Изобретение относится к новым способам получения водорастворимых реагентов для органического синтеза на основе поливалентного иода, которые способны к рециклу и, тем самым, отвечают принципам «зеленой» химии и способам их получения. Реагент может применяться в различных областях техники, в том числе в органической и фармацевтической химии, биохимии.
На основании изучения источников информации можно сделать вывод о том, что в настоящее время не известны реагенты для органического синтеза на основе поливалентного иода, которые можно легко в среде «зеленого» растворителя - воды перевести в активную форму после проведения окислительных процессов с образованием химических связей типа С-С и С-Х (где Х=О, N, S, Se, F, C1, Br, I и т.д.).
В известных источниках информации описано несколько мономерных реагентов на основе поливалентного иода, отвечающих принципам «зеленой» химии.
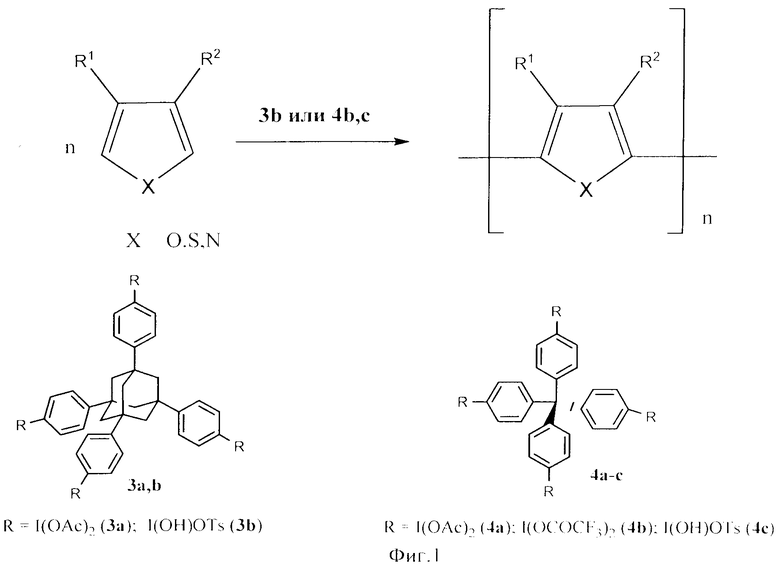
Известны производные поливалентного иода на основе адамантана и тетрафенилметана, предложенные японскими химиками. Так, Кита с соавторами показал, что под действием производных адамантана (3b) и тетрафенилметана (4b, с) протекает окислительная полимеризация ароматических и гетероциклических соединений (фиг.1) [1].
Эти реагенты и их восстановленные формы обладают низкой растворимостью и легко отделяются от продуктов реакции и тем самым отвечают принципам Green Chemistry. В других работах эти же авторы привели способы получения 1,3,5,7-тетракис[4-(диацетоксииодо)фенил]адамантана (3а) и тетракис-[4-ксииодо)фенил]метана (4а)]. Структурные формулы которых: 3a, 3b и 4а, 4b, 4c приведены в Приложении (фиг.1).
К недостаткам способов получения данных реагентов можно отнести, в первую очередь, многостадийность получения. Так, для получения реагента на основе адамантана (3b) необходимо в три стадии получить (3а), который действием пара-толуолсульфокислоты переводят в действующий реагент (3b) [2]. Такие же сложности и в случае (4b, с): в начале через несколько стадий синтезируют (4а), который действием трифторуксусной кислоты или пара-толуолсульфокислоты переводят в (4b, с) [3]. Другим недостатком является уязвимость центральных фрагментов данных реагентов к действию окислителей. Так, возможна окислительная деструкция адамантана и тетрафенилметана действием большинства окислителей, в том числе и самих реагентов (3a, b, и 4а-с) с образованием стабильных карбкатионов или радикалов. Эти реагенты, а также их восстановленные формы практически не растворимы в воде, что также является их недостатком.
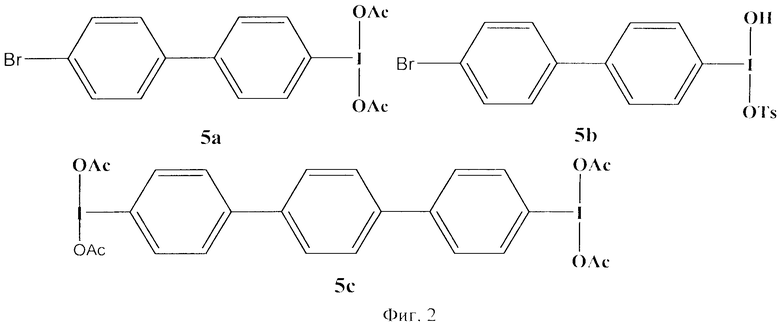
В недавно опубликованном патенте других японских ученых были предложены несколько новых реагентов, соответствующих принципам Green Chemistry [2]. В своей работе японские химики основывались на публикации [5], в которой авторы одновременно с Кита предложили идею синтеза мономерных соединений поливалентного иода, обладающих свойствами полимерных реагентов. Японские химики расширили круг подобного типа реагентов, и были синтезированы новые карбоксилаты (5а) и гидрокситозилаты иоддифенила (5b) и дииодтерфенила (5с) со схожими свойствами, и показана их эффективность в реакциях окисления, перегруппировки, тозилоксилирования [4]. По окончании вышеприведенных реакций, восстановленные формы этих соединений поливалентного иода также отделялись от целевых продуктов фильтрацией, благодаря их чрезвычайно низкой растворимости. Структурные формулы реагентов 5а, b, с приведены в Приложении (фиг.2).
К их недостаткам можно также отнести практическую нерастворимость этих реагентов и их восстановленных форм в воде и многостадийность способов их получения. Эти соединения поливалентного иода (5а-с) не могут быть использованы в проведении окислительных превращений с их участием в водной среде и также невозможно провести отделение остатков непрореагировавшей части реагента и их восстановленной формы через растворимость в воде.
Наиболее близким к предлагаемому способу получения реагента является способ получения орто-карбокси IBX, который выбран в качестве прототипа [4]. Данный реагент 7 на основе поливалентного иода называют модифицированным IBX (modified IBX или mIBX), он хорошо растворим в воде и эффективен при окислении спиртов до карбонильных соединений (Приложение, фиг.3).
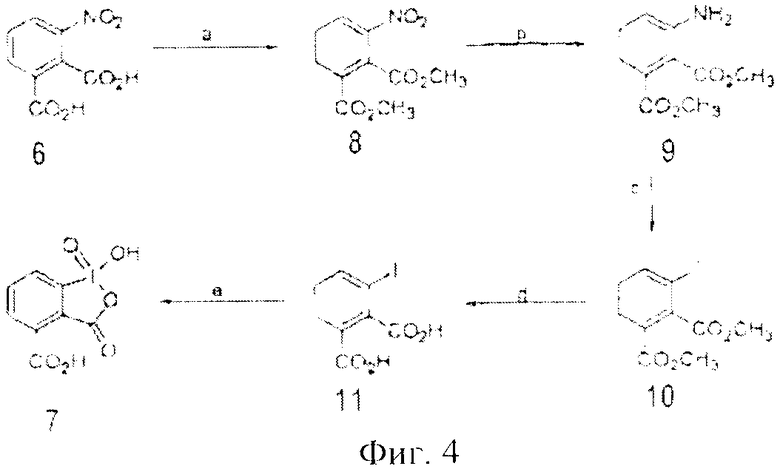
К недостатку способа получения данного реагента 7 можно отнести многостадийность получения из нитрофталевой кислоты 6: через 5 стадий (общий выход 59%) (фиг.4). Хотя авторы приводят количественные выходы 1 и 2 стадий, ясно, что речь идет о техническом выходе этих промежуточных продуктов 8 и 9. Другим недостатком является - нерастворимость в воде восстановленной 1(+3) формы этого реагента, который образуется при окислении спиртов до карбонильных соединений. Также к недостаткам можно отнести соблюдение мер предосторожности при работе с водородом на стадии восстановления соединения 8 в амин 9 и температурного режима (0-5°C) на стадии диазотирования и введения атома иода.
Новая техническая задача - разработка нового водорастворимого реагента для органического синтеза и более простого способа его получения.
Для решения поставленной задачи в качестве водорастворимого реагента для органического синтеза применяют 4-иодбензолсульфонат калия.
Предложен способ получения водорастворимого реагента для органического синтеза, заключающийся в том, что иодобензол сульфируют при перемешивании с концентрированной серной кислотой с концентрацией 94-98% при 40-60°C при соотношении иодбензол - серная кислота - 1:4.6 в течение 30-40 час, при этом каждые 5 ч отфильтровывают выпавшие кристаллы сульфокислоты или же экстрагируют горячим хлороформом, затем полученную 4-иодбензолсульфокислоту окисляют OXONE, (2KHSO5·KHSO4·K2SO4) добавляют порциями (2×6.14 г) через каждые 30 мин, затем реакционную массу нагревают до 50-60°C и выдерживают в течение 2 ч, после чего охлаждают до 0°C, осадок отфильтровывают и промывают на фильтре холодной водой.
4-иодилбензолсульфонат калия (1) и его восстановленная форма - 4-иодбензолсульфонат калия (2а) имеют принципиальные преимущества перед остальными реагентами поливалентного иода - они растворимы в воде и тем самым могут легко удаляться из реакционной массы простым разбавлением водой. Это преимущество позволяет без выделения 2а снова окислить до 1 и тем самым выполнить полный рецикл (фиг.5). Этот реагент может применяться в различных областях техники, в том числе в органической и фармацевтической химии, биохимии. Введение в иодароматическое соединение сульфогруппы увеличивает его водорастворимость и реакционную способность.
Предложенный способ исключает несколько стадий, необходимых в случае других реагентов поливалентного иода:
1. стадию отделения остатком реагента и его восстановленной формы от продукта реакции с помощью специальной лабораторной процедуры (экстракция, фильтрация, хроматографирование, перегонка под вакуумом и т.п.);
2. стадию выделения или регенерации восстановленной формы реагента с помощью химических процедур после отделения от продукта реакции;
3. стадию получения реагента из его восстановленной формы в воде без дополнительной процедуры выделения и очистки.
Экспериментальным путем подобран оптимальный режим температуры, концентрации серной кислоты, соотношение компонентов в реакционной смеси.
Так, при более высокой температуре, выход 4-иодилбензолсульфокислоты 2 не превышал 60%, имело место дополнительное образование побочных продуктов диспропорционирования: повышение температуры выше 60°C приводило к протеканию перегруппировки Якобсона, снижению выхода и образованию побочных продуктов ди-, три- и тетраиодбензолов, а снижение температуры до 30°C увеличивало продолжительность реакции.
Важным является то, что при данных условиях реакция имеет высокую региоселективность, образовывались только продукты пара-замещения, тогда как при использовании олеума наблюдалось образование до 5% и орто-иодбензолсульфокислоты. Более низкие концентрации серной кислоты до 85-90% требуют стадию разбавления и приводят к увеличению продолжительности реакции и повышению температуры, что соприкасается с протеканием перегруппировки Якобсона.
Оптимальным оказалось соотношение иодобензол-серная кислота 1:4,85-5.0, поскольку позволяет при температуре 40-60°C создать достаточную концентрацию иодбензола в серной кислоте для успешного сульфирования.
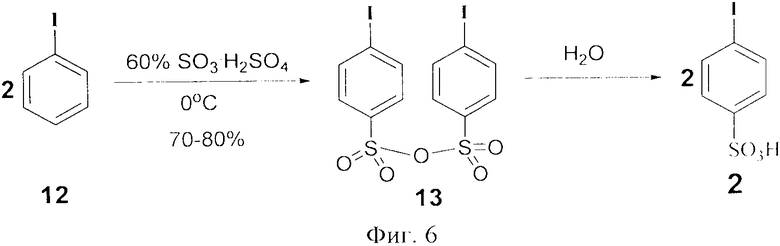
В отличие от известного способа получения 4-иодбензолсульфокислоты 2, где авторы провели синтез через реакцию гидролиза его ангидрида 8 [5], авторами предложен одностадийный метод. В свою очередь, ангидрид 4-иодбензолсульфокислоты был получен в реакции иодбензола и олеума (60% SO3 в серной кислоте) при 0°C с выходом 70-80% (фиг.6).
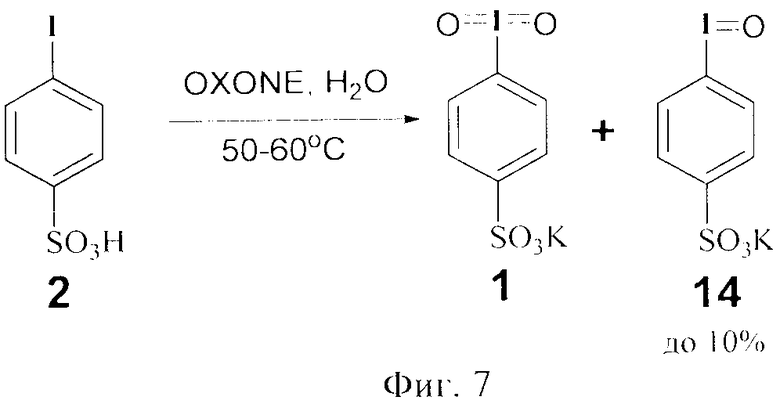
Окисление сульфокислоты 2 до реагента 1 проводят в воде при температуре 50-60°C в течение 2 ч. При этом нет необходимости сульфокислоту 2 обезвоживать и удалять из нее остатки серной кислоты. Остатки серной кислоты до 5% ускоряют окисление соединения 2 до реагента 1. Стадию окисления сульфокислоты 2 в сульфонат 1 проводят в водной среде, что отвечает принципам «зеленой» химии. Соотношение субстрата и окислителя (OXONE) 1:2 является оптимальным, поскольку при добавлении меньших количеств в реакционной массе присутствует и 4-иодозобензолсульфонат калия 14 до 10% по данным ЯМР 1H (фиг.7). Увеличение количества окислителя экономически не целесообразно.
Новым в заявляемом изобретении является то, что для получения целевого водорастворимого реагента 1 используют только две стадии: стадии сульфирования и окисления сульфокислоты с использованием доступных реагентов в простых условиях, не требующих специальной очистки и соблюдения безводных условий, применением взрывоопасных реагентов. На стадии сульфирования авторы разработали удобный метод получения сульфокислоты 2, отличающийся от известного простотой и удобством. Вместо олеума использовали концентрированную серную кислоту, что исключило стадию образования ангидрида 13 и побочного продукта орто(2)-иодбензолсульфокислоты (до 5%). Окисление сульфокислоты 2 проводили действием наиболее удобного окислителя OXONE в водной среде, что исключает добавление 0.73М серной кислоты.
В среде серной кислоты образуется кислая форма реагента 1-4-иодилбензолсульфокислота 1а, для выделения которой необходимо полностью удалить 0.73М серную кислоту, что является достаточно сложной технологической процедурой и требует специальной кислото-стойкой вакуумной установки.
Для демонстрации синтетических возможностей 4-иодилбензолсульфоната калия 1 мы провели окисление спиртов до карбонильных соединений, иодирование аренов и кетонов.
Далее приведены примеры на отработку температурного режима получения 4-иодбензолсульфокислоты и 4-иодилсульфоната калия.
Пример 1. Получение 4-иодбензолсульфокислоты (2) при 60°C.
К 20 мл иодбензола (12) (36.4 г, 0.178 моль) при температуре 5°C и перемешивании добавляли 50 мл 94-98% N2SO4 (84.68 г, 0.86 моль) и далее нагревали до 60°C и выдерживали при этой температуре 30 ч. Уже через 3 ч реакционная масса приобретала розовый цвет. Для удаления остатков иодбензола добавляли 30 мл гексана и перемешивали 5 мин. Гексановый слой удаляли и проводили экстракцию небольшими порциями кипящего хлороформа (100 мл), максимально пытаясь растворить в нем продукт, в результате чего получили систему из 2-х прозрачных слоев. Верхний слой аккуратно переносили пипеткой в колбу, хлороформ отгоняли. Отогнанный хлороформ использовали многократно. Полученные кристаллы отфильтровывали, вновь промывали гексаном от остатков иодбензола и сушили под вакуумом. Выход (2) 48.0 г (95%), т.пл. 66-68°C, (лит. 70°C [8]). Спектр ЯМР 1H (200 MHz, D2O, δ, м.д.): 7.48 (д, J=7.6 Гц, 2Наром.), 7.85 (д, J=7.6 Гц, 2Наром.).
Пример 2. Получение 4-иодбензолсульфокислоты (2) при 60°C.
К 20 мл иодбензола (12) (36.4 г, 0.178 моль) при температуре 5°C и перемешивании добавляли 50 мл 94-98% H2SO4 (84.68 г, 0.82 моль) и далее нагревали до 60°C и выдерживали при этой температуре 30 ч. Реакционную массу охлаждали до 0°C и выпавшие светло кремового цвета кристаллы отфильтровывали, на фильтре промывли холодным гексаном (30 мл) от остатков иодбензола и сушили под вакуумом. Выход (2) 45.5 г (90%), т.пл. 66-68°C.
Пример 3. Получение 4-иодбензолсульфокислоты (2) при 30°C.
К 2.0 мл иодбензола (12) (3.64 г, 0.018 моль) при температуре 5°C и перемешивании добавляли 5.0 мл 94-98% H2SO4 (8.47 г, 0.086 моль) и далее нагревали до 30°C и выдерживали при этой температуре 70 ч. Уже через 3 ч реакционная масса приобретала розовый цвет. Для удаления остатков иодбензола добавляли 30 мл гексана и перемешивали 5 мин. Гексановый слой удаляли и проводили экстракцию небольшими порциями кипящего хлороформа (100 мл), максимально пытаясь растворить в нем продукт, в результате чего получили систему из 2-х прозрачных слоев. Верхний слой аккуратно переносили пипеткой в колбу, хлороформ отгоняли. Отогнанный хлороформ использовали многократно. Полученные кристаллы отфильтровывали, вновь промывали гексаном от остатков иодбензола и сушили под вакуумом. Выход (2) 48.0 г (95%), т.пл. 66-68°C.
Пример 4. Получение 4-иодбензолсульфокислоты (2) при 75°C.
К 2.0 мл иодбензола (12) (3.64 г, 0.018 моль) при температуре 5°C и перемешивании добавляли 50 мл 94-98% H2SO4 (84.68 г, 0.086 моль) и далее нагревали до 75°C и выдерживали при этой температуре 20 ч. Уже через 3 ч реакционная масса приобретала темно-коричневый цвет. Для удаления остатков иодбензола добавляли 30 мл гексана и перемешивали 5 мин. Гексановый слой удаляли и отгоняли на роторном испарителе, получили 5.6 г маслообразную массу, кристаллизирующуюся при охлаждении. Спектр ЯМР 1Н показал наличие сигналов ароматических протонов моно-, ди-, три-тетразамещенных иодбензолов. С оставшейся реакционной массой далее проводили экстракцию небольшими порциями кипящего хлороформа (100 мл), максимально пытаясь растворить в нем продукт, в результате чего получили систему из 2-х прозрачных слоев. Верхний слой аккуратно переносили пипеткой в колбу, хлороформ отгоняли. Отогнанный хлороформ использовали многократно. Полученные кристаллы отфильтровывали, вновь промывали гексаном и холодным хлороформом от остатков иодбензолов и сушили под вакуумом. Выход (2) 40.0 г (79%), т.пл. 64-67°C.
Пример 5. Получение 4-иодилсульфоната калия (1) при соотношении субстрат : окислитель 1:2. В колбу вносили 2.84 г (10 ммоль) 4-иодбензолсульфокислоты 2 и 10.0 мл H2O и перемешивали до полного растворения кислоты. OXONE (2KHSO5·KHSO4·K2SO4) добавляли порциями (2×6.14 г) через каждые 30 мин. Затем реакционную массу нагревали до 60°C и выдерживали в течение 2 ч, после чего охлаждали до 0°C. Осадок отфильтровывали и промывали на фильтре холодной водой. Получили 2.97 г (94%) белых кристаллов. После перекристаллизации из минимального количества воды выход продукта составил 3.15 г (89%) в виде светлых блестящих чешуйчатых кристаллов. Т.пл. 278-280°C (со взрывом). Данные элементного анализа. Найдено: С 20.26; Н 1.10; I 36.07; S 8.86. C6H4IKO5S. Вычислено: С 20.35; Н 1.14; I 35.83; S 9.05. Спектр ЯМР 1Н (400 MГц, D2O, δ, м.д.): 8.11 (с, 2Наром.), 8.12 (с, Гц, 2Наром.). Спектр ЯМР 13С (100 MГц, D2O, δ, м.д.): 118.30, 129.11, 146.87, 151.88. В связи с тем, что 4-иодилсульфоната калия 1 получен впервые, что было подтверждено рентгено-структурным анализом.
Пример 6. Получение 4-иодилсульфоната калия 1 при при соотношении субстрат : окислитель 1:1.5. В колбу вносили 2.84 г (10 ммоль) 4-иодбензолсульфокислоты 2 и 10.0 мл H2O и перемешивали до полного растворения кислоты. OXONE (2KHSO5·KHSO4·K2SO4) добавляли порциями (2×4.61 г) через каждые 30 мин. Затем реакционную массу нагревали до 60°C и выдерживали в течение 2 ч, после чего охлаждали до 0°C. Осадок отфильтровывали и промывали на фильтре холодной водой. Получили 2.6 г белых кристаллов. Спектр ЯМР 1Н (400 MHz, D2O) показал наличие 8% трехвалентного производного - 4-иодозобензолсульфоната калия 14.
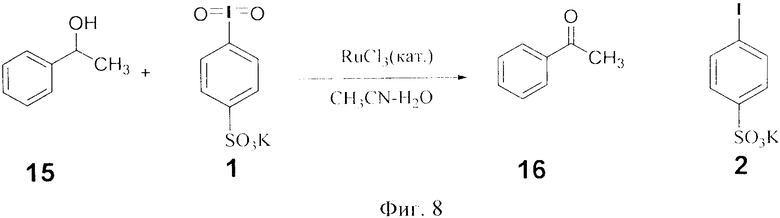
Пример 7. Окисление спиртов на примере 1-фенилэтанола 15 действием 4-иодилсульфоната калия 1 (фиг.8). В колбу внесли 122 мг (1.0 ммоль) 1-фенилэтанола 15, 0.5 мл ацетонитрила, 0.5 мл воды и 195 мг (0. 55 моль) 4-иодилсульфоната калия 1. При перемешивании добавили 10 мкл (1.6.10-3 ммоль) водного раствора RuCl3 и выдерживали при этой температуре 3 ч. Далее добавляли 1.0 мл этилацетата. Органический слой отделяли, промыли водой, отогнали и получили 120 мг (100% по данным ГХ-МС) ацетофенона 16. Водный слой представлял собой смесь остатков реагента 1 и его восстановленной формы 2а.
Пример 8. Иодирование ароматических соединений на примере мезитилена 17 и 4-гидроксиацетофенона 19 действием 4-иодилсульфоната калия 1 (фиг.9 и 10).
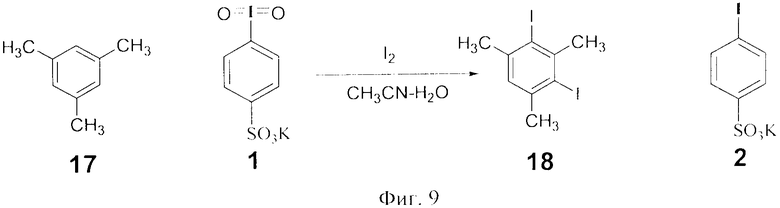
В колбу внесли 120 мг (1.0 ммоль) мезитилена 17, 133 мг (0.52 ммоль) иода, 0.5 мл ацетонитрила, 0.5 мл 5% водного раствора серной кислоты и 88.5 мг (0.25 моль) 4-иодилсульфоната калия 1. Перемешивали при комнатной температуре до полного обесцвечивания реакционной массы (до 12 ч). Далее добавляли 1.0 мл воды. Выпавшие кристаллы отфильтровали, промыли водой и получили 372 мг (100% по данным ГХ-МС и ЯМР 1Н, фиг.11) дииодмезитилена 18 с т.пл. 78-80°C (лит. 79-80.3°C Н. Togo, Т. Nabana, К. Yamaguchi. Preparation and Reactivities of Novel (Diacetoxyiodo)arenes Bearing Heteroaromatics // J. Org. Chem. 2000, Vol.65, №24, P.8391-8394). Полученный дииодид 18 не требовал дополнительной очистки. Водный слой представлял собой смесь остатков реагента 1 и его восстановленной формы 2а.
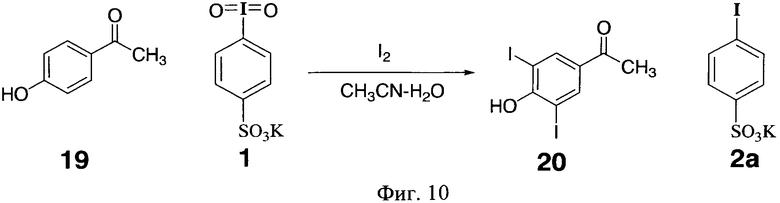
Аналогично проводили иодирование 4-гидроксиацетофенона 19 (фиг.10) и получили с выходом 95% 4-гидрокси-3,5-дииодацетофенон 20 с т.пл. 159-160°C (лит.т.пл. 158-160°C J.N. Moorthy, К. Senapati, S. Kumar // J. Org. Chem. 2009, Vol.74, no. 16. P.6287-6290). Дииодид 20 имел ЯМР-ную чистоту, что подтверждается спектром ЯМР 1Н (фиг.12).
Таким образом, заявляемое изобретение по сравнению с известными аналогами, позволяет посредством двух стадий получить водорастворимый реагент на основе поливалентного иода 4-иодилбензолсульфонат калия с суммарным выходом продукта 80-90%. Так, в отличие от mIBX легко отделяется от продуктов реакции окисления спиртов и иодирования аренов прибавлением воды. Как видно из приведенных спектров ЯМР 1H (фиг.11, 12), иодсодержащие ароматические соединения не содержали следы реагента 1 и его восстановленную форму 2а. Все это позволяет получать целевые продукты с выходами 95-100% с чистотой не менее 98%.
Как видно из экспериментальной части, предлагаемый способ получения не предполагает использование дорогих реагентов и использование сложных манипуляций и позволяет получить реагент на основе поливалентного иода - 4-иодилсульфоната калия в две стадии, при том, что суммарный выход реагента составляет 80-90%. Также при использовании данного реагента в реакции окисления и иодирования не требуется стадия очистки продуктов и максимально упрощенная процедура выделения.
Источники информации
1. Патент JP 2009046653 (A), Publication date: 2009-03-05.
CHIGUSA YASUO; MORIMOTO КОJI; KITA YASUYUKI METHOD FOR PRODUCING POLYMER OF AROMATIC COMPOUND AND HETEROCYCLIC AROMATIC
COMPOUND BY USING HYPER VALENT IODINE REAGENT. Publication number:
JP 2009046653 (A), Publication date: 2009-03-05.
2. Патент JP 2008063314 (A), Publication date: 2008-03-21.
TOGO HIDEO; MOROTA ATSUSHI. ENVIRONMENT-CONSCIOUS HYPERVALENT
IODINE REAGENT. Publication number: JP 2008063314 (A), Publication date: 2008-03-21.
3. M.S. Yusubov, L.A. Drygunova, V.V. Zhdankin, Synthesis, 2004, 2289-2292],
4. Патент US 2004030187 (A1), Publication date: 2004-02-12. VINOD THOTTUMKARA K; THOTTUMKARA ARUN P. User- and eco-friendly hypervalent iodine reagent and method of synthesis.
5. Christensen N.H. The sulfonation of iodobenzene. / N.H. Christensen // Acta Chemica Scandinavica. - 1961. - Vol.15. - P.219-221]
Приложение
Фиг.1 (схема 2) Окислительная полимеризация ароматических и гетероциклических соединений действием производных адамантана и тетрафенилметана.
Фиг.2 (схема 3) Структурные формулы карбоксилата и гидрокситозилата иоддифенила и дииодтерфенила.
Фиг.3 (схема 4) Схема получения орто-карбокси IBX и его возможности при окислении спиртов.
Фиг.4 (схема 5) Полная схема получения орто-карбокси IBX из нитрофталевой кислоты.
Фиг.5 (схема 1) Схема получения 4-иодилбензолсульфокислоты.
Фиг.6 (схема 6) Окисление иодбензола до бензолсульфокислоты.
Фиг.7 (схема 7) Окисление бензолсульфокислоты действием OXONE.
Фиг.8 (схема 8) Окисление 1-фенилэтанола действием 4-иодилбензолсульфокислоты.
Фиг.9 (схема 9) Иодирование мезителена действием 4-иодилбензолсульфокислоты.
Фиг.10 (схема 10) Иодирование 4-гидроксиацетофенона действием 4-иодилбензолсульфокислоты.
Фиг.11 Спектр 1H ЯМР диодмезителена.
Фиг.12 Спектр 1H ЯМР 4-гидрокси-3,5-дииодацетофенона.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ п-ИОДФЕНИЛЖИРНЫХ КИСЛОТ | 2013 |
|
RU2522557C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОМЕГА-ИОДАЛИФАТИЧЕСКИХ КАРБОНОВЫХ КИСЛОТ И ИХ ЭФИРОВ | 2012 |
|
RU2494087C1 |
| СПОСОБ ПОЛУЧЕНИЯ 17β-ГИДРОКСИ-ДЕЗ-А-АНДРОСТ-9, 10-ЕН-5-ОНА | 2016 |
|
RU2715720C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТОПОТЕКАНА | 2010 |
|
RU2447076C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,7-БИС[2-(ДИЭТИЛАМИНО)ЭТОКСИ]ФЛОУРЕНОНА ДИГИДРОХЛОРИДА | 1994 |
|
RU2076097C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИОДПОЛИСТИРОЛА | 2011 |
|
RU2467018C1 |
| ПРОИЗВОДНЫЕ 17-ГАЛОГЕН-4-АЗААНДРОСТЕНА | 1993 |
|
RU2103275C1 |
| СПОСОБ ПОЛУЧЕНИЯ 11БЕТА, 17АЛЬФА, 21-ТРИГИДРОКСИ-16АЛЬФА-МЕТИЛ-9АЛЬФА-ФТОРПРЕГНА-1,4-ДИЕН-3,20-ДИОНА (ДЕКСАМЕТАЗОНА) ИЗ ФИТОСТЕРИНА | 2013 |
|
RU2532902C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАНГИДРИДОВ АРОМАТИЧЕСКИХ ТЕТРАКАРБОНОВЫХ КИСЛОТ | 2017 |
|
RU2682170C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАЦЕТАТА ТРИПЕПТИДА | 2014 |
|
RU2551276C1 |




Изобретение относится к водорастворимому реагенту для органического синтеза 4-иодилбензолсульфонату калия формулы
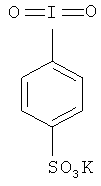 , а также к способу его получения, который заключается в том, что иодбензол сульфируют при перемешивании серной кислотой с концентрацией 94-98% при 40-60°C при соотношении иодбензол - серная кислота - 1:4.6 в течение 30-40 час, при этом, каждые 5 ч отфильтровывают выпавшие кристаллы сульфокислоты или же экстрагируют их горячим хлороформом, затем полученную 4-иодбензолсульфокислоту окисляют с помощью OXONE, добавляя порциями 2×6.14 г через каждые 30 мин, затем реакционную массу нагревают до 50-60°C и выдерживают в течение 2 часов, после чего охлаждают до 0°C, осадок отфильтровывают и промывают на фильтре холодной водой. Технический результат: получено новое соединение - 4-иодилсульфонат калия, который может применяться в органической и фармацевтической химии в качестве реагента для органических синтезов. 2 н.п. ф-лы, 12 ил, 8 пр.
, а также к способу его получения, который заключается в том, что иодбензол сульфируют при перемешивании серной кислотой с концентрацией 94-98% при 40-60°C при соотношении иодбензол - серная кислота - 1:4.6 в течение 30-40 час, при этом, каждые 5 ч отфильтровывают выпавшие кристаллы сульфокислоты или же экстрагируют их горячим хлороформом, затем полученную 4-иодбензолсульфокислоту окисляют с помощью OXONE, добавляя порциями 2×6.14 г через каждые 30 мин, затем реакционную массу нагревают до 50-60°C и выдерживают в течение 2 часов, после чего охлаждают до 0°C, осадок отфильтровывают и промывают на фильтре холодной водой. Технический результат: получено новое соединение - 4-иодилсульфонат калия, который может применяться в органической и фармацевтической химии в качестве реагента для органических синтезов. 2 н.п. ф-лы, 12 ил, 8 пр.
1. Водорастворимый реагент для органического синтеза 4-иодилбензолсульфонат калия формулы
.
2. Способ получения водорастворимого реагента для органического синтеза по п. 1, путем сульфирования иодобензола, отличающийся тем, что иодбензол сульфируют при перемешивании серной кислотой с концентрацией 94-98% при 40-60°C при соотношении иодбензол серная кислота - 1:4.6 в течение 30-40 час, при этом каждые 5 ч отфильтровывают выпавшие кристаллы сульфокислоты или же экстрагируют их горячим хлороформом, затем полученную 4-иодбензолсульфокислоту окисляют OXONE, (2KHSO5·KHSO4·K2SO4) добавляют порциями 2×6.14 г через каждые 30 мин, затем реакционную массу нагревают до 50-60°C и выдерживают в течение 2 часов, после чего охлаждают до 0°C, осадок отфильтровывают и промывают на фильтре холодной водой.
| JP 2008063314 A, 21.03.2008 | |||
| US 20050222417 A1, 06.10.2005 | |||
| US 20040030187 A1, 12.02.2004 | |||
| Способ получения галоидбензолсульфокислот | 1988 |
|
SU1576530A1 |

