Область техники
Настоящее изобретение относится к ингибитору казеинкиназы (CK) 1δ и казеинкиназы 1ε, который содержит в качестве активного ингредиента производное оксазолона, его соль, их сольват или их гидрат.Настоящее изобретение относится к фармацевтическому средству для лечения заболеваний, с патологическими состояниями которых ассоциируют активацию казеинкиназы 1δ или казеинкиназы 1ε. Настоящее изобретение относится к фармацевтическому средству, содержащему такой ингибитор казеинкиназы 1δ и казеинкиназы 1ε, который полезен для лечения и/или предупреждения, в частности, расстройства циркадного ритма (включая расстройство сна), нейродегенеративного заболевания центральной нервной системы и рака среди заболеваний, с патологическими состояниями которых ассоциируют активацию казеинкиназы 1δ или казеинкиназы 1ε.
Предшествующий уровень техники
Казеинкиназа 1 относится к серин/треонинкиназе (которая в некоторых случаях фосфорилирует остаток тирозина). Известны семь типов изоформ, а именно, α, β, y1, y2, y3, δ и ε, в качестве ее изоформ у млекопитающих. Известно, что эти изоформы фосфорилируют разные типы различных белковых субстратов и что данные изоформы способны активировать, инактивировать, стабилизировать или дестабилизировать функции этих белков, и поэтому их ассоциируют с регуляцией функций разных типов различных организмов. Казеинкиназа 1δ или казеинкиназа 1ε млекопитающих в своей структуре имеет киназный домен, который является аналогичным среди других изоформ. Однако, ее N-концевой и C-концевой домены отличаются от таковых среди других изоформ. Другими словами, C-концевой домен имеет множество сайтов аутофосфорилирования, и считается, что он вовлечен в регуляцию аутоферментативной активности. Помимо этого, такой киназный домен содержит последовательность, которая, как полагают, ассоциирована с ядерной транслокацией (NLS: сигналом ядерной локализации) и кинезин-подобным доменом (KHD: кинезин-гомологичным доменом).
Известно, что казеинкиназа 1δ и казеинкиназа 1ε ассоциированы с расстройством циркадного ритма, что казеинкиназа 1δ и казеинкиназа 1ε ассоциированы с нейродегенеративным заболеванием центральной нервной системы и что казеинкиназа 1δ и казеинкиназа 1ε ассоциированы с раком. Подробная информация о связи этих казеинкиназ с патологическими состояниями вышеупомянутых заболеваний становится известной из исследований, касающихся взаимодействия между казеинкиназой 1δ и казеинкиназой 1ε и белками-мишенями, взаимодействующими с казеинкиназой 1δ и казеинкиназой 1ε, такими как белковые субстраты, взаимодействующие с соответствующей казеинкиназой 1δ и казеинкиназой 1ε. Конкретные примеры белкового субстрата, фосфорилируемого под действием казеинкиназы 1δ и казеинкиназы 1ε, включают белок period (Per), тау-белок (tau), р53 и β-катенин.
На сегодняшний день считается, что основа биологических часов, действующих в качестве центрального генератора циркадного ритма, содержит приблизительно 10 типов взаимодействующих генных сетей, называемых "часовыми генами". Среди этих 10 типов групп генов Per 1, 2 и 3 (Period 1, 2 и 3), Cry 1 и 2 (криптохром 1 и 2), Bmal1 (от англ. brain and muscle ARNT-like 1: подобный ядерному транслокатору арилуглеводородного рецептора (aryl hydrocarbon receptor nuclear translocator) белок 1 из головного мозга и мышц) и Clock (circadian locomotor output cycles kaput; "циркадный прерыватель циклов двигательной активности") кодируют транскрипционные факторы. С другой стороны, гены CK1δ и 1ε кодируют казеинкиназу 1δ и казеинкиназу 1ε, которые фосфорилируют эти транскрипционные факторы. Известно, что нарушение функционирования этих часовых генов оказывает влияние на фенотипическое проявление циркадных ритмов у различных типов животных, в том числе людей. В силу того, что молекулярный механизм таких биологических часов является сильно консервативным среди видов, исследования часовых генов, касающиеся фенотипов людей с нарушениями циркадного ритма, могут быть преимущественно проведены в испытаниях in vitro. Clock регулирует путь генерации сигналов активации среди взаимодействующих сетей биологических часов и активирует Per, Cry и другие, расположенные вниз по течению гены-мишени. С другой стороны, Per и Cry, которые регулируют путь генерации регуляторных сигналов, действуют, подавляя активность Clock. Казеинкиназа 1δ и казеинкиназа 1ε фосфорилируют Per и Cry, стимулируя разрушение Per в цитоплазме. Кроме того, результаты такого фосфорилирования ассоциированы с регуляцией ядерной транслокации этих транскрипционных факторов и их стабильностью в ядре. Таким образом считается, что ритм внутренних молекулярных вибраций в живом организме подвержен регуляции. В случае млекопитающих биологические часы находятся в супрахиазматическом ядре (SCN), и эти расположенные в SCN биологические часы работают вместе с управляющими генной экспрессией биологическими часами центральных и периферических тканей, не являющихся SCN.
Per известен в качестве регуляторного белка циркадного ритма в живом организме. Уровни мPHK и белка Per колеблются в условиях циркадного ритма и тесно связаны с регуляцией биологических часов. Например, известно, что если происходит ослабление в фосфорилировании казеинкиназой 1ε или казеинкиназой 1δ, то генетическое заболевание, характеризующееся мутацией (S662G) в сайте фосфорилирования человеческого Рег2, развивается в синдром преждевременного семейного сна (FASPS). Это показывает, что Per играет важную роль в регуляции сна. Известно, что изменение количества белка Per внутри клетки регулируется путем фосфорилирования под действием казеинкиназы 1ε или казеинкиназы 1δ. То есть известно, что если Per фосфорилируется под действием этих киназ, то стабильность данного белка существенно уменьшается.
Xu Y. и др. сообщили об обнаружении того факта, что трансгенные мыши с мутированным (S662G) сайтом фосфорилирования человеческого Per2 демонстрировали те же проявления фенотипа, что и люди с выявленным FASPS. Кроме того, эти исследователи изучили влияние, вызываемое изменением уровня экспрессии казеинкиназы 1δ, используя гибриды мышей между описанными выше трансгенными мышами и мышами WT (дикого типа) по казеинкиназе 1δ или мышами "казеинкиназа 1δ+/-" (гетерозиготными нокаутными мышами). В результате исследователи сообщили о влиянии тем самым на описанный выше фенотип и что нарушение фенотипа циркадного ритма, обнаруженное у гибридов на основе мышей дикого типа, было скорректировано в случае +/- мышей. В этом сообщении описывается статус фосфорилирования Рег2 и важность ассоциации казеинкиназы 1δ с фосфорилированием (непатентная литература 5). Кроме того, Badula Loi и др. сообщили, что фаза циркадного ритма может быть существенно задержана в результате подкожного введения крысам ингибирующего казеинкиназу 1ε соединения, 4-[3-циклогексил-5-(4-фтор-фенил)-3H-имидазол-4-ил]-пиримидин-2-иламина (PF-670462) (непатентная литература 4). Таким образом, статус фосфорилирования Per связан с циркадным ритмом, и применение ингибитора казеинкиназы 1δ или казеинкиназы 1ε предоставляет новый способ такого регулирования циркадного ритма. Можно ожидать, что метод сдвига или перенастройки фазы циркадного ритма будет способствовать лечению расстройства циркадного ритма, в том числе расстройств сна различных типов.
Однако, традиционные ингибиторы, в том числе PF-670462 в качестве типичного примера, оказывают ингибирующее влияние даже в отношении таких киназ (например, p38α), действие которых вызывает озабоченность в связи с проявлением побочных эффектов. Поэтому работа по созданию фармацевтической продукции на основе таких традиционных ингибиторов все еще не доведена до конца.
В методах предшествующего уровня техники не известны практически никакие фармацевтические средства для непосредственного лечения расстройства циркадного ритма. Кроме того, в качестве терапевтических средств для лечения таких расстройств сна разработаны и используются в местах проведения клинических исследований снотворные лекарственные средства. С другой стороны, разработка лекарственных средств для исправления расстройства циркадного ритма сна (расстройства сна, вызванного сменным режимом работы, синдрома десинхронизации физиологических циклов после трансмеридианных перелетов, синдрома преждевременной фазы сна и синдрома задержки фазы сна) и тому подобного, до сих пор не завершена. Кроме того, до сих пор не завершена разработка лекарственной терапии, основанной на методе сдвига или перенастройки фазы циркадного ритма, в отношении других расстройств сна (бессонницы, связанного со сном расстройства дыхания, связанной с нарушениями центральной нервной системы гиперсомнии, парасомнии и связанного со сном нарушения движений).
Далее будет описана корреляция казеинкиназы 1δ или казеинкиназы 1ε с нейродегенеративным заболеванием центральной нервной системы и, в частности, с болезнью Альцгеймера.
Хорошо известно, что агрегация тау-белка в месте поражения болезнью Альцгеймера представляет собой важный маркер патологических состояний. Кроме того, хорошо известно, что избыточное фосфорилирование этого тау-белка тесно ассоциировано с агрегацией. Считается, что в семействе казеинкиназ 1, которые избыточно экспрессируются в месте поражения, имеются киназы-кандидаты в отношении фосфорилирования тау-белка. Так, среди этих казеинкиназ Li Guibon и др. исследовали казеинкиназу 1δ, используя для экспрессии клеточную линию HEK-293 (почки человеческого эмбриона). В результате, используя неселективно ингибирующее казеинкиназу 1 соединение, 3-[(2,3,6-триметоксифенил)метилиденил]-индолин-2-он (IC261), они продемонстрировали, что сначала образуется ассоциат казеинкиназы 1δ с тау-белком in situ и казеинкиназа 1δ непосредственно фосфорилирует тау-белок, и что уровень фосфорилирования в том же самом сайте тау-белка, по которому осуществляется фосфорилированию in vitro, повышается в результате избыточной экспрессии казеинкиназы 1δ (непатентная литература 6). С другой стороны, Hanger Diane Р. и др., используя масс-спектрометрию, провели сравнение между так называемым нерастворимым тау-белком (PHF-тау (парные спиральные филаменты-тау)), представляющим собой чрезмерно фосфорилированный агрегат, полученный из места поражения пациента с болезнью Альцгеймера, и белком здорового человека в отношении сайта фосфорилирования и затем идентифицировали сайт фосфорилирования, характерный для места поражения пациента с болезнью Альцгеймера. При этом, основываясь на характеристиках сайта фосфорилирования, они предположили, что казеинкиназа 1δ, а также киназа 3β гликогенсинтазы, как киназы-кандидаты, с большой долей вероятности ассоциированы с процессом развития поражения (непатентная литература 7).
Далее также будет описана корреляция казеинкиназы 1δ или казеинкиназы 1ε с нейродегенеративным заболеванием центральной нервной системы и, в частности, с болезнью Альцгеймера.
Относительно болезни Альцгеймера считается, что аккумуляция амилоида-β(Aβ), демонстрирующая токсичность в отношении нервных клеток, ассоциирована с их поражением. В то же время известно, что экспрессия казеинкиназы 1 повышена в месте поражения у пациента с болезнью Альцгеймера. Считается, что Aβ образуется в результате расщепления АРР (белка-предшественника амилоида) β-секретазой (аспартильной протеазой β-секретазой) и y-секретазой (пресенилин-зависимой протеазой y-секретазой). Flajolet Маге и др. провели анализ in silico с целью изучения сайта, обычно фосфорилируемого казеинкиназами 1, который, как полагают, присутствует в последовательностях субъединиц указанных АРР, β-секретазы и y-секретазы. Далее, основываясь на полученных результатах, они предприняли попытку проведения избыточной экспрессии казеинкиназы 1ε, конститутивно активной в отношении N2A-клеток (клеток N2A-APP695), стабильно экспрессирующих АРР. В результате, согласно их сообщениям, количества Aβ40 и Aβ42 выросли приблизительно в 2 раза и 2,5 раза, соответственно, относительно контроля. Кроме того, они также сообщили о том, что когда в эту систему добавляли неселективно ингибирующее казеинкиназу 1 соединение IC261, количества Aβ40 и Aβ42 уменьшались и, далее, что те же результаты также могут быть получены с использованием других двух отличающихся типов неселективно ингибирующих казеинкиназу 1 соединений, CKI-7 и D4476 (непатентная литература 8).
Эти сообщения (непатентная литература 6, 7 и 8) убедительно показывают, что казеинкиназа 1 и, в частности, казеинкиназа 1δ или казеинкиназа 1ε ассоциирована с развитием болезни Альцгеймера и что болезнь Альцгеймера можно лечить путем ингибирования активности описанного выше фермента.
Кроме того, хромосома 21, в которой предположительно находится ген, вызывающий болезнь Альцгеймера, оказывается трисомной (триплоидной) в соматических клетках пациента с синдромом Дауна. Так, полагают, что синдром Дауна может служить моделью для исследований генетической предрасположенности или развития болезни Альцгеймера. В частности, аномальное накопление характерных белков, обнаруженная при этих двух типах заболеваний, рассматривается как один из важных патологических и биохимический индикаторов, ассоциированных с их патогенным механизмом, и следовательно подлежит изучению. В самом деле, известно, что у пациентов с синдромом Дауна в зрелом возрасте (приблизительно в возрасте 35 лет) зачастую обнаруживается церебральное нарушение, подобное болезни Альцгеймера. Эти факты убедительно показывают, что даже в случае нейродегенеративного заболевания, ассоциированного с синдромом Дауна, это заболевание можно лечить путем ингибирования ферментативной активности казеинкиназы 1 и, в частности, казеинкиназы 1δ или казеинкиназы 1ε.
В методах предшествующего уровня техники почти неизвестно никаких фармацевтических средств для лечения нейродегенеративных заболеваний центральной нервной системы, в том числе болезни Альцгеймера, которые включают в качестве отправной точки непосредственное ингибирование агрегации тау-белка или амилоида β. Кроме того, в методах предшествующего уровня техники разработка лекарственной терапии для препятствования прогрессированию нейродегенеративных заболеваний центральной нервной системы, основанной на знании рассматриваемого механизма, до сих пор не завершена.
Далее будет описана корреляция казеинкиназы 1δ или казеинкиназы 1ε с раком и, в частности, с раком поджелудочной железы.
Семейство казеинкиназ 1 ассоциируют с регуляцией разных важных физиологических активностей в клетках. Семейство казеинкиназ 1 фосфорилирует большое разнообразие белковых субстратов. Например, опухолеподавляющий фактор ρ53 и онкоген mdm2 оба являются важными белками для контроля появления признаков злокачественности и в то же самое время представляют собой субстраты казеинкиназы 1. Считается, что перерождение клеток в раковые зависит от статуса их фосфорилирования. Наряду с изоформами казеинкиназы 1 большое внимание уделено фосфорилированию ρ53 под действием казеинкиназы 1ε или казеинкиназы 1δ, последующему изменению взаимодействия между ρ53 и mdm2, стабилизации и активации ρ53. Кроме того, также известно, что казеинкиназа 1ε или казеинкиназа 1δ связана с регуляторным белком, ассоциированным с образованием веретена в качестве центросомы во время клеточного деления, и что казеинкиназа 1ε или казеинкиназа 1δ вовлечена в апоптоз, опосредованный TRAIL (ассоциированным с фактором некроза опухолей апоптоз-индуцирующим лигандом) и Fas.
К слову сказать, протоковые аденокарциномы поджелудочной железы (PDAC) считаются рефрактерными формами рака. Brockschmidt С.и др. в своих исследованиях показали, что казеинкиназа 1ε или казеинкиназа 1δ сверхэкспрессирована в PDAC. Основываясь на полученных результатах, к линии клеток рака поджелудочной железы человека in vitro добавляли неселективно ингибирующее казеинкиназу 1 соединение IC261. В результате наблюдали подавление клеточного роста. Одновременно, эту же линию клеток рака поджелудочной железы трансплантировали в подкожный слой мыши и затем этой мыши вводили неселективно ингибирующее казеинкиназу 1 соединение IC261. В результате, Brockschmidt С.и др. сообщили, что полученный эффект подавления роста опухолевых клеток был таким же значительным, как и в случае группы с введением гемцитабина (непатентная литература 9).
В методах предшествующего уровня техники не было известно никакого фармацевтического средства с возможностью его использования в качестве противоракового средства, действие которого было бы основано на ингибировании казеинкиназы 1ε или казеинкиназы 1δ. Кроме того, в методах предшествующего уровня техники разработка лекарственной терапии для лечения рефрактерного рака поджелудочной железы, основанной на знании рассматриваемого механизма, до сих пор не завершена.
Список ссылок
Патентная литература
Патентная литература 1. Публикация патента Японии (Kohyo) №2008-510712 A.
Патентная литература 2. Публикация патента Японии (Kohyo) №2008-510704 A.
Непатентная литература
Непатентная литература 1. Uwe Knippschild et al., Cellular Signaling, 17, 675-689 (2005).
Непатентная литература 2. Takashi Ebisawa, J. Pharmacol. Sci., 103, 150-154 (2007).
Непатентная литература 3. Caroline H. Ко et Joseph S. Takahashi, Hu. Mol. Genetics, 15(2) R271-R277 (2006).
Непатентная литература 4. Lori Badura et al, J. Pharmacol. Exp. Therapy, 322, 730-738 (2007).
Непатентная литература 5. Xu, Y. et al., Cell 128, 59-70 (2007). Непатентная литература 6. Li, Guibin et al., J. Biol. Chem., 279(16), 15938-15945 (2004).
Непатентная литература 7. Hanger, Diane P. et al., J. Biol. Chem., 282(32), 23645-23654 (2007).
Непатентная литература 8. Flajolet, Marc et al., Proc. Nat. Acad. Sci., 104(10), 4159-4164 (2007).
Непатентная литература 9. Brockschmidt, С.et al.: Gut, 57, 799-809 (2008)
Непатентная литература 10. Mashhoon, Neda et al., J. Biol. Chem., 275(26), 20052-20060 (2000).
Непатентная литература 11. Rena, Graham, et al., EMBO Rep., 5(1), 60-65, (2004).
Непатентная литература 12. Godl, Klaus, et al., Proc. Nat. Acad. Sci., 100(26), 15434-15439 (2003).
Непатентная литература 13. Cozza, Giorgio et al., Bioorg. Medicinal Chem. Lett., 18(20), 5622-5675 (2008).
Непатентная литература 14. Банк данных по белкам (он-лайн),<URL:http://www.rcsb.org/pdb/>, ID №: 2CMW (СК1гамма1), 2С47 (СК1гамма2), 2CHL, 2IZR, 2IZS, 2IZT, 2IZU (СК1гамма3), 1CKI, 1CKJ (СК1дельта).
Описание изобретения
Техническая проблема
Настоящим изобретением решается задача разработки ингибитора казеинкиназы 1δ и казеинкиназы 1ε, содержащего в качестве активного ингредиента производное оксазолона, его соль, их сольват или их гидрат.
Помимо этого, другая задача настоящего изобретения заключается в разработке фармацевтического средства, содержащего в качестве фармацевтически активного ингредиента ингибитор по настоящему изобретению, селективный в отношении казеинкиназы 1δ и казеинкиназы 1ε, при этом данное фармацевтическое средство является полезным для лечения и/или предупреждения заболеваний, с патологическими состояниями которых ассоциируют активацию казеинкиназы 1δ или казеинкиназы 1ε, в соответствии с чем функции казеинкиназы 1δ или казеинкиназы 1ε регулируются in vivo. Кроме того, другая задача настоящего изобретения заключается в разработке фармацевтического средства, полезного для лечения и/или предупреждения расстройства циркадного ритма (включая расстройство сна), нейродегенеративного заболевания центральной нервной системы и рака среди заболеваний, с патологическими состояниями которых ассоциируют активацию казеинкиназы 1δ или казеинкиназы 1ε. Помимо всего прочего, другая задача настоящего изобретения заключается в разработке способа лечения и/или предупреждения расстройства циркадного ритма (включая расстройство сна), нейродегенеративного заболевания центральной нервной системы и рака путем введения описанного выше фармацевтического средства субъекту.
К тому же, другая задача настоящего изобретения заключается в разработке нового производного оксазолона, его фармацевтически приемлемых соли и их гидрата.
Решение проблемы
На сегодняшний день известно несколько соединений в качестве исследуемых веществ, обладающих ингибирующим действием в отношении казеинкиназы 1, которые неспецифичны к изоформам казеинкиназы. Репрезентативные примеры таких соединений включают IC261, D4476 и SB203580 (непатентная литература 10, 11 и 12). Эти соединения еще не обладают свойствами, достаточными для решения данных проблем. Сначала планировалось использовать эти соединения просто как оказывающие селективное ингибирующее действие на казеинкиназу 1, и в качестве мишени для них была выбрана казеинкиназа 1δ как одна из изоформ. С другой стороны, имеется соединение PF-670462, разработанное с условием достижения селективного ингибирующего действия на казеинкиназу 1ε. Однако, это соединение также оказывает ингибирующее действие в отношении других киназ. Случайно было обнаружено, что это соединение также оказывает ингибирующее действие на казеинкиназу 1δ, и способность проявлять это ингибирующее действие является фармакологически значимым (непатентная литература 4). Также известны другие соединения, оказывающие такое селективное ингибирующее действие на казеинкиназу 1ε, но селективное ингибирование ими изоформ явным образом не описано (патентная литература 1 и 2). Кроме того, сообщалось, что на основании информации, касающейся пространственной структуры белка-мишени, была разработана модель и что затем был осуществлен так называемый виртуальный скрининг. Однако, действие полученного соединения ограничивалось только ингибирующим действием на казеинкиназу 1δ (непатентная литература 13).
Таким образом, к настоящему времени не известен тот факт, чтобы кто-то специализировался на терапевтической применимости соединения, оказывающего ингибирующее действие с высокой селективностью к казеинкиназе 1, при этом, с учетом селективного ингибирования им изоформ, данное соединение оказывает селективное ингибирующее действие на казеинкиназу 1δ и казеинкиназу 1ε, и чтобы кто-то прежде проводил интенсивные исследования, направленные на поиск представляющего интерес соединения, что и было выполнено авторами настоящего изобретения.
Для решения вышеупомянутой задачи авторы настоящего изобретения разработали сложную модель на основании информации о пространственных структурах казеинкиназы 1δ и других похожих белков с целью обнаружения различных соединений, оказывающих ингибирующее действие в отношении способности казеинкиназы 1δ и казеинкиназы 1ε осуществлять фосфорилирование. Далее авторы изобретения провели виртуальный скрининг, используя DOCK4, при выполнении которого в имеющуюся в продаже базу данных для соединений (где информацию о пространственной структуре казеинкиназы 1δ можно получить после регистрации в банке данных по белкам и т.д. (непатентная литература 14)) была введена консенсусная оценка, так что круг соединений был сужен. Авторы изобретения приобрели или по-иному синтезировали эти соединения и после этого на практике провели скрининг биологической активности соединений.
В результате этого авторы настоящего изобретения обнаружили, что соединение, представленное общей формулой (1), которая показана ниже, оказывает ингибирующее действие в отношении способности казеинкиназы 1δ и казеинкиназы 1ε осуществлять фосфорилирование. Кроме того, авторы изобретения обнаружили, что это соединение проявляет высокоселективное ингибирующее действие, которое к настоящему времени известно не было. Таким образом, авторы изобретения показали, что это соединение является полезным в качестве активного ингредиента фармацевтических средств для лечения вышеупомянутых заболеваний. Настоящее изобретение было выполнено на основании этих данных.
Таким образом, настоящее изобретение относится к производному оксазолона, представленному общей формулой (1), которая показана ниже, оказывающему ингибирующее действие на казеинкиназу 1δ и казеинкиназу 1ε, его фармакологически приемлемой соли, их сольвату или их гидрату, Формула 1
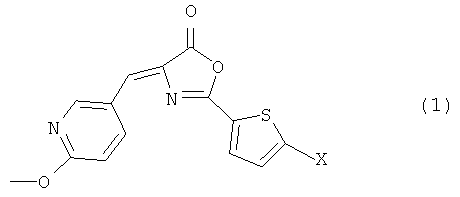
где X представляет собой атом галогена (который может быть любым атомом, выбранным из атома фтора, атома хлора, атома брома и атома йода).
Кроме того, настоящее изобретение относится к ингибитору казеинкиназы 1δ и казеинкиназы 1ε, содержащему в качестве активного ингредиента производное оксазолона, представленное выше общей формулой (1), его фармакологически приемлемую соль, их сольват или их гидрат.
Кроме того, настоящее изобретение относится к фармацевтическому средству, полезному для лечения и/или предупреждения заболеваний, с процессом развития патологических состояний которых ассоциируют активацию казеинкиназы 1δ или казеинкиназы 1ε, причем данное фармацевтическое средство содержит в качестве активного ингредиента производное оксазолона, представленное выше общей формулой (1), его фармакологически приемлемую соль, их сольват или их гидрат. Кроме того, настоящее изобретение относится к фармацевтическому средству для лечения и/или предупреждения расстройства циркадного ритма (включая расстройство сна), нейродегенеративного заболевания центральной нервной системы и рака, причем данное фармацевтическое средство содержит в качестве активного ингредиента производное оксазолона, представленное выше общей формулой (1), его фармакологически приемлемую соль, их сольват или их гидрат.
Кроме того, настоящее изобретение еще относится к способу лечения заболеваний, с патологическими состояниями которых ассоциируют механизм активации казеинкиназы 1δ или казеинкиназы 1ε, включающему введение вышеописанного фармацевтического средства пациенту.
Преимущества изобретения
Соединение по настоящему изобретению может ингибировать активность казеинкиназы 1δ и казеинкиназы 1ε. Таким образом, соединением по настоящему изобретению можно лечить заболевания, с патологическими состояниями которых ассоциируют механизм активации казеинкиназы 1δ или казеинкиназы 1ε.
Соединение по настоящему изобретению и фармацевтическое средство по настоящему изобретению, содержащее вышеупомянутое соединение в качестве фармацевтически активного ингредиента, можно использовать для лечения заболеваний, с патологическими состояниями которых ассоциируют механизм активации казеинкиназы 1δ или казеинкиназы 1ε.
Соединение по настоящему изобретению и фармацевтическое средство по настоящему изобретению, содержащее вышеупомянутое соединение в качестве фармацевтически активного ингредиента, обладают более высокой селективностью в отношении казеинкиназы 1δ и казеинкиназы 1ε, чем традиционные соединения, обладающие ингибирующей казеинкиназу 1 активностью. Таким образом, можно ожидать, что соединение по настоящему изобретению и фармацевтическое средство по настоящему изобретению, содержащее вышеупомянутое соединение в качестве фармацевтически активного ингредиента, будут обладать более высокой клинической эффективностью по сравнению с подобными существующими соединениями при лечении заболеваний, с патологическими состояниями которых ассоциируют механизм активации казеинкиназы 1δ или казеинкиназы 1ε. Одновременно с этим, можно ожидать, что применение соединения по настоящему изобретению и фармацевтического средства по настоящему изобретению, содержащего вышеупомянутое соединение в качестве фармацевтически активного ингредиента, будет характеризоваться большей степенью безопасности, чем применение существующих соединений.
Описание воплощений
Предпочтительное воплощение настоящего изобретения относится к производному оксазолона, представленному общей формулой (1), его фармакологически приемлемой соли, их сольвату или их гидрату. Это соединение оказывает ингибирующее действие на казеинкиназу 1δ или казеинкиназу 1ε.
Соединение, представленное общей формулой (1), синтезировано в настоящем изобретении по-иному. Кроме того, в настоящем изобретении впервые показано, что соединение, представленное общей формулой (1), обладает ингибирующей активностью в отношении казеинкиназы 1δ или казеинкиназы 1ε. Новое производное оксазолона, представленное общей формулой (1) по настоящему изобретению, можно получить, используя метод химического синтеза, описанный в вышеупомянутом разделе Примеры.
Ингибитор казеинкиназы 1δ или казеинкиназы 1ε и производное оксазолона, представленное общей формулой (1), содержащиеся в качестве активного ингредиента в фармацевтическом средстве по настоящему изобретению, включают их таутомеры и геометрические изомеры (например, E-форму, Z-форму и т.д.), если не указано иное. Кроме того, в настоящее изобретение также включены энантиомеры, если они имеются.
Типы соединения, представленного общей формулой (1), его соли и их гидрата не ограничены. Примеры включают следующие соединения:
4-((6-метокси-3-пиридинил)метилен)-2-(5-фтор-2-тиенил)-5(4H)-оксазолон, его приемлемую соль и их гидрат;
4-((6-метокси-3-пиридинил)метилен)-2-(5-хлор-2-тиенил)-5(4H)-оксазолон, его приемлемую соль и их гидрат;
4-((6-метокси-3-пиридинил)метилен)-2-(5-бром-2-тиенил)-5(4H)-оксазолон, его приемлемую соль и их гидрат и
4-((6-метокси-3-пиридинил)метилен)-2-(5-иод-2-тиенил)-5(4H)-оксазолон, его приемлемую соль и их гидрат.
Согласно другому предпочтительному воплощению настоящего изобретения предложен ингибитор казеинкиназы 1δ и казеинкиназы 1ε, содержащий в качестве активного ингредиента производное оксазолона, представленное общей формулой (1), его фармакологически приемлемую соль, их сольват или их гидрат. Согласно следующему предпочтительному воплощению настоящего изобретения предложено фармацевтическое средство для лечения заболеваний, с патологическими состояниями которых ассоциируют активацию казеинкиназы 1δ или казеинкиназы 1ε и, в частности, расстройство циркадного ритма (включая расстройство сна), нейродегенеративное заболевание центральной нервной системы и рак, при этом данное фармацевтическое средство содержит описанное выше соединение в качестве фармакологически активного ингредиента.
Казеинкиназа 1δ в настоящем изобретении может иметь похожие названия или альтернативные названия, такие как "казеинкиназа 1 дельта", "изоформа дельта казеинкиназы 1", "CK1(-)дельта", "CK1d", "HCKID", "казеинкиназа 1δ", "изоформа δ казеинкиназы 1" и "CK1(-)δ". В настоящем изобретении казеинкиназа 1δ означает белок, содержащий аминокислотную последовательность, идентичную или по существу идентичную аминокислотным последовательностям, зарегистрированным под №№регистрации NP_001884.2, NP_620693.1, NP_620690.1 и NP_082150.1 в базе данных ссылочных последовательностей (RefSeq) NCBI, опубликованной Национальным центром биотехнологической информации (NCBI).
В данном описании фраза "белок, содержащий аминокислотную последовательность, по существу идентичную…" означает белок, который содержит аминокислотную последовательность, идентичную приблизительно на 60% или более, предпочтительно приблизительно на 70% или более, более предпочтительно приблизительно на 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97% или 98% и наиболее предпочтительно приблизительно на 99%, на уровне аминокислотной последовательности, описанным выше аминокислотным последовательностям, имеющим RefSeq №№ NP_001884.2, NP_620693.1, NP_620690.1 и NP_082150.1, и который обладает активностью фермента, фосфорилирующего белки.
В других случаях белок, содержащий аминокислотную последовательность, по существу идентичную аминокислотным последовательностям, имеющим RefSeq №№ NP_001884.2, NP_620693.1, NP_620690.1, NP_082150.1 представляет собой белок, аминокислотная последовательность которого содержит делецию, замену или вставку одной или нескольких (предпочтительно примерно 1-30, более предпочтительно примерно 1-10 и еще более предпочтительно 1-5) аминокислот по сравнению с аминокислотными последовательностями, имеющими RefSeq №№ NP_001884.2, NP_620693.1, NP_620690.1 и NP_082150.1, и который обладает активностью фермента, фосфорилирующего белки.
Казеинкиназа 1ε в настоящем изобретении может иметь похожие названия или альтернативные названия, такие как "казеинкиназа 1 эпсилон", "изоформа эпсилон казеинкиназы 1", "CK1(-) эпсилон", "CK1е", "HCKIE", "казеинкиназа 1ε", "изоформа ε казеинкиназы 1" и "CK1(-)ε". В настоящем изобретении казеинкиназа 1ε означает белок, содержащий аминокислотную последовательность, идентичную или по существу идентичную аминокислотным последовательностям, зарегистрированным под №№ регистрации NP_001885.1, NP_689407.1 и NP_038795.3 в базе данных ссылочных последовательностей (RefSeq) NCBI, опубликованной Национальным центром биотехнологической информации (NCBI).
В данном описании фраза "белок, содержащий аминокислотную последовательность, по существу идентичную…" означает белок, который содержит аминокислотную последовательность, идентичную приблизительно на 60% или более, предпочтительно приблизительно на 70% или более, более предпочтительно приблизительно на 80%, 81%, 82%, 83%, 84%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97% или 98% и наиболее предпочтительно приблизительно на 99%, на уровне аминокислотной последовательности, описанным выше аминокислотным последовательностям, имеющим RefSeq №№RefSeq №№ NP_001885.1, NP_689407.1, и который обладает активностью фермента, фосфорилирующего белки.
В других случаях, белок, содержащий аминокислотную последовательность, по существу идентичную аминокислотным последовательностям, имеющим RefSeq №№ NP_001885.1, NP_689407.1 и NP_038795.3, представляет собой белок, аминокислотная последовательность которого содержит делецию, замену или вставку одной или нескольких (предпочтительно примерно 1-30, более предпочтительно примерно 1-10 и еще более предпочтительно 1-5) аминокислот по сравнению с аминокислотными последовательностями, имеющими RefSeq № NP_001885.1, NP_689407.1 и NP_038795.3, и который обладает активностью фермента, фосфорилирующего белки.
Примеры заболеваний, с патологическими состояниями которых ассоциируют механизм активации казеинкиназы 1δ или казеинкиназы 1ε, не ограничены. Примеры таких заболеваний включают расстройство циркадного ритма (включая расстройство сна), нейродегенеративное заболевание и рак.
В описании настоящего изобретения тип расстройства циркадного ритма не ограничен. Расстройство циркадного ритма включает расстройство настроения и расстройство сна. Такое расстройство сна представляет собой расстройство циркадного ритма сна, и расстройство циркадного ритма сна включает заболевание, выбранное из группы, состоящей из расстройства сна, вызванного сменным режимом работы, синдрома десинхронизации физиологических циклов после трансмеридианных перелетов, синдрома преждевременной фазы сна и синдрома задержки фазы сна. Кроме того, расстройство сна включает заболевание, выбранное из группы, состоящей из бессонницы, связанного со сном расстройства дыхания, связанной с нарушениями центральной нервной системы гиперсомнии, парасомнии и связанного со сном нарушения движений. Кроме того, описанное выше расстройство настроения выбрано либо из депрессивного расстройства, либо биполярного расстройства, и данное депрессивное расстройство представляет собой большое депрессивное расстройство. Кроме того, расстройство настроения выбрано либо из депрессивного расстройства, либо биполярного расстройства, и данное биполярное расстройство выбрано из группы, состоящей из биполярного расстройства I типа или биполярного расстройства II типа. Более того, примеры заболевания в настоящем изобретении включают бессонницу, связанное со сном расстройство дыхания, связанную с нарушениями центральной нервной системы гиперсомнию, расстройство циркадного ритма сна, парасомнию, связанное со сном нарушение движений и расстройство сна, вызванное другими причинами.
В описании настоящего изобретения бессонница включает психофизиологическую бессонницу, вызванную стрессом или тому подобными причинами, бессонницу, вызванную заболеванием внутренних органов, и тому подобное. Связанное со сном расстройство дыхания включает синдром апноэ во сне центрального происхождения, синдром обструктивного апноэ во сне, синдром связанной со сном гиповентиляции/гипоксемии и тому подобное. Связанная с нарушениями центральной нервной системы гиперсомния включает нарколепсию, идиопатическую гиперсомнию, рецидивирующую гиперсомнию и тому подобное. Расстройство циркадного ритма сна включает расстройство сна, вызванное сменным режимом работы, синдром десинхронизации физиологических циклов после трансмеридианных перелетов, синдрома преждевременной фазы сна, синдрома задержки фазы и тому подобное. Парасомния включает лунатизм, расстройство поведения в фазе "быстрого сна" и тому подобное. Связанное со сном нарушение движений включает синдром усталых ног, синдром периодических движений конечностей и тому подобное.
В описании настоящего изобретения тип нейродегенеративного заболевания не ограничен. Примеры нейродегенеративного заболевания центральной нервной системы включают: нейродегенеративное заболевание, вызываемое болезнью Альцгеймера, болезнью Паркинсона или синдромом Дауна; дегенеративное нарушение нервной системы, вызванное физическим повреждением нервов (повреждением ткани головного мозга, таким как закрытая травма головного мозга, и повреждением нервов, вызванным травмой головы и тому подобным); и дегенеративное нарушение нервной системы, вызванное физическим повреждением нервов после ишемии или ишемической реперфузии, включая: инсульт, церебральный инфаркт, кровоизлияние в мозг, церебральную ишемию, субарахноидальное кровоизлияние, аневризматическое кровоизлияние, инфаркт миокарда, гипоксию, аноксию и повреждение нервов, вызванное припадком эпилепсии/церебральной ишемией.
Тип рака, происходящего из поджелудочной железы, в описании настоящего изобретения не ограничен. Примеры такого типа рака включают рак протоков поджелудочной железы, инвазивный рак протоков поджелудочной железы, панкреатическую эндокринную опухоль, внутрипротоковую папиллярную муцинозную опухоль, муцинозную цистому, ацинарноклеточный рак и метастатический рак поджелудочной железы.
В качестве активных ингредиентов фармацевтического средства по настоящему изобретению в дополнение к соединениям, представленным выше общей формулой (1), также можно использовать их физиологически приемлемые соли. В том случае, когда присутствует кислотная группа, примеры солей, которые могут быть с ней образованы, включают: соли щелочных металлов и щелочноземельных металлов, таких как литий, натрий, калий, магний и кальций; соли аминов, таких как аммиак, метиламин, диметиламин, триметиламин, дициклогексиламин, трис(гидроксиметил)аминометан, N,Nбис(гидроксиэтил)-пиперазин, 2-амино-2-метил-1-пропанол, этаноламин, N-метил-глюкамин и L-глюкамин; и соли, образованные с основными аминокислотами, такими как лизин, δ-гидроксилизин и аргинин. В том случае, когда присутствует основная группа, примеры солей, которые могут быть с ней образованы, включают: соли с минеральными кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота; соли с органическими кислотами, такими как метансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, уксусная кислота, пропионовая кислота, винная кислота, фумаровая кислота, малеиновая кислота, яблочная кислота, щавелевая кислота, янтарная кислота, лимонная кислота, бензойная кислота, миндальная кислота, коричная кислота, молочная кислота, гликолевая кислота, глюкуроновая кислота, аскорбиновая кислота, никотиновая кислота и салициловая кислота; и соли с кислыми аминокислотами, такими как аспарагиновая кислота и глутаминовая кислота.
Кроме того, в качестве активных ингредиентов фармацевтического средства по настоящему изобретению также можно использовать сольваты или гидраты соединений, представленных выше общей формулой (1), или их солей.
Что касается фармацевтического средства по настоящему изобретению, то соединения, представленные выше общей формулой (1), их фармакологически приемлемые соли, их сольваты или их гидраты, которые содержатся в качестве активных ингредиентов, могут быть введены напрямую. Однако, как правило, желательно вводить фармацевтическое средство по настоящему изобретению в форме фармацевтической композиции, содержащей вышеупомянутое вещество в качестве активного ингредиента и одно или два либо более фармацевтических вспомогательных веществ. В качестве активного ингредиента фармацевтического средства по настоящему изобретению можно использовать два или более типов вышеупомянутого вещества в комбинации. Также можно смешать в вышеописанной фармацевтической композиции активные ингредиенты других фармацевтических средств для лечения и/или предупреждения заболеваний, с патологическими состояниями которых ассоциируют механизм активации казеинкиназы 1δ или казеинкиназы 1ε. Также можно смешать в вышеописанной фармацевтической композиции активные ингредиенты других фармацевтических средств для лечения и/или предупреждения расстройств циркадного ритма (включая расстройство сна), нейродегенеративного заболевания центральной нервной системы и рака, среди вышеупомянутых заболеваний.
Тип фармацевтической композиции конкретно не ограничен. Примеры лекарственной формы включают таблетку, капсулу, гранулу, порошок, сироп, суспензию, суппозиторий, мазь, крем, гель, пластырь, средство для ингаляции и лекарство для инъекции. Эти фармацевтические средства готовят согласно общепринятым способам. Следует отметить, что жидкое средство может принимать вид лекарственной формы, в которой данное средство при его применении находится в растворенном или суспендированном состоянии в воде или подходящем растворителе. Помимо этого, на таблетку или гранулу может быть нанесено покрытие согласно хорошо известным способам. В случае инъекции изготовление осуществляют путем растворения соединения по настоящему изобретению в воде. Соединение по настоящему изобретению также может быть растворено в обычном физиологическом растворе или растворе глюкозы, как будет необходимо. В других случаях к соединению по настоящему изобретению также может быть добавлен буфер или консервант. Фармацевтическое средство по настоящему изобретению предложено в форме того или иного фармацевтического средства для применения при пероральном введении или парентеральном введении. Например, на его основе можно изготовить: фармацевтические композиции для перорального введения, например, в форме гранулы, тонкоизмельченного средства, порошка, твердой капсулы, мягкой капсулы, сиропа, эмульсии, суспензии или жидкости; и фармацевтические композиции для парентерального введения, например в форме лекарства для инъекции с целью внутривенного введения, внутримышечного введения или подкожного введения, в форме капель, трансдермального средства, трансмукозального средства, капель в нос, средства для ингаляции или суппозитория. Лекарство для инъекции, капли или тому подобное могут быть изготовлены в лекарственной форме в виде лиофилизированных порошков, и эти порошки затем при использовании могут быть растворены в подходящей водной среде, такой как обычный физиологический раствор. Кроме того, также возможно непосредственное введение в головной мозг средства с непрерывным высвобождением, покрытого оболочкой из макромолекул и тому подобного.
Типы фармацевтических вспомогательных веществ, используемых в изготовлении фармацевтической композиции, соотношение таких фармацевтических вспомогательных веществ и активного ингредиента или способ изготовления фармацевтической композиции могут быть выбраны соответствующим образом специалистом в данной области в зависимости от формы композиции. В качестве фармацевтических вспомогательных веществ можно использовать неорганические или органические вещества либо твердые или жидкие вещества. В общем случае такие фармацевтические вспомогательные вещества можно смешивать в количестве массовых процентов, составляющем от 1% до 90% из расчета на массу активного ингредиента. Конкретные примеры такого вещества включают лактозу, глюкозу, маннит, декстрин, циклодекстрин, крахмал, сахарозу, алюмометасиликат магния, синтетический силикат алюминия, карбоксиметилцеллюлозы натриевую соль, гидроксипропилкрахмал, карбоксиметилцеллюлозы кальциевую соль, ионообменную смолу, метилцеллюлозу, желатин, аравийскую камедь, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, поливинилпирролидон, поливиниловый спирт, легкую безводную кремниевую кислоту, стеарат магния, тальк, трагакант, бентонит, вигум, диоксид титана, сложный эфир сорбитана и жирной кислоты, лаурилсульфат натрия, глицерин, сложный эфир глицерина и жирной кислоты, очищенный ланолин, глицеринизированный желатин, полисорбат, макрогол, растительное масло, воск, вазелиновое масло, медицинский вазелин, фторированный углеводород, неионное поверхностно-активное вещество, пропиленгликоль и воду.
Чтобы изготовить средство в твердой форме для перорального введения, активный ингредиент смешивают с эксципиентом, таким как лактоза, крахмал, кристаллическая целлюлоза, лактат кальция или ангидрид кремниевой кислоты с образованием средства в виде порошка. В других случаях, если это необходимо, к смеси дополнительно добавляют связующее вещество, такое как сахароза, гидроксипропилцеллюлоза или поливинилпирролидон, разрыхлитель, такой как карбоксиметилцеллюлоза или кальциевая соль карбоксиметилцеллюлозы, и другие вспомогательные вещества и затем полученную смесь обрабатывают методом сухой или влажной грануляции с образованием средства в виде гранул. Кроме того, чтобы изготовить таблетку, такое средство в виде порошка или гранул можно непосредственно подвергнуть таблетированию, либо к средству в виде порошка или гранул можно добавить смазывающее вещество, такое как стеарат магния или тальк, и после этого полученную смесь можно подвергнуть таблетированию. На такое средство в виде гранул или такую таблетку можно нанести основу энтеросолюбильного покрытия, например, из фталата гидроксипропилметилцеллюлозы или полимера метакриловой кислоты-метилметакрилата, с получением средства с энтеросолюбильным покрытием. Альтернативно, на такое средство в виде гранул или такую таблетку можно нанести покрытие из этилцеллюлозы, карнаубского воска, гидрогенизированного масла или тому подобного, с получением средства с непрерывным высвобождением. Кроме того, чтобы изготовить капсулу, средство в виде порошка или гранул помещают в твердую капсулу. В других случаях непосредственно на активный ингредиент наносят желатиновую пленку или его растворяют в глицерине, полиэтиленгликоле, кунжутном масле, оливковом масле или тому подобном и затем наносят желатиновую пленку, получая мягкую капсулу.
Чтобы изготовить лекарство для инъекции, активный ингредиент и при необходимости регулирующий pH агент, такой как соляная кислота, гидроксид натрия, лактоза, молочная кислота, натрий, моногидрофосфат натрия или дигидрофосфат натрия, придающий изотоничность агент, такой как хлорид натрия или глюкоза, и другие вспомогательные вещества, растворяют в дистиллированной воде для инъекций, после этого полученный раствор подвергают фильтрации в асептических условиях и затем его вносят в ампулу. В других случаях к полученному раствору добавляют маннит, декстрин, циклодекстрин, желатин и тому подобное, затем осуществляют сушку сублимацией, получая лекарство для инъекции, которое следует растворить при использовании. Кроме того, к активному ингредиенту добавляют лецитин, полисорбат 80, полиоксиэтилен-гидрогенизированное касторовое масло или тому подобное для его эмульгирования в воде, получая эмульсию для инъекции.
Чтобы изготовить средство для ректального введения, активный ингредиент вместе с суппозиторной основой, такой как масло какао, три-, ди- и моноглицериды жирных кислот или полиэтиленгликоль, растворяют посредством увлажнения и полученный раствор затем выливают в форму с последующим охлаждением. В других случаях активный ингредиент растворяют в полиэтиленгликоле, соевом масле или тому подобном и затем полученный раствор покрывают желатиновой пленкой.
Чтобы изготовить средство для наружного применения на кожу, активный ингредиент добавляют к медицинскому вазелину, пчелиному воску, вазелиновому маслу, полиэтиленгликолю или тому подобному, смесь при необходимости увлажняют и затем смешивают, с тем, чтобы получить мазь. В других случаях, активный ингредиент смешивают вместе со связующим веществом, таким как канифольный или алкилакрилатный полимер, и полученную смесь затем наносят на нетканый материал, например на основе полиалкила, с тем, чтобы получить средство в виде лейкопластыря.
Применяемая доза и количество доз фармацевтического средства по настоящему изобретению конкретно не ограничены. Применяемая доза и количество доз могут быть выбраны, как будет целесообразно, согласно заключению врача в зависимости от разных обстоятельств, таких как цель, заключающаяся в предупреждении и/или лечении ухудшения и прогрессирования целевого заболевания, тип заболевания, масса тела, возраст и другие характеристики пациента и т.д. В общем случае, применяемая доза составляет приблизительно 0,01-1000 мг (по массе активного ингредиента) для взрослого человека в сутки посредством перорального введения. Такую дозу можно ввести однократно или поделить на несколько введений в сутки или в каждые несколько суток. Когда фармацевтическое средство по настоящему изобретению используют в качестве лекарства для инъекции, желательно, чтобы доза 0,01-100 мг (по массе активного ингредиента) вводилась взрослому человеку в течение одних суток непрерывно или периодически.
Используя носитель, способный предотвращать незамедлительное высвобождение средства из организма, фармацевтическое средство по настоящему изобретению можно изготовить в форме средства с замедленным высвобождением, например, в форме имплантированной таблетки или системы доставки, заключенной в микрокапсулу. Например, можно использовать биоразлагаемые биосовместимые полимеры, такие как этиленвинилацетат, ангидрид поликислоты, полигликолевая кислота, коллаген, полиортоэфир и полиуксусная кислота. Специалист в данной области может легко изготовить такие вещества. Помимо этого, в качестве фармацевтически приемлемого носителя также можно использовать липосомальную суспензию. Тип имеющихся в наличии липосом не ограничен. Липосому можно изготовить в виде липидной композиции, содержащей фосфатидилхолин, холестерин и ПЭГ-производное фосфатидилэтанола (ПЭГ-ФЭ), с подходящим для применения размером посредством пропускания ее через фильтр с соответствующим размером пор и дальнейшей очисткой методом обращенно-фазового выпаривания.
Фармацевтическое средство по настоящему изобретению может быть изготовлено в виде фармацевтической композиции, и она может быть помещена в резервуар или упаковку вместе с руководством по введению, с тем чтобы образовать набор. Если фармацевтическая композиция по настоящему изобретению предложена в виде набора, то образующие данную фармацевтическую композицию разные составляющие помещают в разные резервуары и затем их смешивают непосредственно перед применением. Так, составляющие размещают отдельно друг от друга, потому что это позволяет осуществлять длительное хранение без потери функций активных составляющих.
Содержащийся в наборе реагент поставляется в резервуаре определенного типа, в котором составляющие сохраняют свою активность в течение длительного промежутка времени и при этом не адсорбируются на материале данного сосуда резервуара и не разлагаются. Например, герметично закрытая стеклянная ампула содержит буфер, находящийся в атмосфере нейтрального нереакционноспособного газа, такого как газообразный азот. Ампула изготовлена из стекла, органического полимера, такого как поликарбонат или полистирол, керамики, металла, других подходящих материалов, которые обычно используются для хранения данного реагента, и тому подобного. Примеры других подходящих сосудов резервуаров включают обычную бутылку, изготовленную из материала, подобного материалу ампулы, и упаковочный материал, внутренняя сторона которого выложена алюминиевой фольгой или фольгой из алюминиевого сплава. Другие резервуары включают пробирку, флакон, колбу, бутылку, шприц и аналогичные им продукты. Такой резервуар содержит входное отверстие для стерильного доступа, как например бутылка с пробкой, допускающей прокалывание иглой для подкожных инъекций.
Помимо этого к набору приложена инструкция по применению. Инструкция по применению набора, содержащего фармацевтическую композицию по настоящему изобретению, печатается на бумаге или других материалах и/или она может поставляться на электрическом или электромагнитном программоносителе, таком как дискета (зарегистрированного товарного знака), CD-ROM, DVD-ROM, Zip-диск, видеокассета или аудиокассета. Подробная инструкция по применению может быть реально вложена в набор или может быть опубликована на веб-сайте, который определяется производителем или дистрибьютором набора и затем указывается по электронной почте.
Кроме того, согласно настоящему изобретению предложен терапевтический способ лечения целевого заболевания путем введения пациенту фармацевтического средства или фармацевтической композиции, которое(ая) содержит в качестве активного ингредиента соединение, представленное в настоящем изобретении общей формулой (1), его фармакологически приемлемую соль, их сольват или их гидрат.
В настоящем описании термин "лечение" используется для обозначения того случая, когда имеется подавление или уменьшение прогрессирования и ухудшения патологических состояний заболевания у млекопитающего, которое поражено или предполагается, что поражено данным заболеванием, и тем самым термин "лечение" используется для обозначения терапевтического средства, направленного на подавление или уменьшение прогрессирования и ухудшения различных симптомов заболевания.
Кроме того, термин "заболевание" в целом означает заболевания, с патологическими состояниями которых ассоциируют механизм активации казеинкиназы 1δ или казеинкиназы 1ε. Таким образом, тип заболевания конкретно не ограничен. Примеры такого заболевания включают бессонницу, связанное со сном расстройство дыхания, связанную с нарушениями центральной нервной системы гиперсомнию, расстройство циркадного ритма сна, парасомнию и связанное со сном нарушение движений. Расстройство сна представляет собой расстройство циркадного ритма сна, и расстройство циркадного ритма сна включает расстройство сна, вызванное сменным режимом работы, синдром десинхронизации физиологических циклов после трансмеридианных перелетов, синдром преждевременной фазы сна и синдром задержки фазы сна. Расстройство настроения при расстройстве циркадного ритма включает депрессивное расстройство и биполярное расстройство. Помимо этого, рассматриваемое заболевание также включает: нейродегенеративное заболевание, вызываемое болезнью Альцгеймера, болезнью Паркинсона или синдромом Дауна; нейродегенеративное заболевание центральной нервной системы, вызываемое цереброваскулярным расстройством; рак в целом, тип которого не конкретизирован; и в частности рак, происходящий из поджелудочной железы, такой как рак протоков поджелудочной железы, инвазивный рак протоков поджелудочной железы, панкреатическая эндокринная опухоль, внутрипротоковая папиллярная муцинозная опухоль, муцинозная цистома, ацинарноклеточный рак и метастатический рак поджелудочной железы.
Термин "млекопитающее" в качестве субъекта, нуждающегося в лечении, обозначает любое данное животное, входящее в класс млекопитающих, и тип млекопитающего конкретно не ограничен. Примеры таких млекопитающих включают людей, домашних животных, таких как собака, кошка или кролик, и сельскохозяйственных животных, таких как бык, свинья, овца или лошадь. В частности, предпочтительным "млекопитающим" является человек.
Далее настоящее изобретение будет описано в конкретных примерах. Однако подразумевается, что эти примеры не ограничивают объем настоящего изобретения.
Примеры
Пример 1
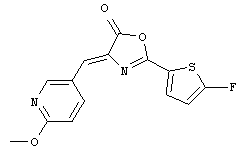
4-((6-Метокси-3-пиридинил)метилен)-2-(5-фтор-2-тиенил)-5(4Н)-оксазолон (Формула 2)
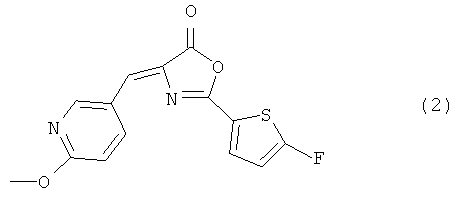
Поскольку, как правило, чрезвычайно трудно ввести группу фторо в тиофеновое кольцо, была сделана попытка синтеза соединения, представленного вышеуказанной формулой (2), способом, показанным на приведенной ниже Схеме 1, содержащим много стадий синтеза по сравнению со способом синтеза, описанным в разделе Примеры заявки на патент Японии №2009-245477. В результате было успешно получено представляющее интерес соединение. Следует отметить, что способ синтеза соединения, представленного формулой (2), не ограничен способом, показанным на Схеме 1.
Формула 3
Схема 1
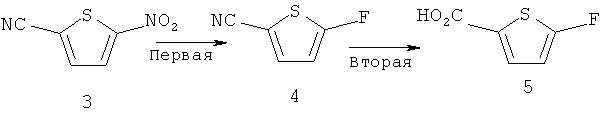
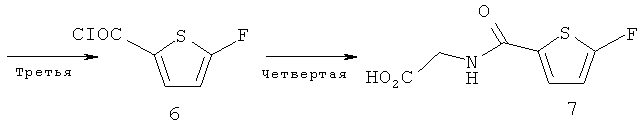
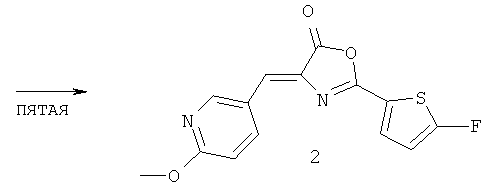
Первая стадия
3,0 г 5-нитро-2-тиофенкарбонитрила (Соединение 3; 19,5 ммоль), 5,66 г (9,75 ммоль) фторида калия, 0,82 г (1,95 ммоль) бромида тетрафенилфосфония, 70 мл сульфолана и 2,81 мл (19,5 ммоль) фталоилдихлорида последовательно добавляли в трехгорлую колбу и затем смесь перемешивали с нагреванием при внешней температуре 180°C в течение 2 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры и далее в него добавляли 300 мл воды, затем три раза экстрагировали, используя по 200 мл диэтилового эфира. После этого органические слои объединяли и объединенный органический слой последовательно промывали, используя дважды по 200 мл 1 н. гидроксида натрия, затем 200 мл воды и затем 100 мл насыщенного рассола. Полученное вещество сушили над сульфатом натрия и затем растворитель удаляли при пониженном давлении. Остаток очищали хроматографией на силикагеле (пентан/диэтиловый эфир=2/1), с тем чтобы получить 150 мг
Соединения 4.
Вторая стадия
138 мг (1,07 ммоль) Соединения 4 и 5 мл 1 н. гидроксида натрия добавляли в колбу баклажаноподобной формы и затем смесь перемешивали с нагреванием при внешней температуре 100°C в течение 2 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры. Затем в реакционный раствор добавляли 5 мл воды и к смеси также добавляли 10 мл 1 н. соляной кислоты, получая в результате pH 1. После этого смесь дважды экстрагировали, используя по 30 мл дихлорметана. Далее органические слои объединяли. Органический слой сушили над сульфатом натрия и затем растворитель удаляли при пониженном давлении, с тем чтобы получить 110 мг Соединения 5.
Третья стадия
100 мг (0,68 ммоль) Соединения 5, 3 мл тионилхлорида и 50 мкл формамида добавляли в колбу баклажаноподобной формы и затем смесь перемешивали с нагреванием при внешней температуре 100°C в течение 2 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры, удаляли тионилхлорид при пониженном давлении, после этого проводили двукратную азеотропную отгонку с толуолом, с тем чтобы получить 112 мг Соединения 6 с количественным выходом.
Четвертая стадия
51 мг (0,68 ммоль) глицина и 1 мл 6 н. гидроксида натрия добавляли в колбу баклажаноподобной формы и затем смесь перемешивали с охлаждением на льду. После этого к реакционному раствору добавляли 5 мл раствора Соединения 6 в диэтиловом эфире и полученную смесь далее перемешивали при комнатной температуре в течение 3 часов. После завершения реакции реакционный раствор охлаждали на льду и затем к полученному раствору добавляли 10 мл воды. Далее к раствору добавляли 10 мл 1 н. соляной кислоты для подведения значения pH до 1 и полученный раствор затем трижды экстрагировали дихлорметаном. После этого органические слои объединяли. Объединенный органический слой промывали 30 мл насыщенного рассола и затем сушили над сульфатом натрия, после чего удаляли растворитель при пониженном давлении, с тем чтобы получить 24 мг Соединения 7.
Пятая стадия
21,7 мг (0,107 ммоль) Соединения 7, 200 мкл уксусного ангидрида и 65 мл пиридина добавляли в колбу баклажаноподобной формы и полученную смесь далее перемешивали с нагреванием при внешней температуре 100°C в течение 1 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры и затем растворитель удаляли при пониженном давлении. Остаток очищали препаративной тонкослойной хроматографией (толуол/диэтиловый эфир=5/1), с тем чтобы получить 14 мг Соединения 2 (0,046 ммоль; общий выход: 0,3%).
MALDI-TOF MS (времяпролетная масс-спектрометрия с ионизацией посредством лазерной десорбции ионов из матрицы): рассчитанная величина для C14H9FN2O3S: 304,03; измеренная величина: 304,23.
В процессе синтеза Соединения 2 было чрезвычайно трудно ввести группу фторо в тиофеновое кольцо на первой стадии и, таким образом для общего выхода и чистоты были получены низкие значения. В результате чистота Соединения 2 как конечного продукта также получилась низкой. Так, ниже приведен, хотя возможно с не очень высокой точностью, пик, полученный в результате анализа Соединения 2, растворенного в DMSO-d6 (диметилсульфоксид-d6) с использованием прибора AV-500 (500 МГц) для ядерного магнитного резонанса (ЯМР) производства Bruker.
1κ-ЯМР (500 МГц, DMSO-d6, δ) 8.73 (s, 1H), 8.19 (d, 1H), 7.72 (m, 1H), 7.36 (d, 1H), 7.26 (s, 1H), 7.19 (s, 1H), 3.91 (s, 3H).
Соединение, представленное формулой (2), может быть синтезировано способом, показанным выше на Схеме 1. Однако, применение этого способа было сопряжено с проблемой, заключающейся в том, что чистота получаемого продукта оказалась низкой.
Соответственно, авторы настоящего изобретения провели исследования, касающиеся других способов синтеза, и в результате им удалось значительно улучшить общий выход и чистоту продукта, следуя способу, показанному на приведенной ниже Схеме 2.
Формула 4 Схема 2
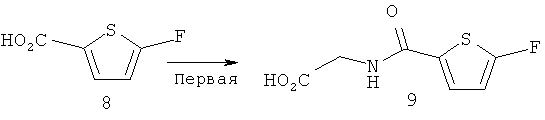
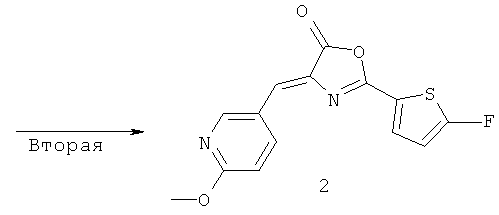
Первая стадия
1,00 г 5-фтортиофен-карбоновой кислоты (чистота: 95%; 5,91 ммоль), 1 мл (13,8 ммоль) тионилхлорида и 10 мл бензола добавляли в трехгорлую колбу и затем смесь перемешивали с нагреванием при температуре бани 105°C в течение 2 часов. После этого реакционный раствор охлаждали до комнатной температуры и затем дистилляцией удаляли тионилхлорид. Остаток подвергали азеотропной отгонке с бензолом и четыреххлористым углеродом, с тем чтобы получить коричневое маслянистое вещество. В отдельно подготовленную трехгорлую колбу добавляли 621,0 мг (8,27 ммоль) глицина и 30 мл 0,5 н. гидроксида натрия и затем смесь перемешивали. К этому реакционному раствору добавляли полученное ранее коричневое маслянистое вещество и полученную смесь далее перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционный раствор охлаждали в ледяной бане и затем к реакционному раствору добавляли 1 мл 6 н. соляной кислоты. Выпавшее в осадок твердое вещество собирали фильтрацией и затем промывали 30 мл холодной воды. Содержащуюся в твердом веществе воду удаляли азеотропной отгонкой с ацетоном и затем остаток сушили при пониженном давлении при комнатной температуре, с тем чтобы получить 432,3 мг Соединения 9 (2,13 ммоль; 36,0%) в виде светло-желтого твердого вещества. В результате проведения анализа продукта с использованием HPLC (высокоэффективной жидкостной хроматографии) было обнаружено, что время элюирования составило 9,04 минуты, а чистота составила 90%. Вторая стадия
100 мг Соединения 9 (чистота: 90,0%; 0,44 ммоль), 80,9 г (0,59 ммоль) 6-метокси-3-пиридинкарбальдегида, 102 мкл (1,08 ммоль) уксусного ангидрида и 100 мкл (1,24 ммоль) пиридина добавляли в трехгорлую колбу и полученную смесь далее перемешивали с нагреванием при 100°C в масляной бане в течение 15 минут. После завершения реакции реакционный раствор охлаждали при 4°C и выпавшее в осадок твердое вещество затем растворяли в 5 мл этанола. Далее нерастворимое вещество собирали фильтрацией. Полученное твердое вещество промывали этанолом, с тем чтобы получить 40,1 мг Соединения 2 (0,13 ммоль; 29,5%) в виде желтого твердого вещества.
HPLC-чистота (длина волны поглощения: 254 нм) Соединения 2 составила 98,7%. Соединение 2 анализировали, используя прибор HPLC/ESI-LIT-TOF MS (TOF-MS с линейной ловушкой (linear ion trap) и электрораспылительной ионизацией (electrospray ionization) в сочетании с HPLC) (NanoFrontier LD), в результате чего было подтверждено значение m/z=355,0; 356,0 [М+СН4O-Н]. Помимо этого ниже приведен пик, полученный в результате анализа Соединения 2 с использованием прибора AV-500 (500 МГц) для ядерного магнитного резонанса производства Bruker.
1H-ЯМР (CDCl3) δ млн-1 (500 МГц): 4.02 (s, 3H), 6.63 (dd, 1H, JHH=8,4 Гц, JHF=1,4 Гц), 6.86 (d, 1H, JHH=8,8 Гц), 7.13 (s, 1H), 7.55 (dd, 1H, JHH=8,4 Гц, JHF=4,1 Гц), 8.60 (dd, 1H, JHH=8,8 Гц, JHH=2,3 Гц), 8.67 (d, 1H, JHH=2,3 Гц).
13C-ЯМР (CDCl3) δ млн-1 (125 МГц): 53.9, 110.0, 110.1, 111.8, 117.4, 117.5, 123.5, 127.6, 130.6, 130.6, 131.7, 140.9, 152.1, 158.2, 165.3, 166.5, 169.4, 171.8.
С учетом HPLC-чистоты, которая составила 98,7%, результатов масс-спектрометрии и вида ЯМР-спектров подтверждено, что синтезированное таким образом соединение представляет собой Соединение 2.
Как описано выше, в результате применения способа синтеза, приведенного на Схеме 2, можно уменьшить общее число стадий с 5 до 3 по сравнению со случаем на Схеме 1, и кроме того, можно повысить общий выход приблизительно в 7 раз и также можно значительно улучшить чистоту Соединения 2 как конечного продукта. На основании этих результатов полагают, что такой путь откроет перспективу стабильным поставкам данного соединения.
Пример 2
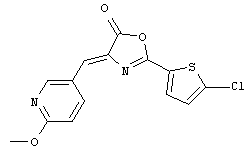
4-((6-Метокси-3-пиридинил)метилен)-2-(5-хлор-2-тиенил)-5(4Н)-оксазолон (Формула 5)
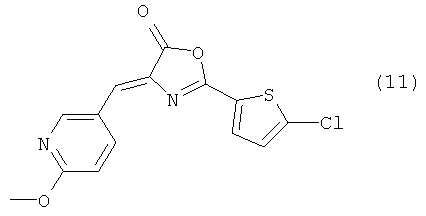
Помимо соединения, представленного формулой (2), в которое введена группа фторо, также было получено соединение, представленное выше формулой (11), в которое введена группа хлоро. Необходимо отметить, что способ синтеза соединения, представленного формулой (11), в которое введена группа хлоро, не ограничен способом, показанным на приведенной ниже Схеме 3.
Формула 6
Схема 3
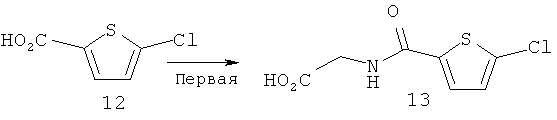
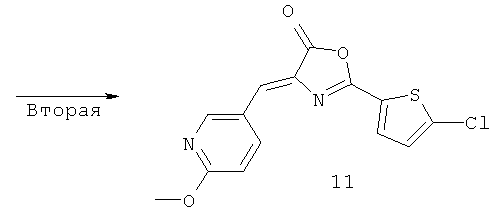
Первая стадия
1,00 г (6,15 ммоль) 5-хлортиофен-карбоновой кислоты, 1 мл (13,8 ммоль) тионилхлорида и 10 мл бензола добавляли в трехгорлую колбу и затем смесь перемешивали с нагреванием при температуре бани 105°C в течение 2 часов. После этого реакционный раствор охлаждали до комнатной температуры и затем дистилляцией удаляли тионилхлорид. Остаток подвергали азеотропной отгонке с бензолом и четыреххлористым углеродом, с тем чтобы получить коричневое маслянистое вещество.
В отдельно подготовленную трехгорлую колбу добавляли 323,2 мг (4,31 ммоль) глицина и 15 мл 0,5 н. гидроксида натрия и затем смесь перемешивали. К этому реакционному раствору добавляли полученное ранее коричневое маслянистое вещество и полученную смесь далее перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционный раствор охлаждали в ледяной бане. Затем к реакционному раствору добавляли 1 мл 6 н. соляной кислоты. Выпавшее в осадок твердое вещество собирали фильтрацией и после этого промывали 30 мл холодной воды. Твердое вещество сушили при пониженном давлении при комнатной температуре, с тем чтобы получить 386,2 мг Соединения 13 (1,76 ммоль; 53%) в виде светло-желтого твердого вещества. В результате проведения HPLC-анализа продукта было обнаружено, что время элюирования составило 10,06 минуты, а чистота составила 99,5% или более.
Вторая стадия
100 мг (0,46 ммоль) Соединения 13, 73,9 г (0,54 ммоль) 6-метокси-3-пиридинкарбальдегида, 102 мкл (1,08 ммоль) уксусного ангидрида, 48 мкл (0,59 ммоль) пиридина и 1 мл 1,4-диоксана добавляли в трехгорлую колбу и полученную смесь далее перемешивали с нагреванием при 100°C в масляной бане в течение 15 минут. После завершения реакции реакционный раствор охлаждали при 4°C и затем выпавшее в осадок твердое вещество растворяли в 5 мл этанола. Далее нерастворимое вещество собирали фильтрацией. Полученное твердое вещество промывали этанолом, с тем чтобы получить 10,1 мг Соединения 11 (0,031 ммоль; 6,0%) в виде желтого твердого вещества.
HPLC-чистота (длина волны поглощения: 254 нм) Соединения 11 составила 98,7%. Соединение 11 анализировали, используя прибор HPLC/ESI-LIT-TOF MS (NanoFrontier LD), в результате чего было подтверждено значение m/z=351,0; 352,0 [М+СН4O-Н]. Помимо этого ниже приведен пик, полученный в результате анализа Соединения 11 с использованием прибора AV-500 (500 МГц) для ядерного магнитного резонанса производства Bruker.
1H-ЯМР (CDCl3) δ млн-1 (500 МГц): 4.02 (s, 3H), 6.86 (d, 1H, JHH=8,8 Гц), 7.03 (s, 1H), 7.65 (d, 1H, JHH=4,1 Гц), 8.62 (dd, 1H, JHH=8,7 Гц, JHH=2,4 Гц), 8.67 (d, 1H, JHH=2,4 Гц).
13С-ЯМР (CDCl3) 5 млн-1 (125 МГц): 54.0, 111.8, 123.6, 126.7,126.9, 128.0, 128.1, 131.7, 132.2, 140.9, 152.2, 157.7, 165.4, 166.5.
С учетом HPLC-чистоты, которая составила 99,5% или более, результатов масс-спектрометрии и вида ЯМР-спектров было подтверждено, что синтезированное таким образом соединение представляет собой Соединение 11.
Пример 3
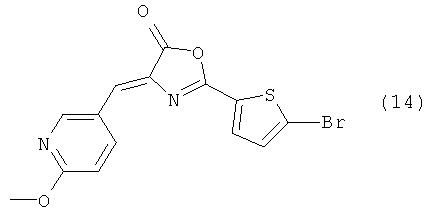
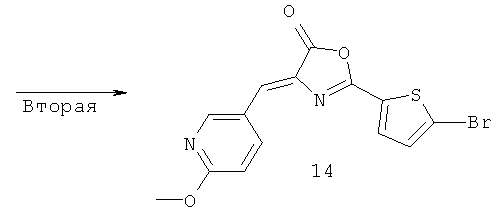
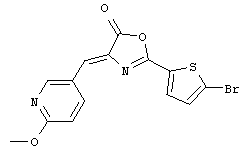
4-((6-Метокси-3-пиридинил)метилен)-2-(5-бром-2-тиенил)-5(4Н)-оксазолон (Формула 7)
Помимо соединения, представленного формулой (2), в которое введена группа фторо, также было получено соединение, представленное выше формулой (14), в которое введена группа бромо. Необходимо отметить, что способ синтеза соединения, представленного формулой (14), в которое введена группа бромо, не ограничен способом, показанным на приведенной ниже Схеме 4.
Формула 8
Схема 4
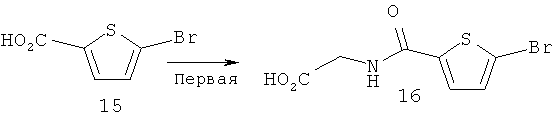
Первая стадия
1,00 г (4,83 ммоль) 5-бромтиофен-карбоновой кислоты, 5 мл (68,9 ммоль) тионилхлорида и 4 мл бензола добавляли в трехгорлую колбу и затем смесь перемешивали с нагреванием при температуре бани 105°C в течение 2 часов. После этого реакционный раствор охлаждали до комнатной температуры и затем дистилляцией удаляли тионилхлорид. Остаток подвергали азеотропной отгонке с бензолом и четыреххлористым углеродом, с тем чтобы получить коричневое маслянистое вещество.
В отдельно подготовленную трехгорлую колбу добавляли 412,9 мг (5,80 ммоль) глицина и 5 мл 6 н. гидроксида натрия и затем смесь перемешивали. К этому реакционному раствору добавляли полученное ранее коричневое маслянистое вещество и полученную смесь далее перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционный раствор охлаждали в ледяной бане. Затем к реакционному раствору добавляли 1 мл 6 н. соляной кислоты. Выпавшее в осадок твердое вещество собирали фильтрацией и после этого промывали 30 мл холодной воды. Содержащуюся в твердом веществе воду удаляли азеотропной перегонкой с ацетоном и остаток сушили при пониженном давлении при комнатной температуре, с тем чтобы получить 492,6 мг Соединения 16 (0,19 ммоль; 39,0%) в виде светло-желтого твердого вещества. В результате проведения HPLC-анализа продукта было обнаружено, что время элюирования составило 10,39 минуты, а чистота составила 99,7%.
Вторая стадия
242,7 мг (0,92 ммоль) Соединения 16, 138,1 мг (1,01 ммоль) 6-метокси-3-пиридинкарбальдегида, 1 мл (10,6 ммоль) уксусного ангидрида и 350 мкл (1,24 ммоль) пиридина добавляли в трехгорлую колбу и полученную смесь далее перемешивали с нагреванием при 100°C в масляной бане в течение 15 минут. После завершения реакции реакционный раствор охлаждали при 4°C и затем выпавшее в осадок твердое вещество растворяли в 5 мл этанола. Далее нерастворимое вещество собирали фильтрацией. Полученное твердое вещество промывали этанолом, с тем чтобы получить 32,2 мг Соединения 14 (0,088 ммоль; 10,0%)) в виде желтого твердого вещества.
HPLC-чистота (длина волны поглощения: 254 нм) Соединения 11 составила 95,8%о. Соединение 14 анализировали, используя прибор HPLC/ESI-LIT-TOF MS (NanoFrontier LD), в результате чего было подтверждено значение m/z=396,9; 397,9 [М+СН4O-Н] Помимо этого ниже приведен пик, полученный в результате анализа Соединения 14 с использованием прибора AV-500 (500 МГц) для ядерного магнитного резонанса производства Bruker.
1H-ЯМР (CDCl3) δ млн-1 (500 МГц): 4.02 (s, 3H), 6.86 (d, 1H, JHH=8,8 Гц), 7.17 (s, 1H), 7.17 (d, 1H, JHH=4,0 Гц), 7.60 (d, 1H, JHH=4,0 Гц), 8.63 (dd, 1H, JHH=8,8 Гц, JHH=2,4 Гц), 8.67 (d, 1H, JHH=2,4 Гц).
13C-ЯМР (CDCl3) 6 млн-1 (125 МГц): 54.0, 111.8, 121.5, 123.6, 128.2, 129.9, 131.7, 131.7, 132.8, 140.9, 152.2, 157.6, 165.4, 166.5.
С учетом HPLC-чистоты, которая составила 95,8%, результатов масс-спектрометрии и вида ЯМР-спектров было подтверждено, что синтезированное таким образом соединение представляет собой Соединение 14.
В отношении растворимости каждого из соединений в растворе DMSO проводили сравнение между соединением, представленным формулой (2), соединениями, представленными формулами (11) и (14), и соединением, у которого в тиофеновое кольцо соединения формулы (2)(4-((6-метокси-3-пиридинил)метилен)-2-(2-тиенил)-5(4Н)-оксазолона) не введен атом галогена или ему подобный атом. Соединение, представленное формулой (2), и соединения, представленные формулами (11) и (14), легко растворялись в растворе DMSO в конечных концентрациях 35 мМ, 10 мМ и 20 мМ, соответственно. С другой стороны, соединение, у которого в тиофеновое кольцо соединения формулы (2) не введен атом галогена или ему подобный атом, растворялось в растворе DMSO в конечной концентрации 10 мМ только при интенсивном перемешивании. Эти результаты могут показать, что соединение, представленное формулой (2), и
соединения, представленные формулами (11) и (14), обладают улучшенной растворимостью по сравнению с селективным ингибитором казеинкиназы 1δ или казеинкиназы 1ε предшествующего уровня техники.
Экспериментальные примеры
1. Ингибирующее действие производного оксазолона в отношении казеинкиназы 1δ и казеинкиназы 1ε
Активность соединения по настоящему изобретению в отношении ингибирования казеинкиназы 1δ и казеинкиназы 1ε измеряли, используя рекомбинантную казеинкиназу 1δ человека (INVITROGEN; № по кат. PV3665) или рекомбинантную казеинкиназу 1ε человека (INVITROGEN; № по кат. PV3500) в качестве источника фермента, а также используя пептид Ser/Thr 11 Z'-LYTE (INVITROGEN; № по кат. PV3671) в качестве субстрата фосфорилирования. Состав смеси (конечная концентрация), примененный в каждом из анализов для измерения ингибирующей активности, приведен ниже.
Анализ для казеинкиназы 1δ
Казеинкиназа 1δ: 3,0 мкг/мл или 1,0 мкг/мл; соединение по настоящему изобретению: 0,3 мкг/мл; 1,0 мкМ пептид; 20 мкМ АТФ; 50 мМ HEPES (N-2-гидроксиэтил-пиперазин-N-2-этансульфоновая кислота) (pH 7,4), 10 мМ MgCl2; 0,01% брий-35 и 0,5% DMSO.
Анализ для казеинкиназы 1ε
Казеинкиназа 1ε: 0,5 мкг/мл; соединение по настоящему изобретению: 0,3 мкг/мл; 1,0 мкМ пептид; 20 мкМ АТФ; 50 мМ HEPES (pH 7,4), 10 мМ MgCl2, 0,01% брий-35 и 0,5% DMSO.
Предварительно осуществляли взаимодействие соединения по настоящему изобретению с ферментом при комнатной температуре в течение 15 минут.Через два часа измеряли оставшуюся фосфорилирующую активность, используя пептид Ser/Thr 11 вместе с набором для анализа киназ Z'-LYTE (INVITROGEN; №по кат.PV3670).
Процент ингибирования соединением по настоящему изобретению рассчитывали относительно фосфорилирующей активности каждой казеинкиназы 1δ и казеинкиназы 1ε, к которой не было добавлено соединение по настоящему изобретению. В результате было установлено, что в вышеупомянутых условиях соединение формулы (2), полученное согласно описанной выше Схеме 1, имело низкую чистоту, но демонстрировало значительную ингибирующую активность в отношении казеинкиназы 1δ и казеинкиназы 1ε. Соединение формулы (2) высокой чистоты, полученное согласно Схеме 2, и соединения, представленные формулами (11) и (14), демонстрировали высокую ингибирующую активность в отношении казеинкиназы 1δ и казеинкиназы 1ε, как показано ниже в Таблице 1.
Специфичность ингибирования соединениями, ингибирующими казеинкиназу 1δ и казеинкиназу 1ε, в отношении различных типов киназ можно исследовать, используя набор Profiler Pro (изготовленный Caliper Life Sciences), согласно способу, описанному в прилагаемой инструкции по применению. Каждое из соединений, представленных формулами (2), (11) и (14), в конечной концентрации 10мкМ, 1 мкМ или 0,1 мкМ оставляли взаимодействовать с различными типами киназ в течение 15 минут. После завершения реакции анализировали ферментативную активность. В результате было установлено, что соединения формул (2), (11) и (14) не обладают значительной ингибирующей активностью в отношении различных типов киназ, действие которых вызывает озабоченность в связи с проявлением побочных эффектов, таких как подавление клеточного роста, обусловленное ингибированием фермента (МАРКАРК2 (протеинкиназа 2, активируемая митоген-активируемой протеинкиназой), AurA (киназа аврора А), РКСζ (протеинкиназа С, зета), RSK1 (рибосомальная S6-киназа 1), PRAK (р38-регулируемая/активируемая протеинкиназа), Erk1 (внеклеточная сигнал-регулируемая киназа 1), PKD2 (протеинкиназа D2), СНК1 (киназа 1 контрольной точки клеточного цикла), ABL (тирозинкиназа Абельсона), FYN, LYN, СНК2, MET, LCK (протеин-тирозинкиназа лимфоцитов), SRC, GSK3β (киназа-3|3 гликогенсинтазы), Erk2, РКА (протеинкиназа А), АКТ2 (протеинкиназа В, тип 2), INSR (инсулиновый рецептор), р38α, АКТ1, MSK1 (митоген- и стресс-активируемая протеинкиназа), РКСβ2 (протеинкиназа С, тип B2, ROCK2 (Rho-ассоциированная протеинкиназа 2), CDK2 (циклинзависимая киназа 2), MST2 (ste20-подобная киназа 2 млекопитающих), PKG1α (протеинкиназа G, тип 1α), РАК2 (р21 -активируемая киназа 2), IGF1R (рецептор инсулиноподобного ростового фактора 1 типа), FGFR1 (рецептор 1 фактора роста фибробластов), MARK1 (киназа 1, регулирующая ассоциированные с микротрубочками белки/сродство белков к микротрубочкам), САМК2δ (Са2+/кальмодулин-зависимая протеинкиназа, тип 2δ), PIM2 (сайт интеграции провируса вируса Молони 2), ВТК (тирозинкиназа Брутона), с-ТАК1 (Сdс25С-ассоциированная протеинкиназа 1), САМК4, АМРК (АМФ-активируемая протеинкиназа), FLT3 (Fms-подобная тирозинкиназа 3), HGK (киназа, подобная киназе клеток-предшественников гепатоцитов/подобная киназе зародышевого центра), VEGFR2 (рецептор 2 сосудистого эндотелиального фактора роста), KDR (рецептор, содержащий домен с киназной вставкой), c-RAF, p70S6K (киназа S6 рибосомального белка р70), IRAK4 (киназа, ассоциированная с рецептором интерлейкина), SGK1 (сыворотка- и глюкокортикоид-индуцибельная киназа) и SYK (тирозинкиназа селезенки)). Следовательно, можно сделать заключение, что соединения по настоящему изобретению являются высокоселективными ингибирующими соединениями в отношении казеинкиназы 1δ и казеинкиназы 1ε.
С другой стороны, авторы настоящего изобретения обнаружили, что когда каждое из соединений формул (2), (11) и (14) добавляли в концентрации 1 мкМ к регулируемой фосфорилированием тирозина киназе 1А с двойной специфичностью (DYRK1A), применяя способ для набора Profiler Pro (изготовленного Caliper Life Sciences), они ингибировали ферментативную активность DYRK1A с процентом ингибирования 65%, 78% и 85%, соответственно, хотя их ингибирующее действие в отношении DYRK1A было слабее проявляемого в отношении казеинкиназы 1δ и казеинкиназы 1ε. Так же, как и казеинкиназа 16, DYRK1A известна в качестве киназы-кандидата в отношении фосфорилирования тау-белков, а также в качестве киназы, контролирующей циркадный ритм. Поскольку соединения по настоящему изобретению ингибируют ферментативную активность каждой из казеинкиназы 1δ, казеинкиназы 1ε и DYRK1A, они могут представлять собой эффективные терапевтические средства для лечения расстройства циркадного ритма и нейродегенеративного заболевания.
2. Касательно эффективности соединения по настоящему изобретению Эффективность ингибирующих казеинкиназу 1δ и казеинкиназу 1ε соединений в отношении лечения расстройства циркадного ритма можно доказать, используя следующие животные модели. Конкретно, акклиматизацию крыс осуществляли в течение одной или более недель в условиях цикла свет/темнота (LD), состоящего из светлого периода в течение 12 часов и темного периода в течение 12 часов. Непосредственно перед началом темного периода соединение, представленное формулой (11), смешивали с солюбилизирующим агентом и смесь вводили внутрибрюшинно каждой крысе в дозе 60 мг/кг.Сразу после введения наблюдали статистически значимое уменьшение количества движения и увеличение продолжительности не быстрого сна у крыс, которым вводили соединение формулы (11), по сравнению с крысами, не обработанными данным соединением. С другой стороны, тот же самый тест, что и описанный выше, проводили для соединения, представленного формулой (2), ((4-((6-метокси-3-пиридинил)метилен)-2-(2-тиенил)-5(4Н)-оксазолона)), в тиофеновое кольцо которого не была введена группа галогено или подобная ей. В результате наблюдали статистически значимое уменьшение количества движения и увеличение продолжительности не быстрого сна у крыс, которым было введено это соединение. Однако, данный медицинский эффект соединения формулы (11) значительно превышал таковой у вышеупомянутого соединения ((4-((6-метокси-3-пиридинил)метилен)-2-(2-тиенил)-5(4Н)-оксазолона)), в тиофеновое кольцо которого не была введена группа галогено или подобная ей. Эти результаты демонстрируют, что соединение формулы (11) является эффективным для лечения расстройства циркадного ритма.
Активность соединения по настоящему изобретению на предмет ингибирования казеинкиназы 1δ, с тау-белком в качестве субстрата, исследовали, используя рекомбинантную казеинкиназу 16 человека (Carna Biosciences; № по кат. 03-103) в качестве источника фермента, а также используя рекомбинантный тау-белок человека (Sigma-Aldrich; № по кат. Т0576) в качестве субстрата фосфорилирования. Состав смеси (конечная концентрация), примененный в каждом из анализов для измерения ингибирующей активности, приведен ниже.
Казеинкиназа 1δ: 25 мкг/мл; тау-белок: 10 мкг/мл, соединение по настоящему изобретению (25 мкМ - 1 мкМ); 20 мкМ АТФ; 50 мМ HEPES (рН 7,4), 10 мМ MgCl2, 0,01% брий-35, 200 мкМ Na3VO4 и 0,25% DMSO.
Предварительно было осуществлено взаимодействие соединения по настоящему изобретению с ферментом при комнатной температуре в течение 15 минут, а затем к реакционной смеси добавляют тау-белок. После этого осуществляют реакцию фосфорилирования в течение 2 часов. По завершении реакции фосфорилирования реакционный раствор подвергают вестерн-блоттингу, чтобы зарегистрировать фосфорилирование рекомбинантного тау-белка человека, перенесенного на мембрану из PVDF (поливинилиденфторида) с использованием Phos-tag (NARD Institute, Ltd.; № по кат. BTL-104) в соответствии с хемилюминесцентным способом (ECL Plus; GE Healthcare; № по кат. RPN 2132). С PVDF-мембраны после детекции реакции фосфорилирования извлекают зонд в соответствии с протоколами для Phos-tag, и после этого по реакции с антителом можно определить общее содержание тау-белка.
Осуществляют взаимодействие мембраны с антителом против тау-белка (DAKO Cytomation; № по кат. А0024), используемым в качестве первичного антитела, и далее осуществляют взаимодействие с вторичным антителом козы против кроличьего антитела, меченым ИК-красителем 680LT (LI-COR; № по кат. 926-68021). Затем детектируют инфракрасную (ИК) флуоресценцию и ее используют в качестве сигнала, соответствующего общему количеству тау-белка.
Для детекции реакции фосфорилирования по хемилюминесценции и для детекции общего тау-белка по инфракрасной флуоресценции используют приборы Typhoon (Amersham Biosciences; модель 9400) и Odyssey (LI-COR; модель 9120), соответственно. Получают отношение интенсивности сигнала фосфорилированного тау-белка к интенсивности сигнала общего тау-белка и полученное отношение обозначают как степень фосфорилирования тау-белка.
Степень фосфорилирования тау-белка в реакционном растворе, содержащем соединение по настоящему изобретению, сравнивали с таковой в реакционном растворе, который не содержал соединение по настоящему изобретению. В результате можно было наблюдать значительное ингибирование в отношении фосфорилирования субстрата в реакционном растворе, содержащем или соединение формулы (2), или соединение формулы (11). Эти результаты демонстрируют, что соединения формул (2) и (11) способны ингибировать аномально избыточное фосфорилирование тау-белка при нейродегенеративных заболеваниях центральной нервной системы (например, болезни Альцгеймера).
Эффективность соединений, ингибирующих казеинкиназу 1δ и казеинкиназу 1ε, в случае нейродегенеративных заболеваний центральной нервной системы можно доказать, используя следующие животные модели. А именно, используют трансгенных мышей, избыточно экспрессирующих мутантный тау-белок, что вызывает у них нейрофибриллярную дегенерацию в головном мозге, и этим мышам вводят ингибирующее соединение, которое примешивают к корму или питьевой воде продолжительный период времени. Эффективность ингибирующего соединения можно доказать тем, что уровень патологии, вызывающей потерю синапсов и неврологический дефицит в группе, получающей соединение, снижен по сравнению с группой, не получающей данное соединение.
Эффективность соединений, ингибирующих казеинкиназу 1δ и казеинкиназу 1ε, в случае рака поджелудочной железы можно доказать, используя следующие животные модели. А именно, линию клеток рака поджелудочной железы человека (приблизительно 5000000 клеток) культивируют in vitro, и эту культуру затем вводят подкожной инъекцией в область дорсальной поверхности мышей с SCID (тяжелым комбинированным иммунодефицитом). Через четырнадцать суток после инъекции ингибирующее соединение смешивают с солюбилизирующим агентом (карбоксиметилцеллюлозой и т.д.) и мышам вводят полученную таким образом смесь посредством перорального или внутрибрюшинного введения. Наблюдение за размером опухоли в подкожном слое проводят в динамике. Кроме того, на 10е сутки после введения опухоль оперативно удаляют из подкожного слоя и измеряют ее массу, чтобы можно было удостовериться в эффективности ингибитора.
Промышленная применимость
Ингибитор казеинкиназы 1δ и казеинкиназы 1ε по настоящему изобретению и фармацевтическое средство по настоящему изобретению, содержащее вышеописанный ингибитор в качестве активного ингредиента, весьма способствуют разработке фармацевтического средства, полезного для лечения и/или предупреждения заболеваний, с патологическими состояниями которых ассоциируют активацию казеинкиназы 1δ или казеинкиназы 1ε. В частности, ингибитор казеинкиназы 1δ и казеинкиназы 1ε по настоящему изобретению и фармацевтическое средство по настоящему изобретению весьма способствуют разработке фармацевтического средства, полезного для лечения и/или предупреждения расстройства циркадного ритма (включая расстройство сна), нейродегенеративного заболевания центральной нервной системы и рака.
Изобретение относится к соединению общей формулы (1):
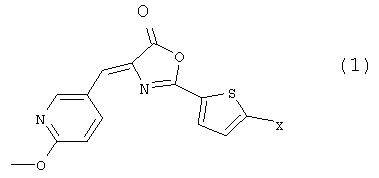
где X представляет собой атом галогена (который может представлять собой атом, выбранный из атома фтора, атома хлора, атома брома и атома йода). Новое производное оксазолона обладает ингибирующей активностью в отношении казеинкиназы 1δ и казеинкиназы 1ε. 4 н. и 7 з.п. ф-лы, 1 табл., 3 пр.
1. Производное оксазолона, представленное следующей общей формулой (1):

где X представляет собой атом галогена (который может представлять собой атом, выбранный из атома фтора, атома хлора, атома брома и атома йода).
2. Производное оксазолона по п.1, где соединение общей формулы (1) представляет собой любое из:
4-((6-метокси-3-пиридинил)метилен)-2-(5-фтор-2-тиенил)-5(4H)-оксазолона;
4-((6-метокси-3-пиридинил)метилен)-2-(5-хлор-2-тиенил)-5(4H)-оксазолона;
4-((6-метокси-3-пиридинил)метилен)-2-(5-бром-2-тиенил)-5(4H)-оксазолона и
4-((6-метокси-3-пиридинил)метилен)-2-(5-йод-2-тиенил)-5(4H)-оксазолона.
3. Ингибитор казеинкиназы 1δ и казеинкиназы 1ε, содержащий в качестве активного ингредиента производное оксазолона по п.1 или 2, его соль, их сольват или их гидрат.
4. Фармацевтическая композиция для лечения заболеваний, с патологическими состояниями которых ассоциируют активацию казеинкиназы 1δ или казеинкиназы 1ε, содержащая в качестве активного ингредиента производное оксазолона по п.1 или 2, его соль, их сольват или их гидрат.
5. Фармацевтическая композиция по п.4, где заболеванием является расстройство циркадного ритма, включая расстройство сна.
6. Фармацевтическая композиция по п.4, где заболеванием является нейродегенеративное заболевание.
7. Фармацевтическая композиция по п.4, где заболеванием является рак.
8. Способ лечения заболеваний, с патологическими состояниями которых ассоциируют активацию казеинкиназы 1δ или казеинкиназы 1ε, включающий введение пациенту фармацевтической композиции, содержащей в качестве активного ингредиента производное оксазолона по п.1 или 2, его соль, их сольват или их гидрат.
9. Способ по п.8, где заболеванием является расстройство циркадного ритма, включая расстройство сна.
10. Способ по п.8, где заболеванием является нейродегенеративное заболевание.
11. Способ по п.8, где заболеванием является рак.
| WO 2009037394, A2,26.03.2009 | |||
| ПРОИЗВОДНЫЕ 1,2-ДИ(ЦИКЛО)ЗАМЕЩЕННОГО БЕНЗОЛА | 2004 |
|
RU2340602C2 |

