ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к 7,7-дифторпроизводному простагландина I2, в котором карбоксигруппа в положении C-1 простагландина (далее по тексту именуемого PG) заменена на тетразольную группу, и в положении C-7 PG находятся два атома фтора, или к его фармацевтически приемлемой соли, его фармацевтической композиции и их применению в качестве лекарственных средств. Более конкретно, настоящее изобретение относится к производному 7,7-дифторпростагландина I2, которое является агонистом EP4, применимому для профилактики или лечения иммунных заболеваний, сердечно-сосудистых заболеваний, сердечных заболеваний, заболеваний дыхательных путей, глазных заболеваний, почечных заболеваний, заболеваний печени, заболеваний костей, заболеваний пищеварительного тракта, неврологических заболеваний, кожных заболеваний и т.п.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Каждый из природных PG связывается со своими специфическими рецепторами и демонстрирует характерные эффекты. Рецепторы для каждого из простагландинов PGI2, PGE2, PGD2, PGF2α и тромбоксана A2 (TXA2) именуются, соответственно, IP, EP, DP, FP и TP. Кроме того рецептор EP дополнительно имеет четыре подтипа, а именно EP1, EP2, EP3 и EP4. Перечисленные рецепторы PG демонстрируют различные модели экспрессии в органах и клетках, и даже если разные рецепторы экспрессируются в одной и той же клетке, оказываемые ими действия различаются.
Хотя производные природных PG испытывают влияние исходного углеродного скелета, они связываются с различными рецепторами, в соответствии с изменениями в их структурах (непатентные документы 1 и 2).
Производные PG, включающие тетразольную группу вместо карбоксигруппы в положении C1 простагландина, были описаны в указанных ниже патентных документах 1-4, непатентном документе 2 и т.п. Кроме того, были описаны 7,7-дифторсодержащие аналоги PGI2 и способы их получения (патентные документы 5 и 6). Помимо этого, сообщалось, что 7,7-дифторсодержащие аналоги PGI2 применимы в качестве профилактических или терапевтических агентов при сердечно-сосудистых заболеваниях (патентный документ 5). 7,7-Дифторсодержащие аналоги PGI2 не только прочно связываются с IP, но также слабо связываются с EP1-4 (непатентные документы 4 и 5). Однако не были опубликованы сообщения о селективном агонисте EP4, который является одним из 7,7-дифторсодержащих аналогов PGI2, демонстрирует слабое сродство к связыванию с IP, EP1, EP2 и EP3, и прочно и селективно связывается только с EP4.
EP4 экспрессируется в иммунных клетках, воспалительных клетках, органах пищеварения, кровеносных сосудах, нейронах, глазах, почках, костях и т.п., и агонисты EP4 выявлены и исследованы в качестве лекарственных средств для лечения иммунных заболеваний, заболеваний пищеварительного тракта, сердечно-сосудистых заболеваний, сердечных заболеваний, неврологических заболеваний, глазных заболеваний, почечных заболеваний, заболеваний печени, заболеваний костей и т.п.
Агонисты EP4 ингибируют выработку TNF-α, содействуют выработке IL-10, подавляют воспаление и иммунную реакцию, и, как считается, подходят для профилактики и/или лечения иммунных заболеваний или воспалительных заболеваний, а именно аутоиммунных заболеваний (например, бокового амиотрофического склероза, множественного склероза, синдрома Шегрена, ревматоидного артрита, системной красной волчанки), отторжения трансплантата после пересадки и подобных состояний, астмы, гибели нервных клеток, артрита, поражений легких, легочного фиброза, эмфиземы, бронхита, хронической обструктивной болезни легких, гепатопатии, острого гепатита, нефритов (острого нефрита, хронического нефрита), почечной недостаточности, синдрома системной воспалительной реакции, сепсиса, гемофагоцитарного синдрома, синдрома активации макрофагов, болезни Стилла, болезни Кавасаки, ожогов, системной гранулемы, язвенного колита, болезни Крона, гиперцитокинемии при диализе, множественного отказа органов, шока и псориаза.
Считается, что агонисты EP4 применимы для профилактики и/или лечения артериосклероза, поскольку они подавляют активацию макрофагов (непатентный документ 6).
Считается, что агонисты EP4 применимы в качестве агентов для профилактики и/или лечения стенокардии или инфаркта миокарда, поскольку они оказывают защитное действие против поражения сердца вследствие ишемии-реперфузии (непатентный документ 7).
Считается, что агонисты EP4 применимы также в качестве агентов для профилактики и/или лечения расстройств мозга, вызванных кровоизлиянием в мозг, инфарктом мозга, субарахноидальным кровоизлиянием и т.п., поскольку они также оказывают защитное действие против поражения мозга, вызванного ишемией-реперфузией (непатентный документ 8).
Считается, что агонисты EP4 применимы также в качестве агентов для профилактики и/или лечения поражения печени, вызванного ишемией-реперфузией (непатентный документ 9).
Считается, что агонисты EP4 применимы в качестве агентов для профилактики и/или лечения глаукомы, поскольку они обладают способностью снижать внутриглазное давление (непатентный документ 10).
Считается, что агонисты EP4 применимы также для профилактики и/или лечения гломерулонефрита и диабетического нефрита, поскольку EP4 в большом количестве экспрессируется в почечных клубочках (непатентный документ 11).
Считается, что агонисты EP4 применимы также для профилактики и/или лечения облысения, алопеции и т.п., поскольку EP4 принимает участие в росте и восстановлении волос (непатентный документ 12).
Считается, что агонисты EP4 применимы в качестве агентов, содействующих созреванию шейки матки, поскольку EP4 вовлечен также в созревание шейки матки (непатентный документ 13).
Считается, что агонисты EP4 применимы в качестве агентов для профилактики и/или лечения остеопороза, или в качестве средства, способствующего заживлению перелома костей, поскольку EP4 вовлечен также в процесс формирования костей (непатентные документы 14 и 15).
Поскольку EP4 экспрессируется в кровеносных сосудах, и агонисты EP4 расслабляют кровеносные сосуды и способствуют увеличению кровотока, считается что эти агонисты применимы для профилактики и/или лечения легочной артериальной гипертензии, сужения периферических артерий (облитерирующего артериосклероза и облитерирующего тромбоангиита) и различных симптомов (перемежающейся хромоты с поясничным спинальным стенозом, онемения ног, синдрома Рейно, эректильной дисфункции, геморроя и т.д.), относящихся к нарушениям периферического кровообращения (непатентные документы 16-20).
EP4 экспрессируется в фибробластах, и считается, что агонист EP4 способствует экспрессии основного фактора роста фибробластов и применим для содействия заживлению пролежней и ран (непатентный документ 21).
Сообщалось, что EP4 экспрессируется в улитке уха, и агонист EP4 применим также для профилактики и/или лечения расстройств слуха, вызванных шумом (непатентный документ 22).
Воспалительные явления в пищеварительном тракте наблюдаются в ротовой полости, пищеводе, желудке, тонком кишечнике, толстом кишечнике и анусе, и включают острое и хроническое воспаление. Если слизистый эпителий подвергается физическому или химическому воздействию, или инфицируется бактериями или вирусами, появляется воспаление и возникают эрозии или язвенные поражения в зависимости от масштаба воспаления. Избыточная секреция желудочной кислоты вследствие стресса вызывает гастрит, язву желудка или язву двенадцатиперстной кишки. Кроме того, избыточное потребление алкоголя вызывает застой крови в слизистых оболочках или рефлюкс желудочной кислоты вследствие уменьшения двигательной функции желудка, что приводит к гастриту, язве желудка, язве двенадцатиперстной кишки или воспалению пищевода. Ортопедические пациенты, пациенты с ревматоидным артритом и подобными заболеваниями, которым осуществляется продолжительное введение нестероидных противовоспалительных средств, страдают от медикаментозной язвы желудка и двенадцатиперстной кишки. Кроме того, у раковых больных под действием радиационной терапии развивается радиационный энтерит или медикаментозный энтерит в результате введения противораковых лекарственных средств. Помимо этого, у пациентов, инфицированных туберкулезом, амебной дизентерией и т.п. развиваются инфекционные энтерогастриты, например, кишечный туберкулез и амебный колит. Кроме того, вследствие ишемии, возникающей из-за затруднения кровотока, развивается ишемический энтерит и подобные заболевания. В случае нарушения иммунитета у пациентов с воспалительным заболеванием пищеварительного тракта, даже при устранении причины, нарушается восстановление пораженного органа и состояние становится хроническим. Из числа указанных воспалительных заболеваний пищеварительного тракта, воспалительными заболеваниями кишечника в широком смысле именуются заболевания, включающие воспаление кишечника.
С другой стороны, существуют воспалительные заболевания кишечника неизвестной природы. Язвенный колит и болезнь Крона являются двумя хорошо известными заболеваниями, которые представляют собой воспалительные заболевания кишечника в узком смысле. Кроме того, эта группа заболеваний включают аналогичные заболевания, например, кишечную болезнь Бехчета и простую язву. Они представляют собой трудноизлечимые хронические желудочно-кишечные заболевания, сопровождающиеся многократными ремиссиями и рецидивами, причем основной причиной этих заболеваний считается ослабление защиты кишечного эпителия или аномальная иммунная реакция кишечника против кишечных бактерий, проникающих в ткани кишечника.
Язвенный колит представляет собой хроническое заболевание толстой кишки, при котором непрерывно образуются эрозии и язвы слизистой оболочки толстого кишечника, начиная от прямой кишки, и симптомы этого заболевания включают боль в животе, диарею, кровянистый стул, повышенную температуру и т.п. С другой стороны, при болезни Крона поражение может возникать на любом участке пищеварительного тракта от полости рта до толстого кишечника и ануса. Это заболевание характеризуется дискретными продольными язвами и синдромом «булыжной мостовой» в желудочно-кишечном тракте, и его симптомы включают боль в животе, диарею, повышенную температуру, плохое усвоение питательных веществ из-за нарушения всасывания питательных веществ в кишечнике, анемию и т.п.
Для профилактики и/или лечения воспаления при воспалительных заболеваниях пищеварительного тракта, если его причина известна, эту причину устраняют или подавляют. Например, против воспаления при гастрите, язве желудка, язве двенадцатиперстной кишки и т.п. применяют антациды, антихолинергические агенты, антагонисты гистаминового рецептора H2, ингибиторы протонного насоса и т.п. с целью подавления секреции и действия желудочной кислоты. В других случаях, производные PGE и подобные средства применяются для пополнения PGE2 при воспалении, вызванном нестероидными противовоспалительными средствами, которые ингибируют выработку PGE2. Однако производные PGI2 не применяются.
С другой стороны профилактика или лечение воспалительных заболеваний кишечника в узком смысле включает лекарственную терапию, пищевую терапию (диету) и хирургическое лечение. Для лекарственной терапии применяются препараты 5-аминосалициловой кислоты (пентаза, салазопирин), стероиды (преднизолон), иммунодепрессанты (азатиопурин, меркаптопурин и такролимус), антитела против TNF-α (инфликсимаб) и т.п. Недавно появилось сообщение, что агонисты EP4 эффективны при воспалительных заболеваниях кишечника (непатентные документы 23-25).
Кроме того, поскольку EP4 также принимает участие в механизме защиты слизистой оболочки, считается, что агонисты EP4 применимы для профилактики и/или лечения поражений желудочно-кишечного тракта, например, язвы желудка, язвы двенадцатиперстной кишки и т.п., а также стоматита (непатентный документ 26).
Документы известного уровня техники
Патентные документы:
Патентный документ 1: DE 2405255
Патентный документ 2: WO 03/103664
Патентный документ 3: WO 00/24727
Патентный документ 4: USP No.7402605
Патентный документ 5: JP-A-7-330752
Патентный документ 6: JP-A-2004-256547
Непатентные документы:
Непатентный документ 1: Biochim. Biophys. Acta, 1483: 285-293 (2000).
Непатентный документ 2: Br. J. Pharmacol., 122: 217-224 (1997).
Непатентный документ 3: J. Med. Chem., 22: 1340-1346 (1979).
Непатентный документ 4: Prostaglandins, 53: 83-90 (1997).
Непатентный документ 5: Br. J. Pharmacol., 134: 313-324 (2001).
Непатентный документ 6: J. Biol. Chem., 283: 9692-9703 (2008).
Непатентный документ 7: Cardiovasc. Res., 81: 123-132 (2009).
Непатентный документ 8: Neurosci. Lett., 438: 210-215 (2008).
Непатентный документ 9: Transplant. Proc., 37: 422-424 (2005).
Непатентный документ 10: Exp. Eye Res., 89: 608-617 (2009).
Непатентный документ 11: Kidney Int., 70: 1099-1106 (2006).
Непатентный документ 12: Biochem. Biophys. Res. Commun., 290: 696-700 (2002).
Непатентный документ 13: Biol. Reprod., 75: 297-305 (2006).
Непатентный документ 14: Proc. Natl. Acad. Sci. U S A., 99: 4580-4585 (2002).
Непатентный документ 15: Expert Opin. Investig Drugs., 18: 746-766 (2009).
Непатентный документ 16: Hypertension, 50: 525-530 (2007).
Непатентный документ 17: Br. J. Pharmacol., 154: 1631-1639 (2008)
Непатентный документ 18: Am. J. Respir. Crit. Care Med., 178: 188-196 (2008).
Непатентный документ 19: Spine, 31: 869-872 (2006).
Непатентный документ 20: Br. J. Pharmacol., 136: 23-30 (2002)
Непатентный документ 21: Kobe J. Med. Sci., 47: 35-45 (2001).
Непатентный документ 22: Neuroscience, 160: 813-819 (2009).
Непатентный документ 23: J. Clin Invest., 109: 883-893 (2002).
Непатентный документ 24: Scand. J. Immunol., 56: 66-75 (2002).
Непатентный документ 25: J. Pharmacol. Exp. Ther., 320: 22-28 (2007).
Непатентный документ 26: World J. Gastroenterol., 15: 5149-5156 (2009).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Проблемы, которые предполагалось решить настоящим изобретением
Цель настоящего изобретения заключалась в разработке соединения, которое является новым производным простагландина I2, обладает превосходной метаболической устойчивостью и селективно связывается с определенным рецептором простагландина.
Средства решения указанных проблем
В попытке решить указанные проблемы, авторы настоящего изобретения синтезировали новые аналоги PG с особыми свойствами, вызванными наличием атомов фтора, и провели исследования для выяснения свойств и физиологической активности этих соединений. В результате авторы изобретения обнаружили, что новые 7,7-дифторпроизводные PGI2, в молекуле которых содержатся два атома фтора, и карбоксигруппа в положении C-1 скелета простаноевой кислоты заменена тетразольной группой, обладают отличным фармакологическим действием и свойствами селективных агонистов EP4, что является неожиданным, даже несмотря на то, что указанные соединения являются производными PGI2, при значительно сниженной агонистической активности в отношении IP, которая наблюдается у соединений с карбоксильной группой в положении C-1, и что указанные соединения, благодаря описанным свойствам являются отличными лекарственными средствами, причем обнаружение перечисленных фактов позволило завершить работу над настоящим изобретением. Селективный агонист EP4 может являться активным ингредиентом лекарственного средства с пониженными побочными эффектами, вызванными действием на другие рецепторы.
Насколько известно авторам настоящего изобретения, до сих пор отсутствовали публикации, освещающие примеры синтеза, свойства, физиологическую активность и т.п. аналогов PGI2, в которых атом C-1 PG замещен тетразольной группой, и в положении C-7 PG имеются два атома фтора.
Таким образом, в настоящем изобретении разработаны 7,7-дифторпроизводные PGI2, представленные приведенной ниже формулой (1), которые являются селективными агонистами EP4 (далее по тексту иногда сокращенно именуемые соединениями формулы (1) по настоящему изобретению), их фармацевтически приемлемые соли и лекарственные средства, содержащие упомянутые соединения и их соли в качестве действующего ингредиента, и сущность изобретения можно выразить следующими пунктами.
[1] Соединение, представленное формулой (1):
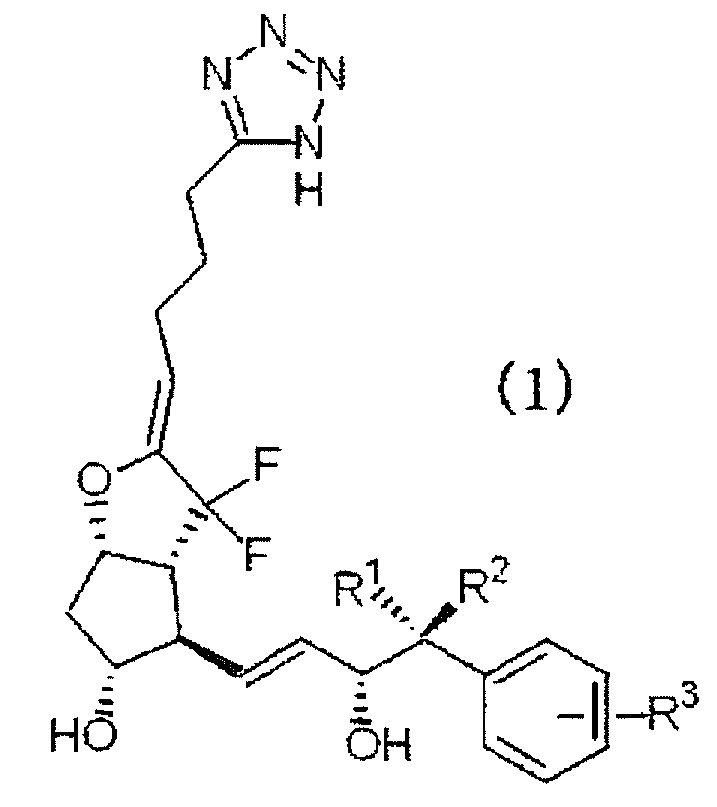
где каждый из заместителей R1 и R2 независимо представляет собой атом водорода или алкильную группу с прямой цепью, включающую от 1 до 3 атомов углерода, и R3 означает атом водорода, алкильную группу, включающую от 1 до 4 атомов углерода, алкоксиалкильную группу, арильную группу, атом галогена или галогеналкильную группу, или его фармацевтически приемлемая соль.
[2] Соединение по п. [1], где R1 означает метильную группу, или его фармацевтически приемлемая соль.
[3] Соединение по п. [1] или [2], где R3 означает метильную группу, или его фармацевтически приемлемая соль.
[4] Соединение по любому из пп. [1]-[3], где R2 является атомом водорода, или его фармацевтически приемлемая соль.
[5] Соединение по любому из пп. [1]-[4], где R1 является метильной группой, и R2 является атомом водорода, или его фармацевтически приемлемая соль.
[6] Соединение по любому из пп. [1]-[5], где R3 является м-метильной группой, или его фармацевтически приемлемая соль.
[7] Соединение по п. [1], где R1 является метильной группой, R2 означает атом водорода, и R3 является метильной группой, или его фармацевтически приемлемая соль.
[8] Соединение по п. [1], где R1 является атомом водорода, R2 означает метильную группу, и R3 является метильной группой, или его фармацевтически приемлемая соль.
[9] 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутан или его фармацевтически приемлемая соль.
[10] 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутан или его фармацевтически приемлемая соль.
[11] 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4S)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутан или его фармацевтически приемлемая соль.
[12] Лекарственное средство, содержащее в качестве действующего ингредиента соединение по любому из пп.[1]-[11] или его фармацевтически приемлемую соль.
[13] Лекарственное средство для профилактики или лечения заболевания пищеварительного тракта, содержащее в качестве действующего ингредиента соединение по любому из пп. [1]-[11] или его фармацевтически приемлемую соль.
[14] Лекарственное средство по п. [13], где заболевание пищеварительного тракта является воспалительным заболеванием или язвенной болезнью пищеварительного тракта.
[15] Лекарственное средство по п. [14], где воспалительное заболевание пищеварительного тракта является воспалительным заболеванием кишечника.
[16] Лекарственное средство по п. [15], где воспалительное заболевание кишечника является язвенным колитом или болезнью Крона.
[17] Лекарственное средство по п. [15], где воспалительное заболевание кишечника является кишечной болезнью Бехчета или простой язвой.
[18] Лекарственное средство по п. [14], где язвенное заболевание пищеварительного тракта представляет собой эзофагит, язву пищевода, гастрит или язву желудка.
[19] Лекарственное средство по п. [18], где гастрит или язва желудка представляют собой медикаментозный гастрит или язву желудка.
[20] Лекарственное средство по п. [19], где медикаментозный гастрит или язва желудка вызваны нестероидным противовоспалительным средством.
[21] Лекарственное средство по п. [18], где гастрит или язва желудка вызваны алкоголем.
[22] Лекарственное средство по п. [14], где язвенное заболевание пищеварительного тракта представляет собой язву тонкого кишечника.
[23] Лекарственное средство по п. [22], где язва тонкого кишечника представляет собой медикаментозную язву тонкого кишечника.
[24] Лекарственное средство по п. [23], где медикаментозная язва тонкого кишечника вызвана нестероидным противовоспалительным средством.
[25] Лекарственное средство по п. [22], где язва тонкого кишечника вызвана алкоголем.
[26] Агонист EP4, содержащий соединение по любому из пп. [1]-[11] или его фармацевтически приемлемую соль.
[27] Лекарственное средство, содержащее агонист EP4 по п.[26] в качестве действующего ингредиента.
[28] Лекарственное средство по п. [27] для профилактики или лечения заболевания, в которое вовлечен EP4.
[29] Лекарственное средство по п. [28] для профилактики или лечения заболевания, симптомы которого могут быть ослаблены действием селективного агониста EP4.
[30] Лекарственное средство по п. [29], где заболевание, симптомы которого могут быть ослаблены действием селективного агониста EP4, представляет собой иммунное заболевание, сердечно-сосудистое заболевание, сердечное заболевание, респираторное заболевание, глазное заболевание, почечное заболевание, заболевание печени, заболевание костей, заболевание пищеварительного тракта, неврологическое заболевание или кожное заболевание.
[31] Лекарственное средство по п. [30], где иммунное заболевание представляет собой боковой амиотрофический склероз, множественный склероз, синдром Шегрена, ревматоидный артрит, системную красную волчанку, отторжение трансплантата после пересадки, артрит, синдром системной воспалительной реакции, сепсис, гемофагоцитарный синдром, синдром активации макрофагов, болезнь Стилла, болезнь Кавасаки, гиперцитокинемию при диализе, множественный отказ органов, шок или псориаз.
[32] Лекарственное средство по п. [30], где сердечно-сосудистое или сердечное заболевание представляет собой артериосклероз, стенокардию, инфаркт миокарда, расстройство мозга, вызванное кровоизлиянием в мозг, расстройство мозга, вызванное инфарктом мозга, расстройство мозга, вызванное субарахноидальным кровоизлиянием, легочную артериальную гипертензию, сужение периферических артерий (облитерирующий артериосклероз и облитерирующий тромбоангиит) или различные симптомы, относящиеся к затруднениям периферического кровообращения (перемежающуюся хромоту или онемение ног, вызванные поясничным спинальным стенозом, синдром Рейно, эректильную дисфункцию, геморрой и т.д.).
[33] Лекарственное средство по п. [30], где респираторное заболевание представляет собой астму, травму легких, легочный фиброз, эмфизему, бронхит или хроническую обструктивную болезнь легких.
[34] Лекарственное средство по п. [30], где глазное заболевание представляет собой глаукому или глазную гипертензию.
[35] Лекарственное средство по п. [30], где почечное заболевание представляет собой гломерулонефрит, диабетическую нефропатию, IgA нефропатию или поражение почек, вызванное ишемией-реперфузией.
[36] Лекарственное средство по п. [30], где заболевание печени представляет собой гепатит, гепатопатию или поражение печени, вызванное ишемией-реперфузией.
[37] Лекарственное средство по п. [30], где заболевание костей представляет собой остеопороз, перелом кости или фазу послеоперационного восстановления после остеотомии.
[38] Лекарственное средство по п. [30], где неврологическое заболевание представляет собой гибель нервных клеток.
[39] Лекарственное средство по п. [30], где заболевание кожи представляет собой пролежень или рану.
[40] Лекарственное средство по п. [28], где заболевание, в которое вовлечен EP4, представляет собой заболевание, выбранное из группы, состоящей из облысения, алопеции, нарушения созревания шейки матки и расстройств слуха.
Эффекты настоящего изобретения
Новые 7,7-дифторпроизводные PGI2, разработанные в настоящем изобретении, позволяют получать лекарственное средство, концентрация которого в крови сохраняется в течение длительного времени, которое демонстрирует фармакологическое действие при парентеральном или пероральном введении, и которое предназначено для профилактики или лечения воспаления пищеварительного тракта или появления диареи или кровянистого стула при воспалительном заболевании кишечника, или для профилактики или лечения гастрита или язвы при язве желудка, язвы тонкого кишечника и т.п. Кроме того, благодаря проявлению агонизма в отношении EP4, можно получить лекарственное средство для профилактики или лечения иммунных заболеваний, сердечно-сосудистых заболеваний, сердечных заболеваний, респираторных заболеваний, глазных заболеваний, почечных заболеваний, заболеваний печени, заболеваний костей, заболеваний пищеварительного тракта, неврологических заболеваний, заболеваний кожи и т.п. Также ожидается, что в клинических случаях соединение по настоящему изобретению проявит эффективность против группы заболеваний, при которых могут иметь эффект агонисты EP4, при том, что снижается опасность побочных эффектов, например, кровотечения, гипотензии, сильного сердцебиения и прилива крови к лицу, благодаря ослаблению свойств агониста IP в кровеносной системе. В частности, соединения по настоящему изобретению благодаря проявлению агонизма в отношении EP4, эффективны при воспалениях пищеварительного тракта, связанных с иммунитетом, медикаментозных поражениях слизистой оболочки пищеварительного тракта, поражениях пищеварительного тракта и задержке выздоровления из-за нарушения регенеративных процессов в слизистой оболочке, при глазных заболеваниях, заболеваниях почек и заболеваниях печени. Конкретно, соединения по настоящему изобретению применимы для лечения воспалительных заболеваний кишечника, таких как язвенный колит и болезнь Крона, алкогольного гастрита, язвы желудка и язвы тонкого кишечника, нефрита, глаукомы, глазной гипертензии, гепатита и т.п.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1A показано влияние на кровяное давление у мышей.
На фиг.1B показано влияние на частоту сердечных сокращений у мышей.
На фиг.2A показано влияние на аномальный стул в мышиной (BALB/c) модели колита, вызванного DSS.
На фиг.2B показано влияние на укорочение толстой кишки в мышиной (BALB/c) модели колита, вызванного DSS.
На фиг.3A показано влияние BPS на аномальный стул в мышиной (C57BL/6) модели колита, вызванного DSS.
На фиг.3B показано влияние на аномальный стул в мышиной (C57BL/6) модели колита, вызванного DSS.
На фиг.3C показано влияние BPS на укорочение толстой кишки в мышиной (C57BL/6) модели колита, вызванного DSS.
На фиг.3D показано влияние на укорочение толстой кишки в мышиной (C57BL/6) модели колита, вызванного DSS.
На фиг.4A показано влияние на аномальный стул в крысиной модели колита, вызванного DSS.
На фиг.4B показано влияние на укорочение толстой кишки в крысиной модели колита, вызванного DSS.
На фиг.4C показано влияние на поражение тканей толстой кишки в крысиной модели колита, вызванного DSS.
На фиг.5 показано влияние на аномальный стул в мышиной модели ремиссии/рецидива колита, вызванного DSS.
На фиг.6A показано влияние на показатели консистенции стула в мышиной модели колита, вызванного введением T-клеток.
На фиг.6B показано влияние на показатели скрытой крови в кале в мышиной модели колита, вызванного введением T-клеток.
На фиг.6C показано влияние на показатели уменьшения массы тела в мышиной модели колита, вызванного введением T-клеток.
На фиг.6D показано влияние на сумму баллов DAI в мышиной модели колита, вызванного введением T-клеток.
На фиг.7 показано влияние на язву желудка в крысиной модели поражения слизистой оболочки желудка, вызванного этанолом.
На фиг.8 показано влияние на язву тонкого кишечника в крысиной модели поражения тонкого кишечника, вызванного индометацином.
На фиг.9A показано влияние на объем мочи в крысиной модели гломерулонефрита, вызванного антителами против Thy-1.
На фиг.9B показано влияние на количество белка в моче в крысиной модели гломерулонефрита, вызванного антителами против Thy-1.
На фиг.9C показано влияние на относительную массу почек в крысиной модели гломерулонефрита, вызванного антителами против Thy-1.
На фиг.9D показано влияние на почечную гистопатологию (общее количество клубочковых клеток) в крысиной модели гломерулонефрита, вызванного антителами против Thy-1.
На фиг.9E показано влияние на почечную гистопатологию (мезангиальную область) в крысиной модели гломерулонефрита, вызванного антителами против Thy-1.
На фиг.9F показано влияние на почечную гистопатологию (количество PCNA-положительных клубочковых клеток) в крысиной модели гломерулонефрита, вызванного антителами против Thy-1.
На фиг.10 показано влияние на внутриглазное давление у кролика.
На фиг.11 показано профилактическое действие в мышиной модели гепатита, вызванного конканавалином A.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Определения
В настоящем описании термин «селективный агонист EP4» означает соединение, которое демонстрирует слабое агонистическое действие (фармакологическую активность) в отношении рецептора PGI2 (IP) по отношению к агонистическому действию, как правило, обнаруживаемому аналогами PGI2, и обладает значительно большим агонистическим действием в отношении рецептора PGE2 подтипа EP4 по сравнению с действием в отношении IP. Агонистическое действие в отношении EP4 можно измерить способом определения агонистической активности, описанным ниже в примере 19. Агонистическое действие в отношении IP можно измерить согласно способу, описанному в примере 20. Произвести оценку того, является ли соединение селективным агонистом EP4, можно путем определения отношения значений констант ингибирования связывания Ki для EP4 и IP (отношения IP/EP4) для одних и тех же образцов в соответствии со способом, описанным в примере 18. Примеры селективных агонистов EP4 включают соединения, имеющие значение указанного соотношения не менее 5, предпочтительно, не менее 10, более предпочтительно, не менее 50, наиболее предпочтительно, не менее 100.
Термин «производное простагландина I2» в настоящем описании означает соединение, имеющее основную структуру PGI2 природного типа и подвергшееся структурной модификации в соответствии с известными способами органической химии. Далее по тексту будет подробно описано соединение по настоящему изобретению.
Определение соединения по настоящему изобретению
В системе обозначений для описания соединений по настоящей заявке, числа, используемые для указания положений в PG скелете, соответствуют номерам положений в скелете простаноевой кислоты. В настоящем описании группа, в которой замещен атом водорода алкильной группы также показана как замещенная алкильная группа. То же самое относится и к другим группам.
Кроме того «низшая» органическая группа, например, алкильная группа и т.п. означает, что число атомов углерода в этой группе составляет 1-6. Предпочтительно, число атомов углерода в «низшей» органической группе составляет 1-4.
«Алкильная группа» может иметь линейную или разветвленную цепь. Если не указано иное, алкильная группа, предпочтительно, является низшей алкильной группой, включающей 1-6 атомов углерода, и особенно предпочтительной является низшая алкильная группа, включающая 1-4 атомов углерода. Примеры алкильной группы включают метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу, изобутильную группу, втор-бутильную группу, терт-бутильную группу, пентильную группу, гексильную группу и т.п.
«Алкоксигруппа», предпочтительно, представляет собой низшую алкоксигруппу, включающую от 1 до 6 атомов углерода, особенно предпочтительно, алкоксигруппу, включающую от 1 до 4 атомов углерода. Алкоксигруппа может иметь линейную или разветвленную цепь. Примеры алкоксигруппы включают метоксигруппу, этоксигруппу, пропоксигруппу, бутоксигруппу и т.п.
«Алкоксиалкильная группа» представляет собой алкильную группу, замещенную алкоксигруппой. Алкоксигруппа алкоксиалкильной группы, предпочтительно, представляет собой низшую алкоксигруппу, включающую от 1 до 4 атомов углерода, и алкильная группа алкоксиалкильной группы, предпочтительно, представляет собой низшую алкильную группу, включающую от 1 до 4 атомов углерода. Алкоксиалкильная группа, предпочтительно, является низшей алкоксиалкильной группой (т.е. общее количество атомов углерода в алкоксиалкильной группе составляет от 1 до 6), более предпочтительно, низшей алкоксиалкильной группой, включающей от 1 до 4 атомов углерода. Примеры алкоксиалкильной группы включают метоксиметильную группу, этоксиметильную группу, пропоксиметильную группу, этоксиэтильную группу и т.п.
«Арильная группа» представляет собой одновалентную ароматическую углеводородную группу, необязательно имеющую заместитель(и). В качестве арильной группы, не имеющей заместителей, предпочтительной является фенильная группа.
Под «замещенной арильной группой» (арильной группой, имеющей заместители), предпочтительно, подразумевается арильная группа, в которой один или несколько атомов водорода замещены низшей алкильной группой, атомом галогена, галогензамещенной (низшей алкильной) группой, низшей алкоксигруппой и т.п. Предпочтительные примеры замещенной арильной группы включают замещенную фенильную группу, конкретные примеры которой включают моногалогенфенильную группу (например, хлорфенильную группу, фторфенильную группу, бромфенильную группу и т.д.), фенильную группу, замещенную галогензамещенным низшим алкилом (например, трифторметилфенильную группу и т.д.), а также фенильную группу, замещенную низшей алкоксигруппой (например, метоксифенильную группу, этоксифенильную группу и т.д.).
«Атом галогена» представляет собой атом фтора, атом хлора, атом брома или атом йода.
«Галогеналкильная группа» является алкильной группой, в которой один или несколько атомов водорода замещены атомами галогена, и предпочтительной является низшая галогеналкильная группа, включающая от 1 до 6 атомов углерода. Примеры галогеналкильной группы включают фторметильную группу, дифторметильную группу, трифторметильную группу, трифторэтильную группу, пентафторэтильную группу, хлорметильную группу, бромметильную группу и т.п.
В качестве соединения формулы (1) по настоящему изобретению с точки зрения фармакологической активности и физических свойств предпочтительны соединения с описанными ниже заместителями.
Так, например, каждый из заместителей R1 и R2 независимо представляет собой атом водорода или алкильную группу с линейной цепью с числом атомов углерода от 1 до 3, и каждый из них независимо, предпочтительно, представляет собой атом водорода или метильную группу. Особенно предпочтительно, один из заместителей R1 и R2 представляет собой атом водорода, и другой является метильной группой.
R3 является атомом водорода, алкильной группой с числом атомов углерода от 1 до 4, алкоксиалкильной группой, арильной группой, атомом галогена или галогеналкильной группой, и предпочтительными являются атом водорода, алкильная группа с числом атомов углерода от 1 до 4, низшая алкоксиалкильная группа, например, метоксиметильная группа и т.п., атом галогена, например, атом хлора, атом фтора и т.п., или низшая галогеналкильная группа, например, низшая фторалкильная группа и т.п. В частности, предпочтительными являются атом водорода, алкильная группа с числом атомов углерода от 1 до 4, атом хлора или галогеналкил с числом атомов углерода от 1 до 4. В качестве алкильной группы с числом атомов углерода от 1 до 4, предпочтительными являются метильная группа и этильная группа, и в качестве галогеналкильной группы с числом атомов углерода от 1 до 4 предпочтительной является трифторметильная группа.
В качестве заместителя R3 наиболее предпочтительны атом водорода, метильная группа или трифторметильная группа.
Кроме того, группа R3 может быть замещена по любому из орто-(о), мета-(м) и пара-(п) положений, по отношению к точке замещения основной цепи простагландинового скелета бензольным циклом. R3, особенно предпочтительно, замещен по мета-(м) положению.
Варианты предпочтительных соединений по настоящему изобретению
Кроме того, предпочтительными являются следующие комбинации R1, R2 и R3 в соединении (1) по настоящему изобретению.
R1 означает атом водорода, R2 означает атом водорода и R3 означает атом водорода.
R1 означает атом водорода, R2 означает атом водорода и R3 означает метильную группу.
R1 означает атом водорода, R2 означает атом водорода и R3 означает атом хлора.
R1 означает атом водорода, R2 означает атом водорода и R3 означает трифторметильную группу.
R1 означает метильную группу, R2 означает атом водорода и R3 означает атом водорода.
R1 означает метильную группу, R2 означает атом водорода и R3 означает метильную группу.
R1 означает метильную группу, R2 означает атом водорода и R3 означает атом хлора.
R1 означает метильную группу, R2 означает атом водорода и R3 означает трифторметильную группу.
R1 означает атом водорода, R2 означает метильную группу и R3 означает атом водорода.
R1 означает атом водорода, R2 означает метильную группу и R3 означает метильную группу.
R1 означает атом водорода, R2 означает метильную группу и R3 означает атом хлора.
R1 означает атом водорода, R2 означает метильную группу и R3 означает трифторметильную группу.
R1 означает метильную группу, R2 означает метильную группу и R3 означает атом водорода.
R1 означает метильную группу, R2 означает метильную группу и R3 означает метильную группу.
R1 означает метильную группу, R2 означает метильную группу и R3 означает атом хлора.
R1 означает метильную группу, R2 означает метильную группу и R3 означает трифторметильную группу.
Кроме того, из числа перечисленных выше комбинаций предпочтительными являются следующие, поскольку они обладают высоким агонистическим действием в отношении рецептора EP4.
R1 означает метильную группу, R2 означает атом водорода и R3 означает метильную группу.
R1 означает атом водорода, R2 означает метильную группу и R3 означает метильную группу. Кроме того, наиболее предпочтительными комбинациями являются следующие.
R1 означает метильную группу, R2 означает атом водорода и R3 означает м-метильную группу.
R1 означает атом водорода, R2 означает метильную группу и R3 означает м-метильную группу.
Способ получения соединений (1) по настоящему изобретению
Соединения (1) по настоящему изобретению могут быть получены, например, на основе способов, описанных в JP-A-07-324081 и JP-A-08-217772, относящихся к изобретениям, сделанным авторами настоящего изобретения. Например, используя лактон Кори в качестве исходного вещества, на первом этапе вводят ω-цепь и путем фторирования полученный лактон превращают в лактон Кори, содержащий два атома фтора в ω-цепи. Затем вводят фрагмент α-цепи реакцией присоединения с металлорганическим реагентом, включающим тетразольную группу в концевом положении и реакцией дегидратации, или реакцией Виттига с использованием соли фосфония, с тетразольной группой в концевом положении, и т.п. и при необходимости снимают защиту с гидроксильной группы, в результате чего может быть синтезировано соединение формулы (1).
В качестве альтернативы дифторзамещенный лактон Кори получают из лактона Кори фторированием. Затем вводят фрагмент α-цепи с помощью реакции присоединения металлоорганического реагента, включающего тетразольную группу в концевом положении, и реакции дегидратации, или с помощью реакции Виттига с использованием соли фосфония с тетразольной группой в концевом положении, и т.п., вводят ω-цепь и, если это необходимо, снимают защиту гидроксильной группы, что позволяет получить соединение формулы (1).
В качестве альтернативы, соединение формулы (1) можно также получить превращением карбоксильной группы производного карбоновой кислоты, описанного в JP-A-07-324081 в цианогруппу с последующим превращением полученного соединения в тетразольное производное.
Далее будут конкретно описаны типовые способы, относящиеся к перечисленным выше способам получения, с использованием следующих химических формул.
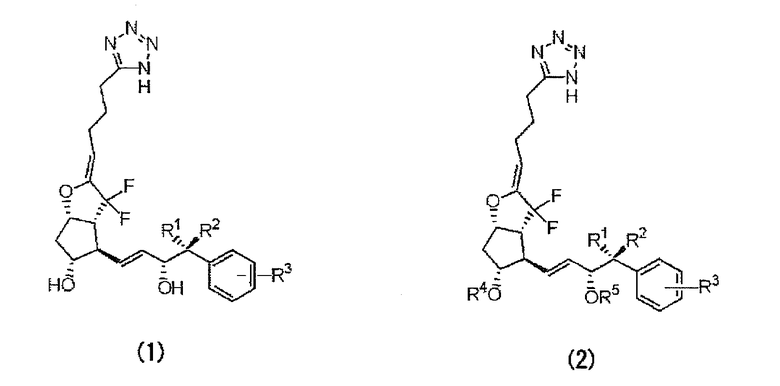
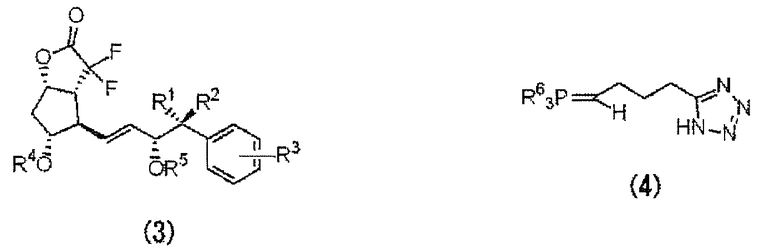
Например, при использовании лактона Кори (7) в качестве исходного вещества, на первой стадии водят ω-цепь, полученное производное лактона Кори, содержащее ω-цепь (6), подвергают фторированию, получая дифторпроизводное лактона Кори, включающего ω-цепь (3), содержащее два атома фтора в α-положении относительно карбонильной группы. Затем полученное производное дифторлактона (3) вводят в реакцию с производным фосфорана (4) для введения в молекулу α-цепи, что позволяет получить производное PGI2 с защищенной гидроксильной группой (2). Удаляют защитную группу гидроксила, получая соединение формулы (1) по настоящему изобретению.
Производное фосфорана формулы (4) можно получить из производного фосфониевой соли (5).
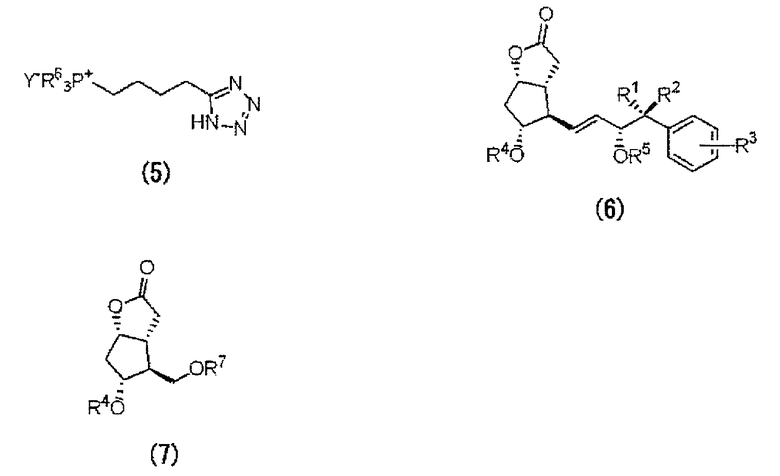
За исключением случаев, когда R1-R3 являются некоторыми определенными заместителями, упомянутое выше производное лактона (6) представляет собой известное соединение. Упомянутые выше новые производные лактона (6), в которых R1-R3 являются определенными заместителями, можно получать способом, аналогичным способу получения известных производных лактона (6). Например, новые производные лактона (6) можно получить взаимодействием диэфира 3-арил-2-оксиалкилфосфоновой кислоты с лактоном Кори, включающим формильную группу. В этом случае алкильные цепи алкилфосфоновой кислоты содержат не менее 3 атомов углерода.
Каждый из заместителей R4, R5 и R7 независимо представляет собой группу, защищающую гидроксил. R4, R5 и R7 могут являться одинаковыми защитными группами. В качестве защитной группы можно применять защитные группы для гидроксила, описанные в “Shin Jikken Kagaku Koza (New Courses in Experimental Chemistry) 14, synthesis and reaction of organic compound (V)” (Maruzen Company, Limited), “Protective Groups in Organic synthesis” (by T.W.Greene, J.Wiley & Sons) и т.п. Конкретно можно упомянуть триорганосилильную группу, алкоксиалкильную группу, одновалентную группу с циклической эфирной структурой и т.п. Предпочтительной триорганосилильной группой является силильная группа, в которой с атомом кремния связаны упомянутые 3 заместителя, выбранные из алкильных групп, арильных групп, аралкильных групп и алкоксигрупп, и группа, в которой с атомом кремния связаны три низшие алкильные группы или арильные группы является особенно предпочтительной. В качестве конкретных примеров защитных групп предпочтительными являются тетрагидропиранильная группа, трет-бутилдиметилсилильная группа, трет-бутилдифенилсилильная группа, триэтилсилильная группа, трифенилсилильная группа или триизопропилсилильная группа и т.п. В частности, предпочтительными являются тетрагидропиранильная группа, трет-бутилдиметилсилильная группа, трет-бутилдифенилсилильная группа и т.п.
Указанную группу, защищающую гидроксил, можно легко удалить. Способы снятия защиты защищенной гидроксильной группы могут представлять собой традиционные способы. Например, можно применять способы, описанные в “Shin Jikken Kagaku Koza (New Courses in Experimental Chemistry) 14, synthesis and reaction of organic compound (I), (II) и (V)” (Maruzen Company, Limited), “Protective Groups in Organic synthesis” (by T.W.Greene, J.Wiley & Sons) и т.п.
Для превращения производного лактона (6) в производное дифторлактона (3) с помощью реакции фторирования, могут использоваться различные известные способы фторирования. Например, можно применять способ, включающий взаимодействие с различными электрофильными фторирующими агентами в инертном растворителе. Кроме того, можно осуществлять фторирование по способу, описанному в заявках JP-A-07-324081 и JP-A-09-110729, поданных авторами настоящего изобретения.
При реакции фторирования лактона (6), предпочтительно, применяются электрофильные фторирующие агенты. В качестве электрофильного фторирующего агента могут использоваться известные или хорошо известные электрофильные фторирующие агенты. Например, можно упомянуть электрофильные фторирующие агенты, описанные в “Chemistry of fluorine” (Kodansha Scientifics Ltd.) by Tomoya Kitazume, Takashi Ishihara и Takeo Taguchi, и т.п. Конкретно можно упомянуть N-фторсульфониламиды, N-сульфонилимидные производные, ацетилгипофторид, газообразный фтор и т.п.
Электрофильный фторирующий агент, предпочтительно, применяют в присутствии инертного растворителя. В качестве инертных растворителей можно упомянуть эфирные растворители, углеводородные растворители, полярные растворители, их смеси и т.п.
Производное дифторлактона (3), полученное реакцией фторирования, вводят во взаимодействие с производным фосфорана (4), получая производное PGI2 (2), гидроксильная группа которого является защищенной. Производное фосфорана (4) получают из соответствующего производного фосфониевой соли (5) в инертном растворителе в присутствии основания, и полученное производное фосфорана (4), предпочтительно, сразу же без промежуточного выделения используют в реакции Виттига с производным дифторлактона (3). Что касается способов получения производного фосфорана (4) и производного фосфониевой соли (5), можно использовать способы, описанные в DE2242239, DE2405255 и т.п. В качестве заместителя R6 в производном фосфорана (4) или производном фосфониевой соли (5), предпочтительными являются арильные группы, например, фенильная группа, толильная группа и т.п., и фенильная группа является особенно предпочтительной. В качестве инертных растворителей можно упомянуть простые эфиры, углеводороды, полярные растворители, водные растворители, спирты, смеси перечисленных растворителей и т.п.
Группу, защищающую гидроксил, удаляют из производного PGI2 (2), полученного описанным выше способом, получая соединение формулы (1).
Поскольку в структуре соединения формулы (1) по настоящему изобретению имеется асимметрический атом углерода, у этих соединений имеются различные стереоизомеры и оптические изомеры. Настоящее изобретение охватывает все эти стереоизомеры, оптические изомеры и их смеси.
Конкретные примеры соединений формулы (1) по настоящему изобретению включают соединения, представленные приведенной ниже формулой (8).
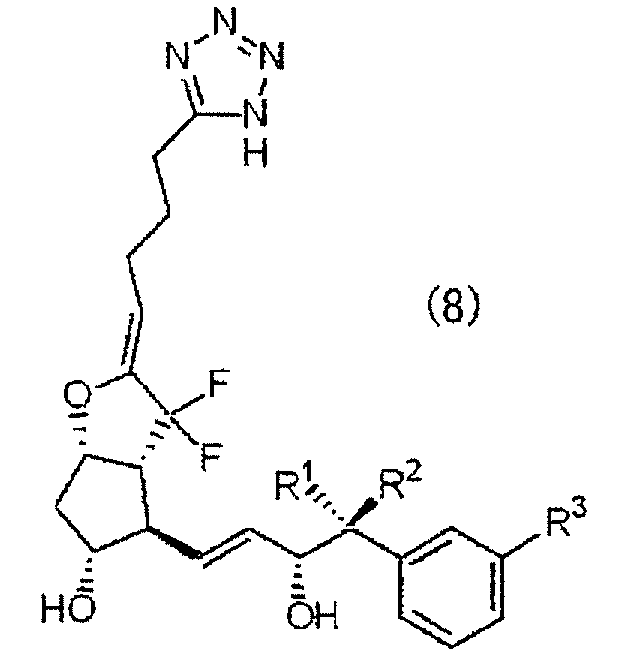
Примеры соединений формулы (1) по настоящему изобретению
Можно упомянуть соединения, в которых заместители R1, R2 и R3 в формуле (8) имеют структуры, показанные в приведенной ниже таблице 1.
Отличительные особенности соединения формулы (1) по настоящему изобретению
Соединение формулы (1) по настоящему изобретению представляет собой производное PGI2, которое с трудом подвергаются метаболизму в организме и обладает повышенной стабильностью. Поскольку карбоксигруппа скелета PG заменена на тетразольную группу, соединение с трудом подвергается метаболизму за счет β-окисления, которое известно в качестве обычного пути метаболизма жирных кислот, в т.ч. простагландинов. Поэтому соединение формулы (1) имеет увеличенное время полужизни в плазме, и эффективная концентрация соединения в плазме может сохраняться в течение более продолжительного времени по сравнению с соединением, несущим на скелете PG карбоксильную группу. Поскольку метаболическая стабильность соединений повышена описанным путем, может быть улучшена биодоступность лекарственного средства.
Соединение формулы (1) по настоящему изобретению или его фармацевтически приемлемая соль проявляют свойства селективного агониста EP4. Примеры предпочтительных соединений формулы (1), демонстрирующих такое действие, представляют собой те же соединения, что и упомянутые выше предпочтительные примеры соединения формулы (1).
Лекарственное средство, содержащее соединение формулы (1) по настоящему изобретению или его фармацевтически приемлемую соль в качестве действующего ингредиента.
Лекарственное средство по настоящему изобретению содержит соединение формулы (1) и/или фармацевтически приемлемую соль соединения формулы (1) и, кроме того, фармацевтически приемлемый носитель и, в некоторых случаях, другие лекарственные компоненты.
Лекарственное средство по настоящему изобретению содержит соединение формулы (1) и/или фармацевтически приемлемую соль соединения формулы (1) или ее гидрат и, кроме того, фармацевтически приемлемый носитель и, в некоторых случаях, другие лекарственные компоненты.
Если профилактический или терапевтический агент по настоящему изобретению вводят пациентам, его дневную дозу меняют в зависимости от возраста и массы тела пациентов, заболевания и его тяжести и т.п. Как правило, желательно вводить 0,0001-10 мг, предпочтительно 0,01-1 мг действующего агента в виде одной или нескольких порций. Например, при пероральном введении, предпочтительно, вводить 0,001-3 мг и, особенно предпочтительно, 0,001-0,5 мг. При внутривенном введении, предпочтительно, применять 0,001-1 мг и, особенно предпочтительно, 0,001-0,1 мг. Дозировку можно менять соответствующим образом в зависимости от заболевания и его состояния. Говоря о схеме введения, продукт для инъекций, включающий соединение по настоящему изобретению, может быть желательно вводить непрерывной капельной инфузией.
Для применения в качестве лекарственного средства, действующий агент по настоящему изобретению можно вводить в организм пероральным путем и парентеральным путем (например, внутрисосудистым (внутривенным, внутриартериальным) путем, подкожным путем, ректальным путем и т.д.). Примеры дозированных форм включают пероральные дозированные формы, например, таблетки, капсулы и сиропы, парентеральные дозированные формы, например, жидкости для инъекции (растворы, эмульсии, суспензии и т.п.), инфузии, суппозитории, назальные препараты, пластыри и ингаляции. Особенно желательно пероральное введение.
Получение упомянутых выше дозированных форм можно осуществить смешиванием соединения формулы (1) по настоящему изобретению или его фармацевтически приемлемой соли с добавками, необходимыми для получения лекарственного состава, например, традиционными носителями, эксципиентами, связующими веществами и стабилизаторами, с превращением этой смеси в желаемый состав стандартными способами. Например, если препарат представляет собой порошок, гранулы, таблетки и т.п. его можно получать с применением любых фармацевтических носителей, предпочтительных для получения твердых дозированных форм, например, эксципиентов, смазывающих средств, дезинтегрирующих средств, связующих средств и т.п.
Этими эксципиентами могут быть, например, инертные разбавители, такие как, карбонат кальция, карбонат натрия, лактоза, фосфат кальция и фосфат натрия; гранулирующие агенты и дезинтегрирующие средства, например, кукурузный крахмал и альгиновая кислота; связующие средства, например, крахмал, желатин и гуммиарабик; и смазывающие средства, например, стеарат магния, стеариновая кислота и тальк. Таблетка может не иметь покрытия или может быть покрыта известными способами для замедления разрушения и абсорбции в желудке и кишечнике, что обеспечивает непрерывное высвобождение в течение более продолжительного периода времени. Например, можно использовать материал, способствующий задержке разрушения, такой как глицерил моностеарат или глицерил дистеарат.
Соединение формулы (1) по настоящему изобретению может быть включено в твердые желатиновые капсулы в виде смеси с твердым инертным разбавителем, например, карбонатом кальция, фосфатом кальция или каолином. В качестве альтернативы, соединение можно включать в состав мягких желатиновых капсул, содержащих смесь с растворителем, который способен смешиваться с водой, например, пропиленгликолем, полиэтиленгликолем и этанолом, или маслами, например, арахисовым маслом, жидким парафином и оливковым маслом.
Если препарат является сиропом или жидкостью, для его получения можно выбрать и применить, например, соответствующие стабилизаторы, суспендирующие агенты, модификаторы, ароматические вещества и т.п. При изготовлении препарата для инъекций, действующий ингредиент растворяют в дистиллированной воде для инъекций, совместно с регулятором pH, например, хлористоводородной кислотой, гидроксидом натрия, лактозой, лактатом натрия, уксусной кислотой, динатрийгидрофосфатом и натрийдигидрофосфатом, и изотоническим агентом, например, хлоридом натрия и глюкозой, и получают препарат для инъекции в асептических условиях. При изготовлении таких препаратов могут применяться неактивные неводные разбавители, например, пропиленгликоль, полиэтиленгликоль, оливковое масло, этанол и полисорбат 80. Кроме того, при получении препарата для инъекций, предназначенного для растворения перед применением, можно добавлять маннит, декстрин, циклодекстрин, желатин и т.п. и подвергать полученную смесь лиофильной сушке в вакууме. Для стабилизации и улучшения доставки лекарственного средства к пораженному участку, помимо перечисленного известными способами можно изготавливать препараты липосом или липидные эмульсии и применять их в качестве препаратов для инъекции.
Кроме того, можно получать ректальные дозированные препараты, применяя основу для суппозиториев, например, масло какао, триглицериды жирных кислот, диглицериды жирных кислот, моноглицериды жирных кислот и полиэтиленгликоль. Кроме того, можно получать мазь для внутриректального введения, добавляя водорастворимую основу, например, полиэтиленгликоль, полипропиленгликоль, глицерин и глицерин-желатин, масляную основу, например, белый вазелин, твердый жир, парафин, жидкий парафин, Plastibase, ланолин и очищенный ланолин и т.п. до достижения подходящей вязкости.
Соединения формулы (1) по настоящему изобретению или их фармацевтически приемлемые соли можно местно наносить на кожу или слизистые оболочки, т.е. осуществлять трансдермальное или трансмукозальное введение. В качестве основных дозированных форм для такого способа введения можно упомянуть гель, гидрогель, лосьон, раствор, крем, мазь, спреи, повязки, пены, пленки, кожные пластыри, облатки, имплантаты, губки, волокнистые материалы, бандажи, микроэмульсии и т.п. В качестве широко применяемых носителей можно упомянуть спирты, воду, минеральное масло, жидкий парафин, белый вазелин, глицерин, полиэтиленгликоль, пропиленгликоль и т.п.
С целью применения в любой из упомянутых выше дозированных форм и улучшения растворимости, скорости растворения, биодоступности и стабильности, соединение формулы (1) по настоящему изобретению может быть смешано с циклодекстрином или его подходящим производным, или растворимым полимером, например, полиэтиленгликольсодержащим полимером. Например, было подтверждено, что комплексы лекарственное средство-циклодекстрин и т.п., как правило, применимы для большинства дозированных форм и путей введения. Можно применять как комплексы включения, так и комплексы, не являющиеся комплексами включения. В качестве другого способа, для непосредственного образования комплексов с лекарственным средством, циклодекстрин можно также применять в виде вспомогательной добавки, т.е. носителя, эксципиента или солюбилизатора. С этой целью, как правило, применяют α-, β- и γ-циклодекстрины и т.п.
Фармацевтически приемлемые соли соединения (1) по настоящему изобретению
Фармацевтически приемлемая соль соединения (1) по настоящему изобретению является солью, образованной тетразольным фрагментом соединения и веществом основного характера, представляющая собой соединение, в котором атом водорода тетразольной группы замещен катионом.
Примеры катиона включают катионы щелочных металлов, например, Na+ и K+, катионы других металлов (отличных от катионов щелочных металлов), например, 1/2 Ca2+, 1/2 Mg2+, 1/2 Zn2+ и 1/3 Al3+, NH4 +, катионы аммония или органических аминов и аминокислот, например, триэтаноламина, диэтаноламина, этаноламина, трометамина, лизина и аргинина и т.п. Предпочтительными катионами являются катионы натрия или калия.
Более конкретно, приемлемой солью является соль, полученная из фармацевтически приемлемого нетоксичного основания, например, неорганического основания и органического основания. В качестве солей, полученных из фармацевтически приемлемого нетоксичного неорганического основания, помимо упомянутых выше солей натрия, калия, кальция, магния, цинка, алюминия, аммония и т.п., можно указать соли лития, меди, железа (III), железа (II), марганца (III), марганца (II) и т.п. Из перечисленных солей предпочтительными являются соли натрия, соли калия, соли кальция, соли магния и соли аммония, причем натриевые соли и калиевые соли являются особенно предпочтительными. Соли, полученные из фармацевтически приемлемых нетоксичных органических оснований включают соли с первичными, вторичными и третичными аминами, замещенными аминами, в т.ч. природными замещенными аминами, циклическими аминами и основными ионообменными смолами. Помимо указанных выше примеров органических аминов и аминокислот, можно упомянуть изопропиламин, диэтиламин, триэтиламин, триметиламин, трипропиламин, этилендиамин, N,N-дибензилэтилендиамин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, морфолин, N-этилморфолин, пиперазин, пиперидин, N-этилпиперидин, бетаин, кофеин, холин, глюкамин, глюкозамин, гистидин, гидрабамин, метилглюкамин, полиаминную смолу, прокаин, пурин, теобромин и т.п.
Применение лекарственного средства, содержащего соединение (1) по настоящему изобретению или его фармацевтически приемлемую соль в качестве действующего ингредиента
Лекарственное средство, содержащее соединение (1) по настоящему изобретению или его фармацевтически приемлемую соль в качестве действующего ингредиента, может применяться при заболеваниях, в развитие которых вовлечен рецептор EP4, предпочтительно, при заболеваниях, симптомы которых могут облегчаться за счет селективного агонистического действия в отношении EP4. Конкретно, такие лекарственные средства применимы при иммунных заболеваниях, заболеваниях пищеварительного тракта, сердечно-сосудистых заболеваниях, сердечных заболеваниях, респираторных заболеваниях, неврологических заболеваниях, глазных заболеваниях, почечных заболеваниях, заболеваниях печени, заболеваниях костей, кожных заболеваниях и т.п.
Иммунные заболевания в контексте настоящего изобретения включают аутоиммунные заболевания, например боковой амиотрофический склероз, множественный склероз, синдром Шегрена, ревматоидный артрит и системную красную волчанку, отторжение трансплантата после пересадки и т.п., и воспалительные заболевания, например, астму, гибель нервных клеток, артрит, травму легких, легочный фиброз, эмфизему, бронхит, хроническую обструктивную болезнь легких, гепатопатию, острый гепатит, нефриты (острый нефрит, хронический нефрит), почечную недостаточность, синдром системной воспалительной реакции, сепсис, гемофагоцитарный синдром, синдром активации макрофагов, болезнь Стилла, болезнь Кавасаки, ожоги, системную гранулему, гиперцитокинемию при диализе, множественный отказ органов, шок и псориаз.
Заболевания пищеварительного тракта в контексте настоящего изобретения включают воспалительные заболевания и язвенные заболевания пищеварительного тракта, которые представляют собой заболевания, вызванные воспалением или образованием язв эпителиальной ткани, ткани слизистых оболочек или подслизистых тканей пищеварительного тракта, или аномальной пролиферацией или дисфункцией слизистого эпителия, и которые вызваны физическими воздействиями, химическими воздействиями, например, действием желудочного сока, действием лекарственных средств, например, нестероидных противовоспалительных средств и стероидов, возникают в результате иммунных и аутоиммунных заболеваний неизвестной этиологии, психических расстройств и т.д.
Воспалительные заболевания пищеварительного тракта включают воспалительные заболевания кишечника, в т.ч. язвенный колит, болезнь Крона, которая представляет собой неспецифичное грануломатозное воспалительное заболевание, сопровождающееся фибриллизацией или изъязвлением, кишечную болезнь Бехчета и простую язву. Язвенные заболевания пищеварительного тракта в контексте настоящего изобретения включают стоматит, афтозный стоматит, воспаление пищевода, язву пищевода, гастрит, язву желудка и язву тонкого кишечника.
Кроме того, гастрит и язва желудка включают медикаментозный гастрит и язву желудка, а также алкогольный гастрит и язву желудка, и медикаментозный гастрит и язва желудка включают гастрит и язву желудка, вызванные нестероидными противовоспалительными средствами.
Язва тонкого кишечника включает медикаментозную язву тонкого кишечника и алкогольную язву тонкого кишечника, и медикаментозная язва тонкого кишечника включает язву тонкого кишечника, вызванную нестероидными противовоспалительными средствами.
Лекарственное средство по настоящему изобретению, в частности, применимо в качестве профилактического или терапевтического агента при язвенном колите, болезни Крона, гастрите, язве желудка или язве тонкого кишечника.
Сердечно-сосудистые и сердечные заболевания включают артериосклероз, стенокардию, инфаркт миокарда, мозговые расстройства, вызванные кровоизлиянием в мозг, мозговые расстройства, вызванные инфарктом мозга, мозговые расстройства, вызванные субарахноидальным кровоизлиянием, легочную артериальную гипертензию, сужение периферических артерий (облитерирующий артериосклероз и облитерирующий тромбоангиит) и различные симптомы (перемежающуюся хромоту с поясничным спинальным стенозом, онемение ног, синдром Рейно, эректильную дисфункцию, геморрой и т.д.), относящиеся к нарушениям периферического кровообращения.
Респираторные заболевания включают астму, поражения легких, фиброз легких, эмфизему, бронхит и хроническую обструктивную болезнь легких.
Неврологические заболевания включают гибель нервных клеток, боковой амиотрофический склероз, множественный склероз и заболевания мозга (расстройства, вызванные кровоизлиянием в мозг, инфарктом мозга и субарахноидальным кровоизлиянием).
Глазные заболевания включают глаукому и глазную гипертензию.
Почечные заболевания включают гломерулонефрит, диабетическую нефропатию, IgA нефропатию и поражение почек, вызванное ишемией-реперфузией.
Заболевания печени включают гепатит, гепатопатию и поражение печени, вызванное ишемией-реперфузией.
Заболевания костей включают остеопороз, перелом кости и фазу послеоперационного восстановления после остеотомии.
Кожные заболевания включают пролежни и раны.
Кроме того, лекарственное средство, содержащее соединение формулы (1) по настоящему изобретению или его фармацевтически приемлемую соль в качестве действующего ингредиента, применимо в качестве профилактического и/или терапевтического агента при алопеции, облысении или расстройствах слуха (например, расстройствах слуха, вызванных шумом) или агента, способствующего созреванию шейки матки.
Ниже по тексту настоящее изобретение подробно объясняется с помощью конкретных примеров, которые не следует истолковывать, как ограничивающие его объем.
Пример 1: Синтез метил (2R)-2-(м-толил)пропионата
Метанол (14,83 г) и концентрированную серную кислоту (6,46 г) добавляли к (2R)-2-(м-толил)пропионовой кислоте и перемешивали смесь при кипячении с обратным холодильником в течение 6 ч. Затем смесь нейтрализовали 10% водным раствором карбоната натрия и экстрагировали гексаном. После высушивания над сульфатом магния, остаток концентрировали при пониженном давлении, получая указанное в заглавии соединение (12,79 г). Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3):δ 1,49(д, J=7,0 Гц, 3H), 2,33(с, 3H), 3,64(с, 3H), 3,69(дд, J=14,4, 7,3 Гц, 1H), 7,06-7,22(м, 4H).
Пример 2: Синтез диметил (3R)-2-оксо-3-(м-толил)бутилфосфоната
Тетрагидрофуран (ТГФ) (25 мл) добавляли к диметилметилфосфонату (1,97 г) и охлаждали смесь до -78°C. Добавляли н-бутиллитий (1,5 М раствор в гексане) (10 мл) и перемешивали смесь в течение 1 ч. Затем при -78°C добавляли раствор метилового эфира (метил (2R)-2-(м-толил)пропионата), синтезированного в примере 1 (1,34 г) в ТГФ (3,8 мл), и перемешивали смесь в течение 2 ч. Полученную реакционную смесь гасили 25 мл насыщенного водного раствора гидрокарбоната натрия и экстрагировали смесь этилацетатом. Экстракт высушивали над сульфатом магния и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (гексан/этилацетат 5:1-1:5), получая указанное в заглавии соединение (1,63 г). Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3): δ 1,39(д, J=6,7 Гц, 3H), 2,34(с, 3H), 2,84(ддд, J=22,3, 14,1, 0,6 Гц, 1H), 3,18(дд, J=22,3, 14,1 Гц, 1H), 3,76(дд, J=19,3, 11,1 Гц, 6H), 4,00(дд, J=13,8, 7,0 Гц, 1H), 7,01-7,24(м, 4H).
Пример 3: Синтез (1S,5R,6R,7R)-6-[(1E,4R)-3-оксо-4-(м-толил)-1-пентенил]-7-бензоилокси-2-оксабицикло[3.3.0]октан-3-она
Гидрид натрия (55%) (8,75 г) диспергировали в 1,2-диметоксиэтане (DME) (300 мл) и охлаждали смесь льдом. Добавляли раствор фосфоната (диметил (3R)-2-оксо-3-(м-толил)бутилфосфоната) (54,7 г), полученного в примере 2, в DME (50 мл) и перемешивали смесь в течение 1 ч. К полученному раствору добавляли раствор (1S,5R,6R,7R)-6-формил-7-бензоилокси-2-оксабицикло[3.3.0]октан-3-она (50,0 г) в DME (400 мл) и перемешивали смесь в течение 1 ч. Реакционную смесь гасили 350 мл 10% раствора соли и экстрагировали смесь этилацетатом. Экстракт высушивали над сульфатом магния и остаток концентрировали при пониженном давлении. Сконцентрированный неочищенный продукт перекристаллизовывали из т-бутилметилового эфира, получая указанное в заглавии соединение (64,7 г). Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3): δ 1,39(д, J=7,0 Гц, 3H), 2,20-2,28(м, 1H), 2,30(с, 3H), 2,34-2,41(м, 1H), 2,49-2,57(м, 1H), 2,76-2,85(м, 3H), 3,80(кв, J=7,0 Гц, 1H), 5,03(т, J=5,3 Гц, 1H), 5,23(кв, J=5,3 Гц, 1H),6,19(д, J=15,5 Гц, 1H), 6,69(дд, J=15,6, 7,6 Гц, 1H), 6,94-7,19(м, 4H), 7,42-7,95(м, 5H).
Пример 4: Синтез (1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-бензоилокси-2-оксабицикло[3.3.0]октан-3-она
Раствор енона {(1S,5R,6R,7R)-6-[(1E,4R)-3-оксо-4-(м-толил)-1-пентенил]-7-бензоилокси-2-оксабицикло[3.3.0]октан-3-она} (147,0 г), синтезированного в примере 3 в ТГФ (1480 мл) охлаждали до -40°C, добавляли (-)-B-хлордиизопинокамфеилборан (1,7 М раствор в гексане) (721 мл) и перемешивали смесь при охлаждении льдом в течение 20 ч. Добавляли ацетон (183 мл) и перемешивали смесь в течение 3 ч. Добавляли водный раствор гидрокарбоната натрия и экстрагировали смесь т-бутилметиловым эфиром. Экстракт высушивали над сульфатом магния и концентрировали при пониженном давлении, получая указанное в заглавии неочищенное соединение (649,9 г).
Пример 5: Синтез (1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-оксабицикло[3.3.0]октан-3-она
Неочищенный спирт {(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-бензоилокси-2-оксабицикло[3.3.0]октан-3-он} (649,9 г), синтезированный в примере 4, растворяли в метаноле (740 г), добавляли карбонат калия (116,3 г) и перемешивали смесь при комнатной температуре в течение 17 ч. Добавляли уксусную кислоту, доводя pH до 7, выпаривали метанол, добавляли воду и экстрагировали смесь этилацетатом. Экстракт очищали колоночной хроматографией на силикагеле (гексан/этилацетат=4/1-0/1), получая указанное в заглавии соединение (22,3 г). Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3): δ 1,33(д, J=7,0 Гц, 3H), 1,70(с, 1H(OH)), 1,86(ддд, J=11,3, 7,8, 3,2 Гц, 1H), 2,07(д, J=4,4 Гц, 1H(OH)), 2,13-2,23(м, 2H), 2,34(с, 3H), 2,35-2,44(м, 3H), 2,47(д, J=3,8 Гц, 1H), 2,56(дд, J=18,2, 9,7 Гц, 1H), 2,80(кв, J=7,0 Гц, 1H), 3,79-3,85(м, 1H), 4,12-4,16(м, 1H), 4,81(дт, J=7,0, 3,2 Гц, 1H), 5,27(ддд, J=15,7, 8,5, 0,6 Гц, 1H), 5,50(дд, J=15,2, 6,8 Гц, 1H), 6,94-7,20(м, 4H).
Пример 6: Синтез (1S,5R,6R,7R)-6-[(1E,3R,4R)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-оксабицикло[3.3.0]октан-3-она
К раствору диола {(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-оксабицикло[3.3.0]октан-3-она} (988 мг), синтезированного в примере 5, в N,N-диметилформамиде (ДМФА) (10 мл) при комнатной температуре добавляли т-бутилдиметилсилилхлорид (1,17 г) и имидазол (1,08 г), и перемешивали смесь в течение 2,5 ч. Реакционную смесь выливали в насыщенный водный раствор гидрокарбоната натрия и экстрагировали смесью гексан/этилацетат=2/1. Экстракт высушивали над сульфатом магния, концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (гексан/этилацетат 20:1-10:1), получая указанное в заглавии соединение (1,56 г). Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3): δ -0,09(д, J=6,4 Гц, 6H), 0,02(д, J=2,4 Гц, 6H), 0,86(с, 9H), 0,89(с, 9H), 1,27(д, J=7,0 Гц, 3H), 1,86-1,92(м, 1H), 1,96-2,02(м, 1H), 2,32(с, 3H), 2,31-2,47(м, 3H), 2,62-2,73(м, 2H), 3,82(кв, J=4,7 Гц, 1H), 4,05(т, J=6,4 Гц, 1H), 4,86(дt, J=8,0, 2,4 Гц, 1H), 5,16(дд, J=15,5, 7,4 Гц,1H), 5,30(дд, J=15,7, 6,3 Гц, 1H), 6,90-7,16(м, 4H).
Пример 7: Синтез (1S,5R,6R,7R)-6-[(1E,3R,4R)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-она
К бромиду марганца (1,48 г) и N-фторбензолсульфонимиду (2,48 г) добавляли тетрагидрофуран (ТГФ) (19 мл), полученную смесь перемешивали в течение 30 мин и охлаждали до -78°C. Добавляли раствор лактона {(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-оксабицикло[3.3.0]октан-3-она} (0,5 г), синтезированного в примере 6, в ТГФ (5 мл), добавляли раствор бис(триметилсилил)амида калия в толуоле (0,5 М, 13 мл), и смесь нагревали до 0°C в течение 3,5 ч. Реакционную смесь выливали в насыщенный водный раствор гидрокарбоната натрия и экстрагировали смесью гексан/этилацетат=1/1. Экстракт высушивали над сульфатом магния, концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (гексан/этилацетат 20:1), получая указанное в заглавии соединение (0,32 г). Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3): δ -0,08-0,03(м, 12H), 0,82(с, 9H), 0,89(с, 9H), 1,28(д, J=7,0 Гц, 3H), 1,70-1,77(м, 1H), 1,96-2,04(м, 1H), 2,31(с, 3H), 2,60-2,91(м, 3H), 3,82-3,87(м, 1H), 3,99-4,23(м, 1H), 5,00(т, J=6,4 Гц, 1H), 5,06(дд, J=15,7, 7,8 Гц, 1H), 5,33(ддд, J=15,9, 6,7, 1,2 Гц, 1H), 6,88-7,16(м, 4H).
19F-ЯМР (CDCl3):δ -113,1(д, J=279,3 Гц), -91,0(дд, J=279,3, 25,9 Гц).
Пример 8: Синтез 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутана
К суспензии 4-(тетразол-5-ил)бутилтрифенилфосфонийбромида (14,0 г) в толуоле (390 мг) добавляли раствор бис(триметилсилил)амида калия в толуоле (0,5М, 120 мл) и перемешивали смесь при 60°C в течение 1 ч. При -10°C добавляли раствор дифторлактона {(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-она}, синтезированного в примере 7 (4,32 г), в толуоле (130 мл) и смесь перемешивали в течение 18 ч, повышая при этом температуру смеси до комнатной. Для гашения реакционной смеси добавляли водный раствор гидрокарбоната натрия и экстрагировали смесью гексан/этилацетат=1/1. Экстракт высушивали над сульфатом магния, концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (гексан/этилацетат=5/1-0/1), получая указанное в заглавии соединение (4,1 г). Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3):δ -0,14-0,01(м, 12H), 0,82(с, 9H), 0,89(с, 9H), 1,23-1,27(м, 3H), 1,82-2,09(м, 5H), 2,21-2,28(м, 1H), 2,31(с, 3H), 2,45-2,53(м, 1H), 2,64-2,73(м, 2H), 2,93-2,97(м, 2H), 3,90(дд, J=11,7, 5,3 Гц, 1H), 4,08-4,09(м, 1H), 4,84-4,87(м, 2H), 5,27(дд, J=15,5, 7,8 Гц, 1H), 5,44(дд, J=15,6, 6,2 Гц, 1H), 6,92-7,16(м, 4H).
19F-ЯМР (CDCl3):δ -112,3(д, J=253,4 Гц), -81,4(дд, J=253,4, 18,7 Гц).
Пример 9: Синтез 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутана
К соединению, синтезированному в примере 8 (4,1 г), добавляли ТГФ (81 мл), воду (81 мл) и уксусную кислоту (244 мл), и перемешивали смесь при 35°C в течение 46 ч. Добавляли воду (500 мл) и экстрагировали смесь хлороформом. Экстракт высушивали над сульфатом магния, концентрировали при пониженном давлении, очищали колоночной хроматографией на силикагеле (гексан/этилацетат=1/5-0/1) и перекристаллизовывали из диэтилового эфира, получая указанное в заглавии соединение (1,1 г). Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3): δ 1,30(д, J=7,0 Гц, 3H), 1,69(дддд, J=14,6, 7,6, 3,0, 2,6 Гц, 1H), 1,82-1,95(м, 2H), 2,10-2,16(м, 2H), 2,29(с, 3H), 2,31-2,41(м, 2H), 2,48-2,56(м, 1H), 2,72(кв, J=7,0 Гц, 1H), 2,93(т, J=7,6 Гц, 2H), 3,78(кв, J=7,6 Гц, 1H), 4,04-4,10(м, 1H), 4,69(дт, J=6,48, 2,96 Гц, 1H), 4,79(дт, J=7,6, 5,0 Гц, 1H), 5,36-5,46(м, 2H), 6,95-7,13(м, 4H).
19F-ЯМР (CDCl3):δ -116,6(д, J=250,5 Гц), -84,8(ддд, J=251,9, 17,3, 14,4 Гц).
Пример 10: Синтез диметил 2-оксо-3-(м-толил)бутилфосфоната
Используя рацемат 2-(м-толил)пропионовой кислоты и методику, аналогичную методике примеров 1-2, синтезировали указанное в заглавии соединение. Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3):δ 1,39(д, J=7,2 Гц, 3H), 2,34(с, 3H), 2,83(дд, J=22,4, 14,4 Гц, 1H), 3,18(дд, J=22,4, 14,0 Гц, 1H), 3,76(дд, J=19,6, 11,2 Гц, 6H), 3,99(дд, J=14,0, 6,8Гц, 1H), 7,01-7,27(м, 4H).
Пример 11: Синтез (1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-оксабицикло[3.3.0]октан-3-она
Используя рацемат диметил 2-оксо-3-(м-толил)бутилфосфоната и методику, аналогичную методике примеров 3-6, синтезировали указанное в заглавии соединение. Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3):δ -0,20-0,10(м, 12H), 0,80-0,90(м, 18H), 1,18-1,28(м, 3H), 1,85-2,20(м, 2H), 2,31(с, 3H), 2,30-2,80(м, 5H), 3,80-4,15(м, 2H), 4,81-4,95(м, 1H), 5,12-5,42(м, 2H), 6,88-7,20(м, 4H).
Пример 12: Синтез (1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-она
Используя (1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-оксабицикло[3.3.0]октан-3-он, синтезированный в примере 11, и методику, аналогичную методике примера 7, синтезировали указанное в заглавии соединение. Структурные свойства соединения приведены ниже.
1H-ЯМР (CDCl3):δ -0,20-0,05(м, 12H), 0,80-0,90(м, 18H), 1,19-1,29(м, 3H), 1,70-2,10(м, 2H), 2,31(с, 3H), 2,60-3,05(м, 3H), 3,84-4,12(м, 2H), 4,95-5,50(м, 3H), 6,85-7,20(м, 4H).
19F ЯМР (CDCl3):δ -113,6 - -112,8(м), -91,7 - -90,6(м).
Пример 13: Синтез 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутана
Используя (1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-он, синтезированный в примере 12, и методику, аналогичную методике примера 8, синтезировали указанное в заглавии соединение. Ниже приведены структурные свойства соединения.
1H-ЯМР (CDCl3): δ -0,15-0,05(м, 12H), 0,80-0,89(м, 18H), 1,20-1,28(м, 3H), 1,80-3,05(м, 14H), 3,90-4,15(м, 2H), 4,85-4,95(м, 2H), 5,23-5,58(м, 2H), 6,90-7,20(м, 4H).
19F-ЯМР (CDCl3):δ -113,0 - -111,3(м), -82,0 - -80,7(м).
Пример 14: Синтез 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутана
Используя 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутан, синтезированный в примере 13, и методику, аналогичную методике примера 9, синтезировали указанное в заглавии соединение. Ниже приведены структурные свойства соединения.
1H-ЯМР (CDCl3): δ 1,15-1,35(м, 3H), 1,80-3,00(м, 11H), 2,29(с, 3H), 4,05-4,20(м, 2H), 4,75-4,85(м, 2H), 5,35-5,70(м, 2H), 6,95-7,25(м, 4H).
19F-ЯМР (CDCl3):δ -114,5 - -112,7(м), -83,5 - -81,8(м).
Пример 15: Синтез 5-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]пентановой кислоты (карбоксилатная форма)
Используя (1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-т-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-т-бутилдиметилсилокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-он, синтезированный в примере 12, и (4-карбоксибутил)трифенилфосфоний бромид, согласно методике, аналогичной методикам примеров 8-9, синтезировали указанное в заглавии соединение. Ниже приведены структурные свойства соединения.
1H-ЯМР (CD3OD):δ 1,17-1,30(м, 3H), 1,63-2,79(м, 11H), 2,29(с, 3H), 3,75-4,12(м, 2H), 4,66-4,85(м, 2H), 5,40-5,58(м, 2H), 6,95-7,15(м, 4H).
19F-ЯМР (CD3OD):δ -118,3 - -117,7(д, J=250,4Гц), -86,1 - -85,3(м).
Пример 16: Метаболическая устойчивость соединений по настоящему изобретению in vitro
Тестировали смесь соединения F и соединения J по настоящему изобретению, описанных в таблице 1 (F:J=52:41), синтезированную в примере 14, и смесь соединений, в которых тетразольная группа в положении C-1 соединения F и соединения J, соответственно, заменена на карбоксильную группу (именуемую «карбоксилатной формой», F:J=54:34, синтезированную в примере 15).
На первом этапе получали фракцию митохондрий из печени крысы по методике приведенной ниже ссылки A. Затем, пользуясь методикой Yamaguchi и соавторов, описанной в приведенных ниже ссылках B и C, исследовали реакцию NADPH-независимого β-окисления. Эту реакцию проводили при 37°C в течение 30 мин и прерывали с помощью метанольного раствора, содержащего вещество, подходящее в качестве внутреннего стандарта. Содержание каждого вещества количественно определяли по методике с использованием внутреннего стандарта, используя прибор для высокоэффективной жидкостной хроматографии/масс спектрометрии (ЖХ-МС/МС). Доли веществ, не вступивших в метаболические реакции, для соединений F, J и обеих карбоксилатных форм во фракции крысиных митохондрий показаны в приведенной ниже таблице 2 в виде среднее значение ± стандартное отклонение для 3 экспериментов.
Остаточные процентные доли исходного соединения после реакции β-окисления
Как видно из приведенной выше таблицы 2, типовые соединения по настоящему соединению F и J не подвергаются β-окислению во фракции митохондрий.
Литература:
A) The Japanese Biochemical Society, ed., Biochemical Experiment Course 12 energy metabolism and biological oxidation (vol. 1), Tokyo Kagaku Dojin, p. 217-218, 1st ed. 2nd printing, published on July 11, 1979.
B) Drug Metabolism And Disposition 23(11): 1195-1201 (1995).
C) Xenobiotica 26(6): 613-626 (1996).
Пример 17: Фармакокинетика в плазме после внутривенного введения крысам
Для подтверждения метаболической устойчивости соединений по настоящему изобретению in vivo, исследовали фармакокинетику в плазме после внутривенного введения крысам. Самцам крыс (возраст 6 недель, масса тела 160-180 г) давали адаптироваться в течение 1 недели, после чего использовали животных, признанных здоровыми. Смесь соединения F и соединения J по настоящему изобретению, описанных в таблице 1 (F:J=52:41), смесь карбоксилатных форм соединения F и соединения J (F:J=54:34) и соединение F (синтезированное в примере 9) растворяли в небольшом количестве этанола и к этим растворам добавляли физиологический солевой раствор, получая растворы тестируемых соединений. Сразу же осуществляли внутривенное введение этих растворов тестируемых соединений в количестве 1 мл/кг в бедренную вену крыс, которые не находились в состоянии голодания, при применении легкой эфирной анестезии. Через 5, 15, 30, 45, 60, 90 и 120 мин после введения отбирали образцы венозной крови из хвостовой вены. Кровь смешивали с гепарином и центрифугировали (3000 об/мин, 4°C, 15 мин), получая плазму. Концентрации соединений в плазме определяли по методике с внутренним стандартом, используя ЖХ-МС/МС. Диапазон концентраций, измеряемых по описанной методике, составлял от 0,1 до 100 нг/мл. Концентрации соединений, полученных для каждой крысы, анализировали модель-независимым способом, используя программу для анализа фармакокинетики WinNonlin (версия 3.3), и получали данные в виде среднее значение ± стандартное отклонение для трех животных в каждой группе. Измеренные значения времени полужизни (t1/2) на стадии выведения показаны в приведенной ниже таблице 3.
Измеренные значения времени полужизни соединений по настоящему изобретению на стадии выведения после внутривенного введения крысам
Как видно из приведенной выше таблицы 3, значения t1/2 соединения F и соединения J по настоящему изобретению составляли примерно 1-2 ч, что существенно превышает соответствующий показатель для карбоксилатных форм, который составляет менее 10 мин. Это дает основание предположить, что соединение F и соединение J по настоящему изобретению обладают отличной устойчивостью к метаболизму.
Пример 18: Сродство к рецепторам
Определяли сродство соединения F и соединения J по настоящему изобретению к рецепторам PG. Использованное соединение F представляло собой продукт, синтезированный в примере 9, и соединение J получали разделением и очисткой продукта, синтезированного в примере 14, с помощью колонки (эти же препараты использовались в следующих примерах). Клетки COS-7 трансфицировали генами EP-4 мыши, EP1-4 человека или IP человека для достижения избыточной экспрессии рецепторов и затем собирали клеточные мембраны. В качестве меченых лигандов использовали меченый тритием PGE2 для EP, и меченый тритием илопрост для IP. Получали значения констант диссоциации Kd, и стандартным способом определяли значения констант ингибирования Ki для каждого тестируемого соединения. В качестве контроля для EP использовали PGE2; для IP использовали берапрост натрия (BPS).
Определенные в результате экспериментов константы диссоциации оказались практически идентичны литературным значениям. Как показано в таблице 4, PGE2 с одинаковым сродством связывается с EP1-4, и его селективность в отношении подтипов EP наблюдать не удалось. BPS селективно связывается с IP. Соединения F и J связываются EP4 мыши и человека. Сродство связывания соединения F к связыванию с EP4 человека в 60 или более раз выше, чем сродство к связыванию с человеческими рецепторами EP1, EP2 и EP3, и в 100 или более раз выше, чем сродство к связыванию с IP. Сродство связывания соединения J к человеческому рецептору EP4 в 40 раз выше сродства к человеческому IP.
Сродство связывания тестируемых соединений к связыванию с различными рецепторами
Пример 19: Агонистическая активность
Агонистическая активность соединения F и соединения J по настоящему изобретению в отношении EP4 определяли по стандартной методике. Вкратце, гены человеческого EP4 и гены репортера CRE-LUC трансфицировали в клетки COS-7. Через один день клетки обрабатывали тестируемым соединением и инкубировали в течение 3 ч. Для определения агонистической активности клетки промывали, добавляли люминесцентный субстрат и измеряли интенсивность люминесценции. В качестве контроля использовали PGE2, и принимали максимальную активность PGE2 за 100%. Рассчитывали концентрации, соответствующие 50% эффективности, и сравнивали агонистическую активность тестируемых соединений.
В результате, как показано в таблице 5, было установлено, что соединения F и J являются агонистами EP4.
Агонистическая активность тестируемых соединений
в отношении EP4
Каждое значение является геометрическим средним трех результатов.
Пример 20: Ингибирующее действие на агрегацию тромбоцитов
Оценивали ингибирующее действие соединений F и J и их карбоксилатных форм на агрегацию тромбоцитов. Карбоксилатные формы получали разделением и очисткой продукта, синтезированного в примере 15 с помощью колонки. Отбирали пробы крови у трех здоровых добровольцев, используя в качестве антикоагулянта цитрат натрия, и получали плазму, обогащенную тромбоцитами. Эту обогащенную тромбоцитами плазму обрабатывали физиологическим солевым раствором или тестируемыми соединениями, через 2 минуты вызывали агрегацию тромбоцитов с помощью аденозин дифосфата (ADP, конечная концентрация 10 мкмоль/л) и регистрировали реакцию с помощью аппарата, измеряющего агрегацию тромбоцитов (нефелометрия). Максимальную агрегацию в группе, обработанной физиологическим солевым раствором, принимали за 100%, рассчитывали концентрацию, необходимую для 50% ингибирования агрегации (IC50), и определяли ингибирующее действие.
В результате, как показано в таблице 6, соединения F и J оказывали значительно более слабое ингибирующее действие на агрегацию по сравнению с агонистами IP (BPS и карбоксилатные формы).
Ингибирующее действие тестируемых соединений на агрегацию тромбоцитов человека
Каждое значение IC50 представляет собой геометрическое среднее данных для трех добровольцев.
Пример 21: Влияние на кровяное давление и частоту сердечных сокращений у мышей
Приобретали самцов мышей ICR (в возрасте 5 недель, Japan SLC), давали адаптироваться в течение 6 дней и использовали в описываемом тесте. Введение препаратов и регистрация результатов осуществлялось при ингаляционной анестезии изофлураном (в начале анестезии содержание изофлурана 2,5%, затем 1,8-2,1%), причем температуру тела поддерживали на уровне 37°C с помощью прибора Heat Controller (ATC-402, Unique Medical Co., Ltd.). Катетер для введения тестируемого соединения вводили в левую бедренную вену, причем для регистрации всех гемодинамических параметров в правую бедренную артерию вводили другой катетер и соединяли с датчиком давления.
Соединение F, карбоксилатную форму соединения F и BPS вводили внутривенно в дозировке 0,01 мг/5 мл/кг и определяли влияние на среднее значение кровяного давления и частоту сердечных сокращений с помощью компьютерной программы для анализа гемодинамики (Fluclet, Dainippon Sumitomo Pharma Co., Ltd.). Контрольной группе аналогичным образом осуществляли внутривенное введение растворителя (1,2% раствор этанола) в концентрации 5 мл/кг. Результаты выражали в виде относительного изменения каждого параметра до и после введения препарата. В каждой группе использовали 3-6 животных и результаты выражали в виде среднее значение ± стандартное отклонение.
В результате проведенных исследований было показано, что растворитель не влияет на среднее значение кровяного давления, но карбоксилатная форма соединения F и BPS значительно снижают кровяное давление через 1 мин после введения, причем в этот момент времени максимальное снижение составляло 45% и 38% соответственно (фиг. 1A). Пониженное кровяное давление восстанавливалось в течение 10 мин, но частота сердечных сокращений увеличивалась и оставалась высокой даже через 10 мин (фиг. 1B). Максимальное уменьшение давления для соединения F составляло 16,2%, что не являлось статистически значимым (фиг. 1A). Кроме того, соединение F оказывало незначительное влияние на частоту сердечных сокращений (фиг. 1B). Таким образом, влияние соединения F на кровяное давление и частоту сердечных сокращений является чрезвычайно слабым по сравнению с карбоксилатной формой и BPS (агонистом IP).
Пример 22: Подавление выработки воспалительных цитокинов
Исследовали противовоспалительное действие соединений F и J in vitro, используя периферическую кровь человека. Отбирали образцы крови у трех здоровых добровольцев и получали CD4 положительные T-клетки. Добавляли антитела против CD3 и антитела против CD28, и через 24 часа с помощью анализа ELISA измеряли содержание выделившихся в среде IL-2 и THF-α. Кроме того, собранную цельную кровь разбавляли средой, обрабатывали индометацином, для ингибирования выработки эндогенного PGE2, и добавляли липополисахарид. С помощью анализа ELISA измеряли количество выделившихся в среде в течение 48 часов IP-10. В обоих случаях за 30 минут до введения стимулирующих добавок добавляли тестируемое соединение. Количество цитокинов, выделившихся в контрольной группе, обработанной растворителем, принимали за 100% и затем определяли концентрацию, необходимую для 50% ингибирования (IC50) выработки цитокинов.
В результате, как показано в таблице 7, PGE2 и соединение F в значительной степени подавляли выработку воспалительных цитокинов IL-2, TNF-α и IP-10 даже при чрезвычайно низких концентрациях. Соединение J также подавляло выработку указанных цитокинов, хотя и слабее, чем соединение F. Действие каждого из соединений в данном эксперименте отражает его сродство к EP4 и агонистическую активность в отношении EP4.
Как было упомянуто выше, хотя соединения по настоящему изобретению, включая соединение F, представляют собой производные PGI2, они являются селективными агонистами EP4 с сильно понижено агонистической активностью в отношении IP, по сравнению с активностью, наблюдаемой у соединений с карбоксильной группой в положении C-1 (карбоксилатных форм). Ожидается, что соединения по настоящему изобретению аналогичным образом проявят клиническую эффективность в отношении тех групп заболеваний, при которых эффективны агонисты EP4. Напротив, соединения по настоящему изобретению вызывают меньше проблем, связанных с побочными эффектами, такими как кровотечение, гипотензия, учащенное сердцебиение и прилив крови к лицу, поскольку его влияние на систему кровообращения также ослаблено, вследствие более слабого агонистического действия в отношении IP. Например, упомянутое агонистическое действие в отношении IP важно ослабить при лечении воспалительных заболеваний кишечника, сопровождающихся кишечным кровотечением. В случае применения соединения F, эффективность соединений по настоящему изобретению была установлена в различных животных моделях, например, моделях воспалительных заболеваний кишечника.
Ингибирующее действие тестируемых соединений на выработку цитокинов
Каждое из приведенных значений является средним геометрическим результатов трех добровольцев.
Пример 23: Профилактическое действие в модели колита, вызванного декстран сульфатом натрия у мышей
Профилактическое действие соединения F на язвенный колит исследовали в модели колита, вызванного декстран сульфатом натрия. В этой животной модели наблюдается воспаление, локализованное в толстом кишечнике, приводящее к диарее и кровянистому калу, что очень похоже на картину клинической патологии при язвенном колите (сравните ссылки D и E).
Приобретали самок мышей BALB/c (в возрасте 6 недель, Japan SLC), давали им адаптироваться в течение 1 недели и использовали для проведения исследования. Мышам, за исключением нормальной группы, давали неограниченно пить раствор декстран сульфата натрия (сокращение DSS, MP Biochemicals, N.W. 36000-50000, партия № 3439J), имеющий концентрацию 2,2% масса/объем, в течение 9 дней, с целью вызвать колит. Соединение F перорально вводили в дозировках 0,1, 0,3 и 1 мг/кг, один раз в день, ежедневно, со дня начала употребления DSS (день 0) и до дня, предшествовавшего вскрытию (день 9). Контрольной группе аналогичным образом перорально вводили растворитель (раствор этанола 1 объемн.%) в количестве 10 мл/кг.
Проведенное авторами предварительное исследование показало, что для стула мышей характерна корреляцию между содержанием воды и формой экскрементов. Поэтому для определения степени диареи, стул подразделяли на 6 категорий; нормальный (0 баллов), экскременты сферической формы составляют не менее 50% (1 балл), экскременты в форме банана составляют менее 50% (2 балла), экскременты в форме банана составляют не менее 50% (3 балла), стул в виде грязи (4 балла), водянистый стул (6 баллов) (шкала оценки консистенции стула). Скрытую кровь в кале (включая прокторрагию) подразделяли на 6 категорий, используя слайд 5 Shionogi II (Shionogi & Co., Ltd.) для выявления скрытой крови в кале: отрицательный (нет отклонения цвета слайда от желтого, 0 баллов), слабо положительный (слабый сине-зеленый цвет, 1 балл), положительный (сине-зеленый цвет, 2 балла), умеренно положительный (отчетливый сине-зеленый, 3 балла), сильно положительный (мгновенное изменение цвета до темно-синего при действии цветного проявителя, 4 балла), и прокторрагия (5 баллов). Сумма баллов консистенции стула и скрытой крови считали суммой баллов, характеризующей стул. В каждой группе было задействовано от 8 до 10 животных и результаты выражали в виде среднее значение ± стандартное отклонение. В день вскрытия, после вскрытия брюшной полости при анестезии эфиром и отбора крови, из животных выпускали кровь до наступления смерти. Затем иссекали толстый кишечник, начиная от участка сразу же за слепой кишкой до ануса, и измеряли его длину.
В результате было установлено, что масса тела постепенно увеличивалась в ходе исследования без каких-либо различий между группами. Контрольная группа продемонстрировала явно худшие показатели качества стула и скрытой крови в кале, начиная с 4 дня приема DSS. В день вскрытия (9 день), длина толстого кишечника у животных этой группы была явно меньше, чем у животных нормальной группы. Соединение F дозозависимым образом подавляло увеличение суммы баллов, характеризующих консистенцию стула, что проявлялось при 0,1 мг/кг и становилось статистически значимым при 0,3 и 1 мг/кг (фиг.2A). Аналогично, соединение F продемонстрировало дозозависимое подавление уменьшения длины толстого кишечника (фиг.2B). Таким образом, соединение F явно препятствует наступлению язвенного колита.
Литература:
D) Lab.Invest. 69(2): 238-249 (1993).
E) Inflamm.Res. 45(4): 181-191 (1996).
Пример 24: Действие агониста IP в модели колита, вызванного декстран сульфатом натрия, у мышей
В ходе исследования выявляли, проявляется ли действие агониста IP в указанной модели колита, используя селективный агонист IP, а именно BPS.
Приобретали самок мышей C57BL/6 (в возрасте 6 недель, Japan SLC), давали им адаптироваться в течение 1 недели и использовали для проведения исследования. Мышам, за исключением животных из нормальной группы, для инициирования колита давали без ограничений пить 3 или 2 масса/объем % раствор DSS (MP Biochemicals, партия № 5653H и 5464H, соответственно) в течение 1 недели. BPS в дозировке 0,3 мг/кг и соединение F в дозировке 0,3 и 1 мг/кг вводили перорально один раз в день, ежедневно, со дня начала употребления DSS (день 0) и до дня, предшествовавшего дню вскрытия (день 9). Контрольной группе аналогичным образом осуществляли пероральное введение растворителя (раствор этанола 1 объемн.%) в количестве 10 мл/кг. Консистенцию стула подразделяли на категории, соответствующие баллам от 0 до 4: нормальный (0), частично жидкий стул (1), жидкий стул (2) и диарея (4). Содержание крови в экскрементах также подразделяли на категории, соответствующие баллам от 0 до 4: нормальное (0), частично кровянистый стул (1), кровянистый стул (2) и кровянистый стул плюс прокторрагия (4). Сумму обоих баллов определяли как сумму баллов, характеризующую стул (максимум 8). Кроме того, по аналогии с примером 23, измеряли длину толстого кишечника. В каждой группе было задействовано от 6 до 10 животных и результаты исследований показаны в виде среднее значение ± стандартное отклонение.
Эксперименты показали, что агонист IP BPS оказался неэффективным, но продемонстрировал тенденцию против ухудшения суммы баллов, характеризующей стул. Кроме того, BPS не оказал влияния на уменьшение длины толстого кишечника (фиг. 3A и 3C). Однако соединение F продемонстрировало прекрасное профилактическое влияние на появление колита, по аналогии с примером 23 (фиг. 3B и 3D). Таким образом, лечебный эффект достигается действием агониста EP4 и в некоторых случаях ослабляется действием агонистов IP. Поэтому важно применение селективных агонистов EP4.
Пример 25: Профилактическое действие на колит, вызванный декстран сульфатом натрия, у крыс
Профилактическое действие соединения F на колит исследовали также у крыс. Приобретали самцов крыс SD в возрасте 7 недель с массой тела примерно 210-240 г (Charles River), давали им адаптироваться в течение 1 недели и использовали в исследовании. Крысам, за исключением животных из нормальной группы, для инициирования колита давали без ограничений пить раствор DSS (MP Biochemicals, М.М. 36000-50000 партия № 4556J) в концентрации 5,5 масса/объем % в течение 8 дней. Соединение F в дозировке 0,3, 1 и 3 мг/кг вводили перорально один раз в день, ежедневно, начиная за один день до начала употребления DSS и до дня, предшествовавшего дню вскрытия (день 7). Контрольной группе осуществляли пероральное введение растворителя (раствор этанола 1 объемн.%) в количестве 5 мл/кг.
На 8-й день после начала приема DSS через хвостовую вену вводили 1,25 масса/объем % раствор синего красителя Эванса в количестве 0,2 мл/100 г. Через 30 мин крысам осуществляли вскрытие брюшной полости под действием эфирной анестезии и выпускали кровь, пока не наступала смерть. После этого иссекали толстый кишечник от участка непосредственно за слепой кишкой до ануса и измеряли его длину мерной лентой. После удаления содержимого толстого кишечника, участок ткани толстой кишки длиной 7 см от ануса три раза промывали физиологическим солевым раствором и высушивали в течение ночи с помощью вакуумного насоса. На следующий день определяли сухую массу, добавляли формамид (2 мл) экстрагировали краситель при 50°C в течение ночи и определяли его количество на длине волны 620 нм. Строили стандартную кривую, используя стандартный раствор синего красителя Эванса и вычисляли содержание (мг) синего Эванса в 1 г ткани толстой кишки для определения степени поражения ткани толстой кишки.
Для определения уровня диареи, форму экскрементов подразделяли на 6 категорий; нормальная (0 баллов), экскременты в форме палочек составляют менее 50% (1 балл), экскременты в форме палочек составляют не менее 50% (2 балла), экскременты в форме палочек и частично стул в виде грязи (3 балла), стул в виде грязи (4 балла), водянистый стул (6 баллов) (шкала оценки консистенции стула). Скрытую кровь в кале определяли тем же способом, который описан в примере 23. Сумму показателей консистенции стула и скрытой крови считали суммой баллов, характеризующей стул. В каждой группе было задействовано от 7 до 10 животных, и результаты выражали в виде среднее значение ± стандартное отклонение.
Как показал эксперимент масса тела животных в контрольной группе стабильно постепенно увеличивалась, но это увеличение было значительно меньшим, чем соответствующее увеличение в нормальной группе. Сумма баллов, характеризующая стул, в контрольной группе значимо увеличивалась, начиная с 1 дня употребления DSS. В день вскрытия (8 день) наблюдалось явное поражение тканей толстого кишечника и его значительное укорачивание. В противоположность этому, введение соединения F в дозировках 1 мг/кг и 3 мг/кг продемонстрировало тенденцию к подавлению или значимый эффект подавления указанных явлений (Фиг. 4A, 4B, 4C). Т.е. соединение F предотвращает развитие язвы в толстом кишечнике и нормализует его деятельность, что приводит к подавлению симптомов диареи и кровянистого стула.
Пример 26: Терапевтическое действие в модели ремиссии/рецидива колита, вызванного декстран сульфатом натрия, у мышей
Далее было исследовано терапевтическое действие соединения F в модели хронического колита. Приобретали самок мышей BALB/c в возрасте 6 недель с массой тела примерно 20 г (Japan SLC), давали им адаптироваться в течение 1 недели и использовали в исследовании. Мышей делили на группу, у которой инициировали колит, и нормальную группу. Мышам в колитной группе разрешали без ограничений пить 2,6% (масса/объем) раствор DSS (MP Biochemicals, ММ 36000-50000, партия № 4556J), с целью вызвать колит. На 8-й день, когда балл оценки стула (определенный в примере 23) в колитной группе достиг примерно 4,5, мышей подразделяли на контрольную группу, группу, получавшую соединение F 1 мг/кг, и группу, получавшую салазосульфапиридин (SIGMA, партия № 085K1930, далее по тексту сокращенно именуемый SASP) 100 мг/кг. Затем мышам вместо раствора DSS давали неограниченно пить дистиллированную воду в течение 9 дней (период ремиссии). После упомянутого разделения на группы, состояние стула оценивали каждые 3-4 дня. Когда балл в контрольной группе достигал примерно 1, мышам вновь давали пить раствор DSS для инициирования рецидива (период рецидива). Период ремиссии и рецидива считали 1 циклом и такой цикл повторяли 5 раз. Однако в 5-м цикле проводили только период ремиссии.
Соединение F в дозировке 1 мг/кг и SASP в дозировке 100 мг/кг вводили перорально, один раз в день, ежедневно в течение 50 дней, начиная с первого периода ремиссии (8 день с начала приема раствора DSS 2,6% масс/объем) до пятого периода ремиссии (57 день с начала приема раствора DSS 2,6% масс/объем). Контрольной группе перорально вводили растворитель (раствор этанола 1% объемн.) в количестве 10 мл/кг. Если у мыши в последний день каждого периода ремиссии были баллы 0 по обоим показателям консистенции стула и скрытой крови в стуле, считалось, что мышь находится «в состоянии ремиссии». Долю мышей, находящихся в состоянии ремиссии (%), вычисляли как отношение количества мышей, находящихся в состоянии ремиссии, к общей численности группы. В каждой группе было задействовано от 8 до 10 мышей, и результаты приведены в виде средних значений.
Исследование показало, что сумма баллов, характеризующая стул, в контрольной группе увеличивалась во время периода рецидива и уменьшалась во время периода ремиссии. Эта сумма была значительно выше соответствующего показателя в нормальной группе практически на протяжении всего периода исследования (Фиг.5). Доля мышей, находящихся в состоянии ремиссии, в этой группе составляла 35,5% в среднем за 5 периодов ремиссии (таблица 8). Соединение F уменьшало сумму баллов, характеризующую стул, в начале периода ремиссии и подавляло увеличение этого показателя в период рецидива. Доля мышей, находящихся в состоянии ремиссии, в этой группе была не менее 60% в каждом периоде ремиссии и в среднем составляла 66,0%, что явно выше соответствующего показателя контрольной группы. С другой стороны SASP не продемонстрировал явного влияния на показатели стула, как в период ремиссии, так и в период рецидива. Доля мышей, находящихся в состоянии ремиссии, в этой группе была несколько выше чем в контрольной группе в 1-м, 3-м и 4-м циклах и, наоборот, ниже во 2-м, и 5-м циклах, и среднее значение было эквивалентно таковому в контрольной группе.
Как показано выше, соединение F обеспечивает не только профилактическое, но также и терапевтическое действие, а также эффект поддержания ремиссии. Кроме того, очевидно, что результаты для этого соединения существенно превосходят результаты SASP при клиническом применении.
Доля мышей, находящихся в состоянии ремиссии, в модели ремиссии/рецидива DSS-индуцированного колита у мышей
Пример 27: Профилактическое действие в модели колита, вызванного введением CD4 + CD25 - T-клеток, у мышей
Исследовали действие соединений на болезнь Крона, т.е. воспалительное заболевание кишечника другого типа. Модель введения T-клеток является хорошо известной моделью болезни Крона, которая приводит к хроническому гастриту или энтериту (смотрите ссылки F, G, H). Кроме того, эту модель можно считать животной моделью кишечной болезни Бехчета или простой язвы, в которой имеется аналогичная язва кишечника, сопровождающаяся активацией Т-клеток (смотрите ссылки I,J).
Приобретали самок мышей BALB/cA Jc1 в возрасте 6 недель с массой тела 19-23 г (CLEA Japan, Inc.) и самок мышей C.B-17/Icr-scid (в возрасте 6 недель, CLEA Japan, Inc.), давали им адаптироваться в течение 1 недели и использовали в исследовании.
После вскрытия брюшной полости под действием эфирной анестезии, из мышей BALB/cA Jc1 выпускали кровь через брюшную аорту и каудальную полую вену, пока не наступала смерть, и удаляли селезенку. Из селезенки выделяли спленоциты и затем получали CD4+CD25- T-клетки с помощью набора для выделения CD4+ T-клеток (No. 130-090-860, Milky Biotech Co., Ltd.) и антител против CD25-биотина (No. 130-092-569, Milky Biotech Co., Ltd.). Клетки отделяли с помощью сепаратора autoMACS (Milky Biotech Co., Ltd.). Выделенные CD4+CD25- T-клетки суспендировали в физиологическом фосфатном буферном растворе и интраперитонеально вводили мышам C.B-17/Icr-scid в количестве 2,5×105 клеток на животное для стимулирования колита.
Вначале, за 5 часов до введения T-клеток CD4+CD25-, вводили соединение F или преднизолон в количестве 1 мг/кг, и затем продолжали пероральное введение один раз в день, ежедневно в течение 20 дней. Контрольной группе перорально вводили растворитель (раствор этанола 1 объемн.%) в количестве 10 мл/кг. В качестве клинических результатов фиксировали сумму показателя консистенции стула (0-5), показателя скрытой крови (0-4) и показателя уменьшения массы тела (0-5), именуемую коэффициентом активности заболевания (далее по тексту сумму DAI: наивысшее значение 14). Показатель консистенции стула подразделялся в зависимости от твердости стула на нормальный (0), слегка жидкий (1), довольно жидкий (2), жидкий (3), очень жидкий (4) и диарею (5). Показатель скрытой крови в стуле оценивали по методике, аналогичной примеру 23. Показатель уменьшения массы тела определялся в зависимости от изменения массы тела, как увеличение (0), уменьшение менее чем на 3% (1), уменьшение не менее, чем на 3% и менее, чем на 6% (2), уменьшение не менее, чем на 6% и менее, чем на 9% (3), уменьшение не менее, чем на 9% и менее, чем на 12% (4), и уменьшение не менее, чем на 12% (5). В каждой группе было задействовано от 8 до 10 мышей, и результаты выражали в виде среднего значения.
В результате эксперимента, показатели консистенции стула и скрытой крови в стуле в контрольной группе продемонстрировали явное увеличение, начиная с 12 дня после введения T-клеток, и показатель уменьшения массы тела продемонстрировал явный прирост на 19 день, причем все цифры достигли практически максимальных значений через 21 день. Соединение F подавляло прирост как показателя консистенции стула, так и показателя скрытой крови в стуле почти наполовину, как показано на Фиг.6A и Фиг.6B соответственно, и почти полностью предотвращало прирост показателя уменьшения массы тела, как показано на Фиг.6C. С другой стороны, хотя преднизолон подавлял увеличение показателя скрытой крови в стуле почти на таком же уровне, как и соединение F, как показано на Фиг.6B, он не смог продемонстрировать явного влияния на показатель консистенции стула на 21 день, как следует из Фиг.6A. Кроме того, показатель уменьшения массы тела остался на более высоких значениях по сравнению с контрольной группой, как показано на Фиг.6C, и преднизолон явно ухудшил этот показатель. Согласно фиг.6D, сумма DAI указывает на то, что соединение F во всех отношениях превосходит преднизолон.
Таким образом, соединение F способно подавлять такие состояния, как болезнь Крона, кишечную болезнь Бехчета и простую язву, а также язвенный колит более эффективно, чем существующие лекарственные средства.
Литература:
F) Immunol Rev. 182: 190-200 (2001).
G) Int. Immunopharmacol. 6(8): 1341-1354 (2006).
H) J. Immunol. 160(3): 1212-1218 (1998).
I) Clin. Exp. Immunol. 139(2): 371-378 (2005).
J) Histopathology. 45(4): 377-383 (2004).
Пример 28: Действие соединений в модели поражения слизистой оболочки желудка, вызванного этанолом, у крыс
Ингибирующее действие соединения F на поражение слизистой оболочки желудка исследовали на модели поражения слизистой оболочки желудка, вызванного этанолом, у крыс. Эта модель часто применяется в качестве животной модели острого гастрита у человека, связанного с застойным поражением слизистой оболочки (ссылка K).
С помощью Oriental BioService Inc. приобретали самцов крыс SD (в возрасте 7 недель, Charles River), давали им адаптироваться в течение 1 недели и использовали в исследовании. За один день до начала исследования крыс разбивали на группы, исходя из массы тела, помещали в чистые клетки, снабженные сетчатым проволочным полом, не давали пищи в течение 19 ч (и воды в течение последних 3 ч) и всем группам перорально вводили этанол (особо чистый, Nacalai Tesque, партия № V8A5862, 1,5 мл), чтобы вызвать поражение слизистой оболочки желудка. За 30 мин до поражения слизистой оболочки желудка перорально вводили соединение F в дозировке 0,01, 0,1 и 1 мг/кг в объеме 5 мл/кг. Контрольной группе аналогичным образом перорально вводили растворитель (раствор этанола 1% объемн.) в объеме 5 мл/кг. В каждой группе были задействованы 8 животных.
Через 1 ч после введения этанола из крыс при анестезии эфиром выпускали кровь через брюшную аорту и каудальную полую вену, пока не наступала смерть, и удаляли желудок. Удаленные желудки немедленно заполняли нейтральным раствором формалина 2% объемн. (6 мл) и фиксировали в течение 15 мин. Желудки разрезали вдоль средней линии большой кривизны от сердечной части до привратниковой части и распрямляли на доске из поливинилхлорида. С помощью стереомикроскопа измеряли длину и ширину каждой язвы, вычисляли площадь, и сумму площадей принимали за общую площадь язвенного поражения.
Как показал эксперимент, общая площадь язвенного поражения в контрольной группе составляла в среднем 103 мм2. Соединение F дозозависимым образом значительно сокращало общую площадь язвенного поражения начиная с дозировки 0,01 мг/кг и уменьшало эту площадь почти до нуля при дозировке 1 мг/кг (Фиг.7). Таким образом, соединение F подавляло поражение слизистой оболочки желудка.
Литература:
K) Dig Dis Sci. 31(2 Suppl), 81S-85S (1986).
Пример 29: Действие соединений в модели поражения тонкого кишечника, вызванного индометацином, у крыс
Способность соединения F подавлять поражение тонкого кишечника исследовали с помощью модели поражения тонкого кишечника, вызванного индометацином, у крыс. Известно, что введение нестероидных противовоспалительных средств (NSAID) вызывает геморрагическое поражение в тонком кишечнике людей. Применяемая в исследовании модель характеризуется поражением слизистой оболочки тонкого кишечника, вызванным введением NSAID, а именно индометацина, крысам, и демонстрирует патологию, сходную с NSAID-индуцированным поражением тонкого кишечника у пациентов с болезнью Крона (ссылки L и M).
Приобретали самцов крыс SD в возрасте 7 недель (Charles River), давали им адаптироваться в течение 1 недели и использовали в исследовании. Крыс разбивали на группы, исходя из массы тела, и всем группам подкожно вводили индометацин (SIGMA партия № 19F0018) в дозировке 15 мг/5 мл/кг, чтобы вызвать поражение тонкого кишечника. За 30 мин до подкожного введения индометацина и через 6 часов после него перорально вводили соединение F в дозировках 0,01, 0,1 и 1 мг/кг в объеме 5 мл/кг. Контрольной группе аналогичным образом перорально вводили растворитель (раствор этанола 1% объемн.) в количестве 5 мл/кг. В каждой группе было задействовано восемь животных.
Через 23,5 часа после введения индометацина крысам под эфирной анестезией внутривенно вводили 2 мл раствора красителя синего Эванса 10 мг/мл. Через 30 мин под эфирной анестезией у крыс выпускали кровь через брюшную аорту и каудальную полую вену, пока не наступала смерть, и удаляли тонкий кишечник. Удаленный тонкий кишечник заполняли необходимым количеством (примерно 35 мл) нейтрального раствора формалина 2% объемн. и фиксировали в течение примерно 15 мин. После этого весь тонкий кишечник разрезали вдоль области прикрепления брыжейки и распрямляли на доске из винилхлорида. С помощью стереомикроскопа измеряли длину и ширину каждой язвы, вычисляли площадь, и сумму площадей принимали за общую площадь язвенного поражения.
В результате проведенного эксперимента было установлено, что общая площадь язвенного поражения тонкого кишечника в контрольной группе составляла примерно 730 мм2. В противоположность этому, в группе, получавшей соединение F, площадь язвенного поражения значительно уменьшалась дозозависимым образом, начиная с дозы 0,1 мг/кг и сокращалась до нуля при дозе 1 мг/кг (фиг.8). Таким образом, соединение F существенно подавляет поражение тонкого кишечника.
Литература:
L) Aliment Pharmacol Ther. 7(1), 29-39 (1993).
M) Acta Gastroenterol Belg. 57(5-6), 306-309 (1994).
Согласно изложенным выше результатам, соединение F продемонстрировало прекрасную способность подавлять прямое поражение слизистой оболочки желудочно-кишечного тракта, вызванное действием спирта и т.п., а также недостаточной регенерацией слизистой оболочки из-за действия NSAID и т.п. Таким образом ожидается, что соединение F продемонстрирует защитное действие и эффект регенерации тканей при поражении слизистых оболочек желудочно-кишечного тракта.
Как показано в упомянутых выше примерах и установлено для соединения F, соединения по настоящему изобретению эффективны при профилактике и лечении поражений желудочно-кишечного тракта, вызванных иммунным воспалением пищеварительного тракта, медикаментозным поражением слизистой оболочки пищеварительного тракта и недостаточной регенерацией слизистой оболочки, вызванной медикаментами. Конкретно, соединения применимы при воспалительных заболеваниях кишечника, например, язвенном колите и болезни Крона, алкогольном гастрите или язве желудка, язве тонкого кишечника и т.п. Это действие основано на агонистическом действии в отношении рецептора EP4 и не ограничивается перечисленными заболеваниями.
Пример 30: Действие соединений в модели гломерулонефрита, вызванного антителами против Thy-1, у крыс
Приобретали самцов крыс Slc:Wistar (в возрасте 6 недель, Japan SLC), давали им адаптироваться в течение 1 недели и использовали в исследовании. Животным, за исключением нормальной группы, один раз внутривенно вводили антитела против Thy-1 (мышиные антитела против CD90 (UK-Serotech Ltd. код:MCA47XZ, клон №: MRC OX-7, партия № 0303)). Со дня введения антител (день 0) и до дня 6, ежедневно, дважды в день (утром и вечером, каждый раз по 0,3 мг/кг) перорально вводили смесь соединения F и соединения J (F:J=52:41, сокращенно обозначаемую соединение F/J). Через три дня после введения антител в течение 1 дня собирали мочу. На 7-й день после введения антител животных вскрывали. Правую почку удаляли, взвешивали и фиксировали с помощью формалина. В каждой группе было задействовано 8 животных и результаты выражали в виде среднее значение ± стандартное отклонение.
Как показал эксперимент, увеличение массы тела в контрольной группе подавлялось по сравнению с нормальной группой через 1 день после введения антител. Однако через 3 дня масса тела увеличивалась как и в нормальной группе. Масса тела в группе, получавшей соединение F/J, продемонстрировала изменение, аналогичное контрольной группе. 24-ч объем мочи в контрольной группе увеличился по сравнению с нормальной группой. Однако объем мочи в группе, получавшей соединение F/J находился на том же уровне, как и в нормальной группе (Фиг.9А). Содержание белка в 24-ч объеме мочи было значительно увеличено в контрольной группе, тогда как в группе, получавшей соединение F/J, было обнаружено меньшее количество белка, чем в контрольной группе (Фиг.9B). Относительная масса почки была заметно увеличена в контрольной группе, но в группе, получавшей соединение F/J, наблюдались меньшие значения массы, близкие к нормальным (Фиг.9C). Гистопатологическое исследование почек позволило установить, что в контрольной группе наблюдалось заметное увеличение общего количества клубочковых клеток, мезангиальной области и PCNA-положительных клеток в клубочках. В группе, получавшей соединения F/J наблюдалось значительное подавление увеличения всех указанных параметров (Фиг.9D, 9E, 9F).
Таким образом, соединения по настоящему изобретению нормализуют объем мочи, уменьшают протеинурию и подавляют иммунную реакцию и реакцию роста клубочков, и эти факты показывают, что соединения эффективны при нефрите.
Пример 31: Влияние соединений на внутриглазное давление у кроликов
Приобретали белых японских кроликов (самцов в возрасте 10 недель, BIOTEC Co., Ltd.), давали им адаптироваться в течение 1 недели и использовали в исследовании. С помощью микропипетки капали раствор соединения F (0,01 масса/объем %) на роговую оболочку обоих глаз в объеме 50 мкл/глаз. Измеряли внутриглазное давление в правом глазу и оценивали местное раздражающее действие на глаза, используя левый глаз. После поверхностной анестезии оксибупранолом (0,4% раствор беноксила для закапывания), измеряли внутриглазное давление перед закапыванием препарата и через 1, 2, 3, 4, 6 и 8 ч после закапывания, используя пневмотонометр (Alcon Ltd.). Кроме того, оценивали местное раздражающее действие на глаза, определяя застой крови в конъюнктиве, отек конъюнктивы, помутнение роговицы, застой крови в радужной оболочке, экскрецию и интенсивность закрытия глаз. Для оценки этих параметров в контрольной группе в качестве растворителя использовали фосфатный буфер, и для сравнения использовали изопропил унопростон (глазные капли Rescula, Santen Pharmaceutical Co., Ltd.). В каждой группе было задействовано шесть животных и результаты выражали в виде средних значений.
Как показали эксперименты, внутриглазное давление в контрольной группе, получавшей растворитель, изменялось в диапазоне ±2 мм рт.ст. от момента времени 0 до 8 часов после закапывания. Группа, получавшая соединение F, продемонстрировала уменьшение внутриглазного давления на 6,9 мм рт.ст. через 1 час после закапывания, на 9,3 мм рт.ст. через 2 часа, и на 6,6 мм рт.ст. через 6 часов, т.е. значительное уменьшение внутриглазного давления. С другой стороны, глазные капли Rescula продемонстрировали профиль уменьшения внутриглазного давления в основном сходный с профилем группы, получавшей соединение F (Фиг.10).
Что касается местного раздражающего действия на глаза, у некоторых животных в группе, получавшей капли соединения F, через 1 час после закапывания наблюдался отек конъюнктивы и закрытие глаз, но позднее эти эффекты исчезали. В группе, получавшей глазные капли Rescula, у некоторых животных наблюдали отек конъюнктивы и закрытие глаз через 1 час после закапывания, и эти явления продолжались до 2 ч после введения. Позднее происходило восстановление животных.
Таким образом, соединение F по настоящему изобретению продемонстрировало способность уменьшать внутриглазное давление и местное раздражающее действие на глаза, эквивалентные соответствующим свойствам глазных капель Rescula, и они применимы в качестве терапевтического агента при глаукоме или высоком внутриглазном давлении.
Пример 32: Профилактическое действие соединений в модели гепатита, вызванного конканавалином A, у мышей
Профилактическое действие соединения F на гепатит исследовали с использованием модели гепатита, вызванного конканавалином A (далее по тексту именуемого Con A). Эта модель является моделью гепатита, в которой происходит поражение паренхимальных гепатоцитов, зависимое от T-клеток, и демонстрирует картину клинической патологии, сходную с аутоиммунным гепатитом или скоротечным гепатитом (смотрите ссылки N, O и P).
Приобретали мышей BALB/c (самки, в возрасте 7 недель, Japan SLC), давали им адаптироваться в течение 1 недели и использовали для исследования. Con A (тип IV, Sigma-Aldrich), растворенный в солевом растворе, вводили в хвостовую вену мышей, за исключением животных группы, в которой не инициировали гепатит, в дозировке 12,5 мг/10 мл/кг с целью стимулирования гепатита. Через двадцать часов под эфирной анестезией вскрывали брюшные полости мышей. Отбирали образцы крови объемом 0,5 мл из каудальной полой вены и получали гепаринизированную плазму для измерения активности ALT и AST в плазме. В число тестируемых соединений входили соединение F (раствор 1 мг/10 мл), преднизолон (Nacalai Tesque, суспензия 5 мг/10 мл (с 0,5 масс./объем% метилцеллюлозы)) и вода для инъекций (вводили контрольной группе), и их вводили перорально в объеме 10 мл/кг за 1 час до инъекции Con A. В каждой группе было задействовано от 7 до 9 животных. Для статистической оценки после преобразования данных к логарифмическому виду проводили однофакторный дисперсионный анализ.
Как показало исследование, активность ALT и AST в плазме контрольной группы заметно повышалась по сравнению с группой, в которой не инициировали гепатит. Соединение F и преднизолон статистически значимо и сильно подавляли это увеличение. На Фиг.11 показаны данные по активности ALT в плазме.
Таким образом, соединение F эффективно при поражении печени, связанном с активацией T-клеток.
Литература:
N) Eur. J. Immunol, 28(12): 4105-4113 (1998).
O) Proc. Natl. Acad. Sci. 97(10): 5498-5503 (2000).
P) J. Exp. Med. 191(1): 105-114 (2000).
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Соединения формулы (1) по настоящему изобретению применимы в качестве действующих ингредиентов лекарственных средств. Лекарственное средство, содержащее в качестве действующего ингредиента соединение формулы (1) по настоящему изобретению, применимо при иммунных заболеваниях, заболеваниях пищеварительного тракта, сердечно-сосудистых заболеваниях, сердечных заболеваниях, респираторных заболеваниях, неврологических заболеваниях, глазных заболеваниях, почечных заболеваниях, заболеваниях печени, заболеваниях костей, кожных заболеваниях и т.п., в развитие которых вовлечен рецептор EP4. В частности, соединения применимы в качестве лекарственного средства для профилактики или лечения язвенного колита, болезни Крона, гастрита или язвы желудка, язвы тонкого кишечника, нефрита, глаукомы или гепатита.
Хотя выше по тексту были подробно описаны некоторые варианты осуществления настоящего изобретения, рядовой специалист в данной области техники имеет возможность осуществить различные модификации и изменения в приведенных выше конкретных вариантах осуществления, не отступая от содержания и преимуществ настоящего изобретения. Эти модификации и изменения соответствуют сути и объему настоящего изобретения, которые изложены в приложенной формуле изобретения.
Настоящая заявка основана на заявках на патент № 2010-51127 и 2010-246208, поданных в Японии, содержание которых в полном объеме включено в настоящую заявку.
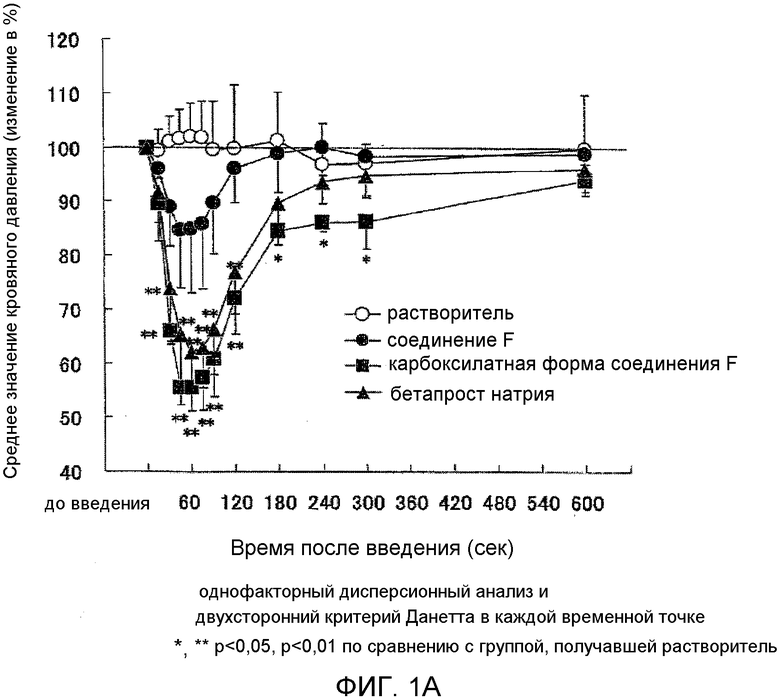
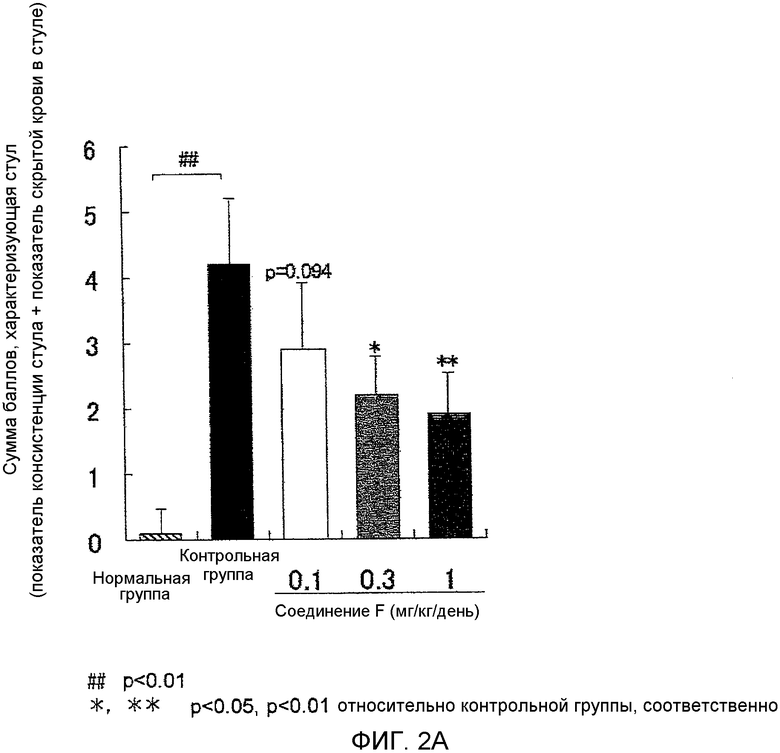
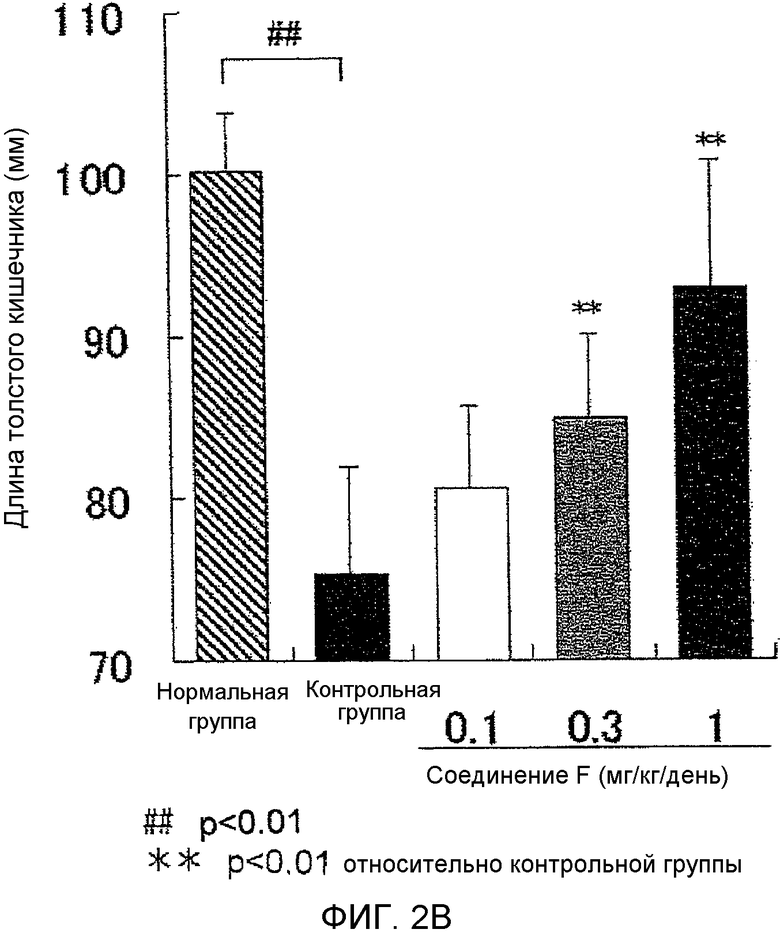
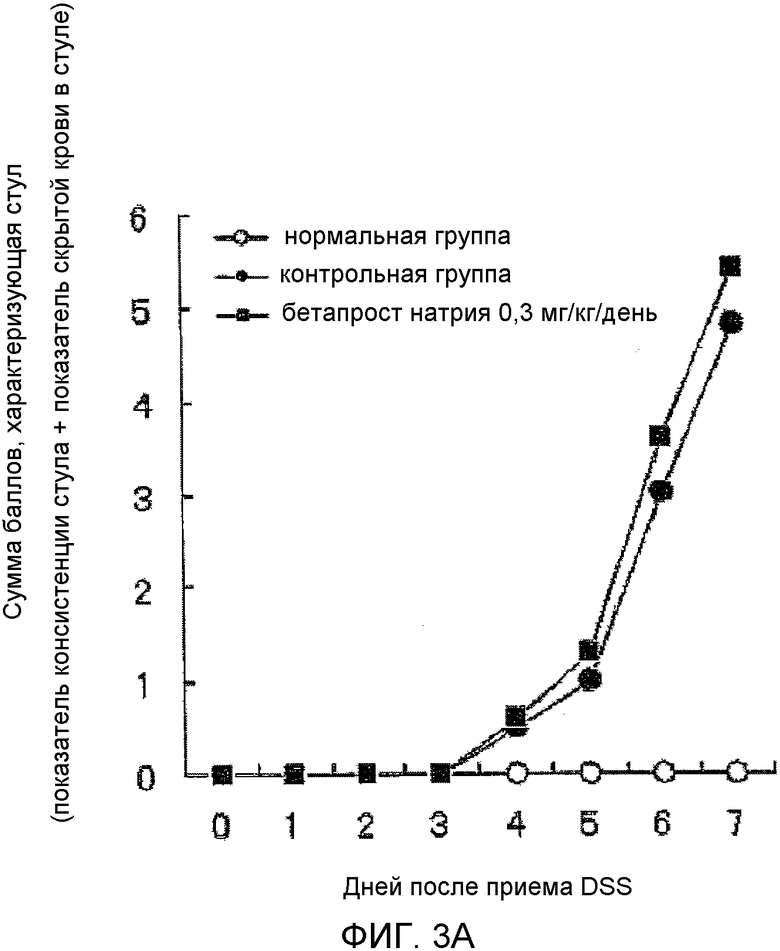
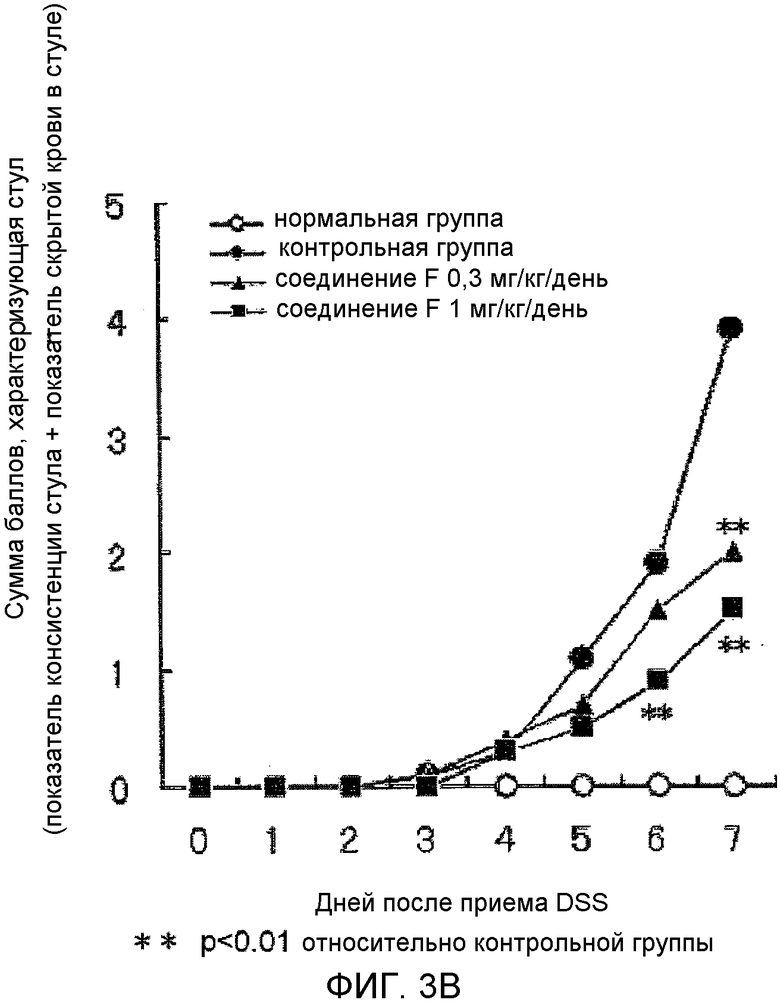
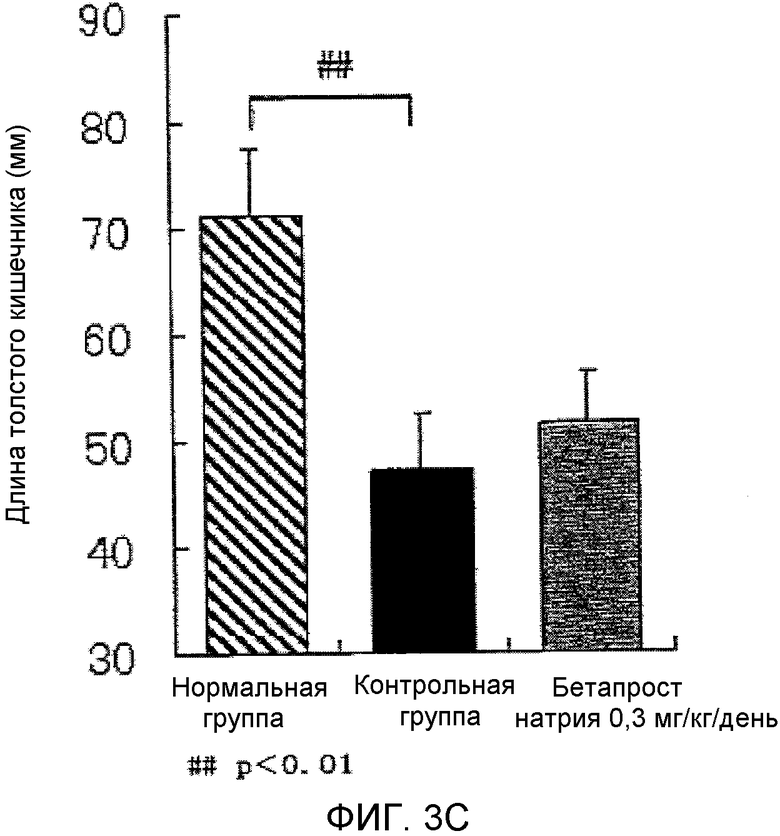
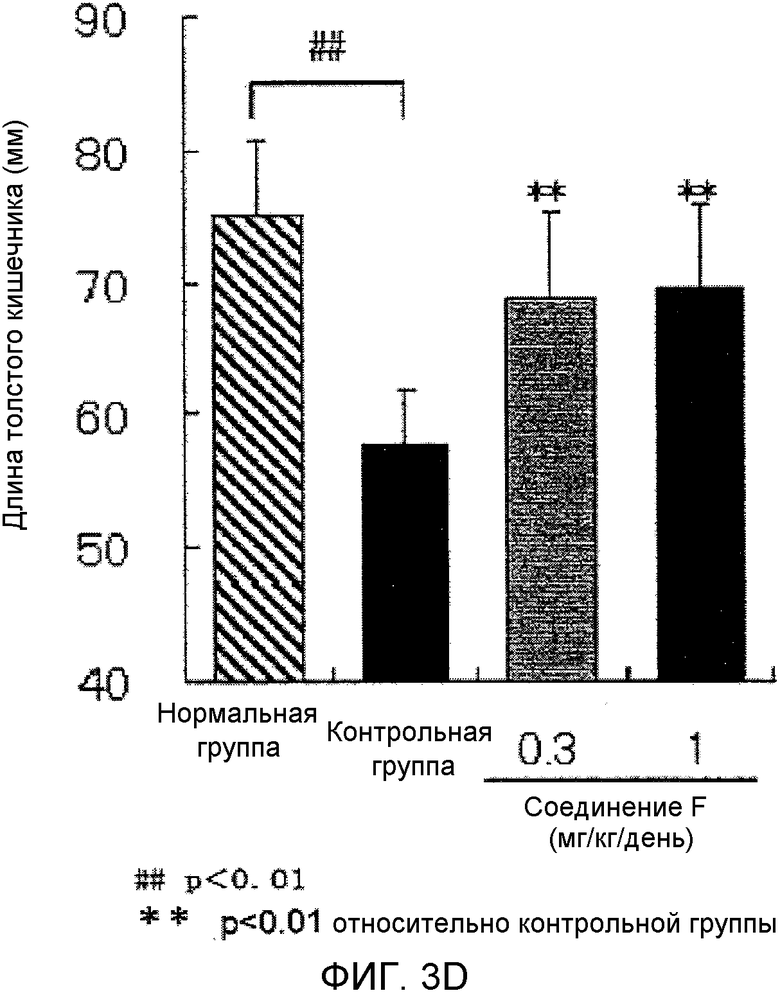
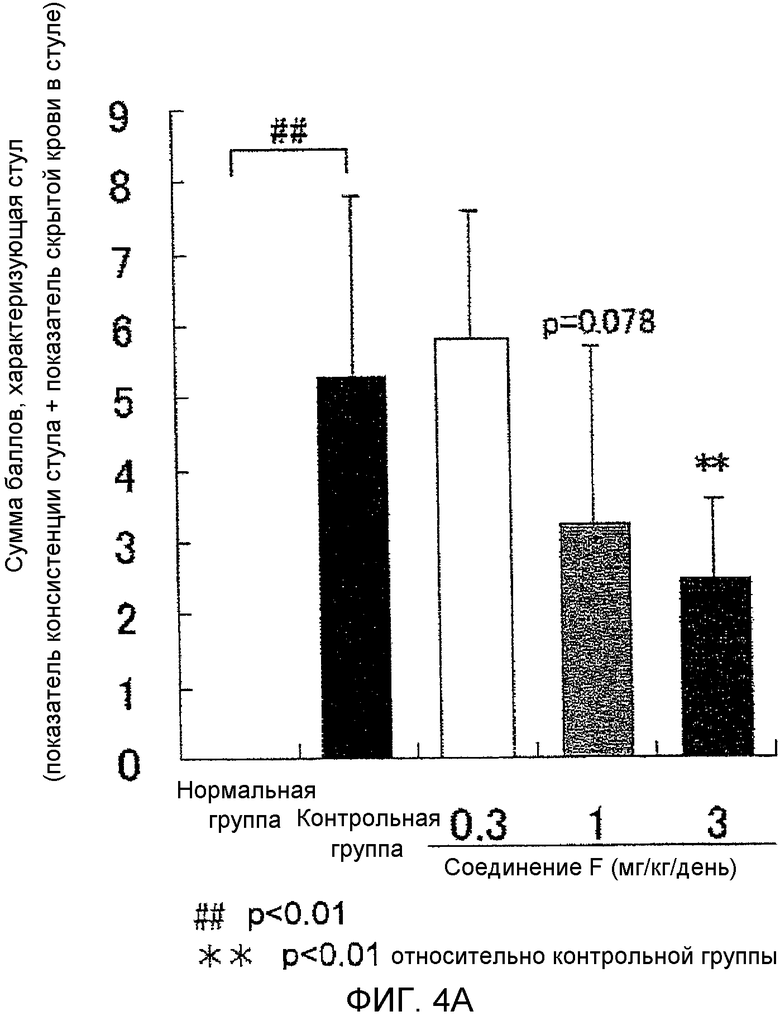
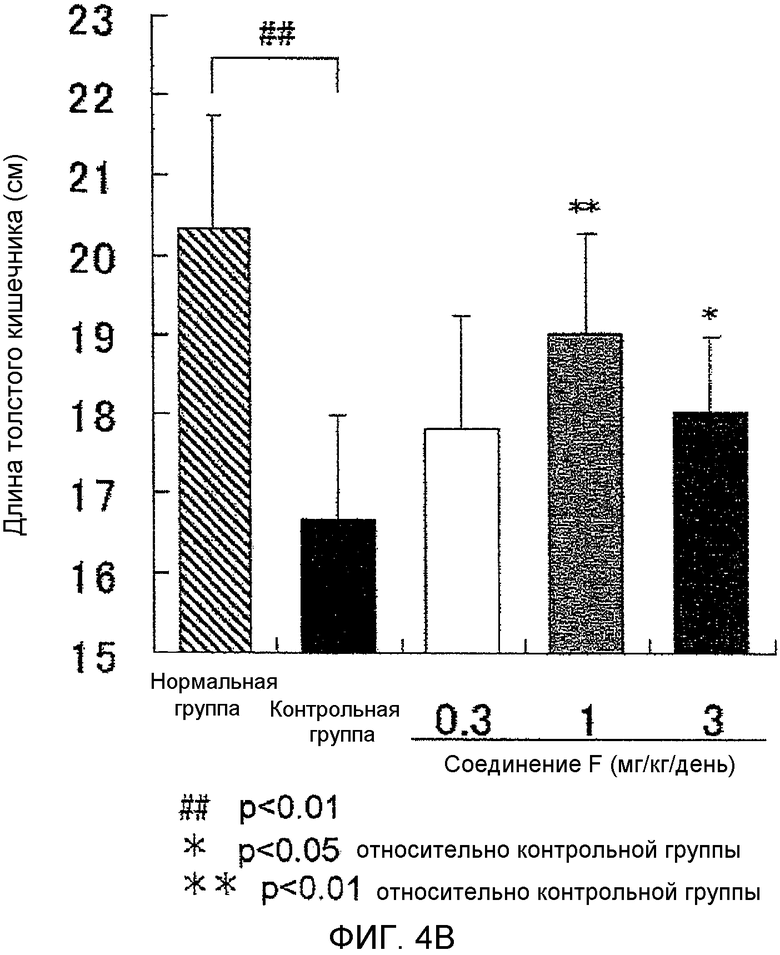
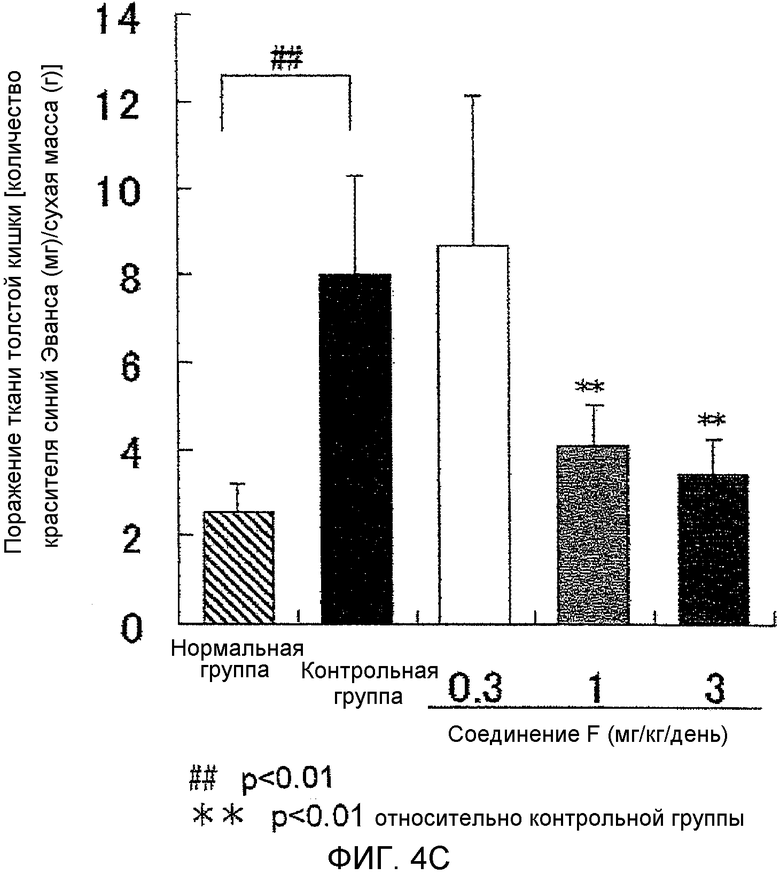
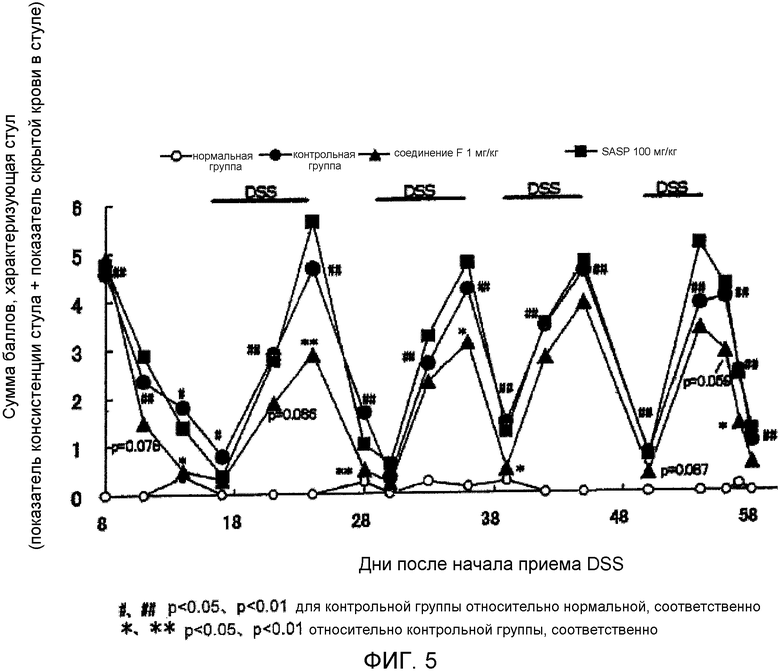
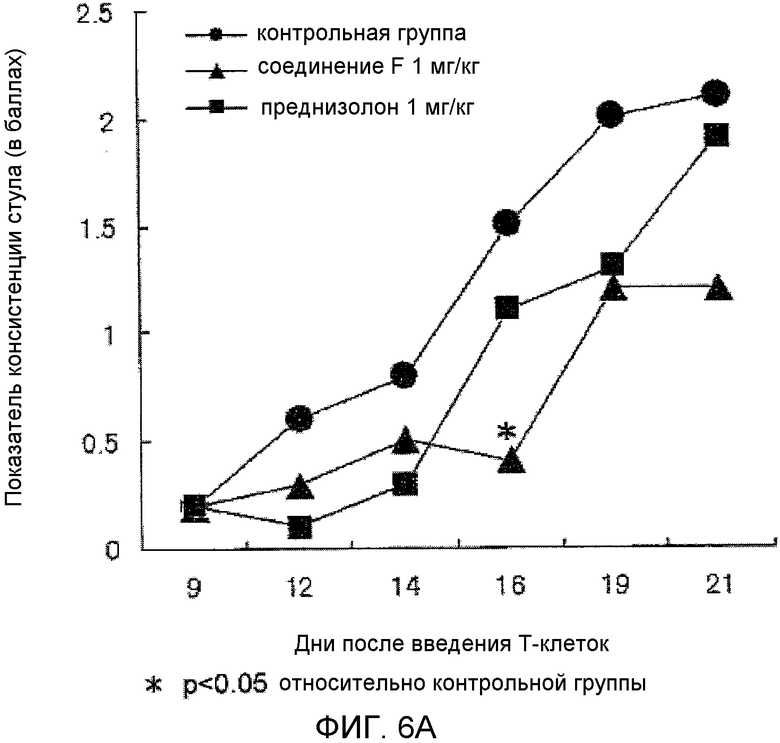
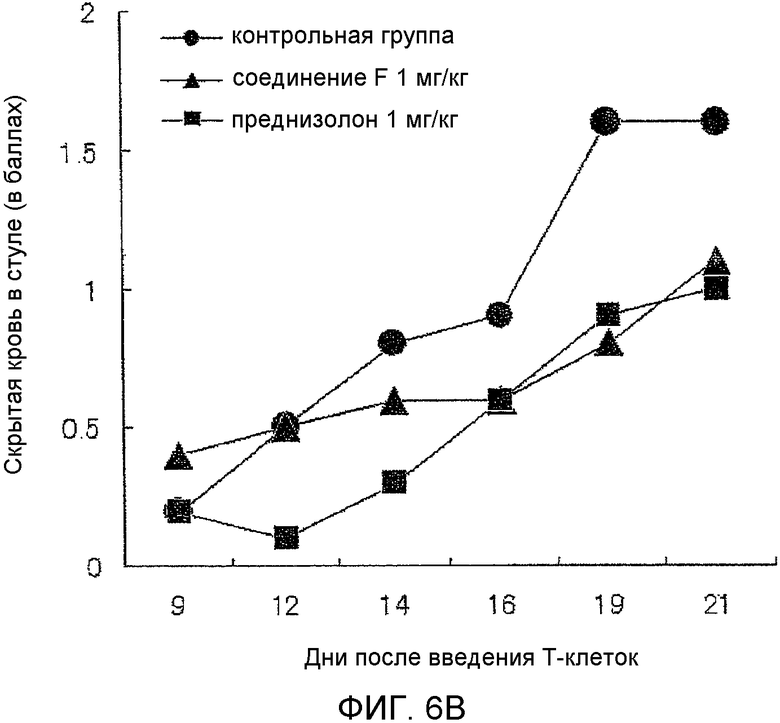
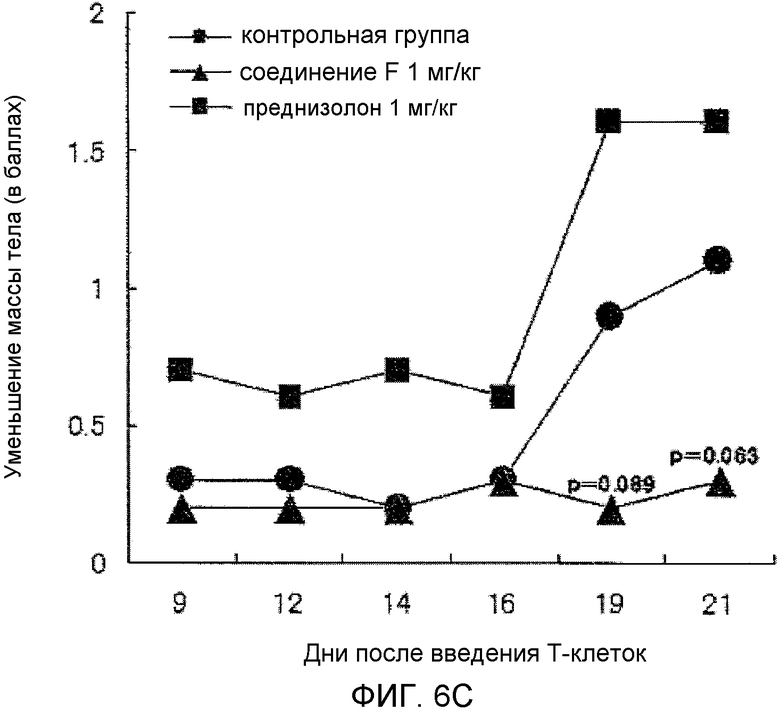
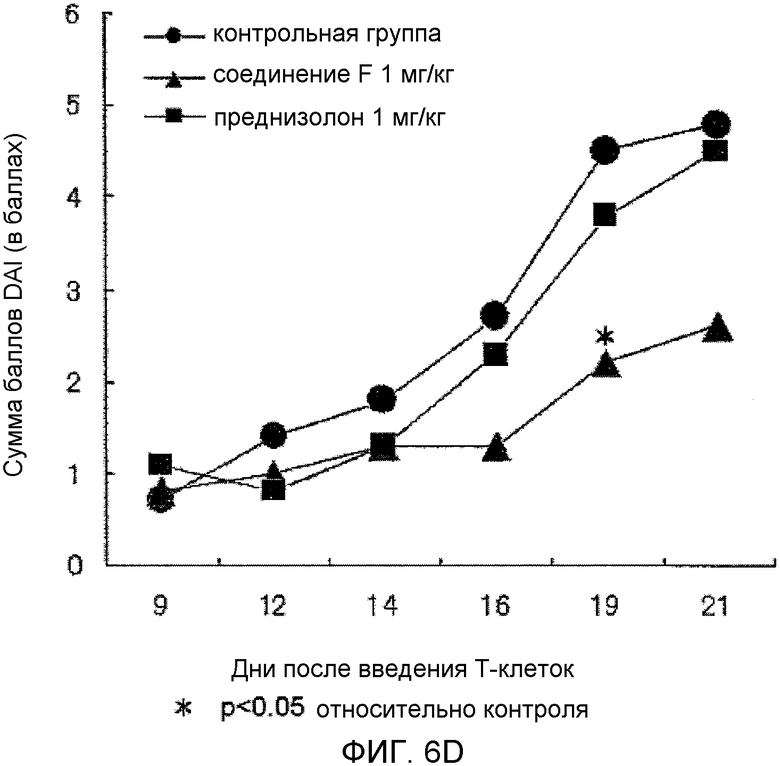
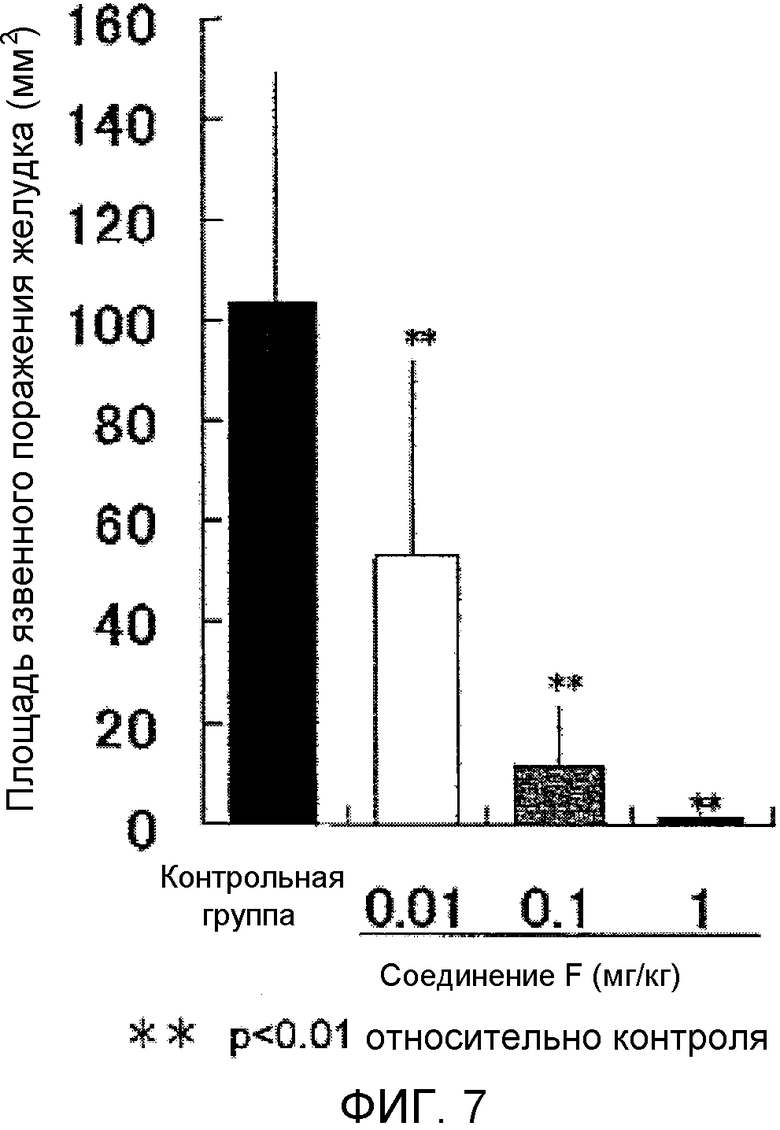
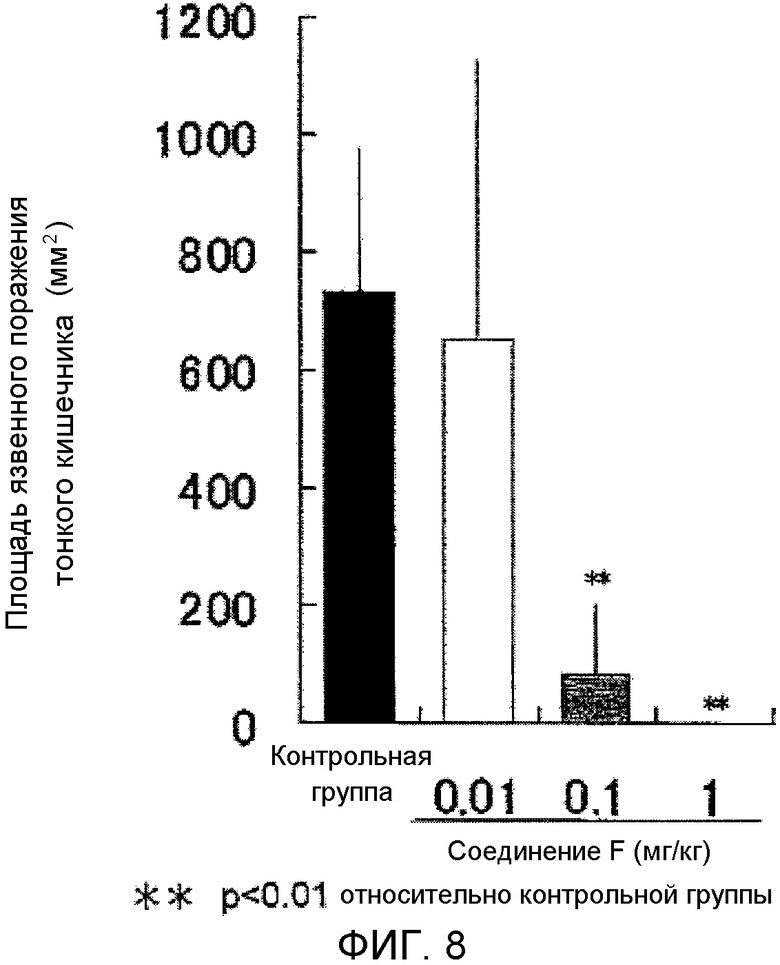
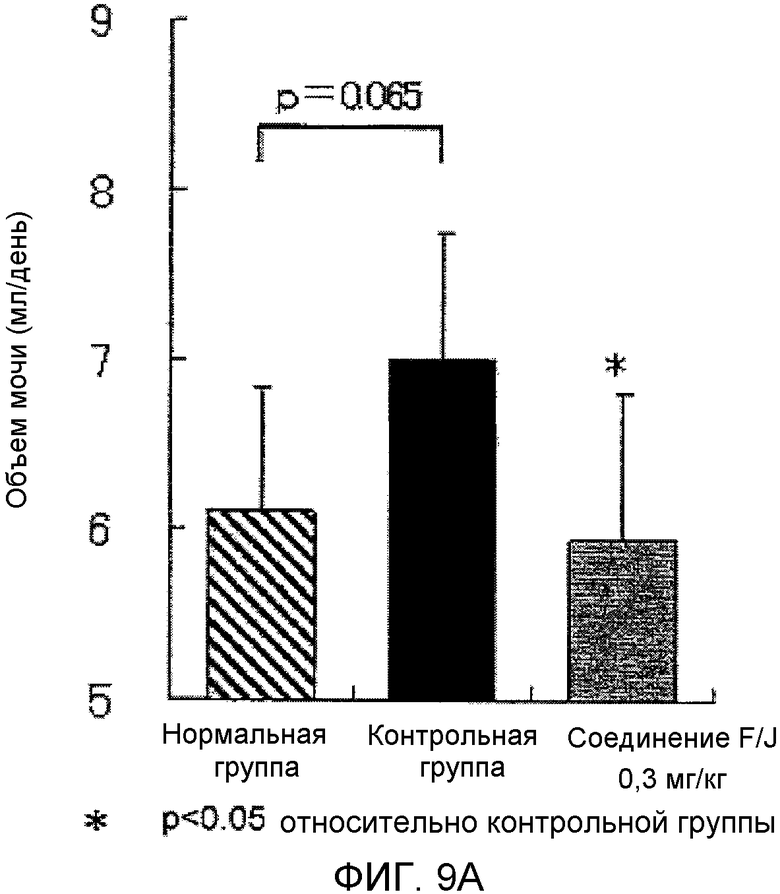
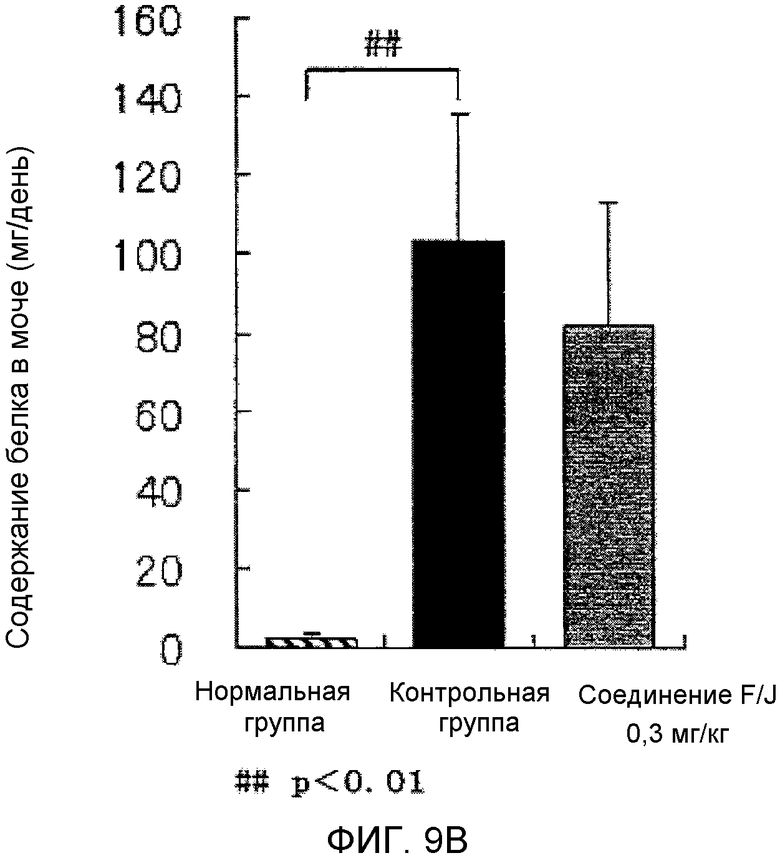
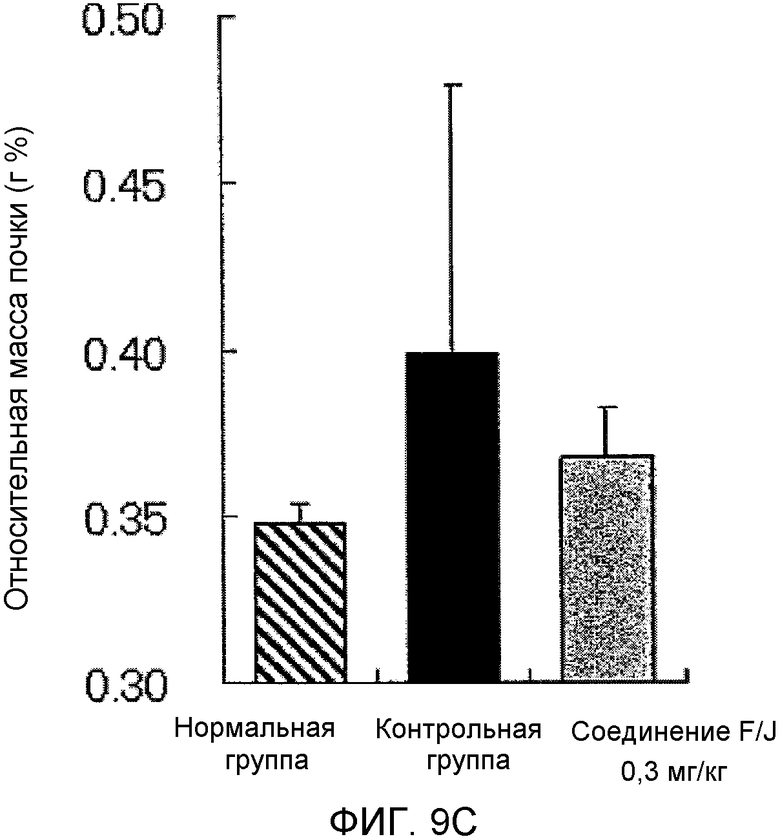
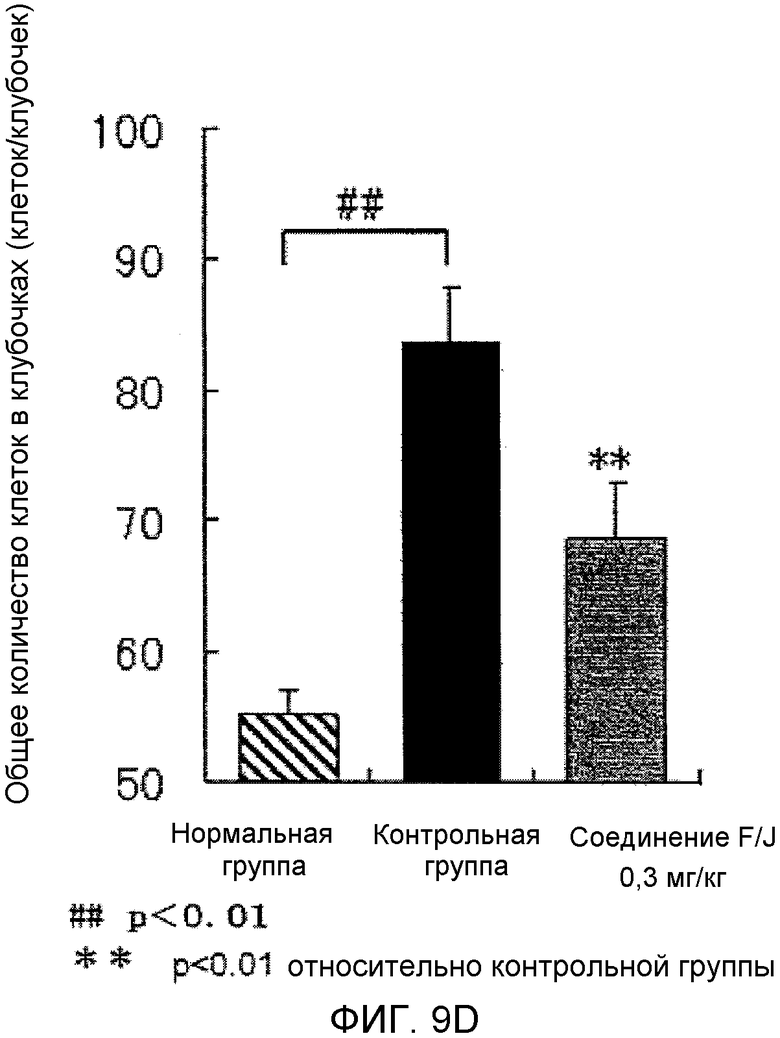
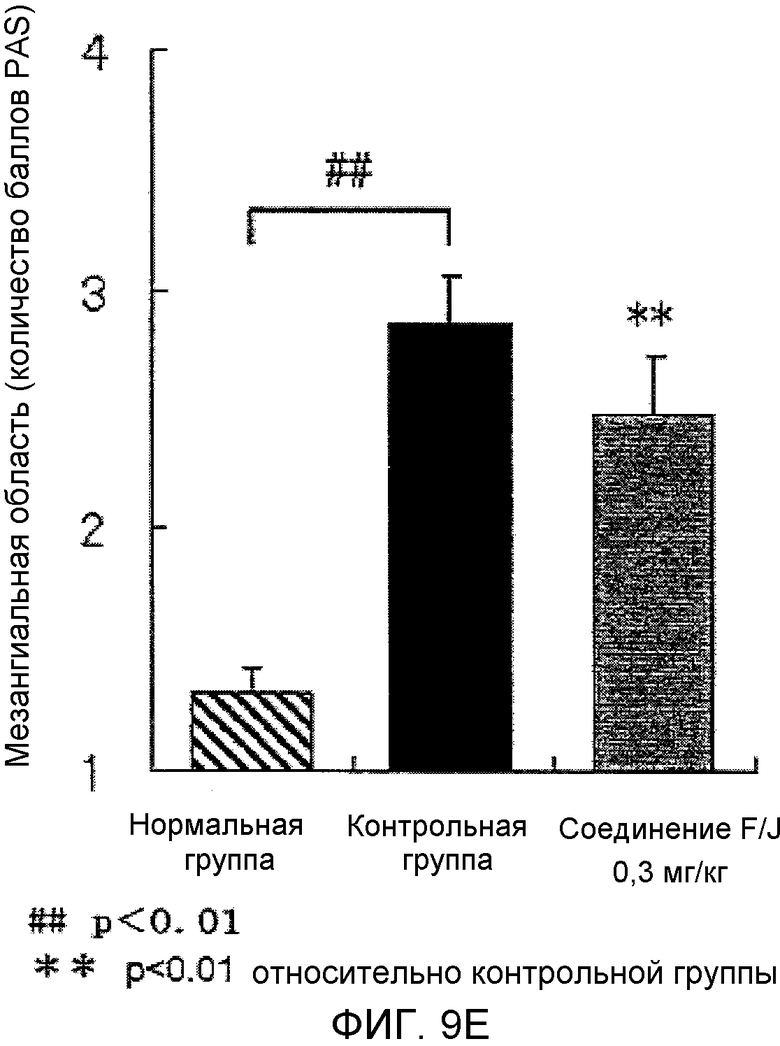
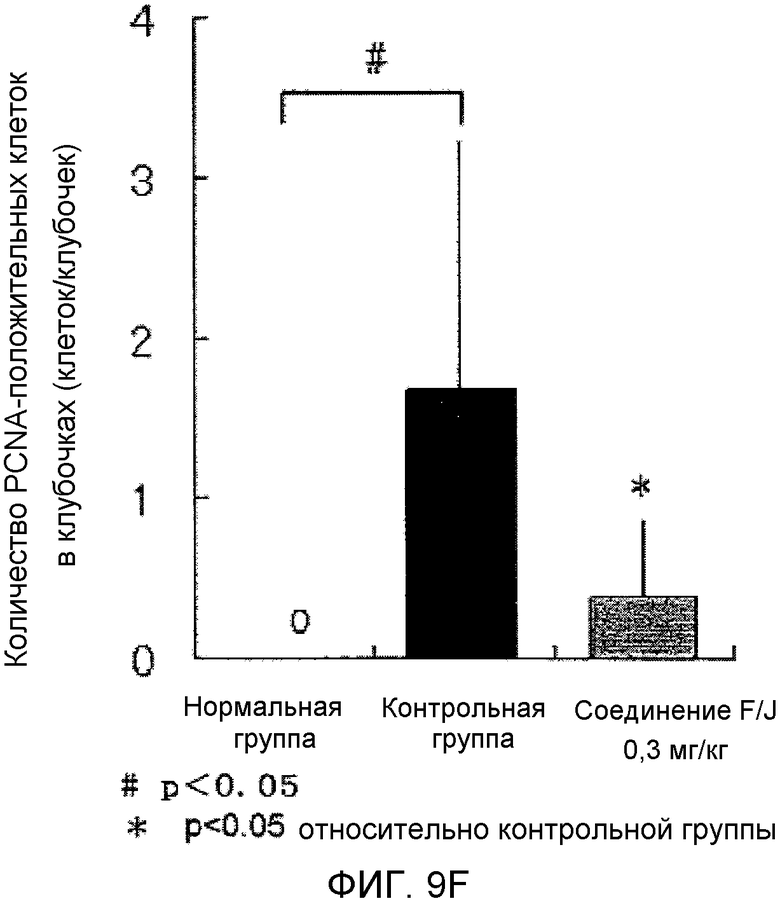
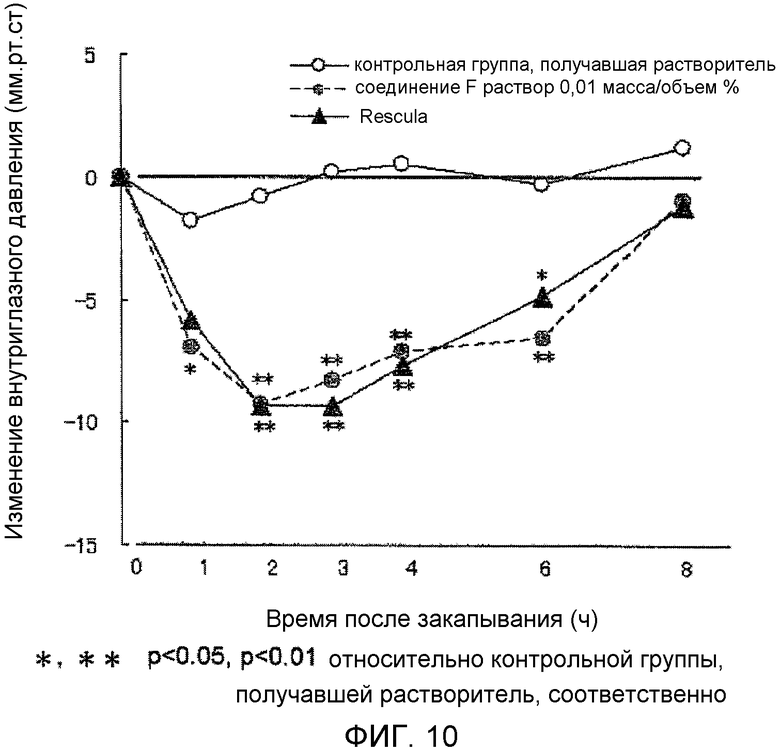
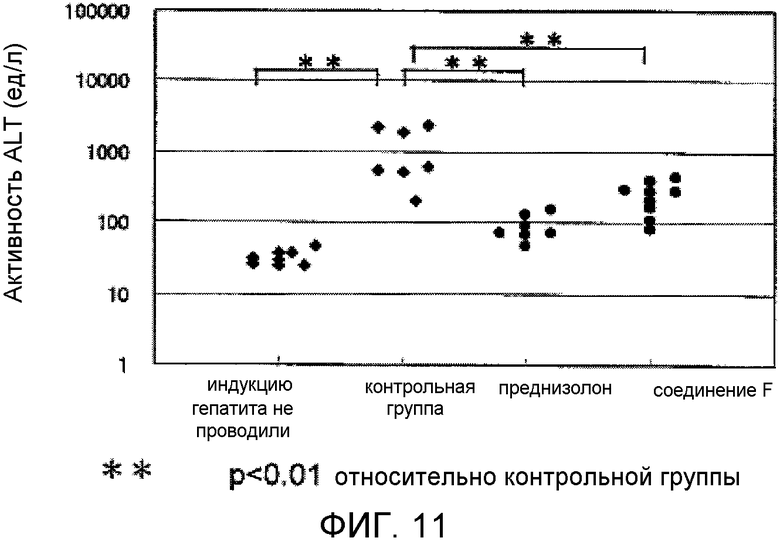
Изобретение относится к соединению, представленному формулой (1), где каждый из заместителей R1 и R2 независимо представляет собой атом водорода или линейную алкильную группу, включающую 1-3 атома углерода, R3 означает алкильную группу, включающую 1-4 атома углерода, или его фармацевтически приемлемой соли, а также изобретение относится к лекарственному средству, включающему это соединение в качестве антагониста EP4, применимое для профилактики и/или лечения иммунных заболеваний, сердечно-сосудистых заболеваний, сердечных заболеваний, респираторных заболеваний, глазных заболеваний, почечных заболеваний, заболеваний печени, заболеваний костей, язвенного заболеваний пищеварительного тракта, язвенного колита или болезни Крона, а также неврологических заболеваний, кожных заболеваний и т.п. 4 н. и 25 з.п. ф-лы, 8 табл., 26 ил., 26 пр.
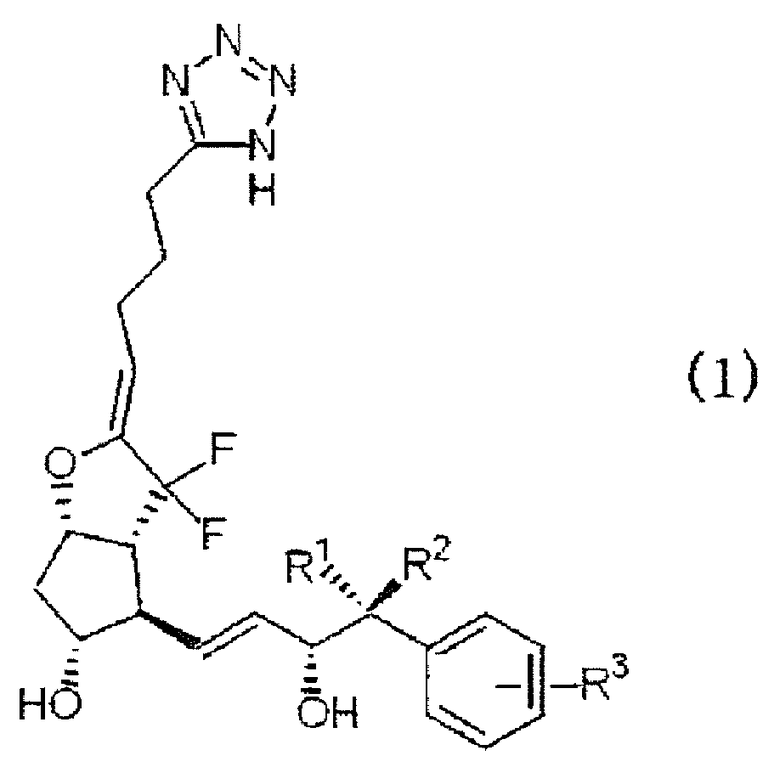
1. Агонист ЕР4, содержащий соединение, представленное формулой (1):
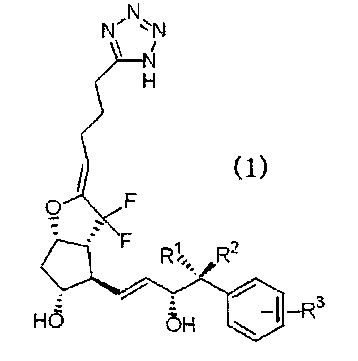
где каждый из заместителей R1 и R2 независимо представляет собой атом водорода или алкильную группу с прямой цепью, включающую от 1 до 3 атомов углерода, и R3 означает алкильную группу, включающую от 1 до 4 атомов углерода, или его фармацевтически приемлемую соль.
2. Агонист ЕР4 по п. 1, где R1 означает метильную группу.
3. Агонист ЕР4 по п. 1 или 2, где R3 означает метильную группу.
4. Агонист ЕР4 по п. 1, где R2 означает атом водорода.
5. Агонист ЕР4 по п. 1, где R1 означает метильную группу и R2 означает атом водорода.
6. Агонист ЕР4 по п. 1, где R3 означает м-метильную группу.
7. Агонист ЕР4 по п. 1, где R1 означает метильную группу, R2 означает атом водорода и R3 означает метильную группу.
8. Агонист ЕР4 по п. 1, где R1 означает атом водорода, R2 означает метильную группу и R3 означает метильную группу.
9. Агонист ЕР4 по п. 1, который представляет собой 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутан или его фармацевтически приемлемую соль.
10. Агонист ЕР4 по п. 1, который представляет собой 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутан или его фармацевтически приемлемую соль.
11. Агонист ЕР4 по п. 1, который представляет собой 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4S)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифторбицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутан или его фармацевтически приемлемую соль.
12. Лекарственное средство, включающее агонист ЕР4 по любому из пп. 1-11 в качестве действующего ингредиента.
13. Лекарственное средство по п. 12 для профилактики или лечения заболевания, в которое вовлечен ЕР4.
14. Лекарственное средство по п. 13 для профилактики или лечения заболевания, симптомы которого могут быть ослаблены действием селективного агониста ЕР4.
15. Лекарственное средство по п. 14, где заболевание, симптомы которого могут быть ослаблены действием селективного агониста ЕР4, представляет собой иммунное заболевание, сердечно-сосудистое заболевание, сердечное заболевание, респираторное заболевание, глазное заболевание, почечное заболевание, заболевание печени, заболевание костей, неврологическое заболевание или кожное заболевание.
16. Лекарственное средство по п. 15, где иммунное заболевание представляет собой боковой амиотрофический склероз, множественный склероз, синдром Шегрена, ревматоидный артрит, системную красную волчанку, отторжение трансплантата после пересадки, артрит, синдром системной воспалительной реакции, сепсис, гемофагоцитарный синдром, синдром активации макрофагов, болезнь Стилла, болезнь Кавасаки, гиперцитокинемию при диализе, множественный отказ органов, шок или псориаз.
17. Лекарственное средство по п. 15, где сердечно-сосудистое или сердечное заболевание представляет собой артериосклероз, стенокардию, инфаркт миокарда, расстройство мозга, вызванное кровоизлиянием в мозг, расстройство мозга, вызванное инфарктом мозга, расстройство мозга, вызванное субарахноидальным кровоизлиянием, легочную артериальную гипертензию, сужение периферических артерий или симптом, относящийся к затруднению периферического кровообращения.
18. Лекарственное средство по п. 15, где респираторное заболевание представляет собой астму, травму легких, легочный фиброз, эмфизему, бронхит или хроническую обструктивную болезнь легких.
19. Лекарственное средство по п. 15, где глазное заболевание представляет собой глаукому или глазную гипертензию.
20. Лекарственное средство по п. 15, где почечное заболевание представляет собой гломерулонефрит, диабетическую нефропатию, IgA нефропатию или поражение почек, вызванное ишемией-реперфузией.
21. Лекарственное средство по п. 15, где заболевание печени представляет собой гепатит, гепатопатию или поражение печени, вызванное ишемией-реперфузией.
22. Лекарственное средство по п. 15, где заболевание костей представляет собой остеопороз, перелом кости или фазу послеоперационного восстановления после остеотомии.
23. Лекарственное средство по п. 15, где неврологическое заболевание представляет собой гибель нервных клеток.
24. Лекарственное средство по п. 15, где заболевание кожи представляет собой пролежень или рану.
25. Лекарственное средство по п. 13, где заболевание, в которое вовлечен ЕР4, представляет собой заболевание, выбранное из группы, состоящей из облысения, алопеции, нарушения созревания шейки матки и расстройств слуха.
26. Лекарственное средство для профилактики или лечения воспалительного заболевания кишечника, включающее агонист ЕР4 по любому из пп. 1-11 в качестве действующего ингредиента.
27. Лекарственное средство по п. 26, где воспалительное заболевание кишечника представляет собой язвенный колит или болезнь Крона.
28. Лекарственное средство для профилактики или лечения язвенного заболевания пищеварительного тракта, включающее агонист ЕР4 по любому из пп. 1-11 в качестве действующего ингредиента.
29. Лекарственное средство по п. 28, где язвенное заболевание пищеварительного тракта представляет собой эзофагит, язву пищевода, гастрит, язву желудка или язву тонкого кишечника.
| Способ измерения временных интервалов между двумя точками синхронизированных сигналов с заданными уровнями сигнала в этих точках | 1977 |
|
SU669329A1 |
| WO 9813356 A1 02.04.1998 | |||
| WO 9717974 A1 22.05.1974 | |||
| WO 9841209 A1 24.09.1998 | |||
| WO 2008116670 A1 02.10.2008 | |||
| WO 8900563 A1 26.01.1989 | |||
| WO 9703973 A1 06.02.1997 | |||
| ПРОИЗВОДНОЕ 8-АЗАПРОСТАГЛАНДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, АГЕНТ ДЛЯ ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЙ | 2003 |
|
RU2306309C2 |

