Изобретение относится к области геохимии, а также к способам разделения веществ (ионов) с использованием ионообменных и адсорбционных механизмов, и может быть использовано для идентификации горных пород.
Известен способ электромагнитного плазменного разделения изотопов по массам в магнитном поле прямого тока [1], по которому рабочее вещество испаряется, смесь изотопов в газообразном состоянии подается в зону ионизации, в которой происходит ионизация данной смеси. Источником ионов является плазменный ускоритель, в котором отсутствует ограничение на ток пучка ионов собственным объемным зарядом, используются все ионы плазменного потока и применяется источник электронов сопровождения. Однако реализация этого способа энергозатратна, трудоемка и экологически не безупречна.
Известен способ обогащения изотопа лития-7 [2], по которому контактируют две жидкие фазы - амальгама лития и водный раствор соединения лития - при циркуляции в противотоке в контактной системе в условиях поддержания высоких значений отношения между скоростью химического обмена лития между фазами и скоростью разложения амальгамы. Такое техническое решение осуществляется с использованием сложной установки, что делает реализацию способа высокозатратным и трудоемким.
Известен способ разделения изотопов лития с использованием неорганического сорбента - дефектного гидроксида алюминия [3]. Навеску дефектного гидроксида алюминия помещают в колбу с раствором гидроксида лития, содержащего литий 6 и 7, которую выдерживают при оптимальной температуре разделения - 60-70°C, после чего отделяют твердую фазу и промывают на фильтре. Фильтрат и промывные воды объединяют и упаривают, а промытую твердую фазу высушивают и из нее получают при температуре не выше 98°C раствор хлорида лития и определяют изотопный состав упаренных растворов. Коэффициент разделения составляет 0,976-1,012. Однако известный способ достаточно сложный и имеет относительно невысокую эффективность разделения изотопов лития.
Известен способ очистки гидроксида лития-7 [4], по которому производят подупаривание раствора гидроксида лития с последующей фильтрацией с использованием тканевых и металлокерамических фильтрующих слоев и сушкой. Весь процесс очистки проводят в герметичном оборудовании в среде инертного газа. На выходе - продукт с содержанием основного вещества 55-56%. Однако известный способ имеет недостаточно высокую степень очистки гидроксида лития-7.
Известен способ разделения изотопов лития методом ионопреципитации [5], который включает изменение одного или более параметров жидкой системы, содержащей соединения лития, до точки, при которой ядра кристаллов соединений лития формируются детектируемыми эффектом Тиндаля; после чего поддерживается неизмененными один или более одного из указанных параметров до окончания разделения созданного таким образом осадка, причем полученная фракция обогащена одним из указанных изотопов, а жидкая система поддерживается гомогенной в отношении указанных одного или более параметров во время реализации способа. Указанный способ позволяет получить несколько фракций, содержащих разное соотношение изомеров лития. Однако этот способ имеет невысокую очистку, является трудоемким и затратным.
Наиболее близким к заявленному изобретению является способ очистки лития [6], который включает абсорбцию на катионообменнике ионов лития и их элюирование (вымывание) с использованием элюента (вымывающего раствора), содержащего раствор водорастворимого соединения металла, другого, чем литий, в воде и/или другом полярном растворителе. В результате абсорбционные метки различных ионов отделяются при элюции и собираются во фракции, причем литиевый компонент выделяется в одну очищенную фракцию или некоторое число очищенных фракций, некоторые из которых обогащены в отношении того или иного из двух изотопов с массовыми числами 6 или 7.
К недостаткам способа, принятого в качестве прототипа, относится недостаточно высокая степень очистки лития и его степень выделения.
Технический результат заявляемого изобретения состоит в более полном выделении лития при минимизации его изотопного фракционирования.
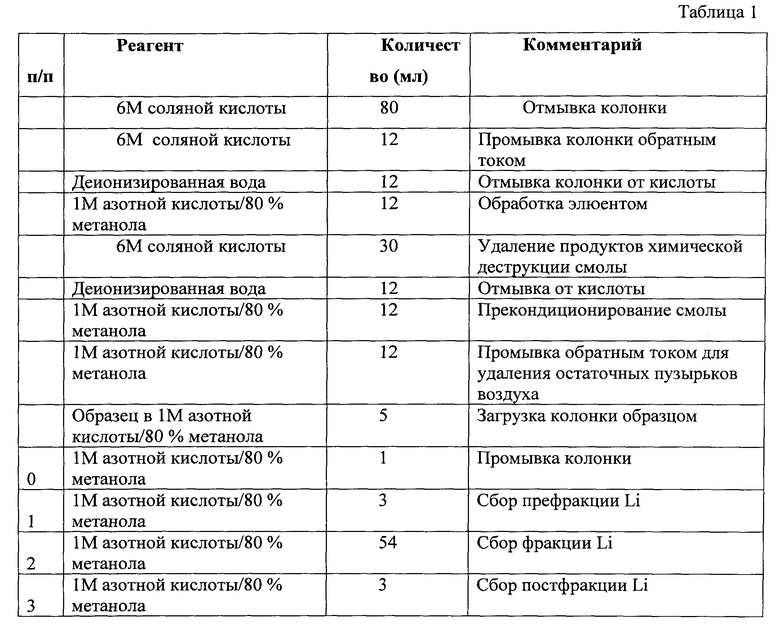
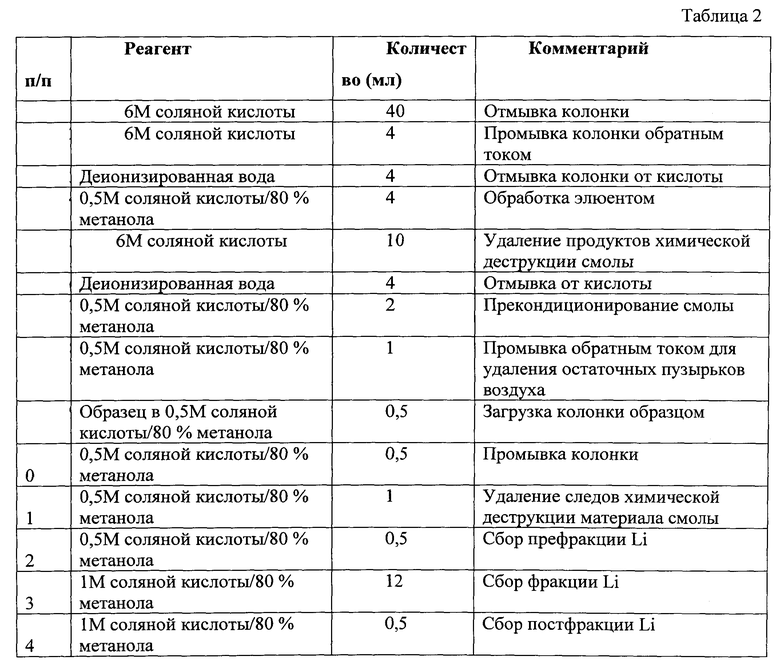
Указанный технический результат достигается тем, что, как и в прототипе, получают раствор солей лития, проводят очистку и выделение лития методом ионообменной хроматографии, его обогащение последующим многократным вымыванием абсорбированных на катионообменнике ионов с использованием вымывающего раствора, содержащего минеральные кислоты и метанол, выделение полученного литиевого компонента в одну очищенную обогащенную фракцию, по которой определяют отношение изотопов лития с массовыми числами 6 и 7, в соответствии с заявленным изобретением используют смеси минеральных кислот; получение раствора солей лития осуществляют многостадийным разложением по меньшей мере до полного растворения испытуемого образца, используют смеси минеральных кислот; на первой стадии в качестве минеральных кислот используют смесь плавиковой кислоты и хлорной кислоты в соотношении 5:1, взятых в количестве 2-3 мл при навеске испытуемого образца 0,1-0,5 г, нагревают до 100°C и выдерживают в течение 30-60 мин, после чего полученную смесь подвергают микроволновому воздействию и одновременно выдерживают при температуре 150-200°C и давлении 5-15 атм; при наличии нерастворимого осадка проводят дополнительное растворение в 3-5 мл 1М азотной кислоты, подвергают ультразвуковому воздействию с частотой 2500 М44Гц в течение 60 мин, а при наличии нерастворимого осадка в жидкой фазе его отделяют центрифугированием в течение 20 мин при 400 об/мин, после чего его растворяют в 5 мл смеси концентрированных плавиковой и азотной кислот в соотношении 1:1; при наличии осадка полученную смесь нагревают до 100°С и выдерживают в течение одного часа до полного растворения осадка, после чего очищают ионообменной смолой Bio-rad AG50W Х8, предварительно очищенной последовательной, не менее трех раз, промывкой деионизованной водой и раствором 6N соляной кислоты и 1N азотной кислоты с 80% метанола; в общем виде последовательность произведенных операций (элюирования лития) приведена в таблице 1. Затем очищают ионообменной смолой Bio-rad AG50W X12, предварительно очищенной 6М соляной кислоты и деионизованной водой и 0,5 моль/л соляной кислоты/80% метанола в количестве 22 мл; в общем виде последовательность произведенных операций (последовательность очистки пробы от натрия) приведена в таблице 2. В пропущенном через ионообменную смолу растворе определяют неоднократно концентрацию ионов лития до достижения максимальной концентрации ионов лития в растворе, после чего определяют изотопный состав методом масс-спектрометрии, по которому идентифицируют горные породы.
Кроме того, указанный технический результат достигается тем, что ионообменную смолу промывают водным раствором, содержащим 6М соляной кислоты, смесь 1М азотной кислоты и 80% метанола, 0,5М соляной кислоты и 80% метанола, 1М соляной кислоты и 80% метанола.
Помимо этого, указанный технический результат достигается тем, что в водном растворе, который используют для промывания ионообменной смолы, все используемые в нем реагенты предварительно подвергают многократной очистке перегонкой.
Сущность заявленного изобретения поясняется Фиг. 1-Фиг. 4 и приведенными ниже примерами конкретной апробации способа по результатам проведенных исследований с таблицами 1-3.
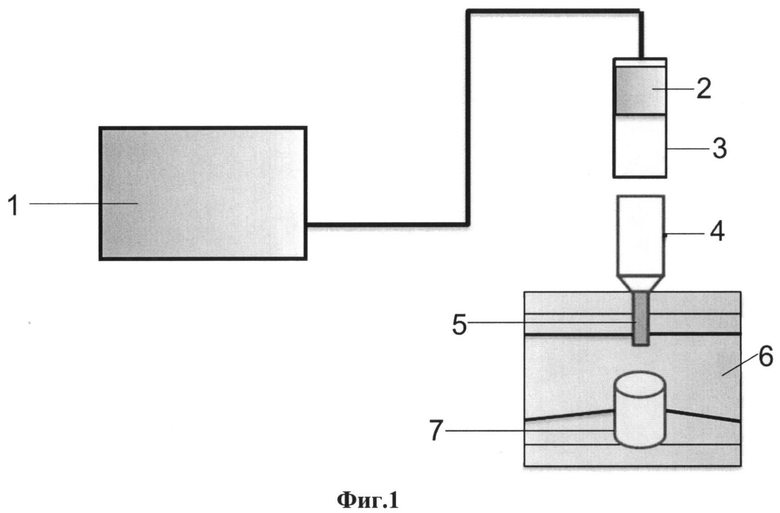
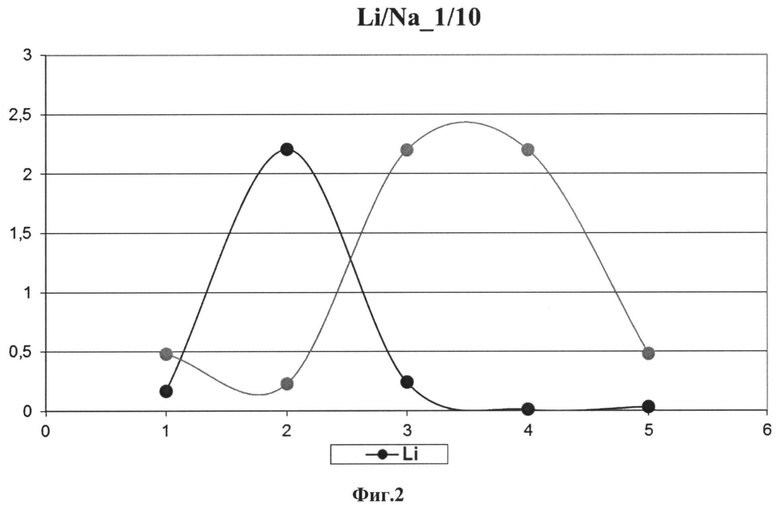
На Фиг. 1 приведена схема установки избыточного давления, которая иллюстрирует внешнее пневматическое давление, оказываемое на образец, для интенсификации просачивания раствора через колонку (1 - аэратор, 2 - фильтр, 3 - переходник, 4 - колонка, 5 - ионообменная смола, 6 - штатив, 7 - сборник элюата); на Фиг. 2 представлен выход лития и натрия при концентрации 1 к 10; на Фиг. 3 показан выход лития и натрия при концентрации 1 к 1000; на Фиг. 4 показана воспроизводимость изотопного отношения 7Li/6Li, скорректированного на масс-дискриминацию при измерении международного стандарта NIST SRM-8545 7Li/6Li=12,02±0,03.
Апробация заявляемого способа была проведена в режиме реального времени на лабораторной базе ФГУП «ВСЕГЕИ» им. А.П. Карпинского; полученные результаты испытаний подтверждают достижение технического результата следующими конкретными примерами реализации.
Ниже приведены результаты апробации заявленного способа на примере трех минералов: хомквистит из Колмозерской коллекции Кольского полуострова; сподумен из Колмозерской полосы пегматитов Кольского полуострова; лепидолит из позднеархейского зеленокаменного пояса Кольского полуострова.
Пример 1
Для подтверждения промышленной применимости заявленного способа идентификации горных пород по изотопному составу лития был взят минерал хомквистит из Колмозерской коллекции Кольского полуострова. Навеска образца из этого минерала 50-100 мг растворялась в 10 мл смеси концентрированных минеральных кислот (HF+HNO3), взятых в соотношении 1:5 соответственно. Затем фторопластовая пробирка с навеской и смесью кислот помещается в микроволновую систему пробоподготовки, где происходит разложение образца при температуре 200°C в течение 30 мин; мощность микроволнового излучения 2500 МГц. Если разложение образца не произошло, то во фторопластовую пробирку добавляют 3 мл концентрированной HCl, и операция в микроволновой системе повторяется до полного разложения образца. Полученный прозрачный раствор выпаривается на электроплитке при температуре 80-90°C до образования сухого остатка. Сухой остаток растворяют в небольшом количестве, до 3 мл, 1М HNO3, переносят в центрифужную пробирку и центрифугируют 15-20 мин при 4000 об/мин. Объем раствора доводят до 5 мл 0,1М азотной кислотой (HNO3).
Для хроматографического выделения изотопов лития были использованы кварцевые колонки диаметром 8 мм для первой ступени отчистки и 3 мм - для второй ступени; бралась ионообменная смола AG50Wx8 и AG50Wx12.
Ионообменную смолу предварительно очищают попеременной промывкой деионизованной водой (сопротивление 18 МОм) и раствором 1N соляной кислоты (не менее трех раз). Прекондицию колонок производят раствором 3% HNO3 и 1N HNO3:80% метанола в количестве двукратного объема колонки.
Для промывки и прекондиционирования колонок использовались водные растворы: 6М соляной кислоты, 1М азотной кислоты/80% метанола, 0,5М соляной кислоты/80% метанола, приготовленные из предварительно очищенных перечисленных выше реагентов и хранившиеся в предварительно подготовленной для испытаний пластиковой таре.
Для отделения литий-содержащей фракции от матричного материала образца использовалась катионная ионообменная смола Bio-rad AG50W Х8 (200-400 mesh), а для дальнейшей очистки лития от продуктов разложения смолы использовалась катионообменная смола Bio-rad AG50W Х12 (200-400 mesh).
Предварительная подготовка ионообменных смол состояла в следующем.
Навеска смолы около 10 грамм помещалась в предварительно отмытую пластиковую 0,5-литровую емкость, которая заполнялась деионизированной водой. Промывка смолы осуществлялась с помощью интенсивного взбалтывания емкости с последующей декантацией наиболее мелких фракций смолы. Процесс повторяли трижды, после чего полученную водную суспензию смолы оставляли в плотно закупоренной емкости на хранение.
После этого проводилась отмывка и заполнение колонок ионообменной смолой.
В процессе разделения использовались тефлоновые колонки с полной емкостью ~4 мл. Перед использованием колонки промывались раствором 80 мл 6М соляной кислоты и в обратную сторону с помощью 12 мл того же раствора. Остатки кислоты со стенок колонок удалялись с помощью промывки их 20 мл деионизованной воды. Отмытые колонки на две трети заполнялись деионизованной водой, куда по капле приливалась водная суспензия ионообменной смолы. После осаждения частиц смолы на фильтре колонки оба ее выхода плотно закрывались парафиновой лентой для предотвращения пересыхания суспензии. Перед использованием консервационный растворитель сливался из колонки, а после завершения разделения колонка снова заполнялась деионизованной водой и плотно запечатывалась.
С целью калибровки колонок, а также для предотвращения фракционирования изотопов лития на стадии прохождения раствора через ионообменную смолу использовался стандарт лития. Стандарт, для которого известны концентрация и соотношения изотопов лития, разводят в 3% HNO3 до концентрации 0,5 ppm. Для стандартизации процесса и для проверки того, что литий полностью элюируется, раствор с известной концентрацией стандарта LSVEC поэтапно пропускают через колонки первой и второй ступени, полностью повторяя схему хроматографического выделения лития, как при использовании образцов пород. Для контроля полного выделения лития изотопный состав измеряют после каждой ступени. Данные процедуры необходимы для того, чтобы избежать эффектов фракционирования на стадии выделения.
Для того чтобы обнаружить причины фракционирования на стадии выделения лития в ионообменной колонке, а также для калибровки колонок был произведен поэтапный сбор элюента по 2 мл. Благодаря этому выявляется процесс фракционирования на обеих ступенях исследования. Вначале с элюентом выходит более тяжелый изотоп, затем легкий. Было обнаружено, что начального количества элюента, предусмотренного заявляемым способом, не хватает для полного смыва лития в колонке. В процессе работы удалось добиться полного элюирования лития в 50 мл смеси 1N HNO3 и 80% метанола. Также велика вероятность возможной потери лития при выпаривании. Дело в том, что по заявляемому способу необходимо несколько раз переходить из одной кислой среды в другую (HNO3 в HCl и обратно); это осуществляется выпариванием, затем добавлением необходимой высококонцентрированной кислоты, вновь повторным выпариванием и уже затем добавлением кислоты требуемой концентрации. Был проделан эксперимент, в котором на масс-спектрометре анализировались два образца - стандарт LSVEC и тот же стандарт, но только после полного ряда процедур (исключая ионообменные колонки), необходимых при постановке методики хроматографического выделения лития. Данная процедура показала, что выпаривание раствора в тефлоновых бюксах при температуре 80°C не влияет на изотопный состав в нем лития.
Для корректного измерения изотопного состава лития необходимо выполнить два главных условия: максимально полное отделение лития от натрия и обеспечение максимально возможного выхода лития из хроматографической колонки, для предотвращения его изотопного фракционирования в ходе химической процедуры.
Выделение литий-содержащей фракции из раствора образца в этом эксперименте осуществлялась следующим образом.
Заполненная ионообменной смолой Bio-rad AG50W Х8 (200-400 mesh) колонка, в которой осуществлялось разделение литий-содержащей фракции образца от материала матрицы, промывалась заранее подготовленным водным раствором 6М соляной кислоты в объеме 80 мл.
Для интенсификации просачивания раствора через колонку использовалась установка для создания внешнего пневматического давления на образец. В качестве воздушного компрессора для создания избыточного давления использовался аквариумный аэратор, оснащенный двумя выходами для нагнетаемого воздуха, как это видно из Фиг. 1, на которой представлена схема установки избыточного давления.
Принцип работы установки создания избыточного давления следующий: атмосферный воздух избыточного давления подается на штуцер переходника 3, подобранного таким образом, чтобы он плотно садился на ионообменную колонку 4. Проходя через волокнистый фильтр 2, очищенный атмосферный воздух создает давление на столб раствора образца, ускоряя его прохождение через колонку 4.
После предварительной промывки колонка промывалась обратным током 12 миллилитрами водного раствора 6М соляной кислоты.
От избытков кислоты колонка отмывалась 12 миллилитрами деионизированной воды. Затем ионообменная смола обрабатывалась 12 миллилитрами элюента, в качестве которого использовался раствор состава 1М азотной кислоты/80% метанола.
Для отмывания ионообменной смолы от следов частичной химической деструкции в ходе обработки элюентом использовалась промывка колонки 30 миллилитрами 6М соляной кислоты. Остатки соляной кислоты смывались со смолы 12 миллилитрами деионизированой воды.
Прекондиционирование ионообменной смолы осуществлялось с помощью ее обработки 12 миллилитрами раствора элюента состава 1М азотной кислоты/80% метанола с последующей промывкой обратным током 6 миллилитрами элюента того же состава для удаления из смолы образовавшихся ранее воздушных пузырьков.
Загрузка смолы колонки образцом производилась путем пропускания через колонку 5 мл раствора состава 1М азотной кислоты/80% метанола, в котором был растворен образец. Далее смола, загруженная испытуемым образцом, промывалась 1 мл раствора 1М азотной кислоты/80% метанола, после чего осуществлялся сбор образца. Сбор префракции лития проводился вместе с 3 мл раствора элюента того же состава (1 моль/л азотной кислоты/80% метанола).
Следующие 54 мл элюента содержали основную фракцию лития, которую собирали в отдельную предварительно отмытую емкость. Постфракция лития собиралась вместе с добавочными 3 мл раствора элюента. Собранные литий-содержащие растворы упаривались и направлялись на доочистку от натрия. В общем виде последовательность произведенных операций (элюирования лития) приведена в таблице 1.
Очистка литий-содержащей фракции от примесей натрия осуществлялась следующим образом.
Перед загрузкой образцом колонки, заполненные 0,6 мл ионообменной смолы Bio-rad AG50W Х12 (200-400 mesh), промывались пропусканием 40 мл 6М соляной кислоты. Затем колонка промывалась обратным током 4 мл 6М соляной кислоты. Остатки кислоты с колонки смывались пропусканием через нее 4 мл деионизованной воды. Обработка смолы элюентом производилась пропусканием через колонку 4 мл раствора состава 0,5М соляной кислоты/80% метанола. Далее смола отмывалась 10 мл 6М соляной кислоты. Остатки кислоты удалялись промывкой 4 мл деионизованной воды. Смола прекондиционировалась 2 мл элюента состава 0,5М соляной кислоты/80% метанола. Образовавшиеся в колонке пузырьки воздуха вымывались обратным током 1 мл элюента 0,5М соляной кислоты/80% метанола. Заполнение колонки осуществлялась из раствора, содержащего образец, полученный на стадии предварительной очистки в 0,5 мл элюента состава 0,5М соляной кислоты/80% метанола. Смола, загруженная образцом, промывается 0,5 мл раствора элюента 0,5М соляной кислоты/80% метанола. Отмывка колонки от следов химического разрушения смолы с помощью ее промывки 2 мл элюента 0,5М соляной кислоты/80% метанола. Сбор префракции лития в 0,5 мл элюента 0,5М соляной кислоты/80% метанола. Сбор основной фракции лития в 12 мл элюента 1М соляной кислоты/80% метанола. Сбор постфракции лития в 0,5 мл элюента 1М соляной кислоты/80% метанола. В общем виде последовательность произведенных операций (последовательность очистки пробы от натрия) приведена в таблице 2.
Для отслеживания динамики выделения лития сбор фракций осуществлялся порциями по 2 мл (0,5 мл в случае отбора предварительной и заключительной фракций) в индивидуальные предварительно отмытые емкости.
Собранные порции литий-содержащего раствора упаривались досуха, перерастворялись и отцентрифугировались от осадка, после чего направлялись на масс-спектрометрический анализ.
Для дальнейшего химически корректного проведения аналитического анализа ионов лития литий-содержащие пробы, находящиеся в солянокислой форме, необходимо перевести в азотнокислую форму. Пробы упаривались до маленькой капли (до объема, равного 0,1-0,2 мл), затем в каждую пробу добавлялось 1,5 мл 3% азотной кислоты. После этого проба помещалась на 20 минут в ультразвуковую ванну (частота 40-60 кГц) и переносилась в 2 мл пробирки. Для отделения твердого осадка от жидкой фазы, ценрифугировали 30 минут (6000 об/мин). Затем происходила процедура отделения жидкой фазы от осадка, для дальнейшего проведения масс-спектрометрического анализа.
На всех этапах растворения и двух ступенях очистки проводился контроль полноты выхода с измерениями концентрации лития и натрия при помощи ICP масс-спектрометра Element-2 (Фиг. 2 и Фиг. 3). Общий выход лития должен составлять не менее 98%, чтобы обеспечивать отсутствие значимых эффектов его изотопного фракционирования при химических процедурах.
При корректном выполнении всех вышеописанных последовательных операций заявленного способа полученный раствор объемом 1-2 мл готов для проведения масс-спектрометрического анализа (прибор Neptune), по определению изотопного соотношения лития можно будет проводить идентификацию горной породы.
В результате двухэтапного выделения на ионообменных колонках был получен выход лития не менее 98%, а содержание Li превышало содержание Na. Изотопный состав анализировался с помощью прибора MC-ICP-MS TERMOFinnigan Neptune, и было получено следующее значение δ7Li=+5.3‰, по которому был идентифицирован минерал исследуемой навески - хомквистит.
Пример 2
В качестве второго примера реализации заявленного способа идентификации горных пород по изотопному составу лития был взят минерал сподумен из Колмозерской полосы пегматитов Кольского полуострова. К измельченной навеске образца весом 20-50 мг добавляют смесь концентрированных минеральных кислот HF+HNO3+HCL в соотношении 1:1:1 и выдерживают в микроволновой системе пробоподготовки при температуре 150°C в течение 60 минут и давлении до 40 атм в следующей последовательности: 10 минут подъем температуры, 20 минут выдержка при указанных температуре и давлении и 30 минут снижение температуры и давления и охлаждение до комнатной температуры. Полученный прозрачный раствор упаривается в химическом боксе в атмосфере азота при температуре 70-90°C до образования аморфного осадка, к которому затем добавляют 10 мл концентрированной HNO3 и выдерживают в микроволновой системе пробоподготовки при температуре 150°C и давлении до 40 атм в течение 40 минут в следующей последовательности: 10 минут подъем температуры, 10 минут выдержка и 20 минут снижение температуры. Азотнокислый раствор центрифугируется 20 минут при 4000 об/мин; в случае образования осадка вся предыдущая процедура повторяется. Прозрачный раствор солей упаривается на электроплите до образования сухого остатка и растворяется в 5 мл 0,1М HNO3. Подготовленный таким образом материал пропускают через ионообменные хроматографические колонки по процедуре, описанной выше в примере 1, и передают для масс-спектрометрического анализа.
В результате измерений образцов минерала сподумен было получено следующее значение δ7Li=+20,3‰, по которому был идентифицирован минерал исследуемой навески - сподумен.
Пример 3
В качестве еще одного примера реализации заявленного способа идентификации горных пород по изотопному составу лития был взят минерал лепидолит из позднеархейского зеленокаменного пояса Кольского полуострова. Измельченную навеску образца весом 1-5 мг помещали во фторопластовую емкость со смесью минеральных кислот HCl+HNO3 (10 мл) в соотношении 3:1 и выдерживали в микроволновой системе пробоподготовки при температуре 180°C и давлении до 60 атмосфер в течение 80 минут в следующей последовательности: 20 минут подъем температуры, 30 минут выдержка при указанных температуре и давлении и 30 минут снижение температуры и давления. Полученный прозрачный раствор упаривался на электроплитке до образования сухого остатка, который растворяли в 2 мл 1М HNO3 и затем общий объем доводили до 5 мл добавлением 0,1М HNO3. Дальнейшие технологические операции повторяют в той же последовательности, как они приведены в примерах 1 и 2.
В результате масс-спектрометрических измерений было получено следующее значение δ7Li=+20,3‰, по которому был идентифицирован минерал исследуемой навески - лепидолит.
Как показывают результаты приведенных примеров, в соответствии с заявленным способом достигается технический результат - наиболее полное выделение лития из хроматографических колонок при минимизации его изотопного фракционирования, как это иллюстрировано на Фиг. 2 и Фиг. 3. В результате общий выход лития на колонках составлял не менее 98%.
Для контроля качества полученных заявленным способом результатов через колонки пропускался раствор изотопного стандарта - синтетический карбонат лития Li2CO3 (NBS L-SVEC, now NIST SRM-8545 7Li/6Li=12,02±0,03) и морской воды (δ7Li=+30…+33‰), что проиллюстрировано в таблице 3 (при измерениях использовались растворы с содержанием лития 0.5 ppm в 3% HNO3). В таблице 3 показана воспроизводимость измерения международного стандарта лития (NIST SRM-8545).

Измерения выполнялись по схеме Стандарт-Проба-Стандарт, коррекция масс-дискриминации в пробе выполнялась по среднему значению двух стандартов.
Отклонения от стандартизованных величин при измерении не превышали ±0.9‰, как это видно из Фиг. 4. Типичная погрешность измерения не превышала ±0.4‰ (2σ). Средняя величина стандартного отклонения (Фиг. 4) составляет 0,081 промилле, что является достаточным для решения абсолютного большинства задач. Корректность измерения подтверждается измерением отношения 7Li/6Li в морской воде. Полученная величина 7Li/6Li имеет сдвиг относительно стандарта NIST SRM-8545=0.5084 промилле, что полностью соответствует стандартным требованиям.
Полученные результаты подтверждают, что величина изотопного сдвига по литию служит не только количественной мерой уровня фракционирования геохимических систем, но и предоставляет новые возможности для оценки перспектив рудоносности и промышленной продуктивности горных пород.
Список использованной литературы
1. RU 2405619 C1, 24.06.2009.
2. RU 2216391 C2, 18.07.2001.
3. RU 2218205 C2, 06.08.2001.
4. RU 2251525 C2, 04.03.2003.
5. GB 866720, 29.04.1957.
6. GB 1247736, 26.09.1968 (прототип).
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ катионообменного выделения радионуклида лютеция-177 из облученного в ядерном реакторе иттербия | 2021 |
|
RU2763745C1 |
| СПОСОБ ВЫДЕЛЕНИЯ СУРЬМЫ-125 ИЗ СМЕСИ ОСКОЛКОВ ДЕЛЕНИЯ, УРАНА, ТРАНСУРАНОВЫХ ЭЛЕМЕНТОВ, ПРОДУКТОВ КОРРОЗИИ И ТЕХНОЛОГИЧЕСКИХ ОТХОДОВ | 1992 |
|
RU2073927C1 |
| СПОСОБ ПОЛУЧЕНИЯ РАДИОНУКЛИДА ВИСМУТ-212 | 2010 |
|
RU2439727C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОЙ СУБСТАНЦИИ НА ОСНОВЕ ЛЮТЕЦИЯ-177 ИЗ ОБЛУЧЕННОГО В НЕЙТРОННОМ ПОТОКЕ ИТТЕРБИЯ-176 | 2023 |
|
RU2823124C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ ЧИСТОГО Ac ПОЛУЧАЕМОГО ИЗ ОБЛУЧЕННЫХ Ra-МИШЕНЕЙ | 2007 |
|
RU2432632C2 |
| СПОСОБ ВЫДЕЛЕНИЯ ЛЮТЕЦИЯ-177 ИЗ ОБЛУЧЕННОГО ИТТЕРБИЯ | 2022 |
|
RU2795790C1 |
| СПОСОБ ВЫДЕЛЕНИЯ И ОЧИСТКИ ИЗОТОПА ИТТРИЯ | 2001 |
|
RU2270170C2 |
| Способ определения органических и неорганических форм мышьяка в слоевищах ламинарии и продуктах на их основе | 2023 |
|
RU2816025C1 |
| СПОСОБ ХИМИЧЕСКОГО РАЗДЕЛЕНИЯ ИЗОТОПОВ УРАНА | 1997 |
|
RU2120329C1 |
| СПОСОБ ПОЛУЧЕНИЯ РАДИОИЗОТОПА ТЕРБИЙ-161 | 2022 |
|
RU2803641C1 |
Изобретение относится к области геохимии, а также к способам разделения веществ с использованием ионообменных и адсорбционных механизмов, и может быть использовано для идентификации горных пород. Способ идентификации горных пород по изотопному составу лития заключается в получении раствора солей лития многостадийным разложением с использованием смеси минеральных кислот до полного растворения испытуемого образца, очистке и выделении лития методом ионообменной хроматографии, его обогащении последующим многократным вымыванием абсорбированных на катионообменнике ионов с использованием вымывающего раствора, содержащего минеральные кислоты и метанол, выделении полученного литиевого компонента в одну очищенную обогащенную фракцию, по которой определяют отношение изотопов лития с массовыми числами 6 и 7. Изобретение обеспечивает полное выделение лития при минимизации его изотопного фракционирования. 4 з.п. ф-лы, 4 ил., 3 табл., 3 пр.
1. Способ идентификации горных пород по изотопному составу лития, заключающийся в получении раствора солей лития, очистке и выделении лития методом ионообменной хроматографии, его обогащении последующим многократным вымыванием абсорбированных на катионообменнике ионов с использованием вымывающего раствора, содержащего минеральные кислоты и метанол, выделении полученного литиевого компонента в одну очищенную обогащенную фракцию, по которой определяют отношение изотопов лития с массовыми числами 6 и 7, отличающийся тем, что получение раствора солей лития осуществляют многостадийным разложением по меньшей мере до полного растворения испытуемого образца с использованием смеси минеральных кислот, на первой из которых используют смеси минеральных кислот плавиковой кислоты и хлорной кислоты в соотношении 5:1, взятых в количестве 2-3 мл при навеске испытуемого образца 0,02-0,5 г, нагревают до 100°С и выдерживают в течение 60 мин, после чего полученную смесь подвергают микроволновому воздействию и одновременно выдерживают при температуре 150-200°С и давлении 5-45 атм, после чего очищают ионообменной смолой Bio-rad AG50W Х8, предварительно очищенной последовательной, не менее трех раз, промывкой деионизованной водой и раствором 6N соляной кислоты и 1N азотной кислоты с 80% метанола, затем очищают ионообменной смолой Bio-rad AG50W X12, предварительно очищенной 6М соляной кислотой и деионизованной водой и 0,5М соляной кислотой/80% метанола в количестве 22 мл, в пропущенном через ионообменную смолу растворе определяют концентрацию ионов лития неоднократно до достижения максимальной концентрации ионов лития в растворе, после чего определяют изотопный состав методом масс-спектрометрии, по которому идентифицируют горные породы.
2. Способ по п.1, отличающийся тем, что ионообменную смолу промывают водным раствором, содержащим 6М соляной кислоты, смесь 1М азотной кислоты и 80% метанола, 0,5М соляной кислоты и 80% метанола, 1М соляной кислоты и 80% метанола.
3. Способ по п.2, отличающийся тем, что в водном растворе использованные реагенты берут после их многократной очистки перегонкой.
4. Способ по п.3, отличающийся тем, что при наличии нерастворимого осадка после стадии микроволнового воздействия проводят дополнительное растворение в 3-5 мл 1М азотной кислоты.
5. Способ по п.4, отличающийся тем, что при наличии нерастворимого осадка в жидкой фазе его отделяют центрифугированием в течение 20 мин при 400 об/мин, после чего его растворяют в 5 мл смеси концентрированных плавиковой и азотной кислот в соотношении 1:1, после чего, при наличии осадка, полученную смесь нагревают до 100°С и выдерживают в течение одного часа до полного растворения осадка.
| Устройство для контроля химического недожога топлива | 1985 |
|
SU1247736A1 |
| СПОСОБ ОЧИСТКИ ГИДРОКСИДА ЛИТИЯ-7 | 2003 |
|
RU2251525C2 |
| СПОСОБ РАЗДЕЛЕНИЯ ИЗОТОПОВ ЛИТИЯ | 2001 |
|
RU2218205C2 |
| СПОСОБ РАЗДЕЛЕНИЯ ИЗОТОПОВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2009 |
|
RU2405619C1 |

