Изобретение относится к молекулярной биологии, биомедицинской химии и биохимии, конкретно к ингибиторам ключевого фермента системы репарации ДНК поли(АДФ-рибозо)полимеразы-1 человека (ПАРП) на основе производных урацила общей формулы (I), (II), (III) и (IV)
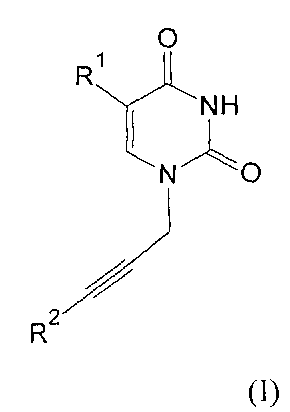
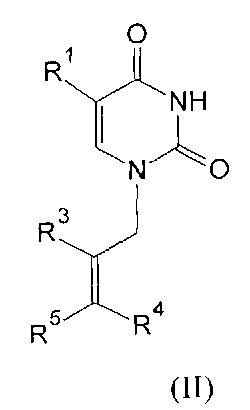
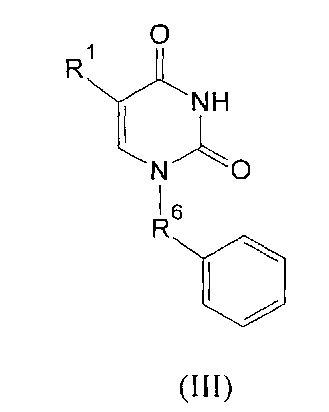
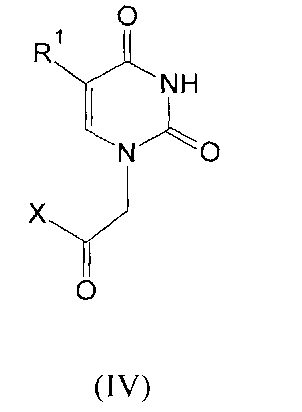
где R1=H, Cl, Br, I, метил, этил, пропил или изопропил; R2, R3, R4, R5=H, метил, этил, пропил или фенил; R6=(CH2)n, где n=1-4; X=OR2 или NR2R3, R2, R3=H, метил, этил, пропил, фенил, 2-гидроксиэтил, 3-гидроксипропил.
Преимущественной областью использования настоящего изобретения является биомедицинская химия, поскольку ингибиторы ПАРП могут быть использованы при лечении таких заболеваний, как инсульт, миокардиальная ишемия, диабет и его осложнения, артриты, колиты, травматическое повреждение ЦНС и ряд других.
Ингибирование ПАРП является также одним из способов повышения активности противоопухолевых агентов (комплексная терапия) за счет ингибирования некроза и активации апоптоза. Ингибиторы ПАРП могут применяться и как самостоятельные агенты терапии некоторых видов опухолей, изначально характеризующихся дефицитом определенных механизмов репарации ДНК. Алкилирующие препараты, вызывающие повреждение ДНК, и ионизирующее излучение применяют в схемах лечения многих форм онкозаболеваний. Системы репарации ДНК противостоят действию агентов, повреждающих ДНК, поэтому терапевтический эффект зависит от эффективности систем репарации ДНК. Селективное воздействие, направленное на ингибирование репарации ДНК, создает предпосылки для эффективной сопровождающей терапии.
К настоящему времени предложен целый ряд ингибиторов ПАРП на основе различных классов химических соединений [Ferraris D.V. Evolution of poly(ADP-ribose)polymerase-1 (PARP-1) inhibitors. From concept to clinic. J. Med. Chem., 53, 4561-4584 (2010)]: никотинамид, 3-аминобензамид и их аналоги [Banasik М., Ueda K. Inhibitors and activators of ADP-ribosylation reactions. Mol. Cell Biochem., 138, 185-197 (1994)]; изохинолиноны и дигидроизохинолиноны (лактамы) [US Patent 5391554; WO 9911645 A1]; бензимидазолы, индолы и родственные соединения [EP 0879820; US Patent 6310082]; изоиндолиноны [Southan G.J., Szabo С. Poly(ADP-ribose)polymeraze inhibitors. Curr. Med. Chem., 10(4), 321-340, (2003)]; фталазиноны и хиназолиноны [Banasik et al. J. Biol. Chem., 267(3), 1569-75 (1992)]; фенантридиноны [WO 9307868 A1].
К недостаткам существующих аналогов настоящего изобретения можно отнести их низкую растворимость в воде, небольшое время жизни в организме, а также низкую специфичность и токсичность для ряда аналогов.
Помимо вышеперечисленных групп ингибиторов известны ингибиторы ПАРП на основе производных нуклеиновых оснований, характеризующиеся высокой растворимостью в воде и специфичностью по отношению к ПАРП. В частности, предложены нуклеозиды [Pivazyan A.D. et al. Inhibition of poly(ADP-ribose)polymerase activity by nucleoside analogs of thymidine. Biochemical Pharmacology, 44, 947-953 (1992)] и дисахаридные нуклеозиды [C.H. Ходырева и др. Средство для ингибирования фермента поли(АДФ-рибозо)полимеразы-1 человека. Патент РФ 2411948 (2009)] с относительно высокой ингибирующей активностью по отношению к ПАРП (IC50 ~50 µM). Эта группа соединений является наиболее близкой по структуре к ингибиторам, предложенным в настоящем изобретении. Следует отметить, однако, что дисахаридные нуклеозиды получают в ходе многостадийного синтеза с использованием относительно малодоступных соединений, что усложняет и удорожает получение ингибиторов этого типа. Репарация ДНК происходит в ядре клетки, активное соединение должно не только проникнуть через клеточную мембрану, но и попасть в ядро. Нуклеозиды и дисахаридные нуклеозиды могут легко превращаться в клетке, например фосфорилироваться, что приводит к уменьшению концентрации ингибитора.
Целью настоящего изобретения является создание эффективных ингибиторов ПАРП на основе производных урацила, которые могут быть получены в результате одностадийного синтеза.
Задача изобретения решена тем, что в качестве ингибиторов ПАРП предложены соединения общей формулы (I-III), которые получают прямым алкилированием замещенных или незамещенных нуклеиновых оснований в диметилформамиде в присутствии 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU). В процессе алкилирования образуется смесь N1-монозамещенных и N1,N3-дизамещенных производных, которые разделяют колоночной хроматографией на силикагеле. Выбранные условия проведения реакции позволяют осуществлять синтез ингибиторов ПАРП в одну стадию в гомогенной среде. Эфиры карбоксиметильных производных урацила (IV) также получают по этой же схеме [Михайлов С.Н. и др. Ациклические аналоги нуклеозидов с амидной связью. Биоорганическая химия, 21(2), 130-132 (1995)]. Эти соединения превращают в амиды реакцией с соответствующими аминами. Активность ингибиторов на основе производных урацила соответствует относительно высокой активности прототипа (IC50 ~50 mM). Значения IC50 для некоторых ингибиторов на основе производных урацила показаны в таблице 1.
Производные урацила (I-IV), рассматриваемые в данном изобретении, впервые предлагаются в качестве ингибиторов ПАРП-1. Некоторые из полученных соединений были ранее описаны в литературе. Соединения формулы (I), где R1=H, F, Cl, Br, I, метил, а R2=H - промежуточные продукты в синтезе антивирусных агентов на основе триазола [Elayadi Н. et al. Preparation of 1,4-disubstituted-1,2,3-triazoloribonucleosides by Na2CuP2O7 catalyzed azide-alkyne 1,3-dipolar cycloaddition. ARKIVOC, 8, 76-89 (2012); Parmenopoulou V. et al. Triazole pyrimidine nucleosides as inhibitors of Ribonuclease A. Synthesis, biochemical and structural evaluation. Bioorganic & Medicinal Chemistry, 20(24), 7184-7193 (2012)]. N1-аллилзамещенные производные 5-фторурацила и тимина (формула (II), R1=F, метил, R3, R4, R5=H) используются в синтезе ряда модифицированных урацилов, обладающих противовирусной и противоопухолевой активностью [Chiacchio U. et al. Enantioselective synthesis of homo-carbocyclic-2′-oxo-3′-aza-nucleosides. Tetrahedron, 62(6), 1171-1181 (2006)]. Известен метод получения урацилкарбоновых кислот, их эфиров и амидов [Baker B.R. et al. Non-classical antimetabolites. XVIII. Simulation of 5′-phosphoribosyl binding. 2. ω-Uracilalkanoic acids related to 2′-deoxyuridylate. Journal of Pharmaceutical Sciences, 54(1), 25-30 (1965)]. Производные N1-бензилурацила (III, R1=H, R6=CH2) [Javaid Z.Z. et al. Pyrimidine nucleobase ligands of orotate phosphribosyltransferase from Toxoplasma gondii. Biochemical Pharmacology, 58(9), 1457-1466 (1999)] используют для ингибирования фосфорибозилтрансферазы, а N1-(карбоксиметил)урацила (IV, R1=OH, метил) [EP 829542 A1], для получения аналогов ДНК.
Структура заявленных соединений подтверждена методами УФ и ЯМР-спектроскопии. ЯМР-спектры регистрировали на приборе Bruker АМХ400 (Германия). Химические сдвиги (δ) измерены относительно внутреннего стандарта - тетраметилсилана (ТМС, δ 0 м.д.) и приведены в миллионных долях (м.д.). Величины констант спин-спинового взаимодействия (J) измерены в герцах (Гц). При описании спектров 1H-ЯМР приняты следующие сокращения: с - синглет, ус - уширенный синглет, д - дублет, т - триплет, м - мультиплет. УФ-спектры регистрировали в воде на приборе Cary 300 UV/VIS (Varian, Австралия). Температуры плавления определяли на приборе Electrotermals (Великобритания). Тонкослойную хроматографию проводили на пластинках Kieselgel 260 F (Merck) с УФ-детекцией (λ=254 нм). Колоночную хроматографию проводили на силикагеле Kieselgel 60 (0.063-0.200 мм, Merck). Очистку растворителей проводили по стандартным методикам.
Пример 1. Синтез 5-йод-1-(пропин-2-ил)урацила
К раствору 0.308 г (2.026 ммоль) 1,8-диазобицикло[5.4.0]ундец-7-ена в 6 мл сухого ДМФА добавляли 0.482 г (2.025 ммоль) 5-йодоурацила и перемешивали в течение 15 мин. К полученной смеси при 20°C в течение 1 мин по каплям добавляли 0.241 г (2.023 ммоль) пропаргилбромида. Реакцию проводили при перемешивании в течение 4,5 ч и температуре 20°C. Смесь оставляли на ночь при комнатной температуре. После окончания реакции растворитель отгоняли в вакууме, остаток разбавляли хлористым метиленом (70 мл), переносили в делительную воронку и промывали водой (40 мл). Водный слой экстрагировали хлористым метиленом (4×15 мл). Органические слои после промывки и экстракции объединяли, промывали водой (4×40 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме досуха. Остаток наносили на колонку с силикагелем (10 г) для дальнейшей хроматографической очистки. Колонку промывали хлористым метиленом, продукты элюировали системами метиленхлорид - этанол - 100:1 и 100:2 (v/v). Фракции, содержащие одинаковые продукты, объединяли, упаривали в вакууме досуха. Выход 5-йодо-1-(пропин-2-ил)урацила 0.244 г (44% в расчете на 5-йодоурацил), Rf 0.44 (метиленхлорид - этанол, 20:1, v/v). 1H ЯМР (400 МГц, ДМСО-D6): 11.73 ус (1H, NH), 8.23 с (1H, 6-H), 4.50 д (2H, 3JCH,CH3=2.6, CH2), 3.41 т (1H, 3JCH,CH2=2.6, CH).
Пример 2. Синтез 5-йод-1-(3-метилбут-2-ен-1-ил)урацила
К раствору 0.451 г (2.963 ммоль) 1,8-диазобицикло[5.4.0]ундец-7-ена в 8 мл сухого ДМФА добавляли 0.690 г (2.890 ммоль) 5-йодоурацила и перемешивали в течение 15 мин. К полученной смеси при 20°C в течение 1 мин по каплям добавляли 0.444 г (2.976 ммоль) изопренилбромида. Реакцию проводили при перемешивании в течение 4,5 ч и температуре 20°C. Смесь оставляли на ночь при комнатной температуре. После окончания реакции растворитель отгоняли в вакууме, остаток разбавляли хлористым метиленом (70 мл), переносили в делительную воронку и промывали водой (35 мл). Водный слой экстрагировали хлористым метиленом (3×30 мл). Органические слои после промывки и экстракции объединяли, промывали водой (4×30 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме досуха. Остаток наносили на колонку с силикагелем (25 г) для хроматографического разделения. Колонку промывали метиленхлоридом, продукты элюировали системами метиленхлорид - этанол, 100:1 и 100:2 (v/v). Фракции, содержащие продукт, объединяли, упаривали в вакууме досуха.
Выход 5-йодо-1-(3-метилбут-2-ен-1-ил)урацила 0.466 г (53% в расчете на 5-йодоурацил), Rf 0.45 (метиленхлорид - этанол, 20:1, v/v). Tпл=146-148°C. УФ (pH 1-7): λmax 291 nm (ε 7849); (pH 13): λmax 281 nm (ε 6046). 1H ЯМР (400 МГц, ДМСО-D6): 11.57 ус (1H, NH), 8.09 с (1H, 6-H), 5.21 м (1H, 3JCH,CH2=6.9, 4JCH3,CH=1.2, CH), 4.26 д (2H, 3JCH2,CH=6.9, CH2), 1.71 д (3H, 4JCH3,CH=1.2, CH3), 1.70 д (3H, 4JCH3,CH=1.2, CH3).
Пример 3. Синтез 5-йод-1-бензилурацила
К раствору 0.152 г (1.0 ммоль) 1,8-диазобицикло[5.4.0]ундец-7-ена (DBU) в 3.0 мл сухого ДМФА добавляли 238 мг (1.0 ммоль) 5-йодурацила и перемешивали при комнатной температуре до образования гомогенного раствора. К реакционной смеси добавляли 0.12 мл (1.0 ммоль) бензилбромида (BnBr). Реакцию проводили при 20°C и перемешивании. Протекание реакции контролировали по ТСХ. После окончания реакции растворитель отгоняли в вакууме, остаток разбавляли хлористым метиленом (30 мл), переносили в делительную воронку и промывали водой (35 мл). Водный слой экстрагировали хлористым метиленом (3×30 мл). Органические слои после промывки и экстракции объединяли, промывали водой (4×30 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме досуха. Остаток наносили на колонку с силикагелем (10 г) для хроматографического разделения. Колонку промывали метиленхлоридом, продукты элюировали системами метиленхлорид - этанол, 100:1, 100:2 и 100:3 (v/v). Фракции, содержащие продукт, объединяли, упаривали в вакууме досуха. Выход продукта - 233 мг (71%). Rf 0.42 (метиленхлорид - этанол, 20:1, v/v). 1H-ЯМР (400 МГц, ДМСО-D6): 11.67 ус (1H, NH), 8.32 с (1H, 6-H), 7.39-7.28 м (5H, Ph), 4.88 с (2H, CH2). 13C-ЯМР (100 МГц, ДМСО-D6): 160.90 (C4), 150.66 (C2), 149.66 (C6), 136.59 (C1 в Ph), 128.58 (C2 и C6 в Ph), 127.67 (C4 в Ph), 127.44 (C3 и C5 в Ph), 68.53 (C5), 50.44 (CH2). Tпл=216-217°C. УФ (pH 1-7): λmax 292 nm (ε 7546); (pH 13): λmax 282 nm (ε 5670).
Пример 4. Синтез 5-йод-1-(3-фенилпропил)урацила
К раствору 0.506 г (3.326 ммоль) 1,8-диазобицикло[5.4.0]ундец-7-ена в 6 мл сухого ДМФА добавляли 0.791 г (3.324 ммоль) 5-йодоурацила и перемешивали в течение 15 мин. К полученной смеси при 20°C в течение 1 мин по каплям добавляли раствор 0.972 г (3.347 ммоль) тозилата 3-фенилпропанола в 3.5 мл сухого ДМФА. Реакцию проводили при перемешивании в течение 4,5 ч и температуре 20°C. Смесь оставляли на ночь при комнатной температуре. После окончания реакции растворитель отгоняли в вакууме, остаток разбавляли хлористым метиленом (70 мл), переносили в делительную воронку и промывали водой (35 мл). Водный слой экстрагировали хлористым метиленом (4×15 мл). Органические слои после промывки и экстракции объединяли, промывали водой (4×40 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме досуха. Продукт подвергали хроматографической очистке на силикагеле (30 г). Колонку промывали метиленхлоридом, продукты элюировали системами метиленхлорид - этанол, 100:1 и 100:2. (v/v). Фракции, содержащие продукт, объединяли, упаривали в вакууме досуха. Выход 5-йодо-1-(3-фенилпропил)урацила 0.760 г (64% в расчете на 5-йодоурацил), Rf 0.46 (метиленхлорид - этанол, 20:1, v/v). 1H ЯМР (400 МГц, ДМСО-d6): 11.53 ус (1H, NH), 8.16 с (1H, 6-Н), 7.30-7.14 м (5H, Ph), 3.71 т (2H, 3JCH2,CH2=7.2, CH2-N), 2.58 т (2H, 3JCH2,CH2=7.8, CH2-Ph), 1.92 м (1H, 2JCHА,CHБ=14.8, 3JCHА,CH2=7.8, 3JCHА,CH2=7.2 Гц, CHА), 1.88 м (1H, 2JCHБ,CHА=14.8, 3JCHБ,CHА=7.8, 3JCHБ,CH2=7.2, CHБ).
Пример 5. Синтез 5-йод-1-(3-фенилэтил)урацила
Вещество получали аналогично примеру 4 с выходом 40%. Rf 0.46 (метиленхлорид - этанол, 20:1, v/v). 1H ЯМР (400 МГц, ДМСО-D6): 11.58 ус (1H, NH), 8.03 с (1H, 6-H), 7.34-7.19 м (5H, Ph), 3.89 т (2H, 3JCH2,CH2=7.6, CH2-N), 2.89 т (2H, 3JCH2,CH2=7.6, CH2-Ph).
Пример 6. Синтез 5-бром-1-(3-метил-бут-2-ен-1-ил)урацила
Вещество получали аналогично примеру 2 с выходом 68%. Rf 0.48 (метиленхлорид - этанол, 20:1, v/v). 1H ЯМР (400 МГц, CDCl3): 11.70 ус (1H, NH), 8.10 с (1H, 6-H), 5.22 м (1H, 3JCH,CH2=6.9, 4JCH3,CH=1.2, CH), 4.27 д (2H, 3JCH2,CH=6.9, CH2), 1.71 д (6H, 4JCH3,CH=1.2, CH3), 1.70 д (6H, 4JCH3,CH=1.2, CH3). Tпл=146-148°C. УФ (pH 1-7): λmax 285 nm (ε 8487); (ph 13): λmax 280 nm (ε 6361).
Пример 7. Синтез 5-бром-1-(2-фенилэтил)урацила
Вещество получали аналогично примеру 4 с выходом 46%. Rf 0.47 (метиленхлорид - этанол, 20:1, v/v). 1Н ЯМР (400 МГц, ДМСО-D6): 11.71 ус (1H, NH), 8.06 с (1H, 6H), 7.34-7.19 м (5H, Ph), 3.90 т (2H, 3JCH2,CH2=7.6, CH2-N), 2.90 т (2H, 3JCH2,CH2=7.6, CH2-Ph).
Пример 8. Синтез 5-бром-1-(3-фенилпропил)урацила
Вещество получали аналогично примеру 4 с выходом 51%. Rf 0.47 (метиленхлорид - этанол, 20:1, v/v). 1Н ЯМР (400 МГц, ДМСО-D6): 11.66 ус (1H, NH), 8.18 с (1H, 6H), 7.31-7.13 м (5H, Ph), 3.72 т (2H, 3JCH2,CH2=7.3, CH2-N), 2.60 т (2H, 3JCH2,CH2=7.8, CH2-Ph), 1.94 м (1H, 2JCHА,CHБ=14.8, 3JCHА,CH2=7.8, 3JCHА,CH2=7.3, CHA), 1.90 м (1H, 2JCHБ,CHА=14.8, 3JCHБ,CH2=7.8, 3JCHБ,CH2=7.3, CHБ).
Пример 9. Синтез 1-(пропин-2-ил)-5-этилурацила
К раствору 0.308 г (2.026 ммоль) 1,8-диазобицикло[5.4.0]ундец-7-ена в 6 мл сухого ДМФА добавляли 0.278 г (1.984 ммоль) 5-этилурацила и перемешивали в течение 15 мин. К полученной смеси при 20°C в течение 1 мин по каплям добавляли 0.238 г (2.005 ммоль) пропаргилбромида и оставляли на ночь при комнатной температуре. После окончания реакции растворитель отгоняли в вакууме, остаток разбавляли хлористым метиленом (60 мл), переносили в делительную воронку и промывали водой (30 мл). Водный слой экстрагировали хлористым метиленом (4×15 мл). Органические слои после промывки и экстракции объединяли, промывали водой (4×30 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме досуха. Остаток наносили на колонку с силикагелем (15 г) для разделения. Колонку промывали метиленхлоридом, продукты элюировали системами метиленхлорид - этанол, 100:1 и 100:2. (v/v). Фракции, содержащие продукт, объединяли, упаривали в вакууме досуха. Выход 1-(пропин-2-ил)-5-этилурацила 0.118 г (33% в расчете на 5-этилурацил), Rf 0.44 (метиленхлорид - этанол, 20:1, v/v). 1Н ЯМР (400 МГц, CDCl3): 9.00 ус (1H, NH), 7.19 ус (1H, 6-H), 4.55 д (2H, 3JCH2,CH=2.5, CH2), 2.46 т (1H, 3JCH,CH2=2.5, CH), 2.39 к (2H, 3JCH2,CH3=7.5, CH2), 1.15 т (3H, 3JCH3,CH2=7.5, CH3). 13C ЯМР (100 МГц, CDCl3): 163.60 (C4), 150.44 (C2), 137.70 (C6), 117.54 (C5), 76.53 (CH≡), 75.24 (CH≡), 36.85 (CH2), 20.21 (CH2), 12.89 (CH3).
Пример 10. Синтез 1-(3-метилбут-2-ен-1-ил)-5-этилурацила
К раствору 0.270 г (1.771 ммоль) 1,8-диазобицикло[5.4.0]ундец-7-ена в 5.2 мл сухого ДМФА добавляли 0.243 г (1.734 ммоль) 5-этилурацила и перемешивали в течение 15 мин. К реакционной смеси при 20°C в течение 1 мин по каплям добавляли 0.258 г (1.734 ммоль) изопренилбромида и оставляли на ночь при комнатной температуре. После окончания реакции растворитель отгоняли в вакууме, остаток разбавляли хлористым метиленом (60 мл), переносили в делительную воронку и промывали водой (30 мл). Водный слой экстрагировали хлористым метиленом (4×15 мл). Органические слои после промывки и экстракции объединяли, промывали водой (4×30 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали в вакууме досуха. Остаток наносили на колонку с силикагелем (15 г) для разделения. Колонку промывали метиленхлоридом, продукты элюировали системами метиленхлорид - этанол, 100:1 и 100:2. (v/v). Фракции, содержащие продукт, объединяли, упаривали в вакууме досуха. Выход продукта 0.133 г (31% в расчете на 5-этилурацил), Rf 0.46 (метиленхлорид - этанол, 20:1, v/v). 1H ЯМР (400 МГц, CDCl3): 8.93 ус (1H, NH), 6.89 д (1H, 4J6-H,CH2=1.1, 6-H), 5.22 м (1H, 3JCH,CH2=7.3, 3JCH,CH3=1.4, CH), 4.31 д (2H, 3JCH2,CH=7.3, CH2), 2.35 дк (1H, 3JCH2,CH3=7.4, 4JCH2,6H=1.1, CH2), 1.78 д (3H, 4JCH3,CH=1.4, CH3), 1.76 ус (3H, CH3), 1.12 т (6H, 4JCH3,CH=7.4, CH3). 13C-ЯМР (100 МГц, CDCl3): 163.89 (C4), 151.03 (C2), 139.27 (C=), 138.96 (C6), 116.64 (C5), 118.10 (CH=), 45.40 (CH2), 25.86 (CH3), 20.19 (CH2), 18.20 (CH3), 13.02 (CH3).
Пример 11. Синтез 5-изопропил-1-(пропин-2-ил)-урацила.
Вещество получали аналогично примеру 9 с выходом 25%. Rf 0.45 (метиленхлорид - этанол, 20:1, v/v). 1H ЯМР (400 МГц, CDCl3): 9.04 ус (1H, NH), 7.15 ус (1H, 6-H), 4.55 д (2H, 3JCH2,CH=2.6, CH2), 2.92 септ (1H, 3JCH,CH3=6.9, CH), 2.46 т (1H, 3JCH2,CH=2.6, CH), 1.17 д (6H, 3JCH3,CH2=6.9, CH3). 13C-ЯМР (100 МГц, CDCl3): 163.27 (C4), 150.32 (C2), 136.82 (C6), 121.94 (C5), 76.54 (C≡), 75.22 (CH≡), 36.94 (CH2), 21.64 (2XCH3), 26.07 (CH).
Пример 12. Синтез 5-изопропил-1-(3-метилбут-2-ен-1-ил)урацила.
Вещество получали аналогично примеру 10 с выходом 31%. Rf 0.47 (метиленхлорид - этанол, 20:1, v/v). 1H ЯМР (400 МГц, CDCl3): 8.80 ус (1H, NH), 6.86 д (1H, 3J6-H,CH=0.9, 6-H), 5.23 м (1H, 3JCH,CH2=7.3, 4JCH,CH3=1.4, CH), 4.31 д (2H, 3JCH2,CH=7.3, CH2), 2.89 м (1H, 3JCH,CH2=7.3, 4JCH,CH3=0.9, CH), 1.77 д (3H, 4JCH3,CH=1.4, CH3), 1.76 ус (3H, CH3), 1.14 д (6Н, 4JCH3,CH=7.3, CH3). 13C ЯМР (100 МГц, CDCl3): 163.43 (C4), 150.80 (C2), 139.25 (C=), 138.01 (C6), 121.03 (C5), 118.11 (CH=), 45.55 (CH2), 25.96 (CH3), 25.85 (CH3), 21.68 (2XCH3), 18.21 (CH=).
Пример 13. Синтез 1-(пропин-2-ил)тимина
К суспензии 1.26 г (10 ммоль) тимина в 16 мл (78 ммоль) гексаметилдисилазана (ГМДС) добавляли каталитическое количество сульфата аммония и кипятили реакционную смесь с обратным водяным холодильником без доступа влаги воздуха до полного растворения гетероциклического основания. Избыток ГМДС удаляли упариванием в вакууме, остаток упаривали с толуолом (2×15 мл) и растворяли в дихлорэтане (ДХЭ). К полученному раствору при 20°C прикапывали 1.1 мл (10 ммоль) 80%-ного раствора пропаргилбромида в толуоле. Реакционную смесь нагревали с обратным водяным холодником до 65°C в течение 8-9 ч без доступа влаги воздуха, после чего проводили нейтрализацию смеси 10%-ным водным раствором бикарбоната натрия (25 мл). Смесь переносили в делительную воронку, вещество экстрагировали из водной фазы хлористым метиленом (4×30 мл). Экстракты объединяли, промывали насыщенным водным NaCl (25 мл), сушили над безводным сульфатом натрия, фильтровали, упаривали в вакууме досуха. Вещество очищали перекристаллизацией из этилового спирта (15 мл). Выход 1-(пропин-2-ил)тимина 808 мг (49%) в виде белых пушистых кристаллов. Rf 0.45 (метиленхлорид - этанол, 20:1, v/v). 1H ЯМР (400 МГц, CDCl3): 8.84 ус (1H, NH3), 7.18 к (1H, 4J6H,5-CH3=1.2, 6H), 4.46 д (2H, 4JCH2,CH=2.6, CH2), 2.39 т (1H, CH), 1.89 д (3H, 4J5-CH3,6H=1.2, 5-CH3).
Пример 14. Синтез 1-бензилтимина
Вещество получали аналогично примеру 3 с выходом 53%. Rf 0.53 (метиленхлорид-этанол, 20:1, v/v). 1Н ЯМР (400 МГц, CDCl3): 8.59 ус (1H, NH3), 7.35-7.17 м (5H, Bn), 6.90 к (1H, 4J6H,5-CH3=1.2, 6H), 4.82 с (2H, CH2 в Bn), 1.81 д (3H, 4J5-CH3,6H=1.2, 5-CH3).
Пример 15. Синтез 1-бензилурацила
Вещество получали аналогично примеру 3 с выходом 54%. Rf 0.50 (метиленхлорид - этанол, 20:1, v/v). 1H ЯМР (400 МГц, CDCl3): 8.73 ус (1H, NH3), 7.34-7.19 м (5H, Bn), 7.08 д (1H, 3J6H,5H=7.9, 6H), 5.62 дд (1H, 3J5H.6H=7.9, 4J5H,N(3)H=1.8, 5H), 4.84 с (2H, CH2, Bn).
Пример 16. Синтез этилового эфира 1-карбоксиметилтимина
Смесь 3.15 г (25 ммоль) тимина, 20 мл гексаметилдисилазана (ГМДС) и 10 мг сульфата аммония кипятили до полного растворения без доступа влаги воздуха, упаривали в вакууме досуха, затем упаривали с сухим 1,2-дихлорэтаном (10 мл). К остатку добавляли 50 мл дихлорэтана (ДХЭ) и 3.0 мл (27 ммоль) бромэтилацетата. Реакционную смесь нагревали 2.5 ч при 80°C без доступа влаги воздуха. После охлаждения до 20°C упаривали в вакууме досуха, к остатку добавляли 100 мл этанола, выпавший осадок отфильтровывали и промывали хлороформом (100 мл). Объединенные фильтраты, содержащие продукт, упаривали досуха; остаток перекристаллизовывали из этанола. Выход 3.9 г (74%). Rƒ 0.41 (дихлорметан:этанол -9:1, v/v). Tпл 170-171°C. 1H-ЯМР (400 МГц, CDCl3): 7.19 к (1H, J6,5=1.2, H-6 Thy), 4.24 с (2H, CH2), 3.98 к (2H, CH2CH3), 1.74 д (3H, CH3 Thy), 1.02 т (3H, CH2CH3).
Пример 17. Синтез 1-(карбоксамидометил)тимина
100 мг (0.472 ммоль) этилового эфира 1-карбоксиметилтимина обрабатывали 5М раствором аммиака в метаноле (47.2 ммоль, 9.4 мл). Реакционную смесь выдерживали при комнатной температуре в течение 20 ч. В процессе протекания аммонолиза продукт реакции выпадал в осадок в виде белых кристаллов. Реакционную смесь упаривали под вакуумом, соупаривали с метанолом (2×5 мл) и сушили над P2O5 в течение 2 ч. Выход 70 мг (81%). Rƒ 0.08 (дихлорметан:этанол - 9:1 v/v). 1H-ЯМР (400 МГц, ДМСО-D6): 10.67 ус (NH3 Thy), 7.55 уш. с. (1H, C(O)NH2), 7.40 к (1H, J6,5=1.1, H-6 Thy), 7.16 уш. с. (1H, C(O)NH2), 4.23 с (2H, CH2), 1.74 д (3H, CH3-Thy).
Пример 18. Синтез 1-(гидроксипропиламинокарбонилметил)Тимина
Смесь 412 мг (2 ммоль) этилового эфира 1-карбоксиметилтимина и 0.76 мл (10 ммоль) 3-аминопропанола в 5 мл этилового спирта кипятили в течение 5 ч. Осадок отфильтровывали, промывали этанолом и сушили над P2O5. Выход - 450 мг (93%). Rƒ 0.66 (хлороформ:этанол - 8:2, v/v). Тпл 244-246°C. 1H-ЯМР (400 МГц, ДМСО-D6): 10.29 ус (1H, NH3 Thy), 7.92 т (1H, JCH-NH=5.0 Гц, C(O)NH), 7.39 к (1H, J6,5=1.2 Гц, H-6 Thy), 4.28 с (2H, CH2), 3.35-2.99 м (4H, C(O)NHCH2CH2CH2OH), 1.76 д (3H, CH3-Thy), 1.63 м (2H, C(O)NHCH2CH2CH2OH). УФ (pH 1-7): λmax 272 nm (ε 9580); (pH 13): λmax 272 nm (ε 7960).
Пример 19. Определение ингибирующей способности производных нуклеиновых оснований в отношении ПАРП с использованием спектрофлуориметрической детекции остаточного НАД+.
Предварительно готовили: 2M раствор КОН в дистиллированной воде; 20% раствор ацетофенона в этиловом спирте; 88% раствор муравьиной кислоты в дистиллированной воде; буфер для проведения ферментативной реакции (ПАРП-буфер): 50 мМ Трис (pH 8.0), 2 мМ MgCl2 в дистиллированной воде (milli-Q); 1,25 мкМ раствор НАД+ в ПАРП-буфере; растворы ингибиторов ПАРП в диметилсульфоксиде в концентрации 5 мМ и разбавляли их в 10 раз ПАРП-буфером.
Далее определяли активность коммерческого препарата ПАРП-1 и измеряли значения IC50. К 40 мкл раствора НАД+ в ПАРП-буфере добавляли 20 мкл активированной ДНК, 20 мкл раствора ингибитора, 1 мкл ПАРП-1 в 19 мкл ПАРП-буфера и инкубировали при 20°C в течение 30 мин. После инкубации добавляли по 40 мкл растворов КОН и ацетофенона, встряхивали и выдерживали 10 мин при 4°C. Затем добавляли 180 мкл раствора муравьиной кислоты, перемешивали на вортексе и выдерживали 10 мин при температуре 110°C. Разбавляли полученную смесь до 1500 мкл ПАРП-буфером и измеряли флуоресценцию раствора при 440 нм.
Флуоресценция раствора в отсутствии ингибиторов ПАРП соответствует минимальному содержанию остаточного НАД+ и максимальной активности фермента.
Флуоресценция раствора, не содержавшего фермента, ингибитора и активированной ДНК, соответствует максимальному содержанию НАД+ и нулевой активности фермента.
Для целей калибровки измеряли флуоресценцию растворов с содержанием НАД+, в 2, 10 и 100 раз меньшим, чем указано выше.
Определение остаточной активности фермента и значений IC50 проводили по данным трех независимых экспериментов.
Для определения значений IC50 использовали растворы ингибитора с различной степенью разбавления.
| название | год | авторы | номер документа |
|---|---|---|---|
| МИМЕТИКИ ПОЛИ (ADP-РИБОЗЫ) И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2559873C2 |
| Средство для ингибирования фермента тирозил-ДНК-фосфодиэстеразы 1 человека на основе производных пентафуранозилнуклеозидов | 2019 |
|
RU2748103C1 |
| АНТИАНДРОГЕННЫЕ СОЕДИНЕНИЯ, ПРЕДШЕСТВЕННИК АНТИАНДРОГЕННОГО СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1993 |
|
RU2141966C1 |
| ИНГИБИТОРЫ ТРОМБИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ЗАЩИЩЕННОЕ ПРОИЗВОДНОЕ | 1996 |
|
RU2176645C2 |
| 1,3-ОКСАТИОЛАН, ЕГО ГЕОМЕТРИЧЕСКИЕ И ОПТИЧЕСКИЕ ИЗОМЕРЫ, СМЕСИ ЭТИХ ИЗОМЕРОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АНТИВИРУСНУЮ АКТИВНОСТЬ | 1990 |
|
RU2092485C1 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2525392C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2466729C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АНТАГОНИСТЫ РЕЦЕПТОРА ВИТРОНЕКТИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ИХ СОДЕРЖАЩИЕ | 2003 |
|
RU2412185C2 |
| СУЛЬФОНАМИДНЫЕ ПЕРИ-ЗАМЕЩЕННЫЕ БИЦИКЛЫ ДЛЯ ЛЕЧЕНИЯ ОККЛЮЗИОННОГО ПОРАЖЕНИЯ АРТЕРИЙ | 2005 |
|
RU2403240C2 |
| НОВЫЕ ГЕМ-ДИФТОРИРОВАННЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2369612C2 |
Изобретение относится к новым ингибиторам поли(АДФ-рибозо)полимеразы-1 человека на основе производных урацила общей формулы (I), (II), (III) и (IV). Ингибиторы поли(АDР-рибозо)полимераз-ферментов участвуют в репарации ДНК. В общей формуле (I), (II), (III) и (IV)
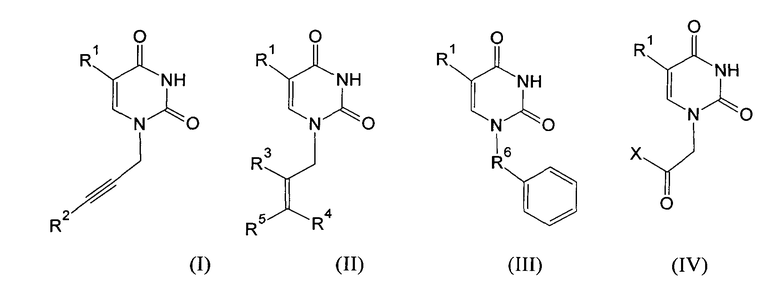
R1=Н, Cl, Br, I, метил, этил, пропил или изопропил; R2, R3, R4, R5=Н, метил, этил, пропил или фенил; R6=(СН2)n, где n=1-4; X=OR2 или NR2R3, R2, R3=Н, метил, этил, пропил, фенил, 3-гидроксипропил. 1 табл., 19 пр.
Ингибиторы поли(АДФ-рибозо)полимеразы-1 человека на основе производных урацила общей формулы (I), (II), (III) и (IV)
где R1=Н, Cl, Br, I, метил, этил, пропил или изопропил; R2, R3, R4, R5=Н, метил, этил, пропил или фенил; R6=(СН2)n, где n=1-4; X=OR2 или NR2R3, R2, R3=Н, метил, этил, пропил, фенил, 3-гидроксипропил.
| PARMENOPOULOU,VANESSA et al.Triazole pyrimidine nucleosides as inhibitors of Ribonuclease A.Synthesis ,biochemical and structural evaluation, Bioorganic & Medicinal Chemistry,20(24),7184-7193 | |||
| ELAYADI, HANANE et al., Preparation of 1,4-disubstituted 1,2,3-triazolo ribonucleosides by Na2CuP2O7 catalyzed azide-alkyne 1,3-dipolar cycloaddition, |

