Изобретение относится к области пептидной химии и конкретно касается получения диацетата трипептида H-β-Ala-Pro-DabNHBzl (Syn-Ake), относящегося к биологически активным соединениям и используемого в качестве активного компонента для косметических средств.
Синтетические пептиды широко используются как в фармацевтической промышленности, так и в современной косметологии. В частности, Syn-Ake вызывает стимуляцию омоложения кожи за счет разглаживания морщин и относится к группе пептидов ботулоподобного действия (блокаторы нейромышечной передачи) наряду с такими синтетическими пептидами, как, например, Аргирелин (Аргирелин NP, ацетил гексапептид-3, ацетил гексапептид-8), SNAP-8 (ацетил глутамил гептапептид-1, EEMQRPA) (DERMAQUEST SKIN THERAPY, http://WWW.Dermaquestinc.Ru), Leuphasyl (пентапептид-18), Vialox (пентапептид-3), Inyline (ацетил гексапептид-25).
Особенности синтеза пептидов определенного строения являются следствием бифункциональности исходных аминокислот и, следовательно, возможности образования пептидных связей как между различными, так и между одинаковыми молекулами аминокислот. Для предотвращения этих осложнений необходимо защищать амино- или карбоксильную группу. Защитная группа должна устанавливаться селективно именно по защищаемой группе и удаляться в таких условиях, в которых не будут затрагиваться остальные функциональные группы в составе молекул. Кроме того, желательно, чтобы защита ставилась и снималась с высоким выходом. Например, один из наиболее распространенных способов защиты (блокировки) аминогруппы заключается во взаимодействии аминокислоты с бензилхлорформиатом или третбутоксикарбонильной группой (БОК) («Химический синтез пептидов», г. Киев, Наук. думка, 1992).
В пептидной химии широко используют ступенчатый способ получения пептидов путем наращивания цепи с C-конца исходной аминокислоты с применением активированных производных аминокислот, например пентафторфениловых эфиров (Боданский М., Клаузнер Я.С. Активированные эфиры и стратегия пептидного синтеза. - Химия полипептидов. Под ред. П. Катосояниса. М., «Мир», 1977, с.30-51).
В WO 2006047900, 11.05.2006 (или US 7964630) описаны соединения, в частности пептиды H-Ala-Pro-Arg-Arg-NHBzl, H-β-Ala-Pro-Dab-NHBzl, H-Dap-Pro-Arg-NHBzl и кислотно-аддитивные соли этих соединений, такие как соли, например, уксусной кислоты, трифторуксусной кислоты и др. Данные соединения - пептиды - пригодны для сглаживания мимических и возрастных морщин в коже человека. Данные соединения (пептиды) могут быть получены в соответствии с известными методами в химии пептидов, включая полностью синтез этих соединений, при необходимости отщепления (снятия) защитной группы, необязательное алкилирование свободной аминогруппы или превращение его в функцию гуанидино и/или этерификацию или амидирование свободной карбоксильной группы, и/или преобразование полученного основного соединения в кислотно-аддитиную соль, и/или получение кислотно-аддитивной соли в другую соль.
Так, в случае синтеза H-β-Ala-Pro-DabNHBzl известный способ состоит из 10 стадий (включая стадии постановки защитных групп) и в качестве исходных аминокислот использует L-пролин, L-2,4-диаминомасляную кислоту (DabOH) и β-аланин. Следует отметить, что L-(DabOH) не является природной аминокислотой и имеет достаточно высокую стоимость.
Диацетат H-β-Ala-Pro-DabNHBz (Syn-Ake) - уксуснокислая соль синтетического пептида, действие которого направлено на непосредственное и длительное расслабление мимических морщин. Это действие подобно действию протеина, содержащегося в яде храмовой куфии (Tropidolaemus wagleri), который препятствует передаче нервных импульсов на мышцы. Применение его в косметике позволяет не только разглаживать мелкие мимические морщины, но и предотвращает их дальнейшее появление (Fragrance Journal V34, N10, h.93-98 (2006). DE 202012002309, Anti-aging cosmetic foam, 2012)
Задачей данного изобретения является упрощение процесса получения, исключение реагентов на постановку и снятие защитных групп, снижение себестоимости производства диацетата H-β-Ala-Pro-DabNHBzl (I).
Заявленный способ относится к области пептидной химии, конкретно к способу получения диацетата H-β-Ala-Pro-DabNHBzl, используемого в качестве биологически активного компонента для косметических средств, формулы I:
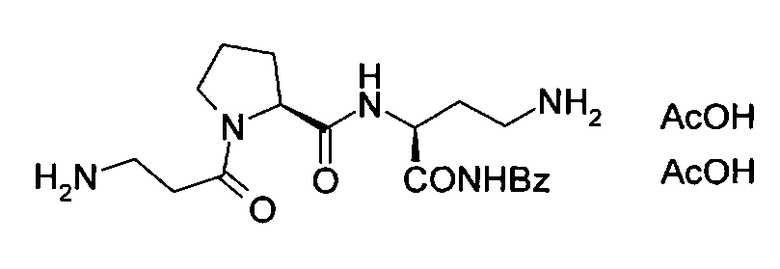
и имеет своей целью разработку простого и удобного способа синтеза целевого соединения. Сущность изобретения заключается в том, что трипептид (I) получают в результате 6 стадийного синтеза без использования стадий постановки и снятия защитных групп и включающей следующие стадии многоступенчатого синтеза пептида:
1. Ацилирование пролина в диоксане β-хлорпропионилхлоридом с использованием избытка пролина в качестве акцептора хлористого водорода дает N-(3-хлорпропионил)пролин (II);
2. Реакция (II) пентафторфенола в этилацетате в присутствии N,N′-дициклогексилкарбодиимида (ДЦК) приводит к петафторфениловому эфиру N-(3-хлорпропионил)пролин (III);
3. Конденсация III с монометиловым эфиром глутаминовой кислоты в диметилформамиде дает N-(β-Хлорпропионил)-Pro-Glu(δ-OMe)OH (IV);
4. Амидирование IV бензиламином с использованием HOBt и ДЦК с образованием N-(β-хлорпропионил)-Pro-Glu(δ-OMe)NHBzl (V);
5. Одновременный аммонолиз и замещение хлора на аминогруппу в V, выполненный в метанольном растворе аммиака, приводит к трипептиду гидрохлориду β-Ala-Pro-Glu(δ-NH2)NHBzl (VI);
6. Перегруппировка по Гофману VI с использованием в качестве реагента диацетата иодбензола дает целевой I.
Используемые выше и ниже сокращения обозначают:
Pro - пролин;
Ala - аланин;
HOBt - 1-гидроксибензотриазол;
ДЦК - N,N′-дициклогексилкарбодиимид;
Bzl - бензил;
Dab - 2,4-диаминомасляная кислота;
AcOH - уксусная кислота;
β-Ala - бета-аланин (3-аминопропионовая кислота).
Итак, решение поставленной задачи достигается 6 стадийным синтезом, не использующим в качестве исходного реагента L-DabOH и не содержащим стадии постановки и снятия защитных групп:
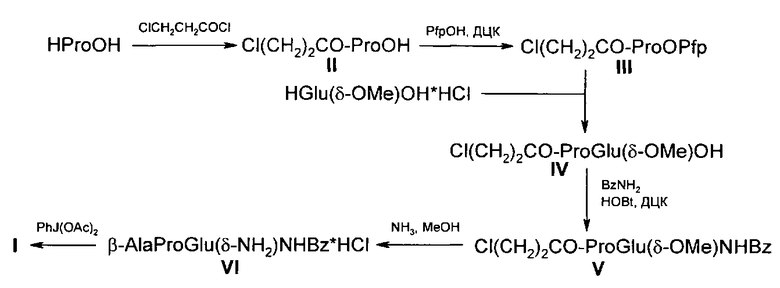
Как видно из нижеприведенных примеров, заявленный в качестве изобретения способ отличается простотой, отсутствием стадий постановки и снятия защитных групп и использует более доступные и дешевые реагенты и исходные продукты.
Разработанный метод позволяет не только упростить процесс получения целевого соединения (уменьшает количество стадий синтеза), но и делает его экономически более эффективным (более доступное и дешевое сырье, отсутствие стадий постановки и снятия защитных групп).
На первой стадии ацилирования L-пролина используют в качестве акцептора выделяющегося хлористого водорода избыток пролина. Процесс проводят при комнатной температуре в диоксане. Выделяющийся гидрохлорид пролина удаляют фильтрованием. Остаток пролина отмывают кислым раствором хлористого натрия (pH 1) и после удаления растворителя в вакууме и промывки остатка серным и петролейным эфиром получают II в виде вязкой прозрачной жидкости (чистота 97.8%).
Пентафторфениловый эфир III получают реакцией II с пентафторфенолом в этилацетате. В качестве конденсирующего агента используют ДНК. Реакцию проводят при комнатной температуре в течение 2 ч. После отделения дициклогексилмочевины фильтрованием, удаления растворителя в вакууме и растирания остатка в петролейном эфире получают кристаллический III с содержанием основного вещества 95.1%.
Взаимодействие III и гидрохлорида монометилового эфира глутаминовой кислоты в диметилформамиде в течение 20 ч при комнатной температуре дает IV. N-Метилморфолин используют для связывания хлористого водорода. Диметилформамид удаляют в вакууме, остаток растворяют этилацетате и отмывают основные примеси лимоннокислым водным раствором хлористого натрия. После отгонки растворителя в вакууме и растирания в системе растворителей хлороформ - диэтиловый эфир 1:1 получают аморфный IV с содержанием основного вещества 94.6%.
Амидирование IV бензиламином проводят в растворе диметилформамида при 5°C с использованием HOBt и ДЦК в течение 18 ч. Увеличение температуры выше 5°C приводит к снижению чистоты конечного продукта за счет побочных реакций: образования бис-бензиламида и нуклеофильного замещения атома хлора. Полученный аморфный V содержит 95.9% основного вещества.
При взаимодействии V с аммиаком в метиловом спирте (20% раствор) происходит одновременный аммонолиз метилового эфира и замена хлора на аминогруппу. Реакцию проводят в герметичном стальном сосуде (для предотвращения испарения аммиака) в течение 70-80 ч. После удаления метилового спирта в вакууме и затирания в тетрагидрофуране остатка получают аморфное вещество с содержанием VI 63%. Дополнительная хроматографическая очистка на силикагеле дает VI с содержанием основного вещества 96.8%.
Целевой I получают перегруппировкой по Гофману VI в смеси диоксан - пиридин с использованием диацетата иодбензола при температуре 5°C. Дробное прибавление диацетата иодбензола позволяет уменьшить побочные процессы и увеличивает выход I. Оптимальное мольное соотношение диацетата иодбензола/VI 4-5/1. Окончательную очистку I и перевод его в диацетатную форму проводят препаративной ВЭЖХ.
Чистота полученного диацетата H-β-Ala-Pro-DabNHBz и его полупродуктов подтверждена ВЭЖХ и ЯМР спектроскопией. Аналитическую ВЭЖХ проводили на хроматографе фирмы Shimadzu. Колонка: Grom-Sil 12J ODS-4HE, 5 µm, 250*4,6 mm. Условия: градиент 20%B (0 мин) 60%B (10 мин) 70%B (15 мин) 70%B (30 мин). A - фосфатный буфер pH 3 (20,4 г KH2PO4 растворяли в 3 л дистиллированной воды и доводили pH до 3 добавлением концентрированной фосфорной кислоты), B - ацетонитрил. Спектры ПМР получены на приборе Bruker AM-360 (рабочая частота 360.13 МГц). Спектры ЯМР 13C получены на приборе Bruker AM-360 (рабочая частота 90.56 МГц). ИК-спектры получены на ИК-фурье спектрометре Nicolet 6700 (Thermo Scientific) в таблетке с KBr (2.5 мг/300 мг KBr). Угол удельного вращения измеряли на автоматическом поляриметре "OTAGO" AP-300. Спектры ESI-MS регестрировали на приборе "Agilent LC/MS 1200" при ионизации пробы электрораспылением в режиме регистрации положительных ионов. Пробы готовили в системе ацетонитрил/вода 1/1, концентрация 2 мг/мл. Условия анализа: поток 1 мл/мин, давление на нибулайзере 20 psi, температура 360°C, скорость потока осушающего газа 9 л/мин, напряжение 3500 B, целевая масса от 100 до 2000.
Описываемый способ иллюстрируется следующими примерами.
Экспериментальная часть
Пример 1. N-(3-Хлорпропионил)пролин (II)
Суспендируют 53 г (0.46 моль) L-пролина в 300 мл безводного диоксана при комнатной температуре. При перемешивании прикапывают 29.5 г (0.18 моль) β-хлорпропионилхлорида (разогрев до 35-45°C). Перемешивают реакционную массу при комнатной температуре 5 ч. Отфильтровывают выпавший осадок гидрохлорида пролина, промывают его на фильтре 50 мл диоксана и объединенные органические фазы отгоняют в вакууме (45°C, 30 мм рт.ст.) до прекращения отгонки. Растворяют остаток в 150 мл этилацетата. Промывают полученный раствор 2 раза по 50 мл насыщенным водным раствором хлорида натрия, подкисленного до pH до 1 концентрированной соляной кислотой. Разбавляют 50 мл гексана, сушат сульфатом натрия и упаривают в вакууме до полной отгонки растворителей (50°C, 3 мм рт.ст.). Вязкий остаток разбавляют 50 мл серного эфира, перемешивают 10 мин и добавляют 50 мл петролейного эфира. Жидкую фазу декантируют, масло промывают 100 мл петролейного эфира и сушат в вакууме до постоянного веса (50°C, 3 мм рт.ст.). Получают 25.5 г (68.9%) бесцветной очень вязкой жидкости. Содержание основного вещества 97.8% (ВЭЖХ). 
Пример 2. Пентафторфениловый эфир N-(3-хлорпропионил)пролина (III)
Растворяют 28.4 г (0.14 моль) II и 28 г (0.15 моль) пентафторфенола в 250 мл этилацетата при комнатной температуре. Охлаждают до 10°C и при этой температуре прикапывают раствор 31 г (0.15 моль) дициклогексилкарбодиимида в 80 мл. Отогревают реакционную массу до комнатной температуры и перемешивают в течение 2 ч. Отфильтровывают выпавшую дициклогексилмочевину и отгоняют этилацетат в вакууме (50°C, 25 мм рт.ст.). Маслообразный остаток растирают 4 раза с 50 мл петролейного эфира до начала кристаллизации. Кристаллический продукт сушат в вакууме до постоянного веса (30°C, 2 мм рт.ст.). Получают 30.5 г (58.6%) бесцветных легкоплавких кристаллов. Содержание основного вещества 95.1% (ВЭЖХ). ТСХ: этилацетат/AcOH 100/1 (алюминиевая пластинка, проявление р-р перманганата калия). Rf(II)=0.20, Rf(III)=0.75. 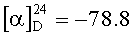
Пример 3. N-(β-Хлорпропионил)-Pro-Glu(δ-OMe)OH (IV)
Растворяют 20 г (0.054 моль) III и 12.8 г (0.065 моль) гидрохлорида метилового эфиром глутаминовой кислоты в 200 мл диметилформамида при комнатной температуре. Охлаждают полученный раствор до 10°C и при этой температуре прикапывают в течение 15 мин 6.5 г (0.064 моль) N-метилморфолина. Перемешивают реакционную массу при комнатной температуре 20 ч. После окончания реакции (ТСХ) отгоняют диметилформамид в вакууме (45°C, 1 мм рт.ст.), маслообразный остаток растворяют в 500 мл этилацетата и полученный раствор промывают 100 и 50 мл насыщенного раствора хлористого натрия с добавкой лимонной кислоты до pH 3. Органическую фазу сушат сульфатом натрия, упаривают в вакууме до полной отгонки растворителя (45°C, 15 мм рт.ст.). Полутвердый остаток растирают 2 раза с 100 мл диэтилового эфира, 2 раза по 40 мл смесью хлороформ-диэтиловый эфир 1/1. Аморфный IV сушат в вакууме до постоянного веса (40-50°C, 2 мм рт.ст.). Получают 15.1 г (80.2%) белого аморфного порошка. Содержание основного вещества 94.6% (ВЭЖХ). ТСХ: BuOH/AcOH/H2O 2/1/1 (алюминиевая пластинка, проявление р-ра перманганата калия). Rf(III)=0.97, Rf(IV)=0.76. 
Пример 4. N-(β-Хлорпропионил)-Pro-Glu(δ-OMe)NHBz (V)
Растворяют 13.8 г (0.040 моль) IV, 6.6 г (0.043) моногидрата N-гидроксибензотриазола и 5.3 г (0.049 моль) бензиламина в 130 мл диметилформамида при комнатной температуре. Охлаждают полученный раствор до 0°C и при этой температуре прикапывают 8.5 г (0.041 моль) дициклогексилкарбодиимида в 20 мл диметилформамида. Перемешивают реакционную массу 18 ч при 5°C. По окончании реакции (ТСХ) отфильтровывают выпавшую дициклогексилмочевину, приливают 500 мл этилацетата. Полученный раствор промывают последовательно 2 раза по 250 мл насыщенным раствором хлористого натрия, 2 раза по 100 мл 1% водного раствора лимонной кислоты, 2 раза по 150 мл 2% водным раствором бикарбоната натрия и 3 раза по 70 мл воды. Органический слой сушат сульфатом натрия и отгоняют растворители в вакууме (50°C, 20 мм рт.ст.). К остатку добавляют 150 мл диэтилового эфира, оставляют полученную суспензию на 12 ч, фильтруют V и сушат в вакууме до постоянного веса (40°C, 2 мм рт.ст.). Получают 12.1 г (69.1%) белого аморфного порошка V. Содержание основного вещества 95.9% (ВЭЖХ). ТСХ: этилацетат/MeOH 5/1 (алюминиевая пластинка, проявление р-ра перманганата калия). Rf(V)=0.06, Rf(VI)=0.71. 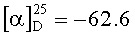
Пример 5. β-Ala-Pro-Glu(δ-NH2)NHBz гидрохлорид (VI)
Растворяют 7.8 г (0.018 моль) V в 300 мл 20% раствора аммиака в метиловом спирте при 10°C. Переливают полученный раствор в герметичный сосуд (стальная пробирка) и выдерживают при комнатной температуре 70-80 ч до окончания реакции (ТСХ). Отгоняют растворитель в вакууме (40°C, 25 мм рт.ст.) до окончания отгонки, к остатку добавляют 20 мл метилового спирта и повторяют отгонку еще раз. Промывают маслообразный остаток 3 раза по 20 мл тетрагидрофурана до затвердевания продукта. Сушат сырой VI в вакууме до постоянного веса (40°C, 2 мм рт.ст.). Получают 6.6 г белого аморфного порошка. Содержание основного вещества 63% (ВЭЖХ). Полученный сырец VI растворяют в 8 мл метилового спирта и хроматографически очищают на силикагеле (колонка: длина 75 см, диаметр 3 см, элюент метиловый спирт/хлороформ 1/1). Целевые фракции (ТСХ) упаривают в вакууме (40°C, 22 мм рт.ст.) и окончательно досушивают при 30-35°C, 1 мм рт.ст. Получают 4.2 г (53%) белого аморфного VI. Содержание основного вещества 96.8% (ВЭЖХ). ТСХ: метанол/уксусная кислота 50/1 (алюминиевая пластинка, проявление нингидрином). Rf(V)=0.87, Rf(VI)=0.23. 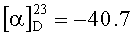
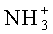
Пример 6. H-β-Ala-Pro-DabNHBz диацетат (I)
Растворяют 8.1 г (0.0183 моль) VI в 150 мл воды при комнатной температуре. Добавляют 100 мл диоксана и 25 мл пиридина. Охлаждают полученный раствор до 5°C и при этой температуре прикапывают 14.1 г (0.0438 моль) диацетата иодбензола в 180 мл диоксана. Перемешивают реакционную массу 3 ч, не снимая охлаждения, и снова прикапывают 10.5 г (0.0327 моль) диацетата иодбензола в 180 мл диоксана. Продолжают перемешивание еще 2 ч (контроль по ТСХ). В реакционную массу присыпают 300 г льда и приливают 500 мл этилацетата. Отделяют водный слой и промывают его сначала 2 раза по 300 мл этилацетата и затем 300 мл смеси этилацетат-бутанол 4:1. Водный слой упаривают (конверсия 81% - ВЭЖХ) и хроматографируют на препаративной ВЭЖХ, после чего переводят продукт в диацетат. Получают 4.92 г (54.3%) маслообразного I. Содержание основного вещества 98.1% (ВЭЖХ). ТСХ: метанол/уксусная кислота 20/1 (алюминиевая пластинка, проявление нингидрином). Rf(VI)=0.35, Rf(I)=0.11. 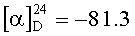
Как видно из вышеприведенного примера, описанный способ отличается простотой, отсутствием стадий постановки и снятия защитных групп и использует более доступные и дешевые реагенты и исходные продукты.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АНАЛОГОВ АДЕНОКОРТИКОТРОПНОГО ГОРМОНА (АКТГ), ПОСЛЕДОВАТЕЛЬНОСТИ (4-10), ОБЛАДАЮЩИХ НЕЙРОТРОПНОЙ АКТИВНОСТЬЮ, И ТЕТРАПЕПТИД ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2315057C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕПТАПЕПТИДА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2303603C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИПЕПТИДОВ | 2006 |
|
RU2303602C2 |
| Способ получения пептида Ac-His-Ala-Glu-Glu-NH | 2021 |
|
RU2767030C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИПЕПТИДА ОБЩЕЙ ФОРМУЛЫ (I) HPyr-His-TrpOH, ИСПОЛЬЗУЕМОГО В КАЧЕСТВЕ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ (1-3 ФРАГМЕНТА) В СИНТЕЗЕ СИНТЕТИЧЕСКИХ АГОНИСТОВ ГОНАДОТРОПИН-РИЛИЗИНГ-ГОРМОНА (LH-RH), МЕТОДОМ ЖИДКОФАЗНОГО ПЕПТИДНОГО СИНТЕЗА БЕЗ ПОСТАНОВКИ И СНЯТИЙ ЗАЩИТНЫХ ГРУПП | 2014 |
|
RU2574392C1 |
| ПРОИЗВОДНЫЕ N-АЦИЛПРОЛИЛДИПЕПТИДОВ | 1993 |
|
RU2119496C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДОДЕКАПЕПТИДА И ТРИПЕПТИД ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2007 |
|
RU2340626C1 |
| ИНГИБИТОРЫ ТРОМБИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2172741C2 |
| N-АЦИЛЬНЫЕ ПРОИЗВОДНЫЕ БИОГЕННЫХ АМИНОВ - МОДУЛЯТОРЫ ПЕРЕКИСНОГО ОКИСЛЕНИЯ ЛИПИДОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2093520C1 |
| ПРОИЗВОДНЫЕ ИНСУЛИНА | 2004 |
|
RU2352581C2 |
Изобретение относится к области пептидной химии и касается получения диацетата трипептида H-β-Ala-Pro-DabNHBzl, относящегося к биологически активному соединению, используемому в косметической промышленности в качестве активного компонента для косметических средств, в частности для стимуляции омоложения кожи, разглаживания морщин, предотвращения появления морщин. Способ основан на 6-стадийном синтезе и не содержит стадий постановки и снятия защитных групп. Способ включает ацилирование пролина β-хлорпропионилхлорида, последующее получение пентафторфенилового эфира N-(3-хлорпропионил)пролина в присутствии N,N′-дициклогексилкарбодиимида. Далее конденсацией полученного пентафторфенилового эфира с монометиловым эфиром глутаминовой кислоты получают N-(β-хлорпропионил)-Pro-Glu(δ-OMe)OH. Осуществляют амидирование бензиламином, одновременный последующий аммонолиз и замещение хлора на аминогруппу. Получают трипептид β-Ala-Pro-Glu(δ-NH2)NHBzl, осуществляют перегруппировку по Гофману с использованием диацетата йодобензола и получают целевой продукт. Способ отличается простотой, эффективностью, является более экономичным, использует более доступные и дешевые реагенты. 6 пр.
Способ получения диацетата трипептида H-β-Ala-Pro-DabNHBzl (I), основанный на 6-стадийном методе пептидного синтеза без использования стадий постановки и снятия защитных групп и включающий следующие стадии:
а) получают сначала N-(3-хлорпропионил)пролин (II) ацилированием пролина (Pro) в диоксане β-хлорпропионилхлоридом;
б) далее получают пентафторфениловый эфир соединения (III) реакцией соединения (II) и пентафторфенола в этилацетате в присутствии N,N′-дициклогексилкарбодиимида;
в) осуществляют конденсацию полученного пентафторфенилового эфира соединения (III) с монометиловым эфиром глутаминовой кислоты в диметилформамиде с получением соединения N-(β-хлорпропионил)-Pro-Glu-(δ-OMe)OH (IV);
г) проводят амидирование соединения (IV) бензиламином в присутствии 1-гидроксибензотриазола и N,N′-дициклогексилкарбодиимида с получением соединения N-(β-хлорпропионил)-Pro-Glu-(δ-OMe)NHBzl (V);
д) проводят одновременный аммонолиз соединения (V) и замещение хлора на аминогруппу с получением трипептида формулы (VI) β-Ala-Pro-Glu(δ-NH2)NHBzl;
е) осуществляют перегруппировку трипептида формулы (VI) по Гофману с использованием диацетата йодобензола и получают целевой продукт диацетат трипептида формулы (I).
| US 20090111731 A1, 30.04.2009 | |||
| WO2009138801 A2, 19.11.2009 | |||
| RU 2009114804 A, 27.11.2010 |

