Изобретение относится к процессам разделения изотопов урана химическими методами и может быть использовано в радиохимическом производстве для корректировки изотопного состава ядерного топлива.
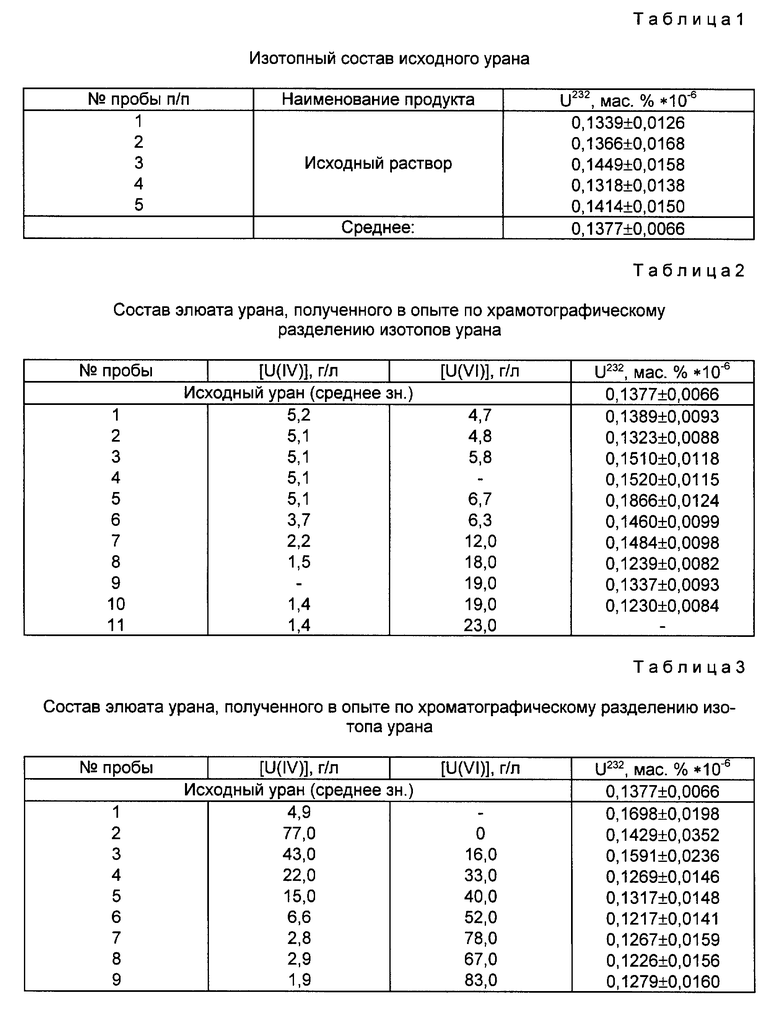
Известны различные способы разделения изотопов урана химическими методами. Из них наиболее эффективными являются методы, основанные на реакции изотопного обмена между двумя валентными формами урана, находящимися в разных фазах - водной и органической. Так, например, известен способ разделения изотопов урана японской фирмы "Асахи Кемикл индастри" (Пат. США N 4118457, кл. B 01 D 59/30, 03.10.78), в основу которого положена реакция изотопного обмена между четырех- и шестивалентным ураном, находящимся соответственно в фазе раствора и анионообменной смолы. Через анионообменную смолу в форме металла-окислителя, например, железа (III), проводят полосу урана (IV), элюируя его солянокислым раствором металла-восстановителя, например, титана (III). Схема ионообменного редокс-хроматографического процесса разделения изотопов урана фирмы "Асахи Кемикл индастри" ("Асахи-процесс) представлена на фиг. 1.
Взаимодействуя с восстановителем, шестивалентный уран восстанавливается до четырехвалентного состояния, десорбируется с анионообменной смолы и с элюатом переносится к зоне окисляющего агента, где окисляется и сорбируется на смоле. Таким образом, происходит движение адсорбционной полосы урана, в ходе которого изотопом уран-238 обогащается уран (IV) по реакции изотопного обмена (р.и.о.) (1):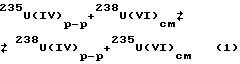
с однократным коэффициентом разделения α = 1,0013 и переносится с ним к фронтальной части полосы. При этом уран в окрестностях тылового фронта полосы урана соответственно обогащается изотопом уран-235.
Так, в одном из экспериментов, приведенных в качестве примера в описании к патенту на описанный способ [1], была использована анионообменная смола, приготовленная хлорметилированием стиролдивинилбензольного сополимера с последующим его аминированием триметиламином. Смола имела степень сшивки 6% и размер частичек от 74 мкм до 149 мкм. Этой смолой заполнили две колонки (каждая длиной 1 м с внутренним диаметром 1 см). Пропустив через две последовательно соединенные колонки раствор, содержащий 0,05 моль/л сульфата трехвалентного железа и 0,6 моль/л серной кислоты, смолу перевели в Fe(III)-форму. Затем подачей раствора состава: уранилхлорида - 0,04 моль/л, серной кислоты - 0,2 моль/л, в первой колонке сформировали на смоле полосу урана (VI) длиной 11,5 см. Элюирование провели раствором, содержащим трихлорида титана 0,05 моль/л и серной кислоты 0,2 моль/л. При этом полоса урана, находящаяся между красной зоной ионов железа (III) и пурпурной зоной титана (III), двигалась со скоростью 20 см/сутки. Таким образом, полоса урана была проведена через две колонки. Масс-спектрометрический анализ первой и последней проб урана, отобранных из элюата, дал следующие значения отношения U235/U238, %: 0,00693 и 0,00759, что соответствует 0,9559 и 1,0469 частям от 0,00725 - отношения изотопов в природном уране.
В промышленном варианте разделения изотопов урана описанным выше способом для получения продукта - урана, обогащенного до 3% изотопом уран-235 и хвостов - урана, содержащего урана-235 0,1%, предполагается полосу урана длиной несколько метров проводить через слой смолы на расстояние от 10 до 1000 м [1]. Такое движение полосы осуществляется с использованием, как минимум, двух колонн, из которых одна занята полосой урана, а вторая в этой время регенерируется.
Наиболее близким к предложенному по технической сущности и достигаемому результату является способ обогащения урана одним из его изотопов в многоступенчатом каскаде путем противоточного контакта экстрагента, содержащего соединения четырехвалентного урана, и водного раствора, содержащего соединения трехвалентного урана (Пат. СССР N 867283, кл. B 01 D 59/28, 23.09.81). В основу метода положена р.и.о. с очень высокой скоростью:
для которой константа равновесия р.и.о., определяемая методом ступенчатого сжатия, равна 1,0012 - 1,0030. Для разделения валентных форм урана предложено использовать несколько экстракционных систем, при этом подбираются условия с минимальным переходом урана (III) и урана (IV) из одной фазы в другую.
Недостатком этих систем является крайняя неустойчивость урана (III) в водных растворах, его тенденция к окислению. Работа с такими соединениями возможна в атмосфере азота при отсутствии ионов металлов III-VIII групп. Контактирование осуществляется при исключении какого-либо соприкосновения фаз с любым твердым электропроводным материалом и с реагентами, выделяющими кислород. Организация процесса разделения изотопов урана экстракционным методом основана на том же методе "движущейся полосы" с противоточным движением фаз, содержащих противопоставляемые ионы урана, и с окислительно-восстановительным рефлаксом в ее фронтальных частях. В качестве органической фазы можно использовать практически все классы экстрагентов, в том числе нейтральные фосфорорганические соединения, кислоты, основания, спирты, амины, кетоны, эфиры, хелаты, а также катионообменные и анионообменные смолы.
В полосе урана его четырехвалентная форма предпочтительно находится в органической фазе, а трехвалентная - в водной фазе, то есть здесь реализуется случай, когда коэффициент разделения валентных форм урана не равен единице. Процесс проводят в растворе хлористоводородной кислоты в присутствии металлов III-IV группы с концентрацией 1 ppm с осуществлением редокс-рефлакса урана (проведением окислительно-восстановительного процесса с переводом урана в противопоставляемую валентную форму и другую фазу) по концам его полосы, а именно с исчерпывающей (полной) реэкстракцией его из органической фазы в водную в головной части полосы урана и восстановлением четырехвалентного урана до трехвалентного состояния с возвратом его в ступень отбора четырехвалентного урана и с окислением трехвалентного урана до четырехвалентного состояния, исчерпывающей экстракцией четырехвалентного урана и возвратом его с экстрагентом в тыловую часть полосы урана (в ступень отбора трехвалентного урана). Запитывают такой экстракционный каскад подачей в среднюю часть каскада исходного водного раствора урана.
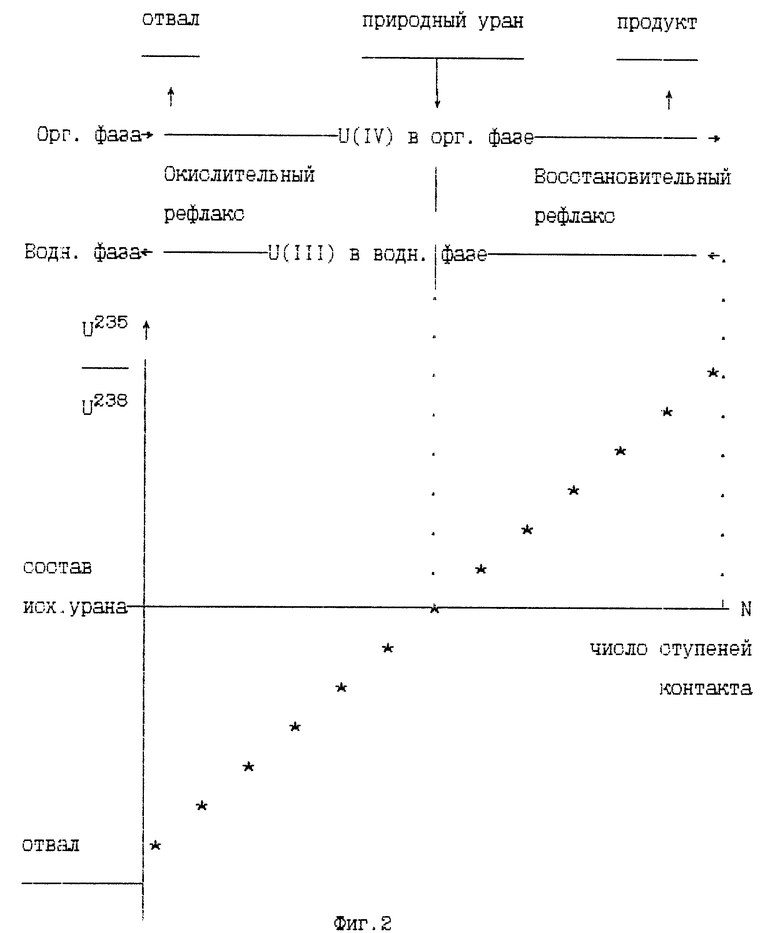
За счет неравномерного перераспределения изотопов урана в его полосе между двумя его валентными формами, находящимися в разных несмешивающихся фазах, осуществляется перенос легких изотопов урана с четырехвалентным ураном и тяжелых изотопов урана - с трехвалентным ураном к противоположным концам полосы урана. При этом чем больше количество ступеней контакта реализуется в полосе урана, тем выше конечная степень разделения изотопов. Схематически процесс разделения изотопов урана с изотопным обменом между ураном (III) и ураном (IV) представлен на фиг. 2.
В одном из примеров реализации указанного способа приведены следующие данные. Разделение изотопов природного урана провели в экстракционном каскаде, состоящем из 70 ступеней. Состав фаз:
- водная фаза на входе каскада имела состав:
[HCl] = 2 N;
[UCl3] = 0,1 M;
- органическая фаза на входе каскада - 50%-ный ТБФ в додекане.
Восстановление четырехвалентного урана проводили электрохимически и диафрагменном электролизе с ртутным катодом, а окисление трехвалентного урана - хлором. Из природного урана, содержащего 0,7194% урана-235, был получен обогащенный продукт, содержащий 0,80% урана-235, и обедненный продукт (отвал), содержащий 0,7047% урана-235.
Недостатками данного способа являются:
необходимость проведения редокс-рефлакса валентных форм урана по концам его полосы (то есть перевода урана в противопоставляемую валентную форму и другую фазу с возвратом его во фронтальную часть полосы урана, где был произведен отбор урана);
крайне низкая химическая устойчивость растворов трехвалентного урана и, как следствие, необходимость использования солянокислой среды с минимальным содержанием примесей металлов III-IV группы. При этом однократный коэффициент разделения изотопов урана не превышает величины 1,0030.
Задача изобретения - увеличение степени разделения изотопов, уменьшение числа используемых реагентов, увеличение химической устойчивости обменной системы.
Поставленная задача решается тем, что в способе разделения изотопов урана, включающем изотопный обмен между двумя его валентными формами, одной из которых является четырехвалентный уран, движущимися в полосе, сформированной в процессе противоточной экстракции, обеспечивающей коэффициент разделения валентных форм урана, не равный единице, с рефлаксом валентных форм урана методами исчерпывающей экстракции и реэкстракции по концам его полосы, в качестве второй валентной формы используют шестивалентный уран, изотопный обмен проводят в растворах азотной кислоты, а рефлакс урана осуществляют без изменения его валентного состояния.
Благодаря тому, что коэффициент разделения валентных форм не равен единице, создается полоса урана с разнесенными по ее концам зонами двух противопоставляемых валентных форм урана: четырех- и шестивалентного урана, и реакция изотопного обмена проходит в месте перекрытия разнесенных зон валентных форм урана, где направление межфазного переноса валентных форм противоположно.
В соответствии с традиционными представлениями в такой системе максимальная степень разделения изотопов не может превысить значения, равного величине однократного коэффициента разделения изотопов, например для пары уран (VI)/уран (IV) α235/238= 1,0013, так как сама операция по разделению валентных форм урана в полосе урана, приведенных в изотопное равновесие в питающем растворе, не должна приводить к увеличению изотопного эффекта.
Однако совершенно неожиданно был получен изотопный эффект, величина которого на два порядка превышала расчетное значение, равное однократному коэффициенту разделения, причем направление изотопного эффекта оказалось обратным аналогу, то есть в этом случае легким изотопом обогатился четырехвалентный уран. Вероятно, это является следствием перераспределения изотопов урана между его валентными формами в процессе встречного межфазного переноса валентных форм урана, которое реализуется в месте перекрытия полос противопоставляемых валентных форм урана. В способе - прототипе и аналоге этот процесс практически отсутствует, так как в них реализуется редокс-рефлакс по концам полосы урана, тогда как в предложенном способе рефлакс урана проводят без изменения его валентности.
Примеры реализации способа
В трех примерах реализации способа приведены данные по разделению смеси изотопов урана-232 и урана-238, присутствующих в высокофоновом уране. Содержание урана-232 определяли по ОСТ 95.999-92, включающем химическое выделение урана и последующее альфа-спектрометрическое определение содержания урана-232 в % к урану-238. Опыты, в которых было организовано движение полосы из шестивалентной и четырехвалентной форм урана, были поставлены в двух вариантах: хроматографическом и в варианте противоточной экстракции.
Пример 1. В эксперименте использовалась хроматографическая колонка, снаряженная тефлоновым порошком с фракцией 0,25 - 0,5 мм, на поверхность которого предварительно нанесен экстрагент 65%-ный ТБФ в РЭД-2 в количестве 5% от веса порошка.
Характеристики колонки:
Высота - 1м;
Диаметр - 0,1 м;
Вес тефлонового порошка (0,25 - 0,5 мм) - 72,14 г;
Вес экстрагента - 3,607 г;
Объем межзернового пространства - 35 см3.
Колонка предварительно была приведена в равновесие с раствором, содержащим азотной кислоты 2 моль/л и гидразина 0,5 г/л. Чтобы сформировать в колонке полосу урана (IV), в нее вводили 2 мл раствора следующего состава:
[U(IV)] = 115,5 г/л;
[HNO3] = 2,0 моль/л;
[N2H4] = 0,5 г/л.
Затем промывали колонку 60 мл раствора U(VI) состава:
[U(VI)] = 31,0 г/л;
[HNO3] = 2,0 моль/л;
[N2H4] = 0,5 г/л.
В конце эксперимента колонка промывалась раствором, содержащим азотную кислоту с концентрацией 0,3 моль/л для реэкстракции урана (VI). Таким образом в эксперименте было реализовано противоточное движение водной и органической фаз с образованием полосы двух валентных форм урана и с исчерпывающей экстракцией и реэкстракцией по концам полосы урана.
Поступающий в колонку шестивалентный уран вытеснял четырехвалентный уран. Движение по колонке полосы четырехвалентного урана, ее передний и задний фронт можно было наблюдать визуально. Продолжительность эксперимента 4 часа. При выходе из колонки заднего фронта четырехвалентного урана (переднего фронта шестивалентного урана) отбирали пробы для анализа на содержание урана четырехвалентного, шестивалентного и изотопного состава. Изотопный состав исходного урана и результаты эксперимента приведены соответственно в табл. 1 и 2.
Из данных табл. 2 следует, что легкими изотопами обогащается уран со стороны заднего фронта полосы четырехвалентного урана в месте ее перекрытия с фронтом шестивалентного урана.
Пример N 2. В эксперименте использовалась та же хроматографическая колонка, что и в первом опыте. Колонка предварительно была приведена в равновесие с раствором, содержащим азотной кислоты 2 моль/л и гидразина 0,5 г/л.
Чтобы сформировать в колонке полосу урана (IV), в нее ввели 1 мл раствора следующего состава:
[U(IV)] = 163,3 г/л;
[U(VI)] = 29,7 г/л;
[HNO3] = 2,13 моль/д;
[N2H4] = 26 г/л,
и затем элюировали четырехвалентный уран раствором шестивалентного урана состава:
[U(VI)] = 100,0 г/л;
[NHO3] = 2,0 моль/л;
[N2H4] = 0,5 г/л.
В процессе элюирования четырехвалентный уран вытеснялся на передний фронт полосы урана. Поступающий в колонку шестивалентный уран вытеснял четырехвалентный уран на передний фронт. Движение по колонке полосы четырехвалентного урана можно было наблюдать визуально.
Продолжительность эксперимента 4 часа. При выходе из колонки заднего фронта четырехвалентного урана (переднего фронта шестивалентного урана) отбирали пробы для анализа на содержание урана четырехвалентного, шестивалентного и изотопного состава. Результаты приведены в табл. 3.
Из данных табл. 3 следует, что легкими изотопами обогащается уран, начиная со стороны заднего фронта полосы четырехвалентного урана в месте ее перекрытия с фронтом шестивалентного урана и кончая передним фронтом полосы четырехвалентного урана.
Пример N 3. В этом примере описан изотопный эффект, который был зафиксирован при экстракционной переработке растворов высокофонового урана, находящегося в шестивалентном состоянии.
Технологией переработки высокофонового урана предусмотрено введение в исходный и промывной растворы четырехвалентного урана, применяемого для восстановления плутония до трехвалентного состояния. В экстракционной колонне в процессе переработки исходного раствора высокофонового урана, содержащего азотной кислоты 30 - 60 г/л, при соотношении фаз O:B = 2,5:1 и насыщении экстрагента шестивалентным ураном 100 - 105 г/л четырехвалентный уран образует концентрационный пик на фронте шестивалентного урана. В зоне перекрытия фронтов четырехвалентного и шестивалентного урана воспроизводится ситуация с противоточным движением валентных форм урана через границу раздела фаз, когда четырехвалентный уран вытесняется из органической фазы шестивалентным ураном и реэкстрагируется в водную фазу, обогащаясь легкими изотопами, а шестивалентный наоборот - экстрагируется в органическую фазу, обогащаясь тяжелыми изотопами.
Частично, в небольших количествах (< 1 г/л), уран уходил с рафинатом. Так как пик четырехвалентного урана находился ближе к выходу рафината из колонны, чем фронт шестивалентного урана, то в рафинате относительное содержание в уране его четырехвалентной формы оказывалось значительно выше, чем в реэкстракте урана, что повлекло изменение изотопного состава урана в рафинате и в реэкстракте.
Результаты анализа исходного урана, урана, ушедшего с рафинатом, и урана в упаренном реэкстракте показали (см. табл. 4), что наблюдается концентрирование урана-232 в уране, теряемом с рафинатом, и обеднение по этому изотопу урана реэкстракта.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОЧИСТКИ УРАНА (VI) ОТ ТЕХНЕЦИЯ (VII) | 2000 |
|
RU2184083C2 |
| СПОСОБ ПЕРЕРАБОТКИ ЯДЕРНОГО ТОПЛИВА, ОБОГАЩЕННОГО ДЕЛЯЩИМСЯ МАТЕРИАЛОМ | 1999 |
|
RU2171507C2 |
| СПОСОБ ПЕРЕРАБОТКИ ВЫСОКООБОГАЩЕННОГО УРАНА | 1998 |
|
RU2131476C1 |
| СПОСОБ ЭКСТРАКЦИОННОЙ ОЧИСТКИ РЕГЕНЕРИРОВАННОГО УРАНА | 2007 |
|
RU2373155C2 |
| СПОСОБ ПЕРЕРАБОТКИ АЗОТНОКИСЛОГО РАСТВОРА РЕГЕНЕРИРОВАННОГО УРАНА С ОЧИСТКОЙ ОТ ТЕХНЕЦИЯ (ВАРИАНТЫ) | 2009 |
|
RU2430175C1 |
| СПОСОБ ОЧИСТКИ РЕГЕНЕРИРОВАННОГО УРАНА | 2010 |
|
RU2447523C2 |
| СПОСОБ ОЧИСТКИ РЕГЕНЕРИРОВАННОГО УРАНА | 2010 |
|
RU2425804C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ УРАНА | 1999 |
|
RU2159741C1 |
| СПОСОБ ПОЛУЧЕНИЯ СМЕШАННЫХ ОКСИДОВ УРАНА И ПЛУТОНИЯ | 2015 |
|
RU2626854C2 |
| СПОСОБ ЭКСТРАКЦИОННОЙ ПЕРЕРАБОТКИ УРАНСОДЕРЖАЩИХ РАСТВОРОВ | 1997 |
|
RU2114469C1 |

Изобретение относится к ядерной энергетике и может быть использовано при корректировке изотопного состава ядерного топлива. В хроматографическую колонну с тефлоновым порошком, на поверхность которого нанесен экстрагент, заливают раствор, содержащий: [U(VI)]-115,5 г/л, [HNO3]-2,0 моль/л, [N2H4] -0,5 г/л. Сформировывают полосу урана (IV). Процесс противоточной экстракции обеспечивает коэффициент разделения, не равный 1. Рефлакс урана осуществляют без изменения валентного состояния методом исчерпывающей экстракции и реэкстракции по концам полосы. Увеличивается степень разделения изотопов U238 и U232, уменьшается число реагентов. Обменная система химически устойчива. 4 табл., 2 ил.
Способ химического разделения изотопов урана, включающий изотопный обмен между двумя его валентными формами, одной из которых является четырехвалентный уран, движущимися в полосе, сформированной в процессе противоточной экстракции, обеспечивающей коэффициент разделения валентных форм урана, не равный единице, с рефлаксом валентных форм урана методами исчерпывающей экстракции и реэкстрации по концам его полосы, отличающийся тем, что в качестве второй валентной формы используют шестивалентный уран, изотопный обмен проводят в растворах азотной кислоты, а рефлакс урана осуществляют без изменения его валентного состояния.
