Данное изобретение относится к новому способу получения антифолатных агентов, имеющих в своей структуре фрагмент глутаминовой кислоты, или их солей общей формулы
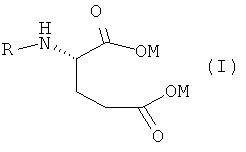
в которой
M обозначает одновалентный или двухвалентный катион, выбранный из группы, состоящей из Na+, K+, 1/2Ca++ или 1/2Mg++; и
R обозначает
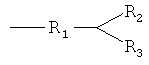
где
R1 представляет собой карбонильную группу; и
R2 и R3 являются одинаковыми или разными и выбраны из:
- линейных или разветвленных, насыщенных или ненасыщенных C1-C20-гетероалкильных групп, которые необязательно могут быть замещены аминогруппами;
- ароматических или алифатических C3-C18-углеводородных колец, которые необязательно могут быть замещены одним или несколькими заместителями, выбранными из группы, состоящей из алкильных, алкенильных, алкинильных, карбокси, гидрокси, аминовых, нитро, тиольных, сульфокси, сульфоновых групп, которые необязательно могут быть замещенными и/или образовывать дополнительные кольца;
- ароматических или алифатических C3-C18-гетероциклов, которые необязательно могут быть замещены одним или несколькими заместителями, выбранными из группы, состоящей из алкильных, алкенильных, алкинильных, карбокси, гидрокси, аминовых, нитро, тиольных, сульфокси, сульфоновых групп, которые необязательно могут быть замещенными и/или образовывать дополнительные кольца;
причем R2 и R3 вместе могут образовывать ароматический или алифатический C3-C18-гетероцикл, который необязательно может быть замещен одним или несколькими заместителями, выбранными из группы, состоящей из алкильных, алкенильных, алкинильных, карбокси, гидрокси, аминовых, нитро, тиольных, сульфокси, сульфоновых групп, которые необязательно могут быть замещенными и/или образовывать дополнительные кольца;
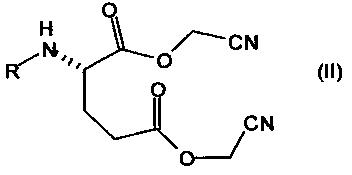
включающему (compressing) проведение реакции соединения следующей формулы
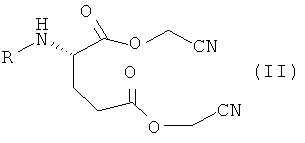
где R имеет такое же значение, как в Формуле (I), с кислотой или основанием в растворе; и
соединения формулы (II) получают в результате проведения реакции глутаминовой кислоты, N-замещенных глутаминовых кислот или их солей с хлорацетонитрилом.
Соединения формулы (I) образуют, в частности, основную цепь ряда известных антифолатных агентов, в которых R представляет собой, например:
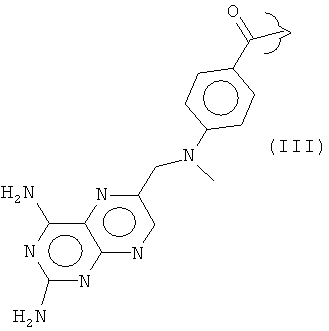
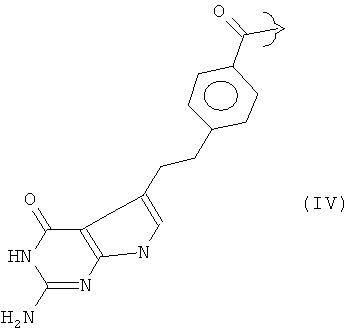
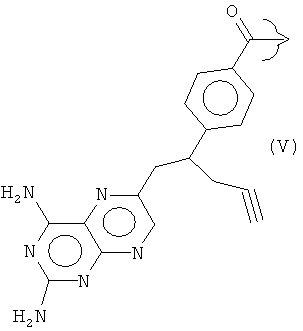
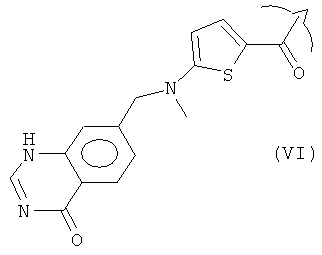
Ингибиторы фолиевой кислоты, содержащие такую гетероциклическую ароматическую основную цепь, принадлежат к группе антиметаболитов, которую в общем называют "антифолатами". Антифолаты, имеющие такую гетероциклическую ароматическую основную цепь, включают метотрексат (R представляет собой III), пеметрексед (R представляет собой IV), пралатрексат (R представляет собой V) и ралтитрексед (R представляет собой VI).
Эти агенты функционируют путем ингибирования действия ключевых ферментов тимидилатсинтазы и дигидрофолатредуктазы и нашли клиническую применимость в качестве противоопухолевых и антинеопластических агентов. Эти агенты ингибируют синтез как пурина, так и пиримидина путем блокирования функций ферментов и вызывают гибель клеток. Они обладают сильным токсичным эффектом на быстро делящиеся клетки, такие как раковые клетки.
Новый способ получения оптически чистых форм активных фармацевтических препаратов может повысить качество лекарственных субстанций.
Соединения Формулы (II) включают прекурсоры вышеуказанных антифолатов, защищенные в виде цианометилового сложного эфира, и могут быть получены путем удаления защитной цианометильной группы глутаматного фрагмента путем использования основного или кислотного агента.
Патент США №4067867 описывает получение хорошо известного сильного антагониста, фолиевой кислоты метотрексата. На последней стадии синтеза метотрексата проводят реакцию диэтил-N-[п-[[(2-амино-3-циано-5-пиризинил)-метил]метиламино]бензоил]глутамата с гуанидинацетатом в ДМФ с образованием диэтилового сложного эфира метотрексата. После очистки было обнаружено, что соединение является полностью рацемическим.
Патент Германии DE 2824011 раскрывает получение п-(N-метил-N-формил)аминобензоил-L-глутаминовой кислоты, которая является прекурсором при синтезе метотрексата. На последней стадии синтеза формильную группу удаляют в основных условиях. Авторы данного изобретения не смогли использовать сильно щелочные условия и высокую температуру с большим временем реакции для удаления формильной группы вследствие риска рацемизации фрагмента глутаминовой кислоты молекулы.
US 4136101 описывает получение диалкил(п-метиламинобензоил)-глутаматов из N-(п-аминобензоил)глутаматов цинка, которые являются промежуточными соединениями в синтезе метотрексата. В патенте указывается, что вследствие кристаллического характера цинковая соль промежуточного соединения является более чистой, чем другие соли металлов. Например, чистота N-(п-аминобензоил)-L-глутамата цинка составляет примерно 80-90% по сравнению с примерно 55-70% для соответствующей динатриевой соли. В данном патенте в качестве растворителя используется низший спирт, такой как метанол, этанол, 2-пропанол и 1-бутанол, и проводится обработка газообразным HCl в безводных условиях для получения соответствующего сложного эфира. Хотя этот подход кажется хорошим, все же существует риск рацемизации глутаматного фрагмента при гидролизе сложноэфирных групп, а использование коррозионноактивного газообразного HCl вместе с дорогими абсолютными спиртами делает этот процесс не пригодным для крупномасштабного синтеза.
Имеются другие патенты и статьи, описывающие получение метотрексата непосредственно из N-(п-аминобензоил)-L-глутаминовой кислоты или ее солей в качестве промежуточного соединения, но в этом случае полученный сырой метотрексат не является чистым и требует многократной кристаллизации. Эти процедуры резко уменьшают выход (например, выход составляет менее 6%, см. J. Am. Chem. Soc. 1949, 71, 1753, или выход составляет 7%, см. патент США №3989703).
В соответствии с литературными источниками, указанными выше, метотрексат сложно получить с хорошим выходом при высокой аналитической и оптической чистоте. Существует потребность в разработке новых путей синтеза для получения антифолатных агентов.
Некоторые соединения с другими алкильными сложными эфирами формулы (II) используются в качестве промежуточных соединений в синтезе соответствующих антифолатов Формулы (I) и требуют удаления защитных групп путем гидролиза при повышенной температуре в сильно основных условиях для получения активных лекарственных субстанций или их солей. Но в таких условиях существует риск рацемизации по альфа-атому углерода фрагмента глутаминовой кислоты и образования продуктов деградации. В случае использования в реакции сочетания при получении антифолатов глутаминовой кислоты или ее металлических солей выходы являются очень низкими, как было указано выше.
Таким образом, целью данного изобретения является описание нового способа получения антифолатных агентов Формулы (I) из соединения Формулы (II) в очень мягких условиях реакции с хорошим выходом при высокой аналитической и оптической чистоте.
Неожиданно было обнаружено, что цианометильная группа может быть успешно использована для защиты карбоксильной группы соединения глутаминовой кислоты Формулы (II), являющегося промежуточным соединением в синтезе антифолатов.
Цианометиловые сложные эфиры могут быть удалены в очень мягких условиях реакции и даже используются в качестве уходящей (living) группы в реакции сочетания неприродных аминокислот с динуклеотидами при получении ошибочно ацилированных транспортных PHK (Arslan et al., J. Am. Chem. Soc. 1997, 119, 10877).
Для получения разных алкильных сложных эфиров формулы (II) из металлической соли глутаминовой кислоты, например из цинковой соли, было необходимо проводить реакцию в абсолютном спирте, который является дорогим, и использовать в качестве источника кислоты очень коррозионноактивный газообразный HCl, что неудобно для крупномасштабного синтеза.
Другим объектом данного изобретения является то, что цианометиловый эфир соединений Формулы (II) или их промежуточные соединения, содержащие фрагмент глутаминовой кислоты, могут быть легко получены, исходя из металлической соли глутаминовой кислоты или из ее N-замещенных производных путем проведения реакции с хлорацетонитрилом в полярном растворителе.
Металлические соли глутаминовой кислоты, используемые в качестве промежуточных соединений в синтезе соединений (I), не могут быть получены в форме чистого соединения, например, чистота N-(п-аминобензоил)-L-глутамата цинка составляет примерно 80-90% по сравнению с примерно 55-70% для соответствующей динатриевой соли при синтезе метотрексата.
Когда цианометиловый сложный эфир N-(п-аминобензоил)-L-глутамата получают и выделяют в соответствии с простой процедурой по данному изобретению, чистота промежуточного соединения составляет более 98% (1Н ЯМР). Реакция сочетания с чистыми промежуточными соединениями дает чистые соединения Формулы (II), в случае метотрексата - с высоким выходом.
Еще одним предметом данного изобретения является то, что цианометильные группы Формулы (II) могут быть гидролизованы в очень мягких основных или кислотных условиях с получением желательных активных веществ или их солей с высоким выходом при высокой аналитической и оптической чистоте.
Цианометиловый сложный эфир выступает в роли защитной группы карбоксильных функциональностей Формулы (II), но легко гидролизуется при более умеренных значениях pH, чем ранее использовавшиеся алкильные сложные эфиры. Они также позволяют получить чистые формы соединений (II), которые дают чистые антифолатные агенты Формулы (I).
В варианте исполнения изобретения, прекурсоры антифолатных агентов, содержащие фрагмент глутаминовой кислоты, защищают в виде дицианометиловых сложных эфиров, реагирующих с хлорацетонитрилом в полярном растворителе, и затем вступающие в реакции сочетания с другими пригодными промежуточными соединениями с получением защищенных антифолатных агентов в виде соединений Формулы (II).
Соединения формулы (II) образуются в качестве промежуточных соединений при синтезе соответствующих антифолатных агентов Формулы (I). На следующей стадии защитные группы должны быть удалены для образования желательных активных соединений Формулы (I).
Предпочтительно, R2 и R3 оба вместе образуют фенильное или тиофеновое кольцо, замещенное алкильной группой или алкильной группой, содержащей гетероатомы, и алкильные группы дополнительно замещены бициклическими или гетероциклическими ароматическими кольцевыми системами, содержащими такие структуры, как пурины или пиримидины.
R1 предпочтительно обозначает карбонил.
Известно, что соединения такой структуры проявляют биологическую активность и потому представляют интерес для синтеза лекарственных субстанций для различных фармацевтических препаратов.
В варианте исполнения изобретения, соединение Формулы (I) представляет собой соединение, проявляющее антифолатную активность, и используется для лечения различных типов рака. Предпочтительно, его выбирают из группы, состоящей из метотрексата, пеметрекседа, пралатрексата и ралтитрекседа, причем метотрексат и пеметрексед являются особенно предпочтительными.
Такие соединения являются противораковыми средствами и потому представляют высокий коммерческий интерес.
В дополнительном варианте исполнения реакции, хлорацетонитрил, являющийся недорогим и коммерчески доступным материалом, используют в реакции эстерификации при получении соединений Формулы (II) или их прекурсоров.
Реакцию эстерификации проводят в полярном растворителе, более предпочтительно в смешивающемся с водой полярном растворителе, особенно растворителе, выбранном из группы, состоящей из диметилформамида, диметилацетамида, диметилсульфоксида, кетона, такого как ацетон или метилизобутилкетон, и ацетонитрила, или их смесей.
Было продемонстрировано, что для такого вида реакции, в частности, ДМФ дает наилучшие результаты по показателю выхода, а также растворимости исходных материалов или их солей. После образования цианометилового эфира Формулы (II) или его промежуточных соединений к реакционной смеси может быть добавлена вода и соответствующий сложный эфир выпадает в осадок и выделяется фильтрацией.
Осаждение является особенно предпочтительным способом выделения образовавшихся цианометиловых сложных эфиров Формулы (II) или их промежуточных соединений, поскольку его можно вызвать простым перемешиванием смеси при комнатной температуре без необходимости использования более сложной методики очистки, такой как хроматография на колонке.
В дополнительном варианте исполнения изобретения, проводят реакцию прекурсоров соединений глутаминовой кислоты Формулы (II) с хлорацетонитрилом при температуре от 20 до 120°C, предпочтительно от 50 до 80°C.
В другом варианте исполнения изобретения, цианометиловый сложный эфир глутаминовой кислоты или его N-замещенные производные вводят в реакцию сочетания с другими промежуточными соединениями, содержащими гетероциклические кольца с галоидалкильными или карбоксильными группами с получением соединения Формулы (II).
Реакция сочетания проводится в воде или в органическом растворителе, особенно полярные растворители обеспечивают наилучшие результаты по показателям выхода, а также растворимости всех используемых агентов. При этом было показано, что диметилформамид, диметилацетамид и вода являются наиболее пригодными растворителями при температуре от 0 до 100°C, предпочтительно от 50 до 75°C.
При использовании воды в качестве растворителя соединения Формулы (II) непосредственно выпадают в осадок из реакционной смеси в интервале pH от примерно 1 до примерно 6, предпочтительно в интервале значений pH от примерно 2 до примерно 5, особенно при pH примерно 4,0 при температуре от 0 до 60°C, предпочтительно от 0 до 25°C.
Было обнаружено, что в вышеуказанных температурных интервалах реакции могут быть проведены за период времени от 0,5 до 4,5 часов, предпочтительно 1 час, с получением хорошего выхода.
В дополнительном варианте исполнения изобретения, проводится реакция соединения Формулы (II) с гидроксидом металла, гидроксидами или карбонатами щелочноземельных металлов, с получением соединения Формулы (I) или их солей.
Реакция гидролиза проводится в водно-спиртовой смеси в присутствии от 1 до 3 эквивалентов, особенно 2 эквивалентов гидроксида металла, гидроксидов или карбонатов щелочноземельных металлов. Полярные растворители дают наилучшие результаты по показателям выхода, а также растворимости всех используемых агентов. Гидролиз цианометильных групп осуществляют при температуре от 0 до 100°C, предпочтительно от 20 до 25°C.
Было обнаружено, что в указанных выше интервалах времени реакция гидролиза протекает практически полностью, что приводит к высоким выходам при высокой аналитической и оптической чистоте соединения Формулы (I) или его фармацевтически пригодных солей.
Солевую форму соединений Формулы (I) получают путем суспендирования соединения Формулы (I) в воде, доведения значения pH до примерно 10 с помощью соответствующего гидроксида металла и затем прибавления их к кетону, такому как ацетон, для осаждения.
Следует понимать, что вышеописанные признаки и признаки, описанные далее, могут быть использованы не только в их описанных комбинациях, но также в других комбинациях, или по отдельности, без выхода за пределы объема изобретения.
Изобретение далее будет проиллюстрировано с помощью примеров. Эти примеры не должны каким-либо образом ограничивать объем изобретения.
ПРИМЕР 1
Получение N-[4-(метиламино)бензоил]-L-глутаминовой кислоты динатриевой соли из N-[4-(метиламино)бензоил]-L-глутаминовой кислоты цинковой соли
В колбу Эрленмейера на 1 л, оснащенную магнитной мешалкой, загружали 500 мл воды и 50 г (0,15 моль) N-[4-(метиламино)бензоил]-L-глутаминовой кислоты цинковой соли (чистота ок. 85%) при комнатной температуре. Значение pH смеси доводили до 8,0 с помощью 0,2М Na2CO3. Осадок оксида цинка выделяли фильтрацией. Значение pH раствора доводили до 6,2 с помощью разбавленного HCl. Растворитель испаряли при пониженном давлении и остаток сушили под вакуумом при 50°C в течение 5-6 часов, получая 36,1 г (0,116 моль) N-[4-(метиламино)бензоил]-L-глутаминовой кислоты динатриевой соли с выходом 90% в виде красной пены.
ПРИМЕР 2
Получение дицианометил-N-[4-(метиламино)бензоил]-L-глутаминовой кислоты из N-[4-(метиламино)бензоил]-L-глутаминовой кислоты динатриевой соли
Колбу на 1 л оснащали магнитной мешалкой, термометром и холодильником. В колбу загружали 250 мл ДМФ, 30 г (0,096 моль) N-[4-(метиламино)бензоил]-L-глутаминовой кислоты динатриевой соли и 20 мл (0,31 моль) хлорацетонитрила при комнатной температуре. Суспензию перемешивали при 60°C в течение 4-5 часов. Раствор охлаждали до комнатной температуры и прибавляли 250 мл воды. Смесь перемешивали в течение 15-20 минут, в результате чего образовывался белый осадок. Твердое вещество выделяли фильтрацией и промывали 20 мл воды. Твердое вещество сушили под вакуумом при 50°C в течение 3 часов и получали дицианометил-N-[4-(метиламино)-бензоил]-L-глутаминовую кислоту 27 г (0,078 моль) в виде белого твердого вещества с выходом 81%. По данным 1Н ЯМР продукт был очень чистым.
1Н ЯМР (ДМСО) δ 2,07 (м, 2H), 2,55 (т, 2H), 2,70 (д, 3H), 4,45 (м, 1H), 4,91 (с, 2H), 4,97 (с, 2H), 6,23 (кв, 1H), 6,53 (д, 2H), 7,65 (д, 2H), 8,43 (д, 1H).
ПРИМЕР 3
Получение дицианометил-N-[4[1(2,4-диамино-6-птеридинил)метил]-метиламино]бензоил]-L-глутамата (метотрексата дицианометиловый сложный эфир)
Колбу на 1 л оснащали магнитной мешалкой, термометром и холодильником. В колбу загружали 143 мл воды и 10 г (0,029 моль) 2,4-диамино-6-(бромметил)птеридина гидробромида при комнатной температуре. К этой суспензии прибавляли 13 г (0,037 моль) дицианометил-N-[4-(метиламино)-бензоил]-L-глутаминовой кислоты при комнатной температуре. Значение pH смеси было равно 2,40. Смесь нагревали до 58-62°C и перемешивали при этой температуре в течение 1 часа. Ход реакции отслеживали методом ТСХ (EtOAc:MeOH, 4:1) до ее завершения. Смесь охладили до комнатной температуры и твердое вещество выделили фильтрацией. Фильтровальную лепешку твердого вещества промывали 15 мл воды и сушили под вакуумом при 50°C в течение 5-6 часов, получая 13,2 г (0,029 моль) метотрексата дицианометилового сложного эфира в виде твердого вещества желтого цвета с выходом 87%. По данным 1H ЯМР продукт был очень чистым.
1H ЯМР (ДМСО) δ 2,08 (м, 2H), 2,55 (т, 2H), 3,23 (с, 3H), 4,46 (м, 1H), 4,82 (д, 2H), 4,94 (с, 2H), 4,98 (с, 2H), 6,82 (д, 2H), 7,35 (м, 2H), 7,72 (д, 2H), 8,30 (с, 1H), 8,52 (д, 2H), 8,64 (с, 1H).
ПРИМЕР 3
Получение N-[4[[(2,4-диамино-6-птеридинил)метил]метиламино]-бензоил]-L-глутаминовой кислоты динатриевой соли из метотрексата дицианометилового сложного эфира
Колбу на 1 л оснащали магнитной мешалкой, термометром и холодильником. В колбу загружали 160 мл метанола, 80 мл воды и 1,75 г (0,03 моль) КОН. Раствор перемешивали при комнатной температуре (rt) в течение 5-10 минут. К этому раствору прибавляли 10 г (0,019 моль) метотрексата дицианометилового сложного эфира при комнатной температуре. Раствор перемешивали в течение 20 мин при комнатной температуре. Ход реакции отслеживали методом ТСХ (EtOAc:MeOH 4:1) до ее завершения. Раствор концентрировали при пониженном давлении. Значение pH раствора доводили до 4,2 разбавленной HCl и желтое вещество выпадало в осадок. Сырой метотрексат выделяли фильтрацией и промывали водой. Влажную фильтровальную лепешку суспендировали в 75 мл воды и pH доводили до 10 с помощью 2N NaOH, получая прозрачный раствор. Прибавляли к раствору 2 г активированного угля, перемешивали в течение 5 мин и фильтровали. Раствор, содержащий метотрексата динатриевую соль, прибавляли при комнатной температуре к 500 мл ацетона при перемешивании. Образовывалось желтоватое твердое вещество, которое выделяли фильтрацией. Твердое вещество сушили под вакуумом при 50°C в течение 8-10 часов, получая 8,3 г (0,017 моль, выход 89%) метотрексата динатрия с чистотой выше 99,8% по результатам анализа методом ВЭЖХ.
1H ЯМР (D2O) δ 1,88 (м, 1H), 1,98 (м, 1H), 2,17 (м, 2H), 2,89 (с, 3H), 4,14 (м, 1H), 4,37 (с, 2H), 6,53 (д, 2H), 7,46 (д, 2H), 8,27 (с, 1H).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 5-ЗАМЕЩЕННЫХ ПИРРОЛО (2,3-α)ПИРИМИДИНОВ | 1993 |
|
RU2127274C1 |
| ПРОИЗВОДНОЕ БЕНЗАЗЕПИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОИЗВОДНОЕ ДИФТОРБЕНЗАЗЕПИНА И ПРОИЗВОДНОЕ (ЗАМЕЩЕННОГО) АМИНОБЕНЗОИЛДИФТОРБЕНЗАЗЕПИНА | 1994 |
|
RU2137760C1 |
| ОДНОСОСУДНЫЙ СПОСОБ ПОЛУЧЕНИЯ ПЕМЕТРЕКСЕДА ДИНАТРИЯ | 2012 |
|
RU2577251C2 |
| ПРОИЗВОДНЫЕ ИНДОЛИЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ТЕСТОСТЕРОН 5α-РЕДУКТАЗЫ | 1992 |
|
RU2120942C1 |
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ БЕНЗАЗЕПИНА, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ДЛЯ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2129123C1 |
| ПРОИЗВОДНЫЕ БИФЕНИЛАМИДИНА | 1998 |
|
RU2197478C2 |
| НОВОЕ ПРОИЗВОДНОЕ БЕНЗОАЗЕПИНА И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2642783C2 |
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИНА, ПРИГОДНЫЕ ДЛЯ ИСПОЛЬЗОВАНИЯ В КАЧЕСТВЕ АНТАГОНИСТОВ ВАЗОПРЕССИНА | 2008 |
|
RU2471784C2 |
| ЗАМЕЩЕННОЕ ПРОПАНАМИДНОЕ ПРОИЗВОДНОЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОЕ ПРОИЗВОДНОЕ | 2006 |
|
RU2394560C2 |
| ПРОИЗВОДНОЕ 4-БЕНЗОИЛИЗОКСАЗОЛА ИЛИ ЕГО ПРИЕМЛЕМАЯ ДЛЯ СЕЛЬСКОГО ХОЗЯЙСТВА СОЛЬ, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ БОРЬБЫ С СОРНЯКАМИ | 1994 |
|
RU2124505C1 |
Изобретение относится к новому способу получения антифолатных агентов, имеющих в своей структуре фрагмент глутаминовой кислоты, или их солей общей формулы (I). Способ включает проведение реакции соединения формулы (II) с кислотой или основанием в растворе. При этом соединения формулы (II) получают в результате реакции N-замещенных глутаминовых кислот или их солей с хлорацетонитрилом. В формуле (I) М обозначает одновалентный или двухвалентный катион, выбранный из группы, состоящей из Na+, К+, 1/2Са++ или 1/2Mg++; R обозначает группу 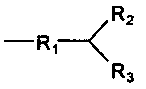 . В указанной группе R1 представляет собой карбонильную группу; R2 и R3 являются одинаковыми или разными и выбраны из линейных или разветвленных, насыщенных или ненасыщенных С1-С20-гетероалкильных групп, которые необязательно могут быть замещены аминогруппами; ароматических или алифатических С3-С18-углеводородных колец, которые необязательно могут быть замещены одним или несколькими заместителями, выбранными из группы, состоящей из алкильных, алкенильных, алкинильных, карбокси, гидрокси, аминовых, нитро, тиольных, сульфокси, сульфоновых групп; ароматических или алифатических С3-С18-гетероциклов, которые необязательно могут быть замещены одним или несколькими заместителями, выбранными из группы, состоящей из алкильных, алкенильных, алкинильных, карбокси, гидрокси, аминовых, нитро, тиольных, сульфокси, сульфоновых групп; причем R2 и R3 оба вместе могут образовывать фенильное или тиофеновое кольцо, которое может быть замещенным алкильной группой или алкильной группой, содержащей гетероатомы, и алкильные группы могут быть дополнительно замещены бициклическими или гетероциклическими кольцевыми системами, содержащими такие структуры, как пурины или пиримидины. В формуле (II) R имеет такое же значение, как в формуле (I). Предлагаемый способ позволяет получать антифолатные агенты формулы (I) с хорошими выходами при высокой аналитической и оптической чистоте. 13 з.п. ф-лы, 4 пр.
. В указанной группе R1 представляет собой карбонильную группу; R2 и R3 являются одинаковыми или разными и выбраны из линейных или разветвленных, насыщенных или ненасыщенных С1-С20-гетероалкильных групп, которые необязательно могут быть замещены аминогруппами; ароматических или алифатических С3-С18-углеводородных колец, которые необязательно могут быть замещены одним или несколькими заместителями, выбранными из группы, состоящей из алкильных, алкенильных, алкинильных, карбокси, гидрокси, аминовых, нитро, тиольных, сульфокси, сульфоновых групп; ароматических или алифатических С3-С18-гетероциклов, которые необязательно могут быть замещены одним или несколькими заместителями, выбранными из группы, состоящей из алкильных, алкенильных, алкинильных, карбокси, гидрокси, аминовых, нитро, тиольных, сульфокси, сульфоновых групп; причем R2 и R3 оба вместе могут образовывать фенильное или тиофеновое кольцо, которое может быть замещенным алкильной группой или алкильной группой, содержащей гетероатомы, и алкильные группы могут быть дополнительно замещены бициклическими или гетероциклическими кольцевыми системами, содержащими такие структуры, как пурины или пиримидины. В формуле (II) R имеет такое же значение, как в формуле (I). Предлагаемый способ позволяет получать антифолатные агенты формулы (I) с хорошими выходами при высокой аналитической и оптической чистоте. 13 з.п. ф-лы, 4 пр.
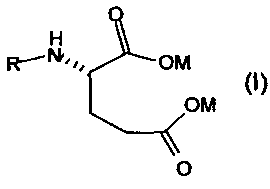
1. Новый способ получения антифолатных агентов, имеющих в своей структуре фрагмент глутаминовой кислоты, или их солей общей формулы
,
в которой
М обозначает одновалентный или двухвалентный катион, выбранный из группы, состоящей из Na+, К+, 1/2Са++ или 1/2Mg++; и
R обозначает
,
где
R1 представляет собой карбонильную группу; и
R2 и R3 являются одинаковыми или разными и выбраны из:
- линейных или разветвленных, насыщенных или ненасыщенных С1-С20-гетероалкильных групп, которые необязательно могут быть замещены аминогруппами;
- ароматических или алифатических С3-С18-углеводородных колец, которые необязательно могут быть замещены одним или несколькими заместителями, выбранными из группы, состоящей из алкильных, алкенильных, алкинильных, карбокси, гидрокси, аминовых, нитро, тиольных, сульфокси, сульфоновых групп;
- ароматических или алифатических С3-С18-гетероциклов, которые необязательно могут быть замещены одним или несколькими заместителями, выбранными из группы, состоящей из алкильных, алкенильных, алкинильных, карбокси, гидрокси, аминовых, нитро, тиольных, сульфокси, сульфоновых групп;
причем R2 и R3 оба вместе могут образовывать фенильное или тиофеновое кольцо, которое может быть замещенным алкильной группой или алкильной группой, содержащей гетероатомы, и алкильные группы могут быть дополнительно замещены бициклическими или гетероциклическими кольцевыми системами, содержащими такие структуры, как пурины или пиримидины;
включающий проведение реакции соединения следующей формулы
,
где R имеет такое же значение, как в формуле (I), с кислотой или основанием в растворе; и
соединения формулы (II) получают в результате проведения реакции N-замещенных глутаминовых кислот или их солей с хлорацетонитрилом.
2. Способ по п.1, отличающийся тем, что соединение формулы (I) представляет собой соединение, проявляющее антифолатную активность, и используется для лечения различных типов рака.
3. Способ по п.2, отличающийся тем, что соединение формулы (I) выбирают из группы, состоящей из метотрексата, пеметрекседа, пралатрексата и ралтитрекседа.
4. Способ по п.3, отличающийся тем, что соединение формулы (I) выбирают из группы, состоящей из метотрексата и пеметрекседа.
5. Способ по п.4, отличающийся тем, что цианометиловый сложный эфир соединений формулы (II) или их промежуточных соединений, содержащих фрагмент глутаминовой кислоты, легко получают из металлической соли N-замещенных производных глутаминовой кислоты путем проведения реакции с хлорацетонитрилом.
6. Способ по п.5, отличающийся тем, что реакцию эстерификации проводят в полярном растворителе, более предпочтительно в смешивающемся с водой полярном растворителе.
7. Способ по п.6, отличающийся тем, что растворитель выбирают из группы, состоящей из диметилформамида, диметилацетамида, диметилсульфоксида, кетона, такого как ацетон или метилизобутилкетон, и ацетонитрила или их смесей.
8. Способ по п.5, отличающийся тем, что цианометиловый сложный эфир N-замещенных производных глутаминовой кислоты вводят в реакцию сочетания с другими промежуточными соединениями, содержащими гетероциклические кольца, содержащие галоидалкильные или карбоксильные группы, с получением соединения формулы (II).
9. Способ по п.8, отличающийся тем, что реакцию сочетания, приводящую к получению соединения формулы (II), проводят в воде или в органическом растворителе, таком как диметилформамид и диметилацетамид.
10. Способ по п.9, отличающийся тем, что реакцию сочетания проводят при температуре от 0 до 100°C, предпочтительно от 50 до 75°C.
11. Способ по п.9, отличающийся тем, что проводят реакцию соединения формулы (II) с гидроксидом металла, гидроксидами или карбонатами щелочноземельных металлов с получением соединений формулы (I) или их солей.
12. Способ по п.11, отличающийся тем, что реакцию гидролиза проводят в водно-спиртовой смеси в присутствии 1-3 эквивалентов, особенно 2 эквивалентов гидроксида металла, гидроксидов или карбонатов щелочноземельных металлов.
13. Способ по п.12, отличающийся тем, что гидролиз цианометильных групп осуществляют при температуре от 0 до 100°C, предпочтительно от 20 до 25°C.
14. Способ по любому из пп.1-13, отличающийся тем, что солевую форму соединений формулы (I) получают путем суспендирования соединений формулы (I) в воде, доведения pH до примерно 10 с помощью соответствующего гидроксида металла и затем прибавления их к кетону, такому как ацетон, для образования осадка.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |

