ОБЛАСТЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к производному 7,7-дифторпростагландин I2, в котором карбоксигруппа простагландина в C-1 замещена тетразольной группой и два атома фтора связаны в C-7 простагландина, к лекарственному средству, содержащему такое производное в качестве активного ингредиента, конкретно к лекарственному средству для профилактики или лечения воспалительного заболевания кишечника.
По данной заявке испрашивается приоритет патентных заявок №. 2008-232133 и 2009-168193, зарегистрированных в Японии, содержание которых включено в настоящий документ посредством данной ссылки.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Воспаление желудочно-кишечного тракта наблюдается в полости рта, пищеводе, желудке, тонком кишечнике, толстом кишечнике и анусе и включает острое и хроническое воспаление. При воздействии на эпителий слизистой оболочки физических или химических факторов или при инфицировании бактериями или вирусами возникает воспаление, и образуются, в зависимости от степени воспаления, участки повреждения с эрозией или изъязвлениями. Повышенная секреция кислоты в желудочном соке обуславливает повреждающее действие, которое приводит к развитию гастрита, язвы желудка или двенадцатиперстной кишки. Кроме того, чрезмерное потребление алкоголя приводит к застою кровотока в слизистой оболочке или к рефлюксу кислоты желудочного сока, вследствие сниженной сократительной способности желудка, что, таким образом, является причиной развития гастрита, язвы желудка, язвы двенадцатиперстной кишки или эзофагита. Пациенты с заболеваниями опорно-двигательного аппарата, с ревматоидным артритом и т.п. при долговременном приеме нестероидного противовоспалительного лекарственного средства страдают от язвы желудка или двенадцатиперстной кишки, обусловленной воздействием лекарственного средства. Кроме того, у пациентов со злокачественной опухолью, при применении химиотерапии, развивается радиационный энтерит или энтерит, обусловленный действием терапии, направленной против злокачественной опухоли. Кроме того, у пациентов, инфицированных туберкулезом, амебной дизентерией и т.п., развивается инфекционный энтерогастрит, такой как туберкулез желудочно-кишечного тракта и амебный колит. Кроме того, под действием ишемии, вследствие нарушения кровотока, развивается ишемический энтерит и т.п. Наличие у пациента с воспалением желудочно-кишечного тракта нарушенного иммунитета, даже при устранении причины заболевания, препятствует восстановлению органа и состояния становятся хроническими. Среди воспалительных заболеваний желудочно-кишечного тракта заболевания с воспалением в кишечном тракте обозначают, в широком смысле, как воспалительное заболевание кишечника.
С другой стороны, существуют воспалительные заболевания кишечника невыясненной этиологии. Язвенный колит и болезнь Крона представляют собой два хорошо известных заболевания, которые представляют собой, в узком смысле, воспалительное заболевание кишечника. Кроме того, воспалительные заболевания кишечника невыясненной этиологии также включают похожие заболевания, такие как болезнь Бехчета, затрагивающая желудочно-кишечный тракт, и простая язва. Они представляют собой трудно поддающиеся лечению хронические кишечные заболевания с многократными ремиссиями и рецидивами, при которых основной причиной заболевания, как полагают, является слабая защита эпителия кишечника или патологический характер иммунного ответа в желудочно-кишечном тракте, направленного против кишечных бактерий, проникающих в ткани кишечника.
Язвенный колит представляет собой хроническое заболевание толстого кишечника, при котором в слизистой оболочке толстого кишечника, непрерывно, начиная с прямого кишечника, образуются эрозии и язвы, и симптомы которого включают боль в животе, диарею, стул с примесью крови, лихорадку и т.п. С другой стороны, при болезни Крона участки поражения образуются в любой области желудочно-кишечного тракта, от полости рта до толстого кишечника и ануса. Это заболевание характеризуется дискретным продольным изъязвлением и появлением в желудочно-кишечном тракте булыжнекоподобного образования, и симптомы заболевания включают боль в животе, диарею, лихорадку, истощение, вследствие пониженного всасывания питательных веществ, анемию и т.п.
Для профилактики и/или лечения воспаления при воспалительных заболеваниях желудочно-кишечного тракта известной этиологии устраняют причину заболевания или ослабляют ее влияние. Для подавления секреции и влияния кислоты желудочного сока при гастрите, язве желудка, язве двенадцатиперстной кишки и т.п. против воспаления используют, например, антацидное средство, антихолинергическое средство, антагонисты рецепторов гистамина H2, ингибитор протонного насоса и т.п. В других случаях, производные простагландина E и т.п. используют в качестве пищевой добавки простагландина E в случае воспаления, вызванного нестероидным противовоспалительным лекарственным средством, которое ингибирует продукцию PGE.
С другой стороны, профилактика или лечение воспалительного заболевания кишечника, в узком смысле, включает терапию лекарственным средством, терапию посредством питательных веществ (диета) и терапию посредством хирургического вмешательства. В случае терапии лекарственным средством используют препараты 5-аминосалициловой кислоты (пентаза, салазопирин), стероидные препараты (преднизолон), иммуносупрессоры (азатиопурин, меркаптопурин и такролимус), антитела против TNF-α (инфликсимаб) и т.п.
В следующих патентных документах 1, 2 и непатентном документе 1 описаны производные PG, имеющие вместо карбоксигруппы в положении C-1 простагландина (далее в настоящем документе, обозначаемый как PG) тетразольную группу.
Кроме того, описаны аналоги 7,7-дифтор PGI2 и способы их получения (патентные документы 3 и 4). Кроме того, описывают, что аналоги 7,7-дифтор PGI2 будут полезны в качестве профилактических или терапевтических средств при сердечно-сосудистых заболеваниях.
Документы известного уровня техники
[патентные документы]
патентный документ 1: DE 2405255
патентный документ 2: WO 03/103664
патентный документ 3: JP-A-7-330752
патентный документ 4: JP-A-2004-256547
[непатентные документы]
непатентный документ 1: J. Med. Chem., 22: 1340 (1979).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Задачи, подлежащие решению посредством изобретения
Целью настоящего изобретения является создание нового производного простагландина I2, которое отличается, как описано выше, от известных аналогов PGI2, или его фармацевтически приемлемой соли.
Средства решения задач
Для решения указанных выше задач, авторы настоящего изобретения синтезировали новые аналоги PG, в соответствии с конкретными свойствами атома фтора, и провели исследования для выяснения их свойств и физиологической активности. В результате, авторы изобретения обнаружили, что новое производное 7,7-дифтор PGI2, где карбоксигруппа в C-1 каркаса простаноевой кислоты замещена тетразольной группой и два атома фтора связаны, является превосходным по свойствам и фармакологическому действию, и что это производное представляет собой превосходное химическое соединение в качестве лекарственного средства, что позволило, в результате, завершить настоящее изобретение.
Насколько авторам настоящего изобретения известно, аналоги PGI2, имеющие вместо карбоксигруппы в C-1 PG тетразольную группу, еще не описаны, кроме того, не описаны примеры синтеза, свойств, физиологической активности и т.п. аналогов PGI2, где C-1 PG представляет собой тетразольную группу и в C-1 PG присутствуют два атома фтора.
Таким образом, настоящее изобретение относится к производному 7,7-дифтор PGI2, представленного следующей формулой (1), его фармацевтически приемлемой соли и лекарственному средству, содержащему это производное в качестве активного ингредиента.
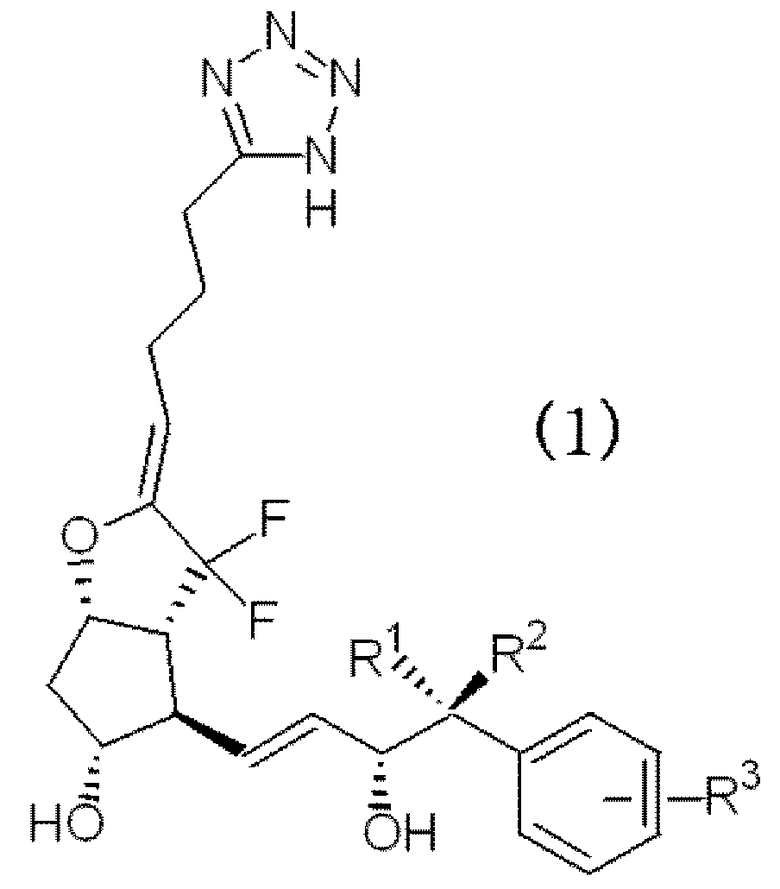
где R1 и R2 независимо представляют собой атом водорода или алкильную группу с неразветвленной цепью с количеством атомов углерода от 1 до 3 и R3 представляет собой атом водорода, алкильную группу с количеством атомов углерода от 1 до 4, алкоксиалкильную группу, арильную группу, атом галогена или галогеналкильную группу.
Эффект изобретения
Новое производное 7,7-дифтор PGI2, представленное настоящим изобретением, может обеспечить лекарственное средство, которое в течение длительного времени сохраняет свою концентрацию в крови и демонстрирует фармакологическое действие при парентеральном введении или пероральном введении и которое создано для профилактики или лечения воспаления желудочно-кишечного тракта, или при появлении диареи или стула с кровью при воспалительном заболевании кишечника, или для профилактики или лечения гастрита, или для профилактики или лечения гастритов или изъязвления при язве желудка, язве тонкого кишечника и т.п.
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ.
На [фиг. 1] показано влияние на аномальный стул на модели колита у мышей, где колит вызван DSS.
На [фиг. 2] показано влияние на процесс укорочения толстого кишечника на модели колита у мышей, где колит вызван DSS.
На [фиг. 3] показано влияние на аномальный стул на модели колита у крыс, где колит вызван DSS.
На [фиг. 4] показано влияние на процесс укорочения толстого кишечника на модели колита у крыс, где колит вызван DSS.
На [фиг. 5] показано влияние на процесс повреждения ткани толстого кишечника на модели колита у крыс, где колит вызван DSS.
На [фиг. 6] показано влияние на аномальный стул на модели ремиссии/рецидива колита у мышей, где колит вызван DSS.
На [фиг. 7] на модели колита у мышей, который вызван переносом T-клеток, показано влияние на величину оценки консистенции стула.
На [фиг. 8] на модели колита у мышей, который вызван переносом Т-клеток, показано влияние на величину оценки наличия скрытой крови в стуле.
На [фиг. 9] на модели колита у мышей, который вызван переносом Т-клеток, показано влияние на величину оценки снижения массы тела.
На [фиг. 10] на модели колита у мышей, который вызван переносом Т-клеток, показано влияние на величину оценки DAI.
На [фиг. 11] на модели повреждения слизистой желудка у крыс, вызванного этанолом, показано влияние на язву желудка.
На [фиг. 12] на модели повреждения тонкого кишечника у крыс, вызванного действием индометацина, показано влияние на язву тонкого кишечника.
Вариант осуществления изобретения
Определение соединений по настоящему изобретению
В номенклатуре соединений по настоящему описанию, номера, используемые для описания положения в каркасе PG, соответствуют номеру в каркасе простаноевой кислоты. В настоящем описании группу, в которой замещен атом водорода алкильной группы, также обозначают как замещенную алкильную группу. То же самое распространяется на другие группы.
Кроме того, понятие «низшие» органические группы, такие как алкильная группа и т.п. обозначает, что число атомов углерода составляет не больше чем 6. Углеродное число «низшей» органической группы предпочтительно составляет не более чем 4.
«Алкильная группа» может представлять собой группу с неразветвленной или разветвленной цепью. Если не указано иначе, алкильная группа представляет собой предпочтительно низшую алкильную группу, имеющую от 1 до 6 атомов углерода, и особенно предпочтительной является низшая алкильная группа, имеющая от 1 до 4 атомов углерода. Примеры алкильной группы включают метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, пентильную группу, гексильную группу и т.п.
«Алкоксигруппа» представляет собой предпочтительно низшую алкокси группу, имеющую от 1 до 6 атомов углерода, особенно предпочтительной является алкоксигруппа, имеющая от 1 до 4 атомов углерода. Алкоксигруппа может представлять собой группу с неразветвленной или разветвленной цепью. Примеры алкоксигруппы включают метокси-группу, этокси-группу, пропокси-группу, бутокси-группу и т.п.
«Алкоксиалкильная группа» представляет собой алкильную группу, замещенную алкоксигруппой. Алкоксигруппа алкоксиалкильной группы представляет собой предпочтительно низшую алкоксигруппу, имеющую от 1 до 4 атомов углерода, и алкильная группа алкоксиалкильной группы представляет собой предпочтительно низшую алкильную группу, имеющую от 1 до 4 атомов углерода. Алкоксиалкильная группа представляет собой предпочтительно низшую алкоксиалкильную группу (т.е. вся алкоксиалкильная группа имеет не более чем 6 атомов углерода), более предпочтительно низшую алкоксиалкильную группу, имеющую не более чем 4 атома углерода. Примеры алкоксиалкильной группы включают метоксиметильную группу, этоксиметильную группу, пропоксиметильную группу, этоксиэтильную группу и т.п.
«Арильная группа» представляет собой моновалентную ароматическую углеводородную группу необязательно с заместителем(ями). В качестве арильной группы без заместителя, предпочтительной является фенильная группа.
В качестве «замещенной арильной группы» (арильная группа с заместителем(ями)), предпочтительной является арильная группа, где один или несколько атомов водорода в арильной группе замещают низшей алкильной группой, атомом галогена, галогенированной (низший алкил) группой, низшей алкоксигруппой и т.п. Предпочтительные примеры замещенной арильной группы включают замещенную фенильную группу и ее конкретные примеры включают моногалофенильную группу (например, хлорфенильную группу, фторфенильную группу, бромфенильную группу и т.д.), (галогенированный низший алкил) замещенная фенильная группа (например, трифторметилфенил группа и т.д.) и (низший алкокси) фенильная группа (например, метоксифенильная группа, этоксифенильная группа и т.д.).
«Атом галогена» представляет собой атом фтора, атом хлора, атом брома или атом йода.
«Галоалкильная группа» представляет собой алкильную группу, где один или несколько атомов водорода в алкильной группе замещены атомом галогена и предпочтительной является низшая галоалкильная группа, имеющая от 1 до 6 атомов углерода. Примеры галоалкильной группы включают фторметильную группу, дифторметильную группу, трифторметильную группу, трифторэтильную группу, пентафторэтильную группу, хлорметильную группу, бромметильную группу и т.п.
В качестве производного 7,7-дифтор PGI2, представленного формулой (1) по настоящему изобретению (далее в настоящем документе, также обозначаемый как производное PGI2 (1) по настоящему изобретению), следующее соединение является предпочтительным исходя из аспектов фармакологической активности и физических свойств.
Т.е. R1 и R2 независимо представляют собой атом водорода или неразветвленную цепь алкильной группы, имеющей от 1 до 3 атомов углерода, и каждый независимо представляет собой предпочтительно атом водорода или метильную группу. Особенно предпочтительно, чтобы один из R1 и R2 представлял собой атом водорода, а другой представляет собой метильную группу.
R3 представляет собой атом водорода, алкильную группу, имеющую от 1 до 4 атомов углерода, алкоксиалкильную группу, арильную группу, атом галогена или галоалкильную группу и атом водорода, алкильную группу, имеющую от 1 до 4 атомов углерода, низшую алкоксиалкильную группу, такую как метоксиметильная группа и т.п., атом галогена, такой как атом хлора, атом фтора и т.п., или низшую галоалкильную группу, такую как низшая фторалкильная группа и т.п., которая является предпочтительной. Конкретно, атом водорода, алкильная группа, имеющая от 1 до 4 атомов углерода, атом хлора или галогеналкильная группа, имеющая от 1 до 4 атомов углерода, которая является предпочтительной. В качестве алкильной группы, имеющей от 1 до 4 атомов углерода, метильная группа и этильная группа являются предпочтительными, а в качестве галогеналкильной группы, имеющей от 1 до 4 атомов углерода, предпочтительной является трифторметильная группа.
В качестве R3, атом водорода, метильная группа или трифторметильная группа, которая является наиболее предпочтительной.
Кроме того, R3 можно замещать бензольным кольцом в любом из положений орто(o), мета(m) и пара(p), относящихся к положению заместителя главной цепи каркаса простагландина. Особенно предпочтительным является R3, замещенный в мета (m) положении.
Вариант осуществления предпочтительного соединения по настоящему изобретению
Кроме того, предпочтительными комбинациями R1, R2 и R3 в соединении по настоящему изобретению являются следующие.
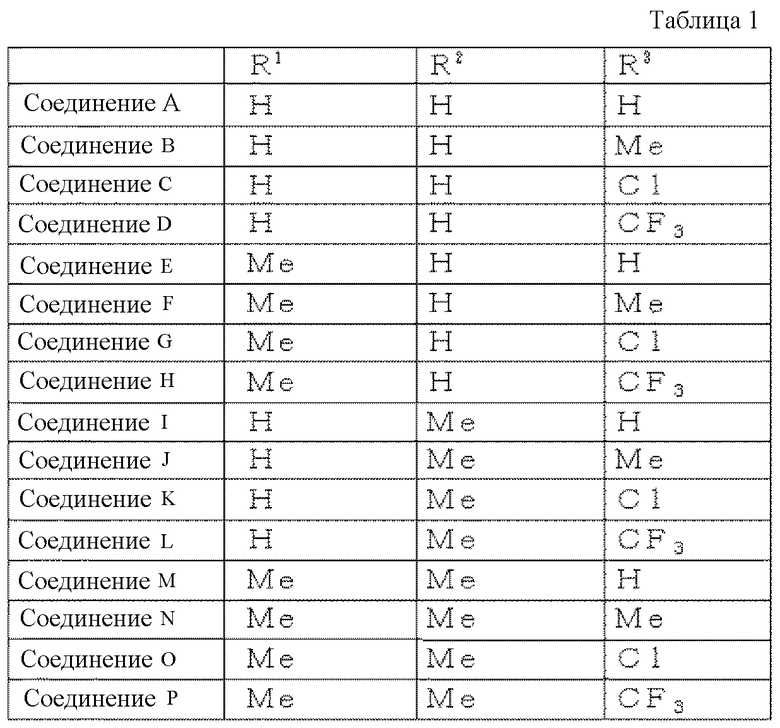
R1 представляет собой атом водорода, R2 представляет собой атом водорода и R3 представляет собой атом водорода.
R1 представляет собой атом водорода, R2 представляет собой атом водорода, а R3 представляет собой метильную группу.
R1 представляет собой атом водорода, R2 представляет собой атом водорода, а R3 представляет собой атом хлора.
R1 представляет собой атом водорода, R2 представляет собой атом водорода, а R3 представляет собой трифторметильную группу.
R1 представляет собой метильную группу, R2 представляет собой атом водорода и R3 представляет собой атом водорода.
R1 представляет собой метильную группу, R2 представляет собой атом водорода и R3 представляет собой метильную группу.
R1 представляет собой метильную группу, R2 представляет собой атом водорода и R3 представляет собой атом хлора.
R1 представляет собой метильную группу, R2 представляет собой атом водорода и R3 представляет собой трифторметильную группу.
R1 представляет собой атом водорода, R2 представляет собой метильную группу и R3 представляет собой атом водорода.
R1 представляет собой атом водорода, R2 представляет собой метильную группу и R3 представляет собой метильную группу.
R1 представляет собой атом водорода, R2 представляет собой метильную группу и R3 представляет собой атом хлора.
R1 представляет собой атом водорода, R2 представляет собой метильную группу и R3 представляет собой трифторметильную группу.
R1 представляет собой метильную группу, R2 представляет собой метильную группу и R3 представляет собой атом водорода.
R1 представляет собой метильную группу, R2 представляет собой метильную группу и R3 представляет собой метильную группу.
R1 представляет собой метильную группу, R2 представляет собой метильную группу и R3 представляет собой атом хлора.
R1 представляет собой метильную группу, R2 представляет собой метильную группу и R3 представляет собой трифторметильную группу.
Кроме того, предпочтительные комбинации из числа тех, которые перечислены выше, представляют собой следующие.
R1 представляет собой метильную группу, R2 представляет собой атом водорода и R3 представляет собой метильную группу.
R1 представляет собой атом водорода, R2 представляет собой метильную группу и R3 представляет собой метильную группу.
Способ получения производного PGI 2 по настоящему изобретению
Производное PGI2 (1) по настоящему изобретению можно получать, например, исходя из способов, описанных в JP-A-07-324081 и JP-A-08-217772, относящихся к изобретениям, которые осуществили авторы настоящего изобретения. Например, используя лактон Кори в качестве исходного материала, сначала вводят ω цепь, и лактон преобразуют фторированием в лактон Кори, содержащий ω цепь с двумя атомами фтора. Затем вводят элемент α цепи посредством дополнительной реакции с металлоорганическим реагентом с тетразольной группой на конце и реакцией дегидратации или реакцией Виттига, с использованием соли фосфония с тетразольной группой на конце и т.п., и, по мере необходимости, снимают защиту с гидроксильной группы, тем самым можно синтезировать производное PGI2 (1).
Альтернативно, лактон Кори с двумя атомами фтора получают посредством фторирования, из лактона Кори в качестве исходного материала. Затем, элемент α цепи вводят посредством дополнительной реакции с металлоорганическим реагентом с тетразольной группой на конце и реакцией дегидратации или реакцией Виттига, с использованием соли фосфония с тетразольной группой на конце и т.п., вводят ω цепь и, по мере необходимости, снимают защиту с гидроксильной группы, тем самым можно синтезировать производное PGI2 (1).
Альтернативно, производное PGI2 (1) можно также синтезировать посредством преобразования карбоксигруппы производного карбоновой кислоты, описанного в JP-A-07-324081, в циано-группу и преобразованием производного в производное тетразола.
Среди этих способов получения, конкретно разъясняют типичные способы, используя следующие химические формулы.
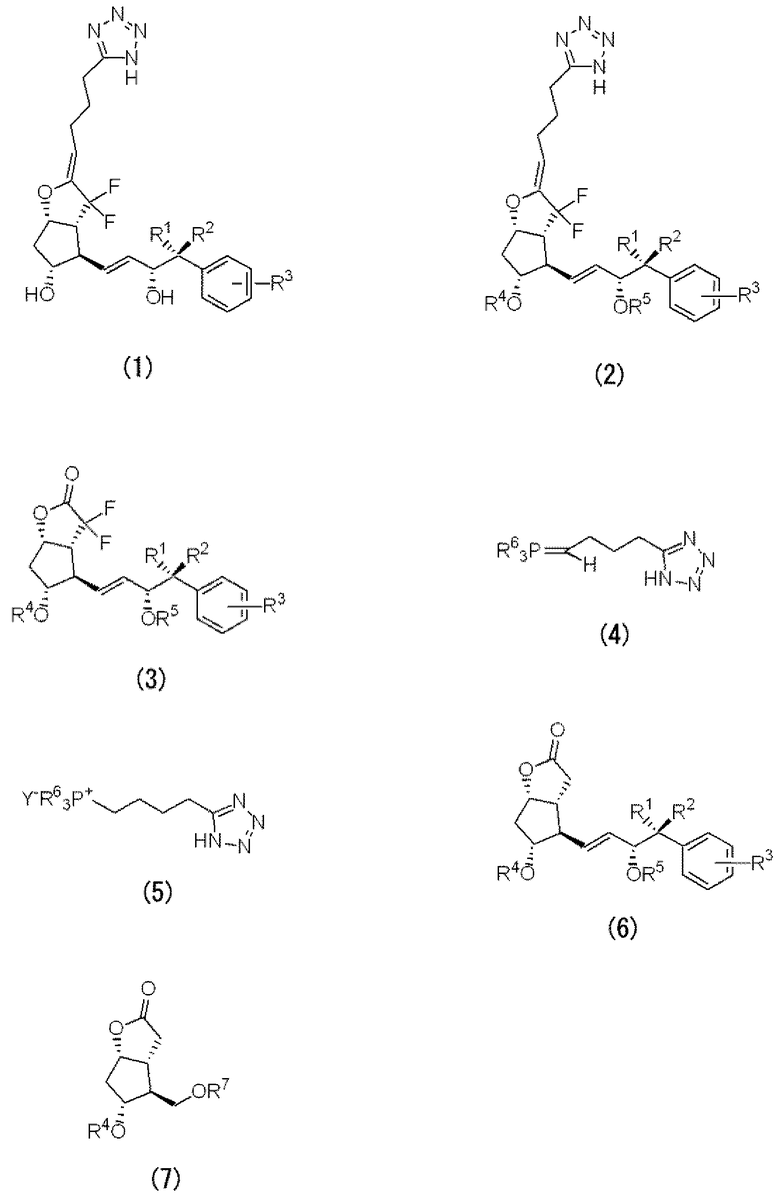
Например, используя лактон Кори (7), в качестве исходного материала, в начале вводят ω цепь, полученное производное лактона Кори (6), содержащее ω цепь, подвергают реакции фторирования для получения ω цепи, содержащей производное лактона Кори (3) с двумя атомами фтора, а именно с двумя атомами фтора в α-положении карбонильной группы. Затем производное лактона с двумя атомами фтора (3) подвергают реакции с производным фосфорана (4) для введения α элемента цепи, тем самым можно получать производное PGI2 (2) с защищенными гидроксильными группами. Группы, защищающие гидроксил, удаляют, для получения производного PGI2 (1) по настоящему изобретению.
Производное фосфорана (4) можно получать из производного соли фосфония (5).
За исключением случаев, когда R1-R3 представляют собой конкретные заместители, указанное выше производное лактона (6) является известным соединением. Указанное выше новое производное лактона (6), где R1-R3 представляет собой конкретные заместители, можно получать способами, аналогичными способам получения известных производных лактона. (6). Например, новые производные лактона (6) можно получать взаимодействием дисложного эфира 3-арил-2-оксоалкилфосфоновой кислоты с лактоном Кори, имеющим формильную группу. В данном изобретении, алкильная цепь алкилфосфоновой кислоты имеет углеродное число не менее чем 3.
R4, R5 и R7 независимо представляют собой защитные группы гидроксила. R4, R5 и R7 могут представлять собой одинаковые защитные группы. В качестве защитной группы можно использовать защитную группу гидроксила, описанную в «Shin Jikken Kagaku Koza (New Courses in Experimental Chemistry) 14, synthesis and reaction of organic compound (V)» (Maruzen Company, Limited), «Protective Groups in Organic synthesis» (by T.W. Greene, J. Wiley & Sons) и т.п. Конкретно, можно назвать триорганосилильную группу, алкоксиалкильную группу, моновалентную группу, имеющую структуру циклического простого эфира, и т.п. В качестве триорганосилильной группы, предпочтительной является силильная группа, где 3 группы, выбранные из алкильной группы, арильной группы, аралкильной группы и алкоксигруппы связывают с атомом кремния, и особенно предпочтительной является группа, где с атомом кремния связывают 3 низшие алкильные группы или арильные группы. В качестве конкретных примеров защитной группы предпочтительными являются тетрагидропиранильная группа, трет-бутилдиметилсилильная группа, трет-бутилдифенилсилильная группа, триэтилсилильная группа, трифенилсилильная группа или триизопропилсилильная группа и т.п. Конкретно, предпочтительными являются тетрагидропиранильная группа, трет-бутилдиметилсилильная группа, трет-бутилдифенилсилильная группа и т.п.
Защитную группу гидроксила можно легко удалять. Способ снятия защиты с защищенной гидроксильной группы может представлять собой традиционный способ. Например, можно применять способы, описанные в «Shin Jikken Kagaku Koza (New Courses in Experimental Chemistry) 14 synthesis and reaction of organic compound (I), (II) и (V) «(Maruzen Company, Limited), «Protective Groups in Organic synthesis» (by T.W. Greene, J. Wiley & Sons) и т.п.
Для преобразования производного лактона (6) в производное лактона с двумя атомами фтора (3), посредством реакции фторирования, можно применять различные известные способы фторирования. Например, можно применять способ, включающий взаимодействие с различными электрофильными фторирующими средствами в инертном растворителе. Фторирование можно также выполнять способами, описанными в JP-A-07-324081 и JP-A-09-110729, относящимися к изобретению, которое осуществили авторы настоящего изобретения.
В реакции фторирования производного лактона (6), предпочтительно используют электрофильное фторирующее средство. В качестве электрофильного фторирующего средства можно использовать известное или широко известное электрофильное фторирующее средство. Например, можно указать электрофильные фторирующие средства, описанные в «Chemistry of fluorine «(Kodansha Scientifics Ltd.) Tomoya Kitazume, Takashi Ishihara, и Takeo Taguchi и т.п. Конкретно можно указать N-фторсульфонил амиды, производное N-сульфонил имида, ацетилгипофторит, фтористый газ и т.п.
Электрофильное фторирующее средство предпочтительно используют в присутствии инертного растворителя. В качестве инертного растворителя можно указать эфирные растворители, углеводородные растворители, полярные растворители, смесь указанных растворителей и т.п.
Производное лактона с двумя атомами фтора (3), полученное реакцией фторирования, затем вступает в реакцию с производным фосфорана (4), что необходимо для получения производного PGI2 (2), где гидроксильная группа защищена. Производное фосфорана (4) получают из соответствующего производного фосфониевой соли (5), в инертном растворителе в присутствии основания, и полученное производное фосфорана (4) предпочтительно используют непосредственно для реакции Виттига с производным лактона с двумя атомами фтора (3) без выделения. В качестве способов получения производного фосфорана (4) и производного фосфониевой соли (5) можно применять способы, описанные в DE2242239, DE2405255 и т.п. В качестве R6 для производного фосфорана (4) или производного фосфониевой соли (5), предпочтительной является арильная группа, такая как фенильная группа, толильная группа и т.п., и особенно предпочтительной является фенильная группа. В качестве инертного растворителя можно указать эфирные растворители, углеводородные растворители, полярные растворители, водные растворители, спиртовые растворители, смесь указанных растворителей и т.п.
Для получения производного PGI2 (1) из производного PGI2 (2) с защищенными гидроксильными группами, полученного способом выше, удаляют защитную группу гидроксила.
Так как производное PGI2 по настоящему изобретению (1) в своей структуре имеет асимметричный атом углерода, то присутствуют различные стереоизомеры и оптические изомеры. Настоящее изобретение относится ко всем таким стереоизомерам, оптическим изомерам и их смесям.
Конкретные примеры производного PGI2 по настоящему изобретению (1) включают соединение, представленное следующей формулой (8).
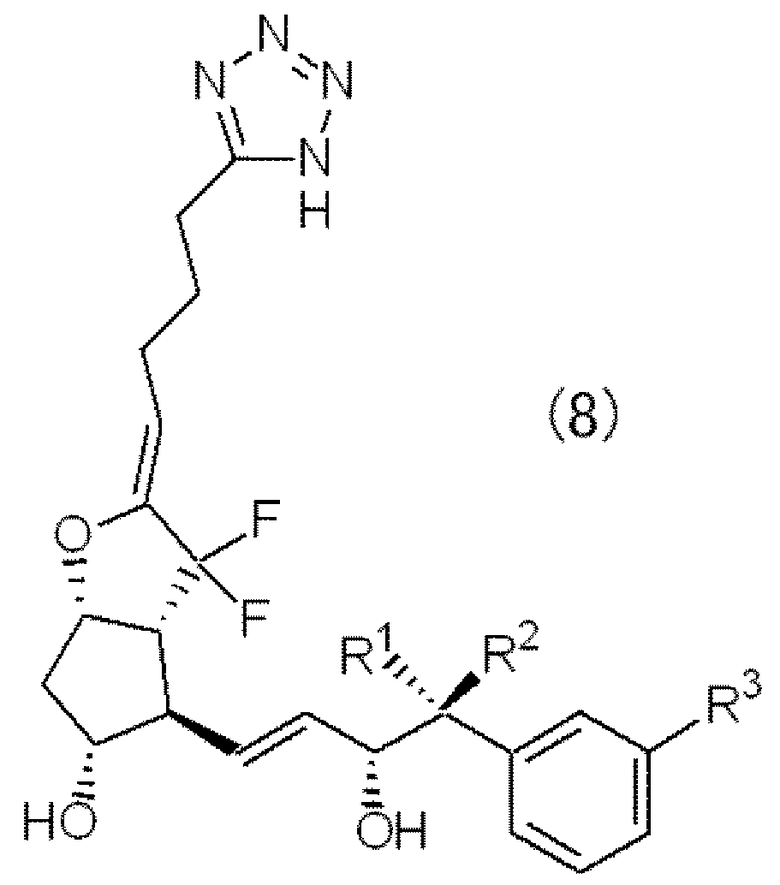
Примеры производного PGI 2 (1) по настоящему изобретению
Можно указать соединение, где в формуле (8) R1, R2 и R3 имеют структуры, представленные в следующей таблице 1.
Характеристики производного PGI 2 (1) по настоящему изобретению
Производное PGI2 (1) по настоящему изобретению представляет собой производное, которое с трудом метаболизируется в организме и обладает повышенной стабильностью. Так как карбоксигруппа каркаса PG преобразована в тетразольную группу, он с трудом метаболизируется посредством β-окисления, которое, как известно, представляет собой общепризнанный метаболический путь жирных кислот, таких как простагландины. Таким образом, производное PGI2 (1) имеет длительный период полувыведения из плазмы и может сохранять эффективную концентрацию в плазме в течение более длительного периода времени, по сравнению с соединением, имеющим карбоксигруппу в каркасе PG. Так как таким способом увеличивают метаболическую стабильность, может быть улучшена и биодоступность лекарственных средств.
Лекарственные средства, содержащие в качестве активного ингредиента производное PGI 2 по настоящему изобретению (1) или его фармацевтически приемлемую соль
Лекарственное средство по настоящему изобретению содержит производное PGI2 (1) и/или фармацевтически приемлемую соль производного PGI2 (1) и дополнительно фармацевтически приемлемый носитель и, в некоторых случаях, другие компоненты для лечения.
Лекарственное средство по настоящему изобретению содержит производное PGI2 (1) и/или фармацевтически приемлемую соль производного PGI2 (1), или его гидрат, и дополнительно фармацевтически приемлемый носитель и, в некоторых случаях, другие компоненты для лечения.
При введении пациентам профилактического или терапевтического средства по настоящему изобретению суточная доза варьирует в зависимости от возраста и массы тела пациентов, вида патологического процесса и его тяжести и т.п. Как правило, желательным является введение 0,0001-10 мг, предпочтительно 0,01-1 мг средства в виде одной или нескольких порций. Например, для перорального введения, предпочтительным является 0,001-3 мг и особенно предпочтительным является 0,01-0,5 мг. Для внутривенного введения, предпочтительным является 0,0001-1 мг и особенно предпочтительным является 0,001-0,1 мг. При необходимости количество дозы можно изменять, в зависимости от заболевания и его состояния. В качестве режима дозирования, может быть желательным введение инъецируемого продукта средства посредством непрерывной капельной инфузии.
Для применения в качестве лекарственного средства, средство можно вводить в организм посредством перорального введения и парентерального введения (например, внутрисосудистого (внутривенного, внутриартериального) введения, подкожного введения, ректального введения и т.д.). Примеры лекарственной формы включают пероральную лекарственную форму, такую как таблетка, капсула и сироп, парентеральную лекарственную форму, такую как инъекция жидкой лекарственной формы (раствор, эмульсия, суспензия и т.п.), инфузию, суппозитории, препараты для назального введения, пластыри и лекарственные формы для ингаляций. Особенно желательным является пероральное дозирование.
Получение в виде указанной выше лекарственной формы можно получать, смешивая производное PGI2 по настоящему изобретению (1) или его фармацевтически приемлемую соль с добавками, необходимыми для состава, такими как общепринятые носители, эксципиенты, связывающие средства и стабилизаторы, и составляя в смесь общепринятым способом. Например, если препарат представляет собой порошок, частицу, таблетку и т.п., его можно получать, используя любые фармацевтические носители, предпочтительно для получения твердой лекарственной формы, например эксципиенты, смазочные средства, разрыхлители, связывающие средства и т.п.
Эти эксципиенты могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция и фосфат натрия; средство для гранулирования и разрыхлитель, такой как кукурузный крахмал и альгиновая кислота; связывающее средство, такое как крахмал, желатин и аравийская камедь, и смазочное средство, такое как стеарат магния, стеариновая кислота и тальк. Таблетка может быть без оболочки или покрыта оболочкой посредством известного способа для замедления процесса разрушения и всасывания в желудке и желудочно-кишечном тракте, обеспечивая, таким образом, замедленное высвобождение в течение более длительного периода времени. Например, можно использовать материал, обеспечивающий задержку во времени, такой как глицерилмоностеарат или глицерилдистеарат.
Производное PGI2 по настоящему изобретению (1) можно получать в виде твердой желатиновой капсулы, содержащей смесь с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином. Альтернативно, производное PGI2 по настоящему изобретению (1) можно получать в виде мягкой желатиновой капсулы, содержащей смесь с растворителем, смешивающимся с водой, таким как пропиленгликоль, полиэтиленгликоль и этанол, или масла, такие как арахисовое масло, парафиновое масло и оливковое масло.
Если препарат представляет собой сироп или жидкость, для получения можно, соответствующим образом, выбрать и использовать, например стабилизаторы, суспендирующие средства, корригирующие средства, ароматические средства и т.п. Для получения инъецируемой формы активный ингредиент растворяют в дистиллированной воде для инъекций вместе с регулятором pH, таким как соляная кислота, гидроксид натрия, лактоза, натрия лактат, уксусная кислота, гидрофосфат динатрия и дигидрогенфосфат натрия, и изотоническом средстве, таком как хлорид натрия и глюкоза, и инъецируемую форму получают в асептических условиях. Неактивный безводный разбавитель, такой как пропиленгликоль, полиэтиленгликоль, оливковое масло, этанол и полисорбат 80, можно использовать для состава препарата. Кроме того, можно добавлять маннит, декстрин, циклодекстрин, желатин и т.п., и смесь высушивают под вакуумом для получения инъекций, подлежащих растворению перед применением. Кроме того, для стабилизации и улучшения доставки лекарственного средства в участок повреждения можно составлять в смесь липосомный препарат или липидную эмульсию посредством известного способа и использовать в качестве инъецируемой формы.
Кроме того, ректальное дозирование препарата можно выполнять, используя суппозитарную основу, такую как масло какао, жирная кислота в виде триглицерида, жирная кислота в виде диглицерида, жирная кислота в виде моноглицерида и полиэтиленгликоль. Кроме того, водорастворимую основу, такую как полиэтиленгликоль, полипропиленгликоль, глицерин и глицерин-желатин, масляную основу, такую как белый вазелин, твердый жир, парафин, парафиновое масло, Plastibase (диспергированный полиэтилен), ланолин и очищенный ланолин и т.п., можно использовать, чтобы придать препарату подходящую вязкость, и также можно получать мазь для внутриректального введения.
Производное PGI2 по настоящему изобретению (1) или его фармацевтически приемлемую соль можно наносить местно на кожу или слизистую оболочку, т.е., трансдермальным или чресслизистым введением. В качестве основных лекарственных форм для этой цели можно назвать гель, гидрогель, лосьон, раствор, крем, мазь, спреи, перевязочное средство, пенный препарат, пленку, трансдермальный пластырь, облатку, имплантат, губку, волокно, бандаж, микроэмульсию и т.п. В качестве широко используемых носителей можно назвать спирт, воду, минеральное масло, парафиновое масло, белый вазелин, глицерин, полиэтиленгликоль, пропиленгликоль и т.п.
Производное PGI2 по настоящему изобретению (1) можно смешивать с циклодекстрином или его подходящим производным или растворимым полимером, таким как полимер, содержащий полиэтиленгликоль, с целью использования в любых указанных выше лекарственных формах и улучшения растворимости, скорости растворения, биодоступности и стабильности. Например, подтверждено, что комплекс лекарственное средство-циклодекстрин и т.п, как правило, будет пригоден для большинства лекарственных форм и способов введения. Можно использовать как комплексы включения, так и комплексы не включения. В качестве другого способа для прямого комплексообразования с лекарственными средствами, также можно использовать циклодекстрин в виде вспомогательного средства, т.е. носителя, эксципиента или солюбилизатора. Как правило, для этих целей используют α-, β- и γ- циклодекстрины и т.п.
Фармацевтически приемлемая соль производного PGI 2 по настоящему изобретению
Фармацевтически приемлемая соль производного PGI2 по настоящему изобретению (1) представляет собой соль молекулы тетразольной группы производного от исходного соединения, которое является соединением, где атом водорода тетразольной группы замещают катионом.
Примеры катиона включают катионы щелочных металлов, таких как Na+ и K+, катионы металлов (отличных от катионов щелочных металлов), таких как 1/2 Ca2+, 1/2 Mg2+, 1/2 Zn2+ и 1/3 Al3+, NH4 +, катионы аммония органического амина и аминокислоты, таких как триэтаноламин, диэтаноламин, этаноламин, трометамин, лизин и аргинин и т.п. Предпочтительным катионом является ион натрия или ион калия.
Более конкретно, подходящая соль представляет собой соль, полученную из фармацевтически приемлемого нетоксического основания, такого как неорганическое основание и органическое основание. В качестве соли, полученной из фармацевтически приемлемого нетоксического неорганического основания, можно назвать литиевую соль, медную соль, соль трехвалентного железа, соль двухвалентного железа, соль трехвалентного марганца, марганцевую соль и т.п. в дополнение к указанным выше натриевой соли, калиевой соли, кальциевой соли, магниевой соли, соли цинка, соли алюминия, аммониевой соли и т.п. Из них являются предпочтительными натриевая соль, калиевая соль, кальциевая соль, магниевая соль и аммониевая соль и особенно предпочтительными являются натриевая соль и калиевая соль. Соль, полученная из фармацевтически приемлемого нетоксического органического основания, включает соли с первичным, вторичным и третичным амином, замещенным амином, включающим природный замещенный амин, циклический амин и основной ион ионообменной смолы. Другими примерами указанных выше органического амина и аминокислоты, можно назвать изопропиламин, диэтиламин, триэтиламин, триметиламин, трипропиламин, этилендиамин, N,N'-дибензилэтилендиамин, 2-диэтиламиноэтанол, 2- диметиламиноэтанол, морфолин, N-этилморфолин, пиперазин, пиперидин, N-этилпиперидин, бетаин, кофеин, холин, глюкамин, глюкозамин, гистидин, гидрабамин, метилглюкамин, полиаминовую смолу, прокаин, пурин, теобромин и т.п.
Применение лекарственного средства, содержащего в качестве активного ингредиента производное PGI 2 по настоящему изобретению или его фармацевтически приемлемую соль
Лекарственное средство, содержащее в качестве активного ингредиента производное PGI2 по настоящему изобретению (1) или его фармацевтически приемлемую соль демонстрирует превосходное действие в качестве лекарственного средства при заболеваниях желудочно-кишечного тракта.
Заболевание желудочно-кишечного тракта в настоящем изобретении относится к воспалительному заболеванию и язвенному заболеванию желудочно-кишечного тракта, которое представляет собой заболевание с наличием воспаления или изъязвлением в эпителиальной, слизистой или подслизистой ткани желудочно-кишечного тракта или патологической пролиферации или дисфункции эпителия слизистой оболочки, и которое вызвано физическими воздействиями, химическими воздействиями, такими как действие желудочного сока, действие лекарственного средства, такого как нестероидные противовоспалительные лекарственные средства и стероидные лекарственные средства, иммунными заболеваниями и аутоиммунными заболеваниями неизвестной этиологии, психическими заболеваниями и т.п.
Воспалительное заболевание желудочно-кишечного тракта включает воспалительное заболевание кишечника, конкретно язвенный колит, болезнь Крона, которое представляет собой неспецифическое гранулематозное воспалительное заболевание, сопровождающееся фибриллированием или изъязвлением, поражение желудочно-кишечного тракта при болезни Бехчета и простая язва. Язвенное заболевание желудочно-кишечного тракта по настоящему изобретению относится к стоматиту, афтозному стоматиту, эзофагиту, язве пищевода, гастриту, язве желудка и язвенной болезни тонкого кишечника.
Кроме того, гастрит и язва желудка включают обусловленные лекарственным средством гастрит, язву желудка, алкогольные гастрит и язву желудка и обусловленные лекарственным средством гастрит и язва желудка включают гастрит и язву желудка, вызванные нестероидным противовоспалительным лекарственным средством.
Язвенная болезнь тонкого кишечника включает язвенную болезнь тонкого кишечника, обусловленную лекарственными средствами, и алкогольную язвенную болезнь тонкого кишечника, и язвенная болезнь тонкого кишечника включает язвенную болезнь тонкого кишечника, обусловленную нестероидным противовоспалительным лекарственным средством.
Конкретно, лекарственное средство по настоящему изобретению является пригодным в качестве профилактического или терапевтического средства для лечения язвенного колита, болезни Крона, гастрита, язвы желудка или язвенной болезни тонкого кишечника.
Настоящее изобретение подробно объясняют далее со ссылками к конкретным примерам, которые не следует истолковывать как ограничивающие.
Пример 1
Синтез метил(2R)-2-(м-толил)пропионата
К (2R)-2-(м-толил)пропионовой кислоте (12,45 г) добавили метанол (14,83 г) и концентрированную серную кислоту (6,46 г) и смесь перемешивали с обратным холодильником в течение 6 часов. Затем смесь нейтрализовали 10% водным раствором карбоната натрия и экстрагировали с гексаном. После сушки над сульфатом магния остаток концентрировали при пониженном давлении для получения указанного в заголовке соединения (12,79 г). Структурное свойство являлось таким, как описано ниже.
1H ЯМР(CDC13):δ 1,49(д, J=7,0 Гц, 3H), 2,33(с, 3H), 3,64(с, 3H), 3.69(дд, J=14,4, 7,3 Гц, 1H), 7,06-7,22(м, 4H).
Пример 2
Синтез диметил(3R)-2-оксо-3-(м-толил)бутилфосфоната
К диметилметилфосфонату (1,97 г) добавили тетрагидрофуран (THF) (25 мл), и смесь охладили до -78°C. Добавили N-бутиллитий (1,5 М раствор гексана) (10 мл), и смесь размешивали в течение 1 часа. Затем, при -78°C добавили раствор сложного метилового эфира {метил(2R)-2-(м-толил)пропионат}, синтезированный в примере 1, (1,34 г) в THF (3,8 мл), и смесь размешивали в течение 2 часов. Реакцию гасили 25 мл насыщенного водного раствора гидрокарбоната натрия, и смесь экстрагировали этилацетатом. Экстракт высушили над сульфатом магния и концентрировали при пониженном давлении. Остаток очистили хроматографией на колонках с силикагелем (гексан/этилацетат 5:1-1:5) для получения указанного в заголовке соединения (1,63 г). Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDC13):δ 1,39 (д, J=6,7 Гц, 3H), 2,34 (с, 3H), 2,84(ддд, J=22,3, 14,1, 0,6 Гц, 1H), 3,18(дд, J=22,3, 14,1 Гц, 1H), 3,76(дд, J=19,3, 11,1 Гц, 6H), 4,00(дд, J=13,8, 7,0 Гц, 1H), 7,01-7,24(м, 4H).
Пример 3
Синтез ((1S,5R,6R,7R)-6-[(1E,4R)-3-оксо-4-(м-толил)-1-пентенил]-7-бензоилокси-2-оксабицикло[3.3.0] октан-3-он
Гидрид натрия (55%) (8,75 г) диспергировали в 1,2-диметоксиэтане (DME) (300 мл), и смесь охладили льдом. Добавили раствор фосфоната {диметил(3R)-2-оксо-3-(м-толил)-бутилфосфонат} (54,7 г), синтезированный в примере 2, в DME (50 мл), и смесь размешивали в течение 1 часа. К указанному выше раствору добавили раствор (1S,5R,6R,7R)-6-формил-7-бензоилокси-2-оксабицикло[3.3.0]октан-3-он (50,0 г) в DME (400 мл), и смесь размешивали в течение 1 часа. Реакцию гасили 350 мл 10% насыщенного солевого раствора, и смесь экстрагировали этилацетатом. Экстракт высушили над сульфатом магния, и остаток концентрировали при пониженном давлении. Сконцентрированный неочищенный продукт перекристаллизовали из трет-бутил метилового эфира для получения указанного в заголовке соединения (64,7 г). Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDCl3):δ 1,39(д, J=7,0 Гц, 3H), 2,20-2,28(м, 1H), 2,30(с, 3H), 2,34-2,41(м, 1H), 2,49-2,57(м, 1H), 2,76-2,85(м, 3H), 3,80(кв, J=7,0 Гц, 1H), 5,03(т, J=5,3 Гц, 1H), 5,23(кв, J=5,3 Гц, 1H), 6,19(д, J=15,5 Гц, 1H), 6,69(дд, J=15,6, 7,6 Гц, 1H), 6,94-7,19(м, 4H), 7,42-7,95(м, 5H).
Пример 4
Синтез (1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-бензоилокси-2-оксабицикло[3.3.0]октан-3-он
Раствор енона {(1S,5R,6R,7R)-6-[(1E,4R)-3-оксо-4-(м-толил)-1-пентенил]-7-бензоилокси-2-оксабицикло[3.3.0]октан-3-он} (147,0 г), синтезированный в примере 3 ,в THF (1480 мл) охладили до -40°C, добавили ((-)-B-хлордиизопинокамфеилборан (1,7 М раствор гексана) (721 мл), и смесь размешивали при охлаждении льдом в течение 20 часов. Добавили ацетон (183 мл), и смесь размешивали в течение 3 часов. Добавили водный раствор гидрокарбоната натрия, и смесь экстрагировали с трет-бутил метиловым эфиром. Экстракт высушили над сульфатом магния и сконцентрировали при пониженном давлении для получения указанного в заголовке неочищенного соединения (649,9 г).
Пример 5
Синтез (1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил) - 1-пентенил]-7-гидрокси-2-оксабицикло[3.3.0]октан-3-он
Неочищенный спирт, {(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-бензоилокси-2-оксабицикло[3.3.0]-октан-3-он} (649,9 г), синтезированный в примере 4, растворили в метаноле (740 мл), добавили карбонат калия (116,3 г), и смесь размешивали при комнатной температуре в течение 17 часов. Добавили уксусной кислоты, чтобы довести pH до уровня 7, метанол выпарили, добавили воду, и смесь экстрагировали с этилацетатом. Экстракт очистили хроматографией на колонках с силикагелем (гексан/этилацетат=4/1-0/1) для получения указанного в заголовке соединения (22, 3 г). Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDCl3):δ 1,33 (д, J=7,0 Гц, 3H), 1,70 (с, 1H(OH)), 1,86(ддд, J=11,3, 7,8, 3,2 Гц, 1H), 2,07(д, J=4,4 Гц, 1H(OH)), 2,13-2,23(м, 2H), 2,34(с, 3H), 2,35-2,44(м, 3H), 2,47(д, J=3,8 Гц, 1H), 2,56(дд, J=18,2, 9,7 Гц, 1H), 2,80(кв, J=7,0 Гц, 1H), 3,79-3,85(м, 1H), 4,12-4,16(м, 1H), 4,81(дт, J=7,0, 3,2 Гц, 1H), 5,27(ддд, J=15,7, 8,5, 0,6 Гц, 1H), 5,50(дд, J=15,2, 6,8 Гц, 1H), 6,94-7,20 (м, 4H).
Пример 6
Синтез (1S,5R,6R,7R)-6-[(1E,3R,4R)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-оксабицикло[3.3.0]октан-3-он
К раствору диола, {(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-оксабицикло[3.3.0]октан-3-он} (988 мг), синтезированного в примере 5, в N,N-диметилформамиде (DMF) (10 мл), добавили при комнатной температуре трет-бутилдиметилсилил хлорид (1,17 г) и имидазол (1,08 г), и смесь размешивали в течение 2,5 часов. Реакционную смесь перелили в насыщенный водный раствор гидрокарбоната натрия, и смесь экстрагировали с гексаном/этилацетат=смесь 2/1. Экстракт высушили над сульфатом магния, сконцентрировали при пониженном давлении, и очистили хроматографией на колонке с силикагелем (гексан/этилацетат 20:1-10:1), для получения указанного в заголовке соединения (1,56 г). Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDCl3): δ-0,09 (д, J=6,4 Гц, 6H), 0,02 (д, J=2,4 Гц, 6H), 0,86(с, 9H), 0,89(с, 9H), 1,27(д, J=7,0 Гц, 3H), 1,86-1,92(м, 1H), 1,96-2,02(м, 1H), 2,32(с, 3H), 2,31-2,47(м, 3H), 2,62-2,73(м, 2H), 3,82(кв, J=4,7 Гц, 1H), 4,05(т, J=6,4 Гц, 1H), 4,86(дт, J=8,0, 2,4 Гц, 1H), 5,16(дд, J=15,5, 7,4 Гц, 1H), 5,30 (дд, J=15,7, 6,3 Гц, 1H), 6,90-7,16(м, 4H).
Пример 7
Синтез (1S,5R,6R,7R)-6-[(1E,3R,4R)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-окса-4,4-дифтор-бицикло[3.3.0]октан-3-он
Тетрагидрофуран (THF) (19 мл) добавили к бромиду марганца (1,48 г) и N-фторбензолсульфонимиду (2,48 г), и смесь размешивали в течение 30 минут и охладили до -78°C. Добавили раствор лактона, {(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-оксабицикло[3.3.0]октан-3-он} (0,5 г), синтезированный в примере 6, в THF (5 мл), добавили раствор (0,5 М, 13 мл) бис(триметилсилил)амида калия в толуоле, и смесь нагревали до 0°C в течение 3,5 часов. Реакционную смесь перелили в насыщенный водный раствор гидрокарбоната натрия, и смесь экстрагировали со смесью гексан/этилацетат=1/1. Экстракт высушили над сульфатом магния, концентрировали при пониженном давлении и очистили хроматографией на колонке с силикагелем (гексан/этилацетат 20:1) для получения указанного в заголовке соединения (0,32 г). Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDCl3):δ-0,08-0,03 (м, 12H), 0,82(с, 9H), 0,89(с, 9H), 1,28(д, J=7,0 Гц, 3H), 1,70-1,77(м, 1H), 1,96-2,04(м, 1H), 2,31 (с, 3H), 2,60-2,91(м, 3H), 3,82-3,87(м, 1H), 3,99-4,23(м, 1H), 5,00 (т, J=6,4 Гц, 1H), 5,06(дд, J=15,7, 7,8 Гц, 1H), 5,33(ддд, J=15,9, 6,7, 1,2 Гц, 1H), 6,88-7,16(м, 4H).
19F-ЯМР(CDCl3): -113,1(д, J=279,3 Гц), -91,0(дд, J=279,3, 25,9 Гц).
Пример 8
Синтез 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-окса-4,4-дифтор-бицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутан
К суспензии 4-(тетразол-5-ил)бутилтрифенилфосфония бромида (14,0 г) в толуоле (390 мл) добавили раствор (0,5 M, 120 мл) бис(триметилсилил)амида калия в толуоле, и смесь размешивали при 60°C в течение 1 часа. При -10°C добавили раствор дифтор-лактон, {(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-окса-4,4-дифтор-бицикло[3.3.0]октан-3-он}, синтезированный в примере 7, (4,32 г) в толуоле (130 мл), и смесь размешивали в течение 18 часов, пока не нагрели смесь до комнатной температуры. Добавили водный раствор гидрокарбоната натрия, чтобы погасить реакцию, и смесь экстрагировали смесью гексан/этилацетат=1/1. Экстракт высушили над сульфатом магния, сконцентрировали при пониженном давлении и очистили хроматографией на колонке с силикагелем (гексан/этилацетат=5/1-0/1) для получения указанного в заголовке соединения (4,1 г). Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDCl3):δ-0,14-0,01(м, 12H), 0,82 (с, 9H), 0,89(с, 9H), 1,23-1,27 (м, 3H), 1,82-2,09 (м, 5H), 2,21-2,28 (м, 1H), 2,31(с, 3H), 2,45-2,53(м, 1H), 2,64-2,73(м, 2H), 2,93-2,97(м, 2H), 3,90(дд, J=11,7, 5,3 Гц, 1H), 4,08-4,09(м, 1H), 4,84-4,87(м, 2H), 5,27(дд, J=15,5, 7,8 Гц, 1H), 5,44(дд, J=15,6, 6,2 Гц, 1H), 6,92-7,16(м, 4H).
19F-ЯМР(CDCl3): -112,3(д, J=253,4 Гц), -81,4(дд, J=253,4, 18,7 Гц).
Пример 9
Синтез 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4R)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифтор-бицикло[3.3.0]-октан-3-илиден]-1-(тетразол-5-ил)бутан
Добавили THF (81 мл), воду (81 мл) и уксусную кислоту (244 мл) к соединению (4,1 г), синтезированному в примере 8, и смесь размешивали при 35°C в течение 46 часов. Добавили воды (500 мл), и смесь экстрагировали хлороформом. Экстракт высушили над сульфатом магния, сконцентрировали при пониженном давлении и очистили хроматографией на колонке с силикагелем (гексан/этилацетат=1/5-0/1) и перекристаллизовали из диэтилового эфира, для получения указанного в заголовке соединения (1,1 г). Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CD3OD):δ 1,30(д, J=7,0 Гц, 3H), 1,69(дддд, J=14,6, 7,6, 3,0, 2,6 Гц, 1H), 1,82-1,95(м, 2H), 2,10-2,16(м, 2H), 2,29(с, 3H), 2,31-2,41(м, 2H), 2,48-2,56(м, 1H), 2,72(кв, J=7,0 Гц, 1H), 2,93(т, J=7,6 Гц, 2H), 3,78(кв, J=7,6 Гц, 1H), 4,04-4,10(м, 1H), 4,69(дт, J=6,48, 2,96 Гц, 1H), 4,79(дт, J=7,6, 5,0 Гц, 1H), 5,36-5,46(м, 2H), 6,95-7,13(м, 4H).
19F-ЯМР(CD3OD): -116,6(д, J=250,5 Гц), -84,8(ддд, J=251,9, 17,3, 14,4 Гц).
Пример 10
Синтез диметил 2-оксо-3-(м-толил)бутилфосфонат
Указанное в заголовке соединение синтезировали таким же способом, как в примерах 1-2, используя рацемат 2-(м-толил) пропионовой кислоты. Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDCl3): δ 1,39(д, J=7,2 Гц, 3H), 2,34(с, 3H), 2,83(дд, J=22,4, 14,4 Гц, 1H), 3,18(дд, J=22,4, 14,0 Гц, 1H), 3,76(дд, J=19,6, 11,2 Гц, 6H), 3,99(дд, J=14,0, 6,8 Гц, 1H), 7,01-7,27(м, 4H).
Пример 11
Синтез (1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-оксабицикло[3.3.0]октан-3-он
Указанное в заголовке соединение синтезировали таким же способом, как в примерах 3-6, используя рацемат диметил 2-оксо-3-(м-толил)бутилфосфоната. Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDCl3):δ -0,20-0,10(м, 12H), 0,80-0,90(м, 18H), 1,18-1,28(м, 3H), 1,85-2,20(м, 2H), 2,31(с, 3H), 2,30-2,80(м, 5H), 3,80-4,15(м, 2H), 4,81-4,95(м, 1H), 5,12-5,42(м, 2H), 6,88-7,20 (м, 4H).
Пример 12
Синтез (1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-окса-4,4-дифтор-бицикло[3.3.0]октан-3-он
Указанное в заголовке соединение синтезировали таким же способом, как в примере 7, используя (1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-оксабицикло[3.3.0]октан-3-он, синтезированный в примере 11. Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDCl3):δ -0,20-0,05(м, 12H), 0,80-0,90(м, 18H), 1,19-1,29(м, 3H), 1,70-2,10(м, 2H), 2,31(с, 3H), 2,60-3,05(м, 3H), 3,84-4,12(м, 2H), 4,95-5,50(м, 3H), 6,85-7,20(м, 4H).
19F-ЯМР(CDCl3): -113,6 - -112,8 (м), -91,7 - -90,6(м).
Пример 13
Синтез 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-окса-4,4-дифтор-бицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутан
Указанное в заголовке соединение синтезировали таким же способом, как в примере 8, используя (1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-окса-4,4-дифтор-бицикло[3.3.0]октан-3-он, синтезированный в примере 12. Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDCl3):δ -0,15-0,05(м, 12H), 0,80-0,89(м, 18H), 1,20-1,28(м, 3H), 1,80-3,05(м, 14H), 3,90-4,15(м, 2H), 4,85-4,95(м, 2H), 5,23-5,58(м, 2H), 6,90-7,20(м, 4H).
19F-ЯМР(CDCl3): -113,0 - -111,3(м), -82,0 - -80,7 (м).
Пример 14
Синтез 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-гидрокси-4-(м-толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифтор-бицикло[3.3.0]-октан-3-илиден]-1-(тетразол-5-ил)бутан
Указанное в заголовке соединение синтезировали таким же способом, как в примере 9, используя 4-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-окса-4,4-дифтор-бицикло[3.3.0]октан-3-илиден]-1-(тетразол-5-ил)бутан, синтезированный в примере 13. Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CDCl3):δ 1,15-1,35(м, 3H), 1,80-3,00(м, 11H), 2,29(с, 3H), 4,05-4,20(м, 2H), 4,75-4,85(м, 2H), 5,35-5,70(м, 2H), 6,95-7,25(м, 4H).
19F-ЯМР(CDCl3): -114,5 - -112,7 (м), -83,5 - -81,8 (м).
Пример 15
Синтез 5-[(Z)-(1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-гидрокси-4-(м- толил)-1-пентенил]-7-гидрокси-2-окса-4,4-дифтор-бицикло[3.3.0]октан-3-илиден] пентановой кислоты (карбоксилатная форма)
Указанное в заголовке соединение синтезировали таким же способом, как в примерах 8-9, используя (1S,5R,6R,7R)-6-[(1E,3R,4RS)-3-трет-бутилдиметилсилокси-4-(м-толил)-1-пентенил]-7-трет-бутилдиметилсилокси-2-окса-4,4-дифтор-бицикло[3.3.0]октан-3-он, синтезированный в примере 12 и (4-карбоксибутил)трифенилфосфония бромид. Структурное свойство являлось таким, как описано ниже.
1H-ЯМР(CD3OD):δ 1,17-1,30(м, 3H), 1,63-2,79(м, 11H), 2,29(с, 3H), 3,75-4,12(м, 2H), 4,66-4,85(м, 2H), 5,40-5,58(м, 2H), 6,95-7,15(м, 4H).
19F-ЯМР(CD3OD): -118,3 - -117,7 (д, J=250,4 Гц), -86,1 - -85,3(м).
Пример 16
Метаболическая стабильность соединения по настоящему изобретению in vitro
Тестировали смесь соединения F и соединения J по настоящему изобретению, описанные в таблице 1 (F:J=52:41, синтезированные в примере 14), и смесь соединений, где тетразольные группы в C-1 соединения F и соединения J соответственно замещены карбоновой кислотой (рассматриваемые в качестве карбоксилатной формы, F:J=54:34, синтезированные в примере 15).
Во-первых, из печени крыс получили митохондриальную фракцию, согласно следующей далее ссылке A. Затем, с применением способа YAMAGUCHI et al., описанного в следующих ссылках B и C, исследовали независимую от NADPH реакцию β-окисления. Реакцию провели при 37°C в течение 30 мин и останавливали раствором метанола, содержащим подходящее в качестве внутреннего стандарта вещество. Количественную оценку каждого соединения проводили способом на основе внутреннего стандарта, используя прибор для высокоэффективной жидкостной хроматографии и масс-спектрометрии (LC-MS/MS). Остаточное отношение соединения после метаболической реакции соединений F, J и каждой их карбоксилатной формы в митохондриальной фракции крыс представлено в таблице 2 в среднем значении, полученном в результате 3 экспериментов ± стандартное отклонение.
Как понятно из указанной выше таблицы 2, типичное соединение F и соединение J по настоящему изобретению не подвергаются β-окислению в митохондриальной фракции.
Ссылки
A) The Japanese Biochemical Society, ed., Biochemical Experiment Course 12 energy metabolism and biological oxidation (vol. 1), Tokyo Kagaku Dojin, p. 217-218, 1st ed. 2nd printing, published on July 11, 1979.
B) Drug Metabolism And Disposition 23(11): 1195-1201 (1995).
C) Xenobiotica 26(6): 613-626 (1996).
Пример 17
Фармакокинетика в плазме после внутривенного введения крысам
Для проверки метаболической стабильности соединения по настоящему изобретения in vivo, оценили фармакокинетику в плазме после внутривенного введения крысам. Самцы крыс (в возрасте 6 недель, масса тела 160-180 г) акклиматизировались в течение 1 недели и в эксперимент отбирали здоровых животных. Смесь соединения F и соединения J по настоящему изобретению, описанных в таблице 1 (F:J=52:41), и смесь карбоксилатных форм соединения F и соединения J (F:J=54:34), и соединение F (синтезированное в примере 9) растворили в небольшом количестве этанола и для получения растворов тестируемого соединения добавили физиологический раствор. Растворы тестируемого соединения немедленно ввели внутривенно из расчета 1 мл/кг в бедренную вену неголодной крысы, находящейся под действием легкой анестезии посредством эфира. Венозную кровь отбирали из хвостовой вены через 5, 15, 30, 45, 60, 90 и 120 мин после введения. Кровь смешивали с гепарином и центрифугировали (3000 об/мин, 4°C, 15 мин) для получения плазмы. Концентрацию соединения в плазме определили посредством способа с внутренним стандартом, используя LC-MS/MS. Диапазон определения посредством этого способа составлял от 0,1 до 100 нг/мл. Концентрации соединений, полученные для каждой крысы, анализировали независимым от модели способом, используя программное обеспечение WinNonlin (версия 3,3) для анализа фармакокинетики, и получили среднее значение ± стандартное отклонение для 3 животных для каждой группы. Период полувыведения (t1/2) представлен в следующей таблице 3.
Как видно из указанной выше таблицы 3, значения t1/2 соединения F и соединения J по настоящему изобретению составили приблизительно 1-2 часа, что явно составило более продолжительный период, по сравнению с менее чем 10 мин для карбоксилатных форм. Это показывает, что соединение F и соединение J по настоящему изобретению обладают великолепной метаболической стабильностью.
Пример 18
Профилактическое действие на модель колита у мышей, вызванного декстраном сульфатом натрия
Профилактическое действие соединения F на язвенный колит изучали на модели колита, вызванного декстраном сульфатом натрия. Модель на животных демонстрирует воспаление, локализованное в толстом кишечнике, которое приводит к развитию диареи и появлению крови в стуле, что имеет близкое сходство с патологическим состоянием при язвенном колите (cf.: References D и E).
Были куплены самки мышей линии BALB/c (возраст 6 недель, Japan SLC), акклиматизация проходила в течение 1 недели и мышей использовали для исследования. За исключением нормальной группы, мышам позволяли в течение 9 суток, для индуцирования колита, без ограничений пить раствор декстрана сульфата натрия (сокращенно как DSS, MP Biochemicals, M.W. 36,000-50,000, Lot №. 3439J), полученный из расчета 2,2 масс./об.%. Соединение F вводили перорально в дозах 0,1, 0,3 и 1 мг/кг, один раз в сутки, ежедневно, с первых суток приема DSS (сутки 0) до суток перед вскрытием (сутки 9). Контрольная группа по той же схеме перорально принимала раствор (1 об.% раствора этанола) из расчета 10 мл/кг.
Наше предварительное исследование выявило, что для экскрементов мыши обнаружена корреляция между содержанием воды и их формой. Таким образом, для определения степени диареи экскременты классифицировали по 6 уровням; нормальный (оценка 0), экскременты сферической формы составляют не менее чем 50% (оценка 1), экскременты в форме банана составляют менее чем 50% (оценка 2), экскременты в форме банана составляют не менее чем 50% (оценка 3), экскременты глинистой консистенции (оценка 4), экскременты водянистой консистенции (оценка 5) (оценка консистенции экскрементов). Наличие скрытой крови в экскрементах классифицировали по 5 уровням, используя предметное стекло 5 для анализа экскрементов на наличие скрытой крови Shionogi II (Shionogi & Co., Ltd.); отрицательный (нет изменения цвета покровного стекла с желтого, оценка 0), слабо положительный (немного сине-зеленый, оценка 1), положительный (сине-зеленый, оценка 2), умеренно положительный (отчетливый сине-зеленый, оценка 3) и отчетливо положительный (мгновенное изменение цвета на темно-синий c проявителем цвета, оценка 4). Сумму оценки консистенции стула и оценки наличия скрытой крови определяют как оценку стула. Для каждой группы использовали от восьми до 10 животных и результаты выражали в виде среднего ± стандартное отклонение.
В результате, масса тела постепенно увеличивалась в течение периода исследования без какого-либо различия среди групп. Контрольная группа явно продемонстрировала наличие жидкого стула и наличие скрытой крови в стуле на 4 сутки приема DSS. В сутки аутопсии (сутки 9) длина толстого кишечника была явно короче, чем длина толстого кишечника нормальной группы. Соединение F дозозависимо снизило увеличение показателя оценки стула, с тенденцией к подавлению при 0,1 мг/кг и значительному подавлению при 0,3 и 1 мг/кг (фиг. 1). Кроме того, соединение F показало дозозависимый подавляющий эффект на укорочение толстого кишечника (фиг. 2). Таким образом, соединение F явным образом предотвратило развитие язвенного колита.
Ссылки
D) Lab. Invest. 69(2): 238-249 (1993).
E) Inflamm. Res. 45(4): 181-191 (1996).
Пример 19
Профилактическое действие на модель колита у крыс, вызванного декстраном сульфатом натрия
Профилактическое действие соединения F на колит также изучено на крысах. Приобрели самцов крыс SD, в возрасте 7 недель, массой тела около 210 г - 240 г (Charles River), подвергли акклиматизации в течение 1 недели и использовали в исследовании. За исключением нормальной группы, крысам позволяли в течение 8 суток, для индуцирования колита, без ограничений пить раствор DSS (MP Biochemicals, M.W. 36,000-50,000, Lot №. 4556J), полученный из расчета 5,5 масс./об.%. Соединение F в дозах 0,3, 1 и 3 мг/кг перорально вводили раз в сутки, ежедневно, в первые сутки до начала приема DSS до суток перед аутопсией (сутки 7). Контрольной группе перорально вводили раствор (1 об.% раствора этанола) из расчета 5 мл/кг.
На 8 сутки с начала приема DSS, в хвостовую вену ввели 1,25 масс./об.% голубого раствора Эванса из расчета 0,2 мл/100. Через 30 мин крысы подверглись лапаротомии под анестезией простым эфиром и обескровливались до наступления смерти. Затем, толстый кишечник иссекли в месте сразу после слепой кишки до ануса и длину измеряли штангенциркулем. Затем содержимое толстого кишечника удаляли, ткань толстого кишечника длиной 7 см от ануса промывали 3 раза физиологическим раствором и высушивали в течение ночи под вакуумным насосом. На следующие сутки, измеряли сухую массу, добавляли формамид (2 мл), удаляли краситель при 50°C в течение ночи, и его количество измеряли при 620 нм. Для оценки степени повреждения ткани толстого кишечника получили калибровочную кривую, используя стандартный голубой раствор Эванса, и рассчитали количество (мг) голубого раствора Эванса в 1 г ткани толстого кишечника.
Для определения степени диареи форму стула классифицировали по 6 уровням, как нормальный (оценка 0), стержневидный стул составляет менее чем 50% (оценка 1), стержневидный стул составляет не менее чем 50% (оценка 2), стержневидный стул и стул частично глинистой консистенции (оценка 3), стул с глинистой консистенцией (оценка 4) и стул водянистой консистенции (оценка 5) (оценка консистенции стула). Оценку наличия скрытой крови в стуле проводили тем же способом, что и описанный в примере 18. Сумму оценки консистенции стула и оценки наличия скрытой крови определяют как оценка стула. От семи до 10 животных использовали для каждой группы и результаты представлены как среднее значение ± стандартное отклонение.
В результате, масса тела контрольной группы неизменно понемногу увеличивалась, но увеличение было значительно меньшим, чем масса тела у животных в нормальной группе. Величина оценки стула контрольной группы значительно повышалась с 1 суток приема DSS. В сутки проведения аутопсии (сутки 8), в контрольной группе наблюдалось явное повреждение ткани в толстом кишечнике и его значительное укорочение. В отличие от этого, при введении соединения F из расчета 1 мг/кг и 3 мг/кг показало значительную тенденцию к подавлению или значимый подавляющий эффект на эти процессы (Фиг. 3, 4, 5). Т.е., соединение F предотвращает развитие язвы в толстом кишечнике и нормализует функцию органов, что, тем самым, приводит к уменьшению симптомов диареи и появлению крови в стуле.
Пример 20
Терапевтический эффект на модель ремиссии/рецидива колита у мышей, вызванного декстраном сульфатом натрия
Затем, терапевтический эффект соединения F на колит исследовали на модели хронического колита. Приобрели самок мышей линии BALB/c, в возрасте 6 недель, массой тела приблизительно 20 г (Japan SLC), акклиматизировались в течение 1 недели и использовались в исследовании. Мышей разделили на группу мышей, у которых индуцировали колит, и нормальную группу. Группе с индуцированным колитом позволяли без ограничения пить раствор 2,6 масс./об.% DSS для индуцирования колита (MP Biochemicals, M.W. 36,000-50,000, Lot №. 4556J). На 8 сутки, когда показатель оценки стула у группы (охарактеризован в примере 18), в которой индуцировали колит, достиг приблизительно 4,5, мышей подразделили на контрольную группу, группу, которой ввели соединение F 1 мг/кг, и группу, которой ввели салазосульфапиридин 100 мг/кг (SIGMA, Lot №. 085K1930, далее в настоящем документе сокращен до SASP). Затем мышам позволили пить дистиллированную воду, взамен раствора DSS в течение 9 суток (период ремиссии). После разделения на группы, проводили оценку стула каждые 3-4 суток. Когда показатель оценки стула контрольной группы достигал приблизительно 1, мышам опять позволяли пить раствор DSS, чтобы спровоцировать рецидив (период рецидива). Периоды ремиссии и рецидива проводились 1 цикл и цикл повторяли 5 раз. Однако, что касается 5-го цикла, осуществляли только период ремиссии.
Соединение F в дозе 1 мг/кг и SASP в дозе 100 мг/кг перорально вводили один раз в сутки, каждые сутки, в течение 50 суток с начала инициации периода ремиссии (8 сутки с начала приема 2,6 масс./об.% DSS) до пятого периода ремиссии (57 сутки с начала приема 2,6 масс./об.% DSS). Контрольной группе перорально вводили раствор (1 об.% раствора этанола) из расчета 10 мл/кг. Если мышь набирала оценку 0 как при оценке консистенции стула, так и при оценке наличия скрытой крови в последние сутки периода ремиссии, мышь рассматривали как находящуюся «в периоде ремиссии». Степень ремиссии (%) рассчитывали как долю мышей в каждой группе, находящихся в периоде ремиссии. В каждой группе использовали от восьми до 10 мышей и результаты выражали в виде среднего значения.
Таким образом, показатель оценки стула контрольной группы увеличивался в период обострения и снижался в период ремиссии. Показатель оценки стула был значимо выше, чем показатель оценки стула нормальной группы, почти на всем протяжении периода исследования (фиг. 6). Степень ремиссии контрольной группы составляла 35,5% в среднем из 5 периодов ремиссии (таблица 4). Соединение F снижало показатель оценки стула в начале периода ремиссии и подавляло увеличение показателя оценки в периоде рецидива. Его степень ремиссии составила не менее чем 60% в любом периоде ремиссии, и среднее значение составило 66,0%, что составляло явно более высокое значение, чем степень ремиссии контрольной группы. С другой стороны, SASP не продемонстрировало явного действия на показатель оценки стула ни в период ремиссии, ни в период рецидива. Его степень ремиссии в 1, 3 и 4 циклах была немного выше, чем степень ремиссии контрольной группы, и, напротив, ниже во 2 и 5 циклах и среднее значение было эквивалентно степени ремиссии контрольной группы.
Как показано выше, соединение F обеспечивает не только профилактическое действие, но также терапевтическое действие, а также эффект поддержания ремиссии. Кроме того, полагают, что действие соединения F далеко превосходит SASP в клиническом применении.
Пример 21
Профилактическое действие переноса CD4 + CD25 - T-клеток на модель колита у мышей
Изучали действие на болезнь Крона, воспалительного заболевания кишечника другого вида. Модель переноса T-клеток является также известной как модель болезни Крона, при которой развивается хронический гастрит или энтерит (см.: ссылки F, G, H). Кроме того, ее также можно расценивать как модель болезни Бехчета с поражением желудочно-кишечного тракта или простой язвы на животных, страдающих от похожей желудочно-кишечной язвы, связанной с активацией T-клеток (см.: ссылки I, J).
Приобретали самок мышей линии BALB/cA Jcl, в возрасте 6 недель, с массой тела 19-23 г (CLEA Japan, Inc.) и самок мышей C.B-17/Icr-scid (возрас 6 недель, CLEA Japan, Inc.), акклиматизировали в течение 1 недели и использовали в исследовании.
После лапаротомии с анестезией простым эфиром, мышей линии BALB/cA Jcl обескровливали до наступления смерти, отбирая кровь из брюшной аорты и полой вены хвоста, и выделяли селезенку. Из селезенки выделяли спленоциты и затем получали CD4+CD25- T-клетки посредством набора для выделения CD4+ T-клеток (№. 130-090-860, Milky Biotech Co., Ltd.) и антитела CD25-Biotin (No. 130-092-569, Milky Biotech Co., Ltd.). Клетки разделяли, используя сепаратор autoMACS (Milky Biotech Co., Ltd.). Разделенные CD4+CD25- T-клетки суспендировали в физиологическом растворе фосфатного буфера и 2,5×105 клеток на животное вводили интраперитонеально мышам линии C.B-17/Icr-scid для индуцирования колита.
Один мг/кг соединения F или преднизолона вводили первоначально за 5 часов до переноса CD4+CD25- T-клеток и в дальнейшем вводили перорально один раз в сутки, каждые сутки, в течение 20 суток. Контрольной группе перорально вводили раствор (1 об.% раствора этанола) из расчета 10 мл/кг. Клинический результат представлял собой сумму показателей оценки консистенции стула (0-5), оценки наличия скрытой крови (0-4) и оценки снижения массы тела (0-5), именуемый как оценка индекса активности болезни (который, далее в настоящем документе сокращают как оценка DAI: самая высокая оценка 14). Оценку консистенции стула проводили, классифицируя стул по твердости как нормальный (0), незначительно неоформленный (1), в некоторой степени неоформленный (2), неоформленный (3), в значительной степени неоформленный (4) и диарея (5). Оценку присутствия скрытой крови в стуле проводили тем же способом, что и в примере 18. Оценку снижения массы тела классифицировали по степени изменения массы организма, как увеличение (0), снижение менее чем на 3% (1), снижение не менее чем на 3% и менее чем на 6% (2), снижение не менее чем на 6% и менее чем на 9% (3), снижение не менее чем на 9% и менее чем на 12% (4) и снижение не менее чем на 12% (5). От восьми до 10 мышей использовали для каждой группы и результаты выражали в виде среднего значения.
Таким образом, на 12 сутки после переноса T-клеток показано явное повышение величины оценки консистенции стула и оценки наличия скрытой крови в стуле контрольной группы, а на 19 сутки показано явное повышение величины оценки снижения масса тела, все величины достигли почти максимального значения спустя 21 сутки. Соединение F подавляло повышение как величины оценки консистенции стула, так и оценки наличия скрытой крови в стуле почти наполовину, как показано на фиг. 7 и 8, соответственно и почти полностью предотвратило повышение величины оценки снижения массы тела, как показано на фиг. 9. С другой стороны, несмотря на то, что преднизолон подавлял повышение величины оценки наличия скрытой крови в стуле почти на том же уровне, что и при введении соединения F, как показано на фиг. 8, это не позволило показать явное влияние на величину оценки консистенции стула на 21 сутки, как показано на фиг. 7. Кроме того, на протяжении периода исследования величина оценки снижения массы тела поддерживалась на более высоком уровне значений, чем величина оценки снижения массы в контрольной группе, как показано на фиг. 9, и преднизолон явно снижал величину оценки. Как показано на фиг. 10, оценка DAI показала, что соединение F превосходит преднизолон во всех отношениях.
Таким образом, соединение F может более эффективно, чем существующие лекарственные средства, облегчать состояние при болезни Крона, болезни Бехчета с поражением желудочно-кишечного тракта и при простой язве, а также язвенном колите.
Ссылки
F) Immunol Rev. 182: 190-200 (2001).
G) Int. Immunopharmacol. 6(8): 1341-1354 (2006).
H) J. Immunol. 160(3): 1212-1218 (1998).
I) Clin. Exp. Immunol. 139(2): 371-378 (2005).
J) Histopathology. 45(4): 377-383 (2004).
Пример 22
Влияние на повреждение слизистой желудка, вызванное этанолом, на модели у крыс
Подавляющее влияние соединения F на повреждение слизистой желудка исследовали на модели повреждения слизистой желудка, вызванного этанолом, у крыс. Эту модель часто использовали в качестве модели на животных острого гастрита человека, связанного с застойным повреждением слизистой оболочки (ссылка K).
Приобрели от Oriental BioService Inc. самцов крыс линии SD (в возрасте 7 недель, Charles River), акклиматизировали в течение 1 недели и брали в исследование. Крыс разделяли по группам в зависимости от массы тела, помещали в чистую клетку с дном из проволочной сетки за одни сутки до исследования, крысы голодали в течение 19 часов (без воды в течение последних 3 часов), и крысам всех групп перорально вводили этанол (особой чистоты, Nacalai Tesque, Lot №. V8A58 62, 1,5 мл) для индуцирования повреждения слизистой желудка. Соединение F перорально вводили в дозах 0,01, 0,1 и 1 мг/кг за 30 мин перед индукцией повреждения слизистой желудка в объеме 5 мл/кг. Контрольной группе перорально вводили раствор (1 об.% раствора этанола) из расчета 5 мл/кг, тем же способом. Восемь животных отбирали в каждую группу.
Крысы были обескровлены до наступления смерти, кровь забирали из брюшной аорты и хвостовой полой вены, под анестезией с простым эфиром, через 1 час после введения этанола, и выделили желудок. Выделенный желудок немедленно наполнили 2 об.% раствором нейтрального формалина (6 мл) и фиксировали в течение 15 мин. Желудок рассекали вдоль средней линии по большой кривизне от кардиальной части до пилорической части и располагали на доске из винилхлорида. Длину и ширину каждой язвы измеряли под стереомикроскопом, рассчитывали площадь и сумму площадей принимали за общую площадь язвы.
Таким образом, общая площадь язвы контрольной группы в среднем составляла 103 мм2. Соединение F значимо снизило общую площадь язвы зависимым от дозы образцов при 0,01 мг/кг, и практически полностью уменьшило площадь в дозе 1 мг/кг (фиг. 11). Таким образом, соединение F уменьшает повреждение слизистой желудка.
Ссылка
K) Dig Dis Sci. 31(2 Suppl), 81S-85S (1986).
Пример 23
Влияние на повреждение тонкого кишечника, вызванное индометацином, на модели у крыс
Подавляющее действие соединения F на повреждение тонкого кишечника исследовали, используя модель повреждения тонкого кишечника, вызванного индометацином, у крыс. Как известно, введение нестероидных противовоспалительных лекарственных средств (NSAID) вызывает геморрагическое повреждение в тонком кишечнике человека. Эта модель характеризуется повреждением слизистой оболочки тонкого кишечника, которое вызвано введением крысам NSAID, индометацина и демонстрирует патологический процесс, схожий с повреждением тонкого кишечника, вызванного NSAID или болезнью Крона у человек (ссылки L и M).
Приобретали самцов крысы SD, в возрасте 7 недель (Charles River), оставляли для акклиматизации в течение 1 недели и использовали в исследовании. Крыс разделяли по группам, в зависимости от массы тела и всем группам подкожно вводили индометацин (SIGMA, Lot №. 19F0018) из расчета 15 мг/5 мл/кг для индуцирования повреждения тонкого кишечника.
Соединение F в дозах 0,01, 0,1 и 1 мг/кг перорально вводили в объеме 5 мл/кг за 30 мин до и через 6 часов после подкожного введения индометацина. Контрольной группе тем же способом перорально вводили раствор (1 об.% раствора этанола) из расчета 5 мл/кг. Для каждой группы использовали восемь животных.
Крысам внутривенно ввели 2 мл 10 мг/мл голубого раствора Эванса под анестезией с простым эфиром через 23,5 часа после введения индометацина. Через 30 мин, крыс обескровливали до наступления смерти, отбирая кровь из брюшной аорты и полой вены хвоста под анестезией простым эфиром и выделяли тонкий кишечник. Выделенный тонкий кишечник заливали надлежащим количеством (приблизительно 35 мл) 2 об.% нейтрального раствора формалина и фиксировали приблизительно 15 мин. Далее, тонкий кишечник полностью рассекали вдоль участка присоединения брыжейки и растягивали на дощечке из винилхлорида. Длину и ширину каждой язвы измеряли под стереомикроскопом, рассчитывали площадь и сумму площадей язв принимали за общую площадь язвы.
Таким образом, в контрольной группе общая площадь язвы в тонком кишечнике составила приблизительно 730 мм2. Напротив, в группе, которой ввели соединение F, площадь язвы значительно уменьшалась зависимым от дозы способом, с величины дозы введения 0,1 мг/кг и язва практически полностью исчезла при величине дозы 1 мг/кг (фиг. 12). Таким образом, соединение F сильно снижает повреждение тонкого кишечника.
Ссылки
L) Aliment Pharmacol Ther. 7(1), 29-39 (1993).
M) Acta Gastroenterol Belg. 57(5-6), 306-309 (1994).
Из сказанного выше, соединение F продемонстрировало превосходное подавляющее действие на прямое поражение слизистой оболочки желудочно-кишечного тракта, вызванное воздействием алкоголя и т.п. и на нарушение регенерации слизистой оболочки из-за NSAID и т.п. Таким образом, как предполагают, при воздействии на пораженные участки слизистой оболочки желудочно-кишечного тракта соединение F демонстрирует защитное действие и эффект восстановления тканей.
Как показано в указанных выше примерах и обнаружено для соединения F, соединение по настоящему изобретению является эффективным соединением при поражении желудочно-кишечного тракта и при задержке в выздоровлении, из-за иммунного воспаления желудочно-кишечного тракта, поражения слизистой оболочки желудочно-кишечного тракта, обусловленного лекарственным средством, и нарушения регенерации слизистой оболочки, обусловленного лекарственным средством. Конкретно, соединение является полезным при воспалительном заболеванием кишечника, таком как язвенный колит и болезнь Крона, алкогольном гастрите или язве желудка, язве тонкого кишечника и т.п., и его польза не ограничивается перечисленными заболеваниями.
ПРИМЕНИМОСТЬ В ПРОИЗВОДСТВЕННЫХ УСЛОВИЯХ
Производное PGI2 по настоящему изобретению является полезным в качестве активного ингредиента лекарственных средств. Лекарственное средство, содержащее производное PGI2 по настоящему изобретению, в качестве активного ингредиента является полезным в качестве лекарственного средства для профилактики или лечения воспалительных заболеваний и язвенных заболеваниях желудочно-кишечного тракта. В частности, производное является полезным в качестве лекарственного средства для профилактики или лечения язвенного колита, болезни Крона, гастрита или язве желудка, язве тонкого кишечника.
Эта заявка основана на патентной заявке Японии №. 2008-232133, поданной 10 сентября 2008 года, и патентной заявке Японии №. 2009-168193, поданной 16 июля 2009 года и их содержание, описываемое в настоящем документе, которое включает описание, пункты формулы изобретения, фигуры и реферат, таким образом, в полном объеме включено в качестве ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ АГОНИСТ ЕР4 | 2011 |
|
RU2564414C2 |
| НОВЫЙ ИНГИБИТОР КИСЛОТНОЙ СЕКРЕЦИИ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2815697C1 |
| ПРЕПАРАТ ПРОТИВ Helicobacter pylori, ИНГИБИРУЮЩИЙ СЕКРЕЦИЮ ЖЕЛУДОЧНОГО СОКА | 2007 |
|
RU2451682C2 |
| АНТАГОНИСТЫ TRPV1, ВКЛЮЧАЮЩИЕ ДИГИДРОКСИГРУППУ В КАЧЕСТВЕ ЗАМЕСТИТЕЛЯ, И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2621708C2 |
| ПРОФИЛАКТИЧЕСКОЕ ИЛИ ТЕРАПЕВТИЧЕСКОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ЛЕЧЕНИЯ ЗАПОРА | 2014 |
|
RU2648467C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ КОЛИТА | 2010 |
|
RU2518416C2 |
| СПОСОБ И КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ПОВРЕЖДЕНИЙ СЛИЗИСТЫХ ОБОЛОЧЕК | 2006 |
|
RU2445961C2 |
| МОНОГИДРАТ 7-КАРБОКСИМЕТИЛОКСИ-3',4' 5-ТРИМЕТОКСИФЛАВОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2004 |
|
RU2302416C2 |
| СОЕДИНЕНИЕ ИМИДАЗОПИРИДИНА | 2005 |
|
RU2373206C2 |
| НОВОЕ ПРОИЗВОДНОЕ 5-ФТОРУРАЦИЛА | 2010 |
|
RU2503673C2 |
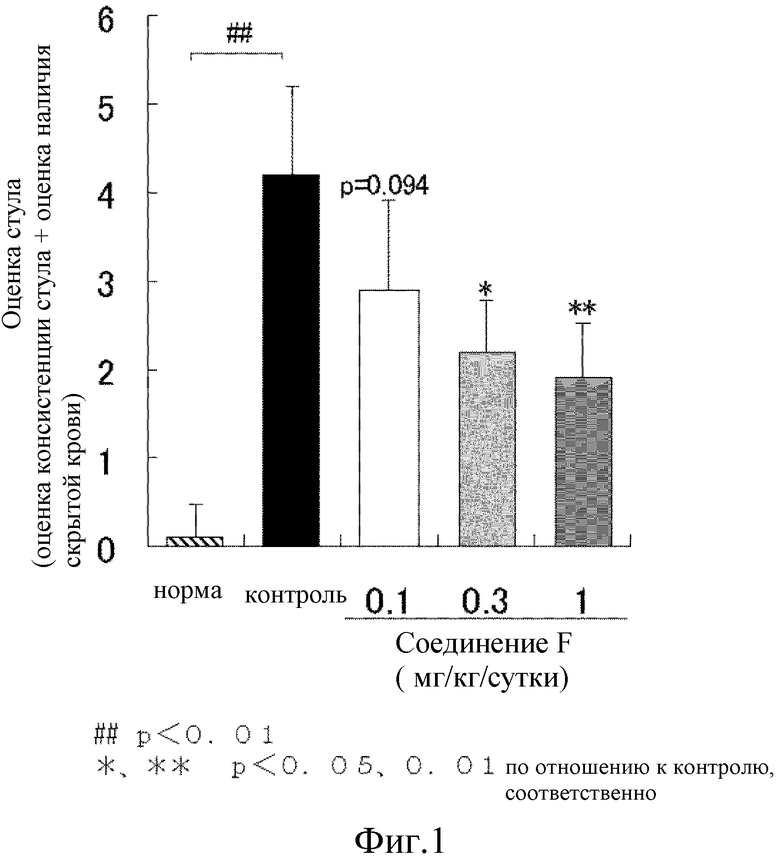
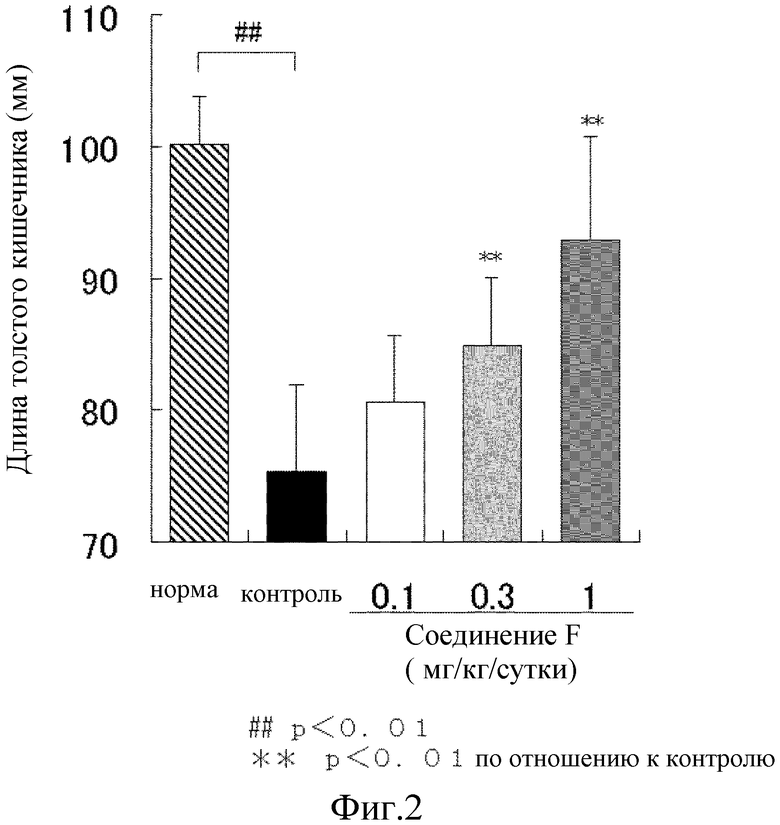
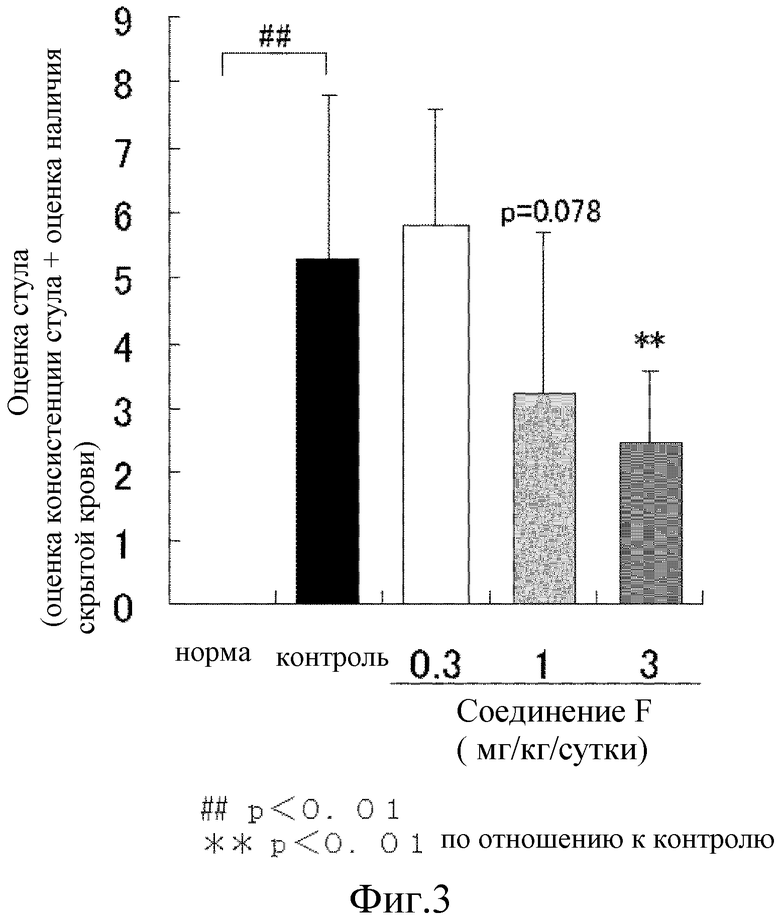
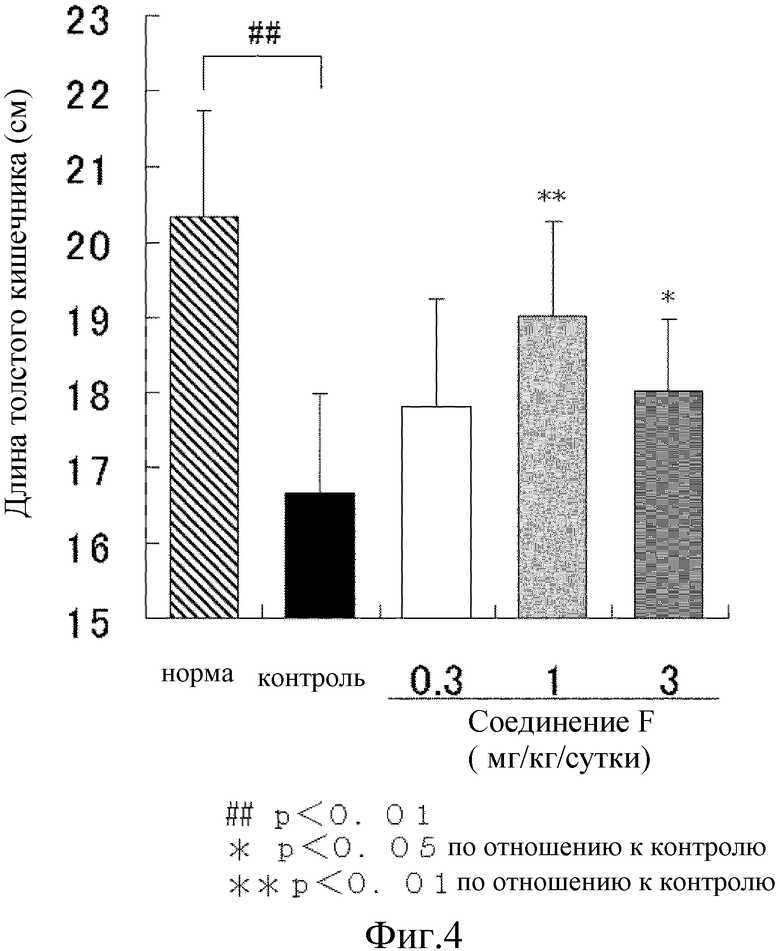
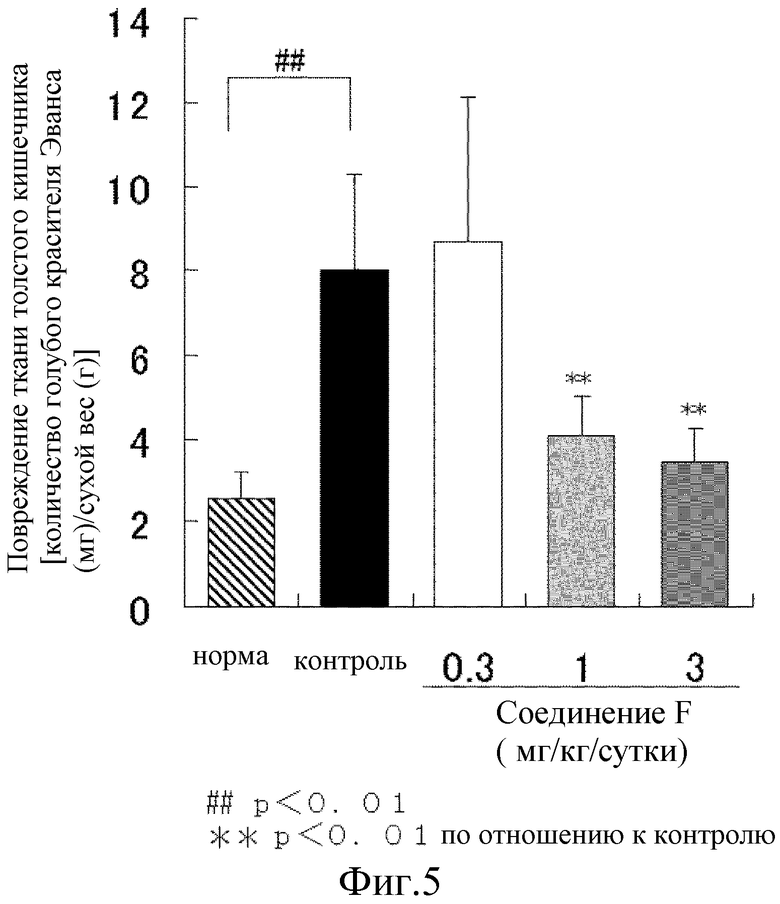
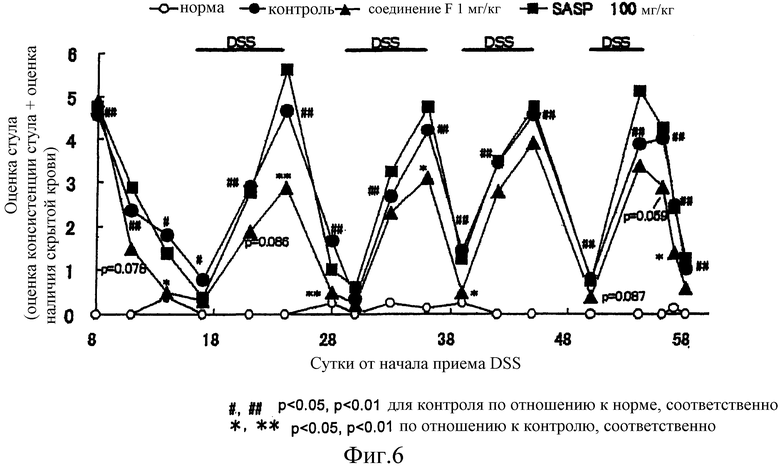
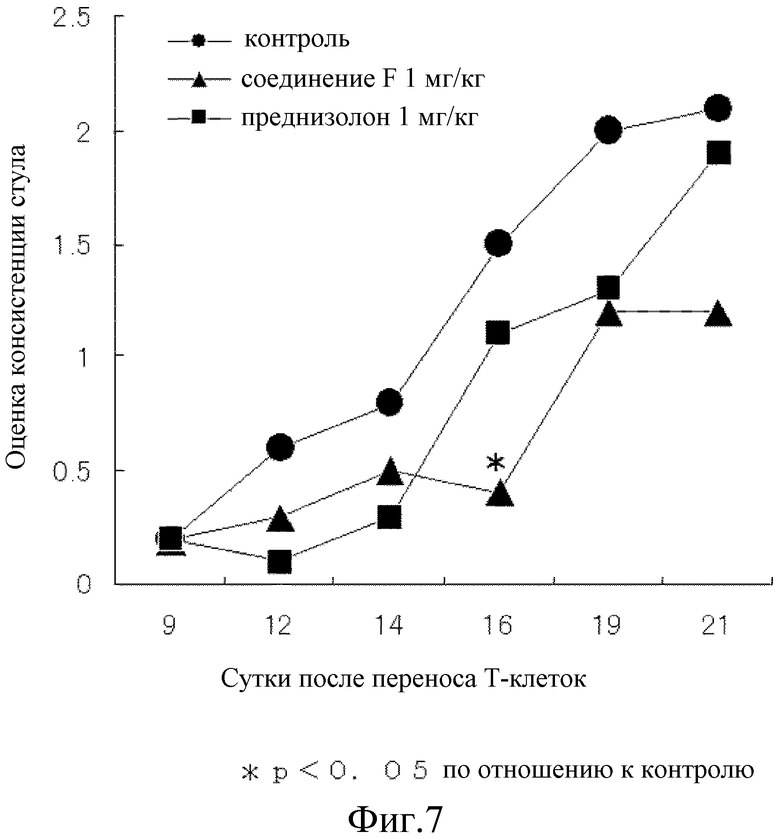
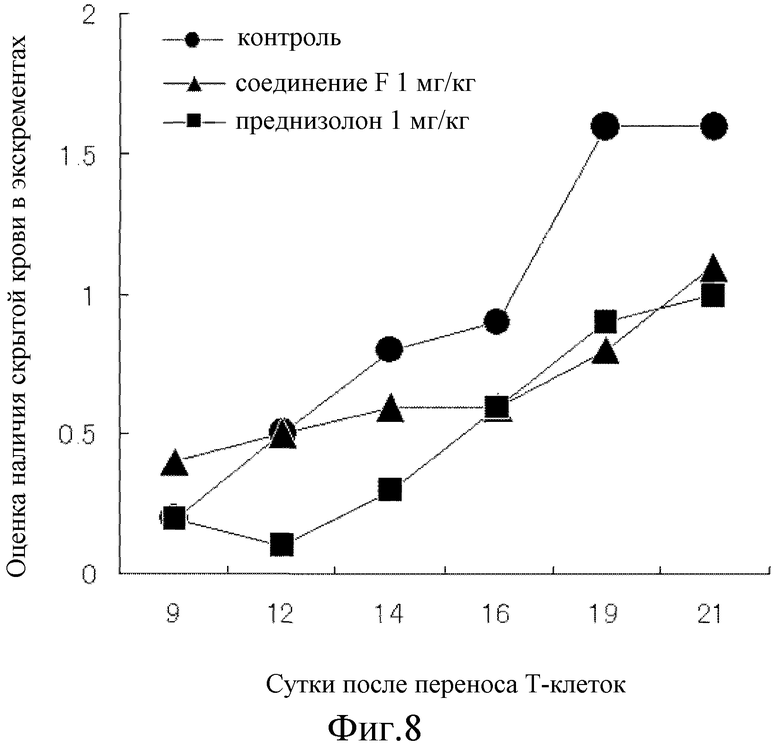
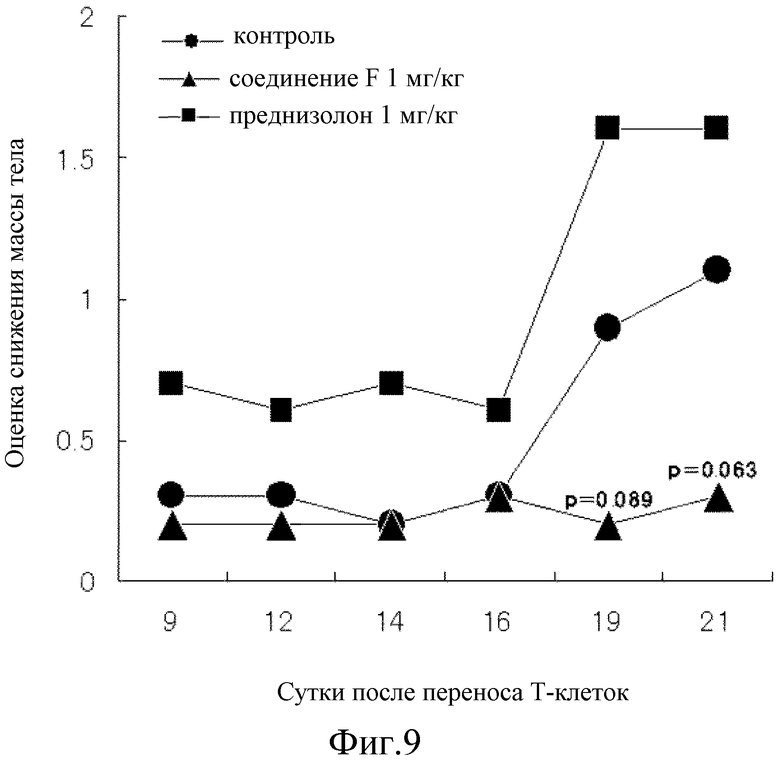
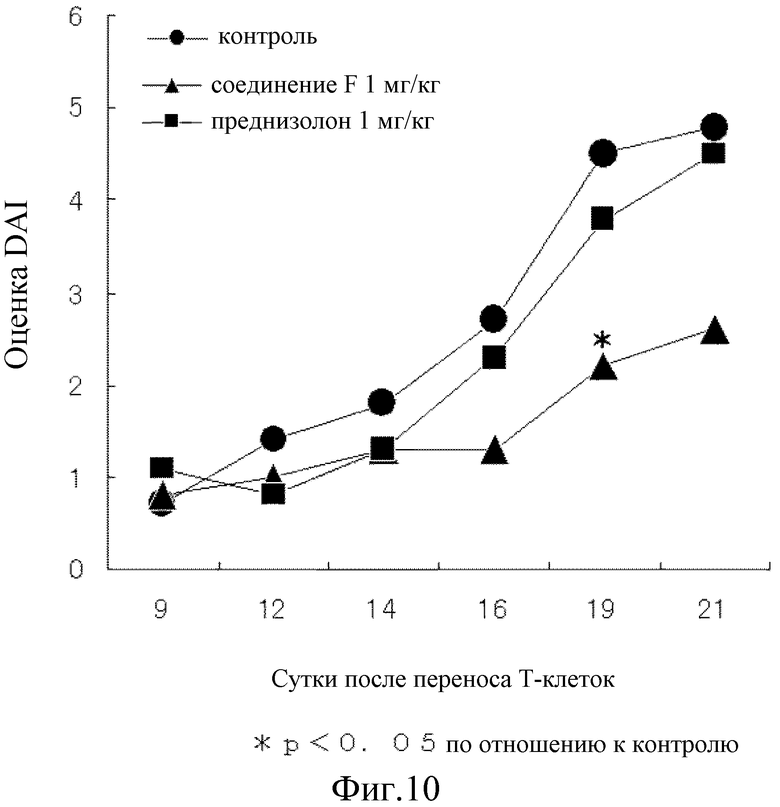
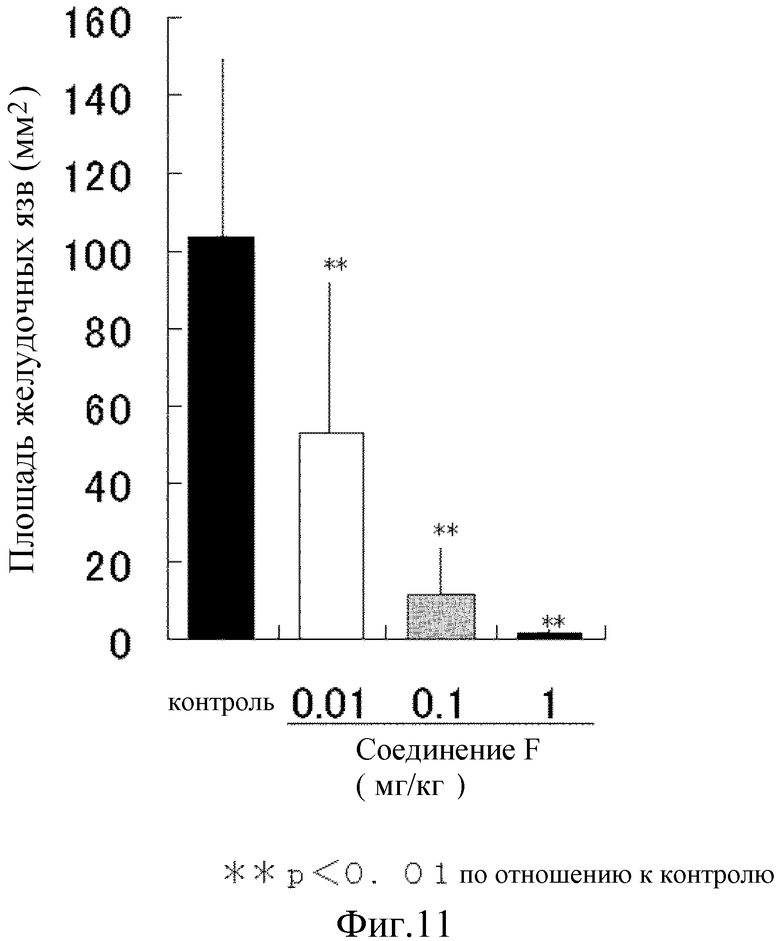
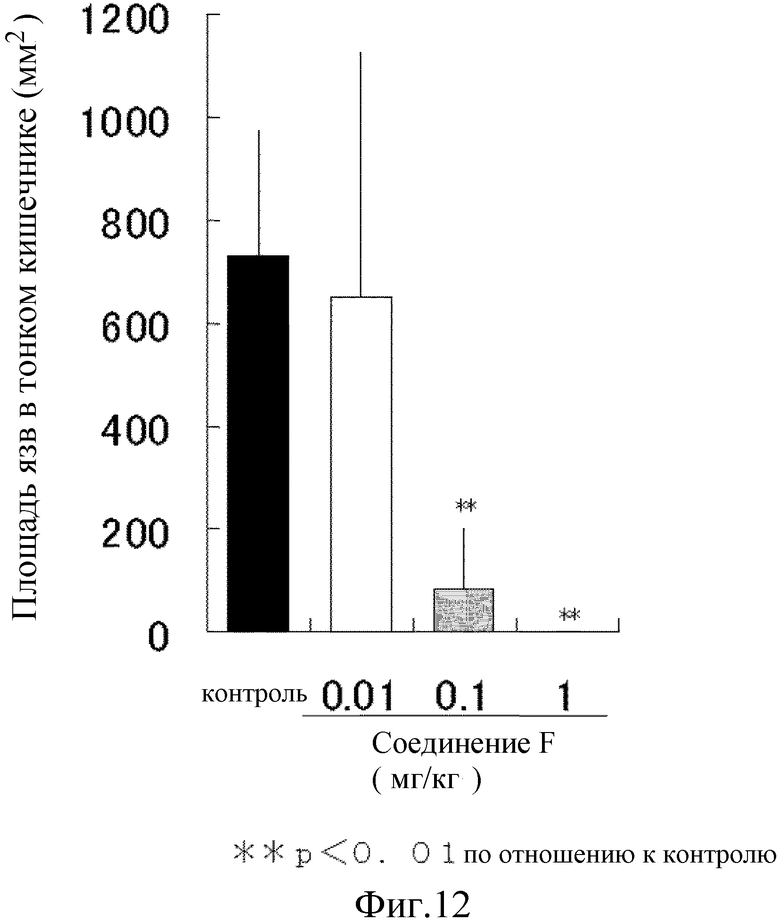
Данное изобретение относится к новому производному простагландина I2 или его фармацевтически приемлемой соли, конкретно к производному 7,7-дифтор-PGI2 (формула (1)), где R1 и R2 каждый независимо представляет собой атом водорода или алкильную группу с неразветвленной цепью, имеющей от 1 до 3 атомов углерода, и R3 представляет собой атом водорода, алкильную группу, имеющую от 1 до 4 атомов углерода, а также к лекарственному средству на основе соединений формулы 1 для лечения и профилактики различных заболеваний желудочно- кишечного тракта. 2н. и 20 з.п. ф-лы, 23 пр.,4 табл.,12 ил.
1. Производное простагландина I2, представленное формулой (1), или его фармацевтически приемлемая соль.
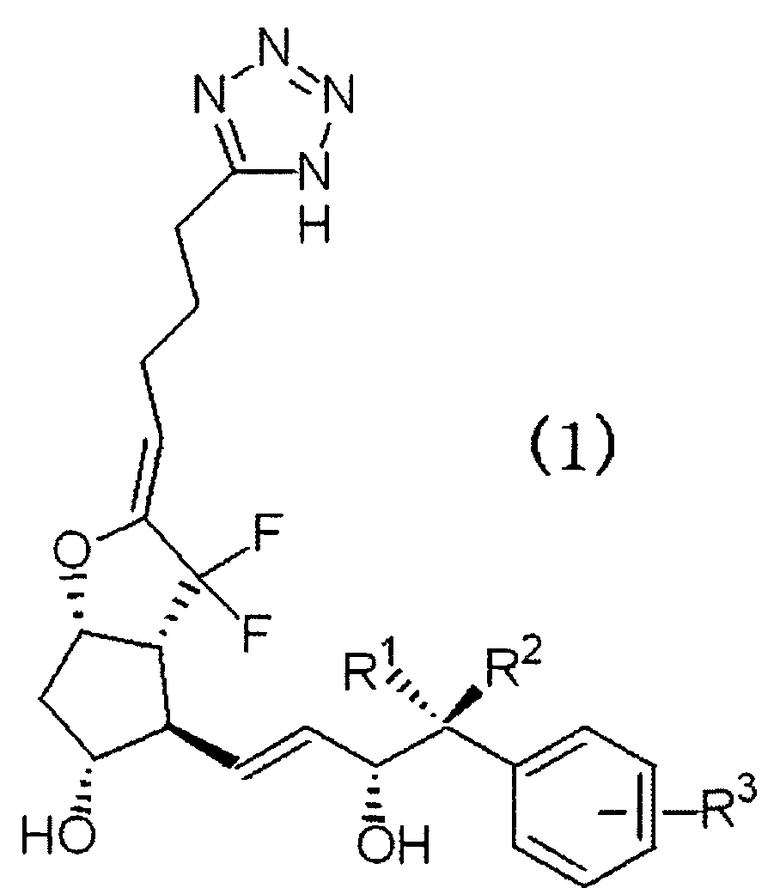
где R1 и R2 каждый независимо представляет собой атом водорода или алкильную группу с неразветвленной цепью, имеющей от 1 до 3 атомов углерода, и R3 представляет собой атом водорода или алкильную группу, имеющую от 1 до 4 атомов углерода.
2. Производное простагландина I2 по п.1, где R1 представляет собой метильную группу, или его фармацевтически приемлемая соль.
3. Производное простагландина I2 по п.1 или 2, где R3 представляет собой метильную группу, или его фармацевтически приемлемая соль.
4. Производное простагландина I2 по п.1, где R2 представляет собой атом водорода, или его фармацевтически приемлемая соль.
5. Производное простагландина I2 по п.1, где R1 представляет собой метильную группу и R2 представляет собой атом водорода, или его фармацевтически приемлемая соль.
6. Производное простагландина I2 по п.1, где R3 представляет собой m-метильную группу, или его фармацевтически приемлемая соль.
7. Производное простагландина I2 по п.1, где R1 представляет собой метильную группу, R2 представляет собой атом водорода и R3 представляет собой метильную группу, или его фармацевтически приемлемая соль.
8. Производное простагландина I2 по п.1, где R1 представляет собой атом водорода, R2 представляет метильную группу и R3 представляет собой метильную группу, или его фармацевтически приемлемая соль.
9. Лекарственное средство для профилактики или лечения заболевания желудочно-кишечного тракта, где лекарственное средство содержит в качестве активного ингредиента производное простагландина I2 по любому из пп.1-8 или его фармацевтически приемлемую соль.
10. Лекарственное средство по п.10, где заболевание желудочно-кишечного тракта представляет собой воспалительное заболевание желудочно-кишечного тракта.
11. Лекарственное средство по п.10, где воспалительное заболевание желудочно-кишечного тракта представляет собой воспалительное заболевание кишечника.
12. Лекарственное средство по п.11, где воспалительное заболевание кишечника представляет собой язвенный колит или болезнь Крона.
13. Лекарственное средство по п.11, где воспалительное заболевание кишечника представляет собой болезнь Бехчета с поражением желудочно-кишечного тракта или простую язву.
14. Лекарственное средство по п.9, где заболевание желудочно-кишечного тракта представляет собой язвенное заболевание желудочно-кишечного тракта.
15. Лекарственное средство по п.14, где язвенное заболевание желудочно-кишечного тракта представляет собой гастрит или язву желудка.
16. Лекарственное средство по п.15, где гастрит или язва желудка представляют собой гастрит или язву желудка, обусловленные лекарственным средством.
17. Лекарственное средство по п.16, где гастрит или язва желудка, обусловленные лекарственным средством, обуславливаются нестероидным противовоспалительным лекарственным средством.
18. Лекарственное средство по п.15, где гастрит или язва желудка обусловлены приемом алкоголя.
19. Лекарственное средство по п.14, где язвенное заболевание желудочно-кишечного тракта представляет собой язву тонкого кишечника.
20. Лекарственное средство по п.19, где язва тонкого кишечника представляет собой язву тонкого кишечника, обусловленную лекарственным средством.
21. Лекарственное средство по п.20, где язва тонкого кишечника обусловлена нестероидным противовоспалительным лекарственным средством.
22. Лекарственное средство по п.19, где язва тонкого кишечника обусловлена приемом алкоголя.
| Способ измерения временных интервалов между двумя точками синхронизированных сигналов с заданными уровнями сигнала в этих точках | 1977 |
|
SU669329A1 |
| 0 |
|
SU351350A1 | |
| БИБЛИОТЕКА ii | 0 |
|
SU351359A1 |
| Konturek S.J | |||
| et al, Prostaglandins, 1984, v.28, no.4, p.443-453 | |||
| АНАЛОГИ ПРОСТАГЛАНДИНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СЕЛЕКТИВНОЙ АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА EP | 2002 |
|
RU2288913C2 |
| ПРОИЗВОДНОЕ 8-АЗАПРОСТАГЛАНДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, АГЕНТ ДЛЯ ПРОФИЛАКТИКИ ЗАБОЛЕВАНИЙ | 2003 |
|
RU2306309C2 |

