ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка представляет собой частичное продолжение предыдущей заявки на патент США №12/144620, поданной 23 июня 2008 г., и испрашивает приоритет предварительной заявки на патент США №61/020930, поданной 14 января 2008 г. Каждая из приведенных ниже заявок полностью включена в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к фармацевтическим лекарственным формам для парентерального введения, содержащим агонисты допамина, отдельно или в комбинации с агентами периферического действия, которые применяют для лечения нарушений метаболизма, а также к способам получения таких лекарственных форм и способам лечения с применением данных лекарственных форм.
УРОВЕНЬ ТЕХНИКИ
Агонисты допамина применяют для лечения различных заболеваний, например мигрени, болезни Паркинсона, акромегалии, гиперпролактинемии, пролактиномы, галактории, аменореи и нарушений обмена веществ. Обычно агонисты допамина предпочитают вводить в форме таблеток или капсул. Однако введение агонистов допамина через кишечник связано с некоторыми проблемами.
Агонисты допамина, всасываемые через слизистую оболочку желудка или кишечника в желудочно-кишечном тракте (ЖКТ), например, обычно подвергаются интенсивному пресистемному метаболизму и разрушению внутренними органами, в первую очередь печенью, что приводит к тому, что только малая часть введенной дозы достигает системного кровотока. Пресистемный метаболизм происходит вследствие инактивации перорально введенного лекарственного средства в кишечнике и печени до того, как лекарственное средство достигнет системного кровотока для доставки в другие органы и ткани. Поэтому дозы соединений для применения в качестве медикамента, которые подвергаются пресистемному метаболизму при применении в качестве медикамента, должны быть достаточно большими, чтобы компенсировать существенную первоначальную потерю лекарственного средства, для того чтобы достаточное количество лекарственного средства достигло системного кровотока для обеспечения терапевтического эффекта.
Всасывание агонистов допамина через слизистую оболочку желудка или кишечника также может быть затруднено, так как агонисты допамина и их метаболиты могут вызывать нежелательные побочные эффекты (например, тошнота, рвота, боли в животе, запор и диарея). Необходимость применения повышенных доз для компенсации пресистемного метаболизма с целью достижения терапевтического эффекта увеличивает вероятность нежелательных побочных эффектов в ЖКТ.
Пресистемного метаболизма и воздействия на внутренние органы можно избежать с помощью лекарственных форм для парентерального введения, которые обеспечивают введение и последующее всасывание агонистов допамина путем или путями, отличными от слизистой оболочки желудка и/или кишечника. Лекарственные формы для парентерального введения также преимущественно обеспечивают механизм снижения общего терапевтического дозируемого количества агонистов допамина, поскольку не требуется преодолевать пресистемный метаболизм.
Тем не менее, получение стабильных лекарственных форм для парентерального введения, содержащих производные агонисты допаминов из спорыньи, особенно затруднительно, так как производные из спорыньи чрезвычайно неустойчивы по отношению к свету и воде. Таким образом, составы производных из спорыньи следует готовить таким образом, чтобы избегать попадания света и предотвратить увлажнение.
Кроме того, для лекарственных форм, применимых для лечения нарушений метаболизма, или их ключевых элементов необходимо создать отдельный фармакокинетический профиль, в котором учитываются ежедневные отклонения уровней различных гормонов. То есть для многих гормонов, участвующих в нарушениях метаболизма, характерны ежедневные циркадные ритмы колебаний их уровней в сыворотке. К таким гормонам относятся стероиды надпочечников, например глюкокортикостероиды, а именно кортизол и пролактин, гормон, секретируемый гипофизом. Указанные дневные ритмы являются ценными показателями для понимания и лечения нарушений метаболизма. Например, пролактин достигает пиковой концентрации в разное время для худых и толстых животных.
Нормальный ежедневный профиль пролактина у здорового человека регулярный и воспроизводимый, для которого характерен низкий и относительно постоянный уровень днем с последующим острым ночным пиком, который возвращается на низкий уровень к началу дня. Смотрите патент США №5679685, содержание которого включено в настоящую заявку посредством ссылки. Изменяя профиль пролактина у субъекта с нарушением метаболизма или его ключевого элемента для того, чтобы данный профиль стал похож на профиль здорового субъекта того же вида и пола, можно обеспечить благотворное терапевтическое действие на субъект. Агонисты допамина применяют для лечения нарушений метаболизма и/или его ключевых элементов, и указанные агонисты можно применять для восстановления ежедневных профилей пролактина у субъектов с нарушениями метаболизма и/или его ключевых элементов до уровня здорового человека.
Введение агонистов допамина может действовать централизованно для исправления нарушенных нейроэндокринных событий, контролирующих периферический метаболизм у субъектов с метаболическим заболеванием. Поэтому терапия агонистами допамина может влиять на этиологические факторы в развитии и поддержании нарушений метаболизма, включая, но не ограничиваясь, нарушения, связанные с ожирением, диабетом 2 типа, преддиабетом, риском кардиометаболического синдрома и/или метаболическим синдромом. Благодаря уникальному централизованному механизму действия, такая терапия может быть эффективно совмещена с различными агентами периферического действия, которые направлены на определенные биохимические процессы, участвующие в проявлении отдельных элементов нарушения метаболизма, которые нельзя полностью облегчить с помощью терапии агонистами допамина, например ингибиторами ГМГКоА-редуктазы для снижения повышенного холестерола в плазме, гипотензивными средствами для снижения кровяного давления, механизм действия которых отличается от терапии агонистами допамина, и противодиабетическими агентами, которые оказывают регулирующее действие агонистов допамина на метаболизм глюкозы, такими как стимуляторы секреции инсулина после принятия пищи или сам инсулин, противовоспалительными агентами и антикоагулянтами.
В данной области существует потребность в улучшенных лекарственных формах для введения агонистов допамина, особенно для лечения метаболических заболеваний. Соответственно приведенные в настоящей заявке улучшенные лекарственные формы, подходящие для парентерального введения агонистов допамина, позволяют избежать проблем и улучшают способы эффективного лечения метаболических заболеваний, характерных для известных лекарственных форм. Приведенные в настоящей заявке лекарственные формы позволяют избежать таких проблем, как, например, пресистемный метаболизм и проявление нежелательных побочных эффектов, влияние на эффективность из-за перорального введения лекарства.
КРАТКОЕ СОДЕРЖАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к лекарственным формам для введения агонистов допамина, включая лекарственные формы, содержащие один или более агонистов допамина и один или более агентов периферического действия, и способам применения таких лекарственных форм для лечения нарушений метаболизма.
Согласно одному из вариантов реализации данного изобретения предложена лекарственная форма, содержащая активный агент, содержащий один или более агонистов допамина и фармацевтически приемлемый наполнитель, при этом указанная лекарственная форма подходит для парентерального введения и имеет фармакокинетический профиль с Tmax в плазме от приблизительно 1 до приблизительно 90 минут после введения, концентрацией лекарства в плазме по меньшей мере 50% Cmax в течение периода по меньшей мере от приблизительно 90 до приблизительно 360 минут и снижением уровня в плазме, что может приблизительно соответствовать кинетике выведения первого порядка.
Согласно другому варианту реализации данного изобретения предложен способ лечения нарушения метаболизма или по меньшей мере его одного ключевого элемента, включающий введение субъекту, нуждающемуся в указанном лечении, терапевтического эффективного количества лекарственной формы, содержащей активный агент, включающий один или более агонистов допамина и фармацевтически приемлемый наполнитель, при этом указанная лекарственная форма подходит для парентерального введения и имеет фармакокинетический профиль с Tmax в плазме приблизительно от 1 до приблизительно 90 минут после введения, концентрацией лекарства в плазме, по меньшей мере, 50% Cmax в течение периода от приблизительно 90 до приблизительно 360 минут и снижением уровня в плазме, что может приблизительно соответствовать кинетике выведения первого порядка.
Согласно другому варианту реализации данного изобретения предложен способ снижения повышенных уровней норепинефрина в плазме, включающий введение субъекту, который в этом нуждается, терапевтически эффективного количества лекарственной формы, содержащей активный агент, включающий один или более агонистов допамина и фармацевтически приемлемый наполнитель, при этом указанная лекарственная форма подходит для парентерального введения и имеет фармакокинетический профиль с Tmax в плазме приблизительно от 1 до приблизительно 90 минут после введения, концентрацией лекарства в плазме по меньшей мере 50% Cmax в течение периода приблизительно от 90 до приблизительно 360 минут и снижением уровня в плазме, что может приблизительно соответствовать кинетике выведения первого порядка.
Согласно другому варианту реализации данного изобретения предложен способ снижения дневного уровня пролактина в плазме при сохранении повышения ночного уровня пролактина по сравнению с дневным уровнем пролактина, включающий введение субъекту, который в этом нуждается, терапевтически эффективного количества лекарственной формы, содержащей активный агент, включающий один или более агонистов допамина и фармацевтически приемлемый наполнитель, при этом указанная лекарственная форма подходит для парентерального введения и имеет фармакокинетический профиль с Tmax в плазме приблизительно от 1 до приблизительно 90 минут после введения, концентрацией лекарства в плазме по меньшей мере 50% Cmax на протяжении приблизительно от 90 до приблизительно 360 минут и снижением уровня в плазме, что может приблизительно соответствовать кинетике выведения первого порядка.
Согласно другому варианту реализации изобретения предложен способ уменьшения повышенных факторов воспаления, связанных с сердечно-сосудистой системой, или сердечно-сосудистых заболеваний или ключевых элементов сердечнососудистых заболеваний, включающий введение субъекту, который в этом нуждается, терапевтически эффективного количества лекарственной формы, содержащей активный агент, включающий один или более агонистов допамина и фармацевтически приемлемый наполнитель, при этом указанная лекарственная форма подходит для парентерального введения и имеет фармакокинетический профиль с Tmax в плазме приблизительно от 1 до приблизительно 90 минут после введения, концентрацией лекарства в плазме по меньшей мере 50% Cmax в течение периода приблизительно от 90 до приблизительно 360 минут и снижением уровня в плазме, что может приблизительно соответствовать кинетике выведения первого порядка.
Согласно одному варианту реализации данного изобретения предложена лекарственная форма, содержащая активный агент, включающий один или более агонистов допамина и фармацевтически приемлемый наполнитель, при этом указанная лекарственная форма подходит для парентерального введения и имеет фармакокинетический профиль, который характеризуется Tmax в плазме приблизительно от 5 до приблизительно 90 минут после введения, пост-Cmax уровнем, составляющим приблизительно одну вторую Cmax в течение периода приблизительно от 30 до приблизительно 150 минут Tmax, причем пост-Cmax уровень составляет приблизительно одну вторую Cmax в течение периода от приблизительно 90 до приблизительно 360 минут, и снижением уровня в плазме, которое может приблизительно соответствовать кинетике выведения первого порядка.
Согласно другому варианту реализации данного изобретения предложен способ лечения нарушения метаболизма или его ключевого элемента посредством введения лекарственной формы для парентерального введения, содержащей один или более агонистов допамина, при этом уменьшаются повышенные уровни норепинефрина или пролактина в плазме и ночной уровень пролактина в плазме повышается по сравнению с заново установленным средним дневным уровнем циркулирующего пролактина.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фигуре 1 представлен график, иллюстрирующий фармакокинетический профиль дозированной лекарственной формы для парентерального введения согласно настоящему изобретению для введения агониста допамина.
На фигуре 2 представлен график, иллюстрирующий другой фармакокинетический профиль дозированной лекарственной формы для парентерального введения согласно настоящему изобретению для введения агониста допамина.
На фигуре 3 представлен график, на котором показано влияние 7-дневного лечения путем парентерального введения лекарственной формы 34Gel (10 мг/кг) на резистентность к инсулину (HOMA-IR) на модели крыс SHR.
На фигуре 4 представлен график, на котором показано влияние 7-дневного лечения путем парентерального введения лекарственной формы 34Gel (10 мг/кг) на уровень инсулина в плазме на модели крыс SHR.
На фигуре 5 представлен график, на котором показано влияние 7-дневного лечения путем парентерального введения лекарственной формы 34Gel (10 мг/кг) на кровяное давление на модели крыс SHR.
На фигуре 6 представлен график, на котором показано влияние7-дневного лечения путем парентерального введения лекарственной гелевой формы 34Gel (10 мг/кг) на изменение массы тела на модели крыс SHR.
На фигуре 7 представлен график, на котором показано влияние 7- дневного лечения путем парентерального введения лекарственной формы 34Gel (10 мг/кг) на массу тела на модели крыс SHR.
На фигуре 8 представлен график, на котором показано влияние 7-дневного лечения путем парентерального введения лекарственной формы 34Gel (10 мг/кг) на уровне эндотелина-1 на модели крыс SHR.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящей заявке представлены лекарственные формы для парентерального введения, например, одного или более допаминового агониста по отдельности или в комбинации с одним или более антигипертензивным, антигиперхолестеринемическим, антигиперглицеридемическим, противовоспалительным, антикоагуляторным или гипогликемическим агентом. Лекарственные формы проявляют физиологические свойства, например фармакокинетический профиль, который оказывает определенное нейроэндокринное действие и позволяет лечить нарушения метаболизма и/или их ключевые элементы. Дозированные лекарственные формы содержат активный агент или активные агенты и один или более наполнителей.
Лекарственные формы особенно подходят для лечения нарушений метаболизма и/или ключевых элементов таких нарушений, включая, но не ограничиваясь: диабет 2 типа, преддиабеты (нарушенный уровень глюкозы натощак или плохая переносимость глюкозы), метаболический синдром или признаки (ключевые элементы) указанных расстройств (увеличенный объем талии, повышенный уровень глюкозы в плазме натощак, повышенный уровень триглицеридов в плазме натощак, пониженный уровень липопротеинов высокой плотности в плазме натощак, повышенное кровяное давление), резистентность к инсулину, гиперинсулинемия, сердечно-сосудистые заболевания (или их ключевые элементы, например артериосклероз, болезнь коронарных артерий, болезнь периферических сосудов или нарушение мозгового кровообращения), застойная сердечная недостаточность, ожирение, повышенный уровень норепинефрина в плазме, повышенный уровень сердечно-сосудистых воспалительных факторов, гиперлипопротеинемия, атеросклероз, гиперфагия, гипергликемия, гиперлипидемия и гипертензия или высокое кровяное давление, повышенный уровень триглицеридов или свободных жирных кислот в плазме после приема пищи, повышенный окислительный стресс в клетках или его признаки в плазме, состояние усиленной гиперсвертываемости крови при циркуляции, заболевание почек, включая почечную недостаточность.
Лекарственные формы, содержащие агонист(ы) допамина и агент(ы), периферического действия, можно применять для отдельных групп пациентов, при необходимости, например, агонист допамина + составы ГМГКоА-редуктазы для пациентов с диабетом 2 типа и гиперхолестеролимией или агонист допамина + лекарственное средство против повышенного давления для пациентов с диабетом 2 типа и очень высоким давлением и другие сочетания.
Более того, такая терапия комбинацией агонистов допамина с агентом периферического действия может иметь большое значение и применение, если они находятся в одной лекарственной форме, которая позволяет надлежащим образом дозировать каждый компонент. В действительности такие лекарственными формы могут представлять собой "полипилюли", которые пытаются создать медицинское и фармацевтическое сообщества для лечения различных нарушений, связанных с распространенными нарушениями метаболизма: диабет 2 типа, ожирение, метаболический синдром и/или риск развития кардиометаболического синдрома с помощью однократного ежедневного применения лекарства. Лекарственные формы для парентерального введения позволяют подобрать оптимальную небольшую дозировку агониста(ов) допамина, как описано в настоящей заявке, а также, согласно некоторым вариантам реализации, ингибиторов ГМГКоА-редуктазы, которые подвергаются пресистемному метаболизму в печени.
Комбинация лекарственных форм для парентерального введения согласно настоящему изобретению также позволят подобрать способ лечения метаболического заболевание у субъекта на индивидуальной основе, который включает централизованно действующий «настраивающий» компонент, который действует на общее метаболическое заболевание (гипертензия, дислипидемия и гипергликемия), и любой из нескольких агентов периферического действия, которые направлены на определенные мишени метаболического заболевания (гипертензия, дислипидемия или гипергликемия), в зависимости от индивидуальных потребностей пациента. В то же время комбинация лекарственных форм для парентерального введения согласно настоящему изобретению также позволит вводить меньшие дозы агонистов допамина и/или агентов периферического действия и благодаря этому смягчить или избежать побочных эффектов, связанных с введением агонистов допамина и периферически действующих агентов. Например, если гипотензивный агент периферического действия достигает пиковой концентрации через несколько часов после агониста допамина, уменьшается или исчезает вероятность возникновения ортостатической гипертензии, а также обморока или потери сознания. В другом примере для комбинаций с ингибиторами НМГСоА-редуктазы можно применять низкие дозы агонистов допамина и ингибиторов ГМГКоА-редуктазы, т.к. и агонист допамина, и ингибитор ГМГКоА-редуктазы подвергаются пресистемному метаболизму в печени (и в случае агонистов допамина из спорыньи в обоих случаях используется один метаболический путь через цитохром Р450-3А). Если ингибитор ГМГКоА-редуктазы высвобождается после агониста допамина, тогда уменьшается вероятность конкурентного взаимодействия в печени при метаболизме, подобное выгодное обстоятельство помогает лучше спрогнозировать циркулирующую дозу для каждого соединения. Это уменьшит возможное побочное действие на мышечную боль, которая возникает при применении каждого из указанных соединений. В другом примере в случае комбинаций агонистов допамина со стимуляторами секреции инсулина (например, стимуляторами непрерывной или, что предпочтительно, секреции инсулина после еды), такие лекарственные формы позволяют вводить дозу раз в день - лекарственные формы согласно настоящему изобретению ускоряют немедленное высвобождение инсулина, за которым следует другое высвобождение инсулина 4 часа спустя, что облегчает высвобождение инсулина в нужное время после завтрака и обеда, при этом сводится к минимуму риск гипогликемии, что очень важно для всех антидиабетических препаратов.
Лекарственные формы для парентерального введения, представленные в настоящей заявке, имеют необходимые свойства по сравнению с пероральными препаратами, включая улучшенную эффективность доставляемого лекарства при лечении нарушения метаболизма и/или его ключевого элемента, введение меньшего количества агониста допамина или агонистов допамина для достижения терапевтического эффекта, пониженный уровень циркулирующих метаболитов лекарства, повышенное соотношение уровней циркулирующего лекарства и метаболитов, лучший терапевтический индекс (т.е. действие лекарства/побочные эффекты), устранение пресистемного метаболизма и возможность избежать побочных эффектов со стороны ЖКТ благодаря взаимодействию лекарства с сайтами связывания агонистов допамина в кишечнике. Также лекарственные формы, представленные в настоящей заявке, обладают преимуществом того, что пациенты сами могут вводить их себе без медицинской помощи.
Применение представленных композиций при лечении нарушений метаболизма позволяет достичь лучшего результата по сравнению с результатом, полученным при введении соответствующей дозы пероральных агонистов допамина. В одном аспекте меньшие дозы лекарственных форм для парентерального введения могут обеспечивать эффективную дозу, эквивалентную большим дозам при пероральном введении того же агониста(ов) допамина. В другом аспекте введение меньших доз агонистов допамина приводит к образованию меньшего количества метаболитов агониста(ов) допамина, особенно в случае агонистов допамина спорыньи. Также в другом аспекте парентеральное введение лекарственных форм приводит к пониженному образованию метаболитов, которые, как считается, обладают биологическими свойствами, которые нейтрализуют действие исходного соединения, по сравнению с пероральным введением лекарственной формы с тем же количеством активного агента. Авторы изобретения также неожиданного обнаружили, что если использовать агонисты допамина, которые применяют при лечении нарушений метаболизма, в соответствующих дозах и в определенное время дня, как описано в настоящей заявке, то они более эффективны, когда уровни активных метаболитов понижены. Таким образом лекарственные формы для парентерального введения обладают большей сравнительной терапевтической эффективностью по сравнению с эквимолярными циркулирующими концентрациями перорально введенных лекарственных форм, в частности, благодаря пониженному относительному уровню активных метаболитов.
Соответственно лекарственная форма для парентерального введения агониста(ов) допамина, которая продуцирует уровень, эквивалентный Tmax для перорально вводимой лекарственной формы агониста(ов) допамина, может увеличивать относительное соотношение между исходным соединением агониста допамина и метаболитом в кровотоке и поэтому увеличивает эффективность агониста(ов) допамина при лечении нарушения метаболизма по сравнению с уровнем, эквивалентным Tmax для перорально вводимого агониста(ов) допамина. Например, терапевтически эффективное количество агониста(ов) допамина, вводимого в пероральной форме для лечения нарушения метаболизма, составляет 1 мг/день и дает 100 мкг агониста(ов) и 900 мкг метаболитов (из-за пресистемного метаболизма) в кровотоке. И наоборот, лекарственная форма для парентерального введения может достигать той же «эффективной» дозы агониста(ов) допамина в кровотоке при введении 120 мкг агониста(ов) допамина, так как при этом не происходит пресистемного метаболизма лекарственного средства, и со временем образуется только 20 мкг метаболитов.
Соответственно, отношение лекарство/метаболит составляет 100/900 при пероральном введении и 100/20 в случае лекарственной формы для парентерального введения. Таким образом, нейтрализующее действие метаболитов на метаболическую активность исходного соединения снижено, особенно при таком введении, как описано в настоящей заявке.
В другом аспекте лекарственные формы, представленные в настоящей заявке, стабильны, и их можно применять после хранения на протяжении длительного периода времени. Необратимая агрегация в лекарственной форме отсутствует или снижена даже при хранении в течение нескольких месяцев.
К активным агентам агонистов допамина для включения в лекарственную форму относятся, например, но не ограничиваясь перечисленными: производные не из спорыньи и из спорыньи. К активным агентам агонистов допамина относятся агонисты рецептора допамина D1 и/или агонисты рецептора допамина D2. Согласно некоторым вариантам реализации пациенту, который нуждается в лечении, вводят агонист допамина D1. Согласно другим вариантам реализации пациенту, который нуждается в лечении, вводят агонист допамина D2. В других вариантах реализации пациенту, который нуждается в лечении, агонист допамина вводят совместно с агонистом допамина D2.
К активным агентам периферического действия которые следует включить в лекарственные формы, представленные в настоящей заявке, относятся, не ограничиваясь перечисленными: антигипертензивные, противоспалительные, антикоагулянты, антигиперхолестеринемические, антигиперглицеридемические и/или антигипергликемические агенты. Согласно некоторым вариантам реализации активный агент периферического действия представляет собой ингибитор ГМГКоА-редуктазы.
Лекарственные формы, представленные в настоящей заявке, могут включать, состоять исключительно или содержать агонист рецептора допамина, в отдельности или в комбинации с агонистом D2 рецептора допамина и, возможно, дополнительно в комбинации с одним или более активными агентами периферического действия.
В настоящей заявке термины «комбинированное» лечение или введение, или лечение и в введение «в комбинации» означает, что пациент получает по меньшей мере первое количество первого активного агента и второе количество второго активного агента. Активные агенты можно вводить в одной лекарственной форме или в отдельных лекарственных формах. Агенты, которые вводят в отдельных лекарственных формах, можно вводить одновременно или в разное время. Например, агонист D1 и агонист D2 можно вводить в одно время (в одной лекарственной форме или двух и более отдельных формах) или последовательно в различное время в различных лекарственных формах.
Терапевтически эффективные количества агониста для человека и позвоночных при введении парентерально в отдельности (без агониста D2), как правило, находятся в пределах от приблизительно 1,0 мкг/кг/день до приблизительно 10,0 мг/кг/день. Предпочтительно терапевтически эффективные количества агониста D1 для человека и позвоночных при введении в отдельности, как правило, находятся в пределах от предпочтительно 1,0 мкг/кг/день до приблизительно 7,0 мг/кг/день. Более предпочтительно терапевтически эффективные количества агониста D1 для человека и позвоночных при введении в отдельности, как правило, находятся в пределах от предпочтительно 1,0 мкг/кг/день до предпочтительно 5,0 мг/кг/день. Наиболее предпочтительно терапевтически эффективные количества агониста D1 для человека и позвоночных при введении в отдельности, как правило, находятся в пределах от предпочтительно 2,0 мкг/кг/день до предпочтительно 3,0 мг/кг/день.
Терапевтически эффективные количества агониста D2 для человека и позвоночных при парентеральном введении по отдельности (без агониста D1), как правило, находятся в пределах от приблизительно 0,5 мкг/кг/день до приблизительно 300 мкг/кг/день. Предпочтительно терапевтически эффективные количества агониста D2 для человека и позвоночных при введении в отдельности (без агониста D1), как правило, находятся в пределах от приблизительно 0,5 мкг/кг/день до приблизительно 250 мкг/кг/день. Более предпочтительно терапевтически эффективные количества агониста D2 для человека и позвоночных при введении в отдельности, как правило, находятся в пределах от приблизительно 0,5 мкг/кг/день до приблизительно 200 мкг/кг/день. Наиболее предпочтительно терапевтически активные количества агониста D2 для человека и позвоночных при введении в отдельности, как правило, находятся в пределах от приблизительно 1 мкг/кг/день до приблизительно 150 мкг/кг/день.
Если терапевтически эффективные количества агониста(ов) D1 и D2 человека и позвоночных вводят в комбинации парентерально, можно использовать приблизительно на 15% меньше каждого агониста. Предпочтительно, если терапевтически эффективные количества агониста(ов) и D2 человека и позвоночных вводят в комбинации парентерально, то используют приблизительно на 17% меньше каждого агониста. Более предпочтительно, если терапевтически эффективные количества агониста(ов) D1 и D2 человека и позвоночных вводят в комбинации парентерально, то используют приблизительно на 20% меньше каждого агониста. Наиболее предпочтительно, если терапевтически эффективные количества агониста(ов) и D2 человека и позвоночных вводят в комбинации парентерально, то используют по меньшей мере на 25% меньше каждого агониста.
В неколлоидной форме агонист допамина обычно формируют в виде частицы размером (d90) приблизительно от 5 до 175 мкм. Предпочтительно, в неколлоидной форме агонист допамина обычно формируют в виде частиц размером приблизительно от 5 до 150 мкм. Более предпочтительно, в неколлоидной форме агонист допамина обычно формируют в виде частиц размером приблизительно от 5 до 125 мкм. Наиболее предпочтительно, в неколлоидной форме агонист допамина обычно формируют в виде частиц размером приблизительно от 10 до 100 мкм.
В коллоидной форме агонист допамина обычно формируют в виде частиц размером приблизительно от 0,1 до 5,0 мкм. Предпочтительно агонист допамина в коллоидной форме обычно формируют в виде частицы размером приблизительно от 0,1 до 3,0 мкм. Более предпочтительно, агонист допамина в коллоидной форме обычно формируют в виде частиц размером приблизительно от 0,1 до 2,0 мкм. Наиболее предпочтительно агонист допамина в коллоидной форме обычно формируют в виде частиц размером приблизительно от 0,1 до 1,0 мкм.
Агонист допамина D1 активирует или усиливает рецепторы допамина D1 или D1 подобные рецепторы, например рецепторы допамина и D5. Агонист также представляет собой селективный агонист для рецепторов D1 по отношению к D2 (т.к. данное соединение для рецептора D1 имеет более низкую Ki или ЕС50, чем для рецептора D2). Согласно одному варианту реализации агонист представляет собой слабый агонист (т.е. Ki или EC5Q больше, чем 1 мкМ или 1 мМ) или частичный агонист (связывающая активность для сайтов D2 меньше, чем для эндогенного допамина) или не является агонистом D2 (т.е. Ki или ЕС50 больше, чем 10 мМ).
В данной области хорошо известны агонисты допамина D1, которые способны активировать или усиливать рецепторы допамина D1. Примеры агонистов D1 включают, но не ограничиваются перечисленными: допамин, апоморфин, SKF38393, дигидрексидин, SKF 75670, SKF 82957, SKF 81297, SKF 82958, SKF 82598, А77636, А68930 и SKF 82526 (фенолдопам) и рацемические смеси транс-10, 11-дигидрокси 5, 6, 6а, 7, 8, 12b-гексагидро- и родственные аналоги бензазепина и агонисты включенные посредством ссылки в настоящую заявку. Предпочтительными агонистами допамина являются SKF 38393 или апоморфин. Смотрите, например, патент США №6855707, включенный в полном объеме в данную заявку посредством ссылки.
Агонисты допамина D2 активируют или усиливают рецепторы допамина D2 (например, рецепторы допамина D2, короткие D2 и длинные рецепторы D2, D4 и рецепторы допамина D4). Согласно одному из вариантов реализации агонист D2 представляет собой селективный агонист рецептора D2, по сравнению с рецептором D1. В другом варианте реализации агонист D2 представляет собой слабый агонист D1 или не является агонистом D1. Примеры агонистов допамина D2 хорошо известны в данной области.
Производные спорыньи агонисты D2 включают, например, но не ограничиваются перечисленными: 2-бром-α-эргокриптин (бромкриптин), тергурид, дигидроэрготоксин (гидергин), эрфотоксин, 6-метил-8-β-карбобензилокси-аминоэтил-10-α-эрголин, 8-ациламиноэрголин, 6-метил-8-α-(N-ацил)амино-9-эрголин, лизурид, дигидро-альфа-эргокриптин, дигидро-альфа-эрготоксин, 6-метил-8-α-(N-фенилацетил)амино-9-эрголин, эргокорнин, 9,10-дигидроэргокорнин, любой D-2-гало-6-алкил-8-замещенный эрголин и D-2-бром-6-метил-8-цианометилэрголин. Наиболее предпочтительными из указанных соединений являются бромкриптин или лизурид или производные спорыньи с небольшой активностью относительно рецептора серотонина 5НТ2В или ее отсутствием.
Примеры агонистов допамина D2, которые не являются производными спорыньи, включают, но не ограничиваются перечисленными: ропинитрол, пирибедил, апоморфин, хинелоран и талипексол.
Примеры агентов периферического действия включают, но не ограничиваются перечисленными: вещества, которые оказывают антигипертензивное, противовоспалительное, антигиперхолестеринемическое, антигипертриглицеридемическое и/или антигипергликемическое действие.
Антигиперотензивные препараты включают, например, и не ограничиваясь перечисленными, например, агенты, которые представляют собой ингибиторы ангиотензинконвертирующего фермента (АСЕ), блокаторы рецептора ангиотензина 2 (ARBs), блокаторы кальциевых каналов, β-блокаторы, α-блокаторы и диуретики. Примеры антигипертензивных агентов включают, например, и не ограничиваясь перечисленными: буметанид, этакриновая кислота, фуросемид, торсемид, хлорталидон, эпитизид, гидрохлоритриазид, хлоротриазид, бендрофлюметазид, индапамид, метолазон, амилорид, триамтерен, спиронолактон, атенолон, метопролол, надолол, окспренолол, пиндолол, пропанолол, тимолол, доксазозин, фентоламин, индорамин, феноксибензамин, празозин, теразозин, толазолин, буциндолол, карведилол, лабеталол, клонидин, метилдопа, амлодипин, фелодипин, израдипин, нифедипин, нимодипин, нитрендипин, дилтиазем, верапамил, каптоприл, эналаприл, фозиноприл, лизиноприл, периндоприл, хинаприл, рамиприл, трандоприл, бензаприл, кандезартан, эпрозартан, ирбезатран, лозатран, олмезатран, телмизатран, валзатран, спиронолактон, нитропруссид натрия, гуанабенз, гуанетидин и резерпин.
Антигиперхолестеринемические агенты включают, например, и не ограничиваясь перечисленными: агенты ингибиторы ГМГКоА-редуктазы (статины) и агенты, которые блокируют всасывание холестерола. Примеры антигиперхолестеринемических агентов включают, но не ограничиваются перечисленными: аторвастатин, церивастатин, флювастатин, ловастатин, мевастатин, правастатин, питавастатин, розувастатин, симвастатин, холестирамин, ситостерол, эзетимиб, гемфиброзил, клофибрат, никотиновая кислота, колестипол и колесевелам. Предпочтительными статиновыми агентами являются аторвастатин, церивастатин, флювастатин, ловастатин, мевастатин, правастатин, питавастатин, розувастатин и симвастатин.
Антигипертриглицеридемические агенты включают, например, и не ограничиваются фибратами. Примеры антигипертриглицеридемических агентов включают, например, и не ограничиваясь перечисленными: гембфиброзил, клофибрат, безафибрат и масло грецкого ореха.
Антигипергликемические агенты включают, например, и не ограничиваясь перечисленными: бигуаниды, стимуляторы секреции инсулина и сенсибилизаторы инсулина. Примеры антигипергликемических агентов включают, например, и не ограничиваясь перечисленными: инсулин, лекарственные средства на основе сульфонилмочевины, метформин, репаглинид, натеглинид, ингибиторы глюкозидазы, триазолидиндионы, аналоги GLP-I и ингибиторы DPP IV.
Лекарственные формы могут включать агонисты допамина или агонисты допамина в составе для получения как ускоренного, так и замедленного высвобождения лекарственного средства в кровоток. Лекарственные средства могут быть твердыми или рассыпчатыми.
В настоящей заявке термин «твердый» обозначает вещество, которое при комнатной температуре находится в твердой или полутвердой форме. Поэтому в настоящей заявке «твердое» вещество может превращаться в жидкость, например, при температуре тела.
Согласно некоторым вариантам реализации могут быть получены лекарственные формы для обеспечения двухфазного высвобождения активного компонента, например, с фазой быстрого высвобождения (a/k/a немедленное высвобождение) и фазой медленного высвобождения (a/k/a отложенное высвобождение). Быстрое и медленное высвобождение активного агента могут быть разделены физически посредством разделения компонентов с различными композициями, при этом для каждой композиции характерно ускоренное или замедленное растворение. Согласно другому варианту реализации фазы быстрого и медленного высвобождения включены в одну комбинированную лекарственную форму, которая может содержать, например, наружный слой, для которого характерно быстрое растворение, и внутренний слой, для которого характерно замедленное растворение.
Согласно другим вариантам реализации лекарственная форма может содержать растворенный агонист допамина, для которого характерно быстрое растворение, и коллоидную суспензию агониста допамина, для которой характерно медленное растворение. Включение агониста допамина с малым размером частиц от приблизительно 0,02 до приблизительно 5,0 мкм в коллоидную суспензию обеспечивает быстрое растворение и всасывание. Однако быстрое растворение и всасывание агониста допамина с малым размером частиц от приблизительно 0,02 до приблизительно 5,0 мкм в коллоидной суспензии происходит более медленно, чем для допаминового агониста уже в растворе. Предпочтительно размер малых частиц агониста допамина составляет от приблизительно 0,1 до приблизительно 3,0 мкм. Более предпочтительно размер малых частиц агониста допамина составляет от приблизительно 0,1 до приблизительно 2,0 мкм. Наиболее предпочтительно размер малых частиц агониста допамина составляет от приблизительно 0,1 до приблизительно 1,0 мкм.
Включение агониста допамина с размером частиц (d90) больше чем приблизительно 5,0 мкм, обеспечивает замедленное растворение и всасывание. Предпочтительно включение агониста допамина с размером частиц (d90) больше чем приблизительно 5-150 мкм обеспечивает замедленное растворение и всасывание. Более предпочтительно включение агониста допамина с размером частиц (d90) больше чем приблизительно 5-125 мкм обеспечивает замедленное растворение и всасывание. Наиболее предпочтительно включение агониста допамина с размером частиц (d90) больше чем приблизительно 10-100 мкм обеспечивает замедленное растворение и всасывание.
Согласно некоторым вариантам реализации частицы агониста(ов) допамина малого и крупного размера присутствуют в лекарственной форме в соотношении приблизительно 50/50. Более предпочтительно частицы агониста(ов) допамина малого и крупного размера присутствуют в лекарственной форме в соотношении приблизительно 60/40. Наиболее предпочтительно частицы агониста(ов) допамина малого и крупного размера присутствуют в соотношении приблизительно 70/30.
Согласно некоторым вариантам реализации компонент частиц малого размера составит 1,0 мкм в таблетке или другой твердой лекарственной форме и размер крупных частиц составляет 1-100 мкм.
Ингибитор пролактина (такой как бромкриптин) можно вводить субъекту-млекопитающему (в частности, человеку) в предопределенное время в течение 24 часов, если у субъекта чрезмерно высокий уровень пролактина в дневное время (по меньшей мере 1 стандартная ошибка среднего больше, чем нормальный дневной уровень пролактина для субъекта того же пола и вида). Введение и режим приема подбирают таким образом, чтобы снизить чрезмерно высокий уровень пролактина в дневное время у субъекта. Однако, возможно, потребуется вводить стимулятор пролактина в другое предопределенное время в течение 24 часов, если у субъекта чрезмерно низкий уровень пролактина ночью, для того чтобы повысить ночной уровень пролактина, чтобы предпочтительно он был не ниже, чем приблизительный уровень пролактина ночью у человека того же пола. Также возможно, что необходимо будет вводить ингибитор и стимулятор пролактина пациенту для того, чтобы обеспечить понижение дневного уровня и повышение ночного уровня пролактина.
Лекарственные формы могут также содержать, не ограничиваясь перечисленными, один или более из следующих: наполнители, безводный растворитель, фармацевтически приемлемая среда для суспендирования, носители или разбавители, поверхностно-активные соединения, регуляторы для корректировки осмолярности, биоадгезивные вещества, полимеры, проникающие агенты, стабилизаторы, безводные агенты, снижающие раздражение слизистой оболочки, наполнители, связующие агенты, разрыхлители, лубриканты, ароматизаторы и подсластители, гелирующие агенты, инертный газ, антиоксиданты, консерванты, увлажняющие агенты, поверхностно-активные вещества, агенты, контролирующие высвобождение вещества, красители, связующие агенты, суспендирующие агенты и диспергирующие агенты, красители, пленкообразующие агенты, пластификаторы или любую комбинацию двух или более из вышеперечисленных агентов.
Наполнители, которые используют в лекарственных формах, могут различаться в зависимости от типа лекарственной формы для парентерального введения. Подходящие наполнители для лекарственных форм хорошо знакомы специалисту в данной области и различаются в зависимости от активного агента, способа введения и необходимого профиля высвобождения активного агента. Неограничивающие примеры подходящих наполнителей для применения в лекарственных формах, приведены ниже.
Термином «фармацевтически приемлемый(ые) наполнитель(ли)» обозначают любой материал, который инертен в том смысле; что он существенно не оказывает терапевтическое и/или профилактическое действие. Такой наполнитель добавляют для того, чтобы фармацевтическая композиция приобрела приемлемые технические свойства.
К примерам безводных растворителей относятся, не ограничиваясь перечисленными: пропиленгликоль, глицерол, замещенные короткоцепочечные или не замещенные спирты, такие как этанол, изопропанол или пропанол. В определенном варанте реализации безводные растворители включают, но не ограничиваются перечисленными: различные гликоли и/или спирты по отдельности или в комбинации, таким образом, что в терапевтической дозе содержится нетоксичные объемы растворителя, например 0,02-0,5 мл.
Примеры фармацевтически приемлемых сред для суспендирования или матриц включают, не ограничиваясь: синтетические, полусинтетические или природные масла, которые предпочтительно можно использовать, триглицериды с цепью средней длины от С8 до С10 в остатке карбоновой кислоты, масло соевых бобов, кунжута, арахиса, оливы, кокосовое масло, касторовое масло, подсолнечное масло, сафлоровое масло или соответствующие гидрогенизированные масла или смеси по меньшей мере двух приведенных выше масел, бентонит, этоксилированные изостеариловые спирты, полиоксиэтиленсорбитол или эфиры сорбитана, микрокристаллическая целлюлоза или ее производные, растительные камеди, полиэтиленгликоли различного размера, метагидроксид алюминия, агар-агар и трагакант, желатины или смесь двух или более из указанных веществ и т.д.
Примеры фармацевтически приемлемых диспергирующих и суспензирующих агентов включают, не ограничиваясь: синтетические и природные камеди, такие как растительная камедь, трагакант, акация, альгинат, декстран, карбоксиметилцеллюлоза натрия, метилцеллюлоза, поливинилпирролидон и желатин.
Примеры фармацевтически приемлемых носителей или растворителей включают, не ограничиваясь: этанол, воду, глицерол, пропиленгликоль, глицерин, моноэтиловый эфир диэтиленгликоля, масла витамина А и Е, минеральное масло, PPG2 миристилпропионат, карбонат магния, фосфат калия, диоксид кремния, растительные масла, например касторовое масло и его производные, растительные камеди, желатин, животные масла, солкетал, карбонат кальция, двухосновный фосфат кальция, трехосновный фосфат кальция, сульфат кальция, микрокристаллическая целлюлоза, порошковая целлюлоза, декстраны, декстрин, декстрозу, фруктозу, каолин, лактозу, маннитол, сорбитол, крахмал, желатинизированный крахмал, сахарозу, сахар и т.д.
Примеры поверхностно-активных веществ включают, но не ограничиваются ими, полиалкиленовые гликоли, например полиэтиленгликоли, полипропиленгликоли или этиленоксид, блокирующие сополимеры пропиленоксида, фосфолипиды, простые или сложные эфиры насыщенных или ненасыщенных жирных спиртов или жирных кислот с полиалкиленгликолями, такими как полиэтиленгликоли или полипропиленгликоли, полисорбаты, такие как моно-, ди- или триэфиры насыщенных или ненасыщенных жирных кислот, особенно предпочтительно, олеиновой кислоты, лауриновой кислоты, пальмитиновой кислоты или стеариновой кислоты, и сорбитол и/или его ангидрид, каждый из которых может содержать до 20 моль единиц этиленоксида на моль сорбитола или ангидрида, предпочтительно полиэтоксисорбитанмонолаурат с 20 единицами этиленоксида, полиэтоксисорбитан монолаурат с 4 единицами этиленоксида, полиэтоксисорбитан монопальмиат с 20 единицами этиленоксида, полиэтоксисорбитанмоностеарат с 20 единицами этиленоксида, полиэтоксисорбитанмоностеарат с 4 единицами этиленоксида, полиэтоксисорбитан тристеарат с 20 единицами этиленоксида, полиэтоксисорбитан моноолеат с 20 единицами этиленоксида, полиэтоксисорбитан моноолеат с 5 единицами этиленоксида или полиэтоксисорбитан триолеат с 20 единицами этиленоксида или смесь по меньшей мере двух указанных выше поверхностно-активных соединений.
Примеры регуляторов осмолярности включают, не ограничиваясь перечисленными: водорастворимые, физиологически приемлемые соединения, такие как неорганические соли, например соли щелочных металлов, предпочтительно хлорид натрия, сахара, например сахароза или декстроза, сахароспирты, например маннитол или полиалкиленовые гликоли, например полиэтиленгликоли, предпочтительно имеющие молекулярную массу от 1,000 до 8,000 г/моль. Также можно использовать смесь по меньшей мере двух представителей различных классов регуляторов или по меньшей мере двух представителей одного класса регуляторов для регуляции осмолярности.
Биоадгезивные вещества входят в состав, например, адгезивных таблеток, растворов, коллоидных суспензий, гелей, мазей, пластырей, пленок, паст или леденцов. Примеры биоадгезивных полимеров включают, не ограничиваясь перечисленными: Бенецел® МР814, Коллидон, хитозан, производные целлюлозы, Карбопол 934Р, Карбопол974Р, IVoveou AA-I, карбополовые смолы, карбомер, ксантановая камедь, поликарбофил и полиэтиленоксид, в комбинации с инертным растворителем и активным ингредиентом, и ионные полисахариды. Некоторые синтетические и полусинтетические биоадгезивные полимеры с разной молекулярной массой и различиями в степени замещения включают, не ограничиваясь перечисленными: гидроксиэтилеллюлозу, поливиниловый спирт, полиакриловую кислоту, карбоксиметилцеллюлозу натрия, поливинилпирролидон, полиэтиленгликоли и другие. Адгезия к слизистой оболочке указанных биоадгезивных лекарственных форм основана на взаимопроникновении гидратированных гидроколлоидных цепей биадгезивных лекарственных форм и гликопротеиновых цепей слизистой полости рта.
Примеры подходящих пленкообразующих агентов включают, но не ограничиваются перечисленными: гидроксипропилметилцеллюлозу, этилцеллюлозу и полиметакрилаты.
Примеры подходящих пластификаторов включают, но не ограничиваются перечисленными: полиэтиленгликоли различной молекулярной массы (например, 200-8000 Да), растительные камеди и пропиленгликоль и триэтилцитрат.
Примеры агентов, улучшающих проницаемость, включают, но не ограничиваются ими: соли желчных кислот, производные жирных кислот, эфиры жирных кислот, например лауриновые, миристиновые и стеариновые моноэфиры полиэтиленгликоля, производные энамина и альфа-кетоальдегиды, холат натрия, гликохолат натрия, дезоксихолат натрия, лаурилсульфат натрия, салицилат натрия, натриевую соль этилендиаминтетраукусной кислоты (EDTA); апротинин, азон, 5-метоксисалицилат натрия, 1-олейлазанциклогептан-2-он и/или оксиды кремния с высоким сродством к водным растворителям, например осажденный диоксид кремния, более известный под товарным знаком Силоид®, мальтодекстрины, β-циклодекстрины, поверхностно-активные вещества, хелаторы, циклодекстрины, хитозан и низшие спирты.
Примеры стабилизаторов включают, но не ограничиваются ими,: лимонную кислоту, аскорбиновую кислоту, олеиновую кислоту, каприловую кислоту, каприновую кислоту, поливинилпирролидон, воски, блокирующие сополимеры, полоксамеры, Полоксамер 188 и 407, полоксамины, Полоксамин 908, поливинилпирролидон, поливиниловый спирт, желатин, полисахарид, гиалуроновую кислоту, хитозан, производные хитозана, полиакриловую кислоту, производные полиакриловой кислоты, поликарбофил, производные целлюлозы, метилцеллюозу, гидроксипропилцеллюлозу, карбоксиметилцеллюлозу, эфиры сахаров, моностеарат сахарозы, цитрат натрия индивидуально, жирные кислоты, жирные спирты, спирты, эфиры длинноцепочечных жирных кислот, длинноцепочеченые эфиры, гидрофильные производные жирных кислот, поливинилэфиры, поливиниловые спирты, углеводы, гидрофобные полимеры, влогопоглощающие полимеры и их комбинации.
Примеры агентов, уменьшающих раздражение обезвоженной слизистой оболочки, включают, но не ограничиваются ими: растительные масла, такие как, не ограничиваясь, оливковое, кукурузное или минеральное масло.
Примеры наполнителей включают, но не ограничиваются ими: ПроСолв, Фармабурст, Кабосил и сахариды, например маннитол, лактоза, ксилитол или их смеси.
Примеры подходящих связующих агентов включают, но не ограничиваются ими, по отдельности или в комбинации, такие связующие агенты, как сахароза, желатин, глюкоза, крахмал, целлюлозные материалы, полиэтиленгликоли, повидон, метилцеллюлоза, карбоксиметилцеллюлоза натрия, альгинат натрия, агар, альгиновая кислота или соли альгиновой кислоты, каррагенан кальция, алюмосиликат магния, полиэтиленгликоль, гуаровая камедь, полисахаридные кислоты, бентониты, поливинилпирролидон (повидон), гидроксиметилполивинилпиролидон, полиметакрилаты (например, Эудрагит), метилцеллюлоза, гидроксипропилметилцеллюлоза (ГПМЦ), гидроксипропилцеллюлоза (Klucel™), этилцеллюлоза (Ethocel™), гидроксипропилметилцеллюлоза, желатинизированный крахмал (например, National™ 1511 и Starch 1500), сахароза, лактоза, паста крахмала, повидон полиэтиленгликоль, Пуллулан и кукурузный сироп, воски и натуральные и синтетические камеди, например акации, трагиканта, растительные камеди, касторовое масло, микрокристаллическая целлюлоза, декстрин, жидкая глюкоза, гуаровая камедь, пектин, ПЭГ, повидон, желатинизированный крахмал и т.д.
Примеры приемлемых разрыхлителей включают, но не ограничиваются ими: крахмал, например куккурузный и рисовый крахмал, N-винил-2-пирролидон с поперечными сшивками (CLPVP), альгиновую кислоту или альгинаты, микрокристаллическую целлюлозу, гидроксипропилцеллюлозу и другие производные целлюлозы, кроскармеллозу натрия, кросповидон, полакриллин калия, крахмал, желатинизированный крахмал, Фармабласт® карбоксиметилкрахмал (например, Примогель® и Эксплотаб® (натрия гликолят крахмала и карбоксиметилкрахмал натрия)), натрия крахмал гликолят и формальдегид казеин. К шипучим разрыхлителям относятся, не ограничиваясь перечисленными, например, крахмал, бикарбонат калия и бикарбонат натрия в комбинации с лимонной или тартаровой кислотами. Дезинтергант представлен в виде внутригранулярного разрыхлителя или экстрагранулярного разрыхлителя.
Примеры приемлемых лубрикантов включают, но не ограничиваются перечисленными: олеат натрия, стеарат натрия, натрия стеарилфумарат, стеариновая кислота, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия, стеарат кальция или другие стеараты металлов, тальк, воски и глицериды, легкое минеральное масло, ПЭГ, бегенат глицерола, коллоидный диоксид кремния, гидрогенизированные растительные масла, кукурузный крахмал, стеарилфумарат натрия, полиэтиленгликоли, алкилсульфаты, бензоат натрия и ацетат натрия.
Примеры ароматизаторов включают, но не ограничиваются перечисленными: ментол, перечную мяту, ваниль, фруктовые ароматизаторы и подсластители, например аспартам или сахаринаты натрия.
Примеры гелеобразующих агентов включают, но не ограничиваются перечисленными: поливинилпирролидон, гидроксипропилметилцеллюлозу, растительные камеди и т.д.
Примеры приемлемых инертных газов включают, но не ограничиваются: азот, гелий и т.д.
Примеры дополнительных добавок включают, не ограничиваясь: сорбитол, тальк и стеариновая кислота.
Примеры приемлемых антиоксидантов включают, но не ограничиваются перечисленными: лимонную кислоту, аскорбиновую кислоту, аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол (ВНТ), монотиоглицерол, метабиссульфид калия, пропилгаллат, токофероловые наполнители.
Примеры приемлемых увлажняющих агентов включают, но не ограничиваются перечисленными: полисорбат, лаурилсульфат натрия, монолаурат сорбитана, моноолеат сорбитана, монопальмитат сорбитана, моностеарат сорбитана.
Примеры соответствующих агентов, контролирующих высвобождение, включают, но не ограничиваются перечисленными: гидроксипропилметилцеллюлозу, гидроксипропилметилцеллюлозу, этилцеллюлозу, гидроксиэтилцеллюлозу.
Примеры поверхностно-активных веществ включают, но не ограничиваются перечисленными: анионные и неионные поверхностно-активные вещества, например лаурилсульфат натрия, полоксамеры (сополимеры полиоксиэтилена и полиоксипропилена), природные или синтетические лецитины, а также эфиры сорбитана и жирных кислот, например Span® (доступный в продаже в Sigma-Aldrich Co., St. Louis, МО), эфиры полиоксиэтиленсорбитана и жирных кислот, например Полисорбаты или Полисорбат® (доступный в Spectrum Chemical, Gardena СА), стеараты полиоксиэтилена, например Myrj® (доступный в Uniqema, New Castle, DE), полиэтоксилированные жирные кислоты, например моно- или диэфиры жирных кислот и полиэтиленгликоля или их смеси, например моно- или диэфиры полиэтиленгликоля и лауриновой кислоты, олеиновой кислоты, стеариновой кислоты, миристиновой кислоты, рициноленовой кислоты, и полиэтиленгликоль выбран из ПЭГ 4, ПЭГ 5, ПЭГ 6, ПЭГ 7, ПЭГ 8, ПЭГ 9, ПЭГ 10, ПЭГ 12, ПЭГ 15, ПЭГ 20, ПЭГ 25, ПЭГ 30, ПЭГ 32, ПЭГ 40, ПЭГ 45, ПЭГ 50, ПЭГ 55, ПЭГ 100, ПЭГ 200, ПЭГ 400, ПЭГ 600, ПЭГ 800, ПЭГ 1000, ПЭГ 2000, ПЭГ 3000, ПЭГ 4000, ПЭГ 5000, ПЭГ 6000, ПЭГ 7000, ПЭГ 8000, ПЭГ 9000, ПЭГ 1000, ПЭГ 10000, ПЭГ 15000, ПЭГ 20000, ПЭГ 35000, эфиры жирных кислот и полиэтиленгликоля и глицерола, например, такие эфиры, как указано выше, но в виде эфиров глицерола и отдельных жирных кислот; эфиры глицерола, пропиленгликоля, этиленгликоля, ПЭГ или сорбитола, например, с растительными маслами, такими как, например, гидрогенизированное касторовое масло, миндальное масло, пальмоядровое масло, касторовое масло, оливковое масло, арахисовое масло, гидрогенизированное пальмоядровое масло и т.д., полиглицериновыми жирными кислотами, например полиглицерол стеарат, полиглицерололеат, полиглицеролрициноолеат, полиглицерол линоолеат, эфиры жирных кислот и пропиленгликоля, например пропиленгликольмонолаурат, пропиленгликольрицинолеат и т.п., моно- и диглицериды, например глицерилмоноолеат, глицерилдиолеат, глицерил моно- и/или диолеат, глицерил каприлат, глицерилкапрат и т.д., стерол и производные стерола, эфиры полиэтиленгликольсорбитана и жирной кислоты (эфиры ПЭГ-сорбитанжирная кислота), например эфиры ПЭГ различной молекулярной массы, приведенные выше, и различные серии Tween(R) (от ICI America, Inc.); алкильные эфиры полиэтиленгликоля, например эфир ПЭГ и олеиновой кислоты и ПЭГ-лауриловые эфиры; эфиры сахаров, например монопальмитат сахарозы и монолаурат сахарозы, алкильные фенолы полиэтиленгликоля, например Tphtoh(R) X и N серий (Union Carbide Chemicals & Plastics Technology Corporation); блокирующие сополимеры полиоксиэтилен-полиоксипропилен, например серия Pluronic(R) (BASF Aktiengesellschaft) серии BASF Aktiengesellschaft, Synperonic(R) (ICI America, Inc., Emkalyx), Lutrol(R) (BASF Aktiengesellschaft), Supronic и т.д.
Количество соединения(ий), которые действуют как поверхностно-активное вещество, регулируется (при использовании в данных целях), для контроля растворимости, пермеабильности и биодоступности агониста(ов) допамина. Предпочтительно соотношение между поверхностно-активным веществом и агонистом(амии) допамина по массе составляет приблизительно от 0,001:1 до приблизительно 1:1, более предпочтительно приблизительно от 0,005:1 до 0,6:1 и наиболее предпочтительно приблизительно от 0,01:1 до приблизительно 0,25:1.
Примеры приемлемых лубрикантов и/или веществ, способствующих скольжению (glidants), включают, не ограничиваясь, в отдельности или в комбинации такие лубриканты и/или вещества, способствующие скольжению, как глицерил бегенат (Компритол™ 888); стеараты металлов (например, стеараты кальция, натрия или соли других длинноцепочечных жирных кислот), стеариновая кислота, гидрогенизированные растительные масла (например, Sterotex™); тальк, воски, Stearowet™; борная кислота, бензоат натрия и ацетат натрия, хлорид натиря, DL-лейцин; полиэтиленгликоли (например, Карбовакс™ 4000 и Карбовакс™ 6000); олеат натрия; бензоат натрия, ацетат натрия, лаурилсульфат натрия, стеарилфумарат натрия (Pruv™) и лаурилсульфат магния.
Дополнительные примеры приемлемых антиадгезивных веществ и веществ, способствующих скольжению, включают, не ограничиваясь, в отдельности или в комбинации, такие антиадгезивные вещества, как тальк, кукурузный крахмал, DL-лейцин, лаурилсульфат натрия и стеараты металлов.
Примеры приемлемых консервантов включают, не ограничиваясь перечисленными: лимонную кислоту, витамин С, витамин Е, 1,1,1-трихлор-2-метил-2-пропанол, фенилэтиловый спирт, сорбиновая кислота, бензильный спирт, алкилбензилдиметиламмония хлорид с длиной цепи от С8 до С18 в алкильной группе, мета-крезол или алкил-4-гидроксибензоат.
Под термином «лекарственная форма для парентерального введения» понимают лекарственную форму, которая обеспечивает всасывание значительного количества, минуя желудок и/или слизистую оболочку желудочно-кишечного тракта.
Способы парентерального введения включают, но не ограничиваются, буккальный, подъязычный, подкожный, назальный, оральный, через уши, через глаза, ректальный, вагинальный, или через слизистую верхних дыхательных путей, или через кожу или легкие. Соответственно, данные лекарственные формы включают, но не ограничиваются перечисленными: формы для инъекции, пероральные, для введения через уши, офтальмологические, или назальные спреи или капли, подъязычные и/или буккальные спреи, капли, таблетки, растворы, коллоидные суспензии и/или мази, твердые капсулы и мягкие капсулы, таблетки, покрытые таблетки, сашеты, леденцы, пленки, жевательные резинки, жевательные таблетки, растворы для полоскания, трансдермальный пластырь, мазь, лосьон или крем, респираторный ингалятор, аэрозоли, ректальные или вагинальные суппозитории.
Лекарственные формы можно вводить посредством инъекции. Инъекции могут представлять собой, например, подкожные, внутрикожные и/или внутрибрюшинные инъекции.
рН раствора или лекарственной формы на основе растворителя согласно изобретению предпочтительно должен находиться в пределах от 3 до 9 во избежание дальнейшего риска повреждения клетки или ткани.
Лекарственные формы не ограничиваются только приведенными ниже формами. Лекарственные формы для назального введения включают назальные спреи и/или капли и/или нанесение назальных мазей. Лекарственные формы для подъязычного или буккального введения включают спреи, капли, растворы, коллоидные суспензии, таблетки, мази, леденцы, пленки, жевательные резинки, жевательные таблетки и/или жидкие растворы для полоскания. Лекарственные формы для аурикулярного введения или введения в глаза включают спреи, капли, мази, лосьоны и/или крем. Лекарственные формы для ректального введения включают суппозитории, спрей, капли, мази, лосьоны и/или крем. Лекарственные формы для вагинального введения включают суппозитории, спрей, капли, мази, лосьоны и/или крем. Лекарственные формы для введения через верхние дыхательные пути или легкие включают респираторные ингаляторы, например ингалятор. Лекарственные формы для введения через кожу включают кожные пластыри, дермальные спреи, капли, мази, лосьоны и/или крем.
Твердые лекарственные формы для парентерального введения предпочтительно включают агонист допамина (предпочтительно, спиртовое производное спорыньи, наиболее предпочтительно, бромкриптин), неакриловый тип мукоадгезивных веществ (например, поливинилпирролидон, Бенецел® и не Карбопол®) и лимонную кислоту для увеличения стабильности и ускорения высвобождения агониста допамина. В отсутствие лимонной кислоты АФИ был нестабилен в растворе на основе акрила (50% лекарства распадается через 90 минут). Лимонная кислота ускоряет стабильность лекарственных препаратов на основе агонистов допамина.
Предпочтительными путями введения являются подкожные инъекции, буккальное, подъязычное, назальное и трансдермальное введение. Более предпочтительными путями введения являются буккальное, подъязычное и назальное введение. Особенно предпочтительными лекарственными формами являются подкожные инъекции, подъязычные или буккальные лекарственные формы и кожные пластыри.
Если лекарственную форму надо инъецировать или вводить с помощью жидкого носителя (например, в случае подъязычного введения), ее можно вводить с помощью двух различных носителей для двух различных растворов в одном шприце. У такого шприца может быть два резервуара и два порта для каждого из двух растворов. В другом случае два различных раствора могут одновременно находиться в одном резервуаре.
Если парентеральное введение представляет собой подкожное введение, приемлемые формы для инъекций могут включать гидрофобную или гидрофильную суспензию.
В одном варианте реализации лекарственной формы, приведенной в настоящей заявке, она содержит суспендированную соль активного ингредиента(ов) или активные ингредиенты в гидрофобной фармацевтически приемлемой суспензии. Такая гидрофобная суспензия предпочтительно может быть основана на фармацевтически приемлемых синтетических, полусинтетических или природных маслах или смесях по меньшей мере двух таких масел.
Суспензию готовят предпочтительно в количествах от 10 до 90% по массе в зависимости от суспензии.
Также возможно использовать лекарственные формы посредством физиологически приемлемой гидрофильной суспензии, и при этом активный ингредиент представляет собой соль агониста D1 и/или агониста D2, который не является производным спорыньи. Гидрофильная суспензия предпочтительно приготовлена на основе воды.
Кроме одного или более регуляторов для поддержания осмолярности, лекарственные формы могут дополнительно включать один или более представителей других приведенных выше наполнителей.
Для того чтобы свести к минимуму или полностью исключить риск разрушения клетки или ткани, осмолярность, то есть концентрация (tonicity) водных лекарственных форм (если они используются) согласно изобретению, которые надо вводить парентерально, предпочтительно доводится до такого значения, что лекарственные формы изотоничны или по меньшей мере приблизительно изотоничны физиологической осмолярности. Осмолярность лекарственных форм согласно данному изобретению, которые можно вводить парентерально, предпочтительно доводят приблизительно до 250-400 мосмоль/кг, в частности предпочтительно 260-320 мосмоль/кг и особенно предпочтительно 280-300 мосмоль/кг.
Также возможно по необходимости использовать регулятор для корректировки различных свойств лекарственных форм. Например, поверхностно-активное соединение также можно использовать для регуляции осмолярности места введения (например, подъязычной или буккальной области).
Также лекарственные формы могут дополнительно содержать одно или более физиологически приемлемых поверхностно-активных соединений.
Лекарственные формы для парентерального введения обычно вводят в объеме приблизительно от 0,01 до 0,75 мл. Предпочтительно, объем для парентерального введения составляет приблизительно от 0,01 до приблизительно 0,5 мл, более предпочтительно от приблизительно 0,01 до приблизительно 0,3 мл и наиболее предпочтительно от приблизительно 0,01 до приблизительно 0,2 мл.
Если лекарственную форму можно вводить перорально, такая лекарственная форма предпочтительно подходит для буккального или подъязычного введения лекарственного средства через слизистую оболочку ротовой полости. Более предпочтительно, лекарственная форма относится к типу форм для подъязычного введения лекарственного средства через слизистую оболочку ротовой полости.
Обычно буккальную лекарственную форму помещают в ротовую полость между челюстью и щекой, где она растворяется в слюне, высвобождая лекарственное средство в ротовую полость в непосредственной близости от капилляров слизистой. Подъязычную лекарственную форму помещают под язык, где она растворяется в слюне, высвобождая лекарственное средство в ротовую полость в непосредственной близости от капилляров слизистой.
Фармацевтически активный агент в данной, пероральной лекарственной форме проникает в кровь в капилляры слизистой посредством диффузии через слизистую и распространяется с током крови по всему телу. Скорость, с которой активный агент распространяется по телу, зависит, помимо прочего, от скорости, с которой лекарственная форма растворяется во рту. Физические свойства лекарственной формы определяют степень контакта со слизистой и впоследствии эффективность всасывания лекарства.
Если парентеральное введение осуществляют через рот, можно предотвратить всасывание через слизистую желудка и/или кишечника, используя определенные компоненты в составе, такие как биоадгезивные агенты, проникающие агенты и стабилизаторы, которые предотвращают и/или снижают проникновение агонистов допамина в желудок и/или слизистую оболочку ЖКТ.
Согласно некоторым вариантам реализации вводимые перорально подъязычно или буккально лекарственные формы для парентерального введения содержат как быстро, так и медленно растворимые компоненты при введении в ротовую полость, как функция двух различных составов в одной лекарственной форме или аппликаторе.
Твердые пероральные лекарственные формы (которые содержат быстро и медленно всасываемые компоненты) можно описать по времени их растворения in vitro.
Твердые пероральные лекарственные формы (которые содержат быстро и медленно всасываемые компоненты) обычно имеют время растворения от 10 секунд до 100 минут. Предпочтительно пероральные лекарственные формы имеют время растворения от 10 секунд до 50 минут. Более предпочтительно пероральные лекарственные формы имеют время растворения от 10 секунд до 30 минут. Наиболее предпочтительно пероральные лекарственные формы имеют время растворения от 10 секунд до 20 минут.
Согласно некоторым вариантам реализации пероральная лекарственная форма образует пленку, например буккальную пленку. Следят за тем, чтобы механические, биоадгезивные свойства и набухаемость пленок подходила для буккального ведения. Пленки для буккального введения предпочтительно гибкие, эластичные, мягкие и в то же время достаточно твердые для того, чтобы выдерживать разворачивание и действие рта, а также они имеют хорошие биоадгезивные свойства, чтобы сохраняться во рту желаемое время. Предпочтительно избегают разбухания пленок или ограничивают его для предотвращения возникновения дискомфорта.
Согласно некоторым вариантам реализации пероральная лекарственная форма представляет собой подъязычную лекарственную форму.
Фармакокинетические профили лекарственной формы контролируются наполнителями. Согласно некоторым вариантам реализации твердая лекарственная форма состоит по меньшей мере из одного агониста допамина (для быстрого и замедленного всасывания), наполнителя (предпочтительно, маннитола, лактозы, ксилитола или их смесей) или матрикса растворителя, связующего агента (например, Коллидона) для одной или двух отобранных по размеру частиц агониста допамина, и разрыхлителя.
Связующий агент предпочтительно используют в минимальном количестве для предотвращения непредвиденного снижения скорости растворения для каждого аспекта «быстрого» и «медленного» растворения лекарственной формы. Предпочтительные связующие агенты растворимы в воде. Предпочтительными связующими агентами являются поливинилпирролидон, гидроксиметилполивинилпирролидон, и также можно использовать желатин.
Содержание разрыхлителя может составлять 0,1-75% гранулы, предпочтительно 1-60%, более предпочтительно 1-40%.
Предпочтительно минимальное содержание лубриканта составляет, например, до 1%, предпочтительно около 0,8%. Предпочтительно использовать экстрагранулярный лубрикант отдельно для минимизирования гидрофобных свойств лекарственной формы.
Таблетки могут включать традиционные наполнители, обычно представленные в количестве приблизительно 10% общей массы. Они могут включать ароматизаторы.
Применяемые ароматизаторы обычно присутствуют в количестве от приблизительно 0,5 до 5% от общей массы таблетки. Подсластители и другие наполнители также могут включать красители, консерванты и наполнители.
Предпочтительно наполнители выбирают из сахаров. Предпочтительны маннитол, лактоза, ксилитол и их смеси благодаря их растворимости и несмотря на содержание воды в лактозе, в частности. Предпочтительно маннитол представлен в количестве 20-40% масс./масс., и более предпочтительно 20-30% масс./масс. Предпочтительно лактоза представлена в количестве 30-60% масс./масс. Предпочтительными наполнителями являются безводные наполнители.
В некоторых вариантах реализации с чресслизистым введением первый активный агент с определенным размером частиц соединяют или вводят совместно со вторым агентом, например с агентом, который облегчает проникновение активного агента в ткань, в клетки или в кровоток. Согласно одному варианту реализации, предложен активный агент совместно с ускорителем проникновения.
К примерам агентов, ускоряющих поглощение активных агентов клетками, относятся жирные кислоты, их производные, липиды или комплексы липидов или содержащие липиды комплексы, например липосомы.
Липосомы представляют собой полые сферические везикулы, состоящие из липидов, организованных таким же образом, что и липиды в клеточной мембране. В липосомах может быть внутреннее водное пространство для поглощения веществ, растворимых в воде, и размер липосом может составлять от 0,05 до нескольких микрон в диаметре. Например, показано, что средство доставки липосом в клетку, которое изначально создавали как инструмент для исследований, Липофектин, доставляет в клетку интактные молекулы. Липосомы обладают несколькими преимуществами: они нетоксичны и биодеградируемые в композиции, имеют длительный период полувыведения из кровотока, и к их поверхности можно без труда прикрепить молекулы для распознавания тканей. Липидные агрегаты можно сформировать из макромолекул с использованием, например, катионных липидов по отдельности или с включением других липидов и амфифильных веществ, например фосфатидилэтаноламина. Для доставки отрицательно заряженных молекул предпочитают липосомы, содержащие катионные липиды.
К другим средствам доставки лекарств, которые можно использовать, относятся гидрогели, циклодекстрины, биоразлагаемые полимеры (хирургические импланты или нанокапсулы) и биоадгезивные микросферы.
Также могут быть получены агенты с механизмом замедленного высвобождения, которые могут включать, например, полимерные микросферы, и другие механизмы, которые знакомы специалисту в данной области, для того, чтобы регулировать скорость высвобождения агента. Соответственно, активный агент можно приготовить совместно с по меньшей мере одним ускорителем проникновения или проницаемости, и/или дополнительно агент может включать по меньшей мере один механизм замедленного высвобождения и/или по меньшей мере одно биоадгезивное вещество. Примеры ускорителей проникновения включают, но не ограничиваются перечисленными: жирные кислоты, Кавитрон, тиомеры, ментол и полиоксиэтилен.
Если лекарственная форма представляет собой трансдермальный пластырь, агонист допамина можно тонко измельчить или растворить и добавить к системе доставки, которые обычно используются в фармацевтических «пластырях» для замедленного высвобождения лекарственного средства в течение длительного периода времени (часов).
Согласно одному варианту реализации, на кожу пациента с нарушением обмена веществ или с наличием его ключевых признаков наносят гелевую композицию, содержащую один или более агонистов допамина. Пероральные композиции можно применять в определенных количествах в виде лосьона или мази. Такие композиции можно наносить, например, на защитный слой для создания лекарственной формы, которая обеспечивает надлежащую адгезию для того, чтобы прикрепить лекарственную форму к субъекту, подвергаемому лечению. Например, защитный слой можно распределить по краям накладываемой композиции гелеобразного агониста допамина и затем распределить горизонтально. На нижнюю сторону полученного кольца можно наносить подходящий адгезивный слой для прикрепления единицы дозирования на кожу субъекта, подвергаемого лечению.
Кожные пластыри могут представлять собой пластыри, которые содержат один слой лекарства в адгезивном слое, несколько слоев лекарства в адгезивном слое, резервуарный пластырь или пластырь матричного типа. Пластырь с одним слоем лекарства в адгезивном слое содержит адгезивный слой, который также имеет один или более агонистов допамина. В пластыре такого типа адгезивный слой служит не только для объединения различных слоев вместе и для крепления всей системы к коже, а также отвечает за механизм высвобождения агониста(ов) допамина. Адгезивный слой окружает временная подкладка и защитный слой. Пластырь с несколькими слоями лекарства в адгезивном слое похож на систему с одним слоем в том, что оба адгезивных слоя отвечают за механизм высвобождения агониста(ов) допамина. Многослойная система отличается тем, что в ней есть дополнительный слой лекарства в адгезивном слое, обычно отделенный мембраной (но не во всех случаях). В таком пластыре также может быть временная подкладка и постоянный защитный слой. Резервуарный пластырь отличается от систем с одним слоем и несколькими слоями тем, что в нем имеется отдельный слой лекарственного средства, содержащий агонист(ы) допамина для двух различных скоростей доставки в кожу. Слой лекарственного средства представляет собой жидкий компартмент, содержащий раствор лекарства или суспензию, отделенную адгезивным слоем. Этот пластырь может иметь защитный слой. Пластырь матричного типа имеет слой лекарства полутвердого матрикса, содержащего раствор или суспензию агониста(ов) допамина. Адгезивный слой в этом пластыре окружает слой лекарства и может частично его перекрывать.
Трансдермальные пластыри могут содержать гелеобразующий агент, предпочтительно насыщенный или высоко насыщенный выбранным агонистом допамина или агонистами допамина. Гелеобразующий агент выбирают таким образом, чтобы он был биосовместимым, совместимым с агонистами допамина и позволял бы агонисту допамина всасываться в кожу.
Также вместо гелеобразующего агента или дополнительно к гелеобразующему агенту можно добавлять гомогенную смесь, включающую агонист допамина, к абсорбенту, который способен поглощать агонист допамина. Подходящий абсорбент можно выбрать из гигроскопической ваты, биосовместимого и подходящего синтетического фиброзного материала, включая материалы, полученные пряжением, и другие абсорбенты, которые может предложить специалист в данной области. Итоговая композиция с агонистом допамина после добавления гелеобразующего агента будет обладать достаточной вязкостью для использования в трансдермальной терапии.
Если лекарственная форма представляет собой аэрозоль, ее можно вводить с помощью двух различных средств для двух различных растворов в одном контейнере. В таком контейнере может быть два различных резервуара и два порта специально для двух растворов. В другом случае два различных раствора можно поместить в один контейнер.
Термин «нарушение метаболизма» включает нарушения (расстройства, заболевания), связанные с нарушенным метаболизмом глюкозы, жиров и/или белков и патологических последствий указанных нарушений. Такие нарушения метаболизма могут быть или не быть связаны с нарушенными паттернами дневных уровней (и отклонений) секреции пролактина.
«Ключевые элементы» указанных нарушений метаболизма включают, но не ограничиваются перечисленными: диабет 2 типа, преддиабеты (нарушенный уровень глюкозы натощак или непероносимость глюкозы), метаболический синдром или его признаки (ключевые элементы) (увеличенный объем талии, повышенный уровень глюкозы в плазме натощак, повышенный уровень триглицеридов в плазме натощак, пониженный уровень липопротеинов высокой плотности, повышенное кровяное давление), резистентность к инсулину, гиперинсулинемию, сердечно-сосудистое заболевание (или его ключевые элементы, такие как артериосклероз, болезнь коронарных артерий, болезнь периферических сосудов, нарушение мозгового кровообращения), сердечная недостаточность с застойными явлениями, ожирение, повышенный уровень норэпинефрина в плазме, повышенный уровень воспалительных факторов, связанных с сердечно-сосудистым заболеванием, повышенный уровень в плазме факторов, усиливающих дисфункцию эндотелия сосудов, гиперлипопротеинемия, артериосклероз или атеросклероз, гиперфагия, гипергликемия, гиперлипидемия или гипертензия или повышенное кровяное давление, повышенный уровень триглицеридов или свободных жирных кислот в плазме после приема пищи, повышенный окислительный клеточный стресс или признаки его в плазме, повышенное гиперсвертывание в кровообращении, болезнь почек, включая отказ почек или почечную недостаточность.
В настоящей заявке термин «профиль растворения» означает растворение агента с течением времени. Растворимость можно определить как относительное количество агента, которое растворяется с течением времени, количество растворенного агента или концентрация растворенного агента в определенное время. Предпочтительный метод определения скорости растворения представляет собой тест на растворение с использованием вращающейся корзины при 100 об/мин в 900 мл водного буфера 0,01 N HCl при 37°C. Также подходят альтернативные методы, включая тест на растворение с использованием лопастной мешалки и другие методы, известные специалисту в данной области.
В настоящей заявке термин «фармацевтически приемлемый» обозначает биологически или фармакологически совместимый лекарственный компонент для использования in vivo и предпочтительно обозначает лекарственный компонент, который одобрен надзорным органом Федерального Правительства или Правительства штата или входит в список Фармакопеи США или других фармакопей для применения на животных и более точно на людях.
Термин «биодоступность» обозначает скорость и степень, с которой агонист допамина абсорбируется биологической системой из введенного лекарственного продукта и становится доступным в месте биологического действия.
В настоящей заявке термин «терапевтически эффективное количество» означает достаточное количество активного агента, которого достаточно для лечения нарушения метаболизма и/или ключевого элемента нарушения метаболизма.
Фармакокинетический профиль b пролактин
Здоровые (нормальные) пациенты, например стройные представители видов, не страдающие от таких нарушений метаболизма и/или их ключевых элементов, имеют предсказуемые профили ежедневного высвобождения пролактина. У человека для таких профилей высвобождения характерен низкий и относительно постоянный уровень пролактина в течение дневного времени, за которым следует острый пик ночью и последующее постепенное понижение до дневного уровня к утру. Один или более агонист допамина можно вводить нуждающемуся субъекту для того, чтобы изменить нарушенные ритмы дневного уровня пролактина таким образом, чтобы они напоминали или больше походили по фазе и амплитуде на нормальный ритм нормального дневного уровня пролактина стройных, молодых и здоровых членов того же вида и пола. Смотрите, например, патенты США №№5468755; 5496803; 5344832, 5585347, 5830895 и 6855707 и заявки РСТ США US 93/12701 и US 95/09061 (содержание которых включено в настоящую заявку посредством ссылки). Такое изменение ритмов пролактина использовали для лечения диабета 2 типа, ожирения, резистентности к инсулину и гиперинсулинемии или гипергликемии, гиперлипопротеинемии, гиперфагии и т.д.
Лекарственные формы для парентерального введения согласно настоящему изобретению могут давать определенный фармакокинетический профиль агониста допамина, который может менять профили пролактина в плазме пациента, снижая избыточный уровень пролактина в плазме до нормального дневного уровня, не приводит к сохранению полученного соответствующего уровня пролактина в течение ночи (или сна), и это не приводит к равному уровню пролактина ночью и днем. Таким образом, можно лечить нарушения метаболизма и/или их ключевые элементы у пациента с такими нарушениями секреции пролактина. Однако следует понимать, что «нормализация» циркадного ритма пролактина сама по себе не обязательно является предпосылкой для улучшения обмена веществ, вызванного агонистом допамина, скорее такая «нормализация» может усиливать улучшения в заболевании нарушения метаболизма или его ключевых элементов, вызванные агонистом допамина.
Лекарственные формы для парентерального введения согласно настоящему изобретению также могут иметь определенный фармакокинетический профиль, который эффективно снижает повышенный уровень концентрации норэпинефрина в плазме, при этом агонист допамина не должен присутствовать в циркуляции на протяжении всего дня. Такое действие парентеральных лекарственных форм, хотя и не является требованием для их влияния на нарушение метаболизма, вызывает улучшения в течении заболевания или его ключевых элементов.
Желательно, чтобы лекарственные формы обладали фармакокинетическим профилем, который усиливает эффективность активного агента.
Фармакокинетические профили показывают абсорбцию и фармакокинетику активных агентов, и они определяются концентрациями в плазме, которые используют для оценки ключевых фармакокинетичских параметров, таких как Tmax, Cmax, AUC и tlag. Tmax - время пиковой концентрации. Cmax - пиковая концентрация. AUC - площадь под кривой (AUC). tlag - время задержки абсорбции.
Процесс абсорбции можно рассматривать как увеличение количества вещества или дозы х, введенной в систему. Исследования абсорбции должны определить вводимое количество, dx/dt дозы х. Например, постоянная скорость введения, R, лекарства может составлять 1 мг/час, при этом интеграл по времени dx/dt представляет собой степень введения лекарства, x(t), т.е. общее количество лекарственного средства х, введенное до этого определенного времени t. Сложные профили абсорбции можно получать с помощью контролируемого, продолжительного или синхронизированного высвобождения лекарства или лекарственной формы.
Фармакокинетика далее подразделяется на исследование всасывания, распределения, метаболизма и выведения или экскреции лекарственного средства, эти параметры обозначаются сокращением ADME.
Процессы распределения можно рассматривать как очищение или избавление от лекарственного средства. Обычно процесс распределения обеспечивает распространение лекарственного средства по системе, его переведение или метаболизирование и выведение лекарства или его метаболитов через мочу, кал, пот, дыхание или другие пути.
Согласно одному варианту реализации парентеральные лекарственные формы согласно настоящему изобретению обеспечивают сохранение ночного подъема уровня пролактина в плазме (нейроэндокринное физиологическое событие) у пациентов, страдающих диабетом 2 типа с ожирением/устойчивостью к инсулину.
Критерий ADME влияет на кинетику и уровни действия лекарственного средства тканям и органам и поэтому влияет на фармакологическую активность лекарства. До того как активный агент окажет действие на ткани, он должен попасть в кровоток. Поэтому активный агент необходимо доставить до его места действия, в большинстве случаев через кровоток. Активные агенты начинают метаболизироваться, как только они попадают в тело. Вещества и их метаболиты нужно вывести из тела через экскрецию, обычно через почки (моча) или с калом. Если выведение происходит неполностью, накопление побочных веществ может влиять на нормальный обмен веществ.
Согласно одному варианту реализации лекарственная форма имеет следующий фармакокинетический профиль: Tmax от приблизительно 1 до приблизительно 90 минут или от приблизительно 5 до приблизительно 90 минут после введения, концентрация лекарства в плазме по меньшей мере 50% Cmax в течение периода приблизительно от 90 до приблизительно 360 минут и снижение уровня в плазме, что может приблизительно соответствовать кинетике выведения первого порядка.
Согласно другому варианту реализации лекарственная форма имеет следующий фармакокинетический профиль: Tmax приблизительно от 1 до приблизительно 90 минут или от приблизительно 5 до приблизительно 90 минут после введения, концентрация лекарства в плазме по меньшей мере 50% Cmax в течение периода приблизительно от 180 до приблизительно 360 минут и снижение уровня в плазме, что может приблизительно быть равно кинетике выведения первого порядка.
Согласно другому варианту реализации лекарственная форма имеет следующий фармакокинетический профиль: Tmax приблизительно от 1 до приблизительно 90 минут или от приблизительно 5 до приблизительно 90 минут после введения, концентрация лекарства в плазме по меньшей мере приблизительно 70-100% Cmax в течение периода приблизительно от 90 до приблизительно 360 минут и снижение уровня в плазме, что может приблизительно соответствовать кинетике выведения первого порядка.
Согласно предпочтительному варианту реализации лекарственная форма имеет следующий фармакокинетический профиль: Tmax приблизительно от 1 до приблизительно 90 минут или от приблизительно 5 до приблизительно 90 минут после введения, концентрация лекарства в плазме по меньшей мере 70-100% Cmax в течение периода приблизительно от 180 до приблизительно 360 минут и снижение уровня в плазме, что может приблизительно соответствовать кинетике выведения первого порядка.
Согласно другому варианту реализации лекарственная форма имеет следующий фармакокинетический профиль: Tmax приблизительно от 1 до приблизительно 90 минут или от приблизительно 5 до приблизительно 90 минут после введения, пост-Cmax уровень приблизительно 35-65% от Cmax в течение периода приблизительно 30-150 минут после Tmax, за которым следует уровень Cmax, равный приблизительно одной второй Cmax в течение приблизительно 60-420 минут, затем следует снижение уровня в плазме, что может приблизительно быть равно кинетике выведения первого порядка.
Предпочтительно фармакокинетический профиль имеет Тmax приблизительно от 15 до приблизительно 90 минут после введения, уровень пост-Cmax приблизительно 35-65% Cmax в течение приблизительно 30-90 минут после Tmax, за которым следует пост-Cmax уровень, который составляет приблизительно одну вторую Cmax в течение приблизительно 60-360 минут, за которым следует снижение уровня в плазме, что приблизительно может соответствовать кинетике выведения первого порядка.
Согласно другому варианту реализации лекарственная форма имеет фармакокинетический профиль с Tmax в плазме приблизительно от 15 до приблизительно 60 минут после введения, уровень пост-Cmax, который составляет приблизительно одну вторую Cmax в течение приблизительно от 30 до приблизительно 150 минут Tmax, за которым следует уровень пост-Cmax, который составляет приблизительно одну вторую Cmax в течение приблизительно 90-360 минут, за которым следует снижение уровня в плазме, что может приблизительно соответствовать кинетике выведения первого порядка.
Согласно еще одному варианту реализации лекарственная форма имеет фармакокинетический профиль с Tmax в плазме приблизительно от 10 до приблизительно 60 минут после введения, уровень пост-Cmax, который составляет приблизительно одну вторую Cmax в течение приблизительно от 30 до приблизительно 150 минут Tmax, за которым следует уровень пост-Cmax, который составляет приблизительно одну вторую Cmax в течение приблизительно от 90 до приблизительно 240 минут, за которым следует снижение уровня в плазме, что может приблизительно соответствовать кинетике выведения первого порядка.
Согласно одному варианту реализации лекарственная форма имеет фармакокинетический профиль с Tmax в плазме приблизительно от 5 до приблизительно 60 минут после введения, Tmax в плазме 10-60 минут после введения, Tmax в плазме 10-90 минут после введения, Tmax в плазме 15-90 минут после введения или Tmax в плазме 15-60 минут после введения.
Согласно одному варианту реализации лекарственная форма имеет фармакокинетический профиль, в котором 90% активного агента выводится из плазмы в течение от приблизительно 240 до приблизительно 480 минут после достижения пост-Cmax уровня.
Согласно одному варианту реализации лекарственная форма имеет фармакокинетический профиль, в котором большая часть активного агента выводится из плазмы через 5 часов после окончания плато.
Согласно одному варианту реализации лекарственная форма имеет фармакокинетический профиль, отличающийся тем, что пост-Cmax плато продолжается 2-8 часа.
Согласно одному варианту реализации более чем приблизительно 10% общего количества активного агента лекарственной формы поступает в плазму. Согласно другому варианту реализации более чем приблизительно 35% общего количества активного агента лекарственной формы поступает в плазму.
Согласно одному варианту реализации усилитель проникновения в одной части лекарственной формы комбинируют с механизмом постоянного высвобождения в другой части формы, чтобы способствовать быстрому пику после продолжительного «хвоста» в фармакокинетическом профиле лекарственной формы.
Приведенные выше фармакокинетические профили способствуют достижению пика агониста допамина в циркуляции, что можно применять для влияния на систему циркадных ритмов в нервной системе (например, супрахиазмальное ядро), чтобы положительно влиять на его регуляцию метаболизма с помощью внешнего контроля других регуляторных центров метаболизма в мозге, посредством чего улучшается периферический метаболизм, за которым непосредственно следует продолжительный низкий уровень выхода агониста допамина в циркуляцию в течение определенного периода времени, что может непосредственно влиять на другие центры регуляции метаболизма в мозге для улучшения метаболизма.
Некоторые из представленных выше фармацевтических профилей способствуют пульсированию пика допаминового агониста в циркуляции, что можно использовать для того, чтобы влиять на систему циркадных ритмов в нервной системе (например, супрахиазмальное ядро), за которым следует продолжительный низкий уровень выхода агониста допамина в циркуляцию в течение определенного периода времени.
Если применять приемлемые дозы в четкое время суток у пациента с нарушением обмена веществ, приведенные выше фармакокинетические профили могут имитировать природный паттерн допамина в центрах, отвечающих за обмен веществ в мозге здоровой нормальной особи того же вида, посредством чего улучшается расстройство метаболизма.
Таким образом, лекарственная форма согласно настоящему изобретению может иметь фармакокинетический профиль с Tmax в плазме, за которым следует уровень пост-Cmax, который представляет собой по меньшей мере 70-100% постоянного периода времени до снижения уровня в плазме, что может приблизительно соответствовать кинетике элиминации первого порядка. Смотрите, например, Фигуру 1. Альтернативно, лекарственная форма согласно настоящему изобретению может иметь фармакокинетический профиль с Tmax в плазме, за которым следует уровень пост-Cmax, который с течением времени снижается приблизительно до одной второй Cmax, пониженный уровень и затем постоянный уровень на период времени до снижения уровня в плазме, что может приблизительно соответствовать кинетике элиминации первого порядка. Смотрите, например, Фигуру 2.
Если один или более агонист допамина вводят вместе с агентом периферического действия, агонист допамина и агент периферического действия, могут иметь одинаковый фармакокинетический профиль или довольно похожие или идентичные профили, например любой из приведенных выше профилей. И наоборот, если один или более агонист допамина вводят вместе с агентом периферического действия, агонист допамина и агент периферического действия могут иметь разные фармакокинетические профили. Согласно некоторым вариантам реализации, например, у одного или более агониста допамина есть фармакокинетический профиль, представленный выше, а агент периферического действия имеет профиль с Tmax 0-90 минут, уровень концентрации в плазме больше или равный приблизительно 25% Cmax от Tmax в течение 12 часов после Tmax.
Определенный фармакокинетический профиль, который дает лекарственная форма согласно настоящему изобретению, будет варьировать, в частности, на основе количества активного агента, включенного в лекарственную форму.
Согласно некоторым вариантам реализации лекарственная форма содержит бромкриптин в качестве активного агента и имеет один из представленных выше фармацевтических профилей, более предпочтительно с Cmax 25-400 пг/мл.
Более того, специалист в данной области должен понимать, что можно получить желаемую скорость растворения in vitro и/или концентрацию агонистов допамина в плазме in vivo, выбирая одну или более форму агонистов допамина, т.е. выбирая одну или более соль, кристаллическую форму (включая одну или более полиморфную форму) или аморфную форму для применения для непосредственного или контролируемого высвобождения композиций согласно настоящему изобретению.
Введение
Количество агониста(ов) допамина, которое надо вводить пациенту, может меняться в зависимости от массы пациента, а также природы или тяжести нарушения метаболизма или его ключевых элементов. Эффективное количество агониста(ов) допамина можно вводить в одной или более лекарственных формах как одновременно, так и в разное время, и агонист допамина можно вводить по отдельности или в комбинации с другим(и) агонистом(ами) допамина.
Предпочтительно лекарственные формы можно вводить в виде одной ежедневной дозы, составляющей от приблизительно 0,01 до приблизительно 50,0 мг активного агента. Предпочтительный диапазон составляет 0,02-50 мг активного агента, более предпочтительный диапазон 0,02-25 мг активного агента, и наиболее предпочтительный диапазон составляет 0,1-25 мг активного агента.
Совместное введение одного или более агониста допамина с одним или более агонистом допамина D2 оказывает синергичное действие по улучшению одного или более показателей метаболизма, связанных с метаболизмом глюкоз или липидов, и поэтому благотворно влияет на метаболизм глюкозы или липидов.
Предпочтительно введение агониста D2 приурочено к определенному времени. Агонист D2 можно вводить в заранее установленное время.
Предпочтительно введение агониста приурочено к определенному времени. Агонист D1 вводят в заранее установленное время. Так как агонист Di усиливает действие соединенного агониста D2, предпочтительно вводить агонист одновременного с соединенным агонистом(ами) D2, так чтобы период действия агониста D1 в кровотоке пациента перекрывался (на самом деле предпочтительно, чтобы он перерывался как можно больше) с периодом действия соединенного агониста(ов) D2. Продолжительность пост-Tmax плато уровня в плазме агониста D1 может сохраняться дольше, чем у агониста D2. для удобства введения и для того, чтобы обеспечить соблюдения режима, агонист можно вводить одновременно с соединенным агонистом(ами) D2.
Предпочтительно лекарственную форму (формы) вводят раз в день. Более предпочтительно лекарственную форму (формы) вводят раз в день по утрам. Наиболее предпочтительно лекарственную форму (формы) вводят раз в день в предопределенное время для биодоступности утром во время пика уровня пролактина в плазме.
Предпочтительно лекарственные формы вводят утром от 4.00 до 12.00. Более предпочтительно лекарственные формы вводят утром от 5.00 до 12.00. Наиболее предпочтительно лекарственные формы вводят утром от 5.00 до 10.00.
Для лечения позвоночных лекарственные формы допаминовых агонистов обычно вводят в течение периода от приблизительно 10 до приблизительно 180 дней или дольше (1 год и дольше). Однако пациентам, например, в особенно тяжелом положении или в зрелом возрасте может потребоваться более продолжительное или даже непрерывное лечение. Может быть желательным период лечения дольше 6 месяцев или даже непрерывное лечение, даже если не требуется.
Введение агонистов D1 и D2 обычно приводит к улучшению по меньшей мере состояния или показателя нарушения метаболизма. Поэтому в некоторых вариантах реализации введение агониста D1 и D2 приводит к уменьшению одного или более нарушения метаболизма и/или его ключевых элементов, таких как отложение жира, масса тела, глюкоза в плазме или крови, циркулирующий инсулин, триглицериды плазмы (TG), свободные жирные кислоты плазмы (FFA), факторы риска сердечно-сосудистых заболеваний, такие как факторы воспаления, связанного с сердечно-сосудистой системой, стимуляторы нарушения работы эндотелия сосудов, вещества, вызывающие гиперсвертывание крови, включая, но не ограничиваясь, PAI-1 или фибриноген, скорость свертывания крови или потенциал, нейроэндокринные факторы, вызывающие устойчивость к инсулину, кровяное давление, нарушение работы почек и/или почечную недостаточность и потребление пищи.
Согласно другим вариантам реализации парентеральные лекарственные формы согласно настоящему изобретению оказывают одно или более из следующих физиологических действий на пациентов с метаболическим синдромом, ожирением, ожирением/устойчивостью к инсулину, преддиабетом или диабетом 2 типа: (1) улучшение гипергликемии, гипертриглицеридемии, нарушенного уровня глюкозы натощак, непереносимости глюкозы или устойчивости к инсулину; (2) улучшение гипертензии; (3) сокращение физиологических показателей сердечно-сосудистого воспаления, эндотелиальной дисфункции, гиперсвертывания или тромбоза (blood clotting); и/или (4) уменьшение отложений жира или массы тела или обоих показателей; (5) улучшение работы почек, (6) улучшение работы сердца.
В отдельном варианте реализации парентеральные лекарственные формы способствуют увеличению уровня пролактина в плазме ночью (2.00-6.00) по меньшей мере на 35% больше по сравнению со средним дневным уровнем (7.00-19.00) циркулирующего гормона после введения рано утром лекарственного препарата, содержащего агонист допамина, измеренный по меньшей мере через 6 месяцев после начала лечения.
В отдельном варианте реализации повышенный (по меньшей мере на 15% от среднего уровня для нормального здорового человека того же пола и возраста) уровень норэпинефрина в плазме снижается на 10% благодаря такому лечению. Согласно одному варианту реализации повышенный уровень норэпинефрина в плазме снижается по меньшей мере на 15%.
В отдельном варианте реализации ночной уровень пролактина в плазме составляет по меньшей мере на 35% больше, чем средний дневной уровень пролактина после лечения агонистом допамина через 6 месяцев после начала лечения.
В отдельном виаранте реализации лекарственные формы для парентерального введения не приводят к появлению побочных эффектов в ЖКТ, таких как рвота, тошнота, боль в брюшной области, копостаз и/или диарея больше чем у 15% пациентов.
В отдельном варианте реализации лекарственная форма содержит бромкриптин и другие производные спорыньи и обеспечивает концентрацию циркулирующих метаболитов не больше чем приблизительно 50% от концентрации метаболитов, которая получается после приема пероральной дозы бромкриптина (или других производных спорыньи), что обеспечивает равный уровень бромкриптина (или других производных спорыньи) в кровотоке.
Приготовление лекарственных форм
Вода и свет могут ускорить разложение веществ спорыньевого типа посредством фотоокисления, фоторедукции, окислительно-восстановительных реакций с участием воды (например, распад бромкриптина до бромкриптинина под действием воды или влажности). Поэтому требуется разработать стабильные лекарственные формы для парентерального введения, содержащие агонисты допамина, производные спорыньи, для того, чтобы свести к минимуму действие света и поглощение воды.
Если приготовление лекарственных форм согласно данному изобретению, которые можно вводить парентерально, происходит не в стерильных условиях, нужно проводить конечную стерилизацию с помощью традиционных методов, известных специалисту в данной области, например с помощью автоклавирования или стерильного фильтрования. Суспензии согласно изобретению, которые можно вводить парентерально, предпочтительно нужно готовить в стерильных условиях.
Другие способы производства можно осуществлять с помощью методов, известных специалисту в данной области. Приведенные ниже способы указаны с целью иллюстрации, но не ограничивают данное изобретение.
В конце к лекарственной форме надо добавить стеарат магния и стеариновую кислоту и перемешивать 2 мин. Следует избегать магния при приготовлении лекарственных форм с агонистами допамина, родственных спорынье, поскольку магний существенно уменьшает их стабильность.
Определенные лекарственные формы можно приготовить с помощью способов, известных в данной области. Во всех вариантах реализации компоненты представлены с процентным содержанием от общей массы. Ниже приведено руководство по приготовлению некоторых типов лекарственных форм.
Лекарственная форма для инъекций или жидкая лекарственная форма
Агонист допамина растворяют в неводном растворителе или в коллоидной суспензии агрегатов малого размера в одном резервуаре, а в жидком носителе во втором резервуаре находится агонист допамина в коллоидной суспензии в виде агрегатов большего размера, чем в первом резервуаре (включая, но не ограничиваясь микронизированным агонистом допамина), в общем количестве 0,02-50,0 мг.
Первый резервуар может содержать различные количества (10-50 мкл) безводных растворителей, таких как этанол, изопропанол или пропанол. К данному раствору добавляют небольшое количество (приблизительно 25% от объема раствора) агента, уменьшающего раздражимость безводной слизистой ткани, например растительные масла, такие как, не ограничиваясь, оливковое масло, кукурузное масло или минеральное масло.
Необязательно в раствор в первом контейнере далее добавляют безводные проникающие агенты, биоадгезивы, полимеры и/или стабилизаторы (например, антиоксиданты, такие как лимонная кислота или аскорбиновая кислота), чтобы общий объем не превышал 100 мкл.
Второй резервуар может содержать водный или безводный растворитель, например этанол, изопропанол или пропанол в объеме 10-50 мкл.
В данный раствор добавляют небольшое количество (25% от объема раствора) агента, уменьшающего раздражимость безводной слизистой ткани, например растительные масла, такие как, не ограничиваясь, оливковое масло, кукурузное масло или минеральное масло.
Дополнительно в раствор во втором контейнере добавляют безводные проникающие агенты, биоадгезивы, полимеры и/или стабилизаторы (например, антиоксиданты, такие как лимонная кислота или аскорбиновая кислота), чтобы итоговый объем не превышал 100 мкл.
Аэрозольная лекарственная форма
Аэрозольные лекарственные формы обычно можно приготовить путем добавления инертного газа (например, азота) в жидкую лекарственную форму.
Аэрозольная лекарственная форма
Агонист допамина растворяют в неводном растворителе, например безводном этаноле, в среде с низкой влажностью, дополнительно совмещенном с агентом, уменьшающим раздражительность слизистой оболочки, и затем помещают в одну камеру или твердую пластиковую канистру, которую обрабатывают под давлением инертным газом, например азотом. Канистра снабжена механизмом дозирования в форме аэрозольного спрея или т.п. в пределах 5-100 мкл на дозу. Необязательно после растворения в этаноле, как описано выше, в раствор агониста допамина добавляют проникающие агенты (например, желчные соли, поверхностно-активные вещества, жирные кислоты и их производные, хелаторы, циклодекстрины, хитозан, низшие спирты), биоадгезивы (например, Карбопол 934Р, 974Р, 1 Voveou AA-I, поливинилпирролидон) и/или стабилизаторы, такие как полиэтиленгликоль, которые, как известно в данной области, ускоряют доставку агониста допамина от места введения в системный кровоток через слизистую оболочку. Дополнительно в раствор агониста допамина в этаноле добавляют безводный полимер, например, полиэтиленгликоль, в таком количестве, чтобы улучшить растворимость растворимых компонентов и снизить концентрацию этанола.
Во второй отдельной камере той же канистры тонко измельчают агонист допамина и добавляют его в соответствующий связующий раствор, например полиэтиленгликоль, для получения коллоидной суспензии. В такую коллоидную суспензию добавляют проникающие агенты, биоадгезивы и/или стабилизаторы, которые, как известно в данной области, растворяются в связующем растворе или образуют коллоидную суспензию. Такую суспензию агониста допамина помещают в металлический или твердый пластиковый контейнер для введения в виде спрея под давлением в атмосфере инертного газа.
Аэрозольная лекарственная форма
Агонист допамина добавляют в растворитель в концентрации 0,1-5,0 мг на приблизительно 10-50 мкл водного или неводного растворителя, такого как этанол. В раствор добавляют небольшое количество (25% от объема раствора) агента, снижающего раздражимость слизистой оболочки, такого как оливковое масло или минеральное масло. Необязательно в смесь можно добавить усилители поглощения слизистой оболочкой, такие как свободные жирные кислоты, и/или биоадгезивы, такие как поливинилпирролидон. Раствор перемешивают до тех пор, пока агонист допамина не растворится полностью, и затем раствор помещают в устройство, в которое не может проникать свет, обрабатывают под давлением инертным газом, например азотом, и снабжают его механизмом дозированной доставки 10-100 мкл при каждом применении для обеспечения дозы 0,1-5,0 мг агониста допамина.
Во вторую изолированную камеру того же устройства к водному или безводному растворителю, например безводному этанолу, добавляют агонист допамина в среде с пониженной влажностью в концентрации 0,1-50,0 мг приблизительно на 5-25 мкл этанола. После полного растворения агониста допамина в растворителе добавляют полимер, например полиэтиленгликоль, жирную кислоту с длинной цепью или растительное масло, например оливковое масло, кукурузное масло или минеральное масло, для получения 70/30 раствора этанола/другого агента, чтобы скорость всасывания агониста допамина в этой лекарственной форме отличалась (была ниже) от лекарственной формы из другой камеры канистры. В этот раствор добавляют небольшой объем (25% от объема раствора) агента, снижающего раздражимость слизистой оболочки, например оливковое масло или минеральное масло. Дополнительно в раствор можно добавить усилители поглощения слизистой оболочкой, например свободные жирные кислоты и/или биоадгезивы, например поливинилпирролидон. Раствор перемешивают до тех пор, пока агонист допамина растворится полностью или станет коллоидной суспензией, а затем раствор помещают в устройство, в которое не может проникать свет, обрабатывают под давлением инертным газом, например азотом, и снабжают его механизмом дозированной доставки 10-50 мкл при каждом применении для обеспечения дозы 0,1-5,0 мг агониста допамина.
Аэрозольная или жидкая лекарственная форма
Агонист допамина измельчают до диаметра 0,1-1,0 мкм, а затем добавляют к полимеру, такому как полиэтиленгликоль, или к жирной кислоте, или растительному маслу, например минеральному маслу или т.п., для получения коллоидной суспензии 0,1-5,0 мг агониста допамина на 10-50 мкл носителя.
К суспензии добавляют небольшое количество (до 25% объема суспензии) агента, снижающего раздражимость слизистой оболочки, например оливковое или минеральное масло. В эту суспензию добавляют усилители поглощения слизистой оболочкой, например жирные кислоты и/или биоадгезивы, например поливинилпирролидон. Суспензию помещают в контейнер, куда не проникает свет, который обрабатывают под давлением инертным газом, например азотом, и снабжают механизмом дозированной доставки 10-100 мкл для обеспечения дозы агониста допамина 0,1-5,0 мг.
В отдельной камере того же устройства добавляют агонист допамина в водный или безводный растворитель, например безводный этанол, в среде с низкой влажностью в концентрации 0,1-50,0 мг на 5-25 мкл этанола. После полного растворения агониста допамина в растворителе добавляют полимер, например полиэтиленгликоль или длинноцепочечные жирные кислоты или растительное масло, например оливковое, кукурузное или минеральное масло, для получения приблизительной пропорции этанол/другой агент 70/30, чтобы скорость всасывания агониста допамина согласно настоящей лекарственной формы отличалась, т.е. была меньше, чем у лекарственной формы из другой камеры канистры. В раствор добавляют небольшое количество (25% объема раствора) агента, уменьшающего раздражимость слизистой оболочки, например оливковое или минеральное масло. В этот раствор добавляют ускорители поглощения слизистой, например свободные жирные кислоты и/или биоадгезивы, например поливинилпирролидон. Этот раствор перемешивают до тех пор, пока агонист допамина полностью не растворится, или готовили коллоидную суспензию, и затем помещают раствор в непроницаемый для света контейнер, который вакуумизируют или не вакуумизируют с помощью азота, и снабжали механизмом постепенной доставки 10-100 мкл спрея для обеспечения дозы 0,1-50,0 мг агониста допамина.
Твердая лекарственная форма
Готовили стабильную твердую парентеральную лекарственную форму, которая содержит: (1) агонист допамина 0,02-50,0 мг, который имеет частицы малого размера и частицы относительно большого размера для медленного растворения и всасывания, смешанный с антиоксидантом, таким как лимонная кислота; (2) смесь комбинируют с носителем, таким как маннитол, а затем с разрыхлителем и биоадгезивом, таким как Бенецел или Коллидон CL, и безводным полимером в качестве связующего агента, таким как целлюлоза или аналоги целлюлозы, полиэтиленгликоль, жирная кислотой или растительное масло; (3) необязательно, небольшое количество безводного агента, уменьшающего раздражительность слизистой оболочки, например оливкового масла или минерального масло; и (4) необязательно добавлены безводные проникающие агенты, биоадгезивы и/или стабилизаторы, а затем лубрикант, такой как стеарат или касторовое масло для получения общей массы не более 200 мг для обеспечения быстро растворимых твердых лекарственных форм с быстрым и медленным постоянным всасыванием агониста допамина, при этом общая доза составляет от 0,02 до 50,0 мг. Данные ингредиенты предпочтительно добавляют в смесь в указанном порядке.
Твердая лекарственная форма
Готовят стабильную твердую парентеральную лекарственную форму согласно настоящему изобретению, которая содержит: (1) измельченный агонист допамина диаметром (а) 0,1-5,0 мкм и (b) 10-200 мкм, итого 0,02-50,0 мг; смешанный с антиоксидантом, например лимонной кислотой; (2) смесь комбинируют с носителем, таким как маннитол, а затем с разрыхлителем и биоадгезивом, например Коллидоном CL, и безводным полимером в качестве связующего агента, таким как целлюлоза или аналоги целлюлозы, полиэтиленгликоль, жирная кислота или растительное масло; (3) необязательно, небольшое количество безводного агента, уменьшающего раздражительность слизистой оболочки, такого как оливковое или минеральное масла; и (4) необязательно добавлены безводные проникающие агенты, биоадгезивы и/или стабилизаторы, а затем лубрикант, такой как стеарат или касторовое масло, для получения общей массы не более 250 мг для получения быстрорастворимых твердых лекарственных форм с быстрым и медленным постоянным всасыванием агониста допамина, при этом общая доза составляет от 0,02 до 50,0 мг. Данные ингредиенты предпочтительно добавляют в смесь в указанном порядке.
Твердая лекарственная форма
Агонист допамина добавляют в водный или безводный растворитель, например этанол, в среде с низкой влажностью в концентрации 0,1-50,0 мг на приблизительно 50-250 мкл растворителя. После полного растворения агониста допамина в растворителе добавляют полимер, такой как полиэтиленгликоль, или жирную кислоту, или растительное масло для получения раствора с соотношением растворитель/другой агент приблизительно 70/30. В раствор добавляют небольшой объем (25% объема растворителя) агента, уменьшающего раздражение безводной слизистой оболочки, например оливковое или минеральное масло. В данный раствор добавляют усилители поглощения слизистой, например жирные кислоты и/или биоадгезивы, такие как поливинилпирролидон. Раствор агониста допамина затем можно объединить со связывающим агентом или матриксом, например растительной камедью, желатином, поливинилпирролидоном, стеаратом магния или касторовым маслом для обеспечения быстрого растворения, и затем получают в одной части твердой лекарственной формы 0,1-50,0 мг на дозу для доставки в слизистую. Вторая часть твердой лекарственной формы состоит из (1) измельченного агониста допамина диаметром 0,1-5,0 мкм или частиц агониста допамина малого размера 10-200 мкм при общей массе 0,02-50,0 мг; смешанных с антиоксидантом, таким как лимонная кислота; (2) смесь объединяют с носителем, например маннитолом, а затем смешивают с разрыхлителем и биоадгезивом, например Коллидоном CL, и безводным полимером в качестве связующего, таким как целлюлоза или аналоги целлюлозы, полиэтиленгликоль, жирная кислота или растительное масло; (3) необязательно, небольшого количества безводного агента, уменьшающего разражение безводной слизистой ткани, такого как оливковое масло или минеральное масло; и (4) необязательно добавлены безводные проникающие агенты, биоадгезивы и/или стабилизаторы, а затем жидкий матричный агент, такой как поливинилпирролидон, желатин или растительная камедь, которые высушивают для получения общей массы не более 250 мг для получения растворимых твердых лекарственных форм с быстрым и медленным постоянным всасыванием агониста допамина, отличающаяся тем, что итоговая доза составляет 0,02-50,0 мг. Данные ингредиенты добавляют в смесь предпочтительно в указанном порядке. Две части лекарственной формы запаивают и упаковывают в алюминиевую фольгу для предотвращения проникновения влаги. Альтернативно, можно объединять две части, заключая одну внутрь другой для доставки, что обеспечивает быстрое всасывание и медленное более постоянное всасывание.
Твердая лекарственная форма
Агонист допамина измельчают до частиц диаметром 0,1-1,0 мкм и затем добавляют в безводный полимер, такой как полиэтиленгликоль, или жирную кислоту, или растительное масло для получения коллоидной суспензии с 0,1-1,0 мг агониста допамина на 10-25 мкл носителя. В суспензию добавляют небольшой объем (25% объема суспензии) агента, уменьшающего раздражимость безводной слизистой, такого как оливковое масло или минеральное масло. В указанную суспензию добавляют усилители поглощения слизистой, например свободную жирную кислоту, и/или биоадгезивы, такие как поливинилпирролидон. Суспензию агониста допамина затем объединяют со связующим агентом или матриксом, например растительной камедью, желатином, маннитолом, поливинилпирролидоном или стеаратом. Агонист допамина - коллоидную суспензию можно потом объединить со связывающим агентом или матриксом, например растительной камедью, желатином, маннитолом, поливинилпирролидоном, стеаратом или касторовым маслом, обеспечивающим быстрое растворение, и затем поместить в одну часть твердой лекарственной формы при 0,1-50,0 мг на дозу для доставки через слизистую.
Вторая часть твердой лекарственной формы состоит из (1) частиц агониста допамина малого размера 10-200 мкм, итого 0,02-50,0 мг, смешанных с антиоксидантом, таким как лимонная кислота; (2) смесь объединяют с носителем, таким как маннитол, а затем смешивают с разрыхлителем и биоадгезивом, таким как Коллидон CL, и безводным полимером в качестве носителя, таким как целлюлоза или аналоги целлюлозы, полиэтиленгликоль, жирная кислота или растительное масло; (3) необязательно, небольшого количества безводного агента, уменьшающего раздражение безводной слизистой ткани, например оливковое или минеральное масло; и (4) необязательно, добавлены безводные проникающие агенты, биоадгезивы и/или стабилизаторы, а затем жидкий матричный агент, такой как поливинилпирролидон, желатин или растительная камедь, которые высушивают для получения общей массы не более 250 мг для получения растворимых твердых лекарственных форм с быстрым и медленным постоянным всасыванием агониста допамина, отличающаяся тем, что итоговая доза составляет 0,02-50,0 мг. Данные ингредиенты добавляют в смесь предпочтительно в указанном порядке. Две части лекарственной формы запаивают и упаковывают в алюминиевую фольгу для предотвращения проникновения влаги. Альтернативно, можно объединять две части, заключая одну внутрь другой для доставки, что обеспечивает быстрое всасывание и медленное более постоянное всасывание.
Твердая таблетированная лекарственная форма
Согласно одному варианту реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 3-50% матрикса высвобождения, приблизительно 0,5-10% вещества, способствующего скольжению (glidant), приблизительно до 70% усилителя растворимости, приблизительно до 25% усилителя биоадгезии, приблизительно до 30% усилителя проникания, приблизительно до 95% разрыхлителя, приблизительно до 95% наполнителя и приблизительно до 65% шипучего агента.
Согласно другому варианту реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 3-20% матрикса высвобождения, приблизительно 0,5-5% вещества, способствующего скольжению (glidant), приблизительно до 30% усилителя растворимости, приблизительно до 10% усилителя биоадгезии, приблизительно до 20% усилителя проникания, приблизительно до 85% разрыхлителя, приблизительно до 80% наполнителя и приблизительно до 45% шипучего агента.
Согласно предпочтительному варианту реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 7-15% матрикса высвобождения, приблизительно 0,5-2,5% вещества, способствующего скольжению, приблизительно 2-20% усилителя растворимости, приблизительно 2-8% усилителя биоадгезии, приблизительно до 15% усилителя проникания, приблизительно до 82% разрыхлителя, приблизительно до 75% наполнителя и приблизительно до 45% шипучего агента.
В другом варианте реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов приблизительно 5-10% матрикса высвобождения, приблизительно 0,5-2% вещества, способствующего скольжению, приблизительно 1-5% усилителя растворимости, приблизительно 2-8% усилителя биоадгезии, приблизительно до 15% усилителя проникания, приблизительно до 12% разрыхлителя и приблизительно до 75% наполнителя.
Согласно другому варианту реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 5-10%) матрикса высвобождения, приблизительно 0,5-2% вещества, способствующего скольжению, приблизительно 1-5% усилителя растворимости, приблизительно 2-8% усилителя биоадгезии, приблизительно до 15% усилителя проникания, приблизительно до 12% разрыхлителя и приблизительно до 75% наполнителя.
Согласно одному варианту реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 5-10% матрикса высвобождения, приблизительно 0,5-2% вещества, способствующего скольжению, приблизительно 1-5% усилителя растворимости, приблизительно 2-8% усилителя биоадгезии, приблизительно 75-85% разрыхлителя, приблизительно до 15% усилителя проникания и приблизительно до 75% наполнителя.
Согласно другому варианту реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 3-20% матрикса высвобождения, приблизительно 0,5-5% вещества, способствующего скольжению, приблизительно 0,5-10% усилителя растворимости, приблизительно 2-15% усилителя биоадгезии, приблизительно 3-25% разрыхлителя, приблизительно до 30% усилителя проникания и приблизительно 3-85% наполнителя.
Согласно другому варианту реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 5-10% матрикса высвобождения, приблизительно 0,5-2% вещества, способствующего скольжению, приблизительно 1-5% усилителя растворимости, приблизительно 2-8% усилителя биоадгезии, приблизительно 60-80% разрыхлителя и приблизительно до 15% усилителя проникания.
В другом варианте реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 3-10% матрикса высвобождения, приблизительно 0,5-5% вещества, способствующего скольжению, приблизительно 1-6% усилителя растворимости, приблизительно 2-6,5% усилителя биоадгезии, приблизительно 60-90% разрыхлителя и приблизительно до 30% усилителя проникания.
Согласно другому варианту реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 3-10% матрикса высвобождения, приблизительно 0,5-5% вещества, способствующего скольжению, приблизительно 1-10% усилителя растворимости, приблизительно 2-10% усилителя биоадгезии, приблизительно 60-90% разрыхлителя и приблизительно до 30% усилителя проникания.
Согласно другому варианту реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 10-20% матрикса высвобождения, приблизительно 0,5-2% вещества, способствующего скольжению, приблизительно 15-25% усилителя растворимости, приблизительно 8-15% усилителя биоадгезии, приблизительно 6-12% разрыхлителя и приблизительно 35-45% шипучего агента.
Согласно другому варианту реализации твердая подъязычная таблетированная лекарственная форма содержит один или более активных агентов, приблизительно 5-35% матрикса высвобождения, приблизительно 0,5-5% вещества, способствующего скольжению, приблизительно 10-40% усилителя растворимости, 5-25% усилителя биоадгезии, приблизительно 3-25% разрыхлителя и приблизительно 10-65% шипучего агента.
Согласно другому варианту реализации твердая подъязычная таблетированная лекарственная форма содержит приблизительно 0,5-5% активных агентов, приблизительно 3-20% матрикса высвобождения, приблизительно 0,5-5% вещества, способствующего скольжению, приблизительно 0,5-10% усилителя растворимости, 2-15% усилителя биоадгезии, приблизительно 3-25% разрыхлителя, приблизительно 40-95% наполнителя и дополнительно приблизительно 5-30% усилителя проникания.
В другом варианте реализации, твердая подъязычная таблетированная лекарственная форма содержит приблизительно 0,5-4,5% активных агентов, приблизительно 3-10% матрикса высвобождения, приблизительно 0,5-5% вещества, способствующего скольжению, приблизительно 1-6% усилителя растворимости, приблизительно 2-6,5% усилителя биоадгезии, приблизительно 60-90% разрыхлителя и дополнительно приблизительно 5-30% усилителя проникания.
Согласно другому варианту реализации твердая подъязычная таблетированная лекарственная форма содержит приблизительно 1-6% активных агентов, приблизительно 3-10% матрикса высвобождения, приблизительно 0,5-5% вещества, способствующего скольжению, приблизительно 1-10% усилителя растворимости, приблизительно 2-10% усилителя биоадгезии, приблизительно 60-90% разрыхлителя и дополнительно приблизительно 5-30% усилителя проникания.
Согласно другому варианту реализации твердая подъязычная таблетированная лекарственная форма содержит приблизительно 0,5-5% активных агентов, приблизительно 5-35% матрикса высвобождения, приблизительно 0,5-5% вещества, способствующего скольжению, приблизительно 10-40% усилителя растворимости, 5-25% усилителя биоадгезии, приблизительно 3-25% разрыхлителя и приблизительно 10-65% шипучего агента.
Для указанных выше лекарственных форм: предпочтительно компонентами матрикса высвобождения являются Карбопол 974, Бенецел® или ксантановая камедь или смесь указанных компонентов; предпочтительными веществами, способствующими скольжению, являются стеарат магния и стеариновая кислота, предпочтительными усилителями растворимости являются лимонная кислота и аскорбиновая кислота, предпочтительным усилителем биоадгезии является поливинилпирролидон; предпочтительными разрыхлителями являются Фармабурст и Эксплотаб (натрия гликолят крахмала и карбоксиметилкрахмал натрия); предпочтительными наполнителями являются Кабосил, гранулярный маннитол и микрокристаллическая целлюлоза, например ПроСолв, и предпочтительным шипучим агентом является Эфферсода-12.
Для указанных выше лекарственных форм: более предпочтительно компонентом матрикса высвобождения является Бенецел®; более предпочтительным веществом, способствующим скольжению, является стеариновая кислота; более предпочтительным усилителем растворимости является лимонная кислота; более предпочтительным усилителем биоадгезии является поливинилпирролидон; более предпочтительным разрыхлителем является Фармабурст; более предпочтительными наполнителями являются гранулярный маннитол и микрокристаллическая целлюлоза, например ПроСолв, и более предпочтительным шипучим агентом является Эфферсода-12.
Согласно одному предпочтительному варианту реализации твердая подъязычная таблетированная лекарственная форма содержит приблизительно 0,5-5% агониста допамина, приблизительно 3-20% гидроксипропилметилцеллюлозы (ГПМЦ), приблизительно 0,5-5% стеариновой кислоты, приблизительно 0,5-10% лимонной кислоты, приблизительно 2-15% поливинилпирролидона (ПВП), приблизительно 3-25% натрия гликолят крахмала и карбоксиметилкрахмала натрия, приблизительно 40-80% маннитола и приблизительно 3-25% ПроСолва.
Согласно другому предпочтительному варианту реализации твердая подъязычная таблетированная лекарственная форма содержит, приблизительно 0,5-5% агониста допамина, приблизительно 3-20% ГПМЦ, приблизительно 0,5-5% стеариновой кислоты, приблизительно 0,5-10% лимонной кислоты, приблизительно 2-15% ПВП, приблизительно 3-25% натрия гликолят крахмала и карбоксиметилкрахмала натрия, приблизительно 40-80% маннитола, приблизительно 3-25% ПроСолва и приблизительно 5-30% циклодекстрина.
Согласно другому предпочтительному варианту реализации твердая подъязычная таблетированная лекарственная форма содержит приблизительно 0,5-4,5% агониста допамина, приблизительно 3-10% ГПМЦ, приблизительно 0,5-5% стеариновой кислоты, приблизительно 1-6% лимонной кислоты, приблизительно 2-6,5% ПВП и приблизительно 60-90% Фармабурста.
Согласно другому предпочтительному варианту реализации твердая подъязычная таблетированная лекарственная форма содержит приблизительно 0,5-4,5% агониста допамина, приблизительно 3-10% ГПМЦ, приблизительно 0,5-5% стеариновой кислоты, приблизительно 1-6% лимонной кислоты, приблизительно 2-6,5% ПВП, приблизительно 60-90% Фармабурста и приблизительно 5-30% циклодекстрана.
Согласно другому предпочтительному варианту реализации твердая подъязычная таблетированная лекарственная форма содержит приблизительно 1-6% агониста допамина, приблизительно 3-10% ГПМЦ, приблизительно 0,5-5% стеариновой кислоты, приблизительно 1-10% лимонной кислоты, приблизительно 2-10% ПВП, и приблизительно 60-90% Фармабурста.
Согласно другому предпочтительному варианту реализации твердая таблетированная лекарственная форма содержит приблизительно 1-6% агониста допамина, приблизительно 3-10% ГПМЦ, приблизительно 0,5-5% стеариновой кислоты, приблизительно 1-10% лимонной кислоты, приблизительно 2-10% ПВП, приблизительно 60-90% Фармабурста и приблизительно 5-30% циклодекстрана.
Согласно другому предпочтительному варианту реализации твердая подъязычная таблетированная лекарственная форма содержит приблизительно 0,5-5% агониста допамина, приблизительно 5-35% ГПМЦ, приблизительно 0,5-5% стеариновой кислоты, приблизительно 10-40% лимонной кислоты, приблизительно 5-25% ПВП, приблизительно 3-25% Фармабурста и приблизительно 10-65% Эфферсода-12.
Трансдермальная гелевая лекарственная форма
Трансдермальные гелевые лекарственные формы согласно настоящему изобретению готовили, растворяя стабилизатор (например, антиоксиданты, такие как лимонная кислота или аскорбиновая кислота) в поверхностно-активном веществе, например лауриловой кислоте, олеиновой кислоте, стеариновой кислоте, миристиновой кислоте, рицинолеиновой кислоте или полиэтиленгликоле. Добавляли неводный растворитель (например, пропиленгликоль, глицерол, короткоцепочечные замещенные или незамещенные спирты, такие как этанол, изопропанол или пропанол) и диспергировали ультразвуком. Возможно в неводный растворитель перед обработкой ультразвуком добавить биоадгезив/матрикс высвобождения активного агониста. Постепенно в раствор при постоянном перемешивании и диспергировании ультразвуком добавляли проникающие агенты (например, желчные соли, производные жирных кислот, эфиры жирных кислот, производные энамина и альфакетоальдегиды, холат натрия, гликохолат натрия, дезоксихолат натрия, лаурилсульфат натрия, салицилат натрия, натриевую соль этилендиаминтетрауксусной кислоты (ЭДТА), апротинин, азон, 5-метоксисалицилат натрия, 1-олеазациклопентан-2-он и/или диоксид кремния с высоким сродством к водным растворителям, такие как осажденный диоксид кремния, более известный под товарным знаком Силоид®, мальтодекстрины, β-циклодекстрины, поверхностно-активные вещества, комплексоны, циклодекстрины, хитозан и низшие спирты).
Полученную суспензию пропускают через сито из нержавеющей стали 40 размера. Молочную кремообразную суспензию (стоковый раствор, постепенно разделившийся через несколько дней хранения в холодильнике) добавляют к полипропиленовую смесь и диспергируют ультразвуком 5 минут. Постепенно в раствор добавляют проникающий агент при постоянном перемешивании и диспергировании ультразвуком.
Трансдермальная гелевая лекарственная форма
Согласно одному варианту реализации трансдермальная гелевая лекарственная форма содержит один или более активных агентов, приблизительно 5-95% растворителей, приблизительно 1-30% загустителя, 0,5-10% стабилизатора и приблизительно до 35% усилителей биоадгезии. Согласно другому варианту реализации трансдермальная гелевая лекарственная форма содержит один или более активных агентов, приблизительно 5-90% растворителей, приблизительно 5-12% загустителей и 0,5-1,5% стабилизаторов.
В другом варианте реализации трансдермальная гелевая лекарственная форма содержит один или более активный агент, приблизительно 5-90% растворителей, приблизительно 3-25% загустителя, приблизительно 0,5-30% усилителей биоадгезии и 0,5-5% стабилизаторов.
В другом варианте реализации трансдермальная гелевая лекарственная форма содержит один или более активный агент, приблизительно 5-90% растворителей, приблизительно 3-25% загустителей и 0,5-5% стабилизаторов.
В другом варианте реализации трансдермальная гелевая лекарственная форма содержит приблизительно 0,5-10% активных агентов, приблизительно 50-95% растворителей, приблизительно 3-25% загустителей и 0,5-5% стабилизаторов.
В другом варианте реализации трансдермальная гелевая лекарственная форма содержит приблизительно 0,5-10% активных агентов, приблизительно 50-95% растворителей, приблизительно 3-25% загустителей, приблизительно 1,5-30% усилителей биоадгезии и 0,5-5% стабилизаторов.
Для всех указанных выше гелевых лекарственных форм: предпочтительными растворителями являются пропиленгликоль и глицерол, предпочтительным загустителем является диоксид кремния 200; предпочтительным стабилизирующим агентом является безводная лимонная кислота, предпочтительными биоадгезивами являются гидроксипропилметилцеллюлоза (ГПМЦ) и поливинилпирролидон (ПВП).
Согласно предпочтительному варианту реализации трансдермальная гелевая лекарственная форма содержит приблизительно 0,5-10% агониста допамина, приблизительно 5-40% ПЭГ, приблизительно 45-85% глицерола, приблизительно 3-25% диоксида кремния и приблизительно 0,5-5% лимонной кислоты.
Согласно другому предпочтительному варианту реализации трансдермальная гелевая лекарственная форма содержит приблизительно 0,5-10% агониста допамина, приблизительно 5-40% ПЭГ, приблизительно 45-85% глицерола, приблизительно 3-25% диоксида кремния, приблизительно 1-15% ГПМЦ, приблизительно 0,5-15% ПВП и приблизительно 0,5-5% лимонной кислоты.
Лекарственная форма в виде трансдермального пластыря
Готовят твердую стабильную лекарственную форму для парентерального введения согласно настоящему изобретению, которая содержит: (1) агонист допамина в растворенном виде, в виде частей с одним или двумя различными размерами частиц от 0,02-5,0 мкм; (2) нетоксичный органический растворитель, такой как этанол, изопропанол, пропанол, в количестве 5-100 мкл, и (3) необязательно добавлен безводный проникающий агент, такой как полиэтиленгликоль, или жирная кислота, или растительное масло.
Затем указанный выше состав добавляют к системе трансдермальной доставки лекарственного средства.
Указанная система трансдермальной доставки включает: (1) матриксную мембрану, контролирующую скорость, содержащую полиэтилен, полиуретан, поливинилхлорид (PVC), полиакрилаты, поликарбонаты, поливинилы, полистирены, полиамиды и их производные, целлюлозу, производные целлюлозы и комбинации вышеуказанных веществ, густоту и пористость которых можно регулировать для того, чтобы управлять скоростью диффузии лекарственного средства из резервуара; и (2) адгезив для прикрепления такого лекарственного матрикса к коже таким образом, что такой адгезив не блокирует высвобождение лекарственного средства из системы доставки в кожу; (3) заднее покрытие, не проницаемое для света, влаги, влажности и содержимого системы доставки; и (4) съемную переднюю часть, не проницаемую для света, влаги, влажности и содержимого системы доставки. Также система доставки характеризуется способностью иметь низкую и высокую скорость высвобождения и прохождения через кожу для относительно медленного и быстрого всасывания телом.
Трансэпителиальная лекарственная форма, содержащая комбинацию агониста допамина и агента периферического действия
К компоненту агониста допамина с медленным высвобождением лекарственных форм можно добавлять агенты, действующие периферически. Затем агенты периферического действия медленно высвобождаются из лекарственной формы для обеспечения постоянного высвобождения в течение 4-12 часов после введения. В некоторых случаях желательно также добавлять агент периферического действия к компоненту с быстрым высвобождением лекарственной формы с агонистом допамина для обеспечения быстрого высвобождения агента периферического действия. В других случаях можно добавлять агент периферического действия к компонентам с быстрым и медленным высвобождением лекарственной формы с агонистом допамина для обеспечения быстрого подъема уровня в плазме, за которым следует постоянный пик, или уровня, который располагается поблизости от пика в течение приблизительно 4-12 часов.
Чресслизистая пленочная лекарственная форма
Готовят твердую стабильную пленку для подъязычного или буккального введения агонистов допамина с поливинилпирролидонами или сополимерами поливинилпирролидона и поливинилацетата. Эти полимеры позволяют использовать безводный растворитель в качестве единственного растворителя в лекарственной форме вместо воды. Это важно при использовании определенных агонистов допамина, например соединений, родственных спорынье, которые неустойчивы по отношению к воде.
Более того, можно усиливать биодоступность и обеспечивать желаемую кривую биодоступности к пикам и плато согласно настоящему изобретению, добавляя дополнительные усилители проникания, например жирные кислоты и биоадгезивы, в пленочную лекарственную форму. Также в пленочную лекарственную форму можно добавлять усилители вкуса для придания приятного вкуса.
Чресслизистая пленочная лекарственная форма
Основу композиции готовят, добавляя поливинилпирролидоны, например Коллидон 90F, Коллидон VA64 и поверхностно-активное вещество, например лауриновую кислоту, олеиновую кислоту, стеариновую кислоту, миристиновую кислоту, рициноленовую кислоту или полиэтиленгликоль, в неводный растворитель, такой как безводный этанол. К основе композиции необязательно может быть добавлен дополнительный безводный растворитель (например, пропиленгликоль, глицерол, короткоцепочечные замещенные или незамещенные спирты, такие как этанол, изопропанол или пропанол). Основу композиции перемешивают на медленной скорости при комнатной температуре в стеклянной бутыли 24 часа.
Дополнительно в перемешанную основу композиции можно добавлять синтетические и полусинтетические биоадгезивные полимеры, такие как гидроксиэтилцеллюлоза, поливиниловый спирт, полиакриловая кислота, карбоксиметилцеллюлоза натрия, поливинилпирролидон или гидроксипропилцеллюлоза (например, KLUCEL® LF), проникающие агенты, например, желчные соли, поверхностно-активные вещества, жирные кислоты и их производные, хелаторные агенты, мальтодекстрины, циклодекстрины или хитозан. Если это осуществляется, основу композиции еще раз перемешивают на медленной скорости при комнатной температуре в стеклянной бутыли 24 часа.
Итоговую лекарственную форму готовят, растворяя стабилизатор (например, антиоксиданты, такие как лимонная кислота или аскорбиновая кислота) в безводном растворителе, таком как безводный этанол в среде с пониженной влажностью. В данный раствор добавляют агонист допамина. Раствор допамина добавляют в основную композицию для получения геля, который можно использовать для отлива пленки. Дополнительно к основной лекарственной форме можно добавлять биоадгезив/матрикс высвобождения активного агониста, например ГПМЦ или неводный растворитель (например, пропиленгликоль, глицерол, короткоцепочечные замещенные или незамещенные спирты, например этанол, изопропанол или пропанол).
Пленку получают, нанося итоговую лекарственную форму на подкладку для пленки, зафиксированную на твердой поверхности, например стеклянной пластине. Оставляют пленку сохнуть до тех пор, пока она не станет тягучей и хорошо сформированной при поддержании температуры поверхности приблизительно 60-70°C.
Пленочная лекарственная форма
Согласно одному варианту реализации пленочная лекарственная форма содержит один или более активных агентов, приблизительно 0,5-10% пленкообразующего агента, приблизительно 5-20% стабилизирующего агента, приблизительно 10-95% усилителя биоадгезии и приблизительно до 50% усилителя растворимости.
Согласно другому варианту реализации пленочная лекарственная форма содержит один или более активных агентов, приблизительно 1-6% пленкообразующего агента, приблизительно 5-10% стабилизирующего агента, приблизительно 50-85% усилителя биоадгезии и приблизительно 0,5-20% усилителя растворимости.
Согласно другому варианту реализации пленочная лекарственная форма содержит один или более активный агент, приблизительно 1-5% пленкообразующего агента, приблизительно 5-10% усилителя стабилизации, приблизительно 50-70% усилителя биоадгезии и приблизительно 15-20% усилителя растворимости.
В другом варианте реализации пленочная лекарственная форма содержит один или более активный агент, приблизительно 0,5-10% пленкообразующего агента, приблизительно 2-20% усилителя стабилизации, приблизительно 10-65% усилителя биоадгезии и приблизительно 3,8-45% усилителя растворимости, содержащего или не содержащего 1-5% олеиновой кислоты.
В другом варианте реализации пленочная лекарственная форма содержит приблизительно 2-20% активных агентов, приблизительно 0,5-10% пленкообразующего агента, приблизительно 2-20% усилителя стабилизации, приблизительно 20-95% усилителя биоадгезии и приблизительно 3,8-45% усилителя растворимости, содержащего или не содержащего 1-5% олеиновой кислоты.
Для всех приведенных выше пленочных лекарственных форм: предпочтительным пленкообразующим агентом является Коллидон VA64; предпочтительным стабилизирующим агентом является лимонная кислота; предпочтительными усилителями биоадгезии являются Коллидон 90F, FLUCEL и ГПМЦ и предпочтительными усилителями растворимости являются ПЭГ400, глицерол и циклодекстрин.
Для всех приведенных выше пленочных лекарственных форм: более предпочтительным пленкообразующим агентом является Коллидон VA64; более предпочтительным стабилизирующим агентом является лимонная кислота; более предпочтительными усилителями биоадгезии являются Коллидон 90F, FLUCEL и более предпочтительными усилителями растворимости являются ПЭГ400, глицерол и циклодекстрин.
Согласно предпочтительному варианту реализации пленочная лекарственная форма содержит приблизительно 2-20% агониста допамина, приблизительно 10-55% Коллидона 90F, приблизительно 0,5-10% Коллидона VA64, приблизительно 0,3-5% ПЭГ400 приблизительно 10-55% KLUCEL, приблизительно 0,5-10% глицерола, 2-20% лимонной кислоты и приблизительно 3-30% циклодекстрина, содержащего или не содержащего 1-5% олеиновой кислоты.
Подкожная лекарственная форма
Активный агент пропускают через сито 40 меш и суспендируют в эмульгирующем агенте. В данный раствор добавляют среды или матрицы (например, синтетические, полусинтетические или натуральные масла, предпочтительно можно использовать триглицериды с цепью средней длины С8-С10 в остатке карбоксильной кислоты, соевое масло, масло кунжута, арахиса, оливы, кокоса, касторовое масло, подсолнечное масло, сафлоровое масло или соответствующие гидрогенизированные масла или смеси по меньшей мере двух из указанных выше масел, бентонит, этоксилированные изостеариловые спирты, полиоксиэтиленсорбитол или эфиры сорбитана, микрокристаллическую целлюлозу или ее производные, растительные камеди, полиэтиленгликоли различного размера, метагидроксид алюминия, агар-агар и трагикант, желатины или смеси двух и более веществ). Образованную прозрачную гомогенную эмульсию активного агента можно использовать для парентерального применения после пропускания через стерильный фильтр. Рекомендуется хорошо встряхнуть смесь непосредственно перед применением.
Подкожная лекарственная форма
Согласно одному варианту реализации подкожная лекарственная форма содержит один или более активных агентов, приблизительно 5-20% эмульгирующего агента и приблизительно 80-95% фармацевтической среды.
В другом варианте реализации подкожная лекарственная форма содержит один или более активных агентов, приблизительно 5-10% эмульгирующего агента и приблизительно 90-95% фармацевтической среды.
Для всех приведенных выше подкожных лекарственных форм: более предпочтительным эмульгирующим агентом является полисорбат 80 и более предпочтительной средой является кунжутное масло.
В другом варианте реализации подкожная лекарственная форма содержит один или более активных агентов, приблизительно 0,01-0,1 бромкриптина, 5-10% полисорбата 80 и приблизительно 90-95% кунжутного масла.
Примеры, приведенные ниже, показывают, что можно изменять компоненты лекарственных форм агонистов допамина, что позволяет получать предсказуемые изменения профиля кривой биодоступности с пиком-плато, и при парентеральном введении эти формы имеют желаемые кривые биодоступности с пиком-плато in vivo. Ежедневное введение в приемлемое время лекарственных форм агонистов допамина, которые имеют желаемые кривые биодоступности с пиком-плато, уменьшают нарушения метаболизма на хорошо известных животных моделях метаболических заболеваний (смотрите Примеры 18-21 и Фигуры 3-8). Более того, можно сделать эти парентеральные лекарственные формы агонистов допамина устойчивыми к действию нагревания и влажности при стандартных условиях разлива по банкам (смотрите Пример 22). Несколько наполнителей могут влиять на профиль растворения активного агента. Например, если увеличивать концентрацию наполнителей ПроСолв (микроцеллюлозный наполнитель) и Бенецел® (биоадгезив/матрикс высвобождения активного агониста, ГПМЦ), то они снижают скорость растворения активного агента. И наоборот, наполнители лимонная кислота и Фармабурст ускоряют раннее и общее растворение агониста допамина, соответственно. Если добавлять лимонную кислоту и понижать уровень Бенецеля®, общая скорость растворения агониста допамина сохраняет желаемый ранний быстрый профиль растворения, за которым следует медленное постоянное растворение. Если далее увеличивать содержание лимонной кислоты (как в случае таблетированной лекарственной формы 11S, описанной ниже), то неожиданно раннее взрывоопасное высвобождение лекарственной формы заметно ускоряется, при этом в течение первых 30 минут высвобождается 40%, а затем следует медленное, но постоянное высвобождение в течение следующих 210 минут. Можно добавлять циклодекстрин для улучшения этого профиля высвобождения при ускорении всасывания лекарственной формы, как в случае лекарственной формы 12S.
Если включать в качестве разрыхлителя Фармабурст вместо Эксплотаба (натрия гликолят крахмала и карбоксиметилкрахмала натрия), то время распада также уменьшается (приблизительно с 15 до 5 минут). Такой ускоренный распад является желаемой характеристикой для введения парентеральной таблетки. Также использование Фармабурста ускоряет общий профиль растворения лекарственной формы. Более того, следует понимать, что изменяя соотношение Эксплотаба и Фармабурста, как и уровней Бенецеля® и ПроСолва в таблетке, можно достичь профиля незамедлительного высвобождения агониста допамина. Такие «гибридные» лекарственные формы позволяют точно корректировать желаемую лекарственную форму агониста допамина для получения желаемого фармакокинетического профиля. Добавление к Фармабурсту Эфферсоды еще больше ускоряет распад и растворение лекарственной формы агониста допамина.
Далее в этих лекарственных формах для парентерального введения можно доводить уровни биоадгезива до максимальной величины для поддержания биоадгезии активного агента при допущении быстрого взрывного растворения активного агента. Повышение уровня биоадгезива приводит к замедлению растворения агента, хотя снижение уровня биоадгезива не влияет на время растворения. Поэтому можно оптимизировать относительные количества биоадгезивного агента, активного агента и других компонентов для получения желаемой кривой с пиком-плато. Далее, можно показать, что повышение содержания активного агента от 1 до 3 мг в таблетке не влияет на свойства растворимости таблетки, поэтому можно допускать диапазон доз парентерального агониста допамина. Однако при повышении уровня активного агента от 1 до 3 мг в таблетке лекарственной формы, которая содержит циклодекстрин или другой проникающий агент вместе с биоадгезивом, можно ускорять высвобождение активного агента, повышая его уровень по отношению к пропорции циклодекстрин/биоадгезив. В этом отношении профиль высвобождения активного агента можно замедлять, включая более сильный биоадгезив, например ксантановую камедь.
ПРИМЕРЫ
Методики
Таблетки можно анализировать на твердость с помощью определителя твердости Hardness Tester (Model # РАН 01, 500N, Pharma Alliance Group). Силу в точке разлома таблетки принимали за твердость таблетки или предел прочности. Величины больше 4 кг обычно считали приемлемыми.
Исследование прочности таблеток на истирание можно проводить с помощью USP <1216> с использованием модели измерителя исследования таблеток на истирание Friability Tester FT-400. Минимум 5 таблеток взвешивали и помещали в барабан. Таблетки вращали при 25 об/мин в течение приблизительно 4 мин (100 вращений). Приемлемой считали потерю массы не более 1% от общей массы.
Тест на распадаемость можно поводить по инструкции USP <701> при 37°C с помощью устройства для распадаемости VanKel Disintegration Tester, модель 10-91 171 В, которая работает при 30 об/мин, и ванную с циркуляцией Lauda М6 Circulating Bath. Таблетки помещают в цилиндр и прикрепляют корзину к тестовому аппарату. В качестве иммерсионной жидкости используют деионизированную воду.
Испытание на растворимость проводят, следуя указаниям USP <711> с помощью системы растворения Distek 2100 В при 37°C. Для каждой лекарственной формы исследуют растворимость 13 таблеток в 450 мл иммерсионной среды. Для высокоэффективной жидкостной хроматографии (ВЭЖХ) в каждый момент времени наблюдения используют аликвоты по 100 мкл. Концентрацию лекарственного средства определяют с помощью калибровочной кривой, подсчитывая площадь пика активного фармацевтического ингредиента АФИ. Благодаря исследованиям NAT исследования нестабильности АФИ проводят в фосфатном буфере (рН 6,8), который является стандартом исследований растворимости, которые мимикрируют слюну. Предпочтительно скорость растворения определяют в цитратном буфере, рН 6,0.
ВЭЖХ
Как правило, все образцы анализировали непосредственно после приготовления/сбора для того, чтобы снизить распад АФИ. Также проводили ВЭЖХ на обратной фазе при следующих условиях.
Установка: автоматический аппарат для ввода проб WISP 712 (Waters) с WISP системой охлаждения образцов с настраиваемым детектором поглощения Waters 484MS и системой доставки мультирастворителей Waters 600Е; Eppendorf СН-30 нагреватель колонки/ТС-50 контроллер и ячейка дегазации растворителя Shodex, модель КТ-375.
Колонка ВЭЖХ: Waters Symmetry Shield RP-18, 4,6×150 мм, 3,5 мкМ. Длина волны: 300 нм. Аналитический метод: Фаза А: вода 95%/ ацетонитрил 5%, 0,1% TFA; Фаза В: ацетонитрил 0,1% TFA; 20-35% градиент B в течение 5 мин с последующим градиентом 35-40% B в течение 15 мин. Время удержания АФИ приблизительно 12,3 мин.
Калибровочная кривая: раствор АФИ в 0,1% лимонной кислоте.
Пример 1: Лекарственные формы на основе акрила
Готовили твердые лекарственные формы для парентерального введения 1S-3S, содержащие:
Состав
В блендер вместимостью 50 мл загружали бромкриптин и Кабосил. Смесь перемешивали при 300 об/мин в течение 10 минут. В случае 1S добавляли лимонную кислоту и перемешивали в течение 15 минут. Добавляли Карбопол и перемешивали в течение 15 минут, после чего добавляли маннитол и далее перемешивали 30 минут. Смесь и стеарат магния пропускали по отдельности через сито 40 меш и затем перемешивали в течение 2 минут. Сухую гранулированную смесь формовали в таблетки (5 мм диаметр, 70-75 мг) с помощью пресса TDP при 4000 Psi (27,58 МПа).
Результаты
Пример 2: Твердые лекарственные формы на основе гидроксипропилметилцеллюлозы/поливинилпирролидона
Твердые лекарственные формы на основе гидроксипропилметилцеллюлозы/поливинилпирролидона (4S, 5S) готовили по следующей схеме:
Состав
В блендер вместимостью 50 мл загружали бромкриптин и, необязательно, Кабосил (4S). Смесь перемешивали при 300 об/мин в течение 10 минут. Добавляли поливинилпиролидон и перемешивали 15 минут, потом добавляли Бенецел® и далее перемешивали 20 минут. Затем добавляли маннитол и перемешивали смесь 30 минут. Смесь и стеарат магния пропускали отдельно через сито 40 меш и затем перемешивали 2 минуты. Сухую гранулированную смесь формовали в таблетки (5 мм диаметр, 70-75 мг) с помощью пресса TDP при 4000 Psi (27,58 МПа).
Пример 3: Цитратный буфер в тесте на растворимость
Для исследования скорости растворения лекарственной формы 6S использовали цитратный буфер вместо фосфатного буфера. Лекарственная форма 6S высвобождает 50% бромкриптина в течение первых 2 часов, после чего концентрация бромкриптина снижается. Снижение концентрации не было связано с разрушением бромкриптина.
Состав
В блендер вместимостью 50 мл загружали бромкриптин и поливинилпирролидон.
Смесь перемешивали при 300 об/мин в течение 10 минут. Добавляли Кабосил и перемешивали 15 минут, потом добавляли Бенецел® и перемешивали еще 20 минут. Затем добавляли маннитол и смесь перемешивали 30 минут. Смесь и стеарат магния пропускали через сито 40 меш и затем перемешивали 2 минуты. Сухую гранулированную смесь формовали в таблетки (5 мм диаметр, 70-75 мг) с помощью пресса TDP при 4000 Psi (27,58 МПа).
Результаты:
Растворение: иммерсионная среда: цитратный буфер рН 6,0 (см. таблицу профиль растворения)
Пример 4: Дополнительные лекарственные формы на основе гидроксипропилметилцеллюлозы (ГПМЦ/поливинилпирролидона
Готовили твердые лекарственные формы для парентерального введения (7S-10S) согласно настоящему изобретению. Лекарственная форма 7S показала хорошую стабильность в процессе экспериментов. Она высвобождала 50% лекарства после 4 часов и 70% лекарства после 6 часов, имела превосходный общий профиль высвобождения. Также данная лекарственная форма позволяла производить таблетки высокого качества, которые имели хорошие текучие свойства и гомогенность и низкую ломкость. На основе 7S, дальнейшие эксперименты использовали другие методы для того, чтобы слегка ускорить высвобождение АФИ и привести его к целевому уровню >80% через 4 часа. Однако повышенные уровни микрокристаллической целлюлозы снижали время высвобождения, и ее можно использовать для снижения времени высвобождения буккальных форм агонистов допамина. Лекарственные формы 9S и 10S увеличивали, добавляя лимонную кислоту, и они содержали стеариновую кислоту в качестве вещества, способствующего скольжению, вместо стеарата магния (для снижения распада). По сравнению с 7S в 9S было 1,4% лимонной кислоты, что приводило к ускоренному высвобождению АФИ, 86% через 3 часа и 100% через 4 часа. 10S содержала меньше ГПМЦ, чем 7S, и высвобождала 70% АФИ через 3 часа и 95% через 4 часа. Обе 9S и 10S имели хорошую стабильность, позволяли создавать превосходные таблетки и демонстрировали пониженное время распада в пределах 13-15 минут.
Лекарственные формы
В блендер вместимостью 50 мл загружали бромкриптин и поливинилпирролидон и, необязательно, лимонную кислоту (9S). Смесь перемешивали при 300 об/мин в течение 10 минут. Добавляли Эксплотаб и перемешивали 15 минут, потом добавляли ПроСолв и Бенецел® и перемешивали еще 15 минут. Затем добавляли маннитол и смесь перемешивали 30 минут. Смесь и стеарат магния пропускали через сито 40 меш и затем перемешивали 2 минуты. Сухую гранулированную смесь формовали в таблетки (5 мм диаметр, 70-75 мг) с помощью пресса TDP при 4000 Psi (27,58 МПа).
Результаты
Растворение
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 5: Лекарственные формы с «взрывным» высвобождением
В следующем эксперименте приготовили лекарственную форму 11S с повышенным уровнем лимонной кислоты, а лекарственную форму 12S дополнили усилителем проникновения (циклодекстрином). Лекарственная форма 11S (повышенное содержание лимонной кислоты 2,9% по сравнению с 9S) проявляла свойства «взрывного высвобождения». Это действие повышения уровня лимонной кислоты до такого количества явилось неожиданным результатом и ранее не описывалось для лекарственных форм с агонистами допамина.
Приготовили лекарственные формы 11S и 12S, которые содержат: (1) агонист допамина, мезилат бромкриптина; (2) гидроксипропилметилцелюлозу (ГПМЦ); (3) поливинилпирролидон (ПВП); (3) повышенный уровень лимонной кислоты и (4) необязательно, усилитель проникновения (12S). Оказалось, что лекарственные формы 11S (2,9% лимонной кислоты по сравнению с 9S) проявляют свойство «взрывного высвобождения» по сравнению с предыдущими лекарственными формами. Общее время высвобождения оставалось похожим на 9S (приблизительно 4 часа), однако на ранних временных точках высвобождалось большее количество 11S (до 36% высвобождалось за первые 30 минут и затем 46% за 60 минут). Для следующей лекарственной формы также использовали повышенный уровень лимонной кислоты, 12S, а также усилитель проникновения из семейства циклодекстринов. Лекарственная форма 12S (2,5% лимонной кислоты и 14% циклодекстрина по сравнению с 9S) оказывала даже более выраженное действие «взрывного высвобождения». В частности 40% бромкриптина высвобождалось за первые 30 минут, а затем скорость высвобождения снижалась (52% через час, 71% через 2 часа, 91% через 3 часа и в итоге полное высвобождение наблюдали через 4 часа).
Лекарственные формы
В блендер вместимостью 50 мл загружали бромкриптин, лимонную кислоту и поливинилпирролидон. Смесь перемешивали при 300 об/мин в течение 10 минут. Добавляли Бенецел® и перемешивали 10 минут. Добавляли Эксплотаб и перемешивали 10 минут, после чего добавляли ПроСолв (1750 мг) и перемешивали 15 минут. В случае лекарственной формы 12S на грануляторе растирали Кавитрон, добавляли его в смесь и перемешивали 20 минут. Затем добавляли маннитол и перемешивали 30 минут. Смесь и отдельно стеарат магния пропускали через сито 40 меш и затем смешивали 2 минуты. Сухую гранулированную смесь формовали в таблетки (5 мм диаметр, 70-75 мг) с помощью пресса TDP при 4000 Psi (27,58 МПа).
Результаты
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 6: Комбинированная таблетка, содержащая агонист допамина и агент, снижающий уровень холестерола
К лекарственной форме 11S добавляли агент, снижающий уровень холестерола, симвастатин для получения лекарственной формы 20S. Оказалось, что лекарственная форма 11S может включать дополнительный агент из семейства статинов без существенного изменения профиля высвобождения агониста допамина.
Добавление к смеси симвастатина существенно снижает текучесть благодаря тому, что симвастатин представляет собой рыхлый порошок, способный забирать статический заряд. Также симвастатин плохо растворяется в воде. В тесте на растворимость измеримая концентрация симвастатина достигает только 6%, что вероятнее всего связано с его плохой растворимостью в воде. Это можно преодолеть, добавляя лиофильные растворяющие агенты. Более того, симвастатин существенно не изменяет профиль высвобождения бромкриптина.
Лекарственная форма 20S
Способ приготовления лекарственной формы был аналогичен способу приготовления 11S с дополнительным этапом после добавления симвастатина в смесь и перемешивания при 300 об/мин в течение 10 минут.
Результаты
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 7: Комбинированная таблетка, содержащая агонист допамина и антигипертензивный агент или агент, понижающий уровень холестерола
Антигипертензивный агент рамиприл добавляли в лекарственную форму 11S для создания лекарственной формы 21S. Оказалось, что в лекарственную форму 11S можно включить дополнительный агент из семейства ингибиторов ангиотензинконвертирующих ферментов без существенного изменения профиля высвобождения агониста допамина.
Лекарственная форма 21S
Процесс приготовления лекарственной формы был аналогичен процессу приготовления 11S с дополнительным этапом после добавления рамиприла в смесь и перемешивания при 300 об/мин в течение 10 минут.
Результаты
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 8: Комбинированная таблетка, содержащая агонист рецептора допамина D2 и агонист рецептора допамина D1
Лекарственная форма 22S представляла собой комбинацию агониста рецептора допамина D1, бромкриптина и агониста рецептора допамина D2, SKF-38393, на основе базовой лекарственной формы 11S с двумя активными агентами. Профили высвобождения для каждого агониста допамина были очень похожи, а профиль растворения был очень похож на лекарственную форму 11S. Проводили небольшое исследование по стабильности SKF-38393. Обнаружили, что не происходит разложения в воде с цитратным буфером в течение 12 часов. И наоборот, в спиртовом растворе АФИ быстро распадается, >5% АФИ теряется уже в течение первого часа. Несмотря на большое количество SKF-38393, лекарственная форма имеет хорошую текучесть и позволяет создавать таблетки хорошего качества.
Лекарственная форма 22S
Способ приготовления лекарственной формы был аналогичен способу приготовления 11S с дополнительным этапом после добавления SKF-38393 в смесь и перемешивания при 300 об/мин в течение 10 минут.
Результаты
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 9: Лекарственные формы с ускоренным «взрывным высвобождением»
На основе результатов, полученных для лекарственных форм 11S и 12S, приготовили следующую серию лекарственных форм (23S, 24S) для того, чтобы еще уменьшить время распада таблеток и время растворения лекарственного средства агониста допамина. Это удалось эффективно выполнить, заменив разрыхлитель Эксплотаб® на Фармабурст®, что снизило время распада с 13-15 минут до 5 минут и ускорило растворение до 100% за 1,0-2,0 часа. Лекарственные формы 23S и 24S имели хорошие текучие свойства и позволили создать очень прочные твердые таблетки с быстрым распадом, при этом 24S распадалась немного быстрее, чем 23S. Эти данные согласуются с нашими предыдущими наблюдениями, что Кавитрон замедляет время распада (например, 11S по сравнению с 12S).
Лекарственные формы
В блендер вместимостью 50 мл загружали бромкриптин, лимонную кислоту и поливинилпирролидон. Смесь перемешивали при 300 об/мин в течение 10 минут. Добавляли Бенецел® и перемешивали 10 минут. В случае лекарственной формы 24S добавляли Кавитрон в смесь и перемешивали 10 минут. Затем добавляли Фармабурст и перемешивали 30 минут. Смесь и отдельно стеарат магния пропускали через сито 40 меш и затем смешивали 2 минуты. Сухую гранулированную смесь формовали в таблетки (5 мм диаметр, 70-75 мг) с помощью пресса TDP при 4000 Psi (27,58 МПа).
Результаты
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 10: Таблетки с двумя слоями для профилей растворения агонистов допамина с пиком-плато
Для обеспечения среднего между 23S и 24S профиля высвобождения разработали таблетки с двумя слоями 30DL. Таблетки создали с помощью резного пресса. Таблетки обладали ожидаемыми свойствами высвобождения. Эти эксперименты подтверждают возможность применения таблеток с двойным слоем для точной модификации других лекарственных форм для того, чтобы достигнуть желаемых профилей высвобождения агониста допамина (и изменить их, ускоряя время достижения пика или замедляя время плато).
Таблетки (70 мг) диаметром 5 мм отштамповали одну за другой на рабочей поверхности резного пресса 20 Ton Carver press с использованием заранее взвешенных количеств двух компонентов А и В (по 35 мг) при давлении в 2000 Psi. Перед применением силы давления лекарственную смесь заранее спрессовали ручным методом. Каждую таблетку проверяли на наличие видимых неровностей и качество наружного слоя, добавив желтый краситель к компоненту А.
Лекарственные формы компонентов А и В
В блендер вместимостью 50 мл загружали 250 мг бромкриптина, лимонную кислоту и поливинилпирролидон. Смесь перемешивали при 300 об/мин в течение 10 минут. Добавляли Бенецел® и дополнительно А1 Lake пигмент и перемешивали 10 минут. В случае компонента В, в смесь добавляли Кавитрон (250 мг) и перемешивали 10 минут. Затем добавляли Фармабурст и перемешивали 30 минут. Смесь и отдельно стеарат магния пропускали через сито 40 меш и затем смешивали 2 минуты.
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 11: Эргокриптин, включенный в лекарственную форму 12S
В случае лекарственной формы 25S, бромкриптин в лекарственной форме 12S заменяли на агонист допамина эргокриптин. Профили высвобождения каждого агониста допамина были очень похожи, и их профили растворения также были похожи на профиль растворения лекарственной формы 12S.
Лекарственная форма 25S
Лекарственную форму готовили тем же способом, что и лекарственную форму 12S, но вместо бромкриптина добавляли эргокриптин.
Результаты
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 12: Лекарственные формы агонистов допамина в виде геля
Разработали серию гелевых лекарственных форм (26S, Гель 31 и Гель 34) для обеспечения введения агонистов допамина с хорошей стабильностью (сроком хранения) через слизистую оболочку, трансдермально и/или подкожно. Поскольку лекарственные формы, основанные на акриле, разрушают агонисты допамина, особенно те, которые относятся к семеству спорыньевых, то разработали различные лекарственные формы, в которых не используют акриловые компоненты и которые обеспечивают приемлемый фармакокинетический профиль с пиком и плато.
Трасдермальная, чресслизистая лекарственная форма с бромкриптином 26S была создана на безводной, содержащей глицерол композиции. Пропиленгликоль (ПГ) обеспечивает высокую растворимость агонистов допамина, например бромкриптина, и также является признанным усилителем трансдермального проникновения и соответствует стандартам FDA и cGMP. Согласно патенту США 4366145, композиции с бромкриптином, содержащие высокий уровень глицерола и полиэтиленгликоля, обладают высокой стабильностью. Дополнительно для увеличения стабильности агонистов допамина АФИ добавляли лимонную кислоту. В итоге, для контроля вязкости использовали диоксид кремния (неорганический материал), поскольку маловероятно, что он влияет на стабильность по сравнению с загустителями, основанными на акриловых производных, и даже ПГ, который, как доказано, ускоряет разрушение агонистов допамина, родственных спорынье.
Для биоадгезивных систем в таблетированные лекарственные формы добавляли смесь гидроксипропилцеллюлозы (Бенецел®) и Кроссповидона в пропорции 2:1 для создания геля с хорошей стабильностью АФИ. Краткосрочное исследование стабильности показало, что за 72 часа хранения геля в холодильнике при 4°С не происходит разложения.
На базе лекарственной формы 26S, основанной на безводной системе, содержащей глицерол и пропиленгликоль, разработали гелевую лекарственную форму Гель 31, в которой вязкость контролировали добавлением диоксида кремния. После предварительных исследований в гелевую форму добавляли биоадгезив ГМПЦ/ПВП. Коллоидная кремнекислота способствует эффективному контролю стабильности, что позволяет создать гель, который имеет хорошую гомогенность после недели хранения. Дополнительно не наблюдали разложения бромкриптина после 3 дней хранения при 5°C. По сравнению с гелевой лекарственной формой 26S требовалось меньше диоксида кремния для достижения аналогичного загущающего действия благодаря добавлению биоадгезивного компонента. Гелевая лекарственная форма Гель 34 была такой же, как 26S, однако она содержала 3% активного агента вместо 1%.
Эти лекарственные формы были стабильны и не содержали ни одного акрилового элемента, которые, как известно, ускоряют деградацию агонистов допамина, основанных на спорынье. Можно регулировать вязкость и биодоступность этих гелей с помощью способов, которые позволяют сохранять профиль биодоступности и поэтому увеличивать уровень всасывания активных агентов из лекарственной формы.
Эти лекарственные формы можно применять трасдермально, подкожно или через слизистую для того, чтобы влиять на парентеральное всасывание.
Лекарственные формы
В случае лекарственных форм 26S и Гель 34 в 100 мл бутыли с завинчивающейся крышкой лимонную кислоту в пропиленгликоле диспергировали ультразвуком 10 мин, что приводило к получению прозрачного бесцветного раствора. Добавляли бромкриптин и диспергировали ультразвуком 10 мин, получали слегка прозрачную жидкость. Добавляли глицерол и обрабатывали смесь ультразвуком еще 10 минут. Постепенно добавляли в раствор диоксид кремния при перемешивании и действии ультразвука. Сначала после добавления получали очень вязкую гетерогенную суспензию, которая постепенно светлела.
В случае лекарственной формы Гель 31, в глицерол добавляли Бенецел® и Повидон 29/32 и полученную суспензию гомогенизировали с помощью гомогенизатора Политрона при 5000 об/мин 5 минут. Затем полученную суспензию пропускали через сито 40 меш для того, чтобы убедиться в отсутствии конгломератов. В смесь полипропилена добавляли молочную кремообразную суспензию (стоковый раствор, слегка разделившийся после нескольких дней хранения в холодильнике) и диспергировали ультразвуком 5 минут. Постепенно добавляли 6 г диоксида кремния в раствор (2×3 г) при постоянном перемешивании и действии ультразвука. Сначала после добавления получили очень вязкую гетерогенную суспензию, которая постепенно светлела. По сравнению с гелевой лекарственной формой 26S, для получения аналогичного загущающего действия требовалось меньше диоксида кремния.
Из-за большого количества выделяемых пузырьков воздуха после выдерживания гелевых лекарственных форм в течение 24 часов в холодильнике при 5°C, из конечной лекарственной формы откачивали газ в вакуумных сушилках в течение 6 часов и получали в результате слегка желтый прозрачный гель. Данный гель упаковали в круглую бутылку с вакуумным насосом.
Пример 13: Влияние уровня биоадгезивной системы в таблетке на профили растворения и распада
В данном примере изменяли количество биоадгезивов ГПМЦ/ПВП в лекарственной форме 23S. Лекарственная форма 27S содержит на 20% больше ГПМЦ/ПВП, чем 23S. По сравнению с 23S, 27S имеет существенно замедленное высвобождение, 60% лекарства высвобождается в 1 час и 94% за 2 часа. Использование повышенных уровней биоадгезивных компонентов, по-видимому, является неприемлемой стратегией обеспечения быстрого пика агониста допамина с последующим замедленным высвобождением.
Лекарственная форма 28F, с другой стороны, имеет на 50% меньше ГПМЦ/ПВП по сравнению с лекарственной формой 23S. Однако профиль высвобождения был очень похож на 23S. Учитывая профиль высвобождения 27S, полученные результаты указывают на то, что пропорция биоадгезивных компонентов для 23S располагается поблизости от точки перехода к медленному начальному высвобождению агониста допамина при повышенных уровнях биоадгезива (т.е. снижение первоначального быстрого пика растворения).
Лекарственная форма 27S
Лекарственные формы готовили таким же образом, что и 23S.
Результаты
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 14: Использование ксантановой камеди в качестве биоадгезивной системы
Лекарственная форма 29S содержала ксантановую камедь вместо ГПМЦ в таком же количестве, что и в лекарственной форме 23S, для исследования влияния таких камедей на распад таблетки и профили растворения агониста допамина. Данное изменение привело к значительно замедленному высвобождению агониста допамина.
Поэтому ксантановую камедь можно считать только альтернативой системе ГПМЦ/ПВП с пониженным уровнем или в комбинации пониженных уровней с «супер быстро» распадающейся таблеткой (см. ниже лекарственную форму 40SUF) для получения кривой растворения с пиком и плато.
Лекарственная форма
Лекарственную форму готовили аналогично лекарственной форме 23S, в качестве биоадгезива использовали ксантановую камедь, а не Бенецел®.
Результаты
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 15: Повышенное содержание активного агента по отношению к биоадгезиву
Исследовали влияние увеличения пропорции агониста допамина по отношению к биоадгезиву. Готовили композиции 32F и 33S по аналогии с 23S и 24S, соответственно, но соотношение между агонистом допамина и системой биоадгезивов составляло 2,5/10 по сравнению с 1/10. Как и ожидалось, лекарственная форма 32F имела профиль высвобождения, похожий на 23S. Несмотря на то что новая композиция 33S содержала в три раза больше бромкриптина, чем 24S, она имела существенно отличающийся от 24S профиль высвобождения. На самом деле профиль высвобождения 33S был больше похож на 23S, чем на 24S, все лекарство высвобождалось в течение 120 минут. Возможно, увеличение пропорции активный агент: биоадгезивная система с добавлением циклодекстрина переполняет резервуар лекарственного средства, что приводит к тому, что вначале высвобождается больше лекарственного средства. Данная лекарственная форма может быть пригодна для доставки большой порции лекарства, за которой следует медленное высвобождение с добавлением усилителя проникновения, например циклодекстрина. Можно регулировать скорость доставки лекарства, увеличивая количество лекарственного средства по отношению к циклодекстрину в лекарственной форме, чтобы изначальное высвобождение не являлось фактором его взаимодействия с циклодекстрином. Уменьшая процент взаимодействующего лекарственного средства с циклодекстрином, можно ускорить начальное высвобождение лекарственного средства из таблетки.
Лекарственные формы
Лекарственные формы готовили, как описано выше для форм 23S и 24S.
Результаты
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 16: Замена лимонной кислоты на аскорбиновую кислоту в таблетированных лекарственных формах
Лекарственная форма 35F представляла собой аналог 23S при наличии 1 мг активного агента в таблетке, и аскорбиновая кислота была замещена лимонной.
Лекарственная форма 36S представляла собой аналог 23S при наличии 3 мг активного агента в таблетке, и аскорбиновая кислота была замещена лимонной.
Лекарственная форма 37F представляла собой аналог 24S при наличии 1 мг активного агента в таблетке, и аскорбиновая кислота была замещена лимонной.
Лекарственная форма 38S представляла собой аналог 24S при наличии 3 мг активного агента в таблетке, и аскорбиновая кислота была замещена лимонной.
Во всех случаях замена аскорбиновой кислоты на лимонную кислоту приводила к замедленному высвобождению лекарственного средства и распаду таблетки, и это можно использовать для осуществления этого без снижения стабильности таблетки.
Лекарственные формы
Лекарственные формы растворяли, как описано для лекарственных форм 23S и 24S.
Результаты.
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 17: Очень быстро распадающиеся таблетки
Очень быстро распадающуюся лекарственную форму (40SUF) готовили с помощью разрыхлителя шипучего типа. В лекарственной форме 40SUF удвоили уровень биоадгезивной системы (до 25% общего ГМПЦ/ПВП). Эта лекарственная форма привела к созданию быстро распадающихся таблеток (4 минуты) с высоким желательным «взрывообразным» высвобождением и практически линейным «хвостом» более медленного высвобождения.
Лекарственная форма 40SUF
Указанную лекарственную форму готовили аналогично лекарственной форме 23S с добавлением Эфферсоды параллельно с добавлением лимонной кислоты.
Результаты
Растворимость
Иммерсионная среда: цитратный буфер, рН 6,0
Пример 18: Исследования биодоступности in vivo твердых лекарственных форм для парентерального введения, содержащих агонисты допамина
Лекарственные формы для парентерального введения согласно настоящему изобретению вводили сирийским хомякам для того, чтобы продемонстрировать in vivo биодоступность агонистов допамина. Большой «карман» для хранения еды у сирийских хомяков является идеальной биологической тканью для исследования слизистого транспорта веществ и лекарственных форм. Сирийские хомячки также имеют дермальную ткань, которую можно использовать для исследования трансдермального транспорта лекарственных средств. Фармацевтические препараты агонистов допамина согласно настоящему изобретению вводили анестезированным сирийским хомячкам (n=2-9 в каждой группе). Образцы крови забирали непосредственно до или точно спустя 30, 60, 90, 120, 180 и дополнительно 240 и 300 минут после введения лекарства и определяли плазменные уровни бромкриптина, агониста допамина в этих лекарственных формах. Из плазмы выделяли бромкриптин и образцы анализировали против стандартов с помощью ВЭЖХ. Данные по биодоступности представлены в форме % от Cmax.
Способ выделения бромкриптина из плазмы
250 мкл плазмы смешивали с 125 мкл 0,5 М NHCl буфера (РН 9.2) и 900 мкл гексана/1-бутанола (5/1). Смесь перемешивали встряхиванием (3 минуты) и центрифугировали (1000×g, 3 минуты). Супернатант переносили в новые пробирки, добавляли в них по 250 мкл 0,025 М H2SO4. Смесь перемешивали встряхиванием (3 минуты) и снова центрифугировали (1000×g, 3 минуты). После отбора верхней органической фазы добавляли 500 мкл дихлорметана и 150 мкл буфера NHCl, затем смесь вортексировали и центрифугировали. Верхнюю водную фазу удаляли и нижний слой выпаривали при 55°C. После выпаривания остатки хранили при -70°C до анализа с помощью ВЭЖХ.
Как показано ниже лекарственные формы согласно настоящему изобретению давали кривые биодоступности с пиком и плато на животной модели. Приведенные примеры также показали, что можно изменять, предсказывать, форму кривой биодоступности, изменяя специфические компоненты лекарственной формы.
Анализ ВЭЖХ
Экстракт растворяли в 50 мкл 50% этанола. Для анализа 10-15 мкл наносили на колонку ВЭЖХ. Условия: подвижная фаза: 0,1 М дигидрофосфат калия (рН 7,5): ацетонитрил (1:1). Скорость потока: 0,4 мл/мин. Колонка: C183ul, 100×2 мм.
Детектор: UV при 300 нм.
Время запуска: 2× время оборота бромкриптина.
Анализ данных по биодоступности
Биодоступность представлена в виде % of Cmax. Данные представляют наилучшую из полученных кривых для пролеченных групп.
Результаты теста на биодоступность лекарственных форм 23S и 24S и комбинации 30DL
Биодоступность лекарственной формы 23S характеризуется пиком уровня агониста допамина в плазме в течение 30 минут после введения лекарства через слизистую оболочку с последующим быстрым снижением его уровня. Если для приготовления лекарственной формы 24S для усиления биоадгезии и проникновения в лекарственную форму 23S добавляли циклодекстрин, биодоступность стала характеризоваться пиком уровня агониста допамина в плазме через 30 минут после введения через слизистую и плато уровня агониста допамина в течение 2,5-4,5 часов при приблизительно >50% Cmax, поэтому кривая биодоступности имела пик-плато с Cmax в 2-3 раза больше по сравнению с лекарственной формой 23S.
Лекарственная форма 30DL представляла собой таблетку, в которой половину составляла лекарственная форма 23S и половину 24S. Такой «гибрид» обладал биодоступностью, которая была очень похожа на 23S, вероятно, из-за того, что соотношение циклодекстрин: агонист допамина было слишком низким для обеспечения биоадгезии и проникновения в ткань агониста допамина.
Результаты анализа биодоступности лекарственных форм 32F и 33S
Биодоступность лекарственной формы 32F характеризовалась пиком уровня агониста допамина в плазме в течение 30-90 минут после введения через слизистую с последующим плато уровня агониста допамина в плазме приблизительно при ≥50% Cmax в течение периода до 3,5 часов. Эта лекарственная форма имела кривую биодоступности, которая представляла собой среднее между 23S и 24S, как и ожидалось исходя из профилей растворения in vitro и свойств компонентов этих лекарственных форма из-за соотношения агонист допамина: биоадгезив (32F vs. 23S).
Лекарственная форма 33S (лекарственная форма 32F с циклодекстрином) имела кривую биодоступности с пиком уровня агониста допамина в плазме в течение 60-90 минут после введения через слизистую и последующим плато уровня агониста допамина приблизительно при ≥50% Cmax в течение 3,5 часов после Tmax. Лекарственная форма 33S также имела Cmax в 2-3 раза больше, чем лекарственная форма 32F. Такие результаты также соответствовали действию включения циклодекстрина в лекарственную форму, т.к. он откладывает по времени растворение таблетки и усиливает проникновение активного агента в ткани in vivo.
Данные представлены в % Cmax
Результаты анализа биодоступности лекарственных форм 35F и 40SUF
Биодоступность лекарственной формы 35F характеризовалась пиком уровня агониста допамина в плазме через 180 минут после введения через слизистую и плато приблизительно при ≥50% Cmax в течение следующих 60 минут со снижением уровня агониста допамина в плазме. Лекарственная форма 35F включает а) увеличение соотношения активного агента и биоадгезива; и б) замену лимонной кислоты на аскорбиновую кислоту из лекарственной формы 23S. Как известно, каждое из таких действий с лекарственной формой 23S замедляет растворение активного агента in vitro, как описано выше в этой заявке, поэтому кривая биодоступности 35F соответствовала его растворимости in vitro. Такие изменения лекарственной формы 35F можно вносить для того, что уравновешивать добавки в лекарственную форму 23S, которые могут слишком ускорять высвобождение и всасывание активного агента in vivo, и биодоступность была бы приблизительно в два раза больше, чем у лекарственной формы 23S.
Лекарственная форма 40SUF характеризовалась пиком уровня агониста допамина в плазме через 30 минут после введения через слизистую и резким снижением уровня агониста допамина в плазме вскоре после этого. Биодоступность лекарственной формы 40SUF была приблизительно в 3-5 раз выше по сравнению с лекарственной формой 23S. Лекарственную форму 40SUF можно применять для снижения in vivo Tmax лекарственных форм, которые имеют временную задержку в достижении Tmax, но которые предпочтительны для формирования кривой биодоступности с пиком-плато и терапевтического действия агониста допамина.
Данные представлены в % Cmax
Пример 19: Лекарственная форма Гель 34 для трансмукозального, трасдермального и подкожного способа доставки
Биодоступность лекарственной формы Гель 34 характеризовалась пиком уровня агониста допамина в течение 60-90 минут после парентерального введения (через слизистую, трасдермально или подкожно) с последующим плато уровня в плазме приблизительно при ≥50% Cmax в течение периода 1,5-3 часов после введения. Данная лекарственная форма демонстрирует профиль биодоступности пик-плато вне зависимости от того, вводится ли она через слизистую, трасдермально или подкожно. Более того, данная лекарственная форма бромкриптина также приводит к высоким желательным и неожиданно действенным улучшениям метаболического заболевания при парентеральном введении в подходящее время по сравнению с традиционной лекарственной формой бромкриптина, которую раньше использовали для лечения нарушений метаболизма на той же животной модельной системе (см. Примеры 30-31). Предыдущая лекарственная форма не подходила для фармацевтического применения по нескольким причинам, включая исключительно низкую стабильность и нежелательные побочные эффекты в месте введения, делая его терапевтическое применение невозможным.
Данные представлены в % Cmax
Пример 20: Уровни бромкриптина в крови после парентерального введения лекарственных форм моделям животных с ожирением, непереносимостью глюкозы и резистентностью к инсулину
Уровень в плазме бромкриптина, введенного интраперитонеально, в этаноле или воде в соотношении 30:70 в дозе, которая приводит к снижению состояния устойчивости к инсулину у сирийских хомячков (5 мг/кг), сравнивали с плазменным уровнем бромкриптина на той же животной модели после парентерального введения (через слизистую, трасдермально или подкожно) лекарственных форм согласно настоящему изобретению, описанных выше. Чресслизистное, кожное или подкожное введение 10-20 мг/кг бромкриптина согласно настоящему изобретению сирийским хомячкам, в частности лекарственных форм 32F, 33S, 26S, гель 34, 35F и 40SUF, приводило к получению такого же уровня бромкриптина в крови, как и при интраперитонеальном введении лекарственных форм (в этаноле/воде) при дозе 5 мг/кг. Поэтому можно доставлять лекарственные формы согласно настоящему изобретению парентерально для достижения терапевтически эффективной дозы агониста допамина, которая необходима для снижения метаболических нарушений на животной модели метаболического заболевания.
Пример 21: Влияние формы Гель 34 in vivo на прибавку в массе, уровень инсулина в плазме, чувствительность к инсулину и кровяное давление на модели 16-недельных крыс SHR
16-недельным самцам крыс со спонтанной гипертензией (SHR) ежедневно вводили Гель 34 с 30% этанолом в качестве носителя посредством парентеральной инъекции 10 мг/кг массы тела (n=100) или 30% этанолом в качестве носителя (n=10) в течение 7 дней при проявлении дневной локомоторной активности животных (при изначально выключенном свете). Измеряли кровяное давление, уровень глюкозы и инсулина в плазме, определяли чувствительность к инсулину на основе уровней глюкозы и инсулина в плазме. По сравнению с контролем обработка гелевой формой 34 приводила к уменьшению устойчивости к инсулину (HOMA-IR) на 12-2,65 (Фигура 1), снижению гиперинсулинемии (2,4-0,5 нг/мл) (Фигура 2), уменьшению систолического и диастолического давления крови (на 25 мм рт.ст.) (Фигура 3), снижению уровня эндотелина-1 в плазме на 47% (Фигура 6), а также масса животных увеличивалась с 50 г на 18 г в контрольной группе (Фигура 4 и 5). Данные результаты показали, что парентеральное введение формы Гель 34 в заранее определенное время дня приводит к получению кривой биодоступности бромкриптина с пиком его уровня в плазме в течение 90 минут с последующим уровнем >50% Cmax в течение 60-90 минут и приводит также к улучшению (сокращению) метаболических расстройств на модели SHR метаболического заболевания.
По сравнению с лекарственной формой, которая не имеет идеальной кривой биодоступности с пиком и плато, как описано в настоящей заявке, при равной дозе парентерально введенная гелевая форма 34 оказывает большее действие на гиперинсулинемию, устойчивость к инсулину и массу тела, если ее вводят в то же время тому же модельному животному с метаболическим заболеванием (Diabetes 57 Suppl 1, А176, 2008). Спонтанное снижение различных факторов риска сердечно-сосудистых заболеваний, таких как устойчивость к инсулину, кровяное давление, набор веса и уровень эндотелина-1, на модели крыс SHR можно осуществить при регулярном парентеральном введении лекарственной формы агониста допамина, которая имеет кривую биодоступности с пиком-плато. Эти результаты подтверждают важность такой терапии для лечения (облегчения) сердечно-сосудистых заболеваний.
Пример 22: Стабильность лекарственных форм для парентерального введения, содержащих бромкриптин
Лекарственные формы с бромкриптином помещали в контейнеры из полиэтилена низкой плотности, снабженные поглощающим влагу диссекантом, и хранили при 50°C и относительной влажности 60% в течение 5 дней. Эти лекарственные формы подготовили для ВЭЖХ и сравнивали содержание бромкриптина и бромкриптинина (главный продукт разложения бромкриптина) в лекарственных формах и стандартных препаратах бромкриптина и бромкриптинина.
Уровень бромкриптина во всех лекарственных формах 24S, 32F и 33S был меньше 2% после выдерживания при 50°C/60% отн. вл. в течение 5 дней. При 4°C из этих лекарственных форм образовывалось менее 1% бромкриптинина. Бромкриптин очень неустойчив под действием тепла и влаги, обычно эти условия вызывают его распад, в результате чего образуется большое количество бромкриптинина. Результаты исследования стабильности лекарственных форм бромкриптинина при 50°C и 60% отн. вл. показывают, что эти лекарственные формы потенциально могут оставаться стабильными в течение долгого периода времени при комнатной температуре (25°C) и влажности.
Обсуждение таблетированных лекарственных форм
Действие нескольких наполнителей на профиль растворения буккальной лекарственной формы агониста допамина можно определить, сравнивая кривые растворимости различных таблеток лекарственных форм (буккальных, подъязычных, слизистых) 7S-24S, приведенных ниже. Во-первых, для достижения in vivo фармакокинетического профиля с быстрым (коротким) Tmax (1-90 минут) с последующим плато на уровне 50%-100% Cmax (60-360 минут) (желательный профиль пик-плато) лекарственная форма способствует быстрому растворению (уклон А) (и всасыванию) с последующим более медленным, но постоянным растворением (уклон <А) (и всасыванием) (желаемый профиль высвобождения). Наполнители Просолв (микроцеллюлозный наполнитель) и Бенецел® (биоадгезив, матрикс высвобождения агониста допамина) замедляют скорость растворения (раннюю и позднюю), т.к. содержание их в таблетке выше. С другой стороны, наполнители лимонная кислота и Фармабурст ускоряют раннюю и общую скорость растворения агониста допамина, соответственно. В этих условиях мы продемонстрировали, что добавление лимонной кислоты и снижение уровня Бенецела в лекарственной форме 7S, как в форме 9S, увеличивает общую скорость растворения агониста допамина, при этом сохраняется желательное раннее быстрое растворение, за которым следует медленное постоянное растворение. Более того, если повысить уровень Просолва в таблетке, общая скорость растворения заметно снизится (лекарственная форма 8S). Если в лекарственную форму 9S добавляют циклодекстрин, как и в 10S, можно далее улучшить желаемый профиль высвобождения при усилении характеристик всасывания лекарственной формы. Если далее повышать уровень лимонной кислоты в таблетке 9S, как в лекарственно форме 11S, то раннее «взрывное» высвобождение лекарственной формы заметно ускоряется: приблизительно 40% высвобождается за первые 30 минут, за этим следует замедленное, но постоянное высвобождение в течение следующих 120 минут. Этот желаемый профиль высвобождения далее улучшают, добавляя циклодекстрин, как и в 12S, который также усиливает всасывание лекарственной формы. Если включать разрыхлитель Эксплотаб вместо Фармабурста, время дезинтеграции уменьшается (от 15 до 5 минут). Это является желаемой характеристикой для буккального/подъязычного/слизистого введения таблетки, что способствует соблюдению режима приема лекарственных средств пациентом. Также применение Фармабурста ускоряет общее растворение лекарственной формы. Следует понимать, что можно получить точный профиль высвобождения, который соответствует общим характеристикам раннего быстрого высвобождения, за которым следует постоянное высвобождение агониста допамина, аккуратно подбирая те наполнители, которые влияют и регулируют кинетику высвобождения (ранне-быстрое или вторичное постоянное медленное высвобождение), как описано выше. Наполнители Просолв (микроцеллюлозный наполнитель) и Бенецел® (биоадгезив, матрикс высвобождения агониста допамина) замедляют скорость распада (ранную и позднюю), т.к. их уровень в таблетке повышен. С другой стороны наполнители лимонная кислота и Фармабурс ускоряют раннюю и общую скорость распада агониста допамина, соответственно. Лекарственные формы 11S и 12S имеют желаемые профили высвобождения лекарственной формы. Более того, было показано, что эта лекарственная форма имеет очень похожий профиль растворения различных агонистов допамина даже при наличии спонтанных комбинаций агонистов допамина, например агонистов рецептора допамина D1 и D2, в одной таблетке лекарственной формы. Также в лекарственную форму агониста допамина можно добавлять другие агенты, которые способны лечить нарушения обмена веществ. Такие добавки могут требовать или не требовать коррекции основы лекарственной формы для улучшения или ускорения профиля высвобождения агониста допамина с помощью методов, описанных ниже.
Хотя лекарственные формы 23S и 24S отличаются от форм 11S и 12S, они также имеют желаемые профили высвобождения. По сравнению с лекарственными формами 11S и 12S, лекарственные формы 23S и 24S, соответственно, обладают преимуществом уменьшенного времени распада, что выражается в повышенной биодоступности активного агента для поглощающей поверхности в течение предпочтительного времени введения (например, наружный слизистый слой или клеточная мембрана) и поэтому повышенной биодоступности в это время. Более быстрый распад должен также способствовать соблюдению пациентом режима приема лекарственного препарата. Более того, следует понимать, что изменяя пропорцию Эксплотаб: Фармабурст, так же как и регулирование уровня Бенецела® и Просолва в таблетке, можно добиться промежуточного между лекарственными формами 11S/12S и 23S/24S профиля высвобождения агониста допамина. Такие гибридные лекарственные формы способствуют точной «настройке» лекарственной формы агониста допамина для получения желаемого профиля высвобождения.
Дальнейшие исследования, проведенные с применением лекарственных форм 23S и 24S, показали, что уровень биоадгезива в таблетке лекарственной формы оптимизируется на максимальном уровне биоадгезива для поддержания биоадгезии активного агента, который по-прежнему способствует быстрому «взрывному» растворению активного агента. Повышение данного уровня (% от общей массы таблетки) приводит к уменьшению времени растворения активного агента, в то время как снижение уровня не влияет на время растворения. Поэтому относительные количества биоадгезивного агента, активного агента и других компонентов лекарственных форм 23S и 24S оптимизируют для получения желаемого профиля биодоступности пик-плато и адгезии на слизистой и для ускорения всасывания тканями. Также можно показать, что повышение уровня активного агента в таблетке 23S от 1 до 3 мг в таблетке не меняет характеристик растворения таблетки, поэтому силу дозы парентеральных агонистов допамина можно рассчитать на основе лекарственной формы 23S. Однако при повышении уровня активного агента от 1 до 3 мг в таблетке лекарственной формы 24S, профиль растворения ускоряется. В лекарственных формах (33S), которые содержат циклодекстрин или другой проникающий агент вместе с биоадгезивом, можно ускорять высвобождение активного агента, повышая его уровень по отношению к уровню циклодекстрина/биоадгезива. В очередной раз новая лекарственная форма (33S) проявляет некоторые желаемые свойства, включая быстрый распад таблетки, наличие оптимального количества биоадгезива, так, что активный агент локализуется в желательном месте всасывания (например, пониженное действие желудка на активный агент при орально-парентеральном введении лекарственных форм), быстрое высвобождение активного агента с последующим линейным медленным высвобождением активного агента (кривая растворения пик-плато) и присутствие проникающего агента для усиленного всасывания активного агента в ткани. В этом смысле профиль высвобождения активного агента в составе таблетки можно замедлять, меняя разрыхлитель с более сильными биоадгезивными свойствами, например на ксантановую камедь.
Для того чтобы больше ускорить высвобождение активного агента из лекарственной формы, создали таблетку с агентом, который образует комбинацию шипучий агент/Фармабурст, вместо Фармабурста в качестве разрыхлителя, но другие компоненты были те же, что и в лекарственной форме 23S. Эта особенная лекарственная форма уменьшала время распада таблетки и время растворения активного агента из таблетки по сравнению с лекарственной формой 23S. Поэтому следует учитывать, что можно регулировать время растворения и распада таблетки лекарственной формы с желаемым профилем биодоступности с пиком-взрывом, за которым следует медленное высвобождение активного агента, меняя разрыхлитель в лекарственной форме 11S. Если в лекарственной форме 11S заменить разрыхлитель (Эксплотаб) на Фармабурст (как в 23S), время распада и растворения уменьшится, если заменить разрыхлитель Фармабурст на комбинацию ЭфферСода/Фармабурст (как в 40SUF), время распада и растворения еще уменьшится. Другой способ ускорения распада и растворения - это добавление лимонной кислоты в лекарственные формы.
Третий способ ускорения распада и растворения - это увеличение соотношения между активным агентом и циклодекстрином в лекарственной форме. В другом случае можно добиться медленной скорости растворения активного агента лекарственной формы, добавляя больше циклодекстрина или меняя лимонную кислоту на аскорбиновую кислоту либо же меняя систему разрыхлитель/биоадгезив Бенецел®, поливинилпирролидон на ксантановую камедь.
Такие препараты имеют желаемый профиль пик-плато высвобождения агониста допамина, являются парентеральными и избегают первого этапа обмена веществ, а также первоначального связывания с рецепторами допамина в ЖКТ, снижая побочные эффекты в ЖКТ, и их можно применять для срочного введения лекарственной формы, поскольку они не являются лекарственными формами для постоянного длительного высвобождения (например, 12-24 часа). Их можно использовать для лечения метаболических заболеваний, если вводить соответствующим образом, т.к. они являются стабильными и способствуют фармацевтическому использованию. Основное открытие этих исследований заключается в том, что изменение профиля растворения определенной лекарственной формы указанным выше образом переходит в аналогичное изменение фармакокинетического профиля in vivo активного агента. Например, если ускорять или замедлять высвобождение активного агента in vitro, это также ускоряет или замедляет всасывание активного агента in vivo и т.д. В итоге приведенные выше примеры обеспечивают способы регуляции профиля растворения и времени распада лекарственной формы при сохранении желаемого быстрого взрывного пикового растворения, за которым следует медленное линейное высвобождение активного агента из лекарственной формы. Такие описанные выше способы можно использовать для регуляции параметров растворения и распада, которые, возможно, надо будет выполнить для восполнения действия дополнительных ингредиентов, например других проникающих агентов, которые используют для ускорения или замедления всасывания активного агента, что позволит изменить профиль биодоступности лекарственной формы. Более того, следует учесть, что приведенные выше примеры позволяют освоить основные принципы естественных наук и основные элементы лекарственных форм и манипуляции со специфическими компонентами лекарственной формы, которые можно использовать, чтобы разрабатывать и готовить другие лекарственные формы, которые обеспечивают желаемую растворимость активного агента in vitro и фармакокинетический профиль in vivo. Другими словами, эти примеры описывают способы регуляции времени и увеличения пика быстрого взрыва активного агента (агониста допамина), а также регуляции медленной фазы растворения лекарственной формы.
Пример 23: Таблетки, усиленные ментолом
На основе лекарственной формы 33S готовили усиленные ментолом таблетки, добавляя ментол в качестве усилителя вкуса или проникновения. Добавление ментола замедляет скорость высвобождения лекарства для агониста допамина, бромкриптина, по сравнению с лекарственной формой 33S. In vivo, однако, замедленному растворению должно противодействовать свойство ментола усиливать проникновение, что приводит к получению желаемой кривой биодоступности с пиком-плато, как для лекарственной формы 33S с дополнительным преимуществом ускоренного всасывания агониста допамина.
Лекарственные формы
В блендер вместимостью 50 мл загружали ментол и лимонную кислоту. Смесь перемешивали при 300 об/мин в течение 10 минут. Добавляли бромкриптин и перемешивали 10 минут, затем добавляли поливинилпирролидон. Добавляли Бенецел® и перемешивали 10 минут. Добавляли Кавитрон и перемешивали 10 минут. Затем добавляли Фармабурст и перемешивали 30 минут. Смесь и отдельно стеарат магния пропускали через сито 40 меш и затем смешивали в течение 2 минут. Сухую гранулированную смесь формовали в таблетки (5 мм диаметром, 70-75 мг) с помощью пресса TDP при 4000 Psi (27,58 МПа).
Результаты
Растворимость
Иммерсионная среда: вода
Примеры чресслизистых пленочных лекарственных форм
Пример 24: Чресслизистые пленочные лекарственные формы на основе поливинилпирролидона с формой гидроксипропилцеллюлозы, растворимой в этаноле (KLUCEL® LF)
Готовили пленки для чресслизистого применения агонистов допамина с сополимерами поливинилпирролидонов. Для того чтобы усилить биоадгезивные свойства пленки использовали KLUCEL® LF. Чресслизистые пленочные лекарственные формы, основанные на поливинилпирролидоне, с KLICEL® LF (Пленка 41 и Пленка 42) готовили по следующей схеме:
Состав
Основную композицию готовили, добавляя Коллидон 90F, Коллидон VA64 и ПЭГ400 в этанол в градуированную бутылку Пирекс с завинчивающейся крышкой вместимостью 2 л. Ингредиенты смешивали на медленном роллере Stovall при средней скорости в течение 24 часов при комнатной температуре. В результате процедуры образовывался прозрачный гомогенный вязкий раствор, который хранили при 4°С в виде стокового раствора.
В основную композицию добавляли KLUCEL® в градуированную бутыль Пирекс с завинчивающейся крышкой вместимостью 200 мл. В случае Пленки 42 также добавляли глицерол и циклодекстрин. Ингредиенты смешивали на медленном роллере Stovall при средней скорости в течение 24 часов при комнатной температуре. В результате образовывался прозрачный гомогенный вязкий раствор, который хранили при 4°C в виде стокового раствора.
Итоговую лекарственную форму готовили, растворяя лимонную кислоту в этаноле при легком нагревании и обработке ультразвуком. В раствор лимонной кислоты добавляли бромкриптин, раствор обрабатывали ультразвуком в течение 5 минут для получения белой суспензии. Суспензию добавляли к основной композиции и обрабатывали ультразвуком в течение 10 минут для получения прозрачного текучего геля, который использовали для приготовления пленки.
Подложку Scotchpack 1022 3М фиксировали на стеклянной пластине (размером приблизительно 8×12 дюймов). Подложку предварительно промывали водой и детергентом для уменьшения смачивающей способности пленки. 20 мл (0,51 мм, толщина во влажном состоянии) пленок наносили на подкладку с помощью ручного аппликатора GARDCO в ламинаре Flow Scientific. Оставляли пленку принять форму и расправиться в течение 20 минут перед применением тока воздуха. Потом действовали потоком воздуха в течение 30 минут. Спустя час, пока пленка все еще была очень вязкой, но хорошо сформированной, обдували ее потоком теплого воздуха в течение 30 минут. Распылитель воздуха регулировали таким образом, что температура у поверхности достигала приблизительно 60-70°C, чтобы минимизировать нагрев и возможное разложение бромкриптина. Для дальнейшего высыхания пленку помещали на 48 часов в вакуумную сушилку, заполненную Drierite®.
Результаты исследования:
Краткосрочные исследования стабильности (в течение 24 часов и 5-10 дней) образцов пластыря с помощью ВЭЖХ показали высокую стабильность бромкриптина и отсутствие продуктов разложения.
Высвобождение лекарственного средства: иммерсионная среда: цитратный буфер, рН 6,0 (смотрите таблицу ниже для профиля растворения)
Пример 25: Чресслизистые пленочные лекарственные формы, основанные на поливинилпирролидоне, с гидрокспропилметилцеллюлозой с большим молекулярным весом (Бенецел® МР844)
В данном примере биоадгезивом, который использовали вместо гидроксипропилцеллюлозы (KLUCEL® LF), был Бенецел® МР844, гидроксипропилметилцеллюлоза с самой большой молекулярной массой. Чресслизистые пленочные лекарственные формы, основанные на поливинилпирролидоне, с Бенецелом МР844 (пленки 43-45) готовили по следующей схеме:
Основную композиции готовили, добавляя Коллидон 90F, Коллидон VA64 и ПЭГ400 в этанол, градуированную бутыль Пирекс с завинчивающейся крышкой вместимостью 2 л. В случае Пленки 45, также добавляли глицерол. Ингредиенты смешивали с помощью роллера Stovall на средней скорости в течение 24 часов при комнатной температуре. В результате образовывался прозрачный гомогенный вязкий раствор, который хранили при 4°C в виде стокового раствора.
Итоговую лекарственную форму готовили, растворяя лимонную кислоту в этаноле при легком нагревании, и диспергировали ультразвуком. В раствор лимонной кислоты добавляли бромкриптин и раствор и диспергировали ультразвуком в течение 5 минут для получения белой суспензии. Суспензию добавляли в основную композицию и диспергировали ультразвуком в течение 10 минут для получения прозрачного лабильного геля. В гель добавляли Бенецел® и диспергировали ультразвуком 10 минут. Полученную суспензию гомогенизировали с помощью гомогенизатора Polytron в течение 3 минут при 5000 об/мин и затем непосредственно использовали для нанесения.
Подложку Scotchpack 1022 3М фиксировали на стеклянной пластине (размером приблизительно 8×12 дюймов). Подложку предварительно промывали водой и детергентом для уменьшения смачивающей способности пленки. 20 мл (0,51 мм, толщина во влажном состоянии) пленок наносили на подложку с помощью ручного аппликатора GARDCO в ламинаре Flow Scientific. Оставляли пленку принять форму и расправиться в течение 20 минут перед применением тока воздуха. Потом действовали потоком воздуха в течение 30 минут. Спустя час, пока пленка все еще была очень вязкой, но хорошо сформированной, обдували ее потоком теплого воздуха в течение 30 минут. Распылитель воздуха регулировали таким образом, что температура у поверхности достигала приблизительно 60-70°C для минимизации нагревания и возможного разложения бромкриптина. Для дальнейшего высыхания пленку на 48 часов помещали в вакуумную сушилку, заполненную Drierite®.
Результаты анализа:
Краткосрочные исследования стабильности (в течение 24 часов и 5-10 дней) образцов пластыря с помощью ВЭЖХ показали высокую стабильность бромкриптина и отсутствие продуктов разложения.
Высвобождение лекарственного средства: иммерсионная среда: цитратный буфер, рН 6,0 (для профиля растворения смотрите таблицу ниже)
Пример 26: Уровни бромкриптина в крови после введения чресслизистых лекарственных форм на животной модели
Чресслизистые лекарственные формы согласно настоящему изобретению (Пленки 41-45) вводили сирийским хомячкам для того, чтобы показать in vivo биодоступность агониста допамина. Большой мешок для пищи сирийских хомячков представляет собой идеальную биологическую ткань для исследования мукозального транспорта соединений лекарственных форм. Каждому сирийскому хомячку вводили 4 мг бромкриптина (n=8 в каждой группе). Образцы крови брали до и непосредственно в определенные интервалы 30 и 300 минут после введения пленки и измеряли уровни бромкриптина в плазме. Из плазмы выделяли бромкриптин и все образцы сравнивали со стандартами с помощью ВЭЖХ, как описано в Примере 18. Данные по биодоступности представлены в виде % от Cmax. Данные представляют собой наилучшие кривые, полученные для каждой группы лечения.
Результаты исследования биодоступности пленочных лекарственных форм Пленки 41-45
Биодоступность чресслизистой пленочной лекарственной пленки характеризовалась пиком бромкриптина в плазме в течение 30 минут после введения лекарства через слизистую с быстрым последующим снижением уровня в плазме. Значения Cmax для пленочных лекарственных форм Пленки 41, 42, 43, 44 и 45 составили 15,2, 36,1, 3,8, 17,6, и 10,7 нг/мл плазмы, соответственно. В пленку 42 добавляли молекулы типа циклодекстрина. Они ускоряют всасывание бромкриптина при неожиданном укорочении Tmax до 60 минут по сравнению с 240 минутами в случае Пленки 41, состав которой похож на Пленку 42, но без молекул типа цилкодекстрина. Этот результат является неожиданным, так как добавление молекул типа циклодекстрина обычно замедляет высвобождение агонистов допамина.
Чресслизистые пленки согласно настоящему изобретению давали желаемую кривую биодоступности агониста допамина с пиком-плато в модели на животных. В частности, лекарственные формы Пленка 42 и Пленка 43 достигали желаемой кривой биодоступности бромкриптина с пиком-плато. Эти примеры биодоступности показывают, что можно изменять, предсказуемым образом, форму кривой биодоступности, меняя некоторые компоненты лекарственной формы. Изменяя соотношение KLUCEL® и Коллидона или добавляя молекулы типа циклодекстрина в пленочную лекарственную форму (например, что сделано в случае пленочной лекарственной формы Пленка 42), можно менять биодоступность агониста допамина для получения пикового уровня агониста допамина в течение 90 минут и плато в течение периода приблизительно от 60 до 240 минут. Такие кривые биодоступности полезно использовать при лечении метаболических нарушений.
Также можно регулировать биодоступность in vivo агонистов допамина в пленочных лекарственных формах, добавляя Бенецел® в лекарственную форму пленку 42 с KLUCEL®/циклодекстрином для того, чтобы достичь медленного всасывания, которое приводит к продленному времени плато после пика всасывания агониста допамина, что Бенецел® делает в случае пленки 44 и пленки 45. Более того, можно усилить биодоступность и достичь желаемой кривой биодоступности с пиком-плато согласно настоящему изобретению, добавляя усилители проникновения, например жирные кислоты и биоадгезивы, в представленные пленочные лекарственные формы.
Данные представлены в виде % Cmax
Пример 27: Чресслизистые пленочные лекарственные формы на основе поливинилпирролидона с олеиновой кислотой
В качестве ускорителя проникновения в лекарственные формы Пленки 42-47 добавляли олеиновую кислоту. Добавление олеиновой кислоты не оказывало существенного влияния на свойства высвобождения лекарственного средства из Пленки 42.
Состав
Лекарственные формы готовили аналогично пленке 42 с добавлением олеиновой кислоты к итоговой лекарственной форме до этапа диспергирования ультразвуком.
Результаты исследования
Краткосрочные исследования стабильности (в течение 24 часов и 5-10 дней) образцов пластыря с помощью ВЭЖХ показали высокую стабильность бромкриптина и отсутствие продуктов разложения.
Высвобождение лекарственного средства: иммерсионная среда: цитратный буфер, рН 6,0 (для профиля растворения смотрите таблицу ниже)
Пример 28: Чресслизистые пленочные лекарственные формы на основе поливинилпирролидона с лизуридом и/или SKF-38393 Пленки для чресслизистого применения лизурида и/или SKF-38393 готовили с поливинилпирролидонами и со-полимерами поливинилпирролидона. Для того чтобы усилить биоадгезивные свойства пленки использовали KLUCEL® LF. Свойства высвобождения лекарственного вещества этих новых лекарственных форм полностью совпадали с пленкой 42, которая содержала бромкриптин в качестве агониста допамина.
Состав
Лекарственные формы готовили аналогично Пленке 42, за исключением добавления лизурида, SKF-38393 или обоих веществ вместо бромкриптина.
Результаты исследования:
Краткосрочные исследования стабильности (в течение 2А часов и 5-10 дней) образцов пластыря с помощью ВЭЖХ показали высокую стабильность бромкриптина и отсутствие продуктов разложения.
Высвобождение лекарственного средства: иммерсионная среда: цитратный буфер, рН 6,0 (для профиля растворения смотрите таблицу ниже)
Пример 29: Подкожная лекарственная форма на основе масла
50 мг бромкриптина пропускали через сито 40 меш, помещенное в сцинтилляционный флакон на 20 мл, и суспендировали в 1 г полисорбата 80. Суспензию обрабатывали ультразвуком в течение 15 минут с периодическим встряхиванием пробирки для того, чтобы прикрепившийся к стенкам материал растворился. Постепенно бромкриптин растворялся до прозрачного раствора с небольшим количеством оставшихся агрегатов. В этот раствор добавляли масло кунжута и обрабатывали ультразвуком 10 минут. Полученную прозрачную эмульсию бромкриптина (приблизительно 0,05%) можно использовать для парентерального применения после пропускания через стерилизующий фильтр.
Рекомендуется непосредственно перед введением встряхнуть раствор. Для доставки 0,5 мг бромкриптина потребуется ввести приблизительно 100 мг эмульсии. Из литературных данных известно, что плотность масла кунжута составляет 0,9 г/см3, а плотность полисорбата 80 равна 1,08 г/см3, что соответствует объему приблизительно 110 мкл. В этот препарат можно добавлять лимонную кислоту для повышения стабильности агониста допамина и ускорения его поступления в кровоток.
Композиция VS-49SC содержит приблизительно 10% полисорбата 80.
Исследование стабильности
Непосредственно после приготовления форм с применением микроскопии не наблюдали капель отдельных фаз масла и полисорбата 80, разрешающая способность 5 мкм.
Но после 2-3 дней хранения в спокойном состоянии при комнатной температуре эмульсия разделилась на отдельные слои из двух компонентов. Можно достичь гомогенизации интенсивным встряхиванием или действием ультразвука.
Настоящее изобретение не должно ограничиваться приведенными в настоящей заявке вариантами реализации. Различные модификации изобретения дополнительно к описанным в настоящей заявке будут очевидны для специалиста в данной области из приведенного выше описания и фигур. Такие модификации входят в объем представленной формулы изобретения.
Следует также понимать, что все значения являются приблизительными и приведены с целью описания. Все размещенные ссылки включены в настоящую заявку в полном объеме в той же степени, как если бы каждая ссылка была бы индивидуально включена в настоящую заявку посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕКАРСТВЕННЫЕ ФОРМЫ И СПОСОБ ЛЕЧЕНИЯ РАССТРОЙСТВА МУЖСКОЙ ЭРЕКТИЛЬНОЙ ФУНКЦИИ | 1998 |
|
RU2204413C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ОСТРЫХ НАРУШЕНИЙ | 1999 |
|
RU2193879C1 |
| СПОСОБ ЛЕЧЕНИЯ ДВИГАТЕЛЬНЫХ РАССТРОЙСТВ БЕФИРАДОЛОМ | 2015 |
|
RU2702101C2 |
| КОМПОЗИЦИИ ТЕТРАГИДРОБИОПТЕРИНА И СПОСОБЫ ЕГО КОЛИЧЕСТВЕННОЙ ОЦЕНКИ | 2008 |
|
RU2486899C2 |
| СЛОИСТЫЕ ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ | 2007 |
|
RU2452471C2 |
| КОМПОЗИЦИИ ДЛЯ ВВЕДЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2009 |
|
RU2554814C2 |
| КОМПОЗИЦИЯ ФЕНТАНИЛА ДЛЯ ЛЕЧЕНИЯ ОСТРОЙ БОЛИ | 1999 |
|
RU2232580C2 |
| РАСТВОРИМЫЕ ДОЗИРОВАННЫЕ ФОРМЫ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ЦЕФЕМА, ПРИЕМЛЕМЫЕ ДЛЯ ПАРЕНТЕРАЛЬНОГО ВВЕДЕНИЯ | 2008 |
|
RU2537237C2 |
| НАНОЧАСТИЦЫ, ВКЛЮЧАЮЩИЕ ЦИКЛОДЕКСТРИН И БИОЛОГИЧЕСКИ АКТИВНУЮ МОЛЕКУЛУ, И ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2460518C2 |
| ОРАЛЬНЫЕ ДОЗИРОВАННЫЕ ФОРМЫ ТОФАЦИТИНИБА С НЕПРЕРЫВНЫМ ВЫСВОБОЖДЕНИЕМ | 2014 |
|
RU2674345C2 |




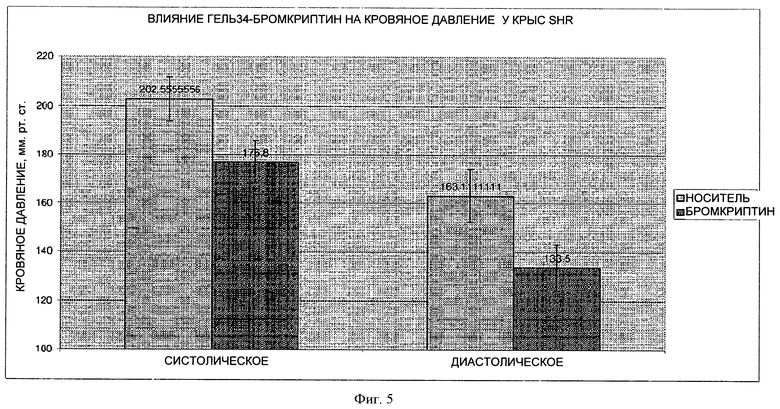
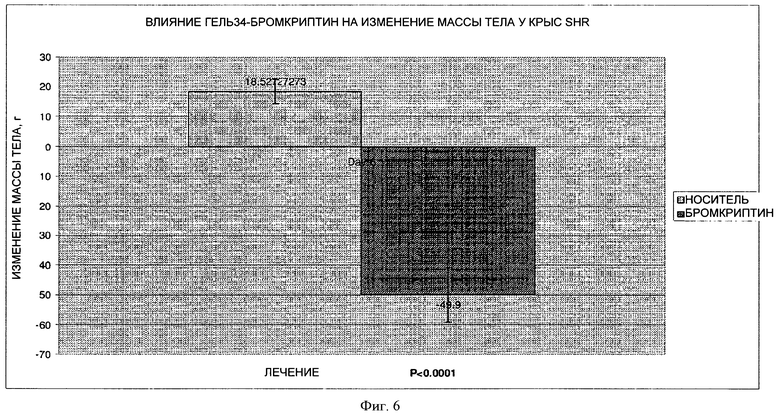
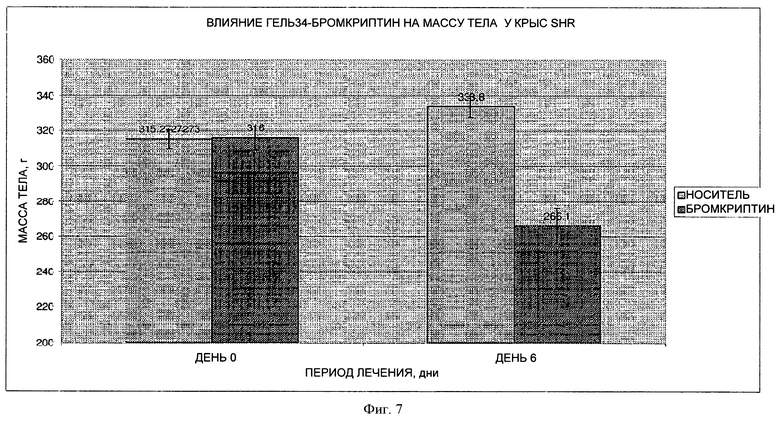
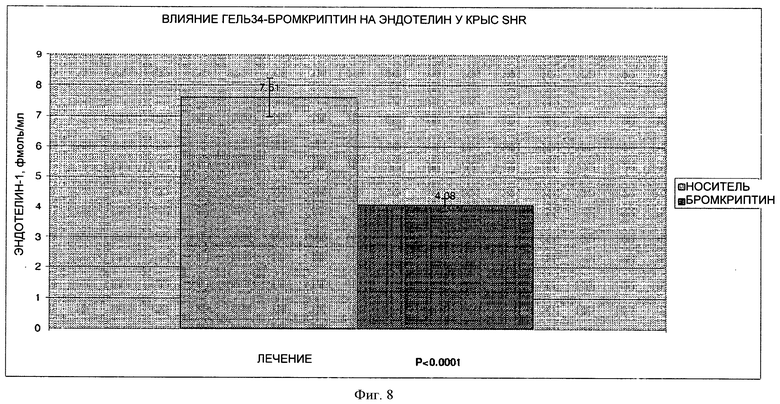
Изобретение относится к стабильным фармацевтическим композициям для парентерального введения, содержащим агонисты допамина и агенты периферического действия, применяемым для лечения нарушений метаболизма. Фармацевтические композиции по изобретению содержат, по меньшей мере, один агонист допамина и фармацевтический наполнитель, включающий фармацевтически приемлемый усилитель проникновения, фармацевтически приемлемый усилитель растворимости и фармацевтически приемлемый усилитель биоадгезии. Лекарственные формы по изобретению имеют фармакокинетический профиль, который характеризуется Тmax до приблизительно 90 минут после введения лекарственной формы и концентрацией лекарственного средства в плазме, по меньшей мере, 50% Сmax в течение периода времени от 90 до 360 минут после достижения Сmax. Указанный специфический фармакокинетический профиль парентеральных лекарственных форм с агонистом допамина обеспечивает улучшенный терапевтический эффект по сравнению с пероральными лекарственными формами агонистов допамина. 2 н. и 63 з.п. ф-лы, 8 ил., 29 пр.
1. Стабильная лекарственная форма для парентерального введения, содержащая по меньшей мере один агонист допамина и фармацевтически приемлемый наполнитель, включающий фармацевтически приемлемый усилитель проникновения, фармацевтически приемлемый усилитель растворимости и фармацевтически приемлемый усилитель биоадгезии, при этом указанная лекарственная форма имеет фармакокинетический профиль, который характеризуется:
а) Tmax приблизительно от 1 до приблизительно 90 минут после введения лекарственной формы для парентерального введения; и
б) концентрацией лекарственного средства в плазме по меньшей мере 50% Cmax в течение периода от приблизительно 90 до приблизительно 360 минут после достижения Cmax.
2. Лекарственная форма для парентерального введения по п.1, имеющая фармакокинетический профиль, в котором по меньшей мере приблизительно 90% агониста допамина удаляется из плазмы в течение от приблизительно 240 до приблизительно 480 минут указанной концентрации лекарственного средства.
3. Лекарственная форма для парентерального введения по п.1, отличающаяся тем, что указанное Tmax составляет от приблизительно 5 до приблизительно 90 минут после введения лекарственной формы для парентерального введения.
4. Лекарственная форма для парентерального введения по п.1, имеющая фармакокинетический профиль, в котором указанное Tmax составляет от приблизительно 5 до приблизительно 90 минут после введения лекарственной формы и указанная концентрация включает пост-Cmax уровень, включающий приблизительно одну вторую Cmax в течение от приблизительно 30 до приблизительно 150 минут после Tmax.
5. Лекарственная форма для парентерального введения по п.1, имеющая фармакокинетический профиль, в котором указанное Tmax составляет от приблизительно 5 до приблизительно 90 минут после введения лекарственной формы и указанная концентрация включает пост-Cmax уровень, включающий приблизительно одну вторую Cmax в течение от приблизительно 90 до приблизительно 360 минут после Tmax.
6. Лекарственная форма для парентерального введения по п.1, содержащая агонист допамина D1, агонист допамина D2 или комбинацию агониста допамина D1 и агониста допамина D2.
7. Лекарственная форма для парентерального введения по п.1, отличающаяся тем, что указанный по меньшей мере один агонист допамина содержит по меньшей мере одно производное спорыньи и одно производное не спорыньи.
8. Лекарственная форма для парентерального введения по п.6, отличающаяся тем, что указанный агонист допамина D2 выбран из группы, состоящей из бромокриптина, лизурида, тергурида, дигидроэрготоксина (гидергина), ропинорола, пирибедила, апоморфина, хинелорана, талипексола, алкалоидных производных спорыньи и производных эрголина.
9. Лекарственная форма для парентерального введения по п.8, отличающаяся тем, что указанные алкалоидные производные спорыньи выбраны из группы, состоящей из дигидро-альфа-эргокриптина, дигидро-альфа-эрготоксина, эргокорнина и 9,10-дигидроэргокорнина.
10. Лекарственная форма для парентерального введения по п.1, отличающаяся тем, что один или более агонистов допамина содержат бромокриптин.
11. Лекарственная форма для парентерального введения по п.6, отличающаяся тем, что указанный агонист D1 выбран из группы, состоящей из допамина, апоморфина, фенолдапама, SKF38393, SKF 75670, SKF 82957, SKF 81297, SKF 82958, SKF 82598, А77636, А68930 и аналогов бензазепина.
12. Лекарственная форма для парентерального введения по п.1 в виде назальной, подъязычной, буккальной, трансдермальной или подкожной лекарственной формы.
13. Лекарственная форма для парентерального введения по п.1, содержащая, в общем, от приблизительно 0,02 до приблизительно 50,0 мг указанного по меньшей мере одного агониста допамина.
14. Лекарственная форма для парентерального введения по любому из пп.1-13, дополнительно содержащая антигипертензивный агент, противовоспалительный агент, антикоагулянт, антигиперхолестеринемический агент, антигипертриглицеридемический агент, антигипергликемический агент или ингибитор ГМГКоА-редуктазы.
15. Лекарственная форма для парентерального введения по п.1, содержащая по массе:
0,5-20% агониста допамина,
3-50% матрикса высвобождения,
0,5-10% вещества, способствующего скольжению,
усилитель растворимости в количестве до 70%,
усилитель биоадгезии в количестве до 25%,
усилитель проникновения в количестве до 30%,
разрыхлитель в количестве до 95%,
наполнитель в количестве до 95% и
шипучее вещество в количестве до 65%,
причем указанная лекарственная форма представляет собой подъязычную таблетированную лекарственную форму.
16. Лекарственная форма для парентерального введения по п.15, содержащая по массе:
0,5-10% агониста допамина,
3-20% матрикса высвобождения,
0,5-5% вещества, способствующего скольжению,
усилитель растворимости в количестве до 30%,
усилитель биоадгезии в количестве до 10%,
усилитель проникновения в количестве до 20%,
разрыхлитель в количестве до 85%,
наполнитель в количестве до 80% и
шипучее вещество в количестве до 45%.
17. Лекарственная форма для парентерального введения по п.15, содержащая по массе:
0,5-5% агониста допамина,
7-15% матрикса высвобождения,
0,5-2,5% вещества, способствующего скольжению,
2-20% усилителя растворимости,
2-8% усилителя биоадгезии,
усилитель проникновения в количестве до 15%,
разрыхлитель в количестве до 82%,
наполнитель в количестве до 75% и
шипучее вещество в количестве до 45%.
18. Лекарственная форма для парентерального введения по любому из пп.15-17, отличающаяся тем, что указанная лекарственная форма дополнительно содержит один или более дополнительных агонистов допамина.
19. Лекарственная форма для парентерального введения по п.15, отличающаяся тем, что указанная лекарственная форма содержит по массе:
0,5-5% агониста допамина,
2-15% поливинилпирролидона,
3-20% гидроксипропилметилцеллюлозы,
3-25% натрия крахмал-гликолята и натрия карбоксиметил-крахмала,
3-25% ПроСолва,
0,5-10% лимонной кислоты,
0,5-5% стеариновой кислоты,
40-80% маннитола и
необязательно 5-30% молекул типа циклодекстрина.
20. Лекарственная форма для парентерального введения по п.15, отличающаяся тем, что указанная лекарственная форма содержит по массе:
0,5-4,5% агониста допамина,
2-6,5% поливинилпирролидона,
3-10% гидроксипропилметилцеллюлозы,
1-6% лимонной кислоты,
0,5-5% стеариновой кислоты,
60-90% Фармабурста (Pharmaburst) и
необязательно 5-30% молекул типа циклодекстрина.
21. Лекарственная форма для парентерального введения по п.15, отличающаяся тем, что указанная лекарственная форма содержит по массе:
1-6% агониста допамина,
2-10% поливинилпирролидона,
3-10% гидроксипропилметилцеллюлозы,
1-10% лимонной кислоты,
0,5-5% стеариновой кислоты,
60-90% Фармабурста (Pharmaburst) и
необязательно 5-30% молекул типа циклодекстрина.
22. Лекарственная форма для парентерального введения по п.15, отличающаяся тем, что указанная лекарственная форма содержит по массе:
0,5-5,0% агониста допамина,
5-25% поливинилпироллидона,
5-35% гидроксипропилметилцеллюлозы,
10-40% лимонной кислоты,
0,5-5% стеариновой кислоты,
3-25% Фармабурста (Pharmaburst) и
10-65% бикарбоната натрия.
23. Лекарственная форма для парентерального введения по любому из пп.15-17 или 19-22, отличающаяся тем, что агонист допамина представляет собой по меньшей мере одно алкалоидное производное спорыньи.
24. Лекарственная форма для парентерального введения по п.23, отличающаяся тем, что по меньшей мере одно алкалоидное производное спорыньи представляет собой бромокриптин.
25. Лекарственная форма для парентерального введения по любому из пп.15-17, 19-22 или 24, дополнительно содержащая агент, понижающий уровень холестерина.
26. Лекарственная форма для парентерального введения по п.25, отличающаяся тем, что агент, понижающий уровень холестерина, выбран из группы, состоящей из аторвастатина, церивастатина, флювастатина, ловастатина, мевастатина, правастатина, питавастатина, розувастатина, симвастатина, холестирамина, ситостерола, эзетимиба, гемфиброзила, клофибрата, никотиновой кислоты, колестипола и колесевелама.
27. Лекарственная форма для парентерального введения по любому из пп.15-17, 19-22 или 24, дополнительно содержащая антигипертензивный агент.
28. Лекарственная форма для парентерального введения по п.27, отличающаяся тем, что антигипертензивный агент выбран из группы, состоящей из буметанида, этакриновой кислоты, фуросемида, торсемида, хлорталидона, эпитизида, гидрохлортиазида, хлортиазида, бендрофлуметиазида, индапамида, метолазона, амилорида, триамтерена, спиронолактона, атенолола, метопролола, надолола, окспренолола, пиндолола, пропранолола, тимолола, доксазозина, фентоламина, индорамина, феноксибензамина, празозина, теразозина, толазолина, буциндолола, карведилола, лабеталола, клонидина, метилдопы, амлодипина, фелодипина, израдипина, нифедипина, нимодипина, нитрендипина, дилтиазема, верапамила, каптоприла, эналаприла, фозиноприла, лизиноприла, периндоприла, хинаприла, рамиприла, трандоприла, бензаприла, кандесартана, эпросартана, ирбесартана, лозартана, олмесартана, телмисартана, валсартана, спиронолактона, нитропруссида натрия, гуанабенза, гуанетидина и резерпина.
29. Лекарственная форма для парентерального введения по п.1, содержащая по массе:
1-3% агониста допамина,
5-95% растворителей,
1-30% загустителя,
0,5-10% стабилизатора и
биоадгезив в количестве до 35%,
причем указанная лекарственная форма представляет собой трансдермальный гель.
30. Лекарственная форма для парентерального введения по п.29, отличающаяся тем, что указанная лекарственная форма дополнительно содержит один или несколько дополнительных агонистов допамина.
31. Лекарственная форма для парентерального введения по п.29, отличающаяся тем, что указанная лекарственная форма содержит по массе:
0,5-10% агониста допамина,
5-40% полиэтиленгликоля,
45-85% глицерина,
3-25% диоксида кремния и
0,5-5% лимонной кислоты.
32. Лекарственная форма для парентерального введения по п.29, отличающаяся тем, что указанная лекарственная форма содержит по массе:
0,5-10% агониста допамина,
5-40% полиэтиленгликоля,
45-85% глицерина,
3-25% диоксида кремния,
0,5-5% лимонной кислоты,
1-15% гидроксипропилметилцеллюлозы,
0,5-15% поливинилпирролидона.
33. Лекарственная форма для парентерального введения по любому из пп.29-32, отличающаяся тем, что агонист допамина представляет собой по меньшей мере одно алкалоидное производное спорыньи.
34. Лекарственная форма для парентерального введения по п.33, отличающаяся тем, что по меньшей мере одно алкалоидное производное спорыньи представляет собой бромокриптин.
35. Лекарственная форма для парентерального введения по любому из пп.29-32 или 34, дополнительно содержащая агент, снижающий уровень холестерина.
36. Лекарственная форма для парентерального введения по п.35, отличающаяся тем, что агент, снижающий уровень холестерина, выбран из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, правастатина, питавастатина, розувастатина, симвастатина, холестирамина, ситостерола, эзетимиба, гемфиброзила, клофибрата, никотиновой кислоты, колестипола и колесевелама.
37. Лекарственная форма для парентерального введения по любому из пп.29-32 или 34, дополнительно содержащая антигипертензивный агент.
38. Лекарственная форма для парентерального введения по п.37, отличающаяся тем, что антигипертензивный агент выбран из группы, состоящей из буметанида, этакриновой кислоты, фуросемида, торсемида, хлорталидона, эпитизида, гидрохлортиазида, хлортиазида, бендрофлуметиазида, индапамида, метолазона, амилорида, триамтерена, спиронолактона, атенолола, метопролола, надолола, окспренолола, пиндолола, пропранолола, тимолола, доксазозина, фентоламина, индорамина, феноксибензамина, празозина, теразозина, толазолина, буциндолола, карведилола, лабеталола, клонидина, метилдопы, амлодипина, фелодипина, израдипина, нифедипина, нимодипина, нитрендипина, дилтиазема, верапамила, каптоприла, эналаприла, фозиноприла, лизиноприла, периндоприла, хинаприла, рамиприла, трандоприла, бензаприла, кандесартана, эпросартана, ирбесартана, лозартана, олмесартана, телмисартана, валсартана, спиронолактона, нитропруссида натрия, гуанабенза, гуанетидина и резерпина.
39. Лекарственная форма для парентерального введения по п.1, содержащая по массе:
5-20% агониста допамина,
1-10% пленкообразующего агента,
5-20% усилителя стабильности,
10-95% усилителей биоадгезии,
0-50% усилителей растворимости и
необязательно 1-10% олеиновой кислоты,
причем указанная лекарственная форма представляет собой чрезслизистую пленку.
40. Лекарственная форма для парентерального введения по п.39, отличающаяся тем, что указанная лекарственная форма дополнительно содержит один или более дополнительных агонистов допамина.
41. Лекарственная форма для парентерального введения по п.39, отличающаяся тем, что указанная лекарственная форма содержит по массе:
2-20% агониста допамина,
10-55% Коллидона 90F,
0,5-10% Коллидона VA64,
0,3-5% полиэтиленгликоля,
10-55% гидроксипропилцеллюлозы,
0,5-10% глицерина,
3-30% циклодекстрина,
2-20% лимонной кислоты и
необязательно 1-5% олеиновой кислоты.
42. Лекарственная форма для парентерального введения по любому из пп.39-41, отличающаяся тем, что агонист допамина представляет собой по меньшей мере одно алкалоидное производное спорыньи.
43. Лекарственная форма для парентерального введения по п.42, отличающаяся тем, что по меньшей мере одно алкалоидное производное спорыньи представляет собой бромокриптин.
44. Лекарственная форма для парентерального введения по любому из пп.39-41 или 43, дополнительно содержащая агент, снижающий уровень холестерина.
45. Лекарственная форма для парентерального введения по п.44, отличающаяся тем, что агент, снижающий уровень холестерина, выбран из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, правастатина, питавастатина, розувастатина, симвастатина, холестирамина, ситостерола, эзетимиба, гемфиброзила, клофибрата, никотиновой кислоты, колестипола и колесевелама.
46. Лекарственная форма для парентерального введения по любому из пп.39-41 или 43, дополнительно содержащая антигипертензивный агент.
47. Лекарственная форма для парентерального введения по п.46, отличающаяся тем, что антигипертензивный агент выбран из группы, состоящей из буметанида, этакриновой кислоты, фуросемида, торсемида, хлорталидона, эпитизида, гидрохлортиазида, хлортиазида, бендрофлуметиазида, индапамида, метолазона, амилорида, триамтерена, спиронолактона, атенолола, метопролола, надолола, окспренолола, пиндолола, пропранолола, тимолола, доксазозина, фентоламина, индорамина, феноксибензамина, празозина, теразозина, толазолина, буциндолола, карведилола, лабеталола, клонидина, метилдопы, амлодипина, фелодипина, израдипина, нифедипина, нимодипина, нитрендипина, дилтиазема, верапамила, каптоприла, эналаприла, фозиноприла, лизиноприла, периндоприла, хинаприла, рамиприла, трандоприла, бензаприла, кандесартана, эпросартана, ирбесартана, лозартана, олмесартана, телмисартана, валсартана, спиронолактона, нитропруссида натрия, гуанабенза, гуанетидина и резерпина.
48. Лекарственная форма для парентерального введения по п.1, содержащая по массе:
0,01-0,10% агониста допамина,
5-20% эмульгирующего агента и
80-95% масла,
причем указанная лекарственная форма представляет собой лекарственную форму для подкожного введения.
49. Лекарственная форма для парентерального введения по п.48, отличающаяся тем, что указанная лекарственная форма дополнительно содержит один или более дополнительных агонистов допамина.
50. Лекарственная форма для парентерального введения по п.48, отличающаяся тем, что агонист допамина представляет собой по меньшей мере одно алкалоидное производное спорыньи.
51. Лекарственная форма для парентерального введения по п.50, отличающаяся тем, что по меньшей мере одно алкалоидное производное спорыньи представляет собой бромокриптин.
52. Лекарственная форма для парентерального введения по п.48, отличающаяся тем, что указанная лекарственная форма содержит по массе:
0,01-0,1% бромокриптина,
5-10% полисорбата 80 и
90-95% масла семян кунжута.
53. Лекарственная форма для парентерального введения по любому из пп.48-52, дополнительно содержащая агент, снижающий уровень холестерина.
54. Лекарственная форма для парентерального введения по п.53, отличающаяся тем, что агент, снижающий уровень холестерина, выбран из группы, состоящей из аторвастатина, церивастатина, флувастатина, ловастатина, мевастатина, правастатина, питавастатина, розувастатина, симвастатина, холестирамина, ситостерола, эзетимиба, гемфиброзила, клофибрата, никотиновой кислоты, колестипола и колесевелама.
55. Лекарственная форма для парентерального введения по любому из пп.48-52, дополнительно содержащая антигипертензивный агент.
56. Лекарственная форма для парентерального введения по п.55, отличающаяся тем, что антигипертензивный агент выбран из группы, состоящей из буметанида, этакриновой кислоты, фуросемида, торсемида, хлорталидона, эпитизида, гидрохлортиазида, хлортиазида, бендрофлуметиазида, индапамида, метолазона, амилорида, триамтерена, спиронолактона, атенолола, метопролола, надолола, окспренолола, пиндолола, пропранолола, тимолола, доксазозина, фентоламина, индорамина, феноксибензамина, празозина, теразозина, толазолина, буциндолола, карведилола, лабеталола, клонидина, метилдопы, амлодипина, фелодипина, израдипина, нифедипина, нимодипина, нитрендипина, дилтиазема, верапамила, каптоприла, эналаприла, фозиноприла, лизиноприла, периндоприла, хинаприла, рамиприла, трандоприла, бензаприла, кандесартана, эпросартана, ирбесартана, лозартана, олмесартана, телмисартана, валсартана, спиронолактона, нитропруссида натрия, гуанабенза, гуанетидина и резерпина.
57. Способ лечения нарушения метаболизма, включающий введение субъекту, нуждающемуся в таком лечении, лекарственной формы по любому из пп.1-56 для лечения указанного нарушения метаболизма.
58. Способ по п.57, отличающийся тем, что указанное нарушение метаболизма выбрано из группы, состоящей из диабета 2 типа, предиабета, метаболического синдрома, резистентности к инсулину, гиперинсулинемии, сердечно-сосудистого заболевания, ожирения, повышенного уровня норэпинефрина в плазме; повышенного уровня факторов воспаления, связанных с сердечно-сосудистыми заболеваниями, или факторов, вызывающих нарушение функции эндотелия сосудов; гиперлипопротеинемии, атеросклероза, гиперфагии, гипергликемии, гиперлипидемии, гипертензии, высокого кровяного давления, метаболического синдрома, повышенного уровня норэпинефрина в плазме, повышенного уровня факторов воспаления, связанных с сердечно-сосудистыми заболеваниями, и гипертензии.
59. Способ по п.57, отличающийся тем, что указанный субъект страдает по меньшей мере одним состоянием, выбранным из диабета 2 типа, ожирения, предиабетов, метаболического синдрома и повышенного уровня факторов воспаления, связанных с сердечно-сосудистой системой.
60. Способ по п.57, включающий введение указанной лекарственной формы для парентерального введения для снижения повышенного уровня факторов воспаления, связанных с сердечно-сосудистой системой, или ослабления сердечно-сосудистого заболевания.
61. Способ по п.57, отличающийся тем, что лекарственную форму для парентерального введения вводят в виде однократной дневной дозы, содержащей в общем от приблизительно 0,02 до приблизительно 50,0 мг указанного по меньшей мере одного агониста допамина.
62. Способ по п.57, отличающийся тем, что лекарственную форму для парентерального введения вводят в заранее определенное время суток.
63. Способ по п.62, отличающийся тем, что указанное заранее определенное время суток представляет собой приблизительный природный дневной пик циркадного ритма центральной допаминергической нейрональной активности в циркадном ритме уровня пролактина в плазме у здоровых особей того же вида и пола, что и указанный нуждающийся субъект.
64. Способ по п.63, отличающийся тем, что лекарственную форму для парентерального введения вводят с приблизительно 04.00 до приблизительно 12.00 часов.
65. Способ по п.57, отличающийся тем, что указанная лекарственная форма содержит бромокриптин.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| US 5877183 A, 02.03.1999 | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |

