ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к ингибиторам 11β-гидроксистероид-дегидрогеназы 1 типа (11β-ГСД1), их лекарственным препаратам и их применениию.
УРОВЕНЬ ТЕХНИКИ
Глюкокортикоиды (например, кортизол (гидрокортизон)) представляют собой стероидные гормоны, обеспечивающие регулировку обмена, функционирования и распределения жиров, а также принимающие участие в обмене углеводов, белков и жиров. Также известно, что глюкокортикоиды оказывают физиологическое воздействие на развитие, нейробиологию, воспалительные процессы, кровяное давление, обмен веществ, а также апоптоз. Кортизол и другие кортикостероиды связываются с глюкокортикоидными рецепторами (ГР) и минералкортикоидными рецепторами (МР), которые являются членами одного суперсемейства ядерных гормональных рецепторов и обусловливают действие кортизола in vivo. Данные рецепторы непосредственно модулируют транскрипцию посредством ДНК-связывающих доменов «цинковые пальцы», а также вызывающих транскрипцию доменов.
До недавнего времени считалось, что действие глюкокортикоидов определяется тремя основными факторами: (1) уровнем циркулирующих глюкокортикоидов (определяемым, в основном, гипоталамо-гипофизарно-надпочечниковой (ГПА) осью); (2) уровнем связывания глюкокортикоидов с белком в кровотоке; а также (3) количеством внутриклеточных рецепторов в тканях-мишенях. Однако недавно был обнаружен четвертый фактор, определяющий действие глюкокортикоидов: тканеспецифический метаболизм активирующими и инактивирующими глюкокортикоиды ферментами на пререцепторном уровне. Данные ферменты, контролирующие действие 11-гидроксистероид-дегидрогеназы (11β-ГСД) на пререцепторном уровне, модулируют активацию ГР и МР путем регулирования глюкокортикоидных гормонов. В настоящее время было осуществлено клонирование и составлена полная характеристика двух различных изоферментов 11-β-ГСД: 11-ГСД1 (также известного как 11-β-ГСД 1 типа, 11βГСД1, ГСД11В1, ЛВП (липопротеин высокой плотности), а также ГСД11L) и 11-ГСД2. 11-ГСД1 представляет собой двустороннюю оксидоредуктазу, обеспечивающую регенерацию активного кортизола из неактивных 11-кето-форм, в то время как 11-ГСД2 представляет собой одностороннюю дегидрогеназу, обеспечивающую инактивацию биологически активного кортизола путем его преобразования в кортизон.
Обе изоформы экспрессируются определенным тканеспецифическим образом в соответствии с различиями в их физиологической роли. 11-ГСД1 широко распределяется в тканях крысы и человека; признаки присутствия ферментов и соответствующие информационные РНК были обнаружены в печени человека, жировых тканях, легких, мужских половых железах, костях и реснитчатом эпителии. В жировой ткани повышенная концентрация кортизола стимулирует дифференциацию адпипоцитов и может играть роль в стимуляции висцерального ожирения. 11-ГСД1 может обеспечивать контроль внутриглазного давления, а также способствовать возникновению глаукомы; некоторые данные свидетельствуют о том, что ингибирование 11-ГСД1 может вызывать снижение внутриглазного давления у пациентов с внутриглазной гипертензией (Kotelevstev et al. (1997), Proc. Natl. Acad, Sci. USA 94(26): 14924-9). Несмотря на то что 11-ГСД1 стимулирует 11-бета-дегидрогенизацию, а также обратную реакцию 11-оксоредукции, 11-ГСД1 действует, в основном, как НАДФН-зависимая оксоредуктаза в здоровых клетках и тканях, стимулируя формирование активного кортизола из инертного кортизола (Low et al. (1994) J. Mol. Endocrin. 13: 167-174). Однако экспрессия 11-ГСД2 обнаружена в основном в тканях-мишенях минералокортикоидов, таких как почки (кортекс и мозговой слой), плацента, сигмовидная и прямая кишка, слюнная железа и клеточные линии прямой кишки. 11-ГСД2 действует как НАД-зависимая дегидрогеназа, которая активизирует преобразование кортизола в кортизон (Albiston et al. (1994) Mol. Cell. Endocrin. 105: R11-R17), а также защищает МР от избытка глюкокортикоида (например, при высоком уровне активного по отношению к рецепторам кортизола) (Blum et al. (2003) Prog. Nucl. Acid Res. Mol. Biol. 75:173-216).
Мутации в генах 11-ГСД1 или 11-ГСД2 приводят к патологии человека. Например, у отдельных пациентов с мутациями в 11-ГСД2 наблюдался дефицит активности по инактивации кортизола, который, в свою очередь, приводил к развитию синдрома кажущегося избытка минералокортикоидов (также упоминается как «СКИМ»), сопровождаемого гипертензией, гипокалиемией и задержкой натрия (Edwards et al. (1988) Lancet 2: 986-989; Wilson et al. (1998) Proc. Natl. Acad. Sci. 95: 10200-10205). Аналогичным образом мутации в 11-ГСД1, а также в гене, кодирующем колокализированный НАДФН-продуцирующий фермент, глюкозо 6-фосфат дегидрогеназе (Г6ДГ), могут вызвать недостаточность кортизон-редуктазы (НКР); у пациентов с данными мутациями наблюдается избыток андрогенного гормона, обусловленный адренокортикотропным гормоном (гирсутизм, нерегулярность менструальных циклов, гиперандрогенизм), а также синдром поликистоза яичников (СПЯ) сходного фенотипа (Draper et al. (2003) Nat. Genet. 34: 434-439).
В значительной степени нарушение гомеостаза в ГПА-оси по причине недостаточной или избыточной секреции или действия приводит к развитию синдрома Иценко-Кушинга и болезни Аддисона, соответственно (Miller and Chrousos (2001) Endocrinology and Metabolism, eds. Felig and Frohman (McGraw-Hill, New York), 4th Ed.: 387-524). У пациентов с синдромом Иценко-Кушинга, а также у пациентов, принимающих глюкокортикоиды, может наблюдаться развитие обратимого висцерального ожирения. Фенотип пациентов, страдающих от синдрома Иценко-Кушинга, обладает значительным сходством с фенотипом метаболического синдрома Равена (также известен как синдром Х или синдром инсулинорезистентности), симптомы которого включают висцеральное ожирение, нарушение толерантности к глюкозе, инсулинорезистентность, гипертензию, диабет 2 типа и гиперлипидемию (Reaven (1993) Ann. Rev. Med. 44: 121-131). Несмотря на отсутствие полной характеристики роли глюкокортикоидов в ожирении человека все большее количество данных указывает на то, что активность 11-ГСД1 играет важную роль в процессе ожирения и развития метаболического синдрома (Bujalska et al. (1997) Lancet 349: 1210-1213); (Livingstone et al. (2000) Endocrinology 131: 560-563; Rask et al. (2001) J. Clin. Endocrinol. Metab. 86: 1418-1421; Lindsay et al. (2003) J. Clin. Endocrinol. Metab. 88: 2738-2744; Wake et al. (2003) J. Clin. Endocrinol. Metab. 88: 3983-3988).
Данные, полученные в ходе проведения испытаний на моделях трансгенных мышей, поддерживают гипотезу о том, что активность 11-ГСД1 в отношении адипоцитов играет основную роль в процессе висцерального ожирения и развития метаболического синдрома (Alberts et al. (2002) Diabetologia. 45(11); 1526-32). Сверхэкспрессия 11-ГСД1 в жировой ткани под контролем промотора аР2 у трансгенных мышей вызвала образование фенотипа, удивительно похожего на фенотип метаболического синдрома человека (Masuzaki et al. (2001) Science 294: 2166-2170; Masuzaki et al. (2003) J. Clinical Invest. 112: 83-90). Более того, повышенная активность 11-ГСД1 у данных мышей обладает значительным сходством с активностью, наблюдаемой при ожирении человека (Rask et al. (2001) J. Clin. Endocrinol. Metab. 86: 1418-1421), Кроме того, данные, полученные в ходе испытаний на мышах с дефицитом 11-ГСД1, вызванным гомологической рекомбинацией, свидетельствуют о том, что нехватка 11-ГСД1 вызывает снижение чувствительности к инсулину и нарушение толерантности к глюкозе по причине тканеспецифического снижения уровня активного глюкокортикоида (Kotelevstev et al. (1997) Proc. Natl. Acad. Sci. 94: 14924-14929; Morton et al. (2001) J. Biol. Chem. 276: 41293-41300; Morton et al. (2004) Diabetes 53: 931-938).
Опубликованные данные поддерживают гипотезу о том, что повышенная экспрессия 11-ГСД1 способствует преобразованию кортизона в кортизол в жировой ткани, вследствие чего 11-ГСД1 играет определенную роль в патогенезе центрального типа ожирения, а также в развитии метаболического синдрома у человека (Engeli et al., (2004) Obes. Res. 12: 9-17). Следовательно, с фармацевтической точки зрения 11-ГСД1 является многообещающим объектом воздействия при лечении метаболического синдрома (Masuzaki et al., (2003) Curr. Drug Targets Immune Endocr. Metabol. Disord. 3: 255-62). Кроме того, ингибирование активности 11-ГСД1 может оказаться благоприятным при лечении многочисленных связанных с глюкокортикоидами нарушений. Например, ингибиторы 11-ГСД1 могут быть эффективны при лечении ожирения и(или) аспектов кластера метаболического синдрома, включающих нарушение толерантности к глюкозе, резистентность к инсулину, гипергликемию, гипертензию и(или) гиперлипидемию (Kotelevstev et al. (1997) Proc. Natl. Acad. Sci. 94: 14924-14929; Morton et al. (2001) J. Biol. Chem. 276: 41293-41300; Morton et al. (2004) Diabetes 53: 931-938). Ингибирование активности 11-ГСД1 может оказывать благоприятное воздействие на поджелудочную железу, включающее усиление секреции инсулина, стимулируемой глюкозой (Billaudel and Sutter (1979) Horm. Metab. Res. 11: 555-560; Ogawa et al. (1992) J. Clin. Invest. 90: 497-504; Davani et al. (2000) J. Biol. Chem. 275: 34841-34844).
Помимо этого, учитывая связь межличностных отличий общей когнитивной функции с различной длительностью воздействия глюкокортикоидами (Lupien et al. (1998) Nat. Neurosci. 1: 69-73), а также то, что дисрегуляция ГПА-оси, вызывающая хронический избыток глюкокортикоидов в определенных подобластях мозга, теоретически, способствует нарушению когнитивной функции (McEwen and Sapolsky (1995) Curr. Opin. Neurobiol. 5: 205-216), можно предположить, что ингибирование 11-ГСД1 позволит уменьшить воздействие глюкокортикоидов на мозг и, следовательно, защитить нейрональную активность от вредного воздействия глюкокортикоидов, вызывающего нарушение когнитивных функций, слабоумие и(или) депрессию. В частности, известно, что стресс и глюкокортикоиды оказывают воздействие на когнитивную функцию (de Quervain et al. (1998) Nature 394: 787-790); также наблюдения показали, что 11-ГСД1 посредством контроля действия глюкокортикоидов на мозг также может оказывать воздействие на нейротоксичность (Rajan et al. (1996) Neuroscience 16: 65-70; Seckl (2000) Neuroendocrinol. 18:49-99).
Кроме того, существуют данные, подтверждающие участие глюкокортикоидов и 11-ГСД1 в изменении уровня внутриглазного давления (ВГД) (Stokes et al. (2000) Invest. Ophthalmol. Vis. Sci. 41: 1629-1683; Rauz et al. (2001) Invest. Ophthalmol. Vis. Sci. 42: 2037-2042); при отсутствии лечения повышенное ВГД может привести к частичной скотоме и последующей слепоте. Таким образом, ингибирование глазного 11-ГСД1 может вызвать снижение местной концентрации глюкокортикоидов и ВГД. Следовательно, 11-ГСД1 теоретически может применяться для лечения глаукомы и прочих нарушений зрения.
У трансгенных мышей с аР2-11-ГСД1 наблюдается высокое кровяное давление, а также повышенная чувствительность к пищевой соли. Кроме того, у трансгенных мышей был отмечен повышенный уровень ангиотензина, ангиотензина II и альдостерона в плазме крови; лечение мышей при помощи антагонистов ангиотензина II способствует снижению гипертензии (Masuzaki et al. (2003) J. Clinical Invest. 112: 83-90). Данный эффект свидетельствует о том, что активность 11-ГСД1 может вызывать или усиливать гипертензию. Следовательно, ингибиторы 11-ГСД1 могут быть полезны при лечении гипертензии, а также связанных с гипертензией сердечно-сосудистых нарушений, Ингибирование 11-ГСД1 в зрелых адипоцитах, предположительно, также может ослаблять секрецию ингибитора активатора плазминогена 1 (ИАП-1), являющегося независимым фактором сердечно-сосудистого риска (Halleux et al. (1999) J. Clin. Endocrinol. Metabl. 84: 4097-4105).
Глюкокортикоиды могут оказывать нежелательное воздействие на ткани скелета; длительное воздействие глюкокортикоидов даже в умеренных дозах может вызвать развитие остеопороза (Cannalis (1996) J. Clin. Endocrinol. Metab. 81: 3441-3447). Помимо этого 11-ГСД1 была обнаружена в первичных культурах остеобластов человека, а также в клетках костей взрослых пациентов (Cooper et al. (2000) Bone 27: 375-381), в то время как ингибитор 11-ГСД1, карбеноксолон, согласно полученным данным, способствует снижению неблагоприятного воздействия глюкокортикоидов на формирование островков окостенения (Bellows et al. (1998) Bone 23: 119-125). Таким образом, ингибирование 11-ГСД1, предположительно, вызывает снижение местной концентрации глюкокортикоидов в остеобластах и остеокластах, тем самым оказывая благоприятное воздействие на различные формы заболеваний костей, включая остеопороз.
Ингибиторы 11-ГСД1 также могут быть полезны при иммуномодуляции. Считается, что глюкокортикоиды оказывают иммуносупрессивное воздействие, на самом деле между ГПА-осью и иммунной системой наблюдается сложное динамическое взаимодействие (Rook (1999) Baillier's Clin. Endocrinol. Metabl. 13: 576-581). Глюкокортикоиды могут принимать участие в модулировании баланса между клеточным иммунным ответом и гуморальным иммунным ответом. При этом высокая активность глюкокортикоидов, как правило, связана с гуморальным ответом. Следовательно, ингибирование 11-ГСД1 может использоваться в качестве средства смещения иммунного ответа в сторону клеточного иммунного ответа. Определенные болезненные состояния, включающие туберкулез, лепру (болезнь Хансена) и псориаз, вызывают иммунный ответ, смещенный в сторону гуморального ответа, однако, наиболее эффективным видом иммунного ответа может быть клеточный иммунный ответ. Таким образом, ингибиторы 11-ГСД1 также могут быть полезны при лечении подобных заболеваний.
Согласно полученным отчетам глюкокортикоиды препятствуют заживлению ран, особенно у пациентов с диабетическими язвами (Bitar et al. (1999) J. Surg. Res. 82: 234-243; Bitar et al. (1999) Surgery 125: 594-601; Bitar (2000) Surgery 127: 687-695; Bitar (1998) Am. J. Pathol. 152: 547-554). Среди пациентов с нарушенной толерантностью к глюкозе и(или) диабетом 2 типа также часто наблюдается нарушение заживления ран. Глюкокортикоиды также способствуют повышению риска заболевания и задержки заживления ран (Anstead (1998) Adv. Wound Care 11:277-285). Кроме того, установлена взаимосвязь между повышением уровня кортизола в раневой жидкости и незаживающими ранами (ЕР Patent App.No, 0902288). Последние опубликованные заявки на патент содержат предположение о благоприятном воздействии определенных ингибиторов 11-ГСД1, способствующем заживлению ран (PCT/US 2006/043,951).
Представленная в данном изобретении информация свидетельствует о возрастающей необходимости в разработке новых и более эффективных лекарственных препаратов, ингибирующих 11-ГСД1. Новые соединения, представленные в данном изобретении, являются эффективными ингибиторами 11-ГСД1.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
На данный момент установлено, что соединения Формулы Im1 или их фармацевтически премлемые соли являются эффективными ингибиторами 11-ГСД1. Изобретение представляет собой соединение Формулы (Im1)
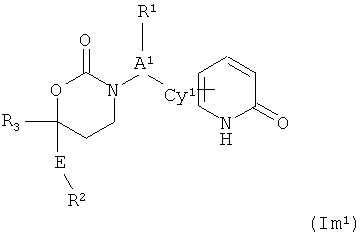 ;
;
или его фармацевтически приемлемую соль, энантиомер или диастереомер.
В первом варианте осуществления настоящего изобретения Формула Im1 и ее компоненты указаны в данном документе в соответствии со следующими определениями:
R1 - (а) отсутствует или (b) выбран из (С1-С6)алкила, (С2-С6)алкенила, (C2-С6)алкинила, (С1-С3)алкокси (С1-С3) алкокси, или (C1-С3) алкокси (С1-С3)алкила, возможные заместители включают в себя не более четырех групп, независимо выбранных из фтора, циано, оксо, R4, R4O-; (R4)2N-, R4O2C-, R4S, R4S(=O)-, R4S(=O)2-, R4S(=O)NR4-, (R4)2NC(=O)-, (R4)2NC(=O)O-, (R4)2NC(=O)NR4-, R4OC(=O)NR4-, (R4) 2NC(=NCN)NR4-, (R4O)2P(=O)O-, (R4O)2P(=O)NR4-, R4OS(=O)2NR4-, (R4)2NS(=O)2O-, (R4)2NS(=O)2NR4-, R4S(=O)2NR4-, R4S(=O)NHC(=O)-, R4S(=O)NHC(=O)O-, R4S(=O)2NHC(=O)NR4-, R4OS(=O)2NHC(=O)-, R4OS(=O)2NHC(=O)O-, R4OS(=O)2NHC(=O)NR4-, (R4)2NS(=O)2NHC(=O)-, (R4)2NS(=O)2NHC(=O)O-, (R4)2NS(=O)2NHC(=O)NR4-, R4C(=O)NHS(=O)2-, R4C(=O)NHS(=O)2O-, R4C(=O)NHS(=O)2NR4-, R4OC(=O)NHS(=O)2-, R4OC(=O)NHS(=O)2O-, R4OC(=O)NHS(=O)2NR4-, (R4)2NC(=O)NHS(=O)2-, (R4)2NC(=O)NHS(=O)2O-, (R4)2NC(=O)NHS(=O)2NR4-, гетероциклила, гетероарила, ариламино и гетероариламино;
А1 представляет собой (а) связь или (b) (С1-С3)алкилен, CH2CH2O, в котором кислород присоединен к Су1, или СН2С(=O), в котором карбонильный углерод присоединен к Су1;
Су1 представляет собой арил, гетероарил, моноциклический циклоалкил или моноциклический гетероциклил, возможные заместители которого включают в себя от 1 до 4 групп, независимо выбранных из фтора, хлора, брома, йода, циано, нитро, амино, гидрокси, карбокси, (С1-С6)алкила, гидрокси(С1-С6)алкила, (С3-С6)циклоалкила, гидрокси(С3-С6)циклоалкила, (С4-С7)циклоалкилалкила, (С2-С6)алкенила, гало(С2-С6)алкенила, гидрокси(С2-С6)алкенила, (С2-С6)алкинила, (С3-С6)циклоалкил(С2-С4)алкинила, гало(С1-С6)алкила, гало(С3-С6)циклоалкила, гало(С4-С7)циклоалкилалкила, (С1-С6)алкокси, (С3-С6)циклоалкокси, (С4-С7)циклоалкилалкокси, гало(С1-С6)алкокси, гало(С3-С6)циклоалкокси, гало(С4-С7)циклоалкилалкокси, (С1-С6)алкилтио, (С3-С6)циклоалкилтио, (С4-С7)циклоалкилалкилтио, гало(С1-С6)алкилтио, гало(С3-С6)циклоалкилтио, гало(С4-С7)циклоалкилалкилтио, (С1-С6)алкансульфинила, (С3-С6)циклоалкансульфинила, (С4-С7)циклоалкилалкансульфинила, гало(С1-С6)алкансульфинила, гало(С3-С6)циклоалкансульфинила, гало(С4-С7)циклоалкилалкансульфинила, (С1-С6)алкансульфонила, (С3-С6)циклоалкансульфонила, (С4-С7)циклоалкилалкансульфонила, гало(С1-С6)алкансульфонила, гало(С3-С6)циклоалкансульфонила, гало(С4-С7)цикло-алкилалкансульфонила, (С1-С6)алкиламино, ди(С1-С6)алкиламино, (С1-С6)алкокси(С1-С6)алкокси, гало(С1-С6)алкокси(С1-С6)алкокси, (С1-С6)алкоксикарбонила, H2NCO, H2NSO2, (С1-С6)алкиламинокарбонила, ди(С1-С6)алкиламинокарбонила, (С1-С3)алкокси(С1-С3)алкиламинокарбонила, гетероциклилкарбонила, (С1-С6)алкиламиносульфонила, ди(С1-С6)алкиламиносульфонила, гетероциклилсульфонила, (С1-С6)алкилкарбониламино, (С2-С6)алкилкарбониламино(С1-С6)алкила, (С1-С6)алкилсульфониламино, (С1-С6)алкил-сульфониламино(С1-С6)алкила, (С1-С6)алкоксикарбонил(С1-С6)алкокси, (С1-С6)алкокси(С1-С6)алкила, гало(С1-С6)алкокси(С1-С6)алкила, гидрокси(С1-С6)алкокси, гетероарил, оксо, амино(С1-С6)алкила, (С1-С6)алкиламино(С1-С6)алкила, ди(С1-С6)алкиламино(С1-С6)алкила амино(С1-С6)алкокси, (С1-С6)алкиламино(С1-С6)алкокси, ди(С1-С6)алкиламино(С2-С6)алкокси, (С1-С6)алкилкарбонила, (С3-С6)циклоалкилкарбонила, (С3-С6)циклоалкиламинокарбонила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонила, ди(С3-С6)циклоалкиламинокарбонила, (С3-С6)циклоалкиламиносульфонила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминосульфонила, ди(С3-С6)циклоалкиламиносульфонила, циано(С1-С6)алкила, аминокарбонил(С1-С6)алкила, (С1-С6)алкиламинокарбонил(С1-С6)алкила, ди(С1-С6)алкиламинокарбонил(С1-С6)алкила, (С3-С6)циклоалкиламинокарбонил(С1-С6)алкила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонил(С1-С6)алкила и ди(С3-С6)циклоалкиламинокарбонил(С1-С6)алкила.
Возможные заместители оксодигидропиридилового кольца Формулы Im1 включают от 1 до 4 групп, независимо выбранных из фтора, хлора, брома, йода, циано, нитро, амино, гидрокси, карбокси, (С1-С6)алкила, гидрокси(С1-С6)алкила, (С3-С6)циклоалкила, гидрокси(С3-С6)циклоалкила, (С4-С7)циклоалкилалкила, (С1-С6)алкенила, гало(С1-С6)алкенила, гидрокси(С1-С6)алкенила, (С2-С6)алкинила, (С3-С6)циклоалкил(С2-С4)алкинила, гало(С1-С6)алкила, гало(С3-С6)циклоалкила, гало(С4-С7)циклоалкилалкила, (С1-С6)алкокси, (С3-С6)циклоалкокси, (С4-С7)циклоалкилалкокси, гало(С1-С6)алкокси, гало(С3-С6)циклоалкокси, гало(С4-С7)циклоалкилалкокси, (С1-С6)алкилтио, (С3-С6)циклоалкилтио, (С4-С7)циклоалкилалкилтио, гало(С1-С6)алкилтио, гало(С3-С6)циклоалкилтио, гало(С4-С7)Циклоалкилалкилтио, (С1-С6)алкансульфинила, (С3-С6)циклоалкансульфинила, (С4-С7)циклоалкилалкансульфинила, гало(С1-С6)алкансульфинила, гало(С3-С6)циклоалкансульфинила, гало(С4-С7)циклоалкилалкансульфинила, (С1-С6)алкансульфонила, (С3-С6)циклоалкансульфонила, (С4-С7)циклоалкилалкансульфонила, гало(С1-С6)алкансульфонила, гало(С3-С6)циклоалкансульфонила, гало(С4-С7)цикло-алкилалкансульфонила, (С1-С6)алкиламино, ди(С1-С6)алкиламино, (С1-С6)алкокси(С1-С6)алкокси, гало(С1-С6)алкокси(С1-С6)алкокси, (С1-С6)алкоксикарбонила, H2NCO, H2NSO2, (С1-C6)алкиламинокарбонила, ди(С1-С6)алкиламинокарбонила, (С1-С3)алкокси(С1-С3)алкиламинокарбонила, гетероциклилкарбонила, (С1-С6)алкиламиносульфонила, ди(С1-С6)алкиламиносульфонила, гетероциклилсульфонила, (С1-С6)алкилкарбониламино, (С1-С6)алкилкарбониламино(С1-С6)алкила, (С1-С6)алкилсульфониламино, (С1-С6)алкилсульфониламино(С1-С6)алкила, (С1-С6)алкоксикарбонил(С1-С6)алкокси, (C1-C6)алкокси(C1-С6)алкила, гало(С1-С6)алкокси(С1-С6)алкила, гидрокси(С1-С6)алкокси, гетероарил, оксо, амино(С1-С6)алкила, (С1-С6)алкиламино(С1-С6)алкила, ди(С1-С6)алкиламино(С1-С6)алкила амино(С2-С6)алкокси, (С1-С6)алкиламино(С2-С6)алкокси, ди(С1-С6)алкиламино(С1-С6)алкокси, (С1-С6)алкилкарбонила, (C3-С6)циклоалкилкарбонила, (С3-С6)циклоалкиламинокарбонила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонила, ди(С3-С6)циклоалкиламинокарбонила, (С3-С6)циклоалкиламиносульфонила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминосульфонила, ди(С3-С6)циклоалкиламиносульфонила, циано(С1-С6)алкила, аминокарбонил(С1-С6)алкила, (С1-С6)алкиламинокарбонил(С1-С6)алкила, ди(С1-С6)алкиламинокарбонил(С1-С6)алкила, (С3-С6)циклоалкиламинокарбонил(С1-С6)алкила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонил(С1-С6)алкила и ди(С3-С6)циклоалкиламинокарбонил(С1-С6)алкила;
Е представляет собой (а) связь или (b) (С1-С3)алкилен или (С1-С2)алкиленилокси, где О присоединен к R2, возможные заместители каждого из которых включают от 1 до 4 групп, независимо выбранных из метила, этила, трифторметила или оксо;
R2 представляет собой (С1-С6)алкил, арил, гетероарил, циклоалкил или гетероциклил, возможные заместители которого включают от 1 до 4 групп, независимо выбранных из фтора, хлора, брома, йода, циано, нитро, амино, гидрокси, карбокси, (С1-С6)алкила, гидрокси(С1-С6)алкила, (С3-С6)циклоалкила, гидрокси(С3-С6)циклоалкила, (C4-С7)циклоалкилалкила, (С2-С6)алкенила, гало(С2-С6)алкенила, гидрокси(С1-С6)алкенила, (С1-С6)алкинила, (С3-С6)циклоалкил(С2-С4)алкинила, гало(С1-С6)алкила, гало(С3-С6)циклоалкила, гало(С4-С7)циклоалкилалкила, (С1-С6)алкокси, (С3-С6)циклоалкокси, (C4-С7)циклоалкилалкокси, гало(С1-С6)алкокси, гало(С3-С6)циклоалкокси, гало(С4-С7)циклоалкилалкокси, (С1-С6)алкилтио, (С3-С6)циклоалкилтио, (C4-С7)циклоалкилалкилтио, гало(С1-С6)алкилтио, гало(С3-С6)циклоалкилтио, гало(С4-С7)циклоалкилалкилтио, (С1-С6)алкансульфинила, (С3-С6)циклоалкансульфинила, (С4-С7)циклоалкилалкансульфинила, гало(С1-С6)алкансульфинила, гало(С3-С6)циклоалкансульфинила, гало(С4-С7)циклоалкилалкансульфинила, (C1-С6)алкансульфонила, (С3-С6)циклоалкансульфонила, (С4-С7)циклоалкилалкансульфонила, гало(С1-С6)алкансульфонила, гало(С3-С6)циклоалкансульфонила, гало(С4-С7)цикло-алкилалкансульфонила, (С1-С6)алкиламино, ди(С1-С6)алкиламино, (С1-С6)алкокси(С1-С6)алкокси, гало(С1-С6)алкокси(С1-С6)алкокси, (С1-С6)алкоксикарбонила, H2NCO, H2NSO2, (С1-С6)алкиламинокарбонила, ди(С1-С6)алкиламинокарбонила, (С1-С3)алкокси(С1-С3)алкиламинокарбонила, гетероциклилкарбонила, (С1-С6)алкиламиносульфонила, ди(С1-С6)алкиламиносульфонила, гетероциклилсульфонила, (С1-С6)алкилкарбониламино, (C1-С6)алкилкарбониламино(С1-С6)алкила, (С1-С6)алкилсульфониламино, (С1-С6)алкилсульфониламино(С1-С6)алкила, (С1-С6)алкоксикарбонил(С1-С6)алкокси, (C1-С6)алкокси(С1-С6)алкила, гало(С1-С6)алкокси(С1-С6)алкила, гидрокси(С1-С6)алкокси, гетероарил, оксо, амино(С1-С6)алкила, (С1-С6)алкиламино(С1-С6)алкила, ди(С1-С6)алкиламино(С1-С6)алкила амино(С2-С6)алкокси, (С1-С6)алкиламино(С2-С6)алкокси, ди(С1-С6)алкиламино(С2-С6)алкокси, (С1-С6)алкилкарбонила, (С3-С6)циклоалкилкарбонила, (С3-С6)циклоалкиламинокарбонила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонила, ди(C3-С6)циклоалкиламинокарбонила, (С3-С6)циклоалкиламиносульфонила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминосульфонила, ди(С3-С6)циклоалкиламиносульфонила, циано(С1-С6)алкила, аминокарбонил(С1-С6)алкила, (С1-С6)алкиламинокарбонил(С1-С6)алкила, ди(С1-С6)алкиламинокарбонил(С1-С6)алкила, (С3-С6)циклоалкиламинокарбонил(С1-С6)алкила, {(С3-С6)циклоалкил}{(С1-C6)алкил}аминокарбонил(С1-С6)алкила и ди(С3-С6)циклоалкиламинокарбонил(С1-С6)алкила;
R3 выбран из (С1-С6)алкила, (С2-С6)алкенила, (С2-С6)алкинила, (С3-С5)циклоалкил(С1-С4)алкила, (С1-С3)алкокси(С1-С3)алкокси или (С1-С3)алкокси(С1-С3)алкила, возможные заместители включают до четырех групп, независимо выбранных из фтора, циано, оксо, R4, R4O-, (R4)2N-, R4O2C-, R4C(=O)O-, R4S, R4S(=O)-, R4S(=O)2-, R4C(=O)NR4-, (R4)2NC(=O)-, (R4)2NC(=O)O-, (R4)2NC(=O)NR4-, R4OC(=O)NR4-, (R4)2NC(=NCN)NR4-, (R4O)2P(=O)O-, (R4O)2P(=O)NR4-, R4OS(=O)2NR4-, (R4)2NS(=O)2O-, (R4)2NS(=O)2NR4-, R4S(=O)2NR4-, R4S(=O)2NHC(=O)-, R4S(=O)2NHC(=O)O-, R4S(=O)2NHC(=O)NR4-, R4OS(=O)2NHC(=O)-, R4OS(=O)2NHC(=O)O-, R4OS(=O)2NHC(=O)NR4-, (R4)2NS(=O)2NHC(=O)-, (R4)2NS(=O)2NHC(=O)O-, (R4)2NS(=O)2NHC(=O)NR4-, R4C(=O)NHS(=O)2-, R4C(=O)NHS(=O)2O-, R4C(=O)NHS(=O)2NR4-, R4OC(=O)NHS(=O)2-, R4OC(=O)NHS(=O)2O-, R4OC(=O)NHS(=O)2NR4-, (R4)2NC(=O)NHS(=O)2-, (R4)NC(=O)NHS(=O)2O-, (R4)2NC(=O)NHS(=O)2NR4-, спироциклоалкила; гетероциклила (возможные заместители которого, в свою очередь, включают алкил, галоалкил, галоген или оксо), гетероарила (возможные заместители которого, в свою очередь, включают алкил, галоалкил, алкокси, алкилтио, алкилсульфонил, галоген, трифторметил, диалкиламино, нитро, циано, CO2H, CONH2, N-моноалкил-замещенный амидо, N,N-диалкил - замещенный амидо или оксо), ариламино (возможные заместители которого, в свою очередь, включают алкил, алкокси, алкилтио, алкилсульфонил, галоген, трифторметил, диалкиламино, нитро, циано, CO2H, CONH2, N-моноалкил - замещенный амидо и N,N-диалкил - замещенный амидо) и гетероариламино (возможные заместители которого, в свою очередь, включают алкил, галоалкил, алкокси, алкилтио, алкилсульфонил, галоген, трифторметил, диалкиламино, нитро, циано, СО2Н, CONH2, N-моноалкил-замещенный амидо, N,N-диалкил- замещенный амидо или оксо); а также
R4 независимо выбран из Н, (С1-С6)алкила, гало(С1-С6)алкила амино(С1-С6)алкила, (С1-С6)алкиламино(С1-С6)алкила, ди(С1-С6)алкиламино(С1-С6)алкила, гидрокси(С1-С6)алкила и (С1-С6)алкокси(С1-С6)алкила.
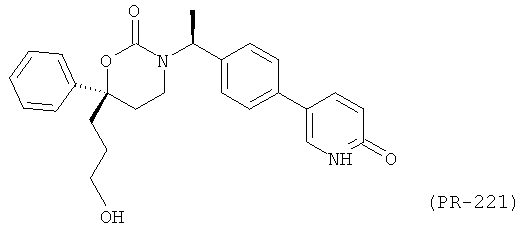
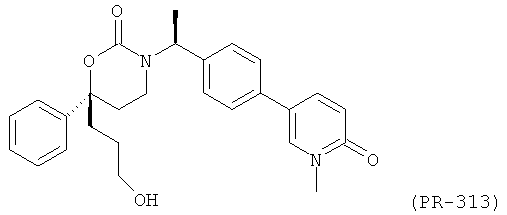
В качестве альтернативы первый вариант осуществления настоящего изобретения, описанный выше, не включает соединения со структурными Формулами PR-221 и PR-313 или их фармацевтически приемлемые соль, энантиомер или диастереомер.
Следующий вариант осуществления настоящего изобретения представляет собой фармацевтическую композицию, включающую i) фармацевтически приемлемый носитель или растворитель, а также ii) соединение с Формулами Ik, 1m1, Im2, Im5, In1, In2, In5, Io1, Io2, Io5, Ip1, Ip3 или его фармацевтически приемлемые соль, энантиомер или диастереомер.
Следующий вариант осуществления настоящего изобретения представляет собой способ ингибирования активности 11β-ГСД1, включающий этап введения млекопитающим, нуждающимся в подобном лечении, эффективного количества соединения с Формулами Ik, Im1, Im2, Im5, In1, In2, In5, Io1, Io2, Io5, Ip1, Ip3 или его фармацевтически приемлемых соли, энантиомера или диастереомера.
Следующий вариант осуществления настоящего изобретения представляет собой способ лечения пациента, страдающего от заболевания, связанного с активностью или экспрессией 11β-ГСД1, включающий этап приема пациентом эффективного количества соединения с Формулами Ik, Im1, Im2, Im5, 1п1, In2, In5, Io1, Io2, Io5, Ip1, Ip3 или его фармацевтически приемлемых соли, энантиомера или диастереомера
Следующий вариант осуществления настоящего изобретения представляет собой способ применения соединения с Формулами Ik, Im1, Im2, Im5, In1, In2, In5, Io1, Io2, Io5, Ip1, Ip3 или его фармацевтически приемлемых соли, энантиомера или диастереомера при производстве медикаментов, предназначенных для ингибирования активности 11β-ГСД1 у млекопитающих, нуждающихся в подобном лечении,
Следующий вариант осуществления настоящего изобретения представляет собой способ применения соединения с Формулами Ik, Im1, Im2, Im5, In1, In2, In5, Io1, Io2, Io5, Ip1, Ip3 или его фармацевтически приемлемых соли, энантиомера или диастереомера при производстве лекарственных препаратов, предназначенных для лечения пациентов, страдающих от заболевания, связанного с активностью или экспрессией 11β-ГСД1.
Следующий вариант осуществления настоящего изобретения представляет собой соединение с Формулами Ik, Im1, Im2, lm5, In1, In2, In5, Io1, Io2, Io5, Ip1, Ip3 или его фармацевтически приемлемые соль, энантиомер или диастереомер, предназначенные для ингибирования активности 11Р-ГСД1 у млекопитающих, нуждающихся в подобном лечении,
Следующий вариант осуществления настоящего изобретения представляет собой соединение с Формулами Ik, lm1, lm2, lm5, In1, In2, In5, Io1, Io2, Io5, Ip1, Ip3 или его фармацевтически приемлемые соль, энантиомер или диастереомер, предназначенные для лечения пациентов, страдающих от заболевания, связанного с активностью или экспрессией 11β-ГСД1.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретения представляет собой соединение Формулы Ik:
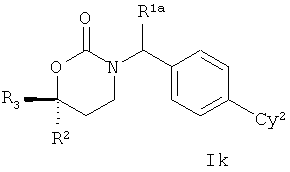 ;
;
или его фармацевтически приемлемые соль, энантиомер или диастереомер; где
R1a отсутствует или представляет собой метил или этил;
Су2 представляет собой 2-оксо-1,2-дигидропиридил, возможные заместители которого включают от 1 до 4 групп, независимо выбранных из гало, гидрокси, метокси, гидроксиметила, метоксикарбонила, амино, карбамоила, метилкарбамоила, диметилкарбамоила, (2-метоксиэтил)аминокарбонила, ацетиламинометила, метилсульфонила, метилсульфониламино, метиламиносульфонила, изопропиламиносульфонила, диметиламиносульфонила, пирролидин-1-сульфонила, метилсульфониламинометила, тетразолила, метила, трифторметила, ацетила, 2-гидроксиэтила и 1-аминоэтила;
R2 представляет собой фенил, тиенил, пиридил или изопропил, возможные заместители каждого из которых включают гало, метил, метилтио или (4-морфолино)метил; а также
R3 представляет собой метил, этил, пропил, бутил, винил, аллил или этоксиэтил, возможные заместители каждого из которых включают до двух групп, независимо выбранных из метила, НО-, MеО-, H2N-, MeC(=O)NH-, MeS(=O)2NH-, H2NC(=O)-, MeNHC(=O)-, HO2C-, (HO)2P(=O)O-, H2NS(=O)2O-, H2NS(=O)2NH-, MeNHC(=O)NH-, MeNHC(=O)O-, оксо, циано, НO2С-, HOCH2CH2NH-, 4-морфолино, HOCH2C(=O)NH-, H2NCH2C(=O)NH-, EtNHC(=O)NH, MeOC(=O)NH-, MeNHC(=NC=N)NH-, Me-, MeS-, MeSO2-MeSO2N(Me)-, MeS(=O)2NHC(=O)-, имидазолиламино-, имидазолила, тетразолила, H2NCONH-, H2NCO2-, НОСН2СН2O-, MeNH-, Me2N- и MeCONMe.
Следующий вариант осуществления настоящего изобретения представляет собой соединение с любой из Формул Im1, Im2 и Im5 или его фармацевтически приемлемые соль, энантиомер или диастереомер;:
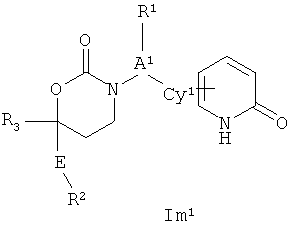
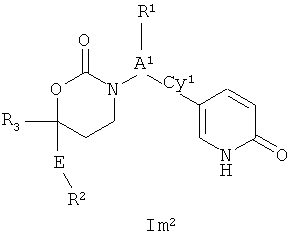
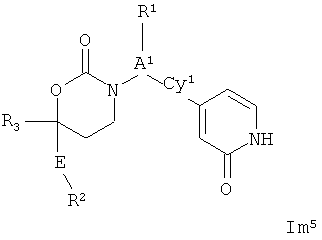 .
.
Возможные заместители оксодигидропиридилового кольца (включая также замещение атомов углерода в кольце, присоединенных к атомам водорода, и атомов азота в кольце, присоединенных к атомам водорода, т.е. включая "замещаемый атом азота в кольце") в Формулах Im1, Im2 и Im5 включают до четырех заместителей, описанных выше. Подходящие заместители оксодигидропиридилового кольца, а также подходящие значения R1, R2, R3, А1, Су1 и Е представлены выше в первом варианте осуществления настоящего изобретения. В качестве альтернативы подходящие заместители Су1 и оксодигидропиридилового кольца в Формулах Im1, Im2 и Im5 могут быть независимо выбраны из фтора, хлора, брома, йода, циано, нитро, амино, гидрокси, карбокси, (С1-С6)алкила, гидрокси(С1-С6)алкила, (С3-С6)циклоалкила, гидрокси(С3-С6)циклоалкила, (C4-С7)циклоалкилалкила, (С1-С6)алкенила, гало(С2-С6)алкенила, гидрокси(С2-С6)алкенила, (С2-С6)алкинила, (С3-С6)циклоалкил(С2-С4)алкинила, гало(С1-С6)алкила, гало(С3-С6)циклоалкила, гало(С4-С7)циклоалкилалкила, (С1-С6)алкокси, (С3-С6)циклоалкокси, (C4-С7)циклоалкилалкокси, гало(С1-С6)алкокси, гало(С3-С6)циклоалкокси, гало(С4-С7)циклоалкилалкокси, (С1-С6)алкилтио, (С3-С6)циклоалкилтио, (C4-С7)циклоалкилалкилтио, гало(С1-С6)алкилтио, гало(С3-С6)циклоалкилтио, гало(С4-С7)циклоалкилалкилтио, (С1-С6)алкансульфинила, (С3-С6)циклоалкансульфинила, (С4-С7)циклоалкилалкансульфинила, гало(С1-С6)алкансульфинила, гало(С3-С6)циклоалкансульфинила, гало(С4-С7)циклоалкилалкансульфинила, (С1-С6)алкансульфонила, (С3-С6)циклоалкансульфонила, (С4-С7)циклоалкилалкансульфонила, гало(С1-С6)алкансульфонила, гало(С3-С6)циклоалкансульфонила, гало(С4-С7)цикло-алкилалкансульфонила, (С1-С6)алкиламино, ди(С1-С6)алкиламино, (С1-С6)алкокси(С1-С6)алкокси, гало(С1-С6)алкокси(С1-С6)алкокси, (С1-С6)алкоксикарбонила, H2NCO, HaNSO2, (С1-С6)алкиламинокарбонила, ди(С1-С6)алкиламинокарбонила, (С1-С3)алкокси(С1-С3)алкиламинокарбонила, гетероциклилкарбонила, (С1-С6)алкиламиносульфонила, ди(С1-C6)алкиламиносульфонила, гетероциклилсульфонила, (С1-С6)алкилкарбониламино, (С1-С6)алкилкарбониламино(С1-С6)алкила, (С1-С6)алкилсульфониламино, (С1-С6)алкилсульфониламино(С1-С6)алкила, (С1-С6)алкоксикарбонил(С1-С6)алкокси, (С1-C6)алкокси(С1-С6)алкила, гало(С1-С6)алкокси(С1-С6)алкила, гидрокси(С1-С6)алкокси, гетероарил, амино(С1-С6)алкила, (С1-С6)алкиламино(С1-С6)алкила, ди(С2-С6)алкиламино(С1-С6)алкила амино(С1-С6)алкокси, (С1-С6)алкиламино(С2-С6)алкокси, ди(С1-С6)алкиламино(С2-С6)алкокси, (С1-С6)алкилкарбонила, (С3-С6)циклоалкилкарбонила, (С3-С6)циклоалкиламинокарбонила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонила, ди(С3-С6)циклоалкиламинокарбонила, (С3-С6)циклоалкиламиносульфонила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминосульфонила, ди(С3-С6)циклоалкиламиносульфонила, циано(С1-С6)алкила, аминокарбонил(С1-С6)алкила, (С1-С6)алкиламинокарбонил(С1-С6)алкила, ди(С1-С6)алкиламинокарбонил(С1-С6)алкила, (С3-С6)циклоалкиламинокарбонил(С1-С6)алкила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонил(С1-С6)алкила и ди(С3-С6)циклоалкиламинокарбонил(С1-С6)алкила; подходяицие значения R1, R2, R3, А1, Су1 и Е представлены выше в первом варианте осуществления настоящего изобретения. В качестве альтернативы подходящие заместители Су1 могут включать (C1-C4)алкил, (C1-C4)алкокси, (C1-C4)галоалкил, (С1-С4)галоалкокси, галоген, циано и нитро; подходящие заместители для замещаемого атома азота в оксодигидропиридиловом кольце в Формулах Im1, Im2 и Im5 включают (C1-С4)алкил, (С3-С4)циклоалкил, (С3-С4) циклоалкил(С1-С2)алкил и (С1-С4)галоалкил; подходящие заместители для замещаемого атома углерода в оксодигидропиридиловом кольце в Формулах Im1, Im2 и Im5 включают фтор, хлор, циано, гидрокси, амино, (С1-С4)алкил, (С3-С4)циклоалкил, (С3-С4)циклоалкил(С1-С2)алкил, гало(С1-С4)алкил, (C1-С4)алкокси, (С1-С4)галоалкокси, CONH2, (С1-С4)алкиламинокарбонил, ди(С1-С4)алкиламинокарбонил и (С1-С4)алкилкарбониламино; подходящие значения R1, R2 R3 А, Су1 и Е представлены выше в первом варианте осуществления настоящего изобретения.
В качестве альтернативы варианты, представленные в данном разделе, не включают следующие соединения:
(R)-6-(3-гидроксипропил)-3-((3)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он.
 ; и
; и
(R)-6-(3-гидроксипропил)-3-((8)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
 ;
;
или его фармацевтически приемлемые соль, энантиомер или диастереомер.
Для каждого из вариантов, описанных в предыдущем разделе, R1, предпочтительно, представляет собой метил или этил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Im1, Im2 и Im5, R1, предпочтительно, представляет собой метил или этил; а R3 представляет собой MeSO2NHCH2CH2CH2, Н2NC(=O)CH2CH2, Н2NC(=O)СМе2СН2, 3-гидроксипропил, 3-гидрокси-3-метилбутил, 2-гидроксиэтил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Im1, Im2 и Im5, R1, предпочтительно, представляет собой метил или этил; а R3 представляет собой H2NC(=O)CMe2CH2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Im1, Im2 и Im5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил, замещаемый 1, 2 или 3 возможными заместителями, выбранными из гало, циано, CONH2, (С1-C4)алкила, (С1-C4)галоалкила, а также SO2Me; a R3 представляет собой MeSO2NHCH2CH2CH2, H2NC(=O)CH2CH2, H2NC(=O)CMe2CH2, 3-гидроксипропил, 3-гидрокси-3-метилбутил, 2-гидроксиэтил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Im1, Im2 и Im5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил, замещаемый 1, 2 или 3 возможными заместителями, выбранными из гало, циано, CONH2, (С1-С4)алкила, (С1-С4)галоалкила, а также SO2Me; a R3 представляет собой H2NC(=O)CMe2CH2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Im1, Im2 и Im5, R1, предпочтительно, представляет собой метил или этил; а R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Im1, Im2 and Im5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил или фторфенил; а R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Im1, Im2 и Im5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил или фторфенил; а R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; заместители замещаемого атома азота в оксодигидропиридиловом кольце в Формулах Im1, Im2 и Im5 включают (C1-C4)алкил, (С3-С4)циклоалкил, (С3-С4)циклоалкил(С1-С2)алкил или (C1-C2)галоалкил; а возможные заместители одного или двух атомов углерода в оксодигидропиридиловом кольце в Формулах Im1, Im2 и Im5 включают метил или этил.
Следующий вариант осуществления настоящего изобретения представляет собой соединение с любой из Формул In1, In2 и In5 или его фармацевтически приемлемые соль, энантиомер или диастереомер:
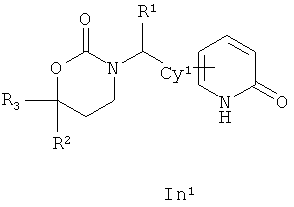
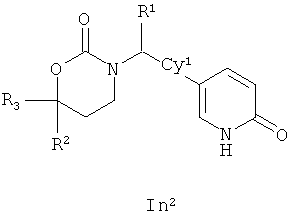
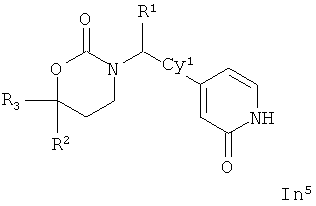 .
.
Возможные заместители оксодигидропиридилового кольца (включая также замещение атомов углерода в кольце, присоединенных к атомам водорода, и атомов азота в кольце, присоединенных к атомам водорода, т.е. включая "замещаемый атом азота в кольце") в Формулах In1, In2 and In5 включают до четырех заместителей, описанных выше для Су2. Подходящие заместители оксодигидропиридилового кольца, а также подходящие значения R1, R2, R3 и Су1 представлены выше в первом варианте осуществления настоящего изобретения. В качестве альтернативы подходящие заместители Су1 и оксодигидропиридилового кольца в Формулах In1, In2 and In5 могут быть независимо выбраны из фтора, хлора, брома, йода, циано, нитро, амино, гидрокси, карбокси, (С1-С6)алкила, гидрокси(С1-С6)алкила, (С3-С6)циклоалкила, гидрокси(С3-С6)циклоалкила, (С4-С7)циклоалкилалкила, (С2-С6)алкенила, гало(С2-С6)алкенила, гидрокси(С2-С6)алкенила, (С2-С6)алкинила, (С3-С6)циклоалкил(С2-С4)алкинила, rano(C1-С6)алкила, гало(С3-С6)циклоалкила, гало(С4-С7)циклоалкилалкила, (С1-С6)алкокси, (С3-С6)циклоалкокси, (С4-С7)циклоалкилалкокси, гало(С1-С6)алкокси, гало(С3-С6)циклоалкокси, гало(С4-С7)циклоалкилалкокси, (С1-С6)алкилтио, (С3-С6)циклоалкитио, (С4-С7)циклоалкилалкилтио, гало(С1-С6)алкилтио, гало(С3-С6)циклоалкитио, гало(С4-С7)циклоалкилалкилтио, (С1-С6)алкансульфинила, (С3-С6)циклоалкансульфинила, (C4-С7)циклоалкилалкансульфинила, гало(С1-С6)алкансульфинила, гало(С3-С6)циклоалкансульфинила, гало(С4-С7)циклоалкилалкансульфинила, (C1-С6)алкансульфонила, (С3-С6)циклоалкансульфонила, (С4-С7)циклоалкилалкансульфонила, гало(С1-С6)алкансульфонила, гало(С3-С6)циклоалкансульфонила, гало(С4-С7)циклоалкилалкансульфонила, (С1-С6)алкиламино, ди(С1-С6)алкиламино, (С1-С6)алкокси(С1-С6)алкокси, гало(С1-С6)алкокси(С1-С6)алкокси, (С1-С6)алкоксикарбонила, H2NCO, H2NSO2, (С1-С6)алкиламинокарбонила, ди(С1-С6)алкиламинокарбонила, (С1-С3)алкокси(С1-С3)алкиламинокарбонила, гетероциклилкарбонила, (С1-С6)алкиламиносульфонила, ди(C1-С6)алкиламиносульфонила, гетероциклилсульфонила, (С1-С6)алкилкарбониламино, (С1-С6)алкилкарбониламино(С1-С6)алкила, (С1-С6)алкилсульфониламино, (C1-С6)алкилсульфониламино(С1-С6)алкила, (С1-С6)алкоксикарбонил(С1-С6)алкокси, (C1-С6)алкокси(С1-С6)алкила, гало(С1-С6)алкокси(С1-С6)алкила, гидрокси(С1-С6)алкокси, гетероарил, амино(С1-С6)алкила, (С1-С6)алкиламино(С1-С6)алкила, ди(С1-С6)алкиламино(С1-С6)алкила амино(С2-С6)алкокси, (С1-С6)алкиламино(С2-С6)алкокси, ди(С1-С6)алкиламино(С2-С6)алкокси, (С1-С6)алкилкарбонила, (С3-С6)циклоалкилкарбонила, (С3-С6)циклоалкиламинокарбонила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонила, ди(С3-С6)циклоалкиламинокарбонила, (С3-С6)циклоалкиламиносульфонила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминосульфонила, ди(С3-С6)циклоалкиламиносульфонила, циано(С1-С6)алкила, аминокарбонил(С1-С6)алкила, (С1-С6)алкиламинокарбонил(С1-С6)алкила, ди(С1-С6)алкиламинокарбонил(С1-С6)алкила, (С3-С6)циклоалкиламинокарбонил(С1-С6)алкила, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонил(С1-С6)алкила и ди(С3-С6)циклоалкиламинокарбонил(С1-С6)алкила; подходящие значения R1, R2, R3 и Су1 представлены выше в первом варианте осуществления настоящего изобретения. В качестве альтернативы подходящие заместители Су1 могут включать (С1-С4)алкил, (С1-C4) алкокси, (C1-C4)галоалкил, (C1-C4)галоалкокси, галоген, циано и нитро; подходящие заместители для замещаемого атома азота в оксодигидропиридиловом кольце в Формулах In1, In2 and In5 включают (C1-С4)алкил, (С3-С4)циклоалкил, (С3-С4) циклоалкил (С1-С4)алкил и (С1-С4)галоалкил; подходящие заместители для замещаемого атома углерода в оксодигидропиридиловом кольце в Формулах In1, In2 и In5 включают фтор, хлор, циано, гидрокси, амино, (С1-С4)алкил, (С3-С4)циклоалкил, (С3-С4)циклоалкил(С1-С6)алкил, гало(С1-С4)алкил, (C1-С4)алкокси, (С1-С4)галоалкокси, CONH2, (С1-С4)алкиламинокарбонил, ди(С1-С4) алкиламинокарбонил и (С1-С4)алкилкарбониламино; подходящие значения R1, R2, R3 и Су1 представлены выше в первом варианте осуществления настоящего изобретения. В качестве альтернативы варианты, представленные в данном разделе, не включают соединения PR-221 и PR-313 или их фармацевтически приемлемые соль, энантиомер или диастереомер
Для каждого из вариантов, описанных в предыдущем разделе, R1, предпочтительно, представляет собой метил или этил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул In1, In2 и In5, R1, предпочтительно, представляет собой метил или этил; а R3 представляет собой MeSO2NHCH2CH2CH2, H2NC(=O)CH2CH2, Н2NC(=O)СМе2СН2, 3-гидроксипропил, 3-гидрокси-3-метилбутил, 2-гидроксиэтил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул In1, In2 и In5, R1, предпочтительно, представляет собой метил или этил; а R3 представляет собой Н2NC(=O)СМе2СН2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул In1, In2 и In5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил, замещаемый 1, 2 или 3 возможными заместителями, выбранными из гало, циано, CONH2, (С1-С4)алкила, (С1-C4)галоалкила, а также SO2Me; a R3 представляет собой MeSO1NHCH2CH2CH2, Н2NC(=O)СН2СН2, H2NC(=O)CMe2CH2, 3-гидроксипропил, 3-гидрокси-3-метилбутил, 2-гидроксиэтил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул In1, In2 и In5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил, замещаемый 1, 2 или 3 возможными заместителями, выбранными из гало, циано, CONH2, (С1-С4)алкила, (С1-С4)галоалкила, а также SO2Me; a R3 представляет собой H2NC(=O)CMe2CH2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил..
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул In1, In2 и In6, R1, предпочтительно, представляет собой метил или этил; а R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул In1, In2 и In5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил или фторфенил; а R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул In1 In2 и In5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил или фторфенил; а R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; заместители замещаемого атома азота в оксодигидропиридиловом кольце в Формулах In1, In2 и In5 включают (С1-C4)алкил, (С3-С4)циклоалкил, (С3-С4)циклоалкил(С1-С2)алкил или (С1-С2)галоалкил; а возможные заместители одного или двух атомов углерода в оксодигидропиридиловом кольце в Формулах In1, In2 и In5 включают метил или этил.
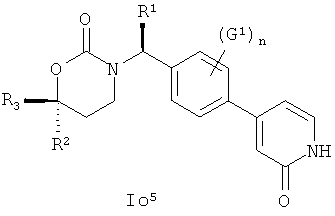
Следующий вариант осуществления настоящего изобретения представляет собой соединение с любой из Формул Io1, Io2 and Io5 или его фармацевтически приемлемую соль:
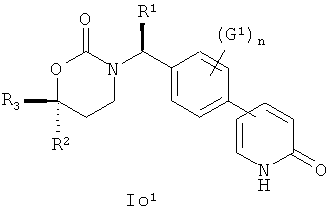
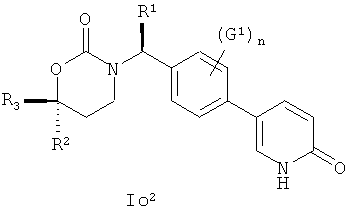
 .
.
Возможные заместители оксодигидропиридилового кольца (включая также замещение атомов углерода в кольце, присоединенных к атомам водорода, и атомов азота в кольце, присоединенных к атомам водорода, т.е. включая "замещаемый атом азота в кольце") в Формулах Io1, Io2 и Io5 включают до четырех заместителей, описанных выше для первого варианта осуществления настоящего изобретения; подходящие значения для G1 включают фтор, хлор, бром, йод, циано, нитро, амино, гидрокси, карбокси, (С1-С6)алкил, гидрокси(С1-С6)алкил, (С3-С6)циклоалкил, гидрокси(С3-С6)циклоалкил, (С4-С7)циклоалкилалкил, (С2-С6)алкенил, гало(С2-С6)алкенил, гидрокси(С2-С6)алкенил, (С2-С6)алкинил, (С3-С6)циклоалкил(С2-С4)алкинил, гало(С1-С6)алкил, гало(С3-С6)циклоалкил, гало(С4-С7)циклоалкилалкил, (С1-С6)алкокси, (С3-С6)циклоалкокси, [С4-С7)циклоалкилалкокси, гало(С1-С6)алкокси, гало(С3-С6)цикпоалкокси, гало(С4-С7)циклоалкилалкокси, (С1-С6)алкилтио, (С3-С6)циклоалкилтио, (C4-С7)циклоалкилалкилтио, гало(С1-С6)алкилтио, гало(С3-С6)циклоалкитио, гало(С4-С7)циклоалкилалкилтио, (С1-С6)алкансульфинил, (С3-С6)циклоалкансульфинил, (С4-С7)циклоалкилалкансульфинил, гало(С1-С6)алкансульфинил, гало(С3-С6)циклоалкансульфинил, гало(С4-С7)циклоалкилалкансульфинил, (С1-С6)алкансульфонил, (С3-С6)циклоалкансульфонил, (С4-С7)цикпоалкилалкансульфонил, гало(С1-С6)алкансульфонил, гало(С3-С6)циклоалкансульфонил, гало(С4-С7)циклоалкилалкансульфонил, (С1-С6)алкиламино, ди(С1-С6)алкиламино, (C1-C6)алкокси(C1-С6)алкокси, гало(С1-С6)алкокси(С1-С6)алкокси, (С1-С6)алкоксикарбонил, H2NCO, H2NSO2, (С1-С6)алкиламинокарбонил, ди(С1-С6)алкиламинокарбонил, (С1-С3)алкокси(С1-С3)алкиламинокарбонил, гетероциклилкарбонил, (С1-С6)алкиламиносульфонил, ди(С1-С6)алкиламиносульфонил, гетероциклилсульфонил, (С1-С6)алкилкарбониламино, (С1-С6)алкилкарбониламино(С1-С6)алкил, (С1-С6)алкилсульфониламино, (C1-С6)алкилсульфониламино(С1-С6)алкил, (С1-С6)алкоксикарбонил(С1-С6)алкокси, (C1-С6)алкокси(С1-С6)алкил, гало(С1-С6)алкокси(С1-С6)алкил, гидрокси(С1-С6)алкокси, гетероарил, амино(С1-С6)алкила, (С1-С6)алкиламино(С1-С6)алкил, ди(C1-С6)алкиламино(С1-С6)алкил, амино(С2-С6)алкокси, (С1-С6)алкиламино(С2-С6)алкокси, ди(С1-С6)алкиламино(С2-С6)алкокси, (С1-С6)алкилкарбонил, (С3-С6)циклоалкилкарбонил, (С3-С6)циклоалкиламинокарбонил, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонил, ди(С3-С6)циклоалкиламинокарбонил, (С3-С6)циклоалкиламиносульфонил, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминосульфонил, ди(С3-С6)циклоалкиламиносульфонил, циано(С1-С6)алкила, аминокарбонил(С1-С6)алкил, (С1-С6)алкиламинокарбонил(С1-С6)алкил, ди(С1-С6)алкиламинокарбонил(С1-С6)алкил, (С3-С6)циклоалкиламинокарбонил(С1-С6)алкил, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонил(С1-С6)алкил и ди(С3-С6)циклоалкиламинокарбонил(С1-С6)алкил; n имеет значения 0, 1, 2 или 3, а подходящие заместители для оксодигидропиридилового кольца и подходящие значения для R1, R2 и R3 представлены в описании первого варианта осуществления настоящего изобретения. В качестве альтернативы n имеет значения 0, 1, 2 или 3; подходящие значения для G1 и заместители для оксодигидропиридилового кольца в Формулах Io1, Io2 и Io5 были независимо выбраны из фтора, хлора, брома, йода, циано, нитро, амино, гидрокси, карбокси, (С1-С6)алкила, гидрокси(С1-С6)алкила, (С3-С6)циклоалкила, гидрокси(С3-С6)циклоалкила, (С4-С7)циклоалкилалкила, (С2-С6)алкенила, гало(С2-С6)алкенила, гидрокси(С2-С6)алкенила, (С2-С6)алкинила, (С3-С6)циклоалкил(С2-С4)алкинила, гало(С1-С6)алкила, гало(С3-С6)циклоалкила, гало(С4-С7)циклоалкилалкила, (С2-С6)алкокси, (С3-С6)циклоалкокси, (C4-С7)циклоалкилалкокси, гало(С1-С6)алкокси, гало(С3-С6)циклоалкокси, гало(С4-С7)циклоалкилалкокси, (С1-С6)алкилтио, (С3-С6)циклоалкитио, (С4-С7)циклоалкилалкилтио, гало(С1-С6)алкилтио, гало(С3-С6)циклоалкитио, гало(С4-С7)циклоалкилалкилтио, (С1-С6)алкансульфинила, (С3-С6)циклоалкансульфинила, (С4-С7)циклоалкилалкансульфинила, гало(С1-С6)алкансульфинила, гало(С3-С6)циклоалкансульфинила, гало(С4-С7)циклоалкилалкансульфинила, (С1-С6)алкансульфонила, (С3-С6)циклоалкансульфонила, (С4-С7)циклоалкилалкансульфонила, гало(С1-С6)алкансульфонила, гало(С3-С6)циклоалкансульфонила, гало(С4-С7)цикло-алкилалкансульфонила, (С1-С6)алкиламино, ди(С1-С6)алкиламино, (С1-С6)алкокси(С1-С6)алкокси, гало(С1-С6)алкокси(С1-С6)алкокси, (С1-С6)алкоксикарбонила, H2NCO, H2NSO2, (С1-С6)алкиламинокарбонила, ди(С1-С6)алкиламинокарбонила, (С1-С3)алкокси(С1-С3)алкиламинокарбонила, гетероциклилкарбонила, (С1-С6)алкиламиносульфонила, ди(С1-С6)алкиламиносульфонила, гетероциклилсульфонила, (С1-С6)алкилкарбониламино, (C1-С6)алкилкарбониламино(С1-С6)алкила, (С1-С6)алкилсульфониламино, (C1-С6)алкилсульфониламино(С1-С6)алкила, (С1-С6)алкоксикарбонил(С1-С6)алкокси, (C1-С6)алкокси(С1-С6)алкила, гало(С1-С6)алкокси(С1-С6)алкила, гидрокси(С1-С6)алкокси, гетероарил, амино(С1-С6)алкила, (С1-С6)алкиламино(С1-С6)алкила, ди(С1-С6)алкиламино(С1-С6)алкила амино(С1-С6)алкокси, (С1-С6)алкиламино(С2-С6)алкокси, ди(С1-С6)алкиламино(С2-С6)алкокси и (С1-С6)алкилкарбонила, значения для R1, R2 и R3 соответствуют указанным в описании первого варианта осуществления настоящего изобретения. В качестве альтернативы n имеет значения 0, 1, 2 или 3; подходящие значения для G1 включают (C1-C4)алкил, (C1-C4)алкокси, (C1-C4)галоалкил, (C1-C4)галоалкокси, галоген, циано и нитро; подходящие заместители для атома азота в оксодигидропиридиловом кольце в Формулах Io1, Io2 и Io5 включают C1-C4 алкил, (С3-С4)циклоалкил, (С3-С4)циклоалкил(С1-С3)алкил и C1-C4 галоалкил; подходящие заместители для атома углерода в оксидигидропиридиловом кольце в Формулах Io1, Io2 и Oo5 включают фтор, хлор, циано, гидрокси, амино, (С1-С4)алкил, (С3-С4)циклоалкил, (С3-С4)циклоалкил(С1-С2)алкил, гало(С1-С4)алкил, (С1-С4)алкокси, (С1-С4)галоалкокси, CONH2, (С1-С4)алкиламинокарбонил, ди(С1-С4)алкиламинокарбонил и (C1-С4)алкилкарбониламино; подходящие значения для R1, R2 и R3 соответствуют представленным в описании первого варианта осуществления настоящего изобретения. В качестве альтернативы варианты, представленные в данном разделе, не включают соединения PR-221 и PR-313 или их фармацевтически приемлемые соль, энантиомер или диастереомер.
Для каждого из вариантов, описанных в предыдущем разделе, R1, предпочтительно, представляет собой метил или этил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Io1, Io2 и Io5, R1, предпочтительно, представляет собой метил или этил; а R3 представляет собой MeSO2NHCH2CH2CH2, H2NC(=O)CH2CH2, H2NC(=O)CMe2CH2, 3-гидроксипропил, 3-гидрокси-3-метилбутил, 2-гидроксиэтил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Io1, Io2 и Io5, R1, предпочтительно, представляет собой метил или этил; а R3 представляет собой Н2NC(=O)СМе2СН2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Io1, Io2 и Io5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил, замещаемый 1, 2 или 3 возможными заместителями, выбранными из гало, циано, CONH2, (С1-С4)алкила, (С1-С4)галоалкила, а также SO2Me; a R3 представляет собой MeSO2NHCH2CH2CH2, H2NC(=O)CH2CH2, H2NC(=O)CMe2CH2, 3-гидроксипропил, 3-гидрокси-3-метилбутил, 2- гидроксиэтил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Io1, Io2 и Io5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил, замещаемый 1, 2 или 3 возможными заместителями, выбранными из гало, циано, CONH2, (С1-C4)алкила, (С1-С4)галоалкила, а также SO2Me; а R3 представляет собой Н2NC(=O)СМе2СН2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Io1, Io2 и Io5, R1, предпочтительно, представляет собой метил или этил; a R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Io1, Io2 и Io5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил или фторфенил; а R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Io1, Io2 и Io5, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил или фторфенил; а R3 представляет собой 2- гидрокси-2-метилпропил или 2-циано-2-метилпропил; заместители замещаемого атома азота в оксодигидропиридиловом кольце в Формулах Io1, Io2 и Io5 включают (С1-С4)алкил, (С3-С4)циклоалкил, (С3-С4)циклоалкил(С1-С2)алкил или (С1-С2)галоалкил; а возможные заместители одного или двух атомов углерода в оксодигидропиридиловом кольце в Формулах Io1, Io2 и Io5 включают метил или этил.
Следующий вариант осуществления настоящего изобретения (именуемый в данном документе как "Первый альтернативный вариант осуществления настоящего изобретения") представляет собой соединение, имеющее структурные Формулы Io1, Io2 и Io5, в которых: n имеет значение 0 или 1, предпочтительно 0; каждая G1 независимо выбрана из (С1-С4)алкила, (C1-C4)алкокси, (C1-C4)галоалкила, (C1-C4)галоалкокси, галогена, циано или нитро; атом азота замещается в оксодигидропиридиловом кольце гидрокси(С1-С6)алкилом, (С1-С6)алкилкарбониламино(С1-С6)алкилом, (С1-С6)алкилсульфониламино(С1-С6)алкилом, (С1-С6)алкокси(С1-С6)алкилом, амино(С1-С6)алкилом, (С1-С6)алкиламино(С1-С6)алкилом, ди(С1-С6)алкиламино(С1-С6)алкилом, циано(С1-С6)алкилом, аминокарбонил(С1-С6)алкилом, (С1-С6)алкиламинокарбонил(С1-С6)алкилом, ди(С1-С6)алкиламинокарбонил(С1-С6)алкилом, (С3-С6)циклоалкиламинокарбонил(С1-С6)алкилом, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонил(С1-С6)алкилом или ди(С3-С6)циклоалкиламинокарбонил(С1-С6)алкилом; один или более атом углерода факультативно замещается в оксодигидропиридиловом кольце группой, независимо выбранной из фтора, хлора, циано, гидрокси, амино, (С1-С4)алкила, (С3-С4)циклоалкила, (С3-С4)циклоалкил(С1-С2) алкила, гало(С1-C4)алкила, (С1-С4)алкокси, (С1-С4)галоалкокси, CONH2, (С1-С4)алкиламинокарбонила, ди(С1-С4) алкиламинокарбонила и (С1-С4)алкилкарбониламино; R1 представляет собой метил или этил; R2 представляет собой фенил, тиенил, пиридил или изопропил, возможные заместители каждого из которых включают до трех групп, независимо выбранных из гало, метила, метилтио, (4-морфолино)метила или циклопропила; R3 представляет собой метил, этил, пропил, бутил, винил, аллил или этоксиэтил, возможные заместители каждого из которых включают до двух групп, независимо выбранных из метила, НО-, МеО-, N2N, MeC(=O)NH-, MeS(=O)2NH-, H2NC(=O)-, MeNHC(=O)-, HO2C-, (HO)2P(=O)O-, Н2NS(=O)2O-, H2NS(=O)2NH-, MeNHC(=O)NH-, MeNHC(=O)O-, оксо, циано, НO2С-, HOCH2CH2NH-, 4-морфолино, HOCH2C(=O)NH-, H2NCH2C(=O)NH-, EtNHC(=O)NH, MeOC(=O)NH-, MeNHC(=NC=N)NH-, Me-, MeS-, MeSO2- MeSC>2N(Me)-, MeS(=O)2NHC(=O)-, имидазолиламино-, имидазолила, тетразолила, H2NCONH-, H2NCO2-, HOCH2CH2O-, MeNH-, Me2N- и MeCONMe,
В качестве альтернативы в Структурных формулах Io1, Io2 и Io5 R2 представляет собой фенил, который может замещаться 1, 2 или 3 заместителяли, независимо выбранными из гало, циано, CONH2, (С1-С4)алкила, (С1-С4)галоалкила и SO2Me; R3 представляет собой MeSO2NHCH2CH2CH2, H2NC(=O)CH2CH2, H2NC(=O)CMe2CH2, 3-гидроксипропил, 3-гидрокси-3-метилбутил, 2-гидроксиэтил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; а остальные переменные соответствуют данным, представленным в описании Первого альтернативного варианта осуществления настоящего изобретения.
В качестве альтернативы в Структурных формулах Io1, Io2 и Io5 R3 представляет собой Н2NC(=O)СМе2СН2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; а остальные переменные соответствуют данным, представленным в описании Первого альтернативного варианта осуществления настоящего изобретения.
В качестве альтернативы в Структурных формулах Io1, Io2 и Io5 R2 представляет собой фенил, который может замещаться 1, 2 или 3 возможными заместителяли, независимо выбранными из гало, циано, CONH2, (С1-С4)алкила, (С1-С4)галоалкила и SO2Me; R3 представляет собой Н2NC(=O)СМе2СН2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; а остальные переменные соответствуют данным, представленным в описании Первого альтернативного варианта осуществления настоящего изобретения.
В качестве альтернативы в Структурных формулах Io1, Io2 и Io5 R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; а остальные переменные соответствуют данным, представленным в описании Первого альтернативного варианта осуществления настоящего изобретения.
В качестве альтернативы в Структурных формулах Io1, Io2 и Io5 R2 представляет собой фенил или фторфенил; R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; а остальные переменные соответствуют данным, представленным в описании Первого альтернативного варианта осуществления настоящего изобретения.
В качестве альтернативы в Структурных формулах Io1, Io2 и Io5 R2 представляет собой фенил или фторфенил; R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; один или два замещаемых атома углерода в оксодигидропиридиловом кольце могут замещаться фтором, метилом или этилом; а остальные переменные соответствуют данным, представленным в описании Первого альтернативного варианта осуществления настоящего изобретения.
Для варианта, представленного в предыдущих семи разделах, n имеет значение 0, а все замещаемые атомы углерода в оксодигидропиридиловом кольце, предпочтительно должны оставаться незамещенными.
Следующий вариант осуществления настоящего изобретения представляет собой соединение с любой из Формул Iр1 и Ip3 или его фармацевтически приемлемую соль:
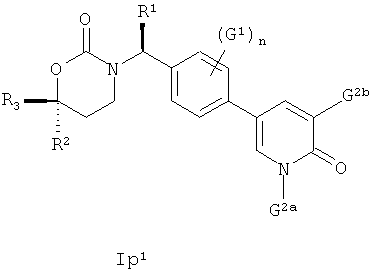
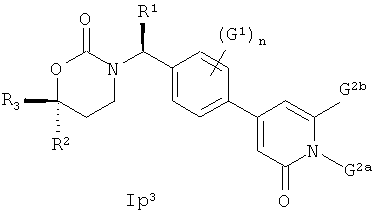
В Формулах Iр1 и Ip3 G1 представляет собой (С1-C4алкил, (С1-С4)алкокси, (C1-С4)галоалкил, (С1-С4)галоалкокси, галоген, циано или нитро; n имеет значение 0, 1 или 2; G2a представляет собой (C1-C4)алкил, (С3-С4)циклоалкил, (С3-С4)циклоалкил(С1-С2)алкил или (С1-С4)галоалкил; G2b представляет собой водород, фтор, хлор, циано, гидрокси, амино, (С1-С4)алкил, (С3-С4)циклоалкил, (С3-С4)циклоалкил(С1-С2)алкил, гало(С1-С4)алкил, (C1-C4) алкокси, (C1-C4) галоалкокси, СОМН2, (С1-С4)алкиламинокарбонил, ди(С1-С4)алкиламинокарбонил или (С1-С4)алкилкарбониламино; подходящие значения для R1, R2 и R3 соответствуют указанным в описании первого варианта осуществления настоящего изобретения. В качестве альтернативы варианты осуществления настоящего изобретения, представленные в данном разделе, не включают соединения PR-221 и PR-313 или их фармацевтически приемлемые соль, энантиомер или диастереомер.
Для каждого из вариантов, описанных в предыдущем разделе, R1, предпочтительно, представляет собой метил или этил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Iр1 и Ip3, R1, предпочтительно, представляет собой метил или этил; a R3 представляет собой MeSO2NHCH2CH2CH2, H2NC(=O)CH2CH2, H2NC(=O)CMe2CH2, 3-гидроксипропил, 3-гидрокси-3-метилбутил, 2-гидроксиэтил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Ip1 и Ip3, R1, предпочтительно, представляет собой метил или этил; а R3 представляет собой Н2NC(=O)СМе2СН2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Ip1 и Ip3, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил, замещаемый 1, 2 или 3 возможными заместителями, выбранными из гало, циано, CONH2, (С1-С4)алкила, (С1-С4)галоалкила, а также SO2Me; a R3 представляет собой MeSO2NHCH2CH2CH2, H2NC(=O)CH2CH2, H2NC(=O)CMe2CH2, 3-гидроксипропил, 3-гидрокси-3-метилбутил, 2- гидроксиэтил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Ip1 и Ip3, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил, замещаемый 1, 2 или 3 возможными заместителями, выбранными из гало, циано, CONH2, (С1-С4)алкила, (С1-С4)галоалкила, а также SO2Me; a R3 представляет собой H2NC(=O)CMe2CH2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил,
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Ip1 и Ip3, R1, предпочтительно, представляет собой метил или этил; а R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Ip1 и Ip3, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил или фторфенил; а R3 представляет собой 2- гидрокси-2-метилпропил или 2-циано-2-метилпропил.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Ip1 и Ip3, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил или фторфенил; а R3 представляет собой 2- гидрокси-2-метилпропил или 2-циано-2-метилпропил; заместитель G2a выбран из (С1-C4)алкила, (С3-С4)циклоалкила, (С3-С4)циклоалкил(С1-С2)алкила и (С1-С2)галоалкила; a G2b может быть выбрана из водорода, метила или этила.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Ip1 и Ip3, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил или фторфенил; а R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; заместитель G2a выбран из галоалкила, (С1-С4)алкила, (С3-С4)циклоалкила, (С3-С4)циклоалкил(С1-С3)алкила и (С1-С2)галоалкила; a G2b может быть выбрана из водорода, метила или этила.
Для каждого из вариантов, описанных в данном разделе непосредственно после Формул Iр1 и Ip3, R1, предпочтительно, представляет собой метил или этил; R2 представляет собой фенил или фторфенил; а R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; заместитель G2a выбран из дифторметила, этила, замещенного 1-3 атомами фтора (предпочтительно, 2-фторэтила или 2,2,2-фторэтила), (С1-С4)алкила, (С3-С4)циклоалкила, (С3-С4)циклоалкил(С1-С2)алкила и (С1-С2)галоалкила; a G2b может быть выбрана из водорода, метила или этила.
Следующий вариант осуществления настоящего изобретения (именуемый в данном документе как "Второй альтернативный вариант осуществления настоящего изобретения") представляет собой соединение, имеющее Структурные формулы Ip1 и Ip3, в которых: n имеет значение 0 или 1, предпочтительно 0; каждая G1 независимо выбрана из (C1-C4)алкила, (С1-С4)алкокси, (С1-С4)галоалкила, (С1-С4)галоалкокси, галогена, циано или нитро; G2a представляет собой гидрокси(С1-С6)алкил, (С1-С4)алкилкарбониламино(С1-С6)алкил, (С1-С6)алкилсульфониламино(С1-С6)алкил, (С1-С6)алкокси(С1-С6)алкил, амино(С1-С6)алкил, (С1-С6)алкиламино(С1-С6)алкил, ди(С1-С6)алкиламино(С1-С6)алкил, циано(С1-С6)алкил, аминокарбонил(С1-С6)алкил, (С1-С6)алкиламинокарбонил(С1-С6)алкил, ди(С1-С6)алкиламинокарбонил(С1-С6)алкил, (С3-С6)циклоалкиламинокарбонил(С1-С6)алкил, {(С3-С6)циклоалкил}{(С1-С6)алкил}аминокарбонил(С1-С6)алкил или ди(С3-С6)циклоалкиламинокарбонил(С1-С6)алкил; С2b представляет собой водород, фтор, хлор, циано, гидрокси, амино, (С1-С4)алкил, (С3-С4)циклоалкил, (С3-С4)циклоалкил(С1-С2)алкил, гало(С1-С4)алкил, (С1-С4)алкокси, (С1-С4)галоалкокси, CONH2, (С1-С4)алкиламинокарбонил, ди(С1-С4)алкиламинокарбонил или (С1-С4)алкилскарбониламино; R1 представляет собой метил или этил; R2 представляет собой фенил, тиенил, пиридил или изопропил, возможные заместители каждого из которых включают до трех групп, независимо выбранных из гало, метила, метилтио или (4-морфолино)метила; R3 представляет собой метил, этил, пропил, бутил, винил, аллил или этоксиэтил, возможные заместители каждого из которых включают до двух групп, независимо выбранных из метила, НО-, МеО-, H2N-, МеС(=O)МН-, MeS(=O)2NH-, H2NC(=O)-, MeNHC(=O)-, HO2C-, (HO)2P(=O)O-, Н2NS(=O)2O-, H2NS(=O)2NH-, MeNHC(=O)NH-, MeNHC(=O)O-, оксо, циано, HO2C-, HOCH2CH2NH-, 4-морфолино, НОСН2С(=O)NH-, H2NCH2C(=O)NH-, EtNHC(=O)NH, MeOC(=O)NH-, MeNHC(=NC=N)NH-, Me-, MeS-, MeSO2- MeSO2N(Me)-, MeS(=O)2NHC(=O)-, имидазолиламино-, имидазолила, тетразолила, H2NCONH-, H2NCO2-, HOCH2CH2O-, MeNH-, Me2N- и MeCONMe.
В качестве альтернативы в Структурных формулах Iр1 и Ip3 R2 представляет собой фенил, который может замещаться 1, 2 или 3 возможными заместителями, независимо выбранными из гало, циано, CONH2, (С1-С4)алкила, (С1-Сд4)галоалкила и SO2Me; R3 представляет собой MeSO2NHCH2CH2CH2, H2NC(=O)CH2CH2, H2NC(=O)CMe2CH2, 3-гидроксипропил, 3-гидрокси-3-метилбутил, 2-гидроксиэтил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; а остальные переменные соответствуют данным, представленным в описании Второго альтернативного варианта осуществления настоящего изобретения.
В качестве альтернативы в Структурных формулах Ip1 и Ip3 R3 представляет собой H2NC(=O)CMe2CH2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; а остальные переменные соответствуют данным, представленным в описании Второго альтернативного варианта осуществления настоящего изобретения.
В качестве альтернативы в Структурных формулах Ip1 и Ip3 R2 представляет собой фенил, который может замещаться 1, 2 или 3 возможными заместителями, независимо выбранными из гало, циано, CONH2, (С1-С4)алкила, (С1-С4)галоалкила и SO2Me; R3 представляет собой H2NC(=O)CMe2CH2, 3-гидрокси-3-метилбутил, 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; а остальные переменные соответствуют данным, представленным в описании Второго альтернативного варианта осуществления настоящего изобретения.
В качестве альтернативы в Структурных формулах Ip1 и Ip3 R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; а остальные переменные соответствуют данным, представленным в описании Второго альтернативного варианта осуществления настоящего изобретения.
В качестве альтернативы в Структурных формулах Ip1 и Ip3 R2 представляет собой фенил или фторфенил; R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; а остальные переменные соответствуют данным, представленным в описании Второго альтернативного варианта осуществления настоящего изобретения.
В качестве альтернативы в Структурных формулах Ip1 и Ip3 R2 представляет собой фенил или фторфенил; R3 представляет собой 2-гидрокси-2-метилпропил или 2-циано-2-метилпропил; один или два замещаемых атома углерода в оксодигидропиридиловом кольце могут в качестве варианта замещаться фтором, метилом или этилом; а остальные переменные соответствуют данным, представленным в описании Второго альтернативного варианта осуществления настоящего изобретения.
Для варианта, представленного в предыдущих семи разделах, n имеет значение 0, a G2b, предпочтительно, представляет собой -Н.
Следующий вариант осуществления настоящего изобретения представляет собой гидрат или моногидрат (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6- дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она, (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она, 3-{(S)-1-[4-(1-циклопропил-2-оксо-1,2-дигидро-пиридин-4-ил)-фенил]-этил}-(S)-6-(2-гидрокси-2-метил-пропил)-6-фенил-[1,3]оксазинан-2-она и их фармацевтически приемлемые соли, включая обе формы гидрата и моногидрата, как нейтральную форма, так и, предпочтительно, фармацевтически приемлемую форму соли.
Описание соединений данного изобретения также содержится в следующих документах: ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ 11β-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ I, предварительная заявка США №61/61/135,933, поданная 25 июля 2008 года (Attorney Docket №4370.1000-000); Циклические ингибиторы 11β-гидроксистероид-дегидрогеназы 1, предварительная заявка США №61/135,933, поданная 1 мая 2008 года; циклические ингибиторы 11β-гидроксистероид-дегидрогеназы 1, предварительная заявка США №61/137,148, поданная 25 июля 2008 года; а также Циклические ингибиторы 11β-гидроксистероид-дегидрогеназы 1, международная заявка № PCT/US2008/009017, поданная 25 июля 2008 года; перечисленные заявки приводятся здесь в качестве ссылки.
ОПРЕДЕЛЕНИЯ
Термин "алкил" относится к группам как с неразветвленными, так и с разветвленными углеводородным радикалам, имеющим 1-10 атомов углерода и включающим, например, метил, этил, n-пропил, изопропил, n-бутил, сек-бутил, изобутил, трет-бутил, n-пентил, n-гексил, n-гептил, n-октил, n-нонил, n-децил и подобные им.
Термин "циклоалкил" относится к моноциклическому, бициклическому или трициклическому насыщенному углеводородному кольцу, имеющему 3-10 атомов углерода, и включает, например, циклопропил (с-Pr), циклобутил, циклопентил, циклогексил, циклогентил, циклооктил, бицикло [2.2.2]октил, бицикло [2.2.1]гентил, спиро[4.4]нонан, адамантил и подобные им.
Термин "арил" относится к ароматическому радикалу, представляющему собой фенильную или нафтильную группу, инданильную или тетрагидронафталиновую группу. Арильная группа может иметь 1-4 возможного заместителя. Представителями заместителей являются алкил, алкокси, алкилтио, алкилсульфонил, галоген, трифторметил, диалкиламино, нитро, циано, СO2Н, CONH2, N-моноалкил-замещенный амидо и N,N-диалкил- замещенный амидо.
Термины "гетероарил" относится к 5- и 6-членному гетероароматическому радикалу, который в качестве варианта может быть слит с насыщенным или ненасыщенным кольцом, содержащим 0-4 гетероатома, выбранных из N, О и S, и может содержать, например, гетероароматический радикал, представляющий собой 2- или 3-тиенил, 2- или 3-фуранил, 2- или 3- пирролил, 2-, 3-, или 4-пиридил, 2-пиразинил, 2-, 4-,или 5-пиримидил, 3- или 4-пиридазинил, 1Н-индол-6-ил, 1Н-индол-5-ил, 1Н-бензимидазол-6-ил, 1Н- бензимидазол - 5-ил, 2-, 4-, 5-, 6-, 7- или 8-хиназолинил, 2-, 3-, 5-, 6-, 7- или 8-хиноксалинил, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолинил, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолинил, 2-, 4-, или 5-тиазолил, 2-, 3-, 4-, или 5-пиразолил, 2-, 3-, 4-, или 5-имидазолил. Гетероарил может быть замещен. Возможные заместители гетероарила включают алкил, алкокси, алкилтио, алкилсульфонил, галоген, трифторметил, диалкиламино, нитро, циано, СO2Н, CONH2, N-моноалкил-замещенный амидо и N,N-диалкил- замещенный амидо или оксо с последующим формированием N-оксила.
Термин "гетероциклил" относится к 4-, 5-, 6- и 7-членному насыщенному или частично ненасыщенному гетероциклическому кольцу, содержащему от 1 до 4 гетероатомов, независимо выбранных из N, О и S. Представителями гетероциклилов являются пирролидин, пирролидин-2-он, 1-метилпирролидин-2-он, пиперидин, пиперидин-2-он, дигидропиридин, тетрагидропиридин, пиперазин, 1-(2,2,2-трифторэтил)пиперазин,
1.2-дигидро-2-оксопиридин, 1,4-дигидро-4-оксопиридин, пиперазин-2-он, 3,4,5,6-тетрагидро-4-оксопиримидин, 3,4-дигидро-4-оксопиримидин, тетрагидрофуран, тетрагидропиран, тетрагидротиофен, тетрагидротиопиран, изоксазолидин, 1,3-диоксолан, 1,3-дитиолан, 1,3-диоксан, 1,4-диоксан, 1,3-дитиан, 1,4-дитиан, оксазолидин-2-он, имидазолидин-2-он, имидазолидин-2,4-дион, тетрагидропиримидин-2(1Н)-он, морфолин, N-метилморфолин, морфолин-3-он, 1,3-оксазинан-2-он, тиоморфлолин, тиоморфлолин 1,1-диоксид, тетрагидро-1,2,5-тиаоксазол 1,1-диоксид, тетрагидро-2Н-1,2-тиазин 1,1-диоксид, гексагидро-1,2,6-тиадиазин 1,1-диоксид, тетрагидро-1,2,5-тиадиазол 1,1-диоксид изотиазолидин 1,1-диоксид, 6-оксо-1,6-дигидропиридазин-3-ил, 6-оксо-1,6-дигидропиридазин-4-ил, 5-оксо-4,5-дигидро-1Н-1,2,4-триазол-3-ил и 5-оксо-4,5-дигидро-1Н-имидазол-2-ил. Гетероциклил имеет 1-4 возможных заместителя, включающих алкил, галоалкил, галоген и оксо.
Термин "спироциклоалкил" относится к циклоалкильной группе, имеющей в кольце один общий атом углерода с другой алкильной или циклоалкильной группой.
Термины "пациент" или "больной", используемые в данном описании, являются взаимозаменяемыми и относятся к млекопитающему, которое подлежит лечению, в том числе домашнему животному (например, кошка, собака и т.д.), сельскохозяйственному животному (например, корова, свинья, лошадь, овца, коза и т.д.), а также лабораторному животному (например, крыса, мышь, морская свинка и т.д.). Как правило, термин "пациент" относится к человеку, нуждающемуся в лечении.
Предполагается, что при упоминании или графическом изображении настоящего соединения или его фармацевтически приемлемой соли также охватываются сольваты или гидраты соединения или его фармацевтически приемлемых солей. Термин "сольваты" относится к кристаллическим формам, в кристаллическую решетку которых во время кристаллизации вошли молекулы растворителя. Сольваты могут включать воду или неводные растворители, такие как этанол, изопропанол, ДМСО, уксусная кислота, этаноламин и этилацетат. Сольваты, образующиеся в случае использования воды в качестве молекулы растворителя, включенной в кристаллическую решетку, как правило, именуются "гидратами". К гидратам относятся стехиометрические соединения, а также композиции, содержащие различное количество воды. Некоторые из соединений, описанных в качестве примеров, могут иметь безводную форму.
Термин "соединение" также охватывает дейтерий-меченые в одной или более позициях соединения. "Дейтерий-меченые в позиции" означает, что количество дейтерия в данной позиции выше, чем в природных условиях. В некоторых случаях количество дейтерия в каждой позиции "соединения" соответствует наблюдаемому в природе.
Некоторые из описанных здесь соединений могут существовать в различных стереоизомерных формах. Стереоизомерами называют соединения, отличающиеся лишь пространственным расположением отдельных атомов при одинаковом порядке их соединения между собой. Энантиомерами называют пары стереоизомеров, зеркальные изображения которых не налагаются друг на друга, в основном, из-за асимметрически замещенного атома углерода, выступающего в качестве хирального центра. Термин "энантиомер" относится к одной из двух молекул, являющихся неналагаемыми зеркальными изображениями. Диастереоизомеры представляют собой стереоизомеры, не являющиеся зеркальными изображениями друг друга, в основном, из-за двух и более асимметричных атомов углерода, выступающих в качестве хиральных центров. Символ "*", представленный в Структурной формуле, указывает на наличие атома углерода, выступающего в качестве хирального центра. Символы "R" и "S" указывают на конфигурацию заместителей вокруг одного или более хиральных центров - атомов углерода. Таким образом, обозначения "R*" и "S*" указывают на относительную конфигурацию заместителей вокруг одного или более хиральных центров - атомов углерода.
Термин "рацемат" или "рацемическая смесь" относится к соединению эквимолярных количеств двух энантиомеров, не обладающему оптической активностью, т.е. не вызывающему вращения плоскости поляризации света.
Термин "геометрический изомер" относится к изомерам, отличающимся друг от друга различным пространственным расположением атомов заместителей относительно плоскости двойной связи углеро-углерод, циклоалкильного кольца или мостиковой бициклической системы. Атомы (помимо Н) с каждой стороны двойной связи углерод-углерод могут иметь конфигурацию Е (заместители расположены на противоположных сторонах двойной связи углерод-углерод) или Z (заместители расположены на одной стороне связи).
Обозначения "R," "S," "S*," "R*," "Е," "Z," "цис" и "транс" указывают на конфигурацию заместителей относительно остова молекулы.
Соединения настоящего изобретения могут быть приготовлены в виде индивидуальных изомеров путем изомер-специфического синтеза или выделения из смеси изомеров. Стандартные методики выделения изомеров включают формирование соли свободного основания каждого изомера изомерной пары с помощью оптически активной кислоты (с последующей фракционной кристаллизацией или восстановлением свободного основания); формирование соли кислотной формы каждого изомера изомерной пары с помощью оптически активного амина (с последующей фракционной кристаллизацией или восстановлением свободной кислоты); формирование эфира или амида каждого изомера изомерной пары с помощью оптически чистой кислоты, амина или спирта (с последующим хроматографическим разделением и удалением хирального вспомогательного элемента); или выделение смеси изомеров из исходного материала либо из готового продукта при помощи широко распространенных хроматографических методик.
При упоминании или графическом изображении стереохимических свойств настоящего соединения предполагается, что упомянутый или изображенный стереоизомер имеет массу чистого вещества не менее 60%, 70%, 80%, 90%, 99% или 99,9% по отношению к другим стереоизомерам. При упоминании или графическом изображении отдельного энантиомера предполагается, что упомянутый или изображенный энантиомер имеет чистоту оптического изомера по массе не менее 60%, 70%, 80%, 90%, 99% или 99,9%. Процентный показатель оптической чистоты по массе является отношением массы энантиомера к общей массе энантиомера и его оптического изомера.
При упоминании или графическом изображении настоящего соединения без указания его стереохимических свойств, а также при условии наличия у соединения, как минимум, одного хирального центра, предполагается, что данное упоминание или графическое изображение охватывает один энантиомер соединения, не содержащий соответствующий оптический изомер, рацемическую смесь соединения, а также смеси, обогащенные одним энантиомером по отношению к соответствующему оптическому энантиомеру настоящего соединения.
При упоминании или графическом изображении настоящего соединения без указания его стереохимических свойств, а также при условии наличия у соединения, как минимум, двух хиральных центров, предполагается, что данное упоминание или графическое изображение охватывает диастереомер, не содержащий других диастереомеров, или пару диастереомеров, не содержащих другие пары диастереомеров, смеси диастереомеров, смеси пар диастереомеров, а также смеси диастереомеров, обогащенные одним диастереомером по отношению к другому/другим диастереомеру (-ам), и смеси пар диастереомеров, обогащенных одной парой диастереомеров по отношению к другой/другим паре(-ам) диастереомеров.
Соединения настоящего изобретения могут иметь форму фармацевтически приемлемых солей. Используемые в составе лекарственных препаратов соли соединений настоящего изобретения относятся к нетоксичным "фармацевтически приемлемым солям". Формы фармацевтически приемлемой соли включают фармацевтически приемлемые кислотные/анионные или основные/катионные соли.
Фармацевтически приемлемые основные/катионные соли включают следующие: натрий, калий, кальций, магний, диэтаноламин, п-метил-О-глюкамин, L-лизин, L-аргинин, аммоний, этаноламин, пиперазин и триэтаноламин.
Фармацевтически приемлемые кислотные/анионные соли включают следующие: ацетат, бензолсульфонат, бензоат, бикарбонат, битартрат, бромид, кальция эдетат, камсилат, карбонат, хлорид, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, фумарат, глицертат, глютамат, гликоллиларсанилат, гексилрезорцинат, гидробромид, гидрохлорид, гидроксинафтоат, йодид, изетионат, лактат, лактобионат, малат, малеат, малонат, манделат, мезилат, метилсульфат, мукат, напсилат, нитрат, памоат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, сульфат, гидрогенсульфат, таннат, тартрат, теоклат, тозилат и триэтиодид.
В таблице ниже представлены используемые сокращения, а также их значения:
ОБЩЕЕ ОПИСАНИЕ СПОСОБОВ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ
Соединения Формулы I* могут быть получены несколькими способами. В представленном ниже материале А1, Су1, Е, R1, R2, R3, Y и n имеют значения, приведенные выше, если не указано иное. Су2 представляет собой в качестве варианта замещенную 2-оксо-1,2-дигидропиридиловую группу. В случаях если описанные ниже промежуточные соединения и конечные продукты Формулы I* содержат потенциально реакционноспособные функциональные группы, представителями которых являются группы амино, гидроксил, тиол и карбоновой кислоты, препятствующие появлению целевой реакции, применение защищенной формы промежуточного соединения может быть более эффективным. Методы выбора, введения и последующего удаления защитных групп хорошо известны специалистам в данной области техники (T.W. Greene и Р.G.M. Wuts "Protective Groups in Organic Synthesis" John Wiley & Sons, Inc., New York 1999). Применение подобных защитных групп учитывается в приведенном ниже материале, однако не описывается подробно. Как правило, реактивы, указанные в схемах реакций, используются в эквимолярных количествах; однако в отдельных случаях может быть желательным применение избыточного количества одного из реактивов для завершения реакции. Подобная мера особенно актуальна в случаях, когда избыточный реактив может быть легко удален путем выпаривания или экстрагирования. Основания, предназначенные для нейтрализации HCl в составе реакционной смеси, как правило, используются в избыточном количестве (от 1,05 до 5 эквивалентов),
В ходе первого процесса соединение Формулы I* может быть получено путем проведения реакции промежуточного аминоспирта Формулы II и реактива Формулы III, где Z1 и Z2 представляют собой уходящие группы, подобные хлориду, 1-имидазолилу или арилоксиду, в инертном растворителе, подобном тетрагидрофурану, CH2Cl2, толуол или MeCN, как правило, в присутствии органического или неорганического основания, аналогичного триэтиламину или NaHCO3 соответственно при температуре от -10°С до 120°С:
Определенные типы реактива III особенно удобны для применения по причине их наличия на рынке. Например, в случае, если Z1 и Z2 представляют собой хлориды, III представляет собой фосген. В случае если Z1 и Z2 представляют собой 1-имидазолил, III представляет собой карбонилдимидазол. В случае если Z1 представляет собой хлорид, а Z2 - p-нитрофеноксид, III представляет собой p-нитрофенил хлорформиат. В случае если Z1 и Z2 представляют собой ОССl3, III представляет собой трифосген и может использоваться в количестве одной трети эквивалента.
Промежуточные аминоспирты Формулы II могут быть получены путем снижения количества амидов в Формуле IV с помощью гидридного реактива, аналогичного ВНз. раствору тетрагидрофурана, ВН3.Ме2S или LiAlH4, в инертном эфирном растворителе, подобном тетрагидрофурану или DME, при температуре от 20°С до 100°С в течение периода длительностью от 1 ч до 48 ч.

Промежуточные соединения Формулы IV могут быть получены путем связывания гидроксикислоты Формулы V и амина Формулы VI с помощью стандартных пептидных связующих веществ, подобных EDC, в присутствии HOBt и N,N-диизопропилэтиламина в инертном растворителе, аналогичном СН2Сl2 при температуре от 0°С до 30°С в течение периода длительностью от 1 ч до 24 ч.
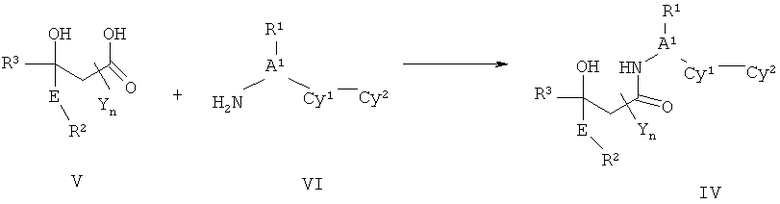
Промежуточные амины Формулы VI, где А1=CH2, a R1 отсутствует, могут быть получены путем снижения количества амидов в Формуле VII с помощью гидридного реактива, аналогичного ВН3. раствору тетрагидрофурана, ВН3.Ме2S или LiAlH4, в инертном эфирном растворителе, подобном тетрагидрофурану или DME, при температуре от 20°С до 100°С в течение периода длительностью от 1 ч до 48 ч.
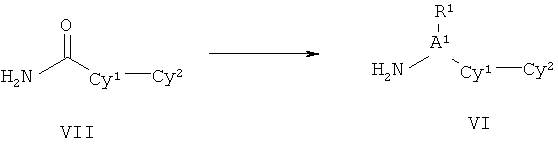
Промежуточные амины Формулы VI, где А1 является связанным, R1 отсутствует, а Су1 не является ароматическим или гетероароматическим кольцом, могут быть получены из кетонов Формулы VIII посредством оксимов Формулы IX или путем восстановительного аминирования кетона Формулы VIII при помощи аммиака:
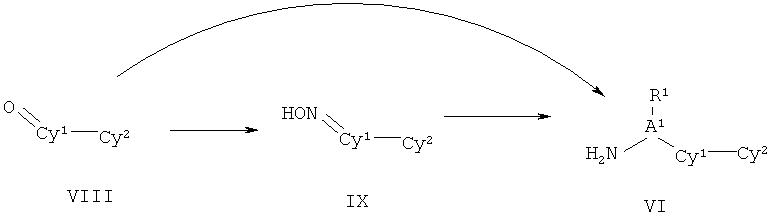
Методы преобразования кетонов в оксимы описаны в публикации Smith, M. В. and March, J. "March's Advanced Organic Chemistry" pp. 1194-1195, 5th Edition, Wiley, New York, NY, 2001. Методы восстановления оксимов до первичных аминов описаны в Smith, M.В. and March, J. "March's Advanced Organic Chemistry" p.1555, 5th Edition, Wiley, New York, NY, 2001. Методы восстановительного аминирования кетонов представлены в публикации Baxter, Е.W. and Reitz, А. В. "Organic Reactions" Volume 59, Ed. Overman, L.E., Wiley Interscience, 2002.
Аналогичным образом могут быть получены промежуточные амины Формулы VI, где А1 представляет собой СН, а R1 - метил или этил, посредством восстановления t-бутилсульфиниламинов Формулы VIIIb, которые в свою очередь, могут быть получены из кетонов Формулы Villa и t-бутилсульфинамидов, или путем добавления металлорганических реактивов Формулы R1M, где R1 представляет собой Me или Et, a M представляет собой Li, MgCl, MgBr или Mgl, к t-бутилсульфинилиминам Формулы VIIId, которые могут быть получены из альдегидов Формулы VIIIc.
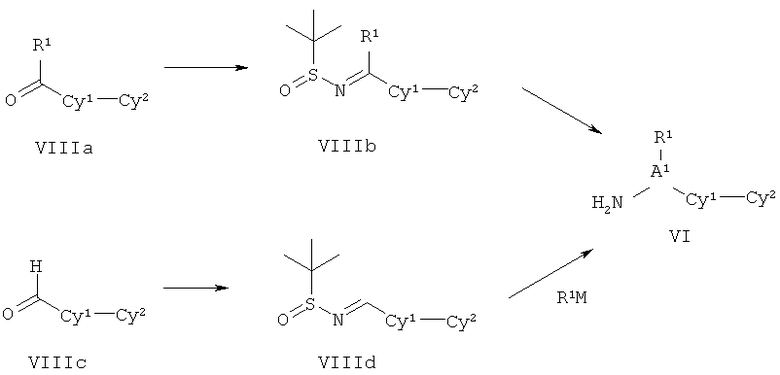
В ходе подобных реакций с применением хиральных t-бутилсульфинилиминов часто достигается высокая стереоселективность.
Промежуточные соединения Формулы II, где n=0, могут быть получены в результате реакции окситанов Формулы Х и аминов Формулы VI, описанной в публикации Smith, М.В. and March, J. "March's Advanced Organic Chemistry" p.505, 5th Edition, Wiley, New York, NY, 2001:
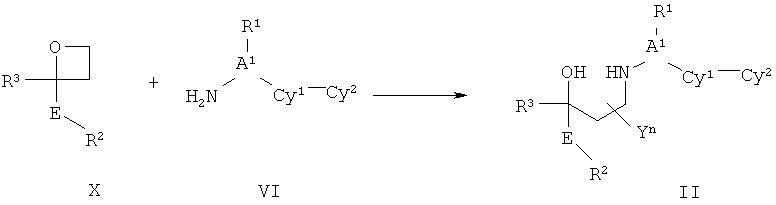
Промежуточные соединения Формулы II также могут быть получены путем восстановительного аминирования гидроксиальдегидов Формулы Ха с помощью аминов Формулы VI. Методы восстановительного аминирования альдегидов описаны в публикации Baxter, E.W. and Reitz, А.В. "Organic Reactions" Volume 59, Ed. Overman, L.E., Wiley Interscience, 2002.
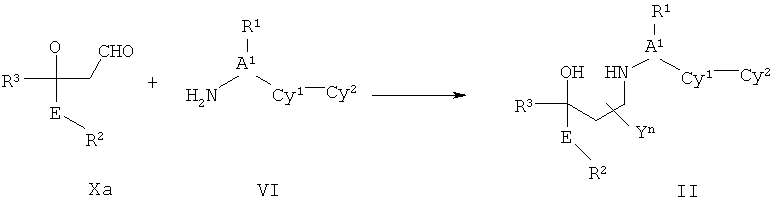
Альдегиды Формулы Ха могут быть получены из гомоаллильных спиртов Формулы XXI путем обработки OsO4 и NalO4.
Промежуточные соединения Формулы II, где А1=CH2, a R1 отсутствует, могут быть получены путем снижения количества промежуточных амидов в Формуле XI с помощью гидридного реактива, аналогичного ВН3. раствору тетрагидрофурана, BH3.Me2S или LiAlH4, в инертном эфирном растворителе, подобном тетрагидрофурану или DME, при температуре от 20°С до 100°С в течение периода длительностью от 1 ч до 48 ч.

Промежуточные амиды Формулы XI могут быть получены в результате реакции промежуточных аминоспиртов Формулы XII и активированной карбоновой кислоты Формулы XIII, где Z3=хлорид или активированный эфир, подобный эфиру N-гидроксисукцинимиду:
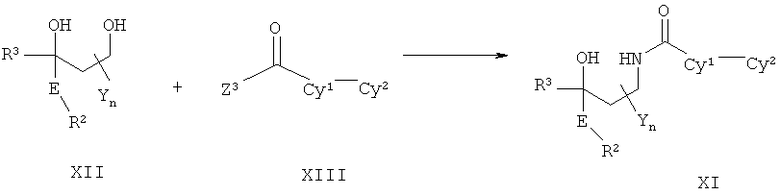
Промежуточные аминоспирты Формулы XII, где n=0, могут быть получены в результате реакции эпоксидов Формулы XIV и цианид-иона с последующим восстановлением полученного гидроксинитрила Формулы XV с помощью газообразного водорода в присутствии катализатора или гидрид-источника, подобного LiAlH4:
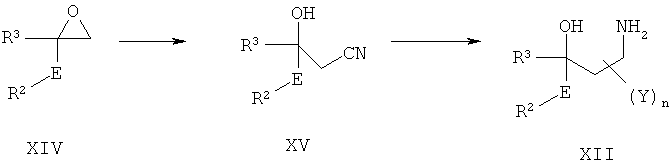
Эпоксидные соединения Формулы XIV, в свою очередь, могут быть получены несколькими способами, включая описанные в публикации Aube, J. "Epoxidation and Related Processes" Chapter 3.2 in Volume 1 of "Comprehensive Organic Synthesis" Edited by B.M. Trost, I. Fleming and Stuart L. Schreiber, Pergamon Press, New York, 1992.
Промежуточные гидроксинитрилы Формулы XV могут быть получены путем обработки кетонов Формулы XVI анионом ацетонитрила, полученным путем обработки n-BuLi или LDA, в инертном, безводном растворителе, подобном тетрагидрофурану при низких температурах:
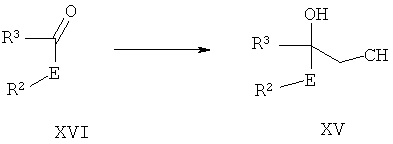
Промежуточные аминоспирты Формулы XII, где значение n равно 0, могут быть получены путем обработки промежуточными сульфонатами Формулы XVII, где R представляет собой, например, метил, трифторметил или р-метилфенил, при помощи аммиака:
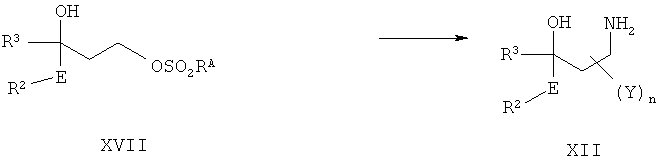
Промежуточные аминоспирты Формулы XII могут быть получены путем обработки промежуточных сульфонатов Формулы XVII азидом натрия для получения промежуточного азида Формулы XVIII с последующим каталитическим гидрированием или восстановлением посредством реакции Штаудингера с помощью PPh3 во влажном тетрагидрофуране:
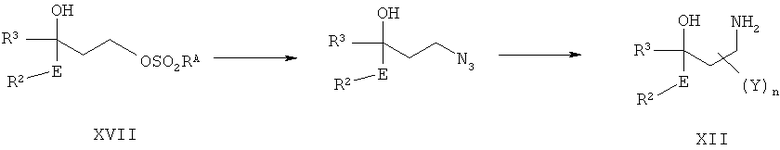
Промежуточные сульфонаты Формулы XVII могут быть получены из промежуточных диолов Формулы XIX с помощью сульфонил хлорида RASO2Cl:
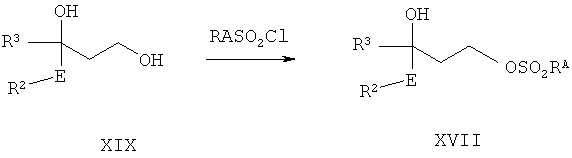
Промежуточные диолы Формулы XIX могут быть получены посредством гидроборирования аллильных спиртов Формулы XX:
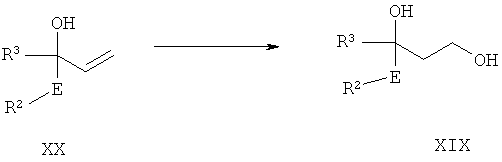
Промежуточные диолы Формулы XIX могут быть получены посредством озонолиза и восстановления гомоаллильных спиртов Формулы XXI:
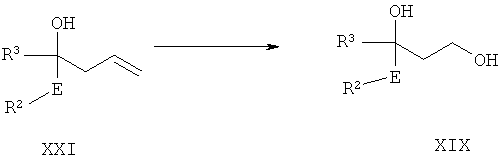
Промежуточные аминоспирты Формулы II, где А1 является связанным, R1 отсутствует, а Су1 представляет собой гетероарильную группу или арильную группу, включающую, как минимум, одну сильную электроноакцепторную группу, подобную CF3, могут быть получены в результате реакции промежуточных аминоспиртов Формулы XII с соединением формулы XXII, где Су1 представляет собой гетероарильную группу или арильную группу, включающую, как минимум, одну сильную электроноакцепторную группу, подобную CF3, a R3 представляет собой уходящую группу, например, фторо, хлоро, бромо или йодо:
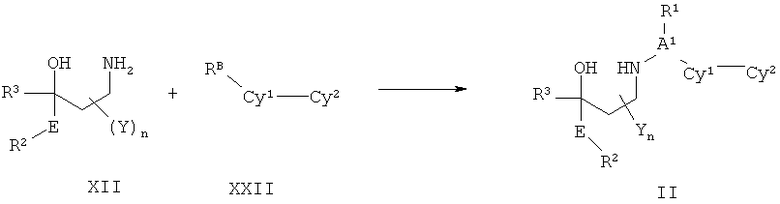
Промежуточные аминоспирты Формулы II, где А1 представляет собой (С1)алкилен, могут быть получены в результате реакции аминоспирта Формулы XII с альдегидом или метиловым кетоном Формулы XII в присутствии восстановителя, подобного NaCNBH3 или Na(OAc)3BH:
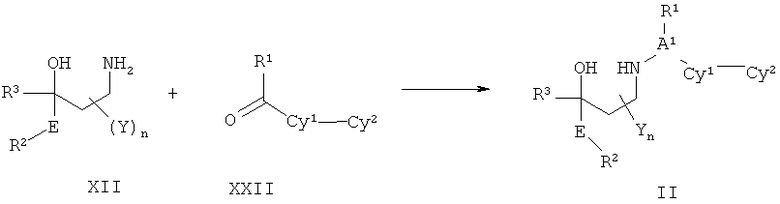
Методы восстановительного аминирования альдегидов и кетонов описаны в публикации Baxter, Е.W. and Reitz, А.В. "Organic Reactions" Volume 59, Ed. Overman, L.E., Wiley Interscience, 2002.
В ходе второго процесса соединение Формулы I* может быть получено в результате реакции кетокарбамата Формулы XXIV, где RD представляет собой алкильную или арилалкильную группу, подобную метилу, t-бутилу или бензилу, с металлорганическими реактивами Формулы XXV, где М включает, помимо прочего, MgCl, MgBr, Mgl или Li:
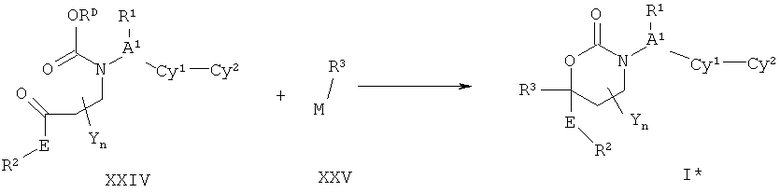
В отдельных случаях металлорганический реактив XXV представляет собой бромид аллилмагния, бромид аллилцинка(II), хлорид (2-метилаллил)магния или бромид (2-метокси-2-оксоэтил)цинка(II). В отдельных случаях, если М представляет собой MgCI, MgBr или Mgl, рекомендуется добавить СеС1з в реакционную смесь.
Кетокарбаматы Формулы XXIV могут быть получены в результате реакции аминокетонов Формулы XXVI с промежуточными соединениями Формулы XXVII, где RE представляет собой уходящую группу, подобную хлориду, сукцинилокси, имидазолилу или t-бутоксикарбоксикарбонилу:
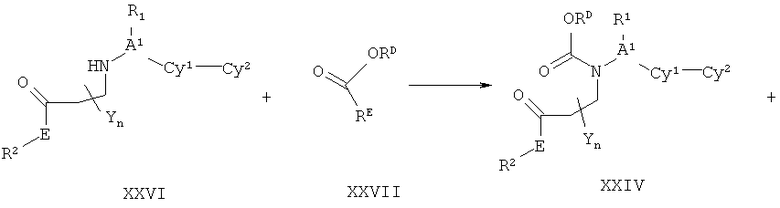
Аминокетоны Формулы XXVI, где n=0, могут быть получены в результате реакции ненасыщенных кетонов Формулы XXVIII с аминами Формулы VI:
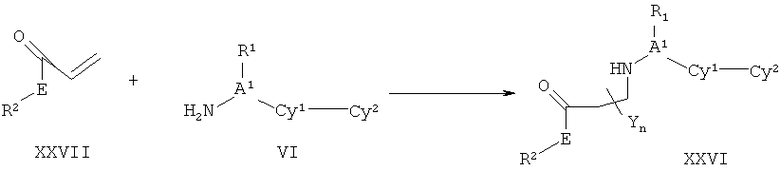
Аминокетоны Формулы XXVI, где n=0, могут быть получены в результате реакции диалкиламирнокетонов Формулы XXVIII, где RF представляет собой низший алкил, в особенности метил, с аминами Формулы VI:
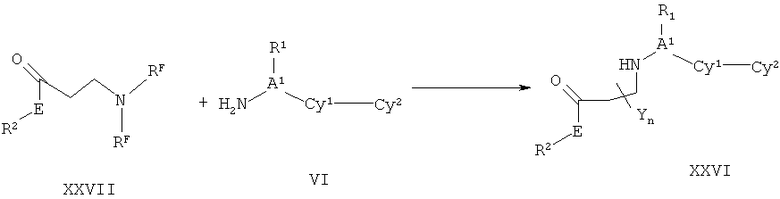
- диалкиламинокетоны Формулы XXVIII, в свою очередь, могут быть получены в результате реакции ненасыщенных кетонов Формулы XXVII с диалкиламинами Формулы RFNHRF.
В ходе третьего процесса соединение Формулы I* может быть получено в результате реакции соединения Формулы XVII с изоцианатом Формулы XXIX в присутствии основания;
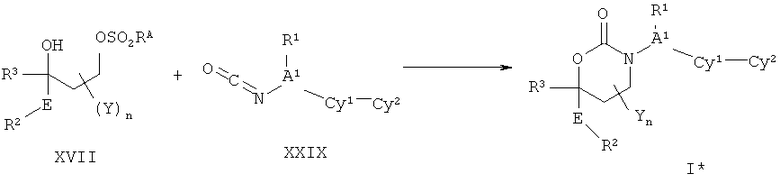
Изоцианаты Формулы XXIX могут быть получены из аминов Формулы VI путем обработки фосгеном, дифосгеном или трифосгеном. Данный третий процесс более подробно описан в предварительной заявке США с серийным номером 61/137,013, поданной 25 июля 2008 года, под названием СИНТЕЗ ИНГИБИТОРОВ 11БЕТА-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ 1 ТИПА (Ид. № пат. повр. 4370.1001-000). Данная заявка приводится здесь в качестве ссылки.
В ходе четвертого процесса соединение Формулы I* может быть получено в результате реакции галогенного соединения Формулы, где Hal представляет собой хлор или бром, с изоцианатом Формулы XXIX в присутствии основания:
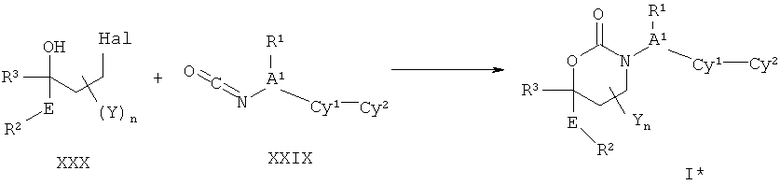
Галогенные соединения Формулы XXX могут быть получены в результате реакции галокетонов Формулы XXXI с металлорганическими реактивами Формулы XXV, где М представляет собой металлосодержащий радикал, включающий MgCl, MgBr, Mgl или Li. Реакция при необходимости может быть проведена в присутствии безводного треххлористого церия:
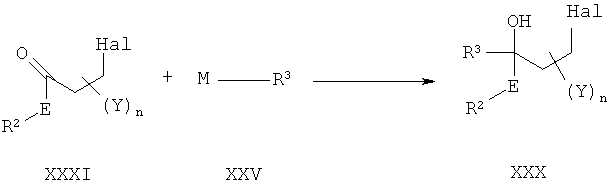
В ходе пятого процесса соединение Формулы I*, где А1 представляет собой CH2 или СН2СН2, a R1 отсутствует, может быть получено в результате реакции соединения Формулы XXXII и соединения Формулы XXXIII, где А1 представляет собой CH2 или CH2CH2, a RG являются уходящей группой, подобной Br, I, OSO2Me, OSO2CF3 или OSO2Ph, в присутствии основания, аналогичного NaH или K2СО3:
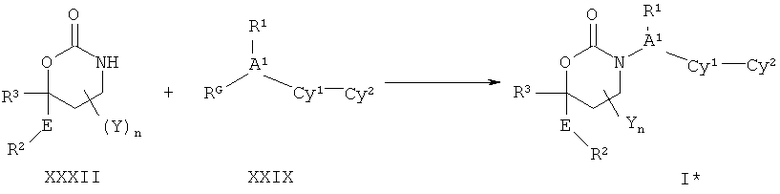
Соединения Формулы XXXII могут быть получены путем обработки соединений Формулы XII различными реактивами Формулы III, где Z1 и Z2 являются уходящими группами, подобными хлориду, 1-имидазолилу или арилоксиду в инертном растворителе, аналогичном тетрагидрофурану, СН2Сl2, толуолу или MeCN, как правило, в присутствии органического или неорганического основания, подобного триэтиламину или NaHCOs соответственно, при температуре от -10°С до 120°С:
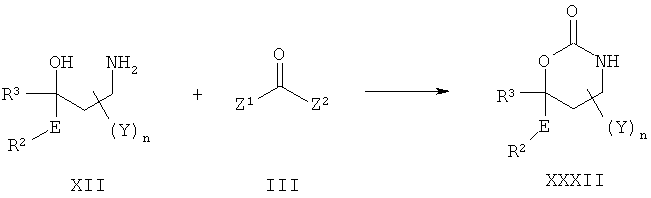
В ходе шестого процесса соединение Формулы I*, где А1 является связанным, a R1 отсутствует, может быть получено в результате реакции соединения Формулы XXXII и соединения Формулы XXII, где RB представляет собой уходящую группу, аналогичную хлоро, бромо, йодо или OSO2CF3, в присутствии основания, подобного 4-(диметиламино)пиридин K2СО3, а также медного или палладиевого катализатора в инертном растворителе, подобном диоксану, DMF или NMP при повышенной температуре:
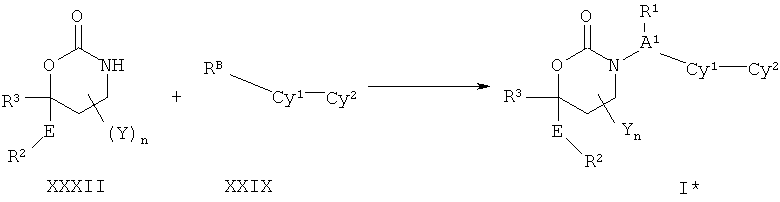
В ходе седьмого процесса соединение Формулы I* может быть получено в результате проведения реакции сочетания Сузуки для соединения Формулы XXXIV, где Су1 представляет собой арил или гетероарил, а Rx представляет собой бромо, йодо или трифторметансульфонилокси, и бориновой кислоты (RY представляет собой водород) или боронатного эфира Формулы XXXV (RY представляет собой (С1-С6)алкил, а две группы RY вместе образуют группу (С1-С12)алкилена).
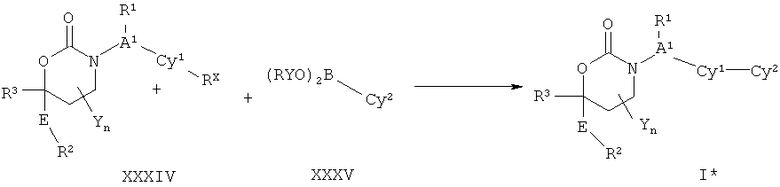
В ходе восьмого процесса из соединения Формулы XXXIV, где Су1 представляет собой арил или гетероарил, а Rx представляет собой бромо, йодо или трифторметансульфонилокси, путем реакции с бис(пинаколато)дибороном в присутствии палладиевого катализатора получают боронатный эфир Формулы XXXVI, из которого, в свою очередь, путем реакции с гетероциклическим соединением Формулы XXXVII, где Rx представляет собой бромо, йодо или трифторметансульфонилокси, также в присутствии палладиевого катализатора, получают соединение Формулы I*.
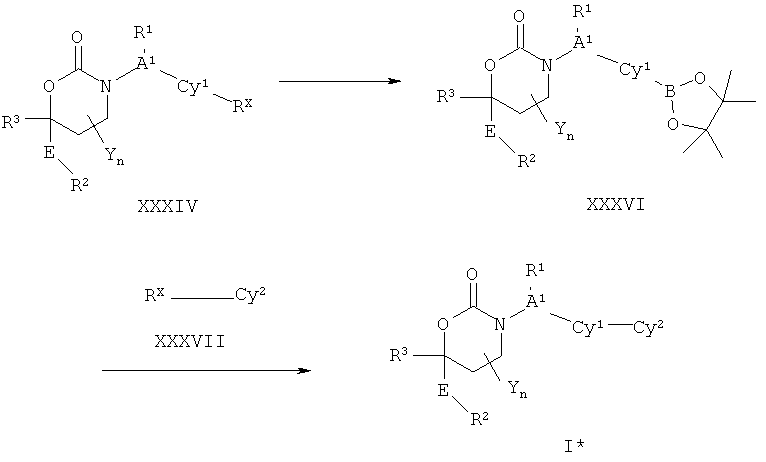
В ходе девятого процесса соединение Формулы I* может быть получено из другого соединения Формулы I*. Например:
(1) соединение Формулы I*, где R1 или R3 представляет собой -гидрокси(С2-С6)алкил, при помощи реактива Джонса может быть окислено до соединения Формулы I*, где R1 или R3 представляет собой -карбокси(С1-С6)алкил.
(2) соединение Формулы I*, где R1 или R3 представляет собой -карбокси(С1-С6)алкил, может быть связано с аммиаком или (С1-С6)алкиламином при помощи стандартного пептидного связующего вещества, аналогичного EDC для получения соединения Формулы I*, где R1 или R3 представляет собой -Н2NC(=O)(С1-С6)алкил или -{(С1-С6)алкилNНС(=O)}(С1-С6)алкил.
(3) соединение Формулы I*, где R1 или R3 представляет собой -гидрокси(С1-С6)алкил, может быть преобразовано в его метансульфонат или трифторметансульфонат посредством обработки азидом натрия с последующим восстановлением для получения соединения Формулы I*, где R1 или R3 представляет собой -амино(С1-С6)алкил.
(4) из соединения Формулы I*, где R1 или R3 представляет собой амино(С1-С6)алкил, путем реакции с уксусным ангидридом или ацетилхлоридом получают соединение Формулы I*, где R1 или R3 представляет собой {ацетиламино}(С1-С6)алкил.
(5) из соединения Формулы I*, где R1 или R3 представляет собой амино(С1-С6)алкил, путем реакции с метансульфонил хлоридом получают соединение Формулы I*, где R1 или R3 представляет собой {метансульфониламино}(С1-С6)алкил.
(6) соединение Формулы I*, где R1 представляет собой (С2-С6)алкенил, при гидроборировании образует соединение Формулы I*, где R1 представляет собой гидрокси(С2-С6)алкил.
(7) соединение Формулы I*, где R3 представляет собой (С2-С6)алкенил, при гидроборировании образует соединение Формулы I*, где R3 представляет собой гидрокси(С2-С6)алкил.
(8) из соединения Формулы I*, где R1 представляет собой (С2-С6)алкенил, путем реакции с осмия тетраоксидом и N-метилморфолин-N-оксидом получают соединение Формулы I*, где R1 представляет собой вицинальный дигидрокси(С2-С6)алкил.
(9) из соединения Формулы I*, где R3 представляет собой (С2-С6)алкенил, путем реакции с осмия тетраоксидом и N-метилморфолин-N-оксидом получают вицинальный диол Формулы I*, где R3 представляет собой вицинальный дигидрокси(С2-С6)алкил.
(10) из соединения Формулы I*, где R1 представляет собой (С2-С6)алкенил, путем реакции с озоном, а затем с NaBH4 получают соединение Формулы I*, где R1 представляет собой -гидрокси(С1-С5)алкил.
(11) из соединения Формулы I*, где R3 представляет собой (С2-С6)алкенил, путем реакции с озоном, а затем с NaBH4 получают соединение Формулы I*, где R1 представляет собой -гидрокси(С1-С5)алкил
(12) из соединения Формулы I*, где R1 или R3 представляет собой амино(С1-С6)алкил, путем реакции с (С1-С6)алкил изоцианатом получают соединение Формулы I*, где R1 или R3 представляет собой (С1-С6)алкиламинокарбониламино(С1-С6)алкил.
(13) из соединения Формулы I*, где R1 или R3 представляет собой амино(C1-С6)алкил, путем реакции с (С1-С6)алкил хлорформиатом получают соединение Формулы I*, где R1 или R3 представляет собой (C1-C6) алкиламинокарбониламино (С1-С6)алкил.
(14) из соединения Формулы I*, где R1 или R3 представляет собой амино(С1-С6)алкил, путем реакции с хлорсульфонилизоцианатом или сульфамидом получают соединение Формулы I*, где R1 или R3 представляет собой аминосульфониламино(С1-С6)алкил.
(15) из соединения Формулы I*, где R1 или R3 представляет собой амино(С1-С6)алкил, путем реакции с (С1-С6)алкилсульфамоил хлоридом получают соединение Формулы I*, где R1 или R3 представляет собой (СгСб)алкиламиносульфониламино(С1-С6)алкил.
(16) из соединения Формулы I*, где R1 или R3 представляет собой гидрокси(С1-С6)алкил, путем реакции с хлорсульфонилизоцианатом получают соединение Формулы I*, где R1 или R3 представляет собой аминосульфонилокси(С1-С6)алкил.
(17) из соединения Формулы I*, где R1 или R3 представляет собой гидрокси(С1-С6)алкил, путем реакции с p-нитрофенилхлорформиатом, пентафторфенилхлорформиатом или карбонилдиимидазолом и последующей реакции с аммиаком, (С1-С6)алкиламином или ди(С1-С6)алкиламином получают соединение Формулы I*, где R1 или R3 представляет собой аминокарбокси(С1-С6)алкил, (С1-С6)алкил аминокарбокси(С1-С6)алкил или ди(С1-С6)алкил аминокарбокси(С1-С6)алкил.
(18) из соединения Формулы I*, где R1 или R3 представляет собой гидрокси(С1-С6)алкил, путем реакции с РОСl3 получают соединение Формулы I*, где R1 или R3 представляет собой (НО)2Р(=O)O(С1-С6)алкил.
(19) из соединения Формулы I*, где R3 представляет собой аллил или гомоаллил, путем реакции с кислородом в присутствии PdCl2 и CuCl получают соединение Формулы I*, где R3 представляет собой 2-оксопропил или 3-оксобутил соответственно.
(20) из соединения Формулы I*, где R3 представляет собой 2-оксопропил или 3-оксобутил, путем реакции с MeMgX, где Х представляет собой Cl, Br или I, получают соединение Формулы I*, где R3 представляет собой 2-гидрокси-2-метилпропил или 3-гидрокси-3-метилпропил соответственно.
(21) соединение Формулы I*, где R3 представляет собой -CH2CO2Me, может быть обработано MeMgX, где Х представляет собой Cl, Br или I, для получения соединения Формулы I*, где R3 представляет собой 2-гидрокси-2-метилпропил.
(22) из соединения Формулы I*, где R3 представляет собой аллил или -СН2С(Ме)=СН2, путем гидроцианирования с помощью TsCN в присутствии трифенилсилана и различных кобальтовых катализаторов могут быть получены соединения Формулы I*, где R3 представляет собой -СН2СН(CN)Ме или -CH2CMe2CN соответственно.
(23) соединение Формулы I*, где R3 представляет собой CH2C(Me)2CN, может быть обработано ацетамидом в присутствии PdCl2 для получения соединения Формулы I*, где R3 представляет собой CH2CMe2CONH2.
(24) соединение Формулы I*, где R3 представляет собой -СН2С(Ме)=СН2, может быть обработано m-СРВА, а затем триэтилборгидридом лития для получения соединения Формулы I*, где R3 представляет собой 2-гидрокси-2-метилпропил.
В ходе десятого процесса определенные соединения Формулы изобретения I** получают следующим образом:
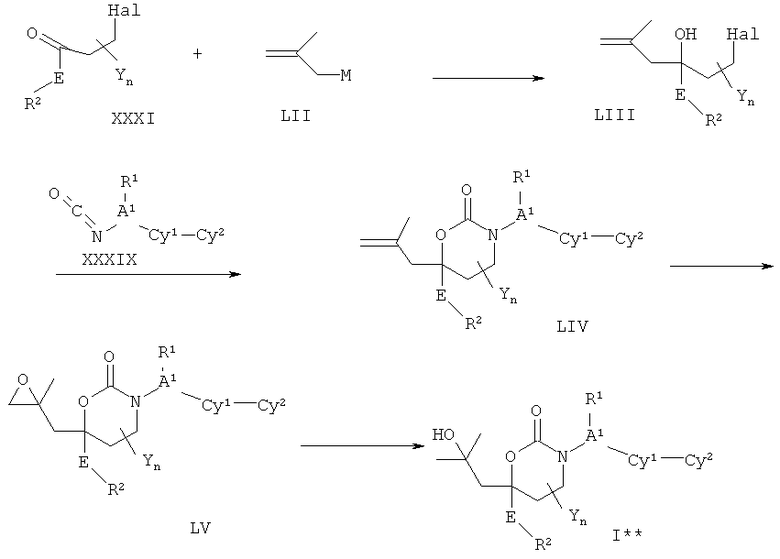
Галогенные соединения Формулы LIII могут быть получены путем обработки β-галокетонов Формулы XXXI металлорганическими реактивами Формулы LII, где М означает MgCl, MgBr, Mgl, ZnBr или Znl. Реакция при необходимости может быть проведена в присутствии безводного треххлористого церия в инертном безводном растворителе, подобном тетрагидрофурану, при температуре от -25 до 0°С в течение 0,5 ч.
Циклические карбаматы Формулы LIV могут быть получены в результате реакции между p-галоспиртами Формулы LIII, где Hal представляет собой хлорид, с изоцианатами Формулы XXXIX в присутствии основания, подобного, помимо прочего, DBU (1,8-диазабицикло[5.4.0]ундец-7-ен), в обратном конденсате инертного растворителя, аналогичного, помимо прочего, тетрагидрофурану.
Третичные спирты Формулы LVII могут быть получены из трехзамещенных алкенов Формулы LIV путем предварительного эпоксидирования алкена при помощи эпоксидирующего вещества, подобного m-СРВА (3-хлорпербензойная кислота), в инертном растворителе, аналогичном дихлорметану, для получения соответствующих эпоксидов Формулы LV. Полученный эпоксид затем подвергается восстановлению с раскрытием кольца для получения соответствующего третичного спирта I* посредством обработки концентрированным гидридным реактивом, подобным триэтилборгидриду, в безводном инертном растворителе, аналогичном тетрагидрофурану.
В ходе одного из вариантов десятого процесса соединение Формулы I*** настоящего изобретения получают путем проведения реакции сочетания Сузуки для боронатного эфира Формулы LIX с помощью галогетероциклического соединения Формулы LX.
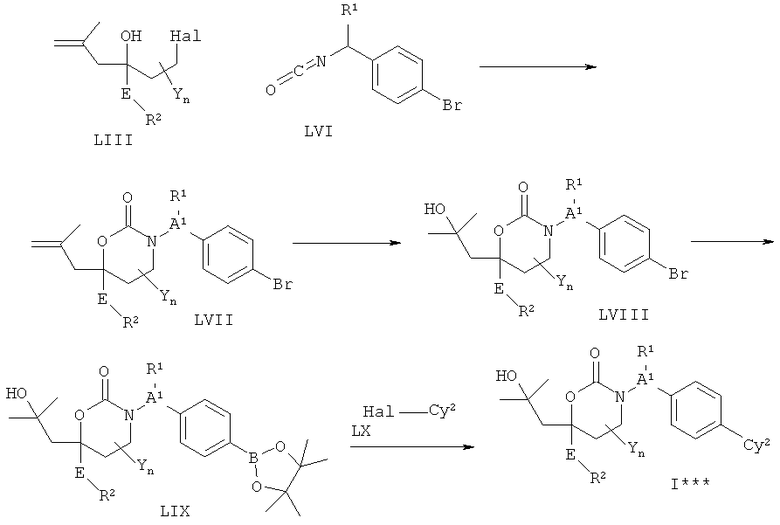
Боронатный эфир Формулы LIX получают в результате реакции бромида Формулы LVIII с бис(пинаколато)дибороном. LVIII получают путем эпоксидирования алкена LVII с последующим восстановлением с раскрытием эпоксидного кольца в соответствии с представленной выше информацией, поскольку 2-метил-2-гидроксипропильную группу получают посредством эпоксидирования и раскрытия гибридного кольца в соответствии с описанным выше преобразованием LIV в I**.
Десятый процесс более подробно описан в предварительной заявке США с серийным номером 61/137,013, поданной 25 июля 2008 года, под названием СИНТЕЗ ИНГИБИТОРОВ 11БЕТА-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ 1 ТИПА (Ид. №пат, повер. 4370.1001-000). Данная заявка приводится здесь в качестве ссылки.
МЕТОДЫ ЖХ-МС
Метод 1 [ЖХ-МС (3 мин метод)]
Колонка: Chromolith SpeedRod, RP-18e, 50 x 4,6 мм; Подвижная фаза: А: 0,01%ТРА/вода, В: 0,01%ТРА/СН3CN; Скорость потока: 1 мл/мин; Градиент:
Метод 2 (10-80)
Метод 3 (30-90)
МЕТОД 4
МЕТОД 5
СПОСОБ ПОЛУЧЕНИЯ 1
(S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
Метод 1
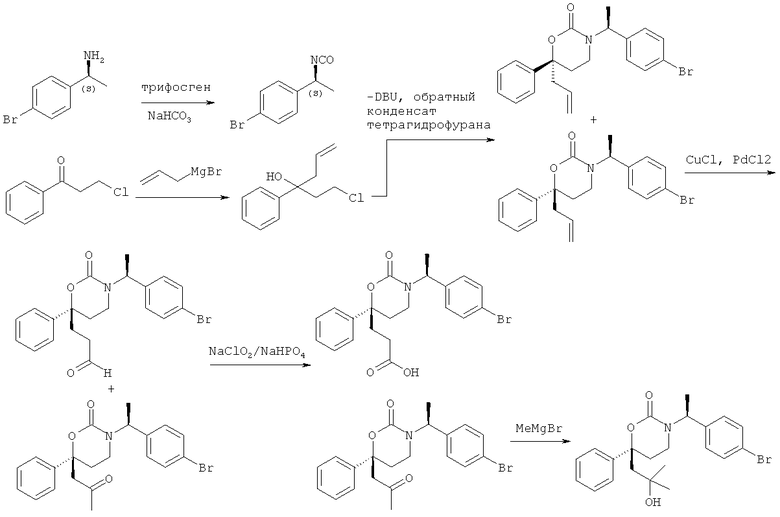
Этап 1: (S)-1-бром-4-(1-изоцианатоэтил)бензол.
В раствор (S)-1-(4-бромфенил)этанамина (240 г, 1,2 моль) в метиленхлориде (3 л) и насыщенного водного раствора NaHCO3 (3 л) был добавлен трифосген (118 г, 0,396 моль) при температуре 0°С. Смесь перемешивалась в течение 15 минут. Затем была отделена органическая фаза, которую затем высушили над Na2SO4 и выпарили для получения 1-бром-4-(1-изоцианатоэтил)-бензола (170 г, 63%).
Этап 2: 1-хлор-3-сренилгекс-5-ен-3-ол
В раствор 3-хлор-1-фенилпропан-1-она (170 г, 1,01 моль) в безводном тетрагидрофуране (1200 мл) был добавлен бромид аллилмагния (1,2 л, 1 моль/л) при температуре -78°С в азотной среде. Полученная смесь перемешивалась в течение 30 минут при температуре -78°С. Реакция была остановлена при помощи водного раствора NaHCO3. Была отделена органическая фаза, которая затем была высушена над Na2SO4 и выпарена для получения неочищенного продукта, позднее очищенного в ходе колоночной хроматографии (петролейный эфир /EtOAc=100:1) для получения 1-хлор-3-фенилгекс-5-ен-3-ола (180 г, 86%). 1H ЯМР (CDCl3): 2,27 (m, 2Н), 2,51 (m, 1H), 2,74 (m, 1H), 3,22 (m, 1H), 3,58 (m, 1H), 5,16 (m, 2Н), 5,53 (m, 1H), 7,23 (m, 1H), 7,39 (m, 4H).
Этап 3: (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-фенил-1,3-оксазинан-2-он
Смесь 1-хлор-3-фенил-гекс-5-ен-3-ол (105 г, 0,050 ммоль), (S)-(-)-1-(-бромфенил)этил изоцианата (170 г, 0,752 моль) и DBU (228 г, 1,5 моль) в тетрагидрофуране (1700 мл) в течение ночи нагревалась для обратного конденсирования. Затем смесь была разбавлена EtOAc и промыта 1N раствором HCl в воде, с последующей экстракцией водной фазы при помощи EtOAc (3×). Объединенная органическая фаза была высушена над Na2SO4. После выпаривания растворителей неочищенный продукт был очищен в ходе колоночной хроматографии (петролейный эфир/EtOAc = от 20:1 до 5:1) для получения (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-фенил-1,3-оксазинан-2-она (100 г, 34%). 1H ЯМР (CDCl3): 1,39 (d, 3Н), 2,14 (m, 1H), 2,24 (m, 2Н), 2,48-2,61 (m, 3Н), 2,82 (m, 2Н), 5,01 (m, 2Н), 5,52 (q, 1H), 5,73 (m, 1H), 6,62 (d, 2Н), 7,12 (m, 2H), 7,28 (m, 2Н).
Этап 4: (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-оксопропил)-6-фенил-1,3-оксазинан-2-он и 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропанал
В раствор (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-фенил-1,3-оксазинан-2-она (31 г, 78 ммоль) и CuCl (19,3 г, 195 ммоль) в безводном DMF (150 мл) были добавлены H2O (50 мл) и PdCl2 (4,10 г, 23 ммоль) при комнатной температуре. После добавления смесь в течение ночи перемешивалась в кислородной среде. После того как с помощью ТСХ было получено подтверждение исчезновения исходного материала, твердое вещество было подвергнуто фильтрации. Затем были добавлены вода (200 мл) и EtOAc (200 мл), органические слои были отделены, а водный слой подвергся экстракции при помощи EtOAc (3×40 мл). Объединенные органические слои были промыты солевым раствором, высушены над Na2SO4, подвержены фильтрации и выпариванию для получения осадка, который затем был очищен в ходе колоночной хроматографии (петролейный эфир/EtOAc = от 5:1 до 1:1) для получения смеси (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-оксопропил)-6-фенил-1,3-оксазинан-2-она и 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропанала, (26 г, 81%).
Этап 5: (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-оксопропил)-6-фенил-1,3-оксазинан-2-он
В смесь (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-оксопропил)-6-фенил-1,3-оксазинан-2-она и 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропанала (20 г, 48,2 ммоль) в t-BuOH (250 мл) и 2-метил-2-бутене (50 мл) был добавлен раствор NaClO2 (19,3 г, 0,213 моль) и NaH2PO4 (28 г, 0,179 моль) в H2O (300 мл) при температуре 0°С. Полученная смесь перемешивалась в течение 1 ч при температуре 0°С. Затем смесь была обработана водой (100 мл) и подверглась экстракции при помощи CH2Cl2. Объединенные органические слои были высушены над Na2SO4, подвержены фильтрации и выпариванию для получения осадка, позднее очищенного в ходе колоночной хроматографии (петролейный эфир/EtOAc = от 5:1 до 2.5:1) для получения (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-оксопропил)-6-фенил-1,3-оксазинан-2-она (10,0 г, 83%). 1Н ЯМР (CDCl3): 1,49 (d, 3Н), 2,12 (s, 3Н), 2,33 (m, 2H), 2,63 (m, 1H), 2,86-3,08 (m, 3H), 5,57 (q, 1H), 6,66 (d, 2H), 7,19 (m, 2H), 7,33 (m, 5H).
Этап 6: (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
В раствор (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-оксопропил)-6-фенил-1,3-оксазинан-2-она (20 г, 46,4 ммоль) в безводном тетрагидрофуране (200 мл) был добавлен по капле бромид метилмагния (31 мл, 144 ммоль) при температуре -78°С в азотной среде. Затем смесь перемешивалась при комнатной температуре в течение 1 ч. Реакция смеси была остановлена раствором NaHCO3 в воде (50 мл) под водяной баней со льдом. Органические слои были отделены, а водный слой подвергся экстракции при помощи EtOAc (150 мл). Объединенные органические слои были промыты, высушены над Na2SO4 и выпарены в вакууме для получения неочищенного продукта, позднее очищенного в ходе колоночной хроматографии (петролейный эфир/EtOAc = от 5:1 до 2:1) для получения (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (13 г, 65%). После повторной кристаллизации из EtOH было получено 4 г чистого соединения. 1H ЯМР (CDCl3): 1,06 (s, 3Н), 1,12 (s, 3Н), 1,44 (d, 3Н), 2,14 (m, 3Н), 2,21 (m, 1H), 2,33 (m, 1H), 2,76 (m, 1H), 5,54 (q, 1H), 6,74 (d, 2H), 7,16 (d, 2H), 7,28 (m, 5H).
Альтернативная процедура для 2 этапа 1 метода
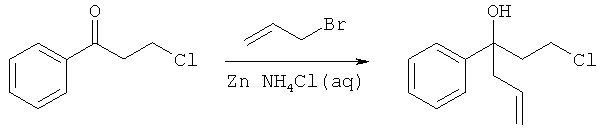
Раствор 3-хлор-1-фенилпропан-1-она (100 г, 0,595 иоль) в тетрагидрофуране (280 мл) были добавлен по капле в тщательно перемешанную смесь цинкового порошка (неактивированного) (40 г, 1,231 моль, насыщ. водного раствора NH4Cl (1500 мл) и тетрагидрофурана (400 мл). Бромид аллила (143 г, 1,19 моль) был растворен в тетрагидрофуране (200 мл) и медленно добавлен в реакционную смесь. Реакция происходила с незначительным выделением тепла и неожиданным обратным конденсированием. После прекращения обратного конденсирования смесь перемешивалась в течение 1 ч. Затем смесь подверглась экстракции при помощи EtOAc, была высушена над безводным Na2SO4 и выпарена для получения 1-хлор-3-фенилгекс-5-ен-3-ола (122 г, 97%). 1H ЯМР: (400 МГц, CDCl3): δ=2,24(s, 1Н), 2,34 (m, 2Н), 2,53 (m, 1H), 2,75 (m, 1H), 3,20 (m, 1H), 3,58 (m, 1H), 5,18 (t, 1H), 5,51 (m, 1H), 7,26 (m, 1H), 7,26-7,39 (m, 3Н).
(R)-6-аллил-3-((S)-1-(4-бромфенил)пропил)-6-фенил-1,3-оксазинан-2-он был получен из (S)-1-(4-бромфенил)пропан-1-амина посредством процедур, аналогичных описанным в рамках 1-3 этапов 1 метода 1 способа получения.
(S)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-он был получен из (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-она посредством процедур, аналогичных описанным в рамках 4 и 6 этапов 1 метода 1 способа получения.
Метод 2
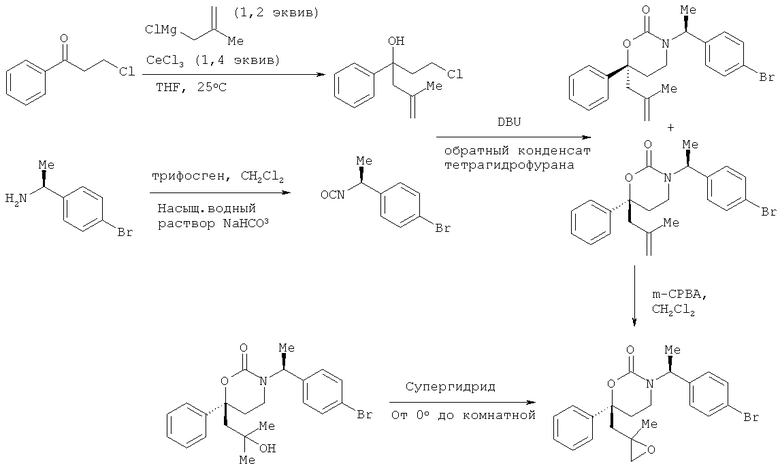
Этап 1. 1-хлор-5-метил-3-фенил-гекс-5-ен-3-ол
В перемешанную суспензию магниевой стружки (46,7 г, 1,94 моль) в 1500 мл тетрагидрофурана (H2O<100 промилле при титровании по методу Карла Фишера) в азотной среде при комнатной температуре было добавлено 53,0 мл раствора 1 М DIBAL-Н в гексане. Затем в полученную смесь, внутренняя температура которой поддерживалась на уровне ниже 30°С, был добавлен 3-хлор-2-метилпроп-1-ен (160 г, 1,77 моль). Полученный раствор встряхивали в течение 2 ч при комнатной температуре, а затем титровали в присутствии 1,1'-бипиридина для получения 0,8 М соответствующего реактива Гриньяра. В сухую колбу, содержащую 307,0 г безводного CeCl3 (1,25 моль), в азотной среде при комнатной температуре было добавлено 1556,8 мл реактива Гриньяра (0,8 М, 1,25 моль). Полученная суспензия была охлаждена до -10°С и ее встряхивали в течение 0,5 ч. В суспензию, внутренняя температура которой поддерживалась на уровне ниже 0°С, был добавлен раствор 200 г 3-хлор-1-фенилпропан-1-она (1,19 моль) в 200 мл тетрагидрофурана, после чего полученная смесь перемешивалась в течение 0,5 ч. Затем в полученную смесь, внутренняя температура которой также поддерживалась на уровне ниже 30°С, было добавлено 1200 мл водного раствора 1 М HCl для получения прозрачного раствора. После разделения фаз была проведена экстракция водного слоя при помощи EtOAc (500 мл). Объединенные органические слои были промыты солевым раствором и высушены над сульфатом натрия. В результате удаления растворителя с помощью вакуума был получен неочищенный 1-хлор-5-метил-3-фенил-гекс-5-ен-3-ол, который растворялся в тетрагидрофуране до получения Н2О<500 промилле при титровании по методу Карла Фишера. Неочищенный продукт (306 г, массовая концентрация в процентах - 83%, выход - 95%) использовался непосредственно в ходе 3 этапа. 1Н-ЯМР спектроскопия (500 МГц, CDCl3) δ 7,38-7,37 (d. J=7,8 Гц, 2Н), 7,33 (t, J=7,9 Гц, 2Н), 7,24 (t, J=7,4 Гц, 1Н), 4,91 (s, 1H), 4,76 (s, 1H), 3,57 (ddd, J=5.6, 10,7, и 10,7, 1Н), 3.13 (ddd, J=4,7, 10,7 и 10,7 Гц, 1H), 2,66 (d, J=13,3 Гц, 1H), 2,54 (d, J=11,3 Гц, 1H), 2,53 (s, 1H), 2,36 (ddd, J=5,4, 10,6 и 13,9 Гц. 1H), 2,29 (ddd, J=5,6, 11,3 и 13,3 Гц, 1H), 1,29 (s, 3Н), 13С-ЯМР спектроскопия (125 МГц, CDCl3) δ 144,3, 141,4, 128,0, 126,6, 124,8, 116,1, 74,2, 51,2, 46,0, 39,9, 23,9.
Этап 2. 1-Бром-4-((S)-1-изоцианато-этил)-бензол
В реактор с рубашкой емкостью 10 л был помещен 241 г бикарбоната натрия (2,87 моль, 2,30 эквив.) и 5 л деионизированной воды. Полученный раствор встряхивался в течение 10-20 мин до полного растворения и получения однородного раствора. В прозрачный раствор затем было добавлено 250 г (1,25 моль, 1,00 эквив.) (S)-(-)-1-(4-бромфенил)этиламина в виде раствора в 1,00 л дихлорметана. После этого в реактор было дополнительно помещено 4 л дихлорметана. Полученный двухфазный раствор встряхивался и охлаждался до Тнач=2-3°С. Трифосген (126 г, 424 ммоль, 0,340 эквив.) добавлялся в реактор двумя приблизительно равными порциями с перерывом в 6 мин. Необходимо отметить, что при добавлении трифосгена наблюдалось незначительное выделение тепла. Полученный мутный раствор встряхивался при Тнач=2-5°С в течение 30 мин, после чего анализ ВЭЖХ показал преобразование >99 А% (220 нм). Слой дихлорметана был отделен и высушен при помощи безводного сульфата. Полученный раствор был пропущен через пробку из целита и выпарен до ~1,5 л с последующим формированием мелких твердых частиц белого цвета. Раствор подвергся фильтрации и выпариванию в вакууме до образования густого масла для получения 239 г 1-бром-4-((S)-1-изоцианато-этил)-бензола (массовая концентрация в процентах - 93,7%, выход -79,4%). 1H-ЯМР спектроскопия (400 МГц, CD2Cl2) δ 7,53 (d, J=11,4 Гц, 2Н), 7,26 (d, J=8,2 Гц, 2Н), 4,80 (q, J=6,7 Гц, 1H), 1,59 (d, J=6,7 Гц, 3Н). Материал использовался в ходе 3 этапа без дополнительной очистки.
Этап 3. (R)-3-((S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-фенил-1,3-оксазинан-2-он
В высушенный реактор с рубашкой емкостью 10 л в азотной среде были помещены 1-хлор-5-метил-3-фенил-гекс-5-ен-3-ол (167 г, массовая концентрация в процентах - 81,7%, 610 ммоль, 1,00 эквив.), 1-бром-4-((S)-1-изоцианато-этил)-бензол (219 г, массовая концентрация в процентах - 93,7%, 911 ммоль, 1,50 эквив.), безводный тетрагидрофуран (3,00 л), а затем 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU, 409 мл, 2,73 моль, 4.50 эквив.). Полученный раствор встряхивался и подвергался обратному конденсированию (Твнутр=67-69°С, Твнеш=75°С) в течение 19 ч, после чего анализ ВЭЖХ показал преобразование ~ 1А% (220 нм) оставшегося 1-хлор-5-метил-3-фенил-гекс-5-ен-3-ола. Полученный раствор темного цвета был охлажден до Твнутр=20-25°С. Два литра тетрагидрофурана были извлечены из раствора посредством вакуумной дистилляции. Оставшийся раствор темного цвета был разбавлен при помощи 4,0 л этилацетата и 1,0 л гексанов. Полученный раствор был промыт 4,0 л 1,0 М водного раствора водорода хлорида (примечание: при промывании наблюдается незначительное выделение тепла). Водный раствор затем был отделен, а оставшийся органический раствор был высушен при помощи безводного сульфата натрия, подвергся фильтрации и выпариванию в вакууме до образования густого масла. Полученный таким образом материал был подвергнут очистке флэш-хроматографией на кремнии (5-30% этилацетата/гексанов, 1,74 кг кремния) для получения 137,8 г материала (массовая концентрация в процентах - 59%, соотношение диастереомеров - 3:1:1 в пользу целевого диастереомера (R)-3-((S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-фенил-1,3-оксазинан-2-она, выход - 32,3%). Материал использовался в ходе 4 этапа без дополнительной очистки.
Данные анализа (R)-3-((S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-фенил-1,3-оксазинан-2-она: 1Н-ЯМР спектроскопия (500 МГц, CD2Cl2) δ 7,42-7,35 (m, 3H), 7,33-7,31 (m, 2Н), 7,25-7,23 (m, 2H), 6,80-6,74 (m, 2), 5,55 (q, J=7,1 Гц, 1Н), 5,37-5,36 (m, 1H), 4,89 (s, 1H), 4,69 (s, 1H), 2,96-2,93 (m, 1H), 2,61 (dd, J=13,8 и 26,4 Гц, 2H), 2,37-2,25 (m, 3H), 1,68 (s, 3H), 1,50 (d, J=7,1 Гц, 3H). 13С-ЯМР спектроскопия (125 МГц, CD2Cl2) δ 152,5, 141,5, 140,1, 138,3, 130,6, 128,1, 128,0, 126,9, 124,4, 120,2, 115,3, 82,4, 52,1, 50,1, 35,6, 29,8, 23,4, 14,5.
Данные анализа (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-фенил-1,3-оксазинан-2-она: 1H-ЯМР спектроскопия (400 МГц, CD2Cl2) δ 7,50-7,48 (m, 2H), 7,43-7,39 (m, 2H), 7,35-7,32 (m, 3H), 7,20-7,18 (m, 2H), 5,60 (q, J=7,1 Гц, 1H), 4,85 (s, 1H), 4,66 (s, 1H), 2,73-2,67 (m, 2H), 2,60 (dd, J=13,9 и 19,4 Гц, 2H), 2,28 (dt, J=3,3 и 13,7 Гц, 1H), 2,14-2,05 (m, 1H), 1,66 (s, 3H), 1,24 (d, J=7,2 Гц, 3Н). 13С-ЯМР спектроскопия (100 МГц, CD2Cl2) δ 153,4, 142,5, 141,0, 140,1, 131,8, 129,3, 128,9, 127,8, 125,3, 121,5, 116,3, 83,9, 53,2, 51,0, 36,6, 31,3, 24,3, 15,4.
Этап 4. (6S)-3-((S)-1-(4-бромфенил)этил)-6-((2-метилоксиран-2-ил)метил)-6-фенил-1,3-оксазинан-2-он
В круглодонную колбу с двумя горлышками емкостью 1,0 л были помещены (R)-3-((S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-фенил-1,3-оксазинан-2-он (135,8 г, массовая концентрация в процентах - 59%, соотношение диастереомеров - 3:1:1, 193 ммоль, 1,00 эквив.), дихлорметан (700 мл), а затем 3-хлорпербензойная кислота (m-СРВА, 70%, 95,3 гр, 386 ммоль, 2,0 эквив,). Полученный раствор встряхивался при комнатной температуре (Тнач=20-25°С) в течение 1 ч, после чего анализ ВЭЖХ показал преобразование >99 А% (220 нм). Полученный раствор был разбавлен 700 мл метил-трет-бутилового эфира (МТВЕ) и промыт 1×500 мл раствора тиосульфата натрия (массовая концентрация в процентах - 30%) и 1×500 мл насыщенного водного раствора бикарбоната натрия. Промывание повторялось в той же последовательности до тех пор, пока пик органического раствора, определенный посредством следового анализа ВЭЖХ, т.е. пик, соответствующий пробе m-СРВА для ВЭЖХ, не составит <2,5 А% (220 нм). В данном случае для получения указанного результата потребовалось повторить промывание три раза. Полученный таким образом органический слой был высушен при помощи безводного сульфата натрия, подвергся фильтрации и выпариванию в вакууме до образования густого масла. Полученный материал был разбавлен 200 мл безводного тетрагидрофурана, а затем выпарен в вакууме до образования густого масла для получения (6S)-3-((S)-1-(4-бромфенил)этил)-6-((2-метилоксиран-2-ил)метил)-6-фенил-1,3-оксазинан-2-она, который использовался в ходе 5 этапа без дополнительной очистки.
Этап 5. (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
В круглодонную колбу с тремя горлышками емкостью 2,0 л, высушенную в сушильном шкафу, были помещены неочищенный (6S)-3-((S)-1-(4-бромфенил)этил)-6-((2-метилоксиран-2-ил)метил)-6-фенил-1,3-оксазинан-2-он и 750 мл безводного тетрагидрофурана. Полученный раствор встряхивали и охлаждался до Тнач=2-3°С. После этого в прозрачный раствор добавлялся 1,0 М раствор триэтилборгидрида в тетрагидрофуране (Супергидрид, 348 мл, 348 ммоль, 1,8 эквив.) В ходе добавления наблюдалось выделение тепла, поэтому температура добавляемого раствора поддерживалась на уровне Тнач=<8°С. Полученный раствор встряхивали при Тнач=2-3°С в течение 1,5 ч, после чего был оставлен на 2,5 ч для нагревания до Тнач=10-13°С. При этом ВЭЖХ анализ показал преобразование ~94 А% (220 нм). Затем к полученному раствору был добавлен раствор перекиси водорода (95,7 мл водного раствора с массовой концентрацией в процентах 35%, разбавленного 400 мл воды, 1,08 моль, 5,60 эквив.). При добавлении наблюдалось значительное выделение тепла, поэтому температура добавляемого раствора поддерживалась на уровне Тнач=<25°С. Полученный раствор был разбавлен 1,00 л метил-терт-бутилового эфира (МТВЕ) и промыт сначала 1,00 л воды, а затем 500 мл раствора тиосульфата натрия с массовой концентрацией в процентах ~30%. Органический раствор был высушен при помощи безводного сульфата натрия, подвергся фильтрации и выпариванию в вакууме. Полученный таким образом материал был подвергнут очистке флэш-хроматографией на кремнии (10-60% этилацетата, 600 г кремния) для получения 68 г материала, состоящего из обоих диастереомеров (соотношение диастереомеров 1,98:1) и 41 г целевого диастереомера (соотношение диастереомеров >99:1). Материал, состоящий из смешанных фракций, был рекресталлизирован из 250 мл изопропил ацетата (IPAC) и 200 мл гептана (антирастворитель) для получения после фильтрации 31,3 г продукта (95,7 А% при 220 нм, соотношение диастереомеров 74:1). Два образца были соединены вместе для получения 72,3 г (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (выход в ходе двухэтапной процедуры составил 83,6%). 1Н-ЯМР спектроскопия (400 МГц, CDCl3) δ 7,37-7,29 (m, 5H), 7,25-7,21 (m, 2H), 6,82-6,79 (m, 2H), 5,61 (q, J=6,9 Гц, 1H), 2,83 (add, J=2,5, 5,4 and 11,6 Гц, 1Н), 2,39 (ddd, J=5,7, 12.0 и 14,1 Гц, 1H), 2,27 (ddd, J=2,6, 4,8 и 14,0 Гц, 1H), 2,21-2,14 (m, 3H), 2,08 (s, 1H), 1,49 (d, J=7,0 Гц, 3H), 1,18 (s, 3H), 1,13 (s, 3H). 13С-ЯМР спектроскопия (100 МГц, CDCl3) δ 153,2, 142,6, 138,5, 131,6, 129,13, 129,10, 128,0, 125,3, 121,6, 84,2, 71,4, 54,1, 53,3, 36,4, 33,6, 32,1, 30,8, 15,6.
СПОСОБ ПОЛУЧЕНИЯ 2
(S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборол-ан-2-ил)фенил)этил)-1,3-оксазинан-2-он
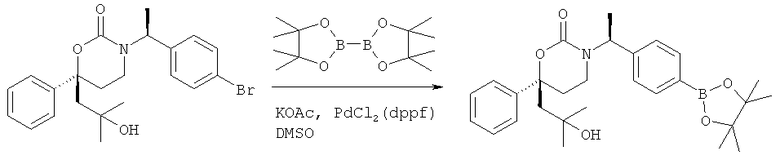
В раствор (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (6,6 г, 15,2 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана (6,1 г, 24,3 ммоль) в сухом DMSO (20 мл) был добавлен КОАс (4,8 г, 48,6 ммоль) и Pd(dppf)cl2 (372 мг, 0.46 ммоль). После добавления смесь была оставлена на 20 ч для нагревания до 100°С. Результаты проведенной ТСХ показали исчезновение исходного материала. Твердое вещество было отфильтровано, затем добавлены вода (60 мл) и EtOAc (20 мл). Было произведено разделение слоев с последующей экстракцией водного слоя при помощи EtOAc (3×15 мл). Объединенный органический слой был промыт солевым раствором и высушен над Na2SO4, после чего подвергся фильтрации и выпариванию для получения осадка, который затем был очищен посредством колоночной хроматографии для получения (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборол-ан-2-ил)фенил)этил)-1,3-оксазинан-2-она (4,4 г, 60%).
(S)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-он был получен из (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-(4-фторфенил)-1,3-оксазинан-2-она в результате аналогичной процедуры.
(S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропил)-1,3-оксазинан-2-он был получен из (S)-3-((S)-1-(4-бромфенил)пропил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она в результате аналогичной процедуры.
(R)-6-Метоксиметил-6-фенил-3-{(S)-1-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-этил}-[1,3]оксазинан-2-он был получен из 3-[1-(4-бром-фенил)-этил]-6-метоксиметил-6-фенил-[1,3]оксазинан-2-она в результате аналогичной процедуры.
СПОСОБ ПОЛУЧЕНИЯ 3
3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрил
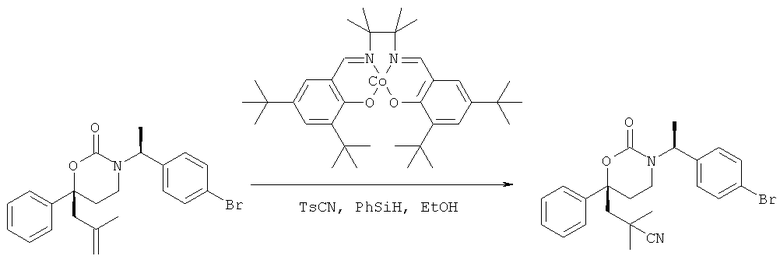
Получение комплекса кобальта(II)
В колбу емкостью 50 мл были помещены N,N'-бис(3,5-ди-трет-бутилсалицилиден)-1,1,2,2-тетраметилендиамин (0,430 г, 0,78 ммоль, 1,0 эквив.), EtOH (17 мл) и Co(OAc)2 (0,139 г, 0,78 ммоль, 1,0 эквив.). Смесь была дегазирована, после чего нагрета в азотной среде в течение 3 ч для обратного конденсирования и охлаждена до комнатной температуры. Полученный осадок подвергся фильтрации, после чего фиолетовое твердое вещество было промыто EtOH (10 мл) и высушено в высоком вакууме для получения 0,353 г (75%) комплекса кобальта(II).
Смесь (R)-3-((S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-фенил-1,3-оксазинан-2-она (490 мг, 1,18 ммоль), комплекса кобальта(II) (описание способа получения представлено в предыдущем абзаце) (8 мг, 0,01 эквив.), TsCN (257 мг, 1,2 эквив.) и PhSiH3 (137 мг, 157 мкл, 1,07 эквив.) в этаноле (10 мл) перемешивалась в течение 4 ч при комнатной температуре. После извлечения в вакууме растворителя остаток смеси был очищен с помощью колоночной хроматографии с применением 40 г силикагеля и подвергся элюированию с помощью 25-80% градиента EtOAc в гексане для получения 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила (267 мг, выход - 51%). Метод ЖХ-МС (3 мин метод) tR=1,89 мин., масса/заряд (m/z) 441, 443 (М+1).
СПОСОБ ПОЛУЧЕНИЯ 4
2,2-диметил-3-((R)-2-оксо-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)пропаннитрил
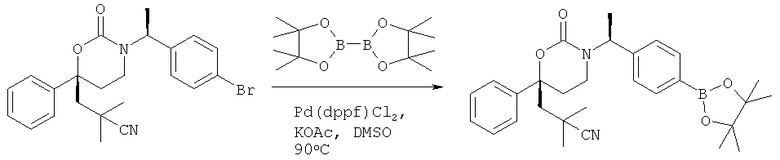
3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрил (467 мг, 1,06 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (538 мг, 2 эквив.), КОАс (333 мг, 3,2 эквив.), PdCl2(dppf)CH2Cl2 (27 мг, 0,033 эквив.) были смешаны с сухим DMSO (6 мл). Смесь подверглась дегазации с повторным насыщением газообразным N2 три раза. Затем смесь в течение ночи нагревалась при температуре 90°С с применением в качестве защитного газа газообразного N2. После охлаждения до комнатной температуры смесь была разбавлена EtOAc (30 мл) и промыта водой (20 мл), после чего водный слой подвергся экстракции при помощи EtOAc (2×15 мл). Объединенный органический слой был промыт водой (15 мл), солевым раствором (2×10 мл) и высушен над Na2SO4, после чего подвергся фильтрации и выпариванию для получения осадка, который затем был очищен посредством колоночной хроматографии для получения 2,2-диметил-3-((R)-2-оксо-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)пропаннитрил (393 мг, выход - 76%).
СПОСОБ ПОЛУЧЕНИЯ 5
3-((R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-2-оксо-1,3-оксазинан-6-ил)-2-метилпропаннитрил
Метод 1
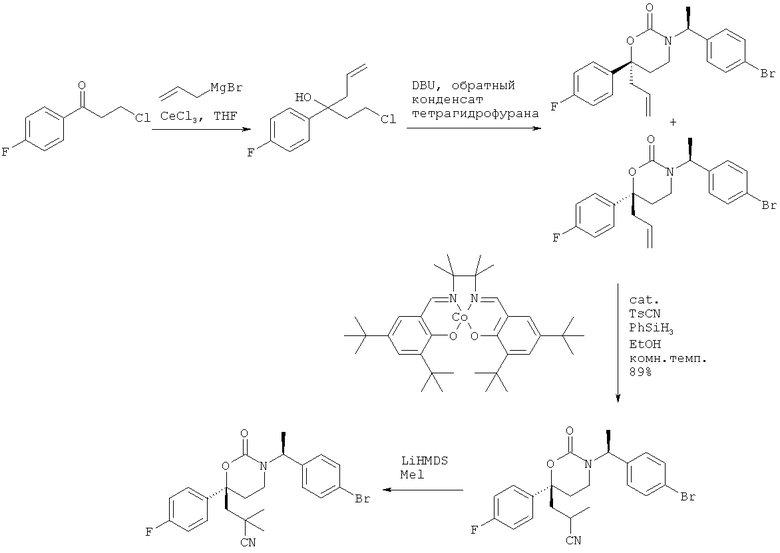
Этап 1. 1-хлор-3-(4-фторфенил)гекс-5-ен-3-ол
В колбу емкостью 250 мл были помещены безводный CeCl3 (5,58 г, 22,6 ммоль) и тетрагидрофуран (40 мл). Смесь была тщательно перемешана в течение 3,5 ч при комнатной температуре Суспензия была охлаждена до -78°С, после чего в нее был добавлен раствор бромида аллилмагния (1,0 М в тетрагидрофуране, 21 мл, 21,0 ммоль). После перемешивания в течение 2 ч при температуре -78°С в суспензию посредством канюли был добавлен раствор 3-хлор-1-(4-фторфенил)пропан-1-она (2,522 г, 13,5 ммоль) в тетрагидрофуране (30 мл). Реакционная смесь была оставлена на ночь (18 ч) для медленного охлаждения до 8°С посредством постоянного перемешивания. Затем реакция была остановлена посредством добавления насыщенного водного раствора NaHCO3 с последующей экстракцией водной фазы при помощи EtOAc и высушиванием над Na2SO4. После выпаривания растворителей осадок был очищен с помощью хроматографии с применением силикагеля и подвергся элюированию с помощью смеси гексанов/EtOAc для получения 1-хлор-3-(4-фторфенил)гекс-5-ен-3-ола (3,0049 г, 97%) в виде масла. Метод ЖХ-МС 1 tR=1,79 мин, масса/заряд 213, 211 (М-ОН)+; 1H ЯМР (400 МГц, CDCl3) 7,37-7,32 (m, 2Н), 7,07-7,02 (m, 2Н), 5,57-5,47 (m, 1H), 5,20-5,19 (m, 1H), 5,16 (m, 1H), 3,59-3,52 (m, 1H), 3,24-3,18 (m, 1H), 2,70 (dd, J=13,8, 5,9 Гц, 1Н), 2,50 (dd, J=13,8, 8,5 Гц, 1H), 2,29 (t, J=7,9 Гц, 2Н), 2,22 (s, 1H); 19F ЯМР (376 МГц, CDCl3) -116,52 (m).
Этап 2. (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-он и (S)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-он.
Смесь 1-хлор-3-(4-фторфенил)гекс-5-ен-3-ола (0,4129 г, 1,8 ммоль, 1,0 эквив.), (S)-(-)-1-(-бромфенил)этил изоцианата (0,5005 г, 2,2 ммоль, 1,2 эквив.), и DBU (0,7375 г, 4,8 ммоль, 2.7 эквив.) в тетрагидрофуране (10 мл) нагревалась для обратного конденсирования в течение 25 ч. Смесь была разбавлена EtOAc и промыта 1 N водным раствором HCl с последующей экстракцией водной фазы при помощи EtOAc (2×). Объединенная органическая фаза была высушена над Na2SO4. Полученный после испарения растворителей неочищенный продукт использовался в ходе следующего этапа без дополнительной очистки.
Аналитическая проба была очищена с помощью хроматографии с применением силикагеля и подверглась элюированию с помощью смеси гексанов/EtOAc для получения двух диастереомеров 6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-она.
Изомер 1: (S)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-он. ЖХ-МС метод 1 tR=2,03 мин, масса/заряд 420, 418 (МН+); 1H ЯМР (400 МГц, CDCl3) 7,46 (d, J=8,2 Гц, 2Н), 7,31-7,28 (m, 2Н), 7,17 (d, J=8,2 Гц, 2Н), 7,07 (t, J=8,5 Гц, 2Н), 5,76-5,66 (m, 2Н), 5,10-4,99 (m, 2Н), 2,75-2,52 (m, 4H), 2,23-2,19 (m, 1H), 2,08-2,00 (m, 1H), 1,24 (d, J=7,0 Гц, 3Н); 19F ЯМР (376 МГц, CDCl3) -115,07 (m).
Изомер 2: (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-он. ЖХ-МС метод 1 tR=1,98 мин, масса/заряд 420, 418 (МН+); 1H ЯМР (400 МГц, CDCl3) 7,25-7,20 (m, 4H), 7,05-7,01 (m, 2Н), 6,71 (d, J=8,5 Гц, 2Н), 5,74-5,64 (m, 1H), 5,58 (q, J=7,0 Гц, 1H), 5,09-4,99 (m, 2Н), 2,92-2,87 (m, 1H), 2,63-2,50 (m, 2Н), 2,33-2,16 (m, 3Н), 1,47 (d, J=7,0 Гц, 3Н); 19F ЯМР (376 МГц, CDCl3) -114,91 (m).
Этап 3
Смесь (R)-6-аллил-3-((S)-1 -(4-бромфенил)этил)-6-(4-фторфенил)-1.3-оксазинан-2-она (1,067 г, 2,55 ммоль, 1,0 эквив.), описанного в способе приготовления 3 катализатора из кобальта(II) (0,016 г, 0,0264 ммоль, 0,010 эквив.), TsCN (0,555 г, 3,06 ммоль, 1,2 эквив.) и PhSiH3 (0,294 г, 2,72 ммоль, 1,07 эквив.) в EtOH (5 мл) перемешивалась при комнатной температуре в течение 4 ч. Полученный в результате удаления в вакууме растворителя осадок был очищен посредством хроматографии с применением силикагеля и подвергся элюированию с помощью смеси гексанов/этилацетата для получения 1,0130 г (89%) 3-((R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-2-оксо-1,3-оксазинан-6-ил)-2-метилпропаннитрила в виде твердого вещества. ЖХ-МС tR=1,83, 1,86 мин, длительность хроматографии 3 мин, масса/заряд 445, 447 (MH+); 1H ЯМР (400 МГц, CDCl3) 7,32-7,22 (m, 4H), 7,13-7,05 (m, 2H), 6,80-6,73 (m, 2H), 5,60-5,56 (m, 1H), 3,00-1,94 (m, 7H), 1,51-1,49 (m, 3H), 1,35-1,32 (m, 1.5H), 1,27-1,24 (m, 1,5H); 19F ЯМР (376 МГц, CDCl3) -113,08 (m), -113,69 (m).
Этап 4
В раствор 3-((R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-2-оксо-1,3-оксазинан-6-ил)-2-метилпропаннитрила (0,332 г, 0,746 ммоль) и Mel (1,40 г, 13 эквив.) в тетрагидрофуране (12 мл) при температуре -78°С было добавлено 2,4 мл (2,4 ммоль, 3,2 эквив.) 1,0 М раствора LiHMDS в тетрагидрофуране. Полученная смесь перемешивалась в течение ночи с медленным повышением температуры до температуры окружающей среды. Реакция реакционной смеси была остановлена с помощью солевого раствора (1 мл), разбавленеа CH2Cl2 и высушена над Na2SO4. После испарения растворителей полученный осадок был очищен посредством обратно-фазовой ВЭЖХ (колонка SunFire™ Prep C18 OBD™ 5 мкм 19×50 мм, 10% →90% CH3CN/H2O, 0,1% CF3COOH в течение 8 мин, затем 90% CH3CN/H2O, 0,1% CF3COOH в течение 2 мин, скорость потока 20 мл/мин) для получения 0,255 г (74%) 3-((R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-2-оксо-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила. Метод ЖХ-МС 1 tR=1,89 мин, масса/заряд 459, 461 (MH+); 1Н ЯМР (400 МГц, CD3OD) 7,31-7,27 (m, 2H), 7,22-7,18 (m, 2H), 7,04-6,99 (m, 2H), 6,83 (d, J=8,2 Гц, 2H), 5,41 (q, J=7,0 Гц, 1H), 3,02-2,97 (m, 1H), 2,42-2,36 (m, 1H), 2,29-2,08 (m, 4H), 1,42 (d, J=7,0 Гц, 3H), 1,30 (s, 3H), 1,22 (s, 3H); 19F ЯМР (376 МГц, CD3OD) -116,50 (m).
Метод 2
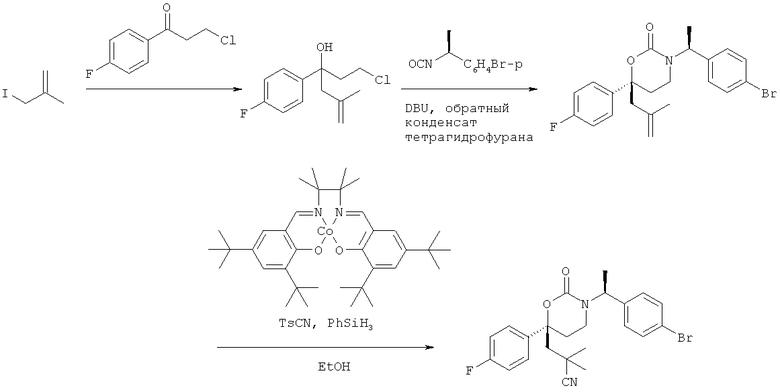
Этап 1
Раствор 3-хлор-1-(4-фторфенил)-пропан-1-она (18,6 г, 0,1 моль) в тетрагидрофуране (50 мл) был добавлен в тщательно перемешанную суспензию цинкового порошка (13 г, 0,2 моль) в смеси водного насыщенного раствора NH4Cl (260 мл) и тетрагидрофурана (65 мл). Затем по капле был добавлен раствор 3-йод-2-метилпроп-1-ен (36,4 г, 0,2 моль) в тетрагидрофуране (50 мл). В ходе реакции наблюдалось незначительное выделение тепла и внезапное обратное конденсирование. После окончания обратного конденсирования смесь перемешивалась в течение 1 ч. ТСХ показала незавершенность реакции 3-хлор-1-(4-фторфенил)пропан-1-она. В смесь был добавлен раствор 3-йод-2-метилпроп-1-ен (18,2 г, 0,1 моль) в тетрагидрофуране (30 мл), после чего смесь перемешивалась при комнатной температуре в течение ночи. Затем смесь подвергалась экстракции при помощи EtOAc (2×500 мл). Объединенный органический слой был высушен и выпарен. Полученный осадок был очищен посредством колоночной хроматографии с применением силикагеля, а затем подвергся элюированию с помощью смеси петролейного эфира/ EtOAc 50:1→30:1→5;1, для получения 1-хлор-3-(4-фторфенил)-5-метилгекс-5-ен-3-ола (17 г, выход - 76%) в виде масла.
Этап 2
Смесь 1-хлор-3-(4-фторфенил)-5-метигекс-5-ен-3-ола (3,15 г, 13 ммоль), (S)-(-)-1-(-бромфенил)этил изоцианата (3,5 г, 16 ммоль) и DBU (8 г, 33 ммоль) в тетрагидрофуране (80 мл) нагревалась для обратного конденсирования в течение 25 ч. Смесь была разбавлена EtOAc и промыта 1N водным раствором HCl. Затем была произведена экстракция при помощи EtOAc (3×). Объединенная органическая фаза была высушена над Na2SO4. После испарения растворителей неочищенный продукт подвергся очистке посредством колоночной хроматографии для получения (R)-3-((S)-1-(4-бромфенил)-этил)-6-(4-фторфенил)-6-(2-метилаллил)-1,3-оксазинан-2-она (2,13 г, выход - 38%).
Этап 3
Смесь (R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-(2-метилаллил)-1,3-оксазинан-2-она (2,13 г, 4,9 ммоль), описанного в способе приготовления 3 катализатора из кобальта(II) (0,032 г, 0,053 ммоль), TsCN (1,11 г, 6,12 ммоль) и PhSiH3 (0,6 г, 5,54 ммоль) в EtOH (10 мл) перемешивалась при комнатной температуре в течение 8 ч. Полученный в результате удаления в вакууме растворителя осадок был очищен посредством колоночной хроматографии для получения 3-((R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-2-оксо-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила (1,84 г, 81,1%).
СПОСОБ ПОЛУЧЕНИЯ 6
3-((R)-6-(4-фторфенил)-2-оксо-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрил
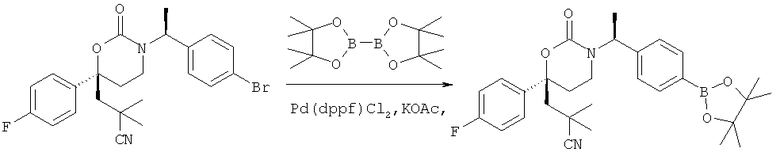
В раствор 3-((R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-2-оксо-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила (730 мг, 1,59 ммоль) в DMSO (8 мл) были добавлены бис(пинаколато)диборон (480 мг, 1,89 ммоль), КОАс (480 мг, 4,89 ммоль) и Pd(dppf)Cl2 (45 мг, 0,042 ммоль) в атмосфере азота. Полученная смесь перемешивалась при температуре 90°С в течение 20 ч. Реакция была остановлена при помощи воды, после чего смесь подверглась экстракции при помощи EtOAc. Объединенная органическая фаза была высушена над Na2SO4 и выпарена для получения неочищенного продукта, который затем был очищен посредством колоночной хроматографии для получения 3-((R)-6-(4-фторфенил)-2-оксо-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила (191 мг, 23,7%).
СПОСОБ ПОЛУЧЕНИЯ 7
(R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-он
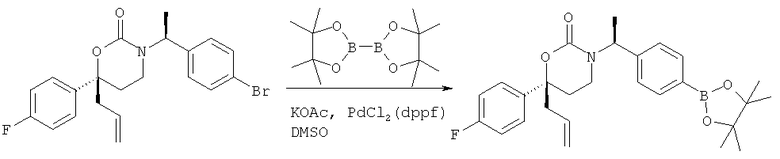
Смесь (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-она (0,4910 г, 1,17 ммоль, 1,0 эквив.), бис(пинаколато)диборона (0,3925 г, 1,55 ммоль, 1,3 эквив.), КОАс (0,3696 г, 3,76 ммоль, 3,2 эквив.) и PdCl2(dppf)·CH2Cl2 (0,0316 г, 0,0386 ммоль, 0,033 эквив.) в DMSO (6 мл) была нагрета при температуре 90°С в N2 среде в течение 20 ч после охлаждения реакционная смесь была разделена между EtOAc и водой. Органическая фаза была промыта солевым раствором и высушена над Na2SO4. Полученный в результате испарения растворителей осадок был очищен посредством хроматографии с использованием силикагеля и подвергся элюированию с помощью смеси гексанов/этилацетата для получения 0,4776 г (87%) (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она в виде твердого вещества белого цвета.
СПОСОБ ПОЛУЧЕНИЯ 8
(R)-3-((S)-1-(4-бромфенил)этил)-6-(3-гидроксипропил)-6-фенил-1,3-оксазинан-2-он
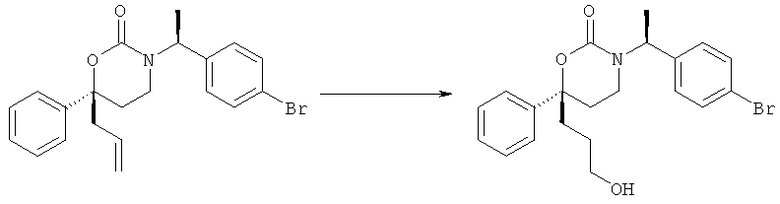
В раствор (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-фенил-1,3-оксазинан- 2-она (5 г, 12,5 ммоль) в тетрагидрофуране (60 мл) был добавлен комплекс ВН3 и тетрагидрофурана (25 мл, 1 моль/л, 25 ммоль) при температуре 0°С в атмосфере азота. Полученная смесь перемешивалась в течение 2 ч. Реакция была остановлена при помощи воды. Затем в описанную выше смесь были добавлены NaOH (3 моль/л, 10 мл) и H2O2 (15 мл). После окончания реакции была проведена экстракция смеси при помощи EtOAc. Объединенная органическая фаза была выпарена для получения неочищенного продукта, который затем был очищен посредством колоночной хроматографии для получения (R)-3-((S)-1-(4-бромфенил)этил)-6-(3-гидроксипропил)-6-фенил l-1,3-оксазинан-2-она (2,5 г, 40%). 1H ЯМР: (400 МГц, CDCl3): δ=1,48 (t, 3Н), 1,53 (m, 1H), 1,73 (m, 1H), 1,93-1,98(m, 2H), 2,17-2,28 (m, 3Н), 3,57 (t, 2H), 5,59 (m, 1H), 6,72 (m, 2H), 7,20 (m, 2H), 7,25-7,37 (m, 5H).
(R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-(3-гидроксипропил)-1,3-оксазинан-2-он был получен из (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-она в результате аналогичной процедуры.
(R)-3-((S)-1-(4-бромфенил)пропил)-6-(3-гидроксипропил)-6-фенил-1,3-оксазинан-2-он был получен из (R)-6-аллил-3-((S)-1-(4-бромфенил)пропил)-6-фенил-1,3-оксазинан-2-она в результате аналогичной процедуры.
СПОСОБ ПОЛУЧЕНИЯ 9
(R)-6-(3-гидроксипропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-он
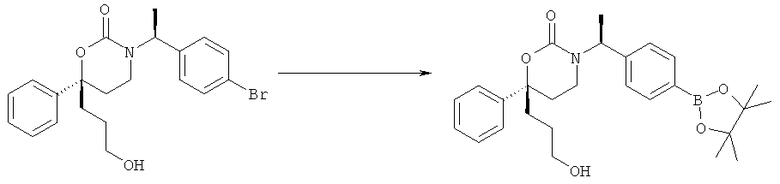
В раствор ((R)-3-((S)-1-(4-бромфенил)этил)-6-(3-гидроксипропил)-6- фенил-1,3-оксазинан-2-она (2 г, 4,8 ммоль) в DMSO (30 мл) были добавлены бис(пинаколато)диборон (1,58 г, 6,3 ммоль), КОАс (1,51 г, 15,4 ммоль) и PdCl2 (130 мг, 0,16 ммоль) в атмосфере азота. Полученная смесь была перемешана при температуре 90°С в течение 20 ч. Реакция была остановлена при помощи воды и подверглась экстракции при помощи EtOAc. Объединенная органическая фаза была выпарена для получения неочищенного продукта, который затем был очищен посредством колоночной хроматографии для получения (R)-6-(3-гидроксипропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (1,7 г, 77%). 1H ЯМР: (400 МГц, CDCl3): δ=1,18 (t, 1Н), 1,33 (S, 11H), 1,43 (m, 2H), 1,48 (m, 3H), 1,71(m, 1H), 1,88(m, 2H), 2,1-2,3 (t, 3H), 2,7 (m, 1H), 3,5 (m, 2H), 5,5 (m, 1H), 6,72 (m, 2H), 7,25-7,37 (m, 5H), 7,48 (m, 2H).
(R)-6-(4-фторфенил)-6-(3-гидроксипропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-он был получен из (R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-(3-гидроксипропил)-1,3-оксазинан-2-она в результате аналогичной процедуры.
(R)-6-(3-гидроксипропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропил)-1,3-оксазинан-2-он был получен из (R)-3-((S)-1-(4-бромфенил)пропил)-6-(3-гидроксипропил)-6-фенил-1,3-оксазинан-2-она в результате аналогичной процедуры.
(R)-6-(3-гидроксипропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропил)-1,3-оксазинан-2-он был получен из (R)-3-((S)-1-(4-бромфенил)пропил)-6-(3-гидроксипропил)-6-фенил-1,3-оксазинан-2-она в результате аналогичной процедуры.
СПОСОБ ПОЛУЧЕНИЯ 10
(R)-3-((S)-1-(4-бромфенил)этил)-6-(метоксиметил)-6-фенил-1,3-оксазинан-2-он
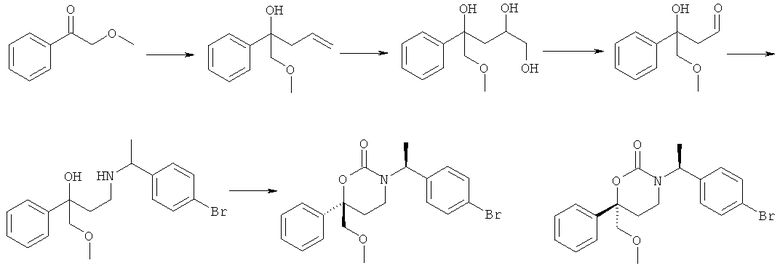
Этап 1. 1-Метокси-2-фенил-пент-4-ен-2-ол
2-Метокси-1-фенил-этанон (5,00 г) был растворен в тетрагидрофуране (50 мл), Затем при комнатной температуре полученный раствор был добавлен в 2 М раствор аллилмагния хлорида в тетрагидрофуране (21 мл). Раствор перемешивался при комнатной температуре в течение 3 ч, после чего в него был добавлен 10% водный раствор NH4Cl (50 мл). Полученная смесь подверглась экстракции с помощью трет-бутилметилового эфира (3×50 мл), после чего объединенные экстракты были промыты водой (50 мл) и солевым раствором (50 мл). Затем растворитель выпаривался для получения титульного соединения в виде бесцветного масла. Выход: 6,40 г (количественный показатель). Масс-спектр (ионизация электрораспылением+): масса/заряд = 175 [М+Н-Н2О]+.
Этап 2. 5-метокси-4-фенил-пентан-1,2,4-триол
OsO4 (4% раствор в воде, 2 мл; в качестве альтернативы может использоваться K2OsO4), а затем N-метил-морфолин-N-оксид (5,20 г) были добавлены в раствор 1-метокси-2-фенил-пент-4-ен-2-ола (1,10 г) в тетрагидрофуране (10 мл), охлажденный в ледяной бане. После извлечения из бани раствор в течение ночи перемешивался при комнатной температуре. Затем был добавлен 10% водный раствор Na2S2O5 (10 мл), после чего полученная смесь снова перемешивалась при комнатной температуре в течение 1,5 ч. После удаления в вакууме органического растворителя оставшаяся смесь подверглась экстракции с помощью этилацетата. Объединенные экстракты были промыты солевым раствором и высушены (MgSO4). Растворитель был выпарен для получения титульного соединения высокой степени чистоты (приблизительно 95%). Выход - 1,20 г (96% теоретического показателя). Масс-спектр (ионизация электрораспылением-): масса/заряд = 225 [М-Н]-.
Этап 3. 3-гидрокси-4-метокси-3-фенил-бутиральдегид
NalO4 (5,20 г) был добавлен в смесь 5-метокси-4-фенил-пентан-1,2,4-триола (1,10 г), дихлорметана (10 мл) и воды (5 мл), охлажденную в ледяной бане. Смесь тщательно перемешивалась в охлаждающей бане для нагревания до температуры окружающей среды, а затем перемешивалась в течение ночи при той же температуре. Затем в смесь были добавлены вода (20 мл) и дихлорметан (50 мл), был отделен органический слой, а водный слой подвергся экстракции с помощью дихлорметана (2×25 мл). Объединенные органические фазы были промыты водой и высушены (MgSO4). После удаления растворителя было получено титульное соединение, которое использовалось в ходе следующего этапа реакции (расщепление гликолей) без дополнительной очистки. Выход: 0,94 г (количественный показатель).
Этап 4. 4-[(S)-1-(4-бром-фенил)-этиламино]-1-метокси-2-фенил-бутан-2-ол
(S)-1-(4-бром-фенил)-этиламин (0,93 г), NaB(OAc)3 (0,98 г) и уксусная кислота (0,27 мл) были добавлены в указанном порядке в раствор 3-гидрокси-4-метокси-3-фенил-бутиральдегида (0,90 г) в тетрагидрофуране (20 мл) при температуре приблизительно 10-15°С. После извлечения из охлаждающей бани смесь перемешивалась при комнатной температуре в течение 2 ч. Затем в смесь были добавлены вода (50 мл) и 1 М водный раствор NaOH (20 мл), после чего полученный раствор снова перемешивался в течение 30 мин. Смесь подверглась экстракции с помощью этилацетата, затем объединенные экстракты были промыты водой и солевым раствором. После высушивания (MgSO4) растворитель был удален для получения титульного соединения, которое использовалось в ходе следующего этапа реакции без дополнительной очистки. Выход: 1,80 г (количественный показатель). Масс-спектр (ионизация электрораспылением+): масса/заряд = 378/380 (Br) [М+Н]+
Этап 5. 3-[(S)-1-(4-бром-фенил)-этил]-(R)-6-метоксиметил-6-фенил-[1,3]оксазинан-2-он и 3-[(S)-1-(4-бром-фенил)-этил]-(S)-6-метоксиметил-6-фенил-[1,3]оксазинан-2-он.
Трифосген (157 мг) был добавлен в ледяной раствор 4-[(S)-1-(4-бром-фенил)-этиламино]-1-метокси-2-фенил-бутан-2-ола (смесь диастереомеров в соотношении 1:1, 200 мг) и EtN/Pr2 (91 мкл) в дихлорметане (5 мл), полученный раствор перемешивался одновременно с охлаждением в течение 2 ч, а также при комнатной температуре в течение ночи. Затем раствор был выпарен в вакууме, в полученный осадок был очищен посредством обратно-фазовой ВЭЖХ (MeCN/H2O/NH3) для получения титульных соединений в составе отдельных фракций.
Изомер 1: 3-[(S)-1-(4-бром-фенил)-этил]-(R)-6-метоксиметил-6-фенил-[1,3]оксазинан-2-он. Выход: 45 мг (21% теоретического показателя). Масс-спектр (ионизация электрораспылением+): масса/заряд = 404 [М+Н]+ 1H ЯМР (400 МГц, DMSO-d6) δ 1,41 (d, J=7,1 Гц, 3Н), 2,19 (fd, J=11,2, 5,2 Гц, 1H), 2,24-2,34 (m, 1H), 2,34-2,41 (m, 1H), 3,02-3,09 (m, 1H), 3,27 (s, 3H), 3,49 (d, В часть сигнала АВ, J=10,6 Гц, 1Н), 3,53 (d, А часть сигнала АВ, J=10,6 Гц, 1Н), 5,34 (q, J=7,0 Гц, 1Н), 6,80 (dm, J=8,4 Гц, 2H), 7,27 (dm, J=8,4 Гц, 2H), 7,32-7,42 (m, 5H).
Изомер 2: 3-[(S)-1-(4-бром-фенил)-этил]-(S)-6-метоксиметил-6-фенил-[1,3]оксазинан-2-он. Выход: 45 мг (21% теоретического показателя). Масс-спектр (ионизация электрораспылением+): масса/заряд = 404 [М+Н]+ 1H ЯМР (400 МГц, DMSO-d6) δ 1,20 (d, J=7,2 Гц, 3H), 2,13-2,23 (m, 1H), 2,32-2,40 (m, 1H), 2,63-2,72 (m, 1H), 2,73-2,81 (m, 1H), 3,26 (s, 3H), 3,48 (d, В часть сигнала АВ, J=10,6 Гц, 1H), 3,55 (d, А часть сигнала АВ, J=10,6 Гц, 1H), 5,35 (q, J=7,2 Гц, 1H), 7,19 (dm, J=8,4 Гц, 2H), 7,32-7,45 (m, 5H), 7,53 (dm, J=8,4 Гц, 2H).
СПОСОБ ПОЛУЧЕНИЯ 11
N-(3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропил)-N-метилацетамид
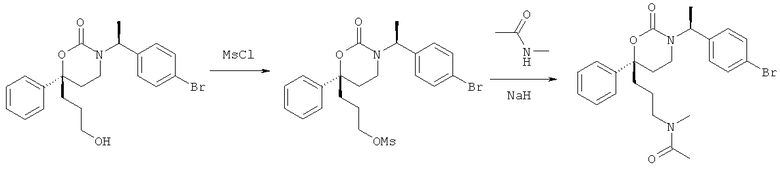
Этап 1
В раствор (R)-3-((S)-1-(4-бромфенил)этил)-6-(3-гидроксипропил)-6-фенил-1,3-оксазинан-2-он (200 мг, 0,48 ммоль) в CH2Cl2 (5 мл) был добавлен Et3N (240 мг, 2,4 ммоль) и метансульфонил хлорид (164 мг, 1,4 ммоль) при температуре 0°С. Реакционная смесь перемешивалась при комнатной температуре в течение 1 ч. Реакция была остановлена при помощи H2O, после чего смесь подверглась экстракции с помощью CH2Cl2. Органическая фаза выпаривалась для получения 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропил метансульфоната (234 мг, 98%), который применялся в ходе следующего этапа без дополнительной очистки.
Этап 2
В раствор 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропил метансульфоната (234 мг, 0,24 ммоль) в CH2Cl2 (3 мл) был добавлен NaH (82 мг, 3,4 ммоль) при температуре 0°С. Смесь перемешивалась при комнатной температуре в течение 30 мин. Затем в полученную смесь был добавлен N-метилацетамид (204 мг, 2,8 ммоль). После этого смесь перемешивалась при температуре 80°С в течение 5 ч. После остановки реакции с помощью воды смесь подверглась экстракции при помощи EtOAc. Объединенная органическая фаза была выпарена для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения N-(3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропил)-N-метилацетамида (150 мг, 68%). Метод ЖХ-МС: 2 tR=1,50 мин, масса/заряд = 497, 495, 475, 473. 1H ЯМР (400 МГц, CDCl3): δ=1,41 (m, 1H), 1,48 (t, 3H), 1,73 (m, 1H), 1,83-1,95 (m, 2H), 2,01 (m, 3H), 2,1-2,3 (m, 3H), 2,71 (m, 1H), 2,81 (s, 3H), 3,1 (m, 1H), 3,2 (m, 1H), 5,5 (m, 1H), 6,72 (m, 2H), 7,10 (m, 2H), 7,20 (m, 2H), 7,37 (m, 3H).
(R)-3-((S)-1-(4-бромфенил)этил)-6-(3-(2-оксопирролидин-1-ил)пропил)-6-фенил-1,3-оксазинан-2-он был получен из (R)-3-((S)-1-(4-бромфенил)этил)-6-(3-гидроксипропил)-6-фенил-1,3-оксазинан-2-она после проведения аналогичной процедуры с применением пирролидин-2-она в ходе 2 этапа.
СПОСОБ ПОЛУЧЕНИЯ 12
(S)-3-((S)-1-(4-бромфенил)этил)-6-(2-(1,-диоксо-изотиазолидин-2-ил)этил)-6-фенил-1,3-оксазинан-2-он
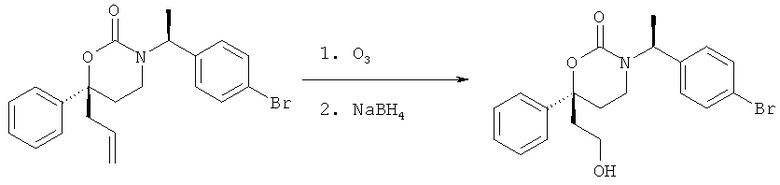
Раствор (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-фенил-1,3-оксазинан-2-она (3 г, 7,5 ммоль) в CH2Cl2 (50 мл) был обработан О3 при температуре -78°С до окрашивания смеси в синий цвет. Затем в раствор были добавлены NaBH4 (285 мг, 75 ммоль) при температуре 0°С, после чего реакционная смесь перемешивалась при комнатной температуре в течение 3 часов. Реакция была остановлена с помощью H2O с последующим проведением экстракции при помощи EtOAc. Объединенная органическая фаза выпаривалась для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (S)-3-((S)-1-(4-бром-фенил)этил)-6-(2-гидроксиэтил)-6-фенил-1,3-оксазинан-2-она (2,5 г, 84%). 1Н ЯМР (CDCl3): 1,48 (t, 3H), 2,05-2,41 (m, 4H), 2,71-2,92 (m, 2H), 3,51 (m, 1H), 3,71 (m, 1H), 5,58 (m, 1H), 6,73 (d, 2H), 7,12 (m, 2H), 7,23-7,45 (m, 6H).
(S)-3-((S)-1-(4-бромфенил)пропил)-6-(2-гидроксиэтил)-6-фенил-1,3-оксазинан-2-он был получен из (R)-6-аллил-3-((S)-1-(4-бромфенил)пропил)-6-фенил-1,3-оксазинан-2-она после проведения процедуры, аналогичной описанной в предыдущем абзаце.
СПОСОБ ПОЛУЧЕНИЯ 13
(S)-3-((S)-1-(4-бромфенил)этил)-6-(2-(1,1-диоксо-изотиазолидин-2-ил)этил)-6-фенил-1,3-оксазинан-2-он
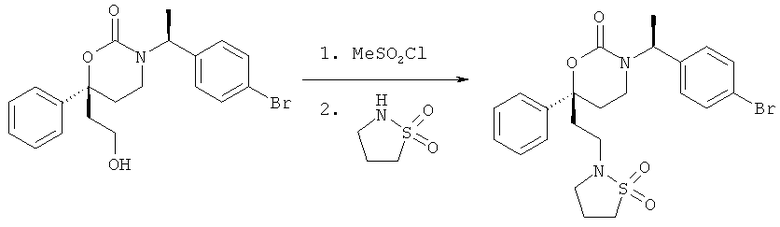
Этап 1
В раствор (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидроксиэтил)-6-фенил-1,3-оксазинан-2-она (300 мг, 0,75 ммоль) в дихлорметане (20 мл) был добавлен Et3N (390 мг, 3,75 ммоль) и метансульфонил хлорид (256 мг, 2,25 ммоль) при температуре 0°С. Затем реакционная смесь перемешивалась при комнатной температуре в течение 1 ч. Реакция была остановлена с помощью Н2О, после чего смесь подверглась экстракции с помощью дихлорметана. Органическая фаза выпаривалась для получения 2-((S)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)этил-метансульфоната (352,8 мг, 98%), который применялся в ходе следующего этапа без дополнительной очистки.
Этап 2
В раствор 2-((S)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)этил-метансульфоната (360 мг, 0,75 ммоль) и K2CO3 (207 мг, 1,5 ммоль) в ацетонитриле (10 мл) был добавлен изотиазолидин 1,1-диоксид (121 мг, 4,6 ммоль), после чего смесь подверглась обратному конденсированию. Затем смесь подверглась фильтрации. Полученный фильтрат выпаривался для получения неочищенного продукта, который был очищен посредством препаративной ВЭЖХ для получения соединения (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-(1,1-диоксо-изотиазолидин-2-ил)этил)-6-фенил-1,3-оксазинан-2-она (2,43 мг, 1%). Метод ЖХ-МС 2 tR=1,37 мин, масса/заряд = 509, 507. 1Н ЯМР (CDCl3): 1,48 (t, 3Н), 2,05-2,41 (m, 7H), 2,71-2,92 (m, 2H), 3,11 (m, 3H), 3,21 (m, 2H), 5,58 (m, 1H), 6,73 (d, 2H), 7,18 (m, 1H), 7,23 (m, 3H); 7,35 (m, 3H).
(R)-3-((S)-1-(4-бромфенил)этил)-6-(3-(1,1-диоксо-изотиазолидин-2-ил)пропил)-6-фенил-1,3-оксазинан-2-он был получен из (R)-3-((S)-1-(4-бромфенил)этил)-6-(3-гидроксипропил)-6-фенил-1,3-оксазинан-2-она после проведения аналогичной процедуры:
СПОСОБ ПОЛУЧЕНИЯ 14
(S)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-((1-гидроксициклопропил)метил)-1,3-оксазинан-2-он
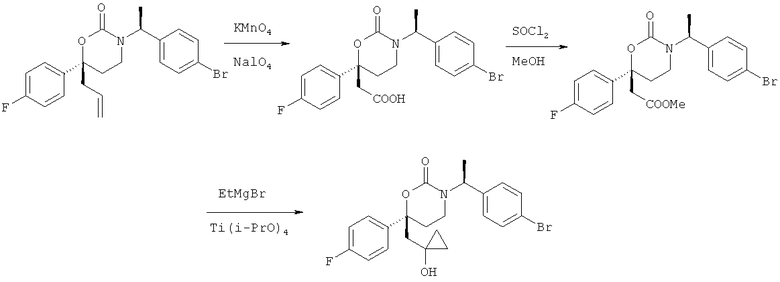
Этап 1
В раствор (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3 оксазинан-2-она (450 мг, 1,01 ммоль) в ацетоне (10 мл) был добавлен раствор KMnO4 (190 мг, 1,2 ммоль) и NalO4 (1,5 г, 7.2 ммоль) в воде (10 мл). Смесь перемешивалась в течение 2 ч при температуре 0°С. Смесь подверглась фильтрации, после чего уровень рН полученного фильтрата был отрегулирован до 5-6 при помощи 1 N водного раствора HCl, Смесь подверглась экстракции при помощи EtOAc. Органическая фаза была промыта солевым раствором, высушена над безводным Na2SO4 и выпарена для получения 2-((S)-3-((S)-1-(4-бромфен-ил)этил)-6(4-фторфенил)-2-оксо-1,3-оксазинан-6-ил)уксусной кислоты (540 мг, неочищенной), которая применялась в ходе следующего этапа без дополнительной очистки.
Этап 2
В раствор 2-((S)-3-((S)-1-(4-бромфен-ил)этил)-6-(4-фторфенил)-2-оксо-1,3-оксазинан-6-ил)уксусной кислоты (540 мг, 1,24 моль) в МеОН (20 мл) был добавлен SOCl2 (5 мл) при температуре 0°С, после чего реакционная смесь перемешивалась при комнатной температуре в течение 2 ч. Затем реакционная смесь выпаривалась с последующей очисткой осадка посредством препаративной ТСХ для получения метил 2-((S)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)-этил)-2-оксо-1,3-оксазинан-6-ил)ацетата (150 мг, 27%). 1H ЯМР (CDCl3): δ=1,49 (d, 3H), 2,19 (m, 1H), 2,44 (m, 1H), 2,60 (m, 1H), 2,77-3,08 (m, 3H), 3,51 (s, 3H), 5,52 (m, 2H), 6,62 (d, 2H), 6,98 (t, 2H), 7,23 (t, 2H), 7,28 (m, 2H).
Этап 3
В раствор метил 2-((S)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)-этил)-2-оксо-1,3-оксазинан-6-ил)ацетата (150 мг, 0,33 ммоль) и тетраизопропоксититана (189 мг, 0,66 ммоль) в тетрагидрофуране (20 мл) при комнатной температуре в атмосфере азота был добавлен 3,0 М этилмагний бромид (4 мл, 12 ммоль). Затем смесь перемешивалась в течение 2 ч. Реакция была остановлена при помощи водного раствора NH4Cl, после чего смесь подверглась фильтрации, а полученный фильтрат - экстракции при помощи EtOAc. Объединенная органическая фаза была промыта солевым раствором, высушена над безводным Na2SO4 и выпарена для получения неочищенного продукта, который затем был очищен посредством препаративной ВЭЖХ для получения (S)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-((1-гидроксициклопропил)метил)-1,3-оксазинан-2-она (2.51 мг, 2%). 1Н ЯМР (CDCl3): 0,03 (m, 1H), 0,18 (m, 1H), 0,49 (m, 1H), 0,60 (m, 1H), 1,43 (m, 3H), 2,08 (s, 2H), 2,26 (m, 1H), 2,37 (m, 2H), 2,88 (m, 1H), 5,53 (m, 1H), 6,66 (d, 2H), 6,97 (t, 2H), 7,16 (m, 2H), 7,26 (m, 2H).
СПОСОБ ПОЛУЧЕНИЯ 15
N-(3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропил)-N-метилметансульфонамид
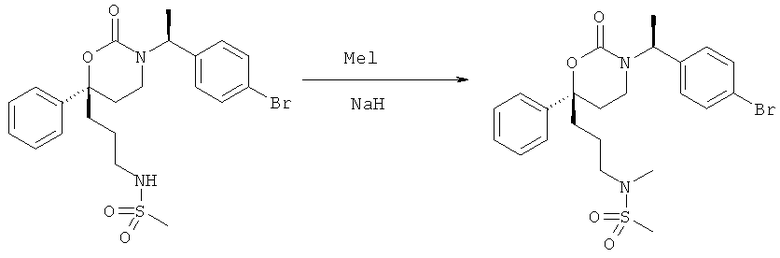
В раствор 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропил метансульфоната (180 мг, 0,36 ммоль) в DMF (5 мл) был добавлен NaH (14.6 мг, 0,36 ммоль) при температуре 0°С. Смесь перемешивалась при комнатной температуре в течение 30 мин. Затем в полученную смесь был добавлен йодометан (153 мг, 1,1 ммоль). Смесь перемешивалась при температуре 40°С в течение 3 ч. После окончания реакции реакция была остановлена раствором NH4Cl, затем смесь подверглась экстракции при помощи EtOAc. Объединенная органическая фаза выпаривалась для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения N-(3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропил)-N-метилметансульфонамида (100 мг, 55%). Метод ЖХ-МС 2 tR=1,41 мин, масса/заряд = 511, 509. 1H ЯМР (400 МГц, CDCl3): δ=1,45 (m, 1H), 1,48 (t, 3H), 1,83-1,97 (m, 3Н), 2,1-2,2 (m, 3H), 2,61 (s, 3H), 2,71 (s, 3H), 2,91 (m, 1H), 3,0 (m, 2H), 5,5 (m, 1H), 6,72 (m, 2H), 7,10 (m, 2H), 7,20 (m, 2H), 7,37 (m, 3H).
ПРИМЕР 1
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
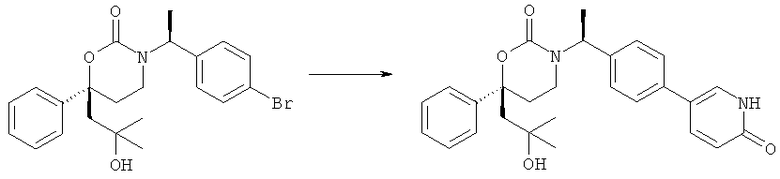
В раствор (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (112 мг, 0,259 ммоль) в 1,4-диоксане (3 мл) были добавлены 6-оксо-1,6-дигидропиридин-3-илбориновая кислота (55 мг, 0,40 ммоль), затем Pd(dppf)Cl2 (11 мг, 0,015 ммоль) и водный раствор Cs2CO3 (0,48 мл, 2М раствор в H2O), После установки обратного конденсатора была произведена дегазация устройства и трехкратное промывание с помощью N2. Реакционная смесь нагревалась до 90°С в течение 24 ч. После охлаждения до комнатной температуры смесь была разбавлена водой и подверглась трехкратной экстракции при помощи EtOAc. Органические слои были промыты солевым раствором, высушены над Na2SO4, подверглись фильтрации и выпариванию. Полученный осадок был очищен посредством препаративной ВЭЖХ для получения титульных соединений (21,6 мг) в виде густого масла. Метод ЖХ-МС 1 tR=1,25 мин, масса/заряд = 447, 389; 1Н ЯМР (CD3OD) 0,96 (s, 3H), 1,28 (s, 3H), 1,57 (d, 3H), 2,16 (s, 2H), 2,21 (m, 1H), 2,46 (m, 2H), 3,03 (m, 1H), 5,57 (q, 1H), 6,66 (d, 1H), 7,02 (d, 2H), 7,25-7,40 (7Н), 7,66 (s,1H), 7,90 (d, 1H).
ПРИМЕР 2
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
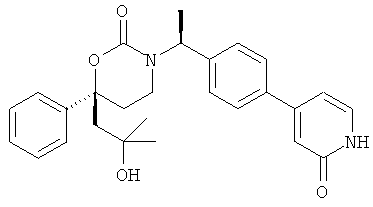
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборол-ан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 4-йодопиридин-2(1Н)-она в результате процедуры, аналогичной описанной в Примере 1. Метод ЖХ-МС 1tR=1,23 мин, масса/заряд = 389, 447 (М+1); 1H ЯМР (CD3OD) 7,40 (d, J=6,7 Гц, 1H), 7,31 (d, J=8,2 Гц, 2H), 7,29-7,20 (m, 5H), 6,96 (d, J=8,2 Гц, 2H), 6,57-6,52 (m, 2H), 5,49 (q, J=7,0 Гц, 1H), 2,98-2,93 (m, 1H), 2,47-2,34 (m, 2H), 2,16-2,09 (m, 1H), 2,07 (s, 2H), 1,45 (d, J=7,0 Гц, 3H), 1,19 (s, 3H), 0,87 (s, 3H).
ПРИМЕР 3
(S)-3-((S)-1-(4-(1-циклопропил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
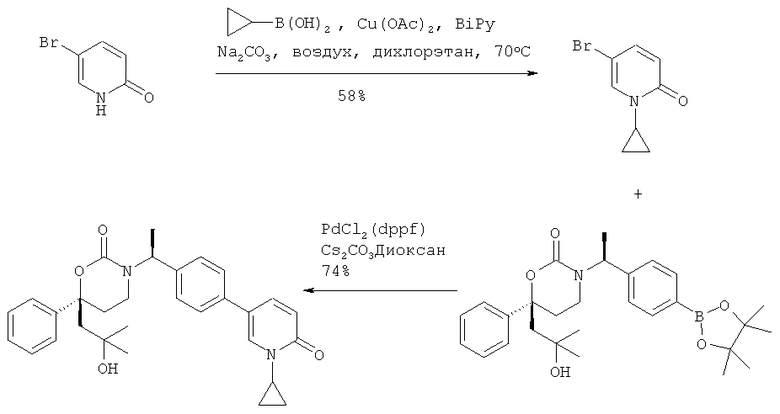
Этап 1. 5-бром-1-циклопропилпиридин-2(1Н)-он
Смесь 5-бром-2-гидроксипиридина (0,8300 г, 4,77 ммоль, 1,0 эквив.), Cu(ОАс)2 (0,902 г, 4,96 ммоль, 1,04 эквив.), бипиридина (0,785 г, 5,03 ммоль, 1,05 эквив.), циклопропилбориновой кислоты (0,846 г, 9,85 ммоль, 2,06 эквив.) и раствора Na2CO3 (1,110 g, 10,47 ммоль, 2,20 эквив.) в дихлорэтане (30 мл) перемешивалась при температуре 70°С в течение 22 ч в воздушной среде. Реакция смеси была остановлена при помощи насыщенного водного раствора NH4Cl, разбавленного CH2Cl2 и высушенного над Na2SO4. После удаления в вакууме растворителя осадок был очищен посредством хроматографии с применением силикагеля и подвергся элюированию с помощью смеси гексанов/EtOAc для получения 0,585 г (58%) 5-бром-1-циклопропилпиридин-2(1H)-она. Метод ЖХ-МС 1 tR=1,05 мин, масса/заряд 214, 216 (MH+); 1H ЯМР (400 МГц, CDCl3) 7,41 (d, J=2,7 Гц, 1H), 7,31 (dd, J=9,7, 2,9 Гц, 1Н), 6,47 (d, J=9,9 Гц, 1Н), 3,33-3,27 (m, 1Н), 1,17-1,12 (m, 2H), 0,89-0,84 (m, 2H); 13С ЯМР (100 МГц, CDCl3) 162,58, 142,29, 137,00, 121,77, 97,92, 32,83, 6,93.
Этап 2. (S)-3-((S)-1-(4-(1-циклопропил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
В раствор (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (0,729 г, 1,52 ммоль) в 1,4-диоксане (16 мл) были добавлены 5-бром-1-циклопропилпиридин-2(1H)-он (0,323 г, 1,51 ммоль), 2 М водный раствор Cs2CO3 (4 мл) и PdC2(dppf)·CH2Cl2 (0,079 г, 0,0964 ммоль). Затем смесь подверглась дегазации и нагреванию в атмосфере азота при температуре 120°С в течение 16 ч, была разбавлена CH2Cl2 и высушена Na2SO4. После испарения растворителей полученный осадок был очищен посредством хроматографии с применением силикагеля и подвергся элюированию с помощью MeOH/CH2Cl2 для получения 0,543 г (74%) (S)-3-((S)-1-(4-(1-циклопропил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она. Метод ЖХ-МС 1 tR=1,41 мин, масса/заряд 487 (МН+); 1H ЯМР (400 МГц, CD3OD) 7,66-7,64 (m, 2H), 7,30-7,19 (m, 7H), 6,94 (d, J=8,2 Гц, 2H), 6,52 (d, J=10 Гц, 1Н), 5,48 (q, J=7,0 Гц, 1Н), 3,32-3,26 (m, 1Н), 2,97-2,92 (m, 1Н), 2,46-2,32 (m, 2H), 2,16-2,09 (m, 1Н), 2,08 (s, 2H), 1,45 (d, J=7,0 Гц, 3Н), 1,19 (s, 3Н), 1,10-1,05 (m, 2H), 0,90-0,86 (m, 5H); 13С ЯМР (100 МГц, CD3OD) 165,59, 155,82, 144,08, 141,05, 139,60, 136,60, 136,30, 129,77, 128,86, 128,64, 126,83, 126,15, 121,93, 120,53, 85,33, 71,67, 55,18, 54,78, 37,46, 34,10, 33, 04, 31,79, 30,00, 15,60, 7,49, 7,47.
ПРИМЕР 4
(S)-3-((S)-1-(4-(1-(дифторметил)-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
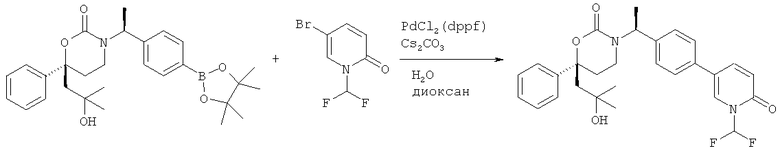
В колбу для обработки микроволнами, оснащенную магнитной мешалкой, были помещены (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-он (20 мг, 0,047 ммоль), 5-бром-1-(дифторметил)пиридин-2(1Н)-он (25 мг, 0,113 ммоль), Cs2CO3 (27 мг, 0,083 ммоль), Н2О (0,1 мл) и сухой диоксан (1 мл). После этого полученная смесь обрабатывалась посредством распыления N2 в течение 10 мин и нагревалась при температуре 110°С в микроволновой печи в течение 0,5 ч. Полученная таким образом смесь была разбавлена ледяной НОАс (0,1 мл) и МеОН (0,5 мл) и подверглась фильтрации. Фильтрат был очищен при помощи препаративной ВЭЖХ для получения (S)-3-((S)-1-(4-(1-(дифторметил)-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (11,8 мг, 57%) в виде масла. Метод ЖХ-МС 1 tR=1,55 мин, масса/заряд = 497, 248; 1H ЯМР (CDCl3) 1,13 (s, 3Н), 1,19 (s, 3Н), 1,56 (d, 3Н), 2,15-2,35 (s, 4H), 2,42(m, 2H), 2,88 (m, 1Н), 5,71 (q, 1Н), 6,64 (d, 1Н), 7,04 (d, 2H), 7,18 (d, 2H), 7,30-7,40 (5H), 7,52 (1Н), 7,60 (m, 1H), 7,75 (t, 1H).
5-бром-1-(дифторметил)пиридин-2(1Н)-он был получен в соответствии с процедурой, описанной в Ando, M.; Wada, Т.; Sato, N. Org. Lett. 2006, 8, 3805-3808.
ПРИМЕР 5 (S)-3-((S)-1-(4-(1-(дифторметил)-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
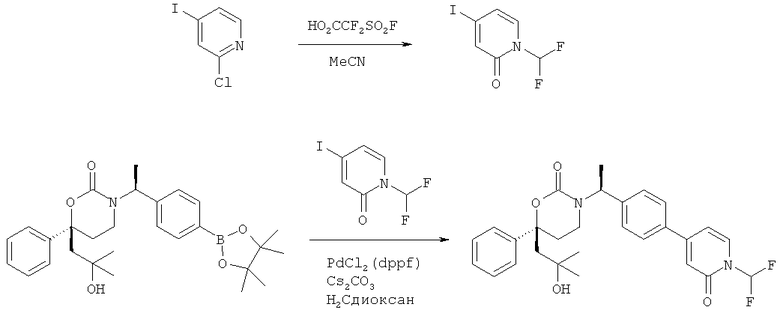
Этап 1
Перемешанная смесь 2-хлор-4-йодопиридина (1,33 г, 5,6 ммоль) и измельченного в порошок NaHCO3 (935 мг, 11,2 ммоль) в MeCN (2 мл) была нагрета до 40°С в масляной бане, после чего в смесь по капле в течение 10 мин добавлялся раствор 2,2-дифтор-2-(фторсульфонил)уксусной кислоты (0,57 мл, 5,6 ммоль) в MeCN (10 мл). Смесь перемешивалась при температуре 40°С в течение 2 ч. В ходе ЖХ-МС наблюдалось частичное преобразование в целевой продукт. Затем в смесь был добавлен измельченный в порошок NaHCO3 (935 мг, 11,2 ммоль), а затем по капле в течение 10 мин - раствор 2,2-дифтор-2-(фторсульфонил)уксусной кислоты (0,57 мл, 5,6 ммоль) в MeCN (10 мл). Полученная смесь перемешивалась при температуре 40°С в течение 2 ч, после чего была разбавлена насыщенным водным раствором NaHCO3 (25 мл) и подверглась выпариванию в вакууме. Водный осадок подвергся экстракции при помощи EtOAc (90 мл). Органический экстракт был промыт солевым раствором (20 мл), высушен над Na2SO4 и подвергся выпариванию для получения янтарного масла (1,14 г). После проведения хроматографии с применением картриджа, содержащего 40 г силикагеля, а также элюирования с помощью градиента - 0-40% смеси EtOAc и гексанов был получен 1-(дифторметил)-4-йодопиридин-2(1Н)-он (255 мг, выход: 16%, предполагаемая чистота: 45%) в виде масла желтого цвета. Метод ЖХ-МС 1 tR=1,23 мин, масса/заряд = 272. Материал использовался без дополнительной очистки.
Этап 2
В колбу для обработки микроволнами, оснащенную магнитной мешалкой, были помещены (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-он (52 мг, 0,11 ммоль), 1-(дифторметил)-4-йодопиридин-2(1Н)-он (29 мг, 0,11 ммоль), Cs2CO3 (71 мг, 0,22 ммоль), H2O (0,1 мл) и сухой диоксан (1 мл). После этого смесь обрабатывалась посредством распыления N2 в течение 5 мин с последующим добавлением PdCl2(dppf) (5 мг, 0,007 ммоль). Смесь обрабатывалась посредством распыления в течение 5 мин и нагревалась в микроволновой печи в течение 1 часа при температуре 110°С. Полученная смесь была разбавлена 5% водным раствором HCl (0,2 мл) и МеОН (2 мл) и подверглась фильтрации. Фильтрат был очищен посредством препаративной ВЭЖХ для получения масла коричневого цвета (16,2 мг), которое было нанесено на картридж SPE, содержащий 2 г силикагеля, и подверглось последовательному элюированию 25 и 50% смесями EtOAc в гексанах (по 15 мл), а также EtOAc (3×15 мл) для получения пяти фракций. Фракции 3 и 4 смешивались вместе и выпаривались для получения (S)-3-((S)-1-(4-(1-(дифторметил)-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (13,4 мг, 25%) в виде бесцветного масла. Метод ЖХ-МС 1 tR=1,57 мин, масса/заряд = 497, 439; 1H ЯМР (CD3OD) 0,96 (s, 3H), 1,27 (s, 3H), 1,56 (d, 3H), 2,15 (s, 2H), 2,21 (m, 1H), 2,40-2,60 (2H), 3,08 (m, 1H), 5,59 (q, 1H), 6,66 (s, 1Н), 6,74 (d, 1H), 7,07 (d, 2H), 7,30-7,40 (5H), 7,45 (d, 2H), 7,77 (1H), 7,79 (t, 1H).
ПРИМЕР 6
2,2-диметил-3-((R)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропанамид
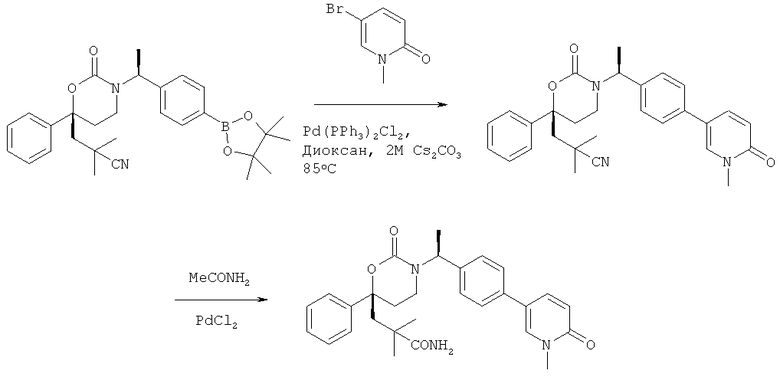
Этап 1
Смесь 2,2-диметил-3-((R)-2-оксо-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)пропаннитрила (392 мг, 0,775 ммоль), 5-бром-1-метилпиридин-2(1Н)-она (220 мг, 1,5 эквив.). 2М водного раствора Cs2CO3 (900 мкл), Pd(PPh3)2Cl2 (40 мг, 7 моль%), а также безводного 1,4-диоксана (8,5 мл) трехкратно подверглась дегазации и повторному насыщению газообразным N2, Затем смесь в течение ночи нагревалась при температуре 85°С с применением в качестве защитного газа газообразного N2. После охлаждения до комнатной температуры смесь была разбавлена EtOAc (20 мл) и промыта водой (20 мл), после чего водный слой подвергся экстракции при помощи EtOAc (2×10 мл). Объединенный органический слой был промыт водой (10 мл), солевым раствором (2×10 мл) и высушен над Na2SO4, после чего подвергся фильтрации и выпариванию для получения осадка, который затем был очищен с помощью хромотографа фирмы Gilson для получения 34 мг продукта (выход: 9%). ЖХ-МС (3 мин метод) tR=1,44 мин, масса/заряд 470 (М+1). 1H ЯМР (CDCl3) δ 7,68 (dd, 1Н), 7,52 (d, 1Н), 7,31 (q, 2H), 7,16 (d, 2H), 7,07 (t, 2H), 6,97 (d, 2H), 6,91 (d, 1H), 5,66 (q, 1H), 3,71 (s, 3Н), 2,99 (dt, 1Н), 2,47 (dd, 2H), 2,27 (m, 1Н), 2,13 (s, 2H), 1,55 (d, 3H), 1,44 (s, 3H), 1,24 (s, 3H).
Этап 2
Раствор 2,2-диметил-3-((R)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропаннитрила (55 мг, 0,12 ммоль), ацетамида (177 мг, 3 ммоль) и PdCl2 (21 мг, 0,12 ммоль) в тетрагидрофуране: H2O (2 мл, 3:1) перемешивался в течение ночи. После удаления растворителя было произведено повторное растворение неочищенного материала в CH3CN с последующей очисткой полученного продукта посредством препаративной ВЭЖХ для получения 2,2-диметил-3-((R)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропанамида (28 мг). Метод ЖХ-МС 1 tR=1,25 мин, масса/заряд = 488 (М+1); 1H ЯМР (CDCl3) 7,55 (dd, 1H, J=9 Гц, 3 Гц), 7,42 (d, 1H, J=3 Гц), 7,32 (d, 1H, J=4 Гц), 7,29 (m, 4H), 7,14 (d, 2H, 8 Гц), 7,00 (d, 2H, J=8 Гц), 6,79 (d, 1H, J=9 Гц), 5,66 (q, 1H, J=8 Гц), 3,62 (s, 3Н), 2,95-2,89 (m, 1H), 2,5 (d, 1H, J=15 Гц), 2,26-2,1 (m, 3Н), 2,2 (d, 1H, J=15 Гц), 2,5 (d, 1H, J=15 Гц), 2,26-2,10 (m, 3Н), 2,2 (d, 1H, J=15 Гц), 1,53 (d, ЗН, J=7 Гц), 1,22(s,3H),1,20(s,3H).
ПРИМЕР 7
(S)-6-(2-амино-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
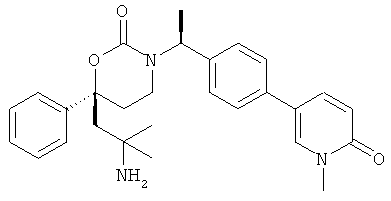
В накрытую фольгой колбу, содержащую смесь 2,2-диметил-3-((R)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропанамида (20 мг, 0,04 ммоль) и 1:1 CH3CN/H2O (1 мл), был помещен Phl(O2CCF3)2 (31 мг, 0,07 ммоль). Окончание реакции наблюдалось спустя 24 ч. После удаления растворителя неочищенный материал подвергся очистке посредством препаративной ВЭЖХ для получения (S)-6-(2-амино-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (16 мг). ЖХ/МС с ионизацией электрораспылением+ = 460 (М+1). Метод ЖХ-МС 1 tR=1,06 мин, масса/заряд = 460 (М+1); 1H ЯМР (CDCl3) 7,60 (dd, 1H, J=9 Гц, 2 Гц), 7,46 (d, 1H, J=3 Гц), 7,39-7,27 (m, 5H), 7,20 (d, 2H, J=7 Гц), 7,1 (d, 2H, J=8 Гц), 6,78 (d, 1H, J=9 Гц, 5,61 (q, 1H, J=7 Гц), 3,65 (s, 3H), 2,87 (m, 1H), 2,80 (d, 1H, J=16 Гц), 2,23 (d, 1H, J=16 Гц), 2,19-2,08 (m, 3Н), 1,54 (d, 3H, J=7 Гц), 1,41 (s, 3Н), 0,96 (s, 3Н).
ПРИМЕР 8
N-(2-метил-1-((S)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропан-2-ил)ацетамид
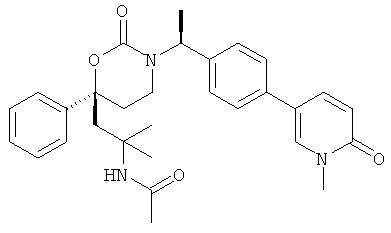
Раствор (S)-6-(2-амино-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-сренил-1,3-оксазинан-2-она (5 мг, 0,009 ммоль) в CH2Cl2 (1 мл) был обработан DMAP (5 мг, 0,04 ммоль), i-Pr2NEt (10 капель) и уксусным ангидридом (20 капель). Полученная смесь перемешивалась в течение ночи, после чего реакционная смесь промывалась водой. Полученный после выпаривания органичесокго слоя неочищенный материал был очищен посредством препаративной ВЭЖХ для получения N-(2-метил-1-((S)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропан-2-ил)ацетамида (0,88 мг). Метод ЖХ-МС 1 tR=1,3 мин, масса/заряд = 502 (М+1).
ПРИМЕР 9
Метил 2-метил-1-((S)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропан-2-илкарбамат
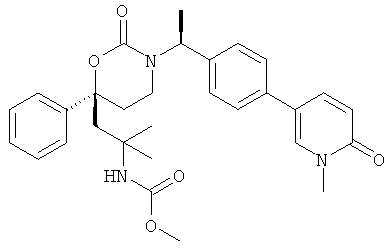
Раствор (S)-6-(2-амино-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (5 мг, 0,009 ммоль) в CH2Cl2 (1 мл) был обработан DMAP (5 мг, 0,04 ммоль), i-Pr2NEt (10 капель) и метилхлорформатом (20 капель). Полученная смесь перемешивалась в течение ночи, после чего реакционная смесь была очищена посредством препаративной ВЭЖХ для получения метил 2-метил-1-((S)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропан-2-илкарбамата (2,58 мг). Метод ЖХ-МС 1 tR=1,45 мин, масса/заряд = 518 (М+1); 1H ЯМР (CDCl3) 7,66 (dd, 1H, J=9 Гц, 2 Гц), 7,48 (d, 1H, J=3 Гц), 7,35-7,27 (m, 5H), 7,15 (d, 2H, J=8 Гц), 7,01 (d, 2H, J=8 Гц), 6,87 (d, 1H, J=9 Гц), 5,67 (q, 1H, J=7 Гц), 3,69 (s, 3H), 2,2 (s, 3H), 1,54 (d, 3H, J=7 Гц), 1,46-1,36 (m, 2H), 1,30 (s, 3H), 1,20 (s, 3H).
ПРИМЕР 10
N-(2-метил-1-((S)-2-оксо-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-6-ил)пропан-2-ил)метансульфонамид
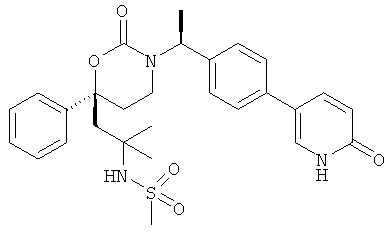
Титульное соединение было получено из N-(1-((S)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2-метилпропан-2-ил)метансульфонамида и 2-оксо-1,2-дигидропиридин-5-илбориновой кислоты после проведения процедуры, аналогичной описанной в Примере 4. Метод ЖХ-МС 1 tR=1,3 мин, масса/заряд = 524 (М+1); 1H ЯМР (CDCl3) 7,81 (d, 1H, J=9 Гц), 7,63 (brs, 1H), 7,39-7,31 (m, 5H), 7,18 (d, 2H, J=8 Гц), 7,03 (d, 2H, J=7 Гц), 6,79 (d, 1H, J=9 Гц), 5,67 (q, 1H, J=6 Гц), 2,93 (s, 3H), 2,90 (m, 1H), 2,49 (d, 1H, J=15 Гц), 2,32 (d, 1H, J=15 Гц), 2,28-2,18 (m, 3H), 1,54 (d, 3H, J=7 Гц), 1,36 (s, 3H), 1,25 (s, 3H).
N-(1-((S)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2-метилпропан-2-ил)метансульфонамид был получен из 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила в результате последовательного проведения процедур, аналогичных описанным в качестве 2 этапа Примера 6, а также в Примерах 7 и 11.
ПРИМЕР 11
N-(2-метил-1-((S)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропан-2-ил)метансульфонамид
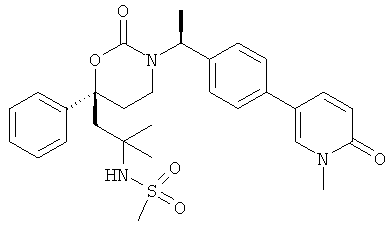
Раствор (S)-6-(2-амино-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (5 мг, 0,009 ммоль) в CH2Cl2 (1 мл) был обработан DMAP (5 мг, 0,04 ммоль), i-Pr2NEt (10 капель) и MsCl (20 капель). Полученная смесь перемешивалась в течение ночи, после чего реакционный раствор промывался водой. Полученный после выпаривания органического слоя неочищенный материал был очищен посредством препаративной ВЭЖХ для получения N-(2-метил-1-((S)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропан-2-ил)метансульфонамида (3,30 мг). Метод ЖХ-МС 1 tR=1,39 мин, масса/заряд = 538 (М+1); 1H ЯМР (CDCl3) 7,61 (dd, 1H, J=9 Гц, 3 Гц), 7,46 (d, 1H, J=2 Гц), 7,40-7,27 (m, 5H), 7,17 (d, 2H, J=8 Гц), 7,04 (d, 2H, J=8 Гц), 6,79 (d, 1H, J=9 Гц), 5,67 (q, 1H, J=7 Гц), 3,66 (s, 3Н), 2,93 (s, 3Н), 2,31-2,22 (m, 2H), 1,55 (d, 3H, J=7 Гц), 1,48-1,36 (m, 2H), 1,33 (s, 3Н), 1,24 (s, 3Н).
ПРИМЕР 12
N-метил-N-(2-метил-1-((S)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропан-2-ил)метансульфонамид
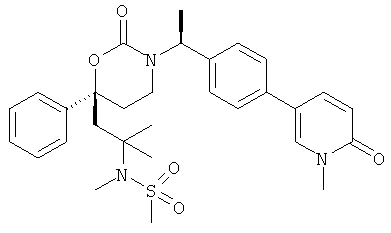
В раствор N-(2-метил-1-((S)-2-оксо-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-6-ил)пропан-2-ил)метансульфонамида (7 мг, 0,013 ммоль) в тетрагидрофуране, имеющий комнатную температуру, был добавлен сначала NaH (10 мг, 0,20 ммоль), а затем метилйодид (30 мкл, 0,080 ммоль). Полученная смесь нагревалась до 60°С в течение 5 ч, после чего колба с раствором была охлаждена сначала до комнатной температуры, а затем до 0°С с последующей остановкой реакции с помощью насыщенного водного раствора NH4Cl. Далее смесь была подвергнута экстракции при помощи EtOAc (3×), объединенный органический слой был высушен над Na2SO4, после чего подвергся фильтрации, выпариванию, а также очистке посредством препаративной ВЭЖХ для получения N-метил-N-(2-метил-1-((S)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропан-2-ил)метансульфонамида (4,63 мг). Метод ЖХ-МС 1 tR=1,45 мин, масса/заряд = 552 (М+1); 1H ЯМР (CDCl3) 7,55 (dd, 1Н, J=9 Гц, 3 Гц), 7,41 (d, 1H, J=3 Гц), 7,35 -7,27 (m, 5H), 7,13 (d, 2H, J=8 Гц), 6,98 (d, 2H, J=8 Гц), 6,79 (d, 1H, J=9 Гц), 6,55 (q, 1H, J=7 Гц), 3,62 (s, 3Н), 2,91-2,86 (m, 1H), 2,86 (s, 3Н), 2,73 (d, 1H, J=15 Гц), 2,74 (s, 3Н), 2,46 (d, 1H, J=15 Гц), 2,39 -2,36 (m, 2H), 2,25-21,6 (m, 1H), 1,53 (d, 3Н, J=7 Гц), 1,53 (s, 3Н), 1,22 (s, 3Н).
ПРИМЕР 13
N-(2-метил-1-((S)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропан-2-ил)метансульфонамид
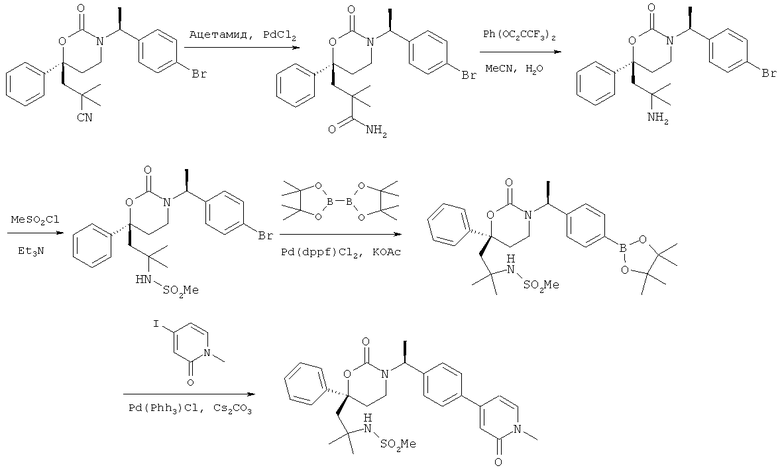
Этап 1
В раствор 3-(R-3-S-1-(4-бромфенил)-этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрил (1 г, 2,27 ммоль) в смеси тетрагидрофурана/H2O (3:1, 10 мл) были добавлены ацетамид (3,35 г, 56,75 ммоль) и PdCl2 (0,402 г, 2,27 ммоль). Полученная смесь перемешивалась в течение ночи. Полученный после удаления растворителя осадок был очищен посредством ТСХ для получения 3-(R-3-S-1-(4-бромфенил)-этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропанамида (0,745 г, 71,6%).
Этап 2
В раствор 3-(R-3-S-1-(4-бромфенил)-этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропанамида (0,74 г, 1,612 ммоль) в смеси CH3CN/H2O (20 мл, 1:1) были добавлен Phl(OC2CCF3)2 (1,178 г, 2,74 ммоль). Смесь перемешивалась в течение ночи, после чего подверглась экстракции при помощи EtOAc (30 мл). Органический слой был промыт солевым раствором и подвергся выпариванию для получения S-6-(2-амино-2-метилпропил)-3-S-1-(4-бромфенил)-этил)-6-фенил-1,3-оксазинан-2-она (0,6 г, 87%).
Этап 3
В раствор S-6-(2-амино-2-метилпропил)-3-S-1-(4-бромфенил)-этил)-6-фенил-1,3-оксазинан-2-она (0,6 г, 1,39 ммоль) в CH2Cl2 (10 мл) был добавлен Et3N (0,84 г, 8,34 ммоль). Смесь была охлаждена при температуре 0°С с последующим добавлением MsCl (0,48 г, 4,17 ммоль). Затем смесь перемешивалась при комнатной температуре в течение 1 ч и выпаривалась для получения неочищенного продукта. Полученный осадок был очищен посредством колоночной хроматографии для получения N-1-S-3-S-1-(4-бромфенил)-этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2-метилпропан-2-ил)-метил-сульфонамида (0,5 г, 70,4%).
Этап 4
В раствор N-1-S-3-S-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2-метилпропан-2-ил)-метил-сульфонамида (0,5 г, 0,98 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би-(1,3,2-диоксаборолана) (0,399 г, 1,57 ммоль) в сухом DMSO (15 мл) были добавлены КОАс (0,31 г, 3,14 ммоль) и Pd(dppf)Cl2 (0,025 г, 0,03 ммоль) в атмосфере N2. После добавления смесь перемешивалась при температуре 90°С в течение ночи. После получения результатов ТСХ, свидетельствовавших об исчезновении исходного материала, твердое вещество было отфильтровано. Затем были добавлены вода (30 мл) и EtOAc (50 мл), после чего смесь подверглась экстракции при помощи EtOAc (3×30 мл). Объединенный органический слой был промыт солевым раствором, высушен над Na2SO4, подвергся фильтрации и выпариванию до получения сухого вещества. Полученный осадок был очищен посредством колоночной хроматографии для получения N-(2-метил-1-S-2-оксо-6-фенил-3-S-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)пропан-2-ил)-метил-сульфонамида (0,2 г, выход: 37%).
Этап 5
В раствор N-(2-метил-1-S-2-оксо-6-фенил-3-S-1-4-4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)пропан-2-ил)-метил сульфонамида (150 мг, 0,27 ммоль) и 4-йод-1-метилпиридин-2(1Н)-она в сухом 1,4-диоксане (5 мл) были добавлены Cs2CO3 (0,3 мл, 6 ммоль) и Pd(PPh3)Cl2 (20 мг). После добавления смесь нагревалась при температуре 110°С в течение 2 мин. После получения результатов ТСХ, свидетельствовавших об исчезновении исходного материала, твердое вещество было отфильтровано. Затем были добавлены вода (20 мл) и EtOAc (30 мл), после чего смесь подверглась экстракции при помощи EtOAc (3×30 мл). Объединенный органический слой был промыт солевым раствором (50 мл), высушен над Na2SO4, подвергся фильтрации и выпариванию до получения неочищенного продукта, который был очищен посредством препаративной ВЭЖХ для получения N-2-метил-1-S-3-S-1-4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)-фенил)-этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил) пропан-2-ил) метил сульфонамида (20 мг, 14%). Метод ЖХ-МС 2 tR=1,154 мин, масса/заряд = 538,1; 1H ЯМР (CDCl3): δ 1,25 (s, 3Н), 1,32 (s, 3Н), 1,55 (d, 3Н), 2,50 (d, 2H), 2,91 (s, 3H), 3,63 (s, 3H), 4,54 (b, 1H), 5,67 (m, 1H), 6,53 (d, 1H), 6,92 (s, 1H), 7,06 (d, 2H), 7,30-7,50 (m, 8H).
ПРИМЕР 14
2,2-диметил-3-((R)-2-оксо-3-((S)-1-(4-(2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-6-ил)пропаннитрил
Титульное соединение было получено из 2,2-диметил-3-((R)-2-оксо-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)пропаннитрила и 4-йодпиридин-2(1Н)-она в результате процедуры, аналогичной описанной в Примере 4. Метод ЖХ-МС 1 tR=1,36 мин, масса/заряд = 456(М+1); 1H ЯМР (CDCl3) 7,77 (d, 1H), 7,43-7,32 (m, 7H), 7,01 (t, 4H), 5,67 (q, 1H), 2,99 (dd, 1H), 2,57-2,43 (m, 2H), 2,32 (m, 1H), 2,17 (s, 2H), 1,57 (d, 3Н), 1,40 (s, 3Н), 1,33 (s, 3H).
ПРИМЕР 15
3-((R)-6-(4-фторфенил)-2-оксо-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрил
Титульное соединение было получено из 3-((R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-2-оксо-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила и 2-оксо-1,2-дигидропиридин-5-илборной кислоты в результате процедуры, аналогичной описанной в Примере 4, Метод ЖХ-МС 1 tR=1,37 мин, масса/заряд = 474(М+1); 1H ЯМР (CDCl3) 7,97, (dd, 1H), 7,73 (s, 1H), 7,33 (m, 2H), 7,20 (d, 2H), 7,17 (t, 2H), 6,98 (m, 3Н), 5,67 (q, 1H), 3,00 (dt, 1H), 2,49 (m, 2H), 2,30 (m, 1H), 2,13 (s, 2H), 1,55 (d, 3Н), 1,45 (s, 3Н), 1,34 (s, 3H).
ПРИМЕР 16
3-((R)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-2-оксо-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрил
Смесь 3-((R)-6-(4-фторфенил)-2-оксо-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила (100 мг, 0,21 ммоль) и 4-йод-1-метил-1Н-пиридин-2-она (40 мг, 0,17 ммоль), Pd(PPh3)2Cl2 (20 мг), а также водного раствора Cs2CO3 (2,0 мл, 2М) в 1,4-диоксане (5 мл) перемешивалась в условиях обратного конденсирования в течение 2 ч. Органическая фаза была отделена и подверглась выпариванию для получения неочищенного продукта, который затем было очищен посредством препаративной ТСХ для получения 3-((R)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-2-оксо-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила (55 мг, 66%). Метод ЖХ-МС 2 tR=1,096 мин, масса/заряд = 488,3; 1Н ЯМР (CDCl3): δ 1,27 (s, 3Н), 1,40 (s, 3Н), 1,48 (d, 3Н), 2,06 (s, 2H), 2,23 (m, 1H), 2,41 (m, 2H), 2,90 (m, 1H), 3,51 (s, 3Н), 5,60 (m, 1H), 6,27 (m, 1H), 6,65 (d, 1H), 6,89 (d, 2H), 6,99 (t, 2H), 7,26 (m, 5H).
ПРИМЕР 17
6-(2-гидрокси-2-метилпропил)-6-изопропил-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-он
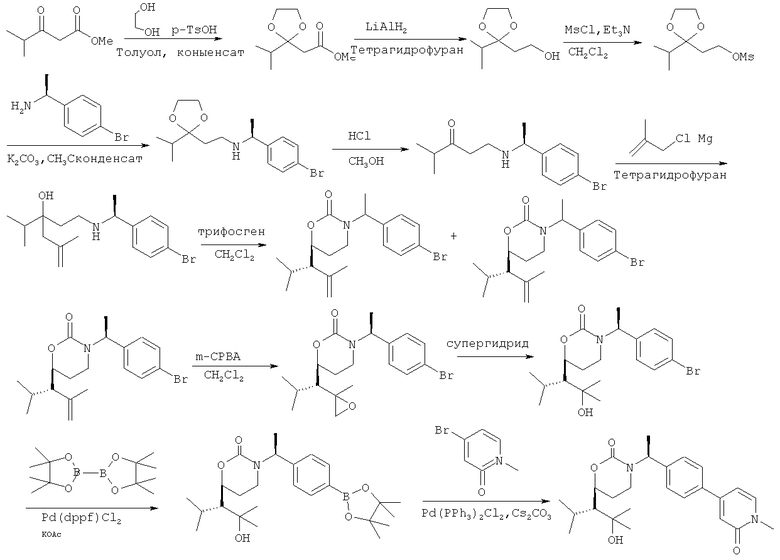
Этап 1
В раствор метил 4-метил-3-оксопентаноата (72 г, 0,5 моль) и этиленгликоля (56 г, 1 моль) в толуоле (500 мл) была добавлена 4-метилбензолсульфоновая кислота (1,9 г, 0,01 моль). Смесь перемешивалась в условиях обратного конденсирования при помощи водоотделителя Дина-Старка для удаления воды. Реакционная смесь затем была промыта небольшим количеством воды и солевого раствора, высушена над безводным Na2SO4 и выпарена в вакууме для получения неочищенного метил 2-(2-изопропил-1,3-диоксолан-2-ил)-ацетата (67 г, выход: 71%), который использовался в ходе следующего этапа без дополнительной очистки.
Этап 2
В высушенную при помощи пламени колбу с тремя горлышками, оснащенную капельной воронкой, магнитной мешалкой, резиновой прокладкой инжектора и клапаном для впрыска азота, были помещены LiAlH4 (3,12 г, 82,1 ммоль) и тетрагидрофуран (700 мл). После охлаждения при температуре 0°С по капле был добавлен раствор метил 2-(2-изопропил-1,3-диоксолан-2-ил) ацетат (12 г, 63,8 ммоль) в тетрагидрофуране (160 мл) с одновременным перемешиванием. Смесь была нагрета до комнатной температуры и перемешивалась в течение 24 часов. Затем была произведена остановка реакции путем медленного добавления воды (5 мл), 15% водного раствора NaOH (10 мл) и снова воды (5 мл). Было произведено отделение органического слоя, после чего осадок был экстрагирован с помощью EtOAc (3×100 мл). Объединенная органическая фаза была высушена над Na2SO4 и выпарена для получения неочищенного продукта, который затем был очищен посредством колоночной хроматографии для получения 2-(2-изопропил-1,3-диоксолан-2-ил)-этанола (6,8 г, 67%). 1H ЯМР (CDCl3): δ 0,90 (d, J=6,8 Гц, 6Н), 1,87-1,96 (m, 3Н), 2,81 (br, 1H), 3,69-3,72 (m, 2H), 3,92-4,01 (m, 4H).
Этап 3
В раствор 2-(2-изопропил-1,3-диоксолан-2-ил)-этанола (8,0 г, 50 ммоль) и триэтиламина (23,5 мл, 170 ммоль) в безводном CH2Cl2 (120 мл) при температуре 0°С был добавлен метансульфонил хлорид (11,6 мл, 150 ммоль), после чего реакционная смесь перемешивалась при комнатной температуре до окончания реакции. Затем реакционная смесь была промыта водой и солевым раствором, высушена над Na2SO4, подверглась фильтрации и выпариванию для получения неочищенного 2-(2-изопропил-1,3-диоксолан-2-ил)этил метансульфоната (12 г, неочищенный), который использовался в ходе следующего этапа без дополнительной очистки.
Этап 4
В раствор 2-(2-изопропил-1,3-диоксолан-2-ил)этил метансульфоната (12 г, 50 ммоль) и (S)-1-(4-метоксифенил)-этил амина (19,9 g, 100 ммоль) в CH3CN (250 мл) был добавлен K2СО3 (8 г, 58 ммоль), после чего смесь подверглась обратному конденсированию в течение 10 ч. Затем раствор подвергся фильтрации с последующим выпариванием полученного фильтрата для получения неочищенного продукта, впоследствии очищенного посредством колоночной хроматографии для получения (S)-1-(4-бромфенил)-N-(2-(2-изопропил-1,3-диоксолан-2-ил)этил)этанамина (6,5 г, выход: 38%).
Этап 5
В раствор (S)-1-(4-бромфенил)-N-(2-(2-изопропил-1,3-диоксолан-2-ил)этил)этанамина (6,5 г, 19 ммоль) в МеОН (60 мл) была добавлена концентрированная HCl (60 мл). Смесь перемешивалась при температуре 65°С до окончания реакции, а затем была охлаждена до 0°С. Уровень рН смеси был отрегулирован до 7 путем добавления насыщенного водного раствора NaHCO3. Далее смесь подверглась выпариванию с последующей экстракцией осадка при помощи EtOAc (3×100 мл). Органический слой был промыт солевым раствором, высушен над Na2SO4 и выпарен для получения (S)-1-(1-(4-бромфенил)этиламино)-4-метилпентан-3-она (5,5 г, выход: 97%), который использовался в ходе следующего этапа без дополнительной очистки. 1Н ЯМР (CDCl3): δ 1,07 (d, J=6,8 Гц, 6Н), 1,29 (d, J=6,4 Гц, 3Н), 1,89 (br, 1H), 2,54-2,62 (m, 4H), 2,66-2,69 (m, 1H), 3,68-3,72 (m, 1H), 7,18-7,20 (m, 2H), 7,41-7,44 (m, 2H).
Этап 6
В суспензию Mg (11 г, 458 ммоль) и 12(0,5 г) в безводном тетрагидрофуране (50 мл) был добавлен 3-хлор-2-метилпроп-1-ен (1 мл) для того, чтобы вызвать реакцию. Затем был добавлен тетрагидрофуран (300 мл), а также дополнительное количество раствора 3-хлор-2-метилпроп-1-ена (15 мл) в тетрагидрофуране (20 мл) по капле при температуре 0°С в атмосфере N2 в течение 30 мин. Далее по капле был добавлен раствор (S)-1-(1-(4-бромфенил)-этил амино)-4-метилпентан-3-она (5 г) в тетрагидрофуране (50 мл) при температуре -78°С в течение 45 мин. Реакционная смесь перемешивалась в течение 2 ч, после чего реакция была осторожно остановлена при помощи насыщенного водного раствора NH4Cl и подверглась фильтрации. Полученный фильтрат подвергся экстракции при помощи EtOAc (3×100 мл), промыванию солевым раствором, высушиванию над безводным Na2SO4, а также выпариванию в вакууме для получения 1-(S-1-(4-бромфениламино)-3-изопропил-5-метилгекс-5-ен-3-ола (6,4 г, выход: 90%), который использовался в ходе следующего этапа без дополнительной очистки.
Этап 7
В раствор 1-(S-1-(4-бромфениламино)-3-изопропил-5-метилгекс-5-ен-3-ола (6,4 г, 16,8 ммоль) и триэтиламина (5,34 г, 52 ммоль) в CH2Cl2 (260 мл) был добавлен трифосген (2,52 г, 8,5 ммоль) при температуре 0°С в атмосфере азота N2, после чего смесь перемешивалась при комнатной температуре в течение ночи. Реакция смеси была остановлена при помощи воды, после чего смесь подверглась экстракции при помощи CH2Cl2 (3×50 мл). Объединенный органический слой был промыт солевым раствором, высушен над Na2SO4, подвергся фильтрации и выпариванию для получения неочищенного продукта, который затем был очищен посредством колоночной хроматографии для получения двух изомеров 3-((S)-1-(4-бромфенил)этил)-6-изопропил-6-(2-метилаллил)-1,3-оксазинан-2-она.
Изомер 1: (1.85 г, выход: 27%) 1H ЯМР (CDCl3): δ 0,83(d, J=7,2 Гц, 3Н), 0,89 (d, J=7,2 Гц, 3Н), 1,45 (d, J=6,8 Гц, 3Н), 1,64-1,70 (m, 2H), 1,79 (s, 3Н), 1,88-1,95 (m, 1H), 2,20-2,34 (m, 2H), 2,59-2,65 (m, 1H), 3,01-3,08 (m, 1H), 4,70 (s, 1H), 4,87 (s, 1H), 5,68-5,77 (m, 1H), 7,14 (d, J=8,4 Гц, 2Н), 7,41 (d, J=8,4 Гц, 2Н).
Изомер 2: (1,25 г, выход: 18%) 1H ЯМР (CDCl3): δ 0,87 (d, J=6,8 Гц, 3Н), 0,92(d, J=6,8 Гц, 3Н), 1,50 (d, J=7,2 Гц, 3Н), 1,60-1,66 (m, 1H), 1,78 (s, 3Н), 1,73-1,79 (m, 1H), 1,78-2,05 (m, 1H), 2,08 (d, J=14,0 Гц, 1H), 2,30 (d, J=14,0 Гц, 1H), 2,62-2,68 (m, 1H), 2,98-3,05 (m, 1H), 4,64 (s, 1H), 4,84 (s, 1H), 5,70-5,75 (m, 1H), 7,13 (d, J=8,4 Гц, 2H), 7,40 (d, J=8,4 ГЦ, 2Н).
Этап 8
В раствор изомера 1 3-((S)-1-(4-бромфенил)этил)-6-изопропил-6-(2-метилаллил)-1,3-оксазинан-2-она (500 мг, 1,32 ммоль) в сухом CH2Cl2 (64 мл) был добавлен при комнатной температуре m-СРВА (455 г, 2,64 ммоль). Реакционная смесь перемешивалась до исчезновения исходного материала (определяется посредством ТСХ). Смесь была разбавлена (СН3)3СОСН3 (70 мл), промыта 30% Na2S3 и водным NaHCO3 (3×), высушена над Na2SO4, подверглась фильтрации и выпариванию для получения изомера 1 3-((S)-1-(4-бромфенил)этил)-6-изопропил-6-((2-метилоксиран-2-ил)метил)-1,3-оксазинан-2-она (520 мг, 99%), который использовался в ходе следующего этапа без дополнительной очистки.
Этап 9
В раствор изомера 1 3-((S)-1-(4-бромфенил)этил)-6-изопропил-6-((2-метилоксиран-2-ил)метил)-1,3-оксазинан-2-она (520 мг, 1,32 ммоль) в тетрагидрофуране (32 мл) при температуре 0°С в атмосфере N2 в течение 30 мин был по капле добавлен LiEt3BH (Супергидрид, 13,6 мл, 13,6 ммоль). Полученный раствор перемешивался при температуре 10-13°С в течение 21,5 ч. В смесь была добавлена H2O2 (40 мл). Полученный раствор был разбавлен (СН3)3СОСН3 (380 мл) и промыт водой, 30% водным раствором Na2S2O3, а также солевым раствором. Органическая фаза была высушена над Na2SO4, после чего подверглась фильтрации. Полученный фильтрат был выпарен для получения неочищенного продукта, позднее очищенного посредством колоночной хроматографии для получения изомера 1 3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-изопропил-1,3-оксазинан-2-она (320 мг, 61%). 1Н ЯМР (CDCl3): δ 0,82 (d, J=6.8 Гц, 3Н), 0,95 (d, J=6,8 Гц, 3Н), 1,31 (s, 3Н), 1,34 (s, 3Н), 1,51 (d, J=10.0 Гц, 3Н), 1,61 (d, J=15,2 Гц, 1Н), 1,78-1,84 (m, 1H), 1,91 (d, J=15,2 Гц, 1Н), 2,02-2,15 (m, 2H), 2,36 (br, 1H), 2,62-2,68 (m, 1Н), 3,03-3,09 (m, 1Н), 5,73 (t, J=7.2 Гц, 1Н), 7,17-7,19 (m, 2H), 7,44-7,48 (m, 2H).
Этап 10
В раствор изомера 1 3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-изопропил-1,3-оксазинан-2-она (315 мг, 0,793 ммоль) в DMSO (10 мл) в атмосфере N2 были добавлены 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолан (602 мг, 2,38 ммоль), CH3CO2K (770 мг, 79,3 ммоль), Pd(dppf)2Cl2 (50 мг, 0,06 ммоль), после чего смесь перемешивалась при температуре 90°С в течение 4 ч. Реакция была остановлена при помощи NH4Cl и подверглась экстракции при помощи EtOAc, а также промыванию водой и солевым раствором. Органическая фаза была высушена над Na2SO4 и подверглась фильтрации. Полученный фильтрат был выпарен для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения изомера 1 6-(2-гидрокси-2-метилпропил)-6-изопропил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (250 мг, 71%).
Этап 11
В раствор изомера 1 6-(2-гидрокси-2-метилпропил)-6-изопропил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (250 мг, 0,39 ммоль), 4-бром-1-метилпиридин-2(1Н)-она (127 мг, 0,68 ммоль) и Cs2CO3 (2N, 4 мл) в 1,4-диоксане (20 мл) в атмосфере N2 был добавлен Pd(PPh3)2Cl2 (54 мг, 0,056 ммоль). Реакционная смесь подверглась обратному конденсированию в течение 2 ч, после чего реакция была остановлена водой, а смесь подверглась экстракции при помощи EtOAc. Органическая фаза была промыта Н2О и солевым раствором, высушена над Na2SO4 и подверглась фильтрации. Фильтрат был выпарен для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ и препаративной ВЭЖХ для получения изомера 1 6-(2-гидрокси-2-метилпропил)-6-изопропил-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-она (79 мг, выход; 47%). Метод ЖХ-МС 2 tR=1,023 мин, масса/заряд = 427,6; 1H ЯМР (CDCl3) 0,85 (d, 3H), 0,96 (d, 3Н), 1,26 (s, 3H), 1,28 (s, 3H), 1,54 (m, 4H), 1,84-1,88 (m, 2H), 2,04 (br, 1H), 2,01-2,18 (m, 2H), 2,75 (m, 1H), 3,10 (m, 1H), 3,52 (s, 3H), 5,80 (t, 1H), 6,37 (m, 1H), 6,74 (m, 1H), 7,28 (m, 1H), 7,25-7,37 (m, 2H), 7,50 (m, 2H).
Изомер 2 6-(2-гидрокси-2-метилпропил)-6-изопропил-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-она был получен из изомера 2 3-((S)-1-(4-бромфенил)этил)-6-изопропил-6-(2-метилаллил)-1,3-оксазинан-2-она в результате процедур, аналогичных описанным для этапов 8-11. Метод ЖХ-МС 2 tR=1,023 мин, масса/заряд = 427,6; 1H ЯМР (CDCl3) 0,79 (d, 3H), 0,92 (d, 3H), 1,27 (s, 3H), 1,30 (s, 3Н), 1,51 (d, 3H), 1,58 (d, 1H), 1,73-1,81 (m, 1H), 1,88 (d, 1H), 2,0 (br, 1H), 2,04-2,08 (m, 2H), 2,65-2,68 (m, 1H),3,04-3,07 (m, 1H), 3,52 (s, 3Н), 5,75 (t, 1H), 6,37 (m, 1H), 6,74 (m, 1H), 7,21-7,35 (m, 3Н), 7,51 (m, 2H).
ПРИМЕР 18
6-циклопропил-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-он
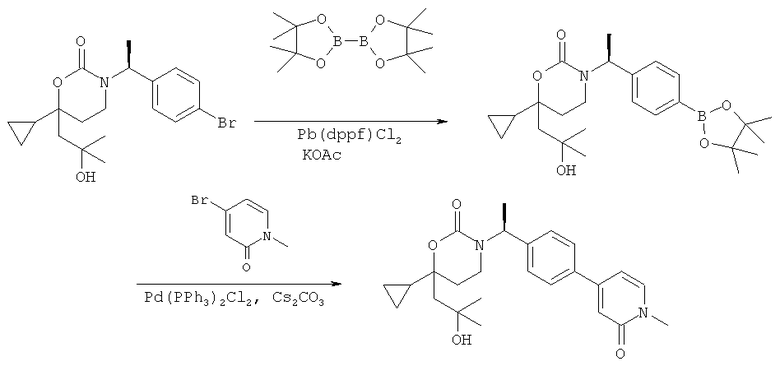
Два диастереомера 3-((S)-1-(4-бромфенил)этил)-6-циклопропил-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-она были получены из метил 3-циклопропил-3-оксопропаноата после применения процедур, аналогичных описанным для этапов 1-7 Примера 17. Титульное соединение было получено следующим образом.
Этап 1
В раствор изомера 1 3-((S)-1-(4-бромфенил)этил)-6-цикпопропил-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-она (230 мг, 0,58 ммоль) в DMSO (15 мл) в атмосфере N2 были добавлены 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолан (450 мг, 1,77 ммоль), СН3СО2К (800 мг, 8,16 ммоль), Pd(pddf)2Cl2 (50 мг, 0,06 ммоль). Смесь перемешивалась при 90°С в течение 34 ч, после чего реакция была остановлена с помощью NH4Cl и подверглась экстракции при помощи EtOAc. Органическая фаза была промыта водой и солевым раствором, высушена над Na2SO4 и подверглась фильтрации. Полученный фильтрат подвергся выпариванию для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения изомера 1 6-циклопропил-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (140 мг, 54,3%).
Этап 2
В раствор изомера 1 6-циклопропил-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (140 мг, 0,316 ммоль), 4-бром-1-метилпиридин-2(1Н)-она (74,3 мг, 0,316 ммоль), а также 2 М водного раствора Cs2CO3 (3 мл) в 1,4-диоксане (20 мл) в атмосфере N2 был добавлен Pd(dppf)2Cl2 (30 мг, 0,043 ммоль). Реакционная смесь подверглась обратному конденсированию в течение 2 ч, после чего реакция была остановлена водой, а смесь подверглась экстракции при помощи EtOAc. Органическая фаза была промыта N2O и солевым раствором, высушена над Na2SO4, а затем подверглась фильтрации. Полученный фильтрат подвергся выпариванию для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ и препаративной ВЭЖХ для получения изомера 1 6-циклопропил-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-она (49,5 мг, 37,7%). Метод ЖХ-МС 2 tR=1,016 мин, масса/заряд = 367,2; 1H ЯМР (CDCl3) 0,50 (m, 2H), 0,62 (m, 2H), 0,97 (m, 1H), 1,32 (m, 6H), 1,58 (d, 3H), 1,97(m, 3Н), 2,28 (m,1H), 2,78 (m, 1H), 3,40 (m, 1H), 3,58 (s,3H), 5,85 (m, 1H), 6,41 (d, 1H), 6,79 (s, 1H), 7,33 (d, 1H), 7,41 (d, 1H), 7,56 (d, 1H)
Изомер 2 6-циклопропил-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-она был получен из изомера 2 3-((S)-1-(4-бромфенил)этил)-6-циклопропил-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-она посредством процедур, аналогичных описанным выше для 1 и 2 этапов. Метод ЖХ-МС 2 tR=0,99 мин, масса/заряд = 367,1; 1H ЯМР (CDCl3) 0,02 (m, 3Н), 0,23 (m, 1H), 0,51 (m,1H), 0,96 (s, 6H), 1,17 (d, 3Н), 1,40-1,60 (m, 4H), 1,94 (m, 1H), 2,55 (m, 1H), 2,73 (m, 1H), 3,20 (s, 3H), 5,41 (m, 1H), 6,03 (d, 1H), 6,40 (s, 1H), 6,98 (m, 1H), 7,03 (m, 2H), 7,18 (m, 2H).
ПРИМЕР 19
(R)-6-(2-(5-метил-1,3,4-оксадиазол-2-ил)этил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
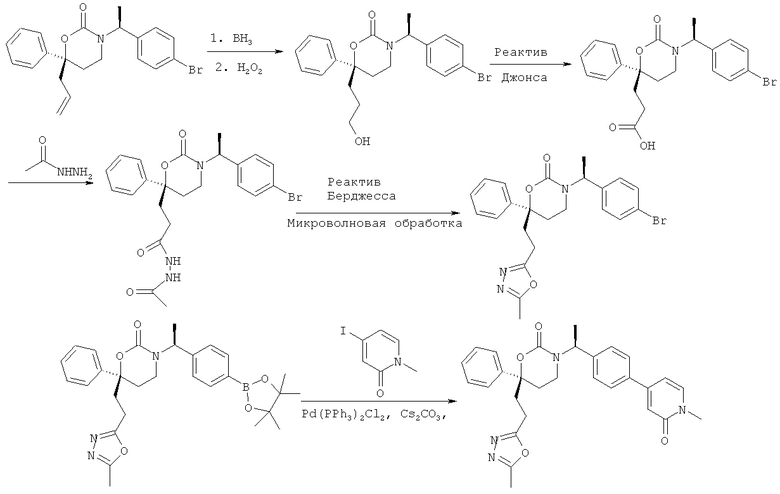
Этап 4
В раствор N'-ацетил-3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропангидразида (0,1 г, 0,21 ммоль) в тетрагидрофуране (2 мл) был добавлен реактив Берджесса (75 мг, 0,315 ммоль). Запечатанный флакон был подвергнут обработке в микроволновой печи при температуре 100°С в течение 15 мин. Полученная смесь подверглась экстракции при помощи EtOAc (3×30 мл). Объединенный органический слой был промыт солевым раствором (50 мл), высушен над Na2SO4, затем подвергся фильтрации и выпариванию. Полученный осадок был очищен посредством препаративной ТСХ для получения (R)-3-((S)-1-(4-бромфенил)этил)-6-(2-(5-метил-1,3,4-оксадиазол-2-ил)этил)-6-фенил-1,3-оксазинан-2-она (58 мг, выход: 59%). 1H ЯМР (CDCl3): 01,49-1,51 (m, 3Н), 2,23-2,26 (m, 2H), 2,30-2,33 (m, 2H), 2,42 (s, 3H), 2,43-2,45 (m, 1H), 2,49-2,54 (m, 1H), 2,87-2,91 (m, 1H), 3,06-3,09 (m, 1H), 5,61-5,63 (m, 1H), 6,76-6,78 (d, 2H), 7,20-7,22 (m, 2H), 7,26-7,33 (m, 2H), 7,35-7,37 (m, 3H).
Этап 5
В раствор (R)-3-((S)-1-(4-бромфенил)этил)-6-(2-(5-метил-1,3,4-оксадиазол-2-ил)этил)-6-фенил-1,3-оксазинан-2-она (490 мг, 1,04 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (424 мг, 1,67 ммоль) в сухом DMSO (20 мл) в атмосфере N2 были добавлены КОАс (326 мг, 3,33 ммоль) и Pd(dppf)Cl2 (25,3 мг, 0,031 ммоль). Смесь нагревалась при температуре 100°С в течение 3 ч. После получения результатов ТСХ, свидетельствующих об исчезновении исходного материала, было отфильтровано твердое вещество, затем добавлены вода (50 мл) и EtOAc (50 мл), после чего смесь подверглась экстракции при помощи EtOAc (3×50 мл). Объединенный органический слой был промыт солевым раствором (50 мл), высушен над Na2SO4, затем подвергся фильтрации и выпариванию. Полученный осадок был очищен посредством препаративной ТСХ для получения (R)-6-(2-(5-метил-1,3,4-оксадиазол-2-ил)этил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (0,395 г, выход: 73,6%).
Этап 6
В раствор (R)-6-(2-(5-метил-1,3,4-оксадиазол-2-ил)этил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (60 мг, 0,12 ммоль) и 4-йод-1-метилпиридин-2(1Н)-она (33 мг, 0,14 ммоль) в сухом 1,4-диоксане (15 мл) были добавлены Cs2CO3 (2М, 1 мл) и Pd(PPh3)Cl2 (7,7 мг, 0,01 ммоль). Смесь нагревалась в условиях обратного конденсирования в течение 2 ч в атмосфере N2, после чего полученное твердое вещество отфильтровывалось, а смесь разбавлялась водой (30 мл) и EtOAc (30 мл). Объединенный органический слой был промыт солевым раствором (50 мл), высушен над Na2SO4, затем подвергся фильтрации и выпариванию. Полученный осадок был очищен посредством препаративной ТСХ для получения (R)-6-(2-(5-метил-1,3,4-оксадиазол-2-ил)этил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (25 мг, выход: 41,8%). Метод ЖХ-МС 2 tR=0,984 мин, масса/заряд = 499,1; 1Н ЯМР (CDCl3): δ 1,48-1,50 (m, 3Н), 2,16-2,20 (m, 1H), 2,23-2,26 (m, 1H), 2,27-2,32 (m, 2H), 2,39 (s, 3Н), 2,40-2,47 (m, 1H), 2,49-2,54 (m, 1H), 2,87-2,90 (m, 1H), 2,98-3,01 (m, 1H), 3,58 (s, 3Н), 5,62-5,64 (m, 1H), 6,45-6,48 (m, 1H), 6,87 (s, 1H), 6,92-6,94 (d, 2H), 7,20-7,24 (m, 4H), 7,25-7,35 (m, 4H).
ПРИМЕР 20
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-(2-гидроксиэтил)-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
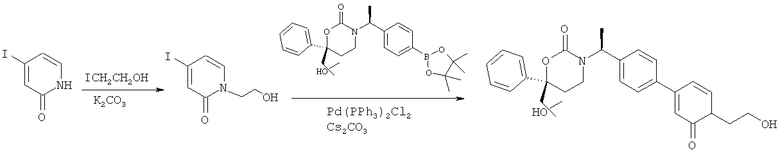
Этап 1
В смесь 4-йодпиридин-2(1Н)-она (50 мг, 0.213 ммоль) и DMF (3 мл) были добавлены 2-йодзтанол (73 мг, 0,426 ммоль), K2СО3 (88 мг, 0,638 ммоль) при комнатной температуре. Смесь перемешивалась в течение 2 ч при комнатной температуре. После окончания реакции смесь была промыта водой и подверглась экстракции при помощи EtOAc. Органическая фаза была промыта солевым раствором, высушена над Na2SO4, затем подверглась фильтрации и выпариванию для получения неочищенного продукта, который был очищен посредством ТСХ для получения 1-(2-гидроксиэтил)-4-йодпиридин-2(1Н)-она (60 мг 100%).
Этап 2
Смесь соединений (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (72 мг, 0,150 ммоль), 1-(2-гидроксиэтил)-4-йодпиридин-2(1Н)-она (48 мг, 0,181 ммоль), Pd(PPh3)2Cl2 (14 мг, 0,020 ммоль), а также Cs2CO3 (2 мл) в 1,4-диоксане (8 мл) перемешивалась в условиях обратного конденсирования в течение 2 ч. После окончания реакции смесь была промыта водой и подверглась экстракции EtOAc. Органическая фаза была промыта солевым раствором, высушена над Na2SO4, затем подверглась фильтрации и выпариванию для получения неочищенного продукта, который был очищен посредством ТСХ для получения соединения (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-(2-гидроксиэтил)-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (19,7 мг, 28%). Метод ЖХ-МС 2 tR=1,065 мин, масса/заряд = 491,2; 1Н ЯМР (CDCl3): δ 1,10 (d, 6H), 1,50 (d, 3H), 2,20 (m, 5H), 2,35 (m, 1H), 3,50 (m, 1H), 3,90 (m, 2H), 4,10 (m, 2H), 5,60 (m, 1H), 6,40 (m, 1H), 6,70 (s, 1H), 6,95 (d, 2H), 7,35 (m, 8H).
ПРИМЕР 21
(6S)-6-(2,3-дигидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
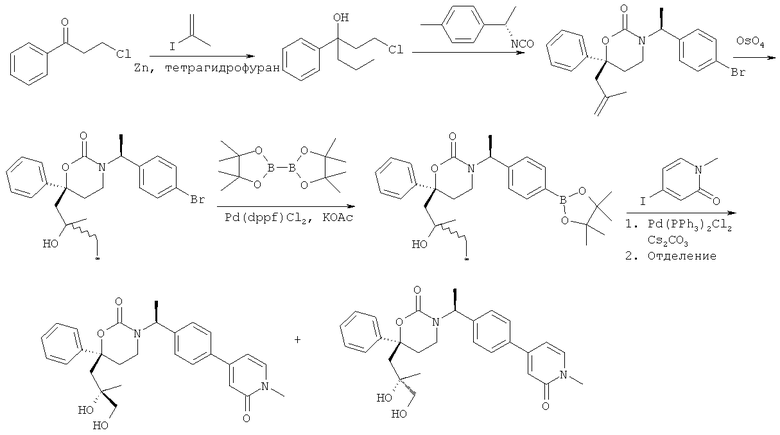
Этап 1
Раствор 3-хлор-1-фенилпропан-1-она (16,8 г, 0.1 моль) в тетрагидрофуране (50 мл) был добавлен в тщательно перемешанную суспензию цинкового порошка (13 г, 0,2 моль) в смеси насыщенного водного раствора NH4Cl (260 мл) и тетрагидрофурана (65 мл). Затем в полученную смесь был по капле добавлен раствор 3-йод-2-метилпроп-1-ена (36,4 г, 0,2 моль) в тетрагидрофуране (50 мл). В ходе реакции наблюдалось незначительное выделение тепла, а также непредвиденное обратное конденсирование. После прекращения обратного конденсирования смесь перемешивалась в течение 1 ч. Результаты ТСХ показали, что реакция 3-хлор-1-фенилпропан-1-она не была полностью окончена. В смесь был добавлен раствор 3-йод-2-метилпроп-1-ена (18,2 г, 0,1 моль) в тетрагидрофуране (30 мл), после чего смесь перемешивалась в течение ночи при комнатной температуре. Затем смесь подвергалась экстракции при помощи EtOAc (2×500 мл). Объединенный органический слой был высушен и выпарен, Полученный осадок был очищен посредством колоночной хроматографии с применением силикагеля, элюированного при помощи петролейного эфира / EtOAc 50:1→30:1→5:1, для получения 1-хлор-5-метил-3-фенилгекс-5-ен-3-ола (17 г, выход 76%) в виде масла. 1Н ЯМР (CDCl3): δ 1,28 (s, 3Н), 2,31 (m, 2Н), 2,54 (m, 2Н), 2,68 (d, 1H), 3,15 (m, 1H), 3,58 (m, 1H), 4,78 (m, 1H), 4,93 (m, 1H), 7,27 (t, 1H), 7,38 (m, 4H).
Этап 2
Смесь 1-хлор-5-метил-3-фенилгекс-5-ен-3-ола (2,9 г, 13 ммоль), (S)-1-бром-4-(1-изоцианатоэтил)бензол (3,5 г, 16 ммоль), а также DBU (8 г, 33 ммоль) в тетрагидрофуране (80 мл) нагревалась в условиях обратного конденсирования в течение ночи. Смесь была разбавлена EtOAc, промыта 1N водным раствором HCl. Водная фаза подверглась экстракции при помощи EtOAc (3×), после чего объъединенная органическая фаза была высушена над Na2SO4. После выпаривания растворителей неочищенный продукт был очищен посредством колоночной хроматографии для получении (R)-3-((S)-1-(4-бромфенил)-этил)-6-(2-метилаллил)-6-фенил-1,3-оксазинан-2-она (1,62 г, выход: 30%).
Этап 3
В раствор (R)-3-((S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-фенил-1,3-оксазинан-2-она (300 мг, 0,726 ммоль), 4-метилморфолин 4-оксида (195 мг, 1,44 ммоль) в смеси H2O (6 мл), тетрагидрофурана (30 мл) и f-BuOH (12 мл) была добавлена окись осмия (VIII) (4%, 0,231 мл) при температуре 0°С. Смесь перемешивалась при комнатной температуре в течение ночи, после чего реакция была остановлена с помощью 3% NaHSO3 (15 мл × 3), а смесь подверглась экстракции при помощи EtOAc. Органический слой был высушен над Na2SO4 и выпарен для получения неочищенного продукта (S)-3-((S)-1-(4-бромфенил)-этил)-6-(2,3-дигидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (242 мг, 74,5%).
Этап 4
В раствор (S)-3-((S)-1-(4-бромфенил)-этил)-6-(2,3-дигидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (235 мг, 0,524 ммоль) в DMSO (5 мл) в атмосфере N2 были добавлены КОАс (771,6 мг, 7,86 ммоль) и Pd(dppf)Cl2 (40 мг). Смесь перемешивалась при температуре 90°С в течение 3 ч. Реакция была остановлена при помощи воды и подверглась экстракции при помощи EtOAc. Органический слой был высушен над Na2SO4 и подвергся выпариванию для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (S)-6-(2,3-дигидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (121 мг, 46,6%).
Этап 5
Смесь (S)-6-(2,3-дигидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (105 мг, 211,9 ммоль), 4-йод-1-метилпиридин-2(1Н)-она (65 мг, 275,5 ммоль), а также Pd(PPh3)2Cl2 (20 мг) в водном растворе Cs2CO3 (3 мл) перемешивалась в условиях обратного конденсирования в течение 2 ч. После окончания реакции смесь была промыта водой и подверглась экстракции при помощи EtOAc. Органический слой был высушен над Na2SO4 и подвергся выпариванию для получения неочищенного продукта, который затем был очищен посредством препаративной ВЭЖХ для получения двух изомеров (6S)-6-(2,3-дигидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она.
Изомер 1 (6,11 мг, 6%): Метод ЖХ-МС 2 tR=0,84 мин, масса/заряд =477,4; 1Н ЯМР (CDCl3): δ 0,97 (s, 3Н), 1,55 (d, 3Н), 2,27 (m, 3Н), 2,38 (m, 3H), 2,91 (m, 1H), 3,34 (d, 1H), 3,58 (s, 3Н), 5,68 (m, 1H), 6,35 (d, 1H), 6,71 (s, 1H), 7,02 (d, 2H), 7,36 (m, 8H).
Изомер 2 (6,78 мг, 6,7%): Метод ЖХ-МС 2 tR=0,832 мин, масса/заряд =477; 1H ЯМР (CDCl3): δ 1,14 (s, 3Н), 1,48 (d, 3Н), 2,09 (m, 1H), 2,14 (m, 2H), 2,31 (m, 2H), 2,81 (m, 1H), 3,24 (d, 1H), 3,27 (d, 1H), 3,50 (s, 3Н), 5,62 (m, 1H), 6,28 (d, 1H), 6,63 (s, 1H), 6,98 (d, 2H), 7,27 (m, 8H).
ПРИМЕР 22
(6S)-6-(2-гидрокси-3-метокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
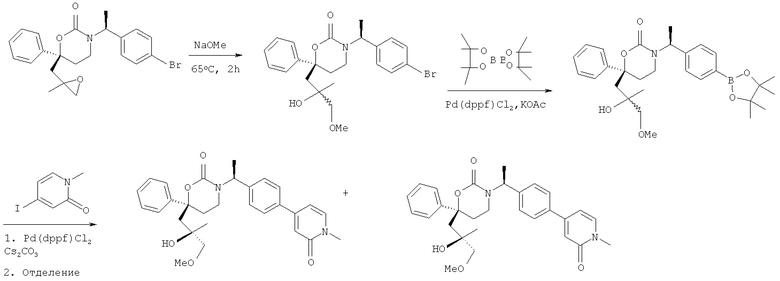
Этап 1
В МеОН (5 мл) был добавлен натрий (90 мг). После исчезновения натрия в смесь был добавлен раствор 6(S)-3-(5-1-(4-бромфенил)этил)-6-((2-метилоксиран-2-ил)-метил)-6-фенил-1,3-оксазинан-2-она (500 мг, 1,16 ммоль). Смесь перемешивалась при температуре 65°С в течение 5 ч, затем в нее была добавлена Н2О, после чего смесь подверглась выпариванию. Полученный осадок подвергся экстракции при помощи EtOAc, органический слой был высушен над Na2SO4 и выпарен для получения неочищенного продукта, который затем был очищен посредством ТСХ (2:1 PE/EtOAc) для получения (S)-3-(5-1-(4-бромфенил)этил)-6-(2-гидрокси-3-метокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (227 мг, 42,3%).
Этап 2
В раствор (S)-3-(S-1-(4-бромфенил)этил)-6-(2-гидрокси-3-метокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (205 мг, 0,443 ммоль) в DMSO (5 мл) в атмосфере N2 были добавлены КОАс (435,13 мг, 4,43 ммоль), Pd(dppf)Cl2 (45 мг). Смесь перемешивалась при температуре 90°С в течение 3 часов. Реакция была остановлена при помощи воды, после чего смесь была подвергнута экстракции при помощи EtOAc. Органический слой был высушен над Na2SO4 и выпарен для получения неочищенного продукта, который был очищен посредством ТСХ (РЕ:ЕА=1:2) для получения (S)-6-(2-гидрокси-3-метокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (113 мг, 50%).
Этап 3
Смесь (S)-6-(2-гидрокси-3-метокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (100 мг, 196,29 ммоль), 4-йод-1-метилпиридин-2(1Н)-она (60,49 мг, 255,2 ммоль), Pd(PPh3)2Cl2 (20 мг) и водного раствора Cs2CO3 (2 моль/л, 3 мл) в 1,4-диоксане (4 мл) перемешивалась в условиях обратного конденсирвоания в течение 2 часов. После окончания реакции смесь была промыта водой и подверглась экстракции при помощи EtOAc. Органический слой был высушен над Na2SO4 и подвергся выпариванию для получения неочищенного продукта, который затем был очищен посредством перпаративной ВЭЖХ для получения двух изомеров (6S)-6-(2-гидрокси-3-метокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она.
Изомер 1 (5,25 мг, 5,6%); Метод ЖХ-МС 2 tR=0,921 мин, масса/заряд=403,2; 1H ЯМР (CDCl3): δ 0,94 (s, 3Н), 1,47 (d, 3Н), 2,28 (m, 4Н), 2,35 (m, 1H), 2,82 (m,1H), 3,11 (d, 1H), 3,16 (s, 3Н), 3,25 (d, 1H), 3,50 (s, 3Н), 5,62 (m, 1H), 6,27 (d, 1H), 6,63 (s, 1H), 6,92 (d, 2H), 7,25 (m, 8H).
Изомер 2 (5,40 мг, 5,8%); Метод ЖХ-МС 2 tR=0,923 мин, масса/заряд=513,1; 1H ЯМР (CDCl3): δ 1,18 (s, 3Н), 1,47 (d, 3Н), 2,11 (m, 2H), 2,23 (m, 2H), 2,45 (m, 1H), 2,81 (d, 2H), 2,96 (d, 1H), 3,15 (s, 3Н), 3,50 (s, 3Н), 5,62 (m, 1H), 6,27 (d, 1H), 6,65 (s, 1H), 6,90 (d, 2H), 7,26 (m, 8H).
ПРИМЕР 23
(S)-6-(2-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-он
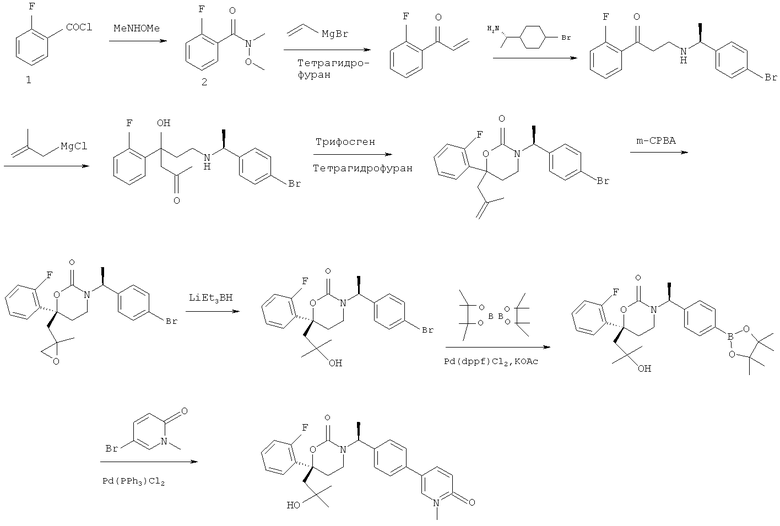
Этап 1
В раствор 2-фторбензоилхлорида (50 г, 0,31 моль) в CH2Cl2 (200 мл) при температуре 0°С был добавлен N,О-диметилгидроксиламин (46 г, 0,47 моль), а также раствор триэтиламина (127 г, 1,26 моль) в CH2Cl2 (100 мл). Реакционная смесь была медленно нагрета до комнатной температуры, после чего перемешивалась в течение 3 ч. Затем реакция была остановлена при помощи ледяной воды, а смесь подверглась экстракции CH2Cl2 (200 мл). Органический слой был высушен над Na2SO4, а также подвергся фильтрации и выпариванию для получения 2-фтор-N-метокси-N-метилбензамида (48 г, выход: 84,6%).
Этап 2
Раствор 2-фтор-N-метокси-N-метилбензамид (16 г, 87,4 ммоль) в тетрагидрофуране (150 мл) был охлажден до -78°С. Затем медленно был добавлен винилмагний бромид (120 мл, 120 ммоль), после чего смесь перемешивалась при -78°С в течение 10 мин, медленно нагревалась до комнатной температуры и дополнительно перемешивалась в течение 3 ч. Реакция была остановлена с помощью 1 N водного раствора HCl (100 мл) при температуре 0°С. Водный слой подвергся экстракции при помощи EtOAc (100 мл). Объединенная органическая фаза была промыта солевым раствором (50 мл), высушена над Na2SO4 и выпарена. Полученный осадок был очищен посредством колоночной хроматографии для получения 1-(2-фторфенил)-проп-2-ен-1-она (7,6 г, выход: 58,4%) в виде бесцветного масла.
Этап 3
В раствор 1-(2-фторфенил)-проп-2-ен-1-она (5,6 г, 37,3 ммоль) в CH3CN (50 мл) был добавлен (S)-1-(4-бромфенил)-этиламин (7,4 г, 37 ммоль), после чего смесь перемешивалась в течение 12 ч при температуре 40°С. Полученный раствор был выпарен для получения неочищенного продукта, который затем был очищен посредством колоночной хроматографии для получения (S)-3-(1-(4-бромфенил)-этиламино)-1-(2-фторфенил)-пропан-1-она (4 г, выход: 30,7%) в виде жидкости желтого цвета.
Этап 4
В суспензию Mg (2,5 г, 104 ммоль) и I2 (0,1 г) в безводном тетрагидрофуране (15 мл) был добавлен 3-хлор-2-метилпроп-1-ен (0,6 мл, 6 ммоль). После добавления 3-хлор-2-метилпроп-1-ена (9 мл, 90 ммоль) в тетрагидрофуране (120 мл) по капле при температуре 0°С в атмосфере N2 в течение 30 мин, а также по капле при температуре -78°С в течение 45 минут добавлялся (S)-3-(1-(4-бромфенил)-этиламино)-1-(2-фторфенил)-пропан-1-он (3 г, 8,6 ммоль) в тетрагидрофуране (50 мл). Реакционная смесь перемешивалась при комнатной температуре в течение 2 ч, после чего реакция была осторожно остановлена путем добавления насыщенного водного раствора NH4Cl. Смесь подверглась экстракции при помощи EtOAc, после чего органический слой был промыт солевым раствором, высушен над безводным Na2SO4 и выпарен в вакууме для получения 1-(5-1-(4-бромфенил)-этиламино)-3-(2-фторфенил)-5-метилгекс-5-ен-3-ола (3,3 г, выход: 94,5%), который затем использовался в ходе следующего этапа без дополнительной очистки.
Этап 5
В смесь 1-(S-1-(4-бромфенил)этиламино)-3-(2-фторфенил)-5-метилгекс-5-ен-3-ола (2 г, 5 ммоль) и раствора триэтиламина (1,5 г, 15 ммоль) в 1,2-дихлорэтане (100 мл) при температуре 0°С в атмосфере N2 был добавлен трифосген (1,46 г, 5 ммоль), после чего смесь нагревалась до температуры 100°С в течение 4 ч. Реакция была остановлена при помощи воды, после чего смесь подверглась экстракции с помощью CH2Cl2 (100 мл). Объединенный органический слой был промыт солевым раствором и высушен над Na2SO4, после чего подвергся фильтрации и выпариванию для получения неочищенного продукта (2,1 г, выход: 99%), который использовался в ходе следующего этапа без дополнительной очистки.
Этап 6
В раствор (S)-3-(1-(4-бромфенил)этил)-6-(2-фторфенил)-6-(2-метилаллил)-1,3-оксазинан-2-она (3,2 г, 7,4 ммоль) в сухом CH2Cl2 (100 мл) при комнатной температуре был добавлен m-СРВА (2,6 г, 14,8 ммоль), после чего смесь перемешивалась в течение ночи. Затем смесь была разбавлена с помощью (СН3)3СОСН3 (100 мл), промыта 30% водным раствором Na2S2O3 и водным раствором NaHCO3, и высушена над Na2SO4, после чего подверглась фильтрации и выпариванию для получения 3-(S-1-(4-бромфенил)-этил)-6-(2-фторфенил)-6-(2-метилоксиран-2-ил-метил)-1,3-оксазинан-2-она (2,8 г, выход: 84,3%), который использовался в ходе следующего этапа без дополнительной очистки.
Этап 7
В раствор 3-(S-1-(4-бромфенил)-этил)-6-(2-фторфенил)-6-(2-метилоксиран-2-ил)-метил)-1,3-оксазинан-2-она (2,2 г, 4,92 ммоль) в тетрагидрофуране (100 мл) по капле при температуре 0°С в атмосфере N2 в течение 30 мин был добавлен LiEt3BH (супергидрид, 50 мл, 50 ммоль). Полученная смесь перемешивалась при температуре 2-3°С в течение 1,5 ч, а затем при температуре 10-13°С в течение 2,5 ч. В смесь по капле была добавлена H2O2 (20 мл), после чего реакционная смесь была разбавлена (СН3)3СОСН3 (280 мл) и промыта водой, 30% водным раствором Na2S2O3 и солевым раствором Органическая фаза была высушена над Na2SO4 и подверглась фильтрации. Полученный фильтрат был выпарен для получения неочищенного продукта, который затем был очищен посредством колоночной хроматографии для получения (S)-3-(S-1-(4-бромфенил)-этил)-6-(2-фторфенил)-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-она (550 мг, 23,9%) в виде твердого вещества белого цвета. 1H ЯМР (CDCl3): δ 1,03 (s, 3Н), 1,14 (s, 3Н), 1,43 (d, J=6,8 Гц, 3Н), 2,08-2,13 (m, 1H), 2,17 (d, J=2,8 Гц, 1Н), 2,21-2,22 (m, 1H), 2,31 (dd, J=0,8, 15,2 Гц, 1H), 2,77-2,81 (m, 1H), 5,56 (q, J=2,8 Гц, 2 Н), 6,82-6,83 (m, 2H), 6,85-6,94 (m, 1H), 7,08-7,13 (m, 1H), 7,18-7,25 (m, 1H), 7,26-7,40 (m, 2H), 7,42-7,44 (m, 1H).
Этап 8
В раствор (S)-3-(S-1-(4-бромфенил)-этил)-6-(2-фторфенил)-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-она (540 мг, 1,2 ммоль) в DMSO (15 мл) в атмосфере N2 были добавлены 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,3,2-диоксаборолан (900 мг, 3,3 ммоль), СН3СО2K (1,5 г, 16 ммоль), Pd(dppf)Cl2 (108 мг, 0,13 ммоль), после чего реакционная смесь перемешивалась при температуре 90°С в течение 2,5 ч. Реакция была остановлена при помощи воды, затем смесь подверглась экстракции при помощи EtOAc (90 мл). Органический слой был промыт водой и солевым раствором, высушен над Na2SO4 и подвергся фильтрации. Полученный фильтрат был выпарен для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (S)-6-(2-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (360 мг, 62%) в виде жидкости желтого цвета.
Этап 9
В раствор (S)-6-(2-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (120 мг, 0,24 ммоль), 5-бром-1-метилпиридин-2-(1Н)-она (54 мг, 0,28 ммоль) и 2N водного раствора Cs2CO3 (2 мл) в диоксане (8 мл) в атмосфере N2 был добавлен Pd(PPh3)2Cl2 (17 мг, 0,024 ммоль). Реакционная смесь подверглась обратному конденсированию в течение 2 ч, после чего в нее была добавлена вода для остановки реакции. Затем смесь подверглась экстракции при помощи EtOAc (30 мл), после которой органический слой был промыт H2O и солевым раствором, высушен над Na2SO4 и подвергся фильтрации. Полученный фильтрат был выпарен для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ и препаративной ВЭЖХ для получения (S)-6-(2-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (14 мг, выход:10%). Метод ЖХ-МС 2 tR=1,2 мин, масса/заряд=473,9; 1H ЯМР (CDCl3): δ 1.21 (s, 3Н), 1.30 (s, 3H), 1,48 (d, J=7,2 Гц, 3Н), 2,15-2,26 (m, 3Н), 2,33 (dd, J=11,2, 26,4 Гц, 1Н), 2,43-2,46 (m, 1H), 2,79-2,85 (m, 1H), 3,63 (s, 3Н), 5,62 (q, J=6,8 Гц, 1H), 6,58-6,60 (m, 1H), 6,89-6,94 (m, 1H), 7,00-7,02 (m, 2H), 7,10-7,24 (m, 3Н), 7,34-7,39 (m, 1H), 7,40-7,45 (m, 1H), 7,48-7,70 (m, 2H).
ПРИМЕР 24
(S)-6-(3-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-он
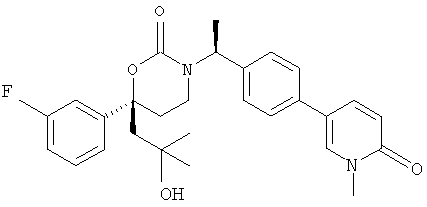
Титульное соединение было получено из (S)-3-(1-(4-бромфенил)этиламино)-1-(3-фторфенил)пропан-1-она после проведения процедур, аналогичных описанным для этапов 4-9 Примера 23. Метод ЖХ-МС 2 tR=1,416 мин, масса/заряд=500,9; 1H ЯМР (CDCl3) 1,18 (s, 3Н), 1,29 (s, 3Н), 1,47 (d, 3Н), 2,05-2,28 (m, 4H), 2,31-2,39 (m, 1H), 2,82-2,87 (m, 1H), 3,58 (s, 3Н), 5,64 (q, 1H), 6,57-6,59 (m, 1H), 6,88-7,19 (m, 5H), 7,21-7,25 (m, 2H), 7,28 (m, 1H), 7,36 (m, 1H), 7,40-7,45 (m, 1H), 7,45-7,48 (m, 1H)
(S)-3-(1-(4-бромфенил)этиламино)-1-(3-фторфенил)пропан-1-он был приготовлен в соответствии с представленными ниже процедурами.
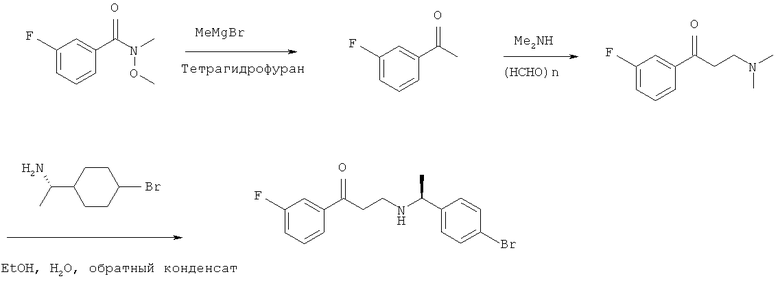
Этап 1
Раствор 3-фтор-N-метокси-N-метилбензамида (16 г, 87,4 ммоль) в тетрагидрофуране (150 мл) был охлажден до -78°С. Затем в раствор медленно был добавлен винилмагний бромид (120 мл, 120 ммоль), после чего смесь перемешивалась при температуре -78°С в течение 10 мин, а также при комнатной температуре в течение 3 ч. Реакция была остановлена при помощи 1 N водного раствора HCl (100 мл) при температуре 0°С. Водный слой подвергся экстракции при помощи EtOAc (100 мл). Объединенный органический слой был промыт солевым раствором (50 мл), высушен над Na2SO4 и выпарен. Полученный осадок был очищен посредством колоночной хроматографии для получения 1-(3-фторфенил)этанона (9,7 г, выход: 75%) в виде бесцветного масла.
Этап 2
1-(3-Фторфенил)этанон (17 г, 0,123 моль), диметиламин (13,7 г, 0,172 моль) и параформальдегид (5,5 г, 0,185 моль) были взвешены в этаноле (50 мл), после чего в суспензию был добавлен концентрированный раствор HCl (0,3 мл). Смесь нагревалась в условиях обратного конденсирования в течение ночи. После удаления в вакууме растворителя полученный осадок был промыт EtOAc (3×) для получения 3-(диметиламино)-1-(3-фторфенил)пропан-1-она (20,7 г, 88%), который использовался в ходе следующего этапа без дополнительной очистки.
Этап 3
Раствор 3-диметиламино-1-(3-фтор-фенил)-пропан-1-она (17 г, 0,087 моль) и (S)-1-(4-бромфенил)-этанамина (17 г, 0,087 моль) в смеси EtOH (50 мл) и H2O (50 мл) был подвергнут обратному конденсированию в течение ночи при температуре 80°С. После удаления растворителя в вакууме полученный осадок был очищен посредством колоночной хроматографии для получения (S)-3-(1-(4-бромфенил)-этиламино)-1-(3-фторфенил)-пропан-1-она (6,2 г, 20%).
ПРИМЕР 25
(S)-3-((S)-1-(4-(1-(2-фторэтил)-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
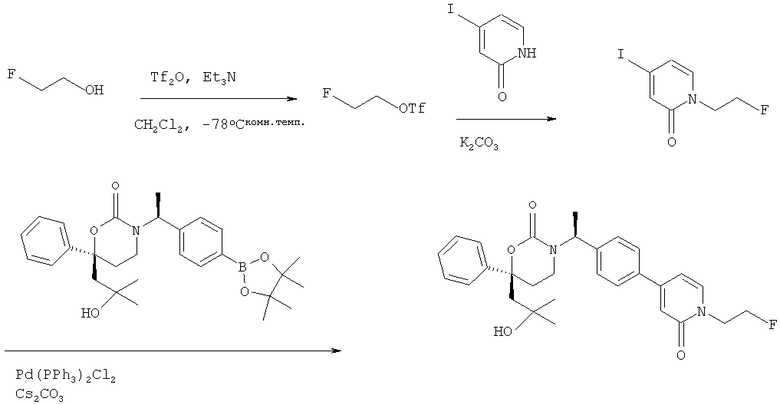
1-(2-фторэтил)-4-йодпиридин-2(1Н)-он был получен из 4-йодпиридин-2(1Н)-она и 2-фторэтил трифторметансульфоната в результате процедуры, аналогичной описанной для 1 этапа Примера 20.
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 1-(2-фторэтил)-4-йодпиридин-2(1Н)-она в результате процедуры, аналогичной описанной для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,09 мин, масса/заряд=515, 493, 475, 435.
ПРИМЕР 26
(S)-6-(2-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-он
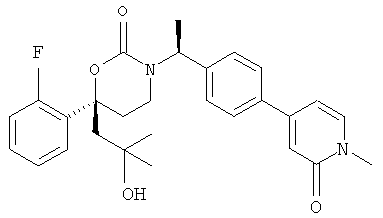
Титульное соединение было получено из (S)-6-(2-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 4-йод-1-метилпиридин-2(1Н)-она в результате процедуры, аналогичной описанной для 9 этапа Примера 23. Метод ЖХ-МС 2 tR=1,58 мин, масса/заряд=501, 479, 421.
ПРИМЕР 27
(S)-6-(3-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-он
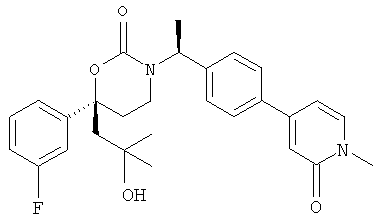
Титульное соединение было получено из (S)-6-(3-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 4-йод-1-метилпиридин-2(1Н)-она в результате процедуры, аналогичной описанной для 9 этапа Примера 23. Метод ЖХ-МС 2 tR=1,57 мин, масса/заряд=501, 479, 421.
ПРИМЕР 28
6-(3-гидроксипропил)-6-13опропил-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-он
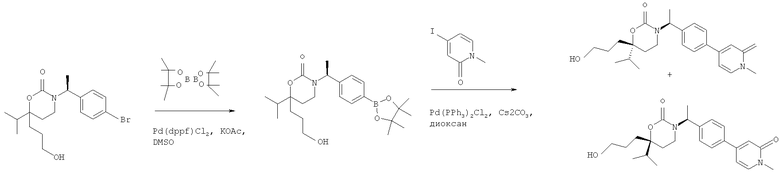
Этап 1
В смесь 3-((S)-1-(4-бромфенил)этил)-6-(3-гидроксипропил)-6-изопропил-1,3-оксазинан-2-она (100 мг, 0,26 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (198 мг, 0,783 ммоль) и ацетата калия (256 мг, 2,61 ммоль) в DMSO (5 мл) в атмосфере N2 был добавлен Pd(dppf)Cl2 (21 мг, 0,0261 ммоль). Полученная смесь перемешивалась при температуре 85°С в течение 3 ч, была обработана EtOAc (50 мл) и водой (50 мл). Органический слой был промыт водой (2×50 мл) и солевым раствором (50 мл), высушен и выпарен для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения 6-(3-гидроксипропил)-6-изопропил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (40 мг, 35%).
Этап 2
Смесь 6-(3-гидроксипропил)-6-изопропил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (40 мг, 0,092 ммоль), 4-йод-1-метилпиридин-2(1Н)-она (20 мг, 0,085 ммоль), Pd(PPh3)2Cl2 (6 мг, 0,0085 ммоль), а также Cs2CO3 (2 N, 0,425 мл, 0,85 ммоль) в 1,4-диоксане (2 мл) была подвергнута обратному конденсированию в атмосфере N2 в течение 3 ч. Реакционная смесь была обработана EtOAc (20 мл) и водой (20 мл), после чего органический слой был высушен и выпарен в вакууме. Полученный осадок был очищен посредством препаративной ВЭЖХ для получения двух изомеров.
Изомер 1: (2,20 мг, 6%). Метод ЖХ-МС 2 tR=1,06 мин, масса/заряд=413;
масса/заряд=1H ЯМР (CD3OD): δ 1,00 (m, 6Н), 1,62 (m, 7Н), 1,82 (m, 1H), 2,10 (m, 2H), 2,85 (m, 1H), 3,29 (m, 1H), 3,54 (t, 2H), 3,63 (s, 3Н), 5,71 (q, 1H), 6,78 (dd, 1H), 6,83 (d, 1H), 7,51 (d, 2H), 7,75 (m, 3Н),
Изомер 2: (2.10 мг, 6%) Метод ЖХ-МС 2 tR=1,03 мин, масса/заряд=413; 1Н ЯМР (CD3OD): δ 0,86 (m, 6Н), 1,53 (m, 5H), 1,71 (m, 4H), 1,92 (m, 1H), 2,82 (m, 1H), 3,25 (m, 1H), 3,49 (t, 2H), 3,52 (s, 3Н), 5,59 (q, 1H), 6,67 (dd, 1H), 6,72 (d, 1H), 7,39 (d, 2H), 7,63 (m, 3Н),
ПРИМЕР 29
(R)-6-(3-гидроксипропил)-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
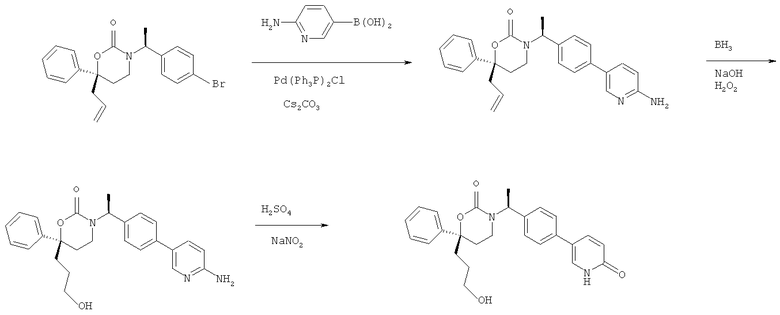
Этап 1
Смесь (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-фенил-1,3-оксазинан-2-она (150 мг, 0,375 ммоль) и 6-аминопиридин-3-илборной кислоты (56 мг, 0,45 ммоль), Pd(Ph3P)2Cl2 (15 мг), а также водного раствора Cs2CO3 (0,5 мл, 2 M) в 1,4-диоксане (10 мл) перемешивалась и нагревалась для обратного конденсирвоания в течение 2 ч. После отделения органическая фаза была выпарена для получения неочищенного продукта, который затем был очищен посредством препаративной ВЭЖХ для получения (R)-6-аллил-3-((S)-1-(4-(6- аминопиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (90 мг, 60%).
Этап 2
В раствор (R)-6-аллил-3-((S)-1-(4-(6-аминопиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (90 мг, 0,23 ммоль) в тетрагидрофуране (10 мл) при температуре 0°С в атмосфере азота был добавлен ВН3 тетрагидрофуран (3,0 мл, 1 моль/л, 4 ммоль). Полученная смесь перемешивалась в течение 2 ч, после чего реакция была остановлена водой. Затем в описанную выше смесь были добавлены NaOH (2 мл, 3 моль/л) и H2O2 (1 мл). После окончания реакции смесь подверглась экстракции при помощи EtOAc. Объединенная органическая фаза была выпарена для получения неочищенного продукта, который затем был очищен посредством препаративной ВЭЖХ для получения (R)-3-((S)-1-(4-(6-аминопиридин-3-ил)фенил)этил)-6-3-гидроксипропил)-6-фенил-1,3-оксазинан-2-она (40 мг, 41%).
Этап 3
(R)-3-((S)-1-(4-(6-аминопиридин-3-ил)фенил)этил)-6-(3-гидроксипропил)-6-фенил-1,3-оксазинан-2-он (40 мг, 0,09 ммоль) был растворен в 3,5 М H2SO4 (10 мл), после чего при температуре 0°С в полученный раствор был добавлен 2 М NaNO2 (10 мл). Реакционная смесь перемешивалась при комнатной температуре в течение 2 ч и обрабатывалась раствором NaOH, после чего смесь подверглась экстракции при помощи EtOAc. Объединенный органический слой был промыт солевым раствором, высушен над безводным Na2SO4 и выпарен для получения осадка, который затем был очищен посредством препаративной ВЭЖХ для получения (R)-6-(3-гидроксипропил)-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (10 мг, 20%). Метод ЖХ-МС 2 tR=1,66, мин, масса/заряд=433, 455; 1H ЯМР (CDCl3): 1,36 (m, 2H), 1,50 (m, 3Н), 1,68 (m, 2H), 1,92 (m, 2H), 2,10-2,30 (m, 3Н), 2,84 (m, 1H), 3,50 (m, 2H), 5,12 (m, 1H), 6,62 (m, 1H), 6,86 (m, 2H), 7,08 (m, 2H), 7,18-7,32 (m, 5H), 7,46 (m, 1H), 7,62 (m, 1H).
ПРИМЕР 30
(R)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
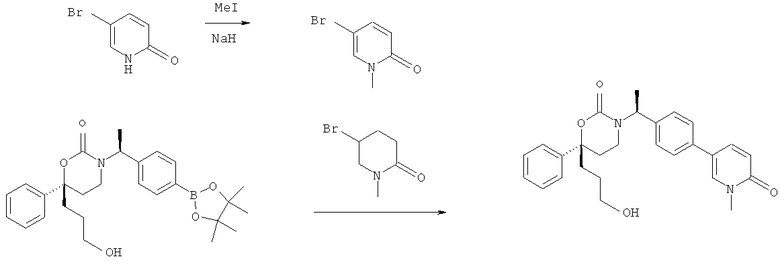
Этап 1
В суспензию NaH (4,8 г, 0,2 моль) в тетрагидрофуране (10 мл) при температуре 0°С был добавлен раствор 5-бромпиридин-2(1Н)-она (8,6 г, 0,05 моль) в тетрагидрофуране (120 мл). Полученная смесь перемешивалась в течение 1 ч с последующим добавлением CH3I (35,5 г, 0.25 моль). Затем смесь перемешивалась еще в течение 3 ч, после чего реакция была остановлена при помощи водного раствора NH4Cl. Органическая фаза была выпарена для получения неочищенного продукта, который затем был очищен посредством колоночной хроматографии для получения 5-бром-1-метилпиридин-2(1Н)-она (8,9 г, 96,78%). 1Н ЯМР (CDCl3): δ=3,5 (S, 3Н), 6,52 (m, 1H), 7,32 (m, 1H), 7,45 (m, 1H).
Этап 2
Смесь (R)-6-(3-гидроксипропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (1,7 г, 3,7 ммоль) и 5-бром-1-метилпиридин-2(1Н)-она (816 мг, 4,4 ммоль), Pd(Ph3P)2Cl2 (200 мг), а также водного раствора Cs2CO3 (4 мл, 2М) в 1,4-диоксане (30 мл) перемешивалась и нагревалась для обратного конденсирования в течение 2 ч. После окончания реакции смесь была промыта водой и подверглась экстракции при помощи EtOAc. Затем органическая фаза была промыта солевым раствором, высушена над Na2SO4, а также подверглась фильтрации и выпариванию для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (R)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (614 мг, 37%). Метод ЖХ-МС 2 tR=1,075 мин, масса/заряд=447,1; 1H ЯМР (CDCl3): δ=1,38 (m, 1Н),1,47 (d, 3Н), 1,73 (m, 2H), 1,98 (m, 2H), 2,20 (m, 1H), 2,31 (m, 2H), 2,94 (m, 1H), 3,51 (m, 2H), 3,56 (s, 3Н), 5,63 (m, 1H), 6,67 (m, 1H), 6,87 (m, 2H), 7,05 (m, 2H), 7,31-7,41(m, 6H), 7,48 (m, 1H).
ПРИМЕР 31
(S)-6-(4-фторфенил)-6-(2-гидроксиэтил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-он
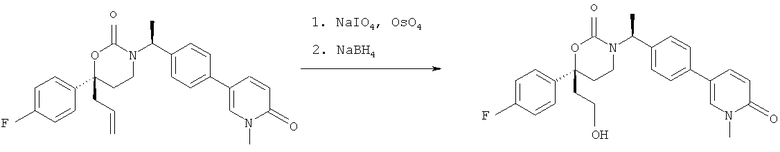
В раствор (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (0,064 г, 0,144 ммоль, 1,0 эквив.) в тетрагидрофуране-H2O (1:1, 6 мл) были добавлены NaIO4 (0,145 г, 0,678 ммоль, 4,7 эквив.) и OsO4 (массовая концентрация в процентах в растворе t-BuOH - 2,5%, 0,048 г, 0,0047 ммоль, 0,033 эквив.), после чего смесь перемешивалась при комнатной температуре в течение 1 ч. Затем смесь была разбавлена EtOAc, высушена над Na2SO4 и выпарена в вакууме. Осадок был растворен в МеОН (3 мл) с последующим добавлением NaBH4 (0,100 г). После перемешивания смеси в течение 0,5 ч при комнатной температуре в смесь был добавлен ацетон. Растворители были удалены в вакууме, после чего осадок был обработан насыщенным солевым раствором, подвергся экстракции CH2Cl2 и высушиванию над Na2SO4. После выпаривания растворителя осадок был очищен посредством обратно-фазовой ВЭЖХ (колонка SunFire™ Prep C18 OBD™ 5 мкм 19×50 мм, 10%→90% CH3CN/H2O, 0,1% CF3COOH в течение 8 мин, затем 90% CH3CN/H2O, 0,1% CF3COOH в течение 2 мин, скорость потока 20 мл/мин) для получения (S)-6-(4-фторфенил)-6-(2-гидроксиэтил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она. Метод ЖХ-МС 1 tR=1,21 мин, масса/заряд=451 (М+1); 1H ЯМР (400 МГц, CD3OD) 7,80 (m, 1H), 7,69 (d, J=9,4 Гц, 1Н), 7,22-7,19 (m, 4H), 7,00-6,92 (m, 4H), 6,52 (d, J=9,4 Гц, 1H), 5,45 (q, J=7,0 Гц, 1H), 3,60-3,52 (m, 1H), 3,52 (s, 3Н), 3,24-3,18 (m, 1H), 3,02-2,98 (m, 1H), 2,39-2,35 (m, 1H), 2,23-2,12 (m, 2H), 2,01 (t, J=7,3 Гц, 2H), 1,43 (d, J=7,0 Гц, 3Н); 19F ЯМР (376 МГц, CD3OD) -117,19 (m).
ПРИМЕР 32
(S)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-он
Метод 1
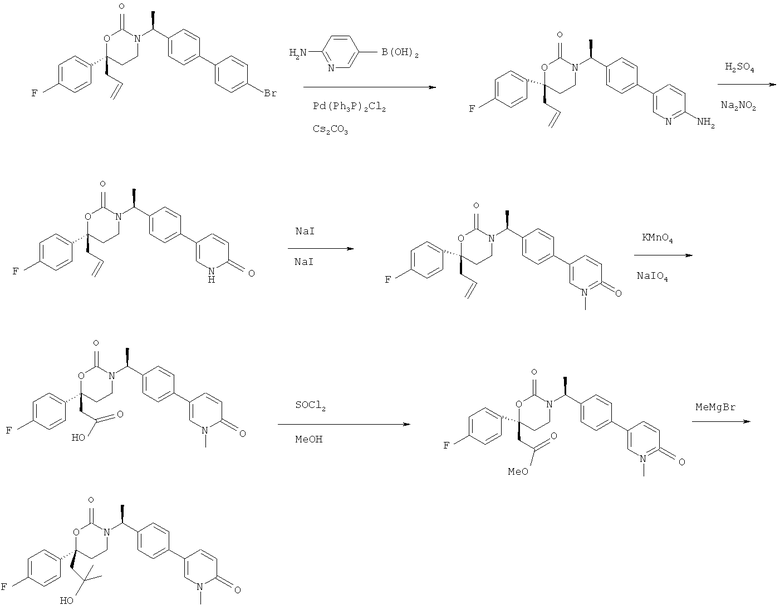
Этап 1
Смесь (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-она (1,6 г, 3,84 ммоль) и 6-аминопиридин-3-илборной кислоты (1,0 г, 4,61 ммоль), Pd(Ph3P)2Cl2 (150 мг), и водного раствора Cs2CO3 (3,84 мл, 2 М) в 1,4-диоксане (150 мл) перемешивалась и нагревалась для обратного конденсирования в течение 2 ч. Затем смесь подверглась фильтрации, а полученный фильтрат - экстракции при помощи EtOAc. Объединенная органическая фаза была промыта солевым раствором, высушена над безводным Na2SO4 и выпарена для получения (R)-6-аллил-3-((S)-1-(4-(6-аминопиридин-3-ил)фенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-она (1,5 г, 90%), который использовался в ходе следующего этапа без дополнительной очистки. 1H ЯМР (CDCl3): δ=1,51 (d, 3Н), 2,17-2,31 (m, 3Н), 2,54-2,60 (m, 2H), 2,90 (m, 1H), 4,46 (s, 2H), 4,99-5,09 (m, 2H), 5,65-5,71 (m, 2H), 6,54 (m, 2H), 6,88 (d, 2H), 7,03 (t, 2H), 7,21-7,27 (m, 3Н), 7,58 (d, 1H), 8,22 (d, 1H).
Этап 2
В раствор (R)-6-аллил-3-((S)-1-(4-(6-аминопиридин-3-ил)фенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-она (1,5 г, 3,47 ммоль) в 3,5 М H2SO4 (25 мл) при температуре 0°С был добавлен 2 М NaNO2 (15 мл). Реакционная смесь перемешивалась при комнатной температуре в течение ночи, после чего была обработана водным раствором NaOH (8%) и подверглась экстракции с помощью CH2Cl2. Объединенный органический слой был промыт солевым раствором, высушен над безводным Na2SO4 и выпарен для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (891 мг, 59%). 1Н ЯМР (CDCl3): δ=1,52 (d, 3Н), 2,15-2,38 (m, 3Н), 2,51-2,60 (m, 2H), 2,94 (m, 1H), 4,99-5,11 (m, 2H), 5,65-5,74 (m, 2H), 6,67 (m, 1H), 6,89 (d, 2H), 7,00 (t, 2H), 7,13-7,20 (m, 2H), 7,20-7,27 (d, 2H), 7,33 (m, 1 Н), 7,46 (m,1H), 7,77 (m, 1H).
Этап 3
В суспензию NaH (330 мг, 8,24 ммоль) в тетрагидрофуране (20 мл) при температуре 0°С был добавлен раствор (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (891 мг, 0,174 ммоль) в тетрагидрофуране (30 мл). Полученная смесь перемешивалась в течение 1 ч, после чего в смесь добавлялась CH3I (2 мл) и смесь перемешивалась в течение ночи. Затем реакция была остановлена при помощи водного раствора NH4Cl, органическая фаза была отделена и выпарена для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (634 мг, 69%). 1H ЯМР (CDCl3): δ=1,52 (d, 3Н), 2,16-2,35 (m, 3Н), 2,52-2,64 (m, 2H), 2,94 (m, 1H), 3,61 (s, 3Н), 5,00-5,11 (m, 2H), 5,66-5,74 (m, 2H), 6,64 (d, 1H), 6,90 (d, 2H), 7,02 (t, 2H), 7,11-7,14 (d, 2H), 7,25-7,28 (m, 2H), 7,41 (m, 1H), 7,53 (m, 1H).
Этап 4
В раствор (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (320 мг, 0,717 ммоль) в ацетоне (20 мл) был добавлен водный раствор KMnO4 и NaIO4 (15 мл), после чего полученная смесь перемешивалась при температуре 0°С в течение 30 мин. Затем смесь подверглась фильтрации с последующим регулированием уровня рН фильтрата до 5-6 с помощью 1 N водного раствора HCl. Полученная смесь подверглась экстракции при помощи EtOAc, после чего полученная объединенная органическая фаза была промыта солевым раствором, высушена над безводным Na2SO4 и выпарена для получения 2-((S)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-1,3-оксазинан-6-ил)уксусной кислоты.
Этап 5
В раствор 2-((S)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-1,3-оксазинан-6-ил)уксусной кислоты (290 мг, 0,625 моль) в МеОН (20 мл) при температуре 0°С был добавлен SOCl2 (2 мл), после чего реакционная смесь перемешивалась при комнатной температуре в течение 2 ч. Реакционная смесь выпаривалась для получения осадка, который затем был очищен посредством препаративной ТСХ для получения метил 2-((S)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-1,3-оксазинан-6-ил)ацетата (130 мг, 43,5%). 1H ЯМР (CDCl3): δ=1,52 (d, 3Н), 2,36-2,55 (m, 3Н), 2,67-2,71 (m, 2H), 2,90-3,04 (m, 3Н), 3,68 (s, 3Н), 3,71 (s, 3Н), 5,66 (m, 2H), 6,66 (d, 1H), 6,90 (d, 2H), 7,03 (t, 2H), 7,13-7,15 (d, 2H), 7,23-7,29 (m, 2H), 7,42 (m, 1H), 7,56 (m, 1H).
Этап 6
В раствор метил 2-((S)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-1,3-оксазинан-6-ил)ацетата (130 мг, 0,22 ммоль) в сухом тетрагидрофуране (20 мл) при температуре -78°С был добавлен MeMgBr (2 мл), после чего смесь перемешивалась при комн. темп. в атмосфере N2 в течение ночи. Реакция была остановлена при помощи воды, а полученная смесь подверглась экстракции при помощи EtOAc. Объединенная органическая фаза была высушена над Na2SO4 и выпарена для получения осадка, который был очищен посредством препаративной ВЭЖХ для получения (S)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (24 мг, 30%). Метод ЖХ-МС 2 tR=1,116 мин, масса/заряд=479,1; 1H ЯМР (CDCl3): 1,1 (m, 6H), 1,18 (m, 1H), 1,48 (d, ЗН), 1,58 (m, 1H), 1,80-2,00 (m, 2H), 2,21 (m, 3Н), 2,86 (m, 1H), 5,55 (m, 1H), 7,72 (m, 2H), 7,00 (m, 2H), 7,18 (m, 4H).
Метод 2
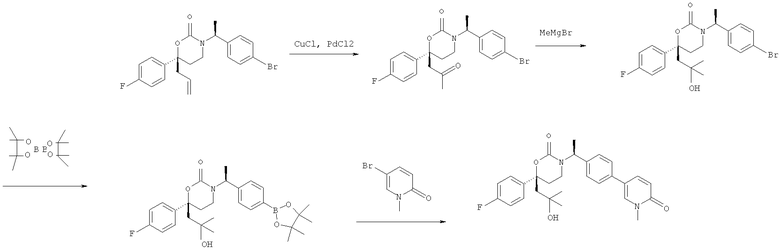
Этап 1
В раствор (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-она (5 г, 12 ммоль) и CuCl (2,75 г, 27,8 ммоль) в сухом DMF (50 мл) при комнатной температуре были добавлены H2O (20 мл) и PdCl2 (950 мг, 3,2 ммоль). Полученная смесь тщательно перемешивалась под колбой с кислородом в течение 24 ч. После получения результатов ТСХ, свидетельствующих об исчезновении исходного материала, твердое вещество было отфильтровано. Затем были добавлены вода (200 мл) и EtOAc (50 мл), произведены разделение слоев и экстракция водного слоя при помощи EtOAc (3×40 мл). Объединенный органический слой был промыт солевым раствором, высушен над Na2SO4, а также подвергся фильтрации и выпариванию для получения (S)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-(2-оксопропил)-1,3-оксазинан-2-она (5,25 г, 92%), который затем был очищен посредством колоночной хроматографии. 1Н ЯМР (CDCl3): 1,47 (s, 3Н), 2,06 (s, 3H), 2,10-2,36 (m, 3H), 2,58 (m, 1H), 2,90 (m, 2H), 5,58 (m, 1H), 6,69 (m, 1H), 6,79 (m, 1H), 7,02 (m, 2H), 7,19-7,33 (m, 4H).
Этап 2
В раствор (S)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-(2-оксопропил)-1,3-оксазинан-2-она (5,25 г, 12,1 ммоль) в безводном тетрагидрофуране (100 мл) по капле при температуре -78°С в атмосфере азота был добавлен метилмагния бромид (20 мл, 60 ммоль). Затем смесь перемешивалась при комнатной температуре в течение 2 ч и охлаждалась в ледяной бане. Реакция была остановлена при помощи водного раствора NH4Cl с последующим разделением слоев. Водный слой подвергся экстракции при помощи EtOAc (15 мл), после чего был промыт солевым раствором (30 мл), высушен над Na2SO4 и выпарен в вакууме для получения неочищенного продукта, который затем был очищен посредством препаративной ВЭЖХ и хиральной ВЭЖХ для получения (S)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-она (2,5 мг, 46%). 1H ЯМР (CDCl3): 1,08 (s, 3H), 1,12 (s, 3H), 1,48 (m, 3H), 1,99 (m, 1H), 2,10-2,24 (m, 4H), 2,35 (m, 1H), 2,85 (m, 1H), 5,61 (m, 1H), 6,80 (m, 2H), 6,99 (m, 2H), 7,15-7,28 (m, 5H).
Этап 3
Смесь (S)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-она (640 мг, 1,42 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (470 мг, 1,85 ммоль), PdCl2dppf (40 мг, 0,047 ммоль) и КОАс (490 мг, 4,97 ммоль) в DMSO (8 мл) нагревалась при температуре 90°С в течение 20 ч. Смесь была разбавлена EtOAc и промыта водой. Органическая фаза была отделена и выпарена для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (S)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (700 мг, 99%). 1H ЯМР (CDCl3): δ=1,08 (s, 3Н), 1,13 (s, 3Н), 1,32 (s, 12H), 1,51 (t, 3Н), 1,94 (m, 2H), 2,16 (m, 5H), 2,33 (m, 1H), 2,83 (m, 1H), 5,69 (m, 1H), 6,99 (m, 4H), 7,25 (m,2H), 7,61 (m, 2H).
Этап 4
Смесь (S)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (700 мг, 1,41 ммоль), 5-бром-1-метилпиридин-2(1Н)-она (398 мг, 2,12 ммоль), PdCl2(Ph3P)2 (70 мг), а также Cs2CO3 (1,5 мл, 3,0 ммоль) в 1,4-диоксане (15 мл) нагревалась в условиях обратного конденсирования в течение 2 ч. Полученная смесь была разбавлена EtOAc и промыта водой с последующим отделением и выпариванием органической фаза для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (S)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (150 мг, 22%). 1H ЯМР (CDCl3): δ=1,12 (s, 3Н), 1,13 (s, 3Н), 1,51 (t, 3Н), 2,16 (m, 2H), 2,21 (m, 2H), 2,41 (m, 1H), 2,92 (m, 1H), 3,63 (s, 3Н), 5,69 (q, 1H), 6,69 (m, 1H), 6,99 (m, 4H), 7,18 (m, 2H), 7,27 (m, 2H), 7,42 (m, 1H), 7,52 (m, 1H).
ПРИМЕР 33
(R)-6-(4-фторфенил)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-он
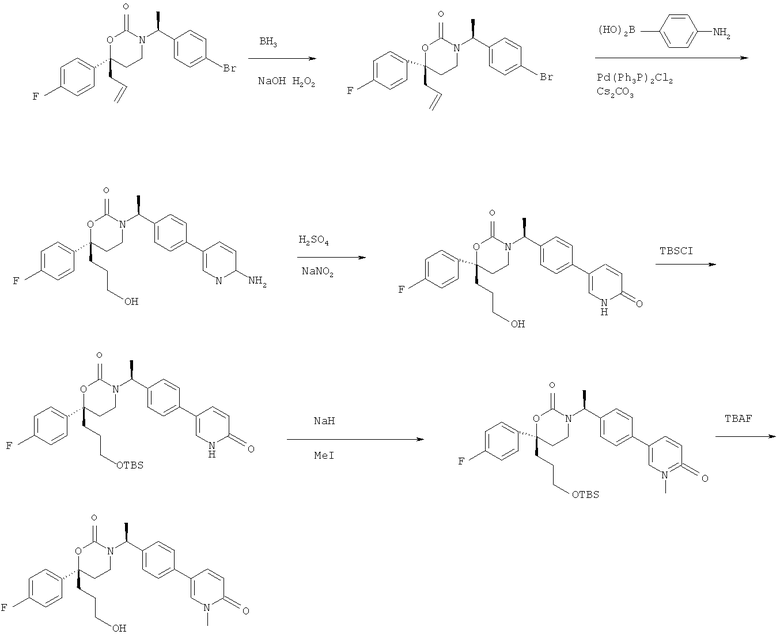
Этап 1
В раствор (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-1,3-оксазинан-2-она (1,19 г, 2,8 ммоль) в тетрагидрофуране (30 мл) при температуре 0°С в атмосфере азота был добавлен ВН3 тетрагидрофуран (8,5 мл, 1 моль/л, 8,5 ммоль). Полученная смесь перемешивалась в течение 2 ч, после чего реакция была остановлена при помощи воды. Затем в указанную выше смесь были добавлены NaOH (1 моль/л, 6 мл) и Н2О2 (5 мл). После окончания реакции смесь подверглась экстракции при помощи EtOAc. Объединенная органическая фаза была выпарена для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-(3-гидроксипропил)-1,3-оксазинан-2-она (1,13 г, 92%).
Этап 2
Смесь (R)-3-((S)-1-(4-бромфенил)этил)-6-(4-фторфенил)-6-(3-гидроксипропил)-1,3-оксазинан-2-она (520 мг, 1,2 ммоль) и 6-аминопиридин-3-илборной кислоты (280 мг, 1,44 ммоль), Pd(Ph3P)2Cl2 (100 мг), а также водного раствора Cs2CO3 (3 мл, 2М) в 1,4-диоксане (20 мл) перемешивалась и нагревалась в условиях обратного конденсирования в течение 2 ч. Затем органическая фаза отделялась и выпаривалась для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (R)-3-((S)-1-(4-(6-аминопиридин-3-ил)фенил)этил)-6-(4-фторфенил)-6-(3-гидроксипропил)-1,3-оксазинан-2-она (400 мг, 74%).
Этап 3
(R)-3-((S)-1-(4-(6-аминопиридин-3-ил)фенил)этил)-6-(4-фторфенил)-6-(3-гидроксипропил)-1,3-оксазинан-2-он (400 мг, 0,88 ммоль) был растворен в 3,5 М H2SO4 (10 мл), после чего при температуре 0°С был добавлен 2 М NaNO2 (6 мл). Реакционная смесь перемешивалась при комнатной температуре в течение 20 мин, после чего была обработана водным раствором NaOH (8%) и подверглась экстракции CH2Cl2. Объединенный органический слой был промыт солевым раствором, высушен над безводным Na2SO4 и выпарен для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (R)-6-(4-фторфенил)-6-(3-гидроксипропил)-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (350 мг, 0,78 ммоль). 1H ЯМР (CDCl3): δ=1,10-1,25 (m, 8H), 1,37 (m, 1H), 1,42-1,55 (m, 2H), 1,78-1,93 (m, 2H), 2,10-2,38 (m, 2H), 2,87 (m, 2H), 3,52-3,58 (m, 1H), 3,31-3,97 (m, 1H), 4,12-4,19 (m, 1H), 5,53-5,63 (m, 1H), 6,85-7,15 (m, 3Н), 7,35-7,55 (m, 1H), 7,75-7,89 (m, 1H), 8,10-8,12 (m, 1H).
Этап 4
Смесь (R)-6-(4-фторфенил)-6-(3-гидроксипропил)-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (100 мг, 0,78 ммоль), имидазола (142,8 мг, 2,1 ммоль), и трет-бутилхлордиметилсилана (350 мг, 2,34 ммоль) в CH2Cl2 (20 мл) перемешивалась в течение ночи. Затем смесь была промыта водой и подверглась экстракции при помощи EtOAc. Объединенная органическая фаза была промыта солевым раствором и высушена над Na2SO4, затем подверглась фильтрации и выпариванию для получения неочищенного (R)-6-(3-(трет-бутилдиметилсилилокси)пропил)-6-(4-фторфенил)-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (120 мг), который использовался в ходе следующего этапа без дополнительной очистки.
Этап 5
В суспензию NaH (18 мг, 0,72 ммоль) в тетрагидрофуране (0,5 мл) при температуре 0°С был добавлен раствор (R)-6-(3-(трет-бутилдиметилсилилокси)пропил)-6-(4-фторфенил)-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (100 мг, 0,18 ммоль) в тетрагидрофуране (10 мл). Полученная смесь перемешивалась в течение 1 ч, затем в нее был добавлен CH3I (613 мг, 43,2 ммоль), после чего смесь перемешивалась в течение 3 ч. Реакция была остановлена при помощи водного раствора NH4Cl. Органическая фаза была отделена и выпарена для получения (R)-6-(3-(трет-бутилдиметилсилилокси)пропил)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (104 мг, 100%), который использовался в ходе следующего этапа без дополнительной очистки.
Этап 6
Смесь (R)-6-(3-(трет-бутилдиметилсилилокси)пропил)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (200 мг, 0,35 ммоль) и TBAF (182 мг, 0,7 ммоль) в CH3CN перемешивалась и нагревалась в условиях обратного конденсирования в течение 15 мин. После окончания реакции смесь была промыта водой и подверглась экстракции при помощи EtOAc. Объединенная органическая фаза была промыта солевым раствором и высушена над Na2SO4, затем подверглась фильтрации и выпариванию для получения неочищенного продукта, который затем был очищен посредством препаративной ВЭЖХ для получения (R)-6-(4-фторфенил)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (5,01 мг, 4%). Метод ЖХ-МС 2 tR=1,065 мин, масса/заряд=464,21; 1H ЯМР (CDCl3): δ=1,38 (m, 1H), 1,47 (d, 3Н), 1,63 (m, 2H), 1,91 (m, 2H), 2,10-2,30 (m, 3Н), 2,87 (m, 1H), 2,84 (m, 1H), 3,51 (m, 2H), 3,56 (s, 3Н), 5,63 (m, 1H), 6,67 (m, 1H), 6,87-6,98 (m, 4H), 7,15 (m, 2H), 7,27 (m, 1H), 7,29 (m, 1H), 7,32 (m, 1H), 7,55 (m, 1H).
ПРИМЕР 34
N-(3-((R)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-1,3-оксазинан-6-ил)пропил)метансульфонамид
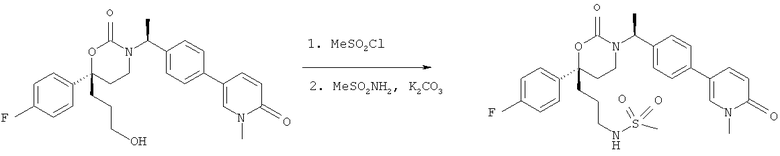
Титульное соединение было получено из (R)-6-(4-фторфенил)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она путем обработки (i) MeSO2Cl и (ii) MeSO2NH2. Метод ЖХ-МС 2 tR=1,02 мин, масса/заряд=542,3; 1H ЯМР (CDCl3) 1,35 (m, 1H), 1,53 (d, 3Н), 1,69 (m, 1H), 1,89 (m, 1H), 2,00 (m, 1H), 2,17-2,33 (m, 3Н), 2,89 (s, 3Н), 2,97 (m, 1H), 3,06 (m, 2H), 3,66 (s, 3Н), 4,38 (s, 1H), 5,67 (m, 1H), 6,82 (d, 1H), 6,99 (m, 4H), 7,15 (m, 2H), 7,22 (m, 2H), 7,47 (s, 1H), 7,63 (d, 1H).
ПРИМЕР 35
3-((R)-2-оксо-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-6-ил)пропанамид
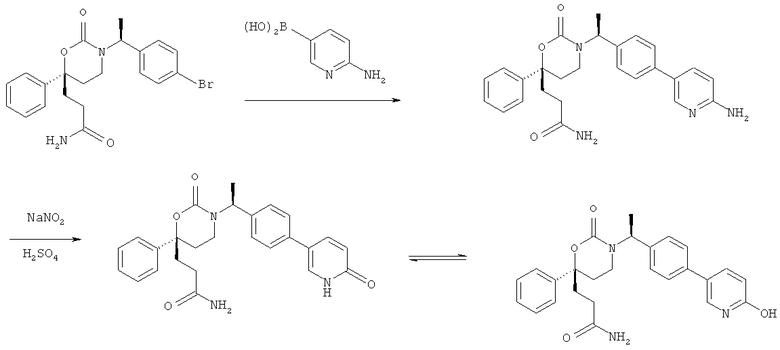
Титульное соединение было получено из 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропанамида в результате проведения процедур, аналогичных описанным для 2 и 3 этапов Примера 33. Метод ЖХ-МС 2 tR=0,999 мин, масса/заряд=446,1; 1H ЯМР (CD3OD) 1,53 (d, 3Н), 1,91-2,01 (m, 1H), 2,18-2,34 (m, 4H), 2,35-2,51 (m, 2H), 3,03-3,12 (m, 1H), 5,56 (m, 1H), 6,62 (d, 2H), 7,03 (d, 2H), 7,24-7,44 (m, 7H), 7,59 (m, 1H), 7,87 (m, 1H).
ПРИМЕР 36
(S)-6-(2-гидроксиэтил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
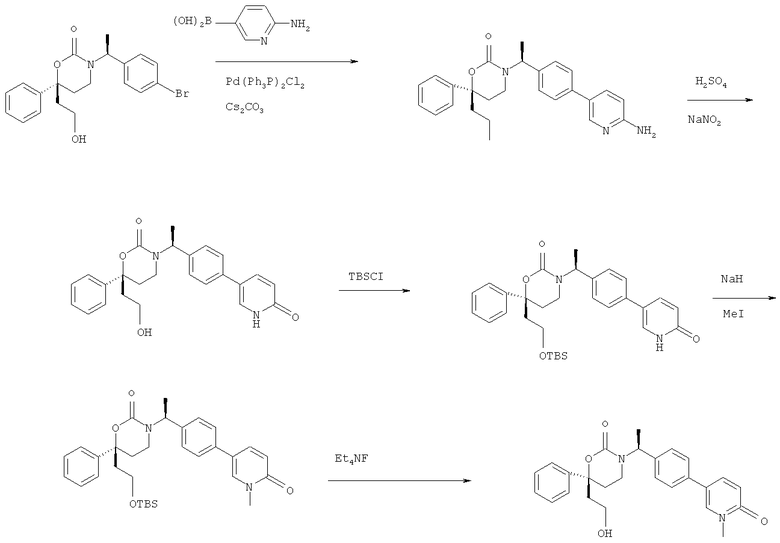
Титульное соединение было получено из (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидроксиэтил)-6-фенил-1,3-оксазинан-2-она в результате проведения процедур, аналогичных описанным для 2-6 этапов Примера 33. Метод ЖХ-МС 2 tR=1,038 мин, масса/заряд=433,1; 1H ЯМР (CDCl3) 1,48 (d, 3Н), 2,06-2,19 (m, 2H), 2,11-2,31 (m, 3H), 2,84 (m, 1H), 3,50 (m, 1H), 3,54 (s, 3Н), 3,72 (m, 1H), 5,62 (m, 1H), 6,60 (d, 1H), 6,86 (d, 2H), 7,06 (d, 2H), 7,26 (m, 3Н), 7,32 (m, 3Н), 7,47 (d, 1H).
ПРИМЕР 37
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
Метод 1
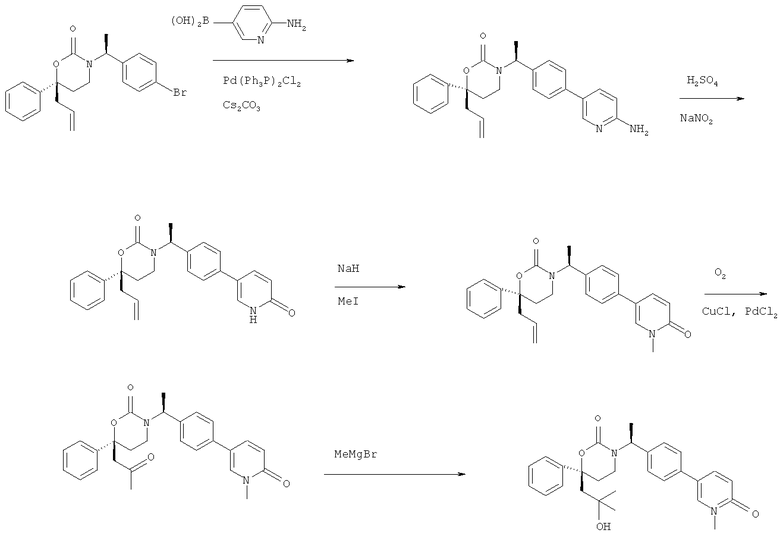
Титульное соединение было получено из (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-фенил-1,3-оксазинан-2-она в результате проведения процедур, аналогичных описанным для 2, 3 и 5 этапов Примера 33 после проведения процедур, аналогичных описанным для 4 и 6 этапов 1 метода Способа получения 1. Метод ЖХ-МС 2 tR=1,116 мин, масса/заряд=461,1; 1H ЯМР (CDCl3) 1,09 (s, 3Н), 1,16 (s, 3Н), 1,51 (m, 3Н), 2,05-2,20 (4Н), 2,40 (m, 1H), 2,84 (m, 1H), 3,59 (s, 3Н), 5,64 (m, 1H), 6,62 (m, 1H), 6,96 (m, 2H), 7,14 (m, 2H), 7,28-7,39 (m, 5H), 7,48 (m, 1H), 7,50 (m, 1H).
Метод 2
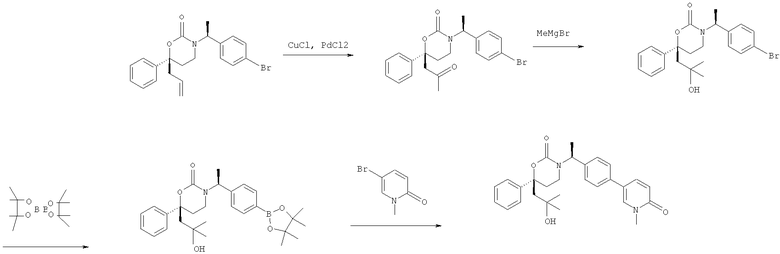
Этап 1
В раствор (R)-6-аллил-3-((S)-1-(4-бромфенил)этил)-6-фенил-1,3- оксазинан-2-она (20 г, 50 ммоль) и CuCl (12,4 г, 125 ммоль) в сухом DMF (50 мл) при температуре 0~-5°С были добавлены H2O (12 мл) и PdCl2 (2,66 г, 15 ммоль). После добавления смесь была оставлена при комнатной температуре на 48 ч в атмосфере O2 для нагревания. После получения результатов ТСХ, свидетельствующих об исчезновении исходного материала, было отфильтровано твердое вещество, к которому затем были добавлены вода (200 мл) и EtOAc (50 мл). После разделения слоев водный слой подвергся экстракции при помощи EtOAc (3×40 мл). Объединенный органический слой был промыт солевым раствором и высушен над Na2SO4, затем подвергся фильтрации и выпариванию для получения осадка, который был очищен посредством колоночной хроматографии для получения (S)-3-((S)-1-(4-бром-фенил)этил)-6-(2-оксопропил)-6-фенил-1,3-оксазинан-2-она (12 г, 58%).
Этап 2
В раствор (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-оксопропил)-6-фенил-1,3-оксазинан-2-она (12 г, 28,8 ммоль) в безводном тетрагидрофуране (100 мл) по капле при температуре -78°С в атмосфере азота был добавлен метилмагния бромид (48 мл, 144 ммоль). Полученная смесь перемешивалась при комнатной температуре в течение 1 ч, после чего реакция была остановлена при помощи водного раствора NH4Cl (50 мл) в водяной бане со льдом. После разделения слоев водный слой подвергся экстракции при помощи EtOAc (150 мл), объединенные органические фазы были промыты солевым раствором (30 мл), высушены над Na2SO4 и выпарены в вакууме для получения неочищенного продукта, который затем был очищен посредством препаративной ВЭЖХ и хиральной ВЭЖХ для получения (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (6,6 г, 53%).
Этап 3
В раствор (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2- метилпропил)-6-сренил-1,3-оксазинан-2-она (6,6 г, 15.2 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолане) (6,1 г, 24,3 ммоль) в сухом DMSO (20 мл) были добавлены KОАс (4,8 г, 48,6 ммоль) и Pd(dppf)Cl2 (372 мг, 0,46 ммоль). После добавления смесь была оставлена на 20 ч при температуре 100°С для нагревания. После получения результатов ТСХ, свидетельствующих об исчезновении исходного материала, твердое вещество было отфильтровано. Затем были добавлены вода (60 мл) и EtOAc (50 мл), произведены разделение слоев и экстракция водного слоя при помощи EtOAc (3×15 мл). Объединенный органический слой был промыт солевым раствором, высушен над Na2SO4, а также подвергся фильтрации и выпариванию для получения осадка, который затем был очищен посредством колоночной хроматографии для получения (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборол-ан-2-ил)фенил)этил)-1,3-оксазинан-2-она (4,4 г, 60%).
Этап 4
В раствор (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (2,2 г, 4,58 ммоль) и 5-бром-1-метилпиридин-2(1Н)-она (1,37 г, 7,33 ммоль) в сухом 1,4-диоксане (4 мл) были добавлены водный раствор CsCO3 (10 мл, 10 ммоль) и Pd(PPh3)2Cl2 (967 мг, 1,38 ммоль). После добавления смесь при помощи микроволновой печи нагревалась при температуре 110°С в течение 30 мин. После получения результатов ТСХ, свидетельствующих об исчезновении исходного материала, твердое вещество было отфильтровано. Затем были добавлены вода (20 мл) и EtOAc (10 мл), произведены разделение слоев и экстракция водного слоя при помощи EtOAc (3×10 мл). Объединенный органический слой был промыт солевым раствором, высушен над Na2SO4, а также подвергся фильтрации и выпариванию для получения осадка, который затем был очищен посредством препаративной ВЭЖХ для получения (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (730 мг, 35%). 1H ЯМР (CDCl3): 1,09 (s, 3Н), 1,16 (s, 3Н), 1,51 (m, 3Н), 2,05-2,20 (4Н), 2,40 (m, 1H), 2,84 (m, 1H), 3,59 (s, 3Н), 5,64 (m, 1H), 6,62 (m, 1H), 6,96 (m, 2H), 7,14 (m, 2H), 7,28-7,39 (m, 5H), 7,48 (m, 1H), 7,50 (m, 1H), Полученное соединение подверглось перекристаллизации в соответствии с двумя методами.
Метод перекристаллизации А
Смесь (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (прибл. 2,94 г) и изопропилацетата (160 мл) тщательно перемешивалась при комнатной температуре или нагревалась при температуре 50°С до растворения большего количества твердого вещества. Полученная смесь подверглась фильтрации через ВЭЖХ фильтр, после чего полученный фильтрат медленно перемешивался при комнатной температуре в течение ночи. Твердые вещества подверглись фильтрации и промыванию изопропилацетатом, а затем были высушены при комнатной температуре в высоком вакууме для получения 1,43 г (49%) кристаллического твердого вещества с температурой плавления 95-101°С. Данная форма была определена как гидрат, при нагревании выделяющий 3,6% воды по массе.
Метод перекристаллизации В
Смесь (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (прибл. 10,03 г) и изопропилацетата (600 мл) нагревалась в условиях обратного конденсирования в масляной бане при температуре 130°С до полного растворения твердого вещества и образования однородного раствора. Затем нагревание было прекращено, а полученная смесь медленно перемешивалась в масляной бане для медленного охлаждения до комнатной температуры. Полученные после охлаждения твердые вещества подверглись фильтрации, затем были промыты изопропилацетатом и высушены при комнатной температуре в высоком вакууме для получения 7,30 г (73%) кристаллического твердого вещества с температурой плавления 180-181°С. Данная форма была определена как безводная.
ПРИМЕР 38
3-((R)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропанамид
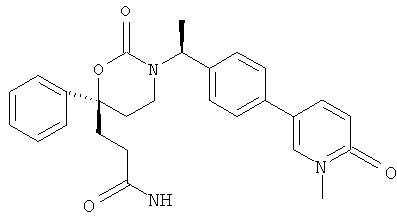
Титульное соединение было получено из (R)-6-аллил-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она в результате проведения процедуры, аналогичной описанной для 2 этапа Примера 29, после обработки (i) реактивом Джонса и (ii) NH3, EDC, HOBt. Метод ЖХ-МС 2 tR=1,028 мин, масса/заряд=460,2; 1H ЯМР (CDCl3) 1,53 (d, 3Н), 1,91-2,01 (m, 1H), 2,11-2,42 (m, 5H), 2,46-2,54 (m, 1H), 2,88-2,96 (m, 1H), 3,60 (s, 3Н), 5,26 (s, 1H), 5,42 (s, 1H), 5,66 (m, 1H), 6,69 (d, 1H), 6,95-7,03 (d, 2H), 7,12-7,20 (m, 2H), 7,24-7,41 (m, 5H), 7,52 (m, 1H).
ПРИМЕР 39
N-(3-((R)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропил)метансульфонамид
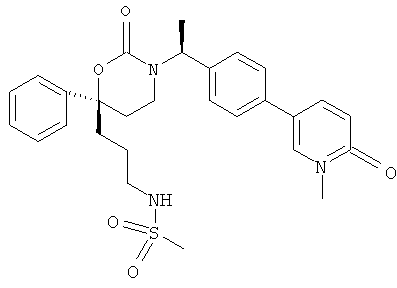
Титульное соединение было получено из (R)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она путем обработки (i) MeSO2Cl и (ii) MeSO2NH2, K2СО3. Метод ЖХ-МС 2 tR=1,095 мин, масса/заряд=524,1; 1H ЯМР (CDCl3) 1,30-1,41 (m, 1Н), 1,52 (d, 3Н), 1,71 (m, 1H), 1,87-2,07 (m, 3H), 2,09-2,20 (m, 3Н), 2,22-2,32 (m, 2H), 2,88 (s, 3Н), 3,06 (m, 2H), 3,60 (s, 3Н), 4,32 (s, 1H), 5,65 (m, 1H), 6,67 (d, 1H), 6,94 (m, 2H), 7,11 (d, 2H), 7,25 (m, 1H), 7,27-7,40 (m, 4H), 7,53 (dd, 1H).
ПРИМЕР 41
(R)-6-(4-фторфенил)-6-(3-гидроксипропил)-3-((S)-1-(4-(2-оксо-1,2-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-он
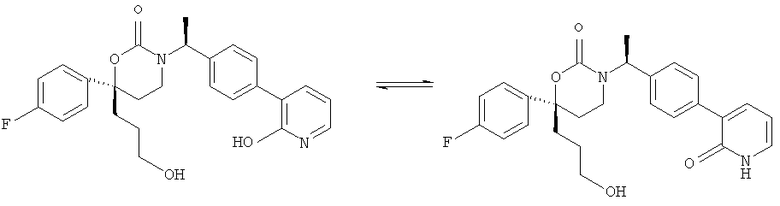
Титульное соединение было получено из (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 3-бром-2-гидроксипиридина в результате применения процедуры, аналогичной описанной для 2 этапа Примера 3, после применения процедуры, аналогичной описанной для 2 этапа Примера 29. Метод ЖХ-МС 1 tR=1,24, масса/заряд=452 (М+1); 1H ЯМР (CDCl3) 7,76 (d, 1H), 7,52 (d, 1H), 7,42 (dd, 2H), 7,24 (m, 2H), 7,08-7,00 (m, 4H), 6,75 (t, 1H), 5,70 (m, 1H), 3,58 (t, 1H), 2,94 (m, 1H), 1,54 (d, 3H).
ПРИМЕР 42
(R)-6-(4-фторфенил)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-он
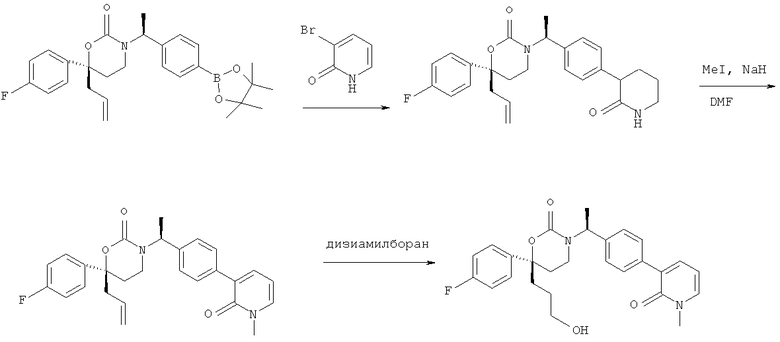
Этап 1
(R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-он (18 мг, 0,039 ммоль), 3-бром-2-гидроксипиридин (14 мг, 2 эквив.), Pd(dppf)Cl2 (3 мг, 10% моль), 2М водный раствор Na2CO3 (800 мкл) и 1,4-диоксан (1,5 мл) перемешивались, затем из смеси откачивался газообразный N2, с последующим повторным насыщением газообразным N2 (3×) перед нагреванием при температуре 85°С в течение ночи. После охлаждения до комнатной температуры смесь подверглась фильтрации и окислению с помощью 5% водного раствора HCl с последующей очисткой посредством препаративной ВЭЖХ для получения (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(2-оксо-1,2-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (7,2 мг, выход: 43%). Метод ЖХ-МС 1 tR=1,57 мин, масса/заряд 433 (М+1).
Этап 2
Раствор (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(2-оксо-1,2-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (15,5 мг, 0,036 ммоль) в сухом DMF (1 мл) был охлажден до 0°С. Затем в раствор был добавлен гидрид натрия (60% в минеральном масле, 3 мг, 2 эквив.). Спустя 20 мин в смесь был добавлен йодметан (4,5 мкл, 2 эквив.). Смесь снова перемешивалась в течение 20 мин, затем медленно нагревалась до комнатной температуры и перемешивалась в течение 2 ч. Полученные после проведения ЖХ-МС результаты свидетельствовали об окончании реакции, которая была остановлена при помощи насыщенного водного раствора NH4Cl (1 мл). Реакционная смесь была очищена посредством препаративной ВЭЖХ для получения (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (13,3 мг, выход: 83%). Метод ЖХ-МС 1 tR=1,63 мин, масса/заряд 447 (М+1).
Этап 3
Раствор (R)-6-аллил-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (13,3 мг, 0,030 ммоль) в сухом тетрагидрофуране (1,5 мл) был охлажден до 0°С. Затем в раствор был добавлен дизиамилборан (0,5М в тетрагидрофуране, 500 мкл, избыток). Спустя 10 мин смесь нагревалсь до комн. темп. и перемешивалась в течение 1 ч. Затем смесь была снова охлаждена до температуры 0°С, после чего реакция была остановлена при помощи воды (1 мл) и NaBO3 (10 мг). Полученная смесь была выпарена и очищена посредством препаративной ВЭЖХ для получения (R)-6-(4-фторфенил)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-3-ил)фенил)этил)-1,3-оксазинан-2-она (4,2 мг, выход: 30%). Метод ЖХ-МС 1 tR=1,33 мин, масса/заряд=487(М+1); 1H ЯМР (CD3Cl) δ 7,47 (dd, 1Н), 7,38 (m, 3Н), 7,24 (m, 2H), 7,07 (t, 2H), 6,96 (d, 2H), 6,39 (t, 1H), 5,65 (m, 1H), 4,26 (t, 1H), 3,66 (s, 3Н), 2,91 (m, 1H), 2,40-2,14 (m, 3Н), 1,54 (d, 3Н).
ПРИМЕР 43
(R)-3-((S)-1-(4-(1-этил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(3-гидроксипропил)-6-фенил-1,3-оксазинан-2-он
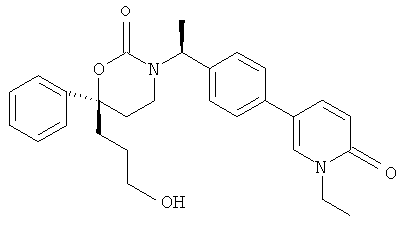
Титульное соединение было получено в результате применения процедур, аналогичных описанным в Примере 30, с применением этилйодида на 1 этапе для получения 5-бром-1-этилпиридин-2(1Н)-она, который использовался в ходе 2 этапа. Метод ЖХ-МС 2 tR=1,297 мин, масса/заряд=461,1; 1Н ЯМР (CDCl3) 1,31 (m, 1H), 1,36 (t, 3Н), 1,51 (d, 3Н), 1,68 (m, 1H), 1,86-2,01 (m, 2H), 2,18 (m, 1H), 2,27 (m, 2H), 2,91 (m, 1H), 3,52 (m, 2H), 4,18 (m, 2H), 5,13 (m, 1H), 5,62 (m, 1H), 6,91 (m, 3Н), 7,08 (m, 2H), 7,18-7,33 (m, 5H), 7,41 (s, 1H), 7,61 (d, 1H).
ПРИМЕР 44
(R)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)пропил)-6-фенил-1,3-оксазинан-2-он
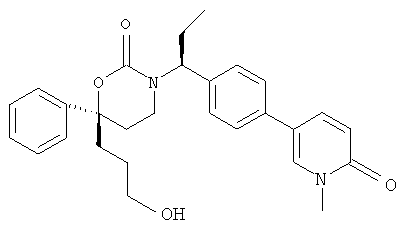
Титульное соединение было получено из (R)-6-(3-гидроксипропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропил)-1,3-оксазинан-2-она в результате применения процедуры, аналогичной описанной для 2 этапа Примера 30. Метод ЖХ-МС 2 tR=1,113 мин, масса/заряд=461,1; 1H ЯМР (CDCl3) 0,95 (t, 3H), 1,30 (m, 1H), 1,68 (m, 1H), 1,81-1,99 (m, 2H), 2,11-2,32 (m, 3H), 2,88 (m, 1H), 3,50 (m, 2H), 3,58 (m, 2H), 5,43 (m, 1H), 6,49 (d, 1H), 6,98 (d, 2H), 7,08 (d, 2H), 7,19 (m, 1H), 7,25 (m, 4H), 7,32 (s, 1H), 7,47 (m, 1H).
ПРИМЕР 45
(R)-6-(3-гидроксипропил)-3-((S)-1-(4-(2-гидроксипиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
Титульное соединение было получено из (R)-6-(3-гидроксипропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 4-бром-2-гидроксипиридина в результате применения процедуры, аналогичной описанной для 2 этапа Примера 30. Метод ЖХ-МС 2 tR=1,019 мин, масса/заряд=865,4; 1H ЯМР (CDCl3) 1,29-1,40 (m, 1H), 1,49 (d, 3H), 1,60-1,72 (m, 1H), 1,83-2,01 (m, 3H), 2,18 (m, 1H), 2,21-2,37 (m, 2H), 2,88 (m, 1H), 3,51 (m, 2H), 5,63 (m, 1H), 6,41 (d, 1H), 6,68 (s, 1H), 6,90 (d, 2H), 7,21-7,33 (m, 7H), 7,39 (d, 1H).
ПРИМЕР 46
(R)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-2-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
Титульное соединение было получено из (R)-6-(3-гидроксипропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 6-бром-1-метилпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 2 этапа Примера 30. Метод ЖХ-МС 2 tR=1,088 мин, масса/заряд=447; 1H ЯМР (CDCl3) 1,38 (m, 1Н), 1,56 (d, 3Н), 1,70 (m, 1H), 1,95-2,08 (m, 2H), 2,23 (m, 1H), 2,37 (s, 2H), 3,05 (m, 1H), 3,33 (s, 3Н), 3,58 (m, 2H), 5,73 (m, 1H), 6,29 (d, 1H), 6,89 (d, 1H), 7,01-7,09 (m, 4H), 7,21-7,39 (m, 5H), 7,53 (t, 1H).
6-Бром-1-метилпиридин-2(1Н)-он был получен из 6-бромпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 59.
ПРИМЕР 47
(R)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
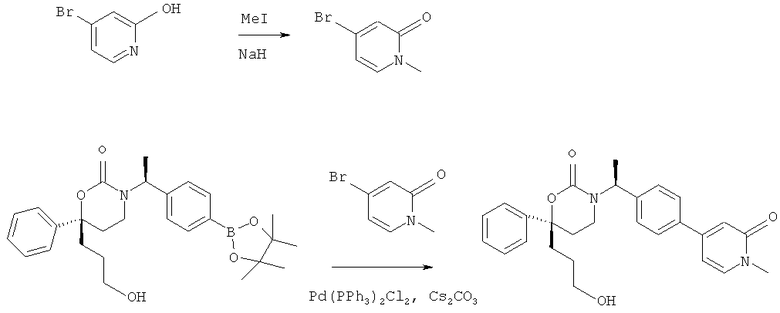
Этап 1
В суспензию NaH (80 мг, 2 ммоль) в тетрагидрофуране (10 мл) при температуре 0°С был добавлен 4-бромпиридин-2-ол (80 мг, 0,46 ммоль). Полученная смесь перемешивалась в течение 1 ч, после чего в нее был добавлен CH3I (355 мг, 2,5 ммоль) и смесь перемешивалась в течение ночи. Реакция была остановлена при помощи водного раствора NH4Cl. Органическая фаза подверглась выпариванию для получения неочищенного продукта, который затем был очищен посредством колоночной хроматографии для получения 4-бром-1-метилпиридин-2(1H)-она (42,3 мг, 50%).
Этап 2
Смесь (R)-6-(3-гидроксипропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (50 мг, 0,11 ммоль) и 4-бром-1-метилпиридин-2(1Н)-она (30 мг, 0,16 ммоль), Pd(Ph3P)2Cl2 (10 мг), а также водного раствора. Cs2CO3 (4 мл, 2 М) в 1,4-диоксане (10 мл) перемешивалась и нагревалась для обратного конденсирования в течение 2 ч. После окончания реакции смесь была промыта водой и подверглась экстракции при помощи EtOAc. Органическая фаза была промыта солевым раствором, высушена над Na2SO4, и подверглась фильтрации для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (R)-6-(3-гидроксипропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (25 мг, 51%). 1Н ЯМР (CDCl3): δ 1,35 (m, 1Н), 1,47 (d, 3Н), 1,63 (m, 2H), 1,94 (m, 2H), 2,18 (m, 1H), 2,39 (m, 2H), 2,86 (m, 1H), 3,51 (m, 5H), 5,63 (m, 1H), 6,31 (m, 1H), 6,70 (m, 1H), 6,91 (m, 2H), 7,20-7,32 (m, 8H).
ПРИМЕР 48
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
Метод 1
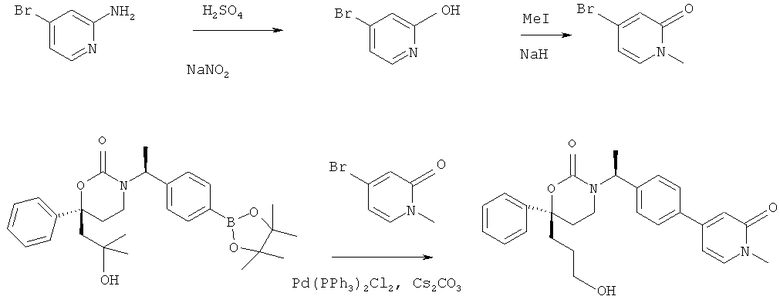
Этап 1
Раствор 4-бромпиридин-2-амина (600 мг, 3,5 ммоль) в смеси 2 M H2SO4 (20 мл) и 2 M Na2NO2 (10 мл) перемешивался при температуре 0-5°С в течение 2 ч. Реакционная смесь подвергалась экстракции CH2Cl2, после чего органический слой промывался насыщенным раствором NaCl, высушивался над безводным Na2SO4 и выпаривался. Полученный осадок был очищен посредством препаративной ТСХ для получения 4-бромпиридин-2-ола (303 мг, 50%).
Этап 2
В суспензию NaH (300 мг, 7,5 ммоль) в тетрагидрофуране (10 мл) при температуре 0°С был добавлен 4-бромпиридин-2-ол (303 мг, 1,73 ммоль). После перемешивания полученной смеси в течение 1 ч в нее был добавлен CH3I (491 мг, 3,46 ммоль), после чего смесь перемешивалась в течение ночи. Реакция была остановлена при помощи водного раствора NH4Cl. Органический слой был выпарен для получения неочищенного продукта, который затем был очищен посредством колоночной хроматографии для получения 4-бром-1-метилпиридин-2(1H)-она (161 мг, 50%).
Этап 3
Смесь (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (200 мг, 0,42 ммоль), 4-бром-1-метилпиридин-2(1Н)-она (118 мг, 0,63 ммоль), Pd(Ph3P)2Cl2 (20 мг), а также 2 М водного раствора Cs2CO3 (5 мл, 10 ммоль) в 1,4-диоксане (20 мл) перемешивалась и нагревалась для обратного конденсирования в течение 2 ч. После окончания реакции смесь была промыта водой и подверглась экстракции при помощи EtOAc. Органическая фаза была промыта солевым раствором, высушена над Na2SO4, а также подверглась фильтрации и выпариванию для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (83 мг, 43%). Метод ЖХ-МС 2 tR=1,16 мин, масса/заряд=921,5; 1H ЯМР (CDCl3) 1,11 (s, 3Н), 1,18 (s, 3Н), 1,22 (t, 1H), 1,52 (m, 3Н), 2,21 (s, 2H), 2,22-2,34 (m, 2H), 2,34-2,46 (m, 1H), 2,85 (m, 1H), 3,57 (s, 3Н), 5,59 (m, 1H), 6,33 (d, 1H), 6,68 (s, 1H), 7,01 (d, 2H), 7,29-7,41 (m, 8H); 1H ЯМР (CD3OD) 0,98 (s, 3Н), 1,29 (s, 3Н), 1,58 (d, 3Н), 2,17 (s, 2H), 2,22 (m, 1H), 2,50 (m, 2H), 3,08 (m, 1H), 3,59 (s, 3Н), 5,59 (m, 1H), 6,61 (d, 1H), 6,66 (s, 1H), 7,08 (m, 2H), 7,30-7,40 (5H), 7,42 (d, 2H), 7,70 (d, 1H).
Метод 2
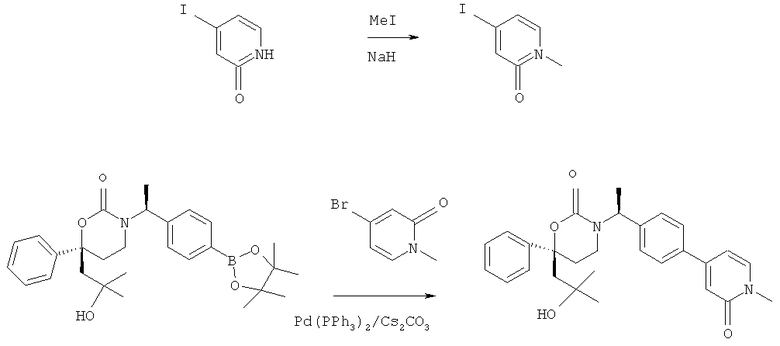
Этап 1
В раствор 4-йодпиридин-2(1H)-она (3 г, 0,013 моль) и K2СО3 (3,55 г, 0,026 моль) в DMF (30 мл) был добавлен йодметан (4,7 г, 0,033 ммоль). Смесь перемешивалась при комнатной температуре в течение ночи, после чего в нее были добавлены вода и EtOAc. Органическая фаза была высушена над Na2SO4 и выпарена для получения 4-йод-1-метилпиридин-2(1H)-она (1,6 г, 53%).
Этап 2
Смесь 4-йод-1-метилпиридин-2(1Н)-она (0,909 г, 3,76 ммоль), (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (1,5 г, 3,13 ммоль), 2 М водного раствора Cs2CO3 (3 мл, 6 ммоль), а также PdCl2(PPh3)2 (0,201 г, 0,282 ммоль) в 1,4-диоксане (15 мл) подверглась обратному конденсированию в атмосфере N2 в течение 2 часов. Полученный после фильтрации реакционной смеси фильтрат подвергся экстракции при помощи EtOAc. Объединенный органичесикий слой был промыт солевым раствором, высушен над Na2SO4 и подвергся выпариванию для получения неочищенного продукта, который затем был очищен посредством препаративной ВЭЖХ и хиральной ВЭЖХ для получения (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (399 мг, 28%). Результаты ЖХ-МС и 1H ЯМР (CD3OD) были аналогичны полученным для продукта, изготовленного посредством Метода 1. Затем соединение подверглось перекристаллизации в соответствии с описанными ниже методами.
Соединение было получено в виде кристаллического моногидрата путем добавления по капле 60 мл воды в раствор 7,6 г соединения в 15 мл метанола. После перемешивания в течение 1 ч твердое вещество подверглось фильтрации путем всасывания, промыванию водой и диэтилэфиром, а также высушиванию в сушильном шкафу над концентрированной серной кислотой/гидроксидом калия. Затем соединение также подверглось перекристаллизации из воды/этанола (80:20) для получения моногидрата. Температура плавления: 118-122°С.
Соединение подверглось перекристаллизации из изопропилацитата. с помощью процедуры, аналогичной описанной для Метода перекристаллизации В Примера 37, для получения кристаллического твердого вещества с температурой плавления 106-116°С. Соединение также подверглось перекристаллизации в соответствии с описанным выше методом из EtOAc (температура плавления 90-93°С, температура плавления 102-122°С), из изобутилацетата (температура плавления 108-126°С), из EtOH/ТВМЕ (температура плавления 108-126°С), а также из 2-бутанона.
Метод 3
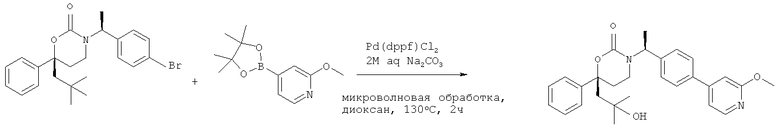
Этап 1
Смесь (S)-3-((S)-1-(4-бромфенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (100 мг, 0,23 ммоль), 2-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (68 мг, 1,25 эквив.), Pd(dppf)Cl2.CH2Cl2 (19 мг, 10% моль), 2М водного раствора Na2CO3 (1 мл), а также 1,4-диоксана (3 мл) подверглась трехкратной дегазации и повторному насыщению газообразным N2 перед обработкой в микроволновой печи в течение 2 ч при температуре 130°С. Полученные результаты ЖХ-МС свидетельствовали об окончании реакции. Смесь была разбавлена EtOAc (50 мл), промыта водой (10 мл) и солевым раствором (8 мл), а также высушена над Na2SO4. После фильтрации и выпаривания осадок был очищен посредством хроматографии с применением колонки, содержащей 12 г силикагеля и элюированной при помощи градиента 0-10% МеОН в CH2Cl2, для получения (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(2-метоксипиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (112 мг, колич. выход). Метод ЖХ-МС 1 tR=1,66 мин, масса/заряд=461 (М+1).
Этап 2
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(2-метоксипиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он (28 мг, 0,061 ммоль), карбонат калия (17 мг, 2 эквив.) и йодметан (40 мкл, 10 эквив.) перемешивались с ацетонитрилом (2,5 мл), после чего смесь нагревалась в условиях обратного конденсирования в течение 4 ч. После охлаждения до комнатной температуры смесь окислялась при помощи 5% водного раствора HCl и очищалась посредством препаративной ВЭЖХ для получения (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (14,4 мг, 52%). Результаты ЖХ-МС и 1Н ЯМР (CD3OD) были аналогичны полученным для продукта, изготовленного в соответствии с Методом 1.
ПРИМЕР 49
2,2-диметил-3-((R)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропаннитрил
Метод 1
Титульное соединение было получено из 2,2-диметил-3-((R)-2-оксо-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)пропаннтрила и 5-бром-1-метилпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,231 мин, масса/заряд=470,1; 1H ЯМР (CDCl3) 1,28 (s, 3Н), 1,40 (s, 3Н), 1,47 (d, 3H), 2,09 (s, 2H), 2,21 (m, 1H), 2,41 (m, 2H), 2,83 (m, 1H), 3,52 (s, 3Н), 5,56 (m, 1H), 6,58 (d, 1H), 6,82 (d, 2H), 7,02 (d, 2H), 7,30 (m, 6H), 7,43 (m, 1H).
Метод 2

Раствор 2,2-диметил-3-((R)-2-оксо-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-6-ил)пропаннитрила (202 мг, 0,444 ммоль) и MeI (110 мкл, 4эквив.) в сухом тетрагидрофуране (5 мл) был охлажден до 0°С. Затем в раствор был добавлен NaH (60% в минеральном масле, 36 мг, 2 эквив.). Спустя 10 мин смесь была медленно нагрета до комнатной температуры и перемешивалась в течение 3 ч. Результаты ЖХ-МС свидетельствовали о приблизительно 50% преобразовании. Смесь нагревалась в течение 1 ч при температуре 60°С, после чего ЖХ-МС показала завершение реакции. После охлаждения до комнатной температуры смесь была охлаждена до 0°С. Реакция была остановлена с помощью насыщенного водного раствора NH4Cl (3 мл), после чего смесь была разбавлена CH2Cl2 (20 мл), промыта 1% водным раствором HCl (5 мл) и солевым раствором (4 мл), а также высушена над Na2SO4. После фильтрации и выпаривания полученный осадок был очищен посредством препаративной ВЭЖХ для получения 2,2-диметил-3-((R)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропаннитрила (177,4 мг, выход: 85%) в виде масла светло-желтого цвета.
Метод 3
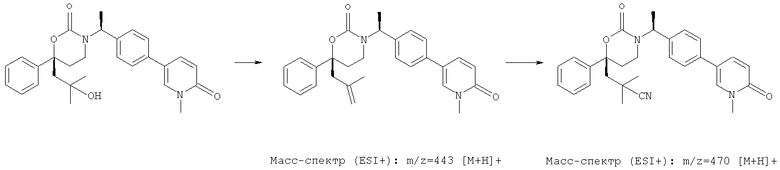
2,2-диметил-3-(3-{(S)-1-[4-(1-метил-6-оксо-1,6-дигидро-пиридин-3-ил)-фенил]-этил}-2-оксо-(S)-6-фенил-[1,3]оксазинан-6-ил)-пропионитрил был получен из (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она в результате применения процедур, аналогичных описанным в Методе 2 Примера 71, для получения 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила. (S)-6-(2-гидрокси-2-метил-пропил)-3-{(S)-1-[4-(1-метил-6-оксо-1,6-дигидро-пиридин-3-ил)-фенил]-этил}-6-фенил-[1,3]оксазинан-2-он был получен путем связывания (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-[(S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил]-1,3-оксазинан-2-она с 5-йод-1-метил-1Н-пиридин-2-оном посредством воздействия Pd(PPh3)4 и 2 М водного раствора Na2CO3 в смеси метанола и диоксана (1:3) при температуре 80°С. Соединение, полученное в виде пены, была растворено в небольшом количестве этилацетата, после чего перемешивалось в течение ночи при комнатной температуре. Полученное твердое вещество подверглось фильтрации путем всасывания, промывалось небольшим количеством диэтилэфира и высушивалось. Температура плавления: 143-145°С.
ПРИМЕР 50
2,2-диметил-3-((R)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропаннитрил
Метод 1
Титульное соединение было получено из 2,2-диметил-3-((R)-2-оксо-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)пропаннитрила и 4-бром-1-метилпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,103, масса/заряд=470,4; 1H ЯМР (CDCl3) 1,26 (s, 3H), 1,41 (s, 3H), 1,49 (d, 3H), 2,09 (s, 2H), 2,24 (m, 1H), 2,53 (m, 2H), 2,88 (m, 1H), 3,56 (s, 3H), 5,59 (m, 1H), 6,38 (d, 1H), 6,78 (s, 1H), 6,84 (d, 2H), 7,19 (m, 2H), 7,31 (m, 6H).
Метод 2
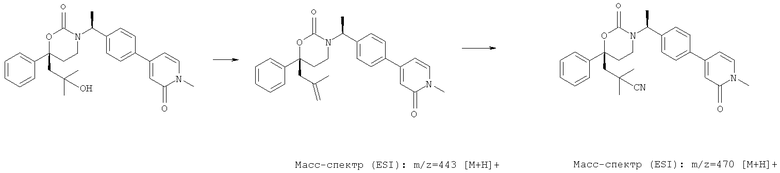
(S)-2,2-диметил-3-(3-{1-[(S)-4-(1-метил-2-оксо-1,2-дигидро-пиридин-4-ил)-фенил]-этил}-2-оксо-6-фенил-[1,3]оксазинан-6-ил)-пропионитрил был получен из (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она в результате применения процедур, аналогичных описанным Методе 2 Примера 71 для последующего получения 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила. Исходное соединение, (5)-6-(2-гидрокси-2-метилпропил)-3-{(S)-1-[4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)-фенил]-этил}-6-фенил-[1,3]оксазинан-2-он, было получено путем связывания (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-[(5)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил]-1,3-оксазинан-2-она и трифтор-метансульфоновой кислоты 1-метил-2-оксо-1,2-дигидропиридин-4-ил эфира в стандартных условиях; Pd(dppf)Cl2*CH2Cl2, 2 M водный раствор Na2CO3, DMF, 90°C, 2 ч. Соединение, полученное в виде смолы, было растворено в небольшом количестве EtOAc и перемешивалось при комнатной температуре в течение ночи. Твердое вещество подверглось фильтрации путем всасывания, промывалось небольшим количеством диэтилэфира и высушивалось. Температура плавления 195-198°С.
ПРИМЕР 51
2,2-диметил-3-((R)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)пропанамид
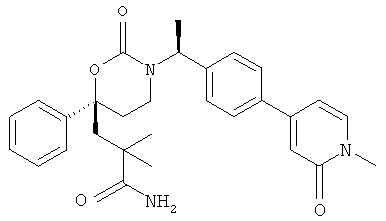
Титульное соединение было получено из 2,2-диметил-3-((R)-2-оксо-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)пропаннитрила и 4-бром-1-метилпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 6, с последующей обработкой H2O2, K2СО3. Метод ЖХ-МС 2 tR=1,133 мин, масса/заряд=488,1; 1H ЯМР (CDCl3) 1,12 (s, 3Н), 1,19 (s, 3Н), 1,49 (d, 3Н), 2,09-2,28 (m, 3Н), 2,32-2,58 (m, 2H), 2,89 (m, 1H), 3,59 (s, 3Н), 5,61 (m, 1H), 6,54 (m, 1H), 6,88 (m, 1H), 6,97-7,10 (m, 2H), 7,28 (m, 6H), 7,42 (m, 1H), 7,53 (m, 1H).
ПРИМЕР 52
(S)-3-((S)-1-(4-(1-этил-6-оксо-1,6-дигидропиридин-3-ил)фенил)пропил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
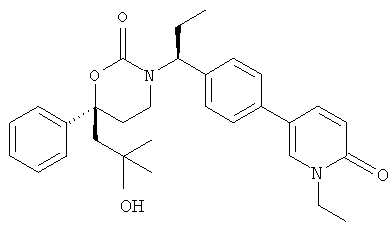
Титульное соединение было получено из (R)-6-аллил-3-((S)-1-(4-бромфенил)пропил)-6-фенил-1,3-оксазинан-2-она в результате применения процедур, аналогичных описанным в Методе 2 Примера 32, с использованием 5-бром-1-этилпиридин-2(1Н)-она в ходе 4 этапа. Метод ЖХ-МС 2 tR=1,732 мин, масса/заряд=475,1; 1H ЯМР (CDCl3) 0,95 (s, 3H), 1,01 (t, 3Н), 1,26 (s, 3Н), 1,38 (t, 3Н), 2,06 (m, 2H), 2,18-2,31 (m, 3Н), 2,36 (m, 1H), 2,55 (m, 1H), 3,04 (m, 1H), 4,11 (m, 2H), 5,37 (m, 1H), 6,66 (d, 1H), 7,11 (m, 2H), 7,20-7,33 (m, 7H), 7,76 (d, 1H), 7,88 (s, 1H).
ПРИМЕР 53
(S)-3-((S)-1-(4-(1-этил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
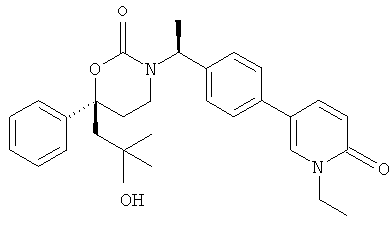
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 5-бром-1-этилпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,224 мин, масса/заряд=475,1; 1Н ЯМР (CDCl3) 1,11 (s, 3Н), 1,19 (s, 3H), 1,39 (t, 3H), 1,56 (d, 3H), 2,20 (s, 2H), 2,26 (m, 1H), 2,36-2,57 (m, 2H), 2,87 (m, 1H), 4,03 (m, 2H), 5,69 (m, 1H), 6,62 (d, 1H), 7,00 (d, 2H), 7,17 (d, 2H), 7,28-7,51 (m, 6H), 7,50 (d, 1H). В результате перекристаллизации из изопропилацетата с применением процедур, аналогичных описанным в Примере 37, Метод перекристаллизации В, было получено кристаллическое твердое вещество с температурой плавления 167-168°С.
ПРИМЕР 54
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)пропил)-6-фенил-1,3-оксазинан-2-он
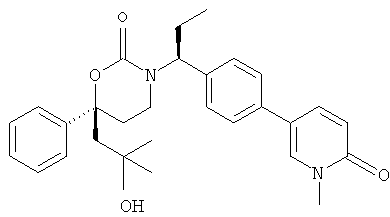
Титульное соединение было получено из (R)-6-аллил-3-((S)-1-(4-бромфенил)пропил)-6-фенил-1,3-оксазинан-2-она в результате применения процедур, аналогичных описанным в Методе 2 Примера 32, с использованием 5-бром-1-метилпиридин-2(1Н)-она в ходе 4 этапа. Метод ЖХ-МС 2 tR=1,746 мин, масса/заряд=475,2; 1H ЯМР (CD3OD) 1,04 (t, 3Н), 1,11 (s, 3Н), 1,24 (s, 3Н), 1,95-2,04 (m, 2H), 2,13-2,26 (m, 4H), 2,44 (m, 1H), 2,91 (m, 1H), 3,61 (s, 3Н), 5,36 (m, 1H), 6,67 (d, 1H), 7,10-7,33 (m, 8H), 7,42 (s, 1H), 7,55 (d, 1H).
ПРИМЕР 55
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)пропил)-6-фенил-1,3-оксазинан-2-он
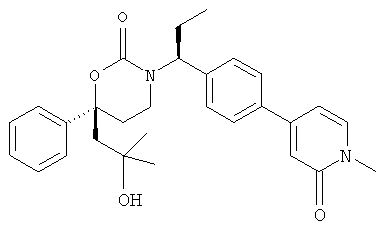
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропил)-1,3-оксазинан-2-она и 4-бром-1-метилпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 2 этапа Примера 59. Метод ЖХ-МС 2 tR=1,203 мин, масса/заряд=971,4; 1Н ЯМР (CDCl3) 0,97 (t, 3Н), 1,12 (s, 3Н), 1,19 (s, 3H), 1,79-2,02 (m, 2H), 2,11-2,24 (m, 4H), 2,29-2,42 (m, 1H), 2,81 (m, 1H), 3,50 (s, 3Н), 5,40 (m, 1H), 6,28 (d, 1H), 6,64 (s, 1H), 7,02 (d, 2H), 7,18 (m, 3Н), 7,20 (m, 2H), 7,28 (m, 3Н).
ПРИМЕР 56
(R)-6-этил-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)пропил)-6-фенил-1,3-оксазинан-2-он
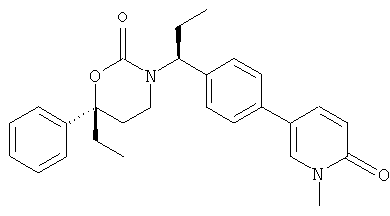
Титульное соединение было получено из (R)-3-((S)-1-(4-бромфенил)пропил)-6-этил-6-фенил-1,3-оксазинан-2-она в результате применения процедур, аналогичных описанным для 3 и 4 этапов Метода 2 Примера 32. Метод ЖХ-МС 1 tR=1,6 мин, масса/заряд=431 (М+1).
(R)-3-((S)-1-(4-бромфенил)пропил)-6-этил-6-фенил-1,3-оксазинан-2-он был получен из 1-хлор-3-фенилпентан-3-ола и (S)-1-(4-бромфенил)пропан-1-амина в результате применения процедуры, аналогичной описанной для 2 этапа Примера 71.
1-хлор-3-фенилпентан-3-ол был получен из 3-хлор-1-фенилпропан-1-она и этилмагния бромида в результате применения процедуры, аналогичной описанной для 2 этапа Метода 1 Способа получения 1.
ПРИМЕР 57
(R)-6-этил-3-((S)-1-(4-(1-этил-6-оксо-1,6-дигидропиридин-3-ил)фенил)пропил)-6-фенил-1,3-оксазинан-2-он
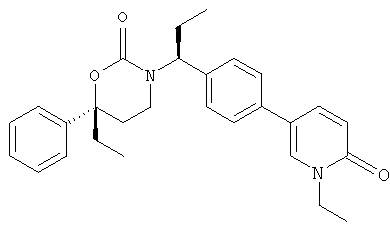
Титульное соединение было получено из (R)-3-((S)-1-(4-бромфенил)пропил)-6-этил-6-фенил-1,3-оксазинан-2-она в результате применения процедур, аналогичных описанным для 3 и 4 этапов Метода 2 Примера 32, с использованием 5-бром-1-этилпиридин-2(1Н)-она в ходе 4 этапа. Метод ЖХ-МС 1 tR=1,68 мин, масса/заряд=445 (М+1).
ПРИМЕР 58
(R)-6-этил-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)пропил)-6-фенил-1,3-оксазинан-2-он
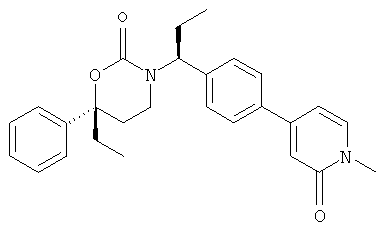
Титульное соединение было получено из (R)-3-((S)-1-(4-бромфенил)пропил)-6-этил-6-фенил-1,3-оксазинан-2-она в результате применения процедур, аналогичных описанным для 3 и 4 этапов Метода 2 Примера 32, с использованием 4-йод-1-метилпиридин-2(1Н)-она в ходе 4 этапа. Метод ЖХ-МС 1 tR=1,58 мин, масса/заряд=431 (М+1); 1H ЯМР (CDCl3) 7,33 (1Н, d, J=7,03 Гц), 7,29-7,21 (7Н, m), 7,01 (2H, d, J=8,20 Гц), 6,75 (1Н, d, J=2,05), 6,39 (1Н, dd, J=2,05, 7,03), 5,48 (1Н, ар dd, J=6,44, 9,66), 3,58 (3H, s), 2,95-2,87 (1Н, m), 2,37-2,14 (3H, m), 2,06-1,81 (m, 4H), 1,00 (3H, t, J=7,32), 082 (3H, t, J=7,61).
ПРИМЕР 59
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-изопропил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
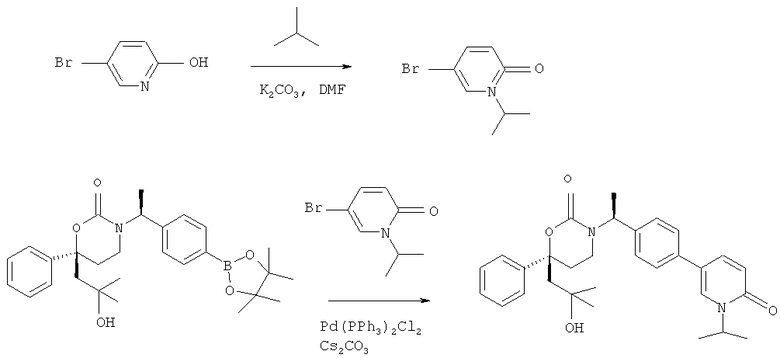
Этап 1
В раствор 5-бромпиридин-2-ола (1 г, 5,75 ммоль) в DMF (10 мл) были добавлены 2-йодпропан (4,9 г, 28,75 ммоль) и K2СО3 (4 г, 28,75 ммоль). Смесь перемешивалась при комнатной температуре в течение ночи, после чего разбавлялась (20 мл) и подвергалась экстракции при помощи EtOAc (3×25 мл). Объединенная органическая фаза была промыта солевым раствором, высушена над Na2SO4, выпарена и очищена посредством препаративной ТСХ для получения 5-бром-1-изопропилпиридин-2(1H)-она (380 мг, 31%). 1H ЯМР (CDCl3): 1,35 (d, 6Н), 5,65-5,75 (m, 1H), 6,48 (d, 1H), 7,30 (m, 1H), 7,41 (d, 1H).
Этап 2
В раствор (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (100 мг, 0,21 ммоль) в 1,4-диоксане (2 мл) был добавлен 5-бром-1-изопропилпиридин-2(1H)-он (54,2 мг, 0,25 ммоль). Затем в смесь были добавлены катализаторы Pd(PPh3)2Cl2 (14 мг, 0,02 ммоль) и Cs2CO3 (1 мл, 2 M). Сосуд был запечатан прижимным колпачком и помещен в микроволновой резонатор. Была применена микроволновое облучение мощностью 100 Вт с повышением температуры от комнатной до 120°С. После достижения указанной температуры реакционная смесь выдерживалась при данной температуре в течение 30 мин. После охлаждения смеси до комнатной температуры была проведена ее фильтрация. Полученный фильтрат подвергся экстракции при помощи EtOAc (20 мл × 4), органический слой был промыт солевым раствором, высушен над Na2SO4 и выпарен для получения неочищенного продукта, который затем был очищен посредством препаративной ВЭЖХ для получения (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-изопропил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (22 мг, 21%). 1H ЯМР (CDCl3): 1,13 (s, 3Н), 1,19 (s, 3Н), 1,40 (6Н), 1,53 (d, 3H), 2,18-2,30 (m, 4Н), 2,40 (m, 1H), 2,88 (m, 1H), 5,31 (m, 1H), 5,70 (m, 1H), 6,73 (d, 1H), 7,02 (d, 2H), 7,15 (d, 2H), 7,27-7,38 (m, 5H), 7,43 (d, 1H), 7,50 (d, 1H).
ПРИМЕР 60
(R)-6-этил-3-((S)-1-(4-(1-изопропил-6-оксо-1,6-дигидропиридин-3-ил)фенил)пропил)-6-фенил-1,3-оксазинан-2-он
Титульное соединение было получено из (R)-3-((S)-1-(4-бромсренил)пропил)-6-этил-6-фенил-1,3-оксазинан-2-она в результате применения процедур, аналогичных описанным для 3 и 4 этапов Метода 2 Примера 32, с использованием 5-бром-1-изопропилпиридин-2(1Н)-она в ходе 4 этапа. Метод ЖХ-МС 1 tR=1,75 мин, масса/заряд=459 (М+1); 1H ЯМР (CDCl3) 7,49, (1H, dd, J=2,34, 9,37 Гц), 7,42 (1H, d, J=2,34 Гц), 7,32-7,24 (5H, m), 7,13 (1H, d, J=8,20), 7,04 (1H, d, J=8,49), 6,66 (1H, d, J=9,37), 5,49 (1H, aq q, J=6,44, 9,37), 5,33 (1H, m), 2,96-2,91 (1H, m), 2,39-2,32 (1H, m), 2,29-2,17 (2H, m), 2,05-1,85 (m, 4Н), 1,41 (6H, dd, J=1,17, 6,73), 1,01 (3H, t, J=7,32 Гц), 0,832 (3Н, t, J=7,32 Гц).
5-бром-1-изопропилпиридин-2(1Н)-он был получен из 5-бромпиридин-2(1Н)-она и изопропилйодида в результате применения процедуры, аналогичной описанной для 1 этапа Примера 59.
ПРИМЕР 61
(S)-3-((S)-1-(4-(1,5-диметил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
Титульное соединение было получено в результате применения процедур, аналогичных описанным в Примере 59 с использованием 1 5-бром-3-метилпиридин-2(1Н)-она и метилйодида в ходе 1 этапа. Метод ЖХ-МС 2 tR=1,197 мин, масса/заряд=475,1; 1H ЯМР (CDCl3) 1,04 (s, 3Н), 1,11 (s, 3Н), 1,46 (d, 3Н), 2,18 (m, 5H), 2,21 (m, 1H), 2,29-2,40 (m, 1H), 2,80 (m, 1H), 3,41 (s, 3Н), 3,56 (s, 3Н), 5,60 (m, 1H), 6,91 (d, 2H), 7,07 (d, 2H), 7,21-7,40 (m, 7H).
ПРИМЕР 62
(S)-3-((S)-1-(4-(1-этил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-он
Титульное соединение было получено в результате применения процедур, аналогичных описанным в Примере 59 с использованием 5-бромпиридин-2(1Н)-она и этилйодида в ходе 1 этапа, а также (S)-6-(4-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она в ходе 2 этапа. Метод ЖХ-МС 2 tR=1,205 мин, масса/заряд=493,2; 1H ЯМР (CDCl3) 1,16 (d, 6Н), 1,39 (t, 3Н), 1,52 (d, 3Н), 2,19 (s, 4H), 2,20-2,31 (m, 2H), 2,38-2,50 (m, 1H), 2,90 (m, 1H), 4,04 (m, 2H), 5,69 (m, 1H), 6,66 (d, 1H), 7,00 (m, 4H), 7,18 (d, 2H), 7,30 (m, 2H), 7,41 (s, 1H), 7,51 (d, 1H). В результате перекристаллизации из изопропилацетата с применением процедур, аналогичных описанным в Примере 37, Метод перекристаллизации В, было получено кристаллическое твердое вещество с температурой 172-173,6°С.
ПРИМЕР 63
(R)-6-метил-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)пропил)-6-фенил-1,3-оксазинан-2-он
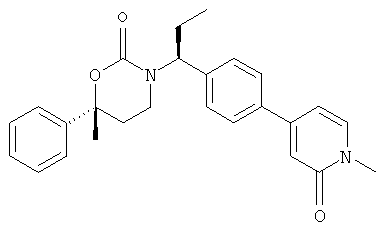
Титульное соединение было получено из (R)-3-((S)-1-(4-бромфенил)пропил)-6-метил-6-фенил-1,3-оксазинан-2-она в результате применения процедур, аналогичных описанным для 3 и 4 этапов Метода 2 Примера 32, с использованием 4-йод-1-метилпиридин-2(1Н)-она в ходе 4 этапа. Метод ЖХ-МС 1 tR=1,55 мин, масса/заряд=417 (М+1); 1H ЯМР (CDCl3) 7,41 (1H, d, J=7,03 Гц), 7,33 (2H, d, J=8,20 Гц), 7,29-7,19 (5Н, m), 7,10 (1H, d, J=8,20), 6,95 (1H, d=1,76), 6,55 (1H, dd, J=2, 7,03 Гц), 5,51 (1H, q, J=6,49, 9,66 Гц), 3,65 (3Н, s), 3,00-2,95 (1H, m), 2,44-2,36 (1H, m), 2,33-2,15 (2H, m), 2,06-1,86 (2H, m), 1,64 (3Н, s), 1,02 (3Н, t, J=7,32 Гц).
(R)-3-((S)-1-(4-бромфенил)пропил)-6-метил-6-фенил-1,3-оксазинан-2-он был получен из 4-хлор-2-фенилбутан-2-ола и (S)-1-(4-бромфенил)пропан-1-амина в результате применения процедуры, аналогичной описанной для 2 этапа Примера 71.
4-хлор-2-фенилбутан-2-ол был получен из 3-хлор-1-фенилпропан-1-она и метилмагния бромида в результате применения процедуры, аналогичной описанной для 2 этапа Метода 1 Способа получения 1.
ПРИМЕР 64
(S)-3-((S)-1-(4-(1,6-диметил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
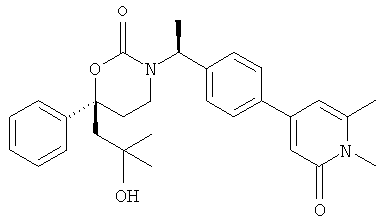
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропил)-1,3-оксазинан-2-она и 4-бром-1,6-диметилпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,173 мин, масса/заряд=475,2; 1H ЯМР (CDCl3) 1,10 (s, 3H),1,16 (s, 3Н), 1,51 (d, 3H), 2,18 (m, 3Н), 2,21 (m, 1H), 2,42 (m, 4H), 2,86 (m, 1H), 3,54 (s, 3Н), 5,66 (m, 1H), 6,21 (s, 1H), 6,60 (s, 1H), 6,97 (m, 2H), 7,23-7,34 (m, 7H).
4-бром-1,6-диметилпиридин-2(1Н)-он был получен путем метилирования 4-бром-6-метилпиридин-2(1Н)-она метилйодидом с использованием K2СО3 в результате применения процедуры, аналогичной описанной для 1 этапа Примера 59.
ПРИМЕР 65
(S)-3-((S)-1-(4-(1-этил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
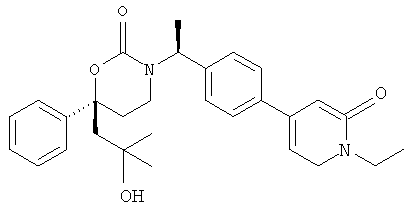
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропил)-1,3-оксазинан-2-она и 1-этил-4-йодпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,228 мин, масса/заряд=971,4; 1Н ЯМР (CDCl3) 1,10 (s, 3H), 1,14 (s, 3H), 1,36 (m, 3H), 1,53 (d, 3H), 2,17 (s, 2H), 2,21-2,32 (m, 2H), 2,32-2,48 (m, 1H), 2,88 (m, 1H), 4,00 (m, 2H), 5,68 (m, 1H), 6,39 (d, 1H), 6,78 (s, 1H), 6,99 (d, 2H), 7,27-7,38 (m, 8H).
1-этил-4-йодпиридин-2(1Н)-он был получен из 4-йодпиридин-2(1Н)-она и этилйодида в результате применения процедуры, аналогичной описанной для 1 этапа Примера 59.
ПРИМЕР 66
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(2-оксо-1-(2,2,2-трифторэтил)-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
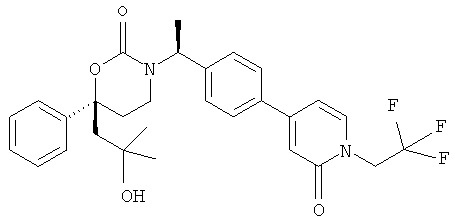
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропил)-1,3-оксазинан-2-она и 4-йод-1-(2,2,2-трифторэтил)пиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,871 мин, масса/заряд=471,1; 1H ЯМР (CDCl3) 1,11 (s, 3H), 1,17 (s, 3H), 1,53 (d, 3H), 2,16-2,33 (m, 4H), 2,35-2,47 (m, 1H), 2,89 (m, 1H), 4,58-4,70 (m, 2H), 5,69 (m, 1H), 6,71 (s, 1H), 7,00 (d, 2H), 7,19-7,38 (m, 8H). В результате перекристаллизации из изопропилацетата с применением процедур, аналогичных описанным в Примере 37, Метод перекристаллизации В, было получено кристаллическое твердое вещество с температурой плавления 144-145,5°С.
4-йод-1-(2,2,2-трифторэтил)пиридин-2(1Н)-он был получен из 4-йодпиридин-2(1Н)-она и 2,2,2-трифторэтил трифторметансульфоната в результате применения процедуры, аналогичной описанной для 1 этапа Примера 59.
ПРИМЕР 67
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(6-оксо-1-(2,2,2-трифторэтил)-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
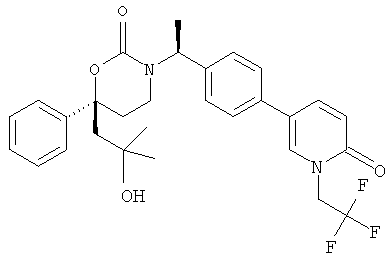
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропил)-1,3-оксазинан-2-она и 5-бром-1-(2,2,2-трифторэтил)пиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,323 мин, масса/заряд=471,1; 1H ЯМР (CDCl3) 1,13 (s, 3Н), 1,19 (s, 3H), 1,53 (d, 3H), 2,19-2,30 (m, 4H), 2,40 (m, 1H), 2,89 (m, 1H), 4,67 (m, 2H), 5,69 (m, 1H), 6,70 (d, 1H), 7,03 (d, 2H), 7,13 (d, 2H), 7,29-7,38 (m, 6H), 7,55 (d, 1H).
5-бром-1-(2,2,2-трифторэтил)пиридин-2(1Н)-он был получен из 5-бромпиридин-2(1Н)-она и 2,2,2-трифторэтил трифторметансульфоната в результате применения процедуры, аналогичной описанной для 1 этапа Примера 59.
ПРИМЕР 68
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-изопропил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
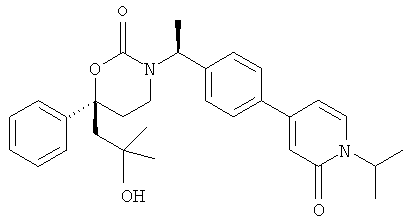
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропил)-1,3-оксазинан-2-она и 4-йод-1-изопропилпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,846 мин, масса/заряд=489,2; 1H ЯМР (CDCl3) 1,10 (s, 3H), 1,24 (s, 3H), 1,39 (d, 6H), 1,52 (d, 3Н), 2,17-2,31 (m, 4H), 2,35-2,46 (m, 1H), 2,88 (m, 1H), 5,27 (m, 1H), 5,69 (m, 1H), 6,49 (d, 1H), 6,88 (s, 1H), 7,00 (d, 2H), 7,29-7,38 (m, 7H), 7,40 (d,1H). В результате перекристаллизации из изопропилацетата с применением процедур, аналогичных описанным в Примере 37, Метод перекристаллизации В, было получено кристаллическое твердое вещество с температурой плавления 134-139°С.
4-йод-1-изопропилпиридин-2(1Н)-он был получен из 4-йодпиридин-2(1Н)-она и изопропилйодида в результате применения процедуры, аналогичной описанной для 1 этапа Примера 59.
ПРИМЕР 69
3-((R)-6-(4-фторфенил)-3-((S)-1-(4-(1-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрил
Титульное соединение было получено из 3-((R)-6-(4-фторфенил)-2-оксо-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила и 5-бром-1-метилпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 2 этапа Примера 3. Метод ЖХ-МС 1 tR=1,45 мин, масса/заряд=488; 1H ЯМР (CDCl3) 7,68 (dd, 1Н), 7,52 (d, 1H), 7,32 (q, 2H), 7,17 (d, 2H), 7,06 (t, 2H), 6,97 (d, 2H), 6,91 (d, 1H), 5,66 (q, 1H), 3,72 (s, 3H), 2,99 (dt, 1H), 2,48 (dd, 2H), 2,27 (m, 1H), 2,11 (s, 2H), 1,55 (d, 3H), 1,44 (s, 3H), 1,34 (s, 3H).
ПРИМЕР 70
(S)-3-((S)-1-(4-(1-этил-5-метил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 5-бром-1-этил-3-метилпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,314 мин, масса/заряд=489; 1H ЯМР (CDCl3) 1,09 (s, 3H), 1,15 (s, 3H), 1,35 (t, 3H), 1,50 (d, 3H), 2,15-2,25 (m, 7H), 2,35 (m, 1H), 2,86 (m, 1H), 4,03 (m, 2H), 5,66 (q, 1H), 6,96 (d, 2H), 7,13 (d, 2H), 7,25-7,36 (m, 7H).
5-бром-1-этил-3-метилпиридин-2(1Н)-он был получен путем алкилирования 5-бром-3-метилпиридин-2(1Н)-она этилйодидом с применением процедуры, аналогичной описанной для 1 этапа Примера 59,
ПРИМЕР 71
2,2-диметил-3-((R)-2-оксо-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-6-ил)пропаннитрил
Метод 1
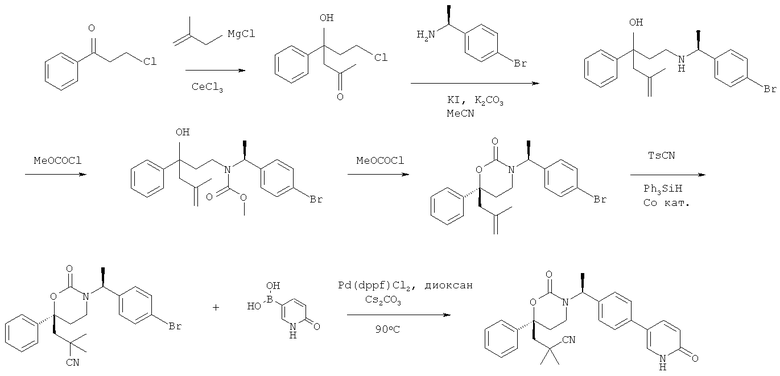
Этап 1
В колбу емкостью 250 мл были помещены безводный CeCl3 (7,1890 г, 29,2 ммоль) и тетрагидрофуран (55 мл). Смесь тщательно перемешивали в течение 2 ч при комнатной температуре, затем полученная суспензия охлаждалась до -78°С, после чего в нее добавлялся раствор 2-метилаллилмагний хлорид (0,5 М в тетрагидрофуране, 56 мл, 28,0 ммоль). После перемешивания в течение 2 ч при температуре -78°С в смесь через канюлю был добавлен раствор 3-хлорпропиофенона (3,350 г, 19,8 ммоль) в тетрагидрофуране (30 мл). Реакционная смесь отстаивалась в течение ночи для медленного нагревания до 8°С и одновременно перемешивалась (18 ч). Реакция была остановлена при помощи насыщенного водного раствора NaHCO3, после чего смесь подверглась экстракции при помощи EtOAc и высушиванию над Na2SO4. После выпаривания растворителей неочищенный 1-хлор-5-метил-3-фенилгекс-5-ен-3-ол использовался в ходе следующего этапа без дополнительной очистки. Метод ЖХ-МС 1 tR =1,91 мин, масса/заряд 248, 207 (М-ОН)+; 1Н ЯМР (400 МГц, CDCl3) 7,39-7,22 (m, 5H), 4,92 (m, 1H), 4,77 (m, 1H), 3,60-3,53 (m, 1H), 3,17-3,10 (m, 1H), 2,67 (d, J=13,2 Гц, 1Н), 2,55 (d, J=13,2 Гц, 1H), 2,41-2,25 (m, 2H), 1,29 (s, 3H); 13С ЯМР (100 МГц, CDCl3) 144,55, 141,72, 128,32, 126,88, 125,07, 116,50, 74,44, 51,46, 46,34, 40,19, 24,22.
Этап 2
1-хлор-5-метил-3-фенилгекс-5-ен-3-ол (1,28 г, 5,7 ммоль), (S)-1-(4-бромфенил)этанамин (1,37 г, 1,2 эквив.), KI (995 мг, 1,05 эквив.) и К2СО3 (1,57 г, 2 эквив.) перемешивались с ацетонитрилом (15 мл) и нагревались для обратного конденсирования (масляная баня 96°С) в течение ночи. После охлаждения до комнатной температуры смесь подверглась фильтрации, выпариванию и очистке посредством хроматографии с применением колонки, содержащей 40 г силикагеля и элюированной при помощи 0~8% МеОН в CH2Cl2, для получения 1-((S)-1-(4-бромфенил)этиламино)-5-метил-3-фенилгекс-5-ен-3-ола (1,33 г, 60%).
Этап 3
В раствор 1-((S)-1-(4-бромфенил)этиламино)-5-метил-3-фенилгекс-5-ен-3-ола (1,33 г, 3,43 ммоль) в CH2Cl2 (100 мл) были добавлены пиридин (277 мкл, 1 эквив.) и триэтиламин (717 мкл, 1,5 эквив.). Смесь охлаждалась до температуры 0°С, после чего в нее медленно был добавлен метилхлорформат (397 мкл, 1,5 эквив.). Спустя 15 мин смесь медленно нагревалась до комнатной температуры и перемешивалась в течение 3 ч. Затем смесь была разбавлена эфиром (200 мл), промыта 5% водным раствором HCl (2×25 мл), насыщенным водным раствором NaHCO3 (25 мл) и солевым раствором (20 мл), а также высушена над Na2SO4. После фильтрации и выпаривания неочищенный метил (S)-1-(4-бромфенил)этил(3-гидрокси-5-метил-3-фенилгекс-5-енил)карбамат использовался в ходе следующих этапов без дополнительной очистки.
Этап 4
Неочищенный метил (S)-1-(4-бромфенил)этил(3-гидрокси-5-метил-3-фенилгекс-5-енил)карбамат, полученный в результате описанной выше процедуры, был растворен в сухом тетрагидрофуране (75 мл), после чего в смесь медленно при комнатной температуре был добавлен NaH (60% в минеральном масле, 274 мг, 2 эквив.). Спустя 10 мин смесь была нагрета для обратного конденсирования в течение 2 ч. После проведения ЖХ-МС, результаты которой свидетельствовали о завершении реакции, смесь охлаждалась до 0°С. Реакция была остановлена при помощи насыщенного водного раствора NH4Cl (10 мл), после чего смесь была разбавлена эфиром (100 мл), промыта 1% водным раствором HCl (25 мл) и солевым раствором (15 мл), а также высушена над Na2SO4. После фильтрации и выпаривания неочищенный продукт был очищен посредством хроматографии с применением колонки, содержащей 40 г силикагеля и элюированной при помощи 10~35% раствора EtOAc в гексанах. Вещество, для которого был получен второй активный пик УФ, было собрано для получения (R)-3-((S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-фенил-1,3-оксазинан-2-она (490 мг, общий выход для 3 и 4 этапа: 34,5%).
Этап 5
Смесь (R)-3-((S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-фенил-1,3-оксазинан-2-она (490 мг, 1,18 ммоль), TsCN (257 мг, 1,2 эквив.), PhSiH3 (157 мкл, 1,07 эквив.), катализатора кобальт N,N'-бис(3,5-ди-трет-бутилсалицилиден)-1,1,2,2-тетраметилэтилендиамина, полученного в соответствии с процедурами, описанными в Способе получения 3 (7,5 мг, 0,01 эквив.), а также этанола (20 мл) перемешивалась в течение 4 ч при комнатной температуре. После проведения ЖХ-МС, результаты которой свидетельствовали об завершении реакции, смесь была выпарена и очищена при помощи системы для флэш-хроматографии ISCO (колонка емкостью 40 г, 25~80% раствор EtOAc в гексанах) для получения 267 мг продукта (выход: 51%). Метод ЖХ-МС 1 tR=1,89 мин, масса/заряд 441/443 (М+1).
Этап 6
В раствор 3-((R)-3-((S)-1-(4-бромфенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила (208 мг, 0,47 ммоль) в 1,4-доксане (5 мл) были добавлены 6-оксо-1,6-дигидропиридин-3-илборная кислота (98 мг, 1,5 эквив.), 2,0 М водный раствор Cs2CO3 (500 мкл), а также Pd(dppf)Cl2 (20 мг, 0,06 эквив.). Полученная смесь подверглась трехкратной дегазации и повторному насыщению газообразным N2, затем была нагрета до 90°С (масляная баня) в течение 3 ч. После проведения ЖХ-МС, результаты которой свидетельствовали об окончании реакции, смесь была охлаждена до комнатной температуры, разбавлена EtOAc (25 мл) и промыта водой (10 мл). Водный слой подвергся экстракции при помощи EtOAc (2×10 мл). Объединенный органический слой был промыт водой (10 мл) и солевым раствором (8 мл), а также высушен над Na2SO4. Полученный после фильтрации и выпаривания осадок был очищен посредством хроматографии (картридж, содержащий 12 г силикагеля, 0~10% раствор МеОН в CH2Cl2, основной УФ пик) для получения 2,2-диметил-3-((R)-2-оксо-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-6-ил)пропаннитрила (202 мг, 94%) в виде масла коричневого цвета. Метод ЖХ-МС 1 tR=1,34 мин, масса/заряд=456 (М+1); 1H ЯМР (CDCl3) 8,01 (d, 1Н), 7,80 (S, 1Н), 7,36 (dt, 6H), 7,19 (d, 2H), 6,98 (m, 3H), 5,65 (d, 1H), 2,98 (d, 1Н), 2,50 (m, 2H), 2,32 (m, 1Н), 2,17 (s, 2H), 1,57 (d, 3H), 1,40 (s, 3H), 1,32 (s, 3H).
Метод 2
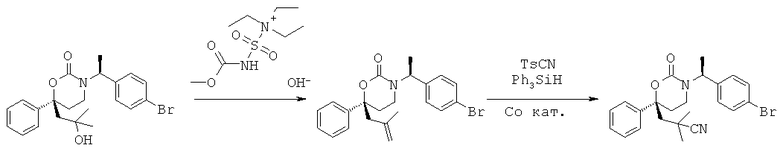
Этап 1. 3-[(S)-1-(4-Бром-фенил)-этил]-(S)-6-(2-метил-аллил)-6-фенил-[1,3]оксазинан-2-он
(Метоксикарбонилсульфамоил)триэтиламмония гидроксид (1,38 г) был добавлен в раствор 3-[(S)-1-(4-бром-фенил)-этил]-(5)-6-(2-гидрокси-2-метил-пропил)-6-фенил-[1,3]оксазинан-2-она (2,0 г) в тетрагидрофуране (30 мл) и толуоле (15 мл). Полученный раствор перемешивался при комнатной температуре в течение 0,5 ч, а также при температуре 75°С в течение 1 ч. После охлаждения при комнатной температуре раствор выпаривался, после к осадку был добавлен этилацетат. Полученная смесь была промыта водным раствором NaHCO3 и солевым раствором, а затем высушена (MgSO4). Титульное соединения было получено после удаления растворителя. Выход: 1,9 г (количественный). Масс-спектр (ионизация электрораспылением+): масса/заряд=414/416 (Br) [M+H]+
Этап 2. 3-{3-[(S)-1-(4-бром-фенил)-этил]-2-оксо-(S)-6-фенил-[1,3]оксазинан-6-ил}-2,2-диметил-пропионитрил
3-[(S)-1-(4-бром-фенил)-этил]-(S)-6-(2-метил-аллил)-6-фенил-[1,3]оксазинан-2-он (0,21 г), р-толуолсульфонил цианид (143 мг), трет-BuOOH (5,5 М раствор в декане, 27 мкл), а также фенилсилан (64 мкл) были добавлены в указанном порядке в колбу, оснащенную магнитной мешалкой и содержащую (1R,2R)-(-)-1,2-циклогександиамино-N,N'-бис(3,5-ди-трет-бутилсалицилиден)кобальт(II) (3 мг) и этанол (15 мл) в атмосфере аргона. Полученный раствор перемешивался при комнатной температуре в течение 3 ч и выпаривался в вакууме. Полученный осадок был очищен посредством хроматографии с применением силикагеля (смесь циклогексана/этилацетата 60:40->0:100) для получения титульного соединения в виде смолоподобного твердого вещества. Выход: 0,16 г (70% теоретического показателя). Масс-спектр (ионизация электрораспылением+): масса/заряд=441/443 (Br) [М+Н]+
ПРИМЕР 72
(S)-3-((S)-1-(4-(1-этил-6-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
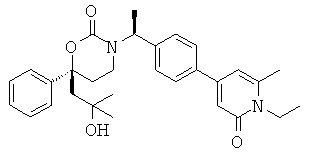
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 4-бром-1-этил-6-метилпиридин-2(1Н)-она в результате применения процедур, аналогичных описанным для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,211 мин, масса/заряд=489,2; 1H ЯМР (CDCl3) 1,10 (s, 3Н), 1,17 (s, 3H), 1,49 (s, 9H), 1,57 (d, 3Н), 2,22 (m, 4H), 2,37 (m, 1H), 2,84 (m, 1H), 5,60 (m, 1H), 5,91 (s, 1H), 7,06 (d, 2H), 7,27-7,40 (m, 5H), 7,68 (d, 1H), 7,24 (d, 2H), 8,09 (d, 1H), 8,90 (s, 1H).
4-бром-1-этил-6-метилпиридин-2(1Н)-он был получен путем алкилирования 4-бром-6-метилпиридин-2(1Н)-она с помощью этилйодида с применением К2СО3 в соответствии с процедурой, аналогичной описанной для 1 этапа Примера 59.
ПРИМЕР 73
(S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(1,5,6-триметил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-он
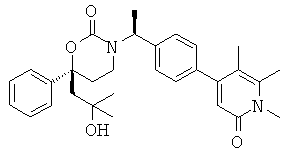
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она and 4-бром-1,5,6-триметилпиридин-2(1Н)-она в результате применения процедур, аналогичных описанным для 1 этапа Примера 6. Метод ЖХ-МС 2 tR=1,187 мин, масса/заряд=489,2; 1H ЯМР (CDCl3) 1,10 (s, 3H), 1,15 (s, 3Н), 1,32 (m, 3H),1,52 (m, 3Н), 1,72 (s, 1H), 2,18 (m, 3Н), 2,19 (m, 1H), 2,42 (m, 4H), 2,86 (m, 1H), 4,12 (m, 2H), 5,66 (m, 1H), 6,16 (s, 1H), 6,53 (s, 1H), 6,98 (m, 2H), 7,23-7,34 (m, 7H).
4-бром-1,5,6-триметилпиридин-2(1Н)-он был получен путем алкилирования 4-бром-5,6-диметилпиридин-2(1Н)-она с помощью метилйодида с применением K2СО3 в соответствии с процедурой, аналогичной описанной для 1 этапа Примера 59. 4-бром-5,6-диметилпиридин-2(1Н)-он был получен в результате применения процедуры, описанной в публикации McElroy, W.Т. and DeShong, P. Org. Lett. 2003, 5, 4779.
ПРИМЕР 74
3-((R)-3-((S)-1-(4-(1-изопропил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрил
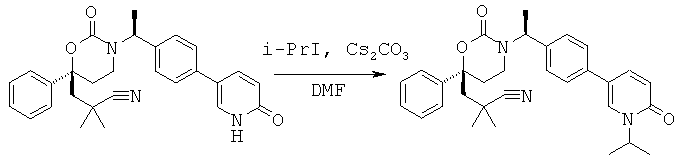
2,2-диметил-3-((R)-2-оксо-3-((S)-1-(4-(6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-6-ил)пропаннитрил (6 мг, 0,013 ммоль) был растворен в DMF (2,5 мл). В полученный раствор были добавлены Cs2CO3 (прибл. 15 мг, избыток) и i-PrI (100 мкл, избыток). Смесь перемешивалась в течение 3 ч при комнатной температуре. После проведения ЖХ-МС, результаты которой свидетельствовали об окончании реакции, смесь была очищена посредством препаративной ВЭЖХ для получения 3-((R)-3-((S)-1-(4-(1-изопропил-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-2-оксо-6-фенил-1,3-оксазинан-6-ил)-2,2-диметилпропаннитрила (1,99 мг, 30%). Метод ЖХ-МС 1 tR=2,03 мин, масса/заряд=498; 1H ЯМР (CDCl3) 8,35 (d, 1Н), 7,80 (dd, 1Н), 7,37 (m, 5H), 7,22 (d, 2H), 6,92 (d, 2H), 6,83 (d, 1Н), 5,66 (q, 1Н), 5,22 (m, 1Н), 2,93 (m, 1Н), 2,16 (s, 2H), 1,55 (d, 3H), 1,46 (s, 3H), 1,40 (d, 6H), 1,33 (s, 3H),
ПРИМЕР 75
3-{(S)-1-[4-(1-Циклопропил-2-оксо-1,2-дигидро-пиридин-4-ил)-фенил]-этил}-(S)-6-(2-гидрокси-2-метил-пропил)-6-фенил-[1,3]оксазинан-2-он
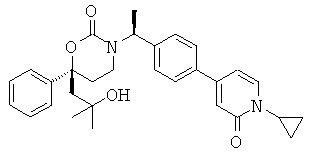
Метод 1
2 M водный раствор Na2CO3 (0,23 мл) был добавлен в раствор (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-[(S)-1-(4-(4,4,5,5- тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил]-1,3-оксазинан-2-она (0,11 г) и трифторметансульфоновой кислоты 1-циклопропил-2-оксо-1,2-дигидро-пиридин-4-ил эфира (74 мг; альтернативный вариант - применение 4-бром-1-циклопропил-1Н-пиридин-2-она) в диметилформамиде (3 мл). В полученную смесь в течение 15 мин распылялся аргон, после чего был добавлен комплекс [1,1'-бис(дифенилфосфино)-ферроцен]-дихлорпалладий(II) дихлорметана (10 мг). Смесь нагревалась до 100°С и перемешивалась при указанной температуре в течение ночи. После охлаждения до температуры окружающей среды в смесь была добавлена вода, после чего смесь подверглась экстракции этилацетатом. Объединенный органический экстракт был промыт солевым раствором, высушен (MgSO4) и выпарен. Полученный осадок был очищен посредством хроматографии с применением силикагеля (CH2Cl2/MeOH 99:1->90:10) для получения титульного соединения, подобного пенообразного твердого вещества, которое подверглось кристаллизации с помощью незначительного количества этилацетата. Выход: 30 мг (27% теоретического показателя); Масс-спектр (ионизация электрораспылением+): масса/заряд=487 [М+Н]+. Соединение (1,3 г) подверглось кристаллизации из 30 мл изопропилацетата. Горячий раствор одновременно перемешивался и медленно охлаждался до комнатной температуры в течение ночи в масляной бане для получения кристаллического моногидрата. Температура плавления 108-110°С.
Кристаллический моногидрат также получали рекристаллизацией 10.6 г соединения по Примеру 75 из 170 мл изопропилацетата, который сатурировали с водой. Горячий раствор одновременно перемешивался и медленно охлаждался до комнатной температуры, перемешивался в течение 2 часов при комнатной температуре и в течение 1 часа на ледяной бане. Твердое вещество отфильтровывали путем всасывания и сушили в течение ночи при температуре 50°С. Выход 10.2 г, темпертаура плавления 112-114°С.
Соединение, полученное в Примере 75 (2,0 г) также подверглось перекристаллизации из смеси 30 мл третбутилметилэфира и 15 мл изопропанола. Полученное твердое вещество подверглось фильтрации путем всасывания, было промыто третбутилметилэфиром и высушено при температуре 45°С, а затем при температуре 65°С в течение ночи. 100 мг полученного твердого вещества перемешивалось в 3 мг воды для образования смолистого вещества, которое затем превращалось в твердое вещество белого цвета. Данное вещество затем перемешивалось еще в течение часа, подвергалось фильтрации путем всасывания и высушиванию при комнатной температуре в течение ночи, а затем при температуре 65°С в течение 3 часов в сушилке с циркулирующим воздухом для получения кристаллического моногидрата. Температура плавления 102-108°С.
Промежуточное соединение XX
1-Циклопропил-4-(4-метокси-бензилокси)-1Н-пиридин-2-он
В емкость, подходящую для микроволновой печи и оснащенную магнитной мешалкой, а также содержащую 4-(4-метокси-бензилокси)-1Н-пиридин-2-она (0,60 г), циклопропилборную кислоту (0,45 г), пиридин (1,50 мл), триэтиламин (1,50 мл), а также толуол (4 мл), был распылен аргон в течение 5 мин. Затем был добавлен Cu(OAc)2 (0,94 г), после чего смесь перемешивалась в микроволновой печи при температуре 140°С в течение 45 мин. Затем было произведено выпаривание растворителя с последующим добавлением воды. Полученная смесь подверглась экстракции этилацетатом, после чего объединенный органический экстракт был промыт водой и водным раствором NaHCO3. После высушивания (MgSO4) и удаления растворителя осадок был очищен посредством хроматографии с применением силикагеля (CH2Cl2/MeOH 99:1->95:5) для получения титульных соединений в виде твердого вещества. Выход: 0,17 г (25% теоретического показателя); Масс-спектр (ионизация электрораспылением+): масса/заряд=272 [М+Н]+.
Промежуточное соединение XXI
1-Циклопропил-4-гидрокси-1Н-пиридин-2-он
Трифторуксусная кислота (1 мл) была добавлена в колбу, оснащенную магнитной мешалкой и содержащую 1-циклопропил-4-(4-метокси-бензилокси)-1Н-пиридин-2-он (0,17 г), после чего колба была охлаждена в бане со льдом/EtOH. Полученная смесь перемешивалась одновременно с охлаждением в течение 1,5 ч, а затем при температуре окружающей среды в течение 4,5 ч. Затем раствор подвергся выпариванию в вакууме, затем полученный осадок были измельчен в порошок вместе с терт-бутилметилэфиром и высушен для получения титульного соединения в виде твердого вещества. Выход: 0,10 г (количественный показатель). Масс-спектр (ионизация электрораспылением+): масса/заряд=152 [М+Н]+.
Промежуточное соединение XXII
Трифтор-метансульфоновой кислоты 1-циклопропил-2-оксо-1,2-дигидро-пиридин-4-ил эфир
Трифторметансульфоновый ангидрид (0,12 мл) был добавлен в колбу, оснащенную магнитной мешалкой и содержащую 1-циклопропил-4-гидрокси-1Н-пиридин-2-он (0,10 г), NEt3 (0,24 мл) и дихлорметан (8 мл), после чего колба была охлаждена в бане со льдом/EtOH. Полученная смесь перемешивалась одновременно с охлаждением в течение 2 ч, а затем при температуре окружающей среды в течение еще 2 ч. Затем раствор был разбавлен дихлорметаном и промыт сначала водой, затем водным раствором NaHCO3 и снова водой. Органический раствор был высушен (MgSO4), растворитель был удален, а осадок был очищен посредством хроматографии с применением силикагеля (дихлорметан/метанол 99:1->90:10) для получения титульного соединения в виде смолоподобного твердого вещества. Выход: 0,07 г (36% теоретического показателя). Масс-спектр (ионизация электрораспылением+): масса/заряд=284 [М+Н]+.
Промежуточное соединение XXIII
4-Бром-1-циклопропил-1Н-пиридин-2-он
Колба, оснащенная магнитной мешалкой и содержащая 4-бром-1 Н-пиридин-2-он (1,80 г), циклопропилборную кислоту (2,00 г), Cu(ОАс)2 (2,00 г), 2,2'-бипиридин (1,70 г), Na2CO3 (2,47 г), а также 1,2-дихлорэтан (75 мл), была нагрета до 70°С, затем смесь перемешивалась с доступом воздуха при указанной температуре в течение ночи. После добавления еще одной части циклопропилборной кислоты (0,50 г) и Na2CO3 (0,55 г) смесь перемешивалась в течение 4 ч при температуре обратного конденсирования. После охлаждения до температуры окружающей среды был добавлен водный раствор NH4Cl, после чего полученная смесь подверглась экстракции дихлорметаном. Объединенный органический экстракт был высушен (MgSO4), затем было произведено выпаривание растворителя. Полученный осадок был очищен посредством хроматографии с применением силикагеля (смесь циклогексана/этилацетата 50:50->35:65) для получения титульного соединения в виде масла, кристаллизирующегося при отстаивании. Выход: 0,82 г (37% теоретичесокго показателя); Масс-спектр (ионизация электрораспылением+): масса/заряд=214/216 (Br) [М+Н]+.
Метод 2
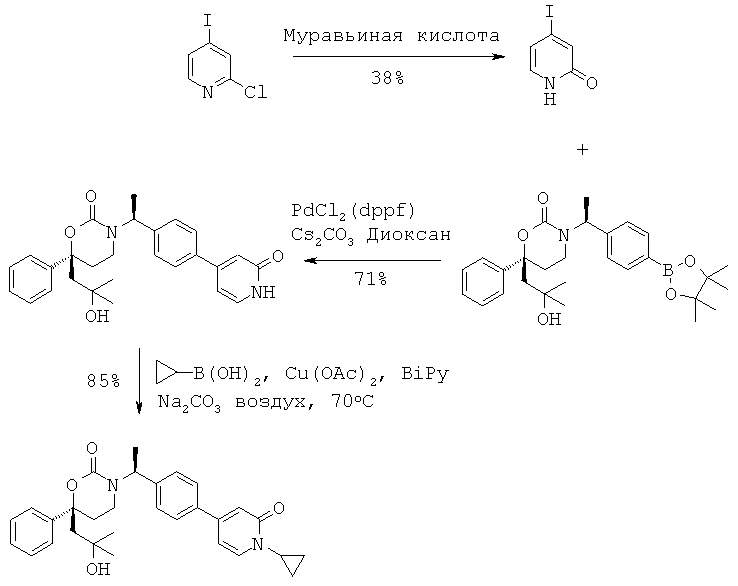
Этап 1. 4-йодпиридин-2(1H)-он
Смесь 2-хлор-4-йодпиридина (4,943 г, 20,6 ммоль) и муравьиной кислоты (88%, 10 мл) перемешивалась при температуре 105°С в течение 21 ч. Избыток муравьиной кислоты был удален в вакууме, реакция смеси была остановлена при помощи 2М водного раствора Na2CO3, после чего сама смесь подверглась экстракции при помощи CH2Cl2 и высушиванию над Na2SO4. После удаления растворителя в вакууме полученный осадок был очищен посредством хроматографии с применением силикагеля, элюированного при помощи CH2Cl2/МеОН, для получения 1,716 г (38%) 4-йодпиридин-2(1Н)-она в виде твердого вещества. Метод ЖХ-МС 1 tR=0,82 мин, масса/заряд=222 (MH+); 1H ЯМР (400 МГц, (CD3)2SO) 7,14 (d, J=6,5 Гц, 1Н), 6,87 (s, 1H), 6,49 (d, J=7,0 Гц, 1Н).
Этап 2. (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
В раствор (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (2,646 г, 5,52 ммоль) в 1,4-доксане (60 мл) были добавлены 4-йодпиридин-2(1Н)-он (1,200 г, 5,43 ммоль), 2 М Cs2CO3 (14,5 мл) и PdCl2(dppf)·CH2Cl2 (0,230 г, 0,28 ммоль). Смесь подверглась дегазации и нагреванию в атмосфере азота при температуре 120°С в течение 15 ч. Смесь была разбавлена CH2Cl2 и высушена над Na2SO4. После выпаривания растворителей осадок был очищен посредством хроматографии с применением силикагеля, элюированного при помощи MeOH/CH2Cl2 для получения 1,717 г (71%) (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она. Метод ЖХ-МС 1 tR=1,23 мин, масса/заряд 389, 447 (MH+); 1H ЯМР (400 МГц, CD3OD) 7,40 (d, J=6,7 Гц, 1Н), 7,31 (d, J=8,2 Гц, 2Н), 7,29-7,20 (m, 5H), 6,96 (d, J=8,2 Гц, 2Н), 6,57-6,52 (m, 2Н), 5,49 (q, J=7,0 Гц, 1Н), 2,98-2,93 (m, 1Н), 2,47-2,34 (m, 2Н), 2,16-2,09 (m, 1Н), 2,07 (s, 2Н), 1,45 (d, J=7,0 Гц, 3Н), 1,19 (s, 3Н), 0,87 (s, 3Н).
Этап 3. (S)-3-((S)-1-(4-(1-циклопропил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он. Смесь (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (1,683 г, 3,77 ммоль, 1,0 эквив.), Cu(ОАс)2 (0,692 г, 3,81 ммоль, 1,01 эквив.), бипиридина (0,599 г, 3,83 ммоль, 1,02 эквив.), циклопропилборной кислоты (0,681 г, 7,93 ммоль, 2,10 эквив.) и Na2CO3 (0,890 г, 8,40 ммоль, 2,23 эквив.) в дихлорэтане (40 мл) перемешивалась в атмосфере воздуха при температуре 70°С в течение 22 ч. Реакция была остановлена при помощи насыщенного водного раствора NH4Cl, после чего смесь была разбавлена CH2Cl2 и высушена над Na2SO4. После удаления растворителя в вакууме осадок был очищен посредством хроматографии с применением силикагеля, элюированного с помощью MeOH/CH2Cl2 для получения 1,560 г (85%) (S)-3-((S)-1-(4-(1-циклопропил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она. ЖХ-МС tR=1,41 мин, 3 мин хроматография, масса/заряд 429, 487 (МН+); 1H ЯМР (400 МГц, CD3OD) 7,52 (d, J=7,0 Гц, 1Н), 7,29-7,18 (m, 7H), 6,92 (d, J=8,2 Гц, 2Н), 6,54 (d, J=1,8 Гц, 1Н), 6,47 (dd, J=7,3, 1,8 Гц, 1Н), 5,47 (q, J=7,0 Гц, 1Н), 3,27-3,21 (m, 1Н), 2,95-2,91 (m, 1Н), 2,48-2,33 (m, 2Н), 2,15-2,08 (m, 1Н), 2,07 (s, 2Н), 1,42 (d, J=7,0 Гц, 3Н), 1,20 (s, 3Н), 1,05-1,00 (m, 2Н), 0,87 (s, 3Н), 0,83-0,79 (m, 2H); 13С ЯМР (100 МГц, CD3OD) 166,17, 155,63, 152,88, 144,03, 142,27, 138,90, 136,91, 129,71, 128,70, 128,58, 127,67, 126,09, 116,08, 107,10, 85,19, 71,49, 55,13, 54,62, 37,44, 33,24, 32,71, 31,86, 30,03, 15,60, 7,27. (S)-3-((S)-1-(4-(1-циклопропил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он (прибл. 1,5 г) и изопропилацетат (30 мл) нагревались в масляной бане при температуре 120°С до образования однородного раствора. Нагревание было прекращено, после чего полученный раствор в течение ночи медленно перемешивался для медленного охлаждения до комнатной температуры в масляной бане. Полученные твердые вещества подверглись фильтрации и промыванию изопропилацетатом, после чего были высушены при комнатной температуре в высоком вакууме для получения кристаллического твердого вещества с температурой плавления 91-94°С.
ПРИМЕР 76
3-{(S)-1-[4-(1-циклопропилметил-6-оксо-1,6-дигидро-пиридин-3-ил)-фенил]-этил}-(S)-6-(2-гидрокси-2-метил-пропил)-6-фенил-[1,3]оксазинан-2-он
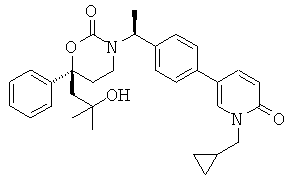
2 М водный раствор Na2CO3 (0,84 мл) был добавлен в раствор (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-[(S)-1-(4-(4,4,5,5- тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил]-1,3- оксазинан-2-она (0,40 г) и 5-бром-1-цикпопропилметил-1Н-пиридин-2-она (0,24 г) в диметилформамиде (4 мл). В полученную смесь в течение 10 мин распылялся аргон перед добавлением комплекса [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II) дихлорметана (34 мг). Смесь нагревалась до 100°С и перемешивалась при указанной температуре в течение 4 ч. После охлаждения до температуры окружающей среды в смесь была добавлена вода, после чего была проведена экстракция полученной смеси этилацетатом. Объединенный органический экстракт был промыт солевым раствором, высушен (MgSO4) и выпарен. Полученный осадок был очищен посредством хроматографии с применением силикагеля (смесь дихлорметана/метанола 99:1->95:5) для получения титульного соединения, которое подверглось кристаллизации при помощи незначительного количества этилацетата. Выход: 0,19 г (46% теоретического показателя); Масс-спектр (ионизация электрораспылением+): масса/заряд=501 [М+Н]+.
Промежуточное соединение XXIV
5-Бром-1-циклопропилметил-1Н-пиридин-2-он
KOtBu (0,68 г) был добавлен в раствор 5-бром-1Н-пиридин-2-она (1,00 г) в тетрагидрофуране (20 мл) при комнатной температуре. После перемешивания в течение 30 мин в суспензию были добавлены циклопропилметилбромид (0,77 мл) и диметилформамид (3 мл), полученная смесь нагревалась до 70°С. После перемешивания смеси при температуре 70°С в течение 2 ч реакция была завершена. Смесь была охлаждена до комнатной температуры, разбавлена этилацетатом (50 мл), а также промыта водой (2×20 мл) и солевым раствором (20 мл). Затем раствор был высушен (MgSO4) с последующим удалением растворителя для получения титульного соединения в виде бесцветного масла. Выход: 1,18 г (90% теоретического показателя). Масс-спектр (ионизация электрораспылением+): масса/заряд=228/230 (Br) [М+Н]+
ПРИМЕР 77
(R)-6-Метоксиметил-3-{(S)-1-[4-(1-метил-6-оксо-1,6-дигидро-пиридин-3-ил)-фенил]-этил}-6-фенил-[1,3]оксазинан-2-он
Титульное соединение было получено из (R)-6-(метоксиметил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 5-бром-1-метилпиридин-2(1Н)-она в результате применения процедур, аналогичных описанным в Примере 76. Масс-спектр (ионизация электрораспылением+): масса/заряд=433 [M+H]+
ПРИМЕР 78
(R)-6-метоксиметил-3-{(S)-1-[4-(1-метил-2-оксо-1,2-дигидро-пиридин-4-ил)-фенил]-этил}-6-фенил-[1,3]оксазинан-2-он
Титульное соединение было получено из (R)-6-(метоксиметил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и трифтор-метансульфоновой кислоты 1-метил-2-оксо-1,2-дигидро-пиридин-4-ил эфира в результате применения процедур, аналогичных описанным в Примере 76. Масс-спектр (ионизация электрораспылением+): масса/заряд=433 [М+Н]+.
ПРИМЕР 79
3-{(S)-1-[4-(5-фтор-1-метил-2-оксо-1,2-дигидро-пиридин-4-ил)-фенил]-этил}-(S)-6-(2-гидрокси-2-метил-пропил)-6-фенил-[1,3]оксазинан-2-он
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-[(S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2- ил)фенил)этил]-1,3-оксазинан-2-она и 4-бром-5-фтор-1-метил-1Н-пиридин-2-она в результате применения процедур, аналогичных описанным в Примере 76. Масс-спектр (ионизация электрораспылением+): масса/заряд=479 [M+H]+. Полученное соединение имело форму масла и кристаллизировалось при отстаивании. Твердое вещество было высушено в вакууме при температуре 80°С. Температура плавления: 120-125°С с выделением газа и последующей кристаллизацией с температурой плавления 183-184°С.
Промежуточное соединение XXV
4-Бром-5-фтор-1-метил-1Н-пиридин-2-он
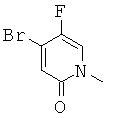
Метилйодид (0,9 мл) при комнатной температуре был добавлен в смесь карбоната калия (2,34 г) и 4-бром-5-фтор-1Н-пиридин-2-она (2,50 г) в диметилформамиде (25 мл). Полученная смесь перемешивалась при комнатной температуре в течение ночи, после чего в нее была добавлена вода. Полученная смесь подверглась экстракции этилацетатом, объединенные экстракты были промыты солевым раствором и высушены (MgSO4). Затем было произведено выпаривание растворителей для получения неочищенного титульного соединения, которое подверглось перекристаллизации из Et2O, Выход: 1,22 г (45% теоретического показателя); Масс-спектр (ионизация электрораспылением+): масса/заряд=206/208 (Br) [М+Н]+.
ПРИМЕР 80
(S)-6-(2-Гидрокси-2-метил-пропил)-3-((S)-1-{4-[1-(2-гидрокси-2-метил-пропил)-2-оксо-1,2-дигидро-пиридин-4-ил]-фенил}-этил)-6-фенил-[1,3]оксазинан-2-он
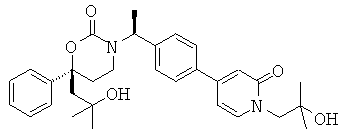
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-[(S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2- ил)фенил)этил]-1,3-оксазинан-2-она и 4-бром-1-(2-гидрокси-2-метил-пропил)-1Н-пиридин-2-она в результате применения процедур, аналогичных описанным в Примере 75. Масс-спектр (ионизация электрораспылением+): масса/заряд=519 [М+Н]+.
Промежуточное соединение XXVI
4-Бром-1-(2-гидрокси-2-метил-пропил)-1Н-пиридин-2-он
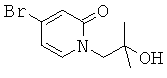
Смесь 4-бром-1Н-пиридин-2-она (0,25 г), 2,2-диметил-оксирана (0,26 мл) и карбоната калия (0,40 г) в диметилформамиде (2,5 мл) перемешивалась под воздействием микроволн при температуре 120°С в течение 30 мин. После охлаждения до температуры окружающей среды смесь была выпарена и очищена посредством ВЭЖХ с обращенными фазами (смесь ацетонитрила/воды) для получения титульного соединения. Выход: 0,34 г (96% теоретического показателя); Масс-спектр (ионизация электрораспылением+): масса/заряд=246/248 (Br) [M+H]+.
ПРИМЕР 81
(S)-6-(2-Гидрокси-2-метил-пропил)-3-((S)-1-{4-[1-(3-метокси-2-метил-пропил)-2-оксо-1,2-дигидро-пиридин-4-ил]-фенил}-этил)-6-фенил-[1,3]оксазинан-2-он
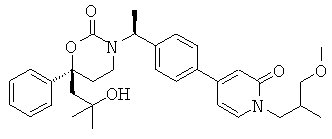
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-[(S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил]-1,3-оксазинан-2-она и 4-бром-1-(3-метокси-2-метил-пропил)-1Н-пиридин-2-она в результате применения процедур, аналогичных описанным в 1 методе Примера 75. Масс-спектр (ионизация электрораспылением+): масса/заряд=533 [М+Н]+.
Промежуточное соединение XXVII
3-(4-бром-2-оксо-2Н-пиридин-1-ил)-2-метил-пропионовой кислоты метилэфир
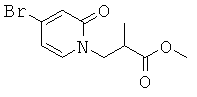
Смесь 4-бром-1Н-пиридин-2-она (0,50 г), метил 2-бромизобутирата (0,45 мл) и карбоната калия (0,68 г) в диметилформамиде (5 мл) перемешивалась при температуре 60°С в течение 3 ч. После охлаждения до температуры окружающей среды в смесь была добавлена вода, затем полученная смесь подверглась экстракции этилацетатом. Объединенный экстракт был промыт солевым раствором, высушен (MgSO4) и выпарен. Полученный осадок был очищен посредством хроматографии с применением силикагеля (смесь циклогексана/этилацетата 70:30->50:50) для получения титульного соединения. Выход: 0,53 г (67% теоретического показателя); Масс-спектр (ионизация электрораспылением+): масса/заряд=274/276 (Br) [M+H]+. Дополнительно был получен 2-(4-бром-пиридин-2-илокси)-2-метил-пропионовой кислоты метилэфир {Выход: 0,15 г; масс-спектр (ионизация электрораспылением+): масса/заряд=274/276 (Br) [М+Н]+}
Промежуточное соединение XXVIII
4-Бром-1-(3-гидрокси-2-метил-пропил)-1Н-пиридин-2-он
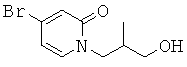
LiAlH4 (1 М раствор в тетрагидрофуране, 1,16 мл) был добавлен в раствор 3-(4-бром-2-оксо-2Н-пиридин-1-ил)-2-метил-пропионовой кислоты метилэфира (0,53 г) в тетрагидрофуране (6 мл), охлажденный в ледяной бане. После перемешивания и одновременного охлаждения раствора в течение 2 ч была добавлена еще одна часть LiAlH4 (1 М в тетрагидрофуране, 0,29 мл). Затем после перемешивания и одновременного охлаждения в течение еще 1 часа реакция была прекращена путем добавления воды. Полученная смесь подверглась экстракции этилацетатом, после чего объединенный органический экстракт был промыт солевым раствором и высушен (MgSO4). Растворитель был удален для получения титульного соединения. Выход: 0,37 г (78% теоретического показателя); Масс-спектр (ионизация электрораспылением+): масса/заряд=246/248 (Br) [М+Н]+.
Промежуточное соединение XXIX
4-бром-1-(3-метокси-2-метил-пропил)-1Н-пиридин-2-он
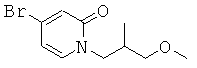
NaH (60% в минеральном масле, 57 мг) был добавлен в раствор 4-бром-1-(3-гидрокси-2-метил-пропил)-1Н-пиридин-2-она (0,53 г) в диметилформамиде (6 мл), охлажденный в ледяной бане. После перемешивания и одновременного охлаждения раствора в течение 0,5 ч в него был добавлен метилйодид (110 мкл). После удаления охлаждающей бани раствор перемешивался при комнатной температуре в течение ночи. Затем раствор выпаривался в вакууме, после чего полученный осадок был разбавлен водой. Полученная смесь подверглась экстракции этилацетатом, после чего объединенный органический экстракт был промыт солевым раствором и высушен (MgSO4). Растворитель был выпарен, а полученный осадок был очищен посредством ВЭЖХ с обращенными фазами (смесь ацетонитрила/воды) для получения титульного соединения в виде масла. Выход: 70 мг (30% теоретического показателя); Масс-спектр (ионизация электрораспылением+): масса/заряд=260/262 (Br) [М+Н]+.
ПРИМЕР 82
(S)-6-(2-гидрокси-2-метил-пропил)-3-((S)-1-{4-[1-(3-гидрокси-2-метил-пропил)-2-оксо-1,2-дигидро-пиридин-4-ил]-фенил}-этил)-6-фенил-[1,3]оксазинан-2-он
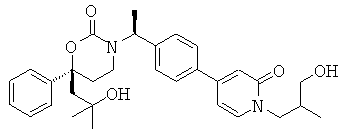
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-[(S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2- ил)фенил)этил]-1,3-оксазинан-2-она и 4-бром-1-(3-гидрокси-2-метил-пропил)-1Н-пиридин-2-она в результате применения процедуры, аналогичной описанной в Примере 75. Масс-спектр (ионизация электрораспылением+): масса/заряд=519 [M+H]+.
ПРИМЕР 83
(S)-6-(2-гидрокси-2-метил-пропил)-3-(1-{4-[1-(2-метокси-2-метил-пропил)-2-оксо-1,2-дигидро-пиридин-4-ил]-фенил}-этил)-6-фенил-[1,3]оксазинан-2-он
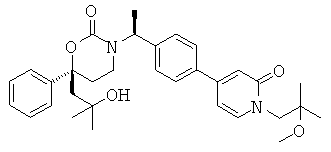
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-[(S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил]-1,3-оксазинан-2-она и 4-бром-1-(2-метокси-2-метил-пропил)-1Н-пиридин-2-она в результате применения процедуры, аналогичной описанной в методе 1 Примера 75. Масс-спектр (ионизация электрораспылением+): масса/заряд=533 [М+Н]+.
Промежуточное соединение XXX
4-Бром-1-(2-метокси-2-метил-пропил)-1Н-пиридин-2-он
Титульное соединение было получено из 4-бром-1-(2-гидрокси-2-метил-пропил)-1Н-пиридин-2-она и метилйодида в результате применения процедуры, аналогичной описанной для Промежуточного соединения XXIX. Масс-спектр (ионизация электрораспылением+): масса/заряд=260/262 (Br) [М+Н]+.
ПРИМЕР 84
6-(3-гидрокси-3-метилбутил)-6-изопропил-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-он
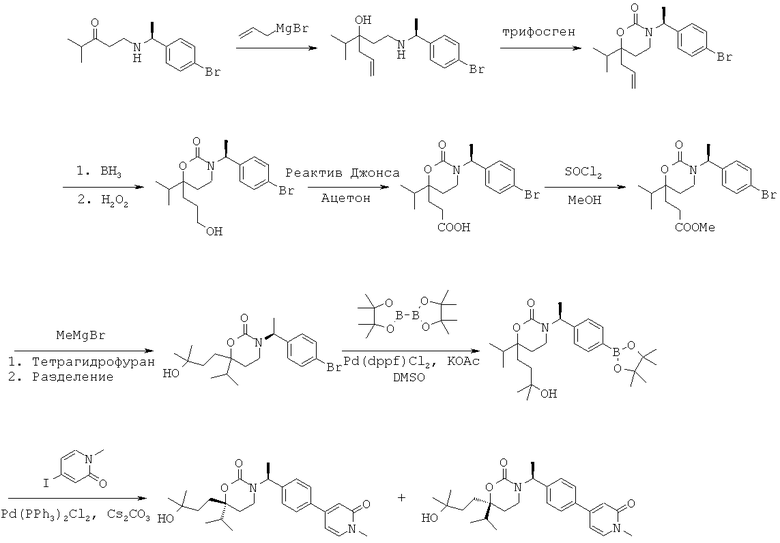
Этап 1
В раствор (S)-1-(1-(4-бромфенил)этиламино)-4-метилпентан-3-она (740 мг, 2,5 ммоль) в тетрагидрофуране (25 мл) был по капле добавлен аллилмагний бромид (25 мл, 25 ммоль) в атмосфере N2 при температуре -78°С. Реакционная смесь перемешивалась при температуре -78°С в течение 2 ч, после чего реакция была остановлена путем добавления насыщенного водного раствора NH4Cl, а смесь подверглась экстракции при помощи EtOAc. Органический слой был промыт солевым раствором, высушен над Na2SO4 и подвергся фильтрации. После удаления растворителя в вакууме был получен 1-((S)-1-(4-бромфенил)этиламино)-3-изопропилгекс-5-ен-3-ол (802 мг, выход: 95%), который использовался в ходе следующего этапа без дополнительной обработки.
Этап 2
В раствор 1-((S)-1-(4-бромфенил)этиламино)-3-изопропилгекс-5-ен-3-ола (802 мг, 2,366 ммоль) и триэтиламина (139 мг, 2,366 ммоль) в CH2Cl2 (20 мл) при температуре 0°С в атмосфере N2 был добавлен трифосген (348 мг, 1,18 ммоль), после чего полученная смесь перемешивалась в течение ночи. После остановки реакции при помощи воды смесь подверглась экстракции с помощью CH2Cl2 Органический слой подвергся промыванию, высушиванию над Na2SO4, фильтрации, выпариванию и очистке посредством колоночной хроматографии для получения 6-аллил-3-((S)-1-(4-бромфенил)этил)-6-изопропил-1,3-оксазинан-2-она (480 мг, выход: 56%).
Этап 3
В раствор 6-аллил-3-((S)-1-(4-бромфенил)этил)-6-изопропил-1,3-оксазинан-2-она (480 мг, 1,315 ммоль) в тетрагидрофуране (5 мл) при температуре 0°С в атмосфере N2 был добавлен ВН3 тетрагидрофуран (5,3 мл, 5,3 ммоль). Реакционная смесь перемешивалась в течение 2 ч, после чего реакция была остановлена при помощи воды, 3 М водного раствора NaOH (1 мл) и H2O2 (5 мл). Полученная смесь перемешивалась в течение 2 ч, затем подверглась экстракции при помощи EtOAc, промыванию солевым раствором, высушиванию над Na2SO4, фильтрации и выпариванию для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ для получения 3-((S)-1-(4-бромфенил)этил)-6-(3-гидроксипропил)-6-изопропил-1,3-оксазинан-2-она. (110 мг, выход: 22%). 1H ЯМР (CDCl3): δ 0,88 (m, 6Н), 1,45 (m, ЗН), 1,60 (m, 4Н), 1,71 (m, 1Н), 1,82 (m, 1Н), 1,99 (m, 1Н), 2,63 (m, 1Н), 3,03 (m, 1Н), 3,59 (m, 2Н), 5,68 (m, 1Н), 7,13 (d, 2Н), 7,40 (d, 2Н),
Этап 4
В смесь 3-((S)-1-(4-бромфенил)этил)-6-(3-гидроксипропил)-6-изопропил-1,3-оксазинан-2-она (41 мг, 0,1 ммоль) и ацетона (10 мл) при температуре 0°С был добавлен реактив Джонса (2,5 М, 1 мл). Полученная смесь перемешивалась при комнатной температуре в течение 1 ч, выпаривалась и подвергалась экстракции при помощи EtOAc. Органический слой был выпарен для получения неочищенного продукта 3-(3-((S)-1-(4-бромсренил)этил)-6-изопропил-2-оксо-1,3-оксазинан-6-ил)пропановой кислоты (51 мг, выход: 95%), который использовался в ходе следующего этапа без дополнительной очистки.
Этап 5
В раствор 3-(3-((S)-1-(4-бромфенил)этил)-6-изопропил-2-оксо-1,3-оксазинан-6-ил)пропановой кислоты (41 мг, 0,1 ммоль) в МеОН (10 мл) при температуре 0°С был добавлен SOCl (5 мл). Реакционная смесь перемешивалась при комнатной температуре в течение 2 ч, выпаривалась и очищалась посредством препаративной ТСХ для получения метил 3-(3-((S)-1-(4-бромфенил)этил)-6-изопропил-2-оксо-1,3-оксазинан-6-ил)пропаноата (42 мг, выход: 96%).
Этап 6
В раствор метил 3-(3-((S)-1-(4-бромфенил)этил)-6-изопропил-2-оксо-1,3-оксазинан-6-ил)пропаноата (42 мг, 0,1 ммоль) в сухом тетрагидрофуране (5 мл) при температуре -78°С был добавлен MeMrBr (2,5 мл, 2,5 ммоль, 1 М раствор в тетрагидрофуране). Смесь перемешивалась при комнатной температуре в течение 0,5 ч. После остановки реакции при помощи насыщенного водного раствора NH4Cl смесь подвергалась экстрации EtOAc. Органический слой был выпарен для получения неочищенного 3-((S)-1-(4-бромфенил)этил)-6-(3-гидрокси-3-метилбутил)-6-изопропил-1,3-оксазинан-2-она.
Посредством препаративной ВЭЖХ могут быть выделены два изомера:
Изомер 1: (1,1 мг, выход: 12%), 1H ЯМР (CDCl3): δ 0,91 (m, 6H), 1,25 (m, 6H), 1,44 (d, ЗН), 1,70 (m, 4H), 1,85 (m, 2H), 2,01 (m, 1H), 2,74 (m, 1H), 3,18 (m, 1H), 5,79 (m, 1H), 7,24 (d, 2H), 7,50 (d, 2H),
Изомер 2: (0,9 мг, выход: 10%), 1H ЯМР (CDCl3): δ 0,89 (m, 6H), 1,15 (s, 6H), 1,45 (m, 5H), 1,55 (m, 3Н), 1,85 (m, 1H), 1,99 (m, 1H), 2,64 (m, 1H), 2,99 (m, 1H), 5,72 (m, 1H), 7,17 (d, 2H), 7,40 (d, 2H),
Этап 7
В раствор соединения 3-((S)-1-(4-бромфенил)этил)-6-(3-гидрокси-3-метилбутил)-6-изопропил-1,3-оксазинан-2-она (105 мг, 0,255 ммоль) в DMSO (8 мл) были добавлены соединение 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (198,5 мг, 0,781 ммоль), КОАс (351,6 мг, 3,587 ммоль) и Pd(dppf)Cl2 (21,9 мг, 0,027 ммоль) в атмосфере N2. Реакционная смесь перемешивалась при температуре 90°С в течение 3,5 ч с последующим добавлением Н2О и экстракцией этилацетатом. Органический слой был промыт водой и солевым раствором, высушен над Na2SO4, выпарен и очищен посредством препаративной ТСХ для получения двух изомеров 6-(3-гидрокси-3-метилбутил)-6-изопропил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она
Изомер 1 (17 мг, 15%).
Изомер 2 (10,3 мг, 9%).
Этап 8
В раствор соединения 4-йод-1-метилпиридин-2(1Н)-она (17 мг, 0,074 ммоль) в DME (4,6 мл) в атмосфере азота был добавлен Pd(PPh3)4 (6,7 мг, 0,007 ммоль). Смесь перемешивалась при комнатной температуре в течение 1 ч, после чего в нее был добавлен раствор соединения изомера 1 6-(3-гидрокси-3-метилбутил)-6-изопропил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (17 мг, 0,037 ммоль) в EtOH (2 мл) и насыщенный водный раствор NaHCO3 (1,5 мл). Смесь перемешивалась при температуре 100°С в течение 2 ч с последующей остановкой реакции при помощи воды и экстракцией при помощи EtOAc. Объединенный органический слой был высушен над безводным Na2SO4 и выпарен для получения соединения изомера 1 6-(3-гидрокси-3-метилбутил)-6-изопропил-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-она (10,73 мг, 65,8%). Метод ЖХ-МС 2 метод 2 tR=1,03 мин, масса/заряд=463, 441; 1H ЯМР (CD3OD): δ 0,89 (m, 6H), 1,11 (s, 6H), 1,42 (m, 2H), 1,51(m, 3Н), 1,60 (m, 2H), 1,82-2,02 (m, 2H), 2,69 (m, 1H), 3,03 (m, 1H), 3,51 (s, 3Н), 5,79 (m, 3Н), 6,35 (d, 1H), 6,72 (s, 1H), 7,28 (d, 1H), 7,39 (d, 2H), 7,49 (m, 2H).
Изомер 2 6-(3-гидрокси-3-метилбутил)-6-изопропил-3-((S)-1-(4-(1-метил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-1,3-оксазинан-2-она был получен из изомера 6-(3-гидрокси-3-метилбутил)-6-изопропил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она в результате применения процедуры, аналогичной описанной выше для 8 этапа. Метод ЖХ-МС 2 Метод 2 tR=1,00 мин, масса/заряд=463, 441; 1H ЯМР (CD3OD): δ 0,89 (m, 6H), 1,18 (m, 6H), 1,43 (m, 1H), 1,51 (m, 3H), 1,63 (m, 2H), 1,76 (m, 2H), 1,92 (m, 1H), 2,61 (m, 1H), 3,12 (m, 1H), 3,51 (s, 3Н), 5,79 (m, 1H), 6,37 (d, 1H), 6,72 (s, 1H), 7,28 (d, 1H), 7,35 (d, 2H), 7,51 (m, 2H).
ПРИМЕР 85
(S)-3-((S)-1-(4-(1 -циклопропил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-изопропил-1,3-оксазинан-2-он
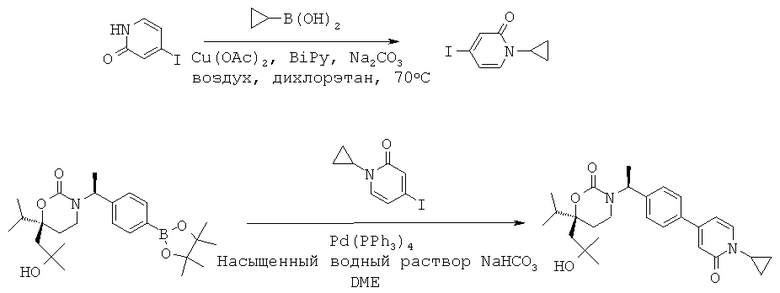
Этап 1
Смесь 4-йодпиридин-2(1Н)-она (0,2425 г, 1,10 ммоль, 1,0 эквив.), Cu(OAc)2 (0,2146 г, 1,18 ммоль, 1,07 эквив.), бипиридина (0,1832 г, 1,17 ммоль, 1,07 эквив.), циклопропилборной кислоты (0,2122 г, 2,47 ммоль, 2,25 эквив.) и Na2CO3 (0,2638 г, 2,49 ммоль, 2,27 эквив.) в дихлорэтане (10 мл) перемешивалась при температуре 70°С в течение 18 ч. Реакция была остановлена при помощи насыщенного водного раствора NH4Cl, после чего смесь была разбавлена CH2Cl2 и высушена над Na2SO4. После удаления растворителя в вакууме полученный осадок был очищен посредством хроматографии с применением силикагеля, элюированного при помощи смеси гексанов/этилацетата, для получения 0,2309 г (81%) 1-циклопропил-4-йодпиридин-2(1Н)-она.
Этап 2
В раствор соединения 1-циклопропил-4-йодпиридин-2(1H)-она (17,60 мг, 0,067 ммоль) в DME (2,5 мл) в атмосфере азота был добавлен Pd(PPh3)4 (6,12 мг, 0,006 ммоль). Смесь перемешивалась при комнатной температуре в течение 1 ч, после чего в нее были добавлены раствор соединения (S)-6-(2-гидрокси-2-метилпропил)-6-изопропил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (15 мг, 0,034 ммоль) в EtOH (1 мл) и насыщенного водного раствора NaHCO3 (1 мл). Полученная смесь перемешивалась при температуре 100°С в течение 2 ч, после чего реакция была остановлена с помощью EtOAc. Объединенный органический слой был высушен над безводным Na2SO4 и выпарен для получения неочищенного готового продукта, который затем был очищен посредством препаративной ВЭЖХ для получения соединения (S)-3-((S)-1-(4-(1 -циклопропил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-изопропил-1,3-оксазинан-2-она (6,50 мг, 43%). Метод ЖХ-МС 2 tR=1,00 мин, масса/заряд=453; 1H ЯМР (CD3OD): δ 0,82 (d, 3Н), 0,89 (m, 2H), 0,99 (d, 3H), 1,17 (m, 2H), 1,35 (m, 6H), 1,58 (d, 3Н), 1,62 (m, 2H), 1,85 (m, 1H), 1,96 (d, 1H), 2,09-2,18 (m, 2H), 2,68-2,78 (m, 1H), 3,11 (m, 1H), 3,37 (m, 1H), 5,81 (m, 1H), 6,40 (d, 2H), 6,78 (s, 1H), 7,31-7,42 (m, 3Н), 7,58 (d, 2H).
ПРИМЕР 86
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-(2-гидроксиэтил)-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
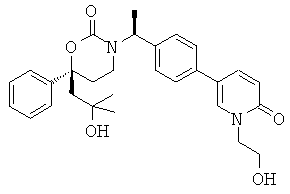
Титульное соединение было получено из (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 5-бром-1-(2-гидроксиэтил)пиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 2 этапа Примера 20. Метод ЖХ-МС 2 tR=1,08 мин, масса/заряд=513, 491; 1H ЯМР (CD3OD) δ 0,95 (s, 3Н), 1,24 (s, 3Н), 1,26 (s, 1H), 1,52 (d, 3Н), 2,12 (s, 2H), 2,18 (m, 1H), 2,40-2,53 (m, 2H), 3,02 (m, 1H), 3,52 (m, 0,5H), 3,64 (m, 0,5H), 3,83 (t, 1H), 4,15 (t, 1H), 5,53 (m, 1H), 6,61 (m, 1H), 7,01 (d, 2H), 7,25-7,40 (m, 7H), 7,79 (m, 2H).
5-бром-1-(2-гидроксиэтил)пиридин-2(1Н)-он был получен из 5-бромпиридин-2(1Н)-она и 2-йодэтанола в результате применения процедуры, аналогичной описанной для 1 этапа Примера 20.
ПРИМЕР 87
(S)-3-((S)-1-(4-(1-(2-фторэтил)-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-он
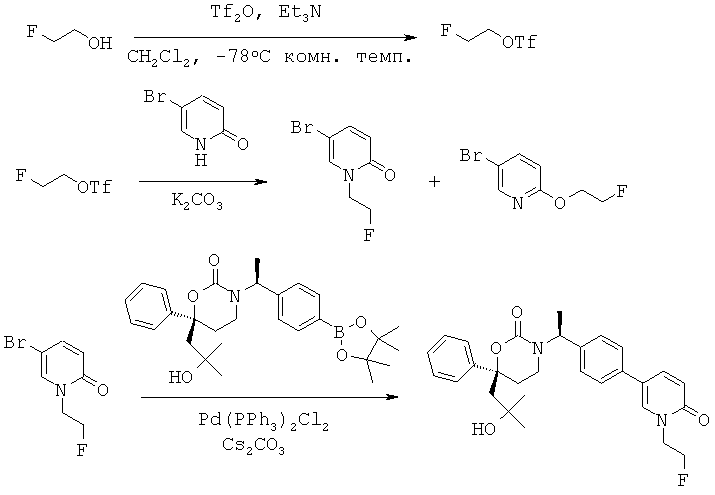
Этап 1
В раствор 2-фторэтанола (3,2 г, 50 ммоль) и триэтиламина (5,5 г, 55 ммоль) в дихлорметане (60 мл) по капле при температуре -78°С в атмосфере N2 был добавлен (CF3SO2)2О (15,5 г, 55 ммоль). Смесь перемешивалась при температуре 10~20°С в течение 1 ч и обрабатывалась водой (100 мл). Затем органический слой был промыт насыщенным водным раствором NaHCO3 (100 мл) и солевым раствором (100 мл), высушен и выпарен для получения трифторметансульфоната (8 г, выход 82%).
Этап 2
Раствор 5-бромпиридин-2(1Н)-она (100 мг, 0,58 ммоль), 2-фторэтил трифторметансульфоната (1,1 г, 5,8 ммоль) и К2СО3 (800 мг, 5,8 ммоль) в DMF (3 мл) перемешивался при комнатной температуре в течение ночи. После добавления 2-Фторэтил трифторметансульфоната (1,1 г, 5,8 ммоль) и К2СО3 (800 мг, 5,8 ммоль), смесь была обработана этилацетатом (20 мл) и водой (20 мл). Органический слой был промыт водой (2×20 мл) и солевым раствором (20 мл), высушен над Na2SO4, выпарен и очищен посредством препаративной ТСХ (1:1 смесь петролейного эфира/EtOAc) для получения двух изомеров.
5-бром-1-(2-фторэтил)пиридин-2(1Н)-он (30 мг, выход: 24%). 1Н ЯМР (CD3OD): δ 4,25 (t, 1Н), 4,32 (t, 1H), 4,62 (t, 1H), 4,74 (t, 1H), 6,52 (d, 1H), 7,61 (dd, 1H), 7,85 (s, 1H).
5-бром-2-(2-фторэтокси) пиридин (30 мг, выход: 24%). 1Н ЯМР (CD3OD): δ 4,46 (t, 1H), 4,53 (t, 1H), 4,64 (t, 1H), 4,76 (t, 1H), 6,79 (d, 1H), 7,79 (dd, 1H), 8,18 (s, 1H),
Этап 3
В раствор (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (20 мг, 0,041 ммоль), 5-бром-1-(2-фторэтил)пиридин-2(1H)-она (9,2 мг, 0,041 ммоль) и Cs2CO3 (2 N, 0,2 мл, 0,41 ммоль) в 1,4-диоксане (2 мл) в атмосфере N2 был добавлен Pd(PPh3)2Cl2 (3 мг, 0,0041 ммоль). Смесь подверглась обратному конденсированию в течение 2 ч, после чего была обработана EtOAc (10 мл) и водой (10 мл). Органический слой был высушен над Na2SO4, выпарен и очищен посредством препаративной ВЭЖХдля получения (S)-3-((S)-1-(4-(1-(2-фторэтил)-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-фенил-1,3-оксазинан-2-она (4,20 мг, 20%). Метод ЖХ-МС 2 tR=1,01 мин, масса/заряд=515, 493; 1Н ЯМР (CD3OD): δ 0,97 (s, 3Н), 1,28 (s, 3H), 1,56 (d, 3H), 2,18 (s, 2H), 2,22 (m, 1H), 2,49 (m, 2H), 3,05 (m, 1H), 4,37 (t, 1H), 4,43 (t, 1H), 4,69 (t, 1H), 4,81 (t, 1H), 5,59 (q, 1H), 6,66 (d, 1H), 7,05 (d, 2H), 7,33 (m, 7H), 7,82 (m, 2H).
ПРИМЕР 88
(S)-3-((S)-1-(4-(1-цикпопропил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-фторфенил)-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-он
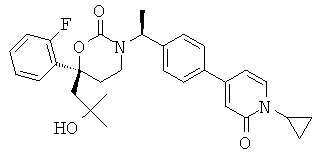
Титульное соединение было получено из (S)-6-(2-фторфенил)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она и 1-циклопропил-4-йодпиридин-2(1Н)-она в результате применения процедуры, аналогичной описанной для 9 этапа Примера 23. Метод ЖХ-МС 2 tR=1,05 мин, масса/заряд=505; 1H ЯМР (CDCl3) δ 0,88 (m, 2H), 1,12 (s, 3Н), 1,15 (s, 1H), 1,17 (s, 1H), 1,21 (s, 3Н), 2,18-2,29 (m, 2H), 2,30-2,34 (m, 1H), 2,42 (d, 1H), 2,54 (d, 1H), 2,90 (m,1H), 3,35 (m, 1H), 5,70 (m, 1H), 6,32 (m, 1H), 6,68 (m, 1H), 6,98 (m, 1H), 7,09 (d, 2H), 7,18 (t, 1H), 7,25-7,36 (m, 4H), 7,50 (t, 1H).
ПРИМЕР 89
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-(2-гидрокси-2-метилпропил)-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-он
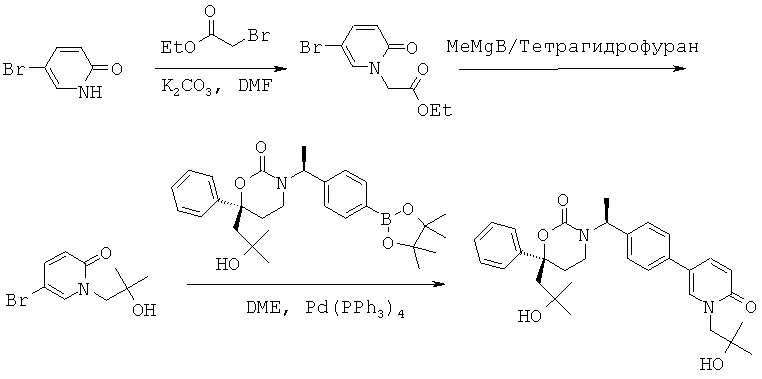
Этап 1
В раствор соединения 5-бромпиридин-2(1Н)-она (348 мг, 2,0 ммоль) и К2СО3 (830 мг, 6,0 ммоль) в DMF (15 мл) был по капле добавлен этилбромацетат. Смесь перемешивалась при комнатной температуре в течение 2 ч, затем подвергалась фильтрации. Полученный фильтрат был выпарен в вакууме, после чего полученный осадок был очищен посредством препаративной ТСХ (1:1 РЕ/EtOAc) для получения этил 2-(5-бром-2-оксопиридин-1(2Н)-ил)ацетата (300 мг, 57,7%). 1H ЯМР CDCl3: δ 7,41-7,26 (m, 2Н), 6,53-6,5 (d, 1Н), 4,59 (s, 2H), 4,28-4,21 (q, 2H), 1,32-1,23 (q, 3H).
Этап 2
В раствор этил 2-(5-бром-2-оксопиридин-1(2Н)-ил)ацетата (130 мг, 0,5 ммоль) в безводном тетрагидрофуране (5 мл) при температуре -78°С был по капле добавлен 1 М раствор MeMgBr (5 мл, 5 ммоль). Одновременно с добавлением раствор перемешивался. Реакционную смесь перемешивали в течение 1 ч при температуре -78°С. Затем реакция была остановлена с помощью водного раствора NH4Cl (5 мл), а полученная смесь подверглась экстракции при помощи EtOAc (3×10 мл). Объединенный органический слой был высушен и выпарен для получения неочищенного готового продукта, который затем был очищен посредством препаративной ТСХ (1:1 РЕ/EtOAc) для получения 5-бром-1-(2-гидрокси-2-метилпропил)пиридин-2(1Н)-она (65 мг, 52,9%).
Этап 3
В раствор 5-бром-1-(2-гидрокси-2-метилпропил)пиридин-2(1Н)-она (20 мг, 81,3 ммоль) в DME (6 мл) в атмосфере азота был добавлен Pd(PPh3)4 (10 мг). Смесь перемешивалась в течение 1 ч при комнатной температуре, затем в нее были добавлены раствор (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-((S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил)-1,3-оксазинан-2-она (38,95 мг, 81,3 ммоль) в EtOH (2 мл) и насыщенный водный раствор NaHCO3 (2 мл). Полученная смесь перемешивалась при температуре 100°С в течение 2 ч и, после остановки реакции, подверглась экстракции при помощи EtOAc. Объединенный органический слой был высушен над безводным Na2SO4 и выпарен для получения неочищенного продукта, который затем был очищен посредством препаративной ТСХ и препаративной ВЭЖХ для получения (S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-(2-гидрокси-2-метилпропил)-6-оксо-1,6-дигидропиридин-3-ил)фенил)этил)-6-фенил-1,3-оксазинан-2-она (6,5 мг, 15,5%). Метод ЖХ-МС 2 tR=0,99 мин, масса/заряд=519; 1H ЯМР (CDCl3): δ 7,60-7,57 (d, 1Н), 7,43 (s, 1H), 7,36-7,26 (m, 5H), 7,15 (d, 2H), 7,01 (d, 2H), 6,70 (d, 1H), 2,85 (m, 1H), 5,69-5,66 (m, 1H), 4,13-4,09 (s, 2H), 4,05-3,98 (s, 1H), 2,89-2,86 (m, 1H), 2,44-2,36 (m, 1H), 2,28-2,16 (m, 5H), 1,58-1,53 (d, 3Н), 1,33-1,30 (s, 6H), 1,19 (s, 3H), 1,12 (s, 3H).
ПРИМЕР 90
3-((S)-1-{4-[1-(3-гидрокси-2,2-диметил-пропил)-2-оксо-1,2-дигидро-пиридин-4-ил]-фенил}-этил)-(S)-6-(2-гидрокси-2-метил-пропил)-6-фенил-[1,3]оксазинан-2-он
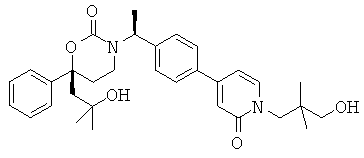
2 М водный раствор Na2CO3 (0,32 мл) был добавлен в смесь 4-бром-1-(3-гидрокси-2,2-диметил-пропил)-1Н-пиридин-2-она (0,13 г) и (S)-6-(2-гидрокси-2-метилпропил)-6-фенил-3-[(S)-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этил]-1,3-оксазинан-2-она (0,15 г) в N,N-диметилформамиде (3 мл). На полученную смесь в течение 5 мин производилось распыление аргона перед добавлением комплекса [1,1'-бис(дифенилфосфино)-ферроцен]дихлор-палладий(II) дихлорметана (26 мг). Смесь нагревалась до 100°С и перемешивалась при указанной температуре на 4 ч. После охлаждения до температуры окружающей среды в смесь была добавлена вода, после чего смесь подверглась экстракции этилацетатом. Объединенный органический экстракт был промыт солевым раствором и высушен (MgSO4). Полученный после выпаривания растворителя осадок был очищен посредством ВЭЖХ с обращенными фазами (метанол/вода/NH4OH) для получения соединения в виде твердого вещества бежевого цвета. Выход: 0,10 г (60% теоретического показателя); Масс-спектр (ионизация электрораспылением+: масса/заряд=533 [М+Н]+.
3-(4-бром-2-оксо-2Н-пиридин-1-ил)-2,2-диметил-пропионовой кислоты метилэфир
3-бром-2,2-диметил-пропионовой кислоты метилэфир (0,75 г) был добавлен в смесь 4-бром-1Н-пиридин-2-она (0,55 г) и карбоната калия (0,75 г) в N,N-диметилформамиде (10 мл) при комнатной температуре. Смесь была нагрета до 60°С, затем перемешивалась при указанной температуре в течение ночи. После дополнительного перемешивания при температуре 80°С в течение 8 ч смесь была охлаждена до комнатной температуры, затем в нее была добавлена вода. Полученная смесь подверглась экстракции этилацетатом, после чего объединенный органический экстракт был промыт солевым раствором и высушен (MgSO4). Полученный после выпаривания растворителя осадок был очищен посредством хроматографии с применением силикагеля (смесь циклогексана/этилацетата 4:1) для получения титульного соединения; в результате данной реакции также был получен 3-(4-бром-пиридин-2-илокси)-2,2-диметил-пропионовой кислоты метилэфир (0,35 г). Выход: 0,29 г (32% теоретического показателя); Масс-спектр (ионизация электрораспылением+): масса/заряд=288/300 (Br) [М+Н]+.
4-бром-1-(3-гидрокси-2,2-диметил-пропил)-1Н-пиридин-2-он
Борогидрид лития (25 мг) был добавлен в раствор 3-(4-бром-2-оксо-2Н-пиридин-1-ил)-2,2-диметил-пропионовой кислоты метилэфира (0,29 г) в тетрагидрофуране (3 мл), охлажденный в ледяной бане. После добавления метанола (45 мкл) смесь перемешивалась в охлаждающей бане в течение 1 ч, а затем при комнатной температуре в течение ночи. Затем смесь была разбавлена тетрагидрофураном с последующим добавлением MgSO4. Фильтрат, полученный после проведения фильтрации, был выпарен, а полученный осадок был очищен посредством хроматографии с применением силикагеля (смесь циклогексана/этилацетата 1:1) для получения титульного соединения в виде бесцветного масла. Выход: 0,13 г (49% теоретического показателя); Масс-спектр (ионизация электрораспылением+: масса/заряд=260/262 (Br) [М+H]+.
ПРИМЕР 91
3-((S)-1-(4-(1-циклопропил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-(тетрагидро-2Н-пиран-4-ил)-1,3-оксазинан-2-он
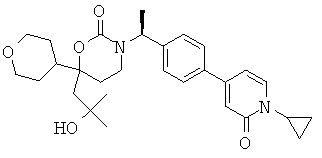
Титульное соединение было получено в результате применения процедур, аналогичных описанным в Примере 23 со следующими изменениями. В ходе 1 этапа тетрагидро-2Н-пиран-4-карбоновая кислота и карбонилдиимидазол использовались вместо 2-фторбензоил хлорида, а в ходе 9 этапа 1-циклопропил-4-йодпиридин-2(1Н)-использовался вместо 5-бром-1-метилпиридин-2(1Н)-она. В ходе реакции были выделены два изомера.
Изомер 1. Метод ЖХ-МС 2 tR=0,95 мин, масса/заряд=495.
Изомер 2. Метод ЖХ-МС 2 tR=0,93 мин, масса/заряд=495.
ПРИМЕР 92
(S)-6-(2-гидрокси-2-метилпропил)-3-((S)-1-(4-(1-тридейтерометил-2-оксо-1,2-дигидропиридин-4-ил)фенил)этил)-6-сренил-1,3-оксазинан-2-он
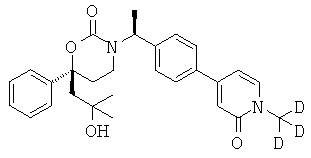
Титульное соединение было получено в результате применения процедур, аналогичных описанным в Методе 2 Примера 48 со следующими изменениями. В ходе 1 этапа тридейтерометил йодид использовался вместо метилйодида, а в ходе 2 этапа PdCl2(dppf) использовался вместо PdCl2(PPh3)2. Метод ЖХ-МС 1 tR=1,30 мин, масса/заряд=464.
ПРИМЕР 93
3-((S)-1-(4-(2-оксо-1-циклопропил-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-(циклопропилметил)-1,3-оксазинан-2-он
Целевое соединение получали по методике, аналогичной описанной в Примере 23, этапы 2-9. На этапе 2 использовали N-метокси-N-метил-2-циклопропилацетамид, полученный конденсацией 2-циклопропилуксусной кислоты с N,O-диметилгидроксиламином в присутствии карбодиимида. Два изомерных 3-((1S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-(циклопропилметил)-1,3-оксазинан-2-она, образующихся на этапе 5, разделяли методом колоночной хроматографии. Полученные изомеры раздельно вводили в этапах 6-9, на этапе 9 применяли 4-йод-1-циклопропилпиридин-2(1Н)-он и получали два изомера целевого соединения.
Изомер 1: Метод 2, ГХ-МС, tR=1.04 мин, m/z=465, 447.
Изомер 2: Метод 2, ГХ-МС, tR=1.06 мин, m/z=465, 447.
ПРИМЕР 94
3-((S)-1-(4-(2-оксо-1-циклопропил-1,2-дигидропиридин-4-ил)фенил)этил)-6-(2-гидрокси-2-метилпропил)-6-неопентил-1,3-оксазинан-2-он
Целевое соединение получали по методике, аналогичной описанной в Примере 23, этапы 2-9. На этапе 2 использовали N-метокси-N-метиламид 3,3-диметилбутановой кислоты, полученный конденсацией 3,3-диметилбутановой кислоты с N,О-диметилгидроксиламином в присутствии HATU [O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата]. Два изомерных 3-((1S)-1-(4-бромфенил)этил)-6-(2-метилаллил)-6-неопентил-1,3-оксазинан-2-она, образующихся на этапе 5, разделяли методом колоночной хроматографии. Полученные изомеры раздельно вводили в этапе 6-9, на этапе 9 применяли 4-йод-1-циклопропил пиридин-2(1Н)-он и получали два изомера целевого соединения.
Изомер 1: Метод 2, ГХ-МС 2 tR=1,09 мин., m/z=503, 481, 423.
Изомер 2: Метод 2, ГХ-МС 2 tR=1.16 мин., m/z=481.
ПРИМЕР 95
3-((S)-1-(4-(2-оксо-1-циклопропил-1,2-дигидропиридин-4-ил)фенил)этил)-6-(3,3-дифторциклобутил)-6-(2-гидрокси-2-метилпропил)-1,3-оксазинан-2-он
Целевое соединение получали по методике, аналогичной описанной в Примере 23, этапы 2-9. На этапе 2 использовали 3,3-дифтор-N-метокси-N-метилциклобутанкарбоксамид, полученный конденсацией 3,3-дифторциклобутанкарбоновой кислоты с N,O-диметилгидроксиамином в присутствии карбодиимида. Два изомерных 3-((S)-1-(4-бромфенил)этил)-6-(3,3-дифторциклобутил)-6-(2-метилаллил)-1,3-оксазинан-2-она, образующихся на этапе 5, разделяли методом колоночной хроматографии. Полученные изомеры раздельно вводили в этапы 6-9, на этапе 9 применяли 4-йод-1-циклопропилпиридин-2(1Н)-он и получали два изомера целевого соединения.
Изомер 1: Метод 2, ГХ-МС 1 tR=1.08 мин., m/z=501.
Изомер 2: Метод 2, ГХ-МС 1 tR=1.07 мин., m/z=501.
Примеры 96-98
Интермедиат 1
(S)-4-Бром-1-(1-изоцианатоэтил)-2-метилбензол
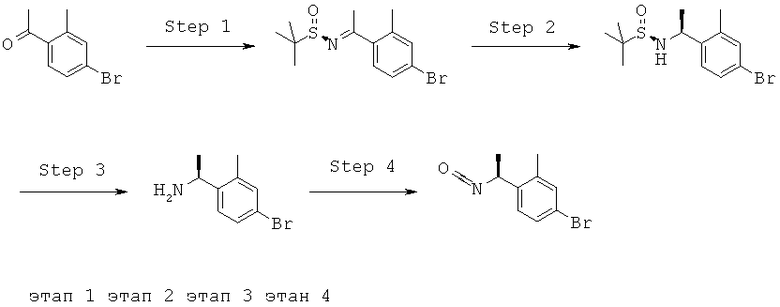
Этап 1: [1-(4-бром-2-метилфенил)этилиден]амид (R)-2-метилпропан-2-сульфиновой кислоты
К раствору 1-(4-бром-2-метилфенил)этанона (4.41 г) и (R)-2-метил-2-пропансульфинамида (2.76 г) в тетрагидрофуране (45 мл) прибавляли тетраэтоксититан(IV) (10.7 мл). Полученный раствор нагревали до 65°С и перемешивали при этой температуре 20 ч. Далее раствор охлаждали до обычной температуры и прибавляли дополнительное количество тетраэтоксититана(IV) (5.4 мл) и (R)-2-метил-2-пропансульфинамида (1.25 г). Раствор перемешивали еще 12 ч при 65°С, охлаждали до комнатной температуры и выливали в водный раствор NaCl. Полученную смесь отфильтровывали через целит, и отделяли органический слой фильтрата. Водный слой фильтрата экстрагировали этилацетатом, и объединяли экстракты с органическим слоем. Объединенную органическую фазу промывали раствором NaCl и сушили (MgSO4), затем растворитель упаривали. Остаток хроматографировали на силикагеле (элюент - циклогексан/этилацетат 9:1→1:1), получая целевое соединение. Выход: 6.24 г (95% от теоретического); Масс-спектр (ЭР+): m/z=316/318 (Br) [M+H+].
Этап 2: [(S)-1-(4-Бром-2-метилфенил)этил]амид (R)-2-метилпропан-2-сульфиновой кислоты
К раствору [1-(4-бром-2-метилфенил)этилиден]амида (R)-2-метилпропан-2-сульфиновой кислоты (6.24 г) в тетрагидрофуране (50 мл) при охлаждении на ледяной бане прибавляли три-втор-бутилборогидрид лития (1 моль/л в тетрагидрофуране, 59.2 мл). Раствор перемешивали 2 ч, давая ему нагреться до комнатной температуры на охлаждающей бане. Затем раствор снова охлаждали на ледяной бане и осторожно добавляли воду. Полученную смесь экстрагировали этилацетатом, и объединенные экстракты промывали водой, насыщенным водным раствором NaHCO3 и водным раствором NaCl. После высушивания (MgSO4) и испарения растворителя получали целевое соединение, которое вводили без очистки в следующий этап синтеза. Выход: 8.85 г (чистота около 75%); Масс-спектр (ЭР+): m/z=318/320 (Br) [М+Н]+.
Этап 3: (S)-1-(4-Бром-2-метилфенил)этиламин
К раствору [(S)-1-(4-бром-2-метилфенил)этил]амида (R)-2-метилпропан-2-сульфиновой кислоты (продукт этапа 2, без очистки, 8.85 г, чистота около 75%) в этилацетате (80 мл) при комнатной температуре прибавляли соляную кислоту (4 моль/л в 1,4-диоксане, 46 мл). Раствор перемешивали 1 час при комнатной температуре и упаривали. К остатку приливали воду, и полученную смесь промывали диэтиловым эфиром. Водную фазу подщелачивали прибавлением 1 М водного NaOH и экстрагировали хлористым метиленом. Объединенные экстракты промывали раствором NaCl, сушили (MgSO4) и упаривали, получая целевое соединение. Выход: 4.04 г (90% от теоретического); Масс-спектр (ЭР-): m/z=214/216 (Br) [М+Н]+.
Этап 4: (S)-4-Бром-1-(1-изоцианатоэтил)-2-метилбензол
К смеси NaHCO3 (3.65 г) в воде (80 мл) и (S)-1-(4-бром-2-метилфенил)этиламина (4.04 г) в хлористом метилене (80 мл) при интенсивном перемешивании и охлаждении на ледяной бане прибавляли одной порцией трифосген (2.24 г). Охлаждающую баню убирали и реакционную массу перемешивали при комнатной температуре еще 30 мин. Затем органический слой отделяли и сушили (MgSO4) и растворитель упаривали, получая изоцианат в виде масла, которое использовали без очистки на следующем этапе. Выход: 4.50 г (99% от теоретического); Масс-спектр (ЭР-): m/z=240/242 (Br) [M-Н]-.
Интермедиат 2
(S)-1-Бром-4-(1-изоцианатоэтил)-2-метилбензол
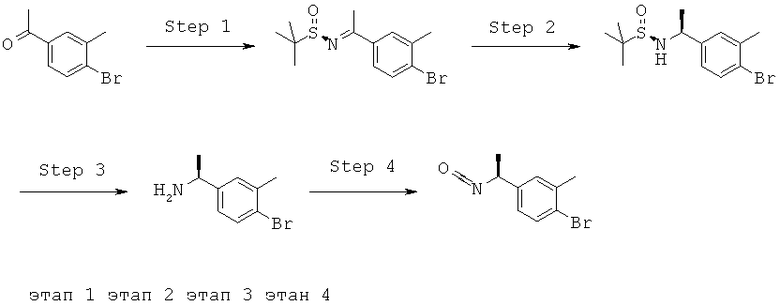
Этап 1: [1-(4-Бром-3-метилфенил)этилиден]амид (R)-2-метилпропан-2-сульфиновой кислоты
Целевое соединение получали из 1-(4-бром-3-метилфенил)этанона и {R)-2-метил-2-пропансульфинамида по методике, аналогичной этапу 1 при получении интермедиата 1. Масс-спектр (ЭР+): m/z=316/318 (Br) [M+H]+.
Этап 2: [(S)-1-(4-бром-3-метилфенил)этил]амид (R)-2-метил-пропан-2-сульфиновой кислоты
Целевое соединение получали из [1-(4-бром-3-метилфенил)этилиден]амида (R)-2-метилпропан-2-сульфиновой кислоты по методике, аналогичной этапу 2 при получении интермедиата 1. ГХ-МС (метод 5): tR=4.04 мин; Масс-спектр (ЭР+): m/z=318/320 (Br) [М+Н]+.
Этап 3: (S)-1-(4-Бром-3-метилфенил)этиламин
Целевое соединение получали из [(S)-1-(4-бром-3-метилфенил)этил]амида (R)-2-метилпропан-2-сульфиновой кислоты по методике, аналогичной этапу 3 при получении интермедиата 1. ГХ-МС (метод 4): tR=3.56 мин; Масс-спектр (ЭР+): m/z=214/216 (Br) [М+Н]+.
Этап 4: (S)-1-Бром-4-(1-изоцианатоэтил)-2-метилбензол
Целевое соединение получали из (S)-1-(4-бром-3-метилфенил)этиламина и трифосгена по методике, аналогичной этапу 4 при получении интермедиата 1, Масс-спектр (ЭР+): m/z=272/274 (Br) [М+Н+МеОН]+.
Интермедиат 3
5-Бром-2-изоцианатометил-1,3-диметилбензол
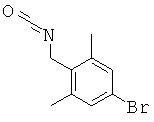
Целевое соединение получали из 4-бром-2,6-диметилбензиламина и трифосгена по методике, аналогичной этапу 4 при получении интермедиата 1. Масс-спектр (EI): m/z=239/241 (Br) [М]+.
Интермедиат 4
4-Бром-1-циклопропил-1Н-пиридин-2-он
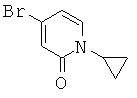
В колбу с магнитной мешалкой загружали 4-бром-1 Н-пиридин-2-он (1.80 г), циклопропилборную кислоту (2.00 г), Cu(O2CCH3)2 (2.00 г), 2,2'-бипиридил (1.70 г), Na2CO3 (2.47 г) и 1,2-дихлорэтан (75 мл) и нагревали до 70°С. Смесь перемешивали на воздухе при этой температуре до утра. Далее прибавляли дополнительное количество циклопропилборной кислоты (0.50 г) и Ма2СО3 (0.55 г) и кипятили с обратным холодильником еще 4 часа. После охлаждения до обычной температуры прибавляли водный раствор NH4Cl, и полученную смесь экстрагировали хлористым метиленом. Объединенные органические экстракты сушили (MgSO4) и растворитель упаривали. Остаток очищали методом хроматографии на силикагеле (элюент - циклогексан/этилацетат 50:50→35:65), получая целевое соединение в виде масла, которое кристаллизовалось при стоянии. Выход: 0.82 г (37% от теоретического); Масс-спектр (ЭР+): m/z=214/216 (Br) [M+H]+.
Пример 96
3-{(S)-1-[4-(1-Циклопропил-2-оксо-1,2-дигидропиридин-4-ил)-2-метилфенил]-этил}-(5)-6-(2-гидрокси-2-метилпропил)-6-фенил-[1,3]оксазинан-2-он
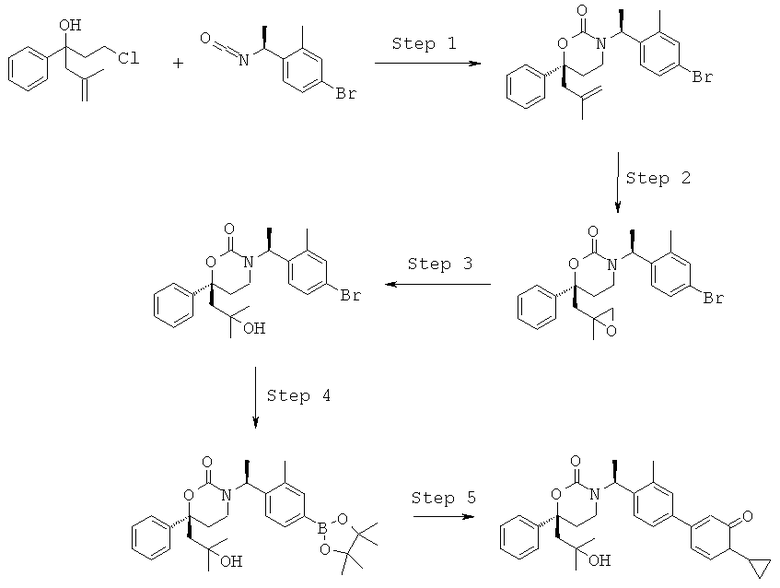
Этап 1: 3-[(S)-1-(4-Бром-2-метилфенил)этил-(R)-6-(2-метилаллил)-6-фенил-[1,3]оксазинан-2-он
К раствору 5-метил-3-фенил-1-хлоргекс-5-ен-3-ола (3.82 г) и (5)-4-бром-1-(1-изоцианатоэтил)-2-метилбензола (4.49 г) в тетрагидрофуране (140 мл) при охлаждении на ледяной бане прибавляли по каплям гексаметилдисилазид лития (1 моль/л в тетрагидрофуране, 18.7 мл) с такой скоростью, чтобы температура раствора оставалась ниже 25°С. Раствор перемешивали 30 мин на охлаждающей бане и еще 60 мин при комнатной температуре. Далее к реакционной смеси медленно прибавляли раствор уксусной кислоты (1.9 мл) в воде (40 мл). Полученную смесь упаривали при пониженном давлении, и к остатку приливали трет-бутилметиловый эфир. Полученный раствор промывали водой, высушивали (MgSO4) и упаривали. Остаток хроматографировали на силикагеле (элюент - циклогексан/этилацетат 90:10→40:60), получая целевое соединение в виде бесцветного твердого вещества. Помимо целевого соединения, был выделен его диастереомер - 3-[(S)-1-(4-бром-2-метилфенил)-этил]-6-(S)-(2-метилаллил)-6-фенил-[1,3]оксазинан-2-он - в виде масла {1,55 г, масс-спектр (ЭР+): m/z=428/430 (Br) [М+Н]+. Выход: 1.34 г (18% от теоретического); Масс-спектр (ЭР+): m/z=428/430 (Br) [М+Н]+; ПМР спектр (400 МГц, ДМСО-d6): 1.40 (д, J=6.8 Гц, 3Н), 1.53 (с, 3Н), 1.71 (с, ЗН), 1.81-190 (м, 1Н), 1.98-2.08 (м, 1Н), ок. 2.43. 2.51 (м, 3Н), 2.86-2.94 (м, 1Н), 4.58 (плохо разрешенный м, 1Н), 4.77 (плохо разрешенный м, 1Н), 5.30 (к, J=6.8 Гц, 1Н), 7.15-7.19 (м, 2Н), 7.24-7.35 (м, 6Н). Отнесение стереогенных центров целевого соединения основано на сравнении данных его ПМР-спектра с данными для известного аналога - 3-[(S)-1-(4-бромфенил)этил]-(R)-6-(2-метилаллил)-6-фенил-[1,3]оксазинан-2-она.
Этап 2: 3-[(S)-1-(4-Бром-2-метил-фенил)-этил]-(S)-6-(2-метил-охiranylметил)-6-фенил-[1,3]оксазинан-2-он
К раствору 3-хлорнадуксусной кислоты (77%, 0.81 г) в хлористом метилене (15 мл) при охлаждении до 5°С прибавляли 3-[(S)-1-(4-бром-2-метилфенил)этил]-(R)-6-(2-метилаллил)-6-фенил-[1,3]оксазинан-2-он (1.34 г) в растворе в хлористом метилене (15 мл). Охлаждающую баню убирали и раствор перемешивали при комнатной температуре до утра. Прибавляли 10%-ный водный раствор Na2S2O3 (10 мл) и насыщенный водный раствор NaHCO3 (25 мл), и полученную смесь перемешивали еще 30 мин. Органический слой отделяли и промывали водным раствором Na2S2O3 в смеси с насыщенным водным раствором NaHCO3, водой и раствором NaCl. Органическую фазу сушили (MgSO4) и упаривали, получая целевое соединение. Выход: 1.55 г (чистота около 85-90%); ГХ-МС (метод 4): tR=4.28 мин.; масс-спектр (ЭР+): m/z=444/446 (Br) [M+H]+.
Этап 3: 3-[(S)-1-(4-Бром-2-метилфенил)этил]-(S)-6-(2-гидрокси-2-метилпропил)-6-фенил-[1,3]оксазинан-2-он
К охлаждаемому льдом раствору 3-[(S)-1-(4-бром-2-метилфенил)этил]-(S)-6-(2-метилоксиранилметил)-6-фенил-[1,3]оксазинан-2-она (1.55 г, чистота около 85-90%) в тетрагидрофуране (15 мл) прибавляли триэтилборогидрид (1 моль/л в тетрагидрофуране, 4,2 мл) с такой скоростью, чтобы температура раствора оставалась ниже 10°С. Полученный раствор перемешивали еще 1 час на охлаждающей бане и 2 часа при комнатной температуре. Далее раствор охлаждали на ледяной бане и прекращали реакцию осторожным прибавлением воды (7 мл). После прибавления водной соляной кислоты и этилацетата (80 мл) органический слой отделяли, промывали раствором NaCl и сушили (MgSO4). Растворитель упаривали, получая целевое соединение. Выход: 1.48 г (95% от теоретического); ГХ-МС (метод 4): tR=4.00 мин; Масс-спектр (ЭР+): m/z=446/448 (Br) [M+H]+.
Этап 4: (S)-6-(2-Гидрокси-2-метилпропил)-3-{(S)-1-[2-метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]этил}-6-фенил-[1,3]оксазинан-2-он
В колбу с магнитной мешалкой загружали ацетат калия (1.14 г), бис(пинаколато)дибор (1.10 г), 3-[(S)-1-(4-бром-2-метилфенил)этил]-(S)-6-(2-гидрокси-2-метилпропил)-6-фенил[1,3]оксазинан-2-он (1.48 г) и диметилсульфоксид (20 мл) и барботировали аргон в течение 10 мин. Далее прибавляли комплекс [1,Г-бис(дифенилфосфино)ферроцен]дихлор-палладия(II) с хлористым метиленом (0.27 г), нагревали смесь до 90°С и перемешивали 2.5 ч при этой температуре. После охлаждения смеси до обычной температуры добавляли воду и этилацетат, и полученную смесь фильтровали через целит. Водную фазу фильтрата отделяли и дважды экстрагировали этилацетатом. Органические экстракты и органическую фазу фильтрата объединяли и промывали водой и раствором NaCl и сушили (MgSO4). Растворитель упаривали и остаток хроматографировали на силикагеле (элюент - циклогексан/этил ацетат 1:1→1:4), получая целевое соединение. Выход: 1.23 г (75% от теоретического); Масс-спектр (ЭР+): m/z=494 [М+Н]+.
Этап 5: 3-{(S)-1-[4-(1-Циклопропил-2-оксо-1,2-дигидропиридин-4-ил)-2-метилфенил]этил}-(S)-6-(2-гидрокси-2-метилпропил)-6-фенил-[1,3]оксазинан-2-он
К смеси 4-бром-1-циклопропил-1Н-пиридин-2-она (0.11 г) и (S)-6-(2-гидрокси-2-метилпропил)-3-{(S)-1-[2-метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]этил}-6-фенил-[1,3]оксазинан-2-она (0.20 г) в N,N-диметилформамиде (2 мл) прибавляли 2 М водный раствор Na2CO3 (0.41 мл). Через полученную смесь барботировали аргон в течение 10 мин., а затем прибавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлор-палладия(II) с хлористым метиленом (33 мг). Смесь нагревали до 100°С и перемешивали при этой температуре до утра. После охлаждения смеси до комнатной температуры прибавляли воду, и полученную смесь экстрагировали этилацетатом. Объединенные органические экстракты промывали водой и раствором NaCl и сушили (MgSO4). Растворитель упаривали и остаток очищали методом обращенно-фазовой ВЭЖХ (элюент - метанол/вода/NH4OH), получая целевое соединение. Выход: 0.13 г (64% от теоретического); ГХ-МС (метод 5): tR=3.43 мин; Масс-спектр (ЭР+): m/z=501 [М+Н]+.
Пример 97
3-{(S)-1-[4-(1-Циклопропил-2-оксо-1,2-дигидропиридин-4-ил)-3-метилфенил]-этил}-(S)-6-(2-гидрокси-2-метилпропил)-6-фенил-[1,3]оксазинан-2-он
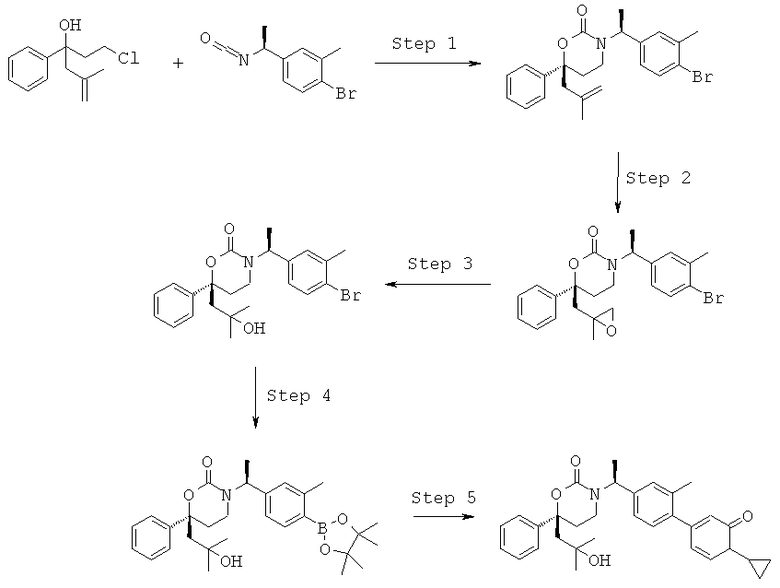
Этап 1: 3-[(S)-1-(4-Бром-3-метилфенил)этил]-(R)-6-(2-метилаллил)-6-фенил-[1,3]оксазинан-2-он
Целевое соединение получали из 5-метил-3-фенил-1-хлоргекс-5-ен-3-ола и (S)-1-бром-4-(1-изоцианатоэтил)-2-метилбензола по методике, аналогичной этапу 1 в примере 96. Масс-спектр (ЭР+): m/z=428/430 (Br) [М+Н]+; Спектр ПМР (400 МГц, ДМСО-d6) 1.40 (д, J=6.9 Гц, 3Н), 1.55 (с, 3Н), 2.13 (с, 3Н) перекрыт 2.07-2.18 (м, 2Н), 2.42-2.47 (м, 1Н), 2.53 (уш.с., 2Н), 2.92-3.03 (м, 1Н), 4.60 (плохо разрешенный м, 1Н), 4.77 (плохо разрешенный м, 1Н), 5.33 (к, J=6.9 Гц, 1Н), 6.62 (дд, J=8.2 Гц, 2.0 Гц, 1Н), 6.76 (плохо разрешенный д, 1Н), 7.26-7.33 (м, 4Н), 7.34-7.40 (м, 2Н). Отнесение стереогенных центров целевого соединения основано на сравнении данных его ПМР-спектра с данными для известного аналога - 3-[(S)-1-(4-бромфенил)этил]-(R)-6-(2-метилаллил)-6-фенил-[1,3]оксазинан-2-он.
Этап 2: 3-[(S)-1-(4-Бром-3-метилфенил)этил]-(S)-6-(2-метил-оксиранилметил)-6-фенил-[1,3]оксазинан-2-он
Целевое соединение получали из 3-[(S)-1-(4-бром-3-метилфенил)этил]-(R)-6-(2-метилаллил)-6-фенил-[1,3]оксазинан-2-она по методике, аналогичной этапу 2 в примере 96. ГХ-МС (метод 4): tR=4.03 мин; Масс-спектр (ЭР+): m/z=444/446 (Br) [М+Н]+.
Этап 3: 3-[(S)-1-(4-Бром-3-метилфенил)этил]-(S)-6-(2-гидрокси-2-метилпропил)-6-фенил-[1,3]оксазинан-2-он
Целевое соединение получали из 3-[(S)-1-(4-бром-3-метилфенил)этил]-(S)-6-(2-метилоксиранилметил)-6-фенил-[1,3]оксазинан-2-она по методике, аналогичной этапу 3 примера 96. ГХ-МС (метод 4): tR=4.03 мин; Масс-спектр (ЭР+): m/z=446/448 (Br) [М+Н]+.
Этап 4: (S)-6-(2-Гидрокси-2-метилпропил)-3-{(S)-1-[3-метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]этил}-6-фенил-[1,3]оксазинан-2-он
Целевое соединение получали из 3-[(S)-1-(4-бром-3-метилфенил)этил]-(S)-6-(2-гидрокси-2-метилпропил)-6-фенил-[1,3]оксазинан-2-она по методике, аналогичной этапу 4 в примере 96. Масс-спектр (ЭР+): m/z=494 [М+Н]+.
Этап 5: 3-{(S)-1-[4-(2-Оксо-1-циклопропил-1,2-дигидропиридин-4-ил)-3-метилфенил]этил}-(S)-6-(2-гидрокси-2-метилпропил)-6-фенил-[1,3]оксазинан-2-он
Целевое соединение получали из (S)-6-(2-гидрокси-2-метилпропил)-3-{(S)-1-[3-метил-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фенил]этил}-6-фенил[1,3]оксазинан-2-она и 4-бром-1-циклопропил-1Н-пиридин-2-она по методике, аналогичной этапу 5 в примере 96. ГХ-МС (метод 5): tR=3.42 мин; Масс-спектр (ЭР+): m/z=501 [М+Н]+.
Пример 98
3-[4-(2-Оксо-1-циклопропил-1,2-дигидропиридин-4-ил)бензил]-6-(2-гидрокси-2-метилпропил)-6-фенил[1,3]оксазинан-2-он
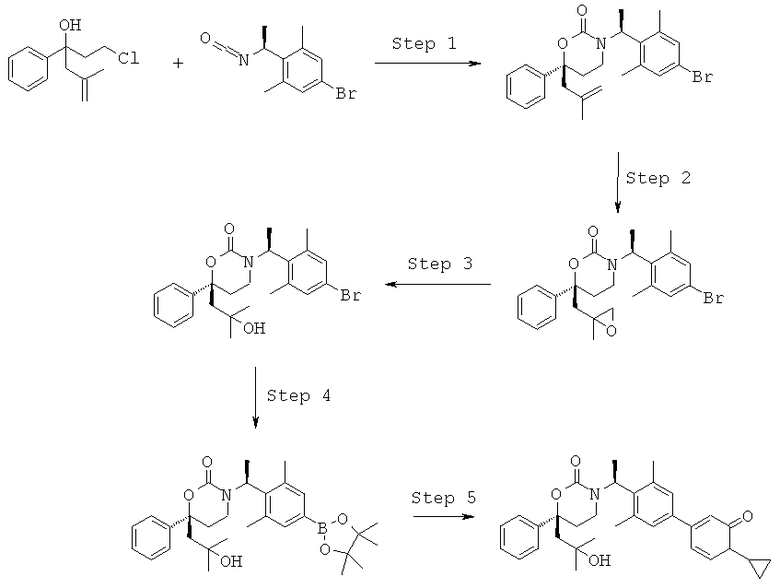
Этап 1: 3-(4-Бром-2,6-диметилбензил)-6-(2-метилаллил)-6-фенил-[1,3]оксазинан-2-он
Целевое соединение получали из 5-метил-3-фенил-1-хлоргекс-5-ен-3-ола и 5-бром-2-изоцианатометил-1,3-диметилбензола по методике, аналогичной этапу 1 в примере 96. ГХ-МС (метод 5): tR=4.95 мин.; Масс-спектр (ЭР+): m/z=428/430 (Br) [М+Н]+.
Этап 2: 3-(4-Бром-2,6-диметилбензил)-6-(2-метилоксиранилметил)-6-фенил[1,3]оксазинан-2-он
Целевое соединение получали из 3-(4-бром-2,6-диметилбензил)-6-(2-метилаллил)-6-фенил-[1,3]оксазинан-2-она по методике, аналогичной этапу 2 в примере 96. ГХ-МС (метод 4): tR=4.05 мин; Масс-спектр (ЭР+): m/z=444/446 (Br) [M+H]+.
Этап 3: 3-(4-Бром-2,6-диметилбензил)-6-(2-гидрокси-2-метилпропил)-6-фенил-[1,3]оксазинан-2-он
Целевое соединение получали из 3-(4-бром-2,6-диметилбензил)-6-(2-метилоксиранилметил)-6-фенил[1,3]оксазинан-2-она по методике, аналогичной этапу 3 в примере 96. ГХ-МС (метод 4): tR=4.05 мин; Масс-спектр (ЭР+): m/z=446/448 (Br) [М+Н]+.
Этап 4: 3-[2,6-Диметил-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)бензил]-6-(2-гидрокси-2-метилпропил)-6-фенил[1,3]оксазинан-2-он
Целевое соединение получали из 3-(4-бром-2,6-диметилбензил)-6-(2-гидрокси-2-метилпропил)-6-фенил[1,3]оксазинан-2-она по методике, аналогичной этапу 4 в примере 96. Масс-спектр (ЭР+): m/z=494 [М+Н]+.
Этап 5: 3-[4-(2-Оксо-1-циклопропил-1,2-дигидропиридин-4-ил)бензил]-6-(2-гидрокси-2-метилпропил)-6-фенил-[1,3]оксазинан-2-он
Целевое соединение получали из 3-[2,6-диметил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)бензил]-6-(2-гидрокси-2-метилпропил)-6-фенил-[1,3]оксазинан-2-она и 4-бром-1-циклопропил-1Н-пиридин-2-она по методике, аналогичной этапу 5 в примере 96, ГХ-МС (метод 5): tR=3.53 мин; Масс-спектр (ЭР+): m/z=501 [М+Н]+.
БИОЛОГИЧЕСКИЙ ТЕСТ, ПРИМЕР 1
Степень ингибирования микросомного препарата 11β-ГСД1 соединениями, рассматриваемыми в изобретении, в основном измерялась методами, описанными выше (К. Solly, S.S. Mundt, H.J. Zokian, G.J. Ding, A. Hermanowski-Vosatka, B. Strulovici, and W. Zheng, High-Throughput Screening of 11-Beta-Hydroxyseroid Dehydrogenase Type 1 in Scintillation Proximity Assay Format. Assay Drug Dev Technol 3 (2005) 377-384). Все реакции проводились при комнатной температуре на 96-луночных прозрачных гибких ПЭТ-планшетах Microbeta (PerkinElmer). Анализ начинается с диспенсирования 49 мкл раствора субстрата (50 мМ N-2-гидроксиэтилпиперазин-N-2-этансульфоновой кислоты (HEPES), уровень рН=7,4, 100 мМ KCI, 5 мМ NaCl, 2 мМ MgCl2, 2 мМ никотинамидадениндинуклеотидфосфата(NADPH) и 160 нМ [3Н] кортизона (1 Ci/ммоль)) и смешивания 1 мкл тестируемых соединений в ДМСО, предварительно разбавлявшегося с половинным приращением (в 8 точках), начиная с 0,1 мМ. После предварительной инкубации в течение 10 минут добавлялось 50 мкл раствора фермента, содержащего микросомы, выделенные из клеток линии СНО, сверхэкспрессирующие 11-ГСД1 человека (общее содержание белка - 10-20 мкг/мл), и пластинки инкубировались при комнатной температуре в течение 90 минут. Реакция останавливалась путем добавления 50 мкл суспензии микрошариков для SPA-анализа, содержащей 10 мкМ 18 - глицирретиновой кислоты, 5 мг/мл микрошариков для YSi SPA, покрытых белком A (GE Healthcare) и 3,3 мкг/мл анти-кортизол-антитела (East Coast Biologies) в буфере Superblock (Bio-Rad). Пластинки встряхивались при комнатной температуре в течение 120 минут, и измерялся сигнал SPA, соответствующий [3H] кортизолу, при помощи устройства чтения пластин Microbeta.
БИОЛОГИЧЕСКИЙ ТЕСТ, ПРИМЕР 2
Ингибирование 11-ГСД1 соединениями, рассматриваемыми в данном изобретении, измерялось во всех клетках, указанным далее способом. Клетки для анализа поступали из двух источников: полностью дифференцированные сальниковые жировые клетки человека (адипоциты) - от компании Zen-Bio Inc.; недифференцированные клетки (преадипоциты) - от компании Lonza Group Ltd. Заранее дифференцированные сальниковые адипоциты Zen-Bio поставлялись в 96-луночных планшетах и использовались в ходе анализа, как минимум, в течение 2 недель после того как произошла дифференциация преадипоцитов. Zen-Bio индуцировали дифференциацию пре-адипоцитов, восполняя среду, содержащую адипогенные и липогенные гормоны (инсулин человека, дексаметазон, изобутилметилксантин, агонист PPAR-гамма). Клетки содержались в полностью адипоцитной среде (DMEM/Ham's F-12 (1:1, об./об.), HEPES с уровнем рН=7,4, фетальная бычья сыворотка, пенициллин, стрептомицин, амфотерицин В, поставляемые Zen-Bio, Inc.) при температуре 37°С, содержание CO2 5%.
Пре-адипоциты, поставленные компанией Lonza Group Ltd. и помещались в среду для выращивания адипоцитов (Preadipocyte Growth Medium-2), дополненную фетальной бычьей сывороткой, пенициллином и стрептомицином (от компании Lonza), где происходило культивирование при температуре 37°С, содержании углекислого газа 5%. Пре-адипоциты дифференциировались путем добавления инсулина, дексаметазона, индометацина и изобутил-метилксантина (от компании Lonza) в среду Preadipocyte Growth Medium-2. Клетки подвергались воздействию дифференцирущих факторов в течение 7 дней; к этому сроку, дифференциация завершалась, и клетки были готовы для анализа. За 1 сутки до его выполнения, дифференцированные сальниковые адипоциты переносились в среду, очищенную от сыворотки и фенолового красного, для инкубации в течение ночи. Общий объем пробы при анализе - 200 мкл. Клетки пре-инкубировались в среде, очищенной от сыворотки и фенолового красного, содержащей 0,1% (об./об.) ДМСО и тестируемые соединения в различных концентрациях, как минимум за 1 час до добавления [3H]-кортизона в этиловом спирте (50 Ci/ммоль, ARC, Inc.), для достижения окончательной концентрации кортизона, 100 нМ. Инкубация клеток происходила в течение 3-4 часов при температуре 37°С, содержании СО2 5%. Отрицательный контроль инкубировался без радиоактивного субстрата и к завершению инкубации принимал в себя аналогичный объем [3H]-кортизона. Образование [3Н] кортизола отслеживалось путем анализа 25 мкл каждого супернатанта методом анализа сближения с сцинтиллятором (SPA). (Solly, К.; Mundt, S.S.; Zokian, H.J.; Ding, G.J.; Hermanowski-Vosatka, A.; Strulovici, В.; Zheng, W. Assay Drug Dev. Technol. 2005, 3, 377-384). Многие соединения, рассматриваемые в данном изобретении, показали заметную активность в этом исследовании.
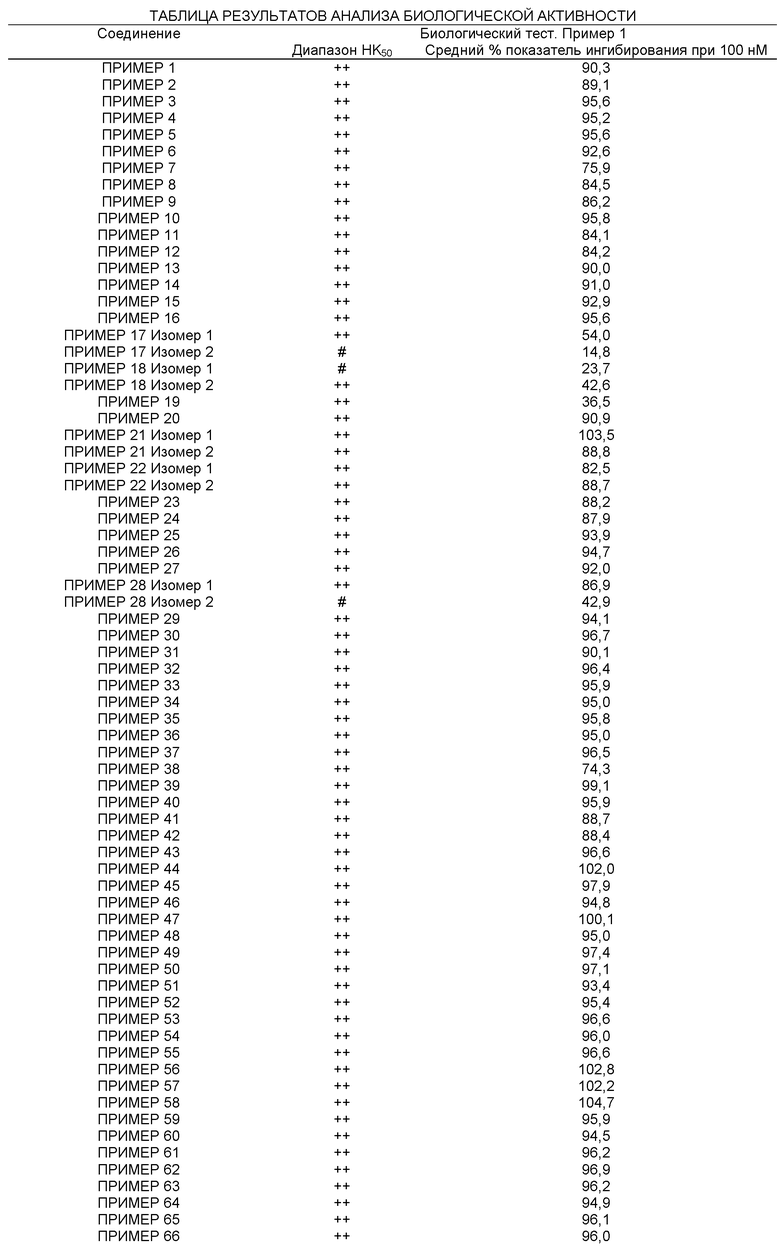

БИОЛОГИЧЕСКИЙ ТЕСТ, ПРИМЕР 3
Степень ингибирования 11β-ГСД1 тестируемыми соединениями определялась методом HTRF (гомогенной флуоресценции с временным разрешением) (cisbio international, Франция); метод позволяет обнаружить кортизол, вырабатываемый из кортистерона микросомами печени человека. Соединения инкубировались в течение 1 часа при температуре 37°С в Трис-буфере (20 мМ трис (гидроксиметиламинометана), 5 мМ ЭДТК с уровнем рН 6.0), содержащем НАДФН (200 мКМ) и кортизон (80 нМ). Кортизол, выработанный в ходе реакции, обнаруживается методом конкурентного иммуноферментного анализа, с использованием двух конъюгатов HTRF: кортизола, связанного с XL665, и анти-кортизол-антитела, маркированного криптатом европия. Период инкубации для реакции обнаружения обычно составлял 2 часа. Количество кортизола определялось по степени флуоресценции с временным разрешением у лунок. (Ех 320/75 нМ; Em 615/8,5 нМ и 665/7,5 нМ). Затем вычислялось соотношение двух сигналов эмиссии (Em665*10000/Em615). Каждый анализ включал инкубирование с контролем растворителем вместо рассматриваемого соединения, в качестве контроля выработки неингибированного кортизола (100% CTL; 'высокие значения') и инкубирования с карбеноксолоном в качестве контроля полностью ингибированного фермента и кортизолового фона (0% CTL; 'низкие значения'). Каждое исследование также включало в себя составление калибровочной кривой, позволяющей соотносить показатели флуоресценции с концентрациями кортизола. Подавление каждого соединения, в пересчете на проценты, определялось относительно сигнала карбеноксолона.
В таблице ниже показано ингибирующее воздействие на 11β-ГСД-1, определенное указанными выше способами; 100% означает отсутствие ингибирования, нулевое или отрицательное значение - полное ингибирование.
БИОЛОГИЧЕСКИЙ ТЕСТ, ПРИМЕР 4
Степень ингибирования микросхемного препарата 11β-ГСД1 соединениями, рассматриваемыми в настоящем изобретении, в присутстви 50% плазмы крови человека, измерялась в большей степени методами, описанными ниже. Микросомы клеток линии СНО, сверхэкспрессирующих 11β-ГСД1 человека, вводились в реакционный буфер, состоящий из 25 мМ N-2-гидроксиэтилпиперазин-N-2-этансульфоновой кислоты (HEPES) с уровнем рН 7,4, 50 мМ KCl, 2,5 мМ NaCl, 1 мМ MgCl2 и 50% (об./об.) плазмы крови человека (BioChemed). Анализ начинается с диспенсирования 49 мкл раствора микросом в 96-луночных полипропиленовых планшетах и добавления 1 мкл тестируемых соединений в ДМСО, предварительно разбавлявшихся с половинным приращением (в 8 точках), начиная с 1,0 мМ. Реакция инициировалась добавлением 50 мкм раствора субстратата, состоящего из реакционного буфера с 2 мМ НАДФН и 160 нМ [3H]кортизона (1 Ci/ммоль). Пластины инкубировались в течение 120 минут при комнатной температуре. Для остановки реации добавлялось 100 мкл ацетонитрила с 20 мМ кортизона и 20 мМ кортизола. Спустя 10-минутного инкубирования при комнатной температуре, 100 мкл содержимого каждой лунки фильтровалось через пластину HV-фильтра Multiscreen HTS (Millipore) и разбавлялось 100 мкл реакционного буфера, очищенного от плазмы крови человека, [3H]кортизон и [3-H]-кортизол отделялись методом ВЭЖХ, на хроматографической колонке Zorbax SB-C8 (4,6×250 мм, изготовитель - Agilent) с изократическим элюированием в 25% ацетонитрила в воде, содержащим 0,01% трифторуксусной кислоты; степень радиоактивности определялась при помощи встроенного детектора β-RAM (IN/US Systems, Inc.).
БИОЛОГИЧЕСКИЙ ТЕСТ, ПРИМЕР 5
(Несвязанные фракции в плазме крови человека)
Степень связывания соединений с белками плазмы определялась методом равновесного диализа плазмы с введенным соединением по сравнению с очищенным от соединения декстранным буфером, через диализную мембрану с проницаемостью 5000 Да. Концентрации соединения в плазме и буфере после инкубирования измерялись посредством ВЭЖХ/Масс спектрометрии.
БИОЛОГИЧЕСКИЙ ТЕСТ, ПРИМЕР 6
(Ингибирование CYP3A4)
Анализ основывался на методе, опубликованном Moody и др. (Xenobiotica 1999). Ингибирование реации N-деметилирования [N-метил-14С]-эритромицина тестируемым соединением, катализированное изоферментом 3А4 цитохрома Р450, исследовалось при температуре 37°С с рекомбинантным цитохромом Р450 3А4. Все анализы выполнялись при помощи роботизированной системы с 96-луночными планшетами. Окончательный объем пробы для инкубирования составлял 200 мкл; в нее входили, трис-буфер (0,1 М), MgCl2 (5 мМ), рекомбинантный белок (40 пмоль/мл), эритромицин (50 мкм) и тестируемое соединение, либо в четырех различных концентрациях в двух повторностях (напр. наивысшая концентрация 10-50 мкМ с последующими серийными разбавлениями 1:5), либо в концентрации 10 мкМ в трех повторностях. После кратковременного доинкубационного периода, реакции инициировались добавлением кофактора (НАДФН, 1 мМ) и останавливались добавлением 50 мкл водного раствора трихлоруксусной кислоты (10% м/об.). Аликвота инкубата переносилась в 96-луночные планшеты для твердофазной экстракции (SPE) и экстрагировалась в картридж. Полученная в результате [14C]-формальдегид/муравьиная кислота не удерживалась в картридже, и ее выделяли из неметаболизированного субстрата, промывая лунки SPE водой. Аликвота элюатов переносилась в луночные планшеты, пригодные для жидкостно-сцинтилляционного измерения активности. Скорость образования [14C]-формальдегида/муравьиной кислоты при этих инкубированиях сравнивалась с контролем активности, не содержащим тестируемого соединения. Если соединение тестировалось с четырьмя концентрациями, вычислялись экспериментальные значения IC50.
БИОЛОГИЧЕСКИЙ ТЕСТ, ПРИМЕР 7
(Ингибирование CYP2C9)
Степень ингибирования цитохрома Р450 изофермента 2С9, катализирующего O-деметилирование [О-метил-14С]-напроксена тестируемым соединением, анализировалась при температуре 37°С с рекомбинантным цитохромом Р450 2С9 человека; процедура аналогична описанной в примере 6. Экспериментальное значение IC50 вычислялось, исходя из процентного содержания контроля при четырех различных концентрациях.
БИОЛОГИЧЕСКИЙ ТЕСТ, ПРИМЕР 8
(Ингибирование CYP2C19)
Степень ингибирования цитохрома Р450 изофермента 2С19, катализирующего N-деметилирование [N-метил-14С]-диазепама тестируемым соединением, анализировалась при температуре 37°С с рекомбинантным цитохромом Р450 2С19 человека; процедура аналогична описанной в примере 6. Экспериментальное значение IC50 вычислялось, исходя из процентного содержания контроля при четырех различных концентрациях.
БИОЛОГИЧЕСКИЙ ТЕСТ, ПРИМЕР 9
(Ингибирование CYP2C9)
Степень ингибирования рекомбинантного цитохрома CYP2C9 соединениями, рассматриваемыми в настоящем изобретении, измерялась при помощи промышленного набора фирмы Invitrogen (кат. №2859), Поставляемые микросомы, выделенные из клеток насекомых, инфицированных бакуловирусами, перестраивающими экспрессию CYP2C9 человека, доводились до объема 10 мМ реакционным буфером (100 мМ натрий-фосфатного буфера, уровень рН=8,0) при помощи системы генерирования НАДФН (3,33 мМ глюкозы-6-фосфата и 0,4 ед/мл глюкозы-6-фосфат дигидрогеназы). 89 мкл данного раствора диспенсировались в каждую лунку 96-луночного планшета из полистирола черного цвета и смешивались с 1 мкл тестируемого соединения, предварительно разбавленного ДМСО с половинным приращением, начиная с 3 мМ. Анализ инициировался путем добавления 10 мкл фторогенного субстрата n-октилоксиметилрезоруфина (OOMR, 20 мкм) с НАДФ (100 мкм), разведенного в реакционном буфере. Планшет немедленно помещался в устройство для считывания Perkin Elmer Fusion. Процесс реакции отслеживался путем замера флуоресценции каждые 2 минуты в течение 20 минут (фильтр возбуждения 530 нМ/фильтр эмиссии 605 нМ).
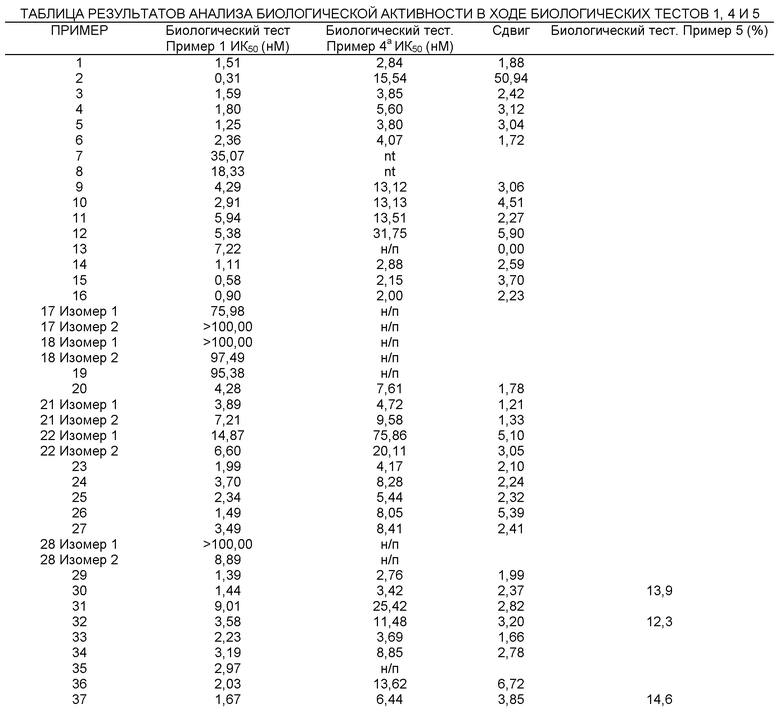
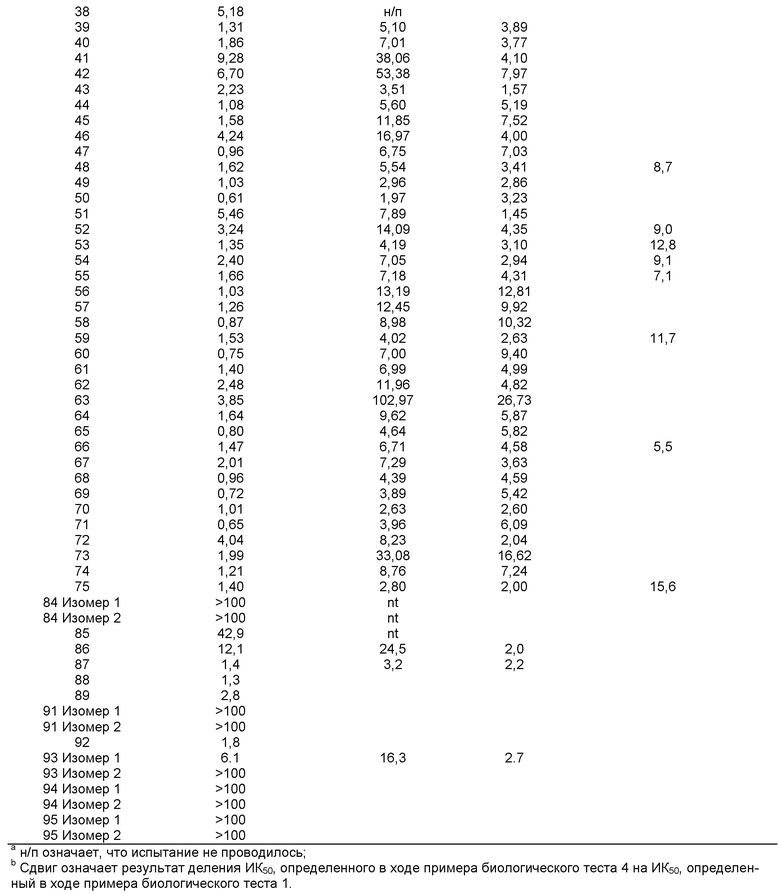


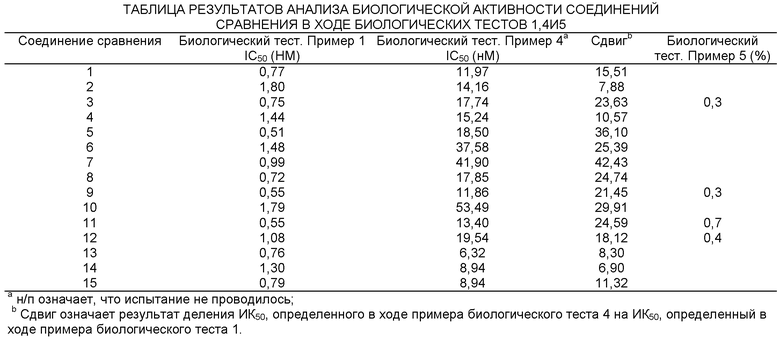
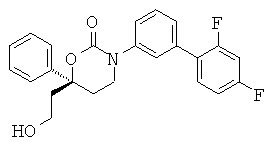
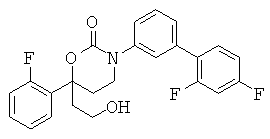
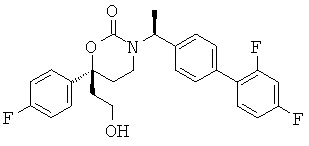
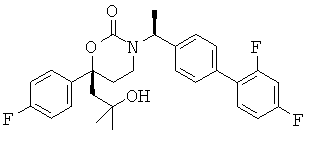
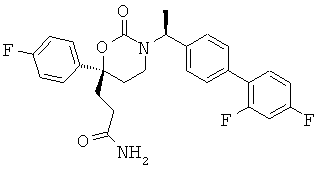
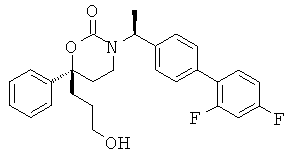
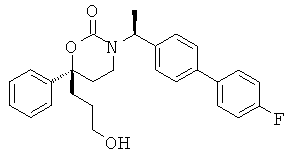
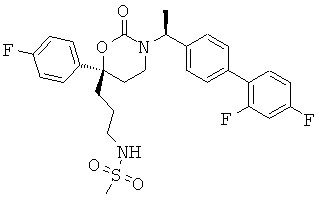
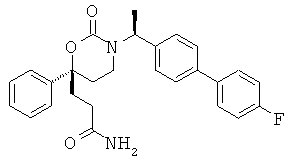
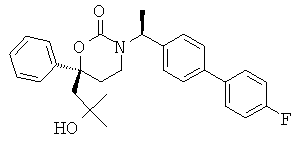
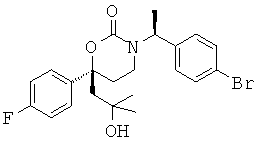
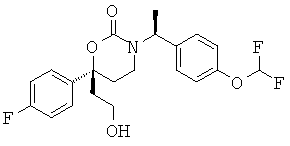
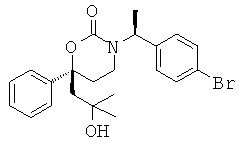
Соединения, представленные в настоящем изобретении, полезны при снижении интенсивности симптомов или при лечении заболеваний или расстройств, при которых снижение уровня содержания кортизола оказывает благоприятное воздействие на течение заболеваня. Таким образом, соединения, представленные в настоящем изобретении, можно использовать при лечении сахарного диабета (напр. диабета II типа), ожирения, симптомов метаболического синдрома, непереносимости глюкозы, гипергликемии, гипертонии, гиперлипидемии, резистентности к инсулину, заболеваний сердечно-сосудистой системы, дислипидемии, атеросклероза, липодистрофии, остеопороза, глаукомы, синдрома Кушинга, болезни Аддисона, висцеральных жировых отложений, связанных с глюкокортикоидной терапией, депрессии, тревожности, болезни Альцгеймера, слабоумия, когнитивного расстройства (в том числе, возрастного), синдрома поликистоза яичника, бесплодия, гипергонадизма. Соединения по настоящему изобретению могут использоваться как лечебные препараты для лечения псевдосиндрома Кушинга, вызванного заболеванием печени при алкоголизме. Помимо этого соединения снижают функции клеток В и Т иммунной системы и, таким образом, могут применяться при лечении туберкулеза, лепры и псориаза. Препарат также можно использовать для ускоренного заживления ран, особенно при диабете.
К иным заболеваниям или расстройствам, относящимся к воздействию 11β-ГСД1, относятся заболевания из группы, включающей липидные расстройства, гипертриглицеридемию, гиперхолестеринемию, низкие уровни содержания HDL-холестерина и высокие уровни содержания LDL-холестерина, сосудистый рестеноз, панкреатит, центральное ожирение, нейродегенеративное заболевание, ретинопатию, нефропатию, нейропатию, диабет, ишемическую болезнь сердца, инсульт, болезнь периферических сосудов, синдром Кушинга, гиперинсулинемию, вирусные заболевания и синдром X. Помимо этого, активность 11β-ГСД1 способна вызвать псевдо-синдром Кушинга, связанный с заболеваниями печени при алкоголизме.
Лекарственный препарат по настоящему изобретению альтернативно или в дополнение к ингибитору 11β-ГСД1 может включать фармацевтически приемлемую соль указанного ингибитора и один или несколько фармацевтически приемлемых носителей. Как вариант, лекарственный препарат может включать непосредственно ингибитор 11β-ГСД1 или его соль в качестве единственного фармацевтически активного вещества в лекарственном препарате. Рассматриваемые ингибиторы 11β-ГСД1 можно использовать в качестве самостоятельного терапевтического средства, либо в сочетании с одним или более дополнительным препаратом при лечении диабета, дислипидемии, сердечно-сосудистой болезни, гипертонии, ожирения, рака или глаукомы.
В составы, рассматриваемые в настоящем изобретении, входят ингибиторы 11β-ГСД1. Указанные составы содержат соединения со средней константой ингибирования (IC50) менее 1,000 нМ по сравнению с 11β-ГСД1; предпочтительно - менее 100 нМ; более предпочтительно - менее 50 нМ; еще более препочтительно - менее 5 нМ; самый предпочтительный вариант - менее 1 нМ.
Настоящее изобретение включает терапевтический способ лечения или улучшения состояния при заболеваниях, опосредованных наличием 11β-ГСД1 у пациентов, нуждающихся в приеме в эффективных количествах ингибитора 1β-ГСД1, либо его энантиомера, диастереомера или фармацевтически приемлемой соли, либо соединения таковых. Понятие «Лечение» далее будет обозначать как профилактическое, так и терапевтическое лечение. Терапевтическое лечение включает сокращение симптомов, ассоциируемых с заболеванием или состоянием и(или) увеличение продолжительности жизни пациента с данным заболеванием или в данном состоянии. Профилактическое лечение обозначает отсрочку в возникновении заболевания или состояния, если имеется риск такого возникновения; либо сокращение вероятности того, что у пациента разовьется такое заболевание или условие, если имеется риск такого возникновения.
Один из вариантов осуществления настоящего изобретения подразумевает прием соединения, ингибирующего 1β-ГСД1, либо состава, в который входит такой ингибитор - при комбинированной терапии с приемом одного или нескольких дополнительных препаратов, для лечения диабета, дислипидемии, сердечно-сосудистого заболевания, гипертонии, ожирения, рака или глаукомы. К числу препаратов для лечения диабета относятся инсулины - такие, как Humulin® (Eli Lilly), Lantus® (Sanofi Aventis), Novolin (Novo Nordisk), и Exubera® (Pfizer); PPAR-гамма агонисты, такие, как Avandia® (розиглитозона малеат, GSK) и Actos® (пиоглитазона гидрохлорид, Takeda/Eli Lilly); сульфонилмочевина, напр. Aмaryl® (глимепирид, Sanofi Aventis), Diabeta® (глибурид, Sanofi Aventis), Micronase®/Glynase® (глибурид, Pfizer), и Glucotrol®/Glucotrol XL® (глипизид, Pfizer); меглитиниды, такие, как Prandin®/NovoNorm® (репаглинид, Novo Nordisk), Starlix® (натеглинид, Novartis), и Glufast® (митиглинид, Takeda); бигуаниды, такие как Glucophase®/Glucophase XR® (метформин HCI, Bristol Myers Squibb) и Glumetza (метформин HCl, Depomed); тиазолидинедионы; аналоги амилина, аналоги GLP-1; ингибиторы DPP-IV; ингибиторы РТВ-1В; ингибиторы протеинкиназы (включая АМР-активированные ингибиторы протеинкиназы); антагонисты глюкагона, бета-ингибиторы киназы глюкоген синтазы-3; ингибиторы глюкозы-6-фосфатазы; ингибиторы глюкоген фосфорилазы; ингибиторы котранспортера натрия глюкозы; ингибиторы альфа-глюкозидазы, такие, как Precose®/Glucobay®/Prandase®/Glucor® (акарбоза, Bayer) и Glyset® (миглитол, Pfizer). Препараты для лечения дислипидемии и сердечно-сосудистых заболеваний включают статины, фибраты и эзетимбе. Препараты для лечения гипертонии включают альфа-блокаторы, бета-блокаторы, блокаторы кальциевых каналов, диуретики, ингибиторы ангиотензин-превращающих ферментов (АПФ), ингибиторы двойного действия, подавляющие АПФ и нейтральную эндопептидазу, блокаторы рецепторов англотендина, ингибиторы синтазы альдостерона, антагонисты рецепторов альдостерона или антагонисты рецепторов эндотелина. Препараты для лечения ожирения включают орлистат, фентермин, сибутрамин и римонабант.
Один из вариантов осуществления настоящего изобретения включает прием соединения, ингибирующего 11β-ГСД1, либо состава, содержащего такое соединение, либо использование комбинированной терапии с приемом одного или нескольких ингибиторов 11β-ГСД1, либо в сочетании с иными продуктами, такими, как Avandamet® (метформин HCl и розиглитазона малеат, GSK); Avandaryl® (глимепирид и розиглитазона малеат, GSK); Metaglip® (глипизид и метформин, Bristol Myers Squibb); и Glucovance® (глибурид и метформин HCl, Bristol Myers Squibb).
Соединения, рассматриваемые в настоящем изобретении, могут быть приготовлены и применяться в широком разнообразии пероральных и парентеральных форм дозировки. Так, соединения, рассматриваемые в настоящем изобретении, могут быть введены путем инъекции - внутривенно, внутримышечно, внутрикожно, подкожно, интрадуоденально, внутрибрюшинно. Помимо этого, соединения, рассматриваемые в настоящем изобретении, могут быть введены через носовую полость или через кожу. Специалистам в данной области техники должно быть очевидно, что указанные ниже формы дозировки могут включать активный компонент, другие компоненты или соответствующую фармацевтически приемлемую соль рассматриваемого соединения.
Для приготовления лекарственных препаратов из компонентов, рассматриваемых в данном изобретении, должны быть использованы фармацевтически приемлемые носители, жидкие или твердые. К препаратам в твердой форме относятся порошки, таблетки, пилюли, капсулы, крахмальные капсулы, суппозитории и диспергируемые гранулы. Твердым носителем может быть вещество, или вещества, которые также могут действовать как разбавители, ароматизаторы, солюбилизаторы, смазывающие вещества, суспендирующие агенты, связующие, консерванты, дезинтеграторы таблеток либо как инкапсулирующий материал. В порошках, носитель представляет собой мелкоизмельченное твердое вещество, смешанное с мелкоизмельченным активным компонентом.
В таблетках, активный компонент смешивается с носителем, что требует добавления связующих в определенных количествах, и компактируется с целью получения желаемой формы и размера.
В состав порошков и таблеток чаще всего входит от 1% до 70% активного компонента. Пригодные для этого носители - карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлоза, натрия карбоксиметил, воск с низкой температурой плавления, масло какао и т.п.Таблетки, порошки, крахмальные капсулы, пастилки, быстрорастворимые полоски, капсулы и пилюли можно использовать как твердые формы дозировки, содержащие активный компонент, пригодный для приема перорально.
Для приготовления суппозиториев, в первую очередь расплавляется воск с низкой температурой плавления, представляющий собой смесь глицеридов жирных кислот или какао-масла, и в него вносится активный компонент; равномерность при внесении обеспечивается, например, за счет непрерывного помешивания. Расплавленная однородная смесь разливается по формам нужного размера, далее ей позволяют остыть и затвердеть.
Препараты в жидкой форме включают в себя растворы, суспензии, удерживающие клизмы и эмульсии - например, водные или водно-пропиленгликолевые растворы. Для парентерального введения жидкие препараты могут быть представлены в виде растворов в составе водного раствора полиэтиленгликоля.
Водные растворы, пригодные для перорального приема, могут быть приготовлены путем растворения активного компонент в воде и добавления пригодных для этого красителей, ароматизаторов, стабилизаторов и загустителей, по желанию. Водные суспензии для перорального приема могут быть приготовлены путем диспергирования активного компонента в воде, содержащей вязкий материал - натуральную или синтетическую камедь, смолы, метилцеллюлозу, натрия карбоксиметилцеллюлозу и другие хорошо известные суспендирующие агенты.
Лекарственный препарат в большинстве случаев представлен в форме дозированных единиц. В такой форме препарат делится на единицы дозировки, содержащие активный компонент в надлежащих количествах. Форма дозированной единицы может быть различной - препарат в упаковке; упаковка, содержащая, например, таблетки, порошки и капсулы во флаконах или ампулах, в дискретных количествах. Форма единицы дозировки также может быть разной - таблетка, крахмальная капсула, обычная капсула, пастилка; либо любое из указанного в необходимых количествах, в упакованном виде.
Содержание активного компонента в препарате, приходящееся на единицу дозировки, может различаться или регулироваться в пределах от 0,1 мг до примерно 1000,0 мг - предпочтительно, от 0,1 мг до 100 мг. Тем не менее дозировки могут различаться в зависимости от требований пациента, тяжести состояния при лечении и используемого состава. Определение нужной дозировки для той или иной ситуации определяется уровнем компетентности специалистов в определенной области техники. Лекарственный препарат, при необходимости, также может включать другие совместимые терапевтические средства.
При терапевтическом лечении, либо в качестве метода использования в роли ингибитора 11β-ГСД1, либо ингибитора при производстве кортизола в клетке, предпочтительнее использовать пероральный прием препарата в твердой дозировочной форме, описанной выше, в объеме примерно 0,1 - 100 мг в сутки; прием производится 1 или более раз в сутки.
Все публикации, патенты и патентные заявки, упомянутые в настоящем документе, включены в него путем ссылки, в такой же степени, как если бы каждая отдельная публикация или патентная заявка была специально и отдельно обозначена как включенная путем ссылки. Описанные примеры и варианты осуществления настоящего изобретения включаются в настоящий документ исключительно в иллюстративных целях, и следует понимать, что возможны модификации и изменения изложенного выше изобретения при сохранении объема и смысла прилагаемой формулы изобретения.
Хотя преимущества настоящего изобретения были частично раскрыты и описаны в данном документе на примере предпочтительных вариантов осуществления, следует понимать, что возможны различные изменения в форме и деталях при сохранении объема изобретения, который приведен в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ 11БЕТА-ГИДРОКСИСТЕРОИД-ДЕГИДРОГЕНАЗЫ 1 | 2009 |
|
RU2531272C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2586974C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА У МЛЕКОПИТАЮЩЕГО | 2000 |
|
RU2263109C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2678305C1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2697090C1 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
Изобретение относится к соединению формулы (Ip1) или (Ip3)или его фармацевтически приемлемой соли, где G1 представляет собой (С1-С4)алкил, (C1-C4)алкокси, (C1-C4)галогеналкил, (С1-С4)галогеналкокси, галоген, циано или нитро; n равен 0; G2a представляет собой (С3-С4)циклоалкил или (С3-С4)циклоалкил(С1-С2)алкил; G2b представляет собой водород; R1 представляет собой метил или этил; R2 представляет собой фенил или фторфенил; и R3 представляет собой 2-гидрокси-2-метилпропил или 2-метил-2-цианопропил. Также изобретение относится к применению соединения формулы (Ip1 и Ip3) для изготовления лекарственного средства или фармацевтической композиции, предназначенных для лечения человека с заболеванием или состоянием, выбранным из сахарного диабета II типа, ожирения, непереносимости глюкозы, гипергликемии, гиперлипидемии, резистентности к инсулину, снижения когнитивных функций и дислипидемии. 3 н. и 2 з.п. ф-лы, 6 табл., 107 пр.
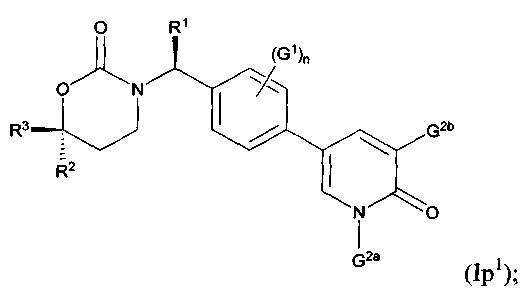
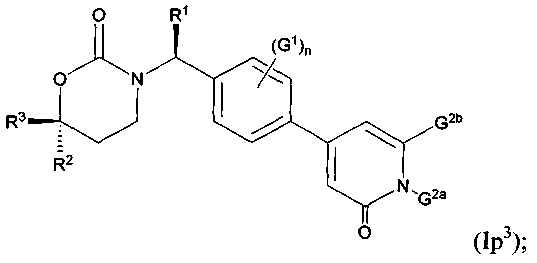
1. Соединение формулы (Ip1) или (Ip3):
или его фармацевтически приемлемая соль;
где
G1 представляет собой (С1-С4)алкил, (C1-C4)алкокси, (C1-C4)галогеналкил, (С1-С4)галогеналкокси, галоген, циано или нитро;
n равен 0;
G2a представляет собой (С3-С4)циклоалкил или (С3-С4)циклоалкил(С1-С2)алкил;
G2b представляет собой водород;
R1 представляет собой метил или этил;
R2 представляет собой фенил или фторфенил; и
R3 представляет собой 2-гидрокси-2-метилпропил или 2-метил-2-цианопропил.
2. Соединение по п.1, которое имеет следующую формулу:
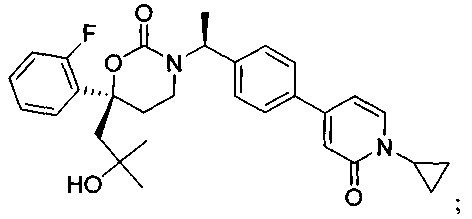
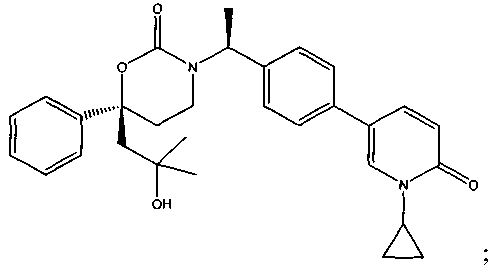 или
или
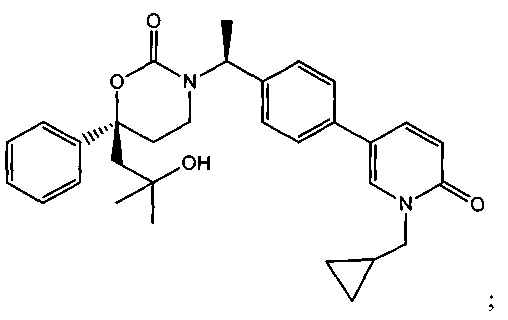
или его фармацевтически приемлемая соль.
3. Соединение по п.1, имеющее формулу:
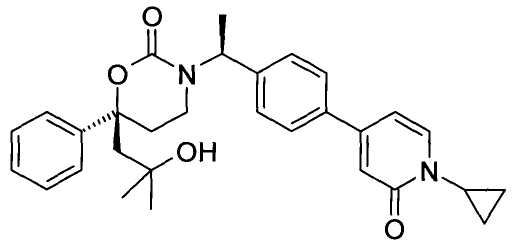
или его фармацевтически приемлемая соль.
4. Фармацевтическая композиция для лечения человека с заболеванием или состоянием, выбранным из сахарного диабета II типа, ожирения, непереносимости глюкозы, гипергликемии, гиперлипидемии, резистентности к инсулину, снижения когнитивных функций и дислипидемии, включающая: i) фармацевтически приемлемый носитель или разбавитель; и ii) соединение по любому из пп.1-3 или его фармацевтически приемлемую соль.
5. Применение соединения по любому из пп.1-3 или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения человека с заболеванием или состоянием, выбранным из сахарного диабета II типа, ожирения, непереносимости глюкозы, гипергликемии, гиперлипидемии, резистентности к инсулину, снижения когнитивных функций и дислипидемии.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| EP 0928789 A, 14.07.1999 | |||
| US 3681349 A, 01.08.1972 | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| ЗАМЕЩЕННЫЕ 1-АМИНОАЛКИЛЛАКТАМЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2241702C2 |

