Область техники
[1] Настоящее изобретение относится к новому ингибитору кислотной секреции и его применению.
Уровень техники
[2] Ингибитор протонного насоса (PPI), примером которого является омепразол, ингибирующий секрецию желудочной кислоты, широко используют в клинических условиях. Однако существующие PPI связаны с проблемами с точки зрения эффективности и побочных эффектов. В частности, существующие PPI часто составлены в форму кишечнорастворимых агентов, что обусловлено их нестабильностью в кислотных условиях, и в таком случае необходимо несколько часов до начала их действия, а до достижения максимального эффекта необходимо около 5 дней непрерывного введения. Кроме того, поскольку существующие PPI проявляют переменный терапевтический эффект вследствие полиморфизма метаболизирующих ферментов и межлекарственного взаимодействия с такими препаратами как диазепам и т.п., необходимо усовершенствование указанных лекарственных средств.
[3] Кроме того, поскольку PPI представляют собой пролекарства, активируемые желудочной кислотой, и действуют только на активный протонный насос, имеют место такие недостатки как задержка времени проявления максимального эффекта лекарственного средства, неэффективность подавления кислотной секреции ночью, необходимость применения до еды и т.п. Кроме того, PPI метаболизируются, главным образом, ферментом CYP2C19, что обусловливает существенное различие эффективности у различных индивидуумов вследствие генетического полиморфизма фермента CYP2C19.
[4] Для преодоления вышеописанных недостатков PPI перспективными являются калий-конкурентные блокаторы кислоты (P-CAB). P-CAB эффективно и быстро ингибируют секрецию желудочной кислоты посредством обратимого и конкурентного связывания с ионами K+ в протонном насосе (H+/K+ АТФаза), который представляет собой фермент, участвующий в последней стадии секреции желудочной кислоты в клетках желудка. Такие лекарственные формы P-CAB демонстрируют эффективное ингибирование при нормальной желудочной кислотности (pH 1-3), в отличие от лекарственных форм PPI. Кроме того, указанные лекарственные формы P-CAB должны обладать фармакологической активностью, снижающей способность ингибирования при увеличении рН, но некоторые препараты P-CAB, согласно отчетам, проявляют фармакологическую активность, сохраняющуюся даже при увеличении рН, что вызывает некоторые побочные эффекты, связанные с данной проблемой. Кроме того, поскольку лекарственные формы P-CAB метаболизируются, главным образом, ферментом CYP3A4, то различие эффективности у различных индивидуумов является относительно низким, и проблемы, связанные с межлекарственными взаимодействиями с препаратами, метаболизируемыми ферментом CYP2C19, являются относительно небольшими.
[5] В международной публикации патента № WO2019/013310 A1 описан вонопразан в качестве калий-конкурентного блокатора кислоты.
[6] Однако подтверждено, что вонопразан вызывает тяжелую гипергастринемию по сравнению с существующим PPI препаратом лансопразолом. Гипергастринемия может вызывать такие проблемы как гиперплазия энтерохромаффиноподобных (ECL) клеток; гиперплазия париетальных клеток; железистые полипы дна желудка; потеря костной массы, нарушение свойств костей, переломы и т.п. Действительно, было описано, что в испытаниях канцерогенности на мышах и крысах вонопразан связан с развитием желудочных нейроэндокринных опухолей. Однако поскольку прекращение введения препаратов на основе P-CAB или PPI, таких как вонопразан, приводит к возобновлению избытка желудочной кислоты и вызывает несварение и т.п., то нельзя просто прекратить введение препаратов ввиду вышеописанных проблем.
[7] С другой стороны, PPI используют для предотвращения язвы желудка и двенадцатиперстной кишки посредством введения нестероидных противовоспалительных препаратов (НСПВП). Однако описано, что вонопразан усугубляет повреждение тонкого кишечника, вызванное различными типами НСПВП. Например, повреждение желудочно-кишечного тракта, вызванное НСПВП, включает отек, эритему, кровоизлияние под слизистую оболочку, эрозию, язву и т.п., и пациенты, которые постоянно применяют НСПВП в течение длительного времени, имеют такие проблемы как множественные повреждения слизистой оболочки тонкого кишечника и т.п. С этой точки зрения, в клиническом аспекте вонопразан может иметь существенные ограничения в комбинации с НСПВП.
[8] В качестве механизма, посредством которого такие препараты как НСПВП или спирты вызывают повреждение слизистой оболочки желудочно-кишечного тракта, известны два основных механизма: местное раздражающее действие и системное раздражающее действие. Местное раздражающее действие вызвано повреждением митохондриальных ионных ловушек, а системное раздражающее действие обусловлено снижением уровня простагландина и NO (оксида азота (II)). Помимо митохондриального повреждения, вызванного окислительным стрессом, в случае повреждения клеток сосудистого эндотелия нарушается микроциркуляция, что обусловливает высокую уязвимость слизистой оболочки желудочно-кишечного тракта к повреждениям и нарушает механизм восстановления слизистой оболочки после повреждения. Вследствие совокупного действия указанных механизмов может возникать или усугубляться повреждение слизистой оболочки желудочно-кишечного тракта, т.е. язва желудка, энтеропатия и т.п.
[9] Соответственно, даже с учетом эффекта ванопразана с точки зрения подавления секреции желудочной кислоты, применение указанного лекарственного средства неизбежно является весьма ограниченным вследствие вышеописанных возможных проблем.
[10] Отдельно следует указать, что в качестве одной из основных причин желудочно-кишечных заболеваний, таких как хронический гастрит, пептическая язва, рак желудка и т.п., является Helicobacter pylori (H. pylori). Даже несмотря на то, что распространенность Helicobacter pylori в Корее постепенно снижается, все еще отмечается встречаемость на уровне 50% или более. В частности, Helicobacter pylori связан с желудочно-кишечными заболеваниями и, следовательно, значение антибактериальных лекарственных агентов растет день ото дня. В частности, как отмечено в нескольких исследованиях, антибактериальное лечение Helicobacter pylori снижает частоту кровотечений при пептической язве, и поэтому в некоторых странах рекомендуется антибактериальное лечение в данной группе пациентов. Для проведения антибактериальной терапии пациентам обычно предлагают принимать кларитромицин, амоксициллин и т.п. в качестве терапии первой линии вместе с ингибиторами желудочной кислоты, такими как PPI. Для мультилекарственного применения PPI и антибиотиков риск межлекарственного взаимодействия (DDI) должен быть низким, и риск такого взаимодействия может быть спрогнозирован на основе in vitro ингибирования CYP, фенотипирования CYP/UGT, испытаний активации CYP и т.п.
[11] Тем не менее, требуется дополнительное или повторное введение различных антибиотиков до второго и третьего лечения, и существуют сообщения о побочных эффектах и переносимости, связанных с ним. Таким образом, существует потребность в разработке лекарственного средства, снижающего желудочную кислотность, для усиления антибактериального эффекта антибиотиков против Helicobacter pylori (H. pylori) и подходящего для длительного применения, например, лекарственного средства, которое проявляет способность ингибировать протонно-калиевый насос и т.п., а также антибактериальную активность в отношении различных штаммов Helicobacter pylori.
[12] Кроме того, для перорального лекарственного средства измеряют биодоступность, которая представляет собой скорость, с которой введенное лекарственное средство поступает в систему кровообращения и используется в организме. Чем выше биодоступность, тем выше скорость и степень абсорбции активного ингредиента или части лекарственного средства и его использования в очаге действия и, следовательно, высокая биодоступность является одним из важнейших элементов пероральных лекарственных средств. Обычно биодоступность увеличивается с усилением абсорбции в желудочно-кишечном тракте и со снижением степени пресистемного метаболизма, и при введении на биодоступность влияет употребление пищи во время приема лекарственного средства, межлекарственные взаимодействия при приеме нескольких препаратов, а также растворимость лекарственного средства, кристаллический полиморфизм, размер и форма частиц, площадь поверхности частиц и т.п. с точки зрения свойств лекарственного средства.
[13] Кроме того, важно поддерживать концентрацию лекарственного средства в органе-мишени, в данном случае в желудочной ткани, а также биодоступность в системе кровообращения. Таким образом, распределение и поддержание лекарственного средства в органе-мишени, желудочной ткани, считается важным фармакокинетическим свойством при разработке лекарственного средства P-CAB.
[14] Вместе с тем, соматостатин, также известный как гормон, ингибирующий гормон роста (GHIH), представляет собой циклический пептид, экспрессируемый в желудочно-кишечном тракте, поджелудочной железе, гипоталамусе и центральной нервной системе. Он секретируется D-клетками желудка и поджелудочной железы и действует как паракринный регулятор секреции желудочной кислоты, и подавляет секрецию желудочной кислоты, ингибируя секрецию гастрина желудочными G-клетками, а также секрецию кислоты париетальными клетками. Активация рецепторов соматостатина аналогами соматостатина и агонистами рецептора соматостатина подавляет секрецию гастрина для регуляции высвобождения гистамина из клеток ECL, а также ингибирует секрецию кислоты. В достоверных животных моделях и у пациентов с гипергастринемией было показано, что аналог соматостатина снижает общую секрецию желудочной кислоты, уменьшая секрецию гастрина и ответ желудочной кислоты.
[15] Подавление желудочной кислоты, вызванное приемом лекарственных средств, таких как PPI и т.п., вызывает гипергастринемию вследствие ингибирования секреции соматостатина D-клетками и усиления секреции гастрина G-клетками по механизму обратной связи. Гастрин ускоряет рост эпителиальных клеток, вызывает гиперплазию обкладочных клеток в теле желудка и увеличивает массу париетальных клеток. Это приводит к пролиферации клеток аденомы и гиперплазии клеток ECL, что может увеличивать риск нейроэндокринных опухолей. Кроме того, распространенность нейроэндокринных опухолей среди опухолей, встречающихся в двенадцатиперстной кишке, является относительно высокой, и известно, что нейроэндокринные опухоли, вызванные секрецией гастрина, являются самой распространенной формой нейроэндокринных опухолей, встречающихся в двенадцатиперстной кишке, на долю которой приходится приблизительно 65% всех случаев. Подтверждено, что в группе, принимающей вонопразан, отмечен более высокий уровень гастрина в крови, чем в группе, принимающей известный препарат PPI, вследствие механизма обратной связи, заключающегося в подавлении избыточной желудочной кислоты. Поскольку гипергастринемия может стимулировать эндокринные клетки кишечника и может увеличивать риск нейроэндокринных опухолей, продолжаются исследования безопасности при длительном применении.
[16] Описано, что ингибирование секреции гастрина посредством активации рецептора соматостатина подавляет гиперпролиферацию клеток ECL. Действительно, было описано, что синтетические пептидные аналоги соматостатина с показаниями для эндокринных заболеваний, таких как акромегалия, нейроэндокринные опухоли (NET) и заболевания пищеварительной системы, такие как кровоизлияния в верхнем отделе желудочно-кишечного тракта и т.п., т.е. Sandostatin® (ацетат октреотида) и Somatuline® в форме депо (ланреотид), ингибируют секрецию гастрина в желудочных нейроэндокринных опухолях, препятствуя гиперпролиферации клеток ECL.
[17] Кроме того, описан противовоспалительный ответ вследствие активации рецептора соматостатина. Соматостатин представляет собой тип нейропептида, который ингибирует неврологическое воспаление и регулирует секрецию гормонов и нейротрансмиттеров. Известно, что соматостатин подавляет нейрогенное воспаление и участвует в ноцицепции, а также высвобождается нервными клетками желудочно-кишечного тракта и нейроэндокринными клетками слизистой оболочки, оказывая противовоспалительное действие. Известно, что соматостатин регулирует секрецию гормонов и нейротрансмиттеров, подавляя нейрогенное воспаление, а также участвует в ноцицепции. Помимо регулирования нейроэндокринной системы, воспалительный соматостатин ингибирует пролиферацию T-лимфоцитов и гранулоцитов. Известно, что аналоги соматостатина усиливают экспрессию противовоспалительного фактора IL-10 и ингибируют экспрессию провоспалительных факторов IFN-γ и TNF-α. Таким образом, противовоспалительная роль соматостатина описана, в основном, в исследованиях, касающихся воспалительной болезни кишечника (ВБК). Известно, что уровни соматостатина в кишечнике снижены у пациентов с ВБК, а также известно, что чем выше степень воспаления кишечного тракта, тем ниже уровень соматостатина. Фактически было описано, что аналог соматостатина октреотид облегчает симптомы ВБК у пациентов и в животных моделях.
[18] На основании известного уровня техники, авторами настоящего изобретения синтезировано новое соединение, обладающее превосходным ингибирующим действием в отношении протонного насоса, которое описано в настоящем документе, и подготовлено настоящее описание.
Сущность изобретения
Техническая задача
[19] В настоящем изобретении предложено соединение, представленное химической формулой 1, или его фармацевтически приемлемая соль.
[20] Кроме того, в настоящем изобретении предложена фармацевтическая композиция, содержащая соединение, представленное химической формулой 1, или его фармацевтически приемлемую соль.
[21] Кроме того, в настоящем изобретении предложена фармацевтическая композиция для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, содержащая соединение, представленное химической формулой 1, или его фармацевтически приемлемую соль.
[22] В настоящем изобретении также предложено соединение, представленное химической формулой 1, или его фармацевтически приемлемая соль для применения для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой.
[23] Кроме того, в настоящем изобретении предложено применение соединения, представленного химической формулой 1, или его фармацевтически приемлемой соли для производства лекарственного средства для лечения заболеваний или симптомов, требующих назначения ингибиторов кислотной секреции, таких как желудочно-кишечные язвы, желудочно-кишечные воспалительные заболевания или заболевания, связанные с желудочной кислотой.
[24] Кроме того, в настоящем изобретении предложен способ лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, включающий: введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения, представленного химической формулой 1, или его фармацевтически приемлемой соли.
[25] В настоящем изобретении также предложен ингибитор секреции желудочной кислоты, содержащий соединение, представленное химической формулой 1, или его фармацевтически приемлемую соль.
[Техническое решение]
[26] Настоящее изобретение относится к соединению, представленному следующей химической формулой 1, или его фармацевтически приемлемой соли:
[27] [Химическая формула 1]
[28] 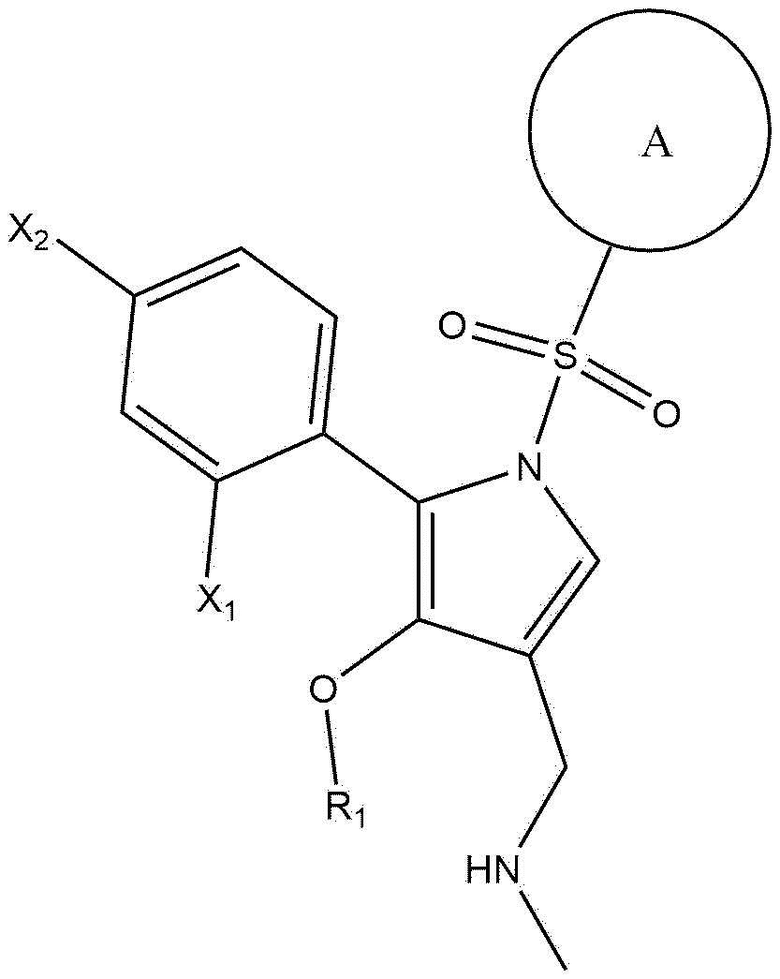
[29] в химической формуле 1
[30]  представляет собой замещенную или незамещенную пиридинильную группу, причем замещенная пиридинильная группа замещена по меньшей мере одним или более из -OH, -O(C1-C4 алкила), -(C1-C4 алкила), галогена или -CN;
представляет собой замещенную или незамещенную пиридинильную группу, причем замещенная пиридинильная группа замещена по меньшей мере одним или более из -OH, -O(C1-C4 алкила), -(C1-C4 алкила), галогена или -CN;
[31] X1 представляет собой галоген, которым является F, Cl, Br или I;
[32] X2 представляет собой водород или галоген, которым является F, Cl, Br или I; и
[33] R1 представляет собой метил или этил.
[34] В соответствии с другим вариантом реализации настоящего изобретения,
[35] может представлять собой, например, замещенный или незамещенный пиридин-3-ил, или замещенный или незамещенный пиридин-2-ил.
[36] Более конкретно, может представлять собой 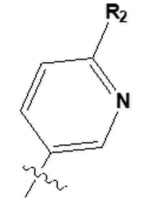 , где R2 представляет собой -O(C1-C4 алкил) или -(C1-C4 алкил).
, где R2 представляет собой -O(C1-C4 алкил) или -(C1-C4 алкил).
[37] В соответствии с другим вариантом реализации настоящего изобретения, соединение, представленное химической формулой 1 согласно настоящему изобретению, или его фармацевтически приемлемая соль может относиться к соединению, представленному следующей химической формулой 2, или его фармацевтически приемлемой соли:
[38] [Химическая формула 2]
[39] 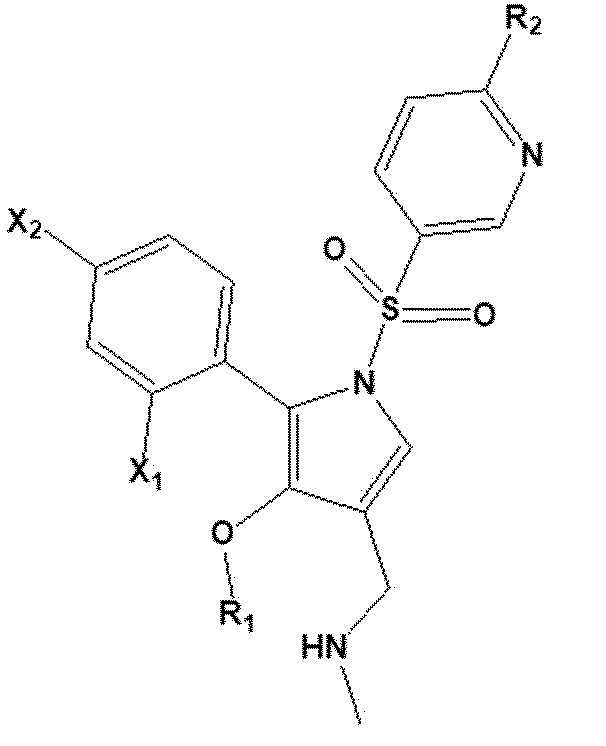
[40] в химической формуле 2
[41] X1 представляет собой F;
[42] X2 представляет собой водород или F;
[43] R1 представляет собой метил или этил; и
[44] R2 представляет собой -O(C1-C4 алкил) или -(C1-C4 алкил).
[45] В соответствии с другим вариантом реализации настоящего изобретения,
[46] в химической формуле 2 -O(C1-C4 алкил) может представлять собой, в частности, метокси или этокси. В частности, -(C1-C4 алкил) может представлять собой метил или этил.
[47] Таким образом, вышеуказанный R2 может представлять собой метокси, этокси, метил или этил. Более предпочтительно, R2 может представлять собой метокси или метил.
[48] Более конкретно, в химической формуле 2
[49] может представлять собой
[50] 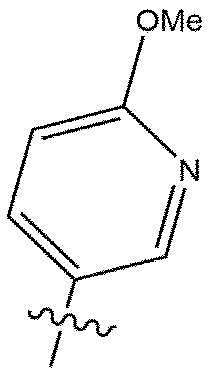 или
или 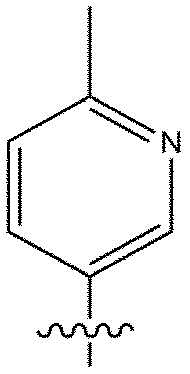 .
.
[51] В соответствии с другим вариантом реализации настоящего изобретения, в химической формуле 2 R1 может представлять собой метил.
[52] В соответствии с другим вариантом реализации настоящего изобретения, в химической формуле 2 R1 может представлять собой метил, а R2 может представлять собой метокси или метил.
[53] В соответствии с другим вариантом реализации настоящего изобретения, в химической формуле 2 X1 может представлять собой F; X2 может представлять собой F; R1 может представлять собой метил; и R2 может представлять собой метокси или метил.
[54] В соответствии с другим вариантом реализации настоящего изобретения, в химической формуле 2 X1 может представлять собой F; X2 может представлять собой водород; R1 может представлять собой метил; и R2 может представлять собой метокси или метил.
[55] В соответствии с другим вариантом реализации настоящего изобретения, в химической формуле 2 X1 может представлять собой F; X2 может представлять собой водород или F; R1 может представлять собой метил; и R2 может представлять собой метокси.
[56] В соответствии с другим вариантом реализации настоящего изобретения, в химической формуле 2 X1 может представлять собой F; X2 может представлять собой водород или F; R1 может представлять собой метил; и R2 может представлять собой метил.
[57] Другой вариант реализации настоящего изобретения относится к соединению, независимо выбранному из одного или из любой комбинации следующих соединений, или к его фармацевтически приемлемой соли:
[58] 1-5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин;
[59] 1-(5-(2-фторфенил)-4-метокси-1-(пиридин-2-илсульфонил)-1H-пиррол-3-ил)-N-метилметанамин;
[60] 1-(5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин;
[61] 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин;
[62] 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин; и
[63] 1-(5-(2-фторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин.
[64] Другой более предпочтительный вариант реализации настоящего изобретения относится к соединению, представленному химической формулой 2, или его фармацевтически приемлемой соли, и относится к соединению, независимо выбранному из любого одного или из любой комбинации следующих соединений, или к его фармацевтически приемлемой соли:
[65] 1-(5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин;
[66] 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин;
[67] 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин; и
[68] 1-(5-(2-фторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин.
[69] Другой, еще более предпочтительный вариант реализации настоящего изобретения относится к соединению, представленному химической формулой 2, или его фармацевтически приемлемой соли, и относится к соединению, независимо выбранному из любого одного или из любой комбинации следующих соединений, или к его фармацевтически приемлемой соли:
[70] 1-(5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин; и
[71] 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин.
[72] В настоящем изобретении фармацевтически приемлемая соль означает соль, обычно используемую в фармацевтической промышленности, и она может представлять собой, например, соли неорганических ионов, полученные из кальция, натрия и т.п., соли неорганических кислот, полученные фосфорной кислоты, бромистоводородной кислоты, иодистоводородной кислоты, серной кислоты и т.п., соли органических кислот, полученные из уксусной кислоты, трифторуксусной кислоты, лимонной кислоты, малеиновой кислоты, молочной кислоты, гликолевой кислоты, аскорбиновой кислоты, угольной кислоты, ванилиновой кислоты и т.п., соли сульфоновых кислот, полученные из метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты и т.п., соли аминокислот, полученные из глицина, аргинина и т.п., и соли аминов, полученные из триметиламина, триэтиламина и т.п., но типы солей согласно настоящему изобретению не ограничены перечисленными солями.
[73] В другом варианте реализации настоящего изобретения предложена фармацевтическая композиция, содержащая соединение, представленное химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемую соль.
[74] В другом варианте реализации настоящего изобретения предложена фармацевтическая композиция, содержащая соединение, представленное химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемую соль; и фармацевтически приемлемый носитель.
[75] В другом варианте реализации настоящего изобретения предложена фармацевтическая композиция для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, содержащая соединение, представленное химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемую соль.
[76] Настоящее изобретение также включает следующие варианты реализации:
[77] соединение, представленное химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемая соль для применения в качестве лекарственного средства;
[78] соединение, представленное химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемая соль для применения для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, рассмотренных в настоящем изобретении;
[79] способ лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, включающий: введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения, представленного химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемой соли;
[80] применение соединения, представленного химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемой соли для производства лекарственного средства для лечения заболеваний или симптомов, требующих назначения ингибиторов кислотной секреции, таких как желудочно-кишечные язвы, желудочно-кишечные воспалительные заболевания или заболевания, связанные с желудочной кислотой;
[81] соединение, представленное химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемая соль для применения для лечения заболеваний или симптомов, требующих назначения ингибиторов секреции кислоты;
[82] фармацевтическая композиция, содержащая соединение, представленное химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемую соль, для лечения заболеваний или симптомов, требующих назначения ингибиторов секреции кислоты; или
[83] ингибитор секреции желудочной кислоты, содержащий соединение, представленное химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемую соль.
[84] Вышеописанное соединение, представленное химической формулой 1, или его фармацевтически приемлемая соль предпочтительно представляет собой соединение, представленное химической формулой 2, или его фармацевтически приемлемую соль.
[85] Все приведенные примеры или их фармацевтически приемлемые соли могут быть заявлены по отдельности или вместе в составе группы, в любой комбинации с любым количеством всех и каждого варианта реализации, описанного в настоящем документе.
[86] Настоящее изобретение также относится к фармацевтической композиции, содержащей соединение, представленное химической формулой 1, определение которого приведено в любом из вариантов реализации, описанных в настоящем изобретении, или его фармацевтически приемлемую соль, для применения для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, рассмотренных в настоящем документе.
[87] В частности, желудочно-кишечная язва относится к язве, которая находится в пищеварительной системе, включая и желудок, и кишечник. Примеры желудочно-кишечной язвы могут включать, но не ограничиваясь ими, пептическую язву, язву желудка, язву двенадцатиперстной кишки, язву, вызванную НСПВП, язву на фоне острого стресса, синдром Золингера-Эллисона и т.п. Если язва принимает тяжелую форму, то может развиваться рак. Например, язва желудка может переходить в рак желудка по мере усугубления тяжести заболевания.
[88] В частности, желудочно-кишечная язва может включать повреждение слизистой оболочки желудка или повреждение слизистой оболочки тонкого кишечника, вызванное лекарственными препаратами, алкоголем или т.п. В частности, она может представлять собой повреждение слизистой оболочки желудка или повреждение слизистой оболочки тонкого кишечника, вызванное НСПВП или алкоголем.
[89] Желудочно-кишечное воспалительное заболевание относится к заболеванию, вызванному воспалением желудочно-кишечного тракта.
[90] Желудочно-кишечное воспалительное заболевание включает, например, но не ограничиваясь ими, инфекцию Helicobacter pylori, гастрит (например, острый геморрагический гастрит, хронический поверхностный гастрит, хронический атрофический гастрит), воспалительную болезнь кишечника, MALT-лимфому желудка и т.п.
[91] Заболевание, связанное с желудочной кислотой, относится к заболеванию, вызванному чрезмерной секрецией желудочной кислоты. Например, заболевание, связанное с желудочной кислотой, включает, но не ограничиваясь ими, эрозивный эзофагит, неэрозивный эзофагит, рефлюкс-эзофагит, симптоматическую гастроэзофагеальную рефлюксную болезнь (симптоматическую ГЭРБ), функциональную диспепсию, повышенную кислотность, кровоизлияние в верхнем отделе желудочно-кишечного тракта вследствие инвазивного стресса и т.п.
[92] В соответствии с настоящим изобретением, желудочно-кишечная язва, желудочно-кишечное воспалительное заболевание или заболевание, связанное с желудочной кислотой, может представлять собой одно или более заболеваний, выбранных из группы, состоящей из пептической язвы, язвы желудка, язвы двенадцатиперстной кишки, язвы, вызванной НСПВП, язвы на фоне острого стресса, синдрома Золингера-Эллисона, инфекции Helicobacter pylori, гастрита, эрозивного эзофагита, неэрозивного эзофагита, рефлюкс-эзофагита, воспалительной болезни кишечника, симптоматической гастроэзофагеальной рефлюксной болезни (симптоматической ГЭРБ), функциональной диспепсии, рака желудка, MALT-лимфомы желудка, повышенной кислотности и кровоизлияния в верхнем отделе желудочно-кишечного тракта вследствие инвазивного стресса.
[93] Соединение, представленное химической формулой 2 согласно настоящему изобретению, или его фармацевтически приемлемая соль прямо и обратимо ингибирует протонный насос, проявляя быстрый фармакологический эффект и слабое межлекарственное взаимодействие, благодаря чему демонстрирует превосходный эффект с точки зрения фармакологической безопасности. Более конкретно, в отношении безопасности соединение, представленное химической формулой 2, или его фармацевтически приемлемая соль не ингибирует фермент CYP, который является основным метаболизирующим ферментом печени, и, таким образом, предположительно с меньшей вероятностью проявляет межлекарственное взаимодействие.
[94] Соединение, представленное химической формулой 2 согласно настоящему изобретению, или его фармацевтически приемлемая соль может иметь высокое внутрижелудочное распределение для поддержания высоких концентраций в желудке, что обеспечивает возможность адекватного регулирования долгосрочной активности желудочной кислоты и, таким образом, его преимуществом является превосходный эффект даже с учетом замедления секреции кислоты в ночное время и текучесть даже во время введения. Кроме того, соединение, представленное химической формулой 2 согласно настоящему изобретению, или его фармацевтически приемлемая соль проявляет высокую биодоступность при пероральном способе введения, демонстрируя отличный эффект с точки зрения фармакокинетики. Другими словами, соединение, представленное химической формулой 2 согласно настоящему изобретению, или его фармацевтически приемлемая соль может иметь превосходную биодоступность при пероральном введении наряду с превосходным внутрижелудочным распределением, демонстрируя достаточно высокий эффект ингибирования секреции желудочной кислоты даже при небольшом количестве лекарственного средства. В частности, концентрация лекарственного средства в желудке сохраняется на уровне выше подходящего уровня и демонстрирует достаточную эффективность одновременно с превосходным эффектом без риска несварения, боли в области живота, гипергастринемии и т.п. вследствие чрезмерного компенсирующего действия.
[95] В частности, соединение, представленное химической формулой 2 согласно настоящему изобретению, или его фармацевтически приемлемая соль проявляет превосходную активность агониста рецептора соматостатина. Соответственно, секрецию кислоты можно контролировать без риска гипергастринемии посредством эффективного ингибирования секреции гастрина. Кроме того, риск гипергастринемии может быть минимизирован посредством регулирования концентрации гастрина в крови. В частности, продемонстрирована превосходная эффективность регулирования секреции кислоты без побочных эффектов или проблем, таких как гиперплазия и нейроэндокринные опухоли, которые могут быть вызваны гипергастринемией, и т.п.
[96] Соединение, представленное химической формулой 2 согласно настоящему изобретению, или его фармацевтически приемлемая соль проявляет превосходный эффект обратимости (при соответствующем рН) восстановления ферментативной активности протонного насоса, в то же время проявляя ингибирующую способность, действуя на протонный насос в течение короткого промежутка времени при низком рН.
[97] Соединение, представленное химической формулой 2 согласно настоящему изобретению, или его фармацевтически приемлемая соль обладает превосходным терапевтическим эффектом в отношении желудочно-кишечного повреждения, включая желудочно-кишечные язвы и желудочно-кишечные воспалительные заболевания, возникающие по причинам, отличным от желудочной кислоты, благодаря превосходной активности агониста рецептора соматостатина. Например, превосходный эффект облегчения воспаления и улучшения слизистой оболочки желудка при повреждении слизистой оболочки желудка или при повреждении слизистой оболочки кишечника, вызванном лекарственным средством. В частности, оно может проявлять превосходный эффект улучшения слизистой оболочки желудочно-кишечного тракта при желудочно-кишечном повреждении и желудочно-кишечных воспалительных заболеваниях, вызванных НСПВП, а также при желудочно-кишечном повреждении и желудочно-кишечных воспалительных заболеваниях, вызванных алкоголем. Кроме того, оно может проявлять превосходный эффект при лечении заболевания посредством заметного улучшения уровней воспалительных цитокинов и активных форм кислорода (ROS) при желудочно-кишечном повреждении и желудочно-кишечных воспалительных заболеваниях, вызванных НСПВП, а также при желудочно-кишечном повреждении и желудочно-кишечных воспалительных заболеваниях, вызванных алкоголем.
[98] В частности, соединение, представленное химической формулой 2 согласно настоящему изобретению, или его фармацевтически приемлемая соль проявляет превосходную терапевтическую эффективность против желудочно-кишечных язв и желудочно-кишечных воспалительных заболеваний благодаря превосходной активности агониста рецептора соматостатина. Более конкретно, превосходный эффект при лечении заболеваний может проявляться в минимизации язвенного поражения и заметном улучшении уровней воспалительных цитокинов и активных форм кислорода (ROS) при язвах пищевода или при язвах двенадцатиперстной кишки.
[99] В частности, соединение, представленное химической формулой 2 согласно настоящему изобретению, или его фармацевтически приемлемая соль может иметь терапевтический эффект без усугубления повреждения тонкого кишечника, вызванного такими препаратами как НСПВП, в отличие от существующих препаратов P-CAB, благодаря превосходной активности агониста рецептора соматостатина и, таким образом, терапевтическая эффективность в отношении кишечных заболеваний, включая воспалительную болезнь кишечника (ВБК), может проявляться вместе с увеличением категории групп пациентов, для которых можно использовать данное лекарственное средство.
[100] Соединение, представленное химической формулой 2 согласно настоящему изобретению, или его фармацевтически приемлемая соль подходит для предотвращения ил лечения заболеваний пищеварительной системы, таких как хронический гастрит, пептическая язва, рак желудка и т.п., вызванных Helicobacter pylori, посредством снижения желудочной кислотности для усиления антибактериального эффекта антибиотиков против Helicobacter pylori (H. pylori).
[101] Термины и символы, использованные в настоящем описании, имеют следующие значения.
[102] PG: защитная группа
[103] ДМФА: диметилформамид
[104] ЭА: этилацетат
[105] ДХМ: дихлорметан
[106] ТФК: трифторуксусная кислота
[107] NaH: гидрид натрия
[108] NaBH4: боргидрид натрия
[109] NaHCO3: бикарбонат натрия
[110] Na2S2O3: тиосульфат натрия
[111] Boc: трет-бутоксикарбонильная защитная группа
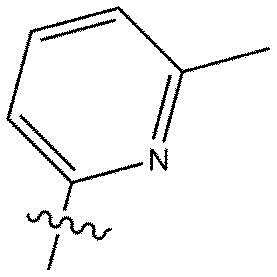
[112] DIBAL-H: гидрид диизобутилалюминия
[113] DMP: периодинан Десс-Мартина
[114] ТГФ: тетрагидрофуран
[115] В данном контексте термин «галоген» относится к фториду, хлориду, бромиду или иодиду.
[116] Термин «алкил» относится к неразветвленной или разветвленной углеводородной группе, имеющей структурную формулу -CnH(2n+1). Неограничивающие примеры включают метил, этил, пропил, изопропил, бутил, 2-метилпропил, 1,1-диметилэтил, пентил, гексил и т.п. Например, «C1-C4 алкил» может относиться к такому алкилу как метил, этил, пропил, бутил, 2-метилпропил или изопропил.
[117] В настоящем описании термин «алкокси» означает «-O-алкил» или «-O-», где алкил является таким, как описано выше.
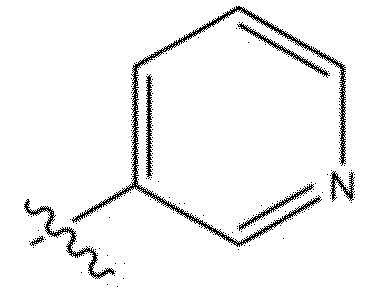
[118] В настоящем описании символ «» может представлять собой замещенную или незамещенную пиридинильную группу.
[119] Замещенная пиридинильная группа является такой, как описано выше.
[120] Термин «пиридинильная группа» в данном контексте относится к 6-членному гетероарильному соединению, содержащему 1 атом азота и 5 атомов углерода. Неограничивающие примеры пиридинильной группы включают:
[121] 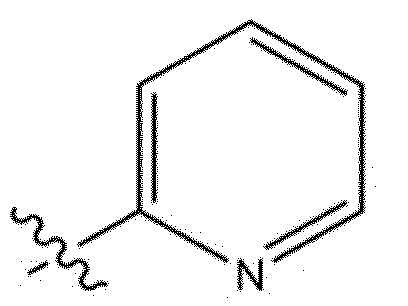 ,
,  ,
,  .
.
[122] В настоящем описании, если символ «» является замещенным, если он представляет собой замещенную пиридинильную группу, она может быть замещена одним или двумя -O(C1-C4 алкилами) или -(C1-C4 алкилами). В частности, -O(C1-C4 алкил) может представлять собой метокси или этокси. В частности, -(C1-C4 алкил) может представлять собой метил или этил.
[123] Более предпочтительно, замещенная пиридинильная группа может представлять собой , где R2 представляет собой -O(C1-C4 алкил) или -(C1-C4 алкил). В частности, -O(C1-C4 алкил) может представлять собой метокси или этокси. В частности, -(C1-C4 алкил) может представлять собой метил или этил.
[124] Например, если символ «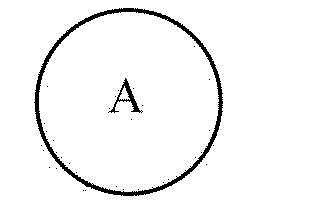 » в настоящем описании является замещенным, то его неограничивающие примеры включают:
» в настоящем описании является замещенным, то его неограничивающие примеры включают:
[125] 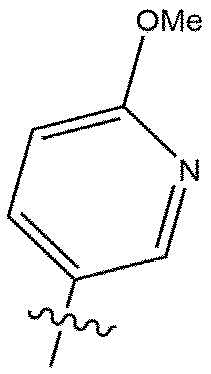 ,
, , .
, .
[126] Если символ «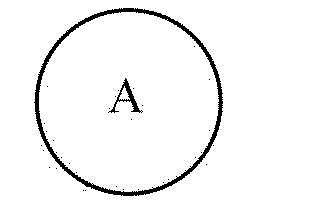 » в настоящем описании является незамещенным, то его неограничивающие примеры включают:
» в настоящем описании является незамещенным, то его неограничивающие примеры включают:
[127] пиридин-3-ил или пиридин-2-ил.
[128] В другом варианте реализации настоящее изобретение включает фармацевтическую композицию.
[129] В настоящем изобретении предложена фармацевтическая композиция для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, содержащая соединение, представленное химической формулой 1, или его фармацевтически приемлемую соль.
[130] Фармацевтическая композиция может содержать соединение согласно настоящему изобретению вместе с фармацевтически приемлемым носителем. Также могут присутствовать другие фармакологически активные ингредиенты. «Фармацевтически приемлемый носитель» в настоящем описании включает любой и все растворители, дисперсионные среды, покрытия, антибактериальные и противогрибковые агенты, изотонические средства и агенты замедления абсорбции, и т.п., которые являются физиологически совместимыми.
[131] Композиция согласно настоящему изобретению может быть в различных формах. Например, композиция включает жидкие, полутвердые и твердые лекарственные формы, такие как жидкие растворы (например, растворы для инъекций и инфузий), дисперсии или суспензии, таблетки, пилюли, порошки, липосомы и суппозитории. Форма зависит от предполагаемого способа введения и терапевтического применения.
[132] Типичная композиция представлена в форме композиций, аналогичных растворам для инъекций и инфузий. Один из способов введения является парентеральным (например, внутривенным, подкожным, интраперитонеальным, внутримышечным).
[133] Пероральное введение твердых лекарственных форм может быть обеспечено, например, в твердых или мягких капсулах, пилюлях, саше, пастилках или таблетках, каждая из которых содержит предварительно определенное количество одного или более соединений согласно настоящему изобретению. В другом варианте реализации пероральное введение может быть обеспечено в порошкообразной или гранулированной форме.
[134] В другом варианте реализации пероральное введение может быть обеспечено в жидкой лекарственной форме. Жидкая лекарственная форма для перорального введения включает, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертный разбавитель (например, воду), обычно используемый в данной области техники.
[135] В другом варианте реализации настоящее изобретение включает парентеральную лекарственную форму. «Парентеральное введение» включает, например, подкожную инъекцию, внутривенную инъекцию, интраперитонеальную инъекцию, внутримышечную инъекцию, интрастернальную инъекцию и инфузию. Препараты для инъекций (т.е. стерильные водные или масляные суспензии для инъекций) могут быть составлены в соответствии с известными технологиями с использованием подходящих диспергирующих, смачивающих агентов и/или суспендирующих агентов.
[136] Также могут быть использованы другие материалы-носители и способы введения, известные в области фармацевтики. Фармацевтические композиции согласно настоящему изобретению могут быть получены любой из общеизвестных фармацевтических технологий, таких как эффективные способы составления и введения лекарственных форм.
[137] Обычно соединение согласно настоящему изобретению вводят в количестве, эффективном для лечения симптомов, описанных в настоящем изобретении. Соединение согласно настоящему изобретению можно вводить в виде соединения как такового или, альтернативно, в виде фармацевтически приемлемой соли. Для описания введения и доз само соединение или его фармацевтически приемлемую соль называют просто соединением согласно настоящему изобретению.
[138] Соединение согласно настоящему изобретению вводят любым подходящим способом в форме фармацевтической композиции, подходящей для такого способа и в дозе, эффективной для предполагаемого лечения. Соединение согласно настоящему изобретению можно вводить перорально, ректально, внутривагинально, парентерально или местно.
[139] Предпочтительно, соединение согласно настоящему изобретению можно вводить перорально. Пероральное введение может включать проглатывание соединения для его попадания в желудочно-кишечный тракт.
[140] В другом варианте реализации соединение согласно настоящему изобретению также можно вводить непосредственно в кровоток, мышцу или внутренние органы. Подходящие способы парентерального введения включают внутривенное, внутриартериальное, интраперитонеальное, внутримышечное и подкожное введение.
[141] Схема введения доз соединения согласно настоящему изобретению и/или композиции, содержащей указанное соединение, зависит от различных факторов, включая тип, возраст, массу, пол и медицинское состояние пациента; тяжесть симптомов; способ введения; и активность конкретного используемого соединения. Соответственно, схема введения доз может значительно варьироваться. В одном варианте реализации общая суточная доза соединения согласно настоящему изобретению обычно составляет от примерно 0,001 до примерно 100 мг/кг [т.е. выраженная по массе соединения согласно настоящему изобретению (мг) на единицу массы тела (кг)] для лечения указанных симптомов, рассмотренных в настоящем описании.
[142] Подходящие субъекты согласно настоящему изобретению включают млекопитающих субъектов. В одном варианте реализации подходящим субъектом является человек. Человек-субъект может быть мужского или женского рода и на любой стадии развития.
[143] Получение
[144] Схемы реакций, описанные ниже, предназначены для обеспечения общего описания методики, использованной для получения соединений согласно настоящему изобретению.
[145] Соединение, представленное химической формулой 1 согласно настоящему изобретению, включает соединения из примеров, получение которых описано ниже. Соединения из приведенных примеров получены или могут быть получены со ссылкой на различные способы, описанные в настоящем документе, а также на обычные общие технические знания, известные специалистам в данной области техники, на основании промежуточных соединений. Соединения из приведенных примеров получены или могут быть получены со ссылкой на следующий способ, представленный на реакционной схеме 1 или схеме 2, описанный в настоящем документе, а также на обычные общие технические знания, известные специалистам в данной области техники, на основании промежуточных соединений.
[146] Реакционные схемы 1 и 2, описанные ниже, раскрывают способ получения химической формулы 1 через промежуточное соединение. Реакционная схема 3, описанная ниже, раскрывает способ получения промежуточного соединения (I), использованного на реакционной схеме 1. Реакционная схема 4, описанная ниже, раскрывает способ получения промежуточного соединения (VI), использованного на реакционной схеме 2.
[147] Способ синтеза
[148] 1. Способ синтеза (1) соединения химической формулы 1
[149] [Реакционная схема 1]
[150] 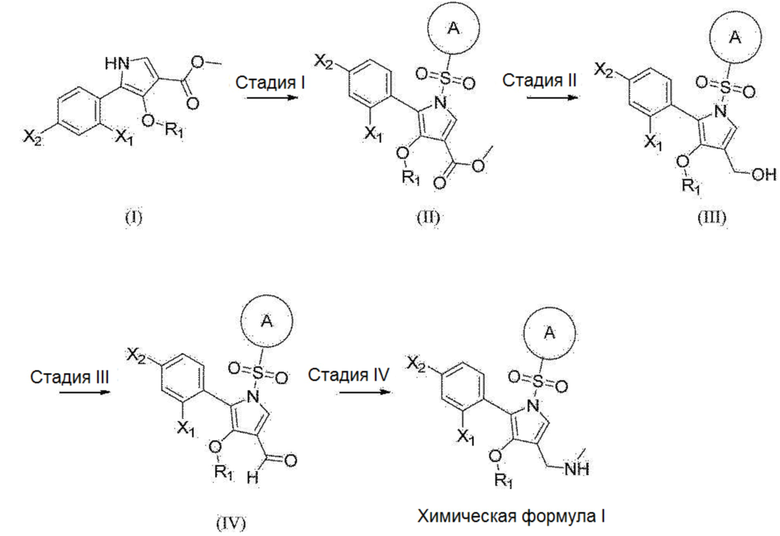
[151] (1) Реакция на стадии (I)
[152] Промежуточное соединение (II) может быть получено по реакции, показанной на стадии (I), с использованием промежуточного соединения (I) и промежуточного соединения (V), как описано ниже. Данная реакция представляет собой процесс внедрения соответствующей гетероарилсульфонильной группы с использованием основания в присутствии инертного растворителя. Растворитель, используемый для реакции на стадии (I), предпочтительно представляет собой углеводороды, такие как толуол и бензол, простые эфиры, такие как тетрагидрофуран и диэтиловый эфир, N,N-диметилформамид или смешанный растворитель из них и т.п., но не ограничиваясь ими. Основание, используемое для данной реакции, предпочтительно представляет собой неорганическую соль, такую как гидроксид натрия, основную соль, такую как карбонат цезия, или соль металла, такую как метоксид натрия, и т.п., но не ограничиваясь ими. Предпочтительная продолжительность данной реакции варьируется в зависимости от соединения, но обычно составляет от 10 минут до 16 часов. Предпочтительная температура реакции для данной реакции варьируется в зависимости от соединения, но обычно составляет от 0 °С до 140 °С. Реакцию можно проводить при добавлении краун-эфира для того, чтобы данная реакция протекала надлежащим образом, и примеры краун-эфира включают 15-краун-5-эфир и т.п.
[153] 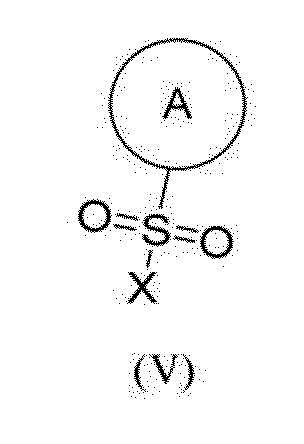
[154] Промежуточное соединение (V) представляет собой вещество, которое доступно в продаже или может быть получено общеизвестным способом, и кольцо A в промежуточном соединении (V) является таким же, как описано выше для химической формулы 1. Символ «X» в промежуточном соединении (V) означает галогенный элемент, например, такой галогенный элемент как F, Cl, Br или т.п.
[155] (2) Реакция на стадии (II)
[156] Промежуточное соединение (III) может быть получено из промежуточного соединения (II) по реакции, представленной на стадии (II). Реакция на стадии (II) представляет собой процесс восстановления с использованием восстановительного агента в присутствии инертного растворителя. Растворитель, используемый для данной реакции, предпочтительно представляет собой углеводороды, такие как толуол и бензол, простые эфиры, такие как тетрагидрофуран и диэтиловый эфир, или смешанный растворитель из них и т.п., но не ограничиваясь ими. Восстановительный агент, используемый для данной реакции, предпочтительно представляет собой гидрид диизобутилалюминия, алюмогидрид лития и т.п., но не ограничиваясь ими. Предпочтительная продолжительность данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от 10 минут до 6 часов. Предпочтительная температура реакции для данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от -78 °С до 25 °С.
[157] (3) Реакция на стадии (III)
[158] Промежуточное соединение (IV) может быть получено из промежуточного соединения (III) по реакции, представленной на стадии (III). Реакция на стадии (III) представляет собой процесс окисления с использованием окислительного агента в присутствии инертного растворителя. Растворитель, используемый для данной реакции, предпочтительно представляет собой органический галогенсодержащий растворитель, такой как дихлорметан, или смешанный с ним растворитель и т.п., но не ограничиваясь ими. Окислительный агент, используемый для данной реакции, предпочтительно представляет собой периодинан Десс-Мартина, хлорхромат пиридиния и т.п., но не ограничиваясь ими. Предпочтительная продолжительность данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от 10 минут до 6 часов. Предпочтительная температура реакции для данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от 0 °С до 25 °С.
[159] (4) Реакция на стадии (IV)
[160] Соединение химической формулы (1) может быть получено из промежуточного соединения (IV) по реакции, показанной на стадии (IV). Реакция на стадии (IV) представляет собой процесс восстановительного аминирования с использованием соответствующего амина и восстановительного агента. Растворитель, используемый для данной реакции, предпочтительно представляет собой простые эфиры, такие как тетрагидрофуран и диэтиловый эфир, спирты, такие как метанол и этанол, или смешанный растворитель из них и т.п., но не ограничиваясь ими. Данную реакцию осуществляют посредством проведения реакции с соответствующим амином, таким как метиламин или т.п., в течение времени, подходящего для образования имина, и добавления подходящего восстановительного агента. Восстановительный агент, используемый для данной реакции, предпочтительно представляет собой боргидрид натрия, цианоборгидрид натрия или триацетоксиборгидрид натрия и т.п., но не ограничиваясь ими. Предпочтительная продолжительность данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от 1 часа до 6 часов. Предпочтительная температура реакции для данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от 0 °С до 60 °С.
[161] 2. Способ синтеза (2) соединения химической формулы 1
[162] Соединение [химической формулы 1] также может быть получено способом, показанным ниже на реакционной схеме 2:
[163] [Реакционная схема 2]
[164] 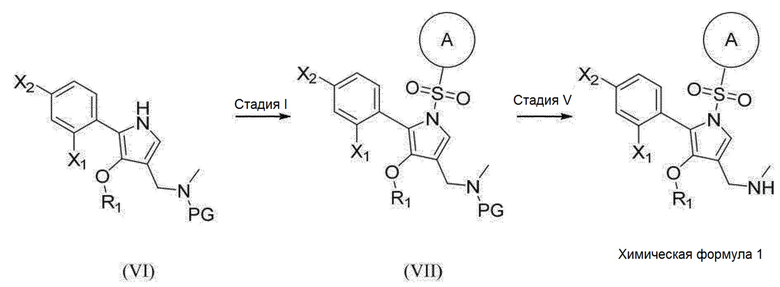
[165] (1) Реакция на стадии (I)
[166] Промежуточное соединение (VII) может быть получено из промежуточного соединения (VI) таким же способом, как способ получения, описанный выше на стадии (I) реакционной схемы 1, или подобным способом.
[167] (2) Реакция на стадии (V)
[168] Соединение химической формулы 1 может быть получено из промежуточного соединения (VII) по реакции, показанной на стадии (V). Реакция на стадии (V) представляет собой процесс снятия защиты, приводящий к удалению защитной группы в соответствующих условиях. Реакция снятия защиты не ограничена конкретными кислотными или основными условиями, например, может быть использован раствор хлороводорода в 1,4-диоксане, трифторуксусная кислота в дихлорметане, раствор карбоната калия в метаноле и т.п., но используемые вещества не ограничены ими. Предпочтительная продолжительность данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от 10 минут до 6 часов. Предпочтительная температура реакции для данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от 0 °С до 25 °С.
[169] 3. Синтетический способ получения промежуточного соединения (I), представленного на реакционной схеме 1
[170] Промежуточное соединение (I) на изображенной выше реакционной схеме 1 может быть получено по реакционной схеме 3, показанной ниже.
[171] [Реакционная схема 3]
[172] 
[173] (1) Реакция на стадии (VI)
[174] Промежуточное соединение (IX) может быть получено из промежуточного соединения (VIII) по реакции, представленной на стадии (VI). Реакция на стадии (VI) представляет собой процесс внедрения защитной группы для аминогруппы промежуточного соединения (VIII). Реакция внедрения защитной группы может быть осуществлена, например, в соответствии с общеизвестными способами, такими как несколько способов, предложенных в публикации T.W. Green (см. Protective Groups in Organic Synthesis, 4е изд., 2007, Wiely & Sons).
[175] (2) Реакция на стадии (VII)
[176] Промежуточное соединение (X) может быть получено из промежуточного соединения (IX) по реакции, представленной на стадии (VII). Реакция на стадии (VII) представляет собой реакцию конденсации Кляйзена для синтеза сложных бета-кетоэфиров из карбоновых кислот. Она представляет собой реакцию активации с помощью соответствующей уходящей группы, такой как карбонилдиимидазол и т.п., с последующей конденсацией с помощью реагента Турбо-Гриньяра, такого как хлорид магния или т.п., с последующим декарбоксилированием при соответствующем уровне кислотности, например, в кислотных условиях. Растворитель, используемый для данной реакции, предпочтительно представляет собой простые эфиры, такие как тетрагидрофуран и диэтиловый эфир, или смешанный растворитель из них и т.п., но не ограничиваясь ими. Время реакции варьируется в зависимости от соединения, но предпочтительно составляет от 3 часов до 24 часов при комнатной температуре, но не ограничиваясь этим.
[177] (3) Реакция на стадии (VIII)
[178] Промежуточное соединение (XI) может быть получено из промежуточного соединения (X) по реакции, представленной на стадии (VIII). Реакция на стадии (VIII) представляет собой реакцию циклизации пиррола, которая протекает при соответствующих условиях. Циклизация представляет собой реакцию, в которой сложный бета-кетоэфирный субстрат образует активированную метиленовую группу, а нуклеофильная атака атома азота в молекуле обеспечивает возможность протекания циклизации в присутствии диметилацеталя N,N-диметилформамида. Растворитель, используемый для данной реакции, предпочтительно представляет собой углеводороды, такие как толуол и бензол, простые эфиры, такие как 1,4-диоксан, или смешанный растворитель из них и т.п., но не ограничиваясь ими. Предпочтительная продолжительность данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от 2 часов до 12 часов. Предпочтительная температура реакции для данной реакции варьируется в зависимости от соединения, но предпочтительно составляет 40 °С или более и в некоторых случаях 100 °С или более.
[179] (4) Реакция на стадии (IX)
[180] Промежуточное соединение (XII) может быть получено из промежуточного соединения (XI) по реакции, представленной на стадии (IX). Реакция на стадии (IX) представляет собой реакцию алкилирования гидроксильной группы в данном соединении. Растворитель, используемый для данной реакции, предпочтительно представляет собой простые эфиры, такие как тетрагидрофуран и диэтиловый эфир, спирты, такие как метанол и этанол, N,N-диметилформамид или смешанный растворитель из них и т.п., но не ограничиваясь ими. Алкилирование можно осуществлять в присутствии соответствующего основания, например, посредством взаимодействия с диэтилсульфатом, диметилсульфатом или т.п., в присутствии карбоната калия или т.п., или с использованием алкилирующего агента, такого как триметилсилилдиазометан или т.п. Предпочтительная продолжительность данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от 3 часов до 24 часов. Предпочтительная температура реакции для данной реакции варьируется в зависимости от соединения, но предпочтительно составляет от 0 °С до 50 °С.
[181] (5) Реакция на стадии (V)
[182] Промежуточное соединение (I) может быть получено из промежуточного соединения (XII) таким же способом, как способ получения, описанный выше на стадии (V) реакционной схемы 2, или подобным способом.
[183] 4. Синтетический способ получения промежуточного соединения (VI), представленного на реакционной схеме 2
[184] Промежуточное соединение (VI) на изображенной выше реакционной схеме 2 может быть получено по реакционной схеме 4, показанной ниже.
[185] [Реакционная схема 4]
[186] 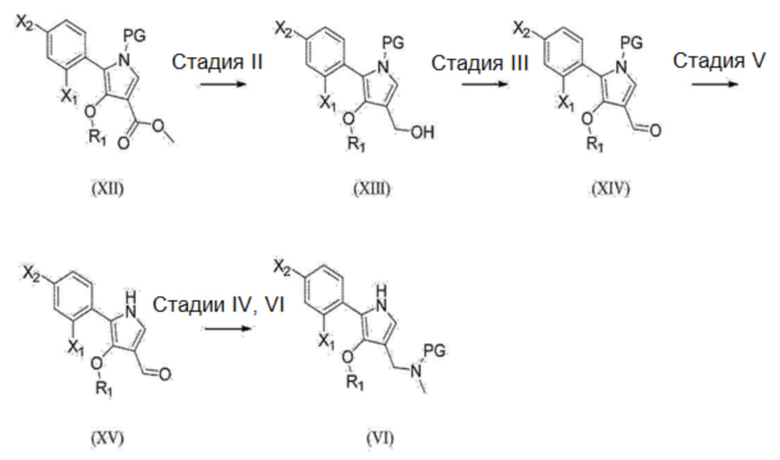
[187] (1) Реакция на стадии (II)
[188] Промежуточное соединение (XIII) может быть получено из промежуточного соединения (XII) таким же способом, как способ получения, описанный выше на стадии (II) реакционной схемы 1, или подобным способом.
[189] (2) Реакция на стадии (III)
[190] Промежуточное соединение (XIV) может быть получено из промежуточного соединения (XIII) таким же способом, как способ получения, описанный выше на стадии (III) реакционной схемы 1, или подобным способом.
[191] (3) Реакция на стадии (V)
[192] Промежуточное соединение (XV) может быть получено из промежуточного соединения (XIV) таким же способом, как способ получения, описанный выше на стадии (V) реакционной схемы 2, или подобным способом.
[193] (4) Реакция на стадиях (IV) и (VI)
[194] Промежуточное соединение (VI) может быть получено из промежуточного соединения (XV) таким же способом, как способ получения в две стадии, описанный на реакционных схемах 1 и 3, т.е. на стадиях (IV) и (VI), или подобным способом.
[Полезные эффекты]
[195] Новое производное согласно настоящему изобретению или его фармацевтически приемлемая соль прямо и обратимо ингибирует протонный насос, проявляя быстрый фармакологический эффект и низкое межлекарственное взаимодействие. Кроме того, соединение согласно настоящему изобретению может иметь высокую биодоступность для проявления высокого фармакологического эффекта даже в низкой дозе, может иметь высокое внутрижелудочное распределение и может сохраняться в желудке на достаточном или более высоком уровне для регулирования активности желудочной кислоты в течение продолжительного периода времени. Кроме того, оно пригодно не только для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, но и для предотвращения и лечения пищеварительных заболеваний, таких как хронический гастрит, пептическая язва, рак желудка и т.п., вызванных Helicobacter pylori, посредством снижения желудочной кислотности для усиления антибактериального эффекта антибиотиков против Helicobacter pylori (H. pylori). Кроме того, соединение согласно настоящему изобретению демонстрирует активность агониста рецептора соматостатина и подавляет секрецию гастрина, эффективно подавляя выработку желудочной кислоты без риска гипергастринемии, в отличие от существующих PPI.
[Наилучший способ реализации]
[196] Далее более подробно описаны способы получения и экспериментальные примеры согласно настоящему изобретению. Однако настоящее изобретение не ограничено представленными способами получения и примерами.
[197] Реагенты и растворители, описанные ниже, приобретали у Sigma-Aldrich, TCI, если не указано иное.
[198] Измерения ЯМР всех соединений проводили на ЯМР спектрометре Bruker Avance™ NEO (400 МГц для 1H, 100 МГц для 13C). Масс-спектрометрию проводили на системе Masslynx и на системе ЖХ/МС на основе СВЭЖХ Waters, а чистоту измеряли методом обращенно-фазовой ВЭЖХ на системе Waters e2695. Если не указано иное, условия для анализа ВЭЖХ были следующими.
[199] [Метод ВЭЖХ I]
[200] Подвижная фаза A: 0,1% ТФК в ацетонитриле, подвижная фаза В: 0,1% ТФК в H2O
[201] Состав градиента элюирования
[202] Исходные условия: A: 10%, B: 90%
[203] A: 10%, B: от 90% A: 100%, B: 0% (от t=0 мин до t=20 мин)
[204] A: 100%, B: 0%, выдерживание (от t=20 мин до t=30 мин)
[205] A: 100%, B: от 0% A: 10%, B: 90% (от t=30 мин до t=30,10 мин)
[206] A: 10%, B: 90%, выдерживание (от t=30,10 мин до t=40 мин)
[207] скорость потока: 1,0 мл/мин, объем ввода: 10 мкл
[208] Спектры 1H ядерного магнитного резонанса (ЯМР) во всех случаях соответствовали предполагаемой структуре. Характеристический химический сдвиг (δ) представлен в миллионных долях (м.д.) для сигнала остаточного протона в дейтерированном растворителе (CDCl3: 7,27 м.д.; CD2HOD: 3,31 м.д.; ДМСО-d6: 2,50 м.д.), и использовали обычные сокращения для обозначения основных пиков: например, с, синглет; д, дублет; т, триплет; к, квартет; м, мультиплет; ш, широкий. Спектры 1H ЯМР записывали при интенсивности электрического поля 400 МГц, если не указано иное.
[209] Синтетический пример
[210] Синтетический пример 1. Синтез промежуточных соединений 1-4
[211] [Промежуточное соединение 1]. Метил-5-(2-фторфенил)-4-метокси-1H-пиррол-3-карбоксилат
[212] Стадия (1). Синтез 2-((трет-бутоксикарбонил)амино)-2-(2-фторфенил)уксусной кислоты
[213] 2-Амино-2-(2-фторфенил)уксусную кислоту (1,0 экв., 2,0 г, 11,82 ммоль) растворяли в ТГФ/H2O=1:1 (70 мл) и затем добавляли гидрокарбонат натрия (3,0 экв., 2,98 г, 35,47 ммоль), затем перемешивали в течение 30 минут. Добавляли ди-трет-бутилдикарбонат (1,2 экв., 3,10 г, 14,18 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. Удаляли ТГФ, понижая давление реакционного раствора, и затем доводили рН до примерно 2,5 с помощью 1 н. водного раствора HCl. Добавляли этилацетат (ЭА) и дважды экстрагировали полученную смесь. Затем сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния с получением 2-((трет-бутоксикарбонил)амино)-2-(2-фторфенил)уксусной кислоты в виде бледно-желтого твердого вещества (3,0 г, 94%).
[214] Стадия (2). Синтез метил-4-((трет-бутокикарбонил)амино)-4-(2-фторфенил)-3-оксобутаноата
[215] 2-((трет-Бутоксикарбонил)амино)-2-(2-фторфенил)уксусную кислоту (1,0 экв., 30,0 г, 111,4 ммоль) и карбонилдиимидазол (1,03 экв., 18,6 г, 114,7 ммоль) растворяли в ацетонитриле (300 мл) и перемешивали при комнатной температуре в течение 1 часа. В другую колбу добавляли монометилмалонат калия (1,03 экв., 17,9 г, 114,7 ммоль), безводный хлорид магния (1,03 экв., 10,94 г, 114,7 ммоль), ацетонитрил (300 мл) и триэтиламин (1,03 экв., 16 мл, 114,7 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Реагенты из двух колб, полученные ранее, смешивали с помощью канюли и кипятили с обратным холодильником при 80 °С в течение 1 часа. После завершения реакции охлаждали смесь до комнатной температуры и добавляли в нее воду. Охлаждали смесь льдом и перемешивали в течение 1 часа. Отфильтровывали полученное твердое вещество, добавляли ЭА и воду и затем доводили рН до примерно 5 с помощью 1 н. HCl. Дважды экстрагировали полученную смесь ЭА, сушили, фильтровали и концентрировали с безводным сульфатом магния с получением метил-4-((трет-бутоксикарбонил)амино)-4-(2-фторфенил)-3-оксобутаноата в виде твердого вещества (19,0 г, 52%).
[216] Стадия (3). Синтез 1-(трет-бутил)-3-метил-5-(фторфенил)-4-гидрокси-1H-пиррол-1,3-дикарбоксилата
[217] Метил-4-((трет-бутоксикарбонил)амино)-4-(2-фторфенил)-3-оксобутаноат (1,0 экв., 15,4 г, 47,33 ммоль) и диметилацеталь N,N-диметилформамида (3,0 экв., 19 мл, 142,00 ммоль) добавляли в толуол (300 мл) и перемешивали при 40 °С в течение 4 часов до завершения реакции. Выпаривали смесь при пониженном давлении для удаления толуола и добавляли ЭА и воду. После нейтрализации до примерно рН 7 с помощью 1 н. HCl полученную смесь дважды экстрагировали ЭП. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния с получением 1-(трет-бутил)-3-метил-5-(фторфенил)-4-гидрокси-1H-пиррол-1,3-дикарбоксилата в виде твердого вещества (14,28 г, 90%).
[218] Стадия (4). Синтез 1-(трет-бутил)-3-метил-5-(2-фторфенил)-4-метокси-1H-пиррол-1,3-дикарбоксилата
[219] 1-(трет-Бутил)-3-метил-5-(фторфенил)-4-гидрокси-1H-пиррол-1,3-дикарбоксилат (1,0 экв., 14,28 г, 42,58 ммоль), карбонат калия (2,0 экв., 11,8 г, 85,17 ммоль) и диметилсульфат (1,13 экв., 4,56 мл, 48,12 ммоль) растворяли в ацетоне (213 мл) и перемешивали при 50 °С в течение ночи. Реакцию завершали добавлением воды и затем удаляли избыток ацетона выпариванием при пониженном давлении. После добавления ЭА и воды нейтрализовали смесь до примерно рН 7 с помощью 1 н. HCl и затем дважды экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией на диоксиде кремния с получением 1-(трет-бутил)-3-метил-5-(2-фторфенил)-4-метокси-1H-пиррол-1,3-дикарбоксилата в виде твердого вещества (14,00 г, 94%).
[220] Стадия (5). Синтез метил-5-(2-фторфенил)-4-метокси-1H-пиррол-3-карбоксилата (промежуточное соединение 1)
[221] 1-(трет-Бутил)-3-метил-5-(2-фторфенил)-4-метокси-1H-пиррол-1,3-дикарбоксилат (1,0 экв., 7,0 г, 20,0 ммоль) и трифторуксусную кислоту (10,0 экв., 15,3 мл, 200,4 ммоль) растворяли в дихлорметане (35 мл) и перемешивали при комнатной температуре в течение 6 часов. После охлаждения до 0-5 °С с помощью ледяной воды добавляли воду и доводили рН до 7,0, используя 50% водный раствор NaOH. После двукратного экстрагирования с помощью ЭА и выпаривания добавляли н-гексан. Затем перемешивали смесь в течение 1 часа и фильтровали с получением метил-5-(2-фторфенил)-4-метокси-1H-пиррол-3-карбоксилата в виде бледно-розового твердого вещества (4,6 г, 92%).
[222] 1H ЯМР (400 МГц, ДМСО-d6) δ 11,46 (с, 1H), 7,64 (дт, J=1,6, 7,8 Гц, 1H), 7,36 - 7,24 (м, 4H), 3,73 (с, 6H).
[223] [Промежуточное соединение 2]. трет-Бутил-((5-(2-фторфенил)-4-метокси-1H-пиррол-3-ил)метил)(метил)карбамат
[224] Стадия (1). Синтез трет-бутил-2-(2-фторфенил)-4-(гидроксиметил)-3-метокси-1H-пиррол-1-карбоксилата
[225] Гидрид диизобутилалюминия (1 М раствор в гексане, 5 экв., 64,4 мл, 64,4 ммоль) растворяли в тетрагидрофуране (200 мл) и медленно, по каплям добавляли 1-(трет-бутил)-3-метил-5-(2-фторфенил)-4-метокси-1H-пиррол-1,3-дикарбоксилат (4,5 г, 12,9 ммоль) при 0 °С, и перемешивали при комнатной температуре в течение 1 часа. По каплям последовательно добавляли воду и 1 н. водный раствор NaOH, сушили над безводным сульфатом магния, фильтровали через целит и концентрировали. Концентрированный остаток очищали колоночной хроматографией с получением трет-бутил-2-(2-фторфенил)-4-(гидроксиметил)-3-метокси-1H-пиррол-1-карбоксилата в виде бесцветного маслянистого вещества (1,7 г, 41,1%).
[226] 1H ЯМР (400 МГц, CDCl3): δ 7,41-7,33(м, 2H), 7,30(с, 1H), 7,19(дт, J= 7,4 Гц, J= 1,2 Гц, 1H), 7,10(дт, J= 9,0 Гц, J= 0,8 Гц, 1H), 4,61(д, J= 4,8 Гц, 2H), 3,60(с, 3H), 1,32(с, 9H)
[227] Стадия (2). Синтез трет-бутил-2-(2-фторфенил)-4-формил-3-метокси-1H-пиррол-1-карбоксилата
[228] трет-Бутил-2-(2-фторфенил)-4-(гидроксиметил)-3-метокси-1H-пиррол-1-карбоксилат (1,7 г, 5,3 ммоль) растворяли в дихлорметане (20 мл), медленно, по каплям добавляли периодинан Десс-Мартина (1 экв., 2,24 г, 5,3 ммоль) и перемешивали при комнатной температуре в течение 1 часа. В реакционную смесь добавляли целит. Полученный продукт концентрировали и очищали колоночной хроматографией с получением (5-(2-фторфенил)-4-метокси-1H-пиррол-3-ил)метанола в виде бесцветного маслянистого вещества (1,23 г, 72,8%).
[229] 1H ЯМР (400 МГц, CDCl3): δ 9,89(с, 1H), 7,92(с, 1H), 7,42-7,37(м, 2H), 7,22(дт, J= 7,5 Гц, J= 0,9 Гц, 1H), 7,12(дт, J= 9,2 Гц, J= 0,9 Гц, 1H), 3,75(с, 3H), 1,38(с, 9H)
[230] Стадия (3). Синтез 5-(2-фторфенил)-4-метокси-1H-пиррол-3-карбальдегида
[231] трет-Бутил-2-(2-фторфенил)-4-формил-3-метокси-1H-пиррол-1-карбоксилат (1,2 г, 3,8 ммоль) растворяли в смеси вода/метанол (1/3, 20 мл), по каплям добавляли карбонат калия (3 экв., 1,6 г, 11,3 ммоль) и затем перемешивали смесь при 100 °С в течение 2,5 часа. Продукт реакции сушили над безводным сульфатом натрия и фильтровали с получением 5-(2-фторфенил)-4-метокси-1H-пиррол-3-карбальдегида в виде желтого твердого вещества (800,0 мг, 97,1%).
[232] 1H ЯМР (400 МГц, CDCl3): δ 9,87(с, 1H), 9,11(шс, 1H), 8,17-8,13(м, 1H), 7,34(д, J= 4,0 Гц, 1H), 7,27-7,23(м, 2H), 7,19-7,16(м, 1H), 3,98(с, 3H)
[233] Стадия (4). Синтез трет-бутил-((5-(2-фторфенил)-4-метокси-1H-пиррол-3-ил)метил)(метил)карбамата (промежуточное соединение 2)
[234] 5-(2-Фторфенил)-4-метокси-1H-пиррол-3-карбальдегид (800 мг, 3,65 ммоль) растворяли в метаноле (50 мл), по каплям добавляли 40% раствор метиламина (2,3 экв., 0,86 мл, 8,4 ммоль) и перемешивали смесь при комнатной температуре в течение 30 минут. Охлаждали реакционную смесь до 0 °С, по каплям добавляли боргидрид натрия (1,5 экв., 207,1 мг, 5,5 ммоль) и затем перемешивали смесь при комнатной температуре в течение 30 минут. В реакционную смесь по каплям добавляли воду (150 мл), полученный продукт перемешивали в течение 1 часа при той же температуре и по каплям добавляли насыщенный солевой раствор, затем экстрагировали ЭА. Экстрагированный органический слой сушили, фильтровали и концентрировали с безводным сульфатом магния. Концентрированный остаток растворяли в ацетонитриле (40 мл), и затем по каплям добавляли ди-трет-бутилдикарбонат (1,2 экв., 955,7 мг, 4,4 ммоль), затем перемешивали при комнатной температуре в течение 2 часов. В реакционную смесь по каплям добавляли воду и ЭА для экстракции и сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния. Концентрированный остаток очищали колоночной хроматографией с получением трет-бутил-((5-(2-фторфенил)-4-метокси-1H-пиррол-3-ил)метил)(метил)карбамата в виде светло-коричневого твердого вещества (946,8 мг, 78,9%).
[235] 1H ЯМР (400 МГц, CDCl3): δ 8,55(шс, 1H), 8,07(дт, J= 7,9 Гц, J= 1,7 Гц, 1H), 7,20-7,06(м, 1H), 6,62(с, 1H), 4,35(с, 2H), 3,72(с, 3H), 2,86(с, 3H), 1,50(с, 9H)
[236] [Промежуточное соединение 3]. Метил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилат
[237] Стадия (1). Синтез 2-((трет-бутоксикарбонил)амино)-2-(2,4-дифторфенил)уксусной кислоты
[238] 2-Амино-2-(2,4-дифторфенил)уксусную кислоту (1,0 экв., 7,22 г, 38,6 ммоль) растворяли в смеси ТГФ/H2O (1:1, 200 мл) и затем охлаждали до 0 °С. Добавляли NaHCO3 (3,0 экв., 9,74 г, 116 ммоль) и Boc2O (1,2 экв., 10,64 мл, 46,3 ммоль) и после перемешивания при комнатной температуре в течение ночи в реакционный раствор добавляли воду, и доводили рН до 2,5. Затем экстрагировали полученную смесь ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния с получением 2-((трет-бутоксикарбонил)амино)-2-(2,4-дифторфенил)уксусной кислоты (17,35 г, 99%) в виде белого твердого вещества без дополнительной очистки. [M+Na]+: 310
[239] Стадия (2). Синтез метил-4-((трет-бутокикарбонил)амино)-4-(2,4-дифторфенил)-3-оксобутаноата
[240] 2-((трет-Бутоксикарбонил)амино)-2-(2,4-дифторфенил)уксусную кислоту (1,0 экв., 38,6 ммоль) и карбонилдиимидазол (1,1 экв., 6,89 г, 42,5 ммоль) растворяли в ацетонитриле (100 мл). В другой колбе растворяли метилмалонат калия (1,1 экв., 6,64 г, 42,5 ммоль), триэтиламин (1,1 экв., 5,97 мл, 42,5 ммоль), хлорид магния (1,1 экв., 4,05 г, 42,5 ммоль) в ацетонитриле (100 мл). После перемешивания каждого раствора при комнатной температуре в течение 1 часа объединяли два раствора, полученных выше, и перемешивали при 80 °С в течение 3 часов. После добавления H2O (100 мл) перемешивали смесь при комнатной температуре в течение 2 часов и отфильтровывали полученное твердое вещество. К отфильтрованному твердому веществу добавляли ЭА и воду и перемешивали смесь при комнатной температуре в течение 10 минут, и нейтрализовали до рН 7 с помощью водн. HCl. Органический слой, экстрагированный ЭА, сушили, фильтровали и концентрировали с безводным сульфатом магния, концентрировали с получением метил-4-((трет-бутоксикарбонил)амино)-4-(2,4-дифторфенил)-3-оксобутаноата (12,64 г, 95%) в виде коричневой жидкости без дополнительной очистки.
[241] 1H ЯМР (400 МГц, CDCl3): δ 7,33-7,28 (м, 1H), 6,97-6,87 (м, 2H), 5,85 (шс, 1H), 5,67 (д, J = 6,8 Гц, 1H), 3,70 (с, 3H), 3,53 (д, J = 16,0 Гц, 1H), 4,75 (д, J = 16,0 Гц, 1H), 1,34 (с, 9H).
[242] Стадия (3). Синтез 1-(трет-бутил)-3-метил-5-(2,4-дифторфенил)-4-гидрокси-1H-пиррол-1,3-дикарбоксилата
[243] Метил-4-((трет-бутоксикарбонил)амино)-4-(2,4-дифторфенил)-3-оксобутаноат (1,0 экв., 12,64 г, 36,8 ммоль) и диметилацеталь N,N-диметилформамида (3 экв., 14,7 мл, 110,4 ммоль) растворяли в толуоле (184 мл) и перемешивали при 40 °С в течение 5 часов. После концентрирования добавляли ЭА и воду и нейтрализовали смесь до рН 7 с помощью 1 н. HCl. Органический слой, экстрагированный ЭА, сушили, фильтровали и концентрировали с безводным сульфатом магния с получением 1-(трет-бутил)-3-метил-5-(2,4-дифторфенил)-4-гидрокси-1H-пиррол-1,3-дикарбоксилата в виде коричневой жидкости без дополнительной очистки.
[244] 1H ЯМР (400 МГц, CDCl3): δ 7,75 (с, 1H), 7,52 (с, 1H), 7,43 (дт, J = 8,4, 6,8 Гц, 1H), 6,98-6,84 (м, 2H), 3,92 (с, 3H), 1,41 (с, 9H).
[245] Стадия (4). Синтез 1-(трет-бутил)-3-метил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-1,3-дикарбоксилата
[246] 1-(трет-Бутил)-3-метил-5-(2,4-дифторфенил)-4-гидрокси-1H-пиррол-1,3-дикарбоксилат (1,0 экв., 36,8 ммоль), карбонат калия (2,0 экв., 10,2 г, 73,6 ммоль) и диметилсульфат (1,2 экв., 4,2 мл, 44,2 ммоль) растворяли в ацетоне (184 мл) и перемешивали при 40 °С в течение ночи. Добавляли ЭА и воду и нейтрализовали смесь до рН 7 с помощью 1 н. HCl. Органический слой, экстрагированный ЭА, сушили, фильтровали и концентрировали с безводным сульфатом магния, очищали хроматографией на диоксиде кремния с получением 1-(трет-бутил)-3-метил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-1,3-дикарбоксилата (12,86 г, 95%) в виде желтой жидкости. [M+H]+: 367
[247] 1H ЯМР (400 МГц, CDCl3): δ 7,90 (с, 1H), 7,36 (дт, J = 8,4, 6,4 Гц, 1H), 6,99-6,86 (м, 2H), 3,90 (с, 3H), 3,70 (с, 3H), 1,41 (с, 9H).
[248] Стадия (5). Синтез метил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилата (промежуточное соединение 3)
[249] 1-(трет-Бутил)-3-метил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-1,3-дикарбоксилат (1,0 экв., 12,8 г, 34,8 ммоль) и трифторуксусную кислоту (10 экв., 26 мл, 348 ммоль) растворяли в дихлорметане (70 мл) и перемешивали при комнатной температуре в течение 5 часов. Добавляли воду (100 мл) при 0 °С и нейтрализовали смесь до рН 7 с помощью 1 н. NaOH. Реакционный раствор экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и переводили в твердое состояние с помощью гексана и ЭА с получением метил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилата в виде бледно-розового твердого вещества (4,98 г, 54%) без дополнительной очистки.
[250] 1H ЯМР (400 МГц, CDCl3): δ 8,82 (шс, 1H), 8,14 (дт, J = 9,1, 6,5 Гц, 1H), 7,33 (д, J = 3,6 Гц, 1H), 7,01-6,87 (м, 2H), 3,89 (с, 3H), 3,88 (с, 3H).
[251] [Промежуточное соединение 4]. трет-Бутил-((5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-ил)метил)(метил)карбамат
[252] Стадия (1). Синтез трет-бутил-2-(2,4-дифторфенил)-4-(гидроксиметил)-3-метокси-1H-пиррол-1-карбоксилата
[253] 1-(трет-Бутил)-3-метил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-1,3-дикарбоксилат (1,0 экв., 10,02 г, 27,3 ммоль) растворяли в ТГФ (137 мл) и охлаждали до 0 °С. Медленно добавляли 1,0 М раствор DIBAL-H (8,0 экв., 219 мл, 219 ммоль) в ТГФ. Перемешивали реакционный раствор при комнатной температуре в течение 2 часов. После охлаждения до 0 °С последовательно добавляли H2O (8,76 мл), 15% NaOH (8,76 мл) и H2O (22 мл). Затем, после перемешивания при комнатной температуре в течение 20 минут, добавляли безводный сульфат магния и перемешивали смесь в течение 20 минут, и фильтровали через целит. После концентрирования осуществляли очистку методом хроматографии на диоксиде кремния с получением трет-бутил-2-(2,4-дифторфенил)-4-(гидроксиметил)-3-метокси-1H-пиррол-1-карбоксилата (5,13 г, 55%).
[254] 1H ЯМР (400 МГц, CDCl3): δ 7,35 (дт, J = 8,4, 6,4 Гц, 1H), 7,29 (с, 1H), 6,97-6,85 (м, 2H), 4,6 (с, 2H), 3,60 (с, 3H), 1,37 (с, 9H).
[255] Стадия (2). Синтез трет-бутил-2-(2,4-дифторфенил)-4-формил-3-метокси-1H-пиррол-1-карбоксилата
[256] трет-Бутил-2-(2,4-дифторфенил)-4-(гидроксиметил)-3-метокси-1H-пиррол-1-карбоксилат (1,0 экв., 5,13 г, 15,1 ммоль) растворяли в дихлорметане (76 мл) и охлаждали до 0 °С. Добавляли DMP (1,1 экв., 7,04 г, 16,6 ммоль), затем перемешивали при комнатной температуре в течение 30 минут. Реакционный раствор промывали водным раствором NaOH и затем экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали хроматографией на диоксиде кремния с получением трет-бутил-2-(2,4-дифторфенил)-4-формил-3-метокси-1H-пиррол-1-карбоксилата (3,56 г, 70%) в виде желтого твердого вещества.
[257] 1H ЯМР (400 МГц, CDCl3): δ 9,88 (с, 1H), 7,91 (с, 1H), 7,36 (дт, J = 8,4, 6,4 Гц, 1H), 7,00-6,90 (м, 2H), 3,75 (с, 3H), 1,41 (с, 9H).
[258] Стадия (3). Синтез 5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбальдегида
[259] трет-Бутил-2-(2,4-дифторфенил)-4-формил-3-метокси-1H-пиррол-1-карбоксилат (1,0 экв., 3,56 г, 10,6 ммоль) и карбонат калия (3 экв., 4,40 г, 31,8 ммоль) растворяли в смеси метанол/H2O (3:1, 104 мл) и перемешивали при 110 °С в течение 1 часа. После концентрирования фильтровали концентрированный продукт с ацетоном и переводили в твердое состояние с помощью дихлорметана и гексана с получением 5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбальдегида (2,17 г, 86%) в виде оранжевого твердого вещества без дополнительной очистки.
[260] 1H ЯМР (400 МГц, CDCl3): δ 9,86 (с, 1H), 8,97 (шс, 1H), 8,12 (дт, J = 9,0, 6,4 Гц, 1H), 7,33 (д, J = 4,0 Гц, 1H), 7,03-6,91 (м, 2H), 3,96 (с, 3H).
[261] Стадия (4). Синтез трет-бутил-((5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-ил)метил)(метил)карбамата (промежуточное соединение 4)
[262] 2,0 М метиламин растворяли в метаноле (90 мл), затем перемешивали при комнатной температуре в течение 30 минут, затем добавляли 5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбальдегид (1,0 экв., 2,17 г, 9,15 ммоль), ТГФ (10 экв., 46 мл, 91,5 ммоль), NaBH4 (5 экв., 1,73 г, 45,8 ммоль) и перемешивали смесь при комнатной температуре в течение 30 минут. Затем добавляли воду и перемешивали смесь еще 30 минут. Промывали реакционный раствор насыщенным солевым раствором и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния. Затем сразу растворяли концентрированный продукт в ацетонитриле (35 мл), медленно добавляли к нему Boc2O (1,2 экв., 2,53 мл, 11,0 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 1 часа. В реакционный раствор добавляли воду, затем экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали хроматографией на диоксиде кремния с получением трет-бутил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-ил)метил)(метил)карбамата (2,46 г, 76%) в виде коричневого твердого вещества.
[263] 1H ЯМР (400 МГц, CDCl3): δ 8,45 (шс, 1H), 8,05 (дт, J = 9,0, 6,5 Гц, 1H), 6,98-6,85 (м, 2H), 6,63 (шс, 1H), 4,37 (шс, 2H), 3,73 (с, 3H), 2,88 (с, 3H), 1,52 (с, 9H).
[264] Соединения из следующих примеров 1-6 синтезировали с использованием синтезированных промежуточных соединений 1-4. Способы их синтеза основаны на реакционных схемах 1 и 2, представленных выше. В качестве примера получения соединений из вышеуказанных примеров ниже описаны, в частности, способы получения примеров 1-6.
[265] Далее подробно описаны способы синтеза примеров 1-6.
[266] Пример синтеза 1. Синтез примера 1
[267] [Пример 1]. 1-(5-(2-Фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин
[268] Стадия (1). Синтез метил-5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбоксилата
[269] Метил-5-(2-фторфенил)-4-метокси-1H-пиррол-3-карбоксилат (промежуточное соединение 1, 1,0 экв., 1,2 г, 4,8 ммоль) растворяли в ТГФ (20,0 мл) и по каплям добавляли NaH (2,0 экв., 384,8 мг, 9,6 ммоль) при 0 °С, затем перемешивали при комнатной температуре в течение 10 минут. Добавляли 6-метоксипиридин-3-сульфонилхлорид (1,5 экв., 1,6 г, 7,2 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. В реакционный раствор добавляли воду и экстрагировали полученный раствор ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией с получением метил-5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбоксилата в виде светло-коричневого твердого вещества (1,85 г, 91,6%).
[270] Стадия (2). Синтез 5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метанола
[271] Метил-5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбоксилат (1,0 экв., 1,0 г, 2,38 ммоль) растворяли в ТГФ (5,0 мл) и по каплям добавляли 1,0 М раствор DIBAL в н-гексане (5,0 экв., 11,9 мл, 11,9 ммоль) при 0 °С, затем перемешивали при комнатной температуре в течение 1 часа. Охлаждали реакционный раствор до 0 °С, завершали реакцию с помощью водного раствора сегнетовой соли и экстрагировали полученный раствор ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией с получением 5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метанола в виде желтого маслянистого вещества (654,8 мг, 70,2%).
[272] Стадия (3). Синтез метил-5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбальдегида
[273] 5-(2-Фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метанол (1,0 экв., 500,0 мг, 1,3 ммоль) и периодинан Десс-Мартина (1,0 экв., 540,4 мг, 1,3 ммоль) растворяли в ДХМ (10,0 мл) и перемешивали при комнатной температуре в течение 1 часа. Продукт реакции концентрировали и очищали колоночной хроматографией с получением 5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбальдегида в виде бледного темно-голубого твердого вещества (388,2 мг, 78,1%).
[274] Стадия (4). Синтез 1-(5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина
[275] 5-(2-Фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбальдегид (1,0 экв., 385,0 мг, 0,99 ммоль) растворяли в ТГФ (5,0 мл) и добавляли 2,0 М раствор метиламина в ТГФ (10 экв., 4,9 мл, 9,9 ммоль). После перемешивания при комнатной температуре в течение 1 часа охлаждали продукт реакции до 0 °С, добавляли NaBH4 (10 экв., 373,4 мг, 9,9 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 1 часа. В реакционный раствор медленно, по каплям добавляли 6,0 н. водный раствор хлороводорода и отфильтровывали полученное твердое вещество. Отфильтрованное твердое вещество растворяли в воде и добавляли к нему 1 н. водный раствор гидроксида натрия, затем экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния с получением 1-(5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина в виде белого твердого вещества (125,8 мг, 28,3%), [M+H]+: 405.
[276] Пример синтеза 2. Синтез примера 2
[277] [Пример 2]. 1-(5-(2,4-Дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин
[278] Стадия (1). Синтез метил-5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбоксилата
[279] Метил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилат (промежуточное соединение 3, 1,0 экв., 802 мг, 3,00 ммоль) и NaH (1,5 экв., 180 мг, 4,5 ммоль) растворяли в безводном ДМФА (15,0 мл) и перемешивали при комнатной температуре в течение 10 минут. Добавляли 6-метоксипиридин-3-сульфонилхлорид (1,5 экв., 934 мг, 4,50 ммоль), затем перемешивали при комнатной температуре в течение 1 часа. После добавления в реакционный раствор дистиллированной воды промывали полученный раствор насыщенным солевым раствором и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией с получением метил-5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбоксилата (1,09 г, 83%).
[280] Стадия (2). Синтез (5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метанола
[281] Метил-5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбоксилат (1,0 экв., 1,09 г, 2,49 ммоль) растворяли в безводном ТГФ (13,0 мл) и затем по каплям добавляли 1,0 М раствор DIBAL в ТГФ (5,0 экв., 12,4 мл, 12,4 ммоль) при 0 °С. Затем перемешивали реакционный раствор при комнатной температуре в течение 4 часов. К полученному реакционному раствору последовательно добавляли 0,50 мл воды, 0,5 мл 1 н. водного раствора гидроксида натрия и 1,25 мл воды. Затем перемешивали полученную смесь в течение 30 минут и добавляли безводный сульфат магния, затем перемешивали в течение 30 минут. Сушили полученный продукт, фильтровали, концентрировали и очищали колоночной хроматографией с получением (5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метанола (869 мг, 85%).
[282] Стадия (3). Синтез 5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбальдегида
[283] 5-(2,4-Дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метанол (1,0 экв., 869 мг, 2,12 ммоль) и периодинан Десс-Мартина (1,1 экв., 988 мг, 2,33 ммоль) растворяли в ДХМ (21,0 мл) и перемешивали при комнатной температуре в течение 30 минут. В реакционный раствор добавляли водный раствор NaHCO3 и промывали полученный раствор водным раствором Na2S2O3, и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией с получением 5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбальдегида (824 мг, 95%).
[284] Стадия (4). Синтез 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина
[285] 5-(2,4-Дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-карбальдегид (1,0 экв., 824 мг, 2,02 ммоль) растворяли в ТГФ (20,0 мл) и добавляли 2,0 М раствор метиламина в ТГФ (20 экв., 20,2 мл, 40,4 ммоль). После перемешивания при комнатной температуре в течение 3 часов добавляли NaBH4 (10 экв., 764 мг, 20,2 ммоль), затем перемешивали в течение 18 часов. После добавления в реакционный раствор дистиллированной воды промывали полученный раствор насыщенным солевым раствором и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния и очищали колоночной хроматографией с получением 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина в виде красного сиропообразного вещества (90,0 мг, 10%), [M+H]+: 423.
[286] Пример синтеза 3. Синтез примера 3
[287] [Пример 3]. 1-(5-(2,4-Дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин
[288] Стадия (1). Синтез метил-5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-карбоксилата
[289] Метил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилат (промежуточное соединение 3, 1,0 экв., 534 мг, 2,0 ммоль) и NaH (1,5 экв., 120 мг, 3,0 ммоль) растворяли в безводном ДМФА (10,0 мл) и перемешивали при 50 °С в течение 50 минут. Добавляли 6-метилпиридин-3-сульфонилхлорид (1,5 экв., 575 мг, 3,0 ммоль), затем перемешивали при 50 °С в течение 16 часов. После добавления в реакционный раствор дистиллированной воды промывали полученный раствор насыщенным солевым раствором и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией с получением метил-5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-карбоксилата (614 мг, 73%).
[290] Стадия (2). Синтез (5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метанола
[291] Метил-5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-карбоксилат (1,0 экв., 614 мг, 1,45 ммоль) растворяли в безводном ТГФ (7,27 мл) и затем по каплям добавляли 1,0 М раствор DIBAL в ТГФ (5,0 экв., 7,27 мл, 7,27 ммоль) при 0 °С. Затем перемешивали полученную смесь при комнатной температуре в течение 30 минут. К полученному реакционному раствору последовательно добавляли 0,29 мл воды, 0,29 мл 15% водного раствора гидроксида натрия и 0,73 мл воды. Затем перемешивали полученную смесь в течение 14 часов. Добавляли безводный сульфат магния, затем перемешивали в течение 30 минут. Сушили полученный продукт, фильтровали, концентрировали и очищали колоночной хроматографией с получением (5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метанола в виде желтого твердого вещества (494 мг, 86%).
[292] Стадия (3). Синтез 5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-карбальдегида
[293] 5-(2,4-Дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метанол (1,0 экв., 494 мг, 1,25 ммоль) и периодинан Десс-Мартина (1,1 экв., 583 мг, 1,38 ммоль) растворяли в ДХМ (12,0 мл) и перемешивали при комнатной температуре в течение 40 минут. В реакционный раствор добавляли водный раствор гидроксида натрия, затем экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией с получением 5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-карбальдегида (422 мг, 86%).
[294] Стадия (4). Синтез 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина
[295] 5-(2,4-Дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-карбальдегид (1,0 экв., 422 мг, 1,07 ммоль) растворяли в MeOH (5,0 мл) и добавляли 2,0 М раствор метиламина в ТГФ (10 экв., 5,2 мл, 10,7 ммоль). После перемешивания при комнатной температуре в течение 30 минут добавляли NaBH4 (5 экв., 204 мг, 5,38 ммоль), затем перемешивали в течение 10 минут. После добавления в реакционный раствор водного раствора NaHCO3 промывали полученный раствор насыщенным солевым раствором и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния и очищали колоночной хроматографией с получением 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина в виде желтого твердого вещества (176 мг, 40%), [M+H]+: 408.
[296] Пример синтеза 4. Синтез примера 4
[297] [Пример 4]. 1-(5-(2-Фторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин
[298] Стадия (1). Синтез трет-бутил-((5-(2-фторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метил)(метил)карбамата
[299] трет-Бутил-((5-(2-фторфенил)-4-метокси-1H-пиррол-3-ил)метил)(метил)карбамат (промежуточное соединение 2, 100,0 мг, 0,3 ммоль), NaH (24,0 мг, 0,6 ммоль) и 15-краун-5-эфир (0,9 мл, 0,5 ммоль) растворяли в безводном ТГФ (1,5 мл) и перемешивали при 50 °С в течение 10 минут. Добавляли 6-метилпиридин-3-сульфонилхлорид (86,0 мг, 0,5 ммоль) и перемешивали при комнатной температуре в течение 30 минут. После добавления в реакционный раствор дистиллированной воды промывали полученный раствор насыщенным солевым раствором и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией с получением трет-бутил-((5-(2-фторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метил)(метил)карбамата в виде бледно-желтого маслянистого вещества (60,9 мг, 42%).
[300] Стадия (2). Синтез 1-(5-(2-фторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина
[301] трет-Бутил-((5-(2-фторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)метил)(метил)карбамат (60,0 мг, 0,1 ммоль) и 1,0 М раствор хлороводорода в этилацетате (2,0 мл) растворяли в этаноле (1,0 мл) и перемешивали при комнатной температуре в течение 4 часов. После добавления в реакционный раствор водного раствора NaHCO3 промывали полученный раствор насыщенным солевым раствором и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния и очищали колоночной хроматографией с получением 1-(5-(2-фторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина в виде светло-желтого твердого вещества (49,8 мг, 62%), [M+H]+: 390.
[302] Пример синтеза 5. Пример 5
[303] [Пример 5]. 1-5-(2,4-Дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин
[304] Стадия (1). Синтез метил-5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-карбоксилата
[305] Метил-5-(2,4-дифторфенил)-4-метокси-1H-пиррол-3-карбоксилат (промежуточное соединение 3, 1,0 экв., 400,0 мг, 1,5 ммоль) и NaH (1,5 экв., 90,0 мг, 2,25 ммоль) растворяли в безводном ДМФА (10,0 мл) и перемешивали при комнатной температуре в течение 30 минут. Добавляли 6-метилпиридин-2-сульфонилхлорид (1,5 экв., 430 мг, 2,25 ммоль), затем перемешивали при комнатной температуре в течение 5 часов. После добавления в реакционный раствор дистиллированной воды промывали полученный раствор насыщенным солевым раствором и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией с получением метил-5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-карбоксилата в виде прозрачного сиропообразного вещества (442,0 мг, 70%).
[306] Стадия (2). Синтез (5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-ил)метанола
[307] Метил-5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-карбоксилат (1,0 экв., 439,0 мг, 1,04 ммоль) растворяли в безводном ТГФ (5,0 мл) и затем по каплям добавляли 1,0 М раствор DIBAL в ТГФ (3,0 экв., 3,12 мл, 3,12 ммоль) при 0 °С. Затем перемешивали реакционный раствор при комнатной температуре в течение 2 часов. В реакционный раствор добавляли MeOH и затем промывали полученный раствор водным раствором сегнетовой соли и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния с получением (5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-ил)метанола в виде желтого сиропообразного вещества (417,0 мг, 102%).
[308] Стадия (3). Синтез 5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-карбальдегида
[309] 5-(2,4-Дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-ил)метанол (1,0 экв., 398,0 мг, 1,01 ммоль) и периодинан Десс-Мартина (1,0 экв., 428,0 мг, 1,01 ммоль) растворяли в ДХМ (10,0 мл) и перемешивали при комнатной температуре в течение 5 часов. В реакционный раствор добавляли водный раствор NaHCO3 и промывали полученный раствор водным раствором Na2S2O3, и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией с получением 5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-карбальдегида в виде желтого сиропообразного вещества (331,0 мг, 84%).
[310] Стадия (4). Синтез 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина
[311] 5-(2,4-Дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-карбальдегид (1,0 экв., 331,0 мг, 0,84 ммоль) растворяли в MeOH (8,5 мл) и добавляли 9,8 М раствор метиламина в MeOH (20 экв., 1,72 мл, 16,9 ммоль). После перемешивания при комнатной температуре в течение 1 часа добавляли NaBH4 (10 экв., 318,0 мг, 8,4 ммоль), затем перемешивали в течение 30 минут. После добавления в реакционный раствор водного раствора NaHCO3 промывали полученный раствор насыщенным солевым раствором и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния и очищали колоночной хроматографией с получением 1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-2-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамина в виде желтого сиропообразного вещества (185,0 мг, 54%), [M+H]+: 407.
[312] Пример синтеза 6. Синтез примера 6
[313] [Пример 6]. 1-(5-(2-Фторфенил)-4-метокси-1-(пиридин-2-илсульфонил)-1H-пиррол-3-ил)-N-метилметанамин
[314] Стадия (1). Синтез трет-бутил-((5-(2-фторфенил)-4-метокси-1-(пиридин-2-илсульфонил)-1H-пиррол-3-ил)метил)(метил)карбамата
[315] трет-Бутил-((5-(2-фторфенил)-4-метокси-1H-пиррол-3-ил)метил)(метил)карбамат (промежуточное соединение 2, 1,0 экв.), NaH (1,5 экв., 90,0 мг, 2,25 ммоль) и 15-краун-5-эфир (каталитическое количество) растворяли в безводном ТГФ (10,0 мл) и перемешивали при комнатной температуре в течение 30 минут. Добавляли пиридин-2-сульфонилхлорид (1,5 экв., 430 мг, 2,25 ммоль), затем перемешивали при комнатной температуре в течение 5 часов. После добавления в реакционный раствор дистиллированной воды промывали полученный раствор насыщенным солевым раствором и экстрагировали ЭА. Сушили органический слой, фильтровали и концентрировали с безводным сульфатом магния, и очищали колоночной хроматографией с получением трет-бутил-((5-(2-фторфенил)-4-метокси-1-(пиридин-2-илсульфонил)-1H-пиррол-3-ил)метил)(метил)карбамата в виде коричневого маслянистого вещества (80 мг, 55%).
[316] Стадия (2). Синтез 1-(5-(2-фторфенил)-4-метокси-1-(пиридин-2-илсульфонил)-1H-пиррол-3-ил)-N-метилметанамина
[317] трет-Бутил-((5-(2-фторфенил)-4-метокси-1-(пиридин-2-илсульфонил)-1H-пиррол-3-ил)метил(метил)карбамат (0,17 экв., 80 мг) и трифторуксусную кислоту (10,0 экв., 0,88 мл, 11,54 ммоль) растворяли в дихлорметане (2,3 мл) и перемешивали при комнатной температуре в течение 6 часов. После удаления растворителя перегонкой при пониженном давлении охлаждали полученный продукт до 0-5 °С с помощью ледяной воды, затем добавляли воду и доводили рН до 7,0 с помощью водного раствора NaHCO3. После двукратной экстракции ЭА и выпаривания добавляли н-гексан и перемешивали полученный продукт в течение 1 часа, и фильтровали с получением 1-(5-(2-фторфенил)-4-метокси-1-(пиридин-2-илсульфонил)-1H-пиррол-3-ил)-N-метилметанамина в виде желтого маслянистого вещества (17 мг, 28%), [M+H]+: 376.
[318] Соединения, перечисленные в таблице 1, синтезировали таким же образом, как описано выше, или подобным образом, используя соответствующие доступные в продаже исходные материалы и промежуточные соединения. Полученные промежуточные соединения и примеры очищали способами, известными специалистам в данной области техники, причем указанные способы не ограничены хроматографией на силикагеле, перекристаллизацией и т.п. Кроме того, конечное соединение, полученное из реакционной смеси, может быть выделено в форме нейтральной, кислотной или основной соли.
[319] [Таблица 1]
[320]
[M + H]+
(Реакционная схема 1)
(Реакционная схема 1)
(Реакционная схема 1)
(Реакционная схема 2)
(Реакционная схема 1)
(Реакционная схема 2)
[321]
[322] В следующих экспериментальных примера эксперименты проводили с использованием любого одного из соединений из примеров 1-4 согласно настоящему изобретению.
[323] [Экспериментальный пример 1]. Ингибирующая активность в отношении протонного насоса (H+/K+-АТФазы)
[324] Ингибирующую активность полученного соединения в отношении протонного насоса (H+/K+-АТФазы) измеряли следующим образом. Желудочные везикулы, выделенные из желудка свиньи, получали в соответствии с описанием, представленным в документе (см. Methods Mol Biol. 2016;1377:19-27). Содержание белка в желудочных везикулах количественно определяли с помощью набора с бицинхониновой кислотой (BCA) (Sigma Aldrich, BCA1). В каждую лунку 96-луночного планшета добавляли 70 мкл 50 мМ буфера Tris-HEPES (рН 6,5), содержащего 125 нг везикул, ДМСО или субстрат для каждой концентрации (конечная концентрация ДМСО 1%), 5 мМ MgCl2 и 10 мМ KCl, и предварительно инкубировали при 37 °С в течение 30 минут. Затем в каждую лунку добавляли 10 мкл 2 мМ АТФ с последующей ферментативной реакцией при 37 °С в течение 40 минут. Реакцию останавливали, добавляя 20 мкл реагента малахитового зеленого (Sigma Aldrich, MAK307) и оставляли полученную смесь стоять при комнатной температуре на 30 минут. Измеряя поглощение при 620 нм с помощью микропланшет-ридера (Biotek, Synergy H4), измеряли количество неорганического фосфора, высвобожденного при разложении АТФ, и активность фермента. Измеряли поглощение образца, прореагировавшего с ферментом, без добавления KCl и вычитали результат измерения из всех результатов вышеописанных измерений. Предполагая, что группа, обработанная 1% ДМСО (контрольная группа с ДМСО), демонстрировала 100% активность фермента H+/K+-АТФазы, и что группа без KCl (контрольная группа KCl) демонстрировала 0% активность фермента H+/K+-АТФазы, рассчитывали % ингибирования по следующему уравнению 1:
[325] [Уравнение 1]
[326] % ингибирование = [1-(ОПэксп. группа - ОПконтр. группа KCl)/(ОПконтр. группа с ДМСО - ОПконтр. группа KCl))]*100.
[327] Значения IC50 получали с помощью нелинейного регрессионного анализа в программе GraphPad Prism7, используя значения % ингибирования для каждой концентрации, и результаты представлены ниже в таблице 2.
[328] [Таблица 2]
[329]
[330] (* Значения IC50, составляющие ровно 0,3 мкМ или менее, обозначены +++)
[331] На основании представленных выше результатов, подтверждена превосходная ингибирующая активность соединений согласно настоящему изобретению в отношении протонного насоса (H+/K+-АТФазы).
[332] [Экспериментальный пример 2]. Ингибирующая активность и оценка обратимости в отношении протонного насоса (H+/K+ АТФаза) в зависимости от рН
[333] Для измерения изменения ингибирующей активности полученного соединения в отношении протонного насоса (H+/K+-АТФазы) в зависимости от рН, проводили такой же эксперимент, как в экспериментальном примере 1, при трех условиях: рН 6,5, рН 7,0 и рН 7,5. Подтверждено, что примеры 1 и 3 имеют более высокую ингибирующую активность в слабокислотной среде по сравнению с нейтральной средой. Это указывает на то, что степень ингибирующей способности в отношении насоса желудочной кислоты выше в кислотной среде, и ингибирующая способность восстанавливается после восстановления рН в желудке.
[334] Кроме того, для подтверждения обратимости ингибирующей способности полученного соединения в отношении протонного насоса (H+/K+-АТФазы) проводили эксперимент методом резкого разбавления. 6,25 мкг желудочных везикул, выделенных из желудка свиньи, и 0,2 мкМ каждого соединения предварительно инкубировали в течение 120 минут и затем сравнивали активность фермента до разбавления и активность фермента после 50-кратного разбавления для каждого времени реакции, и оценивали обратимость. В примере 1 и в примере 3 была подтверждена ингибирующая активность на уровне 50% или более по истечении 20 минут реакции. С другой стороны, при проведении реакции в течение 60 минут после 50-кратного разбавления, активность фермента в примерах 1 и 3 была восстановлена до 90% или более, и были подтверждены обратимые результаты.
[335] Мощное подавление желудочной кислоты приводит к повышению гастрина в сыворотке по компенсаторному механизму, который тесно связан с риском гипергастринемии и т.п.
[336] Однако соединения из примеров 1 и 3 демонстрировали ингибирующую активность, действуя на протонный насос за короткое время при низком рН, а затем демонстрировали обратимость, заключающуюся в быстром восстановлении активности фермента.
[337] Это указывает на обратимость характеристики восстановления секреции кислоты путем простой диссоциации от протонного насоса, и демонстрирует низкую частоту гипергастринемии.
[338] Другими словами, на основании экспериментальных результатов, описанных в настоящем документе, можно ожидать, что соединения согласно настоящему изобретению обладают превосходным эффектом ингибирования секреции кислоты без побочных эффектов в отношении гипергастринемии.
[339] [Таблица 3]
[340]
[341] (* Значения IC50, составляющие ровно 0,1 мкМ или менее, обозначены +++, а значения IC50, составляющие более 0,1 мкМ и менее или ровно 1 мкМ, обозначены ++)
[342] [Таблица 4]
[343]
[344] (* % ингибирования, составляющий 50% или более, обозначен +++, % ингибирования, составляющий 20% или более и менее 50%, обозначен ++, и % ингибирования, составляющий менее 20%, обозначен +)
[345] [Экспериментальный пример 3]. Оценка эффекта агониста SSTR4 (анализ цАМФ)
[346] Эффект агонизма в отношении SSTR4 подтверждали с помощью клеточного функционального анализа цАМФ. Клетки CHO, стабильно экспрессирующие SSTR4 человека, обрабатывали экспериментальными веществами в каждой концентрации и проводили реакцию при 37 °С в течение 30 минут, и измеряли количество выработанной цАМФ методом обнаружения гомогенной флуоресценции с временным разрешением (HTRF). Рассчитывали % ответа по сравнению с эталонным контрольным агонистом (sst-14, 10 нМ) и рассчитывали значения EC50 по кривой зависимости ответа от концентрации. Результаты представлены ниже в таблице 5.
[347] [Таблица 5]
[348]
[349] Представленные выше данные свидетельствуют о том, что соединения согласно настоящему изобретению демонстрируют превосходный эффект как агонисты SSTR4.
[350] [Экспериментальный пример 4]. Оценка ингибирующей способности в отношении базальной секреции желудочной кислоты у крыс с лигированным привратником желудка
[351] Ингибирующую эффективность полученного соединения в отношении базальной секреции желудочной кислоты измеряли на крысиной модели по методу H. Shay [Shay H, et al., Gastroenterology, 1945, 5, 43~61].
[352] Самцов крыс Спрага-Доули (SD) разделяли по 8 крыс на группу и на протяжении 24 часов давали им только воду. Затем, за один час до лигирования привратника желудка, контрольной группе перорально вводили 0,5% раствор метилцеллюлозы, а другим группам перорально вводили соединение из примера в дозе 10 мг/10 мл/кг, суспендированное в 0,5% растворе метилцеллюлозы.
[353] Через 5 часов после лигирования крыс усыпляли под анестезией с золетилом и ксилазином и вылущивали содержимое желудка через разрез брюшной полости. Полученное содержимое центрифугировали при 3000 об./мин в течение 10 минут для отделения надосадочного раствора и собирали желудочный сок. 1 мл собранного желудочного сока помещали в лабораторный стакан и измеряли рН с помощью электродного рН-метра. К 1 мл желудочного сока добавляли по 0,03 мл 0,5% спиртового раствора диметиламиноазобензола и 1% спиртового раствора фенолфталеина для получения красного цвета, а затем добавляли 0,1 н. раствор NaOH, при этом определяли объем до появления розового оттенка определяли как общую кислотность, а общий выход кислоты определяли умножением кислотности желудочного сока на количество желудочного сока. Рассчитывали % ингибирующей активности соединения из примера по следующему уравнению 2, и результаты представлены ниже в таблице 6.
[354] [Уравнение 2]
[355] % ингибирующей активности соединения из примера = [(общая секреция желудочной кислоты в контрольной группе - общая секреция желудочной кислоты в группе, получавшей соединение из примера) / общая секреция желудочной кислоты в контрольной группе] Х 100
[356] [Таблица 6]
[357]
[358] 90% или более: +++, 80% или более, но менее 90%: ++, 70% или более, но менее 80%: +
[359] [Экспериментальный пример 5]. Оценка способности подавления секреции желудочной кислоты у крыс с перфузией брюшной полости (LPR)
[360] Ингибирующую эффективность полученного соединения в отношении гистамин-стимулированной секреции желудочной кислоты измеряли в крысиных моделях с перфузией брюшной полости (LPR) по методу Ghosh и Schild [Ghosh MN, et al., Br J Pharmacol Chemother., 1958, 13(1), 54~61].
[361] Вставляли силиконовую трубку между желудком и пищеводом самцов крыс Спрага-Доули (SD) в состоянии натощак и оставляли для перфузии физиологического солевого раствора с той же скоростью. Помимо этого, вставляли силиконовую трубку между привратником желудка и двенадцатиперстной кишкой для обеспечения возможности выхода перфузата, прошедшего через желудок. Затем с той же скоростью вводили инъекцию гистамина через шприцевой насос для стабилизации рН в желудке на уровне примерно 2,5. После стабилизации рН контрольной группе через яремную вену или двенадцатиперстную кишку вводили только 0,5% метилцеллюлозу, а контрольной группе PPI вводили омепразол, эзомепразол, лансопразол, рабепразол или т.п. Другим группам таким же способом вводили инъекцию соединения из примера. Перфузат собирали аликвотами по 7,5 мл каждые 15 минут после введения лекарственного средства и измеряли рН.
[362] [Экспериментальный пример 6]. Оценка ингибирующей эффективности в отношении желудочного повреждения у крыс с желудочным повреждением, вызванным индометацином.
[363] Эксперимент для оценки эффективности подавления язвы желудка под действием соединения из примера в крысиных моделях повреждения желудка, вызванного индометацином, лекарственным средством из класса НСПВП, проводили следующим образом.
[364] Среди самцов крыс Спрага-Доули в состоянии натощак контрольной группе перорально вводили 0,5% раствор метилцеллюлозы, а другим группам перорально вводили соединение из примера в дозе 10 мг/10 мл/кг, суспендированное в 0,5% растворе метилцеллюлозы.
[365] Через 1 час после перорального введения соединения из примера осуществляли пероральное введение индометацина, а через 5 часов экспериментальных животных усыпляли и вылущивали содержимое желудка. После промывания вылущенной поверхности желудка осуществляли разрез желудка большей кривизны. Вырезанный желудок разворачивали и фиксировали. Затем рассчитывали долю поврежденной площади желудка по общей площади желудка и поврежденной площади на поверхности слизистой оболочки желудка, используя программное обеспечение ImageJ (NIH, Бетесда), и рассчитывали % ингибирующей активности соединения из примера по следующему уравнению 3. Результаты представлены ниже в таблице 7:
[366] [Уравнение 3]
[367] % ингибирующей активности соединения из примера = [(доля поврежденной площади желудка в контрольной группе - доля поврежденной площади желудка в группе, получавшей соединение из примера) / (доля поврежденной площади желудка в контрольной группе)] Х 100
[368] [Таблица 7]
[369]
[370] 97% или более: +++, 90% или более, но менее 97%: ++, 80% или более, но менее 90%: +
[371] [Экспериментальный пример 7]. Оценка эффективности в отношении повреждения желудка, вызванного этанолом, и желудочно-кишечного воспалительного заболевания
[372] Алкоголь может напрямую вызывать повреждение и кровотечение слизистого слоя желудка, а также косвенно ускоряет секрецию воспалительных цитокинов, липополисахаридов, эндотоксинов или свободных радикалов посредством инфильтрации макрофагов и нейтрофилов, вызывая как язву желудка, так и желудочно-кишечное воспаление. Проводили следующий эксперимент для оценки эффективности соединения из примера в отношении подавления язвы желудка и эффективности против воспаления желудочно-кишечного тракта в крысиных моделях повреждения желудка, вызванного алкоголем, и желудочно-кишечного воспалительного заболевания.
[373] Среди самцов крыс Спрага-Доули в состоянии натощак контрольной группе перорально вводили 0,5% раствор метилцеллюлозы, а другим группам перорально вводили соединение из примера, суспендированное в 0,5% растворе метилцеллюлозы.
[374] Через 1 час после перорального введения соединения из примера экспериментальным животным перорально вводили 100% этанол, через 1 час анестезировали и подвергали лапаротомии для сбора крови из задней полой вены. Кровь оставляли стоять при комнатной температуре на примерно 15 минут для коагуляции, а затем центрифугировали для отделения сыворотки. После завершения сбора крови вылущивали желудок. После промывания поверхности вылущенного желудка физиологическим солевым раствором осуществляли разрез желудка большей кривизны. Вырезанный желудок помещали на скобу, разворачивали с помощью щипцов и фиксировали шпилькой. Затем анализировали общую площадь желудка и поврежденную площадь на слизистой оболочке желудка с помощью программного обеспечения Image (NIH, Бетесда). Желудочную ткань гомогенизировали и центрифугировали для получения белка желудочной ткани из надосадочного раствора и измеряли концентрацию воспалительных цитокинов в желудочной ткани. Концентрацию воспалительных цитокинов в крови в выделенной сыворотке измеряли технологией иммуноферментного твердофазного анализа (ИФА).
[375] [Экспериментальный пример 8]. Оценка эффективности в отношении рефлюкс-эзофагита, вызванного кислотным рефлюксом
[376] Следующий эксперимент проводили для оценки эффективности соединения из примера в отношении ингибирования эзофагального повреждения у крыс с рефлюкс-эзофагитом, вызванным кислотным рефлюксом.
[377] Среди самцов крыс Спрага-Доули в состоянии натощак контрольной группе перорально вводили 0,5% раствор метилцеллюлозы, а другим группам перорально вводили соединение из примера, суспендированное в 0,5% растворе метилцеллюлозы.
[378] Через 1 час после перорального введения соединения из примера экспериментальных животных анестезировали и подвергали лапаротомии. Лигировали привратник желудка и дополнительно границу между проксимальным желудком и его телом для обеспечения возможности рефлюкса желудочной кислоты в пищевод. После определенного периода времени желудок и пищевод экспериментальных животных осторожно вылущивали, собирали содержимое желудка и брали желудочный сок, и измеряли рН и количество желудочного сока. Вылущенный пищевод вырезали в продольном направлении и фиксировали для обнажения области слизистой оболочки. Анализировали поврежденную площадь пищевода в программном обеспечении ImageJ (NIH, Бетесда).
[379] [Экспериментальный пример 9]. Оценка эффективности при повреждении двенадцатиперстной кишки, вызванном мепиризолом
[380] Эксперимент для оценки эффективности подавления язвы двенадцатиперстной кишки под действием соединения из примера в крысиных моделях повреждения двенадцатиперстной кишки, вызванного мепиризолом, лекарственным средством из класса НСПВП, проводили следующим образом.
[381] Среди самцов крыс Спрага-Доули контрольной группе перорально вводили 0,5% раствор метилцеллюлозы, а другим группам перорально вводили соединение из примера, суспендированное в 0,5% растворе метилцеллюлозы.
[382] Через 1 час после перорального введения соединения из примера осуществляли пероральное введение мепиризола, а по истечении предварительно определенного периода времени экспериментальных животных усыпляли и вылущивали содержимое двенадцатиперстной кишки. После промывания поверхности вылущенной двенадцатиперстной кишки физиологическим солевым раствором анализировали поврежденную область с помощью программного обеспечения ImageJ (NIH, Бетесда).
[383] [Экспериментальный пример 10]. Измерение изменения гастрина в крови после введения соединения из примера
[384] Эксперимент для наблюдения изменения гастрина в крови после введения соединения из примера согласно настоящему изобретению проводили следующим образом.
[385] Среди самцов крыс Спрага-Доули в состоянии натощак контрольной группе перорально вводили 0,5% раствор метилцеллюлозы, а другим группам перорально вводили соединение из примера, суспендированное в 0,5% растворе метилцеллюлозы.
[386] Собирали примерно 0,5 мл крови из яремной крови экспериментальных животных через 5 часов, 8 часов, 12 часов или 24 часа после перорального введения соединения из примера. В собранной крови измеряли концентрацию гастрина в крови методом ИФА.
[387] [Экспериментальный пример 11]. Оценка противовоспалительной эффективности при воспалении тонкого кишечника, вызванном индометацином
[388] Проводили следующий эксперимент для измерения изменения воспаления после введения соединения из примера у крыс с воспалением тонкого кишечника, вызванным индометацином, препаратом класса НСПВП.
[389] Среди самцов мышей C57BL/6 или самцов крыс Спрага-Доули контрольной группе интраперитонеально ежедневно вводили 0,5% раствор метилцеллюлозы, а другим группам интраперитонеально вводили соединение из примера, суспендированное в 0,5% растворе метилцеллюлозы, на протяжении предварительно определенного периода времени. В последний день введения соединения из примера осуществляли пероральное введение индометацина для инициации воспаления тонкого кишечника.
[390] Через определенный период времени усыпляли экспериментальных животных и вылущивали тонкий кишечник. После промывания поверхности вылущенного тонкого кишечника физиологическим солевым раствором анализировали повреждение тонкого кишечника, такое как кровотечение и воспаление или т.п., с помощью гистологического анализа. Ткань вылущенного тонкого кишечника гомогенизировали и центрифугировали, а затем выделяли общую РНК из ткани тонкого кишечника в надосадочном растворе и измеряли количество мРНК воспалительных цитокинов в ткани тонкого кишечника.
[391] [Экспериментальный пример 12]. Наблюдение желудочно-кишечной нейроэндокринной опухоли после длительного введения соединения из примера
[392] Следующий эксперимент проводили для наблюдения стадии желудочно-кишечных нейроэндокринных опухолей, вызванных изменениями секреции гастрина, после длительного введения соединения из примера.
[393] Среди крыс Спрага-Доули контрольной группе ежедневно перорально вводили 0,5% раствор метилцеллюлозы, а другим группам перорально вводили высокую дозу соединения из примера, суспендированного в 0,5% растворе метилцеллюлозы, в течение 2 лет. Через определенный период времени усыпляли экспериментальных животных, вылущивали желудок и двенадцатиперстную кишку и фиксировали. Затем наблюдали степень гиперплазии клеток ECL и частоту нейроэндокринных опухолей с помощью гистопатологического анализа, и сравнивали с контрольной группой.
[394] [Экспериментальный пример 13]. Испытание внутрижелудочного распределения
[395] После перорального введения здоровым крысам соединения из примера измеряли внутрижелудочное распределение в зависимости от времени следующим образом. Полученное соединение растворяли в дистиллированной воде, содержащей от 0,5% метилцеллюлозы до 0,2 мг/мл, и затем осуществляли пероральное введение в дозе 4 мг/кг. Крыс усыпляли через 1 час, 6 часов, 12 часов и 24 часа после введения. Затем выпускали кровь из сердца и проводили перфузию физиологическим солевым раствором, вылущивали ткань желудка, взвешивали и хранили при -80 °С до проведения анализа. Добавляли буфер PBS в таком количестве, что отношение массы желудочной ткани к буферу PBS составляло 1:4, и экстрагировали соединение из желудочной ткани с помощью гомогенизатора. Брали надосадочный раствор экстракта и осаждали белок ацетонитрилом, и затем измеряли количество соединения из примера методом ЖХ-МС/МС.
[396] Расчетное содержание в желудке AUCконечная, желудок представлено ниже в таблице 8. Пример 1 демонстрировал превосходное внутрижелудочное распределение, и во все моменты времени концентрация в желудке превосходила значение IC50, полученное в анализе ингибирования H+/K+-АТФазы in vitro.
[397] [Таблица 8]
[398] Значение AUCконечная, желудок, полученное после однократного введения соединения из примера 1 в дозе 4 мг/кг
[399]
[400] Приведенные выше результаты подтверждают, что соединения согласно настоящему изобретению обладают превосходным эффектом внутрижелудочного распределения.
[401] [Экспериментальный пример 14]. Фармакокинетическое испытание у крыс и собак породы бигль
[402] Соединение из примера растворяли в PBS, содержащем 5% ДМСО и 20% гидроксипропил(HP)-бета-циклодекстрина, и осуществляли внутривенное введение крысам в дозе 5 мг/кг, и суспендировали соединение из примера в дистиллированной воде, содержащей 0,5% метилцеллюлозы, и осуществляли пероральное введение крысам в дозе 10 мг/кг. Соединение из примера растворяли в PBS, содержащем 5% ДМСО и 20% гидроксипропил(HP)-бета-циклодекстрина, и осуществляли внутривенное введение собака породы бигль в дозе 5 мг/кг, и суспендировали соединение из примера в дистиллированной воде, содержащей 0,5% метилцеллюлозы, и осуществляли пероральное введение собакам породы бигль в дозе 10 мг/кг. Собирали образцы крови в запланированные моменты времени после однократного внутривенного и перорального введения соединения из примера здоровым крысам и собакам породы бигль. В собранные образцы крови добавляли ацетонитрил, содержащий материал внутреннего стандарта, для осаждения белка. Образец, экстрагированный посредством осаждения белка, центрифугировали и затем вводили надосадочный раствор в систему ЖХ-МС/МС для проведения количественного анализа концентрации соединения из примера в крови. Рассчитывали AUC для каждого способа введения на основании профиля зависимости концентрации в крови от времени, полученного в результате вышеописанного эксперимента, и на основании этого рассчитывали биодоступность (F) при пероральном введении.
[403] Результаты представлены в таблицах 9 и 10.
[404] [Таблица 9]
[405] Фармакокинетические параметры, рассчитанные после однократного перорального введения крысам соединений из примеров
[406]
[407] [Таблица 10]
[408] Фармакокинетические параметры, рассчитанные после однократного введения соединений из примеров собакам породы бигль
[409]
[410] В таблицах 9 и 10 показано, что соединение согласно настоящему изобретению обладает превосходной биодоступностью (F) при пероральном введении, демонстрируя выраженный высокий эффект с точки зрения фармакокинетики.
[411[ В настоящем описании опущено подробное описание той информации, которая может быть в достаточной степени распознана и логически выведена специалистами в той области техники, к которой относится настоящее изобретение. Помимо конкретных примеров, описанных в настоящем документе, в пределах объема изобретения могут быть сделаны различные модификации, которые не изменяют техническую сущность или основополагающий принцип настоящего изобретения. Таким образом, настоящее изобретение может быть осуществлено на практике другим образом, отличным от конкретно описанного и проиллюстрированного примерами в настоящем документе, который может быть понятен специалистам в той области техники, к которой относится настоящее изобретение.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ 4-МЕТОКСИПИРРОЛА ИЛИ ИХ СОЛИ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2016 |
|
RU2663895C1 |
| НОВЫЙ СОСТАВ ДЛЯ ИНЪЕКЦИЙ, ВКЛЮЧАЮЩИЙ 1-(5-(2,4-ДИФТОРФЕНИЛ)-1-((3-ФТОРФЕНИЛ)СУЛЬФОНИЛ)-4-МЕТОКСИ-1H-ПИРРОЛ-3-ИЛ)-N-МЕТИЛМЕТАНАМИН | 2022 |
|
RU2829138C2 |
| БЛОКАТОР НАТРИЕВЫХ КАНАЛОВ | 2016 |
|
RU2705578C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 4-МЕТОКСИПИРРОЛА | 2019 |
|
RU2763287C1 |
| НОВЫЕ ФТАЛАЗИНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2636585C2 |
| ИМИДАЗОПИРИДИНИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2020 |
|
RU2833052C2 |
| 1-ГЕТЕРОЦИКЛИЛСУЛЬФОНИЛ, 2-АМИНОМЕТИЛ, 5-(ГЕТЕРО-)АРИЛ ЗАМЕЩЕННЫЕ 1-Н-ПИРРОЛ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ СЕКРЕЦИИ КИСЛОТЫ | 2006 |
|
RU2415838C2 |
| БЕНЗОТИАЗОЛЬНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ РАССТРОЙСТВ | 2018 |
|
RU2778370C2 |
| ПРОИЗВОДНЫЕ 5-ПИРРОЛИЛ-2-ПИРИДИЛМЕТИЛСУЛЬФИНИЛБЕНЗИМИДАЗОЛА И СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1995 |
|
RU2100358C1 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1997 |
|
RU2146254C1 |
Группа изобретений относится к соединению, представленному химической формулой 2, или его фармацевтически приемлемой соли:
Химическая формула 2
 ,
,
также относится к фармацевтической композиции для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, содержащей терапевтически эффективное количество соединения, представленного химической формулой 2 или его фармацевтически приемлемой соли; и одного или более фармацевтически приемлемого носителя, также относится к применению соединения, представленного химической формулой 2, или его фармацевтически приемлемой соли, для производства лекарственного средства для лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, также относится к применению соединения, представленного химической формулой 2, или его фармацевтически приемлемой соли, для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой. Группа изобретений обеспечивает разработку новых соединений, обладающих превосходными ингибирующими действиями в отношении протонного насоса для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой. 6 н. и 14 з.п. ф-лы, 10 табл., 21 пр.
1. Соединение, представленное следующей химической формулой 2, или его фармацевтически приемлемая соль:
[Химическая формула 2]
в Химической формуле 2
X1 представляет собой F;
X2 представляет собой водород или F;
R1 представляет собой метил; и
R2 представляет собой метокси, этокси, метил или этил.
2. Соединение или его фармацевтически приемлемая соль по п. 1,
отличающиеся тем, что R2 представляет собой метокси или метил.
3. Соединение или его фармацевтически приемлемая соль по п. 1,
отличающиеся тем, что R1 представляет собой метил и
R2 представляет собой метокси или метил.
4. Соединение или его фармацевтически приемлемая соль по п. 1,
отличающиеся тем, что
X1 представляет собой F;
X2 представляет собой F;
R1 представляет собой метил; и
R2 представляет собой метокси или метил.
5. Соединение или его фармацевтически приемлемая соль по п. 1,
отличающиеся тем, что
X1 представляет собой F;
X2 представляет собой водород;
R1 представляет собой метил; и
R2 представляет собой метокси или метил.
6. Соединение или его фармацевтически приемлемая соль по п. 1,
отличающиеся тем, что
X1 представляет собой F;
X2 представляет собой водород или F;
R1 представляет собой метил; и
R2 представляет собой метокси.
7. Соединение или его фармацевтически приемлемая соль по п. 1,
отличающиеся тем, что
X1 представляет собой F;
X2 представляет собой водород или F;
R1 представляет собой метил; и
R2 представляет собой метил.
8. Соединение или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что соединение, представленное химической формулой 2, представляет собой любое соединение, выбранное из группы, состоящей из следующих соединений:
1-(5-(2-фторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин;
1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метоксипиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин;
1-(5-(2,4-дифторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин; и
1-(5-(2-фторфенил)-4-метокси-1-((6-метилпиридин-3-ил)сульфонил)-1H-пиррол-3-ил)-N-метилметанамин.
9. Фармацевтическая композиция для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой, содержащая терапевтически эффективное количество соединения по любому из пп. 1–8 или его фармацевтически приемлемой соли и один или более фармацевтически приемлемый носитель.
10. Фармацевтическая композиция по п. 9, отличающаяся тем, что желудочно-кишечная язва, желудочно-кишечное воспалительное заболевание или заболевание, связанное с желудочной кислотой, представляет собой одно или более заболеваний, выбранных из группы, состоящей из пептической язвы, язвы желудка, язвы двенадцатиперстной кишки, язвы, вызванной НСПВП, язвы на фоне острого стресса, синдрома Золингера-Эллисона, инфекции Helicobacter pylori, гастрита, эрозивного эзофагита, неэрозивного эзофагита, рефлюкс-эзофагита, воспалительной болезни кишечника, симптоматической гастроэзофагеальной рефлюксной болезни (симптоматической ГЭРБ), функциональной диспепсии, рака желудка, MALT-лимфомы желудка, повышенной кислотности и кровоизлияния в верхнем отделе желудочно-кишечного тракта вследствие инвазивного стресса.
11. Применение соединения по любому из пп. 1, 3, 5–8 или его фармацевтически приемлемой соли для производства лекарственного средства для лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой.
12. Применение по п. 11, отличающееся тем, что желудочно-кишечная язва, желудочно-кишечное воспалительное заболевание или заболевание, связанное с желудочной кислотой, представляет собой одно или более заболеваний, выбранных из группы, состоящей из пептической язвы, язвы желудка, язвы двенадцатиперстной кишки, язвы, вызванной НСПВП, язвы на фоне острого стресса, синдрома Золингера-Эллисона, инфекции Helicobacter pylori, гастрита, эрозивного эзофагита, неэрозивного эзофагита, рефлюкс-эзофагита, воспалительной болезни кишечника, симптоматической гастроэзофагеальной рефлюксной болезни (симптоматической ГЭРБ), функциональной диспепсии, рака желудка, MALT-лимфомы желудка, повышенной кислотности и кровоизлияния в верхнем отделе желудочно-кишечного тракта вследствие инвазивного стресса.
13. Применение соединения по любому из пп. 1, 3, 5–8 или его фармацевтически приемлемой соли для предотвращения или лечения желудочно-кишечных язв, желудочно-кишечных воспалительных заболеваний или заболеваний, связанных с желудочной кислотой.
14. Применение по п. 13, отличающееся тем, что желудочно-кишечная язва, желудочно-кишечное воспалительное заболевание или заболевание, связанное с желудочной кислотой, представляет собой одно или более заболеваний, выбранных из группы, состоящей из пептической язвы, язвы желудка, язвы двенадцатиперстной кишки, язвы, вызванной НСПВП, язвы на фоне острого стресса, синдрома Золингера-Эллисона, инфекции Helicobacter pylori, гастрита, эрозивного эзофагита, неэрозивного эзофагита, рефлюкс-эзофагита, воспалительной болезни кишечника, симптоматической гастроэзофагеальной рефлюксной болезни (симптоматической ГЭРБ), функциональной диспепсии, рака желудка, MALT-лимфомы желудка, повышенной кислотности и кровоизлияния в верхнем отделе желудочно-кишечного тракта вследствие инвазивного стресса.
| US 20080139639 A1, 12.06.2008 | |||
| US 20160009646 A1, 14.01.2016 | |||
| US 8993598 B2, 31.03.2015 | |||
| АНТАГОНИСТ КИСЛОТНОГО НАСОСА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С ПАТОЛОГИЧЕСКИМ НАРУШЕНИЕМ МОТОРИКИ ЖЕЛУДОЧНО-КИШЕЧНОГО ТРАКТА | 2010 |
|
RU2586276C2 |
| Yasuyoshi Arikawa and at al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Med | |||
| Chem., vol | |||
| Устройство двукратного усилителя с катодными лампами | 1920 |
|
SU55A1 |

