Область техники, к которой относится изобретение
Настоящее изобретение касается области способов синтеза активных ингредиентов для фармацевтического применения, и в частности - способа получения в промышленном масштабе 17β-гидрокси-дез-A-андрост-9,10-ен-5-она-интермедиата, который может применяться в синтезе ретро-прогестеронов.
Предшествующий уровень техники
Ретро-прогестероны представляют собой класс стероидов с гормональной активностью, применяющихся в терапии и лечении функциональных нарушений женских половых путей и при беременности.
Родительским соединением в данном семействе является ретро-прогестерон - соединение, имеющее тетрациклическую стероидную структуру, изображенную на рисунке ниже:

ретропрогестерон
в которой атомы водорода в положениях 8 и 9 имеют бета-пространственную ориентацию, а метил в положении 10 имеет альфа-пространственную ориентацию; эта структура от структуры прогестерона, имеющего конфигурацию, называемую «природной», изображенную на рисунке ниже, отличается противоположной ориентацией атома водорода в положении 9 (альфа) и метила в положении 10 (бета).
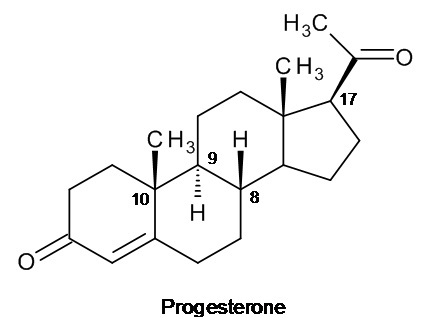
прогестерон
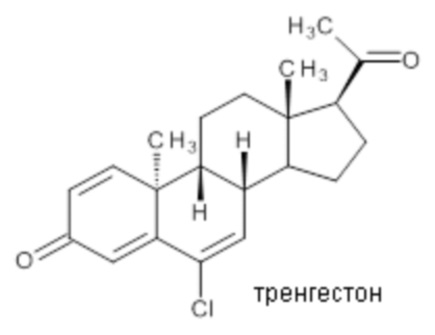
Ретро-прогестеронами, которые могут применяться в области терапии, являются, например, дигидрогестерон и тренгестон, имеющие приведенные ниже структурные формулы:
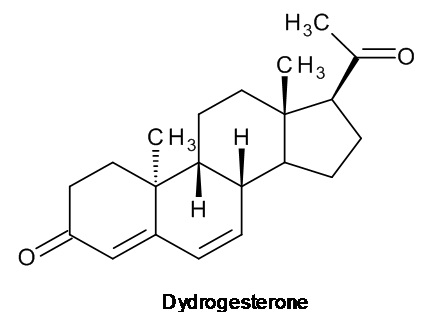
дигидрогестерон
Дигидрогестерон показал свою эффективность в лечении различных состояний, связанных с дефицитом прогестерона, включая бесплодие вследствие недостаточности желтого тела, угроза выкидыша или повторяющиеся выкидыши, нарушения менструации, предменструальный синдром и эндометриоз, а тренгестон применялся для лечения нарушений, связанных с менструальным циклом.
Интермедиатом, применяющимся в синтезе ретро-прогестеронов, является соединение, имеющее приведенную ниже формулу (1), химическое название которого - 17β-гидрокси-дез-A-андрост-9,10-ен-5-он:
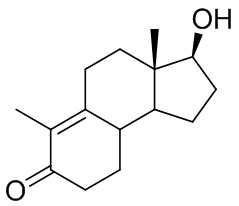
(1)
описанное в J. Org. Chem, 32, 3008 (1967); применение данного соединения в качестве прекурсора в синтезе ретро-прогестеронов описано в J. Org. Chem, 33 (9), 1968.
Соединение (1) можно синтезировать по методикам, приведенным в J. Org. Chem, 32, 3008 (1967), исходя из бициклического интермедиата (2) серией химических реакций, приводящих к рацемической смеси соединения (1):
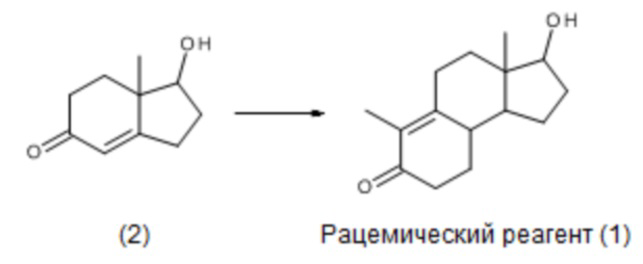
Такую рацемическую смесь нельзя использовать для получения ретро-прогестеронов, поскольку это смесь двух оптических изомеров.
В процитированной статье авторы также сообщают, что соединение (1) было ранее получено в «оптически активной» форме химическим разложением тестостерона ацетата. Этот метод имеет чисто научную ценность, поскольку он позволил впервые получить оптически чистый продукт, но он неприменим для промышленного производства стероидов: фактически, необходимо синтезировать тестостерон ацетат (тетрациклический скелет), и первый цикл (трициклический скелет) необходимо разрушить и затем снова создать.
Получение соединения, имеющего формулу (1), в оптически активной форме описано в Tetrahedron vol. 24, pp. 2039-2046, 1968; согласно описанному методу, исходя из рацемического интермедиата (3) и расщепляя оптические антиподы в ходе синтеза через соль с бруцином, получено соединение (1) в оптически активной форме:
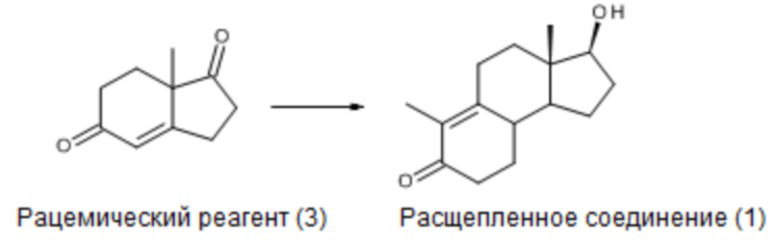
Хотя данный синтез применим для получения ретро-прогестеронов, является недостатком то, что он исходит из рацемического соединения; расщепление рацемата, даже в идеальном случае количественного выхода, дает максимум 50%-ный выход.
В J Med Chem 1985, 28, 1796-1803, схема (I), получали соединения, имеющие общую структуру (5), для изучения и проверки их потенциальной фармакологической активности. Согласно выводам данной статьи, указанные соединения можно получить исходя из прекурсора (4):

В свою очередь, соединения, имеющие общую формулу (4), можно получить как описано в US 3413314, тотальным синтезом согласно схеме:

Данный путь синтеза, однако, также имеет проблему, заключающуюся в том, что одна из стадий (US 3413314, колонка 2, стадия d) заключается в разделении оптических антиподов посредством образования соли с хиральным амином, с соответствующей потерей по меньшей мере половины исходного соединения.
Целью настоящего изобретения является разработка улучшенного пути синтеза для получения 17β-гидрокси-дез-A-андрост-9,10-ен-5-она, в частности более простого, чем процессы, описанные в предшествующем уровне техники, и применимого в промышленном масштабе.
Краткое описание изобретения
Эта и другие цели достигнуты в настоящем изобретении, которое в своем первом аспекте касается способа синтеза 17β-гидрокси-дез-A-андрост-9,10-ен-5-она, соединения (1), включающего следующие стадии:
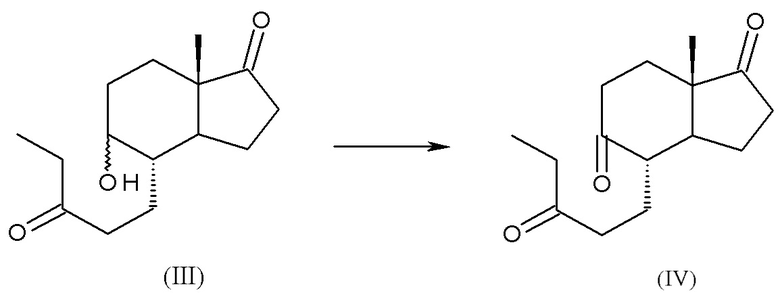
a) реакция (4aR,6aS,9aS,9bS)-декагидро-6a-метил-циклопента[f][1]бензопиран-3,7-диона, соединения (II), с этилмагний бромидом или этилмагний хлоридом с получением смеси изомеров: (4S,5R,7aS)-5-гидрокси-7a-метил-4-(3-оксопентил)октагидро-1H-инден-1-она и (4S,5S,7aS)-5-гидрокси-7a-метил-4-(3-оксопентил)октагидро-1H-инден-1-она, смеси интермедиатов (III):

b) окисление смеси интермедиатов (III) с получением (4S,7aS)-7a-метил-4-(3-оксопентил)гексагидро-1H-инден-1,5(4H)-диона, интермедиата (IV):
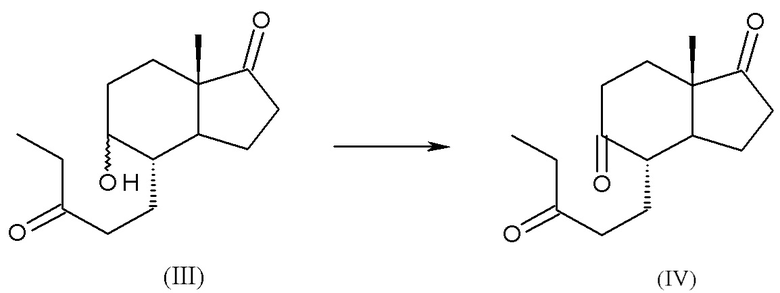
c) циклизация интермедиата (IV) с получением дез-A-андрост-9,10-ен-5,17-диона, интермедиата (V):
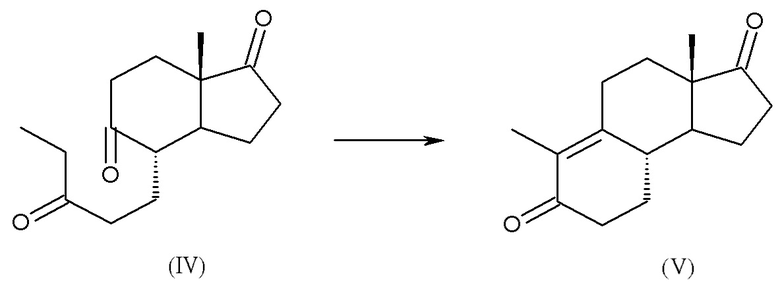
d) восстановление интермедиата (V) до 17β-гидрокси-дез-A-андрост-9,10-ен-5-она (1):
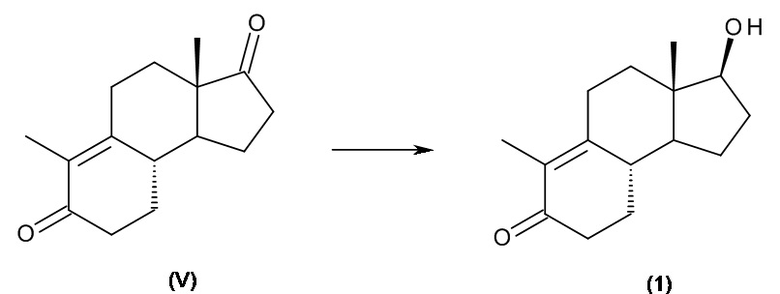 .
.
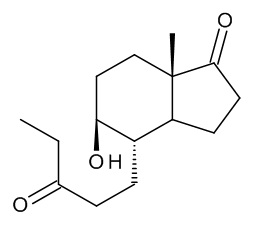
Другим предметом настоящего изобретения является смесь интермедиатов (III), продукт реакции на стадии a) указанного способа.
Краткое описание чертежей
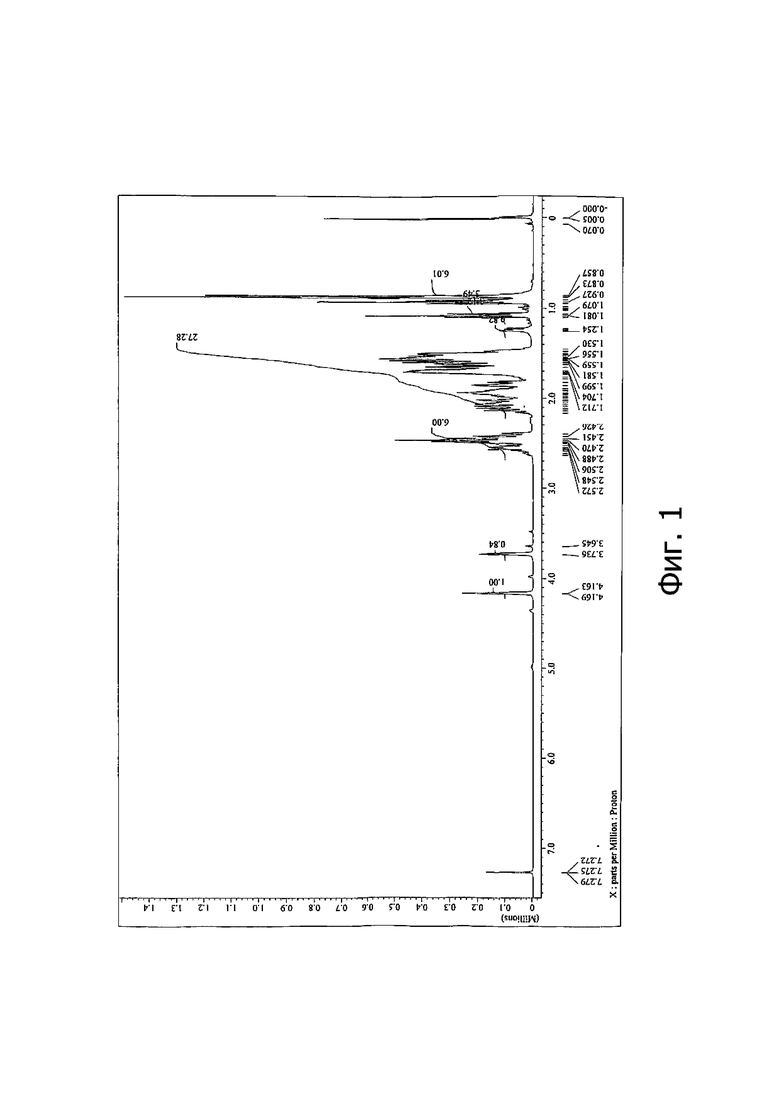
- На Фиг. 1 показан 1H-ЯМР спектр смеси интермедиатов (III);
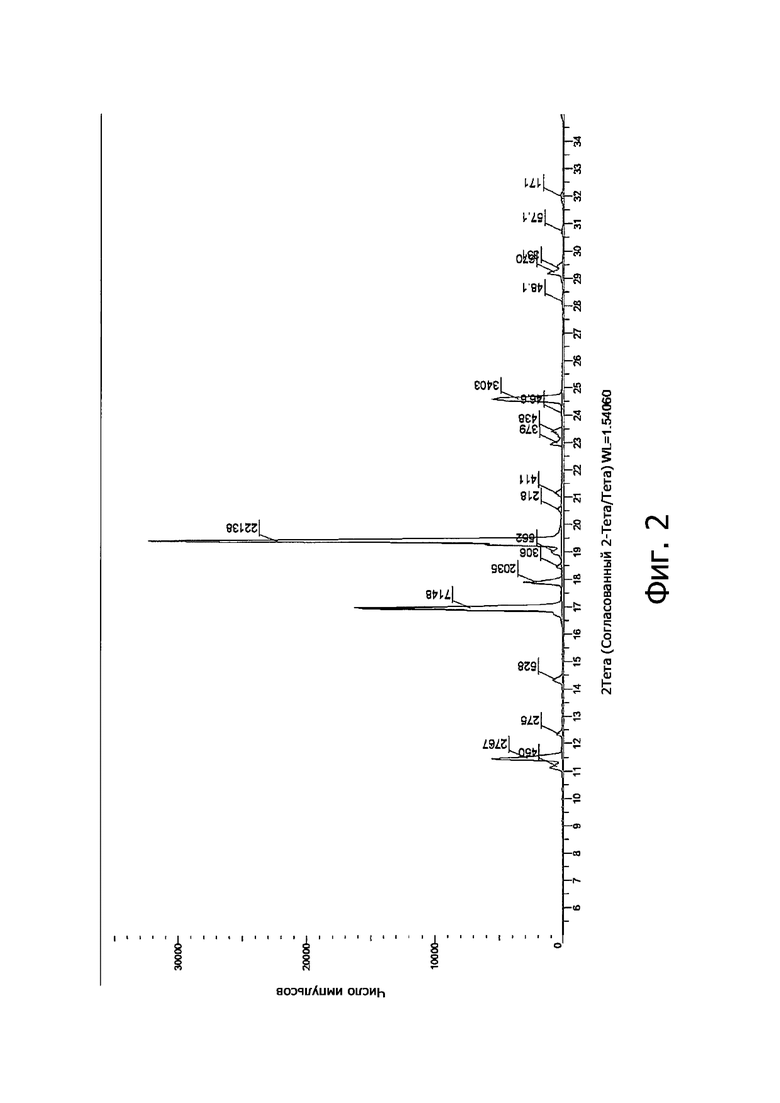
- На Фиг. 2 показана диаграмма рентгеновской дифракции для соединения (1), полученного по настоящему изобретению.
Подробное описание изобретения
В описании и в Формуле изобретения, в случае расхождения между названием соединения и приведенной для него структурной формулой, корректной следует считать формулу.
Исходным веществом в способе по настоящему изобретению является соединение (II), (4aR,6aS,9aS,9bS)-декагидро-6a-метил-циклопента[f][1]бензопиран-3,7-дион], также известный как ситолактон, название которого применяется далее в тексте; структура этого соединения со всей необходимой стереохимической информацией показана ниже:
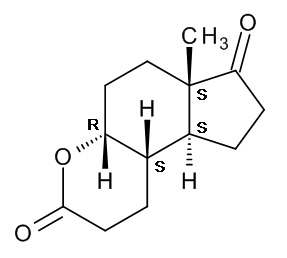
Ситолактон представляет собой коммерчески доступный продукт, полученный микробиологическим разложением фитостеролов, которые являются побочными продуктами переработки сои.
Реакцию на стадии a) проводят в растворителе, выбранном из диэтилового эфира, диизопропилового эфира, тетрагидрофурана или метилтетрагидрофурана, в чистом виде или в виде смеси, в инертной атмосфере при температуре от -50°C до 0°C. Предпочтительно, ее проводят в тетрагидрофуране при температуре от -45°C до -20°C, в токе азота средней силы.
Ситолактон вводят в реакцию с металлорганическим реагентом (реагентом Гриньяра), в котором органическая часть представляет собой этильный радикал, выбранным из этилмагний хлорида и бромида в растворе. Предпочтительно, применяют этилмагний бромид в тетрагидрофуране, добавляя этот раствор порциями в реакционную смесь.
Количество добавляемого реагента и время реакции определяется по контролю реакции, который показывает ее прогресс, например контроль методом ТСХ или ВЭЖХ; смесь интермедиатов (III) подходит для дальнейшей реакции, когда остаток ситолактона составляет ≤ 5% от исходного количества. Мольное соотношение между металлорганическим реагентом и ситолактоном составляет от 1 до 2, предпочтительно от 1.05 до 1.25.
Время реакции между металлорганическим реагентом и ситолактоном обычно составляет от 30 минут до 2 часов, предпочтительно от 40 до 90 минут.
Смесь интермедиатов (III) образована двумя изомерами: (4S,5R,7aS)-5-гидрокси-7a-метил-4-(3-оксопентил)октагидро-1H-инден-1-оном и (4S,5S,7aS)-5-гидрокси-7a-метил-4-(3-оксопентил)октагидро-1H-инден-1-оном, несущими ОН-группу в положении 5 молекулы в двух возможных ориентациях, как показано ниже:
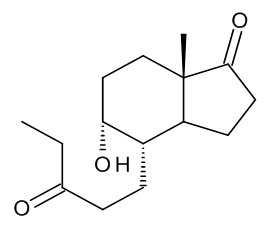
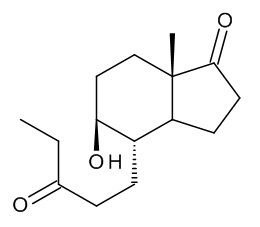
Данную смесь можно использовать без разделения двух изомеров.
Неожиданно было обнаружено, что в применяемых условиях реакции карбонил в положении 7 ситолактона реагирует с металлорганическим реагентом лишь в ничтожной степени, что позволяет избежать необходимости его защищать.
Реакцию на стадии b) проводят в растворителе, выбранном из диэтилового эфира, простого изопропилового эфира, тетрагидрофурана, метилтетрагидрофурана, ацетона, метил-изобутилкетона, толуола, чистого гептана или смеси его изомеров, циклогексана, диметилацетамида, диметилформамида, хлороформа, метиленхлорида, диметилсульфоксида и воды, в чистом виде или в виде смеси.
В качестве окислителя можно применять трихлоризоциануровую кислоту (TCCA) в присутствии органического основания, такого как пиридин или триэтиламин; соединений хрома (VI) в присутствии таких оснований, как пиридин, 3,5-диметилпиразол или триэтиламин, или кислот, таких как серная кислота, перхлорная кислота, уксусная кислота или хлористоводородная кислота; 2,2,6,6-тетраметилпиперидин-1-оксильный радикал, общеизвестный как TEMPO, или его производное, такое как 4-гидрокси-2,2,6,6-тетраметилпиперидин-1-оксильный радикал, 4-метокси-2,2,6,6-тетраметилпиперидин-1-оксильный радикал и 4-(бензоилокси)-2,2,6,6-тетраметилпиперидин-1-оксильный радикал, в присутствии основного водного раствора и соокислителя, такого как кислород и гипохлорит натрия или гипохлорит кальция; гипохлориты как основные окислители, такие как гипохлорит натрия, гипохлорит кальция или гипохлорит тетрабутиламмония; газообразный кислород или кислородно-азотные смеси в присутствии солей меди (I), таких как CuCl; пероксимоносульфат калия KHSO5 доступный на рынке под торговым названием Oxone® (торговая марка, зарегистрированная фирмой E.I. du Pont de Nemours and Company); газообразный хлор, растворенный в галогенированном растворителе, таком как хлороформ или тетрахлорид углерода; изопропилат алюминия в присутствии карбонильного соединения, такого как циклогексанон, бензальдегид, бензофенон или ацетон; диметилсульфоксид и активатор, такой как оксалилхлорид, бензойный ангидрид, трифторуксусный ангидрид, P2O5 и комплекс SO3-пиридин, в присутствии основания, такого как триэтиламин или диизопропилэтиламин; соединения гипервалентного иода, такие как периодинан Десс-Мартина, также известный под аббревиатурой DMP, или IBX, стабилизированный бензойной кислотой и изофталевой кислотой, также известный под аббревиатурой SIBX.
Предпочтительно, реакцию проводят с применением трихлоризоциануровой кислоты (TCCA) в качестве окислителя в ацетоне при температуре от 5 до 40°C в присутствии воды и пиридина.
Окислитель добавляют в реакционный раствор порциями, количество реагента и время реакции определяют, контролируя протекание и прогресс реакции. Качество интермедиата (IV) является подходящим для последующей реакции, когда остаток непрореагировавшей смеси интермедиатов (III) составляет ≤ 5% от исходного количества.
В предпочтительном случае применения TCCA, необходимое число молей данного реагента относительно числа молей интермедиата (III), подвергающегося окислению, находится в диапазоне от 0,5 до 5, предпочтительно от 0,5 до 1,5; время реакции после добавления TCCA составляет от 30 минут до 3 часов, предпочтительно от 45 минут до 1,5 часов.
Реакцию циклизации на стадии c) можно проводить с интермедиатом (IV), который можно получить непосредственно из предыдущей реакции, или с выделенным продуктом.
Данную реакцию проводят в растворителе, выбранном из диэтилового эфира, диизопропилового эфира, тетрагидрофурана, метилтетрагидрофурана, толуола, циклогексана, н-гептана, смеси изомеров гептана, метанола, этанола, изопропанола, уксусной кислоты, ацетонитрила, метиленхлорида, воды, в чистом виде или в виде смеси, в присутствии основных катализаторов, таких как NaOCH3, KOCH3, KOH, NaOH, Na2CO3, K2CO3 или NaNH2, или кислотных катализаторов, таких как HClO4, H2SO4 или HCl.
Температура реакции составляет от 0 до 40°C, предпочтительно от 10 до 30°C.
Реакцию предпочтительно проводят, используя KOH как катализатор в спиртовом или водно-спиртовом растворе, предпочтительно в метаноле и воде. Предпочтительно, ее проводят непосредственно с органическим раствором интермедиата (IV), полученным на предыдущей стадии, без выделения.
Реакция циклизации требует времени в диапазоне от 1 до 4 часов, предпочтительно от 1,5 до 3 часов.
Качество интермедиата (V) является подходящим для проведения следующей реакции, если остаток непрореагировавшего интермедиата (IV) составляет ≤ 5% от исходного количества.
Восстановление на стадии d) проводят с использованием гидридов натрия, лития или калия, таких как боргидрид натрия, боргидрид калия или литий алюминий гидрид; предпочтительным является боргидрид натрия.
Температура реакции составляет от -10 до 40°C, предпочтительно от 10 до 20°C.
Реакцию проводят в растворителе, выбранном из спиртов, в частности в метаноле, этаноле, изопропаноле или циклогексаноле; или в спиртах в смеси с такими растворителями, как диэтиловый эфир, простой изопропиловый эфир, тетрагидрофуран, метилтетрагидрофуран, метиленхлорид, толуол, циклогексан, н-гептан, смесь изомеров гептана, диметилформамид или диметилацетамид. Предпочтительным растворителем является метанол.
Гидрид добавляют в реакционный раствор порциями; количество реагента и время реакции определяют, контролируя прогресс реакции.
Число молей применяемого боргидрида натрия относительно числа молей интермедиата (IV) находится в диапазоне от 0,3 до 1,5, предпочтительно от 0,35 до 0,9; время реакции составляет от 1 до 5 часов, предпочтительно от 2 до 3,5 часов.
Качество соединения (1) считают приемлемым, когда остаток непрореагировавшего интермедиата (V) составляет ≤ 5% от исходного количества.
Настоящее изобретение далее описано с привлечением примеров, которые приведены для иллюстративных, неограничивающих целей.
Применяющиеся в приведенных ниже примерах реагенты доступны из коммерческих источников и применяются без необходимости их очистки для повышения степени чистоты.
Способы и условия реакций
ЯМР:
ЯМР спектрометр JEOL 400 YH (400 МГц);
ЯМР ампулы Aldrich® ColorSpec®;
JEOL Delta Software v5.1.1;
Спектры записывали в дейтерированном хлороформе Sigma-Aldrich: хлороформ-d, D 99.8% атомов, содержащем 0.1% (об/об) тетраметилсилана (ТМС) в качестве внутреннего стандарта; и хлороформ-d, “100%”, D 99.96% атомов, содержащем 0.03% (об/об) ТМС.
Масс-спектры
ВЭЖХ-масс система AB Sciex API 2000 LC/MS/MS;
Образцы вводятся напрямую и химически ионизируются (CI) с муравьиной кислотой.
ДСК
Прибор Perkin Elmer модель Diamond;
Стандартные алюминиевые капсулы и крышки Perkin Elmer, код 02190041;
Скорость сканирования: 10°C/мин;
Диапазон температур: от 20°C до 200°C.
ИК
Спектрометр Thermo Scientific Nicolet 6700;
FT-IR спектры записывали в KBr (тв.) и с устройством smart-iTR для регистрации коэффициента диффузного отражения (ATR);
Бромид калия Sigma-Aldrich Code 221864 (для ИК-анализа).
ВЭЖХ
Хроматографическая система Agilent model 1200
УФ-детектор MODEL 1260 DAD VL и Лазерный детектор 1290 Infinity ELSD
ТСХ
MERCK: ТСХ-пластины с силикагелем 60 F254 на алюминиевой подложке 20 x 20 см, code 1.0554.0001.
ВЭТСХ
MERCK: ТСХ-пластины для ВЭТСХ с силикагелем 60 F254 с зоной концентрирования 10 x 2.5 см, code 1.13727.0001.
ТСХ-обратнофазная
MERCK: ТСХ-пластины с силикагелем 60 RP-18 F254S, code 1.15685.0001.
ТСХ-детектирование
Кислотный раствор фосфомолибдата церия;
Приготовление: 25 г гидрата фосфомолибденовой кислоты (Aldrich P7390), 10 г гидрата сульфата церия (IV) (Aldrich 31606) и 600 мл воды перемешивали до растворения с 60 мл 95-98%-ной серной кислоты (Aldrich 258105); объем раствора доводили до 1000 мл добавлением воды; пластины опрыскивали полученным раствором, затем нагревали до голубого окрашивания.
УФ-свет с длиной волны 254 и 366 нм.
Дифракционный рентгеновский анализ
Bruker D2Phaser;
Источник X-лучей, медная трубка с λ = 1.54184 [Å] электропитание 30 кВ и 10 мА;
Скорость сканирования 0.02° 2θ/сек
Интервал сканирования 5° - 35° 2θ;
Время анализа 1478 сканов за 1704 секунд;
Вращение 10° [1°/мин];
SSD160 детектор (режим 1D) с PSD (позиционно-чувствительный детектор) отверстием 4.6°.
В примерах применяются следующие сокращения:
EtMgBr: этилмагний бромид;
МТБЭ: метил-т-бутиловый эфир;
TCCA: трихлоризоциануровая кислота;
ТГФ: тетрагидрофуран;
Rf: коэффициент подвижности в тонкослойной хроматографии.
Пример 1
Данный пример относится к стадии a) способа по настоящему изобретению.
300 г ситолактона смешивали при перемешивании с 3.6 л ТГФ в инертной атмосфере.
Раствор охлаждали до температуры между -40 и -30°C (ситолактон частично снова выпадает в осадок) при перемешивании.
1.35 л EtMgBr в ТГФ (1 M раствор) добавляли в течение 90 минут при перемешивании, поддерживая температуру реакции в диапазоне -40 < T < -30°C (при добавлении наблюдался разогрев).
После добавления перемешивание продолжали еще 20 минут, поддерживая температуру реакции в диапазоне -40 < T < -30°C.
Прогресс реакции отслеживали методом ВЭТСХ (элюент 100% изопропилацетат), обнаруживая примерно 20% непрореагировавшего ситолактона и образование пятна с Rf большим, чем у ситолактона, а также нескольких пятен с едва детектируемой интенсивностью.
Добавляли еще 300 мл EtMgBr в ТГФ (1 M раствор), поддерживая температуру реакции в диапазоне -40 < T < -30°C.
После добавления продолжали перемешивание еще 20 минут, поддерживая температуру реакции в диапазоне -40 < T < -30°C.
Прогресс реакции отслеживали методом ВЭТСХ (элюент 100% изопропилацетат), обнаруживая примерно 5% непрореагировавшего ситолактона от исходного количества, увеличение интенсивности пятна с большим Rf, и сохранение нескольких пятен с едва детектируемой интенсивностью.
Реакционный раствор затем медленно выливали при перемешивании в 3.4л 4%-ного водного раствора хлорида аммония, предварительно охлажденного до температуры 0 < T < 5°C. Фазы разделяли.
Водную фазу сначала экстрагировали МТБЭ (3 × 1.5л), затем объединенные органические фазы промывали 1.5 литрами водного раствора NaCl и сушили сульфатом натрия. Раствор фильтровали и промывали осадок на фильтре 1 литром МТБЭ, упаривали фильтрат досуха при пониженном давлении, получая 350.7 г желтого остатка, который можно использовать в следующей стадии без дополнительно очистки.
1 г полученного остатка очищали для аналитических целей методом флэш-хроматографии на силикагеле, элюируя 1:1 смесью изопропилацетат/изомеры гептана, и сушили при пониженном давлении до постоянного веса (легкоплавкое бесцветное твердое вещество).
ЯМР (CDCl3): образец представляет собой 54:46 смесь двух изомеров по расположению гидроксила в смеси (III) (уширенный синглет при 4.166 м.д. и уширенный синглет при 3.736 м.д.).
Полный спектр 1H-ЯМР изображен на Фиг .1.
Масс-спектр (CI): [M++1] = 253; [M++1] - H2O = 235; [M++1] - 2H2O = 217;
[M++1] - 3H2O = 199.
FT-IR (ATR): 1735 см-1 и 1702 см-1.
Пример 2
Данный пример относится к стадиям b) и c) способа по настоящему изобретению.
63.42 г смеси интермедиатов (III), полученной как описано в предыдущем примере, растворяли при перемешивании в 286 мл ацетона в инертной атмосфере.
Добавляли 120 мл пиридина и 2.95 мл воды. Температуру доводили до 20°C.
В другом сосуде готовили раствор 29.24 г TCCA в 95.3 мл ацетона, и медленно прикапывали его в раствор смеси интермедиатов (III), поддерживая температуру < 25°C.
По окончании добавления раствор перемешивали в течение 1 часа (осадок образовывался уже в ходе прикапывания), наблюдался слабый разогрев; поддерживали температуру ≤ 25°C.
Прогресс реакции проверяли методом ВЭТСХ (элюент МТБЭ/гептан = 8:2), детектируя полное исчезновение исходного реагента, образование основного пятна с меньшим значением Rf, чем у исходной смеси (III), и несколько пятен с детектируемой интенсивностью.
Смесь охлаждали до T = 0°C и фильтровали, промывая твердый осадок на фильтре 60 мл ацетона. Раствор выдерживали при 0°C в течение 1 часа, выпавший осадок снова отфильтровывали и промывали 60 мл ацетона.
Органическую фазу прикапывали при T ≤ 20°C, в водный раствор, приготовленный из 97.3 г Na2CO3, 117 г Na2SO3 и 672 мл воды.
Полученную суспензию фильтровали, и твердый осадок на фильтре промывали 100 мл ацетона, наблюдая присутствие двух фаз.
Фазы разделяли, органическую фазу упаривали до небольшого объема.
Водную фазу экстрагировали 482 мл толуола, и органическую фазу затем объединяли с органической фазой, отделенной как описано выше (органический раствор С).
В отдельном сосуде готовили раствор из 103.5 г KOH, 92 мл воды и 434 мл метанола, и его медленно прикапывали в органический раствор С, полученный ранее. Полученную смесь перемешивали при T < 25°C в течение 1ч 40 мин.
Протекание реакции отслеживали методом ТСХ на обращенной фазе (элюент ацетон/вода = 7:3), детектируя полное исчезновение интермедиата (IV) и формирование видимого в УФ пятна (основной продукт) и нескольких пятен с едва детектируемой интенсивностью.
Фазы разделяли, и водную фазу экстрагировали 380 мл толуола.
Органические фазы объединяли и промывали сначала 160 мл 10%-ного водного раствора NaCl, затем упаривали при пониженном давлении с получением 51.34 г маслянистого продукта, который со временем самопроизвольно кристаллизовался при комнатной температуре.
В полученный остаток добавляли смесь 100 мл гептана и 5 мл МТБЭ, перемешивали 30 минут и упаривали растворители при пониженном давлении досуха.
Твердый продукт снова получали, добавляя смесь 100 мл гептана и 5 мл МТБЭ и перемешивая 30 минут при комнатной температуре.
Выделенный осадок затем отфильтровывали на воронке Бюхнера и промывали смесью 4:1 гептан/МТБЭ, охлажденной до 0°C. Продукт сушили при 40°C при пониженном давлении, получая 39.4 г желтого твердого вещества (интермедиат V).
3.28 г оранжевого масла получали из маточных растворов после удаления растворителя. Анализ полученного масла методом ТСХ показал присутствие целевого продукта и других пятен.
Анализ:
1H-ЯМР (CDCl3): 1.03 м.д., синглет, 3H; 1.41-1.54 м.д., мультиплет, 2H; 1.54-1.74 м.д., мультиплет, 2H; 1.80-1.82 м.д., 3H, триплет, J = 1.83 Гц; 1.92-1.97 м.д., дублет дублетов, 1H; 2.01-2.1 м.д., мультиплет, 1H; 2.1-2.4 м.д., мультиплет, 4H; 2.4-2.6 м.д., мультиплет, 3H; 2.86-2.91 м.д., 1H, дублет дублетов.
13C-ЯМР (CDCl3): 219.6 м.д., 198.65 м.д.; 156.92 м.д.; 130.89 м.д.; 50.56 м.д.; 47.08 м.д.; 38.32 м.д.; 36.89 м.д.; 35.85 м.д.; 30.61 м.д.; 26.43 м.д.; 21.98 м.д.; 12.99 м.д.; 11.21 м.д..
Масс-спектр (CI): [M++1] = 233.
FT-IR (KBr): 1735 см-1; 1652 см-1; 1604 см-1.
Спектр рентгеновской дифракции показал присутствие как кристаллической фазы (основные пики при 11.178°, 11.435°, 14.501°, 16.332°, 18.671°, 19.579°, 21.553°, 28.738° угла 2θ), так и аморфного продукта.
ДСК: переход начинается при 121°C и достигает пика при 124°C.
Пример 3
Данный пример относится к стадии b) способа по настоящему изобретению.
Повторяли методику из предыдущего примера, получая органический раствор C, который в данном случае упаривали (жидкий остаток).
Образец очищали для аналитических целей методом флэш-хроматографии на силикагеле, элюируя 1:1 смесью изопропилацетат/изомеры гептана, и сушили при пониженном давлении до постоянного веса (получали желтое масло, интермедиат IV).
1H-ЯМР (CDCl3): 1.04-1.08 м.д., 3H, дублет триплетов, J = 7.8 Гц; 1.17 м.д., 3H, синглет; 1.6-1.86 м.д., 5H, мультиплет; 1.96-2.12 м.д., 2H, мультиплет; 2.18-2.28 м.д., 1H, мультиплет; 2.38-2.72 м.д., 8H, мультиплет.
13C-ЯМР (CDCl3): 218.1 м.д., 211.54 м.д.; 211.102 м.д.; 49.59 м.д.; 49.14 м.д.; 47.59 м.д.; 39.84 м.д.; 37.54 м.д.; 36.19 м.д.; 35.99 м.д.; 30.51 м.д.; 22.36 м.д.; 20.28 м.д.; 13.62 м.д.; 7.89 м.д.
Масс-спектр (CI): [M++1] = 251.
FT-IR (ATR): 1730 см-1
Пример 4
Данный пример относится к стадии b) способа по настоящему изобретению.
500 мг интермедиата (III) растворяли при 20°C в 38 мл ацетона.
1 мл реагента Джонса добавляли при перемешивании в течение 50 минут, поддерживая температуру реакции между 15 и 25°C.
Реагент Джонса готовили заранее, растворяя 27 г хромового ангидрида (CrVIO3) в 100 мл воды, и затем добавляли 23 мл 98%-ной серной кислоты.
По окончании добавления полученную смесь перемешивали в течение 10 минут, затем полноту протекания реакции проверяли методом ТСХ (элюент МТБЭ/н-гептан = 8:2).
В реакционную смесь добавляли 20 мл МТБЭ и 40 мл 5%-ного раствора NaHCO3.
Твердый осадок отфильтровывали и промывали 10 мл МТБЭ.
Фазы разделяли, и органическую фазу промывали 20 мл 5%-ного раствора NaHCO3 и 20 мл насыщенного водного раствора NaCl.
Органическую фазу упаривали при пониженном давлении, получая 370 мг интермедиата (IV).
Пример 5
Данный пример относится к стадии b) способа по настоящему изобретению.
100 мг интермедиата (III) растворяли при 20°C в 5 мл дихорметана.
205 мг периодинана Десс-Мартина порциями добавляли при перемешивании в течение 2.5 часов, поддерживая температуру реакции между 15 и 25°C.
Окончание реакции (остаток интермедиата (III) менее 5% от исходного количества) проверяли методом ТСХ (элюент МТБЭ/н-гептан = 8:2).
Реакционную суспензию фильтровали, и твердый осадок промывали 5 мл дихлорметана.
Органическую фазу упаривали при пониженном давлении и хроматографировали на силикагеле (элюент МТБЭ/н-гептан = 8:2), получая, после удаления растворителя, 60 мл интермедиата (IV).
Пример 6
Данный пример относится к стадии d) способа по настоящему изобретению.
5 г интермедиата (V), полученного как описано в Примере 2, растворяли при перемешивании в 50 мл метанола при комнатной температуре.
Раствор охлаждали до T < 15°C и, не превышая этой температуры, добавляли 204 мг NaBH4 при перемешивании в 4 приема в течение 20 минут.
Полученную смесь перемешивали 30 минут при 10 < T < 15°C и затем проверяли протекание реакции методом ВЭТСХ (элюент гептан/изопропилацетат = 1:1), детектируя формирование основного пятна с более низким значением Rf, чем у интермедиата (V), присутствие интермедиата (V) и нескольких пятен с едва детектируемой интенсивностью.
Реакционную смесь перемешивали при 10 < T < 15°C, добавляя 50, 20 и 10 мг NaBH4, соответственно, с интервалами в 30 минут.
Протекание реакции проверяли после каждого добавления методом ВЭТСХ, как описано выше. Через 30 минут после последнего добавления тест показал, что реакция завершена.
Смесь охлаждали до температуры 0 < T < 5°C и добавляли 100 мл воды во время перемешивания.
Полученную смесь перемешивали при температуре 0 < T < 5°C в течение 1 часа, и выпавший твердый осадок отфильтровывали на воронке Бюхнера.
После сушки при пониженном давлении до постоянного веса получали 4.2 г соединения (1) в виде желтого твердого вещества, содержащего менее 5% непрореагировавшего интермедиата (V).
Пример 7
Данный пример относится к стадии d) способа по настоящему изобретению.
294 г интермедиата (V), полученного как описано в Примере 2, суспендировали в 2.5 мл метанола и перемешивали 30 минут при комнатной температуре (неполное растворение).
Полученный раствор охлаждали до температуры T < 15°C и, не превышая этой температуры, добавляли суммарно 18 г NaBH4 в 10 приемов в течение 160 минут, проверяя прогресс реакции методом ВЭТСХ (элюент гептан/изопропилацетат = 1:1).
По окончании реакции, реакционную смесь охлаждали до температуры 0 < T < 5°C и при перемешивании добавляли 5 л воды.
Смесь перемешивали при температуре 0 < T < 5°C в течение 1 часа, и выпавший твердый осадок отфильтровывали на воронке Бюхнера, промывая его водой (600 мл).
После сушки при 45°C при пониженном давлении до постоянного веса получали 241 г соединения (1) (желтое твердое вещество), содержащего менее 5% непрореагировавшего интермедиата (V).
Образец полученного продукта очищали для аналитических целей методом флэш-хроматографии на силикагеле, элюируя 60:40 смесью изомеров гептана/изопропилацетат.
Продукт выделяли, упаривая элюирующий растворитель, и затем продукт суспендировали в МТБЭ при температуре 0 < T < 5°C в течение 30 минут.
После фильтрования и сушки (T = 45°C, P < 1 атм) до постоянного веса, получали твердое белое вещество.
1H-ЯМР (CDCl3): 0.91 м.д., 3H, синглет; 1.11-1.72 м.д., 7H, мультиплет; 1.79-1.80 м.д., 3H, триплет J = 1.6 Гц, J = 2 Гц; 1.9-2.05 м.д., 2H, мультиплет; 2.08-2.2 м.д., 1H, мультиплет; 2.2-2.37 м.д., 3H, мультиплет; 2.45-2.54 м.д., 1H, дублет триплетов; 2.79-2.84 м.д., 1H, дублет дублетов; 3.68-3.72 м.д., 1H, триплет, J = 8.8 Гц.
13C-ЯМР (CDCl3): 199.16 м.д., 158.59 м.д.; 130.10 м.д.; 81.28 м.д.; 50.48 м.д.; 42.44 м.д.; 39 м.д.; 37.1 м.д.; 35.7 м.д.; 30.5 м.д.; 27.04 м.д.; 27 м.д.; 23.5 м.д.; 11.1 м.д.; 10.3 м.д.
Масс-спектр (CI): [M++1] = 235.
FT-IR (KBr): 3451 см-1, 1647 см-1, 1604 см-1
Спектр рентгеновской дифракции показал присутствие кристаллической фазы с характеристичными пиками при значениях 11.482°, 16.958°, 17.899°, 18.483°, 19.401° и 24.583° угла 2θ.
Полная дифракционная диаграмма показана на Фиг. 2.
ДСК: переход начинается при 167.5°C, пик достигается при 169.5°C.
[α]25D=-37.5°(1% CH3Cl).
| название | год | авторы | номер документа |
|---|---|---|---|
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| НОВЫЕ АКТИВАТОРЫ РЕЦЕПТОРОВ ВИТАМИНА D И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2008 |
|
RU2535448C2 |
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ АМИНОДИГИДРОТИАЗИНА | 2009 |
|
RU2476431C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ АМИНОДИГИДРОТИАЗИНА, ПРИГОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВАСЕ | 2012 |
|
RU2593756C2 |
| N-[3-(5-АМИНО-3,3А,7,7А-ТЕТРАГИДРО-1Н-2,4-ДИОКСА-6-АЗА-ИНДЕН-7-ИЛ)-ФЕНИЛ]-АМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ВАСЕ1 И(ИЛИ) ВАСЕ2 | 2012 |
|
RU2597308C2 |
| СПОСОБ ПОЛУЧЕНИЯ (15α,16α,17β)-ЭСТРА-1,3,5(10)-ТРИЕН-3,15,16,17-ТЕТРОЛА (ЭСТЕТРОЛА) И ИНТЕРМЕДИАТЫ В ЭТОМ СПОСОБЕ | 2020 |
|
RU2818561C1 |
| ПРОИЗВОДНЫЕ ПЕРГИДРОИЗОИНДОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2127260C1 |
| ОПТИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ БИСОКСАЗОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2004 |
|
RU2326874C2 |
| ПЕПТИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2106357C1 |
| ПРОИЗВОДНЫЕ ПЕРГИДРОИЗОИНДОЛА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2120438C1 |
Настоящее изобретение относится к способу синтеза 17β-гидрокси-дез-A-андрост-9,10-ен-5-она, который может применяться в качестве интермедиата в синтезе ретро-прогестеронов. Способ включает следующие стадии: a) реакция (4aR,6aS,9aS,9bS)-декагидро-6a-метил-циклопента[f][1]бензопиран-3,7-диона с этилмагний бромидом или этилмагний хлоридом с получением смеси изомеров: (4S,5R,7aS)-5-гидрокси-7a-метил-4-(3-оксопентил)октагидро-1H-инден-1-она и (4S,5S,7aS)-5-гидрокси-7a-метил-4-(3-оксопентил)октагидро-1H-инден-1-она; b) окисление полученной смеси изомеров с получением (4S,7aS)-7a-метил-4-(3-оксопентил)гексагидро-1H-инден-1,5(4H)-диона; c) циклизация (4S,7aS)-7a-метил-4-(3-оксопентил)гексагидро-1H-инден-1,5(4H)-диона с получением дез-A-андрост-9,10-ен-5,17-диона; d) восстановление дез-A-андрост-9,10-ен-5,17-диона до 17β-гидрокси-дез-A-андрост-9,10-ен-5-она. Предлагаемый способ позволяет получить целевой продукт с использованием простой технологии. 2 н. и 8 з.п. ф-лы, 2 ил., 7 пр.
1. Способ синтеза 17β-гидрокси-дез-A-андрост-9,10-ен-5-она, соединения (1), включающий следующие стадии:
a) реакция (4aR,6aS,9aS,9bS)-декагидро-6a-метил-циклопента[f][1]бензопиран-3,7-диона, соединения (II), с этилмагний бромидом или этилмагний хлоридом с получением смеси изомеров: (4S,5R,7aS)-5-гидрокси-7a-метил-4-(3-оксопентил)октагидро-1H-инден-1-она и (4S,5S,7aS)-5-гидрокси-7a-метил-4-(3-оксопентил)октагидро-1H-инден-1-она, смеси интермедиатов (III)

b) окисление смеси интермедиатов (III) с получением (4S,7aS)-7a-метил-4-(3-оксопентил)гексагидро-1H-инден-1,5(4H)-диона, интермедиата (IV)
c) циклизация интермедиата (IV) с получением дез-A-андрост-9,10-ен-5,17-диона, интермедиата (V)

d) восстановление интермедиата (V) до 17β-гидрокси-дез-A-андрост-9,10-ен-5-она, соединения (1)
 .
.
2. Способ по п. 1, в котором стадию a) проводят в растворителе, выбранном из диэтилового эфира, простого изопропилового эфира, тетрагидрофурана, метилтетрагидрофурана или их смесей, в инертной атмосфере и при температуре от -50 до 0°C.
3. Способ по любому из пп. 1 или 2, в котором на стадии a) мольное соотношение между этилмагний бромидом или этилмагний хлоридом и соединением (II) составляет от 1 до 2.
4. Способ по п. 3, в котором указанное соотношение составляет от 1.05 до 1.25.
5. Способ по любому из предшествующих пунктов, в котором стадию b) проводят в растворителе, выбранном из диэтилового эфира, простого изопропилового эфира, тетрагидрофурана, метилтетрагидрофурана, ацетона, метил-изобутилкетона, толуола, чистого гептана или смеси его изомеров, циклогексана, диметилацетамида, диметилформамида, хлороформа, метиленхлорида, диметилсульфоксида, воды и их смесей.
6. Способ по любому из предшествующих пунктов, в котором на стадии b) в качестве окислителя применяют: трихлоризоциануровую кислоту (TCCA) в присутствии органического основания; соединения хрома (VI) в присутствии оснований или кислот; 2,2,6,6-тетраметилпиперидин-1-оксильный радикал или его производное в присутствии основного водного раствора и соокислителя, выбранного из кислорода и гипохлорита натрия или гипохлорита кальция; гипохлорит натрия, гипохлорит кальция или гипохлорит тетрабутиламмония; газообразный кислород или кислородно-азотные смеси в присутствии солей меди (I); пероксимоносульфат калия KHSO5; газообразный хлор, растворенный в галогенированном растворителе; изопропилат алюминия в присутствии карбонильного соединения; диметилсульфоксид и активатор, выбранный из оксалилхлорида, бензойного ангидрида, трифторуксусного ангидрида, P2O5 и комплекса SO3-пиридин, в присутствии основания; соединение гипервалентного иода, выбранное из периодинана Десс-Мартина и иодбензойной кислоты, опционально стабилизированное бензойной кислотой и изофталевой кислотой.
7. Способ по любому из предшествующих пунктов, в котором стадию b) проводят, применяя 0.5-5 молей трихлоризоциануровой кислоты (TCCA) в ацетоне на моль смеси интермедиатов (III) при температуре от 5 до 40°C.
8. Способ по любому из предшествующих пунктов, в котором стадию c) проводят в растворителе, выбранном из диэтилового эфира, простого изопропилового эфира, тетрагидрофурана, метилтетрагидрофурана, толуола, циклогексана, н-гептана, смеси изомеров гептана, метанола, этанола, изопропанола, уксусной кислоты, ацетонитрила, метиленхлорида, воды и их смесей, в присутствии основного или кислотного катализатора, при температуре от 0 до 40°C.
9. Способ по любому из предшествующих пунктов, в котором стадию d) проводят с гидридом металла при температуре от -10 до 40°C в растворителе, выбранном из спиртов или смеси спирта и второго растворителя, выбранного из диэтилового эфира, простого изопропилового эфира, тетрагидрофурана, метилтетрагидрофурана, метиленхлорида, толуола, циклогексана, н-гептана, смеси изомеров гептана, диметилформамида и диметилацетамида.
10. Смесь (III) изомеров: (4S,5R,7aS)-5-гидрокси-7a-метил-4-(3-оксопентил)октагидро-1H-инден-1-она и (4S,5S,7aS)-5-гидрокси-7a-метил-4-(3-оксопентил)октагидро-1H-инден-1-она:

| G | |||
| Saucy et al | |||
| Steroid Total Synthesis, Part II; (-)-17β-Hydroxy-des-A-androst-9-en-5-one | |||
| Helvetica Chimica Acta, 1971, 54(7), 2121-2132 | |||
| G | |||
| Saucy et al | |||
| Totalsynthese von Steroiden | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Печь для сжигания твердых и жидких нечистот | 1920 |
|
SU17A1 |
| Helvetica Chimica Acta, 1971, 54(7), 2034-2042 | |||
| A.R | |||
| Daniewski et al | |||
| Total Synthesis of | |||

