Область техники
Изобретение относится к агенту против Helicobacter pylori, обладающему уникальным свойством ингибировать секрецию желудочного сока.
Уровень техники
Гастрит, язва желудка и язва двенадцатиперстной кишки являются заболеваниями, которые развиваются в результате сложного взаимодействия таких факторов, как стресс, генетическая предрасположенность и стиль жизни. В последние годы большое внимание уделяется бактерии Helicobacter pylori (H. pylori) как одной из главных причин этих заболеваний. Со времени успешного выделения и изучения спиральных бактерий из образцов биопсии желудка, проведенного Warren и Marshall в 1983 г., стали искать связь между присутствием этой бактерии и гастритом, язвой желудка, язвой двенадцатиперстной кишки и раком желудка. В результате было установлено, что если степень инфицирования нормального желудка бактериями Helicobacter pylori (H. pylori) составляет примерно 4%, то при хроническом гастрите степень инфицирования составляет примерно 83%, при язве желудка примерно 69%, при язве двенадцатиперстной кишки примерно 92% и при синдроме неязвенной диспепсии примерно 51% (см. непатентный документ 1). Кроме того, инфицирование бактерией Helicobacter pylori заметно коррелирует с ростом заболеваемости раком желудка, и в 1994 г. Международное Агентство по исследованию рака ВОЗ заявило, что бактерия Helicobacter pylori является канцерогеном.
Основным способом лечения гастрита, язвы желудка, язвы двенадцатиперстной кишки и т.п. была симптоматическая терапия с использованием ингибиторов секреции желудочного сока типа блокаторов H2 и ингибиторов протонного насоса и защитных агентов слизистой оболочки для улучшения субъективных симптомов, таких как эпигастральная боль и ускорение заживления язвы желудка. Однако известно, что хотя под действием этих лекарств повреждения слизистой со временем залечиваются, после прекращения лечения в течение года примерно у 80% пациентов наступает рецидив (см. непатентный документ 1). С другой стороны, также известно, что в случае язвы двенадцатиперстной кишки в результате подавления бактерий Helicobacter pylori степень рецидива составляет 10% или менее и эта степень еще ниже в случае язвы желудка (см. непатентный документ 2).
В настоящее время в качестве способа подавления бактерий Helicobacter pylori проводят лечение, например, ингибиторами протонного насоса (PPI) в сочетании с антибактериальными агентами типа амоксициллина и кларитромицина в больших количествах в течение одной недели или дольше и в некоторых случаях с добавкой метронидазола. Однако введение антибактериальных агентов в больших количествах вызывает также гибель полезных бактерий в кишечнике, и в результате наблюдаются отрицательные эффекты, такие как жидкий стул, диарея и нарушения вкуса, глоссит, стоматит, нарушения функции печени и геморрагический энтерит, а также повышается вероятность стимулирования роста устойчивой к метициллину Staphylococcus aureus (MRSA).
На сегодня подано большое число патентных заявок, относящихся к производным пиридина, используемым в качестве противоязвенных агентов, ингибиторов секреции желудочного сока, антибактериальных агентов против бактерий Helicobacter pylori и т.п. (см. Патенты 1-8). Однако до сих пор не найдено соединение, которое хорошо сбалансированным образом сочетало бы активность против Helicobacter pylori и способность ингибировать секрецию желудочного сока, а также подавлять бактерии Helicobacter pylori при использовании в качестве единственного агента.
Кроме того, было найдено соединение, активное против Helicobacter pylori in vitro и ингибирующее секрецию желудочного сока (см. непатентный документ 3). Однако при исследовании инфицирования когтистой песчанки (Mongolian gerbil) бактериями Helicobacter pylori, которое рассматривается как модель инфицирования человека бактериями Helicobacter pylori, не удалось подтвердить эффективность этого препарата, и его разработка была прекращена.
Несмотря на описанные выше столь экстенсивные попытки в настоящее время широко практикуют тройную терапию для подавления бактерий Helicobacter pylori. Причина заключается в том, что ингибиторы протонного насоса, такие как омепразол, ланзопразол и рабепразол, в сочетании с кларитромицином, очень нестабильны в кислой среде, что затрудняет проявление антибактериальной активности амоксициллина в кислой среде. Таким образом, после мощного ингибирования желудочного сока введенными в кишечник ингибиторами протонного насоса необходимо вводить большие количества указанных выше лабильных в кислой среде антибиотиков.
Патент 1: JP-A No. 61-50979.
Патент 2: JP-A No. 3-173817.
Патент 3: JP-A No. 5-247035.
Патент 4: JP-A No. 59-181277.
Патент 5: JP-A No. 7-69888.
Патент 6: JP-A No. 3-48680.
Патент 7: JP-A No. 2-209809.
Патент 8: JP-A No. 58-39622.
Непатентный документ 1: Martin J. Blaser: Clin. Infectious Disease, 15; 386-393, 1992.
Непатентный документ 2: Graham D.Y., et al.: Ann. Intern. Med., 116; 705-708, 1992.
Непатентный документ 3: Thomas C. Kuehler, et al., J. Med. Chem. 1998, 41, 1777-1788.
Описание изобретения
Проблемы, которые необходимо решить с помощью настоящего изобретения
Настоящее изобретение предлагает новую фармацевтическую композицию для предупреждения и/или лечения заболеваний, связанных с бактерией Helicobacter pylori, и/или заболеваний, связанных с секрецией желудочного сока, которая эффективна для предупреждения и/или лечения заболеваний, связанных с бактериями Helicobacter pylori, и также действует специфично против бактерий Helicobacter pylori и не оказывает влияния на кишечную флору, но при этом устойчива к кислоте и обладает ингибирующей активностью на секрецию желудочного сока; и соединение или его соль, которые используются как активный ингредиент композиции.
Способы решения этих проблем
Заявители предприняли целевой поиск соединения, которое удовлетворяет следующим условиям:
1) должно быть стабильно в кислой среде; 2) должно обладать удовлетворительной антибактериальной активностью против бактерий Helicobacter pylori; 3) должно действовать конкретно против бактерий Helicobacter pylori, но не оказывать влияния на кишечную флору; 4) должно воздействовать даже на те бактерии, которые устойчивы к антибактериальным агентам, используемым при лечении заболеваний, вызванных бактериями Helicobacter pylori; 5) должно ингибировать секрецию желудочного сока и 6) должно оказывать подавляющий эффект на бактерии в модельном инфицировании животных бактериями Helicobacter pylori при использовании в качестве единственного агента (Hirayama et al. J. Gastroenterol., 1996. 31 (Suppl. 9), 24-8).
В результате было обнаружено семейство соединений, которые устойчивы в кислой среде, обладают сильной активностью против Helicobacter pylori при минимальной подавляющей концентрации (MIC) 0,1 мкг/мл или меньше, не действуют на обычные бактерии, присущие самому человеку, проявляют антибактериальное действие конкретно против бактерий Helicobacter pylori, эффективны даже против бактерий, устойчивых к антибиотикам, таким как кларитромицин, который широко используют в клинической практике против бактерий Helicobacter pylori, и также дают конкретные результаты в сочетании с ингибирующей активностью в отношении секреции желудочного сока. Авторами настоящего изобретения также было найдено, что группа соединений обладает активностью против бактерий Helicobacter pylori в модельном исследовании инфицирования животного при использовании в качестве индивидуальных препаратов. На основании этих выводов авторы завершили настоящее изобретение.
Конкретно, настоящее изобретение относится к производному пиридина, представленному формулой (I), или его фармакологически приемлемой соли:
[Химическая формула 1]

в которой R представляет собой линейный алкил с 4-8 атомами углерода.
Настоящее изобретение также относится к фармацевтической композиции, включающей производное пиридина приведенной выше формулы (I) или его фармакологически приемлемую соль, и более конкретно к фармацевтической композиции, включающей производное пиридина приведенной выше формулы (I) или его фармакологически приемлемую соль и фармацевтически приемлемый носитель.
Кроме того, настоящее изобретение относится к применению производного пиридина, представленного формулой (I), или его фрамакологически приемлемой соли для получения препарата для предупреждения или лечения заболевания, вызванного бактериями Helicobacter pylori, и/или заболевания, связанного с секрецией желудочного сока.
Настоящее изобретение также относится к способу предупреждения или лечения заболевания, вызванного бактериями Helicobacter pylori, и/или заболевания, связанного с секрецией желудочного сока, причем данный способ включает введение эффективного количества производного пиридина формулы (I) или его фармакологически приемлемой соли пациенту с заболеванием, вызванным бактериями Helicobacter pylori, и/или заболеванием, связанным с секрецией желудочного сока, или пациенту из группы риска по данному заболеванию.
Настоящее изобретение также относится к способу получения производного пиридина формулы (I) или его фармакологически приемлемой соли, причем способ включает взаимодействие соединения, представленного следующей формулой (II):
[Химическая формула 2]
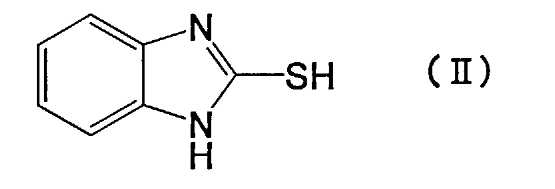
с соединением, представленным следующей формулой (III):
[Химическая формула 3]
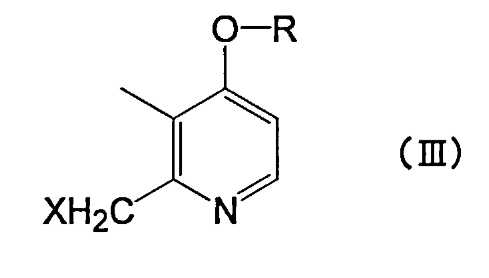
где R представляет собой линейный алкил с 4-8 атомами углерода и X представляет собой атом галогена.
Настоящее изобретение также относится к соединению, представленному формулой (III), приведенной выше.
Настоящее изобретение можно описать более подробно следующим образом.
(1) Производное пиридина, представленное формулой (I), или его фармакологически приемлемая соль.
(2) Производное пиридина по п. (1) или его фармакологически приемлемая соль, причем R в формуле (I) является линейным алкилом с 5-7 атомами углерода.
(3) Фармацевтическая композиция, включающая производное пиридина по пп. (1) или (2) или его фармакологически приемлемую соль и фармацевтически приемлемый носитель.
(4) Фармацевтическая композиция по п. (3), где фармацевтическая композиция предназначена для предупреждения или лечения заболевания, вызванного бактериями Helicobacter pylori, и/или заболевания, связанного с секрецией желудочного сока.
(5) Фармацевтическая композиция по п. (4), причем заболевания включают гастрит, язву желудка, язву двенадцатиперстной кишки, синдром неязвенной диспепсии, лимфому желудка MALT, желудочный гиперпластический полип, рак пищеварительной системы или панкреатит вследствие гипергастринемии, вызванной Helicobacter pylori, воспалительное заболевание кишечника, вызванное Helicobacter pylori, или рак желудка после эндоскопической резекции рака желудка на ранней стадии.
(6) Фармацевтическая композиция по одному из пунктов (3)-(5), дополнительно включающая другой ингибитор секреции желудочного сока и/или антибактериальный агент в качестве активного ингредиента.
(7) Применение производного пиридина по пп. (1) или (2) или его фармакологически приемлемой соли для получения препарата для предупреждения или лечения заболевания, вызванного Helicobacter pylori, и/или заболевания, связанного с секрецией желудочного сока.
(8) Применение по п. (7), при котором заболевание включает гастрит, язву желудка, язву двенадцатиперстной кишки, синдром неязвенной диспепсии, желудочную лимфому MALT, желудочный гиперпластический полип, рак пищеварительной системы или панкреатит вследствие гипергастринемии, вызванной Helicobacter pylori, воспалительное заболевание кишечника, вызванное Helicobacter pylori, или рак желудка после эндоскопической резекции рака желудка на ранней стадии.
(9) Применение по пп. (7) или (8), где препарат для предупреждения или лечения заболевания, вызванного Helicobacter pylori, и/или заболевания, связанного с секрецией желудочного сока, дополнительно включает другой ингибитор секреции желудочного сока и/или антибактериальный препарат в качестве активного ингредиента.
(10) Способ предупреждения или лечения заболевания, вызванного бактериями Helicobacter pylori, и/или заболевания, связанного с секрецией желудочного сока, включающий введение эффективного количества производного пиридина по пп. (1) или (2) или его фармакологически приемлемой соли пациенту с заболеванием, вызванным бактериями Helicobacter pylori, или заболеванием, связанным с секрецией желудочного сока, или пациенту из группы риска по данному заболеванию.
(11) Способ по п. (10), в котором заболевание включает гастрит, язву желудка, язву двенадцатиперстной кишки, синдром неязвенной диспепсии, желудочную лимфому MALT, желудочный гиперпластический полип, рак пищеварительной системы или панкреатит вследствие гипергастринемии, вызванной Helicobacter pylori, воспалительное заболевание кишечника, вызванное Helicobacter pylori, или рак желудка после эндоскопической резекции рака желудка на ранней стадии.
(12) Способ по пп. (10) или (11), в котором вводят эффективное количество производного пиридина по п. (1) или (2) или его фармакологически приемлемой соли и одновременно дополнительно вводят другой ингибитор секреции желудочного сока и/или антибактериальный препарат в качестве другого активного препарата.
(13) Способ получения производного пиридина формулы (I) или его фармакологически приемлемой соли, включающий взаимодействие соединения, представленного приведенной выше формулой (II), с соединением, представленным приведенной выше формулой (III).
(14) Фармацевтическая композиция, включающая производное пиридина по п.п. (1) или (2) или его фармакологически приемлемую соль.
(15) Фармацевтическая композиция по п. (14) для подавления или бактериостаза Helicobacter pylori и ингибирования секреции желудочного сока.
(16) Фармацевтическая композиция по пп. (14) или (15), включающая в качестве активного ингредиента только производное пиридина по п.п. (1) или (2) или его фармакологически приемлемую соль.
(17) Фармацевтическая композиция по любому из приведенных выше пунктов (14)-(16), включающая также другой ингибитор секреции желудочного сока и/или антибактериальный агент в качестве активного ингредиента.
(18) Средство против Helicobacter pylori, включающее производное пиридина по пп. (1) или (2) или его фармакологически приемлемую соль.
(19) Ингибитор секреции желудочного сока, включающий производное пиридина по п.п. (1) или (2) или его фармакологически приемлемую соль.
(20) Профилактическое или терапевтическое средство для лечения заболевания или состояния, вызванного Helicobacter pylori и/или секрецией желудочного сока, включающее производное пиридина по пп. (1) или (2) или его фармакологически приемлемую соль.
(21) Профилактическое или терапевтическое средство по пункту (20), причем заболевание или состояние представляет собой гастрит, язву желудка, язву двенадцатиперстной кишки, синдром неязвенной диспепсии, желудочную лимфому MALT, желудочный гиперпластический полип или рак желудка после эндоскопической резекции рака желудка на ранней стадии.
(22) Профилактическое или терапевтическое средство для лечения заболевания или состояния, вызванного Helicobacter pylori, включающее новое производное пиридина по п.п. (1) или (2) или его фармакологически приемлемую соль.
(23) Профилактическое или терапевтическое средство по пунктам (21) или (22), причем заболевание или состояние представляет собой рак пищеварительной системы или панкреатит вследствие гипергастринемии, вызванной Helicobacter pylori, или воспалительное заболевание кишечника, вызванное Helicobacter pylori.
Эффекты изобретения
Производное пиридина или его соль по данному изобретению не только обладают высокой активностью против Helicobacter pylori, но также чрезвычайно стабильны в кислой среде, так что соединение не разлагается и остается эффективным даже в присутствии желудочного сока. Кроме того, данное соединение при использовании в качестве единственного препарата действует как ингибитор секреции желудочного сока, обладает активностью против бактерий Helicobacter pylori и применяется как профилактическое и/или терапевтическое лекарственное средство при разных заболеваниях, связанных с бактериями Helicobacter pylori, или при различных заболеваниях, связанных с повышенной секрецией желудочного сока, например при гастрите, язве желудка, язве двенадцатиперстной кишки, синдроме неязвенной диспепсии, лимфоме желудка MALT, гиперпластическом полипе желудка, раке желудка после эндоскопической резекции на ранней стадии рака, раке пищеварительной системы или панкреатите вследствие гипергастринемии, вызванной Helicobacter pylori, воспалении кишечника, вызванном Helicobacter pylori, и т.п.
Производное пиридина и его соль по данному изобретению отличаются тем, что эти соединения можно использовать в качестве профилактических и/или терапевтических лекарственных средств, эффективных при этих заболеваниях, в частности при использовании в качестве единственного препарата.
Краткое описание фигур
Фиг.1 представляет графически результаты теста на стабильность соединения по данному изобретению и соединения сравнительного примера в растворе соляной кислоты (pH 2). По оси ординат на фиг.1 представлена стабильность (остаточное соотношение) в %, а по оси абсцисс - время (мин).
Фиг.2 представляет собой графические результаты теста на антибактериальную активность (MIC) против бактерий Helicobacter pylori при наличии или отсутствии окисления тиоэфирной группы соединения по настоящему изобретению и соединения сравнительного примера и в зависимости от разницы в числе атомов углерода в алкиле алкоксильной группы в 4-м положении пиридинового цикла. По оси ординат фиг.2 указаны значения MIC (мкг/мл), а по оси абсцисс - число атомов углерода в алкильной группе. Черные кружки на фиг.2 (темно-синие на первоначальной диаграмме) относятся к SH-форме, а серые кружки (красные на первоначальной диаграмме) относятся к SO-форме.
Наилучший способ осуществления изобретения
Авторы данного изобретения провели подробное исследование соединений 2-(4-алкокси-3-метилпиридин-2-илметилтио)бензимидазола и их сульфинильных производных и получили следующие результаты. Были обнаружены следующие эффекты: (1) в случае сульфинильного производного активность против бактерий Helicobacter pylori характеризуется величиной MIC 3,0 мкг/мл, и хотя производные обладают некоторой активностью, они очень нестабильны в кислой среде (см. таблицу 1 и фиг.1); (2) когда число атомов углерода в алкоксильной группе равно 4-8 и предпочтительно 5-7, активность против бактерий Helicobacter pylori чрезвычайно высока и MIC уменьшается до 1/3-1/10 (см. таблицу 2 и фиг.2); (3) в случае когда атом углерода в α-положении алкоксильной группы образует разветвленную цепь, т.е. в случае когда алкоксильная группа является изоалкоксильной группой, активность против бактерий Helicobacter pylori чрезвычайно понижена (см. таблицу 3 и таблицу 6); (4) соединение по настоящему изобретению проявляет потенциальную антибактериальную активность против устойчивых к кларитромицину штаммов и штаммов, не чувствительных к амоксициллину (см. таблицу 4); (5) соединение по настоящему изобретению, по-видимому, не обладает антибактериальным действием против разных грамотрицательных бактерий и грамположительных бактерий (см. таблицу 5); и (6) соединение по настоящему изобретению ингибирует секрецию желудочного сока (см. таблицу 7).
В результате, соединение по настоящему изобретению оказывается эквивалентным или лучше тройной комбинации омепразол + амоксициллин + кларитромицин, которую широко применяют в мире для терапии против бактерий Helicobacter pylori, и, в частности, соединение по настоящему изобретению проявляет коэффициент эффективности, равный или лучше коэффициента эффективности такой тройной комбинации даже при использовании в качестве единственного агента. Более того, соединение по настоящему изобретению проявляет конкретную антибактериальную активность против бактерий Helicobacter pylori и проявляет антибактериальную активность против бактерий, не чувствительных или устойчивых к амоксициллину и кларитромицину. Далее, соединение оказывает ингибирующее действие на секрецию желудочного сока и также чрезвычайно устойчиво к кислоте, так что не разлагается даже в присутствии желудочного сока и весьма эффективно в клинической практике. Эти характерные эффекты обусловлены тем, что метилтио-группа находится не в виде сульфинильной группы в окисленном состоянии, а в виде тиоэфира, и тем, что алкоксильная группа в 4 положении пиридинового цикла является конкретной алкильной группой.
Кроме того, производное пиридина, представленное приведенной выше формулой (I), или его соль по данному изобретению являются новыми соединениями, которые не описаны в литературе.
R в формуле (I) по настоящему изобретению является линейной алкильной группой с 4-8 и предпочтительно 5-7 атомами углерода. Согласно настоящему изобретению, в качестве линейных алкильных групп предпочтительны н-алкильные группы, среди которых можно упомянуть алкильную группу -CH2-R' (где R' представляет линейный алкил с 3-7 и предпочтительно 4-6 атомами углерода), не разветвленную в α-положении алкоксильной группы. Среди предпочтительных алкильных групп можно назвать н-бутил, н-пентил, н-гексил, н-гептил, н-октил и т.п., а среди более предпочтительных алкильных групп можно указать н-пентил, н-гексил и н-гептил.
Более конкретно в качестве предпочтительных соединений по настоящему изобретению можно назвать
2-[(4-н-бутилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазол;
2-[(4-н-пентилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазол;
2-[(4-н-гексилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазол;
2-[(4-н-гептилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазол и
2-[(4-н-октилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазол.
Кроме того, в качестве более предпочтительных соединений по настоящему изобретению можно назвать
2-[(4-н-пентилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазол;
2-[(4-н-гексилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазол и
2-[(4-н-гептилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазол.
Производное пиридина, представленное формулой (I), или его соль по настоящему изобретению можно получить путем взаимодействия исходных соединений - меркапто-производных, представленных приведенной выше формулой (II), с производным пиридина, представленным приведенной выше формулой (III). В производном пиридина, представленном приведенной выше формулой (III), X не ограничен конкретно по длине, т.к. это уходящая группа, но в качестве предпочтительной уходящей группы можно назвать атом галогена. Среди атомов галогена можно назвать хлор, бром, йод и т.п.
Соединение, представленное формулой (III), является новым соединением, и его используют в качестве промежуточного соединения для получения соединения формулы (I) по настоящему изобретению. Целью настоящего изобретения является предложение соединения, представленного формулой (III).
Эту реакцию предпочтительно проводить в присутствии основания. В качестве основания, используемого в данной реакции, можно назвать, например, гидриды щелочных металлов, такие как гидрид натрия и гидрид калия; алкоголяты натрия, такие как метоксид натрия и этоксид натрия; карбонаты щелочных металлов, такие как карбонат калия и карбонат натрия; органические амины, такие как триэтиламин и т.п. Кроме того, в качестве растворителя данной реакции можно назвать, например, спирты, такие как метанол и этанол, диметилсульфоксид и т.п. Основание, используемое в реакции, обычно берут в небольшом избытке по сравнению с эквивалентным количеством, но можно использовать и большой избыток основания. Например, количество может составлять примерно 2-10 эквивалентов и, более предпочтительно, примерно 2-4 эквивалента. Температура реакции обычно находится в пределах от 0°C до почти температуры кипения используемого растворителя, и, более предпочтительно, можно указать температуру примерно 20-80°C. Время реакции выбирают соответственно, но обычно оно составляет примерно 0,2-24 ч и, более предпочтительно, примерно 0,5-2 ч.
Целевое соединение (I), полученное по описанной выше реакции, можно выделить и очистить традиционными способами, такими как перекристаллизация и хроматография.
Производное пиридина, представленное формулой (I) по настоящему изобретению, можно получить в виде фармацевтически приемлемой соли традиционными способами. Примеры такой соли включают гидрохлорид, гидробромид, гидройодид, фосфат, нитрат, сульфат, ацетат, цитрат и т.п.
Исходное соединение, представленное формулой (III), которое используют для получения производного пиридина, представленного формулой (I) по настоящему изобретению, можно получить по следующей реакции.
[Химическая формула 4]
Способ получения 1
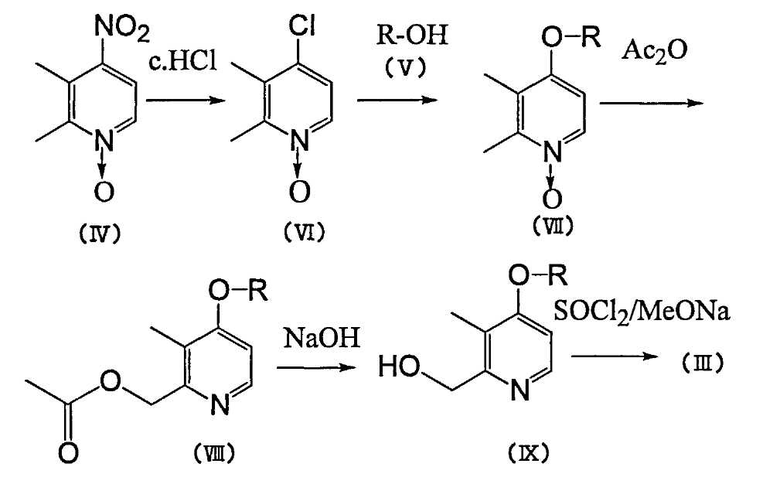
Во-первых, нитросоединение по формуле (IV) вводят в реакцию с концентрированной соляной кислотой с образованием хлорпроизводного (VI), и когда это хлорпроизводное (VI) вводят в реакцию с производным спирта ROH (V) в присутствии основания, можно получить алкокси-производное, представленное формулой (VII). В этом случае основанием могут служить, например, щелочные металлы, такие как литий, натрий и калий; гидриды щелочных металлов, такие как гидрид натрия и гидрид калия; алкоголяты, такие как трет-бутоксид калия, пропоксид натрия, этоксид натрия и метоксид натрия; карбонаты или кислые карбонаты щелочных металлов, такие как карбонат калия, карбонат лития, карбонат натрия, кислый карбонат калия и кислый карбонат натрия; гидроксиды щелочных металлов, такие как гидроксид натрия и гидроксид калия, и т.п. В качестве растворителя реакции можно назвать низшие спирты ROH, а также простые эфиры, такие как тетрагидрофуран и диоксан; кетоны, такие как ацетон и метилэтилкетон; ацетонитрил, диметилформамид, диметилсульфоксид и т.п. Температуру реакции выбирают в интервале от температуры ледяной бани до почти температуры кипения растворителя. Время реакции составляет примерно 1-48 ч.
Полученное таким образом алкоксипроизводное (VII) нагревают до примерно 80-120°C в присутствии только уксусного ангидрида или неорганической кислоты, такой как серная или хлорная кислоты, и получают производное 2-ацетоксиметилпиридина формулы (VIII). Обычно время реакции составляет примерно 0,1-10 ч.
Затем путем щелочного гидролиза соединения (VIII) можно получить производное 2-гидроксиметилпиридина формулы (IX). В этом случае в качестве щелочи можно назвать, например, гидроксид натрия, гидроксид калия, карбонат калия, карбонат натрия и т.п. Растворителем может служить, например, метанол, этанол, вода и т.п. Время реакции составляет примерно 0,1-2 ч.
Затем можно получить производное 2-галоидметилпиридина формулы (III) путем галогенирования соединения (IX) таким галогенирующим агентом, как тионилхлорид. В качестве растворителя можно использовать, например, хлороформ, дихлорметан, тетрахлорэтан и т.п. Температура реакции обычно составляет примерно 20-80°C, а время реакции равно примерно 0,1-2 ч.
Соединение по настоящему изобретению или его соль устойчивы к кислоте и способны подавлять бактерии Helicobacter pylori или способствовать их бактериостазу даже в организме животных из класса млекопитающих (обычно людей). Т.е. соединение по настоящему изобретению или его соль эффективны в качестве агента против Helicobacter pylori.
Соединение по настоящему изобретению или его соль могут также, помимо указанного действия, ингибировать секрецию желудочного сока у животных из класса млекопитающих (обычно людей). Т.е. соединение по настоящему изобретению или его соль являются также эффективными ингибиторами секреции желудочного сока.
Настоящее изобретение также предлагает способ подавления Helicobacter pylori или их бактериостаза у млекопитающих и/или способ ингибирования секреции желудочного сока, который включает введение эффективного количества соединения по настоящему изобретению или его соли млекопитающему, который нуждается в таком способе.
Настоящее изобретение также предлагает использовать соединение по настоящему изобретению или его соль для получения агента против Helicobacter pylori и/или ингибитора секреции желудочного сока.
Настоящее изобретение также предлагает фармацевтическую композицию, сочетающую действие против Helicobacter pylori и ингибирующую активность по отношению к секреции желудочного сока, содержащую соединение по настоящему изобретению или его соль в качестве активного ингредиента. Соединение по настоящему изобретению отличается тем, что обладает действием против Helicobacter pylori и активностью в ингибировании секреции желудочного сока при использовании в качестве единственного агента. Поэтому предпочтительно, чтобы фармацевтическая композиция включала в качестве активного ингредиента только соединение по настоящему изобретению или его соль. Кроме того, фармацевтическая композиция может также включать в качестве активного ингредиента другой ингибитор секреции желудочного сока и/или антибактериальный агент. В качестве другого ингибитора секреции желудочного сока можно назвать H2 блокаторы, ингибиторы протонного насоса и т.п. Примеры H2 блокаторов, которые можно использовать в настоящем изобретении, включают циметидин, фамотидин, ранитидин и т.п., а примеры ингибиторов протонного насоса, которые можно использовать в настоящем изобретении, включают ланзопразол, омепразол, рабепразол, пантопразол и т.п., и примеры этим не ограничиваются.
Лекарственное средство, содержащее соединение по настоящему изобретению или его соль, эффективно для предупреждения или лечения заболевания, вызванного Helicobacter pylori и/или связанного с секрецией желудочного сока (предпочтительно Helicobacter pylori и секрецией желудочного сока). Заболевание, связанное с Helicobacter pylori, относится к заболеванию, которое вызывается или усугубляется инфекцией, выживанием или ростом Helicobacter pylori in vivo. Заболевание, связанное с секрецией желудочного сока, относится к заболеванию, которое вызывается или обостряется секрецией желудочного сока. Примеры таких заболеваний включают гастрит, язву желудка, язву двенадцатиперстной кишки, синдром неязвенной диспепсии, желудочную лимфому MALT, желудочный гиперпластический полип, рак пищеварительной системы или панкреатит вследствие гипергастринемии, вызванной Helicobacter pylori, воспалительное заболевание кишечника, вызванные Helicobacter pylori, рак желудка после эндоскопической резекции рака желудка на ранней стадии и т.п. Что касается рака желудка после эндоскопической резекции рака на ранней стадии, его канцерогенез можно задержать или затормозить с помощью соединения по настоящему изобретению или его соли.
Лекарственное средство, содержащее соединение по настоящему изобретению или его соль, также эффективно для предупреждения или лечения заболевания, вызванного Helicobacter pylori. Примеры таких заболеваний включают рак пищеварительной системы или панкреатит вследствие гипергастринемии, вызванной Helicobacter pylori, или воспалительное заболевание кишечника, вызванное Helicobacter pylori. Что касается рака пищеварительной системы, его развитие можно задержать или затормозить с помощью соединения по настоящему изобретению или его соли.
Настоящее изобретение также предлагает способ предупреждения или лечения заболевания, вызванного Helicobacter pylori и/или связанного с секрецией желудочного сока, который включает введение эффективного количества соединения по настоящему изобретению или его соли млекопитающему, который нуждается в таком способе.
Настоящее изобретение также предлагает использование соединения по настоящему изобретению или его соли для получения профилактического или терапевтического средства для лечения заболевания, связанного с Helicobacter pylori и/или с секрецией желудочного сока.
Настоящее изобретение также предлагает способ предупреждения или лечения заболевания, вызванного Helicobacter pylori, который включает введение эффективного количества соединения по настоящему изобретению или его соли млекопитающему, которое нуждается в таком способе. Настоящее изобретение также предлагает использование соединения по настоящему изобретению или его соли для получения профилактического или терапевтического средства для лечения заболевания, вызванного Helicobacter pylori.
При использовании соединения по настоящему изобретению или его соли в качестве лекарственного средства можно использовать разные лекарственные формы в соответствии с целью предупреждения или лечения, и примеры лекарственных форм могут включать твердые препараты для перорального применения (например, порошки, мелкие гранулы, гранулы, таблетки, таблетки в оболочке и капсулы), жидкие препараты для перорального применения (например, сухие сиропы, сиропы, жидкие препараты для внутреннего употребления и эликсиры), препараты для инъекций (например, подкожных, внутримышечных и внутривенных) и т.п. Эти медикаменты могут содержать наряду с активными ингредиентами соответствующие фармацевтически приемлемые эксципиенты, носители и т.п.
При изготовлении твердого препарата для перорального применения к соединению по настоящему изобретению можно добавить эксципиент и, если нужно, связующее, дезинтегратор, компонент для облегчения проглатывания, краситель, вкусовые добавки и отдушки и т.п. и затем обычными способами изготовить таблетки, таблетки в оболочке, гранулы, порошки, капсулы и т.п. Используют добавки, обычно применяемые на современном уровне техники, и в качестве эксципиента используют, например, зерновой крахмал, лактозу, сахарозу, хлорид натрия, маннит, сорбит, глюкозу, крахмал, карбонат кальция, каолин, микрокристаллическую целлюлозу, кремниевую кислоту и т.п.; в качестве связующего используют воду, этанол, гуммиарабик, трагакант, пропанол, простой сироп, раствор глюкозы, раствор крахмала, раствор желатина, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, желатин, гидроксипропилкрахмал, метилцеллюлозу, этилцеллюлозу, шеллак, фосфат кальция, поливиниловый спирт, поливиниловый эфир, поливинилпирролидон и т.п. В качестве дезинтегратора можно добавить порошок желатина, кристаллическую целлюлозу, сухой крахмал, альгинат натрия, пектин, агаровый порошок, карбоксиметилцеллюлозу, гидрокарбонат натрия, карбонат кальция, цитрат кальция, лаурилсульфат натрия, моноглицерид стеариновой кислоты, лактозу и т.п.; в качестве агента для облегчения проглатывания - оксид кремния, очищенный тальк, соли стеариновой кислоты, боракс, полиэтиленгликоль и т.п.; и в качестве красителя - оксид титана, оксид железа и т.п. Примерами вкусовых добавок и отдушек могут служить сахароза, апельсиновые корки, лимонная кислота, винная кислота и т.п.
При изготовлении жидкого препарата для перорального введения соединение по настоящему изобретению можно дополнить вкусовой добавкой, буфером, стабилизатором, отдушкой и т.п. и приготовить известными способами жидкий препарат для внутреннего применения - сироп, эликсир и т.п. В этом случае в качестве вкусовой добавки/отдушки можно использовать уже указанные вещества, и в качестве буфера можно также назвать цитрат натрия и т.п., а в качестве стабилизатора - трагакант, гуммиарабик, желатин и т.п.
При изготовлении препарата для инъекции к соединению по настоящему изобретению можно добавить pH-регулирующий агент, буфер, стабилизатор, агент для приготовления изотонического раствора, локальный анестетик и т.п., и можно приготовить обычными способами препараты для подкожной, внутримышечной и внутривенной инъекций. В качестве pH-регулирующего агента и буфера в этом случае можно назвать цитрат натрия, ацетат натрия, фосфат натрия и т.п. В качестве стабилизатора можно назвать пиросульфит натрия, EDTA, тиогликолевую кислоту, тиомолочную кислоту и т.п. В качестве локального анестетика можно использовать прокаин гидрохлорид, лидокаин гидрохлорид и т.п. Примерами добавки для приготовления изотонического раствора могут служить хлорид натрия, глюкоза и т.п.
Количество соединения по настоящему изобретению, которое следует ввести в каждую из рассмотренных выше лекарственных форм, не является постоянным и зависит от пациента, которому назначают это соединение, или симптомов пациента, или от препарата; однако желательно установить для препаратов перорального применения количество примерно 1-1200 мг и примерно 0,1-500 мг в случае препаратов для инъекций на единичную лекарственную форму. Кроме того, суточное количество введенного лекарственного средства в указанных лекарственных формах варьируется в зависимости от симптомов, массы тела, возраста, пола и т.п. пациента и не может быть единым для всех пациентов; однако количество может обычно составлять примерно 0,1-5000 мг и предпочтительно 1-1200 мг в день взрослому, и предпочтительно вводить его однократно или делить примерно на две-четыре порции в сутки.
Далее исходные соединения, используемые в способе по настоящему изобретению, и способ получения соединения по настоящему изобретению будут конкретно описаны с помощью соответствующих примеров синтеза и применения, но настоящее изобретение ими не ограничивается.
ВЭЖХ-анализ соединений из приведенных примеров проводили в следующих условиях.
Колонка Inertsil ODS-3 150 мм × 4,6 мм внутр. диаметра
Элюент 0,05 MK H2PO4/ацетонитрил = 50/50 (об./об.)
Скорость потока 1,0 мл/мин
Температура колонки 40°C
Доза инъекции 2 мкл
Длина волны детектирования 254 нм
Пример синтеза 1: получение 4-метокси-2,3-диметилпиридин-1-оксида
К раствору, приготовленному разбавлением 41,6 г (2,0 экв) 28%-ного водного раствора метоксида натрия в 200 мл диметилсульфоксида, добавили по каплям раствор 17,0 г 4-хлор-2,3-диметилпиридин-1-оксида в 70 мл диметилсульфоксида. Смесь нагревали 3 ч при 40-50°C и затем выдержали в течение ночи при комнатной температуре. К реакционной жидкости добавили 15 мл воды, смесь сконцентрировали и получили остаток в виде черной пасты. Затем остаток растворили в 500 мл воды и водный раствор экстрагировали три раза по 670 мл хлороформа. Экстракт высушили над безводным сульфатом магния, сконцентрировали досуха при пониженном давлении и получили 37,2 г 4-метокси-2,3-диметилпиридин-1-оксида.
Пример синтеза 2: получение 2-ацетоксиметил-3-метил-4-метоксипиридина
К 37,2 г 4-метокси-2,3-диметилпиридин-1-оксида добавили 55,0 г (5,0 экв) уксусного ангидрида и смесь нагревали в течение 3 ч при 90-100°C. Уксусный ангидрид отогнали, полученный сконцентрированный остаток очистили на колонке с силикагелем и получили 15,8 г 2-ацетоксиметил-3-метил-4-метоксипиридина в виде масла.
Пример синтеза 3: получение 2-гидроксиметил-3-метил-4-метоксипиридина
15,8 г 2-ацетоксиметил-3-метил-4-метоксипиридина добавили по каплям к 25% водному раствору гидроксида натрия и смесь выдержали в течение одного часа при комнатной температуре. Реакционный раствор разбавили толуолом, толуольный раствор промыли водой, высушили над безводным сульфатом магния, сконцентрировали и получили 5,7 г 2-гидроксиметил-3-метил-4-метоксипиридина в виде масла.
Пример синтеза 4: получение 4-этокси-2,3-диметилпиридин-1-оксида
К раствору, полученному разбавлением 8,6 г (2,0 экв) 60%-ного гидрида натрия в 200 мл диметилсульфоксида, добавили 9,9 г (2,0 экв) безводного этанола и смесь нагревали в течение часа при 60°C. Реакционную жидкость охладили до 30°C и затем по каплям добавили раствор 17,0 г 4-хлор-2,3-диметилпиридин-1-оксида в 70 мл диметилсульфоксида. Смесь нагревали в течение 2 ч при 40-50°C и затем оставили на ночь при комнатной температуре. К реакционной жидкости добавили 16 мл воды, смесь сконцентрировали и получили остаток в виде черной пасты. Остаток растворили в 500 мл воды и водный раствор трижды проэкстрагировали 670 мл хлороформа. Экстракт высушили над безводным сульфатом магния и затем сконцентрировали досуха при пониженном давлении. Полученный остаток очистили на колонке с силикагелем и получили 22,9 г 4-этокси-2,3-диметилпиридин-1-оксида.
Пример синтеза 5: получение 2-ацетоксиметил-3-метил-4-этоксипиридина
К 22,9 г 4-этокси-2,3-диметилпиридин-1-оксида добавили 55,0 г (5,0 экв) уксусного ангидрида и смесь нагревали в течение 6 ч при 90-100°C. Уксусный ангидрид отогнали, полученный сконцентрированный остаток очистили на колонке с силикагелем и получили 28,2 г 2-ацетоксиметил-3-метил-4-этоксипиридина в виде масла.
Пример синтеза 6: получение 2-гидроксиметил-3-метил-4-этоксипиридина
28,2 г 2-ацетоксиметил-3-метил-4-этоксипиридина добавили по каплям к 25% водному раствору гидроксида натрия и смесь выдержали в течение часа при комнатной температуре. Затем реакционную жидкость разбавили толуолом и толуольный раствор промыли водой, высушили над безводным сульфатом магния, сконцентрировали досуха и получили 10,7 г 2-гидроксиметил-3-метил-4-этоксипиридина в виде масла.
Пример синтеза 7: получение 4-пропокси-2,3-диметилпиридин-1-оксида
К раствору, полученному разбавлением 8,6 г (2,0 экв) 60% гидроксида натрия в 200 мл диметилсульфоксида, добавили 13,0 г 1-пропанола (2,0 экв) и смесь нагревали в течение часа при 60°C. Реакционную жидкость охладили до 30°C и затем по каплям добавили раствор 17,0 г 4-хлор-2,3-диметилпиридин-1-оксида в 70 мл диметилсульфоксида. Смесь нагревали в течение 3 ч при 40-50°C и затем выдержали в течение ночи при комнатной температуре. К реакционному раствору добавили 15 мл воды, смесь сконцентрировали и получили остаток в виде черной пасты. Остаток растворили в 500 мл воды и водный раствор проэкстрагировали трижды 670 мл хлороформа. Экстракт высушили над безводным сульфатом магния, сконцентрировали досуха при пониженном давлении, полученный остаток очистили на колонке с силикагелем и получили 20,3 г 4-пропокси-2,3-диметилпиридин-1-оксида.
Пример синтеза 8: получение 2-ацетоксиметил-3-метил-4-пропоксипиридина
К 20,3 г 4-пропокси-2,3-диметилпиридин-1-оксида добавили 44,0 г (4,0 экв) уксусного ангидрида и смесь нагревали в течение 4 ч при 90°C. Уксусный ангидрид отогнали, полученный сконцентрированный остаток очистили на колонке с силикагелем и получили 27,8 г 2-ацетоксиметил-3-метил-4-пропоксипиридина в виде масла.
Пример синтеза 9: получение 2-гидроксиметил-3-метил-4-пропоксипиридина
27,8 г 2-ацетоксиметил-3-метил-4-пропоксипиридина добавили по каплям к 25% водному раствору гидроксида натрия и смесь оставили на один час при комнатной температуре. Затем реакционную жидкость разбавили толуолом, толуольный раствор промыли водой, высушили над безводным сульфатом магния, сконцентрировали досуха и получили 11,0 г 2-гидроксиметил-3-метил-4-пропоксипиридина в виде масла.
Пример синтеза 10: получение 4-бутокси-2,3-диметилпиридин-1-оксида
К раствору, полученному разбавлением 8,6 г (2,0 экв) 60%-ного гидрида натрия в 200 мл диметилсульфоксида, добавили 16,0 г (2,0 экв) 1-бутанола и смесь нагревали в течение часа при 60°C. Реакционную жидкость охладили до 30°C и затем добавили по каплям раствор 17,0 г 4-хлор-2,3-диметилпиридин-1-оксида в 70 мл диметилсульфоксида. Смесь нагревали в течение 3 ч при 40-50°C и затем оставили на ночь при комнатной температуре. К реакционной смеси добавили 15 мл воды, сконцентрировали и получили остаток в виде черной пасты. Остаток растворили в 500 мл воды и водный раствор трижды проэкстрагировали 670 мл хлороформа. Экстракт высушили над безводным сульфатом магния и сконцентрировали досуха при пониженном давлении. Полученный остаток очистили на колонке с силикагелем и получили 20,3 г 4-бутокси-2,3-диметилпиридин-1-оксида.
Пример синтеза 11: получение 2-ацетоксиметил-3-метил-4-бутоксипиридина
К 24,1 г 4-бутокси-2,3-диметилпиридин-1-оксида добавили 44,0 г (4,0 экв) уксусного ангидрида и смесь нагревали в течение 4 ч при 90°C. Уксусный ангидрид отогнали, полученный сконцентрированный остаток очистили на колонке с силикагелем и получили 13,4 г 2-ацетоксиметил-3-метил-4-бутоксипиридина в виде масла.
Пример синтеза 12: получение 2-гидроксиметил-3-метил-4-бутоксипиридина
13,4 г 2-ацетоксиметил-3-метил-4-бутоксипиридина добавили по каплям к 25%-ному водному раствору гидроксида натрия и смесь оставили в течение часа при комнатной температуре. Затем смесь разбавили толуолом, толуольный раствор промыли водой, высушили над безводным сульфатом магния, затем сконцентрировали досуха и получили 8,3 г 2-гидроксиметил-3-метил-4-бутоксипиридина в виде масла.
Пример синтеза 13: получение 4-пентилокси-2,3-диметилпиридин-N-оксида
17,0 г (0,11 моль, 1,0 экв) 4-хлор-2,3-диметилпиридин-N-оксида, 8,6 г (0,225 моль, 2,0 экв) каустической соды и 19,0 г (0,22 моль, 2,0 экв) 1-пентанола добавили к 68 мл толуола, смесь кипятили с обратным холодильником в течение 5 ч и затем охладили до комнатной температуры. К реакционному раствору добавили 15 мл воды, сконцентрировали и получили остаток в виде черной пасты. Остаток растворили в 500 мл воды и водный раствор трижды проэкстрагировали 670 мл хлороформа. Экстракт высушили над безводным сульфатом магния, сконцентрировали досуха при пониженном давлении и получили 28,8 г 4-пентилокси-2,3-диметилпиридин-N-оксида.
Пример синтеза 14: получение 4-пентилокси-2-ацетоксиметил-3-метилпиридина
К 26,7 г (0,11 моль, 1,0 экв) 4-пентилокси-2,3-диметилпиридин-N-оксида добавили 44,0 г (0,43 моль, 4,0 экв) уксусного ангидрида и смесь нагревали в течение 7 ч при 90-100°C. Уксусный ангидрид отогнали, полученный сконцентрированный остаток очистили на колонке с силикагелем и получили 22,0 г 4-пентилокси-2-ацетоксиметил-3-метилпиридина в виде масла (выход 79,6%).
Пример синтеза 15: получение 4-пентилокси-2-гидроксиметил-3-метилпиридина
22,0 г (0,088 моль, 1,0 экв) 4-пентилокси-2-ацетоксиметил-3-метилпиридина добавили по каплям к 25%-ному водному раствору гидроксида натрия и смесь выдержали в течение часа при комнатной температуре. Затем реакционную смесь разбавили толуолом, толуольный раствор промыли водой, высушили над безводным сульфатом магния, сконцентрировали досуха и получили 11,2 г 4-пентилокси-2-гидроксиметил-3-метилпиридина в виде масла (выход 60,8%).
Пример синтеза 16: получение 4-гексилокси-2,3-диметилпиридин-N-оксида
15,8 г (0,1 моль, 1,0 экв) 4-хлор-2,3-диметилпиридин-N-оксида, 8,0 г (0,2 моль, 2,0 экв) каустической соды и 20,4 г (0,2 моль, 2,0 экв) 1-гексанола добавили к 64 мл толуола, смесь кипятили с обратным холодильником в течение 4 ч и затем охладили до комнатной температуры. К реакционной смеси добавили 15 мл воды и затем нейтрализовали концентрированной соляной кислотой (8 мл). Полученную смесь трижды проэкстрагировали 670 мл хлороформа. Экстракт высушили над безводным сульфатом магния, сконцентрировали досуха и получили 33,1 г 4-гексилокси-2,3-диметилпиридин-N-оксида в виде темно-коричневого масла.
Пример синтеза 17: получение 4-гексилокси-2-ацетоксиметил-3-метилпиридина
К 32,6 г (0,1 моль, 1,0 экв) 4-гексилокси-2,3-диметилпиридин-N-оксида добавили 40,8 г (0,4 моль, 4,0 экв) уксусного ангидрида и смесь нагревали в течение 5 ч при 87-93°C. Уксусный ангидрид отогнали, полученный сконцентрированный остаток очистили на колонке с силикагелем и получили 17,6 г 4-гексилокси-2-ацетоксиметил-3-метилпиридина в виде масла (выход 74,3%).
Пример синтеза 18: получение 4-гексилокси-2-гидроксиметил-3-метилпиридина
17,3 г (0,065 моль, 1,0 экв) 4-гексилокси-2-ацетоксиметил-3-метилпиридина добавили по каплям к 25%-ному водному раствору гидроксида натрия при 5-22°C, смесь выдержали в течение часа при комнатной температуре и затем проэкстрагировали хлороформом. Экстракт промыли водой, высушили над безводным сульфатом магния, сконцентрировали досуха и получили 12,7 г 4-гексилокси-2-гидроксиметил-3-метилпиридина в виде коричневого масла (выход 87,6%).
Пример синтеза 19: получение 4-гептилокси-2,3-диметилпиридин-N-оксида
15,8 г (0,1 моль, 1,0 экв) 4-хлор-2,3-диметилпиридин-N-оксида, 8,0 г (0,2 моль, 2,0 экв) каустической соды и 23,2 г (0,2 моль, 2,0 экв) 1-гептанола добавили к 64 мл толуола и кипятили с обратным холодильником в течение 3 ч, затем смесь охладили до комнатной температуры. Добавили 15 мл воды к реакционной смеси и смесь нейтрализовали концентрированной соляной кислотой (8 мл). Полученную смесь трижды проэкстрагировали 670 мл хлороформа. Экстракт высушили над безводным сульфатом магния, сконцентрировали при пониженном давлении и получили 37,3 г 4-гептилокси-2,3-диметилпиридин-N-оксида в виде темно-коричневого масла.
Пример синтеза 20: получение 4-гептилокси-2-ацетоксиметил-3-метилпиридина
К 36,8 г (0,1 моль, 1,0 экв) 4-гептилокси-2,3-диметилпиридин-N-оксида добавили 40,8 г (0,4 моль, 4,0 экв) уксусного ангидрида и смесь нагревали в течение 5 ч при 87-93°C. Уксусный ангидрид отогнали, полученный сконцентрированный остаток очистили на колонке с силикагелем и получили 19,5 г 4-гептилокси-2-ацетоксиметил-3-метилпиридина в виде масла (выход 69,9%).
Пример синтеза 21: получение 4-гептилокси-2-гидроксиметил-3-метилпиридина
19,0 г (0,068 моль, 1,0 экв) 4-гептилокси-2-ацетоксиметил-3-метилпиридина добавили по каплям к 25%-ному водному раствору гидроксида натрия при 13-25°C, смесь выдержали в течение часа при комнатной температуре и затем проэкстрагировали хлороформом. Экстракт промыли водой, высушили над безводным сульфатом магния, сконцентрировали досуха и получили 15,0 г 4-гептилокси-2-гидроксиметил-3-метилпиридина в виде коричневого масла (выход 92,9%).
Пример синтеза 22: получение 4-октилокси-2,3-диметилпиридин-N-оксида
15,8 г (0,1 моль, 1,0 экв) 4-хлор-2,3-диметилпиридин-N-оксида, 8,0 г (0,2 моль, 2,0 экв) каустической соды и 26,0 г (0,2 моль, 2,0 экв) 1-октанола добавили к 64 мл толуола, смесь кипятили с обратным холодильником в течение 3 ч и затем охладили до комнатной температуры. К реакционной смеси добавили 15 мл воды и затем нейтрализовали концентрированной соляной кислотой (8 мл). Полученную смесь трижды проэкстрагировали 670 мл хлороформа. Экстракт высушили над безводным сульфатом магния, сконцентрировали досуха при пониженном давлении и получили 39,2 г 4-октилокси-2,3-диметилпиридин-N-оксида в виде темно-коричневого масла.
Пример синтеза 23: получение 4-октилокси-2-ацетоксиметил-3-метилпиридина
К 38,7 г (0,1 моль, 1,0 экв) 4-октилокси-2,3-диметилпиридин-N-оксида добавили 40,8 г (0,4 моль, 4,0 экв) уксусного ангидрида и смесь нагревали в течение 5 ч при 88-92°C. Уксусный ангидрид отогнали, полученный сконцентрированный остаток очистили на колонке с силикагелем и получили 17,6 г 4-октилокси-2-ацетоксиметил-3-метилпиридина в виде масла (выход 60,0%).
Пример синтеза 24: получение 4-октилокси-2-гидроксиметил-3-метилпиридина
17,3 г (0,059 моль, 1,0 экв) 4-октилокси-2-ацетоксиметил-3-метилпиридина добавили по каплям к 25%-ному водному раствору гидроксида натрия при 11-22°C, смесь выдержали в течение часа при комнатной температуре и затем проэкстрагировали хлороформом. Экстракт промыли водой, высушили над безводным сульфатом магния, сконцентрировали досуха и получили 12,2 г 4-октилокси-2-гидроксиметил-3-метилпиридина в виде светло-коричневого масла (выход 82,3%).
Пример синтеза 25: получение 4-нонилокси-2,3-диметилпиридин-N-оксида
15,8 г (0,1 моль, 1,0 экв) 4-хлор-2,3-диметилпиридин-N-оксида, 8,0 г (0,2 моль, 2,0 экв) каустической соды и 28,9 г (0,2 моль, 2,0 экв) 1-нонанола добавили к 64 мл толуола, смесь нагревали с обратным холодильником в течение 4 ч и затем охладили до комнатной температуры. К реакционной смеси добавили 160 мл воды и нейтрализовали концентрированной соляной кислотой (8,5 г). Полученную смесь трижды проэкстрагировали 160 мл этилацетата. Экстракт высушили над безводным сульфатом магния, сконцентрировали при пониженном давлении и получили 42,4 г 4-нонилокси-2,3-диметилпиридин-N-оксида в виде темно-коричневого масла.
Пример синтеза 26: получение 4-нонилокси-2-ацетоксиметил-3-метилпиридина
К 41,4 г (0,1 моль, 1,0 экв) 4-нонилокси-2,3-диметилпиридина добавили 40,8 г (0,4 моль, 4,0 экв) уксусного ангидрида и смесь нагревали в течение 7 ч при 88-92°C. Уксусный ангидрид отогнали, полученный сконцентрированный остаток очистили на колонке с силикагелем и получили 16,7 г 4-нонилокси-2-ацетоксиметил-3-метилпиридина в виде темно-коричневого масла (общий выход из двух опытов 54,4%).
Пример синтеза 27: получение 4-нонилокси-2-гидроксиметил-3-метилпиридина
15,4 г (0,05 моль, 1,0 экв) 4-нонилокси-2-ацетоксиметил-3-метилпиридина добавили по каплям к 20% водному раствору гидроксида натрия (20,0 г) при 11-21°C, смесь выдержали в течение часа при комнатной температуре и затем проэкстрагировали водой (77 мл) и толуолом (128 мл). Экстракт промыли водой, высушили над безводным сульфатом магния, концентрировали досуха и получили 16,8 г 4-нонилокси-2-гидроксиметил-3-метилпиридина в виде коричневого масла.
Пример синтеза 28: получение 4-децилокси-2,3-диметилпиридин-N-оксида
15,8 г (0,1 моль, 1,0 экв) 4-хлор-2,3-диметилпиридин-N-оксида, 8,0 г (0,2 моль, 2,0 экв) гидроксида натрия и 28,5 г 1-деканола (0,18 моль, 1,8 экв) добавили к 64 мл толуола, смесь кипятили с обратным холодильником в течение 4 ч и затем охладили до комнатной температуры. К реакционной смеси добавили 160 мл воды и нейтрализовали концентрированной соляной кислотой (7,3 г). Полученную смесь проэкстрагировали 100 мл этилацетата. Экстракт высушили над безводным сульфатом магния, сконцентрировали при пониженном давлении и получили 42,2 г 4-децилокси-2,3-диметилпиридин-N-оксида в виде темно-коричневого масла.
Пример синтеза 29: получение 4-децилокси-2-ацетоксиметил-3-метилпиридина
К 41,2 г (0,1 моль, 1,0 экв) 4-децилокси-2,3-диметилпиридин-N-оксида добавили 40,8 г (0,4 моль, 4,0 экв) уксусного ангидрида и смесь нагревали в течение 8,5 ч при 88-92°C. Уксусный ангидрид отогнали, полученный сконцентрированный остаток очистили на колонке с силикагелем и получили 19,8 г 4-децилокси-2-ацетоксиметил-3-метилпиридина в виде темно-коричневого масла (общий выход за два опыта 61,7%).
Пример синтеза 30: получение 4-децилокси-2-гидроксиметил-3-метилпиридина
18,6 г (0,058 моль, 1,0 экв) 4-децилокси-2-ацетоксиметил-3-метилпиридина добавили по каплям к 20%-ному водному раствору гидроксида натрия (23 г) при 11-22°C и смесь выдержали в течение часа при комнатной температуре. Затем добавили воду (93 мл) и проэкстрагировали толуолом (130 мл). Экстракт промыли водой, высушили над безводным сульфатом магния, сконцентрировали досуха и получили 20,6 г 4-децилокси-2-гидроксиметил-3-метилпиридина в виде коричневого масла.
Далее настоящее изобретение будет конкретно описано с помощью примеров согласно настоящему изобретению.
Пример 1
Получение 2-[(4-н-пентилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазола
11,0 г (0,054 моль, 1,0 экв) 4-пентилокси-2-гидроксиметил-3-метилпиридина растворили в 620 мл хлороформа и по каплям добавили 24,9 г (0,281 моль, 4 экв) тионилхлорида при 22-30°C. Затем смесь кипятили с обратным холодильником в течение часа, сконцентрировали и получили 4-пентилокси-2-хлорметил-3-метилпиридин в виде коричневого масла.
К жидкости, приготовленной растворением при перемешивании 7,5 г (0,052 моль, 1,0 экв) 2-меркаптобензимидазола и 61,5 г (0,319 моль, 5,8 экв) 28% раствора метоксида натрия в 120 мл метанола, добавили жидкость, приготовленную растворением всего количества 4-пентилокси-2-хлорметил-3-метилпиридина в 180 мл метанола, при 30°C. Затем смесь кипятили с обратным холодильником в течение часа и охладили, а метанол упарили досуха при пониженном давлении. Полученный желто-коричневый остаток растворили в 250 мл этилацетата и органический слой промыли 100 мл воды. Органический слой высушили над безводным сульфатом магния и сконцентрировали досуха. Полученный остаток очистили колоночной хроматографией на силикагеле, сконцентрировали досуха и получили 6,9 г аморфного твердого продукта. Продукт перекристаллизовали из смеси этилацетат/1-гексан и получили 4,3 г бесцветных кристаллов (ВЭЖХ: 98,5 % площ., выход 10,6%).
1H ЯМР (400 МГц, CDCl3) δ: 0,94 (3H, т, J=6 Гц), 1,37-1,48 (4H, м), 1,80-1,86 (2H, м), 2,26 (3H, с), 4,02 (2H, т, J=6 Гц), 4,37 (2H, с), 6,73 (1H, д, J=6 Гц), 7,17-7,18 (2H, м), 7,53 (2H, м), 8,33 (1H, д, J=6 Гц), 13,04 (1H, шир.с).
MS m/z: 341 (M+).
Пример 2
Получение 2-[(4-н-гексилокси-3-метилпиридин-2-ил)метилтио]-1Н-бензимидазола
12,2 г (0,055 моль, 1,0 экв) 4-гексилокси-2-гидроксиметил-3-метилпиридина растворили в 620 мл хлороформа и добавили по каплям 33,4 г (0,281 моль, 5,1 экв) тионилхлорида при 23-29°C. Затем смесь кипятили с обратным холодильником в течение часа, сконцентрировали и получили 24,7 г 4-гексилокси-2-хлорметил-3-метилпиридина в виде коричневого масла.
К жидкости, полученной растворением при перемешивании 7,8 г (0,052 моль, 0,95 экв) 2-меркаптобензимидазола и 61,5 г (0,319 моль, 5,8 экв) 28% раствора метоксида натрия в 360 мл метанола, добавили жидкость, приготовленную растворением всего количества 4-гексилокси-2-хлорметил-3-метилпиридина в 180 мл метанола, при 30°C. Затем смесь кипятили с обратным холодильником в течение 30 мин и охладили, а метанол упарили досуха при пониженном давлении. Полученный желто-коричневый остаток растворили в 200 мл этилацетата и затем органический слой промыли 80 мл воды. Органический слой высушили над безводным сульфатом магния и сконцентрировали досуха. Полученный остаток очистили колоночной хроматографией на силикагеле, сконцентрировали досуха и получили 15,2 г оранжевого масла. Продукт перекристаллизовали из этилацетата (5 об.) и получили 5,0 г бесцветных кристаллов (ВЭЖХ: 99,1% площ., выход 25,5%).
1H ЯМР (400 МГц, CDCl3) δ: 0,91 (3H, т, J=7 Гц), 1,34-1,37 (4H, м), 1,44-1,50 (2H, м), 1,79-1,86 (2H, м), 2,26 (3H, с), 4,02 (2H, т, J=6 Гц), 4,37 (2H, с), 6,73 (1H, д, J=6 Гц), 7,15-7,20 (2H, м), 7,45-7,62 (2H, м), 8,34 (1H, д, J=6 Гц), 13,04 (1H, шир.с).
MS m/z: 355 (M+).
Пример 3
Получение 2-[(4-н-гептилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазола
14,5 г (0,061 моль, 1,0 экв) 4-гептилокси-2-гидроксиметил-3-метилпиридина растворили в 700 мл хлороформа и добавили по каплям 37,0 г (0,311 моль, 5,1 экв) тионилхлорида при 22-30°C. Смесь кипятили с обратным холодильником в течение 30 мин, сконцентрировали и получили 28,2 г 4-гептилокси-2-хлорметил-3-метилпиридина в виде черного масла.
К жидкости, полученной растворением при перемешивании 8,7 г (0,058 моль, 0,95 экв) 2-меркаптобензимидазола и 68,2 г (0,354 моль, 5,8 экв) 28% раствора метоксида натрия в 400 мл метанола, добавили жидкость, приготовленную растворением всего количества 4-гептилокси-2-хлорметил-3-метилпиридина в 210 мл метанола, при 28°C. Затем смесь кипятили с обратным холодильником в течение часа и охладили, а метанол упарили досуха при пониженном давлении. Полученный желто-коричневый остаток растворили в 250 мл этилацетата, органический слой промыли 100 мл воды, высушили над безводным сульфатом магния и сконцентрировали досуха. Полученный остаток очистили колоночной хроматографией на силикагеле, сконцентрировали досуха и получили 17,8 г оранжевого масла. Оранжевый продукт перекристаллизовали из этилацетата (7 об.) и получили 8,7 г бесцветных кристаллов (ВЭЖХ: 99,0% площ., выход 38,7%).
1H ЯМР (400 МГц, CDCl3) δ: 0,90 (3H, т, J=6 Гц), 1,29-1,40 (6H, м), 1,44-1,51 (2H, м), 1,80-1,86 (2H, м), 2,26 (3H, с), 4,02 (2H, т, J=6 Гц), 4,37 (2H, с), 6,73 (1H, д, J=6 Гц), 7,15-7,20 (2H, м), 7,45-7,62 (2H, м), 8,34 (1H, д, J=6 Гц), 13,04 (1H, шир.с).
MS m/z: 369 (M+).
Пример 4
Получение 2-[(4-н-бутокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазола
8,3 г 2-ацетоксиметил-3-метил-4-бутоксипиридина растворили в хлороформе и добавили 21,6 г (4 экв) тионилхлорида. Смесь кипятили с обратным холодильником в течение часа, сконцентрировали и полученный остаток растворили в метаноле. Раствор сразу добавили к 6,1 г (1 экв) 2-меркаптобензимидазола, 230 мл 28%-ного раствора метоксида натрия и 120 мл метанола и полученную смесь нагревали с обратным холодильником в течение часа. Метанол отогнали, добавили к остатку лед и проэкстрагировали этилацетатом. Этилацетатный слой высушили над безводным сульфатом магния. Затем раствор в этилацетате сконцентрировали досуха, полученный остаток очистили колоночной хроматографией на силикагеле, сконцентрировали досуха и получили 12,2 г желтых кристаллов. Полученные желтые кристаллы перекристаллизовали из смеси этилацетат/1-гексан и получили 4,2 г бесцветных кристаллов. Общий выход 10,8% (чистота по ВЭЖХ: 97,0%).
1H ЯМР (400 МГц, CDCl3) δ: 0,99 (3H, т J=7 Гц), 1,51-1,56 (2H, м), 1,78-1,85 (2H, м), 2,26 (3H, с), 4,03 (2H, т, J=7 Гц), 4,37 (2H, с), 6,74 (1H, д, J=6 Гц), 7,17-7,19 (2H, м), 7,54 (2H, м), 8,34 (1H, д, J=6 Гц), 13,04 (1H, шир.с).
MS m/z: 327 (M+).
Пример 5
Получение 2-[(4-н-октилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазола
11,8 г (0,047 моль, 1,0 экв) 4-октилокси-2-гидроксиметил-3-метилпиридина растворили в 600 мл хлороформа и добавили по каплям 28,6 г (0,240 моль, 5,1 экв) тионилхлорида при 19-27°C. Смесь кипятили с обратным холодильником в течение часа, сконцентрировали и получили 23,8 г 4-октилокси-2-хлорметил-3-метилпиридина в виде черного масла.
К жидкости, полученной растворением при перемешивании 6,7 г (0,045 моль, 0,95 экв) 2-меркаптобензимидазола и 52,5 г (0,273 моль, 5,8 экв) 28% раствора метоксида натрия в 350 мл метанола, добавили жидкость, приготовленную растворением всего количества 4-октилокси-2-хлорметил-3-метилпиридина в 180 мл метанола, при 27°C. Полученную смесь кипятили с обратным холодильником в течение часа и охладили, а метанол упарили досуха при пониженном давлении. Полученный коричневый маслообразный остаток растворили в 250 мл этилацетата, органический слой промыли 100 мл воды, высушили над безводным сульфатом магния и сконцентрировали досуха. Полученный остаток очистили колоночной хроматографией на силикагеле, сконцентрировали досуха и получили 14,9 г оранжевого масла. Оранжевое масло перекристаллизовали из этилацетата (5 об.) и получили 5,8 г бесцветных кристаллов (ВЭЖХ: 98,8% площ., выход 32,2%).
1H ЯМР (400 МГц, CDCl3) δ: 0,89 (3H, т, J=6 Гц), 1,29-1,35 (8H, м), 1,44-1,49 (2H, м), 1,79-1,85 (2H, м), 2,26 (3H, с), 4,02 (2H, т, J=6 Гц), 4,37 (2H, с), 6,73 (1H, д, J=6 Гц), 7,16-7,20 (2H, м), 7,45-7,62 (2H, м), 8,34 (1H, д, J=6 Гц), 13,04 (1H, шир.с).
MS m/z: 383 (M+).
Далее в качестве сравнительных примеров приведены примеры получения ссылочных соединений.
Ссылочный пример 1: получение 2-[(4-метокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазола
5,7 г 2-ацетоксиметил-3-метил-4-метоксипиридина растворили в хлороформе и добавили 18 г (4 экв) тионилхлорида. Смесь кипятили с обратным холодильником в течение часа, сконцентрировали и полученный остаток растворили в метаноле. Раствор сразу добавили к 5,3 г (1 экв) 2-меркаптобензимидазола, 45 мл 28% раствора метоксида натрия и 100 мл метанола и полученную смесь кипятили с обратным холодильником в течение часа. Метанол отогнали, добавили к остатку лед и этилацетат для промывки остатка суспендированием. Полученные кристаллы отфильтровали, отмыли путем суспендирования в воде и получили 7,1 г бесцветных кристаллов. Общий выход 20,9% (чистота по ВЭЖХ: 98,2%).
1H ЯМР (400 МГц, CDCl3) δ: 2,27 (3H, с), 3,89 (3H, с), 4,40 (2H, с), 6,76 (1H, д, J=6 Гц), 7,16-7,20 (2H, м), 7,44-7,63 (2H, м), 8,36 (1H, д, J=6 Гц), 12,96 (1H, шир.с).
MS m/z: 285 (M+).
Ссылочный пример 2: получение 2-[(4-этокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазола
10,7 г 2-ацетоксиметил-3-метил-4-этоксипиридина растворили в хлороформе и добавили 28,2 г (4 экв) тионилхлорида. Смесь кипятили с обратным холодильником в течение часа, сконцентрировали и полученный остаток растворили в метаноле. Раствор сразу добавили к 8,4 г (1 экв) 2-меркаптобензимидазола, 71 мл 28% раствора метоксида натрия и 150 мл метанола и полученную смесь нагревали с обратным холодильником в течение часа. Метанол отогнали, добавили к остатку лед и этилацетат для промывки остатка суспендированием. Полученные кристаллы отфильтровали, растворили в метаноле и раствор высушили над безводным сульфатом магния. Затем метанольный раствор сконцентрировали досуха и остаток растворили в хлороформе. Хлороформенный слой высушили над сульфатом магния, сконцентрировали досуха и получили аморфное твердое вещество. Твердое вещество отмыли суспендированием в этилацетате и получили 2,8 г бесцветных кристаллов. Общий выход 7,9% (чистота по ВЭЖХ: 98,2%).
1H ЯМР (400 МГц, CDCl3) δ: 1,43 (3H, т, J=7 Гц), 2,28 (3H, с), 4,11 (2H, кв., J=7 Гц), 4,37 (2H, с), 6,73 (1H, д, J=6 Гц), 7,16-7,20 (2H, м), 7,44-7,63 (2H, м), 8,33 (1H, д, J=6 Гц), 13,01 (1H, шир.с).
MS m/z: 299 (M+).
Ссылочный пример 3: получение 2-[(4-н-пропокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазола
11,0 г 2-ацетоксиметил-3-метил-4-пропоксипиридина растворили в хлороформе и добавили 30 г (4 экв) тионилхлорида. Смесь кипятили с обратным холодильником в течение часа, сконцентрировали и полученный остаток растворили в метаноле. Раствор сразу добавили к 8,7 г (1 экв) 2-меркаптобензимидазола, 73 мл 28% раствора метоксида натрия и 300 мл метанола и полученную смесь кипятили с обратным холодильником в течение часа. Метанол отогнали, добавили к остатку лед и смесь проэкстрагировали этилацетатом. Этилацетатный слой высушили над безводным сульфатом магния и сконцентрировали досуха. Полученный остаток очистили колоночной хроматографией на силикагеле, сконцентрировали досуха и получили 15,5 г желтых кристаллов. Эти кристаллы перекристаллизовали из этилацетата и получили 6,3 г бесцветных кристаллов. Общий выход 17% (чистота по ВЭЖХ: 98,1%).
1H ЯМР (400 МГц, CDCl3) δ: 1,07 (3H, т, J=7 Гц), 1,83-1,89 (2H, м), 2,27 (3H, с), 3,99 (2H, т, J=7 Гц), 4,37 (2H, с), 6,73 (1H, д, J=6 Гц), 7,17-7,20 (2H, м), 7,44-7,62 (2H, м), 8,35 (1H, д J=6 Гц), 13,02 (1H, шир.с).
MS m/z: 313 (M+).
Ссылочный пример 4: получение 2-[(4-н-нонилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазола
16,0 г (0,05 моль, 1,0 экв) 4-нонилокси-2-гидроксиметил-3-метилпиридина растворили в 320 мл этилацетата и добавили по каплям 6,5 г (0,055 моль, 1,1 экв) тионилхлорида при 8-12°C. Смесь выдержали в течение 0,5 ч при комнатной температуре и затем добавили 7% водный раствор бикарбоната натрия (200 г) для доведения рН реакционного раствора до 7,5. Органический слой промыли 5%-ным водным раствором бикарбоната натрия (160 г), высушили над безводным сульфатом магния, сконцентрировали досуха при пониженном давлении и получили 12,8 г 4-нонилокси-2-хлорметил-3-метилпиридина в виде черного масла.
К жидкости, приготовленной растворением при перемешивании 5,7 г (0,038 моль, 0,9 экв) 2-меркаптобензимидазола и 9,7 г (0,0504 моль, 1,2 экв) 28% раствора метоксида натрия в 120 мл метанола, добавили раствор 12,0 г 4-нонилокси-2-хлорметил-3-метилпиридина в 60 мл метанола при 29°C. Полученную смесь кипятили с обратным холодильником в течение 0,5 ч, охладили и полностью отогнали метанол при пониженном давлении. Полученный коричневый маслообразный остаток растворили в 240 мл этилацетата и органический слой промыли 120 мл воды. К органическому слою добавили при перемешивании 36 г силикагеля, затем силикагель отфильтровали, фильтрат сконцентрировали досуха при пониженном давлении и получили 16,4 г бледно-зелено-коричневых кристаллов. Полученные кристаллы перекристаллизовали из этилацетата (10 об.) и получили 5,5 г бледно-коричневых кристаллов (ВЭЖХ: 98,8% площ., выход 32,9%).
1H ЯМР (400 МГц, CDCl3) δ: 0,89 (3H, т, J=6 Гц), 1,29-1,35 (8H, м), 1,44-1,49 (2H, м), 1,79-1,85 (2H, м), 2,26 (3H, с), 4,02 (2H, т, J=6 Гц), 4,37 (2H, с), 6,73 (1H, д, J=6 Гц), 7,16-7,20 (2H, м), 7,45-7,62 (2H, м), 8,34 (1H, д, J=6 Гц), 13,04 (1H, шир.с).
MS m/z: 397 (M+).
Ссылка 5: получение 2-[(4-н-децилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазола
19,8 г (0,058 моль, 1,0 экв) 4-децилокси-2-гидроксиметил-3-метилпиридина растворили в 300 мл этилацетата и добавили по каплям 7,6 г (0,064 моль, 1,1 экв) тионилхлорида при 12-18°C. Смесь выдержали в течение 0,5 ч при комнатной температуре и затем добавили 7% водный раствор бикарбоната натрия (220 г) для доведения рН реакционного раствора до 7,5. Органический слой промыли 5%-ным водным раствором бикарбоната натрия (160 г), высушили над безводным сульфатом магния, сконцентрировали досуха при пониженном давлении и получили 15,8 г 4-децилокси-2-хлорметил-3-метилпиридина в виде черного масла.
К жидкости, приготовленной растворением при перемешивании 6,8 г (0,045 моль, 0,9 экв) 2-меркаптобензимидазола и 11,6 г (0,06 моль, 1,2 экв) 28% раствора метоксида натрия в 150 мл метанола, добавили раствор 15,0 г 4-децилокси-2-хлорметил-3-метилпиридина в 75 мл метанола при 30°C. Полученную смесь кипятили с обратным холодильником в течение 0,5 ч, охладили и полностью отогнали метанол при пониженном давлении. Полученный оранжевый маслообразный остаток растворили в 300 мл этилацетата и органический слой промыли 150 мл воды. К органическому слою добавили при перемешивании 45 г силикагеля, силикагель отфильтровали, фильтрат сконцентрировали досуха при пониженном давлении и получили 18,5 г бледно-оранжевых кристаллов. Бледно-оранжевые кристаллы перекристаллизовали из этилацетата (10 об.) и получили 9,7 г бледно-оранжевых кристаллов (ВЭЖХ: 99,6% площ., выход 47,1%).
1H ЯМР (400 МГц, CDCl3) δ: 0,89 (3H, т, J=6 Гц), 1,29-1,35 (8H, м), 1,44-1,49 (2H, м), 1,79-1,85 (2H, м), 2,26 (3H, с), 4,02 (2H, т, J=6 Гц), 4,37 (2H, с), 6,73 (1H, д, J=6 Гц), 7,16-7,20 (2H, м), 7,45-7,62 (2H, м), 8,34 (1H, т, J=6 Гц), 13,04 (1H, шир.с).
MS m/z: 411 (M+).
Ссылочный пример 6: получение (±)-2-[(4-н-пентилокси-3-метилпиридин-2-ил)метилсульфинил]-1H-бензимидазола
3,00 г (8,8 ммоль, 1,0 экв) 2-[(4-пентилокси-3-метилпридин-2-ил)метилтио]-1H-бензимидазола растворили в 24,5 мл дихлорметана и смесь охладили до -10°C или ниже в атмосфере азота. Затем по каплям добавили раствор 2,01 г (9,9 ммоль, 1,1 экв) м-хлорнадбензойной кислоты (85% чистоты) в 19,4 мл дихлорметана в течение часа при -10°C или ниже. Смесь перемешивали в течение 2,5 ч при поддержании температуры на уровне -10°C или ниже. Поскольку реакция не завершилась, добавили еще раствор 0,45 г (2,2 ммоль, 0,3 экв) м-хлорнадбензойной кислоты (85% чистоты) в 5,0 мл дихлорметана при -10°C или ниже и смесь продолжали перемешивать еще в течение часа при -10°C или ниже. После того как температура жидкости повысилась до 20°C, добавили 9,41 г 10% водного раствора гидроксида натрия и 36 мл воды для доведения рН водного слоя до 13,04. После удаления слоя дихлорметана в водном слое установили рН 10,49 добавлением 11,21 г 10% водного раствора ацетата аммония и затем проэкстрагировали 50 мл дихлорметана. Органический слой сконцентрировали при пониженном давлении при 40°C или ниже и затем добавили жидкую смесь 1,35 г ацетона и 27,0 г н-гексана. Отфильтровали осадок и получили 1,86 г (±)-2-[(4-н-пентилокси-3-метилпиридин-2-ил)метилсульфинил]-1H-бензимидазола в виде серо-зеленого аморфного порошка (ВЭЖХ: 97,7% площ., выход 59,2%).
1H ЯМР (400 МГц, CDCl3) δ: 0,94 (3H, т, J=7 Гц), 1,35-1,45 (4H, м), 1,76-1,81 (2H, м), 2,13 (3H, с), 3,97 (2H, т, J=6 Гц) 4,71 (1H, д, J=14 Гц), 4,82 (1H, д, J=14 Гц), 6,69 (1H, д, J=6 Гц), 7,33-7,28 (2H, м), 7,63 (2H, м), 8,30 (1H, д, J=6 Гц).
MS m/z: 357 (M+).
Ссылочный пример 7: получение (±)-2-[(4-н-гексилокси-3-метилпиридин-2-ил)метилсульфинил]-1H-бензимидазола
1,00 г (2,8 ммоль, 1,0 экв) 2-[(4-гексилокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазола растворили в 7,8 мл дихлорметана и смесь охладили до -18°C или ниже в атмосфере азота. Затем по каплям добавили раствор 0,63 г (3,1 ммоль, 1,1 экв) м-хлорнадбензойной кислоты (85% чистоты) в 6,2 мл дихлорметана в течение 0,5 ч при -18°C или ниже. Поддерживая температуру на уровне -18°C или ниже, смесь перемешивали в течение часа. После того как температура жидкости повысилась до 20°C, добавили 4,77 г 10% водного раствора гидроксида натрия и 22 мл воды, доведя рН водного слоя до 13,11. После удаления слоя дихлорметана рН водного слоя установили равным 10,47 добавлением 16,56 г 10% водного раствора ацетата аммония и затем проэкстрагировали 30 мл дихлорметана. Органический слой сконцентрировали при пониженном давлении при 40°C или ниже и получили концентрат, к которому для отверждения добавили 20 мл н-гексана. Затем отфильтровали осадок и получили 0,538 г (±)-2-[(4-н-гексилокси-3-метилпиридин-2-ил)метилсульфинил]-1H-бензимидазола в виде серо-зеленого аморфного порошка (ВЭЖХ: 97,3% площ., выход 51,5%).
1H ЯМР (400 МГц, CDCl3) δ: 0,91 (3H, т, J=7 Гц), 1,32-1,47 (6H, м), 1,75-1,82 (2H, м), 2,16 (3H, с), 3,97 (2H, т, J=6 Гц), 4,70 (1H, д, J=14 Гц), 4,82 (1H, д, J=14 Гц), 6,70 (1H, д, J=6 Гц), 7,29-7,33 (2H, м), 7,63 (2H, м), 8,30 (1H, д, J=6 Гц).
MS m/z: 371 (M+).
Ссылочный пример 8: получение (±)-2-[(4-н-гептилокси-3-метилпиридин-2-ил)метилсульфинил]-1H-бензимидазола
1,00 г (2,7 ммоль, 1,0 экв) 2-[(4-гептилокси-3-метилпиридин-2-ил)-метилтио]-1H-бензимидазола растворили в 7,6 мл дихлорметана и смесь охладили до -20°C или ниже в атмосфере азота. Затем по каплям добавили раствор 0,61 г (5,8 ммоль, 2,1 экв) м-хлорнадбензойной кислоты в 6,0 мл дихлорметана в течение 0,5 ч при -20°C или ниже. Поддерживая температуру на уровне -20°C или ниже, смесь перемешивали в течение 40 мин. После того как температура жидкости повысилась до 20°C, добавили 6,0 г 10% водного раствора гидроксида натрия и 30 мл воды, доведя рН водного слоя до 13,16. После удаления слоя дихлорметана водный слой промыли еще 12 мл дихлорметана. Затем добавили свежую порцию дихлорметана (30 мл) и затем 9,42 г 10% водного раствора ацетата аммония, доведя рН водного слоя до 10,44. Органический слой промыли 12 мл воды, сконцентрировали при пониженном давлении при 40°C или ниже и получили концентрат. Для отверждения концентрата добавили 20 мл н-гексана, отфильтровали осадок и получили 0,729 г (±)-2-[(4-н-гептилокси-3-метилпиридин-2-ил)метилсульфинил]-1H-бензимидазола в виде серо-зеленого аморфного порошка (ВЭЖХ: 97,4% площ., выход 69,8%).
Тесты на стабильность
Провели тесты на стабильность тиоэфиров (далее называемых SH-формой) и сульфоксидов (далее называемых SO-формой) при pH 2,0.
Методика
Каждый образец, полученный в примерах 1-3 и в ссылочных примерах 6-8 (сульфинильные соединения в примерах 1-3), выдерживали в растворе при pH 2 с концентрацией HCl 0,5 или 1,0 ммоль/л HCl (37°C) в течение предварительно установленного периода времени. Затем pH раствора довели до 8 с помощью триэтиламина и провели анализ методом ВЭЖХ для расчета остаточного соотношения.
Результаты приведены в таблице 1 в терминах стабильности в кислом растворе соляной кислоты (pH 2).
Стабильность в кислой среде
при pH 2
(-S-)
(-S=О)
В таблице OMZ означает омепразол, LAZ означает ланзопразол, и RAZ означает рабепразол. «Оксид примера 1» представляет собой соединение, приведенное в ссылочном примере 6, «оксид примера 2» - соединение, приведенное в ссылочном примере 7, и «оксид примера 3» - соединение, приведенное в ссылочном примере 8».
По оси ординат фиг.1 представлена стабильность (остаточное соотношение) (%), а на оси абсцисс - время (мин). На фиг.1 черный ромб (темно-синий на исходной диаграмме) означает случай соединения примера 1; черный прямоугольник (красный на исходной диаграмме) относится к соединению примера 2; светлый треугольник (желтый на исходной диаграмме) показывает случай соединения примера 3; серый значок X (голубой на исходной диаграмме) означает соединение ссылочного примера 6; черный значок X (красный на исходной диаграмме) указывает на соединение ссылочного примера 7; черный кружок (красный на исходной диаграмме) означает соединение ссылочного примера 8; серый вертикальный столбик (голубой на исходной диаграмме) относится к омепразолу (OMZ); черный утолщенный сверху значок (темно-голубой на исходной диаграмме) относится к ланзопразолу (LAZ), и серый утолщенный сверху значок (голубой на исходной диаграмме) относится к рабепразолу (RAZ).
По данным настоящего теста, все соединения примеров 1, 2 и 3, которые представляют собой SH-соединения, оказались стабильными к кислоте, и у всех из них остаточное соотношение составляло 100% через 30 мин и 2 ч. С другой стороны, все соединения в SO-форме с окисленными фрагментами SH, т.е. соединения ссылочных примеров 6, 7 и 8, были чрезвычайно нестабильны в кислой среде, и у всех из них остаточное соотношение оказалось равным 0% через 30 мин и 2 ч. Кроме того, омепразол, ланзопразол и рабепразол, которые являются ингибиторами протонного насоса, широко используемыми в настоящее время во всем мире, представляют собой соединения в SO-форме; они не стабильны к кислоте и все характеризуются остаточными соотношениями 0% через 30 мин и 2 ч. Полученные результаты показывают, что соединения по настоящему изобретению в SH-форме неожиданно оказались стабильными в кислой среде.
Пример 1 фармакологического теста
Тест на антибактериальную активность
Тест был проведен для выяснения того, какова разница в активности против Helicobacter pylori в зависимости от числа атомов в линейной алкильной группе заместителя в 4-м положении пиридинового кольца.
Методика
Для проведения тестов in vitro с бактериями Helicobacter pylori использовали стандартный штамм ATCC 43504 в среде колумбийского агара. Каждый образец растворяли в 1% растворе ДМСО и бактерии выращивали при 37°C и pH 7,0 в течение 3 суток. На четвертый день определяли минимальную концентрацию ингибирования роста (MIC, мкг/мл).
Результаты представлены в таблице 2 и на фиг.2.
Число атомов углерода и активность против Helicobacter pylori
На фиг.2 по оси ординат отложены значения MIC (мкг/мл), а по оси абсцисс - число атомов углерода в алкильной группе. Черный кружок (темно-синий на исходной диаграмме) на фиг.2 относится к случаю SH-формы, а серый кружок (красный на исходной диаграмме) относится к случаю SO-формы.
Как видно из таблицы 2 и фиг.2, полученные результаты настоящего теста показали, что у соединений в SH-форме активность против HP возрастает при увеличении числа атомов углерода в данной линейной цепи от 2 до 9 и особенно в интервале 4-8. Кроме того, активность достигает максимума, когда число атомов углерода в линейной цепи равно 5, 6 или 7 и MIC составляет 0,1 мкг/мл. Более того, когда число атомов углерода в линейной цепи возрастает до 8 или 9, активность против Helicobacter pylori снижается, но даже и в этом случае MIC равна 0,3 и 1,0 мкг/мл соответственно, что составляет 1/10 и 1/3 по сравнению со случаем, когда число атомов углерода равно 1. С другой стороны, активность против HP у соединений в SO-форме, соответствующая примерам 1, 2 и 3 с максимальной активностью против Helicobacter pylori, была низкой, составляя 3,0 мкг/мл независимо от числа атомов углерода.
Таким образом, настоящее изобретение раскрывает впервые, что активность против Helicobacter pylori сильно меняется в зависимости от числа атомов углерода в линейной алкильной группе заместителя в 4-положении пиридинового кольца и что эти флуктуации максимальны в случае, когда число атомов углерода в линейной цепи равно 5, 6 или 7, и, таким образом, отсутствует простая зависимость от числа атомов углерода. Максимальная величина даже примерно в 30 раз больше величины, наблюдаемой в том случае, когда число атомов углерода равно 1 или 10. Кроме того, можно также подтвердить, что получен удивительный результат - увеличение примерно в 30 раз даже по сравнению с соответствующими соединениями в SO-форме.
Пример фармакологического теста 2
Методика
Для проведения тестов in vitro с бактериями Helicobacter pylori использовали стандартные штаммы ATCC 43504, ATCC 43629 и ATCC 43570 в среде колумбийского агара. Бактерии выращивали при 37°C и pH 7,0 в течение 3 суток. На четвертый день определяли минимальную концентрацию ингибирования роста (MIC, мкг/мл). Каждый образец растворяли в 1% растворе ДМСО. Далее, в качестве контрольных лекарств антибактериального действия использовали амоксициллин и кларитромицин. Кроме того, синтезировли и использовали для сравнения соединение, описанное в статье Thomas C. Kuehler (J. Med. Chem. 1998, 41, 1777-1788) (сравнительный пример 1), и соединение, описанное в патенте 2 (сравнительный пример 2).
Результаты представлены в таблице 3 (Активность против Helicobacter pylori производных тиоэфиров (MIC: мкг/мл)).
Активность против Helicobacter pylori производных тиоэфиров (MIC: мкг/мл))
AMPC
CAM
43504
483
484
В таблице 3 AMPC означает амоксициллин, и CAM означает кларитромицин. Кроме того, соединение сравнительного примера 1 - это 2-[(4-изобутокси-3-метилпиридин-2-ил)метилтио]-1H-бензимидазол, а соединение сравнительного примера 2 - это 2-[(4-изобутоксипиридин-2-ил)метилтио]-1H-бензимидазол.
По результатам настоящего теста оказалось, что соединения примеров 1, 2 и 3 обладают сильной антибактериальной активностью против соответствующих стандартных штаммов и клинически выделенных штаммов бактерии Helicobacter pylori. Также стало очевидно, что соединения из этих примеров обладают выраженной активностью против Helicobacter pylori по сравнению со сравнительными примерами и проявляют активность против бактерии Helicobacter pylori, эквивалентную активности антибактериальных препаратов амоксициллина или кларитромицина.
Пример фармакологического теста 3
Был проведен тест на антибактериальную активность соединений по настоящему изобретению против штаммов бактерии Helicobacter pylori, устойчивых к кларитромицину, и штаммов, не чувствительных к амоксициллину.
Методика
Был проведен тест на антибактериальную активность соединений по настоящему изобретению с использованием клинически выделенных штаммов бактерии Helicobacter pylori, устойчивых к кларитромицину, и штаммов, не чувствительных к амоксициллину. Для соответствующих штаммов тесты in vitro провели в среде кобумбийского агара. Каждый образец растворяли в 1% растворе ДМСО. Бактерии выращивали при 37°C и pH 7,0 в течение 3 суток, а на четвертый день определяли минимальную концентрацию ингибирования роста (MIC, мкг/мл).
Результаты приведены в таблице 4 (Влияние производных тиоэфиров на устойчивые бактерии Helicobacter pylori (MIC: мкг/мл)).
Влияние производных тиоэфиров на устойчивые бактерии Helicobacter pylori (MIC: мкг/мл)
В таблице 4 CAM означает кларитромицин, и AMPC означает амоксициллин.
Приведенные выше результаты показали, что соединения примеров 1, 2 и 3 обладают сильной антибактериальной активностью против штаммов, устойчивых к кларитромицину, и штаммов, не чувствительных к амоксициллину. Поскольку амоксициллин и кларитромицин широко используют в настоящее время в терапии для подавления бактерий Helicobacter pylori и, в частности, штаммов, устойчивых к кларитромицину, становится все больше, можно видеть, что настоящее изобретение также эффективно против устойчивых бактерий и его клиническая применимость также очень высока.
Пример фармакологического теста 4
Был проведен антибактериальный тест соединений по настоящему изобретению против грамотрицательных и грамположительных бактерий.
Методика
В качестве грамотрицательных бактерий использовали E. coli (ATCC 10536, ATCC 25922), Klebsiella pneumonia (ATCC 10031), Proteus vulgaris (ATCC 13315), Pseudomonas aeruginosa (ATCC 9027) и Salmonella typhimurium (ATCC 13311), а в качестве грамположительных бактерий использовали Staphylococcus aureus, MRSA (ATCC 33591), Staphylococcus epidermidis (ATCC 12228), Streptococcus pneumonia (ATCC 6301), Mycobacterium ranae (ATCC 110) и Enterococcus faecalis (VRE, ATCC 51575). Разные бактерии выращивали при 37°C в течение 20-48 ч традиционными способами и определяли минимальную концентрацию ингибирования роста (MIC, мкг/мл). Каждый образец растворяли в 1% растворе ДМСО. В качестве контрольных лекарственных средств с антибактериальной активностью использовали амоксициллин, кларитромицин и гентамицин.
Результаты представлены в таблице 5 (Влияние производных тиоэфиров на грамотрицательные и грамположительные бактерии (MIC: мкг/мл)).
Влияние производных тиоэфиров на грамотрицательные и грамположительные бактерии (MIC: мкг/мл)
В таблице 5 AMPC означает амоксициллин, CAM означает кларитромицин, и GEM означает гентамицин.
Результаты этого теста показали, что соединения по настоящему изобретению не обладают антибактериальной активностью против различных грамотрицательных и грамположительных бактерий. С другой стороны, амоксициллин, кларитромицин и гентамицин проявили сильное антибактериальное действие против различных грамотрицательных и грамположительных бактерий. Эти данные показывают, что соединения по настоящему изобретению не оказывают влияния на кишечные бактерии.
Пример фармакологического теста 5
В эксперименте когтистые песчанки Mongolian gerbils были инфицированы бактериями Helicobacter pylori, и соединения по настоящему изобретению были протестированы на противоязвенную активность и активность в подавлении бактерий Helicobacter pylori.
Методика
Самцов Mongolian gerbils (30-40 г), по пять животных в каждой группе, перорально заражали бактериями Helicobacter pylori (ATCC 43504) при 1,6×107 CFU/0,5 мл/мышь и содержали в течение недели, после чего вводили лекарственное средство дважды в день в течение двух недель. Крыс содержали в течение трех недель, после чего их не кормили в течение 18 ч и затем вскрывали брюшину. Определяли степень язвенного поражения, количество бактерий Helicobacter pylori в слизистой желудка и титр антител.
Результаты представлены в таблице 6 (Противоязвенное действие и активность в подавлении бактерий у мышей, инфицированных H. pylori).
Противоязвенное действие и активность в подавлении бактерий у мышей, инфицированных H. pylori
2-[(4-изобутокси-3-метилпиридин-2-илметилтио]-1H-бензимидазол
Сравнительный пример 2:
2-[(4-изобутоксипиридин-2-ил)метилтио]-1H-бензимидазол
В таблице 6 соединение сравнительного примера 1 является 2-[(4-изобутокси-3-метилпиридин-2-илметилтио]-1H-бензимидазолом, и соединение сравнительного примера 2 представляет собой 2-[(4-изобутоксипиридин-2-ил)метилтио]-1H-бензимидазол. Далее, OMZ означает омепразол, CAM означает кларитромицин, и AMPC означает амоксициллин.
Приведенные выше результаты показывают, что омепразол, который является ингибитором секреции желудочного сока, кларитромицин и амоксициллин, которые являются антибактериальными препаратами, и соединения сравнительных примеров не проявляют противоязвенной активности и не подавляют бактерии в модели с инфицированием бактериями Helicobacter pylori, когда применяются как единственные препараты. С другой стороны, тройная комбинация (омепразол, кларитромицин и амоксициллин), которую использовали в качестве активного контроля, четко обладает противоязвенным действием и подавляет бактерии Helicobacter pylori. Из этих данных видно, что соединения по данному изобретению столь же эффективны, как и тройная комбинированная терапия, которая в настоящее время широко применяется в клинической практике в качестве терапии подавления бактерий Helicobacter pylori, даже когда используется в качестве единственного препарата.
По результатам данного эксперимента, соединения по настоящему изобретению, применяемые в качестве единственного препарата, обладают противоязвенным действием и подавляют бактерии аналогично активному контролю. В частности, соединение примера 1 ингибировало развитие язвы даже при малых вводимых дозах 10 мг/кг и обладало сильным действием против бактерий Helicobacter pylori. Из этого следует, что обсуждаемый препарат весьма клинически активен в подавлении бактерий Helicobacter pylori.
Пример фармакологического теста 6
Протестировали действие соединений по настоящему изобретению в качестве ингибиторов секреции желудочного сока.
Методика
Взяли группу из пяти животных - самцов крыс SD (примерно 200 г), медикаменты растворили в 0,5% CMC и вводили их в двенадцатиперстную кишку в количестве 30 мг/кг. Крысы голодали в течение ночи накануне дня перед введением тестового вещества до момента введения, и затем накладывали лигатуру на привратник желудка. Через 4 ч извлекали желудок под анестезией и стандартными способами определяли количество желудочного сока и общую кислотность.
Результаты представлены в таблице 7 (Ингибирование секреции желудочного сока новыми производными пиридина (средний %)).
Ингибирование секреции желудочного сока новыми производными пиридина (средний %)
Контрольным лекарственным средством в таблице 7 является циметидин.
Полученные результаты показали, что все соединения по настоящему изобретению ингибировали секрецию желудочного сока и проявляли активность, практически равную активности циметидина.
Обобщение результатов приведенных выше тестов на стабильность и примеров фармакологических тестов представлено в таблице 8 (Обобщение).
Обобщение
(MIC: мкг/мл)
(-S-)
(S=О)
(оксид примера 1)
(оксид примера 2)
(оксид примера 3)
АМРС: амоксициллин, CAM: кларитромицин
В таблице 8 OMZ означает омепразол, LAZ означает ланзопразол, RAZ означает рабепразол, AMPC означает амоксициллин, и CAM означает кларитромицин.
Из приведенного выше обсуждения видно, что в то время как SO-формы (сульфоксидные формы соединений) весьма нестабильны в кислоте, SH-формы (соединения в форме тиоэфиров) стабильны в кислой среде, и особенно высокая активность против бактерий Helicobacter pylori наблюдается тогда, когда число атомов углерода в линейной цепи R составляет 5, 6 или 7. Кроме того, эти три вида соединений подавляют бактерии Helicobacter pylori, когда применяются в качестве единственных препаратов, так же как в модели инфицирования Mongolian gerbil in vivo. Активность была равна или выше активности тройной комбинации омепразол + амоксициллин + кларитромицин, которую широко применяют во всем мире в терапии против бактерий Helicobacter pylori. Кроме того, подтверждено, что соединения по настоящему изобретению обладают конкретной антибактериальной активностью против бактерий Helicobacter pylori и бактерий, которые не чувствительны или резистентны к амоксициллину и кларитромицину, и ингибируют секрецию желудочного сока.
Далее приведены примеры препаратов с использованием соединения по настоящему изобретению.
Пример препарата 1: приготовление таблеток
Таблетку препарата массой 100 мг готовили обычным способом с учетом приведенного выше соотношения в смеси.
Пример препарата 2: препарат в гранулах
Препарат в виде гранулы массой 1000 мг готовили обычным способом при указанном выше соотношении в смеси.
Пример препарата 3: препарат в капсулах
Препарат в виде капсулы массой 96 мг готовили обычным способом при указанном выше соотношении в смеси.
Пример препарата 4: препарат для инъекций
(2 мл на ампулу)
Препарат для инъекций готовили обычным способом при указанном выше соотношении в смеси.
Пример препарата 5: сироп
Сироп готовили обычным способом при указанном выше соотношении смеси.
Пример препарата 6: препарат в таблетках
Препарат в виде таблетки массой 120 мг готовили обычным способом при указанном выше соотношении в смеси.
Возможности промышленного применения
Целью настоящего изобретения является предложение соединения, стабильного к кислоте, активного против бактерий Helicobacter pylori, обладающего антибактериальной активностью при использовании в качестве единственного препарата, не оказывающего влияния на кишечную флору, обладающего антибактериальной активностью даже по отношению к бактериям, устойчивым к антибактериальным препаратам, и ингибирующего секрецию желудочного сока, и фармацевтической композиции с использованием соединения, и изобретение может быть использовано в фармацевтической промышленности и т.п.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИДИНА, ОБЛАДАЮЩИЕ АНТИ-HELICOBACTER PYLORI АКТИВНОСТЬЮ | 2007 |
|
RU2433126C2 |
| ТИОПРОИЗВОДНОЕ ПИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, КОТОРАЯ ЕГО СОДЕРЖИТ И ИМЕЕТ СПОСОБНОСТЬ ДЕЙСТВОВАТЬ ПРОТИВ HELICOBACTER PYLORI | 2010 |
|
RU2484089C1 |
| НОВЫЙ ИНГИБИТОР КИСЛОТНОЙ СЕКРЕЦИИ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2815697C1 |
| НОВЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2184113C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2143899C1 |
| ТИОПИРИДИНЫ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBAKTER | 1995 |
|
RU2142459C1 |
| СПОСОБ ЭРАДИКАЦИИ ИНФЕКЦИИ Helicobacter pylori В ЖЕЛУДКЕ | 2010 |
|
RU2431477C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИНА, ИНГИБИРУЮЩИЕ СЕКРЕЦИЮ КИСЛОТЫ ЖЕЛУДОЧНОГО СОКА | 2009 |
|
RU2509771C2 |
| СПОСОБ ЭНДОСКОПИЧЕСКОЙ БИОПЛАСТИКИ ГАСТРОДУОДЕНАЛЬНЫХ ЯЗВ КОМПЛЕКСНЫМ БИОПЛАСТИЧЕСКИМ МАТЕРИАЛОМ | 2013 |
|
RU2520599C1 |
| НОВОЕ ИМИДАЗОПИРИДИНОВОЕ СОЕДИНЕНИЕ I С ТЕРАПЕВТИЧЕСКИМ ДЕЙСТВИЕМ | 2004 |
|
RU2348634C2 |
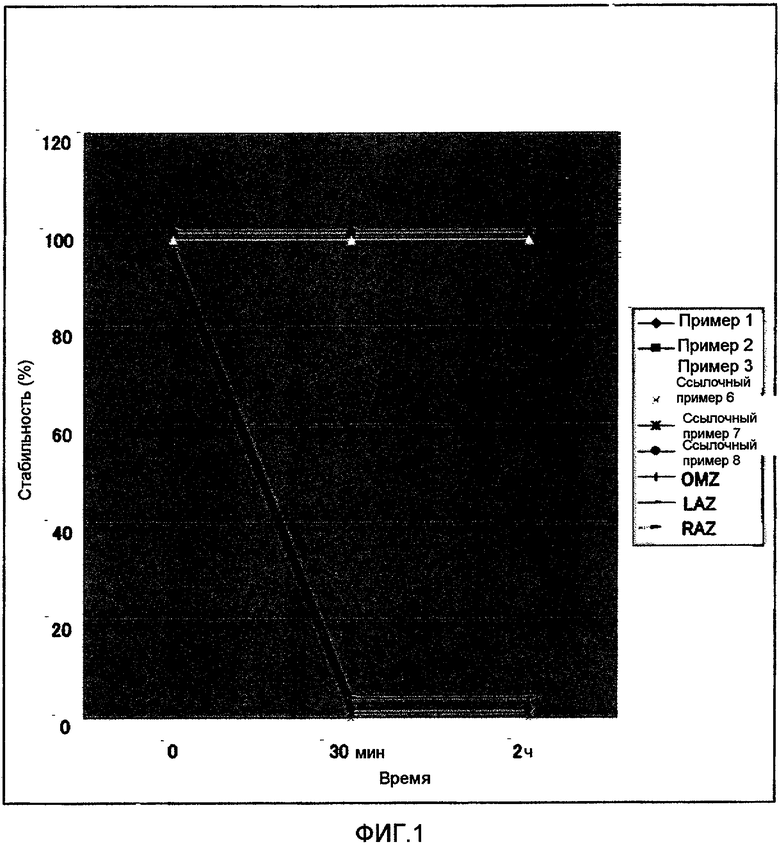
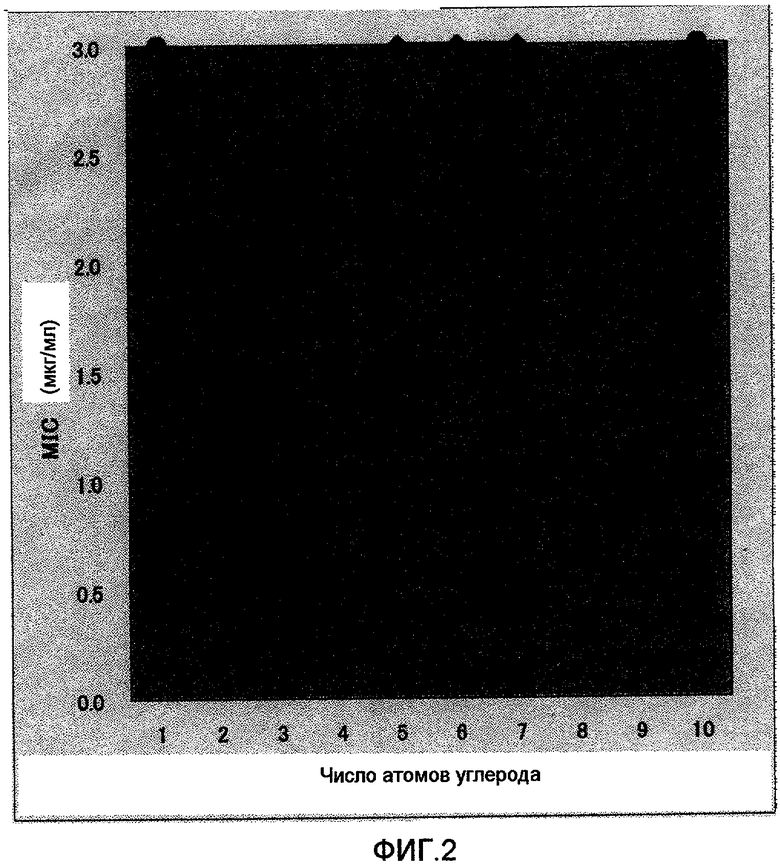
Изобретение относится к новым производным пиридина, представленным формулой (I), где R представляет линейный алкил с 4-8 атомами углерода. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I), применению соединения формулы (I), способу предупреждения или лечения заболевания, вызванного бактериями Helicobacter pylori, и/или заболевания, связанного с секрецией желудочного сока, способу получения производного пиридина формулы (I). Технический результат: получены новые производные пиридина формулы (I), обладающие полезными биологическими свойствами. 5 н. и 4 з.п. ф-лы, 2 ил., 8 табл., 16 пр.
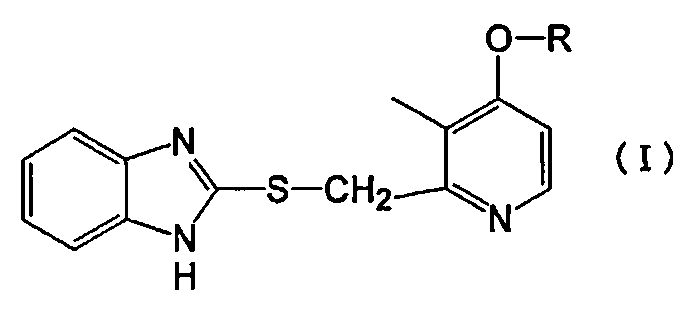
1. Производное пиридина, представленное формулой (I), или его фармацевтически приемлемая соль:
[Химическая формула 5]
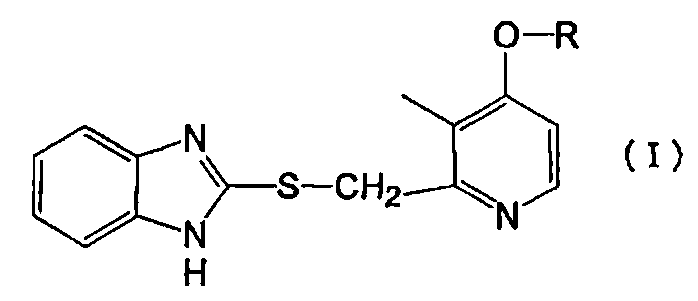
где R представляет линейный алкил с 4-8 атомами углерода.
2. Производное пиридина или его соль по п.1, в котором R в формуле (I) представляет собой линейный алкил с 5-7 атомами углерода.
3. Фармацевтическая композиция для предупреждения или лечения заболевания, вызванного бактериями Helicobacter pylori, и/или заболевания, связанного с секрецией желудочного сока, содержащая производное пиридина по п.1 или 2 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
4. Фармацевтическая композиция по п.3, где заболевание включает гастрит, язву желудка, язву двенадцатиперстной кишки, синдром неязвенной диспепсии, желудочную лимфому MALT, желудочный гиперпластический полип, рак пищеварительной системы или панкреатит вследствие гипергастринемии, вызванной Helicobacter pylori, воспалительное заболевание кишечника, вызванное Helicobacter pylori, или рак желудка после эндоскопической резекции рака желудка на ранней стадии.
5. Применение производного пиридина по п.1 или 2 или его фармацевтически приемлемой соли для получения препарата для предупреждения или лечения заболевания, вызванного бактериями Helicobacter pylori, и/или заболевания, связанного с секрецией желудочного сока.
6. Применение по п.5, причем заболевание включает гастрит, язву желудка, язву двенадцатиперстной кишки, синдром неязвенной диспепсии, желудочную лимфому MALT, желудочный гиперпластический полип, рак пищеварительной системы или панкреатит вследствие гипергастринемии, вызванный Helicobacter pylori, воспалительное заболевание кишечника, вызванное Helicobacter pylori, или рак желудка после эндоскопической резекции рака желудка на ранней стадии.
7. Способ предупреждения или лечения заболевания, вызванного бактериями Helicobacter pylori, и/или заболевания, связанного с секрецией желудочного сока, включающий введение эффективного количества производного пиридина по п.1 или 2 или его фармацевтически приемлемой соли пациенту с заболеванием, вызванным бактериями Helicobacter pylori, и/или заболеванием, связанным с секрецией желудочного сока, или пациенту из группы риска по этим заболеваниям.
8. Способ по п.7, причем заболевание включает гастрит, язву желудка, язву двенадцатиперстной кишки, синдром неязвенной диспепсии, желудочную лимфому MALT, желудочный гиперпластический полип, рак пищеварительной системы или панкреатит вследствие гипергастринемии, вызванной Helicobacter pylori, воспалительное заболевание кишечника, вызванное Helicobacter pylori, или рак желудка после эндоскопической резекции рака желудка на ранней стадии.
9. Способ получения производного пиридина, представленного формулой (I), или его фармацевтически приемлемой соли:
[Химическая формула 8]
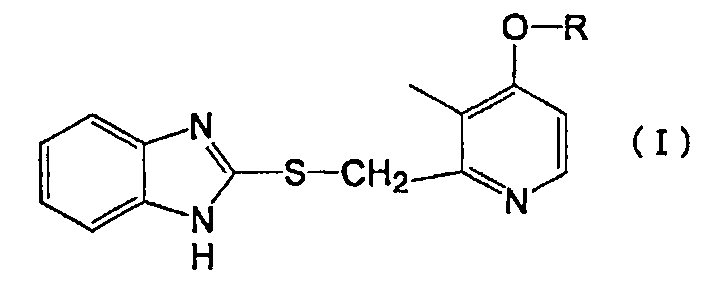
где R представляет собой линейный алкил с 4-8 атомами углерода, включающий взаимодействие соединения формулы (II):
[Химическая формула 6]
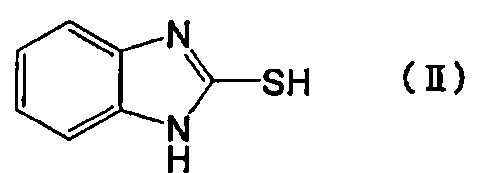
с соединением формулы (III)
[Химическая формула 7]
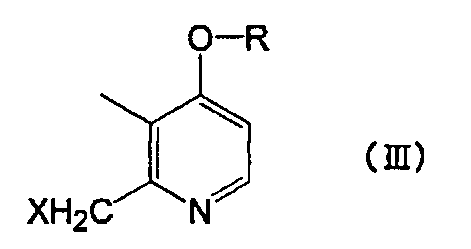
где R представляет собой линейный алкил с 4-8 атомами углерода и X является атомом галогена.
| 0 |
|
SU175464A1 | |
| KUHLER T.C | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Med | |||
| Chem., 1998, v.41, №11, p.1777-1788 | |||
| ПИРИДИЛТИОСОЕДИНЕНИЯ ДЛЯ БОРЬБЫ С БАКТЕРИЯМИ HELICOBACTER | 1995 |
|
RU2149872C1 |

