В общих чертах, настоящее изобретение относится к пептидам, которые являются антагонистами гонадотропинрилизинг-гормона человека (GnRH) и обладают полезными физическими, химическими и биологическими свойствами. Более конкретно, настоящее изобретение относится к декапептидам, которые ингибируют функцию половых желез и способствует высвобождению стероидных гормонов прогестерона и тестостерона в течение продолжительного периода времени, и к способам введения фармацевтических композиций, содержащих указанные декапептиды и предназначенные для этих целей, а в частности к лечению состояний, возникающих в результате гиперсекреции половых стероидных гормонов.
Предпосылки создания изобретения
Фолликулостимулирующий гормон (FSH) и лютеинизирующий гормон (LH), иногда называемые также гонадотропинами или гонадотропными гормонами, высвобождаются из гипофиза, который соединен с гипоталамусом гипофизарной ножкой.
Высвобождение гормона передней долей гипофиза обычно необходимо перед высвобождением гормонов, продуцируемых гипоталамусом, таких как декапептид GnRH.
Введение аналогов GnRH, которые являются антагонистами нормальной функции GnRH, было использовано для подавления секреции гонадотропинов, в основном у млекопитающих, а также для подавления или замедления овуляции.
Поиск лучших антагонистов GnRH привел к получению антида (Antide), то есть [Ac-D-2Nal1, D-4ClPhe2, D-3Pal3, Lys(Nic)5, D-Lys(Nic)6, Ilys8, D-Ala10] -GnRH; и цетрореликса (Cetrorelix), то есть [Ac-D-2Nal1, D-4ClPhe2, D-3Pal3, D-Cit6, D-Ala10]-GnRH. В патенте США N5516887 описаны антагонисты GnRH, которые, как указывается, являются более эффективными в снижении уровней тестостерона в плазме, чем антид, например [Ac-D-2Nal1, D-4ClPhe2, D-3Pal3, D-Nε-карбамоил Lys6, Ilys8, D-Ala10]-GnRH, которые были названы антареликсом.
В патенте США N5296468, изданном 22 марта 1994, описаны создание и синтез ряда антагонистов GnRH, у которых боковые цепи выбранных остатков реагируют с образованием цианогуанидиновых фрагментов некоторые из которых затем спонтанно превращаются в нужный гетероцикл, например 3-амино-1,2,4-триазол(atz). Эти цианогуанидиновые фрагменты происходят от омега-аминогруппы в боковой цепи аминокислоты, такой как лизин, орнитин, 4-аминофенилаланин (4Арh) или ее удлиненный вариант, такой как 4-аминогомофенилаланин (4Aph). Антагонисты GnRH, имеющие такие значительно модифицированные или неприродные аминокислоты, а в 5- и 6-положениях обнаруживают хорошие биологические свойства, и антагонисты, которые происходят от Aph, считаются, в основном, предпочтительными. А особенно предпочтительным является азалин В, то есть [Ac-D-2Nal1, D-4ClPhe2, D-3Pal3, 4Aph(atz)5, D-4Aph(atz)6, Ilys8, D-Ala10] -GnRH. В патенте США N5506207 описаны биологически активные антагонисты GnRH, у которых боковые цепи аминозамещенного фенилаланина в 5- и 6-положениях ацилированы; а особенно эффективным декапептидом является ацилин, то есть, [Ac-D-2Nal1, D-4ClPhe2, D-3Pal3, 4Aph(Ac)5, D-4Aph(Ac)6, Ilys8, D-Ala10] -GnRH.
Несмотря на привлекательные свойства этой группы антагонистов GnRH, поиски еще более сильных антагонистов GnRH все еще продолжаются, особенно таких антагонистов, которые обладают длительным биологическим действием. В большинстве случаев оказывается важным, чтобы пептидный аналог обладал продолжительной активностью по отношению к секреции LH, то есть свойством, которое может быть усилено устойчивостью пептида к деградации протеолитическими ферментами в организме как при кратковременном, так и при длительном курсе лечения. Кроме того, для облегчения введения этих соединений млекопитающим, особенно человеку, без значительного гелеобразования, является крайне желательным, чтобы такие декапептиды-антагонисты GnRH обладали высокой степенью растворимости в воде при нормальных физиологических рН, то есть при рН примерно от 5 до 7,4.
Краткое описание изобретения
Было неожиданно обнаружено, что некоторые другие модификации остатка в 5-положении или остатков в 5- и 6-положениях в антагонистах GnRH того подкласса, к которому относятся центрореликс, антареликс, ацилин, антид и другие, приводят к получению соединений, которые при их подкожном введении обладают особенно преимущественным свойством, заключающимся в продолжительности их биологической активности. Такие модификации были введены в остаток 4аминоРhе или в его эквивалент 4Aph или в 4-аминометилфенилаланин (4Amf), где первичная аминогруппа связана с метильной группой, присоединенной в 4- или параположении. В таких модификациях аминогруппа боковой цепи реагирует с изоцианатом с образованием карбамидной группы или реагирует с гетероциклической карбоновой кислотой, содержащей, по меньшей мере, 2 атома азота, расположенные таким образом, что они составляют карбамидную часть. Предпочтительными гетероциклическими реагентами являются D- или L-гидрооротовая кислота (Ноr) (С4N2H5(0)2СООН) и D- или L-2-имидазолидон-4-карбоновая кислота (Imz) (С3N2H5(0) СООН).
В основном было обнаружено, что декапептиды-антагонисты GnRH, имеющие нижеследующую формулу, и их близкородственные аналоги и фармацевтически приемлемые соли обладают улучшенными фармакологическими свойствами, а в частности пролонгированной биологической активностью:
X-D-Nal-(A)D-Phe-D-Pal-Ser-Xaa5-Xaa6-Leu-Xaa8-Pro-Xaa10,
где Х представляет собой ацильную группу, имеющую 7 атомов углерода или Q, где Q представляет собой 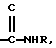 а R представляет собой Н или низший алкил;
а R представляет собой Н или низший алкил;
А представляет собой 4Сl, 4F, 4Br, 4NO2, 4СН3, 4OСН3, 3,4Cl2 или CαMe4Cl;
Xaa5 представляет собой Aph (Q1) или Amf (Q2), где Q1 представляет собой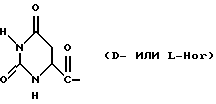
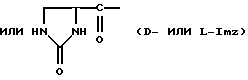
Хаа6 представляет собой D-Aph (Q2), D-Amf (Q2), D-Lys (Nic), D-Cit, D-Hci или D-Pal, где Q2 представляет собой For, Ac, 3-амино-1,2,4-триазол, Q или Q1;
Xaa8 представляет собой Lys(ipr), Arg, Наr, Arg (Et2) или Наr (Et2); и
Хаа10 представляет собой D-Ala-NH2, D-Ala-ол, Ala-ол, NHCH2CH3, Gly-NH3, AzaGly-NH2, Ala-NH2, Agl-NH2, D-Agl-NH2, Agl(Me)-NH2 или D-Agl(Me)-NH3, при условии, однако, что α-аминогруппа Xaa5 может быть, но не обязательно, метилированной; и, кроме того, при условии, что, если Xaa6 содержит D-, или L-Hor, или D-, или L-Imz, то Хаа5 может иметь Ac, For или 3-амино-1,2,4-триазол, в качестве Q1; а если Хаа6 содержит Q, то Хаа5 может также содержать Q.
В другом своем аспекте настоящее изобретение относится к способу in vivo- или in vitro-диагностики состояний, при которых GnRH вызывает избыточную секрецию гормонов или рост опухоли, где указанный способ предусматривает введение пептида-антагониста GnRH вышеописанного типа и мониторинг секреции гормона или пролиферации опухолевых клеток.
В еще одном аспекте настоящее изобретение относится к промежуточному соединению для получения пептида-антагониста GnRH, имеющему формулу
X1D-Nal-(A)D-Phe-D-Pal-Ser (X2)-Xaa5-Xaa6-Leu-Lys(ipr)(X4)-Рrо-Х5,
где X1 - α-аминозащитная группа;
А представляет собой 4Cl или 4F;
X2 представляет собой Н или гидроксилзащитную группу;
Хаа5 представляет собой Aph(Q1) или Amf(Q1), где Q1 представляет собой D-изомер, L-изомер или смесь D/L-изомеров любого из: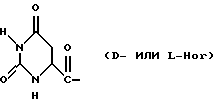
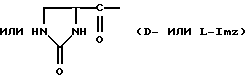
Хаа6 представляет собой D-Aph(Q2), D-Amf(Q2) или D-Pal, где Q2 представляет собой Ac, Q1, карбамоил или метилкарбамоил;
X4 представляет восприимчивую к действию кислоты аминозащитную группу; и
X5 представляет D-Ala-, Gly-, Ala-, Agl-, D-Agl-, Agl(Me)- или D-Agl(Me)-полимерный носитель; N(Et)-полимерный носитель; амид D-Ala, Gly или Ala; этиламид; AzaGly-NH2, или ОН, при условии, однако, что α-аминогруппа Хаа5, может быть, но не обязательно, метилированной.
Эти антагонисты являются особенно эффективными для подавления секреции гонадотропинов и в качестве регуляторов способности к оплодотворению у человека, поскольку они обладают пролонгированной активностью, то есть активностью, продолжающейся вплоть до значительного подавления секреции LH, по меньшей мере, примерно в течение 4 дней. Они обладают улучшенной растворимостью в водных буферах при физиологических рН и допустимыми побочными эффектами в отношении стимуляции высвобождения гистамина, то есть лучшими эффектами по сравнению с суперагонистами GnRH, используемыми в современной клинической практике; и, кроме того, они обладают минимальной способностью к гелеобразованию после их подкожной (п/к) инъекции в эффективной концентрации. Эти антагонисты GnRH могут быть также с успехом использованы в анафилактоидном анализе, вызывая лишь образование относительно небольшого волдыря. В результате этого указанные пептиды являются особенно подходящими для введения млекопитающим, в частности человеку, в качестве регуляторов оплодотворяющей способности, и для лечения патологических состояний, таких как преждевременное половое созревание, гормонзависимая неоплазия, дисменорея, эндометриоз, стероидзависимые опухоли, а также при других показаниях для кратковременного и длительного курса лечения состояний, упомянутых выше. Они могут быть также использованы для диагностики.
Поскольку эти антагонисты GnRH являются легко растворимыми при физиологических значениях рН примерно в пределах от 5 до 7,4, то они могут быть изготовлены и введены в концентрированной форме, особенно при рН примерно от 5 до 7. Благодаря своей полярной природе эти антагонисты являются пригодными для использования в препаратах пролонгированного действия, изготавливаемых на основе известных сополимеров. Поскольку эти антагонисты GnRH оказывают эффективное подавляющее действие на LH и FSH в течение продолжительного периода времени, то они являются также особенно эффективными для контрацептивной обработки млекопитающих-самцов (с необязательным введением тестостерона) и для лечения стероидзависимых опухолей.
Подробное описание изобретения
За последние 10-12 лет были глубоко изучены конкретные свойства каждого из 10 остатков последовательности GnRH с точки зрения создания эффективного антагониста, и в результате этих исследований было обнаружено, что имеются различные эквивалентные остатки, которые могут быть выбраны в качестве замены, и такие замены других остатков одним из этих эквивалентов не оказывают существенного неблагоприятного действия на биологическую активность антагонистов декапептида GnRH. Такие эквивалентные замены могут быть осуществлены в антагонистах GnRH настоящего изобретения.
Так, например, было показано, что в основном включение паразамещенного остатка D-Phe или остатка 2,4-дихлорзамещенного D-Phe или D-CαMe4ClPhe или остатка D-пентаметил (Ме5) Phe в 2-положении значительно усиливает GnRH-антагонистическую активность; однако конкретный выбор кольцевого заместителя из группы, включающей в себя хлор, фтор, бром, нитро, метил и алкокси, имеет лишь относительно небольшое значение. Поэтому такие остатки в 2-положении рассматриваются как эквиваленты D-4ClPhe, которые обычно используются в данном случае. Phe7 считается эквивалентным Leu7. N-конец является предпочтительно N-ацилированным, предпочтительно ацетилом (Ас), но он может быть ацилирован также и другими ацильными группами, имеющими до 7 атомов углерода, например формилом (For), акрилилом (Асr), н-пропионилом (Рn), бутирилом (By), валерилом (Vl), винилацетилом (Vac} и бензоилом (Bz); и альтернативно он может быть модифицирован замещенным или незамещенным карбамоилом. В качестве эквивалентов могут быть также рассмотрены и другие более длинные ацильные группы, но они являются менее предпочтительными. Для повышения водорастворимости α-аминогруппа на остатке в 5-положении может быть, но необязательно, метилирована, как описано в патенте США N5110904, но такая модификация может приводить к сокращению продолжительности подавления секреции LH и к большей вероятности высвобождения гистамина. С-конец представляет собой предпочтительно D-Ala-NH2, D-Ala-ол или Ala-ол; однако вместо них могут быть использованы Gly-NH2, NHCH2CH3, AzaGly-NH2, Ala-NH2, Agl-NH2, D-Agl-NH2, Agl (Me)-NH2, или D-Agl(Me)-NH2, поскольку они считаются известными эквивалентами.
Как указывалось выше, настоящее изобретение относится к семейству антагонистов GnRH, имеющих следующую формулу:
X-D-Nal-(A)D-Phe-D-Pal-Ser-Xaa5-Xaa6-Leu-Xaa8-Pro-Xaa10,
и к их фармацевтически приемлемым солям,
где Х представляет собой For, Ac, Acr, Pn, By, Vl, Vac, Bz и Q, где Q представляет собой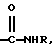
и где R представляет собой Н или низший алкил;
А представляет собой 4Сl, 4F, 4Br, 4NO2, 4СН3, 4OСН3, 3,4Cl2 или CαMe4Cl;
Хаа5 представляет собой Aph(Q1) или Amf(Q1), где Q1 представляет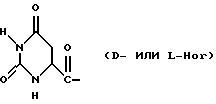
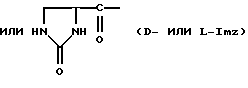
Хаа6 представляет собой D-Aph(Q2), D-Amf(Q2), D-Lys (Nic), D-Cit, D-Hci или D-Pal, где Q2 представляет собой For, Ac, 3-амино-1,2,4-триазол, Q или Q1;
Xaa8 представляет собой Lys(ipr), Arg, Har, Arg(Et2) или Har (Et2); и
Хаа10 представляет собой D-Ala-NH2, D-Ala-ол, Ala-ол, NHCH2СН3, Gly-NH2, AzaGly-NH2, Ala-NH2, Agl-NH2, D-Agl-NH2, Аgl(Ме)-NН2 или D-Agl(Me)-NH2, при условии, однако, что α-аминогруппа Хаа5 может быть, но не обязательно, метилированной.
В близкородственном семействе антагонистов GnRH Xaa5 может иметь либо Ас, либо For, либо 3-амино-1,2,4-триазол в качестве Q1, и в этом случае Хаа6 включает в себя Q2 в форме D-, или L-Hor, или L-Imz.
В другом близкородственном семействе антагонистов GnRH, где Хаа6 содержит Q, Хаа5 может также содержать Q.
D-Nal означает D-изомер аланина, который замещен нафтилом на атоме β-углерода, то есть он также означает 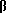 -D-Nal или 3-D-Nal. При этом предпочтительно использовать D-2Nal, где он присоединен к нафталину в 2-положении на кольцевой структуре; однако может быть также использован D-1Nal. D-Cpa представляет собой хлор-D-Phe и D-4ClPhe, то есть предпочтительным является D-4Cpa. D-Pal представляет собой D-изомер аланина, который замещен пиридилом на атоме β-углерода; при этом предпочтительно он связан в 3-положении на пиридиновом кольце, то есть D-3Pal (β-3-пиридил-D-Ala), хотя вместо этого может быть использован D-2Pal (β-2-пиридил-D-Ala). 4Aph означает 4NH2Phe, где аминозаместитель на фенильном кольце находится в 4-положении; причем в этих аналогах 3NH2Phe(3Aph) рассматривается как его эквивалент. Более того, с точки зрения биологической активности, очевидно, что 4NH2Рhе также является эквивалентом. 4Amf означает 4NH2CH2Phe, где имеется метиленовая связь с аминогруппой боковой цепи; 3NH2CH2Phe(3Amf) считается эквивалентом. Hor или L-Hor означает L-гидрооротил, a Imz или L-Imz означает L-2-имидaзoлидoн-4-кapбoнил-, либо каждый из них может быть использован в виде D-изомера или смеси D/L-изомеров. "atz" означает 3-амино-1,2,4-триазол. Aph(atz) также известен под более точным химическим названием 4-(3'-амино-1H-1',2',4'-триазолил-5'-ил)аминофенилаланин. Lys(Nic) означает Nε-никотиноиллизин, то есть ε-аминогруппа Lys ацилирована 3-карбоксипиридином. D-Cit означает D-изомер цитруллина, a D-Hci означает D-изомер гомоцитруллина, который также представляет собой D-Nε-карбамоил-лизин. Ilys или Lys(ipr) означает Nε-изопропиллизин, где ε-аминогруппа Lys алкилирована. Ala-ол означает аланитол, то есть СН3СН(NН2)СН2ОН, a AzaGly-NH2 означает NHNHCONH2. Наr означает гомоаргинин, Аgl означает α-аминоглицин. Cbm означает карбамоил, а MeCbm oзначает метилкарбамоил или -СОNНСН3. Низший алкил означает C1-C5, предпочтительно C1-С3, а более предпочтительно C1- или С2-группу, то есть метил (Me) или этил (Et).
-D-Nal или 3-D-Nal. При этом предпочтительно использовать D-2Nal, где он присоединен к нафталину в 2-положении на кольцевой структуре; однако может быть также использован D-1Nal. D-Cpa представляет собой хлор-D-Phe и D-4ClPhe, то есть предпочтительным является D-4Cpa. D-Pal представляет собой D-изомер аланина, который замещен пиридилом на атоме β-углерода; при этом предпочтительно он связан в 3-положении на пиридиновом кольце, то есть D-3Pal (β-3-пиридил-D-Ala), хотя вместо этого может быть использован D-2Pal (β-2-пиридил-D-Ala). 4Aph означает 4NH2Phe, где аминозаместитель на фенильном кольце находится в 4-положении; причем в этих аналогах 3NH2Phe(3Aph) рассматривается как его эквивалент. Более того, с точки зрения биологической активности, очевидно, что 4NH2Рhе также является эквивалентом. 4Amf означает 4NH2CH2Phe, где имеется метиленовая связь с аминогруппой боковой цепи; 3NH2CH2Phe(3Amf) считается эквивалентом. Hor или L-Hor означает L-гидрооротил, a Imz или L-Imz означает L-2-имидaзoлидoн-4-кapбoнил-, либо каждый из них может быть использован в виде D-изомера или смеси D/L-изомеров. "atz" означает 3-амино-1,2,4-триазол. Aph(atz) также известен под более точным химическим названием 4-(3'-амино-1H-1',2',4'-триазолил-5'-ил)аминофенилаланин. Lys(Nic) означает Nε-никотиноиллизин, то есть ε-аминогруппа Lys ацилирована 3-карбоксипиридином. D-Cit означает D-изомер цитруллина, a D-Hci означает D-изомер гомоцитруллина, который также представляет собой D-Nε-карбамоил-лизин. Ilys или Lys(ipr) означает Nε-изопропиллизин, где ε-аминогруппа Lys алкилирована. Ala-ол означает аланитол, то есть СН3СН(NН2)СН2ОН, a AzaGly-NH2 означает NHNHCONH2. Наr означает гомоаргинин, Аgl означает α-аминоглицин. Cbm означает карбамоил, а MeCbm oзначает метилкарбамоил или -СОNНСН3. Низший алкил означает C1-C5, предпочтительно C1-С3, а более предпочтительно C1- или С2-группу, то есть метил (Me) или этил (Et).
Хотя предпочтительные D-изомеры конкретно описаны для введения в 6-положение этих антагонистов GnRH, однако следует отметить, что в результате интенсивных исследований, проводимых в этой области за последние два десятилетия, стало известно много эквивалентных D-изомеров. Такие известные замены для D-изомеров, которые получают путем специфических замен в 5-положении, описанных в настоящей заявке, могут оказаться приемлемыми и не будут оказывать неблагоприятного действия на биологическую активность, а поэтому они также, но не обязательно, могут быть использованы.
Предпочтительный подвид антагонистов GnRH имеет формулу
X-D-Nal-(A)D-Phe-D-Pal-Ser-Xaa5-Xaa6-Leu-Lys(ipr)-Pro-Хаа10,
и их фармацевтически приемлемые соли, где:
X представляет собой For, Ac, Acr, Pn, By, Vl, Vac, Bz или Q,
где Q представляет собой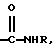
и где R представляет собой Н или низший алкил;
А представляет собой 4Сl или 4F;
Хаа5 представляет собой Aph(Q1) или Amf(Q1), где Q1 представляет собой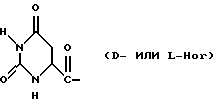
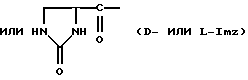
Хаа6 представляет собой D-Aph(Q2), D-Amf(Q2), D-Cit, D-Lys(Nic) или D-Pal, где Q2 представляет собой For, Ac, Q или Q1; и
Хаа10 представляет собой D-Ala-NH2, D-Ala-ол, Ala-ол, NHCH2CH3 или Gly-NH2.
Другой предпочтительный подвид антагонистов GnRH имеет формулу
X-D-Nal-D-4Cpa-D-Pal-Ser-Xaa5-Xaa6-Leu-Lys(ipr)-Pro-Хаа10
и их фармацевтически приемлемые соли, где:
Х представляет собой Ас или Q,
где Q представляет собой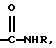
и где R представляет собой Н или метил;
Хаа5 представляет собой Aph(Q1) или Amf(Q1), где Q1 представляет собой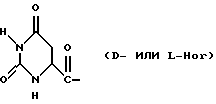
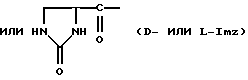
Хаа6 представляет собой D-Арh(Q2), D-Amf(Q2) или D-Pal, где Q2 представляет собой Ac, Q или Q1; и
Xaa10 представляет собой D-Ala-NH2, D-Ala-ол или Ala-ол.
Другой предпочтительный подвид антагонистов GnRH имеет формулу
MeCbm-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(Ноr)-D-Xaa6-Leu-ILys-Pro-Xaa10
и их фармацевтически приемлемые соли,
где D-Хаа6 представляет собой D-4Amf(Q1), D-Aph(Q1) или D-3Раl, где Q1 представляет собой D-Hor или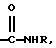
и где R представляет собой Н или низший алкил, а предпочтительно Н или метил;
Xaa10 представляет собой D-Ala-NH2, D-Ala-ол или Ala-ол.
Соединения настоящего изобретения могут быть синтезированы методом классического пептидного синтеза в растворе, и такой синтез является предпочтительным для больших количеств продукта. Для получения ограниченных количеств, например менее 1 кг, может оказаться предпочтительным твердофазный синтез. Защитные группы боковой цепи, хорошо известные специалистам, предпочтительно присутствуют как часть любой аминокислоты, которая имеет особенно реакционноспособную или неустойчивую боковую цепь, при присоединении ее к цепи, присутствующей на полимере. Такой синтез позволяет получить полностью защищенный промежуточный пептидополимер, такой как
X1-D-Nal-(A)D-Phe-D-Pal-Ser(X2)-Xaa5-Xaa6-Leu-Lys(ipr)(X4)-Pro-(X5). Один из примеров химического промежуточного соединения, которое может быть использовано для синтеза антагониста GnRH, имеющего нужный остаток в 5- или 6-положениях, содержащих гидрооротил или ему подобные, представлено формулой
X1-D-Nal-D-4Cpa-D-Pal-Ser-(X2)-Aph(X3)-D-Aph(X3)-Leu-Ilys(X4)-Pro-(X5)
При синтезе пептидных промежуточных соединений, имеющих такую формулу, и других аналогов могут быть использованы группы Х1-Х5, определенные ниже.
X1 представляет собой аминозащитную группу известного типа, которая обычно используется в ступенчатом синтезе полипептидов, и если Х в пептиде нужного состава представлят собой конкретную ацильную группу, то эта группа может быть использована в качестве защитной группы. Классами α-аминозащитных групп, охватываемых X1, являются (1) защитные группы ацильного типа, такие как формил (For), трифторацетил, фталоил, п-толуолсульфонил (Tos), бензоил (Bz), бензолсульфонил, дитиасукциноил (Dts) о-нитрофенилсульфенил (Nps), тритилсульфенил, о-нитрофеноксиацетил, акрилил (Асr), хлорацетил, ацетил (Ас) и у-хлорбутирил; (2) ароматические защитные группы типа уретана, например бензилоксикарбонил {Z}, флуоренилметилоксикарбонил (Fmoc) и замещенный бензилоксикарбонил, такой как п-хлорбензилоксикарбонил (CIZ), п-нитробензилоксикарбонил, п-бромбензилоксикарбонил и п-метоксибензилоксикарбонил; (3) алифатические уретановые защитные группы, такие как трет-бутилоксикарбонил (Воc), диизопропилметоксикарбонил, изопропилоксикарбонил, этоксикарбонил и аллилоксикарбонил; (4) циклоалкильные защитные группы типа уретана, такие как циклопентилоксикарбонил, адамантилоксикарбонил и циклогексилоксикарбонил; (5) защитные группы типа тиоуретана, такие как фенилтиокарбонил; (6) защитные группы типа алкила, такие как аллил (Аlу), трифенилметил (тритил) и бензил (Bzl); (7) триалкилсилановые группы, такие как триметилсилан. Предпочтительной α-аминозащитной группой является Воc.
X2 представляет собой защитную группу для гидроксильной боковой цепи Ser, например Ac, Bz, тритил, 2,6-дихлорбензил (DCB) или бензиловый эфир (Bzl), а предпочтительно Bzl.
X3 представляет собой защитную группу для аминогруппы боковой цепи, которая не удаляется в том случае, если удаляется α-аминозащитная группа или другая аминозащитная группа. Иллюстративными примерами являются (1), восприимчивые к действию основания группы, такие как Fmoc, или некоторые другие обладающие слабыми кислотными свойствами, стабильные защитные ароматические группы типа уретана; (2) восприимчивые к тиолу группы, такие как дитиасукциноил (Dts), которые могут быть удалены или отщеплены путем тиолиза; (3) восприимчивые к гидразину группы, такие как фталоил (Pht), который отщепляется посредством гидразинолиза; (4) нуклеофилвосприимчивые группы, такие как о-нитрофенилсульфенил (Nps) и т.п., которые отщепляются под действием тиоацетамида или слабых кислот или их солей; (5) светочувствительные группы, которые отщепляются посредством фотолиза; и (6) группы, селективно удаляемые путем восстановления, такие как Dts. Для метода твердофазного пептидного синтеза (ТФПС) с использованием Воc предпочтительной является группа Fmoc.
X4 представляет собой восприимчивую к действию кислоты защитную группу для первичной или вторичной аминогруппы боковой цепи, такую как Z или 2ClZ.
X5 может представлять собой D-Ala-, Gly-, Ala-, Agl-, D-Agl-, Agl(Me)- или D-Agl(Me)-NH-[полимерный носитель] или N(Et)-[полимерный носитель]; X5 также может быть амидом любой из Gly, или Ala, или D-Ala, (низший алкид)-замещенный амидом, связанный непосредственно с Pro, AzaGly-NH2 или -ОН (свободная кислота). Если Х5 представляет собой свободную кислоту, то промежуточное соединение представляет собой нонапептидный фрагмент, который был получен так, что он связан с D- или L-аланинолом с образованием декапептида, имеющего спиртовую группу у С-конца.
Критерием для отбора защитных групп Х2-Х4 боковой цепи является то, что на каждой стадии синтеза защитная группа должна быть в основном устойчивой к реагенту в реакционных условиях, выбранных для удаления α-аминозащитной группы (предпочтительно, Воc). Эти защитные группы в основном не должны быть отщеплены в условиях присоединения, но должны быть удаляемыми после завершения синтеза нужной аминокислотной последовательности в реакционных условиях, не изменяющих пептидную цепь. Защитные группы, первоначально используемые для остатков в 5- и 6-положении, предпочтительно удаляют, а селективные реакции проводят перед отщеплением конечного пептида от смолы, как описано ниже. Если промежуточный декапептид синтезирован так, как описано выше, то защитные группы X3 могут быть предпочтительно удалены по отдельности.
Если группа Х5 представляет собой D-Ala-NH-[полимерный носитель], то амидная связь соединяет D-Ala с полимером ВНА или с полимером МВНА; причем это аналогично случаю, когда на С-конце используется Аgl или D-Agl. Если X5 представляет собой N(Et)-[полимерный носитель], то этиламидная связь соединяет Pro с N-алкиламинометиловым полимером (NAAM).
Если необходимо, чтобы N-конец был ацетилирован, то это может быть, например, осуществлено с использованием ацетила в качестве защитной группы X1 для α-аминогруппы β-D-Nal в 1-положении путем ее добавления к аминокислоте перед ее присоединением к пептидной цепи; однако эту реакцию предпочтительно осуществлять с использованием пептидного промежуточного соединения на полимере. После деблокирования α-аминогруппы и в том случае, если желательно оставить группы боковой цепи защищенными, ацетилирование предпочтительно осуществлять посредством реакции с уксусным ангидридом; альтернативно, эта реакция может быть осуществлена с использованием уксусной кислоты в присутствии диизопропила или дициклогексилакарбодиимида (DIC или DCC), либо посредством какой-нибудь другой подходящей реакции ацилирования, известной специалистам. Аналогичную процедуру осуществляют, если необходимо, чтобы на N-конце присутствовала карбамоильная или замещенная карбамоильная группа. Если деблокированные аминогруппы боковой цепи являются модифицированными, а остаток остается частью пептидной цепи, то эта реакция может быть осуществлена с использованием соответствующего изоцианата в присутствии соответствующего основания, например, N, N-диизопропилэтиламина (DIEA), хотя использование такого основания необязательно. Если необходимо, чтобы в конечном продукте присутствовала незамещенная карбамоильная группа, то деблокированная боковая аминоцепь может взаимодействовать с бензилизоцианатом, п-тозилизоцианатом, триметилсилилизоцианатом или трет-бутилизоцианатом, при этом предпочтительным является трет-бутилизоцианат. С использованием такой стратегии, т-бутильную группу удаляют во время отщепления от полимера, что приводит к удалению карбамоильной группы.
Настоящее изобретение также относится к новому способу получения такого антагониста GnRH, имеющего, например, формулу: Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(Ноr)-D-4Aph(Ac)-Leu-ILys-Pro-D-Ala-NH2, где указанный способ предусматривает (а) образование промежуточного пептида, имеющего формулу: Boc-D-4Aph(X3)-Leu-ILys(X4)-Pro-X5, где X3 представляет собой восприимчивую к основанию и восприимчивую к гидразину или восприимчивую к другим соответствующим соединениям защитную группу для аминогруппы; X4 представляет собой восприимчивую к кислоте защитную группу для боковой аминоцепи; и X5 представляет собой D-Ala-NH-[полимерный носитель]; (b) удаление X3 из D-4Aph для деблокирования первичной аминогруппы боковой цепи этого аминокислотного остатка промежуточного пептида; (с) взаимодействие этой деблокированной первичной аминогруппы боковой цепи с уксусным ангидридом; (d) завершение удлинения цепи с получением промежуточного пептида X1-D-2Nal-D-4Cpa-D-3Pal-Ser(X2)-4Aph(X3)-D-4Aph(Ac)-Leu-ILys(X4)-Pro-X5, где X1 представляет собой водород или α-аминозащитную группу, а X2 представляет собой водород или защитную группу для гидроксильной группы остатка Ser; (e) деблокирование α-аминогруппы у N-конца и ацетилирование; (f) удаление X3 из 4Арh, и взаимодействие деблокированной первичной аминогруппы с гидрооротовой кислотой; и (g) отщепление любых остаточных защитных групп и/или отщепление от полимерного носителя, входящего в определение X5.
Конечную очистку пептида осуществляют с помощью хроматографии, а предпочтительно с использованием ОФ-ВЭЖХ, известной специалистам, см. J. Rivier et al. , J. Chromatography, 288, 303-328 (1984) и Miller & J. Rivier, Biopolymers (Peptide Science), 40, 265-317 (1996).
Предполагается, что антагонисты GnRH настоящего изобретения являются эффективными при концентрациях менее 100 микрограммов на килограмм массы тела при их подкожном введении примерно в полдень дня предтечки для предупреждения овуляции у самок крыс. Для длительного подавления овуляции может оказаться необходимым использовать дозы порядка от 0,1 до 2,5 миллиграммов на килограмм массы тела. Эти антагонисты являются также эффективными для прекращения образования спермы при их регулярном введении млекопитающим-самцам, а поэтому они могут быть использованы в качестве противозачаточных средств. Поскольку эти соединения способствуют снижению уровня тестостерона и, таким образом, снижению либидо (нежелательного последствия у нормальных сексуально активных самцов), то, наряду с антагонистами GnRH, может оказаться предпочтительным вводить компенсирующие дозы тестостерона для достижения азооспермии при сохранении либидо. Эти антагонисты могут быть также использованы для регуляции продуцирования гонадотропинов и половых стероидов, а также при других показаниях для кратковременного или длительного курса лечения, как указывалось выше, и, кроме того, они могут быть использованы в ветеринарии в качестве противозачаточных средств для комнатных домашних животных.
Пептиды, полученные в соответствии с настоящим изобретением, являются хорошо растворимыми при физиологических значениях рН и могут быть получены в виде относительно концентрированных растворов для введения, особенно для подкожных инъекций. Эти пептиды хорошо переносятся организмом, и при их подкожном введении в эффективных концентрациях не имеют тенденции к образованию геля. В основном фармацевтические композиции, включающие в себя такие пептиды и подходящий фармацевтически приемлемый носитель, могут быть введены внутривенно (в/в), внутрибрюшинно (в/б), подкожно (п/к) или т.п. в концентрациях примерно от 0,001 мг до 2,5 мг на кг массы тела в день, при этом достаточной дозой является 0,5 мг/кг/день.
Соответствующим образом защищенные D- или L-гидрооротилсодержащие, карбамоилсодержащие и/или D- или L-имидазолидонкарбонилсодержащие аминокислоты могут быть синтезированы, а затем использованы в пептидном синтезе путем удлинения цепи. Однако в равной степени эффективный синтез достигается путем первоначального введения соответствующим образом защищенных остатков Aph, D-Aph, Amf или D-Amf в нужном положении в промежуточном пептиде, и этот синтез может быть осуществлен выбранным лабораторным методом, где желательно сначала проводить синтез лишь в небольших количествах. Эта последняя стратегия достигается путем деблокирования конкретного остатка (либо сразу; либо позже, во время синтеза), а затем посредством реакции деблокирования аминогруппы боковой цепи с нужным реагентом.
Настоящее изобретение, кроме того, представлено нижеследующими примерами.
Пример 1
Было обнаружено, что пептид, имеющий формулу: Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-Lys(Nic)-D-Lys(Nic)-Leu-ILys-Pro-D-Ala-NH2 (антид) обладает очень хорошими биологическими свойствами как антагонист GnRH, поскольку он имеет пептид, который в настоящем описании назван ацилином и который отличается от антида только в 5- и 6-положениях. Авторами изобретения было установлено, что при использовании этих молекул в качестве исходного материала и при создании других заместителей в 5- и 6-положениях или в 5-положении декапептида ацилина могут быть получены антагонисты GnRH, имеющие большую продолжительность биологической активности in vivo. Что касается положений 1-4 и 7-10, то было отмечено, что антид, ацилин и азалин являются абсолютно аналогичными.
Нижеследующий декапептид [4Aph(Hor)5, D-4Aph(Cmb)6]-антид или [Ac-D-2Nal1, D-4Cpa2, D-3Pal3, 4Aph(Hor)5, D-4Aph(Cmb)6, Ilys8, D-Ala10]-GnRH был получен методом твердофазного синтеза. Этот пептид имеет следующую формулу:
Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L-гидрооротил)-D-Aph(карбамоил)-Leu-Lys(изопропил)-Pro-D-Ala-NH2.
Сначала использовали около 0,50 грамма (0,54 ммоль/г) полимера МВНА (Bachem), и Вос-защищенный D-AIB связывали с полимером приблизительно в течение 2 часов в смеси диметилформамида (ДМФ)/СН2Сl2 с использованием около 0,65 ммоль Вос-производного и диизопропилкарбодиимида (DIC) и безводного 1-гидроксибензотриазола (HOBt) н качестве активирующих или связывающих реагентов. Остаток D-Ala связывается с остатком МНВА посредством амидной связи.
После присоединения каждого аминокислотного остатка промывку, деблокирование, а затем присоединение следующего аминокислотного остатка осуществляли в соответствии со схемой 1 лабораторного синтеза, приведенной в конце описания, для получения примерно от 0,5 до 1 грамма исходного полимера.
Вышеуказанную схему использовали для присоединения каждой аминокислоты пептида настоящего изобретения после присоединения первой аминокислоты. NαBoc-защиту использовали для каждой аминокислоты, присоединяемой в процессе синтеза. 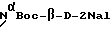 получали способом, известным специалистам, например, как подробно описано в патенте США N4234571, этот пептид также поставляется фирмой Synthe Tech, Oregon, США. Первичные аминогруппы боковой цепи 4Aph в 5-положении и D-4Aph в 6-положении защищены Fmoc. Бензиловый эфир (Bzl) предпочтительно используют в качестве защитной группы боковой цепи для гидроксильной группы Ser; однако Ser может быть присоединен без защиты боковой цепи. NαBoc-Lys (ipr, Z) используют для остатка в 8-положении.
получали способом, известным специалистам, например, как подробно описано в патенте США N4234571, этот пептид также поставляется фирмой Synthe Tech, Oregon, США. Первичные аминогруппы боковой цепи 4Aph в 5-положении и D-4Aph в 6-положении защищены Fmoc. Бензиловый эфир (Bzl) предпочтительно используют в качестве защитной группы боковой цепи для гидроксильной группы Ser; однако Ser может быть присоединен без защиты боковой цепи. NαBoc-Lys (ipr, Z) используют для остатка в 8-положении.
После присоединения D-4Aph для остатка в 6-положении в виде NαBoc-D-4Aph(Fmoc) полученное промежуточное соединение представляло собой: Boc-D-4Aph(Fmoc)-Leu-Lys(ipr, Z)-Pro-D-Ala-NH-[полимерный носитель МВНА]. Затем аминогруппу боковой цепи на остатке в 6-положении модифицируют после первого удаления защиты боковой цепи. Защитную группу Fmoc удаляют путем проведения последовательных обработок 25% пиперидином в ДМФ (10 мл) приблизительно в течение 15 минут (каждую). После промывки пептидополимера ДМФ вновь деблокированную аминогруппу обрабатывают 20-кратным избытком трет-бутил-изоцианата в ДМФ при комнатной температуре в течение около 12 часов, или до полного завершения реакции, которое определяют с использованием нингидринового теста. После этого пептидополимер подвергают стандартной промывке, и Вос удаляют для присоединения следующего остатка.
Затем присоединяют остаток в 5-положении в виде NαBoc-4Aph(Fmoc). После этого боковую цепь деблокируют, как описано ранее, и осуществляют реакцию с 0,10 г (0,66 ммоль) L-гидрооротовой кислоты, 90 мг (0,66 ммоль) HOBt и 0,66 ммоль DIC в 3 мл ДМФ при комнатной температуре в течение около 8 часов, либо до тех пор, пока реакция не будет завершена, на что указывает стандартный нингидриновый тест.
После промывки и удаления NαBoc синтез декапептида завершают посредством последовательной реакции с
После деблокирования α-аминогруппы на N-конце с использованием трифторуксусной кислоты (TFA) ацетилирование проводят с использованием большого избытка уксусного ангидрида в дихлорметане (ДХМ) в течение около 30 минут. Альтернативно Fmoc-защиту 4Aph удаляют лишь после ацетилирования N-конца, а затем осуществляют реакцию с L-гидрооротовой кислотой.
Пептидополимер сушат, и после добавления анизола (0,5 мл) в качестве акцептора пептид отщепляют от полимера и осуществляют деблокирование боковых цепей Ser и Lys при около 0oС с использованием 15 мл HF в течение около 1,5 часа с удалением всех оставшихся трет-бутильных групп. После удаления HF в вакууме полимер два раза промывают 100 мл этилового эфира. Отщепленный пептид экстрагируют 0,2% TFA в 25% СН3СN/Н2О, и процесс повторяют с использованием каждый раз 100 мл. Эти экстракты объединяют и лиофилизуют, в результате чего получают около 600 мг сырого пептидного порошка.
Затем осуществляют очистку пептида с помощью препаративной высокоэффективной жидкостной хроматографии с обращенной фазой (ОФ-ВЭЖХ), известной специалистам и конкретно описанной J. Rivier et al., J Chromatography, 288, 303-328 (1984). При первом разделении с помощью препаративной ОФ-ВЭЖХ использовали буферную систему ТЕАР (фосфат триэтиламмония), а конечное разделение осуществляли с использованием градиента 0,1% TFA (трифторуксусной кислоты), все эти процедуры подробно описаны в статье J.Chromatography.
По проведенной оценке с использованием капиллярного зонального электрофореза (КЗЭ) было установлено, что этот пептид (около 30 мг) (называемый далее пептидом N1) является в основном гомогенным, и его чистота составляет 98%. Аминокислотный анализ очищенного пептида показал соответствие этого пептида формуле для полученной структуры. Молекулярная масса, определенная с помощью жидкостной масс-спектрометрии вторичных ионов (ЖМСВИ), составляла 1631,9 Да, что соответствовало предполагаемой массе 1631,8 Да для этого пептида.
Гидрофильность определяли путем измерения времени удерживания при ОФ-ВЭЖХ с использованием градиента 40% буфера В - 70% буфера B в течение 30 минут, при этом буфером А был ТЕАР, рН 7,0, а буфером В был 70% СН3СN и 30% буфер А. Пептид N1 является более гидрофильным, чем ацилин, и элюировался раньше, чем ацилин. Его растворимость в водных буферах при рН от около 5 до около 7, и его устойчивость к гелеобразованию in vivo, наряду с пролонгированной биологической активностью, направленной на снижение уровней LH в кровотоке, как описано ранее, делают его особенно подходящим для подкожной инъекции по сравнению с другими соединениями со сравнимой биологической эффективностью.
Этот пептид анализировали in vivo для определения его эффективности в подавлении секреции LH у крыс. Измерение уровней LH в кровотоке у кастрированных самцов крыс Sprague-Daw-ley, которым была введена подкожная инъекция этого пептида, осуществляли в соответствии с описанием С. Rivier et al., Biol. Reproduc., 1983, 29, 374-378. Эти пептиды сначала растворяли при концентрации 1,0 или 10 мг/мл в бактериостатической воде, а затем разводили в 0,04 М фосфатном буфере, содержащем 0,1% BSA. Последовательные разведения проводили в фосфатном буфере. Эти пептиды подкожно инъецировали 5 крысам, и брали пробы крови (300 мкл) при анестезии метотаном. Сыворотки (50 мкл) тестировали на уровни LH в дубликатах с использованием реагентов, поставляемых NIDDK в рамках Национальной программы по изучению гипофиза и распределения гормонов. Тесты показали, что доза пептида в 50 мкг на одну крысу подавляет секрецию LH до уровней, значительно меньших, чем 50% от контрольных уровней в течение 96-часового периода времени после инъекции. Более того, уровни, измеренные после этого 96-часового периода составляли только около 30% от уровней LH, обнаруживаемых у крыс, которым было введена аналогичная доза 50 микрограммов ацилина. На этом основании был сделан вывод, что пептид N1 действует очень долго. Исследование крыс показало, что этот пептид очень хорошо переносится животными, причем он не обнаруживал значительного гелеообразования в месте инъекции.
Данные, полученные в результате тестирования большого числа антагонистов GnRH, показали, что пептид, обладающий таким пролонгированным подавлением секреции LH, должен, при анализе in vivo на зрелых самках крыс Sprague-Dawley, полностью блокировать овуляцию при дозе 2,5 микрограммов.
Пример 1А
Повторяли синтез, описанный в Примере 1, заменяя NαBoc-4Aph(Fmoc) на NαBoc-4Аmf(Fmoc). После деблокирования боковой цепи D-4Amf проводили реакцию с т-бутилизоцианатом, как описано ранее. Отщепление от полимера и деблокирование с последующей очисткой осуществляли как описано в Примере 1. Как было оценено, пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L-гидрооротил)-D-Amf(кapбaмоил)-Leu-Lys (изопропил)-Pro-D-Ala-NH2, очищенный с помощью ОФ-ВЭЖХ, был, в основном, гомогенным и имел чистоту более чем 99%. МС-анализ показал, что его масса составляла 1645,9 Да, которая вполне соответствовала ожидаемой массе 1645,8 Да. Из результатов, полученных с помощью ВЭЖХ, можно видеть, что этот пептид является более гидрофильным, чем ацилин.
Оценка этого пептида с использованием стандартного in vivo-теста на подавление секреции LH у крыс, показала, что этот пептид при дозе в 50 микрограммов в первые 1, 2 и 3-й дни является таким же эффективным в подавлении уровней LH, как и ацилин А через 96 часов, уровни LH составляли лишь около 25% от уровней, которые достигались при введении крысам ацилина. Поэтому пептид N1 можно считать пептидом очень продолжительного действия.
Пример 1В
Для создания аналога [4Aph(Ноr)5]-ацилина повторяли процедуру синтеза, описанного в Примере 1, заменяя т-бутилизоцианат на уксусный ангидрид для проведения реакции с боковой цепью, деблокированной в положении 6. Отщепление этого декапептида от полимера и деблокирование с последующей очисткой проводили как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L- гидрооротил)-D-Aph(L-ацетил)Leu-Lys (изопропил) -Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту более чем 99%. МС-анализ показал, что его масса составляла 1630,6 Да, которая соответствовала ожидаемой массе 1630,8 Да.
Оценка этого пептида с использованием стандартного in vivo-теста на подавление секреции LH у крыс, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и в первые 1-4 дня он является почти таким же эффективным, как и ацилин. Поэтому можно считать, что этот пептид обладает очень длительным LH-ингибирующим действием.
Пример 1С
Повторяли синтез, описанный в Примере 1В, заменяя L-гидрооротовую кислоту на D/L-гидрооротовую кислоту с образованием изомерных декапептидов. Отщепление от полимера и деблокирование с последующей очисткой осуществляли как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(D/L-гидрооротил)-D-Aph(ацетил)-Leu-Lys (изопропил) -Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, он представлял собой гомогенную смесь двух соединений без каких-либо примесей. МС-анализ показал, что его масса составляла 1630,6 Да, которая соответствовала ожидаемой массе 1630,8 Да.
Оценка этого пептида с использованием стандартного in vivo-теста на подавление секреции LH у крыс, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и в первые 1-4 дня он является почти таким же эффективным, как и ацилин. Поэтому
можно считать, что этот пептид обладает длительным LH-ингибирующим действием.
Пример 1D
Повторяли синтез, описанный в Примере 1В, заменяя L-гидрооротовую кислоту на D-гидрооротовую кислоту с образованием изомерного декапептида. Отщепление от полимера и деблокирование с последующей очисткой осуществляли как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(D-гидрооротил)-D-Aph(ацетил)-Leu-Lys (изопропил) -Pro-D-Ala-NH2, получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту более чем 98%. МС-анализ показал, что его масса составляла 1630,8 Да, которая соответствовала ожидаемой массе 1630,8 Да.
Оценка этого пептида с использованием стандартного in vivo-теста на подавление секреции LH у крыс, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид обнаруживает пролонгированное биологическое действие, направленное на подавление секреции LH, и в первые 1-4 дня он является почти таким же эффективным, как и ацилин.
Пример 1Е
Повторяли синтез, описанный в Примере 1В, заменяя NαBoc-D-4ClPhe на NαBoc-D-4FPhe с образованием декапептида [D-4FPhe2, 4Aph (Ноr)5]-ацилин. Отщепление от полимера и деблокирование с последующей очисткой осуществляли как описано в Примере 1. Пептид Ac-D-2Nal-D-4Fpa-D-3Pal-Ser-4Aph(L-гидрооротил)-D-Aph(ацетил)-Leu-Lys (изопропил) -Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, это пептид был, в основном, гомогенным и имел чистоту более чем 99%. МС-анализ показал, что его масса составляла 1615,1 Да, которая вполне соответствовала ожидаемой массе 1614,8 Да.
Оценка этого пептида с использованием стандартного in vivo-теста на подавление LH у крыс, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и в первые 1-4 дня он является почти таким же эффективным, как и ацилин. Поэтому можно считать, что этот пептид обладает длительным LH-ингибирующим действием.
Пример 1F
Повторяли синтез, описанный в Примере 1В, заменяя NαBoc-4Aph{Fmoc} на NαBoc-D-4Amf(Fmoc) с образованием декапептида [4Amf (Ноr)5]-ацилин. Отщепление от полимера и деблокирование с последующей очисткой осуществляли как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Amf(L-гидрооротил)-D-Aph(ацетил)-Leu-Lys (изопропил) -Pro-D-Ala-NH2 был получен путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту более чем 98%. МС-анализ показал, что его масса составляла 1644,7 Да, которая соответствовала ожидаемой массе 1644,8 Да.
Оценка этого пептида с использованием стандартного in vivo-теста на подавление LH у крыс, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и в первые 1-4 дня он является почти таким же эффективным, как и ацилин. Поэтому можно считать, что этот пептид обладает длительным LH-ингибирующим действием.
Пример 1G
Повторяли синтез, описанный в Примере 1, однако вместо реакции аминогруппы боковой цепи D-4Aph с т-бутилизоцианатом эту группу и остаток 4Aph одновременно подвергали реакции с гидрооротовой кислотой с образованием декапептида [4Aph(Ног)5, 4Aph(Ноr)6] -антида. Отщепление от полимера и деблокирование с последующей очисткой осуществляли как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Раl-Sеr-4Арh(L -гидрооротил)-D-4Aph (L-гидрооротил) -Leu-Lys (изопропил) -Pro-D-Ala-NH2 получали с помощью очистки ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту более чем 99%. МС-анализ показал, что его масса составляла 1728,4 Да, которая соответствовала вычисленной массе 1728,8 Да. Результаты ОФ-ВЭЖХ показали, что этот пептид является более гидрофильным, чем азалин В, который, в свою очередь, является более гидрофильным, чем ацилин.
Оценка этого пептида с использованием стандартного in vivo-тecтa на подавление LH у крыс, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и в первые 1-4 дня он является почти таким же эффективным, как и ацилин. Поэтому можно считать, что этот пептид обладает длительным LH-ингибирующим действием.
Этот синтез повторяли с заменой NαBoc-4Aph(Fmoc) на NαBoc-D-4Amf(Fmoc) для создания [4Aph(Hor)5, D-Amf(Hor)6]-антида, который, в основном, обладает сильным биологическим действием, направленным на подавление секреции LH.
Пример 1Н
Повторяли синтез, описанный в Примере 1, однако вместо реакции аминогруппы боковой цепи D-4Aph с т-бутилизоцианатом эту группу подвергали реакции с D-гидрооротовой кислотой с образованием декапептида [4Aph(Ноr)5, D-4Aph(Ноr)6] -антида. Отщепление от полимера и деблокирование с последующей очисткой осуществляли как описано в примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L -гидрооротил)-D-4Aph(D- гидрооротил)-Leu-Lys(изопропил)-Pro-D-Ala-NH2 получали с помощью очистки ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту более чем 98%. МС-анализ показал, что его масса составляла 1728,7 Да, которая соответствовала вычисленной массе 1728,8 Да. Результаты ОФ-ВЭЖХ показали, что этот пептид является более гидрофильным, чем азалин В, который, в свою очередь, является более гидрофильным, чем ацилин.
Оценка этого пептида методом стандартного in vivo-теста на подавление LH у крыс, как и в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и в первые 1-3 дня он является почти таким же эффективным, как и ацилин. На 4-й день он обнаруживал значительно большую эффективность, чем ацилин, а поэтому можно считать, что этот пептид обладает очень длительным LH-ингибирующим действием.
Пример 1J
Синтез декапептида [MeCbm-D-2Nal1, 4Aph(Ноr)5]-ацилина осуществляли, в основном, как описано выше в Примере 1В; однако? вместо удаления Fmoс-защитной группы сразу после присоединения NαBoc-4Aph(Fmoc), осуществляли полный синтез декапептида на полимере. Затем после деблокирования N-конца вместо реакции с уксусным ангидридом проводили реакцию с метилизоцианатом с получением метилкарбамоила у N-конца. Затем Fmoc удаляли и аминогруппу боковой цепи 4Aph подвергали реакции с L-гидрооротовой кислотой, как описано в Примере 1В. Отщепление от полимера и деблокирование с последующей чисткой осуществляли как описано в Примере 1. Пептид метилкарбамоил-D-2Nаl-D-4Срa-D-3Раl-Sеr-4Арh(L- гидрооротил)-D-4Aph(ацетил)-Leu-Lys (изопропил)-Pro-D-Ala-NH2 получали с помощью очистки ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту примерно 99%. МС-анализ показал, что его масса составляла 1645,7 Да, которая соответствовала вычисленной массе 1645,8 Да. Результаты ОФ-ВЭЖХ показали, что этот пептид является более гидрофильным, чем азалин В, который, в свою очередь, является более гидрофильным, чем ацилин.
Оценка этого пептида с использованием стандартного in vivo-теста на подавление секреции LH у крыс, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и в первые 1-3 дня он является почти таким же эффективным, как и ацилин, а через 96 часов он является более эффективным почти на 50%. Поэтому можно считать, что этот пептид обладает очень длительным LH-ингибирующим действием.
Пример 1К
Повторяли синтез, описанный в Примере 1, с заменой NαBoc-4Aph(Fmoc) на NαBoc-D-3Pal и без последующего проведения реакции с t-BuNCO, с образованием декапептида [4Aph(Hor)5, D-3Pal6]-антида. Отщепление от полимера и деблокирование с последующей очисткой осуществляли как описано в Примере 1. Пептид ацетил-D-2Nаl-D-4Сра-D-3Раl-Sеr-4Aрh(L- гидрооротил)-D-3Pal-Leu-Lys (изопропил)-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту более чем 99%. МС-анализ показал, что его масса составляла 1574,7 Да, которая соответствовала ожидаемой массе 1574,7 Да.
Оценка этого пептида с использованием стандартного in vivo-теста на подавление LH у крыс, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и в первые три дня он является почти таким же эффективным, как и ацилин, однако через 96 часов наблюдалось снижение уровней LH до значений, составляющих около 35% от уровней, достигаемых с использованием ацилина. Поэтому можно считать, что этот пептид обладает очень длительным LH-подавляющим действием.
Пример 1L
Повторяли синтез, описанный в Примере 1G, заменяя гидрооротовую кислоту на т-бутилизоцианат с образованием декапептида [4Aph(Cmb)5, D-4Aph(Cmb)6]-антида. Отщепление от полимера и деблокирование с последующей очисткой осуществляли как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D- 3Раl-Sеr-4Aрh(карбамоил)-D-4Aрh(карбамоил) -Leu-Lys(изопропил)-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту более чем 99%. МС-анализ показал, что его масса составляла 1534,9 Да, которая вполне соответствовала ожидаемой массе 1534,7 Да.
Оценка этого пептида с использованием стандартного in vivo-теста на подавление LH у крыс, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и в первые четыре дня он являлся почти таким же эффективным, как и ацилин. Поэтому можно считать, что этот пептид обладает длительным LH-ингибирующим действием.
Пример 1М
Повторяли синтез, описанный в Примере 1G, заменяя гидрооротовую кислоту на метилизоцианат с образованием декапептида [4Aph(MeCmb)5, D-4Aph(MeCmb)6] -антида. Отщепление от полимера и деблокирование с последующей очисткой осуществляли как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph (метилкарбамоил)-D-4Aph(метилкарбамоил)-Leu- Lys(изопропил)-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту более чем 99%. МС-анализ показал, что его масса составляла 1562,8 Да, которая соответствовала ожидаемой массе 1562,8 Да.
Оценка этого пептида с использованием стандартного in vivo-теста на подавление LH у крыс, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и в первые два дня он являлся почти таким же эффективным, как и ацилин, а затем его активность в подавлении LH начинала несколько снижаться.
Пример 2
Пептид [4Aph(Hor)5, D-Cit6]-антид, аналог пептида Цетрореликс (Cetrorelix), имеющий формулу Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph (L-гидрооротил)-D-Cit-Leu-ILys-Pro-D-Ala-NH2 синтезировали методом, в основном, описанным в Примере 1. Вместо NαBoc-D-4Aph, в 6-положении присоединяли NαBoc-D-Cit. Альтернативно в 6-положении присоединяли NαBoc-D-Orn(Fmoc), и удлинение цепи временно прекращали после того, как был получен следующий промежуточный пептид: Boc-D-Orn(Fmoc)-Leu-Lys(ipr, Z)-Pro-D-Ala-NH-[полимерный носитель МВНА] . Боковую аминоцепь на остатке Оrn затем деблокировали путем удаления Fmoc-защиты, как описано в Примере 1, и промежуточный пептид обрабатывали избытком т-бутилизоцианата в ДМФ в течение около 6 часов при комнатной температуре для проведения реакции с боковой цепью остатка Оrn. Затем завершение синтеза декапептида проводили как описано в Примере 1.
Затем пептидополимер промывали, расщепляли и деблокировали, после чего его очищали как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L -гидрооротил)-D-Cit-Leu-ILys-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту более чем 99%. ЖМСВИ-анализ показал, что его масса составляла 1583,7 Да, которая соответствовала вычисленной для этого пептида массе 1583,8 Да.
Этот пептид был более гидрофильным, чем цетрореликс, и при тестировании in vivo на подавление секреции LH, описанном в Примере 1, он обнаруживал такое же продолжительное действие, как и цетрореликс. В течение 3 дней этот пептид подавлял секрецию LH с немного более лучшим эффектом, а через 96 часов он давал значительно более лучший эффект.
Пример 2А
Аналог пептида антид, то есть [4Aph(Hor)5]-антид, синтезировали методом синтеза, в основном, как описано в Примере 1 патента США 5169935. После присоединения NαBoc-D-Lys(Fmoc) в 6-положении этот пептид после деблокирования подвергали реакции с избыточным количеством никотиновой кислоты в ДМФ. Затем NαBoc-Aph(Fmoc) присоединяли в 5-положение, после чего боковую аминоцепь на остатке Aph деблокировали как описано в Примере 1. Промежуточный пептид подвергали реакции с L-гидрооротовой кислотой в ДМФ, и синтез промежуточного декапептида завершали как описано в Примере 1.
Последующую стандартную промывку, отщепление от полимера, деблокирование и очистку проводили как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L-гидрооротил)-D-Lys(Nic)-Leu-ILys-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Было установлено, что этот пептид является более гидрофильным, чем цетрореликс и обнаруживает такое же продолжительное LH-подавляющее действие, как и цетрореликс.
Пример 3
Аналог [4Aph(D/L-Imz)5] -ацилин синтезировали методом, в основном, описанным в Примере 1В, за исключением того, что L-Hor заменяли на D/L-Imz. После деблокирования 4Aph в 5-положении промежуточный пептид обрабатывали избыточным количеством В/L-2-имидазолидон-4-карбоновой кислоты, около 90 мг HOBt и около 0,66 ммоль DIC в растворе ДМФ в течение примерно 6 часов при комнатной температуре. Затем завершение синтеза промежуточного декапептида проводили как описано в Примере 1.
Пептидополимер промывали, расщепляли и деблокировали и очищали как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Раl-Sеr-4Арh(D/L-2- имидазолидон-4-карбонил)-D-4Aph(Ac)-Leu-ILys-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ.
Как было оценено, этот пептид представлял собой гомогенную смесь двух соединений без каких-либо других примесей. ЖМСВИ-анализ показал, что его масса составляла 1602,7 Да, которая соответствовала вычисленной для этого пептида массе 1602,8 Да.
Анализ этого пептида, проведенный с использованием стандартного in vivo-теста на крысах, описанного в Примере 1, показал, что при дозе в 50 микрограммов этот пептид обнаруживает продолжительное действие, направленное на подавление секреции LH. В течение 3 дней и в течение 96 часов продолжительность подавления секреции LH была лишь незначительно выше, чем продолжительность действия ацилина.
Пример 3А
Повторяли синтез, описанный в Примере 3, с использованием избытка L-2-имидазолин-4-карбоновой кислоты вместо D/L-Imz. Как было оценено, полученный пептид был, в основном, гомогенным, и его чистота составляла около 99%. ЖМСВИ-анализ показал, что масса этого пептида составляла 1602,5 Да и соответствовала вычисленной для этого пептида массе 1602,8 Да. Этот пептид лучше растворялся в воде, чем ацилин.
Анализ проводили как описано в Примере 1, и в дозе в 50 микрограммов этот пептид обнаруживал продолжительное действие, направленное на подавление секреции LH, причем в течение 96 часов продолжительность его действия была почти такой же, как и у ацилина.
Пример 3В
Повторяли синтез, описанный в Примере 3, с использованием избытка L-2-имидазолин-4-карбоновой кислоты вместо D/L-Imz. Был получен пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(D-Imz)-D-4Aph(Ac)-Leu-ILys-Pro-D-Ala-NH2. ЖМСВИ-анализ показал, что его масса составляла 1602,6 Да, которая соответствовала вычисленной для этого пептида массе 1602,8 Да. Этот пептид лучше растворялся в воде, чем ацилин.
Анализ проводили как описано в Примере 1. Этот пептид обнаруживал биологическую активность, и в дозе в 50 микрограммов он ингибировал секрецию LH.
Пример 3С
Пептид [4Aph(Imz)5, D-4Amf(Cbm)6]-ацилина синтезировали с использованием комбинации методов, описанных в Примерах 1А (для введения D-4Amf(Cbm)6) и 3А (для введения 4Aph(Imz)5). Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L -Imz)-D-4Amf(карбамoил)-Leu-ILys-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и его чистота составляла более чем 98%. ЖМСВИ-анализ показал, что его масса составляла 1617,6 Да, которая соответствовала вычисленной для этого пептида массе 1617,8 Да. Этот пептид анализировали как описано в Примере 1, и при дозе 50 микрограммов он обнаруживал длительное действие, направленное на подавление секреции LH. В течение 3 дней продолжительность подавления секреции LH была, в основном, такой же, как и у ацилина, а в течение 96 часов она была несколько выше.
Пример 4
Пептид [4Aph(Ноr)5, D-4Amf (MeCmb)6]-антид, имеющий формулу Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L -гидрооротил)-D-4Amf(MeCmb)-Leu-ILys-Pro-D-Ala-NH2 синтезировали методом, описанным в Примере 1А. Вместо реакции D-4Amf в 6-положении с избытком т-бутилизоцианата в ДМФ проводили реакцию с метилизоцианатом. Затем завершение синтеза декапептида проводили как описано в Примере 1.
Пептидополимер промывали, расщепляли и деблокировали, после чего его очищали как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L- гидрооротил)-D-4Amf(MeCmb)-Leu-ILys-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и его чистота составляла более чем 99%. ЖМСВИ-анализ показал, что его масса составляла 1659,8 Да, которая соответствовала вычисленной для этого пептида массе 1659,8 Да.
Анализ, проведенный с использованием стандартного in vivo-теста на крысах, показал, что при дозе в 50 микрограммов пептид N4 обнаруживает лучшее подавление секреции LH, чем ацилин, и был сделан вывод, что он обладает биологической активностью очень длительного действия.
Пример 4А
Повторяли синтез, описанный в Примере 4, заменяя метилизоцианат на уксусный ангидрид, в результате чего получали пептид [4Aph(Hor)5, D-4Amf(Ac)6] -антид. Пептид Ac-D-2Nаl-D-4Сра -D-3Раl-Sеr-4Арh(L- гидрооротил)-D-4Amf(Ac)-Leu-ILys-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и его чистота составляла более чем 99%. ЖМСВИ-анализ показал, что его масса составляла 1644,5 Да, которая соответствовала вычисленной для этого пептида массе 1644,8 Да.
Этот пептид анализировали как описано в Примере 1, и при дозе 50 микрограммов он обнаруживал длительное действие, направленное на подавление секреции LH. В течение 3 дней продолжительность подавления секреции LH была такой же, как у ацилина, а в течение 96 часов она была несколько выше.
Пример 4В
Декапептид, синтезированный и протестированный как описано в Примере 1А, модифицировали путем введения в его С-конец D-аланинола вместо D-аланиламида. Сначала синтезировали нонапептидный фрагмент, содержащий у своего С-конца пролин в качестве свободной кислоты, методом синтеза, в основном, описанным в Примере 1А, но с использованием полимера Merrifield (хлорметилированный полистирол с поперечными связями), поставляемого фирмой Bachem, Inc. После отщепления, деблокирования и очистки был получен следующий нонапептид: Ac-D-2Nal-D-4Cpa-D-3Pal-Ser -4Aph(Hor)-D-4Amf(Cmb)-Leu- ILys-Pro-OH. 0,15 ммоль этого пептида полностью деблокировали и ВЭЖХ-очищенный нонапептид растворяли в 3 мл сухого ДМФ вместе с 3,0 ммоль D-аланинола (Lancaster Chemical). Затем добавляли 0,60 ммоль РуВОР (Novabiochem), твердого вещества, используемого в качестве связывающего агента, и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакцию гасили путем добавления 200 мл воды с получением эмульсии, которая была превращена в прозрачный раствор путем доведения рН до 2,5 с использованием ледяной уксусной кислоты. Полученный декапептид, Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph (Ноr)-D-4Amf (Cmb)-Leu-ILys-Pro-D-Ala-oл, был очищен с помощью препаративной ОФ-ВЭЖХ с использованием ТЕАР (рН 2,3) в качестве буфера, с последующей дополнительной очисткой с использованием 0,1% TFA в качестве буфера. Как было оценено, полученный пептид являлся, в основном, гомогенным, и его чистота составляла более чем около 99%. МС-анализ показал, что его масса составляла 1632,9 Да, которая соответствовала вычисленной массе 1632,8 Да.
Оценка этого пептида с использованием стандартного in vivo-теста на крысах, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и эта биологическая активность продолжалась в течение, по крайней мере, 96 часов. Поэтому этот пептид можно считать пептидом с очень продолжительным действием.
Пример 4С
Повторяли синтез, описанный в Примере 4В, с заменой в рекционной смеси D-аланинола на 3,0 ммоль L-аланинола (Aldridge Chemical). Полученный декапептид очищали как описано в Примере 4В, и как было оценено, он был, в основном, гомогенным, и имел чистоту более чем около 99%. МС-анализ показал, что его масса составляла 1632,9 Да, которая соответствовала вычисленной для этого пептида массе 1632,8 Да.
Оценка этого пептида с использованием стандартного in vivo-теста на крысах, описанного в Примере 1, показала, что при дозе в 50 микрограммов этот пептид является биологически активным, и эта биологическая активность продолжается в течение, по крайней мере, 96 часов. Поэтому этот пептид можно считать пептидом с очень продолжительным действием.
Пример 4D
Декапептид, синтезированный и протестированный как описано в Примере 1А, модифицировали путем введения в его С-конец D-аланинола вместо D-аланиламида. Нонапептидный фрагмент, содержащий у своего С-конца пролин в качестве свободной кислоты, синтезировали методом ТФПС на полимере Merrifield, но, в основном, так, как описано в Примере 1. После отщепления, деблокирования и очистки был получен следующий нонапептид: Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph (Ноr)-D-4Aph (Cmb)-Leu-ILys-Pro-OH. Затем этот очищенный нонапептид подвергали реакции с D-аланинолом, как описано в Примере 4В, и полученный декапептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph (Hor)-D-4Aph(Cmb)-Leu-ILys-Pro-D-Ala-oл, очищали с использованием препаративной ОФ-ВЭЖХ, как описано в Примере 4В, за исключением того, что вместо ТЕАР при рН 2,3 использовали ТЕАР при рН 6,5. Как было оценено, полученный пептид являлся, в основном, гомогенным, и его чистота составляла более чем около 99%. МС-анализ показал, что пептид имеет массу 1618,9 Да, которая соответствовала вычисленной массе 1618,8 Да. Анализ пептида in vivo показал, что он является биологически активным.
Пример 4Е
Повторяли синтез, описанный в Примере 4D, заменяя D-аланинол на L-аланинол. Полученный декапептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph (Hor)-D-4Aph(Cmb)-Leu-ILys-Pro-D-Ala-oл, очищали с помощью препаративной ОФ-ВЭЖХ, как описано в Примере 4D, и, как было оценено, этот пептид был, в основном, гомогенным, и его чистота составляла более чем 99%. МС-анализ показал, что его масса составляла 1618,9 Да, которая соответствовала вычисленной для этого пептида массе 1618,8 Да. Анализ пептида in vivo показал, что он является биологически активным.
Пример 5
Пептид [4Aph(Ноr)3, D-4Amf(Cmb)6] -антид, который имел формулу Ас-D-2Nаl-D-4Сра-D-3Ра1-Sеr-4Арh(D-гидрооротил)-D-4Amf(Cmb)-Leu-ILys-Pro-D-Ala-NH2, синтезировали методом, в основном, описанным в Примере 1А. Вместо реакции D-4Aph в 5-положении с L-гидрооротовой кислотой боковую цепь подвергали реакции с D-гидрооротовой кислотой. Затем завершение синтеза промежуточного декапептида проводили как описано в Примере 1А.
Затем пептидополимер подвергали стандартной промывке, расщеплению и деблокированию, после чего его очищали как описано в Примере 1. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(D-гидрооротил)-D-4Amf(Cmb)-Leu-ILys-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и его чистота составляла более чем 98%. ЖМСВИ-анализ показал, что его масса составляла 1645,8 Да, которая соответствовала вычисленной для этого пептида массе 1645,8 Да.
Анализ, проведенный с использованием стандартного in vivo-теста на крысах, показал, что при дозе в 50 микрограммов в течение 2 дней этот пептид обнаруживал почти такую же продолжительность подавления секреции LH, как и ацилин, и это LH-ингибирующее действие, но в несколько меньшей степени продолжалось через 72 и 96 часов.
Пример 5А
Повторяли синтез, описанный в Примере 5, за исключением того, что вместо реакции деблокированной боковой цепи 4Amf с т-бутилизоцианатом проводили реакцию с уксусным ангидридом. Пептид Ас-D-2Nаl-D-4Сра-D-3Раl-Sеr-4Aрh(D -гидрооротил)-D-4Amf(Ac)-Leu-ILys-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным, и его чистота составляла более чем 99%. ЖМСВИ-анализ показал, что его масса составляла 1644,7 Да, которая соответствовала вычисленной для этого пептида массе 1644,8 Да.
Этот пептид анализировали как описано в Примере 1, и при дозе 50 микрограммов в течение 3 дней этот пептид обнаруживал, в основном, такую же продолжительность подавления секреции LH, как и ацилин, а через 96 часов его LH-ингибирующее действие несколько превышало действие ацилина.
Пример 6
Повторяли синтез, в основном, описанный в Примере 1F, за исключением того, что вместо NαBoc-4Aph(Fmoc) в 6-положении использовали NαBoc-D-4Amf(Fmoc) с образованием декапептида [4Amf(Hor)5, D-4Amf(Ac)6]-антида. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Amf(L- гидрооротил)-D-4Amf(ацетил)-Leu-Lys(изопропил)-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и имел чистоту более чем 99%. МС-анализ показал, что его масса составляла 1658,7 Да, которая соответствовала ожидаемой массе 1658,8 Да.
Этот пептид оценивали как описано в Примере 1, и было установлено, что при дозе в 50 микрограммов этот пептид имеет пролонгированное действие, направленное на подавление секреции LH. В течение первых двух дней он обнаруживал почти такую же биологическую активность, как и ацилин, и на 3- и 4-й день, он обнаруживал почти такое же биологическое действие, как и ацилин.
Пример 6А
Повторяли синтез, описанный в Примере 6, за исключением того, что вместо реакции деблокированной боковой цепи D-4Amf с уксусным ангидридом проводили реакцию с т-бутилизоцианатом, как описано в Примере 1, с образованием [4Aph(Hor)5, D-4Aph(Cmb)6]-антида. Декапептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Amf(L-гидрооротил) -D-4Aph (карбамоил) -Leu-Lys (изопропил) -Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным, и его чистота составляла около 99%. МС-анализ показал, что его масса составляла 1659,6 Да, которая соответствовала вычисленной для этого пептида массе 1659,8 Да.
Этот пептид анализировали как описано в Примере 1, и при дозе 50 микрограммов через 1 день этот пептид обнаруживал такую же активность в подавлении секреции LH, как и ацилин, и примерно такую же активность он обнаруживал через 2 дня. Через 3 дня эта активность была несколько меньше, а через 4 дня она было приблизительно такой же, как и активность ацилина.
Пример 6В
Повторяли синтез, описанный в Примере 6А, за исключением того, что реакцию осуществляли с метилизоцианатом вместо т-бутилизоцианата, в результате чего получали пептид [4Amf(Hor)5, D-4Amf(MeCmb)6]-антид. Пептид Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Amf(L -гидрооротил)-D-4Арh(метилкарбамoил)-Leu-Lys(изопропил)-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ.
Как было оценено, этот пептид был, в основном, гомогенным и его чистота составляла около 99%. МС-анализ показал, что его масса составляла 1673,6 Да, которая соответствовала вычисленной для этого пептида массе 1673,8 Да.
Этот пептид анализировали как описано в Примере 1, и при дозе в 50 микрограммов через 1 день этот пептид обнаруживал такую же активность в подавлении секреции LH, как и ацилин, и примерно такую же активность он обнаруживал через 2 дня. На 3- и 4-й дни его активность, ингибирующая секрецию LH, была значительно меньше, чем активность ацилина.
Пример 6С
Повторяли синтез, описанный в Примере 6, заменяя L-гидрооротовую кислоту на D-гидрооротовую кислоту, в результате чего получали пептид [4Amf(Ноr)5, D-4Amf(Ac)6] -антид. Пептид Ас-D-2Nаl-D-4Сра-D-3Раl-Sеr-4Amf(D-гидрооротил)-D-4Amf(ацетил)-Leu-Lys(изопропил)-Pro-D-Ala-NH2 получали путем очистки с помощью ОФ-ВЭЖХ. Как было оценено, этот пептид был, в основном, гомогенным и его чистота составляла более чем 99%. МС-анализ показал, что его масса составляла 1658,7 Да, которая соответствовала вычисленной для этого пептида массе 1658,8 Да.
Этот пептид анализировали как описано в Примере 1, и при дозе в 50 микрограммов, на 1-й и 2-й день, этот пептид обнаруживал, в основном, такую же активность в подавлении секреции LH, как и ацилин. На 3-й день его биологическая активность была существенно ниже, чем активность ацилина, и в дальнейшем она продолжала значительно снижаться.
Пример 7
С использованием методики, в основном, описанной в Примерах 1-5, были также получены пептиды-антагонисты GnRH (I), представленные в конце описания.
Эти пептиды являются биологически активными в ингибировании секреции LH.
Пример 8
С использованием методики, в основном, описанной в Примерах 1-5 и в патенте США N5491217, были также получены следующие пептиды-антагонисты GnRH (II), представленные в конце описания.
Эти пептиды являются биологически активными в ингибировании секреции LH и обладают очень хорошей растворимостью в воде при физиологическом значении рН.
Было показано, что протестированные вышеуказанные соединения обладают биологической активностью в подавлении секреции LH на уровне, который является, по крайней мере, в основном, сравнимым с уровнем подавления секреции LH соответствующим пептидом-антагонистом GnRH, известным под названием антид, аналогами которого являются рассматриваемые пептиды. В результате широких исследований, проводимых в этой области за последнее десятилетие, биологическая активность, определенная в этом широко используемом тесте, который позволяет измерять подавление секреции LH, свидетельствовала о том, что указанные соединения обладают способностью ингибировать секрецию гонадотропина, а поэтому они могут оказывать полезное противогонадное подавляющее овуляцию действие. Исходя из их высокой растворимости, устойчивости к гелеобразованию in vivo, большой продолжительности биологической активности и других свойств, рассматриваемые соединения могут быть использованы в качестве противогонадных агентов, подавляющих секрецию гонадотропинов и ингибирующих высвобождение стероидов половыми железами, например, в качестве подавляющих овуляцию агентов.
Соединения настоящего изобретения, в основном, вводят в форме фармацевтически приемлемых нетоксичных солей, таких как кислотно-аддитивные соли или металлокомплексы, ацетата и памоата, при этом предпочтительной может быть соль памовой кислоты. Если активный ингредиент вводят в виде таблетки, то эта таблетка может содержать фармацевтически приемлемый нетоксичный разбавитель, который включает связующее вещество, такое как трагакант, кукурузный крахмал или желатин; дезинтегрирующий агент, такой как альгиновая кислота; и замасливатель, такой как стеарат магния. Может быть также осуществлено внутривенное введение в изотоническом физиологическом растворе, в фосфатно-буферных растворах или т.п.
Фармацевтические композиции, в основном, содержат эффективное количество пептида в сочетании со стандартным фармацевтически приемлемым носителем или разбавителем. В основном при внутривенном введении доза может включать от около 10 микрограмм до около 2,5 миллиграмма пептида на один килограмм массы тела хозяина. Природа этих соединений может оказаться эффективной для перорального введения; однако пероральная доза может быть выше. В целом, обработку индивидуумов этими пептидами, в основном, осуществляют таким же способом, как и клиническую обработку другими антагонистами GnRH, с использованием подходящего носителя, в котором это соединение является растворимым, и с введением дозы, достаточной для снижения уровней LH и FSH у пациента.
Для доставки аналога-антагониста GnRH в организм в течение продолжительного периода времени, например в течение периода времени от одной недели до одного года после всего лишь одного введения, и для медленного его высвобождения может также оказаться желательным использовать депо-препараты и имплантанты.
Для ингибирования оплодотворяющей способности и/или для ее регуляции, а также в тех целях, когда необходимо обратимое подавление гонадной активности, таких как предотвращение преждевременного полового созревания или во время лучевой терапии или химиотерапии, эти соединения могут быть введены млекопитающим внутривенно, подкожно, внутримышечно, перорально, чрезкожно, интраназально, внутрилегочно, ректально или интравагинально. Они могут быть также использованы для лечения стероидзависимых опухолей. Эффективные дозы могут варьироваться в зависимости от способа введения и конкретного вида млекопитающего, подвергаемого лечению. Некоторые из этих соединений имеют растворимость вплоть до 50 мг/мл, и, в основном, они могут быть использованы в виде 5-10 мг/мл-растворов при рН 5,4. Примером одной из таких типичных лекарственных форм является бактериостатический водный раствор при рН около 6, содержащий пептид, раствор которого вводят парентерально, в дозе порядка от около 0,1 до 2,5 мг/кг массы тела в день. Эти соединения считаются хорошо переносимыми in vivo и устойчивыми к гелеобразованию, и поэтому они могут быть вполне подходящими для подкожных инъекций в форме бактериостатического водного раствора, содержащего около 5% маннита, при рН около 4,9, при подходящих концентрациях выше около 0,75 мг/мл и даже выше около 1,0, без риска гелеобразования в месте инъекции.
Эти пептиды-антагонисты GnRH могут быть также использованы в in vivo и in vitro-диагностике. Эти пептиды могут быть инъецированы in vivo с последующим анализом проб крови пациента для определения степени снижения секреции гормонов, например секреции LH. In vitro-анализы могут быть осуществлены для того, чтобы определить, являются ли некоторые опухолевые клетки восприимчивыми к GnRH. В этих анализах культуры опухолевых клеток обрабатывают пептидами-антагонистами GnRH, а затем прослеживают секрецию гормонов и пролиферацию клеток.
Хотя настоящее изобретение описано на его предпочтительных вариантах осуществления, однако в него могут быть внесены различные изменения и модификации, очевидные для каждого специалиста и не выходящие за рамки объема изобретения, сформулированного в нижеследующей формуле изобретения. N-конец может оставаться незащищенным либо могут быть использованы другие эквивалентные ацилирующие группы, однако предпочтительным является ацетил либо замещенный или незамещенный карбамоил. Вместо Aph или D-Aph могут быть использованы Aph или D-Aph в 5- и 6-положении соответственно. Вместо Aph(Ас) аминоРbе-группа может быть обработана альтернативными ацилирующими агентами, как описано в патенте N 5506207, такими как муравьиная кислота, β-Ala(atz) и гамма-аминомасляная кислота (atz), которые также приводят к получению антагонистов GnRH, обладающих пролонгированным действием, а поэтому полученные остатки рассматриваются как эквиваленты D- и L-4Aph(Ac). Оба Lys(Bu) и Lys(Et2) рассматриваются как эквиваленты Ilys; однако Ilys является наиболее предпочтительным. Другие гидрофобные аминокислотные остатки могут быть также использованы в 1-положении и в 6-положении (как упоминалось выше), предпочтительно в форме D-изомера, и рассматриваются как эквиваленты для вышеуказанных остатков.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПРОИЗВОДСТВА ДЕГАРЕЛИКСА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2011 |
|
RU2602042C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДЕГАРЕЛИКСА | 2010 |
|
RU2515555C2 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ СИНТЕЗА АНТАГОНИСТА РИЛИЗИНГ-ФАКТОРА ЛЮТЕИНИЗИРУЮЩЕГО ГОРМОНА (LHRH), СПОСОБ ИХ ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТА LHRH | 2002 |
|
RU2307125C2 |
| Получение дегареликса | 2013 |
|
RU2657444C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ РАКА ПРЕДСТАТЕЛЬНОЙ ЖЕЛЕЗЫ | 2010 |
|
RU2536245C2 |
| КОМПОЗИЦИИ ПЕПТИДОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2009 |
|
RU2517135C2 |
| АНТАГОНИСТЫ LHRH, ИХ ПОЛУЧЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ПОЛУЧЕНИЕ, СПОСОБ ЛЕЧЕНИЯ ОПУХОЛЕЙ И БЕСПЛОДИЯ У МЛЕКОПИТАЮЩИХ | 2001 |
|
RU2248982C9 |
| ДЕКАПЕПТИД, ОБЛАДАЮЩИЙ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2084458C1 |
| КОМПОЗИЦИЯ КОНТРОЛИРОВАННОГО ВЫСВОБОЖДЕНИЯ, ВКЛЮЧАЮЩАЯ GNRH-II | 1999 |
|
RU2233170C2 |
| АНТАГОНИСТ ЛГ-РИЛИЗИНГ-ФАКТОРА (LHRH), КОМПЛЕКС, СПОСОБ ХИМИЧЕСКОЙ КАСТРАЦИИ И/ИЛИ ЛЕЧЕНИЯ ГОНАДОТРОПИНЗАВИСИМЫХ РАССТРОЙСТВ | 1994 |
|
RU2130464C1 |



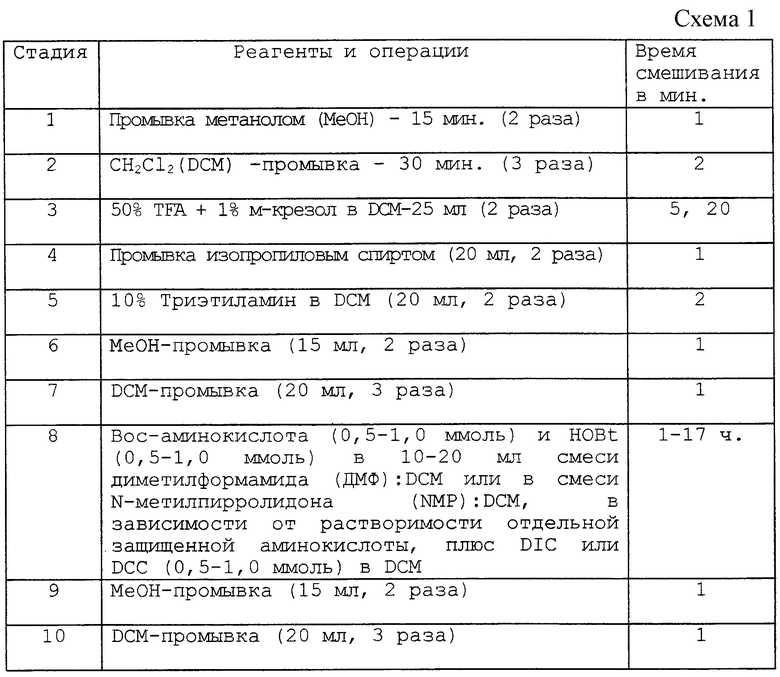
Изобретение относится к пептидам-антагонистам GnRH, имеющим формулу: X-D-Nal-(A)D-Phe-D-Pal-Ser-Xaa5-Xaa6-Leu-Xaa8-Pro-Xaa10, и их фармацевтически приемлемым солям, где Х = ацильная группа, имеющая более 7 атомов углерода; А = 4Cl или 4F; Хаа5 представляет собой Арh(Q1) или Аmf(Q1), где Q1 представляет собой Q или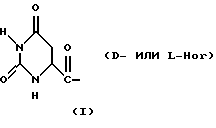
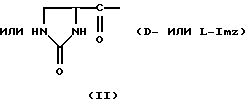
где Q представляет собой 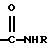 и где R представляет собой Н или низший алкил; Хаа6 представляет собой D-Aph(Q2), D-Amf(Q2), D-Cit или D-Pal, где Q2 представляет собой Аc или Q1; Xaa8 представляет собой Lys(ipr); и Хаа10 представляет собой D-Ala-NH2, D-Ala-ol, Ala-ol, NHCH2CH3, Gly-NH2 или Ala-NH2, при условии, что, если Хаа5 содержит Q, Хаа6 также содержит Q; фармацевтической композиции для ингибирования секреции гонадотропинов у млекопитающих, содержащей в качестве активного ингредиента эффективное количество антагониста по GnRH; способу ингибирования секреции гонадотропинов у млекопитающих и соединению X1-D-Nal-(A)D-Phe-D-Pal-Ser (X2)-Xaa5-Xaa6-Leu-Lys (ipr)(X4)-Pro-X5, гдe Х1 = α-аминозащитная группа; А = 4Сl или 4F; Х2 = Н или гидроксизащитная группа; Хаа5 представляет собой Aph(Q1) или Amf(Q1), где Q1 = структуры I и II, или карбамоил, или метилкарбамоил; Хаа6 = D-Aph(Q2), D-Amf(Q2) или D-Pal, где Q2 = Ас; X4 представляет собой восприимчивую к действию кислоты аминозащитную группу; и Х5 представляет собой D-Ala-, Gly-, Аlа-[полимерный носитель], N(Et)-[полимерный носитель], амид D-Ala, Gly или Аlа; или этиламид, являющемуся промежуточным соединением для получения пептида-антагониста GnRH. 4 с. и 12 з.п.ф-лы.
и где R представляет собой Н или низший алкил; Хаа6 представляет собой D-Aph(Q2), D-Amf(Q2), D-Cit или D-Pal, где Q2 представляет собой Аc или Q1; Xaa8 представляет собой Lys(ipr); и Хаа10 представляет собой D-Ala-NH2, D-Ala-ol, Ala-ol, NHCH2CH3, Gly-NH2 или Ala-NH2, при условии, что, если Хаа5 содержит Q, Хаа6 также содержит Q; фармацевтической композиции для ингибирования секреции гонадотропинов у млекопитающих, содержащей в качестве активного ингредиента эффективное количество антагониста по GnRH; способу ингибирования секреции гонадотропинов у млекопитающих и соединению X1-D-Nal-(A)D-Phe-D-Pal-Ser (X2)-Xaa5-Xaa6-Leu-Lys (ipr)(X4)-Pro-X5, гдe Х1 = α-аминозащитная группа; А = 4Сl или 4F; Х2 = Н или гидроксизащитная группа; Хаа5 представляет собой Aph(Q1) или Amf(Q1), где Q1 = структуры I и II, или карбамоил, или метилкарбамоил; Хаа6 = D-Aph(Q2), D-Amf(Q2) или D-Pal, где Q2 = Ас; X4 представляет собой восприимчивую к действию кислоты аминозащитную группу; и Х5 представляет собой D-Ala-, Gly-, Аlа-[полимерный носитель], N(Et)-[полимерный носитель], амид D-Ala, Gly или Аlа; или этиламид, являющемуся промежуточным соединением для получения пептида-антагониста GnRH. 4 с. и 12 з.п.ф-лы.
X-D-Nal-(A)D-Phe-D-Pal-Ser-Xaa5-Xaa6-Leu-Xaa8-Pro-Xaa10,
и его фармацевтически приемлемые соли,
где Х - ацильная группа, имеющая не более 7 атомов углерода;
А - 4Сl или 4F;
Хаа5 представляет собой Aph(Q1) или Amf(Q1), где Q1 представляет собой Q или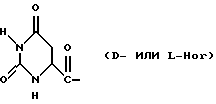
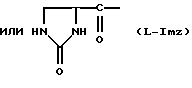
где Q представляет собой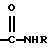
и где R представляет собой Н или низший алкил;
Хаа6 представляет собой D-Aph(Q2), D-Amf(Q2), D-Cit или D-Pal, где Q2 представляет собой Ас или Q1;
Xaa8 представляет собой Lys(ipr); и
Хаа10 представляет собой D-Ala-NH2, D-Ala-ol, Ala-ol, NHCH2CH3, Gly-NH2 или Ala-NH2, при условии, что если Хаа5 содержит Q, Хаа6 также содержит Q.
10. Антагонист GnRH по п.9, где Q1 представляет собой L- или D-Hor и Хаа6 представляет собой D-4Amf(Q), где R представляет собой Н или метил.
Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L-Hor)-Xaa6-Leu-Lys(ipr)-Pro-Xaa10,
где Хаа6 представляет собой D-4Aph(Ac), D-3Pal, D-4Aph(карбамоил), D-4Amf(карбамоил), D-4Аmf(метилкарбамоил) или D-4Aph(D-Hor) и Хаа10 представляет собой D-Ala-NH2, D-Ala-ol или Ala-ol.
Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L-Hor)-D-4Aph(карбамоил)-Leu-Lys(ipr)-Pro-D-Ala-NH2,
или его фармацевтически приемлемая соль.
X1-D-Nal-(A)D-Phe-D-Pal-Ser(X2)-Xaa5-Xaa6-Leu-Lys(ipr)(X4)-Pro-X5, гдe Х1 представляет собой α-амино-защитную группу;
А представляет собой 4Сl или 4F;
Х2 представляет собой Н или гидроксил-защитную группу;
Хаа5 представляет собой Aph(Q1) или Amf(Q1), где Q1 представляет собой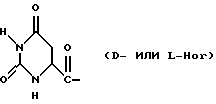
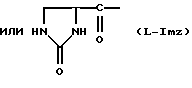
Хаа6 представляет собой D-Aph(Q2), D-Amf(Q2) или D-Pal, где Q2 представляет собой Ас, Q1, карбамоил или метилкарбамоил;
X4 представляет собой восприимчивую к действию кислоты аминозащитную группу; и
Х5 представляет собой D-Ala-, Gly-, А1а-[полимерный носитель], N(Et)-[полимерный носитель], амид D-Ala, Gly или А1а; или этиламид.
| ДЕКАПЕПТИД, ОБЛАДАЮЩИЙ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 1993 |
|
RU2084458C1 |
| RU 93004994/04, 10.03.1996 | |||
| Аналоги люлиберина, проявляющие ингибирующее действие на ход процессов овуляции у животных | 1981 |
|
SU1028663A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| US 5296468 А, 22.03.1994. | |||

