Предлагаемое изобретение относится к способам получения диангидридов ароматических тетракарбоновых кислот с бифенильными или бинафтильными фрагментами, которые могут быть применены для получения полиимидных материалов, используемых при изготовлении матриц для радиационностойких композиционных материалов, применяемых в различных областях техники.
В области создания материалов с высокой температурой эксплуатации и стойкостью к радиационному излучению, в качестве термоустойчивой полимерной основы, сочетающей при нагреве высокую деформационную устойчивость (теплостойкость), химическую устойчивость (термостойкость), огнестойкость, могут быть эффективно использованы полиимиды, которые являются циклоцепными полимерами из чередующихся ароматических и гетероциклических циклов (полигетероариленами). Предельная тепло- и термостойкость характерна для полигетероариленов, цепи которых построены непрерывно чередующимися ароматическими и гетероциклами.
Высокая термостойкость (химическая устойчивость при нагреве, Тд) полиимидов связана со стабилизацией структуры и упрочнением связей за счет эффектов сопряжения, благодаря наличию неподеленной пары электронов у гетероатома в цикле (у азота) и атомов с высокой электроотрицательностью (кислород в карбонильных группах) и стабилизация имидного цикла за счет сопряжения связей C-N.
Радиационная стойкость полиимидов связана с высокой прочностью связей в имидном цикле, а также с тем, что облучение полиимидов сопровождается конкурирующими процессами - разрывом макроцепей и молекулярным сшиванием. При облучении в них появляется гель-фракция. Поэтому снижение прочности за счет деструкции в значительной степени компенсируется ее увеличением за счет структурирования.
Таким образом основными требованиями, предъявляемыми к таким полиимидам, являются высокая механическая прочность в сочетании с химической и термической и радиационной стабильностью.
Качество полиимидных материалов напрямую зависит от выбора тех или иных исходных ароматических и гетероциклических соединений. К ароматическим соединениям, используемым при получении полиимидных материалов, относятся ангидриды ароматических тетракарбоновых кислот с бифенилными и бинафтильными фрагментами. Таковыми соединениями являются: диангидрид 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты (1), диангидрид 2,2',3,3'-бифенил тетракарбоновой кислоты (2), диангидрид 3,3',4,4'-бифенил тетракарбоновой кислоты (3), структурные формулы которых представлены ниже:
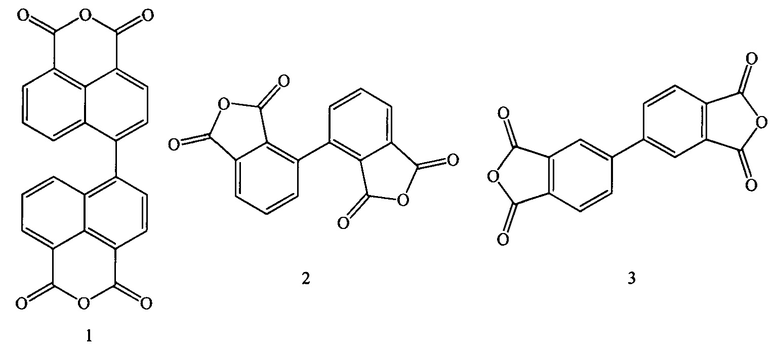
Как видно из приведенных на рисунке структур, все они являются ароматическими соединениями, а, следовательно, они склонны к схожим типам химических превращений.
Из ранее опубликованных информационных источников следует, что данные соединения могут быть получены общим методом синтеза, в основе которого лежит образование С-С связи между двумя ароматическими фрагментами.
Так, например, в статье [Tong Y. et. al. // J. Polym. Sci., Polym. Chem. Ed. 1999. V. 37. P. 1425-1433., McLoughlin V.C.R., Thrower J. // Tetrahedron. 1969. V. 25. P. 5921-5940.] предлагают 2,2',3,3'-бифенил тетракарбоновую кислоту (предшественник соответствующего ангидрида) получать из тетраэфира обработкой KOH в спирте (получение соли) и последующим подкислением соляной кислотой. В свою очередь тетраметиловый эфир данной кислоты получают медь-катализируемой конденсацией диметил 3-иодфталата по реакции Ульмана при 240-260°С по описанной схеме:
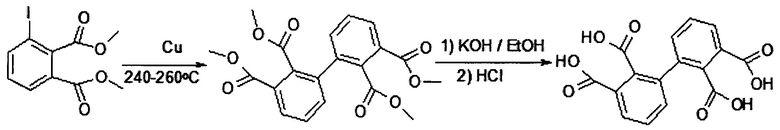
При этом исходный диметил 3-иодфталат получают путем диазотирования диметил 3-аминофталата NaNO2 в кислой среде с последующим замещением диазо-группы на атом иода с помощью KI [Thottumkara А.Р., Vinod Т.K. // Tetrahedron Lett. 2002. V. 43. P. 569-572.], a диметил 3-аминофталат получают восстановлением соответствующего нитро-соединения водородом на палладиевом катализаторе. Возможны и другие способы восстановления нитросоединения: железом в среде электролита, металлами в кислой или щелочной среде растворами сульфидов. Но лучшим способом, как по выходу целевых продуктов, так и по отсутствию большого количества неорганического шлама, является восстановление водородом на палладиевом катализаторе [Лисицын В.Н. Химия и технология промежуточных продуктов. М.: Химия. 1987]. Исходное соединение - диметил-3-нитрофталат получают из соответствующей 3-нитрофталевой кислоты этерификацией соответствующего дихлорангидрида, а 3-нитрофталевую кислоту получают нитрованием коммерчески доступного фталевого ангидрида. При этом получается смесь 3- и 4-нитрофталевых кислот (в соотношении 2:1) [Методы получения химических реактивов и препаратов. Выпуск N. 15. М.: ИРЕА. 1967.].
Таким же образом путем восстановления нитрогруппы в положении 4 и получением диметил-4-иодфталата через реакцию диазотирования, и с последующей затем конденсацией, гидролизом и сушкой удается получить диангидрид 3,3',4,4'-бифенил тетракарбоновой кислоты так же, как это описано выше для его изомера - диангидрида 2,2',3,3'-бифенил тетракарбоновой кислоты. Однако известные способы получения диангидридов 2,2',3,3' и 3,3',4,4'-бифенил тетракарбоновой кислоты многостадийны и, кроме того, включают в себя реакции, проходящие при высоких температурах и с агрессивными реагентами, что осложняет их промышленное применение.
Способ синтеза диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты описан в ряде информационных источников. Впервые синтез диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты описан в 1973 году. [L.A. Jones, R. Watson // Can. J. Chem. 1973. V. 51. P. 1833-1837.] В основе этого синтеза лежит реакция конденсации коммерчески доступного 5-бромаценафтена с 5-магнийбромаценафтеном, предварительно полученным из того же 5-бромаценафтена и магниевых стружек в тетрагидрофуране, протекающая в присутствии CoCl2 с суммарным выходом (45%). Получение диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты (или 5,5'-бинафтил) с использованием реактива Гриньяра в данном процессе протекает по следующей схеме:
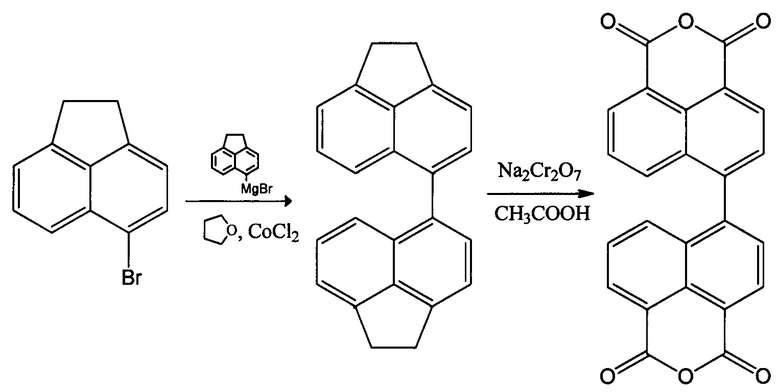
Основным недостатком этого способа является необходимость использования хроматографии для отделения 4,4'-диаценафтена от образующегося в результате отщепления брома аценафтена и исходного 5-бромаценафтена. Использование колоночной хроматографии серьезно ограничивает возможности для промышленного осуществления данного процесса. Существенным недостатком также является невысокий выход конечного продукта (43%), а также использование в процессе легко воспламеняющихся и химически агрессивных веществ (стадию окисления осуществляют бихроматом натрия в ледяной уксусной кислоте).
Во всех последующих работах, касающихся синтеза производных бинафтила, ключевой стадией также является образование С-С связи между двумя ароматическими фрагментами, осуществляемой с использованием катализаторов, в качестве которых используются различные металлы и соли металлов. Так, например, в работе [Jaworek W.,  // European Journal of Inorganic Chemistry. 1991. V. 124. №. 2. P. 347-352.] описывается способ получения 4,4'-диаценафтена (1) из аценафтена в присутствии тетраацетата свинца и BF3*(С2Н5)2O в ацетонитриле при комнатной температуре, осуществляемый с выходом 47%.
// European Journal of Inorganic Chemistry. 1991. V. 124. №. 2. P. 347-352.] описывается способ получения 4,4'-диаценафтена (1) из аценафтена в присутствии тетраацетата свинца и BF3*(С2Н5)2O в ацетонитриле при комнатной температуре, осуществляемый с выходом 47%.
В качестве катализатора для синтеза производных бинафтила также применяется палладий, например, в способе получения диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты, описанном в китайском патенте [CN 101397287, 2009]. Данный способ осуществляют в несколько стадий. Сначала исходный диангидрид 4-гало-1,8-нафталиндикарбоновой кислоты смешивают с 4-10 кратным молярным избытком гидроксида калия, добавляют дистиллированную воду и нагревают для полного растворения. Затем к раствору добавляют катализатор Pd/C (палладий на углероде) и восстановитель и реакционную массу при температуре 80-120°С кипятят с обратным холодильником в течение от 3-40 часов. После этого отработанный катализатор - Pd/C и примеси отфильтровывают, а затем в фильтрат добавляют хлористоводородную кислоту или азотную кислоту до установления рН 1-3. Затем осажденная желтая эмульсия промывается дистиллированной водой в течение 3-5 раз и сушится в вакуумной печи в течение 10-20 часов при температуре от 180-200°С. В результате получается твердый порошок желтого цвета, который затем перекристаллизовывают из диметилформамида, и получают 4,4',5,5'-бинафтил-тетракарбоновый диангидрид(син. диангидрид 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты). Преимущество использования катализатора- палладия на углероде, не смотря на его дороговизну, заключается в том, что за счет использования восстановителя катализатор не теряет своей активности и может быть повторно использован в синтезе.
В другой публикации [Li N. et al. // Polymer. 2007. V. 48. №.25. P. 7255-7263.] диангидрид 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты получают через образование эфира 4-хлор-1,8-нафталиндикарбоновой кислоты с последующей конденсацией. Для этого ангидрид 4-хлор-1,8-нафталиндикарбоновой кислоты обрабатывают смесью PCl5/POCl3, а затем метанолом и полученный диметиловый эфир данной кислоты конденсируют, затем гидролизуют раствором KOH в этиленгликоле и высушивают в вакууме при 200°С в течение 12 часов. Суммарный выход конечного продукта-диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты, составляет 75%. Данный способ, не смотря на свою эффективность, имеет существенные недостатки, которые заключаются в использовании на первой стадии процесса в больших количествах высокотоксичных и реакционноспособных хлорангидридов фосфора. Работа с данными соединениями, как известно, требует применения особых мер безопасности и использования коррозионностойкого оборудования.
В ряде работ синтез производных бинафтила предлагается проводить с использованием в качестве катализаторов безводных солей никеля, применяемых часто в присутствии цинкового порошка и трифенилфосфина в диметиацетамиде. Исходным продуктом для синтеза производных бинафтила является, например, 5-бромаценафтен, получение которого, в свою очередь, может быть осуществлено бромированием аценафтена, как это описано в работах [Liu L., Zhang С., Zhao J. // Dalton Transactions. 2014. V. 43. №. 35. P. 13434-13444.; Constantine P.R., Deady L.W., Topsom R.D. // The Journal of Organic Chemistry. 1969. V. 34. №. 4. P. 1113-1115.].
С применением никелевых солей в качестве катализатора был предложен способ получения диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты из 5-бромаценафтена, описанный в статье [Sun F. et al. // Polymer. 2010. V. 51. №. 17. P. 3887-3898.], протекающий по следующей схеме:
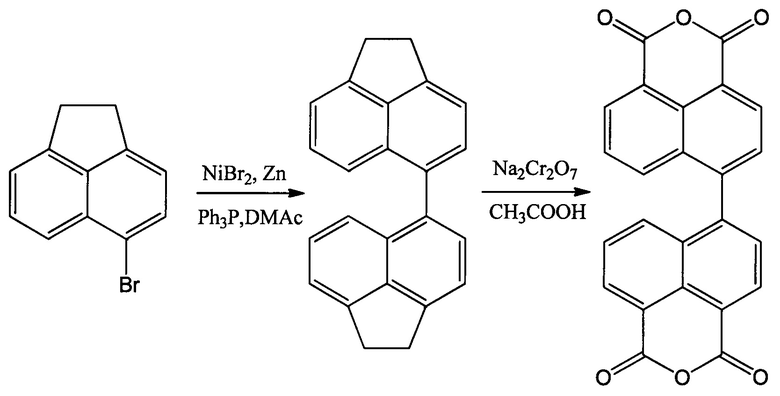
Целевой диангидрид, согласно этой схемы, получают из 4,4'-диаценафтена путем его окисления бихроматом натрия в ледяной уксусной кислоте, а 4,4'-диаценафтен получают из коммерчески доступного 5-бромаценафтена путем его конденсации в присутствии бромида никеля(II), цинкового порошка и трифенилфосфина в диметиацетамиде. Основной недостаток применения 4,4'-диаценафтена в синтезе диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты заключается в необходимости применения хроматографических методов выделения 4,4'-диаценафтена, что ограничивает возможность его промышленной применимости. Кроме того, данный способ осуществляется со сравнительно невысоким суммарным выходом реакции (35%).
Наиболее близким аналогом предлагаемого способа является известный способ получения диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты из ангидрида 4-хлор-1,8-нафталиндикарбоновой кислоты [Gao J.P., Wang Z.Y. // Journal of Polymer Science Part A: Polymer Chemistry. 1995. V. 33. №. 10. P. 1627-1635], который осуществляют по следующей схеме:
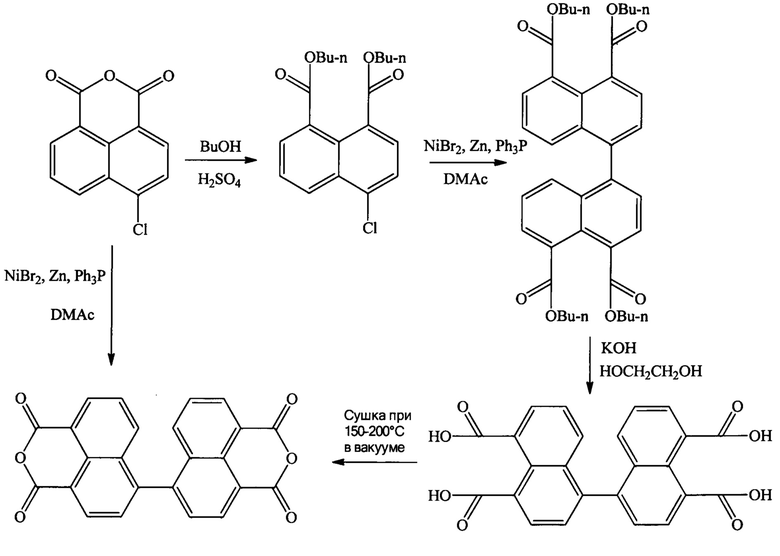
В этой работе приводятся два варианта получения диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты. Первый вариант-это получение непосредственно из ангидрида 4-хлор-1,8-нафталиндикарбоновой кислоты путем конденсации, осуществляемой в присутствии бромида никеля(II), цинкового порошка и трифенилфосфина в диметиацетамиде. Второй вариант включает стадию образования бутилового эфира 4-хлор-1,8-нафталиндикарбоновой кислоты, после чего осуществляется его конденсация с превращением сложного эфира в диангидрид 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты.
Второй вариант заключается в следующем: на первой стадии из ангидрида 4-хлор-1,8-нафталиндикарбоновой кислоты получают сложный эфир путем кипячения в течение 16 часов в избытке бутилового спирта (на 100 г диангидрида берут 800 мл н-бутанола с каталитическим количеством серной кислоты (5 мл), с последующим упариванием спирта, и полным растворением сухого остатка в 150 мл диэтилового эфира и 150 мл толуола, подщелачиванием 150 мл 1Н раствором гидроокиси натрия, отделением органического слоя, высушиванием над сульфатом натрия, упариванием растворителя и перегонкой остатка в вакууме.
Полученный таким образом эфир затем вступает в реакцию конденсации. При этом реакцию конденсации проводят в атмосфере инертного газа в амидном органическом растворителе и присутствии каталитической смеси, содержащей, в расчете на 12,4 ммоль дибутилового эфира 4-хлор-1,8-нафталинкарбоновой кислоты, следующие компоненты: 0,6 ммоля безводного галогенида никеля(II), 4,3 моля трифенилфосфина и 17,6 ммоля активированного цинкового порошка. Раствор полученного сложного эфира в диметиацетамиде добавляют по каплям к раствору с каталитической смесью, имеющую температуру 90°С, образовавшаяся реакционная смесь перемешивается при этой температуре в течение 12 часов, после чего раствор отфильтровывают от катализатора в 200 мл воды, выпавший липкий осадок отфильтровывают и высаживается водой, после чего полученный тетраэфир отфильтровывается, кипятится в этиленгликоле в присутствии 5 кратного мольного избытка по отношению к исходному эфиру кислоты твердого KOH в течение 24 часов, охлаждается, и осадок отфильтровывается, промывается ацетоном и водой, и сушится в вакууме в течение 10 часов при 200°С до образования целевого продукта.
Никель выступает в роли катализатора образования С-С связи. Избыток цинка восстанавливает никель до нулевой степени окисления и поддерживает его в этом состоянии все время реакции, а за счет комплексообразования с трифенилфосфином никель остается в растворе.
Основным недостатком первого варианта (конденсация диангидрида 4-хлор-1,8-нафталиндикарбоновой кислоты осуществляется так же как и дибутилового эфира) являются трудности, возникающие на стадии очистки конечного продукта, связанные с образованием неидентифицируемого красноватого побочного продукта, отделять который от целевого диангидрида удается только многократным переосаждением метанолом из щелочного раствора. Это в свою очередь сказывается на невысоком выходе конечного продукта (43,8%). Второй вариант осуществления рассматриваемого процесса, не смотря на большее количество стадий, более технологичен чем первый вариант, поскольку исключает трудоемкую очистку, а также более эффективен, так как проходит с суммарным выходом 66%.
Второй вариант осуществления рассмотренного выше способа по своей технической сущности является наиболее близким аналогом заявленному способу и выбран в качестве его прототипа.
Целью предлагаемого изобретения является разработка технологичного процесса получения диангидридов ароматических тетракарбоновых кислот, расширяющего ассортимент исходных продуктов и обеспечивающего получение чистого продукта, удовлетворяющего требованиям, предъявляемым к функциональным мономерам для изготовления полиимидных материалов.
Для достижения указанной цели предлагается Способ получения диангидридов ароматических тетракарбоновых кислот конденсацией по С-С-связям в ароматических фрагментах сложных эфиров ароматических галогендикарбоновых кислот, предварительно полученных из диангидридов ароматических галогендикарбоновых кислот, выбранных из следующей группы: ангидрид 4-хлор-1,8-нафталиндикарбоновой кислоты, ангидрид 4-бром-1,8-нафталиндикарбоновой кислоты, ангидрид 3-иод-фталевой кислоты, ангидрид 4-иод-фталевой кислоты, каждый из которых переводят в соответствующий сложный эфир кипячением с С1-С4 одноатомным спиртом, взятым в 5-7 кратном избытке по массе от эквимолярного количества при кипячении в присутствии каталитических количеств серной кислоты течение 8-16 часов, с последующим выделением сложного эфира упариванием спирта, полным растворением сухого остатка в толуоле, подщелачиванием раствором гидроокиси натрия до нейтрального рН при перемешивании, отделением органического слоя, высушиванием, упариванием растворителя и перегонкой остатка в вакууме, после чего раствор полученного сложного эфира ароматической галогендикарбоновой кислоты в сухом амидном органическом растворителе вводится медленно в предварительно полученную каталитическую смесь, имеющую температуру 80-100°С и содержащую 0,03-0,07 моля безводного галогенида никеля(II), 0,2-0,4 моля трифенилфосфина и 2-4 моля активированного цинкового порошка на 1 моль диангидрида ароматической галогендикарбоновой кислот, и образовавшаяся реакционная масса перемешивается при 80-100°С в течение 6-12 часов, после чего органический амидный растворитель упаривается и к оставшейся реакционной массе добавляется низкокипящий хлорсодержащий органический растворитель, и затем полученный раствор при охлаждении и перемешивании приливается к водному раствору соляной кислоты, фильтруется, органический слой отделяется, растворитель упаривается, а остаток растворяется и полученный диангидрид ароматической тетракарбоновой кислоты сушится в вакууме в течение 5-10 часов при 150-200°С.
Способ осуществляется с применением в составе каталитической смеси галогенидов никеля(II), выбранных из группы: хлорид никеля(II), бромид никеля(II), иодид никеля(II).
На стадии конденсации способ осуществляется с использованием сухих амидных органических растворителей, выбранных из группы: формамид, ацетамид, N,N-диметилформамид, N,N-диметилацетамид.
К реакционной массе после упаривания амидного растворителя добавляется низкокипящий хлорсодержащий органических растворитель, выбранный из группы: хлористый метилен, хлороформ, тетрахлорметан.
Образование сложных эфиров осуществляется с использованием С1-С4 одноатомных спиртов, выбранных из группы: метанол, этанол, 1-пропанол, 2-пропанол 1-бутанол.
Реакция взаимодействия диангидридов ароматических галогендикарбоновых кислот и низших одноатомных спиртов осуществляется при кипячении в присутствии каталитических количеств серной кислоты, взятых из расчета 0,05 г кислоты на 1 г исходного диангидрида.
Предлагаемый способ, как и прототип протекает по одной схеме:
из диангидридов бифенил тетракарбоновых кислот на первой стадии получается сложный эфир, который затем вступает в реакцию каталитической конденсации с образованием С-С связи, затем полученный таким образом тетраэфир гидролизуют и высушивают в вакууме, где одновременно происходит процесс образования диангидрида.
Основное отличие предлагаемого способа от прототипа и известных аналогов заключается в возможности осуществления способа с применением широкой группы исходных продуктов, включающих как нафтильные фрагменты, так и фенильные на примере таких соединений как: ангидрид 4-хлор-1,8-нафталиндикарбоновой кислоты, ангидрид 4-бром-1,8-нафталиндикарбоновой кислоты, ангидрид 3-иод-фталевой кислоты, ангидрид 4-иод-фталевой кислоты. Кроме того, расширяется и ассортимент других используемых в процессе соединений. Так в предлагаемом способе, кроме используемого в составе каталитической смеси бромида никеля(II), дополнительно используются также хлорид никеля(II) и иодид никеля(II), а в качестве растворителей из группы низкокипящих хлорсодержащих органических растворителей используются соединения, выбранные из группы: хлористый метилен, хлороформ, тетрахлорметан, и в качестве амидных органических растворителей используются соединения, выбранные из группы: формамид, ацетамид, N,N-диметилформамид, N,N-диметилацетамид. В предлагаемом способе расширяется и ассортимент низших спиртов, используемых для получения сложных эфиров, а именно образование сложных эфиров осуществляется с использованием С1-С4 одноатомных спиртов, выбранных из группы: метанол, этанол, 1-пропанол, 2-пропанол 1-бутанол.
Существенными признаками процесса также являются: выбранные весовые соотношения исходных соединений, а также последовательность стадий обработки и временно-температурные регламенты процесса на всех его стадиях.
Как и в прототипе в обеих вариантах используется каталитическая смесь, содержащая безводный галогенид никеля(II), трифенилфосфин и активированный цинковый порошок. Однако в предлагаемом способе предлагаются экспериментально подобранные молярные соотношения реагентов, а именно берутся следующие соотношения исходных компонентов в расчете на 1 моль исходного диангидрида ароматической галоген-дикарбоновой кислоты: 0,03-0,07 моля безводного галогенида никеля(II), 0,2-0,4 моля трифенилфосфина и 2-4 моля активированного цинкового порошка. В прототипе соотношения реагентов составляют 1:0,05:0,35:1,4.
На основании экспериментальных исследований, наилучшие результаты достигнуты при получении диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты при проведении процесса конденсации при использовании безводного бромида никеля(II) (0,07 моль на 1 моль производного нафталина), трифенилфосфина (0,35 моль на 1 моль производного нафталина), и предварительно активированного цинкового порошка (4 моль на 1 моль производного нафталина) и использовании в качестве растворителя диметилацетамида и при выдержке в течение 12 часов при 90°С. При таком температурно-временном режиме основная реакция протекает наиболее полно и с максимальным выходом.
Важным условием осуществления процесса является проведение его в атмосфере инертного газа, например, такого как азот, или аргон, что объясняется, тем, что в воздушной среде при нагревании могут происходить нежелательные процессы окисления вступающих в реакцию диаминов.
Важным условием осуществления процесса является длительная сушка (не менее 5 часов) в вакууме при температуре не менее 150°С, что позволяет получать продукт, не содержащий примесей трудноудаляемых высококипящих органических растворителей и воды.
Рассмотренные выше режимы осуществления процесса подобраны экспериментально при сравнении выхода и чистоты получаемых продуктов методами 1Н ЯМР и элементного CHNS-анализа.
Основным преимуществом предлагаемого способа перед известными аналогами является тот факт, что он не содержит процессов, ограничивающих его масштабирование, таких как, например, колоночная хроматография, и не предполагает использования дорогих катализаторов.
Получаемые продукты являются функциональными мономерами для получения полиимидных материалов, обладающих высокой механической прочностью в сочетании с химической и термической стабильностью, обладающих ионной (протонной) проводимостью, применяемых, например, при изготовлении мембран топливных элементов а также при изготовлении электрохимических сенсоров, аккумуляторов и суперконденсаторов / Изготовление мембран на основе полиимидов описано в работе [Sun F. et al. // Polymer. 2010. V. 51. №. 17. P. 3887-3898.]. Области применения полимерных электролитов охарактеризованы, например [Sammes N. (ed.). Fuel cell technology: reaching towards commercialization. - Springer Science & Business Media, 2006.].
Ниже изобретение иллюстрируется следующими примерами:
Активация цинка.
Перед тем как цинк вносится в каталитическую смесь его обрабатывают раствором соляной кислоты для удаления пленок оксидов. Для этого к порошку цинка добавляют 5-10% водный раствора соляной кислоты и после перемешивания в течение 10 минут раствор декантируют. Затем осадок промывают водой декантацией и отфильтровывают, после чего промывают осадок цинка ацетоном и высушивают в вакууме водоструйного насоса.
Пример 1.
Смесь 20 г (0,086 моль) ангидрида 3-иод-1-фталевой кислоты и 160 г этанола и 1 г 98% серной кислоты кипятят в колбе с насадкой Дина-Старка в течение 8 часов. После спирт упаривают и к сухому остатку добавляют 100 мл толуола, а затем 100 мл 1М раствора NaOH и перемешивают. Органический слой отделяют, промывают еще 150 мл 1М раствором NaOH, высушивают над безводным сульфатом магния и упаривают растворитель. Остаток перегоняют в вакууме. Получают 23,95 г диэтилового эфира 3-иод-1-фталевой кислоты (85% чистоты). 1Н-ЯМР (CDCl3, δ, м.д.): 1.36 (t, 6Н), 4.36 (q, 4 Н), 7.22 (t, 1Н), 4.30 (t, 4 Н), 7.87-7,91 (d, 2 Н), 7.96-7,99 (d, 1 Н).
В колбу, снабженную магнитной мешалкой, баней с силиконовым маслом, капельной воронкой, обратным холодильником и вводом/выводом инертного газа помещают безводный хлорид никеля(II) 0,22 г (0,00171 моль), трифенилфосфин 2,9 г (0,0114 моль), и предварительно активированный цинковый порошок 7,41 г (0,114 моль) и 105 мл сухого ацетамида. Смесь нагревают при перемешивании в токе аргона до 100°С выдерживают 30 мин. После этого медленно прибавляют раствор диэтилового эфира 3-иод-1-фталевой кислоты 20 г (0,057 моль) в 60 мл сухого ацетамида. Смесь выдерживают при 100°С в течение 12 часов. По окончании из реакционной смеси упаривают весь объем ацетамамида и добавляют 200 мл хлороформа.
Соотношение диагидрида, галогенида никеля, трифенилфосфина и цинкового порошка составляет 1:0,03:0,2:2
Полученный раствор осторожно, при охлаждении перемешивании приливают к 180 мл 10% водного раствора HCl. По окончании выделения газов фильтруют смесь через слой кизельгура марки Celite, отделяют органический слой и экстрагируют водный слой 100 мл хлороформа. Объединенные экстракты высушивают над сульфатом магния. Хлористый метилен упаривают. Осадок растворяют в 50 мл метанола. Кипятят раствор при перемешивании 15 минут. Отфильтровывают не растворившейся осадок, и сушат в вакууме в течение 10 часов при 200°С. Получают 5,2 г диангидрида 2,2',3,3'-бифенил тетракарбоновой кислоты. Выход: 31% 1Н ЯМР (DMSO d6, δ, м.д.): 7,77 (t, 2Н); 8,31 (m, 4Н). ЭА: Рассчитано для C12H6O6: С: 65.3%; Н: 2.06%. Найдено: С:65.2%; Н: 2.1%.
Пример 2
Смесь 35 г (0,128 моль) ангидрида 4-иод-фталевой кислоты и 280 г 1-пропанола и 1,8 г 98% серной кислоты кипятят в колбе с насадкой Дина-Старка в течение 8 часов. После спирт упаривают и к сухому остатку добавляют 180 мл толуола, а затем 180 мл 1М раствора NaOH и перемешивают. Органический слой отделяют, промывают еще 180 мл 1М раствором NaOH, высушивают над безводным сульфатом магния и упаривают растворитель. Остаток перегоняют в вакууме. Получают 54 г дипропилового эфира (83% чистоты). 1Н-ЯМР (CDCl3, δ, м.д.):1,04 (t, 6Н), 1.80 (m, 4Н), 4.28 (t, 4 Н), 7,58-7,62 (dd, 1Н), 7.69-7.72 (d, 3Н).
В колбу, снабженную магнитной мешалкой, баней с силиконовым маслом, капельной воронкой, обратным холодильником и вводом/выводом инертного газа помещают безводный иодид никеля(II) 2,23 г (0,007 моль), трифенилфосфин 9,35 г (0,0357 моль), и предварительно активированный цинковый порошок 31 г (0,357 моль) и 200 мл сухого диметилформамида. Смесь нагревают при перемешивании в токе аргона до 90°С и выдерживают в течение 30 мин. После этого медленно прибавляют раствор дипропилового эфира 4-иодфталевой кислоты (0,119 моль) в 120 мл сухого диметилформамида. Смесь выдерживают при 90°С в течение 10 часов. По окончании из реакционной смеси упаривают весь объем органического растворителя и добавляют 300 мл хлороформа.
Соотношение диагидрида, галогенида никеля, трифенилфосфина и цинкового порошка составляет 1:0,06:0,3:3.
Полученный раствор осторожно, при охлаждении перемешивании приливают к 250 мл 10% водного раствора HCl. По окончании выделения газов фильтруют смесь через слой кизельгура марки Celite, отделяют органический слой и экстрагируют водный слой 100 мл хлороформа. Объединенные экстракты высушивают над сульфатом магния. Хлороформ упаривают. Остаток растворяют в 100 мл технического ацетона и высаживают водой, прибавляя небольшими порциями полученный тетропропиловый эфир диаценафтена. Отфильтровывают осадок и кипятят в 200 мл этиленгликоля с 30,2 г KOH в течение 18 часов. После охлаждения, выпавший осадок отфильтровывают, промывают ацетоном и растворяют в воде. Подкисляют концентрированной HCl до рН=2, выпавший осадок отфильровывают и промывают несколько раз водой, затем и сушат в вакууме в течение 10 часов 200°С. Получают таким образом 12,9 г диангидрида 3,3',4,4'-бифенил тетракарбоновой кислоты. Выход: 37%. 1Н ЯМР (DMSO d6, δ, м.д.): 7.62-7.65 (dd, 2Н); 8.20 (s, 2Н); 8.23-8.26 (d, 2Н) ЭА: Рассчитано для C16H6O6: С, 65.32%; Н, 2.06%. Найдено: С, 65.4%; Н, 2.03%.
Пример 3:
Смесь 30 г (0,108 моль) ангидрида 4-бром-1,8-нафталин дикарбоновой кислоты и 150 г 1-бутанола и 2 г 98% серной кислоты кипятят в колбе с насадкой Дина-Старка в течение 8 часов. После спирт упаривают и к сухому остатку добавляют 150 мл толуола, а затем 150 мл 1М раствора NaOH и перемешивают. Органический слой отделяют, промывают еще 150 мл 1М раствором NaOH, высушивают над безводным сульфатом магния и упаривают растворитель. Остаток перегоняют в вакууме. Получают 34,8 г бутилового эфира (80% чистоты). 1Н-ЯМР (CDCl3, δ, м.д.):0,91 (t, 6Н), 1.51 (m, 4Н), 1.65 (m, 4 H), 4.19 (t, 4H), 7.55 (t, 3Н), 7,92-7,94 (d, 1H), 8.08-8,10 (d, 1H), 8.81-8,83 (d, 1H).
В колбу, снабженную магнитной мешалкой, баней с силиконовым маслом, капельной воронкой, обратным холодильником и вводом/выводом инертного газа помещают безводный бромид никеля(II) 1,04 г (0,00476 моль), трифенилфосфин 6,24 г (0,0238 моль), и предварительно активированный цинковый порошок 17,7 г (0,272 моль) и 150 мл сухого диметилацетамида. Смесь нагревают при перемешивании в токе аргона до 100°С и выдерживают в течение 30 мин. После этого медленно прибавляют раствор дибутилового эфира 4-бром-1,8-нафталин дикарбоновой кислоты (0,068 моль) в 90 мл сухого диметиформамида. Смесь выдерживают при 90°С в течение 12 часов. По окончании из реакционной смеси упаривают весь объем диметиацетамида и добавляют 160 мл тетрахлорметана.
Соотношение диагидрида, галогенида никеля, трифенилфосфина и цинкового порошка составляет 1:0,07:0,35:4.
Полученный раствор осторожно, при охлаждении перемешивании приливают к 225 мл 10% водного раствора HCl. По окончании выделения газов фильтруют смесь через слой кизельгура марки Celite, отделяют органический слой и экстрагируют водный слой 75 мл тетрахлорметана. Объединенные экстракты высушивают над сульфатом магния. Тетрахлорметан упаривают. Остаток растворяют в 75 мл технического ацетона и высаживают водой, прибавляя небольшими порциями полученный тетрабутиловый эфир диаценафтена. Отфильтровывают тетраэфир и кипятят в 200 мл этиленгликоля с 30,2 г KOH в течение 18 часов. После охлаждения, выпавший осадок отфильтровывают, промывают ацетоном и растворяют в воде. Подкисляют концентрированной НС1 до рН=2, выпавший осадок отфильровывают и промывают несколько раз водой, затем и сушат в вакууме в течение 10 часов 200°С. Получают таким образом 26,2 г диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты. Выход: 49%. 1Н ЯМР (DMSO d6, δ, м.д.): 8.70-8.72 (d, 2H); 8.59-8.61 (d, 2H); 7.98-8.00 (d, 2H); 7.79-7.83 (t, 2H); 7.73-7.75 (d, 2H). ЭА: Рассчитано для C24H10O6: С, 73.1%; Н, 2.56%. Найдено: С, 72.8%; Н, 2.9%.
Пример 4:
Смесь 20 г (0,086 моль) ангидрида 4-хлор-1,8-нафталин дикарбоновой кислоты и 160 г метанола и 2 г 98% серной кислоты кипятят в колбе с насадкой Дина-Старка в течение 8 часов. После спирт упаривают и к сухому остатку добавляют 100 мл толуола, а затем 100 мл 1М раствора NaOH и перемешивают. Органический слой отделяют, промывают еще 150 мл 1М раствором NaOH, высушивают над безводным сульфатом магния и упаривают растворитель. Остаток перегоняют в вакууме. Получают 23,95 г диметилового эфира (85% чистоты). 1Н-ЯМР (CDCl3, δ, м.д.): 3.53 (s, 6Н), 7.66 (m, 2 Н), 7.90 (d, 1 Н), 8.05 (d, 1 Н), 8.50 (d, 1 Н).
В колбу, снабженную магнитной мешалкой, баней с силиконовым маслом, капельной воронкой, обратным холодильником и вводом/выводом инертного газа помещают безводный хлорид никеля(II) 1,11 г (0,0051 моль), трифенилфосфин 7,65 г (0,0292 моль), и предварительно активированный цинковый порошок 19 г (0,292 моль) и 100 мл сухого диметилацетамида. Смесь нагревают при перемешивании в токе аргона до 80°С и выдерживают в течение 10 мин. После этого медленно прибавляют раствор диметилового эфира 4-хлор-1,8-нафталин дикарбоновой кислоты (0,073 моль) в 60 мл сухого диметиацетамида. Смесь выдерживают при 80°С в течение 6 часов. По окончании из реакционной смеси упаривают весь объем органического растворителя и добавляют 160 мл хлористого метилена.
Соотношение диагидрида, галогенида никеля, трифенилфосфина и цинкового порошка составляет 1:0,07:0,4:4.
Полученный раствор осторожно, при охлаждении перемешивании приливают к 150 мл 10% водного раствора HCl. По окончании выделения газов фильтруют смесь через слой кизельгура марки Celite, отделяют органический слой и экстрагируют водный слой 50 мл хлористого метилена. Объединенные экстракты высушивают над сульфатом магния. Хлористый метилен упаривают. Остаток растворяют в 50 мл технического ацетона и высаживают водой, прибавляя небольшими порциями полученный тетраметиловый эфир диаценафтена. Отфильтровывают тетраэфир и кипятят в 160 мл этиленгликоля с 14,4 г KOH в течение 18 часов. После охлаждения, выпавший осадок отфильтровывают, промывают ацетоном и растворяют в воде. Подкисляют концентрированной HCl до рН=2, выпавший осадок отфильровывают и промывают несколько раз водой, затем и сушат в вакууме в течение 10 часов 150°С. Получают таким образом 7,2 г диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты. Выход: 21%. 1Н ЯМР (DMSO d6, δ, м.д.): 8.70-8.72 (d, 2Н); 8.59-8.61 (d, 2Н); 7.98-8.00 (d, 2Н); 7.79-7.83(t, 2Н); 7.73-7.75 (d, 2Н). ЭА: Рассчитано для C24H10O6: С, 73.1%; Н, 2.56%. Найдено: С, 73.13%; Н, 2.59%.
Пример 5
Пример 5 приводится аналогично примеру 3, с теми же самыми количествами, но с использованием другого одноатомного спирта, а именно 2-пропанола
Оптимальными параметрами являются: галогенид никеля (0,07:1), цинк (4:1), трифенилфосфин (0,35:1) время выдержки 12 часов при температуре 90°С. Сушка в вакууме 12 часов при 200°С.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ДИАНГИДРИДА 4,4´-БИНАФТИЛ-1,1´,8,8´-ТЕТРАКАРБОНОВОЙ КИСЛОТЫ ИЗ ГАЛОГЕНАЦЕНАФТЕНОВ | 2017 |
|
RU2671579C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИИМИДНЫХ СОПОЛИМЕРОВ, СОДЕРЖАЩИХ КРАУН-ЭФИРНЫЕ И ПОЛИСИЛОКСАНОВЫЕ ФРАГМЕНТЫ | 2016 |
|
RU2644152C1 |
| СПОСОБ ПОЛУЧЕНИЯ КУБОВЫХ КРАСИТЕЛЕЙ И ПИГМЕНТОВ, СОДЕРЖАЩИХ ПЕРИЛЕНОВЫЙ ФРАГМЕНТ | 1997 |
|
RU2128200C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИИМИДНОГО КОМПОЗИТНОГО ПЛЕНОЧНОГО ПОКРЫТИЯ, АРМИРОВАННОГО НАНОСТРУКТУРИРОВАННЫМ КАРБИДОМ КРЕМНИЯ (ВАРИАНТЫ) | 2015 |
|
RU2620122C2 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАНГИДРИДА 3,3-БИС-(3,4-ДИКАРБОКСИФЕНИЛ)ФТАЛИДА | 2018 |
|
RU2698914C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИИМИДНОГО КОМПОЗИТНОГО ВОЛОКНА НА УГЛЕРОДНОЙ ОСНОВЕ, АРМИРОВАННОГО НАНОСТРУКТУРИРОВАННЫМ КАРБИДОМ КРЕМНИЯ | 2016 |
|
RU2644906C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИИМИДНОГО КОМПОЗИТНОГО МАТЕРИАЛА, АРМИРОВАННОГО НАНОСТРУКТУРИРОВАННЫМ КАРБИДОМ БОРА (ВАРИАНТЫ) | 2016 |
|
RU2656045C2 |
| ЦИКЛИЧЕСКОЕ КАРБОДИИМИДНОЕ СОЕДИНЕНИЕ | 2009 |
|
RU2503669C2 |
| {4,4′-[БИС-(4,4′-ДИБЕНЗИЛИЛЕН)-БИС-(КАРБОНИЛ)]}ДИФТАЛЕВЫЙ АНГИДРИД В КАЧЕСТВЕ МОНОМЕРА ДЛЯ ПОЛУЧЕНИЯ ТЕРМОСТОЙКИХ ПОЛИМЕРОВ | 1991 |
|
RU1804064C |
| Способ получения диангидрида 2,3,7,8-тетракарбокси | 1971 |
|
SU418037A1 |
Изобретение относится к способам получения диангидридов ароматических тетракарбоновых кислот с бифенильными или бинафтильными фрагментами, которые могут быть применены для получения полиимидных материалов, используемых при изготовлении матриц для радиационностойких композиционных материалов, применяемых в различных областях техники. Предлагаемый способ получения диангидридов ароматических тетракарбоновых кислот осуществляют конденсацией по С-С-связям в ароматических фрагментах сложных эфиров ароматических галогендикарбоновых кислот, предварительно полученных реакцией взаимодействия соответствующих диангидридов ароматических галогендикарбоновых кислот и низших одноатомных спиртов, осуществляемой при кипячении в присутствии каталитических количеств серной кислоты с последующим выделением полученных продуктов, причем последующая реакция конденсации эфиров ароматических галогендикарбоновых кислот проводится в атмосфере инертного газа, в амидном органическом растворителе в присутствии каталитической смеси, содержащей безводный галогенид никеля, трифенилфосфин и активированный цинковый порошок, после которой осуществляется последовательные стадии обработки реакционной массы: фильтрация, высаживание водой, кипячение в присутствии щелочного агента, охлаждение, промывка и сушка, где в качестве исходных ангидридов ароматических галогендикарбоновых кислот используются соединения, выбранные из следующей группы: ангидрид 4-хлор-1,8-нафталиндикарбоновой кислоты, ангидрид 4-бром-1,8-нафталиндикарбоновой кислоты, ангидрид 3-иод-1-фталевой кислоты, ангидрид 4-иод-1-фталевой кислоты, которые переводят в соответствующие сложные эфиры кипячением с С1-С4 одноатомным спиртом, взятым в 5-7-кратном избытке по массе от эквимолярного количества в течение 8-16 часов, с последующим упариванием спирта, и полным растворением сухого остатка в толуоле, подщелачиванием раствором гидроокиси натрия при перемешивании, отделением органического слоя, его промывкой, высушиванием, упариванием растворителя и перегонкой остатка в вакууме, после чего раствор полученного сложного эфира ароматической галогендикарбоновой кислоты в сухом амидном органическом растворителе медленно вводится в предварительно полученную каталитическую смесь, имеющую температуру 80-100°С и содержащую 0,03-0,07 моля безводного галогенида никеля(II), 0,2-0,4 моля трифенилфосфина и 2-4 моля активированного цинкового порошка на 1 моль диангидрида ароматической галогендикарбоновой кислот, и образовавшаяся реакционная масса перемешивается при 80-100°С в течение 6-12 часов, после чего органический амидный растворитель упаривается и к оставшейся реакционной массе добавляется низкокипящий хлорсодержащий органический растворитель, и затем полученный раствор при охлаждении и перемешивании приливается к водному раствору соляной кислоты, фильтруется, органический слой отделяется, растворитель упаривается, а остаток растворяется и полученный диангидрид ароматической тетракарбоновой кислоты сушится в вакууме в течение 5-10 часов при 150-200°С. Выход конечных продуктов 21-49%. Получение конечных продуктов (диангидрида 2,2',3,3'-бифенил тетракарбоновой кислоты, диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты, диангидрида 4,4'-бинафтил-1,1',8,8'-тетракарбоновой кислоты, диангидрида 3,3',4,4'-бифенил тетракарбоновой кислоты) подтверждено методами 1Н ЯМР и элементного CHNS-анализа. 5 з.п. ф-лы, 5 пр.
1. Способ получения диангидридов ароматических тетракарбоновых кислот конденсацией по С-С-связям в ароматических фрагментах сложных эфиров ароматических галогендикарбоновых кислот, предварительно полученных реакцией взаимодействия соответствующих диангидридов ароматических галогендикарбоновых кислот и низших одноатомных спиртов, осуществляемой при кипячении в присутствии каталитических количеств серной кислоты с последующим выделением полученных продуктов, при чем последующая реакция конденсации эфиров ароматических галогендикарбоновых кислот проводится в атмосфере инертного газа, в амидном органическом растворителе в присутствии каталитической смеси, содержащей безводный галогенид никеля, трифенилфосфин и активированный цинковый порошок, после которой осуществляется последовательные стадии обработки реакционной массы: фильтрация, высаживание водой, кипячение в присутствии щелочного агента, охлаждение, промывка и сушка, отличающийся тем, что в качестве исходных ангидридов ароматических галогендикарбоновых кислот используются соединения, выбранные из следующей группы: ангидрид 4-хлор-1,8-нафталиндикарбоновой кислоты, ангидрид 4-бром-1,8-нафталиндикарбоновой кислоты, ангидрид 3-иод-1-фталевой кислоты, ангидрид 4-иод-1-фталевой кислоты, которые переводят в соответствующие сложные эфиры кипячением с С1-С4 одноатомным спиртом, взятым в 5-7-кратном избытке по массе от эквимолярного количества в течение 8-16 часов, с последующим упариванием спирта, и полным растворением сухого остатка в толуоле, подщелачиванием раствором гидроокиси натрия при перемешивании, отделением органического слоя, его промывкой, высушиванием, упариванием растворителя и перегонкой остатка в вакууме, после чего раствор полученного сложного эфира ароматической галогендикарбоновой кислоты в сухом амидном органическом растворителе медленно вводится в предварительно полученную каталитическую смесь, имеющую температуру 80-100°С и содержащую 0,03-0,07 моля безводного галогенида никеля(II), 0,2-0,4 моля трифенилфосфина и 2-4 моля активированного цинкового порошка на 1 моль диангидрида ароматической галогендикарбоновой кислот, и образовавшаяся реакционная масса перемешивается при 80-100°С в течение 6-12 часов, после чего органический амидный растворитель упаривается и к оставшейся реакционной массе добавляется низкокипящий хлорсодержащий органический растворитель, и затем полученный раствор при охлаждении и перемешивании приливается к водному раствору соляной кислоты, фильтруется, органический слой отделяется, растворитель упаривается, а остаток растворяется и полученный диангидрид ароматической тетракарбоновой кислоты сушится в вакууме в течение 5-10 часов при 150-200°С.
2. Способ по п. 1, отличающийся тем, что осуществляется с применением в составе каталитической смеси галогенидов никеля(II), выбранных из группы: хлорид никеля(II), бромид никеля(II), иодид никеля(II).
3. Способ по п. 1, отличающийся тем, что на стадии конденсации осуществляется с использованием сухих амидных органических растворителей, выбранных из группы: формамид, ацетамид, N,N-диметилформамид, N,N-диметилацетамид.
4. Способ по п. 1, отличающийся тем, что осуществляется с использованием низкокипящих хлорсодержащих органических растворителей, выбранных из группы: хлористый метилен, хлороформ, тетрахлорметан.
5. Способ по п. 1, отличающийся тем, что осуществляется с использованием С1-С4 одноатомных спиртов, выбранных из группы: метанол, этанол, 1-пропанол, 2-пропанол, 1-бутанол.
6. Способ по п. 1, отличающийся тем, что реакция взаимодействия диангидридов ароматических галогендикарбоновых кислот и низших одноатомных спиртов осуществляется при кипячении в присутствии каталитических количеств серной кислоты, взятых из расчета 0,05 г кислоты на 1 г исходного диангидрида.
| Jian Ping Gao et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Электрический насос | 1923 |
|
SU1627A1 |
| Sun F | |||
| et al | |||
| "Synthesis and characterization of sulfonated polyimides bearing sulfonated aromatic pendant group for DMFC applications" Polymer, 2010, 51, 17, р | |||
| ПРИСПОСОБЛЕНИЕ К СТАНКУ ДЛЯ НАНЕСЕНИЯ ДЕЛЕНИЙ НА ЛИНЕЙКАХ, ШКАЛАХ И Т. П. | 1925 |
|
SU3887A1 |
| ЖАРИНОВА МАРИНА Юрьевна, "СИНТЕЗ И СВОЙСТВА НОВЫХ СУЛЬФИРОВАННЫХ ПОЛИНАФТОИЛЕНИМИДОВ И ПОЛИТРИАЗОЛОВ ДЛЯ ПРОТОНПРОВОДЯЩИХ МЕМБРАН НИЗКОТЕМПЕРАТУРНЫХ ТОПЛИВНЫХ ЭЛЕМЕНТОВ", ДИССЕРТАЦИЯ на соискание ученой степени кандидата химических наук, Москва, 2017 | |||
| Liu L | |||
| et al | |||
| "The effect of the regioisomeric naphthalimide acetylide ligands on the photophysical properties of N,N Pt(II) bisacetylide complexes" Dalton Trans., 2014, 43, р.13434-13444. | |||

