Область техники, к которой относится изобретение
Настоящее изобретение относится к определенным новым альдегидам, моющим спиртам, поверхностно-активным веществам и потребительским продуктам, таким как продукты для стирки, продукты личной гигиены, продукты для ухода за посудой, шампуни и продукты для очистки твердых поверхностей и подобные продукты, содержащие указанные композиции поверхностно-активных веществ. Также описаны способы получения новых альдегидов, спиртов и поверхностно-активных веществ.
Уровень техники
Поверхностно-активные вещества даже в настоящее время являются единственными важными чистящими ингредиентами в продуктах для стирки и бытовых продуктах для очистки. Анионные поверхностно-активные вещества как класс являются наиболее распространенными в терминах применения во всем мире и их типично применяют на уровнях от 30 до 40% моющей композиции. Развитие данного класса, который иногда имеет название "основные поверхностно-активные вещества", всегда было замедленным из-за длительных периодов разработок и высокой стоимости капитальных вложений в данной индустрии продуктов, требующей ежегодных многомиллиардных вложений фунтов стерлингов. Изменения часто вызваны изменениями потребностей потребителей или их привычек, такими как разработка новых типов тканей, которые могут требовать более низких температур стирки и циклов щадящей стирки или в динамичном обществе, в котором мы теперь живем, все более короткие периоды стирки становятся нормой. Все вышеуказанные факторы сыграли роль в последних разработках новых анионных поверхностно-активных веществ. В результате потребности в поверхностно-активных веществах, обладающих свойствами, которые придают им высокую устойчивость к осаждению кальцием и магнием в жесткой воде, а также повышенными очистительными свойствами при более холодных температурах стирки и при более коротких циклах стирки, в последние годы было осуществлено несколько химических разработок, которые привели к введению конкретных метильных и этильных разветвленных поверхностно-активных веществ. Примеры таких разработок описаны в статье J. Scheibel, Journal of Surfactants and Detergents, "The Evolution of Anionic Surfactant Technology to Meet the Requirements of the Laundry Detergent Industry", volume 7, number 4, October, 2004 ("Scheibel JSD Article" в данной заявке), где определены потребность и разработки таких технологий разветвленных поверхностно-активных веществ. Технологии указывают на необходимость в минимизации разветвления с обеспечением эффективных поверхностно-активных веществ с хорошей биораспадаемостью.
Сильно разветвленные поверхностно-активные вещества были получены из тетрапропилена и имеют название алкилбензолсульфонаты или "ТВЕРДЫЕ ABS". Твердые ABS имеют очень сложные разветвленные структуры, состоящие из 3 или 4 разветвлений на молекулу. Структура, приведенная ниже, иллюстрирует один пример твердой ABS молекулы. Иллюстрация демонстрирует четыре разветвления с метильным и этильными разветвлениями в четвертичном, а также геминальном разветвлениях.
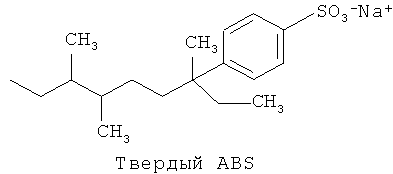
Твердые ABS, как было найдено, имеют значительно более низкую биораспадаемость, чем линейные альтернативы. Спиртовые сульфаты, полученные из такого высокоразветвленного тетрапропиленового исходного вещества, имеют аналогичные проблемы, что и твердые ABS, включая ингибированную биораспадаемость. Как таковые, твердые ABS и родственные спиртовые сульфаты имеют ограниченное применение в продуктах для стирки или других потребительских продуктах.
Один пример продающихся в данное время разветвленных поверхностно-активных веществ, которые используют в потребительских продуктах, представляет собой слегка разветвленный алкилсульфат и имеет название "HSAS" для высокорастворимого алкилсульфата. HSAS проиллюстрирован в статье Scheibel JSD и других внешних статьях. HSAS получают из нефтяного сырья. Легкие разветвления вещества обеспечивают высокую растворимость, сопротивляемость механическим воздействиям и хорошие эксплуатационные характеристики.
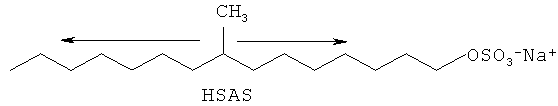
Таким образом, хотя данное поверхностно-активное вещество и другие разработаны для удовлетворения потребностей потребителей сегодня в очистке в холодной воде, остается проблема обеспечения альтернативных разветвленных поверхностно-активных веществ из ненефтяных источников для дальнейшей экологической безопасности при производстве моющих средств, а также в других производствах, которые основаны на технологии поверхностно-активных веществ и предпочитают разветвленные вещества со свойствами HSAS.
В данной заявке описаны способы получения новых альдегидов, спиртов и поверхностно-активных веществ, полезных в композиции потребительских продуктов, таких как продукты личной гигиены и продукты для стирки и чистки.
Сущность изобретения
В данной заявке описан ациклический альдегид, имеющий 16 или 21 атомов углерода, содержащий, по меньшей мере, три разветвления и три или менее углерод-углеродные двойные связи, который является полезным исходным веществом для получения моющих поверхностно-активных веществ, и его конкретные осуществления.
Также описана композиция моющих спиртов, содержащая, по меньшей мере, один ациклический спирт, имеющий 16 атомов углерода, содержащий, по меньшей мере, три разветвления, при этом разветвления представляют собой метил, этил или их смеси.
Также описана композиция поверхностно-активного вещества, содержащая одно или более поверхностно-активных производных изомеров ациклического моющего спирта, содержащего 11, 16 или 21 атомов углерода и два, три, четыре или пять метальных или этильных разветвлений или их смесей.
Способ получения смеси моющих спиртов, включающий стадии, на которых (а) обеспечивают один или более полиразветвленных полиолефинов, при этом полиразветвленные полиолефины должны содержать один неразветвленный концевой олефин и один или более дополнительных разветвленных олефинов в молекуле; (b) гидроформилируют указанные полиразветвленные полиолефины с получением полиразветвленного олефина, содержащего альдегидный продукт с одним или более олефинами или их смесью, с применением катализатора, выбранного из группы, состоящей из модифицированных или немодифицированных переходных металлов группы IX, и условий процесса, включающих температуру процесса в диапазоне от приблизительно 50°С до приблизительно 130°С, молярное соотношение водорода и моноксида углерода в диапазоне от приблизительно 0,25:1 до приблизительно 4:1 и общее давление в диапазоне от приблизительно 300 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм; (с) восстанавливают альдегидный продукт, полученный на стадии (b) в присутствии водорода и катализатора гидрогенизации, с применением условий процесса, включающих температуру процесса в диапазоне от приблизительно 20°С до приблизительно 130°С и давление водорода в диапазоне от 100 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм с образованием смеси полиразветвленных моющих спиртов; и (d) удаляют указанную смесь полиразветвленных моющих спиртов из указанного катализатора.
Способ получения смеси моющих спиртов, при этом указанный способ включает стадии, на которых (а) обеспечивают полиразветвленные полиолефины, содержащие один неразветвленный концевой олефин и один или более дополнительных разветвленных олефинов в молекуле; (b) гидроформилируют и восстанавливают указанный полиразветвленный полиолефин с применением катализатора, выбранного из конкретных модифицированных переходных металлов группы IX и условий процесса, включающих температуру процесса в диапазоне от приблизительно 90°С до приблизительно 200°С, молярное соотношение водорода и моноксида углерода в диапазоне от приблизительно 2 к 1 до приблизительно 5 к 1 и общее давление в диапазоне от приблизительно 300 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм; и (с) удаляют указанную спиртовую композицию из указанного катализатора.
Настоящее изобретение основано на неожиданном открытии того, что моющие спирты и производные с двумя или более разветвлениями могут иметь хорошую биораспадаемость, высокую растворимость в холодной жесткой воде, являются высокоэффективными в комбинации с другими моющими ингредиентами, такими как со-поверхностно-активные вещества, ферменты, модифицирующие компоненты, хелаторы и чистящие полимеры. Дополнительно, будут определены способы, обеспечивающие улучшенную синтетическую эффективность по сравнению с получением других разветвленных поверхностно-активных веществ на основе нефтяного исходного вещества.
Подробное описание изобретения
Настоящее изобретение относится к способу получения смеси моющих спиртов, включающему стадии, на которых:
а. обеспечивают один или более полиразветвленных полиолефинов, при этом полиразветвленные полиолефины должны содержать один неразветвленный концевой олефин и один или более дополнительных разветвленных олефинов в молекуле;
b. гидроформилируют полиразветвленные полиолефины с получением полиразветвленного олефина, содержащего альдегидный продукт с одним или более олефинами или их смесью;
с. восстанавливают альдегидный продукт, полученный на стадии (b) в присутствии водорода и катализатора гидрогенизации; и
d. удаляют полученную в результате полиразветвленную спиртовую смесь из указанного катализатора.
Одно осуществление данного способа включает стадию гидроформилирования и стадию восстановления, которые проводят одновременно.
Полиразветвленные полиолефиновые структуры
Ключевым элементом способа в соответствие с настоящим изобретением является исходное вещество - Полиразветвленные полиолефины. Для того чтобы лучше проиллюстрировать возможную сложность предпочтительного полиразветвленного полиолефинового исходного вещества для настоящего изобретения, ниже показаны структуры (а)-(j). Это только несколько из сотен возможных предпочтительных структур, которые составляют возможное исходное вещество, и они не должны рассматриваться как ограничивающие настоящее изобретение.
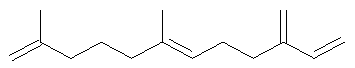
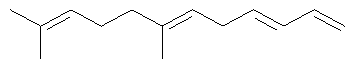
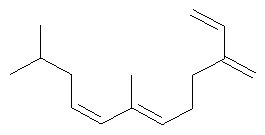
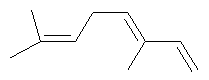
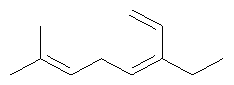
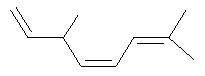
Молекула, представленная структурой (d), может потенциально быть получена из диизопрена и является иллюстрацией полезности способа применения других исходных веществ, кроме тех, которые являются исключительно описанными исходными веществами для предпочтительных изобретений.
Соединение (а), (b), (с) и (е) может быть получено из:
i. фарнезена природного происхождения, экстрагированного из уже существующих растений и организмов;
ii. фарнезена, полученного посредством генетически модифицированных организмов;
iii. синтетически полученных тримеров изопрена;
iv. их смесей.
Другие проиллюстрированные примеры полиразветвленных полиолефинов показаны для того, чтобы документально показать полезность способов в соответствие с настоящим изобретением для функционирования с другими олефинами, которые не могут быть получены по способам i, ii, iii или iv. Эти примеры являются менее предпочтительными.
Высокопредпочтительным олефином в соответствие с настоящим изобретением является (k), который может быть использован для превращения предпочтительного спирта 1 в соответствие с настоящим изобретением.
i. Фарнезен природного происхождения, экстрагированный из уже существующих растений и организмов:
Примеры фарнезенов природного происхождения и возможных других проиллюстрированных структур могут относиться к классу природных веществ под названием терпены. Терпены представляют собой большой и разнообразный класс углеводородов, полученных главным образом из широкого разнообразия растений, в особенности хвойных и других сосновых, хотя также при помощи некоторых насекомых, например, бабочки-кавалера. Поскольку многие из таких веществ выделены из растений и других природных организмов, часто присутствующих как макросмеси, может быть желаемой очистка компонентов перед применением в способах в соответствие с настоящим изобретением. См. патент США №4,605,783.
Термин "фарнезен" относится к набору из шести близко родственных химических соединений, все из которых являются сесквитерпенами. α-Фарнезен и β-фарнезен являются изомерами, которые отличаются расположением одной двойной связи, α-Фарнезен (структура (b) выше) является 3,7,11-триметил-1,3,6,10-додекатетраеном, а β-фарнезен (структура (а) выше) является 7,11-диметил-3-метилен-1,6,10-додекатриеном. Альфа форма может существовать в виде четырех стереоизомеров, которые отличаются геометрией двух из трех внутренних двойных связей (стереоизомеры третьей внутренней двойной связи являются идентичными). Бета изомер существует в виде двух стереоизомеров вокруг геометрии центральной двойной связи.
О двух из стереоизомеров а-фарнезена сообщено в Nature. (Е,Е)-α-Фарнезен является наиболее распространенным изомером. Он найден в кожуре яблок и других фруктов. (7,Е)-α-Фарнезен был выделен из периллового масла.
β-Фарнезен имеет один встречающийся в природе изомер. Е изомер является составляющей различных эфирных масел. Было показано, что некоторые растения, включая сорта картофеля, синтезируют данный изомер.
ii. Фарнезен. полученный посредством генетически модифицированных организмов:
Некоторые недавние примеры теперь позволяют доставку фарнезена и других производных изопрена посредством генетически модифицированных организмов. Примеры таких источников могут быть найдены в патенте США №7,399,323 В2. В данной ссылке описано возможное применение фарнезана в качестве топлива, полученного посредством генетически сконструированного фарнезена. Другой источник генетически сконструированного фарнезена и изопренов описан в патенте США №6,872,556 В2.
iii. Синтетически полученные тримеры изопрена:
Синтетически полученные тримеры могут быть получены из различных источников, два из которых показаны в японских патентах JP 52031841 и JP 48040705. В JP 48040705 описан способ получения соединения (b), как проиллюстрировано выше. Способ включает олигомеризацию изопрена в присутствии двухвалентного Ni, производных фосфина и органомагниевых соединений, с получением высоких выходов, т.е., 75% соединения (b). Доступны другие синтетические способы получения тримеров.
Смеси любых описанных выше неограничивающих исходных веществ могут быть применены в способах в соответствие с настоящим изобретением, так же, как и изомерные формы.
Способ получения моющей спиртовой смеси
Первый способ осуществления настоящего изобретения представляет собой способ получения смеси моющих спиртов, включающий стадии, на которых:
а. обеспечивают один или более полиразветвленных полиолефинов, при этом полиразветвленные полиолефины должны содержать один неразветвленный концевой олефин и один или более дополнительных разветвленных олефинов в молекуле;
b. гидроформилируют указанные полиразветвленные полиолефины с получением полиразветвленного олефина, содержащего альдегид с одним или более олефинами или их смесью, с применением катализатора, выбранного из переходных металлов группы IX, модифицированных или немодифицированных, и условий процесса, включающих: температуру процесса в диапазоне от приблизительно 50°С до приблизительно 130°С, молярное соотношение водорода и моноксида углерода в диапазоне от приблизительно 0,25 к 1 до приблизительно 4 к 1, общее давление в диапазоне от приблизительно 300 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм;
с. восстанавливают альдегидный продукт, полученный на стадии (b) в присутствии водорода и катализатора гидрогенизации, с применением условий процесса, включающих: температуру процесса в диапазоне от приблизительно 20°С до приблизительно 130°С, давление водорода в диапазоне от 100 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм; и
d. удаляют указанную полиразветвленную спиртовую композицию из указанного катализатора.
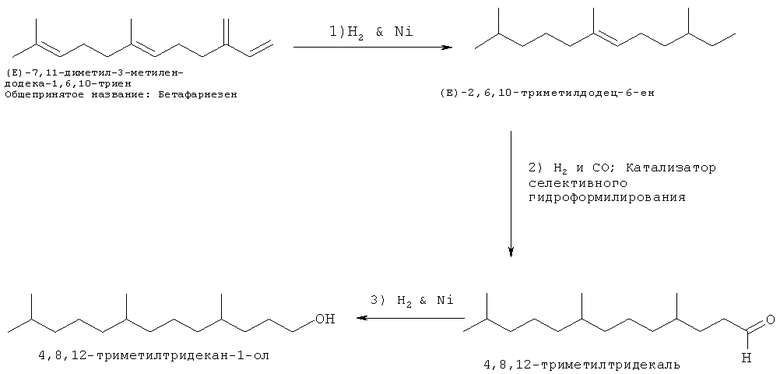
Такое первое осуществление способа может быть проиллюстрировано при помощи следующей СХЕМЫ ПРОЦЕССА I, где использован, как неограничивающеий пример, альфа-фарнезен в качестве исходного вещества.
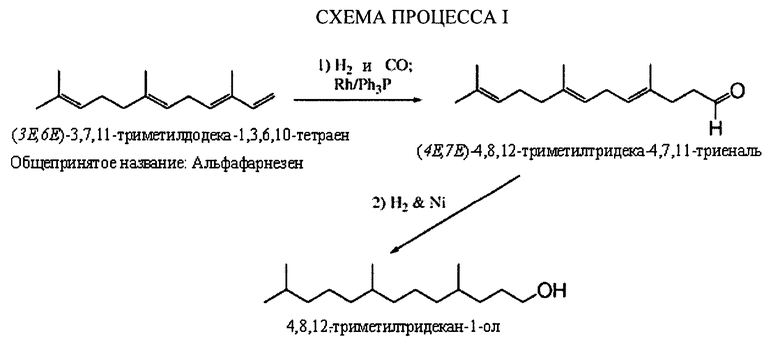
Выбор олефина на стадии а способа предварительно проиллюстрирован выше. Могут быть применены любая смесь или одно вещество из списка структур или другие, содержащие элементы, являющиеся полиразветвленными и полиолефиновыми, где один олефин не разветвлен в концевом положении цепи.
Стадия 1 - Гидроформилирование - Один или более полиразветвленных полиолефинов (альфа-фарнезен показан в данной заявке) может реагировать в присутствии водорода, моноксида углерода и родиевого/трифенилфосфинового катализатора с получением желаемых полиразветвленных полиолефиновых альдегидов. Другие металлы группы IX, такие как кобальт, могут быть также использованы на данной стадии способа. Кобальт и родий являются предпочтительными, но иридий также приемлем для данного способа. Карбонилгидридотрис(трифенилфосфин)родий(1) является металлокомплексом, который может быть приобретен у Aldrich Chemical и других источников, для использования вместе с трифенилфосфином. Поскольку некоторые катализаторы гидроформилирования являются пирофорными, то желательно использовать стандартные способы получения и обработки для поддержания уровней кислорода ниже 40 ppm, в среднем ниже 1 ppm.
Перемешивание получают путем применения покрытой ПТФЕ магнитной мешалки, помещенной в стеклянную рубашку 300 мл реактора. Реактор, в свою очередь, помещен на магнитную пластину для перемешивания, которая магнитным образом соединена с магнитной мешалкой. Скорости перемешивания до 200 об/мин являются возможными без утраты магнитного соединения.
Также может быть применен немодифицированный Rh, но он может требовать применения высоких температур и давлений при применении из-за более низкой селективности. HRh(CO)(PPh3)2 является катализатором, обеспечивающим хорошую селективность, в особенности при применении на Стадии 1 при 25°С, 90-200 фунтов на кв. дюйм и соотношениях смесей моноксида углерода и водорода, составляющих 1:1. Другие катализаторы, такие как НКА(СО)(РРh3)2, могут также обеспечивать хорошую селективность при применении в условиях реакции, например, от 80 до 100 фунтов на кв. дюйм и 90°С и соотношениях смесей моноксида углерода и водорода, составляющих 1:1, и высоких соотношениях избыточного трифенилфосфина при приблизительно 800:1 по отношению к родию. Применение родия с избытком фосфинового лиганда создает активную, селективную и стабильную каталитическую систему при 80-100 фунтов на кв. дюйм и 90°С.
Температура, давление и соотношение моноксида углерода и водорода необходимы для того, чтобы контролировать реакцию с получением моноальдегида на стадии b данного способа изобретения (СХЕМА ПРОЦЕССА 1, Стадия 2). Могут быть использованы температуры в диапазоне от 60 до 90°С с давлениями от 300 до 600 фунтов на кв. дюйм и соотношениями моноксида углерода и водорода, составляющие 2:1. Как указано выше, модифицированный родий является предпочтительным, однако, при желании использовать немодифицированный катализатор для стадии b способа, необходимо использовать вместо этого кобальт, ввиду его высокой реакционной способности и возможности изомеризовать олефины с получением большего количества желаемых продуктов концевого присоединения. Необходимо также применять более высокие соотношения водорода с кобальтом для того, чтобы избежать внутреннего гидроформилирования, приводящего к менее желаемым продуктам, которые не входят в объем настоящего изобретения.
Образование полиальдегида может быть достигнуто путем проведения процесса при температурах выше 90°С. Более высокие соотношения моноксида углерода и водорода могут быть также применены для максимизации диальдегидов и других полиальдегидов.
Стадия 2 - Восстановление - На стадии 2, полученные полиразветвленные полиолефиновые альдегиды реагируют с водородом в присутствии катализатора восстановления, такого как никель, с обеспечением, по существу, триметилзамещенного ненасыщенного спирта. Никель на кизельгуре является одним неограничивающим примером восстановительной каталитической системы. Родий на кремнеземе, палладий на кизельгуре являются другими примерами катализаторов, которые могут быть применены для восстановления полиразветвленных полиолефиновых альдегидов.
Стадию с способа проводят с различными катализаторами в диапазоне от никеля на кизельгуре, родия на кремнеземе, палладия на кизельгуре, которые являются другими примерами катализаторов, которые могут быть применены для восстановления полиразветвленных полиолефиновых альдегидов. Условия реакции варьируют от 20°С до приблизительно 130°С, давление водорода в диапазоне от 100 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм водорода и загрузки катализаторов могут типично находиться в диапазоне от 1 до 5% на субстрате относительно полиразветвленного полиолефинового альдегида. Таким образом, высокоэффективный процесс определен обеспечением специфичного поверхностно-активного спирта и спиртовых смесей для применения при получении поверхностно-активных веществ. Времена реакции будут варьироваться в соответствии с соотношением катализаторов, выбранной температурой и давлением водорода. Типичные условия составляют 150°С при 1000 фунтов на кв. дюйм в течение 16 часов при периодическом режиме. Способ не органичен периодическими процессами. Непрерывная реакция может быть также применена для настоящего изобретения. Образование парафина может наблюдаться во время последовательности процессов, но он легко удаляется путем перегонки полиразветвленного полиолефинового альдегида после проведения стадии с способа или он может быть также удален из полиразветвленного спирта после проведения стадии d способа в случае необходимости. Таким образом, определен высокоэффективный процесс для обеспечения специфичного поверхностно-активного спирта и спиртовых смесей для применения при получении поверхностно-активных веществ. Полиразветвленные спиртовые композиции могут быть превращены посредством ряда традиционных средств в композиции поверхностно-активных веществ, например, моющего спиртового этоксилата, моющего спиртового сульфата и моющего этоксилированного спиртового сульфата, которые проиллюстрированы в примерах синтеза.
ПРИМЕР СИНТЕЗА I: применение СХЕМЫ ПРОЦЕССА I:
Синтез полученного из фарнезена полиразветвленного полиолефина, содержащего альдегид, и его смесей
1,6 грамма карбонилгидридотрис(трифенилфосфин)родия(I) [17185-29-4], 3,0 грамма трифенилфосфина [603-35-0] и 336 грамм смеси изомеров альфа-фарнезена [502-61-4] загружали в сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании. Реактор продували воздухом при помощи циклов вакуум-азот с последующей загрузкой смеси моноксида углерода и водорода в соотношении 2:1 до начального давления, составляющего 300 фунтов на кв. дюйм. Реактор нагревали до 85°С при перемешивании при помощи магнитной мешалки при 500 об/мин и давление доводили до 600 фунтов на кв. дюйм при помощи смеси моноксида углерода и водорода в соотношении 2:1. По мере расходования моноксида углерода и водорода в реакции давление поддерживали путем использования смеси моноксида углерода и водорода в соотношении 1:1. Содержимое реактора отбирали по времени и анализировали при помощи газовой хроматографии ("ГХ") для контроля хода реакции. Когда проба ГХ анализа указывала на то, что исходный альфа-фарнезен полностью израсходован, реакционную смесь охлаждали до комнатной температуры и смесь моноксид углерод: водород отводили. В зависимости от чистоты альфа-фарнезена процесс может проходить от нескольких часов до 70 часов. Перед тем как перейти к следующей стадии реакции, остаток моноксида углерода удаляли при помощи циклов вакуум-азот. Альдегидную смесь не удаляли из реактора до превращения в спирт в ПРИМЕРЕ II, хотя альдегид может быть очищен при желании или при применении в других реакциях.
ПРИМЕР СИНТЕЗА II: применение стадий c,d СХЕМЫ ПРОЦЕССА I.
Синтез полученного из фарнезена полиразветвленного спирта и его смесей
20 грамм никеля на кизельгуре (60 масс.% загрузка) и 200 мл тетрагидрофурана загружали в сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании. Реактор продували воздухом при помощи циклов вакуум-азот с последующей загрузкой водородом до начального давления, составляющего приблизительно 600 фунтов на кв. дюйм. Смесь нагревали до приблизительно 150°С с перемешиванием при 500 об/мин. Водород загружали до конечного давления, составляющего приблизительно 1000 фунтов на кв. дюйм и поддерживали такое давление в течение 16 часов. Содержимое реактора затем охлаждали до комнатной температуры и давление снижали до приблизительно 50 фунтов на кв. дюйм.
Смесь, которую получали в Примере синтеза I, затем загружали в реактор, исключая поступление воздуха из атмосферы при непрерывном перемешивании содержимого реактора. Катализатор гидроформилирования из Примера синтеза 1 может оставаться в альдегидной смеси или может быть удален из альдегидной смеси перед применением. Реактор затем герметизировали водородом до начального давления, составляющего приблизительно 600 фунтов на кв. дюйм и нагревали до приблизительно 125°С при перемешивании при приблизительно 500 об/мин при помощи магнитной мешалки. Давление водорода затем повышали до 1000 фунтов на кв. дюйм и данное давление поддерживали. Ход реакции контролировали при помощи ГХ, пока не переставал образовываться дополнительный продукт. Время реакции будет варьироваться в соответствии с условиями реакции.
Очистка неочищенной спиртовой смеси может быть достигнута при помощи стандартных известных процедур, таких как перегонка, или другие способы очистки, известные из уровня техники.
ПРИМЕР СИНТЕЗА III: применение СХЕМЫ ПРОЦЕССА I:
Синтез полученной из фарнезена смеси, в основном состоящей из 4,8,12-триметил-тридекан-1-ола (спирт 1) и 3-этил-7,11-диметил-до декан- 1-ола (спирт 2) и их смесей
Сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании при помощи магнитной мешалки использовали в качестве реактора №1, используя вакуум для вытягивания веществ, избегая попадания воздуха. 1,80 грамма карбонилгидридотрис(трифенилфосфин)родия(I) [17185-29-4] и 5,84 грамма ксантофоса [161265-03-8] суспендировали в 77 граммах пентана и загружали в реактор №1. Пентан удаляли, используя вакуум без нагревания, затем добавляли 50 мл толуола. Реактор продували воздухом при помощи циклов вакуум-азот с последующей загрузкой 10 атм смеси моноксида углерода и водорода в соотношении 1:1 и нагревали до 60°С в течение двух часов с последующим охлаждением до 30°С.
Реактор помещали в вакуум, затем 100,86 грамма транс-бета-фарнезена [18794-84-8] плюс 50 мл толуола загружали в реактор, исключая попадание воздуха. Реактор продували воздухом при помощи циклов вакуум-азот с последующей загрузкой приблизительно 44 атм смеси моноксида углерода и водорода в соотношении 2:1. Реактор сначала нагревали до 45°С и поддерживали при данной температуре в течение 19 часов. По мере расходования моноксида углерода и водорода в реакции давление поддерживали путем использования смеси моноксида углерода и водорода в соотношении 1:1.
Содержимое реактора отбирали по времени и анализировали при помощи ГХ для контроля хода реакции. Через 19 часов температуру реакции повышали до 85°С, продолжая реакцию в течение дополнительных 54 часов при поддержании давления. Перед тем как перейти к следующей стадии реакции, остаток моноксида углерода удаляли путем использования нагревания и вакуума. В то же время, толуол выпаривали до менее чем 15%, как определено при помощи ГХ анализа.
Сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании использовали в качестве реактора №2. Никель на кремнеземе (10 грамм 64% никеля на кремнеземе, восстановленного и стабилизированного) суспендированного в 50 мл пентана, загружали в реактор №2 с добавлением дополнительных 50 мл пентана для промывания линий. Пентан выпаривали при помощи нагревания и вакуума. Реактор нагревали от 270 до 275°С в вакууме с последующей загрузкой водородом до 150-250 фунтов на кв. дюйм через нижнее дренажное отверстие для поддержания данной зоны, свободной от катализатора, и предотвращения засорения дренажного отверстия. Реактор оставляли на 15 минут. Водород отводили и реактор затем помещали в вакуум при помощи водяного аспиратора. Реактор загружали водородом второй раз, оставляли на 15 минут, затем продували, а затем применяли вакуум. Это повторяли еще дважды. Реактор затем загружали водородом до приблизительно 250 фунтов на кв. дюйм (всегда через нижнее дренажное отверстие) и реактор оставляли на всю ночь при температуре (270-275°С) и давлении (приблизительно 250 фунтов на кв. дюйм H2). Реактор затем продували, применяли вакуум в течение 15 минут, затем повторно загружали водородом (150-250 фунтов на кв. дюйм) в течение 15 минут. Это повторяли еще дважды. Реактор загружали водородом до 250 фунтов на кв. дюйм, а затем охлаждали до менее чем 40°С.
Дренажную линию реактора №1 подсоединяли к реактору №2. Содержимое реактора №1 загружали в реактор №2, исключая попадание воздуха, путем герметизации реактора №1 водородом и выталкивания жидкости из реактора №1 в реактор №2 при поддержании перемешивания реактора при приблизительно 200 об/мин. Дополнительный водород загружали в реактор через нижнее дренажное отверстие для очистки зоны катализатора. Реактор затем загружали водородом до 150 фунтов на кв. дюйм (всегда через нижнее дренажное отверстие) и реактор перемешивали при приблизительно 500 об./мин. Реакцию продолжали пока не переставал расходоваться водород и пробы, которые выводили из реактора, не указывали на завершение реакции. Реактор нагревали в течение 24 часов при 125°С при поддержании давления водорода от 450 до 500 фунтов на кв. дюйм H2. Смесь продуктов выводили из реактора. Катализатор удаляли путем фильтрования и летучие вещества удаляли при помощи роторного испарителя. Анализ конечной смеси при помощи газовой хроматографии показал, что смесь содержала приблизительно 39% 4,8,12-триметил-тридекан-1-ола, 34% 3-этил-7,11-диметил-додекан-1-ола, 10% общего парафина и смешанных олефинов и 10% общих смешанных диокисленных веществ.
ПРИМЕР СИНТЕЗА IV с использованием стадий a, b СХЕМЫ ПРОЦЕССА I:
Синтез полученного из бета-мирцена (С11) полиразветвленного полиолефина, содержащего альдегид, и его смесей
1,6 грамма карбонилгидридотрис(трифенилфосфин)родия (I) [17185-29-4], 3,0 грамма трифенилфосфина [603-35-0] и 336 грамм бета-мирцена [84776-26-1], смесь изомеров загружали в сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании. Реактор продували воздухом при помощи циклов вакуум-азот с последующей загрузкой смеси моноксида углерода и водорода в соотношении 2:1 до начального давления, составляющего 300 фунтов на кв. дюйм. Реактор нагревали до 85°С при механическом перемешивании при 500 об/мин и давление доводили до 600 фунтов на кв. дюйм при помощи смеси моноксида углерода и водорода в соотношении 2:1. По мере расходования моноксида углерода и водорода в реакции давление поддерживали путем использования смеси моноксида углерода и водорода в соотношении 1:1. Содержимое реактора отбирали по времени и анализировали при помощи ГХ для контроля хода реакции. Когда проба ГХ анализа указывала на то, что исходный бета-мирцен полностью израсходован, реакционную смесь охлаждали до комнатной температуры и смесь моноксид углерод: водород отводили через вентиляционную систему. В зависимости от чистоты бета-мирцена, время процесса может варьироваться. Перед тем как перейти к следующей стадии реакции, остаток моноксида углерода удаляли при помощи циклов вакуум-азот. Альдегидную смесь не удаляли из реактора до превращения в спирт в ПРИМЕРЕ V, хотя альдегид может быть очищен при желании или при применении в других реакциях.
ПРИМЕР СИНТЕЗА V: Применение стадий c,d СХЕМЫ ПРОЦЕССА I.
Синтез полученного из бета-мирцена полиразветвленного спирта и его смесей
Никель на кизельгуре (20 грамм 60-мас.% загрузка) плюс тетрагидрофуран (200 мл) загружали в сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании. Реактор продували воздухом при помощи циклов вакуум-азот с последующей загрузкой водородом до начального давления, составляющего приблизительно 600 фунтов на кв. дюйм. Смесь нагревали до приблизительно 150°С с перемешиванием при 500 об/мин. Водород загружали до конечного давления, составляющего приблизительно 1000 фунтов на кв. дюйм, и поддерживали такое давление в течение 16 часов Содержимое реактора затем охлаждали до комнатной температуры и давление снижали до приблизительно 50 фунтов на кв. дюйм.
Альдегидную смесь, полученную в ПРИМЕРЕ СИНТЕЗА IV, затем загружали в реактор, исключая поступление воздуха из атмосферы при непрерывном перемешивании содержимого реактора. Катализатор гидроформилирования оставался в альдегидной смеси. При желании катализатор может быть удален из альдегидной смеси перед применением. Смесь затем герметизировали водородом при начальном давлении, составляющем приблизительно 600 фунтов на кв. дюйм, и нагревали до приблизительно 125°С при перемешивании при приблизительно 500 об/мин. Давление водорода затем повышали до 1000 фунтов на кв. дюйм и данное давление поддерживали при периодическом отборе содержимого реактора для анализа при помощи ГХ. Ход реакции контролировали при помощи ГХ, пока не переставал образовываться дополнительный продукт. Время реакции будет варьироваться в соответствии с условиями реакции. Очистка неочищенной спиртовой смеси может быть достигнута при помощи стандартных известных процедур, таких как перегонка, или других способов очистки, известных из уровня техники.
ПРИМЕР СИНТЕЗА VI: с использованием СХЕМЫ ПРОЦЕССА I:
Синтез полученной из бета-мирцена смеси, в основном состоящей из 4,8-диметил-нона-1-ола и 3-этил-7-метил-окта-1-ола и их смесей
1,80 грамма карбонилгидридотрис(трифенилфосфин)родия(I) [17185-29-4] и 5,84 грамма ксантофоса [161265-03-8] суспендировали в 77 граммах пентана и загружали в реактор №1, сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании магнитной мешалкой при 300-500 об/мин использовали для всего процесса, используя вакуум для вытягивания веществ, избегая попадания воздуха. Пентан удаляли, используя вакуум без нагревания. Добавляли 50 мл толуола. Реактор продували воздухом при помощи циклов вакуум-азот и затем загружали 10 атм смеси моноксида углерода и водорода в соотношении 1:1. Его нагревали до 60°С в течение двух часов с последующим охлаждением до 30°С. Реактор помещали в вакуум. 100,86 грамма бета-мирцена [18794-84-8] плюс 50 мл толуола загружали в реактор, исключая попадание воздуха. Реактор продували воздухом при помощи циклов вакуум-азот с последующей загрузкой приблизительно 44 атм смеси моноксида углерода и водорода в соотношении 2:1. Реактор сначала нагревали до 45°С и поддерживали при данной температуре в течение 19 часов. По мере расходования моноксида углерода и водорода в реакции давление поддерживали путем использования смеси моноксида углерода и водорода в соотношении 1:1.
Содержимое реактора отбирали по времени и анализировали при помощи ГХ для контроля хода реакции. Через 19 часов температуру реакции повышали до 85°С, продолжая реакцию в течение дополнительных 54 часов при поддержании давления.
Перед тем, как перейти к следующей стадии реакции, остаток моноксида углерода удаляли путем использования нагревания и вакуума. В то же время толуол выпаривали до менее, чем 15% при помощи ГХ анализа.
Никель на кремнеземе (10 грамм 64% никеля на кремнеземе, восстановленного и стабилизированного) суспендированного в 50 мл пентана и загружали в сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании с добавлением дополнительных 50 мл пентана для промывания линий. Пентан выпаривали при помощи нагревания и вакуума. Реактор нагревали от 270 до 275°С в вакууме, с последующей загрузкой водородом от 150 до 250 фунтов на кв. дюйм водорода через нижнее дренажное отверстие для поддержания данной зоны, свободной от катализатора, и предотвращения засорения дренажного отверстия. Реактор оставляли на 15 минут. Водород отводили и реактор затем помещали в вакуум при помощи водяного аспиратора. Реактор загружали водородом, оставляли на 15 минут, затем продували, затем применяли вакуум. Это повторяли еще дважды. Реактор затем загружали водородом до приблизительно 250 фунтов на кв. дюйм (всегда через нижнее дренажное отверстие) и реактор оставляли на всю ночь при температуре (270-275°С) и давлении (приблизительно 250 фунтов на кв. дюйм H2).
Реактор отводили через вентялиционную систему и применяли вакуум в течение 15 минут. Затем реактор повторно загружали водородом (150-250 фунтов на кв. дюйм) в течение 15 минут. Это повторяли еще дважды. Реактор загружали водородом до 250 фунтов на кв. дюйм, затем охлаждали до <40°С.
Дренажную линию реактора №1 соединяли с реактором №2. Содержимое реактора №1 загружали в реактор №2, исключая попадание воздуха, путем герметизации реактора №1 водородом и выталкивания жидкости из реактора №1 в реактор №2 при поддержании перемешивания реактора при приблизительно 200 об/мин. Дополнительный водород загружали в реактор через нижнее дренажное отверстие для очистки зоны катализатора. Реактор затем загружали водородом до 150 фунтов на кв. дюйм (всегда через нижнее дренажное отверстие) и реактор перемешивали при приблизительно 500 об/мин. Реакцию продолжали пока не переставал расходоваться водород и пробы, которые выводили из реактора, не указывали на завершение реакции. Смесь продуктов выводили из реактора, катализатор удаляли путем фильтрования и летучие вещества удаляли при помощи роторного испарителя.
Второе осуществление способа, представленное при помощи СХЕМЫ ПРОЦЕССА II, включает стадию селективной гидрогенизации полиразветвленного полиолефина перед гидроформилированием.
СХЕМА ПРОЦЕССА II
Соответственно данное осуществление включает:
а. обеспечение полиразветвленных полиолефинов в реакторе;
b. селективную гидрогенизацию всех, кроме одного олефина, смеси полиразветвленных полиолефинов с получением смеси полиразветвленных моноолефинов;
с. гидроформилирование продукта смеси полиразветвленных моноолефинов, полученного на стадии (b), в присутствии селективного катализатора гидроформилирования и условий процесса, включающих: температуру процесса в диапазоне от приблизительно 50°С до приблизительно 130°С, молярное соотношение водорода и моноксида углерода в диапазоне от приблизительно 0,25 к 1 до приблизительно 5 к 1, общее давление в диапазоне от приблизительно 300 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм; с получением смеси полиразветвленных альдегидов;
d. восстановление полиразветвленного альдегидного продукта, полученного на стадии (с) в присутствии водорода и металлического катализатора; и
е. удаление указанной композиции полиразветвленного спирта из указанного катализатора.
В некоторых случаях стадия d данного осуществления может быть минимизирована или даже пропущена, поскольку некоторые катализаторы гидроформилирования могут превращать моноолефин непосредственно в спирт с только небольшими количествами альдегидного промежуточного вещества. В данном эквивалентном способе все еще может оставаться потребность в применении стадии d как стадии доочистки для превращения небольшого количества альдегида в спирт, поскольку данный альдегид может быть опасен для реакций, включающих превращение в поверхностно-активные вещества. Примеры таких катализаторов описаны в патенте США №3420898.
Если полиразветвленные моноолефины получены из других биологических или синтетических средств, стадии реакции а и b могут быть пропущены и стадии c и d осуществляют непосредственно.
Селективная гидрогенизация - Катализаторы и системы, которые могут быть применены для стадии b способа СХЕМА ПРОЦЕССА II с получением селективной гидрогенизации в моноолефины, описаны в патенте США №6627778 В2, выданном Xu et аl. В нем описаны специфичные катализаторы и условия реакции превращения диолефинов в моноолефины. Данный способ может быть применен к реакционной последовательности полиразветвленных полиолефинов в данном осуществлении способа. Другие приемлемые катализаторы и системы описаны в патентах США №4695560, 4523048, 4520214, 4761509 и китайском патенте CN 1032157. Некоторые осуществления катализатора в данном способе могут отличаться тем, что он содержит от 1,0 до 25 мас.% никеля, от 0,05 до 1,5 мас.% серы и подложка представляет собой небольшие шарики Аl2О3, полученные масляно-капельным способом, где шарики имеют объем пор от 1,44 до 3,0 см3/г, площадь поверхности, превышающую 150 м2/г, и не содержат драгоценных металлов и, по существу, не содержат галогены, щелочноземельные металлы и щелочные металлы (<0,1 мас.%). Поскольку основным активным элементом катализатора, используемого в данном процессе, является никель, селективная гидрогенизация должна быть проведена при температуре, превышающей 200°С для придания определенной активности. Дополнительно, для повышения селективности диолефинов в моноолефины необходимо часто сульфатировать катализатор так, чтобы подавить его активность.
Другим подходом к обеспечению промежуточного моноолефина, при желании, из стадии b данного осуществления способа является не контроль гидрогенизации, но применение стандартных катализаторов гидрогенизации и позволение образования смеси моноолефина и парафина. Реакционная смесь может быть затем проведена через последовательность процесса гидроформилирования с и восстановления d, а парафин может быть удален из конечного разветвленного спирта после процесса d при помощи стандартной перегонки.
Для данного осуществления стадии с способа гидроформилирования, температура, давление и соотношение водорода и моноксида углерода необходимы для контроля реакции для минимизации образования парафина в данном случае. Предпочтительные температуры находятся в диапазоне от 60 до 90°С при давлениях от 300 до 600 фунтов на кв. дюйм и более высокие соотношения смеси моноксида углерода 2:1 или выше являются предпочтительными или более низкими для минимизации гидрогенизации олефинов в парафины. Как указано выше, модифицированный кобальт является предпочтительным ввиду его более высокой реакционной способности и возможности изомеризовать олефины с получением большего количества желаемых продуктов концевого присоединения. При желании использовать немодифицированный кобальт, необходимо также применять более низкие соотношения водорода для того, чтобы избежать внутреннего гидроформилирования, приводящего к менее желаемым продуктам, которые не входят в объем настоящего изобретения.
Стадию d способа проводят с различными катализаторами в диапазоне от никеля на кизельгуре, родия на кремнеземе, палладия на кизельгуре, которые являются другими примерами катализаторов, которые могут быть применены для восстановления полиразветвленных альдегидов. Условия реакции варьируются от 20°С до приблизительно 130°С, давление водорода в диапазоне от 100 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм водорода и загрузки катализатора могут типично находиться в диапазоне от 1 до 5% на субстрате относительно полиразветвленного полиолефинового альдегида. Таким образом, высокоэффективный процесс определен обеспечением конкретного поверхностно-активного спирта и спиртовых смесей для применения при получении поверхностно-активных веществ. Времена реакции будут варьироваться в соответствии с соотношением катализатора, выбранной температурой и давлением водорода. Типичные условия представляют собой 150°С при 1000 фунтов на кв. дюйм в течение 16 часов в периодическом режиме. Способ не органичен периодическими реакциями, но непрерывная реакция может быть также применена для настоящего изобретения. Образование парафина может наблюдаться во время последовательности процессов, но он легко удаляется путем перегонки полиразветвленного полиолефинового спирта после стадии d способа, или он может быть также удален из полиразветвленного спирта после осуществления стадии е способа в случае необходимости.
ПРИМЕР СИНТЕЗА VII: (СХЕМА ПРОЦЕССА II):
Синтез полученного из фарнезена полиразветвленного моноолефина и его смесей
Катализатор никель на кремнеземе (5 грамм 64% никеля на кремнеземе, восстановленного и стабилизированного) суспендировали в 50 мл пентана и загружали в сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании с добавлением дополнительных 50 мл пентана для промывания линий. Пентан выпаривали при помощи нагревания и вакуума. Реактор нагревали от 270 до 275°С в вакууме с последующей загрузкой водородом от 150 до 250 фунтов на кв. дюйм водорода через нижнее дренажное отверстие для поддержания данной зоны, свободной от катализатора, и предотвращения засорения дренажного отверстия. Реактор оставляли на 15 минут. Водород отводили, а реактор затем помещали в вакуум при помощи водяного аспиратора. Реактор загружали водородом, оставляли на 15 минут, затем продували и применяли вакуум. Это повторяли еще дважды. Реактор затем загружали водородом до приблизительно 250 фунтов на кв. дюйм (всегда через нижнее дренажное отверстие) и реактор оставляли на всю ночь при температуре (270-275°С) и давлении (приблизительно 250 фунтов на кв. дюйм H2). Реактор затем продували, применяли вакуум к реактору в течение 15 минут, а затем реактор повторно загружали водородом (150-250 фунтов на кв. дюйм) в течение 15 минут. Это повторяли еще дважды. Реактор загружали водородом до 250 фунтов на кв. дюйм, а затем охлаждали до <40°С.
Транс-бета-фарнезен [18794-84-8] (100 грамм) загружали в цилиндр для хранения проб на 300 мл с добавлением 50 мл пентана до метки. Цилиндр для хранения проб подсоединен к реактору на 600 мл системой трубок и клапанов. Цилиндр для хранения проб продували в атмосферу, используя циклы вакуум-водород. Водород вводили через нижнее отверстие цилиндра для хранения проб и через жидкую смесь для того, чтобы способствовать разбрызгиванию жидкости при удалении нижних слоев воздуха. Всего проводили четыре цикла вакуум-водород. Транс-бета-фарнезеновую смесь затем загружали в реактор на 600 мл, исключая попадание воздуха, путем герметизации цилиндра для хранения проб водородом и выталкивания жидкости в реактор при перемешивания реактора при приблизительно 200 об/мин. Дополнительный водород загружали в реактор через нижнее дренажное отверстие для очистки зоны катализатора. Реактор затем загружали водородом до 150 фунтов на кв. дюйм (всегда через нижнее дренажное отверстие) и реактор перемешивали при приблизительно 500 об/мин. Реакцию продолжали, пока не переставал расходоваться водород и пробы, которые выводили из реактора, не указывали на завершение реакции. Смесь продукта и пентана выводили из реактора. Катализатор удаляли путем фильтрования, а пентан удаляли при помощи роторного испарителя.
ПРИМЕР СИНТЕЗА VIII: СХЕМА ПРОЦЕССА II С ИСПОЛЬЗОВАНИЕМ ПРОДУКТА, ПОЛУЧЕННОГО В ПРИМЕРЕ VII:
Синтез полученных из фарнезена полиразветвленных спиртов и их смесей
1,17 ммоль октакарбонила дикобальта и 4,7 ммоль ейкозилфобана (смесь изомеров [13887-00-8] и [13886-99-2]) соединяли в 48 мл высушенного дегазированного 2-пропилового спирта в сосуде высокого давления из нержавеющей стали на 300 мл, имеющем стеклянную рубашку и покрытую ПТФЕ магнитную мешалку. 47,7 ммоль полученной из фарнезена смеси парафин/моноолефин, полученной в ПРИМЕРЕ СИНТЕЗА VII, предварительно высушенной над ХА молекулярными ситами и отфильтрованной, добавляли к подающей пробирке реактора. Рубашки реактора продували воздухом при помощи циклов вакуум-азот. Реактор на 300 мл затем продували смесью моноксида углерода и водорода в соотношении 1:1.
Реактор, содержащий смесь октакарбонила дикобальта, ейкозилфобана и 2-пропилового спирта, загружали до начального давления, составляющего приблизительно 150 фунтов на кв. дюйм, смесью моноксида углерода и водорода в соотношении 1:1. Реактор нагревали от 60 до 65°С при механическом перемешивании при 150-200 об/мин при поддержании давления от 150 до 200 фунтов на кв. дюйм с использованием смеси моноксида углерода и водорода в соотношении 1:1. Через 1-2 часа реактор охлаждали до ниже 40°С.
Реактор продували и полученную из фарнезена смесь парафин/моноолефин загружали в реактор. Реактор затем загружали смесью моноксида углерода и водорода в соотношении 1:2. Реактор затем нагревали от 160 до 165°С при поддержании давления от 500 до 700 фунтов на кв. дюйм при помощи газовой смеси СО:Н2 в соотношении 1:2. Содержимое реактора отбирали по времени и анализировали при помощи ГХ для контроля хода реакции. Когда проба ГХ анализа указывала на завершение реакции, реакционную смесь охлаждали до комнатной температуры и смесь моноксид углерод-водород отводили через вентиляционную систему.
Спиртовый продукт может образовываться непосредственно при помощи такого катализатора и только стадия доочистки гидрогенизации необходима для обеспечения стабильных спиртовых продуктов.
ПРИМЕР СИНТЕЗА IX: с использованием стадии с СХЕМЫ ПРОЦЕССА II посредством коммерческого концевого моноолефина фарнезена:
Синтез 4,8,12-триметил-тридеканаля и его смесей
1,22 грамма карбонилгидридотрис(трифенилфосфин)родия(I) [17185-29-4] и 3,11 грамма Ксантофоса [161265-03-8] суспендированных в 53 граммах гексана загружали в сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании при приблизительно 300-500 об/мин, используя вакуум для вытягивания проб, избегая попадания воздуха. Реактор продували воздухом при помощи циклов вакуум-азот, затем загружали 10 атм смеси моноксида углерода и водорода в соотношении 1:1 и нагревали до 60°С в течение двух часов, с последующим охлаждением до 30°С. Реактор помещали в вакуум. 27,4 грамма 3,7,11-триметил-1-додецена [1189-36-2] плюс 85 грамм толуола загружали в реактор, исключая попадание воздуха. Реактор продували воздухом при помощи циклов вакуум-азот, затем загружали от 10 до 15 атм смеси моноксида углерода и водорода в соотношении 2:1. Реактор нагревали до 45°С. По мере расходования моноксида углерода и водорода в реакции давление поддерживали при помощи смеси моноксида углерода и водорода в соотношении 1:1. Содержимое реактора отбирали по времени и анализировали при помощи ГХ для контроля хода реакции. Когда проба ГХ анализа указывала на завершение реакции, реакционную смесь охлаждали до комнатной температуры и смесь моноксид углерод: водород отводили.
В зависимости от чистоты 3,7,11-триметил-1-додецена, процесс может проходить от нескольких часов до 120 часов. Перед тем как перейти к следующей стадии реакции, остаток моноксида углерода удаляли при помощи циклов вакуум-азот. Альдегидную смесь не обязательно удалять из реактора до превращения в спирт в ПРИМЕРЕ IX, хотя альдегид может быть очищен при желании или при применении в других реакциях.
ПРИМЕР СИНТЕЗА X: использование стадии d СХЕМЫ ПРОЦЕССА II
Синтез 4,8,12-триметил-тридекан-1-ола и его смесей
Никель на кизельгуре (20 грамм 60-мас.% загрузка) плюс тетрагидрофуран (200 мл) загружали в сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании. Реактор продували воздухом при помощи циклов вакуум-азот с последующей загрузкой водородом до начального давления, составляющего приблизительно 600 фунтов на кв. дюйм. Смесь нагревали до приблизительно 150°С с перемешиванием при приблизительно 500 об/мин. Водород загружали до конечного давления, составляющего приблизительно 900 фунтов на кв. дюйм, и поддерживали такое давление в течение 16 часов. Содержимое реактора затем охлаждали до комнатной температуры и снижали давление до приблизительно 50 фунтов на кв. дюйм.
Альдегидную смесь, полученную в ПРИМЕРЕ СИНТЕЗА VI, затем загружали в реактор, исключая поступление воздуха из атмосферы при непрерывном перемешивании содержимого реактора. Катализатор гидроформилирования может оставаться в альдегидной смеси. При желании, катализатор может быть удален из смеси перед применением. Смесь затем герметизировали водородом при начальном давлении, составляющем приблизительно 600 фунтов на кв. дюйм, и нагревали до приблизительно 125°С при перемешивании при приблизительно 500 об/мин. Давление водорода затем повышали до приблизительно 900 фунтов на кв. дюйм и данное давление поддерживали при периодическом отборе содержимого реактора для анализа при помощи ГХ. Ход реакции контролировали при помощи ГХ, пока не переставал образовываться дополнительный продукт. Время реакции будет варьироваться в соответствии с условиями реакции.
Очистка неочищенной спиртовой смеси может быть достигнута при помощи стандартных известных процедур, таких как перегонка, или других способов очистки, известных из уровня техники.
Другое осуществление способа в соответствие с натоящим изобретением проиллюстрировано при помощи СХЕМЫ ПРОЦЕССА III:
СХЕМА ПРОЦЕССА III
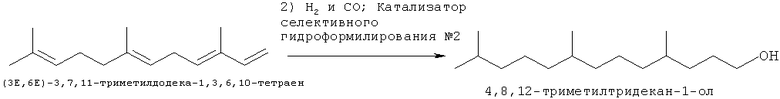
Данное осуществление представляет собой способ в соответствии с первым осуществлением, в котором, однако, стадии гидроформилирования и восстановления проводят одновременно в одной стадии. Соответственно способ включает стадии, на которых:
а. обеспечивают полиразветвленные полиолефины, при этом полиразветвленные полиолефины должны содержать один неразветвленный концевой олефин и один или более дополнительных разветвленных олефинов в молекуле; и
b. гидроформилируют и восстанавливают указанный полиразветвленный полиолефин с применением катализатора, выбранного из конкретных модифицированных переходных металлов группы IX и условий процесса, включающих: температуру процесса в диапазоне от приблизительно 90°С до приблизительно 200°С, молярное соотношение водорода и моноксида углерода в диапазоне от приблизительно 2 к 1 до приблизительно 5 к 1, общее давление в диапазоне от приблизительно 300 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм; и
с. удаляют указанную спиртовую композицию из указанного катализатора.
В последовательностях третьего осуществления способа, приведенного в данной заявке выше, выбор исходных веществ для а является таким же, как и для других осуществлений. В случае стадии b реакции необходим специализированный катализатор гидроформилирования и условия процесса, для достижения максимального образования спирта без выделения альдегида. Дополнительно, ключевым результатом данного процесса также является одновременная гидрогенизация непрореагировавших олефинов в полиразветвленных полиолефиновых исходных веществах. Это наиболее эффективный способ. Однако он осложнен необходимостью избегания образования больших количеств парафинов. Катализаторы типа, проиллюстрированного в патенте США №3420898, являются приемлемыми катализаторами для такого третьего осуществления. Условия процесса для стадии b требуют температуру в диапазоне от приблизительно 50°С до приблизительно 130°С, молярное соотношение водорода и моноксида углерода в диапазоне от приблизительно 2:1 до приблизительно 5:1 и общее давление в диапазоне от приблизительно 300 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм.
Катализаторы, предпочтительные для данного способа, получены на основе кобальта и модифицированы трифенилфосфином. Добавление небольших количеств Ph2PCH2CH2CH2CH2PPh2 может способствовать данной реакции.
Наконец, стадию с проводят для удаления разветвленной спиртовой композиции из катализатора путем перегонки или другими способами, которые традиционно применяют в данной области. Парафины образуются легче в данном способе и перегонка требуется как таковая для очистки спирта.
ПРИМЕР СИНТЕЗА XI: СХЕМА ПРОЦЕССА III:
Синтез полученных из фарнезена полиразветвленных спиртов и их смесей
В устройстве для эксплуатации герметизированного оборудования 1,17 ммоль октакарбонила дикобальта и 4,7 ммоль эйкозилфобана (смесь изомеров [13887-00-8] и [13886-99-2]) соединяли в 48 мл высушенного дегазированного 2-пропилового спирта в сосуде высокого давления из нержавеющей стали на 300 мл, имеющем стеклянную рубашку и покрытую ПТФЕ магнитную мешалку. 47,7 ммоль транс-бета фарнезена (предварительно высушенного над ХА молекулярными ситами и отфильтрованного), добавляли в подающую пробирку, присоединенную к реактору. Рубашки реактора продували воздухом при помощи циклов вакуум-азот. Реактор на 300 мл затем продували смесью моноксида углерода и водорода в соотношении 1:1.
Реактор на 300 мл, содержащий смесь октакарбонила дикобальта, ейкозилфобана и 2-пропилового спирта, загружали до начального давления, составляющего приблизительно 150 фунтов на кв. дюйм смесью моноксида углерода и водорода в соотношении 1:1. Реактор нагревали от приблизительно 60 до 65°С при перемешивании при 150-200 об/мин при поддержании давления от 150 до 200 фунтов на кв. дюйм с использованием смеси моноксида углерода и водорода в соотношении 1:1. Через 1-2 часа реактор охлаждали до ниже 40°С.
Реактор продували и транс-бета-фарнезен загружали в реактор. Подающую трубку отделяли от реактора и в реактор затем загружали смесь моноксида углерода и водорода в соотношении 1:2. Реактор на 300 мл затем нагревали от 160 до 165°С при поддержании давления от 500 до 700 фунтов на кв. дюйм при помощи смеси моноксида углерода и водорода в соотношении 1:2. Содержимое реактора отбирали по времени и анализировали при помощи ГХ для контроля хода реакции. Когда проба ГХ анализа указывала на завершение реакции, реакционную смесь охлаждали до комнатной температуры и смесь моноксид углерод: водород отводили через вентиляционную систему. Полученный в результате неочищенный продукт содержал спирт 1 и спирт 2.
Полиразветвленные ациклические альдегиды
Другое осуществление в соответствии с настоящим изобретением представляет собой образование новых ациклических альдегидов, имеющих 16 или 21 атомов углерода и содержащих, по меньшей мере, три разветвления и три или менее двойных связи углерод-углерод. Такие новые альдегиды могут быть применены в отдушках и ароматизаторах. Примеры таких ациклических альдегидов включают, но не ограничиваясь приведенным, 3-этил-7,11-диметилдодеканаль; 2,3,7,11-тетраметил-додеканаль; 7,11-диметил-3-винилдодека-6,10-диеналь; 8,12-диметилтридека-4,7,11-триеналь. Другими осуществлениями являются ациклические альдегиды, содержащие одну, две или три углерод-углеродные двойные связи, где разветвления представляют собой метил, этил или оба из них. Другое осуществление представляет собой то, где ациклический альдегид насыщен, а разветвления представляют собой метил, этил или оба из них. Ациклические альдегиды могут быть смешаны с другими веществами с получением полезных композиций.
Неограничивающие примеры структур нового полиразветвленного полиолефина, содержащего альдегиды в соответствии с настоящим изобретением показаны ниже:
Четыре альдегида, показанные ниже (а1-а4), являются структурами, образованными путем реакции бета-фарнезена в соответствии с осуществлением процесса один.

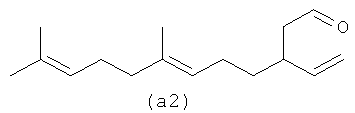
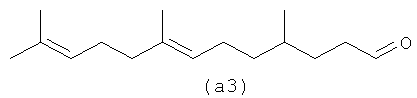
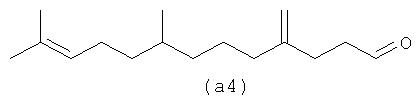
Ниже также приведены возможные полиразветвленные полиальдегидные структуры, которые могут быть получены из бета-фарнезена путем контролирования условий реакции для максимизации их получения.
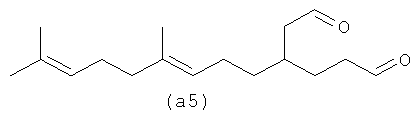
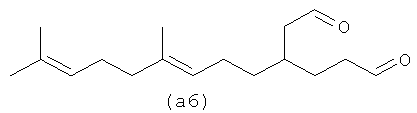
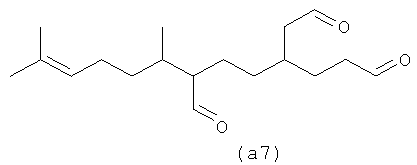
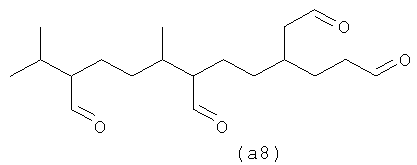
Полиальдегиды превращают в полиспирты и затем в полифункционализованные поверхностно-активные вещества. Считают, что полиразветвленные полизамещенные (например, дианионные) поверхностно-активные вещества имеют хорошие суспендирующие загрязнения возможности без тенденции к кристаллизации и имеют плохую растворимость, тенденцию к демонстрации которой имеют линейные дианионные поверхностно-активные вещества.
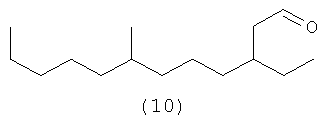
4,8,12-триметилтридеканаль (а9) является возможным альдегидом из СХЕМЫ процесса II посредством второго осуществления способа. (а10) является также другим полученным в результате альдегидом в соответствии с настоящим изобретением, а также смесями их обоих.
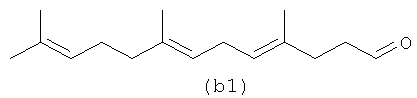
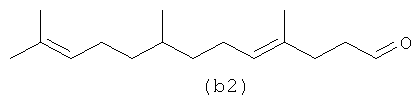
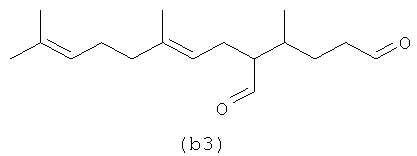
Ниже (b1-b2) показан полиразветвленный полиолефин, содержащий альдегид, который может быть получен из альфа-фарнезена. (b3) является диальдегидом, который может быть образован в определенных условиях процесса, если желаемым является получение диальдегида.
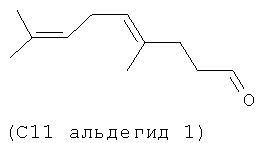
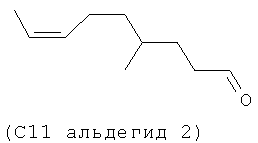
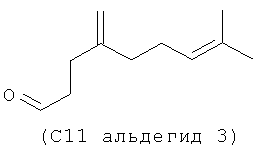
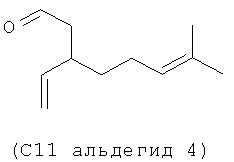
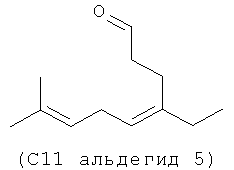
Следующие (С11 альдегиды 1-4) также являются примерами альдегидов в соответствии с данным способом изобретения в соответствии со СХЕМОЙ ПРОЦЕССА I и подробными элементами процесса в длинах цепи С11 и C21. Они могут быть образованы по реакции в соответствии с процессом, где используют оцимен (1-2) и мирцен (3-4) с (альдегидом 5), полученным из (Z)-3-этил-7-метилокта-1,3,6-триена (С11 полиразветвленный полиолефин).
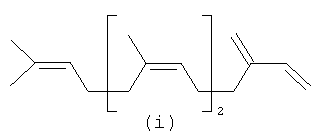
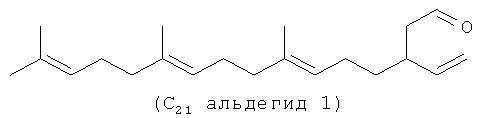
Ниже представлен пример C21 полиразветвленного полиолефинового альдегида, который может быть получен из С20 терпенов, таких как олефин (i).
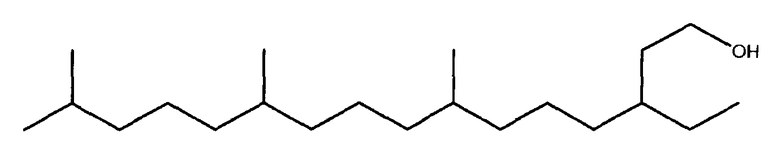
Полиразветвленные моющие спирты
Другим осуществлением в соответствие с настоящим изобретением являются полиразветвленные моющие спирты, образованные при помощи данного способа, содержащие 11, 16 или 21 атомов углерода.
Определенные осуществления полиразветвленных моющих спиртов в соответствии с настоящим изобретением включают С11 и C21 моющие спирты, содержащие два, три, четыре или пять метильных или этильных разветвлений или их смеси. Их можно получить посредством структур диизопренов и тетраизопренов или других полиразветвленных полиолефиновых исходных веществ. Они могут быть применены в шампунях, средствах для мытья посуды и/или очистителях твердых поверхностей после превращения в соответствующие композиции поверхностно-активных веществ. Примеры таких спиртов показаны ниже. Полезные осуществления будут иметь высокие уровни метильного разветвления и будут содержать более чем 70% двух, трех или четырех метильных групп или их смесей.


Другие полезные осуществления включают композиции полиразветвленных моющих спиртов, которые являются ациклическими и имеют длину углеродной цепи, составляющую 16 атомов. Осуществления могут иметь более чем 10% триметильных разветвлений или более чем 30% триметильных разветвлений или даже 70% или более триметильных разветвлений.
Было найдено, что осуществление полиразветвленных моющих спиртов, полученных из фарнезена природного происхождения, экстрагированного из уже существующих растений и организмов, фарнезена, полученного посредством генетически модифицированных организмов, синтетически полученных тримеров изопрена, их смесей, является полезным в чистящих композициях. Полиразветвленные моющие спирты и их смеси могут быть получены из смесей изомеров фарнезена.
Хотя должно быть понятно, что любой олефин на основе изопрена любой длины цепи может быть применен для получения смеси моющих спиртов при помощи способа в соответствии с настоящим изобретением пока производные произведены из олигомеров, которые получают из веществ, подобных ациклическому изопрену, любыми способами, описанными в данной заявке выше. Примеры C16 полиразветвленных моющих спиртов проилюстированы ниже.
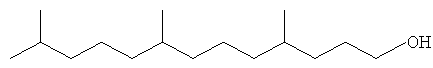
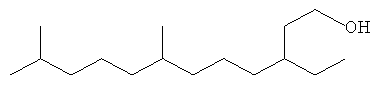
Полиразветвленные моющие спирты в соответствии с настоящим изобретением включают спирты, содержащие одну или более спиртовых групп. Способы в соответствие с настоящим изобретением могут быть оптимизированы для контроля минимизированного или максимизированного образования полиспирта (ди, три и тетра спиртов) в отличие от моноспирта.
ПРИМЕР СИНТЕЗА XII: с использованием СХЕМЫ ПРОЦЕССА I:
Синтез полученных из фарнезена полиразветвленных полиспиртов
1,17 ммоль октакарбонила дикобальта и 4,7 ммоль эйкозилфобана (смесь изомеров [13887-00-8] и [13886-99-2]) объединяли в 48 мл высушенного дегазированного 2-пропилового спирта в сосуде высокого давления из нержавеющей стали на 300 мл, имеющем стеклянную рубашку и покрытую ПТФЕ магнитную мешалку. 47,7 ммоль транс-бета-фарнезена (предварительно высушенного над молекулярными ситами и отфильтрованного) добавляли в подающую трубку, присоединенную к реактору. Рубашки реактора продували воздухом при помощи циклов вакуум-азот. Реактор затем продували смесью моноксида углерода и водорода в соотношении 1:1. Реактор, содержащий смесь октакарбонила дикобальта, ейкозилфобана и 2-пропилового спирта, загружали до начального давления, составляющего приблизительно 150 фунтов на кв. дюйм, смесью моноксида углерода и водорода в соотношении 1:1. Реактор нагревали до температуры от 60 до 65°С при перемешивании при 150-200 об/мин при поддержании давления от 150 до 200 фунтов на кв. дюйм с использованием смеси моноксида углерода и водорода в соотношении 1:1. Через 1-2 часа реактор охлаждали ниже 40°С.
Реактор продували и содержимое подающей трубки (транс-бета-фарнезен) загружали в реактор путем открывания клапанов, отделяющих два контейнера. Реактор затем загружали новой смесью моноксид углерода-водорода, состоящей из смеси моноксида углерода и водорода в соотношении 1:2. Реактор затем нагревали от 160 до 165°С при поддержании давления от 500 до 700 фунтов на кв. дюйм при помощи смеси моноксида углерода и водорода в соотношении 1:2.
Содержимое реактора отбирали по времени и анализировали при помощи ГХ для контроля хода реакции. Когда проба ГХ анализа указывала на завершение реакции, реакционную смесь охлаждали до комнатной температуры и смесь моноксид углерод: водород отводили. Катализатор удаляли и полученная в результате смесь содержала более чем 30% диолов и высших полиолов. Диолы и высшие полиолы отделяли от парафинов и моноспиртов при помощи стандартной процедуры перегонки.
Полиразветвленные поверхностно-активные вещества
Другие осуществления в соответствии с настоящим изобретением включают композиции поверхностно-активных веществ, полученные из полиразветвленных моющих спиртов. Они могут иметь длины цепи, составляющие С11, C16 или C21, и быть полиразветвленными, где разветвления представляют собой метил, этил или их смеси. Поверхностно-активные вещества могут быть образованы путем процесса дериватизации любого спирта в поверхностно-активное вещество, известного в данной области. Они могут включать этоксилаты спирта, спиртовые сульфаты или этоксилированные спиртовые сульфаты или их смеси.
Примерами С11 и С21 полиразветвленных поверхностно-активных веществ являются:
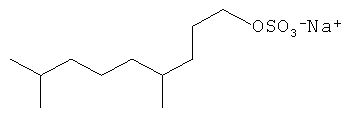

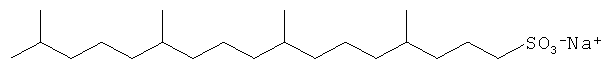
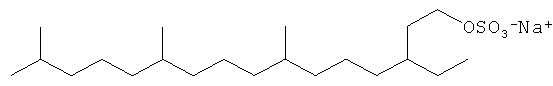
Другие композиции поверхностно-активных веществ могут быть получены из ациклических C16 полиразветвленных моющих спиртов и могут включать ациклические C16 моющие спиртовые этоксилаты, сульфаты или этоксилированные сульфаты. Неорганичивающие примеры структур предпочтительных C16 полиразветвленных спиртовых поверхностно-активных веществ проиллюстрированы ниже:
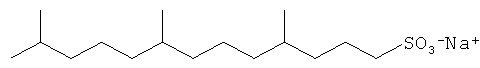
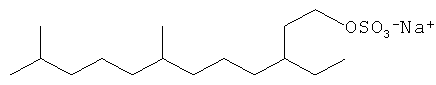
Смеси поверхностно-активных веществ могут быть также желаемыми.
Спирты в соответствии с настоящим изобретением могут быть алкоксилированы при помощи стандартных коммерческих и лабораторных методов и/или сульфированы с использованием любого подходящего агента, например хлорсульфоновой кислоты, SО3/воздуха или олеума, с получением целевых полученных из спирта композиций поверхностно-активных веществ.
Следующие примеры подробно определяют синтез полиразветвленных композиций поверхностно-активных веществ, шестое осуществление в соответствии с настоящим изобретением:
ПРИМЕР СИНТЕЗА XIII
Синтез полученного из фарнезена полиразветвленного спиртового сульфата и его смесей
Реакционный сосуд, при перемешивании и продувании азота для исключения воздуха, использовали для соединения 96 грамм полиразветвленного спиртового вещества, полученного в любом из ПРИМЕРОВ СИНТЕЗА II, V или X, и 149 грамм диэтилового эфира. Смесь охлаждали до -5°С, затем 50 грамм хлорсульфоновой кислоты [7790-94-5] добавляли по каплям при поддержании температуры смеси ниже 10°С. Применяли вакуум для удаления выделяющегося газа НС1, давая смеси нагреваться до ~30°С. Диэтиловый эфир замещали дважды, поскольку он испаряется, при непрерывном перемешивании в течение двух часов. Затем простой эфир удаляли с помощью вакуума до проведения следующей стадии.
Полученную в результате смесь медленно добавляли, при перемешивании в стакан из нержавеющей стали, содержащий 287 грамм 9% метоксида натрия в метаноле, который охлаждали на ледяной бане. Смесь перемешивали в течение часа и затем погружали на поддон из нержавеющей стали. Растворители затем испаряли и пробу дополнительно высушивали при помощи вакуумной печи.
ПРИМЕР СИНТЕЗА XIV
Синтез полученного из фарнезена 7-моль этоксилата полиразветвленного спирта (АЕ7) и его смесей
200 грамм полученных из фарнезена спиртов (и других спиртов, полученных из них любыми способами или в ПРИМЕРЕ СИНТЕЗА II) плюс достаточное количество катализатора для того, чтобы способствовать реакции спирта с этиленокисидом в течение приемлемого периода времени и при контроле загружали в сосуд высокого давления из нержавеющей стали на 600 мл при перемешивании с охлаждающим змеевиком.
Приемлемый катализатор представлял собой 1,1 грамма раствора, состоящего из 50% гидроксида калия в воде. Другие типы и количества катализатора могут быть применены в зависимости от требования процесса.
Реактор нагревали при применении вакуума для удаления веществ, которые могут привести к образованию побочных продуктов, например воды, которые могут быть введены с катализатором, при температуре, которая не позволит потерю фарнезеновых спиртов, в общем, от 40°С до 90°С, но предпочтительно от приблизительно 60°С до приблизительно 80°С, при помощи водяного аспиратора как источника вакуума. Удаление воды производили при помощи низкоскоростного перемешивания, в общем, при приблизительно 50 об/мин, барботируя смесь малоинтенсивным (струйкой) потоком инертного газа через нижний дренажный клапан, или через дисперсионную фритту из нержавеющей стали, или любую инертную погружную трубку, или фриттованное вещество из спеченного металла, или путем разворачивания области выше смеси инертным газом. Пробы могут быть вытянуты из реактора и проанализированы на содержание воды соответствующим аналитическим способом, например по методу Карла-Фишера.
После завершения стадии удаления воды этиленоксид добавляли в реактор. Этиленоксид может быть добавлен весь сразу, если система реактора надлежащим образом разработана для предотвращения неконтролируемой скорости реакции. Однако наилучший контроль реакции достигали путем изначального нагревания реактора в статическом вакууме (или необязательно при избыточном давлении инертного газа, такого как азот) до температуры, приемлемой для реакции смеси спирт-катализатор с этиленоксидом для получения минимального количества побочных продуктов и цветообразования, в общем от 85° до 150°С, но предпочтительно от приблизительно 110°C до 130°C.
После достижения реактором желаемой температуры добавляли 254 грамма этиленоксида при скорости, которую можно будет контролировать при помощи системы охлаждения, в общем в течение периода от 30 до 60 минут. После завершения добавления этиленоксида перемешивание и нагревание продолжали до окончания расходования этиленоксида реакцией.
ПРИМЕР СИНТЕЗА XV
Синтез полученного из фарнезена 10-моль этоксилата полиразветвленного спирта (АЕ10) и его смесей
Использовали оборудование и процедуру из ПРИМЕРА XIII, но количество использованного этиленоскида составляло 363 грамма для получения полученных из фарнезена 10-моль этоксилата полиразветвленного спирта.
ПРИМЕР СИНТЕЗА XVI
Синтез полученного из фарнезена 3-моль этоксилата полиразветвленного спирта (АЕ3) и его смесей
Использовали оборудование и процедуру из ПРИМЕРА XIII, но количество использованного этиленоскида составляло 109 грамм для получения полученного из фарнезена 3-моль этоксилата полиразветвленного спирта.
ПРИМЕР СИНТЕЗА XVII
Синтез полученного из фарнезена этоксилата полиразветвленного спиртового сульфата (AE3S) и его смесей
Реакционный сосуд при перемешивании и продувании азота для исключения воздуха использовали для объединения 62 грамм вещества, полученного в ПРИМЕРЕ XV, и 149 грамм диэтилового эфира. Смесь охлаждали до -5°С, затем 50 грамм хлорсульфоновой кислоты [7790-94-5] добавляли по каплям при поддержании температуры смеси ниже 10°С. Применяли вакуум для удаления выделяющегося газа НСl, давая смеси нагреваться до ~30°С. Диэтиловый эфир замещали дважды, поскольку он испаряется, при непрерывном перемешивании в течение двух часов. Затем простой эфир удаляли с помощью вакуума перед проведением следующей стадии.
Полученную выше смесь медленно добавляли при перемешивании в стакан из нержавеющей стали, содержащий 287 грамм 9% метоксида натрия в метаноле, который охлаждали на ледяной бане. Смесь перемешивали в течение часа и затем погружали на поддон из нержавеющей стали. Растворители затем испаряли и пробу дополнительно высушивали при помощи вакуумной печи.
ПРИМЕР СИНТЕЗА XVIII
Синтез 3-этил-7,11-диметил-додеканаля и его смесей
Использовали оборудование и процедуру из ПРИМЕРА I, однако исходный фарнезен, который использовали, представлял собой 336 грамм транс-бета-фарнезена [18794-84-8]. Продукт может быть использован непосредственно в ПРИМЕРЕ XIX.
ПРИМЕР XIX
Синтез 3-этил-7,11-диметил-додекан-1-ола и его смесей
Использовали оборудование и процедуру из ПРИМЕРА СИНТЕЗА II. Однако исходные вещества для реакции получали в процессе, приведенном в ПРИМЕРЕ СИНТЕЗА XVIII выше, где использовали транс-бета-фарнезен. Конечную реакционную смесь фильтровали через фильтр на 0,5 микрон для удаления катализатора. Полученную в результате смесь выпаривали из всех нелетучих загрязнителей, включая остатки катализатора, при помощи колонки для молекулярной перегонки при температурах до 250°С и источника вакуума 1 Торр. Неочищенный дистиллят затем фракционно перегоняли на колонке Oldershaw (WILMAD-LABGLASS деталь№: G-2008-015J), собирая небольшой объем отогнанных фракций от 30 до 45 грамм каждая при температурах до 350°С и при помощи источника вакуума 5 Торр. Данные фракции анализировали при помощи ГХ с использованием капиллярной ГХ колонки Restek RTX-5 (Кат. №:10244).
ПРИМЕР СИНТЕЗА XX
Синтез 3-этил-7,11-диметил-додекан-1-олового спиртового сульфата и его смесей
Использовали оборудование и процедуру из ПРИМЕРА СИНТЕЗА III. Однако спирт, который использовали, представлял собой 3-этил-7,11-диметил-додекан-1-ол, который получали в ПРИМЕРЕ XDC выше. Продукт анализировали при помощи ЯМР и масс-спектрометрии и полученный в результате анализ согласовался с прогнозируемым продуктом 3-этил-7,11-диметил-до декан-1-оловым спиртовым сульфатом.
КОМПОЗИЦИИ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ И ПРОДУКТЫ. ИСПОЛЬЗУЮЩИЕ ПРОИЗВОДНЫЕ ПОЛИРАЗВЕТВЛЕННЫХ МОЮЩИХ СПИРТОВ И КОМПОЗИЦИИ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ
Композиция полиразветвленного поверхностно-активного вещества, содержащая одно или более производных моющего спирта, выбранного из сульфата, алкоксилированного или алкоксилированного сульфата или их смесей в соответствии с настоящим изобретением, является чрезвычайно приемлемой в качестве добавок, способствующих отделению загрязнений для моющих средств для стирки и для чистящих композиций. Они проявляют высокую растворяющую способность, особенно в случае жирных загрязнений. Особое преимущество состоит в том, что они проявляют способность к отделению загрязнений даже при низких температурах стирки.
Композиция поверхностно-активного вещества, содержащая одно или более производных новых полиразветвленных моющих спиртов, выбранных из сульфата, алкоксилированного или алкоксилированного сульфата или их смесей в соответствии с настоящим изобретением, является чрезвычайно приемлемой в качестве добавок, способствующих отделению загрязнений для моющих средств для стирки и для чистящих композиций. Они проявляют высокую растворяющую способность, особенно в случае жирных загрязнений. Особое преимущество состоит в том, что они проявляют способность к отделению загрязнений даже при низких температурах стирки.
Композиции полиразветвленных поверхностно-активных веществ в соответствии с настоящим изобретением могут быть добавлены к моющим средствам для стирки и чистящим композициям в количествах, составляющих в общем от 0,05 до 70% по массе, предпочтительно от 0,1 до 40% по массе и более предпочтительно от 0,25 до 10% по массе, исходя из всей конкретной композиции.
Дополнительно, моющие средства для стирки и чистящие композиции в общем содержат поверхностно-активные вещества и, если подходит, другие полимеры в качестве моющих веществ, модифицирующих компонентов и дополнительных стандартных ингредиентов, например сомодифицирующих компонентов, комплексообразователей, отбеливателей, стандартизаторов, ингибиторов приобретения серого оттенка, ингибиторов переноса красителя, ферментов и отдушек.
Новые композиции поверхностно-активных веществ в соответствии с настоящим изобретением могут быть применены в моющих средствах для стирки или чистящих композициях, содержащих систему поверхностно-активных веществ, содержащую C10-C15 алкилбензолсульфонаты (LAS) и одно или более со-поверхностно-активных веществ, выбранных из неионных, катионных, анионных веществ или их смесей. Выбор со-поверхностно-активного вещества может зависеть от желаемого полезного эффекта. В одном осуществлении, со-поверхностно-активное вещество выбирают в качестве неионного поверхностно-активного вещества, предпочтительно C12-C18 алкилэтоксилатов. В другом осуществлении, со-поверхностно-активное вещество выбирают в качестве анионного поверхностно-активного вещества, предпочтительно С10-C18 алкилалкокси сульфатом (AExS) где х имеет значение 1-30. В другом осуществлении со-поверхностно-активное вещество выбирают в качестве катионного поверхностно-активного вещества, предпочтительно диметил гидроксиэтиллауриламмоний хлорид. Если система поверхностно-активных веществ содержит C10-C15 алкилбензолсульфонаты (LAS), LAS применяют на уровнях в диапазоне от приблизительно 9% до приблизительно 25%, или от приблизительно 13% до приблизительно 25%, или от приблизительно 15% до приблизительно 23% по массе композиции.
Система поверхностно-активных веществ может содержать от 0% до приблизительно 7%, или от приблизительно 0,1% до приблизительно 5%, или от приблизительно 1% до приблизительно 4% по массе композиции со-поверхностно-активного вещества, выбранного из неионного со-поверхностно-активного вещества, катионного со-поверхностно-активного вещества, анионного со-поверхностно-активного вещества и их любой смеси.
Неограничивающие примеры неионных со-поверхностно-активных веществ включают: C12-C18 алкилэтоксилаты, например, NEODOL® неионные поверхностно-активные вещества от Shell; C6-C12 алкилфенолалкоксилаты, где алкоксилатные звенья представляют собой смесь этиленокси и пропиленокси звеньев; C12-C18 спиртовые и C6-C12 алкилфенольные конденсаты с этиленоксидными/пропиленоксидными блок алкилполиаминэтоксилатами, например, PLURONIC® от BASF; C14-C22 разветвленные спирты средней цепи, ВА, как обсуждено в патенте США №6150322; С14-С22 разветвленные алкилалкоксилаты средней цепи, ВАЕх, где х имеет значение 1-30, как обсуждено в патенте США №6153577, патенте США №6020303 и патенте США №6093856; алкилполисахариды, как обсуждено в патенте США №4565647 Llenado, выданном 26 января 1986 г.; в особенности, алкилполигликозиды, как обсуждено в патенте США №4483780 и патенте США №4483779; моющие амиды полигидроксикислот, как обсуждено в патенте США №5332528; и имеющие концевые простые эфиры поли(оксиалкилированных) спиртовых поверхностно-активных веществ, как обсуждено в патенте США №6482994 и WO 01/42408.
Неограничивающие примеры полуполярных неионных со-поверхностно-активных веществ включают: водорастворимые аминоксиды, содержащие один алкильный фрагмент, состоящий из от приблизительно 10 до приблизительно 18 атомов углерода, и 2 фрагмента, выбранные из группы, состоящей из алкильных фрагментов и гидроксиалкильных фрагментов, содержащих от приблизительно 1 до приблизительно 3 атомов углерода; водорастворимые фосфиноксиды, содержащие один алкильный фрагмент, состоящий из от приблизительно 10 до приблизительно 18 атомов углерода, и 2 фрагмента, выбранные из группы, состоящей из алкильных фрагментов и гидроксиалкильных фрагментов, содержащих от приблизительно 1 до приблизительно 3 атомов углерода; и водорастворимые сульфоксиды, содержащие один алкильный фрагмент, состоящий из от приблизительно 10 до приблизительно 18 атомов углерода, и фрагмент, выбранный из группы, состоящей из алкильных фрагментов и гидроксиалкильных фрагментов, содержащих от приблизительно 1 до приблизительно 3 атомов углерода. См. WO 01/32816, патент США №4,681,704 и патент США №4,133,779.
Неограничивающие примеры катионных со-поверхностно-активных веществ включают: четвертичные аммониевые поверхностно-активные вещества, которые могут содержать до 26 атомов углерода, включающие: алкоксилатные четвертичные аммониевые (AQA) поверхностно-активные вещества, как обсуждено в патенте США №6136769; диметил гидроксиэтил четвертичный аммоний, как обсуждено в патенте США №6004922; диметил гидроксиэтиллаурил аммоний хлорид; полиаминные катионные поверхностно-активные вещества, как обсуждено в WO 98/35002, WO 98/35003, WO 98/35004, WO 98/35005 и WO 98/35006; катионные сложноэфирные поверхностно-активные вещества, как обсуждено в патентах США №4228042, 4239660 4260529 и патенте США №6022844; и аминные поверхностно-активные вещества, как обсуждено в патенте США №6221825 и WO 00/47708, в особенности амидопропилдиметиламин (АРА).
Неограничивающие примеры анионных со-поверхностно-активных веществ, полезных в данной заявке, включают: С10-C20 первичные, с разветвленной цепью и произвольные алкил сульфаты (AS); C10-C18 вторичные (2,3) алкил сульфаты; C10-C18 алкилалкоксисульфаты (AExS), где х имеет значение 1-30; C10-C18 алкилалкоксикарбоксилаты, содержащие 1-5 этоксизвеньев; разветвленные алкил сульфаты средней цепи, как обсуждено в патенте США №6020303 и патенте США №6060443; разветвленные алкилалкоксисульфаты средней цепи, как обсуждено в патенте США №6008181 и патенте США №6020303; модифицированный алкилбензолсульфонат (MLAS), как обсуждено в WO 99/05243, WO 99/05242 и WO 99/05244; метилэфирсульфонат (MES); и альфа-олефинсульфонат (AOS).
Настоящее изобретение может также относится к композициям, содержащим композицию поверхностно-активного вещества в соответствии с настоящим изобретением, с его шестым осуществлением и седьмым осуществлением, композицию поверхностно-активного вещества, содержащую C8-C18 линейное алкилсульфонатное поверхностно-активное вещество и со-поверхностно-активное вещество. Композиции могут быть в любой форме, а именно, в форме жидкости; твердого вещества, например порошка, гранул, агломерата, пасты, таблетки, пакетов, бруска, геля; эмульсии; форм, которые доставляют в двухкомпонентных контейнерах; спрея или пенного моющего средства; предварительно увлажненных салфеток (то есть, чистящей композиции в комбинации с нетканым материалом, таким как, например, описано в патенте США №6121165, Mackey, et al.); сухих салфеток (то есть, чистящей композиции в комбинации с неткаными материалами, такими, как, например описано в патенте США №5980931, Fowler, и др.) таких, которые потребитель активирует водой; и других гомогенных или мультифазных формах потребительских продуктов.
В седьмом осуществлении, чистящая композиция в соответствии с настоящим изобретением является жидкой или твердой композицией моющего средства для стирки. В другом седьмом осуществлении, чистящая композиция в соответствии с настоящим изобретением представляет собой чистящую композицию для твердых поверхностей, где предпочтительно чистящая композиция для твердых поверхностей пропитывает нетканый субстрат. Как используют в данной заявке, "пропитывает" означает, что чистящую композицию для твердых поверхностей приводят в контакт с нетканым субстратом таким образом, что, по меньшей мере, в часть нетканого субстрата проникает чистящая композиция для твердых поверхностей, предпочтительно чистящая композиция для твердых поверхностей насыщает нетканый субстрат. Чистящая композиция может быть также применена в композициях по уходу за автомобилями, для очистки различных поверхностей, например твердой древесины, кафельной плитки, керамики, пластика, кожи, металла, стекла. Такая чистящая композиция может быть также разработана для применения в композициях для личной гигиены и для ухода за домашними животными, таких как композиция шампуня, композиция для мытья тела, жидкое или твердое мыло и другие чистящие композиции, в которых поверхностно-активное вещество контактирует со свободными твердыми и всеми композициями, которые требуют твердой приемлемой системы поверхностно-активного вещества, например композиции бурового раствора.
В другом седьмом осуществлении чистящая композиция является чистящей композицией для мытья посуды, например жидкими композициями для ручного мытья посуды, твердыми композициями для машинного мытья посуды, жидкими композициями для машинного мытья посуды и формы таблетки/стандартной дозы композиции для машинного мытья посуды.
Вполне типично, чистящие композиции в данной заявке, такие как моющие средства для стирки, добавки к моющим средства для стирки, очистители твердых поверхностей, синтетические бруски и бруски на основе мыла для стирки, смягчители тканей и жидкости для ухода за тканями, твердые композиции и средства по уходу всех видов, требующие нескольких вспомогательных веществ, посредством определенных просто оставляемых продуктов, например отбеливающих добавок, могут требовать только, например, кислородного отбеливателя и поверхностно-активного вещества, как описано в данной заявке. Полный список приемлемых веществ для стирки или чистящих вспомогательных веществ может быть найден в WO 99/05242.
Традиционные чистящие вспомогательные средства включают модифицирующие компоненты, ферменты, полимеры, которые не были обсуждены в данной заявке выше, отбеливатели, активаторы отбеливания, каталитические вещества и т.п., за исключением любых веществ, уже определенных в данной заявке выше. Другие чистящие вспомогательные средства в данной заявке могут включать усилители пенообразования, подавители пенообразования (противовспениватели) и т.п., другие активные ингредиенты или специализированные вещества, например диспергирующие полимеры (например, от BASF Corp. или Rohm & Haas), отличные от описанных в данной заявке выше, средства против возникновения пятен краски, средства по уходу за серебряными изделиями, средства против потускнения и/или средства от коррозии, красители, наполнители, бактерициды, источники щелочности, гидротропные вещества, антиоксиданты, средства, стабилизирующие ферменты, проотдушки, отдушки, солюбилизаторы, носители, технологические добавки, пигменты и для жидких композиций - растворители, хелаторы, ингибиторы переноса красителя, диспергирующие средства, осветлители, подавители пенообразования, красители, агенты придания эластичности структурам, смягчители для тканей, противоабразивные агенты, гидротропные вещества, технологические добавки, и другие средства по уходу за тканями, средства по уходу за поверхностями и кожей. Приемлемые примеры таких других чистящих вспомогательных средств и уровней применения приведены в патентах США №5576282, 6306812 В1 и 6326348 B1.
Способ применения
Настоящее изобретение включает способ очистки целевой поверхности. Как используют в данной заявке, "целевая поверхность" может включать такие поверхности, как ткань, посуда, стеклянные и другие варочные поверхности, твердые поверхности, волосы или кожу. Как используют в данной заявке, "твердая поверхность" включает твердые поверхности, которые присутствуют в типичном доме, например твердую древесину, кафельную плитку, керамику, пластик, кожу, металл, стекло. Такой способ включает стадии контактирования композиции, содержащей модифицированное полиольное соединение, в неразбавленном виде или разбавленную промывным раствором с, по меньшей мере, частью целевой поверхности с последующим необязательным промыванием целевой поверхности. Предпочтительно целевую поверхность подвергают стадии мытья перед проведением указанной выше необязательной стадией промывания. Для целей настоящего изобретения, мытье включает, но не ограничиваясь приведенным, очистку скребком, протирание и механическое перемешивание.
Как будет оценено специалистом в данной области техники, чистящие композиции в соответствии с настоящим изобретением идеально подходят для применения в быту (чистящие композиции для твердых поверхностей) и/или применений для стирки.
Значение рН растора композиции выбирают так, чтобы оно наиболее соответствовало целевой поверхности, которая подлежит очистке, широкий диапазон значения рН, составляет от приблизительно 5 до приблизительно 11. Для личной гигиены, например ухода за кожей и мытья волос, значение рН такой композиции предпочтительно составляет от приблизительно 5 до приблизительно 8, для чистящих композиций для стирки значение рН составляет от приблизительно 8 до приблизительно 10. Композиции предпочтительно применяют при концентрациях от приблизительно 200 ppm до приблизительно 10000 ppm в растворе. Температуры воды предпочтительно находятся в диапазоне от приблизительно 5°С до приблизительно 100°С.
Для применения в чистящих композициях для стирки, композиции предпочтительно применяют при концентрациях от приблизительно 200 ppm до приблизительно 10000 ppm в растворе (или моющем растворе). Температуры воды предпочтительно находятся в диапазоне от приблизительно 5°С до приблизительно 60°С. Соотношение воды и ткани предпочтительно составляет от приблизительно 1:1 до приблизительно 20:1.
Способ может включать стадию контактирования нетканого субстрата, пропитанного осуществлением композиции в соответствии с настоящим изобретением. Как используют в данной заявке, "нетканый субстрат" может включать любое традиционно разработанные нетканое полотно или ткань, имеющие приемлемую основную массу, толщину (плотность), впитывающую способность и характеристики прочности. Примеры приемлемых коммерчески доступных нетканых субстратов включают субстраты, которые продают под торговым названием SONTARA® от DuPont и POLYWEB® от James River Corp.
Как будет оценено специалистом в данной области техники, чистящие композиции в соответствии с настоящим изобретением идеально подходят для применения в жидких композициях для мытья посуды. Способ применения жидкой композиции для мытья посуды в соответствии с настоящим изобретением включает стадии контактирования загрязненной посуды с эффективным количеством, типично от приблизительно 0,5 мл до приблизительно 20 мл (на 25 обрабатываемых тарелок) жидкой композиции для мытья посуды в соответствии с настоящим изобретением, разбавленной водой.
Составы композиций Пример XXI - Гранулированное моющее вещество для стирки

Натрий триполифосфат К1
Пример XXII - Гранулированное моющее вещество для стирки
Водная композиция суспензии.
Получение высушенного распылением порошка.
Водную суспензию, имеющую композицию, описанную выше, получают при содержании влаги 25,89%. Водную суспензию нагревают до 72°С и прокачивают при высоком давлении (от 5,5×106 Hм-2 до 6,0×106 Нм-2) в противоточную колонну распылительной сушки при температуре входящего воздуха от 270°С до 300°С. Водную суспензию распыляют и распыленную суспензию высушивают с получением твердой смеси, которую затем охлаждают и просеивают с удалением чрезмерно крупных веществ (>1,8 мм) с образованием высушенного распылением порошка, который является сыпучим. Тонкоизмельченное вещество (<0,15 мм) декантируют с выпуском выходящего воздуха в колонну распылительной сушки и собирают в систему удерживания после колонны. Высушенный распылением порошок имеет содержание влаги 1,0 мас.%, объемную плотность 427 г/л и распределение размеров частиц такое, что 95,2 мас.% высушенного распылением порошка имеет размер частиц от 150 до 710 микрометров. Композиция высушенного распылением порошка приведена ниже.
Композиция высушенного распылением порошка.
Получение частицы анионного поверхностно-активного вещества 1
Частицу анионного моющего поверхностно-активного вещества 1 получают из загрузки 520 г с использованием смесителей Tilt-A-Pin, а затем Tilt-A-Plow (оба произведены Processall). 108 г сульфата натрия добавляют в смеситель Tilt-A-Pin вместе с 244 г карбоната натрия. 168 г 70% пасты активного С25Е3S (натрий этоксисульфат на основе C12/15 спирта и этиленоксида) добавляют в смеситель Tilt-A-Pin. Затем компоненты смешивают при 1200 об/мин в течение 10 секунд. Полученный в результате порошок затем переносят в смеситель Tilt-A-Plow и смешивают при 200 об/мин в течение 2 минут с образованием частиц. Частицы затем высушивают в сушилке с псевдоожиженным слоем при скорости 2500 л/мин, при 120°С до достижения равновесной относительной влажности частиц менее чем 15%. Высушенные частицы затем просеивают и оставляют фракцию от 1180 мкм до 250 мкм. Композиция частицы анионного моющего поверхностно-активного вещества 1 является следующей:
25,0% мас./мас. С25Е3S натрий этоксисульфата
18,0% мас./мас. сульфата натрия
57,0% мас./мас. карбоната натрия
Получение частицы катионного моющего поверхностно-активного вещества 1
Частицу катионного поверхностно-активного вещества 1 получают из загрузки 14,6 кг в смесителе Morton FM-50 Loedige. 4,5 кг микронизованого сульфата натрия и 4,5 кг микронизованого карбоната натрия предварительно смешивают в смесителе Morton FM-50 Loedige. 4,6 кг 40% водного раствора активного моно-С12-14 алкилмоногидроксиэтилдиметил четвертичного аммоний хлорида (катионного поверхностно-активного вещества) добавляют в смеситель Morton FM-50 Loedige при работающих главном приводе и измельчителе. Через приблизительно две минуты смешивания в смеситель добавляют 1,0 кг смеси микронизованного сульфата натрия и микронизованного карбоната натрия в массовом соотношении 1:1. Полученный в результате агломерат собирают и высушивают при помощи сушилки с псевдоожиженным слоем при потоке воздуха 2500 л/мин, при 100-140°С в течение 30 минут. Полученный в результате порошок просеивают и фракцию 1400 мкм собирают как частицу катионного поверхностно-активного вещества 1. Композиция частицы катионного поверхностно-активного вещества 1 является следующей:
15% мас./мас. моно-С12-14 алкилмоногидроксиэтилдиметил четвертичный аммоний хлорид
40,76% мас./мас. карбоната натрия
40,76% мас./мас. сульфата натрия
3,48% мас./мас. влаги и различных веществ
Получение гранулированной моющей композиции для стирки
10,84 кг высушенного распылением порошка, полученного в примере 6, 4,76 кг частицы анионного моющего поверхностно-активного вещества 1, 1,57 кг частицы катионного моющего поверхностно-активного вещества 1 и 7,83 кг (общее количество) другого отдельно дозированного добавляемого в сухом виде вещества дозируют в смеситель для перемешивания бетона диаметром 1 м при 24 об/мин. После дозирования всех веществ в смеситель смесь перемешивают в течение 5 минут с образованием гранулированной моющей композиции для стирки. Композиция гранулированной моющей композиции для стирки описана ниже:
Гранулированная моющая композиция для стирки.
Пример XXII - Жидкие моющие средства для стирки
2диэтилентриаминпентакисметиленфосфониевая кислота, натриевая соль
3этилендиаминтетрауксусная кислота, натриевая соль
4Acusol OP 301
2Alco 725 (стирол/акрилат)
Пример XXIII - Жидкие моющие средства для ручного мытья посуды
21,3,BAC представляет собой 1,3 бис(метиламин)-циклогексан.
3(N,N-димeтилaминo)этилмeтaкpилaтный гомополимер
Пример 11 - Моющее средство для машинного мытья посуды
2Например, ACUSOL® 445N, доступный от Rohm & Haas или ALCOSPERSE® от Alco.
СПОСОБЫ АНАЛИЗА
Следующие два аналитических способа для характеристики разветвления являются полезными в композициях поверхностно-активных веществ в соответствии с настоящим изобретением:
Отделение и идентификация компонентов в моющих спиртах (выполняют перед алкоксилированием или после гидролиза спиртового сульфата для аналитических целей). Положение и длина разветвления, которые находят в прекурсорах - моющих спиртовых веществах, определяют при помощи методов ГХ/MS [см.: D.J.Harvey, Biomed, Environ. Mass Spectrom (1989), 18(9), 719-23; D.J.Harvey, J.M.Tiffany, J. Chromatogr. (1984), 301(1), 173-87; К.A.Karlsson, В.Е.Samuelsson, G.О.Steen, Chem. Phys. Lipids (1973), 11(1), 17-38].
Идентификация разделенных моющих спиртовых алкоксисульфатных компонентов при помощи MS/MS. Положение и длину разветвления также можно определять при помощи ионно-распылительного MS/MS или ББА-MS/MS методов на предварительно выделенных моющих спиртовосульфатных компонентах.
Среднее общее количество атомов углерода разветвленных первичных алкильных поверхностно-активных веществ в данной заявке может быть рассчитано по гидроксильному числу смеси прекурсоров - моющих спиртов или по гидроксильному числу спиртов, восстановленных путем экстракции после гидролиза смеси спиртовых сульфатов в соответствии с традиционными процедурами, такими, которые описаны в "Bailey's Industrial Oil and Fat Products", Volume 2, Fourth Edition, edited by Daniel Swern, p.440-441.
Если не указано иное, все уровни компонента или композиции приведены относительно активного уровня такого компонента или композиции и не содержат примесей, например, остаточных растворителей или побочных продуктов, которые могут присутствовать в коммерчески доступных источниках.
Все процентные содержания и соотношения рассчитывают по массе, если не указано иное. Все процентные содержания и соотношения рассчитывают исходя их общей композиции, если не указано иное.
Должно быть понятно, что каждое максимальное численное ограничение, данное по всему описанию, включает каждое более низкое численное ограничение, как будто такие более низкие численные ограничения выражены в данной заявке в письменном виде. Каждое минимальное численное ограничение, приведенное по всему описанию, будет включать любое более высокое численное ограничение, как будто такие более высокие численные ограничения выражены в данной заявке в письменном виде. Каждый численный диапазон, приведенный по всему данному описанию, будет включать каждый более узкий численный диапазон, который подпадает под такой более широкий численный диапазон, как будто все такие более узкие численные диапазоны выражены в данной заявке в письменном виде.
Размеры и значения, описанные в данной заявке, не должны быть истолкованы как строго ограниченные приведенными точными численными значениями. Вместо этого, если не указано иное, каждый такой размер предназначен для обозначения как приведенного значения, так и функционально эквивалентного диапазона около данного значения. Например, размер, описанный как "40 мм", предназначен для обозначения "приблизительно 40 мм."
Все документы, процитированные в подробном описании настоящего изобретения, являются, в их релевантных частях, включенными в данную заявку путем ссылки; цитирование какого-либо документа не должно быть истолковано как допущение того, что оно представляет собой уровень техники в отношении настоящего изобретения.
В то время как были проиллюстрированы и описаны конкретные осуществления настоящего изобретения, специалисту в данной области техники будет очевидно, что различные другие изменения и модификации могут быть осуществлены не выходя за суть и объем настоящего изобретения. Поэтому формула, которая прилагается к данной заявке, предназначена для схватывания всех таких изменений и модификаций, которые входят в объем настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛУЧЕНИЕ РАЗВЕТВЛЕННЫХ АЛИФАТИЧЕСКИХ СПИРТОВ С ПРИМЕНЕНИЕМ ТЕХНОЛОГИЧЕСКОГО ПОТОКА ИЗ УСТАНОВКИ ИЗОМЕРИЗАЦИИ С РЕЦИКЛИРОВАНИЕМ В УСТАНОВКУ ДЕГИДРИРОВАНИЯ | 2004 |
|
RU2360899C2 |
| ПРОСТЫЕ ПОЛИЭФИРАМИНЫ НА ОСНОВЕ 1,3-ДИСПИРТОВ | 2014 |
|
RU2678325C2 |
| МОЮЩИЕ СРЕДСТВА ДЛЯ СТИРКИ С ОПТИМИЗИРОВАННЫМ ВЫСВОБОЖДЕНИЕМ ОТБЕЛИВАЮЩИХ СИСТЕМ | 2011 |
|
RU2569297C2 |
| РАЗВЕТВЛЕННЫЙ ПЕРВИЧНЫЙ СПИРТ И ЕГО ПРОИЗВОДНЫЕ | 2001 |
|
RU2281279C2 |
| Моющие средства для стирки и чистящие композиции, содержащие полимеры с карбоксильными группами | 2012 |
|
RU2614765C2 |
| Композиция моющего средства для стирки | 2015 |
|
RU2659398C1 |
| ПРИМЕНЕНИЕ НЕИОННЫХ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ, РАСТВОРИМЫХ В ДИОКСИДЕ УГЛЕРОДА, ДЛЯ ПОВЫШЕНИЯ НЕФТЕДОБЫЧИ | 2012 |
|
RU2612756C2 |
| ЧИСТЯЩИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ pH-ЗАВИСИМЫЕ АМИННЫЕ ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА | 2013 |
|
RU2575130C1 |
| СПОСОБЫ И СИСТЕМЫ ДЛЯ ПОЛУЧЕНИЯ МНОГОАТОМНЫХ СПИРТОВ | 2007 |
|
RU2454391C2 |
| Производство химических веществ и топлив из биомассы | 2012 |
|
RU2616620C2 |
Изобретение относится к новому ациклическому альдегиду, имеющему 16 атомов углерода, содержащему, по меньшей мере, три разветвления и выбранному из группы, состоящей из: 3-этил-7,11-диметилдодеканаля, 2,3,7,11-тетраметил-додеканаля, 7,11-диметил-3-винилдодека-6,10-диеналя и 4,8,12-триметилтридека-4,7,11-триеналя, к композиции веществ, пригодной для использования в качестве исходного материала для получения поверхностно-активных веществ и содержащей, по меньшей мере, один из заявленных ациклических альдегидов, к композиции моющих спиртов, пригодной для получения композиции поверхностно-активных веществ и содержащей, по меньшей мере, один ациклический спирт, конвертированный из заявленного ациклического альдегида, и к композиции поверхностно-активного вещества, пригодной для использования в моющей или чистящей композиции и содержащей одно или более поверхностно-активных производных изомеров ациклического моющего спирта, конвертированного из заявленного ациклического альдегида. Также изобретение относится к вариантам чистящей композиции и к вариантам способа получения спиртовой смеси для композиции моющих спиртов. 8 н. и 11 з.п. ф-лы, 10 табл., 24 пр.
1. Ациклический альдегид, имеющий 16 атомов углерода, содержащий, по меньшей мере, три разветвления и выбранный из группы, состоящей из: 3-этил-7,11-диметилдодеканаля; 2,3,7,11-тетраметил-додеканаля; 7,11-диметил-3-винилдодека-6,10-диеналя; 4,8,12-триметилтридека-4,7,11-триеналя.
2. Композиция веществ, пригодная для использования в качестве исходного материала для получения поверхностно-активных веществ, содержащая, по меньшей мере, один ациклический альдегид по п.1.
3. Композиция веществ по п.2, отличающаяся тем, что ациклический альдегид является насыщенным и разветвления представляют собой метил, этил или их смеси.
4. Композиция моющих спиртов, пригодная для получения композиции поверхностно-активных веществ, содержащая, по меньшей мере, один ациклический спирт, конвертированный из ациклического альдегида по п.1, имеющий 16 атомов углерода, и содержащий также, по меньшей мере, три разветвления, при этом разветвления представляют собой метил, этил или их смеси.
5. Композиция по п.4, отличающаяся тем, что более чем приблизительно 10% разветвленных спиртов являются триметилразветвленными спиртами.
6. Композиция по п.4, отличающаяся тем, что более чем приблизительно 30% разветвленных спиртов являются триметилразветвленными спиртами.
7. Композиция по п.4, отличающаяся тем, что более чем приблизительно 70% разветвленных спиртов являются триметилразветвленными спиртами.
8. Композиция по п.4, отличающаяся тем, что спирт получен из исходного полиразветвленного полиолефина, выбранного из группы, состоящей из:
a. фарнезена природного происхождения, экстрагированного из уже существующих растений и организмов;
b. фарнезена, полученного посредством генетически модифицированных организмов;
c. синтетически полученных тримеров изопрена; и
d. их смесей.
9. Композиция поверхностно-активного вещества, пригодная для использования в моющей или чистящей композиции, содержащая одно или более поверхностно-активных производных изомеров ациклического моющего спирта, конвертированного из ациклического альдегида по п.1 и содержащего 16 атомов углерода и два, три или четыре метильных или этильных разветвлений или их смесей.
10. Композиция поверхностно-активного вещества по п.9, отличающаяся тем, что более чем 70% производных ациклического поверхностно-активного вещества содержат две, три или четыре метильные группы.
11. Композиция поверхностно-активного вещества по п.9, отличающаяся тем, что поверхностно-активные производные изомеров ациклических моющих спиртов представляют собой сульфатные производные, этоксилированные производные, этоксилированные и сульфонатные производные или их смеси.
12. Чистящая композиция, содержащая композицию поверхностно-активного вещества по п.9.
13. Чистящая композиция, содержащая композицию поверхностно-активного вещества по п.10.
14. Способ получения спиртовой смеси для композиции моющих спиртов по п.4, включающий стадии, на которых:
a. обеспечивают один или более полиразветвленный полиолефин, при этом полиразветвленные полиолефины должны содержать один неразветвленный концевой олефин и один или более дополнительных разветвленных олефинов в молекуле;
b. гидроформилируют указанные полиразветвленные полиолефины с получением полиразветвленного олефина, содержащего альдегидный продукт с одним или более олефинами или их смесью, с применением катализатора, выбранного из группы, состоящей из модифицированных или немодифицированных переходных металлов группы IX, и условий процесса, включающих температуру процесса в диапазоне от приблизительно 50°C до приблизительно 130°C, молярное соотношение водорода и моноксида углерода в диапазоне от приблизительно 0,25:1 до приблизительно 4:1 и общее давление в диапазоне от приблизительно 300 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм;
c. восстанавливают альдегидный продукт, полученный на стадии (b) в присутствии водорода и катализатора гидрогенизации, с применением условий процесса, включающих температуру процесса в диапазоне от приблизительно 20°C до приблизительно 130°C, и давление водорода в диапазоне от 100 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм с образованием смеси полиразветвленных спиртов; и
d. удаляют указанную смесь полиразветвленных спиртов из указанного катализатора.
15. Способ по п.14, отличающийся тем, что полиразветвленные полиолефины выбирают из группы, состоящей из:
a. фарнезена природного происхождения и смесей фарнезенов, экстрагированных из природных растений и организмов;
b. фарнезена и смесей фарнезенов, полученных посредством генетически модифицированных организмов;
c. синтетически полученных тримеров изопрена; и
d. их смесей.
16. Способ по п.14, отличающийся тем, что дополнительно включает стадию, на которой селективно гидрогенизируют все соединения кроме одного олефина смеси полиразветвленных полиолефинов с получением смеси полиразветвленных моноолефинов перед проведением стадии гидроформилирования b.
17. Способ по п.16, отличающийся тем, что полиразветвленные полиолефины выбирают из группы, состоящей из:
e. фарнезена природного происхождения и смесей фарнезенов, экстрагированных из природных растений и организмов;
f. фарнезена и смесей фарнезенов, полученных посредством генетически модифицированных организмов;
g. синтетически полученных тримеров изопрена; и
a. их смесей.
18. Способ получения спиртовой смеси для композиции моющих спиртов по п.4, включающий стадии, на которых:
a. обеспечивают полиразветвленные полиолефины, содержащие один неразветвленный концевой олефин и один или более дополнительных разветвленных олефинов в молекуле;
b. одновременно гидроформилируют и восстанавливают указанный полиразветвленный полиолефин с применением катализатора, выбранного из конкретных модифицированных переходных металлов группы IX и условий процесса, включающих температуру процесса в диапазоне от приблизительно 90°C до приблизительно 200°C, молярное соотношение водорода и моноксида углерода в диапазоне от приблизительно 2 к 1 до приблизительно 5 к 1 и общее давление в диапазоне от приблизительно 300 фунтов на кв. дюйм до приблизительно 2000 фунтов на кв. дюйм; и
c. удаляют указанную спиртовую композицию из указанного катализатора.
19. Способ по п.18, отличающийся тем, что дополнительно включает стадию, на которой селективно гидрогенизируют все соединения кроме одного олефина смеси полиразветвленных полиолефинов с получением смеси полиразветвленных моноолефинов перед проведением стадии гидроформилирования b.
| Устройство для сигнализации о состояниии контролируемого объекта | 1975 |
|
SU542217A1 |
| US 6153577A1, 28.11.2000 | |||
| Francis J | |||
| McQuillin et al “Complexing of Terpenes with | |||
| Transition Metals | |||
| Part V | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Thallium(III)” J.C.S.Perkin I, 1975, 19, 2092-2096 | |||
| Hiroshi Ohrui et al “Development of | |||

