Область техники, к которой относится изобретение
Настоящее изобретение касается материалов и способов для лечения диабета (типа 1 и типа 2) и метаболического синдрома.
Уровень техники
Во всем мире примерно 250 миллионов человек больны диабетом и это количество по прогнозам удвоится в ближайшие два десятилетия. Более 90% из этих людей страдают от сахарного диабета 2 типа (T2DM). По оценкам в настоящее время диагноз поставлен только 50-60% человек, которые болеют T2DM или находятся на стадии, предшествующей открытому проявлению T2DM.
T2DM представляет собой гетерогенное заболевание, которое характеризуется нарушениями в метаболизме углеводов и жиров. Причины возникновения T2DM являются многофакторными и включают как генетические, так и связанные с окружающей средой элементы, нарушающие функцию β-клеток и чувствительность к инсулину в тканях, таких как мышцы, печень, поджелудочная железа и жировая ткань. Как следствие, наблюдается пониженная секреция инсулина, которая происходит параллельно с прогрессирующим снижением функции β-клеток и хронической резистентностью к инсулину. Неспособность эндокринных клеток поджелудочной железы компенсировать периферическую резистентность к инсулину приводит к гипергликемии и началу развития клинических проявлений диабета. Резистентность тканей к опосредованному инсулином поглощению глюкозы в настоящее время считается основным патофизиологическим фактором в развитии T2DM.
Критерием успеха для оптимального лечения T2DM является снижение уровней глюкозы в крови, которое может представлять собой как хроническое снижение уровней глюкозы в крови, так и повышение способности переносить высокие уровни глюкозы после приема пищи, что описывается как снижение пиковых уровней глюкозы и более быстрый клиренс. Обе эти ситуации обеспечивают меньшую нагрузку на продукцию инсулина β-клетками и их функционирование.
Одним из подходов к контролю T2DM является применение инкретиновых гормонов - глюкагон-подобного пептида 1 (GLP-1) и зависимого от глюкозы инсулинотропного полипептида (GIP), которые продуцируются эндокринными клетками кишечника после приема пищи и стимулируют продукцию инсулина. GLP-1 является неэффективным в клиническом применении для лечения диабета, поскольку он имеет очень короткое время полужизни in vivo. Фармацевтическими синтетическими примерами инкретинов являются Экзенатид (Exenatide) и Лираглютид (Liraglutide), которые обладают биологическими свойствами, сходными с GLP-1 человека, но имеют более длительное время полужизни. Однако эти аналоги GLP-1 имеют некоторые побочные эффекты, такие как редкие, но опасные случаи панкреатита и воздействие на сердечно-сосудистую систему.
ЕР 0309100, зарегистрированный в 1988 году, показывает, что амилин может секретироваться вместе с инсулином, и в силу этого предлагает парентеральную композицию для лечения диабета типа 1, включающую агонист амилина, такой как, амилин сам по себе и инсулин. Предполагаемая роль амилина закючается в предотвращении гипогликемии, наблюдаемой при лечении инсулином. Указывается, что амилин снижает скорость синтеза гликогена.
Gomez-Foix et al.. сообщали, что амилин и CGRP проявляют антиинсулиновые эффекты на изолированных гепатоцитах крысы, дополняя более ранние сообщения о том, что амилин и CGRP ингибируют секрецию инсулина.
WO 89/06135 сообщает, что соединения, блокирующие действие амилина (антагонисты амилина), являются пригодными для лечения диабета 2 типа. Соединения, которые могут применяться в качестве антагонистов амилина, представляют собой поперечно сшитые варианты агонистов амилина. Указывается, что амилин обладает эффектами, приводящими к меньшему высвобождению инсулина р-клетками поджелудочной железы и вызывающими также значительное снижение как базального, так и инсулин-стимулируемого синтеза гликогена в скелетных мышцах, обеспечивая игнорирование мышечными клетками инсулинового сигнала.
WO 93/10146 раскрывает некоторые агонисты амилина в качестве парентеральных агентов для лечения диабета типа 1 и для лечения гипогликемии. Указывается, что при диабете типа 1 уровни амилина резко понижены или амилин отсутствует. Также предполагается, что агонисты амилина будут предотвращать низкий уровень глюкозы в крови, вызванный лечением инсулином.
EP 0717635B1/WO 95/07098 подтверждает, что амилин обладает гипергликемическим действием, но указывает, что он также может снижать моторику желудка и замедлять освобождение желудка, таким образом, скорее снижая, чем повышая уровни глюкозы в плазме после приема пищи. В соответствии с этим, данный документ рассматривает применение агонистов амилина для этой цели, но специально исключает кальцитонины.
US 7399744 раскрывает применение амилина или агонистов амилина для регуляции жировой массы тела. В экспериментах амилин, который вводился крысам с помощью осмотического насоса, вызывал снижение роста массы тела у крыс, находящихся на диете с высоким содержанием жиров. Кальцитонины, включая кальцитонины костных рыб, даются в качестве примера пригодного агониста, но не приводится данных, относящихся к их применению.
Таким образом, амилин для инъекций сейчас рассматривается в качестве жизненно важного подхода для гликемического контроля при T2DM, поскольку он мощно ингибирует секрецию глюкагона после приема пищи и освобождение желудка, снижает потребление пищи и, следовательно, физиологически регулирует поглощение углеводов. Один из аналогов амилина, Pramlintide (или Symlin), разрешен к применению для лечения пациентов с диабетом типа 1 и типа 2, которые также принимают инсулин. Лечение аналогом Pramlintide снижает средний уровень сахаров в крови и значительно снижает патологическое увеличение уровня сахара в крови у диабетиков после приема пищи. Лечение с применением Pramlintide также приводит к снижению веса и позволяет пациентам вводить меньше инсулина. US 2009/0018053 кратко предлагает композиции с покрытием, рассасывающимся в кишечнике, содержащие Pramlintide, для его высвобождения в желудочно-кишечном тракте для лечения диабета.
Амилин является одним из представителей семейства кальцитонина, состоящего из кальцитонина, пептида, относящегося к гену α-кальцитонина (αCGRP), βCGRP, адреномодуллина и амилина. Эта уникальная группа пептидов имеет консервативную третичную структуру с образованным дисульфидной связью кольцом на N-конце [Hay 2003 Br J Pharmacol]. У млекопитающих эти пептиды действуют через два близкородственных типа рецепторов II GPCRs (Calcitonin Receptor and Calcitonin Receptor-like Receptor) и через три уникальных белка, модулирующих активность рецепторов (RAMPs) [Hay Regul Pept 2003]. Таким образом, применение любой из этих маленьких сигнальных молекул может вызвать больше плейотропных эффектов во множестве органов вследствие совместного использования рецепторов и дивергентной эволюции.
Кальцитонин (СТ) представляет собой природный пептидный гормон, продуцируемый парафолликулярными клетками (С-клетками) в щитовидной железе и секретируемый в ответ на избыток кальция в сыворотке крови. СТ снижает резорбцию остеокластов за счет прямого связывания с его рецепторами на поверхности клеток остеокластов. СТ разрешен к применению для лечения остеопороза, связанной со злокачественным ростом гиперкальциемии, болезни Паджета (экземоподобного рака молочной железы), поскольку все эти заболевания связаны с ускоренной перестройкой костной ткани. В литературе недавно описана пероральная форма кальцитонина.
Имеются многочисленные сообщения о диабетогенном действии, вызываемом введением кальцитонина, у животных и у человека, за исключением, вероятно, пациентов, страдающих диабетом типа 1.
Анализируя вначале исследования на животных и in vitro, можно отметить, что Lupulescu сообщил в 1974, что высокие дозы (600 MRC U в месяц или 50 MRC U в виде однократной дозы) синтетического кальцитонина лосося при введении кроликам путем инъекций вызывают значительное снижение уровня глюкозы, измеряемого после голодания, при введении либо только однократно, либо три раза в день в течение одного месяца. Механизм этого эффекта не был понят, и не проводилось исследований влияния на уровни глюкозы после теста на толерантность к глюкозе или во время/после нормального кормления. Они подтвердили, что подобные результаты не наблюдались в работе Aldred et al.. 1968, когда кальцитонин свиньи вводился крысам, кроликам или мышам.
Greeley в 1989 сообщил, что введение кальцитонина лосося в желудочки головного мозга крыс увеличивало стимулируемое глюкозой высвобождение инсулина, однако уровни глюкозы в плазме не зависят исключительно от инсулина, и другие исследования (смотри ниже) показывают, что помимо увеличения уровня инсулина, кальцитонин также увеличивает уровни глюкагона, которые превалируют над увеличением уровня инсулина, что приводит к повышенному уровню глюкозы.
Pittner в 1997 сообщил, что наблюдается незначительный эффект при исследовании действия амилина, пептида, связанного с геном кальцитонина, или кальцитонина крысы или лосося на гепатоциты крысы и другие клетки.
Young et al.. в 1995 сообщили, что у крыс и мышей кальцитонин лосося при внутривенной болюсной инъекции вызывает повышение уровня глюкозы в плазме при голодании, кальцитонин крысы обладал менее выраженным эффектом. У гипогликемической крысы кальцитонин обеспечивал более значительное восстановление гликемии, чем глюкагон, и в комбинации с ним синергитично увеличивал восстановление гликемии, вызываемое глюкагоном.
Young в 2005 подробно проанализировал эффекты амилина и в меньшей степени кальцитонина лосося на уровни глюкозы и лактата у животных. Он сообщил, что как амилин, так и кальцитонин лосося при парентеральном введении голодным животным вызывают повышение уровней глюкозы, при этом кальцитонин обеспечивает более сильный эффект на повышение уровня глюкозы, но что эти эффекты у людей были умеренными или отсутствовали. Он также сообщил, что амилин, введенный крысам парентерально незадолго перед проведением теста на толерантность к глюкозе, снижает увеличение уровня глюкозы. Таким образом, в этом отношении ясно, что амилин и кальцитонин лосося у крыс ведут себя по-разному, поскольку твердо установлено, что кальцитонин, введенный парентерально, вызывает увеличение максимального уровня глюкозы при пероральном тесте на толерантность к глюкозе.
Chelikani et al.. в 2007 сообщили, что хроническое введение анорексигенных веществ животным путем инфузии или неоднократных инъекций не оказывает продолжительного влияния на потребление пищи или вес тела. Хотя острое парентеральное введение кальцитонина лосося сильно индуцировало кратковременное снижение потребления пищи у крыс или мышей, эффект сохранялся только в течение 3 дней.
Однако Bello et al.. в 2008 сообщили, что кальцитонин лосося, введенный парентерально, снижает потребление пищи макаками-резусами во время 5-дневного периода введения.
Анализируя далее работы на человеке, можно отметить, что Ziegler et al.. (1972) сообщили, что у здоровых волонтеров инфузия синтетического кальцитонина человека вызывает достоверное ухудшение ассимиляции глюкозы и продукции инсулина.
Blahos et al. в 1976 сообщили, при внутримышечном введении здоровым волонтерам кальцитонин лосося ингибирует снижение уровня глюкозы в крови при утреннем голодании и ухудшает тест на толерантность к глюкозе.
Petralito et al. (1979) сообщили, что в случае латентного диабета у людей инфузия кальцитонина лосося способна достоверно снижать уровень инсулина в сыворотке и увеличивать уровень сахара в крови.
Giugliano et al. 1980 сообщили, что хотя ранее было показано, что острое введение кальцитонина ухудшает толерантность к глюкозе у нормальных, страдающих ожирением и находящихся в пре-диабетическом состоянии людей, кальцитонин лосося, введенный путем внутривенной инфузии инсулинозависимым диабетическим пациентам, устранял повышение уровня глюкозы, обычно наблюдаемое в тесте на толерантность к аргинину, с немедленным восстановлением уровня глюкозы сразу после прекращения инфузии кальцитонина.
Gattereau et al. (1980) сообщили, что инъекция либо кальцитонина лосося, либо кальцитонина человека пациентам страдающим костной формой болезни Паджета, вызывала умеренный немедленный рост уровня глюкозы в сыворотке и небольшое снижение уровня инсулина в сыворотке.
Passariello et al. (1981) сообщили, что внутримышечное введение синтетического кальцитонина лосося людям вызывало ухудшение в ответе в тесте на толерантность к глюкозе у нормальных пациентов и вызывало дополнительное ухудшение в тесте на толерантность к глюкозе у субъектов с IGT. Было обнаружено, что интегрированная площадь под кривой уровня глюкозы в плазме увеличивалась примерно вдвое.
Starke et al. в 1981 сообщили, что у нормальных волонтеров инфузия кальцитонина лосося вызывает снижение уровней циркулирующего глюкагона и снижение уровня инсулина в сыворотке, эффект в целом заключался в ожидаемом снижении уровня глюкозы, так как снижение уровня глюкагона маскировалось эффектом снижения уровня инсулина. Однако у инсулинозависимых диабетиков кальцитонин лосося вызывал снижение уровня глюкозы параллельно со снижением уровня глюкагона.
Giugliano et al. (1982) сообщили о результатах длительного (2 месяца) введения 100 MRC единиц в день кальцитонина лосося пациентам с костной формой болезни Паджета или сильным остеопорозом. При таком лечении толерантность к глюкозе достоверно не ухудшалась (хотя наблюдался недостоверный рост пиковой концентрации глюкозы), но было обнаружено достоверное увеличение базального уровня глюкозы в плазме. В первые десять минут ответ инсулина на введение глюкозы ослаблялся кальцитонином.
Giustina et al. (1985) исследовали эффект кратковременного (15 дней) внутримышечного введения 100 MRC единиц дважды в день кальцитонина лосося пациентам с костной формой болезни Паджета, идиопатическим остеопорозом или остеодистрофией Зудека. Некоторые из пациентов были не инсулинозависимыми диабетиками и также получали антидиабетическую терапию. Было установлено, что кратковременное внутримышечное введение СТ лосося не вызывает значительного изменения в метаболизме углеводов после стимулирования смешанной пищей. У трех диабетических пациентов, участвующих в исследовании, наблюдалось недостоверное снижение уровня глюкозы в ночное время.
Zofkova (1987 - Exp. Clin. Endocrinol.) обнаружила, что 100 U кальцитонина при внутривенном введении в случае OGTT вызывали устойчивость к гипергликемии после начального медленного роста. Это объяснялось влиянием [кальцитонина] на поглощение глюкозы и метаболизм в печени. Также было обнаружено ингибиторное действие кальцитонина на секрецию инсулина.
Zofkova (1987 - Horm. Metabol. Res.) исследовала влияние двух дозировок (50 и 100 U) кальцитонина лосося на уровни глюкозы в крови у здоровых волонтеров и получила результаты, сходные с только что описанными выше при обоих уровнях дозировки.
Mangiafico (1988) обнаружил, что комбинация нифедипина с кальцитонином лосося при введении в дозе 100 U/день вызывает статистически достоверное увеличение уровня сахара в крови у субъектов с гипертензией, или с не инсулинозависимым диабетом, или со сниженной толерантностью к глюкозе постоянно в течение 3-недельного исследования.
Jonderko (1989) обнаружил, что высвобождение инсулина после приема пищи прекращается при введении кальцитонина лосося в дозе 62,26 пмоль/кг у пациентов с язвой двенадцатиперстной кишки, одновременно с параллельным ростом уровня глюкозы в сыворотке во время инфузии кальцитонина.
Young (2005) сообщил, что в плане приема пищи, введение амилина подавляет увеличение уровня глюкозы в плазме крови у крыс, собак и людей, но в отсутствие пищи введение амилина приводит к увеличению уровня глюкозы в плазме у грызунов, но не у людей.
Таким образом, процитированные выше исследования влияния парентерального введения кальцитонина людям, страдающим от не инсулинозависимого диабета или сниженной толерантностью к глюкозе, убедительно показывают, что также введение кальцитонина не приносит терапевтической пользы.
Раскрытие изобретения
Авторы настоящего изобретения провели исследование, может ли введение СТ через кишечник улучшить гипергликемическое состояние и обеспечить улучшенный гликемический контроль. Мы сравнили предполагаемый эффект пероральной композиции СТ лосося с таковым для хорошо охарактеризованного PPAR-γ агониста росиглитазона (rosiglitazone) при индуцированном диетой ожирении (diet induced obese, DIO) у крыс, которые проявляют определенные характеристики T2DM. Наконец мы исследовали влияние кальцитонина на обмен глюкозы у здоровых взрослых крыс. Неожиданно мы обнаружили, что пероральное введение кальцитонина вызывает эффект, в значительной степени противоположный тому, который ранее раскрывался для инъекции или инфузии кальцитонина, как описано и показано ниже. В противоположность этому, инсулин, например, который доступен для перорального введения или для инъекций, обеспечивает один и тот же эффект независимо от пути его введения.
В соответствии с этим настоящее изобретение предоставляет фармацевтическую композицию для введения через кишечник для лечения диабета типа 1, диабета типа 2, или метаболического синдрома, или для ослабления резистентности к инсулину, или для снижения нежелательно высокого уровня глюкозы в сыворотке при голодании, или для снижения нежелательно высокого пикового уровня глюкозы в сыворотке, или для снижения нежелательно высокого пикового уровня инсулина в сыворотке, где композиция включает активное соединение, которое является членом семейства кальцитонина, но не амилином, модифицированным членом семейства кальцитонина, но не модифицированным амилином, или агонистом рецептора кальцитонина. Композиция может также включать носитель, обеспечивающий эффективное введение через кишечник указанного активного соединения.
Предпочтительно указанная композиция готовится для перорального введения через пищеварительный тракт.
Предпочтительно активное соединение представляет собой кальцитонин, наиболее предпочтительно кальцитонин лосося.
Данное активное соединение может представлять собой модифицированный представитель семейства кальцитонина, имеющий, по меньшей мере, 75% аминокислотной гомологии с членом семейства кальцитонина, но не амилином, и который модифицирован по отношению к указанному представителю семейства кальцитонина путем вставки, замены или делеции аминокислот и сохраняет способность к связыванию и к активации рецептора кальцитонина.
Предпочтительно указанный носитель представляет собой 5-CNAC.
Настоящее изобретение включает способ лечения диабета типа 1, диабета типа 2 или метаболического синдрома, включающий введение через кишечник пациенту, который в этом нуждается для лечения указанного состояния, фармацевтически эффективного количества фармацевтической композиции, включающей активное соединение, которое является членом семейства кальцитонина, но не амилином, модифицированным представителем семейства кальцитонина, но не модифицированным амилином, или агонистом рецептора кальцитонина, и необязательно носитель, обеспечивающий эффективное введение через кишечник указанного активного соединения. Указанный способ может включать предварительную стадию для выяснения того, страдает ли пациент от указанного состояния, и/или последующую стадию для определения, до какой степени указанное лечение является эффективным для облегчения состояния указанного пациента, например, в каждом случае путем проведения перорального теста на толерантность к глюкозе или уровня сахара в крови в покое.
Для улучшенного контролирования веса пациента, чтобы обеспечить снижение веса или избежать увеличения веса, активное соединение предпочтительно вводится, по меньшей мере, дважды в день, например 2-4 раза в день. Композиции активного соединения могут содержать единичные дозировки, пригодные для такой схемы введения. Активные соединения могут вводиться с целью контролирования веса пациента, получающего лечение от диабета или метаболического синдрома.
Пероральные композиции для кишечного введения предназначены для употребления путем пропитывания для последующего высвобождения в тонком кишечнике уже после желудка и, следовательно, доставки через портальную вену в печень, в отличие от композиций, которые рассасываются во рту для обеспечения переноса в кровоток через подъязычные или буккальные пути.
Не будучи связанными теорией, мы предполагаем, что резкое различие в эффекте кальцитонина, связанное с тем, вводится ли он парентерально, или вводится ли он через кишечник, о котором мы здесь сообщаем, может быть связано с тем, что введение через кишечник обеспечивает немедленную доставку кальцитонина в печень через портальную вену после всасывания в кишечнике, в результате чего рецепторы в печени экспонируются в присутствии гораздо более высоких концентраций кальцитонина, чем когда кальцитонин вводится внутривенно или внутримышечно. Прямой путь в печень после введения кальцитонина через кишечник делает возможным достижение антигипергликемического эффекта несмотря на короткое время полужизни кальцитонина (подобное таковому для GLP-1). Механизм, лежащий в основе противоположного эффекта кальцитонина при его введении путем инъекции и при пероральном введении, может заключаться в том, что в достаточно высокой концентрации кальцитонин может выступать как агонист рецепторов, на которые обычно действует амилин, и, вследствие этого, может обеспечивать амилин-подобный эффект, тогда как более низкие концентрации кальцитонина являются эффективными только в отношении других рецепторов, которые обеспечивают гипергликемический эффект. Однако возможны и другие объяснения. Например, может быть при пероральном введении эти агенты действуют непосредственно на рецепторы кальцитонина, отличающиеся от таковых, на которые действует кальцитонин при инъекции, например, на рецепторы самого кишечного тракта.
Кальцитонины являются высококонсервативными у большого числа видов. Последовательности примеров кальцитонинов приведены ниже:

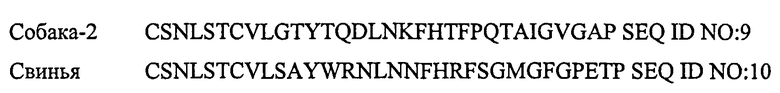
В соответствии с этим, предпочтительное соединение для применения в настоящем изобретении имеет следующую формулу:
CXlX2LSTCX3LX4X5X6X7X8X9Xl0XllX12Xl3X14X15X16X17X18X19X20X21GX22X23X24P
SEQ ID NO:11,
где X1 представляет собой A, G или S; предпочтительно S;
X2 представляет собой N или S; предпочтительно N;
X3 представляет собой М или V; предпочтительно V;
X4 представляет собой G или S; предпочтительно G;
X5 представляет собой Т, K или А; предпочтительно Т или K; наиболее предпочтительно K;
X6 представляет собой L или Y; предпочтительно L;
X7 представляет собой Т, S или W; предпочтительно Т или S; наиболее предпочтительно S;
X8 представляет собой Q, K или R; предпочтительно Q;
X9 представляет собой D, Е или N; предпочтительно D или Е; наиболее предпочтительно Е;
X10 представляет собой F или L; предпочтительно L;
X11 представляет собой N или Н; предпочтительно Н;
X12 представляет собой К или N; предпочтительно K;
X13 представляет собой F или L; предпочтительно L;
X14 представляет собой Н или Q; предпочтительно Q;
X15 представляет собой Т или R; предпочтительно Т;
X16 представляет собой F или Y; предпочтительно Y;
X17 представляет собой Р или S; предпочтительно Р;
X18 представляет собой Q, G или R; предпочтительно Q или R; наиболее предпочтительно R;
X19 представляет собой Т, I или М; предпочтительно Т;
X20 представляет собой А, N, D, S или G; предпочтительно N;
X21 представляет собой I, Т, V или F; предпочтительно Т,
X22 представляет собой V, S, А или Р; предпочтительно V, S, или А; наиболее предпочтительно S;
X23 представляет собой G или Е, предпочтительно G;
X24 представляет собой А или Т; предпочтительно Т.
В соответствии с этим, предпочтительно, чтобы соединение имело формулу
CSNLSTCVLGX5LX7QX9LHKLQTYPX18TNTGX22GTP
SEQ ID NO:12
где X5 представляет собой Т или K; более предпочтительно K;
X7 представляет собой Т или S; более предпочтительно S;
X9 представляет собой D или Е; более предпочтительно Е;
X18 представляет собой Q или R; более предпочтительно R;
X22 представляет собой V, S или А; более предпочтительно S.
Кальцитонин лосося является наиболее предпочтительным.
Кроме того, кальцитонин является представителем семейства пептидных гормонов, включающего амилин, относящийся к гену кальцитонина пептид, адреномедуллин, интермедии, относящийся к гену кальцитонина пептид II и стимулирующие рецептор кальцитонина пептиды (calcitonin receptor stimulating peptide, CRSP) 1, 2, 3, 4 и 5. Среди них CRSP-1 является предпочтительным для применения в настоящем изобретении среди других пептидов, стимулирующих рецептор кальцитонина.
Эти представители семейства кальцитонина имеют высокую степень гомологии аминокислотных последовательностей, а также структурное сходство. Это сходство включает образованную дисульфидным мостиком петлю из 6 или 7 аминокислот на N-конце, амидированный по С-концу ароматический остаток, находящийся на С-конце, и участок с предсказанной амфипатической α-спиральной структурой из остатков 8-18 или 8-22.
Аминокислотные последовательности других представителей семейства кальцитонина из различных видов показаны ниже:
АМИЛИН
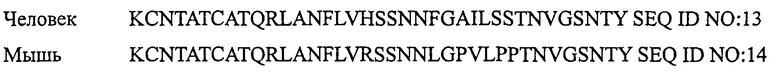
ПЕТИД, ОТНОСЯЩИЙСЯ К ГЕНУ КАЛЬЦИТОНИНА
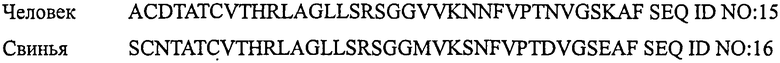
АДРЕНОМЕДУЛЛИН
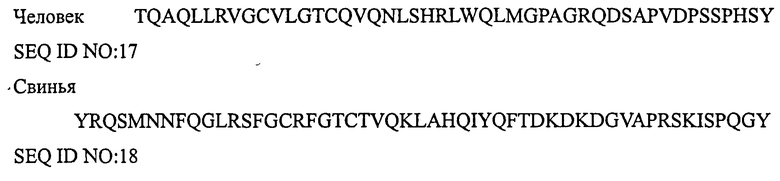
ИНТЕРМЕДИН
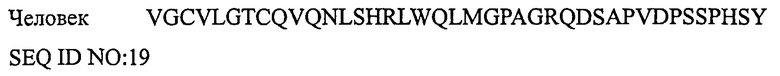
ПЕПТИД II, ОТНОСЯЩИЙСЯ К ГЕНУ КАЛЬЦИТОНИНА
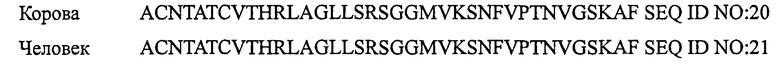
ПЕПТИД-1, СТИМУЛИРУЮЩИЙ РЕЦЕПТОР КАЛЬЦИТОНИНА

ПЕПТИД-2, СТИМУЛИРУЮЩИЙ РЕЦЕПТОР КАЛЬЦИТОНИНА

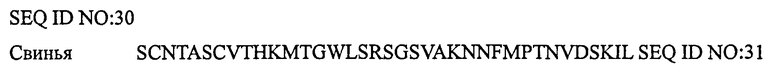
ПЕПТИД-3, СТИМУЛИРУЮЩИЙ РЕЦЕПТОР КАЛЬЦИТОНИНА

ПЕПТИД-4, СТИМУЛИРУЮЩИЙ РЕЦЕПТОР КАЛЬЦИТОНИНА
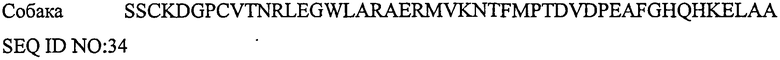
ПЕПТИД-5, СТИМУЛИРУЮЩИЙ РЕЦЕПТОР КАЛЬЦИТОНИНА
Можно отметить, что хотя они экспрессируются разными генами и образуются из разных пептидов-предшественников, зрелые пептиды CRSP-2, -3 и -4 у собаки являются одинаковыми.
Последовательности некоторых представителей семейства кальцитонина также показаны на фигурах 6 и 7. На фигуре 6 пустые места указывают, что соответствующая аминокислота является такой же, как показано в последовательности амилина крысы, напечатанный однобуквенный код аминокислоты указывает, что последовательность содержит данную аминокислоту в указанном положении, и заштрихованный квадрат указывает, что кальцитонины не содержат аминокислот, соответствующих остаткам 23-27 в амилине. На фигуре 7 участки в рамке являются полностью гомологичными (за исключением делеций) и заштрихованные области указывают, что в этих положениях в данных последовательностях аминокислоты отсутствуют.
Разные представители семейства способны в значительной степени связываться и активировать те же самые рецепторы, которые активирует кальцитонин, то есть способны действовать как агонисты рецептора кальцитонина (СТ-рецептора). В отличие от большинства рецепторов, сопряженных с G-белками, СТ-рецептор может модифицироваться при связывании одного из трех белков, модифицирующих активность рецептора (receptor activity modifying proteins, RAMPs), которые пронизывают мембрану один раз.
В настоящем изобретении термин «агонист СТ рецептора» обозначает любое соединение, но особенно пептид, способный связываться и активировать СТ рецептор способом, который может быть продемонстрирован с помощью, по меньшей мере, одного из «протоколов исследования», как показано ниже.
Термин «представитель семейства кальцитонина» обозначает любой один из кальцитонина, амилина, относящегося к гену кальцитонина пептида, адреномедуллина, интермедина, относящегося к гену кальцитонина пептида II и стимулирующего рецептор кальцитонина пептида-1, которые встречаются в природе у любых видов.
Термин «модифицированный представитель семейства кальцитонина» обозначает соединение, имеющее аминокислотную последовательность любого представителя семейства кальцитонина, о котором идет речь, модифицированную по сравнению с нативной последовательностью, но таким образом, чтобы данное соединение представляло собой агонист СТ рецептора. Модификация может быть проведена для разных целей, включая увеличение эффекта данного соединения как агониста СТ рецептора, для увеличения биологического времени полужизни данного соединения, или для того, чтобы обеспечить для композиции с данным соединением, приготовленной для фармацевтического применения, увеличения его стабильности при хранении.
Названия индивидуальных представителей семейства кальцитонина, например «кальцитонин», относятся к любому встречающемуся в природе такому представителю семейства из любого вида, включая каждый из видов, для которых последовательности кальцитонина приведены выше, если не указано иначе.
Термин «модифицированный», стоящий перед названием индивидуального представителя семейства кальцитонина, обозначает соединение, имеющее аминокислотную последовательность представителя семейства кальцитонина, о котором идет речь, модифицированную по сравнению с нативной последовательностью, но таким образом, чтобы соединение, о котором идет речь, представляло собой агонист СТ рецептора.
Модифицированные представители семейства кальцитонина для применения согласно настоящему изобретению не модифицированного включают амилина и, следовательно, не должны иметь более чем 50% гомологии с амилином, предпочтительно не более чем 30%. Из фигуры 5 можно видеть, что кальцитонин лосося содержит только 10 из 37 аминокислот амилина в своей последовательности из 32 аминокислот, и таким образом имеет 10/32*100% гомологию с амилином, то есть 27%. Модификации кальцитонина до такой степени, что они делают его «слишком похожим на амилин», исключаются.
Способом модификаций указанных выше аминокислотных последовательностей в модифицированных представителях семейства кальцитонина может быть вставка, деления или замена на природные или не встречающиеся в природе аминокислоты. Предпочтительно модифицированная последовательность является, по меньшей мере, на 75% гомологичной, более предпочтительно, по меньшей мере, на 90%, более предпочтительно, по меньшей мере, на 95% гомологичной нативной последовательности представителя семейства кальцитонина, о котором идет речь.
В соответствии с этим настоящее изобретение включает композицию, в которой активное соединение представляет собой модифицированный представитель семейства кальцитонина, имеющий, по меньшей мере, 75% аминокислотной гомологии с представителем семейства кальцитонина, но не амилином, и который модифицирован по отношению к указанному представителю семейства кальцитонина путем вставки, замены или делеции аминокислот и сохраняет способность связываться и активировать рецептор кальцитонина.
Среди видов, порядок предпочтения для представителей семейства кальцитонина для применения согласно настоящему изобретению таков: костные рыбы > птицы > млекопитающие, но не человек > человек.
Представитель семейства кальцитонина, если встречается в природе, может быть натуральным или может быть синтезированным (включая рекомбинантные), и если не встречается в природе, может быть синтезированным.
Среди встречающихся в природе кальцитонинов кальцитонин лосося является особенно предпочтительным.
Протоколы испытаний
Для выяснения того, является ли соединение-кандидат агонистом рецептора кальцитонина, было разработано четыре протокола испытаний.
Клетки COS-7 выращивали до 80% конфлуентности в колбах на 75 см2. Для трансфекции клеток последовательностями, кодирующими рецептор, использовали pcDNA3.1(+) от Invitrogen. Клетки трансфицировали, используя 300 нг конструкции pcDNA-CTR самой по себе (Протокол испытаний 1), или ко-трансфицировали, используя 300 нг конструкции pcDNA-CTR и 1 мкг pcDNA-RAMP-1 (Протокол испытаний 2), или pcDNA-RAMP-2 (Протокол испытаний 3), или pcDNA-RAMP-3 (Протокол испытаний 4), с использованием 7,8 мкл реагента FuGene 6.
Последовательность, включенная в конструкцию pcDNA-CTR, является такой, которая дана для cDNA рецептора кальцитонина человека в банке генов под номером CALCR: NM_001742. Последовательности RAMP I DNA, RAMP 2 DNA и RAMP 3 DNA, включенные в конструкции pcDNA-RAMP, представляют собой последовательности RAMP человека, представленные в банке генов под следующими специфическими номерами:
RAMPL NM_005855
RAMP2: NM 005854
RAMP3: NM 005856
Через 48 часов клетки снимали обработкой трипсином, центрифугировали при 500 × g и ресуспендировали в циклазном буфере (DMEM, содержащий 0,1% (вес/объем) БСА и 1 мМ IBMX). Аликвоты клеток (5×105) вносили в 1,5 мл пробирки Eppendorf и преинкубировали в течение 20 мин при 37°С. Далее клетки инкубировали в течение 18 мин в отсутствие (базальный уровень) или в присутствии возрастающих концентраций агонистов: 100 микромолярная, 1 микромолярная, 0,01 микромолярная. В качестве положительного контроля для этой системы проводили инкубацию в присутствии 1 мМ форсколина для определения максимального накопления цАМФ.
После инкубации в реакционных средах количественно измеряли внутриклеточный вторичный мессенджер цАМФ согласно инструкции к набору cAMP-EIA (Amersham Biosciences, US).
Исследуемое соединение рассматривалось в качестве агониста CTR, если оно в концентрации 10 мкМ индуцировало образование на 50% больше цАМФ в любом одном из четырех протоколов испытаний, чем носитель, используемый как контроль (DMEM, содержащий 0,1% (вес/объем) БСА и 1 мМ изобутилметилксантин (IBMX)). sCT обеспечивает (по меньшей мере, в протоколе испытаний 1) более чем 10-кратную индукцию по сравнению с отрицательным контролем, даже при использовании в концентрации 1 мкМ. Таким образом, предпочтительно, чтобы соединение для применения согласно настоящему изобретению обеспечивало, по меньшей мере, 100% (то есть 2-кратную) индукцию, по меньшей мере, в одном из протоколов испытаний, предпочтительно, по меньшей мере, 5-кратную индукцию. Альтернативно этому, соединение для применения согласно настоящему изобретению обеспечивает, по меньшей мере, 25% от индукции, обеспечиваемой sCT, по меньшей мере, в одном из указанных протоколов испытания, предпочтительно, по меньшей мере, 50% от индукции, обеспечиваемой sCT.
Предпочтительные агонисты CTR дают положительный результат, по меньшей мере, в двух из указанных протоколов (предпочтительно в протоколах испытаний 1 и 2 или 1 и 4), предпочтительно в трех протоколах испытаний (предпочтительно в 1, 2 и 4) и наиболее предпочтительно во все четырех протоколах испытаний. Альтернативно этому, предпочтительно, чтобы ответ, обеспечиваемый тестируемым соединением в протоколе испытаний 1, был, по меньшей мере, на 25%, более предпочтительно, по меньшей мере, на 50%, еще более предпочтительно, по меньшей мере, на 100% больше, чем ответ, обеспечиваемый тестируемым соединением в любом из протоколов испытаний 2-4.
Примеры модифицированных кальцитонинов, которые могут применяться в настоящем изобретении, можно найти в US 5536812. Таким образом, С-концевой пролинамид кальцитонина может быть замещен (заменен) на гомосеринамид (Hse-NH^).
Модифицированные таким образом кальцитонины лосося или угря являются наиболее предпочтительными.
Кальцитонины или другие представители семейства кальцитонина могут быть модифицированы так, как указано в GB 1590645, путем замены кольцевого элемента Cys-Cys на более стабильную структуру, такую, которая обеспечивается путем замены первого и второго из этих остатков Cys (обычно первой и седьмой аминокислот) на аминосубериновую кислоту, в результате чего sCT приобретает следующую структуру:
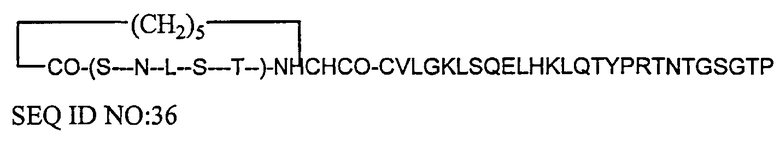
Как описано в ЕР 0464549 пептиды-аналоги кальцитонина, действующие как агонисты рецептора кальцитонина, могут быть представлены общей формулой:
CX1NLSTCX2LGX3X4X5GX6X7X8LX9X10TX11PX12TX13X14GX15GX16P-X17
SEQ ID N0:37
где X1 представляет собой Ser, Gly или Ala; X2 представляет собой Val или Met; X3 представляет собой Thr или Lys; X4 представляет собой Tyr или Leu; X5 представляет собой Thr или Ser; X6 представляет собой Asp или Glu; X7 представляет собой Phe или Leu; X8 представляет собой Asn или His; X9 представляет собой Tyr, Phe или Leu; X10 представляет собой His или Gln; X11 представляет собой Tyr или Phe; X12 представляет собой Gln или Arg; X13 представляет собой Ala, Ser, Asn или Asp; X14 представляет собой Ile, Thr или Val; X15 представляет собой Val, Ser или Ala; X16 представляет собой Ala, Thr или Val; и X17 представляет собой амидированный гомосерин, гомосеринлактон, прореагировавший с первичным алкиламином, содержащим 1-20 атомов углерода, или необязательно с полипептидной цепью, и содержащий амидированный гомосерин на С-конце.
Альтернативно этому, аналог кальцитонина может иметь последовательность РО-1 (CGNLSTCMLGKLSQELHKLQTYPQTAIGVGAP-NH2 SEQ ID NO:38), имеющую аминокислотную последовательность обоих, N- и С-концевых, десяти аминокислот, такую же, как у кальцитонина человека, и 12 аминокислот центральной части таких, как в кальцитонине лосося, или РО-23 ([цикло-Asp1, Lys7]-[des-Gly2]-[Leu8]-PO-1), или РО-29 ([Asp15, Asn17, Phe19, His20]-PO-23). РО-23 имеет N-концевую Cys-Cys S-S связь из РО-1, замещенную кольцевой структурой, образованной из Asp-Lys пептидной связью, для усиления физико-химической стабильности. РО-29 представляет собой центральную часть молекулы РО-23, модифицированную для более точной имитации кальцитонина человека.
В литературе известно много других модифицированных кальцитонинов, обладающих свойствами агонистов рецептора кальцитонина.
Агонисты рецептора кальцитонина, включенные для применения в настоящем изобретении, включают «низкомолекулярные» (не пептидные) агонисты.
Они являются предпочтительно такими, которые удовлетворяют классическим правилам применимости в качестве лекарства (druggability) и предпочтительно имеют, по меньшей мере, 4 свойства из следующих 5: молекулярный вес MW≤500, logP (логарифм коэффициента его распределения между н-октанолом и водой log(соктанол/cвода))≤5, атомов-доноров Н-связей≤5, атомов-акцепторов Н-связей (сумма атомов N и О)≤10, и необязательно одно или оба из следующих свойств: площадь полярной поверхности ≤140 А2 (или сумма атомов, доноров и акцепторов Н-связей, ≤12) и число связей, вокруг которых происходит вращение, ≤10.
Приемлемые лекарственные формы для применения согласно настоящему изобретению включают таблетки, минитаблетки, капсулы, гранулы, драже, порошки, твердые шипучие и твердые жевательные композиции. Такие композиции могут включать желатин, который предпочтительно представляет собой гидролизованный желатин или низкомолекулярный желатин. Такие композиции могут быть получены путем высушивания замороженного гомогенного водного раствора, включающего кальцитонин, или его фрагмент, или конъюгат и гидролизованный желатин или низкомолекулярный желатин, и дополнительной обработки полученного твердого материала для получения указанной фармацевтической композиции, и в которых желатин может иметь средний молекулярный вес от 1000 до 15000 дальтон. Такие композиции могут включать соединение, выполняющее роль защитного носителя, такое как 5-CNAC или другие, которые раскрываются в настоящем изобретении.
Хотя пероральные композиции, такие как таблетки и капсулы, являются предпочтительными, композиции для применения согласно настоящему изобретению могут применяться в форме суппозиториев или тому подобного. Пероральная доставка кальцитонина, например кальцитонина лосося, обычно является предпочтительным способом доставки вследствие своего удобства, относительной легкости и общей безболезненности, что приводит к лучшему соблюдению пациентами инструкций по приему лекарства по сравнению с другими способами доставки. Однако биологические, химические и физические барьеры, такие как изменение рН в желудочно-кишечном тракте, мощные пищеварительные ферменты и непроницаемые для активного агента желудочно-кишечные мембраны, делают проблематичной пероральную доставку кальцитонинов, например кальцитонина лосося, млекопитающим, например, пероральную доставку кальцитонинов, которые представляют собой длинноцепочечные полипептидные гормоны, секретируемые парафолликулярными клетками щитовидной железы у млекопитающих и ультимобранхиальной железой птиц и рыб, как изначально показано, она является трудной вследствие, по меньшей мере, отчасти недостаточной стабильности кальцитонина в желудочно-кишечном тракте, а также вследствие неспособности кальцитонина легко переносится через кишечные стенки в кровоток. Тем не менее, пригодные пероральные композиции описаны ниже.
Кальцитонин и другие представители семейства могут быть приготовлены для энтерального, особенно перорального введения путем смешивания с пригодным соединением-носителем. Введенный перорально либо сам по себе, либо в виде водного раствора/суспензии, агент является неэффективным для обеспечения эффектов сохранения костей. Пригодные соединения-носители включают таковые, описанные в US 5773647 и US 5866536, и среди них 5-CNAC (N-(5-хлорсалицилоил)-8-аминокаприловую кислоту, обычно в виде ее динатриевой соли), которая является особенно эффективной. Другими предпочтительными носителями или средствами доставки являются SNAD (натриевая соль 10-(2-гидроксибензамидо)декановой (каприновой) кислоты) и SNAC (натриевая соль N-(8-[2-гидроксибензоил]амино)каприловой кислоты).
Кроме того, WO 00/059863 раскрывает динатриевые соли с формулой I
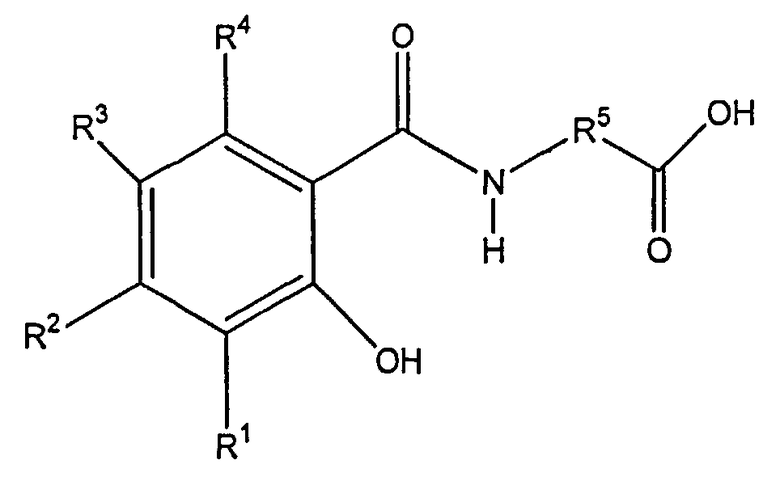
где R1, R2, R3, и R4 независимо друг от друга представляют собой водород, -ОН, -NR6R7, галоген, С1-С4алкил, или C1-C4алкокси;
R5 представляет собой замещенный или незамещенный C2-C16алкилен, замещенный или незамещенный C1-C12алкилен, замещенный или незамещенный C1-C12алкил(арилен), или замещенный или незамещенный арил(С1-С12алкилен); и
R6 и R7 независимо друг от друга представляют собой водород, кислород, или C1-C4 алкил; и их гидраты и сольваты, в качестве особенно эффективных для пероральной доставки активных агентов, таких как кальцитонины, например кальцитонин лосося, и они могут применяться в настоящем изобретении.
Предпочтительные композиции для кишечного всасывания кальцитонина лосося и необязательно тонкодисперсного 5-CNAC могут быть такими, как описано в WO 2005/014031.
Кальцитонин и другие представители семейства могут быть приготовлены в виде композиций для перорального введения с помощью способов, применяемых для продукта марки Capsitonin компании Bone Medical Limited. Они могут включать способы, применяемые в композициях Axcess. Более конкретно, активный ингредиент может быть инкапсулирован в капсулу для рассасывания в кишечнике, которая способна выдержать прохождение через желудок. Капсула может содержать активное соединение вместе с гидрофильным ароматическим спиртом, усиливающим всасывание, например, как описано в WO 02/028436. Известно, что покрытие для кишечного рассасывания может становиться проницаемым рН-зависимым способом, например, при повышении рН от 3 до 7. WO 2004/091584 также описывает пригодные способы приготовления композиций с применением ароматических спиртов, усиливающих всасывание.
Кальцитонин или другие представители семейства могут быть приготовлены в виде композиций с применением способов, используемых для продуктов компании Unigene Enteripep®. Способы могут включать таковые, как описано в US 5912014, US 6086918 или US 6673574. В частности, это может включать применение конъюгации кальцитонина или других представителей семейства с переносчиком через мембрану, таким как белок-трансдуцирующий домен белка HIV ТАТ, одновременное введение в композицию одного или более протеазных ингибиторов, и/или агента, снижающего рН, и/или устойчивого к кислоте защитного носителя, и/или усилителя всасывания, который может представлять собой сурфактант.
Кальцитонин или другие представители семейства могут быть приготовлены в виде композиций с применением способов, используемых для продуктов компании Oramed, которые могут включать приготовление композиции с омега-3 жирной кислотой, как описано в WO 2007/029238 или как описано в US 5102666.
Как правило, могут применяться фармацевтически приемлемые соли (особенно моно- и динатриевые соли), сольваты (например, спиртовые сольваты) и гидраты этих носителей или агентов для доставки.
Фармацевтические композиции настоящего изобретения обычно содержат эффективное для доставки количество носителя, такого как 5-CNAC, то есть количество, достаточное для того, чтобы доставить кальцитонин для желаемого эффекта. Как правило, носитель, такой как 5-CNAC, присутствует в количестве от 2,5% до 99,4% (по весу), более предпочтительно от 25% до 50% (по весу) от всей композиции. Пероральное введение фармацевтических композиций согласно настоящему изобретению может производиться регулярно, например один или более раз на дневной или недельной основе; периодически, например, нерегулярно в течение дня или недели; или циклически, например, регулярно в течение некоторого количества дней или недель с последующим периодом без введения. Лекарственная форма фармацевтических композиций настоящего изобретения может представлять собой любую известную форму, например жидкие или твердые лекарственные формы. Жидкие лекарственные формы включают растворы эмульсий, суспензий, сиропов и эликсиров. В дополнение к кальцитонину и носителю, такому как 5-CNAC, жидкие композиции могут также включать инертные наполнители, обычно применяемые в данной области техники, такие как солюбилизирующие агенты, например этанол; масла, такие как хлопковое масло, касторовое масло и кунжутное масло; увлажняющие агенты; эмульгирующие агенты; суспендирующие агенты; подсластители; ароматизаторы; и растворители, такие как вода. Твердые лекарственные формы включают капсулы, мягкие гелевые капсулы, таблетки, таблетки, покрытые оболочкой, порошки, гранулы или другие пероральные твердые лекарственные формы, все из которых могут быть приготовлены способами, хорошо известными в данной области техники. Фармацевтические композиции могут дополнительно включать добавки в обычно применяемых количествах, включая, но ими не ограничиваясь, регулятор рН, консервант, ароматизатор, маскирующий вкус агент, аромат(изатор), увлажнитель, тонизирующий агент (tonicifier), краситель, сурфактант, пластификатор (смягчитель), смазочное вещество, такое как стеарат магния, регулятор текучести, регулятор сжатия, солюбилизирующий агент, наполнитель, растворитель, такой как микрокристаллическая целлюлоза, например Avicel РН 102, поставляемый корпорацией FMC, или любую их комбинацию. Другие добавки могут включать буферы на основе фосфатных солей, лимонную кислоту, гликоли и другие диспергирующие агенты. Композиция может также включать один или более ингибиторов ферментов, таких как актинонин или эпиактинонин и их производные; апротинин, Trasylol и ингибитор Боумена-Берка (Bowman-Birk inhibitor). Кроме того, в композиции настоящего изобретения может присутствовать ингибитор транспорта, то есть, [rho]-гликопротеин, такой как Ketoprofin. Твердые фармцевтические композиции настоящего изобретения могут быть приготовлены обычными способами, например перемешиванием смести кальцитонина, носителя, такого как 5-CNAC, и любых других ингредиентов, растиранием и заполнением данной смесью капсул, или вместо заполнения капсул, прессованием с последующим таблетированием, или формованием под давлением для получения таблеток. Кроме того, твердая дисперсия может формироваться известными способами и последующим приготовлением в форме таблетки или капсулы. Предпочтительно ингредиенты в фармацевтических композициях настоящего изобретения являются гомогенно или однородно смешанными по всему объему твердой лекарственной формы.
Альтернативно этому активное соединение может быть приготовлено в виде конъюгата с указанным носителем, который может представлять собой олигомер, как описано в US 2003/0069170, например,
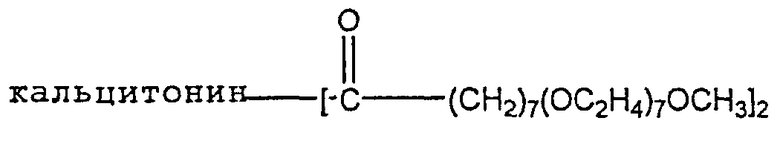
который в случае конъгирования с кальцитонином лосося известен как СТ-025. Такие конъюгаты могут вводиться в комбинации с жирной кислотой и солью желчной кислоты, как здесь описано.
Могут применяться конъюгаты с полиэтиленгликолем (ПЭГ), как описано, например, в работе Mansoor et al..
Альтернативно этому, активные соединения могут быть смешаны с нитрозо-N-ацетил-D,L-пеницилламином (SNAP) и раствором карбопола (Carbopol) или с таурохолатом и раствором карбопола для получения микроадгезивной эмульсии.
Активное соединение может быть приготовлено путем помещения в нанокапсулы из хитозана, как это раскрывается в работе Prego et al. (необязательно, модифицированные ПЭГ, как в работе Prego Prego С, Torres D, Femandez-Megia E, Novoa-Carballal R, Quinoa E, Alonso MJ.), или покрытые хитозаном или ПЭГ липидные наночастицы, как раскрывается в работе Garcia-Fuentes et al.. Хитозановые наночастицы для этой цели могут быть модифицированы иминотиоланом, как описано в работе Guggi et al.. Они могут быть приготовлены в эмульсиях вода/масло/вода, как описано в работе Dogru et al.. Биодоступность активных соединений может быть увеличена путем применения тауродезоксихолата или лауроилкарнитина, как описано в работе Sinko et al. или в работе Song et al.. В целом, пригодные наночастицы в качестве носителей обсуждаются в работе de la Fuente et al. и могут применяться в настоящем изобретении.
Другие подходящие стратегии для пероральной композиции включают применение системы временного усилителя проницаемости (transient permeability enhancer, TPE), как описано в WO 2005/094785, принадлежащем Chiasma Ltd. TPE обеспечивает применение масляной суспензии твердых гидрофильных частиц в гидрофобной среде для защиты молекулы лекарства от инактивации в недружелюбном окружении желудочно-кишечного тракта (ЖКТ), и в то же время действует на стенку ЖКТ, индуцируя проницаемость доставляемых им молекул лекарства.
Дополнительно включено применение глутатиона или соединений, содержащих многочисленные тиоловые группы, как описано в US 2008/0200563, для ингибирования работы выкачивающих насосов в мембране слизистой оболочки. Практические примеры таких способов описаны также в работе Caliceti, P. Salmaso, S., Walker, G. and Bemkop-Schnurch, A. (2004) 'Development and in vivo evaluation of an oral insulin-PEG delivery system.' Eur. J. Pharm. Sci., 22, 315-323, в работе Guggi, D., Krauland, A.H., and Bemkop-Schnurch, A. (2003) 'Systemic peptide delivery via the stomach: in vivo evaluation of an oral dosage form for salmon calcitonin'. J. Control. Rel. 92, 125-135, и в работе Bernkop-Schnürch, A., Pinter, Y., Guggi, D., Kahlbacher, H., Schöffmann, G., Schuh, M., Schmerold, I., Del Curto, M.D., D'Antonio, M., Esposito, P. and Huck, Ch. (2005) "The use of thiolated polymers as carrier matrix in oral peptide delivery' - Proof of concept. J. Control. Release, 106,26-33.
Активное соединение может быть помещено в бесшовные микросферы, как описано в WO 2004/084870, где активный фармацевтический ингредиент солюбилизируется в виде эмульсии, микроэмульсии или суспензии, помещается внутрь минисфер и покрывается разными способами либо с помощью обычных, либо с помощью новых способов нанесения покрытий. Результат представляет собой инкапсулированное лекарство в «заранее солюбилизированной» форме, которое при пероральном введении обеспечивает заранее заданное немедленное или длительное высвобождение активного лекарства в специфических местах и со специфическими скоростями по длине желудочно-кишечного тракта. По существу, предварительная солюбилизация лекарства увеличивает предсказуемость его кинетического профиля, одновременно увеличивая проницаемость и стабильность лекарства.
Можно применять покрытые хитозаном нанокапсулы, как описано в US 2009/0074824. Активная молекула, вводимая таким способом, является защищенной внутри нанокапсул, поскольку они являются устойчивыми к действию желудочного сока. Кроме того, микроадгезивные свойства системы увеличивают время адгезии к кишечным стенкам (было подтверждено, что наблюдается задержка в продвижении этих систем по желудочно-кишечному тракту), облегчая более эффективное поглощение активной молекулы.
Могут применяться способы, разработанные компанией TSR1 Inc. Эти способы включают технологию гидрофильной солюбилизации (Hydrophilic Solubilization Technology, HST), в которой желатин, природный экстракт коллагена, несущий как положительные, так и отрицательные заряды, покрывает частицы активного ингредиента, содержащегося в лецитиновых мицеллах, и предотвращает их агрегацию или слипание. Это приводит к улучшенной смачиваемости частиц гидрофобного лекарства за счет полярных взаимодействий. Кроме того, амфифильный лецитин снижает поверхностное натяжение между растворяющей жидкостью и поверхностью частиц.
Активный ингредиент может быть приготовлен в виде композиции с кукурбитурилами в качестве наполнителей.
Альтернативно этому, может применяться технология GIPET компании Merrion Pharmaceuticals для получения покрытых таблеток для кишечного рассасывания, содержащих активный ингредиент с усилителем всасывания, который может представлять собой жирную кислоту со средней длиной цепи, производное жирной кислоты со средней длиной цепи, как описано в US 2007/0238707, или транслоцируемый через мембрану пептид, как описано в US 7268214.
Можно применять технологию GIRES™, которая состоит из лекарственной формы с контролируемым высвобождением внутри надуваемого мешка, который помещается в лекарственную капсулу для перорального введения. При растворении капсулы генерирующая газ система надувает мешок в желудке. В клинических испытаниях было показано, что мешок остается в желудке в течение 16-24 часов.
Альтернативно этому, активное соединение может быть конъюгировано с защитным модификатором, что позволяет ему избежать ферментативной деградации в желудке, и облегчает его всасывание. Активное соединение может быть ковалентно конъюгировано с монодисперсным производным короткоцепочечных полиэтиленгликоль-гликолипидов, которые кристаллизуются и лиофилизируются в сухой активный фармацевтический ингредиент после очистки. Такие способы описаны в US 5438040 и на сайте www.biocon.com.
Для доставки активного соединения также можно применять направленную на печень везикулу (hepatic-directed vesicle, HDV). HDV может состоять из липосом (диаметр ≤150 нм), содержащих активное соединение, которые также содержат в своем липидном бислое молекулу, мишенью которой являются гепатоциты. Данная адресующая молекула обеспечивает доставку инкапсулированного активного соединения в клетки печени, вследствие чего для обеспечения действия требуется относительно небольшое количество активного соединения. Такая технология описана в US 2009/0087479 и дополнительно на сайте www.diasome.com.
Активное соединение может быть включено в композицию, дополнительно содержащую в значительной степени не водную гидрофильную среду, включающую спирт и ко-растворитель в комбинации с неполным глицеридом со средней длиной цепей, необязательно в смеси с длинноцепочечными видами ПЭГ, как описано в US 2002/0115592 в отношении инсулина.
Альтернативно этому, можно применять кишечные пластыри, как описано в работе Shen Z, Mitragotri S, Pharm Res. 2002 Apr; 19 (4): 391-5 'Intestinal patches for oral drug delivery'.
Активное соединение может быть заключено в разрушающуюся матрицу, сделанную из гидрогеля, смешанного с гидрофобным полимером, как описано в US 7189414.
Пригодные уровни пероральной лекарственной формы кальцитонина для взрослых людей, нуждающихся в лечении, могут находиться в диапазоне от 0,05 до 5 мг, предпочтительно примерно от 0,1 до 2,5 мг.
Дозировки агонистов рецептора кальцитонина могут быть такими, как описано выше для кальцитонина, необязательно с коррекцией на относительную эффективность в качестве агониста данного конкретного агониста по сравнению с самим кальцитонином в Протоколах испытаний, описанных выше.
Частота приема лекарства пациентами может составлять от 1 до шести раз в день, например, от двух до четырех раз в день. Лечение желательно должно проводиться в течение длительного периода времени, по меньшей мере, 6 недель, предпочтительно, по меньшей мере, 6 месяцев, предпочтительно, по меньшей мере, год, и необязательно пожизненно.
Комбинированное лечение при соответствующих состояниях может проводиться с применением композиции согласно настоящему изобретению и с отдельным введением одного или более других лекарств. Альтернативно этому, композиция согласно настоящему изобретению может включать одно или более других лекарств для совместного введения.
Комбинированное лечение согласно настоящему изобретению включает комбинации активного соединения, как описано, с инсулином, GLP-2, GLP-1, GIP или амилином или обычно с другими анти-диабетическими препаратами. Таким образом, комбинированное лечение, включая применение совместных композиций, может проводиться с применением сенсибилизирующих агентов для инсулина, включая бигуаниды, такие как метформин, буформин и фенформин, тиазолидинедионы (TZD's) (PPAR), такие как пиоглитазон, ривоглитазон, розиглитазон и троглитазон, двойные агонисты PPAR, такие как алеглитазар, мураглитазар и тезаглитазар, или стимуляторы секреции, включая препараты сульфонилмочевины, такие как карбутамид, хлорпропамид, гликлазид, толбутамид, толазамид, глипизид, глибенкламид, глибурид, гликвидон, гликлопирамид и глимепририд, меглитиниды/глиниды (K+), такие как натеглинид, репаглинид и митиглинид, аналоги GLP-1, такие как эксенатид, лираглутид и албиглутид, ингибиторы DPP-4, такие как алоглиптин, линаглиптин, саксаглиптин, ситаглиптин и вилдаглиптин, аналоги инсулина или специфические композиции, такие как (быстрого действия) инсулин лиспро, инсулин аспарт, инсулин глулизин, (длительного действия) инсулин гларгин, инсулин детемир), вдыхаемый инсулин - эксубра и NPH инсулин, и другие, включая ингибиторы альфа-глюкозидазы, такие как акарбоза, миглитол и воглибоза, аналоги амилига, такие как прамлинтид, ингибиторы SGLT2, такие как дапаглифлозин, ремоглифлозин и серглифлозин, а также агенты смешанного действия, включая бенфлуорекс и толрестат.
Мы предполагаем, что эффект введенных через кишечник представителей семейства кальцитонина, особенно самого кальцитонина, для улучшения уровней глюкозы у пациентов, страдающих от резистентности к инсулину, может быть объяснен на основе того, что кальцитонин вызывает противоположные эффекты на скелетные мышцы и на печень в отношении увеличения или уменьшения уровня глюкозы в сыворотке соответственно, и что введение через кишечник преимущественно обеспечивает доставку кальцитонина в печень, тогда как парентеральное введение кальцитонина обеспечивает его преимущественное действие на скелетные мышцы. Мы предполагаем, что при действии на скелетные мышцы кальцитонин стимулирует потребление глюкозы с образованием лактата, который попадает в печень и стимулирует печень к образованию глюкозы. С другой стороны, кальцитонин действует в печени как активатор образования гликогена для запасания глюкозы.
Эта теория может объяснить аномальные результаты, о которых сообщал Lupulescu, где очень высокие дозы кальцитонина при инъекции приводили к снижению уровня глюкозы в плазме, в противоположность результатам, полученным во всех других исследованиях на животных. Мы предполагаем, что используемые очень высокие дозы могли быть достаточными для того, чтобы активировать рецепторы для кальцитонина в печени, в результате чего был преодолен ответ скелетных мышц, который наблюдался в других исследованиях.
Активное соединение может представлять собой агонист рецептора кальцитонина. В данной области техники известно много агонистов, которые по своей природе не являются пептидами, а скорее представляет собой синтетические низкомолекулярные соединения. Другими известными агонистами рецептора кальцитонина являются пептиды, имитирующие кальцитонин. Примеры обоих типов агонистов обсуждаются ниже.
Агонисты рецептора кальцитонина, которые описаны для применения согласно настоящему изобретению, включают таковые, которые описаны в JP 2001294574, включая таковые с общей формулой
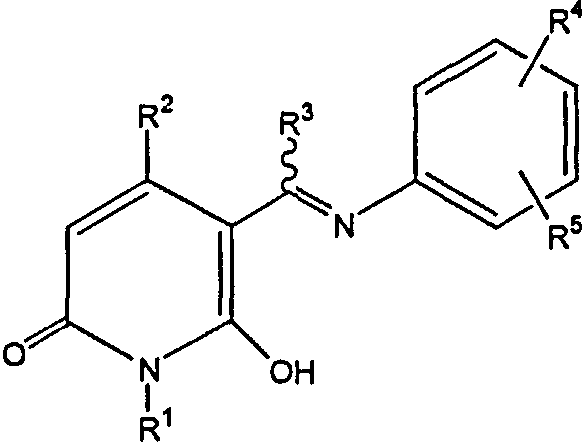
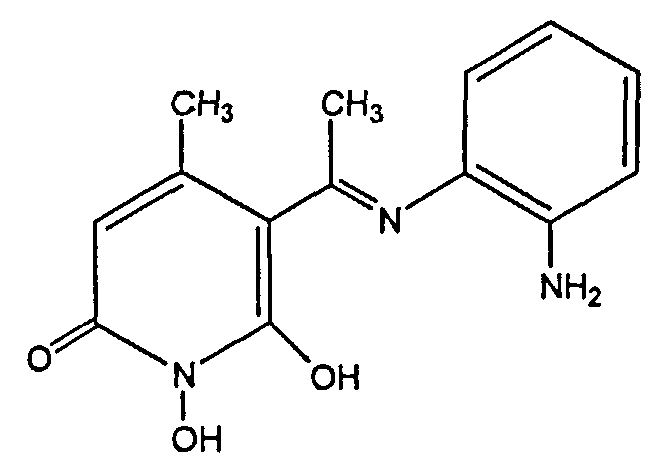
где R1 представляет собой Н, гидроксильную группу, 1-4С алкилоксигруппу или 7-10С аралкилоксигруппу; R2 и R3 каждый представляют собой 1-4С алкильную группу; и R4 и R5 каждый представляют собой Н, атом галогена, гидроксильную группу, 1-4С алкильную группу, 7-10С аралкильную группу, 1-4С алкилокси группу, 7-10С аралкилоксигруппу, 1-7С ацилоксигруппу, аминогруппу, 1-4С алкиламино группу, 17-10С аралкиламино группу, 1-7С ациламиногруппу, карбоксильную группу, 1-4С алкилоксикарбонильную группу, 7-10С аралкилоксикарбонильную группу, карбамоильную группу, которая может иметь, по меньшей мере, один заместитель, ацильную группу или сульфамоимльную группу и фармакологические приемлемые их соли.
Предпочтительное соединение (SUN B8155) представляет собой соединение с формулой
Миметики кальцитонина для применения согласно настоящему изобретению включают таковые, описанные в WO 99/37604. Там описываются соединения с формулой
где R1 и R2 каждый представляют собой членов, независимо выбираемых из группы, состоящей из водорода, алкилов, имеющих от 1 до 6 атомов углерода, алкенилов, имеющих от 1 до 6 атомов углерода, арила, замещенного арила, алкиларила, замещенного алкиларила, карбоциклического кольца, замещенного карбоциклического кольца, гетероциклического кольца, замещенного гетероциклического кольца, и их комбинаций, комбинации слиты или ковалентно связаны и заместители выбираются из группы, состоящей из галогена, галоалкила, гидрокси, арилокси, бензилокси, алкокси, галоалкокси, амино, моноалкиламино, диалкиламино, ацилокси, ацила, алкила и арила;
R3 представляет собой 2,5 двузамещенный арил;
R4 и R5 каждый независимо выбираются из группы, состоящей из водорода и алкилов, имеющих от 1 до 6 атомов углерода, или вместе выбираются из кольца, выбираемого из группы, состоящей из насыщенных или ненасыщенных пятичленных колец, насыщенных или ненасыщенных шестичленных колец и насыщенных или ненасыщенных семичленных колец;
Z и Х каждый независимо выбираются из группы NH, 0, S, или NR, где R представляет собой низшую алкильную группу, содержащую от 1 до 6 атомов углерода; и пит каждый независимо представляют собой целое число от 0 до 6.
Эти соединения включают соединения формулы
где R1 и R2 каждый независимо выбираются из группы, состоящей из водорода, алкилов, имеющих от 1 до 6 атомов углерода, алкенилов, имеющих от 1 до 6 атомов углерода, арила, замещенного арила, алкиларила, замещенного алкиларила, карбоциклического кольца, замещенного карбоциклического кольца, гетероциклического кольца, замещенного гетероциклического кольца, и их комбинаций, комбинации слиты или ковалентно связаны и заместители выбираются из группы, состоящей из галогена, галоалкила, гидрокси, арилокси, бензилокси, алкокси, галоалкокси, амино, моноалкиламино, диалкиламино, ацилокси, ацила, алкила и арила;
S1, S3 и S4 каждый независимо выбираются из группы, состоящей из водорода, галогена, галоалкила, гидрокси, арилокси, бензилокси, алкокси, галоалкокси, амино, моноалкиламино, диалкиламино, ацилокси, ацила, алкила и арила; и S2 и S5 каждый независимо представляют собой алкил или ар ил.
В частности, подходящий R1 представляет собой 4-этоксибензил, 1-этил-индолилметил, бензил, 4-аллоксибензил, 1-аллил-индолилметил, 4-хлорбензил, 4-фторбензил, 4-йодбензил, 2-нафтилметил или фенил;
R2 представляет собой этил, аллил, бензил или 2-нафтилметил;
и S2 и S5 представляют собой трет-бутил.
Дополнительные примеры соединений, которые могут применяться, можно найти в US 7396936. Они включают соединения формулы:
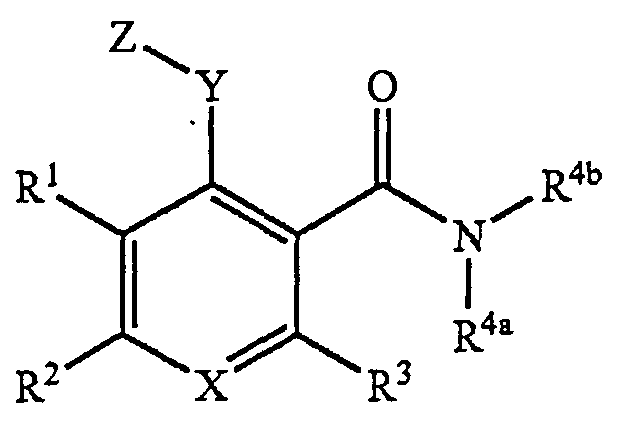
их стереоизомеры, их таутомеры, их сольваты, их пролекарства и их фармацевтически приемлемые соли,
где Х представляет собой N или NO;
Y представляет собой двухвалентный замещенный или незамещенный арил, гетероциклил, или циклоалкильную группу;
Z представляет собой -C(O)OR5, -C(O)NR6R7, -NR6C(O)R5, -NR6C(O)NR6R7, -C(O)R5, -NR6R7, -OR8, -SO2NR6R7, -NR6SO2R5, или -S(O)mR5;
R1 представляет собой -H, -C(O)OR9, -С(O)NR10R11, -CN, -C(O)R9, или -NR10C(O)R9;
R2 представляет собой -(C1-2алкил)-R12, где C1-2 алкил является замещенным или незамещенным;
R3 представляет собой С2-6 разветвленную или неразветвленную алкильную группу, необязательно замещенную одним или более F;
R4a и R4b каждый независимо представляют собой -H или замещенную или незамещенную С1-4алкильную группу;
R5 представляет собой замещенный или незамещенный аралкил, гетероаралкил, гетероциклилалкил, или циклоалкильную алкильную группу;
R6 и R7 каждый независимо представляют собой -Н или замещенный или незамещенный аралкил, гетероаралкил, гетероциклилаликл, или циклоалкильную алкильную группу; или
R6 и R7, когда присоединены к одному и тому же атому, вместе образуют замещенную или незамещенную гетероциклическую группу;
R8 представляет собой замещенный или незамещенный, разветвленный или неразветвленный С2-6алкил, С2-6алкенил, С2-6алкинил, или C7-10аралкильную группу;
R9 представляет собой -Н или замещенный или незамещенный алкил, гетероалкил, алкенил, алкинил, арил, аралкил, гетероциклил, или гетероциклилалкильную группу;
R10 и R11 каждый независимо представляют собой -Н или замещенный или незамещенный алкил, гетероалкил, алкенил, алкинил, арил, аралкил, гетероциклил, или гетероциклилалкильную группу; или R10 и R11, когда присоединены к одному и тому же атому, вместе образуют замещенную или незамещенную гетероциклическую группу;
R12 представляет собой замещенный или незамещенный циклоалкил, арил, гетероарильную группу, или представляет собой незамещенную алкильную группу; и m равно 0, 1 или 2.
Примеры включают этиловый эфир 5-Карбамоил-2-[2-(4-фтор-фенил)-этил]-4-{4-[(фуран-2-илметил)-карбамоил]-фенил}-6-пропил-никотиновой кислоты;
этиловый эфир 5-Карбамоил-2-(2-циклогексил-этил)-4-{4-[(фуран-2-илметил)-карбамоил]-фенил} -6-изобутил-никотиновой кислоты;
этиловый эфир 5-Карбамоил-2-[2-(4-фтор-фенил)-этил]-4-{4-[(фуран-2-илметил)-карбамоил]-фенил}-6-изобутил-никотиновой кислоты;
этиловый эфир 5-Карбамоил-2-(2-циклогексил-этил)-4-{4-[(фуран-2-илметил)-карбамоил]-фенил}-6-пропил-никотиновой кислоты;
этиловый эфир 5-Карбамоил-2-[2-(4-фтор-фенил)-этил]-6-пропил-4-{4-[(пиридин-3-илметил)-карбамоил]-фенил}-никотиновой кислоты;
этиловый эфир 5-Карбамоил-2-(2-циклогексил-этил)-6-пропил-4-{4-[(пиридин-3-илметил)-карбамоил]-фенил}-никотиновой кислоты;
диамид 2-[2-(4-Фтор-фенил)-этил]-4-{4-[(фуран-2-илметил)-карбамоил]-фенил}-6-изобутил-пиридин-3,5-дикарбоновой кислоты;
этиловый эфир 5-Карбамоил-6-этил-2-[2-(4-фтор-фенил)-этил]-4- {4-[(фуран-2-илметил)-карбамоил]-фенил}-никотиновой кислоты; и
этиловый эфир 5-Карбамоил-2-[2-(4-фтор-фенил)-этил]-6-изобутил-4-{4-[(пиридин-3-илметил)-карбамоил]-фенил}-никотиновой кислоты.
Кроме того, активное соединение может быть таким, как описано в WO 98/37077.
Следовательно, оно может иметь формулу
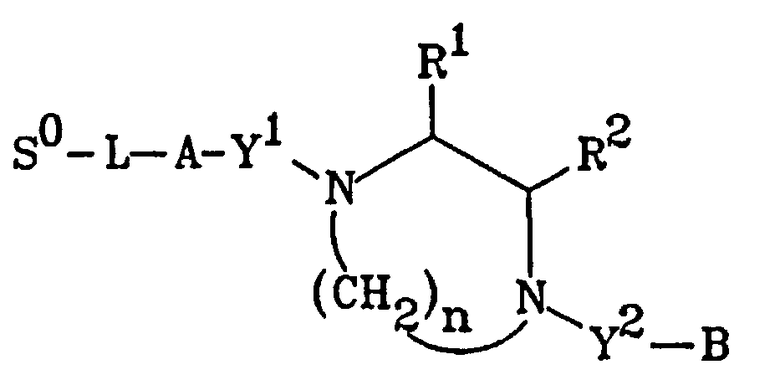
где А и В каждый представляют собой членов, независимо выбираемых из группы, которая включает арил, замещенный арил, карбоциклическое кольцо, замещенное карбоциклическое кольцо,
гетероциклическое кольцо, замещенное гетероциклическое кольцо, и их комбинацию, указанные
комбинации являются слитыми или ковалентно связанными и указанные заместители выбираются из группы, состоящей из галогена, галоалкила, гидрокси, арилокси, бензилокси, алкокси, галоалкокси, амино, моноалкиламино, диалкиламино, ацилокси, ацила, алкила и арила; R1 и R2 каждый независимо выбираются из группы, состоящей из водорода и алкильных групп, имеющих от 1 до 6 атомов углерода, или взятые вместе образуют кольцо, выбираемое из группы, состоящей из насыщенных или ненасыщенных пятичленных колец, насыщенных или ненасыщенных шестичленных колец и насыщенных или ненасыщенных семичленных колец; Y1 и Y2 каждый независимо представляют собой связь или двухвалентный радикал, выбираемый из группы, состоящей из -СН2-, -NHC(O)-, -NRC(O)-, -NHC(S)-, -NRC(S)-, -NHC(=NH)-, -OC(O)-, -C(O)-, и -C(S)-, в которых R представляет собой низшую алкильную группу, содержащую от одного до шести атомов углерода; и n является целым числом от нуля до четырех.
Подходящие примеры включают
Среди миметиков на основе пептидов, которые могут применяться, имеются таковые, которые описаны в US 5,698,521, включая любой из
Ацетил-Trp-Xaa1-Gln-Xaa2-Ile-Thr-Хаа3-Leu-Xaa4-Pro-Gln-Xaa5-Pro-Хаа6-Хаа7-Phe-Gly-COOH и
Ацетил-Trp-Xaa1-Gln-Xaa2-Ile-Thr-Хаа3-Leu-Xaa4-Pro-Gln-Xaa5-Pro-Xaa6-Хаа7-Phe-COOH (SEQ ID NO.2); где Ацетил это СН3 СО-, Xaa1 это изовалин, Хаа2,3,4,5, и 6 это 2-аминоизомасляная кислота и Хаа7 это 4-метилпролин.
Настоящее изобретение далее будет описано и проиллюстрировано с помощью следующих примеров, в которых сделаны ссылки на прилагаемые чертежи.
Краткое описание чертежей
Фигура 1 показывает изменение веса во всех группах, получающих лечение, в течение исследования; а) - группы, получающие носитель и росиглитазон; б) - группы, получающие кальцитонин и 5-CNAC;
фигура 2 показывает (на панелях от А до F) результаты перорального теста на толерантность к глюкозе (oral glucose tolerance test, OGTT) в течение 120 минут для А) групп, получавших носитель и росиглитазон; и D) групп, получавших кальцитонин и 5-CNAC; и (на панелях В и Е) относительное изменение в уровнях глюкозы в течение 120 минут OGTT, нормализованные к t=0 для В) групп, получавших носитель и росиглитазон, и Е) групп, получавших кальцитонин и 5-CNAC. Столбики (на панелях С и F) показывают интегральную площадь под кривой (AUC);
фигура 3 показывает (на панелях от А до F) общие уровни инсулина, измеренные у крыс DIO в группах, получавших А) носитель и росиглитазон и D) 5-CNAC и кальцитонин в течение 120 минут в тесте OGTT; и (на панелях В и Е) изменение уровней инсулина у крыс DIO от момента времени t=0 в группах, получавших В) носитель и росиглитазон и Е) 5-CNAC и кальцитонин в течение 120 минут в тесте OGTT. Столбики (на панелях С и F) показывают интегральную площадь под кривой (AUC);
фигура 4 показывает результаты теста OGTT у здоровых контрольных животных, получавших кальцитонин и 5-CNAC. А) Общие уровни глюкозы, регистрируемые в группах, получавших 5-CNAC и кальцитонин. В) Относительное изменение уровней глюкозы в течение 120 минут теста OGTT в группах, получавших 5-CNAC и кальцитонин, нормализованное к t=0. С) Интегральная AUC;
фигура 5 показывает уровни инсулина в тесте OGTT, приведенном на фигуре 4. А) Общие уровни инсулина, регистрируемые в группах, получавших 5-CNAC и кальцитонин. В) Относительное изменение уровней инсулина, измеренное в течение 120 минут теста OGTT в группах, получавших 5-CNAC и кальцитонин, нормализованное к t=0. С) Интегральная AUC;
фигура 6 показывает аминокислотные последовательности нескольких амилин-подобных пептидов по сравнению с амилином (SEQ ID NOs 1, 2, 6, 10, 48-56);
фигура 7 показывает аминокислотные последовательности нескольких агонистов рецепторов амилина по сравнению с амилином (SEQ ID NOs:39-47);
фигура 8 показывает результаты, полученные в примере 4, демонстрирующие влияние перорального введения кальцитонина лосося на увеличение веса у нормальных крыс в течение 8-недельного периода обработки. Данные представлены как среднее значение +/-SEM;
фигура 9 показывает результаты, полученные в примере 4, демонстрирующие эффект перорального введения кальцитонина лосося, частично защищающего против OVX-индуцированного увеличения веса в течение 7-недельного периода обработки. Данные представлены как среднее значение +/-SEM; и
фигура 10 показывает результаты, полученные в примере 4, демонстрирующие, что пероральное введение кальцитонина лосося в виде однократной ежедневной дозы не защищает против OVX-индуцированного увеличения веса в течение 8-недельного периода обработки. Данные представлены как среднее значение +/-SEM и нормализованы к исходному весу.
фигура 11 на панелях от А до D показывает результаты, полученные в примере 5, демонстрирующие биодоступность перорально введенной композиции sCT в комбинации со SNAC.
фигура 12 далее иллюстрирует результаты, полученные в примере 5, демонстрирующие биодоступность перорально введенной композиции sCT в комбинации со SNAC.
Осуществление изобретения
Пример 1: Эффект кальцитонина лосося в комбинации с 5-CNAC на ответ в тесте пероральной толерантности к глюкозе у крыс DIO по сравнению с Росиглитазоном
МАТЕРИАЛЫ И МЕТОДЫ
Животные
В эксперименте было использовано всего 48 избирательно выведенных самцов крыс с индуцированным диетой ожирением (DIO). В начале эксперимента возраст животных составлял 35 недель, из которых 31 неделю они содержались на диете, богатой жирами. Росиглитазон суспендировали в 10% Гидроксипропил-бета-циклодекстрине (Номер по каталогу А0367.0100). Кальцитонин лосося (СТ) и 5-CNAC (N-(5-хлорсалицилоил)-8-аминокаприловая кислота) были суспендированы в воде MilliQ. Животные получали один раз в день дозировки либо 5-CNAC 150 мг/кг/день или такое же количество 5-CNAC в комбинации с кальцитонином 2 мг/кг/день в течение 5 недель, или 3 мг/кг или 10 мг/кг росиглитазона (известное анти-диабетическое средство, использовался в качестве положительного контроля).
В дни 7, 21 и 42 на голодный желудок собирали образцы сыворотки и мочи и общее состояние тканей всего тела оценивали MR-сканированием (магнитно-резонансная томография). В день 42 проводился пероральный тест на толерантность к глюкозе (OGTT) для оценки у крыс гомеостаза глюкозы. В день -1 животных разделяли на группы, получавшие разное лечение, в соответствии с весом и массой жира всего тела (оценивалась с помощью сканера 4-в-1 EchoMRI). Группы рандомизировали на три команды по весу тела.
Первым днем введения был день 0. Животным перорально вводили дозы в объеме 5 мл/кг. Раствор соединения вводился один раз в день от 7:00 до 14:00 в течение всего исследования (дни 0-42) через пероральный зонд с использованием желудочной трубки, соединенной со шприцем на 5 мл с наконечником Люэра (luer lock™. Becton).
Увеличение веса
Поскольку известно, что глитазоны индуцируют увеличение веса, животных постоянно взвешивали и, как показано на фигуре 1.А и 1.В, животные, получавшие носитель, сохраняли стабильный вес тела в течение всего исследования. В противоположность этому, крысы, получавшие росиглитазон в дозе 3 мг/кг, демонстрировали достоверное (р=0,05) увеличение веса их тела во время проведения исследования, начиная сразу после начала введения препарата. Животные, получавшие росиглитазон в дозе 10 мг/кг, также имели достоверное увеличение веса тела по сравнению с животными, получавшими только носитель (р=0,0001). Ни группа, получавшая СТ, ни группа, получавшая 5-CNAC, не показали никаких изменений веса тела (р=0,8).
Для оценки распределения нарастающего веса во время программы лечения животных после окончания эксперимента подвергали MR-анализу. Анализировались два параметра: 1) изменение в массе жира и 2) изменение в общем содержании воды.
Обе дозировки росиглитазона привели к достоверному увеличению (р<0,0001) общего содержания жира по сравнению с крысами в группе, получавшей носитель, то же наблюдалось и в группе, получавшей 5-CNAC. Однако группа, получавшая СТ, имела достоверно меньшее увеличение в содержании жира по сравнению с группой, получавшей росиглитазон (р<0.0001) (смотри таблицу 2), но достоверное увеличение по сравнению с группами, получавшими 5-CNAC и носитель. Ни одно из проводимых лечений, похоже, не приводит к накоплению воды. Регистрация потребления пищи показала, что увеличение веса тела не было вызвано увеличением потребления пищи (данные не показаны).
ПЕРОРАЛЬНЫЙ ТЕСТ ТОЛЕРАНТНОСТИ К ГЛЮКОЗЕ (OGTT)
Для исследования метаболизма глюкозы в группах, получавших разное лечение, проводился пероральные тест на толерантность к глюкозе (OGTT). Базовые уровни глюкозы у животных, получавших только носитель, были достоверно выше, чем таковые в группах, получавших росиглитазон (ANOVA р=0,0005; скорректированные по Даннетту: носитель vs, росиглитазон 3 мг/кг р=0,006; носитель vs, росиглитазон 10 мг/кг р=0,0003), что показывает, что росиглитазон действительно снижает базальные уровни глюкозы (фигура 2А). Во время теста OGTT уровни глюкозы в плазме в группе, получавшей носитель, увеличивались до 10,1 ммоль/л глюкозы и оставались повышенными во время всего периода OGTT, устанавливая состояние гипергликемии, как и ожидалось у крыс DIO, демонстрирующих недостаточный клиренс глюкозы как одну из характеристик данной модели. Наоборот, как и ожидалось, животные, получавшие лечение Росиглитазоном, демонстрировали более хорошую регуляцию глюкозы на уровне базовой линии и во время теста OGTT (фигура 2А и 2В). При сравнении животных, получавших СТ, с группой, получавшей 5-CNAC, показано, что СТ значительно снижает пиковые уровни глюкозы (фигура 2С и 2D). В группе, получавшей СТ, уровни глюкозы в крови увеличивались максимум до 8,2 мМ через 15 минут после теста OGTT, уровня, который сохранялся во время всего теста OGTT, тогда как в группе, получавшей 5-CNAC, уровень глюкозы достигал пиковых уровней в 9,8 мМ через 60 минут и оставался повышенным в течение всего теста OGTT.
Площадь под кривой (AUC) чистого изменения в клиренсе глюкозы в плазме применялась для сравнения эффектов различных стратегий лечения. Лечение как росиглитазоном, так и СТ вызывало достоверно увеличенный клиренс глюкозы во время теста OGTT. Лечение 10 мг/кг росиглитазона демонстрировало достоверно более быстрый клиренс глюкозы по сравнению с эффектом лечения 3 мг/кг росиглитазона. Эффект СТ на изменение AUC был значительно больше, чем изменения, наблюдаемые в группах, получавших росиглитазон.
Кривые ответа инсулина в плазме были рассчитаны для всех животных в группах, получавших разное лечение, отражая их уровни инсулина в плазме в разные временные точки во время теста OGTT. Группа, получавшая носитель, по сравнению с группами, получавшими росиглитазон, имела более высокие базальные уровни инсулина, составляющие примерно 5,34 мкг/л и очень большой ответ инсулина с пиком в 11,97 мкг/л через 15 минут после вливания глюкозы. После первоначального ответа уровни инсулина снижались до примерно 8,40 мкг/л и это гиперинсулинемическое состояние оставалось неизменным в течение оставшегося времени теста OGTT (фигура 3). Росиглитазон дозо-зависимым образом снижал как базальный, так и индуцированный OGTT уровни инсулина (фигура 3А и В), по сравнению с группой, получавшей носитель. Лечение СТ приводило к ослабленному ответу инсулина при сравнении с группой, получавшей 5-CNAC, с пиковыми уровнями инсулина, наблюдаемыми через 15 минут после вливания глюкозы, после чего уровни инсулина возвращались к базальным уровням 4,53 мкг/л, тогда как в группе, получавшей 5-CNAC, пиковые значения также наблюдались через 15 минут, но уровни оставались в гиперинсулинемическом состоянии в течение всего теста OGTT.
Описанные выше результаты дополнительно показаны на Фигуре ЗЕ, иллюстрирующей изменение AUC, где лечение СТ приводит к самому низкому изменению в уровнях инсулина, к эффекту, который значительно больше, чем таковой росиглитазона.
Пример 2: Влияние кальцитонина лосося в комбинации с 5-CNAC на ответ в пероральном тесте толерантности к глюкозе у крыс Sprague Dawley
OGTT НА ЗДОРОВЫХ КРЫСАХ
Для дальнейшего исследования того, обладает ли СТ способностью снижать уровни глюкозы, был проведен тест OGTT на 20 (подобранных по возрасту и полу) контрольных (без ожирения) крысах Sprague Dawley, 10 из которых были включены в группу, получавшую 5-CNAC, и 10 - в группу, получавшую СТ. Крысы получали лечение дважды в день в течение 5 недель с использованием тех же дозировок, которые получали крысы DIO. Введение СТ снижало базальные уровни глюкозы у этих животных (фигура 4А). Во время теста OGTT группа, получавшая носитель, показала пиковый уровень глюкозы 10,1 ммоль/л через 15 минут, после чего уровни глюкозы снизились почти до базовой линии. Введение СТ предотвращало этот резкий пик и поддерживало более низкие уровни глюкозы, более быстрый клиренс, демонстрируя полезный эффект на гликемический контроль и клиренс глюкозы в крови в группе, получавшей СТ. Нужно отметить, что животные, получавшие как носитель, так и СТ, достигали базальных уровней глюкозы в крови к концу эксперимента t=120 мин. Расчеты изменения AUC во время OGTT подтвердили, что введение СТ значительно снижает уровни глюкозы (фигура 4С).
Кроме того, также исследовались эффекты СТ на уровни инсулина во время теста OGTT. Во время теста OGTT, группа, получавшая носитель, отвечала увеличением уровней инсулина до 2,5 мкг/л, и этот уровень сохранялся примерно 30 минут. Через 120 минут уровни инсулина в группе, получавшей носитель, возвращались к базальным значениям. Животные, получавшие СТ, отвечали на OGTT небольшим увеличением примерно на 0,5 мкг/л, и уровень инсулина возвращался к базальным значениям уже через 60 мин. Изменение уровней инсулина у животных, получавших носитель, по сравнению с контролем от времени 0 до 120 минут было недостоверным (р=0,53), тогда как изменение AUC в этих группах было достоверным (р=0,08). Результаты представлены на фигурах 5А и 5В.
Пример 3: Скрининг агонистов рецептора кальцитонина
ИССЛЕДОВАНИЯ АГОНИСТА РЕЦЕПТОРОВ КАЛЬЦИТОНИНА И АМИЛИНА
Для идентификации агониста рецептора кальцитонина (CTR) и рецептора амилина были использованы клетки COS-7, трансфецированные CTR в отсутствие или в присутствии рецептор-амплифицирующих белков (Receptor amplifying proteins, RAMPs). CTR представляет собой рецептор, сопряженный с G-белком (GPCR), и существует много способов (1-12) для анализа агонистов GPCRs, среди которых индукция образования цАМФ является возможным кандидатом для анализа. Клетки COS-7 были выбраны в связи с тем, что в них отсутствуют фенотипически значительные уровни эндогенных RAMPs, рецепторов СТ и CL (13-15). В отсутствие значительной базовой экспрессии таких компонентов рецептора, можно аккуратно сравнивать определенные подтипы рецептора.
Было разработано 4 способа для анализа.
1. Анализ, который был специфичным для CTR за счет суперэкспресии CTR в отсутствие RAMP1, RAMP2 или RAMP3.
2. Анализ, который был специфичным для активации рецептора амилина за счет суперэкспресии CTR с RAMP1, но в отсутствие RAMP2 или RAMP3.
3. Анализ, который был специфичным для активации рецептора амилина за счет суперэкспресии CTR с RAMP2, но в отсутствие RAMP1 или RAMP3.
4. Анализ, который был специфичным для активации рецептора амилина за счет суперэкспресии CTR с RAMP3, но в отсутствие RAMP1 или RAMP2.
Соединения рассматривались в качестве специфических индукторов специфического рецептора, если они индуцировали образование цАМФ более чем на 50%, по сравнению с контролем-носителем (либо в присутствии, либо в отсутствие 1 мМ IBMX).
Последовательность рецептора кальцитонина и последовательность рецептора амилина уже известны (16; 16-35), для разный видов в разный вариантах сплайсинга.
Для анализа действия агонистов клетки выращивали до 80% конфлуентности в колбах объемом 75 см2. Клетки были ко-трансфецированы с использованием 300 нг конструкции pcDNA-CTR и 1 мкг либо pcDNA-RAMP-1, либо pcDNA-RAMP-2, либо pcDNA-RAMP-3 с использованием 7,8 мкл реагента FuGene 6. Через 48 часов клетки снимали обработкой трипсином, центрифугировали при 500×g и ресуспендировали в циклазном буфере (DMEM, содержащий 0,1% (вес/объем) БСА и 1 мМ IBMX). Аликвоты клеток (5×105) вносили в 1,5 мл пробирки Eppendorf проинкубировали в течение 20 мин при 37°С. Далее клетки инкубировали в течение 18 мин в отсутствие (базальный уровень) или в присутствии возрастающих концентраций агонистов. В качестве положительного контроля использовался пептид форсколин, инкубацию с которым проводили для определения максимального накопления цАМФ в этой системе.
После инкубации реакционную смесь центрифугировали на микроцентрифуге Beckman при 12000×g в течение 1 мин при 4°С. Клетки промывали PBS и снова центрифугировали. цАМФ экстрагировали 0,5 мл абсолютного этанола. Образцы упаривали досуха и растворяли в буфере (50 мМ ацетат натрия, 1 мМ теофиллин). Уровни цАМФ определяли с использованием специального набора для определения цАМФ. Количественное измерение содержания внутриклеточного вторичного мессенджера цАМФ проводили согласно инструкции к набору EIA (Amersham Biosciences, US) или с помощью метода RIA после ацетилирования образцов. Несвязавшуюся радиоактивность экстрагировали в течение 15 мин при 4°С 1 мл буфера для разделения (100 мМ двузамещенный фосфат натрия, 100 мМ фосфат калия, рН 7,4, содержащий 0,25% (вес/объем) БСА и 0,2% (вес/объем) активированного угля) и отделяли от связанной с антителами радиоактивности центрифугированием в течение 15 мин при 4000×g. Супернатант отсасывали, а радиоактивность осадка подсчитывали на γ-счетчике Packard.
Альтернативные методы для идентификации агонистов GPCR широко описаны в литературе, включая новые методы (1-12), и могут быть осуществлены специалистом в данной области техники.
Пример 4: Введенный перорально кальцитонин лосося снижает увеличение веса как у здоровых крыс Sprague-Dawley, так и у крыс Sprague-Dawley с индуцированным удалением яичников увеличением веса.
Крысы в количестве 40 штук были разделены на 4 группы: две группы с двусторонним удалением яичников (OVX) и две нормальные группы. Животные в обеих группах, нормальной и OVX, получали дважды в день либо 5-CNAC 150 мг/кг/день, либо такое же общее количество 5-CNAC в комбинации с 2 мг/кг/день кальцитонина лосося в течение 7 или 8 недель, в течение которых их вес измерялся один раз в неделю.
Первый день дозировки был днем 0. Животным перорально вводили 5 мл/кг. Раствор соединения вводили дважды в день, первую дозу в 7:00 часов утра и вторую дозу через 8 часов (между 15:00 и 16:00 часами) в течение всего исследования. Введение проводили через пероральный зонд с использованием кишечной трубки, соединенной со шприцем на 5 мл. Кормление осуществлялось ad libitum.
Как видно из фигуры 8, пероральное введение нормальным крысам дважды в день кальцитонина лосося приводило к снижению нормального увеличения веса, наблюдаемого в контрольной группе во время эксперимента. Кроме того, как видно из фигуры 9, в группах OVX пероральное введение кальцитонина лосося частично защищало от OVX-индуцированного увеличения веса тела. Таким образом, пероральное введение кальцитонина лосося дважды в день приводит к снижению увеличения веса, вероятно, этот эффект опосредован через регуляцию аппетита.
Проведенное для сравнения такое же исследование, в котором введение проводилось один раз в день, не показало снижения веса по сравнению с контролем. Всего у 20 крыс было проведено двустороннее удаление яичников (OVX). Животным вводили один раз в день либо только 5-CNAC 150 мг/кг/день, либо такое же общее количество 5-CNAC в комбинации с 2 мг/кг/день кальцитонина лосося в течение 8 недель, в течение которых их вес измерялся один раз в неделю. Кормление осуществлялось ad libitum.
Первый день дозировки был днем 0. Животным перорально вводили 5 мл/кг. Раствор соединения вводили один раз в день между 7:00 и 8:00 часами в течение всего исследования через пероральный зонд с использованием кишечной трубки, соединенной со шприцем на 5 мл.
Как видно из фигуры 10, не наблюдалось никакого эффекта на OVX-индуцированное увеличение веса, когда животным вводили дозировки один раз в день, что хорошо согласуется с данными из примера 1, в котором показано отсутствие увеличения веса у крыс DIO, которым вводили дозировки один раз в день.
Пример 5: Композиция кальцитонина лосося с SNAC является биодоступной, как показано по снижению резорбции коллагена типа II
Для выяснения того, являются ли наблюдаемые эффекты при пероральном введении sCT специфически зависимыми от пероральной композиции с носителем 5-CNAC, было проведено сравнительное исследование с двумя носителями для пероральной композиции - 5-CNAC и SNAC. Для этого в качестве суррогатного маркера для исследования биодоступности при пероральном введении и, следовательно, эффективности в контроле за уровнем сахара в крови использовали снижение резорбции хряща, измеряемое с применением CTX-II.
Голодным крысам вводили пероральные дозировки одного носителя (5-CNAC или SNAC), одного sCT, или комбинации sCT с носителем (5-CNAC или SNAC). Образцы крови брали на базовой линии, через один, три и шесть часов после перорального введения дозировок и получали сыворотку. После этого в образцах сыворотки измеряли концентрации CTX-II.
Крыс рандомизировали в соответствии с весом на пять групп и выдерживали голодными в течение ночи. Крысам вводили пероральные дозировки только носителей (150 мг/кг) (5-CNAC или SNAC), только кальцитонина лосося (2 мг/кг), или комбинации носителя (150 мг/кг) и кальцитонина лосося (2 мг/кг). Образцы крови брали из хвостовой вены на базовой линии, через один, три и шесть часов после перорального введения. Из образцов крови выделяли сыворотку и в неразведенных образцах сыворотки измеряли концентрации CTX-II. Фигура 11, панели A-D, показывает концентрации CTX-II в указанных временных точках. Фигура 12 показывает комбинированные результаты во всех обрабатываемых группах в течение времени. Каждая группа содержала 6 животных. Статистический анализ проводили с помощью одностороннего теста ANOVA и теста Бонферрони для множественных сравнений для внесения поправки на множественные сравнения. Величина разброса показывает стандартную ошибку среднего (SEM). Звездочки обозначают: **=р<0,01, ***=р<0,001.
Было установлено, что через один час после перорального введения сам sCT, sCT с 5-CNAC и sCT с SNAC во всех случаях достоверно снижают концентрацию CTX-II по сравнению с двумя группами, которым вводили только носители (фигура 11, панель В). Через три и шесть часов после перорального введения было установлено, что введение sCT самого по себе приводило к возвращению концентраций CTX-II на уровень базовой линии (фигура 11, панели С и D и фигура 12). В противоположность этому, когда sCT вводился в комбинации либо с 5-CNAC, либо с SNAC, продолжительное снижение концентраций CTX-II наблюдалось как через три, так и через шесть часов после перорального введения (фигура 11, панели С и D и фигура 12).
Важным является наблюдение, что уровни CTX-II снижаются одинаково хорошо в случае введения как sCT с 5-CNAC, так и sCT с SNAC, и не наблюдается достоверных различий между этими двумя группами в любой из указанных временных точек. Эти результаты позволяют считать, что сообщаемые эффекты sCT не являются специфически зависимыми от 5-CNAC. Можно заключить, что достоверные длительные эффекты при пероральном введении sCT зависят от пероральной композиции, содержащей носитель. Однако, как видно из фигур 11 и 12, два разных носителя для пероральных композиций (5-CNAC и SNAC) в комбинации с sCT дают эквивалентные результаты. Это означает, что наблюдаемые эффекты sCT не ограничиваются одной специфической пероральной композицией, и можно ожидать, что это наблюдение может распространяться на снижающие уровни сахара в крови эффекты sCT.
В этом описании, если специально не оговорено по-другому, слово «или» («либо») применяется в смысле оператора, который подтверждает истинное значение, когда любое или оба из указанных состояний выполняются, в противоположность оператору «исключительно», который означает, что выполняется только одно из состояний. Слово «включает» употребляется скорее в смысле « включает в себя», чем в смысле «состоит из». Все приведенные материалы из уровня техники, указанные выше, включаются в настоящее изобретение путем отсылки. Отсутствие здесь ссылок на любой ранее опубликованный документ должно рассматриваться как признание или принятие того, что знание этого в настоящее время является общеизвестным фактом в Австралии или где-либо еще.
ССЫЛКИ:
Albrandt K; Mull E, Brady ЕМ, Herich J, Moore CX, Beaumont K. Molecular cloning of two receptors from rat brain with high affinity for salmon calcitonin. FEBS Lett 1993; 325 (3): 225-32.
Aldred J.P., Clements G.R., Schlueter R.J., Bastian J.W., Dailey J.P. and Wazeteer F.X.; Symposium on Thyrocalcitonin and C-cells, ed by S.Taylor, pp.379-389, W.Heinemann, London, 1968.
Arvinte T, Drake AF. Comparative study of human and salmon calcitonin secondary structure in solutions with low dielectric constants. J Biol Chem 1993; 268 (9): 6408-14.
Bello NT, Kemm MH, Moran TH. Am J Physiol Regul Integr Comp Physiol. 2008 Jul; 295 (1): R76-81. Epub 2008 May 14.
Bemkop-Schnurch, A., Pinter, Y., Guggi, D., Kahlbacher, H., Schoffmann, G., Schuh, M., Schmerold, I., Del Curto, M.D., D'Antonio, M., Esposito, P. and Huck, Ch. (2005) The use of thiolated polymers as carrier matrix in oral peptide delivery - Proof of concept. J. Control. Release, 106, 26-33.
Beukers MW, Ijzerman AP. Techniques: how to boost GPCR mutagenesis studies using yeast. Trends Pharmacol Sci 2005; 26 (10): 533-9.
Blahos J, Svoboda Z, Höschl C. Endokrinologie. 1976; 68 (2): 226-30.
Caliceti, P. Salmaso, S., Walker, G. and Bemkop-Schnurch, A. (2004) Development and in vivo evaluation of an oral insulin-PEG delivery system. Eur. J. Pharm. Sci., 22, 315-323.
Chelikani PK, Haver AC, Reidelberger RD. Am J Physiol Regul Integr Comp Physiol. 2007 Nov; 293 (5): R1798-808. Epub 2007 Aug 29.
Cornish J, Reid IR. Effects of amylin and adrenomedullin on the skeleton. J Musculoskelet Neuronal Interact 2001; 2 (1): 15-24.
Comish J, Callon KE, Bava U, Kamona SA, Cooper GJ, Reid IR. Effects of calcitonin, amylin, and calcitonin gene-related peptide on osteoclast development. Bone 2001; 29(2); 162-8.
Dacquin R, Davey RA, Laplace C, Levasseur R, Morris HA, Goldring SR et al.. Amylin inhibits bone resorption while the calcitonin receptor controls bone formation in vivo. J Cell Biol 2004; 164 (4): 509-14.
de la Fuente M, Csaba N, Garcia-Fuentes M, Alonso MJ. Nanomed. 2008 Dec; 3 (6): 845-57.
Dogru ST, Calls S, Oner F. J Clin Pharm Ther. 2000 Dec; 25 (6): 435-43
Eglen RM. Assessing GPCR activation using protein complementation: a novel technique for HTS. Biochem Soc Trans 2007; 35 (Pt 4): 746-8.
Garcia-Fuentes M, Prego С, Torres D, Alonso MJ. Eur J Pharm Sci. 2005 May; 25 (1): 133-43.
Garcia-Fuentes M, Torres D, Alonso MJ Int J Pharm. 2005 May 30; 296 (1-2): 122-32. Epub 2005 Apr 7.
Gattereau A, Bielmann P, Durivage J, Davignon J, Larochelle P. J Clin Endocrinol Metab. 1980 Aug; 51 (2): 354-7. Effect of acute and chronic administration ofcalcitonin on serum glucose in patients with Paget's disease of bone.
Giugliano D, Passariello N, Sgambato S, D'Onofrio F. Lancet. 1980 Mar 22; 1 (8169): 653.
Giugliano D, Passariello N, Sgambato S, Torella R., Ceriello A. and D'Onofrio F., Diabete & Metabolisme (Paris) 1982, 8, 213-216.
Giustina G, Cerudelli B, Cimino A, Rigosa C, Rotondi A, Radaeli E. J Endocrinol Invest. 1985 Feb; 8 (1): 19-23.
Greeley GH Jr, Cooper CW, Jeng YJ. Eldridge JC, Thompson JC., Regul Pept. 1989 Mar; 24 (3): 259-68.
Guggi D, Kast CE, Bemkop-Schnurch A. Pharm Res. 2003 Dec; 20 (12): 1989-94.
Guggi, D., Krauland, A.H., and Bemkop-Schnürch, A. (2003) Systemic peptide delivery via the stomach: in vivo evaluation of an oral dosage form for salmon calcitonin. J. Control. Rel. 92, 125-135.
Hay DL, Howitt SG, Conner AC, Doods H, Schindler M, Poyner DR. A comparison of the actions of BIBN4096BS and CGRP (8-37) on CGRP and adrenomedullin receptors expressed on SK-N-MC, L6, Col 29 and Rat 2 cells. Br J Pharmacol 2002; 137 (1): 80-6.
Hay DL, Howitt SG, Conner AC, Schindler M, Smith DM, Poyner DR. CL/RAMP2 and CL/RAMP3 produce pharmacologically distinct adrenomedullin receptors: a comparison of effects of adrenomedullin 22-52, CGRP8-37 and BIBN4096BS. Br J Pharmacol 2003; 140 (3): 477-86.
Hay DL, Poyner DR, Smith DM. Desensitisation of adrenomedullin and CGRP receptors. Regul Pept 2003; 112 (1-3): 139-45.
Hay DL, Poyner D, Dickerson I. CGRP receptor heterogeneity: a role for receptor component protein? Trends Endocrinol Metab 2003; 14 (1): 3-4.
Hay DL, Christopoulos G, Christopoulos A, Sexton PM. Determinants of 1-piperidinecarboxamide, N-[2-[[5-amino-1-[[4-(4-pyridinyl)-1-piperazinyl]carbonyl]pentyl]amino]-1-[(3,5-dibromo-4-hydroxyphenyl)methyl]-2-oxoethyl]-4-(l,4-dihydro-2-oxo-3(2H)-quinazolinyl) (BIBN4096BS) affinity for calcitonin gene-related peptide and amylin receptors- the role of receptor activity modifying protein 1. Mol Pharmacol 2006; 70 (6): 1984-91.
Hay DL, Christopoulos G, Christopoulos A, Sexton PM. Amylin receptors: molecular composition and pharmacology. Biochem Soc Trans 2004; 32 (Pt 5): 865-7.
Hay DL, Christopoulos G, Christopoulos A, Poyner DR, Sexton PM. Pharmacological discrimination of calcitonin receptor: receptor activity-modifying protein complexes. Mol Pharmacol 2005; 67 (5): 1655-65.
Heilker R. High content screening to monitor G protein-coupled receptor intemalisation. Ernst Sobering Found Symp Proc 2006; (2): 229-47.
Heilker R, Zemanova L, Valler MJ, Nienhaus GU. Confocal fluorescence microscopy for high-throughput screening of G-protein coupled receptors. Curr Med Chem 2005; 12 (22): 2551-9.
Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. The 7 TM G-protein-coupled receptor target family. ChemMedChem 2006; 1 (8): 761-82.
Jonderko К. Gut. 1989 Apr; 30 (4): 430-5.
Kostenis E. G proteins in drug screening: from analysis of receptor-G protein specificity to manipulation of GPCR-mediated signalling pathways. Curr Pharm Des 2006; 12 (14): 1703-15.
Kuestner RE, Elrod RD, Grant FJ, Hagen FS, Kuijper JL, Matthewes SL et al.. Cloning and characterization of an abundant subtype of the human calcitonin receptor. Mol Pharmacol 1994; 46 (2): 246-55.
Lee SK, Goldring SR, Lorenzo JA. Expression of the calcitonin receptor in bone marrow cell cultures and in bone: a specific marker of the differentiated osteoclast that is regulated by calcitonin. Endocrinology 1995; 136 (10): 4572-81.
Leifert WR, Aloia AL, Bucco 0, Glatz RV, McMurchie EJ. G-protein-coupled receptors in drug discovery: nanosizing using cell-free technologies and molecular biology approaches. J Biomol Screen 2005; 10 (8): 765-79.
Lin HY, Harris TL, Flannery MS, Aruffo A, Kaji EH, Gom A et al.. Expression cloning of an adenylate cyclase-coupled calcitonin receptor. Science 1991; 254 (5034): 1022-4.
Mangiafico С., Firoe C.E., Foti R., Lunetta M., Fargetta C., Grimaldi D.R. and Petralito A., La Riforma Medica, Vol.103, No 12, 563-566.
Mansoor S, Youn YS, Lee КС.Pharm Dev Technol. 2005; 10 (3): 389-96.
Nishikawa T, Ishikawa H, Yamamoto S, Koshihara Y. A novel calcitonin receptor gene in human osteoclasts from normal bone marrow. FEBS Lett 1999; 458 (3): 409-14.
Notoya et al.; European Journal of Pharmacology, 560,2007, 234-239
Nussenzveig DR, Mathew S, Gershengom MC. Alternative splicing of a 48-nucleotide exon generates two isoforms of the human calcitonin receptor. Endocrinology 1995; 136 (5): 2047-51.
Oh DY, Kim K, Kwon HB, Seong JY. Cellular and molecular biology of orphan G protein-coupled receptors. Int Rev Cytol 2006; 252: 163-218.
Passariello N, Giugliano D, Sgambato S, Torella R, D'Onofrio F. J Clin Endocrinol Metab. 1981 Aug; 53 (2): 318-23. Calcitonin, a diabetogenic hormone?
Petralito A, Lunetta M, Liuzzo A, Fiore CE, Heynen G. J Endocrinol Invest. 1979 Apr-Jun; 2 (2): 209-11. Effects of salmon calcitonin on blood glucose and insulin levels under basal conditions and after intravenous glucose load.
Pittner RA., Eur J Pharmacol. 1997 May 1; 325 (2-3): 189-97.
Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W et al.. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev 2002; 54 (2): 233-46.
Prego C, Fabre M, Torres D, Alonso MJ Pharm Res. 2006 Mar; 23 (3): 549-56. Epub 2006 Mar 16.
Prego C, Torres D, Alonso MJ. JNanosci Nanotechnol. 2006 Sep-Oct; 6 (9-10): 2921-8.
Prego C, Garcia M, Torres D, Alonso MJ. J Control Release. 2005 Jan 3; 101 (1-3): 151-62.
Prego C, Torres D, Femandez-Megia E, Novoa-Carballal R, Quinoa E, Alonso MJ. J Control Release. 2006 Apr 10; 111 (3): 299-308. Epub 2006 Feb 14.
Purdue BW, Tilakaratne N, Sexton PM. Molecular pharmacology of the calcitonin receptor. Receptors Channels 2002; 8 (3-4): 243-55.
Qi T, Christopoulos G, Bailey Ю, Christopoulos A, Sexton PM, Hay DL. Identification of N-terminal receptor activity-modifying protein residues important for calcitonin gene-related peptide, adrenomedullin, and amylin receptor function. Mol Pharmacol 2008; 74 (4): 1059-71.
Shen Z, Mitragotri S, Pharm Res. 2002 Apr; 19 (4): 3 91-5 'Intestinal patches for oral drug delivery'.
Sinko PJ, Lee YH, Makhey V, Leesman GD, Sutyak JP, Yu H, Perry B, Smith CL, Hu P, Wagner EJ, Falzone LM, McWhorter LT, Gilligan JP, Stem W. Pharm Res. 1999 Apr; 16 (4): 527-33.
Smith DM, Coppock HA, Withers DJ, Owji AA, Hay DL, Choksi TP et al.. Adrenomedullin: receptor and signal transduction. Biochem Soc Trans 2002; 30 (4): 432-7.
Song KH, Chung SJ, Shim CK J Control Release. 2005 Sep 2; 106 (3): 298-308.
Starke A, Keck E, Berger M, Zimmermann H. Diabetologia. 1981 May; 20 (5): 547-52.
Takahashi S, Goldring S, Katz M, Hilsenbeck S, Williams R, Roodman GD. Downregulation of calcitonin receptor mRNA expression by calcitonin during human osteoclast-like cell differentiation. J Clin Invest 1995; 95 (1): 167-71.
Thomson W, Frazer J, Unett D. Functional assays for screening GPCR targets. Curr Opin Biotechnol 2005; 16 (6): 655-65.
Waller A, Simons PC, Biggs SM, Edwards BS, Prossnitz ER, Sklar LA. Techniques: GPCR assembly, pharmacology and screening by flow cytometry. Trends Pharmacol Sci 2004; 25 (12): 663-9.
Xiao SH, Reagan JD, Lee PH, Fu A, Schwandner R, Zhao X et al.. High throughput screening for orphan and liganded GPCRs. Comb Chem High Throughput Screen 2008; 11 (3): 195-215.
Yamin M, Gom AH, Flannery MR, Jenkins NA, Gilbert DJ, Copeland NG et al.. Cloning and characterization of a mouse brain calcitonin receptor complementary deoxyribonucleic acid and mapping of the calcitonin receptor gene. Endocrinology 1994; 135 (6): 2635-43.
Young AA, Wang MW, Gedulin B, Rink TJ, Pittner R, Beaumont K. Metabolism. 1995 Dec; 44 (12): 1581-9. Diabetogenic effects of salmon calcitonin are attributable to amylin-like activity.
Young A; Advances in Pharmacology, Volume 52, 2005, pp.193-207.
Ziegler R, Bellwinkel S, Schmidtchen D, Minne H. Horm Metab Res. 1972 Jan; 4 (1): 60. Effects of hypercalcemia, hypercalcemia and calcitonin on glucose stimulated insulin secretion in man.
Zofková I, Nedvidkova J, Starka L, Zamrazil V. Exp Clin Endocrinol. 1987 Mar; 89 (1): 91-6.
Zofková I, Zamrazil V. Horm Metab Res. 1987 Dec; 19 (12): 656-60.
Zolnierowicz S, Cron P, Solinas-Toldo S, Fries R, Lin HY, Hemmings BA. Isolation, characterization, and chromosomal localization of the porcine calcitonin receptor gene. Identification of two variants of the receptor generated by alternative splicing. J Biol Chem 1994; 269 (30): 19530-8.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДНЫЕ АНАЛОГИ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ И РАССТРОЙСТВ | 2012 |
|
RU2616511C2 |
| МИМЕТИК КАЛЬЦИТОНИНА ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНЕЙ И НАРУШЕНИЙ | 2014 |
|
RU2689551C1 |
| ПЕПТИД, ОБЛАДАЮЩИЙ СВОЙСТВАМИ АМИЛИНА (ВАРИАНТЫ), И ЕГО ПРИМЕНЕНИЕ (ВАРИАНТЫ) | 2005 |
|
RU2385878C2 |
| ПРИМЕНЕНИЕ КАЛЬЦИТОНИНА ПРИ ОСТЕОАРТРИТЕ | 2004 |
|
RU2368390C2 |
| СПОСОБЫ РЕГУЛИРОВАНИЯ МОТОРИКИ ЖЕЛУДОЧНО-КИШЕЧНОГО ТРАКТА | 1994 |
|
RU2177331C2 |
| ПРИМЕНЕНИЕ КАЛЬЦИТОНИНА ДЛЯ ЛЕЧЕНИЯ РЕВМАТОИДНОГО АРТРИТА | 2006 |
|
RU2453330C2 |
| СПОСОБ ЛЕЧЕНИЯ ОЖИРЕНИЯ | 1998 |
|
RU2207871C2 |
| ЛЕЧЕНИЕ САХАРНОГО ДИАБЕТА ТИПА II АГОНИСТАМИ АМИЛИНА | 1996 |
|
RU2166958C2 |
| СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ОЖИРЕНИЯ | 2002 |
|
RU2314121C9 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2006 |
|
RU2469709C2 |










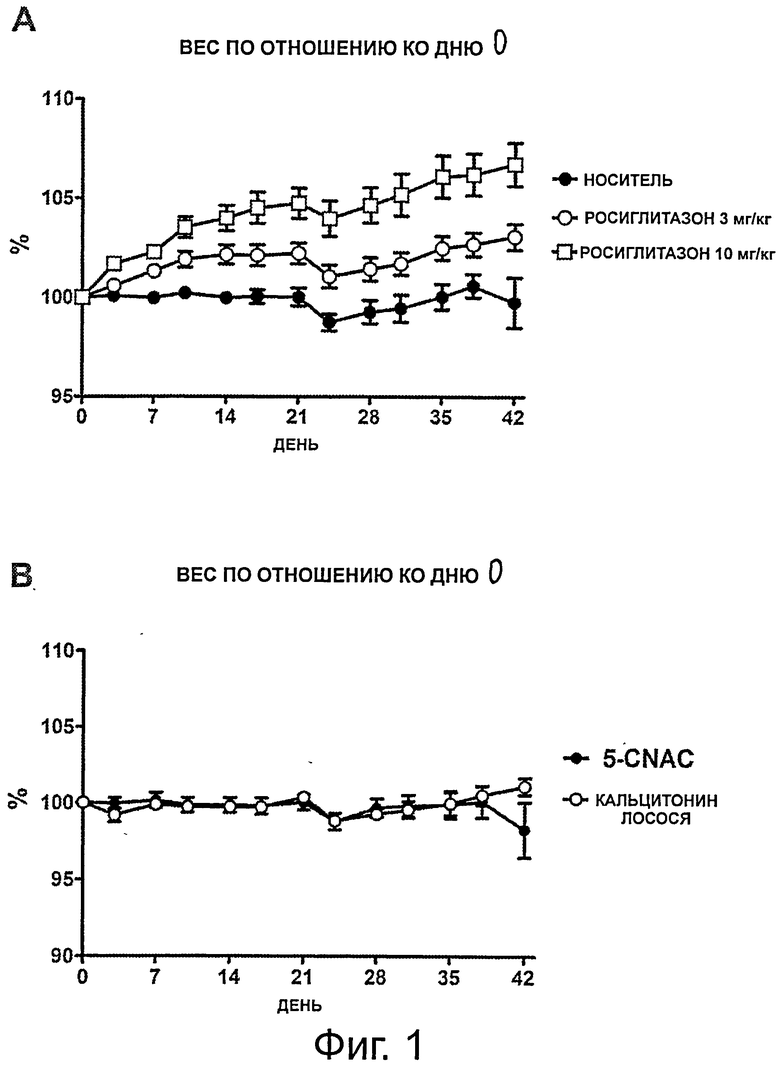
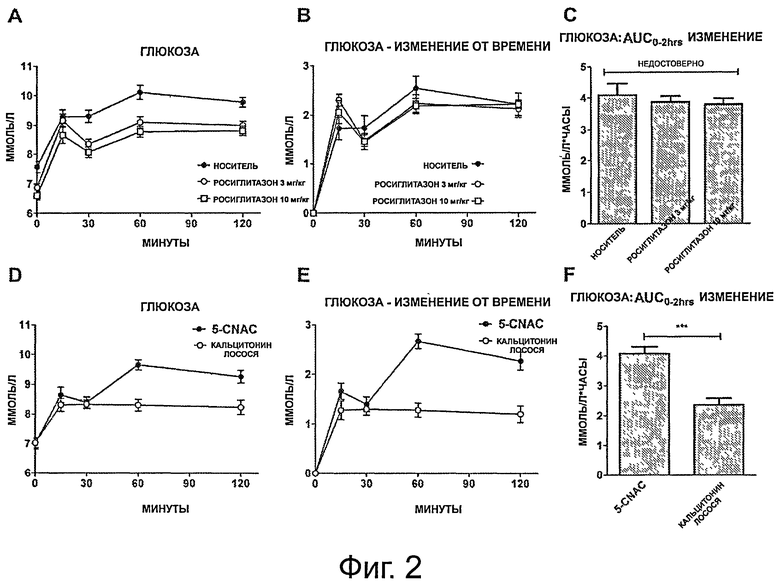
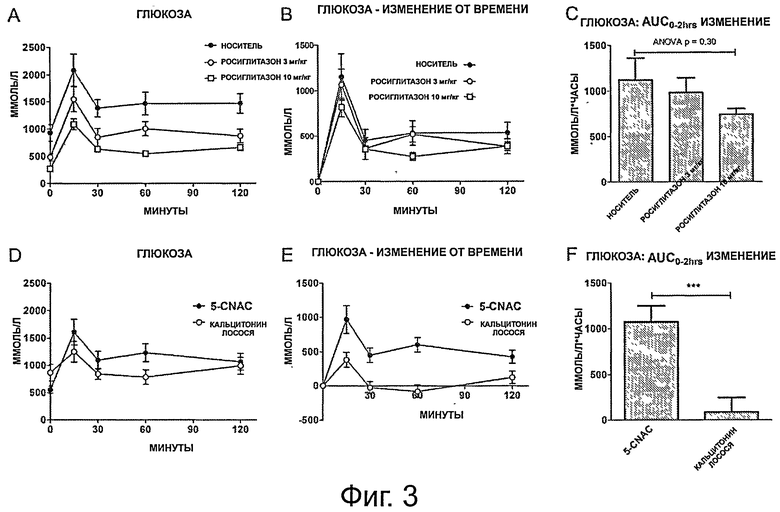
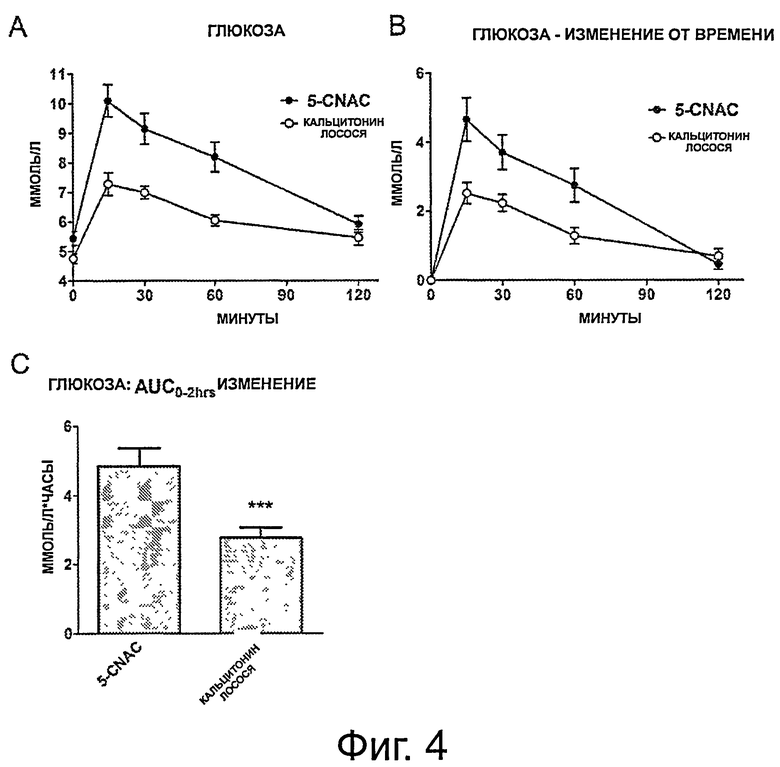
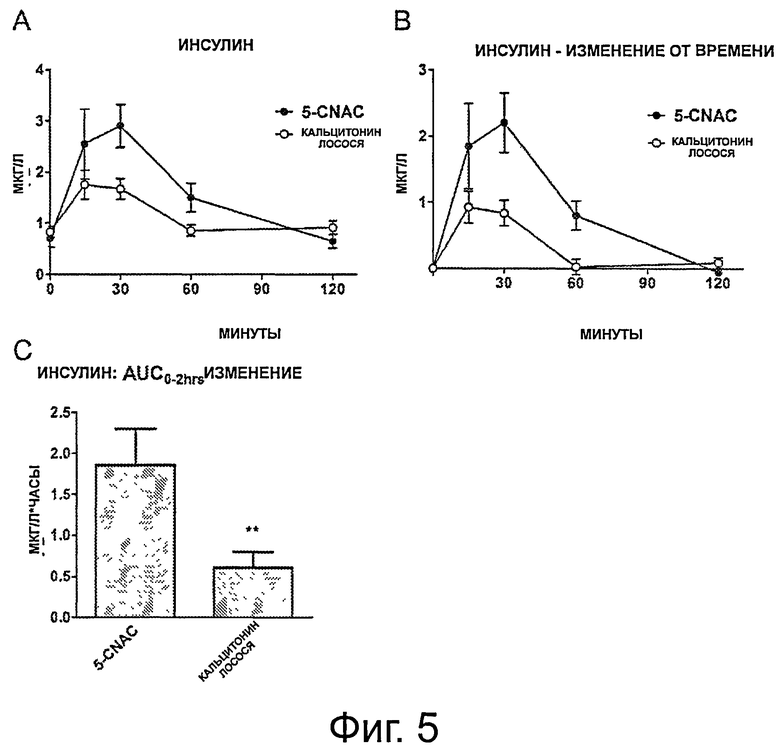
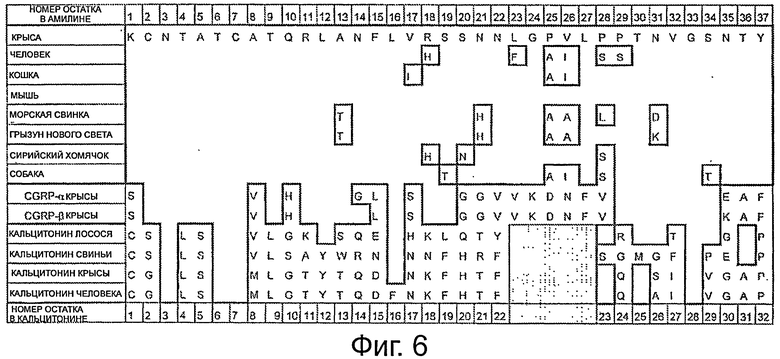
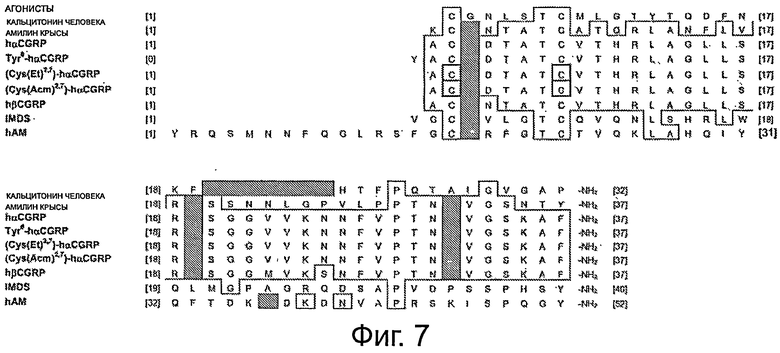
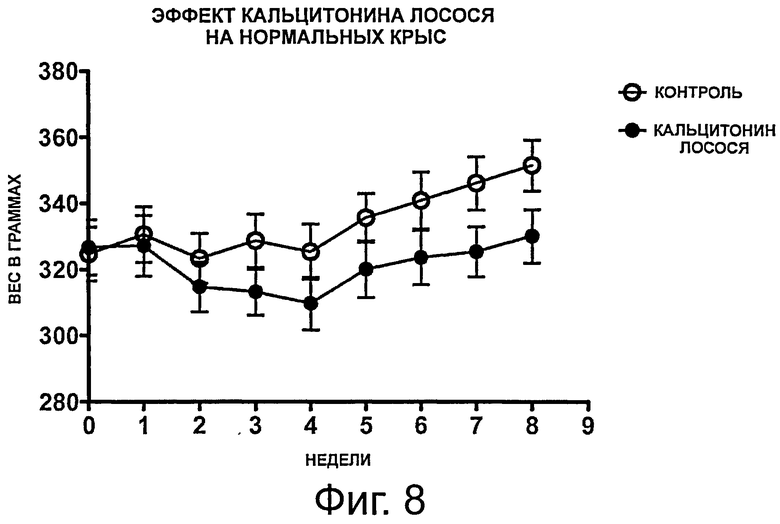
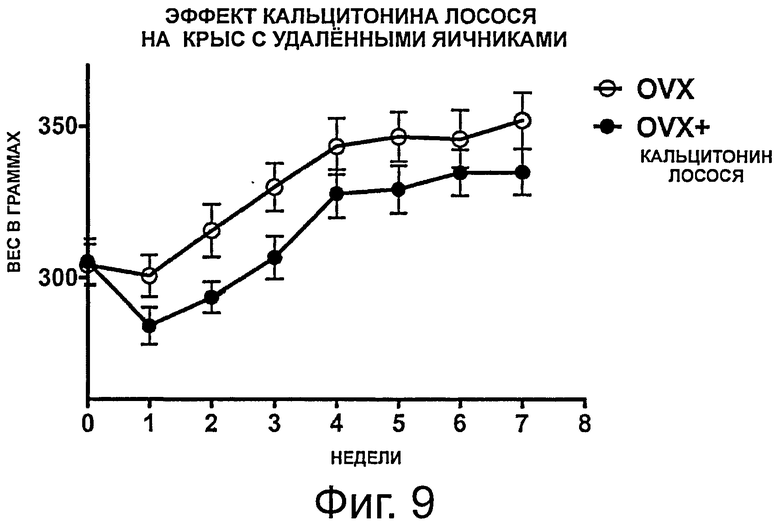
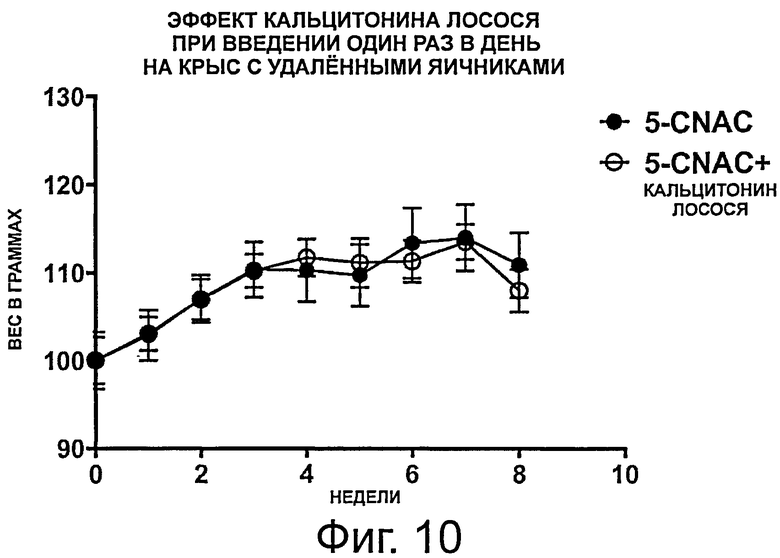
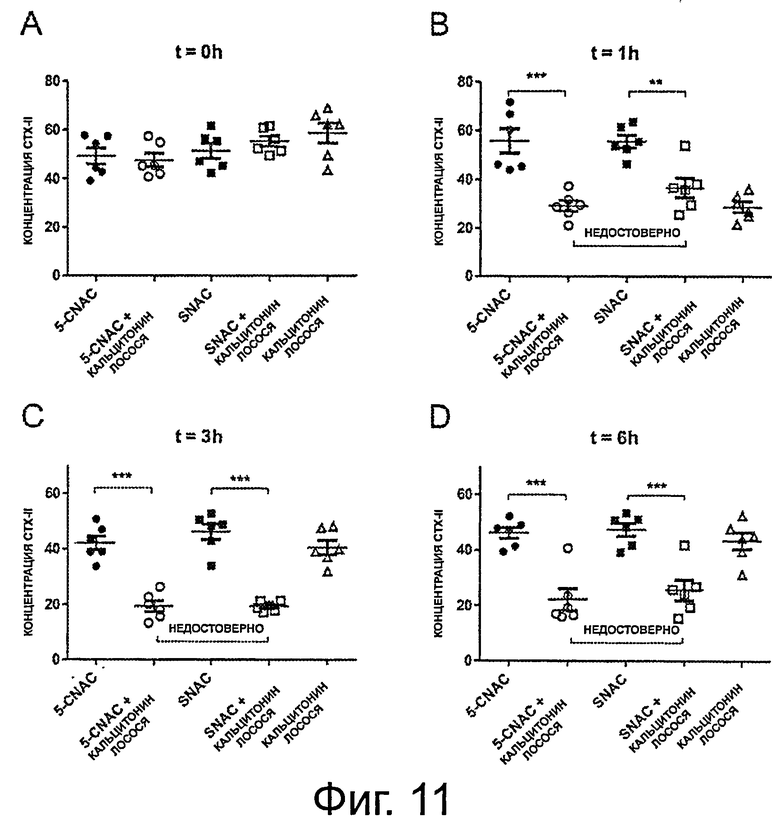
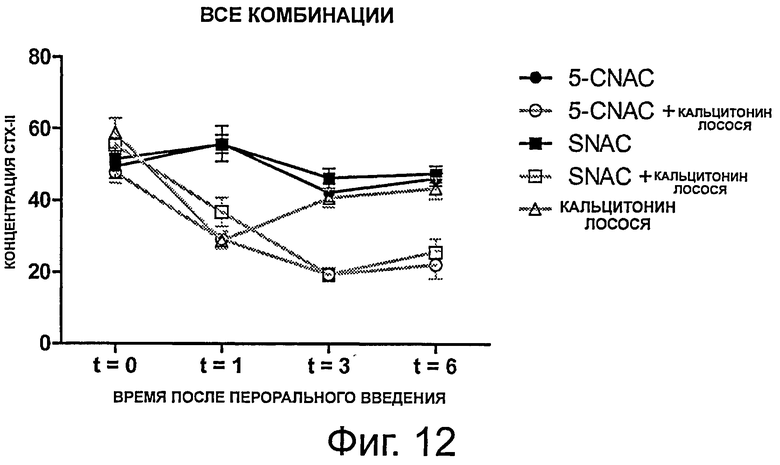
Изобретение относится к области фармацевтики и медицины и касается фармацевтической композиции для энтерального введения для снижения нежелательно высокого уровня глюкозы в сыворотке или для снижения нежелательно высокого пикового уровня инсулина в сыворотке, которая включает активное соединение, которое является представителем семейства кальцитонина, но не амилин, и которое является модифицированным представителем семейства кальцитонина, который имеет аминокислотную гомологию, по меньшей мере равную 75% с кальцитонином, но не амилином, и который модифицирован путем добавления, замены или делеции аминокислот и сохраняющий способность к связыванию и активации кальцитонинового рецептора, или не пептидную «низкомолекулярную» молекулу агониста рецептора кальцитонина. Изобретение позволяет уменьшить нежелательно высокую концентрацию глюкозы в сыворотке. 8 з.п. ф-лы, 12 ил., 5 пр.
1. Фармацевтическая композиция для энтерального введения для снижения нежелательно высокого уровня глюкозы в сыворотке или для снижения нежелательно высокого пикового уровня инсулина в сыворотке, которая включает активное соединение, которое является представителем семейства кальцитонина, но не амилином, и которое является модифицированным представителем семейства кальцитонина, который имеет аминокислотную гомологию, по меньшей мере равную 75% с кальцитонином, но не амилином и который модифицирован путем добавления, замены или делеции аминокислот и сохраняющий способность к связыванию и активации кальцитонинового рецептора, или не пептидную «низкомолекулярную» молекулу агониста рецептора кальцитонина.
2. Композиция по п.1, предназначенная для перорального введения в пищеварительный тракт.
3. Композиция по п.1 или 2, в которой активное соединение представляет собой кальцитонин.
4. Композиция по п.3, в которой кальцитонин представляет собой кальцитонин лосося.
5. Композиция по п.1 или 2, в которой активное соединение имеет общую формулу:
CX1X2LSTCX3LX4X5X6X7X8X9X10X11X12X13X14X15X16X17X18X19X20X21GX22X23X24P
SEQID NO: 11
где X1 представляет собой A, G, или S;
X2 представляет собой N или S;
X3 представляет собой М или V;
X4 представляет собой G или S;
X5 представляет собой Т, К или А;
X6 представляет собой L или Y;
X7 представляет собой Т, S или W;
X8 представляет собой Q, К или R;
X9 представляет собой D, Е или N;
X10 представляет собой F или L;
X11 представляет собой N или Н;
Х12 представляет собой К или N;
X13 представляет собой F или L;
X14 представляет собой Н или Q;
Х15 представляет собой Т или R;
X16 представляет собой F или Y;
X17 представляет собой Р или S;
X18 представляет собой Q, G или R;
X19 представляет собой Т, I или М;
X20 представляет собой A, N, D, S или G;
X21 представляет собой I, Т, V или F;
X22 представляет собой V, S, А или Р;
X23 представляет собой G или Е;
X24 представляет собой А или Т.
6. Композиция по п.5, в которой активное соединение имеет формулу
CSNLSTCVLGX5LX7QX9LHKLQTYPX18TNTGX22GTP
SEQ ID NO:12
где X5 представляет собой Т или К;
X7 представляет собой Т или S;
X9 представляет собой D или Е;
X18 представляет собой Q или R;
X22 представляет собой V, S или А.
7. Композиция по любому из пп.1, 2, 4 или 6, в которой указанное активное соединение комбинируется с носителем для перорального введения.
8. Композиция по п.7, в которой носитель увеличивает пероральную биодоступность активного соединения.
9. Композиция по п.7, в которой носитель включает 5-CNAC, SNAD или SNAC.
| WO2006083254 A1, 10.08.2006 | |||
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ ФАРМАКОЛОГИЧЕСКИ ДЕЙСТВУЮЩИХ ВЕЩЕСТВ | 2001 |
|
RU2287999C2 |
| Прибор для разметки расстояний между двумя точками на равные части | 1926 |
|
SU11029A1 |
| WO2005025504 A2, 24.03.2005 | |||
| WO2007134151 A2, 22.11.2007 | |||

