Настоящее изобретение относится к ненуклеозидным соединениям формулы I и некоторым их производным, которые являются ингибиторами РНК-зависимой РНК вирусной полимеразы. Это соединения применимы для лечения РНК-зависимой РНК вирусной инфекции. Они являются особенно подходящими для применения в качестве ингибиторов NS5B полимеразы вируса гепатита С (HCV), в качестве ингибиторов репликации HCV и для лечения инфицирования вирусом гепатита С.
Вирус гепатита С является главной причиной хронических заболеваний печени во всем мире (Boyer, N. et al., J. Hepatol. 2000 32:98-112). Для пациентов, инфицированных посредством HCV, существует опасность развития цирроза печени и последующей гепатоцеллюлярной карциномы и, следовательно, наличие HCV является основным показанием для трансплантации печени.
HCV классифицирован, как представитель семейства вирусов Flaviviridae, которое включает виды флавивирусов, пестивирусов и гепацивирусов, которые включают вирусы гепатита С (Rice, С.М., Flaviviridae: The viruses and their replication. In: Fields Virology, Editors: B. N. Fields, D. М. Knipe and P. М. Howley, Lippincott-Raven Publishers, Philadelphia, Pa., Chapter 30, 931-959, 1996). HCV представляет собой оболочечный вирус, содержащий геном (+)-смысловой однотяжевой РНК, содержащий примерно 9,4 тысяч пар нуклеотидов. Вирусный геном содержит высококонсервативную 5'-нетранслированную область (НТО), длинную открытую рамку считывания, кодирующую предшественник полипротеина, содержащий примерно 3011 аминокислот, и короткую 3'-НТО.
С помощью генетического анализа HCV обнаружено 6 основных генотипов, которые отличаются друг от друга более чем на 30% последовательности ДНК. Охарактеризовано более 30 подтипов. В США примерно 70% инфицированных индивидуумов страдают от инфекции типа 1а и 1b. Инфекция типа 1b является наиболее распространенной в Азии (X. Forns and J. Bukh, Clinics in Liver Disease 1999 3:693-716; J. Bukh et al., Semin. Liv. Dis. 1995 15:41-63). К сожалению, инфекции типа 1 более устойчивы к лечению по сравнению с генотипами типа 2 или 3 (N. N. Zein, Clin. Microbiol. Rev., 2000 13:223-235).
Вирусные структурные белки содержат нуклеокапсидный белок (С) и 2 оболочечных гликопротеина, Е1 и Е2. HCV также кодирует 2 протеазы, цинк-зависимую металлопротеиназу, кодируемую в области NS2-NS3, и серинпротеазу, кодируемую в области NS3. Эти протеазы необходимы для расщепления конкретных областей предшественника полипротеина с образованием зрелых белков. Карбоксильная часть неструктурированного белка 5, NS5B, содержит РНК-зависимую РНК полимеразу. Функция остальных неструктурированных белков, NS4A и NS4B, и части NS5A (аминный концевой участок неструктурированного белка 5) остается неизвестной. Предполагается, что большинство неструктурированных белков, кодируемых геномом HCV РНК, вовлечены в репликацию РНК.
В настоящее время количество существующих утвержденных к применению способов лечения инфекции HCV ограничено. Новые и существующие терапевтические способы лечения инфекции HCV и ингибирования HCV NS5B полимеразы описаны в обзорах: R. G. Gish, Sem. Liver. Dis., 1999 19:5; Di Besceglie, A. М. and Bacon, B. R., Scientific American, October: 1999 80-85; G. Lake-Bakaar, Current and Future Therapy for Chronic Hepatitis C Virus Liver Disease, Curr. Drug Targ. Infect Dis. 2003 3(3):247-253; P. Hoffmann et al., Recent patent on experimental therapy for hepatitis С virus infection (1999-2002), Exp. Opin. Ther. Patents 2003 13(11):1707-1723; M. P. Walker et al., Promising Candidates for the treatment of chronic hepatitis C, Exp.Opin. Investing. Drugs 2003 12(8): 1269-1280; S.-L. Tan et al., Hepatitis C Therapeutics: Current Status and Emerging Strategies, Nature Rev. Drug Discov. 2002 1:867-881; J. Z. Wu and Z. Hong, Targeting NS5B RNA-Dependent RNA Polymerase for Anti-HCV Chemotherapy, Curr. Drug Targ. - Infect. Dis. 2003 3(3):207-219.
Рибавирин (амид 1-((2R,3R,5R)-3,4-дигидрокси-5-гидроксиметилтетрагидрофуран-2-ил)-1Н-[1,2,4]триазол-3-карбоновой кислоты; виразол®) является синтетическим неиндуцирующим интерферон противовирусным аналогом нуклеозида, обладающим широким спектром действия. Рибавирин in vitro обладает активностью по отношению к различным ДНК и РНК вирусам, включая Flaviviridae (Gary L. Davis. Gastroenterology 2000 118:S104-S114). Несмотря на то что при использовании в монотерапии рибавирин снижает содержание аминотрансферазы в сыворотке до нормального у 40% пациентов, он не снижает содержание HCV-PHK в сыворотке. Кроме того, рибавирин обладает значительной токсичностью и известно, что он вызывает анемию. Вирамидин является пролекарством рибаривина, которое в гепатоцитах превращается в рибаривин с помощью аденозиндеаминазы (J. Z. Wu, Antivir. Chem. Chemother. 2006 17(1):33-9).
Интерфероны (IFN) применялись для лечения хронического гепатита в течение примерно 10 лет. IFN представляют собой гликопротеины, продуцирующиеся иммунными клетками в ответ на вирусную инфекцию. Обнаружены два различных типа интерферона: тип 1 включает несколько интерферонов-альфа и один интерферон-бета, тип 2 включает интерферон-гамма. Интерфероны типа 1 в основном продуцируются инфицированными клетками и защищают соседние клетки от нового инфицирования. IFN подавляют вирусную репликацию многих вирусов, включая HCV, и если их используют в качестве единственного средства для лечения инфицирования гепатитом С, то IFN снижает содержание HCV-PHK в сыворотке до необнаруживаемого уровня. Кроме того, IFN нормализует содержания аминотрансферазы в сыворотке. К сожалению, действия IFN являются временными. Прекращение лечения приводит к рецидивам, частота которых составляет 70%, и только в 10-15% случаях обнаруживается постоянный ответ вирусов на лечение и нормальный уровень аланинтрансферазы в сыворотке (Davis, Luke-Bakaar, см. выше).
Одним из ограничений в использующихся ранее способах лечения с помощью IFN являлось быстрое выведение белка из крови. Химическое модифицирование IFN полиэтиленгликолем (ПЭГ) привело к получению белков со значительным образом улучшенными фармакокинетическими характеристиками. PEGASYS® представляет собой конъюгат интерферона α-2а с разветвленным монометокси-ПЭГ с молекулярной массой, равной 40 кДа, и PEG-INTRON® представляет собой конъюгат интерферона α-2а с монометокси-ПЭГ с молекулярной массой, равной 12 кДа (В. A. Luxon et al., Clin. Therap. 2002 24(9):13631383; A. Kozlowski and J. M. Harris, J. Control. Release 2001 72:217-224).
В настоящее время оптимальным способом лечения инфекции HCV является комбинированная терапия HCV с использованием рибаривина и иинтерферона-α. Комбинирование рибаривина и ПЭГ-IFN (см. ниже) приводит к постоянному ответу вирусов (ПОВ) на лечение у 54-56% пациентов, инфицированных посредством HCV типа 1. ПОВ достигает 80% для HCV типа 2 и 3 (Walker, см. выше). К сожалению, комбинированная терапия также приводит к побочным эффектам, которые приводят к затруднениям при клиническом использовании. Депрессия, симптомы, сходные с гриппом, и кожные реакции связаны с подкожным введением IFN-α, и гемолитическая анемия связана с длительным лечением рибаривином.
В настоящее время идентифицирован целый ряд возможных молекулярных мишеней для разработки лекарственных средств, использующихся в качестве анти-HCV средств, включая, но не ограничиваясь только ими, NS2-NS3 аутопротеазу, NS3 протеазу, NS3 геликазу и NS5B полимеразу. РНК-зависимая РНК полимераза совершенно необходима для репликации генома однотяжевой (+)-смысловой РНК. Этот фермент вызывает значительный интерес у медицинских химиков.
Нуклеозидные ингибиторы могут действовать в качестве агентов, обрывающих цепь, или в качестве конкурентных ингибиторов, которые препятствует связыванию нуклеотидов с полимеразой. Для воздействия в качестве агентов, обрывающих цепь, аналог нуклеозида необходимо in vivo включить в клетку и превратить in vivo в его трифосфат, чтобы он выступал в роли конкурента в качестве субстрата для центра связывания полимеразы с нуклеотидом. Это превращение в трифосфат обычно опосредуется клеточными киназами, которые налагают дополнительные ограничения на структуру любых нуклеозидов. Кроме того, такая необходимость фосфорилирования ограничивает прямую оценку действия нуклеозидов как ингибиторов репликации HCV в исследованиях, основанных на использовании клеток (J. A. Martin et al., патент U.S. №6846810; С.Pierra et al., J. Med. Chem. 2006 49(22):6614-6620; J. W. Tomassini et al., Antimicrob. Agents and Chemother. 2005 49(5):2050; J. L. dark et al.,J. Med. Chem. 2005 48(17):2005).
Соединения, предлагаемые в настоящем изобретении, их изомерные формы и фармацевтически приемлемые соли также применимы для лечения и предупреждения вирусных инфекций, в частности инфицирования гепатитом С, и заболевания живых реципиентов, при их применении в комбинации друг с другом и с другими биологически активными средствами, включая, но не ограничиваясь только ей, группу, включающую интерферон, пэгилированный интерферон, рибаривин, ингибиторы протеазы, ингибиторы полимеразы, небольшие соединения, мешающие РНК, антисмысловые соединения, аналоги нуклеотидов, аналоги нуклеозидов, иммуноглобулины, иммуномодуляторы, гепатозащитные средства, противовоспалительные средства, антибиотики, противовирусные и противоинфекционные соединения. Такая комбинированная терапия также может включать введения соединения, предлагаемого в настоящем изобретении, одновременно или последовательно с другими лекарственными средствами или усиливающими воздействие средствами, такими как рибаривин и родственные соединения, амантадин и родственные соединения, различные интерфероны, такие как, например, интерферон-альфа, интерферон-бета, интерферон-гамма и т.п., а также альтернативные формы интерферонов, такие как пэгилированные интерфероны. Кроме того, в качестве дополнительной комбинированной терапии комбинации рибаривина и интерферона можно вводить по меньшей мере с одним из соединений, предлагаемых в настоящем изобретении.
Другие интерфероны, которые в настоящее время разрабатываются, включают альбинтерферон-α-2b (альбуферон), IFN-омега с ДУРОС, ЛОКТЕРОНТМ и интерферон-α-2b XL. Ожидается, что эти и другие интерфероны поступят на рынок для применения в комбинированной терапии с соединениями, предлагаемыми в настоящем изобретении.
Ингибиторы HCV полимеразы являются другой мишенью для создания лекарственных средств и соединения, которые разрабатываются, включают R-1626, R-7128, IDX184/IDX102, PF-868554 (Pfizer), VCH-759 (ViroChem), GS-9190 (Gilead), A-837093 и А-848837 (Abbot), MK-3281 (Merck), GSK949614 и GSK625433 (Glaxo), ANA598 (Anadys), VBY 708 (ViroBay).
Ингибиторы HCV NS3 протеазы также идентифицированы как потенциально применимые для лечения инфекции HCV. Ингибиторы протеазы, использующиеся в клинических испытаниях, включают VX-950 (телапревир, Vertex), SCH503034 (боцепревир, Schering), TMC435350 (Tibotec/Medivir) и ITMN-191 (Intermune). Другие ингибиторы протеазы, находящиеся на начальных стадиях разработки, включают МК.7009 (Merck), BMS-790052 (Bristol Myers Squibb), VBY-376 (Virobay), IDXSCA/IDXSCB (Idenix), BI12202 (Boehringer), VX-500 (Vertex), PHX1766 Phenomix).
Другие мишени анти-HCV терапии, находящиеся в стадии исследования, включают ингибиторы циклофилина, которые ингибируют связывание РНК с NS5b, нитазоксанид, целгосивир (Migenix), ингибитор α-глюкозидазы-1, ингибиторы каспазы, агонисты Toll-подобного рецептора и иммуностимуляторы, такие как задаксин (SciClone).
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В настоящее время не существует способов предупредительного лечения вируса гепатита С (HCV), и в настоящее время количество существующих утвержденных к применению способов лечения только HCV ограничено. Поэтому необходимы создание и разработка новых фармацевтических соединений.
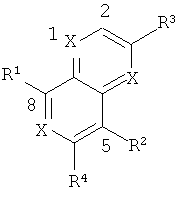
Одним объектом настоящего изобретения является соединение формулы I:
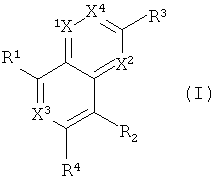
в которой
Х обозначает N и Х2, Х3 и Х4 обозначают CR5; или
Х1 и Х2 обозначают N и Х3 и Х4 обозначают CR5; или
Х1, Х2 и Х4 обозначают CR5, и Х3 обозначает N; или
Х1 и Х4 обозначают N и Х2 и Х3 обозначают CR5; или
X1, X2, X3 и X4 обозначают CR5;
R1 обозначает (а) гетероарильный радикал, выбранный из группы, включающей пиридинил, 2-оксо-1,2-дигидропиридин-3-ил, 3-оксо-3,4-дигидропиразин-2-ил, 3-оксо-2,3-дигидропиридазин-4-ил, 2-оксо-1,2-дигидропиримидин-4-он-5-ил, 6-оксо-1,6-дигидро-[1,2,4]триазин-5-ил, 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, 2-оксо-2(Н)-пиридин-1-ил, 6-оксо-6Н-пиридазин-1-ил, 6-оксо-6Н-пиримидин-1-ил и 2-оксо-2Н-пиразин-1-ил, указанный гетероарил необязательно содержит в качестве заместителей галоген, С1-С6-алкил, C1-С3-галогеналкил, С1-С6-гидроксиалкил, С1-С3-алкокси-С1-С3-алкил, C1-С6-алкоксигруппу, Х1(CH2)16СО2Н или Xl-(CH2)2-6NRgRh, или
(b) гетероциклический радикал, выбранный из группы, включающей 2-оксотетрагидропиримидин-1-ил, 2-оксоимидазолидин-1-ил, 2-оксопиперидин-1-ил, 2-оксопирролидин-1-ил, 2,6-диоксотетрагидропиримидин-1-ил, 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил и 2,5-диоксоимидазолидин-1-ил и 2,4-диоксотетрагидропиримидин-1-ил;
R2 обозначает водород, C1-С6-алкоксигруппу, C1-С6-алкил, C1-С6-галогеналкил, C1-С6-галогеналкоксигруппу или галоген;
R3 обозначает (а) арил,
(b) гетероарил, где указанный арил или указанный гетероарил независимо необязательно содержат 1-3 заместителя, выбранных из группы, включающей гидроксигруппу, C1-С6-алкоксигруппу, C1-С6-алкил, C1-С6-гидроксиалкил, галоген, (CH2)nNRcRd, цианогруппу, C1-С6-алкоксикарбонил, карбамоил, N-алкилкарбамоил, N,N-диалкилкарбамоил, (СН2)0-3CO2H, SO2NH2, C1-С6-алкилсульфинил и C1-С6-алкилсульфонил
(c) NRaRb,
(d) водород,
(e) галоген
или (f)-X(R7)[C(R6)2-6NReRf, где Х обозначает О или NR7, или
R7 обозначает водород или С2-С4-алкил,
R6 в каждом случае независимо обозначает водород, C1-С3-алкил, или
два фрагмента R6, присоединенные к одному и тому же атому углерода, образуют С2-С5-алкилен или
два фрагмента R6, присоединенные к разным атомам углерода, образуют C1-C4-алкилен;
Ra и Rb вместе с атомом азота, к которому они присоединены, образуют циклический амин, независимо замещенный 1-3 группами, независимо выбранными из группы, включающей C1-С6-алкил, галоген или (CH2)nNReRf;
Rc и Rd независимо обозначают водород, C1-С6-алкил, C1-С6-галогеналкил, C1-С6-ацил, SO2R8, где R8 обозначает (а) C1-С6-алкил, (b) C1-С6-галогеналкил, (с) С3-С7-циклоалкил, (d) С3-С7-пиклоалкил-С1-С3-алкил, (е) С1-С6-алкокси-С1-С6-алкил или (f) SO2[C(R9)2]0-6NRkR1, C1-С3-алкилкарбамоил или C1-С3-диалкилкарбамоил;
Re и Rf независимо обозначают водород, C1-С6-алкил, C1-С6-галогеналкил, C1-С6-ацил, SO2R8, где R8 обозначает (а) C1-С6-алкил, (b) C1-С6-галогеналкил, (с) С3-С7-циклоалкил, (d) С3-С7-циклоалкил-С1-С3-алкил, (е) С1-С6-алкокси-С1-С6-алкил или (f) SO2[C(R9)2]0-6NRkR1;
Ri и Rj (i) независимо обозначают водород, C1-С3-алкил или (CH2)2-6NRgRh, или (ii) вместе с атомом азота, к которому они присоединены, образуют (CH2)2Х5(CH2)2, где X5 обозначает О или NRk и Rk обозначает водород, C1-С3-алкил, C1-С3-ацил или C1-С3-алкилсульфонил;
R4 обозначает водород, CF3, CH2CF3, С3-С5-циклоалкил, галоген, C1-С6-алкоксигруппу, C1-С3-галогеналкоксигруппу, CHR4aR4b или CR4aR4bR4c,
где (i) R4a, R4b и R4c независимо выбраны из группы, включающей C1-С3-алкил, CD3, С1-С2-алкоксигруппу, С1-С2-фторалкил, C1-С3-гидроксиалкил, цианогруппу или гидроксигруппу; или
(ii) взятые вместе R4a и R4b образуют С2-С4-алкилен и R4c обозначает водород, C1-С3-алкил, С1-С2-алкоксигруппу, галоген, C1-С3-гидроксиалкил, цианогруппу или С1-С2-фторалкил, или R4a и R4b вместе с атомом углерода, к которому они присоединены, образуют 3-оксетанил или тетрагидрофуран-2-ил;
R5 в каждом случае независимо обозначает водород, галоген, C1-C6-алкоксигруппу или C1-С6-алкил;
R8, Rg и Rh в каждом случае независимо обозначают водород или C1-С3-алкил;
Rk и R1 (i) в каждом случае независимо обозначают водород или C1-С6-алкил, или
(ii) Rk и R1 вместе с атомом азота, к которому они присоединены, образуют циклический амин;
n в каждом случае независимо равно 0-3; или
его фармацевтически приемлемая соль.
Соединения общей формулы I могут являться нейтральными соединениями или их фармацевтически приемлемыми солями.
Настоящее изобретение также относится к способу лечения инфицирования вирусом гепатита С (HCV) путем введения нуждающемуся в нем пациенту соединения формулы I в терапевтически эффективном количестве. Соединение можно вводить по отдельности или можно вводить совместно с другими противовирусными соединениями или иммуномодуляторами.
Настоящее изобретение также относится к способу ингибирования репликации HCV в клетке путем введения соединения формулы I в количестве, эффективном для подавления HCV.
Настоящее изобретение также относится к применению соединения формулы I для лечения инфицирования вирусом гепатита С (HCV) или для приготовления лекарственного средства, предназначенного для лечения инфицирования вирусом гепатита С (HCV). Соединение можно вводить по отдельности или можно вводить совместно с другими противовирусными соединениями или иммуномодуляторами.
Настоящее изобретение также относится к применению соединения формулы I для подавления репликации HCV в клетке или для приготовления лекарственного средства, предназначенного для подавления репликации HCV в клетке.
Настоящее изобретение также относится к фармацевтической композиции, включающей соединение формулы I и по меньшей мере один фармацевтически приемлемый носитель, разбавитель или инертный наполнитель.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
При использовании в настоящем изобретении объект в форме единственного числа относится к одному или большему количеству объектов; например, "соединение" означает одно или большее количество соединений или не менее одного соединения. Форма единственного числа и выражения "один или большее количество" и "не менее чем один" в настоящем изобретении можно использовать как взаимозаменяемые.
Выражение "определенная выше в настоящем изобретении" относится к самому широкому определению каждой группы, приведенному в Кратком изложении сущности изобретения. Для всех остальных вариантов осуществления, приведенных ниже, заместители, которые могут использоваться в каждом варианте осуществления и которые не определены явно, соответствуют самому широкому определению, приведенному в Кратком изложении сущности изобретения.
При использовании в настоящем описании, в промежуточной фразе или в содержании пункта формулы изобретения термины "включает" и "включающий" следует интерпретировать как допускающие изменения значения. Таким образом, термины следует интерпретировать как синонимичные с выражениями "содержащий по меньшей мере" или "включающий по меньшей мере". При использовании в контексте способа термин "включающий" означает, что способ включает по меньшей мере указанные стадии, но может включать дополнительные стадии. При использовании в контексте соединения или композиции термин "включающий" означает, что соединение или композиция включает по меньшей мере указанные характеристики или компоненты, но также может включать дополнительные характеристики или компоненты.
Термин "независимо" при использовании в настоящем изобретении означает, что переменную используют в любом случае независимо от присутствия или отсутствия в том же соединении переменной, обладающей таким же или другим определением. Таким образом, в соединении, в котором R" содержится дважды и определен как "независимо обозначает углерод или азот", оба R" могут обозначать углерод, оба R" могут обозначать азот или один R" может обозначать углерод и другой может обозначать азот.
Если любая переменная (например, R1, R4a, Ar, X или Het) содержится более одного раза в любом фрагменте или формуле, изображающей и описывающей соединения, использованные или заявленные в настоящем изобретении, ее определение в каждом случае не зависит от ее определения в каждом другом случае. Кроме того, комбинации заместителей и/или переменных допустимы только если такие соединения приводят к стабильным соединениям.
Значок "*" в конце связи или "……", проведенный через связь, означают положение присоединения функциональной группы или другого химического фрагмента к остальной части молекулы, частью которой она является. Так, например:
MeC(=O)OR4, где 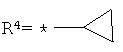 или
или 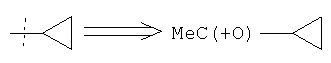 .
.
Связь, направленная внутрь кольцевой системы (в противоположность к присоединенной к определенному положению), означает, что связь может быть присоединена к любому подходящему атому кольца.
Термин "необязательный" или "необязательно" при использовании в настоящем изобретении означает, что последующее описанное событие или обстоятельство может, но не должно осуществиться, и что описание включает случаи, когда событие или обстоятельство осуществляется, и случаи, когда оно не осуществляется. Например, "необязательно замещенный" означает, что необязательно замещенный фрагмент может включать водород или заместитель.
Термин "примерно" при использовании в настоящем изобретении означает:
приблизительно, в области, ориентировочно или около. Если термин "примерно" используется в связи с числовым диапазоном, то он изменяет этот диапазон путем расширения границ за пределы верхней и нижней границы указанного числового диапазона. Обычно термин "примерно" при использовании в настоящем изобретении изменяет числовое значение выше и ниже указанного значения с отклонением на 20%.
При использовании в настоящем изобретении указание числового диапазона переменной означает, что настоящее изобретение можно осуществить при значении переменной, равном любым значениям, находящимся в этом диапазоне. Так, переменная, которая по определению является дискретной, может быть равна любому целому числу в этом числовом диапазоне, включая конечные значения диапазона. Аналогичным образом, переменная, которая по определению является непрерывной, может быть равна любому вещественному числу в этом числовом диапазоне, включая конечные значения диапазона. Например, переменная, которая описана как обладающая значениями, равными от 0 до 2, может быть равна 0, 1 или 2 в случае, если она по определению является дискретной, и может быть равна 0,0, 0,1, 0,01, 0,001 или любому другому вещественному числу, если она по определению является непрерывной.
Соединения формулы I обладают таутомерией. Таутомерные соединения могут существовать в виде двух взаимопревращающихся форм. Прототропные таутомеры образуются вследствие переноса ковалентно связанного атома водорода между двумя атомами. Таутомеры обычно находятся в виде равновесной смеси и попытки выделить индивидуальные таутомеры обычно приводят к смеси, химические и физические характеристики которой соответствуют характеристиками смеси соединений. Положение равновесия зависит от химического строения молекулы. Например, для многих алифатических альдегидов и кетонов, таких как ацетальдегид, преобладает кетоформа, тогда как для фенолов преобладает енольная форма. Обычно прототропные таутомеры включают таутомеры кетон/енол (-С(=O)-СН-↔-С(-ОН)=СН-), амид/имидиновая кислота (-C(=O)-NH-↔-C(-OH)=N-) и амидины (-C(=NR)-NH-↔-C(-NHR)=N-). Два последних являются особенно распространенными для гетероарильных и гетероциклических колец и в объем настоящего изобретения входят все таутомерные формы соединений.
Специалист в данной области техники должен понимать, что некоторые соединения формулы I могут содержать один или большее количество хиральных центров и поэтому могут существовать в виде двух или большего количества стереоизомерных форм. Рацематы этих изомеров, отдельные изомеры и смеси, обогащенные одним энантиомером, а также диастереоизомеры, в которых содержатся два хиральных центра, и смеси, частично обогащенные конкретными диастереоизомерами, входят в объем настоящего изобретения. Специалист в данной области техники также должен понимать, что замещение тропанового кольца может происходить в эндо- или экзо-конфигурации, и настоящее изобретение включает обе конфигурации. Настоящее изобретение включает все отдельные стереоизомеры (например, энантиомеры), рацемические смеси и частично разделенные смеси соединений формулы I и, если это является подходящим, их отдельные таутомерные формы.
Рацематы можно использовать без обработки или их можно разделять на отдельные изомеры. Разделение может привести к получению стереохимически чистых соединений или смесей, обогащенных одним или большим количеством изомеров. Методики разделения изомеров хорошо известны (см. публикацию Allinger N. L. and Eliel E. L. in "Topics in Stereochemistry", Vol.6, Wiley Interscience, 1971) и включают физические методики, такие как хроматография с использованием хирального сорбента. Отдельные изомеры можно получить в хиральной форме из хиральных предшественников. Альтернативно, отдельные изомеры можно выделить из смеси химическим путем посредством образования диастереоизомерных солей с хиральной кислотой, такой как отдельные энантиомеры 10-камфорсульфоновой кислоты, камфорной кислоты, альфа-бромкамфорной кислоты, виннокаменной кислоты, диацетилвиннокаменной кислоты, яблочной кислоты, пирролидон-5-карбоновой кислоты и т.п., фракционной кристаллизации солей и последующего выделения одного или обоих разделенных оснований, необязательного повторения процедуры для получения одного из них или обоих, в основном не содержащего другого; т.е. формы, обладающей оптической чистотой >95%. Альтернативно, рацематы можно ковалентно связать с хиральным соединением (вспомогательным) с получением диастереоизомеров, которые можно разделить с помощью хроматографии или фракционной кристаллизации, после чего хиральное вспомогательное вещество удаляется химическим путем с получением чистых энантиомеров.
Соединения формулы I могут содержать основный центр и образуют подходящие соли присоединения с кислотами, которые образуют нетоксичные соли. Примеры солей неорганических кислот включают гидрохлорид, гидробромид, гидройодид, хлорид, бромид, йодид, сульфат, бисульфат, нитрат, фосфат, гидрофосфат. Примеры солей органических кислот включают ацетат, фумарат, памоат, аспартат, безилат, карбонат, бикарбонат, камзилат, D- и L-лактат, D- и L-тартрат, эзилат, мезилат, малонат, оротат, глюцептат, метилсульфат, стеарат, глюкуронат, 2-напзилат, тозилат, гибензат, никотинат, изетионат, малат, малеат, цитрат, глюконат, сукцинат, сахарат, бензоат, эзилат и памоат. Обзор подходящих солей приведен в публикации Berge et al., J. Pharm. Sci„ 1977 66:1-19 и G. S. Paulekuhn et al., J. Med. Chem. 2007 50:6665.
Если не приведено другое определение, то все технические и научные термины, использованные в настоящем изобретении, обладают такими же значениями, которые обычно подразумевает специалист в области техники, к которой относится настоящее изобретение. В настоящем изобретении приведены ссылки на различные методологии и материалы, известные специалистам в данной области техники. Стандартные справочники, в которых изложены общие положения фармакологии, включают публикацию Goodman and Oilman's The Pharmacological Basis of Therapeutics, 10th Ed., McGraw Hill Companies Inc., New York (2001). Исходные вещества и реагенты, использующиеся для получения этих соединений, обычно можно приобрести у промышленных поставщиков, таких как Aldrich Chemical Co, или получить по методикам, известным специалистам в данной области техники, описанным в литературе. Материалы, реагенты и т.п., на которые приведены ссылки в последующем описании и примерах, можно получить их коммерческих источников, если не указано иное. Некоторые методики синтеза описаны в монографиях, таких как Fieser and Fieser's Reagents for Organic Synthesis; Wiley & Sons: New York, Volumes 1-21; R. C. LaRock, Comprehensive Organic Transformations, 2nd edition Wiley-VCH, New York, 1999; Comprehensive Organic Synthesis, B. Trost and I. Fleming (Eds.) vol. 1-9 Pergamon, Oxford, 1991; Comprehensive Heterocyclic Chemistry, A. R. Katritzky and C. W. Rees (Eds) Pergamon, Oxford, 1984, vol. 1-9; Comprehensive Heterocyclic Chemistry II, A. R. Katritzky and C. W. Rees (Eds) Pergamon, Oxford, 1996, vol. 1-11; и Organic Reactions, Wiley & Sons: New York, 1991, Volumes 1-40, и знакомы специалистам в данной области техники.
Термин "изотополог" используют для определения соединений, которые различаются только своим изотопным составом (IUPAC Compendium of Chemical Terminology 2" Edition 1997). Изотопологи могут различаться степенью изотопного обогащения в одном или большем количестве положений и/или в положении (положениях) изотопного обогащения.
Отклонения от уровня содержания изотопа в природе могут произойти в синтезированных соединениях в зависимости от источника химических предшественников, использующихся для синтеза, и вследствие изотопного обмена в ходе синтеза. Таким образом, коэффициент изотопного обогащения для каждого содержащегося атома дейтерия, находящегося в положении, обозначенном как положение дейтерирования, не зависит от дейтерирования в других положениях, и возможны некоторые различия содержания дейтерия в других необозначенных положениях, и эти различия могут привести к образованию изотопологов, которые заявлены в объеме настоящего изобретения. Коэффициент обогащения дейтерием в положениях, необозначенных как "дейтерий" или "D", составляет менее 49,5%, и обычно значительно менее 49,5%, и чаще всего менее 20%.
Поскольку содержание дейтерия в природе составляет 0,015%, эти отличия от уровня содержания дейтерия, наблюдаемого в природе, не будут существенно влиять на наблюдающиеся биологические характеристики соединений.
Если не указано иное, когда специально указано "Н" или "водород", это означает водород, обладающий природным изотопным составом при условии, что в результате синтеза возможны некоторые случайные отклонения.
Термин "коэффициент изотопного обогащения" при использовании в настоящем изобретении означает отношение содержания изотопа D в определенном положении в соединении, предлагаемом в настоящем изобретении, к содержанию этого изотопа в природе. В одном варианте осуществления настоящее изобретение относится к соединению формулы I, в которой коэффициент изотопного обогащения трет-бутильного фрагмента равно не менее 3300 (49,5%). Во избежание двусмысленности, коэффициент изотопного обогащения для трет-бутила относится к совокупности трех метальных групп и метальные группы не оценены независимо.
В других вариантах осуществления настоящее изобретение относится к соединению формулы I с коэффициентом изотопного обогащения для каждого атома дейтерия, находящегося в положении, обозначенном как потенциальное положение дейтерирования соединения, равным не менее 4000 (содержание дейтерия равно 60%), не менее 4500 (содержание дейтерия равно 67,5%), не менее 5000 (содержание дейтерия равно 75%), не менее 5500 (содержание дейтерия равно 82,5%), не менее 6000 (содержание дейтерия равно 90%), не менее 6333,3 (содержание дейтерия равно 95%), не менее 6466,7 (содержание дейтерия равно 97%), не менее 6600 (содержание дейтерия равно 99%) или не менее 6633,3 (содержание дейтерия равно 99,5%).
В одном варианте осуществления настоящее изобретение относится к соединению формулы I
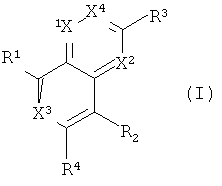
в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении.
В одном варианте осуществления настоящее изобретение относится к соединению формулы I, в которой Х1 обозначает N и Х2, Х3 и Х4 обозначают CR5; или Х1 и Х2 обозначают N и Х3 и Х4 обозначают CR5; или Х1, Х2 и Х4 обозначают CR5, и Х3 обозначает N; или Х1 и Х4 обозначают N и Х2 и Х3 обозначают CR5; или Х1, Х2, Х3 и Х4 обозначают CR5; предпочтительно, если Х1 обозначает N и Х2, Х3, Х4 обозначают CR5, или Х1 и Х2 обозначают N и Х3 и Х4 обозначают CR5;
R1 обозначает (а) гетероарильный радикал, выбранный из группы, включающей пиридинил, 2-оксо-1,2-дигидропиридин-3-ил, 3-оксо-3,4-дигидропиразин-2-ил, 3-оксо-2,3-дигидропиридазин-4-ил, 2-оксо-1,2-дигидропиримидин-4-он-5-ил, 6-оксо-1,6-дигидро-[1,2,4]триазин-5-ил, 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, 2-оксо-2(Н)-пиридин-1-ил, 6-оксо-6Н-пиридазин-1-ил, 6-оксо-бН-пиримидин-1-ил; 2-оксо-2Н-пиразин-1-ил, предпочтительно пиридинил, 2-оксо-1,2-дигидропиридин-3-ил, 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил и 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, указанный гетероарил необязательно содержит в качестве заместителей галоген, C1-С6-алкил, C1-С3-галогеналкил, C1-С6-алкоксигруппу, X-(CH2)1-6СО2Н или Х-(СН2)2-6NRgRh, предпочтительно галоген, C1-С6-алкил или С1-С6-алкоксигруппу; или (b) гетероциклический радикал, выбранный из группы, включающей 2-оксотетрагидропиримидин-1-ил, 2-оксоимидазолидин-1-ил, 2-оксопиперидин-1-ил, 2-оксопирролидин-1-ил, 2,6-диоксотетрагидропиримидин-1-ил и 2,5-диоксоимидазолидин-1-ил;
R2 обозначает водород, C1-С6-алкоксигруппу, C1-С6-алкил, C1-С6-галогеналкил, C1-С6-галогеналкоксигруппу или галоген;
R3 обозначает (а) арил, (b) гетероарил, (с) NRaRb, (d) водород, (е) галоген, предпочтительно (а) арил, (с) NRaRb, (d) водород, (е) галоген, где указанный арил или указанный гетероарил независимо необязательно содержат 1-3 заместителя, выбранных из группы, включающей гидроксигруппу, C1-С6-алкоксигруппу, C1-С6-алкил, C1-С6-гидроксиалкил, галоген, (CH2)nNRcRd, цианогруппу, С1-С6-алкоксикарбонил, карбамоил, N-алкилкарбамоил, N,N-диалкилкарбамоил, (СН2)nCO2H, SO2NH2, C1-С6-алкилсульфинил и C1-С6-алкилсульфонил, предпочтительно галоген, (CH2)nNRcRd, где n равно 1, или (f) -Х(Rg)[C(R6)]pNReRf, где R6 в каждом случае независимо обозначает водород, C1-С3-алкил, или два фрагмента R6, присоединенные к одному и тому же атому углерода, образуют С2-С5-алкилен, или два фрагмента R6, присоединенные к разным атомам углерода, образуют С1-С4-алкилен; Ra и Rb вместе с атомом азота, к которому они присоединены, образуют циклический амин, независимо содержащий в качестве заместителей С1-С6-алкил, галоген или (CH2)nNReRf, предпочтительно (CH2)nNReRf, где n равно 0-2; Rc и Rd независимо обозначают водород, C1-С6-алкил, C1-С6-галогеналкил, C1-С6-ацил, C1-С6-сульфонил, C1-С6-галогеналкилсульфонил, С3-С7-циклоалкилсульфонил, С3-С7-циклоалкил-С1-С3-алкилсульфонил, С1-С6-алкокси-С1-С6-алкилсульфонил, -SO2-NRiRj, C1-С3-алкилкарбамоил или C1-С3-диалкилкарбамоил, предпочтительно, если Rc обозначает водород и Rd обозначает C1-С6-сульфонил; Re и Rf независимо обозначают водород, C1-С6-алкил, C1-С6-галогеналкил, C1-С6-ацил, C1-С6-сульфонил, C1-С6-галогеналкилсульфонил, С3-С7-циклоалкилсульфонил, С3-С7-циклоалкил-С1-С3-алкилсульфонил, С1-С6-алкокси-С1-С6-алкилсульфонил, -SO2-NRiRj; Ri и Rj (i) независимо обозначают водород, C1-С3-алкил или (СН2)2-6NRgRh, или (ii) вместе с атомом азота, к которому они присоединены, образуют (СН2)2Х5(СН2)2, где Х5 обозначает О или NRk и Rk обозначает водород, C1-С3-алкил, C1-С3-ацил или С1-С3-алкилсульфонил; Rg и Rh в каждом случае независимо обозначают водород или С1-С3-алкил, предпочтительно, если Re обозначает водород и Rf обозначает C1-С6-сульфонил;
R4 обозначает водород, C1-С6-алкил, C1-С6-галогеналкил, С3-С5-циклоалкил, галоген, C1-С6-алкоксигруппу, C1-С3-галогеналкоксигруппу или CR4aR4bR4c, где: (i) R4a, R4b и R4c независимо выбраны из группы, включающей С1-С3-алкил, C1-С2-алкоксигруппу, С1-С2-фторалкил, C1-С3-гидроксиалкил, цианогруппу или гидроксигруппу; или (ii) взятые вместе R4a и R4b образуют С2-С4-алкилен и R4c обозначает водород, С1-С3-алкил, С1-С2-алкоксигруппу, галоген, C1-С3-гидроксиалкил, цианогруппу или С1-С2-фторалкил, или R4a и R4b вместе с атомом углерода, к которому они присоединены, образуют 3-оксетанил или тетрагидрофуран-2-ил, предпочтительно, если R4 обозначает CR4aR4bR4c, где R4a, R4b и R4c обозначают метил или R4 обозначает трифторметил, 3,3,3-трифторэтил или CR4aR4bR4c, где R4a, R4b и R4c обозначают СН3 или CD3,
R5 в каждом случае независимо обозначает водород, галоген, C1-С6-алкоксигруппу или C1-С6-алкил;
Х в каждом случае независимо обозначает водород O или NRg;
n в каждом случае независимо равно 0-3; или
его фармацевтически приемлемой соли.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R3 обозначает (а) арил или (b) гетероарил, где указанный арил или указанный гетероарил независимо необязательно содержат 1-3 заместителя, выбранных из группы, включающей гидроксигруппу, C1-С6-алкоксигруппу, C1-С6-алкил, C1-С6-гидроксиалкил, галоген, цианогруппу, С1-С6-алкоксикарбонил, карбамоил, N-алкилкарбамоил, N,N-диалкилкарбамоил, карбоксигруппу, SO2NH2, C1-С6-алкилсульфинил и C1-С6-алкилсульфонил, где n равно 0-3.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой Х1 обозначает N и Х2, Х3 и Х4 обозначают CR5 или X1 и X2 обозначают N и X3 и X4 обозначают CR5;
R1 обозначает гетероарильный радикал, выбранный из группы, включающей пиридинил, 2-оксо-1,2-дигидропиридин-3-ил, 2,4-диоксо-1, 2,3,4-тетрагидропиримидин-5-ил и 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, указанный гетероарил необязательно содержит в качестве заместителей галоген, C1-С6-алкил или C1-С6-алкоксигруппу; R2 обозначает водород или С1-С6-алкоксигруппу; или R1 обозначает 2-оксотетрагидропиримидин-1-ил; R3 обозначает арил, NRaRb, водород или галоген, где указанный арил независимо необязательно содержит 1-3 заместителя, выбранных из группы, включающей галоген, (CH2)nNRcRd, где n равно 1; Ra и Rb вместе с атомом азота, к которому они присоединены, образуют циклический амин, необязательно замещенный 1-3 группами, независимо выбранными из группы, включающей (CH2)nNReRf, где n равно 0-2, C1-С6-алкил или галоген; Re обозначает водород и Rf обозначает C1-С6-сульфонил; Rc обозначает водород и Rd обозначает C1-С6-сульфонил; R4 обозначает трифторметил, 3,3,3-трифторэтил или CR4aR4b R4c, где R4a, R4b и R4c обозначают СН3 или CD3, R5 в каждом случае независимо обозначает водород, галоген, С1-С6-алкоксигруппу или C1-С6-алкил. Этот вариант осуществления включает фармацевтически приемлемую соль соединения, входящего в объем настоящего изобретения.
В одном варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R3 обозначает (а) арил или (b) гетероарил, где указанный арил или указанный гетероарил независимо необязательно содержат 1-3 заместителя, выбранных из группы, включающей гидроксигруппу, C1-С6-алкоксигруппу, С1-С6-алкил, C1-С6-гидроксиалкил, галоген, цианогруппу, C1-С6-алкоксикарбонил, карбамоил, N-алкилкарбамоил, N,N-диалкилкарбамоил, карбоксигруппу, SO2NH2, C1-С6-алкилсульфинил и C1-С6-алкилсульфонил, где n равно 0-3.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой Х1 обозначает N и Х2, Х3 и Х4 обозначают CR5; R3 обозначает (а) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0, или (b) NRaRb. Выражение "фенил, замещенный по меньшей мере с помощью (CH2)nNRaRb в положении 4", означает (i), где незамещенные положения дополнительно могут быть необязательно замещены. Выражение "фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4", означает (i), где незамещенные положения дополнительно могут быть необязательно замещены.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R1 обозначает 2-оксо-1,2-дигидропиридин-3-ил или 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, необязательно содержащий в качестве заместителей галоген, C1-С6-алкил, C1-С3-галогеналкил или C1-С6-алкоксигруппу; Х1 обозначает N и Х2, Х3 и Х4 обозначают CR5, и R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R1 обозначает 2-оксо-1,2-дигидропиридин-3-ил, 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, необязательно содержащий в качестве заместителей галоген, С1-С6-алкил, C1-С3-галогеналкил или C1-С6-алкоксигруппу; Х1 обозначает N и Х2, Х3 и Х4 обозначают CR5; R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0, и R4 обозначает CR4aR4bR4c, где (a) R4a, R4b и R4c обозначают СН3, CD3 или фтор, или R4a и R4b вместе образуют С2-алкилен, и (b) R4c обозначает C1-С3-алкил, C1-C2-алкоксигруппу, галоген, C1-С3-гидроксиалкил, цианогруппу или C1-C2-фторалкил.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R1 обозначает 2-оксо-1,2-дигидропиридин-3-ил, 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, необязательно содержащий в качестве заместителей галоген, C1-С6-алкил, C1-С3-галогеналкил или C1-С6-алкоксигруппу; Х1 обозначает N и X2, X3 и X4 обозначают CR5; R3 обозначает NRaRb; и R4 обозначает CR4aR4bR4c, где (а) R4a, R4b и R4c обозначают СН3, CD3 или фтор, или R4a и R4b вместе образуют С2-алкилен, и (b) R4c обозначает C1-С3-алкил, C1-C2-алкоксигруппу, галоген, C1-С3-гидроксиалкил, цианогруппу или C1-C2-фторалкил.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R1 обозначает 2-оксо-1,2-дигидропиридин-3-ил, 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, необязательно содержащий в качестве заместителей галоген, С1-С6-алкил, C1-С3-галогеналкил или C1-С6-алкоксигруппу; Х1 обозначает N и X2, X3 и X4 обозначают CR5; R3 обозначает NRaRb, где NRaRb вместе образует циклический амин, замещенный с помощью (CH2)nNReRf, где n равно 0-2; и Re и Rf независимо обозначают водород, C1-С6-алкил, C1-С6-галогеналкил, SO2R8, где R8 обозначает (а) C1-С6-алкил, (b) C1-С6-галогеналкил, (с) С3-С7-циклоалкил, (d) С3-С7-циклоалкил-С1-С3-алкил, (е) C1-С6-алкокси-С1-С6-алкил, и R4 обозначает CR4aR4bR4c, и (a) R4a, R4b и R4c обозначают СН3, CD3 или фтор, или R4a и R4b вместе образуют С2-алкилен, и (b) R4c обозначает C1-С3-алкил, С1-С2-алкоксигруппу, галоген, C1-С3-гидроксиалкил, цианогруппу или С1-С2-фторалкил.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R1 обозначает 6-оксо-1,6-дигидро-[1,2,4]триазин-5-ил; Х1 обозначает N и Х2, Х3 и Х4 обозначают CR5, и R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R1 обозначает 2-оксотетрагидропиримидин-1-ил; Х1 обозначает N и Х2, Х3 и Х4 обозначают CR5, и R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой Х1 и Х2 обозначают N и Х3 и Х4 обозначают CR5; R3 обозначает (а) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0, или (b) NRaRb.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R1 обозначает 2-оксо-1,2-дигидропиридин-3-ил или 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, необязательно содержащий в качестве заместителей галоген, C1-С6-алкил, C1-С3-галогеналкил или C1-С6-алкоксигруппу; Х1 и Х2 обозначают N и Х3 и Х4 обозначают CR5, и R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0 или 1.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R1 обозначает 2-оксо-1,2-дигидропиридин-3-ил или 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, необязательно содержащий в качестве заместителей галоген, С1-С6-алкил, С1-С3-галогеналкил или C1-С6-алкоксигруппу; Х1 и Х2 обозначают N и Х3 и Х4 обозначают CR5, и R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0 или 1.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R1 обозначает 2-оксо-1,2-дигидропиридин-3-ил, 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, необязательно содержащий в качестве заместителей галоген, С1-С6-алкил, С1-С3-галогеналкил или C1-С6-алкоксигруппу; Х1 и Х2 обозначают N и Х3 и Х4 обозначают CR5, и R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0, и R4 обозначает CR4aR4bR4c, и (а) R4a, R4b и R4c обозначают СН3, CD3 или фтор, или R4a и R4b вместе образуют С2-алкилен, и (b) R4c обозначает С1-С3-алкил, C1-C2-алкоксигруппу, галоген, С1-С3-гидроксиалкил, цианогруппу или C1-С2-фторалкил.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой R1 обозначает 2-оксо-1,2-дигидропиридин-3-ил, 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, необязательно содержащий в качестве заместителей галоген, C1-С6-алкил, C1-С3-галогеналкил или C1-С6-алкоксигруппу; Х1 и Х2 обозначают N и Х3 и Х4 обозначают CR5; R3 обозначает NRaRb, где NRaRb вместе образует циклический амин, замещенный с помощью (CH2)nNReRf, где n равно 0-2; и Re и Rf независимо обозначают водород, C1-С6-алкил, C1-С6-галогеналкил, SO2R8, где R8 обозначает (а) C1-С6-алкил, (b) C1-С6-галогеналкил, (с) С3-С7-циклоалкил, (d) С3-С7-циклоалкил-С1-С3-алкил, (е) C1-С6-алкокси-С1-С6-алкил и R4 обозначает CR4aR4bR4c, и (а) R4a, R4b и R4c обозначают СН3, CD3 или фтор, или R4a и R4b вместе образуют С2-алкилен, и (b) R4c обозначает С1-С3-алкил, С1-С2-алкоксигруппу, галоген, C1-С3-гидроксиалкил, цианогруппу или С1-С2-фторалкил.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой Х1, Х2 и Х3 обозначают CR5, и Х3 обозначает N.
В другом варианте осуществления, в котором Х1, Х2 и Х4 обозначают CR5, и Х3 обозначает N; R3 обозначает (а) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0, или (b) NRaRb. Выражение "фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4", означает (i) где незамещенные положения дополнительно могут быть необязательно замещены. Выражение "фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4", означает (i) где незамещенные положения дополнительно могут быть необязательно замещены.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой Х1 и Х4 обозначают N и Х2 и Х3 обозначают CR5.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой Х1 и Х4 обозначают N и Х2 и Х3 обозначают CR5, R3 обозначает (а) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0, или (b) NRaRb. Выражение "фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4" означает (i), где незамещенные положения дополнительно могут быть необязательно замещены. Выражение "фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4" означает (i), где незамещенные положения дополнительно могут быть необязательно замещены.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой Х1, Х2, Х3 и Х4 обозначают CR5.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой Х1, Х2, Х3 и Х4 обозначают CR5; R3 обозначает (а) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0, или (b) NRaRb. Выражение "фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4" означает (i), где незамещенные положения дополнительно могут быть необязательно замещены. Выражение "фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4" означает (i), где незамещенные положения дополнительно могут быть необязательно замещены.
В другом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой Х1 обозначает N и Х2, Х3 и Х4 обозначают CR5, R1 обозначает 2,6-диоксотетрагидропиримидин-1-ил, 2,5-диоксоимидазолидин-1-ил или 2,4-диоксотетрагидропиримидин-1-ил; и R3 обозначает (а) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0, или (b) NRaRb.
В другом варианте осуществления настоящее изобретение относится к соединению, выбранному из числа соединений I-1-I-60, приведенных в таблице I, и соединений II-1-II-2, приведенных в таблице II, предпочтительно выбранному из числа соединений I-1-I-33, приведенных в таблице I, предпочтительно выбранному из числа соединений I-1-I-31, приведенных в таблице I.
В другом варианте осуществления настоящее изобретение относится к способу лечения инфекции HCV у нуждающегося в нем пациента, включающему введение соединения формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, в терапевтически эффективном количестве.
В другом варианте осуществления настоящее изобретение относится к способу лечения инфекции HCV у нуждающегося в нем пациента, включающему совместное введение соединения формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, в терапевтически эффективном количестве и по меньшей мере одного модулятора иммунной системы и/или по меньшей мере одного противовирусного средства, которое подавляет репликацию HCV.
В другом варианте осуществления настоящее изобретение относится к способу лечения заболевания, вызванного посредством HCV у пациента, нуждающегося в таком лечении, включающему совместное введение соединения формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, в терапевтически эффективном количестве и по меньшей мере одного модулятора иммунной системы, выбранного из группы, включающей интерферон, интерлейкин, фактор некроза опухоли или колониестимулирующий фактор.
В другом варианте осуществления настоящее изобретение относится к способу лечения инфекции HCV у нуждающегося в нем пациента, включающему совместное введение соединения формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, в терапевтически эффективном количестве и интерферона или химического производного интерферона.
В другом варианте осуществления настоящее изобретение относится к способу лечения инфекции HCV у нуждающегося в нем пациента, включающему совместное введение соединения формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, в терапевтически эффективном количестве и другого противовирусного соединения, выбранного из группы, включающей ингибитор протеазы HCV, другой ингибитор полимеразы HCV, ингибитор геликазы HCV, ингибитор примазы HCV и ингибитор слияния HCV.
В другом варианте осуществления настоящее изобретение относится к способу ингибирования репликации вируса в клетке, включающему введение соединения формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, в терапевтически эффективном количестве в смеси по меньшей мере с одним фармацевтически приемлемым носителем, разбавителем или инертным наполнителем.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, для лечения инфекции HCV.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, для приготовления лекарственного средства, предназначенного для лечения инфекции HCV.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, и по меньшей мере одного модулятора иммунной системы и/или по меньшей мере одного противовирусного средства, которое подавляет репликацию HCV, для лечения инфекции HCV.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, и по меньшей мере одного модулятора иммунной системы и/или по меньшей мере одного противовирусного средства, которое подавляет репликацию HCV, для приготовления лекарственного средства, предназначенного для лечения инфекции HCV.
В другом варианте осуществления настоящее изобретение относится к композиции, включающей соединение формулы I, в которой R1, R2, R3, R4, X1, X2, X3 и Х4 являются такими, как определено выше в настоящем изобретении, вместе с одним фармацевтически приемлемым носителем, разбавителем или инертным наполнителем.
Термин "алкил" при использовании в настоящем изобретении без дополнительных ограничений по отдельности или в комбинации с другими группами означает обладающий неразветвленной или разветвленной цепью насыщенный одновалентный углеводородный остаток, содержащий от 1 до 10 атомов углерода. Термин "низш. алкил" означает обладающий неразветвленной или разветвленной цепью углеводородный остаток, содержащий от 1 до 6 атомов углерода. "С1-С6-Алкил" при использовании в настоящем изобретении означает алкил, содержащий от 1 до 6 атомов углерода. Примеры алкильных групп включают, но не ограничиваются только ими, низшие алкильные группы, включая метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил, неопентил, гексил и октил. Без отклонения от объема настоящего изобретения любая связь углерод-водород может быть заменена связью углерод-дейтерий.
Термины, определенные в настоящем изобретении, можно объединять с образованием химически возможных комбинаций, например "гетероалкиларил", "галогеналкилгетероарил", "арилалкилгетероциклил", "алкилкарбонил", "алкоксиалкил" и т.п. Если термин "алкил" используется в качестве суффикса после другого термина, как в терминах "фенилалкил" или "гидроксиалкил", это означает, что алкильная группа, определенная выше, замещена одним или двумя заместителями, выбранными из числа других конкретных названных групп.Так, например, "фенилалкил" означает алкильную группу, содержащую 1 или 2 фенильных заместителя, и таким образом включает бензил, фенилэтил и бифенил. "Алкиламиноалкил" означает алкильную группу, содержащую 1 или 2 алкиламиновых заместителя. "Гидроксиалкил" включает 2-гидроксиэтил, 2-гидроксипропил, 1-(гидроксиметил)-2-метилпропил, 2-гидроксибутил, 2,3-дигидроксибутил, 2-(гидроксиметил), 3-гидроксипропил и т.п. Соответственно при использовании в настоящем изобретении термин "гидроксиалкил" используют для определения подмножества гетероалкильных групп, определенных ниже. Термин (арил)алкил означает незамещенный алкил или арилалкильную группу. Термин (гетеро)арил означает арильную или гетероарильную группу.
Термин "алкилен" при использовании в настоящем изобретении означает двухвалентный насыщенный линейный углеводородный радикал, содержащий от 1 до 10 атомов углерода (например, (СН2)n), или разветвленный насыщенный двухвалентный углеводородный радикал, содержащий от 2 до 10 атомов углерода (например, -СНМе- или -CH2CH(i-Pr)CH2-), если не указано иное. C0-С4-Алкилен означает линейный или разветвленный насыщенный двухвалентный углеводородный радикал, содержащий 1-4 атома углерода, или в случае С0 алкиленовый радикал отсутствует. За исключением метилена свободные валентности алкиленовой группы не выходят из одного и того же атома. Примеры алкиленовых радикалов включают, но не ограничиваются только ими, метилен, этилен, пропилен, 2-метилпропилен, 1,1-диметилэтилен, бутилен, 2-этилбутилен.
Термин "алкоксигруппа" при использовании в настоящем изобретении означает группу -O-алкил, в которой алкил является таким, как определено выше, такую как метоксигруппу, этоксигруппу, н-пропилоксигруппу, изопропилоксигруппу, н-бутилоксигруппу, изобутилоксигруппу, трет-бутилоксигруппу, пентилоксигруппу, гексилоксигруппу, включая их изомеры. "Низш. алкоксигруппа" при использовании в настоящем изобретении означает алкоксигруппу, содержащую "низш. алкильную" группу, определенную выше. "C1-С10-Алкоксигруппа" при использовании в настоящем изобретении означает -O-алкил, в котором алкил представляет собой С1-С10.
Термин "галогеналкил" при использовании в настоящем изобретении означает обладающую неразветвленной или разветвленной цепью алкильную группу, определенную выше, в которой 1, 2, 3 или большее количество атомов водорода замещены на галоген. Примерами являются 1-фторметил, 1-хлорметил, 1-бромметил, 1-йодметил, дифторметил, трифторметил, трихлорметил, 1-фторэтил, 1-хлорэтил, 1 2-фторэтил, 2-хлорэтил, 2-бромэтил, 2,2-дихлорэтил, 3-бромпропил или 2,2,2-трифторэтил. Термин "фторалкил" при использовании в настоящем изобретении означает галогеналкильный фрагмент, в котором галогеном является фтор.
Термин "галогеналкоксигруппа" при использовании в настоящем изобретении означает группу -OR, в которой R обозначает галогеналкил, определенный выше. Термин "галогеналкилтиогруппа" при использовании в настоящем изобретении означает группу -SR, в которой R обозначает галогеналкил, определенный выше.
Термин "циклоалкил" при использовании в настоящем изобретении означает насыщенное карбоциклическое кольцо, содержащее от 3 до 8 атомов углерода, т.е. циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил. "С3-С7-Циклоалкил" при использовании в настоящем изобретении означает циклоалкил, содержащий в карбоциклическом кольце от 3 до 7 атомов углерода.
Термин "галоген" при использовании в настоящем изобретении означает фтор, хлор, бром или йод.
Термины "гидроксиалкил" и "алкоксиалкил" при использовании в настоящем изобретении означают алкильный радикал, определенный в настоящем изобретении, в котором от 1 до 3 атомов водорода у различных атомов углерода замещены на гидроксигруппы или алкоксигруппы соответственно. С1-С3-Алкокси-С1-С6-алкильный фрагмент означает C1-С6-алкильный заместитель, в котором от 1 до 3 атомов водорода замещены на C1-С3-алкоксигруппу и положением присоединения алкоксигруппы является атом кислорода.
Термины "алкоксикарбонил" и "арилоксикарбонил"при использовании в настоящем изобретении означают группу формулы -C(=O)OR, в которой R обозначает алкил или арил соответственно, и алкил и арил являются такими, как определено в настоящем изобретении.
Термин "цианогруппа" при использовании в настоящем изобретении означает атом углерода, связанный с атомом азота тройной связью, т.е. -C=N. Термин "нитрогруппа" при использовании в настоящем изобретении означает группу -NO2. Термин "карбоксигруппа" при использовании в настоящем изобретении означает группу -СО2Н.
Термин "оксогруппа" означает атом кислорода, связанный двойной связью (=O), т.е. карбонильную группу.
Термин "ацил" (или "алканоил") при использовании в настоящем изобретении означает группу формулы -C(=O)R, в которой R обозначает водород или низш. алкил, определенный в настоящем изобретении. Термин "алкилкарбонил" при использовании в настоящем изобретении означает группу формулы C(=O)R, в которой R обозначает алкил, определенный в настоящем изобретении. Термин C1-С6-ацил или "алканоил" означает группу -C(=O)R, содержащую от 1 до 6 атомов углерода. C1-Ацильная группа означает формильную группу, в которой R=Н, и С6-ацильная группа означает гексаноил, если алкильная цепь является неразветвленной. Термин "арилкарбонил" или "ароил" при использовании в настоящем изобретении означает группу формулы C(=O)R, в которой R обозначает арильную группу; термин "бензоил" при использовании в настоящем изобретении означает "арилкарбонильную" или "ароильную" группу, в которой R обозначает фенил.
Термин "циклический амин" при использовании в настоящем изобретении означает насыщенное углеродное кольцо, содержащее от 3 до 6 атомов углерода, определенное выше, и в котором по меньшей мере один из атомов углерода заменен на гетероатом, выбранный из группы, включающей N, О и S, например пиперидин, пиперазин, морфолин, тиоморфолин, диоксотиоморфолин, пирролидин, пиразолин, имидазолидин, азетидин, где атомы углерода цикла необязательно содержат один или большее количество заместителей, выбранных из группы, включающей галоген, гидроксигруппу, фенил, низш. алкил, низш. алкоксигруппу, или 2 атома водорода у атома углерода оба замещены на оксогруппу (=O). Если циклическим амином является пиперазин, то один атом азота может быть необязательно замещен C1-С6-алкилом, С1-С6-ацилом, C1-С6-алкилсульфонилом.
Термины "алкилсульфонил" и "арилсульфонил" при использовании в настоящем изобретении означают группу формулы S(=O)2R, в которой R обозначает алкил или арил соответственно, и алкил и арил являются такими, как определено в настоящем изобретении. Термин C1-С3-алкилсульфониламиногруппа при использовании в настоящем изобретении означает группу RSO2NH-, в которой R обозначает С1-С3-алкильную группу, определенную в настоящем изобретении. Термины C1-С6-галогеналкилсульфонил, С3-С7-циклоалкилсульфонил, С3-С7-циклоалкил-С1-С3-алкилсульфонил или C1-С6-алкокси-С1-С6-алкилсульфонил означают соединение S(=O)2R, в котором R обозначает C1-С6-галогеналкил, С3-С7-циклоалкил, С3-С7-циклоалкил-С1-С3-алкил и С1-С6-алкокси-С1-С6-алкил соответственно.
Термин "сульфамоил" при использовании в настоящем изобретении означает радикал -S(O)2NH2. Термины "N-алкилсульфамоил" и "N,N-диалкилсульфамоил" при использовании в настоящем изобретении означают радикал -S(O)2NR'R", в котором R' и R" обозначают водород и низш. алкил и R' и R" независимо обозначают низш. алкил соответственно. Примеры N-алкилсульфамоильных заместителей включают, но не ограничиваются только ими, метиламиносульфонил, изопропиламиносульфонил. Примеры N,N-диалкилсульфамоильных заместителей включают, но не ограничиваются только ими, диметиламиносульфонил, изопропилметиламиносульфонил.
Термин "карбамоил" при использовании в настоящем изобретении означает радикал -CONH2. Приставки "N-алкилкарбамоил" и "N,N-диалкилкарбамоил" означают радикалы CONHR' или CONR'R" соответственно, в которых группы R' и R" независимо обозначают алкил, определенный в настоящем изобретении. Приставка "N-арилкарбамоил" означает радикал CONHR', в котором R' обозначает арильный радикал, определенный в настоящем изобретении.
Термин "бензил" при использовании в настоящем изобретении означает радикал C6H5CH2, в котором фенильное кольцо может необязательно содержать один или большее количество заместителей, предпочтительно 1 или 3 заместителя, независимо выбранных из группы, включающей гидроксигруппу, тиогруппу, цианогруппу, алкил, алкоксигруппу, низш. галогеналкоксигруппу, алкилтиогруппу, галоген, галогеналкил, гидроксиалкил, нитрогруппу, алкоксикарбонил, аминогруппу, алкиламиногруппу, диалкиламиногруппу, аминоалкил, алкиламиноалкил и диалкиламиноалкил, алкилсульфонил, арилсульфинил, алкиламиносульфонил, ариламиносульфонил, алкилсульфониламиногруппу, арилсульфониламиногруппу, карбамоил, алкилкарбамоил и диалкилкарбамоил, арилкарбамоил, алкилкарбониламиногруппу, арилкарбониламиногруппу, если не указано иное.
Термин "гетероарил" при использовании в настоящем изобретении без дополнительных определений или ограничений означает "пиридинильные", "пиразинильные" и "пиридазинильные" кольца. Термин "пиридин" ("пиридинил") означает 6-членное гетероароматическое кольцо, содержащее 1 атом азота. Термины "пиримидин" (пиримидинил), "пиразин" ("пиразинил") и "пиридазин" ("пиридазинил") означают 6-членное неконденсированное гетероароматическое кольцо, содержащее 2 атома азота, находящихся в положениях 1,3, 1,4 и 1,2 соответственно. Соответствующие названия радикалов приведены в скобках.
Термины "оксетан" (оксетанил), "тетрагидрофуран" (тетрагидрофуранил) и "тетрагидропиран" (тетрагидропиранил") означают 4-, 5- и 6-членные неконденсированные гетероароматические кольца соответственно, каждое из которых содержит 1 атом кислорода.
Термин "арил" при использовании в настоящем изобретении означает фенил.
Термины (i) 3-оксо-3,4-дигидропиразин-2-ил, (ii) 3-оксо-2,3-дигидропиридазин-4-ил, (iii) 2-оксо-1,2-дигидропиримидин-4-он-5-ил, (iv) 2-оксо-1,2-дигидропиридин-3-ил, (v) 6-оксо-1,6-дигидро-[1,2,4]триазин-5-ил и (vi) 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил означают следующие фрагменты:
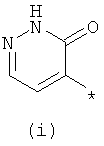
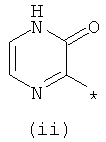
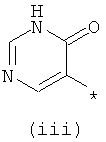
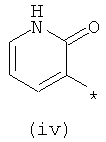
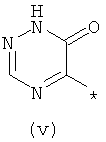
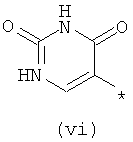 .
.
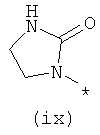
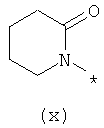
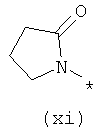
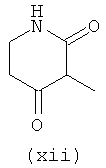
Термины (viii) 2-оксотетрагидропиримидин-1-ил, (ix) 2-оксоимидазолидин-1-ил, (х) 2-оксопиперидин-1-ил, (xi) 2-оксопирролидин-1-ил (xii) 2,6-диоксотетрагидропиримидин-1-ил и (xiii) 2,5-диоксоимидазолидин-1-ил означают следующие фрагменты:
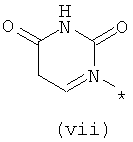
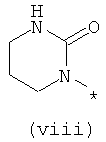
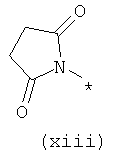
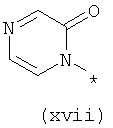
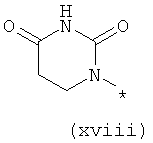
Термины (xiv) 2-оксо-2(Н)-пиридин-1-ил, (xv) 6-оксо-бН-пиридазин-1-ил, (xvi) 6-оксо-6Н-пиримидин-1-ил, (xvii) 2-оксо-2Н-пиразин-1-ил и (xviii) 2,4-диоксотетрагидропиримидин-1-ил означают следующие фрагменты:
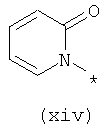
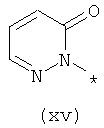
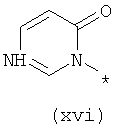
Соединения, предлагаемые в настоящем изобретении, их изомерные формы и фармацевтически приемлемые соли также применимы для лечения и предупреждения вирусных инфекций, в частности инфицирования гепатитом С, и заболевания живых реципиентов, при их применении в комбинации друг с другом и с другими биологически активными средствами, включая, но не ограничиваясь только ей, группу, включающую интерферон, пэгилированный интерферон, рибаривин, ингибиторы протеазы, ингибиторы полимеразы, небольшие соединения, мешающие РНК, антисмысловые соединения, аналоги нуклеотидов, аналоги нуклеозидов, иммуноглобулины, иммуномодуляторы, гепатозащитные средства, противовоспалительные средства, антибиотики, противовирусные и противоинфекционные соединения. Такая комбинированная терапия также может включать введения соединения, предлагаемого в настоящем изобретении, одновременно или последовательно с другими лекарственными средствами или усиливающими воздействие средствами, такими как рибаривин и родственные соединения, амантадин и родственные соединения, различные интерфероны, такие как, например, интерферон-альфа, интерферон-бета, интерферон-гамма и т.п., а также альтернативные формы интерферонов, такие как пэгилированные интерфероны. Кроме того, в качестве дополнительной комбинированной терапии комбинации рибаривина и интерферона можно вводить по меньшей мере с одним из соединений, предлагаемых в настоящем изобретении.
В одном варианте осуществления соединения формулы I, предлагаемые в настоящем изобретении, применяют в комбинации с другими активным терапевтическими ингредиентами или средствами для лечения пациентов, инфицированных вирусом HCV. В контексте настоящего изобретения активным терапевтическим ингредиентом, который используют в комбинации с соединением, предлагаемым в настоящем изобретении, может быть любое средство, обладающее терапевтическим воздействием при его использовании в комбинации с соединением, предлагаемым в настоящем изобретении. Например, активными средствами, использующимися в комбинации с соединением, предлагаемым в настоящем изобретении, могут являться интерфероны, аналоги рибаривина, ингибиторы HCV NS3 протеазы, нуклеозидные ингибиторы HCV полимеразы, ненуклеозидные ингибиторы HCV полимеразы и другие лекарственные средства, предназначенные для лечения инфекции HCV, или их смеси.
Примеры нуклеозидных ингибиторов NS5b полимеразы включают, но не ограничиваются только ими, NM-283, валопицитабин, R1626, PSI-6130 (R1656), IDX184 и IDX102 (Idenix) BILB 1941.
Примеры ненуклеозидных ингибиторов NS5b полимеразы включают, но не ограничиваются только ими, HCV-796 (ViroPharma and Wyeth), МК-0608, МК-3281 (Merck),NM-107, R7128 (R4048), VCH-759, GSO625433 и GSK625433 (Glaxo), PF-868554 (Pfizer), GS-9190 (Gilead), A-837093 и А848837 (Abbot Laboratories), ANA598 (Anadys Pharmaceuticals); GL100597 (GNLB/NVS), VBY 708 (ViroBay), производные бензимидазола (H. Hashimoto et al. WO 01/47833, H. Hashimoto et al. WO 03/000254, P. L. Beaulieu et al. WO 03/020240 A2; P. L. Beaulieu et al. US 6448281 Bl; P. L. Beaulieu et al. WO 03/007945 Al), производные бензо-1,2,4-тиадиазина (D. Dhanak et al. WO 01/85172 Al, поданная 5/10/2001; D. Chai et al., WO 2002098424, поданная 6/7/2002, D. Dhanak et al. WO 03/037262 A2, поданная 10/28/2002; К. J. Duffy et al. WO 03/099801 Al, filed 5/23/2003, M. G. Darcy et al. WO 2003059356, filed 10/28/2002; D.Chai et al. WO 2004052312, поданная 6/24/2004, D.Chai et al. WO 2004052313, поданная 12/13/2003; D. M. Fitch et al., WO 2004058150, поданная 12/11/2003; D. K. Hutchinson et al., WO 2005019191, поданная 8/19/2004; J. К. Pratt et al. WO 2004/041818 Al, поданная 10/31/2003), производные 1,1-диоксо-4Н-бензо[1,4]тиазин-3-ила (J. F. Blake et al. в публикации патента US 20060252785 и 1,1-диоксобензо[d]изотиазол-3-ильные соединения (J. F. Blake et al. в публикации патента US 2006040927).
Примеры ингибиторов HCV NS3 протеазы включают, но не ограничиваются только ими, SCH-503034 (Sobering, SCH-7), VX-950 (телапревир. Vertex), BILN-2065 (Boehringer-Ingelheim, BMS-605339 (Bristol Myers Squibb) и ITMN-191 (Intermune).
Примеры интерферонов включают, но не ограничиваются только ими, пэгилированный rIFN-альфа 2b, пэгилированный rIFN-альфа 2а, rIFN-альфа 2b, rIFN-альфа 2а, консенсусный IFN-альфа (инферген), ферон, реаферон, интермакс-альфа, r-IFN-бета, инферген и актиммун, IFN-омега с ДУРОС, альбуферон, локтерон, альбуферон, ребиф, предназначенный для перорального введения интерферон-альфа, 1IFN-альфа-2b XL, AVI-005, PEG-инферген и пэгилированный IFN-бета.
Аналоги рибаривина и пролекарство рибаривина, вирамидин (тарибавирин). вводят с интерферонами для борьбы с инфекцией HCV.
Общепринятые аббревиатуры включают: ацетил (Ас), атмосфера (атм), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (БИНАФ), трет-бутоксикарбонил (Воc), ди-трет-бутилпирокарбонат или boc-ангидрид (BOC2O), бензил (Bn), бутил (Bu), регистрационный № в журнале Chemical Abstracts (регистрационный № CAS), бензилоксикарбонил (CBZ или Z), карбонилдиимидазол (КДИ), 1,5-диазабицикло[4.3.0]нон-5-ен (ДБН), 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), N,N'-дициклогексилкарбодиимид (ДЦК), 1,2-дихлорэтан (ДХЭ), дихлорметан (ДХМ), диэтилазодикарбоксилат (ДЭАД), диизопропилазодикарбоксилат (ДИАД), диизобутилалюминийгидрид (ДИБАЛ или ДИБАЛ-Н), диизопропилэтиламин (ДИПЭА), N,N'-диметилацетамид (ДМА), 4-N,N-диметиламинопиридин (ДМАП), N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), этил (Et), этанол (EtOH), 1,1'-бис-(дифенилфосфино)этан (dppe), 1,1'-бис-(дифенилфосфино)ферроцен (dppf), 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид (EDCI), этилацетат (EtOAc), этиловый эфир 2-этокси-2Н-хинолин-1-карбоновой кислоты (EEDQ), диэтиловый эфир (Et2O), O-(7-азабензотриазол-1-ил)-N, N,N'N'-тетраметилуронийгексафторфосфат (HATU), уксусная кислота (HOAc), 1-N-гидроксибензотриазол (HOBt), высокоэффективная жидкостная хроматография (ВЭЖХ), изопропанол (ИПА), метанол (МеОН), температура плавления (т.пл.), MeSO2-(мезил или МС), метил (Me), ацетонитрил (MeCN), мета-хлорпербензойная кислота (МХПБК), масс-спектр (МС), метил-трет-бутиловый эфир (МТБЭ), N-метилморфолин (NMM), N-метилпирролидон (NMP), фенил (Ph), пропил (Pr), изопропил (i-Pr), пиридин (pyr), комнатная температура (КТ), трет-бутилдиметилсилил или t-BuMe2Si (ТБДМС), триэтиламин (ТЭА или Et3N), трифлат или CF3SO2- (Tf), трифторуксусная кислота (ТФК), О-бензотриазол-1-ил-N,N,N',N'-тетраметилуронийтетрафторборат (TBTU), тонкослойная хроматография (ТСХ), тетрагидрофуран (ТГФ), тетраметилэтилендиамин (ТМЭДА), триметилсилил или MeaSi (ТМС), моногидрат п-толуолсульфоновой кислоты (TsOH или pTsOH), 4-Ме-C6H4SO2- или тозил (Ts), N-уретан-N-карбоксиангидрид (UNCA). При использовании для алкильного фрагмента общепринятая номенклатура, включая приставки нормальный (н), изо (i-), вторичный (втор-), третичный (трет-) и нео, обладает обычными значениями (J. Rigaudy and D. P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford.).
Примеры типичных соединений, входящих в объем настоящего изобретения, приведены в представленной ниже таблице. Эти примеры и приведенные ниже методики синтеза приведены для того, чтобы специалисты в данной области техники смогли лучше понять и осуществить настоящее изобретение. Их следует рассматривать не в качестве ограничивающих объем настоящего изобретения, а только в качестве его иллюстративных и типичных примеров.
Обычно номенклатура, использованная в настоящей заявке, основана на AUTONOM v.4.0, компьютерной системе института Beilstein для генерации систематической номенклатуры ИЮПАК (Международный союз теоретической и прикладной химии). Если имеется расхождение между изображенной структурой и названием, присвоенным этой структуре, более точной является изображенная структура. Кроме того, если стереохимическая конфигурация структуры или части структуры не указана, например, жирными или штриховыми линиями, то структуру или часть структуры следует интерпретировать, как включающую все ее стереоизомеры.
В настоящем изобретении используют следующую систему нумерации.
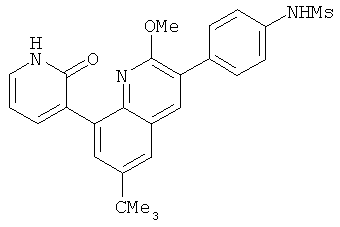

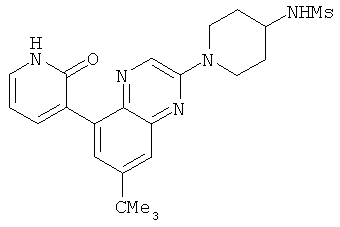
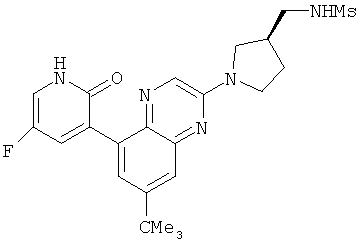
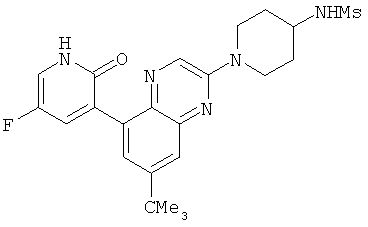
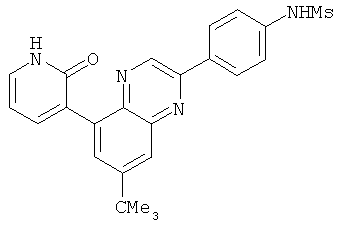
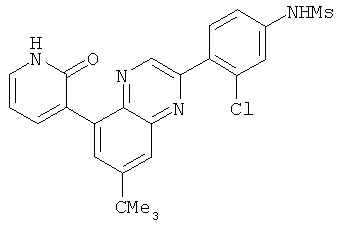
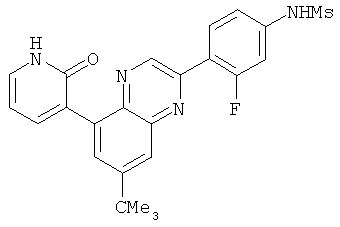
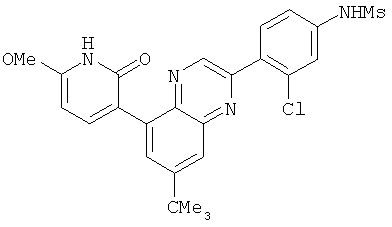
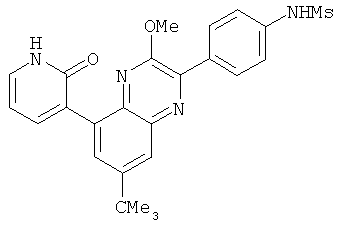
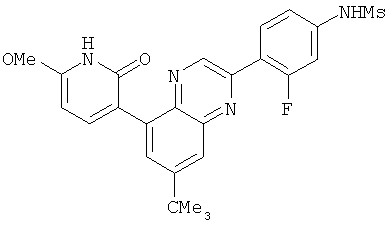
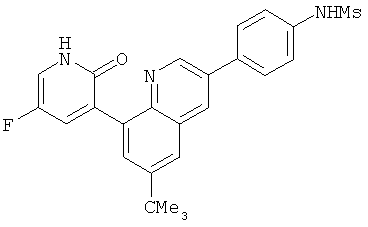
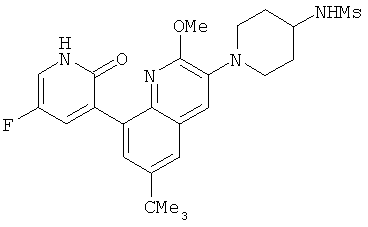
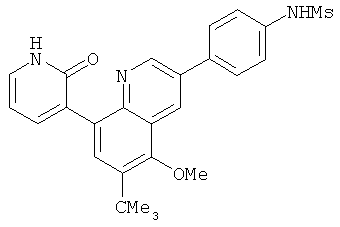
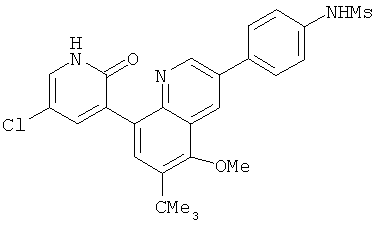
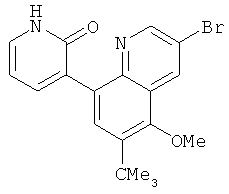
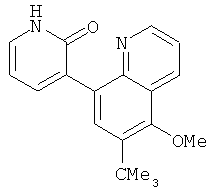
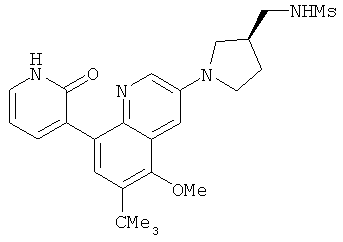
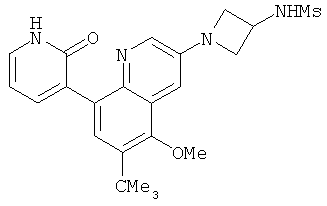
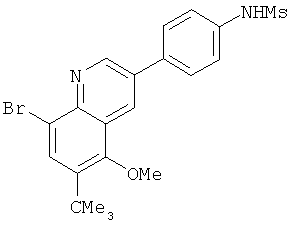
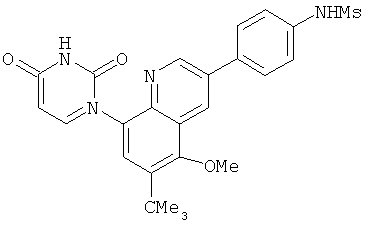
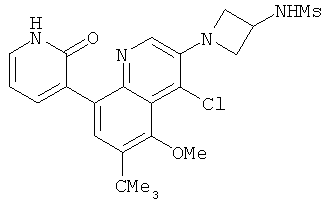
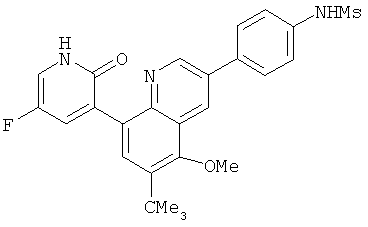
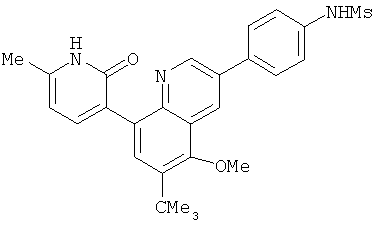
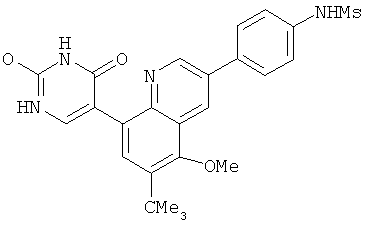
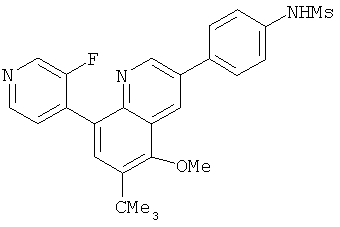
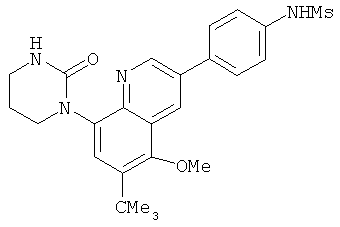
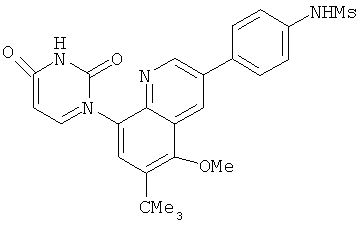
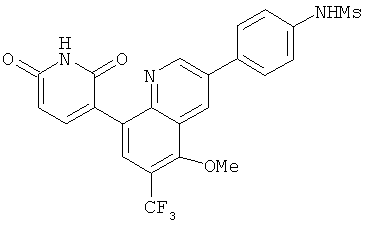
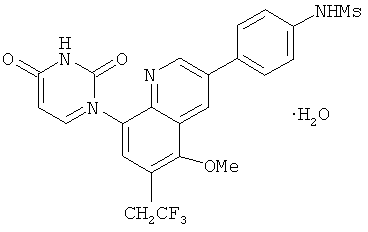
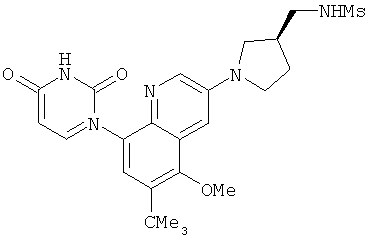
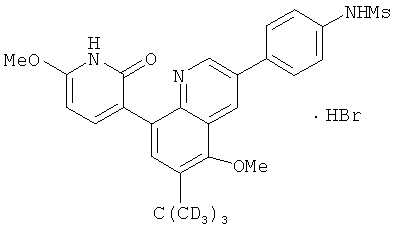
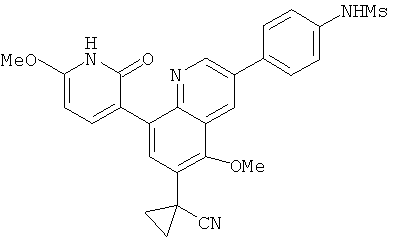
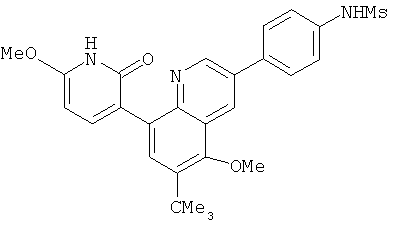
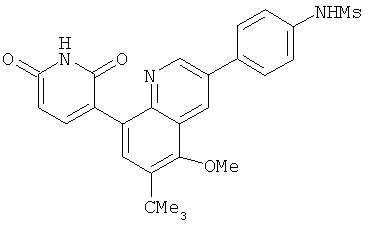

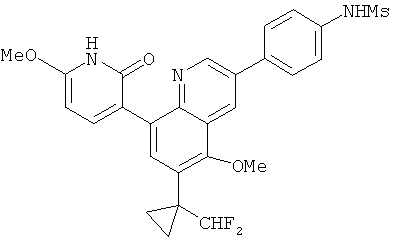
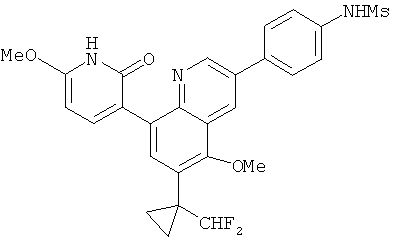
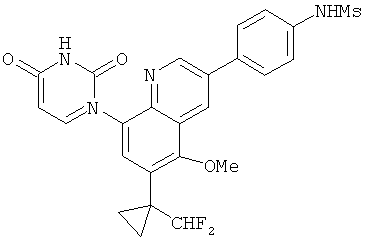
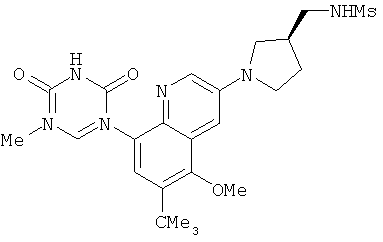
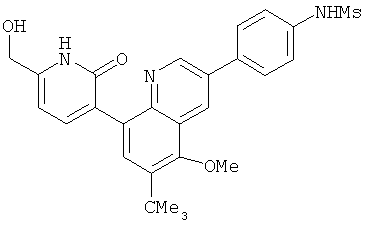
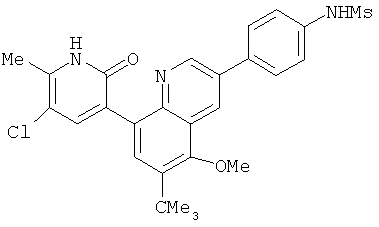
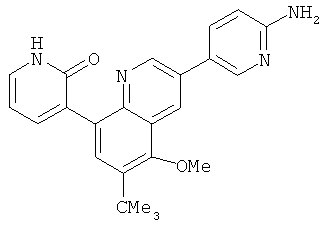
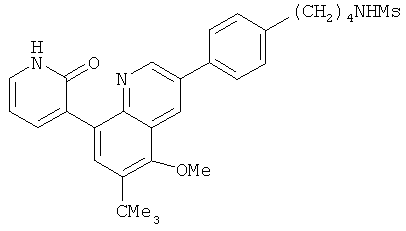
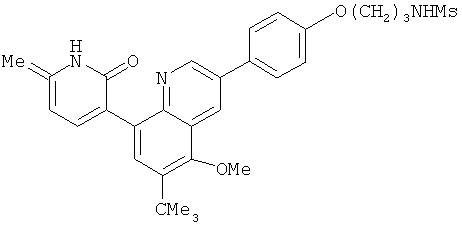
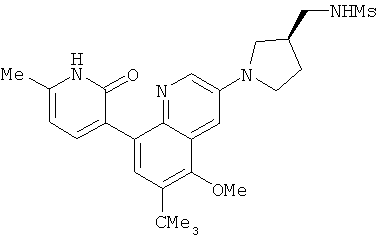
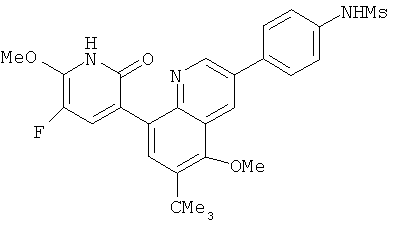
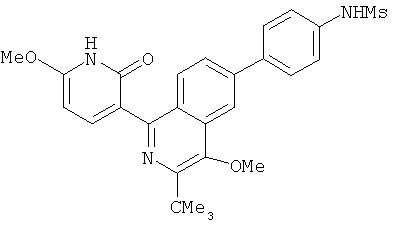
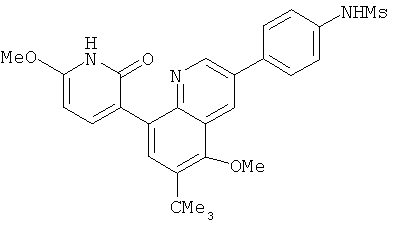
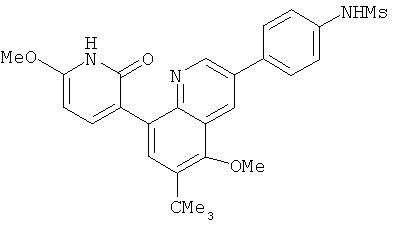
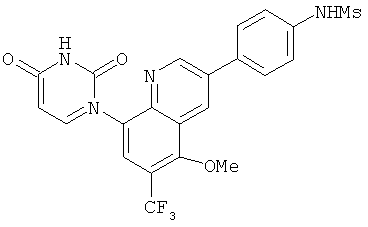
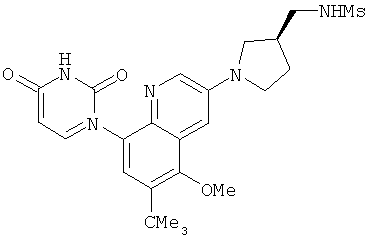
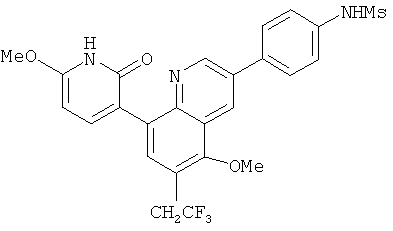
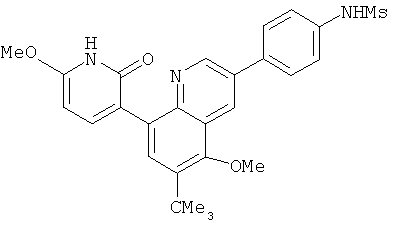
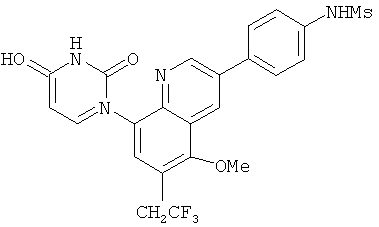
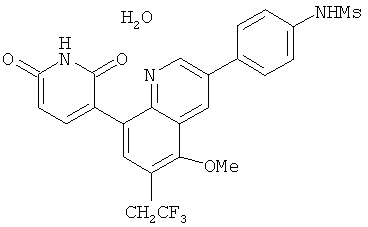
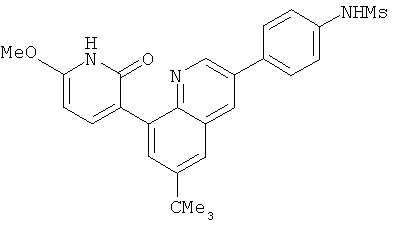
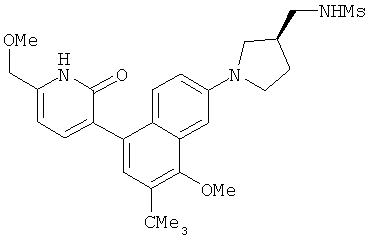
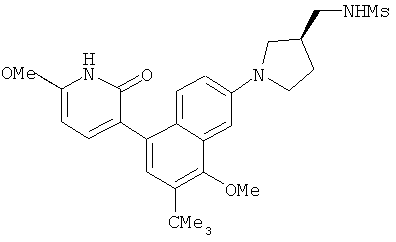
2. 1Н ЯМР: δ (CDCCl3) 1,486 (трет-Bu), 2,97 (SO2Me), 3,438 (NMe), 3,932 (OMe)
Соединения, приведенные в таблице II, являются дополнительными примерами соединений, входящих в объем настоящего изобретения.
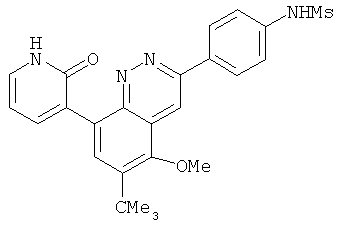
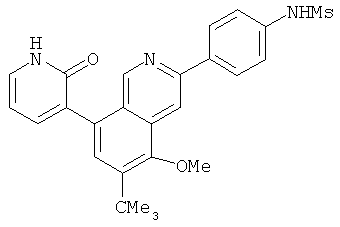
Соединения, предлагаемые в настоящем изобретении, можно получить с помощью множества методик, представленных на иллюстративных схемах реакций синтеза и описанных ниже. Исходные вещества и реагенты, использующиеся при получении этих соединений, обычно приобретают у поставщиков, таких как Aldrich Chemical Co., или их получают по методикам, известным специалистам в данной области техники по процедурам, описанным в литературе, такой как Fieser and Fieser's Reagents/or Organic Synthesis', Wiley & Sons: New York, Volumes 1-21; R. C. LaRock, Comprehensive Organic Transformations, 2" edition Wiley-VCH, New York 1999; Comprehensive Organic Synthesis, B. Trost and I. Fleming (Eds.) vol. 1-9 Pergamon, Oxford, 1991;
Comprehensive Heterocyclic Chemistry, A. R. Katritzky and C. W. Rees (Eds) Pergamon, Oxford 1984, vol. 1-9; Comprehensive Heterocyclic Chemistry II, A. R. Katritzky and С.W. Rees (Eds) Pergamon, Oxford 1996, vol. 1-11; и Organic Reactions, Wiley & Sons: New York, 1991, Volumes 1-40. Приведенные ниже схемы реакций синтеза являются лишь иллюстрациями некоторых методик, по которым можно синтезировать соединения, предлагаемые в настоящем изобретении, и в эти схемы реакций синтеза можно внести различные изменения, которые должен предложить специалист в данной области техники на основании раскрытия, приведенного в настоящей заявке.
Исходные вещества и промежуточные продукты для схем реакций синтеза при необходимости можно выделить и очистить по обычным методикам, включая, но не ограничиваясь только ими, фильтрование, перегонку, кристаллизацию, хроматографию и т.п. Такие вещества можно охарактеризовать с помощью обычных средств, включая определение физических характеристик и получение спектральных данных.
Если не указано иное, то реакции, описанные в настоящем изобретении, предпочтительно проводить в инертной атмосфере при атмосферном давлении и в диапазоне температур от примерно -78 до примерно 150°С, более предпочтительно от примерно 0 до примерно 125°С, и наиболее предпочтительно и удобно при температуре, близкой к комнатной (или температуре окружающей среды), например, примерно при 20°С.
На приведенных ниже схемах некоторые соединения изображены с заместителями, указанными в общем виде; однако специалист в данной области техники должен сразу же понять, что природа групп R может меняться и приводить к различным соединениям, входящим в объем настоящего изобретения. Кроме того, условия проведения реакций являются типичными и хорошо известны альтернативные условия. Последовательности реакций, использованные в приведенных ниже примерах, не означают ограничения объема настоящего изобретения, описанного в формуле изобретения.
Схема A
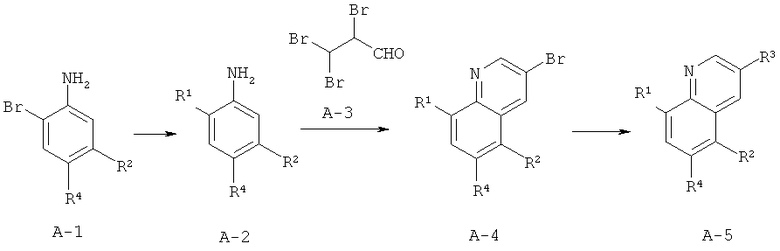
Производные хинолина, входящие в объем настоящего изобретения, получают по модифицированной реакции Скраупа для синтеза хинолина, где катализируемая кислотой реакция конденсации анилина А-2 и 1,2,2-трибромакролеина (А-3) дает бромхинолин А-4. Конденсацию обычно проводят с использованием анилина, в котором R обозначает гетероарильный фрагмент, описанный в Кратком изложении сущности изобретения, или его защищенную форму, которую в заключение превращают в указанный гетероарильный фрагмент. Введение гетероарильного фрагмента с получением соединения А-2, в котором R1 обозначает гетероарил, легко осуществить путем катализируемой палладием реакции сочетания орто-броманилина А-1 и гетероарилбороновой кислоты.
Бороновые кислоты, которые применимы для получения соединений, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, 2-метоксипиридин-3-илбороновую кислоту (регистрационный №CAS 163105-90-6), 2-бензилокси-3-пиридинбороновую кислоту, 2-оксо-1,2-дигидропиридин-3-бороновую кислоту (регистрационный № CAS 951655-49-5), 5-фтор-2-метокси-3-пиридинбороновую кислоту (регистрационный № CAS 957120-32-0), 2-метокси-6-метилпиридин-3-илбороновую кислоту (регистрационный № CAS 1000802-75-4), 5-хлор-2-метоксипиридин-3-илбороновую кислоту (регистрационный № CAS 943153-22-8), 2,6-диметоксипиридин-3-илбороновую кислоту (115, регистрационный №CAS 221006-70-8, В-(2,3-дигидро-3-оксо-4-пиридазинил)-бороновую кислоту (пример 16) или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-илбороновую кислоту (регистрационный №CAS 70523-22-7). Специалист в данной области техники должен понимать, что бороновые кислоты и сложные эфиры бороновых кислот, такие как 4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ильный радикал, в реакции сочетания Судзуки можно использовать как взаимозаменяемые. Оксогруппу можно маскировать путем образования простого алкилового эфира, что делает необходимым проведение последующей стадии деалкилирования с получением оксогруппы, которую легко провести путем нагревания в смеси HBr/НОАс.
После обработки хинолина вторая реакция сочетания Судзуки с арилбороновой кислотой, такой как 4-(метансульфонамидо)-фенилбороновая кислота или 4-нитрофенилбороновая кислота, позволяет прямо ввести 3-арильный заместитель в качестве R и дает соединение А-5. Специалист в данной области техники должен понимать, что наличие самых различных арилбороновых кислот позволяет в широких пределах менять последовательности превращений функциональных групп и природу заместителя R3.
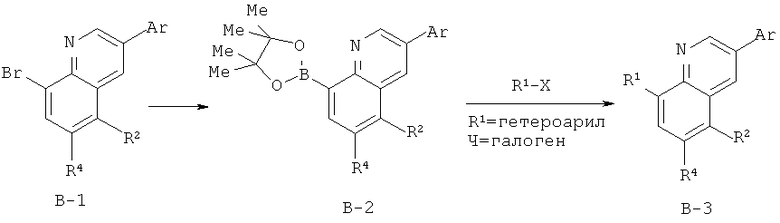
Реакцию конденсации Скраупа также можно провести с использованием броманилина и получить соединение А-4, в котором R обозначает бром, и затем провести реакцию сочетания Судзуки для введения гетероарильного заместителя R с использованием хинолина. Как показано в примере 13, сочетание предпочтительно происходит в положении 3. Использование 8-бромпроизводного (В-1) позволяет вносить в синтез дополнительные изменения. Металлирование 8-бромхинолина, в котором Ar является нереакционноспособным при условиях проведения реакции, позволяет осуществить введение бороновой кислоты в хинолиновое кольцо (В-2), что позволяет провести реакцию сочетания Судзуки с гетероарильными соединениями, замещенными содержащими галоген или трифторметилсульфонилоксигруппу заместителями, такими как 2-хлор-3-метоксипиразин (регистрационный №CAS 40155-28-0), полученное соединение можно деметилировать и получить 3-оксо-3,4-дигидропиразин-2-ильный фрагмент.
Катализируемая палладием реакция сочетания Судзуки бороновой кислоты (R-B(OH)2), где R обозначает арил или винил) с арил- или винилгалогенидом, или трифлатом (R'Y, где R'=арил или винил; Y=галогенид или -OSO2CF3) дает соединение R-R'. Типичные катализаторы включают Pd(PPh3)3, Pd(OAc)2 и PdCl2(dppf). С использованием Pd(Cl2(dppf) первичные алкилбораны можно ввести в реакцию сочетания с арил- или винилгалогенидом, или трифлатом без проведения β-элиминирования. Идентифицированы обладающие высокой активностью катализаторы (см., например, J. P. Wolfe et al., J. Am. Chem. Soc. 1999 121(41):9550-9561 и A. F. Littke et al.. J. Am. Chem. Soc. 2000 122(17):4020-4028). Реакцию можно проводить в различных органических растворителях, включая толуол, ТГФ, диоксан, ДХЭ, ДМФ, ДМСО и MeCN, водных растворителях и в двухфазных системах. Реакции обычно проводят при температурах в диапазоне от примерно комнатной температуры до примерно 150°С. Реакцию сочетания часто ускоряют добавки (например, CsF, KF, T10H, NaOEt и КОН). В реакции сочетания Судзуки существует большое количество параметров, включая источник палладия, лиганд, добавки и температуру, и для выбора оптимальных условий иногда необходимо оптимизировать параметры для определенной пары реагентов. В цитированной выше публикации А. F. Littke et al. раскрыты условия проведения реакции перекрестного сочетания Судзуки с арилбороновыми кислотами с получением высоких выходов, которые проводят при КТ с использованием Pd2(dba)3/P(трепт-bu)3, и условия проведения реакции перекрестного сочетания арил- и винилтрифлатов при КТ с использованием Pd(OAc)2/P(C6H11)3. В цитированной выше публикации J. Р. Wolfe/ al. раскрыты условия эффективного проведения реакции перекрестного сочетания Судзуки с использованием Pd(OAc)2/o-(ди-трет-бутилфосфино)бифенила или о-(дициклогексилфосфино)бифенила. Специалист в данной области техники может определить оптимальные условия без чрезмерного объема экспериментов.
СХЕМА С
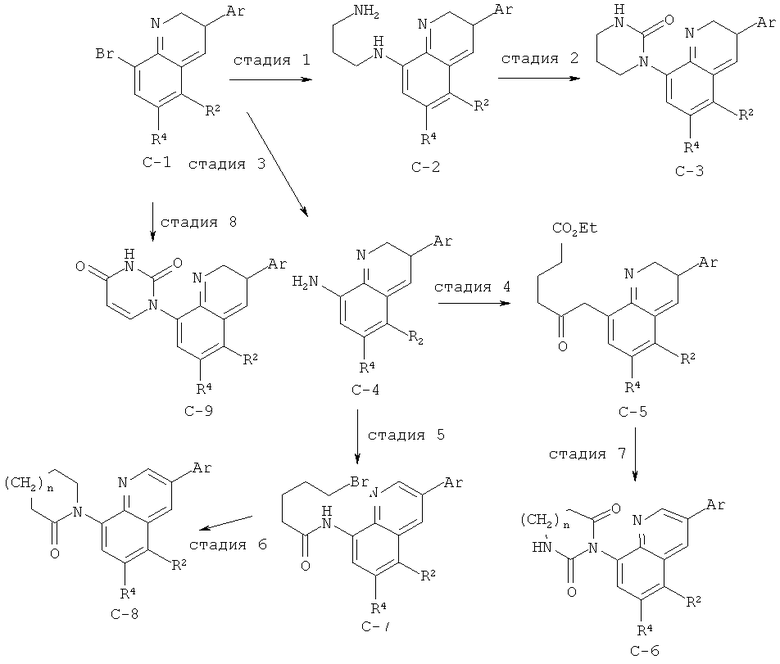
Соединения общей формулы I, в которой R обозначает гетероцикл, присоединенный связью углерод-азот, можно получить по катализируемой медью или палладием реакции аминирования арила. Методики аминирования арила описаны. Введение заместителей, 2-оксотетрагидропиримидин-1-ила или 2-оксоимидазолидин-1-ила, можно провести по катализируемой с помощью Cul реакции аминирования арила, бромхинолина с помощью 1,3-диаминопропана (стадия 1) или 1,2-диаминоэтана (D. Ma et al., Org. Lett. 2003 5(14):2453) с последующей реакцией внутримолекулярной циклизации с использованием карбонилдиимидазола (стадия 2). Альтернативно, гетероциклическое кольцо можно получить из первичного амина С-4. Описаны многочисленные методики введения первичного амина в арильное кольцо путем замещения галогена (стадия 3). (J.P. Wolfe et al. Tetrahedron Lett. 1997 38(36):6367; C.-Z. Tao et al., Tetrahedron Lett. 2008 49:70; Q. Shen and J.F. Hartwig, J. Am. Chem. Soc. 2006 128:10028; S. S. Surry and S. L. Buchwald, J. Am. Chem. Soc. 2007 129:10354). Ацилирование соединения С-4 5-бромпентановой кислотой (стадия 5) или 4-броммасляной кислотой с последующей реакцией внутримолекулярной циклизации (стадия 6) дают заместители пиперидон (С-8, n=1) и пирролидон (С-8, n=0) соответственно. Реакция конденсации соединения С-4 и этил-3-изоцианатопропаноата (регистрационный №CAS 5100-34-5) или этил-4-изоцианатоацетата (регистрационный №CAS 2949-22-6) (стадия 4) и последующей реакцией внутримолекулярного ацилирования (стадия 7) дают 2,5-диоксоимидазолидин-1 -ильный (С-6, n=0) или 2,6-диоксотетрагидропиримидин-1-ильный (С-6, n=1) фрагменты, соответственно. 2,4-Диоксо-3,4-дигидро-2Н-пиримидин-1-ильный фрагмент (С-9) можно ввести путем катализируемого с помощью CuI замещения брома на урацил (стадия 8) (R. Wagner et al. W02009/039127).
Соединения общей формулы А-5, в которой связь арил-R представляет собой связь углерод-азот, можно получить путем катализируемого палладием замещения брома в соединении А-3 на необязательно замещенный циклический амин, как это описано в примерах 11 и 12 (J.F. Hartwig et al., J. Org. Chem. 1999 64:5575).
Введение заместителей в двух положениях проводят через получение хинолона (пример 1), который получают по катализируемой палладием реакции перекрестного сочетания метилакрилата и соединения 20а по методике Хека и катализируемой кислотой реакции циклизации лактама, O-алкилирование лактама дает соединение с 2-метоксигруппой. Реакция хинолона и оксигалогенида фосфора дает 2-галогенхинолин, который можно заместить и ввести другую функциональную группу.
Введение ациклических заместителей в положении R3 проводят по реакции сочетания Хека гетероарилгалогенида и подходящего замещенного алкена или алкина (см., например, пример 29). Реакция Хека (или реакция Мицороки-Хека) представляет собой катализируемую палладием реакцию сочетания арил-, алкенил-, алкинил- или бензилгалогенида, или трифлата с алкенстиролом, эфиром акриловой кислоты, акрилонитрилом, простым эфиром енола или простым тиоэфиром енола (A. se Meijere and F. E. Meyer, Angew Chem. Int. Ed. English 1994 33:2379-2411; W. Cabri and I. Cand'iani, Ace. Chem. Res. 1995 28(1):2-7), содержащим по меньшей мере один протон и часто являющимся электронодефицитным, таким как эфир акриловой кислоты или MeCN. Обычно использующиеся палладиевые катализаторы включают Pd(PPh3)4, Pd(OAc)2, PdCl2, Pd2(dba)3. Фосфиновые лиганды, такие как PPh3, Р(о-Tol)3 и БИНАФ обычно вводят в реакционную смесь в виде предварительно полученных комплексов фосфина или в виде свободных фосфинов, которые могут образовывать комплексы in situ. Обычно необходимо использование оснований, таких как ТЭА, 1,2,2,6,6-пентаметилпиперидин, ДБУ, K2CO3, KОАс, Ag2CO3 и KO-трет-Bu. Реакцию обычно проводят в апротонных растворителях, часто в ДМФ, ДМСО, NMP или MeCN; однако также можно использовать менее полярные растворители и водные сорастворители. Поскольку существует несколько переменных в условиях проведения реакции, условия определены, и специалист в данной области техники может определить подходящие условия без чрезмерного объема экспериментов.
Аналогичные превращения приводят к получению соединений формулы I, в которой R4 отличается от трет-бутила. Дибромирование 4-(1-метилциклопропил)бензоламида (регистрационный №CAS 114833-72-6) или 1-(4-аминофенил)циклопропанкарбонитрила (CASRN108858-86-2) дает промежуточные продукты, которые можно ввести в совершенно аналогичную последовательность реакций. Альтернативно, метил-4-амино-5-бром-2-метоксибензоат (регистрационный №CAS 111049-68-4) можно ввести в реакцию Скраупа, которая дает метил-3,8-дибром-5-метоксихинолин-6-карбоксилат (122а), который превращают в соответствующий циклопропанкарбонитрил (124а) по обычным методикам (см., например, пример 25). Нитрильную группу можно легко превратить в соответствующую альдегидную группу (и поэтому также в соответствующую кислотную или сложноэфирную группу) и затем в дифторметильную (фторирование с помощью ДАТС (диэтиламинотрифторид серы)) или гидроксиметильную (восстановление с помощью ВН3) группу. Аналогичные превращения можно провести и получить соответствующие аналоги, не содержащие метоксигруппу. Аналогичным образом 4-трифторметиланилин (регистрационный №CAS 455-14-1) и 3-метокси-4-трифторметиланилин можно превратить в хинолины и хиназолины.
СХЕМА D
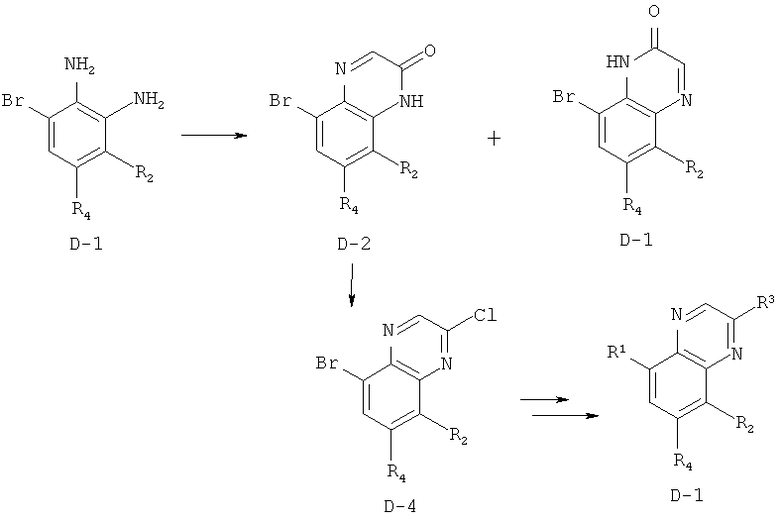
Хиназолины, входящие в объем настоящего изобретения, получают по реакции конденсации подходящего замещенного орто-диаминобензола с 1,2-дикарбонильным соединением. Например, этилглиоксилат и соединение 32 конденсируют с получением смеси 5-бром-7-трет-бутил-1Н-хиноксалин-2-она и 8-бром-6-трет-бутил-1Н-хиноксалин-2-она. Введение заместителей в положения С-3 и С-8 можно провести путем последовательно проводимых реакций замещения, как это описано выше (см., например, пример 2).
СХЕМА Е
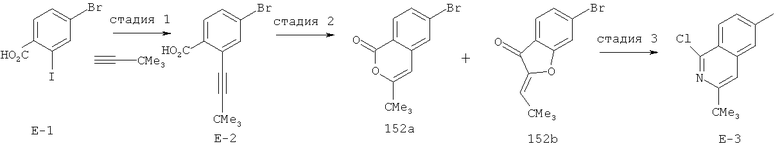
Соединения общей формулы I, в которой Х3 обозначает N и Х1, Х2 и Х4 обозначают CR6, получают из соединения Е-3 путем последовательно проводимых катализируемых палладием реакций сочетания для введения заместителей R1 и R3 с использованием условий, аналогичных описанным выше. Превращение соединения Е-1 в соединение Е-2 проводят с использованием методики катализируемой палладием реакции сочетания Хека. Катализируемая палладием внутримолекулярная реакция лактонизации ацетиленовой кислоты приводит к образованию смеси 6-эндо-диг и 5-экзо-диг продуктов 152а и 152b соответственно (Н. Sashida and A. Kawamuki, Synthesis 1999 1145). Обработка соединения 152а аммиаком дает соответствующий изохинолин, который превращают в соединение Е-3 с помощью POCl3. Получение соединения Е-1 описано в публикации G.C. Colossi et al., W02008/087057.
СХЕМА F
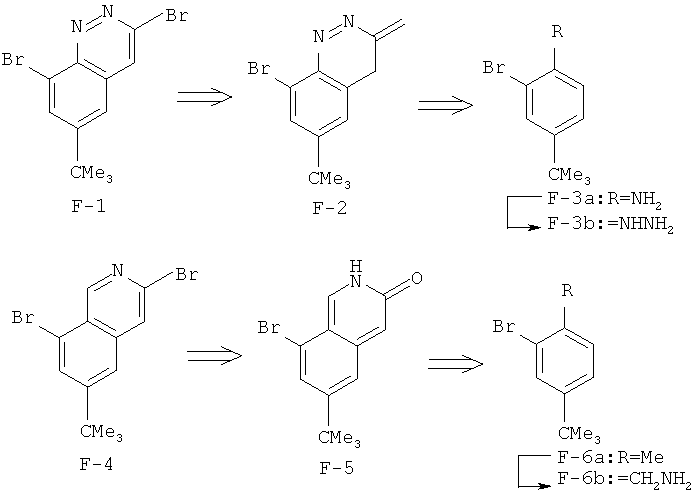
Соединения общей формулы I, в которой Х3 и Х2 обозначают CR6, и Х1 и Х4 обозначают N (производные циннолина), получают аналогичным образом из соединения F-1 путем последовательно проводимых катализируемых палладием реакций сочетания для введения заместителей R1 и R3 с использованием условий, аналогичных описанным выше. Превращение соединения F-2 в соединение F-1 проводят с использованием оксибромида фосфора. Соединение F-2 получают путем обработки соединения F-3b диэтоксиацетилхлоридом для ацилирования гидразина и введения его в реакцию внутримолекулярного ацилирования Фриделя-Крафтса. Соединения общей формулы I, в которой Х1, Х2 и Х3 обозначают CR6, получают аналогичным образом из соединения F-4, которое получают из соединения F-5. Соединение F-5 получают по аналогичной реакции внутримолекулярного ацилирования Фриделя-Крафтса соединения F-6a. Во избежание двусмысленности следует понимать, что стрелки, приведенные на схеме F ("⇒"), означают ретроразмыкание. (E.J. Corey Angew. Chem. Intl. Ed. Engl. 1991 30:455).
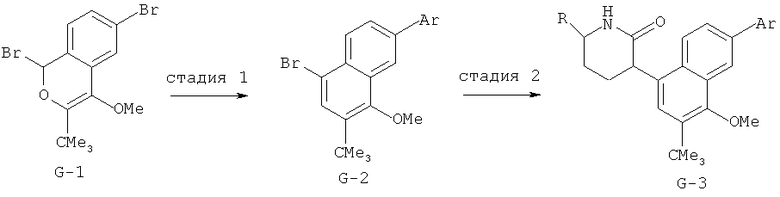
Соединения общей формулы I, в которой Х1, Х2, Х3 и Х4 обозначают CR5, получают из дибромнафталина G-1 путем последовательно проводимых катализируемых палладием реакций сочетания для введения заместителей R1 и R3 с использованием условий, аналогичных описанным выше. Получение соединения G-1 описано в примере 22 вместе с последовательно проводимыми реакциями сочетания Судзуки для введения фрагментов R1 и R3.
Активность соединений, предлагаемых в настоящем изобретении, как ингибиторов активности HCV, можно определить с помощью любых подходящих методик, известных специалистам в данной области техники, включая исследования in vivo и in vitro. Например, ингибирующую активность соединений формулы I по отношению к HCV NS5B можно определить по стандартным методикам анализа, описанным в публикациях Behrens et al., EMBO J. 1996 15:12-22, Lohmann et al.. Virology 1998 249:108-118 и Ranjith-Kumar et al., J. Virology 2001 75:8615-8623. Если не указано иное, то в таких стандартных исследованиях соединения, предлагаемые в настоящем изобретении, обладают ингибирующей активностью по отношению к HCV NS5B in vitro. Условия проведения исследования HCV полимеразы, использующиеся для соединений, предлагаемых в настоящем изобретении, описаны в примере 8. Разработаны системы репликонов с использованием клеток для HCV, в которых неструктурированные белки стабильно реплицируют субгеномную вирусную РНК в клетках Huh7 (V. Lohmann et al., Science 1999 285:110 и K. J. Blight et al., Science 2000 290:1972. Условия исследования репликонов с использованием клеток, использующиеся для соединений, предлагаемых в настоящем изобретении, описаны в примере 4. В отсутствие очищенной функциональной HCV репликазы, состоящей из вирусных неструктурированных белков и белков хозяина, наше понимание синтеза Flaviviridae РНК основано на исследованиях с использованием рекомбинантных РНК-зависимых РНК полимераз и подтверждения этих исследований на системах репликонов HCV. Ингибирование рекомбинантной очищенной HCV полимеразы соединениями в биохимических исследованиях in vitro можно подтвердить с помощью системы репликонов, где полимераза существует в комплексе с репликазой, связанной с другими вирусными и клеточными полипептидами при соответствующей стереохимической конфигурации. Подтверждение ингибирования репликации HCV с использованием клеток можно лучше прогнозировать с помощью воздействия in vivo, чем подтверждение ингибирования активности HCV NS5B в биохимических исследованиях in vitro.
Соединения, предлагаемые в настоящем изобретении, можно приготовить в виде самых различных дозированных форм, предназначенных для перорального введения, и с использованием различных носителей. Пероральное введение можно осуществлять с помощью таблеток, таблеток с покрытием, драже, капсул из мягкого и твердого желатина, растворов, эмульсий, сиропов или суспензий. Соединения, предлагаемые в настоящем изобретении, эффективны при введении другими путями, включая, наряду с другими путями введения, непрерывное (внутривенное капельное), местное, парентеральное, внутримышечное, внутривенное, подкожное, чрескожное (которое может включать агент, увеличивающий проницаемость), трансбуккальное, назальное, ингаляционное и проводимое с помощью суппозиториев введение. Предпочтительным путем введения обычно является пероральный с использованием обычного суточного дозировочного режима, который можно подобрать в соответствии с тяжестью заболевания и реакцией пациента на активный ингредиент.
Соединение или соединения, предлагаемые в настоящем изобретении, а также их фармацевтически применимые соли, совместно с одним или большим количеством обычных инертных наполнителей, носителей или разбавителей можно приготовить в виде фармацевтических композиций и разовых дозированных форм. Фармацевтические композиции и разовые дозированные формы могут включать обычные ингредиенты в обычных количествах с добавлением или без добавления дополнительных активных соединений или действующих веществ, и разовые дозированные формы могут содержать любое подходящее эффективное количество активного ингредиента, соответствующего применяемому суточному дозировочному диапазону. Фармацевтические композиции можно использовать в виде твердых веществ, таких как таблетки или заполненные капсулы, полужидких веществ, порошков, композиций пролонгированного действия, или в виде жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или заполненные капсулы для перорального введения; или в виде суппозиториев для ректального или вагинального введения;
или в виде стерильных растворов для инъекций, предназначенных для парентерального введения. Типичный препарат будет содержать от примерно 5 до примерно 95% (мас./мас.) активного соединения или соединений. Подразумевается, что термин "препарат" или "дозированная форма" включает и твердые, и жидкие композиции активного соединения, и специалист в данной области техники должен понимать, что активный ингредиент может находиться в разных препаратах в зависимости от органа-мишени или ткани-мишени, от требуемой дозы и фармакокинетических параметров.
Термин "инертный наполнитель" при использовании в настоящем изобретении означает соединение, которое является пригодным для приготовления фармацевтической композиции, обычно безопасное, нетоксичное и биологически, и в других отношениях не являющееся нежелательным, и включает инертные наполнители, которые приемлемы для применения в ветеринарии, а также для фармацевтического применения для людей. Соединения, предлагаемые в настоящем изобретении, можно вводить по отдельности, но обычно их вводят в смеси с одним или большим количеством фармацевтических инертных носителей, разбавителей или носителей, выбранных в соответствии с предполагаемым режимом введения и стандартной фармацевтической практикой.
"Фармацевтически приемлемое" означает применимое для приготовления фармацевтический композиции, обычно безопасное, нетоксичное и биологически, и в других отношениях не являющееся нежелательным.
"Фармацевтически приемлемой соли" активного ингредиента может быть изначально придана желательная фармакокинетическая характеристика активного ингредиента, которая отсутствует у несолевой формы, и она даже может благоприятно влиять на фармакодинамику активного ингредиента с точки зрения его терапевтической активности в организме. Выражение "фармацевтически приемлемая соль" соединения означает соль, которая является фармацевтически приемлемой и которая обладает необходимой фармакологической активностью исходного соединения. Такие соли включают:
(1) соли присоединения с кислотами, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п.; или образованные с органическими кислотами, такими как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло [2.2.2]-окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п.; или (2) соли, образованные с кислым протоном, содержащимся в исходном соединении, замещенным ионом металла, например ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; или координацией с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п.
Твердые формы препаратов включают порошки, таблетки, пилюли, капсулы, облатки, суппозитории и диспергирующиеся гранулы. Твердым носителем может быть одно или большее количество веществ, которые также могут выступать в качестве разбавителей, вкусовых веществ, солюбилизаторов, смазывающих веществ, суспендирующих веществ, связующих, консервантов, веществ, обеспечивающих распадаемость таблеток, или капсулирующего материала. В порошках носитель обычно представляет собой тонкоизмельченное твердое вещество, которое образует смесь с тонкоизмельченным активным компонентом. В таблетках активный компонент обычно в подходящих соотношениях смешан с носителем, обладающим необходимой связывающей способностью, и спрессован с приданием необходимой формы и размера. Подходящие носители включают, но не ограничиваются только ими, карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакантовую камедь, метилцеллюлозу, натриевую соль карбоксиметилцеллюлозы, легкоплавкий воск, масло какао и т.п. Твердые формы препаратов в дополнение к активному компоненту могут содержать красители, ароматизаторы, стабилизаторы, буферные вещества, искусственные и натуральные подсластители, диспергирующие вещества, загустители, солюбилизирующие агенты и т.п.
Жидкие композиции, также пригодные для перорального введения, включают жидкую композицию, включая эмульсии, сиропы, эликсиры, водные растворы, водные суспензии. Они включают твердые формы препаратов, которые предназначены для проводимого незадолго до введения превращения в жидкие формы препаратов. Эмульсии можно приготовить в растворах, например в водных растворах пропиленгликоля, или они могут содержать эмульгирующие вещества, например, такие как лецитин, сорбитанмоноолеат или камедь акации. Водные растворы можно приготовить путем растворения активного компонента в воде и добавления подходящих красителей, ароматизаторов, стабилизаторов и загущающих веществ. Водные суспензии можно приготовить путем диспергирования тонкоизмельченного активного компонента в воде, содержащей вязкое вещество, такое как натуральные или синтетические камеди, смолы, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы и другие хорошо известные суспендирующие вещества.
Соединения, предлагаемые в настоящем изобретении, можно приготовить для парентерального введения (например, путем инъекции, например инъекции ударной дозы вещества или непрерывного вливания) и можно приготовить в виде разовой дозированной формы в ампулах, предварительно заполненных шприцах, предназначенных для вливания контейнерах небольшого объема или содержащих множество доз контейнерах с прибавленным консервантом.
Композиции могут находиться в таких формах, как суспензии, растворы или эмульсии в масляных или водных разбавителях, например, в виде растворов в водном растворе полиэтиленгликоля. Примеры масляных и неводных носителей, разбавителей, растворителей и наполнителей включают пропиленгликоль, полиэтиленгликоль, растительные масла (например, оливковое масло) и пригодные для инъекции органические сложные эфиры (например, этилолеат) и они могут содержать образующие композиции вещества, такие как консервирующие, смачивающие, эмульгирующие или суспендирующие, стабилизирующие и/или диспергирующие вещества. Альтернативно, активный ингредиент может находиться в порошкообразном виде, полученном асептическим выделением стерильного твердого вещества или лиофилизацией из раствора для проводимого перед применением восстановления с помощью подходящего разбавителя, например стерильной апирогенной воды.
Соединения, предлагаемые в настоящем изобретении, можно приготовить для местного введения в эпидермис в виде мазей, кремов или лосьонов или в виде чрескожного пластыря. Мази и кремы можно, например, приготовить на водной или масляной основе путем добавления подходящих загущающих и/или желирующих агентов. Лосьоны можно приготовить на водной или масляной основе и они также обычно содержат один или большее количество эмульгирующих агентов, стабилизирующих агентов, диспергирующих агентов, суспендирующих агентов, загущающих агентов или окрашивающих агентов. Препараты, применимые для местного введения в полость рта, включают лепешки, содержащие активные средства в придающей вкус основе, обычно сахарозе, камеди акации или трагакантовой камеди; пастилки содержат активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и камедь акации; и средства для полоскания рта содержат активный ингредиент в подходящем жидком носителе.
Соединения, предлагаемые в настоящем изобретении, можно приготовить для введения в виде суппозиториев. Сначала расплавляют низкоплавкий воск, такой как смесь глицеридов жирных кислот, или масло какао, и активный компонент равномерно диспергируют, например, путем перемешивания. Затем расплавленную однородную смесь выливают в формы соответствующего размера, дают ей охладиться и затвердеть.
Соединения, предлагаемые в настоящем изобретении, можно приготовить для вагинального введения. В данной области техники известно, что подходящими являются пессарии, тампоны, кремы, гели, пасты, пены и аэрозольные препараты, содержащие такие носители в дополнение к активному ингредиенту.
Соединения, предлагаемые в настоящем изобретении, можно приготовить для назального введения. Растворы или суспензии вводят непосредственно в полость рта с помощью обычных средств, например капельницы, пипетки или путем распыления. Препараты можно приготовить в виде разовой дозы или в многодозовой форме. В последнем случае использования капельницы или пипетки это можно выполнить путем введения пациенту подходящего заранее заданного объема раствора или суспензии. В случае распыления его можно выполнить, например, с помощью дозирующего распыляющего насоса.
Соединения, предлагаемые в настоящем изобретении, можно приготовить для введения в виде аэрозоля, в особенности в дыхательные пути и, включая внутриназальное введение. Соединение обычно обладает частицами небольшого размера, например, порядка пяти (5) мкм или менее. Частицы такого размера можно получить по методикам, известным в данной области техники, например, путем микронизации. Активный ингредиент поставляется в упаковке под давлением с подходящим пропеллентом, таким как хлорфторуглерод (ХФУ), например, дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, или диоксид углерода, или другой подходящий газ. Аэрозоль обычно также может содержать поверхностно-активное вещество, такое как лецитин. Дозу лекарственного средства можно регулировать с помощью дозирующего клапана. Альтернативно, активные ингредиенты могут поставляться в виде сухого порошка, например, порошкообразной смеси соединения с подходящей порошкообразной основой, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилметилцеллюлоза и поливинилпирролидон (ПВП). Порошкообразный носитель будет образовывать гель в полости носа. Порошкообразная композиция может поставляться в виде разовой дозы, например в капсулах или картриджах, например, желатиновых, или в блистерных упаковках, из которых порошок можно вводить с помощью ингалятора.
При необходимости композиции можно приготовить с энтеросолюбильным покрытием, приспособленным для пролонгированного или регулируемого введения активного ингредиента. Например, соединения, предлагаемые в настоящем изобретении, можно поместить в устройства для чрескожной или подкожной доставки лекарственных препаратов. Такие устройства доставки являются предпочтительными, когда необходимо пролонгированное действие соединения и когда принципиально важным является соблюдение пациентом режима лечения. Системы чрескожной доставки, содержащие соединения, часто прикрепляют к приклеивающейся к коже твердой подложке. Необходимое соединение также можно объединять со средством, увеличивающим проницаемость, например, азоном (1-додецилазациклогептан-2-оном). Системы доставки пролонгированного действия вводят подкожно в субдермальный слой оперативным путем или путем инъекции. Подкожный имплантат содержит соединение, капсулированное в растворимой в липидах мембране, например, силиконовом каучуке, или в биологически разрушающемся полимере, например, полимолочной кислоте.
Подходящие композиции, а также фармацевтические носители, разбавители и инертные наполнители описаны в публикации Remington: The Science and Practice of Pharmacy 1995, edited by E. W. Martin, Mack Publishing Company, 19th edition, Easton, Pennsylvania. После изучения настоящего изобретения специалист по составлению композиций может внести в композиции изменения и получить многочисленные композиции, предназначенные для конкретного пути введения, не делая композиции, предлагаемые в настоящем изобретении, нестабильными и не ухудшая их терапевтическую активность.
Модификацию соединений, предлагаемых в настоящем изобретении, например, для того, чтобы сделать их лучше растворимыми в воде или другом разбавителе, можно без труда осуществить путем внесения небольших изменений (состава соли, этерификации и т.п.), которые входят в компетенцию специалиста с общей подготовкой в данной области техники. В компетенцию специалиста с общей подготовкой в данной области техники также входит изменение пути введения и дозировочного режима введения конкретного соединения, так чтобы управлять фармакокинетикой соединений, предлагаемых в настоящем изобретении, для обеспечения максимального благоприятного воздействия на пациента.
Термин "терапевтически эффективное количество" при использовании в настоящем изобретении означает количество, необходимое для ослабления симптомов заболевания у индивидуума. Дозу следует подбирать в каждом конкретном случае в соответствии с индивидуальными потребностями. Эта дозировка может меняться в широких пределах в зависимости от множества факторов, таких как тяжесть подвергающегося лечению заболевания, возраст и общее состояние здоровья пациента, другие лекарственные средства, которыми лечится пациент, путь и форма введения и предпочтения и опыт практикующего врача. При пероральном введении суточная доза, составляющая от примерно 0,01 до примерно 1000 мг/(кг массы тела) в сутки, должна являться подходящей для монотерапии и/или комбинированной терапии. Предпочтительная суточная доза составляет от примерно 0,1 до примерно 500 мг/(кг массы тела), более предпочтительная - от 0,1 до примерно 100 мг/(кг массы тела) и наиболее предпочтительная - от 1,0 до примерно 10 мг/(кг массы тела) в сутки. Таким образом, для введения лицу массой 70 кг дозировочный диапазон должен составлять от примерно 7 мг до 0,7 г в сутки. Суточную дозу можно вводить в виде одной дозы или разделенных доз, обычно от 1 до 5 доз в сутки. Обычно лечение начинают с меньших доз, которые меньше оптимальной дозы соединения. Затем дозу увеличивают небольшими шагами, пока не будет достигнут оптимальный для данного конкретного пациента эффект. Специалист с общей подготовкой в данной области техники при лечении заболеваний, описанных в настоящем изобретении, без чрезмерного экспериментирования и на основании личной подготовки, опыта и раскрытия настоящей заявки должен быть способен определить терапевтически эффективное количество соединений, предлагаемых в настоящем изобретении, необходимое для лечения данного заболевания и пациента.
В вариантах осуществления настоящего изобретения активное соединение или соль можно вводить в комбинации с другим противовирусным агентом, таким как рибаривин, ингибитор нуклеозидной полимеразы HCV, другой ингибитор ненуклеозидной полимеразы HCV или ингибитор протеазы HCV. Если активное соединение или его производное или соль вводится в комбинации с другим противовирусным агентом, то активность по сравнению с исходным соединением может увеличиться. Если методикой лечения является комбинированная терапия, такое введение может быть совместным или последовательным по отношению к нуклеозидным производным. Таким образом, "совместное введение" при использовании в настоящем изобретении включает введение агентов в одно и то же время или в разные моменты времени. Введение двух или большего количества агентов в одно и то же время можно осуществить с помощью одной композиции, содержащей два или большее количество активных ингредиентов, или путем в основном совместного введения двух или большего количества дозированных форм, содержащих по одному активному агенту.
Следует понимать, что в настоящем изобретении указания на лечение распространяются на профилактику, а также на лечение имеющихся патологических состояний. Кроме того, термин "лечение" инфекции HCV при использовании в настоящем изобретении также включает лечение или профилактику заболевания или патологического состояния, сопутствующего инфекции HCV или опосредуемого ею, или его клинических симптомов.
Термин "терапевтически эффективное количество" при использовании в настоящем изобретении означает количество, необходимое для ослабления симптомов заболевания у индивидуума. Дозу следует подбирать в каждом конкретном случае в соответствии с индивидуальными потребностями. Эта дозировка может меняться в широких пределах в зависимости от множества факторов, таких как тяжесть подвергающегося лечению заболевания, возраст и общее состояние здоровья пациента, другие лекарственные средства, которыми лечится пациент, путь и форма введения и предпочтения и опыт практикующего врача. При пероральном введении суточная доза, составляющая от примерно 0,01 до примерно 1000 мг/(кг массы тела) в сутки, должна являться подходящей для монотерапии и/или комбинированной терапии. Предпочтительная суточная доза составляет от примерно 0,1 до примерно 500 мг/(кг массы тела), более предпочтительная - от 0,1 до примерно 100 мг/(кг массы тела) и наиболее предпочтительная - от 1,0 до примерно 10 мг/(кг массы тела) в сутки. Таким образом, для введения лицу массой 70 кг дозировочный диапазон должен составлять от примерно 7 мг до 0,7 г в сутки. Суточную дозу можно вводить в виде одной дозы или разделенных доз, обычно от 1 до 5 доз в сутки. Обычно лечение начинают с меньших доз, которые меньше оптимальной дозы соединения. Затем дозу увеличивают небольшими шагами, пока не будет достигнут оптимальный для данного конкретного пациента эффект. Специалист с общей подготовкой в данной области техники при лечении заболеваний, описанных в настоящем изобретении, без чрезмерного экспериментирования и на основании личной подготовки, опыта и раскрытия настоящей заявки должен быть способен определить терапевтически эффективное количество соединений, предлагаемых в настоящем изобретении, необходимое для лечения данного заболевания и пациента.
Обычно терапевтически эффективное количество соединения, предлагаемого в настоящем изобретении, и необязательно одного или большего количества дополнительных противовирусных средств является количеством, эффективным для снижения вирусной нагрузки или обеспечения постоянного ответа вирусов на лечение. Полезными индикаторами постоянного ответа вирусов в дополнение к вирусной нагрузке включают, но не ограничиваются только ими, фиброз печени, повышение концентрации трансаминазы в сыворотке и некровоспалительной активности в печени. Одним обычным примером, который является типичным и не ограничивающим, маркера является аланинтрансаминаза (АЛТ) сыворотки, концентрацию которой определяют с помощью стандартных клинических анализов. В некоторых вариантах осуществления настоящего изобретения эффективным режимом лечения является такой, который приводит к уменьшению концентрации АЛТ в сыворотке до значения, равного менее примерно 45 МЕ/мл.
Модификацию соединений, предлагаемых в настоящем изобретении, например, для того, чтобы сделать их лучше растворимыми в воде или другом разбавителе, можно без труда осуществить путем внесения небольших изменений (состава соли, этерификации и т.п.), которые входят в компетенцию специалиста с общей подготовкой в данной области техники. В компетенцию специалиста с общей подготовкой в данной области техники также входит изменение пути введения и дозированого режима введения конкретного соединения, так чтобы управлять фармакокинетикой соединений, предлагаемых в настоящем изобретении, для обеспечения максимального благоприятного воздействия на пациента.
Представленные ниже примеры иллюстрируют получение и исследование биологических характеристик соединений, входящих в объем настоящего изобретения. Эти примеры и методики получения, приведенные ниже, предоставлены для того, чтобы специалист в данной области техники мог яснее понять и осуществить настоящее изобретение. Их следует считать не ограничивающими объем настоящего изобретения, а просто иллюстративными и типичными.
Пример 1
Н-{4-[6-трет-Бутил-2-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид (I-1)
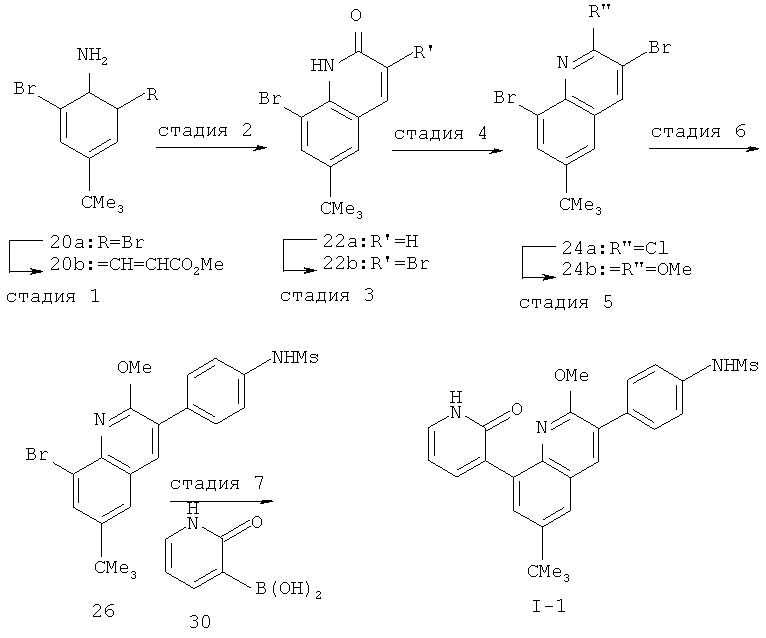
Стадия 1 - К раствору соединения 20а (10,0 г) в MeCN (200 мл) добавляли три-(о-толил)фосфин (1,33 г), Pd(II)(OAc)2 (0,730 г), ТЭА (6,8 мл) и метилакрилат (2,35 мл). Реакционную смесь перемешивали при 100°С в течение ночи. Реакционную смесь охлаждали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (0% EtOAc - 0-5 мин, 20% EtOAc - 5,5-15 мин и 40% EtOAc - 15,5-30 мин) и получали 3,02 г соединения 20b.
Стадия 2 - К раствору соединения 20b (6,73 г) в ТГФ (150 мл) добавляли 6 н. раствор HCl (150 мл) и полученный раствор нагревали при 100°С в течение ночи. Раствор охлаждали и концентрировали в вакууме. Реакционную смесь подщелачивали твердым NaHCO3 и трижды экстрагировали с помощью EtOAc (3×150 мл). Объединенные экстракты промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 в градиентном режиме, описанном на стадии 1. Объединенные фракции выпаривали и растирали с Et2O и получали соединение 22а в виде почти белого твердого вещества.
Стадия 3 - К раствору соединения 22а (0,500 г, 1,78 ммоля) в ДХМ (10 мл), охлажденному до 0°С (баня со льдом), шприцем медленно добавляли Br2 (90 мкл, 1,78 ммоля). Реакционную смесь перемешивали в течение 3 ч, нагревая ее до КТ, и затем концентрировали. Неочищенную смесь очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (в ступенчатом режиме: 0, 20 и 40% EtOAc) и получали 0,273 г (42%) соединения 22b.
Стадия 4 - К раствору соединения 22b (0,273 г, 0,76 ммоля) в MeCN (10 мл) добавляли POCl3 (0,14 мл, 1,52 ммоля). Реакционную смесь нагревали при 100°С в течение 8 ч, затем концентрировали и подвергали распределению между EtOAc и водой (25 мл/25 мл). Водный слой отделяли и дважды промывали с помощью EtOAc (2×25 мл). Объединенные экстракты промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали, и получали 0,287 г (100%) соединения 24а.
Стадия 5 - К раствору соединения 24а (0,242 г, 0,64 ммоля) в ДМФ (2 мл) добавляли метоксид натрия (1,54 мл, 0,5М раствор в МеОН, 0,769 ммоля). Реакционную смесь нагревали при 90°С в течение 30 мин, затем концентрировали и подвергали распределению между EtOAc и Н2О (25 мл/25 мл). Водный слой отделяли и экстрагировали с помощью EtOAc (2×25 мл). Объединенные органические слои промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали, и получали 0,239 г (100%) соединения 24b.
Стадия 6 - В сосуд, содержащий соединение 24b (0,100 г, 0,26 ммоля) и смесь МеОН/ДХМ (3 мл/1 мл), добавляли Na2CO3 (0,082 г, 0,78 ммоля), 4-метилсульфониламинофенилбороновую кислоту (25, 0,056 г, 0,26 ммоля) и Pd(PPh3)4 (0,030 г, 0,26 ммоля). Реакционную смесь облучали в микроволновом синтезаторе при 115°С в течение 30 мин. Реакционную смесь охлаждали, концентрировали и подвергали распределению между EtOAc и Н2О (25 мл/25 мл). Водный слой отделяли и промывали с помощью EtOAc (2×25 мл). Объединенные органические экстракты промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (в ступенчатом режиме: 0% EtOAc - 0-5 мин, 20% EtOAc 5,5-15 мин и 40% EtOAc - 15,5-30 мин) и получали 0,068 г (57%) соединения 26.
Стадия 7- К раствору соединения 26 (0,061 г, 0,13 ммоля) в смеси МеОН/ДХМ (3 мл/1 мл) добавляли Na2CO3 (0,041 г, 0,40 ммоля), соединение 30 (0,017 г, 0,16 ммоля) и Pd(PPh3)4 (0,015 г, 0,013 ммоля). Реакционную смесь облучали в микроволновом синтезаторе при 115°С в течение 30 мин. Реакционную смесь охлаждали, концентрировали и подвергали распределению между EtOAc и Н2О (25 мл/25 мл). Водный слой отделяли и промывали с помощью EtOAc (2×25 мл). Объединенные экстракты промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт очищали с помощью препаративной ТСХ на пластинках с SiO2, обрабатывая смесью 40% EtOAc/гексан, затем сушили и повторно элюировали с помощью 100% EtOAc и получали 0,013 г (21%) соединения I-1.
Пример 2
N-{4-[7-трет-Бутил-5-(5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-фенил}-метансульфонамид (1-2)
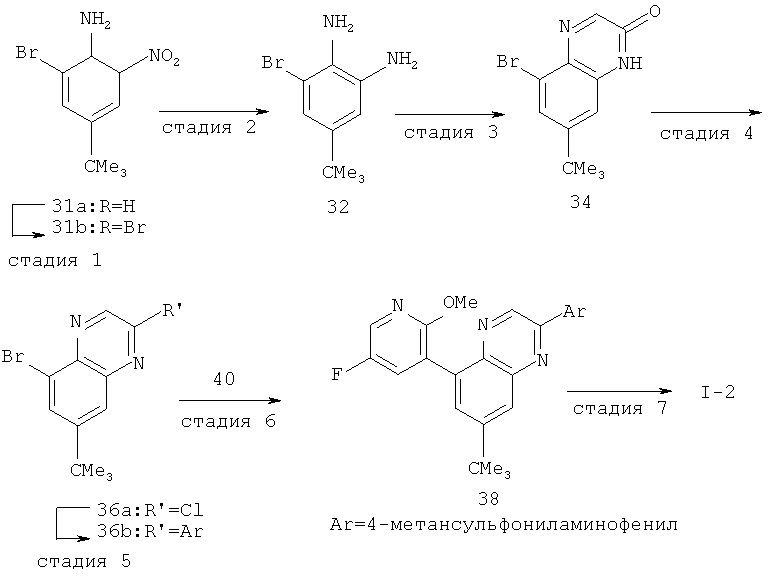
Стадия 1 - К 4-трет-бутил-2-нитроанилину (31а, 5,0 г, 25,74 ммоля) добавляли НОАс (40 мл). Реакционную смесь нагревали при 50°С до образования прозрачного оранжево-коричневого раствора. Колбонагреватель удаляли и шприцем осторожно добавляли бром (1,46 мл, 28,32 ммоля). Реакционную смесь перемешивали в течение еще 45 мин, охлаждая до КТ, затем выливали на лед (100 мл). Взвесь перемешивали стеклянной палочкой, и после того, как лед растаял, осаждалось дополнительное количество твердого вещества. Твердое вещество собирали на фильтре из пористого стекла и сушили, и получали 6,95 г (99%) соединения 31b.
Стадия 2 - К раствору соединения 31b (2,5 г, 9,15 ммоля) в смеси МеОН/Н2О (100 мл/25 мл) добавляли электролитическое железо (1,53 г, 27,46 ммоля) и NH4Cl (1,47 г, 27,46 ммоля). Реакционную смесь кипятили с обратным холодильником в течение 4 ч, затем для удаления железа фильтровали через стекловолоконный фильтр на воронке Бюхнера. Твердое вещество промывали с помощью МеОН и фильтрат концентрировали и подвергали распределению между EtOAc и H2O (50 мл/50 мл). Водный слой отделяли и промывали с помощью EtOAc (2×50 мл). Объединенные органические экстракты промывали рассолом (50 мл), сушили (NO2SO4), фильтровали и концентрировали, и получали 2,23 г (100%) соединения 32.
Стадия 3 - К раствору соединения 32 (0,500 г, 1,8 ммоля) в EtOH добавляли этилглиоксалат (50 мас.% в толуоле, 0,54 мл, 2,7 ммоля). Реакционную смесь кипятили с обратным холодильником в течение ночи. Добавляли дополнительное количество этилглиоксалата (0,54 мл, 2,7 ммоля) и реакционную смесь повторно кипятили с обратным холодильником в течение ночи. Почти белый осадок собирали на фильтре из пористого стекла и получали 0,280 г (51%) соединения 34. (В неочищенной смеси также присутствовал другой изомер, но его не выделяли).
Стадия 4 - К соединению 34 (0,269 г, 0,96 ммоля), суспендированному в MeCN (10 мл), добавляли POCl3 (0,52 мл, 5,74 ммоля). Реакционную смесь нагревали при 100°С в течение 3 ч, затем концентрировали и подвергали распределению между EtOAc и Н2О (25 мл/25 мл). Водный слой отделяли и промывали с помощью EtOAc (2×25 мл). Объединенные экстракты промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали, и получали 0,286 г (количественный выход) соединения 36а.
Стадия 5 - К раствору соединения 36а (0,050 г, 0,17 ммоля) в МеОН и ДХМ (3 мл/1 мл) добавляли Na2CO3 (0,053 г, 0,50 ммоля), 4-метилсульфониламинофенилбороновую кислоту (0,028 г, 0,13 ммоля) и Pd(PPh3)4 (0,019 г, 0,017 ммоля). Реакционную смесь облучали в микроволновой печи при 115°С в течение 30 мин. Реакционную смесь охлаждали, концентрировали и подвергали распределению между EtOAc и Н2О (25 мл/25 мл). Водный слой отделяли и промывали с помощью EtOAc (2×25 мл). Объединенные органические экстракты промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (в ступенчатом режиме: 0% EtOAc - 0-5 мин, 20% EtOAc -5,5-15 мин и 40% EtOAc - 15,5-30 мин) и получали 0,042 г (58%) соединения 36b.
Стадия 6 - К соединению 36b (0,056 г, 0,13 ммоля), растворенному в МеОН и ДХМ (3 мл/1 мл), добавляли Na2CО3 (0,041 г, 0,39 ммоля), 5-фтор-2-метокси-3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-пиридин (соединение 40, 0,026 г, 0,16 ммоля, регистрационный №CAS 1083168-95-9) и Pd(PPh3)4 (0,015 г, 0,013 ммоля). Реакционную смесь облучали в микроволновом синтезаторе при 115°С в течение 30 мин. Реакционную смесь охлаждали, концентрировали и подвергали распределению между EtOAc и Н2О (25 мл/25 мл). Водный слой отделяли и промывали с помощью EtOAc (2×25 мл). Объединенные органические экстракты промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (в ступенчатом режиме: 0% EtOAc - 0-5 мин, 20% EtOAc - 5,5-15 мин и 40% EtOAc - 15,5-30 мин) и получали 0,061 г (100%) соединения 38.
Стадия 7 - К раствору соединения 38 (0,058 г, 0,148 ммоля) в НОАс (2 мл) добавляли HBr (0,1 мл, 50% раствор в воде). Реакционную смесь нагревали в герметизированной пробирке на песчаной бане при 70°С в течение ночи. Реакционную смесь охлаждали, выливали на лед (25 мл) и медленно добавляли насыщенный водный раствор NaHCO3 (25 мл). Льду давали растаять и полученный осадок собирали на фильтре из пористого стекла и получали 0,034 г (61%) соединения I-2.
Соединение 1-6 получали аналогичным образом, за исключением того, что на стадии 6 соединение 40 заменяли на соединение 30. Неочищенный продукт адсорбировали на SiO2 и очищали с помощью хроматографии на SiO2 при элюировании смесью 10% МеОН/ДХМ.
Пример 3
N-{1-[7-трет-Бутил-5-(2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-пиперидин-4-ил}-метансульфонамид (I-3)
Стадия 1 - К раствору соединения 36а (0,100 г, 0,33 моля) и соли N-пиперидин-4-илметансульфонамида с HCl (соединение 37, 0,143 г. 0,66 ммоля, регистрационный №CAS 70724-72-0) в ДМФ (2 мл) добавляли ДИПЭА (0,2 мл, 1,00 ммоля). Реакционную смесь облучали в микроволновом синтезаторе при 140°С в течение 30 мин. Реакционную смесь охлаждали, концентрировали и адсорбировали на SiO2 и очищали с помощью хроматографии на SiO2 при элюировании смесью 5% МеОН/ДХМ и получали 0,102 г (69%) N-[1-(5-бром-7-трет-бутилхиноксалин-2-ил)-пиперидин-4-ил]-метансульфонамида (42).
Стадия 2 - К раствору соединения 42 (0,040 г, 0,10 ммоля) в МеОН и ДХМ (3 мл/1 мл) добавляли Na2CO3 (0,029 г, 0,27 ммоля), соединение 30 (0,012 г, 0,11 ммоля) и Pd(PPh3)4 (0,010 г, 0,010 ммоля). Реакционную смесь облучали в микроволновом синтезаторе при 115°С в течение 30 мин. Реакционную смесь охлаждали, концентрировали и подвергали распределению между EtOAc/H2O (25 мл/25 мл). Водный слой отделяли и промывали с помощью EtOAc (2×25 мл). Объединенные органические экстракты промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт адсорбировали на SiO2 и очищали с помощью хроматографии на SiO2 при элюировании смесью 10% МеОН/ДХМ и получали 0,030 г (73%) соединения I-3.
Соединение 1-5 получали аналогичным образом, за исключением того, что на стадии 2 соединение 30 заменяли на соединение 40 и деметилирование проводили так, как описано на стадии 7 примера 2. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью 10% МеОН/ДХМ.
Пример 4
N-{(S)-1-[7-трет-Бутил-5-(5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-пирролидин-3-илметил}-метансульфонамид (I-4)
Стадия 1 - К раствору соединения 36а (0,050 г, 0,13 ммоля) и соли N-(S)-1-пирролидин-3-илметилметансульфонамида с HCl (44, 0,053 г. 0,25 ммоля, регистрационный №CAS 1064048-61-8) в ДМФ (2 мл) добавляли ДИПЭА (0,1 мл, 0,50 ммоля). Реакционную смесь облучали в микроволновом синтезаторе при 140°С в течение 30 мин. Реакционную смесь охлаждали, концентрировали и адсорбировали на диоксиде кремния и очищали с помощью хроматографии на SiO2 при элюировании смесью 5% МеОН/ДХМ и получали 0,050 г (68%) N-[(S)-1-(5-бром-7-трет-бутилхиноксалин-2-ил)-пирролидин-3-илметил]-метансульфонамида (46).
Стадия 2 - Реакцию перекрестного сочетания Судзуки соединений 46 и 40 проводили так, как описано на стадии 6 примера 2. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью 5% МеОН/ДХМ (500 мл), затем смесью 10% МеОН/ДХМ (500 мл) и получали 0,047 г (89%) N-{(S)-1-[7-трет-бутил-5-(5-фтор-2-метоксипиридин-3-ил)-хиноксалин-2-ил]-пирролидин-3-илметил}-метансульфонамида (48).
Стадия 3 - Деметилирование соединения 48 проводили так, как описано на стадии 7 примера 2. Неочищенный продукт очищали с помощью препаративной ТСХ на пластинках с SiO2, последовательно обрабатывая с помощью 100% EtOAc, затем смесью 5% МеОН/ДХМ, и получали соединение I-4.
Пример 5
N-{4-[7-трет-Бутил-5-(2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-фенил}-метансульфонамид (I-6)
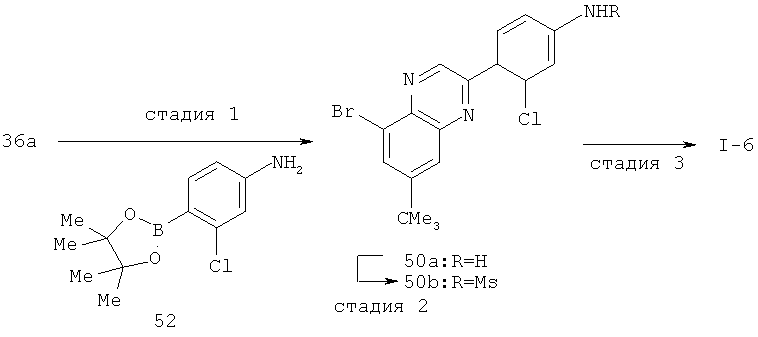
Стадия 1 - К соединению 36а (0,216 г, 0,72 ммоля), растворенному в МеОН и ДХМ (9 мл/3 мл), добавляли Na2CO3 (0,229 г, 2,2 ммоля), соединение 52 (0,146 г, 0,58 ммоля, регистрационный №CAS 877160-63-9) и Pd(PPh3)4 (0,083 г, 0,072 ммоля). Реакционную смесь облучали в микроволновом синтезаторе при 115°С в течение 30 мин. Реакционную смесь охлаждали, концентрировали и подвергали распределению между EtOAc и H2O (25 мл/25 мл). Водный слой отделяли и промывали с помощью EtOAc (2×25 мл). Объединенные органические экстракты промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (в ступенчатом режиме: 0, 20 и 40% EtOAc) и получали 0,142 г (51%) соединения 50а.
Стадия 2 - К раствору соединения 50а (0,140 г, 0,36 ммоля) в ДХМ добавляли пиридин (21 мкл, 0,39 ммоля) и раствор охлаждали до 0°С (баня со льдом). Добавляли метансульфонилхлорид (46 мкл, 0,39 ммоля) и через 15 мин баню со льдом удаляли и реакционную смесь нагревали до КТ и перемешивали в
течение 3 ч. Реакционную смесь концентрировали и подвергали распределению между EtOAc (25 мл) и насыщенным водным раствором NaHCO3 (25 мл). Водный слой отделяли и промывали с помощью EtOAc (2х25 мл). Объединенные органические экстракты промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (в ступенчатом режиме: 0, 20 и 40% EtOAc) и получали 0,110 г (66%) соединения 50b.
Стадия 3 - В сосуд помещали соединение 50b (0,110 г, 0,24 ммоля), МеОН и ДХМ (3 мл/1 мл), затем добавляли Na2CO3 (0,075 г, 0,71 ммоля), соединение 30 (0,030 г, 0,28 ммоля) и Pd(PPh3)4 (0,027 г, 0,024 ммоля). Реакционную смесь облучали в микроволновом синтезаторе при 115°С в течение 30 мин. Реакционную смесь охлаждали, концентрировали и подвергали распределению между EtOAc и Н2О (25 мл/25 мл). Водный слой отделяли и промывали с помощью EtOAc (2×25 мл). Объединенные органические экстракты промывали рассолом (25 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт адсорбировали на SiO2 и очищали с помощью хроматографии на SiO2 при элюировании смесью 5% МеОН/ДХМ и получали 0,043 г (38%) соединения I-6.
Соединение I-8 получали аналогичным образом, за исключением того, что на стадии 1 соединение 52 заменяли на пинаколовый эфир 4-амино-2-фторфенилбороновой кислоты (регистрационный №CAS 819057-45-9).
Соединение I-9 получали аналогичным образом, за исключением того, что на стадии 3 соединение 30 заменяли на 2,6-диметоксипиридин-3-бороновую кислоту и простой метиловый эфир расщепляли по методике, описанной на стадии 7 примера 1.
Соединение I-11 получали аналогичным образом, за исключением того, что на стадии 1 соединение 52 заменяли на пинаколовый эфир 4-амино-2-фторфенилбороновой кислоты, на стадии 3 соединение 30 заменяли на 2,6-диметоксипиридин-3-бороновую кислоту и простой метиловый эфир расщепляли по методике, описанной на стадии 7 примера 2.
Пример 6
N-{4-[7-трет-Бутил-3-метил-5-(2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-фенил}-метансульфонамид (I-10)
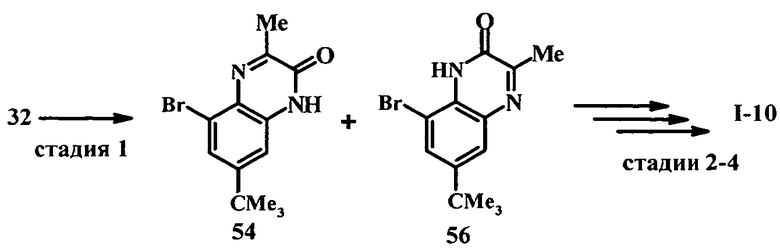
Стадия 1 - К раствору соединения 32 (2,5 г, 10,28 ммоля) в EtOH (40 мл) добавляли пировиноградную кислоту (0,86 мл, 12,34 ммоля). Реакционную смесь нагревали при 100°C (кипячение с обратным холодильником) и перемешивали в течение 2 ч. Раствор в течение ночи медленно охлаждали до КТ, после чего происходило образование осадка. Кристаллы собирали на фильтре из пористого стекла, но они содержали оба изомера и поэтому их объединяли с маточным раствором и концентрировали. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью 20% EtOAc/гексан и получали 0,694 г соединения 54 (23%) и 0,931 г соединения 56 (31%).
Соединение 54 превращали в соединение I-10 в соответствии со стадиями 4-6 примера 2, за исключением того, что на стадии 6 соединение 40 заменяли на соединение 30 и стадию 7 исключали. Неочищенный продукт адсорбировали на SiO2 и очищали с помощью хроматографии на SiO2 при элюировании смесью 5% МеОН/ДХМ и получали соединение I-10.
Пример 7
N-{4-[6-трет-Бутил-8-(5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид (I-12)
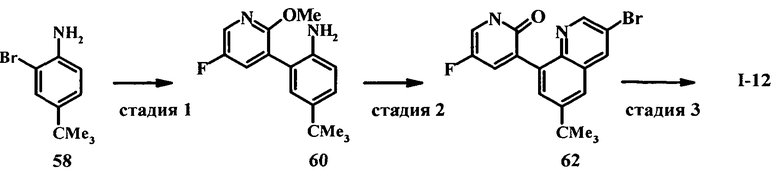
Стадия 1 - В сосуд для микроволновой печи помещали соединение 58 (587 мг, 2,57 ммоля), 5-фтор-2-метоксипиридин-3-илбороновую кислоту (соединение 59, 660 мг, 3,86 ммоля), Pd(PPh3)4 (148 мг, 0,12 ммоля), Na2CO3 (818 мг, 7,8 ммоля) и смесь МеОН (0,7 мл)/ДХМ (3,5 мл), сосуд герметизировали и облучали в микроволновом синтезаторе при 115°С в течение 2 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали водой, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме и получали 460 мг (65%) соединения 60 в виде коричневого масла.
Стадия 2 - Раствор Br2 (191 мг, 1,6 ммоля) в НОАс (5 мл) при КТ добавляли к раствору α-бромакролеина (230 мг, 1,71 ммоля) в НОАс (5 мл) до появления слабой красноватой окраски избыточного брома. После перемешивания при КТ в течение 15 мин добавляли раствор соединения 60 (437 мг, 1,59 ммоля) в НОАс (5 мл). Реакционную смесь нагревали при 100°С в течение 2 ч. Реакционную смесь осторожно выливали в холодный насыщенный водный раствор NaHCO3 и затем экстрагировали с помощью EtOAc. Органический слой промывали рассолом, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью гексаны/этилацетат состава 1:1 и получали 260 мг (43%) соединения 62 в виде коричневого масла.
Стадия 3 - В сосуд для микроволновой печи помещали соединение 62 (60 мг, 0,16 ммоля), соединение 25 (52 мг, 0,241 ммоля), Pd(PPh3)4 (10 мг, 0,008 ммоля) и Na2CO3 (51 мг, 0,48 ммоля) в смеси МеОН (0,1 мл) и ДХМ (0,5 мл), сосуд герметизировали и облучали в микроволновом синтезаторе при 115°С в течение 2 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали водой, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании с помощью EtOAc и получали 32 мг (42%) соединения I-12 в виде почти белого твердого вещества: МС (ЭР) (электрораспыление) (М+Н)+=466.
Пример 8
N-{1-[6-трет-Бутил-2-метокси-8-(5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-пиперидин-4-ил}-метансульфонамид (I-13)
В сосуд помещали соединение 62 (70 мг, 0,187 ммоля), соединение 37 (44 мг, 0,205 ммоля), Pd(OAc)2 (4 мг, 0,018 ммоля), NaO-трет-Bu (72 мг, 0,75 ммоля) и Р(трет-Bu)3 (4 мг, 0,018 ммоля) в толуоле (3 мл), сосуд герметизировали и нагревали при 100°C в течение 4 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали водой, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью препаративной ТСХ на пластинках с SiO2, обрабатывая смесью ДХМ/МеОН состава 9:1, и получали 28 мг (32%) соединения I-13 в виде коричневого масла: МС (ЭР) (М+Н)+=473.
Пример 9
N-{4-[6-трет-Бутил-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид (1-14)
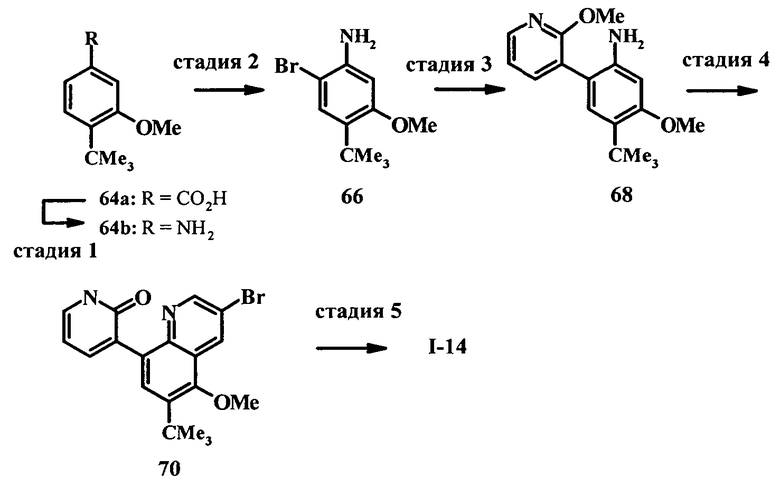
Стадия 1 - Смесь соединения 64а (6 г, 28,84 ммоля, регистрационный № CAS 79822-46-1), дифенилфосфорилазида (8 г, 29,09 ммоля) и ТЭА (4,32 мл, 30,99 ммоля) в трет-бутаноле (500 мл) кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали до КТ и летучие вещества выпаривали. Неочищенное вещество при 0°C обрабатывали с помощью смеси ТФК и ДХМ состава 1:1 (20 мл). После перемешивания при КТ в течение 3 ч реакционную смесь охлаждали до 0°C и обрабатывали 2М водным раствором NaOH. Реакционную смесь разбавляли гексанами, разделяли, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан состава 1:4 и получали 3,4 г (66%) соединения 64b в виде коричневого масла.
Стадия 2 - NBS (498 мг, 2,7 ммоля) при КТ добавляли к раствору соединения 64b (500 мг, 2,7 ммоля) в MeCN (10 мл). Реакционную смесь перемешивали при КТ в течение 3 ч. Реакционную смесь разбавляли с помощью EtOAc и промывали 1 н. раствором NaOH, сушили (MgSO4), фильтровали и концентрировали и получали 700 мг соединения 66 в виде коричневого масла.
Стадия 3 - В сосуд для микроволновой печи помещали соединение 66 (700 мг, 2,71 ммоля), соединение 30 (612 мг, 4 ммоля), Pd(PPh3)4 (231 мг, 0,2 ммоля) и Na2CO3 (636 мг, 6 ммолей), МеОН (1 мл) и ДХМ (9 мл), сосуд герметизировали и облучали в микроволновом синтезаторе при 115°С в течение 1 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали водой, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме и получали 420 мг (54%) соединения 68 в виде коричневого масла.
Стадия 4 - Раствор Br2 (191 мг, 1,6 ммоля) в НОАс (5 мл) при КТ добавляли к раствору α-бромакролеина (230 мг, 1,71 ммоля) в НОАс (5 мл) до появления устойчивой слабой красноватой окраски брома. После перемешивания при КТ в течение 15 мин добавляли раствор соединения 68 (420 мг, 1,47 ммоля) в НОАс (5 мл). Реакционную смесь нагревали при 100°С в течение 2 ч. Реакционную смесь осторожно выливали в холодный насыщенный водный раствор NaHCO3 и затем экстрагировали с помощью EtOAc. Органический слой промывали рассолом, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью гексаны/EtOAc состава 1:1 и получали 150 мг (26%) соединения 70 в виде коричневого масла: МС (ЭР) (M+H)+=388.
Стадия 5 - В сосуд для микроволновой печи помещали соединение 70 (150 мг, 0,387 ммоля), соединение 25 (125 мг, 0,581 ммоля), Pd(PPh3)4 (45 мг, 0,038 ммоля), Na2CO3 (123 мг, 1,16 ммоля), МеОН (0,2 мл) и ДХМ (1,5 мл), сосуд герметизировали и облучали в микроволновом синтезаторе при 115°С в течение 1 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали водой, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью ДХМ/МеОН состава 95:5 и получали 40 мг (21%) соединения 1-14 в виде почти белого твердого вещества.
Пример 10
N-{4-[6-трет-Бутил-8-(5-хлор-2-оксо-1,2-дигидропиридин-3-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид (I-15)
NCS (15 мг, 0,112 ммоля) добавляли к раствору соединения I-14 (48 мг, 0,1 ммоля) в MeCN (5 мл) и ДМФ (2 мл), нагретому до 70°С. Реакционную смесь перемешивали при 70°С в течение 5 ч, затем охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали с помощью Н2О, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме и получали 25 мг (49%) соединения I-15 в виде белого твердого вещества.
Пример 11
N-{1-[6-трет-Бутил-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-азетидин-3-илметил}-метансульфонамид (1-19) и N-{1-[6-трет-бутил-4-хлор-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-азетидин-3-илметил}-метансульфонамид (I-22)
Стадия 1 - В сосуд помещали соединение 70 (301 мг, 0,77 ммоля), N-азетидин-3-илметилметансульфонамидгидрохлорид (200 мг, 0,934 ммоля), Pd(OAc)2 (18 мг, 0,08 ммоля), NaO-трет-Bu (298 мг, 3,1 ммоля) и Р(трет-Bu)3 (16 мг, 0,079 ммоля) в толуоле (3 мл), сосуд герметизировали и нагревали при 100°С в течение 20 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали насыщенным водным раствором NaHCO3, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью ДХМ/МеОН в градиентном режиме и получали 95 мг (26%) соединения 1-19 в виде почти белого твердого вещества.
Стадия 2 - NCS (14 мг, 0,105 ммоля) при 70°С добавляли к раствору соединения I-19 (45 мг, 0,095 ммоля) в MeCN (5 мл). Реакционную смесь перемешивали при 70°С в течение 2 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали 1 н. раствором NaOH, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью препаративной ТСХ на пластинках с SiO2, обрабатывая смесью ДХМ/МеОН состава 9:1, и получали 13 мг (27%) соединения 1-22 в виде полужидкого вещества.
Пример 12
N-{(8)-1-[6-трет-Бутил-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-пирролидин-3-илметил}-метансульфонамид (I-18)
N-Пирролидин-3-илметилметансульфонамид (соединение 72) и ТЭА (1,05 мл, 7,5 ммоля) при 0°С добавляли к раствору (R)-3-(аминометил)-1-N-Вос-пирролидина (1 г, 5 ммолей) в ДХМ (25 мл). Затем добавляли метансульфонилхлорид (0,43 мл, 5,5 ммоля). После перемешивания при 0°С в течение 2 ч реакционную смесь разбавляли водой. Органическую фазу отделяли, сушили (MgSO4), фильтровали и концентрировали. Неочищенное вещество при КТ обрабатывали 1М раствором HCl в МеОН (25 мл) и перемешивали при КТ в течение 20 ч. Летучие вещества удаляли при пониженном давлении и получали 0,95 г соединения 72 в виде белого твердого вещества.
Стадия 1 - В сосуд помещали соединение 70 (301 мг, 0,77 ммоля), соединение 72 (200 мг, 0,778 ммоля), Pd(OAc)2 (18 мг, 0,08 ммоля), NaO-трет-Bu (298 мг, 3,1 ммоля) и Р(трет-Bu)3 (16 мг, 0,079 ммоля) в толуоле (3 мл), сосуд герметизировали и нагревали при 100°С в течение 20 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали насыщенным водным раствором NaHCO3, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью ДХМ/МеОН состава 9:1 и получали 60 мг (16%) соединения 1-18 и 60 мг (25%) 3-(6-трет-бутил-5-метоксихинолин-8-ил)-1Н-пиридин-2-она [(М+Н+)=309] в виде белых твердых веществ.
Пример 13
N-{4-[6-трет-Бутил-8-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид (I-21)
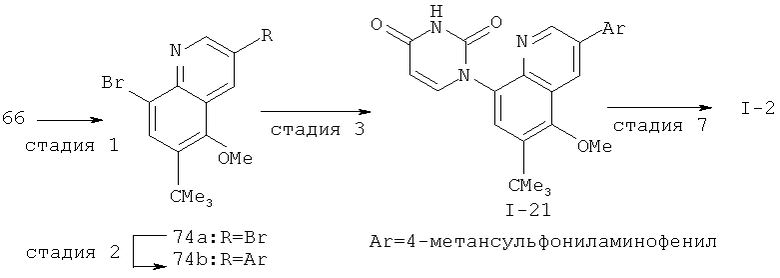
Стадия 1 - Раствор Br2 (126 мг, 1,05 ммоля) в НОАс (5 мл) при КТ добавляли к раствору α-бромакролеина (148 мг, 1,1 ммоля) в НОАс (5 мл) до появления устойчивой слабой красноватой окраски брома. После перемешивания при КТ в течение 15 мин добавляли раствор соединения 66 (260 мг, 1,01 ммоля) в НОАс (5 мл). Реакционную смесь нагревали при 100°С в течение 4 ч. Реакционную смесь осторожно выливали в холодный насыщенный водный раствор NaHCO3 и затем экстрагировали с помощью EtOAc. Органический слой промывали рассолом, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью гексан/EtOAc в градиентном режиме и получали 170 мг (45%) соединения 74а в виде коричневого масла.
Стадия 2 - В сосуд помещали соединение 74а (850 мг, 2,27 ммоля), соединение 25 (539 мг, 2,5 ммоля), Pd(PPh3)4 (263 мг, 0,227 ммоля), Na2CO3 (725 мг, 6,83 ммоля), МеОН (8 мл) и ДХМ (5 мл), сосуд герметизировали и облучали в микроволновом синтезаторе при 120°С в течение 1 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали водой, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании с помощью EtOAc и получали 600 мг (57%) соединения 74b в виде почти белого твердого вещества: МС (ЭР) (М+Н+)=464.
Стадия 3 - В сосуд помещали соединение 74b (200 мг, 0,431 ммоля), урацил (72 мг, 0,642 ммоля), N-(2-цианофенил)пиколинамид (19 мг, 0,085 ммоля), Cul (8 мг, 0,042 ммоля), K3PO4 (183 мг, 0,86 ммоля), ДХМ и МеОН и затем добавляли дегазированный ДМСО (1,5 мл). Сосуд герметизировали и облучали в микроволновом синтезаторе при 150°С в течение 5 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали 1 н. раствором NaHSO4, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании с помощью EtOAc и получали 17 мг (8%) соединения I-21 в виде полужидкого вещества.
N-{(S)-1-[6-трет-Бутил-5-метокси-8-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-пирролидин-3-илметил}-метансульфонамид (I-55) получали из соединения 74а по реакции сочетания Судзуки с соединением 115 и с помощью деметилирования по методикам, описанным на стадиях 2 и 3 примера 24.
Пример 14
N-{4-[6-трет-Бутил-8-(2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид (I-25)
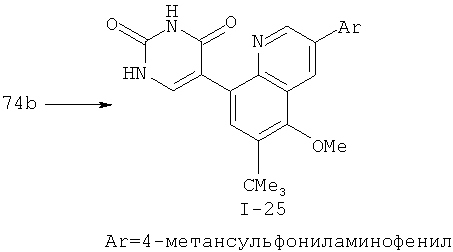
В сосуд для микроволнового синтезатора объемом 5 мл помещали соединение 74b (98,7 мг, 0,213 ммоля), 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-илбороновую кислоту (67,9 мг, 0,436 ммоля, регистрационный №CAS 70523-22-7), Na2CO3 (119,8 мг, 1,13 ммоля) и Pd(PPh3)4 (27,8 мг, 0,024 ммоля), МеОН (1,6 мл) и ДХМ (0,4 мл), сосуд герметизировали и облучали в микроволновом синтезаторе при 115°С в течение 60 мин. После охлаждения до КТ реакционную смесь выливали в насыщенный водный раствор NaHCO3 (30 мл) и экстрагировали с помощью EtOAc (3×20 мл). Объединенные экстракты промывали насыщенным водным раствором NaHCO3 (2×40 мл), рассолом (40 мл), сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью МеОН/ДХМ в градиентном режиме (от 2 до 10% МеОН) и получали 15 мг (14%) соединения I -25 в виде светло-желтого твердого вещества.
Соединение I-23 получали аналогичным образом, за исключением того, что 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-илбороновую кислоту заменяли на соединение 59 и простой метиловый эфир расщепляли по методике, описанной на стадии 7 примера 2. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью МеОН/ДХМ в градиентном режиме (от 2 до 5% МеОН) и получали соединение I-23 в виде почти белого твердого вещества.
Соединение I-24 получали аналогичным образом, за исключением того, что 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-илбороновую кислоту заменяли на 2-метокси-6-метилпиридин-3-илбороновую кислоту (регистрационный №CAS 1000802-75-4) и простой метиловый эфир расщепляли по методике, описанной на стадии 7 примера 2. Неочищенный продукт осаждался в виде желтых кристаллов.
Соединение I-26 получали аналогичным образом, за исключением того, что 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-илбороновую кислоту заменяли на 3-фторпиридин-4-илбороновую кислоту. Неочищенный продукт осаждался в виде желтых кристаллов. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью МеОН/ДХМ в градиентном режиме (от 2 до 5% МеОН) и получали соединение 1-26 в виде почти белого твердого вещества.
Соединение I-34 получали аналогичным образом, за исключением того, что 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-илбороновую кислоту заменяли на соединение 115 и простой метиловый эфир расщепляли по методике, описанной на стадии 7 примера 2.
Пример 15
N-{4-[6-трет-Бутил-5-метокси-8-(2-оксотетрагидропиримидин-1-ил)-хинолин-3-ил]-фенил}-метансульфонамид (I-27)
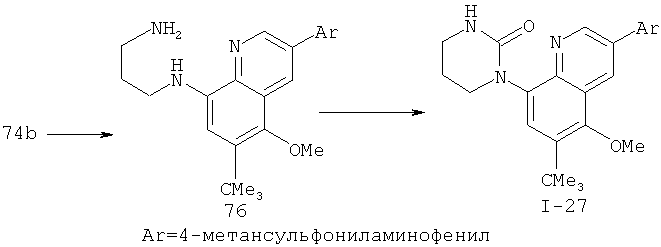
В сосуд помещали соединение 74b (80 мг, 0,172 ммоля), 1,3-пропандиамин (300 мкл), D,L-пролин (8 мг, 0,069 ммоля), Cul (8 мг, 0,036 ммоля), K2CO3 (96 мг, 0,695 ммоля) и добавляли дегазированный ДМСО (0,5 мл). Реакционную смесь нагревали при 150°С в течение 48 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали водой, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток растворяли в ТГФ (5 мл) и обрабатывали карбонилдиимидазолом (350 мг). После перемешивания при КТ в течение 4 ч реакционную смесь разбавляли с помощью EtOAc. Органический слой промывали водой, сушили (MgSO4), фильтровали и концентрировали. Амин циклизовался неполностью. Поэтому вещество растворяли в диоксане (15 мл) и облучали в микроволновом реакторе при 150°С в течение 30 мин. Реакционную смесь охлаждали до КТ и концентрировали. Неочищенный остаток очищали с помощью препаративной ТСХ на пластинках с SiO2, обрабатывая смесью ДХМ/МеОН состава 95:5, и получали 15 мг (18%) соединения I-27 в виде светло-желтого твердого вещества.
Пример 16
N-{4-[6-трет-Бутил-5-метокси-8-(3-оксо-2,3-дигидропиридазин-4-ил)-хинолин-3-ил]-фенил}-метансульфонамид (76)
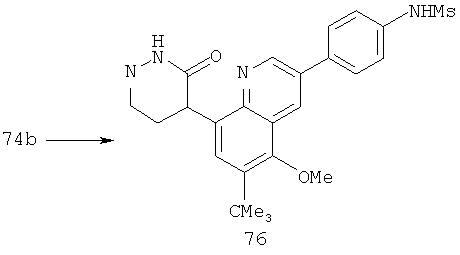
В-(2,3-дигидро-3-оксо-4-пиридазинил)-бороновая кислота (соединение 78):
Стадия а - В круглодонную колбу объемом 1 л помещали 4-хлор-5-гидразинил-3(2Н)-пиридазинон (8,0 г, 50 ммолей, регистрационный №CAS 6959-56-4), CuSO4·5H2O (26,12 г, 10,5 ммоля) и Н2О (300 мл) и смесь перемешивали и кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали до 0°С и добавляли водный раствор NaOH до установления рН 4. Водный слой трижды экстрагировали с помощью EtOAc (по 500 мл). Объединенные экстракты сушили (Na2SO4), фильтровали и выпаривали. Значение рН оставшейся водной фазы 37% раствором HCl доводили до 2 и раствор 6 раз экстрагировали с помощью EtOAc. Экстракты объединяли, сушили (Na2SO4), фильтровали и выпаривали и получали 4,75 г 4-хлор-2Н-пиридазин-3-она (75).
Стадия b - В сосуд для микроволновой печи помещали соединение 75 (0,400 г, 3 ммоля), бис-(пинаколято)дибор (0,934 г, 4 ммоля), дициклогексил[2',4',6'-трис(1-метилэтил)[1,1'-бифенил]-2-ил]-фосфин (X-Phos, 0,058 г, 0,12 ммоля), Pd2(dba)3 (0,056 г, 0,061 ммоля) и КОАс (0,902 г, 9 ммолей) и колбу откачивали и повторно заполняли с помощью Ar и герметизировали. Добавляли диоксан (6 мл) и реакционную смесь нагревали при 110°С в течение ночи. Реакционную смесь охлаждали до КТ и экстрагировали с помощью EtOAc (120 мл). Органический экстракт последовательно промывали с помощью Н2О (10 мл) и рассолом (10 мл), сушили (Na2SO4), фильтровали и выпаривали. Неочищенный продукт растирали с Et2O и получали 0,217 г соединения 78.
Катализируемую палладием реакцию сочетания соединений 74b и 78 проводили, как это описано в примере 14, и получали соединение 76.
Пример 17
N-{4-[6-трет-Бутил-8-(2,4-диоксотетрагидропиримидин-1-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид (I-28)
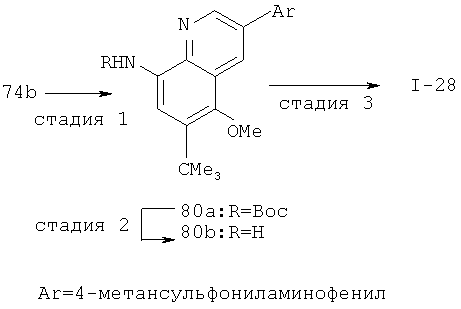
Стадия 1 - Высушенную пробирку для микроволновой печи продували аргоном и в нее помещали Pd2(dba)3·CHCl3 (0,11 г, 0,106 ммоля), 2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил (0,136 г, 0,321 ммоля), трет-бутоксид натрия (0,10 г, 1,041 ммоля), соединение 74b (0,33 г, 0,712 ммоля) и трет-бутилкарбамат (0,10 г, 0,855 ммоля). Добавляли толуол (3,5 мл) и полученную смесь дегазировали путем пропускания через раствор аргона в течение 1 ч. Пробирку закрывали и реакционную смесь перемешивали при КТ в течение 2 дней, затем разбавляли с помощью EtOAc и промывали насыщенным водным раствором NH4Cl. Водный слой подвергали обратной экстракции с помощью EtOAc. Объединенные органические слои сушили (Na2SO4), фильтровали, и выпаривали. Остаток очищали с помощью флэш-хроматографии на SiO2 при элюировании смесью гексаны/EtOAc (7,5/2,5) и получали 0,315 г (89% выход) соединения 80а.
Стадия 2 - HCl (6 мл, 4М раствор в диоксане) при КТ добавляли к раствору соединения 80а (0,315 г, 0,631 ммоля) в ДХМ (5 мл). Полученную смесь перемешивали при КТ в течение 4 ч, затем выпаривали. Остаток подвергали распределению между насыщенным водным раствором NaHCO3 и EtOAc. Водный слой дважды подвергали обратной экстракции с помощью EtOAc. Объединенные органические слои сушили (Na2SO4), фильтровали, и выпаривали и получали соединение 80b, которое использовали на следующей стадии без очистки.
Стадия 3 - Акриловую кислоту (0,09 мл, 1,304 ммоля) при КТ тремя порциями (0,02, 0,02, 0,05 мл) добавляли к раствору соединения 80b (теоретическое значение 0,631 ммоля) в толуоле (2,5 мл). После добавления первой порции реакционную смесь перемешивали при 120°С в течение 2 ч. После добавления второй порции реакционную смесь перемешивали при 120°С в течение 1 ч и после добавления третьей порции реакционную смесь перемешивали при 120°С в течение ночи. Реакционную смесь охлаждали до КТ и выпаривали. Остаток переносили в ледяную НОАс (2 мл) и добавляли мочевину (0,095 г, 1,576 ммоля). Реакционную смесь перемешивали при 120°С в течение 6 ч, затем охлаждали до КТ и выпаривали. Темно-коричневый остаток подвергали распределению между ЕОАс и насыщенным водным раствором NaHCO3. Водный слой дважды подвергали обратной экстракции с помощью EtOAc. Объединенные органические слои сушили (Na2SO4), фильтровали, и выпаривали. Остаток адсорбировали на SiO2 и очищали с помощью хроматографии на SiO2 (12 г SiO2) при элюировании смесью ДХМ/EtOAc в градиентном режиме (от 50 до 80% EtOAc) и получали 0,07 г соединения I-28 в виде зеленовато-серого порошкообразного вещества. Порошкообразное вещество переносили в минимальное количество ДХМ и нерастворимое вещество отфильтровывали и промывали с помощью небольшого количества ДХМ и получали 0,04 г соединения I-28 в виде светло-серого порошкообразного вещества.
Пример 18
N-{4-[8-(2,4-Диоксотетрагидропиримидин-1-ил)-5-метокси-6-трифторметилхинолин-3-ил]-фенил}-метансульфонамид (I-29)
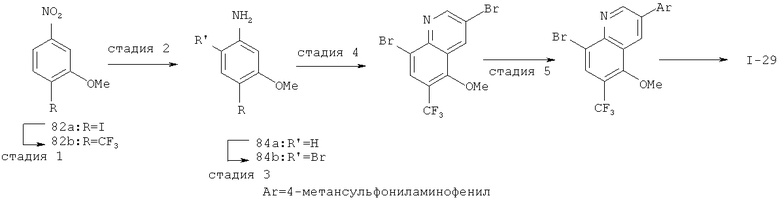
Стадия 1 - В перчаточной камере в атмосфере азота смесь Cu(I) (10,03 г) и CsF (21,40 г) тонко измельчали в ступке и получали сыпучее порошкообразное вещество и его переносили в высушенную в сушильном шкафу круглодонную колбу объемом 250 мл, снабженную стержнем для перемешивания и мембраной. Затем в колбу помещали 2-йод-5-нитроанизол (15,17 г) и сульфолан (30 мл) и энергично перемешивали при 45°С. К смеси в шприцевым насосом течение 4 ч по каплям добавляли триметил(трифторметил)силан (20 мл) и полученную смесь перемешивали при КТ в течение ночи. Реакционную смесь разбавляли с помощью EtOAc (500 мл) и перемешивали с некоторым количеством целита® 512. Реакционную смесь фильтровали через слой целита. Фильтрат разбавляли с помощью EtOAc до объема, равного 1 л, и промывали с помощью 1 л 10% водного раствора NH4OH, 1 л 1,0 М раствора HCl и 500 мл рассола. Органическую фазу сушили (MgSO4), фильтровали и концентрировали в вакууме. Янтарный остаток разбавляли с помощью ДХМ и очищали с помощью флэш-хроматографии (колонка Supeico VersaPak™, 770 г SiO2) и элюировали в градиентном режиме смесью ДХМ/гексан (от 0 до 40% ДХМ) в количестве, равном 10 объемам колонки, и получали 8,61 г соединения 82b в виде желтого кристаллического твердого вещества.
Стадия 2 - В колбу для гидрирования Парра объемом 500 мл помещали соединение 82b (8,60 г) и 10% Pd/C (1,75 г). Колбу продували азотом и осторожно добавляли EtOH (150 мл). Смесь продували азотом в течение 5 мин. Восстановление проводили в аппарате Парра со встряхивающим устройством при давлении водорода, равном 57 фунт-сила/дюйм, при 55°С в течение 18 ч. Реакционную смесь охлаждали и катализатор отфильтровывали через стекловолоконный фильтр и промывали с помощью ИПС (изопропиловый спирт). Растворитель удаляли в вакууме. Неочищенное вещество разбавляли с помощью ДХМ, очищали с помощью флэш-хроматографии (колонка Analogix™ SF65, 200 г SiO2) и элюировали в градиентном режиме смесью EtOAc/гексан (от 0 до 40% EtOAc) в количестве, равном 15 объемам колонки, и получали 7,18 г соединения 84а в виде почти белого воскообразного твердого вещества.
Стадия 3 - В круглодонную колбу объемом 100 мл, снабженную стержнем для перемешивания и мембраной, помещали соединение 84а (3,01 г) и в ней поддерживали атмосферу азота. В колбу добавляли безводный диоксан (25 мл) и НОАс (7,5 мл). Раствор энергично перемешивали в бане со льдом и шприцевым насосом в течение 30 мин по каплям добавляли раствор брома в диоксане (20 мл, 0,135 г Br2/мл). Образовывался бежевый осадок и смесь загустевала. Баню со льдом удаляли и реакционную смесь перемешивали при КТ в течение 1 ч.
Реакционную смесь выливали в смесь 1,0 М раствора NaOH (150 мл) и 2,0 М раствора Na2CO3 (150 мл) и экстрагировали с помощью ДХМ (3×100 мл). Объединенные экстракты последовательно промывали 0,5 М раствором Na2CO3 (150 мл) и рассолом (150 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученная бледно-желтая жидкость медленно затвердевала и темнела, и получали соединение 84b (4,10 г) в виде черного кристаллического твердого вещества.
Стадия 4 - В круглодонную колбу объемом 100 мл, снабженную стержнем для перемешивания и мембраной, помещали НОАс (50 мл), затем добавляли 2-бромакролеин (1,91 г). При перемешивании к смеси по каплям добавляли Br2 до появления устойчивой красной окраски (примерно 720 мкл). К этому раствору добавляли раствор соединения 84b (3,48 г) в НОАс (15 мл). Образовывался густой осадок и перемешивание продолжали при 100°С в течение 1 ч. Осадок растворялся и через 10 мин получали прозрачный темно-янтарный раствор. Через 30 мин повторно происходило образование осадка. Реакционную смесь охлаждали до КТ и при перемешивании выливали в охлажденный льдом 2,0 М водный раствор NaOH (550 мл). К смеси медленно добавляли 2,0 М водный раствор Na2CO3 [сильное вспенивание] до установления значения рН раствора, равного примерно 8, и полученный раствор экстрагировали с помощью ДХМ (3×250 мл). Объединенные экстракты промывали рассолом (500 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме и получали темную смолу, которую очищали с помощью флэш-хроматографии (колонка Supeico VersaPak™, 385 г SiO2) и элюировали в градиентном режиме смесью ДХМ/гексан (от 0 до 100% ДХМ) в количестве, равном 10 объемам колонки. Анализ с помощью ЖХ-МС и ТСХ показывал, что соединение не являлось чистым. Продукт повторно хроматографировали (колонка Supeico VersaPak, 100 г SiO2) и элюировали в градиентном режиме смесью EtOAc/гексан (от 0 до 100% EtOAc) в количестве, равном 30 объемам колонки, и получали 938 мг соединения 86 в виде почти белого твердого вещества.
Стадия 5 - В колбу объемом 20 мл, снабженную стержнем для перемешивания и крышкой с мембраной, помещали соединение 86 (850 мг) и N-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенилметансульфонамид (661 мг, регистрационный №CAS 616880-14-9), диоксан (10 мл) и водный раствор Cs2CO3 (2,3 мл, 0,956 г Cs2CO3/мл). Реакционную смесь продували азотом в течение 10 мин, затем добавляли (dppf)PdCl2·ДХМ (74 мг). Реакционную смесь продували азотом в течение 5 мин, колбу герметизировали и перемешивали при 65°С в течение 110 мин. Реакционную смесь охлаждали и выливали в ДХМ (100 мл) и 0,5 М водный раствор Na2CO3 (50 мл). Фазы разделяли и последовательно промывали с помощью H2O (50 мл) и рассолом (50 мл) сушили (MgSO4), фильтровали и концентрировали в вакууме. Продукт очищали с помощью флэш- хроматографии (колонкам Supeico VersaPak®, 100 г SiO2) при элюировании смесью EtOAc/ДХМ в градиентном режиме (от 0 до 100% EtOAc) в количестве, равном 15 объемам колонки, и получали 538 мг соединения 88 в виде светло-янтарного твердого вещества.
Превращение соединения 88 в соединение 1-29 проводили в соответствии со стадиями I-3 примера 17.
N-{4-[5-Метокси-8-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-трифторметилхинолин-3-ил]-фенил}-метансульфонамидгидробромид (1-49) получали аналогичным образом, за исключением того, что на стадии 5 N-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенилметансульфонамид заменяли на 2-метокси-6-метилпиридин-3-илбороновую кислоту. Деметилирование пиридинил-O-метилового простого эфира проводили в соответствии с методикой, описанной на стадии 7 примера 2.
N-{4-[5-Метокси-8-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-6-трифторметилхинолин-3-ил]-фенил}-метансульфонамид получали аналогичным образом, за исключением того, что на стадии 5, N-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенилметансульфонамид заменяли на соединение 115. Деметилирование пиридинил-O-метилового простого эфира проводили в соответствии с методикой, описанной на стадии 7 примера 2.
N-{4-[8-(2,4-Диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-5-метокси-6-трифторметилхинолин-3-ил]-фенил}-метансульфонамид (соединение I-51) получали из соединения 88 по методике, описанной на стадии 3 примера 13.
Пример 19
N-{(S)-3-[6-трет-Бутил-8-(диоксотетрагидропиримидин-1-ил)-5-метоксихинолин-3-ил]-циклопентил}-метансульфонамид (I-31)
Стадия 1 - Высушенную пробирку для микроволновой печи продували аргоном и в нее помещали соединение 86 (0,47 г, 1,26 ммоля), трет-бутиловый эфир (S)-1-пирролидин-3-илметилкарбаминовой кислоты (0,38 г, 1,897 ммоля, регистрационный №CAS 173340-26-6), трет-бутоксид натрия (0,18 г, 1,873 ммоля), xantphos (0,146 г, 0,252 ммоля) и Pd2(dba)3(0) (0,115 г, 0,126 ммоля). Пробирку продували аргоном и добавляли 1 мл толуола. Реакционную смесь дегазировали в течение 15 мин путем пропускания через смесь аргона и полученную смесь перемешивали при 100°С в течение ночи, охлаждали до КТ и подвергали распределению между EtOAc и насыщенным водным раствором NH4Cl. Водный слой дважды подвергали обратной экстракции с помощью EtOAc. Объединенные органические экстракты сушили (Na2SO4), фильтровали, и выпаривали. Остаток очищали с помощью хроматографии на SiO2 (40 г SiO2) при элюировании смесью EtOAc/гексан в градиентном режиме (от 10 до 30% EtOAc) и получали 0,28 г (45%) соединения 90а.
Стадия 2 - К раствору соединения 90а (0,32 г, 0,65 ммоля) в ДХМ (2 мл) при КТ добавляли HCl (2 мл, 4 М раствор в диоксане). Полученную оранжевую смесь перемешивали при КТ в течение ночи, затем выпаривали. Ярко-оранжевое твердое вещество подвергали распределению между EtOAc и насыщенным водным раствором NaHCO3. Водный слой дважды подвергали обратной экстракции с помощью EtOAc. Объединенные органические экстракты сушили (Na2SO4), фильтровали, и выпаривали и получали 0,25 г неочищенного соединения 90b, которое использовали в следующей реакции без дополнительной очистки.
Стадия 3 - К смеси неочищенного соединения 90b (0,25 г, 0,637 ммоля) и пиридина (0,062 г, 0,765 ммоля) в 3 мл ДХМ, охлажденной до 0°С, добавляли метансульфонилхлорид (0,055 мл, 0,716 ммоля). Полученную смесь перемешивали при КТ в течение 2 ч, затем подвергали распределению между EtOAc и 1М водным раствором NaOH. Водный слой дважды подвергали обратной экстракции с помощью EtOAc. Объединенные органические экстракты сушили (Na2SO4), фильтровали, и выпаривали. Остаток очищали с помощью хроматографии на SiO2 (22 г SiO2) при элюировании смесью EtOAc/гексан (4/1) и получали 0,12 г (выход 40%) соединения 90с.
Введение диоксотетрагидропиримидин-1-ильного фрагмента проводили в соответствии с методиками, описанными на стадиях 1-3 примера 18, и получали соединение I-31.
N-{(S)-1-[6-трет-Бутил-8-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-5-метоксихинолин-3-ил]-пирролидин-3-илметил}-метансульфонамид (I-52) получали из соединения 90с по методикам введения 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ильного фрагмента, описанным на стадиях 1-3 примера 23.
Соль N-{(S)-1-[6-трет-бутил-5-метокси-8-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-пирролидин-3-илметил}-метансульфонамид с HBr получали аналогичным образом, за исключением того, что 6-метил-2-оксо-1,2-дигидропиридин-3-ильный фрагмент вводили в положение 8 соединения 90с, как это описано на стадии 7 примера 1.
Пример 20
N-{4-[8-(2,4-Диоксотетрагидропиримидин-1-ил)-5-метокси-6-(2,2,2-трифторэтил)-хинолин-3-ил]-фенил}-метансульфонамид (I-30)
Стадия 1 - В круглодонную колбу объемом 250 мл, снабженную стержнем для перемешивания и крышкой, помещали соединение 92а (9,93 г) и безводный ДМЭ (диметоксиэтан) (100 мл) и перемешивали и получали прозрачный желтый раствор. К этому раствору последовательно добавляли CF3SiMe3 (9,0 мл) и CsF (792 мг). Реакционную смесь обрабатывали ультразвуком в течение 20 мин и затем перемешивали при КТ в течение еще 40 мин. Добавляли 2,0 М раствор HCl (100 мл) и полученную смесь перемешивали при КТ в течение 1 ч. Добавляли EtOAc (200 мл) и фазы разделяли. Органическую фазу промывали насыщенным водным раствором NaHCO3 (150 мл) и рассолом (150 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью флэш-хроматографии (колонка Supeico VersaPak™, 385 г SiO2) при элюировании смесью ДХМ/гексан в градиентном режиме (от 0 до 100% ДХМ) в количестве, равном 5 объемам колонки. Выделенный продукт растворяли в ДХМ (100 мл) и добавляли гексан (200 мл). Примерно 2/3 растворителя медленно удаляли в роторном испарителе. Полученный осадок отфильтровывали, промывали гексаном и сушили в высоком вакууме и получали 13,10 г соединения 92b в виде белого твердого вещества.
Стадия 2 - В круглодонную колбу объемом 1 л, снабженную стержнем для перемешивания, обратным холодильником и патрубком для подачи азота, помещали соединение 92b (13,01 г) и в ней поддерживали атмосферу азота. Добавляли безводный ТГФ (300 мл) и смесь перемешивали и получали прозрачный желтый раствор. К раствору при КТ добавляли NaH (2,25 г, 60 мас.% дисперсия в минеральном масле). Смесь обрабатывали ультразвуком в течение 20 мин, перемешивали при КТ в течение еще 10 мин. При КТ добавляли раствор п-толуолсульфонилхлорида (11,86 г) в сухом ТГФ (100 мл) и перемешивали при 50°С в течение 90 мин. Раствор охлаждали и выливали в 0,5 М водный раствор NaHCO3 (1 л). Реакционную смесь разбавляли с помощью EtOAc (500 мл) и органическую фазу отделяли, промывали рассолом (500 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт растворяли в ДХМ и хроматографировали (колонка Supeico VersaPak™, 385 г SiO2) при элюировании смесью ДХМ/гексан в градиентном режиме (от 0 до 100% ДХМ) в количестве, равном 10 объемам колонки. Светло-желтую смолу медленно кристаллизовали и получали 20,36 г соединения 92с в виде белого твердого вещества.
Стадия 3 - В колбу для гидрирования Парра помещали соединение 92с (20,35 г) и растворяли в горячем EtOH (200 мл) и твердое вещество смывали в колбу с помощью 50 мл EtOH. Раствор продували азотом в течение 5 мин, поддерживая раствор теплым. К раствору добавляли 10% Pd/C (4,02 г) и колбу продували азотом. Раствор гидрировали в аппарате Парра со встряхивающим устройством при давлении водорода, равном 55 фунт-сила/дюйм2, при 50°С в течение 1,5 ч. Катализатор удаляли путем фильтрования через стекловолоконный фильтр и промывали горячим EtOH (100 мл). Фильтрат концентрировали в роторном испарителе. Тозилат осаждался в виде белого твердого вещества. Остаток подвергали распределению между 1,0 М водным раствором NaOH (500 мл) и Et2O (300 мл). Фазы разделяли и водную фазу экстрагировали с помощью Et2O (3×300 мл). Объединенные органические экстракты промывали рассолом (450 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии (колонка Supeico VersaPak™, 385 г SiO2) при элюировании с помощью ДХМ и получали 9,04 г соединения 94а в виде почти белого твердого вещества.
Стадия 4 - В круглодонную колбу объемом 250 мл, снабженную стержнем для перемешивания и мембраной, помещали соединение 94а (8,16 г) и в ней поддерживали атмосферу азота. В колбу добавляли сухой диоксан (100 мл) и НОАс (23 мл) и раствор охлаждали в бане со льдом до 5-10°С. Шприцевым насосом в течение 30 мин по каплям добавляли раствор Br2 (7,03 г) в диоксане (45 мл). Образовывался бежевый осадок. Раствор нагревали при КТ и перемешивали в течение 1 ч, затем выливали в 1,0 М раствор NaOH (500 мл) и экстрагировали с помощью ДХМ (3×300 мл). Объединенные экстракты промывали рассолом (450 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме и получали 11,42 г соединения 94b в виде бледно-оливкового твердого вещества.
Стадия 5 - В сосуд объемом 40 мл, снабженную стержнем для перемешивания и мембраной, помещали 2-бромакролеин (2,71 г) и добавляли НОАс (25 мл). Раствор охлаждали в бане из воды со льдом и по каплям добавляли Br2 до появления устойчивой красной окраски (примерно 1,0 мл). Добавляли несколько капель 2-бромакролеина и снова получали бесцветный раствор. Этот раствор при перемешивании выливали в раствор соединения 94b (5,67 г) в НОАс (25 мл) и полученную смесь перемешивали при 100°С в течение 2 ч. Раствор охлаждали, разбавляли с помощью Н2О (250 мл) и экстрагировали с помощью EtOAc (3×100 мл). Объединенные экстракты последовательно промывали 2,0 М водным раствором NaOH (200 мл) и рассолом (150 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии (колонка Supeico VersaPak™, 385 г SiO2) при элюировании с помощью ДХМ и получали 2,69 г соединения 96 в виде светло-оранжевого твердого вещества.
Введение 4-метансульфониламинофенильного заместителя в положение 3 проводили в соответствии с методикой, описанной на стадии 5 примера 19 и получали N-{4-[8-бром-5-метокси-6-(2,2,2-трифторэтил)-хинолин-3-ил]-фенил}-метансульфонамид (97). Введение диоксотетрагидропиримидин-1-ильного фрагмента в положение 8 соединения 97 проводили в соответствии с методиками, описанными на стадиях I-3 примера 18, и получали соединение I-30.
N-{4-[5-Метокси-8-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(2,2,2-трифторэтил)-хинолин-3-ил]-фенил}-метансульфонамидгидробромид (I-53) получали из соединения 96 по методике введения метансульфониламинофенильного фрагмента, описанной на стадии 5 примера 19, с последующей последовательностью реакций сочетания Судзуки/деметилирования, описанной на стадиях 3 и 4 примера 9, за исключением того, что на стадии 3 2-метоксипиридин-3-илбороновую кислоту заменяли на 2-метокси-6-метилпиридин-3-илбороновую кислоту.
N-{4-[5-Метокси-8-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-6-(2,2,2-трифторэтил)-хинолин-3-ил]-фенил}-метансульфонамид (I-54) получали аналогичным образом, за исключением того, что на стадии 3 2-метоксипиридин-3-илбороновую кислоту заменяли на соединение 115. Деметилирование в обоих случаях давало соли с НВ2.
N-{4-[8-(2,4-Диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-5-метокси-6-(2,2,2-трифторэтил)-хинолин-3-ил]-фенил}-метансульфонамид (I-56) получали аналогичным образом, за исключением того, что введение 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ильного фрагмента в положение 8 соединения 97 проводили в соответствии с методиками, описанными на стадиях I-3 примера 23.
Пример 21
N-{4-[6-[1,1-ди(Метил-d3)этил-2,2,2-аз]-5-метокси-8-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид (I-32)
Стадия 1 - Раствор CD3OD (25 мл) и 4-бромфенола (8,5 г, 49,2 ммоля) перемешивали при КТ в течение 30 мин для обмена фенольного протона и затем CD3OD удаляли в вакууме. Полученное твердое вещество растворяли в CDCl3 (10 мл) и (CD3)3COD (4 мл) и нагревали при 60°С. Пятью порциями по 2 мл в течение 50 мин добавляли концентрированную D2SO4 (10 мл). Реакционную смесь выдерживали при 60°С в течение ночи и затем выливали на лед (50 мл) и экстрагировали с помощью EtOAc (2×75 мл). Объединенные органические вещества экстрагировали 2 н. водным раствором КОН (3×300 мл), промывали 1 н. водным раствором HCl (75 мл) и рассолом (25 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 0 до 10% EtOAc за 40 мин) и получали 5,83 г соединения 98а в виде коричневого масла: ЭР МС (М-Н) 236,1.
Стадия 2 - К раствору 2-(D9-трет-бутил)-4-бромфенола (соединение 98а, 35,1 г, 147 ммолей) и ТЭА (17,9 г, 177 ммолей) в Et2O (285 мл), выдерживаемому при 0°С в бане со льдом, в течение 10 мин по каплям добавляли этилхлорформиат (18,4 г, 16,3 мл, 169 ммолей). Примерно через 5 мин происходило образование белого осадка. Реакционную смесь выдерживали при энергичном перемешивании при 0°С в течение 3 ч. Смесь разбавляли насыщенным водным раствором NH4Cl (100 мл) и слои разделяли. Водный слой промывали с помощью Et2O (100 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 0 до 10% EtOAc за 45 мин) и получали 36,5 г соединения 98b в виде коричневого масла: ЭРМС(М+Н)310,1.
Стадия 3 - К раствору соединения 98b (36 г, 116 ммолей) в концентрированной серной кислоте (147 г, 80 мл, 1,5 моля), охлажденному до 0°С, в течение 10 мин по каплям добавляли 70% раствор HNO3 (8,77 г, 6,22 мл, 139 ммолей). Реакционную смесь выдерживали при 0°С в течение 2 ч и затем выливали на лед (примерно 500 г). Водный раствор экстрагировали смесью EtOAc/гексаны состава 1:1 (3×200 мл) и объединенные органические экстракты промывали рассолом, сушили (MgSO4), фильтровали и концентрировали на SiO2 (100 г). Продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 0 до 10% EtOAc за 45 мин) и получали 35,8 г соединения 100а в виде желтого твердого вещества: ЭР МС (М+Н) 355,1.
Стадия 4 - Твердые гранулы КОН (8,29 г, 148 ммолей) в течение 1 мин добавляли к раствору соединения 100а (35,0 г, 98,5 ммоля) в МеОН (800 мл) и полученный раствор помещали в баню с водой при КТ на ночь. МеОН удаляли в вакууме и остаток растворяли в ДХМ (150 мл). Органический раствор промывали 2 н. раствором HCl (200 мл) и водный слой подвергали обратной экстракции с помощью ДХМ (50 мл). Объединенные экстракты промывали рассолом, сушили (MgSO4), фильтровали и концентрировали в вакууме и получали 27,8 г соединения 100b в виде вязкой оранжевой жидкости, которую использовали на следующей стадии без какой-либо дополнительной очистки: ЭР МС(М-Н) 281,1.
Стадия 5 - Йодметан (17,4 г, 7,67 мл, 123 ммоля) при КТ по каплям добавляли к суспензии соединения 100b (27,8 г, 98,2 ммоля) и K3CO3 (20,4 г, 147 ммолей) в ацетоне (110 мл). Красную суспензию энергично перемешивали при КТ в течение 16 ч. Добавляли воду со льдом (500 мл), что приводило к образованию мелкодисперсного желтого осадка. Смесь перемешивали в течение 30 мин, фильтровали и твердое вещество промывали с помощью Н2О (150 мл).
Твердое вещество сушили в вакууме при 40°С в течение ночи и получали 24,8 г соединения 100с, которое использовали без дополнительной очистки: ЭР МС (М+Н) 297,1.
Стадия 6 - В трехгорлую колбу объемом 1 л помещали соединение 100с (24 г, 80,8 ммоля), порошкообразное железо (22,5 г, 404 ммоля) и NH4Cl (21,6 г, 404 ммоля), EtOH (200 мл) и Н2О (200 мл). Колбу снабжали холодильником и желтую суспензию нагревали до 70°С и энергично перемешивали с помощью механической мешалки в течение 15 ч. Реакционную смесь охлаждали до КТ и фильтровали через целит. Слой целита промывали с помощью МеОН (примерно 100 мл). Большую часть МеОН и EtOH удаляли в вакууме. Водную смесь экстрагировали с помощью EtOAc (3×100 мл). Объединенные экстракты сушили (MgSO4), фильтровали и концентрировали в вакууме и получали 21,1 г соединения 102 в виде светло-коричневого масла, которое затвердевало при выдерживании и которое использовали без какой-либо дополнительной очистки: ЭР МС (М+Н) 267,1.
Превращение соединения 102 в соединение 1-32 проводили по методикам, описанным на стадиях 2-5 примера 9 за исключением того, что на стадии 3 вместо соединения 59 использовали 2,6-метоксипиридин-3-илбороновую кислоту: 1Н ЯМР (300 МГц, ДМСО-d6) δ d 10,0 (br s, 1H), 9,12 (d, J=2,3 Гц, 1Н), 8,54 (d, J=2,3 Гц, 1H), 7,88 (d, J=8,6 Гц, 2 H), 7,60 (s, 1H), 7,36 (d, J=8,6 Гц, 1H), 6,31 (br s, 1H), 4,00 (s, 3H), 3,87 (s, 3H), 3,04 (s, 3H).
Пример 22
N-{4-[7-трет-Бутил-8-метокси-5-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-нафталин-2-ил]-фенил}-метансульфонамид (I-34)
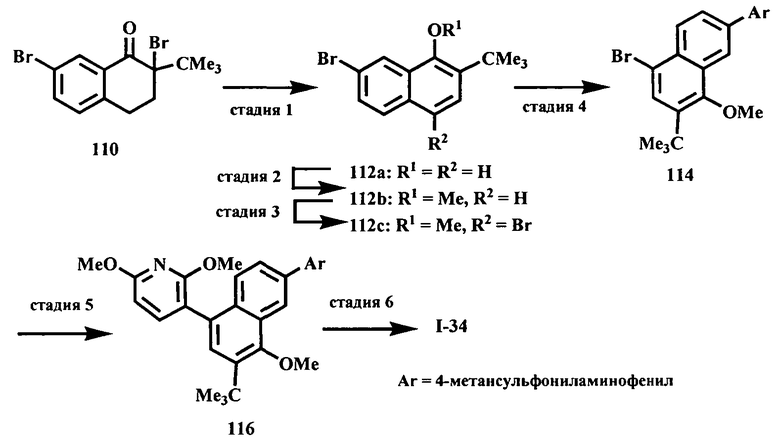
2,7-Дибром-2-трет-бутил-3,4-дигидро-2Н-нафталин-1-он (соединение 110):
Стадия а - Раствор (7-бром-3,4-дигидронафталин-1-илокси)триметилсилана (6,85 г, 11,5 ммоля, регистрационный № CAS 309929-09-7) и ДХМ (23,0 мл) охлаждали до -40°C. Добавляли 2-хлор-2-метилпропан (1,12 г, 1,32 мл, 12,1 ммоля) и раствор перемешивали в атмосфере азота. По каплям добавляли раствор TiCl4 (2,19 г, 1,27 мл, 11,5 ммоля) в ДХМ (6 мл), поддерживая температуру раствора, равной -40°C. Вскоре после завершения добавления ТСХ указывала на 50% превращение в продукт. Реакционную смесь перемешивали при КТ в течение выходных дней, затем выливали на лед. Смесь подвергали распределению между EtOAc и Н2О и водный слой нейтрализовывали насыщенным водным раствором NaHCO3. Смесь фильтровали через слой целита для удаления комкообразного белого осадка. Органический слой отделяли, промывали рассолом, сушили (MgSO4), фильтровали и концентрировали. Неочищенный продукт помещали в колонку с SiO2, приведенную в равновесие с гексаном, и элюировали в градиентном режиме смесью EtOAc/гексан (от 0 до 5% EtOAc) и получали 3,26 г (количественный выход) 7-бром-2-трет-бутил-3,4-дигидро-2Н-нафталин-1-она (соединение 109) в виде желтого твердого вещества.
Стадия b - К раствору соединения 109 (3 г, 10,7 ммоля) в НОАс (40 мл) в атмосфере N2 при перемешивании при КТ через канюлю в течение 20 мин по каплям добавляли раствор брома (1,88 г, 605 мкл, 11,7 ммоля) в НОАс (20,0 мл). Раствор перемешивали при КТ в течение 1 ч, затем реакционную смесь нагревали до 50°С и перемешивали в течение 1 ч. Добавляли аликвоту неразбавленного брома (100 мкл) и нагревание продолжали. Общее время нагревания составляло 2,5 ч. Реакционную смесь выливали на лед, подвергали распределению между EtOAc и Н2О и водную фазу нейтрализовывали насыщенным водным раствором NaHCO3. Органический слой отделяли, промывали рассолом, сушили (MgSO4), фильтровали и концентрировали. Продукт очищали с помощью хроматографии на SiO2 при элюировании гексанами и получали 3,8 г (количественный выход) соединения 110.
Стадия 1 - В круглодонную колбу помещали соединение 110 (3,8 г, 10,6 ммоля), LiBr (275 мг, 3,17 ммоля), Li2CO3 (780 мг, 10,6 ммоля) и ДМФ (44,0 мл) и через белую суспензию в течение 10 мин пропускали Ar. Суспензию нагревали в атмосфере N2 при 100°С в течение 1 ч. Реакционную смесь охлаждали до КТ, разбавляли с помощью EtOAc, трижды промывали водой, затем рассолом, сушили (MgSO4), фильтровали и концентрировали и получали соединение 112а в виде светло-коричневого вязкого масла, которое использовали без дополнительной очистки.
Стадия 2 - К раствору соединения 112а (2,9 г, 10,4 ммоля) и K2CO3 (3,59 г, 26,0 ммоля) в ДМФ (29,7 мл) добавляли Ме1 (1,77 г, 779 мкл, 12,5 ммоля) и сосуд закрывали и смесь перемешивали при 25°С в течение ночи. Смесь разбавляли с помощью EtOAc и водой и водную фазу нейтрализовывали 1 н. раствором HCl. Органический слой отделяли, промывали рассолом, сушили (MgSO4), фильтровали и концентрировали и получали соединение 112b, которое использовали без дополнительной очистки.
Стадия 3 - К раствору соединения 112b (1,6 г, 5,46 ммоля) в НОАс (30 мл), который выдерживали в атмосфере азота, через капельную воронку по каплям добавляли раствор брома (872 мг, 281 мкл, 5,46 ммоля) в НОАс (20 мл). Смесь перемешивали при КТ в течение ночи. Смесь разбавляли с помощью EtOAc и водой и водную фазу нейтрализовывали насыщенным водным раствором NaHCO3. Органический слой отделяли, промывали рассолом, сушили (MgSO4), фильтровали и концентрировали и получали соединение 112с, которое использовали без дополнительной очистки.
Стадия 4 - В пробирку для микроволновой печи объемом 2-5 мл помещали соединение 112 с (1 г, 2,69 ммоля), соединение 25 (578 мг, 2,69 ммоля), Na2CO3 (855 мг, 8,06 ммоля) МеОН (7,14 мл), толуол (3,57 мл) и Н2О (1,79 мл). Смесь дегазировали аргоном в течение 10 мин, затем добавляли Pd(PPh3)4 (155 мг, 134 мкмоля). Дегазирование продолжали в течение еще 5 мин, затем сосуд герметично закрывали и термически нагревали при 115°С в течение 1,5 ч. Смесь охлаждали, подвергали распределению между EtOAc и водой и водную фазу нейтрализовывали 1 н. раствором HCl. Органический слой отделяли, промывали рассолом, сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 и элюировали в градиентном режиме смесью EtOAc/гексан (от 20 до 50% EtOAc) и получали 0,67 г (54%) соединения 114 в виде белого твердого вещества.
Стадия 5 - В пробирку для микроволнового реактора объемом 2-5 мл помещали соединение 114 (0,08 г, 173 мкмоля), 2,6-диметоксипиридин-3-илбороновую кислоту (соединение 115, 34,8 мг, 190 мкмолей) и Na2CO3 (55,0 мг, 519 мкмолей), МеОН (1,5 мл), толуол (750 мкл) и Н2О (65 мкл). Смесь дегазировали аргоном в течение 10 мин, затем добавляли Pd(PPh3)4 (10,0 мг, 8,65 мкмоля) и дегазирование продолжали в течение еще 5 мин. Сосуд герметизировали и облучали в микроволновом реакторе при 115°С в течение 20 мин. Смесь охлаждали, разбавляли с помощью EtOAc и Н2О и водную фазу нейтрализовывали 1 н. раствором HCl. Органический слой отделяли, промывали рассолом, сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью препаративной ТСХ на пластинках с SiO2, обрабатывая смесью 40% EtOAc/гексан, и получали 82 мг (92%) соединения 116 в виде светло-коричневого вспененного вещества.
Стадия 6 - В сосуд помещали соединение 116 (0,082 г), HBr (53,1 мг, 35,6 мкл, 315 мкмолей) и НОАс (0,75 мл), сосуд продували аргоном и герметизировали. Герметизированный сосуд со смесью нагревали при 55°С в течение 6 ч. Реакционную смесь охлаждали, разбавляли с помощью EtOAc и выливали на лед. Полученный раствор нейтрализовывали насыщенным водным раствором NaHCO3. Органическую фазу промывали рассолом, сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали на пластинках с SiO2, обрабатывая смесью 50% EtOAc/гексан, и получали 44 мг соединения I-34.
Пример 23
N-{4-[7-трет-Бутил-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-8-метоксинафталин-2-ил]-фенил}-метансульфонамид (I-35)
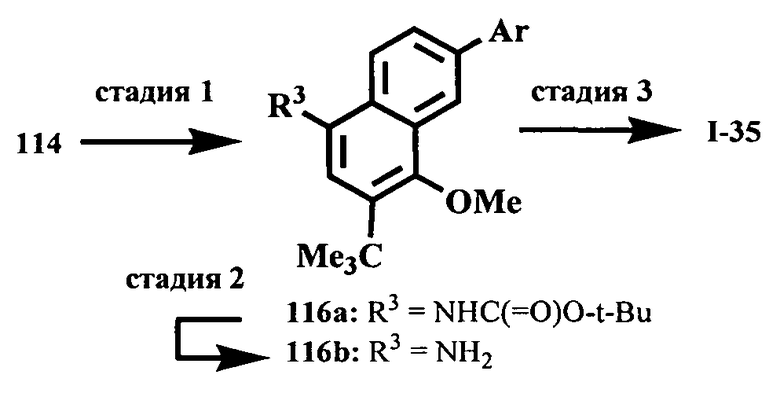
Стадия 1 - В пробирку для микроволновой печи объемом 10-20 мл помещали соединение 114 (0,35 г, 0,757 ммоля), трет-бутилкарбамат (124 мг, 1,06 ммоля) и трет-бутоксид натрия (107 мг, 1,11 ммоля) и толуол (6,00 мл) и получали белую суспензию. Смесь продували аргоном в течение 10 мин. Очень вязкую смесь разбавляли толуолом (4 мл), затем добавляли Pd2(dba)3 (104 мг, 114 мкмолей) и 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенил (145 мг, 341 мкмоль) и через смесь в течение 5 мин пропускали аргон. Реакционную смесь перемешивали в герметизированном сосуде при КТ в течение выходных дней. Смесь подвергали распределению между EtOAc и Н2О и водный раствор нейтрализовывали 1 н. раствором HCl. Органический слой отделяли, промывали рассолом, сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (20-60% EtOAc) и получали 196 мг (52%) соединения 116а.
Стадия 2 - В колбу грушевидной формы объемом 25 мл помещали соединение 116а (0,196 г, 197 мкмолей), ДХМ (1,5 мл) и 4 М раствор HCl в диоксане (491 мкл, 1,97 ммоля), затем перемешивали при КТ в течение 3 ч. После израсходования исходного вещества раствор разбавляли, выливали на лед и нейтрализовывали насыщенным водным раствором NaHCO3. Смесь концентрировали и очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (20-50% EtOAc) и получали соединение 116b, которое использовали без дополнительной очистки.
Стадия 3 - Небольшую колбу закрывали фольгой и в нее помещали цианат серебра (135 мг, 903 мкмоля) и нагревали в высоком вакууме при 50°С в течение ночи. К полученному твердому веществу последовательно добавляли сухой толуол (1,29 мл), (Е)-3-метоксиакрилоилхлорид (65,3 мг, 542 мкмоля). Полученную взвесь нагревали в атмосфере азота при 120°С в течение 30 мин. Смесь охлаждали до КТ, затем помещали в баню со льдом и осаждалось твердое вещество. В отдельной сухой колбе соединение 116b (0,072 г, 181 мкмоль) растворяли в ДМФ (1,03 мл) и охлаждали до 0°С. К этому раствору в ДМФ в течение 10 мин по каплям добавляли надосадочную жидкость из колбы, содержащей цианат серебра. После добавления получали светло-коричневую неоднородную смесь, которую перемешивали в бане со льдом в течение 30 мин. Смесь разбавляли с помощью EtOAc, последовательно промывали с помощью Н2О и рассолом. По данным ЯМР в смеси присутствовала промежуточная мочевина в виде смеси цис- и транс-изомеров. Мочевину переносили в EtOH (1,03 мл) и добавляли 11% раствор H2SO4 в Н2О (1,03 мл). Полученную смесь герметизировали в сосуде и нагревали при 120°С в течение 1,5 ч до получения гомогенного раствора. Смесь охлаждали, выливали на лед и разбавляли с помощью EtOAc. Органическую фазу разбавляли с помощью EtOAc, последовательно промывали с помощью Н2О и рассолом, сушили (MgSO4), фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 50 до 100% EtOAc) и получали примерно 60 мг соединения I-35 в виде желтого твердого вещества.
Пример 24
N-{(S)-1-[7-трет-Бутил-8-метокси-5-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-нафталин-2-ил]-пирролидин-3-илметил}-метансульфонамид (I-36)
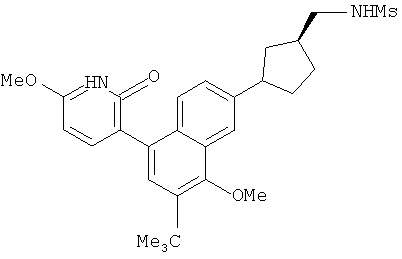
Стадия 1 - В пробирку для микроволновой печи объемом 10-20 мл помещали соединение (S)-72 (184 мг, 1,03 ммоля) и толуол (4,69 мл) и получали коричневый раствор. К нему добавляли соединение 112 с (349 мг, 938 мкмолей) и толуол (4,69 мл). Раствор продували аргоном в течение 10 мин, затем добавляли Pd2(dba)3 (85,9 мг, 93,8 мкмоля) и XANTPHOS (109 мг, 188 мкмолей). Дегазирование продолжали в течение 5 мин, затем быстро добавляли трет-бутоксид натрия (135 мг, 1,41 ммоля), раствор продували аргоном и герметизировали. Раствор облучали в микроволновом синтезаторе при 100°С в течение 10 мин, затем перемешивали при КТ в течение ночи. Для растворения твердых веществ добавляли небольшое количество ДМСО и сосуд термически нагревали в течение 3 ч. Смесь охлаждали, разбавляли с помощью EtOAc и водой и водный слой нейтрализовывали 1 н. раствором HCl. Органический слой отделяли, промывали рассолом, сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью препаративной ТСХ на пластинках с SiO2, дважды обрабатывая смесью 50% EtOAc/гексан, и получали 92 мг соединения 118. Продукт использовали без дополнительной очистки.
Стадия 2 - В пробирку для микроволнового реактора объемом 2-5 мл помещали соединение 118 (85 мг, 181 мкмоль), соединение 115 (39,8 мг, 217 мкмолей), Na2CO3 (57,6 мг, 543 мкмоля), МеОН (0,4 мкл), толуол (0,2 мкл) и Н2О (0,2 мкл). Смесь продували аргоном в течение 10 мин, затем добавляли Pd(PPh3)4 (10,5 мг, 9,05 мкмоля). Раствор продували аргоном в течение еще 5 мин. Сосуд герметизировали и облучали в микроволновом реакторе при 115°С в течение 20 мин. Смесь охлаждали, разбавляли с помощью EtOAc и водой и водный слой нейтрализовывали 1 н. раствором HCl. Органический слой отделяли, промывали рассолом, сушили (MgSCO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (25-50% EtOAc) и получали 56,6 мг соединения 120.
Стадия 3 - Деметилирование простого эфира с получением пиридона проводили так, как описано на стадии 6 примера 22. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 50 до 75% EtOAc) и получали соединение I-36 в виде почти белого твердого вещества.
Пример 25
N-{4-[6-(1-Дифторметилциклопропил)-5-метокси-8-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид (I-37)
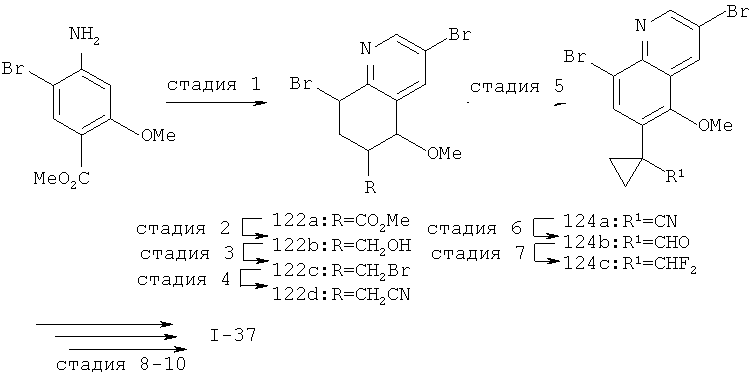
Стадия 1 - Бром (4,48 мл, 86,9 ммоля) в НОАс (50 мл) при КТ добавляли к раствору 2-бромакролеина (11,7 г, 86,9 ммоля, регистрационный №CAS 111049-68-4) в НОАс (100 мл) до появления слабой окраски Br3. К этому раствору добавляли метил-4-амино-5-бром-2-метоксибензоат (22,6 г, 86,9 ммоля) и полученный раствор постепенно нагревали до 100°С. После установления температуры, равной 100°С, перемешивание продолжали в течение 15 мин, затем раствор охлаждали и концентрировали в вакууме. Реакционную смесь нейтрализовывали насыщенным водным раствором NaHCO3 и полученное твердое вещество отфильтровывали и промывали водой. Твердое вещество промывали эфиром, затем смесью 10% МеОН/ДХМ и получали 11,04 г соединения 122а. Фильтрат адсорбировали на SiO2 и очищали с помощью флэш-хроматографии при элюировании смесью ДХМ/гексан в градиентном режиме (от 50 до 100% ДХМ и дополнительно получали 3,46 г соединения 122а.
Стадия 2 - К гетерогенному раствору соединения 122а (11,05 г, 29,5 ммоля) в ДХМ (550 мл) при 0°С по каплям добавляли ДИБАЛ (9,22 г, 64,38 ммоля). После завершения добавления реакция завершалась. Реакцию останавливали водным раствором соли Рошеле и смесь подвергали распределению между Н2О и ДХМ. Органический слой промывали с помощью Н2О и сушили (Na2SCO4), фильтровали и концентрировали в вакууме. Неочищенный продукт адсорбировали на SiO2 и очищали с помощью флэш-хроматографии при элюировании смесью ДХМ/МеОН в градиентном режиме (от 0 до 10% МеОН) и получали 9,5 г соединения 122b.
Стадия 3 - Раствор соединения 122b (7,82 г, 22,5 ммоля), CBr4 (8,97 г, 1,2 экв.) и Ph3P (7,09 г, 1,2 экв.) в ДХМ (250 мл) перемешивали при КТ в течение ночи. На следующее утро добавляли по 0,5 экв. CBr4 и PPh3. Через 1 ч реакция завершалась. Неочищенную реакционную смесь концентрировали в вакууме. Неочищенный продукт адсорбировали на SiO2 и очищали с помощью флэш-хроматографии при элюировании смесью ДХМ/гексан в градиентном режиме (от 0 до 100% ДХМ) и получали 7,62 г соединения 122с в виде белого твердого вещества.
Стадия 4 - Раствор 122с (8,00 г, 19,1 ммоля), KCN (12,4 г, 191 ммоль), ДХМ (156 мл) и H2O (140 мл) кипятили с обратным холодильником в течение 15 мин. Реакционную смесь разбавляли с помощью Н2О и экстрагировали с помощью ДХМ. Органические слои сушили (Na2SO4), и концентрировали в вакууме. Остаток в сухом виде загружали в колонку с SiO2 для проведения флэш-хроматографии и очищали с помощью флэш-хроматографии при элюировании смесью МеОН/ДХМ в градиентном режиме (от 0 до 5% МеОН) и получали соединение 122d в виде белого твердого вещества.
Стадия 5 - Смесь соединения 122d (5,02 г, 14,1 ммоля), 1,2-дибромэтана (3,18 г, 1,46 мл, 16,9 ммоля) и ДМФ (60 мл) охлаждали до 0°С и добавляли NaH (1,69 г, 42,3 ммоля, 60% дисперсия в минеральном масле). Смесь нагревали до КТ и перемешивали в течение 1 ч. Реакционную смесь разбавляли с помощью Н2О и экстрагировали с помощью ДХМ. Органический слой дважды промывали с помощью Н2О, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт в сухом виде загружали в колонку с SiO2 и очищали с помощью флэш-хроматографии при элюировании смесью ДХМ/гексан в градиентном режиме (от 0 до 100% ДХМ) и получали 1,91 г соединения 124а.
Стадия 6 - К раствору соединения 124а (0,28 г, 0,733 ммоля) в ДХМ (14 мл) при -78°С по каплям добавляли ДИБАЛ (870 мкл, 0,879 ммоля, 1М раствор в ДХМ). Реакционную смесь перемешивали при -78°С в течение 1 ч. Реакцию останавливали водным раствором соли Рошеле и смесь экстрагировали с помощью ДХМ. Органический слой промывали с помощью Н2О, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенное вещество очищали с помощью флэш-хроматографии и получали 0,22 г соединения 124b в виде белого твердого вещества.
Стадия 7 - К раствору соединения 124b (0,63 г, 1,64 ммоля) в ДХМ (14,3 мл) добавляли ДАТС (1,05 г, 6,54 ммоля) и полученный раствор перемешивали при в течение 72 ч. Реакционную смесь разбавляли с помощью Н2О и экстрагировали с помощью ДХМ. Органический слой промывали с помощью Н2О и сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью ДХМ/гексан в градиентном режиме (от 50 до 100% ДХМ) и получали 0,60 г соединения 124с в виде белого твердого вещества.
Стадии 8-10 проводили в соответствии с методиками, описанными на стадиях 4-6 примера 22, за исключением того, что на стадии 5 соединение 115 заменяли на соединение 75.
N-{4-[6-(1-Дифторметилциклопропил)-5-метокси-8-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид (I-38) получали аналогичным образом, за исключением того, что на стадии 5 соединение 115 заменяли на соединение 75.
N-{4-[6-(1-Дифторметилциклопропил)-8-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид (I-39) получали из соединения 124с в соответствии со стадиями 1-3 примера 23 для введения урацильного фрагмента.
Пример 26
N-{(8)-1-[7-трет-Бутил-8-метокси-5-(1-метил-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил)-нафталин-2-ил]-пирролидин-3-илметил}-метансульфонамид (I-40)
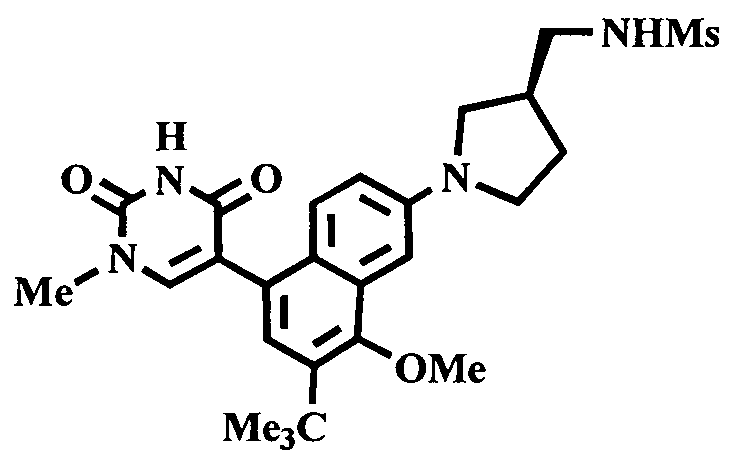
2,4-Диметокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин (117) - В круглодонную колбу объемом 250 мл помещали 5-бром-2,4-диметодкипиримидин (1,23 г, 5,62 ммоля), PdCl2(dppf)·CH2Cl2 (229 мг, 281 мкмоль), бис-(пинаколято)дибор (1,71 г, 6,74 ммоля), KOAc (1,65 г, 16,8 ммоля) и ДМФ. Светло-желтый раствор нагревали до 100°C и перемешивали в течение 1 ч. Реакционную смесь выливали в 50 мл Н2О и экстрагировали смесью EtOAc/толуол (1:1, 3×50 мл). Органические слои объединяли, промывали с помощью Н2О (1×50 мл), насыщенным водным раствором NaCl (50 мл). Органический слой сушили (Na2SO4), сушили, фильтровали и концентрировали в вакууме и получали смесь соединения 117 (1,67 г, 78%) и извлеченного бромида состава 7:3. Темное твердое вещество использовали на следующей стадии без дополнительной очистки.
Стадия 1 - В пробирку с винтовой крышкой объемом 10 мл помещали N-[(8)-1-(5-бром-7-трет-бутил-8-метоксинафталин-2-ил)-пирролидин-3-илметил]-метансульфонамид (118, 0,188 г, 400 мкмолей), PdCl2(dppf)·CH2Cl2 (16,3 мг, 20,0 мкмоля), Cs2CO3 (391 мг, 1,2 ммоля) и соединение 117 (182 мг, 480 мкмолей), диоксан (3,2 мл) и Н2О (799 мкл) и получали темно-коричневый раствор. Реакционную смесь нагревали до 100°C и перемешивали в течение 30 мин. Реакционную смесь выливали в 50 мл Н2О и экстрагировали с помощью EtOAc (3×50 мл). Органические слои объединяли, промывали с помощью Н2О (50 мл) и рассолом (50 мл). Органические слои сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенное вещество очищали с помощью хроматографии на SiO2 при элюировании смесью МеОН/ДХМ в градиентном режиме (от 0 до 5% МеОН) и получали 0,077 г (36%) соединения 120 в виде твердого вещества.
Стадия 2 - В пробирку с винтовой крышкой объемом 10 мл помещали соединение 120 (0,055 г, 104 мкмоля), Mel (250 мг, 0,11 мл, 1,76 ммоля) и ДХМ (0,11 мл). Светло-желтый раствор перемешивали в течение 5 ч и затем выпаривали. Неочищенное вещество очищали с помощью хроматографии на SiO2 при элюировании смесью ДХМ/гексан в градиентном режиме (от 0 до 6% МеОН) и получали 0,022 г (40%) (8)-N-((1-(6-трет-бутил-5-метокси-8-(4-метокси-1-метил-2-оксо-1,2-дигидропиримидин-5-ил)хинолин-3-ил)пирролидин-3-ил)метил)метансульфонамида (соединение 122) в виде твердого вещества.
Стадия 3 - В пробирку с винтовой крышкой объемом 10 мл помещали соединение 122 (0,021 г, 39,6 мкмоля), HBr (16,0 мг, 10,8 мкл, 198 мкмолей) и НОАс. Через 4 ч реакционную смесь выливали в насыщенный водный раствор NaHCO3 (50 мл) и экстрагировали с помощью EtOAc (3×20 мл). Органический экстракт сушили (Na2SO4), фильтровали и выпаривали и получали 0,020 г (98%) соединения I-40 в виде желтого твердого вещества.
N-{4-[6-трет-Бутил-5-метокси-8-(1-метил-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил)-хинолин-3-ил]-фенил}-метансульфонамид получали из соединения 114 по методикам, описанным на стадиях 1-3 этого примера.
Пример 27
N-{4-[6-трет-Бутил-8-(5-хлор-6-метил-2-оксо-1,2-дигидропиридин-3-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид (I-42)
Нейтрализация гидробромида соединения 1-24 (34 мг, 0,059 ммоля) насыщенным водным раствором МаНСО3 давала свободное основание, которое экстрагировали с помощью EtOAc. Экстракт, содержащий EtOAc, сушили (MgSO4), фильтровали и концентрировали. Остаток растворяли в MeCN (1 мл) и ДМФ (1 мл) и нагревали при 60°С. Затем к реакционной смеси добавляли NCS (8 мг, 0,06 ммоля). После перемешивания при 60°С в течение 1,5 ч реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали водой, сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью ДХМ/МеОН состава 9:1 и получали 8 мг (49%) соединения 1-42 в виде белого твердого вещества. МС m/z (ЭР): 527 (М+Н)+.
Пример 28
3-[3-(6-Аминопиридин-3-ил)-6-трет-бутил-5-метоксихинолин-8-ил]-1Н-пиридин-2-он (I-43)
Стадия 1 - В пробирку помещали соединение 70 (208 мг, 0,582 ммоля), 2-аминопиридин-5-илбороновую кислоту (227 мг, 0,84 ммоля), Pd(PPh3)4 (61 мг, 0,052 ммоля), Na2CO3 (286 мг, 2,69 ммоля), МеОН (1,6 мл) и ДХМ (0,5 мл), герметизировали и облучали в микроволновом реакторе при 115°С в течение 1 ч. Реакционную смесь охлаждали до КТ и разбавляли с помощью EtOAc. Органический слой промывали насыщенным водным раствором NaHCO3 (30 мл). Органическую фазу отделяли и водную фазу повторно экстрагировали с помощью EtOAc (3×30 мл). Объединенные органические экстракты сушили (MgSO4), фильтровали и концентрировали. Неочищенный остаток очищали с помощью хроматографии на SiO2 при элюировании смесью ДХМ/МеОН в градиентном режиме и получали 173 мг (71%) соединения I-43 в виде белого твердого вещества.
5-{5-[6-трет-Бутил-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-пиридин-2-ил}метансульфонамид (124) можно получить путем сульфонилирования соединения I-43 метансульфонилхлоридом в соответствии с методикой, описанной на стадии 2 примера 5.
Пример 29
N-{4-[6-трет-Бутил-5-метокси-8-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-бутил}-метансульфонамид (I-44)
3-(3-Бром-6-трет-бутил-5-метоксихинолин-8-ил)-6-метил-1Н-пиридин-2-он (128) можно получить в соответствии с методиками, использованными на стадиях 1-4 примера 9 для получения соединения 70, за исключением того, что на стадии 3 соединение 30 заменяли на соединение 75.
Стадия 1 - Смесь 1-бутинола (0,5 г, 7,13 ммоля), MsNHBoc (2,09 г, регистрационный №CAS 147741-16-4), PPh3 (2,8 г) в ТГФ (30 мл) охлаждали в бане со льдом и добавляли ДЭАД (1,86 г). Смесь перемешивали в течение ночи, затем концентрировали в вакууме. Остаток растирали с EtsO и твердое вещество отфильтровывали. Фильтрат концентрировали и процедуру повторяли. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 0 до 10% EtOAc) и получали 1,0 г соединения 126.
Стадия 2 - В пробирку помещали соединение 128 (0,25 г, 0,62 ммоля), 126 (0,3 г, 1,25 ммоля), Cul (12 мг), Pd(PPh3)4 (0,072 мг), ТЭА (0,5 мл) и ДМФ, ее герметизировали и нагревали при 90°С в течение ночи. Пробирку охлаждали и реакционную смесь разбавляли с помощью EtOAc, последовательно промывали с помощью H2O и рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 50 до 100% EtOAc). Остаток растворяли в ДХМ и добавляли ТФК (1 мл) и полученный раствор перемешивали при КТ в течение ночи. Растворители выпаривали, и остаток очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 50 до 100% EtOAc), затем повторно хроматографировали при элюировании смесью MeOH/EtOAc в градиентном режиме (от 0 до 8% МеОН) и получали 80 мг соединения 130.
Стадия 3 - В колбу Парра помещали соединение 130 (60 мг, 128 мкмолей), Pd/C (13,7 мг) и смесь EtOAc и МеОН и гидрировали в атмосфере водорода при давлении, равном 50 фунт-сила/дюйм, в течение 20 ч. Добавляли дополнительную аликвоту Pd/C (13 мг) и гидрирование продолжали в течение 72 ч. Реакционную смесь фильтровали через целит, промывали с помощью ДХМ и фильтрат концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью MeOH/EtOAc в градиентном режиме (от 0 до 10% МеОН) и получали 30 мг соединения I-44.
N-{3-[6-трет-Бутил-5-метокси-8-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-пропил}-метансульфонамид получали аналогичным образом, за исключением того, что на стадии 2 катализируемое палладием аминирование проводили с помощью 3-пропинилметансульфонамида вместо соединения 126.
N-{(Е)-4-[6-трет-Бутил-5-метокси-8-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-бут-3-енил}-метансульфонамид получали аналогичным образом, за исключением того, что на стадии 1 бутинол заменяли на бут-3-ен-1-ол и исключали стадию гидрирования (стадия 3).
Пример 30
N-{3-[6-трет-Бутил-5-метокси-8-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-илокси]-пропил}-метансульфонамид (I-45)
3-(6-Бром-3-трет-бутил-4-метоксинафталин-1-ил)-6-этил-1Н-пиридин-2-он (131) получали в соответствии со стадиями 1-4 примера 8 за исключением того, что на стадии 3 соединение 75 использовали вместо соединения 80.
Стадия 1 - Раствор соединения 70 в ДМФ и триметилоксонийтетрафторборат перемешивали при КТ в течение 3 дней. Полученный раствор последовательно промывали с помощью Н2О и рассолом, сушили (Na2SO4), фильтровали и выпаривали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiCO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 0 до 5% EtOAc) и получали соединение 132а.
Стадия 2 - В круглодонную колбу помещали соединение 132а (0,1 г, 241 мкмоль), КОН (135 мг, 2,41 ммоля), 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2,'4,'6' триизопропил-1,1'-бифенил (23,2 мг, 48,2 мкмоля), диоксан и воду и получали бесцветную суспензию. Смесь продували азотом в течение 30 мин и добавляли Pd2(dba)3 и продувание с помощью N2 продолжали в течение еще 10 мин. Сосуд для проведения реакции закрывали и нагревали при 100°С в течение 20 ч. Реакционную смесь выливали в EtOAc (50 мл) и последовательно промывали с помощью Н2О (20 мл) и рассолом (20 мл). Органические экстракты сушили (Na2SO4), фильтровали и концентрировали. Неочищенное вещество очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан (от 30 до 60% EtOAc) и получали 70 мг (82,5%) соединения 132b.
Стадия 3 - В колбу помещали соединение 132b (120 мг, 340 мкмолей), 2-бромэтанол (213 мг, 1,7 ммоля), K2CO3 (94,1 мг, 681 мкмоль) и MeCN (5 мл) и получали бесцветный раствор. Реакционную смесь нагревали до 70°С и перемешивали в течение 20 ч. Реакционную смесь выливали в EtOAc (50 мл) и промывали с помощью Н2О (20 мл). Слой, содержащий EtOAc, промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенное вещество очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме и получали 100 мг (74,1%) соединения 134а.
Стадия 4 - В колбу помещали соединение 134а (100 мг, 252 мкмоля), трет-бутилметилсульфонилкарбамат (73,9 мг, 378 мкмолей), PPh3 (99,2 мг, 378 мкмолей) и ТГФ (5 мл) и раствор охлаждали в бане со льдом. К раствору добавляли ДЭАД и реакционную смесь перемешивали при КТ в течение 20 ч.
Неочищенную реакционную смесь концентрировали в вакууме. Неочищенное вещество очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 20 до 60% EtOAc). Верхнее пятно представляло собой смесь (120 мг) продукта 134b и исходного вещества BocNHMs состава 1:1 (не обнаруживаются при УФ-освещении, трудно отделить друг от друга) и извлекали 40 мг соединения 134а.
Стадия 5 - В круглодонную колбу помещали соединение 134b (60 мг, 105 мкмолей), полученное на предыдущей стадии, HBr (74,5 мг, 921 мкмоль) и НОАс (0,5 мл). Смесь нагревали при 60°C в течение 3 ч. Раствор охлаждали, разбавляли с помощью Н2О (5 мл) и 4 н. раствором NaOH (1 мл). Полученный раствор экстрагировали с помощью EtOAc (50 мл). Органический слой последовательно промывали с помощью Н2О и рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенное вещество очищали с помощью хроматографии на SiO2 при элюировании смесью EtOc/гексан в градиентном режиме (от 50 до 100% EtOAc), затем с помощью хроматографии на SiO2 при элюировании смесью 10% MeOH/EtOAc и получали 30 мг (62,4%) соединения I-45.
Пример 31
N-{(S)-1-[7-трет-Бутил-8-метокси-5-(б-метоксиметил-2-оксо-1,2-дигидропиридин-3-ил)-нафталин-2-ил]-пирролидин-3-илметил}-метансульфонамид (I-59)
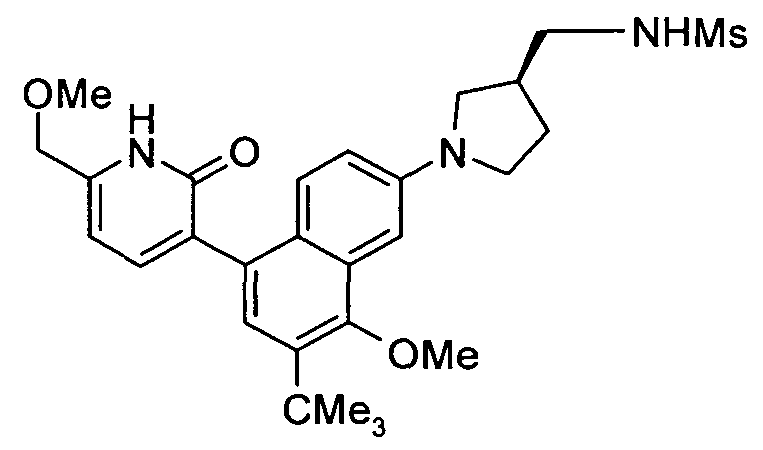
Стадия 1 - Дисперсию NaH (226 мг, 5,64 ммоля, 60% дисперсия в минеральном масле) растирали с гексанами (3×10 мл) и сушили в токе N2, затем суспендировали в ТГФ (23,5 мл) и охлаждали до 0°C. По каплям добавляли раствор 3-бром-2-метокси-6-(гидроксиметил)пиридина (0,88 г, 3,79 ммоля) в ТГФ (10 мл) и смесь перемешивали в течение 30 мин. К раствору добавляли MeI (1,00 г, 441 мкл, 7,05 ммоля) и смесь нагревали до КТ. Через 1 ч неочищенную реакционную смесь концентрировали в вакууме и смесь выливали в Н2О (100 мл) и экстрагировали с помощью EtOAc (3×50 мл). Объединенные экстракты последовательно промывали с помощью Н2О, рассолом, сушили, фильтровали и концентрировали и получали 3-бром-2-метокси-6-метоксиметилпиридин (соединение 136) в виде светло-желтого масла.
Стадия 2 - В круглодонную колбу помещали соединение 136 (0,88 г, 3,79 ммоля), PdCl2(dppf)·CH2Cl2 (155 мг, 190 мкмолей), бис-(пинаколято)дибор (1,16 г, 4,55 ммоля), KOAc (1,12 г, 11,4 ммоля) и ДМФ. Светло-желтый раствор нагревали до 100°C и перемешивали в течение 1 ч. Реакционную смесь охлаждали, выливали в 50 мл Н2О и экстрагировали смесью EtOAc/толуол (1:1, 3×50 мл). Органические экстракты объединяли, последовательно промывали с помощью Н2О (50 мл) и рассолом (50 мл). Экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме и получали 2-метокси-6-(метоксиметил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (соединение 138), которое использовали без дополнительной очистки.
Стадия 3 - В пробирку с винтовой крышкой объемом 10 мл помещали соединение 118 (0,103 г, 219 мкмолей), PdCl2(dppf)·CH2Cl2 (8,94 мг, 10,9 мкмоля), Cs2CO3 (214 мг, 657 мкмолей), соединение 138 (183 мг, 263 мкмоля), диоксан (1,75 мл) и Н2О (438 мкл), ее герметизировали и нагревали при 100°C в течение 30 мин. Реакционную смесь охлаждали, выливали в Н2О (50 мл) и экстрагировали с помощью EtOAc (3×50 мл). Объединенные экстракты последовательно промывали с помощью Н2О (50 мл) и рассолом (50 мл). Органические слои сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенное вещество очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 10 до 100% EtOAc) и получали 0,073 г (61%) (8)-N-((1-(6-трет-бутил-5-метокси-8-(2-метокси-6-(метоксиметил)пиридин-3-ил)хинолин-3-ил)пирролидин-3-ил)метил)метансульфонамида (138) в виде желтого вспененного вещества.
Стадия 4 - Деметилирование соединения 138 с получением соединения 1-32 проводили в соответствии с методикой, описанной на стадии 7 примера 2.
Пример 32
N-{4-[6-трет-Бутил-8-(5-фтор-6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид (I-47)
Стадия 1 - В колбу, содержащую 2,3,6-трифторпиридин (2 г, 15 ммолей), добавляли МеОН (5 мл), затем метанольный раствор NaOMe (5 мл, 25% раствор NaOMe в МеОН). Происходила экзотермическая реакция и образовывалось небольшое количество осадка. Реакционную смесь перемешивали в течение 10 мин и разбавляли с помощью Н2О. Твердое вещество отфильтровывали и промывали с помощью Н2О. Твердое вещество растворяли в EtOAc, последовательно промывали водой и рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме и получали 1,5 г (69%) 3,6-дифтор-2-метоксипиридина (соединение 140). Извлеченное вещество использовали без дополнительной очистки.
Стадия 2 - К раствору бензилового спирта (1,12 г, 10,3 ммоля) в ТГФ добавляли NaH (0,413 г, 10,3 ммоля, 60% дисперсия в минеральном масле) и перемешивали в течение 30 мин. К этому раствору добавляли соединение 140 (1,5 г, 10,3 ммоля) и полученный раствор облучали в микроволновом синтезаторе при 100°C в течение 1 ч. Реакционную смесь охлаждали, разбавляли с помощью Н2О, экстрагировали с помощью EtOAc (2×50 мл). Объединенные экстракты промывали рассолом, сушили (Na2SO4), фильтровали и концентрировали. Неочищенный продукт очищали с помощью хроматографии на SiO2 и получали 1 г (41%) 6-бензилокси-3-фтор-2-метоксипиридина (соединение 142).
Стадия 3 - Раствор соединения 142 (1 г, 4,29 ммоля) в ДМФ (10 мл) охлаждали в бане со льдом и добавляли NBS (0,763 г, 4,29 ммоля). Бесцветную смесь перемешивали в течение 2 ч и затем реакцию останавливали с помощью Н2О. Смесь экстрагировали с помощью EtOAc (2×50 мл). Объединенные экстракты последовательно промывали с помощью Н2О и рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 и получали 0,5 г 6-бензилокси-5-бром-3-фтор-2-метоксипиридина (соединение 144).
Стадия 3 - N-{4-[6-трет-Бутил-5-метокси-8-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-хинолин-3-ил]-фенил}-метансульфонамид (соединение 146) можно получить путем конденсации соединения 74b и бис-(пинаколято)дибора в соответствии с методикой, описанной на стадии b примера 16.
Стадия 4 - В сосуд для микроволновой печи помещали соединение 146 (1 экв.), соединение 144 (1 экв.), Pd(PPh3)4 (0,1 экв.), Na2СО3 (3 экв.), NeOH (9 мл) и ДХМ (3 мл), его герметизировали и облучали в микроволновом синтезаторе при 115°C в течение 15 мин. Реакционную смесь разбавляли с помощью EtOAc, последовательно промывали с помощью Н2О и рассолом. Экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 0 до 20% EtOAc) и получали 0,100 г N-(4-(8-(2-(бензилокси)-5-фтор-6-метоксипиридин-3-ил)-6-трет-бутил-5-метоксихинолин-3-ил)фенил)-метансульфонамида (соединение 148).
Стадия 5 - Смесь соединения 148 (0,100 г, 0,162 ммоля), Pd/C (30 мг) и EtOAc перемешивали при давлении водорода, равном 1 атм, в течение 20 ч. Катализатор отфильтровывали и промывали с помощью EtOAc. Фильтрат концентрировали и неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 30 до 80% EtOAc) и получали 25 мг (29,3%) соединения I-47.
Пример 33
N-{4-[3-трет-Бутил-4-метокси-1-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-изохинолин-6-ил]-фенил}-метансульфонамид (I-48)
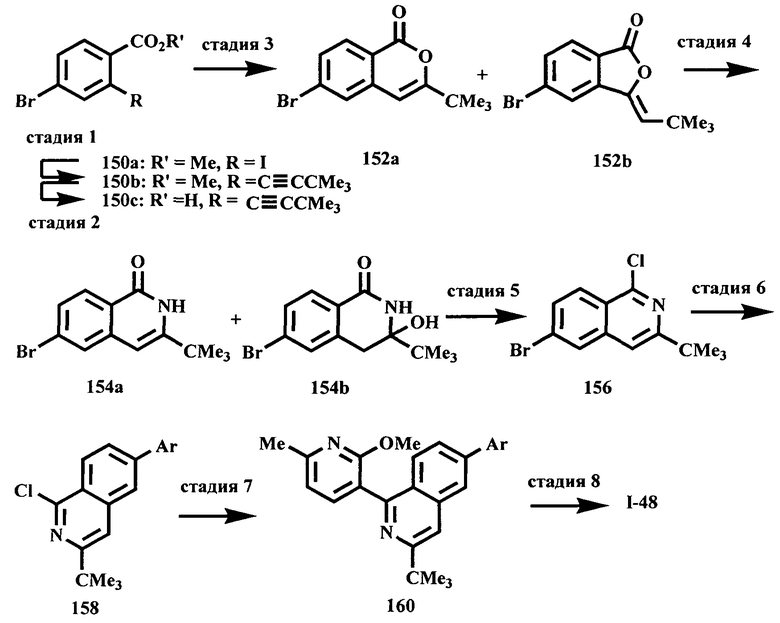
Стадия 1 - К раствору соединения 150а (5,93 г, 17,4 ммоля), Pd(PPh3)2Cl2 (610 мг, 870 мкмолей), CuI (0,331 г, 1,74 ммоля) и ТГФ (178 мл) добавляли ТЭА (14,1 г, 19,4 мл, 139 ммолей). Реакционную смесь дегазировали с помощью Ar и затем к смеси добавляли 3,3-диметилбут-1-ин. Смесь перемешивали при КТ в течение ночи, затем разбавляли эфиром и промывали с помощью Н2О. Органический экстракт промывали 2 н. раствором HCl и насыщенным водным раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали. Неочищенное вещество очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан (от 0 до 10% EtOAc) и получали соединение 150b. Данные ЯМР указывали на содержание в продукте 10% исходного вещества.
Стадия 2 - Гидролиз соединения 150b метанольным раствором NaOH при стандартных условиях давал соединение 150с.
Стадия 3 - Раствор соединения 150с (4,58 г, 16,3 ммоля), PdCl2(MeCN)2 (1,63 ммоля), ТЭА (5,88 г, 8,1 мл, 58,1 ммоля) и ТГФ (320 мл) перемешивали при КТ в течение ночи. Реакционную смесь разбавляли с помощью Et2O и последовательно промывали 10% раствором HCl, Н2О и насыщенным раствором NaHCO3. Органический экстракт сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью ДХМ/гексан в градиентном режиме и получали 2,47 г (53,9%) соединения 152а и вторую фракцию, которая представляла собой смесь соединения 152а и соединения 152b.
Стадия 4 - В колбу помещали EtOH и ее насыщали аммиаком. К раствору добавляли соединение 152а (2,47 г, 8,79 ммоля) и раствор облучали в микроволновом синтезаторе при 130°C в течение 5 ч. Реакционную смесь охлаждали до КТ и выпадал белый осадок, который отфильтровывали и сушили и получали 1,849 г соединения 154а. Фильтрат концентрировали и получали 0,562 г смеси соединения 154а и соединения 154b состава 1:1.
Стадия 5 - Смесь соединения 154а (0,5 г, 1,78 ммоля) и РОСl3 (5 мл) нагревали при 120°C в течение 10 мин. После охлаждения до КТ смесь нейтрализовывали насыщенным водным раствором NaHCO3. Реакционную смесь экстрагировали с помощью EtOAc и объединенные экстракты промывали насыщенным водным раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенное вещество в сухом виде загружали в колонку с SiO2 и элюировали в градиентном режиме смесью EtOAc/гексан (от 0 до 5% EtOAc) и получали 0,5 г (93,8%) соединения 156.
Стадия 6 - В сосуд помещали соединение 156 (0,5 г 1,67 ммоля) и соединение 25 (0,395 г, 1,84 ммоля), Na2CO3 (532 мг, 5,02 ммоля), Pd(PPh3)4 (0,193 г, 0,167 мкмоля) диоксан (3 мл) и Н2О (1 мл). Реакционную смесь нагревали до 80°C и перемешивали в течение ночи. Реакционную смесь фильтровали через стекловолоконный фильтр и фильтрат подвергали распределению между Н2О и EtOAc. Органический слой промывали рассолом и сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 0 до 50% EtOAc) и получали 0,38 г (58,4%) соединения 158 (Ar=4-метансульфониламинофенил).
Стадия 7 - В сосуд помещали соединение 158 (0,38 г, 977 мкмолей), соединение 75 (326 мг, 1,95 ммоля), Na2CO3 (311 мг, 2,93 ммоля), Pd(PPh3)4 (0,113 г, 97,7 мкмоля) и ДМЭ. Реакционную смесь нагревали до 80°C и перемешивали в течение ночи. Через 18 ч в смеси все еще присутствовало некоторое количество соединения 158. Реакционную смесь фильтровали через бумагу из стекловолокна и фильтрат концентрировали в вакууме. Неочищенное вещество очищали с помощью хроматографии на SiO2 при элюировании смесью EtOAc/гексан в градиентном режиме (от 0 до 50% EtOAc) и получали 0,120 г (25,8%) соединения 160.
Стадия 8 - Деметилирование соединения 160 (0,12 г) проводили в соответствии с методикой, описанной на стадии 7 примера 2 и получали 0,10 г (85,9%) соединения I-48. Продукт осаждался и его очищали путем промывки с помощью Н2О и Et2O.
Пример 34
N-{4-[6-трет-Бутил-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-циннолин-3-ил]-фенил}-метансульфонамид (161)
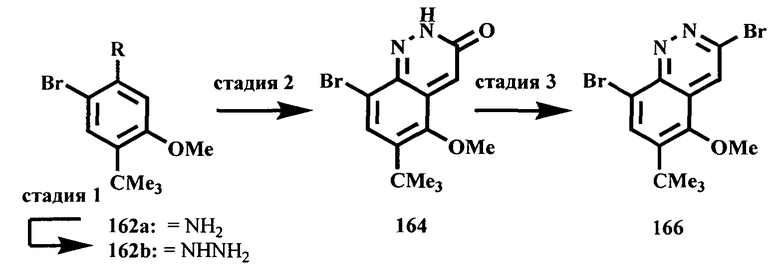
Стадия 1 - По методике, описанной в литературе (J. Organomet. Chem. 2009 694:2493), соединение 162а (1 экв.) при 0°C объединяли с NaNO2 (3 экв.) и HCl в H2O. Реакционной смеси давали нагреться до КТ и ее перемешивали в течение 1 ч. Реакционную смесь повторно охлаждали до 0°C и добавляли SnCl2 (5 экв.). Реакционную смесь в течение ночи нагревали до КТ. Реакционную смесь разбавляли с помощью ДХМ и промывали 2 н. раствором KOH для удаления солей олова. Органический слой сушили и концентрировали в вакууме. Затем остаток очищали путем пропускания через тонкий слой SiO2 и получали соединение 162b.
Стадия 2 - Раствор соединения 162b (1 экв.), диэтоксиацетилхлорида (1 экв., получали из диэтоксиуксусной кислоты и SOCl2) и ТЭА (2 экв.) в ДХМ выдерживали при КТ в течение ночи. Реакционную смесь промывали насыщенным водным раствором NH4Cl, сушили и концентрировали в вакууме. Полученный остаток при 0°C растворяли в концентрированной H2SO4 и затем реакционную смесь нагревали до КТ и перемешивали в течение 20 ч. Реакционную смесь нейтрализовывали насыщенным водным раствором NaHCO3 и экстрагировали с помощью ДХМ. Органический слой сушили и концентрировали в вакууме и остаток очищали с помощью хроматографии на SiO2 и получали соединение 164.
Стадия 3 - По методике, описанной на стадии 3 примера 36, из раствора соединения 164 (1 экв.) и POBr3 (3 экв.) в ДМФ получали дибромциннолин 166.
Превращение соединения 166 в соединение 161 проводили с помощью последовательного катализируемого палладием сочетания с соединением 25 и соединением 30.
Пример 35
N-{4-[6-трет-Бутил-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-изохинолин-3-ил]-фенил}-метансульфонамид (174)
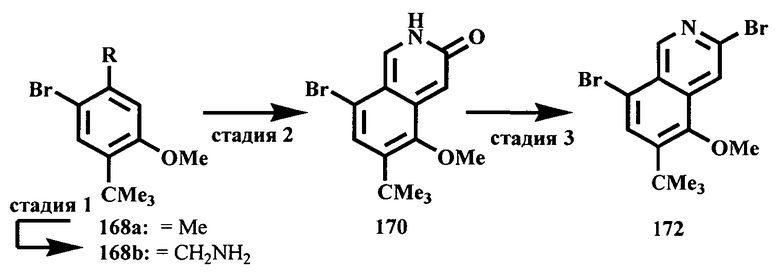
Стадия 1 - Раствор соединения 168а (1 экв. Bull. Soc. Chim. Fr. 1969 6:2129), NBS (1 экв.), АИБН (2-2-азоизобисбутиронитрил) (0,005 экв.) и бензола кипятили с обратным холодильником в течение 14 ч. Реакционную смесь охлаждали до КТ и твердый осадок удаляли фильтрованием. Бензол удаляли в вакууме и полученный остаток растворяли в смеси NH3/МеОН (4 М) и перемешивали при КТ в течение ночи. Растворители удаляли в вакууме и остаток очищали с помощью хроматографии на SiO2 и получали соединение 168b.
Стадия 2 - Раствор соединения 168b (1 экв.), диэтоксиацетилхлорида (1 экв., получали из диэтоксиуксусной кислоты и SOCl2) и Et3N (2 экв.) в ДХМ перемешивали при КТ в течение 3 ч. Реакционную смесь промывали насыщенным водным раствором NH4Cl, сушили и концентрировали в вакууме. Полученный остаток при 0°C растворяли в концентрированной H2SO4 и затем реакционной смеси давали нагреться до КТ и ее выдерживали в течение 28 ч. Реакционную смесь нейтрализовывали насыщенным водным раствором NaHO3 и экстрагировали с помощью ДХМ. Органический слой сушили и концентрировали в вакууме и остаток очищали с помощью хроматографии на SiO2 и получали соединение 170.
Стадия 3 - По методике, описанной в литературе (Chem. Lett. 2007 36(8): 1036), раствор соединения 170 (1 экв.) и POBr3 (3 экв.) в ДМФ выдерживали при 90°C в течение 2 ч. Раствор охлаждали до КТ, подщелачивали 1 н. раствором КОН, экстрагировали с помощью ДХМ. Объединенные экстракты промывали с помощью Н2О, сушили, фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на SiO2 и получали соединение 172.
Превращение соединения 172 в соединение 174 проводили с помощью последовательного катализируемого палладием сочетания с соединением 25 и соединением 30.
Пример 36
N-{4-[6-трет-Бутил-8-(6-гидроксиметил-2-оксо-1,2-дигидропиридин-3-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид (1-41)
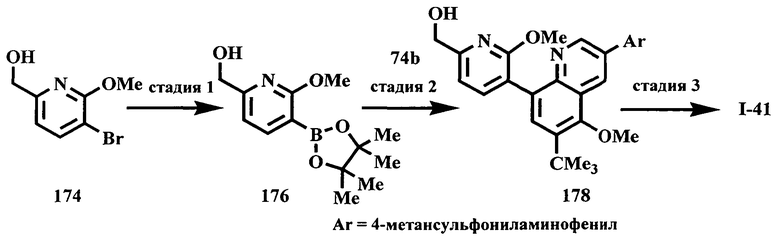
Стадия 1 - 6-Гидроксиметил-2-метокси-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (соединение 176, регистрационный № CAS 1206776-83-1) получали из соединения 174 в соответствии с методикой, описанной в примере 26, за исключением того, что соединение 174 использовали вместо 2,4-диметокси-5-бромпиримидина.
Стадия 2 - В сосуд помещали соединение 74b (0,125 г, 0,27 ммоля) соединение 176 (71 мг, 0,27 ммоля), PdCl2(dppf)·CH2Cl2 (0,011 г, 0,05 ммоля), Cs2CO3 (0,269 г, 0,809 ммоля), диоксан (2 мл) и H2O (0,5 мл), дегазировали, герметизировали и нагревали при 100°C в течение 1 ч. Продукт охлаждали, подвергали распределению между EtOAc и Н2О. Органический экстракт промывали рассолом, сушили (MgSO4), фильтровали и концентрировали в вакууме и получали соединение 178, которое использовали без дополнительной очистки.
Стадия 3 - Деметилирование соединения 178 проводили в соответствии с методикой, описанной на стадии 7 примера 2. Неочищенный продукт очищали на пластинках для препаративной ТСХ, обрабатывая смесью 10% МеОН/ДХМ, и получали 3 мг соединения I-41.
Пример 37
N-{(S)-1-[7-трет-Бутил-5-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-нафталин-2-ил]-пирролидин-3-илметил}-метансульфонамид (I-60)
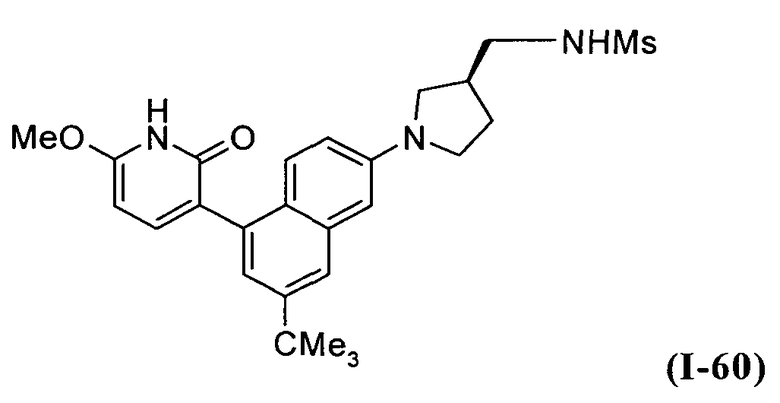
6-трет-Бутил-3,8-дибромхинолин (180) получали из 2-бром-4-трет-бутилнафталина с использованием акролеина и брома в соответствии с методикой, описанной на стадии 2 примера 7. Введение 1-пирролидин-3-илметилметансульфонамидного фрагмента можно провести с использованием трет-бутилового эфира (S)-1-пирролидин-3-илметилкарбаминовой кислоты в соответствии с методиками, описанными на стадиях 1-3 примера 19. Введение пиридинового кольца проводили путем конденсации по Судзуки промежуточного бромхинолина с соединением 115 в соответствии с методикой, описанной на стадии 2 примера 24, и последующее деметилирование метилпиридинилового простого эфира в соответствии с методикой, описанной стадии 6 примера 22, давало соединение I-60.
Пример 38
N-{4-[6-трет-Бутил-8-(диоксотетрагидропиримидин-1-ил)-хинолин-3-ил]-фенил}-метансульфонамид (I-61)
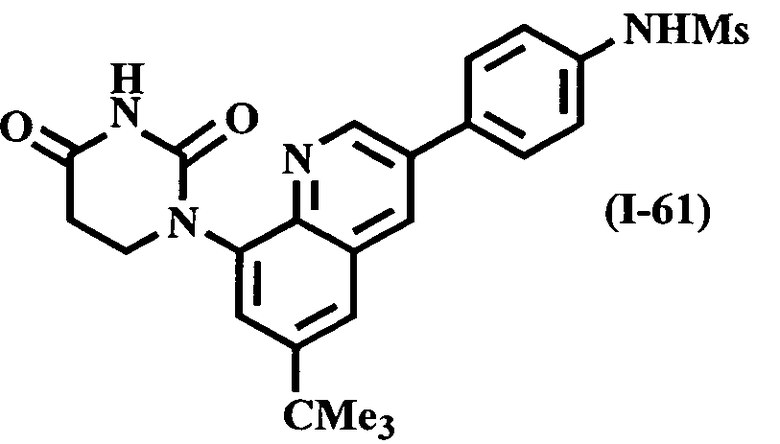
Искомое соединение получали из соединения 180 путем введения метансульфониламинофенильного фрагмента по реакции сочетания Судзуки в соответствии с методикой, описанной на стадии 2 примера 13. Введение диоксотетрагидропиримидин-1-ильного заместителя проводили в соответствии с методиками, описанными на стадиях 1-3 примера 17, и получали соединение I-61.
Пример 39
Активность HCV NS5B РНК полимеразы
Ферментативную активность полимеразы HCV (NS5B570n-Con1) определяли путем включения радиоактивно меченых нуклеотидмонофосфатов в нерастворимые в кислоте продукты РНК. Невключенный радиоактивно меченый субстрат удаляли фильтрованием и в промытые и высушенные фильтрующие планшеты, содержащие радиоактивно меченый продукт РНК, добавляли сцинтилляционную жидкость. Содержание продукта РНК, образовавшегося с помощью NS5B570-Con1 в конце реакции, обратно пропорционально интенсивности излучения сцинтилляционной жидкости.
HCV полимераза с N-концевой 6-гистидиновой меткой, полученная из штамма HCV Con1, генотип 1b (NS5B570n-Con1), содержит делецию 21 аминокислоты на С-конце по сравнению с полноразмерной HCV полимеразой и ее выделяли путем очистки из штамма Е. coli BL21(DE) pLysS. Конструкцию, содержащую кодирующую последовательность HCV NS5B Con1 (GenBank accession number AJ242654) вставляли в плазмидную конструкцию рЕТ17b в прямом направлении от кассеты экспрессии промотора Т7 и трансформировали в Е. coli. Одну колонию выращивали в течение ночи в качестве исходной культуры и затем ее использовали для инокуляции 10 л среды LB, к которой добавлено 100 мкг/мл ампициллина при 37°C. Экспрессирование белка индуцировали путем добавления 0,25 мМ изопропил-β-D-тиогалактопиранозида (IPTG), при этом оптическая плотность культуры при 600 нм составляла от 0,6 до 0,8 и клетки собирали через 16-18 ч при 30°C. NS5B570n-Con1 очищали до однородности по трехстадийной методике, включая последующую хроматографию на смолах Ni-NTA, SP-Sepharose HP и Superdex 75.
Каждые 50 мкл ферментной реакционной смеси содержали 20 нМ матрицы РНК, полученной из комплементарной последовательности сайта внутреннего рибосомного входа (cIRES), 20 нМ фермента NS5B570n-Con1, 0,5 мкКи тритиированного UTP (Perkin Elmer catalog no. TRK-412; удельная активность: от 30 до 60 Ки/ммоль; концентрация исходного раствора от 7,5×10-5 М до 20,6×10-6 М), по 1 мкМ АТФ (аденозинтрифосфат), ЦТФ (цитидинтрифосфат) и ГТФ (гуанозинтрифосфат), 40 мМ Tris-HCl [Tris=трис(гидроксиметиламинометан)] pH 8,0, 40 мМ NaCl, 4 мМ ДТТ (дитиотреитол), 4 мМ MgCl2 и 5 мкл соединения серийно разведенного в ДМСО. Реакционные смеси помещали в 96-луночные фильтрующие планшеты (cat # MADVNOB, Millipore Co.) и инкубировали при 30°C в течение 2 ч. Реакции останавливали путем проводимого в конце добавления 10% (об./об.) трихлоруксусной кислоты и инкубировали при 4°C в течение 40 мин. Реакционные смеси фильтровали, промывали с помощью равного 8 объемам реакционной смеси количества 10% (об./об.) трихлоруксусной кислоты, равного 4 объемам реакционной смеси количества 70% (об./об.) этанола, сушили воздухом и в каждую лунку добавляли 25 мкл сцинтиллирующей жидкости (Microscint 20, Perkin-Elmer).
Интенсивность света, испускаемого сцинтиллирующей жидкостью, пересчитывали в количество импульсов в минуту (КИМ) с помощью устройства считывания планшетов Topcount® (Perkin-Elmer, Диапазон энергий: низкий, режим эффективности: нормальный, длительность подсчета: 1 мин, вычитание фона: не проводили, уменьшение переходных воздействий: отключено).
Данные анализировали с помощью программ Excel® (Microsoft®) и ActivityBase® (idbs®). Реакцию при отсутствии фермента использовали для определения фонового сигнала, который вычитали из данных для ферментных реакций. Для положительного контроля реакции проводили при отсутствии соединения, и скорректированную на фон активность считали 100% активностью полимеразы. Все данные представляли в процентах от значения для положительного контроля. Концентрацию соединения, при которой скорость катализируемой ферментом реакции синтеза РНК уменьшалась на 50% (IС50), рассчитывали путем аппроксимации данных уравнением (i),
в котором ″Y″ означает относительную активность фермента (в %), ″%Min″ означает остаточную относительную активность при концентрации соединения, соответствующей насыщению, ″%Мах″ означает относительную максимальную активность фермента, ″X″ означает концентрацию соединения и ″S″ означает коэффициент Хилла (или наклон).
Пример 40
Исследование репликона HCV
В этом исследовании определяли способность соединений формулы I подавлять репликацию HCV РНК и, следовательно, их потенциальную пригодность для лечения инфекций HCV. В исследовании использовали репортер в качестве простого считывающего устройства для уровня репликона HCV внутриклеточной РНК. Ген Renilla luciferase помещали в первую открытую рамку считывания конструкции NK5.1 репликона генотипа 1b (N. Krieger et al., J. Virol. 2001 75(10):4614) непосредственно после последовательности сайта внутреннего рибосомного входа (IRES) и объединяли с геном неомицинфосфотрансферазы (NPTII) путем саморасщепления белка 2А, взятого из вируса ящура (M.D. Ryan & J. Drew, EMBO 1994 13(4):928-933). После транскрипции in vitro РНК электропорировали в клетки гепатомы человека Huh7 и выделяли и размножали устойчивые к G418 колонии. Выбранная стабильная клеточная линия 2209-23 содержала репликационную HCV субгеномную РНК и активность Remlla luciferase, экспрессированного с помощью репликона, отражает его уровень РНК в клетках. Это исследование проводили в двух одинаковых планшетах, один был непрозрачным белым и один - прозрачным, для одновременного определения противовирусной активности и цитотоксичности химического соединения, что гарантировало, что наблюдаемая активность не является следствием сниженной пролиферации клеток или гибели клеток.
Клетки репликона HCV (2209-23), которые экспрессируют репортер Renilla luciferase, культивировали в модифицированной Иглом среде (МИС) Дульбекко (Invitrogen cat no. 10569-010), содержащей 5% фетальной бычьей сыворотки (ФБС, Invitrogen cat. no. 10082-147), и высевали в 96-луночный планшет по 5000 клеток/лунка, и инкубировали в течение ночи. Через 24 ч к клеткам добавляли разные разведения химических соединений в среде для выращивания и затем инкубировали при 37°С в течение 3 дней. В конце инкубационного периода клетки, находящиеся в белых планшетах, собирали и измеряли активность люциферазы с помощью системы анализа R. luciferase (Promega cat no. E2820). Все реагенты, описанные в приведенном ниже абзаце, были включены в набор, предоставленный изготовителем, и реагенты готовили в соответствии с инструкциями изготовителя. Клетки один раз промывали с помощью 100 мкл/лунка забуференного фосфатом физиологического раствора (рН 7,0) (ЗФФ) и подвергали лизису с помощью 20 мкл 1×литического буфера для исследования R. Luciferase, затем инкубировали при комнатной температуре в течение 20 мин. Затем планшеты вставляли в люминометр для микропланшетов Centre LB 960 (Berthold Technologies) и в каждую лунку добавляли по 100 мкл буфера для исследования R. luciferase и измеряли значение сигнала с помощью программы:
2 с - задержка, 2 с - измерение. Концентрацию лекарственного средства, необходимую для снижения содержания репликона на 50% по сравнению со значением для необработанных клеток, являющихся контрольными, IC50, можно рассчитать по зависимости выраженного в процентах уменьшения активности люциферазы от концентрации лекарственного средства, как это описано выше.
Для исследования цитотоксичности использовали реагент WST-1, выпускающийся фирмой Roche Diagnostic (cat no. 1644807). 10 мкл Реагента WST-1 добавляли в каждую лунку прозрачных планшетов, включая лунки, которые содержали только среду, в качестве холостых проб. Затем клетки инкубировали при 37°С в течение 2 ч и значения оптической плотности (ОП) измеряли с помощью считывающего устройства для планшетов для микротитрования MRX Revelation (Lab System) при длине волны 450 нм (эталонный фильтр при 650 нм). Затем концентрацию лекарственного средства, необходимую для снижения пролиферации клеток на 50% по сравнению со значением для необработанных клеток, являющихся контрольными, СС50, можно рассчитать по зависимости уменьшения значения WST-1 от концентрации лекарственного средства, как это писано выше.
Пример 43
Фармацевтические композиции из соединений, предлагаемых в настоящем изобретении, предназначенные для введения различными путями, готовили так, как это описано в этом примере.
Композиция для перорального введения (А)
Ингредиенты смешивают и дозируют в капсулы, содержащие примерно по 100 мг каждая; одна капсула содержит приблизительно полную суточную дозу. Композиция для перорального введения (В)
Ингредиенты объединяют и гранулируют с использованием растворителя, такого как метанол. Затем композицию сушат и формуют в таблетки (содержащие примерно 20 мг активного соединения) на соответствующей таблетирующей машине.
Композиция для пепопального введения (C)
Ингредиенты смешивают с получением суспензии для перорального введения.
Композиция для парентерального введения (D)
Активный ингредиент растворяют в порции воды для инъекций. Затем при перемешивании прибавляют количество хлорида натрия, достаточное для получения изотонического раствора. Раствор доводят до необходимой массы путем добавления оставшейся воды для инъекций, фильтруют через мембранный фильтр с размером пор 0,2 мкм и упаковывают в стерильных условиях.
Отличительные признаки, раскрытые в приведенном выше описании или в приведенной ниже формуле изобретения, выраженные в конкретных формах или с помощью средств для осуществления раскрытого назначения, или методика или способ для достижения раскрытого результата, если это целесообразно, могут по отдельности или в виде любой комбинации таких отличительных признаков быть использованы для осуществления настоящего изобретения в его различных формах.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОТИВОВИРУСНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2011 |
|
RU2572558C2 |
| ПРОИЗВОДНЫЕ (4-МЕТИЛСУЛЬФОНИЛАМИНОФЕНИЛ)-ХИНОЛИНА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ ГЕПАТИТА С | 2011 |
|
RU2572835C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2654068C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2678305C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2689777C1 |
| БЕНЗОКСАЗЕПИНОВЫЕ ИНГИБИТОРЫ PI3K И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2600927C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2010 |
|
RU2542585C2 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ МОНОАЦИЛГЛИЦЕРИНЛИПАЗЫ | 2019 |
|
RU2801190C2 |
Изобретение относится к области органической химии, а именно к новым гетероциклическим соединениям общей формулы (I) или к их фармацевтически приемлемым солям, где X1 обозначает N и X2, X3 и X4 обозначают CR5; или X1 и X2 обозначают N и X3 и X4 обозначают CR5; или X1, X2 и X4 обозначают CR5, и X3 обозначает N; или X1, X2, X3 и X4 обозначают CR5; R1 обозначает (a) гетероарильный радикал, выбранный из группы, включающей пиридинил, 2-оксо-1,2-дигидропиридин-3-ил, 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, указанный гетероарил необязательно содержит в качестве заместителей галоген, C1-C6-алкил, C1-C6-гидроксиалкил, C1-C3-алкокси-C1-C3-алкил, C1-C6-алкоксигруппу, или (b) гетероциклический радикал, выбранный из группы, включающей 2-оксотетрагидропиримидин-1-ил, 2,6-диоксотетрагидропиримидин-1-ил, 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил и 2,4-диоксотетрагидропиримидин-1-ил; R2 обозначает водород или C1-C6-алкоксигруппу; R3 обозначает (a) фенил, (b) пиридин, где указанный фенил или указанный пиридин независимо необязательно содержат 1-3 заместителя, выбранных из группы, включающей галоген, (CH2)nNRcRd, или (с) NRaRb, (d) водород, (e) галоген; Ra и Rb вместе с атомом азота, к которому они присоединены, образуют циклический амин, содержащий от 3 до 5 атомов углерода, независимо замещенный группой (CH2)nNReRf; Rc и Rd независимо обозначают водород, SO2R8, где R8 обозначает C1-C6-алкил; Re и Rf независимо обозначают водород, SO2R8, где R8 обозначает C1-C6-алкил; R4 обозначает CF3, CH2CF3 или CR4aR4bR4c, где (i) R4a, R4b и R4c независимо выбраны из группы, включающей C1-C3-алкил, CD3; или (ii) взятые вместе R4a и R4b образуют C2-C4-алкилен и R4c обозначает цианогруппу или C1-C2-фторалкил; R5 в каждом случае независимо обозначает водород, галоген, C1-C6-алкоксигруппу или C1-C6-алкил; n в каждом случае независимо равно 0-3. Также изобретение относится к способу лечения инфицирования вирусом гепатита С, способу ингибирования репликации HCV, применению соединения формулы (I) и фармацевтической композиции на его основе. Технический результат: получены новые гетероциклические соединения формулы (I), обладающие полезной биологической активностью. 6 н. и 18 з.п. ф-лы, 3 табл., 43 пр.
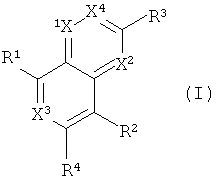
1. Соединение формулы I, в которой:
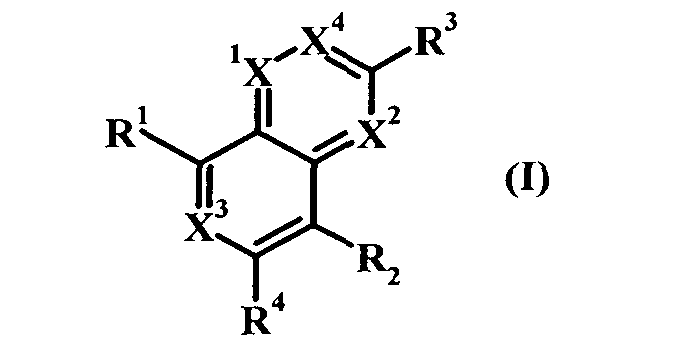
X1 обозначает N и X2, X3 и X4 обозначают CR5; или
X1 и X2 обозначают N и X3 и X4 обозначают CR5; или
X1, X2 и X4 обозначают CR5, и X3 обозначает N; или
X1, X2, X3 и X4 обозначают CR5;
R1 обозначает (a) гетероарильный радикал, выбранный из группы, включающей пиридинил, 2-оксо-1,2-дигидропиридин-3-ил, 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, указанный гетероарил необязательно содержит в качестве заместителей галоген, C1-C6-алкил, C1-C6-гидроксиалкил, C1-C3-алкокси-C1-C3-алкил, C1-C6-алкоксигруппу, или
(b) гетероциклический радикал, выбранный из группы, включающей 2-оксотетрагидропиримидин-1-ил, 2,6-диоксотетрагидропиримидин-1-ил, 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил и 2,4-диоксотетрагидропиримидин-1-ил;
R2 обозначает водород или C1-C6-алкоксигруппу;
R3 обозначает (a) фенил, (b) пиридин, где указанный фенил или указанный пиридин независимо необязательно содержат 1-3 заместителя, выбранных из группы, включающей галоген, (CH2)nNRcRd, или (с) NRaRb, (d) водород, (e) галоген;
Ra и Rb вместе с атомом азота, к которому они присоединены, образуют циклический амин, содержащий от 3 до 5 атомов углерода, независимо замещенный группой (CH2)nNReRf;
Rc и Rd независимо обозначают водород, SO2R8, где R8 обозначает C1-C6-алкил;
Re и Rf независимо обозначают водород, SO2R8, где R8 обозначает C1-C6-алкил;
R4 обозначает CF3, CH2CF3 или CR4aR4bR4c, где
(i) R4a, R4b и R4c независимо выбраны из группы, включающей C1-C3-алкил, CD3; или
(ii) взятые вместе R4a и R4b образуют C2-C4-алкилен и R4c обозначает цианогруппу или C1-C2-фторалкил;
R5 в каждом случае независимо обозначает водород, галоген, C1-C6-алкоксигруппу или C1-C6-алкил;
n в каждом случае независимо равно 0-3;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором X1 обозначает N и X2, X3 и X4 обозначают CR5, и R3 обозначает (a) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, и где n равно 0, или (b) NRaRb.
3. Соединение по п.2, в котором R1 обозначает 2-оксо-1,2-дигидропиридин-3-ил или 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, необязательно содержащий в качестве заместителей галоген, C1-C6-алкил или C1-C6-алкоксигруппу, или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, и R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0 или 1.
4. Соединение по п.3, в котором R4 обозначает CR4aR4bR4c, и (a) R4a, R4b и R4c обозначают CH3, CD3, или R4a и R4b вместе образуют C2-алкилен, и (b) R4c обозначает цианогруппу или C1-C2-фторалкил.
5. Соединение по п.2, в котором R3 обозначает NRaRb и R4 обозначает CR4aR4bR4c, и (a) R4a, R4b и R4c обозначают CH3, CD3, или R4a и R4b вместе образуют C2-алкилен, и (b) R4c обозначает цианогруппу или C1-C2-фторалкил.
6. Соединение по п.5, в котором NRaRb вместе образует циклический амин, замещенный с помощью (CH2)nNReRf, где n равно 0-2; и Re и Rf независимо обозначают водород, SO2R8, где R8 обозначает C1-C6-алкил.
7. Соединение по п.2, в котором R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, и где n равно 0.
8. Соединение по п.2, в котором R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, и где n равно 0, и R1 обозначает 2-оксотетрагидропиримидин-1-ил.
9. Соединение по п.1, в котором X1 и X2 обозначают N и X3 и X4 обозначают CR5, и R3 обозначает (a) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, и где n равно 0, или (b) NRaRb.
10. Соединение по п.9, в котором R1 обозначает 2-оксо-1,2-дигидропиридин-3-ил или 2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил, необязательно содержащий в качестве заместителей галоген, C1-C6-алкил или C1-C6-алкоксигруппу, или 2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил, и R3 обозначает фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, где n равно 0 или 1.
11. Соединение по п.10, в котором R4 обозначает CR4aR4bR4c, и (a) R4a, R4b и R4c обозначают CH3, CD3, или R4a и R4b вместе образуют C2-алкилен, и (b) R4c обозначает цианогруппу или C1-C2-фторалкил.
12. Соединение по п.9, в котором NRaRb вместе обозначает циклический амин, замещенный с помощью (CH2)nNReRf, где n равно 0-2; и Re и Rf независимо обозначают водород, SO2R8, где R8 обозначает C1-C6-алкил.
13. Соединение по п.1, в котором X1, X2 и X4 обозначают CR5 и X3 обозначает N.
14. Соединение по п.13, в котором R3 обозначает (a) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, и где n равно 0, или (b) NRaRb.
15. Соединение по п.1, в котором X1, X2, X3 и X4 обозначают CR5.
16. Соединение по п.15, в котором R3 обозначает (a) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, и где n равно 0, или (b) NRaRb.
17. Соединение по п.1, в котором X1 обозначает N и X2, X3 и X4 обозначают CR5, R1 обозначает 2,6-диоксотетрагидропиримидин-1-ил или 2,4-диоксотетрагидропиримидин-1-ил; и R3 обозначает (a) фенил, замещенный по меньшей мере с помощью (CH2)nNRcRd в положении 4, и где n равно 0, или (b) NRaRb.
18. Соединение по п.1, выбранное из группы, включающей:
N-{4-[6-трет-бутил-2-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[7-трет-бутил-5-(5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-фенил}-метансульфонамид;
N-{1-[7-трет-бутил-5-(2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-пиперидин-4-ил}-метансульфонамид;
N-{(S)-1-[7-трет-бутил-5-(5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-пирролидин-3-илметил}-метансульфонамид;
N-{1-[7-трет-бутил-5-(5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-пиперидин-4-ил}-метансульфонамид;
N-{4-[7-трет-бутил-5-(2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-фенил}-метансульфонамид;
N-{4-[7-трет-бутил-5-(2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-3-хлорфенил}-метансульфонамид;
N-{4-[7-трет-бутил-5-(2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-3-фторфенил}-метансульфонамид;
N-{4-[7-трет-бутил-5-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-3-хлорфенил}-метансульфонамид;
N-{4-[7-трет-бутил-3-метил-5-(2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-фенил}-метансульфонамид;
N-{4-[7-трет-бутил-5-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-хиноксалин-2-ил]-3-фторфенил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид;
N-{1-[6-трет-бутил-2-метокси-8-(5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-пиперидин-4-ил}-метансульфонамид;
N-{4-[6-трет-бутил-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(5-хлор-2-оксо-1,2-дигидропиридин-3-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид;
3-(3-бром-6-трет-бутил-5-метоксихинолин-8-ил)-1Н-пиридин-2-он;
3-(6-трет-бутил-5-метоксихинолин-8-ил)-1Н-пиридин-2-он;
N-{(S)-1-[6-трет-бутил-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-пирролидин-3-илметил}-метансульфонамид;
N-{1-[6-трет-бутил-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-азетидин-3-илметил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид;
N-{1-[6-трет-бутил-4-хлор-5-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-азетидин-3-илметил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(5-фтор-2-оксо-1,2-дигидропиридин-3-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-5-метокси-8-(6-метил-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(3-фторпиридин-4-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-5-метокси-8-(2-оксотетрагидропиримидин-1-ил)-хинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(2,4-диоксотетрагидропиримидин-1-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[8-(2,4-диоксотетрагидропиримидин-1-ил)-5-метокси-6-трифторметилхинолин-3-ил]-фенил}-метансульфонамид;
N-{(S)-1-[6-трет-бутил-8-(2,4-диоксо-3,4-дигидро-2H-пиримидин-1-ил)-5-метоксихинолин-3-ил]-пирролидин-3-илметил}-метансульфонамид;
N-{4-[6-[1,1-ди(метил-d3)этил-2,2,2-d3]-5-метокси-8-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид; и
N-{4-[8-(2,4-диоксотетрагидропиримидин-1-ил)-5-метокси-6-(2,2,2-трифторэтил)-хинолин-3-ил]-фенил}-метансульфонамид;
или его фармацевтически приемлемая соль.
19. Соединение по п.1, выбранное из группы, включающей:
N-{4-[6-трет-бутил-2-метокси-8-(2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-5-метокси-8-(6-метокси-2-оксо-1,2-дигидропиридин-3-ил)-хинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(6-гидроксиметил-2-оксо-1,2-дигидропиридин-3-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(2,4-диоксотетрагидропиримидин-1-ил)-хинолин-3-ил]-фенил}-метансульфонамид;
N-{4-[6-трет-бутил-8-(5-фтор-6-метокси-2-оксо-1,6-дигидропиридин-3-ил)-5-метоксихинолин-3-ил]-фенил}-метансульфонамид; и
N-{(S)-1-[7-трет-бутил-8-метокси-5-(6-метоксиметил-2-оксо-1,2-дигидропиридин-3-ил)-нафталин-2-ил]-пирролидин-3-илметил}-метансульфонамид;
или его фармацевтически приемлемая соль.
20. Способ лечения инфицирования вирусом гепатита C (HCV) путем введения нуждающемуся в нем пациенту соединения по п.1 в терапевтически эффективном количестве.
21. Способ ингибирования репликации HCV в клетке путем введения соединения по п.1.
22. Применение соединения формулы I по п.1 для лечения инфекции HCV.
23. Применение соединения формулы I для приготовления лекарственного средства, предназначенного для лечения инфекции HCV.
24. Фармацевтическая композиция, обладающая активностью в качестве ингибиторов РНК-зависимой РНК вирусной полимеразы, включающая соединение по п.1 в смеси по меньшей мере с одним фармацевтически приемлемым носителем, разбавителем или инертным наполнителем.
| WO 2001085172 A1, 15.11.2001 | |||
| WO 2000009543 A3, 24.02.2000 | |||
| ПРОИЗВОДНЫЕ УРАЦИЛА, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ БОРЬБЫ С СОРНЯКАМИ | 1990 |
|
RU2029472C1 |

