Настоящее изобретение относится к лечению аутоиммунных заболеваний. Изобретение относится к средству, такому как гуманизированное моноклональное антитело с длительным эффектом, которое при лечении можно вводить пациентам менее часто, чем описано ранее. Средство особенно полезно для пациентов, имеющих заболевания или признаки, которые не могут быть устранены за короткий период времени или требуют неопределенного периода времени лечения для эффективного купирования симптомов. Изобретение предусматривает применения и способы лечения с использованием композиций и лекарственных средств, содержащих такое средство.
Аутоиммунитет означает неспособность организма распознавать свои собственные составные части (ниже субмолекулярных уровней) как «свое», что приводит к иммунной реакции против своих собственных клеток и тканей. Любое заболевание, которое возникает в результате такой аномальной иммунной реакции, называют аутоиммунным заболеванием. Аутоиммунные заболевания включают рассеянный склероз (MS), ревматоидный артрит (RA), псориаз, псориатический артрит, язвенный колит, болезнь Крона, тяжелую псевдопаралитическую миастению (MG), аутоиммунный полигландулярный синдром типа II (APS-II), тиреоидит Хашимото (НТ), диабет типа-1 (T1D), системную красную волчанку (SLE) и аутоиммунный лимфопролиферативный синдром (ALS).
Аутоиммунное заболевание возникает в том случае, когда Т-клетки узнают и взаимодействуют с «собственными» молекулами, то есть молекулами, продуцируемыми клетками хозяина. Активация «аутореактивных» T-клеток в результате презентации аутоантигенов, процессированных антигенпрезентирующими клетками (APC), приводит к их клональной экспансии и миграции в специфичные ткани, где они индуцируют воспаление и разрушение тканей.
В норме T-клетки толерантны по отношению к аутологичной ткани и реагируют только на презентацию гетерологичных структур. Центральная толерантность и периферическая толерантность включают в себя два механизма, посредством которых иммунная система препятствует индукции вредных функций аутореактивных T-клеток. Центральная толерантность опосредована негативной селекцией. Указанный процесс вызывает опосредованную клональной делецией элиминацию аутореактивных T-клеток во время онтогенетического развития в тимусе.
Периферическая толерантность является резервом, действующим в том случае, если центральная толерантность недостаточна и аутореактивные клетки покидают тимус. Такой механизм толерантности работает непрерывно в течение жизни, удерживая аутореактивные клетки под контролем посредством иммунологического игнорирования (анергии), периферической делеции и/или активной супрессии.
Регуляторные T-клетки (T-рег, ранее также называемые «супрессорными клетками») в качестве части активной супрессии поддерживают периферическую толерантность и регулируют аутоиммунитет (Suri-Payer et al., J Immunol. 157: 1799-1805 (1996); Asano et al., J Exp. Med. 184:387-396 (1996); Bonomo et al., J. Immunol. 154: 6602-6611 (1995); Willerford et al., Immunity 3: 521-530 (1995); Takahashi et al., Int. Immunol. 10: 1969-1980 (1998); Salomon et al., Immunity 12: 431-440 (2000); Read et al., J Exp. Med. 192: 295-302 (2000). В общем, регуляторные T-клетки ингибируют активацию и/или функцию эффекторных клеток T-хелперов типа 1 (TH1) и TH2. Дисрегуляция частоты или функционирования клеток T-рег может приводить к инвалидизирующим аутоиммунным заболеваниям (Baecher-Allan et al., Immunol. Review 212: 203-216 (2006); Shevach, Annu. Rev. Immunol. 18: 423-449 (2000); Salomon et al., Immunity 12: 431-440 (2000); Sakaguchi et al., Immunol. Rev. 182: 18-32 (2001)).
Охарактеризовано несколько подгрупп регуляторных T-клеток. Семейство T-рег состоит из двух ключевых подгрупп: естественно появляющиеся клетки, например, T-рег CD4+CD25+, и периферически индуцируемые, T-рег Tr1 и Th3. Кроме того, описаны NK-T-рег и CD8+-T-рег у человека и грызунов (Fehervari et al., J. Clin. Investigation 114: 1209-1217 (2004)).
Полученные из тимуса клетки T-рег (естественные CD4+CD25+ T-рег) являются основными регуляторными клетками, вовлеченными в регуляцию аутоиммунитета или патогенных иммунных реакций.
i) они представляют собой T-клетки CD4+ и составляют 5-10% периферических T-клеток CD4+;
ii) они созревают в тимусе;
iii) они обычно характеризуются совместной экспрессией рецептора IL-2 (CD25), низкомолекулярной изоформы молекулы CD45, CD152 (CTLA-4) и фактора транскрипции FoxP3.
Роль T-рег лучше всего проиллюстрирована в экспериментах, которые заключаются в восстановлении иммунодефицитных мышей nude с использованием клеток CD4+, которые были истощены по клеткам CD25+. У восстановленных мышей CD4+CD25- развивались различные органоспецифичные аутоиммунные заболевания, такие как гастрит, оофорит, орхит и тиреоидит (Suri-Payer et al., J. Immunol. 160: 1212-1218 (1998)).
Включение подгруппы CD4+CD25+ в эксперименты по восстановлению мышей nude предотвращает появление таких заболеваний (Sakaguchi et al., J. Immunol. 155: 1151-1164 (1995)). Защитное значение клеток CD4+CD25+ против органоспецифичного аутоиммунитета также показано на нескольких других моделях аутоиммунитета (например, аутоиммунный гастрит, простатит, оофорит, гломерулонефрит, эпидидимит и тиреоидит), вызванного неонатальной тимэктомией, осуществляемой через 3 дня после рождения (d3Tx), или воспалительного заболевания кишечника, вызванного путем разведения мышей SCID с использованием клеток T-клеток CD45RBhigh, CD4+CD25-. Введение анти-CD25-антитела in vivo у мышей также индуцирует органоспецифичное локализованное аутоиммунное заболевание.
Открытие важного значения транскрипционного регулятора FoxP3 в функционировании регуляторных T-клеток CD4+D25+ мышей и предыдущие наблюдения того, что пациенты с синдромом IPEX (X-сцепленный синдром иммунной дисрегуляции с полиэндокринопатией и энтеропатией), тяжелым воспалительным заболеванием, сходным с заболеванием, наблюдаемым у мышей с дефицитом регуляторных клеток CD4+CD25+ (синдром scurfy), имеют мутации в FoxP3, показали прямую корреляцию между аутоиммунной моделью на животных, мышиными регуляторными T-клетками и аутоиммунным заболеванием человека (Sakaguchi et al., J. Immunol. 155: 1151-1164 (1995)).
Фармацевтический механизм регуляторных T-клеток не полностью выяснен. T-рег CD4+CD25+ ингибируют активацию поликлональных и антигенспецифичных T-клеток. Супрессия опосредована зависимым от клеточных контактов механизмом, который требует активации T-рег CD4+CD25+ посредством TCR, но T-рег не дают пролиферативного ответа при активации TCR или стимуляции митогенными антителами (анергические) (Shevach, Nature Rev. Immunol. 2: 389 (2002). После стимуляции они становятся компетентными в отношении супрессии независимым от антигенов образом ответа T-клеток CD4+ и T-клеток CD8+, а также ингибирования активации B-клеток и их клональной экспансии.
Имеются дополнительные данные, свидетельствующие о том, что супрессорная активность T-рег CD4+CD25+ отчасти также основана на противовоспалительных цитокинах, подобных TGF-β (Kingsley et al., J. Immunol. 168: 1080 (2002); Nakamura et al., J. Exp. Med. 194: 629-644 (2001)). Функциональная значимость секреции TGF-β дополнительно подтверждена данными о том, что у мышей, дефицитных по TGF-β, развивается аутоиммунное заболевание и что введение нейтрализующих антител к TGF-β отменяет in vivo предотвращение аутоиммунитета или индуцирующей толерантность активности T-клеток CD4+ в некоторых моделях.
В подгруппе T-клеток CD4+ могут существовать по меньшей мере 2 различающихся типа клеток с супрессорной функцией, которые индуцируются после воздействия специфичного экзогенного антигена (называемые «адаптивными или индуцируемыми регуляторными T-клетками»): регуляторные T-клетки типа 1 (Tr1) клетки и клетки Th3. Указанные типы клеток, по-видимому, можно отличить от T-рег CD4+CD25+ на основании профилей продукции цитокинов. Однако взаимосвязь между указанными разными типами неясна и способы их действия перекрываются.
Клетки Tr1 индуцировали многократной стимуляцией TCR в присутствии IL-10, и было показано, что в основном осуществляют понижающую регуляцию иммунных реакций посредством продукции высоких уровней IL-10 вместе с умеренными количествами TGF-β (Chen et al., J. Immunol. 171: 733-744 (2003)).
Клетки Th3 (идентифицированные в модели EAE после пероральной доставки антигена) продуцируют высокие количества TGF-β и различные количества IL-4 и IL-10. Показано, что IL-4, сам по себе, является ключевым фактором дифференцировки клеток Th3, в отличие от клеток Tr1, которые дифференцируются IL-10 (Chen et al., Science 265: 1237-1240 (1994)).
Супрессия функции T-клеток в результате применения иммунодепрессивных лекарственных средств является основной терапевтической методикой, которую успешно применяли для лечения аутоиммунных заболеваний. Однако такие лекарственные средства индуцируют общую иммуносупрессию вследствие их плохой избирательности, что приводит к ингибированию не только опасных функций иммунной системы, но также и полезных функций. Как следствие могут возникать некоторые риски, подобные инфекции, злокачественной опухоли и лекарственной токсичности.
Средства, создающие помехи для функционирования T-клеток, являются терапевтической основой различных аутоиммунных заболеваний.
Способ применения средств с целью активации регуляторных T-клеток для терапии аутоиммунных заболеваний, как было показано до настоящего времени, чрезвычайно сложен. Активация T-рег посредством TCR с использованием агонистического анти-CD3-антитела OKT-3 (Abramowicz et al, N Engl. J Med. 1992 Sep 3;327(10):736) или посредством костимулирующей молекулы CD28 с использованием суперагонистического анти-CD28-антитела TGN 1412 приводит к полному истощению популяции регуляторных T-клеток, а также других подходящих T-клеток и системной индукции и высвобождению избыточных количеств провоспалительных цитокинов, включая IFN-γ, TNF-α, IL-1 и IL-2, приводя к клинически выраженному синдрому выброса цитокинов (CRS) у человека (Suntharalingam et al., N. Engl. J. Med. 2006 Sep 7; 355(10): 1018-28).
После первых двух-трех инъекций 5 мг моноклонального антитела OKT3 у большинства пациентов развивается синдром выброса цитокинов с высокими уровнями фактора некроза опухолей-альфа, интерлейкина-2 и гамма-интерферона, появляющихся в пределах 1-2 часов в кровообращении у реципиентов почечных трансплантатов. (Abramowicz et al., Transplantation, 1989 Apr; 47(4): 606-8). Это приводит к узкому терапевтическому окну, которое ограничивает применимость указанного антитела для лечения аутоиммунного заболевания.
Лечение суммарной дозой 5-10 мг TGN1412 (0,1 мг анти-CD28 на килограмм массы тела) приводит к системной воспалительной реакции с полиорганной недостаточностью в течение 90 минут после получения однократной внутривенной дозы TGN 1412 (Suntharalingam et al., N. Engl. J. Med. 2006 Sep 7; 355(10): 1018-28).
В общем, установлено, что T-клетки CD4 играют главную роль в инициации и поддержании аутоиммунитета. Соответственно было предложено применение мАт против поверхностных молекул T-клеток CD4 и, в частности анти-CD4-мАт, в качестве иммунодепрессантов. Хотя многочисленные клинические исследования подтвердили возможную пользу такого способа, в таких исследованиях также выявлено несколько проблем, связанных с получением анти-CD4-мАт, более подходящих для применения в обычной клинической практике.
Было предложено несколько разных механизмов действия CD4-мАт, включая: (1) антагонизм взаимодействий CD4-MHC II, приводящий к ингибированию активации T-клеток, (2) модулирование рецепторов CD4, которое определяют по снижению экспрессии CD4 на клеточной поверхности, (3) частичную передачу сигнала через рецептор CD4 в отсутствие перекрестного связывания T-клеточных рецепторов, которая может супрессировать последующую активацию T-клеток и запускать апоптозную гибель T-клеток CD4, (4) Fc-опосредованную зависимую от комплемента цитотоксичность (CDC) или зависимую от антител клеточную цитотоксичность (ADCC), приводящую к истощению T-клеток CD4, и (5) стимуляцию регуляторных T-клеток.
Fc-опосредованная зависимая от комплемента цитотоксичность (CDC) или зависимая от антител клеточная цитотоксичность (ADCC), приводящая к истощению T-клеток CD4, является основным наблюдаемым механизмом и, в частности, показана для антител подкласса IgG1. Только несколько CD4-антител были отнесены к другим механизмам, подобные TRX-1, TNX-355, IDEC-151, OKTcdr4A, при этом только TRX-1 является IgG1 (Schulze-Koops et al., J. Rheumatol. 25(11): 2065-76 (1998); Mason et al., J. Rheumatol. 29(2): 220-9 (2002); Choy et al., Rheumatology 39(10): 1139-46 (2000); Herzyk et al., Infect Immun. 69(2): 1032-43 (2001); Kon et al., Eur Respir J. 18(1): 45-52 (2001); Mourad et al., Transplantation 65(5): 632-41 (1998); Skov et al., Arch Dermatol. 139(11): 1433-9 (2003); Jabado et al., J. Immunol. 158(1): 94-103 (1997)).
Зависимое от дозы истощение T-клеток CD4+ при «высоких» дозах (несколько циклов с использованием доз >100 мг) и кратковременную секвестрацию (краткосрочное истощение) при «более низких» дозах (несколько циклов с использованием доз >10 мг) наблюдают при использовании нескольких CD4-антител (Mason et al., J. Rheumatol. 29 (2): 220-229 (2002); Kon et al., Eur. Respir J. 18(l):45-52 (2001)) и HuMax-CD4 (Skov et al., Arch Dermatol. 139(11): 1433-1439 (2003), Choy et al., Rheumatology 41 (10): 1142-8 (2002)). Несмотря на свою активность в истощении популяций клеток, мАт к CD4 не способны обеспечить клиническую пользу и соответствующую эффективность при исследованных аутоиммунных заболеваниях, например ревматоидном артрите (Strand et al., Nature Reviews 6: 75-92 (2007)). Кроме того, обычно считают, что истощение T-клеток CD4+ представляет собой сценарий, который может вызывать тяжелую иммуносупрессию.
Антитело B-F5 (мышиный IgG1 против CD4 человека) испытывали при разных аутоиммунных заболеваниях.
Небольшое количество пациентов с тяжелым псориазом лечили мышиным B-F5-антителом, и были описаны некоторые положительные эффекты (Robinet et al., Eur. J. Dermatol. 1996: 6: 141-6, и Robinet et al., J. Am. Acad. Dermatol. 1997; 36: 582-8).
У пациентов с ревматоидным артритом наблюдаемые результаты в плацебоконтролируемом испытании с использованием ежедневной дозы B-F5 не показали значимого улучшения (Wendling et al. J. Rheumatol.; 25(8): 1457-61, 1998).
У пациентов с рассеянным склерозом (MS) наблюдали некоторые позитивные эффекты после 10-дневного лечения у пациентов с рецидивирующими-ремитирующими формами, некоторые из которых оставались без рецидивов на 6 месяцев после терапии (Racadot et al., J. Autoimmun., 6(6): 771-86, 1993). Сходные эффекты наблюдали Rumbach с соавторами (Mutt. Scler.; 1(4): 207-12, 1996).
При тяжелом заболевании Крона не наблюдали значимого улучшения у пациентов, получающих B-F5 в течение 7 последовательных дней (Canva-Delcambre et al., Aliment Pharmacol. Ther. 10(5): 721-7, 1996).
В случае профилактики отторжения аллотрансплантата сообщалось, что биодоступность B-F5 была недостаточной, чтобы обеспечить его применение для профилактики отторжения аллотрансплантата (Dantal et al. Transplantation, 27; 62(10): 1502-6, 1996).
На основании вышесказанного очевидно, что первой проблемой, которую необходимо решить, является потребность в применении более длительных доз мАт, чтобы получить клиническое улучшение. В общем, длительное применение модифицирующих заболевание терапевтических средств приводит к увеличению числа неблагоприятных событий или утрате клинических эффектов, что делает необходимым увеличение дозы («нарастание дозы») для поддержания ответа (Strand et al., Nature Reviews Drug Discovery 6: 75-92, (2007)) вследствие иммуногенности. Чем меньше и/или менее часто нужно вводить модифицирующие заболевание терапевтические средства, тем ниже будут такие неблагоприятные эффекты, плюс будет иметь место немедленный полезный эффект для пациентов, которым требуются более низкие и/или меньшее количество доз терапевтических средств.
Другой недостаток терапии моноклональными антителами у человека заключается в том, что такие антитела обычно получают из клеток мышей, и они вызывают реакции против Ig мышей у людей-реципиентов. Это приводит не только к более низкой эффективности данного лечения и еще более низкой эффективности любого лечения мышиными моноклональными антителами в будущем, но также и к повышенному риску анафилаксии.
В принципе такого недостатка можно избежать благодаря применению гуманизированных антител, получаемых в результате прививки определяющих комплементарность областей (CDR) мышиного моноклонального антитела, которые определяют специфичность связывания антигена, в каркасные области (FR) молекулы иммуноглобулина человека. Целью гуманизации является получение рекомбинантного антитела, обладающего такими же антигенсвязывающими свойствами, как и мышиное моноклональное антитело, из которого получены области CDR, и намного менее иммуногенного у человека.
В некоторых случаях замена областями CDR из мышиного антитела CDR человека в каркасах человека достаточна для переноса антигенсвязывающих свойств (включая не только специфичность, но также и аффинность по отношению к антигену). Однако во многих антителах некоторые остатки FR важны для связывания антигена, поскольку они непосредственно контактируют с антигеном в комплексе антитело-антиген или поскольку они влияют на конформацию CDR и, следовательно, их эффективность в связывании антигена.
Таким образом, в большинстве случаев также необходимо заменять одним или несколькими остатками каркаса из мышиного антитела соответствующие остатки FR человека. Так как количество заменяемых остатков должно быть небольшим, насколько это возможно, чтобы предотвратить реакции против Ig мыши, то проблема состоит в том, чтобы определить, какой аминокислотный остаток (остатки) важен для сохранения антигенсвязывающих свойств. Были предложены различные способы прогнозирования более подходящих сайтов для замены. Хотя такие способы предлагают общие принципы, которые могут оказать некоторую помощь на первых этапах гуманизации, конечный результат варьирует от одного антитела к другому. Таким образом, для данного антитела очень трудно предсказать, какие замены будут обеспечивать требуемый результат.
Ранее были предприняты попытки гуманизации мышиного B-F5, и был достигнут успех в получении гуманизированного B-F5 (далее называемого hB-F5), обладающего свойствами связывания CD4, сходными со свойствами исходного мышиного B-F5.
Таким образом, в заявке WO 2004/083247 указано, что было обнаружено, что гуманизированное антитело BT061 (гуманизированное B-F5 или просто hB-F5) применимо для лечения аутоиммунных заболеваний, таких как псориаз и ревматоидный артрит. Указанное антитело представляет собой антитело, которое способно активировать регуляторные T-клетки CD4+CD25+. В заявке на выдачу патента раскрыты композиции для парентерального введения, приготовленные так, чтобы обеспечить введение дозы 0,1-10 мг, предпочтительно 1-5 мг. Предполагаемые схемы дозирования представляют собой внутривенное введение дозы 1 мг в сутки и дозы 5 мг через день пациентам с ревматоидным артритом в течение периода времени 10 дней.
Исследование также описано Wijdenes с соавторами в реферате и стендовом докладе, представленном на конференции EULAR в июне 2005. Описано лечение 11 пациентов, страдающих ревматоидным артритом. Пациентов лечили с использованием 5 внутривенных инфузий по 5 мг BT061 через день с сопутствующим лечением 150 мг диклофенака.
Антитело, описанное в настоящем исследовании, не раскрыто как подходящее для применения в более высоких дозах или более длительных дозах, и все еще требуется найти средства лечения с использованием более длительно действующих доз, так чтобы лечить большее число пациентов.
Регуляторные T-клетки CD4+CD25+ способны поддерживать периферическую толерантность и регулировать аутоиммунитет, ингибируя пролиферацию и продукцию цитокинов антигенспецифичных T-клеток CD4+ и CD8+.
Предыдущие исследования на мышах показали, что мышиные T-клетки CD4+CD25+ супрессируют как пролиферацию, так и продукцию IFN-γ T-клетками CD8+, индуцированными либо поликлональными, либо Аг-специфичными стимулами. Кроме того, T-клетки CD4+CD25+ ингибируют активацию клеток-респондеров CD8+ как посредством ингибирования продукции IL-2, так и посредством повышающей регуляции экспрессии IL-2Rα-цепи (CD25) (Piccirillo et al., J. Immunol. 167: 1137-1140 (2001)). В отличие от регуляторных T-клеток обычные T-клетки экспрессируют CD25 только при активации иммуногенным стимулом.
У человека T-клетки CD4+CD25+, предварительно активированные анти-CD3- или анти-CD28-антителом, влияют на T-клетки CD8+, приводя к пониженной пролиферации в ответ на поликлональную и аллогенную стимуляцию и к пониженной продукции IFN-γ. Супрессированные T-клетки CD8+ сохраняют свой анергический фенотип, несмотря на многократные повторные стимуляции родственным антигеном и IL-2 в течение периода времени, составляющего до 3 недель (Camara et al., Eur. J. Immunol. 33: 3473-3483 (2003), Dieckman et al., Immunology. 115(3): 305-14 (2005)).
Учитывая описанный выше уровень техники, целью настоящего изобретения является лечение пациентов, имеющих аутоиммунные заболевания, меньшим количеством и/или более низкими дозами. В частности, целью настоящего изобретения является поиск средств лечения, которые можно применять на пациентах менее часто в ходе длительной терапии, чтобы минимизировать количество инъекций, необходимых для получения клинической пользы.
Удивительно, что авторы изобретении наблюдали, что BT061 специфично стимулирует T-рег CD4+CD25+, которые подавляют пролиферацию T-клеток CD8+ посредством ингибирования продукции IL-2 и IFN-γ аллореактивными T-клетками CD8+. Кроме того, предварительно BT061-активированные T-рег CD4+CD25+ делают супрессированные T-клетки CD8+ неспособными экспрессировать CD25 при рестимуляции вплоть до 10 дней, впервые обеспечивая возможность долговременного терапевтического эффекта при лечении пациентов таким антителом, так как экспрессия CD25 является одним из признаков активации таких T-клеток CD8+. Следовательно, неспособность экспрессировать CD25 приводит к уменьшению нежелательной активации иммунной системы.
Соответственно, изобретение относится к фармацевтической композиции для лечения аутоиммунного заболевания, содержащей фармацевтически приемлемый носитель и средство, способное активировать регуляторные T-клетки CD4+CD25+, при этом композицию вводят субъекту не чаще, чем каждые 3 дня.
Предпочтительно период между введениями составляет по меньшей мере 4 недели, по меньшей мере 12 недель, по меньшей мере двадцать четыре недели, по меньшей мере 6 календарных месяцев или по меньшей мере год. В одном варианте период лечения составляет более чем один год, и композицию вводят ежегодно в течение периода времени, составляющего 1, 2, 3, 4 или 5 лет.
В частности, композицию необходимо вводить не больше чем каждые 6 дней, предпочтительно не больше чем каждые 10 дней. Обычно лекарственное средство можно вводить субъекту каждые 3-31 день, более предпочтительно каждые 3-10 дней или каждые 3-6 дней.
Долговременный характер терапии особым образом не ограничен при условии, что терапия продолжается не дольше, чем период времени, необходимый для немедленного/острого лечения данного пациента. Однако предусмотрены периоды времени, составляющие по меньшей мере 6 месяцев, 1 год, 18 месяцев, 2 года и 5 лет, а также неопределенные периоды (например, в течение жизни пациента, или вплоть до исцеления пациента, или до тех пор, пока симптомы больше не будут проявляться).
При лечении пациентов средством, способным активировать регуляторные T-клетки CD4+CD25+, может быть продемонстрирован как немедленный эффект, так и долговременный терапевтический эффект. Не имея намерения быть связанными с какой-либо теорией, предполагают, что это является следствием того факта, что средство стимулирует T-рег CD4+CD25+, которые не только непосредственно ингибируют T-клетки CD8+, но также делают T-клетки CD8+ не способными экспрессировать CD25 при рестимуляции. Читается, что долговременные эффекты дополнительно опосредованы сочетанием эффективности при низких уровнях и медленным снижением эффективной концентрации антитела, получаемого в клинической ситуации, приводящим к более длительным периодам наличия эффективного уровня средства.
Из приведенных выше схем будет понятно, что авторы изобретения неожиданно обнаружили, что гуманизированное антитело BT061 (гуманизированное B-F5 или просто hB-F5) по существу не модулирует и не индуцирует высвобождение провоспалительных цитокинов по сравнению с другими взаимодействующими с T-клетками антителами (например, анти-CD3-антителами), а стимулирует T-рег CD4+CD25+, которые непосредственно ингибируют T-клетки CD8+ плюс также делают T-клетки CD8+ не способными экспрессировать CD25 при рестимуляции. На основании проведенного авторами изобретения исследования дозы могут быть введены с большими интервалами и в течение намного более длительного периода, чем описано ранее в WO 2004/083247.
Концентрация средства особым образом не ограничена, при условии что оно присутствует в концентрации, которая является подходящей для пациента. Однако в случае высоких доз предпочтительно концентрация средства составляет от 10 до 150 мг/мл, от 15 до 150 мг/мл, от 15 до 100 мг/мл, от 15 (или больше, чем 10) до 75 мг/мл или от 20 до 60 мг/мл. Наиболее предпочтительно концентрация средства имеет (примерно) любое одно из следующих значений: 10 мг/мл, 12,5 мг/мл, 20 мг/мл, 25 мг/мл, 50 мг/мл, 60 мг/мл, 70 мг/мл, 80 мг/мл, 90 мг/мл или 100 мг/мл.
В альтернативных предпочтительных вариантах в случае более низких доз концентрация средства составляет от 0,1 мкг/мл до 30 мг/мл или от 0,1 до 1000 мкг/мл и более предпочтительно 1-500 мкг/мл и 2-250 мкг/мл. Наиболее предпочтительно концентрация средства имеет (примерно) любое одно из следующих значений 15 мкг/мл, 25 мкг/мл, 125 мкг/мл, 250 мкг/мл или 500 мкг/мл, 1 мг/мл, 12,5 мг/мл или 25 мг/мл. Изобретение также относится к применению средства, которое определено в настоящем описании, для производства лекарственного средства для лечения аутоиммунного заболевания, при этом средство необходимо вводить субъекту по схеме дозирования, описанной в настоящей публикации. Кроме того, изобретение относится к средству, которое определено в настоящем описании, для применения при лечении аутоиммунного заболевания, при этом средство необходимо вводить субъекту по схеме дозирования, описанной в настоящей публикации.
Объем дозы, применяемый для субъекта при использовании композиции, особым образом не ограничен при условии, что ее доставляют в течение длительного периода времени по сравнению с уже известными схемами, и, следовательно, она подходит для лечения людей, которым может быть полезна длительная терапия, таким как, без ограничения, пациенты с тяжелыми случаями с длительной историей болезни и неудовлетворительной реакцией на современную терапию. Концентрация средства в определенных объемах доз может варьировать, чтобы обеспечить требуемые дозы, которые описаны в настоящей заявке.
Объем дозы будет варьировать в зависимости от способа введения. Предпочтительно парентеральное введение. Примерами парентерального введения являются внутримышечное введение, внутривенное введение или подкожное введение. В том случае, когда композицию необходимо ввести посредством внутривенной инфузии, объем дозы может составлять от 0,1 или 0,5 мл до 500 мл, предпочтительно от 15 до 25 мл и обычно около 20 мл. В том случае, когда композицию необходимо ввести путем подкожной или внутримышечной инъекции, объем дозы может составлять от 0,1 до 3 мл, предпочтительно от 0,5 до 1,5 мл и, обычно, около 1 мл.
Однако в некоторых вариантах композиция может быть представлена в концентрированной форме и разбавлена в такой степени, которая необходима для пациентов, подвергаемых лечению. Предпочтительно, в таких ситуациях композиция представлена в сравнительно небольших объемах около 1, 2, 3, 4 или 5 мл. В альтернативных вариантах композиция представлена в требуемой концентрации и объеме дозы, описанной выше (т.е., в готовой для введения форме). В одном конкретном варианте фармацевтические композиции для подкожного введения приготовлены в готовой для введения форме, которая не требует разбавления, так что их без труда может ввести немедицинский персонал.
Как указано ранее, не было известно, что средства, способные лечить аутоиммунные заболевания, можно вводить в ходе длительной терапии с низкой частотой, как предполагается в настоящем изобретении. Хотя известные дозы средств, способных лечить аутоиммунное заболевание, эффективны у некоторых людей или при определенных типах заболевания, реализация того, что они могут быть эффективны в течение более длительных периодов времени и переносимы в течение более длительных периодов, открыла путь для более эффективного лечения некоторых аутоиммунных заболеваний и классов пациентов.
Изобретение будет проиллюстрировано только в виде примера со ссылкой начертежи, на которых показано следующее:
На фигуре 1 показано влияние BT061 на синтез цитокинов в культуре цельной крови, полученной от 3 здоровых доноров. Культуры стимулировали в нескольких экспериментах 4 разными типами активаторов: CD3 = анти-CD3-антитела; LPS = липополисахарид; ФГА = фитогемагглютинин + анти-CD28-антитела; SEB = стафилококковый энтеротоксин B + анти-CD28-антитела. Определяли разные цитокины, чтобы измерить влияние на разные субпопуляции лейкоцитов: T-рег-клетки (CD3: TGF-β, IL-10); моноциты/макрофаги (LPS: IL-10, TNFα, IL-1β); Th2-клетки (PHA: IL-4, IL-5, IL-13); Th1-клетки (SEB: IL-2, IFNγ).
На фигуре 2 показано влияние BT061 в культурах цельной крови человека (доноры с ревматоидным артритом) на синтез цитокинов, запускаемый разными стимулами.
На фигуре 3 показана нуклеотидная последовательность, кодирующая VH-область мышиного B-F5 (SEQ ID NO: 5).
На фигуре 4 показана нуклеотидная последовательность, кодирующая Vκ-область мышиного B-F5 (SEQ ID NO: 6).
На фигуре 5 показана нуклеотидная последовательность (SEQ ID NO: 3) фрагмента плазмиды, кодирующего VH-область гуманизированного BF-5. Последовательность, кодирующая V-область, подчеркнута, и соответствующая полипептидная последовательность (SEQ ID NO: 17) показана под нуклеотидной последовательностью.
На фигуре 6 показана нуклеотидная последовательность (SEQ ID NO: 4) фрагмента плазмиды, кодирующей VK-области гуманизированного BF-5. Последовательность, кодирующая V-область, подчеркнута, и соответствующая полипептидная последовательность (SEQ ID NO: 2) показана под нуклеотидной последовательностью.
На фигуре 7 показано, что активированные BT061 (анти-CD4-мАт) клетки T-рег CD25+ подавляют синтез цитокинов и реэкспрессию CD25 T-клеток CD8+. Вверху: CD4-активированные T-рег CD25+ подавляют синтез цитокинов коактивированных T-клеток CD8+. Внизу: CD4- активированные T-рег. CD25+ подавляют реэкспрессию CD25 T-клеток CD8+. На диаграмме показаны только пропускаемые при сортировке T-клетки CD8+. Показан типичный результат из трех. Числами указано процентное содержание позитивных клеток.
На фигурах 8A и 8B соответственно показано высвобождение TFNα и IL-6, наблюдаемое в клиническом испытании BT061 (однократная внутривенная инфузия или подкожная инъекция) у здоровых добровольцев по сравнению с уровнями, сообщенными для случая использования моноклональных анти-CD3-антител. На фигурах представлены уровни доз и время для восстановления. Результаты для TRX4, обозначенные на фигурах в виде «2)», описаны в Keymeulen et al., 2005 N. Engl. J. Med. Пациенты с диабетом типа 1. Результаты для теплизумаба, обозначенные на фигурах в виде «3)», описаны в Herold et al., 2002 N. Engl. J. Med. Пациенты с диабетом типа I. Нормальные значения, указанные на фигурах в виде «4)», описаны в Straub et al., 2007, Athr. & Rheumat. «*)» означает однократную дозу, «**)» означает суммарную дозу, инъецированную вплоть до достижения пиковой концентрации.
На фигуре 9 показаны уровни в плазме IL-2 и IFN-γ после введения однократной внутривенной или подкожной дозы BT061 у здоровых добровольцев. ULN = верхняя граница нормы; LLN = нижняя граница нормы.
На фигуре 10 показана кинетика подсчета клеток CD4 (клеток в миллилитре плазмы) у добровольцев, которых лечили с использованием однократной внутривенной дозы BT061. Показаны средние значения для 3 пациентов на группу дозирования. Пунктирные линии показывают верхнюю границу нормы (ULN) и нижнюю границу нормы (LLN).
На фигуре 11 показана кинетика подсчета клеток CD4 (клеток в миллилитре плазмы) у добровольцев, которых лечили с использованием однократной подкожной дозы BT061. Показаны средние значения для 3 пациентов на группу дозирования. Пунктирные линии показывают верхнюю границу нормы (ULN) и нижнюю границу нормы (LLN).
На фигуре 12, части A-H, представлены графики, показывающие данные клинических испытаний на пациентах с псориазом группы дозирования I, которые описаны в примере 8, в которых пациентов лечили с использованием внутривенной инъекции 0,5 мг BT061 или плацебо. Части A-H фигуры 8 представляют собой графики оценки индекса охвата и тяжести псориаза (PASI) у пациентов 1-8 группы дозирования I, соответственно.
На фигуре 13, части A-H, представлены графики, показывающие данные клинических испытаний на пациентах с псориазом группы дозирования II, которые описаны в примере 8, в которых пациентов лечили с использованием внутривенной инъекции 2,5 мг BT061 или плацебо. Части A-H фигуры 9 представляют собой графики оценки PASI у пациентов 1-8 группы дозирования II, соответственно.
На фигуре 14, части A и B, представлены фотографии, сделанные при клинических испытаниях на пациентах с псориазом, которые описаны в примере 8. Фотографии одного и того же пациента, который был представителем группы дозирования II. Фотография, показанная в части A, была сделана до лечения. Фотография, показанная в части B, была сделана через 28 дней после лечения.
На фигуре 15 представлены результаты клинического испытания на пациентах с ревматоидным артритом, которое описано в примере 9. На фигуре показана гистограмма процента пациентов из групп дозирования, подкожно получавших 1,25 мг, 6,25 мг, 12,5 мг и 25 мг BT061, достигающих, по меньшей мере, ответа ACR20. Шесть пациентов в каждой группе получали дозу антитела, а два пациента получали плацебо.
На фигуре 16A и 16B представлены результаты клинического испытания на пациентах с ревматоидным артритом, которое описано в примере 9. На фигуре 16A показана гистограмма количества болезненных суставов у пациентов из группы дозирования, получавшей 25 мг BT061 подкожно. На фигуре 16B показана гистограмма количества распухших суставов у пациентов из той же самой группы дозирования. Шесть пациентов в каждой группе получали дозу антитела, а два пациента получали плацебо.
На фигуре 17A и 17B представлены результаты клинического испытания на пациентах с ревматоидным артритом, которое описано в примере 9. На фигурах показаны изменения индивидуальных параметров (в %) для одного отвечающего пациента (фигура 17A) и одного не отвечающего пациента (фигура 17B) из группы, получавшей дозу 25 мг подкожно. На фигурах обозначения «ГО пациента» и «ГО врача» относятся к глобальной оценке пациентом и глобальной оценке врачом соответственно. Термин «ОП боли» относится к оценке боли пациентом.
На фигуре 18A и 18B представлены дополнительные результаты клинического испытания на пациентах с ревматоидным артритом, которое описано в примере 9. На фигурах показано количество болезненных суставов у пациентов из группы, получавшей дозу 1,25 мг подкожно (фигура 18A), и из группы, получавшей дозу 6,25 мг подкожно (фигура 18B).
На фигуре 19A и 19B представлены дополнительные результаты клинического испытания на пациентах с ревматоидным артритом, которое описано в примере 9. На фигурах показано количество болезненных суставов у пациентов из группы, получавшей подкожную дозу 50 мг (фигура 19A), и из группы, получавшей внутривенную дозу 6,25 мг (фигура 19B).
На фигуре 20 показано выравнивание полипептидных последовательностей VK мышиного B-F5 (SEQ ID NO: 8), FK-001 (SEQ ID NO: 9, 10, 11 и 12), L4L (SEQ ID NO: 18) и L4M (SEQ ID NO: 2) при конструировании гуманизированной формы B-F5 (т.е. BT061).
На фигуре 21 показано выравнивание полипептидных последовательностей VH мышиного B-F5 (SEQ ID NO: 7), M26 (SEQ ID NO: 13, 14, 15 и 16), H37L (SEQ ID NO: 1) и H37V (SEQ ID NO: 17) при конструировании гуманизированной формы B-F5.
Далее изобретение будет описано более подробно.
Средствами, которые являются подходящими для применения в настоящем изобретении, являются средства, которые способны активировать регуляторные T-клетки CD4+CD25+. Средство может представлять собой полипептид, белок или антитело. В том случае, когда средство представляет собой антитело, оно может быть моноклональным антителом. Предпочтительно антитело является моноклональным анти-CD4-антителом. Антитело также предпочтительно может представлять собой IgG1-антитело и может быть немодифицированным IgG1-антителом.
В предпочтительном аспекте изобретения средство не вызывает значимого увеличения уровня провоспалительных цитокинов в плазме крови субъектов после введения по сравнению с анти-CD3-антителами. В частности, уровни IFN-γ, TNF-α, IL-6 и/или IL-2 после введения средства значимо не повышаются по сравнению с уровнями в плазме, измеряемыми у здоровых субъектов (смотри таблицу A1). В частности, если ULN (верхнюю границу нормы) для данного конкретного цитокина, указанного в таблице A1, принять за X, то в течение 96 часов после введения средства согласно изобретению может быть менее чем 20-кратное увеличение X. Предпочтительно, может быть менее чем 10-кратное увеличение X. Более предпочтительно, такие уровни имеют место в течение периода времени от 10 минут после начала введения до 96 часов после завершения введения.
Вероятно, что у аутоиммунных пациентов уровни цитокинов перед введением средства уже выше, чем уровни, наблюдаемые у здоровых людей (ULN приведены в таблице A1), например, вследствие модифицированного статуса активации иммунных клеток по сравнению со статусом активации клеток у здоровых субъектов. В указанных случаях концентрацию конкретного цитокина непосредственно перед введением средства принимают за X, и в течение 96 часов после введения средства согласно изобретению может быть менее чем 20-кратное увеличение X. Предпочтительно, может быть менее чем 10-кратное увеличение X. Более предпочтительно, такие уровни имеют место в течение периода времени от 10 минут после начала введения до 96 часов после завершения введения.
Уровни цитокинов, измеренные в плазме здоровых добровольцев. ULN (верхнюю границу нормы) вычисляют на основе средних значений, измеренных у 39 людей + 2 × стандартное отклонение
В следующем предпочтительном аспекте изобретения средство не вызывает значимого длительного снижения количества клеток лимфоцитов CD4+ в плазме крови субъектов. В частности, в течение периода времени от 72 до 96 часов после введения количество лимфоцитов CD4+ в плазме крови субъектов может быть выше 250 клеток/мкл (или, по меньшей мере, 250 клеток/мкл).
Предпочтительно, эффекты, наблюдаемые в отношении цитокинов и лимфоцитов CD4+, описанные выше, наблюдаются по меньшей мере у 80% пациентов, подвергаемых лечению.
В данной области известно, что для того, чтобы предотвратить негативное влияние на иммунную систему, например снижение количества лимфоцитов или индукцию выброса цитокинов, используют антитела (в частности, взаимодействующие с T-клетками антитела) подкласса IgG2, IgG3 или IgG4, так как в случае антител подкласса IgG1 наблюдают более высокие уровни взаимодействий с Fc-рецептором. Также в данной области известно, что антитела (в частности, взаимодействующие с T-клетками антитела) можно модифицировать посредством Fc-мутации, дегликозилирования, гликомодификации или гликоконструирования, чтобы уменьшить взаимодействия с Fc-рецепторами.
Авторы настоящего изобретения обнаружили, что избегание антител подкласса IgG1 и модификации не являются обязательными в случае средства согласно настоящему изобретению. В частности, средство согласно настоящему изобретению не вызывает значимого длительно истощения клеток CD4+ или не индуцирует значимого выброса цитокинов по сравнению с анти-CD3-антителами.
Соответственно, в предпочтительном аспекте изобретения средство представляет собой немодифицированное IgG1-антитело, т.е. антитело, которое не содержит Fc-мутацию, и не было подвергнуто дегликозилированию, гликомодификации или гликоконструированию, чтобы уменьшить взаимодействия с Fc-рецепторами, или его фрагмент или производное.
Антитела, которые являются наиболее подходящими для применения в настоящем изобретении, представляют собой гуманизированные анти-CD4-антитела или их фрагменты или производные, которые способны активировать регуляторные T-клетки CD4+CD25+. Примеры антител, которые способны активировать регуляторные T-клетки CD4+CD25+, обсуждаются в Becker et al. (European Journal of Immunology (2007), Vol. 37: 1217-1223).
В общем, антитело, используемое в изобретении, дополнительно содержит константную область человека (Fc). Такая константная область может быть выбрана из константных доменов из любого класса иммуноглобулинов, включая IgM, IgG, IgD, IgA и IgE, и любого изотипа, включая IgG1, IgG2, IgG3 и IgG4. Предпочтительные константные области выбраны из константных доменов IgG, в частности IgG1.
Настоящее изобретение также относится к любому фрагменту антитела, содержащему его V-области. В частности, относится к фрагментам Fab, Fab', F(ab)'2, Fv и scFv.
В особенно предпочтительном аспекте настоящего изобретения антитело представляет собой гуманизированное анти-CD4-антитело, или его фрагмент, или производное, полученное из мышиного моноклонального анти-CD4-антитела B-F5. Примером такого антитела является антитело BT061.
Антитело BT061, его фрагменты и производные
Гуманизированное антитело BT061 (hB-F5) получено из мышиного мАт B-F5 и имеет V-домены, определенные следующими полипептидными последовательностями:
- V-домен H-цепи: EEQLVESGGGLVKPGGSLRLSCAASGFSFSDCRMYWLRQA PGKGLEWIGVISVKSENYGANYAESVRGRFTISRDDSKNTVYLQMNSLKTEDTAVYYCSAS YYRYDVGAWFAYWGQGTLVTVSS (SEQ ID NO: 1)
- V-домен L-цепи:
DIVMTQSPDSLAVSLGERATINCRASKSVSTSGYSYIYWYQQ KPGQPPKLLIYLASILESGVPDRFSGSGSGTDFTLTISSLQAEDVAVYYCQHSRELPWTFG QGTKVEIK (SEQ ID NO: 2).
Производные такого антитела также подходят для применения в настоящем изобретении. Производные включают производные с V-доменами, определенными полипептидными последовательностями, имеющими по меньшей мере 80%, предпочтительно по меньшей мере 90%, наиболее предпочтительно по меньшей мере 95% идентичность последовательности с последовательностью SEQ ID NO: 1 или SEQ ID NO: 2.
Особенно предпочтительными антителами являются антитела, которые содержат определяющие комплементарность области (CDR) мышиного мАт B-F5 и сохраняют способность hB-F5 активировать регуляторные T-клетки CD4+CD25+. Положение CDR в доменах VH и VK показано на фигурах 20 и 21. Такие антитела необязательно могут иметь изменения в последовательности CDR, которые по существу не влияют на специфичность и/или аффинность связывания.
В общем, антитело hB-F5, используемое в изобретении, дополнительно содержит константную область человека (Fc). Как указано выше, такая константная область может быть выбрана из константных доменов из любого класса иммуноглобулинов, включая IgM, IgG, IgD, IgA и IgE, и любого изотипа, включая IgG1, IgG2, IgG3 и IgG4. Предпочтительные константные области выбраны из константных доменов IgG, в частности IgG1.
Настоящее изобретение также относится к любому фрагменту антитела BT061, содержащему его V-области. В частности относится к фрагментам Fab, Fab', F(ab)'2, Fv и scFv.
Полинуклеотид, кодирующий V-домен H-цепи или L-цепи антитела BT061, может быть слит с полинуклеотидом, кодирующим константную область цепи H или L человека, в целях экспрессии полных цепей H и L, полученных таким образом; также может быть добавлена последовательность, кодирующая сигнальный пептид, обеспечивающий возможность секреции белка.
В изобретении также используют кассеты экспрессии, в которых полинуклеотид, который описан выше, связан с соответствующими регуляторными последовательностями, обеспечивающими регуляцию его транскрипции и трансляции в выбранной клетке-хозяине, и рекомбинантные векторы, содержащие полинуклеотид или кассету экспрессии согласно изобретению.
Такие рекомбинантные ДНК-конструкции могут быть получены и введены в клетки-хозяева хорошо известными способами получения рекомбинантной ДНК и генетической инженерии.
В изобретении также используют клетку-хозяина, трансформированную полинуклеотидом согласно изобретению. Применимыми клетками-хозяевами в рамках настоящего изобретения могут быть прокариотические или эукариотические клетки. Среди подходящих эукариотических клеток в качестве примера можно указать растительные клетки, клетки дрожжей, таких как Saccharomyces, клетки насекомых, таких как Drosophila или Spodoptera, и клетки млекопитающих, такие как HeLa, CHO, 3T3, C127, BHK, COS и т.д.
Конструирование экспрессирующих векторов, используемых в изобретении, и трансформацию клеток-хозяев можно осуществить стандартными способами молекулярной биологии.
Антитело BT061 (hB-F5), используемое в изобретении, может быть получено в результате культивирования клетки-хозяина, содержащей экспрессирующий вектор, содержащий последовательность нуклеиновой кислоты, кодирующей указанное антитело, в условиях, подходящих для его экспрессии, и извлечения указанного антитела из культуры клеток-хозяев.
Конструирование гуманизированного B-F5
Конструкция гуманизированных V H - и V K -областей B-F5
Последовательности ДНК, кодирующие VH- и VK-области мышиного B-F5, соответственно показаны на фигуре 3 и фигуре 4 с идентификационными номерами последовательностей SEQ ID NO: 5 и SEQ ID NO: 6. VH и VK человека, в которые привиты мышиные CDR, выбирали с помощью поиска в базах данных в отношении VH человека, наиболее сходной с VH и VK исходного мышиного B-F5. VH-область антитела человека (M26; номер доступа A36006) имела наиболее высокую степень гомологии с VH B-F5. VK-область другого антитела человека (FK-001; Nakatani et al., Biotechnology, 7 (1989), 805-810)) имела наиболее высокую степень гомологии с VK B-F5.
Конструировали два типа VK, отличающиеся между собой тем, что 4-м остатком является лейцин или метионин, и обозначили L4L и L4M. Конструировали два типа VH, отличающихся между собой тем, что 37-м аминокислотным остатком является лейцин или валин, и обозначили H37L и H37V. Выравнивание полипептидных последовательностей B-F5, FK-001, L4L и L4M показано на фигуре 20. Выравнивание полипептидных последовательностей B-F5, M26, H37L и H37V показано на фигуре 21. Остатки FR, которые, как сообщалось ранее, являются важными для упаковки CDR (Chothia et al., Nature 342(1989), 877; Foote et al., J. Mol. Biol., 224 (1992), 487), указаны в прямоугольниках.
Комбинируя такие VH и VK, конструировали 4 варианта V-областей.
Экспрессия гуманизированного B-F5
Последующие стадии получения гуманизированного B-F5 были такими же, как стадии, раскрытые в патенте США 5886152 для гуманизированного B-B10.
Коротко, по отдельности конструировали экспрессирующие плазмиды для H-цепи (гуманизированная область VH, слитая с константной областью y-1-цепи человека (Takahashi et al., Cell, 29 (1982), 671-679)) и L-цепи (гуманизированная область VK, слитая с константной областью K-цепи FK-001) гуманизированного B-F5. В таких плазмидах экспрессия гуманизированного B-F5 управляется промотором/энхансером гена моноклонального IgM человека, FK-001. На фигурах 5 и 6 соответственно показаны фрагменты плазмид, кодирующих VH- и VK-области гуманизированного BF-5. Последовательности, кодирующие V-область, подчеркнуты, и соответствующие полипептидные последовательности указаны под нуклеотидной последовательностью. Обе плазмиды и pSV2neo одновременно вводили в миелому мышей Sp2/0 (ATCC CRL-1581), используя липофектин. Трансфектомы, продуцирующие IgG человека, отбирали с помощью ELISA, используя антитело против IgG человека (y-цепи) и антитело против K-цепи Ig человека.
Характеристика разных вариантов гуманизированного B-F5
Оценка активности связывания CD4
Надосадки культур трансфектом, продуцирующих четыре варианта hB-F5, собирали и концентрировали. Разные антитела очищали из надосадков культур аффинной хроматографией, используя белок A-сефарозы, и оценивали в отношении их способности к связыванию CD4 посредством измерения с помощью конкурентного ELISA их ингибирующих активностей, направленных против связывания биотинилированного mB-F5 с растворимым CD4, которым покрывали планшеты для микротитрования. Инкубацию осуществляли в течение 2 часов при 37°C и в течение ночи при 4°C.
Относительные активности связывания hB-F5 (активность связывания mB-F5 принимали за 100%) показаны в таблице A.
(% от mB-F5)
Из результатов, показанных в таблице A, ясно, что 37-й остаток лейцина является важным для сохранения активности связывания CD4 антителом hB-F5, так как активность связывания CD4 в несколько раз снижается в случае превращения 37Leu в 37Val. Напротив, обнаружено, что 4-й остаток VK не так важен для активности связывания CD4. Так как структурное различие между вариантами VH c 37Leu и 37Val ясно не продемонстрировано при молекулярном моделировании, преимущество H37L по сравнению с H37V для активности связывания CD4 было неожиданным.
H37L/L4L и H37L/L4M были выбраны для оценки.
Исследование биологических активностей гуманизированного B-F5 in vitro
Оценивали биологические активности in vitro мышиного B-F5 и гуманизированных B-F5 (H37L/L4M IgG1 и H37L/L4L IgG1). Также тестировали гуманизированные B-F5 IgG2-типа (H37L/L4M IgG2 и H37L/L4L IgG2).
Биологические активности in vitro mB-F5 и четырех типов hB-F5 оценивали, используя мононуклеарные клетки периферической крови (PBMC) здоровых доноров. PBMC активировали ConA (2,5 пг/мл, 3 дня) или PPD (10 пг/мл, 4 дня) в присутствии мышиного или hB-F5 и исследовали в отношении их пролиферативных ответов по включению 3H-тимидина.
Мышиное и hB-F5 могли умеренно ингибировать ConA-индуцированную пролиферацию, но активности варьировали от антитела к антителу и/или от донора к донору. Также мышиное и hB-F5 были способны ингибировать Аг-специфичную пролиферацию PBMC, индуцированную PPD.
hB-F5 IgG1-типа ингибировало PPD-индуцированную пролиферацию более эффективно (ингибирование до 70%), чем mB-F5. IgG1-тип по-видимому является более эффективным, чем IgG2-тип, ингибирующая активность которого была почти такой же, как mB-F5. В случае IgG1-типа H37L/L4M был более эффективным, чем H37L/L4L. H37L/L4M и H37L/L4L IgG2-типа имели почти одинаковые ингибирующие активности. Коротко, ингибирующие активности B-F5, направленные против PPD-индуцированной пролиферации PBMC, были следующими: H37L/L4M IgG1 > H37L/L4L IgG1 > H37L/L4M IgG2 = H37L/L4L IgG2 = mB-F5.
С учетом эффективности биологической активности in vitro и меньшего количества мышиных аминокислот для дальнейшей оценки выбрали H37L/L4M IgG1, и было выбрано антитело, которое названо BT061, и его использовали для демонстрации настоящего изобретения в примерах, приведенных в настоящей заявке.
Композиции и применения
Как было указано, фармацевтическая композиция и лекарственные средства, используемые в настоящем изобретении, предпочтительно способны лечить аутоиммунное заболевание у пациентов, которым полезна длительная терапия.
В некоторых вариантах (более высокие дозы) предпочтительными дозами средства являются дозы от 10 мг до 100 мг, от 10 мг до 80 мг, от 15 мг до 80 мг, от 20 мг до 75 мг, предпочтительно, от 20 мг до 60 мг и, наиболее предпочтительно, от 25 мг до 60 мг.
Альтернативно дозу можно определить на основе кумулятивной дозы по сравнению с множеством доз. В частности, в случае множества доз в течение периода времени 10 суток, предпочтительно, кумулятивная доза больше 25 мг, но меньше или равна 200 мг, более предпочтительно, доза составляет от 28 мг до 100 мг и, наиболее предпочтительно, от 30 мг до 100 мг. Кроме того, кумулятивная доза в течение периода времени 5 суток, предпочтительно, больше 15 мг, но меньше или равна 100 мг, более предпочтительно, доза составляет от 18 мг до 100 мг и, наиболее предпочтительно, от 20 мг до 100 мг. В указанном аспекте изобретения особенно предпочтительно дозы вводят подкожно.
В альтернативном варианте (более низкие дозы) предпочтительными дозами являются дозы от 0,2 мг до 30 мг, от 0,2 до 20 мг на дозу, от 0,3 мг до 7,5 мг, от 0,3 до 5 мг на дозу или, предпочтительно, от 0,3 до 1 мг на дозу.
Альтернативно доза может быть определена на основе кумулятивной дозы по сравнению с множеством доз. В частности, в случае множества доз в течение периода времени 10 суток, предпочтительно, кумулятивная доза больше 0,2 но меньше 25 мг, более предпочтительно, от 0,2 до 20 мг и, наиболее предпочтительно, больше 0,2, но меньше 10 мг. Кроме того, кумулятивная доза в течение периода времени 5 суток должна быть больше 0,2, но меньше 15 мг, предпочтительно, от 0,2 до 12 мг и, более предпочтительно, больше 0,2, но меньше 5 мг.
В указанном аспекте изобретения в случае введения дозы внутривенно предпочтительно дозы в течение периода времени 10 суток составляют от 0,2 до 10 мг, наиболее предпочтительно от 0,2 до 7,5 мг. Альтернативно в случае введения доз подкожно или внутримышечно, предпочтительно, дозы в течение периода времени 10 суток составляют от 1 мг до 30 мг, более предпочтительно, от 5 мг до 30 мг.
Доза также может быть рассчитана на основе массы тела субъекта или площади поверхности тела (ППТ) субъекта. Площадь поверхности тела (ППТ) может быть рассчитана любым известным способом. Примерами способов расчета ППТ являются следующие способы:
Формула Мостеллера: (ППТ (м2)=([Рост (см) × масса (кг)]/3600)1/2
(Mosteller RD: Simplified Calculation of Body Surface Area. N. Engl. J. Med. 1987 Oct. 22; 317(17): 1098)
Формула Дюбуа-Дюбуа: ППТ (м2)=0,20247 × рост (м)0,725 × масса (кг)0,425
(DuBois D; DuBois EF: A formula to estimate the approximate surface area if height and weight be known. Arch. Int. Med. 1916 17: 863-71.
Формула Хейкука: ППТ (м2)=0,024265 × рост (см)0,3964 × масса (кг)0,5378
(Haycock G.B., Schwartz G.J.,Wisotsky D.H. Geometric method for measuring body surface area: A height weight formula validated in infants, children and adults. The Journal of Pediatrics 1978 93:1:62-66)
Формула Гехана и Джорджа: ППТ (м2)=0,0235 × рост (см)0,42246 × масса (кг)0,51456
(Gehan EA, George SL, Estimation of human body surface area from height and weight. Cancer Chemother Rep. 1970 54: 225-35)
Формула Бойда: ППТ (м2) = 0,0003207 × рост (см)0,3 × масса (граммы)(0,7285-(0,0188×LOG(грамм)).
Согласно вариантам осуществления изобретения с более высокими дозами доза средства для субъекта в расчете на массу тела составляет от 0,1 до 2 мг/кг, предпочтительно, от 0,15 до 1,5 мг/кг и, наиболее предпочтительно, от 0,2 до 1 мг/кг. В расчете на ППТ доза средства для субъекта составляет от 5 до 60 мг/м2 ППТ, предпочтительно, от 6 до 50 мг/м2 и, наиболее предпочтительно, от 8 до 40 мг/м2.
В указанных аспектах изобретения, когда доза основана на площади поверхности тела или массе тела субъекта, предпочтительно, дозы в течение периода времени 10 суток составляют от 10 мг/м2 до 120 мг/м2, более предпочтительно, от 16 мг/м2 до 120 мг/м2 или от 0,2 мг/кг до 4 мг/кг, более предпочтительно, от 0,4 мг/кг до 4 мг/кг. Особенно предпочтительно, дозы вводят подкожно.
Согласно вариантам осуществления изобретения с более низкими дозами доза средства для субъекта в расчете на массу тела составляет от 1 до 500 мкг/кг, предпочтительно, от 2 до 400 мкг/кг, более предпочтительно, от 2 до 250 мкг/кг и, наиболее предпочтительно, от 2,5 до 20 мкг/кг. В расчете на ППТ доза средства для субъекта составляет от 0,12 до 15 мг/м2, более предпочтительно, от 0,20 до 10 мг/м2 и, наиболее предпочтительно, от 0,30 до 0,50 мг/м2.
В указанных аспектах изобретения, когда доза основана на площади поверхности тела или массе тела субъекта, в том случае, когда дозу вводят внутривенно, предпочтительно, дозы в течение периода времени 10 суток составляют от 0,20 до 10 мг/м2, более предпочтительно, от 0,20 до 4 мг/м2 или от 2 до 250 мкг/кг, более предпочтительно, от 2 до 100 мкг/кг. Альтернативно, в том случае, когда дозы вводят подкожно или внутримышечно, предпочтительно, дозы в течение периода времени 10 суток составляют от 0,30 до 20 мг/м2, более предпочтительно, от 0,5 до 20 мг/м2 или от 2,5 до 500 мкг/кг, более предпочтительно, от 20 до 500 мкг/кг.
Частота введения особым образом не ограничена при условии, что она обеспечивает преимущества при длительном лечении. Согласно изобретению предпочтительно вводят множество доз, по меньшей мере, на следующей основе: раз в 3 дня, еженедельно, каждые четыре недели, каждые 6 недель, каждые 12 недель, каждые 24 недели, каждый календарный месяц, каждые 3 календарных месяца, каждые шесть календарных месяцев или ежегодно. Таким образом, дозы могут быть разделены по меньшей мере тремя днями или альтернативно по меньшей мере одной неделей, по меньшей мере 10 днями, по меньшей мере одним месяцем, по меньшей мере 3 месяцами, по меньшей мере 6 месяцами или по меньшей мере одним годом (это значит, что дозы принимают по меньшей мере каждые 3 дня, каждую неделю, каждые 10 дней, каждый месяц, каждые 3 месяца, каждые 6 месяцев или каждый год). В следующем альтернативном варианте множество доз принимают каждые 3-31 день, каждые 3-10 дней, каждые 3-6 дней или каждые 1-12 месяцев.
Изобретение также относится к набору для применения, которое определено выше, при этом набор содержит множество лекарственных доз, которые определены выше, для одновременного, последовательного или раздельного введения субъекту.
Также предлагается способ лечения аутоиммунного заболевания, при этом способ включает в себя введение субъекту лекарственного средства, которое определено выше, описанным выше способом.
Как указано выше, фармацевтическая композиция и лекарственные средства, применяемые согласно настоящему изобретению, предпочтительно способны лечить аутоиммунное заболевание у пациентов, которым полезна длительная терапия. Такими пациентами являются без ограничения пациенты с тяжелыми случаями заболевания с длительной историей болезни.
В общем, фармацевтическая композиция и лекарственные средства, применяемые согласно настоящему изобретению, предназначены для лечения аутоиммунного заболевания. Предпочтительно, аутоиммунное заболевание выбрано из псориаза, ревматоидного артрита, рассеянного склероза, диабета типа-1, воспалительного заболевания кишечника, болезни Крона, тиреоидита Хашимото, аутоиммунного тиреоидита, аутоиммунной тяжелой псевдопаралитической миастении, системной красной волчанки, язвенного колита, атопического дерматита, миокардита и связанных с трансплантацией заболеваний, таких как реакции «трансплантат против хозяина» или «хозяин против трансплантат» или проблемы, связанные с общей органной толерантностью.
В особенно предпочтительном аспекте изобретения фармацевтические композиции предназначены для лечения аутоиммунного заболевания псориаза. В частности, такие фармацевтические композиции следует вводить внутривенно или подкожно в дозах, указанных в настоящем описании.
Псориаз является расстройством, которое вызывает псориатические повреждения или бляшки на коже пациента.
Подсчет индекса охвата и тяжести псориаза (PASI) обычно используют для оценки и регистрации уровня псориаза, наблюдаемого у пациентов. Подсчет PASI заключается в оценке эритемы (E), инфильтрации (I) и десквамации (D) и вовлечении площади поверхности тела (A) в 4 областях тела (голова (h), туловище (t), верхние (u) и нижние (l) конечности). В таблице B ниже показано, как система оценки работает.
Система оценки PASI
Так как голова, верхние конечности, туловище и нижние конечности соответствуют примерно 10, 20, 30 и 40% площади поверхности тела соответственно, то оценку PASI вычисляют по формуле
PASI=0,1(Eh+Ih+Dh)Ah+0,2(Eu+Iu+Du)Au+0,3(Et+It+Dt)At+0,4(El+Il+Dl)Al
Оценка PASI находится в диапазоне 0-72. Оценка 0 означает отсутствие псориаза, тогда как оценка 72 означает наиболее тяжелый псориаз.
В предпочтительном варианте данного аспекта фармацевтическая композиция согласно настоящему изобретению способна лечить псориаз, обеспечивая, по меньшей мере, 40% и, предпочтительно, по меньшей мере, 50% улучшение оценки PASI у пациента. Предпочтительно, до лечения субъект имеет оценку PASI, составляющую, по меньшей мере, 10. Такие эффекты можно наблюдать, по меньшей мере, 56 дней после введения однократной дозы антитела, более предпочтительно, по меньшей мере, 75 дней после введения однократной дозы антитела. В частности, такие эффекты можно наблюдать, по меньшей мере, у 80% пациентов.
В следующем аспекте настоящего изобретения фармацевтические композиции предназначены для лечения ревматоидного артрита.
Ревматоидный артрит является аутоиммунным заболеванием, которое вызывает хроническое воспаление суставов и окружающих тканей, и также может влиять на другие ткани и органы тела.
Улучшение при ревматоидном артрите, наблюдаемое у подвергаемого лечению пациента, обычно оценивают, используя базовый набор критериев Американской коллегии ревматологии (ACR) (Felson et al., Arthritis and Rheumatism, 1995, 38(6), 727-735). В такой системе определяют значение ACR20 как 20% улучшение оценки болезненных и распухших суставов и 20% улучшение по 3 из 5 остальных измерений базового набора ACR: глобальные оценки пациентом и врачом, боли, нетрудоспособности и реактанта острой фазы, такого как C-реактивный белок (CRP).
В частности, фармацевтические композиции для лечения ревматоидного артрита предпочтительно следует вводить внутримышечно или подкожно в дозах, указанных в настоящем описании.
Современное лечение артрита включает в себя применение лекарственных средств первой линии для контролирования боли и воспаления, классифицируемых как нестероидные противовоспалительные лекарственные средства (НПВС), например, аспирина, ибупрофена, напроксена и т.д. Вторая линия лечения артрита включает в себя применение кортикостероидов (например, преднизона и дексаметазона), медленно действующих противоревматических средств (SAARD) или модифицирующих заболевание противоревматических средств (DMARD), например, метотрексата, пенициллинамина, циклофосфамида, солей золота, азотиоприна, лефлуномида и т.д.
Кортикостероиды, синтетические варианты гормона кортизона в организме, используют для ингибирования прогрессирования RA (например, преднизон и дексаметазон).
Также разработана другая группа лекарственных средств, названных модификаторами биологического ответа (BRM) для лечения RA, включая антагонисты TNF-альфа (адалимумаб, инфликсимаб, этанерцепт), которые работают посредством связывания с рецептором или непосредственного связывания с белком TNF-альфа.
В одном варианте данного аспекта изобретения композиции следует вводить в сочетании с лекарственными средствами, используемыми в настоящее время для лечения ревматоидного артрита. В частности, композиции необходимо вводить с одним из лекарственных средств, указанных выше, предпочтительно с метотрексатом.
Известные лекарственные средства, такие как метотрексат, и фармацевтическую композицию согласно настоящему изобретению можно вводить одновременно, последовательно или раздельно.
Далее изобретение будет описано дополнительно в связи со следующими конкретными вариантами.
ПРИМЕРЫ
ПРИМЕР 1 - Исследование иммуномодулирующей способности BT061 в отношении пролиферации T-клеток
Способ
Культивирование цельной крови осуществляли, используя свежие образцы периферической крови. Коротко, используя иглы 19G собирали кровь от трех здоровых добровольцев в гепаринизированные шприцы. Кровь высевали в 96-луночные планшеты для культивирования не позднее чем через 60 мин после отбора крови.
Антитело, используемое в изобретении (BT061, партия 40588, или партия 70A0013B), добавляли к культурам перед стимуляцией лейкоцитов в 5 разных концентрациях (смотри «Тестируемые вещества» ниже). Клеткам давали возможность взаимодействовать с антителом в течение 90 минут при 37°C, 5% CO2 во влажной атмосфере, затем к отдельным культурам добавляли четыре разных стимулятора:
(a) анти-CD3-антитела (R&D Systems; 50 нг/мл);
(b) фитогемагглютинин (ФГА, Biochrom KG; 3 пг/мл) вместе с анти-CD28-антителами (Becton-Dickinson; 1 мкг/мл);
(c) липополисахарид (ЛПС, подтип 055:B15, из Sigma Aldrich; 1 мкг/мл);
(d) SE-B (Bernhard-Nocht-Institut; 25 нг/мл) вместе с анти-CD28-антителами (Becton-Dickinson; 1 мкг/мл).
Все культуры цельной крови инкубировали в течение 24 часов при 37°C, 5% CO2 (влажная атмосфера). Затем собирали надосадки культур для определения конечных данные о цитокинах, за исключением культур, стимулированных ФГА/антиCD28, которые инкубировали в течение 48 часов, чтобы получить достаточную стимуляцию клеток Th2.
Результаты указаны на фигуре 1.
Результаты
BT061 не оказывало значимого влияния на основные активности моноцитов/макрофагов и на активности Th1, а также Th2 в культурах цельной крови здоровых добровольцев. Имело место зависимое от концентрации влияние на клетки T-рег (наблюдаемое в виде увеличения высвобождения TGF-бета).
В частности, результаты подтверждают, что имеет место:
- отсутствие модуляции воспалительного цитокина IL-2 при концентрациях до 50 мкг/мл, соответствующих применению высоких доз до 150 мг у пациентов;
- отсутствие индукции воспалительного цитокина IFN-гамма при концентрациях до 50 мкг/мл, соответствующих применению высоких доз до 150 мг у пациентов;
- отсутствие модуляции цитокинов Th1/Th2 при концентрациях до 50 мкг/мл, соответствующих применению высоких доз у пациентов;
- повышающая регуляция IL-6 с минимальными приращениями происходит только спорадически, значимость которой ставится под сомнение;
- увеличение высвобождения TGF-бета (клетки T-рег);
- отсутствие влияния на основные регуляторные активности моноцитов/макрофагов и на активности Th1 и Th2.
ПРИМЕР 2 - Тестирование BT061 в отношении его влияния на культивируемые PBMC человека, примированные для ответа против столбнячного токсоида, и анализ цитокинов
Способ
Анализ пролиферации
Свежевыделенные PBMC культивировали в 96-луночных планшетах для микротитрования с плоским дном в объеме 200 мкл/лунку (4 × 105 клеток/лунку). Тестируемый объект (анти-CD4-мАт BT061) использовали в концентрациях 20 мкг/мл, 4 мкг/мл и 0,8 мкг/мл (дополнительно 40 мкг/мл в предварительном тесте); столбнячный токсоид использовали в концентрациях 25 мкг/мл, 5 мкг/мл и 1 мкг/мл. В качестве негативного контроля клеточной культуры брали среду. Все культуры получали в трех повторах.
В случае ConA-стимуляции использовали концентрацию 2,5 мкг/мл и объем 200 мкл/лунку. PBMC доводили до плотности 1 × 106/мл и диспергировали в объеме 100 мкл на лунку.
В конце периода культивирования выявляли пролиферацию клеток добавлением 0,4 мккюри 3H-тимидина на лунку на шестнадцать часов. В конце периода культивирования клетки открепляли от поверхности, используя раствор EDTA, и собирали на фильтрах из стекловолокна, используя устройство для сбора клеток Scatron. Количество радиоактивности, включенной в ДНК, в каждой лунке измеряли в сцинтилляционном счетчике, и оно пропорционально количеству пролиферирующих клеток, которое в свою очередь является функцией количества лейкоцитов, которые были стимулированы для вхождения в S-фазу клеточного цикла. Регистрируемым параметром было количество импульсов в минуту (имп./мин), и индекс стимуляции (SI) для каждой концентрации определяли как имп./минсоединение/имп./минконтроль
Анализ цитокинов
Все цитокины количественно оценивали в надосадках культур, используя коммерческие наборы ELISA, согласно соответствующим инструкциям производителя. Используемые реагенты указаны в таблице 1.
Уровни интерлейкина (IL)-1 в надосадках культур определяли, используя набор для тестирования ELISA Set A для выявления IL-1 человека (Bender) согласно инструкциям производителей. Диапазон тестирования, предварительно определяемый с использованием стандарта, поставляемого в наборе, установлен от 1,3 до 130 пг/мл для неразбавленных образцов.
Уровни IL-4, 5, 6 и 10 в надосадках культур, а также трансформирующий фактор роста (TGF) β1 и фактор некроза опухолей (TNF) α определяли, используя набор для тестирования OptEIA (BD biosciences) согласно инструкциям производителей. Диапазон тестирования, предварительно определяемый с помощью стандартов, поставляемых в наборах, установлен от 3,8 до 330 пг/мл в случае IFN-γ, от 6,3 до 616 пг/мл (в случае IL-4, 5, 10 и TNF) при измерении неразбавленных образцов и от 7,6 до 660 пг/мл в случае IL-6 при использовании двукратных разведений образцов.
Для расчета среднего значения и стандартного отклонения использовали два (в случае культур моноцитов) или восемь (в случае культур PBMC) определений цитокинов в анализе ELISA, каждое из которых осуществляли для независимых микрокультур. Титры выше верхнего значения диапазона тестирования (например, 616 пг/мл в случае TNF) для расчета приравнивали к такому значению. Нижнее значение диапазона тестирования вычитали из каждого среднего значения перед вычислением.
Результаты
Антитело BT061 (также называемое гуманизированным B-F5 или просто hB-F5) способно подавлять зависимым от дозы образом специфичную для столбнячного токсоида пролиферацию T-клеток; не наблюдали влияния на общее количество T-клеток. Показана общая супрессия высвобождения цитокинов.
В таблице 2 показано влияние анти-CD4-мАт BT061 в анализе специфичной для столбнячного токсоида пролиферации T-клеток, измеряемой в трех повторах.
Показаны средние значения и SD включения 3H-Tdr, измеряемого в трех повторах, а также индекс стимуляции (SI, определяемый как имп./минсоединение/имп./минноль) для каждой концентрации и уровень значимости при использовании непарного двухстороннего t-критерия против контрольной среды (n.s.: не значимые; *: p<0,05; **: p<0,01; ***: p<0,001).
В данной таблице показано влияние анти-CD4-мАт BT061 на продукцию цитокинов в стимулированных токсоидом титана культурах PBMC. Приведены средние значения и SD для восьми отдельных микрокультур.
В настоящей таблице показано влияние анти-CD4-мАт BT061 на продукцию воспалительных цитокинов в LPS-стимулированных культурах моноцитов. Приведены средние значения и SD для двух отдельных микрокультур.
Данные, приведенные в таблицах 2-4, показывают следующее:
- Зависимая от дозы супрессия индуцированной столбнячным токсоидом пролиферации T-клеток (вторичный ответ) показана даже при высоких дозах BT061, что свидетельствует о том, что BT061 не отменяет все иммунные ответы. Используемые дозы соответствуют применению высоких доз до 60 мг у пациентов.
- Общая супрессия высвобождения цитокинов (зависимое от дозы снижение уровней IFN-гамма, IL-5 и TNF-альфа, без изменения IL-1, IL-4, IL-6, IL-10) и отсутствие влияния на баланс Th1/Th2.
- Увеличение высвобождения TGF-бета.
ПРИМЕР 3 - Исследование индуцированной BT061 (анти-CD4-мАт) ADCC (зависимой от антител опосредованной клетками цитотоксичности) с помощью проточной цитометрии
Клетки-мишени HuT 78 метили BT061 (hB-F5) и инкубировали с клетками PBMC в качестве эффекторов. Погибшие клетки могли быть выявлены вследствие поглощения красителя ДНК йодида пропидия после 30-минутного периода инкубации. Результаты показаны в таблице 5.
средняя интенсивность флуоресценции
средняя интенсивность флуоресценции
Данные, представленные в таблице, показывают, что не было индукции ADCC антителом BT061 (hB-F5) даже при высоких концентрациях.
ПРИМЕР 4 - Апоптоз
При тестировании с использованием проточной цитометрии индуцированного BT061 (анти-CD4-мАт) апоптоза PBMC из цельной крови инкубировали с BT061 или позитивным контролем.
После 7-дневного периода инкубации осуществляли регистрацию апоптозных клеток посредством окрашивания апоптозных клеток аннексином-V-флуоресцеином. Результаты показаны в таблице 6.
[мкг/мл]
Данные показывают, что индукции апоптоза не было даже при высоких концентрациях BT061.
ПРИМЕР 5 - Связывание комплемента
При тестировании с использованием проточной цитометрии связывания фактора комплемента C1q выделяли PBMC и инкубировали с BT061 (анти-CD4-мАт) с последующей инкубацией в очищенным рекомбинантным C1q.
ATG (тецелак) служил в качестве позитивного контроля.
Регистрацию осуществляли, используя меченое ФИТЦ регистрационное антитело против C1q. Результаты показаны в таблице 7.
[мкг/мл]
BT061
Данные показывают, что связывание комплемента не наблюдали даже при высоких концентрациях.
ПРИМЕР 6 - CD4-активированные T-рег CD25 + подавляют синтез цитокинов и реэкспрессию CD25 T-клетками CD8 +
На фигуре 7 показано, что CD4-активированные T-рег CD25+ подавляют синтез цитокинов и реэкспрессию CD25 T-клетками CD8+. Вверху: CD4-активированные T-рег CD25+ подавляют синтез цитокинов коактивированными T-клетками CD8+.
Способ
T-рег CD25+ стимулировали в присутствии/отсутствие анти-CD3-мАт или анти-CD4-мАт (B-F5, 1 мкг/мл каждого) в течение 48 часов. Затем клетки культивировали совместно с облученными истощенными по T-клеткам сингенными PBMC и аллогенными T-клетками CD8+ (соотношение 1:1). Продукцию цитокинов анализировали на 7 день. Аллореактивные T-клетки CD8+ повторно стимулировали ФГА/PMA в течение 5 часов в присутствии моненсина, фиксировали и красили анти-CD8 и специфичными для цитокинов мАт согласно соответствующей инструкции производителя (все BD PharMingen). Синтез цитокинов анализировали с помощью проточной цитометрии, выбирая гейт лимфоцитов CD8+. Повторную экспрессию CD25 T-клетками CD8+ измеряли согласно следующему способу: T-рег CD25+ стимулировали в присутствии/отсутствие анти-CD3-мАт или анти-CD4-мАт (B-F5, 1 мкг/мл каждого) в течение 48 часов. Затем клетки культивировали совместно с истощенными по T-клеткам сингенными PBMC и аллогенными T-клетками CD8+. На 10 день T-клетки CD8+ повторно стимулировали аллогенными истощенными по CD3 PBMC того же донора, которого использовали для первой стимуляции (соотношение 1:1) и анализировали повторную экспрессию CD25 T-клетками CD8+ через 24 часа после повторной стимуляции с помощью проточной цитометрии.
Результаты
Активированные T-рег CD25+ не только подавляли пролиферацию обычных T-клеток, но также ингибировали продукцию цитокинов и способность T-клеток CD8+ экспрессировать a-цепь рецептора IL-2, CD25. Как показано на фигуре 7, CD4-активированные T-рег CD25+ эффективно ингибировали продукцию IL-2 и IFN-γ аллореактивными T-клетками CD8+, подобно анти-CD3-стимулированным T-рег CD25+. Одновременно они делали супрессированные T-клетки CD8+ неспособными экспрессировать CD25 при повторной стимуляции (фигура 7). Полученные данные вместе взятые показывают, что стимуляция CD4 полностью активирует супрессорные свойства T-рег CD25+.
ПРИМЕР 7 - Безопасность и переносимость возрастающих доз BT061
Исследование проводили для того, чтобы проверить безопасность и переносимость BT061, используя возрастающие дозы антитела, на здоровых добровольцах мужского и женского пола в возрасте ≥18, но ≤75 лет.
Тридцать добровольцев получали BT061 посредством внутривенного введения в 10 группах дозирования, по 3 добровольца на группу. Кроме того, 15 добровольцев получали BT061 посредством подкожного введения в группах дозирования, также по 3 добровольца на группу. Введение BT061 внутривенно проиллюстрировано в таблице 8.
Внутривенная доза BT061
Каждую дозу разбавляли 0,9% хлоридом натрия для инъекций до общего объема 20 мл. Дозу вводили в виде одной непрерывной внутривенной инфузии в течение 2 часов.
Введение BT061 подкожно проиллюстрировано в таблице 9.
Подкожная доза BT061
Каждую дозу инъецировали в виде одной болюсной инъекции.
Добровольцев оценивали в течение 3-месячного периода времени после инъекции.
В случае подкожного применения образцы плазмы брали перед введением и через 3, 6, 12, 24, 36, 48, 56, 72, 88, 96, 120, 144 и 168 часов после введения и на 75 день.
В случае внутривенного применения образцы плазмы брали перед введением и через 30 минут, 1, 2, 3, 6, 12, 24, 36, 48, 72, 96, 120, 144 и 168 часов после введения.
Образцы плазмы анализировали с использованием стандартной методики ELISA, чтобы установить уровни цитокинов. Соответствующие анализируемые цитокины включали: IFN-γ, TNF-α, IL-6 и IL-2.
Образцы плазмы также анализировали, используя стандартные способы проточной цитометрии, чтобы измерить количество лимфоцитов CD4+.
Результаты
Было обнаружено, что внутривенные и подкожные дозы до 60 мг обычно были хорошо переносимыми.
Уровни цитокинов
Индукция высвобождения цитокинов является обычным непосредственным осложнением, возникающим в случае применения взаимодействующих с T-клетками терапевтических антител, таких как ATG, OKT3, CAMPATH-1H и гуманизированное анти-CD3-мАт (TRX4, визилизумаб и теплизумаб). Симптомы главным образом включают в себя умеренное повышение температуры, головные боли и самопроизвольно проходящие желудочно-кишечные проявления. Побочные эффекты, взаимосвязанные с индукцией цитокинов после введения антитела, требуют применения дополнительных лекарственных средств, таких как антигистаминный препарат гидрохлорид дифенгидрамина и/или противовоспалительное средство ибупрофен.
При использовании OKT3 (муромонаб-CD3), мышиного, специфичного к CD3 терапевтического моноклонального антитела, сообщалось даже о смертельных случаях, и тяжелые побочные эффекты ограничивают клиническое применение такого антитела главным образом пациентами с подавленным иммунитетом.
Хотя гуманизированные не связывающие FcR CD3-специфичные моноклональные антитела, которые применяют в настоящее время в клинике для лечения аутоиммунного заболевания (теплизумаб и TRX4) дают более низкие побочные эффекты, индуцируемые активацией T-клеток и/или активацией экспрессирующих Fc-рецептор клеток после первой дозы, по сравнению со связывающими FcR CD3-специфичными антителами, такими как OKT3, все еще наблюдается некоторая степень активации T-клеток и активации экспрессирующих Fc-рецептор клеток, которая приводит к высвобождению цитокинов, обычно связанному с зависимыми от цитокинов побочными эффектами.
В настоящем исследовании неожиданно обнаружено, что индукция цитокинов, наблюдаемая у здоровых добровольцев после внутривенного или подкожного применения BT061, была низкой и кратковременной по сравнению с анти-CD3-антителами. Индукция цитокинов обычно возрастала с увеличением дозы. Однако даже при наиболее высоких дозах от 40 до 60 мг индукция цитокинов намного ниже, чем индукция, наблюдаемая в случае других взаимодействующих с T-клетками моноклональных антител (фигура 8A и B).
Медианные максимальные концентрации цитокинов, наблюдаемые в любой временной точке в пределах 96 часов после введения с использованием наиболее высоких доз (от 40 мг до 60 мг BT061), показаны на фигурах 8 и 9.
Медианную максимальную концентрацию каждого цитокина вычисляют следующим образом: медиана наиболее высоких концентраций цитокинов, наблюдаемых после введения антитела.
На фигуре 8A и B показано высвобождение TNFα и IL-6, наблюдаемое у здоровых добровольцев после внутривенного или подкожного введения BT061, по сравнению с TNFα и IL-6, высвобождаемыми после введения моноклональных анти-CD3-антител, теплизумаба и TRX4. Нормальные значения таких цитокинов взяты из публикации Straub et al. (2007, Arthr. and Rheumat). На фигуре 9 показаны уровни в плазме IL-2 и IFN-γ после внутривенного или подкожного введения BT061. Медианные максимальные уровни вычисляли для группы, получавшей от 40 до 60 мг, в пределах 4 дней после инъекции антитела. Верхнюю границу нормы (ULN) вычисляли на основе уровней цитокинов, измеренных у 39 здоровых субъектов, где ULN = среднее значение + 2× стандартное отклонение.
По сравнению с теплизумабом и TRX4 (результаты взяты из публикаций Herold et al., 2002, New Engl. J. Med, и Keymeulen et al., 2005 New Engl. J. Med, соответственно) BT061 индуцировал только незначительное и кратковременное высвобождение цитокинов. Уровни TNF-α и IL-6 были слегка увеличены. На фигуре 8 показано, что медианные максимальные значения уровней цитокинов IL-6 и TNFα, выявленные в плазме после применения BT061 (40 и 60 мг), ниже, чем уровни, наблюдаемые после лечения CD3-специфичными терапевтическими антителами теплизумабом и TRX4.
Кроме того, в отличие от анти-CD3-мАт, BT061 не приводил к значимо повышенным уровням IFN-γ и IL-2 (фигура 9), которые сообщались в случае применения TRX4 (Keymeulen et al. 2005).
Лимфоциты CD4 +
Кроме того, в испытание также включали исследование количества CD4-позитивных лимфоцитов в собранных образцах плазмы.
Результаты внутривенного введения показаны в таблицах 10, 11 и 12. В таблице 13 показаны результаты испытания с использованием подкожного введения. Результаты показаны графически на фигурах 10 и 11.
Количество клеток CD4+ у отдельных здоровых добровольцев после внутривенного введения от 3,5 мкг до 2,5 мг BT061
Количество клеток CD4+ у отдельных здоровых добровольцев после внутривенного введения от 2,5 мг до 20 мг BT061
В частности на фигуре 10 показано количество клеток CD4 (клеток в мл плазмы) у добровольцев, получавших однократную внутривенную дозу BT061. Точки данных представляют средние значения для 3 пациентов в каждой группе дозирования. Пунктирные линии указывают верхнюю границу нормы (ULN) и нижнюю границу нормы (LLN). ULN и LLN (среднее значение + (или -) стандартное отклонение) были вычислены на основе количества клеток у здоровых добровольцев и составляют 443 CD4-клетки в мкл (нижняя граница нормы; LLN) и 1324 CD4-клетки с мкл (верхняя граница нормы; ULN).
На фигуре 11 показано количество CD4-клеток (клеток в мл плазмы) у добровольцев, получавших однократную подкожную дозу BT061. Как и на фигуре 10, точки данных представляют средние значения для 3 пациентов в каждой группе дозирования. Пунктирные линии показывают верхнюю границу нормы (ULN) и нижнюю границу нормы (LLN).
Количество клеток CD4+ у отдельных здоровых добровольцев после внутривенного введения от 20 мг до 60 мг BT061
Количество клеток CD4+ у отдельных здоровых добровольцев после подкожного введения BT061
Многие CD4-специфичные моноклональные антитела, известные в данной области (такие как антитела, обзор которых приведен в Strand et al., 2007), достигают иммуносупрессии посредством истощения популяции CD4-позитивных лимфоцитов. Недостаток таких антител заключается в том, что у людей, подвергаемых лечению, возникает иммунная недостаточность, и они становятся чувствительными к другим инфекциям.
Напротив, настоящее исследование показало, что BT061 не индуцировало массивного длительного истощения CD4-позитивных клеток. Однако наблюдали временное снижение количества CD4-позитивных лимфоцитов с восстановлением до нормальных значений в периферической крови в пределах 72 часов после введения антитела.
Во временной точке 72 часа после применения BT061 подсчет CD4-клеток у четырех добровольцев в группе, получавшей внутривенные дозы, показал уровни CD4, которые были ниже нормальных значений, а именно: у 1 добровольца после внутривенной дозы 100 мкг: 400 CD4-клеток в мкл; у 1 добровольца из группы, получавшей 5 мг: 419 CD4-клеток в мкл; у 1 добровольца из группы, получавшей 10 мг: 440 CD4-клеток в мкл; и у 1 добровольца из группы, получавшей 20 мг: 392 CD4-клеток в мкл.
Однако такие значения были только не намного ниже нормальных значений. Количество клеток CD4 у остальных 26 добровольцев из групп, получавших внутривенные дозы, было в пределах нормальных значений через 72 часа после введения BT061.
В группах, получавших подкожные дозы, через 72 часа только у одного из 15 добровольцев наблюдали количество CD4-клеток ниже нормальных значений.
В заключение, в отличие от истощающих CD4-специфичных мАт, даже при высоких дозах антитело BT061 индуцировало только временное снижение количества CD4-позитивных клеток с последующим общим восстановлением. На основании временного снижения и быстрого общего восстановления был сделан вывод о том, что имело место временное перераспределение CD4-позитивных клеток, а не истощение популяции таких клеток.
Таким образом, данные подтверждают возможность применения BT061 в качестве средства, вводимого в виде множества доз в течение длительного периода времени.
ПРИМЕР 8 - Клиническое испытание BT061 у пациентов с хроническим псориазом от формы умеренной тяжести до тяжелой формы
Способность hB-F5 BT061 лечить аутоиммунное заболевание подвергают тестированию на 56 пациентах, страдающих псориазом от формы умеренной тяжести до тяжелой формы. Испытание включает в себя исследование повышения однократной дозы, чтобы оценить безопасность и эффективность hB-F5.
Использовали следующие условия испытания:
56 пациентов делили на семь групп дозирования, при этом каждая группа включает в себя восемь человек. Пять групп дозирования (группы дозирования I-V) получали антитело или плацебо посредством внутривенного введения, и две группы дозирования (группы дозирования VI и VII) получали антитело или плацебо посредством подкожного введения. Два пациента в каждой группе дозирования получали плацебо, тогда как остальные шесть пациентов в каждой группа дозирования получали дозу BT061. В группе дозирования I шесть пациентов внутривенно получали 0,5 мг BT061. В группах дозирования II-V шесть пациентов получали 2,5 мг, 5 мг, 10 мг или 20 мг BT061 соответственно. В группах дозирования VI и VII, в которых введение осуществляли подкожно, шесть пациентов получали 12,5 мг или 25 мг BT061 соответственно.
В случае внутривенного введения антитело/плацебо вводили в виде инфузии в вену предплечья одобренными с медицинской точки зрения способами. В данном случае общий объем вводят в виде однократной внутривенной инфузии в течение периода времени, составляющего 2 часа, с помощью перфузора (Fresenius Pilot C, Fresenius AG, Germany). Каждую дозу антитела разбавляли в 0,9% растворе хлорида натрия для инъекций (B. Braun Melsungen AG, Germany) до общего объема 20 мл.
В случае подкожного введения антитело вводят в виде однократной подкожной инъекции. Такую же процедуру осуществляли в случае плацебо.
Уровень псориаза, наблюдаемый у каждого пациента, регистрировали, используя индекс охвата и тяжести псориаза (PASI). Как описано выше, более высокие оценки PASI соответствуют более высокому уровню псориаза. Пациенты, включенные в испытание, имеют псориаз от умеренного от тяжелого, т.е. оценка PASI составляет 10 или выше.
Оценку PASI для пациента определяют до испытания, чтобы получить значение «исходного уровня» в 0 день, и многократно в ходе испытания на 5, 7, 14, 21, 28, 42, 56 и 75 день.
Группа дозирования I
Шесть пациентов из группы дозирования I получают однократную внутривенную дозу 0,5 мг BT061, а два пациента из группы дозирования 1 получают плацебо. Доза в расчете на массу и доза в расчете на площадь поверхности тела (ППТ) для каждого пациента показаны в таблице 14. Площадь поверхности тела вычисляли согласно формуле Мостеллера, описанной в настоящей публикации.
Оценки PASI для пациентов в группе дозирования I показаны в таблице 14 вместе с улучшением в процентах оценки PASI по сравнению с исходным уровнем.
Группа дозирования II
Шесть пациентов из группы дозирования II получают однократную внутривенную инъекцию 2,5 мг BT061, тогда как два пациента из группы дозирования 2 получают плацебо. Доза в расчете на массу и доза в расчете на площадь поверхности тела (ППТ) для каждого пациента показаны в таблице 15.
Оценки PASI для пациентов в группе дозирования II показаны в таблице 15 вместе с улучшением в процентах оценки PASI по сравнению с исходным уровнем.
Группа дозирования III
Шесть пациентов из группы дозирования III получают однократную внутривенную инъекцию 5,0 мг BT061, тогда как два пациента из группы дозирования III получают плацебо. Доза в расчете на массу и доза в расчете на площадь поверхности тела (ППТ) для каждого пациента показаны в таблице 15B.
Оценки PASI для пациентов в группе дозирования III показаны в таблице 15B вместе с улучшением в процентах оценки PASI по сравнению с исходным уровнем.
Группа дозирования IV
Шесть пациентов из группы дозирования IV получают однократную внутривенную инъекцию 10,0 мг BT061, тогда как два пациента из группы дозирования IV получают плацебо. Доза в расчете на массу и доза в расчете на площадь поверхности тела (ППТ) пациентов показаны в таблице 15C.
Оценки PASI для пациентов в группе дозирования IV показаны в таблице 15C вместе с улучшением в процентах оценки PASI по сравнению с исходным уровнем.
Оценки PASI для пациентов в группе дозирования I (внутривенная доза 0,5 мг) в ходе испытания
Оценки PASI для пациентов в группе дозирования II (внутривенная доза 2,5 мг) в ходе испытания
Оценки PASI для пациентов в группе дозирования III (внутривенная доза 5,0 мг) в ходе испытания
Оценки PASI для пациентов в группе дозирования IV (внутривенная доза 10,0 мг) в ходе испытания
Кроме того, оценки PASI в зависимости от времени для отдельных пациентов показаны в графической форме на фигурах 12A-12H и на фигурах 13A-13H. Графики, показанные на фигурах 12A-12H, представляют оценки PASI для пациентов из группы дозирования I, тогда как графики, показанные на фигурах 13A-13H, представляют оценки PASI для пациентов из группы дозирования II.
Как можно видеть из результатов, показанных в таблицах 14 и 15, у 75% всех пациентов из группы дозирования I и группы дозирования II наблюдается явное улучшение оценок PASI, т.е. по меньшей мере 40% улучшение по сравнению со значением исходного уровня после однократной дозы. Следует отметить, что 25% пациентов в группе дозирования I и группе дозирования II получали плацебо.
Действительно, в обеих группах дозирования у 50% пациентов наблюдали, по меньшей мере, 50% улучшение оценок PASI, при этом у одного пациента в группе дозирования II наблюдали 88% улучшение оценки PASI на 56 день (т.е. пациент 3 в таблице 15). Кроме того, терапевтический эффект был длительным даже при таких низких дозах, при этом улучшение у многих пациентов все еще продолжали наблюдать в конце испытания через 75 дней после введения.
У пациентов в группе дозирования III также наблюдали улучшение оценки PASI, при этом у шести из восьми пациентов наблюдали более чем 20% улучшение, и у двух из этих шести наблюдали более чем 30% улучшение после лечения. Однако улучшение не было таким значительным, как улучшение, наблюдаемое у пациентов из группы дозирования I и группы дозирования II, которые получали более низкую дозу антитела. Некоторую эффективность также наблюдали у пациентов группы дозирования IV. В частности, у пациентов 1, 4, 5 и 8 в данной группе дозирования (как показано в таблице 15C) наблюдали явное улучшение оценок PASI, хотя такое улучшение было ограниченным по сравнению с пациентами групп дозирования I-III.
Количество пациентов, у которых наблюдали, по меньшей мере, 40%, 50%, 60% и 75% улучшение оценки PASI, показано в таблице 16.
Суммарные результаты для групп дозирования I-III
0,5 мг BT-061
2,5 мг BT-061
5,0 мг BT061
На фигурах 14A и 14B представлены фотографии, доказывающие улучшение уровня псориаза до и после лечения. На фигуре 14A показана площадь псориаза на коже пациента в группе дозирования II перед введением. На фигуре 14B показана та же площадь псориаза через 28 дней после введения. Площади улучшения отмечены на фигуре 14B темными прямоугольниками.
На основании таких результатов можно ясно видеть, что BT061 обеспечивает эффективное лечение умеренного и тяжелого хронического псориаза.
Результаты данного исследования в сочетании с результатами, описанными выше в примере 7, которые показывают, что даже высокие дозы антитела согласно изобретению обычно хорошо переносимы людьми, свидетельствуют о способности фармацевтических композиций согласно изобретению обеспечивать эффективное длительное лечение аутоиммунных заболеваний с использованием схем дозирования, описанных в настоящей публикации.
ПРИМЕР 9 - Клиническое испытание BT061 у пациентов с ревматоидным артритом
Способность BT061 обеспечивать лечение ревматоидного артрита подвергают тестированию на пациентах, страдающих таким заболеванием. Испытание включает в себя исследование многократных доз с участием 96 пациентов, разделенных на 12 групп. В каждой группе два пациента получают плацебо, тогда как 6 пациентов получают BT061. Пациенты получают дозу один раз в неделю в течение периода времени 6 недель.
Пациентов делят на пациентов, получающих антитело подкожно, и пациентов, получающих антитело внутривенно. Группы, получающие подкожные дозы: 1,25 мг, 6,25 мг, 12,5 мг, 25 мг, 50 мг, 75 мг и 100 мг. Группы, получающие внутривенные дозы: 0,5 мг, 2 мг, 6,25 мг, 12,5 мг и 25 мг.
В группе, получающей подкожную дозу 1,25 мг, пациенты имели номера 101, 102, 103, 104, 105, 106, 107 и 108. В группе, получающей подкожную дозу 6,25 мг, пациенты имели номера 201-208. В группе, получающей подкожную дозу 12,5 мг, пациенты имели номера 301-308. В группе, получающей подкожную дозу 25 мг, пациенты имели номера 401-408. В группе, получающей подкожную дозу 50 мг, пациенты имели номера 501-508. В группе, получающей внутривенную дозу 6,25 мг, пациенты имели номера 601-608.
Способ внутривенного и подкожного введения был таким же, как способ, описанный в примере 8, используемый в испытании на пациентах с псориазом.
Уровень ревматоидного артрита регистрировали еженедельно, оценивая параметры ACR, и, в частности, исследуя количество болезненных и распухших суставов и затем уровни C-реактивного белка (CRP) и скорость оседания эритроцитов (СОЭ). Указанные параметры оценивали перед испытанием, чтобы получить значение «исходного уровня» в 0 день, и многократно во время периода испытания, и затем на 8, 22 и 43 день после окончания периода введения (т.е. последующее наблюдение на 8 день, наблюдение на 22 день и наблюдение на 43 день).
В таблицах представлены данные, полученные в таком испытании. В частности, в таблицах 17-22 представлено количество болезненных и распухших суставов на протяжении испытания.
Количество болезненных и распухших суставов в группе, получавшей дозу 1,25 мг подкожно
Неделя 1
Неделя 2
Неделя 3
Неделя 4
Неделя 5
Неделя 6
День 43 неделя 7
День 57 неделя 9
День 78 неделя 12
Количество болезненных и распухших суставов в группе, получавшей дозу 6,25 мг подкожно
Неделя 1
Неделя 2
Неделя 3
Неделя 4
Неделя 5
Неделя 6
День 43 неделя 7
День 57 неделя 9
День 78 неделя 12
Количество болезненных и распухших суставов в группе, получавшей дозу 12,5 мг подкожно
Неделя 1
Неделя 2
Неделя 3
Неделя 4
Неделя 5
Неделя 6
День 43 неделя 7
День 57 неделя 9
День 78 неделя 12
Количество болезненных и распухших суставов в группе, получавшей дозу 25 мг подкожно
Неделя 1
Неделя 2
Неделя 3
Неделя 4
Неделя 5
Неделя 6
День 43 неделя 7
День 57 неделя 9
День 78 неделя 12
Количество болезненных и распухших суставов в группе, получавшей дозу 50 мг подкожно
Неделя 1
Неделя 2
Неделя 3
Неделя 4
Неделя 5
Неделя 6
День 43 неделя 7
День 57 неделя 9
День 78 неделя 12
Количество болезненных и распухших суставов в группе, получавшей дозу 6,25 мг внутривенно
Неделя 1
Неделя 2
Неделя 3
Неделя 4
Неделя 5
Неделя 6
День 43 неделя 7
День 57 неделя 9
День 78 неделя 12
На фигуре 15 показан процент пациентов из групп дозирования, получавших подкожно 1,25 мг, 6,25 мг, 12,5 мг и 25 мг BT061, у которых было достигнуто по меньшей мере 20% улучшение соответствующих параметров ACR в ходе испытания, и процент пациентов, у которых достигнут по меньшей мере ответ ACR20 на 7 неделе.
В частности, можно видеть, что у 50% пациентов в группе, получавшей подкожную дозу 25 мг (т.е. у 4 из 8 пациентов, при этом 2 пациента получали плацебо), достигали по меньшей мере 20% улучшения соответствующих параметров ACR на 6 неделе. Указанные числовые значения возрастали до 5 из 8 пациентов на 7 неделе, т.е. у 5 из 8 пациентов достигали по меньшей мере ACR20. У одного пациента в данной группе дозирования достигали более чем 50% улучшения соответствующих параметров ACR на 5 и 6 неделе (полный набор данных не показан).
Позитивные результаты также получены у пациентов в других группах дозирования. У одного пациента в группе, получавшей дозу 6,25 мг подкожно, достигнуто по меньшей мере 50% улучшение соответствующих параметров ACR на 4 неделе, тогда как у другого достигнуто по меньшей мере 70% улучшение соответствующих параметров ACR на 3 неделе (полный набор данных не показан).
На фигурах 16A и 16B показаны результаты определения количества болезненных и распухших суставов, выявленных у пациентов из группы, получавшей дозу 25 мг BT061 подкожно на протяжении шестинедельного периода. У нескольких пациентов наблюдали уменьшение количества болезненных и распухших суставов в течение периода лечения. Результаты для одного отвечающего на лечение пациента и одного не отвечающего на лечение пациента из данной группы дозирования показаны на фигурах 17A и 17B соответственно. У отвечающего пациента выявлено значимое улучшение по количеству болезненных и распухших суставов и уровням боли.
Уменьшение количества болезненных и распухших суставов также наблюдали у пациентов из других групп дозирования. На фигурах 18A, 18B, 19A и 19B показано количество болезненных суставов в группах дозирования, получавших 1,25 мг подкожно, 6,25 мг подкожно, 50 мг подкожно и 6,25 мг внутривенно соответственно в ходе испытания и в течение нескольких недель после испытания.
Полученные результаты демонстрируют эффективность средства согласно настоящему изобретению в лечении ревматоидного артрита в диапазонах доз, представленных в данном описании.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛЕЧЕНИЯ АУТОИММУННОГО ЗАБОЛЕВАНИЯ (ВАРИАНТЫ) | 2009 |
|
RU2539110C2 |
| СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 2009 |
|
RU2540013C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 2009 |
|
RU2531548C2 |
| ГУМАНИЗИРОВАННОЕ АНТИ-CD4 АНТИТЕЛО С ИММУНОДЕПРЕССИВНЫМИ СВОЙСТВАМИ | 2004 |
|
RU2375375C2 |
| СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 2010 |
|
RU2598719C2 |
| АНТИТЕЛА ПРОТИВ ОХ40 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2562874C1 |
| ЛИГАНД GITR И СВЯЗАННЫЕ С ЛИГАНДОМ GITR МОЛЕКУЛЫ И АНТИТЕЛА И ВАРИАНТЫ ИХ ПРИМЕНЕНИЯ | 2004 |
|
RU2369636C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВОЛЧАНКИ | 2010 |
|
RU2607022C2 |
| ГУМАНИЗИРОВАННЫЕ АНТИТЕЛА ПРОТИВ IL-10 ДЛЯ ЛЕЧЕНИЯ СИСТЕМНОЙ КРАСНОЙ ВОЛЧАНКИ (SLE) | 2010 |
|
RU2587622C2 |
| КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ РАССЕЯННОГО СКЛЕРОЗА | 2008 |
|
RU2492234C2 |








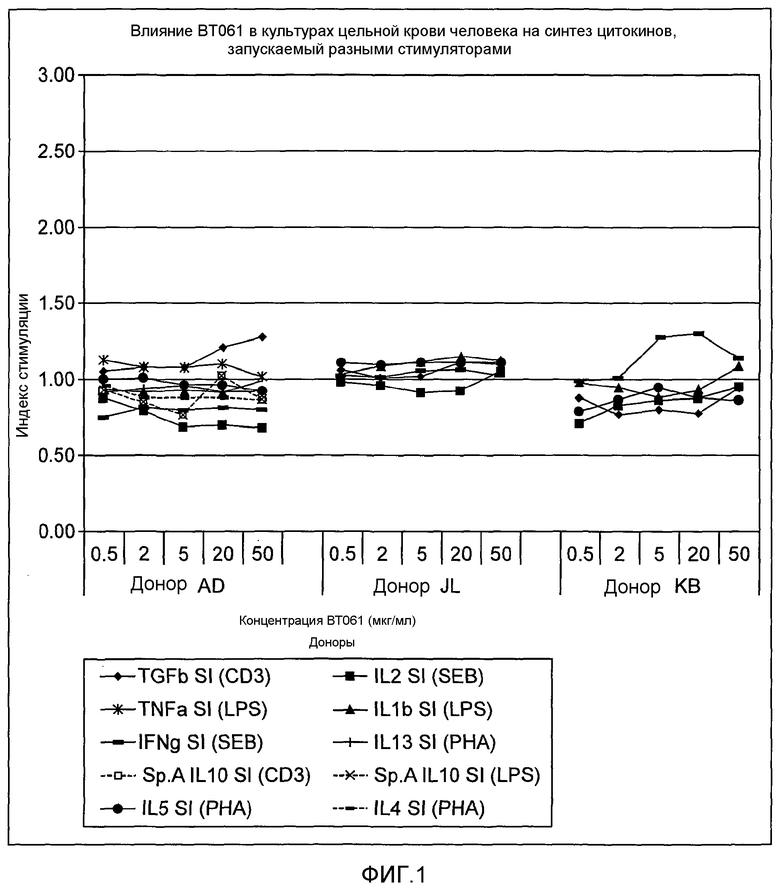





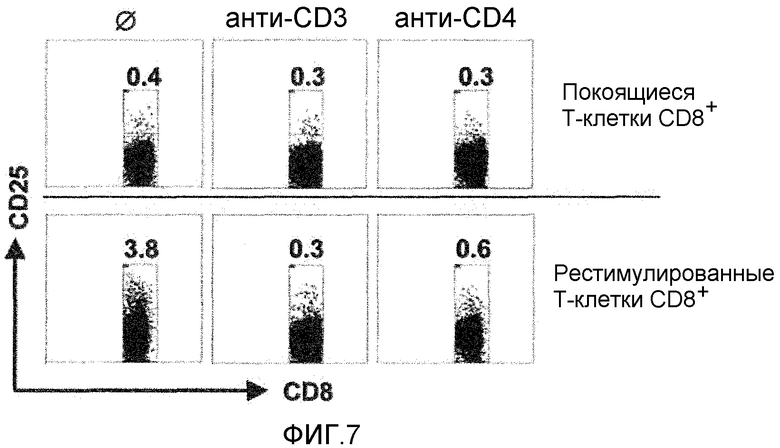
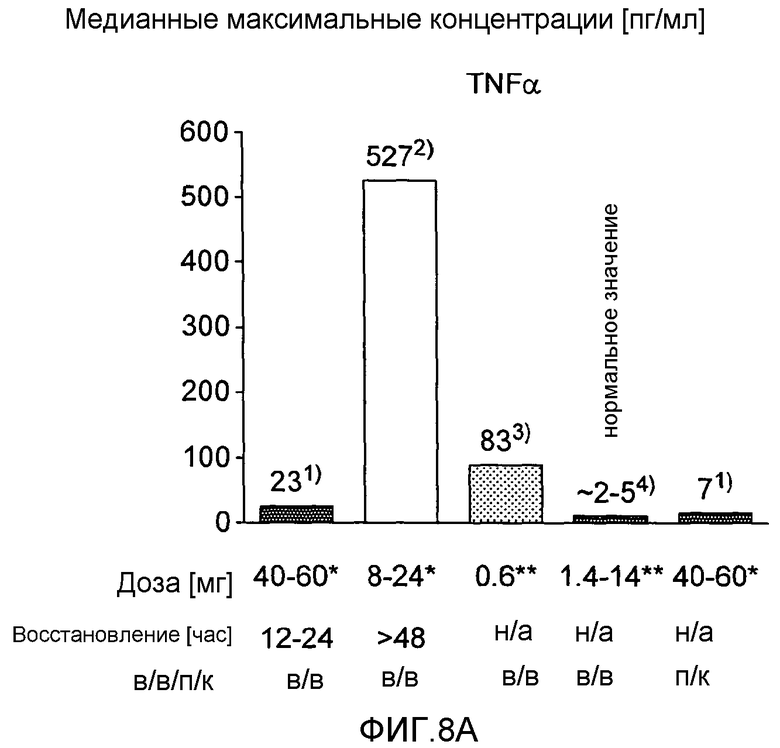
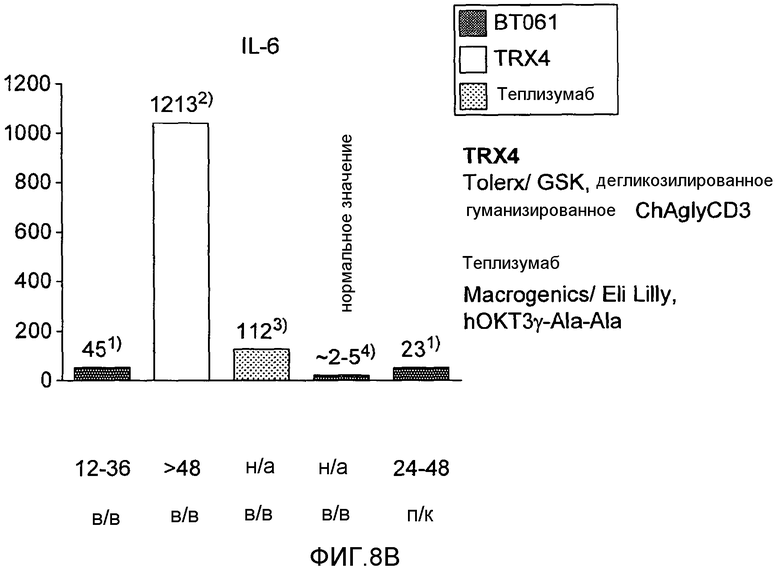
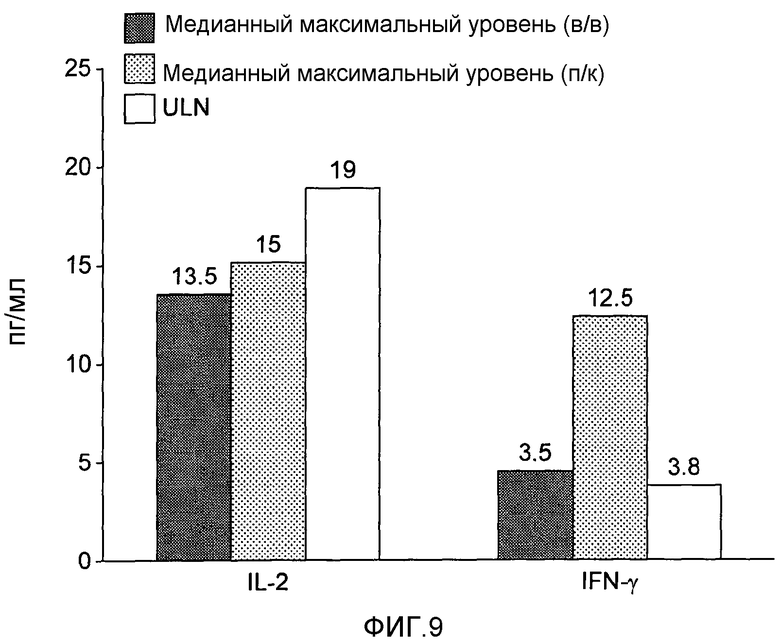
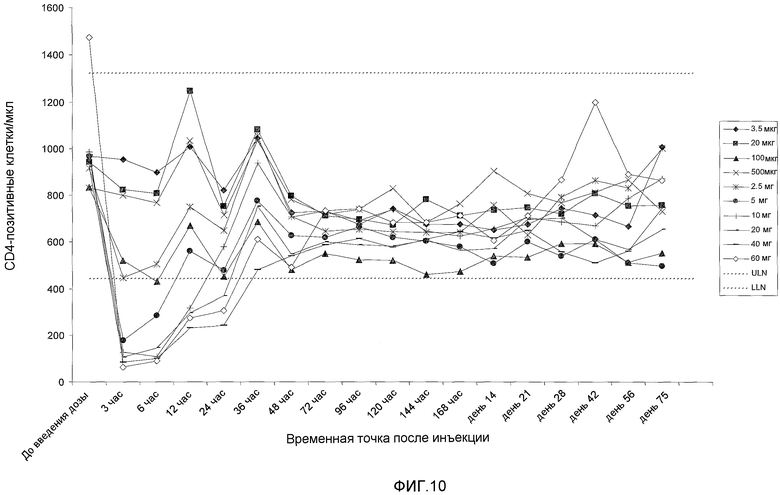
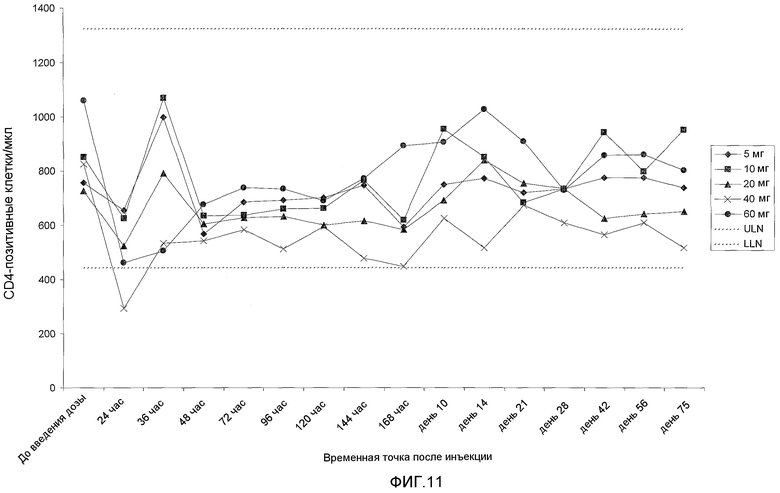

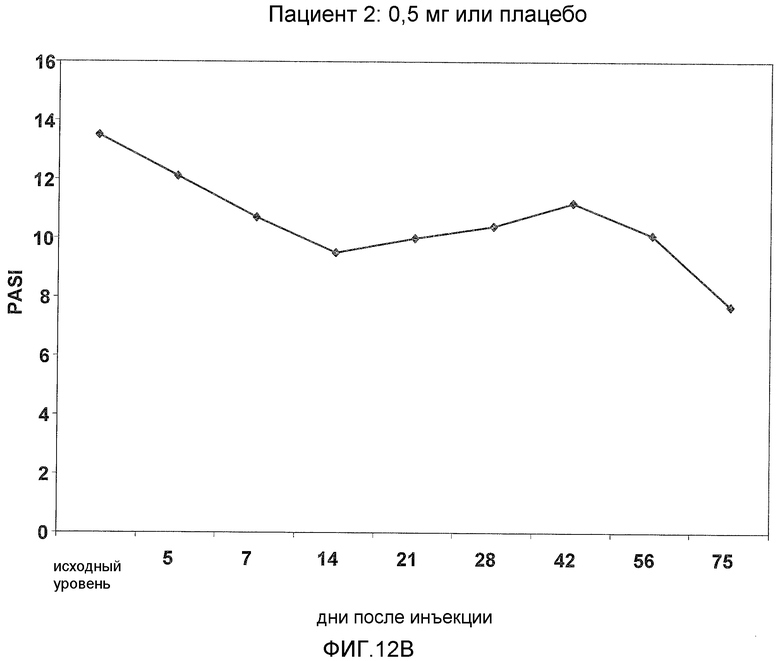
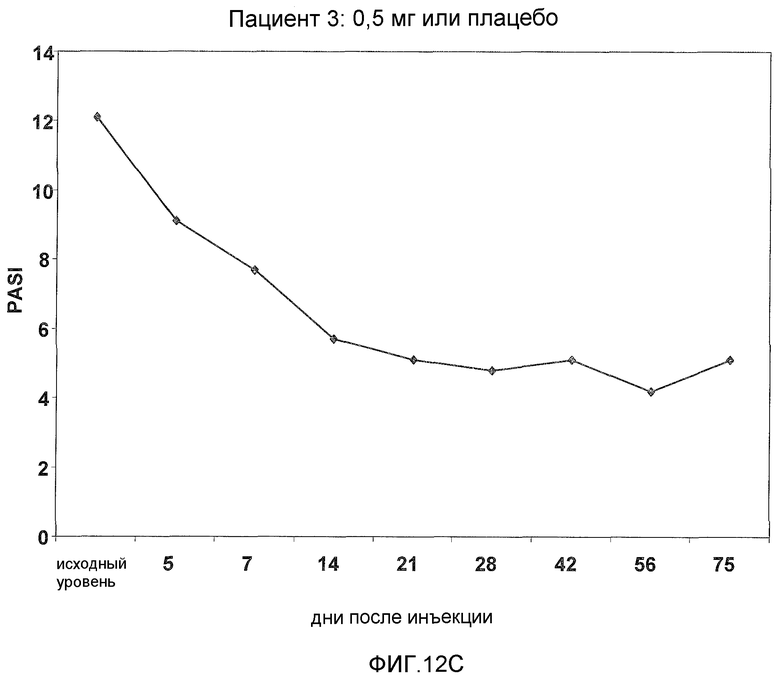

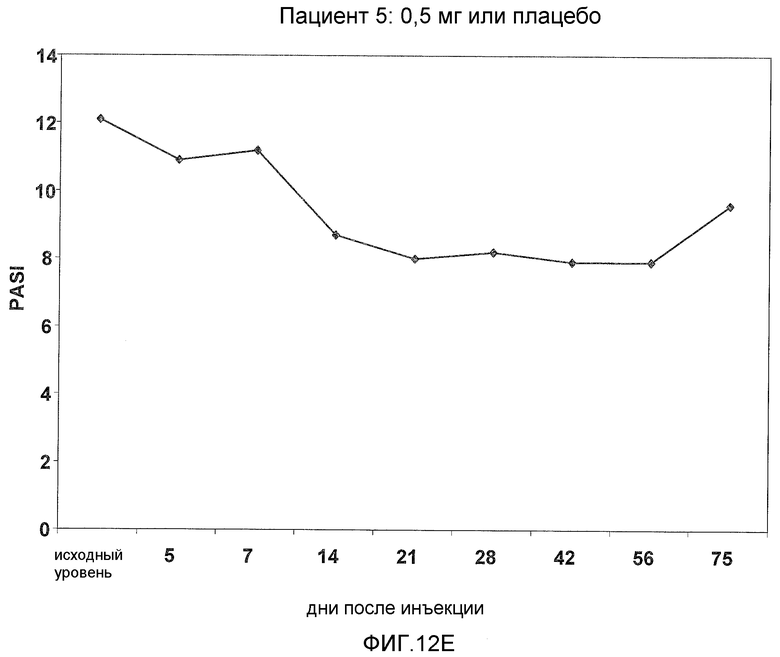
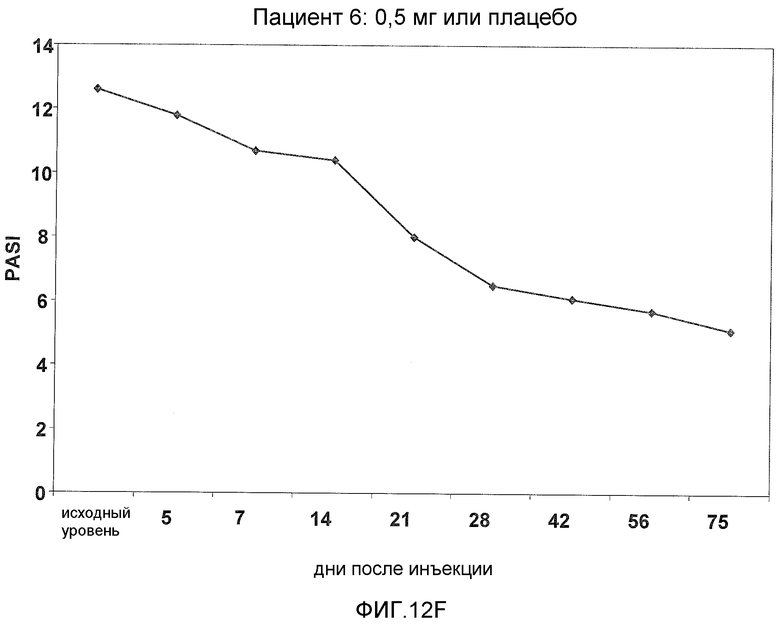
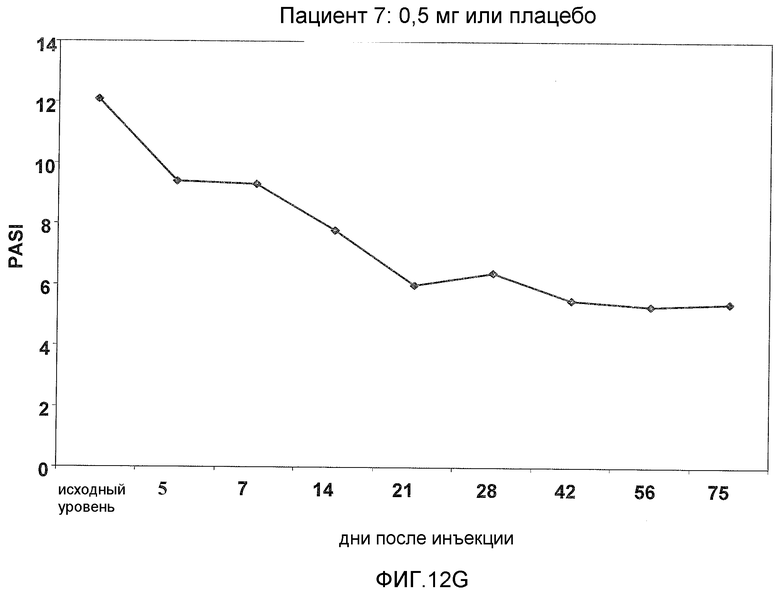

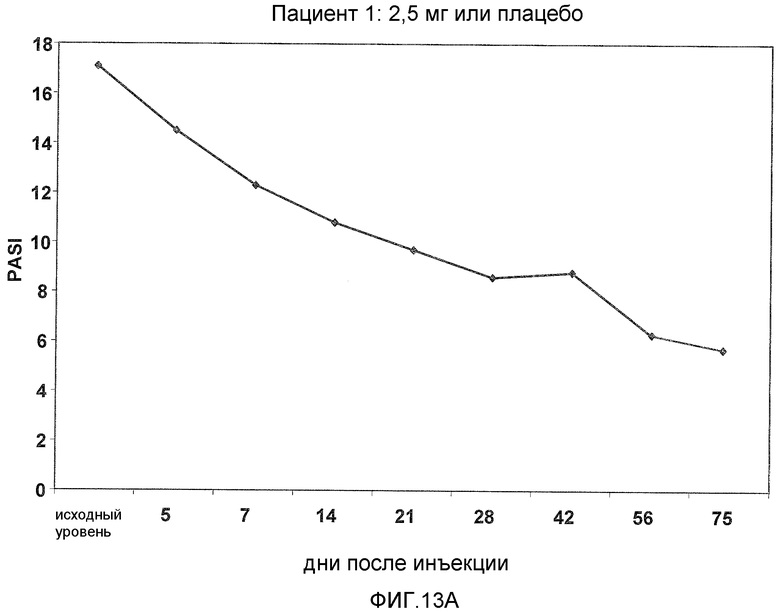
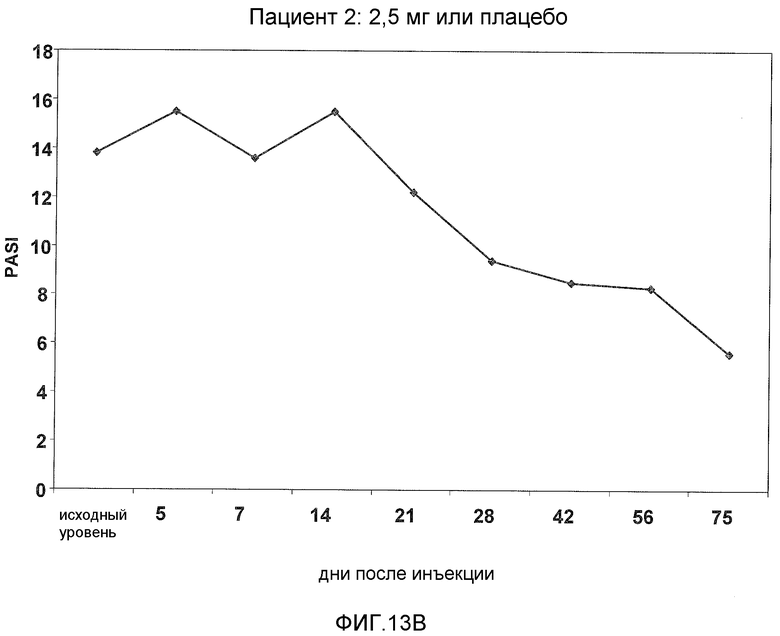
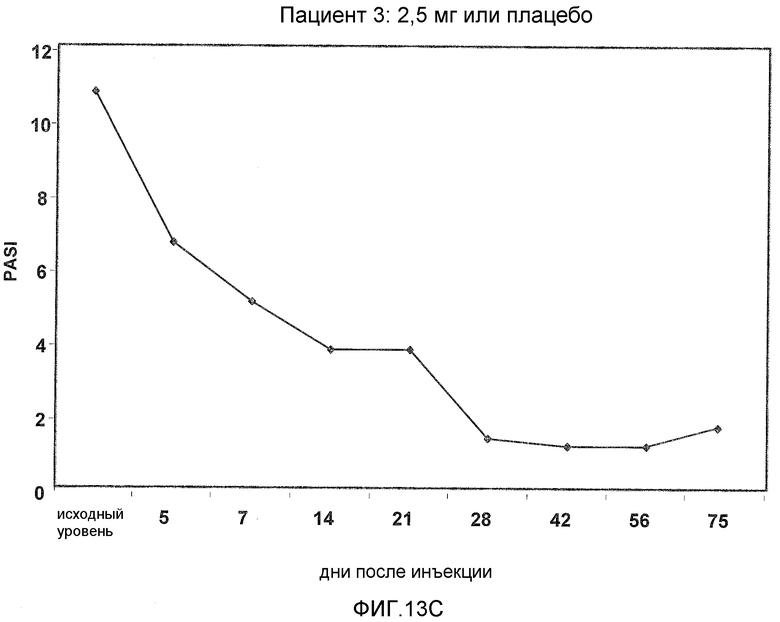
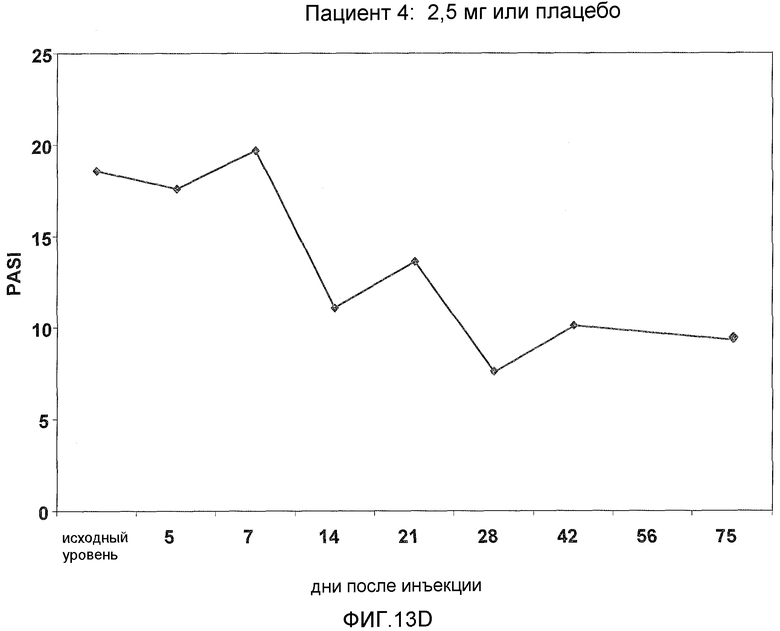
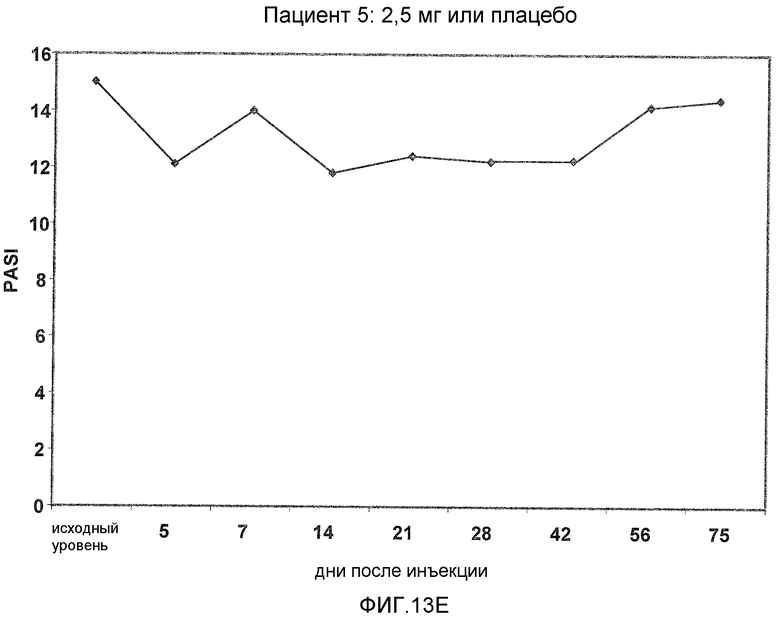
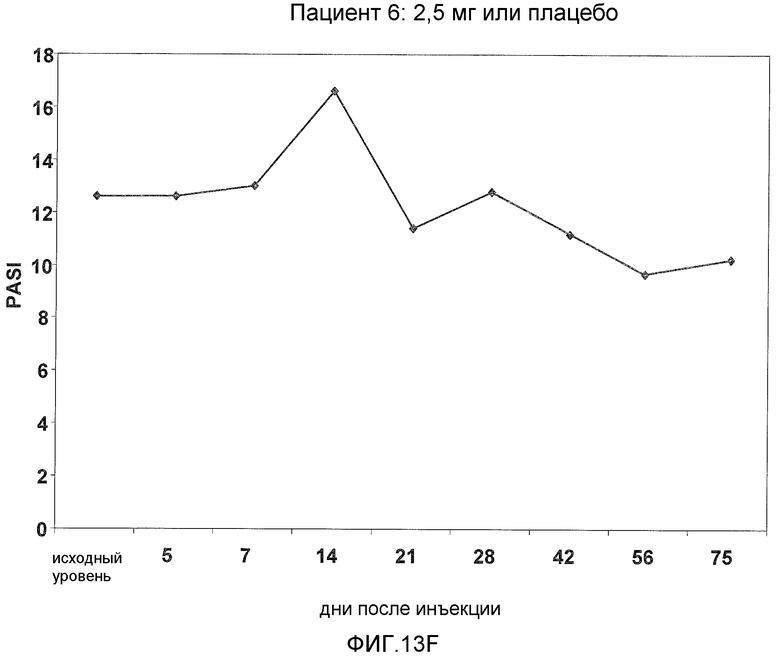
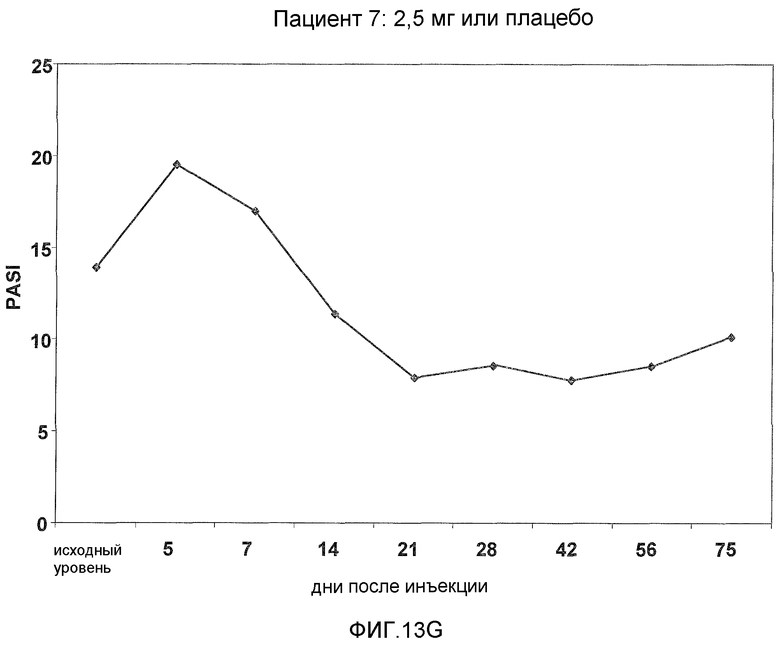
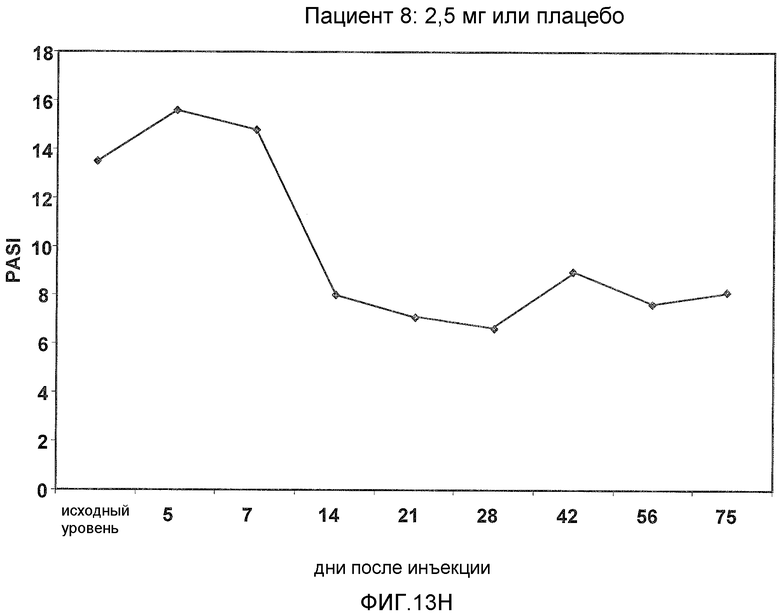
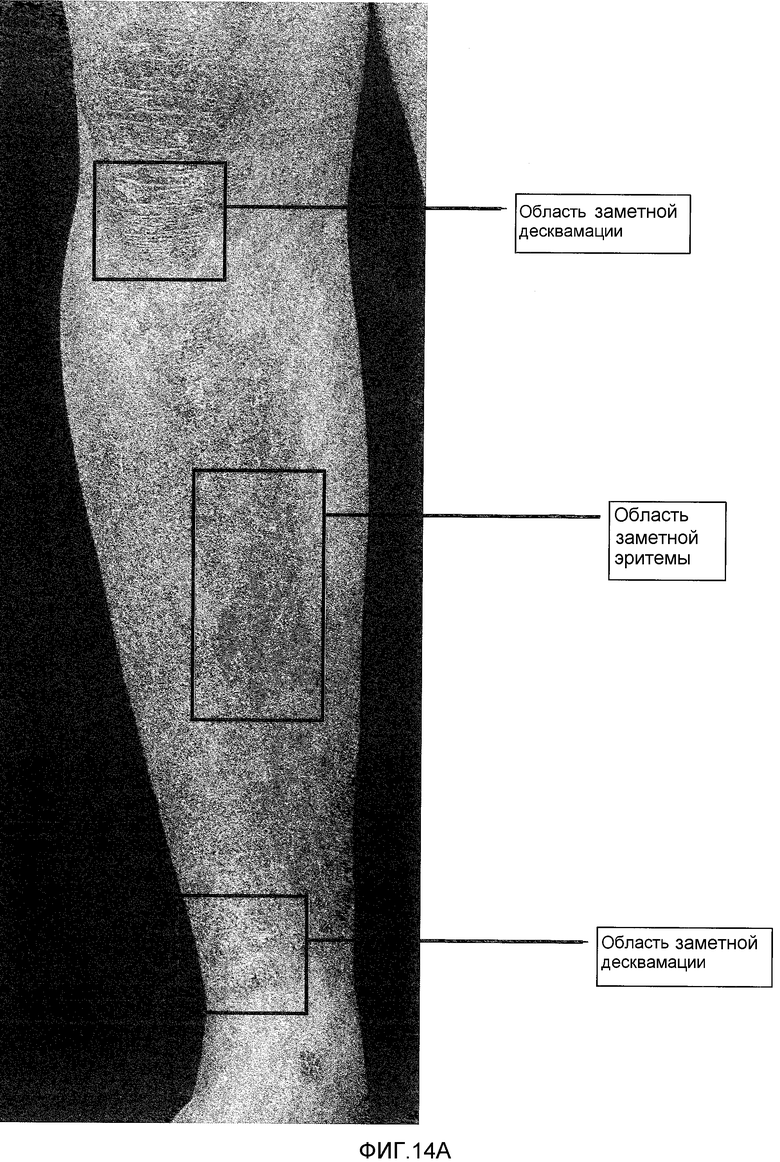
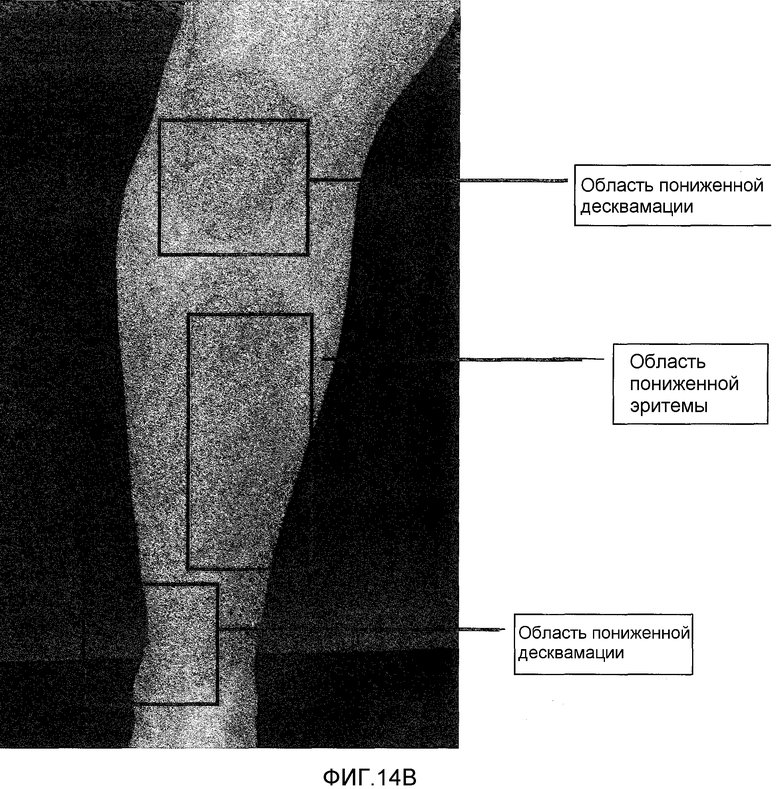
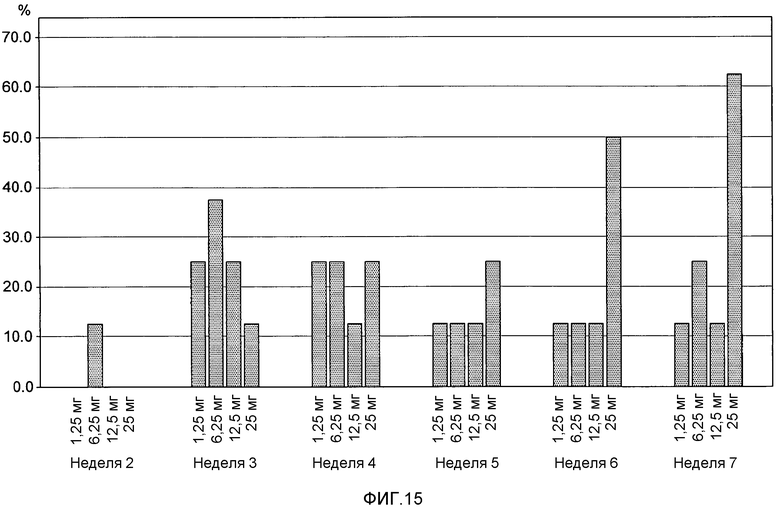
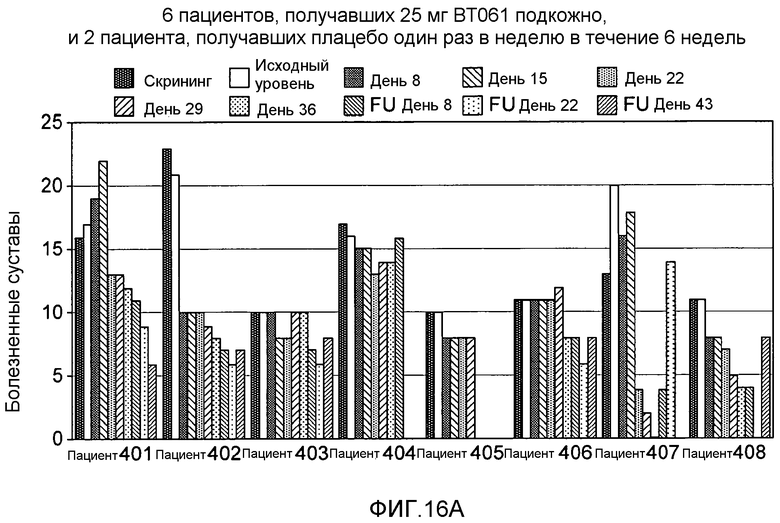
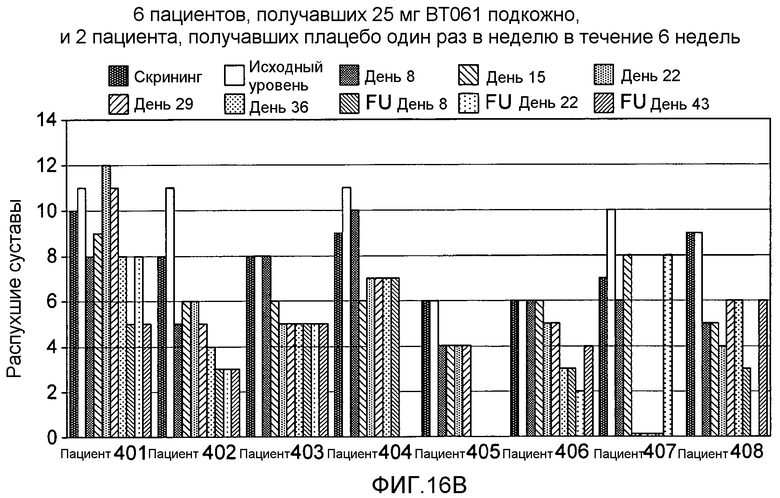
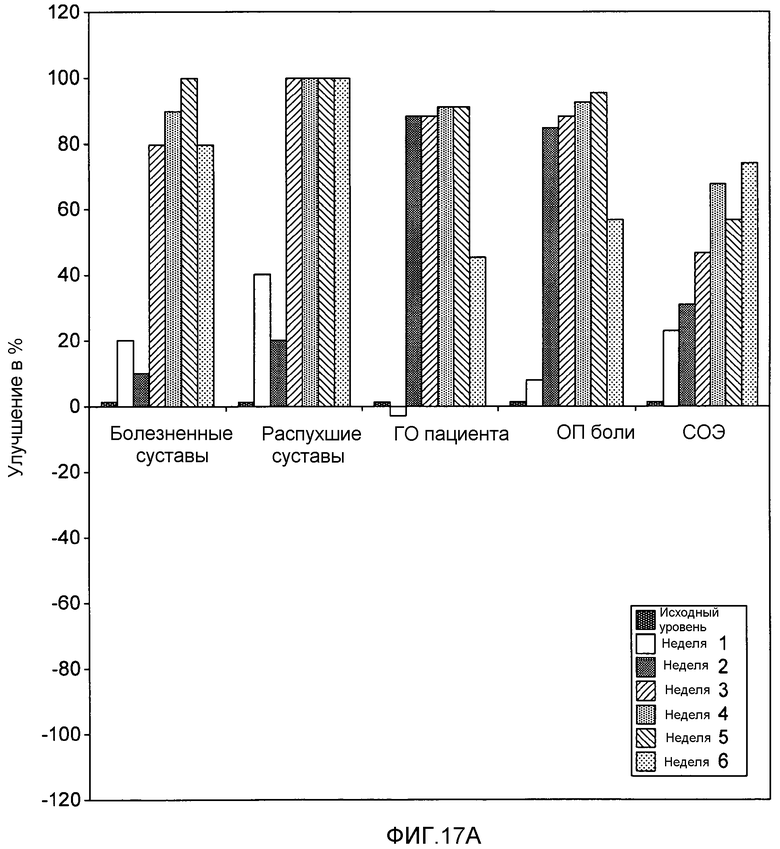


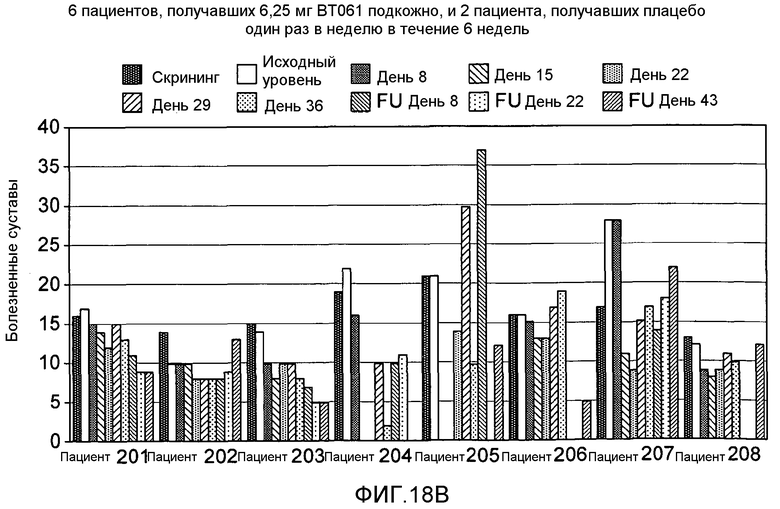
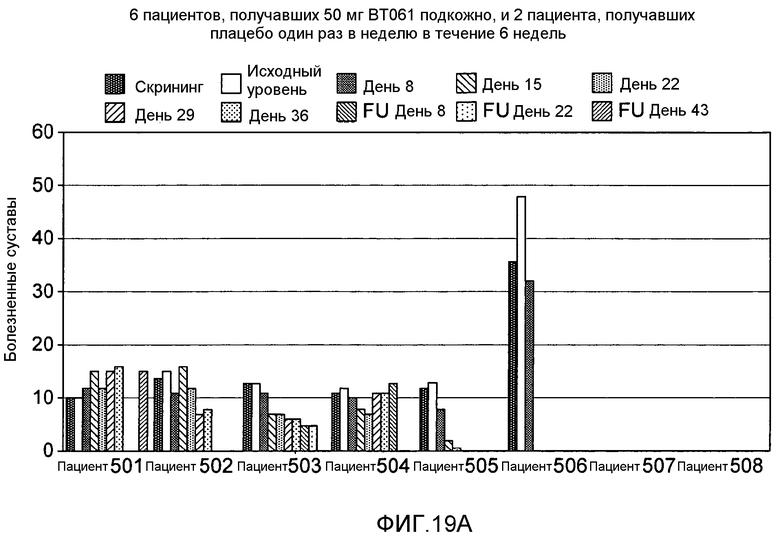

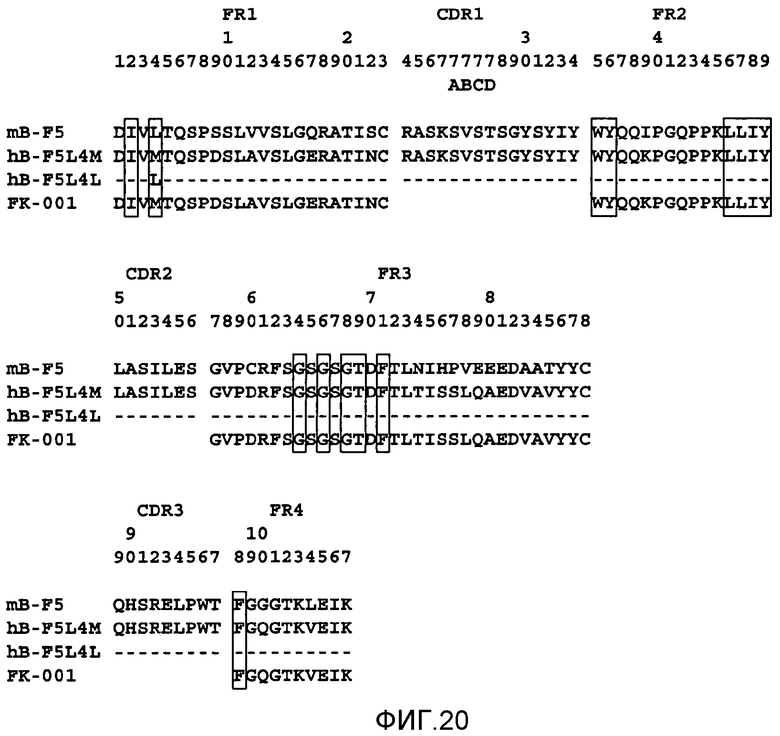
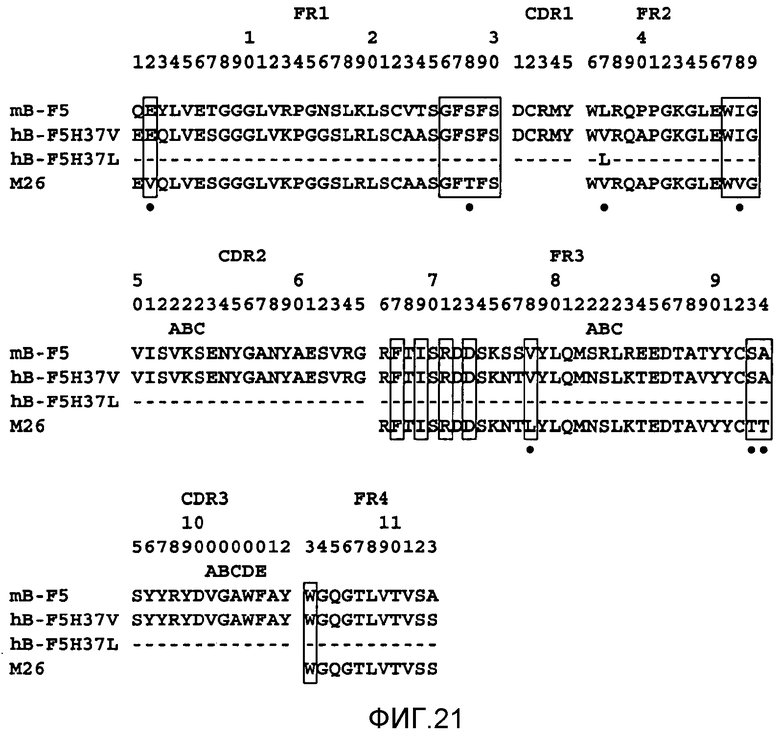
Изобретение относится к биотехнологии и представляет собой способы лечения аутоиммунного заболевания, включающие введение субъекту фармацевтической композиции, содержащей фармацевтически приемлемый носитель и гуманизированное анти-CD4-антитело, способное активировать регуляторные T-клетки CD4+CD25+, где указанное антитело содержит V-домен H-цепи, содержащий последовательности DCRMY, VISVKSENYGANYAESVRG и SYYRYDVGAWFAY, и V-домен L-цепи, содержащий последовательности RASKSVSTSGYSYIY, LASILES и QHSRELPWT, при этом указанную композицию вводят субъекту с частотой от еженедельного приема до введения через каждый 31 день и с дозой гуманизированного анти-CD4-антитела от 20 до 200 мг, либо от 8 до 60 мг/м2 площади поверхности тела субъекта, либо от 0,2 до 2 мг/кг. Настоящее изобретение также раскрывает набор для лечения аутоиммунного заболевания, который содержит множество доз указанной выше фармацевтической композиции. Настоящее изобретение позволяет вводить фармацевтическую композицию, содержащую гуманизированное анти-CD4-анитело, с более продолжительными интервалами дозирования и в более высоких дозах, не теряя при этом терапевтического эффекта и не вызывая увеличения побочных эффектов. 6 н. и 53 з.п. ф-лы, 41 ил., 27 табл., 9 пр.
1. Способ лечения аутоиммунного заболевания, включающий введение субъекту фармацевтической композиции, содержащей фармацевтически приемлемый носитель и гуманизированное анти-CD4-антитело, способное активировать регуляторные Т-клетки CD4+CD25+, где гуманизированное анти-CD4-антитело содержит V-домен Н-цепи, V-домен L-цепи и константный домен IgG1, где
(a) V-домен Н-цепи содержит последовательности DCRMY, VISVKSENYGANYAESVRG и SYYRYDVGAWFAY, и V-домен L-цепи содержит последовательности RASKSVSTSGYSYIY, LASILES и QHSRELPWT; и/или
(b) V-домен Н-цепи имеет по меньшей мере 80% идентичность последовательности SEQ ID No: 1, и V-домен L-цепи имеет по меньшей мере 80% идентичность последовательности SEQ ID No: 2,
при этом композицию необходимо вводить субъекту с частотой от еженедельного приема до ведения через каждый 31 день, и где композицию вводят субъекту с дозой гуманизированного анти-CD4-антитела от 20 до 200 мг.
2. Способ по п.1, где указанное введение осуществляют не чаще одного раза в каждые четыре недели.
3. Способ по п.1, где указанное введение осуществляют каждую неделю.
4. Способ п.1, указанное введение осуществляют каждые 10 дней.
5. Способ по п.1, где указанное введение осуществляют каждый месяц.
6. Способ по п.1, где доза гуманизированного анти-CD4-антитела составляет от 20 до 60 мг.
7. Способ по п.1, где доза гуманизированного анти-CD4-антитела составляет 20 мг, 60 мг или 100 мг.
8. Способ по п.1, где композицию вводят в дозе гуманизированного анти-CD4-антитела от 20 мг до 100 мг на дозу.
9. Способ по п.1, где гуманизированное анти-CD4 антитело присутствует в концентрации от 10 мкг/мл до 150 мкг/мл.
10. Способ по любому из пп.1-9, где аутоиммунное заболевание выбрано из псориаза, ревматоидного артрита, рассеянного склероза, диабета типа 1, воспалительных заболеваний кишечника, болезни Крона, тиреоидита Хашимото, аутоиммунного тиреоидита, аутоиммунной тяжелой псевдопаралитической миастении, системной красной волчанки и связанных с трансплантацией заболеваний, таких как реакции «трансплантат против хозяина» или проблемы, связанные с общей органной толерантностью.
11. Способ по п.1, где аутоиммунным заболеванием является псориаз.
12. Способ по п.1, где заболеванием является ревматоидный артрит.
13. Способ по любому из пп.1-9, где композиция предназначена для парентерального введения.
14. Способ по п.13, где введение является внутримышечным, внутривенным или подкожным.
15. Способ по п.14, где заболеванием является псориаз, и введение является внутривенным или подкожным.
Способ по п.14, где заболеванием является ревматоидный артрит, и введение является внутривенным или подкожным.
17. Способ по любому из пп.1-9, где в течение периода времени от 10 минут после начала введения до 96 часов после завершения введения уровень цитокина в плазме крови субъекта увеличен менее чем в 20 раз по сравнению с уровнем непосредственно перед введением, и где цитокин выбран из IFN-γ, TNF-α, IL-6 и IL-2.
18. Способ по любому из пп.1-9, где в течение периода времени от 10 минут после начала введения до 96 часов после завершения введения уровень цитокина в плазме крови субъекта увеличен менее чем в 20 раз по сравнению со значением верхней границы нормы (ULN), при этом цитокин выбран из IFN-γ, TNF-α, IL-6 и IL-2, и при этом значение ULN составляет 3,8, 2,8, 4,4 и 19,4 пг/мл, соответственно.
19. Способ по любому из пп.1-9, где в пределах периода времени от 72 до 96 часов после введения количество клеток лимфоцитов CD4+ в плазме крови субъекта составляет, по меньшей мере, 200 клеток/мкл.
20. Способ п.19, где в пределах периода времени от 72 до 96 часов после введения количество клеток лимфоцитов CD4+ в плазме крови субъекта составляет, по меньшей мере, 250 клеток/мкл.
21. Способ по любому из пп.1-9, где гуманизированное анти-CD4-антитело содержит V-домен Н-цепи, имеющий, по меньшей мере, 95% идентичность последовательности SEQ ID No: 1, и V-домен L-цепи, имеющий, по меньшей мере, 95% идентичность последовательности SEQ ID No: 2.
22. Способ по любому из пп.1-9, где гуманизированное анти-CD4-антитело получено из мышиного моноклонального анти-CD4-антитела B-F5.
23. Способ по любому из пп.1-9, где в течение периода времени от 3 до 6 часов после введения количество клеток лимфоцитов CD4+ в плазме крови субъекта составляет ниже 250 клеток/мкл.
24. Способ по любому из пп.1-9, где в течение периода времени от 72 до 96 часов после введения количество клеток лимфоцитов CD4+ в плазме крови у субъекта равно или выше 50% количества клеток у субъекта непосредственно перед введением.
25. Способ по п.11, где лечение обеспечивает, по меньшей мере, 40% улучшение оценки индекса охвата и тяжести псориаза (PASI) у субъекта.
26. Способ по п.11, где лечение обеспечивает, по меньшей мере, 50% улучшение оценки индекса охвата и тяжести псориаза (PASI) у субъекта.
27. Способ лечения аутоиммунного заболевания, включающий введение субъекту фармацевтической композиции, содержащей фармацевтически приемлемый носитель и гуманизированное анти-CD4-антитело, способное активировать регуляторные Т-клетки CD4+CD25+, где гуманизированное анти-CD4-антитело содержит V-домен Н-цепи, V-домен L-цепи и константный домен IgG1, где
(a) V-домен Н-цепи содержит последовательности DCRMY, VISVKSENYGANYAESVRG и SYYRYDVGAWFAY, и V-домен L-цепи содержит последовательности RASKSVSTSGYSYIY, LASILES и QHSRELPWT; и/или
(b) V-домен Н-цепи имеет по меньшей мере 80% идентичность последовательности SEQ ID No: 1, и V-домен L-цепи имеет по меньшей мере 80% идентичность последовательности SEQ ID No: 2, при этом композицию необходимо вводить субъекту с частотой от еженедельного приема до ведения через каждый 31 день, и где композицию вводят субъекту с дозой гуманизированного анти-CD4-антитела от 8 до 60 мг/м2 площади поверхности тела субъекта.
28. Способ по п.27, где аутоиммунное заболевание выбрано из псориаза, ревматоидного артрита, рассеянного склероза, диабета типа 1, воспалительных заболеваний кишечника, болезни Крона, тиреоидита Хашимото, аутоиммунного тиреоидита, аутоиммунной тяжелой псевдопаралитической миастении, системной красной волчанки и связанных с трансплантацией заболеваний, таких как реакции «трансплантат против хозяина» или проблемы, связанные с общей органной толерантностью.
29. Способ по п.27, где композиция предназначена для парентерального введения.
30. Способ по п.29, где введение является внутримышечным, внутривенным или подкожным.
31. Способ по п.30, где заболеванием является псориаз, и введение является внутривенным или подкожным.
32. Способ по п.30, где заболеванием является ревматоидный артрит, и введение является внутривенным или подкожным.
33. Способ по п.27, где в течение периода времени от 10 минут после начала введения до 96 часов после завершения введения уровень цитокина в плазме крови субъекта увеличен менее чем в 20 раз по сравнению с уровнем непосредственно перед введением, и где цитокин выбран из IFN-γ, TNF-α, IL-6 и IL-2.
34. Способ по п.27, где в течение периода времени от 10 минут после начала введения до 96 часов после завершения введения уровень цитокина в плазме крови субъекта увеличен менее чем в 20 раз по сравнению со значением верхней границы нормы (ULN), при этом цитокин выбран из IFN-γ, TNF-α, IL-6 и IL-2, и при этом значение ULN составляет 3,8, 2,8, 4,4 и 19,4 пг/мл, соответственно.
35. Способ по п.27, где в пределах периода времени от 72 до 96 часов после введения количество клеток лимфоцитов CD4+ в плазме крови субъекта составляет, по меньшей мере, 200 клеток/мкл.
36. Способ по п.35, где в пределах периода времени от 72 до 96 часов после введения количество клеток лимфоцитов CD4+ в плазме крови субъекта составляет, по меньшей мере, 250 клеток/мкл.
37. Способ по п.27, где гуманизированное анти-CD4-антитело содержит V-домен Н-цепи, имеющий, по меньшей мере, 95% идентичность последовательности SEQ ID No: 1, и V-домен L-цепи, имеющий, по меньшей мере, 95% идентичность последовательности SEQ ID No: 2.
38. Способ по п.27, где гуманизированное анти-CD4-антитело получено из мышиного моноклонального анти-CD4-антитела B-F5.
39. Способ по п.27, где в течение периода времени от 3 до 6 часов после введения количество клеток лимфоцитов CD4+ в плазме крови субъекта составляет ниже 250 клеток/мкл.
40. Способ по п.27, где в течение периода времени от 72 до 96 часов после введения количество клеток лимфоцитов CD4+ в плазме крови у субъекта равно или выше 50% количества клеток у субъекта непосредственно перед введением.
41. Способ лечения аутоиммунного заболевания, включающий введение субъекту фармацевтической композиции, содержащей фармацевтически приемлемый носитель и гуманизированное анти CD4 антитело, способное активировать регуляторные Т-клетки CD4+CD25+, где гуманизированное анти-CD4-антитело содержит V-домен Н-цепи, V-домен L-цепи и константный домен IgG1, где
(а) V-домен Н-цепи содержит последовательности DCRMY, VISVKSENYGANYAESVRG и SYYRYDVGAWFAY, и V-домен L-цепи содержит последовательности RASKSVSTSGYSYIY, LASILES и QHSRELPWT; и/или
(b) V-домен Н-цепи имеет по меньшей мере 80% идентичность последовательности SEQ ID No: 1, и V-домен L-цепи имеет по меньшей мере 80% идентичность последовательности SEQ ID No: 2, при этом композицию необходимо вводить субъекту с частотой от еженедельного приема до ведения через каждый 31 день, и где композицию вводят субъекту с дозой гуманизированного анти-CD4-антитела от 0,2 до 2 мг/кг.
42. Способ по п.41, где аутоиммунное заболевание выбрано из псориаза, ревматоидного артрита, рассеянного склероза, диабета типа 1, воспалительных заболеваний кишечника, болезни Крона, тиреоидита Хашимото, аутоиммунного тиреоидита, аутоиммунной тяжелой псевдопаралитической миастении, системной красной волчанки и связанных с трансплантацией заболеваний, таких как реакции «трансплантат против хозяина» или проблемы, связанные с общей органной толерантностью.
43. Способ по п.41, где композиция предназначена для парентерального введения.
44. Способ по п.43, где введение является внутримышечным, внутривенным или подкожным.
45. Способ по п.44, где заболеванием является псориаз, и введение является внутривенным или подкожным.
46. Способ по п.44, где заболеванием является ревматоидный артрит, и введение является внутривенным или подкожным.
47. Способ по п.41, где в течение периода времени от 10 минут после начала введения до 96 часов после завершения введения уровень цитокина в плазме крови субъекта увеличен менее чем в 20 раз по сравнению с уровнем непосредственно перед введением, и где цитокин выбран из IFN-γ, TNF-α, IL-6 и IL-2.
48. Способ по п.41, где в течение периода времени от 10 минут после начала введения до 96 часов после завершения введения уровень цитокина в плазме крови субъекта увеличен менее чем в 20 раз по сравнению со значением верхней границы нормы (ULN), при этом цитокин выбран из IFN-γ, TNF-α, IL-6 и IL-2, и при этом значение ULN составляет 3,8, 2,8, 4,4 и 19,4 пг/мл, соответственно.
49. Способ по п.41, где в пределах периода времени от 72 до 96 часов после введения количество клеток лимфоцитов CD4+ в плазме крови субъекта составляет, по меньшей мере, 200 клеток/мкл.
50. Способ по п.49, где в пределах периода времени от 72 до 96 часов после введения количество клеток лимфоцитов CD4+ в плазме крови субъекта составляет, по меньшей мере, 250 клеток/мкл.
51. Способ по п.41, где гуманизированное анти-CD4-антитело содержит V-домен Н-цепи, имеющий, по меньшей мере, 95% идентичность последовательности SEQ ID No: 1, и V-домен L-цепи, имеющий, по меньшей мере, 95% идентичность последовательности SEQ ID No: 2.
52. Способ по п.41, где гуманизированное анти-CD4-антитело получено из мышиного моноклонального анти-CD4-антитела B-F5.
53. Способ по п.41, где в течение периода времени от 3 до 6 часов после введения количество клеток лимфоцитов CD4+ в плазме крови субъекта составляет ниже 250 клеток/мкл.
54. Способ по п.41, где в течение периода времени от 72 до 96 часов после введения количество клеток лимфоцитов CD4+ в плазме крови у субъекта равно или выше 50% количества клеток у субъекта непосредственно перед введением.
55. Способ лечения аутоиммунного заболевания, предусматривающий введение лекарственного средства субъекту, при этом лекарственное средство содержит гуманизированное анти-CD4-антитело, содержащее константный домен IgG1, V-домен Н-цепи, имеющий SEQ ID NO:1, и V-домен L-цепи, имеющий SEQ ID NO:2, и при этом лекарственное средство вводят субъекту еженедельно, каждый 10 дней или каждый месяц, и где композицию вводят субъекту с количеством гуманизированного анти-CD4-антитела от 20 мг до 200 мг.
56. Способ лечения по п.55, в котором аутоиммунное заболевание определено в любом из пп.10-12.
57. Способ лечения аутоиммунного заболевания, предусматривающий введение лекарственного средства субъекту, при этом лекарственное средство содержит гуманизированное анти-CD4-антитело, содержащее константный домен IgG1, V-домен Н-цепи, имеющий SEQ ID NO:1, и V-домен L-цепи, имеющий SEQ ID NO:2,и при этом лекарственное средство вводят субъекту с частотой от еженедельного приема до ведения через каждый 31 день, и где композицию вводят подкожно субъекту с количеством гуманизированного анти-CD4-антитела от 20 мг до 200 мг.
58. Способ лечения по п.57, в котором аутоиммунное заболевание определено в любом из пп.10-12.
59. Набор для лечения аутоиммунного заболевания, содержащий множество доз фармацевтической композиции, причем фармацевтическая композиция содержит гуманизированное анти-CD4-антитело, способное активировать регуляторные Т-клетки CD4+CD25+, и фармацевтически приемлемый носитель, где фармацевтическая композиция содержит от 20 до 200 мг гуманизированного анти-CD4-антитела, где гуманизированное анти-CD4-антитело содержит V-домен Н-цепи, V-домен L-цепи и константный домен IgG1, где
(a) V-домен Н-цепи содержит последовательности DCRMY, VISVKSENYGANYAESVRG и SYYRYDVGAWFAY, и V-домен L-цепи содержит последовательности RASKSVSTSGYSYIY, LASILES и QHSRELPWT; и/или
(b) V-домен Н-цепи имеет по меньшей мере 80% идентичность последовательности SEQ ID No: 1, и V-домен L-цепи имеет по меньшей мере 80% идентичность последовательности SEQ ID No: 2.
| Способ оценки шелконосности коконов | 1987 |
|
SU1460088A1 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ С ИСПОЛЬЗОВАНИЕМ КОМБИНАЦИИ ЭЛИМИНИРУЮЩЕГО В-КЛЕТКИ/ИММУНОРЕГУЛЯТОРНОГО АНТИТЕЛ | 2001 |
|
RU2285541C2 |
| US 6037454 A, 14.03.2000 | |||

