Изобретение относится к области медицины и химической технологии, может быть использовано в производстве лекарственных средств для лечения сахарного диабета 2-го типа.
Сахарный диабет представляет собой группу метаболических заболеваний, характеризующихся патологически повышенным уровнем глюкозы в крови, который возникает вследствие дефектов в секреции инсулина, действии инсулина, или обоих дефектах. Число больных сахарным диабетом в мире в конце 1999 года составляло 120-140 млн человек, и это число по прогнозам к 2025 году может удвоиться [1].
Лечение сахарного диабета заключается в поддержании нормального уровня глюкозы в крови. Для этой цели используются различные лекарственные средства, например сульфонилмочевины, метформин и инсулин. Эксенатид является одним из лекарственных средств, эффективность лечения которым инсулинонезависимого сахарного диабета является доказанной в клинических испытаниях на людях.
Эксенатид представляет собой полипептид, известный первоначально под названием экзендин-4 (exendin-4), способ выделения которого из яда ящерицы Heloderma suspectum и аминокислотная последовательность HGEGTFTSDL SKQMEEEAVR LFIEWLKNGG PSSGAPPPS-NH2 (1), впервые были опубликованы в апреле 1992 [2].
Эксенатид обладает сахаропонижающим действием и является лекарственным препаратом против диабета второго типа [3].
Известно [4], что полипептид формулы (1) может быть получен твердофазным методом с использованием амидных смол Ринка (Rink amide, Rink amide AM, Rink amide MBHA) по Fmoc/t-Bu стратегии последовательным наращиванием цепи по одной аминокислоте. Недостатком известного способа является использование дорогостоящих полимерных носителей, а также длительное время синтеза последовательным наращиванием цепи.
Известен способ получения полипептида формулы (1) [5] твердофазным методом с использованием амидной смолы Зибера по Fmoc/t-Bu стратегии последовательным наращиванием цепи по одной аминокислоте. Недостатками данного способа являются использование дорогостоящего полимерного носителя и длительное время синтеза последовательным наращиванием цепи, а также применение дорогостоящих реагентов (HATU и HOAt) для присоединения первой аминокислоты.
Недостатками способа получения эксенатида последовательным наращиванием цепи [4, 5] является возможность удвоения и/или исключения одной из аминокислот последовательности, а также побочные процессы по боковым функциональным группам, что приводит к усложнению очистки целевого продукта, и, как следствие, низкому выходу [6]. К недостаткам известного способа относится и то, что при последовательном наращивании цепи длинных пептидов возможно образование упорядоченных структур [6] и/или агрегация растущих цепей, что приводит к усложнению синтеза целевого продукта.
Известен способ получения полипептида формулы (1) [7] комбинированным методом с использованием как твердофазного синтеза по Fmoc/t-Bu стратегии, так и классического в растворе с использованием псевдопролина. В патенте представлена синтетическая схема (10) для получения эксенатида из первого пептидного фрагмента (12), второго пептидного фрагмента (14) и третьего пептидного фрагмента (16). Четвертый пептидный фрагмент (20) получают из третьего фрагмента реакцией конденсации с амидом серина. Затем четвертый фрагмент объединяют со вторым фрагментом, чтобы получить пятый фрагмент (22). В конечном итоге, пятый фрагмент объединяют с первым для получения целевого пептида. Известно [7], что ключевая проблема в твердофазном и растворном синтезах эксенатида заключается в наличии последовательности из трех остатков глутаминовой кислоты в положениях 15, 16, 17. Особенно трудно провести химический синтез пептидных фрагментов при условии наличия в них нескольких подряд идущих остатков глутаминовой кислоты, так как последовательность повторяющихся остатков глутаминовой кислоты склонна образовывать регулярную структуру. Это значительно усложняет эффективное продолжение построения фрагмента через глутаминовую цепь.
В патенте [7] указано, что длинные пептидные фрагменты, включающие в себя повторяющуюся глутаминовую последовательность, могут быть легко получены, если фрагменты также будут содержать один или более псевдопролиновых остатков как замену двум соответствующим аминокислотным остаткам. Эти предлагаемые псевдопролины являются дипептидами, содержащими как минимум одну аминокислоту, выбранную из группы серин-треонин, где боковая цепь защищена оксазолидиновым циклом.
В патенте [7] указано, что фрагмент (12) имеет как минимум один псевдопролин и является Xj,k эксенатидом (1-m), где 1 Xj,k обозначает псевдопролиновый остаток, m колеблется от 15-20 и C-концевая аминокислота фрагмента (12) пронумерована в соответствии с нумерацией аминокислотных остатков в эксенатиде.
Второй пептидный фрагмент (14) является эксенатидом (n-q), где n - это m+1 (m определено ранее в соответствии с первым фрагментом от 15 до 20), и q имеет номер от 25 до 30. Третий пептидный фрагмент (16) рассмотрен как экзенатид (q+1-38).
Недостатком известного способа является использование псевдопролинов в синтезе фрагментов эксенатида, что значительно увеличивает стоимость целевого продукта.
Наиболее близким по технической сущности и достигаемому результату к предложенному способу является способ получения полипептида (1) комбинированным методом с использованием как твердофазного синтеза по Fmoc/t-Bu стратегии, так и классического в растворе [6]. Согласно данному патенту вся последовательность эксенатида разбивается на четыре более коротких фрагмента: 1-10, 11-21, 22-29 и 30-39 (N- и C-концевые аминокислоты фрагментов пронумерованы в соответствии с нумерацией аминокислотных остатков в эксенатиде).
Известный фрагмент 1-10 Boc-His1(Trt)-Gly-Glu(OtBu)-Gly-Thr(tBu)-Phe-Thr(tBu)-Ser(tBu)-Asp(OtBu)-Leu10-OH (последовательность №11) получен с помощью твердофазного синтеза по Fmoc/t-Bu стратегии на 2-хлор-тритилхлоридной смоле с использованием Fmoc-Asp(OtBu)-OPfp и таких конденсирующих агентов, как DIC/HOBt и PyBOP. Для удаления временной Fmoc-защитной группы использовалась смесь пиперидин/DBU в DMF или в NMP. Для отщепления полученного фрагмента от полимерного носителя использовали 3.0-0.5% раствор TFA в дихлорметане. Целевой пептид последовательности №11 был получен с выходом 99% и чистотой 95%.
Известный фрагмент 11-21 Fmoc-Ser(tBu)-Lys(Boc)-Gln(Trt)-Met-Glu15(OtBu)-Glu(OtBu)-Glu(OtBu)-Ala-Val-Arg20(Pbf)-Leu-OH (последовательность №8) получен аналогично фрагменту 1-10 с использованием TOTU в качестве конденсирующего агента при присоединении Fmoc-Arg(Pbf)-OH и DIC/HOBt для присоединения других аминокислот. Целевой пептид последовательности №8 был получен с выходом 104% и чистотой 94%.
Известный фрагмент 22-29 Fmoc-Phe-Ile-Glu(OtBu)-Trp(Boc)-Leu-Lys(Boc)-Asn(Trt)-Gly-OH (последовательность №3) получен аналогично фрагменту 1-10 с использованием Fmoc-Asn(Trt)-OPfp и DIC/HOBt для присоединения других аминокислот. Целевой пептид последовательности №3 был получен с выходом 93% и чистотой 95%.
Известный фрагмент 30-39 Fmoc-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-Ala-Pro-Pro-Pro-Ser(tBu)-NH2 (последовательность №4) получен двумя различными методами.
Первый способ: известный фрагмент 30-38 Fmoc-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-Ala-Pro-Pro-Pro-OH (последовательность №12) получен на 2-хлор-тритилхлоридной смоле с использованием дипептида Fmoc-Pro-Pro-OH для присоединения к полимерному носителю, DIC/HOBt в качестве конденсирующего агента и раствора 25% пиперидина с 2.5% HOBt в DMF для удаления временной Fmoc-защитной группы. Для отщепления полученного фрагмента от полимерного носителя использовали 3.0-0.5% раствор TFA в дихлорметане. Целевой пептид последовательности №11 был получен с выходом 106% и чистотой 97%.
Конденсацию известного фрагмента 30-38 с H-Ser(tBu)-NH2 проводили в растворе с использованием TOTU в качестве конденсирующего агента. Целевой пептид последовательности №4 был получен с выходом 92% и чистотой 97%.
Второй способ: известный фрагмент 30-39 Fmoc-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-Ala-Pro-Pro-Pro-Ser(tBu)-NH2 (последовательность №4) получен твердофазным методом с использованием амидной смолы Зибера по Fmoc/t-Bu стратегии. В качестве конденсирующего агента применяли TCTU и Cl-HOBt, при наращивании цепи использовали дипептидный фрагмент Fmoc-Pro-Pro-OH. Целевой пептид последовательности №4 был получен с выходом 60% и чистотой 78%.
Известный фрагмент 22-39 Fmoc-Phe-Ile-Glu(OtBu)-Trp(Boc)-Leu-Lys(Boc)-Asn(Trt)-Gly-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-Ala-Pro-Pro-Pro-Ser(tBu)-NH2 (последовательность №6) получен в растворе NMP конденсацией фрагментов №3 и №4 в молярном соотношении 1:1 с предварительным деблокированием Fmoc-защитной группы фрагмента №4 (выход 99%, чистота 91%). В качестве конденсирующего агента использовали TBTU. Целевой пептид последовательности №6 был получен с выходом 100% и чистотой 89%.
Известный фрагмент 11-39 Fmoc-Ser(tBu)-Lys(Boc)-Gln(Trt)-Met-Glu15(OtBu)-Glu(OtBu)-Glu(OtBu)-Ala-Val-Arg20(Pbf)-Leu-Phe-Ile-Glu(OtBu)-Trp(Boc)-Leu-Lys(Boc)-Asn(Trt)-Gly-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-Ala-Pro-Pro-Pro-Ser(tBu)-NH2 (последовательность №9) получен в растворе конденсацией фрагментов №8 и №6 в молярном соотношении 1:1 с предварительным деблокированием Fmoc-защитной группы фрагмента №6 (выход 100%, чистота 87%). В качестве конденсирующего агента использовали TOTU. Целевой пептид последовательности №6 был получен с выходом 96% и чистотой 80%.
Известный фрагмент 1-39 Boc-His1(Trt)-Gly-Glu(OtBu)-Gly-Thr(tBu)-Phe-Thr(tBu)-Ser(tBu)-Asp(OtBu)-Leu10-Ser(tBu)-Lys(Boc)-Gln(Trt)-Met-Glu15(OtBu)-Glu(OtBu)-Glu(OtBu)-Ala-Val-Arg20(Pbf)-Leu-Phe-Ile-Glu(OtBu)-Trp(Boc)-Leu-Lys(Boc)-Asn(Trt)-Gly-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-Ala-Pro-Pro-Pro-Ser(tBu)-NH2 (последовательность №1) получен в растворе конденсацией фрагментов №11 и №9 в молярном соотношении 1:1 с предварительным деблокированием Fmoc-защитной группы фрагмента №9 (выход 98%, чистота 78%). В качестве конденсирующего агента использовали PyBOP. Целевой пептид последовательности №6 был получен с выходом 100% и чистотой 79%.
Деблокирование временной защитной Fmoc-группы проводилось в подходящих растворителях, таких как CH2Cl2, NMP или DMF с добавлением диэтиламина. Выделение фрагментов осуществляли с помощью DIPE.
Полное деблокирование проводили в растворе TFA, содержащем воду, TIS, NH4I и DMS. Финальная смесь получена с общим выходом 42% и содержанием целевого пептида 33%.
Недостатком известного способа получения смеси, содержащей полипептид формулы (1) является невысокий выход (42%) на последней стадии синтеза и низкое содержание (33%) целевого пептида в продукте. Также недостатком известного способа является использование различных конденсирующих агентов, таких как TOTU, TBTU, TCTU, PyBOP, а также пентафторфениловьгх эфиров аспарагиновой кислоты и аспарагина, усложняющих производственный процесс и выделяющих в процессе реакции аллергены и канцерогены [8, 9]. Использование таких реагентов и дипептидного фрагмента Fmoc-Pro-Pro-OH увеличивает стоимость производства препарата. Также нежелательно использование взрывоопасного DIPE в нескольких стадиях синтеза (таблица 1).
Кроме того, использование четырех длинных пептидных фрагментов (8, 10 и 11 аминокислотных остатков) в качестве исходных соединений для синтеза всей последовательности может приводить к возникновению проблем, существующим в методе последовательного наращивания цепи по одной аминокислоте, а именно: длительное время синтеза, возможные удвоения и/или исключения одной из аминокислот последовательности, побочные процессы по боковым функциональным группам.
Техническим результатом заявляемого изобретения является повышение выхода (42%-60%), чистоты (58%-75%) и содержания целевого продукта в смеси (33%-42%), использование более дешевого конденсирующего агента (DIC) на всех стадиях конденсации (таблица 1) и ускорение процесса. Указанный технический результат достигается способом, заключающимся в конденсации пептидных фрагментов соединения формулы (1) по схеме 34+5, согласно которому защищенный полипептид формулы (2) Boc-His1(Trt)-Gly-Glu(OtBu)-Gly-Thr(tBu)-Phe-Thr(tBu)-Ser(tBu)-Asp(OtBu)-Leu10-Ser(tBu)-Lys(Boc)-Gln(Trt)-Met-Glu15(OtBu)-Glu(OtBu)-Glu(OtBu)-Ala-Val-Arg20(Pbf)-Leu-Phe-Ile-Glu(OtBu)-Trp(Boc)-Leu-Lys(Boc)-Asn(Trt)-Gly-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-OH конденсируют в растворе с пентапептидом формулы (3) H-Ala-Pro-Pro-Pro-Ser-NH2, полученным деблокированием защищенного пентапептида формулы (4) Х-Ala-Pro-Pro-Pro-Ser(tBu)-NH2, где X может быть Fmoc, Boc или Z защитной группой. При конденсации полипептида (2) и пентапептида (3) могут быть использованы обычные в пептидном синтезе методы конденсации, например карбодиимидный метод.
Полипептид формулы (2) может быть получен обычными методами твердофазного синтеза, например постадийной конденсацией аминокислот или фрагментной конденсацией с использованием карбодиимидов или других конденсирующих агентов. В данном случае полипептид формулы (2) разбивают на 5 более коротких фрагментов 1-9 Fmoc-His(Trt)-Gly-Glu(OtBu)-Gly-Thr(tBu)-Phe-Thr(tBu)-Ser(tBu)-Asp(OtBu)-OH (формула 5), 10-14 Fmoc-Leu10-Ser(tBu)-Lys(Boc)-Gln(Trt)-Met-OH (формула 6), 15-20 Fmoc-Glu15(OtBu)-Glu(OtBu)-Glu(OtBu)-Ala-Val-Arg20(Pbf)-OH (формула 7), 21-24 Fmoc-Leu-Phe-Ile-Glu(OtBu)-OH (формула 8) и 25-34 Fmoc-Trp(Boc)-Leu-Lys(Boc)-Asn(Trt)-Gly-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-OH (формула 9). Все фрагменты могут быть получены известными способами, например твердофазным синтезом с использованием различных полимерных носителей и активирующих агентов. В данном случае фрагменты получены на 2-хлор-тритилхлоридной смоле с использованием DIC/HOBt в качестве конденсирующего агента. Полипептид формулы (2) получен конвергентным твердофазным методом путем конденсации вышеуказанных фрагментов на 2-хлор-тритилхлоридной смоле с использованием DIC/HOBt в качестве конденсирующего агента с высоким выходом и чистотой.
Небольшая длина пептидных фрагментов (от 4 до 10 аминокислотных остатков) позволяет использовать полимерный носитель с высокой нагрузкой и не применять дорогостоящих конденсирующих агентов без потерь в качестве получаемых пептидов. Благодаря использованию 6 фрагментов, синтез которых можно проводить параллельно, при сравнимых затратах (таблица 2), уменьшается время полного синтеза, финальную смесь удается получить с более высоким выходом и содержанием полипептида формулы (1).
Качественные реакции
Тест Кайзера. Небольшое количество смолы промыли несколько раз этанолом. К смоле добавили по 2 капли следующих растворов: 5% нингидрина в этаноле, 80% фенола в этаноле и KCN в пиридине (2 мл 0,001 М KCN в 98 мл пиридина). Смесь нагрели до 120°C. Синее окрашивание смолы указывало на присутствие свободной аминогруппы.
Полноту протекания конденсации в твердофазном методе синтеза контролировали также с помощью индикатора бромфенолового синего. В реакционную смесь добавляли 3 капли 1% раствора бромфенолового синего в диметилацетамиде (DMA). По мере протекания реакции окраска раствора изменялась с синей на желтую.
Оценка посадки первой аминокислоты на смолу
Посадку оценивали по количеству выделяемого в результате деблокирования Fmoc-группы дибензофульвена. Для этого хроматографически анализировали раствор дибензфульвена в N,N′-диметилформамиде, получившегося в результате деблокирования и промывки полимерного носителя. Концентрацию дибензфульвена вычисляли по площади его пика с помощью калибровочной кривой.
Аналитическую высокоэффективную жидкостную хроматографию проводили на хроматографе KNAUER Smartline 2500, оснащенном спектрофотометрическим детектором при длине волны 214 нм. При проведении аналитической ВЭЖХ использовали колонку Waters X-Bridge C18, 5 мкм, 4,5×10 мм при скорости потока элюента 1 мл/мин.
Пример 1.
Твердофазный синтез заявляемого фрагмента 1-9 Fmoc-His(Trt)-Gly-Glu(OtBu)-Gly-Thr(tBu)-Phe-Thr(tBu)-Ser(tBu)-Asp(OtBu)-OH (формула 5).
В твердофазном реакторе суспендировали 2 г 2-хлортритилхлоридной смолы (емкость 1,5 ммоль/г) в 15 мл DCM, выдерживали в течение 5 мин и отфильтровывали смолу. К смоле добавляли раствор 1,42 г (3,3 ммоль) С-концевой аминокислоты FmocAsp(OtBu)OH и 2 мл (12,0 ммоль) DIPEA в 10 мл DCM, перемешивали в течение 2 ч при комнатной температуре. Смолу отфильтровывали и промывали 2×10 мл DCM. Смолу обрабатывали 2×10 мл смеси DСМ/метанол/DIPEA (17:2:1) в течение 10 мин, промывали 2×10 мл DCM и 3×10 мл DMF. В реактор загружали 10 мл 20%-ного раствора диэтиламина в DMF, перемешивали в течение 5 мин и отфильтровывали; данную операцию повторяли еще раз в течение 20 мин, после чего смолу промывали 5×10 мл DMF. (Операция 4) К смоле добавляли охлажденный раствор 6,0 ммоль следующей защищенной аминокислоты, 0,98 г (7,2 ммоль) HOBt и 1,12 мл (7,2 ммоль) DIC в 10 мл DMF, перемешивали при комнатной температуре. Полноту протекания реакции контролировали по изменению окраски бромфенолового синего. Смолу отфильтровывали, промывали 6×10 мл DMF. (Операция 5) Операции 4 и 5 повторяли до получения требуемой последовательности. Затем смолу промывали 2×10 мл DCM, обрабатывали 30×10 мл 1%-ным раствором TFA в DCM; получаемые растворы объединяли в колбе, содержащей 40 мл 10%-ного раствора пиридина в метаноле. Смесь концентрировали на ротационном испарителе, продукт высаживали водой, отфильтровывали, промывали большим количеством воды, сушили на воздухе. Целевой пептид формулы 5 был получен с выходом (по посадке первой аминокислоты) 99% - 4,71 г и чистотой 99+%.
Пример 1a
Загрузка была увеличена в лабораторных условиях до 15 г смолы в реакторе. Присоединение первой аминокислоты Fmoc-Asp(OtBu)-OH к 2-хлортритилхлоридной смоле проводили в присутствии DIPEA в хлористом метилене. Посадку оценивали однократно хроматографически по количеству дибензофульвена, образующегося при деблокировании. Fmoc-группу удаляли 20% раствором диэтиламина в диметилформамиде. Конденсации следующих Fmoc-аминокислот были осуществлены с помощью реагентов DIC/HOBt в диметилформамиде с полуторократными избытками активируемых аминокислот. Фрагмент был отщеплен от подложки многократной обработкой 1% раствором TFA в хлористом метилене. Целевой пептид формулы 5 был получен с выходом 99% (по посадке первой аминокислоты) и чистотой 99+%.
Пример 2.
Твердофазный синтез заявляемого фрагмента 10-14 Fmoc-Leu10-Ser(tBu)-Lys(Boc)-Gln(Trt)-Met-OH (формула 6).
В твердофазном реакторе суспендировали 1,5 г 2-хлортритилхлоридной смолы (1,5 ммоль/г) в 15 мл DCM, выдерживали в течение 5 мин и отфильтровывали смолу. К смоле добавляли раствор 0,81 г (2,3 ммоль) C-концевой аминокислоты Fmoc-Met-OH и 1,5 мл (9,0 ммоль) DIPEA в 10 мл DCM, перемешивали в течение 2 ч при комнатной температуре. Смолу отфильтровывали и промывали 2×10 мл DCM. Смолу обрабатывали 2×10 мл смеси DCM/метанол/DIPEA (17:2:1) в течение 10 мин, промывали 2×10 мл DCM и 3×10 мл DMF. В реактор загружали 10 мл 20%-ного раствора диэтиламина в DMF, перемешивали в течение 5 мин и отфильтровывали; данную операцию повторяли еще раз в течение 20 мин, после чего смолу промывали 5×10 мл DMF. К смоле добавляли охлажденный раствор 4,5 ммоль следующей аминокислоты, 0,73 г (5,4 ммоль) HOBt и 0,85 мл (5,4 ммоль) DIC в 10 мл DMF, перемешивали при комнатной температуре. Полноту протекания реакции контролировали по изменению окраски бромфенолового синего. Смолу отфильтровывали, промывали 6×10 мл DMF. Операции 4 и 5 повторяли до получения требуемой последовательности. Затем смолу промывали 2×10 мл DCM, обрабатывали 20×10 мл 1%-ным раствором TFA в DCM; получаемые растворы объединяли в колбе, содержащей 40 мл 10%-ного раствора пиридина в метаноле. Смесь концентрировали на ротационном испарителе, продукт высаживали водой, отфильтровывали, промывали большим количеством воды, сушили на воздухе. Целевой пептид формулы 6 был получен с выходом 2,84 г - 105% и чистотой 96%.
Пример 2a
Загрузка была увеличена в лабораторных условиях до 15 г смолы в реакторе. Фрагмент формулы 6 получен аналогично фрагменту 1-9 с использованием DIC/HOBt в качестве конденсирующего агента. Целевой пептид формулы 6 был получен с выходом 106% и чистотой 95%.
Пример 3.
Твердофазный синтез заявляемого фрагмента 15-20 Fmoc-Glu15(OtBu)-Glu(OtBu)-Glu(OtBu)-Ala-Val-Arg20(Pbf)-OH (формула 7).
В твердофазном реакторе суспендировали 2 г 2-хлортритилхлоридной смолы (1,5 ммоль/г) в 15 мл DCM, выдерживали в течение 5 мин и отфильтровывали смолу. К смоле добавляли раствор 1,80 г (3,3 ммоль) C-концевой аминокислоты FmocArg(Pbf)OH и 2 мл (12,0 ммоль) DIPEA в 10 мл DCM, перемешивали в течение 2 ч при комнатной температуре. Смолу отфильтровывали и промывали 2×10 мл DCM. Смолу обрабатывали 2×10 мл смеси DCM/метанол/DIPEA (17:2:1) в течение 10 мин, промывали 2×10 мл DCM и 3×10 мл DMF. В реактор загружали 10 мл 20%-ного раствора диэтиламина в DMF, перемешивали в течение 5 мин и отфильтровывали; данную операцию повторяли еще раз в течение 20 мин, после чего смолу промывали 5×10 мл DMF. К смоле добавляли охлажденный раствор 6,0 ммоль следующей аминокислоты, 0,98 г (7,2 ммоль) HOBt и 1,12 мл (7,2 ммоль) DIC в 10 мл DMF, перемешивали при комнатной температуре. Полноту протекания реакции контролировали по изменению окраски бромфенолового синего. Смолу отфильтровывали, промывали 6×10 мл DMF. Операции 4 и 5 повторяли до получения требуемой последовательности. Затем смолу промывали 2×10 мл DCM, обрабатывали 30×10 мл 1%-ным раствором TFA в DCM; получаемые растворы объединяли в колбе, содержащей 40 мл 10%-ного раствора пиридина в метаноле. Смесь концентрировали на ротационном испарителе, продукт высаживали водой, отфильтровывали, промывали большим количеством воды, сушили на воздухе. Целевой пептид формулы 7 был получен с выходом 2,13 г - 98% и чистотой 99%.
Пример 3a
Загрузка была увеличена в лабораторных условиях до 15 г смолы в реакторе. Фрагмент формулы 7 получен аналогично фрагменту 1-9 с использованием DIC/HOBt в качестве конденсирующего агента. Целевой пептид формулы 7 был получен с выходом 97% и чистотой 99%.
Пример 4.
Твердофазный синтез заявляемого фрагмента 21-24 Fmoc-Leu-Phe-Ile-Glu(OtBu)-OH (формула 8).
В твердофазном реакторе суспендировали 2 г 2-хлортритилхлоридной смолы (1,5 ммоль/г) в 15 мл DCM, выдерживали в течение 5 мин и отфильтровывали смолу. К смоле добавляли раствор 1,92 г (4,5 ммоль) C-концевой аминокислоты FmocGlu(OtBu)OH и 2 мл (12,0 ммоль) DIPEA в 10 мл DCM, перемешивали в течение 2 ч при комнатной температуре. Смолу отфильтровывали и промывали 2×10 мл DCM. Смолу обрабатывали 2×10 мл смеси DCM/метанол/DIPEA (17:2:1) в течение 10 мин, промывали 2×10 мл DCM и 3×10 мл DMF. В реактор загружали 10 мл 20%-ного раствора диэтиламина в DMF, перемешивали в течение 5 мин и отфильтровывали; данную операцию повторяли еще раз в течение 20 мин, после чего смолу промывали 5×10 мл DMF. К смоле добавляли охлажденный раствор 6,0 ммоль следующей аминокислоты, 0,98 г (7,2 ммоль) HOBt и 1,12 мл (7,2 ммоль) DIC в 10 мл DMF, перемешивали при комнатной температуре. Полноту протекания реакции контролировали по изменению окраски бромфенолового синего. Смолу отфильтровывали, промывали 6×10 мл DMF. Операции 4 и 5 повторяли до получения требуемой последовательности. Затем смолу промывали 2×10 мл DCM, обрабатывали 30×10 мл 1%-ным раствором TFA в смеси DCM/THF (2:1); получаемые растворы объединяли в колбе, содержащей 40 мл 10%-ного раствора пиридина в метаноле. Смесь концентрировали на ротационном испарителе, продукт высаживали водой, отфильтровывали, промывали большим количеством воды, сушили на воздухе. Целевой пептид формулы 8 был получен с выходом 2,38 г - 100% и чистотой 99+%.
Пример 4a.
Загрузка была увеличена в лабораторных условиях до 15 г смолы в реакторе. Фрагмент формулы 8 получен аналогично фрагменту 1-9 с использованием DIC/HOBt в качестве конденсирующего агента. Целевой пептид формулы 8 был получен с выходом 100% и чистотой 99+%.
Пример 5.
Твердофазный синтез заявляемого фрагмента 25-34 H-Trp(Boc)-Leu-Lys(Boc)-Asn(Trt)-Gly-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-OH (формула 9).
В твердофазном реакторе суспендировали 1 г 2-хлортритилхлоридной смолы (1,5 ммоль/г) в 8 мл DCM, выдерживали в течение 5 мин и отфильтровывали смолу. К смоле добавляли раствор 0,51 г (2,1 ммоль) C-концевой аминокислоты FmocGlyOH и 1 мл (6,0 ммоль) DIPEA в 8 мл DCM, перемешивали в течение 2 ч при комнатной температуре. Смолу отфильтровывали и промывали 2×10 мл DCM. Смолу обрабатывали 2×10 мл смеси DCM/метанол/DIPEA (17:2:1) в течение 10 мин, промывали 2×10 мл DCM и 3×10 мл DMF. В реактор загружали 10 мл 20%-ного раствора диэтиламина в DMF, перемешивали в течение 5 мин и отфильтровывали; данную операцию повторяли еще раз в течение 20 мин, после чего смолу промывали 5×10 мл DMF. К смоле добавляли охлажденный раствор 3,0 ммоль следующей аминокислоты, 0,49 г (3,6 ммоль) HOBt и 0,56 мл (3,6 ммоль) DIC в 10 мл DMF, перемешивали при комнатной температуре. Полноту протекания реакции контролировали по изменению окраски бромфенолового синего. Смолу отфильтровывали, промывали 6×10 мл DMF. Операции 4 и 5 повторяли до получения требуемой последовательности. Затем в реактор загружали 10 мл 20%-ного раствора диэтиламина в DMF, перемешивали в течение 5 мин и отфильтровывали; данную операцию повторяли еще раз в течение 20 мин, после чего смолу промывали 5×10 мл DMF. Аналитический образец смолы, содержащей декапептид формулы 9, промывали 3×1 мл DCM и обрабатывали смесью TFA:TIS:H2O (95:0,25:0,25) в течение двух часов. Полученный раствор концентрировали на роторном испарителе, продукт высаживали МТВЕ, отфильтровывали, промывали большим количеством МТВЕ, сушили на воздухе. Чистота деблокированного пептида составила 97%.
Пример 5a
Загрузка была увеличена в лабораторных условиях до 15 г смолы в реакторе. Фрагмент формулы 9 получен аналогично фрагменту 1-9 с использованием DIC/HOBt в качестве конденсирующего агента.
Пример 6
Твердофазный синтез заявляемого фрагмента 1-34 Boc-His-(Trt)-Gly-Glu(OtBu)-Gly-Thr(tBu)-Phe-Thr(tBu)-Ser(tBu)-Asp(OtBu)-Leu-Ser(tBu)-Lys(Boc)-Gln(Trt)-Met-Glu(OtBu)-Glu(OtBu)-Glu(OtBu)-Ala-Val-Arg(Pbf)-Leu-Phe-Ile-Glu(OtBu)-Trp(Boc)-Leu-Lys(Boc)-Asn(Trt)-Gly-Gly-Pro-Ser(tBu)-Ser(tBu)-GlyOH (формула 2).
Пример 6а: Последовательным наращиванием цепи
В твердофазном реакторе суспендировали 2 г 2-хлортритилхлоридной смолы (1,5 ммоль/г) в 15 мл DCM, выдерживали в течение 5 мин и отфильтровывали смолу. К смоле добавляли раствор 0,18 г (0,6 ммоль) С-концевой аминокислоты FmocGlyOH и 2 мл (12,0 ммоль) DIPEA в 6 мл DCM, перемешивали в течение 2 ч при комнатной температуре. Смолу отфильтровывали и промывали 2×10 мл DCM. Смолу обрабатывали 2×10 мл смеси DCM/метанол/DIPEA (17:2:1) в течение 10 мин, промывали 2×10 мл DCM и 3×10 мл DMF. В реактор загружали 10 мл 20%-ного раствора диэтиламина в DMF, перемешивали в течение 5 мин и отфильтровывали; данную операцию повторяли еще раз в течение 20 мин, после чего смолу промывали 5×10 мл DMF. К смоле добавляли охлажденный раствор 1,2 ммоль следующей аминокислоты, 0,19 г (1,4 ммоль) HOBt и 0,22 мл (1,4 ммоль) DIC в 8 мл DMF, перемешивали при комнатной температуре. Полноту протекания реакции контролировали по изменению окраски бромфенолового синего. Смолу отфильтровывали, промывали 6×10 мл DMF.
Операции 4 и 5 повторяли до получения требуемой последовательности. Затем смолу промывали 2×10 мл DCM, обрабатывали 30×10 мл 1%-ным раствором TFA в DCM; получаемые растворы объединяли в колбе, содержащей 40 мл 10%-ного раствора пиридина в метаноле. Смесь концентрировали на ротационном испарителе, продукт высаживали водой, отфильтровывали, промывали большим количеством воды, сушили на воздухе. Целевой пептид формулы 2 был получен с выходом 1,12 г - 51% и чистотой 79%.
Пример 6б: Конвергентным методом
К смоле, содержащей декапептид формулы 9, добавляли охлажденный раствор 1,8 ммоль следующего фрагмента 21-24, 0,50 г (3,6 ммоль) HOBt и 0,55 мл (3,6 ммоль) DIC в 8 мл DMF, перемешивали при комнатной температуре. Полноту протекания реакции контролировали по изменению окраски бромфенолового синего. Смолу отфильтровывали, промывали 6×10 мл DMF. Затем в реактор загружали 10 мл 20%-ного раствора диэтиламина в DMF, перемешивали в течение 5 мин и отфильтровывали; данную операцию повторяли еще раз в течение 20 мин, после чего смолу промывали 5×10 мл DMF.
Операции 4 и 5 повторяли с последующими защищенными фрагментами 15-20, 10-14 и 1-9. Затем смолу промывали 2×10 мл DCM, обрабатывали 30×10 мл 1%-ным раствором TFA в DCM; получаемые растворы объединяли в колбе, содержащей 40 мл 10%-ного раствора пиридина в метаноле. Смесь концентрировали на ротационном испарителе, продукт высаживали водой, отфильтровывали, промывали большим количеством воды, сушили на воздухе. Целевой защищенный пептид формулы 2 был получен с выходом 1,4 г - 62% и чистотой 95+%.
Пример 6c
Загрузка была увеличена в лабораторных условиях до 15 г смолы в реакторе. Фрагмент формулы 2 получен с использованием DIC/HOBt в качестве конденсирующего агента с выходом 87% и чистотой 95%.
Пример 7 Синтез фрагмента H-Ala-Pro-Pro-Pro-Ser-NH2, (формула 3)
Пример 7a: Твердофазным конвергентным методом
В твердофазном реакторе суспендировали 1 г 2-хлортритиламинной смолы (1,5 ммоль/г) в 8 мл DCM, выдерживали в течение 5 мин и отфильтровывали смолу. К смоле добавляли охлажденный раствор 3 ммоль первой Fmoc-аминокислоты, 3,6 ммоль HOAt и 3,6 ммоль DIC в 8 мл DMF, перемешивали в течение 5 дней при комнатной температуре. Смолу отфильтровывали, промывали 10 мл DMF (5×1 мин). К смоле добавляли раствор 4,5 ммоль Ac2O и 4,5 ммоль DIPEA в 8 мл DMF, перемешивали в течение 1 ч и отфильтровывали. Промывали смолу 10 мл DMF (5×1 мин). К смоле добавляли 10 мл 20% раствора Et2NH в DMF, перемешивали в течение 5 мин и отфильтровывали. Операцию повторяли в течение 20 мин. Завершенность реакции деблокирования проверяли с помощью теста Кайзера. Промывали смолу D 10 мл DMF (5×1 мин). Посадку первой аминокислоты оценивали хроматографически по количеству дибензофульвена, образующегося при удалении Fmoc-группы. К смоле добавляли раствор 4,5 ммоль трет-бутилоксикарбонил-пролил-пролил-пролина, 9 ммоль HOBt и 9 ммоль DIC в 8 мл DMF, перемешивали в течение 160 мин при комнатной температуре; отфильтровывали смолу. Полноту протекания конденсации наблюдали по окраске индикатора бромфенолового синего. Промывали смолу 10 мл DMF (5×1 мин) и 10 мл DCM (3×1 мин).
К смоле добавляли 11 мл раствора TFA в DCM (3:8), перемешивали в течение 1 ч при комнатной температуре. Смолу отфильтровывали, промывали 10 мл DCM (3×1 мин), 10 мл DMF (5×1 мин), 10 мл 10% раствора DIPEA в DMF (3×2 мин). Завершенность реакции деблокирования проверяли с помощью теста Кайзера. Промывали смолу 10 мл DMF (5×1 мин). К смоле добавляли охлажденный раствор 6 ммоль последней Fmoc-аминокислоты, 6,6 ммоль HOBt и 6,6 ммоль DIC в 8 мл DMF, перемешивали в течение 160 мин при комнатной температуре; смолу отфильтровывали. Полноту протекания конденсации наблюдали по окраске индикатора бромфенолового синего. Промывали смолу 10 мл DMF (5×1 мин). К смоле добавляли 10 мл чистой TFA, перемешивали в течение 12 ч при комнатной температуре. Раствор отфильтровывали в сухую колбу. Оставшийся пептид отмывали от смолы 10 мл чистой TFA (3×2 мин). TFA отгоняли на ротационном испарителе, остаток заливали МТВЕ. Выпавший осадок отфильтровывали, промывали МТВЕ, сушили на воздухе. Целевой пептид формулы 3 был получен с выходом 0, 30 г - 30% и чистотой 94%.
Пример 7б: Классическим методом в растворе
Синтез Boc-Pro-Pro-OH
К раствору 12,9 г (0,041 моль) сукцинимидного эфира трет-бутилоксикарбонил-пролина в 37 мл диоксана при перемешивании прибавляли раствор 9,47 г (0,082 моль) пролина и 3,29 г (0,041 моль) гидроксида натрия в мл воды, реакционную смесь оставляли на ночь. Раствор упаривали на 2/3, подкисляли 1 М серной кислотой до рН~4. Продукт высаливали хлоридом натрия, экстрагировали этилацетатом. Этилацетатный раствор промывали насыщенным раствором хлорида натрия, сушили сульфатом натрия. Отгоняли растворитель, остаток кристаллизовали из 40 мл этилацетата. Выход 12.3 г (96%).
Синтез Boc-Pro-Pro-OSu
К раствору 8,35 г (0,027 моль) Boc-Pro-Pro-ОН и 3,41 г (0,030 моль) N-гидроксисукцинимида в 110 мл тетрагидрофурана при перемешивании добавляли охлажденный раствор 6,13 г (0,030 моль) дициклогексилкарбодиимида в 15 мл тетрагидрофурана. Через 2 часа отфильтровывали выпавшую мочевину. Раствор упаривали, остаток кристаллизовали из изопропилового спирта. Выход 10.1 г (91%).
Синтез Boc-Pro-Pro-Pro-OH
К раствору 6,75 г (0, 017 моль) Boc-Pro-Pro-OSu в 25 мл диоксана прибавляли раствор 3,83 г (0,034 моль) пролина в 17 мл 1М гидроксида натрия; реакционную смесь оставляли перемешиваться при комнатной температуре на ночь. Реакционную смесь промывали диэтиловым эфиром, подкисляли 1М серной кислотой до рН~4. Продукт высаливали хлоридом натрия, экстрагировали этилацетатом. Этилацетатный раствор промывали насыщенным раствором хлорида натрия, сушили сульфатом натрия. Отгоняли растворитель, остаток кристаллизовали из 40 мл этилацетата. Выход 6,68 г (95%).
Синтез TFA*Pro-Pro-Pro-OH
3 г (0,007 моль) Boc-Pro-Pro-Pro-OH растворяли в 10 мл трифторуксусной кислоты, через полчаса отгоняли растворитель, заливали 30 мл изопропилового спирта, снова упаривали. Остаток кристаллизовали из 40 мл MTBE. Выход 3,01 г (94%).
Синтез Z-Ala-Pro-Pro-Pro-OH
К раствору 17,8 г (0,042 моль) трифторацетата пролил-пролил-пролина и 15,3 мл (0,092 моль) диизопропилэтиламина в 90 мл диоксана добавили раствор 15,5 г (0,048 моль) N-бензилоксикарбонил-аланина в 90 мл диоксана, оставили перемешиваться при комнатной температуре на ночь. Полноту протекания реакции проверяли с помощью ВЭЖХ. Диоксан упарили досуха, остаток растворили в 210 мл воды. Раствор промывали диэтиловым эфиром. Продукт высаливали, экстрагировали н-бутанолом. Бутанольный раствор промывали насыщенным водным раствором хлорида натрия, затем - дистиллированной водой. Раствор упарили; остаток высушили упариванием с изопропиловым спиртом, кристаллизовали из метилтретбутилового эфира. Продукт отфильтровывали, промывали метилтретбутиловым эфиром, сушили на воздухе. Выход 19.6 (91%).
Синтез Z-Ala-Pro-Pro-Pro-Ser(tBu)-NH2
К охлажденному раствору 17,5 г (0,034 моль) бензилоксикарбонил-аланил-пролил-пролил-пролина и 5,8 г (0,036 моль) O-трет-бутил-серил-амида в 100 мл тетрагидрофурана добавляли охлажденный раствор 8,2 г (0,040 моль) N,N′-дициклогексилкарбодиимида в 40 мл тетрагидрофурана. Реакционную смесь оставляли на два дня при комнатной температуре, полноту протекания реакции проверяли с помощью ВЭЖХ. Выпавшую дициклогексилмочевину отфильтровывали, промывали тетрагидрофураном, растворитель отгоняли на ротационном испарителе. Остаток кристаллизовали из метилтретбутилового эфира. Продукт отфильтровывали, промывали метилтретбутиловым эфиром, сушили под вакуумом. Выход 20.7 г (92%).
Синтез H-Ala-Pro-Pro-Pro-Ser(tBu)-NH2
К раствору 16,2 г (0,025 моль) Z-Ala-Pro-Pro-Pro-Ser(tBu)-NH2 в 500 мл метанола добавили катализатор - 10% палладия на угле, при перемешивании пропускали водород. За протеканием реакции следили с помощью ВЭЖХ. После фильтрования растворитель отгоняли на ротационном испарителе, остаток высушивали упариванием с изопропиловым спиртом. Выход 12.7 г (99%), чистота 97%.
Синтез Z-Ala-Pro-Pro-Pro-Ser-NH2
К раствору 0,66 г (0,0017 моль) трифторацетата TFA*Pro-Pro-Pro-Ser-NH2 и 0,64 г (0,0020 моль) Z-Ala-OSu в 20 мл DMF добавляли 0,37 мл (0,0034 моль) N-метилморфолина, оставляли на ночь при комнатной температуре. Полноту протекания реакции проверяли с помощью ВЭЖХ. Растворитель отгоняли на ротационном испарителе, остаток заливали водой, экстрагировали н-бутанолом. Раствор упарили, остаток кристаллизовали из метилтретбутилового эфира. Осадок отфильтровывали, промывали метилтретбутиловым эфиром, сушили на воздухе. Выход 0,92 г (92%).
Синтез Fmoc-Ala-Pro-Pro-Pro-Ser-NH2
К раствору 0,57 г (0,0014 моль) TFA*Pro-Pro-Pro-Ser-NH2 и 0,65 г (0,0016 моль) Fmoc-Ala-OSu в 20 мл диметилформамида добавляли 0,31 мл (0,0028 моль) N-метилморфолина, оставляли на ночь при комнатной температуре. Полноту протекания реакции проверяли с помощью ВЭЖХ. Растворитель отгоняли на ротационном испарителе, остаток высаживали из воды, экстрагировали н-бутанолом. Раствор упарили, остаток кристаллизовали из метилтретбутилового эфира. Осадок отфильтровывали, промывали метилтретбутиловым эфиром, сушили на воздухе. Выход 0,91 г (95%).
Синтез Boc-Ala-Pro-Pro-Pro-Ser-NH2
К раствору 0,70 г (0,0018 моль) TFA*Pro-Pro-Pro-Ser-NH2 и 0,56 г (0,0020 моль) Boc-Ala-OSu в 20 мл DMF добавляли 0,40 мл (0,0036 моль) N-метилморфолина, оставляли на ночь при комнатной температуре. Полноту протекания реакции проверяли с помощью ВЭЖХ. Растворитель отгоняли на ротационном испарителе, остаток высаживали из воды, экстрагировали н-бутанолом, высаливали из водного слоя и снова экстрагировали н-бутанолом. Раствор упаривали, остаток кристаллизовали из MTBE. Осадок отфильтровывали, промывали МТВЕ, сушили на воздухе. Выход 0,98 г (98%).
Синтез H-Ala-Pro-Pro-Pro-Ser-NH2
0,80 г (0,0014 моль) Boc-Ala-Pro-Pro-Pro-Ser-NH2 растворяли в смеси ТFА:дихлорметан 1:1; через час раствор упаривали, остаток кристаллизовали из MTBE, продукт сушили под вакуумом. Выход 0,65 г (99%), чистота 98%).
Пример 8
Конденсация в растворе фрагмента формулы (2) и H-Ala-Pro-Pro-Pro-Ser(tBu)-NH2
320 мг (55,2 мкмоль) фрагмента формулы (2) и 140 мг (221 мкмоль) фрагмента Н-Ala-Pro-Pro-Pro-Ser(tBu)-NH2 растворили в 5 мл N-метилпирролидона, 30 мг (221 мкмоль) HOBt и 35 мкл (221 мкмоль) DIC. За протеканием реакции следили с помощью ВЭЖХ. Реакционную смесь заливали водой, выпавший осадок отфильтровывали, промывали водой и сушили на воздухе. Выход 0.32 г (92%).
Пример 8a
Конденсация в растворе фрагмента формулы (2) и TFA*H-Ala-Pro-Pro-Pro-Ser-NH2 (формула 3)
9.0 г (0.00156 М) фрагмента формулы (2) и 1.97 г (0.0031 М) фрагмента TFA*H-Ala-Pro-Pro-Pro-Ser-NH2 растворили в 100 мл N-метилпирролидона, 0.42 г (0.0031 M) HOBt и 1.02 мл (0.0031 M) DIC. За протеканием реакции следили с помощью ВЭЖХ. Реакционную смесь заливали водой, выпавший осадок отфильтровывали, промывали водой и сушили на воздухе. Выход 9.98 г (99%).
Пример 9. Удаление защитных групп
10.58 г пептида обрабатывали в течение 2 ч 200 мл смеси TFA:TIS:EDT:H2O 37:1:1:1. Реакционную смесь отфильтровывали и упаривали на ротационном испарителе. Остаток растирали в MTBE; твердый осадок отфильтровывали, сушили под вакуумом. Продукт растворяли в 100 мл водного раствора 1% уксусной кислоты, выдерживали сутки, лиофилизовали. Неочищенный пептид формулы 1 был получен с выходом 10.02 г - 60% (в расчете на содержание пептида), чистотой 75% и содержанием целевого продукта 42%.
Как показывают примеры 1-9, в результате предлагаемого синтеза повышается выход неочищенного пептида формулы 1 (42%-60%), также увеличивается чистота (58%-75%) и содержание целевого продукта в смеси (33%-42%). Кроме того, на всех стадиях конденсации используется более дешевый конденсирующий агент (DIC) (таблица 1). Одновременный синтез нескольких коротких фрагментов позволяет ускорить процесс получения целевого пептида. Полученный в ходе синтеза пептид может быть использован в производстве лекарственных средств для лечения сахарного диабета 2-го типа.
Формулы пептидов:
H-His1-Gly-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Leu10-Ser-Lys-Gln-Met-Glu15-Glu-Glu-Ala-Val-Arg20-Leu-Phe-Ile-Glu-Trp25-Leu-Lys-Asn-Gly-Gly30-Pro-Ser-Ser-Gly-Ala35-Pro-Pro-Pro-Ser-NH2 (формула 1)
Boc-His1(Trt)-Gly-Glu(OtBu)-Gly-Thr(tBu)-Phe-Thr(tBu)-Ser(tBu)-Asp(OtBu)-Leu10-Ser(tBu)-Lys(Boc)-Gln(Trt)-Met-Glu15(OtBu)-Glu(OtBu)-Glu(OtBu)-Ala-Val-Arg20(Pbf)-Leu-Phe-Ile-Glu(OtBu)-Trp(Boc)-Leu-Lys(Boc)-Asn(Trt)-Gly-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-OH (формула 2)
H-Ala-Pro-Pro-Pro-Ser-NH2 (формула 3)
X-Ala-Pro-Pro-Pro-Ser(tBu)-NH2, (формула 4), где X=Fmoc, Boc, Z
Fmoc-His(Trt)-Gly-Glu(OtBu)-Gly-Thr(tBu)-Phe-Thr(tBu)-Ser(tBu)-Asp(OtBu)-OH (формула 5)
10-14 Fmoc-Leu10-Ser(tBu)-Lys(Boc)-Gln(Trt)-Met-OH (формула 6)
15-20 Fmoc-Glu15(OtBu)-Glu(OtBu)-Glu(OtBu)-Ala-Val-Arg20(Pbf)-OH (формула 7)
21-24 Fmoc-Leu-Phe-Ile-Glu(OtBu)-OH (формула 8)
25-34 Fmoc-Trp(Boc)-Leu-Lys(Boc)-Asn(Trt)-Gly-Gly-Pro-Ser(tBu)-Ser(tBu)-Gly-OH (формула 9)
Список сокращений
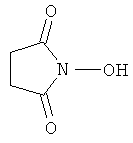

Источники информации
1. Diabetes mellitus, WHO information, Fact Sheet №138, Reviewed November (1999).
2. Eng, J. et al., Isolation and Characterization of Exendin-4, an Exendin-3 Analogue, from Heloderma suspectum Venom. J. Biol. Chem., Vol.267, Issue 11, 7402-7405, Apr, 1992.
3. US 5424286 Exendin-3 and exendin-4 polypeptides, and pharmaceutical compositions comprising same.
4. CN 101357938A. Method for synthesizing Exenatide from solid phase polypeptide.
5. CN 101538324 A. Method for preparing Exenatide.
6. WO 201100664 A2. Process for the production of Exenatide and of an Exenatide analogue (прототип).
7. US 2009149628 A1. Insulinotropic peptide synthesis using solid and solution phase combination techniques.
8. H.Gausepohl, et al. in "Peptides, Chemistry and Biology, Proc. 12th American Peptide Symposium".
9. A.Smith and J.E.Rivier (Eds.) ESCOM, Lieden, 1992, pp.523.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИД ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА 2-ГО ТИПА И ЕГО ОСЛОЖНЕНИЙ | 2014 |
|
RU2573933C1 |
| ПЕПТИД GLP-1 С ПРИСОЕДИНЕННОЙ ОЛИГОСАХАРИДНОЙ ЦЕПЬЮ | 2008 |
|
RU2539829C2 |
| АНАЛОГ ЭКСЕНАТИДА | 2020 |
|
RU2823623C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЛИКОПЕПТИДА, ИМЕЮЩЕГО СИАЛИРОВАННУЮ САХАРНУЮ ЦЕПЬ, И СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО СИАЛИЛГЛИКОАСПАРАГИНА | 2012 |
|
RU2586524C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДОВ, СОДЕРЖАЩИХ ЛИПОФИЛЬНО МОДИФИЦИРОВАННУЮ БОКОВУЮ ЦЕПЬ ЛИЗИНА | 2017 |
|
RU2755543C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА | 2008 |
|
RU2478105C2 |
| Сайт-специфически монопегилированные аналоги эксендина и способ их получения | 2012 |
|
RU2625015C2 |
| ПРОИЗВОДНОЕ АНАЛОГА GLP-1 ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ИХ ПРИМЕНЕНИЕ | 2010 |
|
RU2565536C2 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ НА ОСНОВЕ УНДЕКАПЕПТИДА, СОДЕРЖАЩЕГО D-ОКТААРГИНИНОВЫЙ ВЕКТОР | 2013 |
|
RU2541127C1 |
| ГЛИКОЗИЛИРОВАННЫЙ ПЕПТИД GLP-1 | 2009 |
|
RU2543157C2 |
Изобретение относится к области медицины и химической технологии. Предложен способ получения пептида эксенатида. Технический результат изобретения заключается в повышении выхода неочищенного пептида (42%-60%), а также увеличении чистоты (58%-75%) и содержания целевого продукта в смеси (33%-42%). Кроме того, на всех стадиях конденсации используется более дешевый конденсирующий агент (DIC), а одновременный синтез нескольких коротких фрагментов позволяет ускорить процесс получения целевого пептида. Полученный в ходе синтеза пептид эксенатида может быть использован в производстве лекарственных средств для лечения сахарного диабета 2-го типа. 2 з.п. ф-лы, 2 табл., 8 пр.
1. Способ получения пептида эксенатида (1)
пептид (1) имеет формулу (I)

отличающийся тем, что пептид (1) получен конденсацией двух фрагментов (А) и (В) классическим синтезом в растворе; при этом пептидный фрагмент (А) является модифицированной частью пептида (1); в пептидном фрагменте (А) N-терминальной аминокислотой является аминокислота, занимающая положение 1 в пептиде (1), C-терминальной аминокислотой является аминокислота, занимающая положение 34 в пептиде (1);
пептидный фрагмент (А) имеет N-терминальную защитную группу PGA уретанового типа в пептидном фрагменте (А) защищены боковые функциональные группы;
пептидный фрагмент (А) имеет формулу (II)

пептидный фрагмент (А) получен конденсацией пяти пептидных фрагментов (С), (D), (Е), (F) и (G) твердофазным конвергентным синтезом; пептидный фрагмент (С) является модифицированной частью пептида (1); в пептидном фрагменте (С) N-терминальной аминокислотой является аминокислота, занимающая положение 25 в пептиде (1), C-терминальной аминокислотой является аминокислота, занимающая положение 34 в пептиде (1);
пептидный фрагмент (С) присоединен к полимерному носителю,
пептидный фрагмент (С) не имеет N-терминальную защитную группу,
в пептидном фрагменте (С) защищены боковые функциональные группы; пептидный фрагмент (С) имеет формулу (IX)

пептидный фрагмент (D) является модифицированной частью пептида (1);
в пептидном фрагменте (D) N-терминальной аминокислотой является аминокислота, занимающая положение 21 в пептиде (1), C-терминальной аминокислотой является аминокислота, занимающая положение 24 в пептиде (1);
пептидный фрагмент (D) имеет N-терминальную защитную группу PGD уретанового типа в пептидном фрагменте (D) защищены боковые функциональные группы;
пептидный фрагмент (D) имеет формулу (VIII)
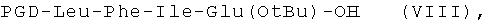
пептидный фрагмент (Е) является модифицированной частью пептида (1);
в пептидном фрагменте (Е) N-терминальной аминокислотой является аминокислота, занимающая положение 15 в пептиде (1), C-терминальной аминокислотой является аминокислота, занимающая положение 20 в пептиде (1);
пептидный фрагмент (Е) имеет N-терминальную защитную группу PGE уретанового типа в пептидном фрагменте (Е) защищены боковые функциональные группы;
пептидный фрагмент (Е) имеет формулу (VII)
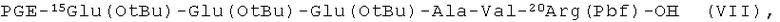
пептидный фрагмент (F) является модифицированной частью пептида (1);
в пептидном фрагменте (F) N-терминальной аминокислотой является аминокислота, занимающая положение 15 в пептиде (1), C-терминальной аминокислотой является аминокислота, занимающая положение 20 в пептиде (1);
пептидный фрагмент (F) имеет N-терминальную защитную группу PGF уретанового типа в пептидном фрагменте (F) защищены боковые функциональные группы;
пептидный фрагмент (F) имеет формулу (VI)
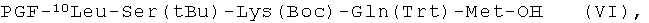
пептидный фрагмент (G) является модифицированной частью пептида (1);
в пептидном фрагменте (G) N-терминальной аминокислотой является аминокислота, занимающая положение 15 в пептиде (1), C-терминальной аминокислотой является аминокислота, занимающая положение 20 в пептиде (1);
пептидный фрагмент (G) имеет N-терминальную защитную группу PGA уретанового типа в пептидном фрагменте (G) защищены боковые функциональные группы;
пептидный фрагмент (G) имеет формулу (V)
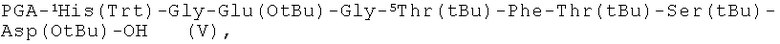
затем пошагово конденсируют заявленные пять пептидных фрагментов в следующем порядке:
шаг первый: пептидный фрагмент (D) конденсируют с пептидным фрагментом (С), результатом чего является пептидный фрагмент (DC), имеющий N-терминальную защитную группу PGD и присоединенный к полимерному носителю;
пептидный фрагмент (DC) имеет формулу (X)
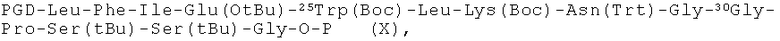
шаг второй: деблокируют N-терминальную защитную группу PGD пептидного фрагмента (D);
шаг третий: пептидный фрагмент (Е) конденсируют с пептидным фрагментом (DC), результатом чего является пептидный фрагмент (ЕС), имеющий N-терминальную защитную группу PGE и присоединенный к полимерному носителю;
пептидный фрагмент (ЕС) имеет формулу (XI)

шаг четвертый: деблокируют N-терминальную защитную группу PGE пептидного фрагмента (Е);
шаг пятый: пептидный фрагмент (F) конденсируют с пептидным фрагментом (ЕС), результатом чего является пептидный фрагмент (FC), имеющий N-терминальную защитную группу PGF и присоединенный к полимерному носителю;
пептидный фрагмент (FC) имеет формулу (XII)

шаг шестой: деблокируют N-терминальную защитную группу PGF пептидного фрагмента (FC);
шаг седьмой: пептидный фрагмент (G) конденсируют с пептидным фрагментом (FC), результатом чего является пептидный фрагмент (А), имеющий N-терминальную защитную группу PGA и присоединенный к полимерному носителю;
после чего пептидный фрагмент (А) отделяют от полимерного носителя; и затем пептидный фрагмент (А) конденсируют с пептидным фрагментом (В), результатом чего является пептид (1) имеющий N-терминальную защитную группу PGA;
после этого деблокируют N-терминальную защитную группу PGA пептида (1) и на этой стадии деблокируют защитные группы боковых функциональных групп пептида (1),
при этом пептидный фрагмент (В) является модифицированной частью пептида (1); в пептидном фрагменте (В) N-терминальной аминокислотой является аминокислота, занимающая положение 35 в пептиде (1), C-терминальной аминокислотой является аминокислота, занимающая положение 39 в пептиде (1);
пептидный фрагмент (В) имеет формулу (III)
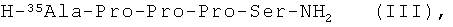
пептидный фрагмент (В) получен классическим синтезом в растворе, твердофазным синтезом или комбинацией растворного и твердофазного синтезов.
2. Способ по п.1, отличающийся тем, что пептидный фрагмент (А) или пептидный фрагмент (В) получены твердофазным пептидным синтезом.
3. Способ по п.1, отличающийся тем, что пептидный фрагмент (А) получен твердофазным пептидным синтезом, а пептидный фрагмент (В) классическим синтезом в растворе.
| СПОСОБЫ ПОДАВЛЕНИЯ ГЛЮКАГОНА | 2000 |
|
RU2247575C9 |
| US 2011046349 A1, 24.02.2011 | |||
| US 2009149628 A1, 11.06.2009 | |||
| US 5424286 A, 13.06.1995. | |||

