Изобретение относится к новому производному нуклеиновой кислоты, а более конкретно к 3'-замещенному производному нуклеозида или его фармацевтически приемлемой соли, которое(ая) проявляет высокую противоопухолевую активность и полезно(а) как лекарственное средство, такое, как противоопухолевое средство, и к применению такого соединения в медицине.
Пиримидиновые соединения, такие, как 5-фтороурацил, тегафур, UFT, доксифлуридин, кармофур, цитарабин и эноцитабин, уже известны как противоопухолевые средства, являющиеся антиметаболитами нуклеиновых кислот.
С другой стороны, из Tetrahedrom 47, 1727-1736 (1991) известен 1-(2-O-(трет-бутилдиметилсилил)-3-C-этинил- β- D-рибофуранозил)тимин как пиримидиновый или пуриновый нуклеотид, имеющий алкинильную группу в положении 3 сахарной составляющей. Однако не описано применение этого соединения в качестве лекарственного средства, в частности, противоопухолевого действия. Там же описаны также 1-(3-C-этинил -β- D-ксилофуравнозил)тимин и 1-(2-O-(трет-бутилдиметилсилил)-3-C-этинил -β- D-ксилофуранозил)тимин. Сахарные составляющие этих двух соединений состоят из ксилозы и отличаются по конфигурации в положении 3 от рибозы в соединениях по настоящему изобретению. Кроме того, в указанной работе ничего не сказано о противоопухолевом действии этих соединений. Соединения, имеющие алкильную группу в положении 3 сахарной составляющей, описаны в публикациях N 11908/1970 и N 4376/1971 патентов Японии. Но их противоопухолевое действие крайне слабо и потому они при данных обстоятельствах не имеют никакой ценности как противоопухолевые средства.
Таким образом, задачей настоящего изобретения является создание нового производного нуклеиновой кислоты, проявляющего высокую противоопухолевую активность и полезного в качестве лекарственного вещества, и лекарственного средства, содержащего такое соединение.
Ввиду вышесказанного создатели настоящего изобретения провели обширные исследования. В результате было обнаружено, что производное нуклеиновой кислоты, в положение 3 сахарной составляющей которого был введен заместитель, проявляет высокую противоопухолевую активность и полезно как противоопухолевое средство, и это привело к созданию настоящего изобретения.
В соответствии с настоящим изобретением предлагается 3'-замещенное производное нуклеозида, представленное следующей общей формулой (1):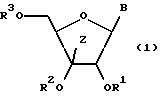
где B представляет собой основание нуклеиновой кислоты, которое может иметь заместитель, Z представляет собой низшую алкинильную или низшую алкенильную группу, которые могут быть замещены группой, представленной общей формулой (2):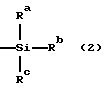
в которой Ra, Rb, и Rc могут быть одинаковыми или различными и каждый представляет низшую алкильную или фенильную группу, или оксиранильную группу, которые могут быть замещены, по крайней мере, одной низшей алкильной группой, R1 и R2, по отдельности, представляют собой атом водорода или образующий сложный эфир остаток, способный легко отщепляться в живом организме, и R3 представляет собой атом водорода, остаток моно- или полифосфоновой кислоты или образующий сложный эфир остаток, способный легко отщепляться в живом организме, при условии, что сахарная составляющая является рибозой, или его фармацевтически приемлемая соль.
Соединение по настоящему изобретению, представленное общей формулой (1), проявляет высокую противоопухолевую активность и полезно как лекарственное средство, такое, как средство для лечения различных опухолей.
Поэтому в соответствии с настоящим изобретением предлагается также лечебная композиция, содержащая соединение общей формулы (1) или его фармацевтически приемлемую соль и фармацевтический носитель.
В соответствии с настоящим изобретением предлагается также лекарственное средство, в частности противоопухолевое средство, содержащее соединение общей формулы (1) или его фармацевтически приемлемую соль в качестве активного компонента.
Далее, в соответствии с настоящим изобретением предлагается применение соединения общей формулы (1) или его фармацевтически приемлемой соли в медицине.
Кроме того, в соответствии с настоящим изобретением предлагается способ лечения или профилактики рака у млекопитающего, включающий введение млекопитающему эффективного количества соединения общей формулы (1) или его фармацевтически приемлемой соли.
Кроме того, в соответствии с настоящим изобретением предлагается способ получения соединения общей формулы (1) или его фармацевтически приемлемой соли.
Примеры остатка нуклеиновой кислоты, обозначенного символом B в общей формуле (1), включают пиримидиновые основания, такие, как цитозин, тимин и урацил, и пуриновые основания, такие, как аденин и гуанин.
Примеры заместителя, которым может быть замещено основание нуклеиновой кислоты, включают атомы галогена, низшие алкильные группы, ацильные группы, такие, как алифатические или ароматические ацильные группы, и замещенные оксикарбонильные группы, такие, как низшие алкоксикарбонильные, низшие алкенилоксикарбонильные или аралкилоксикарбонильные группы.
Примеры атомов галогена включают атомы фтора, хлора, брома и иода.
Примеры низших алкильных групп включают неразветвленные или разветвленные алкильные группы, имеющие 1-6 атомов углерода, такие, как метильная, этильная, н-пропильная, изопропильная, н-бутильная, изобутильная, втор-бутильная, трет-бутильная, пентильная и гексильная группы.
Примеры алифатических ацильных групп включают неразветвленные или разветвленные ацильные группы, имеющие 1-6 углеродных атомов, такие, как формильная, ацетильная, пропионильная, бутирильная, изобутирильная, пентаноильная и гексаноильная группы. Примеры ароматических ацильных групп включают бензоил, α- нафтоил и β- нафтоил. Эти группы могут также иметь низшую алкильную группу, низшую алкоксигруппу, атом галогена, нитрогруппу или подобные в качестве заместителя.
В качестве примеров низшей алкильной группы и атома галогена можно назвать те же самые группы и атомы, которые указаны выше.
Примеры низшей алкоксигруппы включают неразветвленные и разветвленные алкоксигруппы, имеющие 1-6 углеродных атомов, такие, как метокси-, этокси-, н-пропокси, изопропокси, н-бутокси, изобутокси-, втор-бутокси-, трет-бутокси-, пентил-окси-, и гексилоксигруппа.
Примеры низших алкоксикарбонильных групп включают неразветвленные и разветвленные алкоксикарбонильные группы, имеющие 2-7 углеродных атомов, такие, как метоксикарбонильная, этоксикарбонильная, н-пропоксикарбонильная, изопропоксикарбонильная, н-бутоксикарбонильная, изобутоксикарбонильная, вторбутоксикарбонильная, трет-бутоксикарбонильная, пентилоксикарбонильная и гексилоксикарбонильная группы.
Примеры низших алкенилоксикарбонильных групп включают неразветвленные или разветвленные алкенилоксикарбонильные группы, имеющие 3-7 углеродных атомов, такие, как винилоксикарбонильная, аллилоксикарбонильная, изопропенилоксикарбонильная, 1-бутенилоксикарбонильная и 2-бутенилоксикарбонильная группы.
Примеры аралкилоксикарбонильных групп включают аралкилоксикарбонильные группы, имеющие 8-12 углеродных атомов, такие, как бензилоксикарбонильная, фенетилоксикарбонильная, α- нафтилметилоксикарбонильная и β- нафтилметилоксикарбонильная группы. Эти группы могут иметь низшую алкильную группу, низшую алкоксигруппу, атом галогена, нитрогруппу или подобные в качестве заместителя.
Примеры низшей алкинильной группы, представленной символом Z, включают алкинильные группы, имеющие 2-6 углеродных атомов, такие, как этинильная, пропинильная (1-пропинил, 2-пропинил), бутинильная (1-бутинил, 2-бутинил и т. д.), пентинильная (1-пентинил и т.д.) и гексинильная (1-гексинил и т.д.), а примеры низшей алкенильной группы включают алкенильные группы, имеющие 2-6 углеродных атомов, такие, как этенильная, пропенильная (1-пропенил, 2- пропенил, изопропенил), бутенильная (1-бутенил, 2-бутенил, 3-бутенил и т.д. ), пентенильная (1-пентенил и т. д.), и гексенильная (1-гексенил и т.д.) группы. Примеры оксиранильной группы, имеющей по крайней мере одну низшую алкильную группу, включают оксиранильные группы, замещенные одной или двумя низшими алкильными группами, такие, как 3-метилоксиранильная, 3-этилоксиранильная, 3-пропилоксиранильная, 3-изопропилоксиранильная, 3-бутилоксиранильная, 3-трет-бутилоксиранильная, З,3-диметилоксиранильная и 3,3-диэтилоксиранильная группы.
Примеры группы, представленной общей формулой (2), включают силильные группы, замещенные тремя неразветвленными или разветвленными алкильными группами, имеющими 1-6 углеродных атомов, такие, как триметилсилильная, триэтилсилильная, трипропилсилильная, триизопропилсилильная, три-трет-бутилсилильная, 3-гексилсилильная, диметилэтилсилильная, диметилизопропилсилильная, диэтилизопрописилильная, диизопропилмиетилсилильная, ди-трет-бутилметилсилильная и трет-бутилдиметилсилильная группы и дифенилметилсилильная, диметилфенилсилильная, трет-бутилдифенилсилильная и трифенилсилильная группы.
Образующие сложный эфир остатки, способные легко отщепляться в живом организме, которые представлены символами R1, R2 и R3, означают нетоксичные сложноэфирные остатки, легко расщепляющиеся в крови и тканях млекопитающих, включая человека, с высвобождением соответствующих им гидроксильных соединений (а именно соединений, в которых R1, R2 и/или R3 превращаются в атом водорода). Могут быть использованы любые эфирообразующие остатки, которые, как это широко известно, защищают гидроксильные группы нуклеозида и образуют сложный эфир. Их примеры включают ацильные группы, такие, как алифатические ацильные группы, которые могут иметь заместитель, и ароматические ацильные группы, которые могут иметь заместитель, низшие алкилкарбамоильные группы и аминокислотные остатки.
Примеры алифатических или ароматических ацильных групп, которые может иметь заместитель, включают низшие алканоильные, арилкарбонильные, гетероциклические карбонильные, арилоксикарбонильные, низшие алкоксикарбонильные и ацилоксиацильные группы.
Примеры низших алканоильных групп включают алканоильные группы, которые могут иметь атом галогена, низшую алкоксигруппу или тому подобное в качестве, по крайней мере, одного заместителя и имеют 1-6 углеродных атомов, такие, как формильная, ацетильная, пропионильная, бутирильная, изобутирильная, пентаноильная, гексаноильная, хлорацетильная, дихлорацетильная, трихлорацетильная, трифторацетильная, метоксиацетильная и этоксиацетильная группы.
Примеры арилкарбонильных групп включают бензоильные и нафтилкарбонильные группы, которые могут иметь низшую алкильную группу, низшую алкоксигруппу, атом галогена, карбоксильную группу, нитрогруппу, цианогруппу и тому подобное в качестве, по крайней мере, одного заместителя, такие, как бензоильная, α- нафтилкарбонильная, β- нафтилкарбонильная, 2-метилбензоильная, 3-метилбензоильная, 4-метилбензоильная, 2,4-диметилбензоильная, 4-этилбензоильная, 2-метоксибензоильная, 3-метоксибензоильная, 4-метоксибензоильная, 2,4-диметоксибензоильная, 4-этоксибензоильная, 2-метокси-4-этоксибензоильная, 4-пропоксибензоильная, 2-хлорбензоильная, 3-хлорбензоильная, 4-хлорбензоильная, 2,3-дихлорбензоильная, 2-бромбензоильная, 4-фторбензоильная, 2-карбоксибензоильная, 3-карбоксибензоильная, 4-карбоксибензоильная, 2-цианбензоильная, 4-цианбензоильная, 2-нитробензоильная, 4-нитробензоильная и 2,4-динитробензоильная группы.
Примеры гетероциклических карбонильных групп включают 2-фуранилкарбонильную, 4-тиазолилкарбонильную, 2-хинолилкарбонильную, 2-пиразинилкарбонильную, 2-пиридилкарбонильную, 3-пиридилкарбонильную и 4-пиридилкарбонильную группы.
Примеры арилоксикарбонильных групп включают феноксикарбонильную; α- нафтилоксикарбонильную, β- нафтилоксикарбонильную, 2-метилфеноксикарбонильную, 3-метилфеноксикарбонильную, 4-метилфеноксикарбонильную, 2,4-диметилфеноксикарбонильную, 4-этилфеноксикарбонильную, 2-метоксифеноксикарбонильную, 3-метоксифеноксикарбонильную, 4-метоксифеноксикарбонильную, 2,4-диметоксифеноксикарбонильную, 4-этоксифеноксикарбокси-, 2-метокси-4-этоксифеноксикарбонильную, 2-хлорфеноксикарбонильную, 3-хлорфеноксикарбонильную, 4-хлорфеноксикарбонильную, 2,3-дихлорфеноксикарбонильную, 2-бромфеноксикарбонильную; 4-фторфеноксикарбонильную, β- метил -α- нафтилоксикарбонильную и β- хлор -α- нафтилоксикарбонильную группы.
Примеры низших алкоксикарбонильных групп включают алкоксикарбонильные группы, имеющие 2-6 углеродных атомов, такие, как метоксикарбонильная, этоксикарбонильная, н-пропоксикарбонильная, изопропоксикарбонильная, н-бутоксикарбонильная, изобутоксикарбонильная, втор-бутоксикарбонильная, третбутоксикарбонильная и пентилоксикарбонильная группы.
Примеры ацилоксиацильных групп включают ацетилоксиацетильную, пропионилоксиацетильную, α- (ацетилокси)пропионильную и β- (пропионилокси)пропионильную группы.
Примеры низших алкилкарбамоильных групп включают карбамоильные группы, моно- или дизамещенные низшими алкильными группами, имеющими 1-6 углеродных атомов, такие, как метилкарбамоильная, этилкарбамоильная, пропилкарбамоильная, бутилкарбамоильная, пентилкарбамоильная, гексилкарбамоильная, диметилкарбамоильная и диэтилкарбамоильная группы.
Аминокислотные остатки представляют собой группы, образованные удалением гидроксильной группы из карбоксильной группы аминокислоты, и могут быть производными как от природных, так и от синтетических аминокислот. Примеры таких аминокислот включают глицин, аланин, β- аланин, валин и изолейцин. Но могут быть включены любые аминокислотные остатки, если это аминокислотные остатки, описанные в выложенной заявке N 104093/1989 на патент Японии.
В качестве других образующих сложный эфир остатков можно назвать любые из обычных эфирообразующих остатков, описанных, например, у THEODORA W.GREENE, "PROTECTIVE GROUPS IN ORGANIC SYNTESIS", Second Edition, JOHN WILEY & SONS. INC. (1991); "Shin Jikken Kagaku Koza 4 (New Experimental Chemistry Course 4)" edited by The Chemical Societ of Japan, "Synthesis and Reaction of Organic Compounds (V)", глава 11, стр. 2495, Maruzen (1983); и в выложенных заявках N 106593/1986, N 149696/1987 и N 153696/1989 на патент Японии и традиционно используемые в качестве эфирообразующих остатков.
Примеры остатков моно- или полифосфорных кислот, представленных символом R3, включают монофосфатную, дифосфатную и трифосфатную группы и их радикалы с защищенными гидроксильными группами. Примеры защитных групп включают низшие алкильные группы, которые могут быть замещены атомом галогена или цианогруппой; бензильную группу, которая может иметь заместитель, и фенильную группу, которая может иметь заместитель. Кроме того, этот остаток может быть 3',5-циклической фосфатной группой, которая образует циклическую структуру с основанием нуклеиновой кислоты.
Предпочтительные примеры B включают цитозин, тимин, урацил, аденин, гуанин, 5-фторцитозин, 5-фторурацил, N6-бензоиладенин, 2-ацетилгуанин и 2-хлораденин. Более предпочтительные примеры их включают цитозин, урацил, аденин, 5-фторцитозин и 5-фторурацил.
Предпочтительные примеры Z включают низшие алкинильные или низшие алкенильные группы, которые могут быть замещены группой, представленной общей формулой (2). Более предпочтительные их примеры включают этинильную, пропинильную, бутинильную, этенильную, триметилсилилэтинильную, триэтилсилилэтинильную, триизопропилсилилэтинильную и трифенилсилилэтинильную группы. Особо предпочтительные примеры их включают этинильную и триметилсилилэтинильную группы.
Предпочтительным примером R1 и R2 является атом водорода.
Предпочтительные примеры R3 включают атом водорода и остатки моно- и полифосфорных кислот. Более предпочтительные примеры их включают атом водорода и дифосфатную группу.
Предпочтительные примеры образующих сложный эфир остатков, способных легко уходить в живом организме, которые представлены символами R1, R2 и R3, включают ацильные группы. Более предпочтительные примеры их включают ацетильную и бензоильную группы.
Предпочтительными соединениями по настоящему изобретению являются 3'-замещенные производные нуклеозидов, в которых B в общей формуле (1) представляет цитозин, тимин, урацил, аденин, гуанин, 5-фторцитозин, 5-фторурацил, N6-бензоиладенин, N2-ацетилгуанин или 2-хлораденин, Z представляет низшую алкинильную или низшую алкенильную группу, которая может быть замещена группой, представленной общей формулой (2), R1 и R2 представляют атомы водорода и R3 представляет атом водорода или остаток моно- или полифосфорной кислоты.
Более предпочтительными соединениями являются 3'-замещенные производные нуклеозидов, в которых В в общей формуле (1) представляет цитозин, урацил, аденин, 5-фторцитозин или 5-фторурацил, Z представляет этинильную, пропинильную, бутинильную, этенильную, триметилсилилэтинильную, триэтилсилилэтинильную, триизопропилсилилэтинильную или трифенилсилилэтинильную группу, R1 и R2 представляют атомы водорода и R3 представляет атом водорода или дифосфатную группу.
Особо предпочтительными соединениями являются 3'-замещенные производные нуклеозидов, в которых B в общей формуле (1) представляет цитозин или урацил, Z представляет этинильную или триметилсилилэтинильную группу и R1, R2 и R3 представляют атомы водорода.
Соединения по настоящему изобретению включают также соединения в виде соли. Могут быть использованы любые соли, которые являются фармацевтически приемлемыми солями, например, в случае, когда R3 представляет атом водорода, можно привести в качестве примеров соли присоединения кислоты, такие как соли неорганических кислот (гидрохлораты, гидроброматы и сульфаты) и соли органических кислот, такие, как органические сульфонаты (метансульфонаты и бензолсульфонаты) и соли алифатических карбоновых кислот (ацетаты, пропионаты и трифторацетаты). В случае, когда R3 представляет остаток моно- или полифосфорной кислоты, в качестве примеров можно привести соли щелочных металлов, такие, как соли натрия, калия и лития, соли щелочноземельных металлов, такие, как соли кальция, и соли аммония. Соединения по настоящему изобретению включают также их гидраты.
Соединения по настоящему изобретению, представленные общей формулой (1), могут быть получены по, например, следующей схеме реакций 1 или 2.
Схема 1 реакций: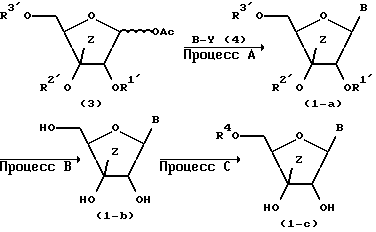
где B и Z представляют то же, что указано выше, R1', R2' и R3' представляют по отдельности защитную группу для гидроксильной группы, Y представляет силильную защитную группу и R4 представляет атом водорода или остаток моно- или полифосфорной кислоты.
Можно использовать любые защитные группы для гидроксильных групп, представленные символами R1, R2 и R3, пока они не могут быть использованы как обычные защитные группы для нуклеозидов. Примеры их включают ацильные группы, такие, как алифатические ацильные группы, которые могут иметь заместитель, и ароматические ацильные группы, которые могут иметь заместитель, низшие алкоксикарбонильные группы, низшие алкилкарбамоильные группы, низшие алкильные группы, аралкильные группы, силильные группы защитные группы и аминокислотные остатки.
В качестве ацильных групп, таких, как, алифатические или ароматические ацильные группы, низшие алкоксикарбонильные группы, низшие алкилкарбамоильные группы и аминокислотные остатки, могут быть использованы те, что описаны выше. В качестве низших алкильных групп могут быть использованы те, что описаны выше, причем, могут быть включены алкильные группы, имеющие атом галогена, низшую алкоксигруппу или тому подобное в качестве заместителя, такие, как хлорметильная, метоксиметильная, этоксиметильная, метоскиэтильная и этоксиэтильная группы.
Примеры арилалкильных групп включают бензильную, бензгидрильную и тритильную группы. Указанные группы могут иметь низшую алкильную группу, низшую алкоксигруппу, атом галогена, нитрогруппу или тому подобное в качестве заместителя.
Примеры силильных защитных групп включают триметилсилил, трет-бутилдиметилсилил, метиддиизопропилсилил, триизопропилсилил и тетраизопропилдисилоксил (TIPDS). То же самое можно сказать относительно силильной защитной группы, представленной символом Y.
(Способ А)
Соединение, представленное общей формулой (3), подвергают взаимодействию с силилированным основанием нуклеиновой кислоты, представленным общей формулой (4), с получением соединения по настоящему изобретению, представленного общей формулой (1-a).
Соединение, представленное общей формулой (3), является известным соединением или соединением, полученным любым известным способом. В частности, соединение может быть получено по схеме реакций 3, которая будет описана далее.
Силилированное основание нуклеиновой кислоты, представленное общей формулой (4), является известным соединением или полученным любым известным способом. В общем, соединение может быть получено, например, способом, описанным by Vorbruggen et al. (Chem. Ber. 114, 1234 (1982)). В частности, из основания нуклеиновой кислоты и силилирующего агента, такого, как гексаметилдисилазан, получают суспензию. К суспензии в необходимом количестве добавляют триметилсилилхлорид и смесь нагревают с обратным холодильником в атмосфере аргона, в результате чего получают целевое соединение.
Реакцию в способе А проводят в присутствии кислоты Льюиса в неполярном растворителе.
Может быть использована любая кислота Льюиса. Но примеры ее включения триметилсилилтрифторметансульфонат, тетрахлорида олова и тетрахлорид титана. В качестве неполярного растворителя может быть использован любой растворитель, если он не участвует в реакции. Примеры его включают хлороформ, дихлорметан и ацетонитрил.
Что касается пропорций реагентов в реакции, то предпочтительно использовать соединение общей формул (4) и кислоту Льюиса в пропорциях соответственно 1-10 моль (предпочтительно 1-5 моль) и 1-10 моль (предпочтительно 1-5 моль) на моль соединения общей формулы (3). Что касается температуры реакции, то кислоту Льюиса добавляют при 0oC и реакцию проводят при 0-100oC, предпочтительно при температуре близкой к комнатной. Что касается времени реакции, то реакцию надлежащим образом проводят за 0,1 - 50 часов, предпочтительно 1 - 24 часа.
(Способ B)
Для удаления защитных групп, представленного общей формулой (1-а) соединения, полученного по способу A, используют способы, обычно применяемые для удаления защитных групп, например можно использовать кислотный гидролиз, щелочной гидролиз, обработку аммонием и каталитическое восстановление. Соединение по настоящему изобретению, представленное общей формулой (1-b), может быть получено гидролизом соединения (1-a) с щелочью, такой, как гидроксид натрия, гидроксид калия или производное аммония, в низшем спирте, например метаноле, в случае, когда защитные группы являются, например, ацильными группами, или путем обработки соединения (1-a) производным аммонийхлорида в случае, когда защитные группы являются силильными группами.
Что касается пропорций реагентов в реакции, то является предпочтительным использовать основное соединение в каталитическом количестве по отношению к соединению, представленному общей формулой (1-a), в случае, когда защитные группы являются ацильными группами. Температура реакции находится в диапазоне 0-150oC, предпочтительно в диапазоне от комнатной температуры до 100oC. Что касается времени реакции, то реакцию надлежащим образом проводят за 0,1 - 100 часов, предпочтительно 1-60 часов.
(Способ C)
Полученное способом B соединение, представленное общей формулой (1-b), фосфорилируют фосфорилирующим агентом в присутствии растворителя или без какого-либо растворителя с получением соединения по настоящему изобретению, представленного общей формулой (1-c). Примеры фосфорилирующего агента включают фосфорилирующие агенты, обычно используемые при избирательном фосфорилировании в 5'-положении, такие, как оксигалогениды фосфора (такие, как оксихлорид фосфора и оксибромид фосфора), безводные фосфорные кислоты (такие, как пирофосфатная и полифосфорная кислоты), фосфорная кислота, сложные моноэфиры фосфорной кислоты (такие, как п-нитрофенилфосфат), тетрахлорпирофосфорная кислота и триалкиламмонийпирофосфаты. Из них предпочтительными являются оксихлорид фосфора и трибутиламмонийпирофосфат. В качестве растворителя может быть использован любой растворитель, если он не принимает участия в реакции. Примеры его включают пиридин, гексаметилфосфорный триамид, дихлорметан, хлороформ, бензол, толуол, триметилфосфат и триэтилфосфат. Содержание используемого в реакции фосфорилирующего агента предпочтительно составляет 1-5 моль на моль соединения общей формулы (1-b). Температура реакции находится в пределах от -80oC до 100oC, предпочтительно от -20oC до 50oC. Что касается времени реакции, то обычно реакцию должным образом проводят за примерно 0,5 - 12 часов.
Между прочим, при фосфорилировании можно использовать 1,1'-карбонилдиимидазол, тетразол, производное 1,2,4-триазола или тому подобное в качестве ускорителя реакции.
Схема 2 реакций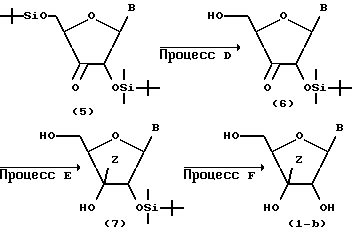
где B и Z имеют такое же значение, как указано выше.
(Процесс D)
Соединение, представленное общей формулой (5), частично гидролизуют, например, способом, описанным в J.Org. Chem, 55, 410-412 (1990), а именно реакцией соединения (5) при 0oC в смеси трифторуксусная кислота вода, тем самым проводя избирательное десилилирование в 5'-положении с получением соединения, представленного общей формулой (6).
Соединение, представленное общей формулой (5), является известным соединением или соединением, полученным любым известным способом, например способом, описанным в J. Org. Chem, как указано выше: SYNTHESIS 283-288 (1991); или Tetrahedron, 47, 1727-1736 (1991).
(Способ E)
В 3-положение соединения, представленного общей формулой (6), вводят заместитель, обозначенный символом Z, с получением соединения, представленного общей формулой (7). Этот процесс может быть проведен, например, 1) способом, в котором соединение (которое может быть газообразным), представленное формулой ZH или комплексом хлорида церия и ZH, подвергают взаимодействию с соединением (6) в присутствии н-бутиллития в тетрагидрофуране, или 2) способом, в котором осуществляют взаимодействие реактива Гриньяра (ZMgBr) с соединением (6) в тетрагидрофуране.
Что касается пропорций реагентов, предпочтительно использовать реагент (ZH) и н-бутиллитий в пропорциях соответственно 1-10 моль (предпочтительно 1-5 моль) и 1-10 моль (предпочтительно 1-5 моль) на моль соединения общей формулы (6). В случае, когда используют хлорид церия, количество используемого хлорида церия предпочтительно почти эквимолярно указанному реагенту. Реактив Гриньяра предпочтительно используют в соотношении 1-5 моль (более предпочтительно 1-2 моль) на моль соединения формулы (6). Температуру реакции предпочтительно поддерживают на уровне -70oC или ниже в случае способа 1), в котором используют н-бутиллитий, или в пределах от -20oC до 50oC (предпочтительно от -10oC до 10oC) в случае способа 2), в котором реакцию осуществляют с реактивом Гриньяра. Что касается времени реакции, то реакцию должным образом проводят за 0,1-50 часов, предпочтительно 1/24 часа.
(Способ F)
Соединение, представленное общей формулой (7), гидролизуют, например, реакцией соединения (7) в смеси хлороводородная кислота/метанол с получением соединения по настоящему изобретению, представленного общей формулой (1-b).
Температуру реакции поддерживают в диапазоне 0-100oC, предпочтительно близкой к комнатной температуре. Что касается времени реакции, то реакцию должным образом проводят за 1-100 часов.
Кроме того, полученное по этому способу соединение, представленное общей формулой (1-b), повергают такой же реакции, как в способе C схемы 1, в результате чего может также быть получено соединение, представленное общей формулой (1-c).
Эфирообразующие остатки могут быть введены в гидроксильные группы в 2'-, 3'- и 5'-положениях соединений общей формулы (1-b), полученных по схемам 1 и 2 реакций, или в гидроксильные группы в 2'- и 3'- положениях соединения общей формулы (1-c) любым известным традиционным способом, например способом, раскрытым в описанной выше литературе "PROTECTIVE GROUPS IN ORGANIC SYNTHESIS", Second Edition или "Shin Jikken Kagaku Koza 4 (New Experimental Chemistry Cours 4)" edited by The Chemical Society of Japan, "Synthesis and Reaction of Organic Compounds (V)", или способом, описанным в вышеизложенной заявке N 152898/1983, N 56996/1985, N 106593/1986, N 149696/1987 или N 153696/1689 на патент Японии, с получением других соединений по настоящему изобретению из указанных соединений.
Соединения по настоящему изобретению, полученные в результате описанных выше реакций, могут быть превращены в соли известным традиционным способом, например способом, в котором они взаимодействуют с любой из вышеописанных неорганических или органических кислот в подходящем растворителе. Примеры растворителя включают воду, метанол, этанол, дихлорметан, тетрагидрофуран, этилацетат и гексан. Реакцию предпочтительно проводят при температуре 0-50oC. Кроме того, соединения по настоящему изобретению, полученные в результате осуществления описанных выше реакций, могут быть превращены в соли известным традиционным способом, например способом, в котором они взаимодействуют с сильным основанием, таким, как гидроксид щелочного или щелочноземельного метала (такой, как гидроксид натрия или гидроксид калия), или сильным основанием, таким как метоксид натрия, метоксид калия или гидроксид натрия, в подходящем растворителе.
Вышеописанное исходное соединение (3) может быть получено, например, по следующей схеме реакций.
Схема 3 реакций: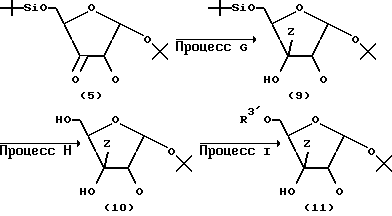
где Z, R1, R2 и R3 имеют значения, указанные выше.
(Способ G)
Известное соединение, представленное общей формулой (8), реагирует так же, как в способе E в описанной выше схеме 2, с получением соединения, представленного общей формулой (9).
(Способ H)
Соединение, представленное общей формулой (9), взаимодействует с тетрабутиламмонийфторидом в тетрагидрофуране с получением соединения, представленного общей формулой (10).
Что касается пропорций реагентов в реакции, то является предпочтительным использовать тетрабутиламмонийфторид в соотношении 1-10 моль (предпочтительно 1-5 моль) на моль соединения общей формулы (9). Реакцию проводят при температуре 0-100oC, предпочтительно при температуре, близкой к комнатной. Что касается времени реакции, то реакцию должным образом проводят за 0,1-2 часа, предпочтительно 5-50 минут.
(Способ I)
Соединение, представленное общей формулой (10), взаимодействует с реакционноспособным веществом, которое защищает гидроксильную группу, в подходящем растворителе с получением соединения, представленного общей формулой (11).
В качестве растворителя может быть использован любой растворитель без какого-либо особого ограничения, который не принимает участия в реакции. В случае, когда защитная группа является ацильной группой, необходимо лишь вводить в реакцию ацилирующий агент, такой, как ангидрид кислоты или галогенангидрид, в пиридине. В реакцию с этим ацилирующим агентом может быть добавлен амин, такой, как диметиламинопиридин или триэтиламин, в качестве катализатора.
Что касается пропорций реагентов в реакции, то является предпочтительным использовать реакционноспособное вещество, которое защищает гидроксильную группу, в соотношении 1-10 моль (предпочтительно 1-5 моль) на моль соединения общей формулы (10). В случае использования катализатора его предпочтительно используют в каталитическом количестве. Реакцию проводят при температуре в пределах от - 20oC до 100oC, предпочтительно при температуре, близкой к комнатной. Что касается времени реакции, то реакцию должным образом проводят за 0,1-10 часов, предпочтительно за время от 30 минут до 5 часов.
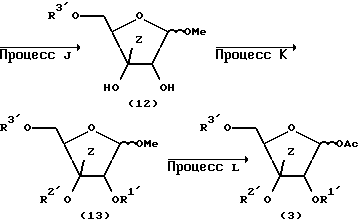
(Способ J)
Соединение, представленное общей формулой (11), подвергают кислотному алкоголизу с получением соединения, представленного общей формулой (12).
В качестве спирта предпочтительно использовать низший спирт, такой, как метанол или этанол. Можно использовать смесь растворителей спирт/вода.
Примеры кислотного соединения включают карбоновые кислоты, такие, как муравьиная и уксусная кислоты, ангидриды кислоты, такие, как уксусный ангидрид, галогенангидриды, такие, как ацетилхлориды, и неорганические кислоты, такие, как хлороводородная, бромоводородная и серная кислоты.
Что касается пропорций реагентов в реакции, то предпочтительно использовать кислотное соединение в соотношении 10-50 моль (предпочтительно 20-40 моль) на моль соединения общей формулы (11). Реакцию проводят при температуре 0-100oC, предпочтительно при температуре, близкой к комнатной. Что касается времени реакции, то реакцию должным образом проводят за время от 1 минуты до 10 часов, предпочтительно от 5 минут до 5 часов.
(Способ K)
Соединение, представленное общей формулой (12), взаимодействует с реакционноспособным веществом, которое защищает гидроксильную группу, в подходящем растворителе с получением соединения, представленного общей формулой (13).
В качестве растворителя может быть использован любой растворитель без какого-либо особого ограничения, который не принимает участия в реакции.
В случае, когда защитная группа является ацильной группой, соединение (13) получают введением в реакцию ацилирующего агента, такого, как ангидрид кислоты или галогенангидрид, в пиридине. В реакцию с этим ацилирующим агентом может быть добавлен амин, такой, как диметиламинопиридин или триэтиламин, в качестве катализатора.
Что касается пропорций реагентов в реакции, то предпочтительно использовать реакционноспособное вещество, которое защищает гидроксильную группу, в соотношении 1-20 моль (предпочтительно 1-15 моль) на моль соединения общей формулы (12). В случае использования катализатора его предпочтительно используют в каталитическом количестве, предпочтительно в соотношении 1-5 моль на моль соединения (12). Температура реакции находится в пределах от 0oC до 200oC, предпочтительно от комнатной температуры до 150oC. Что касается времени реакции, то реакцию должным образом проводят за 0,1-50 часов, предпочтительно 1-30 часов.
(Способ L)
Соединение, представленное общей формулой (13), ацетилируют добавлением концентрированной серной кислоты к соединению (13) в уксусной кислоте и/или уксусном ангидриде с получением соединения, представленного общей формулой (3).
Реакцию проводят при температуре 0-100oC, предпочтительно при температуре, близкой к комнатной. Что касается времени реакции, то реакцию должным образом проводят за 0,1-24 часа, предпочтительно за время от 10 минут до 5 часов.
Соединения по настоящему изобретению, полученные в результате осуществления вышеописанных реакций, и отдельные соединения могут быть выделены и очищены с использованием известных традиционных способов выделения и очистки, например концентрирования, экстрагирования растворителями, фильтрования, перекристаллизации, различных видов хроматографии и т.д.
Соединения по настоящему изобретению могут быть использованы для получения лечебных композиций с подходящими фармацевтическими носителями способом, известным в данной области техники.
В качестве носителей могут быть использованы различные виды носителей, обычно используемых в лекарственных средствах, например наполнители, связывающие вещества, разрыхляющие вещества, смазывающие вещества, красители, вкусовые вещества, корригенты запаха, поверхностно-активные вещества и т.д.
Может быть использована любая лекарственная форма для использования лекарственного средства или лечебной композиции по настоящему изобретению в качестве средства для лечения опухоли у млекопитающих, включая человека. Лекарственная форма может быть правильно выбрана в соответствии с объектом лечения. Конкретные примеры формы включают парентеральные препараты, такие, как растворы для инъекции, суппозитории, препараты наружного применения (мази, пластыри и т.д.) и аэрозольные препараты, и пероральные препараты, такие, как таблетки с покрытиями, порошки, гранулы, капсулы, драже и растворы (суспензии, эмульсии и т.д.).
Различные композиции, описанные выше, изготавливают способами, по существу, известными в данной области техники.
Когда композицию изготавливают в форме раствора для инъекции, в качестве носителей могут быть использованы, например, растворитель, такой, как вода, этиловый спирт, макрогель (полигликоль), пропиленгликоль, этоксилированный изостеариловый спирт, полиоксилированный изостеариловый спирт или полиоксиэтиленсорбитановый эфир жирной кислоты, регулятор pH и буфер, такой, как цитрат натрия, ацетат натрия или фосфат натрия, стабилизатор, такой, как пиросульфит натрия, этилендиаминтетрауксусная кислота, тиогликолевая кислота или тиомолочная кислота, и тому подобное. В этом случае лекарственный препарата может содержать хлорид натрия, глюкозу или глицерин в количестве, достаточном для получения изотонического раствора. Кроме того, могут быть добавлены также традиционные солюбилизирующие вспомогательные вещества, аналгетики, местные анестетики и тому подобное. Указанные носители могут быть добавлены для получения растворов для подкожных, внутримышечных и внутривенных инъекций способом, известным в данной области техники perse.
При изготовлении композиции в форме суппозитория в качестве носителей могут быть использованы полиэтиленгликоль, масло какао, ланолин, высшие спирты, сложные эфиры высших спиртов, желатин, полусинтетические глицериды, Витепсол (торговая марка, продукт Ф. "Dynamit Nobel Co.") и т.п. с добавлением к ним подходящего вещества, обладающего свойством абсорбции.
Когда композицию изготавливают в форме мазей, например пасты, крема и геля, то включают основу, стабилизатор, смачивающее вещество, консервант и тому подобное, которое обычно используют в необходимых количествах, и компоненты смешивают для изготовления требуемых препаратов в соответствии со способом, известным в данной области техники perse. В качестве основы можно использовать, например, белый вазелин, парафин, глицерин, производное целлюлозы, полиэтиленгликоль, силикон или бентонит. В качестве консерванта можно использовать метил-пара-гидроксибензоат, этил-пара-гидроксибензоат, пропил-пара-гидроксибензоат или тому подобное.
При изготовлении пластыря необходимо лишь нанести описанную выше мазь, крем, гель или пасту на подложку, обычно используемую в способе, известном в данной области техники perse. В качестве подложки годится тканый или нетканый материал из хлопка, гидратцеллюлозного или химического волокна или пленка или листовой вспененный материал из мягкого поливинилхлорида, полиэтилена или полиуретана.
При изготовлении композиции в форме твердых препаратов для перорального применения, таких, как таблетки, порошок и гранулы, в качестве носителей могут быть использованы наполнители, такие, как лактоза, сахароза, хлорид натрия, глюкоза, мочевина, крахмал, карбонат кальция, каолин, кристаллическая целлюлоза, кремневая метилцеллюлоза, глицерин, альгинат натрия и аравийская камедь; связывающие вещества, такие, как простой сироп, раствор глюкозы, раствор крахмала, раствор желатина, поливиниловый спирт, простой поливиниловый эфир, поливинилпирролидон, карбоксиметилцеллюлоза, шеллак, метилцеллюлоза, этилцеллюлоза, вода, этанол и фосфат калия; разрыхлители, такие, как сухой крахмал, альгинат натрия, агаровый порошок, ламинарана, бикарбонат натрия, карбонат кальция, сложные полиоксиэтиленсорбитановые эфиры жирных кислот, лаурилсульфат натрия, моноглицерид стеариновой кислоты, крахмал и лактоза; препятствующие распаду вещества, такие, как сахароза, стеариновая кислота, масло какао и гидрогенизированные масла; вещества, вызывающие абсорбцию, такие, как четвертичное аммониевое основание и лаурилсульфат натрия; увлажнители, такие, как глицерин и крахмал; адсорбенты, такие, как крахмал, лактоза, каолин, бентонит и коллоидный кремнезем; смазывающие вещества, такие, как очищенный тальк, соли стеариновой кислоты, порошкообразная борная кислота и полиэтиленгликоль, и тому подобное. Таблетки могут быть снабжены обычными покрытиями, как, например, таблетки, покрытые сахаром, покрытые желатином, с энтеросолюбильным покрытием, с пленочным покрытием, с двухслойным покрытием, с многослойным покрытием и тому подобное.
Препараты в виде капсул изготавливают путем смешивания соединения по настоящему изобретению с различными носителями, примеры которых приведены выше, и помещения смеси в твердые желатиновые капсулы, мягкие капсулы и тому подобное.
Когда композицию изготавливают в форме драже, то в качестве носителей можно использовать наполнители, такие, как глюкоза, лактоза, крахмал, масло какао, отвержденные растительные масла, каолин и тальк; связывающие вещества, такие, как порошкообразная аравийская камедь, трагакант, желатин и этанол; разрыхлители, такие, как ламинаран и агар, и тому подобное.
Жидкие лекарственные формы могут представлять собой водные или масляные суспензии, растворы, сиропы или эликсиры. Их изготавливают, используя обычные добавки, способом, известным так таковым в данной области техники.
Количество соединения по настоящему изобретению, которое должно содержать вышеописанные композиции, изменяется в соответствии с типом композиции, способом введения, схемой дозирования и тому подобное и, следовательно, не может быть точно названо, а выбирается в соответствии с потребностью из широкого диапазона. Однако, композиция может предпочтительно содержать примерно 1-70 мас.% соединения от веса композиции.
Может быть использован любой способ введения лекарственной формы и способ введения, такой как парэнтеральное, пероральное, ректальное, внутриоральное или подкожное введение, который определяют в соответствии с типом композиции, возрастом, полом и другими состояниями принимающего объекта, такого, как больной, болезненным состоянием больного и тому подобное. Например, таблетки, драже, растворы, суспензии, эмульсии, гранулы и капсулы вводят перорально, а суппозитории - интраректально. Растворы для инъекции вводят внутривенно отдельно или в сочетании с обычным заменителем жидкости, содержащим глюкозу, аминокислоты и тому подобное, а также, когда требуется, внутриартериально, внутримышечно, внутрикожно или подкожно. Мази наносят на кожу, слизистую оболочку рта и т.д.
Дозу соединения по настоящему изобретению выбирают в соответствии со способом введения, возрастом, полом, болезненным состоянием и типом опухоли, принимающего объекта, такого, как больной, видом дозируемого соединения и другими условиями. Но обычно является желательным дозировать соединение в соответствии примерно 1-1000 мг для пероральной композиции раствора или примерно 5-1000 мг для суппозитория на композицию. Кроме того, суточную дозу лекарства в виде любой из вышеописанных лекарственных форм предпочтительно назначают на основе количества в пределах обычно от примерно 0,1 до 200 мк/кг веса в день, предпочтительно от примерно 0,5 до 100 мг/кг веса в день. Указанные композиции по настоящему изобретению могут вводиться за один раз или примерно 2-4 раза в день.
Виды злокачественных опухолей, которые могут быть излечены препаратом, содержащим соединение по настоящему изобретению не ограничены. Примеры их включают рак головы и шеи, рак пищевода, рак желудка, рак толстой кишки, рак прямой кишки, рак печени, рак желчного пузыря и желчного протока, рак поджелудочной железы, рак легких, рак молочной железы, рак яичника, рак мочевого пузыря, рак предстательной железы, опухоль яичника, остеохондросаркому, злокачественную лимфому, лейкоз, рак шейки матки, рак кожи, опухоль мозга и тому подобное.
Примеры
Далее настоящее изобретение проиллюстрировано более подробно на следующих ссылочных примерах, примерах и примерах фармакологических испытаний. Однако следует иметь в виду, что эти примеры не ограничивают настоящее изобретение.
Ссылочный пример 1
Синтез 5-O-трет-бутилдиметилсилил-1,2-O-изопропилиден- 3-C-(2-триметилсилилэтинил) -α- рибофуранозы
В 60 мл тетрагидрофурана растворяли 6,3 мл (45 ммоль) триметилсилилацетилена в атмосфере аргона и раствор перемешивали при -78oC. Поддерживая температуру реакционного раствора на уровне -70oC или ниже, добавляли по каплям в течение 30 минут н-бутиллитий (раствор в н-гексане, 1,62 моль/литр; 27,8 мл, 45 ммоль). Спустя 30 минут после добавления по каплям добавляли по каплям 4,5 г (15 ммоль) 5-O-трет-бутилдиметилсилил-1,2-O-изопропилиден -α- D-этритропентофураноз-3-улозы, растворенного в 30 мл тетрагидрофурана в течение 10 минут, и смесь перемешивали еще 3 часа. Затем в реакционной смеси добавляли 60 мл 1 н. водного раствора хлорида аммония и температуру смеси повышали до комнатной. После этого реакционную смесь экстрагировали этилацетатом (3 x 35 мл) и полученный органический слой промывали насыщенным водным раствором (3 x 20 мл) хлорида натрия и сушили над сульфатом натрия. Затем высушенный органической слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5% этилацетата-н-гексан) с получением 5,92 г (выход 99%) целевого соединения в виде белого порошкообразного вещества.
Т.пл.: 84 - 86oC.
FAB-MC: m/z 401 (MH+), 383 (M+-OH)
1H-ЯМР (CDCl3) δ: 5,85 (д, 1H, H-1, J1,2 = 3,8 Гц), 4,56 (д, 1H, H-2, J2,1 = 3,8 Гц), 4,01-3,94 (м, 3H, H-4, H-5), 3,05 (с, 1H, 3-OH, обмениваемый с D2O), 1,56, 1,37 (с, каждый 3H, изопропил, 0,91 (с, 9H, B), 0,18 (с, 9Р, Me), 0,06, 0,03 (с, каждый 3H, Me).
Элементный анализ:
Вычислено (для C19H36O5Si2): C, 56,96; H, 9,06.
Найдено: C 56,82; H 9,25.
Ссылочный пример 2
Синтез 5-O-бензоил-1-C-этинил-1,2-O-изопропилиден -α -D-рибофуранозы
В 15 мл тетрагидрофурана растворяли 1,44 г (3,6 ммоль) соединения, полученного в ссылочном примере 1 и добавляли 5,4 мл (5,4 ммоль) 1 н. раствора тетрабутиламмонийфторида в тетрагидрофуране с последующим перемешиванием в течение 10 минут при комнатной температуре. Отгоняли растворитель, получали 5-O-трет-бутилдиметилсилил-3-C-этинил-1,2-O-изопропилиден -α- D-рибофуранозу в виде сиропообразного вещества. Это соединение растворяли в 30 мл пиридина и добавляли при охлаждении ледяной водой 0,92 мл (7,8 ммоль) бензоилхлорида с последующим перемешиванием при комнатной температуре в течение 2 часов. Отгоняли под пониженным давлением растворитель и остаток подвергали трехкратной азеотропной перегонке с толуолом. Полученный остаток растворяли в 50 мл этилацетата и раствор подвергали жидкостному разделению с использованием 25 мл воды и насыщенного водного раствора гидрокарбоната натрия (3 x 25 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 0-5-10% этилацетата-н-гексан) с получением 1,07 г (выход 93%) целевого соединения в виде желтого сиропообразного вещества.
FAB-MC: m/z 319 (MH+), 303 (M+-Me).
1H-ЯМР (CDCl3) δ: 8,12-7,42 (м, 5H, бензоил),
5,97 (д, 1H, E-1, J1,2=3,8 Гц),
4,76 (дд, 1H, H-5a, J5a,4=3,8 Гц, J5a,5b=12,0 Гц),
4,61 (дд, 1H, H-5, J5b,4=7,4 Гц, J5b,5b=12,0 Гц),
4,60 (д, 1H, H-2, J2,1=3,8 Гц),
4,23 (дд, 1H, H-4, J4,5a=3,8 Гц, J4,5b=7,4 Гц),
3,2 (с, 1H, 3-OH, обмениваемый с D2O), 2,63 (с, 1H, 3-C=CH),
1,62, 1,41 (с, каждый 3H, изопропил).
Элементный анализ:
Вычислено (для C17H18O6): C, 64,14; H, 5,70.
Найдено: C, 64,08; H, 5,73.
Ссылочный пример 3
Синтез метил 2,3,5-три-O-бензоил-3-C-этинил -α,β- D-рибофуранозы
1) В 27 мл абсолютного метанола растворяли 637 мг (2,0 ммоль) соединения, полученного в ссылочном примере 2, и добавляли 1,25 г (5,0 ммоль) пиридиний п-толуолсульфоната, после чего смесь нагревали с обратным холодильником в течение 3 дней в атмосфере аргона. После охлаждения реакционной смеси до комнатной температуры отгоняли при пониженном давлении растворитель, остаток растворяли в 30 мл этилацетата и раствор подвергали жидкостному разделению с использованием 15 мл воды и насыщенного водного раствора гидрокарботана натрия (3 x 15 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем осушенный органический слой фильтровали и отгоняли растворитель, получая в результате метил 5-O-бензоил-3-C-этинил -α,β- D-рибофуранозу в виде сиропообразного вещества. Это соединение подвергали три раза азеотропной перегонке с пиридином и затем растворяли в 30 мл пиридина. К раствору добавляли, при охлаждении ледяной водой, 2,32 мл (20 ммоль) бензоилхлорида и 367 мг (3 ммоль) диметиламинопиридина и перемешивали смесь при 100oC в течение 24 часов. После охлаждения реакционной смеси до комнатной температуры отгоняли при пониженном давлении растворитель и остаток подвергали трехкратной азеотропной перегонке с толуолом. Полученный остаток растворяли в 20 мл этилацетата и раствор подвергали жидкостному разделению с использованием 10 мл воды и насыщенного водного раствора гидрокарбоната натрия (3 x 10 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 0-10% этилацетата-н-гексан) и получением 825 мг (выход 83%) целевого соединения в виде желтого сиропообразного вещества.
E1-MC: m/z 500 (M+).
1H-ЯМР (CDCl3) δ: 8,12-7,30 (м, 15H, бензоил 1x3),
6,03 (д, 0,66H, β- H-1, J1,2 = 1,2 Гц),
5,82 (д, 0,33H, α- H-1, J1,2=4,4 Гц),
5,47 (д, 0,33H, α- H-2, J2,1=4,4 Гц),
5,17 (д, 0,66H, β- H-2, J2,1=1,2 Гц),
5,10-4.78 (м, 3H, α,β- H-4, H-5), 3,53 (с, 1,98, β- OMe),
3,45 (с, O, 99H, α- OMe), 2.86 (с, O, 66H, β- 3-C=CH),
2,78 (с, O, 38H, α-3-C≡CH).
Соотношение номеров α:β = 1:2 (при определении по спектру 1H-ЯМР).
Элементный анализ:
Вычислено (для C29H24O8): C, 69,59; H, 4,83
Найдено: C, 69,32: H, 4,76.
2) К 5,0 мл абсолютного метанола добавляли при охлаждении ледяной водой 0,15 мл (2 ммоль) ацетилхлорида и смесь перемешивали при комнатной температуре в течение 20 минут. К этому раствору добавляли по каплям 837 мг (2,0 ммоль) полученного в ссылочном примере 2 соединения, растворенного в 2,0 мл абсолютного метанола, после чего смесь перемешивали при комнатной температуре в течение 2 дней. Реакционную смесь нейтрализовали 165 мл триэтиламина и отгоняли при пониженном давлении растворитель. Остаток подвергали три раза азеотропной перегонке с пиридином и затем растворяли в 30 мл пиридина. К раствору добавляли при охлаждении ледяной водой 2,32 мл (20 ммоль) бензоилхлорида и 367 мг (3 ммоль) диметиламинопиридина и перемешивали смесь при 100oC в течение 24 часов. После охлаждения реакционной смеси до комнатной температуры отгоняли при пониженном давлении растворитель и остаток подвергали трехкратной азеотропной перегонке с толуолом. Полученный остаток растворяли в 20 мл этилацетата и раствор подвергали жидкостному разделению с использованием 10 мл воды и насыщенного водного раствора гидрокарбоната натрия (3 x 10 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали путем колоночной хроматографии на силикагеле (элюирование смесью 0-10% этилацетата-н-гексан) с получением 718 мг (выход 72%) целевого соединения в виде желтого сиропообразного вещества. Значения его физических свойств были идентичны значениям соединения, полученного способом (1), за исключением того, что число протонов в ЯМР-спектре было другим.
Соотношение номеров α:β = 1:1 (при определении по спектру 1H-ЯМР).
3) К 3,2 мл воды и 8,86 мл абсолютного метанола добавляли при охлаждении ледяной водой 1,94 мл (27,3 ммоль) ацетилхлоридом и смесь перемешивали при комнатной температуре в течение 20 минут. К этому раствору добавляли по каплям 318 мг (1 ммоль) полученного в ссылочном примере 2 соединения, растворенного в 2,0 мл абсолютного метанола, после чего смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь нейтрализовали 4,5 мл триэтиламина и отгоняли при пониженном давлении растворитель с получением метил 5-O-бензоил-3-C-этинил -α,β- D-рибофуранозы в виде сиропообразного вещества. Это соединение подвергали три раза азеотропной перегонке с пиридином и затем растворяли в 30 мл пиридина. К раствору добавляли при охлаждении ледяной водой 1,16 мл (10 ммоль) бензоилхлорида и 184 мг (1,5 ммоль) диметиламинопиридина и перемешивали смесь при 100oC в течение 24 часов. После охлаждения реакционной смеси до комнатной температуры отгоняли при пониженном давлении растворитель. Остаток подвергали трехкратной азеотропной перегонке с толуолом. Полученный остаток растворяли в 10 мл этилацетата и раствор подвергали жидкостному разделению с использованием 5 мл воды и насыщенного водного раствора гидрокарбоната натрия (3 x 5 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 0-10% этилацетат/н-гексан) с получением 435 мг (выход 87%) целевого соединения в виде желтого сиропообразного вещества. Значения его физических свойств были идентичны значениям соединения, полученного способом (1).
Соотношение номеров α:β= 1:2 (при определении по спектру 1H-ЯМР).
Ссылочный пример 4
Синтез 1-O-ацентил-2,3,5-три-O-бензоил-3-C-этинил -α,β- D-рибофуранозы
В 1,75 мл уксусной кислоты и 0,22 мл уксусного ангидрида растворяли 264 мг (0,53 ммоль) соединения, полученного в ссылочном примере 3 (3). Добавляли при охлаждении ледяной водой 0,11 мл концентрированной серной кислоты и смесь перемешивали при комнатной температуре 4 часа. Добавляли к реакционной смеси 4 мл хлороформа и полученную смесь подвергали жидкостному разделению с использованием 0,4 мл воды, насыщенного водного раствора гидрокарбоната натрия (3 x 1,2 мл) и воды (2 x 0,4 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование хлороформом) с получением 259 мг (выход 93%) целевого соединения в виде сиропообразного вещества.
E1-MC: m/z 528 (M+, 485 (M+-Ac).
1H-ЯМР (CDCl3) δ: 8,17-7,32 (м, 15H, бензоил 1 x 3),
6,77 (д, 0,33H, α- H-1, J1,2=4,6 Гц),
6,39 (д, 0,66H, β- H-1, J1,2=1,5 Гц),
6,19 (д, 0,66H, β- H-2, J2,1=1,5 Гц),
6.07 (д, 0,33H, α- H-2, J2,1=4,6 Гц),
5,07-4,79 (м, 3H, α,β- H-4, H-5), 2,89 (с, 0,66H, β- 3-C=CH),
2,81 (с, 0,33H, α- 3-C=CH), 2,14 (с, 1,98H, β- ацетил),
2,00 (с, 0,99H, α- ацетил)
Соотношение номеров α:β = 1:2 (при определении по спектру 1H-ЯМР).
Элементный анализ:
Вычислено (для C30H24O9): C, 68,18: H, 4,58
Найдено: C, 68,01; H, 4,64.
Ссылочный пример 5
Синтез 5-O-трет-бутилдиметилсилил-1,2-O-изопропилиден- 3-C(1-пропинил) -α- D-рибофуранозы
Газообразный пропин сжижали при -30oC в атмосфере аргона и, накопив примерно 0,5 мл жидкого пропина в трехгорлой колбе, добавляли туда 5 мл тетрагидрофурана и смесь перемешивали при -78oC. Поддерживая температуре реакционной смеси на уровне -70oC или ниже, добавляли по каплям в течение 30 минут н-бутиллитий (раствор в н-гексане 1,63 моль/литр; 1,84 мл, 3,0 ммоль). По истечении 30 минут после капельного добавления добавляли по каплям в течение 10 минут 302 мг (1,0 ммоль) 5-O-трет-бутилдиметилсилил-1,2-O-изопропилиден -α-D -эритропентофураноз-3-улозы, растворенной в 2,5 мл тетрагидрофурана, и смесь перемешивали еще 2 часа. Затем к реакционной смеси добавляли 5 мл 1 н.водного раствора аммонийхлорида и температуру смеси повышали до комнатной температуры. После этого реакционную смесь экстрагировали этилацетатом (3 х 5 мл) и полученный органический слой промывали насыщенным водным раствором (3 х 3 мл) хлорида натрия и затем сушили над сульфатом натрия. Затем органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5% этилацетат/н-гексан) с получением 320 мг (выход 93%) целевого соединения в виде сиропообразного вещества.
FAB-MC: m/z 327 (M+-Me).
ИК (чистый): 2255 см-1(-C=C-)
1H-ЯМР (CDCl3) δ: 5,83 (д, 1H, H-1, J1,2 = 3,6 Гц), 4,51 (д, 1H, H-2, J2,1 = 3,6 Гц), 4.00-3.91 (м, 3H, H-4, H-5), 2,97 (с, 1H, 3-OH, обмениваемый с D2O), 1,87 (с, 3H, 3-C=C-CH3), 1.56, 1.36 (с, каждый 3H, изопропил), 0,91 (с, 9H, tBu), 0,10, 0.09 (с, каждый 3H, Me).
Элементный анализ:
Вычислено (для C17H30O5): C, 59,61; H, 8,83
Найдено: C, 59,38; H, 8,94.
Ссылочный пример 6
Синтез 5-O-бензоил-3-C-(1-пропинил)-1,2-O-изопропилиден -α- D-рибофуранозы
В 30 мл тетрагидрофурана растворяли 3,42 г (10,0 ммоль) соединения, полученного в ссылочном примере 5, и добавляли 10,0 мол (10,0 ммоль) 1 н. раствора тетрабутиламмонийфторида в тетрагидрофуране с последующим перемешиванием при комнатной температуре в течение 20 минут. Отгоняли растворитель и получали 3-C-(1-пропинил)-1,2-O-изопропилиден -α- D-рибофуранозу в виде сиропообразного вещества. Это соединение растворяли в 50 мл пиридина и к раствору добавляли при охлаждении ледяной водой 2,90 мл (25,0 ммоль) бензоилхлорида с последующим перемешиванием при комнатной температуре в течение 4 часов. Отгоняли при пониженном давлении растворитель и остаток подвергали три раза азеотропной перегонке с толуолом. Полученный остаток растворяли в 100 мл этилацетата и раствор подвергали жидкостному разделению с использованием 50 мл воды и насыщенного водного раствора гидрокарбоната натрия (3 х 50 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5-10-15% этилацетат/н-гексан) с получением 2,47 г (выход 74%) целевого соединения в виде белого порошкообразного вещества.
Т.пл.: 120-122oC.
FAB-MC: m/z 333 (MH+.
1H-ЯМР (CDCl3) δ: 8,09-7,43 (м, 5H, бензоил),
5,92 (д, 1H, H-1, J1,2=3,6 Гц),
4,72 (дд, 1H, H-5, J5a,4=3,4 Гц, J5a,5b=11,9 Гц),
4,56 (дд, 1H, H-5a, J5b,4=7,7 H, J5b,5a=11,9 Гц),
4,53 (д, 1H, H-2, J2,1=3,6 Гц),
4,17 (дд, 1H, H-4, J4,5a=3,4 Гц, J4,5b=7,7 Гц),
2,90 (с, 1H, 3-OH, обмениваемый с D2O),
1.89 (с, 3H, 3-C -α,β C-CH30, 1,60,1,39 (с, каждый 3H, изопропил).
Элементный анализ:
Вычислено: (для C18H20O6): C, 65,05; H, 6,07
Найдено: C, 64,92; H, 6,19 .
Ссылочный пример 7
Синтез метил 2,3,5-три-O-бензоил-3-C-(1-пропинил) -α,β- D-рибофуранозы
К 22, 1 мл воды и 74,9 мл метанола добавляли при охлаждении ледяной водой 13,4 мл (179 ммоль ацетилхлорида и смесь перемешивали при комнатной температуре в течение 20 минут. К этому раствору добавляли 2,3 г (6,9 ммоль) соединения, полученного в ссылочном примере 6, после чего смесь перемешивали при комнатной температуре в течение 8 часов. Реакционную смесь нейтрализовали 30 мл триэтиламина и отгоняли при пониженном давлении растворитель с получением метил 5-O-бензоил-3-C-(1-пропинил) -α,β D-рибофуранозы в виде сиропообразного вещества. Это соединение подвергали три раза азеотропной перегонке с пиридином и затем растворяли в 110 мл пиридина. К раствору добавляли при охлаждении ледяной водой 8,0 мл (69 ммоль) бензоилхлорида и 1,27 (10,4 ммоль) диметиламинопиридина и перемешивали смесь при 100oC в течение 24 часов. После охлаждения реакционной смеси до комнатной температуры отгоняли при пониженном давлении растворитель. Остаток подвергали трехкратной азеотропной перегонке с толуолом. Полученный остаток растворяли в 150 мл этилацетата и раствор подвергали жидкостному разделению с использованием 50 мл воды и насыщенного водного раствора гидрокарбоната натрия (3 х 50 мл) в указанном порядке, после чего полученный органический слой сушили на сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 0-10% этилацетата-н-гексан) с получением 2,8 г (выход 80%) целевого соединения в виде желтого сиропообразного вещества.
FAB-MC: m/z 515 (MH+, 483 (M+-OMe).
1H-ЯМР (CDCl3) δ: 8,16-7,29 (м, 15H, бензоил х 3), 5,95 (с, 0,6H, β- H-1), 5,79 (д, 0,4H, α- H-1, J1,2 =4,4 Гц), 5,45 (д, 0,4H, α- H-2, J2,1=4,4 Гц), 5,13 (с, 0,6H, β- H-2), 5,03-4,72 (м, 3H, α,β- H-4, H-5), 3,51 (с, 1,8H, β- OMe), 3,43 (с, 1,2H, α- OMe), 1,90 ( 1,8H, β-3-C≡ C-CH3 ), 1,87 (с, 1,2H, 
Соотношение номеров α:β= 2:3 (при определении по спектру 1H-ЯМР).
Элементный анализ:
Вычислено (для C30H26O8): C, 70,03; H 5,09
Найдено: C, 69,77; H, 4,86.
Ссылочный пример 8
Синтез 1-O-ацетил-2,3,5-три-O-бензоил-3-C-(1-пропинил) -α,β- D-рибофуранозы
В 16,55 мл уксусной кислоты и 2,08 мл уксусного ангидрида растворяли 2,57 г (5,0 ммоль) соединения, полученного в ссылочном примере 7. Добавляли при охлаждении ледяной водой 1,04 мл концентрированной серной кислоты и смесь перемешивали при комнатной температуре 30 минут. Добавляли к реакционной смеси 50 мл хлороформа и полученную смесь подвергали жидкостному разделению с использованием 5 мл воды, насыщенного водного раствора гидрокарбоната натрия (3 x 15 мл) и воды (2 x 5 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 10 - м20% этилацетат/н-гексан) с получением 2,64 г (выход 98%) целевого соединения в виде сиропообразного вещества.
FAB-MC: m/z 543 (MH+).
1H-ЯМР (CDCl3) δ: 8,16 - 7,34 (м, 15H, бензоил • 3), 6,75 (д, 0,4H, α- H-1, J1,2 = 4,4 Гц), 6,35 (д, 0,6H, β- H-1, J1,2 = 1,5 Гц), 6,11 (д, 0,6H, β- H-2, J2,1 = 1,5 5 Гц), 6,02 (д, 0,4H, -α- H-2, J2,1 = 4,4 Гц), 4,97 - 4.79 (м, 3H, α,β- H-4, H - 5), 2,13 (с, 1,8H, β- ацетил), 1,97 (с, 1,2H, α- ацетил), 1,92 (с, 1,8H,  ), 1,86 (с, 1,2H,
), 1,86 (с, 1,2H, 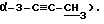
Соотношение номеров α:β = 2 : 3 (при определении по спектру 1H ЯМР).
Элементный анализ:
Вычислено (для C31H26O9): C, 68,63; H, 4,83
Найдено: C, 68,45; H, 4,71.
Ссылочный пример 9
Синтез 5-O-трет-бутилдиметилсилил-1,2-O-изопропилиден-3- C-(1-бутинил) _α- D-рибофуранозы
Газообразный бутин сжижали при -30oC в атмосфере аргона и, накопив примерно 0,4 мл жидкого бутина в трехгорлой колбе, добавляли туда 5 мл тетрагидрофурана и смесь перемешивали при -78oC. Поддерживая температуру реакционной смеси на уровне -70oC или ниже, добавляли по каплям в течение 30 минут н-бутиллитий (раствор в н-гексане, 1,63 моль/литр; 1,84 мл, 3,0 ммоль). По истечении 30 минут после капельного добавления добавляли по каплям в течение 10 минут 302 мг (1,0 ммоль) 5-O-трет-бутилдиметилсилил-1,2-O-изопропилиден -α- D-эритропентофураноз-3-улозы, растворенной в 2,5 мл тетрагидрофурана, и смесь перемешивали еще 2 часа. Затем к реакционной смеси добавляли 5 мл 1 н. водного раствора аммоний-хлорида и температуру смеси повышали до комнатной температуры. После этого реакционную смесь экстрагировали этилацетатом (3 x 5 мл) и полученный органический слой промывали насыщенным водным раствором (3 x 3 мл) хлорида натрия и затем сушили над сульфатом натрия. Затем органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5% этилацетат/н-гексан) с получением 259 мг (выход 73%) целевого соединения в виде сиропообразного вещества.
FAB-MC: m/z 341 (M+-Me).
ИК (чистый): 2245 см-1 (-C=CC-).
1H-ЯМР (CDCl3) δ: 5,83 (д, 1H, H-J1,2 = 3,5 Гц), 4,51 (д, 1H, H-2, J2,1 = 3,3 Гц), 4,06 - 3,92 (м, 3H, H-4, H-5), 2,97 (с, 1H, 3-OH, обмениваемый с D2O), 2,24 (м, 2H, 3- 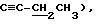 1,60, 1,37 (с, каждый, 3H, изопропил), 1,15 (т, 3H, J = 7,4 Гц,
1,60, 1,37 (с, каждый, 3H, изопропил), 1,15 (т, 3H, J = 7,4 Гц, 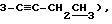 0,91 (с, 9H, tBu), 0,11o, 0,09 (с, каждый, 3H, Me).
0,91 (с, 9H, tBu), 0,11o, 0,09 (с, каждый, 3H, Me).
Элементный анализ:
Вычислено (для C18H32O5): C, 60,64; H, 9,05.
Найдено: C, 60,21; H, 9,12.
Ссылочный пример 10
Синтез 5-O-бензоил-3-C-(1-бутинил)-1,2-O-изопропилиден -α- D-рибофуранозы
В 30 мл тетрагидрофурана растворяли 3,56 г (10,0 ммоль) соединения, полученного в ссылочном примере 9, и добавляли 10,0 мл (10,0 ммоль) 1 н. раствора тетрабутиламмонийфторида в тетрагидрофуране с последующим перемешиванием при комнатной температуре в течение 20 минут. Отгоняли растворитель и получали 3-C-(1-бутинил)-1,2-O-изопропилиден -α_ D-рибофуранозу в виде сиропооразного вещества. Это соединение растворяли в 50 мл пиридина и к раствору добавляли при охлаждении ледяной водой 2,55 мл (22,0 ммоль) бензоилхлорида с последующим перемешиванием при комнатной температуре в течение 4 часов. Отгоняли при пониженном давлении растворитель и остаток подвергали три раза азеотропной перегонке с толуолом. Полученный остаток растворяли в 100 мл этилацетата и раствор подвергали жидкостному разделению с использованием 50 мл воды и насыщенного водного раствора гидрокарбоната натрия (3 x 50 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5 - 10 - 20% этилацетат/н-гексан) с получением 2,98 г (выход 86%) целевого соединения в виде белого порошкообразного вещества.
Т.пл.: 110 - 113oC.
FAB-MC: m/z 347 (MH+).
1H-ЯМР (CDCl3) δ: 8,09 - 7,42 (м, 5H, бензоил), 5,93 (д, 1H, H-1, J1,2 = 3,6 Гц), 4,73 (дд, 1H, H-5a, J5a,4 = 3,4 Гц, J5a,5b = 12,0 Гц), 4,56 (дд, 1H, H-5b, J5b,4 = 7,7 Гц, J5b,5a = 12,0 Гц), 4,53 (д, 1H, H-2, J2,1 = 3,6 Гц), 4,18 (дд, 1H, H-4, J4,5a = 3,4 Гц, J4,5b = 7,7 Гц), 2,90 (с, 1H, 3-OH, обмениваемый с D2O), 2,26 (м, 2H,  ) 1,60, 1,39 (с, каждый 3H, ipr, изопропил), 1,16 (т, 3H, J = 7,5 Гц,
) 1,60, 1,39 (с, каждый 3H, ipr, изопропил), 1,16 (т, 3H, J = 7,5 Гц, 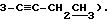 )
)
Элементный анализ:
Вычислено (для C19H22O6): C, 65,88; H, 6,40.
Найдено: C, 65,69; H, 6,52.
Ссылочный пример 11
Синтез 2,3,5-три-O-трет-бензоил-3-C-(1-бутинил) -α,β- D-рибофуранозы
К 20,3 мл воды и 69,0 мл метанола добавляли при охлаждении ледяной водой 12,3 мл (164 ммоль) ацетилхлорида и смесь перемешивали при комнатной температуре в течение 20 минут. К этому раствору добавляли 2,2 г (6,4 ммоль) соединения, полученного в ссылочном примере 10, после чего смесь перемешивали при комнатной температуре в течение 10 часов. Реакционную смесь нейтрализовали 30 мл триэтиламина и отгоняли при пониженном давлении растворитель с получением метил 5-O-бензоил-3-C-(1-бутинил) -α,β- D-рибофуранозы в виде сиропообразного вещества. Это соединение подвергали три раза азеотропной перегонке с пиридином и затем растворяли в 110 мл пиридин. К раствору добавляли при охлаждении ледяной водой 7,4 мл (64 ммоль) бензоилхлорида и 1,2 г (9,5 ммоль) диметиламинопиридина и перемешивали смесь при 100oC в течение 24 часов. После охлаждения реакционной смеси до комнатной температуры отгоняли при пониженном давлении растворитель. Остаток подвергали трехкратной азеотропной перегонке с толуолом. Полученный остаток растворяли в 150 мл этилацетата и раствор подвергали жидкостному разделению с использованием 50 мо воды и насыщенного водного раствора гидрокарбоната натрия (3 x 50 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 0 - 10% этилацетат/н-гексан) с получением 3,0 г (выход 88%) целевого соединения в виде желтого сиропообразного вещества.
FAB-MC: m/z 529 (M+), 497 (M+-OMe).
1H-ЯМР (CDCl3) δ: 8,16 - 7,31 (м, 15H, бензоил • 3), 5,96 (д, 0,6H, β- H-1, J1,2 = 0,9 Гц), 5,80 (д, 0,4H, α- H-1, J1,2 = 4,4 Гц), 5,45 (д, 0,4H, α- H-2, J2,1 = 4,4 Гц), 5,14 (д, 0,6H, β- H-2, J2,1 = 0,9 Гц), 5,01 - 4,73 (м, 3H, α,β- H-4, H-5), 3,51 (с, 1,8H, β- OMe), 3,43 (с, 1,2H, α- OMe), 2,28 - 2,21 (м, 2H, 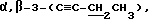 ), 1,14 - 1,10 (м, 3H,
), 1,14 - 1,10 (м, 3H, 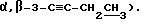
Соотношение номеров α:β = 2 : 3 (при определении по спектру 1H-ЯМР).
Элементный анализ:
Вычислено (для C31H28O8): C, 70,44; H, 5,34.
Найдено: C, 70,05; H, 5,11.
Ссылочный пример 12
Синтез 1-O-ацетил-2,3,5-три-O-бензоил-3-C-(1-бутинил) α,β- D-рибофуранозы
В 16,89 мл уксусной кислоты и 2,13 мл уксусного ангидрида растворяли 2,69 г (5,1 ммоль) соединения, полученного в ссылочном примере 11. Добавляли при охлаждении ледяной воды 1,06 мл концентрированной серной кислоты и смесь перемешивали при комнатной температуре 30 минут. Добавляли к реакционной смеси 50 мл хлороформа и полученную смесь подвергали жидкостному разделению с использованием 5 мл воды, насыщенного водного раствора гидрокарбоната натрия (3 х 15 мл) и воды (2 х 5 мл) в указанном порядке, после чего полученный органический слоя сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 10% этилацетат/н-гексан) с получением 2,37 г (выход 83%) целевого соединения в виде сиропообраного вещества.
FAB-МС: m/z 557 (MH+), 513 (M+-Ac), 497 (N+-OAc).
1H-ЯМР (CDCl3) δ: 8,16-7,34 (м, 15H, бензоил х 3),
6,75 (д, 0,4H, α- H-J1,2 = 4,5 Гц),
6,36 (д, 0,6H, β- H-1, J1,2 = 1,1 Гц),
6,12 (д, 0,6H, β- H-2, J2,1 = 1,1 Гц),
6,03 (д, 0,4H, α- H-2, J2,1 = 4,5 Гц),
5,02-4,77 (м, 3H, α,β- H-4, H-5),
2,31-2,28 (м, 1,2H, 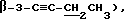
2,25-2,19 (м, P, 8H, α-3-C≡C-CH2CH3),
2,14 (с, 1,8H, β- ацетил), 1,99 (с, 1,2H, α- ацетил),
1,18-1,15 (м, 1,8H, 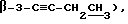
1,10-1,07 (м, 1,2H, 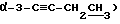
Соотношение номеров α:β = 2:3 (при определении по спектру 1H ЯМР).
Элементный анализ:
Вычислено: (для C32H28O9): C, 69,06; H, 5,07.
Найдено: C, 68,88; H, 5,15.
Ссылочный пример 13
Синтез 5-O-трет-бутилдиметилсилил-1,2-O-изопропилиден-3-C-этенил -α- D-рибофуранозы
Винилмагнийбромид (1 н. тетрагидрофурановый раствор, 30,0 мл, 30,0 ммоль) растворяли в 30 мл тетрагидрофурана в атмосфере аргона и раствор перемешивали при -15oC. Поддерживая температуру реакционного раствора на уровне -10oC или ниже, добавляли по каплям в течение 30 минут 3,02 г (10,0 ммоль) 5-O-трет-бутилдиметилсилил-1,2-O-изопропилиден -α- D-эритропентофураноз-3-улозы, растворенной в 40 мл тетрагидрофурана, и смесь перемешивали еще 2 часа. Затем к реакционной смеси добавляли 50 мл 1 н. водного раствора аммонийхлорида и температуру смеси повышали до комнатной температуры. После этого реакционную смесь экстрагировали этилацетатом (3 x 35 мл) и полученный органический слой промывали насыщенным водным раствором (3 x 30 мл) хлорида натрия и затем сушили над сульфатом натрия. Затем органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5% этилацетата-н-гексан) с получением 2,11 г (выход 64%) целевого соединения в виде сиропообразного вещества.
FAB-МС: m/z 315 (M+-Me).
1H-ЯМР (CDCl3) δ: 5,83 (д, 1H, H-1, J1,2 = 3,8 Гц),
5,77 (дд, 1H, 3-CHc=CHaCHb, Jc,a = 17,2 Гц, Jc,b = 11,0 Гц),
5,53 (дд, 1H, 3-CHc=CHaCHb, Ja,c = 17,2 Гц, Ja,b = 1,5 Гц),
5,28 (дд, 1H, 3-CHc=CHaCHb, Jb,c = 11,0 Гц, Jb,a = 1,5 Гц),
4,22 (д, 1H, H-2, J2,1 = 3,8 Гц), 3,98 (т, 1H, H-4, J = 5,6 Гц), 3,69-3,67 (м, 2H, H-6),
2,76 (с, 1H, 3-OH, обмениваемый с D2O),
1,61, 1,35 (с, каждый 3H, изопропил), 0,88 (с, 9H, tBu),
0,06, 0,05 (с, каждый, 3H, Me).
Ссылочный пример 14
Синтез 5-O-бензоил-3-C-этенил-1,2-O-изопропилиден -α- D-рибофуранозы
В 20 мл тетрагидрофурана растворяли 1,67 г (5,1 ммоль) соединения, полученного в ссылочном примере 13, и добавляли 5,1 мл (5,1 ммоль) 1 н. раствора тетрабутиламмонийфторида в тетрагидрофуране с последующим перемешиванием при комнатной температуре в течение 20 минут. Отгоняли растворитель и получали 5-O-трет-бутилдиметилсилил-3-C-этенил-1,2-O-изопропилиден -α- D-рибофуранозу в виде сиропообразного вещества. Это соединение растворяли в 35 мл пиридина и к раствору добавляли при охлаждении ледяной водой 1,72 мл (15,0 ммоль) бензоилхлорида с последующим перемешиванием при комнатной температуре в течение 4 часов. Отгоняли три раза азеотропной перегонкой с толуолом. Полученный остаток растворяли в 65 мл этилацетата и раствор подвергали жидкостному разделению с использованием 25 мл воды и насыщенного водного раствора гидрокарбоната натрия (3 x 25 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5-10-20% этилацетат/г-гексан) с получением 1,46 г (выход 90%) целевого соединения в виде белого порошкообразного вещества.
Т.пл.: 110-111oC.
FAB-МС: m/z 321 (MH+), 305 (M+-Me).
1H-ЯМР (CDCl3) δ: 8,07-7,42 (м, 5H, бензоил),
5,91 (д, 1H, H-1, J1,2 = 3,8 Гц),
5,81 (дд, 1H, 3-CHc-CHaCHb, Jc,a = 17,3 Гц, Jc,b = 11,0 Гц),
5,60 (дд, 1H, 3-CHc=CHaCHb, Ja,c = 17,3 Гц, Ja,b = 1,3 Гц),
5,36 (дд, 1H, 3-CHc=CHaCHb, Jb,c = 11,0 Гц, Jb,a = 1,3 Гц),
4,45 (дд, 1H, H-5a, J5a,4 = 2,9 Гц, J5a,5b = 12,1 Гц),
4,30 (дд, 1H, H-5b, J5b,4 = 8,1 Гц, J5b,5a = 12,1 Гц),
4,26 (д, 1H, H-2, J2,1 = 3,8 Гц),
4,21 (дд, 1H, H-4, J4,5a = 2,9 Гц, H4,5b = 8,1 Гц),
2,84 (с, 1H, 3-OH, обмениваемый с D2O),
1,62, 1,38 (с, каждый, 3H, изопропил).
Элементный анализ:
Вычислено (для C17H20O6): C, 63,74; H, 6,29.
Найдено: C, 63,75; H, 6,21.
Ссылочный пример 15
Синтез метил 2,3,5-три-O-бензоил-3-C-этенил -α,β- D-рибофуранозы
К 17,0 мл воды и 57,6 мл метанола добавляли при охлаждении ледяной водой 10,3 мл (137 ммоль) ацетилхлорида и смесь перемешивали при комнатной температуре в течение 20 минут. К этому раствору добавляли 1,7 г (5,3 ммоль) соединения, полученного в ссылочном примере 14, после чего смесь перемешивали при комнатной температуре в течение 5 часов. Реакционную смесь нейтрализовали 20 мл триэтиламина и отгоняли при пониженном давлении растворитель с получением метил 5-O-бензоил-3-C-этенил -α,β- D-рибофуранозы в виде сиропообразного вещества. Это соединение подвергали три раза азеотропной перегонке с пиридином и затем растворяли в 85 мл пиридина. К раствору добавляли при охлаждении ледяной водой 6,2 мл (53 ммоль) бензоилхлорида и 0.97г (8,0 ммоль) диметиламинопиридина и перемешивали полученную смесь при 100oC в течение 24 часов. После охлаждения реакционной смеси до комнатной температуры отгоняли при пониженном давлении растворитель. Остаток подвергали трехкратной азеотропной перегонке с толуолом. Полученный остаток растворяли в 120 мл этилацетат и раствор подвергали жидкостному разделению с использованием 35 мл воды и насыщенного водного раствора гидрокарбоната натрия (3 х 35 м) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 0-10% этилацетата-н-гексан) с получением 1,7 (выход 66%) целевого соединения в виде желтого сиропообразного вещества.
FAB-MC: m/z 487 (M+-Me), 471 (M+-OMe).
1H-ЯМР (CDCl3) δ: 8,14-7,24 (м, 15H, бензоил • 3), 6,36 (дд, 0,75 H, β- 3-CHc-CHaCHb, Jc,a = 17,6 Гц, Jc,b = 11,2 Гц), 6.22 (дд, 0,25H, -3-CHc-CHaCHb, Jc,a = 17,6 Гц, Jc,b = 11,2 Гц), 5,86 (с, 0,75H, β- H-1), 5,72 (д, 0,25H, α- H-1, J1,2 = 4,7 Гц), 5,45 (д, 0,25H, α- H-2, J2,1 = 4,7 Гц), 5,40 - 5,33 (м, 2H, α,β- 3-CHc=CHaCHb), 5,15 (c, 0,75H β- H-2), 4.95 - 4,93 (м, 1H, α,β- H-4), 4,75 (дд, 0,25 H, α- H-5a, J5a,4 = 3,9 Гц, J5a,5b = 11,9 Гц), 4,69 (дд, 0,75H, β- H-5a, J5a,4 =4,2 Гц, J5a,5b = 11,8 Гц), 4,57 (дд, 0,75H, α- H-5b, J5b,4 = 6,2 Гц, J5b,5a = 11,9 Гц), 4,51 (дд, 0,75H, β- H-5b, J5b,4 = 7,2 Гц, J5b,5a = 11,8 Гц), 3,53 (с, 2,25H, β- OMe), 3,44 (с, 0,75H, α- OMe).
Соотношение аномеров α:β = 1:3 (при определении по спектру 1H-ЯМР.
Элементный анализ:
Вычислено (для C29H26O8): C, 69,31; H, 5,21.
Найдено: C, 69,45; H, 5,00.
Ссылочный пример 16
Синтез 1-O-ацетил-2,3,5-три-O-бензоил-3-C-этенил -α,β- D-рибофуранозы
В 9,86 мл уксусной кислоты и 1,24 мл уксусного ангидрида растворяли 1,50 г (2,98 ммоль) соединения, полученного в ссылочном примере 15. Добавляли при охлаждении ледяной водой 0,62 мл концентрированной серной кислоты и смесь перемешивали при комнатной температуре 30 минут. Добавляли к реакционной смеси 23 мл хлороформа и полученную смесь подвергали жидкостному разделению с использованием 3 мл воды, насыщенного водного раствора гидрокарбоната натрия (3х7 мл) и воды (2х3 мл) в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 10-20% этилацетат/н-гексан) с получением 2.27 г (выход 80%) целевого соединения в виде сиропообразного вещества.
FAB-MC: m/z 531 (MH+).
1H-ЯМР (CDCl3) δ: 8,15 - 7,31 (м, 15H, бензоил • 3),
6,73 (д, 0,33H, α- H-1, J1,2 = 4,7 Гц),
6,40 (д, 0,66H, β- H-1, J1,2 = 1,3 Гц),
6,33 (дд, 0,66H, β- B-CHc=CHaCHb, Jc,a = 17,4 Гц, Jc,b = 11,3 Гц),
6,26 (дд, 0,33H, α- 3-CHc=CHaCHb, Jc,a = 17,5 Гц, Jc,b = 11,4 Гц),
6,04 (д, 0,66H, β- H-2, J2,1 = 1,3 Гц),
5,97 (д, 0,33H, α- H-2, J2,1 = 4,7 Гц),
5,48 - 5,41 (м, 2H, α,β- 3-CHc=CHaCHb),
5,27 - 5,03 (м, 1H, α,β- H-4),
4,78-4,50 (м, 2H, α,β-H-5),
2,14 (c, 1,98H, β- ацетил), 1,97 (с, 0,99H, α- ацетил).
Соотношение аномеров α:β = 1:2 (при определении по спектру 1H-ЯМР).
Элементный анализ:
Вычислено (для C30H26O9): C, 67,92; H, 4,94.
Найдено: C, 67,75; H, 4,83.
Пример 1
Синтез 1-(2,3,5-три-O-бензоил-3-C-этинил -α- C-рибофуранозил)цитозина (Соединение 1)
К 222 мг (2,0 ммоль) цитозина добавляли 2,0 мл гексаметилдисилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения цитозина. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 264 мг (0,5 ммоль) полученного в ссылочном примере 4 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,29 мл (2,5 ммоль) тетрахлорида олова при 0oC. Перемешивали смесь при комнатной температуре 18 часов. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия и перемешивали смесь при комнатной температуре в течение 30 минут. Затем отфильтровывали через целит образовавшийся осадок. Фильтрат подвергали жидкостному разделению с использованием воды (2 х 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5% метанол/хлороформ) с получением 235 мг (выход 81%) целевого соединения 1 в виде вспененного вещества.
FAB-MC: m/z 580 (MH+)
1H-ЯМР (CDCl3) δ: 8,15 - 7,51 (м, 15H, бензоил • 3),
7,76 (д, 1H, H-6, J6,5 = 7 Гц),
6,60 (д, 1H, H-1, J1',2' = 5,2 Гц),
6,06 (д, 1H, H-2', J2',1' = 5,2 Гц),
5,72 (д, 1H, H-5, J5,6 = 7,5 Гц), 4,96 - 4,89 (м, 3H, H-4', H-5'), 2,88 (c, 1H, 3′-C≡CH).
Пример 2
Синтез 1-(3-C-этинил -β- рибофуранозил)цитозина (Соединение 2)
В 9 мл раствора аммония в метаноле растворяли 200 мг (0,35 ммоль) соединения 1, полученного в примере 1, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5-20% метанол/хлороформ), получая в результате 83.0 мг (выход 90%) целевого соединения 2 в виде белого порошкоообразного вещества.
Т.пл.: 233-235oC,
E1-MC: m/z 267 (M+), 250 (M+-OH).
ИК (нуйор): 2115 см-1 (-C≡C-).
УФ: λmax (H2O) 270 нм (ε 9100):
λmax (0,5 н. хлороводородная кислота) 280 нм (ε 13400);
λmax (0,5 н. гидроксид натрия) 272 нм (ε 9300).
1H-ЯМР (DMCO-d6: 7,86 (д, 1H, H-6, J6.5 = 7,7 Гц), 7,25, 7.18 (шир.с, каждый 1H, -NH, обмениваемый c D2O), 5,88 (д, 1H, H-1', J1',2' = 6,6 Гц), 5.84 (с, 1H, 3'-OH, обмениваемый с D2O), 5,79 (д, 1H, 2-OH, J2'-CH,2' = 6,6 Гц, обмениваемый с D2O), 5,78 (д, 1H, H-5, J5,6 = 7,7 Гц), 5,06 (дд, 1H, 5'-OH, J5'-OH,5'a = 4,4 Гц, J5'-OH ,5'b= 5,5 Гц, обмениваемый с D2O), 4,16 (т, 1H, H-2', J= 6,6 Гц). 3,92- 3,89 (м, 1H, H-4'), 3,74 - 3,71 (м, 2H, H-5'), 3,55 (c, 1H, 3′-C≡CH).
Элементный анализ:
Вычислено (для C11H133O5): C, 49,45; H, 4,90; N, 15,72.
Найдено: C, 49,55; H, 4.76; N, 15,70.
Пример 3
Синтез 1-(2,3,5-три-O-бензоил-3-C-этинил -β- D-рибофуранозил)-5-фторцитозина (Соединение 3)
К 258 мг (2,0 ммоль) 5-фторцитозина добавляли 2,0 мл гексаметилдисилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения 5-фторцитозина. Затем реакционную смесь охлаждали до комнатной температуре, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 264 мг (0,5 ммоль) полученного в ссылочном примере 4 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,29 мл (2,5 ммоль) тетрахлорида олова при 0oC. Перемешивали смесь при комнатной температуре 18 часов. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия и перемешивали смесь при комнатной температуре в течение 30 минут. Затем отфильтровывали через целит образовавшийся осадок. Фильтрат подвергали жидкостному разделению с использованием воды (2 х 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5% метанол/хлороформ) с получением 224 мг (выход 81%) целевого соединения 3 в виде вспененного вещества.
FAB-MC: m/z 598 (MH+).
1H-ЯМР (CDCl3) δ: 8,15 - 7,31 (м, 15H, бензоил • 3), 7,7 (д, 1H, H-6, J6,5-Г = 6,1 Гц), 6,54 (д, 1H, H-1', J1',2'= 5,1 Гц), 6,03 (д, 1H, H-2', J2',1' = 5,1 Гц), 4,97 - 4,96 (м, 2H, H-5'), 4,91 - 4,89 (м, 1H, H-4'), 2,90 (c, 1H, 3′-C≡CH).
Пример 4
Синтез-1-(3-C-этинил -β- D-рибофуранозил)-5-фторцитозина (Соединение 4)
В 8 мл растворе аммония метанола растворяли 189 мг (0,32 ммоль) соединения 3, полученного в примере 3, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5-20% метанол/хлороформ), получая в результате 81.0 мг (выход 89%) целевого соединения 4 в виде белого порошкообразного вещества.
Т.пл.: 242oC (разложение).
E1-MC: m/z 285 (M+).
ИК (нуйор): 2115 см-1 ( (-C≡C-).
1H-ЯМР (ДМСО-d6) δ: 8,11 (д, 1H, H-6, J6,5-F = 7,2 Гц),
7,82, 7,57 (шир.с., каждый 1H, -NH, обмениваемый с D2O),
5,84 (дд, 1H, H-1', J1',2' = 6,9 Гц, J1',5-F = 1,9 Гц).
5,81 (с, 1H, 3'-OH, обмениваемый c D2O),
5,72 (д, 1H, 2'-OH, J2'-OH,2' = 6,6 Гц, обмениваемый с D2O),
5,14 (т, 1H, 5'-OH, J = 4,5 Гц, обмениваемый с D2O),
4,13 (дд, 1H, H-2', J2',1' = 6,9 Гц, J2',2'-OH = 6,6 Гц,
3,88 - 3,87 (м, 1H, H-4'), 3,75 - 3,60 (м, 2H, H-5'),
3,53 (c, 1H, 3′-C≡CH).
Элементный анализ:
Вычислено (для C11H12FN3O5):
C, 46,32; H, 4,24; F, 6,66; N, 14,73.
Найдено: C, 46,12; H, 4,28; F, 6,61; N, 14,69.
Пример 5
Синтез 1-(2,3,5-три-O-бензоил-/-C-этинил -β-D- рибофуранозил)урацила (Соединение 5) и 3-(2,3,5-три-O-бензоил-3-C-этинил- -β- D-рибофуранозил)урацила (Соединение 6)
К 225 мг (2,0 ммоль) урацила добавляли 2,0 мг гексаметилилдисилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения урацила. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 264 мг (0,5 ммоль) полученного в ссылочном примере 4 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,23 мл (2,0 ммоль) тетрахлорида олова при 0oC. Перемешивали смесь при комнатной температуре 2 дня. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия и перемешивали смесь при комнатной температуре в течение 30 минут. Затем отфильтровывали через целит образовавшийся осадок. Фильтрат подвергали жидкостному разделению с использованием воды (2 x 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 0 - 5% метанола-хлороформ) с получением 167 мг целевого соединения 5 (элюирование хлороформом, выход 58%) и 57 мг целевого соединения 6 (элюирование смесью 5% метанола/хлороформ, выход 20%), оба в виде вспененных веществ.
Соединение 5:
FAB-MC: m/z 581 (MH+).
1H-ЯМР (CDCl3) δ: 8,13 - 7,30 (м, 15H, бензоил • 3),
8,05 (шир. 1H, - NH, обмениваемый с D2O),
7,71 (д, 1H, H-6, J6,5 = 8,2 Гц),
6,38 (д, 1H, H-1', J1',2' = 5,0 Гц),
6,01 (д, 1H, H-2', J2',1' = 5,0 Гц),
5,74 (дд, 1H, H-5, J5,6 = 8,2 Гц, J5,NH = 2,0 Гц),
4,98 - 4,87 (м, 3H, H-4', H-5'), 2,92 (с, 1H, 3' -C≡CH).
Соединение 6:
FAB-MC: m/z 581 (MH+).
1H-ЯМР (CDCl3) δ: 9,26 (шир. 1H, - NH, обмениваемый с D2O),
8,14 - 7,29 (м, 16H, бензоил • 3, H-6),
6,92 (д, 1H, H-1', J1',2' = 6,8 Гц),
6,69 (д, 1H, H-2', J2',1' = 6,8 Гц),
5,82 (д, 1H, H-5, J5,6 = 7,6 Гц), 5,04 - 4,88 (м, 3H, H-4', H-5'),
2,82 (с, 1H, 3' -C≡CH).
Пример 6
Альтернативный синтез 1-(2,3,5-три-O-бензоил-3-C-этинил -β- D-рибофуранозил)урацила (Соединение 5)
К 225 мг (2,0 ммоль) урацила добавляли 2,0 мл гексаметилдисилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения урацила. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 264 мг (0,5 ммоль) полученного в ссылочном примере 4 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,39 мл (2,0 ммоль) триметилсилилтрифторметансульфоната при 0oC. Перемешивали смесь при комнатной температуре 5 часов. Затем добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия и, перемешав смесь при комнатной температуре в течение 30 минут, подвергали реакционную смесь жидкостному разделению с использованием воды (2 x 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование хлороформом) с получением 274 мг (выход 95%) целевого соединения 5 в виде вспененного вещества.
Пример 7
Синтез 1-(3-C-этинил -β D/рибофуранозил)урацила (Соединение 7)
В 10 мл раствора аммония метанола растворяли 150 мг (0,26 ммоль) соединения 5, полученного в примере 5 или 6, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5 - 15% метанонал/хлороформ), получая в результате 59,4 мг (выход 85%) целевого соединения 7 в виде белого порошкообразного вещества.
Т. пл.: 226 - 228oC.
E1-MC: m/z 268 (M+).
ИК (нуйор): 2110 см-1 (-C ≡ C-).
УФ: λmax (H2O, 0,5 н. хлороводородная кислота) 261 нм (ε 10200);
λmax (0,5 н. гидроксид натрия) 262 нм (ε 7500).
1H-ЯМР (ДМСО-d6) δ: : 11,35 (шир.с, 1H, - NH, обмениваемый с D2O), 7,99 (д, 1H, H-6, J6,5 = 8,2 Гц), 5,93 (с, 1H, 3'-OH, обмениваемый с D2O), 5,86 (д, 1H, 2'-OH, J2'-OH,2' = 6,7 Гц, обмениваемый с D2O), 5,83 (д, 1H, H-1', J1',2' = 7,3 Гц), 5,69 (д, 1H, H-5, J5,6 = 8,2 Гц), 5,13 (т, 1H, 5'-OH, J = 4,5H, обмениваемый с D2O), 4,18 (дд, 1H, H-2', J2',1' = 7,3 Гц, J2',2'-OH = 6,7 Гц), 3,90 - 3,88 (м, 1H, H-4'), 3,74 - 3,60 (м, 2H, H-5'), 3,55 (с, 1H, 3' -C≡CH).
Элементный анализ:
Вычислено (для C11H12N2O6):
C, 49,26; H, 4,51; N, 10,44.
Найдено: C, 49,00; H, 4,84; N, 10,52.
Пример 8
Синтез 3-(3-C-этинил -β- D-рибофуранозил)урацила (Соединение 8)
В 1,5 мл раствора аммония в метаноле растворяли 55 мг (0,10 ммоль) соединения 6, полученного в примере 5, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5-15% метанол/хлороформ), получая в результате 17,1 мг (выход 67%) целевого соединения 8 в виде белого порошкообразного вещества.
Т. пл.: 255oC (разложение).
E1-MC: m/z 250 (M+-OH), 237 (M+-CH2OH).
ИК (нуйор): 2230 см-1 (-C ≡ C-).
УФ: λmax (H2O) 264 нм (ε 7000);
λmax (0,5 н. хлороводородная кислота) 264 нм ( ε 7200);
λmax (0,5 н. гидроксид натрия) 293 нм (ε 9900).
1H-ЯМР (ДМСО-d6) δ: 11,20 (шир.с, 1H, - NH, обмениваемый с D2O), 7,47 (д, 1H, H-6, J6,5 = 7,6 Гц), 6,09 (д, 1H, H-1', J1',2' = 8,3 Гц), 5,67 (с, 1H, 3'-OH, обмениваемый с D2O), 5,62 (д, 1H, 2'-OH, J2'-OH,2' = 7,0 Гц, обмениваемый с D2O) 5,60 (д, 1H, H-5, J5,6 = 7,6 Гц) 5,06 (дд, 1H, H-2', J2',1' = 8,3 Гц, J2',2'-OH = 7,0 Гц), 4,48 - 4,46 (м, 1H, 5'-OH, обмениваемый с D2O) 3,86 - 3,84 (м, 1H, H-4'), 3,68 - 3,61 (м, 2H, H-5') 3,37 (с, 1H, 3′-C≡CH).
Элементный анализ:
Вычислено (для C11H12N2O6 • 1/2H2O):
C, 47,66; H, 4,73; N, 10,10.
Найдено: C, 47,99; H, 4,66; N, 10,19.
Пример 9
Синтез 1-(2,3,5-три-O-бензоил-3-C-этинил -β- D-рибофуранозил)-5-фторурацила (Соединение 9)
К 262 мг (2,0 ммоль) 5-фторурацила добавляли 2,0 мл гексаметилдисилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения 5-фторурацила. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 264 мг (0,5 ммоль) полученного в ссылочном примере 4 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,23 мл (2,0 ммоль) тетрахлорида олова при 0oC. Перемешивали смесь при комнатной температуре 6,5 часов. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия и перемешивали смесь при комнатной температуре в течение 30 минут. Затем отфильтровывали через целит образовавшийся осадок. Фильтрат подвергали жидкостному разделению с использованием воды (2 x 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование хлороформом) с получением 283 мг (выход 95%) целевого соединения 9 в виде вспененного вещества.
FAB-MC: m/z 599 (MH+).
1H-ЯМР (CDCl3) δ: 8,16 (шир.с, 1H, - NH, обмениваемый с D2O), 8,14 - 7,30 (м, 15H, бензоил • 3), 7,83 (д, 1H, H-6, J6,5-F = 5,8 Гц), 6,35 (дд. 1H, H-1', J1',2' = 4,9 Гц, J1',5-F = 1,7 Гц), 5,79 (д, 1H, H-2', J2',1' = 4,9 Гц), 4,96 - 4,95 (м, 2H, H-5'), 4,89 - 4,87 (м, 1H, H-4') 2,95 (с, 1H, 3′-C≡CH).
Пример 10
Синтез 1-(3-C-этинил -β- D-рибофуранозил)-5-фторурацила (Соединение 10)
В 12 мл раствора аммония в метаноле растворяли 276 мг (0,46 ммоль) соединения 9, полученного в примере 9, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5-20% метанола-хлороформ), получая в результате 121 мг (выход 92%) целевого соединения 10 в виде белого порошкообразного вещества.
E1-MC: m/z 286 (M+), 251 (M+-OH)
1H-ЯМР (ДМСО-d6) δ: 11,87 (шир.с, 1H, - NH, обмениваемый с D2O),
8,33 (д, 1H, H-6, J6,5-F = 7,2 Гц),
5,93 (с, 1H, 3'-OH, обмениваемый с D2O),
5,83 (дд, 1H, H-1', J1',2' = 7,2 Гц, J1',5-F = 1,8 Гц),
5,82 (д, 1H, 2'-OH, J2'-OH2' = 6,4 Гц, обмениваемый с D2O)
5,27 (т, 1H, 5'-OH, J = 4,1 Гц, обмениваемый с D2O),
4,18 (дд, 1H, H-2', J2',1' = 7,2 Гц, J2',2'-OH = 6,4 Гц),
3,92 - 3,91 (м, 1H, H-4'), 3,77 - 3,64 (м, 2H, H-5'),
3,56 (с, 1H, 3'-C ≡ CH).
Пример 11
Синтез 1-(2,3,5-три-O-бензоил-3-C-этинил -β- D-рибофуранозил)тимина (Соединение 11)
К 252 г (2,0 ммоль) тимина добавляли 2,0 мл гексаметилдисилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения тимина. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 264 мг (0,5 ммоль) полученного в ссылочном примере 4 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,23 мл (2,0 ммоль) тетрахлорида олова при 0oC. Перемешивали смесь при комнатной температуре 27 часов. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия и перемешивали смесь при комнатной температуре в течение 30 минут. Затем образовавшийся осадок отфильтровывали через целит. Фильтрат подвергали жидкостному разделению с использованием воды (2 x 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем высушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование хлороформом) с получением 290 мг (выход 98%) целевого соединения 11 в виде вспененного вещества.
FAB-MC: m/z 595 (MH+).
1H-ЯМР (CDCl3): δ 8,17 - 7,34 (м, 17H, бензоил • 3,6-6,-NH), 6,55 (д, 1H, H-1', J1',2' = 5,9 Гц). 6,06 (д, 1H, H-2', J2',1' = 5,9 Гц), 4,98 - 4,95 (м, 1H, H-4'), 4,93 - 4,90 (м. 2H, H-5'), 2,91 (C, 1H, 3′-C≡CH), 1,72 (с, 3H, 5-Me).
Пример 12
Синтез 1-(3-C-этинил -β- D-рибофуранозил)тимина (Соединение 12)
В 12 мл раствора аммония в метаноле растворяли 290 мг (0,49 ммоль) соединения 11, полученного в примере 11, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесь. 5-20% метанола-хлороформ), получая в результате 115 мг (выход 83%) целевого соединения 12 в виде белого порошкообразного вещества.
Т. пл.: 113 - 118oC.
E1-MC: m/z 282 (M+)
ИК (нуйор): 2115 см-1 (-C≡C-)
УФ: λmax (H2O) 266 нм (ε 8800):
λmax (0,5 н. хлороводородная кислота) 265 нм (ε 8600):
λmax (0,5 н. гидроксид натрия) 267 нм (ε 7000).
1H-ЯМР (ДМСО-d6) δ: 11,30 (шир.с, 1H, - NH, обмениваемый с D2O),
7,85 (д, 1H, H-6, J6,5-Me = 1,0 Гц),
5,90 (с, 1H, 3'-OH, обмениваемый с D2O),
5,83 (д, 1H, H-1', J1',2' = 7,5 Гц),
5,81 (д, 1H, 2'-OH, J2'-OH,2' = 6,7 Гц, обмениваемый с D2O),
5,13 (т, 1H, 5'-OH, J = 4,4 Гц обмениваемый с D2O),
4,19 (дд, 1H, H-2', J2',1' = 7,5 Гц, J2',2'-OH = 6,7 Гц),
3,88 - 3,86 (м, 1H, H-4'), 3,74 - 3,65 (м, 2H, H-5'),
3,55 (с, 1H, 3′-C≡CH), 1,73 ( , 3H, 5-Me, J5-Me,6 = 1,0 Гц).
Элементный анализ:
Вычислено (для C12H14N2O6 • MeOH):
C, 49,68; H, 5,77; N, 8,91.
Найдено: C, 49,62; H, 5,81; N, 8,97.
Пример 13
Синтез 9-(2,3,5-три-O-бензоил-3-C-этинил -β- D-рибофуранозил)-N6-бензоиладенина (Соединение 13)
К 478 мг (2,0 ммоль) N6-бензоиладенина добавляли 6,0 мл гексаметилдисилазана и 2 мл пиридина в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения N6-бензоиладенина. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 264 мг (0,5 ммоль) полученного в ссылочном примере 4 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,35 мл (3,0 ммоль) тетрахлорида олова при 0oC. Перемешивали смесь при комнатной температуре 7 часов. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия и перемешивали смесь при комнатной температуре в течение 30 минут. Затем образовавшийся осадок отделяли фильтрованием через целит. Фильтрат подвергали жидкостному разделению с использованием воды (2 x 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем осушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование хлороформом) с получением 261 мг (выход 74%) целевого соединения 13 в виде вспененного вещества.
FAB-MC: m/z 708 (MH+).
1H-ЯМР (CDCl3) δ: 8,99 (шир. с, 1H, -NHBz, обмениваемый с D2O), 8,75 (с, 1H, H-8), 8,49 (с, 1H, H-2) 8,16 - 7,31 (м, 15H, бензоил • 3), 6,58 (д, 1H, H-1', J1',2' = 4,8 Гц), 6,56 (д, 1H, H-2', J2',1' = 4,8 Гц), 5,07 - 5,03 (м, 2H, H-5'), 4,98 - 4,94 (м, 1H, H-4'), 2,95 (с, 1H, 3′-C≡C).
Пример 14
Синтез 9-(3-C-этинил -β- D-рибофуранозил)аденина (Соединение 14)
В 5 мл раствора аммония в метаноле растворяли 234 мг (0,33 ммоль) соединения 13, полученного в примере 13, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5-20% метанола-хлороформ), получая в результате 80 мг (выход 83%) целевого соединения 14 в виде белого порошкообразного вещества.
E1-MC: m/z 291 (M+), 274 (M+-OH).
1H-ЯМР (ДМСО-d6) δ: 8,35 (с, 1H, H-8), 8,14 (с, 1H, H-2), 7,36 (шир.с, 2H, - NH2, обмениваемый с D2O) 5,99 (с, 1H, 3-OH, обмениваемый с D2O), 5,88 (д, 1H, 2-OH, J2'-OH,2' = 7,2 Гц обмениваемый с D2O), 5,85 (д, 1H, H-1', J1',2' = 7,8 Гц), 5,60 (дд, 1H, 5'-OH, J5'-OH,5'a = 7,4 Гц, J5'-OH,5'b = 4,0 Гц, обмениваемый с D2O). 4,81 (дд, 1H, H-2', J2',1' = 7,8 Гц, J2',2'-OH = 7,2 Гц), 4,01 - 3,80 (м, 1H, H-4'), 3,79 - 3,69 (м, 2H, H-5'), 3,54 (с, 1H, 3′-C≡CH).
Пример 15
Синтез 9-(2,3,5-три-O-бензоил-3C-этинил -β- D-рибофуранозил)-N2-ацетилгуанина (Соединение 15)
К 386 мг (2,0 ммоль) N2-ацетилгуанина добавляли 6,0 мл гексаметилдисилазана и 2 мл пиридина в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения N2-ацетилгуанина. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 264 мг (0,5 ммоль) полученного в ссылочном примере 4 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,35 (3,0 ммоль) тетрахлорида олова при 0oC. Перемешивали смесь при комнатной температуре в течение 2 часов. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия и перемешивали смесь при комнатной температуре в течение 30 минут. Затем образовавшийся осадок отделяли фильтрованием через целит. Фильтрат подвергали жидкостному разделению с использованием воды (2 х 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем осушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование хлороформом) с получением 327 мг (выход 99%) целевого соединения 15 в виде вспененного вещества.
FAB-МС: m/z 662 (MH+).
1H-ЯМР (CDCl3) δ: 12,26 (шир.с, 1H, 1-NH, обмениваемый с D2O), 10,36 (шир. с, 1H, 2-NHAc, обмениваемый с D2O), 8,38 (с, 1H, H-8), 8,15-7,28 (м, 15H, бензоил • 3), 6,85 (д, 1H, H-1', J1',2' = 3,9 Гц), 6,32 (д, 1H, H-2', J2',1' = 3,9 Гц), 5,09-5,05 (м, 1H, H-4'), 5,04-4,86 (м, 2H, H-5'), 2,97 (с, 1H, 3′-C≡CH), 2,36 (с, 3H, 2-HAc).
Пример 16
Синтез 9-(3-C-этинил -β- D-рибофуранозил)гуанина (Соединение 16)
В 30 мл метанола суспендировали 320 мг (0,49 ммоль) соединения 15, полученного в примере 15, и к суспензии добавляли 0,90 мл (0,25 ммоль) метоксида натрия. Полученную смесь перемешивали при комнатной температуре 2 часа. После нейтрализации реакционной смеси Амберлитом 1RC-50S (карбоновая смола) ее подвергали фильтрованию посредством всасывания и фильтрат перегоняли под пониженном давлении. Полученный остаток растворяли в 150 мл воды и раствор подвергали жидкостному разделению с использованием хлороформа (2 х 100 мл). Водный слой отгоняли при пониженном давлении с получением 95 мг (выход 63%) целевого соединения 16 в виде белого порошкообразного вещества.
Т.пл.: 225oC (разложение).
E1-MC: m/z 3 307 (M+).
1H-ЯМР (ДМСО-d6) δ: 10,95 (шир. с, 1H, 1-H), 8,28 (с, 1H, H-8), 6,25 (шир.с, 2H, -NH2), 6,03 (д, 1H, H-1', J1',2' = 7,4 Гц), 5,92 (с, 1H, 3'-OH), 5,86 (д, 1H, 2'-OH, H2'-OH,2' = 6,5 Гц), 4,93 (т, 1H, 5'-OH, J = 5,0 Гц), 4,62 (дд, 1H, H-2', J2',1' = 7,4 Гц, J2'2'-OH = 6,5 Гц), 3,94-3,92 (м, 1H, H-4'), 3,75-3,66 (м, 2H, H-5'), 3,53 (с, 1H, 3′-C≡CH).
Элементный анализ:
Вычислено (для C12H13N5O5): C, 46,90; H, 4,26; N, 22,80.
Найдено: C, 46,80; H 4,40; N, 22,85.
Пример 17
Синтез 1-(2,3,5-три-O-бензоил-3-C-(1-пропинил) -β- D-рибофуранозил)цитозина (Соединение 17)
К 222 мг (2,0 ммоль) цитозина добавляли 2,0 мл гексаметилдисилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения цитозина. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 271 мг (0,5 ммоль) полученного в ссылочном примере 8 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,29 (2,5 ммоль) тетрахлорида олова при 0oC. Перемешивали смесь при комнатной температуре 2,5 часа. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия и перемешивали смесь при комнатной температуре в течение 30 минут. Затем отфильтровывали через целит образовавшийся осадок. Фильтрат подвергали жидкостному разделению с использованием воды (2 х 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натри. Затем осушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5% метанол/хлороформ) с получением 214 мг (выход 72%) целевого соединения 17 в виде вспененного вещества.
FAB-МС: m/z 594 (MH+)
1H-ЯМР (CDCl3) δ: 8,15-7,31 (м, 15H, бензоил • 3), 7,82 (д, 1H, H-6, J6,5 = 7,5 Гц), 6,52 (д, 1H, H-1', J1',2' = 4,6 Гц), 6,03 (д, 1H, H-2', H2',1' = 4,6 Гц), 5,74 (д, 1H, H-5, J5,6 = 7,5 Гц), 4,96-4,86 (м, 3H, H-4', H-5'), 1,86 (с, 3H, 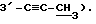
Пример 18
Синтез 1-(3-C-(1-пропинил) -β- D-рибофуранозил)цитозина (Соединение 18)
В 6 мл раствора аммония в метаноле растворяли 150 мг (0,25 ммоль) соединения 17, полученного в примере 17, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5-20% метанол/хлороформ), получая в результате 63,0 мг (выход 89%) целевого соединения 18 в виде бледно-желтого порошкообразного вещества.
Т.пл.: 162-165oC.
EI-МС: m/z 281 (M+).
1H-ЯМР (ДМСО-d6+D2O) δ: 8,26 (д, 1H, H-6, J6,5 = 7,9 Гц), 6,15 (д, 1H, H-5, J5,6 = 7,9 Гц), 5,78 (д, 1H, H-1', J1',2' = 5,7 Гц), 4,08 (д, 1H, H-2', J2',1' = 5,7 Гц), 3,95-3,94 (м, 1H, H-4'), 3,75 (дд, 1H, H-5'a, J5'a,4' = 4,7 Гц, J5'a,b = 12,0 Гц), 3,67 (дд, 1H, H-5'b, J5'b,4' = 2,5 Гц, J5'b,a = 12,0 Гц), 1,81 (с, 3H, 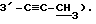
Элементный анализ:
Вычислено (для C12H15N3O5• HCl•HCl•1/5H2O): C, 44,85; H, 5,14; N, 13,08.
Найдено: C, 44,72; H, 5,10; N, 12,93.
Пример 19
Синтез 1-(2,3,5-три-O-бензоил-3-C-(1-бутинил) -β- D-рибофуранозил)цитозина (Соединение 19)
К 444 мг (4,0 ммоль) цитозина добавляли 4,0 мл гексаметилдисилазана и 14 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения цитозина. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 556 мг (1,0 ммоль) полученного в ссылочном примере 12 соединения, растворенного в 8 мл безводного ацетонитрила, с последующим добавлением 0,59 мл (5,0 ммоль) тетрахлорида олова при 0oC. Перемешивали смесь при комнатной температуре 19 часов. Добавляли к реакционной смеси 25 мл хлороформа и 10 мл насыщенного водного раствора гидрокарбоната натрия и перемешивали смесь при комнатной температуре в течение 30 минут. Затем отфильтровывали через целит образовавшийся осадок. Фильтра подвергали жидкостному разделению с использованием воды (2 х 10 мл) и 10 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем осушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5% метанол/хлороформ) с получением 412 мг (выход 68%) целевого соединения 19 в виде вспененного вещества.
FAB-МС: m/z 608 (MH+).
1H-ЯМР (CDCl3) δ: 8,15-7,31 (м, 15H, бензоил х 3), 7,84 (д, 1H, H-6, H6,5 = 7,4 Гц), 6,54 (д, 1H, H-1', J1',2' = 4,6 Гц), 6,04 (д, 1H, H-2', J2',1' = 4,6 Гц), 5,72 (д, 1H, H-5, J5,6 = 7,4 Гц), 4,97-4,85 (м, 3H, H-4', H-5'), 2,25-2,20 (м, 2H, 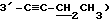 1,08 (т, 3H, J = 7,5 Гц, 3′-C≡C-CH2CH3)
1,08 (т, 3H, J = 7,5 Гц, 3′-C≡C-CH2CH3)
Пример 20
Синтез 1-(3-C-(1-бутинил) -β- D-рибофуранозил)цитозина (Соединение 20)
В 12 мл раствора аммония в метаноле растворяли 336 мг (0,55 ммоль) соединения 19, полученного в примере 19, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5-20% метанол/хлороформ), получая в результате 154 мг (выход 95%) целевого соединения 20 в виде бледно-желтого порошкообразного вещества.
Т.пл.: 181-184oC.
E1-MC: m/z 295(M+).
1H-ЯМР (ДМСО-D6+D2O) δ: 8,27-(д, 1H, H-6, J6,5=7,8 Гц), 6,18 (д, 1H, H-5, J5,6= 7,8 Гц), 5,77 (д, 1H, H-1', J1',2'=5,5 Гц), 4,08 (д, 1H, H-2', J2',1'= 5,5 Гц), 3,96-3,95 (м, 1H, H-4), 3,77-3,67 (м, 2H, H-5'), 2,27-2,17 (м, 2H, 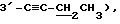 1,05 (т, 3H, J=7,5 Гц,
1,05 (т, 3H, J=7,5 Гц, 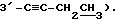
Элементный анализ:
Вычислено (для C19H17N3O5 • HCl): C, 47,07; H, 5,47; N 12,67.
Найдено: C, 46,91; H, 5,61; N 12.50.
Пример 21
Синтез 1-(2,3,5-три-O-бензоил-3-C-этенил -β- D-рибофуранозил)цитозина (Соединение 21)
К 222 мг (2,0 ммоль) цитозина добавляли 2,0 мл гексаметилдисилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения цитозина. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 265 мг (0,5 ммоль) полученного в ссылочном примере 16 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавление 0,29 мл (2,5 ммоль) тетрахлорида олова при 0oC. Перемешивали смесь при комнатной температуре 18 часов. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната и перемешивали смесь при комнатной температуре в течение 30 минут. Затем отфильтровывали образовавшийся осадок через цеолит. Фильтрат подвергали жидкостному разделению с использованием воды (2 х 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем осушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографий на силикагеле (элюирование смесью 5% метанол/хлороформ) с получением 201 мг (выход 69%) целевого соединения 21 в виде вспененного вещества.
FAB-MC: m/z 582 (MH+).
1H-ЯМР (CDCl3) δ: 8,12-7,38 (м, 16H, бензоил x 3, H-6),
6,61 (д, 1H, H-1', J1',2'=7 Гц),
6,35 (дд, 1H, 3'-CHC=CHaCHb, Jc,a=17,4 Гц, Jc,b=11,0 Гц,
6,03 (д, 1H, H-2', J2',1'-7,6 Гц),
5,61 (д, 1H, -5, J5,6=6,9 Гц),
5,40-5,33 (м, 2H, 3'-CHc=CHaCHb),
5,20-5,12 (м, 1H, H-4'),
4,84-4,65 (м, 2H, H-5').
Пример 22
Синтез 1-(3-C-этенил -β- D-рибофуравнозил)цитозина (Соединение 22)
В 13 мл раствора аммония в метаноле растворяли 323 мг (0,55 ммоль) соединение 21, полученного в примере 21, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли под пониженным давлением растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5-20% метанол/хлороформ), получая в результате 156 мг (выход 96%) целевого соединения 22 в виде белого порошкообразного вещества.
Т.пл.: 194-197oC.
E1-MC: m/z 296 (M+).
1H-ЯМР (ДМСО-d6) δ: 7,93 (д, 1H, H-6, J6,5=7,5 Гц),
7,20, 7,17 (шир.с, каждый 1H, NH),
6,05 (дд, 1H, 3'-CHc=CHaCH, Jc,a=17 Гц, Jc,a=10,6 Гц),
5,93 (д, 1H, H-1', J1',2'=7,9 Гц),
5,75 (д, 1H, H-5, J5,6=7,5 Гц),
5,46 (дд, 1H, 3'-CHc=CHaCHb, Ja,c=17,2 Гц, Ja,b=2,0 Гц),
5,31 (д, 1H, 2'-OH, J2'-OH,2'=6,7 Гц),
5,23 (дд, 1H, 3'-CHc=CHaCHb, Jb,c=10,6 Гц, Jb,a=2,0 Гц),
5,21 (т, 1H, 5'-OH, J=4,5 Гц), 4,77 (с, 1H, 3'-OH),
4,13 (дд, 1H, H-2', J2',1'=7,9 Гц, J2'-2'-OH=6 Гц),
3,76-3,75 (м, 1H, H-4'), 3,55-3,39 (м, 2H, H-5').
Элементный анализ:
Вычислено (для C11H15N3O5): C, 49,07; H, 5,62; N, 15,61.
Найдено: C, 49,17; H, 5,48; N, 15,59.
Пример 23
Синтез 1-(2,3,5-три-O-бензоил-3-C-(1-пропинил) -β- D-рибофуранозил)урацила (Соединение 23)
К 225 мг (2,0 ммоль) урацила добавляли 2,0 мл гексаметилсилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения урацила. Затем реакционную смесь охлаждали до комнатной температуры, и отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 271 мг (0,5 ммоль) полученного в ссылочном примере 8 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,39 мл (2,0 ммоль) триметилсилилтрифторметансульфоната при 0oC. Перемешивали смесь при комнатной температуре 8 часов. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия и, перемешав смесь при комнатной температуре в течение 30 мин, подвергали ее жидкостному разделению с использованием воды (2 х 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем осушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование хлороформом) с получением 241 мг (выход 81%) целевого соединения 23 в виде вспененного вещества.
FAB-MC: m/z 595 (MH+).
1H-ЯМР (CDCl3) δ: 8,14-7,30 (м, 15H, бензоил х 3),
7,97 (шир. 1H, -NH, обмениваемый с D2O),
7,78 (д, 1H, H-6, J6,5=8,0 Гц),
6,32 (д, 1H, H-1', J1',2'=4,5 Гц),
5,96 (д, 1H, H-2', J2',1'=4,5 Гц),
5,75 (дд, 1H, H-5, J5,6=8,0 Гц, J5,NH=2,0 Гц),
4,93-4,84 (м, 3H, H-4', H-5',), 1,91 (с, 3H, 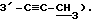
Пример 24
Синтез 1-(3-C-(1-пропинил) -β- D-рибофуранозил)урацила (Соединение 24)
В 10 мл раствора аммония в метаноле растворяли 236 мг (0,40 ммоль) соединения 23, полученного в примере 23, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 0-5% метанол/хлороформ), получая в результате 86,6 мг (выход 77%) целевого соединения 24 в виде белого порошкообразного вещества.
Т.пл.: 211-213oC.
E1-MC: m/z 283 (MH+).
ИК (нуйор): 2240 см-1 (-C≡C-).
1H-ЯМР (ДМСО-d6) δ: 11,33 (шир.с., 1H, -NH, обмениваемый с D2O), 7,97 (д, 1H, H-6, J6,5 = 8,2 Гц),
5,80 (д, 1H, H-1', J1',2' = 6,9 Гц),
5,75 (д, 1H, 2'-OH, J2'-OH,2' = 6,5 Гц, обмениваемый с D2O),
5,70 (с, 1H, 3'-OH, обмениваемый с D2O),
5,68 (дд, 1H, H-5, J5,6 = 8,2 Гц, J5,NH = 2,0 Гц),
5,04 (т, 1H, 5'-OH, J = 4,6 Гц, обмениваемый с D2O),
4,10 (дд, 1H, H-2', J2',1' = 6,9 Гц, J2',2'-OH = 6,5 Гц),
3,87 - 3,85 (м, 1H, H-4'), 3,73 - 3,62 (м, 2H, H-5'),
1,83 (с, 3H, 3′-C≡C-CH3).
Элементный анализ:
Вычислено (для C12H14N2O6 • 1/5 H2O);
C, 50,42; H, 5,08; N, 9,80.
Найдено: C, 50,51; H, 4,86; N, 9,78.
Пример 25
Синтез 1-(2,3,5-три-O-бензоил-3-C-(1-бутинил) -β- D-рибофуранозил)урацила (Соединение 25)
К 225 мг (2,0 ммоль) урацила добавляли 2,0 мл гексаметилдисилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения урацила. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 278 мг (0,5 ммоль) полученного в ссылочном примере 12 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,39 мл (2,0 ммоль) триметилсилилтрифторметансульфоната при температуре 0oC. Перемешивали смесь при комнатной температуре. Через два дня добавляли 0,19 мл (1,0 ммоль) триметилсилилтрифторметансульфоната и смесь перемешивали еще 5 часов при комнатной температуре. Затем добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия, перемешивали смесь при комнатной температуре в течение 30 минут и подвергали ее жидкостному разделению с использованием воды (2 x 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем осушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование хлороформом) с получением 294 мг (выход 97%) целевого соединения 25 в виде вспененного вещества.
FAB-MC: m/z 609 (MH+).
1H-ЯМР (CDCl3) δ: 8,15 - 7,29 (м, 15H, бензоил • 3), 8,02 (шир. 1H, NH), 7,82 (д, 1H, H-6, J6,5 = 8,2 Гц), 6,34 (д, 1H, H-1', J1',2' = 4,4 Гц), 5,97 (д, 1H, H-2', J2',1' = 4,4 Гц), 5,75 (дд, 1H, H-5, J5,6 = 8,2 Гц, J5,NH = 2,3 Гц), 4,93 - 4,84 (м, 3H, 3H, H-4', H-5'), 2,30 - 2,25 (м, 2H, 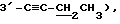 1,12 (т, 3H, J = 7,5 Гц,
1,12 (т, 3H, J = 7,5 Гц, 
Пример 26
Синтез 1-(3-C-(1-бутинил) -β- D-рибофуранозил)урацила (Соединение 26)
В 10 мл раствора аммония в метаноле растворяли 286 мг (0,47 ммоль) соединения 25, полученного в примере 25, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 0 - 5% метанол/хлороформ), получая в результате 129 мг (выход 93%) целевого соединения 26 в виде белого порошкообразного вещества.
Т.пл.: 139 - 142oC.
E1-MC: m/z 297 (MH+).
1H-ЯМР (ДМСО-d6) δ: 11,31 (шир.с, 1H, NH), 7,96 (д, 1H, H-6, J6,5 = 7,8 Гц), 5,79 (д, 1H, H=1', J1',2' = 6,7 Гц), 5,74 (д, 1H, 2'- OH), 5,67 (шир.с, 1H, 3'-OH), 5,66 (д, 1H, H-5, J5,6 = 7,8 Гц), 4,99 (т, 1H, 5'-OH), 4,08 (дд, 1H, H-2', J1',2' = 6,7 Гц, J2',OH = 6,4 Гц), 3,87-3,86 (м, 1Н, Н-4'), 3,74-3,64 (м, 2Н, Н-5'), 2,23 - 2,19 (м, 2H, 3'-C=C-CH2CH3), 1,07 (т, 3H, J = 7,5 Гц, 3'-C=C-CH2CH3).
Элементный анализ:
Вычислено (для C13H16N2O6 • 3/5 H2O):
C, 50,85; H, 5,65; N, 9,12.
Найдено: C, 50,72; H, 5,47; N, 9,20.
Пример 27
Синтез 1-(2,3,5-три-O-бензоил-3-C-этенил- -β- D-рибофуранозил)урацила (Соединение 27)
К 225 мг (2,0 ммоль) урацила добавляли 2,0 мл гексаметилдисилазана и 7 мг сульфата аммония в атмосфере аргона и смесь нагревали с обратным холодильником до полного растворения урацила. Затем реакционную смесь охлаждали до комнатной температуры, отгоняли при пониженном давлении растворитель, сохраняя смесь сухой, и остаток подвергали три раза азеотропной перегонке с толуолом. К полученному остатку добавляли 265 мг (0,5 ммоль) полученного в ссылочном примере 16 соединения, растворенного в 4 мл безводного ацетонитрила, с последующим добавлением 0,39 мл (2,0 ммоль) триметилсилилтрифторметансульфоната при 0oC. Перемешивали смесь при комнатной температуре 21 час. Добавляли к реакционной смеси 12 мл хлороформа и 5 мл насыщенного водного раствора гидрокарбоната натрия, перемешивали смесь при комнатной температуре в течение 30 минут и подвергали ее жидкостному разделению с использованием воды (2 x 5 мл) и 5 мл насыщенного водного раствора гидрокарбоната натрия в указанном порядке, после чего полученный органический слой сушили над сульфатом натрия. Затем осушенный органический слой фильтровали, отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование хлороформом) с получением 284 мг (выход 98%) целевого соединения 27 в виде вспененного вещества.
FAB-MC: m/z 583 (MH+).
1H-ЯМР (CDCl3) δ: 8,17 - 7,44 (м, 16H, бензоил • 3, H-6), 8,05 (шир. 1H, -NH, обмениваемый с D2O), 6,52 (д, 1H, H-1', J1',2' = 7,7 Гц), 6,41 (дд, 1H, 3'-CHc= CHaCHb, Jc,a = 17,4 Гц, Jc,b = 11,1 Гц), 6,03 (д, 1H, H-2', J2',1' = 7,7Гц), 5,54 (дд, 1H, H-5, J5,6 = 8,2 Гц, J5,NH = 2,2 Гц), 5,43 - 5,41 (м, 2H, 3'-CHc=CHaCHb), 5.25 (дд, 1H, H-4', J4',5'a = 3,2 Гц, J4',5'b = 3,7 Гц), 4.83 (дд, 1H, H-5'a, J5'a,4' = 3,2 Гц, J5'a,5'b = 12,6 Гц), 4,71 (дд, 1H, H-5'b, J5'b,4' = 3,7 Гц, J5'b,5'a = 12,6 Гц).
Пример 28
Синтез 1-(3-C-этенил -β- D-рибофуранозил)урацила (Соединение 28)
В 13 мл раствора аммония в метаноле растворяли 279 мг (0,48 ммоль) соединения 27, полученного в примере 27, и раствор перемешивали при комнатной температуре 2 дня. Отгоняли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (элюирование смесью 5 - 10% метанол/хлороформ), получая в результате 121 мг (выход 93%) целевого соединения 28 в виде бледно-желтого порошкообразного вещества.
Т.пл.: 129 - 222oC.
E1-MC: m/z 271 (MH1).
1H-ЯМР (ДМСО-d6) δ: 11,32 (шир.с, 1H-NH, обмениваемый с D2O),
8,08 (д, 1H, H-6, J6,5 = 8,0 Гц),
6,05 (дд, 1H, 3'-CHc=CHaCHb, Jc,a = 17,2 Гц, Jc,b = 10,7 Гц),
5,95 (д, 1H, H-1', J1',2' = 8,0 Гц),
5,70 (дд, 1H, H-5, J5,6 = 8,0 Гц, J5,NH = 2,1 Гц), 5,48 (дд, 1H, 3'-CHc= CHaCHb, Ja,c= 17,2 Гц, Ja,b= 1,9 Гц), 5,46 (д, 1H, 2'-OH, J2'-OH,2' = 7,0 Гц, обмениваемый с D2O), 5,26 (дд, 1H, 3'-CHc=CHaCHb, Jb,c = 10,7 Гц, Jb,a = 1,9 Гц), 5,24 (т, 1H, 5'-OH, J=4,2 Гц, обмениваемый с D2O), 4,90 (с, 1H, 3'-OH, обмениваемый с D2O), 4,11 (дд, 1H, H-2', J2',1' = 8,0 Гц, J2',2'-OH = 7,0 Гц), 3,78 - 3,76 (м, 1H, H-4'), 3,57 - 3,41 (м, 2H, H-5').
Элементный анализ:
Вычислено (для C11H14N2O6):
C, 48,89; H, 5,22: N, 10,37.
Найдено: C, 48,81; H, 5,18; N, 10,34.
Пример 29
Синтез 1-(3-C-этинил -β- D-рибофуранозил)цитозин-5-монофосфата (Соединение 29)
В 14 мл триметилфосфата растворяли 267 мг (1,0 ммоль) соединения 2, полученного в примере 2, и к раствору при -10o добавляли 466 мл (5,0 ммоль) оксихлорида фосфора с последующим перемешиванием при 4oC в течение 24 часов. После подтверждения прохождения реакции электрофорезом на бумаге (буфер 0,05 н. бикарбонат триэтиламмония, 700 B, 1 час) к реакционной смеси добавляли 100 мл воды и 100 мл хлороформа для осуществления жидкостного разделения. Полученный водный слой промывали три раза хлороформом. К водному слою добавляли воду до объема 400 мл и в этот объем добавляли активированный уголь до тех пор, пока абсорбционность (оптическая плотность) при 270 нм водного раствора не достигнет 0,5 или ниже. Активированный уголь помещали в колонку (5 см в поперечнике x 17 см) и промывали 1500 мл воды, после чего элюировали 4500 мл этанола. Фракции (каждая около 20 мл) водного раствора, абсорбционность при 270 нм которых составляли 0,5 или выше, концентрировали и растворяли в небольшом количестве этанола. Этот раствор добавляли к диэтиловому эфиру, в результате чего образовался порошок, который собирали фильтрованием, получая в результате 299 мг (выход 86%) соединения 29 в виде белого порошкообразного вещества.
FAB-MC (отрицательный): m/z 246 (M-).
1H-ЯМР (D2O) δ: 8,25 (д, 1H, H-6, J6,5 = 8,0 Гц), 6,32 (д, 1H, H-5, J5,6 = 8,0 Гц), 6,05 (д, 1H, H-1', J1',2' = 6,4 Гц), 4,47 (д, 1H, H-2', J2',1' = 6,4 Гц), 4,39 - 4,37 (м, 1H, H-4'), 4,31 - 4,13 (м, 2H, H-5'), 3,13 (с, 1H, 3′-C≡CH).
31P-ЯМР (D2O, 85% H3PO4 (в качестве внутреннего стандарта)) δ: 0,24 (с).
Пример 30
Синтез 1-(3-С-этинил -β- D-рибофуранозил)цитозин-5'-дифосфата (Соединение 30) и 1-(3-C-этинил -β- D-рибофуранозил)-цитозин-5'-трифосфата (Соединение 31)
В 3 мл безводного диметилформамида растворяли 36,8 мг (0,106 ммоль) соединения 29, полученного в примере 29, в атмосфере аргона и к раствору добавляли 65,7 мг (0,405 ммоль) 1,1'-карбонилдиимидазола с последующим перемешиванием при комнатной температуре в течение 16 часов. После подтверждения прохождения реакции электрофорезом на бумаге (буфер 0,05 н.бикарбонат триэтиламмония, 700 B, 1 час) к реакционной смеси добавляли 27 мл (0,65 ммоль) метанола с последующим перемешиванием при комнатной температуре в течение 30 минут. К полученной реакционной смеси добавляли 0,67 мл (0,67 ммоль) 1M раствора пирофосфата трибутиламмония в диметилформамиде и перемешивали смесь при комнатной температуре 16 часов. После подтверждения прохождения реакции электрофорезом на бумаге (буфер 0,05 г. бикарбонат триэтиламмония, 700 B, 1 час) отфильтровывали образовавшийся осадок и фильтрат промывали 3 мл диметилформамида и 3 мл этанола и перегоняли при пониженном давлении. Затем к остатку добавляли воду до объема 100 мл и, адсорбировав остаток на колонке DEAE-Cellulofine A-200 (3 см в поперечнике x 21 см), промывали его 500 мл воды, элюировали 0-0,5 н. буфером бикарбонат триэтиламмония (pH около 7,9, 1700 мл) с линейным градиентом концентрации и в результате получили целевое соединение 30 (элюированное при 300 OD, 0,10-0,15 н., выход 31%) и соединение 31 (элюированное при 80 OD, 0,18-0,22 н., выход 8%) в виде сиропообразных веществ.
Чистота соединений 30 и 31 была подтверждена ВЭЖХ с линейным градиентом концентрации при использовании 0,2 - 0,4 M фосфатного буфера (pH 7,0) на колонке YMC-Pack IES-Ax.
Условия:
Колонка: YMC-Pack IES-Ax.
Элюент: 0,2 M (0,1 сек.)-0,4 M (25 мин.) фосфатный буфер (pH 7,0).
Скорость потока: 1,0 мл/мин.
Длина волны определения: 254 нм.
Вводимый объем: 20 мкл (0,5 мг/мл).
Температура: комнатная.
Соединение 30 - время удерживания: 9,8 мин.
Соединение 31 - время удерживания: 18,1 мин.
Пример 31
Синтез 1-(3-C-этинил -β- D-рибофуранозил)урацил-5'-монофосфата (Соединение 32)
После трехкратной азеотропной перегонки 100 мг соединения 7, полученного в примере 7 с диоксаном, его растворяли в 10 мл триметилфосфата и к раствору добавляли при 0oC в атмосфере аргона 250 мкл оксихлорида фосфора с последующим перемешиванием при 4oC в течение 24 часов. После подтверждения прохождения реакции электрофорезом на бумаге (буфер 0,05 н. бикарбонат триэтиламмония (pH 8), 700 В, 45 минут) к реакционной смеси добавляли 100 мл 0,2 н. буфера бикарбонат триэтиламмония и 100 мл хлороформа для осуществления жидкостного разделения. Полученный водный слой промывали два раза хлороформом. Водный слой концентрировали при пониженном давлении и остаток растворяли в 500 мл воды для адсорбирования производных нуклеиновой кислоты в водном растворе на активированном угле (активированный уголь добавляли до тех пор, пока абсорбционность при 260 нм не достигла 0,2 или ниже). Активированный уголь помещали в колонку (5 см в поперечнике x 13 см) и промывали 500 мл воды, после чего элюировали 3000 мл этанола. Полученный раствор этанола концентрировали при пониженном давлении и остаток растворяли в 500 мл воды для введения раствора в колонку с (диэтиламино)этилцеллюлозой. После промывки 500 мл воды остаток очищали, используя 0 - 0,15 н.буфер бикарбонат триэтиламмония с линейным градиентом концентрации, и в результате получили целевое соединение 32 (25000 OD260нм, выход 62,5%).
FAB-MC: m/z 347 (M-H).
1H-ЯМР (D2O) δ: 8,05 - д, 1H, J = 8,2 Гц), 6,08 (д, 1H, J = 6,9 Гц), 5,96 (д, 1H, J = 8,2 Гц), 4,47 (д, 1H, J = 6,9 Гц), 4,35 (м, 1H), 4,28 (м, 1H), 4,16 (м, 1H), 3,15 (с, 1H).
32P-ЯМР (D2O, H3PO4 (в качестве внутреннего стандарта)) δ: 3,79 (с).
Пример 32
Синтез 1-(3-C-этинил -β- D-рибофуранозил)урацил-5'-дифосфата (Соединение 33) и 1-(3-C-этинил -β- D-рибофуранозил)-урацил-5'-трифосфата (Соединение 34)
После азеотропной перегонки соединения 32 (550 OD260нм, 0,055 ммоль), полученного в примере 31, три раза с диоксаном его растворяли в 2 мл безводного диметилформамида и к раствору добавляли 30 мг 1,1'-карбонилдиимидазола с последующим перемешиванием при комнатной температуре в течение 10 часов в атмосфере аргона. После подтверждения прохождения реакции электрофорезом на бумаге (буфер 0,05 н.бикарбонат триэтиламмония (pH 8), 700 В, 45 минут) к реакционной смеси добавляли 1 мл диметилформамида, в котором было растворено 4,2 мкл метанола, с последующим перемешиванием при комнатной температуре в течение 30 минут. К полученной реакционной смеси добавляли 300 мкл 1 М раствора пирофосфата трибутиламмония в диметилформамиде и перемешивали смесь при комнатной температуре 12 часов. После подтверждения прохождения реакции электрофорезом на бумаге (буфер 0,05 н. бикарбонат триэтиламмония (pH 8), 700 В 45 минут) к реакционной смеси добавляли воду до общего объема 500 мл и полученную смесь вводили в колонку с (диэтиламино)этилцеллюлозой. Промыв смесь 500 мл воды, проводили элюирование 0-0,25 н.буфером бикарбонат триэтиламмония с линейным градиентом концентрации и в результате получили целевое соединение 33 (элюированное при 230 OD260нм, 0,15 - 0,16 н., выход 42%) и соединение 34 (элюированное при 120 OD260нм, 0,18 - 0,21 н., выход 22%).
Получение соединений 33 и 34 было подтверждено ионообменной ВЭЖХ (YMC-Pack IES-Ax) с использованием 0,2 - 0,4 M фосфатного буфера (pH 7,0) при скорости потока 1,0 мл/мин. В результате их время удерживания было 8 минут и 16 минут соответственно.
Соединение 33:
FAB-MC: m/z 427 (M-H).
1H-ЯМР (D2O) δ: 8,06 (д, 1H, J = 8,0 Гц), 6,06 (д, 1H, J = 6,9 Гц), 6,00 (д, 1H, J = 8,0 Гц), 4,50 (д, 1H, J = 6,9 Гц), 4,35 (м, 1H), 4,32 (м, 1H), 4,28 (м, 1H), 3,15 (с, 1H).
32P-ЯМР (D2O), H3PO4 (в качестве внутреннего стандарта)) δ: -9,7 (м).
Соединение 34:
FAB-MC: m/z 507 (M-H).
1H-ЯМР (D2O) δ: 8,06 (д, 1H, J = 8,0 Гц), 6,08 (д, 1H, J= 6,1 Гц), 6,00 (д, 1H, J = 8,0 Гц), 4,50 (д, 1H, J = 6,1 Гц), 4,35 (м, 2H), 4,31 (м, 1H), 3,15 (с, 1H).
32P-ЯМР (D2O, H3PO4 (в качестве внутреннего стандарта)) δ: -8,58 (м), -9,76 (м), -11,12 (д).
Пример 33
Синтез 1-(3-C-триметилсилилэтинил -β- D-рибофуранозил)-урацила (Соединение 35)
Охладив смесь 30 мл трифторуксусной кислоты и 3,0 мл воды до 0oC, добавляли к ней 4,70 г 1-(2,5-бис-0-(трет-бутилдиметилсилил) -β- D-эритропентофуран-3-улозил)урацила. После перемешивания при 0oC в течение 20 минут смесь концентрировали досуха. Затем остаток очищали препаративной колоночной хроматографией при среднем давлении (SiO2, элюирование смесью н-гексан:этилацетат = 1:3), добавляли смесь н-гексанэтилацетат (10:1), чтобы кристаллизовать продукт, и в результате получили 2,40 г (выход 67,3%) 1-(2-0-(трет-бутилдиметилсилил) -β- D-эритропентофуран-3-улозил)урацила.
Нагревали затем 2,24 г гептагидрата хлорида церия при 140oC в течение 5 часов при пониженном давлении 0,2-0,3 мм ртутного столба, охлаждали его до комнатной до комнатной температуры и возвращали давление до обычного с помощью азота. Затем обработанный описанным образом хлорид церия охлаждали ледяной водой, добавляли к нему 7 мл тетрагидрофурана в перегнанном виде и смесь перемешивали всю ночь при комнатной температуре. Полученную при этом суспензию охлаждали до -78oC.
С другой стороны, к 4 мл тетрагидрофурана добавляли в атмосфере азота 0,85 мл триметилсилилацетилена и смесь охлаждали до -78oC и затем добавляли к ней 3,61 мл 1,66 M раствора н-бутиллития в н-гексане. Эту смесь перемешивали при -78oC в течение 30 минут и затем с помощью шприца добавляли по каплям к охлажденной до -78oC суспензии хлорида церия. Затем полученную смесь перемешивали при -78oC в течение 1 часа, добавляли к ней раствор 356 мг полученного ранее 1-(2-O-(трет-бутилдиметилсилил)- -β- -D-эритропентофуран-3-улозил)урацила в 2 мл тетрагидрофурана и смесь перемешивали 2,5 часа. Добавляли 0,9 мл уксусной кислоты, смесь нагревали до комнатной температуры, добавляли этилацетат и промывали два раза рассолом. Этилацетатный слой сушили над сульфатом магния и затем концентрировали досуха. К остатку добавляли для его кристаллизации 4 мл смеси этилацетат/н-гексан с получением 358 г (выход 69,6%) кристаллов 1-(2-O-(трет-бутилдиметилсилил)-3-C-триметилсилилэтинил -β- D-рибофуранозил)урацила в виде моноацетата.
Т.пл.: 188-189oC.
FAB-MC: m/z 455 (MH+).
1H-ЯМР (ДМСО-d6) δ: 11,96 (шир. 1H, обмениваемый с D2O), 11,39 (д, 1H, J = 2,2 Гц, обмениваемый с D2O), 8,06 (д, 1H, J = 8.1 Гц), 5.89 (д, 1H, J = 7,2 Гц), 5,83 (с, 1H, обмениваемый c D2O), 5,72 (дд, 1H, J = 8,1 Гц, 2,2 Гц), 5,12 (т, 1H, J = 3,9 Гц, обмениваемый с D2O), 4,35 (д, 1H, J = 7,2 Гц), 3,95 (т, 1H, J = 3,2 Гц), 3,64 - 3,77 (м, 2H), 1,91 (c, 3H), 0,82 (c, 9H), 0,15 (c, 9H), 0,09 (c, 3H), -0,04 (c, 3H).
К 2 мл метанола добавляли 309 мг полученного 1-(2-O-(трет-бутилдиметилсилил)-3-C-триметилсилилэтилнил -β- D-рибофуранозил)урацила и 6 мл 2,5%-ной хлороводородной кислоты в метаноле. После перемешивания смеси при комнатной температуре в течение 64 часов его концентрировали при пониженном давлении досуха. К остатку добавляли, чтобы кристаллизовать его, этилацетат с получением 163 мг (выход 80%) целевого соединения 35.
Т.пл.: 175-177oC.
FAB-MC: m/z 341 (MH+).
1H-ЯМР (ДМСО-d6) δ: 11,36 (шир., 1H, обмениваемый с D2O), 7,94 (д, 1H, J = 8,2 Гц), 5,92 (с, 1H, обмениваемый с D2O), 5,85 (д, 1H, J = 6,6 Гц, обмениваемый с D2O), 5,79 (д, 1H, J = 6,6 Гц), 5,66 (д, 1H, J = 8,2 Гц), 5,02 (т, 1H, J = 4,5 Гц, обмениваемый с D2O), 4.13 (т, 1H, J = 6,6 Гц), 3,88 (т, 1H, J = 3,7 Гц), 3,63 - 3,73 (м, 2H), 0,16 (с, 9H).
Пример 34
Синтез 1-(3-C-триэтилсилилэтинил) -β- D-рибофуранозил)-урацила (Соединение 36).
После высушивания 9,9 г гептагидрата хлорида церия при 140oC в течение 40 часов при пониженном давлении возвращали давление до обычного введением газообразного аргона. Охлаждали хлорид церия ледяной водой и интенсивно перемешивая его, добавляли к нему за один раз 31 мл тетрагидрофурана и смесь перемешивали всю ночь при комнатной температуре.
К 18 мл тетрагидрофурана добавляли 4,77 мл триэтилсилилацетилена и смесь охлаждали до -78oC в атмосфере аргона, после чего добавляли по каплям в течение 20 минут 15,8 мл 1,68 M раствора н-бутиллития в н-гексане. После капельного добавления смесь перемешивали еще 30 минут и посредством канюли добавляли по каплям в течение примерно 10 минут к охлажденной до -78oC с суспензии хлорида церия. После завершения капельного добавления смесь перемешивали еще 60 минут. С помощью канюли добавляли к смеси по каплям в течение примерно 5 минут раствор 1,58 г 1-(2-O-(трет/бутилдиметилсилил) -β- D-эритропентофуран-3-улозил)урацила в 9 мл тетрагидрофурана. Через 75 минут к реакционной смеси добавляли 4,8 мл уксусной кислоты и полученную смесь нагревали до комнатной температуры. Смесь подвергали жидкостному разделению с использованием 220 мл этилацетата и 180 мл воды. Промыв полученный органический слой 180 мл воды и 100 мл насыщенного рассола, его сушили над безводным сульфатом натрия. После осушения удаляли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (гексан: этилацетат = 3: 2) с получением 1,85 г (выход 83%) 1-(2-O-(трет-бутилдиметилсилил)-3-C-триэтилсилилэтинил -β- D-рибофуранозил)урацила в виде белого вспененного вещества.
E1-MC: m/z 496 (M+), 481 (M+ - Me), 439 (M+ - tBu).
1H-ЯМР (ДМСО-d6) δ: 11,30 (шир.с., 1H), 7,97 (д, 1H, J = 8,1 Гц), 5,80 (д, 1H, J = 7,3 Гц), 5,72 (с, 1H), 5,64 (дд, 1H, J = 8,1 Гц), 5,02 (т, 1H), 4,27 (д, 1H, J = 7,3 Гц), 3,88 (т, 1H), 3,63 (м, 2H), 0.89 (т, 9H), 0,72 (c, 9H), 0,50 (кв. 6H), -0,08 (с, 3H), -0,12 (c, 3H).
К 1,8 г полученного 1-(2-O-(трет-бутилдиметилсилил)-3- C-триэтилсилилэтинил -β- D-рибофуранозил)урацила добавляли 50 мл 2,6%-ного (масса/объем) раствора хлороводородной кислоты в метаноле и смесь перемешивали при комнатной температуре. Через 80 минут отгоняли при пониженном давлении растворитель и остаток подвергали два раза азеотропной перегонке с этанолом и очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 50:1 - 15:1), получая в результате 1,3 г (выход 91%) целевого соединения в виде белого порошкообразного вещества.
Т.пл.: 194-196oC.
E1-MC: m/z 382 (M+), 353 (+-Et).
1H-ЯМР (ДМСО-d6) δ: 9,35 (шир.с, 1H), 7,91 (д, 1H, J = 8,2 Гц); 5,92 (с, 1H), 5,87 (д, 1H), 5,78 (д, 1Н), 5,66 (д, 1H, J = 8,2 Гц), 5,00 (т, 1H), 4.12 (т, 1H), 3,88 (т, 1H), 3,70 (м, 2H), 0,96 (т, 9H), 0,57 (кв. 6H).
Пример 35
Синтез 1-(3-C-триизопропилсилилэтинил- -β- D-рибофуранозил)урацила (Соединение 37)
После высушивания 22,4 г гептагидрата хлорида церия при 140oC в течение 7 часов при пониженном давлении возвращали давление до обычного введения газообразного аргона. Охлаждали хлорид церия ледяной водой и интенсивно перемешивали его, добавляли к нему за один раз 70 мл тетрагидрофурана и смесь перемешивали 4 дня при комнатной температуре.
К 18 мл тетрагидрофурана добавляли 6,2 мл триизопропилсилилацетилена и смесь охлаждали до -78oC в атмосфере аргона, после чего добавляли по каплям в течение 20 минут 16,3 мл 1,68 М раствора н-бутиллития в н-гексане. В результате раствор затвердевал и поэтому твердое вещество нагревали до -70oC и перемешивали еще 30 минут. Посредством шприца полученный раствор добавляли по каплям в течение примерно 5 минут к охлажденной до -78oC суспензии хлорида церия. После завершения капельного добавления смесь перемешивали еще 60 минут. С помощью канюли добавляли к смеси по каплям в течение примерно 30 минут раствор 1,63 г 1-(2-O-(трет-бутилдиметилсилил) -β- D-эритропентофуран-3-улозил)урацила в 15 мл тетрагидрофурана. Через 150 минут к реакционной смеси добавляли 4,1 мл уксусной кислоты и полученную смесь нагревали до комнатной температуры. Смесь подвергали жидкостному разделению с использованием 500 мл этилацетата и 300 мл воды. Промывали полученный органический слой 300 мл воды и 100 мл насыщенного рассола, его сушили над безводным сульфатом натрия. После сушки удаляли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат=2:1 - 3: 2) с получением 2,05 г (выход 83%) 1-(2-O-(трет-бутилдиметилсилил)-3-C-триизопропилсилилэтинил -β- D-рибофуранозил)урацила в виде белого вспененного вещества.
E1-MC: m/z 538 (M+), 523 (M+-Me), 495 (M+-iPr), 481 (M+-tBu).
1H-ЯМР (ДМСО-d6) δ: 9,32 (ширс., 1H), 8,01 (д, 1H), 5,90 (д, 1H, J = 7,3 Гц), 5,73 (с, 1H), 5,72 (д, 1H), 5,06 (т, 1H), 4,37 (д, 1H, J = 7,3 Гц), 3,96 (шир.с, 1H), 3,68 - 3,80 (м, 2H), 1,06 (м, 21H), 0,81 (с, 9H), 0,10 (с, 3H), -0,04 (c, 3H).
К 2,0 г полученного 1-(2-O-(трет-бутилдиметилсилил)- 3-C-триизопропилсилиэтинил -β- D-рибофуранозил)урацила добавляли 50 мл 2,6%-ного (масса/объем) раствора хлороводородной кислоты в метаноле и смесь перемешивали при комнатной температуре. Через 180 минут отгоняли при пониженном давлении растворитель и остаток подвергали азеотропной перегонке с этанолом, очищали колоночной хроматографией на силикагеле (хлороформ:метанол = 50:1 - 15:1) и затем суспендировали в гексане. Суспензию фильтровали, получая в результате 1,4 г (выход 89%) целевого соединения в виде белого порошкообразного вещества.
Т.пл.: 199-202oC.
E1-MC: m/z 424 (M+), 381 (M+-iPr).
1H-ЯМР (ДМСО-d6) δ: 9,35 (шир.с, 1H), 7,90 (д, 1H, J = 8,1 Гц), 5,87 (с, 1H), 5,86 (д, 1H), 5,78 (д, 1H, J = 6,1 Гц), 5,64 (д, 1H, J = 8,1 Гц), 4,95 (т, 1H), 4,12 (т, 1H, J = 6,1 Гц), 3,89 (т, 1H), 3,73 (м, 2H), 1,06 (с, 21H).
Пример 36
Синтез 1-(3-C-трифенилсилилэтинил -β- D-рибофуранозил)-урацила (Соединение 38)
После высушивания 6,6 г гептагидрадата хлорида церия при 140oC в течение 7 часов при пониженном давлении, доводили давление до обычного введением газообразного аргона. Охлаждали хлорид церия ледяной водой и, интенсивно перемешивая его, добавляли к нему за один раз 21 мл тетрагидрофурана и смесь перемешивали всю ночь при комнатной температуре.
К 12 мл тетрагидрофурана добавляли 5 мл трифенилсилилацетилена и смесь охлаждали до -78oC в атмосфере аргона, после чего добавляли по каплям в течение 10 минут 10,5 мл 1,68 М раствора н-бутиллития в н-гексане. В результате раствор затвердел и поэтому твердое вещество постепенно нагревали до 0oC для получения раствора. Этот раствор перемешивали еще 30 минут и посредством канюли добавляли по каплям в течение примерно 30 минут к охлажденной до -78oC суспензии хлорида церия. После завершения капельного добавления смесь перемешивали еще 60 минут. С помощью канюли добавляли к смеси по каплям в течение примерно 15 минут раствор 1,04 г 1-(2-O-(трет-бутилдиметилсилил) -β- D-эритропентофуран-3-улозил)урацила в 6 мл тетрагидрофурана. Через 60 минут к реакционной смеси добавляли 2,7 мл уксусной кислоты и полученную смесь нагревали до комнатной температуры. Смесь подвергали жидкостному разделению с использованием 300 мл этилацетата и 150 мл воды. Промывали полученный органический слой 150 мл воды и 150 мл насыщенного рассола и сушили его над безводным сульфатом натрия. После сушки удаляли при пониженном давлении растворитель и остаток очищали колоночной хроматографией на силикагеле (хлороформ: метанол = 25:1) с получением 1,3 г (выход 72% 1-(2-O-(трет-бутилдиметилсилил)-3-C-трифенилсилилэтинил -β- D-рибофуранозил)урацила в виде белого вспененного вещества.
E1-MC: m/z 583 (M+ - tBu).
1H-ЯМР (ДМСО-d60 δ: 9,31 (шир.с, 1H), 8,07 (д, 1H, J = 8,1 Гц), 7,62 (д, 6H), 7,40 - 7,50 (м, 9H), 6,12 (с, 1H), 5,96 (д, 1H, J = 7,5 Гц), 5,68 (дд, 1H, J = 8,1 Гц), 5,35 (т, 1H), 4,50 (д, 1H, J = 7,5 Гц), 3,74 - 3,83 (м, 2H), 0,73 (с, 9H), -0,08 (с, 3H), -0,14 (с, 3H).
К 1,3 г полученного 1-(2-O-(трет-бутилдиметилсилил)-3- C-трифенилсилилэтинил -β- D-рибофуранозил)урацила добавляли 30 мл, 2,6%-ного (масса/объем) раствора хлороводородной кислоты в метаноле и смесь перемешивали при комнатной температуре. Через 110 минут отгоняли при пониженном давлении растворитель и остаток подвергали азеотропной перегонке с этанолом, очищали колоночной хроматографией на силикагеле (хлороформ: метанол = 20:1 = 15:1) и затем суспендировали в гексане. Суспензию фильтровали, получая в результате 940 мг (выход 86%) целевого соединения в виде бледно-желтого порошкообразного вещества.
Т.пл.: 114-116oC.
E1-MC: m/z 526 (M+), 449 (M+-Ph).
1H-ЯМР (ДМСО-d6) δ: 9,37 (шир.с, 1H), 7,96 (д, 1H, J = 8,1 Гц), 7,62 (д, 6H), 7,42 - 7,51 (м, 9H), 6,23 (с, 1H), 5,98 (д, 1H), 5,87 (д, 1H, J = 6,9 Гц), 5,48 (дд, 1H, J = 8,1 Гц), 5,19 (т, 1H), 4,33 (т, 1H, J = 6,9 Гц), 4,01 (т, 1H), 3,74 - 3,85 (м, 2H).
Пример 1 испытаний.
Выживаемость при мышином лейкозе P388
Клетки (1х106 клеток) мышиного лейкоза P388 имплантировали внутрибрюшинно трем женским особям мышей CDF1 (возраст 8 недель) на группу. День спустя после имплантации и на пятый день мышам соответствующих групп вводили внутрибрюшинно испытуемые соединения в разных концентрациях. Определяя среднее число дней выживания каждой группы, находили коэффициент выживаемости (T/C %), в соответствии с приведенным ниже уравнением. Результаты показаны в таблице 1.
Коэффициент выживаемости (T/C, %) = Среднее число дней выживания обработанной группы/Среднее число дней выживания контрольной группы х 100
Пример 2 испытаний.
Цитотоксичность.
Человеческие клетки KB распределяли по 1•105 клеток на ячейку в планшете с 96 ячейками. Затем растворяли в дистиллированной воде соединение по настоящему изобретению, раствор разбавляли до разных концентраций средой RPM1 1640 и добавляли в каждую ячейку. После инкубирования при 37oC в течение 3 дней в инкубаторе с 5% CO2, подсчитывали число клеток по МТТ методу.
Цитотоксичность каждого из испытуемых соединений выражали через концентрацию (IC50) соединения, при которой число клеток уменьшалось на 50% по сравнению с контрольным. Результаты показаны в таблице 2.
Как видно из результатов, соединения по настоящему изобретению проявляли очень высокую цитотоксическую активность по сравнению с уже известным соединением 1-(3-C-этил -β-D- рибофуранозил)урацилом.
Далее описаны примеры композиций, в которых использованы соединения по настоящему изобретению.
Пример 1 препаративной формы: Капсула
Капсулу изготавливали, используя следующий состав композиции и способ, известный per se в данной области техники.
Соединение 2 - 200 мг
Лактоза - 30 мг
Кукурузный крахмал - 50 мг
Кристаллическая целлюлоза - 10 мг
Стеарат магния - 3 мг
Одна капсула содержит - 293 мг
Пример 2 препаративной формы: Таблетка
Таблетку изготавливали, используя следующий состав и способ, известный per se в данной области техники.
Соединение 7 - 100 мг
Лактоза - 47 мг
Кукурузный крахмал - 50 мг
Кристаллическая целлюлоза - 50 мг
Гидроксипропилцеллюлоза - 15 мг
Тальк - 2 мг
Стеарат магния - 2 мг
Этилцеллюлоза - 30 мг
Глицерид ненасыщенной жирной кислоты - 2 мг
Диоксид титана - 2 мг
Одна таблетка содержит - 300 мг
Пример 3 препаративной формы: Гранула
Гранулы изготавливали, используя следующий состав и способ, известный per se в данной области техники.
Соединение 14 - 200 мг
Маннит - 540 мг
Кукурузный крахмал - 100 мг
Кристаллическая целлюлоза - 100 мг
Гидроксипропилцеллюлоза - 50 мг
Тальк - 10 мг
Одна упаковка содержала - 1000 мг
Пример 4 препаративной формы: Мелкие гранулы
Мелкие гранулы изготавливали, используя следующий состав и способ, известный per se в данной области техники.
Соединение 35 - 200 мг
Маннит - 520 мг
Кукурузный крахмал - 100 мг
Кристаллическая целлюлоза - 100 мг
Гидроксипропилцеллюлоза - 70 мг
Тальк - 10 мг
Одна упаковка содержала - 1000 мг
Пример 5 препаративной формы: Препаративная форма для инъекций
Препаративную форму для инъекций изготавливали, используя следующий состав и способ, известный per se в данной области техники.
Соединение 4 - 100 мг.
Одна ампула дистиллированной воды для инъекции содержит 2 мл.
Пример 6 препаративной формы: Суппозиторий
Суппозиторий изготавливали используя следующий состав и способ известный per se в данной области техники.
Соединение 10 - 200 мг
Витепсол S-55 (смесь моно-, ди- и триглицеридов насыщенных жирных кислот от лауриновой до стеариновой, продукт. - 1300 мг
Одна препаративная форма содержит - 1500 мг
3'-замещенные производные нуклеозидов по настоящему изобретению проявляют высокую противоопухолевую активность и потому полезны для лечения и предупреждения различных видов рака путем введения их в различных формах.
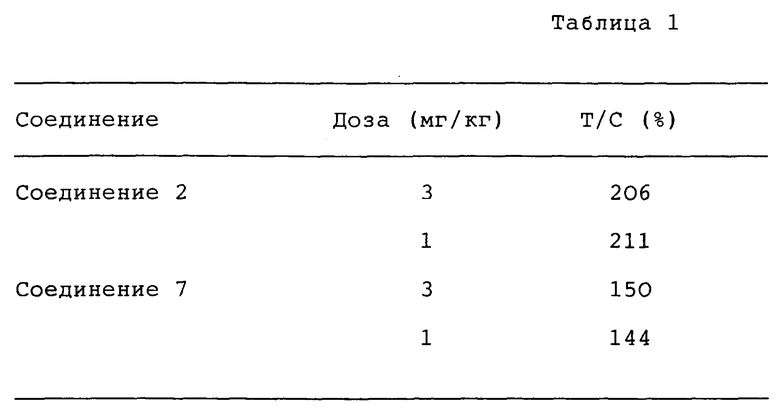

Изобретение относится к 3'-замещенному производному нуклеозида, представленному общей формулой (1), где Б представляет собой основание нуклеиновой кислоты, которое может иметь заместитель, Z представляет собой низшую алкинильную или низшую алкенильную группу, которые могут быть замещены группой, представленной общей формулой (2), в которой Ra, Rb и Rc одинаковы и представляют собой низшую алкильную или фенильную группу, R1 и R2, по отдельности, представляют собой атом водорода или бензоил и R3 представляет собой атом водорода, остаток моно- или полифосфорной кислоты или бензойной кислоты, при условии, что сахарная составляющая является рибозой, или его фармацевтически приемлемой соли. 3'-Замещенное производное нуклеозида по настоящему изобретению проявляет высокую противоопухолевую активность и потому полезно для лечения и профилактики разных видов рака. Предложены также способы получения соединения общей формулы (1), заключающиеся 1) во взаимодействии производного сахара общей формулы (3), где R1', R2' и R3' представляют собой, по отдельности, защитные группы для гидроксильной группы, Ас представляет ацетильную группу с производным нуклеиновой кислоты В-У, где В указано выше, а У представляет собой силильную защитную группу, и при необходимости, удаление защитной группы; 2) в частичном гидролизе соединения формулы (5) с образованием соединения формулы (6) и взаимодействии образованного соединения (6) с соединением формулы Z - X, где Х представляет атом водорода, или галогена, или MgBr, с получением соединения (7), которое гидролизуют и затем получают, при необходимости, сложный эфир фосфорной кислоты с одной из гидроксильных групп. Лекарственная композиция, имеющая антиопухолевую активность, и способ лечения или профилактики рака у млекопитающего. 5 с. и 12 з. п. ф-лы, 2 табл.
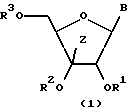
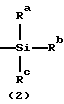
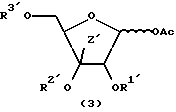
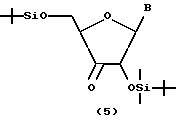
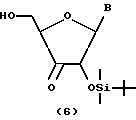
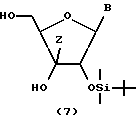
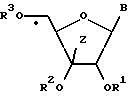
где В представляет собой основание нуклеиновой кислоты, которое может иметь заместитель;
Z представляет собой низшую алкильнильную или низшую алкенильную группу, которые могут быть замещены группой, представленной общей формулой 2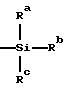
в которой Ra, Rb, Rc одинаковы и представляют собой низшую алкильную или фенильную группы;
R1 и R2, по отдельности, представляют собой атом водорода или бензоил;
R3 представляет собой атом водорода, остаток моно- или полифосфорной кислоты или бензойной кислоты, при условии, что сахарная составляющая является рибозой,
или его фармацевтически приемлемая соль.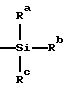
в которой Ra, Rb, Rc одинаковы и представляют собой низшую алкильную группу или фенильную группу;
R1 и R2 представляют собой атом водорода;
R3 представляет собой атом водорода, или остаток моно- или полифосфорной кислоты.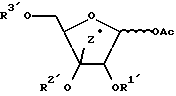
где R1', R2' и R3' представляют собой, по отдельности, защитную группу для гидроксильной группы;
Ас представляет собой ацетильную группу;
Z имеет такое же значение, как указано в п.1,
с производным нуклеиновой кислоты, представленным общей формулой 4
B - Y,
где В имеет такое же значение, как указано в п.1;
Y представляет собой силильную защитную группу,
и, при необходимости, удаление защитной группы по крайней мере одной из гидроксильных групп с образованием сложного эфира, способного легко отщепляться в живом организме, или сложного эфира фосфорной кислоты с указанной гидроксильной группой.
где В имеет такое же значение, как указано в п.1,
с образованием соединения, представленного общей формулой 6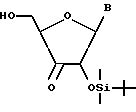
где В имеет такое же значение, как указано в п.1,
взаимодействие этого соединения с соединением, представленным формулой
Z - X,
где Z имеет такое же значение, как указано в п.1;
X представляет собой атом водорода или галогена или MgBr,
с получением соединения, представленного общей формулой 7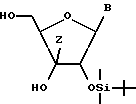
где В и Z имеют такое же значение, как указано в п.1,
которое, в свою очередь, гидролизуют и затем получают, при необходимости, сложный эфир фосфорной кислоты с по крайней мере одной из гидроксильных групп.
Приоритет по пунктам:
13.12.94 - по пп.1 - 17 (где Z - незамещенный низший алкинил и низший алкенил);
16.08.95 - по пп.1 - 17 (где Z низший алкинил и низший алкенил, замещенные группой общей формулы (2)).
| Г. М. Мильнер, Н. А. Пожилов, И. М. Самовский, А. Н. Соколов, М. Н. Сластенин, А. В. Тихомиров, О. И. Фаминский, А. И. Хануков и А. И. Шапиро | 0 |
|
SU278497A1 |
| Sophie Huss at al, synthesis of 3'-c-ethyl-nucleo-sides of thymyne, J.Tetrahedron, vol | |||
| Способ очищения сернокислого глинозема от железа | 1920 |
|
SU47A1 |
| Телефонная трансляция с катодным реле | 1920 |
|
SU1727A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРИДИНОВЫХ НУКЛЕОЗИДОВ | 1989 |
|
RU2036199C1 |
| Способ получения фосфонатных эфиров нуклеозидов | 1976 |
|
SU729196A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1985, ч.2, с.493, 598. | |||
