Изобретение относится к молекулярной биологии, медицинской химии, биохимии и биотехнологии, конкретно к средствам регуляции метаболизма поли(ADP-рибозы) (ПАР), представляющим собой полимерные производные дисахаридных нуклеозидов и их аналогов, включая их линейные и разветвленные сополимеры с нуклеозидами и их производными.
Дисахаридным нуклеозидом называют нуклеозид с присоединенным дополнительным углеводным остатком. Дополнительный остаток соединен с пентафуранозным циклом нуклеозида О-гликозидной связью. Аналоги дисахаридных нуклеозидов, представленные в настоящем изобретении, представляют собой нуклеозиды, модифицированные по 2′, 3′ или 5′ положению О-гидроксиалкильными, О-гидроксиалкилоксиметильными, N-гидроксиалкилкарбоксамидными, О-гидроксиалкил(тио)фосфорильными или O-полиалкиленгликоль(тио)фосфорильными фрагментами.
Поли-ADP-рибозилирование - посттрансляционная модификация белков в эукариотических клетках. Процесс катализируется поли(ADP-рибозо)полимеразами (ПАРП). Эти ферменты осуществляют превращение никотинамидадениндинуклеотида (NAD+) в поли(ADP-рибозу) (ПАР) с высвобождением никотинамида.
В обзорах (D.D′ Amours, et al. Biochem J. 342, 249-268 1999; P.O.Hassa, et al., Microbiol. Mol. Biol. Rev. 70, 789-829, 2006) рассмотрена многообразная биологическая роль этого полимера. К настоящему времени механизм реакции поли(ADP-рибозил)ирования детально не изучен. Активный центр фермента достаточно протяженный, в нем связывается субстрат NAD+ и ′′праймер′′ поли(ADP-рибоза), присоединенный к белку. Ключевой стадией является образование О-гликозидной связи, с образованием никотинамида. Многоточечная фиксация NAD+ по остаткам никотинамида, пирофосфатного остатка и аденозина ориентирует 2′-O-гидроксильную группу для присоединения к С′-1 атому углерода с высвобождением никотинамида (Фиг. 1).
Известно, что ПАР вовлечен в процессы модуляции структуры хроматина, репликации, транскрипции, репарации ДНК и дифференцировки клеток. Детальное изучение внутриклеточных функций поли(ADP-рибозы) продолжается и в настоящее время. Молекулы полимера, как выделенные из природных источников, так и синтезированные in vitro с использованием поли(ADP-рибозо)полимеразы, могут содержать более 200-300 мономерных звеньев. Линейные участки длиной 20-50 звеньев чередуются с разветвленными фрагментами, которые, по-видимому, стабилизируют конформацию поли(ADP-рибозы). Структура поли(АDР-рибозы) была установлена с помощью ЯМР-спектроскопии и ферментативного гидролиза фосфодиэстеразой змеиного яда и щелочной фосфатазой, который приводил к дифосфату α-D-рибофуранозил-(1′′→2′)-аденозина и соответствующему дисахаридному нуклеозиду α-RibAdo, структуру которых установили с помощью ЯМР-спектроскопии (A.M. Ferro, N.J.Oppenheimer Proc. Natl. Acad. Sci. USA. 75, 809-813, 1978). Структура трисахаридного нуклеозида, расположенного в месте разветвления, также подтверждена физико-химическими методами (T. Sugimura, M. Miwa Mol. Cell. Biochem. 138, 5-12, 1994).
В процессе деградации поли(ADP-рибозы) участвуют ADP-рибозилпротеинлиаза и поли(ADP-рибозо)-гликогидролаза (ПАРГ). ADP-рибозилпротеинлиаза ответственна за гидролиз связи между белком и ближайшим остатком ADP-рибозы. ПАРГ, обладающая экзо- и эндогликозидазной активностью, расщепляет связи между аденозином и рибофуранозным остатком с образованием ADP-рибозы (D′Amours D., et al Biochem J. 342, 249-268, 1999), поэтому в клетке присутствуют ковалентно связанный с белком ПАР и свободный полимер. Недавно было показано, что свободный ПАР, попадая из ядра в цитоплазму, является токсичным и может вызывать смерть клеток (Y. Wang, V.L. Dawson, Т.М. Dawson. Exp Neurol. 218, 193-202, 2009).
Поли(ADP-рибозил)ирование белков катализируется несколькими полимеразами, которые постоянно и в значительном количестве имеются в клетке. Все они используют в качестве субстрата NAD+ и переносят ADP-рибозу на остатки глутамата и лизина в составе белков. В клетках эукариот найдены ПАРП-2, ПАРП-3, а также танкиразы 1 и 2 и др. (R. Krishnakumar and W.L. Kraus. Molecular Cell 39, 6-24, 2010). Из ферментов, осуществляющих поли(ADP-рибозил)ирование, ПАРП-1 является основным, и именно он ответственен за синтез 90% поли(ADP-рибозы) в клетке. Этот фермент специфично узнает разрывы в ДНК, активируется и осуществляет синтез ПАР, ковалентно присоединенной к некоторым белкам, причем главным акцептором является сам фермент. Поли(ADP-рибозил)ирование белков рассматривается как главный механизм, обеспечивающий формирование внуктриклеточного сигнала о повреждении ДНК и модулирование функций белков в ответ на генотоксические воздействия. Чрезмерная активация ПАРП при массированных разрывах ДНК, сильно снижая содержание внутриклеточного NAD+, ведет к подавлению гликолиза и митохондриального дыхания и вызывает гибель клетки по варианту некроза. (М.В. Суханов, О.И. Лаврик, С.Н. Ходырева, Мол. биол. 38, 834-847, 2004).
До настоящего времени не разработано химических методов синтеза поли(ADP-рибозы) (Е.В. Ефимцева, И.В. Куликова, С.Н. Михайлов, Молекулярная биология, 43, 327-338, 2009) и только недавно синтезирован ее структурный элемент 2′-O-α-D-рибофуранозиладенозин (S.N. Mikhailov et al Tetrahedron, 64, 2871-2876, 2008). На основе дисахаридных нуклеозидов могут быть созданы новые эффективные регуляторы биосинтеза ПАР.
Возможны несколько путей регуляции метаболизма поли(ADP-рибозы), связанных с подавлением (ингибирование ПАРП, активация ПАРГ) или, наоборот, с активацией ее биосинтеза (активация ПАРП, ингибирование ПАРГ). В последние годы ведутся активные поиски ингибиторов поли(ADP-рибозо)полимераз (ПАРП), которые рассматриваются как перспективные ферменты-мишени для создания лекарственных препаратов при лечении инсульта, ишемии, диабета, артритов, колитов и других воспалительных заболеваний. Ингибиторы ПАРП также могут использоваться как потенциальные противораковые и противовирусные агенты (P. Jagtap, C. Szabo, Nature Rev. Drug Discovery, 4, 421-440, 2005). Основное внимание исследователей сосредоточено на аналогах никотинамида. Было показано, что 3-аминобензамид ингибирует ПАРП в концентрациях 10-6 М. Дальнейшие исследования привели к обнаружению мощных ингибиторов гетероциклической природы с наномолярными константами ингибирования. Ряд перспективных соединений проходят клинические испытания: в комбинации с алкилирующими агентами в противоопухолевой терапии, а также для лечения инфаркта (D.V. Ferraris. J. Med. Chem. 53, 4561-4584, 2010). Применение алкилирующих агентов в химиотерапии онкологических заболеваний приводит к повреждению ДНК как в опухолевых, так и нормальных клетках, при этом активируется система репарации ДНК. Использование ингибиторов ПАРП в комплексной химиотерапии позволит снизить концентрацию алкилирующих агентов и, следовательно, понизить общую токсичность лечения. Также проводится поиск ингибиторов поли(ADP-рибозо)-гликогидролазы (ПАРГ), фермента ответственного за гидролиз ПАР в клетке и определяющего его уровень (D.W. Koh, et al. J. Med. Chem. 46, 4322-4332, 2003).
В настоящее время клинические испытания проходят семь гетероциклических ингибиторов ПАРП, которые являются аналогами никотинамида (D.V. Ferraris. J. Med. Chem. 53, 4561-4584, 2010). К недостаткам существующих ингибиторов ПАРП-1 в первую очередь следует отнести плохую растворимость гетероциклических соединений в воде, а также небольшое время жизни их в организме.
В ряду нуклеозидов ингибиторами ПАРП являются 2′-дезоксинуклеозиды (A.D. Pivazyan et al., Biochem. Pharm., 44, 947-953, 1992; Preiss et al., FEBS Lett., 19, 244-246, 1971) и дисахаридные нуклеозиды (С.Η. Ходырева, О.И. Лаврик, А.Л. Захаренко, С.Н. Михайлов, И.В. Куликова, Е.В. Ефимцева. Средство для ингибирования фермента поли(АДФ-рибозо)полимеразы 1 человека. Патент РФ 2411948). Природные 2′-дезоксинуклеозиды являются структурными элементами нуклеиновых кислот и участвуют в многочисленных процессах в клетке, что исключает специфичность их воздействия на определенный фермент. Дисахаридные нуклеозиды являются более мощными ингибиторами ПАРП-1 с IC50 0.20 - 0.05 mM, однако очевидно, что и тот уровень ингибирования не является достаточным для создания на их основе лекарственных препаратов.
Второй путь регуляции метаболизма ПАР - это активация ее биосинтеза. В ядре клетки фермент ПАРП активируется, связываясь с повреденной ДНК. Связывание поли(ADP-рибозы) с доменом автомодификации ПАРП (AMD-PARP) обеспечивает регуляцию каталитической активности фермента (R. Krishnakumar et al. Molecular Cell., 39, 8-24, 2010). Одноцепочечные олигодезоксинуклеотиды обладают разнообразной биологической активностью, не связанной с образованием комплементарных комплексов с нуклеиновыми кислотами (М. Markosian and R.Μ. Hyde, Antiviral Chem. Chemotherapy 16, 91-102, 1995). Одноцепочечные гуанозин-богатые олигодезоксинуклеотиды вызывают гибель клеток, по механизму активации каспаз (US Patent Application 2009/0312399), обладают выраженной анти-ВИЧ активностью (US Patent 6323185, US Patent 6355785). Олигонуклеотиды, содержащие тиофосфатные группы, препятствуют проникновению вирусов через клеточную мембрану (D.I. Bernstein et al, Antimicrob.Agents Chemotherapy, 52, 2727-2733, 2008; Т. Matsumara et al, Gastroenterology, 137, 673-681, 2009). Описаны полимеры, в которых 1,2-диоксипропильные производные нуклеиновых оснований связаны фосфодиэфирными связями (US Patent 6448373). Однако в этих соединениях формальные расстояния между мономерными остатками существенно меньше, чем в ПАР.
Задачей изобретения является получение миметиков ПАР, обладающих следующими отличительными свойствами:
1. В модифицированных ПАР сохраняются все основные функциональные группы и расстояния между ними, что необходимо для обеспечения потенциальных взаимодействий с белками; согласно расчетным данным поли(ADP-рибоза) и ее миметики обладают значительной конформационной подвижностью и формальные расстояния между С1′-С1′ в двух соседних нуклеозидных остатках составляет 12-14 Å.
2. Перспективным является получение миметиков ПАР, в которых пирофосфатная группа заменена на фосфатный остаток. В этом случае можно использовать модифицированную технологию автоматического олигонуклеотидного синтеза с использованием фосфорамидитных производных. Эта технология позволяет включать коммерчески доступные фосфорамидитные производные нуклеозидов, а также остатки этиленгликоля, пропан- и бутандиолов, гидрофобные и флуоресцентные группы для увеличения стабильности и эффективной доставки аналогов ПАР в клетку. При этом один или несколько модифицированных остатков могут быть расположены в заданном положении ОН цепи.
3. Защитные группы, используемые при синтезе дисахаридных нуклеозидов и их производных, должны сочетаться с группами, применяемыми для автоматического синтеза, что существенно сократит время получения миметиков ПАР; селективное блокирование пяти гидроксильных групп (две первичные и три вторичные группы) в 2′-O-α-D-рибофуранозиладенозине и его аналогах, является важным этапом получения фосфорамидитных производных.
4. Для увеличения стабильности миметиков ПАР к ПАРГ предлагается изменить конфигурацию О-гликозидной связи в остатках 2′-O-α-D-рибофуранозиладенозина или использовать его производные, такие как 2′-O-пентафуранозиладенозин и 2′-O-гидроксиалкоксиметилнуклеозиды. Также возможно получение различных сополимеров на основе этих соединений.
Миметики ПАР, связываясь с ПАРП и активируя его, могут замещать ПАР в различных клеточных процессах (модуляция структуры хроматина и репарция поврежденной ДНК, транскрипция, дифференцировка клеток и др.) и представляют заметный интерес как прототипы новых препаратов для молекулярной медицины, а также в качестве новых объектов при исследованиях в молекулярной биологии, медицинской химии и биохимии. Важно отметить, что к настоящему времени структурные миметики ПАР не описаны в литературе и не используются в промышленности.
Задача получения миметиков ПАР с указанными характеристиками решена тем, что в качестве заявляемых соединений предлагаются линейные и (или) разветвленные полимеры общей формулы
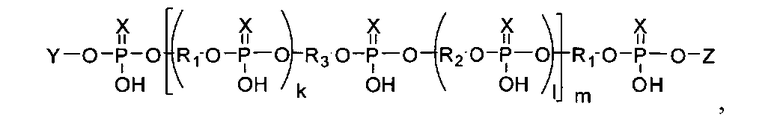
где в качестве концевых фрагментов Υ, Ζ могут быть использованы рибо- и 2′-дезоксирибонуклеозиды, присоединенные к полимеру фосфодиэфирной связью (Υ: дезоксиаденозин-5′-ил, дезоксигуанозин-5′-ил, дезоксицитидин-5′-ил, дезокситимидин-5′-ил; Ζ: аденозин-3′-ил, гуанозин-3′-ил, цитидин-3′-ил, уридин-3′-ил), аминопроизводное разветвленного алифатического соединения (С7-6-амино-(2-гидроксиметил)-гексан-1-ил), производные трифенилметана (Tr - трифенилметил, MMTr - 4-метокситрифенилметил, DMTr - 4,4′-диметокситрифенилметил) флуоресцентные красители трифенилметанового ряда (флуоресцеин, эозин, родамин), связанные с миметиком сложноэфирной связью, либо амидной связью с линкером С7, присоединенным к миметику. Общая длина главной цепи полимера (k+1)*m может варьироваться от 1 до 100, а фосфодиэфирные группы (X=О) могут сочетаться или быть полностью заменены на тиофосфорильные (X=S). В качестве мономерных звеньев R1 и R2 могут быть использованы дисахаридные нуклеозиды 2′-O-β-D-рибофуранозиладенозин или 2′-О-β-D-рибофуранозилгуанозин или 2′-O-β-D-рибофуранозилцитидин или 2′-O-β-D-рибофуранозилуридин или 2′-O-α-D-арабинофуранозиладенозин или 2′-O-α-D-арабинофуранозилгуанозин или 2′-O-α-D-арабинофуранозилцитидин или 2′-O-α-D-арабинофуранозилуридин или 2′-O-α-L-арабинофуранозиладенозин или 2′-O-α-L-арабинофуранозилгуанозин или 2′-O-α-L-арабинофуранозилцитидин или 2′-O-α-L-арабинофуранозилуридин или 2′-O-α-D-рибофуранозиладенозин или 2′-O-α-D-рибофуранозилгуанозин или 2′-O-α-D-рибофуранозилцитидин или 2′-O-α-D-рибофуранозилуридин или 2′-O-β-D-глюкопиранозиладенозин или 2′-O-β-D-глюкопиранозилгуанозин или 2′-O-β-D-глюкопиранозилцитидин или 2′-O-β-D-глюкопиранозилуридин, соединенные между собой 5′-5′′-фосфодиэфирными связями, или их аналоги общей формулы:
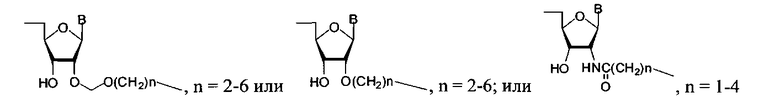
где В=Ade (аденин-9-ил); В=Ura (урацил-1-ил); В=Cyt (цитозин-1-ил); В=Gua (гуанин-9-ил).
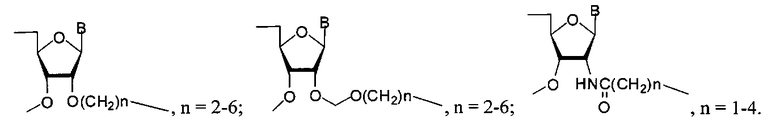
Также в качестве мономерных звеньев R1 и R2 могут быть использованы фрагменты общей формулы -((CH2)nO)m-(P=X(OH))O-N-, где n=1-6, m=1-6, а N = остаток нуклеозида, или общей формулы
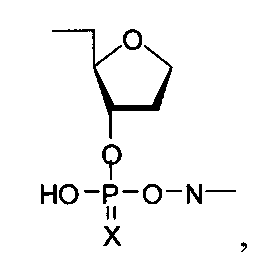 где N = остаток нуклеозида.
где N = остаток нуклеозида.
В главную цепь полимеров - миметиков ПАР могут быть введены разветвители R3 общей формулы
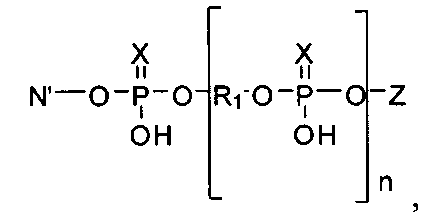 где N′ остаток дисахаридного нуклеозида - 2′-O-β-D-рибофуранозиладенозина или 2′-O-β-D-рибофуранозилгуанозина или 2′-O-β-D-рибофуранозилцитидина или 2′-O-β-D-рибофуранозилуридина, соединенного с основной цепью миметика фосфодиэфирными связями по 3' и 5′ положениям аденозинового фрагмента, и с боковой цепью по 5′′-гидроксильной группе β-D-рибофуранозного остатка; или
где N′ остаток дисахаридного нуклеозида - 2′-O-β-D-рибофуранозиладенозина или 2′-O-β-D-рибофуранозилгуанозина или 2′-O-β-D-рибофуранозилцитидина или 2′-O-β-D-рибофуранозилуридина, соединенного с основной цепью миметика фосфодиэфирными связями по 3' и 5′ положениям аденозинового фрагмента, и с боковой цепью по 5′′-гидроксильной группе β-D-рибофуранозного остатка; или
(нуклеозидные звенья соединены между собой 3′-5′-фосфодиэфирной связью, боковая цепь миметика присоединена к основной цепи через ациклический фрагмент - (СН2)n) Представленные мономерные звенья вводятся в состав полимера в виде фосфорамидитных производных.
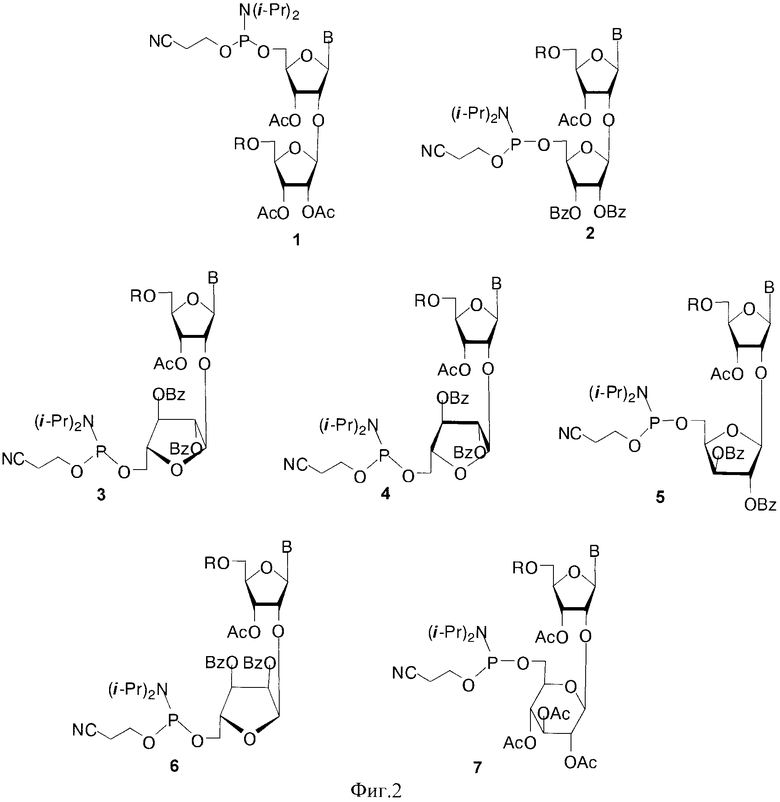
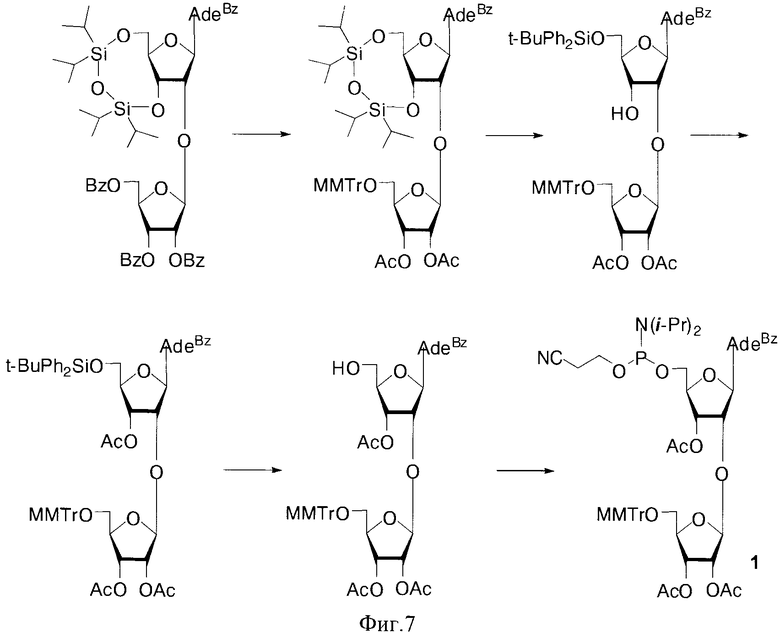
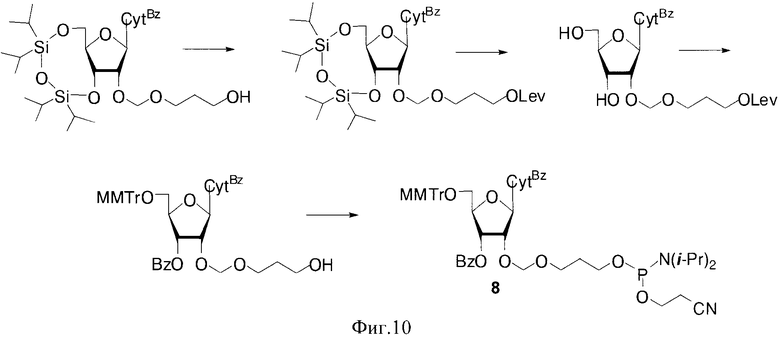
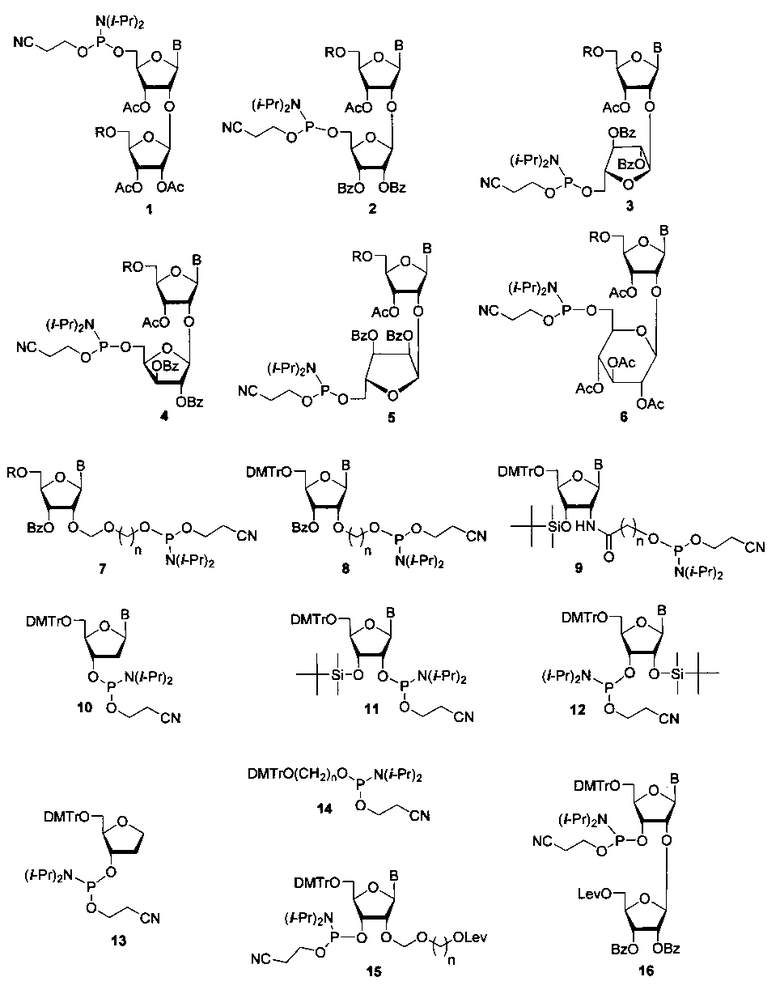
Для получения полимеров используются фосфорамидитные синтоны с монометокситритильной (MMTr) или диметокситритильной (DMTr) защитными группами. Соединения 1-6, где В=AdeBz (Н6-бензоиладенин-9-ил); В=Ura (урацил-1-ил); В=CytBz (N4-бензоилцитозин-1-ил); В=GuaiBu (N2-изобутироилгуанин-9-ил). R=MMTr или DMTr, были получены исходя из дисахаридных нуклеозидов (Фиг. 2).
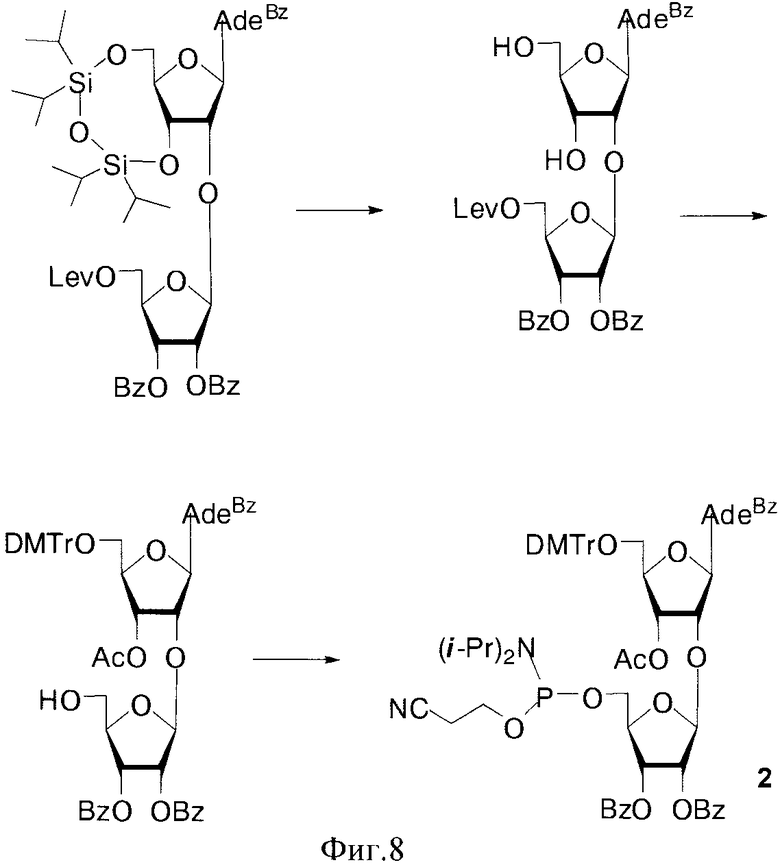
В случае соединения 1 монометокситритильная группа блокирует 5′′-гидроксильную группу дополнительного 2′-O-β-D-рибофуранозного остатка. Также были получены изомерные фосфорамидитные производные 2, в которых ди(моно)метокситритильная группа блокирует 5′-ОН остатка аденозина. В этом случае гликозилирование осуществлялось 1,2,3-три-O-бензоил-5-O-левулинил-O-рибофуранозой (S.N. Mikhailov et al. Chemistry & Biodiversity, 2, 1153-1163, 2005), что позволило существенно сократить количество стадий и увеличить общий выход продуктов 2. Для синтеза полимеров были использованы фосфорамидитные производные с монометокситритильной и диметокситритильной группами, для защиты экзоциклических групп гетероциклических оснований использовали ацильные группы: бензоильную (ΒZ) и изобутироильную (iBu) группы.
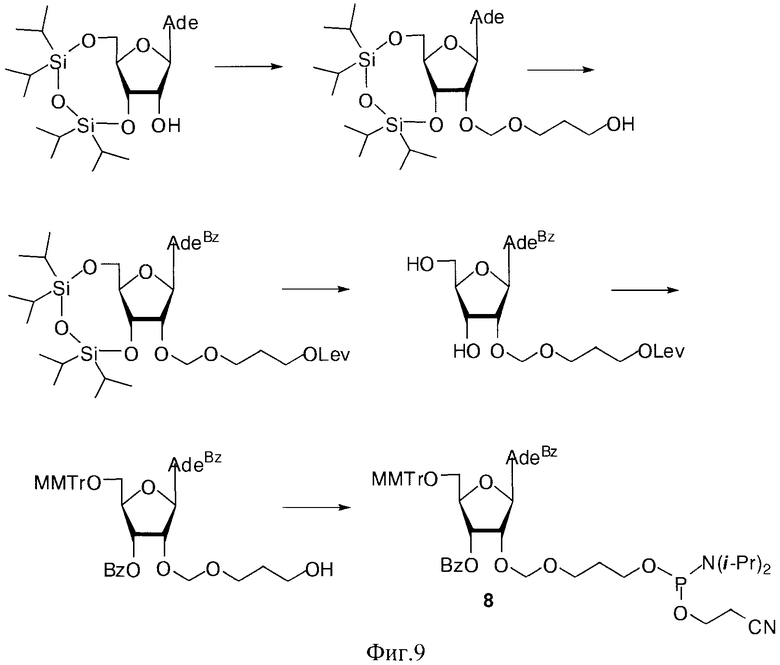
Были синтезированы фосфорамидитные производные на основе 2′-O-гидроксиалкоксинуклеозидов 7-9, где В=AdeBz, Ura, CytBz, GuaiBu, R=MMTr или DMTr, n=2-4 (Фиг 3).
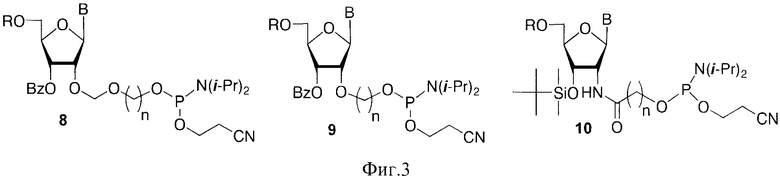
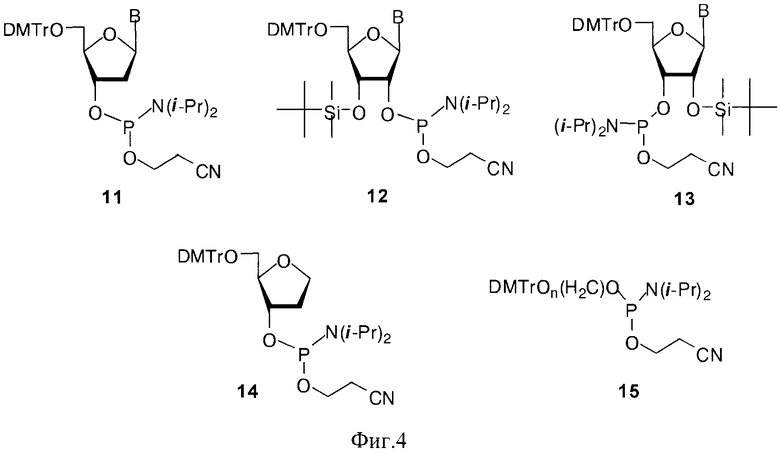
Наряду с дисахаридными нуклеозидами и их производными для получения миметиков ПАР был использован ряд коммерчески доступных фосфорамидитных синтонов 10-14, где В=AdeBz, Ura, CytBz, GuaiBu (Фиг.4).
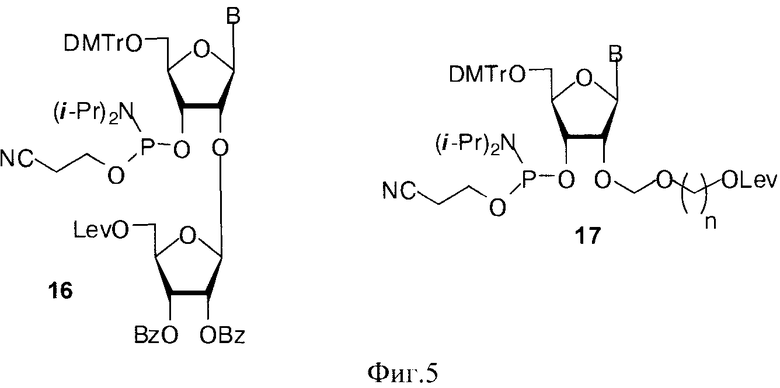
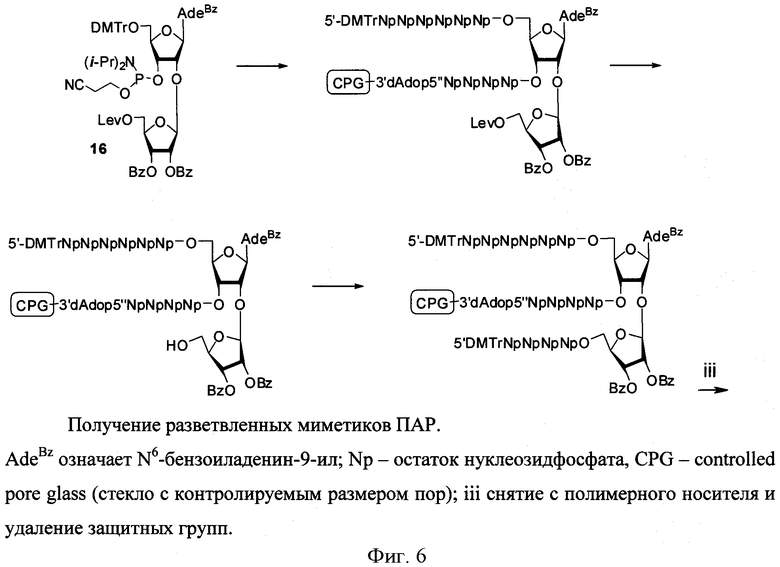
Для получения разветвленных полимеров использовали синтоны 15 и 16, где В=AdeBz, Ura, CytBz, GuaiBu, n=2-4 (Фиг. 5).
Фосфорамидитные синтоны включались в состав миметиков ПАР с использованием технологии автоматизированного олигонуклеотидного синтеза (Фиг. 6, 11)
После синтеза полимера на полимерном носителе проводили селективное удаление левулинильной группы (Lev=С(O)СН2СН2С(O)СН3) гидразин гидратом. Последующее наращивание цепи и удаление цианэтильной и ацильных защитных групп приводило к разветвленным миметикам ПАР (Фиг.6).
Синтез миметиков ПАР осуществляется с использованием описанных выше фосфорамидитных блоков 1-16 в автоматическом режиме на нерастворимом силикатном, полистирольном или полиакрилатном носителе методами олигонуклеотидной химии при температуре 20°C. В ходе автоматического синтеза происходит последовательное присоединение мономерных блоков к 5′- или 5′′-концу синтезируемого олигомера до получения полимера необходимой длины. Каждый цикл присоединения состоит из 4 стадий:
1. Деблокирование (удаление трифенилметильной (MMTr или DMTr) защитной группы с 5′ гидроксильной группы олигонуклеотидной цепи трихлоруксусной кислотой).
2. Конденсация (присоединение амидофосфитного блока в присутствии катализатора -1H-тетразола или 2-этилтиотетразола).
3. Кэпирование (ацетилирование непрореагировавших гидроксильных групп смесью пропионового ангидрида и пиридина в соотношении от 1:1 до 1:3 в тетрагидрофуране).
4. Окисление (превращение межнуклеотидного фосфита в фосфотриэфир или тиофосфодиэфир).
На заключительном этапе синтеза полимерных миметиков ПАР проводят удаление ацильных защитных групп в стандартных условиях (конц. водный аммиак, 6 часов, 55°C).
Рассмотренные ранее модифицированные фосфорамидиты различаются между собой как по реакционной способности амидофосфитной группы, так и по типу тритильной защиты на гидроксильной группе.
Наиболее реакционноспособными являются синтоны с амидофосфитом на первичной гидроксильной группе.
Стандартной защитной группой по 5′ положению амидофосфитных блоков является 4,4′-диметокситритильная. Помимо этого может быть использована монометокситритильная защитная группа. Скорость удаления ее стандартным деблокирующим раствором (3% трихлоруксусная кислота в хлористом метилене) ниже по сравнению с диметокситритильной группой. Ее применение целесообразно при синтезе набора олигомеров, различающихся между собой одним мономерным звеном: Ν, Ν-1, Ν-2 и т.д. При этом в конечном наборе каждый олигомер будет содержать монометокситритильную защитную группу.
Стандартные подходы в олигонуклеотидной химии позволяют вводить как фосфодиэфирные, так и тиофосфорильные межнуклеотидные связи. Таким образом, могут быть получены тиофосфорильные производные олигомерных миметиков ПАР.
Автоматический синтез олигомерных миметиков ПАР позволяет получать не только линейные, но и разветвленные олигомеры. Это достигается благодаря использованию соответствующих коммерчески доступных амидофосфитных блоков, например дендримерных фосфорамидитов.
Функциональность рассмотренных миметиков ПАР может быть существенно расширена за счет введения реакционноспособных групп, флуоресцентных меток, тушителей флуоресценции, фрагментов, обеспечивающих транспорт олигомеров к мишени. В качестве реакционноспособных групп могут выступать аминогруппы, альдегидные, тиольные, фосфатные группы, биотин, фрагмент ЭДТА. В олигомеры также могут быть введены флуоресцентные репортерные фрагменты на основе производных флуоресцеина, цианиновых красителей, акридина, фенантрена, пирена, а также тушители флуоресценции, например, на основе производных азобензола.
Наличие гидрофобной диметокситритильной группы на 5′ конце синтезированного олигомера существенно облегчает хроматографическое выделение целевого полноразмерного продукта. Благодаря этому, олигомерные миметики ПАР могут быть получены с высокой степенью очистки при использовании обращенно-фазной ВЭЖХ.
Разработанная методология позволяет получить регулярные и нерегулярные структуры со степенью полимеризации до 100 мономерных звеньев, что сравнимо с природными ПАР. Число (k+1)·m определяется техническими возможностями ОН-синтезатора. Разработанная технология позволяет синтезировать полимеры на основе диэфиров фосфорной кислоты с повторяющимся мономерным звеном, полимеры с регулярной структурой с повторяющимися несколькими звеньями, а также нерегулярные полимеры с несколькими мономерными звеньями. Отличительной чертой данной технологии является получение полимеров с заданным молекулярным весом, а следовательно с определенными свойствами.
Настоящее изобретение проиллюстрировано примерами синтеза фосфорамидитных блоков и миметиков ПАР, имеющих на концах полимерной цепи и в середине цепи различные функциональные и репортерные группы.
Структура заявленных соединений подтверждена методами УФ-, ЯМР- и масс-спектрометрии. ЯМР-спектры регистрировали на спектрометре Bruker АМХ400 (Германия). Химические сдвиги (δ) измерены относительно внутреннего стандарта - тетраметилсилана (δ 0 м.д.) и приведены в миллионных долях. Величины констант спин - спинового взаимодействия (J) измерены в герцах. При описании спектров 1H-ЯМР приняты следующие сокращения: с - синглет, уш. с - уширенный синглет, д - дублет, т - триплет, м - мультиплет. Модифицированные олигонуклеотиды были получены на автоматическом синтезаторе Applied Biosystems (США). Тонкослойную хроматографию проводили на пластинках Kieselgel 260 F (Merck), пятна проявляли в ультрафиолетовом свете (λ=254 нм). Колоночную хроматографию проводили на силикагеле Kieselgel 60 (0.063-0.200 мм, Merck). Очистку растворителей проводили по стандартным методикам.
Пример 1. Синтез N6-Бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-монометокситритил-β-D)-рибофуранозил)-3-O-ацетил-5-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-β-D-рибофуранозил]аденина (1, В=AdeBz, R=MMTr. Фиг. 7).
а) N6-Бензоил-9-[2-O-(β-D)-рибофуранозил)-3,5-O(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил]аденин.
Раствор 1.66 г (1.56 ммоль) N6-бензоил-9-[2-O-(2,3,5-три-O-бензоил-β-D-рибофуранозил)-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D)-рибофуранозил]аденина (S.N. Mikhailov et al J. Carbohydr. Chem. 16, 75-92, 1997) в 0.15 Μ растворе метилата натрия в сухом метаноле (42 мл) выдерживали 40 мин. при 20°C, добавляли 10% уксусную кислоту в метаноле до рН 7.0, упаривали в вакууме досуха. Остаток растворяли в 100 мл дихлорметана и последовательно промывали 30 мл насыщенного раствора бикарбоната натрия и водой (2×30 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали досуха. Остаток хроматографировали на колонке, содержащей 35 г силикагеля. Вещество элюировали системой 1.5% этанол/дихлорметан (500 мл). Фракции, содержащие продукт, объединяли и упаривали в вакууме досуха. Выход 680 мг (58% пена). Rƒ 0.28 в системе 5% этанол/дихлорметан. 1H-ЯМР (CDCl3): 9.28 уш. с (1Н, NH), 8.76 с (1Н, Н-8), 8.43 с (1Н, Н-2), 8.03 д (2Н, Bz), 7.60 т (1H, Bz), 7.52 т (2H, Βz), 6.11 с (1H, Н-1′ Ado), 5.33 с (1H, Н-1′ Rib), 4.76 дд (1H, J3′,2′=4.6, J3′,4=7.6, Η-3′ Rib), 4.50 дд (1H, J3′,2′ 4.2, J3′,4=9.2, H-3′ Ado), 4.47 д (1H, H-2′ Ado), 4.29 д (1H, J5′a5′b=- 13.2, Η-5′а Ado), 4.17 м (2H, Η-2′ Rib, Η-4′ Ado), 4.03 м (3H, H-4′,5′a Rib, Η-5′b Ado), 3.76 д (1H, J5′b,5′a=-10.7, Η-5′b Rib), 1.14-0.95 м (28H, i-Pr). 13С-ЯМР (CDCl3): 164.58 (C=O), 152.17 (C-2), 149.89 (C-6), 149.66 (C-4), 140.55 (C-8), 132.73, 128.72, и 127.85 (Bz), 123.65 (C-5), 106.11 (С-1′, Rib), 89.17 (С-1′, Ado), 83.79, 81.67 (C-4′), 76.62, 75.49 (C-2′), 68.98, 68.91 (C-3′), 60.21 (C-5′, Rib), 59.42 (C-5′, Ado), 17.32, 17.25, 17.15, 17.07, 16.89, 13.27, 12.78, 12.78 и 12.57 (i-Pr).
б) N6-Бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил]аденин.
Получали из 518 мг (0.7 ммоль) N6-Бензоил-9-[2-О-(β-D-рибофуранозил)-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил]аденина по стандартной методике введения тритильной защитной группы с последующим ацилированием. Продукт подвергали хроматографической очистке на колонке, содержащей 30 г силикагеля. Вещество элюировали системой 1.5% этанол/дихлорметан. Фракции, содержащие продукт, объединяли и упаривали в вакууме досуха. Выход 640 мг (83% пена). Rƒ0.41 в системе 5% этанол/дихлорметан. 1H-ЯМР (CDCl3): 8.98 уш. с (1Н, NH), 8.59 с (1Н, Н-8), 8.02-7.15 м (18Н, Н-2, Bz, Ph), 6.74 д (2Н, Ph), 5.76 с (1H, Н-1′ Ado), 5.73 дд (1H, J3′,2′ 3.3, J3′,4′ 6.9, Η-3′ Rib), 5.54 с (1H, Н-1′ Rib), 5.53 д (1H, Η-2′ Rib), 5.11 дд (1H, J3′,2′ 4.8, J3′,4′ 8.8, H-3′ Ado), 4.87 д (1H, H-2′ Ado), 4.27 ддд (1H, J4′,5′a 4.8, J4′,5′b 3.7, H-4′ Rib), 4.05 м (3H, H-4′,5′a,5′b Ado), 3.73 с (3H, OMe), 3.36 дд (1H, J5′a,5′b - 10.3, Η-5′а Rib), 3.23 дд (1H, Η-5′b Rib), 2.09 с (3H, Ac), 2.03 с (3H, Ac), 1.11-1.04 м (28H, i-Pr). 13С-ЯМР (CDCl3): 169.75, 169.42, 164.56 (C=O), 158.70 (Ph), 152.63 (C-2), 150.60 (C-6), 149.42 (C-4), 142.44 (C-8), 135.06, 132.75, 130.45, 128.90, 128.47, 128.38, 127.81 и 127.02 (Bz, Ph), 123.36 (C-5), 113.03 (Ph), 105.45 (С-1′, Rib), 88.84 (С-1′, Ado), 86.17 (Ph3C), 81.22, 79.82 (C-4′), 78.64, 74.97 (C-2′), 71.36, 69.96 (C-3′), 62.95 (C-5′, Rib), 59.85 (C-5′, Ado), 55.15 (OMe), 20.46 и 20.37 (Ac), 17.33, 17.28, 17.18, 17.00, 16.82, 13.23, 12.95, 12.84 и 12.60 (i-Pr).
в) N6-Бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-β-D-рибофуранозил]аденин.
К раствору 324 мг (0.29 ммоль) N6-бензоил-9-[2-О(2,3-ди-О-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил]аденина в тетрагидрофуране (4 мл) добавляли 280 мг (0.88 ммоль) тетрабутиламмоний фторида тригидрата в тетрагидрофуране (3 мл). Реакция протекает в течение 15 мин при 20°C. Реакционную смесь упаривали в вакууме, а затем переупаривали с хлороформом (2×10 мл). Остаток хроматографировали на колонке, содержащей 30 г силикагеля, элюируя вещество системой 1.5% этанол/дихлорметан. Фракции, содержащие продукт, объединяли и упаривали в вакууме досуха. Выход 230 мг (92% в виде пены), Rƒ0.30 в системе 5% этанол/дихлорметан. 1H-ЯМР (CDCl3): 9.11 ус (1Н, NH), 8.77 с (1Н, Н-8), 8.01-7.28 м (18Н, Н-2, Bz, Ph), 6.88 д (2Н, PhOMe), 5.90 д (1Н, J1′,2′=7.0, Н-1′ Ado), 5.42 дд (1Н, J3′,2′ =5.0, J3′,4′=5.2, Η-3′ Rib), 5.35 дд (1H, J2′,1′=2.2, Η-2′ Rib), 5.01 дд (1H, J2′,3′=4.8, H-2′ Ado), 4.81 д (1H, Н-1′ Rib), 4.64 дд (1H, J3′,4′=1.1, H-3′ Ado), 4.29 д (1H, H-4′ Ado), 4.04 ддд (1H, J4′,5′a=3.6, J4′,5′b=4.2, H-4′ Rib), 3.96 д (1H, J5′a,5′b-=13.2, H-5′a Ado), 3.73 с (3H, OMe), 3.70 д (1H, Η-5′b Ado), 3.32 дд (1H, J5′a5′b=-10.6, H-5′a Rib), 3.23 дд (1H, Η-5′b Rib), 2.05 с (3H, Ac), 2.02 с (3Н, Ac). 13С-ЯМР (CDCl3): 169.90, 164.53 (C=O), 158.92 (Ph), 152.12 (C-2), 150.36 (C-6), 149.78 (C-4), 141.42 (C-8), 134.79, 132.94, 130.48, 128.96, 128.47, 128.11, 127.87 и 127.32 (Bz, Ph), 124.16 (C-5), 113.39 (Ph), 106.62 (С-1′, Rib), 89.72 (С-1′, Ado), 86.93 (Ph3C), 86.83 (C-4′), 81.04, 76.36 (C-2′), 75.00, 72.02 (C-3′), 71.14 (C-5′, Rib), 63.01 (C-5′, Ado), 55.21 (OMe), 20.43, 19.87 (Ac).
г) N6-Бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-5-O-трет-бутилдифенилсилил-β-D-рибофуранозил]аденин.
К раствору 370 мг (0.43 ммоль) N6-Бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-β-D-рибофуранозил]аденина и имидазола 35 мг (0.51 ммоль) в сухом пиридине (4 мл) добавляли 0.19 мл (0.75 ммоль) трет-бутилдифенилсилилхлорида, выдерживали 48 ч при 20°C. Смесь упаривали досуха, остаток растворяли в хлороформе (20 мл) и промывали водой (10 мл), насыщенным раствором бикарбоната натрия (10 мл), а затем водой (10 мл). Органический слой сушили безводным сульфатом натрия, фильтровали, упаривали досуха, переупаривали с толуолом (2×10 мл). Остаток хроматографировали на колонке, содержащей 20 г силикагеля. Вещество элюировали системой 1% этанол/дихлорметан (400 мл). Фракции, содержащие продукт, объединяли и упаривали в вакууме досуха. Выход 420 мг (88%, пена). Rƒ 0.37 в системе 5% этанол/дихлорметан. 1Н-ЯМР (CDCl3): 9.21 ус (1Н, NH), 8.60 с (1Н, Н-8), 8.04-7.18 м (28Н, Н-2, Bz, Ph), 6.83 д (2Н, PhOMe), 6.13 д (1Н, J1′,2′=2.8, Н-1′ Ado), 5.49 дд (1H, J3′,2′ 5.0, J3′,4′=5.2, Η-3′ Rib), 5.46 дд (1H, J2′,1′=2.4, H-2′ Rib), 5.20 д (1H, Н-1′ Rib), 4.95 дд (1H, J2′,3′=5.6, H-2′ Ado), 4.76 дд (1H, J3′,4′=5.9, H-3′ Ado), 4.22 д (1H, H-4′ Rib), 4.05 ддд (1H, J4′,5′a=3.6, J4′,5′b=3.4, Η-4′ Ado), 4.03 д (1H, J5′a,5′b-=11-2, Η-5′а Ado), 3.86 д (1H, Η-5′b Ado), 3.74 с (3H, OMe), 3.36 дд (1H, J5′a,5′b=-9.9, Η-5′а Rib), 3.30 дд (1H, Η-5′b Rib), 2.09 с (3H, Ac), 2.06 с (3H, Ac), 1.04 с (9H, t-Bu). 13С-ЯМР (CDCl3): 165.94, 162.53 (C=O), 157.92 (Ph), 151.12 (C-2), 151.26 (C-6), 148.76 (C-4), 143.41 (C-8), 134.79, 132.94, 130.48, 128.96, 128.47, 128.11, 127.87 и 127.32 (Bz, Ph), 124.16 (C-5), 113.39 (Ph), 107.42 (С-1′, Rib), 88.41 (С-1′, Ado), 87.05 (Ph3C), 84.42, 81.52 (C-4′), 81.29, 76.36 (C-2′), 75.00, 71.80 (C-3′), 71.14 (C-5′, Rib), 63.08 (C-5′, Ado), 55.34 (OMe), 26,97 (t-Bu), 20.43, 19.43 (Ac).
д) N6-Бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-3-O-ацетил-5-O-трет-бутилдифенилсилил-β-D-рибофуранозил]аденин.
К раствору 350 мг (0.32 ммоль) N6-бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-5-O-трет-бутилдифенилсилил-β-D-рибофуранозил]аденина в сухом пиридине (20 мл) добавляли 50 мкл (0.5 ммоль) уксусного ангидрида и выдерживали 2 ч при 20°C, после чего добавляли 0.3 мл метанола, и выдерживали 30 мин. Реакционную смесь упаривали в вакууме. Остаток растворяли в хлороформе (30 мл), промывали 10% водным раствором бикарбоната натрия (15 мл), водой (2×10 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали, упаривали, а затем упаривали с толуолом (2×10 мл), Остаток хроматографировали на колонке, содержащей 20 г силикагеля. Вещество элюировали системой 1.5% этанол/дихлорметан. Фракции, содержащие продукт, объединяли и упаривали в вакууме досуха. Выход 277 мг (76% пена). Rƒ 0.40 в системе 5% этанол/дихлорметан. 1H-ЯМР (CDCl3): 9.06 ус (1Н, NH), 8.54 с (1H, Н-8), 8.04-7.18 м (28Н, Н-2, Bz, Ph), 6.76 д (2Н, PhOMe), 6.10 д (1Н, J1′,2′=3.8, Н-1′ Ado), 5.43 дд (1H, J3′,2′=4.9, J3′,4′=5.3, H-3′ Ado), 5.34 дд (1H, J3′,2′ 4.2, J3′,4′=5.6, H-3′ Rib), 5.32 дд (1H, J2′,1′=1.4, H-2′ Rib), 5.09 д (1H, J1′,2′=3.8, Н-1′ Ado), 4.99 д (1H, Н-1′ Rib), 4.28 д (1H, H-4′ Ado), 4.10 ддд (1H, J4′,5′a=3.6, J4′,5′b=3.4, H-4′ Rib), 3.96 д (1H, J5′a,5′b-=11.2, Η-5′а Ado), 3.86 д (1H, Η-5′b Ado), 3.73 с (3H, OMe), 3.36 м (2H, Η-5′а, Η-5′b Rib), 2.11 с (3H, Ac), 2.05 с (3H, Ac), 2.02 с (3H, Ac), 1.05 с (9H, t-Bu). 13C-ЯМР (CDCl3): 165.94, 162.53 (C=O), 157.92 (Ph), 151.12 (C-2), 151.26 (C-6), 148.76 (C-4), 143.41 (C-8), 134.79, 132.94, 130.48, 128.96, 128.47, 128.11, 127.87 и 127.32 (Bz, Ph), 124.16 (C-5), 113.39 (Ph), 107.42 (С-1′, Rib), 88.41 (С-1′, Ado), 87.05 (Ph3C), 84.42, 81.52 (C-4′), 81.29, 76.36 (C-2′), 75.00, 71.80 (C-3′), 71.14 (C-5′, Rib), 63.08 (C-5′, Ado), 55.34 (OMe), 26.97 (f-Bu), 20.43, 20.87, 19.43 (Ac).
е) N6-Бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-3-О-ацетил-β-D-рибофуранозил]аденин.
К раствору 250 мг (0.22 ммоль) N6-Бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-3-O-ацетил-5-O-трет-бутилдифенилсилил-β-D-рибофуранозил]аденина в тетрагидрофуране (5 мл) добавляли 105 мг (0.33 ммоль) тетрабутиламмоний фторида тригидрата в тетрагидрофуране (5 мл) и выдерживали 15 мин при 20°C. Реакционную смесь упаривали в вакууме, переупаривали с хлороформом (2×10 мл). Остаток хроматографировали на колонке, содержащей 20 г силикагеля. Вещество элюировали системой 5% этанол/дихлорметан (500 мл). Фракции, содержащие продукт, объединяли и упаривали в вакууме досуха. Выход 177 мг (89% пена). Rƒ0.34 в системе 5% этанол/дихлорметан. 1H ЯМР (CDCl3): 9.31 ус (1Н, NH), 8.87 с (1Н, Н-8), 8.01-7.28 м (18Н, Н-2, Bz, Ph), 6.79 д (2Н, PhOMe), 5.83 д (1Н, J1′,2′=5.2, Н-1′ Ado), 5.45 дд (1Н, J2′,1′=2.2, Η-2′ Rib), 5.32 дд (1H, J3′,2′=5.0, J3′,4′=4.2, Η-3′ Rib), 5.21 д (1H, Н-1′ Rib), 5.06 дд (1H, J2′,3′=6.8, H-2′ Ado), 4.75 дд (1H, J3′,4′=3.1, H-3′ Ado), 4.29 д (1H, H-4′ Rib), 4.04 ддд (1H, J4′,5′a=2.6, J4′,5′b=3.2, H-4′ Ado), 3.96 д (1H, J5′a,5′b-=11.2, Η-5′а Ado), 3.84 д (1H, Η-5′b Ado), 3.73 с (3H, OMe), 3.35 дд (1H, J5′a,5′b=-10.2, Η-5′а Rib), 3.28 дд (1H, Η-5′b Rib), 2.11 с (3H, Ac), 2.07 с (3H, Ac), 2.01 с (3Н, Ac). 13С-ЯМР (CDCl3): 169.90, 167.57, 164.53 (C=O), 158.92 (Ph), 153.12 (C-2), 151.36 (C-6), 148.78 (C-4), 141.35 (C-8), 134.79, 132.94, 130.48, 128.96, 128.47, 128.11, 127.87 и 127.32 (Bz, Ph), 123.15 (C-5), 113.43 (Ph), 106.72 (С-1′, Rib), 88.76 (С-1′, Ado), 87.95 (Ph3C), 81.88 (C-4′), 81.04, 76.36 (C-2′), 75.00, 72.02 (C-3′), 71.14 (C-5′, Rib), 63.01 (C-5′, Ado), 55.76 (OMe), 21.15, 20.46, 19.97 (Ac).
ж) N6-Бензоил-9-[2-O(2,3-ди-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-3-O-ацетил-5-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-β-D-рибофуранозил]аденин (1, B=AdeBz, R=MMTr).
К раствору 150 мг (0.17 ммоль) N6-Бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил)-3-O-ацетил-β-D-рибофуранозил]аденина в 10 мл дихлорэтана, добавляли диизопропилэтиламин (0.08 мл, 0.5 ммоль) и 2-цианоэтил-Ν,Ν-диизопропилхлорофосфорамидит (0.07 мл, 0.35 ммоль). Через 2 часа добавляли 10% водный раствор гидрокарбоната натрия (3 мл), перемешивали 10 мин. Добавляли раствор гидрокарбоната натрия (20 мл) и дихлорметан (20 мл). Органический слой отделяли, промывали водой (2×20 мл), сушили безводным сульфатом натрия, фильтровали, упаривали досуха. Остаток хроматографировали на колонке, содержащей 15 г силикагеля. Вещество элюировали системой этилацетат/гексан/триэтиламин (70/29/1).
Фракции, содержащие продукт, объединяли, упаривали в вакууме досуха. Выход 140 мг (74% пена). Rƒ0.42 в системе 5% этанол/дихлорметан. 31Р-ЯМР (CDCl3): 151.47, 150.12.
Пример 2. Синтез N6-Бензоил-9-[2-O-(2,3-ди-O-ацетил-5-O-диметокситритил-β-D-рибофуранозил)-3-O-ацетил-5-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-β-D-рибофуранозил]аденина (1, B=AdeBz, R=DMTr).
Вещество получали аналогично примеру 1 с суммарным выходом 17%. Rƒ0.44 в системе 5% этанол/дихлорметан. 31Р-ЯМР (CDCl3): 151.45, 150.11.
Пример 3. Синтез N6-Бензоил-9-{2-O-[2,3-ди-O-бензоил-5-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-β-D-рибофуранозил]-3-O-ацетил-5-O-диметокситритил-β-D-рибофуранозил}аденина (2, B=AdeBz, R=DMTr. Фиг. 8).
а) N6-Бензоил-9-[2-O-(2,3-ди-O-бензоил- β-D-рибофуранозил)-3-O-ацетил-5-O-диметокситритил-β-D-рибофуранозил]аденин.
Из 595 мг (0.73 ммоль) N6-Бензоил-9-[2-O-(2,3-ди-O-бензоил-5-O-левулинил-β-D-рибофуранозил)-β-D-рибофуранозил]аденина (S.N. Mikhailov et al Chemistry & Biodiversity, 2, 1153-1163, 2005) получали N6-Бензоил-9-[2-O-(2,3-ди-O-бензоил-5-O-левулинил-β-D-рибофуранозил)-3-O-ацетил-5-O-диметокситритил-β-D-рибофуранозил]аденин по стандартной методике тритилирования-ацилирования. N6-Бензоил-9-[2-O-(2,3-ди-O-бензоил-5-O-левулинил-β-D-рибофуранозил)-3-O-ацетил-5-O-диметокситритил-β-D-рибофуранозил]аденин без предварительной хроматографической очистки растворяли в 10 мл пиридина, охлаждали до 0°C и добавляли 1.89 мл (33 ммоль, 45 эквивалентов по отношению к исходному N6-Бензоил-9-[2-(9-(2,3-ди-O-бензоил-5-О-левулинил-β-D-рибофуранозил)-β-D-рибофуранозил]аденину) уксусной кислоты. Далее при 0°C прикапывали 0.214 мл (4.41 ммоль, 6 эквивалентов по отношению к исходному N6-Бензоил-9-[2-O-(2,3-ди-O-бензоил-5-O-левулинил-β-D-рибофуранозил)-β-D-рибофуранозил]аденину) гидразина моногидрата и выдерживали в течение 40 мин при 0°C. Реакционную смесь разбавляли этилацетатом (30 мл) и промывали последовательно насыщенным водным раствором бикарбоната натрия (20 мл) и водой (4×20 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали под вакуумом. Остаток хроматографировали на колонке, содержащей 15 г силикагеля. Вещество элюировали системой этилацетат/гексан/триэтиламин (66/33/1 по объему). Выход продукта на 3 стадии 578 мг (75%) в виде белой пены. Rƒ0.26 в системе этилацетат/гексан/триэтиламин (49/49/2 по объему). 1H- ЯМР (DMSO-D6): 11.29 ус (1Н, ΝΗ), 8.68 с (1Η, H-8), 8.65 с (1Н, Н-2), 8.10-7.20 м (24Н, Bz, Ph), 6.88 д (2Н, J=3.5, mPhOMe), 6.86 д (2Н, J=3.3, mPhOMe), 6.40 д (1Н, J1′,2′=5.4, Н-1′ Ado), 5.59 дд (1H, J2′,3′=6.0, J3′,4′=4.6, H-3′ Ado), 5.56-5.54 м (2H, Η-2′, H-3′ Rib), 5.46 дд (1H, J1′,2′=5.4, J2′,3′=6.0, H-2′ Ado), 5.31 д (1H, J4′,3′a=1.2, Н-1′ Rib), 5.12 т (1H, J5′,OH=5.2, 5′-OH), 4.37 ддд (1H, J4′,5′a=4.3, J5′a,5′b=-9.0, Η-5′а Ado), 4.35-4.30 м (1H, Η-5′b Ado), 3.79 с (6H, OMe), 3.42-3.38 м (2H, Η-4′ Ado, Η-4′ Rib), 3.36-3.32 м (2H, Η-5′а, Η-5′b Rib), 2.07 с (3Н, Ac). 13С-ЯМР (DMSO-D6): 170.15 (C=O-Ac), 166.19, 165.28, 164.81 (C=O-Bz), 158.44 (Ph), 152.25 (С-2), 152.01 (С-6), 150.73 (С-4), 144.94 (С-8), 144.23, 135.69, 135.62, 134.28, 134.14, 133.56, 132.95, 130.02, 129.50, 129.09, 128.91, 128.77, 128.16, 127.93, 127.14 (Bz, DMTr), 126.04 (С-5), 113.48 (m-MeOPh), 105.60 (С-1′, Rib), 87.52 (Ph3C), 86.10 (С-1′, Ado), 82.59, 81.19 (С-41), 76.89, 75.14 (С-2′), 72.40, 71.15 (С-3′), 63.09, 61.24 (С-5′, Ado), 55.34 (OMe), 20.58 (Ас).
б) N6-Бензоил-9-{2-O-[2,3-ди-O-бензоил-5-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-β-D-рибофуранозил]-3-O-ацетил-5-O-диметокситритил-β-D-рибофуранозил}аденин (2, B=AdeBz, R=DMTr).
К раствору 500 мг (0.47 ммоль) N6-Бензоил-9-[2-O-(2,3-ди-О-бензоил-β-D-рибофуранозил)-3-O-ацетил-5-O-диметокситритил-β-D-рибофуранозил]аденина и 36 мг (0.52 ммоль) 1H-тетразола в 6 мл смеси ацетонитрил/пиридин (5/1 по объему) прикапывали 0.254 мл (0.80 ммоль) 2-цианоэтил-N,N,N′,N′-тетраизопропилфосфорамидита. Реакцию проводили в инертной атмосфере азота. Через 1 ч добавляли 10% водный раствор гидрокарбоната натрия (3 мл), перемешивали 10 мин. Добавляли раствор гидрокарбоната натрия (20 мл) и этилацетат (20 мл). Органический слой отделяли, промывали водой (2×20 мл), сушили безводным сульфатом натрия, фильтровали, упаривали досуха. Остаток хроматографировали на колонке, содержащей 15 г силикагеля. Вещество элюировали системой этилацетат/гексан/триэтиламин (69/29/2). Фракции, содержащие продукт, объединяли, упаривали в вакууме досуха. Выход 502 мг (85% пена). Rƒ0.66 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (DMSO-D6): 150.92, 150.69.
Пример 4. Синтез N6-Бензоил-9-{2-O-[2,3-ди-O-бензоил-5-O-(2-цианоэтил)-N,N-диизопропиламидофосфинил-β-D-рибофуранозил]-3-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил}аденина (2, B=AdeBz, R=MMTr).
Вещество было получено согласно примеру 3, исходя из N6-Бензоил-9-[2-O-(2,3-ди-O-бензоил-5-О-левулинил-β-D-рибофуранозил)-β-D-рибофуранозил]аденина. Общий выход продукта на четыре стадии составляет 67%. Rƒ0.65 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (DMSO-D6): 150.91, 150.67.
Пример 5. Синтез 1-{2-O-[2,3-ди-O-бензоил-5-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-β-D-рибофуранозил]-3-O-ацетил-5-O-диметокситритил-β-D-рибофуранозил}урацила (2, B=Ura, R=DMTr).
Вещество было получено согласно примеру 3, исходя из 1-[2-O-(2,3-ди-O-бензоил-5-O-левулинил-β-D-рибофуранозил)-β-D-рибофуранозил]урацила. Общий выход продукта на четыре стадии составляет 71%. Rƒ0.75 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (DMSO-D6): 150.90, 150.65.
Пример 6. Синтез 1-{2-O-[2,3-ди-O-бензоил-5-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-β-D-рибофуранозил]-3-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил}урацила (2, B=Ura, R=MMTr).
Вещество было получено согласно примеру 3, исходя из 1-[2-О(2,3-ди-O-бензоил-5-O-левулинил-β-D-рибофуранозил)-β-D-рибофуранозил]урацила. Общий выход продукта на четыре стадии составляет 77%. Rƒ0.75 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (DMSO-D6): 150.87, 150.61.
Пример 7. Синтез N6-Бензоил-9-{2-O-[2,3-ди-O-бензоил-5-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-α-D-арабинофуранозил]-3-O-ацетил-5-O-диметокситритил-β-D-рибофуранозил}аденина (3, B=AdeBz, R=DMTr).
Вещество было получено согласно примеру 3, исходя из N6-Бензоил-9-[2-O-(2,3-ди-O-бензоил-5-О-левулинил-α-D-арабинофуранозил)-β-D-рибофуранозил]аденина. Общий выход продукта на четыре стадии составляет 63%. Rƒ0.67 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (DMSO-D6): 150.90, 150.70.
Пример 8. Синтез N6-Бензоил-9-{2-O-[2,3-ди-O-бензоил-5-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-α-D-арабинофуранозил]-3-O-ацетил-5-O-монометокситритил-β-D-рибофуранозил}аденина (3, Β=ΑdeΒz, R=MMTr).
Вещество было получено согласно примеру 3, исходя из N6-Бензоил-9-[2-O-(2,3-ди-O-бензоил-5-O-левулинил-α-D-арабинофуранозил)-β-D-рибофуранозил]аденина. Общий выход продукта на четыре стадии составляет 63%. Rƒ0.67 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (DMSO-D6): 150.90, 150.71.
Пример 9. Синтез N6-Бензоил-9-{2-O-[2,3-ди-O-бензоил-5-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-α-D-рибофуранозил]-3-O-ацетил-5-O-диметокситритил-β-D-рибофуранозил}аденина (5, B=AdeBz, R=DMTr).
Вещество было получено исходя из 2′-O-α-D-рибофуранозиладенозина (S.N. Mikhailov et al Tetrahedron, 64, 2871-2876, 2008). Rƒ0.70 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (DMSO-D6): 150.87, 150.68.
Пример 10. Синтез N6-Бензоил-9-{2-O-[(3-(2-цианоэтил-N,N-диизопропиламидофосфинил)оксипропокси)метил]-3-O-бензоил-5-O-монометокситритил-β-D-рибофуранозил}аденина (7, B=AdeBz, R=MMTr, n=3. Фиг. 9).
а) 9-{2-O-[(3-Ацетоксипропокси)метил]-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}аденин.
К раствору, содержащему 1.02 г (2 ммоль) 9-[3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил]аденина и 0.76 г (4 ммоль) 3-(гидроксиметокси)пропан-1-ол диацетата в 10 мл 1,2-дихлорэтана в атмосфере азота при перемешивании и охлаждении до -10°C добавляли 0.749 мл (6.4 ммоль) тетрахлорида олова. Раствор выдерживали 40 минут при -10°C. К раствору добавляли 20 мл насыщенного раствора бикарбоната натрия и 30 мл дихлорметана, перемешивали 20 минут и фильтровали через Hyflo Super-cel (целит). Органический слой отделяли, промывали водой (2×50 мл), высушивали над безводным сульфатом натрия, фильтровали и упаривали в вакууме досуха. Остаток хроматографировали на колонке, содержащей 35 г силикагеля. Вещество элюировали системой 1.5% этанол/дихлорметан. Выход 0.73 г (56%) в виде белой пены. Rƒ0.23 в системе 5% этанол/дихлорметан. 1Н-ЯМР (CDCl3): 8.30 с (1H, Н-8), 8.12 с (1H, Н-2), 6.05 с (1Н, Н-1′), 5.66 с (2Н, NH2), 4.98 с (2Н, OCH2O), 4.73 дд (1H, J3′,2′=4.4, J3′,4′=9.3, Η-3′), 4.50 д (1Н, Н-2′), 4.23 д (1H, J5′a,5′b=-13.2, Η-5′а), 4.12-4.04 м (3Н, 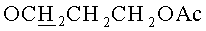
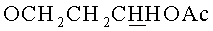
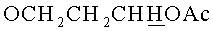
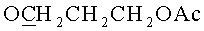
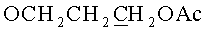
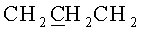
б) 9-{2-O-[(3-Гидроксипропокси)метил]-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}аденин.
9-{2-O-[(3-Ацетоксипропокси)метил]-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}аденин (1.38 г, 2.16 ммоль) обрабатывали 15 мл 4М раствора метиламина в этаноле (MeNH2/EtOH) и оставляли на сутки при комнатной температуре. Через 24 часа содержимое колбы упаривали под вакуумом, переупаривали с дихлорметаном (2×15 мл) и наносили на колонку, содержащую 30 г силикагеля. Вещество элюировали системой 5% этанол/дихлорметан. Выход 1.10 г (85% пена). Rƒ0.19 в системе 5% этанол/дихлорметан. 1Н-ЯМР (CDCl3): 8.27 с (1H, Н-8), 8.22 с (1Н, Н-2), 6.33 с (2Н, NH2), 6.08 с (1Н, Н-1′), 5.00 д (1H, J=-7.0, 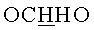
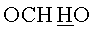
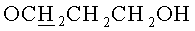
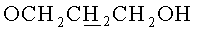
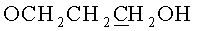
в) N6-Бензоил-9-{2-O-[(3-гидроксипропокси)метил]-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}аденин.
К суспензии 9-{2-O-[(3-Гидроксипропокси)метил]-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}аденина (0.75 г, 1.26 ммоль) в сухом пиридине (30 мл) добавляли триметилхлорсилан (0.8 мл, 6.33 ммоль). Суспензию перемешивали при комнатной температуре 40 мин. Добавляли бензоилхлорид (0.8 мл, 6.89 ммоль), оставляли при 20°C и перемешивании на 2 ч. Реакционную смесь охлаждали до 0°C, добавляли воду (10 мл), через 5 мин добавляли 25% раствор водного аммиака (10 мл). Выдерживали смесь 30 мин при 20°C и упаривали под вакуумом. Упаривали с толуолом (2×15 мл). Остаток растворяли в этилацетате (50 мл), полученный раствор промывали последовательно насыщенным раствором бикарбоната натрия (20 мл), водой (2×20 мл), сушили над безводным сульфатом натрия, фильтровали, упаривали в вакууме досуха. Остаток хроматографировали на колонке, содержащей 30 г силикагеля. Вещество элюировали системой 2% этанол/дихлорметан. Выход 0.64 г (72%) в виде белой пены. Rƒ0.30 в системе 5% этанол/дихлорметан. 1H-ЯМР (CDCl3): 9.32 ус (1Н, NHBz), 8.77 с (1Н, Н-8), 8.38 с (1Н, Н-2), 8.03-7.42 м (5Н, Bz), 6.14 с (1Н, Н-1′), 5.02 д (1H, J=-6.9, 
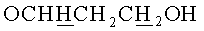
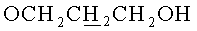
г) N6-Бензоил-9-{2-O-[(3-левулинилоксипропокси)метил]-3,5-(9-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}аденин.
К раствору, содержащему 0.64 г (0.92 ммоль) N6-Бензоил-9-{2-O-[(3-гидроксипропокси)метил]-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}аденина, 0.27 г (2.3 ммоль) левулиновой кислоты в 15 мл 1,2-дихлорэтана при 20°C добавляли 0.76 г (3.6 ммоль) N,N′-дициклогексилкарбодиимида. Раствор выдерживали 1.5 ч. при 20°C. Выпавший осадок отфильтровывали. Органический слой отделяли, промывали водой (2×50 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали под вакуумом. Остаток хроматографировали на колонке, содержащей 35 г силикагеля. Вещество элюировали системой 1.5% этанол/дихлорметан. Выход 0.57 г (78%) в виде белой пены. Rƒ0.34 в системе 5% этанол/дихлорметан. 1Н-ЯМР (CDCl3): 9.41 ус (1Н, NHBz), 8.72 с (1H, Н-8), 8.31 с (1H, Н-2), 8.01-7.44 м (5Н, Bz), 6.10 с (1H, Н-1′), 4.96 д (1H, J=-6.7,
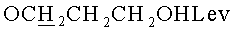
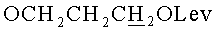
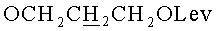
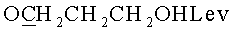
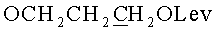
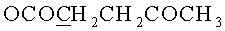
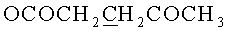
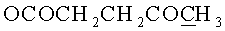
д) N6-Бензоил-9-{2-O-[(3-левулинилоксипропокси)метил]-β-D-рибофуранозил}аденин.
1.42 г (1.78 ммоль) N6-Бензоил-9-{2-O-[(3-левулинилоксипропокси)метил]-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}аденина обрабатывали 2.5 мл 0.5 Μ раствора тетрабутиламмоний фторида тригидрата в тетрагидрофуране и выдерживали 10 мин при 20°C. Реакционную смесь упаривали под вакуумом, переупаривали с хлороформом (2×10 мл). Остаток хроматографировали на колонке содержащей 30 г силикагеля. Вещество элюировали системой 4% этанол/дихлорметан. Выход 803 мг (81%) в виде белой аморфной пены R/ 0.35 в системе 10% этанол/дихлорметан. 1Н- ЯМР (CDCl3): 9.53 ус (1Н, NHBz), 8.76 с (1Н, Н-8), 8.22 с (1Н, Н-2), 8.03-7.48 м (5Н, Bz), 6.03 д (1Н, J1′,2′=7.2, Н-1′), 4.94 дд (1Н, J2′,3′=4.7, Η-2′), 4.67 д (1Н, J=-6.5,
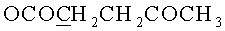
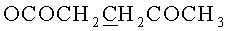
е) N6-Бензоил-9-{2-O-[(3-гидроксипропокси)метил]-3-O-бензоил-5-O-монометокситритил-β-D-рибофуранозил}аденин.
Раствор 450 мг (0.81 ммоль) N6-Бензоил-9-{2-O-[(3-левулинилоксипропокси)метил]-β-D-рибофуранозил}аденина и 426 мг (1.38 ммоль)монометокситритилхлорида в сухом пиридине (5 мл) выдерживали 16 ч при 20°C. Реакционную смесь охлаждали до 0°C и при перемешивании приливали 0.140 мл (1.21 ммоль) бензоилхлорида. Бензоилирование проводили при 0°C, контролируя протекание реакции по ТСХ до полного исчезновения исходного вещества. Затем к реакционной смеси добавляли метанол (2 мл) и выдерживали в течение 30 мин при комнатной температуре, после чего разбавляли дихлорметаном (20 мл) и промывали последовательно насыщенным водным раствором бикарбоната натрия (20 мл) и водой (2×20 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали, упаривали. Остаток растворяли в 10 мл пиридина, охлаждали до 0°C и добавляли 2.08 мл (36.4 ммоль, 45 эквивалентов по отношению к N6-Бензоил-9-{2-О-[(3-левулинилоксипропокси)метил]-β-D-рибофуранозил}аденину)уксусной кислоты. Далее приливали 0.236 мл (4.86 ммоль, 6 эквивалентов по отношению к исходному нуклеозиду) гидразина моногидрата и выдерживали в течение 1 ч при 0°C. Реакционную смесь разбавляли этилацетатом (30 мл) и промывали последовательно насыщенным водным раствором бикарбоната натрия (20 мл), водой (4×20 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и упаривали под вакуумом. Остаток хроматографировали на колонке, содержащей 15 г силикагеля. Вещество элюировали системой 2% этанол/дихлорметан. Выход на 3 стадии 406 мг (60%) в виде белой пены. Rf0.35 в системе 5% этанол/дихлорметан. 1H-ЯМР (CDCl3): 9.31 ус (1H, NHBz), 8.73 с (1Н, Н-8), 8.30 с (1Н, Н-2), 8.12-7.22 м (22Н, Ph), 6.81 д (2Н, 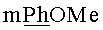
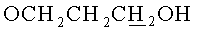
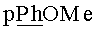
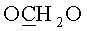
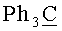
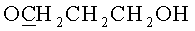
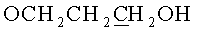
ж) N6-Бензоил-9-{2-O-[(3-(2-цианоэтил-N,N-диизопропиламидофосфинил)оксипропокси)метил]-3-O-бензоил-5-O-монометокситритил-β-D-рибофуранозил}аденин (7, B=AdeBz, R=MMTr, n=3).
К раствору 400 мг (0.48 ммоль) N6-Бензоил-9-{2-O-[(3-гидроксипропокси)метил]-3-O-бензоил-5-O-монометокситритил-β-D-рибофуранозил}аденина и 37 мг (0.53 ммоль) 1Н-тетразола в 6 мл смеси ацетонитрил/пиридин (5/1 по объему) прикапывали 0.229 мл (0.72 ммоль) 2-цианоэтил-N,N,N′,N′-тетраизопропилфосфорамидита. Реакцию проводили в инертной атмосфере азота. Через 30 мин добавляли 10% водный раствор гидрокарбоната натрия (3 мл), перемешивали 10 мин. Добавляли раствор гидрокарбоната натрия (20 мл) и этилацетат (20 мл). Органический слой отделяли, промывали водой (2×20 мл), сушили безводным сульфатом натрия, фильтровали, упаривали досуха. Остаток хроматографировали на колонке, содержащей 15 г силикагеля. Вещество элюировали системой этилацетат/гексан/триэтиламин (69/29/2). Фракции, содержащие продукт, объединяли, упаривали в вакууме досуха. Выход 303 мг (61% пена). Rƒ0.35 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (CDCl3): 150.68, 149.31.
Пример 11. Синтез N6-Бензоил-9-{2-O-[(2-(2-цианоэтил-N,N-диизопропиламидофосфинил)оксиэтокси)метил]-3-O-бензоил-5-O-монометокситритил-β-D-рибофуранозил}аденина (7, B=AdeBz, R=MMTr, n=2).
Вещество было получено аналогично примеру 10 с суммарным выходом 9.8%. Rƒ0.34 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (DMSO-D6): 150.18, 150.14.
Пример 12. Синтез 1-{2-O-[(2-(2-цианоэтил-N,N- диизопропиламидофосфинил)оксиэтокси)метил]-3-O-бензоил-5-O-монометокситритил-β-D-рибофуранозил}урацила (7, B=Ura, R=MMTr, n=2).
Вещество было получено аналогично примеру 10 с суммарным выходом 16%. Rƒ0.71 в системе этилацетат/гексан/триэтиламин (69/29.5/1.5). 31Р-ЯМР (DMSO-D6): 150.17, 150.12.
Пример 13. Синтез 1-{2-O-[(2-(2-цианоэтил-N,N-диизопропиламидофосфинил)оксиэтокси)метил]-3-O-бензоил-5-O-диметокситритил-β-D-рибофуранозил}урацила (7, B=Ura, R=DMTr, n=2).
Вещество было получено аналогично примеру 10 с суммарным выходом 14%. Rƒ0.74 в системе этилацетат/гексан/триэтиламин (69/29.5/1.5). 31Р-ЯМР (DMSO-D6): 150.15, 150.09.
Пример 14. Синтез N6-Бензоил-9-{2-O-[(4-(2-цианоэтил-N,N-диизопропиламидофосфинил)оксибутокси)метил]-3-O-бензоил-5-O-монометокситритил-β-D-рибофуранозил}аденина (7, B=AdeBz, R=MMTr, n=4).
Вещество было получено аналогично примеру 10 с суммарным выходом 8%. Rƒ0.50 в системе этилацетат/гексан/триэтиламин (69/29.5/1.5). 31Р-ЯМР (DMSO-D6): 149.10.
Пример 15. Синтез N4-Бензоил-1-{2-O-[(3-левулинилоксипропокси)метил]-3-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-5-O-монометокситритил-β-D-рибофуранозил}цитозина (15, B=CytBz, R=MMTr, n=3. Фиг. 10).
а) N4-Бензоил-1-{2-O-[(3-левулинилоксипропокси)метил]-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}цитозин.
К раствору, содержащему 0.80 г (1.18 ммоль) N4-Бензоил-1-{2-O-[(3-гидроксипропокси)метил]-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}цитозина (G.V. Bobkov et al. Tetrahedron, 64, 6238-6251, 2008) и 0.27 г (2.36 ммоль) левулиновой кислоты в 15 мл диоксана при 20°C добавляли 11 мг (0.091 ммоль) Ν,Ν-диметиламинопиридина (ДМАП) и 0.48 г (2.36 ммоль) Ν,Ν-дициклогексилкарбодиимида. Раствор выдержали в течение 1.5 ч. при 20°C. Выпавший белый осадок Ν,Ν′-дициклогексилмочевины отфильтровывали. Органический слой отделяли, промывали водой (2×50 мл), сушили над безводным сульфатом натрия, фильтровали и упаривали под вакуумом. Остаток хроматографировали на колонке, содержащей 30 г силикагеля. Вещество элюировали системой 1.5% этанол/дихлорметан. Выход 0.75 г (82%) в виде белой пены. Rƒ0.43 в системе 5% этанол/дихлорметан. 1H-ЯМР (CDCl3): 10.20 ус (1Н, NHBz), 8.34 д (1H, J6,5=7.5, Н-6), 7.97-7.45 м (6Н, Bz, Н-5), 5.83 с (1Н, Н-1′), 5.07 д (1H, J=-6.5,
б) N4-Бензоил-1-{2-O-[(3-левулинилоксипропокси)метил]-β-D-рибофуранозил}цитозин.
0.75 г (0.98 ммоль) N4-Бензоил-1-{2-O-[(3-левулинилоксипропокси)метил]-3,5-O-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-рибофуранозил}цитозина обрабатывали 6 мл 0.1 Μ раствора тригидрата фторида тетрабутиламмония в тетрагидрофуране и выдерживали 10 мин при 20°C. Реакционную смесь упаривали под вакуумом, переупаривали с хлороформом (2×10 мл). Остаток хроматографировали на колонке, содержащей 30 г силикагеля. Вещество элюировали системой 5% этанол/дихлорметан. Выход 0.37 г (71%) в виде белой аморфной пены. Rƒ0.20 в системе 5% этанол/дихлорметан. 1H-ЯМР (CDCl3): 10.20 ус (1Н, NHBz), 8.46 д (1H, J6,5=7.5, Н-6), 7.93-7.44 м (6Н, Bz, Н-5), 5.90 д (1Н, J1′2′=2.5, Н-1′), 5.00 д (1H, J=-6.5,
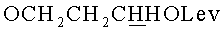
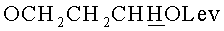
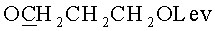
в) N4-Бензоил-1-{2-O-[(3-левулинилоксипропокси)метил]-5-O-монометокситритил-β-D-рибофуранозил}цитозин.
Раствор 0.37 г (0.69 ммоль) N4-Бензоил-1-{2-O-[(3-левулинилоксипропокси)метил]-β-D-рибофуранозил}цитозина и 0.36 мг (1.17 ммоль) монометокситритилхлорида в сухом пиридине (10 мл) выдерживали в течение 16 ч. при 20°C. К реакционной смеси добавляли метанол (2 мл) и выдерживали в течение 30 мин при комнатной температуре, после чего упаривали. Остаток разбавляли дихлорметаном (20 мл) и промывали последовательно насыщенным водным раствором бикарбоната натрия (20 мл) и водой (20 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали, упаривали под вакуумом. Остаток хроматографировали на колонке, содержащей 15 г силикагеля. Вещество элюировали системой 2% этанол/дихлорметан. Выход 0.5 г (90%) в виде белой пены. Rƒ0.41 в системе 5% этанол/дихлорметан. 1Н-ЯМР (CDCl3): 10.20 ус (1H, NHBz), 8.47 д (1Н, J6,5=7.5, Н-6), 8.00-7.10 м (17Н, Bz, Н-5, MMTr), 6.86 д (2Н, J=9.0, m-PhOMe), 5.99 с (1Н, Н-1′), 5.15 д (1H, J=-6.5,
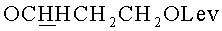
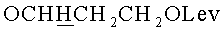
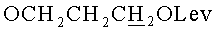
г) N4-Бензоил-1- {2-O-[(3-левулинилоксипропокси)метил]-3-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-5-O-монометокситритил-β-D-рибофуранозил}цитозин (15, B=CytBz, R1=MMTr, n=3).
К раствору 500 мг (0.62 ммоль) N4-Бензоил-1-{2-O-[(3-левулинилоксипропокси)метил]-5-O-монометокситритил-β-D-рибофуранозил}цитозина в 10 мл дихлорэтана, добавляли диизопропилэтиламин (0.32 мл, 1.86 ммоль) и 2-цианоэтил-N,N-диизопропилхлорофосфорамидит (0.28 мл, 1.24 ммоль). Через 2 часа добавляли 10% водный раствор гидрокарбоната натрия (3 мл), перемешивали 10 мин. Добавляли раствор гидрокарбоната натрия (20 мл) и дихлорметан (20 мл). Органический слой отделяли, промывали водой (2×20 мл), сушили безводным сульфатом натрия, фильтровали, упаривали досуха. Остаток хроматографировали на колонке, содержащей 15 г силикагеля. Вещество элюировали системой этилацетат/гексан/триэтиламин (69/29/2). Фракции, содержащие продукт, объединяли, упаривали в вакууме досуха. Выход 405 мг (65% пена). Rƒ0.54 в системе дихлорметан/метанол/гексан (9:1:10). 31Р-ЯМР (DMSO-d6): 152.48, 151.03.
Пример 16. Синтез N6-Бензоил-9-[2-O-(2,3-ди-O-бензоил-5-O-левулинил-β-D-рибофуранозил)-3-O-(2-цианоэтил-N,N-диизопропиламидофосфинил)-5-O-диметокситритил-β-D-рибофуранозил]аденина (16, B=AdeBz).
Вещество было получено согласно литературным данным (S.N. Mikhailov et al. Chemistry & Biodiversity, 2, 1153-1163, 2005). Белый порошок. Rƒ0.43 в системе гексан/ацетон/триэтиламин (49/49/2). 31Р-ЯМР: 150.94, 150.81.
Пример 17. Синтез N6-Бензоил-9-{2-дезокси-2-[2-(2-цианоэтил-N,N-диизопропиламидофосфинил)оксиэтил-1-карбоксамидо]-3-O-трет-бутилдиметилсилил-5-О-диметокситритил-β-D-рибофуранозил}аденина (9, B=AdeBz, R=DMTr, n=2).
Получали из 2′-амино-2′-дезоксиаденозина. Rƒ0.31 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (CDCl3): 150.63, 149.29.
Пример 18. Синтез N2-Изобутироил-9-{2-дезокси-2-[2-(2-цианоэтил-N,N-диизопропиламидофосфинил)оксиэтил-1-карбоксамидо]-3-О-трет-бутилдиметилсилил-5-O-диметокситритил-β-D-рибофуранозил}гуанина (9, B=GuaiBu, R=DMTr, n=2) Получали из 2′-дезокси-2′-аминогуанозина. Rƒ0.30 в системе этилацетат/гексан/триэтиламин (69/29/2). 31Р-ЯМР (CDCl3): 150.40, 149.10.
Пример 19. Синтез олигомерного миметика ПАР C7H16NO2-(5′′pAraAdo5′)11 (Фиг. 11)
Твердофазный синтез олигомера проводили на автоматическом синтезаторе ABI 3400 (Applied Biosystems, USA) с использованием в качестве твердого носителя стекла с контролируемым размером пор (500Å) с ковалентно пришитым защищенным аминолинкером С-7. Температурный режим синтеза - 20°C. Масштаб синтеза составлял 0,2 мкмоль. Использовалась стандартная программа синтеза. Цикл присоединения одного мономерного звена состоял из 4 стадий (Фиг. 11):
1. Конденсация (присоединение амидофосфитного блока, растворенного в ацетонитриле или дихлорметане в концентрации 0.5-0.05М, в присутствии 1H-тетразола), осуществляли с использованием амидофосфита дисахаридного нуклеозида 3 при 20°C (стадия i). Время конденсации устанавливалось равным 300 сек.
2. Кэпирование (стадия ii - ацилирование непрореагировавших гидроксильных групп смесью пропионового ангидрида и пиридина в соотношении от 1:1 до 1:3 в тетрагидрофуране) - 20-60 сек при 20°C.
4. Окисление 0.05-0.1 Μ раствором иода в смеси тетрагидрофуран-пиридин-вода (стадия iii - превращение межнуклеотидного фосфита в фосфотриэфир, Х=O)-15-20 сек при 20°C. Фосфордитиоаты олигонуклеотидов (стадия iii, X=S) получают по реакции межнуклеотидных фосфитов с 3-((диметиламинометилен)амино)-3Н-1,2,4-дитиазол-3-тионом в смеси тетрагидрофуран-пиридин (концентрация сульфирующего реагента 0.05-0.1М, время реакции - 360-600 сек при 20°C).
5. Деблокирование (стадия iv - удаление тритильной защитной группы с 5' гидроксильной группы олигонуклеотидной цепи 2-5% раствором ди- или трихлоруксусной кислоты) - 60 сек при 20°C. Удаление монометокситритильной защиты (MMTr) перед стадией конденсации производилось в ручном режиме с использованием функции ′′Hold′′.
Последовательное присоединение звеньев проводили, повторяя стадии i-iv (Фиг 11). Последнюю MMTr группу не удаляли. Удаление ацетильных защитных групп и отщепление олигомера с носителя проводили в стандартных условиях (конц. водный аммиак, 6 часов, 55°C, стадия v, AraAdo - обозначение дисахаридного нуклеозида, Tfa-трифторацетил, CEt - β-цианоэтил, pr (сокр. protecting groups)-защитные группы, подлежащие удалению). Частично деблокированный олигомер выделяли с помощью обращенно-фазной ВЭЖХ (Hypersil С18, 5mkm, 4.6×250 mm) в 0.05 Μ ТЕАА и градиенте ацетонитрила (10-50% за 30 мин). После удаления MMTr защитной группы 80% водной уксусной кислотой в течение 30 мин при комнатной температуре (стадия vi) проводили повторную очистку олигомера в указанном буфере и градиенте ацетонитрила от 0 до 25% (30 мин). Выход олигомера после выделения составил 20 о.е. (260 нм).
Пример 20. Синтез олигомерного миметика ПАР Эозин-C7H16NO2-(5′′pAraAdo5′)11.
Олигомер C7H16NO2-(5′′pAraAdo5′)11 (2 о.е.) растворяли в 50 мкл 0.1 Μ Na-бикарбонатного буфера (рН 9) и добавляли при 0°C 50 мкл раствора сукцинимидного эфира эозина (1 мг) в безводном ДМФА. Смесь оставляли на 16 ч при 4°C. Затем доводили объем раствора до 1 мл и экстрагировали непрореагировавший краситель бутанолом до конечного объема водной фазы 100 мкл. Олигомер высаживали 2% LiClO4 в ацетоне. Конъюгат выделяли с помощью обращеннофазной ВЭЖХ (Hypersil С18, 5mkm, 4.6×250 mm) в 0.05 Μ ТЕАА и градиенте ацетонитрила (10-50% за 30 мин). Выход олигомера после выделения составил 1 о.е. (260 нм).
Пример 21. Синтез разветвленного олигомерного миметика ПАР 3′-dAdo(5′′pAraAdo5′)5-X-[(5′′pAraAdo5′)5]2, Х = несимметричный разветвитель).
Синтез олигомера проводили по стандартной схеме, как описано в примере 19, с незначительными изменениями. В качестве твердого носителя использовали стекло с контролируемым размером пор (1000Å) и ковалентно пришитым защищенным дезоксиаденозином. Время конденсации амидофосфита несимметричного разветвителя устанавливали равным 15 мин. По завершении синтеза основной ветви олигомера, MMTr защитную группу удаляли и ацилировали свободную гидроксильную группу кэпирующей смесью 30 мин при 20°C. Затем удаляли Fmoc защиту разветвителя 1М DBU в безводном MeCN 5 мин при комнатной температуре. Затем проводили синтез боковой цепи олигомера. Выход олигомера после выделения составил 10 о.е. (260 нм).
Пример 22. Активация ПАРП-1 миметиками ПАР.
К 40 мкл раствора НАД+ в ПАРП-буфере (50 мМ Трис-HCl (рН 8.0), 2 мМ MgCl2) добавляли 20 мкл миметика ПАР (1-10 мкМ) или активированной ДНК, 20 мкл воды или раствора ингибитора (при использовании ингибитора в качестве контроля), 1 мкл ПАРП-1 в 19 мкл ПАРП-буфера и инкубировали при 20°C в течении 30 мин. После инкубации добавляли по 40 мкл растворов 2М КОН и ацетофенона (20% в спирте), встряхивали и выдержали 10 мин при 4°C. Затем добавляли 180 мкл 88% раствора муравьиной кислоты, перемешивали на вортексе и выдержали 10 мин при температуре 110°C. Разбавляли полученную смесь до 1500 мкл ПАРП-буфером и измеряли флуоресценцию раствора при 440 нм. Флуоресценция раствора в отсутствии ингибитора ПАРП-1 соответствовала минимальному содержанию остаточного НАД+ и максимальной активности фермента. Флуоресценция раствора, не содержавшего фермента, ингибитора и активированной ДНК соответствует максимальному содержанию НАД+ и нулевой активности фермента. Для целей калибровки проводили измерение флуоресценции растворов с содержанием НАД+, в 2, 10 и 100 раз меньшим, чем указано выше. Специфическая активность ПАРП-1 составляла не менее 600 ед/мг. Одна единица ПАРП-1 представляет собой количество фермента, необходимое для синтеза 1 нмоль поли(АDР-рибозы) в минуту при 25°C и рН 7.5. Концентрация фермента составляла 1 мг/мл в соответствии с сертификатом продукции.

| название | год | авторы | номер документа |
|---|---|---|---|
| СРЕДСТВО ДЛЯ ИНГИБИРОВАНИЯ ФЕРМЕНТА ПОЛИ(АДФ-РИБОЗО)ПОЛИМЕРАЗЫ-1 ЧЕЛОВЕКА | 2009 |
|
RU2411948C1 |
| БЕТА-L-2' ДЕЗОКСИНУКЛЕОЗИДЫ ДЛЯ ЛЕЧЕНИЯ ГЕПАТИТА В | 1999 |
|
RU2424016C2 |
| Средство для ингибирования фермента тирозил-ДНК-фосфодиэстеразы 1 человека на основе производных пентафуранозилнуклеозидов | 2019 |
|
RU2748103C1 |
| β-L-2'-ДЕЗОКСИНУКЛЕОЗИДЫ ДЛЯ ЛЕЧЕНИЯ ГЕПАТИТА В | 1999 |
|
RU2300381C2 |
| 3'-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ НУКЛЕОЗИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ | 1995 |
|
RU2130029C1 |
| НОВЫЕ БИЦИКЛИЧЕСКИЕ НУКЛЕОЗИДЫ И ОЛИГОМЕРЫ, ПОЛУЧЕННЫЕ ИЗ НИХ | 2017 |
|
RU2824141C2 |
| L-НУКЛЕОЗИДЫ, ОБЛАДАЮЩИЕ АНТИ-HBV ИЛИ АНТИ-EBV АКТИВНОСТЬЮ, СПОСОБ ИНГИБИРОВАНИЯ HBV ИЛИ EBV ИНФЕКЦИИ | 1995 |
|
RU2171809C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛЗАМЕЩЕННЫХ 2-ДЕЗОКСИ-2-ФТОР-D-РИБОФУРАНОЗИЛ-ПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2407747C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ДЕЗОКСИ-2', 2'-ДИФТОРЦИТИДИНА | 2005 |
|
RU2360919C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ФТОР-2'-АЛКИЛЗАМЕЩЕННЫХ ИЛИ ДРУГИХ ЗАМЕЩЕННЫХ РИБОФУРАНОЗИЛПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2433124C2 |
Настоящее изобретение относится к миметикам ПАР и способу их получения для создания новых медицинских препаратов общей формулы

где Y - остаток нуклеозида, аминопроизводного алифатического соединения, флуоресцентного красителя; Z - остаток нуклеозида, (k+1)·m=1-200; X является О или S; R1 и R2 являются остатком дисахаридного нуклеозида или остаток формул




где n=2-6; 2-6 или 1-4 соответственно, N=остаток нуклеозида, или -((CH2)nO)m-(P=X(OH))O-N-, где n=2-6, m=1-6, R3 представляет собой разветвитель формулы

где N′- остаток дисахаридного нуклеозида, n число до 100, или остаток формул
 n=2-6; или
n=2-6; или  n=2-6; или
n=2-6; или  n=1-4
n=1-4
где В=аденин-9-ил, урацил-1-ил, цитозин-1-ил или гуанин-9-ил. Предложены новые миметики ПАР, сохраняющие все основные функциональные группы и расстояния между ними, необходимые для взаимодействий с белками, но модифицированные для облегчения их синтеза и увеличения стабильности. 2 н.п. ф-лы, 21 пр., 2 табл., 10 ил.
1. Миметики ПАР общей формулы
где Y представляет собой остаток нуклеозида, такого как дезоксиаденозин, дезоксигуанозин, аденозин, гуанозин, цитидин, уридин; аминопроизводного линейного или разветвленного алифатического соединения С7, такого как 6-амино-(2-гидроксиметил)-гексан-1-ил, флуоресцентного красителя, такого как флуоресцеин или эозин,
где Z представляет собой остаток нуклеозида, такого как дезоксиаденозин, дезоксигуанозин, аденозин, гуанозин, цитидин, уридин, необязательно с присоединенным по 5′-гидроксильной группе остатком монометокситритила или диметокситритила,
(k+1)·m=1-200; X представляет собой О или S;
R1 и R2 представляют собой остаток дисахаридного нуклеозида, который является нуклеозидом с присоединенным дополнительным углеводным остатком, соединенным с пентафуранозным циклом нуклеозида О-гликозидной связью, или остаток формул
n=2-6; или n=2-6; или n=1-4 или -((CH2)nO)m-(P=X(OH))O-N-, где n=2-6, m=1-6, или
где N=остаток нуклеозида, такого как дезоксиаденозин, дезоксигуанозин, аденозин, гуанозин, цитидин, уридин,
R3 представляет собой разветвитель где N′- остаток дисахаридного нуклеозида; n - число мономерных звеньев со степенью полимеризации до 100 мономерных звеньев, или остаток одной из формул
n=2-6; или n=2-6; или n=1-4
где В=аденин-9-ил, В=урацил-1-ил, В=цитозин-1-ил, В=гуанин-9-ил.
2. Способ получения миметиков ПАР, охарактеризованных в п. 1, на нерастворимом силикатном, полистирольном или полиакрилатном носителе при температуре 20-40°C с последовательным в ходе синтеза присоединением амидофосфитных мономерных блоков 1-16 до получения полимера необходимой длины:
где R=MMTr, DMTr, В=N6-бензоиладенин-9-ил, N2-изобутироилгуанин-9-ил, N4-бензоилцитозин-1-ил, урацил-1-ил, n=2-6 для амидофосфитов 7-8, n=1-4 для амидофосфита 9, в ручном или автоматическом режиме, где каждый реакционный цикл включает в себя: 1) деблокирование 2-5% раствором ди- или трихлоруксусной кислоты в дихлорметане, 2) стадию конденсации с использованием 0,05-0,5 М раствора амидофосфита в ацетонитриле или дихлорметане; 3) ацилирование непрореагировавших гидроксильных групп смесью пропионового ангидрида и пиридина в соотношении от 1:1 до 1:3 в тетрагидрофуране; 4) окисление полученного на предыдущей стадии межнуклеотидного фосфита в фосфотриэфир 0,05-0,1 М раствором иода в смеси тетрагидрофуран-пиридин-вода; с последующим деблокированием полученного миметика в стандартных условиях водным аммиаком и очисткой продукта с использованием обращенно-фазной ВЭЖХ.
| Michailov Sergey N et al, Tetrahedron, 2008, 64 (12), 2871-2876 | |||
| СРЕДСТВО ДЛЯ ИНГИБИРОВАНИЯ ФЕРМЕНТА ПОЛИ(АДФ-РИБОЗО)ПОЛИМЕРАЗЫ-1 ЧЕЛОВЕКА | 2009 |
|
RU2411948C1 |
| Kulikova Irina V | |||
| et al, Collection Symposium Series, 2008, 155-158 | |||
| Mikhailova E.S | |||
| et al, Doklady Akademii Nauk SSSR, 1966, 167(2), 444-447 | |||
| СПОСОБ РАБОТЫ ИГРОВОГО АВТОМАТА | 2003 |
|
RU2260852C2 |
| Nauwelaerts Koen et al, Nucleic Acids Research, 2003, 31(23), 6758-6769 | |||
| US 2002014994 | |||

