Область техники
Настоящее изобретение относится к производственному способу получения дегареликса.
Предшествующий уровень техники
Рак предстательной железы представляет собой главную причину заболеваемости и смертности мужчин в промышленно развитых странах. Дегареликс, также известный как FE 200486, является антагонистом рецептора гонадотропин-рилизинг-фактора (GnRH) (блокатором GnRH) третьего поколения, который был разработан и одобрен к применению пациентами с диагнозом рак предстательной железы, нуждающимися в терапии посредством андрогенной абляции (Doehn et al., Drugs, 2006, vol. 9, No. 8, pp. 565-571; WO 09846634). Дегареликс действует путем непосредственного и конкурентного блокирования рецепторов GnRH в гипофизе и, подобно другим антагонистам GnRH, не приводит к изначальной стимуляции выработки лютеинизирующего гормона посредством системы гипоталамус-гипофиз-половые железы и поэтому не приводит к выбросу тестостерона или «клиническому сигналу» (Van Poppel, Cancer Management and Research, 2010, 2, 39-52; Van Poppel et al., Urology, 2008, 71(6), 1001-1006); James E.F. et al., Drugs, 2009, 69(14), 1967-1976).
Дегареликс представляет собой синтетический линейный декапептид, содержащий семь неприродных аминокислот, пять из которых являются D-аминокислотами. В остове этого декапептида имеется десять хиральных центров. Аминокислотный остаток в положении 5 последовательности имеет дополнительный хиральный центр в заместителе боковой цепи, поэтому в общей сложности получается одиннадцать хиральных центров. Его регистрационный номер в CAS (химическая реферативная служба) 214766-78-6 (для свободного основания), и он доступен на рынке под товарным знаком Firmagon™. Эта лекарственная субстанция имеет химическое обозначение D-аланинамид, N-ацетил-3-(2-нафталинил)-D-аланил-4-хлор-D-фенилаланил-3-(3-пиридинил)-D-аланил-L-серил-4-[[[(4S)-гексагидро-2,6-диоксо-4-пиримидинил]карбонил]амино]-L-фенилаланил-4-[(аминокарбонил)амино]-D-фенилаланил-L-лейцил-N6-(1-метилэтил)-L-лизил-L-пролил- и представлена приведенной ниже химической структурой (далее также обозначаемой как формула I):

Структура дегареликса также может быть представлена в виде: Ac-D-2Nal-D-4Cpa-D-3Pal-Ser-4Aph(L-Hor)-D-4Aph(Cbm)-Leu-Lys(iPr)-Pro-D-Ala-NH2, где Ас представляет собой ацетил, 2Nal представляет собой 2-нафтилаланин, 4Сра представляет собой 4-хлорфенилаланин, 3Pal представляет собой 3-пиридилаланин, Ser представляет собой серии, 4Aph представляет собой 4-аминофенилаланин, Hor представляет собой гидрооротил, Cbm представляет собой карбамоил, Leu представляет собой лейцин, Lys(iPr) представляет собой N6-изопропиллизин, Pro представляет собой пролин, и Ala представляет собой аланин.
При описании данного изобретения для каждой аминокислоты в дегареликсе будет приведено сокращенное обозначение, указанное ниже:
AA1 представляет собой D-2Nal, АА2 представляет собой D-4Cpa, АА3 представляет собой D-3Pal, АА4 представляет собой Ser, АА5 представляет собой 4Aph(L-Hor), АА6 представляет собой D-Aph(Cbm), АА7 представляет собой Leu, АА8 представляет собой Lys(iPr), АА9 представляет собой Pro, и АА10 представляет собой D-Ala.
Таким образом, дегареликс может быть представлен в виде Ас-АА1-АА10-NH2.
Раньше дегареликс получали с использованием Вос(трет-бутилоксикарбонил)-методологии твердофазного пептидного синтеза (SPPS), как изложено в WO 98/46634 и Jiang et al., J. Med. Chem., 2001, 44, 453-467.
В WO 2010/12835 и WO 2011/066386 описано получение дегареликса с использованием Fmoc(флуоренилметоксикарбонил)-стратегии. В WO 2012/055905 и WO 2012/055903 описаны способы синтеза дегареликса в жидкой фазе.
Сущность изобретения
Определение физико-химических характеристик дегареликса показало, что этот декапептид обладает способностью к самоассоциации и, в конечном счете, к образованию гелей в водном растворе. Способность к самоагрегации способствует тому, что это соединение образует депо in situ при введении подкожной или внутримышечной инъекцией. Было показано, что депо дегареликса обеспечивает длительное высвобождение активного вещества в течение нескольких месяцев в зависимости от дозировки. В настоящее время это лекарственное средство вводят в дозировках 120 мг (40 мг/мл) для первой инъекции и 80 мг (20 мг/мл) для длительного высвобождения в течение одного месяца.
Авторы настоящего изобретения неожиданным образом обнаружили, что вязкость и, следовательно, свойства, обуславливающие длительное высвобождение, и биодоступность повторно разведенного лекарственного продукта можно регулировать в процессе обработки неочищенного пептида (например, полученного с использованием Fmoc-стратегии, синтеза в жидкой фазе или другого способа) с целью получения лекарственной субстанции. Вязкость, обусловленная лекарственной субстанцией, неожиданным образом коррелирует с вязкостью, обусловленной лекарственным продуктом, даже после дальнейшего повторного разведения и лиофилизации. Вязкость лекарственного продукта необходимо регулировать в диапазоне до 15 мПа⋅с включительно, предпочтительно в диапазоне от 2 до 12 мПа⋅с, чтобы произошло образование желаемого депо, и таким образом было реализовано длительное высвобождение. Согласно настоящему изобретению предложены способы, которые позволяют осуществить изготовление лекарственных продуктов, демонстрирующих такую вязкость, определенную при повторном разведении с использованием жидкости для повторного разведения в концентрации 20 мг дегареликса в одном мл жидкости для повторного разведения.
Таким образом, в первом аспекте согласно настоящему изобретению предложен способ регулирования вязкости продукта на основе дегареликса, так чтобы она составляла не более 15 мПа⋅с, предпочтительно в диапазоне 2-12 мПа⋅с, как определено при повторном разведении водой для инъекций из расчета 20 мг дегареликса в виде свободного основания/мл, включающий стадии:
1) предоставления лиофилизированной лекарственной субстанции дегареликса, которая демонстрирует вязкость до 3,2 мПа⋅с включительно, определенную после растворения в воде, содержащей маннит (2,5% масс./об.), в количестве 20 мг дегареликса в виде свободного основания/мл;
2) растворения лиофилизированной лекарственной субстанции дегареликса в маннит-содержащей воде с получением водной смеси дегареликс-маннит;
3) лиофилизации водной смеси дегареликс-маннит с получением лекарственного продукта на основе дегареликса.
Во втором аспекте согласно настоящему изобретению предложен способ изготовления лиофилизированного продукта на основе дегареликса, который демонстрирует вязкость до 15 мПа⋅с включительно, предпочтительно в диапазоне от 2 до 12 мПа⋅с, определенную при повторном разведении водой для инъекций в количестве 20 мг дегареликса в виде свободного основания/мл, включающий стадии:
1) предоставления лиофилизированной лекарственной субстанции дегареликса, которая демонстрирует вязкость до 3,2 мПа⋅с включительно, определенную после растворения в воде, содержащей маннит (2,5% масс./об.), в количестве 20 мг дегареликса в виде свободного основания/мл;
2) растворения лиофилизированной лекарственной субстанции дегареликса в маннит-содержащей воде с получением водной смеси дегареликс-маннит; и
3) лиофилизации водной смеси дегареликс-маннит с получением лекарственного продукта на основе дегареликса.
В третьем аспекте согласно настоящему изобретению предложен способ изготовления лекарственного продукта на основе дегареликса, включающего лиофилизированный лекарственный продукт на основе дегареликса и жидкость для повторного разведения (среду для повторного разведения), который, при повторном разведении указанной жидкостью в количестве 20 мг дегареликса в виде свободного основания/мл, демонстрирует вязкость до 15 мПа⋅с включительно, предпочтительно в диапазоне 2-12 мПа⋅с, включающий стадии:
1) растворения лиофилизированной лекарственной субстанции дегареликса в маннит-содержащем водном растворе с получением смеси дегареликс-маннит;
2) лиофилизации смеси дегареликс-маннит с получением лекарственного продукта на основе дегареликса,
где к смеси дегареликс-маннит перед лиофилизацией добавляют снижающий вязкость агент.
Согласно настоящему изобретению также предложены лиофилизированная лекарственная субстанция дегареликса, которая, после растворения в воде, содержащей 2,5 масс. % маннита, в количестве 20 мг дегареликса в виде свободного основания/мл, демонстрирует вязкость до 3,2 мПа⋅с включительно, и способы получения этой лиофилизированной лекарственной субстанции дегареликса.
Кроме того, согласно настоящему изобретению предложен лекарственный продукт на основе дегареликса, содержащий лиофилизированный лекарственный продукт на основе дегареликса и жидкость для повторного разведения, где продукт при повторном разведении указанной жидкостью для повторного разведения в количестве 20 мг дегареликса в виде свободного основания/мл, демонстрирует вязкость до 15 мПа⋅с включительно, предпочтительно в диапазоне от 2 до 12 мПа⋅с, и содержит снижающий вязкость агент в количестве 0,001-5 мг/мл.
Графические материалы
На Фиг. 1 показано соотношение между вязкостью лекарственной субстанции и вязкостью готовой лекарственной формы.
На Фиг. 2 и 3 показано соотношение между динамической вязкостью суспензии с дозированным количеством дегареликса (20 мг/мл) и концентрациями дегареликса в плазме крови (у крыс) на 3-и сутки (Фиг. 2) и 28-е сутки (Фиг. 3).
Подробное описание изобретения
Способы изготовления лиофилизированного продукта на основе дегареликса как по первому, так и по второму аспекту начинаются с использования лекарственной субстанции дегареликса, как будет описано более подробно.
Лекарственная субстанция дегареликса
Декапептид дегареликс может быть получен твердофазным пептидным синтезом, как описано в WO 98/46634, WO 2010/12835 и WO 2011/066386, или пептидным синтезом в жидкой фазе, как описано в WO 2012/055905 или WO 2012/055903. В результате этого пептидного синтеза получают неочищенный дегареликс, который далее очищают и затем лиофилизируют с получением лиофилизированного продукта, состоящего из дегареликса, уксусной кислоты, остаточного количества воды и незначительных количеств примесей, если они имеются, обусловленных способом изготовления. Этот продукт обозначается в настоящем изобретении как лекарственная субстанция дегареликса или просто лекарственная субстанция. Лекарственная субстанция дегареликса предпочтительно содержит дегареликс, 4,5-10 масс. % уксусной кислоты (масс./масс.) и до 10 масс. % включительно воды (масс./масс.).
Процесс получения лекарственной субстанции можно разделить на первую стадию (А), обеспечивающую получение очищенного дегареликса в растворе, и вторую стадию (Б), обеспечивающую получение лекарственной субстанции дегареликса.
Стадия (А): очистка
Стадия (А) включает в себя очистку неочищенного дегареликса за одну или более стадий, предпочтительно две стадии, возможно с последующей стадией концентрирования на колонке и/или стадией замены соли.
Неочищенный дегареликс, полученный с использованием жидкофазного (LPPS) или твердофазного (SPPS) пептидного синтеза, сначала подвергают очистке. Очистку предпочтительно осуществляют путем нанесения раствора пептида, полученного с использованием SPPS или LPPS, на колонку с носителем для обращенно-фазовой хроматографии, которую предварительно уравновешивают буфером. Эта первая стадия очистки предпочтительно обеспечивает чистоту по меньшей мере 95%, как определено посредством HPLC.
В предпочтительном воплощении обращенно-фазовую колоночную хроматографию повторяют, получая продукт с чистотой по меньшей мере 97,5%, как определено посредством HPLC.
В особо предпочтительном воплощении раствор очищенного дегареликса с чистотой по меньшей мере 95%, предпочтительно по меньшей мере 97,5%, подвергают дополнительной стадии колоночной хроматографии для предварительного концентрирования раствора дегареликса и/или для замены соли (в частности, если на последней стадии очистки для подведения pH элюентов используют уксусную кислоту).
Эту стадию предварительного концентрирования и/или замены соли также предпочтительно осуществляют на колонке для обращенно-фазовой хроматографии. Очищенный раствор дегареликса разбавляют водой (предпочтительно в 1,5-2,5 раза) и наносят на колонку, предварительно уравновешенную буфером. Предпочтительно, колонку сначала промывают этанолом (в низкой концентрации, как правило, ниже 20%) и водным раствором ацетата аммония и после этого смесью этанол (в низкой концентрации, как правило, ниже 20%)/уксусная кислота/вода. Затем колонку элюируют, например смесью этанол(в высокой концентрации, как правило, 20-60%)/уксусная кислота/вода, получая более концентрированный раствор дегареликса по сравнению с растворами после стадии(ий) очистки. Данный способ не ограничивается этанолом в качестве органического модификатора в элюенте. Также можно использовать и другие растворители, такие как ацетонитрил.
После стадии (А) получают очищенный дегареликс в растворе.
Стадия (Б)
Последующую обработку очищенного дегареликса в растворе с получением лекарственной субстанции дегареликса можно осуществить разными путями. Далее проиллюстрировано четыре предпочтительных пути (стадии (Б1), (Б2), (Б3) и (Б4)).
Стадия (Б1): концентрирование - дезагрегация - лиофилизация
Очищенный дегареликс в растворе сначала подвергают стадии концентрирования, на которой этанол или другой органический модификатор, такой как ацетонитрил, удаляют путем выпаривания. Эту стадию предпочтительно осуществляют с использованием роторного вакуумного испарителя. Предпочтительная максимальная температура при выпаривании составляет 40°С. Полученный сильно концентрированный вязкий продукт на основе дегареликса (агрегированный продукт, который обычно находится в форме геля) затем обрабатывают уксусной кислотой (стадия дезагрегации), предпочтительно фильтруют и лиофилизируют.
Стадия дезагрегации является важной для регулирования вязкости лекарственной субстанции. Поэтому, стадию дезагрегации предпочтительно осуществляют с применением одного или более, наиболее предпочтительно всех следующих условий:
- конечная концентрация уксусной кислоты: 6-40%, предпочтительно 15-35% (об./об.);
- температура 0-35°С, предпочтительно 2-30°С, наиболее предпочтительно 5-15°С;
- концентрация дегареликса (свободного основания) 5-35 г/л, предпочтительно 10-20 г/л;
- продолжительность 1-15 часов.
В этих границах подходящие комбинации параметров способа могут быть найдены простым экспериментированием.
Стадию лиофилизации предпочтительно осуществляют при толщине слоя льда 1,2-2,4 см и продолжительности вторичной сушки 1-17 часов. Температура вторичной сушки обычно составляет около 20°С (15-25°С).
Таким образом, в предпочтительном воплощении на стадии (Б1) получают лиофилизированную лекарственную субстанцию, которая демонстрирует вязкость менее 3,2 мПа⋅с, предпочтительно от 1,15 до 2 мПа⋅с (определенную после растворения в количестве 20 мг дегареликса в виде свободного основания в 1 мл воды, содержащей 2,5% (масс./об.) маннита). Метод измерения вязкости описан в экспериментальном разделе. Лекарственная субстанция, отвечающая такому требованию к вязкости, далее обозначается как лекарственная субстанция (1), тогда как лекарственная субстанция, не отвечающая такому требованию к вязкости, далее обозначается как лекарственная субстанция (2). Лекарственная субстанция (2) имеет предпочтительную вязкость 3,2-15 мПа⋅с после растворения в количестве 20 мг в 1 мл воды. Лекарственная субстанция (2) даже может быть гелеобразно, при условии существенного превышения 3,2 мПа⋅с, даже если ее вязкость точно измерить невозможно.
В связи с этим необходимо отметить, что фраза "после растворения в количестве 20 мг в 1 мл воды" относится главным образом к условиям для измерения вязкости и не означает, что лекарственная субстанция присутствует в растворе в концентрации 20 мг/мл. Наиболее предпочтительно, лекарственная субстанция присутствует в лиофилизированной форме.
Предпочтительно, лекарственная субстанция, в частности лекарственная субстанция (1), дополнительно характеризуется содержанием уксусной кислоты 4,5-10,0% (масс./масс.) и/или содержанием воды 10% или меньше (масс./масс.). Кроме того, лекарственная субстанция, в частности лекарственная субстанция (1), предпочтительно демонстрирует оптическую плотность 0,10 AU (единицы оптической плотности) или меньше (в концентрации 20 мг дегареликса в виде свободного основания в одном мл 2,5%-ного маннита (водн.)). Способ измерения оптической плотности описан в экспериментальном разделе.
Таким образом, согласно изобретению предложен способ получения лекарственной субстанции дегареликса (1), включающий стадии:
а) очистки дегареликса, полученного жидкофазным или твердофазным пептидным синтезом, с получением раствора дегареликса с чистотой по меньшей мере 95%;
б) выпаривания растворителя с целью концентрирования раствора дегареликса с получением агрегированного дегареликса;
в) дезагрегации агрегированного дегареликса с использованием уксусной кислоты; и
г) лиофилизации дезагрегированного дегареликса с получением лекарственной субстанции дегареликса.
Согласно изобретению также предложен способ модулирования вязкости дегареликса, так что после лиофилизации и повторного разведения водой вязкость раствора дегареликса в концентрации 20 мг/мл в 2,5%-ном (масс./об.) манните составляет не более 15 мПа⋅с, включающий:
- обработку агрегированного дегареликса уксусной кислотой; и
- лиофилизацию смеси дегареликса и уксусной кислоты.
В некоторых воплощениях условиями для добавления уксусной кислоты и лиофилизации являются такие, как описано выше. В конкретных воплощениях содержание уксусной кислоты после лиофилизации составляет 4,5-10,0% (масс./масс.).
Стадия (Б2): концентрирование в колонке - лиофилизация
Раствор дегареликса, полученный на стадии А (предпочтительно после осуществления метода одно- или двустадийной очистки, например, без предварительного концентрирования/замены соли), загружают на хроматографическую колонку, осуществляют ионный обмен и колонку промывают разбавленной уксусной кислотой (примерно 1%-ной). Дегареликс элюируют с колонки, используя водный раствор уксусной кислоты с концентрацией АсОН 20-50 масс. %, предпочтительно 23-37 масс. %, предпочтительно 23-27 масс. %, предпочтительно 27-37 масс. %, предпочтительно 33-37 масс. %, предпочтительно 35 масс. %, и после этого возможно разбавляют до соответствующей концентрации АсОН, фильтруют. Затем раствор дегареликса в смеси АсОН/вода лиофилизируют. Неподвижная фаза в колонке может быть разных типов. Неподвижная фаза может содержать функциональные группы типа групп углеводородов (алифатических и ароматических), спиртов, нитрилов, групп с соответствующими свойствами кислот/оснований и ионообменных групп, но не ограничивается этим типом групп. В результате осуществления этого способа получают лекарственную субстанцию, предпочтительно лекарственную субстанцию (1).
Таким образом, согласно изобретению также предложен способ получения лекарственной субстанции дегареликса, включающий стадии:
а) очистки дегареликса, полученного жидкофазным или твердофазным пептидным синтезом, с получением раствора дегареликса с чистотой по меньшей мере 95%;
б) загрузки раствора дегареликса на хроматографическую колонку;
в) элюирования дегареликса с колонки с использованием уксусной кислоты с получением элюированного дегареликса;
г) лиофилизации элюированного дегареликса с получением лекарственной субстанции дегареликса.
Стадия (Б3): выделение посредством лиофилизации - повторное разведение в смеси АсОН/вода - лиофилизация
Очищенный дегареликс в растворе, полученный со стадии А, выделяют посредством лиофилизации; полученный лиофилизированный продукт растворяют в концентрации от 10 до 20 г/л в 2%-ной уксусной кислоте и снова лиофилизируют, получая лекарственную субстанцию дегареликса (1).
Стадия (Б4): распылительная сушка
Очищенный дегареликс в растворе (в смеси EtOH/(вода, содержащая АсОН) или ACN/(вода, содержащая АсОН), как получено после стадии А или как описано на стадии Б1, за исключением лиофилизации, или Б2, за исключением лиофилизации) подвергают распылительной сушке, получая лекарственную субстанцию дегареликса (1).
В предпочтительном воплощении очищенный раствор дегареликса, полученный на стадии А, непосредственно подвергают процессу распылительной сушки. Концентрацию АсОН в указанном водном растворе дегареликса, подвергаемом распылительной сушке, подводят до 6-40% (об./об.), предпочтительно 15-35% (об./об.).
После изложения процесса изготовления лекарственной субстанции, далее будет описано изготовление лекарственного продукта на основе дегареликса. Лиофилизированный лекарственный продукт на основе дегареликса содержит лекарственную субстанцию дегареликса и маннит, т.е. он содержит (и предпочтительно состоит из) дегареликс, уксусную кислоту, маннит, остаточное количество воды и незначительные количества примесей, если они имеются, обусловленных способом изготовления.
Способ А для изготовления лекарственного продукта на основе дегареликса
Способ изготовления А представляет собой первый аспект изобретения, упомянутый выше, т.е. способ изготовления лекарственного продукта на основе дегареликса, который, при повторном разведении водой для инъекций в количестве 20 мг дегареликса в виде свободного основания/мл, демонстрирует вязкость до 15 мПа⋅с включительно, предпочтительно в диапазоне 2-12 мПа⋅с, включающий стадии:
а) предоставления лиофилизированной лекарственной субстанции дегареликса, предпочтительно лекарственной субстанции дегареликса (1);
б) растворения лиофилизированной лекарственной субстанции дегареликса в маннит-содержащей воде с получением водной смеси дегареликс-маннит;
в) лиофилизации водной смеси дегареликс-маннит с получением лекарственного продукта на основе дегареликса.
Использованная в настоящем изобретении фраза "при повторном разведении водой для инъекций в количестве 20 мг/мл" относится к условиям для измерения вязкости и не означает, что лекарственный продукт присутствует в растворе в концентрации 20 мг/мл. Наиболее предпочтительно, лекарственный продукт присутствует в лиофилизированной форме, возможно в комбинации с жидкостью для повторного разведения. Предпочтительные количества на один флакон находятся в диапазоне 60-300 мг (например, 120 мг, 80 мг и 240 мг). Альтернативно, он может быть предложен в виде повторно разведенного лекарственного продукта с предпочтительными концентрациями в диапазоне от 2 до 100 мг/мл, предпочтительно от 10 до 70 мг/мл (как например, 40 мг/мл, 20 мг/мл и 60 мг/мл).
Стадия б) также может быть названа стадией смешивания. В предпочтительном воплощении фильтрацию и розлив во флаконы осуществляют после смешивания и перед сублимационной сушкой, таким образом, в целом предпочтительный способ изготовления А включает стадии:
- смешивания с получением нефильтрованного нерасфасованного лекарственного продукта;
- фильтрации (стерильной);
- розлива во флаконы;
- сублимационной сушки/лиофилизации.
Смешивание с получением нерасфасованного лекарственного продукта
Лекарственную субстанцию подвергают стадии смешивания, которую обычно осуществляют так, как описано ниже.
Чтобы изготовить нефильтрованный нерасфасованный лекарственный продукт, лекарственную субстанцию и маннит растворяют в воде (чистой воде; обычно в воде Milli-Q), и то и другое в количествах 10-60 г; из расчета на партию размером 1000 г. Типичные количества составляют от 20 до 50 г лекарственной субстанции (по содержанию дегареликса в виде свободного основания, как определено посредством HPLC) и от 10 до 50 г маннита на 1000 г. Фактическое количество зависит от конечной концентрации дегареликса в лекарственном продукте и объема жидкости для повторного разведения (маннит предпочтительно добавляют так, чтобы после повторного разведения был получен изотонический раствор с осмоляльностью 300 мОсм +/- 30 мОсм).
При изготовлении в сосуд для смешивания добавляют воду (обычно приблизит 80% от общего количества воды). Добавляют маннит, и растворение выполняют при перемешивании. Затем к перемешиваемому раствору маннита добавляют лекарственную субстанцию, и массу приготовленного нерасфасованного продукта (партии) подводят до конечного значения, добавляя оставшуюся воду. Это смешивание осуществляют таким образом, чтобы избежать значительного увеличения вязкости. Таким образом, вязкость нерасфасованного продукта предпочтительно остается ниже 5 мПа⋅с, предпочтительно ниже 3,2 мПа⋅с, в ходе стадии смешивания (вязкость определяли по окончании фильтрации после растворения в количестве 20 мг в 1 мл 2,5%-ного (масс./об.) водного раствора маннита). Этого можно добиться путем увлажнения пептида при высокой скорости перемешивания в течение относительно короткого промежутка времени (до 30 минут включительно) и затем растворения пептида при сниженной скорости перемешивания с целью избегания вспенивания и агрегации (в общем случае, от 30 до 90 минут). Температуру обычно поддерживают в диапазоне 6-15°С. Предпочтительно используют такую мешалку, которая обеспечивает турбулентное перемешивание без вихреобразования
Фильтрация
Затем нерасфасованный лекарственный продукт предпочтительно подвергают стерильной фильтрации, например, через два стерилизующих фильтра, расположенных последовательно, подавая под давлением азота приготовленный нерасфасованный продукт.
Розлив
Осуществляют розлив профильтрованного нерасфасованного лекарственного продукта в стерилизованные флаконы, и их укупоривают пробкой наполовину (положение для сублимационной сушки) в асептических условиях.
Сублимационная сушка
Установку для сублимационной сушки перед применением предпочтительно стерилизуют паром. Флаконы помещают на полки сублимационной сушилки. Последующий процесс сублимационной сушки предпочтительно включает стадии замораживания, основной сушки (сублимации) и вторичной сушки. Предпочтительные условия приведены ниже.
Процесс сублимационной сушки предпочтительно включает или даже состоит из трех основных стадий, т.е. замораживания, основной сушки (сублимации) и вторичной сушки.
- Замораживание
Флаконы загружают на охлаждаемые полки, температуру которых поддерживают при 2-10°С, например, 5°С.
Полки охлаждают от, например, 5°С до температуры от -30 до -40°С, например, -35°С. Температуру на полках поддерживают, например, при -35°С в течение минимум двух часов, чтобы гарантировать осуществление полного замораживания всей партии перед началом первичной сушки.
- Основная сушка (сублимация)
Основную сушку осуществляют путем снижения давления в камере (предпочтительно до 0,100 мбар (10 Па) или ниже) и повышения температуры на полках (предпочтительно до 10-20°С, например, +17°С).
Продолжительность основной сушки составляет по меньшей мере 15 часов.
- Вторичная сушка
По завершении процесса первичной сушки давление в камере снижают (предпочтительно до 0,01 мбар (1 Па) или меньше), а температуру на полках повышают (предпочтительно до 20-30°С, например, 25°С). Обычно вторичная сушка завершается в пределах 7 часов.
Лиофилизированный лекарственный продукт затем снабжают этикеткой, упаковывают и объединяют с соответствующим количеством жидкости для повторного разведения.
Жидкость для повторного разведения выбирают в зависимости от вязкости лекарственной субстанции. Если в качестве исходного вещества для способа изготовления А используют лекарственную субстанцию (1), т.е. лиофилизированную лекарственную субстанцию, которая демонстрирует вязкость менее 3,2 мПа⋅с, предпочтительно от 1,15 до 2 мПа⋅с (после растворения в количестве 20 мг в 1 мл 2,5%-ного (масс./об.) водного раствора маннита), то полученный лиофилизированный лекарственный продукт предпочтительно объединяют с водой для инъекций (WFI) в качестве жидкости для повторного разведения. Если лиофилизированный лекарственный продукт повторно разводят, используя WFI, то вязкость обычно находится в диапазоне 2-15 мПа⋅с (по результатам измерений после растворения 20 мг дегареликса (свободного основания) в 1 мл WFI). Было обнаружено, что лекарственный продукт с вязкостью в пределах этого диапазона обеспечивает образование депо, достаточного для высвобождения дегареликса in vivo.
Способ Б для изготовления лекарственного продукта на основе дегареликса
Способ изготовления Б представляет собой способ изготовления лекарственного продукта на основе дегареликса, содержащего лиофилизированный лекарственный продукт на основе дегареликса и жидкость для повторного разведения (среду для повторного разведения), который, при повторном разведении указанной жидкостью в количестве 20 мг дегареликса в виде свободного основания/мл, демонстрирует вязкость до 15 мПа⋅с включительно, предпочтительно в диапазоне 2-12 мПа⋅с, включающий стадии:
а) растворения лиофилизированной лекарственной субстанции дегареликса в маннит-содержащем водном растворе с получением смеси дегареликс-маннит;
б) лиофилизации смеси дегареликс-маннит с получением лекарственного продукта на основе дегареликса,
где к смеси дегареликс-маннит перед лиофилизацией добавляют снижающий вязкость агент.
Стадия а) также может быть названа стадией смешивания. В предпочтительном воплощении фильтрацию и розлив во флаконы осуществляют после смешивания и перед сублимационной сушкой, таким образом, в целом предпочтительный способ изготовления Б включает стадии:
- смешивания с получением нефильтрованного нерасфасованного лекарственного продукта;
- фильтрации (стерильной);
- розлива во флаконы;
- сублимационной сушки/лиофилизации.
Способ изготовления Б идентичен способу изготовления А за исключением того, что на стадии смешивания перед лиофилизацией добавляют снижающий вязкость агент, предпочтительно неионное поверхностно-активное вещество. Неионное поверхностно-активное вещество предпочтительно добавляют в количестве 0,0003-1,5 мг/мл к нерасфасованному раствору, что соответствует количеству 0,001-5 мг в одном мл, более предпочтительно 0,1-1 мг в одном мл повторно разведенного лекарственного продукта (например, при повторном разведении до концентрации дегареликса, составляющей 60 мг дегареликса в виде свободного основания/мл). Предпочтительными неионными поверхностно-активными веществами являются вещества с линейной алкильной цепью, имеющей по меньшей мере 8 атомов углерода (предпочтительно без двойных связей), и углеводной группировкой. В частности, предпочтительными являются вещества, одобренные к применению для подкожных инъекций, такие как твин 20 (полисорбат 20). Другие подходящие неионные поверхностно-активные вещества включают токоферил-полиэтилен-гликоль-1000-сукцинат (TPGS) и другие твины.
В качестве исходного вещества для способа изготовления Б предпочтительно используют лекарственную субстанцию (2), т.е. лиофилизированную лекарственную субстанцию, которая демонстрирует вязкость по меньшей мере 3,2 мПа⋅с (после растворения в количестве 20 мг дегареликса в виде свободного основания в 1 мл 2,5 масс. %-ного водного раствора маннита). Полученный лиофилизированный лекарственный продукт обычно объединяют с WFI в качестве жидкости для повторного разведения. Если лиофилизированный лекарственный продукт повторно разводят, используя WFI, вязкость обычно находится в диапазоне 2-15 мПа⋅с (по результатам измерений после растворения 20 мг дегареликса в виде свободного основания в 1 мл WFI). Было обнаружено, что лекарственный продукт с вязкостью в пределах этого диапазона обеспечивает образование депо, достаточного для реализации задержанного высвобождения дегареликса in vivo.
Способ изготовления Б особенно предпочтителен для продуктов на основе дегареликса, которые при повторном разведении имеют относительно высокую концентрацию дегареликса, как например, 50 мг дегареликса в виде свободного основания/мл или более, например 60 мг дегареликса в виде свободного основания/мл (240 мг лекарственного продукта).
Новый лекарственный продукт на основе дегареликса
Способ изготовления Б обеспечивает получение нового лекарственного продукта на основе дегареликса, отличающегося от известных лекарственных продуктов тем, что такой повторно разведенный лекарственный продукт содержит снижающий вязкость агент. Снижающий вязкость агент представляет собой агент, используемый на стадии Б, предпочтительно неионное поверхностно-активное вещество.
Таким образом, согласно настоящему изобретению предложен лекарственный продукт на основе дегареликса, содержащий лиофилизированный лекарственный продукт на основе дегареликса и жидкость для повторного разведения, который, при повторном разведении указанной жидкостью в количестве 20 мг дегареликса в виде свободного основания/мл, демонстрирует вязкость до 15 мПа⋅с включительно, предпочтительно в диапазоне 2-12 мПа⋅с, и содержит снижающий вязкость агент в концентрации 0,001-5 мг/мл, более предпочтительно 0,1-1 мг/мл.
Экспериментальный раздел
Пример 1. Очистка, дезагрегация и лиофилизация
Неочищенный дегареликс синтезировали, как описано в WO 2012/055905 А1 до стадии 12 в примере 5. Стадия 13, описанная в WO 2012/055905 А1, была заменена следующими стадиями. Кратко, способ очистки неочищенной лекарственной субстанции дегареликса, полученной по окончании последней стадии удаления защиты, состоит из трех стадий препаративной обращенно-фазовой хроматографии (RPC), где третья стадия RPC представляет собой главным образом стадию обессоливания.
Стадия 13 (стадия очистки)
Неочищенный раствор дегареликса со стадии 12 в WO 2012/055905 A1 наносили на колонку, заполненную носителем для обращенно-фазовой хроматографии, предварительно уравновешенную буфером (90% 0,12%-ной TFA (трифторуксусная кислота) и 10% EtOH). Загрузка: не более 30 г/л объема колонки. Колонку промывали и элюировали градиентом: буфер (EtOH от 29% до 50% и 0,12%-ная TFA (водн.) от 71% до 50%). Полученные фракции анализировали и объединяли таким образом, чтобы чистота основного пула отвечала критерию приемлемости для контроля технологического процесса.
По данным контроля процесса элюирования с использованием «метода HPLC» чистота составляет не менее 95%.
Стадия 14 (стадия очистки)
Основной пул, полученный на стадии 13, разбавляли в два раза водой и наносили на колонку, заполненную носителем для обращенно-фазовой хроматографии, предварительно уравновешенную буфером (90% 1%-ной АсОН и 10% EtOH). Загрузка: не более 25 г/л объема колонки. Колонку сначала промывали первым буфером (10% EtOH и 90% AcONH4 в концентрации 0,5 моль/л) и затем вторым буфером (90% 1%-ной АсОН и 10% EtOH).
Затем колонку элюировали смесью буфера и этанола (76% 1%-ной АсОН и 24% EtOH). Полученные фракции анализировали и объединяли таким образом, чтобы чистота основного пула отвечала критерию приемлемости для контроля технологического процесса.
По данным контроля процесса элюирования с использованием «метода HPLC» чистота составляет не менее 97,5%.
Стадия 15 (предварительное концентрирование/замена соли)
Основной пул, полученный на стадии 14, разбавляли в два раза водой и наносили на колонку, заполненную носителем для обращенно-фазовой хроматографии, предварительно уравновешенную буфером (90% 1%-ной АсОН и 10% EtOH). Загрузка: не более 18 г/л объема колонки. Колонку сначала промывали первым буфером (10% EtOH и 90% AcONH4 в концентрации 0,5 моль/л) и затем вторым буфером (90% 1%-ной АсОН и 10% EtOH). Затем колонку элюировали смесью буфера и этанола (50% 1%-ной АсОН и 50% EtOH.
В качестве альтернативы дегареликс может быть элюирован раствором AcOH/MeCN/вода, таким как 12% АсОН и 22% MeCN в воде.
Стадия 16 (концентрирование - дезагрегация - лиофилизация)
Перед лиофилизацией пул растворов очищенного дегареликса со стадии 15 концентрировали при температуре ниже 40°С. К концентрированному раствору добавляли водный раствор уксусной кислоты и воду, получая концентрацию ниже 15 г/л и концентрацию уксусной кислоты 30%. Этот раствор затем фильтровали и лиофилизировали, получая лекарственную субстанцию дегареликса.
Результаты
Вязкость лекарственной субстанции, полученной в результате выполнения последовательности стадий, составляла меньше 2,5 мПа⋅с, как определено для концентрации 20 мг/мл в 2,5%-ном (масс./об.) растворе маннита.
Пример 2. Способ измерения вязкости для лекарственной субстанции и лекарственного продукта
Определение вязкости раствора лекарственной субстанции и лекарственного продукта основано на методике Европейской фармакопеи (Ph. Eur.) и Фармакопеи США (USP) в текущем издании, в которой используется ротационный вискозиметр, оснащенный измерительная системой «конус-плоскость» с D=60 мм и 1°. Скорость сдвига увеличивают от 0 до 500 с-1 за 20 шагов с использованием пошаговой программы для вращения с регулируемой скоростью (CR) при постоянной температуре 20±0,2°С, убеждаясь, что система достигает равновесия прежде, чем вязкость регистрируется при скорости сдвига 500 с-1.
Пример 3. Способ измерения оптической плотности для лекарственной субстанции и лекарственного продукта
Оборудование и материалы
- УФ-спектрофотометр.
- Кюветы, пропускающие ультрафиолетовое (УФ) излучение, длина оптического пути 10 мм.
- Крышки для кювет.
- Вода для инъекций (используемая для повторного разведения лекарственного продукта на основе дегареликса и в кювете сравнения).
Проведение анализа
В промежутке между повторным разведением и измерением повторно разведенные образцы должны храниться при 22°С±1°С.
Во флакон с лекарственной субстанцией вносят 2,5%-ный раствор маннита в воде (масс./об.) для повторного разведения. Во флакон с лекарственным продуктом вносят воду для инъекций для повторного разведения. Во флакон отмеряют растворитель, и содержимое флакона перемешивают путем его вращения до полного растворения или в течение нескольких секунд с применением вортекса. Жидкость должна быть прозрачной и не должна содержать видимого нерастворенного порошка или частиц. Необходимо хранить флакон в вертикальном положении и не встряхивать. Измерение для образца выполняют при 350 нм, через 120 минут после добавления растворителя.
За четыре минуты до измерения образец необходимо гомогенизировать путем осторожного переворачивания кюветы пять раз туда и обратно приблизительно через 180 градусов. Такая четырехминутная задержка позволяет удалить все пузырьки воздуха перед снятием показаний.
Поглощение, обусловленное кюветой и водой для инъекций, необходимо вычесть из показаний для образца.
Пример 4. Распылительная сушка
Подготовка загрузочного материала
При подготовке загрузочного материала (feed) взвешенное количество дегареликса, показанное ниже в таблице, растворяли с использованием магнитной мешалки. Разные растворы загрузочного материала, W-VI и AI-IV, готовили путем добавления воды (W) Milli-Q или чистой ледяной уксусной кислоты (А) (99,9%-ной) следующим образом:
а) партия W-IV: к взвешенному количеству дегареликса добавляли соответствующий объем воды;
б) партия от А-I до A-III: для каждого эксперимента готовили растворы уксусной кислоты путем разведения 99,0%-ной ледяной уксусной кислоты водой Milli-Q, получая 30-, 5-, 2-процентные растворы, к которым добавляли взвешенное количество дегареликса.
Перед всеми циклами распылительной сушки раствор повторно разведенного пептида фильтровали в мерную посуду через фильтр Ministar, 0,20 мкм, от Sartoriuos перед распылительной сушкой.
Распылительная сушка
Перед распылительной сушкой регулировали температуру на входе и скорость подачи загрузочного материала. Подводящие шланги насоса помещали в раствор загрузочного материала и начинали сушку. После завершения сушки температуру на входе оставляли опускаться до 700°С, после чего циклон и приемник для сбора снимали, чтобы осуществить сбор порошка. Порошок собирали щеточками в чашки Петри, которые взвешивали до и после сбора для определения выхода.
Описание параметров настройки, применяемых для распылительной сушки

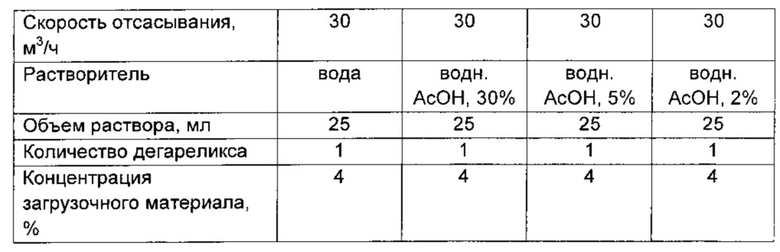
Вязкость повторно разведенного дегареликса, высушенного распылением из растворов уксусной кислоты четырех разных концентраций (от А-I до A-III), в сравнении с одной партией дегареликса, высушенного распылением из воды (W-IV)

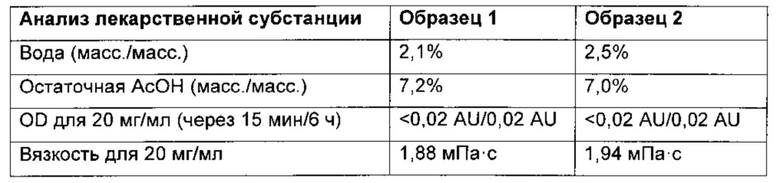
Пример 5. Концентрирование в колонке - лиофилизация
Пул со стадии 14 примера 1 разбавляли водой и наносили на колонку, упакованную носителем для обращенно-фазовой хроматографии. После промывания колонки 1%-ной АсОН в воде дегареликс элюировали 35%-ной АсОН в воде. Фракции регулировали таким образом, чтобы они содержали 35 г дегареликса/л, 27% АсОН (образец 1), и 15 г дегареликса/л, 30% АсОН (образец 2). После доведения фракции подвергали сублимационной сушке и анализировали. Результаты: см. приведенную ниже таблицу.
Пример 6. Изготовление лекарственной субстанции дегареликса и исследования дезагрегации
- Материалы
Исследования дезагрегации очищенной сырой лекарственной субстанции
- Очищенный сырой дегареликс поставлялся в концентрации 57,4 мг/мл.
- Уксусная кислота, 100%-ная, поставлялась фирмой Merck.
- Оборудование для исследования дезагрегации:
- сосуд для смешивания: стеклянный сосуд с двойными стенками емкостью 300 мл,
- бутылки с голубыми крышками, 50 или 100 мл,
- магнитный стержень,
- магнитная мешалка.
- Первичное фасование:
- бесцветные стеклянные флаконы марки 20R. Флаконы промывали и сушили в термошкафу,
- пробки для сублимационной сушки, 20 мм, тип I, соответствующие обжимным съемным колпачкам типа flip-off, 20 мм, по Ph. Eur./USP.
Изготовление лекарственных продуктов с использованием дезагрегированных лекарственных субстанций
- Использовали лекарственные субстанции из двух экспериментов. В качестве контроля используют имеющуюся в продаже лекарственную субстанцию.
- Маннит: D-(-)-маннит (PF-05-0232).
- Первичное фасование:
- бесцветные стеклянные флаконы марки 10R (согласно DIN ISO 8362).
Флаконы промывают и сушат в термошкафу,
- пробки для сублимационной сушки, 20 мм, тип I, согласно Ph. Eur./USP (тип 1319, из резины W4416/50/серые, The West Company),
- обжимные съемные колпачки типа flip-off, 20 мм (The West Company).
- Способы
Эксперименты по дезагрегации с использованием концентрированного (stock) раствора лекарственной субстанции
Температура в ходе экспериментов:
в процессе исследований дезагрегации температуру устанавливали на 5, 20, 25 или 35°С.
Концентрация лекарственной субстанции дегареликса в концентрированных растворах в ходе экспериментов:
концентрация лекарственной субстанции дегареликса составляла 5, 15, 25 или 35 мг/мл.
Концентрация уксусной кислоты в ходе экспериментов:
концентрация уксусной кислоты составляет 13, 15, 18, 20, 22, 26, 30, 35 или 40%.
Анализ, выполненный в ходе экспериментов:
вязкость: отобрать образец приблизительно 1,2 мл в моменты времени 1, 60, 120 и 240 мин.
Если по регламенту удобнее отобрать образец в другой момент времени, то это можно сделать лишь при условии, что в протоколе технологии изготовления записано корректное время отбора. Тем не менее, точка отбора образца на момент времени 1 мин должна быть сохранена.
Оптическая плотность (OD): в конце каждого эксперимента необходимо отобрать образец (приблизительно 1 мл) для измерения конечной оптической плотности.
Протокол экспериментов по дезагрегации, общий объем 20 или 69 мл:
1) взвесить необходимое количество концентрированного раствора в химическом стакане. Уравновесить до желаемой температуры;
2) взвесить необходимое количество уксусной кислоты (100%-ной) в химическом стакане;
3) смешать уксусную кислоту (100%-ную) с 3 или 10 мл воды milli-Q и уравновесить до желаемой температуры;
4) смешать концентрированный раствор дегареликса с раствором уксусная кислота/вода milli-Q;
5) дополнить водой milli-Q до 20 или 69 мл;
6) тщательно перемешать;
7) отобрать образцы для определения вязкости и оптической плотности, как описано выше. В ходе эксперимента делать пометки относительно внешнего вида раствора.
Параметры для дезагрегации двух концентрированных растворов дегареликса по 69 мл:
концентрация лекарственной субстанции дегареликса в обоих экспериментах 25 мг/мл;
уксусная кислота: 10% или 13%; температура: 5°С.
Розлив растворов двух тестируемых дезагрегированных лекарственных субстанций (69 мл) во флаконы и сублимационная сушка: дезагрегированные растворы немедленно разливали во флаконы марки 20R. В каждый флакон разливали по 5 мл. В процессе розлива флаконы и нерасфасованную субстанцию следует держать на холоде (предпочтительно в диапазоне 5-10°С). Как только розлив во флаконы был закончен, их помещали в сублимационную сушилку и запускали программу.
После сублимационной сушки и закрывания флаконов их оставляли в сублимационной сушилке при 5°С до выгрузки.
Для двух подвергнутых сублимационной сушке лекарственных субстанций проводили следующие анализы:
- вязкость измеряли для образцов свободного основания в концентрации 20 мг/мл,
- оптическую плотность измеряли для образцов в концентрации 20 мг/мл,
- содержание дегареликса,
- содержание ацетата.
- Результаты

Пример 7. Изготовление лекарственного продукта без снижающего вязкость агента
Стадия смешивания для лабораторных партий лекарственного продукта с использованием двух дезагрегированных лекарственных субстанций
Во всех экспериментах со смешиванием используют эксципиенты, перечисленные ниже в Таблице 1.

Оборудование для проведения смешивания:
- сосуд для смешивания: стеклянный сосуд с двойными стенками емкостью 300 мл,
- бутылки с голубыми крышками, 100 мл,
- магнитный стержень,
- магнитная мешалка.
Первичное фасование:
- бесцветные стеклянные флаконы марки 10R. Флаконы промывали и сушили в термошкафу,
- пробки для сублимационной сушки, 20 мм, тип I,
- обжимные съемные колпачки типа flip-off, 20 мм.
Описание исследования
Нерасфасованные растворы дегареликса, содержащие дегареликс в концентрации 20 мг/г и маннит в концентрации 25 мг/г, смешивают, используя разные партии лекарственной субстанции и разные настройки температуры.
Вязкости нерасфасованных растворов измеряют в ходе/после смешивания для двух случаев (после растворения и при t=120 минут).
Состав нерасфасованных растворов
Размер партии и состав двух нерасфасованных растворов представлены в Таблице 1.


План эксперимента
Параметры смешивания приведены ниже в Таблице 2.

Смешивание
Смешивание проводят в центральном положении мешалки. Перед началом экспериментов необходимо зафиксировать центральное положение.
1. Взвесить необходимые ингредиенты:
- 40 г воды milli-Q в сосуде для смешивания,
- 1,25 г маннита в подходящем контейнере,
- взвесить точное количество лекарственной субстанции дегареликса (см. Таблицу 2) в подходящем контейнере,
- взвесить оставшееся количество воды milli-Q в подходящем контейнере.
2. К охлаждающему циркулятору присоединить сосуд с двойными стенками для смешивания, содержащий воду milli-Q. Присоединить последовательно второй сосуд с двойными стенками. Контейнер, содержащий оставшуюся воду, поместить во второй сосуд с двойными стенками, чтобы подвести его температуру до корректной температуры, после чего добавить к нерасфасованному раствору (стадия 9).
3. Осторожно поместить магнитный стержень для перемешивания в сосуд для смешивания. Установить скорость перемешивания 50 об/мин, используя тахометр.
4. Установить корректную температуру в охлаждающем устройстве, начать перемешивание и добавить маннит в сосуд для смешивания. Перемешивать до полного растворения маннита.
5. Оставить систему достигать заданной температуры.
6. Измерить температуру в растворе маннита.
7. Удалить мешалку. Включить таймер и незамедлительно добавить лекарственную субстанцию. Затем вновь установить мешалку.
8. При t=5 минут добавить остаток воды milli-Q, равномерно над поверхностью лекарственной субстанции, используя стеклянную пипетку Пастера.
9. Когда остается лишь незначительное количество комков, смыть лекарственную субстанцию со стенок сосуда средой для растворения.
10. После полного растворения лекарственной субстанции сделать отметку времени и измерить температуру. Отобрать образец для измерения вязкости нерасфасованного раствора.
11. Продолжить эксперимент до t=120 минут. Отобрать образец для измерения вязкости нерасфасованного раствора.
Фильтрование
Нерасфасованные растворы не будут подвергнуты фильтрованию, поскольку ожидается, что по меньшей мере в одной из партий будет образовываться много агрегатов, и поэтому ее фильтрование будет сильно затруднено. Кроме того, эти партии не будут использованы в тех случаях, когда необходима стерильность.
Розлив
Провести розлив по 6,40 г нерасфасованного раствора во флаконы марки 10R. В процессе розлива флаконы и нерасфасованный раствор следует держать на холоде (предпочтительно в диапазоне 5-10°С). Как только розлив во флаконы завершен, их необходимо поместить в сублимационную сушилку и запустить программу. В результате для каждого нерасфасованного раствора должно получиться приблизит. 6-7 флаконов.
- Результаты
18 разных партий лекарственной субстанции (DS, drug substance) с разными значениями вязкости, которые указаны ниже в таблице, обрабатывали, как описано выше. Вязкость для соответствующих лекарственных продуктов (DP, drug products) определяли так, как приведено ниже.
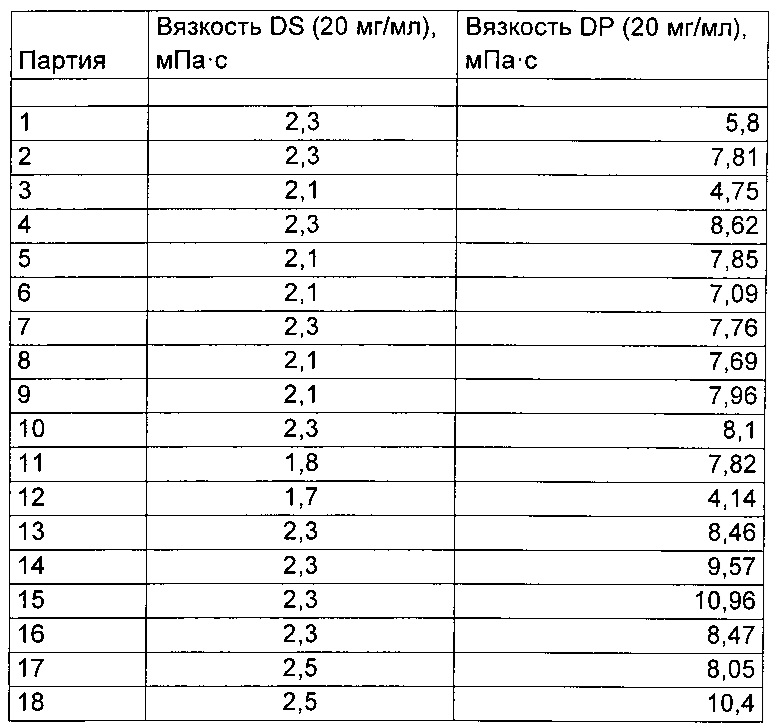
Эти результаты проиллюстрированы графически на Фиг. 1. На Фиг. 1 показана корреляция между вязкостью лекарственной субстанции и вязкостью лекарственного продукта.
Пример 8. Изготовление лекарственного продукта со снижающим вязкость агентом
Эксперимент 1. Тестируемые поверхностно-активные вещества (повторное разведение раствором WFI/поверхностно-активного вещества)
Методика:
- партия дегареликса, повторно разведенная с использованием WFI (в качестве контроля) и с использованием WFI, содержащей поверхностно-активные вещества. Выполняли повторное разведение содержимого флаконов до концентрации дегареликса 60 мг/мл. Партия лекарственного продукта, использованная для этих экспериментов, в виде нерасфасованного раствора имела вязкость приблизит.3,8 мПа⋅с;
- готовили растворы TPGS и твина 20 с содержанием 1 мг/мл (0,1%-ные растворы);
- OD (поглощение) измеряли при 350 нм;
- поглощение регистрировали через 30 мин, 60 мин, 120 мин и 24 часа.
Результат
Как TPCG, так и твин 20 снижали оптическую плотность до уровня ниже, чем в контроле.
Эксперимент 2. Повторное разведение дегареликса во флаконах с использованием [WFI + твин 20] не влияет на профиль высвобождения in vitro (диагностика in vitro (IVD)) при концентрации дегареликса 40 мг/мл
Тестируемые концентрации твина 20:
1 мг/мл; 0,5 мг/мл; 0,25 мг/мл;
контроль: WFI.
Результаты: профили высвобождения in vitro не изменяются при добавлении твина-20.
Пример 9. Вязкость и биодоступность лекарственного продукта
Краткое изложение
Многомерный анализ данных выполняли, используя набор данных от 38 партий лекарственного продукта на основе дегареликса, в том числе фармакокинетические данные in vivo (модель на крысах).
Соотношение между физико-химическими характеристиками и фармакокинетическими параметрами in vivo устанавливали на крысиной модели. В результате проведения этого исследования было выявлено, что вязкость повторно разведенного продукта на основе дегареликса, по-видимому, является главным и единственным параметром, по которому можно до некоторой степени предсказывать свойства депо in vivo.
- Введение
Лекарственный продукт на основе дегареликса изготавливают в виде высушенного сублимационной сушкой продукта, содержащего маннит. Такие продукты используют в качестве исследуемых лекарственных продуктов в клинических исследованиях. Готовили несколько композиций, содержащих различные количества дегареликса на один флакон, а именно, 10 мг, 20 мг, 40 мг, 88 мг, 128 мг, 120 мг (40 мг/мл), 180 мг (60 мг/мл) и 240 мг (60 мг/мл) и характеризующихся разными соотношениями дегареликс/маннит.
- Материалы
Было изготовлено приблизительно 40 разных партий дегареликса с использованием разных партий субстанции и композиций дегареликса. Стандартная концентрация дегареликса в нерасфасованном растворе составляет 20 г/л, если не указано иное.
- Методы
- Физико-химические методы
Для характеристики агрегации были выбраны разные методы из многофакторного анализа данных. Собирали данные от 38 партий. Однако, для некоторых методов использовали меньшее количество, и эти методы осуществляли только для некоторых композиций (например, измерения при 40 мг/мл не проводили для композиции в 20 мг).
- Биологический анализ на крысах
Использовали стандартный анализ на крысах, заключающийся в отслеживании фармакокинетического профиля дегареликса в течение 28 суток. Чтобы избежать местных побочных эффектов, крысам вводили дегареликс в виде суспензии 20 мг/мл, при этом вводимый объем составлял 100 мкл. Использовали группы из 8 крыс, при этом всех обрабатывали суспензией, приготовленной повторным разведением из одного единственного флакона. Сначала концентрации в плазме крови измеряли на момент времени 2 ч, на 1-е сутки, 7-е сутки и 28-е сутки и площадь под кривой по частям (partial AUC) рассчитывали в виде AUC0-7 сут и AUC7-28 сут. Затем схему эксперимента изменяли, пропуская измерение концентрации в плазме крови (Ср) на момент времени 2 ч и заменяя на Ср на 3-и сутки. Аналогичным образом, AUC0-7 сут заменяли на AUC1-7 сут. Это объясняет неоднородность в объемах совокупности некоторых из переменных параметров in vivo.
- Многомерный анализ данных
С учетом размера набора данных задействовали подход с использованием многомерного анализа данных (метода главных компонент (РСА, principal components analysis) и метода частных наименьших квадратов (PLS, partial least squares)) с программным обеспечением Simca-P, версия 10 (Umetrics AB, SE-Umeå). Данные обрабатывали, как и ранее, включая мягкое шкалирование блока (soft block scaling) (корень 4-й степени (1/4 root)), приведение к единичной дисперсии (scaling to unit variance) (UV) и центрирование. Для данных, относящихся к мутности, выполняли логарифмическое преобразование для улучшения распределения данных. Для данных, относящихся к Ср in vivo, также выполняли логарифмическое преобразование для стабилизации дисперсии.
Соотношение между физико-химическими характеристиками лекарственного продукта и свойствами in vivo в крысиной модели
- Соотношение между физико-химическими характеристиками и свойствами in vivo
Первую модель рассчитывали, беря за основу 2 компоненты. Критерий согласия (R2 составляет 0,58) был относительно низким с учетом высокой вариабельности биологических данных (20-30%), но точность прогноза (Q2 составляет 0,35) была удовлетворительной (разница R2-Q2 должна быть в диапазоне от 0,2 до 0,3). Из диаграммы разброса данных по нагрузке (loading scatter plot) (не показана) можно видеть, что такие переменные, как мутность, измеренная при 20 мг/мл, и содержание ацетата, не оказывали существенного влияния.
Поэтому создавали новую модель, исключая эти 3 переменные (2 относящиеся к мутности переменные, измеренной при 20 мг/мл, и одну к ацетату), беря за основу 2 главные компоненты и получая аналогичное описание данных (R2 составляет 0,53), но улучшенный прогноз (Q2 составляет 0,42). Самыми обоснованными и прогнозируемыми переменными в крысиной модели были концентрации в плазме крови на 3-й сутки и 28-е сутки (Ср3 cут, Ср28 сут) и площадь под кривой в интервале от 1-х суток до 7-х суток (AUC1-7 сут).
Диаграмма анализа коэффициентов (coefficient overview plot) указывала на то, что более сильное влияние оказывали данные для вязкости, соответственно, при 20 мг/мл, а затем при 40 мг/мл, все были статистически значимыми при доверительном уровне 0,95 для каждой биологической переменной. Характерная площадь под кривой и мутность несомненно оказывали меньшее влияние и не были статистически значимыми даже при доверительном уровне 0,90.
Соотношение между динамической вязкостью, измеренной при 20 мг/мл, и наилучшим образом аппроксимированными биологическими переменными показано на Фиг. 2 и 3.
- Заключение
Было установлено устойчивое соотношение (R2 составляет 0,53) между вязкостью полученного после разведения продукта и свойствами in vivo, что изучали на крысиной модели. В обоих случаях более высокое значение вязкости приводит к ослаблению высвобождения из депо. Другие физико-химические переменные оказались нерелевантными, за исключением содержания ацетата для растворения in vitro.
Таким образом, вязкость полученного после разведения продукта на основе дегареликса, по-видимому, является главным параметром, по которому можно до некоторой степени предсказывать высвобождение in vitro и функциональные свойства депо in vivo.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения лиофилизата бортезомиба и фармацевтическая композиция, содержащая бортезомиб в форме стабильного лиофилизованного продукта, полученная указанным способом | 2018 |
|
RU2696854C1 |
| Фармацевтическая композиция нейропротекторного действия для парентерального применения на основе гексаметилендиамида бис-(N-моносукцинил-L-глутамил-L-лизина) в лиофилизированной лекарственной форме | 2017 |
|
RU2678203C2 |
| Способ получения лиофилизата бортезомиба и фармацевтическая композиция, содержащая бортезомиб в форме стабильного лиофилизованного продукта, полученная указанным способом | 2017 |
|
RU2659160C1 |
| НОВЫЙ СПОСОБ СТАБИЛИЗАЦИИ БИОФАРМАЦЕВТИЧЕСКОГО ЛЕКАРСТВЕННОГО ПРОДУКТА ПРИ ПРОИЗВОДСТВЕ | 2017 |
|
RU2744630C2 |
| ЛИОФИЛИЗИРОВАННЫЕ ПРЕПАРАТЫ БЕНДАМУСТИНА ГИДРОХЛОРИДА | 2014 |
|
RU2679614C2 |
| Препарат гормона роста человека длительного типа | 2014 |
|
RU2683823C2 |
| Фармацевтический состав слитого белка taci-fc | 2020 |
|
RU2779387C1 |
| ЛИОФИЛИЗИРОВАННЫЕ КОМПОЗИЦИИ CCI-779 | 2004 |
|
RU2345772C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭКТИНЭСАЙДИН И ДИСАХАРИД | 2005 |
|
RU2382647C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОДУКТА ГЛАТИРАМЕРА АЦЕТАТА | 2015 |
|
RU2669769C2 |

Группа изобретений относится к медицине и касается дегареликса и способов изготовления лекарственного продукта, содержащего субстанцию дегареликса. Субстанция включает дегареликс, уксусную кислоту в количестве от 4,5 до 10,0% (масс./масс.), воду в количестве 10% (масс./масс.). После растворения в воде 20 мг дегареликса в виде свободного основания и маннита в количестве 2,5 масс. % субстанция демонстрирует вязкость до 3,2 мПа⋅с. Вязкость измеряют вискозиметром с измерительной системой «конус-плоскость» с D=60 мм и 1°. Скорость сдвига увеличивают от 0 до 500 с-1 за 20 шагов при постоянной температуре 20±0,2°С. Также предложены способы получения лекарственной субстанции дегареликса и изготовления лиофилизированного лекарственного продукта дегареликса. Изобретения позволяют осуществить изготовление лекарственных продуктов, демонстрирующих такую вязкость и биодоступность, которая обеспечивает длительное высвобождение активного вещества в течение нескольких месяцев в зависимости от дозировки. 5 н. и 7 з.п. ф-лы, 3 ил., 2 табл., 8 пр.
1. Лиофилизированная лекарственная субстанция дегареликса, состоящая из дегареликса, уксусной кислоты в количестве от 4,5 до 10,0% (масс./масс.), воды в количестве 10% (масс/масс.) или менее и незначительного количества примесей, если они обусловлены способом изготовления, которая после растворения в воде в количестве 20 мг дегареликса в виде свободного основания/мл воды, содержащей 2,5 масс. % маннита, демонстрирует вязкость до 3,2 мПа⋅с включительно, где вязкость измеряют посредством ротационного вискозиметра, оснащенного измерительной системой «конус-плоскость» с D=60 мм и 1°, при этом скорость сдвига увеличивают от 0 до 500 с-1 за 20 шагов с использованием пошаговой программы для вращения с регулируемой скоростью (CR) при постоянной температуре 20±0,2°С.
2. Лиофилизированная лекарственная субстанция дегареликса по п. 1, которая после растворения в воде в количестве 20 мг дегареликса в виде свободного основания/мл воды, содержащей 2,5 масс. % маннита, демонстрирует вязкость от 1,15 до 2,0 мПа⋅с.
3. Способ получения лекарственной субстанции дегареликса по п. 1 или 2, включающий стадии:
а) очистки дегареликса, полученного жидкофазным или твердофазным пептидным синтезом, с получением раствора дегареликса с чистотой по меньшей мере 95%;
б) выпаривания растворителя для концентрирования раствора дегареликса с получением агрегированного дегареликса;
в) дезагрегации агрегированного дегареликса с использованием уксусной кислоты, где концентрация уксусной кислоты в конце стадии (в) находится в диапазоне от 15 до 35% (об./об.), и
г) лиофилизации дезагрегированного дегареликса с получением лекарственной субстанции дегареликса, которая после растворения в воде в количестве 20 мг дегареликса в виде свободного основания/мл воды, содержащей 2,5 масс. % маннита, демонстрирует вязкость до 3,2 мПа⋅с включительно.
4. Способ по п. 3, где температура на стадии (в) находится в диапазоне от -5 до 30°С.
5. Способ по п. 3, где концентрация дегареликса (в виде свободного основания) находится в диапазоне от 10 до 35 г/л.
6. Способ получения лекарственной субстанции дегареликса по п. 1 или 2, включающий стадии:
а) очистки дегареликса, полученного жидкофазным или твердофазным пептидным синтезом, с получением раствора дегареликса с чистотой по меньшей мере 95%;
б) загрузки раствора дегареликса на хроматографическую колонку;
в) элюирования дегареликса с колонки уксусной кислотой, имеющей концентрацию от 27 до 37 масс. %, с получением элюированного дегареликса;
г) лиофилизации элюированного дегареликса с получением лекарственной субстанции дегареликса, которая после растворения в воде в количестве 20 мг дегареликса в виде свободного основания/мл воды, содержащей 2,5 масс. % маннита, демонстрирует вязкость до 3,2 мПа⋅с включительно.
7. Способ по п. 6, где элюированный дегареликс фильтруют перед лиофилизацией.
8. Способ получения лекарственной субстанции дегареликса по п. 1 или 2, включающий стадии:
а) очистки дегареликса, полученного жидкофазным или твердофазным пептидным синтезом, с получением раствора дегареликса с чистотой по меньшей мере 95%;
б) доведения концентрации уксусной кислоты в очищенном растворе дегареликса до 6-40% (масс./масс.);
в) распылительной сушки раствора дегареликса с получением лекарственной субстанции дегареликса, которая после растворения в воде в количестве 20 мг дегареликса в виде свободного основания/мл воды, содержащей 2,5 масс. % маннита, демонстрирует вязкость до 3,2 мПа⋅с включительно.
9. Способ изготовления лиофилизированного лекарственного продукта на основе дегареликса, содержащего лекарственную субстанцию дегареликса и маннит, где продукт при повторном разведении водой для инъекций в количестве 20 мг дегареликса в виде свободного основания/мл воды, содержащей 2,5 масс. % маннита, демонстрирует вязкость до 15 мПа⋅с включительно, включающий стадии:
а) предоставления лиофилизированной лекарственной субстанции дегареликса по п. 1 или 2;
б) растворения лиофилизированной лекарственной субстанции дегареликса в маннитсодержащей воде с получением водной смеси дегареликс-маннит;
в) лиофилизации водной смеси дегареликс-маннит с получением лиофилизированного лекарственного продукта на основе дегареликса.
10. Способ по п. 9, где стадию растворения осуществляют так, что вязкость водной смеси дегареликс-маннит остается на уровне 3,2 мПа⋅с или менее.
11. Способ по п. 9, где за стадией растворения следует стадия фильтрации и стадия розлива перед лиофилизацией.
12. Способ по п. 9, где стадия лиофилизации включает стадию сублимации при температуре ниже температуры коллапса.
| WO 2011004260 A2,13.01.2011 | |||
| US 2005282731 A1, 22.12.2005 | |||
| Приспособление для опоражнивания открытых котлов | 1926 |
|
SU12183A1 |
| ЖИДКИЕ ДЕПО-ПРЕПАРАТЫ | 2005 |
|
RU2390331C2 |

