Уровень техники изобретения
Область изобретения
[0001]
Настоящее изобретение относится к соединениям, ингибиторам казеинкиназы 1δ и/или активин-рецептор-подобной киназы 5, терапевтическим лекарственным средствам для лечения расстройств циркадного ритма сна, терапевтическим лекарственным средствам для лечения деменции альцгеймеровского типа, терапевтическим лекарственным средствам для лечения дистрофии роговицы, терапевтическим лекарственным средствам для лечения рака и терапевтическим лекарственным средствам для лечения андрогенетической алопеции.
Описание предшествующего уровня техники
[0002]
Ритм сна-бодрствования регулируется биологическими часами и соответствует примерно однодневному ритму. Такой ритм называется циркадным ритмом. В то время как цикл биологических часов человека составляет около 25 часов, цикл одного дня на Земле составляет 24 часа, что приводит к смещению примерно на один час. Обычно это одночасовое смещение изменяется в ответ на различные раздражители (такие как свет, физические упражнения и прием пищи). Однако если состояние, в котором это смещение не может быть изменено и продолжается в течение длительного времени, это нарушает сон и бодрствование в соответствующее время. Попытки принудительно изменить это смещение могут вызвать физические расстройства, такие как утомляемость, анорексия и в некоторых случаях головная боль. Такие нарушения сна, связанные с циркадными ритмами, называются расстройства циркадного ритма сна.
[0003]
Известно, что семейный синдром фазового опережения сна, одно из расстройств циркадного ритма сна, является заболеванием, вызванным точечной мутацией гена казеинкиназы 1δ (CK1δ) человека (Nature 2005, 434, 640-644), и предполагается, что циркадный ритм человека колеблется за счет модуляции CK1δ. Также сообщается, что CK1δ участвует в контроле циркадных ритмов грызунов, таких как мыши, и негрызунов, таких как обезьяны (Proc. Natl. Acad. Sci. USA, 2010, 107, 15240-15245).
[0004]
Существует также несколько сообщений в неклинических исследованиях о том, что ингибиторы CK1δ изменяют циркадный ритм in vitro и in vivo, что возлагает надежды на ингибиторы CK1δ как на лекарственные средства для лечения расстройств циркадного ритма сна.
[0005]
Также сообщается о связи между CK1δ и деменцией альцгеймеровского типа (Японский перевод публикации международной заявки PCT No. 2014-503527). В частности, считается, что деменции альцгеймеровского типа вызывается внутриклеточной агрегацией гиперфосфорилированного тау-белка (нейрофибриллярный клубок), и предполагается, что фосфорилирование тау-белка вызывается CK1δ. По этой причине ожидается, что деменцию альцгеймеровского типа лечится путем ингибирования CK1δ.
[0006]
Также сообщается о связи между дистрофией роговицы (международная публикация No. WO 2015/064768) или раком (Anticancer Res. 2007, 27, 4149-4158) и активин-рецептор-подобной киназой 5 (ALK5). ALK5 правильно называют активин-рецептор-подобной киназой, хотя ее также называют активиноподобной киназой в более коротком названии. Сообщается, что дистрофия роговицы вызывается индукцией гибели клеток эндотелиальных клеток роговицы посредством стрессовой реакции эндоплазматического ретикулума денатурированным белком, чрезмерно накопленным в тканях роговицы, а стресс эндоплазматического ретикулума вызывается активацией сигнала ALK5 (рецептор TGF-β типа I). Хотя известно, что TGF-β обладает эффектом подавления клеточной пролиферации, сообщается, что TGF-β способствует пролиферации и метастазированию раковых клеток на более поздних стадиях рака. По этой причине предполагается, что дистрофия роговицы и рак будут лечиться путем ингибирования ALK5. Например, соединения согласно японскому переводу публикации международной заявки PCT No. 2004-517068 и международной публикации No. WO 2001/062756 описаны как ингибиторы ALK5.
Литература предшествующего уровня техники
[0007]
Патентная литература 1: Японский перевод публикации международной заявки PCT No. 2014-503527
Патентная литература 2: международная публикация No. WO 2015/064768
Патентная литература 3: Японский перевод публикации международной заявки PCT No. 2004-517068
Патентная литература 4: международная публикация No. WO 2001/062756
[0008]
Непатентная литература 1: Nature 2005, 434, 640-644
Непатентная литература 2: Proc. Natl. Acad. Sci. USA, 2010, 107, 15240-15245
Непатентная литература 3: Anticancer Res. 2007, 27, 4149-4158
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Техническая задача
[0009]
Целью настоящего изобретения является создание соединения, обладающего ингибирующей активностью в отношении CK1δ и/или ингибирующей активностью в отношении ALK5, и ингибитора CK1δ и/или ALK5, терапевтического лекарственного средства для лечения расстройств циркадного ритма сна, терапевтического лекарственного средства для лечения деменции альцгеймеровского типа, терапевтического лекарственного средства для лечения дистрофии роговицы, терапевтического лекарственного средства для лечения рака, терапевтического лекарственного средства для лечения андрогенетической алопеции, которые содержат соединение.
Решение задачи
[0010]
Авторы настоящего изобретения, которые провели обширные исследования, обнаружили, что соединение, имеющее заданную структуру, обладает ингибирующей активностью в отношении CK1δ и/или ингибирующей активностью в отношении ALK5, и выполнили настоящее изобретение.
[0011]
Настоящее изобретение включает варианты осуществления, указанные ниже.
[1]
Соединение, представленное следующей формулой (1):
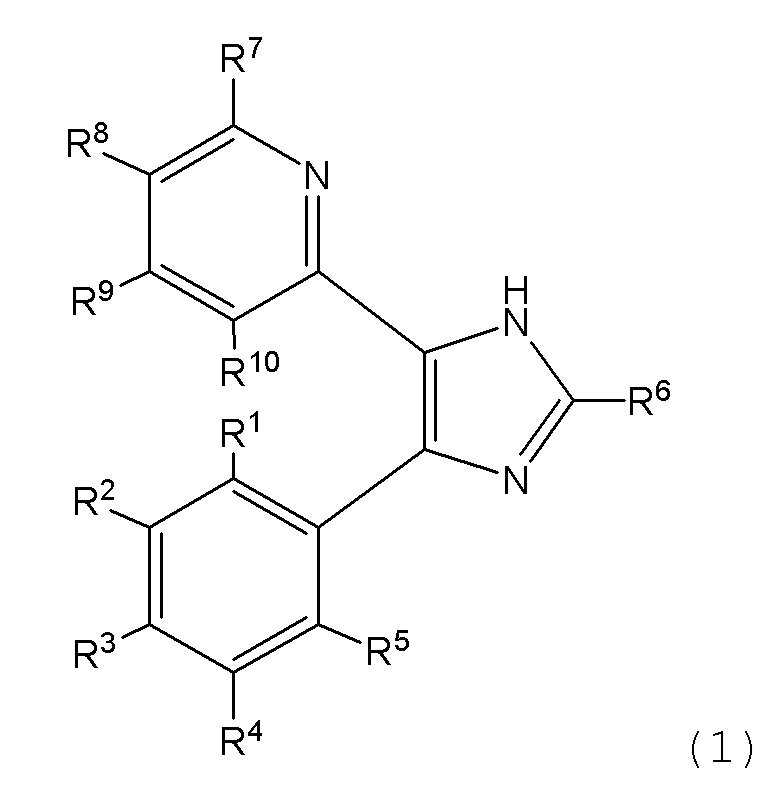
где
R1 - R10 каждый независимо представляет собой водород, алкил, циклоалкил или галоген, где
R2 и R3 или R4 и R5 вместе с двумя атомами углерода, к которым они присоединены, образуют 5-членное кольцо, содержащее один гетероатом, выбранный из группы, состоящей из атома кислорода, атома азота и атома серы, необязательно замещенное алкилом;
или его фармацевтически приемлемая соль, где соединение исключает 2-[4-(2,3-дигидро-5-бензофуранил)-2-(1,1-диметилэтил)-1H-имидазол-5-ил]-6-метилпиридин.
[2]
Соединение по [1] или его фармацевтически приемлемая соль, где
R1 - R10 каждый независимо представляет собой водород, алкил или галоген, где
R2 и R3 или R4 и R5 вместе с двумя атомами углерода, к которым они присоединены, образуют 5-членное кольцо, содержащее один гетероатом, выбранный из группы, состоящей из атома кислорода, атома азота и атома серы, необязательно замещенное алкилом.
[2-1]
Соединение по [1] или [2] или его фармацевтически приемлемая соль, где R1 - R5 представляют собой водород, за исключением R2 и R3 или R4 и R5, которые образуют 5-членное кольцо.
[2-2]
Соединение по любому из [1] - [2-1] или его фармацевтически приемлемая соль, где R6 представляет собой водород, алкил или циклоалкил.
[2-3]
Соединение по любому из [1] - [2-2] или его фармацевтически приемлемая соль, где R6 представляет собой водород или алкил.
[2-4]
Соединение по любому из [1] - [2-3] или его фармацевтически приемлемая соль, где R6 представляет собой водород.
[2-5]
Соединение по любому из [1] - [2-4] или его фармацевтически приемлемая соль, где R7 представляет собой водород или алкил.
[2-6]
Соединение по любому из [1] - [2-5] или его фармацевтически приемлемая соль, где R7 представляет собой водород.
[2-7]
Соединение по любому из [1] - [2-6] или его фармацевтически приемлемая соль, где R8 представляет собой водород или галоген.
[2-8]
Соединение по любому из [1]-[2-7] или его фармацевтически приемлемая соль, где R8 представляет собой водород.
[2-9]
Соединение по любому из [1]-[2-8] или его фармацевтически приемлемая соль, где R9 представляет собой водород.
[2-10]
Соединение по любому из [1]-[2-9] или его фармацевтически приемлемая соль, где R10 представляет собой водород.
[3]
Соединение по любому из [1]-[2-10] или его фармацевтически приемлемая соль, где R2 и R3 или R4 и R5 вместе с двумя атомами углерода, к которым они присоединены, образуют тетрагидрофурановое кольцо, необязательно замещенное алкилом.
[3-1]
Соединение по [3] или его фармацевтически приемлемая соль, где R4 и R5 вместе с двумя атомами углерода, к которым они присоединены, образуют тетрагидрофурановое кольцо, необязательно замещенное алкилом.
[4]
Соединение по [3] или его фармацевтически приемлемая соль, где R2 и R3 вместе с двумя атомами углерода, к которым они присоединены, образуют тетрагидрофурановое кольцо, необязательно замещенное алкилом.
[5]
Соединение по [4] или его фармацевтически приемлемая соль, где R2 и R3 вместе с двумя атомами углерода, к которым они присоединены, образуют незамещенное тетрагидрофурановое кольцо.
[5-1]
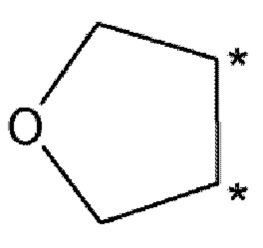
Соединение по любому из [3] - [5] или его фармацевтически приемлемая соль, где тетрагидрофурановое кольцо, образованное R2 и R3 или R4 и R5, имеет следующую структуру:
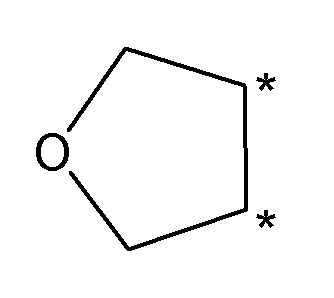
где атомы углерода со звездочкой (*) представляют собой атомы углерода бензольного кольца, связанные с R2 и R3 или R4 и R5.
[6]
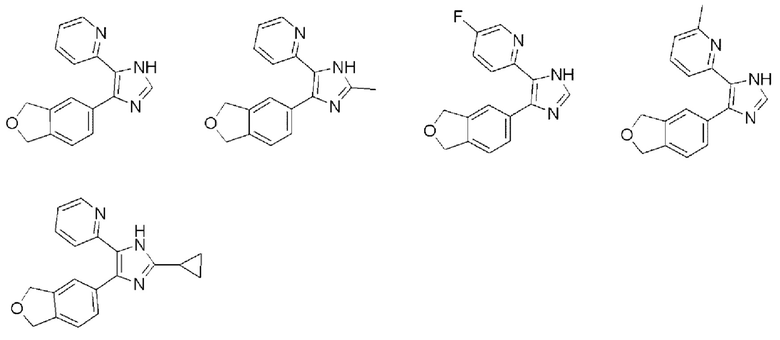
Соединение по [1], выбранное из группы, состоящей из следующих соединений:
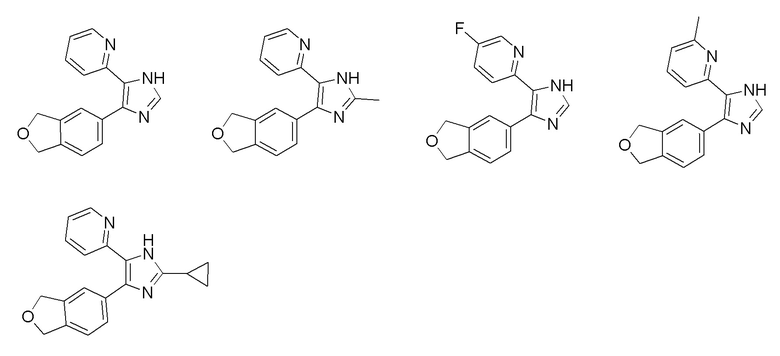
или его фармацевтически приемлемая соль.
[7]
Ингибитор казеинкиназы 1δ, включающий соединение по любому из [1] - [6] или его фармацевтически приемлемую соль.
[8]
Терапевтическое лекарственное средство для лечения расстройств циркадного ритма сна, включающее соединение по любому из [1]-[6] или его фармацевтически приемлемую соль.
[9]
Терапевтическое лекарственное средство по [8], где расстройства циркадного ритма представляют собой нерегулярное нарушение ритма сон-бодрствование или вечернее обострение, сопровождающееся деменцией альцгеймеровского типа.
[10]
Терапевтическое лекарственное средство для лечения деменции альцгеймеровского типа, включающее соединение по любому из [1] - [6] или его фармацевтически приемлемую соль.
[11]
Ингибитор активин-рецептор-подобной киназы 5, включающий соединение по любому из [1] - [6] или его фармацевтически приемлемую соль.
[12]
Терапевтическое лекарственное средство для лечения рака, включающее соединение по любому из [1] - [6] или его фармацевтически приемлемую соль.
[13]
Терапевтическое лекарственное средство по [12], где рак представляет собой опухоль головного мозга, рак печени, рак мочевого пузыря, миелодиспластические синдромы, рак толстой кишки или рак поджелудочной железы.
[14]
Терапевтическое лекарственное средство по [12] или [13] для применения в комбинации с лекарственным средством для лечения рака, отличным от терапевтического лекарственного средства для лечения рака по [12] или [13], и/или радиотерапией.
[15]
Терапевтическое лекарственное средство для лечения дистрофии роговицы, включающее соединение по любому из [1]-[6] или его фармацевтически приемлемую соль.
[16]
Терапевтическое лекарственное средство для лечения андрогенетической алопеции, включающее соединение по любому из [1]-[6] или его фармацевтически приемлемую соль.
[17]
Ингибитор казеинкиназы 1δ и активин-рецептор-подобной киназы 5, включающий соединение по любому из [1] - [6] или его фармацевтически приемлемую соль.
[0012]
Настоящее изобретение также включает следующие варианты осуществления.
[A1]
Способ ингибирования казеинкиназы 1δ и/или активин-рецептор-подобной киназы 5, включающий введение эффективного количества соединения по любому из [1] - [6] или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
[A2]
Способ лечения расстройств циркадного ритма сна, включающий введение эффективного количества соединения по любому из [1] - [6] или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
[A3]
Способ лечения деменции альцгеймеровского типа, включающий введение эффективного количества соединения по любому из [1] - [6] или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
[A4]
Способ лечения дистрофии роговицы, включающий введение эффективного количества соединения по любому из [1] - [6] или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
[A5]
Способ лечения рака, включающий введение эффективного количества соединения по любому из [1] - [6] или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
[A6]
Способ лечения андрогенетической алопеции, включающий введение эффективного количества соединения по любому из [1] - [6] или его фармацевтически приемлемой соли пациенту, нуждающемуся в этом.
[B1]
Соединение по любому из [1] - [6] или его фармацевтически приемлемая соль для применения для ингибирования казеинкиназы 1δ и/или активин-рецептор-подобной киназы 5.
[B2]
Соединение по любому из [1] - [6] или его фармацевтически приемлемая соль для применения в лечении расстройств циркадного ритма сна.
[B3]
Соединение по любому из [1] - [6] или его фармацевтически приемлемая соль для применения в лечении деменции альцгеймеровского типа.
[B4]
Соединение по любому из [1] - [6] или его фармацевтически приемлемая соль для применения в лечении дистрофии роговицы.
[B5]
Соединение по любому из [1] - [6] или его фармацевтически приемлемая соль для применения в лечении рака.
[B6]
Соединение по любому из [1] - [6] или его фармацевтически приемлемая соль для применения в лечении андрогенетической алопеции.
[C1]
Применение соединения по любому из [1] - [6] или его фармацевтически приемлемой соли для ингибирования казеинкиназы 1δ и/или активин-рецептор-подобной киназы 5.
[C2]
Применение соединения по любому из [1] - [6] или его фармацевтически приемлемой соли для лечения расстройств циркадного ритма сна.
[C3]
Применение соединения по любому из [1] - [6] или его фармацевтически приемлемой соли для лечения деменции альцгеймеровского типа.
[C4]
Применение соединения по любому из [1] - [6] или его фармацевтически приемлемой соли для лечения дистрофии роговицы.
[C5]
Применение соединения по любому из [1] - [6] или его фармацевтически приемлемой соли для лечения рака.
[C6]
Применение соединения по любому из [1] - [6] или его фармацевтически приемлемой соли для лечения андрогенетической алопеции.
[D1]
Применение соединения по любому из [1] - [6] или его фармацевтически приемлемой соли для получения ингибитора казеинкиназы 1δ и/или ингибитора активин-рецептор-подобной киназы 5.
[D2]
Применение соединения по любому из [1]-[6] или его фармацевтически приемлемой соли для получения терапевтического лекарственного средства для лечения расстройств циркадного ритма сна.
[D3]
Применение соединения по любому из [1]-[6] или его фармацевтически приемлемой соли для получения терапевтического лекарственного средства для лечения деменции альцгеймеровского типа.
[D4]
Применение соединения по любому из [1]-[6] или его фармацевтически приемлемой соли для получения терапевтического лекарственного средства для лечения дистрофии роговицы.
[D5]
Применение соединения по любому из [1]-[6] или его фармацевтически приемлемой соли для получения терапевтического лекарственного средства для лечения.
[D6]
Применение соединения по любому из [1]-[6] или его фармацевтически приемлемой соли для получения терапевтического лекарственного средства для лечения андрогенетической алопеции.
Полезные эффекты изобретения
[0013]
Настоящее изобретение может обеспечить соединение, обладающее ингибирующей активностью в отношении CK1δ и/или ингибирующей активностью в отношении ALK5, и ингибиторы CK1δ и/или ALK5, терапевтические лекарственные средства для лечения расстройств циркадного ритма сна, терапевтические лекарственные средства для лечения деменции альцгеймеровского типа, терапевтические лекарственные средства для лечения дистрофии роговицы, терапевтические лекарственные средства для лечения рака и терапевтические лекарственные средства для лечения андрогенетической алопеции, которые содержат соединение.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
[0014]
Далее будут конкретно описаны варианты осуществления настоящего изобретения, но эти варианты осуществления не должны рассматриваться как ограничения настоящего изобретения, которые могут быть подвергнуты различным модификациям без отхода от сущности изобретения.
[0015]
<Соединение>
Один вариант осуществления настоящего изобретения относится к соединению, представленному следующей формулой (1):
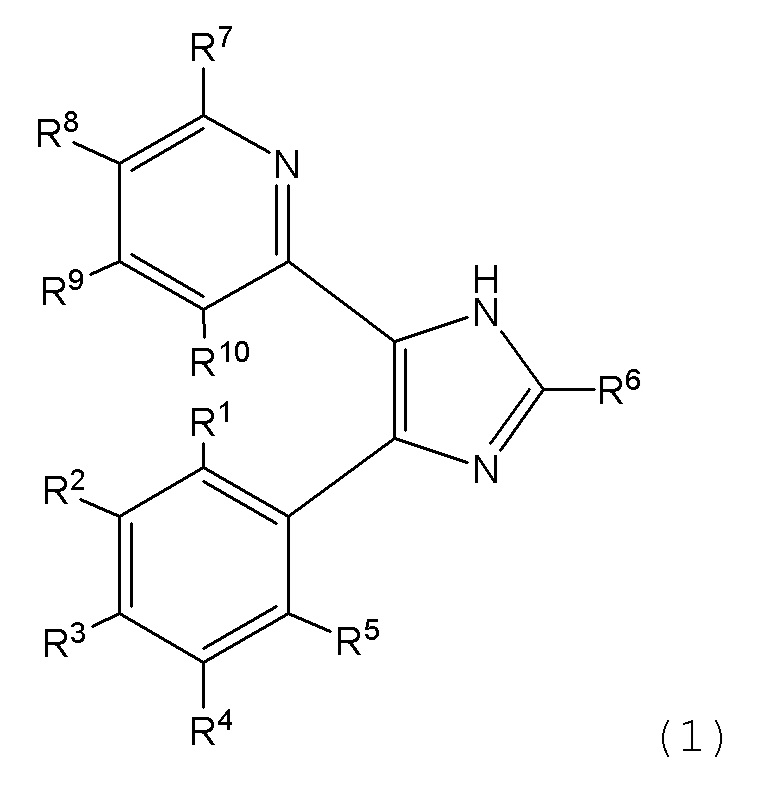
где
R1 - R10 каждый независимо представляет собой водород, алкил, циклоалкил или галоген, где
R2 и R3 или R4 и R5 вместе с двумя атомами углерода, к которым они присоединены, образуют 5-членное кольцо, содержащее один гетероатом, выбранный из группы, состоящей из атома кислорода, атома азота и атома серы, необязательно замещенное алкилом;
или его фармацевтически приемлемой соли, где соединение исключает 2-[4-(2,3-дигидро-5-бензофуранил)-2-(1,1-диметилэтил)-1H-имидазол-5-ил]-6-метилпиридин). Следует отметить, что формула (1) также охватывает таутомер, имеющий имидазольный фрагмент в формуле (1), где протон и двойная связь перемещены.
[0016]
В формуле (1), предпочтительно, R2 и R3 или R4 и R5 с двумя атомами углерода, к которым они присоединены, образуют 5-членное кольцо, содержащее один атом кислорода (тетрагидрофурановое кольцо), где 5-членное кольцо может иметь заместитель, выбранный из алкильных групп.
[0017]
В формуле (1), более предпочтительно, R2 и R3 с двумя атомами углерода, к которым они присоединены, образуют тетрагидрофурановое кольцо, необязательно замещенное алкилом.
[0018]
В формуле (1) R4 и R5 с двумя атомами углерода, к которым они присоединены, могут образовывать тетрагидрофурановое кольцо, необязательно замещенное алкилом.
[0019]
В формуле (1), еще более предпочтительно, R2 и R3 с двумя атомами углерода, к которым они присоединены, образуют незамещенное тетрагидрофурановое кольцо.
[0020]
В формуле (1), предпочтительно, тетрагидрофурановое кольцо, образованное R2 и R3 или R4 и R5, имеет следующую структуру:
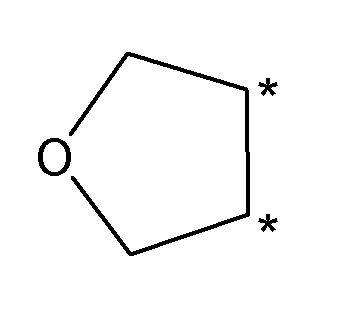
где атомы углерода со звездочкой (*) представляют собой атомы углерода бензольного кольца, связанные с R2 и R3 или R4 и R5.
[0021]
В формуле (1), R1 - R5 предпочтительно представляют собой водород, за исключением R2 и R3 или R4 и R5, которые образуют 5-членное кольцо.
[0022]
В формуле (1), R6 предпочтительно представляет собой водород, алкил или циклоалкил, более предпочтительно водород или алкил, еще более предпочтительно водород.
[0023]
В формуле (1), R7 предпочтительно представляет собой водород или алкил, более предпочтительно водород.
[0024]
В формуле (1), R8 предпочтительно представляет собой водород или галоген, более предпочтительно водород.
[0025]
В формуле (1), R9 и R10 предпочтительно представляют собой водород.
[0026]
В данном описании алкил предпочтительно представляет собой алкил, содержащий от 1 до 6 атомов углерода, более предпочтительно алкил, содержащий от 1 до 3 атомов углерода, еще более предпочтительно метил. Алкил также включает линейные и разветвленные алкильные группы.
[0027]
В данном описании, циклоалкил предпочтительно представляет собой циклоалкил, содержащий от 3 до 6 атомов углерода, более предпочтительно циклоалкил, содержащий от 3 до 5 атомов углерода.
[0028]
В данном описании, галоген предпочтительно представляет собой фтор, хлор, бром или йод, более предпочтительно фтор.
[0029]
Хотя конкретно не ограничено, соединение, представленное формулой (1), предпочтительно представляет собой соединения, перечисленные ниже:
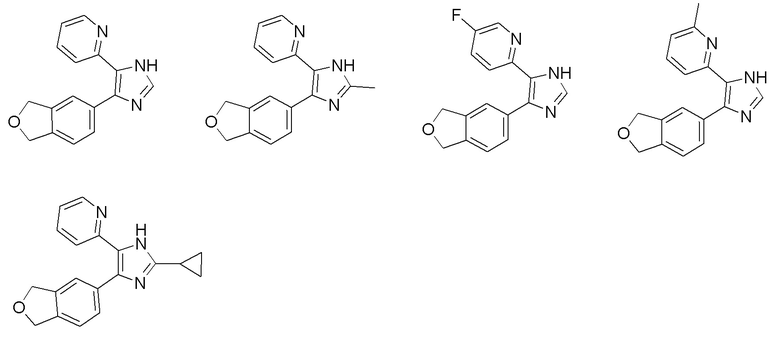
[0030]
Фармацевтически приемлемая соль соединения, представленного формулой (1), может быть любым соединением, которое можно использовать в качестве лекарственного средства. Их примеры включают соли неорганических кислот, такие как хлористоводородные соли, сульфатные соли, нитратные соли, фосфатные соли и бромистоводородные соли; и соли органических кислот, такие как фумаратные соли, малеатные соли, малатные соли, тартратные соли, сукцинатные соли, цитратные соли, метансульфонатные соли, п-толуолсульфонатные соли, ацетатные соли, лактатные соли и пальмитатные соли.
[0031]
Соединение, представленное формулой (1) или его фармацевтически приемлемая соль может образовывать сольват, такой как гидрат. В данном описании, сольват включен в соединение, представленное формулой (1), или его фармацевтически приемлемую соль.
[0032]
<Ингибитор киназы>
Один вариант осуществления настоящего изобретения относится к ингибитору CK1δ и/или ALK5, включающему соединение или его фармацевтически приемлемую соль.
[0033]
(Ингибитор казеинкиназы 1δ)
Один вариант осуществления настоящего изобретения относится к ингибитору CK1δ. Существующий ингибитор CK1δ PF-670462 может вызывать побочные эффекты, поскольку концентрация для ингибирования CK1δ близка к концентрации для ингибирования p38α. Напротив, концентрация ингибирования CK1α ингибитора CK1α в соответствии с настоящим вариантом осуществления достаточно далека от концентрации ингибирования p38α, что делает возможным селективное ингибирование CK1α.
[0034]
В частности, концентрация ингибирования p38α (IC50)/концентрация ингибирования CK1δ (IC50) предпочтительно составляет 10 или более, более предпочтительно 20 или более, еще более предпочтительно 40 или более, еще более предпочтительно 80 или более, особенно предпочтительно 150 или более. Хотя конкретно не ограничено, верхний предел концентрации ингибирования p38α (IC50)/концентрации ингибирования CK1δ (IC50) может составлять, например, 10000, 1000 или 500. Концентрацию ингибирования p38α и концентрацию ингибирования CK1δ можно измерить способом согласно примеру тестирования 1, описанному ниже.
[0035]
Концентрация ингибирования CK1δ (IC50) ингибитора CK1δ согласно настоящему варианту осуществления предпочтительно составляет 200 нМ или менее, более предпочтительно 160 нM или менее, еще более предпочтительно 120 нM или менее, еще более предпочтительно 80 нM или менее, особенно предпочтительно 60 нM или менее. Хотя конкретно не ограничено, нижний предел концентрации ингибирования CK1δ (IC50) может составлять, например, 0,1 нМ, 1 нМ или 10 нМ.
[0036]
С помощью ингибитора CK1δ в соответствии с настоящим вариантом осуществления можно лечить заболевания, связанные с CK1δ.
[0037]
(Ингибитор активин-рецептор-подобной киназы 5)
Один вариант осуществления настоящего изобретения относится к ингибитору ALK5. Концентрация ингибирования ALK5 (IC50) ингибитора ALK5 согласно настоящему варианту осуществления предпочтительно составляет 400 нM или менее, более предпочтительно 300 нM или менее, еще более предпочтительно 200 нM или менее, еще более предпочтительно 100 нM или менее, особенно предпочтительно 50 нM или менее. Хотя конкретно не ограничено, нижний предел концентрации ингибирования ALK5 (IC50) может составлять, например, 0,1 нМ, 1 нМ или 10 нМ. Концентрацию ингибирования ALK5 можно измерить способом согласно примеру тестирования 3, описанному ниже.
[0038]
С помощью ингибитора ALK5 в соответствии с настоящим вариантом осуществления можно лечить заболевания, связанные с ALK5.
[0039]
<Терапевтические лекарственные средства>
Один вариант осуществления настоящего изобретения относится к терапевтическим лекарственным средствам для лечения расстройств циркадного ритма сна, деменции альцгеймеровского типа, дистрофии роговицы, рака и/или андрогенетической алопеции, которые содержат соединение или его фармацевтически приемлемую соль.
[0040]
Примеры расстройств циркадного ритма сна включают нарушения сна, вызванные смещением биологических часов в течение короткого промежутка времени по антропогенным или социальным причинам, эндогенные расстройства сна, вызванные нарушением функции согласования биологических часов с внешним циклом. В частности, их примеры включают синдром смены часовых поясов, расстройство сна при сменной работе, синдром поздней фазы сна, синдром отсроченной фазы сна, синдром непродолжительного сна-бодрствования. Более конкретно, их примеры включают синдром смены часовых поясов, расстройство сна при сменной работе, синдром фазового опережения сна, синдром отсроченной фазы сна, синдром не 24-часового цикла сон-бодрствование, нерегулярное нарушение ритма сон-бодрствование, нарушения циркадных ритмов, сопровождающиеся деменцией альцгеймеровского типа (такие как вечернее обострение, сопровождающееся деменцией альцгеймеровского типа), и нарушения циркадных ритмов, сопровождающиеся болезнью Паркинсона. Хотя конкретно не ограничено, один вариант осуществления согласно настоящему изобретению предпочтительно используется при лечении нерегулярного нарушения ритма сон-бодрствование или вечернего обострения, сопровождающегося деменцией альцгеймеровского типа.
[0041]
Примеры деменции альцгеймеровского типа включают деменцию альцгеймеровского типа, сопровождающуюся накоплением амилоида β в головном мозге, и деменцию альцгеймеровского типа, сопровождающуюся накоплением тау-белка.
[0042]
Примеры дистрофии роговицы включают эпителиальные, паренхиматозные и энтодермальные дистрофии роговицы. Хотя конкретно не ограничено, один вариант осуществления по настоящему изобретению предпочтительно используется для лечения энтодермальной эндотелиальной дистрофии роговицы Фукса.
[0043]
Примеры рака включают опухоли головного мозга (например, глиому и мультиформную глиобластому), рак печени (например, рак гепатоцитов), рак мочевого пузыря, миелодиспластические синдромы, рак толстой кишки и рак поджелудочной железы. Хотя конкретно не ограничено, один вариант осуществления согласно настоящему изобретению предпочтительно используется для лечения опухоли головного мозга или рака печени.
[0044]
При лечении рака можно использовать в комбинации еще одно лекарственное средство для лечения рака (далее именуемое «второе лекарственное средство для лечения рака») и/или радиотерапию. Вторым лекарственным средством для лечения рака, которое следует использовать, могут быть существующие лекарственные средства для лечения рака. Хотя конкретно не ограничено, примеры второго лекарственного средства для лечения рака включают блокаду иммунных контрольных точек, вакцины для терапии рака, лекарственные средства на основе антител для лечения рака, лекарственные средства для генной терапии и другие противоопухолевые лекарственные средства (такие как темозоломид, гемцитабин, помалидомид и паклитаксел). Эти вторые лекарственные средства для лечения рака можно использовать отдельно или в комбинации. Лекарственное средство для лечения рака и второе лекарственное средство для лечения рака в соответствии с настоящим вариантом осуществления могут быть предоставлены в виде комбинированного лекарственного средства или могут быть предоставлены по отдельности.
[0045]
Терапевтические лекарственные средства согласно настоящему варианту осуществления можно вводить перорально или парентерально. Примеры лекарственных форм для перорального введения включают пилюли, шарики, гранулы, порошки, капсулы, сиропы, эмульсии и суспензии. Примеры лекарственных форм для парентерального введения включают инъекции, инфузии, капельницы, глазные капли и суппозитории.
[0046]
Терапевтические лекарственные средства в соответствии с настоящим вариантом осуществления могут содержать эксципиент, связующее вещество, смазывающее вещество, дезинтегрант, подсластитель, поверхностно-активное вещество, суспендирующий агент, эмульгатор, краситель, консервант, отдушку, вкусовое вещество, стабилизатор и загуститель.
[0047]
Количество вводимого терапевтического лекарственного средства в соответствии с настоящим вариантом осуществления, которое зависит от состояния пациента, его веса, типа соединения, типа заболевания, пути введения и т.п., может быть соответствующим образом определено врачом. В качестве одного примера, при лечении расстройств циркадного ритма сна, терапевтическое лекарственное средство в соответствии с настоящим вариантом осуществления можно вводить взрослому (масса: около 60 кг) в количестве 0,1-3000 мг для перорального введения и в количестве 0,01-1000 мг для парентерального введения. При лечении деменции альцгеймеровского типа, терапевтическое лекарственное средство в соответствии с настоящим вариантом осуществления можно вводить взрослому (масса: около 60 кг) в количестве 0,1-3000 мг для перорального введения и в количестве 0,01-1000 мг для парентерального введения. При лечении эндотелиальной дистрофии роговицы терапевтическое лекарственное средство согласно настоящему варианту осуществления можно вводить взрослому (масса: около 60 кг) в количестве 0,1-3000 мг для перорального введения и в количестве 0,001-1000 мг для парентерального введения. При лечении рака терапевтическое лекарственное средство согласно настоящему варианту осуществления может быть введено взрослому (масса: около 60 кг) в количестве 0,1-3000 мг для перорального введения и в количестве 0,01-1000 мг для парентерального введения.
[0048]
<Способ получения соединения>
Соединение или его фармацевтически приемлемая соль могут быть синтезированы соответствующим образом с использованием известного способа. Один из примеров метода синтеза включает приведенную ниже схему А:
<Схема А>
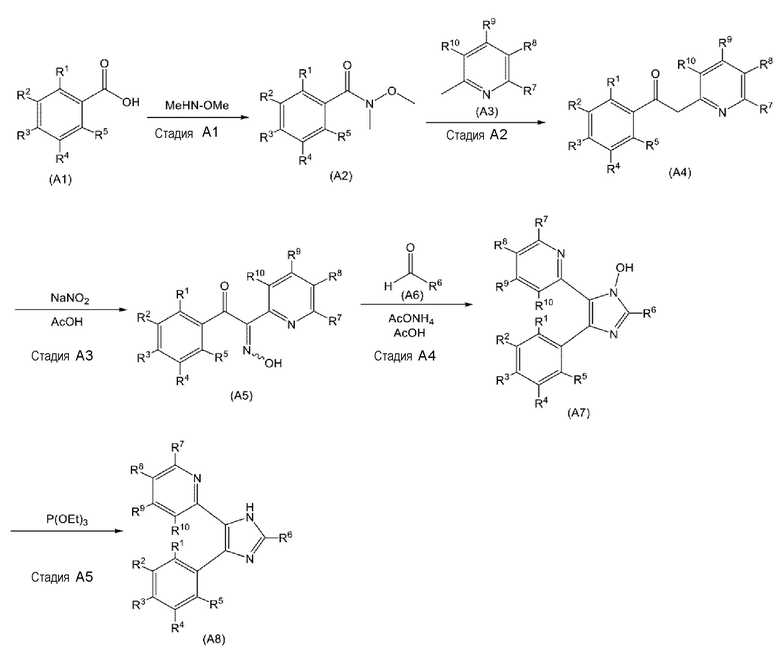
где R1 - R10 имеют значение, как определено выше.
[0049]
(Стадия A1)
На стадии А1 соединение (А1) подвергают взаимодействию с N, O-диметилгидроксиламином в присутствии конденсирующего агента с получением соединения (А2). Соединение (А1) может быть коммерческим продуктом или может быть получено известным способом.
[0050]
Примеры конденсирующего агента, используемого на стадии A1, включают, но не ограничиваются ими, 1,1'-карбонилдиимидазол (CDI), водорастворимый карбодиимид (WSC), 1-гидроксибензотриазол (HOBT), 1,3-дициклогексанкарбодиимид (DCC), 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (EDC), 2-хлор-1-метилпиридиния йодид, O-(бензотриазол-1-ил)-N, N,N',N'-тетраметилурония гексафторфосфат (HBTU) и O-(7-азабензотриазол-1-ил)-N, N,N',N'-тетраметилурония гексафторфосфат (HATU).
[0051]
(Стадия A2)
На стадии А2 соединение (А3) подвергают взаимодействию с органическим соединением лития и затем подвергают взаимодействию с соединением (А2) с получением соединения (A4). Соединение (A3) может быть коммерческим продуктом или может быть получено известным способом.
[0052]
Примеры органического соединения лития, используемого на стадии A2, включают, но не ограничиваются ими, диизопропиламид лития, бис(триметилсилил)амид лития, метиллитий, н-бутиллитий, втор-бутиллитий, трет-бутиллитий и фениллитий.
[0053]
(Стадия A3)
На стадии A3 соединение (A4) подвергают взаимодействию с нитритом натрия в присутствии уксусной кислоты с получением соединения (A5).
[0054]
(Стадия A4)
На стадии A4 соединение (A5) подвергают взаимодействию с соединением (A6) в присутствии ацетата аммония и уксусной кислоты с получением соединения (A7).
[0055]
(Стадия A5)
На стадии A5 соединение (A7) подвергают взаимодействию с триэтилфосфитом с получением соединения (A8).
[0056]
Примеры другого способа синтеза соединения или его фармацевтически приемлемой соли включают приведенную ниже схему B:
<Схема В>
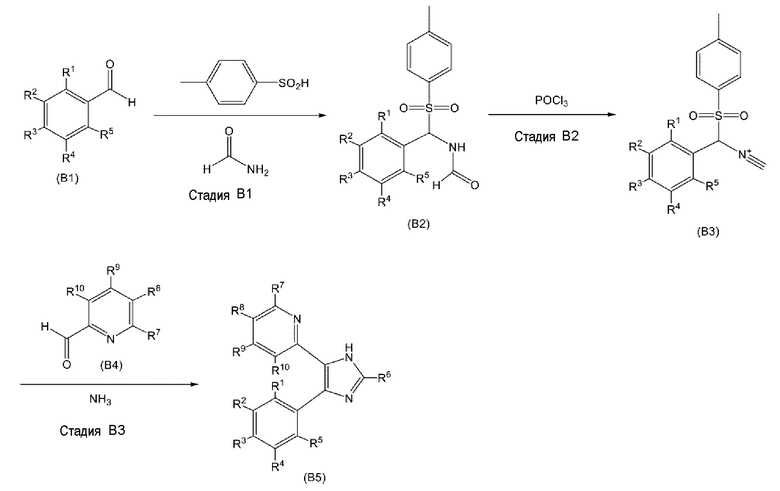
где R1 - R10 имеют значение, как определено выше.
[0057]
(Стадия B1)
На стадии B1 соединение (B1) подвергают взаимодействию с 4-метилбензолсульфиновой кислотой и формамидом с получением соединения (B2). Соединение (B1) может быть коммерческим продуктом или может быть получено известным способом.
[0058]
(Стадия B2)
На стадии B2 соединение (B2) подвергают взаимодействию с оксихлоридом фосфора с получением соединения (B3).
[0059]
(Стадия B3)
На стадии B3 соединение (B4) подвергают взаимодействию с аммиаком и затем подвергают взаимодействию с соединением (B3) с получением соединения (B5). Соединение (B4) может быть коммерческим продуктом или может быть получено известным способом.
[0060]
Способ синтеза не ограничивается приведенными выше схемами A и B. Например, синтез может быть выполнен другим путем синтеза со ссылкой на приведенный ниже пример получения.
Примеры
[0061]
Настоящее изобретение теперь будет описано более подробно посредством примеров, но технический объем настоящего изобретения не ограничивается этими примерами.
[0062]
[Пример получения 1-1]
N-Метокси-N-метил-1,3-дигидроизобензофуран-5-карбоксамид
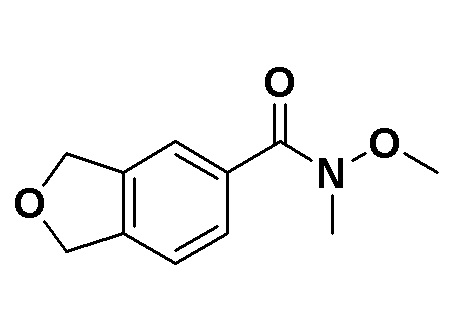
При 0°C, 1,3-дигидроизобензофуран-5-карбоновую кислоту (12 г, 76 ммоль) добавляли к смеси 1,1'-карбонилдиимидазола (16 г, 98 ммоль) и N, N-диметилформамида (DMF) (160 мл), затем перемешивали при комнатной температуре в течение двух часов. Реакционную смесь поддерживали так, чтобы она имела температуру 0°C, и добавляли N, O-диметилгидроксиламин гидрохлорид (9,6 г, 98 ммоль) при той же температуре, затем перемешивали при комнатной температуре в течение 12 часов. Затем к реакционной смеси добавляли воду с последующей экстракцией этилацетатом и тетрагидрофураном (2:1) три раза (400 мл х 3 раза). Органический слой последовательно промывали водой (50 мл х два раза) и насыщенным солевым раствором. Органический слой сушили над сульфатом магния и фильтровали. Затем растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (этилацетат:гептан=1:1) с получением указанного выше в заголовке соединения (11 г).
1H-ЯМР спектр (CDCl3) δ (ppm): 3,37 (с, 3H), 3,56 (с, 3H), 5,14 (с, 4H), 7,25-7,29 (м, 1H), 7,56 (д, J=1,10 Гц, 1H), 7,60 (дд, J=7,68, 1,46 Гц, 1H)
[0063]
[Пример получения 1-2]
1-(1,3-Дигидроизобензофуран-5-ил)-2-(гидроксиимино)-2-(пиридин-2-ил)этанон
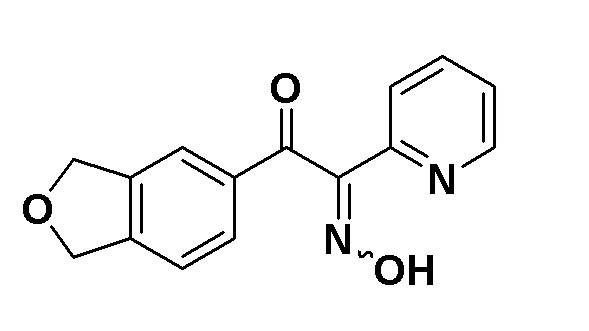
При -78°C, н-бутиллитий (5,0 мл, 13 ммоль) по каплям добавляли к смеси диизопропиламина (1,9 мл, 14 ммоль) и тетрагидрофурана (THF) (50 мл). Раствор перемешивали при той же температуре в течение 30 минут и затем по каплям добавляли 2-пиколин (1,5 мл, 15 ммоль) при той же температуре. Реакционную смесь перемешивали при 0°C в течение 30 минут, и затем охлаждали до -78°C. После этого по каплям добавляли смесь N-Метокси-N-метил-1,3-дигидроизобензофуран-5-карбоксамида (2,5 г, 12 ммоль), полученного в примере получения 1-1, и THF (10 мл) при той же температуре. Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над сульфатом натрия и фильтровали. Затем, растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на NH-силикагеле (этилацетат:гептан=0:1-2:3 в градиенте), и затем снова очищали в тех же условиях с получением неочищенного продукта 1-(1,3-дигидроизобензофуран-5-ил)-2-(пиридин-2-ил)этанона (600 мг).
[0064]
Смесь нитрита натрия (0,52 г, 7,6 ммоль) и воды (4 мл) по каплям добавляли к смеси неочищенного продукта 1-(1,3-дигидроизобензофуран-5-ил)-2-(пиридин-2-ил)этанона (1,5 г), THF (10 мл) и уксусной кислоты (15 мл) при 0°C, затем перемешивали при комнатной температуре в течение 3 часов. Растворитель отгоняли при пониженном давлении, и затем добавляли этилацетат и насыщенный водный раствор бикарбоната натрия. Органический слой отделяли и последовательно промывали насыщенным водным раствором бикарбоната натрия, водой и насыщенным солевым раствором. Затем органический слой, сушили над сульфатом натрия и фильтровали. Затем растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на NH-силикагеле (гептан:этилацетат:метанол=1:1:0 до 0:1:0 в градиенте, и затем 0:1:0 до 0:9:1 в градиенте) с получением указанного в заголовке соединения (1,3 г) в виде смеси формы Е и формы Z. Ниже показаны данные ЯМР смеси формы E и формы Z.
1H-ЯМР спектр (CDCl3) δ (ppm): 5,14 (с, 4H), 5,16 (с, 4H), 7,28-7,30 (м, 1H), 7,36 (д, J=7,68 Гц, 1H), 7,36 (д, J=7,68 Гц, 1H), 7,52-7,55 (м, 1H), 7,33-7,91 (м, 5H), 7,33-7,91 (м, 4H), 7,94-8,02 (м, 1H), 8,48-8,56 (м, 1H), 8,58-8,64 (м, 1H)
[0065]
[Пример 1]
2-(4-(1,3-Дигидроизобензофуран-5-ил)-1H-имидазол-5-ил)пиридин
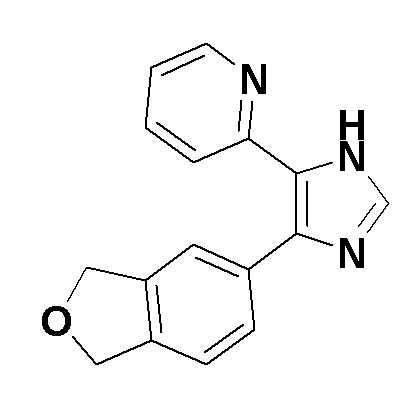
Параформальдегид (50 мг, 0,56 ммоль) добавляли к смеси 1-(1,3-дигидроизобензофуран-5-ил)-2-(гидроксиимино)-2-(пиридин-2-ил)этанона (300 мг, 1,1 ммоль), полученного в примере получения 1-2, ацетата аммония (520 мг, 6,7 ммоль) и уксусной кислоты (6 мл) при комнатной температуре, затем перемешивали при той же температуре в течение 1 часа, и затем перемешивали при 100°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и растворитель отгоняли при пониженном давлении. Триэтилфосфит (0,38 мл, 2,2 ммоль) добавляли к смеси остатка (310 мг) и N-метилпирролидинона (NMP) (7 мл) при комнатной температуре, затем перемешивали при 120°C в течение 3 часов. Реакционную смесь поддерживали так, чтобы она имела комнатную температуру, и добавляли воду и этилацетат. Затем добавляли насыщенный водный раствор бикарбоната натрия. Органический слой отделяли, и водный слой экстрагировали этилацетатом и тетрагидрофураном (2:1). Два органических слоя объединяли и полученный органический слой последовательно промывали водой и насыщенным солевым раствором. Органический слой сушили над сульфатом магния и фильтровали. Растворитель затем отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на NH-силикагеле (этилацетат:метанол=20:1) с получением указанного выше в заголовке соединения (44 мг).
1H-ЯМР спектр (CDCl3) δ (ppm): 5,15 (с, 2H), 5,18 (д, J=1,46 Гц, 2H), 7,12 (ддд, J=6,59, 4,76, 2,20 Гц, 1H), 7,29 (д, J=8,05 Гц, 1H), 7,48-7,57 (м, 4H), 7,75 (с, 1H), 8,55 (д, J=4,76 Гц, 1H), 10,39 (ушир. с., 1H)
[0066]
[Пример 2]
2-(4-(1,3-Дигидроизобензофуран-5-ил)-2-метил-1H-имидазол-5-ил)пиридин
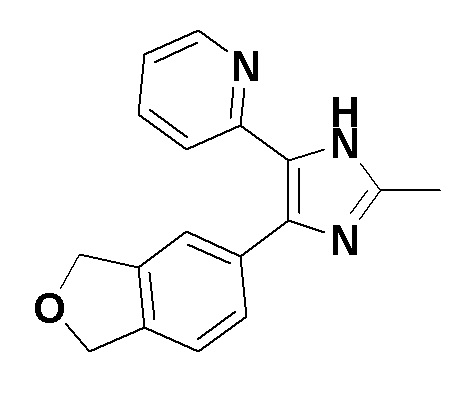
Ацетальдегид (13 мг, 0,27 ммоль) добавляли к смеси 1-(1,3-дигидроизобензофуран-5-ил)-2-(гидроксиимино)-2-(пиридин-2-ил)этанона (61 мг, 0,22 ммоль), полученного в примере получения 1-2, ацетата аммония (110 мг, 1.4 ммоль) и уксусной кислоты (1,5 мл) при комнатной температуре, затем перемешивали при той же температуре в течение 15 минут и затем перемешивали при 115°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и растворитель отгоняли при пониженном давлении. Триэтилфосфит (0,38 мл, 2,2 ммоль) добавляли к смеси остатка (67 мг) и NMP (1,5 мл) при комнатной температуре, затем перемешивали при 120°C в течение 6 часов. Реакционную смесь поддерживали так, чтобы она имела комнатную температуру. Добавляли воду и этилацетат, и затем добавляли насыщенный водный раствор бикарбоната натрия. Органический слой отделяли, и водный слой экстрагировали этилацетатом и тетрагидрофураном (2:1). Два органических слоя объединяли и полученный органический слой последовательно промывали водой и насыщенным солевым раствором. Органический слой сушили над сульфатом магния и фильтровали. Растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на NH-силикагеле (этилацетат:метанол=40:1) с получением указанного выше в заголовке соединения (44 мг).
1H-ЯМР спектр (CDCl3) δ (ppm): 2,50 (с, 3H), 5,12 (с, 2H), 5,15 (д, J=1,36 Гц, 2H), 7,04-7,09 (м, 1H), 7,22-7,28 (м, 1 H), 7,43-7,54 (м, 4H), 8,50 (дт, J=4,98, 1,36 Гц, 1H), 9,97 (ушир.д, J=3,17 Гц, 1H)
[0067]
[Пример получения 3-1]
N-((1,3-дигидроизобензофуран-5-ил)(тозил)метил)формамид
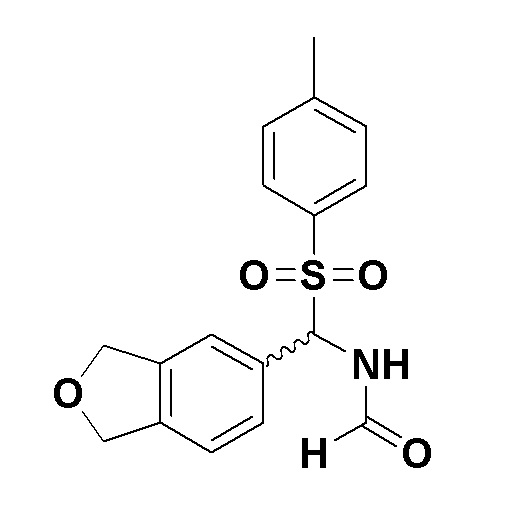
Хлортриметилсилан (3,1 мл, 25 ммоль) добавляли к смеси 1,3-дигидроизобензофуран-5-карбальдегида (3,3 г, 22 ммоль), 4-метилбензолсульфиновой кислоты (5,3 г, 34 ммоль), формамида (2,2 мл, 56 ммоль), ацетонитрила (30 мл), и толуола (30 мл) при 0°C, затем перемешивали при комнатной температуре в течение 30 минут. Затем полученный раствор перемешивали при 50°C в течение 8 часов 20 минут, и реакционную смесь охлаждали до комнатной температуры. Нерастворимое содержимое удаляли фильтрованием и растворитель отгоняли при пониженном давлении из фильтрата. Остаток очищали хроматографией на колонке с силикагелем (этилацетат:гептан=4:1) с получением указанного выше в заголовке соединения (2,9 г).
1H-ЯМР спектр (CDCl3) δ (ppm): 2,38-2,43 (м, 3H), 5,01 (т J=5,47 Гц, 4H), 6,36-6,43 (м, 1H), 7,34-7,39 (м, 1H), 7,40-7,51 (м, 4H), 7,69-7,76 (м, 2H), 7,90-7,95 (м, 1H), 9,72-9,80 (м, 1H)
[0068]
[Пример 3]
2-(4-(1,3-Дигидроизобензофуран-5-ил)-1H-имидазол-5-ил)-5-фторпиридин
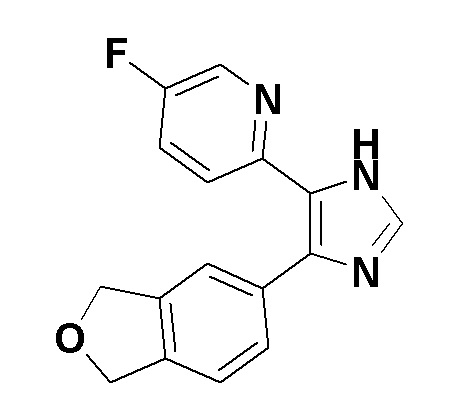
Оксихлорид фосфора (0,12 мл, 1,2 ммоль) добавляли к смеси N-((1,3-дигидроизобензофуран-5-ил)(тозил)метил)формамида (210 мг, 0,62 ммоль), полученного в примере получения 3-1, и THF (2 мл) при комнатной температуре, затем перемешивали при той же температуре в течение 20 минут. Реакционную смесь охлаждали до 0°C и добавляли триэтиламин (0,52 мл, 3,7 ммоль) при той же температуре, затем перемешивали при той же температуре в течение 2 часов. К реакционной смеси добавляли воду при 0°C, с последующей экстракцией этилацетатом. Органический слой промывали насыщенным солевым раствором и затем растворитель отгоняли при пониженном давлении. Остаток фильтровали с помощью колоночной хроматографии через NH-силикагель (этилацетат). Растворитель отгоняли при пониженном давлении с получением неочищенного продукта 5-(изоциано(тозил)метил)-1,3-дигидроизобензофурана (190 мг). 49 мг неочищенного продукта массой 190 мг использовали в следующей реакции.
[0069]
Смесь 5-фтор-2-формилпиридина (19 мг, 0,16 ммоль) и 28% водного раствора аммиака (0,50 мл) перемешивали при 50°C в течение 1 часа, и затем растворитель отгоняли при пониженном давлении. К остатку добавляли смесь неочищенного продукта 5-(изоциано(тозил)метил)-1,3-дигидроизобензофурана (49 мг) и DMF (1 мл) при комнатной температуре, и затем добавляли карбонат калия (54 мг, 0,39 ммоль), затем перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли воду, с последующей экстракцией этилацетатом. Органический слой промывали насыщенным солевым раствором и растворитель отгоняли при пониженном давлении. Остаток очищали с помощью LC-MS (система растворителей ацетонитрил/вода, содержащая 0,1% трифторуксусной кислоты), и затем очищали с помощью колоночной хроматографии на NH-силикагеле (этилацетат:метанол=20:1) с получением указанного выше в заголовке соединения (1,2 мг).
1H-ЯМР спектр (CDCl3) δ (ppm): 5,13 (с, 2H), 5,16 (д, J=1,36 Гц, 2H), 7,26 (ушир.с, 1H), 7,28 (с, 1H), 7,49 (ушир.с, 1H), 7,51 (ушир.с, 2H), 7,73 (с, 1H), 8,39 (ушир.д, J=2,27 Гц, 1H), 9,98-10,35 (м, 1H)
[0070]
[Пример 4]
2-(4-(1,3-Дигидроизобензофуран-5-ил)-1H-имидазол-5-ил)-6-метилпиридин
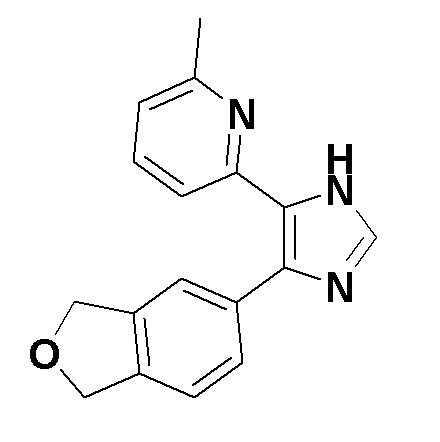
Смесь 6-метил-2-пиридинкарбоксальдегида (19 мг, 0,16 ммоль) и 28% водного раствора аммиака (0,50 мл) перемешивали при 50°C в течение 1 часа, и затем растворитель отгоняли при пониженном давлении. Смесь неочищенного продукта 5-(изоциано(тозил)метил)-1,3-дигидроизобензофурана (49 мг), полученного в примере 3, и DMF (1 мл) добавляли к остатку при комнатной температуре, и затем добавляли карбонат калия (54 мг, 0,39 ммоль), затем перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли воду, с последующей экстракцией этилацетатом. Органический слой промывали насыщенным солевым раствором и затем растворитель отгоняли при пониженном давлении. Остаток очищали с помощью LC-MS (система растворителей ацетонитрил/вода, содержащая 0,1% трифторуксусной кислоты), и затем очищали с помощью колоночной хроматографии на NH-силикагеле (этилацетат:метанол=20:1) с получением указанного выше в заголовке соединения (2,0 мг).
1H-ЯМР спектр (CDCl3) δ (ppm): 2,54 (с, 3H), 5,13 (с, 2H), 5,15 (д, J=1,36 Гц, 2H), 6,96 (д, J=7,25 Гц, 1H), 7,26-7,33 (м, 2H), 7,37-7,43 (м, 1H), 7,51-7,55 (м, 2H), 7,72 (с, 1H), 10,22-10,57 (м, 1H)
[0071]
[Пример получения 5-1]
N-Метокси-N-метил-1,3-дигидроизобензофуран-5-карбоксамид
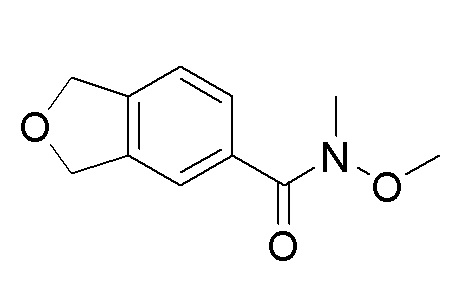
N, N-диизопропилэтиламин (4,05 мл, 21,9 ммоль) медленно добавляли к смеси 1,3-дигидроизобензофуран-5-карбоновой кислоты (1,20 г, 7,31 ммоль), N, O-диметилгидроксиламин гидрохлорида (1.42 г, 14,6 ммоль) и дихлорметана (20 мл) при 0°C, затем перемешивали при той же температуре в течение 10 минут. Пропилфосфоновый ангидрид (50% раствор в этилацетате, 9,20 мл, 30,6 ммоль) медленно добавляли к реакционной смеси, затем перемешивали при комнатной температуре в течение 16 часов. К реакционной смеси добавляли ледяную воду с последующей экстракцией дихлорметаном. Органический слой промывали насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (гексан/этилацетат) с получением указанного выше в заголовке соединения (0,90 г).
1H NMR (400 MГц, DMSO-d6) δ 7,51-7,48 (м, 2H), 7,38-7,36 (м, 1H), 5,02 (с, 4H), 3,53 (с, 3H), 3,25 (с, 3H).
[0072]
[Пример получения 5-2]
1-(1,3-Дигидроизобензофуран-5-ил)-2-(пиридин-2-ил)этан-1-он
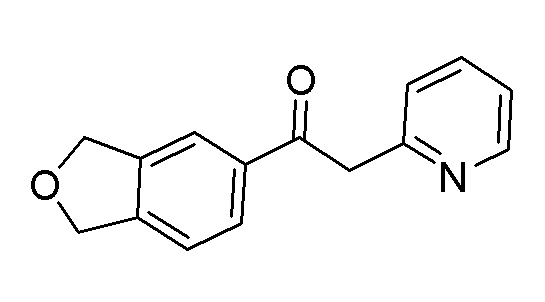
В атмосфере аргона, диизопропиламид лития (2 М раствор тетрагидрофурана, 2,51 мл, 5,02 ммоль) медленно добавляли к смеси 2-метилпиридина (0,757 мл, 7,72 ммоль) и тетрагидрофурана (8,0 мл) при -78°C, затем перемешивали при той же температуре в течение 30 минут. Смесь N-метокси-N-метил-1,3-дигидроизобензофуран-5-карбоксамида (800 мг, 3,86 ммоль) и тетрагидрофурана (4 мл) медленно добавляли к реакционной смеси, затем перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом. Органический слой промывали насыщенным солевым раствором и сушили над сульфатом натрия. Затем растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (гексан/этилацетат) с получением указанного выше в заголовке соединения (0,60 г).
ESI-MS: m/z 240,10 [M+1]+
[0073]
[Пример получения 5-3]
1-(1,3-Дигидроизобензофуран-5-ил)-2-(гидроксиимино)-2-(пиридин-2-ил)этан-1-он
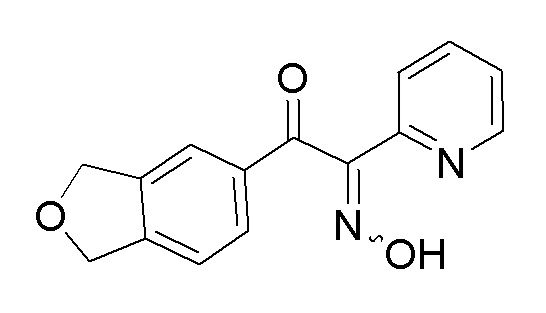
Нитрит натрия (260 мг, 3,76 ммоль), содержащий небольшое количество воды, медленно добавляли к смеси 1-(1,3-дигидроизобензофуран-5-ил)-2-(пиридин-2-ил)этан-1-она (600 мг, 2,51 ммоль), тетрагидрофурана (10 мл), и уксусной кислоты (10 мл) при 0°C, затем перемешивали при комнатной температуре в течение 2 часов. Растворитель отгоняли из реакционной смеси при пониженном давлении. К остатку добавляли насыщенный водный раствор бикарбоната натрия, с последующей экстракцией этилацетатом. Органический слой промывали насыщенным солевым раствором и сушили над сульфатом натрия. Затем растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (гексан/этилацетат) с получением указанного выше в заголовке соединения (0,50 г).
ESI-MS: m/z 266,91 [M-1]-
[0074]
[Пример получения 5-4]
2-Циклопропил-4-(1,3-дигидроизобензофуран-5-ил)-5-(пиридин-2-ил)-1H-имидазол-1-ол
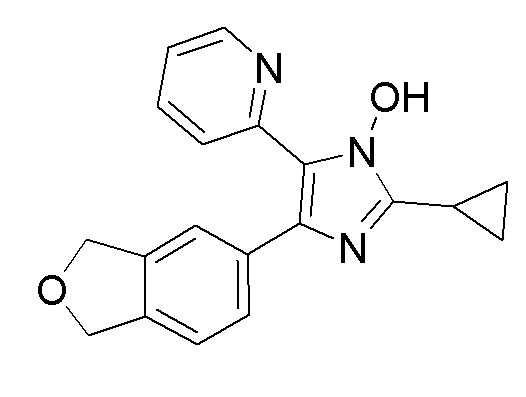
Ацетат аммония (460 мг, 5,96 ммоль) и циклопропан карбальдегид (209 мг, 2,98 ммоль) добавляли к смеси 1-(1,3-дигидроизобензофуран-5-ил)-2-(гидроксиимино)-2-(пиридин-2-ил)этан-1-она (800 мг, 2,98 ммоль) и ацетонитрила (12 мл) при 0°C, затем перемешивали при комнатной температуре в течение 10 минут. К реакционной смеси добавляли трифторуксусную кислоту (34,0 мг, 0,298 ммоль), затем перемешивали при 50°C в течение 16 часов. Растворитель отгоняли из реакционной смеси при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем (дихлорметан/метанол) с получением указанного выше в заголовке соединения (0,20 г).
ESI-MS: m/z 319,13 [M+1]+
[0075]
[Пример 5]
2-(2-Циклопропил-4-(1,3-дигидроизобензофуран-5-ил)-1H-имидазол-5-ил)пиридин
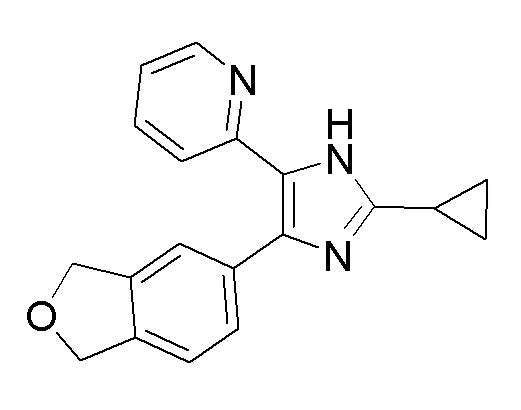
Раствор хлорида титана(III) (12% раствор хлористоводородной кислоты, 2,0 мл) медленно добавляли к смеси 2-циклопропил-4-(1,3-дигидроизобензофуран-5-ил)-5-(пиридин-2-ил)-1H-имидазол-1-ола (200 мг, 0,626 ммоль) и метанола (2 мл) при 0°C, затем перемешивали при комнатной температуре в течение 16 часов. Растворитель отгоняли из реакционной смеси при пониженном давлении. К остатку добавляли насыщенный водный раствор бикарбоната натрия, и остаток отфильтровывали с использованием церита, промывая 20% метанол/дихлорметан. Органический слой, отделенный от фильтрата, промывали насыщенным солевым раствором и сушили над сульфатом натрия. Затем растворитель отгоняли при пониженном давлении. Остаток очищали с помощью высокоэффективной жидкостной хроматографии (X Bridge Shield (19 х 250 мм) 10 мкм, 5 мM ацетата аммония/вода) с получением указанного выше в заголовке соединения (0,035 г).
ESI-MS: m/z 304,29 [M+1]+
1H NMR (400 MГц, DMSO-d6) δ 12,39, 12,13 (с, 1H), 8,57, 8,33 (д, 1H, J=4,0 Гц), 7,76-7,15 (м, 6H), 5,01, 4,99 (с, 4H), 2,08-1,97 (м, 1H).
[0076]
[Пример тестирования 1]
Оценка ингибирующей активности в отношении CK1δ и ингибирующего действия в отношении p38α
1. Получение раствора исследуемого вещества
Исследуемое вещество растворяли в диметилсульфоксиде (DMSO). Раствор дополнительно разбавляли DMSO для получения раствора с концентрацией, в 100 раз превышающей испытуемую концентрацию. Раствор дополнительно разбавляли в 25 раз буфером для анализа для получения раствора исследуемого вещества. Вещество положительного контроля также обрабатывали, как указано выше, для получения раствора вещества положительного контроля.
[0077]
2. Получение протеинкиназы
CK1δ: Использовали человеческий CK1δ, полученный путем слияния GST (61 кДа) с N-концом домена активности фермента CK1δ человека (участок аминокислотной последовательности 1-294 с номером доступа NP_001884.2), с последующей экспрессией в E. сoli и затем очисткой с помощью системы хроматографии на глутатион-сефарозе.
p38α: Использовали человеческий p38α, полученный следующим образом: GST (66 кДа) сливали с N-концом участка аминокислотной последовательности 9-352 под номером доступа NP_620581.1, с последующей экспрессией в E. сoli и затем очисткой с помощью системы хроматографии на глутатион-сефарозе, и продукт активировали с помощью His-меченого MAP2K6 и снова очищали с помощью системы хроматографии на глутатион-сефарозе.
[0078]
3. Реагент и способ тестирования
5 мкл 4-кратного раствора исследуемого вещества, полученного с буфером для анализа (20 мМ HEPES, 0,01% Triton X-100, 2 мМ DTT, рН 7,5), 5 мкл 4-кратного раствора субстрат/ATP/металл и 10 мкл 2-кратного раствора киназы смешивали в лунках 384-луночного планшета из полипропилена и подвергали взаимодействию при комнатной температуре в течение 1 часа. 70 мкл буфера для прекращения реакции (QuickScout Screening Assist MSA; Carna Biosciences) добавляли для прекращения реакции. Субстрат-пептид и фосфорилированный пептид в реакционном растворе разделяли с помощью системы LabChip (Perkin Elmer) и определяли количество. Киназную реакцию оценивали на основе отношения продукта (P/(P+S)), рассчитанного из высоты пика субстрата-пептида (S) и высоты пика фосфорилированного пептида (P).
[0079]
4. Условия реакции
[Таблица 1]
Таблица 1
[0080]
5. Анализ данных
Средний сигнал от контрольных лунок, содержащих все компоненты реакции, определяли как 0% ингибирования, и средний сигнал от фоновых лунок (без добавления фермента) определяли как 100% ингибирование. Степень ингибирования рассчитывали по среднему сигналу тест-лунок, содержащих каждое исследуемое вещество. Значение IC50 определяли путем аппроксимации графика зависимости концентрации исследуемого вещества и степени ингибирования к 4-параметрической логистической кривой методом нелинейных наименьших квадратов. Результаты показаны в таблице 2. Соединения в примерах обладали хорошей ингибирующей активностью в отношении CK1δ. Была обнаружена большая разница между концентрацией ингибирования CK1δ и концентрацией ингибирования p38α.
[Таблица 2]
Таблица 2
[0081]
[Пример тестирования 2]
Оценка ингибирующего действия в отношении ALK5
1. Получение раствора исследуемого вещества
Исходный раствор 10% DMSO (концентрация соединения: 10 мМ), содержащий 0,1 мг/мл BSA (бычий сывороточный альбумин), получали в виде раствора исследуемого вещества.
[0082]
2. Киназа
ALK5: ALK5 человека, GenBank ID=BC071181.
[0083]
3. Реагент, способ тестирования и условия реакции
Анализ киназы проводили с использованием набора для анализа ADP-Glo(TM), доступного от Promega Corporation, в соответствии со следующим способом анализа реакции:
Компонент 1: 1 мкл разведенной активной протеинкиназы
Компонент 2: 1 мкл субстрата
Компонент 3: 1 мкл буфера для анализа киназы
Компонент 4: 1 мкл соединения (10 концентраций) или 10% DMSO
Компонент 5: 1 мкл исходного ATP (25 мкМ конечная концентрация в лунке)
[0084]
Анализ начинали с инкубации реакционной смеси в 384-луночном планшете при комнатной температуре в течение 40 минут. После инкубации добавляли 5 мкл реагента ADP-Glo(TM). Планшет встряхивали, затем инкубировали при комнатной температуре в течение 40 минут. После этого добавляли 10 мкл реагента для выявления киназы и планшет встряхивали, затем инкубировали при комнатной температуре в течение 30 минут. Планшет измеряли на планшете-ридере GloMax с использованием протокола считывания люминесценции ADP-Glo(TM). Для холостого контроля анализ проводили с использованием всех компонентов анализа, за исключением того, что добавляли соответствующий субстрат (замененный таким же количеством буфера для разведения для анализа). Скорректированное значение активности рассчитывали путем вычитания значения пустого контроля из измеренного значения.
[0085]
4. Анализ данных
Относительную единицу люминесценции (RLU) измеряли для 10 соединений с концентрацией от 0,3 нМ до 10000 нМ. Степень ингибирования рассчитывали следующим образом:
{(RLU контроля-RLU исследуемого вещества)/(RLU контроля- RLU фоновая)} х 100
Анализ методом нелинейной регрессии выполнен с использованием GraphPad Prism version 5.01 для определения значения IC50. Результаты показаны в таблице 3. Выявлено, что соединение в примере 1 обладает хорошей ингибирующей активностью в отношении ALK5.
[Таблица 3]
Таблица 3
[0086]
[Пример тестирования 3]
Оценка ингибирующего действия в отношении ALK5
Используя человеческий рекомбинантный GST-меченый TGFB, доступный от SignalChem (Catalog No. T07-11G), субстрат-пептид TGFBR1 Peptide (Catalog No. T36-58) и набор для анализа ADP-Glo (Promega Corporation), измерения проводили в реакционной системе 5-мкл в 384-луночном планшете (GloMax планшет-ридер) в соответствии с документом, приложенным к набору для анализа ADP-Glo. Для используемых условий реакции концентрация фермента составляла 5 нг/мкл, концентрация субстрата составляла 200 нг/мкл, концентрация ATP составляла 25 мкМ и время реакции составляло 2 часа. Значение IC50 рассчитывали с использованием GraphPad Prism version 5.01 на основе степени ингибирования при добавлении исследуемого вещества (8-10 концентраций, разбавленных в 3 раза от 3000 нМ).
[Таблица 4]
Таблица 4
[0087]
[Пример тестирования 4]
Оценка ингибирующего действия в отношении CK1δ
Используя человеческий рекомбинантный GST-меченый CK1δ, доступный от SignalChem (Catalog No. C65-10G), субстрат-пептид казеин дефосфорилированный (Catalog No. C03-54BN) и набор для анализа ADP-Glo (Promega Corporation), измерения проводили в реакционной системе 5-мкл в 384-луночном планшете (GloMax планшет-ридер) в соответствии с документом, приложенным к набору для анализа ADP-Glo. Для используемых условий реакции концентрация фермента составляла 2 нг/мкл, концентрация субстрата составляла 200 нг/мкл, концентрация ATP составляла 25 мкМ и время реакции составляло 40 минут. Значение IC50 рассчитывали с использованием GraphPad Prism version 5.01 на основе степени ингибирования при добавлении исследуемого вещества (8-10 концентраций, разбавленных в 3 раза от 3000 нМ).
[Таблица 5]
Таблица 5
[0088]
[Пример тестирования 5]
Оценка ингибирующего действия в отношении p38α
Используя человеческий рекомбинантный GST-меченый p38α, доступный от SignalChem (Catalog No. M39-10 BG), субстрат-пептид p38 Substrate (Catalog No. P03-58) и набор для анализа ADP-Glo (Promega Corporation), измерения проводили в реакционной системе 5-мкл в 384-луночном планшете (GloMax планшет-ридер) в соответствии с документом, приложенным к набору для анализа ADP-Glo. Для используемых условий реакции концентрация фермента составляла 2 нг/мкл, концентрация субстрата составляла 100 нг/мкл, концентрация ATP составляла 25 мкМ и время реакции составляло 40 минут. Значение IC50 рассчитывали с использованием GraphPad Prism version 5.01 на основе степени ингибирования при добавлении исследуемого вещества (8-10 концентраций, разбавленных в 3 раза от 3000 нМ).
[Таблица 6]
Таблица 6
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР КАЗЕИНКИНАЗЫ 1 ДЕЛЬТА И КАЗЕИНКИНАЗЫ 1 Е | 2011 |
|
RU2562833C2 |
| ПРОИЗВОДНЫЕ 3-АРИЛТИОИНДОЛ-2-КАРБОКСАМИДОВ И ИХ АНАЛОГИ КАК ИНГИБИТОРЫ КАЗЕИНКИНАЗЫ Iε | 2005 |
|
RU2391098C2 |
| АМИДЫ 3-ЗАМЕЩЕННОЙ 5- И 6-АМИНОАЛКИЛИНДОЛ-2-КАРБОНОВОЙ КИСЛОТЫ И РОДСТВЕННЫЕ АНАЛОГИ КАК ИНГИБИТОРЫ КАЗЕИНКИНАЗЫ IΕ | 2005 |
|
RU2369599C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2709786C2 |
| ЗАМЕЩЕННЫЕ АМИДЫ ТИЕНОПИРРОЛКАРБОНОВОЙ КИСЛОТЫ, АМИДЫ ПИРРОЛОТИАЗОЛКАРБОНОВОЙ КИСЛОТЫ И РОДСТВЕННЫЕ АНАЛОГИ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЗЕИНКИНАЗЫ Iε | 2005 |
|
RU2403257C2 |
| СПОСОБЫ ЛЕЧЕНИЯ РАССТРОЙСТВ ЦИРКАДНОГО РИТМА СНА | 2017 |
|
RU2763493C2 |
| ИНГИБИТОРЫ ФЕРМЕНТА ФОСФОДИЭСТЕРАЗЫ 10 | 2013 |
|
RU2667058C2 |
| АГЕНТЫ, ИНГИБИРУЮЩИЕ Р38 КИНАЗУ | 2010 |
|
RU2532376C2 |
| ПРОИЗВОДНОЕ ФЕНИЛПИРРОЛА | 2012 |
|
RU2618228C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, АКТИВНЫЕ В ОТНОШЕНИИ РЕЦЕПТОРА СВ1 | 2005 |
|
RU2377238C2 |
Настоящее изобретение относится к соединению, представленному формулой (1) или его фармацевтически приемлемой соли. В формуле (1): R1, R4 и R5 представляют собой водород, R6, R7, R9 и R10 каждый независимо представляет собой водород, C1-С6 алкил или С3-С6 циклоалкил, R8 представляет собой водород, C1-С6 алкил, С3-С6 циклоалкил или галоген, где R2 и R3 или R4 и R5 вместе с двумя атомами углерода, к которым они присоединены, образуют тетрагидрофурановое кольцо, имеющее следующую структуру:
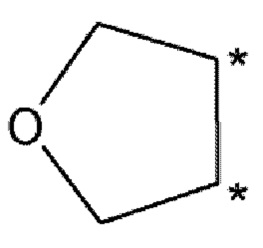
где атомы углерода со звездочкой (*) представляют собой атомы углерода бензольного кольца, связанные с R2 и R3, где тетрагидрофурановое кольцо необязательно замещено C1-С6 алкилом. Также предложены ингибитор казеинкиназы 1δ и активин-рецептор-подобной киназы 5 (ALK5), включающий указанное соединение, терапевтические лекарственные средства, содержащие указанное соединение. Предложенное соединение обладает ингибирующей активностью в отношении CK1δ и/или ингибирующей активностью в отношении ALK5 и может быть использовано для лечения расстройств циркадного ритма сна, для лечения деменции альцгеймеровского типа, для лечения рака, для лечения дистрофии роговицы, для лечения андрогенетической алопеции. 8 н. и 5 з.п. ф-лы, 6 табл., 17 пр.
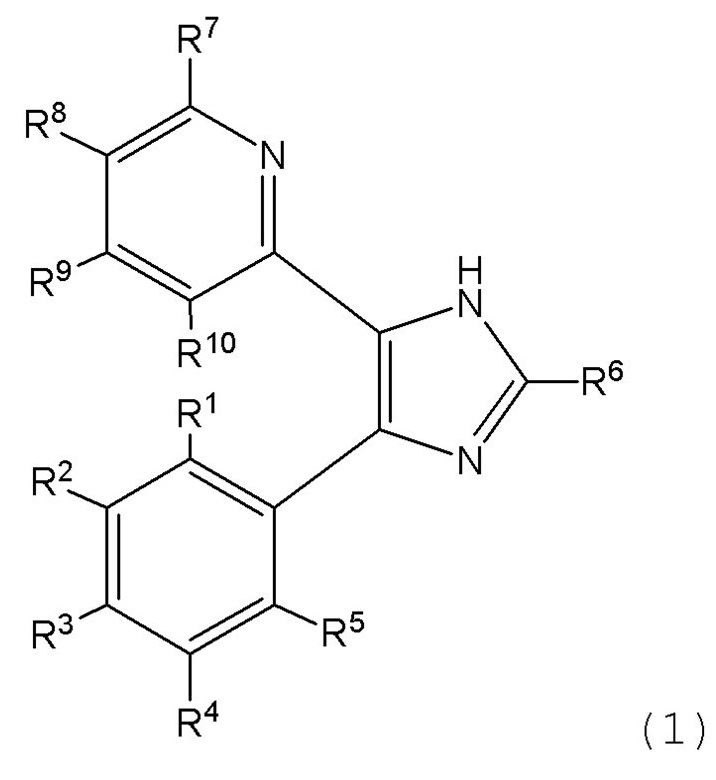
1. Соединение, представленное следующей формулой (1):
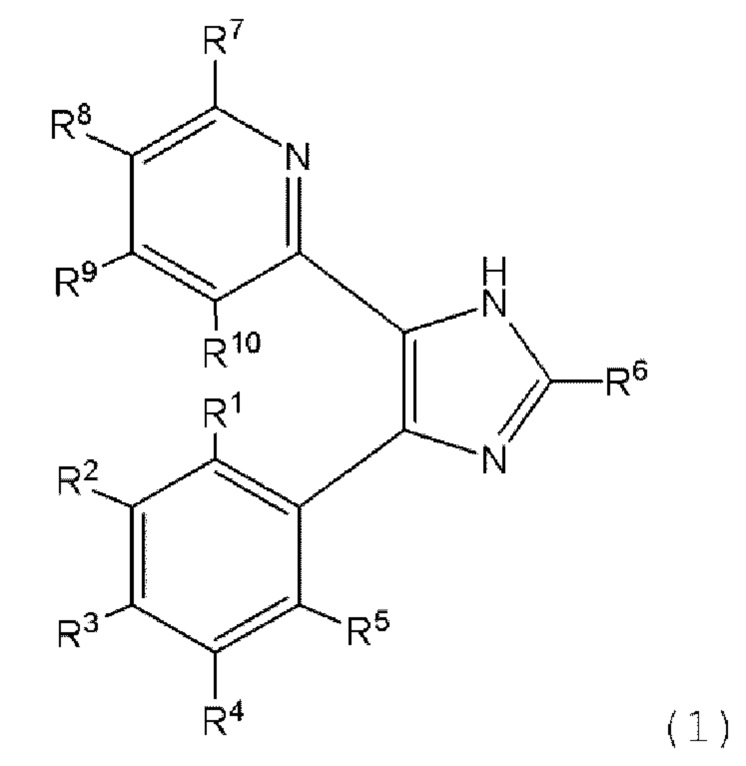
где
R1, R4 и R5 представляют собой водород,
R6, R7, R9 и R10 каждый независимо представляет собой водород, C1-С6 алкил или С3-С6 циклоалкил,
R8 представляет собой водород, C1-С6 алкил, С3-С6 циклоалкил или галоген,
где
R2 и R3 или R4 и R5 вместе с двумя атомами углерода, к которым они присоединены, образуют тетрагидрофурановое кольцо, имеющее следующую структуру:
где атомы углерода со звездочкой (*) представляют собой атомы углерода бензольного кольца, связанные с R2 и R3, где тетрагидрофурановое кольцо необязательно замещено C1-С6 алкилом;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где
R2 и R3 вместе с двумя атомами углерода, к которым они присоединены, образуют незамещенное тетрагидрофурановое кольцо.
3. Соединение по п. 1, выбранное из группы, состоящей из следующих соединений:
или его фармацевтически приемлемая соль.
4. Ингибитор казеинкиназы 1δ, включающий соединение по любому из пп. 1-3 или его фармацевтически приемлемую соль.
5. Терапевтическое лекарственное средство для лечения расстройств циркадного ритма сна, ассоциированных с казеинкиназой 1δ, включающее соединение по любому из пп. 1-3 или его фармацевтически приемлемую соль.
6. Терапевтическое лекарственное средство по п. 5, где расстройства циркадного ритма сна представляют собой нерегулярное нарушение ритма сон-бодрствование или вечернее обострение, сопровождающееся деменцией альцгеймеровского типа.
7. Терапевтическое лекарственное средство для лечения деменции альцгеймеровского типа, ассоциированной с казеинкиназой 1δ, включающее соединение по любому из пп. 1-3 или его фармацевтически приемлемую соль.
8. Ингибитор активин-рецептор-подобной киназы 5, включающий соединение по любому из пп. 1-3 или его фармацевтически приемлемую соль.
9. Терапевтическое лекарственное средство для лечения рака, ассоциированного с активин-рецептор-подобной киназой 5, включающее соединение по любому из пп. 1-3 или его фармацевтически приемлемую соль.
10. Терапевтическое лекарственное средство по п. 9, где рак представляет собой опухоль головного мозга, рак печени, рак мочевого пузыря, миелодиспластические синдромы, рак толстой кишки или рак поджелудочной железы.
11. Терапевтическое лекарственное средство для лечения дистрофии роговицы, ассоциированной с активин-рецептор-подобной киназой 5, включающее соединение по любому из пп. 1-3 или его фармацевтически приемлемую соль.
12. Терапевтическое лекарственное средство для лечения андрогенетической алопеции, ассоциированной с активин-рецептор-подобной киназой 5, включающее соединение по любому из пп. 1, 2 и 4-6 или его фармацевтически приемлемую соль.
13. Ингибитор казеинкиназы 1δ и активин-рецептор-подобной киназы 5, включающий соединение по любому из пп. 1-3 или его фармацевтически приемлемую соль.
| US 20030166633 A1, 04.09.2003 | |||
| WO 2009047163 A1, 16.04.2009 | |||
| WO 2008071605 A2, 19.06.2008 | |||
| WO 2012080727 A2, 21.06.2012 | |||
| WO 2016149756 A1, 29.09.2016 | |||
| 2-ПИРИДИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ КАК ИНГИБИТОРЫ РЕЦЕПТОРОВ ALK5 И/ИЛИ ALK4 | 2004 |
|
RU2348626C2 |
