Описание
Настоящее изобретение относится к химическому классу пери-замещенных бициклических ацилсульфонамидов, которые полезны для лечения и профилактики окклюзионного поражения артерий и родственных простагландин-опосредованных расстройств.
Атеросклероз представляет собой патологию, которая лежит в основе ряда заболеваний, характеризующихся наивысшей смертностью, таких как инфаркт миокарда и периферийное окклюзионное поражение артерий (PAOD). PAOD представляет собой атеросклероз больших и средних артерий конечностей, в частности нижних конечностей, а также затрагивает аорту и подвздошные артерии. Это заболевание зачастую протекает одновременно с поражением коронарной артерии и цереброваскулярным расстройством. Пациенты с PAOD подвержены повышенному риску возникновения других сосудистых заболеваний, таких как инфаркт миокарда или инсульт [Waters, RE, Terjung RL, Peters KG & Annex BH. J. Appl. Physiol. 2004; Ouriel K. Lancet, 2001, 258:1257-64; Kroger, K. Angiology, 2004, 55:135-138]. Клинически значимые повреждения могут постепенно сужать периферийные артерии, что приводит к возникновению боли при ходьбе, обычно снижающейся в состоянии покоя (хромоте), ишемическим язвам, гангрене, а иногда и ампутации конечности. Терапевтическое лечение обычно неэффективно, но операции обвода или замены повреждения искусственными или венозными трансплантатами дистально улучшают кровоток, по меньшей мере, до тех пор, пока артерии снова не станут суженными [Haustein, K.O., Int. J. Clin. Pharmacol. Ther., 35:266 (1997)]. В последнее время в процессе генетического анализа групп сцепления человека было обнаружено, что изменения в ДНК гена PTGER3, который кодирует простагландиновый E2 рецептор подтипа 3 (известный как EP3), повышают риск развития у индивидуума PAOD (см. публикацию патентной заявки США 2003/0157599). Таким образом, антагонисты простагландина E2 (PGE2), связывающиеся с EP3 рецептором, могут обеспечить эффективное лечение или профилактику PAOD.
В ответ на различные внеклеточные раздражители простагландины легко вырабатываются из свободной арахидоновой кислоты посредством последовательного действия циклооксигеназ и синтаз. Простагландины проявляют свое действие в непосредственной близости от сайта, где они синтезируются. До настоящего времени было клонировано и охарактеризовано восемь простаноидных рецепторов. Эти рецепторы являются представителями увеличивающегося класса G-белок-связывающих рецепторов. PGE2 связывается предпочтительно с рецепторами EP1, EP2, EP3 и EP4; PGD2 - с рецепторами DP и FP рецепторами; PGF2a - с рецепторами FP и EP3 рецепторами; PGI2 - с IP рецептором, и TXA2 - с TP рецептором. Было установлено, что PGE2, связывающийся с EP3 рецептором, играет ключевую роль в регулировании транспорта ионов, сокращении гладких мышц ЖК-тракта, секреции кислоты, сокращении матки в процессе оплодотворения и имплантации, при лихорадке (fever generation) и гипералгезии. EP3 рецептор был обнаружен во многих органах, таких как почки, желудочно-кишечный тракт, матка и мозг. В сердечно-сосудистой системе EP3 экспрессирован сосудистым эндотелием и гладкой мышцей, и, по меньшей мере, четыре изоформы EP3 экспрессированы на тромбоцитах человека [Paul, B.Z., B. Ashby. S.B. Sheth. Distribution of prostaglandin IP and EP receptor subtypes and isoforms in platelets and human umbilical artery smooth muscle cells. British Journal of Haematology, 1998. 102(5): p. 1204-11].
Простаноиды, действующие посредством специфических мембранных рецепторов, принадлежащих к надсемейству G-белок-связывающих рецепторов (GPCR), играют существенную роль в сосудистом гомеостазе, включая функцию регулирования тромбоцитов. Среди простаноидов тромбоксан А2 (ТхА2) является мощным стимулятором агрегации тромбоцитов, в то время как простагландин (PG)I2 ингибирует их активацию. С другой стороны, сообщалось, что простагландин Е2(PGE2) обладает двухфазным эффектом на ответную реакцию тромбоцитов, потенцируя их агрегацию при низких концентрациях и ингибируя ее при более высоких концентрациях. Было показано, что стимулирующие эффекты PGE2 на агрегацию тромбоцитов осуществляются, главным образом, через ЕР3 рецептор, один из четырех подтипов рецепторов, активируемых PGE2.
Локализованный синтез простагландидов в стенке артериального сосуда может играть важную роль в развитии атеросклероза. Несмотря на то, что в стенке здорового сосуда присутствует только СОХ-1, в атеросклеротической бляшке присутствуют как СОХ-1, так и СОХ-2 [Schonbeck, U., et al., Augmented expression of cyclooxygenase-2 in human atherosclerotic lesions. Am J Pathol, 1999. 155(4): p. 1281-91; Cipollone, F., et al., Overexpression of functionally coupled cyclooxygenase-2 and prostaglandin E synthase in symptomatic atherosclerotic plaques as a basis of PGE2-dependent plaque instability. Circulation, 2001. 104(8): p. 921-7]. Их повышенная экспрессия в сочетании с повышенной экспрессией прогландин-Е-синтазы может объяснять повышенное продуцирование PGE2, отмеченное выше. В организме генетически модифицированной мыши с недостатком рецепторов липопротеина низкой плотности (LDL-R) образование атеросклеротической бляшки может быть снижено лечением с помощью рофекоксиба, селективного ингибитора СОХ-2, через снижение продуцирования PGE2 и других простагландинов [Burleigh M.E., Babaev V.R., Oates J.A., Harris R.C., Gautam S., Riendeau D., Mamett L.J., Morrow J.D., Fazio S., Linton M.F. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation. 2002 Apr 16; 105(15):1816-23].
Было показано, что внутри атеросклеротической бляшки клетки гладкой мышцы экспрессируют ЕР3 рецептор, и PGE2 стимулирует их пролиферацию и миграцию, что является показателем формирования атеросклеротической бляшки [Blindt R., Bosserhoff A.K., vom Dahl J., Hanrath P., Schror K., Hohlfeld T., Meyer-Kirchrath J. Activation of IP and EP(3) receptors alters cAMP-dependent cell migration. Eur. J. Pharmacol. 2002 May 24; 444 (1-2): 31-7]. Следовательно, можно смело предположить, что хронически воспаленные сосуды продуцируют PGE2 в количестве, достаточном для активации EP3 рецепторов на клетках сосудов гладких мышц (способствуя формированию атеросклеротического повреждения) и на тромбоцитах (способствуя тромбозу). Локально продуцированный PGE2 (непосредственно из тромбоцитов, из компонентов стенки сосуда и из воспалительных клеток) усиливает агрегацию тромбоцитов с помощью субоптимальных количеств протромботических тканевых факторов, которые сами по себе не могут вызывать их агрегацию в процессе сенсибилизации протеинкиназы C. Внутриклеточные события, инициированные активацией EP3 рецептора, могут повышать агрегацию тромбоцитов, противодействуя эффекту PGI2 и повышая эффекты агентов, вызывающих первичную агрегацию, таких как коллаген. Таким образом, активация EP3 рецептора может способствовать развитию атеросклероза и риску развития тромбоза, наблюдаемого при патологических состояниях, таких как васкулит и PAOD.
Применяемые в настоящее время способы лечения РАOD направлены на снижения риска болезненного проявления сердечно-сосудистых расстройств, таких как инфаркт миокарда и инсульт, или обеспечивают симптоматическое облегчение хромоты. Все эти способы лечения действуют на функции тробмоцитов. Способы лечения, снижающие риск болезненного проявления сердечно-сосудистых расстройств, включают введение аспирина в низких дозах (достаточных для снижения агрегации тромбоцитов, но позволяющих стенке сосуда продуцировать PGE2) и введение ингибиторов аденозиндифосфатного рецептора тромбоцитов (клопидогрел). Связывание аденозиндифосфата с тромбоцитным аденозиндифосфатным рецептором вызывает снижение тромбоцитной сАМР с последующей активацией и агрегацией тромбоцитов. Способы лечения, обеспечивающие симптоматическое облегчение хромоты, включают ингибиторы тромбоцитной фосфодиэстеразы 3 типа, такие как цилостазол, который вызывает повышение внутриклеточных уровней сАМР. Ингибиторы тромбоцитного аденозиндифосфатного рецептора или тромбоцитной фосфодиэстеразы 3 типа прямо или косвенно вызывают повышение содержания сАМР в тромбоцитах, ингибируя, таким образом, активацию и последующую агрегацию тромбоцитов с образованием тромбов. PGE2 связывание с ЕР3 снижает сАМР, поэтому ожидается, что антагонист PGE2 связывания с ЕР3 рецептором, противодействуя PGE2-зависимому снижению сАМР, необходимому для индуцирования активации тромбоцитов и последующей агрегации, или противодействуя PGE2-зависимому снижению клеточной сАМР сосудов гладких мышц, необходимому для стимулирования миграции, может обеспечить полезное терапевтическое действие при PAOD. Такой антагонист может также модифицировать заболевание ингибированием или снижением образования бляшек.
Таким образом, простагландины вовлечены в широкий спектр болезненных состояний, включая боль, лихорадку или воспаление, связанное с ревматической атакой, грипп или другие вирусные инфекции, насморк, поясничную боль и боль в области шеи, скелетную боль, послеродовую боль, дисменорею, головную боль, мигрень, зубную боль, боль, связанную с растяжениями и деформациями связок, миозит, невралгию, синовит, артрит, включая ревматоидный артрит, дегенеративный артроз (остеоартрит), подагру и анкилозирующий спондилит, бурсит, ожоги, включая радиационные (лучевые) и коррозивные химические повреждения, а также солнечные ожоги, боль после хирургических операций и стоматологических процедур, иммунные и аутоиммунные заболевания; перерождение нормальных клеток в клетки опухоли или метастатический рост опухоли; диабетическую ретинопатию, ангиогенез опухоли; простаноид-индуцированное сокращение гладких мышц, связанное с дисменореей, преждевременными родами, астмой или расстройствами, связанными с эозинофильными гранулоцитами; болезнь Альцгеймера; глаукому; потерю костной массы; остеопороз; болезнь Педжета; пептические язвы, гастрит, региональный энтерит (болезнь Крона), неспецифический язвенный колит, дивертикулит или другие желудочно-кишечные расстройства; желудочно-кишечное кровотечение; расстройства, связанные в нарушением коагуляции, выбранные из гипопротромбинемии, гемофилии и других расстройств, ассоциированных с кровотечением; и заболевание почек.
Хотя уровни содержания циркулирующих простагландинов в здоровых организмах являются предельно низкими [FitzGerald G.A., Brash A.R., Falardeau P. & OatesJ A. JCI 1981 68:12472-1275], местная концентрация PGE2 может резко повышаться в областях воспаления. Например, было показано, что местное продуцирование PGE2 in vitro повышается более чем в 30 раз при окклюзионном поражении аорты [Reilly J., Miralles M., Wester W. & Sicard G. Surgery, 1999, 126:624-628]. Следовательно, можно смело предположить, что хронически воспаленные сосуды продуцируют достаточные количества PGE2 для активации ЕР3 рецепторов на тромбоцитах. В такой среде внутриклеточные события, инициированные активацией ЕР3 рецептора, могут повышать агрегацию тромбоцитов противодействием эффекту PGI2 и повышением эффектов первичных ферментов, вызывающих агрегацию, таких как ADP. Таким образом, активация ЕР3 рецептора может способствовать тромбозу, наблюдаемому при патологических состояниях, таких как васкулит и атеросклероз. Периферийное окклюзионное поражение артерии (PAOD) представляет собой атеросклеротическое заболевание, которое встречается, прежде всего, у пожилых людей вследствие окклюзии просвета периферийных артерий, главным образом бедренной артерии, и это связано с повышенным риском сосудистых событий, таких как инфаркт миокарда и инсульт [Waters, R.E., Terjung R.L., Peters K.G. & Annex B.H. J. Appl. Physiol. 2004; OurielK. Lancet, 2001, 258:1257-64; Kroger K. Angiology, 2004, 55:135-138]. Результаты некоторых клинических исследований показали, что лечение простагландинами улучшает симптомы PAOD [Reiter M, Bucek R, Stumpflen A & Minar E. Cochrane Database Syst. Rev. 2004, 1:CD000986; Bandiera G., Forletta M., Di Paola F.M., Cirielli С. Int. Angiol. 2003, 22:58-63; Matsui K., Ikeda U., Murakami Y., Yoshioka T., Shimada K. Am. Heart J. 2003, 145:330-333], подтверждая связь между PAOD и функцией простаноидного рецептора.
Орто-замещенные фенилацилсульфонамиды и их применение для лечения простагландин-опосредованных расстройств описаны в патенте США № 6242493 и двух публикациях: Juteau et al. BioOrg. Med. Chem. 9, 1977-1984 (2001); Gallant et al. BioOrg. Med. Chem. Let. 12, 2583-2586 (2002), содержание которых введено в данное описание в виде ссылок.
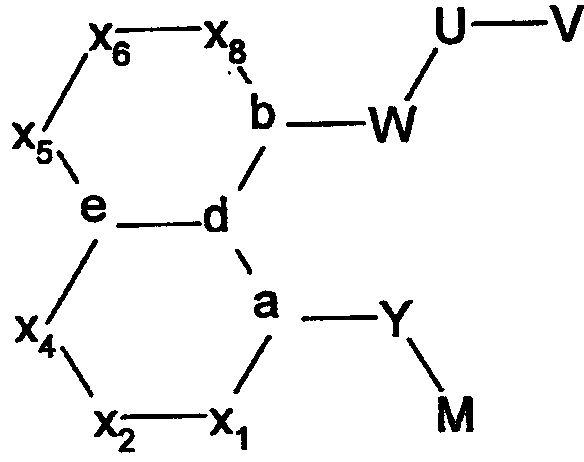
В соответствии с одним аспектом, изобретение относится к соединениям формулы
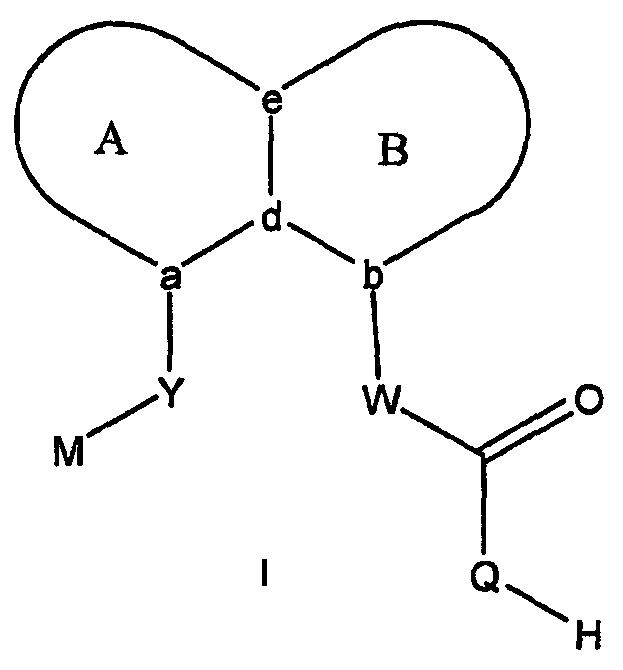
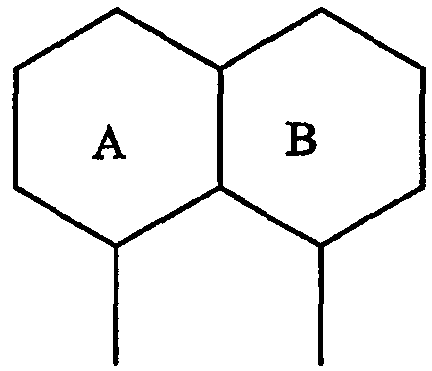
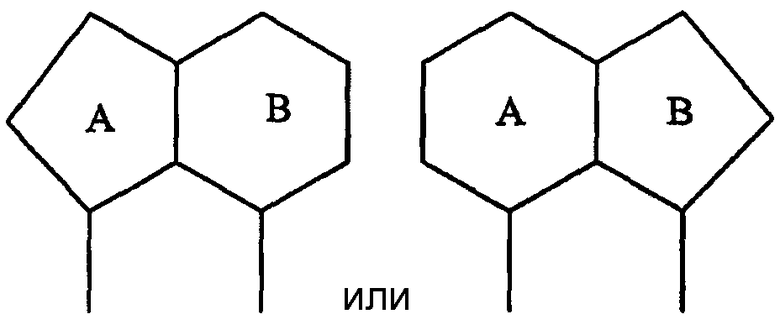
где А и В представляют собой два конденсированных 5-, 6- или 7-членных кольца. Конденсированная А/В кольцевая система может содержать от 0 до четырех гетероатомов, выбранных из атомов азота, кислорода и серы, и может быть дополнительно замещена от 0 до четырех заместителями, независимо выбранными из галогена, -ОН, низшего алкила, -О-низшего алкила, низшего фторалкила, -О-низшего фторалкила, метилендиокси, этилендиокси, алкокси-низшего алкила, гидрокси-низшего алкила, оксо, оксида, -CN, нитро, -S-низшего алкила, амино, низшего алкиламино, ди-низшего алкиламино, ди-низшего алкиламиноалкила, карбокси, карбоалкокси, ацила, ацилалкила, карбоксамидо, низшего алкилсульфоксида, ациламино, фенила, бензила, спиротиазолидинила, фенокси и бензилокси. Разветвления, обозначенные как «а» и «b», представляют собой точки присоединения остатков Y и W, соответственно, и именно точки «a» и «b» на конденсированной А/В кольцевой системе по отношению друг к другу находятся в пери-положении. Разветвления, обозначенные как «d» и «е», представляют собой точки конденсации кольца А и кольца B в конденсированной А/В кольцевой системе. Каждое из разветвлений а, b, d и е может представлять собой атом углерода или азота.
W и Y представляют собой связующие звенья, содержащие от нуля до 8 атомов в цепи.
М выбран из арила, замещенного арила, гетероциклила, замещенного гетероциклила, С6-С20алкила и замещенного С6-С20алкила.
В одной подгруппе соединений (Ia) Q выбран из -N(SО2R1)-, -N(COR1)-, -N[PO(O-алкил)2]-, -NHNR10(SO2Rl) и, когда W представляет собой -CF2- или -CH2CF2-, Q может дополнительно представлять собой -NH-; R1 выбран из арила, замещенного арила, гетероарила, замещенного гетероарила, С3-С20алкила и фторалкила; и R10 выбран из алкила, арила и гетероарила. В другой подгруппе (Ib) Q представляет собой -О-, и соединения представляют собой карбоновые кислоты. Формула изобретения, представленная ниже, относится к соединениям подгруппы (Ia). Формула изобретения связанной заявки, озаглавленной «Бициклические пери-замещенные карбоновые кислоты для лечения окклюзионного поражения артерий», поданной на регистрацию после подачи настоящей заявки, относится к подгруппе соединений Ib.
Другие родственные соединения, полезные для лечения окклюзионного поражения артерий и родственных простагландин-опосредованных расстройств, включают соединения формулы Ic:
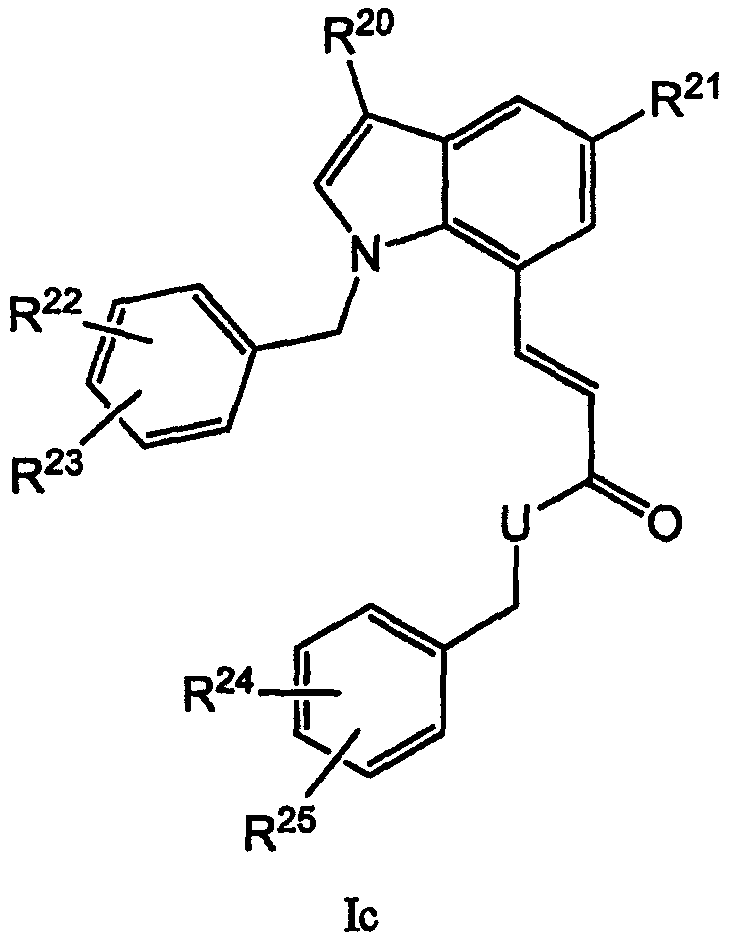
где U выбран из -О- и -NH-; и
R20-R25 независимо выбраны из водорода, галогена и метила.
В соответствии со вторым аспектом, изобретение относится к фармацевтическим препаратам, содержащим фармацевтически приемлемый носитель и соединение, как описано выше, или его сложный эфир, фармацевтически приемлемую соль или гидрат.
В соответствии с третьим аспектом, изобретение относится к способам лечения или профилактики простагландин-опосредованного заболевания или состояния. Способы включают введение млекопитающему терапевтически эффективного количества соединения, описанного в настоящем изобретении.
Заболевание или состояние может представлять собой, например, боль, лихорадку или воспаление, связанное с ревматизмом, гриппом или другими вирусными инфекциями, насморком, поясничную боль и боль в области шеи, скелетную боль, послеродовую боль, дисменорею, головную боль, мигрень, зубную боль, боль, связанную с растяжениями и деформациями связок, миозит, невралгию, синовит, артрит, включая ревматоидный артрит, дегенеративный артроз (остеоартрит), подагру и анкилозирующий спондилит, бурсит, ожоги, включая радиационные (лучевые) и коррозивные химические повреждения, а также солнечные ожоги, боль после хирургических операций и стоматологических процедур, иммунные и аутоиммунные заболевания. Соединения согласно настоящему изобретению являются антагонистами ЕР3, проникающие в ЦНС и особенно подходят для устранения боли.
Соединения согласно настоящему изобретению, которые ингибируют агрегацию тромбоцитов и повышают региональный кровоток, полезны для лечения первичной тромбоэмболии, тромбоза и окклюзионного поражения сосудов. Соединения могут преимущественно применяться в сочетании с другими ингибиторами агрегации тромбоцитов и с ингибиторами биосинтеза или поглощения холестерина. Соединения могут также успешно применяться в сочетании с ингибиторами циклооксигеназы-2 для лечения воспалительных состоянии.
Другими заболеваниями или состояниями, которые также могут быть подвергнуты лечению, являются, например, перерождение клеток с образованием опухоли или метастатический рост опухоли; диабетическая ретинопатия, ангиогенез опухоли; простаноид-индуцированное сокращение гладких мышц, связанное с дисменореей, преждевременными родами, астмой или расстройствами, связанными с эозинофильными гранулоцитами; болезнь Альцгеймера; глаукома; потеря костной массы; остеопороз или болезнь Педжета; пептические язвы, гастрит, региональный энтерит (болезнь Крона), дивертикулит или другие желудочно-кишечные расстройства; желудочно-кишечное кровотечение; нарушения коагуляции, выбранные из гипопротромбинемии, гемофилии и других расстройств, ассоциированных с кровотечением; и заболевание почек. Аспект изобретения, связанный со способом, включает также способы ускорения образования кости, цитопротекции и уменьшения бляшки при лечении склероза.
В соответствии с четвертым аспектом, настоящее изобретение относится к способам cкрининга селективных простаноидных рецепторов, в особенности ЕР3 лигандов. Способ скрининга может представлять собой cкрининг в условиях in vitro.
Соединения класса, представленного приведенными выше формулами Ia и Ic, являются антагонистами ЕР2 рецептора. Они полезны для лечения и профилактики простагландин-опосредованных состояний, которые описаны выше, в особенности таких состояний, как окклюзионное поражение сосудов.
Композиции согласно настоящему изобретению содержат эффективную дозу или терапевтически эффективное количество соединения, описанного выше, и может дополнительно содержать другие терапевтические средства, такие как ингибиторы агрегации тромбоцитов (тирофибан, дипиридамол, клопидогрел, тиклопидин и т.п.); ингибиторы HMG-CoA редуктазы (ловастатин, симвастатин, правастатин, росувастатин, мевастатин, аторвастатин, церивастатин, питавастатин, флувастатин и т.п.); и ингибиторы циклооксигеназы. Дополнительный перечень неограничивающих примеров антигиперлипидемических средств, которые могут применяться в сочетании с соединениями согласно настоящему изобретению, можно найти в колонках 5-6 патента США № 6498156, содержание которого введено в настоящее описание посредством ссылки. Предпочтительными ингибиторами циклооксигеназы-2 являются ингибиторы, которые селективны в отношении циклооксигеназы-2 относительно циклооксигеназы-1. Предпочтительные ингибиторы циклооксигеназы-2 включают рофекоксиб, мелоксикам, целекоксиб, эторикоксиб, лумиракоксиб, валдекоксиб, парекоксиб, цимикоксиб, диклофенак, сулиндак, этодолак, кеторалак, кетопрофен, пироксикам и LAS-34475, хотя изобретение не ограничено этими или другими известными ингибиторами циклооксигеназы-2.
Способы согласно изобретению в равной степени относятся к композициям и препаратам. Способы включают введение пациенту при необходимости лечения терапевтически эффективного количества пери-замещенного конденсированного А/В-циклического соединения согласно изобретению. Настоящее изобретение относится также к способам скрининга in vitro селективных агонистов и антагонистов простаноидных рецепторов. Простаноидные рецепторы включают ЕP1, EP2, EP3, EP4, соединение IP и FР рецепторы. Селективные ЕР3 лиганды представляют огромный интерес для способа, включающего контактирование меченого соединения согласно настоящему изобретению с клонированным ЕР3 рецептором человека и количественное измерение его замены тестируемым соединением.
Класс соединений согласно настоящему изобретению включает соединения формулы Ia:
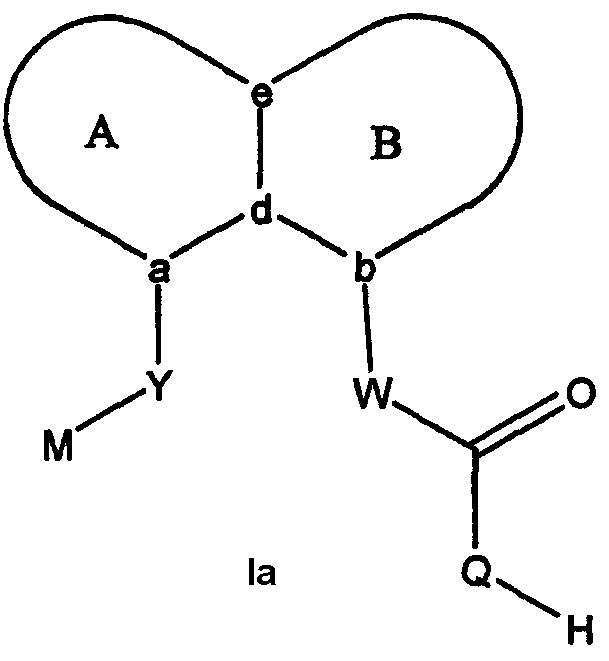
где Q выбран из -N(SO2R1)-, -N(COR1)- и -N[PO(O-алкил)2]- и, когда W представляет собой CF2-, Q дополнительно может представлять собой -NН-. Заместители на Q выбраны для придания водороду, к которому присоединен Q, кислотных свойств. В одном подклассе соединений Q представляет собой -N[PO(О-алкил)2]. В другом подклассе Q представляет собой -N(COR1)-. В третьем подклассе Q представляет собой -N(SО2Rl). R1 выбран из арила, замещенного арила, гетероарила, замещенного гетероарила и CF3. В одном варианте осуществления изобретения R1 выбран из фенила, замещенного фенила, 5-членного гетероарила, замещенного 5-членного кольцевого гетероарила и CF3.
Каждый из А и В независимо представляет собой 5-, 6- или 7-членное кольцо. Конденсированная А/В кольцевая система содержит от нуля до четырех гетероатомов, выбранных из атомов азота, кислорода и серы, и кольца дополнительно замещены от 0 до четырех заместителями. Подходящие заместители включают галоген, -ОН, низший алкил, -О-низший алкил, низший фторалкил, -О-низший фторалкил, метилендиокси, этилендиокси, алкокси-низший алкил, гидрокси-низший алкил, оксо, оксид, -CN, нитро, -S-низший алкил, амино, низший алкиламино, ди-низший алкиламино, ди-низший алкиламиноалкил, карбокси, карбоалкокси, сложные ортоэфиры, ацил, карбоксамидо, низший алкилсульфоксид, ациламино, фенил, бензил, спиротиазолидинил, фенокси и бензилокси. Поскольку конденсированная А/В кольцевая система может включать атом азота или серы, заместители могут включать оксиды, например N→O и S→O.
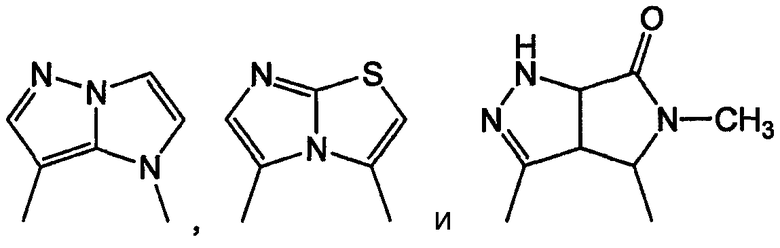
В одном подклассе А/В кольцевая система представляет собой пару конденсированных 5-членных колец:
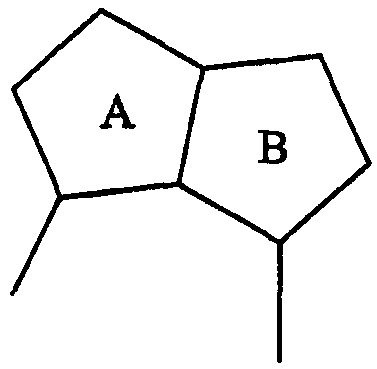
Примерами таких 5/5 кольцевых систем являются
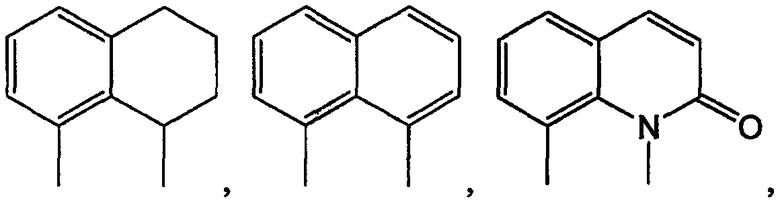
В еще одном подсемействе А/В кольцевая система представляет собой пару конденсированных 6-членных колец:
Примерами таких 6/6 кольцевых систем являются:
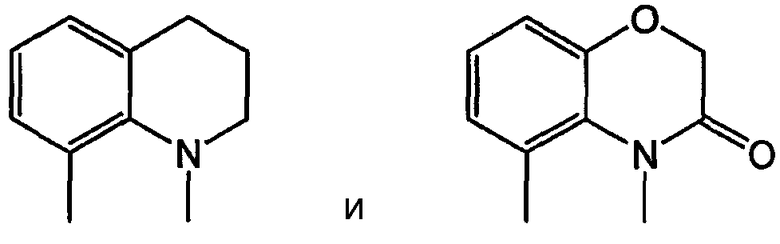
В еще одном подсемействе А/В кольцевая система представляет собой конденсированную пару 5- и 6-членного кольца
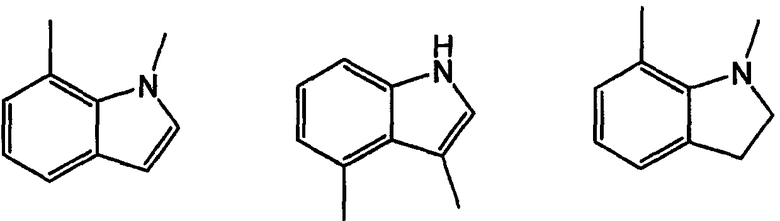
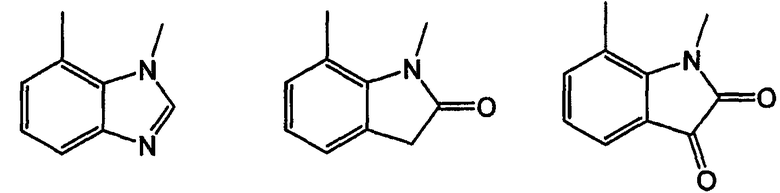
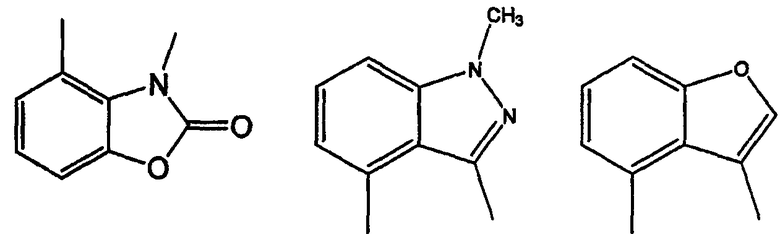
Примерами таких 5/6 кольцевых систем являются индолы, индолины, индолоны, изатины, бензимидазолы, бензоксазолиноны, бензофураны и индазолоны:
Как указано выше, кольцевые системы могут быть замещенными, например:
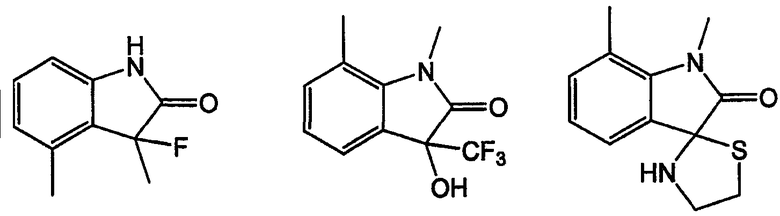
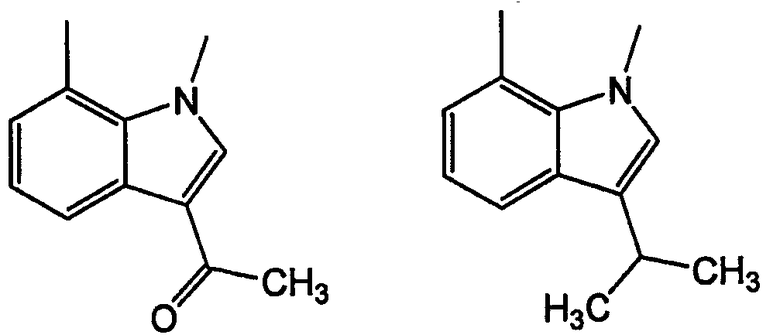
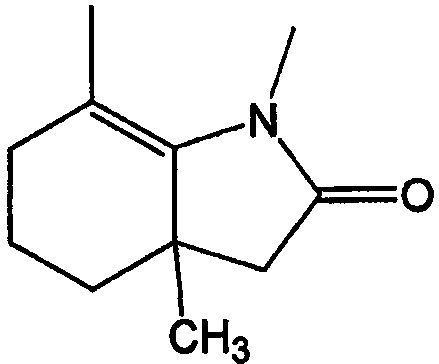
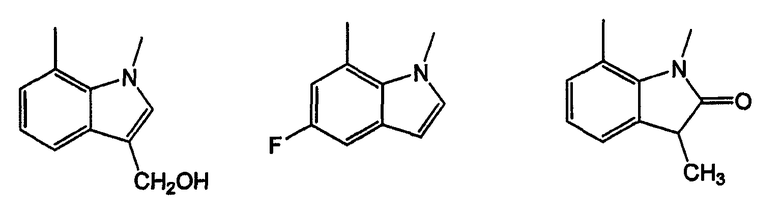
W и Y представляют собой мостиковые связи, содержащие от 0 до 8 атомов в цепи. Предпочтительно, они представляют собой С1-С8алкил, в котором один или оба -СН2- могут быть заменены -О-, -С(=О)-, -СН=СН-, -CF2-, -S-, -SO-, -SO2-, -NH- или -N(алкил)-. Более предпочтительно, W и Y представляют собой двухатомные цепи, т.е. С1 или С2алкил, в которых один или оба -СН2- могут быть заменены группами, указанными выше. В одном варианте осуществления изобретения W выбран из -СН2-СН2-, -ОСН2-, -С(=О)-, -CH2O-, -OCF2-, -OC(CH3)2-, -ОСН(СН3)-, -CH=CH-, -NHC(=O)- и -NHCH2-; и Y выбран из -CH2-, -O-, -OCH2-,=N-, -S-, -SO- и -SO2-. Левая связь показывает точку присоединения к кольцу А или В.
М выбран из арила, замещенного арила, гетероциклила, замещенного гетероциклила, C6-C20алкила, замещенного C6-C20алкила. В одном предпочтительном варианте осуществления изобретения М выбран из арила, замещенного арила, гетероциклила и замещенного гетероарила, более предпочтительно из фенила, замещенного фенила, нафтила, замещенного нафтила, гетероарила и замещенного гетероарила.
В одном варианте осуществления изобретения А/В кольцевая система представляет собой индол. В другом дополнительном варианте осуществления изобретения Q представляет собой -N(SО2R1)- и R1 выбран из фенила, замещенного фенила, 5-членного гетероарила, замещенного 5-членного гетероарила и CF3. В другом варианте осуществления изобретения М выбран из замещенного фенила, нафтила и бициклического азотсодержащего гетероарила. В еще одном варианте осуществления изобретения Y представляет собой -СН2- и W представляет собой -CH=CH-.
Подкласс, который включает все вышеуказанные элементы, представляет собой подкласс дизамещенных индолов формулы:
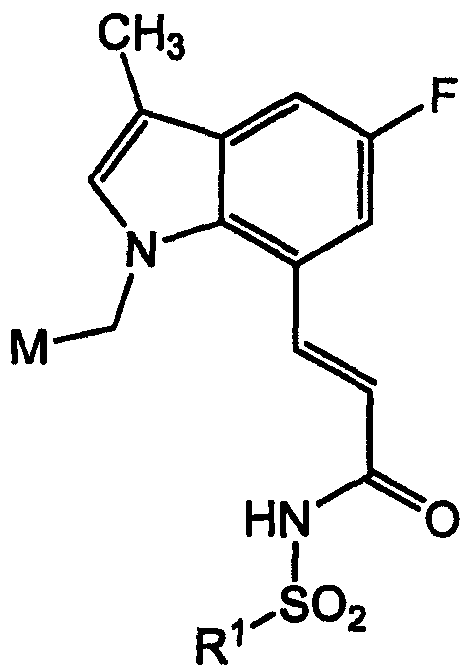
Предпочтительное соединение в данном подсемействе представляет собой соединение, где М представляет собой 2,4-дихлорфенил и R1 представляет собой дихлортиен-2-ил (пример Р067).
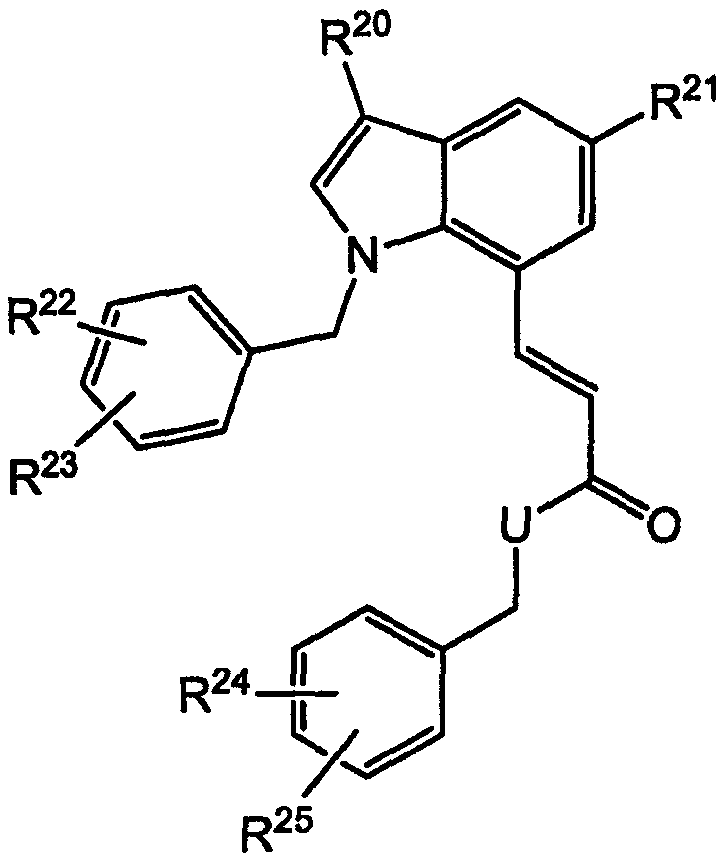
где U выбран из -О- и -NH-; и
R20-R25 выбраны из водорода, галогена и метила.
В некоторых вариантах осуществления изобретения U представляет собой О. В других вариантах U представляет собой О, и R22, R23, R24 и R25 представляют собой галогены. В конкретных вариантах осуществления изобретения R22, R23, R24 и R25 все являются хлором.
В других вариантах осуществления изобретения U представляет собой -NH-. В некоторых вариантах осуществления U представляет собой -NH- и R22 и R23 представляют собой галоген.
Соединения согласно настоящему изобретению являются кислотными, что дает возможность представлять их в виде солей. Термин «фармацевтически приемлемая соль» относится к солям, противоион которых получен из фармацевтически приемлемых нетоксичных кислот и оснований. Подходящие фармацевтически приемлемые основно-аддитивные соли соединений настоящего изобретения включают, но без ограничения, соли металлов, полученные из солей алюминия, кальция, лития, магния, калия, натрия и цинка, или органические соли, полученные из лизина, N,N-диалкиламинокислотных производных (например, N,N-диметилглицина, пиперидин-1-уксусной кислоты и морфолин-4-уксусной кислоты), N,N'-дибензилэтилендиамина, хлорпрокаина, холина, диэтаноламина, этилендиамина, меглумина (N-метилглюкамина) и прокаина. Когда соединения содержат основной остаток, подходящие фармацевтически приемлемые основно-аддитивные соли соединений согласно настоящему изобретению включают неорганические кислоты и органические кислоты. Примеры таких солей включают ацетат, бензолсульфонат (безилат), бензоат, бикарбонат, бисульфат, карбонат, камфорсульфонат, цитрат, этансульфонат, фумарат, глюконат, глутамат, бромид, хлорид, изетионат, лактат, малеат, малат, манделат, метансульфонат, мукат, нитрат, памоат, пантотенат, фосфат, сукцинат, сульфат, тартрат, п-толуолсульфонат и т.п.
Определения
Используемые далее в настоящем описании термины и заместители имеют следующие определения.
Как использовано в данном описании, термин «алкил» включает линейные, разветвленные и циклические углеводородные структуры и их комбинации. Термин «низший алкил» относится к алкильным группам, содержащим от 1 до 6 атомов углерода. Примеры низших алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил и т.п. Предпочтительными алкильными и алкиленовыми группами являются группы от С20 и ниже. Циклоалкил представляет подгруппу алкила и включает циклические углеводородные группы, содержащие от 3 до 8 атомов углерода. Примеры циклоалкильных групп включают циклопропил, циклобутил, циклопентил, норборнил, адамантил и т.п.
Термин «С1-С20 углеводород» включает алкил, циклоалкил, алкенил, алкинил, арил и их комбинации. Примеры включают бензил, фенетил, циклогексилметил, камфорил и нафтилэтил.
Термины «алкокси» или «алкоксил» относятся к группам, содержащим от 1 до 8 атомов углерода в прямой, разветвленной, кольцевой конфигурации и их комбинациях, присоединенных к основной структуре через кислород. Примеры включают метокси, этокси, пропокси, изопропокси, циклопропилокси, циклогексилокси и т.п. Термин «низший алкокси» относится к группам, содержащим от одного до четырех атомов углерода.
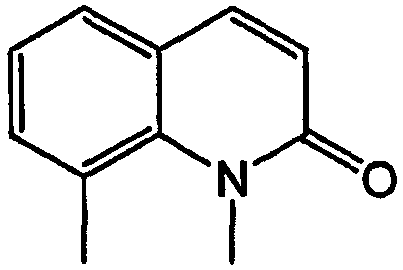
Термин «оксаалкил» относится к алкильным остаткам, в которых один или несколько атомов углерода (и связанные с ними атомы водорода) заменены кислородом. Примеры таких групп включают метоксипропокси, 3,6,9-триоксадецил и т.п. Подразумевается, что смысл термина общеизвестен в данной области [см. Naming and Indexing of Chemical Substances for Chemical Abstracts, published by the American Chemical Society, 196, но без ограничения l27(a)], т.е. относится к соединениям, где кислород соединен с соседним атомом через одинарную связь (образуя простые эфирные связи). Аналогично, термины «тиаалкил» и «азаалкил» относятся к алкильным радикалам, в которых один или несколько атомов углерода заменены атомом серы и азота, соответственно. Примеры таких радикалов включают этиламиноэтил и метилтиопропил. Подразумевается, что термин «оксо» относится к заместителю, который присоединяется посредством двойной связи и представляет собой кислород (карбонил). Таким образом, например, 2-оксохинолин согласно изобретению должен иметь структуру:
Термин «ацил» относится к группе, содержащей от 1 до 8 атомов углерода в прямой, разветвленной, кольцевой конфигурации, насыщенной, ненасыщенной и ароматической и их различных комбинациях, которые присоединяются к основной структуре через карбонильную функциональную группу. Один или несколько атомов углерода в ацильном остатке могут быть заменены атомами азота, кислорода или серы, при условии, что точка присоединения к основной структуре остается при карбониле. Примеры таких групп включают формил, ацетил, пропионил, изобутирил, трет-бутоксикарбонил, бензоил, бензилоксикарбонил и т.п. Термин «низший ацил» относится к группе, содержащей от одного до четырех атомов углерода. Термин «ацилалкил» относится к остатку, в котором ацильная группа соединена с алкильной группой, которая в свою очередь соединена с основной частью молекулы. Примером такой группы может служить СН3С(=О)СН2-. Такие остатки могут характеризоваться как «оксоалкильные» остатки.
Термины «арил» и «гетероарил» означает 5- или 6-членное ароматическое и гетероароматическое кольцо, содержащее 0-3 гетероатома, выбранных из атомов О, N или S; бициклическую 9- или 10-членную ароматическую или гетероароматическую кольцевую систему, содержащую 0-3 гетероатома, выбранных из атомов О, N или S; или трициклическую и 13- или 14-членную ароматическую или гетероароматическую кольцевую систему, содержащую 0-3 гетероатома, выбранных из атомов 0, N или S. Ароматические 6-14-членные карбоциклические кольца включают, например, бензол, нафталин, индан, тетралин и флуорен, и 5-10-членные ароматические гетероциклические кольца включают, например, имидазол, пиридин, индол, тиофен, бензопиранон, тиазол, фуран, бензимидазол, хинолин, изохинолин, хиноксалин, пиримидин, пиразин, тетразол и пиразол.
Термин «ариалалкил» означает алкильный остаток, присоединенный к арильному кольцу. Примерами таких групп являются бензил, фенетил и т.п.
Термины «замещенный алкил», «замещенный арил», «замещенный циклоалкил», «замещенный гетероциклил», относятся к алкилу, арилу, циклоалкилу или гетероциклилу, соответственно, в которых до трех атомов Н заменены галогеном, низшим алкилом, галогеналкилом, гидроксигруппой, низшей алкоксигруппой, карбокси, карбоалкокси (который также называется алкоксикарбонилом), карбоксамидом (который также называется алкиламинокарбонилом), циано, карбонилом, нитро, амино, алкиламино, диалкиламино, меркапто, алкилтио, сульфоксидом, сульфоном, ациламино, амидино, фенилом, бензилом, гетероарилом, фенокси, бензилокси или гетероарилокси. В приведенной ниже формуле изобретения метилендиокси и этилендиокси указаны в качестве заместителей. Хотя метилендиокси присоединен к соседним атомам углерода кольца, этилендиокси может быть присоединен либо к соседним атомам углерода кольца, либо к одному атому углерода, образуя спиродиоксоль (кеталь), аналогично спиротиазолидинилу. Различные варианты такого присоединения показаны в соединениях 114, 144 и 160.
Термин «галоген» означает фтор, хлор, бром или йод.
Термин «пролекарство» относится к соединению, которое более активно «in vivo». Активация in vivo может осуществляться посредством химического воздействия или при воздействии ферментов. Микрофлора в ЖК-тракте может также вносить вклад в активацию пролекарства in vivo.
При характеристике переменных указано, что А и В представляют пару конденсированных 5-, 6- или 7-членных колец и что конденсированная А/В кольцевая система может содержать от 0 до четырех гетероатомов, выбранных из атомов азота, кислорода и серы. Подразумевается, что эти кольца могут быть представлены в различной степени ненасыщенности от полностью насыщенного до ароматического. Ароматические и частично ненасыщенные кольца предпочтительны.
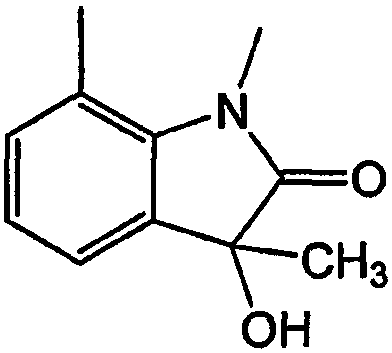
При характеристике переменных указано, что кольца могут быть дополнительно замещены от 0 до четырех заместителями, независимо выбранными из перечня различных определений. Структура, приведенная ниже, иллюстрирует способ присоединения. В этом примере конденсированные кольца замещены тремя заместителями: CH3, -OH и оксо.
Следует понимать, что соединения согласно изобретению могут существовать в меченых радиоактивным изотопом формах, т.е. соединения могут содержать один или несколько атомов с атомной массой или массовым числом, отличным от атомной массы или массового числа, обычно встречающегося в природе. Радиоизотопы водорода, углерода, фосфора, фтора и хлора включают 2Н, 3Н, 13С, 14С, 15N, 35S, 18F и 36Cl, соответственно. Соединения, которые содержат данные радиоизотопы и/или другие радиоизотопы других атомов, входят в объем данного изобретения. Тритий, т.е. 3Н, и углерод-14, т.е. 14С-радиоизотоп, особенно предпочтительные из-за их легкого получения и обнаружения. Меченые радиоактивными изотопами соединения формулы I и формулы Ic согласно изобретению и их пролекарства обычно могут быть легко получены с помощью способов, хорошо известных специалистам данной области. Такие меченые радиоизотопами соединения удобно получать с помощью методик, представленных в примерах и на схемах, заменяя обычные реагенты доступными мечеными радиоизотопами реагентами.
Специалисту в данной области понятно, что термин «соединение», который используется в настоящем описании, включает соли, сольваты, со-кристаллы и комплексы включения данного соединения.
Термин «сольват» относится к соединению формулы I в твердом состоянии, где в кристаллическую решетку введены молекулы подходящего растворителя. Подходящим растворителем для терапевтического введения является растворитель, который во вводимых дозах является физиологически толерантным. Примерами подходящих растворителей для терапевтического введения являются этанол и вода. Когда растворителем является вода, сольват называется гидратом. Обычно сольваты могут быть получены растворением соединения в подходящем растворителе и выделением сольвата охлаждением или с использованием антирастворителя. Сольват обычно сушат или подвергают азеотропной перегонке в стандартных условиях. Со-кристаллы представляют собой сочетание двух или нескольких различных видов молекул, расположенных таким образом, что они образуют уникальную кристаллическую форму, физические свойства которой отличаются от физических свойств этих веществ в чистом состоянии. Фармацевтические со-кристаллы давно представляют интерес с точки зрения улучшенной стабильности, способа получения препарата и биодоступности таких лекарственных средств как итраконазол [см. Remenar et al. J. Am. Chem. Soc. 125. 8456-8457 (2003)] и флуоксетин. Комплексы включения описаны в публикации: Remington: The Science и Practice of Pharmacy 19th Ed. (1995) volume 1, page 176-177]. Наиболее часто применяемые комплексы представляют собой комплексы с циклодекстринами, и все такие циклодекстриновые комплексы, природные и синтетические, с добавлением различных добавок и полимеров или без них, как описано в патентах США 5324718 и 5472954, включены в объем настоящего изобретения. Указанные публикация Remingtona и патенты США также введены в данное описанные посредством ссылки.
Термин «способы лечения и профилактики» означает улучшение состояния, профилактику или облегчение симптомов и/или эффектов, связанных с липидным расстройствами. Термин «профилактика» в данном описании относится к введению лекарственного средства для предупреждения болевой чувствительности. Специалист в данной области (для которого предлагаемый способ предназначен) понимает, что термин «профилактика» не является абсолютным термином. В медицинской области признано назначать профилактическое введение лекарственного средства для снижения вероятности возникновения заболевания или снижения серьезности состояния, и этот смысл подразумевается в формуле изобретения. Как использовано в данном описании, термин «лечение» пациента включает профилактику. В данном описании различные ссылки сделаны на данный термин. Описания данных публикаций включены в настоящее описание посредством ссылки во всей своей полноте.
Термин «млекопитающее» используется в общепринятом смысле. Люди включены в группу млекопитающих, и люди должны быть предпочтительными пациентами способов лечения.
Стереоизомеры
Соединения, описанные в данном описании, могут содержать асимметрические центры и могут, таким образом, давать энантиомеры, диастереомеры и другие изомерные формы. Каждый хиральный центр может быть определен как (S)- или (R)- в соответствии с терминами стереохимии. Настоящее изобретение включает все такие возможные изомеры, а также их рацемические и оптически активные формы. Оптически активные (R)- и (S)- или (D)- и (L)-изомеры могут быть получены с использованием хиральных способов или реагентов или с использованием стандартных методов растворения. Когда соединения, описанные в данном описании, содержат олефиновые двойные связи или другие центры геометрической асимметрии, если не оговорено особо, то подразумевается, что соединения согласно изобретению включают как Е, так и Z геометрические изомеры. Таким образом, все таутомерные формы также считаются включенными.
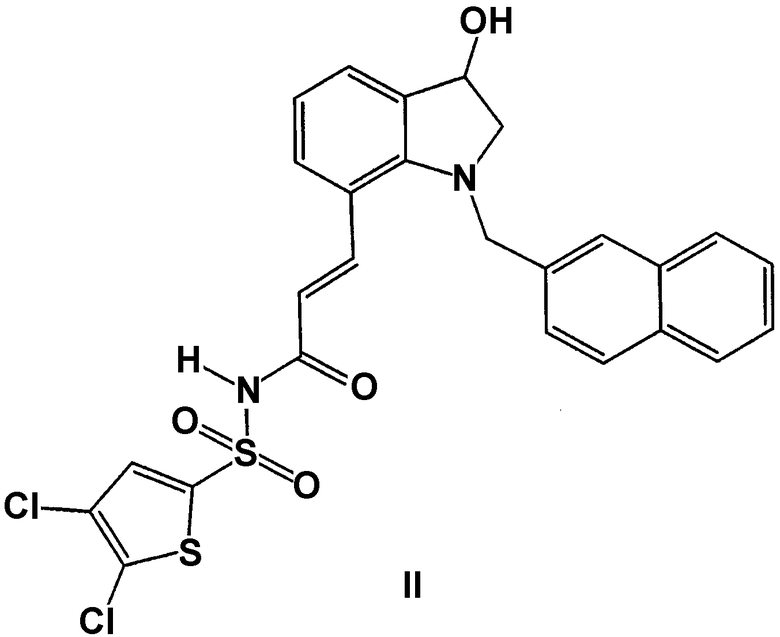
Графическое представление рацемических, амбисалемических и скалемических или энантиомерно чистых соединений, используемое в настоящем описании, взято из публикации Maehr. J. Chem. Ed. 62. 114-120 (1985), черные и светлые клинья используются для показа абсолютной конфигурации хирального элемента, волнистые линии и одна тонкая линия показывают отказ от любого стереохимического затруднения, которое связь может создавать, и черные контуры и светлые контуры не только представляют относительную геометрическую конфигурацию, но и определяют рацемический характер соединения; и клинья, обведенные черными и полыми или пунктирными линиями, обозначают энантиомерно чистые промежуточные продукты абсолютной конфигурации. Таким образом, подразумевается, что формула II включает все чистые энантиомеры, а также рацемические смеси и любые промежуточные продукты смесей энантиомеров.
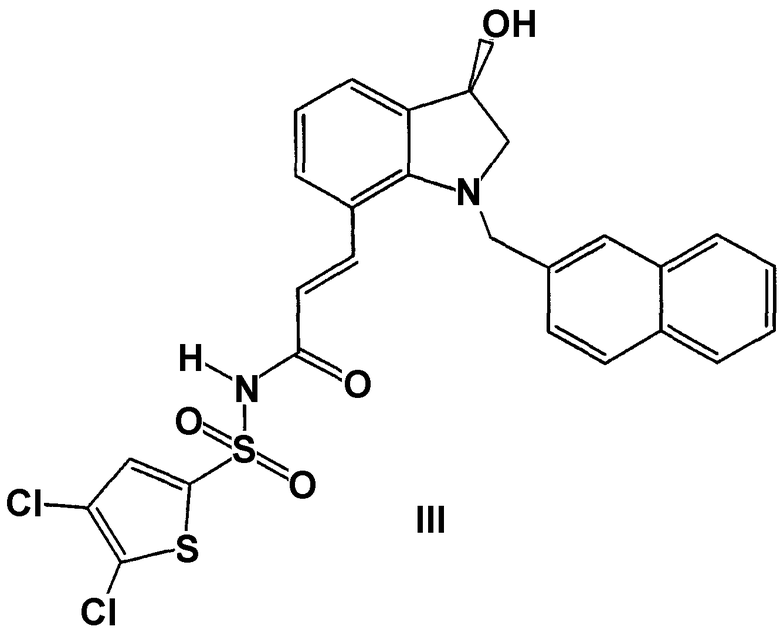
в то время как формула III включает любые чистые энантиомеры данной структуры:
и формула IV представляет чистый, единственный специфический (S)-энантиомер:
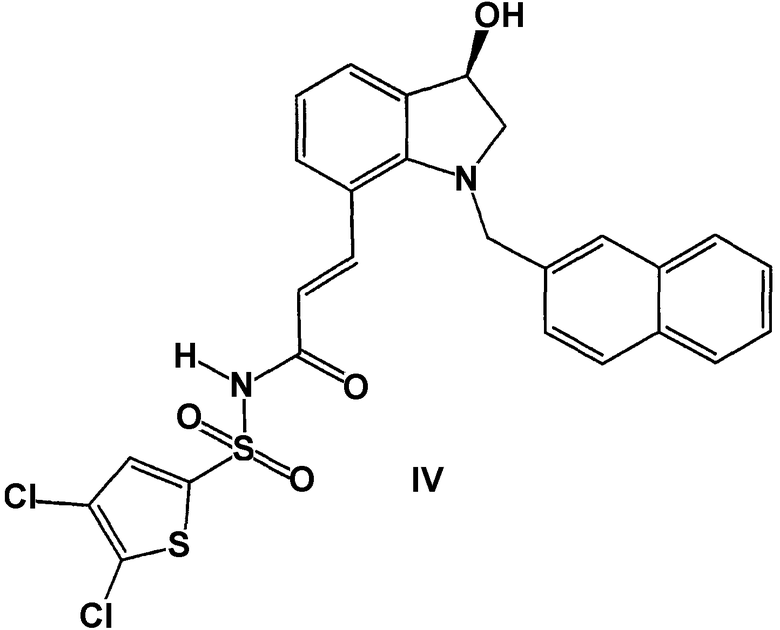
Конфигурация любой углерод-углеродной двойной связи, показанная в настоящем описании, выбрана только для удобства и, если не оговорено иное, не предназначена для выделения особой конфигурации. Так углерод-углеродная двойная связь, изображенная произвольно выше как Е, может представлять собой Z, E или смеси обоих в любой пропорции.
В данном описании используется терминология, связанная с «защитой» «снятием защиты» и «защищенными соединениями». Такая терминология хорошо известна специалисту в данной области и используется в сложных способах, которые включают последовательную обработку серии реагентов. В таком комплексе термин «защитная группа» относится к группе, которая используется для маскировки функциональных групп на данной стадии синтеза, которая в противном случае подвергается нежелательному взаимодействию. Защитная группа предотвращает реакцию на данной стадии, но впоследствии может быть удалена для открытия исходной функциональной группы. Удаление или «снятие защиты» осуществляется после завершения реакции или реакций, в которых функциональные группы могут быть подвергнуты нежелательной реакции. Таким образом, когда последовательность реагентов является специфической, как в способе настоящего изобретения, специалист в данной области без труда определит группы, которые подходят в качестве «защитных групп». Подходящие группы для этой цели описаны в стандартном справочнике, известном в области химии, таком как Protective Groups in Organic Synthesis by T.W.Greene [John Wiley & Sons, New York, 1991], который включен в данное описание посредством ссылки. Особое внимание уделено главам "Protection for the Hydroxyl Group, Including 1,2- и 1,3-Diols" (pages 10-86).
Аббревиатуры Ме, Et, Ph, Tf, Ts и Mf означают метил, этил, фенил, трифтометансульфонил, толуолсульфонил и метансульфонил, соответственно. Полный список аббревиатур, используемых химиками-органиками (т.е. специалистом в данной области) выходит в первом номере каждого тома издания Journal of Organic Chemistry. Этот перечень, который обычно представлен в виде таблицы, озаглавленной «Standard List of Abbreviations», включен в данное описание посредством ссылки.
Фармацевтические препараты
Хотя соединения формулы I или формулы Ic можно вводить и непосредственно, предпочтительно их предоставлять в виде фармацевтической композиции. Согласно дополнительному аспекту, настоящее изобретение предоставляет фармацевтическую композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль или сольват и один или несколько фармацевтических носителей и, необязательно, один или несколько других терапевтических ингредиентов. Согласно еще одному дополнительному аспекту настоящее изобретение предоставляет фармацевтическую композицию, содержащую соединение формулы (Ic) или его фармацевтически приемлемую соль или сольват и один или несколько фармацевтически приемлемых носителей и, необязательно, один или несколько других терапевтических ингредиентов. Носитель(и) должен(ны) быть «приемлемыми» с точки зрения совместимости с другими ингредиентами препарата и не оказывать вредного влияния на них.
Препараты включают композиции, подходящие для перорального, парентерального (включая подкожное, интрадермальное, внутримышечное, внутривенное и внутрисуставное), ректального и местного (включая дермальное, буккальное, подъязычное и глазное) введения. Наиболее подходящий способ введения может выбираться в зависимости от состояния и заболевания реципиента. Препараты могут удобно вводиться в единичной дозированной форме и могут приготавливаться любым из способов, хорошо известных в данной области фармакологии. Все способы включают стадию контактирования соединения формулы I или формулы Ic или его фармацевтически приемлемой соли или сольвата («активный ингредиент») с носителем, который составляют один или несколько вспомогательных ингредиентов. Обычно препараты получают однородным и тщательным смешением активного ингредиента с жидким носителем или тонко измельченным твердым носителем, или с обоими такими носителями и затем, если необходимо, формованием продукта в желаемый препарат.
Препараты согласно настоящему изобретению, подходящие для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, саше и таблетки, каждая из которых содержит предопределенное количество активного ингредиента; в виде порошка (включая микронизированный и тонко измельченный порошок) или в виде гранул; в виде растворов или суспензий в водной жидкости или неводной жидкости; или в виде эмульсии типа «масло в воде» или типа «вода в масле». Активный ингредиент также может быть представлен в виде болюса, эликсира или пасты.
Таблетка может быть изготовлена прессованием или плавлением, необязательно с одним или несколькими вспомогательными ингредиентами. Пересованные таблетки могут быть получены прессованием в подходящей машине активного ингредиента в свободно текучей форме, такой как порошок или гранулы, необязательно в смеси со связующим веществом, лубрикантом, инертным разбавителем, поверхностно-активным веществом или дисперсантом. Плавленые таблетки могут быть получены в подходящей машине плавлением смеси порошкообразного соединения, увлажненной подходящим жидким инертным разбавителем. Таблетки необязательно могут быть покрыты оболочкой или иметь насечку или могут быть изготовлены таким образом, чтобы обеспечивать поддерживаемое, замедленное или контролируемое высвобождение из нее активного ингредиента.
Фармацевтические композиции могут включать «фармацевтически приемлемый инертный носитель», и подразумевается, что данный термин включает один или несколько инертных эксципиентов, которые включают крахмалы, многоатомные спирты, гранулирующие вещества, микрокристаллическую целлюлозу, разбавители, лубриканты, связующие вещества, дезинтегрирующие агенты и т.п. При необходимости, дозированные таблетки описываемых композиций могут быть покрыты оболочкой стандартными водными или неводными методами. Термин «фармацевтически приемлемый носитель» включает также контролируемое высвобождение активного вещества.
Композиции согласно изобретению могут также необязательно содержать другие терапевтические ингредиенты, добавки, предотвращающие спекание, консерванты, подслащивающие вещества, пигменты, вкусовые добавки, влагопоглотители, пластификаторы, красители и т.п. Любой такой необязательный ингредиент должен, несомненно, быть совместим с соединением согласно изобретению для гарантии стабильности препарата.
Интервал доз для взрослых людей обычно составляет от 0,1 мкг до 10 г в день перорально. Таблетки и другие формы, представленные в дискретных единицах, могут удобно содержать количество соединения согласно настоящему изобретению, которое эффективно в такой дозировке или в нескольких таких дозировках, например в единичных дозах, содержащих от 0,5 до 500 мг, обычно от около 5 мг до 200 мг. Точное количество соединения, вводимого пациенту, будет определяться лечащим врачом. Однако применяемая доза будет зависеть от ряда факторов, включая возраст и пол пациента, точный диагноз расстройства, которое подлежит лечению, и его тяжесть. Частота введения будет зависеть от фармакодинамики отдельного соединения и препарата в дозированной форме, которая может быть оптимизирована способами, хорошо известными в данной области (например, контролируемым или длительным высвобождением действующего вещества, энтеросолюбильным покрытием и т.д.).
Комбинированная терапия может достигаться введением двух или нескольких лекарственных средств, каждое из которых приготавливается и вводится больному отдельно, или введением двух или нескольких лекарственных средств в одном препарате. Другие комбинации также входят в понятие комбинированная терапия. Например, два лекарственных средства могут быть объединены в одном препарате и введены больному вместе с третьим лекарственным средством, которое представлено в отдельном лекарственном препарате. Хотя такие два или несколько лекарственных средств в комбинированной терапии могут вводиться одновременно, они не должны быть вместе.
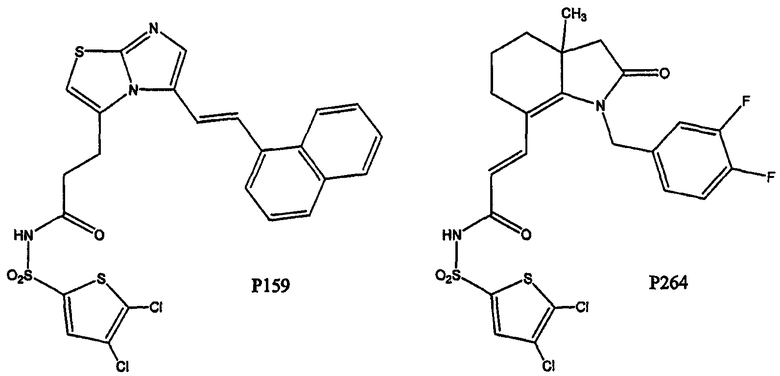
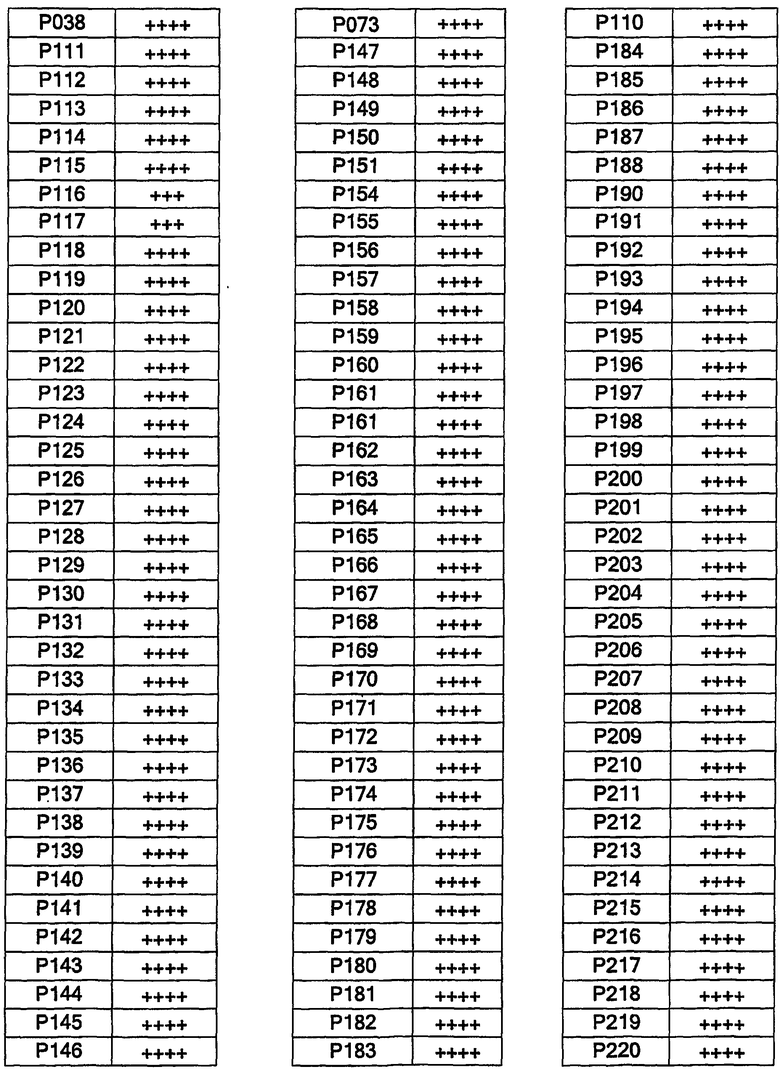
Было синтезировано примерно три сотни соединений. Их структуры представлены в таблицах 1-2 ниже. В данных таблицах тире означает прямую связь. Таким образом, например, соединение Р159, где Х2 и Х6 показывают связи, представляет собой имидазолотиазол (т.е. представляет собой пару конденсированных пятичленных колец). Насыщение и ненасыщение показано водородной связью; таким образом, соединение Р264 представляет собой гексагидроиндол-2-он:
Соединения в таблице 1 содержат «d», означающий [C], и U, означающий -NHSO
2
-, за исключением соединения Р159, где «d» означает N и U означает -NHSO
2
, и соединения Р153, где «d» означает [X] и U означает NH
2
.
пиридинил
пиридинил
пиридинил
пиридинил
пиридинил
пиридинил
пиридинил
пиридинил
пиридинил
пиридинил
пиридинил
пиридинил
Где 'a' означает [N], и 'b'='c'='d' означает [C]
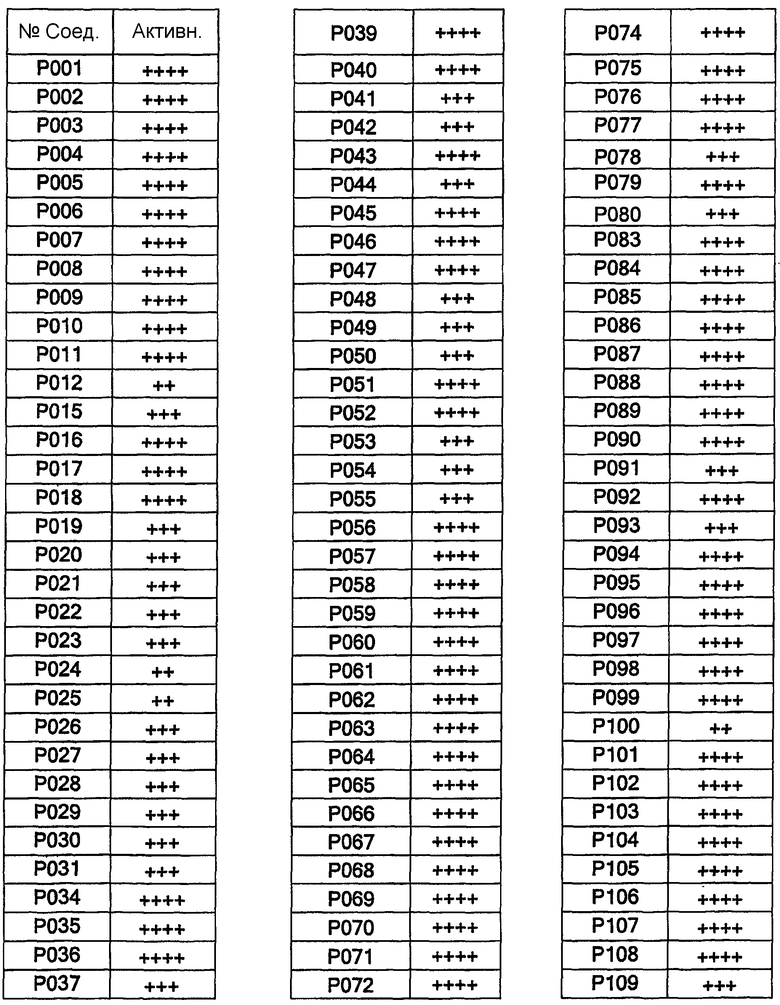
Соединения согласно изобретению были испытаны на их связывание c простаноидными ЕР3 рецепторами в соответствии с методом Abramovitz et al. [Bioch. Biophys. Acta, 1473, 286-293 (2000)]. В диаграмме 1 в колонке 2 представлена активность соединений. Соединения с IC50<1 мкМ представлены как +++++; соединения с IC50 1-10 мкМ представлены как +++; соединения с IC50>10 мкМ представлены как ++. Все примеры соединений, представленные в таблицах 1 и 2, были синтезированы, охарактеризованы и испытаны на связывание с ЕР3 рецептором.
Соединения согласно настоящему изобретению были испытаны на их действие в отношении агрегации тромбоцитов in vitro. В экспериментах на тромбоцитах человека цельную кровь отбирали у человеческих доноров, голодающих в течение ночи. Каждый эксперимент проводили с кровью от одного индивидуума. В экспериментах с тромбоцитами грызунов цельную кровь отбирали из сердца самок мышей или самцов крыс под изофлурановой анестезией (Abbott). В каждом эксперименте с кровью крыс или мышей кровь объединяли от двух или десяти индивидуумов, соответственно. Во всех случаях кровь собирали в пробирки с 3,8% цитрата натрия (Greiner Bio-one). Обогащенную тромбоцитами плазму (PRP) получали центрифугированием при 100×g в течение 15 минут при 25°С для крови человека, при 150×g для крови крыс или при 80×g в течение 10 минут для крови мышей. Плазму с низким содержанием тромбоцитов получали центрифугированием оставшейся крови при 2400×g в течение 10 минут при 25°С. После подсчета на автосчетчике (Model 920 EO, Swelab) тромбоциты разводили, когда это было необходимо, до желаемых исходных концентраций (200000-300000 тромбоцитов/мкл) с использованием 0,9% изотонического раствора NaCl (Braun).
Агрегацию тромбоцитов определяли с помощью световой абсорбции с использованием тромбоцитного агрегометра с постоянной магнитной мешалкой (Model 490, Chronolog Cop., Havertown, Pennsylvania, USA) и объемом 500 мкл на кювету. В процессе выполнения экспериментов раствор тромбоцитов непрерывно перемешивали при умеренном горизонтальном встряхивании. Коллаген (Sigma) и PGE2 или сулпростон (Cayman Chemicals) использовали в качестве ускорителей агрегации тромбоцитов. Соединения, используемые в данном анализе, растворяли и хранили в 100% ДМСО растворе. После разведения конечная концентрация ДМСО в данном анализе была ниже 0,1% об./об. Было установлено, что данная концентрация ДМСО не ингибирует агрегацию тромбоцитов в данном анализе. Ускорители и ЕР3 испытуемые соединения разводили в изотоническом растворе до нужной концентрации. Для вычисления концентрации испытуемого соединения, необходимой для ингибирования агрегации тромбоцитов на 50%, использовали сигмоидальную нелинейную регрессию. Значения IC50 испытуемых соединений вычисляли с использованием программного обеспечения Graph.Pad. Prism 3,02 для Windows (GraogPad Software, San Diego California USA). Полученные данные представлены в таблице 3.
Соединения были испытаны в отношении PGE2 (940 нМ), или в отношении сулпростона (100 нМ), или коллагена (0,125 мкг мл) для человека, которые продуцировали 90% агрегацию.
Соединения были испытаны в отношении PGE2 (940 нМ), или сулпростона (100 нМ), или коллагена (2,0 мкг мл) для крысы, которые продуцировали 60% агрегацию.
Соединения согласно настоящему изобретению оценивались также на их влияние на агрегацию тромбоцитов in vivo. В испытании in vivo активация тромбоцитов представляет собой введение легочной тромбоэмболии посредством арахидоновой кислоты, предшественника образования простагландинов. Ингибиторы синтеза простагландинов, например СОХ-1 ингибитор, такой как аспирин, обладают защитным действием в данном анализе. Для анализа легочной тромбоэмболии женским особям мыши C57BL/6 перорально вводили испытуемые соединения и спустя 30 минут вводили тромбоэмболию инъекцией анахидоновой кислоты в хвостовую вену в дозе 30 мг на кг массы тела. Количество выживших особей подсчитывали через час после введения арахидоновой кислоты, поскольку мыши, которые выжили в течение этого периода времени, как правило, полностью выживали. Инъекцию арахидоновой кислоты проводили через латеральную хвостовую вену у мыши, которая в течение непродолжительного времени согревалась под теплой лампой (расширение хвостовых вен с использованием тепловой интенсификации, способствующей инъекции). Для дозировки использовали шприц для инъекций инсулина объемом 0,5 мл (от Becon Dickinson). Данный объем дозы испытуемого соединения и арахидоновой кислоты приводят в соответствии с массой мыши (объем дозы перорально для испытуемых соединений и внутривенно для раствора арахидоновой кислоты составлял 10 мкл и 5 мкл на грамм массы тела, соответственно). Доля выживших мышей, обработанных арахидоновой кислотой, составила только 1 особь на 10 испытанных или 10%. Доли выживших мышей, обработанных испытуемыми соединениями (100 мг/кг, перорально) и затем анахидоновой кислотой, представлены в таблице 4 ниже.
Обычно соединения согласно настоящему изобретению могут быть получены способами, проиллюстрированными на общих реакционных схемах, как, например, описано ниже, или посредством их модификаций с использованием легко доступных исходных веществ, реагентов и стандартных методик синтеза. В указанных реакциях можно также применить их варианты, которые известны как таковые, но не указаны в данном описании. Исходные вещества в случае подходящим образом замещенных соединений с конденсированными А/В кольцами либо коммерчески доступны, либо могут быть получены способами, хорошо известными специалисту данной области.
Диаграмма 1


Химический синтез
Обычно соединения формулы I могут быть получены из соответствующим образом функционализированных замещенных бициклических ядер, как показано на схемах 1-17. В частности, когда узел «а» представляет собой атом азота, введение любой функциональной группы у атома углерода, узел «b», бициклического ядра соединения G1 (где на схеме 1 показан атом галогена), можно провести посредством реакции сочетания с использованием палладиевого катализатора Хека с получением углерод-связанного сложного эфира (G2) или амида (G4). Другим путем связывания углерода узла «b» может являться истинный гетероатом (соединение G6), как показано на схеме 2. Промежуточные продукты G2 и G6 затем могут быть функционализированы на узле «а» (атом азота бициклического ядра) с получением полузамещенных сложных эфиров G3 и G7, как показано на схемах 1 и 2, соответственно. Введение азотных заместителей может достигаться либо до (соединения G3 или G7), либо после функционализации карбоновой кислоты (соединения G4 или G8) с получением ациламида/ацилсульфонамидов G5 и G9, которые охватываются формулой I.
Схема 1
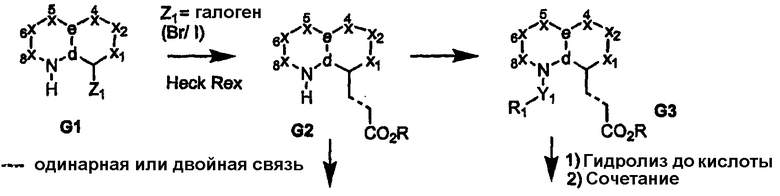
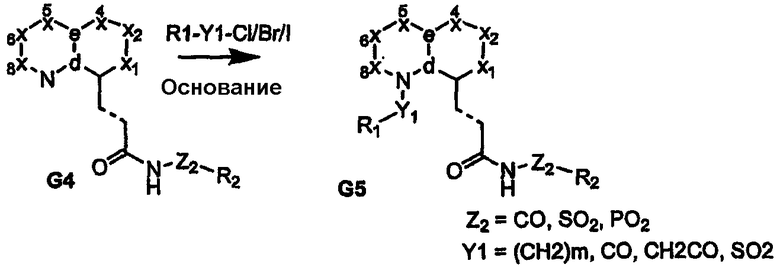
Схема 2
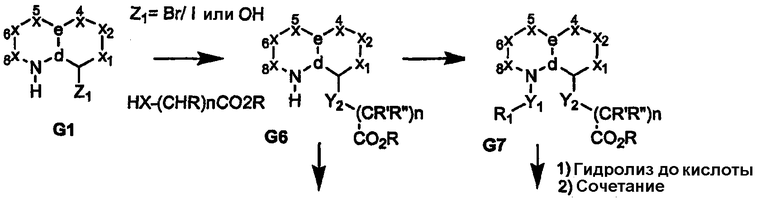
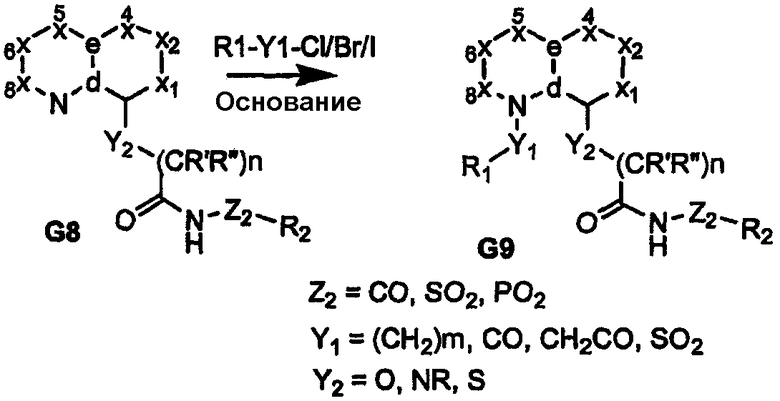
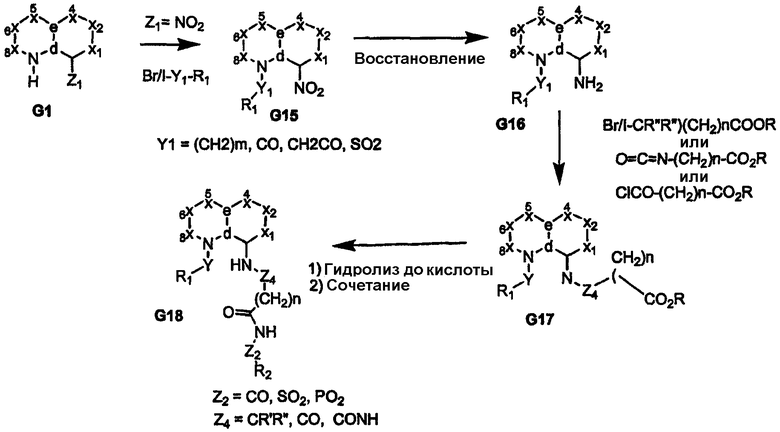
Когда узел «b» представляет собой атом углерода, несущий сложную эфирную или нитрильную функциональную группу, восстановление приводит к получению соответствующего спирта или амина G10, как показано на схемах 3 и 4. Спирт или амин может быть последовательно алкилирован, ацилирован или подвергнут взаимодействию с изоцианатом с получением пери-замещенного бициклического промежуточного продукта G11, который, в свою очередь, может быть преобразован в соединения формулы I, где сульфонамидный фрагмент и др. содержат различные заместители, как показано в соединении G12. Альтернативно, амин G10 может быть подвергнут взаимодействию с циклическими (насыщенным или арильным/гетероарильным) изоцианатами, несущими группу сложного эфира карбоновой кислоты с получением более жесткой циклической связи, разделяющей бициклическое ядро, и ацилсульфонамидной функциональной группы, как показано в соединении G14 (схема 4). Аналогично, производные, где углерод бициклического ядра соединения G1 непосредственно несет атом азота (т.е. нитро/амин, G15/G16), дают соответствующие амиды или мочевины в качестве пространственного заместителя для ацилсульфонамидов G18, как показано на схеме 5.
Схема 3
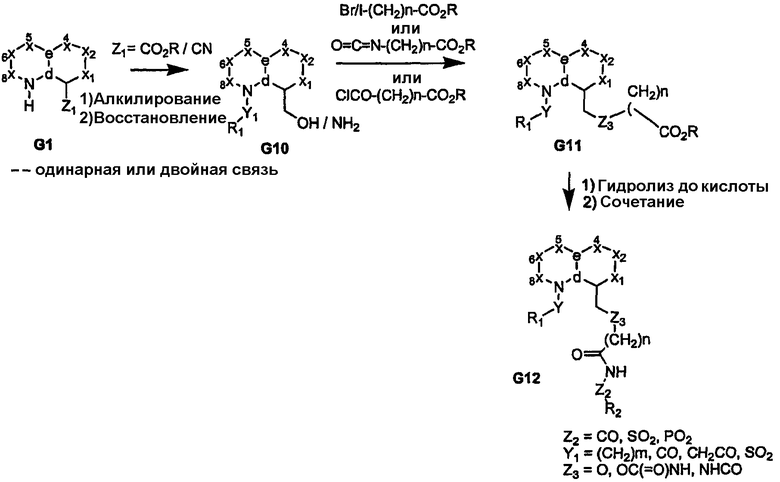
Схема 4
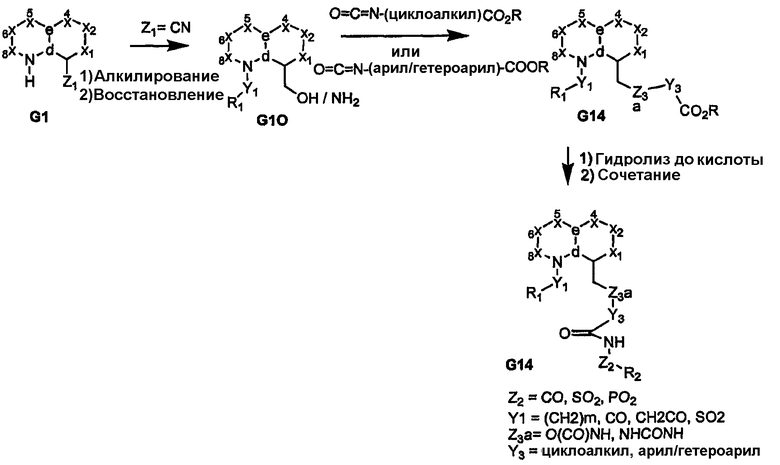
Схема 5
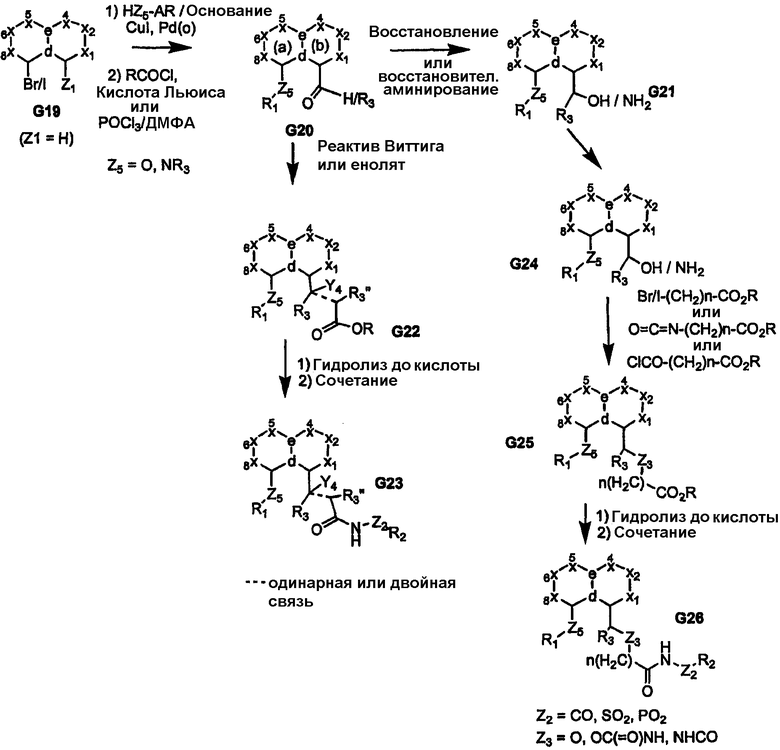
Бициклические ядра, в которых как узел «а», так и «b» представляют собой атомы углерода, могут быть получены из такого исходного вещества, как соединение G19. Функционализация атома углерода, несущего галоген, через палладий-опосредуемое образование простого эфира или амина в соответствии с химией Бухвальда с последующим введением ацильной или формильной группы посредством электрофильной реакции приводит к получению ключевого пери-функционализированного промежуточного продукта G20. Последняя реакция особенно подходит, когда кольцо (b) бициклического ядра богато электронами. Реакция кетона или альдегида с использованием реагента Виттига приводит к получению желаемого олефинсвязанного сложного эфира, который может быть восстановлен с получением соответствующей насыщенной связи, при необходимости. Альтернативно, кетоны или альдегиды могут быть подвергнуты взаимодействию с соответствующим енолятом (или даже гомо-енолятом) с получением дополнительных функциональных групп (например, Y4=OH и т.п.) в линкерной части с получением соединения G22. Функциональная группа Y4 может быть далее видоизменена или удалена с получением олефинового мостика. В дополнение, бензиловый спирт G21 может быть преобразован в галогенид (например, Br), и бензилгалогенид, таким образом, может быть преобразован посредством реакции Хека или, альтернативно, взаимодействием с ICH2CH2COOR [Higuchi K. et al. Org. Letters 2003, 3704] с получением продукта G24. Альдегид/кетон G20 в результате реакции с гомоенолатом дает сложный эфир G22. Последующее преобразование соединений G22 и G24 приводит к получению продуктов G23 и G24, соответственно.
Схема 6
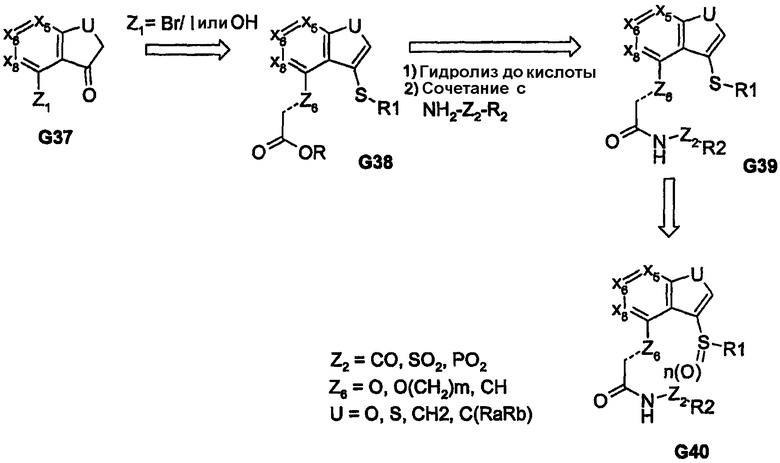
Дополнительные примеры высокореакционноспособных/электрофильных бициклических ядер, в которых введение функциональных групп, связанных гетероатомом, обеспечивает обе углерод-связанные пери-функциональные группы, показаны на схемах 7 и 8. Такие способы синтеза обеспечиваются путем введения ацильной части фрагмента, содержащего различные линкеры. Такая химия обеспечивает введение арильных и гетероарильных групп, связанных через серу, и дает возможность регулировать степень окисления серы, а также получать, таким образом, аналоги, представленные соединениями G31 и G36. Альтернативно, используя кетоны G37 можно получить соединения, родственные соединениям G31/G36, которые обеспечивают такие ядра, как бензофуран и бензотиофен, соединения G40.
Схема 7
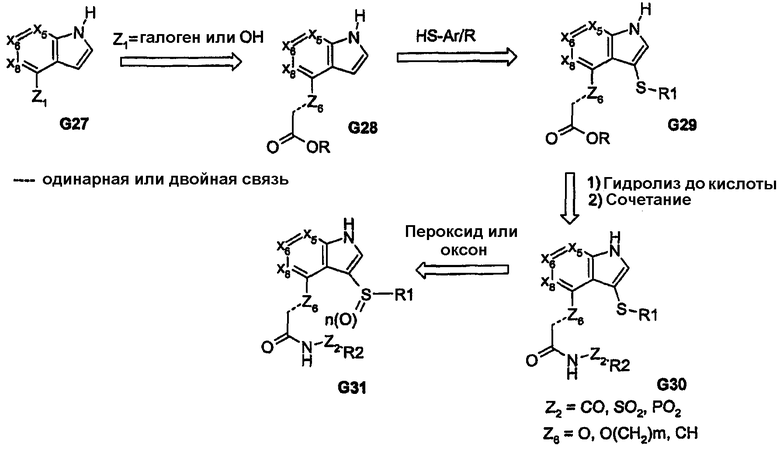
Схема 8
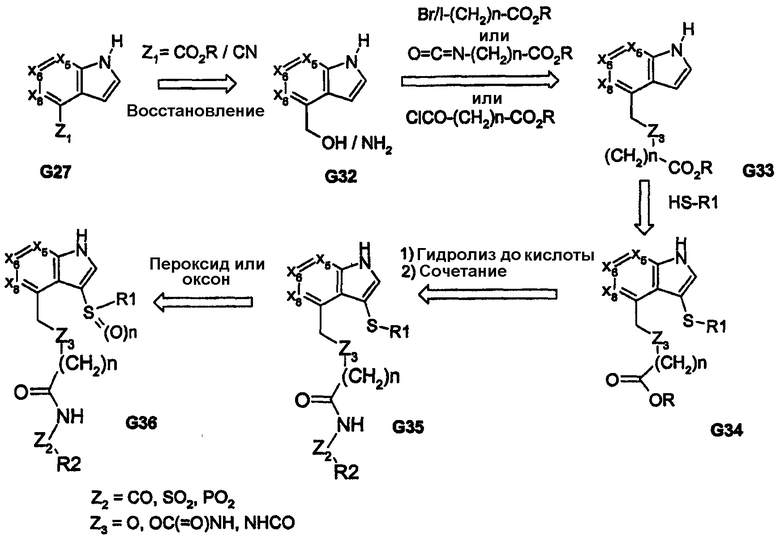
Схема 9
Высоконуклеофильные тиолы дают возможность использовать ядра, подобные соединению G27, как показано при преобразовании соединения G33 в соединение G34 на схеме 8. Для получения соответствующих аза-(или окса)-связанных арильной/гетероарильной/алкильной групп (R1) можно использовать промежуточные продукты, родственные изатину, как показано на примере соединения G43, которое получено из соединения G41, на схеме 10. Как видно на схеме 10, промежуточный продукт G43 обеспечивает различные аза-связанные соединения, которые образуются в результате углерод-связанного присоединения к бициклическому ядру. Другое на основе изатина производное (показано на схеме 11) приводит к получению пери-замещенных бициклических соединений; такой способ обеспечивает функциональные группы, которые связаны через атомы углерода и азота ядра бициклической системы. В дополнение, обеспечение ключевого промежуточного продукта G56, который содержит реакционноспособный карбонил, отдаленно напоминающий концевые группы пери-заместителей R1 и R2, дает возможность применить широкий спектр химических методов, представленных на схеме 11. Такие химические превращения, например получение кеталей, в дополнение к карбонилу и реакции с DAST обеспечивают аналоги cамых различных функциональных групп, как показано на примерах соединений G56-G60. Аналоги на схемах 10 и 11 также обеспечивают бициклические ядра, содержащие одно или два кольца, которые не являются ароматическими.
Схема 10
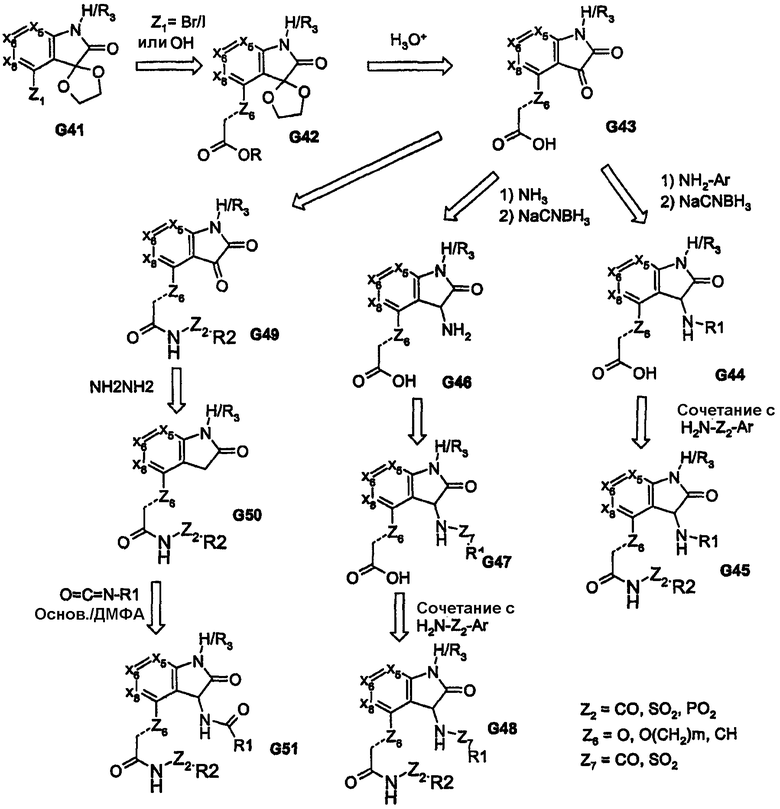
Схема 11
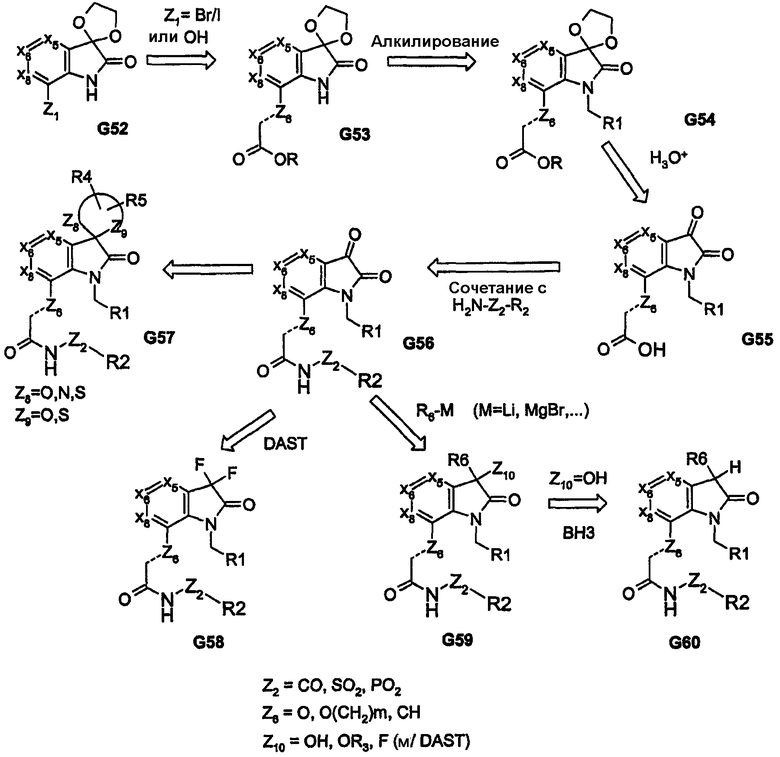
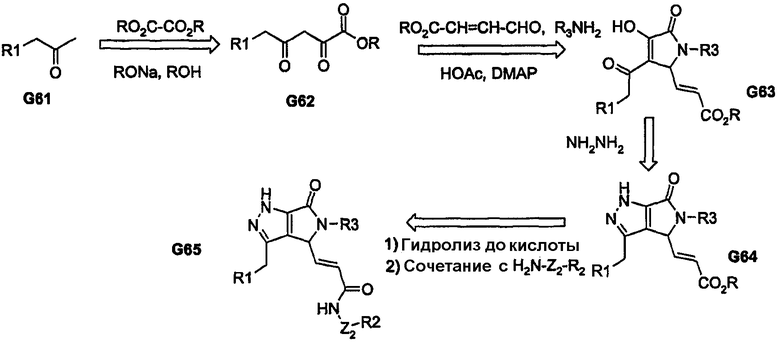
Во всех представленных выше способах синтеза по существу используется бициклическое ядро, которое подвергается подходящему видоизменению для получения соединений описанной выше формулы I. В приведенных далее химических превращениях показан способ введения, по меньшей мере, пери-фрагментов, как часть конструкции бициклического ядра. Последовательность химических превращений, представленная на схеме 12, включает трехкомпонентную реакцию конденсации, посредством которой сложный α,γ-дикетоэфир (G62) в результате реакции с альдегидом и первичным амином приводит к получению моноциклического продукта G63. Продукт G63 в процессе реакции, например, с гидразином (или монозамещенным гидразином) дает пери-замещенное бициклическое ядро (в данном случае 5-5-кольцевую систему, как показано соединением G64), которое затем приводит к получению аналога G65.
Схема 12
Другие примеры химических превращений, которые включают образование бициклических ядер, представлены на схемах 13 и 14, которые представляют синтез бензимидазольных ядер. Для получения пери-замещенной системы группу R1 вводят региоспецифично на стадии соединений G67-G68, которая после закрытия кольца обеспечивает наличие желаемого пери-замещенного производного G69. На схеме 14 желаемое региоспецифичное введение группы R1 проводят посредством миграции О к N ацила с последующим восстановлением амида до вторичного амида. В этом случае закрытие кольца также приводит к получению целевых пери-заместителей, таких как в соединении G77.
Схема 13
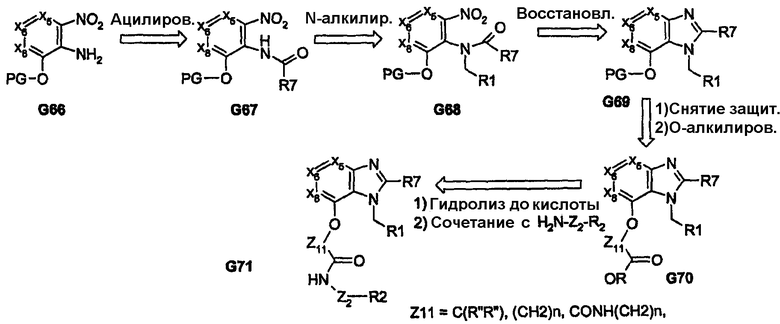
Схема 14
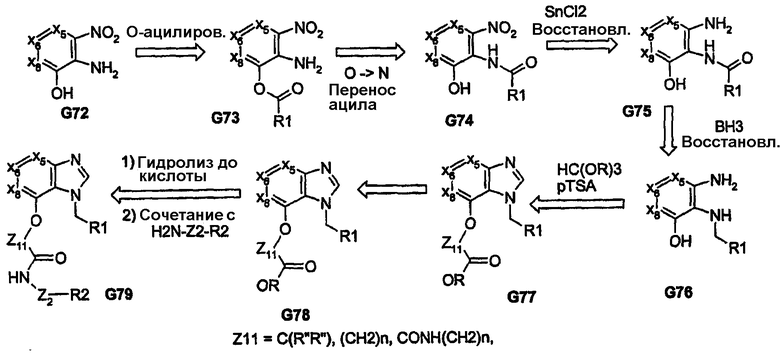
Другой пример химических превращений, включающий образование бициклических ядер с желаемыми пери-функциональными группами, представлен на схеме 15. Здесь термическая циклизация амина с циклической пространственно ориентированной γ-кетокислотой G82 приводит к получению желаемого бициклического промежуточного продукта G83. Бромирование с последующей, например, реакцией Хека приводит к получению целевого пери-бициклического производного G85, которое в результате последующих превращений приводит к получению соединения G87. Такая последовательность химических реакций дает возможность синтезировать по существу неароматические кольцевые системы, а также дает возможность создать бициклические кольцевые системы, в которых кольцо (а) является 5-членным. Кольцо (а) образуется в результате реакции циклизации, в то время как размер кольца (b) контролируется использованием циклического кетона на начальной стадии синтеза и, таким образом, дает возможность получать «5-N»-бициклические системы. Помимо возможности оказывать влияние на размер, заместитель и присутствие гетероатомов в циклическом кетоне также дает возможность придать большую пластичность. Природа третичной группы может также изменяться, и ее можно вводить на стадии циклизации кетона, что дает возможность в значительной степени контролировать его региохимию. В положениях Х5/Х6 также могут находиться гетероатомы и/или дополнительные заместители.
Схема 15
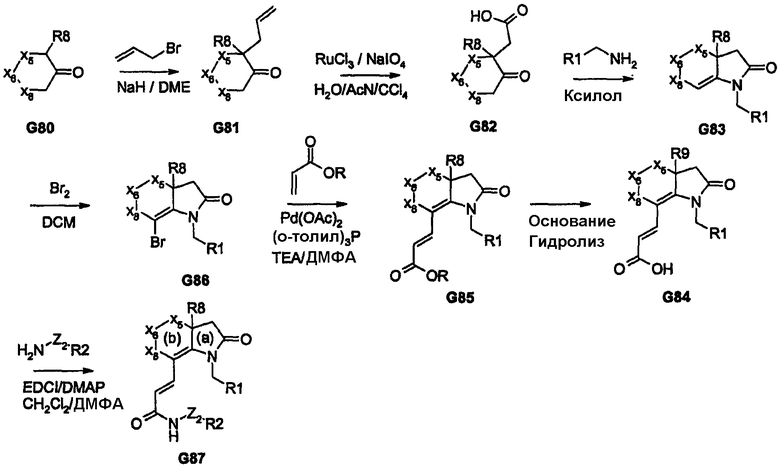
Пример, который дает возможность ввести ацильный фрагмент (несущий R2) посредством электрофильной реакции, представлен на схеме 16. Такая последовательность приводит к получению аналогов соединений G90 и G91. Бензильная карбонильная группа, присутствующая в соединении G90 и G91, может быть подвергнута дальнейшим преобразованиям, например восстановлению до спирта или СН2, образованию оксима, имина или гидразина, кеталей и т.д. Последняя стадия восстановления также дает возможность вводить радиоактивный атом углерода (14С) или трития (3Н) для создания аналогов, используемых в различных in vivo и in vitro исследованиях.
Схема 16
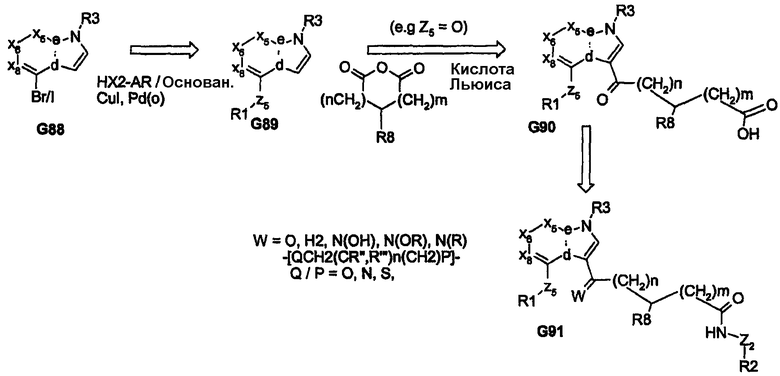
Соединения общей структуры G92 (представлена ниже) являются либо коммерчески доступными, либо легко могут быть получены из коммерчески доступных веществ с использованием методик, описанных в литературе. Замена Y (галоген/трифлат) в соединении G92 может быть осуществлена с получением простых ариловых эфиров, сульфидов или анилидов, соответственно (G93). В соединении G93, когда W представляет собой NH, N-производные соединения G94 могут быть образованы либо через N-алкилирование, либо через реакцию присоединения Михаэля по акрилату (или пропионату). Сложный эфир G94 последовательно синтезируют как показано на схемах выше для соединения G95. Однако когда соединение G93 представляет собой бициклический гетероцикл, где W означает СН, введение боковой цепи, несущей функциональную группу сложного эфира, приводит к получению соединения G97. Получение производных из соединений G93 может быть осуществлено через формилирование (G96, X=CHO) с последующей реакцией Виттига или посредством других химических преобразований с получением соответствующих сложных эфиров G97. Альтернативно, соединение G93 может быть галогенировано с получением соединения G96 (X=галоген). Замена данного галогена гетероатомными нуклеофилами (гидроксилом, меркапто- или сложными аминоэфирами) приводит к простому эфиру, тиоэфиру или аминопроизводному продукта G97 (где Z7=O, N или S). Полученные соединения затем подвергают тщательной химической обработке с получением целевых ацилсульфонамидов или родственных продуктов, G98, как описано выше. Соединение G98 может быть подвергнуто дальнейшим преобразованиям с получением соединения G99. В дополнение, производные карбоновых кислот соединений G94 и/или G97 могут быть подвергнуты реакции сочетания с Н2N-Z3, где Z3 означает NH2-Z2-R2 (когда Z2 означает NHSO2, NHCO или NHPO) с получением ацилгидразида (RC(=O)NHNHC(=O)R), aцилсульфгидразида (RC(=O)NHNHS(=O)2R) или ацилфосфоргидразида (RC(=O)NHNHP(=O)R1R2), соответственно. В действительности такие превращения могут быть осуществлены с карбоновыми кислотами, которые получены в результате химических преобразований, представленных на схемах 1-16, выше.
Схема 17
И, наконец, некоторые подходящим образом функционализированные бициклические ядра являются коммерчески доступными либо их синтез описан в литературе или может быть очевиден для специалиста данной области. Примеры некоторых из них кратко описаны в экспериментальной части.
Для бициклических систем, в которых один из узлов представляет собой атом азота, индол-производные служат легко доступным и полезным ядром. 4-Бром- и 4-гидроксииндолы являются коммерчески доступными. 7-Замещенные индолы, например 7-СО2R, 7-алкокси, 7-бензилокси и т.д., могут быть получены в соответствии с химией Батчо-Леймгрубера (Batcho-Leimgruber) из подходящим образом замещенного 2-нитротолуола (Org. Synthesis Co., Vol. 7). Такой путь также обеспечивает 7-Ме-, 7-СНО-, 7-CN- и 7-ОН-индолы посредством манипуляций функциональными группами. Альтернативно, 7-галогениндолы доступны из 2-галогенанилинов в соответствии с химией Бартоли (Bartoli, G. et al. Tett. Letters, 1989, 30, 2129-2132). Различные 7-замещенные индолы могут быть также получены посредством селективной функционализации индола через направленное орто-металлирование в соответствии с методикой Снекуса [Snieckus V. et al. Org. Letters 2003, 1899-1902]. Такие различные подходы также обеспечивают другие замещенные производные индола. 8-Гидрокситетрагидрохинолины с [6:6]-ядром могут быть получены из коммерчески доступного 8-гидроксихинолина восстановлением. 8-ОН-1Н-Хинолин-2-он, 8-ОН-3,4-дигидро-1Н-хинолин-2-он, 2,6-дигидроанилины и родственные гетероциклы могут быть преобразованы в 5-гидрокси-4Н-бензо[1,4]оксазин-3-он, 5-гидрокси-4Н-бензо[1,4]оксазин-2,3-дион, 4-гидрокси-3Н-бензооксазол-2-он, бициклические производные. Окисление 1,7-дизамещенного производного индола или 3,4-дизамещенного бициклического аналога индола приводит к получению соответствующих оксииндол-производных. Различные анилины могут быть преобразованы в аналоги изатина с использованием методик, описанных в литературе, и примеры таких преобразований описаны ниже. Синтез ряда производных [5:5]-бициклических ядер (например, имидазотиазола и пирролопиразолона) описан в конкретных примерах. Другая группа [6:5]-бициклический ядер может быть получена методами, аналогично описанными в литературе для синтеза таких ядер, как имидазопиридин и имидазопиримидин [Katritzky A.R. et al. JOC 2003, 68, 4935-37], пирролопиримидины [Norman M. et al. JMC 2000, 43, 4288-4312]. Эти разнообразные бициклические ядра могут затем быть преобразованы с получением аналогов соединений формулы I.
В целом, спектр химических превращений, представленный выше, дает возможность получить мощные простеноидные антагонисты/агонисты. Химия дает возможность манипулировать структурой ядра и вводить оптимальные функциональные группы для достижения желаемого баланса гидрофобности-гидрофильности; она дает возможность вводить донор водородной связи и акцепторы с желаемой типологией; она дает возможность регулировать желаемые физические характеристики, подходящие для достижения желаемых фармацевтических свойств и свойств всасывания, распределения, обмена, выделения (например, мембранной проницаемости, низкого плазменного белка связывания, желаемого профиля метаболизма и т.д.). Способность регулировать физико-химические характеристики дает возможность изготавливать подходящий препарат для пероральной биодоступности, который, в свою очередь, дает возможность контролировать размер и частоту дозы, вводимой млекопитающим для достижения желаемого фармакологического ответа. Возможность регулировать метаболический профиль дает возможность снижать до минимума возможные взаимодействия между лекарствами. Таким образом, объем настоящего изобретения предоставляет не только сильные простеноидные антагонисты с изоферментной селективностью в качестве полезных инструментов для исследования, но и соединения, представляющие ценность в терапии.
Примеры
Приведенные далее примеры не ограничивают область данного изобретения и являются только иллюстративными примерами.
Пример 1. Получение соединения P001
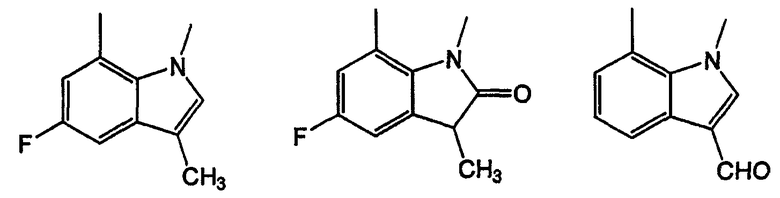
Индол-7-карбоксальдегид (I-1). Этилиндол-7m-карбоксилат получают в соответствии с методикой, приведенной в литературе [Batcho B. и Leimgruber, K., Org. Syn. Vol IIV, page 34-40]. К раствору метил-7-индолкарбоксилата (13 г, 74,2 ммоль) в 250 мл безводного ТГФ добавляют LiA1H4 (10,9 г, 0,288 моль) несколькими порциями и реакционную смесь кипятят с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры избыток гидрида гасят добавлением воды (12 мл), 15% NaOH (12 мл) и воды (26 мл). Твердый осадок удаляют фильтрованием через слой целита и фильтрат упаривают в вакууме, получая (1H-индол-7-ил)метанол (10,7 г, 98%). 1Н-ЯМР (CDCl3). К раствору спирта (1H-индол-7-ил)метанола (8,0 г, 54,3 ммоль) в 400 мл метиленхлорида добавляют активированный оксид марганца (IV) (85%, 41,0 г, 0,40 моль) и смесь перемешивают при температуре окружающей среды в течение 72 часов. В реакционную смесь добавляют 200 мл метиленхлорида и 400 мл метанола и смесь фильтруют через слой силикагеля для удаления твердых веществ. Фильтрат концентрируют, получая неочищенный продукт, который очищают колоночной хроматографией на силикагеле, получая 1H-индол-7-карбальдегид, I-1 (6,55 г, 83%). 1Н-ЯМР (CDCl3).
Этиловый эфир 3-(1Н-индол-7-ил)акриловой кислоты(I-2). В круглодонную колбу (100 мл), которая содержит суспензию NaH (60% в минеральном масле, 320 мг, 8 ммоль) в ТГФ (20мл) добавляют триэтилфосфоноацетат (1,5 г, 6,6 ммоль) при 0°C. Полученной смеси дают возможность нагреться до комнатной температуры и смесь перемешивают в течение 2 часов, затем охлаждают до 0°C. К полученному раствору добавляют индол-7-карбоксальдегид I-1 (450 мг, 3 ммоль) при 0°C. Полученной реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают в течение 2 часов, затем нагревают до 78°C и перемешивают при 78°C в течение 14 часов. Реакционную смесь охлаждают до 5°C и гасят добавлением водного NH4Cl (насыщенный, 15 мл) с последующей экстракцией EtOAc (3×30мл). Объединенные органические слои промывают насыщенным раствором соли (2×20мл), сушат (Na2SO4) и растворитель удаляют при пониженном давлении. Остаток очищают флэш-хроматографией (силикагель, EtOAc/гексан=1:20-1:8) получая целевой этиловый эфир 3-(1H-индол-7-ил)акриловой кислоты I-2 (450 мг, 68%) в виде белого твердого вещества. МС(ESI) m/z (216,3,100%). 1H-ЯМР (CDCl3), 13C ЯМР(CDCl3).
3-(1H-индол-7-ил)акриловая кислота (I-3). В круглодонную колбу (500 мл), которая содержит раствор NaOH (1,2 г, 30 ммоль) в EtOH (100 мл) и H2O (30 мл), добавляют этиловый эфир 3-(1Н-индол-7-ил)акриловой кислоты 2 (3,2 г, 15 ммоль) при 5°C. Полученной смеси дают возможность нагреться до комнатной температуры и смесь перемешивают в течение 10 минут, затем нагревают до 78°C и перемешивают в течение 4 часов. Реакционную смесь охлаждают до 5°C и подкисляют добавлением водного HCl (10%) до pH=1, затем экстрагируют CH2Cl2/MeOH (95/5, 3×150 мл). Объединенные органические слои промывают насыщенным раствором соли (2×20 мл), сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают перекристаллизацией из смеси ацетон/EtOAc/гексан, получая целевую 3-(1H-индол-7-ил)акриловую кислоту I-3 (2,4 г, 86%) в виде белого твердого вещества. МС(APCI-) m/z(186,2, 100%). ЖХМС(APCI-)>95%.
((E)-3-1H-индол-7-илакрилоил)амид тиофен-2-сульфоновой кислоты (I-4). В круглодонную колбу (500 мл), которая содержит раствор тиофенсульфонамида (1,05 г, 6 ммоль), 4-диметиламинопиридин (DMAP, 1,56 г, 13 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCl, 2,4 г, 13 ммоль) в CH2Cl2 (150 мл) добавляют 3-(1Н-индол-7-ид)акриловой кислоты I-3 (1,2 г, 6 ммоль) при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение 72 часов, затем охлаждают до 5°C и подкисляют до pH=1 добавлением водного HCl (10%) с последующей экстракцией CH2Cl2/MeOH (9/1, 3×100 мл). Объединенные органические слои сушат над безводным Na2SO4 и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2; CH2Cl2/EtOAc/гексан=1:10:20-1:20:10), получая целевой (3-1H-индол-7-илакрилоил)амид тиофен-2-сульфоновой кислоты I-4 (1,2 г, 56%) в виде белого твердого вещества. МС(ESI) m/z (331,1, 100%). ЖХМС(ESI-)>95%.
Общая методика N-алкилирования (3-1H-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (A-1). К суспензии NaH (60% в минеральном масле, 5 мг, 0,11 ммоль) в ДМФА (5мл) добавляют (3-1H-индол-7-илакрилоил)амид тиофен-2-сульфоновой кислоты I-4 (20 мг, 0,066 ммоль) при 0°C. [За исключением случая, когда Ar/R-CH2Br(Сl) используется в форме соли, например HCl соли, используют дополнительное количество NaH]. Полученной смеси дают возможность нагреться до комнатной температуры, перемешивают в течение 2 часов и затем охлаждают до 0°C. К полученному раствору добавляют ArCH2Br(или Cl) (0,072 ммоль, 1,1 экв.) при 0°C, полученной реакционной смеси дают возможность нагреться до комнатной температуры и перемешивание продолжают в течение 16-48 часов. Реакционную смесь охлаждают до 5°C и подкисляют добавлением водного HCl (10%) до pH=1 с последующей экстракцией CH2Cl2/MeOH (9/1, 3×10мл). Объединенные органические слои сушат над безводным Na2SO4 и растворитель удаляют при пониженном давлении, получая неочищенный продукт который, очищают флэш-хроматографией (силикагель, CH2Cl2; EtOAc/гексан=1:8-1:2); перекристаллизацией или растиранием в эфире, получая целевые N-алкилированные (3[CH2R]-1H-индол-7-илакрилоил)амиды тиофен-2-сульфоновой кислоты.
Пример 2. Получение соединения P002.
Общую методику (A-1) используют для алкилирования (3-1H-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) бензилбромидом, получая соединение P002, МС(ESI) m/z 421,2, (100%). ЖХМС(ESI-)>80%.
Пример 3.Получение соединения P006
Общую методику (A-1) используют для алкилирования (3-1H-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) 2-трифторметилбензилбромидом, получая соединение P006 МС(ESI-)m/z=489,4, (100%), ЖХМС(ESI-)>85%.
Пример 4. Получение соединения P007
Общую методику (A-1) используют для алкилирования (3-1H-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) 3-трифторметилбензилбромидом, получая соединение P007.
1H ЯМР (500 МГЦ, ацетон-d6); 5,75 (2H, с), 6,47 (д, J=15 Гц, 1H), 6,65 (д, J=3,0 Гц, 1H), 7,08 (т, J=7,5 Гц, 1H), 7,24-7,34 (м, 4H), 7,45 (т, J=8,0 Гц, 1H), 7,52-7,57 (м, 2H), 7,70 (д, J=8,0 Гц, 1H), 7,92 (м, 1H), 8,00 (м, 1H), 8,23 (д, J=15 Гц, 1H). ЖХ/МС (86%) ESI- Вычислено: 490,5; m/z найдено: 489,4 m/z (M-1).
Пример 5. Получение соединения P008
Общую методику (A-1) используют для алкилирования (3-1H-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) 2,5-диметилбензилбромидом, получая соединение P008. МС(ESI) m/z 449,3 (100%), ЖХМС(ESI-)>70%.
Пример 6. Получение соединения P009
Общую методику (A-1) используют для алкилирования (3-1H-индол-7-акрилоил)амида тиофен-2-сульфоновой кислоты (I-4) 3,4-диметилбензилбромидом, получая соединение P009. МС(ESI-) m/z=449,4(100%), ЖХМС(ESI-) >91%.
Пример 7. Получение соединения P010
Общую методику (A-1) используют для алкилирования 3-1Н-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) 2,6-дихлорбензилбромидом, получая соединение P010. МС(ESI-) m/z=489,3, (100%), ЖХМС (ESI-) >70%.
Пример 8. Получение соединения P011.
Общую методику (A-1) используют для алкилирования (3-1Н-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) (100 мг, 0,3 ммоль) 3,4-дихлорбензилбромидом (50 мг, 0,33 ммоль), получая неочищенный продукт (75 мг, 51%), который далее очищают перекристаллизацией из эфира с получением 53 мг (>90%) соединения P011 в виде светло-желтого твердого вещества. МС(ESI-) m/z=489,4, (100%). ЖХМС(ESI-) >90%. 1H ЯМР (CDCl3).
Пример 9. Получение соединения P017.
Общую методику (A-1) используют для алкилирования (3-1Н-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) c помощью 4-метоксибензилбромида, получая соединение P017.
1H ЯМР (500 MHz, метанол-d4); 6,62 (д, J=16 Гц, 1H), 7,01 (д, J=3,5 Гц, 1H), 7,12 (т, J=7,5 Гц, 1H), 7,19 (т, J=7,5 Гц, 1H), 7,37 (дд, J=8,5, 2,0 Гц, 1H), 7,60 (т, J=7,5 Гц, 1H), 7,65-7,68 (м, 2H), 7,73 (м, 1H), 7,80 (1H, уш.), 7,86-7,89 (м, 2H), 7,93 (д, J=8,5 Гц, 1H), 8,06 (с, 1H), 8,19 (д, J=8,0 Гц, 1H). ЖХ/МС (65%) ESI- Вычислено: 524,6; m/z, найдено: 523,5 m/z (М-1).
Пример 10. Получение соединения P035
Общую методику (A-1) используют для алкилирования (3-1H-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) 5-бромметилбензо[1,3]диоксолом, получая соединение P035. МС(ESI-) m/z=465,3, (100%), ЖХМС(ESI-)>81%.
Пример 11. Получение соединения P036
Общую методику (A-1) используют для алкилирования (3-1Н-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) 3,5-диметоксибензилбромидом, получая соединение P36. МС(ESI-)m/z=481,2, (100%), ЖХМС(ESI-)>77%.
Пример 12. Получение соединения P043.
Общую методику (A-1) используют для алкилирования (3-1H-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) (2-фенил)бензилбромидом, получая соединение P043. МС(ESI-) m/z=497,6 (100%), ЖХМС(ESI) >85%.
Пример 13. Получение соединения P054.
Общую методику (A-1) используют для алкилирования (3-1Н-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) гидробромидом 3-пиридилметилбромида, получая соединение P054. МС(ESI-)m/z=422,3, (100%), ЖХМС(ESI-)>95%.
Пример 14. Получение соединения P055.
Общую методику (A-1) используют для алкилирования (3-1Н-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты (I-4) гидробромидом 2-(3,5-диметил-4-метокси)пиридилметилбромида, получая соединение P055. МС(ESI-) m/z=480,3, (100%), ЖХМС(ESI-) >80%.
Пример 15. Получение соединения P056.
Получение 7-бром-3-метил-1H-индола (I-5). 2-Бромнитробензол подвергают взаимодействию с аллилмагнийбромидом в соответствии с методикой, описанной в литературе (Dobbs A. J.Org Chem. 2001, 66, 638-641), получая 7-бром-3-метил-1H-индол.
Получение метилового эфира (E)-3-(3-метил-1H-индол-7-ил)акриловой кислоты (I-6). К смеси соединения I-5 (300 мг, 1,42 ммоль) и метилакрилата (183 мг, 2,13 ммоль) в триэтиламине (1 мл) добавляют ацетат палладия (H)(31 мг, 0,14 ммоль) и три-o-толилфосфин (129 мг, 0,42 ммоль) в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивают при 100oC в течение 4 часов в герметичной трубе под давлением и затем охлаждают до комнатной температуры. Смесь разбавляют метилeнхлоридом (50 мл), промывают водой (3×30 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя EtOAc/гексан в качестве элюента, получая 250 мг соединения I-6 в виде желтого твердого вещества. 1H-ЯМР (500 MГц, CDCl3) МС(ESI-) 214,5 (M-1).
Получение (E)-3-(3-метил-1H-индол-7-ил)акриловой кислоты (I-7). К раствору метилового эфира (E)-3-(3-метил-1H-индол-7-ил)акриловой кислоты (2,180 мг, 0,84 ммоль) в ТГФ (5 мл) и метанола (4 мл) добавляют при комнатной температуре NaOH водный (4 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем водной 2н. HCl рН доводят до кислотного. Реакционную смесь экстрагируют EtOAc (2×30 мл). Объединенные органические фазы промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 170 мг соединения I-7. 1H-ЯМР (500 MГц, ДМСО-d6). Получение [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8). Смесь кислоты I-7 (170 мг, 0,84 ммоль), 2-тиофенсульфонамида (163 мг, 1 ммоль), 4-диметиламинопиридина (207 мг, 1,7 ммоль) м EDCl (325 мг, 1,7 ммоль) в дихлорметане (20 мл) и ДМСО (0,5 мл) перемешивают в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. Растворитель удаляют и остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь метанол/CH2Cl2 и получая 170 мг соединения I-8. 1H-ЯМР (500 MГц, ДМСО-d6).
Общая методика (A-2) N-алкилирования I-8. К раствору ацилсульфонамида (I-8) в ДМФА при комнатной температуре добавляют 2,8 водного NaH (60% суспензия в минеральном масле) [за исключением тех случаев, когда Ar/R-CH2Br(Cl) используется в форме соли, например HCl соли, используют дополнительное количество водного NaH]. Добавляют соответствующий арилгалогенид и смесь перемешивают при комнатной температуре в течение ночи. Смесь разбавляют EtOAc и подкисляют 10% HCl. Органические фазы промывают водой (3×), насыщенным раствором соли и сушат над безводным Na2SO4. Раствор фильтруют, концентрируют в вакууме и остаток очищают флэш-хроматографией на SiO2, используя дихлорметан в качестве элюента, или перекристаллизовывают, получая указанное в заголовке соединение.
Пример 16. Получение соединения P057
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) 2-(брометил)нафталином, получая соединение P057. 1H-ЯМР (500 MГц, ДМСО-d6). МС (ESI-): 485,5 (M-1), ЖХ-МС: чистота 97%.
Пример 17. Получение соединения P084
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) бензилбромидом, получая соединение P084. ESI- MС (M-1): 435,4 с 91% 1H-ЯМР (500 MГц, CDCl3).
Пример 18. Получение соединения P086
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) 2-хлорфенилхлоридом, получая соединение P086.
Пример 19. Получение соединения P087
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) 3,4-дихлорбензилбромидом, получая соединение P087. ESI- MС дает (M-1): 503,3, ЖХМС=84%; 1H-ЯМР (500 MГц, CDCl3).
Пример 20. Получение соединения P088.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) 3,5-дифторбензилбромидом, получая соединение P088. ESI- MС дает (М-1): 471,4 ЖХМС 84%. 1H-ЯМР (500 MГц, CDCl3).
Пример 21. Получение соединения P089
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) 5-хлор-6-хлорметилбензо[1,3]диоксолом, получая соединение P089. ESI-MС дает (M-1): 513,6, ЖХМС=80%. 1H-ЯМР (500 MГц, CDCl3).
Пример 22. Получение соединения P099.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) 2-хлорметилхинолином, получая соединение P099. ESI- MС (M-1): 486,5, ЖХМС=92%.
Пример 23. Получение соединения P100.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) гидробромидом 2-пиридинилметилбромида, получая соединение P100. ESI MС (M-1)=436,4, ЖХМС=86%.
Пример 24. Получение соединения P101
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) 5-бромметилбензо[1,3]диоксолом, получая соединение P101. ESI- MС (М-1): 479,3, ЖХМС=87%. 1H-ЯМР (500 MГц, CDCl3).
Пример 25. Получение соединения P102
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1Н-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) 5-бромметил-7-фтор-2,3-дигидробензо[1,4]диоксином, получая соединение P102. 1H-ЯМР (500 MГц, CDCl3) 2,35 (с, 3H), 4,93 (с, 2H), 5,34 (с, 2H), 5,40 (с, 2H), 5,98 (дд, J=8,5, 2,5 Гц, 1H), 6,15 (д, J=15,0 Гц, 1H), 6,57 (дд, J=8,5, 2,5 Гц, 1H), 6,91 (с, 1H), 7,12 (м, 2H), 7,23 (д, J=7,5 Гц, 1H), 7,66 (д, J=7,5 Гц, 1H), 7,70 (дд, J=5,0, 1,5 Гц, 1H), 7,90 (дд, J=4,0,1,5 Гц, 1H), 7,96 (д, J=15,0 Гц, 1H), 7,97 (уш. с, 1H). ЖХ/МС=85% чистота, МС (ESI-) Вычислено (M-H) 511,6; найдено: 511,5.
Пример 26. Получение соединения P103
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) 3,4-дифторбензилбромидом, получая соединение P103. ESI- MС (M-1)=471,4, ЖХМС=84%. 1H-ЯМР (500 MГц, CDCl3).
Пример 27. Получение соединения P109
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) 3-пиридинилметилбромидом, получая соединение P109. ESI MС (M-1)=436,4, ЖХМС=88%; 1H-ЯМР (500 MГц, CDCl3).
Получение [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9). К (E)-3-(3-метил-1H-индол-7-ил)акриловой кислоте (I-7) (1 ммоль), 4-диметиламинопиридину (DMAP, 2 ммоль) и гидрохлориду 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 2 ммоль) в CH2Cl2 (30 мл) добавляют амид 4,5-дихлортиофен-2-сульфоновой кислоты (1,1 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 24-40 часов. Реакционную смесь разбавляют 30 мл дихлорметана и затем подкисляют 10% водным раствором HCl до pH~1-2. Органические фазы промывают насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2CI2; CH2Cl2/EtOAc/гексан=1:10:20-1:20:10), получая целевой продукт, I-9. 1H-ЯМР (500 MГц, CDCl3)ESI- MС (М-1): 436,4, 88%.
Пример 28. Получение соединения P118
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 3,4-дихлорбензилбромидом, получая соединение P118. ESI- МС (M-1)=573,1, ЖХМС=97%. 1H-ЯМР (ДМСО-d6) подтверждает структуру.
Пример 29. Получение соединения P119
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 3-трифторметилбензилбромидом, получая соединение P119. ESI МС (M-1)=573,0 ЖХМС=96%. 1H-ЯМР (500 MГц, CDCl3).
Пример 30. Получение соединения P120
Общую методику (A-2) используют для алкилирования [(E)-3-(1,3-диметил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 4-фторбензилбромидом, получая соединение P120. ESI МС (M-1)=523,1; ЖХМС=88%. 1H-ЯМР (500 MГц, CDCl3).
Пример 31. Получение соединения P121
Общую методику (A-2) используют для алкилирования [(E)-3-(1,3-диметил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 3,4-диметокси-2-пиридилметилбромидом, получая соединение P121. ESI- МС (M-1)=568,3; ЖХМС=96%. 1H-ЯМР (500 MГц, CDCl3).
Пример 32. Получение соединения P122
Общую методику (A-2) используют для алкилирования [(E)-3-(1,3-диметил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) бензилбромидом, получая соединение P122. ESI- МС (М-1)=505,0, ЖХМС=91%. 1H-ЯМР (ДМСО-d6) подтверждает структуру.
Пример 33. Получение соединения P123
Общую методику (A-2) используют для алкилирования [(E)-3-(1,3-диметил-1H-индол-7-ил)акрилоил]амида 4,5-дихллортиофен-2-сульфоновой кислоты (I-9) 2-хлорбензилбромидом, получая соединение P123. ESI МС (M-1)=539,3, ЖХМС=94%. 1H-ЯМР (ДМСО-d6) подтверждает структуру.
Пример 34. Получение соединения P124
Общую методику (A-2) используют для алкилирования 4,5-дихлортиофен-2-сульфоновой кислоты [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида (I-9) 2,6-дихлорбензилбромидом, получая соединение P124. ESI- МС (M-1)=573,2, ЖХМС=99%. 1H-ЯМР(ДМСО-d6) подтверждает структуру.
Пример 35. Получение соединения P125
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 2-бромметилбифенилом, получая соединение P125. ESI МС (M-1)=579,5, ЖХМС=88%. 1H-ЯМР(ДМСО-d6) подтверждает структуру.
Пример 36. Получение соединения P135.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 3,4-дифторбензилбромидом, получая соединение P135. ESI- МС (M-1)=541,2, ЖХМС=96%. 1H-ЯМР (500 MГц, CDCl3).
Пример 37. Получение соединения P136.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 3,5-дифторбензилбромидом, получая соединение P136. ESI МС (M-1)=541,2, ЖХМС=96%. 1H-ЯМР (500 MГц, CDCl3).
Пример 38. Получение соединения P137.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 4-хлорбензилбромидом, получая соединение P137. ESI MС (M-1): 539,2, 99%.
1H-ЯМР(ДМСО-d6) подтверждает структуру.
Пример 39. Получение соединения P138.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 2,5-диметилбензилбромидом, получая соединение P138. ESI МС (M-1)=531,3, ЖХМС=99%. 1H-ЯМР(ДМСО-d6) подтверждает структуру.
Пример 40. Получение соединения P139.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 5-бромметилбензо[1,3]диоксолом, получая соединение P139. 1H-ЯМР (500 MГц, ДМСО-d6) 2,27 (с, 3H), 5,40 (с, 2H), 5,93 (с, 2H), 6,33 (д, J=15,0 Гц, 1H), 6,42 (д, J=7,5 Гц, 1H), 6,47 (с, 1H), 6,68 (д, J=8,0 Гц, 1H), 7,08 (дд, J=8,0, 7,5 Гц, 1H), 7,24 (д, J=7,5 Гц, 1H), 7,29 с, 1H), 7,61 (д, J=7,5 Гц, 1H), 7,90 (с, 1H), 8,17 (д, J=15,0 Гц, 1H), 12,6 (уш. с, 1H). ЖХ/МС=95% чистота, МС (ESI-) Вычислено (M-H+2)549; Найдено: 549.
Пример 41. Получение соединения P140.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 3-трифторметоксибензилбромидом, получая соединение P140. ESI- МС (M-1)=587,3, ЖХМС=98%. 1H-ЯМР(ДМСО-d6) подтверждает структуру.
Пример 42. Получение соединения P141
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 3,5-диметоксибензилбромидом, получая соединение P141. ESI- МС (M-1)=563,3, ЖХМС=93%. 1H-ЯМР(ДМСО-d6) подтверждает структуру.
Пример 43. Получение соединения P142.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 3-метоксибензилбромидом, получая соединение P142. ESI- МС (M-1)=535,2, ЖХМС=86%. 1H-ЯМР(ДМСО-d6) подтверждает структуру.
Пример 44. Получение соединения P143.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 4-трифторметоксибензилбромидом, получая соединение P143. ESI- МС (M-1)=587,5, ЖХМС=99%. 1H-ЯМР(ДМСО-d6) подтверждает структуру.
Пример 45. Получение соединения P144.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1Н-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 4-бромметилтетрагидропираном, получая соединение P144. 1H-ЯМР(ДМСО-d6) 1,02 (м, 2H), 1,39 (м, 2H), 1,89 (м, 1H), 2,21 (с, 3H), 3,10 (м, 2H), 3,77 (м, 2H), 4,10 (д, J=5,6 Гц, 2H), 6,39 (д, J=12,4 Гц, 1H), 7,05 (дд, J=6,4, 6,0 Гц, 1H), 7,10 (с, 1H), 7,28 (д, J=5,6 Гц, 1H), 7,55 (д, J=6,4 Гц, 1H), 7,76 (с, 1H), 8,27(д, J=12 Гц, 1H). ЖХ/МС (96%) ESI- Вычислено 513,5 m/z; найдено: 511,5 m/z.
Пример 46. Получение соединения P145.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1Н-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 4-дифторметоксибензилбромидом, получая соединение P145. 1H-ЯМР (500 МГц, ДМСО-d6) 2,28 (с, 3H), 5,51 (с, 2H), 6,28 (д, J=15,0 Гц, 1H), 6,99 (м, 4H), 7,08 (т, J=7,5 Гц, 1H), 7,19 (т, J=74 Гц, 1H), 7,22 (д, J=7,5 Гц, 1H), 7,33 (с, 1H), 7,62 (д, J=7,5 Гц, 1H, 7,91 (с, 1H), 8,12 (д, J=15,0 Гц). ЖХ/МС=95% чистота, МС (ESI-) Вычислено: (M-H) 569; найдено: 569.
Пример 47. Получение соединения P146.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1Н-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты (I-9) 2-бромметилхинолином, получая соединение P146 ESI- МС (M-1)=556,0, ЖХМС=96%. 1H-ЯМР(ДМСО-d6) подтверждает структуру.
Пример 48. Получение соединения P072.
Метиловый эфир 3-(3-метил-1Н-индол-7-ил)пропионовой кислоты, (I-10). Смесь метилового эфира (E)-3-(3-метил-1Н-индол-7-ил)акриловой кислоты (I-6a) (190 мг, 0,88 ммоль) и Pd/C (5%, 100 мг) в метаноле (15 мл) и EtOAc (5 мл) гидрируют водородом при давлении 40 фунтов на кв. дюйм при комнатной температуре в течение ночи. Реакционную смесь фильтруют через слой целита и слой целита промывают EtOAc и метанолом. Фильтрат концентрируют, получая 170 мг продукта I-10 (метилового эфира 3-(3-метил-1Н-индол-7-ил)пропионовой кислоты) 1H-ЯМР (500 МГц, CDCl3).
3-(3-Метил-1Н-индол-7-ил)пропионовая кислота (I-11). К раствору сложного эфира I-10 (170 мг, 0,78 ммоль) в ТГФ (8 мл) и метаноле (8 мл) добавляют 2н. водный NaOH (3 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем pH подкисляют добавлением водной 2н. HCl. Реакционную смесь экстрагируют EtOAc (2×40 мл). Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После фильтрования и удаления растворителя остаток промывают смесью эфир/гексан, получая 75 мг 3-(3-метил-1Н-индол-7-ил)пропионовой кислоты (I-11). 1H-ЯМР (500 МГц, ДМСО-d6).
[3-(3-Метил-1Н-индол-7-ил)пропионил]амид 4,5-дихлортиофен-2-сульфоновой кислоты, I-12: Смесь кислоты I-11 (75 мг, 0,37 ммоль), 4,5-дихлор-2-тиофенсульфонамида (103 мг, 0,44 ммоль), 4-диметиламинопиридина (90 мг, 0,74 ммоль) и EDCI (141 мг, 0,74 ммоль) в дихлорметане (10 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой и сушат над сульфатом натрия. После фильтрования и удаления растворителя остаток промывают эфиром, получая 60 мг ацилсульфонамида I-12. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P072. К раствору сульфонамида I-12 (35 мг, 0,084 ммоль) в ДМФА (3 мл) добавляют NaH (60% в масле, 15 мг, 0,37 ммоль) при 0oC. После перемешивания при комнатной температуре в течение 20 минут добавляют 2,4-дихлорбензилхлорид (47 мг, 0,24 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем разбавляют метиленхлоридом (12 мл). Реакционную смесь промывают разбавленной водной HCl (2×8 мл), водой (4×8 мл), насыщенным раствором соли и сушат над сульфатом натрия. После фильтрования и удаления растворителя остаток промывают смесью эфир/гексан, получая 33 мг соединения P072. МС (ESI-)=575,2 (M-1), ЖХМС: 86%. 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 49. Получение соединения P073
К раствору сульфонамида I-12 (35 мг, 0,084 ммоль) в ДМФА (3 мл) добавляют NaH (60% в масле, 15 мг, 0,37 ммоль) при 0°C. После перемешивания при комнатной температуре в течение 20 минут добавляют 2-(бромметил)нафталин (53 мг, 0,24 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем разбавляют метилeнхлоридом (12 мл). Реакционную смесь промывают разбавленной водной HCl (2×8 мл), водой (4×8 мл), насыщенным раствором соли и сушат над сульфатом натрия. После фильтрования и удаления растворителя остаток промывают смесью эфир/гексан, получая 35 мг соединения P073. МС(ESI-)=557,0 (M-1), ЖХМС: 70%. 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 50. Получение соединения P078 и P079
Синтез метилового эфира (E)-3-[1-(2,4-дихлорбензил)-3-метил-1Н-индол-7-ил]акриловой кислоты, (I-13). К раствору метилового эфира (I-6a) (730 мг, 3,4 ммоль) в ДМФА (15 мл), добавляют NaH (60% в масле, 272 мг, 6,8 ммоль) при 0°C. После перемешивания при комнатной температуре в течение 30 минут добавляют 2,4-дихлорбензилхлорид (1326 мг, 6,8 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем разбавляют метилeнхлоридом (150 мл). Реакционную смесь промывают разбавленной водной HCl (2×50 мл), водой (4×100 мл), насыщенным раствором соли и сушат над сульфатом натрия. После фильтрования и удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя гексан и EtOAc/гексан в качестве элюента и получая 60 мг метилового эфира I-13 и 470 мг сложного эфира I-14 (2,4-дихлорбензиловый эфир 3-[1-(2,4-дихлорбензил)-3-метил-1Н-индол-7-ил]акриловой кислоты). 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез (E)-3-[1-(2,4-дихлорбензил)-3-метил-1Н-индол-7-ил]акриловой кислоты, I-15. К раствору сложного эфира I-13 (300 мг, 0,8 ммоль) в ТГФ (6 мл) и метаноле (6 мл) при комнатной температуре добавляют водный 2н. NaOH (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем pH подкисляют добавлением водной 2н. HCl. Реакционную смесь экстрагируют EtOAc (2×40 мл). Объединенные органические фазы промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После фильтрования и удаления растворителя остаток промывают эфиром, получая 280 мг кислоты I-15. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P079. Смесь кислоты I-15 (54 мг, 0,15 ммоль), 4,5-дихлор-2-тиофенсульфонамида (42 мг, 0,18 ммоль), 4-диметиламинопиридина (37 мг, 0,3 ммоль) и EDCI (57 мг, 0,3 ммоль) в дихлорметане (5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl (2×10 мл), водой (4×10 мл) и насыщенным раствором соли и сушат над сульфатом натрия. После фильтрования и удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан и смесь метанол/дихлорметан в качестве элюентов и получая 45 мг соединения P079. МС(ESI-)=573,1 (M-1), ЖХМС: 93%. 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 51. Получение соединения P090
Смесь кислоты I-15 (54 мг, 0,15 ммоль), бензолсульфонамида (28 мг, 0,18 ммоль), 4-диметиламинопиридина (37 мг, 0,3 ммоль) и EDCI (57 мг, 0,3 ммоль) в дихлорметане (5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl (2×10 мл), водой (4×10 мл), насыщенным раствором соли и сушат над сульфатом натрия. После фильтрования и удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан и метанол/дихлорметан в качестве элюентов и получая 40 мг соединения P090. МС (ESI-)=499,3 (M-1), ЖХМС: 92%. 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 52. Получение соединения P074
В круглодонную колбу (50 мл) помещают раствор P001 (40 мг, 0,8 ммоль) в EtOH (5 мл), добавляют Pd/C (60 мг) при комнатной температуре. В колбе создают вакуум и заполняют H2 (3×) и полученную реакционную смесь перемешивают при комнатной температуре в атмосфере H2 в течение 72 часов. Катализатор отфильтровывают и промывают EtOH (3×10 мл). Растворитель удаляют в вакууме, получая остаток, который очищают перекристаллизацией из смеси ацетон/EtOAc/гексаны, получая целевой P074 (25 мг, 60% выход) в виде твердого, не совсем белого вещества. МС (ESI-)=473,4 (M-1), ЖХМС (ESI-)>90%. 1H-ЯМР (500 МГц).
Пример 53. Получение соединения P005
К суспензии NaH (60% в минеральном масле, 5 мг, 0,122 ммоль) в безводном ДМФА (1 мл) при 0°C добавляют раствор (3-1Н-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты I-8 (20 мг, 0,061 ммоль) в ДМФА (1 мл). Полученную смесь перемешивают при комнатной температуре в течение 0,5 часа, охлаждают до 0°C и добавляют 2-нафтоилхлорид (13 мг, 0,067 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Затем добавляют насыщенным водным раствор NH4Cl (1 мл) и смесь экстрагируют смесью эфир/EtOAc 8:2 (2×15 мл). Объединенные органические слои сушат над MgSO4, смесь фильтруют, растворитель удаляют и неочищенный продукт очищают колоночной хроматографией, используя метиленхлорид, 2% MeOH/метиленхлорид, получая 4,5 мг соединения P005.
Пример 54. Получение соединения P016.
Синтез трифтор-N-(3-1Н-индол-7-илпропионил)метансульфонамида, I-17. К суспензии NaH (60% в минеральном масле, 360 мг, 9,0 ммоль) в ДМФА (30 мл) добавляют 3-(1Н-индол-7-ил)акриловую кислоту, I-7 (560 мг, 3,0 ммоль) при 0°C. Полученной смеси дают возможность нагреться до комнатной температуры, перемешивают в течение 2 часов и затем снова охлаждают до 0°C. К полученному раствору добавляют 2-бромметилнафталин (680 мг, 3,1 ммоль) при 0°C, полученной смеси дают возможность нагреться до комнатной температуры и перемешивают в течение 16 часов, затем реакционную смесь охлаждают до 5°C и подкисляют, добавляя водную 10% HCl до достижения pH 1. Полученную смесь экстрагируют CH2Cl2/MeOH(9/1,3×50 мл). Объединенные органические экстракты сушат (Na2SO4), фильтруют и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2/EtOAc/гексан=1:8-1:2), получая целевую акриловую кислоту, I-17 (400 мг, 41%) в виде белого твердого вещества. МС(ESI-) m/z (326, 100%), ЖХМС (ESI-)>95%. 1H-ЯМР (CDCl3), 13C-ЯМР (CDCl3).
Синтез соединения P016. К раствору трифторметилсульфонамида (17 мг, 0,11 ммоль), 4-диметиламинопиридина (DMAP, 20 мг, 0,18 ммоль) и гидрохлорида 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 32 мг, 0,18 ммоль) в CH2Cl2 (5 мл) при перемешивании при комнатной температуре добавляют промежуточный продукт I-17 (33 мг, 0,1 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 72 часов, затем охлаждают до 5°C, подкисляют водной HCl (10%) до рH=1 и экстрагируют CH2Cl2/MeOH (9/1, 3×10 мл). Объединенные органические слои сушат (Na2SO4), растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2; EtOAc/гексан=1:4-2:1), получая соединение P016 (26 мг, 56%) в виде ярко-желтого твердого вещества. 1H ЯМР (500 МГц, CDCl3); 6,62 (д, J=16,0 Гц, 1H), 7,01 (д, J=3,5 Гц, 1H), 7,12 (т, J=7,5 Гц, 1H), 7,19 (т, J=8,0 Гц, 1H), 7,25-7,30 (м, 3H), 7,43-7,48 (м, 2H), 7,70 (м, 1H), 7,78-7,81 (м, 3H), 8,45 (д, J=15 Гц, 1H). ЖХ/МС (99%) ESI- Вычислено: 458,5 m/z, найдено: 457,4 m/z (M-1).
Пример 55. Получение соединения P003
К суспензии ((E)-3-1Н-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты I-8 (20 мг, 0,062 ммоль) в ледяной уксусной кислоте (1 мл) небольшими порциями при 0°C добавляют цианоборогидрид натрия (95%, 8 мг, 0,124 ммоль). Смеси дают возможность нагреться до комнатной температуры и перемешивают в течение 4 часов. Реакционную смесь концентрируют и затем гасят водой (1 мл). Значение рН смеси доводят до 5 добавлением водного 5% NaHCO3 и смесь экстрагируют метилeнхлоридом (2×5 мл). Объединенные органические слои промывают водой и сушат (MgSO4). Раствор фильтруют и растворитель удаляют при пониженном давлении, получая продукт, I-18. Полученный продукт используют далее без дополнительной очистки. К суспензии карбоната калия (17 мг, 0,123 ммоль) в безводном ДМФА (1 мл) добавляют индолин I-18 (20 мг, 0,061 ммоль), 2-бромметилнафталин (13,5 мг, 0,061 ммоль) и йодид калия (10 мг, 0,061 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 дней. Добавляют воду (6 мл) и смесь экстрагируют эфиром (2×10 мл). Объединенные органические слои промывают водой (2 мл), насыщенным раствором соли (2 мл), сушат над MgSO4 и концентрируют в вакууме. Неочищенный продукт очищают препаративной хроматографией, получая продукт P003.
Пример 56. Получение соединения P045
К суспензии ((E)-3-1Н-индол-7-илакрилоил)амида тиофен-2-сульфоновой кислоты I-4 (120 мг, 0,39 ммоль) в уксусной кислоте (ледяная, 10 мл) небольшими порциями при комнатной температуре добавляют цианоборогидрид натрия (95%, 150 мг, 2,5 ммоль). Полученную смесь нагревают и перемешивают при 70°C в течение 4 часов. Реакционную смесь охлаждают до 0 °C и реакцию гасят водой (10 мл) при 0°C. Смесь подкисляют водной HCl (10%) до рH=1, затем экстрагируют CH2Cl2/MeOH (20:1,3×30 мл). Объединенные органические слои сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, EtOAc/гексан=1:5-1:1) с получением целевого индолина I-18 (80 мг, 65%) в виде белого твердого вещества. МС(ESI-)m/z=335,2. К раствору полученного индолина I-18 (37 мг, 0,11 ммоль) в CH2Cl2 (7 мл) при перемешивании добавляют триэтиламин (21 мг, 0,22 ммоль) и затем 2-нафталинсульфонилхлорид (25 мг, 0,11 ммоль) при 0°C. Полученной смеси дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 24 часов. Реакционную смесь охлаждают до 0°C, затем подкисляют водной HCl (10%) до рH=1, затем экстрагируют CH2Cl2/MeOH (10:1, 3×20 мл). Объединенные органические слои сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, EtOAc/гексан=1:5-1:1), получая целевой продукт P045 (12 мг, 21%) в виде светло-желтого твердого вещества. МС(ESI-) m/z=523,5, 100%, ЖХМС(ESI)>70%. 1H ЯМР (500 МГц, CD3OD) 2,11 (т, J=8,0 Гц, 2H), 4,08 (т, J=8,0 Гц, 2H), 6,62 (д, J=16,0 Гц, 1H), 7,01 (дд, J=7,5, 1,0 Гц, 1H) 7,11 (дд, J=5,0, 4,0 Гц, 1H), 7,19 (дд, J=7,5, 7,5 Гц, 1H), 7,36 (дд, J=8,5, 2,0 Гц, 1H), 7,60 (м, 1H), 7,67 (м, 2H), 7,73 (дд, J=5,5, 1,5 Гц, 1H), 7,79 (дд, J=3,5, 0,5 Гц, 1H), 7,87 (м, 2H), 7,93 (д, J=8,5 Гц, 1H), 8,06 (с, 1H), 8,19 (д, J=16,0 Гц, 1H).
Пример 57. Получение соединения P085.
Метиловый эфир (E)-3-(3-ацетил-1Н-индол-7-ил)акриловой кислоты, I-19. В предварительно высушенную круглодонную колбу (100 мл) загружают метиловый эфир I-2a (645 мг, 3 ммоль) в ДМФА (5 мл) и по каплям добавляют POCl3 (330 мкл) при 0-5°C. Полученной реакционной смеси дают возможность нагреться до комнатной температуры, смесь перемешивают при комнатной температуре в течение 20 минут, затем нагревают до 40°C и перемешивают при 40°C в течение 1 часа. Реакционную смесь охлаждают до 0°C и выливают в водную ледяную суспензию (50 мл), затем добавляют водный NaOH (0,5 г в 10 мл воды). Смесь экстрагируют EtOAc (3×50 мл), объединенные органические слои промывают водой (3×50 мл), сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая целевой I-19 (450 мг, 58% выход) в виде желтого твердого вещества. МС (ESI-)=256,3 (M-1). 1H-ЯМР (500 МГц, CDCl3).
Метиловый эфир (E)-3-[3-ацетил-1-(2,4-дихлорбензил)-1Н-индол-7-ил]акриловой кислоты, I-20. В круглодонную колбу (250 мл) загружают суспензию соединения I-19 (400 мг, 1,6 ммоль), KI (300 мг) и CsCO3 (600 мг) в ДМФА (40 мл) при комнатной температуре, при перемешивании добавляют 2,4-дихлорбензилхлорид (350 мг, 1,8 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 72 часов. Смесь охлаждают до 0°C, добавляют водный NH4Cl (насыщенный, 10 мл), затем экстрагируют EtOAc (2×100 мл), объединенные органические слои промывают водой (3×100 мл), сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2; EtOAc/гексан=1:8-1:2), получая соединение I-20 (480 мг, 74% выход) в виде желтого твердого вещества. МС (ESI+)=416,7 (M). 1H-ЯМР (500 МГц, CDCl3).
(E)-3-[3-Ацетил-1-(2,4-дихлорбензил)-1Н-индол-7-ил]акриловая кислота, I-21. В круглодонную колбу (50 мл) загружают раствор NaOH (300 мг, 7,5 ммоль) в EtOH (20 мл) и H2O (10 мл), добавляют соединение I-20 (300 мг, 0,7 ммоль) при 5°C. Полученную реакционную смесь нагревают до 50°C и перемешивают при 50°C в течение 5 часов. Реакционную смесь охлаждают до 0°C и подкисляют водной HCl (10%) до pH=1, затем добавляют воду (100 мл) и экстрагируют смесью дихлорметан-MeOH (10:1, 3×20 мл). Объединенные органические слои сушат над Na2SO4 и растворитель удаляют в вакууме, получая неочищенный продукт, который очищают перекристаллизацией из смеси ацетон/EtOAc/Hex, получая кислоту I-21 (260 мг, 90% выход) в виде желтого твердого вещества. МС (ESI-)=386,4 (M-2). ЖХМС(ESI-)>90%. 1H-ЯМР (500 МГц).
Синтез соединения P085. В круглодонную колбу (25 мл), в которую добавлены раствор 2-тиофенсульфонамида (28 мг, 0,17 ммоль), 4-диметиламинопиридин (DMAP, 23 мг, 0,3 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 40 мг, 0,4 ммоль) в (5 мл) при комнатной температуре добавляют акриловую кислоту I-21 (60 мг, 0,16 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 72 часов, затем охлаждают до 0°C. После добавления MeOH (1 мл) и воды (5 мл) реакционную смесь подкисляют водным HCl (10%) до pH=1, затем экстрагируют CH2Cl2/MeOH (9/1, 3×5 мл). Объединенные органические слои сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2/EtOAc/гексан=1:1:4, EtOAc), получая целевой продукт P085 (35 мг, 40% выход) в виде ярко-желтого твердого вещества. 1H-ЯМР (500 МГц, CDCl3); 2,53 (c, 3H), 5,53 (c, 2H), 6,20 (д, J=15,0 Гц, 1H), 6,42 (д, J=8,5 Гц, 1H), 7,07 (дд, J=8,0, 3,0 Гц, 1H), 7,16 (т, J=4,5 Гц, 1H), 7,26-7,30 (м, 2H), 7,41 (д, J=2,0 Гц, 1H), 7,69 (с, 1H), 7,74 (дд, J=5,5, 1,5 Гц, 1H), 7,91-7,94 (м, 2H), 8,57 (дд, J=7,0, 2,0 Гц, 1H). ЖХ/МС (78%) ESI-: вычислено: 533,5 m/z, найдено: 535,1 m/z (M).
Пример 58. Получение соединения P091
Метиловый эфир (E)-3-[1-(2,4-дихлорбензил)-3-изопропенил-1Н-индол-7-ил] акриловой кислоты, I-22. В предварительно высушенную круглодонную колбу (250 мл) загружают раствор Ph3PCH3Br (1300 мг, 0,6 ммоль) в ТГФ (55 мл), при перемешивании при 0°С добавляют BuLi (2,0 мл, 1,6М в эфире). Полученной реакционной смеси дают возможность нагреться до комнатной температуры, затем смесь нагревают до 40°C и перемешивают при 40°C в течение 2 часов. Смесь охлаждают до 0°C и к смеси добавляют соединение I-20 (460 мг, 1,1 ммоль). Смесь нагревают до комнатной температуры, затем нагревают до 40°C и перемешивают при 40°C в течение 3 часов. После охлаждения до 0°C реакцию гасят водным NH4Cl (насыщенный, 5 мл), затем экстрагируют EtOAc (2×100 мл), объединенные органические слои промывают водой (3×100 мл), сушат (Na2SO4), растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, EtOAc/гексан=1:8-1:4), получая целевой I-22 (380 мг, 85% выход)) в виде ярко-желтого твердого вещества. МС (ESI+): 414,8 (M). 1H-ЯМР (500 МГц, CDCl3).
Метиловый эфир 3-[1-(2,4-дихлорбензил)-3-изопропил-1Н-индол-7-мл]пропионовой кислоты, I-23. В круглодонную колбу (50 мл) загружают раствор соединения I-22 (260 мг, 0,6 ммоль) в EtOH (55 мл) и добавляют водный Pd/C (30 мг) при комнатной температуре. Затем в колбе создают вакуум и загружают H2 3 раза, полученную реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Катализатор отфильтровывают и промывают EtOAc (3×50 мл), фильтрат промывают водой (3×50 мл), сушат над Na2SO4 и растворитель удаляют в вакууме, получая соединение I-23 (240 мг, 95% выход) в виде твердого, не совсем белого вещества. 1H-ЯМР (500 МГц).
3-[1-(2,4-дихлорбензил)-3-изопропил-1Н-индол-7-ил]пропионовая кислота, I-24. В круглодонную колбу (50 мл) загружают раствор NaOH (200 мг, 5 ммоль) в EtOH (30 мл) и H2O (20 мл) при 5°C добавляют соединение I-23 (180 мг, 0,4 ммоль). Полученную реакционную смесь нагревают до 50°C и перемешивают при 50°C в течение 5 часов. Реакционную смесь охлаждают до 0°C и подкисляют с водной HCl (10%) до pH=1, затем добавляют воду (100 мл) и экстрагируют смесью дихлорметан-MeOH (10:1, 3×20 мл). Объединенные органические слои сушат над Na2SO4 и растворитель удаляют в вакууме, получая неочищенный продукт, который очищают перекристаллизацией из смеси ацетон/EtOAc/Hex, получая целевую I-24 (150 мг, 90% выход) в виде желтого твердого вещества. МС (ESI-)=388,3 (M-1). ЖХМС(ESI-)>85%. 1H-ЯМР (500 МГц).
Синтез соединения P091. В круглодонную колбу (25 мл), в которой находится раствор 2-тиофенсульфонамида (28 мг, 0,16 ммоль), 4-диметиламинопиридина (DMAP, 30 мг, 0,3 ммоль) и 1-[3-(диметиламино)пропил]-3-этилкарбодиимида гидрохлорида (EDCI, 40 мг, 0,4 ммоль) в CH2Cl2 (12 мл) добавляют пропионовую кислоту I-24 (60 мг, 0,15 ммоль) при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение 48 часов, затем охлаждают до 0 °C. После добавления MeOH (1 мл) и воды (5 мл) реакционную смесь подкисляют водным HCl (10%) до pH=1, затем экстрагируют CH2C12 (3×5 мл). Объединенные органические слои сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2, EtOAc/гексан=1:1, EtOAc), получая целевой P091 (40 мг, 40% выход) в виде твердого, не совсем белого вещества. МС(ESI-) m/z=535,2, (M+); ЖХМС(ESI-) >95%. 1H-ЯМР (500 МГц).
Пример 59. Получение соединения P092.
В круглодонную колбу (25 мл) загружают раствор 4,5-дихлортиофенсульфонамида (21 мг, 0,09 ммоль), 4-диметиламинопиридин (DMAP, 15 мг, 0,2 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 20 мг, 0,2 ммоль) в CH2Cl2 (7 мл) при комнатной температуре добавляют акриловую кислоту I-24 (30 мг, 0,08 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 48 часов, затем охлаждают до 0°C. После добавления MeOH (1 мл) и воды (5 мл) реакционную смесь подкисляют водным HCl (10%) до pH=1, затем экстрагируют CH2Cl2 (3×5 мл). Объединенные органические слои сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2, EtOAc/гексан=1:1, EtOAc) с получением целевого P092 (15 мг, 32% выход) в виде твердого не совсем белого вещества. МС(ESI-)m/z=603,2, (M-1); ЖХМС(ESI-)> 95%. 1H-ЯМР (500 МГц).
Пример 60. Получение соединения P093
В емкость (10 мл) при перемешивании загружают раствор P091 (3 мг, 0,05 ммоль) в ДМСО-d6 (1 мл), медленно при 0°С добавляют HCl (концентрированная, 3 мл). Полученной смеси дают возможность нагреться до комнатной температуры и смесь перемешивают при комнатной температуре в течение 4 часов, затем охлаждают до 0°C и добавляют воду (10 мл). Образовавшийся твердый продукт отфильтровывают, промывают водой и сушат на воздухе и затем в вакууме, получая оксииндол-производное P093 (1,5 мг, 48% выход) в виде твердого, не совсем белого вещества. 1H ЯМР (500 МГц, ацетон-d6); 0,98 (д, J=7,0 Гц, 3H), 1,06 (д, J=7,0 Гц, 3H), 2,48-2,52 (м, 2H), 2,68-2,71 (м, 2H), 5,15 (м, 2H), 6,94 (д, J=8,0 Гц, 1H), 6,97 (д, J=8,0 Гц, 1H), 7,03 (д, J=7,5 Гц, 1H), 7,18 (т, J=3,5 Гц, 1H), 7,23 (д, J=7,5 Гц, 1H), 7,28 (дд, J=8,0, 3,0 Гц, 1H), 7,45 (д, J=2,5 Гц, 1H), 7,75 (д, J=3,0 Гц, 1H), 7,90 (д, J=5,0 Гц, 1H). ЖХ/МС (97%) ESI-. Вычислено: 551,5 m/z, найдено: 551,2 m/z (M).
Пример 61. Получение соединения P069
Этиловый эфир (E)-3-(3-формил-1-метил-1Н-индол-7-ил)акриловой кислоты, I-23. В предварительно высушенную круглодонную колбу (100 мл) загружают раствор соединения I-2 (2,6 г, 13 ммоль) в ДМФА (5,2 мл) и по каплям добавляют POCl3 (1250 мкл) при 0-5°C. Полученной реакционной смеси дают возможность нагреться до комнатной температуры, перемешивают при комнатной температуре в течение 20 минут и затем нагревают до 40°C и перемешивают при 40°C в течение 1 часа. Реакционную смесь охлаждают до 0°C и выливают в суспензию воды со льдом (50 мл), затем добавляют водный NaOH (1,5 г в 20 мл воды). Смесь экстрагируют EtOAc (3×100 мл), объединенные органические слои промывают водой (3×100 мл), сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая целевой I-23 (2,0 г, 75%) в виде желтого твердого вещества. МС (ESI-): 242,2 (M-1). 1H-ЯМР (500 МГц, CDCl3).
Этиловый эфир (E)-3-[1-(2,4-дихлорбензил)-3-формил-1Н-индол-7-ил]акриловой кислоты, I-24. В круглодонную колбу (250 мл) загружают суспензию соединения I-23 (480 мг, 2 ммоль), KI (500 мг) и CsCO3 (1 г) в ДМФА (40 мл) при перемешивании добавляют 2,4-дихлорбензилхлорид (440 мг, 24 ммоль) при комнатной температуре. Полученную реакционную смесь перемешивают при комнатной температуре в течение 72 часов. Смесь охлаждают до 0°C и добавляют водный NH4Cl (нас. 10 мл), затем экстрагируют EtOAc (2×100 мл), объединенные органические слои промывают водой (3×100 мл), сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2; EtOAc/гексан=1:4-1:2), получая целевой I-24 (400 мг, 50%) в виде желтого твердого вещества. МС (ESI+): 402,3 (M+1). 1H-ЯМР (500 МГц, CDCl3).
(E)-3-[1-(2,4-Дихлорбензил)-3-формил-1Н-индол-7-ил]акриловая кислота, I-25: В круглодонную колбу (50 мл) загружают раствор NaOH (80 мг, 2 ммоль) в EtOH (5 мл) и H2О (3 мл), добавляют соединение I-24 (80 мг, 0,2 ммоль) при 5°C. Полученную реакционную смесь нагревают до 50°C и перемешивают при 50°C в течение 4 часов. Реакционную смесь охлаждают до 0°C и подкисляют водным HCl (10%) до pH=1, затем добавляют воду (20 мл) и экстрагируют смесью дихлорметан-MeOH (10:1, 3×20 мл). Объединенные органические слои сушат над Na2SO4 и растворитель удаляют в вакууме, получая неочищенный продукт, который очищают перекристаллизацией из смеси ацетон/EtOAc/Hex, получая соединение I-25 (60 мг, 80% выход) в виде желтого твердого вещества. МС (ESI-): 372,1 (M-2). 1H-ЯМР (500 МГц).
Синтез соединения P069. В круглодонную колбу (25 мл) загружают раствор 2-тиофенсульфонамида (9 мг, 0,05 ммоль), 4-диметиламинопиридин (DMAP, 20 мг, 0,2 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 25 мг, 0,3 ммоль) в (5 мл) и при комнатной температуре добавляют акриловую кислоту I-25 (9 мг, 0,03 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 72 часов, затем охлаждают до 0°C. После добавления MeOH (1 мл) и воды (5 мл) реакционную смесь подкисляют водным HCl (10%) до pH=1, затем экстрагируют CH2Cl2/MeOH (9/1, 3×5 мл). Объединенные органические слои сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2/EtOAc/гексан=1:1:4, EtOAc), получая целевой P069 (6,3 мг, 45% выход) в виде ярко-желтого твердого вещества. 1H ЯМР (500 МГц, метанол-d4); 5,70 (с, 2H), 6,13 (д, J=15,0 Гц, 1H), 6,36 (д, J=8,5 Гц, 1H), 7,09-7,11 (м, 2H), 7,23 (м, 1H), 7,29-7,35 (м, 2H), 7,60 (м, 1H), 7,70 (м, 1H), 7,89-7,94 (м, 2H), 8,34 (дд, J=9,0, 1,5 Гц, 1H), 9,95 (с, 1H). ЖХ/МС (71%) ESI- Вычислено: 519,4 m/z, найдено: 517,4 m/z (M-2).
Пример 62. Получение соединения P062
Этиловый эфир (E)-3-[1-(2,4-дихлорбензил)-3-гидроксиметил-1Н-индол-7-ил]акриловой кислоты, I-26: В круглодонную колбу (100 мл) загружают суспензию соединения I-24 (100 мг, 0,25 ммоль) в EtOH (5 мл) и c перемешиванием добавляют NaBH4 (100 мг, избыток) при 0°C. Полученной реакционной смеси дают возможность нагреться до комнатной температуры и смесь перемешивают при комнатной температуре в течение 4 часов. Смесь охлаждают до 0°C, добавляют водный NH4Cl (насыщенный, 5 мл), затем экстрагируют CH2Cl2 (2×30 мл), объединенные органические слои промывают водой (3×30 мл), сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2; EtOAc/гексан (элюирование с градиентом от 1:4 до 1:1), получая спирт I-26 (85 мг, 84% выход) в виде твердого не совсем белого вещества. МС (APCI+): 406,2 (M+2). 1H-ЯМР (500 МГц, CDCl3).
(E)-3-[1-(2,4-дихлорбензил)-3-гидроксиметил-1Н-индол-7-ил]акриловой кислоты, I-27. В круглодонную колбу (50 мл) загружают раствор соединения I-26 (80 мг, 0,2 ммоль) в MeOH (15 мл) при комнатной температуре добавляют водный NaOH (2н., 0,5 мл). Полученную реакционную смесь нагревают до 80°C и перемешивают при 80°C в течение 48 часов. Реакционную смесь охлаждают до 0°C и подкисляют водной HCl (10%) до достижения pH=1, затем добавляют воду (20 мл) и экстрагируют смесью дихлорметан-MeOH (10:1, 3×20 мл). Объединенные органические слои сушат над Na2SO4 и растворитель удаляют в вакууме, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2; EtOAc/гексан=1:4-1:1, EtOAc), получая кислоту I-27 (65 мг, 80% выход) в виде твердого не совсем белого вещества. МС (ESI)=374,3 (M-2). ЖХМС(ESI-)>90%. 1H-ЯМР(500 МГц).
Синтез соединения P062. В круглодонную колбу (25 мл) загружают раствор 2-тиофенсульфонамида (35 мг, 0,15 ммоль), 4-диметиламинопиридин (DMAP, 45 мг, 0,5 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 60 мг, 0,6 ммоль) в CH2Cl2 (5 мл) и добавляют акриловую кислоту I-27 (25 мг, 0,07 ммоль) при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение 48 часов, затем охлаждают до 0°C. После добавления MeOH (1 мл) и воды (5 мл) реакционную смесь подкисляют водным HCl (10%) до достижения pH=1, затем экстрагируют CH2Cl2 (3×5 мл). Объединенные органические слои сушат над безводным Na2SO4 и растворитель удаляют при пониженном давлении, получая неочищенный продукт который очищают флэш-хроматографией (силикагель, CH2Cl2, EtOAc/гексан=1:1, EtOAc), получая соединение P062 (16 мг, 35% выход) в виде твердого не совсем белого вещества. МС (ESI-) m/z=519,3, (M-2); ЖХМС(ESI) >85%. 1H-ЯМР (500 МГц).
Пример 63. Получение соединения P070
Этиловый эфир (E)-3-[1-(2,4-дихлорбензил)-3-метоксиметил-1Н-индол-7-ил]акриловой кислоты, I-28. В круглодонную колбу (25 мл) загружают суспензию NaH (15 мг, 0,25 ммоль) в ДМФА (5 мл), при перемешивании при 0°С добавляют соединение I-26 (30 мг, 0,08 ммоль). Полученной смеси дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 90 минут, затем охлаждают до 0°C и добавляют МeI (1 мл). Полученной реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 72 часов, охлаждают до 0°C и добавляют водный NH4Cl (насыщенный, 1 мл), затем экстрагируют EtOAc (2×5 мл), объединенные органические слои промывают водой (2×5 мл), сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, EtOAc/гексан=1:8), получая соединение I-28 (17 мг, 50% выход) в виде твердого не совсем белого вещества. 1H-ЯМР (500 МГц, CDCl3).
(E)-3-[1-(2,4-Дихлорбензил)-3-метоксиметил-1Н-индол-7-ил]акриловая кислота, I-29. В круглодонную колбу (50 мл) загружают раствор NaOH (50 мг, 1,2 ммоль) в MeOH (1 мл) в воде (2 мл) и добавляют сложный эфир I-28(17 мг, 0,04 ммоль) при комнатной температуре. Полученную реакционную смесь нагревают до 50°C и перемешивают при 50°C в течение 4 часов, затем охлаждают до 5°C, подкисляют водной HCl (10%) до pH=1. После разбавления водой (10 мл) смесь экстрагируют со смесью дихлорметан-MeOH (10:1,3×5 мл). Объединенные органические слои сушат над Na2SO4 и растворитель удаляют в вакууме, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2; EtOAc/гексан=1:4-1:1, EtOAc) с получением кислоты I-29 (15 мг, 90% выход) в виде твердого не совсем белого вещества. МС (ESI-)=388,3 (M-2). 1H-ЯМР (500 МГц).
Синтез соединения P070. В круглодонную колбу (25 мл) загружают раствор 2-тиофенсульфонамида (15 мг, 0,09 ммоль), 4-диметиламинопиридина (DMAP, 20 мг, 0,2 ммоль) и гидрохлорида 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 25 мг, 0,25 ммоль) в CH2Cl2 (5 мл), при комнатной температуре добавляют акриловую кислоту I-29 (10 мг, 0,025 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 48 часов, затем охлаждают до 0°C. После добавления MeOH (1 мл) и воды (5 мл) реакционную смесь подкисляют добавлением водного HCl (10%) до достижения рH=1, и экстрагируют CH2Cl2 (3×5 мл). Объединенные органические слои сушат над безводным Na2SO4 и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2, EtOAc/гексан=1:1), получая соединение P070 (1,5 мг, 10% выход) в виде твердого не совсем белого вещества. 1H ЯМР (500 МГц, метанол-d4); 3,39 (с, 3H), 4,67 (с, 2H), 5,70 (с, 2H), 6,16 (д, J=15,5 Гц, 1H), 6,36 (д, J=9,5 Гц, 1H), 7,05 (дд, J=8,0, 3,0 Гц, 1H), 7,12 (т, J=8,0 Гц, 1H), 7,20 (м, 1H), 7,25 (м, 1H), 7,31 (м, 1H), 7,74 (д, J=7,5 Гц, 1H), 7,84 - 7,87 (м, 2H), 7,91 (д, J=15,0 Гц, 1H). ЖХ/МС (78%) ESI- Вычислено: 535,5 m/z, найдено: 535,0 m/z (M-1).
Пример 64. Получение соединения P060
Синтез метилового эфира (E)-3-(5-фтор-1Н-индол-7-ил)акриловой кислоты метил, I-30. К смеси 7-бром-5-фтор-1Н-индола [который получают в соответствии с известным способом (Dobbs, A., J. Org. Chem., 66, 638-641 (2001)] (400 мг, 1,87 ммоль) и метилакрилата (241 мг, 2,8 ммоль) в триэтиламине (1,5 мл) в атмосфере аргона добавляют ацетат палладия(II) (43 мг, 0,19 ммоль) и три-o-толилфосфин (170 мг, 0,56 ммоль), реакционную смесь перемешивают при 100°C в течение 4 часов в герметичном реакторе и затем охлаждают до комнатной температуры, разбавляют CH2Cl2 (50 мл), промывают водой (3×30 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя EtOAc/гексан в качестве элюента, в результате получают 300 мг соединения I-30 в виде желтого твердого вещества. 1H-ЯМР (500 МГц, CDCl3).
Синтез (E)-3-(5-фтор-1Н-индол-7-ил)акриловой кислоты, I-31. К раствору соединения I-30 (219 мг, 1 ммоль) в ТГФ (5 мл) и метаноле (4 мл) добавляют NaOH водный (4 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем значение pH доводят до кислотного добавлением 2н. HCl водной. Реакционную смесь экстрагируют EtOAc (2×30 мл). Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 180 мг кислоты I-31. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 204,2 (M-1).
Синтез тиофен-2-сульфоновой кислоты [(E)-3-(5-фтор-1Н-индол-7-ил)акрилоил]амида, I-32. Смесь кислоты I-31 (180 мг, 0,88 ммоль), 2-тиофенсульфонамида (172 мг, 1,2 ммоль), 4-диметиламинопиридина (215 мг, 1,77 ммоль) и EDCI (336 мг, 1,77 ммоль) в дихлорметане (20 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя смесь метанол/CH2Cl2 в качестве элюента и получая 150 мг сульфонамида I-32. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P060. К раствору соединения I-32 (45 мг, 0,13 ммоль) в ДМФА (2 мл) добавляют NaH (60% в масле, 16 мг, 0,4 ммоль) при 0°C, полученную смесь перемешивают при комнатной температуре в течение 1 часа и затем добавляют 2-(бромметил)нафталин (57 мг, 0,26 ммоль). Полученную смесь перемешивают при комнатной температуре в течение ночи и разбавляют CH2Cl2 (10 мл). Реакционную смесь промывают разбавленной водной HCl (2×8 мл), водой (4×8 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя смесь метанол/CH2Cl2 в качестве элюента и получая 30 мг соединения P060. 1Н-ЯМР (500 МГц, ДМСО-d6) МС (ESI-): 489,4 (M-1), ЖХ-МС: 80%.
Пример 65. Получение соединения P061.
К раствору соединения I-32 (45 мг, 0,13 ммоль) в ДМФА (2 мл) добавляют NaH (60% в масле, 16 мг, 0,4 ммоль) при 0°C, смесь перемешивают при комнатной температуре в течение 1 часа и затем добавляют 2,4-дихлорбензилхлорид (51 мг, 0,26 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и разбавляют CH2Cl2 (10 мл). Реакционную смесь промывают разбавленной водной HCl (2×8 мл), водой (4×8 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя метанол/CH2Cl2 в качестве элюента и получая 55 мг соединения P061. 1H-ЯМР (500 МГц, ДМСО-d6) МС (ESI-): 509,3 (M-1), ЖХ-МС: 93%.
Пример 66. Получение соединения P063.
Синтез метилового эфира (E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акриловой кислоты, I-33. К смеси 7-бром-5-фтор-3-метил-1Н-индола [который получают аналогично соединению I-30 в соответствии с методикой, описанной Dobbs, A., J. Org. Chem., 66, 638-641 (2001)], (870 мг, 3,8 ммоль) и метилакрилата (819 мг, 9,5 ммоль) в триэтиламине (4 мл) добавляют ацетат палладия(II) (112 мг, 0,5 ммоль) и три-o-толилфосфин (428 мг, 1,42 ммоль) в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивают при 100oC в течение 3 часов в герметичном реакторе под давлением и затем охлаждают до комнатной температуры. Реакционную смесь разбавляют CH2Cl2 (70 мл), промывают водой (3×40 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя смесь метанол/дихлорметан в качестве элюента и получая 690 мг соединения I-33 в виде твердого вещества. 1H-ЯМР (500 МГц, CDCl3).
Синтез (E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акриловой кислоты, I-34. К раствору соединения I-33 (233 мг, 1 ммоль) в ТГФ (5 мл) и метаноле (5 мл) добавляют водный NaOH (3 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем значение pH доводят до кислотного добавлением водного 2н. HCl. Реакционную смесь экстрагируют EtOAc (2×40 мл). Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 210 мг кислоты I-34. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез [(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты, I-35. Смесь кислоты I-34 (100 мг, 0,46 ммоль), 2-тиофенсульфонамида (90 мг, 0,55 ммоль), 4-диметиламинопиридина (112 мг, 0,92 ммоль) и EDCI (176 мг, 0,92 ммоль) в дихлорметане (10 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl и водой. Образовавшееся твердое вещество отфильтровывают и промывают водой, дихлорметаном, получая 55 мг соединения 4. Оставшуюся дихлорметановую маточную жидкость очищают колоночной хроматографией на силикагеле, используя смесь метанол/CH2Cl2 в качестве элюента и получая 45 мг ацилсульфонамида I-35. Получают 100 мг соединения I-35. 1H-ЯМР (500 МГц, ДМСО-d6).
Получение соединения P063.
К раствору соединения I-35 (45 мг, 0,12 ммоль) в ДМФА (3 мл) добавляют NaH (60% в масле, 15 мг, 037 ммоль) при 0oC. Полученную смесь перемешивают при комнатной температуре в течение 1 часа и затем добавляют 2,4-дихлорбензилхлорид (47 мг, 0,24 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и разбавляют CH2Cl2 (12 мл). Реакционную смесь промывают разбавленной водной HCl (2×8 мл), водой (4×8 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая 55 мг соединения P063. 1H ЯМР (ДМСО-d6) 2,25 (с, 3H), 5,52 (с, 2H), 6,14 (д, J=8,0 Гц, 1H), 6,25 (д, J=15,0 Гц, 1H), 7,02 (дд, J=10,0, 2,0 Гц, 1H), 7,22 (м, 2H), 7,33 (с, 1H), 7,43 (дд, J=10,2,0 Гц, 1H), 7,51 (м, 1H), 7,66 (д, J=15,0 Гц, 1H), 7,76 (м, 1H), 8,01 (д, J=5,0 Гц, 1H), 12,4 (уш., 1H). ЖХ/МС (94%) ESI-Вычислено: 521,4 m/z;найдено: 521,6 m/z.
Пример 67. Получение соединения P065
К раствору соединения P063 (10 мг, 0,02 ммоль) в ДМСО (1 мл) по каплям добавляют концентрированную хлористоводородную кислоту (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 5 часов, затем разбавляют CH2Cl2, промывают водой (4×10 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя EtOAc/гексан в качестве элюента и получая 4 мг соединения P065. 1H-ЯМР (500 МГц, CDCl3), МС (ESI-): 537,3 (M-1), ЖХ-МС: 77%.
Пример 68. Получение соединения P064.
К раствору соединения I-35 (45 мг, 0,12 ммоль) в ДМФА (3 мл) добавляют NaH (60% в масле, 15 мг, 037 ммоль) при 0°C, полученную реакционную смесь перемешивают при комнатной температуре в течение 1 часа и затем добавляют 2-(бромметил)нафталин (53 мг, 0,24 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и разбавляют CH2Cl2 (12 мл). Реакционную смесь промывают разбавленной водной HCl (2×8 мл), водой (4×8 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают смесью эфир/гексан, получая 55 мг соединения P064. 1H-ЯМР (500 МГц, ДМСО-d6) МС (ESI-): 503,4 (М-1), ЖХ-МС: 95%.
Пример 69. Получение соединения P066.
К раствору соединения P064 (10 мг, 0,02 ммоль) в ДМСО (1 мл) по каплям добавляют концентрированную хлористоводородную кислоту (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 5 часов, разбавляют CH2Cl2 и промывают водой (4×10 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя EtOAc/гексан в качестве элюента и получая 4 мг соединения P066. 1H-ЯМР(CDCl3) 1,61 (д, J=7,5 Гц, 3H), 3,62 (кв., J=7,5 Гц, 1H), 5,26 (с, 2H), 6,01 (д, J=15,0 Гц, 1H), 6,83 (дд, J=10,2,0 Гц, 1H), 7,04 (м, 1H), 7,13 (м, 1H), 7,29 (дд, J=8, 2,0 Гц, 1H), 7,43 (м, 2H), 7,69 (м, 3H), 7,76 (м, 2H), 7,91 (м, 1H), 8,01 (д, J=15,0 Гц, 1H). ЖХ/МС (80%) ESI- Вычислено: 519,6 m/z; найдено: 519,4 m/z.
Пример 70. Получение соединения P067.
Синтез [(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]амида 4,5-дихлортиофен-2-сульфоновой кислоты, I-36. Смесь кислоты I-34 (100 мг, 0,46 ммоль), 4,5-дихлор-2-тиофенсульфонамида (128 мг, 0,55 ммоль), 4-диметиламинопиридина (112 мг, 0,92 ммоль) и EDCI (176 мг, 0,92 ммоль) в дихлорметане (10 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной НCl и водой. Образовавшийся твердый продукт отфильтровывают и промывают водой, дихлорметаном, получая 140 мг соединения I-36. 1H-ЯМР (500 МГц, ДМСО-d6) МС (ESI-): 433,1 (M-1)ЖХ-МС: 97.
Синтез соединения P067. К раствору сульфонамида I-36 (52 мг, 0,12 ммоль) в ДМФА (3 мл) при 0°С добавляют NaH (60% в масле, 15 мг, 037 ммоль), смесь перемешивают при комнатной температуре в течение 1 часа и затем добавляют 2,4-дихлорбензил (47 мг, 0,24 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и разбавляют CH2Cl2 (12 мл). Реакционную смесь промывают разбавленной HCl водной (2×8 мл), водой (4×8 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, элюируя смесью метанол/дихлорметан и получая 35 мг соединения P067. 1H ЯМР (ДМСО-d6) 2,25 (с,3H), 5,54 (с,2H), 6,13 (д, J=8,0 Гц, 1H), 6,21 (д, J=15,0 Гц, 1H), 7,04 (дд, J=10, 2,0 Гц, 1H), 7,23 (м, 1H), 7,38 (м, 2H), 7,45 (м, 1H), 7,74 (д, J=15,0 Гц, 1H), 7,90 (с, 1H), 12,5 (уш. с, 1H). ЖХ/МС (98%) ESI- Вычислено: 591,3 m/z; найдено: 591,1 m/z.
Пример 71. Получение соединения P068
К раствору соединения I-36 (52 мг, 0,12 ммоль) в ДМФА (3 мл) добавляют NaH (60% в масле, 15 мг, 0,37 ммоль) добавляют при 0°C. Смесь перемешивают при 0 °С в течение 1 часа и затем при комнатной температуре в течение 1 часа, затем добавляют 2-(бромметил)нафталин (53 мг, 0,24 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и разбавляют CH2Cl2 (12 мл). Реакционную смесь промывают разбавленной водной HCl(2×8 мл), водой (4×8 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, элюируя смесью метанол/дихлорметан и получая 44 мг соединения P068. 1H-ЯМР (500 МГц, ДМСО-d6) МС (ESI-): 571,3 (M-1) ЖХ-МС: 97%.
Пример 72. Получение соединения P077.
Смесь соединения P064 (25 мг, 0,05 ммоль) и Pd/C (5%, 50 мг) в метаноле (5 мл) и ТГФ (5 мл) гидрируют водородом при давлении 40 фунтов на кв. дюйм в течение ночи при комнатной температуре. Смесь фильтруют через целит и промывают метанолом. После удаления растворителя остаток промывают эфиром, получая 16 мг соединения P077 в виде твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 505,4 (M-1), ЖХ-МС: 86%.
Пример 73. Получение соединения P076.
К смеси соединения P068 (10 мг, 0,017 ммоль) и Cs2CO3 (17 мг, 0,052 ммоль) в ДМСО (1 мл) добавляют йодметан (2 капли). Реакционную смесь перемешивают при комнатной температуре в течение ночи, разбавляют EtOAc и затем промывают разбавленной водной HCl (2×8 мл), водой (4×8 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан в качестве элюента и получая 5 мг соединения P076. 1H ЯМР (ДМСО-d6) 2,33 (с, 3H), 3,18 (с, 3H), 5,57 (с, 2H), 6,89 (д, 15,0 Гц, 1H), 7,00 (дд, J=10,0, 2,0 Гц, 1H), 7,05 (с, 1H), 7,13 (м, 1H), 7,31 (м, 1H), 7,41 (м, 2H), 7,50 (м, 2H), 7,67 (м, 1H), 7,73 (д, J=8,5 Гц, 1H), 7,76 (м, 1H), 8,27 (д, J=15,0 Гц, 1H).
Пример 74. Получение соединения P058.
К раствору соединения P056 (7 мг, 0,014 ммоль) в ДМСО (1 мл) по каплям добавляют концентрированную хлористоводородную кислоту (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, затем разбавляют дихлорметаном (10 мл), промывают водой (4×6 мл) и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя смесь метанол/CH2Cl2 в качестве элюента и получая 6 мг соединения P058. 1H-ЯМР (500 МГц, CDCl3)МС(ESI-): 519,4 (M-1), ЖХ-МС: 76%.
Пример 75. Получение соединения P059.
К раствору соединения P057 (33 мг, 0,067 ммоль) в ДМСО (5 мл) по каплям добавляют концентрированную хлористоводородную кислоту (2 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, разбавляют EtOAc (40 мл), промывают водой (3×30 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя смесь метанол/CH2Cl2 в качестве элюента и получая 15 мг соединения P059. 1H ЯMР (ДМСО-d6) 1,49(д, J=7,5 Гц, 3H), 3,79 (кв., J=7,5 Гц, 1H), 5,22 (м, 2H), 6,18 (д, J=15,0 Гц, 1H), 7,07 (т, J=7,5 Гц, 1H), 7,20 (д, J=7,5 Гц, 1H), 7,27 (м, 2H), 7,46 (м, 3H), 7,66 (м, 2H), 7,73 (д, J=8,5 Гц, 1H), 7,85 (м, 2H), 7,97 (д, J=15,0 Гц, 1H), 8,13 (д, J=5,5 Гц, 1H), 12,3 (уш.с, 1H). ЖХ/МС (99%). ESI- Вычислено: 501,6 m/z; найдено: 501,4 m/z.
Пример 76. Получение соединения P107
Синтез (E)-3-[1-(2,4-дихлорбензил)-3-метил-2-оксо-2,3-дигидро-1Н-индол-7-ил]акриловой кислоты (I-37): К раствору соединения I-15 (135 мг, 0,375 ммоль) в ДМСО (3 мл) медленно при комнатной температуре добавляют 40 мл смеси AcOH/концентрированная HCl (4:1). Реакционную смесь перемешивают при комнатной температуре в течение 5 часов, разбавляют EtOAc, промывают водой (4×200 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая 105 мг соединения I-37. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P107. Смесь кислоты I-37 (56 мг, 0,15 ммоль), 4,5-дихлор-2-тиофенсульфонамида (42 мг, 0,18 ммоль), 4-диметиламинопиридина (37 мг, 0,3 ммоль) и EDCI (57 мг, 0,3 ммоль) в дихлорметане (8 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl (2×10 мл), водой (4×10 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан и метанол/дихлорметан в качестве элюентов и получая 40 мг соединения P107. 1H-ЯМР (500 МГц, ДМСО-d6) МС (ESI-): 589,0 (M-1), ЖХ-МС: 87%.
Пример 77. Получение соединения P112.
Синтез соединения (E)-3-(3-метил-1-нафталин-2-илметил-1Н-индол-7-ил)акриловой кислоты (I-39): Гидролиз I-39. К раствору кислоты I-7 (300 мг, 1,5 ммоль) в ДМФА (15 мл) при 0°С по каплям добавляют NaH (60% в масле, 180 мг, 4,5 ммоль) и перемешивают при комнатной температуре в течение 20 минут, затем при 0°С добавляют 2-(бромметил)нафталин (365 мг, 1,65 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и разбавляют CH2Cl2 (100 мл). Реакционную смесь промывают разбавленной водной HCl (2×100 мл), водой (4×100 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя твердый остаток промывают CH2Cl2, получая 165 мг соединения I-39. Выход: 32%. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез (E)-3-(3-метил-1-нафталин-2-илметил-2-оксо-2,3-дигидро-1Н-индол-7-ил)акриловой кислоты, (I-40). К раствору соединения I-39 (102 мг, 0,3 ммоль) в ДМСО (3 мл) медленно при комнатной температуре добавляют 40 мл смеси AcOH/концентрированная HCl (4:1). Реакционную смесь перемешивают при комнатной температуре в течение 4 часов, разбавляют EtOAc, промывают водой (4×200 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая 80 мг соединения I-40. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P112. Смесь кислоты I-40 (53 мг, 0,15 ммоль), 4,5-дихлор-2-тиофенсульфонамида (42 мг, 0,18 ммоль), 4-диметиламинопиридина (37 мг, 0,3 ммоль) и EDCI (57 мг, 0,3 ммоль) в дихлорметане (8 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl (2×10 мл), водой (4×10 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан и метанол/дихлорметан в качестве элюентов, получая 40 мг соединения P112. 1H-ЯМР (500 МГц, ДМСО-d6) МС (ESI-): 569,4 (М-1), ЖХ-МС: 82%.
Пример 78. Получение соединения P110.
К смеси соединения P057 (160 мг, 0,33 ммоль) в ТГФ (15 мл) и 95% трет-BuOH/H2O (30 мл) при комнатной температуре добавляют NBS (125 мг, 0,7 ммоль) в 95% трет-BuOH/H2O (4 мл) и ТГФ (2 мл). Реакционную смесь перемешивают при комнатной температуре в течение 4 часов, добавляют NBS (125 мг, 0,7 ммоль) и полученную смесь перемешивают при комнатной температуре в течение ночи. После удаления растворителя остаток растворяют в EtOAc и промывают водой (3×50 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая 135 мг соединения P 110. МС (ESI-): 581,1 (M-1), ЖХ-МС: 82%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 79. Получение соединения P111.
К раствору соединения P057 (58 мг, 0,1 ммоль) в ТГФ (6 мл) и воде (3 мл) при комнатной температуре добавляют NaOH водный (2н., 0,15 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем подкисляют до кислотной pH добавлением 2н. HCl (водн.). Реакционную смесь экстрагируют EtOAc. Органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя, остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан и метанол/дихлорметан в качестве элюентов и получая 6 мг соединения P111. 1H ЯМР (CDCl3) 1,73 (с, 3H), 3,01 (с, 1H), 5,21 (с, 2H), 6,02 (д, J=15,0 Гц, 1H), 6,99 (т, J=8,0 Гц, 1H), 7,13 (м, 2H), 7,27 (м, 1H), 7,43 (м, 3H), 7,71 (м, 5H), 7,91 (м, 1H), 8,00 (д, J=15,0 Гц, 1H), 8,39 (с, 1H). ЖХ/МС (81%) ESI+ Вычислено: 517,6 m/z; найдено: 517,4 m/z.
Пример 80. Получение соединения P161
Синтез (E)-3-(3-фтор-3-метил-1-нафталин-2-илметил-2-оксо-2,3-дигидро-1Н-индол-7-ил)акриловой кислоты, (I-41). К смеси соединения I-39 (160 мг, 0,47 ммоль) в ацетонитриле (8 мл) и воды (3 мл) добавляют Selectfluor (500 мг, 1,41 ммоль) и полученную смесь перемешивают при комнатной температуре в течение ночи, раствор разбавляют EtOAc и затем промывают разбавленной водной HCl, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан и метанол/дихлорметан в качестве элюентов и получая 65 мг соединения I-41. 1H-ЯМР (500 МГц, CDCl3), 19F-ЯМР (400 МГц, CDCl3).
Синтез соединения P161. Cмесь кислоты I-41 (32 мг, 0,085 ммоль), 4,5-дихлор-2-тиофенсульфонамида (23 мг, 0,1 ммоль), 4-диметиламинопиридин (21 мг, 0,17 ммоль) и EDCI (33 мг, 0,17 ммоль) в дихлорметане (5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl (2×10 мл), водой (4×10 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан и метанол/дихлорметан в качестве элюентов и получая 12 мг соединение P161. МС (ESI-): 589,0 (M-1), ЖХ-МС: 91%. 1H-ЯМР (500 МГц, Acetone-d6).
Пример 81. Получение соединения P160.
Смесь кислоты I-41 (32 мг, 0,085 ммоль), 2-тиофенсульфонамида (16 мг, 0,1 ммоль), 4-диметиламинопиридина (21 мг, 0,17 ммоль) и EDCI (33 мг, 0,17 ммоль) в дихлорметане (5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl (2×10 мл), водой (4×10 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан и метанол/дихлорметан в качестве элюентов и получая 20 мг соединения P160. 1H-ЯМР (CDCl3) 1,90 (д, J=22 Гц, 3H), 5,20 (д, J=16 Гц, 1H), 5,33 (д, J=16 Гц, 1H), 6,02 (д, J=15,0 Гц, 1H), 7,09 (т, J=8,0 Гц, 1H), 7,15 (м, 1H), 7,23 (д, J=8,0 Гц, 1H), 7,26 (с, 1H), 7,30 (д, J=8,0 Гц, 1H), 7,44 (м, 2H), 7,0 (д, J=7 Гц, 1H), 7,70 (м, 2H), 7,75 (м, 2H), 7,87 (с, 1H), 7,92 (м, 1H), 8,00 (д, J=15,0 Гц, 1H). ЖХ/МС (93%) ESI- Вычислено: 519,6 m/z; найдено: 519,3 m/z.
Пример 82. Получение соединения P044.
Синтез метилового эфира 1-нафталин-2-илметил-1Н-индол-7-карбоновой кислоты, (I-41). Метиловый эфир 1Н-индол-7-карбоновой кислоты, полученный в соответствии с методикой, описанной в литературе [Batcho B., Leimgruber, K., Org. Syn. Vol. IIV, page 34-40], (1 г, 5,7 ммоль) в ДМФА (10 мл), добавляют NaH (60% в масле, 275 мг, 6,9 ммоль) при 0°C, полученную смесь перемешивают при комнатной температуре в течение 1 часа и затем добавляют 2-(бромметил)нафталин (1,26 г, 5,7 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. ДМФА удаляют и остаток разбавляют EtOAc (25 мл). Реакционную смесь промывают водой (2×15 мл), сушат над сульфатом натрия, фильтруют и растворитель удаляют. В результате получают 1,59 г соединения I-41. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез (1-нафталин-2-илметил-1Н-индол-7-ил)метанола, (I-42). К охлажденной на ледяной бане суспензии LiAlН4 (216 мг, 5,7 ммоль) в 15 мл безводным ТГФ в атмосфере N2 по капле добавляют раствор сложного эфира I-41 (940 мг, 2,85 ммоль) в 8 мл безводным ТГФ. Реакционную смесь перемешивают при 0°C в течение 1 часа, затем дают ей нагреться до комнатной температуры. ТСХ (25% EtOAc/гексан) показывает отсутствие исходного вещества. Смесь гасят при 0°C медленным добавлением 1 мл воды, 1 мл NaOH 1N, 1,5 мл воды. Смесь перемешивают в течение 10 минут при комнатной температуре. Суспензию фильтруют и твердый осадок промывают несколько раз ТГФ и EtOAc. Объединенные органические слои промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют, получая 800 мг неочищенного продукта. Очистка колоночной хроматографией с использованием 5%-15% смеси EtOAc/гексан приводит к получению 450 мг спирта I-42. 1H-ЯМР (500 МГц, CDCl3).
Синтез соединения P044. К раствору спирта I-42 (57,8 мг, 0,2 ммоль) в 1 мл безводного метиленхлорида добавляют при 0°C диэтоксифосфинилизоцианат (34 мг, 0,191 ммоль). Реакционную смесь перемешивают при 0°C в течение 2 часов, затем при комнатной температуре в течение 3 часов. Смесь обрабатывают водным раствором, затем очищают колоночной хроматографией, используя в качестве элюента CH2Cl2 до 2% MeOH/CH2Cl2 и получая в результате 79 мг соединения P044. 1H ЯМР (400 МГц, CDCl3) 1,21-1,24 (т, J=7,2 Гц, 6H), 4,01-4,10 (м, 4H), 5,23 (с, 2H), 5,73 (с, 2H), 6,67 (д, J=3,6 Гц, 1H), 7,10 (т, J=7,2 Гц, 1H), 7,17 (м, 2H), 7,2 (д, J=3,6 Гц, 1H), 7,24 (уш.с, 1H), 7,42-7,45 (м, 3H), 7,65 (м, 1H), 7,72 (дд, J=8,0, 1,2 Гц, 1H), 7,8 (д, J=8,0 Гц, 2H) ЖХ-МС (93%): ESI- Вычислено: 466 m/z; Найдено: 465.
Пример 83. Получение соединения P108
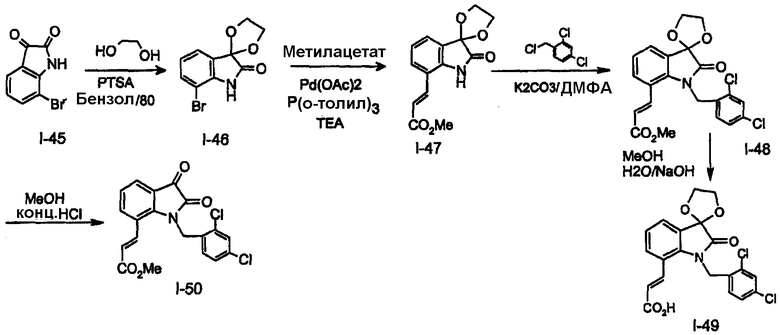
Синтез 7-бром-1Н-индол-2,3-диона, I-45. В круглодонную колбу (500 мл) при перемешивании загружают раствор хлоральгидрата (12 г, 66 ммоль) в воде (150 мл), добавляют Na2SO4 (14 г, 100 ммоль) и суспензию 2-броманилина (9 г, 50 ммоль) в 2н. водной HCl (60 мл) при комнатной температуре. Полученную реакционную смесь кипятят с обратным холодильником в течение 30 минут, затем охлаждают до комнатной температуры. Образовавшийся твердый осадок отфильтровывают, промывают водой (3×100 мл) и сушат в вакууме при 50°C, получая неочищенный N-(2-бромфенил)-2-гидроксииминоацетамид, (I-44) (9 г), который сразу используют на следующей стадии без дополнительной очистки. В круглодонную колбу (500 мл) помещают предварительно нагретый (до 50°C) раствор концентрированной H2SO4 (80 мл) и при перемешивании одной порцией добавляют промежуточный продукт I-44 при 50°C. Полученную реакционную смесь нагревают до 80°C и перемешивают при 80°C в течение 30 минут, затем охлаждают до комнатной температуры, выливают при перемешивании в 600 мл смеси воды со льдом. Образовавшееся твердое вещество отфильтровывают, промывают водой (3×100 мл) и сушат в вакууме при 50°C, получая целевой I-45 (5,2 г, 44% для двух стадий) МС (ESI+): 227 (M+1). 1Н-ЯМР: (500 МГц, CDCl3).
Синтез соединения I-46. В круглодонную колбу (500 мл) загружают раствор 7-броминдол-2,3-диона I-45 (5 г, 22 ммоль) и моногидрат п-толуолсульфоновой кислоты (500 мг, 10 моль.%) в сухом бензоле (200 мл) и добавляют этиленгликоль (5 г, 82 ммоль, 3,8 экв.). Полученную реакционную смесь кипятят с обратным холодильником в течение 23 часов с азеотропной отгонкой воды (ловушка Дина-Старка). Реакционную смесь охлаждают до комнатной температуры, промывают 10% водным NaHCO3 (100 мл) и затем водой (100 мл). После удаления растворителя получают 6 г неочищенного продукта, который очищают перекристаллизацией из смеси CH2Cl2/EtOAc/гексан, получая целевой ацеталь, I-46 (5,4 г, 90% выход) в виде твердого не совсем белого вещества. МС (ESI+): 270 (M+1). 1H-ЯМР (500 МГц, CDCl3).
Синтез соединения I-47. В пробирку при комнатной температуре загружают раствор соединения I-46 (5,4 г, 20 ммоль), три-o-толилфосфин (2,2, 7 ммоль, 0,3 экв.) и ацетат палладия (500 мг, 2 ммоль, 0,1 экв.) в триэтиламине (20 мл) и добавляют метилaкрилат (5 г, 70 ммоль, 3,5 экв.). Пробирку герметично закрывают, реакционную смесь нагревают и перемешивают при 100°C в течение 6 часов, затем охлаждают до комнатной температуры, выливают при перемешивании в 600 мл смеси воды со льдом, экстрагируют CH2Cl2 (3×100 мл). Органические слои промывают водой (2×100 мл), насыщенным раствором соли (100 мл) и сушат (Na2SO4). После удаления растворителя получают 6 г неочищенного продукта, который очищают перекристаллизацией из смеси CH2Cl2/EtOAc/гексан и флэш-хроматографией (силикагель, CH2Cl2; CH2Cl2/EtOAc/гексан, об./об./об.=1:10:20-1:20:10; EtOAc), получая целевой продукт I-47 (всего 4,5 г, 81%) в виде твердого не совсем белого вещества. МС (APCI-): 274 (M-1). 1H-ЯМР (500 МГц, CDCl3).
Синтез соединения I-48. В круглодонную колбу (250 мл) загружают суспензию соединения I-47 (3 г, 11 ммоль) и K2CO3 (10 г, 55 ммоль, 5 экв.) в ДМФА (40 мл), добавляют 2,4-дихлорбензилхлорид (2,4 г, 12 ммоль, 1,05 экв.). Полученную реакционную смесь нагревают до 50°C, перемешивают при 50°C в течение 3 часов и перемешивают при комнатной температуре в течение ночи. Смесь выливают при перемешивании в 600 мл смеси воды со льдом. Образовавшееся твердое вещество отфильтровывают, промывают водой (3×100 мл) и сушат в вакууме при 50°C, получая целевой I-48 (2,8 г) в виде твердого не совсем белого вещества. Дополнительный 1 г соединения I-48 получают из остатка после экстракции EtOAc (2×100 мл) и затем после очистки флэш-хроматографией (силикагель, CH2Cl2; CH2Cl2/EtOAc/гексан, об./об./об.=1:10:20-1:20:10; EtOAc). Всего получают 3,8 г соединения I-48 (80%). МС (APCI+): 434,3 (M), 436,3 (M+2). 1H-ЯМР (500 МГц, CDCl3).
Синтез (E)-3-[1-(2,4-дихлорбензил)-2,3-диоксо-2,3-дигидро-1Н-индол-7-ил]акриловой кислоты, I-49. В круглодонную колбу (200 мл) загружают раствор NaOH (500 мг, 12 ммоль) в MeOH (40 мл) и H2O (40 мл) и при 5°С добавляют соединение I-48 (450 мг, 10,3 ммоль). Полученной реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают в течение 10 минут, нагревают до 50°С, перемешивают при 50°C в течение 2 часов и затем при 75°C в течение 2 часов. Реакционную смесь охлаждают до 0°C и подкисляют водным HCl (2н.) до pH=1, затем добавляют воду (150 мл). Образовавшийся твердый продукт отфильтровывают, промывают водой (3×60 мл) и сушат в вакууме при 50°C, получая целевой I-49 (430 мг, 95%) в виде твердого не совсем белого вещества. МС (APCI-): 418,2 (M-2). 1H-ЯМР (500 МГц, CDCl3).
Синтез соединения P108. В круглодонную колбу (200 мл) загружают раствор 4,5-дихлортиофен-2-сульфонамида (243 мг, 1,1 ммоль), 4-диметиламинопиридин (DMAP, 350 мг, 3 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 450 мг, 3 ммоль) в CH2Cl2 (50 мл) добавляют акриловую кислоту I-49 (420 мг, 1,0 ммоль) при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение 4 дней, затем охлаждают до 0°C и подкисляют водным HCl (10%) до pH=1, затем экстрагируют CH2Cl2/MeOH (9/1, 3×100 мл). Объединенные органические слои сушат (Na2SO4), растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2; EtOAc/гексан=1:2-1:0) с получением целевого P108 (350 мг, 55%) в виде белого твердого вещества. 1H ЯМР (500 МГц, ацетон-d6); 4,36-4,38 (м, 2H), 4,48-4,51 (м, 2H), 5,05 (с, 2H), 6,17 (д, J=15,5 Гц, 1H), 6,97 (д, J=8,0 Гц, 1H), 7,11 (т, J=8,0 Гц, 1H), 7,25 (дд, J=8,0, 2,0 Гц, 1H), 7,35-7,45 (м, 2H), 7,45 (дд, J=7,5, 1,0 Гц, 1H), 7,53 (д, J=15,5 Гц, 1H), 7,63 (с, 1H). ЖХ/МС (99,5%) ESI- Вычислено: 634,3 m/z, найдено: 633,2 m/z (M-1).
Пример 84. Получение соединения P113.
Синтез метилового эфира (E)-3-[1-(2,4-дихлорбензил)-2,3-диоксо-2,3-дигидро-1Н-индол-7-ил]акриловой кислоты, I-50. К суспензии I-48 (2,1 г, 5 ммоль) в MeOH (50 мл) при комнатной температуре при перемешивании добавляют концентрированную HCl (50 мл). Полученную реакционную смесь нагревают до 90°C, перемешивают при 90°C в течение 3 часов, охлаждают до комнатной температуры и при перемешивании выливают в 200 мл смеси воды со льдом. Образовавшийся твердый продукт отфильтровывают, промывают водой (3×100 мл) и сушат в вакууме при 50°C, получая 1,7 г (83%) целевого изатин-производного, I-50, в виде оранжевого твердого вещества. МС (APCI+): 390,3 (M), 392,2 (M+2).1H-ЯМР (500 МГц, CDCl3).
Синтез (E)-3-[1-(2,4-дихлорбензил)-2,3-диоксо-2,3-дигидро-1Н-индол-7-ил]акриловой кислоты, I-51. В круглодонную колбу (500 мл) загружают раствор NaOH (1 г, 25 ммоль) в MeOH (50 мл) и H2O (50 мл) и добавляют соединение I-50 (1,6 г, 15 ммоль) при 5°C. Полученной реакционной смеси дают возможность нагреться до комнатной температуры, смесь перемешивают при комнатной температуре в течение 10 минут, затем нагревают до 50°C и перемешивают при 50°C в течение 4 часов. Реакционную смесь охлаждают до 5°C и подкисляют добавлением водного HCl (10%) до pH=1, затем добавляют воду (250 мл). Образовавшееся твердое вещество отфильтровывают, промывают водой (3×100 мл) и сушат в вакууме при 50°C, получая целевую кислоту I-51 (1,34 г, 85%) в виде оранжевого твердого вещества. МС (APCI-): 374,1(M-2), 376,2 (M). 1H-ЯМР (500 МГц, CDCl3).
Синтез соединения P113.
В круглодонную колбу (250 мл) загружают раствор 4,5-дихлортиофен-2-сульфонамида (1,28 г, 5,5 ммоль), 4-диметиламинопиридин (DMAP, 1,5 г, 15 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 1,8 г, 15 ммоль) в CH2Cl2 (150 мл) и добавляют акриловую кислоту I-51 (1,9 г, 5 ммоль) при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение 72 часов, затем охлаждают до 0°C и подкисляют водной HCl (10%) до pH=1. Добавляют 150 мл смеси воды со льдом, смесь энергично перемешивают в течение 30 минут. Образовавшееся твердое вещество отфильтровывают, промывают водой (3×150 мл) и сушат в вакууме при 50°C, получая целевой P113 (2,3 г, 77%) в виде оранжевого твердого вещества. 1H ЯМР (500 МГц, ДМСО-d6); 4,96 (с, 2H), 6,16 (д, J=15,5 Гц, 1H), 7,22 (т, J=7,5 Гц, 1H), 7,30-7,33 (м, 3H), 7,55 (д, J=8,5 Гц, 1H), 7,58 (д, J=8,0 Гц, 1H), 7,70 (д, J=7,5 Гц, 1H), 7,90 (с, 1H). ЖХ/МС (99%) ESI- Вычислено: 590,3 m/z, найдено: 589,1 m/z (M-1).
Пример 85. Получение соединения P128.
В круглодонную колбу (25 мл) загружают раствор P113 (50 мг, 0,09 ммоль) в ацетоне (5 мл), добавляют Et2NH (0,5 мл) при 0°C. Полученной смеси дают возможность нагреться до комнатной температуры и смесь перемешивают при комнатной температуре в течение 72 часов, затем охлаждают до 0°C и подкисляют водным HCl (2н.) до pH=1. К смеси добавляют смесь воды со льдом 200 мл, образовавшийся твердый продукт отфильтровывают, промывают водой (3×20 мл) и сушат в вакууме при 50°C, получая целевой P128 (30 мг, 55%) в виде твердого ярко-желтого вещества. МС (ESI-) m/z (647, M-1). ЖХМС (ESI-)>95%. 1H-ЯМР.
Пример 86. Получение соединения P134.
В круглодонную колбу (25 мл) загружают раствор P113 (50 мг, 0,09 ммоль) в CH3NO2 (5 мл) и при 0°С добавляют Et2NH (0,5 мл). Полученную смесь перемешивают при комнатной температуре в течение 72 часов, затем охлаждают до 0°C и подкисляют водным HCl (2н.) до pH=1. Добавляют 200 мл смеси воды со льдом. Образовавшееся твердое вещество отфильтровывают, промывают водой (3×20 мл) и сушат в вакууме при 50°C, получая целевой продукт P134 (30 мг, 55%) в виде ярко-желтого твердого вещества. 1H ЯМР (500 МГц, ацетон-d6); 5,08-5,20 (м, 2H), 5,25-5,32, (м, 2H), 6,33 (д, J=15,0 Гц, 1H), 7,17-7,21 (м, 2H), 7,28 (дд, J=8,5, 2,0 Гц, 1H), 7,39 (д, J=2,0 Гц, 1H), 7,42 (д, J=8,0 Гц, 1H), 7,56 (д, J=15,0 Гц, 1H), 7,70 (д, J=7,5 Гц, 1H), 7,78 (д, J=2,0 Гц, 1H). ЖХ/МС (99%) ESI- Вычислено: 651,3 m/z, найдено: 650,2 m/z (M-1).
Пример 87. Получение соединения P129.
В круглодонную колбу (25 мл) загружают раствор P113 (50 мг, 0,09 ммоль) в EtOH (10 мл) и при 0°С добавляют NaBH4 (50 мг). Полученной смеси дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 2 часов, затем охлаждают до 0°C, гасят добавлением 20 мл смеси воды со льдом и подкисляют водным HCl (2н.) до pH=1. Смесь экстрагируют дихлорметаном (3×30 мл). Объединенные органические слои сушат над Na2SO4 и растворитель удаляют в вакууме, получая смесь, которую разделают флэш-хроматографией (силикагель, CH2Cl2; CH2Cl2/EtOAc/гексан, об./об./об.=1:10:20-1:20:10; EtOAc), получая 6 мг соединения P129 (10%) в виде белого твердого вещества. МС(ESI-) m/z(559, M-1). ЖХМС(ESI-) 95%. 1H-ЯМР.
Пример 88. Получение соединения P130.
В круглодонную колбу (25 мл) загружают раствор P113 (50 мг, 0,09 ммоль) в EtOH (10 мл) и при -78°С добавляют NaBH4 (50 мг). Полученную смесь перемешивают при -78 °C в течение 6 часов, затем гасят добавлением водного HCl (2н.) до pH=1. Смесь разбавляют 100 мл, смесь экстрагируют EtOAc(3×30 мл). Объединенные органические слои промывают водой (2×30 мл), сушат над Na2SO4 и растворитель удаляют в вакууме, получая смесь, которую очищают флэш-хроматографией (силикагель, CH2Cl2; CH2Cl2/EtOAc/гексан, об./об./об.=1:10:20-1:20:10; EtOAc) с получением 20 мг соединения P130 (70%) в виде белого твердого вещества. 1H-ЯМР (500 МГц, ацетон-d6); 5,02-5,14 (м, 2H), 5,23 (с, 1H), 6,33 (д, J=15,5 Гц, 1H), 7,07 (д, J=8,5 Гц, 1H), 7,14 (т, J=8,0 Гц, 1H), 7,24 (дд, J=8,5, 2,0 Гц, 1H), 7,34 (д, J=7,5 Гц, 1H), 7,39 (д, J=2,0 Гц, 1H), 7,54 (д, J=7,0 Гц, 1H), 7,58 (д, J=15,5 Гц, 1H), 7,79 (с, 1H). ЖХ/МС (96%) ESI- Вычислено: 592,3 m/z, найдено: 591,0 m/z (M-1).
Пример 89. Получение соединения P133.
В круглодонную колбу (50 мл) загружают суспензию P113 (50 мг, 0,09 ммоль) в ТГФ (10 мл) и по каплям добавляют MeMgBr (3н. в эфире, 0,5 мл) при -78°C. Полученной смеси дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 30 минут и затем охлаждают до 0°C, гасят добавлением водного HCl (2н.) до достижения pH=1, затем разбавляют 100 мл воды и смесь экстрагируют дихлорметаном (3×30 мл). Объединенные органические слои сушат над Na2SO4 и растворитель удаляют в вакууме, получая смесь, которую разделают флэш-хроматографией (силикагель, CH2Cl2; CH2Cl2/EtOAc/гексан, об./об./об.=1:10:20-1:20:10; EtOAc), получая соединение P133 (20 мг, 40%) в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6); 1,51 (с, 3H), 4,99 (д, J=5,0 Гц, 2H), 6,13 (д, J=15,0 Гц, 1H), 6,98 (д, J=8,5 Гц, 1H), 7,16 (т, J=8,0 Гц, 1H), 7,27 (д, J=8,0 Гц, 1H), 7,32 (дд, J=8,0, 2,0 Гц, 1H), 7,35 (д, J=2,0 Гц, 1H), 7,44 (д, J=15,5 Гц, 1H), 7,51 (д, J=6,0 Гц, 1H), 7,87 (с, 1H). ЖХ/МС (95%) ESI- Вычислено: 606,3 m/z, найдено: 605,3 m/z (M-1).
Пример 90. Получение соединения P132
Синтез метилового эфира (E)-3-[1-(2,4-дихлорбензил)-3,3-дифтор-2-оксо-2,3-дигидро-1H-индол-7-ил]акриловой кислоты, I-52. В круглодонную колбу (25 мл) загружают раствор соединения I-50 (200 мг, 0,5 ммоль) в CH2Cl2 (8 мл) и при 5°С добавляют трифторид диэтиламиносеры (DAST, 0,5 мл, избыток). Полученной реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 3 дней, затем охлаждают до 5°C. Реакцию гасят добавлением MeOH (1 мл) при 0°C и перемешивают при 0°C в течение 20 минут, затем при комнатной температуре в течение 10 минут. Реакционную емкость охлаждают до 5°C, добавляют воду (10 мл) и дают возможность смеси нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 30 минут, затем экстрагируют дихлорметаном (2×30 мл). Объединенные органические слои промывают водой (2×20 мл), сушат над Na2SO4 и растворитель удаляют в вакууме, получая неочищенный продукт I-52 (200 мг, 95%) в виде ярко-желтого твердого вещества, который используют на следующей стадии без дополнительной очистки. МС (APCI-): (410,3, M-2). 1H-ЯМР (400 МГц, CDCl3).
Синтез (E)-3-[1-(2,4-дихлорбензил)-3,3-дифтор-2-оксо-2,3-дигидро-1Н-индол-7-ил]акриловой кислоты, I-53. В круглодонную колбу (50 мл) загружают раствор NaOH (120 мг, 3 ммоль) в MeOH (7,5 мл) и H2O (7,5 мл), смесь охлаждают до 5°С и добавляют соединение I-52 (120 мг, 0,3 ммоль). Полученной реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 2 часов, затем нагревают до 50°C и перемешивают при 50°C в течение 2 часов. Реакционную смесь охлаждают до 0°C и подкисляют водным HCl (10%) до pH=1, затем добавляют воду (50 мл) и экстрагируют смесью дихлорметан-MeOH (10:1, 5×30 мл). Объединенные органические слои сушат над Na2SO4 и растворитель удаляют в вакууме, получая неочищенный продукт, который очищают перекристаллизацией из смеси ацетон/EtOAc/гексан, получая целевую кислоту I-53 (90 мг, 75% выход) в виде твердого не совсем белого вещества. МС (ESI-): (396,1, M-2; 398,0, M). ЖХМС(ESI-) 95%. 1H-ЯМР (500 МГц, ДМСО-d6)).
Синтез соединения P132. В круглодонную колбу (100 мл) загружают раствор 4,5-дихлортиофен-2-сульфонамида (60 мг, 0,26 ммоль), 4-диметиламинопиридин (DMAP, 60 мг, 0,6 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 70 мг, 0,6 ммоль) в CH2Cl2 (20 мл) и добавляют акриловую кислоту I-53 (80 мг, 0,21 ммоль) при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение 2 дней, затем охлаждают до 0°C. После добавления MeOH (5 мл) и воды (20 мл) реакционную смесь подкисляют добавлением водного HCl (10%) до pH=1 и перемешивают при комнатной температуре в течение 30 минут. Образовавшийся твердый продукт отфильтровывают, промывают водой (3×20 мл) и сушат в вакууме при 50°C, получая неочищенный продукт 19 (80 мг), который очищают флэш-хроматографией (силикагель, CH2Cl2; EtOAc/гексан=1:4-1:2), получая целевой продукт P132 (55 мг, 45%) в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6); 5,05 (с, 2H), 6,13 (д, J=15,5 Гц, 1H), 7,03 (д, J=8,0 Гц, 1H), 1,29-135 (м, 3H), 7,41 (д, J=15,0 Гц, 1H), 7,87 (д, J=7,5 Гц, 1H), 7,89 (с, 1H). ЖХ/МС (99%) ESI- Вычислено: 612,3 m/z, найдено: 611,0 m/z (M-1).
Пример 92. Получение соединения P216
В круглодонную колбу (25 мл) загружают раствор тиофен-2-сульфонамида (17 мг, 0,11 ммоль), 4-диметиламинопиридин (DMAP, 20 мг, 0,2 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 23 мг, 0,3 ммоль) в CH2Cl2 (2 мл) и при комнатной температуре добавляют акриловую кислоту I-53 (40 мг, 0,1 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 2 дней, затем охлаждают до 0°C. После добавления MeOH (2 мл) и воды (10 мл) реакционную смесь подкисляют добавлением водного HCl (10%) до pH=1 и экстрагируют смесью CH2Cl2/MeOH (9/1, 3×10 мл). Объединенные органические слои сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2/EtOAc/гексан=1:1:4, EtOAc) с получением целевого ацилсульфонамида P216 (15 мг, 25%) в виде белого твердого вещества. 1H ЯМР (500 МГц, ацетон-d6); 5,04 (с, 2H), 6,15 (д, J=15,0 Гц, 1H), 7,01 (д, J=8,5 Гц, 1H), 7,25-7,27 (м, 2H), 7,33 (т, J=8,6 Гц, 1H), 7,38 (д, J=15,5 Гц, 1H), 7,41 (д, J=2,0 Гц, 1H), 7,53 (д, J=8,0 Гц, 1H), 7,81 (д, J=4,0 Гц, 1H), 7,85 (д, J=8,0 Гц, 1H), 8,10 (д, J=4,0 Гц, 1H). ЖХ/МС (92%) ESI- Вычислено: 543,4 m/z, найдено: 541,4 m/z (M-2).
Пример 93. Получение соединения P127.
Синтез (E)-3-[1-(2,4-дихлорбензил)-2-оксо-2,3-дигидро-1Н-индол-7-ил]акриловой кислоты, I-54. В баростойкую колбу (350 мл) загружают соединение I-51 (430 мг, 1,2 ммоль) и при комнатной температуре добавляют NH2NH2H2O (20 мл). Полученную смесь герметично закрывают, нагревают до 130°C и перемешивают при 130°C в течение 1 часа, затем охлаждают до 0°C. К смеси добавляют смесь воды со льдом (300 мл), реакционную смесь подкисляют добавлением водного HCl (10%) до pH=1 и перемешивают при комнатной температуре в течение 30 минут. Образовавшийся твердый продукт отфильтровывают, промывают водой (3×50 мл) и сушат в вакууме при 50°C, получая целевое оксииндол-производное, I-54 (300 мг, 73%) в виде белого твердого вещества. МС(ESI-)m/z=360,1, (M-2); 1Н-ЯМР (500 МГц, ацетон-d6).
Синтез соединения P127. В круглодонную колбу (100 мл) загружают раствор 4,5-дихлортиофен-2-сульфонамида (130 мг, 0,55 ммоль), 4-диметиламинопиридина (DMAP, 150 мг, 1,5 ммоль) и гидрохлорида 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 180 мг, 1,5 ммоль) в CH2Cl2 (20 мл) добавляют акриловую кислоту P-54 (180 мг, 0,5 ммоль) при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение 2 дней, затем охлаждают до 0°C. После добавления MeOH (5 мл) и воды (120 мл) реакционную смесь подкисляют добавлением водного HCl (10%) до pH=1 и перемешивают при комнатной температуре в течение 30 минут. Образовавшееся твердое вещество отделяют фильтрованием, промывают водой (3×50 мл) и сушат в вакууме при 50°C, получая целевой продукт P127 (143 мг, 40%) в виде белого твердого вещества. МС(APCI-) m/z=574,9 (M-1); ЖХМС(ESI-)>95%. ВЭЖХ>95%. 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 94. Получение соединения P219
В круглодонную колбу (25 мл) загружают раствор тиофенсульфонамида (17 мг, 0,11 ммоль), 4-диметиламинопиридин (DMAP, 20 мг, 0,2 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (EDCI, 23 мг, 0,3 ммоль) в CH2Cl2 (2 мл) и при комнатной температуре добавляют акриловую кислоту I-54 (26 мг, 0,1 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 2 дней, затем охлаждают до 0°C. После добавления MeOH (2 мл) и воды (10 мл) реакционную смесь подкисляют добавлением водной HCl (10%) до pH=1, затем экстрагируют смесью CH2Cl2/MeOH (9/1,3×10 мл). Объединенные органические слои сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2/EtOAc/гексан=1:1:4, EtOAc) с получением целевого продукта P219 (23 мг, 45%) в виде белого твердого вещества. МС(ESI-) m/z (505,2, M-2); ЖХМС(ESI-)>95%. 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 95. Получение соединения P131.
Синтез метилового эфира (E)-3-[1-(2,4-дихлорбензил)-2-оксо-2,3-дигидро-1Н-индол-7-ил]акриловой кислоты, I-55. В баростойкую колбу (50 мл) загружают суспензию соединения I-54 (100 мг, 0,3 ммоль) в MeOH (15 мл) при комнатной температуре и при перемешивании добавляют концентрированную HCl (0,5 мл), реакционную смесь герметично закрывают, нагревают до 85°C, перемешивают при 85°C в течение 5 часов, охлаждают до 0°C, нейтрализуют смесь добавлением водного NH4Cl (насыщенный, 2 мл), разбавляют водой (30 мл) и экстрагируют EtOAc (3×20 мл). Объединенные органические слои промывают водой (2×20 мл), сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая соединение I-55 (103 мг, 99%) в виде белого твердого вещества. МС (ESI-): (374,2, M-2). 1H-ЯМР (500 МГц, CDCl3).
Синтез метилового эфира (E)-3-[1-(2,4-дихлорбензил)-3,3-диметил-2-оксо-2,3-дигидро-1Н-индол-7-ил]акриловой кислоты, I-56. В круглодонную колбу (20 мл) загружают суспензию соединения I-55 (45 мг, 0,12 ммоль) и при перемешивании при 0°С добавляют K2CO3 (45 мг) в ДМФА (2 мл) и метилйодид (0,5 мл, избыток). Полученной реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 3 дней. Смесь выливают в 30 мл смеси воды со льдом и экстрагируют EtOAc (3×20 мл). Объединенные органические слои промывают водой (2×20 мл), сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт 25, который очищают флэш-хроматографией (силикагель, CH2Cl2; EtOAc/гексан=1:5), получая целевой продукт I-56 (35 мг, 75%) в виде белого твердого вещества. МС (APCI-): (404,2, M). 1H-ЯМР (500 МГц, CDCl3).
(E)-3-[1-(2,4-дихлорбензил)-3,3-диметил-2-оксо-2,3-дигидро-1Н-индол-7-ил]акриловая кислота, I-57. В круглодонную колбу (50 мл) загружают раствор NaOH (25 мг, 0,6 ммоль) в MeOH (5 мл) и H2O (5 мл), затем охлаждают до 5°С и добавляют соединение I-56 (25 мг, 0,05 ммоль). Полученной реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь охлаждают до 0°C и подкисляют добавлением водного HCl (10%) до pH=1, добавляют воду (10 мл) и затем экстрагируют дихлорметаном (3×20 мл). Объединенные органические слои сушат над Na2SO4 и растворитель удаляют в вакууме, получая неочищенный продукт I-57 (20 мг, 80% выход) в виде твердого не совсем белого вещества. МС (ESI-): (388,3, M-2). 1H-ЯМР (500 МГц, CDCl3).
Синтез соединения P131. В круглодонную колбу (25 мл) загружают раствор 4,5-дихлортиофен-2-сульфонамида (7 мг, 0,03 ммоль), 4-диметиламинопиридин (DMAP, 9 мг, 0,9 ммоль), гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимид (EDCl 12 мг, 0,9 ммоль) в CH2Cl2 (3 мл) и к смеси добавляют акриловую кислоту I-57 (10 мг, 0,025 ммоль) при комнатной температуре. Полученную смесь перемешивают при комнатной температуре в течение 2 дней, затем охлаждают до 0°C. После добавления MeOH (5 мл) и воды (20 мл), реакционную смесь подкисляют добавлением водного HCl (10%) до pH=1, смесь экстрагируют CH2Cl2/MeOH (9/1, 3×10 мл). Объединенные органические слои сушат (Na2SO4) и растворитель удаляют при пониженном давлении, получая неочищенный продукт, который очищают флэш-хроматографией (силикагель, CH2Cl2/EtOAc/гексан=1:1:4, EtOAc), получая соединение P131 (6,5 мг, 45%) в виде белого твердого вещества. 1Н-ЯМР (500 МГц, ДМСО-d6); 1,40 (с, 6H), 5,03 (с, 2H), 6,12 (д, J=15,5 Гц, 1H), 6,81 (д, J=8,5 Гц, 1H), 7,12-7,16 (м, 1H), 7,23 (д, J=8,5 Гц, 1H), 7,28 (дд, J=8,5, 2,0 Гц, 1H), 7,37 (д, J=2,0 Гц, 1H), 7,47 (д, J=15,5 Гц, 1H), 7,51 (д, J=7,0 Гц, 1H), 7,86 (с, 1H). ЖХ/МС (85%) ESI- Вычислено: 604,4 m/z, найдено: 603,0 m/z (M-1).
Пример 96. Получение соединения P019.
Синтез 5-нитро-4H-бензо[1,4]оксазин-3-она, I-58. Смесь 2-амино-3-нитрофенола (1,54 г, 10 ммоль), этилбромацетата (1,67 г, 10 ммоль), карбоната калия (1,54 г, 11 ммоль) и ДМФА (5,0 мл) перемешивают при комнатной температуре в течение 20 часов. Реакционную смесь разбавляют водой (100 мл) и экстрагируют EtOAc (100 мл×3). Объединенные слои EtOAc промывают водой (50 мл×2), сушат (насыщенным раствором соли, сульфатом натрия), концентрируют и сушат в высоком вакууме, получая 5-нитро-4H-бензо[1,4]оксазин-3-он, I-58(1,6 г).
Синтез 4-нафталин-2-илметил-5-нитро-4H-бензо[1,4]оксазин-3-она, I-59. Гидрид натрия (94 мг, 3,0 ммоль) добавляют порциями к раствору 5-нитро-4H-бензо[1,4]оксазин-3-она, I-58 (388 мг, 2 ммоль) в ДМФА. Спустя 30 минут добавляют 2-бромметилнафталин (442 мг, 2 ммоль) и смесь перемешивают при комнатной температуре в течение 20 часов. Реакционную смесь разбавляют водой (100 мл) и экстрагируют EtOAc (100 мл×3). Объединенные EtOAc слои промывают водой (50 мл×2), сушат (насыщенным раствором соли, сульфатом натрия), концентрируют и сушат в высоком вакууме, получая 4-нафталин-2-илметил-5-нитро-4H-бензо[1,4]оксазин-3-он, I-59 (653 мг).
Синтез 5-амино-4-нафталин-2-илметил-4H-бензо[1,4]оксазин-3-она, I-60. A раствор 4-нафталин-2-илметил-5-нитро-4H-бензо[1,4]оксазин-3-она, I-59 (0,65 г) в метаноле (30 мл) и диоксане (7,0 мл) гидрируют в присутствии 10% Pd-C водородом при давлении 45 фунтов на кв. дюйм в течение 22 часов. Реакционную смесь фильтруют и фильтрат концентрируют, получая 5-амино-4-нафталин-2-илметил-4H-бензо[1,4]оксазин-3-он, I-60 (620 мг).
Синтез метилового эфира N-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-ил)щавелевой кислоты, I-61. К раствору 5-амино-4-нафталин-2-илметил-4H-бензо[1,4]оксазин-3-она, I-60 (120 мг, 04 ммоль) и этилглиоксилилхлорида (52 мг, 0,4 ммоль) в ТГФ (3,0 мл) по каплям при комнатной температуре добавляют триэтиламин (0,1 мл, 1,0 ммоль) и полученную смесь перемешивают в течение 18 часов. Реакционную смесь концентрируют и остаток очищают на силикагеле, используя смесь хлороформ:метанол (97:3) в качестве элюента, получая метиловый эфир N-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-ил)щавелевой кислоты, I-61 (102 мг).
Синтез N-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-ил)щавелевой кислоты, I-62. К суспензии метилового эфира N-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-ил)щавелевой кислоты, I-61 (95 мг), в метаноле (2,0 мл) добавляют 1,0М NaOH (0,5 мл) и затем ТГФ (2,0 мл), получая прозрачный раствор. Реакционную смесь перемешивают при комнатной температуре в течение 30 минут и затем концентрируют для удаления растворителей. Остаток переносят в воду (2,0 мл), подкисляют добавлением 1,0М HCl и экстрагируют EtOAc (5,0 мл×4). Объединенные экстракты промывают водой (5,0 мл), сушат (насыщенным раствором соли, Na2SO4) и концентрируют, получая N-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-ил)щавелевую кислоту, I-62 (69 мг). APCI m/z 375 (M-H)+.
Синтез N-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-ил)-2-оксо-2-(тиофен-2-сульфониламино)ацетамида, P019: Смесь N-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-ил)щавелевой кислоты, I-62 (52 мг, 0,14 ммоль), тиофенсульфонамида (28 мг, 0,17 ммоль), EDCI (33 мг, 0,17 ммоль), DMAP (22 мг, 0,17 ммоль) в метиленхлориде перемешивают при комнатной температуре в течение 24 часов. Реакционную смесь разбавляют хлороформом (10 мл) и промывают 6,0М HCl (3,0 мл×4), водой (3,0 мл). Слой хлороформа концентрируют и остаток очищают на силикагеле, используя смесь хлороформом:метанол (90:10) в качестве элюента и получая N-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-ил)-2-оксо-2-(тиофен-2-сульфониламино)ацетамид, P019 (7,0 мг). 1H-ЯМР (500 МГц, ДМСО-d6) 4,59 (с, 2H), 5,35 (с, 2H), 6,84 (дд, J=8,0, 1,5 Гц, 1H), 6,90 (дд, J=8,0, 8,0 Гц, 1H), 7,02 (м, 2H), 7,06 (дд, J=5,0, 3,5 Гц, 1H), 7,44 (м, 1H), 7,56 (с, 1H), 7,61 (дд, J=3,5, 1,5 Гц, 1H), 7,71-7,65 (м, 3H), 7,78 (м, 1H), 10,05 (с, 1H). МС (ESI-) Вычислено: (M+) 521,6; найдено: 520,7 (M-1).
Пример 97. Получение соединения P049.
Синтез этилового эфира (4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-иламино)уксусной кислоты, I-63: Смесь 5-амино-4-нафталин-2-илметил-4H-бензо[1,4]оксазин-3-она, I-60 (152 мг, 0,5 ммоль), этилглоксилата (50% в толуоле, 370 мг) и безводного Na2SO4 (520 мг) в толуоле (4,0 мл) нагревают до 110°C и выдерживают при данной температуре в течение 4 часов. Реакционную смесь фильтруют. Фильтрат концентрируют и полученный остаток переносят в метанол (4,0 мл) и при комнатной температуре добавляют борогидрид натрия (40 мг). Смесь перемешивают в течение 18 часов, затем метанол выпаривают. К остатку добавляют воду (10 мл) и экстрагируют хлороформом (10,0 мл×2). Объединенные органические слои сушат (насыщенным раствором соли, Na2SO4), концентрируют и сушат, получая этиловый эфир (4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-иламино)уксусной кислоты, I-63 (105 мг).
Синтез (4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-иламино)уксусной кислоты, I-64. Смесь этилового эфира (4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-иламино)уксусной кислоты, I-63 (105 мг), метанола (3,0 мл), ТГФ (2,0 мл) и 2,0М NaOH (0,5 мл) перемешивают при комнатной температуре в течение 18 часов. Метанол выпаривают и остаток переносят в воду (3,0 мл) и подкисляют до pH 1 добавлением 2,0М HCl, в результате выпадает твердый белый осадок. Твердый продукт экстрагируют хлороформом (10 мл×2), и объединенные экстракты сушат (насыщенным раствором соли, безводным сульфатом натрия), концентрируют и сушат в высоком вакууме, получая (4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-иламино)уксусную кислоту, I-64 (102 мг).
Синтез соединения P049. [2-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-иламино)ацетил]амид тиофен-2-сульфоновой кислоты. Раствор (4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-иламино)уксусной кислоты, I-64 (102 мг, 0,281 ммоль), тиофен-2-сульфонамида (46 мг, 0,281 ммоль), EDCI (40 мг, 0,33 ммоль), DMAP (40 мг, 0,33 ммоль) в CH2Cl2 перемешивают при комнатной температуре в течение 48 часов. Реакционную смесь разбавляют CH2Cl2 (30,0 мл) и промывают 6н. HCl (5 мл×3), водой (5 мл), сушат (насыщенным раствором соли, Na2SO4) концентрируют и полученный остаток очищают на силикагеле, используя в качестве элюента смесь хлоформ:метанол (95:5), получая [2-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-иламино)ацетил]амид тиофен-2-сульфоновой кислоты, P049 (16 мг). 1H-ЯМР (500 МГц, ДМСО-d6) 3,76 (с, 2H), 4,48 (с, 2H), 5,39 (с, 2H), 5,52 (уш. с, 1H), 6,11 (дд, J=8,5, 1,0 Гц, 1H), 6,32 (дд, J=8,5, 1,0 Гц, 1H), 6,74 (т, J=8,5 Гц, 1H), 7,12 (м, 2H), 7,44 (м, 2H), 7,60 (с, 1H), 7,73-7,67 (м, 2H), 7,79 (м, 1H), 7,90 (д, J=4,5 Гц, 1H), 12,45 (уш. с, 1H). ЖХ/МС=93,8% чистота, МС (ESI-) Вычислено: (M+) 511,6; найдено: 511,5 (M-1).
Пример 98. Получение соединения P018.
Синтез метилового эфира (3-метоксикарбонилметокси-2-нитрофенокси)уксусной кислоты, I-65. Метилбромацетат (23 г, 150 ммоль) добавляют при перемешивании к смеси 2-нитробензол-1,3-диола (9,3 г, 60 ммоль), K2CO3 (24,8 г, 180 ммоль) в ацетоне (250 мл) и воде (5 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение недели. Растворитель удаляют в вакууме и остаток распределяют между EtOAc (200 мл) и водой (200 мл). Твердый осадок отфильтровывают и промывают водой, EtOAc и эфиром. После сушки получают 9,5 г соединения 2 в виде белого твердого вещества. Водный слой экстрагируют EtOAc (2×100 мл). Объединенные органические фазы промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают EtOAc, получая 4 г соединения I-65. Всего получают 13,5 г соединения 2. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез метилового эфира 2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)пропионовой кислоты, I-66. Смесь соединения I-65 (3,9 г, 13 ммоль) и 1,4 г Pd/C (5%) в ТГФ (300 мл и метанола (100 мл) гидрируют при комнатной температуре в течение 2 дней. Реакционную смесь фильтруют через целит и целит промывают EtOAc и метанолом. После удаления растворителя в вакууме твердый продукт промывают эфиром, получая 3,07 г соединения I-66 в виде белого твердого вещества. 1H-ЯМР (500 МГц, CDCl3).
Синтез метилового эфира 2-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)пропионовой кислоты, I-67. Смесь соединения I-66 (350 мг, 1,5 ммоль), 2-(бромметил)нафталина (500 мг, 2,25 ммоль), KI (374 мг, 2,25 ммоль) и K2CO3 (310 мг, 2,25 ммоль) в ДМФА (8 мл) и воде (10 капель) перемешивают при комнатной температуре в течение ночи и затем распределяют между дихлорметаном и водой. Водный слой экстрагируют дихлорметаном (3×30 мл). Объединенную органическую фазу промывают водой (4×30 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя смесь EtOAc/гексан в качестве элюента и получая 300 мг соединения I-67. 1H-ЯМР (500 МГц, CDCl3).
Синтез 2-(4-нафталин-2-илметил-3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)пропионовой кислоты, I-68. К раствору соединения I-67 (300 мг, 0,8 ммоль) в EtOH (5 мл) и ТГФ (5 мл) при комнатной температуре при перемешивании добавляют водный NaOH (2н., 5 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем значение pH доводят до кислотного добавлением 2н. водной HCl. Реакционную смесь экстрагируют EtOAc (2×30 мл). Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток растворяют в эфире и твердый осадок отфильтровывают. После удаления эфира получают 140 мг соединения I-68. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P018. К смеси кислоты I-68 (140 мг, 0,38 ммоль), 2-тиофенсульфонамида (76 мг, 0,45 ммоль), 4-диметиламинопиридина (94 мг, 0,77 ммоль) в дихлорметане (8 мл) добавляют EDCl (147 мг, 0,77 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя полученный твердый остаток промывают эфиром, получая 135 мг соединения P018. МС (ESI-): 507,5 (M-1), ЖХ-МС: 83%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 99. Получение соединения P020.
Синтез 3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)уксусной кислоты, I-69. К раствору соединения I-66: (1,5 г, 6,3 ммоль) в EtOH (15 мл) и ТГФ (35 мл) при комнатной температуре добавляют водный NaOH (2н., 10 мл). Реакционную смесь перемешивают при комнатной температуре в течение недели и затем значение pH доводят до кислотного добавлением 2н. водной HCl. Образовавшийся твердый продукт отфильтровывают и промывают водой, EtOAc и эфиром. После сушки получают 1,3 г соединения I-69 в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70. К смеси кислоты I-69 (1,11 г, 5 ммоль), 2-тиофенсульфонамида (913 мг, 5,5 ммоль), 4-диметиламинопиридина (1,22 г, 10 ммоль) в дихлорметане (200 мл) добавляют EDCI (1,91 г, 10 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Раствор разбавляют водой и значение pH доводят до кислотного добавлением водного HCl (2н.). Образовавшийся твердый осадок отфильтровывают и промывают разбавленной водной HCl, CH2Cl2 и эфиром. После сушки получают 1,6 г соединения I-70 в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6). МС (ESI-): 367,2 (M-1), ЖХ-МС: 96%.
Общая методика (A-3) N-алкилирования соединения I-70.
Смесь соединения I-70 (50 мг, 0,136 ммоль), замещенного бензилбромида (или хлорида) (0,27 ммоль), KI (45 мг, 0,27 ммоль) и K2CO3 (38 мг, 0,27 ммоль) в ДМФА (3 мл) и воде (3 капли) перемешивают при комнатной температуре в течение 5 дней. Раствор разбавляют водой и значение pH доводят до кислотного добавлением водного HCl (2н.). Образовавшийся твердый осадок отфильтровывают и промывают разбавленной водной HCl и водой. Твердый продукт растворяют в CH2Cl2 и нерастворимый осадок отфильтровывают. После удаления растворителя остаток промывают эфиром с получением продукта.
Синтез соединения P020. Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 3,4-диметилбензилбромидом с получением соединения P020. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI): 485,4 (M-l) ЖХ-МС: 85%.
Пример 100. Получение соединения P021.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты I-70 2,5-диметилбензилбромидом, получая соединение P021. 1H-ЯМР(500 МГц, ДМСО-d6) МС (ESI-): 485,4 (M-1), ЖХ-МС: 95%.
Пример 101. Получение соединения P022.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, бензилбромидом, получая соединение P022. 1Н-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 457,4 (M-1), ЖХ-МС: 93%.
Пример 102. Получение соединения P023.
Общую методику (A-3) используют для алкилирования тиофен-2-сульфоновой кислоты [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амид, I-70, 4-метилбензилбромидом, получая соединение P023. 1H-ЯМР (500 МГц, ДМСО-d6) МС (ESI): 471,4 (M-l), ЖХ-МС: 87%.
Пример 103. Получение соединения P024.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 4-фторбензилбромидом, получая соединение P024. 1H-ЯМР (500 МГц, ДМСО-d6) МС (ESI-): 475,2 (M-1), ЖХ-МС: 87%.
Пример 104. Получение соединения P025.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 4-хлорбензилхлоридом, получая соединение P025. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 491,3 (M-1), ЖХ-МС: 90%.
Пример 105. Получение соединения P026.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 1-бромметил-4-дифторметоксибензолом, получая соединение P026. 1H-ЯМР (ДМСО-d6) 4,57 (с, 2H), 4,59 (с, 2H), 5,35 (с, 2H), 6,45 (д, J=8,5 Гц, 1H), 6,67 (д, J=8,5 Гц, 1H), 6,91 (т, J=8,0 Гц, 1H), 6,99 (м, 2H), 7,02 (м, 2H), 7,14 (с, 1H), 7,20 (м, 1H), 7,79 (с, 1H), 8,02 (с, 1H), 12,5 (уш.с, 1H). ЖХ/МС (91%) ESI- Вычислено: 523,5 m/z; Найдено: 523,4 m/z.
Пример 106. Получение соединения P027.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 4-трифторметоксибензилбромидом, получая соединение P027. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 541,5 (M-1), ЖХ-МС: 90%.
Пример 107. Получение соединения P028.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 3-трифторметоксибензилбромидом, получая соединение P028. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 542,9 (M-1), ЖХ-МС: 81%.
Пример 108. Получение соединения P029.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 3-трифторметилбензилбромидом, получая соединение P029. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 526,9 (M-1), ЖХ-МС: 83%.
Пример 109. Получение соединения P030.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 3-метоксибензилбромидом, получая соединение P030. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 487,4 (M-1), ЖХ-МС: 83%.
Пример 110. Получение соединения P031.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 2-трифторметилбензилбромидом, получая соединение P301. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 525,5 (M-1), ЖХ-МС: 90%.
Пример 111. Получение соединения P032.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 4-метилсульфонбензилбромидом, получая соединение P032. 1H-ЯМР (ДМСО-d6) 3,15 (с,3H), 4,53 (с,2H), 4,65 (с, 2H), 5,40 (с, 2H), 6,46 (д, J=8,5Гц, 1H), 6,71 (д, J=8,0 Гц, 1H), 6,93 (т, J=8,5Гц, 1H), 7,21 (т, J=4 Гц, 1H), 7,29 (д, J=8,5 Гц, 2H), 7,75 (д, J=8,5 Гц, 2H), 7,79 (м, 1H), 8,04 (м, 1Н). ЖХ/МС (98%) ESI- Вычислено: 535,6 m/z; найдено: 535,3 m/z.
Пример 112. Получение соединения P039.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 3,4-дихлорбензилбромидом, получая соединение P039. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 525,5 (M-1), ЖХ-МС: 90%.
Пример 113. Получение соединения P040.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 2,4-дихлорбензилбромидом, получая соединение P040. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 525,4 (M-1), ЖХ-МС: 78%.
Пример 114. Получение соединения P041.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 3,5-диметоксибензилбромидом, получая соединение P041. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 517,5 (M-1), ЖХ-МС: 94%.
Пример 115. Получение соединения P042.
Общую методику (A-3) используют для алкилирования [2-(3-оксо-3,4-дигидро-2H-бензо[1,4]оксазин-5-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, I-70, 5-бромметилбензо[1,2,5]оксадиазолом, получая соединение P042. 1H-ЯМР (ДМСО-d6) 4,55 (с, 2H), 4,69 (с, 2H), 5,35 (с, 2H), 6,48 (д, J=8,5 Гц, 1H), 6,74 (д, J=8,0 Гц, 1H), 6,94 (т, J=8,0 Гц, 1H), 7,21 (т, J=4,0 Гц, 1H), 7,30 (д, J=9,0 Гц, 1H), 7,60 (с, 1H), 7,77 (м, 1H), 7,90 (д, J=9,0 Гц, 1H), 8,07 (д, J=5,0 Гц, 1H), 12,5 (уш.с, 1H). ЖХ/МС (92%) ESI-; Вычислено: 499,5 m/z; найдено: 499,8 m/z.
Пример 116. Получение соединения P004.
Синтез метилового эфира (2-оксо-1,2-дигидрохинолин-8-илокси)уксусной кислоты, I-71. Метилбромацетат (367 мг, 2,4 ммоль) добавляют при перемешивании к смеси 8-гидрокси-1Н-хинолин-2-она [который получают в соответствии с методикой, приведенной в литературе (Wang, T.C. et al., Synthesis, 1997, 87-90)], (322 мг, 2 ммоль) и K2CO3 (414 мг, 3 ммоль) в ДМФА (10 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем распределяют между дихлорметаном и водой. Водный слой экстрагируют дихлорметаном (2×50 мл). Объединенную органическую фазу промывают водой (3×50 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая 250 мг соединения I-71.
Синтез метилового эфира (1-нафталин-2-илметил-2-оксо-1,2-дигидрохинолин-8-илокси)уксусной кислоты, I-72. Смесь соединения I-71 (233 мг, 1 ммоль), 2-(бромметил)нафталина (332 мг, 1,5 ммоль), KI (250 мг, 1,5 ммоль) и K2CO3 (207 мг, 1,5 ммоль) в ДМФА (8 мл) и воде (2 капли) перемешивают при комнатной температуре в течение недели и затем распределяют между дихлорметаном и водой. Водный слой экстрагируют дихлорметаном (2×50 мл). Объединенную органическую фазу промывают водой (4×50 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя, остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента 2% метанол в дихлорметане и получая 120 мг соединения I-72 и 50 мг O-алкилированного побочного продукта: метилового эфира (1-нафталин-2-илметил-2-оксо-1,2-дигидрохинолин-8-илокси)уксусной кислоты. Для соединения I-72: 1H-ЯМР (500 МГц, CDCl3), 13C-ЯМР (125 МГц, CDCl3).
Синтез (1-нафталин-2-илметил-2-оксо-1,2-дигидрохинолин-8-илокси)уксусной кислоты, I-73. К раствору соединения I-72 (120 мг, 0,32 ммоль) в EtOH (8 мл) при комнатной температуре добавляют водный NaOH (2н., 5 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем значение pH доводят до кислотного добавлением водного 2н. HCl. Реакционную смесь экстрагируют EtOAc (2×10 мл). Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая 100 мг соединения I-73. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P004. К смеси кислоты I-73 (36 мг, 0,1 ммоль), 2-тиофенсульфонамида (20 мг, 0,12 ммоль) и 4-диметиламинопиридина (25 мг, 0,2 ммоль) в дихлорметане (5 мл) добавляют EDCI (38 мг, 0,2 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя твердый остаток промывают эфиром, получая 35 мг соединения P004. 1H-ЯМР (ДМСО-d6) 4,36 (с, 2H), 6,06 (с, 2H), 6,74 (д, J=9 Гц, 1H), 6,92 (д, J=8,0 Гц, 1H), 7,11 (т, J=7,5 Гц, 1H), 7,16 (м, 2H), 7,34 (д, J=7 Гц, 1H), 7,38-7,42 (м, 3H), 7,63-7,72 (м, 3H), 7,81 (м, 1H), 7,95 (м, 2H). ЖХ/МС (90%) ESI- Вычислено: 503,6 m/z; найдено: 503,4 m/z
Пример 117. Получение соединения P012.
Синтез 8-(нафталин-2-илметокси)-1Н-хинолин-2-она, I-74. 2-(бромметил)нафталин (530 мг, 2,4 ммоль) при комнатной температуре добавляют при перемешивании к смеси 8-(нафталин-2-идметокси)-1Н-хинолин-2-она (322 мг, 2 ммоль) и K2CO3 (414 мг, 3 ммоль) в ДМФА (10 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем распределяют между дихлорметаном и водой. Водный слой экстрагируют дихлорметаном (2×50 мл). Объединенную органическую фазу промывают водой (3×50 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая 470 мг соединения I-74. 1Н-ЯМР (500 МГц, CDCl3).
Синтез метилового эфира [8-(нафталин-2-илметокси)-2-оксо-2H-хинолин-1-ил]уксусной кислоты, I-75. Смесь соединения I-74 (210 мг, 0,7 ммоль), метилбромацетата (230 мг, 1,5 ммоль), KI (250 мг, 1,5 ммоль) и K2CO3 (207 мг, 1,5 ммоль) в ДМФА (8 мл) и воде (2 капли) перемешивают при комнатной температуре в течение ночи и затем распределяют между дихлорметаном и водой. Водный слой экстрагируют дихлорметаном (2×50 мл). Объединенную органическую фазу промывают водой (4×50 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента гексан, смесь гексан/дихлорметан (1:1), дихлорметан, 1% метанол в дихлорметане, получая 160 мг соединения I-75 и 30 мг O-алкилированного побочного продукта метилового эфира [8-(нафталин-2-илметокси)хинолин-2-илокси]уксусной кислоты. Для соединения I-75: 1H-ЯМР (500 МГц, CDCl3); 13C-ЯМР (125 МГц, CDCl3).
Синтез [8-(нафталин-2-илметокси)-2-оксо-2H-хинолин-1-ил]уксусной кислоты, I-76. К раствору соединения I-75 (30 мг, 0,08 ммоль) в ТГФ (3 мл) и MeOH (3 мл) при комнатной температуре добавляют водный NaOH (2н., 3 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи и значение рН доводят до кислотного добавлением водного 2н. HCl. Реакционную смесь экстрагируют EtOAc (2×15 мл). Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая 25 мг кислоты I-76. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P012. К смеси кислоты I-76 (25 мг, 0,07 ммоль), 2-тиофенсульфонамида (14 мг, 0,084 ммоль) и 4-диметиламинопиридина (18 мг, 0,15 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) добавляют EDCI (29 мг, 0,15 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя твердый остаток промывают эфиром, получая 30 мг соединения P-12. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 503,3 (M-1), ЖХ-МС: 91%.
Пример 119. Получение соединения P015.
Синтез 8-(нафталин-2-илметокси)хинолина, I-81. 2-(Бромметил)нафталин (663 мг, 3 ммоль) при комнатной температуре при перемешивании добавляют к смеси 8-гидроксихинолина (435 мг, 3 ммоль) и K2CO3 (621 мг, 4,5 ммоль) в ацетоне (20 мл). Реакционную смесь перемешивают при комнатной температуре в течение недели и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc (2×100 мл). Объединенную органическую фазу промывают водой (2×100 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая 650 мг соединения I-81. 1H-ЯМР (500 МГц, CDCl3).
Синтез 8-(нафталин-2-илметокси)-1,2,3,4-тетрагидрохинолина, I-82. К раствору соединения I-81 (285 мг, 1 ммоль) в AcOH (15 мл) при комнатной температуре добавляют NaCNBH3 (252 мг, 4 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и затем при 60°C в течение 1 часа. Реакционную смесь перемешивают при комнатной температуре в течение ночи и растворитель удаляют в вакууме. Остаток распределяют между дихлорметаном и водой и значение pH доводят до 8-8 добавлением водного раствора NH4OH. Водную фазу экстрагируют дихлорметаном (3×50 мл). Объединенную органическую фазу промывают насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента дихлорметан и 2% смесь метанол/дихлорметан и получая 200 мг соединения I-82. 1H-ЯМР (500 МГц, CDCl3).
Синтез метилового эфира [8-(нафталин-2-илметокси)-3,4-дигидро-2H-хинолин-1-ил]уксусной кислоты, I-83. Смесь соединения I-82 (200 мг, 0,7 ммоль), метилбромацетата (153 мг, 1 ммоль), KI (233 мг, 1,4 ммоль) и K2CO3 (193 мг, 1,4 ммоль) в ДМФА (10 мл) и воде (0,2 мл) перемешивают при комнатной температуре в течение ночи и затем распределяют между дихлорметан и водой. Водный слой экстрагируют дихлорметаном (2×50 мл). Объединенную органическую фазу промывают водой (4×50 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 250 мг неочищенного соединения I-83. 1H-ЯМР (500 МГц, CDCl3).
Синтез [8-(нафталин-2-илметокси)-3,4-дигидро-2H-хинолин-1-ил]уксусной кислоты, I-84. К раствору соединения I-83 (250 мг, 0,7 ммоль) в ТГФ (3 мл) и EtOH (6 мл) при комнатной температуре добавляют водный NaOH (2н., 4 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем pH доводят до кислотного значения добавлением водного 2н. HCl. Реакционную смесь экстрагируют EtOAc (2×15 мл). Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая 120 мг соединения I-84. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединение P015. К смеси кислоты I-84 (25 мг, 0,07 ммоль), 2-тиофенсульфонамида (16 мг, 0,1 ммоль), 4-диметиламинопиридина (24 мг, 0,2 ммоль) в дихлорметане (6 мл) и ДМСО (0,5 мл) добавляют EDCI (38 мг, 0,2 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан и 2% метанол/дихлорметан в качестве элюента и получая 7 мг соединения P015. 1H ЯМР (ДМСО-d6) 1,81 (м, 2H), 2,78 (т, J=6,5 Гц, 2H), 3,03 (м, 2H), 3,62 (с, 2H), 5,12 (с, 2H), 6,66 (д, J=7,5 Гц, 1H), 6,72 (д, J=7,5Гц, 1H), 6,79 (м, 1H), 6,87 (м, 1H), 7,19 (м, 1H), 7,35 (м, 1H), 7,43 (м, 1H), 7,49 (м, 2H), 7,62 (м, 1H), 7,72 (м, 2H), 7,82 (м, 3H). ЖХ/МС (97%) ESI- Вычислено: 491,6 m/z; найдено: 491,2 m/z.
Пример 120. Получение соединения P046.
Синтез 2-аминобензол-1,3-диола, I-85. Смесь 2-нитробензола-1,3-диола (3,1 г, 20 ммоль) и 1 г Pd/C (5%) в метаноле (100 мл) гидрируют водородом при давлении 30 фунтов на кв. дюйм при комнатной температуре в течение 2 дней. Реакционную смесь фильтруют через целит, целит промывают этанолом, водой и метанолом. После удаления растворителя в вакууме получают 2,6 г соединения I-85 в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез 2-[(нафталин-2-илметил)амино]бензол-1,3-диола, I-86. Смесь соединения I-85 (250 мг, 2 ммоль) и 2-нафтальдегида (343 мг, 2,2 ммоль) в метаноле (20 мл) перемешивают при комнатной температуре в течение 2 часов, затем охлаждают до -10°C. В реакционную смесь медленно при температуре в интервале от -10°С до 0°С добавляют NaBH4 (304 мг, 8 ммоль). Смесь перемешивают при 0°C в течение 30 минут и при комнатной температуре в течение 2 часов, затем охлаждают до 0°C и гасят водой. Значение рН реакционной смеси доводят до ~4 и экстрагируют EtOAc (2×100 мл). К водному слою добавляют хлорид натрия и полученную смесь экстрагируют дихлорметаном. Из смеси вода/CH2Cl2 выпадает твердый осадок. Твердое вещество отфильтровывают и промывают водой и CH2Cl2, получая 140 мг соединения I-86. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез 4-гидрокси-3-нафталин-2-илметил-3H-бензоксазол-2-она, I-87. Смесь соединения I-86 (140 мг, 0,46 ммоль), триэтиламина (46 мг, 0,46 ммоль) и 1,1'-карбонилдиимидазола (80 мг, 0,5 ммоль) в ТГФ (20 мл) кипятят с обратным холодильником в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 140 мг соединения I-87 в виде белого твердого вещества. 1H-ЯМР (500 МГц, ДМСО-d6). 13C-ЯМР (125 МГц, ДМСО-d6).
Синтез трет-бутилового эфира (3-нафталин-2-илметил-2-оксо-2,3-дигидробензоксазол-4-илокси)уксусной кислоты, I-88. Смесь соединения I-87 (140 мг, 0,48 ммоль), трет-бутилбромацетата (136 мг, 0,7 ммоль) и K2CO3 (95 мг, 0,7 ммоль) в ацетоне (10 мл) и ДМФА (5 мл) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 160 мг соединения I-88. 1H-ЯМР (500 МГц, CDCl3).
Синтез (3-нафталин-2-илметил-2-оксо-2,3-дигидробензоксазол-4-илокси)уксусной кислоты, I-89. К раствору соединения I-88 (150 мг, 0,37 ммоль) в дихлорметане (5 мл) при комнатной температуре добавляют ТФУК (10 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 130 мг соединения I-89. МС (ESI-): 348,2 (M-1), ЖХ-МС: 99%, 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P046. Смесь кислоты I-89 (35 мг, 0,1 ммоль), 2-тиофенсульфонамида (20 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют с дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя твердый остаток промывают эфиром, получая 30 мг соединения P046. МС (ESI-): 493,4 (M-1), ЖХ-МС: 81%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 121. Получение соединения P047.
Смесь кислоты I-89 (35 мг, 0,1 ммоль), 2-метокси-5-бромфенилсульфонамида (32 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь метанол/дихлорметан и получая 24 мг соединения P047. МС (ESI-): 597,2 (M-1), ЖХ-МС:94%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 122. Получение соединения P048.
Смесь кислоты I-89 (35 мг, 0,1 ммоль), трифторметилсульфонамида (18 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCl (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 30 мг соединения P048 в форме DMAP-соли. МС (ESI-): 479,3 (M-1), ЖХ-МС: 96%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 123. Получение соединения P050.
Синтез 4-гидрокси-3H-бензоксазол-2-она, I-90. Смесь 2-аминобенз-1,3-диола (2,03 г, 16,2 ммоль) и 1,1'-карбонилдиимидазола (2,63 г, 16,2 ммоль) в ТГФ (200 мл) кипятят с обратным холодильником в течение ночи. После удаления ТГФ в вакууме остаток растворяют в EtOAc и промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя твердый остаток промывают эфиром, получая 1,9 г соединения I-90. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез трет-бутилового эфира 2-оксо-2,3-дигидробензоксазол-4-илового эфира, I-91. К раствору соединения I-90 (1 г, 6,6 ммоль) в ТГФ (20 мл) и воде (8 мл) при комнатной температуре при перемешивании добавляют водный NaOH (2н., 13 мл) и ди-трет-бутилдикарбонат (3,16 г, 14,5 ммоль) в ТГФ (15 мл). После перемешивания при комнатной температуре в течение ночи реакционную смесь разбавляют водой и EtOAc и охлаждают льдом, затем значение pH доводят до примерно 2-3 добавлением водного HCl (2н.). Водный слой экстрагируют EtOAc (2×100 мл). Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 2,3 г соединения I-91. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез 3-бензо[1,3]диоксол-5-илметил-4-гидрокси-3H-бензоксазол-2-она, I-92. Смесь соединения I-91 (251 мг, 1 ммоль), 3,4-метилендиоксибензилхлорид (255 мг, 1,5 ммоль), K2CO3 (207 мг, 1,5 ммоль) и KI (249 мг, 1,5 ммоль) в ацетоне (10 мл) и воде (5 капли) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя промежуточное O-BOC-соединение (3-бензо[1,3]диоксол-5-илметил-2-оксо-2,3-дигидробензоксазол-4-иловый эфир трет-бутилового эфира карбоновой кислоты) растворяют в дихлорметане (6 мл) и при комнатной температуре добавляют ТФУК (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 15 минут и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 100 мг соединения I-92. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез трет-бутилового эфира (3-бензо[1,3]диоксол-5-илметил-2-оксо-2,3-дигидробензоксазол-4-илокси)уксусной кислоты, I-93. Смесь соединения I-92 (100 мг, 0,35 ммоль), трет-бутилбромацетата (136 мг, 0,7 ммоль) и K2CO3 (95 мг, 0,7 ммоль) в ацетоне (8 мл) и ДМСО (2 мл) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 100 мг соединения I-93. 1H-ЯМР (500 МГц, CDCl3).
Синтез (3-бензо[1,3]диоксол-5-илметил-2-оксо-2,3-дигидробензоксазол-4-илокси)уксусной кислоты, I-94. К раствору соединения I-93 (100 мг, 0,25 ммоль) в дихлорметане (4 мл) при комнатной температуре добавляют ТФУК (8 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 80 мг соединения I-94. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 342,2 (M-1), ЖХ-МС:>90%.
Синтез соединения P050. Смесь кислоты I-94 (35 мг, 0,1 ммоль), 2-тиофенсульфонамида (20 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь метанол/дихлорметан и получая 35 мг соединения P050. МС (ESI): 487,4 (M-1), ЖХ-МС: 87%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 124. Получение соединения P051.
Синтез 3-(3,4-дихлорбензил)-4-гидрокси-3H-бензоксазол-2-она, I-95. Смесь соединения I-91 (251 мг, 1 ммоль), 3,4-дихлорбензилхлорида (292 мг, 1,5 ммоль), K2CO3 (207 мг, 1,5 ммоль) и KI (249 мг, 1,5 ммоль) в ацетоне (10 мл) и воде (5 капли) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя O-BOC-продукт (трет-бутиловый эфир 3-(3,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-илового эфира карбоновой кислоты) растворяют в дихлорметане (6 мл), затем при комнатной температуре добавляют ТФУК (3 мл), реакционную смесь перемешивают при комнатной температуре в течение 15 минут и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 80 мг соединения I-95. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез трет-бутилового эфира [3-(3,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-илокси]уксусной кислоты, I-96. Смесь соединения I-95 (80 мг, 0,26 ммоль), трет-бутилбромацетата (136 мг, 0,7 ммоль) и K2CO3 (95 мг, 0,7 ммоль) в ацетоне (8 мл) и ДМСО (2 мл) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 90 мг соединения I-96. 1H-ЯМР(500 МГц,CDCl3).
Синтез [3-(3,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-илокси]уксусной кислоты, I-97:20. К раствору соединения I-96 (90 мг, 0,21 ммоль) в дихлорметане (4 мл) при комнатной температуре добавляют ТФУК (8 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 70 мг кислоты I-97. МС (ESI-): 367,9 (M-1), ЖХ-МС: >90%, 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P051. Смесь кислоты I-97 (37 мг, 0,1 ммоль), 2-тиофенсульфонамида (20 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь метанол/дихлорметан и получая 32 мг соединения P051. МС (ESI-): 511,4 (M-1), ЖХ-МС:89%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 125. Получение соединения P052.
Синтез 3-(2,4-дихлорбензил)-4-гидрокси-3H-бензоксазол-2-она, I-98. Смесь соединения I-91 (251 мг, 1 ммоль), 2,4-дихлорбензилхлорида (292 мг, 1,5 ммоль), K2CO3 (207 мг, 1,5 ммоль) и KI (249 мг, 1,5 ммоль) в ацетоне (10 мл) и воде (5 капли) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя O-BOC-производное (3-(2,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-иловый эфир трет-бутилового эфира карбоновой кислоты) растворяют в дихлорметане (6 мл), при комнатной температуре добавляют ТФУК (3 мл), полученную смесь перемешивают при комнатной температуре в течение 15 минут и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 110 мг соединения I-98. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез трет-бутилового эфира 3-(2,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-илокси]уксусной кислоты, I-99. Смесь соединения I-98 (80 мг, 0,26 ммоль), трет-бутилбромацетата (136 мг, 0,7 ммоль) и K2CO3 (95 мг, 0,7 ммоль) в ацетоне (8 мл) и ДМСО (2 мл) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 140 мг соединения I-99. 1H-ЯМР (500 МГц, CDCl3).
Синтез трет-бутилового эфира (3-нафталин-2-илметил-2-оксо-2,3-дигидробензоксазол-4-илокси)уксусной кислоты, I-88. Смесь соединения I-87 (140 мг, 0,48 ммоль), трет-бутилбромацетата (136 мг, 0,7 ммоль) и K2CO3 (95 мг, 0,7 ммоль) в ацетоне (10 мл) и ДМФА (5 мл) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 160 мг соединения I-88. 1H-ЯМР (500 МГц, CDCl3).
Синтез (3-нафталин-2-илметил-2-оксо-2,3-дигидробензоксазол-4-илокси)уксусной кислоты, I-89. К раствору соединения I-88 (150 мг, 0,37 ммоль) в дихлорметане (5 мл) при комнатной температуре добавляют ТФУК (10 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 130 мг соединения I-89. МС (ESI-): 348,2 (M-1), ЖХ-МС: 99%, 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P046. Смесь кислоты I-89 (35 мг, 0,1 ммоль), 2-тиофенсульфонамида (20 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют с дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя твердый остаток промывают эфиром, получая 30 мг соединения P046. МС (ESI-): 493,4 (M-1), ЖХ-МС: 81%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 121. Получение соединения P047.
Смесь кислоты I-89 (35 мг, 0,1 ммоль), 2-метокси-5-бромфенилсульфонамида (32 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь метанол/дихлорметан и получая 24 мг соединения P047. МС (ESI-): 597,2 (M-1), ЖХ-МС:94%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 122. Получение соединения P048.
Смесь кислоты I-89 (35 мг, 0,1 ммоль), трифторметилсульфонамида (18 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCl (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 30 мг соединения P048 в форме DMAP-соли. МС (ESI-): 479,3 (M-1), ЖХ-МС: 96%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 123. Получение соединения P050.
Синтез 4-гидрокси-3H-бензоксазол-2-она, I-90. Смесь 2-аминобенз-1,3-диола (2,03 г, 16,2 ммоль) и 1,1'-карбонилдиимидазола (2,63 г, 16,2 ммоль) в ТГФ (200 мл) кипятят с обратным холодильником в течение ночи. После удаления ТГФ в вакууме остаток растворяют в EtOAc и промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя твердый остаток промывают эфиром, получая 1,9 г соединения I-90. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез трет-бутилового эфира 2-оксо-2,3-дигидробензоксазол-4-илового эфира карбоновой кислоты, I-91. К раствору соединения I-90 (1 г, 6,6 ммоль) в ТГФ (20 мл) и воде (8 мл) при комнатной температуре при перемешивании добавляют водный NaOH (2н., 13 мл) и ди-трет-бутилдикарбонат (3,16 г, 14,5 ммоль) в ТГФ (15 мл). После перемешивания при комнатной температуре в течениеночи реакционную смесь разбавляют водой и EtOAc и охлаждают льдом, затем pH доводят до примерно 2-3 добавлением водного HCl (2н.). Водный слой экстрагируют EtOAc (2×100 мл). Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 2,3 г соединения I-91. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез 3-бензо[1,3]диоксол-5-илметил-4-гидрокси-3H-бензоксазол-2-она, I-92. Смесь соединения I-91 (251 мг, 1 ммоль), 3,4-метилендиоксибензилхлорид (255 мг, 1,5 ммоль), K2CO3 (207 мг, 1,5 ммоль) и KI (249 мг, 1,5 ммоль) в ацетоне (10 мл) и воде (5 капли) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя промежуточное O-BOC-соединение (3-бензо[1,3]диоксол-5-илметил-2-оксо-2,3-дигидробензоксазол-4-иловый эфир трет-бутилового эфира карбоновой кислоты) растворяют в дихлорметане (6 мл) и при комнатной температуре добавляют ТФУК (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 15 минут и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 100 мг соединения I-92. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез трет-бутилового эфира (3-бензо[1,3]диоксол-5-илметил-2-оксо-2,3-дигидробензоксазол-4-илокси)уксусной кислоты, I-93. Смесь соединения I-92 (100 мг, 0,35 ммоль), трет-бутилбромацетата (136 мг, 0,7 ммоль) и K2CO3 (95 мг, 0,7 ммоль) в ацетоне (8 мл) и ДМСО (2 мл) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 100 мг соединения I-93. 1H-ЯМР (500 МГц, CDCl3).
Синтез (3-бензо[1,3]диоксол-5-илметил-2-оксо-2,3-дигидробензоксазол-4-илокси)уксусной кислоты, I-94. К раствору соединения I-93 (100 мг, 0,25 ммоль) в дихлорметане (4 мл) при комнатной температуре добавляют ТФУК (8 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 80 мг соединения I-94. 1H-ЯМР (500 МГц, ДМСО-d6), МС (ESI-): 342,2 (M-1), ЖХ-МС:>90%.
Синтез соединения P050. Смесь кислоты I-94 (35 мг, 0,1 ммоль), 2-тиофенсульфонамида (20 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь метанол/дихлорметан и получая 35 мг соединения P050. МС (ESI): 487,4 (M-1), ЖХ-МС: 87%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 124. Получение соединения P051
Синтез 3-(3,4-дихлорбензил)-4-гидрокси-3H-бензоксазол-2-она, I-95. Смесь соединения I-91 (251 мг, 1 ммоль), 3,4-дихлорбензилхлорида (292 мг, 1,5 ммоль), K2CO3 (207 мг, 1,5 ммоль) и KI (249 мг, 1,5 ммоль) в ацетоен (10 мл) и воде (5 капли) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя O-BOC-продукт (3-(3,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-иловый эфир трет-бутилового эфира карбоновой кислоты) растворяют в дихлорметане (6 мл), затем при комнатной температуре добавляют ТФУК (3 мл), реакционную смесь перемешивают при комнатной температуре в течение 15 минут и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 80 мг соединения I-95. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез трет-бутилового эфира [3-(3,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-илокси]уксусной кислоты, I-96. Смесь соединения I-95 (80 мг, 0,26 ммоль), трет-бутилбромацетата (136 мг, 0,7 ммоль) и K2CO3 (95 мг, 0,7 ммоль) в ацетоне (8 мл) и ДМСО (2 мл) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 90 мг соединения I-96. 1H-ЯМР(500 МГц,CDCl3).
Синтез [3-(3,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-илокси]уксусной кислоты, I-97:20. К раствору соединения I-96 (90 мг, 0,21 ммоль) в дихлорметане (4 мл) при комнатной температуре добавляют ТФУК (8 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 70 мг кислоты I-97. МС (ESI-): 367,9 (M-1), ЖХ-МС: >90%, 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P051. Смесь кислоты I-97 (37 мг, 0,1 ммоль), 2-тиофенсульфонамида (20 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь метанол/дихлорметан и получая 32 мг соединения P051. МС (ESI-): 511,4 (M-1), ЖХ-МС:89%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 125. Получение соединения P052.
Синтез 3-(2,4-дихлорбензил)-4-гидрокси-3H-бензоксазол-2-она, I-98. Смесь соединения I-91 (251 мг, 1 ммоль), 2,4-дихлорбензилхлорида (292 мг, 1,5 ммоль), K2CO3 (207 мг, 1,5 ммоль) и KI (249 мг, 1,5 ммоль) в ацетоне (10 мл) и воде (5 капли) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя O-BOC-производное (3-(2,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-иловый эфир трет-бутилового эфира карбоновой кислоты) растворяют в дихлорметане (6 мл), при комнатной температуре добавляют ТФУК (3 мл), полученную смесь перемешивают при комнатной температуре в течение 15 минут и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 110 мг соединения I-98. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез трет-бутилового эфира 3-(2,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-илокси]уксусной кислоты, I-99. Смесь соединения I-98 (80 мг, 0,26 ммоль), трет-бутилбромацетата (136 мг, 0,7 ммоль) и K2CO3 (95 мг, 0,7 ммоль) в ацетоне (8 мл) и ДМСО (2 мл) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 140 мг соединения I-99. 1H-ЯМР (500 МГц,CDCl3).
Синтез 3-(2,4-дихлорбензил)-2-оксо-2,3-дигидробензоксазол-4-илокси]уксусной кислоты, I-100. К раствору соединения I-99 (90 мг, 0,33 ммоль) в дихлорметане (4 мл) при комнатной температуре добавляют ТФУК (8 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 105 мг соединения I-100. МС (ESI-): 366,2 (M-1), ЖХ-МС: >90%, 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P052. Смесь кислоты I-100 (37 мг, 0,1 ммоль), 2-тиофенсульфонамида (20 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя, остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь метанол/дихлорметан и получая 34 мг соединения P052. МС (ESI-): 511,4 (M-1), ЖХ-МС: 89%, 1H-ЯМР (500 МГц, ДМСО-d6).
Пример 126. Получение соединения P053.
Синтез 3-(2,5-диметилбензил)-4-гидрокси-3H-бензоксазол-2-она, I-101. Смесь соединения I-91 (251 мг, 1 ммоль), 2,5-диметилбензилхлорида (233 мг, 1,5 ммоль), K2CO3 (207 мг, 1,5 ммоль) и KI (249 мг, 1,5 ммоль) в ацетоне (10 мл) и воде (5 каплей) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя O-BOC-производное (3-(2,5-диметилбензил)-2-оксо-2,3-дигидробензоксазол-4-иловый эфир трет-бутилового эфира карбоновой кислоты) растворяют в дихлорметане (6 мл) и затем при комнатной температуре добавляют ТФУК (3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 15 минут и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 110 мг соединения I-101.
Синтез трет-бутилового эфира 3-(2,5-диметилбензил)-2-оксо-2,3-дигидробензоксазол-4-илокси]уксусной кислоты, I-102. Смесь соединения I-101 (110 мг, 0,4 ммоль), трет-бутилбромацетата (136 мг, 0,7 ммоль) и K2CO3 (95 мг, 0,7 ммоль) в ацетоне (8 мл) и ДМСО (2 мл) перемешивают при комнатной температуре в течение ночи и затем распределяют между EtOAc и водой. Водный слой экстрагируют EtOAc. Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 150 мг соединения I-102. 1H-ЯМР (500 МГц,CDCl3).
Синтез [3-(2,5-диметилбензил)-2-оксо-2,3-дигидробензоксазол-4-илокси]уксусной кислоты, I-103. К раствору соединения I-102 (150 мг, 0,39 ммоль) в дихлорметане (4 мл) при комнатной температуре добавляют ТФУК (8 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и растворитель удаляют в вакууме. Остаток промывают эфиром, получая 120 мг соединения I-103. МС(ESI-): 326,4 (M-1), ЖХ-МС: >90%, 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P053. Cмесь кислоты I-103 (33 мг, 0,1 ммоль), 2-тиофенсульфонамида (20 мг, 0,12 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (0,5 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя в качестве элюента смесь метанол/дихлорметан и получая 37 мг соединения P053. 1H-ЯМР (ДМСО-d6) 2,11 (с, 3H), 2,24 (с, 3H), 4,58 (с, 2H), 5,13 (с, 2H), 6,69 (с, 2H), 6,92 (д, J=8,0 Гц, 1H), 7,01 (д, J=8,0 Гц, 1H), 7,04 (м, 2H), 7,18 (м, 1H), 7,73 (м, 1H), 8,01 (м, 1H), 12,5 (уш. с, 1H). ЖХ/МС (88%) ESI- Вычислено: 471,5 m/z; найдено: 471,4 m/z.
Пример 127. Получение соединения P083.
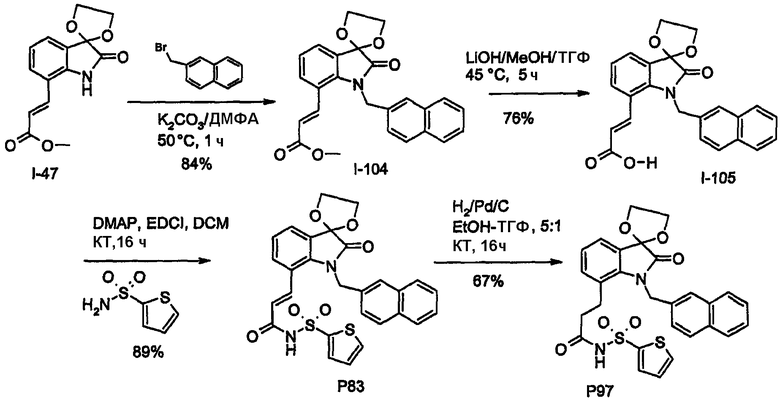
Синтез соединения I-104. Смесь соединения I-47 (0,7 г, 2,54 ммоль, 1 экв.), 2-бромметилнафталина (0,59 г, 2,67 ммоль, 1,05 экв.) и K2CO3 (1,76 г, 12,72 ммоль, 5 экв.) в ДМФА (10 мл) выдерживают при 50°C в течение 1 часа. Суспензию фильтруют, промывают водой, насыщенным раствором соли, сушат над MgSO4, получая продукт I-104, 0,89 г (84%) в виде желтого твердого вещества. МС (ESI-): 416 (M+1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез соединения I-105. Раствор сложного эфира I-104 (600 мг, 1,44 ммоль,l экв.), LiOH H2O (70 мг, 1,6 ммоль, 1,15 экв.) в ТГФ/MeOH/H2O (3:1:1, 22 мл) перемешивают при 45°C в течение 5 часов. Реакционную смесь концентрируют в вакууме и к остатку добавляют нас. NH4Cl (10 мл). Осадок экстрагируют CH2Cl2. Полученный раствор промывают насыщенным NH4Cl, насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме. Остаток растирают в эфире (8 мл) с получением кислоты I-105 [362 мг (62%)] в виде белого твердого вещества. МС (ESI-): 400 (M-1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез соединения P083. К смеси кислоты, I-105 (71 мг, 0,177 ммоль, 1 экв.) в 2 мл дихлорметана добавляют DMAP (43 мг, 0,354 ммоль, 2 экв.), 2-тиофенсульфонамид (37 мг, 0,225 ммоль) и EDCI (82 мг, 0,425 ммоль). Смесь перемешивают при комнатной температуре в течение 16 часов и затем гасят добавлением NH4Cl (6 мл). Смесь экстрагируют EtOAc (6 мл). Экстракт промывают насыщенным раствором соли и затем сушат над MgSO4. Растворитель удаляют в вакууме. Остаток растирают в эфире (2 мл) с получением 86 мг (86%) сульфонамида P083 в виде твердого не совсем белого вещества. ЖХ-МС (ESI-): 545 (M-1) (81%). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Пример 128. Получение соединения P097.
Сульфонамид P083 (57 мг, 0,104 ммоль) растворяют в смеси EtOH-ТГФ (24 мл, 5:1) и раствор насыщают водородом. Баллон с водородом присоединяют к аппарату и реакционную смесь перемешивают при комнатной температуре в течение 20 часов, затем при 60 C в течение 22 часов. Полученный раствор фильтруют через фильтр из ватмана 0,45 мкм. Растворитель удаляют в вакууме. Растирание остатка в эфире (2×2 мл) приводит к получению 44 мг (67%) сульфонамида P097 в виде твердого не совсем белого вещества. ЖХ-МС (ESI-): 547 (M-1) (95%). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Пример 129. Получение соединения P096.
Синтез (E)-3-(1-нафталин-2-илметил-2,3-диоксо-2,3-дигидро-1Н-индол-7-ил)акриловой кислоты, I-106. Раствор соединения I-105 (48 мг, 0,12 ммоль) в смеси концентрированной HCl и i-PrOH (1:1, 4 мл) выдерживают при 100°C в течение 30 минут. Реакционную смесь охлаждают до комнатной температуры, фильтруют и промывают водой, получая изатин-производное I-106, 28 мг (66%) в виде ярко-оранжевого твердого вещества, которое нерастворимо в эфире, EtOAc или CH2Cl2. МС(ESI-) 356 (M-1).
Синтез соединения P096. К смеси соединения I-106 (21 мг, 0,059 ммоль, 1 экв.) в 0,5 мл дихлорметана добавляют DMAP (19 мг, 0,157 ммоль, 2,6 экв.), 2-тиофенсульфонамид (13 мг, 0,078 ммоль, 1,3 экв.) и EDCI (30 мг, 0,157 ммоль, 2,6 экв.). Смесь перемешивают при комнатной температуре в течение 16 часов, затем гасят 10% HCl и экстрагируют смесью EtOAc-CH2Cl2. Экстракт промывают водой, насыщенным раствором соли и затем сушат над MgSO4. Растворитель удаляют в вакууме. Остаток растворяют в CH2Cl2 (2 мл) и фильтруют. Хроматография на SiO2 (5 г) с использованием EtOAc/гексан (1:1) приводит к получению 10 мг (33%) сульфонамида P096 в виде твердого, не совсем белого вещества. ЖХ-МС (ESI-): 501 (M-1) (98%). 1Н-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Пример 130. Получение соединения P126.
Раствор изатин-производного P113 (64 мг, 0,108 ммоль, 1 экв.), 2-меркаптоэтиленамина (8,3 мг, 0,108 ммоль, 1 экв.) в AcOH (1 мл) нагревают до 100°C и выдерживают при этой температуре в течение 20 минут. Добавляют другую часть 2-меркаптоэтиленамина (4,3 мг, 0,056 ммоль, 0,5 экв.) и нагревание продолжают в течение 10 минут. Реакционную смесь упаривают, полученное масло растирают в смеси ТГФ-эфир (1:3), раствор фильтруют, получая тиазолидин-производное P126 (28,3 мг, 40%) в виде красного твердого вещества. 1H ЯМР (ДМСО-d6) 3,5-3,7 (м, 3H), 3,99 (дд, J=11, 8,0 Гц, 1H), 4,93 (д, J=18,0 Гц, 1H), 5,01 (д, J=18,0 Гц, 1H), 6,03 (д, J=15,2 Гц, 1H), 6,85 (д, J=8,4 Гц, 2H), 7,11 (т, J=4,0 Гц, 1H), 7,19 (д, J=15,0 Гц, 2H), 7,30-7,34 (м, 2H), 7,36 (с, 1H), 7,36 (д, J=2,0 Гц, 1H), 7,51 (дд, J=7,2, 1,2 Гц, 1H). ЖХ-МС (99%): ESI- Вычислено: 547 m/z; найдено: 547.
Пример 131. Получение соединения P037
Синтез 2-бензилокси-6-нитрофениламина, I-107: Смесь 2-амино-3-нитрофенола (1,54 г, 0,01 моль), бензилбромида (1,71 г, 0,01 моль) и K2CO3 (1,54 г, 0,011 моль) в ДМФА (5,0 мл) перемешивают при комнатной температуре. Спустя 20 часов реакционную смесь разбавляют водой (100 мл) и экстрагируют EtOAc (100 мл×3). Объединенные органические слои промывают 2н. NaOH (20 мл×3), водой (100 мл), насыщенным раствором соли (100 мл), сушат над безводным сульфатом натрия и концентрируют, получая 2-бензилокси-6-нитрофениламин, I-107 (2,01 г).
Синтез N-(2-бензилокси-6-нитрофенил)ацетамида, I-108. К раствору 2-бензилокси-6-нитрофениламина (I-107, 1,52 г, 0,0062 моль) в уксусном ангидриде (1,27 г, 0,0125 моль) добавляют 4 капли концентрированной H2SO4 при комнатной температуре и перемешивают в течение одной минуты, затем реакционную колбу переносят на масляную баню при 90°C. Спустя 2 минуты, затвердевшую реакционную смесь переносят в воду (20 мл) и твердые вещества отфильтровывают. Твердый осадок дополнительно промывают водой (20 мл×2) и сушат в вакууме, получая N-(2-бензилокси-6-нитрофенил)ацетамид, I-108 (1,59 г).
Синтез N-(2-бензилокси-6-нитрофенил)-N-нафталин-2-илметилацетамида, I-109. К раствору N-(2-бензилокси-6-нитрофенил)ацетамида (0,576 г, 2,0 ммоль) в ДМФА (2,0 мл) небольшими порциями в течение 10 минут при комнатной температуре добавляют NaH в виде 60% дисперсии в минеральном масле (160 мг, 4,0 ммоль). В реакционную смесь добавляют 2-(бромметил)нафталин (442 мг, 2,0 ммоль) и смесь перемешивают при комнатной температуре в течение 1,5 часов. ДМФА удаляют в вакууме, остаток переносят в CH2Cl2 (60 мл) и промывают водой (20 мл) насыщенным раствором соли (20 мл), сушат над безводным сульфатом натрия и концентрируют. Полученный темно-коричневый маслянистый сироп переносят в 40 мл смеси эфир/гексан (1:1) и перемешивают в течение 1 часа, образовавшийся осадок отфильтровывают и сушат, получая N-(2-бензилокси-6-нитрофенил)-N-нафталин-2-илметилацетамид, I-109 (478 мг).
Синтез 7-бензилокси-2-метил-1-нафталин-2-илметил-1Н-бензоимидазола, I-110. Fe (470 мг) порциями при перемешивании добавляют к суспензии N-(2-бензилокси-6-нитрофенил)-N-нафталин-2-илметилацетамида (517 мг) в MeOH (15 мл) и концентрированной HCl (1,5 мл). Реакционную смесь кипятят с обратным холодильником в течение 17 часов и затем фильтруют для удаления твердых веществ. Фильтрат концентрируют, полученный остаток переносят в воду (5 мл), доводят значение pH до 14 добавлением 6н. NaOH и затем экстрагируют метилeнхлоридом (10 мл×3). Объединенные фракции промывают водой (10 мл), насыщенным раствором соли (10 мл), сушат над безводным сульфатом натрия и концентрируют. Полученный остаток очищают на силикагеле, элюируя смесью хлороформ/метанол (97:3 об./об.) с получением 7-бензилокси-2-метил-1-нафталин-2-илметил-1Н-бензимидазола, I-110 (212 мг).
Синтез 2-метил-3-нафталин-2-илметил-1H-бензимидазол-4-ола, I-111. 7-Бензилокси-2-метил-1-нафталин-2-илметил-1H-бензимидазол (211 мг) в метаноле (12 мл) и уксусной кислоте (1,0 мл) гидрируют в присутствии 10% Pd-C водородом при давлении 45 фунтов на кв. дюйм в течение 20 часов. Реакционную смесь фильтруют через слой целита. Фильтрат концентрируют, получая 2-метил-3-нафталин-2-илметил-1Н-бензимидазол-4-ол, I-111 (211 мг).
Синтез 2-метил-3-нафталин-2-илметил-1Н-бензимидазол-4-илокси)уксусной кислоты, I-112. Смесь 2-метил-3-нафталин-2-илметил-1Н-бензимидазол-4-ола (200 мг), метилбромацетата (162 мг), карбоната калия (530 мг) и ацетона (20 мл) кипятят с обратным холодильником в течение 36 часов. Реакционную смесь фильтруют. Полученный фильтрат концентрируют и полученный остаток переносят в метанол (1,0 мл), ТГФ (10 мл) и 2н. NaOH (1,0 мл) и перемешивают при комнатной температуре в течение 48 часов. Реакционную смесь концентрируют и остаток переносят в воду (5,0 мл) и подкисляют 1,0н. HCl до pH 1. Смесь экстрагируют этилацетатом (10 мл×4), объединенные органические фракции сушат (насыщенный раствор соли, сульфат натрия), концентрируют и сушат, получая 2-метил-3-нафталин-2-илметил-1Н-бенозимидазол-4-илокси)уксусную кислоту, I-112 (77 мг).
Синтез [2-(2-метил-3-нафталин-2-илметил-3H-бензимидазол-4-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, P037. 2-Метил-3-нафталин-2-илметил-1Н-бензимидазол-4-илокси)уксусную кислоту (77 мг, 2,2 ммоль), тиофен-2-сульфонамид (36 мг, 2,2 ммоль), EDCI (47 мг, 0,25 ммоль), DMAP (30 мг, 2,5 ммоль) в метиленхлориде (2,0 мл) перемешивают при комнатной температуре в течение 48 часов. Реакционную смесь концентрируют и остаток очищают на силикагеле, используя смесь хлороформ:метанол (97:3) в качестве элюента, с получением [2-(2-метил-3-нафталин-2-илметил-3H-бенозимидазол-4-илокси)ацетил]амида тиофен-2-сульфоновой кислоты, P037 (5,2 мг). 1Н-ЯМР (500 МГц, CD3OD) 2,47 (с, 3H), 4,50 (с, 2H), 5,86 (с, 2H), 6,60 (д, J=8,0 Гц. 1H), 6,1 (дд, J=5,0, 4,0 Гц, 1H), 7,06 (дд, J=8,5, 8,0 Гц, 1H), 7,26 (м, 2H), 7,32 (с, 1H), 7,36 (дд, J=5,0,1,0 Гц, 1H), 7,45-7,40 (м, 3H), 7,51 (дд, J=3,5, 1,0 Гц, 1H), 7,68 (м, 1H), 7,72 (д, J=8,5 Гц, 1H), 7,70 (м, 1H). ЖХ/МС=95,7% чистота, МС (ESI-) Вычислено: (M+) 491,5; Найдено: 490,5 (M-l).
Пример 132. Получение соединения P149
Синтез 2-амино-3-нитрофенилового эфира 2,4-дихлорбензойной кислоты, I-113. В круглодонную колбу объемом 500 мл с одной горловиной, снабженную магнитной мешалкой и перегородкой, добавляют 2-амино-3-нитрофенол (4,25 г, 27,6 ммоль), безводный CH2Cl2 (140 мл), 4-(диметиламино)пиридин (3,37 г, 27,6 ммоль) и 2,4-дихлорбензоилхлорид (5,78 г, 3,87 мл, 27,6 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. ТСХ анализ показывает полный расход исходного вещества. Реакционную смесь разбавляют CH2Cl2 (300 мл) и смесь промывают H2O (2×200 мл), сушат (Na2SO4) и концентрируют, получая 8,91 г (98,7%) продукта в виде желтовато-оранжевого твердого вещества. 1Н-ЯМР анализ показывает, что вещество, I-113, достаточно чисто для его использования на следующей стадии. 1Н-ЯМР (500 МГц, CDCl3).
Синтез 2,4-дихлор-N-(2-гидрокси-6-нитрофенил)бензамида, I-114. Круглодонную одногорлую колбу, объемом 500 мл, содержащую I-113 (8,91 г, 27,2 ммоль), снабжают магнитной мешалкой и вносят безводный ТГФ (300 мл) и реакционную емкость помещают в атмосферу N2. Осторожно небольшими порциями при перемешивании в течение 2 минут добавляют гидрид натрия (1,08 г, 44,9 ммоль, 60% масляная дисперсия). По истечении дополнительных 2 минут наблюдают выделение газообразного H2 (довольно быстро), а также небольшое выделение теплоты. Смесь перемешивают при комнатной температуре в течение 1 часа. ТСХ анализ в это время показывает, что реакция полностью завершена. Смесь перемешивают при комнатной температуре в течение ночи. Смесь осторожно гасят медленным добавлением воды сначала по каплям (50 мл), а затем небольшими порциями. Смесь выливают в EtOAc (1 л) и воду (200 мл). Водный слой подкисляют 1н. HCl до pH~1 и экстрагируют. Слои разделяют и водный слой экстрагируют EtOAc (100 мл). Объединенные органические экстракты сушат (Na2SO4), фильтруют и концентрируют, получая соединение I-114 (9,36 г) в виде желтовато-коричневого твердого вещества. 1Н-ЯМР (500 МГц, CDC13).
Синтез N-(2-амино-6-гидроксифенил)-2,4-дихлорбензамида I-115. В реактор для гидрирования объемом 250 мл добавляют водную суспензию никеля Ренея (700 мг) и осторожно разбавляют EtOH (60 мл). Соединение I-114 (700 мг, 2,14 ммоль) добавляют в виде твердого вещества. Боковые стенки реактора споласкивают EtOH (10 мл) и смесь гидрируют на шейкере Пара газообразным водородом при давлении 50 фунтов на кв. дюйм при комнатной температуре в течение ночи. Реакционную смесь фильтруют через слой целита и слой целита промывают EtOH (400 мл). Фильтрат концентрируют, получая с количественным выходом темно-коричневое твердое вещество, I-115. 1Н-ЯМР (400 МГц, ДМСО-d6).
Синтез 3-амино-2-(2,4-дихлорбензиламино)фенола, I-116. В круглодонную колбу, объемом 250 мл с одним горлом, снабженную магнитной мешалкой, обратным холодильником и помещенную в атмосферу N2, добавляют соединение I-115 (635 мг, 2,14 ммоль). Добавляют безводный ТГФ (31 мл), затем по каплям добавляют 1M BH3 в ТГФ (8,6 мл, 8,6 ммоль). Реакционную смесь кипятят с обратным холодильником в течение ночи. Охлажденную реакционную смесь осторожно гасят, по каплям добавляя метанол (50 мл). Полученную смесь концентрируют на роторном испарителе. Остаток снова растворяют в метаноле (50 мл) и снова концентрируют. Такое растворение остатка в метаноле и концентрирование повторяют еще два раза, получая с количественным выходом коричневое масло, I-116. 1Н-ЯМР (500 МГц, ДМСО-d6).
Синтез 3-(2,4-дихлорбензил)-3H-бензимидазол-4-ол, I-117. В реактор объемом 20 мл, снабженный магнитной мешалкой, добавляют соединение I-116 (980 мг, 3,46 ммоль) и абсолютный EtOH (8 мл). К полученной суспензии при перемешивании добавляют триэтилортоформиат (0,634 мл, 3,81 ммоль) и моногидрат п-толуолсульфоновой кислоты (33 мг, 0,173 ммоль). Реактор закрывают и выдерживают на масляной бане при 75°C в течение 1 часа. В это время крышку с реактора удаляют, температура масляной бани возрастает до 95-100°С, и растворитель отгоняют. Последние следы растворителя удаляют в высоком вакууме. Остаток дважды растирают со смесью 1:1 гексаны/ацетон (6 мл каждый раз) и образовавшееся темно-коричневое твердое вещество отфильтровывают и сушат, получая соединение I-117, 570 мг (56%). 1Н-ЯМР (500 МГц, ДМСО-d6).
Синтез метилового эфира [3-(2,4-дихлорбензил)-3H-бензимидазол-4-илокси]уксусной кислоты, I-118. В емкость объемом 5 мл, снабженную магнитной мешалкой и содержащую соединение I-117 (60 мг, 0,204 ммоль), добавляют безводный ДМФА (0,8 мл), безводный карбонат калия (34 мг, 0,246 ммоль) и метилбромацетат (24 мкл, 0,246 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют, получая остаток, который растворяют в смеси 1:1 гексаны/ацетон (1 мл) и очищают колоночной хроматографией на флэш-силикагеле (6 г), используя в качестве элюента сначала смесь 4:1 гексаны/ацетон, затем 7:3 гексаны/ацетон и получая соединение I-118 (40 мг, 54%) в виде полутвердого вещества. 1Н-ЯМР (500 МГц, CDCl3).
Синтез [3-(2,4-дихлорбензил)-3H-бенозимидазол-4-илокси]уксусной кислоты, I-119. В круглодонную колбу объемом 50 мл с одним горлом, содержащую соединение I-118 (32 мг, 0,088 ммоль), добавляют абсолютный этанол (0,5 мл), воду (0,5 мл) и 15% водный гидроксид натрия (0,025 мл, 0,093 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Смесь концентрируют, получая твердый продукт. Твердый продукт растворяют в воде (3 мл) и раствор подкисляют добавлением 1н. HCl (0,25 мл). pH раствора проверяют лакмусовой бумажкой, которая показывает рН 2-3. Образовавшийся осадок отфильтровывают и сушат. Водный фильтрат экстрагируют EtOAc (3×1 мл) и органические экстракты концентрируют, получая твердый продукт. Твердый продукт объединяют с выделенным ранее осадком, получая 28 мг (91%), I-119, в виде твердого не совсем белого вещества. 1Н-ЯМР (500 МГц, ДМСО-d6, ЖХ/МС=96%, ESI/- 349,2.
Синтез соединения P149. В реактор объемом 3 мл, снабженный магнитной мешалкой и содержащий соединение I-119 (21 мг, 0,060 ммоль), добавляют безводный CH2Cl2 (2 мл) и затем DMAP (14,7 мг, 0,120 ммоль), в результате образуется гомогенный раствор. В раствор добавляют 4,5-дихлортиофен-2-сульфонамид (15,5 мг, 0,66 ммоль) и затем EDCI (23 мг, 0,12 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение в течение 4 часов и затем разбавляют CH2Cl2 и водой (5 мл каждого растворителя). Водный слой подкисляют добавлением 1н. HCl до достижения pH 2-3 (лакмусовая бумажка). Слои разделяют, и водный слой экстрагируют CH2Cl2 (5 мл). Объединенные органические слои концентрируют и сушат в вакууме. Полученный твердый продукт растирают с горячим CH2Cl2 (3 мл) и охлажденный раствор фильтруют и сушат, получая 26 мг (71%) P149 в виде твердого, не совсем белого вещества. 1Н-ЯМР (500 МГц, ДМСО-d6) 4,59 (с, 2H), 5,91 (с, 2H), 6,85 (д, J=8,0 Гц, 1H), 7,04 (д, J=8,5 Гц, 1H), 7,30 (т, J=8,0 Гц, 1H), 7,33 (дд, J=8,5, 2,0 Гц, 1H), 7,36 (д, J=8,0 Гц, 1H), 7,54 (с, 1H), 7,65 (д, J=2,0 Гц, 1H), 8,98 (с, 1H). ЖХ/МС=97,8% чистота, МС (ESI+) Вычислено: (M+H) 564,4; найдено: 564,4.
Пример 133. Получение соединения P152
Синтез 2-амино-3-нитрофенилового эфира нафталин-2-карбоновой кислоты, I-120. Соединение I-119 синтезируют из 2-амино-3-нитрофенола (99%) по методике, аналогично синтезу соединения I-113. 1Н-ЯМР (500 МГц, CDCl3).
Синтез (2-гидрокси-6-нитрофенил)амида нафталин-2-карбоновой кислоты, I-121. Соединение I-121 синтезируют из соединения I-120 (количественный выход) аналогично синтезу соединения I-114 из соединения I-113. 1Н-ЯМР (500 МГц, CDCl3).
Синтез (2-амино-6-гидроксифенил)амида нафталин-2-карбоновой кислоты, I-122. В круглодонную колбу объемом 50 мл с одним горлом, снабженную магнитной мешалкой, обратным холодильником и устройством для ввода/вывода N2, добавляют соединение I-121 (100,7 мг, 0,327 ммоль) и абсолютный этанол (5 мл). Реакционную смесь помещают на масляную баню при 70°C в атмосфере N2. Добавляют хлорид олова (II) дигидрат (738 мг, 3,27 ммоль) и затем по каплям добавляют 6н. HCl (2,18 мл, 13,1 ммоль). После выдерживания при 70°C в течение 1 часа реакционную смесь охлаждают до комнатной температуры, разбавляют водой и EtOAc (по 50 мл каждого) и подщелачивают (pH~8) осторожным добавлением насыщенного водного раствора NaHCO3. Раствор фильтруют для удаления осадка солей олова и органический слой отделяют, сушат (Na2SO4), фильтруют и концентрируют, получая соединение I-122, 81 мг (89%). 1Н-ЯМР (500 МГц, ДМСО-d 6), МС; AP- 277,0.
Синтез 3-амино-2-[(нафталин-2-илметил)амино]фенола, I-123. Соединение I-122 синтезируют из соединения I-121 (количественный выход) по методике, аналогично синтезу соединения I-116 из соединения I-115. 1Н-ЯМР (500 МГц, ДМСО-d6).
Синтез 3-нафталин-2-илметил-3H-бензимидазол-4-ола, I-124. Соединение I-124 синтезируют из соединения I-123 (35%) по методике, аналогично синтезу соединения I-117 из соединения I-116. 1Н-ЯМР (500 МГц, ДМСО-d6).
Синтез метилового эфира (3-нафталин-2-илметил-3H-бенозимидазол-4-илокси)уксусной кислоты, I-125. Соединение I-125 синтезируют из соединения I-124 (55%) по методике, аналогично синтезу соединения I-118 из соединения I-117. 1Н-ЯМР (500 МГц, ДМСО-d6).
Синтез (3-нафталин-2-илметил-3H-бенозимидазол-4-илокси)уксусной кислоты, I-126. Соединение I-126 синтезируют из соединения I-125 (55%) по методике, аналогично синтезу соединения I-119 из соединения I-118. ЖХ/МС=95,7%, ESI/- 331,1, 1Н-ЯМР (500 МГц, ДМСО-d6).
Синтез соединения P152. Соединение P152 синтезируют из соединения I-126 (52%) по методике, аналогично синтезу соединения P149 из соединения I-119. 1Н-ЯМР (500 МГц, ДМСО-d6) 4,65 (с, 2H), 6,00 (с, 2H), 6,86 (м, 1H), 7,31 (м, 2H), 7,54-7,44 (м, 3H), 7,60 (дд, J=9,0, 2,0 Гц, 1H), 7,79 (дд, J=9,0, 2,0 Гц, 1H), 7,88-7,82 (м, 2H), 8,09 (с, 1H), 9,36 (уш. с, 1H). ЖХ/МС=96% чистота, МС (ESI+) Вычислено: (M+H) 546,5; Найдено: 546,7.
Пример 134. Получение соединения P253
Общая методика синтеза гексагидро-2-оксииндол-производных:
Синтез 2-метил-2-аллилциклогексанона, I-127: К раствору гидрида натрия (1 экв.; 60% дисперсия в минеральном масле) в диметоксиэтиленгликоле при 5°C в атмосфере азота по каплям добавляют 2-метилциклогексанон. Раствору дают возможность нагреться до комнатной температуры, затем нагревают до 80°C и выдерживают при этой температуре в течение 1,5 часов. Затем раствор охлаждают до комнатной температуры и затем до 5°C. К смеси по каплям добавляют аллилбромид (1 экв.), реакционную смесь нагревают до 80°С и выдерживают при этой температуре в течение 1,5 часов. Реакционную смесь охлаждают до комнатной температуры и к смеси по каплям добавляют воду (~14 экв.). Водный слой дважды экстрагируют этиловым эфиром и сушат над сульфатом натрия. После концентрирования неочищенный продукт очищают хроматографией на силикагеле с использованием 2,5% этилового эфира в гексанах в качестве элюента с получением соединения I-127 с 35% выходом. 1Н-ЯМР (CDCl3) подтверждает структуру.
Синтез (1-метил-2-оксоциклогексил)уксусной кислоты, I-128: В двухфазный раствор 1-метил-1-аллилциклогексанона, I-127, в H2O/AcN/CCl4 в атмосфере азота добавляют NaIO4 (20 экв.), затем RuCl3-H2O. Реакционную смесь перемешивают при комнатной температуре в течение ночи. К смеси по каплям добавляют 2-пропанол (~88 экв.), что вызывает почернение реакционной смеси. Смесь разбавляют водой и этиловым эфиром, фильтруют через слой целита и промывают этиловым эфиром. Водный слой экстрагируют дихлорметаном и EtOAc. Объединенные органические фракции сушат над сульфатом натрия и концентрируют в вакууме, получая соединение I-128 с количественным выходом. 1Н-ЯМР (CDCl3) подтверждает структуру.
Общая методика (A-4) получения гексагидроиндол-2-онов, I-129x. Раствор (1-метил-2-оксоциклогексил)уксусной кислоты, I-128 (1 экв.) и подходящего ариламина (1 экв.) в м-ксилоле кипятят с обратным холодильником при 145°C в течение 3 часов. Реакционную смесь концентрируют в вакууме и остаток либо используют неочищенным на следующей стадии, либо очищают хроматографией на силикагеле, используя гексаны в дихлорметане (10-20%) в качестве элюента с получением целевого продукта, I-129x. Структуру полученного продукта анализируют 1Н-ЯМР.
Общая методика (A-5) бромирования гексагидроиндол-2-онов, I-129x для получения винилбромидов I-130x: К раствору подходящего гексагидроиндол-2-она, I-129, в дихлорметане при 0°C по каплям добавляют бром (1 экв.). Реакционную смесь перемешивают до исчезновения окраски брома и затем в течение дополнительных 5 минут. Одной порцией к смеси добавляют триэтиламин (3 экв.) и смесь перемешивают при комнатной температуре в течение 10 минут. Реакционную смесь промывают водой (3×) и сушат над сульфатом магния. Дихлорметановый раствор концентрируют в вакууме. Остаток либо используют неочищенным на следующей стадии, либо очищают хроматографией на силикагеле, используя дихлорметан в качестве элюента с получением подходящего винилбромида, I-130x. Структуру продукта подтверждают 1Н-ЯМР.
Общая методика (A-6) реакции сочетания Хека l-130x для получения сложного акрилатного эфира I-131x: В круглодонную колбу, снабженную обратным холодильником и устройством для ввода/вывода азота, помещают раствор подходящего 7-бромгексагидроиндол-2-она и триэтиламина (10 экв.) в ДМФА. К раствору добавляют в указанном порядке: метилакрилат (1,1 экв.), ацетат палладия (II) (0,1 экв.) и три-o-толилфосфина (0,3 экв.). Реакционную смесь нагревают до 100°C, выдерживают при этой температуре в течение 16 часов и затем дают возможность смеси охладиться до комнатной температуры. Реакционную смесь фильтруют через целит и промывают дихлорметаном, затем разбавляют дихлорметаном и водой и слои разделяют. Объединенные органические фазы промывают водой (2×) и насыщенным раствором соли и сушат над сульфатом магния. Органические фазы концентрируют в вакууме. Остаток либо используют неочищенным, либо очищают хроматографией на силикагеле, используя 15% гексаны в дихлорметане в качестве элюента с получением акрилатного эфира I-131x. Структуру продукта подтверждают 1Н-ЯМР.
Общая методика (A-7) гидролиза метиловых эфиров для получения акриловой кислоты, I-132x: К раствору подходящего метилового эфира, I-131x, в ТГФ/MeOH (2:1) добавляют водный NaOH (3 экв.) и реакционную смесь перемешивают в течение 24-72 часов при комнатной температуре. Смесь промывают 2 порциями диэтилового эфира, разбавляют EtOAc и pH доводят до 2-3 добавлением 1н. HCl. Органические фракции промывают насыщенным раствором соли, сушат над сульфатом магния и концентрируют с получением кислоты I-132x. Структуру продукта подтверждают 1Н-ЯМР.
Общая методика (A-8) реакции сочетания l-132x для получения ацилсульфонамидов: К раствору подходящей кислоты, I-132x, добавляют подходящий сульфонамид (1,2 экв.) и DMAP (2,4 экв.) в CH2Cl2 и затем добавляют EDCI (2 экв.). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь промывают 1н. HCl (водн.), водой и насыщенным раствором соли, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают либо хроматографией на силикагеле с использованием метанола (0-3%) в дихлорметане в качестве элюента, либо растиранием со смесью дихлорметан/гексаны с получением целевого ацилсульфонамидного продукта. Структуру продукта подтверждают 1Н-ЯМР, чистоту определяют с помощью ESI ЖХ/МС.
Синтез соединения P253. Синтез 3a-метил-1-нафталин-2-илметил-l,3,3a,4,5,6-гексагидроиндол-2-она, I-129A: В соответствии с общей методикой A-4, (1-метил-2-оксоциклогексил)уксусную кислоту, I-128 преобразовывают в соединение I-129A. Структуру подтверждают 1Н-ЯМР.
Синтез 7-бром-3a-метил-1-нафталин-2-илметил-1,3,3a,4,5,6-гексагидроиндол-2-она, I-130A: В соответствии с общей методикой A-5, 3a-метил-1-нафталин-2-илметил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-129A, преобразовывают в соединение I-130A. Структуру подтверждают 1Н-ЯМР.
Синтез метилового эфира (E)-3-(3a-метил-1-нафталин-2-илметил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил)акриловой кислоты, I-131A: В соответствии с общей методикой A-6, 7-бром-3a-метил-1-нафталин-2-илметил-l,3,3a,4,5,6-гексагидроиндол-2-она, I-130A, преобразовывают в соединение I-131A. Структуру продукта подтверждают 1Н-ЯМР.
Синтез (E)-3-(3a-метил-1-нафталин-2-илметил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил)акриловой кислоты, I-132A: В соответствии с общей методикой A-7, метиловый эфир (E)-3-(3a-метил-1-нафталин-2-илметил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил)акриловой кислоты, I-131A, преобразовывают в соединение I-132A. Структуру подтверждают 1Н-ЯМР.; ЖХМС (M-1=362,6).
Синтез соединения P253.
В соответствии с общей методикой A-8, (E)-3-(3a-метил-1-нафталин-2-илметил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил)акриловую кислоту, I-132A, преобразовывают в соединение P253. Структуру подтверждают 1Н-ЯМР.; ЖХ-МС (M-l=575,2).
Пример 135. Получение соединения P269.
В круглодонную колбу, содержащую I-132A, растворенный в пиридине (1 экв.) и дихлорметане, в атмосфере азота добавляют цианурфторид (8 экв.). Полученную смесь кипятят с обратным холодильником в течение 2 часов. Реакционную смесь промывают охлажденной (0°C) водой и сушат над сульфатом натрия. Раствор фильтруют и концентрируют в вакууме. Остаток растворяют в дихлорметане, к полученному раствору добавляют 4-диметиламинопиридин (1,4 экв.) и 4-трифторметоксибензолсульфонамид (1,7 экв.), и полученный раствор перемешивают при комнатной температуре в течение ночи. Реакционную смесь промывают 1н. HCl, водой и насыщенным раствором соли и сушат над сульфатом магния. Раствор фильтруют и концентрируют и остаток очищают хроматографией на силикагеле, используя смесь метанол/дихлорметан в качестве элюента, с получением остатка, который растворяют в уксусной кислоте, осаждают водой и фильтруют с получением соединения P269. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-1=583,5)
Пример 136. Получение соединения P262.
Синтез 1-(3-фторбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-она, I-129B: В соответствии с общей методикой A-4, (1-метил-2-оксоциклогексил)уксусную кислоту, I-128, преобразовывают в соединение I-129B. Структуру продукта подтверждают 1Н-ЯМР.
Синтез 7-бром-1-(3-фторбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-она, I-130B: В соответствии с общей методикой A-5, 1-(3-фторбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-129B, преобразовывают в соединение I-130B. Структуру подтверждают 1Н-ЯМР.
Синтез метилового эфира (E)-3-[1-(3-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-131B: В соответствии с общей методикой A-6, 7-бром-1-(3-фторбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-130B, преобразовывают в соединение I-131B. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[1-(3-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-132B: В соответствии с общей методикой A-7, метиловый эфир (E)-3-[1-(3-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-131B, преобразовывают в соединение I-132B. Структуру подтверждают 1Н-ЯМР; ЖХМС (M-1=330,6).
Синтез соединения P262. В соответствии с общей методикой A-8, (E)-3-[1-(3-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132B, преобразовывают в соединение P262. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-1=541,5).
Пример 137. Получение соединения P263.
Синтез 1-(4-фторбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-она, I-129C: В соответствии с общей методикой A-4, (1-метил-2-оксоциклогексил)уксусную кислоту, I-128, преобразовывают в соединение I-129C. Структуру подтверждают 1Н-ЯМР.
Синтез 7-бром-1-(4-фторбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-она, I-130C: В соответствии с общей методикой A-5, 1-(4-фторбензил)-3a-метил-l,3,3a,4,5,6-гексагидроиндол-2-он, I-129C, преобразовывают в соединение I-130C. Структуру подтверждают 1Н-ЯМР.
Синтез метилового эфира (E)-3-[1-(4-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-131C: В соответствии с общей методикой A-6, 7-бром-1-(4-фторбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-130C, преобразовывают в соединение I-131C. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[1-(4-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-132C: В соответствии с общей методикой A-7, метиловый эфир (E)-3-[1-(4-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-131C, преобразовывают в соединение I-132C. Структуру подтверждают 1Н-ЯМР; ЖХМС (M-l=330,5).
Синтез соединения P263. В соответствии с общей методикой A-8, (E)-3-[1-(4-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132C, преобразовывают в соединение P263. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-1=543,3).
Пример 138. Получение соединения P294.
В соответствии с общей методикой A-8, (E)-3-[1-(4-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132C, преобразовывают в соединение P294. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-1=503,7).
Пример 139. Получение соединения P295
В соответствии с общей методикой A-8, (E)-3-[1-(4-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132C, преобразовывают в соединение P295. 1Н-ЯМР (CDC13) 1,18 (с, 3H), 1,84 (м, 3H), 2,18 (м, 3H), 2,44 (м, 2H), 4,74 (д, J=16,4 Гц, 1H), 5,26 (д, J=16,0 Гц, 1H), 5,52 (д, J=14,8 Гц, 1H), 6,97 (т, J=8,8 Гц, 2H), 7,11 (м, 1H), 7,18 (дд, J=8,8, 5,6 Гц, 2H), 7,62 (м, 2H), 7,77 (д, J=15,2 Гц, 1H). ЖХ/МС (98%) ESI- Вычислено: 504,5 m/z; найдено: 503,7 m/z.
Пример 140. Получение соединения P300.
В соответствии с общей методикой A-8, (E)-3-[1-(4-фторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132C, преобразовывают в соединение P300. Структуру подтверждают 1Н-ЯМР; ЖХ-МС(M-1=521,8).
Пример 141. Получение соединения P264.
Синтез 1-(3,4-дифторбензил)-3a-метил-l,3,3a,4,5,6-гексагидроиндол-2-она, I-129D: В соответствии с общей методикой A-4, (1-метил-2-оксоциклогексил)уксусную кислоту, I-128, преобразовывают в соединение I-129D. Структуру подтверждают 1Н-ЯМР.
Синтез 7-бром-1-(3,4-дифторбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-она, I-130D: В соответствии с общей методикой A-5, 1-(3,4-дифторбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-129D, преобразовывают в соединение I-130D. Структуру подтверждают 1Н-ЯМР.
Синтез метилового эфира (E)-3-[1-(3,4-дифторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловой кислоты, I-131D: В соответствии с общей методикой A-6, 7-бром-1-(3,4-дифторбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-130D, преобразовывают в соединение I-131D. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[1-(3,4-дифторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-132D: В соответствии с общей методикой A-7, метиловый эфир (E)-3-[1-(3,4-дифторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-131D, преобразовывают в соединение I-132D. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-1=348,5).
Синтез соединения P264. В соответствии с общей методикой A-8, (E)-3-[1-(3,4-дифторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132D, преобразовывают в соединение P264. Структуру подтверждают 1Н-ЯМР; ЖХ-МС(M-l=561,3).
Пример 142. Получение соединения P266.
В соответствии с общей методикой A-8, (E)-3-[1-(3,4-дифторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132D, преобразовывают в соединение P266. Структуру подтверждают 1Н-ЯМР; ЖХ-МС(М+1=557,0).
Пример 143. Получение соединения P267.
В соответствии с общей методикой A-8, (E)-3-[1-(3,4-дифторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132D, преобразовывают в соединение P267. Структуру подтверждают 1Н-ЯМР; ЖХ-МС(M+1=541,9).
Пример 144. Получение соединения P304.
В соответствии с общей методикой A-8, (E)-3-[1-(3,4-дифторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132D, преобразовывают в соединение P304. 1Н-ЯМР (CDCl3) 1,19 (с, 3H), 1,57 (м, 1H), 1,87 (м, 3H), 2,19 (д, J=6,8 Гц, 2H), 2.,4 (д, J=1,2 Гц, 2H), 4,75 (д, J=16,4 Гц, 1H), 5,17 (д, J=16,0 Гц, 1H), 5,53 (д, J=14,8 Гц, 1H), 7,04 (м, 3H), 7,35 (ддд, J=16,8, 9,2, 7,6 Гц, 1H), 7,72 (д, J=14,8 Гц, 1H), 7,88 (м, 2H), 7,93 (ддд, J=9,2, 7,2, 2,4 Гц, 1H). ЖХ/МС (95%) ESI- Вычислено: 522,5 m/z; найдено: 521,6 m/z.
Пример 145. Получение соединения P305.
В соответствии с общей методикой A-8, (E)-3-[1-(3,4-дифторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132D, преобразовывают в соединение P305. Структуру подтверждают 1Н-ЯМР.
Пример 146. Получение соединения P274.
Синтез 1-(2,4-дихлорбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-она, I-129E: В соответствии с общей методикой A-4, (1-метил-2-оксоциклогексил)уксусную кислоту, I-128, преобразовывают в соединение I-129E. Структуру подтверждают 1Н-ЯМР.
Синтез 7-бром-1-(2,4-дихлорбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-она, I-130E: В соответствии с общей методикой A-5, 1-(2,4-дихлорбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-129E, преобразовывают в соединение I-130E. Структуру подтверждают 1Н-ЯМР.
Синтез метилового эфира (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-131E: В соответствии с общей методикой A-6, 7-бром-1-(2,4-дихлорбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-130E преобразовывают в соединение I-131E. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-132E: В соответствии с общей методикой A-7, метиловый эфир (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-131E, преобразовывают в соединение I-132E. Структуру подтверждают 1Н-ЯМР; ЖХ-МС(M-1=380,3).
Синтез соединения P274. В соответствии с общей методикой A-8, (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132E, преобразовывают в соединение P274. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-1=536,4).
Пример 147. Получение соединения P275.
В соответствии с общей методикой A-8, (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132E, преобразовывают в соединение P275. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-1=573,3).
Пример 148. Получение соединения P276.
В соответствии с общей методикой A-8, (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132E, преобразовывают в соединение P276. 1Н-ЯМР (ДМСО-d6) 1,20 (с, 3H), 1,61 (м, 1H), 1,80 (м, 3H), 2,11 (м, 1H), 2,19 (м, 1H), 2,34 (д, J=16,0 Гц, 1H), 2,65 (д, J=16,4 Гц, 1H), 4,75 (д, J=17,6 Гц, 1H), 4,91 (д, J=17,6 Гц, 1H), 5,73 (д, J=14,8 Гц, 1H), 7,01 (д, J=8 Гц, 1H), 7,19 (д, J=14,8 Гц, 1H), 7,19 (дд, J=4,8, 3,6 Гц, 1H), 7,30 (дд, J=8,0, 2,4 Гц, 1H), 7,67 (д, J=2,4 Гц, 1H), 7,70 (дд, J=4,0, 2,4 Гц, 1H), 8,03 (дд, J=4,8, 1,2 Гц, 1H), 12,0 (с, 1H). ЖХ/МС (93%) ESI- Вычислено: 525,5 m/z; найдено: 525,5 m/z.
Пример 149. Получение соединения P278.
В соответствии с общей методикой A-8, (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132E, преобразовывают в соединение P278.
Пример 150. Получение соединения P279.
В соответствии с общей методикой A-8, (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132E, преобразовывают в соединение P279. 1Н-ЯМР (CDCl3) 1,26 (с, 3H), 1,59 (м, 1H), 1,81 (м, 2H), 1,89 (дд, J=10,4, 2,4 Гц, 1H), 2,18 (м, 1H), 2,29 (м, 1H), 2,45 (с, 2H), 4,99 (д, J=4 Гц, 2H), 5,67 (д, J=14,8 Гц, 1H), 6,94 (дд, J 4,0, 0,4 Гц, 1H), 6,97 (д, J=8,4 Гц, 1H), 7,16 (дд, J=8,4, 2,4 Гц, 1H), 7,34 (д, J=14,8, 1H), 7,46 (д, J=2,0 Гц, 1H), 7,62 (д, J=4,0 Гц, 1H). ЖХ/МС (96%) ESI- Вычислено: 559,9 m/z; найдено: 559,3 m/z.
Пример 151. Получение соединения P280.
В соответствии с общей методикой A-8, (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132E, преобразовывают в соединение P280. Структуру подтверждают 1Н-ЯМР; ЖХ-МС(M-1=587,1).
Пример 152. Получение соединения P281.
В соответствии с общей методикой A-8, (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132E, преобразовывают в соединение P281. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-1=553,3).
Пример 153. Получение соединения P282.
В соответствии с общей методикой A-8, (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132E, преобразовывают в соединение P282.
1Н-ЯМР (400 МГц, CDCl3) 1,26 (с, 3H), 1,61 (м, 2H), 1,89 (м, 2H), 2,18 (м, 2H), 2,46 (с, 2H), 4,95 (м, 2H), 5,58 (д, J=15,2 Гц, 1H), 6,92 (м, 1H), 7,06 (м, 1H), 7,10 (м, 1H), 7,16 (м, 1H), 7,28 (м, 1H), 7,58 (м, 2H). ЖХ/МС=99% чистота, МС (ESI-) Вычислено: (M/Z) 555; Найдено: 555.
Пример 154. Получение соединения P283.
В соответствии с общей методикой A-8, (E)-3-[1-(2,4-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132E, преобразовывают в соединение P283. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-1=555,3).
Пример 155. Получение соединения P285.
Синтез 1-(3-хлорбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-она, I-129F: В соответствии с общей методикой A-4, (1-метил-2-оксоциклогексил)уксусную кислоту, I-128, преобразовывают в соединение I-129F. Структуру подтверждают 1Н-ЯМР.
Синтез 7-бром-1-(3-хлорбензил)-3а-метил-1,3,3а,4,5,6-гексагидроиндол-2-она, I-130F; В соответствии с общей методикой А-5, 1-(3-хлорбензил)-3а-метил-1,3,3а,4,5,6-гексагидроиндол-2-он, I-129F, преобразовывают в соединение I-130F. Структуру подтверждают 1Н-ЯМР.
Синтез метилового эфира (E)-[1-(3-хлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловой кислоты, I-131F: В соответствии с общей методикой A-6, 7-бром-1-(3-хлорбензил)-3a-метил-l,3,3a,4,5,6-гексагидроиндол-2-он, I-130F, преобразовывают в соединение I-13IF. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[1-(3-хлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловой кислоты, I-132F: В соответствии с общей методикой A-7, метиловый эфир (E)-3-[l-(3-хлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловой кислоты, I-131F преобразовывают в соединение I-132F. Структуру подтверждают 1Н-ЯМР; ЖХ-МС.
Синтез соединения P285. В соответствии с общей методикой A-8, (E)-3-[1-(3-хлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132F, преобразовывают в соединение P285. 1Н-ЯМР (ДМСО-d6) 1,24 (с, 1H), 1,7 (с, 5H), 2,14 (с, 2H), 2,32 (с, 1H), 2,56 (с, 1H), 4,73 (д, J=16,4, 1H), 5,12 (д, J=41,6, 1H), 5,65 (д, J=14,8, 1H), 7,16 (м, 1H), 7,29 (м 3H). 7,57 (д, J=15,5, 1H), 7,65 (с, 1H). ЖХ/МС (96%) ESI-Вычислено: 559,91; Найдено: 559,3 m/z.
Пример 156. Получение соединения P296.
Синтез 1-(2,3-дихлорбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-она, I-129G: В соответствии с общей методикой A-4, (1-метил-2-оксоциклогексил)уксусную кислоту, I-128, преобразовывают в соединение I-129G. Структуру подтверждают 1Н-ЯМР.
Синтез 7-бром-1-(2,3-дихлорбензил)-3a-метил-l,3,3a,4,5,6-гексагидроиндол-2-она, I-130G: В соответствии с общей методикой A-5, l-(2,3-дихлорбензил)-3a-метил-l,3,3a,4,5,6-гексагидроиндол-2-он, I-129G, преобразовывают в соединение I-130G. Структуру подтверждают 1Н-ЯМР.
Синтез метилового эфира (E)-3-[1-(2,3-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловой кислоты, I-131G: В соответствии с общей методикой A-6,7-бром-1 -(2,3-дихлорбензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-130G преобразовывают в соединение I-131G. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[1-(2,3-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловой кислоты, I-132G: В соответствии с общей методикой A-7, метиловый эфир (E)-3-[l-(2,3-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловой кислоты, I-13IG преобразовывают в соединение I-132G. Структуру подтверждают 1Н-ЯМР; ЖХ-МС.
Синтез соединения P296: В соответствии с общей методикой A-8, (E)-3-[1-(2,3-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132G, преобразовывают в соединение P296. Структуру подтверждают 1Н-ЯМР.; ЖХ-МС (M-1=573,3).
Пример 157. Получение соединения P297.
В соответствии с общей методикой A-8, (E)-3-[1-(2,3-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132G, преобразовывают в соединение P297. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-1=553,4).
Пример 158. Получение соединения P298.
В соответствии с общей методикой A-8, (E)-3-[1-(2,3-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132G, преобразовывают в соединение P298. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-l=553,4).
Пример 159. Получение соединения P299.
В соответствии с общей методикой A-8, (E)-3-[1-(2,3-дихлорбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132G, преобразовывают в соединение P299.
Структуру подтверждают 1Н-ЯМР.; ЖХ-МС (M-l=593,3).
Пример 160. Получение соединения P306.
Синтез 1-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидроиндол-2-она, I-129H: В соответствии с общей методикой A-4, (1-метил-2-оксоциклогексил)уксусную кислоту, I-128, преобразовывают в соединение I-129H. Структуру подтверждают 1Н-ЯМР.
Синтез 7-бром-1-(3-метоксибензил)-3a-метил-l,3,3a,4,5,6-гексагидроиндол-2-она, I-130H: В соответствии с общей методикой A-5, 1-(3-метоксибензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-129H, преобразовывают в соединение I-130H. Структуру подтверждают 1Н-ЯМР.
Синтез метилового эфира (E)-3-[1-(3-метоксибензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловой кислоты, I-131H: В соответствии с общей методикой A-6, 7-бром-1-(3-метоксибензил)-3a-метил-1,3,3a,4,5,6-гексагидроиндол-2-он, I-130H, преобразовывают в соединение I-131H. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[1-(3-метоксибензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловой кислоты, I-132H. В соответствии с общей методикой A-7, метиловый эфир (E)-3-[1-(3-метоксибензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловой кислоты, I-131H, преобразовывают в соединение I-132H. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P306. В соответствии с общей методикой A-8, (E)-3-[1-(3-метоксибензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132H, преобразовывают в соединение P306. Структуру подтверждают 1Н-ЯМР.
Пример 161. Получение соединения P307
В соответствии с общей методикой A-8, (E)-3-[1-(3-метоксибензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]акриловую кислоту, I-132H, преобразовывают в соединение P307. 1Н-ЯМР (400 МГц, CDCl3) 1,23 (с, 3H), 1,59 (м, 2H), 1,87 (м, 2H), 2,16 (м, 2H), 2,45 (с, 2H), 3,77 (с, 3H), 4,75 (д, J=16,4 Гц, 1H), 5,24 (д, J=16,4 Гц, 1H), 5,52 (д, J=15,2 Гц, 1H), 6,79 (м, 3H), 7,07 (м, 1H), 7,23 (м, 1H), 7,58 (м, 2H), 7,71 (д, J=15,2 Гц, 1H). ЖХ/МС=96% чистота, МС (ESI -) Вычислено (M/Z) 516; найдено: 515.
Пример 162. Получение соединения P308В в соответствии с общей методикой A-8, (E)-3-[1-(3-метоксибензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловую кислоту, I-132H, преобразовывают в соединение P308. Структуру подтверждают 1Н-ЯМР.
Пример 163. Получение соединения P260.
В соответствии с общей методикой A-8, (E)-3-[l-(3-метоксибензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]акриловой кислоты, I-132H, преобразовывают в соединение P260. Структуру подтверждают 1Н-ЯМР; ЖХ-МС (M-l=553,4).
Пример 164. Получение соединения P151.
Синтез метилового эфира (1Н-индол-4-илокси)уксусной кислоты, I-138. К смеси 4-гидроксииндола (1,33 г, 10 ммоль) и K2CO3 (2,07 г, 15 ммоль) в ацетоне (50 мл) при комнатной температуре добавляют метилбромацетат (1,84 г, 12 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и твердый продукт удаляют. После удаления растворителя в вакууме остаток растворяют в EtOAc и промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя твердый остаток промывают смесью эфир/гексан, получая 2,02 г соединения I-138. Структуру подтверждают 1Н-ЯМР.
Синтез (1Н-индол-4-илокси)уксусной кислоты, I-139. К раствору соединения I-138 (410 мг, 2 ммоль) в ТГФ (6 мл) и метаноле (6 мл) при комнатной температуре добавляют водный NaOH (2н., 3 мл). Реакционную смесь перемешивают при комнатной температуре в течение 4 часов и затем значение pH доводят до кислотного добавлением 2н. водной HCl. Реакционную смесь экстрагируют EtOAc (2×30 мл). Объединенные органические фазы промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 370 мг соединения I-139. Структуру подтверждают 1Н-ЯМР.
Общая методика (A-9) реакции сочетания I-139 с сульфонамидами.
Смесь кислоты I-139 (250 мг, 1,3 ммоль), подходящего сульфонамида (1,57 ммоль), 4-диметиламинопиридина (317 мг, 2,6 ммоль) и EDCI (497 мг, 2,6 ммоль) в дихлорметане (20 мл) и ДМСО (8 мл) перемешивают в течение выходных. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя EtOAc в качестве элюента и получая соединение I-140x.
Общая методика (A-10) получения сульфидов.
К смеси соединения I-140x (0,12 ммоль) и подходящего тиола (0,18 ммоль) в этаноле (5 мл) и воде (2 мл) добавляют 0,25 мл йода-йодида калия (1н. в этанол/H2O, 1:1) и перемешивают при комнатной температуре в течение выходных. Раствор разбавляют EtOAc, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя твердый остаток промывают CH2Cl2/гексан (1:1), получая целевой сульфид.
Общая методика (A-11) получения сульфона.
К смеси сульфида [из общей методики (A-10)] (0,07 ммоль) в метаноле (2 мл) и этаноле (2 мл), добавляют ОXONE (44 мг, 0,07 ммоль в 0,5 мл воды) и смесь перемешивают при комнатной температуры в течение 6 часов. Реакционную смесь разбавляют EtOAc, промывают разбавленной водной HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя дихлорметан, EtOAc/гексан в качестве элюента и получая целевое сульфоновое соединение.
Синтез 4,5-дихлортиофен-2-сульфоновой кислоты [2-(1Н-индол-4-илокси)ацетил]амида, I-140A: В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с сульфонамидом амид 4,5-дихлортиофен-2-сульфоновой кислоты, получая соединения I-140A. Структуру подтверждают 1H-ЯМР.
Синтез соединения P151. В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с нафталин-2-тиолом с получением соединения P151. Структуру подтверждают 1Н-ЯМР.
Пример 165. Получение соединения P150.
В соответствии с общей методикой A-11, сульфид P151 подвергают окислению до сульфона P150. Структуру подтверждают 1Н-ЯМР.
Пример 166. Получение соединения P164.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с хинолин-2-тиолом с получением соединения P164. 1Н-ЯМР (500 МГц, ДМСО-d6) 4,26 (с, 2H), 6,28 (д, J=8,0 Гц, 1H), 7,03 (м 3H), 7,10 (д, J=8,0 Гц, 1H), 7,46 (м, 2H), 7,66 (м, 2H), 7,81 (м, 2H), 8,02 (д, J=8,5 Гц, 1H), 11,74 (с, 1H). ЖХ/МС=95% чистота, MС (ESI-) Вычислено (M/Z) 564; найдено: 562.
Пример 167. Получение соединения P169
В соответствии с общей методикой A-11, сульфид P164 подвергают окислению до сульфона P169. 1Н-ЯМР (500 МГц, ДМСО-d6) 4,31 (с, 2H), 6,34 (с, 2H), 7,07 (м, 2H), 7,49 (с, 1H), 7,71 (м, 1H), 7,80 (м, 1H), 7,90 (м, 1H), 8,04 (м, 1H), 8,23 (с, 1H), 8,21 (с, 1H), 8,60 (с, 1H), 12,21 (с, 1H). ЖХ/МС=95% чистота, MС (ESI-) Вычислено: (M/Z) 596; найдено: 594.
Пример 168. Получение соединения P165.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 2,4-диметилбензолтиолом с получением соединения P165. Структуру подтверждают 1Н-ЯМР.
Пример 169. Получение соединения P172.
В соответствии с общей методикой A-11, сульфид P165 подвергают окислению до сульфона P172. Структуру подтверждают 1Н-ЯМР.
Пример 170. Получение соединения P167.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 3,4-диметоксибензолтиол с получением соединения P167. Структуру подтверждают 1Н-ЯМР.
Пример 171. Получение соединения P170.
В соответствии с общей методикой A-11, сульфид P167 подвергают окислению до сульфона P170. Структуру подтверждают 1Н-ЯМР.
Пример 172. Получение соединения P168.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 2-хлор-4-фторбензолтиол с получением соединения P168. Структуру подтверждают 1Н-ЯМР.
Пример 173. Получение соединения P234.
В соответствии с общей методикой A-11, сульфид P168 подвергают окислению до сульфона P234. Структуру подтверждают 1Н-ЯМР.
Пример 174. Получение соединения P173.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 4-хлорбензолтиолом с получением соединения P173. Структуру подтверждают 1Н-ЯМР.
Пример 175. Получение соединения P190.
В соответствии с общей методикой A-11, сульфид P173 подвергают окислению до сульфона P190. Структуру подтверждают 1Н-ЯМР.
Пример 176. Получение соединения P178.
В соответствии с общей методикой A-10, сульфонамид, I-140A подвергают взаимодействию с 3,4-дихлорбензолтиолом с получением соединения P178. Структуру подтверждают 1Н-ЯМР.
Пример 177. Получение соединения P204.
В соответствии с общей методикой A-11, сульфид P178 подвергают окислению до сульфона P204. Структуру подтверждают 1Н-ЯМР.
Пример 178. Получение соединения P181.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с бензоксазол-2-тиолом с получением соединения P181. Структуру подтверждают 1Н-ЯМР.
Пример 179. Получение соединения P182.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с бензотиазол-2-тиол с получением соединения P182. Структуру подтверждают 1Н-ЯМР.
Пример 180. Получение соединения P196.
В соответствии с общей методикой A-11, сульфид P182 подвергают окислению до сульфона P196. 1H-ЯМР (ДМСО-d6) 4,54 (с, 2H), 6,51 (д, J=7,0, 1H), 7,14 (м, J=7,5 2H), 7,58 (м, J=6,0, 3H), 7,96 (м, J=3,5, 1H), 8,35 (д, J=5,0, 1H), 12,58. ЖХ/МС (80%) ESI- Вычислено: 602,52; найдено: 602,0 m/z.
Пример 181. Получение соединения P194.
В соответствии с общей методикой A-10, сульфонамид I-140 A подвергают взаимодействию с 2,5-диметоксибензолтиол с получением соединения P194. Структуру подтверждают 1Н-ЯМР.
Пример 182. Получение соединения P202.
В соответствии с общей методикой A-11, сульфид P194 подвергают окислению до сульфона P202. 1Н-ЯМР (500 МГц, ДМСО-d6) 3,60 (с, 3H), 3,69 (с, 3H), 4,67 (с, 2H), 6,46 (д, J=7,5 Гц, 1H), 6,98 (д, J=9,0 Гц, 1H), 7,06 (м, 2H), 7,11 (д, J=8,0 Гц, 1H), 7,48 (д, J=3,0 Гц, 1H), 7,83 (с, 1H), 8,07 (д, J=3,0 Гц, 1H), 12,19 (с, 1H). ЖХ/МС=97% чистота, MС (ESI-) Вычислено (M+) 605,5; Найдено: 605,2.
Пример 183, Получение соединения P195.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 1-метил-1Н-бензимидазол-2-тиолом с получением соединения P195. Структуру подтверждают 1Н-ЯМР.
Пример 184. Получение соединения P197.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 2,4-дифторбензолтиолом с получением соединения P197. Структуру подтверждают 1Н-ЯМР.
Пример 185. Получение соединения P206.
В соответствии с общей методикой A-11, сульфид P197 подвергают окислению до сульфона P206. Структуру подтверждают 1Н-ЯМР.
Пример 186. Получение соединения P198.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с бензолтиолом с получением соединения P198. Структуру подтверждают 1Н-ЯМР.
Пример 187. Получение соединения P207.
В соответствии с общей методикой A-11, сульфид P198 подвергают окислению до сульфона P207. Структуру подтверждают 1Н-ЯМР.
Пример 188. Получение соединения P200.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 4-метоксибензолтиолом с получением соединения P200. Структуру подтверждают 1Н-ЯМР.
Пример 189. Получение соединения P210.
В соответствии с общей методикой A-l1, сульфид P200 подвергают окислению до сульфона P210. Структуру подтверждают 1Н-ЯМР.
Пример 190. Получение соединения P201.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с пиридин-2-тиолом с получением соединения P201. Структуру подтверждают 1Н-ЯМР.
Пример 191. Получение соединения P221.
В соответствии с общей методикой A-11, сульфид P201 подвергают окислению до сульфона P221. 1Н-ЯМР (500 МГц, ДМСО-d6) 4,58 (с, 2H), 6,8 (д, J=7,5 Гц, 1H), 7,09 (м, 1H), 7,13 (м, 1H), 7,54 (м, 1H), 7,82 (с, 1H), 8,0 (с, 1H), 8,13 (с, 1H), 8,17 (м, 1H), 8,52, (м, 1H), 12,35 (с, 1H). ЖХ/МС=96% чистота, MС (ESI-) Вычислено (M/Z) 546; найдено: 544.
Пример 192. Получение соединения P205.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 2,4-дихлорбензолтиол с получением соединения P205. Структуру подтверждают 1Н-ЯМР.
Пример 193. Получение соединения P238
В соответствии с общей методикой A-11, сульфид P205 подвергают окислению до сульфона P238. Структуру подтверждают 1Н-ЯМР.
Пример 194. Получение соединения P208.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 3-метоксибензолтиолом с получением соединения P208. Структуру подтверждают 1Н-ЯМР.
Пример 195. Получение соединения P209
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 3,4-дифторбензолтиолом с получением соединения P209. Структуру подтверждают 1Н-ЯМР.
Пример 196. Получение соединения P211
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с пиримидин-2-тиолом с получением соединения P211. 1Н-ЯМР (500 МГц, ДМСО-d6) 4,45 (с, 2H), 6,27 (д, J=8,0 Гц, 1H), 6,98 (т, J=8,0 Гц, 2H), 7,08 (м, 2H), 7,54 (с, 1H), 7,84 (с, 1H), 8,46 (с, 2H), 11,59 (с, 1H). ЖХ/МС=95% чистота, MС (ESI-) Вычислено (M/Z) 515; найдено: 513.
Пример 197. Получение соединения P212.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 2-метоксибензолтиолом с получением соединения P212. Структуру подтверждают 1Н-ЯМР.
Пример 198. Получение соединения P222.
В соответствии с общей методикой A-11, сульфид P212 подвергают окислению до сульфона P222. Структуру подтверждают 1Н-ЯМР.
Пример 199. Получение соединения P213
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 2-хлорбензолтиолом с получением соединения P213. Структуру подтверждают 1Н-ЯМР.
Пример 200. Получение соединения P232.
В соответствии с общей методикой A-11, сульфид P213 подвергают окислению до сульфона P232. Структуру подтверждают 1Н-ЯМР.
Пример 201. Получение соединения P233.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с N-(4-меркаптофенил)ацетамидом с получением соединения P233. Структуру подтверждают 1Н-ЯМР.
Пример 202. Получение соединения P235.
В соответствии с общей методикой A-11, сульфид P233 подвергают окислению до сульфона P235. 1Н-ЯМР (500 МГц, ДМСО-d6) 2,05 (с, 3H), 4,72 (с, 2H), 6,47 (д, J=7,5 Гц, 1H), 7,08 (д, J=8,0 Гц, 1H), 7,12 (д, J=8,5 Гц, 1H), 7,67 (д, J=9,0 Гц, 2H), 7,82 (д, J=9,0 Гц, 2H), 10,25 (с, 1H), 12,24 (с, 1H). ЖХ/МС=90% чистота, MС (ESI-) Вычислено (M/Z) 602; найдено 600.
Пример 203. Получение соединения P220
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 1Н-имидазол-2-тиолом с получением соединения P220. Структуру подтверждают 1Н-ЯМР.
Пример 204. Получение соединения P227
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 1-метил-1Н-тетразол-5-тиолом с получением соединения P227. Структуру подтверждают 1Н-ЯМР.
Пример 205. Получение соединения P239. В соответствии с общей методикой A-11, сульфид P227 подвергают окислению до сульфона P239. 1Н-ЯМР (ДМСО-d6) 4,26 (с, 3H), 4,58 (с, 2H), 6,53 (д, J=7,5, 1H), 7,17 (м, 2H), 7,80 (с, 1H), 8,45 (с, 1H), 12,74 (с, 1H); ЖХ/МС (89%) ESI- Вычислено: 551,41; Найдено: 549,2 m/z.
Пример 206. Получение соединения P228.
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 4H-[1,2,4]триазол-3-тиолом с получением соединения P228. 1Н-ЯМР (ДМСО-d6) 4,65 (с, 2H), 6,34 (д, J=8,0), 6,97 (т, J=8,0, 1H), 7,05 (д, J=3,0, 1H), 7,58 (д, J=3,0, 1H), 7,81 (с, 1H), 8,25 (с, 1H) и 11,60 (с, 1H). ЖХ/МС (94%) ESI+ Вычислено: 504,40; найдено: 506,1 m/z.
Пример 207. Получение соединения P240.
В соответствии с общей методикой A-11, сульфид P228 подвергают окислению до сульфона P240. Структуру подтверждают 1Н-ЯМР.
Пример 208. Получение соединения P230
В соответствии с общей методикой A-10, сульфонамид I-140A подвергают взаимодействию с 5-метил[l,3,4]тиадиазол-2-тиолом с получением соединения P230. Структуру подтверждают 1Н-ЯМР.
Пример 209. Получение соединения P231
В соответствии с общей методикой A-11, сульфид P230 подвергают окислению до сульфона P231. 1Н-ЯМР (500 МГц, ДМСО-d6) 2,71 (с, 3H), 4,67 (с, 2H), 6,53 (м, 1H), 7,16 (м, 2H), 7,83 (с, 1H), 8,35 (д, J=3,5 Гц, 1H), 12,62 (с, 1H). ЖХ/МС=96% чистота, MС (ESI-) Вычислено (M/Z) 567; найдено: 567.
Пример 210. Получение соединения P166.
Синтез тиофен-2-сульфоновой кислоты [2-(1Н-индол-4-илокси)ацетил]амида, I-140B:
В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с амидом тиофен-2-сульфоновой кислоты с получением соединения I-140B. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P166. В соответствии с общей методикой A-10, сульфонамид I-140B подвергают взаимодействию с нафталин-2-тиолом с получением соединения P166. Структуру подтверждают 1Н-ЯМР.
Пример 211. Получение соединения P163.
В соответствии с общей методикой A-11, сульфид P166 подвергают окислению до сульфона P163. Структуру подтверждают 1Н-ЯМР.
Пример 212. Получение соединения P174.
Синтез 5-хлортиофен-2-сульфонгвой кислоты [2-(1Н-индол-4-илокси)ацетил]амида, I-140C. В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с амидом 5-хлортиофен-2-сульфоновой кислоты с получением соединения I-140C. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P174. В соответствии с общей методикой A-10, сульфонамид I-140C подвергают взаимодействию с нафталин-2-тиолом с получением соединения P174. Структуру подтверждают 1Н-ЯМР.
Пример 213. Получение соединения P187.
В соответствии с общей методикой A-1, сульфид P174 подвергают окислению до сульфона P187. Структуру подтверждают 1Н-ЯМР.
Пример 214. Получение соединения P175.
Синтез N-[2-(1Н-индол-4-илокси)ацетил]бензолсульфонамида, I-140D. В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с бензолсульфонамидом с получением соединения I-140D. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P175. В соответствии с общей методикой A-10, сульфонамид I-140D подвергают взаимодействию с нафталин-2-тиолом с получением соединения P175. Структуру подтверждают 1Н-ЯМР.
Пример 215. Получение соединения P188.
В соответствии с общей методикой A-11, сульфид P175 подвергают окислению до сульфона P188. Структуру подтверждают 1Н-ЯМР.
Пример 216. Получение соединения P176.
Синтез N-[2-(1Н-индол-4-илокси)ацетил]-2,5-диметокси-бензолсульфонамид, I-140E: В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с 2,5-диметокси-бензолсульфонамид с получением соединения I-140E. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P176. В соответствии с общей методикой A-10, сульфонамид I-140E подвергают взаимодействию с нафталин-2-тиолом с получением соединения P176. Структуру подтверждают 1Н-ЯМР.
Пример 217. Получение соединения P189.
В соответствии с общей методикой A-11, сульфид P176 подвергают окислению до сульфона P189. 1Н-ЯМР (ДМСО-d6) 3,75 (с, 3H), 3,84 (с, 3H), 4,71 (с, 2H), 6,32 (д, J=7,5 Гц, 1H), 7,04 (т, J=8,0 Гц, 1H), 7,10 (д, J=8,0 Гц, 1H), 7,25 (м, 2H), 7,35 (м, 1H), 7,59 (т, J=7,0 Гц, 1H), 7,66 (т, J=7,5 Гц, 1H), 7,82 (д, J=8,0 Гц, 1H), 7,94 (м, 2H), 8,03 (д, J=8,0 Гц, 1H), 8,17 (д, J=3,0 Гц, 1H), 8,65 (с, 1H), 12,05 (с, 1H), 12,29 (с, 1H). ЖХ/МС (95%) ESI- Вычислено: 579,6 m/z; найдено: 579,5 m/z.
Пример 218. Получение соединения P177.
Синтез 3,5-дихлор-N-[2-(1Н-индол-4-илокси)ацетил]-бензолсульфонамида, I-140F: В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с 3,5-дихлорбензолсульфонамидом с получением соединения I-140F. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P177. В соответствии с общей методикой A-10, сульфонамид I-140F подвергают взаимодействию с нафталин-2-тиолом с получением соединения P177. Структуру подтверждают 1Н-ЯМР.
Пример 219. Получение соединения P186.
В соответствии с общей методикой A-11, сульфид P177 подвергают окислению до сульфона P186. Структуру подтверждают 1Н-ЯМР.
Пример 220. Получение соединения P183.
Синтез 2-хлор-N-[2-(1Н-индол-4-илокси)ацетил]бензолсульфонамида, I-140G. В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с сульфонамидом 2-хлорбензолсульфонамидом с получением соединения I-140G. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P183. В соответствии с общей методикой A-10, сульфонамид I-140G подвергают взаимодействию с нафталин-2-тиолом с получением соединения P183. Структуру подтверждают 1Н-ЯМР.
Пример 221. Получение соединения P191.
В соответствии с общей методикой A-11, сульфид P183 подвергают окислению до сульфона P191. Структуру подтверждают 1Н-ЯМР.
Пример 222. Получение соединения P184.
Синтез 3-хлор-N-[2-(1Н-индол-4-илокси)ацетил] бензолсульфонамида, I-140H: В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с 3-хлорбензолсульфонамидом с получением соединения I-140H. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P184. В соответствии с общей методикой A-10, сульфонамид I-140H подвергают взаимодействию с нафталин-2-тиолом с получением соединения P184. Структуру подтверждают 1Н-ЯМР.
Пример 223. Получение соединения P192.
В соответствии с общей методикой A-11, сульфид P184 подвергают окислению до сульфона P192. Структуру подтверждают 1Н-ЯМР.
Пример 224. Получение соединения P185.
Синтез N-[2-(1Н-индол-4-илокси)ацетил]-4-метоксибензолсульфонамид, I-140I: В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с 4-метоксибензолсульфонамидом с получением соединения I-140I. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P185. В соответствии с общей методикой A-10, сульфонамид I-140I подвергают взаимодействию с нафталин-2-тиолом с получением соединения P185. Структуру подтверждают 1Н-ЯМР.
Пример 225. Получение соединения P193
В соответствии с общей методикой A-11, сульфид P185 подвергают окислению до сульфона P193. Структуру подтверждают 1Н-ЯМР.
Пример 226 Получение соединения P199.
Синтез [2-(1Н-индол-4-илокси)ацетил]амида 3,5-диметилизоксазол-4-сульфоновой кислоты, I-140J: В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с амидом 3,5-диметилизоксазол-4-сульфоновой кислоты с получением соединения I-140J. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P199. В соответствии с общей методикой A-10, сульфонамид I-140J подвергают взаимодействию с нафталин-2-тиолом с получением соединения P199. Структуру подтверждают 1Н-ЯМР.
Пример 227. Получение соединения P203.
В соответствии с общей методикой A-11, сульфид P199 подвергают окислению до сульфона P203. Структуру подтверждают 1Н-ЯМР.
Пример 228. Получение соединения P214.
Синтез 3,5-дифтор-N-[2-(1Н-индол-4-илокси)ацетил]бензолсульфонамида, I-140K. В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с сульфонамидом 3,5-дифторбензолсульфонамидом с получением соединения I-140K. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P214. В соответствии с общей методикой A-10, сульфонамид I-140K подвергают взаимодействию с нафталин-2-тиолом с получением соединения P214. Структуру подтверждают 1Н-ЯМР.
Пример 229. Получение соединения P223.
В соответствии с общей методикой A-11, сульфид P214 подвергают окислению до сульфона P223. Структуру подтверждают 1Н-ЯМР.
Пример 230. Получение соединения P215.
Синтез 3,4-дифтор-N-[2-(1Н-индол-4-илокси)ацетил]бензолсульфонамида, I-140L: В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения I-140L. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P215. В соответствии с общей методикой A-10, сульфонамид I-140L подвергают взаимодействию с нафталин-2-тиолом с получением соединения P215. Структуру подтверждают 1Н-ЯМР.
Пример 231. Получение соединения P224.
В соответствии с общей методикой A-11, сульфид P215 подвергают окислению до сульфона P224. Структуру подтверждают 1Н-ЯМР.
Пример 232. Получение соединения P225.
Синтез 4-фтор-N-[2-(1Н-индол-4-илокси)ацетил]бензолсульфонамида, I-140M: В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с 4-фторбензолсульфонамидом с получением соединения I-140M. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P225. В соответствии с общей методикой A-10, сульфонамид I-140M подвергают взаимодействию с нафталин-2-тиолом с получением соединения P225. Структуру подтверждают 1Н-ЯМР.
Пример 233. Получение соединения P236.
В соответствии с общей методикой A-11, сульфид P225 подвергают окислению до сульфона P236. Структуру подтверждают 1Н-ЯМР.
Пример 234. Получение соединения P226
Синтез 2,4,5-трифтор-N-[2-(1Н-индол-4-илокси)ацетил]бензолсульфонамида, I-140N: В соответствии с общей методикой A-9, соединение I-139 подвергают взаимодействию с 2,4,5-трифторбензолсульфонамид с получением соединения I-140N. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P226. В соответствии с общей методикой A-10, сульфонамид I-140N подвергают взаимодействию с нафталин-2-тиолом с получением соединения P226. Структуру подтверждают 1Н-ЯМР.
Пример 235. Получение соединения P237.
В соответствии с общей методикой A-11, сульфид P226 подвергают окислению до сульфона P237. Структуру подтверждают 1Н-ЯМР.
Пример 236. Получение соединения P243
Синтез метилового эфира [3-(нафталин-2-илсульфанил)-1Н-индол-4-илокси]уксусной кислоты, I-141: К смеси соединения I-138 (102 мг, 0,5 ммоль) и нафталин-2-тиола (160 мг, 1 ммоль) в этаноле (10 мл) и воде (5 мл) добавляют 1 мл йода-йодида калия (1н. в этаноле/H2O, 1:1) и полученную смесь перемешивают при комнатной температуре в течение выходных. К реакционной смеси добавляют воду (~5 мл) и перемешивание продолжают в течение 30 минут при комнатной температуре. Твердые вещества отфильтровывают и промывают водой, смесью вода/этанол (1:1), CH2Cl2/гексан (5 мл/5 мл), получая 50 мг соединения I-141. Структуру подтверждают 1Н-ЯМР.
Синтез [3-(нафталин-2-илсульфанил)-1Н-индол-4-илокси]уксусной кислоты, I-142. К раствору соединения I-141 (50 мг, 13 ммоль) в ТГФ (3 мл) и метаноле (3 мл) добавляют водный NaOH (2н., 1,5 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем pH доводят до кислотного значения добавлением 2н. водной HCl. Реакционную смесь экстрагируют EtOAc (2×20 мл). Объединенные органические фазы промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 35 мг соединения I-142. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P243. Cмесь кислоты I-142 (35 мг, 0,1 ммоль), трифторметилсульфонамида (30 мг, 0,2 ммоль), 4-диметиламинопиридина (25 мг, 0,2 ммоль) и EDCI (38 мг, 0,2 ммоль) в дихлорметане (5 мл) и ДМСО (1 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленной водной HCl, водой и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя EtOAc/гексан с качестве элюента и получая 23 мг соединения P243. MС (ESI-): 479,2 (M-l), ЖХ-МС: 94% чистота, 1H-ЯМР (500 МГц, ДМСО-d6) ВЭЖХ с обращенной фазой: 92% чистота.
Пример 237. Получение соединения P277.
Синтез 4-бром-1-метил-1Н-индола, I-133. К раствору NaH (60% в масле, 600 мг, 15 ммоль) в ДМФА (20 мл) при -10 °С добавляют 4-броминдол (1,96 г, 10 ммоль), полученную смесь перемешивают при комнатной температуре в течение 10 минут и затем добавляют йодметан (6,7 г, 50 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и разбавляют CH2Cl2 (200 мл). Реакционную смесь промывают водой (3×200 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают 3 г неочищенного соединения I-133, которое используют на следующей стадии без дополнительной очистки. Структуру подтверждают 1Н-ЯМР.
Общая методика (A-12) реакции сочетания 4-броминдола с фенолом.
Смесь соединения I-133 (420 мг, 2 ммоль), CuI (38 мг, 0,2 ммоль, 0,1 экв.), HCl соли N,N-диметилглицина (84 мг, 0,6 ммоль, 0,3 экв.), фенола (3 ммоль, 1,5 экв.) и Cs2CO3 (1,3 г, 4 ммоль, 2 экв.) в диоксане (4 мл) перемешивают в атмосфере Ar при 105°C в течение 3 дней. Реакционную смесь разбавляют EtOAc и промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя EtOAc/гексан в качестве элюента и получая соединение I-134.
Синтез l-метил-4-(нафталин-2-илокси)-1Н-индола, I-134A. В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с нафталин-2-олом с получением соединения I-134A. Структуру подтверждают 1Н-ЯМР.
Синтез l-метил-4-(нафталин-2-илокси)-1Н-индол-3-карбальдегида, I-135A. Общая методика (A-13) превращения индола в 3-формальдегидиндол. К раствору соединения I-134 (1,6 ммоль) в ДМФА (8 мл) по каплям при комнатной температуре добавляют POCl3 (1,8 ммоль, 1,1 экв.). Реакционную смесь перемешивают при 50°C в течение 1 часа и выливают в ледяную воду. Значение pH полученной смеси доводят до 8-9 добавлением NaOH (водн.) (2н.) и затем полученную смесь перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь экстрагируют EtOAc (2×40 мл). Органическую фазу промывают водой (3×50 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают I-135 в виде твердого вещества.
В соответствии с общей методикой A-13, соединение I-134A преобразовывают в формальдегид I-135A. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[l-метил-4-(нафталин-2-илокси)-1Н-индол-3-ил]акриловой кислоты, I-136A. Общая методика (A-14) превращения 3-формальдегидиндола в этиловый эфир 3-индолакриловой кислоты. К раствору NaH (60% в масле, 100 мг, 2,5 ммоль, 2,5 экв.) в ТГФ (20 мл) при -10°С добавляют триэтилфосфоноацетат (493 мг, 2,2 ммоль, 2,2 экв.), полученную смесь перемешивают при комнатной температуре в течение 10 минут и затем при -10°С добавляют соединение I-135 (1 ммоль), растворенный в ТГФ (10 мл). Реакционную смесь перемешивают при 60°C в течение ночи и затем разбавляют EtOAc и промывают NH4Cl водным, водой (2×100 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают неочищенный продукт I-136, который используют на следующей стадии без дополнительной очистки.
В соответствии с общей методикой A-14, соединение I-135A преобразовывают в этиловый эфир акриловой кислоты I-136A. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[l-метил-4-(нафталин-2-илокси)-1Н-индол-3-ил]акриловой кислоты, I-137A.
Общая методика (A-7A). Гидролиз этилового эфира 3- акриловой кислоты до 3-акриловой кислоты. Смесь неочищенного соединения I-136 (1 ммоль) в ТГФ (10 мл), MeOH (10 мл) и NaOH (водн.) (2н., 10 мл) перемешивают при 50°C в течение 3 часов. После удаления ТГФ и MeOH реакционную смесь разбавляют водой (~20 мл), доводят pH до кислотного добавлением водного HCl (2н.) и экстрагируют EtOAc (2×30 мл). Объединенные органические фазы промывают водой (2×30 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток промывают эфиром, получая соединение I-137.
В соответствии с общей методикой A-7А, соединение I-136A гидролизуют до акриловой кислоты I-137A. Структуру подтверждают 1Н-ЯМР.
Общая методика (A-8A) сочетания кислоты до арилсульфонамида. Смесь кислоты I-137 (0,16 ммоль), сульфонамида (0,20 ммоль, 1,2 экв.), 4-диметиламинопиридина (41 мг, 0,33 ммоль, 2 экв.) и EDCI (63 мг, 0,33 ммоль, 2 экв.) в дихлорметане (10 мл) перемешивают при комнатной температуре в течение ночи. Реакционную смесь промывают разбавленной водной HCl, водой и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, используя смесь метаноле/дихлорметан в качестве элюента с получением продукта.
Синтез соединения P277. В соответствии с общей методикой A-8A, акриловую кислоты I-137A подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P277. Структуру подтверждают 1Н-ЯМР.
Пример 238. Получение соединения P343.
В соответствии с общей методикой A-8A, акриловую кислоту I-137A подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P343. 1Н-ЯМР (ДМСО-d6) 3,89 (с, 3H) 6,43 (д, J=15,0 Гц, 1H), 6,70 (д, J=7,5 Гц, 1H), 7,23 (т, J=8,0 Гц, 1H), 7,27 (м, 1H), 7,35-7,48 (м, 4H), 7,73 (д, J=8,0 Гц, 1H), 7,83-7,98 (м, 5H), 7,97 (д, J=15,0 Гц, 1H), 12,6 (с, 1H). ВЭЖХ (97%) МС ESI- Вычислено: 535,5 m/z; найдено 535,7 m/z.
Пример 239. Получение соединения P334.
В соответствии с общей методикой A-8A, акриловую кислоту I-137A подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения P334. 1Н-ЯМР (ДМСО-d6) 3,89 (с, 3H), 6,39 (д, J=15,0 Гц, 1H), 6,70 (д, J=7 Гц, 1H), 7,23 (т, J=8,0 Гц, 1H), 7,30 (м, 1H), 7,35 (м, 1H), 7,37 (д, J=8,0 Гц, 1H), 7,44 (м, 2H), 7,6-7,8 (м, 3H), 7,89-7,97 (м, 4H), 7,96 (д, J=15,0 Гц, 1H), 12,2 (с, 1Н). ВЭЖХ (99%) MС ESI- Вычислено: 517,5 m/z; найдено: 517,7 m/z.
Пример 240. Получение соединения P290.
Синтез 4-(3,4-дихлорфенокси)-1-метил-1Н-индола, I-134B: В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 3,4-дихлорфенолом с получением соединения I-134B. Структуру подтверждают 1Н-ЯМР.
Синтез 4-(3,4-дихлорфенокси)-1-метил-1Н-индол-3-карбальдегида, I-135B: В соответствии с общей методикой A-13, соединение I-134B преобразовывают в формальдегид I-135B. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[4-(3,4-дихлорфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-136B: В соответствии с общей методикой A-14, соединение I-135B, преобразовывают в этиловый эфир акриловой кислоты I-136B. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[4-(3,4-дихлорфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137B. В соответствии с общей методикой A-7А, соединение I-136B гидролизуют до акриловой кислоты I-137B. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P290. В соответствии с общей методикой A-8A, акриловую кислоту I-137B подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P290. Структуру подтверждают 1Н-ЯМР.
Пример 241. Получение соединения P292.
В соответствии с общей методикой A-8A, акриловую кислоту I-137B подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P292. Структуру подтверждают 1Н-ЯМР.
Пример 242. Получение соединения P293.
В соответствии с общей методикой A-8A, акриловую кислоту I-137B подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения P293. Структуру подтверждают 1Н-ЯМР.
Пример 243. Получение соединения P291.
Синтез 4-(2,3-дихлорфенокси)-1-метил-1Н-индола, I-134C. В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 2,3-дихлорфенолом с получением соединения I-134C. Структуру подтверждают 1Н-ЯМР.
Синтез 4-(2,3-дихлорфенокси)-1-метил-1Н-индол-3-карбальдегида, I-135C. В соответствии с общей методикой A-13, соединение I-134C преобразовывают в формальдегид I-135C. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[4-(2,3-дихлорфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-136C. В соответствии с общей методикой A-14, соединение I-135C преобразовывают в этиловый эфир акриловой кислоты I-136C. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[4-(2,3-дихлорфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137C. В соответствии с общей методикой A-7А, соединение I-136C гидролизуют до акриловой кислоты I-137C. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P291. В соответствии с общей методикой A-8A, акриловую кислоту I-137C подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P291. Структуру подтверждают 1Н-ЯМР.
Пример 244. Получение соединения P303.
Синтез 4-(2,4-дихлорфенокси)-1-метил-1Н-индола, I-134D: В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 2,4-дихлорфенолом с получением соединения I-134D. Структуру подтверждают 1Н-ЯМР.
Синтез 4-(2,4-дихлорфенокси)-1-метил-1Н-индол-3-карбальдегида, I-135D: В соответствии с общей методикой A-13, соединение I-134D преобразовывают в формальдегид I-135D. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[4-(2,4-дихлорфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-136D. В соответствии с общей методикой A-14, соединение I-135D преобразовывают в этиловый эфир акриловой кислоты I-136D. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[4-(2,4-дихлорфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137D. В соответствии с общей методикой A-7А, соединение I-136D гидролизуют до акриловой кислоты I-137D. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P303. В соответствии с общей методикой A-8A, акриловую кислоту I-137D подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P303. Структуру подтверждают 1Н-ЯМР.
Пример 245. Получение соединения P301.
В соответствии с общей методикой A-8A, акриловую кислоту I-137D подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P301. Структуру подтверждают 1Н-ЯМР.
Пример 246. Получение соединения P302.
В соответствии с общей методикой A-8A, акриловую кислоту I-137D подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения P302. Структуру подтверждают 1Н-ЯМР.
Пример 247. Получение соединения P311.
Синтез 4-(4-хлорфенокси)-1-метил-1Н-индола, I-134E: В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 4-хлорфенолом с получением соединения I-134E. Структуру подтверждают 1Н-ЯМР.
Синтез 4-(4-хлорфенокси)-1-метил-1Н-индол-3-карбальдегида, I-135E: В соответствии с общей методикой A-13, соединение I-134E преобразовывают в формальдегид I-135E. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[4-(4-хлорфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-136E. В соответствии с общей методикой A-14, соединение I-135E преобразовывают в этиловый эфир акриловый кислоты I-136E. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[4-(4-хлорфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137E. В соответствии с общей методикой A-7А, соединение I-136E гидролизуют до акриловой кислоты I-137E. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P311. В соответствии с общей методикой A-8A, акриловую кислоту I-137 подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P311. 1Н-ЯМР (ДМСО-d6) 3,87 (с, 3H), 6,36 (д, J=15,0 Гц, 1H), 6,69 (д, J=8,0 Гц, 1H), 6,99 (д, J=7 Гц, 2H), 7,23 (д, J=7 Гц, 1H), 7,38 (м, 1H), 7,39 (д, J=7 Гц, 2H), 7,86 (с, 1H), 7,94 (д, J=15,0 Гц, 1H), 7,99 (с, 1Н), 12,5 (уш., 1H). ВЭЖХ (97%) MС ESI- Вычислено: 41,8 m/z; найдено: 541,4 m/z.
Пример 248. Получение соединения P309.
В соответствии с общей методикой A-8A, акриловую кислоту I-137E подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P309. Структуру подтверждают 1Н-ЯМР.
Пример 249. Получение соединения P310.
В соответствии с общей методикой A-8A, акриловую кислоту I-137E подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения P310. Структуру подтверждают 1Н-ЯМР.
Пример 250. Получение соединения P320.
Синтез 4-(3,4-дифторфенокси)-1-метил-1Н-индола, I-134F: В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 3,4-дифторфенолом с получением соединения I-134F. Структуру подтверждают 1Н-ЯМР.
Синтез 4-(3,4-дифторфенокси)-1-метил-1Н-индол-3-карбальдегида, I-135F: В соответствии с общей методикой A-13, соединение I-134F преобразовывают в формальдегид I-135F. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[4-(3,4-дифторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-136F: В соответствии с общей методикой A-14, соединение I-135F преобразовывают в этиловой эфир акриловой кислоты I-136F. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[4-(3,4-дифторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137F. В соответствии с общей методикой A-7А, соединение I-136F гидролизуют до акриловой кислоты I-137F. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P320. В соответствии с общей методикой A-8A, акриловую кислоту I-137F подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P320. Структуру подтверждают 1Н-ЯМР.
Пример 251. Получение соединения P318.
В соответствии с общей методикой A-8A, акриловую кислоту I-137F подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P318. Структуру подтверждают 1Н-ЯМР.
Пример 252. Получение соединения P319.
В соответствии с общей методикой A-8A, акриловую кислоту I-137F подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения P319. Структуру подтверждают 1Н-ЯМР.
Пример 253. Получение соединения P326.
Синтез 4-(2,4-дифторфенокси)-1-метил-1Н-индола, I-134G: В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 2,4-дифторфенолом с получением соединения I-134G. Структуру подтверждают 1Н-ЯМР.
Синтез 4-(2,4-дифторфенокси)-1-метил-1Н-индол-3-карбальдегида, I-135G. В соответствии с общей методикой A-13, соединение I-134G преобразовывают в формальдегид I-135G. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[4-(2,4-дифторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-136G. В соответствии с общей методикой A-14, соединение I-135G преобразовывают в этиловый эфир акриловой кислоты I-136G. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[4-(2,4-дифторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137G. В соответствии с общей методикой A-7А, соединение I-136G гидролизуют до акриловой кислоты I-137G. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P326. В соответствии с общей методикой A-8A, акриловую кислоту I-137G подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P326. Структуру подтверждают 1Н-ЯМР.
Пример 254. Получение соединения P324.
В соответствии с общей методикой A-8A, акриловую кислоту I-137G подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P324. Структуру подтверждают 1Н-ЯМР.
Пример 255. Получение соединения P325.
В соответствии с общей методикой A-8A, акриловую кислоту I-137G подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения P325. 1H-ЯМР (ДМСО-d6) 6,12 (д, J=8,0 Гц, 1H), 6,21 (д, J=15,2 Гц, 1H), 7,04 (дд, J=10,0, 2,4 Гц, 1H), 7,23 (дд, J=8,0, 2,4 Гц, 1H), 7,36 (с, 1H), 7,39 (д, J=2,4 Гц, 1H), 7,45 (дд, J=9,2, 2,4 Гц, 1H), 7,72 (д, J=15,2 Гц, 1H), 7,88 (с, 1H) ЖХ/МС (96%) (ESI-) Вычислено: 594,34 m/z; найдено 593,3 m/z.
Пример 256. Получение соединения P330.
Синтез 4-(3-хлор-4-фторфенокси)-1-метил-1Н-индола, I-134H: В соответствии с общей методикой А-12, соединение I-133 подвергают взаимодействию с 2,4-дифторфенолом с получением соединения I-134G. Структуру подтверждают 1Н-ЯМР.
Синтез 4-(3-хлор-4-фторфенокси)-1-метил-1Н-индол-3-карбальдегида, I-135H. В соответствии с общей методикой A-13, соединение I-134H преобразовывают в формальдегид I-135H. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[4-(3-хлор-4-фторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-136H. В соответствии с общей методикой A-14, I-135H преобразовывают в этиловый эфир акриловой кислоты I-136H. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[4-(3-хлор-4-фторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137H. В соответствии с общей методикой A-7А, соединение I-136H гидролизуют до акриловой кислоты I-137H. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P330. В соответствии с общей методикой A-8A, акриловую кислоту I-137H подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P330. Структуру подтверждают 1Н-ЯМР.
Пример 257. Получение соединения P328.
В соответствии с общей методикой A-8A, акриловую кислоту I-137H подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P328. Структуру подтверждают 1Н-ЯМР.
Пример 258. Получение соединения P329.
В соответствии с общей методикой A-8A, акриловую кислоту I-137H подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения P329. Структуру подтверждают 1Н-ЯМР.
Пример 259. Получение соединения P333.
Синтез 4-(4-хлор-3-фторфенокси)-1-метил-1Н-индола, I-134I. В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 4-хлор-3-фторфенолом с получением соединения I-134L. Структуру подтверждают 1Н-ЯМР.
Синтез 4-(4-хлор-3-фторфенокси)-1-метил-1Н-индол-3-карбальдегида, I-1351. В соответствии с общей методикой A-13, соединение I-134I преобразовывают в формальдегид I-135I. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[4-(4-хлор-3-фторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-1361. В соответствии с общей методикой A-14, соединение I-135I, преобразовывают в этиловый эфир акриловой кислоты I-136I. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[4-(4-хлор-3-фторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137I. В соответствии с общей методикой A-7А, соединение I-136I гидролизуют до акриловой кислоты I-1371. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P333. В соответствии с общей методикой A-8A, акриловую кислоту I-137I подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P333. Структуру подтверждают 1Н-ЯМР.
Пример 260. Получение соединения P331.
В соответствии с общей методикой A-8A, акриловую кислоту I-137 подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P331. 1Н-ЯМР (ДМСО-d6) 3,88 (с, 3H), 6,38 (д, J=15,0 Гц, 1H), 6,72 (м, 1H), 6,78 (д, J=7 Гц, 1H), 7,09 (дд, J=10 и 3 Гц, 1H), 7,26 (т, J=8,0 Гц, 1H), 7,42 (д, J=8,0 Гц, 1H), 7,50 (т, J=9 Гц, 1H), 7,80 (д, J=15,0 Гц, 1H), 7,88 (м, 1H), 7,98 (м, 2H), 12,6 (с, 1H). ВЭЖХ (93%) MС ESI- Вычислено: 537,9 m/z; найдено: 537,4 m/z.
Пример 261. Получение соединения P332.
В соответствии с общей методикой A-8A, акриловую кислоту I-137I подвергают взаимодействию с 3,4-дифторбензолсульфонамид с получением соединения P332, Структуру подтверждают 1Н-ЯМР.
Пример 262. Получение соединения P339.
Синтез 4-(4-хлор-2-фторфенокси)-1-метил-1Н-индола, I-134J. В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 4-хлор-2-фторфенол с получением соединения I-134J. Структуру подтверждают 1Н-ЯМР.
Синтез 4-(4-хлор-2-фторфенокси)-1-метил-1Н-индол-3-карбальдегидом, I-135J. В соответствии с общей методикой A-13, соединение I-134J преобразовывают в формальдегид I-135J. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[4-(4-хлор-2-фторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-136J. В соответствии с общей методикой A-14, соединение I-135J преобразовывают в этиловый эфир акриловой кислоты I-136J. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[4-(4-хлор-2-фторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137J. В соответствии с общей методикой A-7A, соединение I-136J гидролизуют до акриловой кислоты I-137J. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P339. В соответствии с общей методикой A-8A, акриловую кислоту I-137J подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P339. 1Н-ЯМР(ДМСО-d6) 3,87 (с, 3H), 6,40 (д, J=15,0 Гц, 1H), 6,49 (д, J=8,0 Гц, 1H), 7,17 (м, 2H), 7,32 (м, 2H), 7,65 (дд, J=10 и 2 Гц, 1H), 7,86 (с, 1H), 8,02 (с, 1H), 8,04 (д, J=15,0 Гц, 1H), 12,5 (уш., 1H). ВЭЖХ (89%) MС ESI- Вычислено: 558,8 m/z; найдено: 559,1 m/z.
Пример 263. Получение соединения P337.
В соответствии с общей методикой A-8A, акриловую кислоту I-137J подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P337. Структуру подтверждают 1Н-ЯМР.
Пример 264. Получение соединения P338.
В соответствии с общей методикой A-8A, акриловую кислоту I-137J подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения P338. Структуру подтверждают 1Н-ЯМР.
Пример 265. Получение соединения P342.
Синтез 4-(2-хлор-4-фторфенокси)-1-метил-1Н-индола, I-134K. В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 2-хлор-4-фторфенолом с получением соединения I-134K. Структуру подтверждают 1Н-ЯМР.
Синтез 4-(2-хлор-4-фторфенокси)-1-метил-1Н-индол-3-карбальдегида, I-135K. В соответствии с общей методикой A-13, соединение I-134K, преобразовывают в формальдегид I-135K. Структуру подтверждают 1Н-ЯМР.
Синтез этилового эфира (E)-3-[4-(2-хлор-4-фторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-136K. В соответствии с общей методикой A-14, соединение I-135K преобразовывают в этиловый эфир акриловой кислоты I-136K. Структуру подтверждают 1Н-ЯМР.
Синтез (E)-3-[4-(2-хлор-4-фторфенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137K. В соответствии с общей методикой A-7А, соединение I-136K гидролизуют до акриловой кислоты I-137K. Структуру подтверждают 1Н-ЯМР.
Синтез соединения P342. В соответствии с общей методикой A-8A, акриловую кислоту I-137K подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P342. 1Н-ЯМР (ДМСО-d6) 3,87 (с, 3H), 6,31 (д, J=8,0 Гц, 1H), 6,41 (д, J=15,0 Гц, 1H), 7,13 (т, J=8,0 Гц, 1H), 7,25 - 7,30 (м, 3H), 7,64 (дд, J=8 и 2 Гц, 1H), 7,86 (с, 1H), 8,.01 (с, 1H), 8,11 (д, J=15,0 Гц, 1H), 12,5 (уш., 1H). ВЭЖХ (86%) MС ESI- Вычислено: 558,8 m/z; найдено: 559,1 m/z.
Пример 266. Получение соединения P340.
В соответствии с общей методикой A-8A, акриловую кислоту I-137K подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P340. Структуру подтверждают 1Н-ЯМР.
Пример 267. Получение соединения P341.
В соответствии с общей методикой A-8A, акриловую кислоту I-137K подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения P341. Структуру подтверждают 1Н-ЯМР.
Пример 268. Получение соединения P034.
Синтез этилового эфира 5-амино-1-(2,2-диэтоксиэтил)-1Н-пиразол-4-карбоновой кислоты, I-141. К раствору гидразингидрата (17,5 мл, 0,355 молей) в 60 мл абсолютного этанола при кипячении с обратным холодильником при перемешивании по каплям добавляют бромацетальдегид диэтилацеталя (20 г, 0,101 молей). Смесь кипятят с обратным холодильником в течение ночи. Охлажденную реакционную смесь концентрируют в вакууме, к остатку добавляют 25 мл 35% NaOH в смеси с 3 г NaCl и полученную смесь экстрагируют эфиром (2×100 мл). Объединенные органические фазы сушат над MgSO4 и концентрируют в вакууме, получая 12,3 г масла (I-138). Масло используют на следующей стадии без дополнительной очистки. К раствору соединения I-138 в (2,2-диэтоксиэтил)гидразине (12,3 г, 0,0831 молей) в 25 мл абсолютного этанола добавляют раствор этилэтоксиметилена цианоацетата (14 г, 0,0831 моль) в 75 мл абсолютного этанола. Смесь перемешивают при комнатной температуре в течение 3 дней. Растворитель удаляют, получая маслянистый остаток этиловый эфир (5-амино-1-(2,2-диэтоксиэтил)-1Н-пиразол-4-карбоновой кислоты, I-141), который используют на следующей стадии без дополнительной очистки.
Синтез этилового эфира 1Н-имидазо[1,2-b]пиразол-7-карбоновой кислоты, I-142. К раствору неочищенного соединения I-141 (2 г, в 10 мл абсолютного этанола) добавляют водный 20% раствор серной кислоты (12 мл). Смесь кипятят с обратным холодильником в течение 1 часа. Реакционную смесь охлаждают, растворитель удаляют, смесь выливают на лед и рН доводят до 8 добавлением раствора гидрокарбоната натрия. Нерастворимые вещества отфильтровывают и фильтрат экстрагируют метилeнхлоридом (2×60 мл). Объединенные органические фракции сушат над MgSO4. Фильтрование и упаривание смеси приводит к получению 1,2 г остатка. Остаток очищают колоночной хроматографией с использованием CH2Cl2 до 2% MeOH/ CH2Cl2 приводит к получению 280 мг продукта I-142.Н
Синтез 1-нафталин-2-илметил-1Н-имидазо[1,2-b]пиразол-7-карбоновой кислоты, I-143. К суспензии NaH (60% в минеральном масле, 65 мг, 1,6 ммоль) в 5 мл ДМФА при 0°C добавляют соединение I-142 (240 мг, 1,34 ммоль). Смесь перемешивают при 0°C в течение 30 минут, затем при комнатной температуре в течение 2 часов. Реакционную смесь охлаждают до 0°C и добавляют 2-бромметилнафталин. Смесь перемешивают при комнатной температуре в течение ночи. Реакцию гасят добавлением насыщенного раствора NH4C1 (10 мл) к смеси и полученный раствор экстрагируют CH2Cl2 (2×50 мл). Объединенные органические экстракты промывают насыщенным раствором соли и сушат над MgSO4. После выпаривания растворителя получают 300 мг неочищенного продукта. Очистка колоночной хроматографией с использованием 5%-20% этилацетат/гексан приводит к получению 208 мг соединения I-143.
Синтез (1-нафталин-2-идметил-1Н-имидазо[1,2-b]пиразол-7-ил)метанола, I-144. К раствору соединения I-143 (100 мг, 0,313 ммоль) в безводном метиленхлориде (5 мл), охлажденному до -70°C, по каплям добавляют DIBAL (1M раствор в CH2Cl2, 0,94 мл, 3 экв.) Реакционную смесь перемешивают при указанной температуре в течение 4 часов. Смесь гасят МeOH, затем добавляют 50% насыщенный раствором тартрата натрия калия и дают возможность смеси нагреться до комнатной температуры. Смесь экстрагируют CH2Cl2 (2×20 мл), объединенные органические слои промывают насыщенным раствором соли и сушат над MgSO4. После удаления растворителя получают 70 мг неочищенного спирта, I-144. Продукт используют на следующей стадии без дополнительной очистки.
Синтез 1-нафталин-2-илметил-1Н-имидазо[1,2-b]пиразол-7-карбальдегида, I-145. Активированный MnO2 (110 мг, 1,26 ммоль) добавляют к суспензии спирта (70 мг, 0,25 ммоль) в 10 мл безводного метиленхлорида. Реакционную смесь перемешивают при комнатной температуре в течение ночи. ТСХ показывает наличие некоторого количества непрореагированного спирта, поэтому реакцию продолжают еще один день. После обработки смесь очищают колоночной хроматографией с использованием от 100% CH2Cl2 до 5% MeOH/CH2Cl2, получая 70 мг соединения I-145.
Синтез этилового эфира 3-(1-нафталин-2-илметил-1Н-имидазо[l,2-b]пиразол-7-ил)акриловой кислоты, I-146. К суспензии NaH (60% в минеральном масле, 21 мг, 0,525 ммоль) в безводном ТГФ (3 мл) при 0°C добавляют триэтилфосфоноацетат (92 мкл, 0,462 ммоль). Реакционную смесь нагревают до комнатной температуры и выдерживают при указанной температуре в течение 1 часа. Затем смесь охлаждают до 0°C и добавляют раствор соединения I-145 (60 мг, 0,21 ммоль) в 2 мл безводного ТГФ. Смеси дают возможность нагреться до комнатной температуры и перемешивают при комнатной температуре в течение 1 часа, затем смесь нагревают до 70°C и перемешивают при указанной температуре в течение ночи. Реакционную смесь охлаждают до комнатной температуры, растворитель удаляют, к смеси добавляют 10 мл CH2Cl2 и смесь гасят 1 мл насыщенного раствора NH4Cl. Органический слой промывают насыщенным раствором соли и сушат над MgSO4. Растворитель удаляют, получая 70 мг неочищенного соединения I-146, структуру которого подтверждают 1Н-ЯМР. Продукт используют для последующего гидролиза без дополнительной очистки.
Синтез 3-(1-нафталин-2-илметил-1Н-имидазо[1,2-b]пиразол-7-ил)акриловой кислоты, I-147. К раствору соединения I-146 (70 мг, 0,2 ммоль) в MeOH/ТГФ (2 мл/1 мл) добавляют 1 мл 1н. раствора NaOH. Реакционную смесь перемешивают при комнатной температуре в течение 3 дней. Растворитель удаляют, к остатку добавляют 1 мл 10% водного раствора HCl и смесь экстрагируют EtOAc (3×10 мл). Объединенные органические слои промывают насыщенным раствором соли и сушат над MgSO4. После удаления растворителя получают 50 мг неочищенный кислоты. Продукт очищают с использованием небольшого слоя силикагеля I-147 (25 мг).
Синтез 3-(l-нафталин-2-илметил-1Н-имидазо[l,2-b]пиразол-7-ил)акриловой кислоты, P034. К суспензии кислоты I-147 (18,6 мг, 0,0586 ммоль) в безводном метиленхлориде (1,5 мл) добавляют DMAP (14,3 мг, 0,117 ммоль), 2-тиофенсульфониламид (9,6 мг, 0,0586 ммоль) и EDCI (22,5 мг, 0,117 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 дней. Смесь гасят добавлением 10% водного раствора HCl (1 мл) и смесь экстрагируют EtOAc. Органический слой сушат над MgSO4 и после удаления растворителя 10 мг неочищенного остатка очищают колоночной хроматографией с использованием CH2Cl2 до 3% MeOH/CH2Cl2, получая 5 мг соединения P034. 1Н-ЯМР (500 МГц, CDCl3) 5,4 (с, 2H), 5,98 (д, J=14,5 Гц, 1H), 6,78 (с, 1H), 7,05 (т, 1H), 7,30 (дд, J=8,0, 1,0 Гц, 1H), 7,5-7,53 (м, 3H), 7,62 (м, 1H), 7,68 (уш., 1H), 7,70 (м, 1H), 7,76 (д, J=14,5 Гц, 1H), 7,82-7,85 (м, 2H), 7,89 (с, 1H). ЖХ-МС (92%): APCI: Вычислено 462 m/z; найдено: 461 (M-1).
Пример 269. Получение соединения P013.
Синтез 1-нафталин-2-илметил-1Н-имидазо[l,2-b]пиразол-7-карбоновой кислоты, I-148. К раствору соединения I-145 (24 мг, 0,075 ммоль) в 1,4-диоксане (1 мл) добавляют 1M водный LiOH (0,4 мл). Реакционную смесь нагревают до 80°C и перемешивают при указанной температуре до исчезновения исходного вещества (примерно 5 часов). Растворитель удаляют и остаток переносят в EtOAc (10 мл). Добавляют 10% водную HCl до pH=3. Суспензию фильтруют и твердый остаток растирают в эфире, получая 20 мг кислоты, I-148. ЖХ-МС (95%).
Синтез соединения P013. К суспензии кислоты I-148 (20 мг, 0,068 ммоль) в метиленхлориде (2 мл), добавляют DMAP (17 мг, 0,137 ммоль). Суспензия превращается в гомогенный раствор. К полученному раствору добавляют тиофен-5-сульфониламид (11 мг, 0,068 ммоль) и затем EDCI (26 мг, 0,137 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют, остаток растворяют в EtOAc (5 мл) и промывают 10% водным раствором HCl (1 мл). Органический слой промывают насыщенным раствором соли и сушат над MgSO4. Растворитель выпаривают, получая 20 мг неочищенного продукта. Очистка колоночной хроматографией с использованием в качестве элюента от 100% метилeнхлорида до 3% раствора метанола в метилeнхлориде приводит к получению 10 мг соединения P013.
Пример 270. Получение соединения P033.
К раствору п-толуолсульфонилизоцианата (27 мг, 0,135 ммоль) в безводном ТГФ (1 мл) при 0°C добавляют раствор спирта I-144 (25 мг, 0,09 ммоль) в ТГФ (1 мл). Смесь перемешивают при 20°C в течение 3 часов. Растворитель удаляют и остаток очищают колоночной хроматографией, используя CH2Cl2 и 3% MeOHCH2Cl2 с получением 5 мг соединения P033. 1Н-ЯМР (500 МГц, CDC13) 2,38 (с, 3H), 3,96 (д, 6,0 Гц, 1H), 4,27 (т, J=6,0 Гц, 1H), 5,35 (с, 2H), 6,78 (д, J=3,5 Гц, 1H), 7,24-7,31 (м, 5H), 7,49-7,51 (м, 2H), 7,66-7,68 (м, 3H), 7,80-7,84 (м, 3H). ЖХ-МС (95%): APCI- Вычислено: 430 m/z; найдено: 429 (M-l).
Пример 271. Получение соединения P116.
Метиловый эфир 5-(2,4-дихлорфенил)-2,4-диоксопентановой кислоты, I-149. Метанольный 25% мас. раствор метоксида натрия (11 мл, 48 моль, 2 экв.) осторожно (в течение 4 минут) добавляют к раствору 2,4-дихлорфенилацетона (4,88 г, 24 ммоль, 1 экв.) и диметилоксалата (4,8 г, 40 ммоль, 1,7 экв.) в метаноле (55 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Добавляют 10% водный раствор HCl, выделившееся масло отделяют, промывают водой. Масло кристаллизуют добавлением гексана. Осадок отфильтровывают и перекристаллизовывают из MTBE (12 мл), получая 0,78 г (11%) I-149. MС (AP-): 288 (M-1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Этиловый эфир (E)-3-{3-[2-(2,4-дихлорфенил)ацетил]-4-гидрокси-1-метил-5-оксо-2,5-дигидро-1Н-пиррол-2-ил}акриловой кислоты, I-150. 40% водный раствор метиламина (28 мг, 0,363 ммоль, 1,1 экв.) добавляют к раствору пирувата I-149 (96 мг, 0,33 ммоль, 1 экв.) и этилового эфира (E)-4-оксобутеновой кислоты (45 мг, 0,35 ммоль, 1,05 экв.) в AcOH-диоксане, 1:1 (0,4 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи. К смеси добавляют эфир и полученную смесь фильтруют. Маточную жидкость концентрируют в вакууме, получая неочищенный пирролидион I-150 (117 мг, 87%) в виде масла. ЖХ-МС (ESI-): 397 (M-1) (87%). 1H-ЯМР (500 МГц, CDC13) подтверждает структуру.
Этиловый эфир (E)-3-[3-(2,4-дихлорбензил)-5-метил-6-оксо-1,4,5,6-тетрагидропирроло[3,4-c]пиразол-4-ил]акриловой кислоты, I-151. Гидразингидрат (18 мг, 0,35 ммоль, 1,2 экв.) одной порцией добавляют к раствору пирролдиона I-150 (117 мг, 0,294 ммоль, 1 экв.) в AcOH (1,2 мл), реакционную смесь нагревают до 80°C и выдерживают при этой температуре в течение 2 часов. Реакционную смесь гасят насыщенным водным раствором NaHCO3 с последующей экстракцией EtOAc. Органическую фазу промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют с получением остатка (120 мг). Остаток хроматографируют на SiO2 (5 г) с использованием в качестве элюента EtOAc, и получая пиразолпирролдион I-151 (20 мг, 17%) в виде желтого масла. MС(ESI-): 392 (M-l). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
(E)-3-[3-(2,4-дихлорбензил)-5-метил-6-оксо-1,4,5,6-тетрагидропирроло[3,4-c]пиразол-4-ил]акриловой кислоты, I-152. LiOH/H2O (6,2 мг, 0,146 ммоль, 1,2 экв.) добавляют раствору пиразолпирролдиона I-151 (48 мг, 0,122 ммоль, 1 экв.) в смеси MeOH-вода-ТГФ, 1:1:1 (1,2 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи. К смеси добавляют водный насыщенный раствор NH4Cl (4 мл) и реакционную смесь экстрагируют CH2Cl2. Органическую фазу промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют, получая кислоту I-152 (18,5 мг, 42%) в виде масла. МС (ESI+): 367 (М+1).
Синтез соединения P116. К смеси кислоты I-152 (18,5 мг, 0,0505 ммоль, 1 экв.) в дихлорметане (0,5 мл) добавляют DMAP (12,4 мг, 0,01010 ммоль, 2 экв.), 4,5-дихлор-2-тиофенсульфонамид (11,7 мг, 0,0505 ммоль, 1 экв.), и EDCI (19,4 мг, 0,1010 ммоль, 2 экв.). Смесь перемешивают при комнатной температуре в течение 3 дней и затем гасят 10% водной HCl. Затем смесь экстрагируют EtOAc. Экстракт промывают насыщенным раствором соли, сушат над MgSO4. Растворитель удаляют в вакууме и остаток хроматографируют на SiO2 (1 г), используя смесь MeOH-EtOAc (19:1) в качестве элюента. Получают 2 мг (7%) сульфонамида P116 в виде масла. ЖХ-МС (ESI-): 579 (M-1) (86%). 1Н-ЯМР (500 МГц, ДМСО) подтверждает структуру.
Пример 272. Получение соединения P117.
3-{3-[2-(2,4-Дихлорфенил)ацетил]-4-гидрокси-1-метил-5-оксо-2,5-дигидро-1Н-пиррол-2-ил}пропионовая кислота, I-153. 2,0М ТГФ-раствор метиламина (0,2 мл, 0,4 ммоль, 1,1 экв.) добавляют в раствор пируват I-149 (100 мг, 0,35 ммоль, 1 экв.) и 15% водный раствор полуальдегида янтарной кислоты (250 мг, 2,42 ммоль, 7 экв.) в AcOH (0,5 мл). Реакционную смесь перемешивают при температуре 80°C в течение 30 минут, добавляют еще одну порцию метиламина (0,1 мл, 0,2 ммоль, 0,55 экв.) и реакционную смесь перемешивают при температуре 80°C в течение 30 минут. Реакционную смесь концентрируют, остаток растворяют в EtOAc, раствор промывают водой, насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют, получая пирролидион I-153 (147 мг, 114%) в виде желтого масла. Масло превращают в бежевое твердое вещество (104 мг, 81%) растиранием в смеси MTBE-гексан, 1:1. МС (ESI-): 371 (M-l). 1Н-ЯМР (500 МГц, ДМСО) подтверждает структуру.
3-[3-(2,4-Дихлорбензил)-5-метил-6-оксо-1,4,5,6-тетрагидропирроло[3,4-c]пиразол-4-ил]пропионовая кислота, I-154. К раствору пирролидиона I-153 (50 мг, 0,134 ммоль, 1 экв.) в AcOH (0,5 мл) одной порцией добавляют гидразингидрат (23 мг, 0,46 ммоль, 3,4 экв.), реакционную смесь нагревают до 80°C и выдерживают при этой температуре в течение 2,5 часов. Реакционную смесь разбавляют водой и затем экстрагируют EtOAc. Органический слой промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют, получая неочищенный продукт (60 мг) в виде масла. Масло растирают с эфиром, получая пирразолпрролидион I-154 (40 мг, 80%) в виде желтого твердого вещества. МС(AP-): 366 (M-1). 1H-ЯМР (500 МГц, ДМСО) подтверждает структуру.
Синтез соединения P117. К смеси пиразолпирролдиона I-154 (30 мг, 0,0815 ммоль, 1 экв.) в дихлорметане (0,8 мл) добавляют DMAP (20 мг, 0,163 ммоль, 2 экв.), 2-тиофенсульфонамид (19 мг, 0,0815 ммоль, 1 экв.) и EDCI (31 мг, 0,163 ммоль, 2 экв.). Смесь перемешивают при комнатной температуре в течение 3 часов и затем гасят 10% водной HCl. Смесь экстрагируют EtOAc. Экстракт промывают насыщенным раствором соли, затем сушат над MgSO4, фильтруют и концентрируют, получая 6,5 мг (14%) сульфонамида P117 в виде белого твердого вещества. 1Н-ЯМР (ДМСО-d6) 1,4-2,0 (м, 4H), 2,77 (с, 3H), 4,08 (с, 2H), 4,26 (с, 1H), 7,37 (с, 2H), 7,49 (с, 1H), 7,81 (с, 1H), 12,4 (уш., 1H), 13,3 (уш., 1H). ЖХ-МС (89%): ESI- Вычислено 580 m/z; найдено: 580.
Пример 273. Получение соединения P344.
Этиловый эфир 1-аллил-2-оксоциклогексанкарбоновой кислоты, I-155: В круглодонную колбу объемом 1 л, снабженную обратным холодильником и магнитной мешалкой, в атмосфере азота добавляют этиловый эфир 2-оксоциклогексанкарбоновой кислоты (19,0 г, 112 ммоль), аллилбромид (14,2 г, 117 ммоль), ТГФ (223 мл) и трет-бутоксид калия (13,2 г, 117 ммоль). Смесь кипятят с обратным холодильником на масляной бане (67°C) в течение 18 часов. Реакционную смесь охлаждают и растворитель удаляют на роторном испарителе. К смеси добавляют 1М водную HCl, пока смесь не превратится из пасты в мутный раствор. Смесь экстрагируют CH2Cl2 (3×250 мл). Объединенные экстракты промывают водой (2×250 мл) и насыщенным раствором соли (250 мл), сушат (MgSO4) и концентрируют при пониженном давлении, получая 21,8 г (93%) соединения I-155 в виде бледно-желтого масла. 1Н-ЯМР анализ показывает, что вещество достаточно чисто для того, чтобы его использовать на следующей стадии. 1Н-ЯМР (400 МГц, CDCl3).
Этиловый эфир l-карбоксиметил-2-оксоциклогексан карбоновой кислоты, I-156: В трехгорлую колбу объемом 2 л, снабженную верхнеприводной мешалкой и двумя свободно закрывающимися крышками, помещают соединение I-155 (21,8 г, 104 ммоль). В колбу Эрленмейера, снабженную стержневой мешалкой, добавляют периодат натрия (182 г, 850 ммоль), перманганат калия (3,28 г, 20,7 ммоль), карбонат калия (10,9 г, 78,8 ммоль) и воду (430 мл) при комнатной температуре. Окислительную смесь перемешивают в течение 5 минут и затем одной порцией добавляют в колбу, содержащую олефин I-155. Смесь перемешивают при комнатной температуре в течение 24 часов. Затем в реакционную смесь добавляют дополнительную порцию перманганата калия (0,820 г, 5,19 ммоль). Содержимое колбы перемешивают в течение дополнительных 2,5 часов. Реакцию гасят медленным добавлением водного раствора гидросульфита натрия до тех пор, пока смесь не превратится из темно-коричневого раствора в прозрачный желтый раствор. Водную смесь экстрагируют CH2Cl2 (3×250 мл). Объединенные экстракты промывают водой (2×250 мл) и насыщенным раствором соли (250 мл), сушат (MgSO4) и концентрируют при пониженном давлении, получая 18,6 г (79%) I-156 в виде бледно-желтого масла. 1Н-ЯМР анализ показывает, что продукт достаточно чист для использования на следующей стадии. 1Н-ЯМР (400 МГц, CDCl3).
Этиловый эфир 1-(2,4-дихлорбензил)-2,4-диоксо-1,2,3,4,5,6-гексагидроиндол-3a-карбоновой кислоты, I-157. В круглодонную колбу, снабженную обратным холодильником и мешалкой, загружают соединение I-156 (5,00 г, 21,9 ммоль), м-ксилол (45 мл) и 2,4-дихлорбензиламин (2,95 мл, 21,9 ммоль). Смесь кипятят с обратным холодильником на масляной бане (138°C) в течение 6 часов. Затем реакционную смесь охлаждают до комнатной температуры. Растворитель удаляют выпариванием, получая 7,92 г (98%) соединения I-157 в виде красновато-апельсинового масла. 1Н-ЯМР анализ показывает, что вещество достаточно чисто для использования его на следующей стадии. 1Н-ЯМР (400 МГц, CDCl3).
Этиловый эфир 7-бром-1-(2,4-дихлорбензил)-2-оксо-1,2,3,4,5,6-гексагидроиндол-3a-карбоновой кислоты, I-158: В емкость объемом 40 мл, снабженную мешалкой и крышкой, помещают соединение I-157 (600 мг, 1,63 ммоль), растворенное в CH2Cl2 (16 мл). Раствор охлаждают до 0°C на ледяной бане и добавляют бром (416 мкл, 8,15 ммоль). Смесь перемешивают в течение 2 часов, в течение которых смесь медленно нагревают до комнатной температуры и затем добавляют триэтиламин (750 мкл, 5,38 ммоль). Смесь перемешивают в течение дополнительных 45 минут, затем реакцию гасят водой (15 мл). После перемешивания в течение 30 минут органическую часть экстрагируют и затем сушат MgSO4. Растворитель удаляют выпариванием, получая коричневое масло, которое очищают хроматографией на силикагеле. Система растворителя: 20% EtOAc/гексан. После очистки получают 431 мг соединения I-158 в виде желтовато-коричневого твердого вещества (59%). 1Н-ЯМР анализ показывает, что вещество достаточно чисто для использования его на следующей стадии. 1H-ЯМР (400 МГц, CDCl3).
Этиловый эфир 7-((E)-2-карбоксивинил)-1-(2,4-дихлорбензил)-2-оксо-1,2,3,4,5,6-гексагидроиндол-3a-карбоновой кислоты I-159: В емкость объемом 18 мл, снабженную мешалкой и крышкой, помещают соединение I-158 (431 мг, 0,964 ммоль), безводный ДМФА (4,8 мл) и триэтиламин (1,34 мл, 9,64 ммоль). Смесь перемешивают в течение 10 минут, в течение которых дегазируют азотом. После дегазации добавляют трет-бутилакрилат (423 мкл, 2,89 ммоль), ацетат палладия (21,6 мг, 0,0964 ммоль) и три(о-толил)фосфин (88,0 мг, 0,289 ммоль). После добавления смесь дегазируют азотом в течение дополнительных 3 минут и затем емкость герметично закрывают крышкой. Емкость нагревают на масляной бане до 100°C и выдерживают при этой температуре в течение 22 часов. Затем реакционную смесь охлаждают и фильтруют через целит. Смесь разбавляют CH2C12 (75 мл) и затем промывают водой (2×75 мл) и насыщенным раствором соли (75 мл). Органическую часть сушат MgSO4 и концентрируют при пониженном давлении, получая трет-бутиловый эфир в виде коричневого масла. Неочищенный продукт растворяют в CH2C12 (4,4 мл) и охлаждают до 0°C. К полученному раствору добавляют трифторуксусную кислоту (440 мл, 5,9 экв.) и смесь перемешивают при комнатной температуре. После перемешивания в течение 4,5 часов вводят дополнительную порцию трифторуксусной кислоты (440 мкл, 5,9 экв.) и смесь перемешивают в течение 20 минут. Растворитель удаляют продувкой азотом и продукт, растворенный в эфире, промывают насыщенным водным раствором NaHCO3 (4×30 мл). В процессе второй промывки NaHCO3, добавляют небольшую порцию 3M NaOH (20 мл) для лучшего разделения слоев. Водные слои объединяют и подкисляют 3M водной HCl до pH 1. Водный слой экстрагируют CH2C12 (3×100 мл). Органические фракции объединяют, сушат (MgSO4) и концентрируют на роторном испарителе, получая 182 мг соединения I-159 в виде бледно-желтого твердого вещества (43% выход реакции I-158 в соединение I-159). 1Н-ЯМР анализ показывает, что чистота продукта достаточна для использования его на следующей стадии. 1Н-ЯМР (400 МГц, CDCl3) ЖХ/МС чистота=81,0%.
Синтез соединения P344. В емкость объемом 8 мл, снабженную мешалкой, помещают кислоту I-159 (50,0 мг, 0,114 ммоль), CH2Cl2 (2,4 мл), 2,4,5-трифторфенилсульфонамид (28,9 мг, 0,137 ммоль), DMAP (33,5 мг, 0,274 ммоль) и EDCI (54,6 мг, 0,285 ммоль). Смесь перемешивают при комнатной температуре в течение 18 часов. Смесь разбавляют CH2Cl2 (30 мл) и органическую часть промывают 1M водной HCl (30 мл), водой (3×30 мл) и насыщенным раствором соли (30 мл). Органическую часть сушат (MgSO4) и концентрируют на роторном испарителе, получая продукт в виде светло-желтого твердого вещества. После сушки получают 40,2 мг соединения P344 в виде бледно-желтого твердого вещества (56%). 1Н-ЯМР (400 МГц, CDCl3) 1,26 (т, J=7,2 Гц, 3H), 1,53-1,68 (м, 2H), 1,92 (м, 1H), 2,20-2,26 (м, 2H), 2,54 (м, 1H), 2,78 (AB кв., J=16,8 Гц, 2H), 4,19 (кв., J=7,2 Гц, 2H), 4,95 (AB кв., J=17,2, 17,6 Гц, 2H), 5,68 (д, J=15,2 Гц, 1H), 7,03-7,08 (м, 2H), 7,10 (д, J=8,0 Гц, 1H), 7,17 (дд, J=8,4, 2,0 Гц, 1H), 7,22 (с, 1H), 7,94 (м, 1H). ЖХ/МС (95%), МС (ESI-) Вычислено: 630,5 m/z; найдено: 631,3 m/z.
Пример 274. Получение соединения P345.
Синтез соединения P345. В соответствии с методикой синтеза P344, исходя из кислоты I-159 (50,0 мг, 0,114 ммоль), CH2Cl2 (2,4 мл), 4,5-дихлортиофен-2-сульфонамида (31,8 мг, 0,137 ммоль), DMAP (33,5 мг, 0,274 ммоль) и EDCI (54,6 мг, 0,285 ммоль), получают 49,0 мг соединения P345 в виде бледно-желтого твердого вещества (66%). 1Н-ЯМР (400 МГц, CDCl3) 1,27 (т, J=6,8 Гц, 3H), 1,53-1,65 (м, 2H), 1,92 (м, 1H), 2,20-2,29 (м, 2H), 2,54 (м, 1H), 2,79 (AB кв., J=16,8 Гц, 2H), 4,20 (кв., J=7,2 Гц, 2H), 4,99 (AB кв., J=17,2, 17,6 Гц, 2H), 5,65 (д, J=15,2 Гц, 1H), 7,12 (д, J=8,4 Гц, 2H), 7,20 (дд, J=8,4, 2,0 Гц, 1H), 7,29 (с, 1H), 7,49 (д, J=2,0 Гц, 1H), 7,62 (с, 1H). Сульфонамидная часть на спектре не видна. ЖХ/МС (95%), МС (ESI-) Вычислено: 651,4 m/z; найдено: 651,4 m/z.
Пример 275. Получение соединения P346.
Синтез соединения P346. В соответствии с методикой синтеза P344, исходя из кислоты I-159 (35,0 мг, 0,0799 ммоль), CH2Cl2 (1,7 мл), 3,4-дифторфенилсульфонамида (18,5 мг, 0,0959 ммоль), DMAP (23,5 мг, 0,192 ммоль) и EDCI (38,3 мг, 0,200 ммоль), получают 30,8 мг соединения P346 в виде бледно-желтого твердого вещества (63%). 1Н-ЯМР (400 МГц, CDCl3) ЖХ/МС чистота=86,5%.
Пример 276. Получение соединения P075.
1Н-Индол-4-иловый эфир уксусной кислоты, I-160. Уксусный ангидрид (16,75 мл, 169 ммоль, 1,5 экв.) медленно (в течение 7 минут) добавляют к раствору 4-гидроксииндола (15 г, 113 ммоль, 1 экв.) в пиридине (113 мл) при комнатной температуре (температура возрастает с 25 до 32°C). После перемешивания в течение дополнительных 5 минут реакционную смесь охлаждают ледяной водой и добавляют 10% водную HCl (340 мл), затем EtOAc (565 мл). Органическую часть отделяют и водный слой экстрагируют EtOAc (100 мл). Объединенные органические слои промывают 10% водной HCl (2×50 мл), водой (2×300 мл), насыщенным раствором соли (300 мл) и сушат над MgSO4. Фильтрование и удаление растворителя выпариванием приводят к получению 19,3 г (98%) I-160 в виде светло-коричневого твердого вещества. 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру. 1Н-ЯМР (500 МГц, CDCl3) 2,39 м.д. (с, 3H), 6,43 м.д. (уш. с, 1H), 6,86 (д, 1H, J=16,0 Гц), 7,14 (м, 2H), 7,24 (м, 1H), 8,20 (уш. с, 1H).
3-(Нафталин-2-карбонил)-1Н-индол-4-иловый эфир уксусной кислоты, I-161. Метилмагнийбромид (26 мл, 78 ммоль, 1,05 экв., 3,0М раствор в эфире) медленно через шприц (в течение 10 минут) добавляют к раствору ацетата I-160 (13 г, 74 ммоль, 1 экв.) в безводном CH2Cl2 (260 мл) при комнатной температуре. Смесь перемешивают в течение 5 минут при комнатной температуре, к смеси с помощью шприца добавляют в течение 5 минут при комнатной температуре хлорид цинка (223 мл, 3,0 экв., 1,0М раствор в эфире). Смесь перемешивают в течение 10 минут при комнатной температуре, затем при комнатной температуре в течение 3 минут добавляют раствор 2-нафтоилхлорида (14,85 г, 1,05 экв.) в безводном CH2Cl2 (87 мл). Реакционную смесь перемешивают в течение 3 часов при комнатной температуре и затем выливают в насыщенный водный раствор NH4Cl (866 мл) и cмесь разбавляют CH2Cl2 (200 мл). Водный слой экстрагируют CH2Cl2 (100 мл). Объединенные органические слои промывают водой (500 мл×2), насыщенным раствором соли (500 мл), сушат над MgSО4, фильтруют и концентрируют, получая 24,4 г (100%) в виде коричневого твердого вещества. К твердому продукту добавляют MTBE (100 мл), смесь кипятят с обратным холодильником, охлаждают до 30-40°C и фильтруют, остаток промывают на фильтре MTBE (50 мл, 25 мл, 5 мл×2), затем эфиром (5 мл×2), получая 16,0 г (65%) I-161 в виде светло-коричневого твердого вещества. МС (ESI-): 329. 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру. 1H-ЯМР (500 МГц, CDCl3) 2,41 (с, 3H), 6,89 (д, 1H), 7,03 (дд, 1H, J=16,5 Гц), 7,09 (м, 2H), 7,52 (т, J=15,0 Гц, 1H), 7,56 (т, 1H, J=15,0 Гц), 7,79 (м, 3H), 7,88 (д, 1H, J=16,0 Гц), 8,12 (уш. с, 1H), 9,35 (уш. с, 1H).
3-Нафталин-2-илметил-1Н-индол-4-ол, I-162 BH3ТГФ (1M в ТГФ, 120 мл, 120 ммоль, 3,3 экв.) медленно добавляют к раствору соединения I-161 (12,0 г, 36,4 ммоль, 1 экв.) при 0°C в атмосфере азота. Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают в течение ночи. Реакционную смесь гасят медленным добавлением метанола (120 мл), упаривают и подвергают совместному испарению с MeOH (3×120 мл). Остаток очищают хроматографией на силикагеле (150 г, CH2Cl2), получая соединение I-162 (6,2 г, 62%). МС (ESI-): 271(M-1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Метиловый эфир (3-нафталин-2-илметил-1Н-индол-4-илокси)уксусной кислоты, I-163. Раствор метилбромацетата (3,10 г, 1,9 мл, 20,3 ммоль, 1,05 экв.) в ДМФА (20 мл) медленно добавляют к смеси соединения I-162 (5,29 г, 19,4 ммоль, 1 экв.) и K2CO3 (3,21 г, 23,2 ммоль, 1,2 экв.) в ДМФА (51 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи (17 часов). Добавляют смесь вода:насыщенный раствор соли (5:1, 180 мл) и полученный раствор экстрагируют EtOAc (180 мл, 140 мл, 70 мл). Органический слой промывают насыщенным раствором соли (2×210 мл), сушат над MgSO4, фильтруют и концентрируют, получая 7,8 г в виде твердого вещества. Остаток промывают эфиром (20 мл) и фильтруют, получая 5,0 г (75%) I-163 в виде твердого не совсем белого вещества. МС (ES+): 346 (М+1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
(3-Нафталин-2-илметил-1Н-индол-4-илокси)уксусная кислота, I-164. Раствор 2н. NaOH (9,5 мл, 19 ммоль, 2 экв.) добавляют к раствору соединения I-163 (3,3 г, 9,55 ммоль, 1 экв.) в смеси ТГФ-MeOH, 2:1 (132 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь концентрируют до ~20 мл, добавляют 10% водную HCl (10 мл) и затем воду (50 мл). Смесь экстрагируют EtOAc (250 мл) и органическую фазу сушат над MgSO4, фильтруют и концентрируют, получая соединение I-164 (2,95 г, 92%) в виде твердого, не совсем белого вещества. 1H-ЯМР (500 МГц, ДМСО) подтверждает структуру.
Синтез соединения P075. К раствору соединения I-164 (2,87 г, 8,65 ммоль, 1 экв.) в дихлорметане (80 мл) добавляют DMAP (2,11 г, 17,30 ммоль, 2 экв.), 4,5-дихлор-2-тиофенсульфонамид (2,11 мг, 9,08 ммоль, 1,05 экв.) и EDCI (3,22 г, 17,30 ммоль, 2 экв.). Смесь перемешивают при комнатной температуре в течение 16 часов и затем гасят 10% водной HCl (5 мл), затем EtOAc (100 мл). Водный слой экстрагируют EtOAc (2×50 мл). Объединенные органические слои промывают насыщенным водным NH4Cl (100 мл), насыщенным раствором соли (2×100 мл), сушат над MgSO4, фильтруют и концентрируют, получая остаток (3,31 г, 63%). Остаток растирают с MeOH (12 мл), кипятят с обратным холодильником, затем охлаждают до 0°C и отфильтровывают, получая 2,97 г (71%) в виде полунеочищенного сульфонамида 4. ВЭЖХ: 96,2%.: ESI- Вычислено: 544 m/z; найдено: 544. 1Н-ЯМР (ДМСО-d6) подтверждает структуру. 1Н-ЯМР (ДМСО-d6) 4,32 (с, 2H), 4,68 (с, 2H), 6,18 (д, J=7,5 Гц, 1H), 6,87 (т, J=8,0 Гц, 1H), 6,94 (д, J=8,5 Гц, 1H), 7,01 (д, J=3,5 Гц, 1H), 7,41 (м, 2H), 7,46 (дд, J=8,5, 1,5 Гц, 1H), 7,72 (д, J=8,5 Гц, 2H), 7,79-7,81 (м, 2H), 7,89 (с, 1H), 10,9 (уш.с, 1H).
Пример 277. Получение соединения P162.
Метиловый эфир 2-(3-нафталин-2-илметил-1Н-индол-4-илокси)пропионовой кислоты, I-165. Раствор индола I-162 (125 мг, 0,46 ммоль, 1 экв.), метилбромпропионат (80 мг, 0,48 ммоль, 1,05 экв.) и K2CO3 (76 мг, 0,55 ммоль, 1,2 экв.) в ДМФА (1,7 мл) перемешивают при 50°C в течение 16 часов. Добавляют смесь воды и насыщенного раствора соли, 5:1, суспензию экстрагируют EtOAc, органический слой промывают водой, насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют, получая сложный эфир I-165 (150 мг, 91%) в виде зеленого масла. МС (AP+): 360 (М+1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
2-(3-Нафталин-2-илметил-1Н-индол-4-илокси)пропионовая кислота, I-166. 2н. раствор NaOH (0,44 мл, 0,88 ммоль, 2,1 экв.) по каплям добавляют к раствору сложного эфира I-165 (150 мг, 0,42 ммоль, 1 экв.) в ТГФ-MeOH (2:1, 6 мл). Реакционную смесь перемешивают в течение 2 часов при комнатной температуре. Реакционную смесь концентрируют в вакууме и затем к остатку добавляют 10% водную HCl (4 мл) и воду (6 мл). Осадок экстрагируют EtOAc (6 мл, 4 мл). Раствор промывают насыщенным раствором соли, сушат над MgSO4, получая кислоту I-166 (149 мг) в виде масла. МС (ESI-): 344 (M-1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез соединения P162. К кислоте I-166 (134 мг, 0,39 ммоль, 1 экв.) в дихлорметане (4 мл) добавляют DMAP (95 мг, 0,77 ммоль, 2 экв.), 4,5-дихлор-2-тиофенсульфонамид (94 мг, 0,41 ммоль, 1,05 экв.), и EDCI (148 мг, 0,77 ммоль, 2 экв.). Смесь перемешивают при комнатной температуре в течение 2 часов и гасят 10% водной HCl (1 мл) и затем водой (4 мл). Водный слой экстрагируют EtOAc (4 мл). Объединенные органические слои промывают насыщенным раствором соли и затем сушат над MgSO4. Полученный остаток очищают хроматографией на SiO2 (5 г) CH2Cl2, получая сульфонамид P162 (101 мг, 47%) в виде зеленоватого твердого вещества. ЖХ-МС (ESI-): 558 (M-1) (99%). 1Н-ЯМР (500 МГц, ДМСО) подтверждает структуру.
Пример 278. Получение соединения P171.
Метиловый эфир 2-метил-2-(3-нафталин-2-илметил-1Н-индол-4-илокси)пропионовой кислоты, I-167. Раствор индола I-162 (300 мг, 1,1 ммоль, 1 экв.), метилбромизобутират (609 мг, 3,29 ммоль, 3 экв.), K2CO3 (607 мг, 4,39 ммоль, 4 экв.) и MgSO4 (132 мг, 1,1 экв.) в ДМФА (13,3 мл) при перемешивании нагревают до 75°C и выдерживают при этой температуре в течение 24 часов. Реакционную смесь фильтруют, концентрируют, получая ~0,4 г неочищенного продукта. Хроматография на SiO2 (20 г) с использованием в качестве элюента смеси EtOAc/гексан (1:4) приводит к получению соединения I-167 (96 мг, 23%) в виде масла. R f =0,31 (EtOAc/гексан, 1:3). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
2-Метил-2-(3-нафталин-2-илметил-1Н-индол-4-илокси)пропионовая кислота, I-168. 2н. раствор NaOH (0,53 мл, 1,06 ммоль, 4,1 экв.) по каплям добавляют к раствору эфира I-167 (96 мг, 0,26 ммоль, 1 экв.) в смеси ТГФ-MeOH (2:1,3 мл). Реакционную смесь перемешивают при температуре 75°C в течение 1,5 часа. Реакционную смесь концентрируют в вакууме и к полученному остатку добавляют 10% HCl (0,5 мл). Смесь экстрагируют EtOAc (6 мл). Раствор промывают насыщенным раствором соли, сушат над MgSO4, получая кислоту I-168 (92 мг, 100%) в виде масла. R f =0,61 (MeOH/CH2Cl2, 1:7). МС (ESI+): 358 (М+1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез соединения P171. К кислоте I-168 (88 мг, 0,24 ммоль, 1 экв.) в 4 мл дихлорметана добавляют DMAP (60 мг, 0,49 ммоль, 2 экв.), 4,5-дихлор-2-тиофенсульфонамид (60 мг, 0,26 ммоль, 1,05 экв.) и EDCI (94 мг, 0,49 ммоль, 2 экв.). Смесь перемешивают при комнатной температуре в течение 3 дней и гасят добавлением 10% раствора HCl (1 мл) и затем водой (4 мл). Водный слой экстрагируют EtOAc (4 мл). Объединенные органические слои промывают насыщенным раствором соли и сушат над MgSO4. Раствор концентрируют и остаток экстрагируют эфиром, получая сульфонамид P171 (120 мг, 87%) в виде желтого твердого вещества. R f =0,56 (MeOH/CH2Cl2, 1:7). 1H-ЯМР (CDCl3) 1,34 (с, 6H), 4,42 (с, 2H), 5,89 (д, J=8,0 Гц, 1H), 6,81 (т, J=8,0 Гц, 1H), 6,86 (д, J=2,0 Гц, 1H), 7,06 (д, J=8,0 Гц, 1H), 7,33 (дд, J=8,0, 3,0 Гц, 1H), 7,39 (с, 1H), 7,40-7,46 (м, 2H), 7,57 (уш., 1H), 7,70-7,83 (м, 2H), 7,79 (д, 8,5 Гц, 1H), 8,09 (уш., 1H), 8,51 (уш., 1H). ЖХ-МС (99%): ESI- Вычислено: 572 m/z; найдено: 572.
Пример 279. Получение соединения P259.
3-(l-Этил-1Н-бензимидазол-2-илсульфанил)бензофуран-4-ол, I-169. Раствор 2,0М HCl в эфире (3,75 мл, 7,5 ммоль, 5 экв.) добавляют к предварительно нагретой (40°C) суспензии 4-гидроксибензофурана (225 мг, 1,5 ммоль, 1 экв.) и 1-метил-1Н-бензимидазол-2-тиола (Aldrich, 246 мг, 1,5 ммоль, 1 экв.) в этаноле (3,75 мл). Раствор перемешивают при комнатной температуре в течение ночи, затем концентрируют до ~0,5 мл. К остатку добавляют смесь EtOAc/дихлорметан (8 мл, 1:1), промывают смесь водой (8 мл×2), насыщенным раствором соли (6 мл), сушат над MgSO4, концентрируют. Добавляют эфир (4 мл), твердый продукт отфильтровывают и маточную жидкость концентрируют, получая 277 мг неочищенного продукта в виде красного масла. Масло растирают с MTBE (c метил-трет-бутиловым эфиром) и отфильтровывают, получая 260 мг продукта. Масло очищают хроматографией на SiO2 (10 г), используя смесь 3:7 дихлорметана/гексан, 1:1 дихлорметана/гексан и 100% дихлорметан и получая сульфид I-169 (52 мг, 11%) в виде желтого масла. R f 0,28 (дихлорметана). МС (AP-): 309 (M-1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Метиловый эфир [3-(1-этил-1Н-бензимидазол-2-илсульфанил)бензофуран-4-илокси]уксусной кислоты, I-170. Раствор метилбромацетата (27 мг, 0,173 ммоль, 1,2 экв.) в ацетоне (0,2 мл) добавляют к суспензии сульфида I-169 (45 мг, 0,1445 ммоль, 1 экв.) и K2CO3 (30 мг, 0,217 ммоль, 1,5 экв.) в ацетоне (0,2 мл). Реакционную смесь герметично закрывают в емкости объемом 4 мл, нагревают до 50°C и выдерживают при данной температуре в течение 5 часов. К смеси добавляют воду и затем EtOAc, эмульсию гасят 10% HCl до достижения pH=2. Органический слой отделяют, промывают водой, насыщенным раствором соли, сушат над MgSO4, получая 50 мг продукта в виде оранжевого масла, которое частично кристаллизуется на воздухе. Продукт растирают в MTBE, затем в смеси MTBE/гексан (1:1), получая эфир I-170 (36 мг, 65%) в виде оранжевого твердого вещества. Rf 0,49 (дихлорметан). МС (ESI-): 381 (M-l), 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
[3-(1-Этил-1Н-бензимидазол-2-илсульфанил)бензофуран-4-илокси]уксусная кислота, I-171. 2н. раствор NaOH (0,1 мл, 0,21 ммоль, 2,5 экв.) по каплям добавляют к раствору эфира I-170 (32 мг, 0,084 ммоль, 1 экв.) в смеси ТГФ-MeOH (1:1, 0,5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 15 минут. Реакционную смесь концентрируют в вакууме, к остатку добавляют воду, затем EtOAc. Суспензию гасят 10% HCl. Органический слой промывают водой, насыщенным раствором соли, сушат над MgSO4, получая кислоту I-171 (27 мг, 88%) в виде твердого не совсем белого вещества. Rf =0,37 (MeOH/дихлорметан, 1:4). МС (ESI-): 367 (M-1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез соединения P259. К кислоте I-171 (24 мг, 0,065 ммоль, 1 экв.) в 0,5 мл дихлорметана добавляют DMAP (16 мг, 0,130 ммоль, 2 экв.), 4,5-дихлор-2-тиофенсульфонамид (15 мг, 0,065 ммоль, 1,05 экв.) и EDCI (25 мг, 0,130 ммоль, 2 экв.). Смесь перемешивают при комнатной температуре в течение ночи, затем гасят 10% HCl и водой. Водный слой экстрагируют EtOAc. Объединенные органические слои промывают насыщенным раствором соли и сушат над MgSO4. Раствор концентрируют и твердый остаток экстрагируют смесью MTBE/гексан (1:1), затем эфиром. Раствор концентрируют, получая 24,5 мг продукта в виде желтого масла. Масло хроматографируют на SiO2 (элюирование: EtOAc/гексан, 1:3, 2:3), получая сульфонамид P259 (10 мг, 26%) в виде желтого масла. R f=0,28 (EtOAc). 1Н-ЯМР (CDCl3) 1,47 (м, J=7,2 Гц, 3H), 4,29 (кв., J=7,2 Гц, 2H), 4,76 (с, 2H), 6,71 (дд, J=6,4, 1,8 Гц, 1H), 6,91 (дд, J=11,6, 0,4 Гц, 1H), 7,16 (дд, J=11,6, 0,4 Гц, 1H), 7,26-7,32 (м, 3H), 7,73 (м, 1H), 8,20 (д, J=4,8 Гц, 2H). ЖХ-МС (94%): ESI- Вычислено: 581 m/z; найдено: 581.
Пример 280. Получение соединения P153.
Соединение I-162 (0,50 г, 0,0018 моль) растворяют в ДМФА (8 мл). К раствору добавляют карбонат калия (1,00 г, 0,0072 моль) и смесь перемешивают в течение 15 минут. Добавляют этилбромдифторацетат (2,5 мл, 0,087 моль) и полученную смесь перемешивают при 70°С в течение ночи, концентрируют и распределяют между CH2Cl2 и водой. Слой CH2Cl2 промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют, концентрируют и хроматографируют на силикагеле (элюирование с градиентом; EtOAc/гексан), получая фракцию, обогащенную этиловым эфиром дифтор(3-нафталин-2-илметил-1Н-индол-4-илокси)уксусной кислоты (~30%, 1H-ЯМР, ЖХ-МС), которую используют на следующей стадии. Дифторэфирную фракцию (с содержанием примерно 30 мг) обрабатывают 7M NH3/MeOH (2 мл) при комнатной температуре в течение ночи в герметично закрытой емкости. Хроматография с обращенной фазой (C18-силикагель; элюирование с градиентом MeCN/вода) приводит к получению соединения P153, 5,8 мг. 1Н-ЯМР (400 МГц, ДМСО-d6) 4,29 (с, 2H), 6,83 (д, J=8,0 Гц, 1H), 7,03 (д, J=8,0 Гц, 1H), 7,06 (м, 1H), 7,27 (дд, J=8,0, 0,8 Гц, 1H), 7,39-7,48 (м, 3H), 7,72 (с, 1H), 7,76-7,86 (м, 3H), 8,24 (уш., 1H), 8,48 (уш., 1H), 11,15 (уш., 1H). ЖХ/МС (86,3%) ESI- Вычислено: M=366,4; найдено: 365,3 m/z.
Пример 281. Получение соединения P159.
Синтез этилового эфира 5-бром-4-оксопентановой кислоты, I-171. Бром (22 г, 7,2 мл, 138,8 ммоль) в потоке N2 в течение получаса добавляют к раствору этиллевулината (20 г, 138,8 ммоль) в 250 мл EtOH при комнатной температуре. После завершения добавления реакционную смесь перемешивают при комнатной температуре в течение еще получаса и затем кипятят с обратным холодильником в течение 1,5 часа. 1H-ЯМР анализ образца показывает наличие трех продуктов c соотношением SM: монобром:дибром=1:2,5:0,5. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. Остаток переносят в простой эфир, промывают насыщенным раствором NaHCO3 (3×50 мл), водой, насыщенным раствором соли, сушат над MgSO4 и концентрируют, получая 20,3 г соединения I-171 в виде темно-коричневого масла, которое используют на следующей стадии синтеза без дополнительной очистки. 1Н-ЯМР подтверждает структуру.
Синтез этил-2-амино-4-тиазолил-3-пропионата, I-172. Раствор неочищенного бромида I-171 (8 г, 35,8 ммоль) в 50 мл этанола добавляют к раствору тиомочевины (2,85 г, 37,59 ммоль) в 20 мл этанола. Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и кипятят с обратным холодильником в течение 4 часов. Растворитель удаляют; остаток переносят в 100 мл EtOAc, промывают водой, насыщенным раствором NaHCO3, насыщенным раствором соли, сушат над MgSO4 и концентрируют, получая неочищенный масляный продукт. Неочищенный продукт очищают колоночной хроматографией, используя в качестве элюента от 100% метиленхлорида до 5% MeOH/метиленхлорид и получая 3,75 г соединения I-172. 1Н-ЯМР, МС.
Синтез этилового эфира 3-имидазо[2,1-b]тиазол-3-илпропионовой кислоты, I-173. Диацеталь бромацетальдегида (8 г, 40,31 ммоль) в 60 мл водной 3н. HCl кипятят с обратным холодильником в течение 1 часа. Раствор охлаждают до комнатной температуры и экстрагируют эфиром (3×30 мл). Объединенные органические слои сушат над MgSO4 и по каплям добавляют к кипящему раствору 3,75 г этил-2-амино-4-тиазолил-3-пропионата, I-172, в 100 мл EtOH. Отгоняющийся эфир собирают. После окончания сбора эфира реакционную смесь кипятят с обратным холодильником в течение 8 часов. Растворитель удаляют и остаток промывают насыщенным раствором NaHCO3. Смесь экстрагируют метилeнхлоридом (3×50 мл). Объединенные органические слои промывают водой, насыщенным раствором соли, сушат над MgSO4, концентрируют с получением 3 г неочищенного продукта. Неочищенный продукт очищают колоночной хроматографией, используя в качестве элюента смесь от 20% до 50% EtOAc/гексан и получая 1 г чистого продукта I-173. 1Н-ЯМР.
Этиловый эфир 3-(5-формилимидазо[2,1-b]тиазол-3-ил)пропионовой кислоты, I-174. Реагент Вилсмейера получают при 0°C-5°C добавлением по каплям POCl3 (0,085 мл, 0,892 ммоль) при перемешивании в раствор 0,1 мл ДМФА. Смесь перемешивают при температуре 0°C в течение Ѕ часа, затем по каплям добавляют имидазотиазола I-173 (100 мг, 0,446 ммоль) в 2 мл CHCl3. Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, затем кипятят с обратным холодильником в течение 24 часов. Смесь охлаждают до комнатной температуры и гасят водой, перемешивают в течение Ѕ часа, затем экстрагируют метиленхлоридом. Органический слой сушат над MgSO4 и концентрируют, получая 30 мг продукта, I-174. 1Н-ЯМР.
Этиловый эфир 3-[5-((E)-2-нафталин-2-илвинил)имидазо[2,1-b]тиазол-3-ил]пропионовой кислоты, I-175. К суспензии NaH (60% в минеральном масле, 10 мг, 0,25 ммоль) в 3 мл безводного ТГФ добавляют бромид (2-нафтил)метилтрифенилфосфония (80 мг, 0,158 ммоль). Смесь нагревают до 60°C и выдерживают при этой температуре в течение 1 часа, затем добавляют альдегид, I-174 (40 мг, 0,158 ммоль) в 2 мл ТГФ. Реакционную смесь перемешивают при температуре 60°C в течение ночи. Смесь охлаждают до комнатной температуры, гасят насыщенным раствором NH4Cl и экстрагируют метилeнхлоридом. Органический слой сушат над MgSO4, концентрируют и полученный неочищенный продукт очищают колоночной хроматографией, используя в качестве элюента от 20% до 50% EtOAc/гексан и получая 30 мг чистого продукта, I-175. 1Н-ЯМР.
3-[5-((E)-2-Нафталин-2-илвинил)имидазо[2,1-b]тиазол-3-ил]пропионовая кислота, I-176. К раствору этилового эфира I-175 (30 мг, 0,079 ммоль) в смеси ТГФ:MeOH (1,5 мл/0,5 мл) добавляют 0,16 мл LiOH (1н.). Смесь перемешивают при температуре 40°C в течение 2 часов. ТСХ (5% MeOH/CH2Cl2) показывает, что реакция завершена. Реакционную смесь концентрируют в вакууме и к полученному остатку добавляют 10% раствор HCl до достижения pH=5-6. Смесь экстрагируют CH2Cl2 (2×5 мл). Объединенные органические слои промывают насыщенным раствором соли, сушат (MgSO4) и концентрируют, получая 20 мг неочищенного продукта, I-176. 1Н-ЯМР (500 МГц, CD3OD) подтверждает структуру, ЖХМС (ESI-): 348 (M-1), 90%.
Синтез соединения P159. К суспензии кислоты I-176 (10 мг, 0,028 ммоль) в CH2Cl2 (0,5 мл) последовательно добавляют 2,3-дихлортиофен-5-сульфонамид (7 мг, 0,028 ммоль), DMAP (7 мг, 0,057 ммоль) и EDCI (11 мг, 0,057 ммоль). Смесь перемешивают при комнатной температуре в течение ночи. Раствор подкисляют 10% HCl до pH=5-6 и экстрагируют EtOAc (2×5 мл). Органический слой промывают водой, сушат над MgSO4 и концентрируют в вакууме, получая 20 мг неочищенного продукта. Очистка колоночной хроматографией с использованием CH2Cl2 приводит к получению 7 мг чистого продукта, P153. 1Н-ЯМР (500 МГц, CDCl3) подтверждает структуру, ЖХМС (ESI-): 562 (M-1), 60%.
Пример 282. Получение соединения P244.
Синтез 4-бром-1Н-индол-2,3-диона (I-177) и 6-бром-1Н-индол-2,3-диона (I-178) (2a). К раствору хлоральгидрата (50,0 г, 0,247 моль) в воде (237 мл) последовательно добавляют Na2SO4 (69,0 г, 0,486 моль), 3-броманилин (40,0 г, 0,233 моль) в смеси с 37% HCl (25 мл, 0,302 моль) и водой (632 мл) при энергичном перемешивании. После завершения добавления полученную реакционную смесь кипятят с обратным холодильником в течение 10 минут и дают возможность охладиться до комнатной температуры. Выпавший осадок отделяют фильтрованием, промывают водой (3×100 мл) и сушат в вакууме, получая неочищенный изонитрозоацетанилид. Данный продукт добавляют одной порцией при энергичном перемешивании к концентрированной H2SO4 (790 мл) с такой скоростью, чтобы температура реакционной смеси находилась в интервале от 50 до 70°C. Реакционную смесь нагревают до 80°C, выдерживают при этой температуре в течение 20 минут и дают возможность смеси охладиться до комнатной температуры. Охлажденную смесь выливают в измельченный лед (примерно 3200 г). Смесь оставляют на 1 час. Выпавший оранжевый осадок отделяют фильтрованием, промывают водой и сушат, получая смесь соединения I-177 и I-178 (40 г, 83%). МС (ESI+): 227 (М+1). 1H-ЯМР (ДМСО-d6).
Синтез этиленкеталя I-179. Смесь соединения I-177 и I-178 (25,0 г, 0,111 моль), этилeнгликоля (27,5 г, 0,442 моль) и моногидрата п-толуолсульфоновой кислоты (2,5 г, 11,3 ммоль) в бензоле (500 мл) кипятят с обратным холодильником с ловушкой Дина-Старка для удаления образующейся воды. Реакционной смеси дают возможность охладиться до комнатной температуры, промывают ее 10% водным раствором NaHCO3 и затем водой. После концентрирования получают неочищенный продукт (25 г), который очищают перекристаллизацией из смеси EtOAc/гексан, получая 4-бромизотин-3-этиленкеталь, I-179 (8,2 г, 27%), в виде твердого, не совсем белого вещества. МС (ESI+): 270 (М+1). 1H-ЯМР (ДМСО-d6).
Синтез этиленацеталя I-180 (N-H-производное). Смесь соединения I-179 (5,4 г, 20 ммоль), три-o-толилфосфина (2,2 г, 7 ммоль) и ацетата палладия (0,5 г, 2 ммоль) в триэтилaмине (20 мл) и метилакрилате (5 г, 70 ммоль) в герметично закрытой пробирке нагревают до 100°C и перемешивают при температуре 100°C в течение 6 часов, затем охлаждают. Реакционную смесь выливают при перемешивании в 600 мл смеси воды со льдом, экстрагируют CH2Cl2. Объединенные органические слои промывают водой, насыщенным раствором соли (100 мл) и сушат над Na2SO4. После удаления растворителя неочищенный продукт очищают хроматографией на силикагеле, получая соединение I-180 (всего 4,5 г, 81%) в виде твердого, не совсем белого вещества. 1H-ЯМР (ДМСО-d 6).
Синтез этиленацеталя I-181 (N-CH3-производное): Смесь кеталя I-180 (3 г, 11 ммоль), метилйодида (г, ммоль) и K2CO3 (10 г, 55 ммоль) в ДМФА (40 мл) перемешивают при комнатной температуре в течение ночи. Смесь выливают в 600 мл смеси воды со льдом при перемешивании. Продукт выделяют экстракцией этилацетатом и неочищенный продукт очищают хроматографией на силикагеле, получая N-метил-производное I-181 в виде твердого не совсем белого вещества (2,8 г, 80%). 1Н-ЯМР(ДМСО-d6).
Гидролиз до I-182 (N-H-производное). К раствору сложного эфира I-180 (2,0 г, 5,58 ммоль) в MeOH (20 мл) добавляют раствор NaOH (0,45 г, 11,2 ммоль) в воде и полученную смесь перемешивают при комнатной температуре в течение 16 часов. После удаления метанола водный остаток охлаждают до -5°C и подкисляют 10% HCl до pH~2. Осадок отделяют фильтрованием, промывают водой и сушат, получая соединение I-182 (1,8 г, 86%).1H-ЯМР (ДМСО-d6).
Гидролиз до I-183 (N-CH3-производное). Соединение I-183 получают из I-181 аналогично получению соединения I-182 (95% выход). 1H-ЯМР (ДМСО-d6).
(E)-3-(2,3-Диоксо-2,3-дигидро-1Н-индол-4-ил)акриловая кислота, I-184. К суспензии I-182 (2,1 г, 5 ммоль) в MeOH (50 мл) при перемешивании при комнатной температуре добавляют концентрированную HCl (50 мл). Полученную реакционную смесь нагревают до 50°C и выдерживают при этой температуре в течение 3 часов, охлаждают до комнатной температуры и выливают при перемешивании в 200 мл смеси воды со льдом. Образовавшийся осадок отделяют фильтрованием, промывают водой и сушат, получая смесь соединения I-184 в виде оранжевого твердого вещества (1,6 г, 90%). 1H-ЯМР (ДМСО-d6 ).
(E)-3-(1-Метил-2,3-диоксо-2,3-дигидро-1Н-индол-4-ил)акриловая кислота, I-185. Соединение I-185 получают с 90% выходом из I-183 аналогично получению соединения I-184. 1H-ЯМР (ДМСО-d6).
[(E)-3-(2,3-Диоксо-2,3-дигидро-1Н-индол-4-ил)акрилоил]амид 4,5-дихлортиофен-2-сульфоновой кислоты, I-186. К раствору кислоты I-184 (1,4 г, 6,06 ммоль) в CH2Cl2 (28 мл) последовательно при комнатной температуре добавляют 4-диметиламинопиридин (1,48 г, 12,11 ммоль), 3,4-дихлортиофенсульфонамид (1,56 г, 6,67 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (2,32 г, 12,11 ммоль) и полученную смесь перемешивают в течение 20 часов. После удаления растворителя остаток растворяют в уксусной кислоте (l г/1 мл) и разбавляют водой (10 мл). Образующийся осадок отделяют фильтрованием, промывают водой и сушат в вакууме, получая соединение I-186 (2,1 г, 78%). 1H-ЯМР (ДМСО-d6 ).
[(E)-3-(1-Метил-2,3-диоксо-2,3-дигидро-1Н-индол-4-ил)акрилоил]амид 4,5-дихлортиофен-2-сульфоновой кислоты, I-187. Соединение I-187 получают из I-185 аналогично получению соединения I-186, c 79% выходом. 1H-ЯМР (ДМСО-d6).
Общая методика получения аналогов «3-аминооксииндола».
Общая методика A-15. Смесь соединения I-187 (1 экв.), анилина (1,2 экв.) и моногидрата p-TsA (0,05 экв.) в MeOH нагревают до 65°С, выдерживают при данной температуре в течение 6 часов и затем дают возможность смеси охладиться до комнатной температуры. После концентрирования в вакууме остаток очищают хроматографией на силикагеле, получая имин. К раствору имина (1 экв.) в ледяной уксусной кислоте добавляют цианоборогидрид натрия (3 экв.) при комнатной температуре и перемешивание продолжают в течение 2 часов. Полученную реакционную смесь гасят водой. Смесь перемешивают в течение 30 минут при комнатной температуре. Осадок отделяют фильтрованием, промывают водой и сушат, получая 3-амино-2-оксииндол-производные.
Общая методика A-16. Смесь соединения I-187 (1 экв.), анилина (1,2 экв.) и моногидрата p-TsA (0,05 экв.) в толуоле кипятят с обратным холодильником в течение 16 часов, затем дают возможность смеси охладиться до комнатной температуры. Реакционную смесь разбавляют метилeнхлоридом. К полученной смеси несколькими порциями добавляют Na(CN)BH3. После перемешивания при комнатной температуре в течение 2 часов реакцию гасят водой. Образовавшийся осадок отделяют фильтрованием, промывают водой и сушат, получая 3-амино-2-оксииндол-производные.
Синтез соединения P244. В соответствии с методикой A-15, соединение I-186 подвергают взаимодействию с 2-нафтиламином с получением соединения P244 с 76% выходом. 1H-ЯМР (ДМСО-d6) 6,78 (д, J=16,0 Гц, 1H), 6,94 (д, J=8,0 Гц, 1H), 7,22 (дд, J=9,0, 2,5 Гц, 1H), 7,37 (д, J=29,0 Гц, 1H), 7,4-7,5 (м, 4H), 7,80-7,90 (м, 5H), 8,66 (д, J=16,0 Гц, 1H), 11,00 (с, 1H) ЖХ/МС (78%) (ESI-) Вычислено: 556,46 m/z; найдено 556,0 m/z.
Пример 283. Получение соединения P241.
В соответствии с методикой A-16, соединение I-186 подвергают взаимодействию с 2-нафтиламином с получением соединения P241 с 82% выходом. 1H-ЯМР (ДМСО-d6) 5,40 (с, 1H), 6,54 (д, J=16 Гц, 1H), 6,96 (дд, J=20,5, 8,0 Гц, 2H), 7,12 (т, J=7,5 Гц, 1H), 7,21-7,37 (м, 5H), 7,61 (дд, J=26,5, 8,5 Гц, 2H), 7,85 (с, 1H), 7,94 (д, J=16,5 Гц, 1H), 10,74 (с, 1H) ЖХ/МС (82%) (ESI-) Вычислено: 558,46 m/z; найдено 558,0 m/z.
Пример 284. Получение соединения P245.
В соответствии с методикой A-15, соединение I-186 подвергают взаимодействию с 2-нафтиламином, получая соединение P245 c 71% выходом; ЖХ/МС 94%. 1H-ЯМР (ДМСО- d6) 3,17 (с, 3H), 6,77 (д, J=16 Гц, 1H), 6,93 (д, J=8 Гц, 1H), 7,24 (дд, J=9,0, 2,5 Гц, 1H), 7,40 (д, J=29,0 Гц, 1H), 7,40-7,50 (м, 4H), 7,80-7,90 (м, 5H), 8,68 (д, J=16,0 Гц, 1H), 11,03 (с, 1H) ЖХ/МС (71%) (ESI-) Вычислено: 570,48 m/z; найдено 568,5 m/z.
Пример 285. Получение соединения P242.
В соответствии с методикой A-16, соединение I-187 подвергают взаимодействию с 2-нафтиламином, получая соединение P242 c 94% выходом. 1Н-ЯМР (ДМСО-d6).
Пример 286. Получение соединения P252.
В соответствии с методикой A-16, соединение I-187 подвергают взаимодействию с 2,4-дихлоранилином, получая соединение P252 с 99% выходом. 1H-ЯМР (ДМСО-d6) 3,18 (с, 3H), 5,52 (д, J=8,4 Гц, 1H), 6,18 (с, 1H), 6,51 (д, J=15,6 Гц, 2H), 7,01 (дд, J=8,8, 2,8 Гц, 1H), 7,24 (д, J=8,4 Гц, 1H), 7,29 (д, J=2,4 Гц, 2H), 7,42 (т, J=8 Гц, 1H), 7,84 (д, J=16 Гц, 1H), 7,88 (с, 1H) ЖХ/МС (99%) (APCI-) Вычислено: 589 m/z; найдено 588 m/z.
Пример 287. Получение соединения P268.
В соответствии с методикой A-16, соединение I-186 подвергают взаимодействию с 2,4-дихлоранилином, получая соединение P268 с 99% выходом. 1Н-ЯМР (ДМСО-d6).
Пример 288. Получение соединения P270.
В соответствии с методикой A-16, соединение I-186 подвергают взаимодействию с 3,4-дифторанилином, получая соединение P270 с 99% выходом. 1H-ЯМР (ДМСО-d6) 3,15 (с, 3H), 5,48 (д, J=8,0 Гц, 1H), 6,24 (уш. д, J=8,8 Гц, 1H), 6,48 (м, 1H), 6,57 (д, J=16 Гц, 1H), 6,71 (уш. д, J=9,6 Гц, 1H), 7,07 (кв., J=9,2 Гц, 1H), 7,14 (д, J=8,0 Гц, 1H), 7,30 (д, J=8,0 Гц, 1H), 7,45 (т, J=8,0 Гц, 1H), 7,83 (д, J=16 Гц, 1H), 7,87 (с, 1H). ЖХ/МС (95%) (ESI-). Вычислено: 558,41 m/z; найдено 557 m/z.
Пример 289. Получение соединения P247.
[(E)-3-(3-Амино-1-метил-2-оксо-2,3-дигидро-1Н-индол-4-ил)акрилоил]амид 4,5-дихлортиофен-2-сульфоновой кислоты, I-188. К раствору изатин-производного I-187 (350 мг, 0,8 ммоль) в MeOH (8 мл) при ~5°C добавляют NH4OH (1 мл). Реакционную смесь перемешивают при комнатной температуре в течение 16 часов и затем разбавляют 50 мл воды. Образовавшийся осадок отделяют фильтрованием, сушат на воздухе и суспендируют в ледяной уксусной кислоте (8 мл). К полученной смеси добавляют цианоборогидрид натрия (150 мг) при комнатной температуре и перемешивание продолжают в течение 2 часов. Полученную реакционную смесь гасят водой. Смесь перемешивают в течение 30 минут при комнатной температуре. Осадок отделяют фильтрованием, промывают водой и сушат, получая амино-производное I-188 (300 мг, 85%). 1Н-ЯМР; МС.
Синтез соединения P247. К раствору амина I-188 (45 мг, 0,1 ммоль) и DMAP (45 мг) в CH2Cl2 (5 мл) добавляют нафталин-2-карбонилхлорид (21 мг, 0,12 ммоль) при 0°C. Реакционную смесь перемешивают при комнатной температуре в течение 24 часов. После удаления растворителя остаток растворяют в уксусной кислоте (3 мл) и раствор разбавляют ледяной водой (50 мл). Образовавшийся осадок отделяют фильтрованием, промывают водой и сушат, получая неочищенный продукт (60 мг), который очищают хроматографией на силикагеле, получая соединение P247 (25 мг, 40%) в виде твердого, не совсем белого вещества. 1H-ЯМР (500 МГц, ДМСО-d6); 3,20 (с, 3H), 6,40 (с, 1H), 6,46 (д, J=16,0 Гц, 1H), 7,04 (д, J=7,5 Гц, 1H), 7,31-7,39 (м, 2H), 7,52 (д, J=16,0 Гц, 1H), 7,59 (т, J=7,0 Гц, 1H), 7,67 (т, J=7,0 Гц, 1H), 7,92 (д, J=7,5 Гц, 1H), 7,99 (д, J=9,0 Гц, 1H), 8,14 (д, J=8,0 Гц, 1H), 8,63 (с, 1H). ЖХ/МС (97%) ESI- Вычислено: 600,5 m/z; найдено: 599,3 m/z (M-1).
Пример 290. Получение соединения P265.
[(E)-3-(3-амино-1-метил-2-оксо-2,3-дигидро-1Н-индол-4-ил)акрилоил]амид 4,5-дихлортиофен-2-сульфоновой кислоты, I-188 (0,110 г, 0,25 ммоль) растворяют в хлороформе (3 мл). В раствор добавляют ледяную уксусную кислоту (0,043 мл, 0,75 ммоль), затем изоамилнитрил (0,34 мл, 0,2525 ммоль). Смесь перемешивают при 60°C в течение 30 минут, затем дают возможность смеси охладиться до комнатной температуры, выдерживают при комнатной температуре в течение 1 часа, затем концентрируют в атмосфере азота, промывают (3×) водой декантированием (без отходов) и сушат в атмосфере азота, получая соль диазония. Полученную соль используют на следующей стадии без дополнительной очистки. ИК (чистый) показывает значительную абсорбцию при 2100 см-1.
2-Нафтол (0,0540 г, 0,375 ммоль) растворяют в 3 мл сухого толуола. К раствору добавляют тетраацетат диродия (II) (0,0055 г, 0,0125 ммоль) и смесь нагревают в атмосфере азота до 65°C, в это время к смеси добавляют раствор 4-[(E)-3-(4,5-дихлортиофен-2-сульфониламино)-3-оксопропенил]-1-метил-2-оксо-2,3-дигидро-1Н-индол-3-диазония (0,250 ммоль) в 1 мл безводного толуола. Полученную смесь перемешивают в течение 30 минут при температуре более 65°C, затем смесь охлаждают до комнатной температуры и выдерживают при комнатной температуре в течение 1 часа, концентрируют и хроматографируют на силикагеле (элюирование с градиентом, MeOH/CH2Cl2). Фракция 81 содержит 6,5 мг {(E)-3-[1-метил-3-(нафталин-2-илокси)-2-оксо-2,3-дигидро-1Н-индол-4-ил]акрилоил}амида 4,5-дихлортиофен-2-сульфоновой кислоты, P265. 1H-ЯМР (500 МГц, ДМСО-d6) 3,30 (с, 3H), 5,75 (с, 1H), 6,23 (д, J=16,0 Гц, 1H), 6,57 (д, J=8,4 Гц, 1H), 6,69 (д, J=16,0 Гц, 1H), 6,9-7,8 (м, 9H), 8,05 (д, J=16,0 Гц, 1H), 10,44 (с, 1H), ЖХ/МС(8,3%) ESI- Вычислено: M=573,5; найдено: 573,3 m/z.
Пример 291. Получение соединения P284.
Синтез 2-фтор-6-(нафталин-2-илокси)бензонитрила, I-189. В емкость объемом 20 мл, снабженную магнитной мешалкой, добавляют гидрид натрия (103,7 мг, 4,32 ммоль, 60% в минеральном масле), затем безводный ДМФА (2 мл), что приводит к выделению газа. К полученной смеси при перемешивании небольшими порциями в течение 5 минут добавляют 2-нафтол (623 мг, 4,32 ммоль) в виде твердого вещества. Смесь перемешивают при комнатной температуре в течение 3 минут и затем одной порцией добавляют 2,6-дифторбензонитрил (601 мг, 4,32 ммоль) в безводном ДМФА. Реакционную смесь нагревают на масляной бане до 100°C и выдерживают при этой температуре в течение 2 часов, затем дают возможность смеси охладиться до комнатной температуры. Реакционную смесь разбавляют водой (15 мл) и экстрагируют EtOAc (2×20 мл). Объединенные органические экстракты промывают водой (3×20 мл), сушат (Na2SО4), фильтруют и концентрируют, получая соединение I-189 в виде коричневого твердого вещества (1,216 г), которое является достаточно чистым для использования на следующей стадии (содержит 2-нафтол). 1H-ЯМР.
Синтез 1-метил-4-(нафталин-2-илокси)-1Н-индазол-3-иламина, I-190. В емкость объемом 20 мл, снабженную магнитной мешалкой, загружают 2-фтор-6-(нафталин-2-илокси)бензонитрил (966 мг, 3,67 ммоль) и безводный N,N-диметилацетамид и смесь перемешивают до получения раствора. К смеси добавляют метилгидразин (390 мкл, 7,34 ммоль), емкость закрывают, реакционную смесь помещают на масляную баню, нагревают до 120°C и перемешивают при данной температуре в течение ночи. Реакционную смесь нагревают до 130°C и выдерживают при этой температуре в течение дополнительных 3 часов. Охлажденную реакционную смесь разбавляют водой (20 мл) и экстрагируют EtOAc (2×30 мл). Объединенные органические фракции промывают водой (3×15 мл), сушат (Na2SO4), фильтруют и концентрируют, получая 1,03 г продукта в виде желтовато-коричневого твердого вещества. Остаток очищают флэш-хроматографией на силикагеле (120 г), используя сначала CH2Cl2 и затем смесь CH2Cl2/EtОAc (9:1) в качестве элюента и получая 770 мг соединения I-190 в виде не совсем белого твердого вещества. Полученный продукт имеет достаточную степень чистоты для использования в последующих реакциях. 1H-ЯМР, MС.
Синтез [1-метил-4-(нафталин-2-илокси)-1Н-индазол-3-ил]амида 4,5-дихлортиофен-2-сульфоновой кислоты, P284. В емкость объемом 5 мл, снабженную магнитной мешалкой, добавляют соединение I-190 (37 мг, 0,13 ммоль), безводный CH2Cl2 (1 мл), N,N-диметиламинопиридин (17,2 мг, 0,141 ммоль) и 4,5-дихлортиофен-2-сульфонилхлорид. Реакционную смесь перемешивают при комнатной температуре в течение 36 часов, образовавшийся твердый осадок отфильтровывают и промывают CH2Cl2, получая после сушки 18,5 мг соединения P284 в виде белого твердого вещества. 1H-ЯМР (400 МГц, ДМСО-d6) 4,16 (с, 3H), 6,46 (д, J=7,6 Гц, 1H), 7,08 (дд, J=8,8, 2,4 Гц, 1H), 7,41-7,35 (м, 2H), 7,47 (д, J=8,0 Гц, 1H), 7,53 (м, 2H), 7,87 (уш. д, J=8,4 Гц, 1H), 8,01-7,95 (м, 3H). МС (ESI+) Вычислено: (M+H) 504,4; найдено: 504,4.
Пример 292. Получение соединения P056.
Общую методику (A-2) используют для алкилирования [(E)-3-(3-метил-1H-индол-7-ил)акрилоил]амида тиофен-2-сульфоновой кислоты (I-8) с использованием 2,4-дихлорбензилбромида с получением соединения P056. 1H-ЯМР (500 МГц, ДМСО-d6). МС (ESI-): 505,1 (M-1), ЖХ-МС: 96% чистота
Пример 293, Получение соединения P347.
В соответствии с общей методикой A-8, акриловую кислоту I-137B подвергают взаимодействию с 4,5-дихлортиофен-2-сульфонамидом с получением соединения P347. Структуру подтверждают 1H-ЯМР.
Пример 294. Получение соединения P350.
Синтез 7-бром-1-(2,4-дихлорбензил)-5-фтор-3-метил-1H-индола, I-191. NaH (60% в масле, 526 мг, 13,15 ммоль, 1,5 экв.) добавляют к раствору 7-бром-5-фтор-3-метил-1H-индола, [полученному аналогично соединению I-30 в соответствии с методикой публикации Dobbs, A., J. Org. Chem., 66, 638-641 (2001)], (2 г, 8,77 ммоль, 1 экв.) в ДМФА (30 мл) при -10°C. Реакционную смесь нагревают до комнатной температуры и перемешивают при комнатной температуре в течение 30 минут. Раствор 2,4-дихлорбензилхлорида (2,06 г, 10,52 ммоль, 1,2 экв.) в ДМФА (10 мл) осторожно добавляют в течение 2,5 минут при -10°C. Реакционной смеси дают возможность нагреться до комнатной температуры и смесь перемешивают в течение 1 часа. Реакционную смесь при перемешивании выливают в 10% HCl/вода/эфир (1:1:2, 40 мл) смесь и водный слой экстрагируют эфиром (2×10 мл). Объединенные органические слои промывают водой (3×75 мл), насыщенным раствором соли (75 мл), сушат над MgSО4, фильтруют и концентрируют с получением неочищенного продукта в виде коричневого твердого вещества. К неочищенному продукту добавляют эфир (4 мл), смесь охлаждают до -78°C и фильтруют, получая соединение I-191 (2,49 г, 73%) в виде твердого, не совсем белого вещества. 1Н-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез этилового эфира 1-(2,4-дихлорбензил)-5-фтор-3-метил-1Н-индол-7-карбоновой кислоты, I-192. К раствору соединения I-191 (400 мг, 1,03 ммоль, 1 экв.) в эфире (7 мл) при -78°C в атмосфере Ar медленно в течение 7 минут добавляют BuLi (1,6М в гексанах, 0,97 мл, 1,55 ммоль, 1,5 экв). Реакционную смесь перемешивают при -78°C в течение дополнительных 30 минут. К реакционной смеси медленно добавляют этилхлорформиат (0,2 мл, 2,07 ммоль, 2 экв.), реакционной смеси дают возможность нагреться до комнатной температуры и смесь перемешивают при комнатной температуры в течение 30 минут. Реакционную смесь гасят добавлением 10% водной HCl (5 мл). Органический слой промывают водой (2×10 мл), насыщенным раствором соли (10 мл), сушат над MgSO4, фильтруют и концентрируют с получением соединения I-192 мг (98%) в виде коричневого масла. МС (AP+): 380,382 (М+1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез 1-(2,4-дихлорбензил)-5-фтор-3-метил-1Н-индол-7-карбоновой кислоты, I-193. 2н. водный раствор NaOH (2,5 мл, 5 ммоль, 5 экв.) добавляют к раствору сложного эфира I-192 (381 мг, 1 ммоль, 1 экв.) в смеси MeOH/ТГФ (1:1,4 мл). Реакционную смесь нагревают до 75°C и перемешивают при этой температуры в течение 1 часа. Реакционную смесь концентрируют, охлаждают до -70°C, гасят добавлением 10% водного раствора HCl (6 мл) и экстрагируют EtOAc (4 мл). Органический слой промывают водой (2×4 мл), насыщенным раствором соли (4 мл), сушат над MgSO4, фильтруют и концентрируют. Полученный остаток растирают в гексане (2×2 мл) и сушат, получая соединение I-193 (262 мг, 74%) в виде твердого, не совсем белого вещества. Структуру подтверждают 1H-ЯМР (500 МГц, CDCl3).
Синтез соединения P350. 4,5-Дихлор-2-тиофенсульфонамид (23,2 мг, 0,1 ммоль, 1 экв.) добавляют при комнатной температуре к раствору соединения I-193 (35 мг, 0,1 ммоль, 1 экв.) в DMAP (24,5 мг, 0,2 ммоль, 2 экв.) и безводном CH2Cl2 (0,5 мл) и затем добавляют EDCI (38 мг, 0,2 ммоль, 2 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и затем оставляют при 0°C на 2 дня. Реакционную смесь нагревают до комнатной температуры, гасят добавлением 10% HCl (водн.) (1 мл) и экстрагируют EtOAc (1 мл). Органический слой промывают смесью воды и насыщенного раствора соли (1:1, 3×1 мл), сушат над MgSО4, фильтруют и концентрируют с получением неочищенного продукта (51 мг) в виде светло-коричневого твердого вещества. Неочищенный продукт растирают в гексане (2×1 мл), затем в смеси гексан-MeOH (30:1, 1 мл), получая соединение P350 (45 мг, 79%) в виде твердого не совсем белого вещества. ЖХ-МС (95%): ESI- Вычислено: 564 m/z; найдено: 564. 1H-ЯМР (ДМСО-d6) 2,24 (д, J=0,8 Гц, 3H), 5,39 (с, 2H), 6,01 (д, J=8,4 Гц, 1H), 7,15 (дд, J=8,4, 2,4 Гц, 1H), 7,28 (дд, J=8,4, 2,4 Гц, 1H), 7,29 (с, 1H), 7,25 (д, J=2,4 Гц, 1H), 7,60-7,64 (м, 1H), 7,64 (с, 1H), 7,95 (уш. с, 1H).
Пример 295. Получение соединения P417.
3,4-Дифторбензолсульфонамид (19,3 мг, 0,1 ммоль, 1 экв.) при комнатной температуре добавляют к раствору соединения I-193 (35 мг, 0,1 ммоль, 1 экв.) и DMAP (24,5 мг, 0,2 ммоль, 2 экв.) в безводном CH2Cl2 (Aldrich, 0,5 мл), затем добавляют EDCI (38 мг, 0,2 ммоль, 2 экв.) аналогично методике, описанной для получения соединения P350, с получением соединения P417 (42 мг, 79%) в виде бежевого твердого вещества. ЖХ-МС (98%): ESI- Вычислено: 526 m/z; найдено: 526. lН-ЯМР (ДМСО-d6) 2,22 (д, J=1,2 Гц, 3H), 5,22 (с, 2H), 5,96 (д, J=8,4 Гц, 1H), 7,14 (дд, J=8,2, 1,8 Гц, 1H), 7,24 (с, 1H), 7,29 (дд, J=9,6, 2,4 Гц, 1H), 7,36 (д, J=2,4 Гц, 1H), 7,54 (уш. с, 1H), 7,61-7,66 (м, 2H), 7,67-7,70 (м, 1H), 7,76-7,81 (м, 1H).
Пример 296. Получение соединения P354.
Синтез этилового эфира [1-(2,4-дихлорбензил)-5-фтор-3-метил-1Н-индол-7-ил]оксоуксусной кислоты, I-194. Раствор n-BuLi (2,5М в гексанах, 0,31 мл, 0,78 ммоль, 1,5 экв.) медленно добавляют при комнатной температуре к раствору бромида I-191 (200 мг, 0,517 ммоль, 1 экв.) в безводном Et2О (4 мл) при -78°C в атмосфере аргона. Реакционную смесь перемешивают при -78°C в течение 20 минут. К смеси медленно в течение 2 минут добавляют этилоксалат (0,146 мл, 1,034 ммоль, 2,0 экв.) при -78°C, затем реакционную смесь нагревают до 0°C на бане со смесью воды со льдом. Реакционную смесь нагревают до комнатной температуры, гасят добавлением 10% HCl (водн.) (3 мл) и экстрагируют эфиром (3 мл). Органический слой промывают водой (2×6 мл), насыщенным раствором соли (6 мл), сушат над MgSO4, фильтруют и концентрируют с получением соединения I-194 (215 мг, 100%) в виде оранжевого масла. 1Н-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез [1-(2,4-дихлорбензил)-5-фтор-3-метил-1Н-индол-7-ил]оксоуксусной кислоты, I-195. Водный раствор 2н. NaOH (0,29 мл, 0,58 ммоль, 1,1 экв.) медленно добавляют в течение 2 минут к раствору соединения I-194 (215 мг, 0,53 ммоль, 1 экв.) в смеси ТГФ-MeOH (1:1, 4 мл) при 0°C. Реакционную смесь перемешивают при 0°C в течение 30 минут и затем нагревают до комнатной температуры. К смеси одной порцией добавляют 2н. водный NaOH (0,24 мл, 0,48 ммоль, 0,9 экв.) и реакционную смесь перемешивают в течение 15 минут. Реакционную смесь концентрируют, к остатку добавляют 10% HCl (водная, 1 мл), затем полученную смесь экстрагируют эфиром (2 мл). Органический слой промывают водой (3×2 мл), насыщенным раствором соли (2 мл), сушат над MgSO4, фильтруют и концентрируют с получением соединения I-195 (141 мг, 95%) в виде желтого твердого вещества. ЖХ-МС (98%): ESI- Вычислено: 380,2 m/z; найдено: 380,2. 1Н-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез соединения P354. К раствору соединения I-195 (40 мг, 0,11 ммоль, 1 экв.) и DMAP (26 мг, 0,2 ммоль, 2 экв.) в безводном CH2Cl2 (0,6 мл) при комнатной температуре добавляют 2,4,5-трифторбензолсульфонамид (22 мг, 0,11 ммоль, 1 экв.), затем EDCI (40 мг, 0,2 ммоль, 2 экв.) по методике, аналогично описанной для соединения P350, получая неочищенный продукт в виде желтого твердого вещества. Неочищенный продукт растирают с MTBE (2 мл), кипятят в течение одной минуты, раствор фильтруют и промывают MTBE (2×0,5 мл), гексаном (2 мл) при комнатной температуре, получая соединение P354 (28 мг, 46%) в виде желтого твердого вещества. ЖХ-МС (100%): AP- Вычислено: 572 m/z; найдено: 572. 1H-ЯМР (ДМСО-d6) 2,27 (д, J=0,8 Гц, 3H), 5,57 (с, 2H), 6,14 (д, J=8,4 Гц, 1H), 7,15-7,19 (м, 2H), 7,35 (с, 1H), 7,54 (д, J=2,0 Гц, 1H), 7,73 (дд, J=8,6, 2,6 Гц, 1H), 7,76-7,82 (м, 2H).
Пример 297. Получение соединения P351.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 4-фторбензолсульфонамидом с получением соединения P351. Структуру подтверждают 1Н ЯМР.
Пример 298. Получение соединения P352.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 2-хлорбензолсульфонамидом с получением соединения P352. Структуру подтверждают 1Н-ЯМР.
Пример 299. Получение соединения P353.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 3-хлорбензолсульфонамидом с получением соединения P353. Структуру подтверждают 1Н-ЯМР.
Пример 300. Получение соединения P355.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 3,4-дихлорбензолсульфонамидом с получением соединения P355. Структуру подтверждают 1Н-ЯМР.
Пример 301. Получение соединения P356.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 2,4-дихлорбензолсульфонамидом с получением соединения P356. Структуру подтверждают 1Н-ЯМР.
Пример 302. Получение соединения P357.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 3,5-дихлорбензолсульфонамидом с получением соединения P357. Структуру подтверждают 1Н-ЯМР.
Пример 303. Получение соединения P358.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 2,4-дифторбензолсульфонамидом с получением соединения P358. Структуру подтверждают 1Н-ЯМР.
Пример 304. Получение соединения P359.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 2,5-дифторбензолсульфонамидом с получением соединения P359. Структуру подтверждают 1Н-ЯМР.
Пример 305. Получение соединения P360.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 2,6-дифторбензолсульфонамидом с получением соединения P360. Структуру подтверждают 1Н-ЯМР.
Пример 306. Получение соединения P361.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 3,5-дифторбензолсульфонамидом с получением соединения P361. Структуру подтверждают 1Н-ЯМР.
Пример 307. Получение соединения P363.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 3-фторбензолсульфонамидом с получением соединения P363. Структуру подтверждают 1Н-ЯМР.
Пример 308. Получение соединения P364.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 2-фторбензолсульфонамидом с получением соединения P364. Структуру подтверждают 1Н-ЯМР.
Пример 309. Получение соединения P365.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 4-хлорбензолсульфонамидом с получением соединения P365. Структуру подтверждают 1Н-ЯМР.
Пример 310. Получение соединения P366.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 4-метоксибензолсульфонамидом с получением соединения P366. Структуру подтверждают 1Н-ЯМР.
Пример 311. Получение соединения P373.
В соответствии с общей методикой A-8, акриловую кислоту I-137A подвергают взаимодействию с 2,3,4,5,6-пентафторбензолсульфонамидом с получением соединения P373. Структуру подтверждают 1Н-ЯМР.
Пример 312. Получение соединения P367.
Синтез 2,4,5-трифтор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]бензолсульфонамида, I-196. В емкость объемом 8 мл, снабженную мешалкой, при комнатной температуре загружают кислоту I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 2,4,5-трифторбензолсульфонамид (346 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и EDCI (314 мг, 1,64 ммоль). Емкость герметично закрывают крышкой и реакцию проводят в течение 17 часов при комнатной температуре. Содержимое емкости переносят в делительную воронку и разбавляют CH2Cl2 (20 мл). Затем в воронку добавляют воду (20 мл) и 1M HCl (20 мл) и органическую часть удаляют. Водный слой экстрагируют EtOAc (2×20 мл). Органические части объединяют, сушат (MgSO4) и концентрируют, получая коричневое масло. Неочищенный продукт растирают с охлажденным льдом CH2Cl2 (2 мл), получая нерастворимое твердое вещество. Твердый продукт отделяют фильтрованием с подсосом и промывают охлажденным льдом CH2Cl2 (4 мл), получая 135 мг соединения I-196 в виде желтого твердого вещества (24%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС (87%).
Общая методика (A-17) алкилирования ацилсульфонамидов.
К раствору ацилсульфонамида в ТГФ при комнатной температуре добавляют 3 экв. трет-бутоксида калия. К смеси добавляют соответствующий арилгалогенид и смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют EtOAc и подкисляют 10% HCl. Органические фракции промывают водой (3×), насыщенным раствором соли и сушат над Na2SО4. Раствор фильтруют, концентрируют и остаток очищают растиранием с дихлорметаном при 40°C или хроматографией на силикагеле, используя смесь метанол/дихлорметан в качестве элюента.
Синтез соединения P367. в соответствии с общей методикой (A-17), ацилсульфонамид I-196 алкилируют 3-метоксибензилбромидом с получением соединения P367 (30%). ЖХ/МС (95%) ESI- Вычислено: 532,5 m/z; найдено: 531,5 m/z. 1H-ЯМР(ДМСО-d6).
Пример 313. Получение соединения P368.
Синтез 3,4-дифтор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]бензолсульфонамида, I-197. В соответствии с методикой, описанной для синтеза соединения I-196, соединение I-34 подвергают взаимодействию с 3,4-дифторбензолсульфонамидом, получая соединение I-197. После фильтрования выделяют 223 мг соединения I-197 в виде желтого твердого вещества (41%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС (99%).
В соответствии с общей методикой (A-17), ацилсульфонамид I-197 алкилируют 3-метоксибензилбромидом с получением соединения P368 (19%). ЖХ/МС (97%) ESI- Вычислено: 514,5 m/z; найдено: 513,7 m/z. 1H-ЯМР(ДМСО-d6).
Пример 314. Получение соединения P369.
В соответствии с общей методикой (A-17), ацилсульфонамид I-197 алкилируют 4-(хлорметил)-3,5-диметилизоксазолом с получением соединения P369 (31%). ЖХ/МС (92%) ESI- Вычислено: 503,5 m/z; найдено: 502,4 m/z. 1H-ЯМР(ДМСО-d6).
Пример 315. Получение соединения P370.
В соответствии с общей методикой (A-17), ацилсульфонамид I-196 алкилируют 4-(хлорметил)-3,5-диметилизоксазолом с получением соединения P370 (18%). ЖХ/МС (96%) ESI- Вычислено: 521,5 m/z; найдено: 520,6 m/z. 1H-ЯМР (ДМСО-d6) 1,68 (с, 3H), 1,75 (с, 3H), 2,21 (с, 3H), 5,29 (с, 2H), 6,40 (д, J=15,6 Гц, 2H), 7,09 (дд, J=10,4, 2,4 Гц, 1H), 7,21 (с, 1H), 7,44 (дд, J=8,8, 2,4 Гц, 1H), 7,93 (ддд, J=10,0, 10,0, 6,0 Гц, 1H), 8,02 (д, J=14,8 Гц, 1H), 8,03 (дд, J=18,0, 6,4 Гц, 1H), 12,8 (с, 1H).
Пример 316. Получение соединения P374.
В соответствии с общей методикой (A-17), ацилсульфонамид I-197 алкилируют 3,5-диметоксибензилбромидом с получением соединения P374 (20%). ЖХ/МС (97%) ESI- Вычислено: 544,6 m/z; найдено: 543,6 m/z. 1H-ЯМР(ДМСО-d6)
Пример 317. Получение соединения P375.
В соответствии с общей методикой (A-17), ацилсульфонамид I-196 алкилируют 3,5-диметоксибензилбромидом с получением соединения P375 (37%). ЖХ/МС (98%) ESI- Вычислено: 562,5 m/z; найдено: 561,5 m/z. 1Н-ЯМР(ДМСО-d6).
Пример 318. Получение соединения P378.
Синтез (5-фтор-3-метил-7-(нафталин-2-илокси)-1Н-индола (I-198A). Общая методика A-18: В емкость объемом 40 мл, снабженную стержневой мешалкой, при комнатной температуре загружают 7-бром-5-фтор-3-метил-1Н-индол [полученный аналогично соединению I-30 в соответствии с методикой, приведенной в публикации Dobbs, A., J. Org. Chem., 66, 638-641 (2001)] (1,10 г, 4,78 ммоль), безводный диоксан (9,5 мл), 2-нафтол (1,03 г, 7,17 ммоль), CuI (91,0 мг, 0,478 ммоль), гидрохлорид N,N-диметилглицина (200 мг, 1,43 ммоль) и Cs2CO3 (3,12 г, 9,56 ммоль). Смесь дегазируют аргоном в течение 15 минут, герметично закрывают крышкой, помещают на масляную баню, нагревают до 100°C и выдерживают при этой температуре в течение 65 часов. Реакцию гасят водой (50 мл) и экстрагируют EtOAc (3×50 мл). Органические фракции объединяют, промывают водой (2×50 мл), насыщенным раствором соли (50 мл), сушат (MgSO4) и концентрируют, получая темно-коричневое масло. Масло очищают колоночной хроматографией на силикагеле, используя систему растворителей 5% EtOAc/гексаны в качестве элюента и получая 791 мг соединения I-198A в виде желтого твердого вещества (57%). 1H-ЯМР (400 МГц, CDCl3).
Синтез 3-[5-фтор-3-метил-7-(нафталин-2-илокси)индол-1-ил]пропионовой кислоты (I-200A): Общая методика A-19. В емкость объемом 18 мл, снабженную стержневой мешалкой, при комнатной температуре загружают соединение Ib-1 (500 мг, 1,72 ммоль), метилакрилат (3,10 мл, 34,4 ммоль) и DBU (257 мкл, 1,72 ммоль). Емкость дегазируют N2 в течение 1 минуты, герметично закрывают крышкой и реакцию проводят в течение 18 часов при комнатной температуре. Реакцию гасят 1M водным раствором HCl (8 мл) и полученную смесь перемешивают в течение 10 минут. После переноса смеси в делительную воронку к смеси добавляют дополнительное количество 1M водного раствора HCl (50 мл) и смесь экстрагируют CH2Cl2 (2×100 мл). Органические фракции объединяют, промывают водой (60 мл) и насыщенным раствором соли (60 мл), сушат (MgSО4) и концентрируют, получая 749 мг соединения I-199A в виде желтого масла. В емкость объемом 40 мл, снабженную стержневой мешалкой, при комнатной температуре загружают неочищенное соединение 1b-2 (600 мг, 1,59 ммоль), ТГФ (7,40 мл), MeOH (3,70 мл) и 50% водный раствор NaOH (3,34 мл, 1,67 ммоль). По истечении 10 минут реакцию гасят насыщенным раствором NaHCO3 (75 мл) и экстрагируют Et2О (100 мл). Слой Et2О промывают насыщенным раствором NaHCО3 (2×75 мл). Водные фракции объединяют, подкисляют 3M HCl до pH=1 и экстрагируют CH2Cl2 (3×80 мл). Органические фракции объединяют, промывают водой (100 мл), насыщенным раствором соли (100 мл), сушат (MgSО4) и концентрируют, получая 338 мг соединения I-200A в виде оранжевого масла (54% выход: из соединения I-198A до I-200A). 1H-ЯМР (400 МГц, CDCl3).
Синтез соединения P378. 2,4,5-Трифтор-N-{3-[5-фтор-3-метил-7-(нафталин-2-илокси)индол-1-ил]пропионил}бензолсульфонамида. Общая методика A-20. В емкость объемом 8 мл, снабженную стержневой мешалкой, загружают соединение I-200A (65,0 мг, 0,179 ммоль), CH2Cl2 (1,2 мл), 2,4,5-трифторфенилсульфонамид (45,4 мг, 0,215 ммоль), DMAP (52,5 мг, 0,430 ммоль) и EDCI (85,9 мг, 0,448 ммоль). Смесь перемешивают при комнатной температуре в течение 18 часов. Реакцию гасят 1M водным раствором HCl (3 мл) и полученную смесь перемешивают в течение 10 минут. Смесь переносят в делительную воронку, добавляют 1M водный раствор HCl (30 мл), затем экстрагируют CH2Cl2 (2×30 мл). Органические части объединяют, промывают водой (35 мл), насыщенным раствором соли (35 мл), сушат (MgSО4) и концентрируют, получая 78,3 мг соединения P378 в виде белого твердого вещества (79%). 1H-ЯМР(400 MГц, CDCl3). ЖХ/МС чистота: 92%.
Пример 319. Получение соединения P380.
Соединение I-200A (65,0 мг, 0,179 ммоль) подвергают взаимодействию с 4,5-дихлортиофен-2-сульфонамидом (49,9 мг, 0,215 ммоль), DMAP (52,5 мг, 0,430 ммоль) и EDCI (85,9 мг, 0,448 ммоль) в безводном CH2Cl2 (1,2 мл) по методике, аналогично описанной для соединения P378. Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей 20% EtOAc/гексаны, 1% AcOH. После концентрирования и азеотропной перегонки с толуолом (3×75 мл) выделяют 64,5 мг соединения P380 в виде белого твердого вещества (63%). 1H-ЯМР (400 МГц, ДМСО-d6) 2,16 (с, 3H), 2,78 (т, J=6,8 Гц, 2H), 4,36 (т, J=6,8 Гц, 2H), 6,54 (дд, J=10,4, 2,4 Гц, 1H), 7,00 (с, 1H), 7,10 (дд, J=9,2, 2,4 Гц, 1H), 7,32 (дд, J=8,8, 2,4 Гц, 1H), 7,38 (д, J=2,4 Гц, 1H), 7,44-7,52 (м, 2H), 7,80 (уш. с, 1H), 7,81 (д, J=9,6 Гц, 1H), 7,93 (д, J=8,0 Гц, 1H), 7,96 (д, J=8,8 Гц, 1H). ВЭЖХ чистота: 99%, МС (ESI-) Вычислено: 576,5 m/z; найдено: 577,1 m/z.
Пример 320. Получение соединения P379.
Соединение I-200A (65,0 мг, 0,179 ммоль) подвергают взаимодействию с 3,4-дифторфенилсульфонамидом (49,9 мг, 0,215 ммоль), DMAP (52,5 мг, 0,430 ммоль) и EDCI (85,9 мг, 0,448 ммоль) в безводном CH2Cl2 (1,2 мл) аналогично методике, описанной для получения P378. Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей 30% EtOAc/гексаны, 1% AcOH. Смесь концентрируют и отгоняют азеотроп с толуолом (3×75 мл), получая 48,3 мг соединения P379 в виде белого твердого вещества (50%). 1H-ЯМР (400 МГц, ДМСО-d6), ВЭЖХ чистота: 97%.
Пример 321. Получение соединения P381.
Соединение I-200A (65,0 мг, 0,179 ммоль) подвергают взаимодействию с 3-хлорфенилсульфонамидом (49,9 мг, 0,215 ммоль), DMAP (52,5 мг, 0,430 ммоль) и EDCI (85,9 мг, 0,448 ммоль) в безводном CH2Cl2 (1,2 мл) по методике, аналогично описанной для соединения P378. Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей: 30% EtOAc/гексаны, 1% AcOH. После концентрирования и азеотропной перегонки с толуолом (3×75 мл) получают 86,7 мг соединения P381 в виде белого твердого вещества (90%). 1H-ЯМР (400 МГц, ДМСО-d6), ВЭЖХ чистота: 95%. МС: Вычислено 537,0; найдено 535,3.
Пример 322. Получение соединения P382.
Синтез 7-(2,4-дихлорфенокси)-5-фтор-3-метил-1Н-индола, I-198B. В соответствии с общей методикой A-18, 7-бром-5-фтор-3-метил-1Н-индол (1,60 г, 7,02 ммоль) подвергают взаимодействию с 2,4-дихлорфенолом (1,72 г, 10,5 ммоль), CuI (134 мг, 0,702 ммоль), гидрохлоридом N,N-диметилглицина (293 мг, 2,10 ммоль) и Cs2CO3 (4,57 г, 14,0 ммоль) в безводном диоксане (14 мл). Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей 5% EtOAc/гексаны и получая 746 мг соединения I-198B в виде желтого масла (34%). 1H-ЯМР (400 МГц, CDCl3).
Синтез 3-[7-(2,4-дихлорфенокси)-5-фтор-3-метилиндол-1-ил]пропионовой кислоты (I-200B): В соответствии с общей методикой A-19, соединение I-198B (690 мг, 2,22 ммоль) подвергают взаимодействию с метилакрилатом (4,00 мл, 44,4 ммоль) и DBU (348 мкл, 2,33 ммоль). После концентрирования получают 882 мг соединения I-199B в виде оранжевого масла. Неочищенный I-199B (880 мг, 2,22 ммоль) омыляют с использованием ТГФ (10,3 мл), MeOH (5,17 мл) и 50% водного раствора NaOH (4,66 мл, 2,33 ммоль). Первая обработка приводит к получению 320 мг соединения I-200B в виде белого твердого вещества. Эфирный слой концентрируют, промывают 1M водным раствором HCl (35 мл) и экстрагируют CH2Cl2 (2×60 мл). Органические части объединяют, промывают водой (40 мл), насыщенным раствором соли (40 мл) и сушат (MgSО4). После концентрирования получают 279 мг соединения I-200B в виде оранжевого твердого вещества. Две части соединения I-200B идентифицируют 1H-ЯМР и объединяют, получая 599 мг соединения I-200B (70% исходя из I-198B до I-200B). 1H-ЯМР (400 МГц, CDCl3).
Синтез соединения P382. Соединение I-200B (70,0 мг, 0,183 ммоль) подвергают взаимодействию с 2,4,5-трифторфенилсульфонамидом (46,5 мг, 0,220 ммоль), DMAP (53,6 мг, 0,439 ммоль), и EDCI (87,8 мг, 0,458 ммоль) в безводном CH2Cl2 (1,2 мл) по методике, аналогично описанной для соединения P378. Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей 30% EtOAc/гексаны, 1% AcOH. После концентрирования и азеотропной перегонки с толуолом (3×75 мл) получают 73,0 мг соединения P382 в виде желтого масла (70%). 1H-ЯМР (400 МГц, ДМСО-d6). Чистота после ВЭЖХ: 85%.
Пример 323. Получение соединения P384.
Соединение I-200B (70,0 мг, 0,183 ммоль) подвергают взаимодействию с 4,5-дихлортиофен-2-сульфонамидом (51,1 мг, 0,220 ммоль), DMAP (53,6 мг, 0,439 ммоль) и EDCI (87,8 мг, 0,458 ммоль) в безводном CH2Cl2 (1,2 мл) аналогично методике получения P378. Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей 30% EtOAc/гексаны, 1% AcOH. После концентрирования и азеотропной перегонки с толуолом (3×75 мл) получают 84,8 мг соединения P384 в виде желтовато-коричневого твердого вещества (78%). 1H-ЯМР (400 МГц, ДМСО-d6). ВЭЖХ чистота: 94%.
Пример 324. Получение соединения P383.
Соединение I-200B (70,0 мг, 0,183 ммоль) подвергают взаимодействию с 3,4-дифторфенилсульфонамидом (42,5 мг, 0,220 ммоль), DMAP (53,6 мг, 0,439 ммоль) и EDCI (87,8 мг, 0,458 ммоль) в безводном CH2Cl2 (1,2 мл) по методике, аналогично описанной для соединения P378. Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей 30% EtOAc/гексаны, 1% AcOH. После концентрирования и азеотропной перегонки с толуолом (3×75 мл) выделяют 66,5 мг соединения P383 в виде белого твердого вещества (65%). 1H-ЯМР (400 МГц, ДМСО-d6) 2,13 (с, 3H), 2,74 (т, J=6,8 Гц, 2H), 4,32 (т, J=6,8 Гц, 2H), 6,38 (дд, J=10,4, 2,4 Гц, 1H), 6,97 (с, 1H), 7,07 (дд, J=9,2, 2,4 Гц, 1H), 7,11 (д, J=8,8 Гц, 1H), 7,44 (дд, J=9,2, 2,8 Гц, 1H), 7,67-7,78 (м, 3H), 7,87 (м, 1H), 12,25 (уш. с, 1H). ВЭЖХ чистота: 88%. МС (ESI-) Вычислено: 556,4 m/z; найдено: 555,1 m/z.
Пример 325. Получение соединения P385.
Соединение I-200B (70,0 мг, 0,183 ммоль) подвергают взаимодействию с 3-хлорфенилсульфонамидом (42,2 мг, 0,220 ммоль), DMAP (53,6 мг, 0,439 ммоль) и EDCI (87,8 мг, 0,458 ммоль) в безводном CH2Cl2 (1,2 мл) по методике, аналогично описанной для соединения P378. Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей 30% EtOAc/гексаны, 1% AcOH. После концентрирования и азеотропной перегонки с толуолом (3×75 мл) выделяют 72,2 мг соединения P385 в виде белого твердого вещества (71%). 1H-ЯМР (400 МГц, ДМСО-d6). ВЭЖХ чистота: 90%.
Пример 326. Получение соединения P386.
Синтез 7-(3,4-дихлорфенокси)-5-фтор-3-метил-1Н-индола (I-198C): В соответствии с общей методикой A-18,7-бром-5-фтор-3-метил-1Н-индол (727 мг, 3,19 ммоль) подвергают взаимодействию с 3,4-дихлорфенолом (779 мг, 4,78 ммоль), CuI (60,8 мг, 0,319 ммоль), гидрохлоридом N,N-диметилглицина (134 мг, 0,957 ммоль) и Cs2CO3 (2,07 г, 6,38 ммоль) в безводном диоксане (6,4 мл). Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей 5% EtOAc/гексаны и получая 506 мг соединения I-198C в виде желтого масла (34%). 1H-ЯМР (400 МГц, CDCl3).
Синтез 3-[7-(3,4-дихлорфенокси)-5-фтор-3-метилиндол-1-ил]пропионовой кислоты (I-200C): В соответствии с общей методикой A-19, соединение I-198C (450 мг, 1,45 ммоль) подвергают взаимодействию с метилакрилатом (2,61 мл, 29,0 ммоль) и DBU (227 мкл, 1,52 ммоль). После концентрирования выделяют 584 мг соединения I-199C в виде оранжевого масла. Неочищенный I-199C (580 мг, 1,46 ммоль) омыляют с использованием ТГФ (6,80 мл), MeOH (3,40 мл) и 50% водного раствора NaOH (3,07 мл, 1,53 ммоль). Эфирный слой упаривают, промывают 1M водным раствором HCl (35 мл) и экстрагируют CH2Cl2 (2×60 мл). Органические части объединяют, промывают водой (40 мл), насыщенным раствором соли (40 мл) и сушат (MgSО4). После концентрирования выделяют 240 мг соединения I-200C в виде желтого масла (43% выход, исходя из I-198C до I-200C). 1H-ЯМР (400 MГц, CDCl3).
Синтез соединения P386. Соединение I-200C (65,0 мг, 0,170 ммоль) подвергают взаимодействию с 2,4,5-трифторфенилсульфонамидом (43,1 мг, 0,204 ммоль), DMAP (49,8 мг, 0,408 ммоль) и EDCI (81,5 мг, 0,425 ммоль) в безводном CH2Cl2 (1,2 мл) по методике, аналогично описанной для соединения P378. Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей 30% EtOAc/гексаны, 1% AcOH. После концентрирования и азеотропной перегонки с толуолом (3×75 мл) выделяют 80,6 мг соединения P386 в виде зеленого твердого вещества (82%). 1H-ЯМР(400 МГц, ДМСО-d6), ВЭЖХ чистота: 93%.
Пример 327. Получение соединения P388.
Соединение I-200C (75,0 мг, 0,196 ммоль) подвергают взаимодействию с 4,5-дихлортиофен-2-сульфонамидом (54,5 мг, 0,235 ммоль), DMAP (57,4 мг, 0,470 ммоль) и EDCI (93,9 мг, 0,490 ммоль) в безводном CH2Cl2 (1,2 мл) по методике, аналогично описанной для соединения P378. Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей 20% EtOAc/гексаны, 1% AcOH. После концентрирования и азеотропной перегонки с толуолом (3×75 мл) выделяют 93,0 мг соединения P388 в виде зеленого твердого вещества (79%). 1H-ЯМР (400 МГц, ДМСО-d6). ВЭЖХ чистота: 93%.
Пример 328. Получение соединения P387.
Соединение I-200C (65,0 мг, 0,170 ммоль) подвергают взаимодействию с 3,4-дифторфенилсульфонамидом (39,4 мг, 0,204 ммоль), DMAP (49,8 мг, 0,408 ммоль) и EDCI (81,5 мг, 0,425 ммоль) в безводном CH2Cl2 (1,2 мл) по методике, аналогично описанной для соединения P378. Неочищенный продукт очищают колоночной хроматографией на силикагеле, используя систему растворителей: 30% EtOAc/гексаны, 1% AcOH. После концентрирования и азеотропной перегонки с толуолом (3×75 мл) выделяют 62,7 мг соединения P387 в виде зеленого твердого вещества (66%). 1H-ЯМР (400 МГц, ДМСО-d6) 2,13 (с, 3H), 2,69 (т, J=6,8 Гц, 2H), 4,27 (т, J=6,8 Гц, 2H), 6,58 (дд, J=10,4, 2,4 Гц, 1H), 6,95 (уш. с, 1H), 7,00 (дд, J=8,8, 2,8 Гц, 1H), 7,10 (дд, J=9,2, 2,4 Гц, 1H), 7,31 (д, J=2,8 Гц, 1H), 7,63 (д, J=8,8 Гц, 1H), 7,65-7,76 (м, 2H), 7,86 (м, 1H), 12,24 (уш. с, 1H). ВЭЖХ чистота: 95%, МС (ESI-) Вычислено: 556,4 m/z; найдено: 556,9 m/z.
Пример 329. Получение соединения P389.
В емкость объемом 8 мл, снабженную стержневой мешалкой, при комнатной температуре загружают соединение I-36 (250 мг, 0,576 ммоль) и безводный ТГФ (2,8 мл). Полученный желтый раствор охлаждают до 0°C на ледяной бане, затем медленно, небольшими порциями добавляют NaH (60%) (69,2 мг, 1,73 ммоль). Раствор из желтого превращается в ярко-красный. Смеси дают возможность нагреться до комнатной температуры, затем снова охлаждают до 0°C, в процессе чего добавляют 2-(бромметил)бензонитрил (192 мг, 0,981 ммоль). Содержимое емкости оставляют для взаимодействия в течение 18 часов. Реакцию осторожно гасят водой (1 мл) и 1M водным раствором HCl (3 мл) и полученную смесь перемешивают в течение 10 минут. Смесь переносят в делительную воронку, промывают водой (15 мл) и 1M водным раствором HCl (15 мл), затем экстрагируют EtOAc (3×40 мл). Органические части объединяют, промывают водой (40 мл) и насыщенным раствором соли (40 мл), сушат (MgSO4) и концентрируют, получая желтый твердый продукт. Неочищенный продукт растирают с охлажденным льдом CH2Cl2 (30 мл) и отделяют фильтрованием с подсосом, затем промывают дополнительным количеством охлажденного льдом CH2Cl2 (2×30 мл). После фильтрования выделяют 193 мг соединения P389 в виде бледно-желтого твердого вещества (61%). 1H-ЯМР (400 МГц, d6-ДМСО). ВЭЖХ чистота: 95%.
Пример 330. Получение соединения P390.
{(E)-3-[1-(3-цианобензил)-5-фтор-3-метил-1Н-индол-7-ил]акрилоил}амид 4,5-дихлортиофен-2-сульфоновой кислоты. Соединение I-36 (250 мг, 0,576 ммоль) алкилируют, используя NaH (60%) (69,2 мг, 1,73 ммоль) и 3-(бромметил)бензонитрил (192 мг, 0,981 ммоль) в безводном ТГФ (2,8 мл), по методике, аналогично описанной для соединения P389. После фильтрования выделяют 186 мг соединения P390 в виде светло-зеленого твердого вещества (59%). 1H-ЯМР (400 МГц, ДМСО-d6) 2,26 (с, 3H), 5,59 (с, 2H), 6,32 (д, J=15,6 Гц, 1H), 7,05 (дд, J=10,0, 2,4 Гц, 1H), 7,22 (д, J=7,6 Гц, 1H), 7,37 (уш. с, 1H), 7,40-7,47 (м, 3H), 7,69 (д, J=7,6 Гц, 1H), 7,90 (с, 1H), 7,94 (д, J=15,6 Гц, 1H). ВЭЖХ чистота: 87%. МС (ESI-) Вычислено: 547,5 m/z; найдено: 546,2 m/z.
Пример 331. Получение соединения P391.
Соединение I-36 (250 мг, 0,576 ммоль) алкилируют, используя NaH (60%) (69,2 мг, 1,73 ммоль) и 4-(бромметил)бензонитрил (192 мг, 0,981 ммоль) в безводном ТГФ (2,8 мл) по методике, аналогично описанной для соединения P389. После фильтрования выделяют 90,8 мг соединения P391 в виде светло-зеленого твердого вещества (29%). 1H-ЯМР (400 МГц, d6-ДМСО). ВЭЖХ чистота: 95%.
Пример 332. Получение соединения P396.
К раствору P067 (4,0 г, 6,75 ммоль) в ТГФ (32 мл) добавляют воду (1,6 мл). К полученной смеси одной порцией добавляют 2,3-дихлор-5,6-дициано-1,4-бензохинон (4,6 г, 20,3 ммоль) и полученный раствор перемешивают в течение ночи при комнатной температуре. Смесь концентрируют в вакууме для удаления органических растворителей и добавляют этанол (60 мл). Полученную смесь кипятят с обратным холодильником в течение 1 часа, затем дают возможность остыть до комнатной температуре в течение ночи. Полученную суспензию фильтруют и промывают этанолом до тех пор, пока промывные жидкости не станут бесцветными. Полученный твердый продукт кипятят с ацетонитрилом для удаления остаточного P067 и затем дают возможность смеси охладиться до комнатной температуры в течение ночи. Полученный твердый продукт обрабатывают кипящим этанолом, охлаждают до комнатной температуры, фильтруют и сушат при 55°C в вакууме в течение ночи, получая 1,72 г P396 (42%). МС (ESI-) Вычислено: 606,3 m/z; найдено: 605,1 m/z. 1H-ЯМР (ДМСО-d6)
Пример 333. Получение соединения P397.
К раствору P396 (1,3 г, 2,16 ммоль) в ТГФ (66 мл) добавляют воду (0,52 мл) и раствор охлаждают до 0-5°C. Несколькими порциями в течение 5 минут добавляют борогидрид натрия (409 мг, 10,8 ммоль) и полученную смесь перемешивают в течение 30 минут. Реакционную смесь разбавляют EtOAc (75 мл) и промывают полунасыщенным раствором хлорида аммония в воде (2×80 мл), водой (2×40 мл) и насыщенным раствором соли (50 мл). Раствор концентрируют в вакууме и образовавшийся твердый осадок растирают в EtOAc, фильтруют и сушат в вакууме. Образующихся твердый продукт растирают дважды с диэтиловым эфиром и сушат в вакууме, получая 1,07 г (82%) P397. 1H-ЯМР(ДМСО-d6).
Пример 334. Получение соединения P398
К раствору P396 (300 мг, 0,495 ммоль) и 2-метил-2-бутена (495 мкл, 0,99 ммоль) в ТГФ (15 мл) в течение 5 минут добавляют раствор хлорида натрия (246 мг, 2,72 ммоль) и моноосновного фосфата натрия (416 мг, 3,46 ммоль) в воде (3,6 мл). Смесь перемешивают при комнатной температуре в течение 2 часов, добавляют дополнительное количество хлорида натрия (246 мг, 2,72 ммоль) и моноосновного фосфата натрия (416 мг, 3,46 ммоль) в воде (3,6 мл) и полученную реакционную смесь перемешивают при комнатной температуре в течение дополнительных 2 часов. Слои разделяют и органические фракции промывают 1н. водным раствором хлористоводородной кислоты (15 мл), водой (2×15 мл) и насыщенным раствором соли (15 мл). Полученный раствор концентрируют в вакууме и образовавшийся твердый осадок обрабатывают дихлорметаном при 40°C в течение 2 часов, затем смеси дают возможность самопроизвольно охладиться до комнатной температуры в течение ночи. Полученную суспензию фильтруют, твердый продукт промывают дихлорметаном и сушат при 35°C в вакууме, получая 151 мг соединения P398 (50%). 1H-ЯМР(ДМСО-d6); ЖХ/МС (93%) ESI- Вычислено: 622,3 m/z; найдено: 621,3 m/z.
Пример 335. Получение соединения P402
Синтез соединения P402. В емкость объемом 8 мл, снабженную стержневой мешалкой, при комнатной температуре загружают соединение I-196 (100 мг, 0,243 ммоль) и безводный ДМФА (500 мкл). Полученный желтый раствор охлаждают до 0°C на ледяной бане и затем добавляют NaH (60%) (29,2 мг, 0,729 ммоль). Полученную красную смесь нагревают до комнатной температуры и затем снова охлаждают до 0°C, в процессе чего добавляют 2-хлорметилимидазо[1,2-a]пиридин (48,7 мг, 0,292 ммоль). Смеси дают возможность нагреться до комнатной температуры и взаимодействовать в течение 17 часов. Реакцию гасят водой (2 мл) и 1M HCl (4 мл). Образовавшееся твердое вещество отделяют фильтрованием с отсосом, промывают охлажденным на ледяной бане CH2Cl2 (4 мл) и сушат в печи с высоким вакуумом при 70°C в течение 5 часов. После сушки выделяют 14,3 мг соединения P402 в виде желтовато-коричневого твердого вещества (11%). 1H-ЯМР (400 МГц, ДМСО-d6). ЖХ/МС: 74%.
Пример 336. Получение соединения P395.
Соединение I-36 (250 мг, 0,576 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (115 мг, 0,692 ммоль) и NaH (60%) (138 мг, 3,46 ммоль) в безводном ДМФА (2,8 мл) по методике, аналогично описанной для соединения P402. После сушки выделяют 233 мг соединения P395 в виде желтовато-коричневого твердого вещества (72%). 1H-ЯМР (400 МГц, ДМСО-d6) 2,22 (с, 3H), 5,49 (с, 2H), 6,24 (д, J=15,6 Гц, 1H), 6,87 (м, 1H), 7,04 (дд, J=10,8, 2,8 Гц, 1H), 7,19-7,26 (м, 2H), 7,36 (уш. с, 1H), 7,47 (с, 1H), 7,48 (д, J=8,4 Гц, 1H), 8,22 (д, J=15,2 Гц, 1H), 8,27 (с, 1H), 8,43 (д, J=6,8 Гц, 1H). ВЭЖХ чистота: 89%, МС (ESI-) Вычислено: 562,5 m/z, найдено: 563,1 m/z.
Пример 337. Получение соединения P399.
Соединение I-197 (100 мг, 0,254 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (50,8 мг, 0,305 ммоль) и NaH (60%) (30,5 мг, 0,762 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 16,2 мг соединения P399 в виде желтовато-коричневого твердого вещества (12%). 1H-ЯМР (400 МГц, ДМСО-d6), ЖХ/МС: 94%.
Пример 338. Получение соединения P400.
Синтез 3-хлор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]бензолсульфонамида, I-201. Соединение I-201 синтезируют по методике, аналогично описанной для соединения I-196, используя соединение I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 3-хлорбензолсульфонамид (314 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и EDCI (314 мг, 1,64 ммоль). После фильтрования выделяют 286 мг соединения I-201 в виде желтого твердого вещества (53%). 1H-ЯМР (400 МГц, ДМСО-d6). ЖХ/МС: 98%.
Синтез соединения P400. Соединение I-201 (100 мг, 0,255 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (51,0 мг, 0,306 ммоль) и NaH (60%) (30,7 мг, 0,765 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 83,0 мг соединения P400 в виде желтого твердого вещества (62%). 1H-ЯМР (400 МГц, ДМСО-d6) 2,24 (с, 3H), 5,73 (с, 2H), 6,40 (д, J=15,2 Гц, 1H), 7,08 (дд, J=2,4, 10,0 Гц, 1H), 7,26-7,32 (м, 1H), 7,38 (с, 1H), 7,46 (дд, J=2,4, 9,2 Гц, 1H), 7,68-7,74 (м, 3H), 7,80 (с, 1H), 7,82-7,85 (м, 1H), 7,89-7,94 (м, 2H), 8,10 (д, J=15,2 Гц, 1H), 8,60 (д, J=6,8 Гц, 1H). ЖХ/МС: 95%. МС (ESI-) Вычислено: 522,0 m/z; найдено: 521,5 m/z.
Пример 339. Получение соединения P401.
Синтез 3-фтор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]бензолсульфонамида, I-202. Соединение I-202 синтезируют по методике, аналогично описанной для соединения I-196, используя соединение I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 3-фторбензолсульфонамид (287 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и EDCI (314 мг, 1,64 ммоль). После фильтрования выделяют 212 мг соединения I-202 в виде желтого твердого вещества (41%). 1H-ЯМР (400 МГц, ДМСО-d6). ЖХ/МС: 97%.
Синтез соединения P401. Соединение I-202 (100 мг, 0,266 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (53,1 мг, 0,319 ммоль) и NaH (60%) (32,0 мг, 0,798 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 74,9 мг соединения P401 в виде желтовато-коричневого твердого вещества (55%). 1H-ЯМР (400 МГц, ДМСО-d6). ЖХ/МС: 97%.
Пример 340. Получение соединения P413.
Синтез 2,4-дифтор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)-акрилоил]бензолсульфонамида, I-203. Соединение I-203 синтезируют по методике, аналогично описанной для соединения I-196, используя соединение I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 2,4-дифторбензолсульфонамид (317 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и EDCI (314 мг, 1,64 ммоль). После фильтрования выделяют 297 мг соединения I-203 в виде желтого твердого вещества (55%). 1H-ЯМР (400 МГц, ДМСО-d6). ЖХ/МС: 97%.
Синтез соединения P413. Соединение I-203 (100 мг, 0,254 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (50,8 мг, 0,305 ммоль) и NaH (60%) (30,5 мг, 0,762 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 103 мг соединения P413 в виде желтовато-коричневого твердого вещества (77%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: 99%.
Пример 341. Получение соединения P403.
Синтез 4-фтор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]бензолсульфонамида, I-204. Соединение I-204 синтезируют по методике, аналогично описанной для соединения I-196, используя соединение I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 4-фторбензолсульфонамид (287 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и EDCI (314 мг, 1,64 ммоль). После фильтрования выделяют 273 мг соединения I-204 в виде желтого твердого вещества (53%). 1H-ЯМР (400 МГц, ДМСО-d6). ЖХ/МС (96%).
Синтез соединения P403. Соединение I-204 (100 мг, 0,266 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (53,1 мг, 0,319 ммоль) и NaH (60%) (32,0 мг, 0,798 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 90,9 мг соединения P403 в виде желтовато-коричневого твердого вещества (67%). 1H-ЯМР (400 МГц, ДМСО-d6). ЖХ/МС: 95%.
Пример 342. Получение соединения P404.
Синтез 3,5-дифтор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]бензолсульфонамида, I-205. Соединение I-205 синтезируют по методике, аналогично описанной для соединения I-196, используя соединение I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 3,5-дифторбензолсульфонамид (317 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и ЕDCI (314 мг, 1,64 ммоль). После фильтрования выделяют 278 мг соединения I-205 в виде желтого твердого вещества (51%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: 98%.
Синтез соединения P404. Соединение I-205 (100 мг, 0,54 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (50,8 мг, 0,305 ммоль) и NaH (60%) (30,5 мг, 0,762 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 90,2 мг соединения P404 в виде желтовато-коричневого твердого вещества (68%). 1H-ЯМР (400 МГц, ДМСО-d6) 2,24 (с, 3H), 5,77 (с, 2H), 6,44 (д, J=15,2 Гц, 1H), 7,09 (дд, J=2,8, 10,4 Гц, 1H), 7,34-7,37 (м, 1H), 7,40 (с, 1H), 7,47 (дд, J=2,4, 8,8 Гц, 1H), 7,62-7,65 (м, 2H), 7,72-7,80 (м, 3H), 7,86 (с, 1H), 8,07 (д, J=14,8 Гц, 1H), 8,66 (д, J=6,8 Гц, 1H). ЖХ/МС: 95%, МС (ESI-) Вычислено: 523,5 m/z, найдено: 523,6 m/z.
Пример 343. Получение соединения P405.
Синтез 4-хлор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]бензолсульфонамида, I-206. Соединение I-206 синтезируют по методике, аналогично описанной для соединения I-196 используя соединение I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 4-хлорбензолсульфонамид (314 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и EDCI (314 мг, 1,64 ммоль). После фильтрования выделяют 213 мг соединения I-206 в виде желтого твердого вещества (40%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС (98%).
Синтез соединения P405. Соединение I-206 (100 мг, 0,255 ммоль) подвергают взаимодействию с 2-хлорметил-имидазо[1,2-a]пиридином (51,0 мг, 0,306 ммоль) и NaH (60%) (30,7 мг, 0,765 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 110 мг соединения P405 в виде желтовато-коричневого твердого вещества (83%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: 96%.
Пример 344. Получение соединения P406.
3,4-Дихлор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]бензолсульфонамид, I-207. Соединение I-207 синтезируют по методике, аналогично описанной для соединения I-196, используя соединение I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 3,4-дихлорбензолсульфонамид (371 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и EDCI (314 мг, 1,64 ммоль). После фильтрования выделяют 321 мг соединения I-207 в виде желтого твердого вещества (55%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: 99%.
Синтез соединения P406. Соединение I-207 (100 мг, 0,234 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (46,8 мг, 0,281 ммоль) и NaH (60%) (28,0 мг, 0,702 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 95,1 мг соединения P406 в виде желтого твердого вещества (73%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: 95%.
Пример 345. Получение соединения P407.
Синтез 2,5-дифтор-N-[(E)-3-(5-фтор-3-метил-1H-индол-7-ил)акрилоил]бензолсульфонамида, I-208. Соединение I-208 синтезируют по методике, аналогично описанной для соединения I-196, используя соединение I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 2,5-дифторбензолсульфонамид (317 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и EDCI (314 мг, 1,64 ммоль). После фильтрования выделяют 284 мг соединения I-208 в виде желтого твердого вещества (53%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: 99%.
Синтез соединения P407. Соединение I-208 (100 мг, 0,254 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (50,8 мг, 0,305 ммоль) и NaH (60%) (30,5 мг, 0,762 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 106 мг соединения P407 в виде желтого твердого вещества (80%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: 97%.
Пример 346. Получение соединения P408.
Синтез 3,5-дихлор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]бензолсульфонамида, I-209. Соединение I-209 синтезируют по методике, аналогично описанной для соединения I-196, используя соединение I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 3,5-дихлорбензолсульфонамид (371 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и EDCI (314 мг, 1,64 ммоль). После фильтрования выделяют 215 мг соединения I-209 в виде желтого твердого вещества (37%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: 98%.
Синтез соединения P408. Соединение I-209 (100 мг, 0,234 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (46,8 мг, 0,281 ммоль) и NaH (60%) (28,0 мг, 0,702 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 117 мг соединения P408 в виде желтовато-коричневого твердого вещества (90%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: (96%).
Пример 347. Получение соединения P409.
Синтез 2-фтор-N-[(E)-3-(5-фтор-3-метил-1Н-индол-7-ил)акрилоил]бензолсульфонамида, I-210. Соединение I-210 синтезируют по методике, аналогично описанной для соединения I-196, используя соединение I-34 (300 мг, 1,37 ммоль), безводный CH2Cl2 (4 мл), 2-фторбензолсульфонамид (287 мг, 1,64 ммоль), DMAP (200 мг, 1,64 ммоль) и EDCI (314 мг, 1,64 ммоль). После фильтрования выделяют 275 мг соединения I-210 в виде желтого твердого вещества (53%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: 99%.
Синтез соединения P409. Соединение I-210 (100 мг, 0,266 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (53,1 мг, 0,319 ммоль) и NaH (60%) (32,0 мг, 0,798 ммоль) в безводном ДМФА (500 мкл) по методике, аналогично описанной для соединения P402. После сушки выделяют 98,1 мг соединения P409 в виде желтовато-коричневого твердого вещества (73%). 1H-ЯМР (400 МГц, ДМСО-d6); ЖХ/МС: 98%.
Пример 348. Получение соединения P395.
Соединение I-36 (250 мг, 0,576 ммоль) подвергают взаимодействию с 2-хлорметилимидазо[1,2-a]пиридином (115 мг, 0,692 ммоль) и NaH (60%) (138 мг, 3,46 ммоль) в безводном ДМФА (2,8 мл) по методике, аналогично описанной для соединения P402. После сушки выделяют 233 мг соединения P395 в виде желтовато-коричневого твердого вещества (72%). 1H-ЯМР (400 МГц, ДМСО-d6) 2,22 (с, 3H), 5,49 (с, 2H), 6,24 (д, J=15,6 Гц, 1H), 6,85-6,89 (м, 1H), 7,04 (дд, J=2,8, 10,8 Гц, 1H), 7,19-7,26 (м, 2H), 7,36 (уш.с, 1H), 7,47 (с, 1H), 7,48 (д, J=8,4 Гц, 1H), 8,22 (д, J=15,2 Гц, 1H), 8,27 (с, 1H), 8,43 (д, J=6,8 Гц, 1H). ВЭЖХ чистота: 89%, МС (ESI-) Вычислено: 562,5 m/z; найдено: 563,1 m/z.
Общая методика (A-21) гидрирования.
К раствору соединения I-131D в этаноле добавляют 10% Pd/C (0,1 г/г соединения 1). Полученную смесь дегазируют и атмосферу заменяют водородом (3 раза). Смесь перемешивают в атмосфере водорода при атмосферном давлении в течение 5 дней. Реакционную смесь фильтруют через целит, осадок на фильтре промывают этанолом и фильтрат концентрируют. Неочищенную реакционную смесь очищают хроматографией на силикагеле, используя дихлорметан в качестве элюента, затем хроматографией на силикагеле, используя элюирование с градиентом смеси EtOAc/гексаны, получая соединения I-211 и I-212. Соединение I-211: метиловый эфир 3-[l-(3,4-дифторбензил)-3a-метил-2-оксооктагидроиндол-7-ил]пропионовой кислоты; МС (ESI+) Вычислено: 365,4 m/z; найдено: 366,0 m/z. 1H-ЯМР(CDCl3). Соединение I-212: метиловый эфир 3-[1-(3,4-дифторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1Н-индол-7-ил]пропионовой кислоты; МС (ESI+) Вычислено: 363,4 m/z; найдено: 364 m/z; 1H-ЯМР(CDCl3).
Общая методика (A-22) гидролиза метиловых эфиров
К раствору подходящего метилового эфира в ТГФ/MeOH (2:1) добавляют водный раствор NaOH (3 экв.) и полученную реакционную смесь перемешивают в течение 24-72 часов при комнатной температуре. Смесь концентрируют для удаления органических растворителей, разбавляют водой и промывают 2 порциями дихлорметана. Значение pH доводят до 2-3 (лакмус) добавлением 1н. HCl и смесь экстрагируют EtOAc. Органические фракции промывают водой и насыщенным раствором соли, сушат над сульфатом магния и концентрируют с получением целевого соединения.
Общая методика (A-23) получения ацилсульфонамидов
К раствору подходящей исходной кислоты, подходящего сульфонамида (1,05 экв.) и DMAP (2,4 экв.) в CH2Cl2 добавляют EDCI (2,5 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 1-5 дней. Реакционную смесь промывают 1н. HCl (водн.), водой и насыщенным раствором соли, сушат над сульфатом магния и концентрируют в вакууме. Остаток очищают либо хроматографией на силикагеле, используя элюирование с градиентом (0-5% метанол в дихлорметане), либо растирая в смеси дихлорметан/гексаны, получая указанное в заголовке соединение.
Пример 349. Получение соединения P410.
Синтез 3-[1-(3,4-дифторбензил)-3a-метил-2-оксооктагидроиндол-7-ил]пропионовой кислоты I-213.
В соответствии с общей методикой (A-22), соединения I-211 преобразовывают в соединение I-213 с количественным выходом. ЖХ/МС: 100%; APCI- Вычислено: 351,4 m/z; найдено: 350,2 m/z; 1H-ЯМР(CDCl3).
Синтез соединения P410. В соответствии с общей методикой A-23, соединение I-213 подвергают реакции сочетания с 2,4,5-трифторбензолсульфонамидом с получением соединения P410 с 46% выходом. ЖХ/МС: 84%; ESI- Вычислено: 544,5 m/z; найдено: 543,5 m/z; 1H-ЯМР (СDCl3).
Пример 350. Получение соединения P411.
Синтез 3-[1-(3,4-дифторбензил)-3a-метил-2-оксо-2,3,3a,4,5,6-гексагидро-1H-индол-7-ил]пропионовой кислоты, I-214
В соответствии с общей методикой (A-22), соединение I-212 преобразовывают в соединение I-214 с выходом 61%. ЖХ/МС (98%) ESI+ Вычислено: 349,4 m/z; найдено: 350,9 m/z; 1H-ЯМР(CDCl3)
Синтез соединения P411. В соответствии с общей методикой A-23, соединение I-214 подвергают реакции сочетания с 2,4,5-трифторбензолсульфонамидом с получением соединения P411 с выходом 81%. ЖХ/МС: 81%; ESI- Вычислено: 542,5 m/z; найдено: 542,3 m/z; 1Н-ЯМР(CDCl3).
Пример 351. Получение соединения P415.
В соответствии с общей методикой A-23, соединение I-213 подвергают реакции сочетания с 4,5-дихлортиофен-2-сульфонамидом с получением соединения P415 с выходом 70%. ЖХ/МС: 93%; ESI- Вычислено: 563,5 m/z; найдено: 563,4 m/z 1Н-ЯМР(СDCl3).
Пример 352. Получение соединения P414.
В соответствии с общей методикой A-21, P306 подвергают гидрированию, получая соединение P414 с выходом 40%. ЖХ/МС: 98,9%; ESI- Вычислено: 537,5 m/z; найдено: 537,5 m/z; 1H-ЯМР(CDCl3).
Пример 353. Получение соединения P412.
Синтез 3-[1-метил-4-(нафталин-2-илокси)-1H-индол-3-ил]пропионовой кислоты, I-215. В соответствии с общей методикой A-21, I-137A подвергают гидрированию, получая соединение I-215 с выходом 40%. 1H-ЯМР(CDCl3).
Синтез соединения P412. В соответствии с общей методикой A-23, соединение I-215 подвергают реакции сочетания с 2,4,5-трифторбензолсульфонамидом с получением соединения P412 с выходом 46%. ЖХ/МС: 90%; ESI- Вычислено: 538,4 m/z; найдено: 537,8 m/z. 1H-ЯМР(CDCl3).
Пример 354. Получение соединения P362.
В соответствии с общей методикой A-23, соединение I-215 подвергают реакции сочетания с 3,4,-дифторбензолсульфонамидом с получением соединения P362 с 54% выходом. 1H-ЯМР (400 МГц, ДМСО-d6), МС(ESI-): 519,6 (M-1), ВЭЖХ c обращенной фазой: 94%.
Пример 355. Получение соединения P257.
Синтез 4-йод-1-метил-1H-индазол-3-иламина, I-216. В емкость объемом 330 мл под давлением, снабженную магнитной мешалкой, добавляют 2-фтор-6-йодбензонитрил (4,5 г, 18,2 ммоль), N,N-диметилацетамид (45 мл) и метилгидразин (1,2 мл, 23,7 ммоль). Емкость продувают газообразным N2, герметично закрывают, нагревают до 87°C и выдерживают при этой температуре в течение ночи, затем до 120°C и выдерживают при этой температуре в течение 5 часов. Реакционную смесь охлаждают, разбавляют EtOAc (300 мл) и промывают водой. Водный слой экстрагируют и объединенные EtOAc экстракты промывают водой (3×150 мл), насыщенным раствором соли, сушат (Na2SO4), фильтруют и концентрируют, получая коричневое твердое вещество. Твердый продукт растирают в смеси EtOAc/гексаны (1:1, 10 мл), фильтруют, промывают смесью EtOAc/гексаны (1:1) и сушат, получая 4,04 г (81%) I-216 в виде бежевого твердого вещества. 1H-ЯМР (400 МГц, ДМСО-d6).
Синтез (4-йод-1-метил-1Н-индазол-3-ил)амида нафталин-2-карбоновой кислоты, I-217. К соединению I-216 (470 мг, 1,71 ммоль), растворенному в безводном ТГФ (7 мл), добавляют триэтиламин (477 мкл, 346 мг, 3,42 ммоль) и полученную смесь при перемешивании охлаждают до 0°C в атмосфере азота. К раствору при перемешивании одной порцией добавляют 2-нафтоилхлорид (326 мг, 1,71 ммоль), дают возможность реакционной смеси нагреться до комнатной температуры и перемешивают в течение ночи. Реакционную смесь фильтруют и образовавшийся твердый осадок промывают ТГФ и CH2Cl2. Полученный твердый продукт смешивают с этанолом (15 мл), фильтруют, образовавшийся твердый осадок промывают этанолом и сушат, получая 476 мг (65%) I-217 в виде белого твердого вещества. 1H-ЯМР (400 МГц, ДМСО-d6).
Синтез метилового эфира (E)-3-{1-метил-3-[(нафталин-2-карбонил)амино]-1H-индазол-4-ил}акриловой кислоты, I-218. В трубку высокого давления, снабженную магнитной мешалкой, загружают соединение I-217 (200 мг, 0,468 ммоль), метиламин (10 мл), ацетат палладия (II) (10,5 мг, 0,047 ммоль) и три-o-толилфосфин (43 мг, 0,14 ммоль). Раствор дегазируют, барботируя газообразный N2 через раствор в течение 30 минут. Добавляют метилакрилат (956 мг, 1 мл, 36 ммоль), раствор дегазируют в течение дополнительных 5 минут и затем трубку герметично закрывают, нагревают до 75°C и выдерживают при указанной температуре в течение 30 минут, затем при 100°C в течение 6 часов. Реакционную смесь охлаждают до комнатной температуре, добавляют безводный ДМФА (20 мл) и дополнительную порцию ацетата палладия (II) (11 мг, 0,049 ммоль). Раствор снова дегазируют, герметично закрывают, нагревают на масляной бане до 105°C и выдерживают при указанной температуре в течение 6 часов, затем дают возможность охладиться до комнатной температуры. Реакционную смесь разбавляют EtOAc и промывают водой. Водный слой экстрагируют EtOAc и объединенные органические экстракты промывают водой (3×), насыщенным раствором соли и сушат (Na2SO4). Раствор фильтруют и концентрируют, получая 210 мг соединения I-218 со степенью чистоты, достаточной для использования на следующей стадии синтеза. 1H-ЯМР (400 МГц, ДМСО-d6).
Синтез (E)-3-{1-мeтил-3-[(нафталин-2-карбонил)амино]-1Н-индазол-4-ил}акриловой кислоты, I-219. В круглодонную колбу объемом 25 с одним горлом, снабженную магнитной мешалкой, добавляют соединение I-218 (210 мг, 0,468 ммоль), метанол и TГФ (по 5 мл каждого). К полученному раствору при перемешивании добавляют 2M водный раствор NaOH (0,85 мл, 1,70 ммоль). Спустя 2 часа добавляют дополнительную порцию 2M водного раствора NaOH (0,54 мл, 1,08 ммоль), реакционную смесь нагревают до 40°C и выдерживают при указанной температуре в течение ночи. Охлажденную реакционную смесь подкисляют 1M водным раствором HCl и экстрагируют EtOAc (3×). Объединенные органические экстракты промывают насыщенным раствором соли и сушат (Na2SO4). Раствор упаривают, остаток очищают колоночной хроматографией, используя смесь 2:2:1 CH2Cl2/ТГФ/гексан и затем 6:6:1 CH2Cl2/ТГФ/метанол, получая 95 мг соединения I-219. 1H-ЯМР (400 МГц, ДМСО-d6).
Синтез соединения P257. В соответствии с общей методикой A-8, акриловую кислоту I-219 подвергают взаимодействию с амидом 4,5-дихлортиофен-2-сульфоновой кислоты с получением соединения P257. 1H-ЯМР (ДМСО-d6) 4,06 (с, 3H), 6,61 (д, J=20,0), 7,47 (м, 2H), 7,56 (с, 1H), 7,66 (м, 2H), 7,72 (м, 1H), 8,01 (м, 3H), 8,16 (м, 1H), 8,67 (с, 1H), 10,74 (с, 1H). ЖХ/МС (96%) ESI+ Вычислено: 585,49; найдено: 585,4 m/z.
Пример 356. Получение соединения P246.
Синтез 1,3-бис-триметилсиланилокси-2-(l-триметилсиланилоксивинил)бензола, I-220.
1M раствор LiHMDS (52 мл, 51,8 ммоль, 3,15 экв.) в ТГФ осторожно добавляют к раствору 2,6-дигидроацетофенона (2,5 г, 16,4 ммоль, 1 экв.) в ТГФ (100 мл) и реакционную смесь перемешивают в течение 15 минут при комнатной температуре. К смеси одной порцией добавляют хлортриметилсилан (7,3 мл, 57,4 ммоль, 3,5 экв.) (экзотермическая реакция) и реакционную смесь перемешивают в течение 5 минут. Реакционную смесь концентрируют примерно до 11 г. Добавляют безводный CH2Cl2 (50 мл), осадок отфильтровывают, промывают CH2Cl2 и маточную жидкость концентрируют, получая соединение I-220 (6,04 г, 100%) в виде коричневой жидкости. 1Н-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез 4-гидроксибензофуран-3-она, I-221. Раствор N-бромсукцинимида (3,2 г, 18,02 ммоль, 1,1 экв.) в ацетонитриле (22 мл) осторожно добавляют к раствору соединения I-220 (6,04 г, 16,4 ммоль, 1 экв.) в ацетонитриле (72 мл) и реакционную смесь перемешивают в течение 20 минут. Неочищенную смесь концентрируют, разбавляют CH2Cl2 (50 мл). CH2Cl2 раствор промывают последовательно насыщенным водным раствором NaHCО3 (30 мл), 1н. водным раствором NaOH (20 мл×2), водой (50 мл), насыщенным раствором соли (50 мл) и концентрируют. Добавляют MeOH (34 мл) и воду (12 мл). Медленно добавляют концентрированную HCl (2 мл), раствор перемешивают в течение 15 минут, затем добавляют воду (50 мл) и смесь концентрируют в вакууме. Добавляют воду (50 мл) и смесь экстрагируют EtOAc (3×15 мл). Объединенные органические экстракты промывают водой, насыщенным раствором соли, сушат над MgSO4, получая желтое твердое вещество (3,21 г). Добавляют ацетон (80 мл), затем K2CO3 (4,8 г) при комнатной температуре. Суспензию перемешивают при комнатной температуре в течение 1 часа, затем фильтруют и концентрируют. Добавляют CH2Cl2, затем 10% HCl. Органический слой промывают водой, насыщенным раствором соли, сушат над MgSO4, получая неочищенный I-221 (2,0 г, 84% выход неочищенного продукта) в виде оранжевого твердого вещества. Добавляют MTBE (6 мл), суспензию охлаждают до 0°C и твердый продукт отфильтровывают, получая чистое соединение I-221 (1,24 г, 50%) в виде оранжевого твердого вещества. МС (AP+): 151 (M+1). 1H-ЯМР (500 МГц, CDCl3) подтверждает структуру.
Синтез 3-(нафталин-2-илсульфанил)бензофуран-4-ола, I-222. Раствор HCl (2,0 M, в эфире, 3,75 мл, 5 экв.) добавляют к предварительно нагретому до 40°C раствору соединения I-221 (225 мг, 1,5 ммоль, 1 экв.) и 2-нафталинтиола (240 мг, 1,5 ммоль, 1 экв.) в EtOH (3,75 мл) и перемешивают при комнатной температуре в течение 23 часов. Реакционную смесь концентрируют в вакууме. Остаток растворяют в EtOAc, раствор промывают водой, насыщенным раствором соли, сушат над MgSО4, получая 470 мг в виде желтого твердого вещества. Твердый продукт растирают в гексане (4 мл) при комнатной температуре, затем кипятят с обратным холодильником с гексаном (2 мл) и фильтруют, получая соединение I-222 (291 мг, 66%) в виде желтого твердого вещества.
Синтез метилового эфира [3-(нафталин-2-илсульфанил)бензофуран-4-илокси]уксусной кислоты, I-123. К раствору метилбромацетата (156 мг, 1,02 ммоль, 1,2 экв.) в ацетоне (1 мл) добавляют суспензию соединения I-222 (249 мг, 0,85 ммоль, 1 экв.) и K2CO3 (176 мг, 1,275 ммоль, 1,5 экв.) в ацетоне (2 мл). Реакционную смесь герметично закрывают в емкости объемом 20 мл, нагревают до 70°-80°C и выдерживают при указанной температуре в течение 3 часов. Добавляют воду (8 мл), затем EtOAc (4 мл) и смесь подкисляют 10% HCl до pH=5. Органический слой отделяют, промывают водой, насыщенным раствором соли, сушат над MgSО4 получая соединение I-223 (310 мг, 100%) в виде красного масла, которое кристаллизуется. МС (ESI+): 366 (M+l). Структуру подтверждают 1H-ЯМР (500 МГц, CDCl3).
Синтез [3-(нафталин-2-илсульфанил)бензофуран-4-илокси]уксусной кислоты, I-224. 2н. водный раствор NaOH (1,05 мл, 2,09 ммоль, 2,5 экв.) по каплям добавляют к раствору соединения I-223 (305 мг, 0,834 ммоль, 1 экв.) в смеси ТГФ-MeOH (1:1, 5 мл). Реакционную смесь перемешивают при комнатной температуре в течение 15 минут. Смесь концентрируют в вакууме, добавляют воду, затем EtOAc. Смесь подкисляют 10% HCl. Органический слой промывают водой, насыщенным раствором соли, сушат над MgSO4 получая соединение I-224 (293 мг, 100%) в виде масла. МС (ESI-): 349 (M-1). Структуру подтверждают 1H-ЯМР (500 МГц, CDCl3).
Синтез соединения P246. К раствору соединения I-224 (22 мг, 0,063 ммоль, 1 экв.) в 0,5 мл дихлорметана добавляют DMAP (15 мг, 0,126 ммоль, 2 экв.), 4,5-дихлор-2-тиофенсульфонамид (31 мг, 0,132 ммоль, 2,1 экв.) и EDCI (24 мг, 0,126 ммоль, 2 экв.). Смесь перемешивают при комнатной температуре в течение 3 часов, затем гасят 10% HCl и водой. Водный слой экстрагируют EtOAc. Объединенные органические экстракты промывают водой, насыщенным раствором соли и затем сушат над MgSO4. Раствор концентрируют и остаток хроматографируют на силикагеле, элюируя смесью EtOAc/гексан (3:7, затем 1:1), получая соединение P246 (13 мг, 37%) в виде желтого масла. ЖХ-МС (ESI-): 563 (M-l) (92%). 1H-ЯМР (CDCl3) 4,53 (с, 2H), 6,55 (м, 1H), 7,33 (уш., 2H), 7,35 (дд, J=11,0, 2,0 Гц, 1H), 7,40-7,47 (м, 3H), 7,60 (д, J=2,0 Гц, 1H), 7,64 (ущ.д, J=8,5 Гц, 1H), 7,73-7,78 (м, 2H), 7,85 (уш. с, 1H), 10,10 (с, 1H). ЖХ-МС (99%): ESI- Вычислено: 563 m/z; найдено: 563,3.
Пример 357. Получение соединения P256.
Раствор оксона (131 мг, 0,21 ммоль, 3 экв.) в воде (0,7 мл) осторожно при перемешивании добавляют к раствору P246 (40 мг, 0,071 ммоль, 1 экв.) в смеси диоксан/MeOH (1:1, 2 мл) при комнатной температуре. Реакционную смесь перемешивают в течение ночи при комнатной температуре. Добавляют воду (5 мл), осадок отфильтровывают, промывают водой (2×1,5 мл) и этанолом (2 мл) и сушат, получая соединение P256 (32 мг, 78%) в виде белого твердого вещества. ЖХ-МС (98%): ESI- Вычислено: 579 m/z; найдено: 579. 1Н-ЯМР (CDCl3) 4,48 (д, J=14,8 Гц, 1H), 4,52 (д, J=14,8 Гц, 1H), 6,60 (дд, J=8,0, 0,8 Гц, 1H), 7,25 (дд, J=8,4, 0,4 Гц, 1H), 7,32 (д, J=8,4 Гц, 1H), 7,50 (дд, J=8,8, 2,0 Гц, 1H), 7,59-7,63 (м, 2H), 7,73 (д, J=4,8 Гц, 1H), 7,88-7,93 (м, 4H), 8,32 (д, J=2,0 Гц, 1H), 12,70 (уш. с, 1H).
Пример 358. Получение соединения P254.
Синтез соединения P254. Перуксусную кислоту (0,45 мл, 32%, 2,13 ммоль) осторожно c перемешиванием при комнатной температуре добавляют к раствору P246 (40 мг, 0,071 ммоль, 1 экв.) в CH2Cl2 (2 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 дней. Реакцию гасят добавлением смеси вода/CH2Cl2 (по 4 мл каждого). Органический слой отделяют и промывают водой, насыщенным раствором соли, сушат над MgSО4, получая соединение P254 (32 мг, 76%) в виде масла, которое кристаллизуется с получение белого твердого вещества. ЖХ-МС (92%): ESI- Вычислено: 595 m/z; найдено: 595. 1H-ЯМР (CDCl3) 4,67 (с, 2H), 6,94 (дд, J=8,0, 0,4 Гц, 1H), 7,28 (дд, J=8,4, 0,8 Гц, 1H), 7,51 (дд, J=8,4, 0,8 Гц, 1H), 7,62-7,71 (м, 2H), 7,72 (с, 1H), 7,88-7,94 (м, 3H), 7,97 (д, J=9,2 Гц, 1H), 8,08 (с, 1H), 8,62 (д, J=0,8 Гц, 1H), 12,70 (уш. с, 1H).
Пример 359. Получение соединения P317
Соединение I-205 (78 мг, 0,15 ммоль) подвергают взаимодействию с 3,4-дифторбензилбромидом (37 мг, 0,18 ммоль) и NaH (60%) (13 мг, 0,33 ммоль) в безводном ДМФА (2 мл) по методике, аналогично описанной для соединения P402. После сушки выделяют 57 мг соединения P317 в виде желтого твердого вещества (73%). 1Н-ЯМР (CDCl3) 2,29 (д, J=1,2 Гц, 3H), 5,34 (с, 2H), 6,11 (д, J=15,2 Гц, 1H), 6,69 (м, 1H), 6,81 (м, 1H), 6,95 (с, 1H), 6,97 (дд, J=9,6, 2,4 Гц, 1H), 7,03 (м, 1H), 7,12 (м, 1H), 7,29 (дд, J=8,4, 2,4 Гц, 1H), 7,64-7,70 (м, 2H), 8,09 (дд, J=15,6, 1,2 Гц, 1H), 8,23 (уш.с, 1H). ЖХ-МС (95%): ESI- Вычислено: 520,5 m/z; найдено: 520,5.
Пример 360. Получение соединения P321.
Соединение I-36 (70 мг, 0,16 ммоль) алкилируют, используя NaH (60%) (8,5 мг, 0,36 ммоль) и 3,4-дифторбензилбромид (50 мг, 0,24 ммоль) в безводном ДМФА (2,8 мл) по методике, аналогично описанной для соединения P068. После фильтрования выделяют и колоночной хроматографии выделяют 50 мг соединения P321 в виде желтого твердого вещества (55%). 1H-ЯМР (ДМСО-d6) 5,51 (с, 2H), 6,26 (д, J=16 Гц, 1H), 6,66 (м, 1H), 6,99 (м, 1H), 7,04 (дд, J=10, 2,4 Гц, 1H), 7,41 (с, 1H), 7,23 (м, 1H), 7,43 (дд, J=12, 2,4 Гц, 1H), 7,87 (с, 1H), 7,98 (д, J=11,6 Гц, 1H), ЖХ/МС (92%) (ESI-) Вычислено: 558 m/z; найдено: 557 m/z.
Пример 361. Получение соединения P258.
Синтез 4-метокси-1-метил-1Н-индол-2,3-диона, I-225. 4-Метоксиизатин (полученный в соответствии с методикой публикации Hewaam, P., Meanwell, N. A. Tet. Lett. 1994, 35, 7303-7306, 1,5 г, 8,47 ммоль) растворяют в ацетоне (30 мл) и добавляют карбонат калия (2,9 г, 21,2 ммоль) и йодметан (1,3 мл, 3,0 г, 21,2 ммоль). Полученный раствор перемешивают при комнатной температуре в течение ночи. Реакционную смесь фильтруют и твердый осадок промывают ацетоном, получая 1,5 г твердого вещества. Полученный твердый продукт нагревают в этаноле (13 мл) и 33% водном растворе KOH (5 мл) до 70°C и выдерживают при данной температуре в течение 1,5 часов. Смесь концентрируют и затем подкисляют разбавленным водным раствором HCl. Выпавший в осадок красный твердый продукт отфильтровывают и сушат. получая 0,8 г соединения I-225. 1H-ЯМР (500 МГц, ДМСО-d6).
Синтез 4-гидрокси-1-метил-1Н-индол-2,3-диона, I-226. Соединение I-225 (0,446 г, 0,0023 моль) растворяют в безводном CH2Cl2 (20 мл) и охлаждают до 0°C в атмосфере азота. К полученному раствору по каплям добавляют раствор 1M BBr3 в CH2Cl2 (9,2 мл, 0,0092 моль). Полученную смесь перемешивают в течение 1 часа при комнатной температуре, затем концентрируют, переносят в смесь ТГФ/EtOAc (2:1), промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая соединение I-226 (0,300 г). МС (ESI-)=176,3 (M-l). ЖХ/МС(ESI-) 91,3%. 1H-ЯМР (400 МГц, ДМСО-d6).
Синтез метилового эфира (l-метил-2,3-диоксо-2,3-дигидро-1Н-индол-4-илокси)уксусной кислоты, I-227. Соединение I-226 (0,300 г, 0,0017 моль) растворяют в EtCOMe (10 мл). К полученному раствору добавляют карбонат калия (0,2573 г, 0,00186 моль) и метилбромацетат (0,21 мл, 0,0022 моль). Смесь перемешивают при комнатной температуре в течение 3 дней и затем концентрируют. Остаток разбавляют EtOAc, промывают насыщенным раствором соли, органический слой сушат над безводным сульфатом натрия, фильтруют и концентрируют, получая соединение I-227 (0,300 г). МС (ESI+)=250,2 (M+l). ЖХ/МС (ESI+) 92,8%.
Синтез (1-метил-2,3-диоксо-2,3-дигидро-1H-индол-4-илокси)уксусной кислоты, I-228.
I-227 (0,300 г, 0,0012 моль) растворяют в ТГФ (10 мл). К раствору добавляют водный 1M раствор LiOH (2,4 мл, 0,0024 моль) и смесь перемешивают в течение 1 часа при комнатной температуре. Смесь упаривают, остаток растирают с 2M водным раствором HCl, водой и сушат, получая соединение I-228 (0,280 г). МС (ESI+)=236,2 (M+l). ЖХ/МС (ESI+) 86,4%. 1H-ЯМР (400 МГц, ДМСО-d6).
Синтез [2-(l-метил-2,3-диоксо-2,3-дигидро-1Н-индол-4-илокси)ацетил]амида 4,5-дихлортиофен-2-сульфоновой кислоты, I-229. Соединение I-228 (0,280 г, 0,0012 моль) растворяют в ТГФ (2 мл). К раствору добавляют CH2Cl2 (1 мл), EDCI (0,4601 г, 0,0024 моль), DMAP (0,2928 г, 0,0024 моль) и 2,3-дихлортиофен-5-сульфонамид (0,3062 г, 0,00132 моль), смесь перемешивают в течение двух дней при комнатной температуре, концентрируют, растворяют в ТГФ/EtOAc (2:1) и распределяют в 2M водном растворе HCl. Органический слой промывают насыщенным раствором соли, сушат над безводный сульфатом натрия, фильтруют, концентрируют и растирают в смеси эфир/EtOAc (2:1), получая соединение I-229 (0,140 г). МС (ESI-)=447,2 (M-l). 1H-ЯМР (400 МГц, ДМСО-d6).
Синтез соединения P258. Соединение I-229 (0,100 г, 0,223 ммоль) растворяют в метаноле (2 мл). К полученному раствору добавляют 3,4-дихлоранилин (0,043 г, 0,267 ммоль), затем гидрат п-толуолсульфоновой кислоты (0,002 г, 0,010 ммоль). Полученную смесь перемешивают в закрытой емкости при 80°C в течение ночи, затем охлаждают до комнатной температуры, концентрируют, снова растворяют в уксусной кислоте (1 мл) и к раствору добавляют цианоборогидрид натрия (0,100 г, 1,591 ммоль). Смесь перемешивают при комнатной температуре в течение 4 часов, затем концентрируют, промывают водой и хроматографируют на силикагеле (элюирование с градиентом, MeOH/CH2Cl2 от 0,5% до 20%), получая соединение P258 (9 мг). 1H-ЯМР (500 МГц, ДМСО-d6) 3,07 (с, 3H), 3,50-3,70 (м, 1H), 4,05-4,40 (м, 2H), 4,98 (д, J=7,0 Гц, 1H), 5,75 (с, 1H), 6,46 (д, J=8,5 Гц, 1H), 6,62 (д, J=7,5 Гц, 1H), 6,70 (с, 1H), 6,73 (м, 1H), 7,18 (дд, J=8,5, 8,0 Гц, 1H), 7,23 (с, 1H), 7,34 (с, 1H). ЖХ/МС (90,2%) ESI+ Вычислено: M=595,3; найдено: 596,1 m/z.
Пример 362. Получение соединения P249.
Используя методику A-15 и заменяя 2-нафтилсульфонамид анилином, соединение I-186 преобразовывают в соединение P249 с 70% выходом; 1H-ЯМР (ДМСО-d6) 3,15 (с, 3H), 6,81 (д, J=16 Гц, 1H), 7,15 (д, J=8 Гц, 1H), 7,39 (д, J=8 Гц, 1H), 7,44 (с, 1H), 7,68 (м, 3H), 7,81 (с, 1H), 7,88 (дд, J=8,8, 2,0 Гц, 1H), 8,04 (м, 2H), 8,13 (т, J=9,2 Гц, 2H), 8,22 (д, J=16,0 Гц, 1H), 8,43 (м, 1H)
Пример 363. Получение соединения P251.
Используя методику A-15 и заменяя 2-нафтилсульфонамид анилином, соединение I-186 преобразовывают в соединение P242 с 80% выходом. 1H-ЯМР (ДМСО-d6) 3,09 (с, 3H), 6,66 (д, J=16 Гц, 1H), 7,03 (д, J=7,6 Гц, 1H), 7,28 (д, J=8 Гц, 1H), 7,38 (т, J=7,6 Гц, 1H), 7,44 (с, 1H), 7,64 -7,71 (м, 2H), 7,81 (с, 1H), 7,87 - 7,93 (м, 2H), 8,02 (д, J=7,6 Гц, 1H), 8,11 (т, J=9,2 Гц, 2H), 8,43 (д, J=1,6 Гц, 1H).
Пример 364. Получение соединения P323.
Синтез метилового эфира (E)-3-[1-(2,4-дихлорбензил)-5-фтор-3-метил-1Н-индол-7-ил]акриловой кислоты, I-230. К раствору соединения I-191 (320 мг, 0,826 ммоль) в безводном ДМФА (2 мл) добавляют ацетат палладия (II) (4,6 мг, 0,0206 ммоль), три-o-толилфосфин (25,2 мг, 0,0824 ммоль) и триэтиламин (0,574 мл, 4,12 ммоль). Смесь дегазируют, барботируя через раствор газообразный N2 в течение 20 минут. К смеси добавляют метилакрилат (370 мкл, 4,12 ммоль), раствор дегазируют в течение дополнительных 5 минут, затем емкость закрывают, помещают на масляную баню и нагревают до 100°C. После выдерживания при указанной температуре в течение 40 часов реакционную смесь охлаждают до комнатной температуры, разбавляют EtOAc и водой (по 4 каждого растворителя) и экстрагируют. Водный слой экстрагируют этилацетатом (2×4 мл), и объединенные органические экстракты промывают водой (3×10 мл) и сушат (Na2SO4). Раствор фильтруют и концентрируют, получая 337 мг соединения I-230 со степенью чистоты, достаточной для использования на следующей стадии. 1H-ЯМР (400 МГц, CDCl3).
Синтез (E)-3-[l-(2,4-дихлорбензил)-5-фтор-3-метил-1Н-индол-7-ил]акриловой кислоты, I-231. В емкость объемом 18 мл, снабженную стержневой мешалкой, при комнатной температуре загружают сложный эфир I-XXX (337 мг, 0,859 ммоль), ТГФ (4 мл), MeOH (2 мл) и 15% водный раствор NaOH (1,28 мл, 4,81 ммоль). Смесь перемешивают при комнатной температуре в течение 24 часов. Реакционную смесь концентрируют, промывают Et2O (3×20 мл), подкисляют 1M раствором HCl до pH 1 и экстрагируют CH2Cl2 (2×35 мл). Органические части объединяют, сушат (MgSO4) и концентрируют, получая 259 мг кислоты I-XXX в виде светло-зеленого твердого вещества (80%). 1H-ЯМР (400 МГц, CDCl3).
Синтез соединения P323. В емкость объемом 8 мл, снабженную стержневой мешалкой, при комнатной температуре загружают соединение I-231 (41 мг, 0,108 ммоль), 2,4,5-трифторбензолсульфонамид (23,2 мг, 0,110 ммоль), DMAP (31,6 мг, 0,259 ммоль), безводный CH2Cl2 (2 мл) и EDCI (51,8 мг, 0,270 ммоль). Емкость герметично закрывают крышкой и дают возможность смеси взаимодействовать в течение 22 часов при комнатной температуре. Содержимое емкости промывают 1M раствором HCl (5 мл), водой (3×5 мл) и насыщенным раствором соли (5 мл). Органическую часть сушат (MgSO4) и концентрируют, получая соединение P323 в виде светло-зеленого твердого вещества. Неочищенный продукт растворяют в кипящем Et2O (1 мл), затем добавляют гексаны (1 мл). Выпавший осадок отделяют фильтрованием с подсосом, получая 23,9 мг соединения P323 в виде светло-зеленого твердого вещества (39%). 1H-ЯМР (400 МГц, CDCl3) 2,29 (с, 3H), 5,37 (с, 2H), 6,18 (д, J=15,2 Гц, 1H), 6,31 (д, J=8,4 Гц, 1H), 6,87 (с, 1H), 6,82 (дд, J=2,8, 10,0 Гц, 1H), 7,04 (дд, J=2,0, 8,4 Гц, 1H), 7,07-7,13 (м, 1H), 7,31 (дд, J=2,8, 8,8 Гц, 1H), 7,41 (д, J=2,4 Гц, 1H), 7,82 (д, J=14,8 Гц, 1H), 7,98-8,04 (м, 1H). ЖХ/МС (96%); МС (ESI-) Вычислено: 570,4 m/z; найдено: 569,4 m/z.
Пример 365. Получение соединения P371.
Синтез 4-(3-метоксифенокси)-1-метил-1Н-индола, I-134L: В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 3-метоксифенолом с получением соединения I-134L. 1Н-ЯМР подтверждает структуру.
Синтез 4-(3-метоксифенокси)-1-метил-1Н-индол-3-карбальдегида, I-135L: В соответствии с общей методикой A-13, соединение I-134L преобразовывают в формальдегид I-135L. 1Н-ЯМР подтверждает структуру.
Синтез этилового эфира (E)-3-[4-(3-метоксифенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-136L. В соответствии с общей методикой A-14, соединение I-135L преобразовывают в этиловый эфир акриловой кислоты I-136L. 1Н-ЯМР подтверждает структуру.
Синтез (E)-3-[4-(3-метоксифенокси)-1-метил-1Н-индол-3-ил]акриловой кислоты, I-137L. В соответствии с общей методикой A-7, соединение I-136L гидролизуют до акриловой кислоты I-137L. 1Н-ЯМР подтверждает структуру.
Синтез соединения P371. В соответствии с общей методикой A-8, акриловую кислоту I-137L подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P371. 1Н-ЯМР подтверждает структуру.
Пример 366. Получение соединения P372.
Синтез соединения P372. В соответствии с общей методикой A-8, акриловую кислоту I-137L подвергают взаимодействию с 2,3,4,5,6-пентафторбензолсульфонамидом с получением соединения P372. 1Н-ЯМР подтверждает структуру.
Пример 367. Получение соединения P376.
Синтез 6-(1-мeтил-1Н-индол-4-илокси)хинолина, I-134M: В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 6-гидроксихинолином с получением соединения I-134M. 1Н-ЯМР подтверждает структуру.
Синтез 1-метил-4-(хинолин-6-илокси)-1H-индол-3-карбальдегида, I-135M: В соответствии с общей методикой A-13, соединение I-134M преобразовывают в формальдегид I-135M. 1Н-ЯМР подтверждает структуру.
Синтез этилового эфира (E)-3-[l-метил-4-(хинолин-6-илокси)-1Н-индол-3-ил]акриловой кислоты, I-136M. В соответствии с общей методикой A-14, соединение I-135M преобразовывают в этиловый эфир акриловой кислоты I-136M. 1Н-ЯМР подтверждает структуру.
Синтез (E)-3-[1-метил-4-(хинолин-6-илокси)-1Н-индол-3-ил]акриловой кислоты, I-137M. В соответствии с общей методикой A-7, соединение I-136M гидролизуют до акриловой кислоты I-137M. 1Н-ЯМР подтверждает структуру.
Синтез соединения P376. В соответствии с общей методикой A-8, акриловую кислоту I-137M подвергают взаимодействию с 3,4-дифторбензолсульфонамидом с получением соединения P376. 1Н-ЯМР подтверждает структуру.
Пример 367. Получение соединения P377.
Синтез соединения P377. В соответствии с общей методикой A-8, акриловую кислоту I-137M подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P377. 1Н-ЯМР подтверждает структуру.
Пример 368. Получение соединения P393.
Синтез 2-(1-метил-1Н-индол-4-илокси)хиноксалина I-134N: В соответствии с общей методикой A-12, соединение I-133 подвергают взаимодействию с 6-гидроксихинолином с получением соединения I-134N. 1Н-ЯМР подтверждает структуру.
Синтез 1-метил-4-(хиноксалин-2-илокси)-1Н-индол-3-карбальдегида, I-135N: В соответствии с общей методикой A-13, соединение I-134N преобразовывают в формальдегид I-135N. 1Н-ЯМР подтверждает структуру.
Синтез этилового эфира (E)-3-[l-метил-4-(хиноксалин-2-илокси)-1Н-индол-3-ил]акриловой кислоты, I-136N. В соответствии с общей методикой A-14, соединение I-135N преобразовывают в этиловый эфир акриловой кислоты I-136N. 1Н-ЯМР подтверждает структуру.
Синтез (E)-3-[l-Метил-4-(хиноксалин-2-илокси)-1Н-индол-3-ил]акриловой кислоты, I-137N. В соответствии с общей методикой A-7, соединение I-136N гидролизуют до акриловой кислоты I-137N. 1Н-ЯМР подтверждает структуру.
Синтез соединения P393. В соответствии с общей методикой A-8, акриловую кислоту I-137N подвергают взаимодействию с 3,4-дифторбензолсульфонамид с получением соединения P393. 1Н-ЯМР подтверждает структуру.
Пример 369. Получение соединения P394.
Синтез соединения P372. В соответствии с общей методикой A-8, акриловую кислоту I-137N подвергают взаимодействию с 2,4,5-трифторбензолсульфонамидом с получением соединения P394. 1Н-ЯМР подтверждает структуру.
Пример 371. Получение соединения P349.
Синтез a,a-ди-[2H]-2,4-дихлорбензилового спирта, I-238. К раствору метил-2,4-дихлорбензоата (2,0 г, 9,75 ммоль) в эфире (20 мл) добавляют LiAlD4 (0,41 г, 9,75 ммоль) при 0°C в атмосфере N2. Смесь перемешивают при 0°C в течение 1 часа, реакцию гасят водой, NaOH (15% водный), водой (1:1:3). Образовавшийся твердый осадок отфильтровывают, фильтрат концентрируют в вакууме. Неочищенный продукт очищают колоночной хроматографией на силикагеле, получая соединение I-238 (1,2 г, 69%). 1H-ЯМР (CDCl3).
Синтез a,a-ди-[2H]-2,4-дихлорбензилбромида, I-239. К раствору соединения I-238 (0,6 г, 3,35 ммоль) и трифенилфосфина (0,97 г, 3,69 ммоль) в CH2Cl2 (10 мл), охлажденному до 0°С, в атмосфере азота добавляют N-бромсукцинимид (0,66 г, 3,69 ммоль). Реакционную смесь перемешивают при 0°C в течение 10 минут и при комнатной температуре в течение 30 минут. После удаления растворителя остаток растирают в 10% растворе эфира в гексане и фильтруют через слой силикагеля, получая соединение I-239 (0,62 г, 77%).1H-ЯМР(CDCl3).
Синтез соединения I-240. К раствору хлоральгидрида (11,3 г, 68,4 ммоль) в воде (143 мл) при энергичном перемешивании добавляют Na2SO4 (15,7 г, 0,486 моль), 2-бром-4-фторанилин (10,0 г, 52,6 ммоль) в смеси 37% HCl (1,2 мл, 57,9 ммоль) и воды (54 мл). После завершения добавления полученную реакционную смесь кипятят с обратным холодильником в течение 10 минут и охлаждают до комнатной температуры. Образовавшийся осадок отделяют фильтрованием, промывают водой (3×100 мл) и сушат в вакууме, получая неочищенный изонитрозоацетанилид. Полученный продукт при энергичном перемешивании добавляют небольшими порциями к концентрированной H2SO4 (178 мл) с такой скоростью, чтобы температура реакционной смеси находилась в интервале от 50 до 70°С. Реакционную смесь нагревают до 80°C и выдерживают при этой температуре в течение 20 минут, затем охлаждают до комнатной температуры. Охлажденную смесь выливают в измельченный лед (~3200 г). Смесь оставляют на 1 часа. Образовавшийся осадок твердого вещества отфильтровывают, водный слой экстрагируют CH2Cl2, промывают насыщенным раствором соли и сушат над Na2SO4. После концентрирования в вакууме остаток растирают со смесью 20% эфира в гексане, получая соединение I-240 (2 г, 17%). 1H-ЯМР(ДМСО-d6); МС (APCI-): 243 (M-1).
Синтез соединения I-241. К Mg (0,44 г/атом) в безводном эфире (4 мл) при комнатной температуре в течение 3 часов добавляют CD3I в безводном эфире (100 мл). После завершения добавления реакционную смесь перемешивают в течение дополнительных 30 минут.
К раствору соединения I-240 (1,65 г, 6,76 ммоль) в безводном ТГФ (85 мл) добавляют CD3MgI, полученный как описано выше, при -78°C в течение 30 минут, и перемешивание продолжают в течение 1 часа. После нагревания смеси до -10°C реакцию гасят водой, экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над Na2SО4 и концентрируют в вакууме. Остаток очищают колоночной хроматографией на силикагеле, получая соединение I-241 (1,5 г, 84%). 1H-ЯМР (ДМСО-d6); МС (APCI-): 262 (M-l).
Синтез соединения I-242. К раствору соединения I-241 (1,2 г, 4,61 ммоль) в ТГФ (60 мл) добавляют BH3 (1M в ТГФ, 11 мл, 11 ммоль) при 0°C и полученную смесь перемешивают при комнатной температуре в течение 5 часов. Реакцию гасят добавлением 1н. водного раствора HCl, смесь экстрагируют EtOAc, промывают насыщенным раствором соли, сушат над Na2SО4 и концентрируют в вакууме. Остаток очищают колоночной хроматографией на силикагеле, получая соединение I-242 (l г, 95%). 1H-ЯМР (ДМСО-d6).
Синтез соединения I-243. Смесь соединения I-242 (1,0 г, 4,33 ммоль) и метилакрилата (0,75 г, 8,66 ммоль) в триэтиламине (6 мл), ацетата палладия (II) (97 мг, 0,43 ммоль) и три-o-толилфосфина (0,26 г, 0,866 ммоль) в ДМФА (20 мл) перемешивают при 100°C в течение 4 часов, затем охлаждают до комнатной температуры. Реакционную смесь разбавляют CH2Cl2 (70 мл), промывают водой (3×40 мл), насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле, получая соединение I-243 (0,9 г, 90%). 1H-ЯМР (CDCl3); МС (APCI-): 235 (M-1).
Синтез соединения I-244. К раствору соединения I-243 (1,0 г, 4,23 ммоль) в ТГФ (5 мл) и метаноле (5 мл) при комнатной температуре добавляют 1н. водный раствор NaOH (0,51 г, 12,7 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем pH доводят до кислотного добавлением 2н. водного раствора HCl. Реакционную смесь экстрагируют EtOAc (2×40 мл). Объединенную органическую фазу промывают водой, насыщенным раствором соли и сушат над сульфатом натрия. После удаления растворителя получают I-244 (0,9 г, 96%). 1Н-ЯМР (500 МГц, ДМСО-d6)
Синтез соединения I-245. Смесь соединения I-244 (0,9 г, 4,1 ммоль), 3,4-дихлор-2-тиофенсульфонамида (1,0 г, 4,45 ммоль), 4-диметиламинопиридин (0,99 г, 8,1 ммоль) и EDCI (1,55 г, 8,1 ммоль) в дихлорметане (20 мл) перемешивают при комнатной температуре в течение ночи. Раствор разбавляют дихлорметаном, промывают разбавленным водным раствором HCl и водой. Неочищенный продукт очищают колоночной хроматографией на силикагеле, получая соединение I-245 (1,6 г, 88%).
Синтез соединения P349. К раствору соединения I-245 (0,8 г, 1,83 ммоль) в ДМФА (30 мл) при 0°С добавляют NaH (60% в минеральном масле, 97 мг, 4,03 ммоль). Смесь перемешивают при комнатной температуре в течение 1 часа, затем добавляют соединение I-239 (0,43 г, 2,2 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи и разбавляют CH2Cl2 (12 мл). Реакционную смесь промывают разбавленным водным раствором HCl, водой, насыщенным раствором соли и сушат над сульфатом натрия. Растворитель удаляют, остаток очищают колоночной хроматографией на силикагеле, получая соединение P349 (0,34 г, 34%).1H & 13C-ЯМР (ДМСО-d6 ); ВЭЖХ: 99% чистота. Элементный анализ. Вычислено для C23H15C14FN2O3S2: C 46,64; H 2,55; N 4,73; Cl 23,94; S 10,83. Найдено: C 46,31; H 2,63; N 4,70; Cl 23,78; S 10,72.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2711502C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2528046C2 |
| СПОСОБ МОДУЛЯЦИИ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2525115C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2382779C2 |
| МОДУЛЯТОРЫ ТРАНСПОРТЕРОВ АТФ-СВЯЗЫВАЮЩЕЙ КАССЕТЫ | 2005 |
|
RU2556984C2 |
| ПЕРИ-ЗАМЕЩЕННЫЕ АРИЛСУЛЬФОНАМИДНЫЕ БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ПРЕДНАЗНАЧЕННЫЕ ДЛЯ ЛЕЧЕНИЯ ОККЛЮЗИОННОГО ЗАБОЛЕВАНИЯ АРТЕРИЙ | 2005 |
|
RU2407736C2 |
| ФОСФОИНДОЛЫ КАК ИНГИБИТОРЫ ВИЧ | 2005 |
|
RU2393163C2 |
| ИНГИБИТОРЫ КИНАЗ, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ МИЕЛОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ И ДРУГИХ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2482112C2 |
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЙ АМИНО-ПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ПРОСТАГЛАНДИНА D2 | 2005 |
|
RU2417990C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2681849C2 |
Описываются новые ацилсульфонамидные пери-замещенные конденсированные бициклические соединения общей формулы (I), значения радикалов указаны в формуле изобретения. Соединения могут использоваться для ингибирования связывания простогландина Е2 с рецептром ЕР3. Описывается также фармацевтическая композиция на основе соединения формулы (I). 2 н. и 28 з.п. ф-лы, 4 табл.
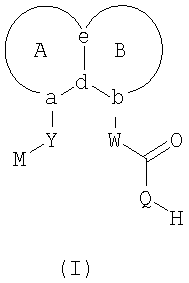
1. Соединение формулы
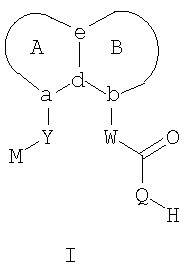
где А и В представляют собой два конденсированных 5- или 6-членных кольца, причем указанная конденсированная А/В кольцевая система содержит от одного до трех гетероатомов, выбранных из атомов азота, кислорода и серы, и указанные кольца дополнительно замещены от 0 до трех заместителями, независимо выбранными из галогена, -ОН, низшего алкила, низшего фторалкила, низшего алкокси-низшего алкила, гидрокси-низшего алкила, оксо, карбокси, карбо-низшего алкокси и ацила, содержащего от 1 до 8 атомов углерода;
«а» и «b» представляют собой точки присоединения остатков Y и W соответственно и «а» и «b» на конденсированной А/В кольцевой системе по отношению друг к другу находятся в пери-положении;
«d» и «е» представляют собой точки слияния колец А и В указанной конденсированной А/В кольцевой системе;
W представляет собой С2-С8алкил, в котором один или два СН2 каждый может быть замещен -O-, -С(=O)- или -NH- или два СН2 могут вместе быть замещены -СН=СН-;
Y представляет собой С1-С8алкил, в котором один или два СН2 каждый могут быть замещены -O-, -С(=O)-, -S-, -SO-, -SO2-, =N- или -NH- или два СН2 могут вместе быть замещены -СН=СН- или =NSO2-;
М выбран из фенила, нафтила, фенила, замещенного до трех раз галогеном, низшим алкилом, низшим галогеналкилом, низшей алкоксигруппой, низшей галогеналкоксигруппой, циано, фенилом, метилендиокси, ациламино, содержащим от 1 до 8 атомов углерода, гетероарила, означающего 5- или 6-членное гетероароматическое кольцо, содержащее от 1 до 4 гетероатомов, выбранных из О, N или S, бициклическую 9- или 10-членную гетероароматическую кольцевую систему, содержащую от 1 до 2 гетероатомов, выбранных из О, N или S, гетероарила, замещенного низшим алкилом, низшей алкоксигруппой, тетрагидропиранила, имидазопиридинила;
Q выбран из -N(SO2R1)- или -N[РО(O-низший алкил)2]-, Q может дополнительно представлять собой -NH-;
R1 выбран из фенила, фенила, замещенного до пяти раз галогеном, низшей алкоксигруппой, низшим алкилом, тиофена, изоксазола и тиофена, замещенных до 2 раз галогеном, низшим алкилом, низшего фторалкила.
2. Соединение по п.1, где W представляет собой C1-2алкил, в котором один или два
-СН2- могут быть замещены -O-, -С(=O) или -NH- или два СН2 могут вместе быть замещены -СН=СН-; и
Y представляет собой С1-С2алкил, в котором один или два -СН2- могут быть замещены -O-, -С(O)-, -S-, -SO-, -SO2-, -NH- или два СН2 могут вместе быть замещены
-СН=СН-.
3. Соединение по п.1, где W выбран из -СН2СН2-, -ОСН2-, -С(=O)-, -CH2O-,
-ОС(СН3)2-, -ОСН(СН3)-, -СН=СН-, и -NHCH2-; Y выбран из -СН2-, -O-, -ОСН2-, -N-,
-NH-, -NSO2, -NHSO2, -NHC(=O)-, -S-, -SO- и -SO2-, где левая связь показывает точку присоединения к кольцу А или кольцу В.
4. Соединение по любому из пп.1-3, где Q представляет собой -N[РО(O-низший алкил)2]-.
5. Соединение по любому из пп.1-3, где Q представляет собой -N(SO2R1)-.
6. Соединение по п.5, где R1 выбран из фенила, фенила, замещенного до пяти раз галогеном, низшим алкилом, низшей алкоксигруппой, тиофена, изоксазола и тиофена, замещенных до 2 раз галогеном, низшим алкилом и CF3.
7. Соединение по п.6, где R1 представляет собой фторфенил.
8. Соединение по любому из пп.1-3, где М представляет собой фторфенил.
9. Соединение по любому из пп.1-3, где А/В кольцевая система представляет собой пару конденсированных 5-членных колец:
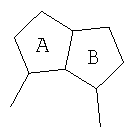
10. Соединение по п.9, где А/В кольцевая система выбрана из
 и
и 
11. Соединение по любому из пп.1-3, где А/В кольцевая система представляет собой пару конденсированных 6-членных колец:
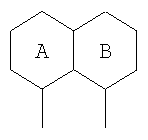
12. Соединение по п.11, где А/В кольцевая система выбрана из
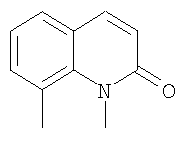
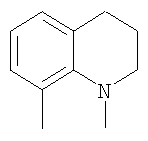 и
и 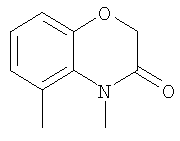
13. Соединение по любому из пп.1-3, где А/В кольцевая система представляет собой конденсированную пару 5-членного и 6-членного колец:
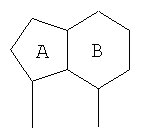 или
или 
14. Соединение по п.13, где А/В кольцевая система выбрана из индола, индолона, пергидроиндолона, изатина, бензимидазола, бензоксазолинона, бензофурана и индазола.
15. Соединение по п.3, где А/В кольцевая система представляет собой индол или индолон.
16. Соединение по п.15, где А/В кольцевая система представляет собой индол.
17. Соединение по п.16, где Q представляет собой -N(SO2R1)- и R1 выбран из фенила, фенила, замещенного от двух до трех раз галогеном, низшей алкоксигруппой, низшим алкилом, тиофена, изоксазола и тиофена, замещенных до 2 раз галогеном, низшим алкилом, и CF3.
18. Соединение по п.17, где М выбран из фенила, замещенного до трех раз галогеном, низшим алкилом, низшим галогеналкилом, низшей алкоксигруппой, низшей галогеналкоксигруппой, циано, фенилом, метилендиоксигруппой, нафтила, хиналинила и имидазопиридинила.
19. Соединение по п.18, где, по меньшей мере, один из R1 и М представляет собой фторфенил.
20. Соединение по п.18, где Y представляет собой -СН2- и W представляет собой
-СН=СН-.
21. Соединение по п.20, которое представляет собой
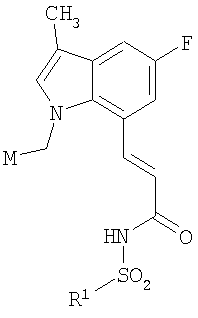
22. Соединение по п.21, где М представляет собой 2,4-дихлорфенил и R1 представляет собой 4,5-дихлортиен-2-ил.
23. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и соединение по любому из пп.1-3 или 14-22 для ингибирования связывания простагландина Е2 с рецептором ЕР3.
24. Соединение по п.1 формулы
где А и В представляют собой 6-членное кольцо карбоцикла, конденсированное с 5-членным кольцом карбоцикла, содержащего азот, где А/В кольцевая система выбрана из индола, восстановленного индола, 2-оксоиндола и восстановленного 2-оксииндола.
25. Соединение по п.24, где указанная А/В кольцевая система представляет собой индол формулы

где R3 и R4 являются заместителями в одном из двух или обоих кольцах, независимо выбранными из галогена, -ОН, низшего алкила, низшего фторалкила, низшего алкокси-низшего алкила, гидрокси-низшего алкила, оксо, карбокси, карбоалкокси и ацила, содержащего от 1 до 8 атомов углерода.
26. Соединение по п.24, где указанная А/В кольцевая система представляет собой 2-оксоиндол формулы
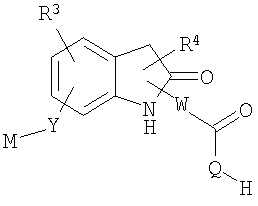
где R3 и R4 являются заместителями в одном из двух или обоих кольцах, независимо выбранными из галогена, -ОН, низшего алкила, низшего фторалкила, низшего алкокси-низшего алкила, гидрокси-низшего алкила, оксо, карбокси, карбо-низшего алкокси и ацила, содержащего от 1 до 8 атомов углерода.
27. Соединение по п.24, где указанная А/В кольцевая система представляет собой восстановленный 2-оксоиндол формулы
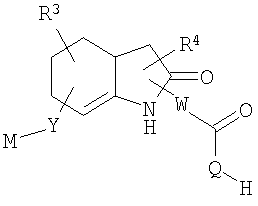
где R3 и R4 являются заместителями в одном из двух или обоих кольцах, независимо выбранными из галогена, -ОН, низшего алкила, низшего фторалкила, низшего алкокси-низшего алкила, гидрокси-низшего алкила, оксо, карбокси, низшего карбо-низшего алкокси и ацила, содержащего от 1 до 8 атомов углерода.
28. Соединение по п.24, где М выбран из фенила, нафтила, фенила, замещенного до трех раз галогеном, низшим алкилом, низшим галогеналкилом, низшей алкоксигруппой, низшей галогеналкоксигруппой, циано, фенилом, метилендиокси, ациламино, содержащим от 1 до 8 атомов углерода, гетероарила, означающего 5- или 6-членное гетероароматическое кольцо, содержащее от 1 до 4 гетероатомов, выбранных из О, N или S, бициклическую 9- или 10-членную гетероароматическую кольцевую систему, содержащую от 1 до 2 гетероатомов, выбранных из О, N или S, гетероарила, замещенного низшим алкилом, низшей алкоксигруппой, тетрагидропиранила, имидазопиридинила.
29. Соединение по п.28, где W представляет собой мостиковую связь, содержащую два атома в цепи и Y представляет собой мостиковую связь, содержащую один или два атома в цепи.
30. Соединение по п.29, где W выбрано -СН2О-, -OCF2-, ОС(СН3)2-, -ОСН(СН3)-,
-СН=СН- и -NHCH2-;
Y выбран из -СН2-, -O-, -ОСН2-, -S-, -SO- и -SO2-, где левая связь показывает точку присоединения к кольцу А или кольцу В.
| Устройство для электрохимического снятия заусенцев | 1979 |
|
SU882718A2 |
| US 6348474 B1, 19.02.2002 | |||
| US 6348032 B1, 19.02.2002 | |||
| Способ подготовки массы для изготовления бумажного диэлектрика | 1975 |
|
SU539117A1 |
| ЕР 0620122 А1, 19.10.1994 | |||
| ЕР 1431267 А1, 26.03.2004 | |||
| 3-Метокси-4-[1-метил-5-(2-метил-4,4,4-трифторбутилкарбамоил)индол-3-илметил]-N-(2-метилфенилсульфонил)бензамид или его фармацевтически приемлемые соли в качестве антагонистов лейкотриена и полупродукты для их получения | 1990 |
|
RU2002740C1 |

