4-([2-(5-Гидантоил)]ацетиламино)-фенилаланин в продукте выявлен не был (HPLC).
ПРИМЕР 4
Синтез и очистка дегареликса с использованием Fmoc-Rink amide MBHA
Осуществляли по существу так же как синтез и очистку в Примере 1. Отличия от способа из Примера 1:
а) вместо Fmoc-D-Aph(tBu-Cbm)-OH использовали Fmoc-D-Aph(Сbm)-ОН;
б) ацетилирование N-конца Н-D-2-Nal-пептид-смолы проводили с использованием уксусного ангидрида вместо ацетилимидазола;
в) при очистке вместо этанола использовали ацетонитрил. При HPLC 4-([2-(5-гидантоил)]ацетиламино)-фенилаланин в продукте выявлен не был.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПРОИЗВОДСТВА ДЕГАРЕЛИКСА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2602042C2 |
| АНТАГОНИСТЫ GNRH, МОДИФИЦИРОВАННЫЕ В ПОЛОЖЕНИЯХ 5 И 6 | 1998 |
|
RU2199549C2 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ СИНТЕЗА АНТАГОНИСТА РИЛИЗИНГ-ФАКТОРА ЛЮТЕИНИЗИРУЮЩЕГО ГОРМОНА (LHRH), СПОСОБ ИХ ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТА LHRH | 2002 |
|
RU2307125C2 |
| Получение дегареликса | 2013 |
|
RU2657444C2 |
| СИНТЕЗ ЛИРАГЛУТИДА | 2017 |
|
RU2766331C2 |
| СПОСОБ СИНТЕЗА ТЕРАПЕВТИЧЕСКИХ ПЕПТИДОВ | 2012 |
|
RU2625793C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДОДЕКАПЕПТИДА И ТРИПЕПТИД ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2340626C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДОВ, СОДЕРЖАЩИХ ЛИПОФИЛЬНО МОДИФИЦИРОВАННУЮ БОКОВУЮ ЦЕПЬ ЛИЗИНА | 2017 |
|
RU2755543C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА ЭКСЕНАТИДА | 2011 |
|
RU2458066C1 |
| АГОНИСТЫ ОКСИТОЦИНОВЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2539692C2 |
Изобретение относится к способу получения дегареликса. Предложен постадийный синтез дегареликса, содержащего 0,3% по массе или менее аналога 4-([2-(5-гидантоил)]ацетиламино)-фенилаланина, на твердой подложке, содержащей аминогруппу, который включает стадии: обеспечение раствора аминокислоты или пептида, где α-аминогруппа защищена флуоренилметилоксикарбонильной группой (Fmoc); приведение подложки в контакт с этим раствором в присутствии реагента для образования пептидной связи между карбоксильной группой аминокислоты или пептида и твердой подложкой, содержащей аминогруппу, связанную с этой подложкой; удаление Fmoc путем приведения подложки в контакт с органическим основанием, в частности пиперидином, в органическом растворителе. 2 н. и 8 з.п. ф-лы, 4 пр.
1. Способ получения дегареликса, Ac-D-2Nal-D-Phe(4Cl)-D-3Pal-Ser-4Aph(Hor)-D-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-NH2, содержащего 0,3% по массе или менее, в частности 0,1% по массе или менее, особенно 0,01% по массе или менее Ac-D-2Nal-D-Phe(4Cl)-D-3Pal-Ser-X-D-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-NH2, где X представляет собой 4-([2-(5-гидантоил)]ацетиламино)-фенилаланин, включающий постадийный синтез на твердой подложке, содержащей аминогруппу, связанную с этой подложкой, где стадия включает: обеспечение раствора аминокислоты или пептида, где α-аминогруппа защищена флуоренилметилоксикарбонильной группой (Fmoc); приведение подложки в контакт с этим раствором в присутствии реагента для образования пептидной связи между карбоксильной группой растворенной аминокислоты или пептида и аминогруппой, связанной с подложкой, на время, достаточное для образования указанной пептидной связи; удаление Fmoc путем приведения подложки в контакт с органическим основанием, выбранным из пиперидина и пиперидина, замещенного C-алкилом, в частности 2-алкилпиперидина, 3-алкилпиперидина, 2,4-диалкилпиперидина, 2,5-диалкилпиперидина, 2,6-диалкилпиперидина, где алкил представляет собой разветвленную или неразветвленную цепь из 1-6 атомов углерода, в частности метил или этил, особенно метил, в органическом растворителе.
2. Способ по п.1, где органическое основание представляет собой пиперидин.
3. Способ по п.1, где органический растворитель представляет собой диметилформамид.
4. Способ по п.1, где реагент для образования пептидной связи включает N,N'-диизопропилкарбодиимид.
5. Способ по п.1, где аминогруппа, связанная с подложкой, представляет собой α-аминогруппу фрагмента дегареликса, связанного с подложкой.
6. Способ по п.1, где пептид, защищенный Fmoc, представляет собой фрагмент дегареликса.
7. Способ по п.1, где подложка выбрана из смолы Rink amide AM и смолы Rink amide MBHA.
8. Способ по любому из пп.1-7, включающий отделение дегареликса от подложки путем обработки кислотой.
9. Способ получения дегареликса, содержащего 0,3% по массе или менее, особенно 0,01% по массе или менее Ac-D-2Nal-D-Phe(4Cl)-D-3Pal-Ser-X-D-4Aph(Cbm)-Leu-ILys-Pro-D-Ala-NH2, где X представляет собой 4-([2-(5-гидантоил)]ацетиламино)-фенилаланин, включающий постадийный синтез на твердой подложке, содержащей аминогруппу, связанную с этой подложкой, где стадия включает: обеспечение раствора аминокислоты или пептида, где α-аминогруппа защищена флуоренилметилоксикарбонильной группой (Fmoc); приведение подложки в контакт с этим раствором в присутствии реагента для образования пептидной связи между карбоксильной группой растворенной аминокислоты или пептида и аминогруппой, связанной с подложкой, на время, достаточное для образования указанной пептидной связи; удаление Fmoc путем приведения подложки в контакт с пиперидином в растворителе, выбранном из диметилформамида, диэтилформамида, N,N-диметилацетамида, N-метилпирролидона.
10. Способ по п.9, в котором указанным реагентом является N,N'-диизопропилкарбодиимид.
| АНТАГОНИСТЫ GNRH, МОДИФИЦИРОВАННЫЕ В ПОЛОЖЕНИЯХ 5 И 6 | 1998 |
|
RU2199549C2 |
| KANETI J ET AL: "Thorpe-Ingold effects in cyclizations to five-membered and six-membered rings containing planar segments | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| CESCATO RENZO ET AL: | |||

