РОДСТВЕННЫЕ ПАТЕНТНЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет предварительной заявки на патент Индии №2424/MUM/2011, поданной 30 августа 2011 г., описание которой полностью включено в настоящую заявку путем ссылки, как если бы она была полностью переписана в ней. Все ссылки, включая патенты, патентные заявки и библиографические ссылки, приведенные в описании, полностью специально включены в настоящее описание путем ссылки.
ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к азотсодержащим гетероциклическим соединениям, их получению и их применению при предотвращении и/или лечении инфекций.
УРОВЕНЬ ТЕХНИКИ
Появление бактериальной устойчивости к известным антибактериальным средствам становится основной проблемой при лечении бактериальных инфекций. Одним путем направления на лечение бактериальных инфекций и, в частности, тех, которые вызваны устойчивыми бактериями, является разработка новых антибактериальных средств, которые могут преодолеть бактериальную устойчивость. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) провели обзор новых подходов к разработке новых антибиотиков. Однако разработка новых антибактериальных средств является проблематичной задачей. Например, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) провели обзор проблем при разработке антибактериальных средств.
В уровне техники были описаны несколько антибактериальных средств (например, см. заявки на международные патенты PCT №№ PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178 и PCT/US2009/041200). Однако остается потребность в активных антибактериальных средствах для предотвращения и/или лечения бактериальных инфекций, включая те, которые вызваны бактериями, устойчивыми к известным антибактериальным средствам.
Заявители, к удивлению, обнаружили азотсодержащие гетероциклические соединения с антибактериальными свойствами.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Соответственно, настоящее изобретение относится к азотсодержащим гетероциклическим соединениям, способам получения этих соединений, фармацевтическим композициям, содержащим эти соединения, и способам предотвращения или лечения бактериальной инфекции у субъекта с использованием этих соединений.
В одном общем аспекте изобретение относится к соединениям формулы (I):
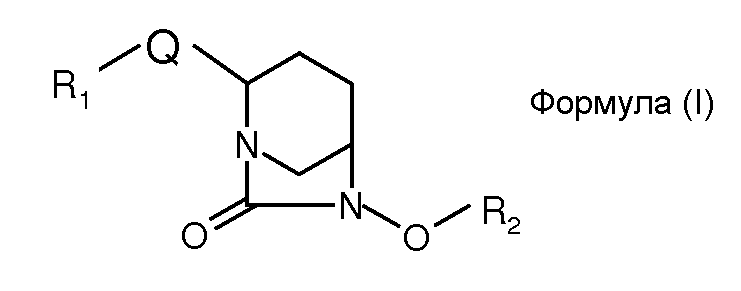
или их стереоизомеру или фармацевтически приемлемой соли; где:
Q обозначает гетероарил;
R1 обозначает:
(a) водород,
(b) (CO)n-R3 или
(c) COOR4,
n=0, 1 или 2;
R2 обозначает:
(a) SO3M,
(b) SO2NH2,
(c) PO3M,
(d) CH2COOM,
(e) CF2COOM,
(f) CHFCOOM или
(g) CF3;
M обозначает водород или катион;
R3 обозначает:
(a) водород,
(b) C1-C6-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, OR5, CN, COOR5, CONR6R7, NR6R7, NR5COR8, NR5CONR6R7, гетероциклила, гетероарила, циклоалкила или арила,
(c) CN,
(d) NR6R7,
(e) CONR6R7,
(f) NHCONR6R7,
(g) арил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(h) гетероциклил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(i) гетероарил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(j) циклоалкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(k) циклоалкил, необязательно замещенный C1-C6-алкилом, причем C1-C6-алкил дополнительно замещен одним или несколькими заместителями, независимо выбранными из OR5, NR6R7, галогена, CN или CONR6R7, или
(l) OR8;
R4 обозначает:
(a) водород,
(b) C1-C6-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, OR5, CN, COOR5, CONR6R7, NR6R7, NR5COR8, гетероциклила, гетероарила, циклоалкила или арила,
(c) арил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(d) гетероциклил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(e) гетероарил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7, или
(f) циклоалкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7;
R5 и R8, каждый независимо, обозначают:
(a) водород или
(b) C1-C6-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, CN, CONR6R7, NR6R7, гетероциклила, гетероарила, циклоалкила или арила;
R6 и R7, каждый независимо, обозначают:
(a) водород,
(b) C1-C6-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, OR5, CN, COOR5, CONR5R8, NR5R8, NR5COR8, гетероциклила, гетероарила, циклоалкила или арила,
(c) арил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR5R8, галогена, CN, CONR5R8, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR5R8,
(d) гетероциклил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR5R8, галогена, CN, CONR5R8, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR5R8,
(e) гетероарил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR5R8, галогена, CN, CONR5R8, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR5R8,
(f) циклоалкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR5R8, галогена, CN, CONR5R8, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR5R8, или
(g) R6 и R7 соединены вместе для образования от четырех- до семичленного кольца.
В другом общем аспекте изобретение относится к фармацевтическим композициям, включающим соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль.
В другом общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли.
В другом общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли.
В другом общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль.
В еще одном общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль.
В другом общем аспекте изобретение относится к фармацевтические композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль и (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
В другом общем аспекте изобретение относится к фармацевтические композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль и (b) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В другом общем аспекте изобретение относится к фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (c) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В другом общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль и (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
В еще одном общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, и (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
В другом общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, и (b) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В еще одном общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, и (b) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В другом общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (c) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В еще одном общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (c) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В другом общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
В еще одном общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
В другом общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или их стереоизомера или фармацевтически приемлемой соли, и (b) по меньшей мере одного антибактериального средства или его фармацевтически приемлемой соли.
В еще одном общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного антибактериального средства или его фармацевтически приемлемой соли.
В другом общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (c) по меньшей мере одного антибактериального средства или его фармацевтически приемлемой соли.
В еще одном общем аспекте изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (c) по меньшей мере одного антибактериального средства или его фармацевтически приемлемой соли.
В другом общем аспекте изобретение относится к способам увеличения антибактериальной эффективности антибактериального средства у субъекта, причем указанный способ включает совместное введение указанного антибактериального средства или его фармацевтически приемлемого производного с фармацевтически эффективным количеством соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли.
Более подробно один или более варианты осуществления изобретения изложены ниже в описании. Другие признаки, цели и преимущества изобретения станут очевидными из следующего описания, включая формулу изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Теперь будут описаны иллюстративные варианты осуществления изобретения, и для их описания будут использоваться конкретные формулировки. Тем не менее, следует понимать, что они не предназначены для ограничения объема изобретения. Изменения и другие модификации признаков изобретения и дополнительные применения иллюстрируемых в настоящем описании принципов, которые возникли бы у специалиста в данной области и относятся к настоящему описанию, должны рассматриваться как включенные в объем изобретения. Необходимо отметить, что пока содержание ясно не требует иного, используемые в настоящем описании и прилагаемой формуле изобретения формы единственного числа включают их соответствующие формы множественного числа. Все ссылки, включая патенты, патентные заявки и библиографические источники, приведенные в описании, специально полностью включены в него путем ссылки.
Заявители, к удивлению, обнаружили новые азотсодержащие гетероциклические соединения, имеющие антибактериальные свойства.
Используемый в настоящем описании термин «C1-C6-алкил» относится к разветвленному или неразветвленному ациклическому углеводородному радикалу с 1-6 атомами углерода. Обычно неограничивающие примеры «C1-C6-алкила» включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, изопентил, н-гексил и тому подобные. «C1-C6-алкил» может быть незамещенным или замещенным одним или несколькими заместителями. Типичные неограничивающие примеры таких заместителей включают галоген, алкокси, CN, COOH, CONH2, OH, -NH2, -NHCOCH3, циклоалкил, гетероциклил, гетероарил, арил и тому подобные.
Используемый в настоящем описании термин «циклоалкил» относится к от трех- до семичленным циклическим углеводородным радикалам. Циклоалкильная группа необязательно включает одну или несколько двойных или тройных связей или комбинацию двойных связей и тройных связей, но которая не является ароматической. Типичные неограничивающие примеры циклоалкильных групп включают циклопропан, циклобутан, циклопентан, циклогексан и циклогептан. Циклоалкил может быть незамещенным или замещенным одним или несколькими заместителями. Типичные неограничивающие примеры таких заместителей включают C1-C6-алкил, галоген, алкокси, CN, COOH, CONH2, OH, NH2, NHCOCH3, гетероциклил, гетероарил, арил, SO2-алкил, SO2-арил, OSO2-алкил, -OSO2-арил и тому подобные.
Используемый в настоящем описании термин «гетероциклил» относится к от четырех- до семичленной циклоалкильной группе, содержащей один или несколько гетероатомов, выбранных из азота, кислорода или серы. Гетероциклоалкильная группа необязательно включает одну или несколько двойных или тройных связей или комбинацию двойных связей или тройных связей, но которая не является ароматической. Типичные неограничивающие примеры гетероциклоалкильных групп включают азетидин, пирролидин, 2-оксо-пирролидин, имидазолидин-2-он, пиперидин, оксазин, тиазин, пиперазин, пиперазин-2,3-дион, морфолин, тиаморфолин, азапан и тому подобные. Гетероциклоалкил может быть незамещенным или замещенным одним или несколькими заместителями. Типичные неограничивающие примеры таких заместителей включают C1-C6-алкил, галоген, алкокси, CN, COOH, CONH2, OH, NH2, NHCOCH3, гетероциклил, гетероарил, арил, SO2-алкил, SO2-арил, OSO2-алкил, OSO2-арил и тому подобные.
Используемый в настоящем описании термин «арил» относится к моноциклическому или полициклическому ароматическому углеводороду. Типичные неограничивающие примеры арильных групп включают фенил, нафтил, антраценил, флуоренил, фенантренил и тому подобные. Арильная группа может быть незамещенной или замещенной одним или несколькими заместителями. Типичные неограничивающие примеры таких заместителей включают C1-C6-алкил, галогена, алкокси, CN, COOH, CONH2, OH, NH2, NHCOCH3, гетероциклил, гетероарил, арил, SO2-алкил, SO2-арил, OSO2-алкил, OSO2-арил и тому подобные.
Используемый в настоящем описании термин «гетероарил» относится к моноциклической или полициклической ароматической углеводородной группе, где один или несколько атомов углерода были замещены гетероатомами, выбранными из азота, кислорода и серы. Если гетероарильная группа содержит более чем один гетероатом, то гетероатомы могут быть одинаковыми или различными. Типичные неограничивающие примеры гетероарильных групп включают 1,2,4-оксадиазол, 1,3,4-оксадиазол, 1,3,4-тиадиазол, 1,2,3,4-тетразол, 1,3-оксазол, 1,3-тиазол, пиридин, пиримидин, пиразин, пиридазин, фуран, пиррол, тиофен, имидазол, пиразол, бензофуран, бензотиофен, бензимидазол, бензоксазол, бензотиазол, тиазол и тому подобные. Гетероарильная группа может быть незамещенной или замещенной одним или несколькими заместителями. Типичные неограничивающие примеры таких заместителей включают C1-C6-алкил, галоген, алкокси, CN, COOH, CONH2, OH, NH2, NHCOCH3, гетероциклил, гетероарил, арил, SO2-алкил, SO2-арил, OSO2-алкил, OSO2-арил и тому подобные.
Используемый в настоящем описании термин «стереоизомеры» относится к соединениям, которые имеют идентичный химический состав, но отличаются в отношении расположения их атомов или групп в пространстве. Соединения формулы (I) могут содержать асимметричные или хиральные центры и, поэтому, существуют в различных стереоизомерных формах. Предполагается, что пока нет иных определений, все стереоизомерные формы соединения формулы (I), а также их смеси, включая рацемические смеси, составляют часть настоящего изобретения. Кроме того, настоящее изобретение включает все геометрические и позиционные изомеры (включая цис- и транс-формы), а также их смеси включены в объем изобретения. В целом, ссылка на соединение предназначена для охвата его стереоизомеров и смесей различных стереоизомеров.
Используемый в настоящем описании термин «необязательно замещенные» означает, что замещение является необязательным и поэтому включает и незамещенные, и замещенные атомы и части. «Замещенный» атом или часть указывает на то, что любой водород на обозначенном атоме или части может быть замещен выбором из указанной группы заместителей, при условии, что не превышается нормальная валентность обозначенного атома или части, и что замещение приводит к получению устойчивого соединения.
Используемый в настоящем описании термин «фармацевтически приемлемая соль» относится к одной или нескольким солям данного соединения, которые обладают желательной фармакологической активностью свободного соединения, и которые не являются ни биологически, ни иным образом нежелательными. В целом, «фармацевтически приемлемые соли» относятся к солям, которые подходят для применения в контакте с тканями человека и животных без нежелательной токсичности, раздражения, аллергической реакции и тому подобного, и соответствуют приемлемому соотношению выгоды/риска. Фармацевтически приемлемые соли хорошо известны в данной области. Например, в публикации S.M. Berge, et al. (J. Pharmaceutical Sciences, 66: 1-19 (1977)), полностью включенной в настоящее описание путем ссылки, подробно описаны различные фармацевтически приемлемые соли.
В целом, соединения в соответствии с изобретением содержат основные (например, атомы азота), а также кислотные фрагменты (например, соединения формулы (I), где M обозначает водород). Специалисту в данной области понятно, что такие соединения, поэтому, могут образовывать кислотные соли (образованные с неорганическими и/или органическими кислотами), а также основные соли (образованные с неорганическими и/или органическими основаниями). Такие соли могут быть получены с использованием процедур, описанных в данной области. Например, основная часть может быть превращена в ее соль обработкой соединения подходящим количеством кислоты. Типичные неограничивающие примеры таких подходящих кислот включают хлористоводородную кислоту, трифторуксусную кислоту, метансульфоновую кислоту и тому подобные. Альтернативно, кислотная часть может быть превращена в ее соль обработкой подходящим основанием. Типичные неограничивающие примеры таких оснований включают карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или тому подобные. В случае соединений, содержащих более чем одну функциональные группы, способные превращаться в соль, каждая такая функциональная группа может быть независимо превращена в соль. Например, в случае соединений, содержащих два основных атома азота, один основный азот может образовывать соль с одной кислотой, тогда как другой основный азот может образовывать соль с другой кислотой. Некоторые соединения в соответствии с изобретением содержат и кислотные, а также основные части и, таким образом, могут образовывать внутренние соли или соответствующие цвиттерионы. В целом, предусматривается, что все фармацевтически приемлемые солевые формы соединений формулы (I) в соответствии с изобретением, включая кислотно-аддитивные соли, основно-аддитивные соли, цвиттерионы или тому подобные, входят в объем настоящего изобретения и в целом именуются фармацевтически приемлемыми солями.
Используемый в настоящем описании термин «галоген» или «галоид» относится к хлору, брому, фтору и иоду.
Используемый в настоящем описании термин «инфекция» или «бактериальная инфекция» включает присутствие бактерий в организме или на теле субъекта, ингибирование роста которых было бы благоприятно для субъекта. В сущности, используемый в настоящем описании термин «инфекция», в дополнение к ссылке на присутствие бактерий, также относится к нормальной флоре, которая нежелательна. Используемый в настоящем описании термин «инфекция» включает инфекцию, вызванную бактериями.
Используемый в настоящем описании термин «лечить» или «лечение» относится к введению лекарственного препарата, включая фармацевтическую композицию или один или несколько фармацевтически активных ингредиентов, в профилактических и/или терапевтических целях. Используемый в настоящем описании термин «профилактическое лечение» относится к лечению субъекта, у которого еще нет инфекции, но который восприимчив или иным образом подвержен риску инфекции (предотвращение бактериальной инфекции). Используемый в настоящем описании термин «терапевтическое лечение» относится к назначению лечения субъекту, уже имеющему инфекцию. Используемый в настоящем описании термин «лечить» или «лечение» также относится к введению композиций или одного или нескольких фармацевтически активных ингредиентов, описанных в настоящем описании, с или без дополнительных фармацевтически активных или инертных ингредиентов для: (i) уменьшения или устранения бактериальной инфекции или одного или нескольких симптомов бактериальной инфекции, или (ii) задержки прогрессирования бактериальной инфекции или одного или нескольких симптомов бактериальной инфекции, или (iii) уменьшения тяжести бактериальной инфекции или одного или нескольких симптомов бактериальной инфекции, или (iv) подавления клинического проявления бактериальной инфекции, или (v) подавления побочных симптомов бактериальной инфекции.
Используемый в настоящем описании термин «фармацевтически эффективное количество» или «терапевтически эффективное количество» или «эффективное количество» относится к количеству, которое оказывает терапевтический эффект или представляет собой количество, требуемое для получения терапевтического эффекта у субъекта. Например, терапевтически или фармацевтически эффективное количество антибактериального средства или фармацевтической композиции представляет собой количество антибактериального средства или фармацевтической композиции, требуемое для получения желательного терапевтического эффекта, о котором можно судить по результатам клинического испытания, исследований на модели инфекции у животных и/или исследований in vitro (например, на агаровых или бульонных средах). Фармацевтически эффективное количество зависит от нескольких факторов, включая без ограничения вовлеченный микроорганизм (например, бактерию), характеристики субъекта (например, рост, массу тела, пол, возраст и медицинский анамнез), тяжесть инфекции и конкретный тип применяемого антибактериального средства. Для профилактического лечения терапевтически или профилактически эффективное количество представляет собой то количество, которое было бы эффективным при предотвращении микробной (например, бактериальной) инфекции.
Используемый в настоящем описании термин «введение» включает подачу субъекту композиции или одного или нескольких фармацевтически активных ингредиентов, включая, например, подачу любым целесообразным способом, который служит для доставки композиции или ее активных ингредиентов или других фармацевтически активных ингредиентов в участок инфекции. Способ введения может варьироваться в зависимости от различных факторов, таких как, например, компоненты фармацевтической композиции или природа фармацевтически активных или инертных ингредиентов, участка потенциальной или действительной инфекции, вовлеченного микроорганизма, тяжести инфекции, возраста и физического состояния субъекта и тому подобного. Некоторые неограничивающие примеры путей введения композиции или фармацевтически активного ингредиента субъекту в соответствии с настоящим изобретением включают пероральное, внутривенное, топическое, интрареспираторное, внутрибрюшинное, внутримышечное, парентеральное, сублингвальное, трансдермальное, интраназальное, аэрозольное, внутриглазное, интратрахеальное, подоболочечное, вагинальное введение, введение с использованием генной пушки, трансдермальной системы, глазных капель, ушных капель или композиции для полоскания ротовой полости. В случае фармацевтической композиции, включающей несколько ингредиентов (активных или инертных), один путь введения такой композиции представляет собой смешивание ингредиентов (например, в виде подходящей стандартной лекарственной формы, такой как таблетка, капсула, раствор, порошок и тому подобные) и затем введение лекарственной формы. Альтернативно, ингредиенты могут также вводиться отдельно (одновременно или друг за другом), пока эти ингредиенты достигают благоприятного терапевтического эффекта, так что композиция в целом обеспечивает синергический и/или желательный эффект.
Используемый в настоящем описании термин «рост» относится к росту одного или нескольких микроорганизмов и включает репродукцию или распространение популяции микроорганизма (например, бактерии). Термин также включает поддержание продолжающихся метаболических процессов микроорганизма, включая процессы, которые сохраняют жизнеспособность микроорганизма.
Используемый в настоящем описании термин «эффективность» относится к способности лечения или композиции или одного или нескольких фармацевтически активных ингредиентов вызывать желательный биологический эффект у субъекта. Например, используемый в настоящем описании термин «антибактериальная эффективность» композиции или антибактериального средства относится к способности композиции или антибактериального средства предотвратить или вылечить микробную (например, бактериальную) инфекцию у субъекта.
Используемый в настоящем описании термин «синергический» или «синергия» относится к взаимодействию двух или более средств с тем, чтобы их комбинированный эффект был больше, чем их отдельные эффекты.
Используемый в настоящем описании термин «антибактериальное средство» относится к любому веществу, соединению или комбинации веществ, способных: (i) ингибировать, уменьшать или предотвращать рост бактерий; (ii) ингибировать или уменьшать способность бактерий вызвать инфекцию у субъекта или (iii) ингибировать или уменьшать способность бактерий к размножению или сохранению инфекционной способности в окружающей среде. Используемый в настоящем описании термин «антибактериальное средство» также относится к соединениям, способным уменьшать инфекционную способность или вирулентность бактерий.
Используемый в настоящем описании термин «бета-лактамовое антибактериальное средство» относится к соединениям с антибактериальными свойствами и содержащим бета-лактамовое ядро в их молекулярной структуре.
Используемый в настоящем описании термин «бета-лактамаза» относится к любому ферменту или белку или любому другому субстрату, который разрушает кольцо бета-лактама. Используемый в настоящем описании термин «бета-лактамаза» включает ферменты, которые продуцируются бактериями и обладают способностью гидролизировать кольцо бета-лактама в бета-лактамовом соединении или частично, или полностью.
Используемый в настоящем описании термин «ингибитор бета-лактамазы» относится к соединению, способному ингибировать активность одного или нескольких ферментов бета-лактамазы или частично, или полностью.
Используемый в настоящем описании термин «фармацевтически инертный ингредиент», или «носитель», или «эксципиент» относится к соединению или материалу, используемому для содействия введению соединения, включая, например, увеличение растворимости соединения. Типичные неограничивающие примеры твердых носителей включают крахмал, лактозу, дикальций фосфат, сахарозу и каолин и т.д. Типичные неограничивающие примеры жидких носителей включают стерильную воду, солевой раствор, буферы, неионные поверхностно-активные вещества и пищевые масла, такие как масло, арахисовое и кунжутное масла и т.д. Кроме того, могут быть включены различные адъюванты, обычно используемые в данной области. Эти и другие такие соединения описаны в литературе, например, в указателе Merck Index (Merck & Company, Rahway, N.J.). Соображения для включения различных компонентов в фармацевтические композиции описаны, например, в руководстве Gilman et al. (Eds.) (1990); Goodman and Gilman's: The Pharmacological Basis of Therapeutics, 8th Ed., Pergamon Press., которое полностью включено в настоящее описание путем ссылки.
Используемый в настоящем описании термин «субъект» относится к позвоночному или беспозвоночному, включая млекопитающее. Используемый в настоящем описании термин «субъект» включает человека, животное, птицу, рыбу или амфибию. Типичные неограничивающие примеры «субъекта» включают людей, кошек, собак, лошадей, овец, быков, коров, свиней, баранов, крыс, мышей и морских свинок.
Используемый в настоящем описании термин «фамацевтически приемлемое производное» включает и относится к любому из фармацевтически приемлемых солей, пролекарств, метаболитов, сложных эфиров, простых эфиров, гидратов, полиморфов, сольватов, комплексов, энантиомеров или аддуктов соединений, описанных в настоящей заявке, которое после введения субъекту способно обеспечить (прямо или опосредованно) образование материнского соединения. Например, используемый в настоящем описании термин «антибактериальное средство или его фамацевтически приемлемое производное включает все производные антибактериального средства (такие как соли, пролекарства, метаболиты, сложные эфиры, простые эфиры, гидраты, полиморфы, сольваты, комплексы, энантиомеры или аддукты), которое, после введения субъекту, способно обеспечить (прямо или опосредованно) получение антибактериального соединения.
В целом, используемый в настоящем описании термин «катион» включает Na, K, Mg, Ca, NH4 +, (CH3CH2)3N+ и т.д.
В одном общем аспекте изобретение относится к соединениям формулы (I):
или их стереоизомеру или фармацевтически приемлемой соли; где:
Q обозначает гетероарил;
R1 обозначает:
(a) водород,
(b) (CO)n-R3 или
(c) COOR4,
n=0, 1 или 2;
R2 обозначает:
(a) SO3M,
(b) SO2NH2,
(c) PO3M,
(d) CH2COOM,
(e) CF2COOM,
(f) CHFCOOM или
(g) CF3;
M обозначает водород или катион;
R3 обозначает:
(a) водород,
(b) C1-C6-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, OR5, CN, COOR5, CONR6R7, NR6R7, NR5COR8, NR5CONR6R7, гетероциклила, гетероарила, циклоалкила или арила,
(c) CN,
(d) NR6R7,
(e) CONR6R7,
(f) NHCONR6R7,
(g) арил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(h) гетероциклил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(i) гетероарил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(j) циклоалкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(k) циклоалкил, замещенный C1-C6-алкилом, причем C1-C6-алкил дополнительно замещен одним или несколькими заместителями, независимо выбранными из OR5, NR6R7, галогена, CN или CONR6R7, или
(l) OR8;
R4 обозначает:
(a) водород,
(b) C1-C6-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, OR5, CN, COOR5, CONR6R7, NR6R7, NR5COR8, гетероциклила, гетероарила, циклоалкила или арила,
(c) арил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(d) гетероциклил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7,
(e) гетероарил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7, или
(f) циклоалкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR6R7, галогена, CN, CONR6R7, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR6R7;
R5 и R8, каждый независимо, обозначает:
(a) водород или
(b) C1-C6-алкил, необязательно замещенный одним или несколькими заместителями, необязательно выбранными из галогена, CN, CONR6R7, NR6R7, гетероциклила, гетероарила, циклоалкила или арила;
R6 и R7, каждый независимо, обозначает:
(a) водород,
(b) C1-C6-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, OR5, CN, COOR5, CONR5R8, NR5R8, NR5COR8, гетероциклила, гетероарила, циклоалкила или арила,
(c) арил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR5R8, галогена, CN, CONR5R8, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR5R8,
(d) гетероциклил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR5R8, галогена, CN, CONR5R8, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR5R8,
(e) гетероарил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR5R8, галогена, CN, CONR5R8, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR5R8,
(f) циклоалкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C1-C6-алкила, OR5, NR5R8, галогена, CN, CONR5R8, SO2-алкила, SO2-арила, OSO2-алкила, OSO2-арила или NHCONR5R8, или
(g) R6 и R7 соединены вместе для образования семичленного кольца.
Типичные неограничивающие примеры соединений в соответствии с изобретением включают:
сложный моно-[2-(5-аминометил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-иловый] эфир транс-серной кислоты;
сложный моно-[2-(5-((S)-1-аминоэтил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-иловый] эфир транс-серной кислоты;
сложный моно-[2-(5-((R)-1-аминоэтил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-иловый] эфир транс-серной кислоты;
сложный моно-[2-(5-(пиперидин-4-ил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-иловый] эфир транс-серной кислоты;
сложный моно-[2-(5-((S)-пирролидин-2-ил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-иловый] эфир транс-серной кислоты;
сложный моно-[2-(5-(пиперазин-1-илметил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-иловый] эфир транс-серной кислоты;
сложный моно-[2-(5-((RS)-1-амино-1-фенилметил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-иловый] эфир транс-серной кислоты;
моно-[2-(5-(пиперидин-4-ил)[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан транс-серной кислоты;
моно-[2-(5-((R)пиперидин-3-ил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан транс-серной кислоты;
натриевую соль транс-6-(сульфоокси)-2-(5-метил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-этил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-трифторметил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-карбоксамидо[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-(изооксазол-3-ил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-(фуран-2-ил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-фенил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-(пиридин-2-ил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-(6-карбоксамидо-пиридин-2-ил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-(5,6-дигидро-8H-имидазо[2,1-c][1,4]оксазин-2-ил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-(морфолино-4-метил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль сложного моно-[2-(5-(морфолин-4-илкарбонил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-илового] эфира транс-серной кислоты;
натриевую соль транс-6-(сульфоокси)-2-(5-метил[1,3,4]тиадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-метил[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-этоксикарбонил[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(2-метил-2H-[1,2,3,4]-тетразол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(1-метил-1H-[1,2,3,4]-тетразол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(3-этоксикарбонил-[1,2,4]оксадиазол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевую соль транс-6-(сульфоокси)-2-(5-карбоксамидо[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
или их стереоизомер, или фармацевтически приемлемую соль.
В целом, соединения по изобретению могут быть получены в соответствии со следующими процедурами. Специалисту в данной области понятно, что описанные способы могут дополнительно варьироваться или оптимизироваться для получения желательных и родственных соединений. В описанных ниже процедурах все переменные величины представляют собой, как определено выше.
(A) Синтез соединений, содержащих 1,3,4-оксадиазольные группы
В целом, соединения в соответствии с изобретением, содержащие 1,3,4-оксадиазольные группы, получали с использованием процедуры, представленной на схеме 1.
Обычно проводили реакцию транс-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (1a, которая описана в документе WO 2009/091856 A2), с алкилом или арилом или подходяще замещенными гидразидами арил- или алкил кислот в присутствии подходящего связывающего агента (например, гидрохлорида EDC (1-этил-3-(3-диметиламинопропил)карбодиимида), дициклогексилкарбодиимида (DCC) или пивалоилхлорида) в подходящем растворителе (например, N,N-диметилформамиде, N,N-диметилацетамиде, 1,4-диоксане или хлороформе), и в присутствии подходящего основания (например, N-метилморфолина, триэтиламина или диизопропилэтиламина) и N-гидроксибензотриазола (HOBt) при температуре в диапазоне от примерно -15°C до 60°C, в течение примерно от 1 до 24 часов для получения промежуточного соединения (1b).
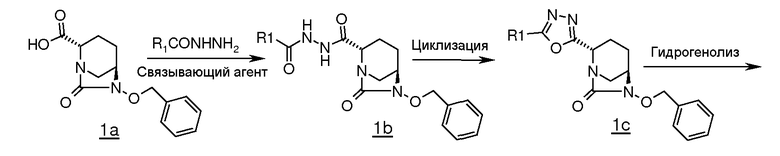
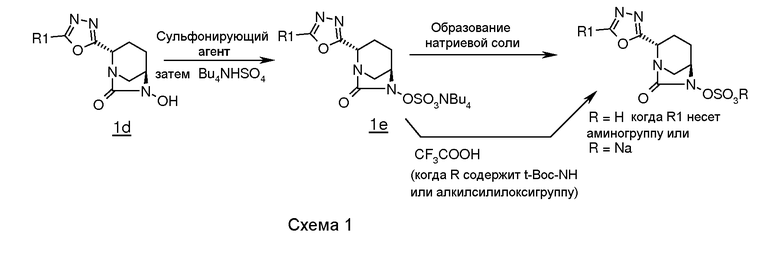
Циклизацию промежуточного соединения (1b) осуществляли обработкой промежуточного соединения (1b) подходящим реагентом, таким как п-толуолсульфонилхлорид, п-нитробензолсульфонилхлорид или метансульфонилхлорид, в подходящем растворителе (например, толуоле, хлороформе, дихлорметане или Ν,Ν-диметилформамиде) при температуре в диапазоне от 25°C до 110°C в течение примерно от 1 до 24 часов для получения 1,3,4-оксадиазольного промежуточного соединения (1c). Альтернативно, промежуточное соединение (1c) может также быть получено кипячением промежуточного соединения (1b) в сосуде с обратным холодильником в толуоле в присутствии молекулярных сит 4Е.
1,3,4-оксадиазольное промежуточное соединение (1c) подвергали гидрогенолизу c использованием подходящего катализатора (например, 5% или 10% палладия на углероде или 20% гидроксида палладия на углероде) в присутствии подходящего источника водорода (например, газообразного водорода, формиата аммония или циклогексена) в подходящем растворителе (например, метаноле, этаноле, смеси метанола-дихлорметана или смеси N,N-диметилформамида-дихлорметана) при температуре в диапазоне примерно от 25°C до 60°C в течение примерно от 1 до 24 часов для получения промежуточного соединения (1d).
Промежуточное соединение (1d) сульфонировали подходящим сульфонирующим реагентом (например, комплексом пиридина с триоксидом серы или комплексом Ν,Ν-диметилформида с триоксидом серы) в подходящем растворителе (например, пиридине, N,N-диметилформамиде, дихлорметане или их смеси) при температуре в диапазоне примерно от 25°C до 60°C в течение примерно от 1 до 24 часов для получения пиридиновой соли сульфоновой кислоты, которую в последующем обрабатывали сульфатом тетрабутиламмония для получения соли сульфоновой кислоты тетраметиламмония, промежуточного соединения (1e).
Некоторые соединения по изобретению выделяли в виде натриевой соли пропусканием промежуточного соединения (1e) через натриевую форму смолы Аберлит 200C в водном тетрагидрофуране с последующим выпариванием фракций растворителя в вакууме.
Некоторые другие соединения, когда R содержит трет-бутоксикарбонильную группу или алкилсилильную группу, выделяли в виде цвиттерионов обработкой промежуточного соединения (1e) трифторуксусной кислотой в отсутствие растворителя или в присутствии растворителя (например, дихлорметана, хлороформа или ацетонитрила) при температуре в диапазоне примерно от -10°C до 40°C в течение примерно от 1 до 14 часов.
(B) Синтез соединений, содержащих 1,3,4-тиадиазольные группы
В целом, соединения в соответствии с изобретением, содержащие 1,3,4-тиадиазол, получали с использованием общей процедуры, описанной на схеме 2.
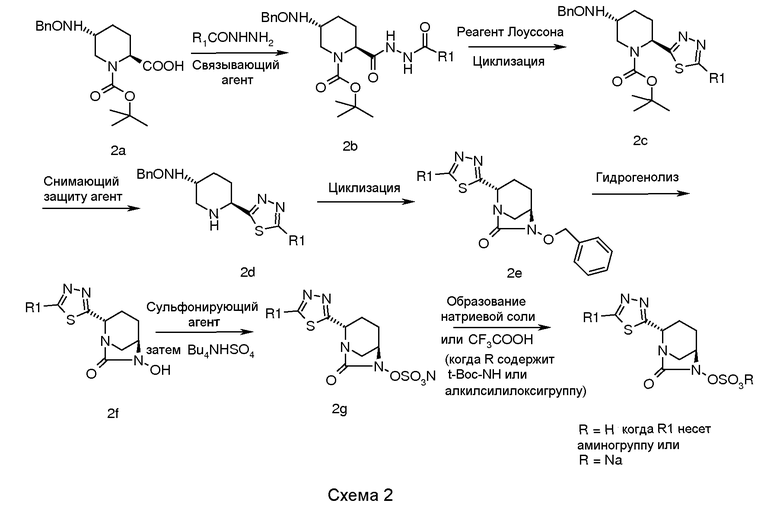
В соответствии со схемой 2, сложный 1-трет-бутиловый эфир транс-5-бензилоксиаминопиперидин-1,2-дикарбоновой кислоты (2a) взаимодействовал с алкилом или арилом или подходящим образом замещенными гидразидами арил- или алкиловых кислот в присутствии подходящего связывающего агента (например, гидрохлорида EDC, дициклогексилкарбодиимида (DCC) или пивалоилхлорида) в подходящем растворителе (например, N,N-диметилформамиде, N,N-диметилацетамиде, 1,4-диоксане или хлороформе), в присутствии подходящего основания (например, N-метилморфолина, триэтиламина или диизопропилэтиламина) и N-гидроксибензотриазола (HOBt) при температуре в диапазоне от примерно -5°C до 60°C в течение примерно от 1 до 24 часов для получения промежуточного соединения (2b).
Циклизацию промежуточного соединения (2b) осуществляли обработкой промежуточного соединения (2b) реагентом Лоуссона в подходящем растворителе (например, толуоле, хлороформе, тетрагидрофуране или Ν,Ν-диметилформамиде) при температуре в диапазоне от 25°C до 110°C в течение примерно от 1 до 24 часов для получения 1,3,4-тиадиазольного промежуточного соединения (2c).
Защиту промежуточного соединения (2c) снимали для получения промежуточного соединения (2d), используя снимающий защиту агент, такой как трифторуксусная кислота или гидрохлорид в присутствии растворителя, такого как дихлорметан, хлороформ, ацетонитрил или вода, при температуре в диапазоне примерно от -5°C до 50°C в течение примерно от 1 до 24 часов.
Циклизацию промежуточного соединения (2d) достигали обработкой промежуточного соединения (2d) с использованием подходящего реагента (например, раствора фосгена, дифосгена или трифосгена) в подходящем растворителе (например, толуоле, хлороформе, ацетонитриле, и в присутствии подходящего основания (например, триэтиламина или диизопропилэтиламина, N,N-диметиламинопиридина) при температуре в диапазоне примерно от -5°C до 50°C в течение примерно от 1 до 24 часов для получения циклизованного промежуточного соединения (2e).
Циклизованное промежуточное соединение (2e) подвергали гидрогенолизу с использованием подходящего катализатора (например, 5% или 10% палладия на углероде, или 20% гидроксида палладия на углероде) в присутствии подходящего источника водорода (например, газообразного водорода, формиата аммония или циклогексана) в подходящем растворителе (например, метаноле, этаноле, смеси метанола-дихлорметана или смеси N,N-диметилформамида и дихлорметана), при температуре в диапазоне примерно от 25°C до 60°C в течение примерно от 1 до 24 часов для получения N-гидрокси промежуточного соединения (2f).
Промежуточное соединение (2f) сульфонировали взаимодействием с подходящим сульфонирующим реагентом (например, комплексом пиридина и триоксида серы, или комплексом N,N-диметилформамида и триоксида серы) в подходящем растворителе (например, пиридине, N,N-диметилформамиде, дихлорметане или их смеси) при температуре в диапазоне примерно от 0°C до 50°C в течение примерно от 1 до 24 часов для получения пиридиновой соли сульфоновой кислоты, которую в последующем обрабатывали ацетатом тетрабутиламмония для получения тетрабутиламмониевой соли сульфоновой кислоты, промежуточного соединения (2g).
Соединение по изобретению выделяли в виде натриевой соли, пропуская промежуточное соединение (2g) через натриевую форму смолы Аберлит SR-L в водном тетрагидрофуране с последующим выпариванием фракций растворителя в вакууме. Альтернативно, когда R содержит трет-бутоксикарбонильную группу или алкилсилильную группу, то соединение по изобретению выделяют в виде цвиттерионов обработкой промежуточного соединения (2g) трифторуксусной кислотой в отсутствие растворителя или в присутствии подходящего растворителя (например, дихлорметана, хлороформа или ацетонитрила) при температуре в диапазоне примерно от -10°C до 40°C в течение примерно от 1 до 14 часов.
(C) Синтез соединений, содержащих 1,2,4-оксадиазол-3-ильные группы
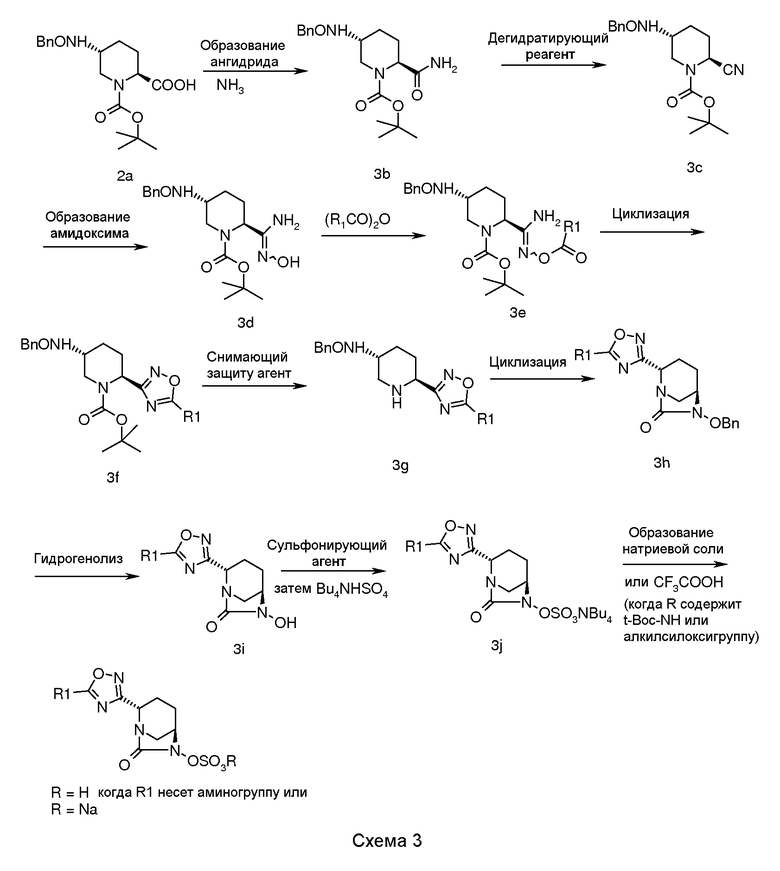
В целом, соединение по изобретению, содержащее 1,2,4-оксадиазол-3-ильные группы, получали, используя общую процедуру, описанную на схеме 3.
В соответствии со схемой 3, 1-трет-бутиловый эфир транс-5-бензилоксиаминопиперидин-1,2-дикарбоновой кислоты (2a) взаимодействовал с подходящим хлоридом кислоты (например, пивалоилхлоридом) в присутствии подходящего основания (например, N-метилморфолина, триэтиламина, диизопропилэтиламина) в подходящем растворителе (например, дихлорметане, тетрагидрофуране, 1,4-диоксане или хлороформе), при температуре в диапазоне примерно от -5°C до 35°C, в течение примерно от 1 до 2 часов для получения ангидрида, который в последующем обрабатывали газообразным аммиаком при температуре в диапазоне примерно от -50°C до 5°C в течение примерно от 0,5 до 2 часов для получения амида, промежуточного соединения (3b).
Дегидратацию промежуточного соединения (3b) осуществляли обработкой промежуточного соединения (3b) трифторуксусным ангидридом в подходящем растворителе (например, толуоле, хлороформе, тетрагидрофуране или дихлорметане), при температуре в диапазоне примерно от -5°C до 35°C, в течение примерно от 1 до 24 часов для получения нитрила, промежуточного соединения (3c).
Промежуточное соединение (3c) подвергали взаимодействию с гидрохлоридом гидроксиламина в подходящем растворителе (например, метаноле, воде, этаноле или их смеси) при температуре в диапазоне примерно от -5°C до 35°C, в течение примерно от 1 до 24 часов для получения амидоксима, промежуточного соединения (3d).
Промежуточное соединение (3d) подвергали взаимодействию с подходяще замещенным алкилангидридом в подходящем растворителе (например, дихлорметане, хлороформе, тетрагидрофуране или их смеси), и в присутствии подходящего основания (например, N-метилморфолина, триэтиламина или диизопропилэтиламина) при температуре в диапазоне примерно от -5°C до 35°C в течение примерно от 1 до 24 часов для получения O-ацилированного амидоксима, промежуточного соединения (3e).
Циклизацию промежуточного соединения (3e) осуществляли кипячением в сосуде с обратным холодильником промежуточного соединения (3e) в пиридине в течение примерно от 1 до 24 часов для получения циклизованного промежуточного соединения (3f).
Промежуточное соединение (3f) подвергали снятию защиты для получения промежуточного соединения (3g), используя подходящий снимающий защиту агент (например, трифторуксусную кислоту или хлористоводородную кислоту) в подходящем растворителе (например, дихлорметане, хлороформе, ацетонитриле или воде), при температуре в диапазоне примерно от -25°C до 50°C в течение примерно от 1 до 24 часов.
Циклизацию промежуточного соединения (3g) достигали обработкой промежуточного соединения (3g) подходящим реагентом (например, раствором фосфгена, дифосгеном или трифосгеном) в подходящем растворителе (например, толуоле, хлороформе или ацетонитриле) и в присутствии подходящего основания (например, триэтиламина или диизопропилэтиламина) при температуре в диапазоне примерно от -5°C до 50°C в течение примерно от 1 до 24 часов для получения циклизованного промежуточного соединения (3h).
Циклизованное промежуточное соединение (3h) подвергали гидрогенизации с использованием подходящего катализатора (например, 5% или 10% палладия на углероде или 20% гидроксида палладия на углероде) в присутствии подходящего источника водорода (например, газообразного водорода, формиата аммония или циклогексена) в подходящем растворителе (например, метаноле, этаноле, смеси метанола-дихлорметана или смеси N,N-диметилформамида с дихлорметаном) при температуре в диапазоне примерно от 25°C до 60°C, в течение примерно от 1 до 24 часов для получения N-гидрокси промежуточного соединения (3i).
Промежуточное соединение (3i) сульфонировали его взаимодействием с подходящим сульфонирующим реагентом (например, комплексом пиридина с триоксидом серы или комплексом Ν,Ν-диметилформамида с триоксидом серы) в подходящем растворителе (например, пиридине, N,N-диметилформамиде, дихлорметане или их смеси) при температуре в диапазоне примерно от -5°C до 50°C в течение примерно от 0,5 до 24 часов для получения пиридиновой соли сульфоновой кислоты, которую в последующем обрабатывали ацетатом тетрабутиламмония для получения соли сульфоновой кислоты тетрабутиламмония, промежуточного соединения (3j).
Соединение по изобретению выделяли в виде натриевой соли пропусканием промежуточного соединения (3j) через натриевую форму смолы Dowex 50WX8 200 в водном тетрагидрофуране с последующим выпариванием фракций растворителя в вакууме.
Альтернативно, когда R содержит трет-бутоксикарбонильную группу или алкилсилильную группу, то соединение по изобретению выделяют в виде цвиттерионов обработкой промежуточного соединения (3j) трифторуксусной кислотой в отсутствие растворителя или в присутствии растворителя (например, дихлорметана, хлороформа или ацетонитрила) при температуре в диапазоне от -10°C до 40°C в течение примерно от 1 до 14 часов.
(D) Синтез соединений, содержащих 1,2,3,4-тетразольные группы
В соответствии со схемой 4, соединение сложного 1-трет-бутилового эфира транс-5-бензилоксиамино-2-цианопиперидин-1-карбоновой кислоты (3c) подвергали взаимодействию с азидом натрия в присутствии гидрохлорида триэтиламина в подходящем растворителе (например, толуоле или ксилоле) при температуре кипячения в сосуде с обратным холодильником в течение примерно от 1 до 12 часов для получения тетразола, промежуточного соединения (4b).
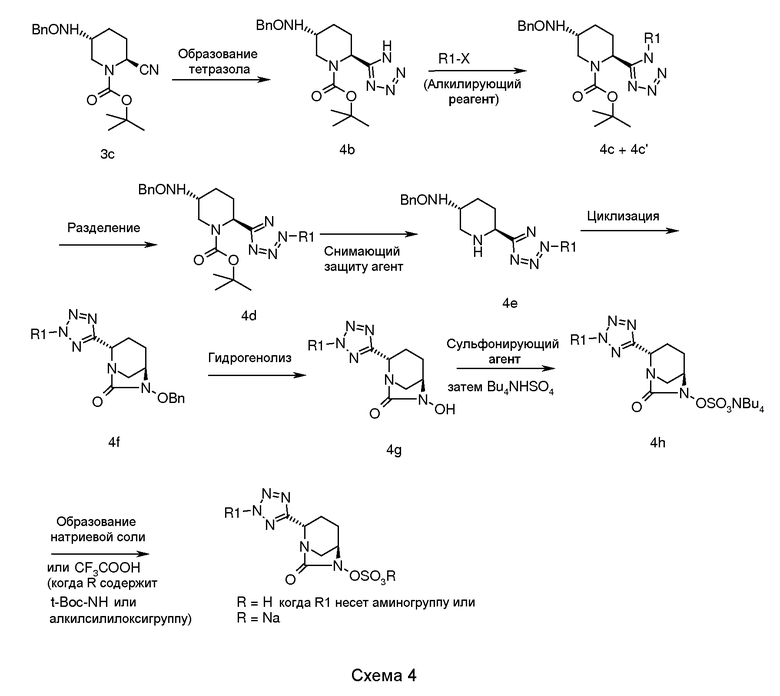
Алкилирование промежуточного соединения (4b) осуществляли обработкой промежуточного соединения (4b) подходящим алкилгалидом в подходящем растворителе (например, N,N-диметилформамиде, Ν,Ν-диметилацетамиде, тетрагидрофуране) и в присутствии подходящего основания (например, карбоната цезия, карбоната калия или карбоната натрия) при температуре в диапазоне примерно от -5°C до 35°C в течение примерно от 1 до 24 часов для получения изомерной смеси N-алкилтетразола, промежуточных соединений (4c) и (4c′), которые разделяли с использованием технологии колоночной хроматографии для получения изомерно чистого соединения (4c) и (4c′), и пока чистое промежуточное соединение именуется соединением (4d).
Промежуточное соединение (4d) подвергали снятию защиты для получения промежуточного соединения (4e), используя подходящий снимающий защиту агент (трифторуксусную кислоту или хлористоводородную кислоту), в подходящем растворителе (например, дихлорметане, хлороформе, ацетонитриле или воде), при температуре в диапазоне примерно от -25°C до 50°C, в течение примерно от 1 до 24 часов.
Циклизацию промежуточного соединения (4e) достигали обработкой промежуточного соединения (4e) с использованием подходящего реагента (например, раствора фосгена, дифосгена или трифосгена) в подходящем растворителе (например, толуоле, хлороформе или ацетонитриле) в присутствии подходящего основания (например, триэтиламина или диизопропилэтиламина) и N,N-диметиламинопиридина при температуре в диапазоне примерно от -5°C до 50°C в течение примерно от 1 до 24 часов для получения циклизованного промежуточного соединения (4f).
Циклизованное промежуточное соединение (4f) подвергали гидрогенолизу использованием подходящего катализатора (например, 5% или 10% палладия на углероде или 20% гидроксида палладия на углероде) в присутствии подходящего источника водорода (например, газообразного водорода, формиата аммония или циклогексена) в подходящем растворителе (например, метаноле, этаноле, смеси метанола-этилацетата или смеси N,N-диметилформамида-дихлорметана) при температуре в диапазоне примерно от 25°C до 60°C в течение от 1 до 24 часов для получения N-гидрокси промежуточного соединения (4g).
Промежуточное соединение (4g) сульфонировали взаимодействием с подходящим сульфонирующим реагентом, таким как (например, комплекс пиридина и триоксида серы или комплекс N,N-диметилформамида и триоксида серы) в подходящем растворителе (например, пиридине, N,N-диметилформамиде, дихлорметане или их смеси) при температуре в диапазоне примерно от -5°C до 50°C в течение примерно от 0,5 до 24 часов для получения пиридиновой соли сульфоновой кислоты, которую в последующем обрабатывали ацетатом тетрабутиламмония для получения тетрабутиламмониевой соли сульфоновой кислоты, промежуточного соединения (4h).
Соединение по изобретению выделяли в виде натриевой соли пропусканием промежуточного соединения 4h через натриевую форму смолы Dowex 50WX8 200 в водном тетрагидрофуране с последующим выпариванием фракций растворителя в вакууме. Альтернативно, когда R содержит трет-бутоксикарбонильную группу или алкилсилильную группу, то соединение по изобретению выделяли в виде цвиттерионов обработкой промежуточного соединения 4h трифторуксусной кислотой в отсутствие растворителя или в присутствии подходящего растворителя (например, дихлорметана, хлороформа или ацетонитрила) при температуре в диапазоне примерно от -10°C до 40°C в течение примерно от 1 до 14 часов.
(E) Синтез соединений, содержащих 1,2,4-оксадиазол-5-ильные группы
В целом, соединения в соответствии с изобретением, содержащие 1,2,4-оксадиазол-5-ильные группы, получали, используя общую процедуру, описанную на схеме 5.
В соответствии со схемой 5, транс-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновую кислоту, соединение (1a), подвергали взаимодействию с подходяще замещенным арил- или алкиламидоксимом в присутствии подходящего связывающего агента (например, EDC гидрохлорида, дициклогексилкарбодиимида (DCC) или пивалоилхлорида) в подходящем растворителе (например, N,N-диметилформамиде, N,N-диметилацетамиде, 1,4-диоксане или хлороформе) и в присутствии подходящего основания (например, N-метилморфолина, триэтиламина или диизопропилэтиламина) и N-гидроксибензотриазола (HOBt) при температуре в диапазоне примерно от -5°C до 60°C в течение примерно от 1 до 24 часов для получения промежуточного соединения (5b).
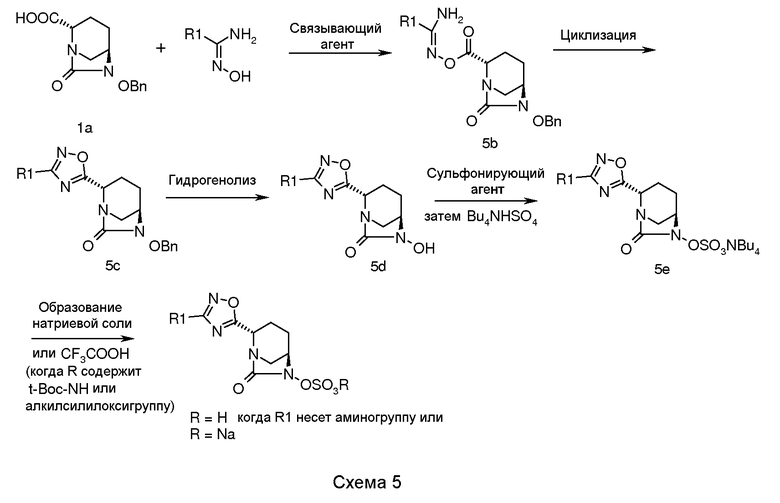
Циклизацию промежуточного соединения (5b) достигали кипячением в сосуде с обратным холодильником промежуточного соединения (5b) в пиридине в течение примерно от 1 до 24 часов для получения циклизованного промежуточного соединения (5c).
Циклизованное промежуточное соединение (5c) подвергали гидрогенолизу с использованием подходящего катализатора (например, 5% или 10% палладия на углероде или 20% гидроксида палладия на углероде) в присутствии подходящего источника водорода (например, газообразного водорода, формиата аммония или циклогексена) в подходящем растворителе (например, метаноле, этаноле, смеси метанол-этилацетат, смеси N,N-диметилформамида и дихлорметана) при температуре в диапазоне примерно от 25°C до 60°C в течение примерно от 1 до 24 часов для получения N-гидрокси промежуточного соединения (5d).
Промежуточное соединение (5d) сульфонировали взаимодействием его подходящим сульфонирующим реагентом (например, комплексом пиридина и триоксида серы, или комплексом N,N-диметилформамида и триоксида серы) в подходящем растворителе (например, пиридине, N,N-диметилформамиде, дихлорметане или их смеси) при температуре в диапазоне примерно от -5°C до 50°C в течение примерно от 0,5 до 24 часов для получения пиридиновой соли сульфоновой кислоты, которую в последующем обрабатывали ацетатом тетрабутиламмония для получения тетрабутиламмониевой соли сульфоновой кислоты, промежуточного соединения (5e).
Соединение по изобретению выделяли в виде натриевой соли пропусканием промежуточного соединения 5e через натриевую форму смолы Амберлит 200C в водном тетрагидрофуране с последующим выпариванием фракций растворителя в вакууме. Альтернативно, когда R содержит трет-бутоксикарбонильную группу или алкилсилильную группу, то соединение по изобретению выделяют в виде цвиттерионов обработкой промежуточного соединения (5e) трифторуксусной кислотой в отсутствие растворителя или в присутствии подходящего растворителя (например, дихлорметана, хлороформа или ацетонитрила) при температуре в диапазоне примерно от -10°C до 40°C в течение примерно от 1 до 14 часов.
В некоторых вариантах осуществления изобретение относится к фармацевтическим композициям, содержащим соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества соединения формулы (I) или его стереоизомера или фармацевтически приемлемой соли.
В некоторых вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль.
В некоторых вариантах осуществления изобретение относится к фармацевтическим композициям, содержащим: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль и (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
В некоторых других вариантах осуществления изобретение относится к фармацевтические композиции содержащей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, и (b) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В некоторых других вариантах осуществления изобретение относится к фармацевтическим композициям, содержащим: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (c) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I) или его стереоизомер, или фармацевтически приемлемую соль, и (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, и (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, и (b) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, и (b) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (c) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции, включающей: (a) соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль, (b) по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (c) по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного антибактериального средства или его фармацевтически приемлемого производного.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного антибактериального средства или его фармацевтически приемлемого производного.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (c) по меньшей мере одного антибактериального средства или его фармацевтически приемлемого производного.
В некоторых других вариантах осуществления изобретение относится к способу предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества: (a) соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли, (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (c) по меньшей мере одного антибактериального средства или его фармацевтически приемлемого производного.
В некоторых вариантах осуществления изобретение относится к способам увеличения антибактериальной эффективности антибактериального средства у субъекта, причем указанный способ включает совместное введение указанного бактериального средства или его фармацевтически приемлемого производного с фармацевтически эффективным количеством соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли.
В некоторых вариантах осуществления в композициях и способах в соответствии с изобретением используются соединения формулы (I), или их стереоизомер, или фармацевтически приемлемая соль в комбинации по меньшей мере с одним антибактериальным средством или его фармацевтически приемлемом производным. Может применяться широкое разнообразие антибактериальных средств. Типичные неограничивающие примеры антибактериальных средств включают одно или несколько антибактериальных соединений, в целом классифицируемых как аминогликозиды, ансамицины, карбацефемы, цефалоспорины, цефамицины, линкозамиды, липопептиды, макролиды, монобактамы, нитрофураны, пенициллины, полипептиды, хинолоны, сульфонамиды, тетрациклины, оксазолидинон и тому подобные.
Типичные неограничивающие примеры аминогликозидных антибактериальных средств включают амикацин, гентамицин, канамицин, неомицин, нетилмицин, тобрамицин, паромомицин, арбекацин, стрептомицин, апрамицин и тому подобные.
Типичные неограничивающие примеры ансамициновых антибактериальных средств включают гелданамицин, гербимицин и тому подобные.
Типичные неограничивающие примеры карбацефемовых антибактериальных средств включают лоракарбеф и тому подобные.
Типичные неограничивающие примеры карбапенемовых антибактериальных средств включают эртапенем, дорипенем, имипенем, меропенем и тому подобные.
Типичные неограничивающие примеры цефалоспориновых и цефамициновых антибактериальных средств включают цефазолин, цефацетрил, цефадроксил, цефалексин, цефалоглицин, цефалоний, цефалоридин, цефалотин, цефапирин, цефатризин, цефазедон, цефазафлур, цефрадин, цефроксадин, цефтезол, цефаклор, цефамандол, цефминокс, цефоницид, цефоранид, цефотиам, цефпрозил, цефбуперазон, цефуроксим, цефузонам, цефамицин, цефокситин, цефотетан, цефметазол, карбацефем, цефиксим, цефтазидим, цефтриаксон, цефкапен, цефдалоксим, цефдинир, цефдиторен, цефетамет, цефменоксим, цефодизим, цефоперазон, цефотаксим, цефпимизол, цефпирамид, цефподоксим, цефсулодин, цефтерам, цефтибутен, цефтиолен, цефтизоксим, оксацефем, цефепим, цефозопран, цефпиром, цефквином, цефтобипрол, цефтиофур, цефквином, цефовецин, CXA-101, цефтаролин, цефтобипрол и т.д.
Типичные неограничивающие примеры линкозамидных антибактериальных средств включают клиндамицин, линкомицин и тому подобные.
Типичные неограничивающие примеры макролидных антибактериальных средств включают азитромицин, кларитромицин, диритромицин, эритромицин, рокситромицин, тролеандомицин, телитромицин, спектиномицин, солитромицин и тому подобные.
Типичные неограничивающие примеры монобактамных антибактериальных средств включают азтреонам и тому подобные.
Типичные неограничивающие примеры нитрофурановых антибактериальных средств включают фуразолидон, нитрофурантоин и тому подобные.
Типичные неограничивающие примеры пенициллиновых антибактериальных средств включают амоксициллин, ампициллин, азлоциллин, карбенициллин, клоксациллин, диклоксациллин, флуклоксациллин, мезлоциллин, метициллин, нафциллин, оксациллин, пенициллин G, пенициллин V, пиперациллин, темоциллин, тикарциллин и тому подобные.
Типичные неограничивающие примеры полипептидных антибактериальных средств включают бацитрацин, колистин, полимиксин B и тому подобные.
Типичные неограничивающие примеры хинолоновых антибактериальных средств включают ципрофлоксацин, эноксацин, гатифлоксацин, левофлоксацин, ломефлоксацин, моксифлоксацин, налидиксиновую кислоту, левонадифлоксацин, норфлоксацин, офлоксацин, тровафлоксацин, грепафлоксацин, спарфлоксацин, темафлоксацин и тому подобные.
Типичные неограничивающие примеры сульфонамидных антибактериальных средств включают мафенид, сульфонамидохризоидин, сульфацетамид, сульфадиазин, сульфаметизол, сульфаметоксазол, сульфасалазин, сульфисоксазол, триметоприм и тому подобные.
Типичные неограничивающие примеры тетрациклиновых антибактериальных средств включают демеклоциклин, доксициклин, миноциклин, окситетрациклин, тетрациклин, тигециклин и тому подобные.
Типичные неограничивающие примеры оксазолидиноновых антибактериальных средств включают тедизолид, линезолид, ранбезолид, торезолид, радезолид и т.д.
Фармацевтические композиции в соответствии с изобретением могут включать один или несколько фармацевтически приемлемых носителей или эксципиентов или тому подобных. Типичные неограничивающие примеры таких носителей или эксципиентов включают маннит, лактозу, крахмал, стеарат магния, сахарин натрия, тальк, целлюлозу, кросскармелозу натрия, глюкозу, желатин, сахарозу, карбонат магния, смачивающие агенты, эмульгирующие агенты, солюбилизирующие агенты, агенты, забуферивающие pH, смазывающие вещества, стабилизирующие агенты, связывающие агенты и т.д.
Фармацевтические композиции в соответствии с настоящим изобретением могут существовать в различных формах. В некоторых вариантах осуществления фармацевтическая композиция представлена в форме порошка или раствора. В некоторых других вариантах осуществления фармацевтические композиции в соответствии с изобретением представлены в форме порошка, влагосодержание которого может быть восстановлено добавлением совместимого восстанавливающего влагосодержание растворителя перед парентеральным введением. Неограничивающий пример такого восстанавливающего влагосодержание растворителя включает воду.
В некоторых других вариантах осуществления, фармацевтические композиции в соответствии с изобретением представлены в форме замороженной композиции, которая может быть разведена совместимым разбавителем перед парентральным введением.
В некоторых других вариантах осуществления фармацевтические композиции в соответствии с изобретением представлены в форме, готовой к применению для парентерального введения.
В способах в соответствии с изобретением фармацевтическая композиция и/или другие фармацевтически активные ингредиенты, описанные в настоящей заявке, могут вводиться любым целесообразным способом, который служит для доставки композиции или ее составляющих компонентов или активных ингредиентов в желательный участок. Способ введения может варьироваться в зависимости от различных факторов, таких как, например, компоненты фармацевтической композиции и природа активных ингредиентов, участок потенциальной или действительной инфекции, вовлеченный микроорганизм (например, бактерии), тяжесть инфекции, возраст и физическое состояние субъекта. Некоторые неограничивающие примеры введения композиции субъекту в соответствии с настоящим изобретением включают пероральное, внутривенное, топическое введение, введение в дыхательные пути, внутрибрюшинное, внутримышечное, парентеральное, сублингвальное, трансдермальное, интраназальное, аэрозольное, внутриглазное, интратрахеальное, интраректальное, вагинальное введение, введение с использованием генной пушки, трансдермальной системы, глазных капель, ушных карель или композиции для полоскания ротовой полости.
Композиции в соответствии с изобретением могут составляться в различные лекарственные формы, в которых активные ингредиенты и/или эксципиенты могут присутствовать или вместе (например, в виде смеси), или в виде отдельных компонентов. Когда различные ингредиенты в композиции включаются в состав в виде смеси, то такая композиция может доставляться введением такой смеси. Композиция или лекарственная форма, в которых ингредиенты не поступают в виде смеси, но включаются в виде отдельных компонентов, то такая композиция/лекарственная форма может вводиться несколькими путями. При одном возможном пути ингредиенты могут смешиваться в желательных пропорциях, и затем смесь вводится по необходимости. Альтернативно, компоненты или ингредиенты (активные или инертные) могут вводиться отдельно (одновременно или друг за другом) в соответствующей пропорции с тем, чтобы достичь такого же или эквивалентного терапевтического уровня или эффекта, который мог бы быть достигнут введением эквивалентной смеси.
Аналогичным образом, в способах в соответствии с изобретением активные ингредиенты, описанные в настоящей заявке, могут вводиться субъекту несколькими путями в зависимости от требований. В некоторых вариантах осуществления активные ингредиенты смешиваются в соответствующих количествах, и затем смесь вводится субъекту. В некоторых других вариантах осуществления активные ингредиенты вводятся отдельно. Поскольку изобретение предусматривает, что активные ингредиенты и средства могут вводиться отдельно, то изобретение дополнительно относится к комбинированию отдельных фармацевтических композиций в форме набора. Набор может включать одну или несколько отдельных фармацевтических композиций, причем каждая включает один или несколько активных ингредиентов. Каждая из таких отдельных композиций может присутствовать в отдельном контейнере, таком как флакон, ампула, шприцы, коробочки, мешочки и тому подобные. Обычно набор включает инструкции по введению отдельных компонентов. Форма набора особенно предпочтительна, когда отдельные компоненты предпочтительно вводятся в различных лекарственных формах (например, пероральных и парентеральных), или вводятся через различные интервалы времени. Когда активные ингредиенты вводятся отдельно, они могут вводиться одновременно или последовательно.
Фармацевтическая композиция или активные ингредиенты в соответствии с настоящим изобретением могут составляться в разнообразные лекарственные формы. Типичные неограничивающие примеры лекарственных форм включают твердые, полутвердые, жидкие и аэрозольные лекарственные формы; такие как таблетки, капсулы, порошки, растворы, суспензии, суппозитории, аэрозоли, гранулы, эмульсии, сиропы, эликсиры и тому подобные.
В целом, фармацевтические композиции и способы, описанные в настоящей заявке, могут применяться при предотвращении или лечении бактериальных инфекций. Преимущественно, композиции и способы, описанные в настоящей заявке, также эффективны при предотвращении или лечении инфекций, вызванных бактериями, которые считаются менее восприимчивыми или невосприимчивыми к одному или нескольким из известных антибактериальных средств или их известным композициям. Некоторые неограничивающие примеры таких бактерий, известных как имеющие развившуюся устойчивость к различным антибактериальным средствам, включают Acinetobacter, E. coli, Pseudomonas aeruginosa, Staphylococcus aureus, Enterobacter, Klebsiella, Citrobacter и тому подобные. Другие неограничивающие примеры инфекций, которые можно предотвратить или лечить с использованием композиций и/или способов по изобретению, включают: инфекции кожи и мягких тканей, фебрильную нейтропению, инфекцию мочевыводящих путей, внутрибрюшные инфекции, инфекции дыхательных путей, пневмонию (внутрибольничную), бактеремический менингит, хирургические инфекции и т.д.
К удивлению, соединения, композиции и способы в соответствии с изобретением также эффективны при предотвращении или лечении бактериальных инфекций, которые вызваны бактериями, продуцирующими один или несколько бета-лактамазных ферментов. Способность композиций и способов в соответствии с настоящим изобретением лечить такие устойчивые бактерии типичными бета-лактамными антибиотиками представляет значимое усовершенствование в данной области.
В целом, соединения формулы (I) или их стереоизомер или фармацевтически приемлемая соль в соответствии с изобретением могут также применяться для увеличения антибактериальной эффективности антибактериального средства у субъекта. Антибактериальная эффективность одного или нескольких антибактериальных средств может быть увеличена, например, совместным введением указанного антибактериального средства или его фармацевтически приемлемой соли с фармацевтически эффективным количеством соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли в соответствии с изобретением.
Для специалиста в данной области вполне очевидно, что в описанное в настоящей заявке изобретение могут быть внесены варьирующиеся замещения и модификации без отхода от объема и сущности изобретения. Например, специалистам в данной области будет понятно, что изобретение может осуществляться с использованием разнообразных других соединений в пределах описанных общих прописей.
ПРИМЕРЫ
Следующие примеры иллюстрируют варианты осуществления изобретения, которые лучше всего известны в настоящее время. Однако следует понимать, что следующие варианты являются лишь примерными или иллюстративными в отношении применения принципов настоящего изобретения. Специалисты в данной области могут разработать многочисленные модификации и альтернативные композиции, способы и системы без отхода от сущности и объема настоящего изобретения. Прилагаемая формула изобретения предназначена для охвата таких модификаций и систематизаций. Таким образом, хотя настоящее изобретение было описано выше в конкретной форме, следующие примеры представляют дополнительные подробности в связи с теми вариантами, которые в настоящее время считаются наиболее практичными и предпочтительными вариантами осуществления изобретения.
Пример 1
Натриевая соль транс-6-(сульфоокси)-2-(5-метил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
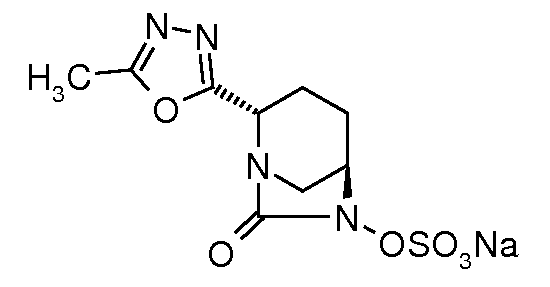
Стадия 1: Получение N′-ацетилгидразида транс-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты
К смеси гидразида уксусной кислоты (1,47 г, 19,9 ммоль) и транс-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (5 г, 18,1 ммоль) в N,N-диметилформамиде (50 мл), добавляли EDC гидрохлорид (5,19 г, 27,1 ммоль), DIPEA (диизопропилэтиламин) (9,50 мл, 54,3 ммоль) и HOBt (3,66 г, 27,1 ммоль). Реакционную смесь перемешивали при температуре от 25°C до 35°C в течение 14 часов. Смесь разбавляли водой (250 мл) и экстрагировали этилацетатом (75 мл × 3). Объединенный органический экстракт промывали водой, насыщенным водным раствором хлорида натрия (100 мл) и слои разделяли. Органический слой концентрировали в вакууме с получением неочищенного соединения, которое подвергали колоночной хроматографии на силикагеле с использованием метанолхлороформа с получением продукта стадии 1 (2,2 г) при выходе 33% в виде порошка.
Анализ: MS (масс спектроскопия) (ES+) C16H20N4О4 = 333,2 (M+1);
1H ЯМР (CDCl3) = 8,52 (с, 1H), 7,67 (с, 1H), 7,35-7,43 (м, 5H), 5,05 (д, 1H), 4,91 (д, 1H), 4,01 (д, 1H), 3,21 (шир.д, 1H), 3,04-3,15 (м, 2H), 2,30-2,35 (м, 1H), 2,02 (с, 3H), 1,89-2,05 (м, 3H).
Стадия 2: Получение транс-6-бензилокси-2-(5-метил-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
К продукту, полученному на стадии 1 (2 г, 6,0 ммоль) в хлороформе (30 мл), добавляли диизопропилэтиламин (3 мл, 17,4 ммоль) с последующим добавлением п-толуолсульфонилхлорида (1,72 г, 9,0 ммоль). Реакционную смесь кипятили в сосуде с обратным холодильником до тех пор, пока ТСХ (тонкослойная хроматография) не показывала полное потребление исходного материала. Когда реакция завершалась, добавляли воду (50 мл), и смесь экстрагировали хлороформом (50 мл × 2) и слои разделяли. Органический слой концентрировали в вакууме с получением неочищенного соединения, которое подвергали колоночной хроматографии на силикагеле для получения циклизованного продукта стадии 2 в виде порошка (1,25 г) при выходе 80%.
Анализ:
MS (ES+) C16H18N4O3 = 315,2 (M+1);
1H ЯМР (CDCl3) = 7,25-7,44 (м, 5H), 4,08 (д, 1H), 4,23 (д, 1H), 4,69 (т, 1H), 3,36 (шир.т, 1H), 2,90-2,94 (м, 1H), 2,80 (д, 1H), 2,54 (с, 3H), 2,26-2,31 (м, 2H), 2,10-2,12 (м, 1H), 1,94-1,98 (м, 1H).
Стадия 3: Получение транс-6-гидрокси-2-(5-метил-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
Продукт стадии 2 (1,2 г, 3,8 ммоль) и 10% палладий на углероде (300 мг) в метаноле (25 мл) перемешивали под атмосферным давлением водорода при температуре 30°С в течение 4 часов. Когда ТСХ указывала на завершение реакции, катализатор фильтровали через слой целлита, и содержащий катализатор слой промывали дополнительным метанолом. Фильтрат концентрировали в вакууме с получением соединения в виде порошка белого цвета в количестве 0,82 г в виде продукта стадии 3 при выходе 80%.
Анализ:
MS (ES+) C9H12N4O3 = 225,1 (M+1);
1H ЯМР (ДМСО-d6) = 9,84 (с, 1H), 4,54 (д, 1H), 3,63 (шир.c, 1H), 2,92 (шир.д, 1H), 2,69 (д, 1H), 2,48 (с, 3H), 1,98-2,16 (м, 3H), 1,81-1,88 (м, 1H).
Стадия 4: Получение тетрабутиламмониевой соли транс-6-сульфоокси-2-(5-метил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
Продукт, полученный на стадии 3 (0,82 г, 3,66 ммоль), растворяли в пиридине (8,2 мл) и к прозрачному раствору добавляли комплекс пиридина и триоксида серы (2,91 г, 18,3 ммоль). Суспензию перемешивали при температуре 35°С в течение ночи. Суспензию фильтровали и твердые вещества промывали дихлорметаном (10 мл × 2). Фильтрат выпаривали в вакууме, и остаток растворяли в 0,5н водном растворе дигидрофосфата калия (50 мл) в течение 0,5 часа. Раствор промывали дихлорметаном (30 мл) и слои разделяли. К водному слою добавляли сульфат тетрабутиламмония (1,23 г, 3,66 ммоль) и перемешивали в течение двух часов при 25°С. Когда ТСХ показывала завершение реакции, раствор экстрагировали дихлорметаном (50 мл × 2). Объединенный органический слой сушили на Na2SO4 и выпаривали в вакууме с получением твердого вещества белого цвета в качестве продукта стадии 4 (1,95 г) при выходе 80%.
Анализ:
MS (ES-) C9H12N4О6S.N(C4H9)4 в форме соли = 303,2 (M-1) в виде свободной сульфоновой кислоты;
1H ЯМР (CDCl3) = 4,65 (д, 1H), 4,39 (шир.c, 1H), 3,23-3,31 (м, 8H), 2,84 (д, 1H), 2,54 (с, 3H), 2,21-2,34 (м, 3H), 1,98-2,01 (м, 1H), 1,49-1,70 (м, 8H), 1,29-1,49 (м, 8H), 0,95 (т, 12H).
Стадия 5: Натриевая соль транс-6-сульфоокси-2-(5-метил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан
Соединение (0,546 г), полученное в виде продукта стадии 4, загружали в смесь тетрагидрофурана и воды 1:9 (10 мл) и медленно пропускали через свежеактивированную натриевую форму смолы Амберлит 200C (50 г). Фракции анализировали на ТСХ, и объединенные фракции выпаривали для удаления растворителя с более низкой температурой кипения в вакууме при температуре ниже 40°С. Затем водный слой промывали дихлорметаном (10 мл × 2), и слои разделяли. Водный слой концентрировали в вакууме при температуре ниже 40°С для получения остатка, который растирали в порошок с ацетоном. Твердое вещество получали в качестве соединения примера 1 по изобретению (0,3 г) при выходе 90%.
Анализ:
MS (ES-) C9H11N4O6SNa = 303,3 (M-1) в виде свободной сульфоновой кислоты;
1H ЯМР (ДМСО-d6) = 4,59 (д, 1H), 4,04 (шир.c, 1H), 2,92 (шир.д, 1H), 2,73 (д, 1H), 2,5 (с, 3H), 2,10-2,16 (м, 1H), 1,92-2,02 (м, 2H), 1,80-1,88 (м, 1H).
Соединения со 2 по 12 (таблица 1) получали, используя процедуру, описанную в примере 1, и используя соответствующее соединение R1CONHNH2 вместо гидразида уксусной кислоты.
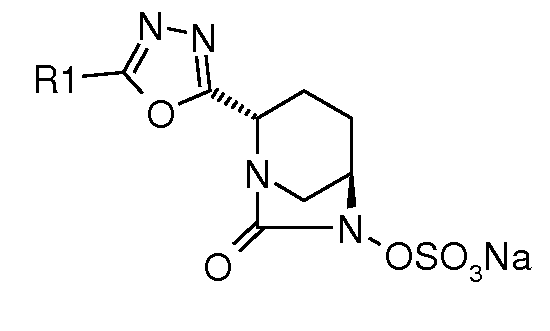
(C10H13N4О6S.Na)
(C9H10N5O7S.Na)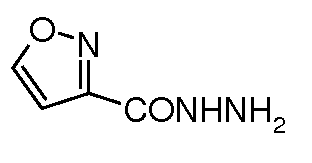
(C11H10N5O7S.Na)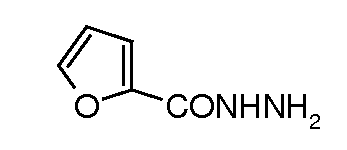
(C12H11N4O7S.Na
(C14H13N4O6S.Na)
(C13H12N5O6S.Na)
(C9H11N4O6S.Na)
(C14H15N6O7S.Na)
(C13H18N5O7S.Na)
(C13H16N5O8S.Na)
Пример 13
Сложный моно-[2-(5-аминометил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-иловый] эфир транс-сульфоновой кислоты
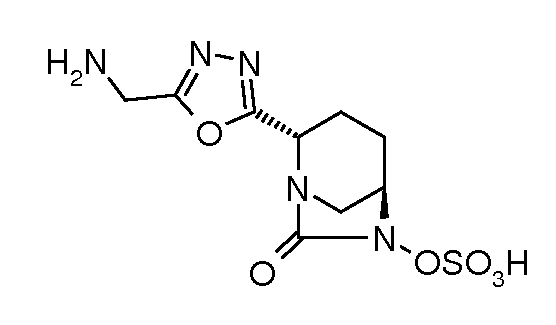
Тетрабутиламмониевую соль сложного моно-[2-(5-аминометил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-илового] эфира транс-сульфоновой кислоты (600 мг, полученную следующей процедурой со стадии 1 по стадию 4, как описано в примере 1, и путем использования трет-бутоксикарбониламиногидразида уксусной кислоты вместо гидразида уксусной кислоты) медленно шприцем добавляли раствор трифторуксусной кислоты (1,2 мл) в дихлорметане (1,2 мл) при -5°С в течение периода 5 минут. Смесь поддерживали в условиях перемешивания в течение 1 часа. Растворители удаляли при температуре ниже 40°С в высоком вакууме для получения остатка, который растирали в порошок с простым диэтиловым эфиром (50 мл × 5), и каждый раз простой диэтиловый эфир удаляли. Полученное твердое вещество белого цвета дополнительно растирали в порошок с ацетонитрилом (50 мл × 2). Полученное твердое вещество перемешивали в дихлорметане (50 мл) и суспензию фильтровали. Твердое вещество сушили в вакууме с получением указанного в заголовке соединения по изобретению в количестве 0,265 г.
Анализ:
MS (ES-) C9H13N5O6S.CF3COOH = 318,2 (M-1) в виде свободной сульфоновой кислоты;
1H ЯМР (ДМСО-d6 после обмена D2O) = 4,69 (д, 1H), 4,45 (с, 2H), 4,09 (шир.c, 1H), 2,97 (шир.д, 1H), 2,74 (д, 1H), 2,17-2,19 (м, 1H), 1,94-2,10 (м, 2H), 1,80-2,00 (м, 1H).
Соединения 14-19 (таблица 2) получали, используя процедуру, описанную в примере 13, и используя соответствующий R1CONHNH2, причем функция амина защищена BOC (трет-бутоксикарбонильной) группой.
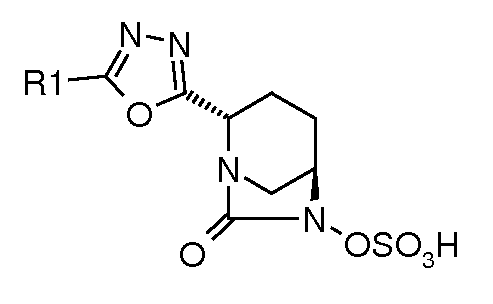
(C10H15N5O6S)
(C10H15N5O6S)
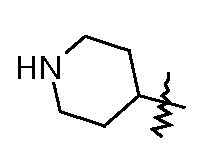
(C13H18N5O6S)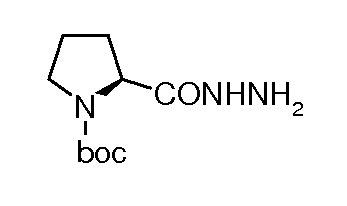
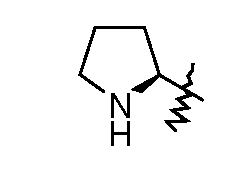
(C12H17N5O6S)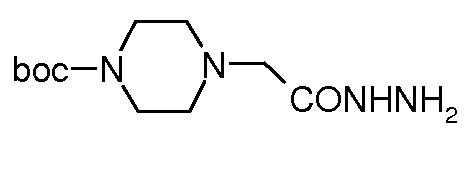
(C13H20N6О6S)
(C15H17N5O6S)
Пример 20
Натриевая соль транс-6-(сульфоокси)-2-(5-метил[1,3,4]тиадиазол-2-ил)-7-оксо-1,6-диазабицикло [3.2.1]октана
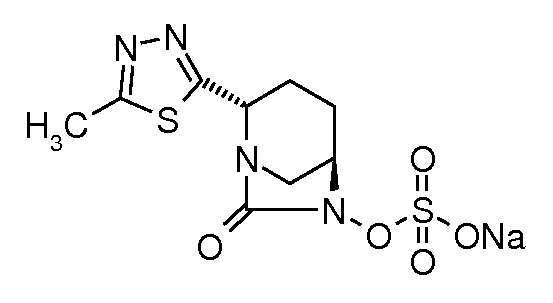
Стадия 1: Получение сложного трет-бутилового эфира транс-2-(N"-ацетилгидразинокарбонил)-5-бензилоксиаминопиперидин-1-карбоновой кислоты
К раствору 1-трет-бутилового эфира транс-5-бензилоксиаминопиперидин-1,2-дикарбоновой кислоты (12 г, 0,034 моль) в Ν,Ν-диметилформамиде (60 мл), последовательно добавляли EDC-HCl (9,82 г, 0,051 моль) и N-метилморфолин (11,4 мл) при 10°С-15°С при перемешивании. К реакционной смеси добавляли ацетилгидразид (2,79 г, 0,0377 моль) и HOBt (4,62 г, 0,034 моль). Полученной смеси давали возможность нагреться до 25°С-35°С и перемешивали в течение 16 часов. Реакционную смесь выливали в воду (600 мл) и перемешивали в течение 30 мин. Отделенное твердое вещество фильтровали и фильтрат экстрагировали этилацетатом (3×400 мл). Объединенный органический экстракт сушили над сульфатом натрия, и растворитель выпаривали в вакууме с получением остатка. Остаток очищали колоночной хроматографией с получением продукта стадии 1 в виде бледно-желтого густого масла в количестве 9,8 г (выход 70%).
Анализ:
MS: 407 (M+H); MF: C20H30N4O5; MW (молекулярная масса): 406,49.
Стадия 2: Получение сложного трет-бутилового эфира транс-2-(5-метил[1,3,4]тиадиазол-2-ил)-5-бензилоксиаминопиперидин-1-карбоновой кислоты
К раствору продукта стадии 1 (9,6 г, 0,0236 моль) в толуоле (240 мл) добавляли Реагент Лоуссона (12,4 г, 0,0307 моль), и полученную смесь нагревали до 60°С-65°С в течение 2 часов при перемешивании. Реакционной смеси давали возможность охладиться до 25°С-35°С и промывали насыщенным раствором бикарбоната натрия (2×150 мл). Органический слой отделяли, сушили, и растворитель выпаривали в вакууме с получением остатка. Остаток очищали колоночной хроматографией с получением продукта стадии 2 в виде бледно-желтого масла в количестве 9,5 г.
Анализ:
MS: 405 (M+H); MF: C20H28N4O3S; MW: 404,54;
Стадия 3: Получение транс-2-(5-метил[1,3,4]тиадиазол-2-ил)-5-бензилоксиаминопиперидина
К раствору продукта стадии 2 (9,5 г, 0,0236 моль) в дихлорметане (19 мл) добавляли трифторуксусную кислоту (38 мл) при 10-15°С в течение периода 5 мин при перемешивании. Полученному раствору давали возможность перемешиваться при 25°С-35°С в течение 30 мин и раствор концентрировали в вакууме с получением остатка. Остаток разбавляли водой (50 мл) и водным раствором бикарбоната натрия. Остаток экстрагировали этилацетатом (2×100 мл). Объединенный органический слой сушили над сульфатом натрия и выпаривали в вакууме с получением остатка. Остаток очищали колоночной хроматографией с получением продукта стадии 3 в виде твердого вещества не совсем белого цвета в количестве 4,4 г (выход 62%).
Анализ:
MS: 305 (M+H); MF: C15H20N4OS; MW: 305.
Стадия 4: Получение транс-2-(5-метил[1,3,4]тиадиазол-2-ил)-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октана
К раствору продукта стадии 3 (4,4 г, 0,0144 моль) и триэтиламина (6,02 мл, 0,0432 моль) в ацетонитриле (66 мл) добавляли раствор трифосгена (2,57 г, 0,00865 моль) в ацетонитриле (22 мл) при 10°С-15°С в течение период 10 минут при перемешивании. Через 30 мин к реакционной смеси добавляли N,N-диметиламинопиридин (176 мг, 0,00144 моль), и смеси затем давали возможность нагреться до 25°С-35°С и перемешивали в течение 16 часов. Полученную смесь гасили насыщенным раствором бикарбоната натрия (30 мл). Растворители выпаривали в вакууме с получением остатка. Остаток очищали колоночной хроматографией с получением продукта стадии 4 в виде твердого вещества не совсем белого цвета в количестве 1,1 г (выход 23%).
Анализ:
MS: 331 (M+H); MF: C16H18N4О2S; MW: 330.
Стадия 5: Получение транс-2-(5-метил[1,3,4]тиадиазол-2-ил)-6-гидрокси-7-оксо-1,6-диазабицикло[3.2.1]октана
К раствору продукта стадии 4 (1,1 г, 0,0033 моль) в метаноле (100 мл) добавляли формиат аммония (10 г) с последующим добавлением 10% Pd/C (2,2 г, 2 раза масс./масс.). Суспензию перемешивали в течение 4 часов при 25°С-35°С. Катализатор фильтровали через целит, и фильтрат концентрировали в вакууме с получением продукта стадии 5 в количестве 0,7 г (выход 90%), который использовали без дополнительной обработки для следующей реакции.
Анализ:
MS: 241 (M+H); MF: C9H12N4O2S; MW: 240
Стадия 6: Получение тетрабутиламмониевой соли транс-6-(сульфоокси)-2-(5-метил[1,3,4]тиадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
К раствору продукта стадии 5 (630 мг) в смеси Ν,Ν-диметилформамид:дихлорметан (3 мл: 3 мл) добавляли комплекс Ν,Ν-диметилформамида с триоксидом серы (480 мг) при 0°С-5°С при перемешивании. Реакционную смесь перемешивали в течение 30 мин. Ее концентрировали в вакууме для удаления дихлорметана, и к оставшемуся раствору добавляли раствор ацетата тетрабутиламмония (956 мг) в воде (3 мл). Реакционную смесь перемешивали в течение 16 часов при 25°С-35°С. Ее концентрировали под пониженным давлением (4 мм рт.ст.) для получения остатка. К остатку добавляли воду (10 мл), и смесь экстрагировали дихлорметаном (2×15 мл). Объединенный органический слой концентрировали в вакууме с получением продукта стадии 6 в виде твердого вещества белого цвета в количестве 590 мг (выход 54%).
Анализ:
MS: 562 (M+H); MF: C25H47N5O5S2; MW: 561
Стадия 7: Натриевая соль транс-6-(сульфоокси)-2-(5-метил[1,3,4]тиадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
Продукт, полученный как на стадии 6, растворяли в 20% тетрагидрофуране в воде (1 мл), и указанный выше раствор пропускали через колонку смолы Амберлит SR01-L-Na (15 г). Колонку сначала элюировали ТГФ-водой (10:1, 50 мл) с последующим добавлением воды (50 мл). Фракции объединенного водного слоя выпаривали в вакууме с получением соединения по изобретению в виде твердого вещества не совсем белого цвета в количестве 250 мг (выход 70%).
Анализ:
MS: 319 (M-H свободной сульфоновой кислоты); MF: C9H11N4O5S2Na; MW: 342,33
1H ЯМР (D2О) = 4,85 (д, 1H), 4,122 (шир.c, 1H), 3,11 (шир.д, 1H), 2,88 (д, 1H), 2,66 (с, 3H), 2,42-2,37 (м, 1H), 2,16-2,05 (м, 2H), 1,89-1,82 (м, 1H).
Пример 21
Натриевая соль транс-6-(сульфоокси)-2-(5-метил[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
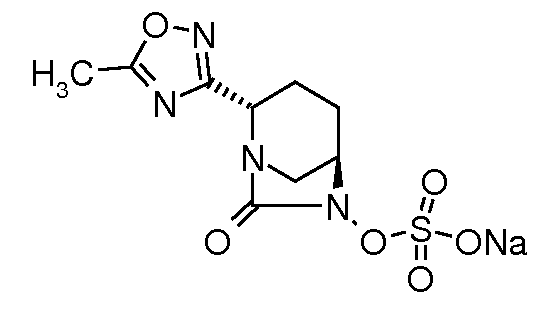
Стадия 1: Получение сложного 1-трет-бутилового эфира транс-5-бензилоксиамино-2-карбоксамидопиперидин-1-карбоновой кислоты
К перемешанному раствору сложного 1-трет-бутилового эфира транс-5-бензилоксиаминопиперидин-1,2-дикарбоновой кислоты (21 г, 0,06 моль) и триэтиламина (25,12 мл, 0,18 моль) в дихлорметане (210 мл) по каплям добавляли пивалоилхлорид (11,07 мл, 0,09 моль) при температуре 0°С. Полученную смесь перемешивали в течение 1,5 часов. Реакционную смесь охлаждали до -40°С и сухой аммиачный газ барботировали через нее в течение 30 мин. Смеси давали возможность нагреться до 25°С-35°С, и полученную суспензию отфильтровывали. Фильтрат выпаривали под пониженным давлением для получения остатка, и остаток хроматографировали на колонке с силикагелем с получением продукта стадии 1 в виде густого масла в количестве 10,2 г (выход: 49%).
Анализ:
MS: 348 [M+]; MF: C19H28N2O4; MW: 348.
Стадия 2: Получение сложного 1-трет-бутилового эфира транс-5-бензилоксиамино-2-цианопиперидин-1-карбоновой кислоты
К раствору соединения стадии 1 (10,2 г, 0,0286 моль) и триэтиламина (17,99 мл, 1,289 моль) в дихлорметане (306 мл) добавляли трифторуксусный ангидрид (12,08 г, 0,0573 моль) при 0°С при перемешивании. Полученному раствору давали возможность нагреться до 25°С-35°С и перемешивали в течение 6 часов. Реакционную смесь промывали последовательно водой (3×100 мл), насыщенным водным раствором хлорида аммония (100 мл) и солевым раствором (100 мл). Объединенный органический слой сушили над сульфатом натрия и растворитель выпаривали в вакууме с получением остатка. Остаток хроматографировали на колонке с силикагелем, используя смесь ацетон:гексан (1:19), с получением продукта стадии 2 в виде твердого вещества белого цвета в количестве 9,7 г (выход - количественный).
Анализ:
MS: 331 (M+); MF: C18H25N3O3; MW: 331.
Стадия 3: Получение сложного 1-трет-бутилового эфира транс-5-бензилоксиамино-2-(N-гидроксикарбамимидоил)пиперидин-1-карбоновой кислоты
К охлажденному раствору продукта стадии 2 (9,7 г, 0,0293 моль) в метаноле (145,5 мл) добавляли последовательно гидрохлорид гидроксиламина (3,05 г, 0,0439 моль) и бикарбонат натрия (5,53 г, 0,0659 моль) при 0°С при перемешивании. Реакционной смеси давали возможность нагреться до 25°С-35°С и перемешивали в течение 24 часов. Полученную суспензию отфильтровывали и фильтрат концентрировали в вакууме при температуре ниже 40°С для получения остатка. Остаток очищали колоночной хроматографией через силикагель смесью ацетон : гексан (1:5) с получением продукта стадии 3 в виде бесцветного масла в количестве 7,6 г (выход: 72%).
Анализ:
MS: 364 (M+); MF: C18H28N4O4; MW: 364.
Стадия 4: Получение сложного 1-трет-бутилового эфира транс-5-бензилоксиамино-2-(N-ацетилоксикарбамимидоил)пиперидин-1-карбоновой кислоты
К раствору продукта стадии 3 (7,6 г, 0,0208 моль) в дихлорметане (76 мл) добавляли триэтиламин (5,82 мл, 0,0417 моль) с последующим добавлением по каплям уксусного ангидрида (2,34 г, 0,02293 моль) при 0°С. Полученной смеси давали возможность нагреться до 25°С-35°С и перемешивали в течение 4 часов. Реакционную смесь промывали последовательно водой (2×75 мл), насыщенным водным раствором хлорида аммония (75 мл) и солевым раствором (35 мл). Органический слой сушили над сульфатом натрия и выпаривали в вакууме с получением остатка. Остаток хроматографировали на колонке с силикагелем смесью 1:10 ацетон : гексан для выхода продукта стадии 4 в виде твердого вещества белого цвета в количестве 7,5 г (выход: 88%).
Анализ:
MS: 405 (M+); MF: C21H31N3O5; MW: 405.
Стадия 5: Получение сложного 1-трет-бутилового эфира транс-5-бензилоксиамино-2-(5-метил[1,2,4]оксадиазол-3-ил)пиперидин-1-карбоновой кислоты
Раствор продукта стадии 4 (7,5 г) в пиридине (112,5 мл) нагревали при температуре кипения в сосуде с обратным холодильником в течение 6 часов. Растворитель выпаривали в вакууме при температуре ниже 40°С, и остаток разбавляли 10% водным раствором KHSO4 (100 мл). Смесь экстрагировали этилацетатом (3×75 мл). Объединенные органические экстракты промывали водой (2×75 мл) с последующим промыванием солевым раствором (37,5 мл). Органический слой сушили над сульфатом натрия и растворитель выпаривали в вакууме с получением остатка. Остаток очищали колоночной хроматографией смесью 1:20 ацетон : гексан с получением продукта стадии 5 в виде твердого вещества белого цвета в количестве 6,0 г (выход: 84%).
Анализ:
MS: 387 (M+); MF: C21H29N3O4; MW: 387.
Стадия 6: Получение транс-6-бензилоксиамино-2-(5-метил[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
К раствору продукта стадии 5 (6 г) в дихлорметане (150 мл) добавляли трифторуксусную кислоту (12 мл) при -15°С при перемешивании, и полученной смеси давали возможность нагреться до 25°С-35°С. Смесь перемешивали в течение 4 часов. Растворитель выпаривали в вакууме при температуре ниже 40°С. Полученную массу разбавляли насыщенным водным раствором бикарбоната натрия (60 мл), и смесь экстрагировали дихлорметаном (2×60 мл). Объединенные органические экстракты промывали водой (60 мл), сушили над сульфатом натрия и выпаривали в вакууме при температуре ниже 40°С для получения 4,2 г соединения пиперидина с удаленной защитой трет-бутоксикарбонильной группы.
К раствору соединения пиперидина с удаленной защитой трет-бутоксикарбонильной группы (4,2 г) в ацетонитриле (63 мл) добавляли триэтиламин (5.28 мл) с последующим добавлением раствора трифосгена (1,9 г) в ацетонитриле (16,8 мл) при 0°С-5°С при перемешивании. Раствор перемешивали в течение 30 мин и добавляли Ν,Ν-диметиламинопиридин (0,178 г). Реакционной смеси давали возможность нагреться до 25°С-35°С и перемешивали в течение 16 часов. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия (33,6 мл), и полученную смесь перемешивали в течение 30 мин. Смесь концентрировали до 1/3 ее объема в вакууме, и ее разбавляли водой (42 мл), и полученную смесь экстрагировали дихлорметаном (2×40 мл). Объединенный органический слой выпаривали в вакууме с получением остатка, и остаток очищали через колонку с силикагелем, используя смесь 1:4 ацетон : гексан с получением продукта стадии 6 в виде твердого вещества белого цвета в количестве 2,3 г (выход: 48%).
Анализ:
MS: 314 (M+); MF: C16H18N4O3; MW: 314.
Стадия 7: Получение тетрабутиламмониевой соли транс-6-сульфоокси-2-(5-метил[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
К раствору продукта стадии 6 (2,3 г, 0,00732 моль) в смеси дихлорметана и N,N-диметилформамида (по 11,5 мл каждого) добавляли 10% палладий на углероде (575 мг), и суспензию перемешивали в атмосфере водорода (1 атм) при 25°С-35°С в течение 5 часов. Катализатор фильтровали и фильтрат концентрировали под пониженным давлением для получения дебензилированного продукта. Его использовали без дополнительной обработки для следующей реакции. Дебензилированный продукт, полученный как описано выше, растворяли в N,N-диметилформамиде (11,5 мл), и для осветления раствора добавляли одной порцией комплекс Ν,Ν-диметилформамида и триоксида серы (1,34 г, 0,00878 моль) в атмосфере аргона при 0°С. Реакционную смесь перемешивали в течение 1 часа. К реакционной смеси добавляли сульфат тетрабутиламмония (2,66 г, 0,00878 моль), растворенный в воде (8,8 мл), и полученной смеси давали возможность нагреться до 25°С-35°С и перемешивали в течение 1 часа. Растворители удаляли в вакууме при температуре ниже 40°С для получения остатка, и его растирали в порошок с ксилолом (25 мл) для удаления следов Ν,Ν′-диметилформамида. Остаток разделяли между смесью 1:1 воды (23 мл) и дихлорметана (23 мл), и слои разделяли. Водный слой повторно экстрагировали дихлорметаном (23 мл). Объединенные органические экстракты последовательно промывали водой (23 мл) и солевым раствором (23 мл). Органический слой сушили над сульфатом натрия и концентрировали в вакууме с получением продукта стадии 7 в количестве 2,05 г (выход - 51%).
Анализ:
MS: 304 (M-H) сульфата; M.F: C25H47N5O6S; M.W: 545,7.
Стадия 8: Натриевая соль транс-6-сульфоокси-2-(5-метил[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
Продукт стадии 7 (1,7 г) растворяли в смеси 5% тетрагидрофурана в воде (2 мл), и раствор загружали на колонку со смолой Dowex 50WX8 200 Na+ (25 см × 3,0 см). Колонку элюировали 5% тетрагидрофураном в воде (50 мл), а затем водой (150 мл). Выпаривание фракций в вакууме при температуре ниже 40°С давало указанное в заголовке соединение по изобретению в виде твердого вещества белого цвета в количестве 860 мг (выход 85%).
Анализ:
MS: 304 (M-H) сульфата; M.F: C9H11N4NaO6S; M.W: 326,27.
1H ЯМР (ДМСО-d6 после обмена D2O) = 4,46 (т, 1H), 4,02 (шир.с, 1H), 2,90 (шир.с, 2H), 2,59 (с, 3H), 2,06-1,93 (м, 3H), 1,83-1,77 (м, 1H).
Пример 22
Натриевая соль транс-6-(сульфоокси)-2-(5-этоксикарбонил[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
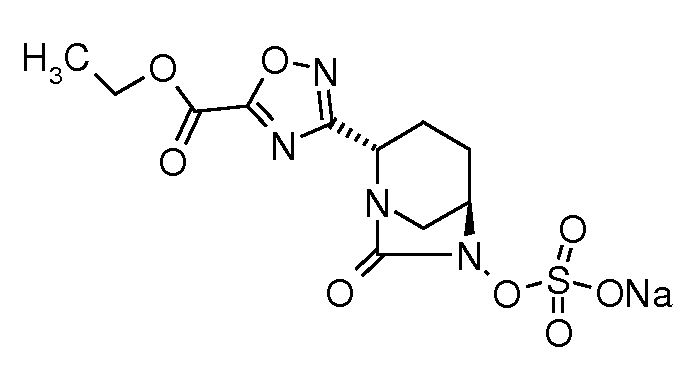
Соблюдая процедуру, описанную в примере 21, и используя простой этиловый эфир оксалилхлорида вместо уксусного ангидрида на стадии 4, получали указанное в заголовке соединение в количестве 731 мг.
Анализ:
MS: 333 (M-H) сульфата; M.F: C9H10N5NaO7S; M.W: 355.
1H ЯМР (ДМСО-d6 после обмена D2O) = 4,61 (д, 1H), 4,41 (кв, 2H), 4,04 (шир.c, 1H), 2,92-2,82 (м, 2H), 2,09-2,03 (м, 2H), 1,99-1,96 (м, 1H), 1,83-1,81 (м, 1H), 1,33 (т, 3H).
Пример 23
Натриевая соль транс-6-(сульфоокси)-2-(2-метил-2H-[1,2,3,4]-тетразол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
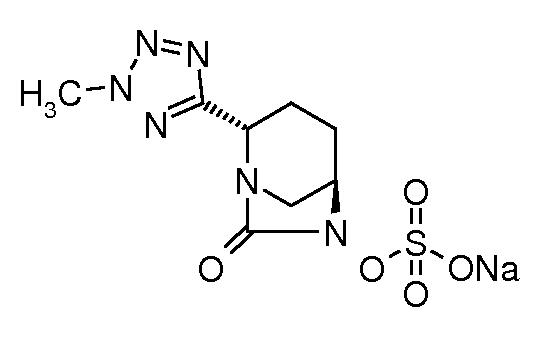
Стадия 1: Получение сложного 1-трет-бутилового эфира транс-5-бензилоксиамино-2-(1H-тетразол-5-ил)пиперидин-1-карбоновой кислоты
К раствору сложного 1-трет-бутилового эфира транс-5-бензилоксиамино-2-цианопиперидин-1-карбоновой кислоты (6 г, 0,018 моль) в толуоле (120 мл) добавляли азид натрия (5,88 г, 0,090 моль), гидрохлорид триэтиламина (12,42 г, 0,090 моль), и полученную смесь нагревали при 100°С при перемешивании в течение 2 часов. Реакционную смесь охлаждали до 25°С-35°С и выливали в насыщенный водный раствор хлорида аммония (150 мл). Смесь перемешивали в течение 15 мин, и водный слой экстрагировали этилацетатом (1×120 мл). Объединенный органический слой сушили над сульфатом натрия с получением продукта стадии 1 в виде бесцветного масла в количестве 6,0 г (выход 89%).
Анализ:
MS: 375 (M-H); FW: C18H26N6О3; MW: 376.
Стадия 2: Получение измеров: изомер A 1-трет-бутилового эфира транс-5-бензилоксиамино-2-(1-метил-1H-тетразол-5-ил)пиперидин-1-карбоновой кислоты и изомер B транс-5-бензилоксиамино-2-(2-метил-2H-тетразол-5-ил)пиперидин-1-карбоновой кислоты
К смеси продукта стадии 1 (6,0 г, 0,0168 моль) и карбоната цезия (7,84 г, 0,0240 моль) в N,N-диметилформамиде (60 мл) добавляли метилиодид (3,41 г, 0,0240 моль) при температуре от 0°С до 5°С при перемешивании. Реакционную смесь перемешивали в течение 30 мин, согревали до 25°С-35°С и перемешивали в течение 2 часов. Ее выливали на ледяную воду (600 мл) и экстрагировали этилацетатом (3×150 мл). Объединенный органический слой сушили над сульфатом натрия и концентрировали в вакууме с получением остатка. Остаток очищали колоночной хроматографией, используя смеси 5-20% этилацетата в гексане для получения двух изомеров: изомера A при Rf 0,50 и изомера B при Rf 0,42. Эти изомеры были идентифицированы отдельно. Общее количество полученных продуктов составило 5,7 г (выход 91%).
Анализ:
MS: 389 (M-H); FW: C19H28N6О3; MW: 390
Стадия 3: Получение транс-5-бензилоксиамино-2-(2-метил-2H-тетразол-5-ил)пиперидин-1-карбоновой кислоты
К раствору изомера B, полученного на стадии 2 (1,35 г, 0,00347 моль) в дихлорметане (13,5 мл), добавляли трифторуксусную кислоту (2,7 мл) при 0°С при перемешивании. Раствору давали возможность нагреться до 25°C-35°С и перемешивали в течение 4 часов. Реакционную смесь концентрировали в вакууме с получением остатка, и остаток разбавляли водой (20 мл) и обрабатывали насыщенным водным раствором бикарбоната натрия (5 мл). Полученную смесь экстрагировали дихлорметаном (2×20 мл). Объединенные экстракты сушили над сульфатом натрия и концентрировали в вакууме с получением продукта стадии 3 в виде бесцветного масла в количестве 900 мг (выход 90%).
Анализ:
MS: 288 (M+); FW: C14H20N6О; MW: 288.
Стадия 4: Получение транс-6-(бензилокси)-2-(2-метил-2H-[1,2,3,4]тетразол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
К раствору продукта стадии 3 (900 мг, 0,00321 моль) и триэтиламина (1,3 мл, 0,00937 моль) в ацетонитриле (13,5 мл) добавляли раствор трифосгена (400 мг, 0,00137 моль) в ацетонитриле (3,6 мл) при 0єC при перемешивании. Через 30 минут к смеси добавляли N,N-диметиламинопиридин (38 мг, 0,00031 моль), и реакционной смеси давали возможность нагреться до 25°С-35°С. Смесь перемешивали в течение 16 часов. Реакционную смесь разбавляли насыщенным водным раствором бикарбоната натрия (9 мл), и органический растворитель выпаривали под пониженным давлением для получения массы. Массу экстрагировали дихлорметаном (2×10 мл). Объединенный органический слой концентрировали в вакууме с получением остатка, и остаток очищали колоночной хроматографией, используя смеси 15-20% этилацетата в гексане с получением продукта стадии 4 в виде бесцветного масла в количестве 300 мг (выход 32%).
Анализ:
MS: 315 (M+H): FW: C15H18N6O2; MW: 314,35.
Стадия 5: Получение транс-6-(гидрокси)-2-(2-метил-2H-[1,2,3,4]-тетразол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
К раствору продукта стадии 4 (300 мг, 0,00095 моль) в смеси этилацетат : метанол (3 мл:0,6 мл) добавляли 10% Pd/C (60 мг, 2 раза масс./масс.), и смесь перемешивали в атмосфере водорода при 25°С-35°С в течение 2 часов. Катализатор фильтровали через целит, и фильтрат выпаривали в вакууме с получением продукта стадии 5 в виде бесцветного масла в количестве 200 мг (выход 93%). Его использовали без дополнительной обработки для следующей реакции.
Анализ:
MS: 224 (M+); FW: C8H12N6О2; MW: 224.
Стадия 6: Получение тетрабутиламмониевой соли транс-6-(сульфоокси)-2-(2-метил-2H-[1,2,3,4]-тетразол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
К раствору продукта стадии 5 (200 мг) в Ν,Ν-диметилформамиде (1 мл) добавляли комплекс Ν,Ν-диметилформамида и триоксида серы (488 мг) одной партией при 0°С при перемешивании. Полученной смеси давали возможность нагреться до 25°С-35°С и перемешивали в течение 2 часов. К реакционной смеси добавляли раствор ацетата тетрабутиламмония (636 мг) в воде (2,1 мл). Смесь перемешивали еще в течение 16 часов. Растворитель выпаривали в вакууме при температуре ниже 40°С для получения остатка, и остаток разбавляли в воде (10 мл). Полученную смесь экстрагировали дихлорметаном (2×10 мл). Объединенный органический слой концентрировали в вакууме с получением продукта стадии 6 в виде твердого вещества белого цвета в количестве 275 мг (выход 51%).
Анализ:
MS: 599 (M+H); FW: C25H49N7O5S; MW: 599.
Стадия 7: Натриевая соль транс-6-(сульфоокси)-2-(2-метил-2H-[1,2,3,4]тетразол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
Продукт стадии 6 (270 мг) растворяли в 20% тетрагидрофуране в воде (2 мл), и полученный раствор загружали на колонку с активированной смолой Dowex 50 WX8-200-Na (длина 45 см × диаметр 3 см). Колонку элюировали тетрагидрофураном (100 мл) с последующей обработкой 50% смесью тетрагидрофурана и воды. Фракции, содержащие продукт, выпаривали в вакууме при температуре ниже 40°С, для получения указанного в заголовке соединения в виде гигроскопичного твердого вещества белого цвета в количестве 100 мг (выход 67%).
Анализ:
MS: 303 (M-H в виде свободной сульфоновой кислоты); FW для C8H11N6NaO5S; MW: 326,27.
1H ЯМР (ДМСО-d6 после обмена D2O) = 4,64 (т, 1H), 4,36 (с, 3H), 4,03 (шир.c, 3H), 2,85 (шир.c, 2H), 2,07-2,04 (м, 2H), 2,00 (м, 1H), 1,91-1,85 (м, 1H).
Пример 24
Натриевая соль транс-6-(сульфоокси)-2-(1-метил-1H-[1,2,3,4]тетразол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
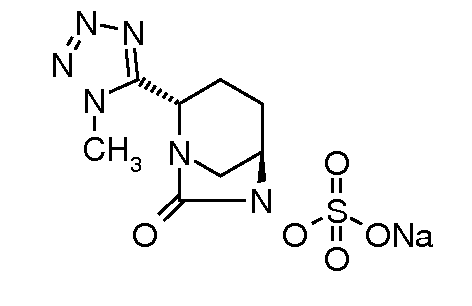
Указанное в заголовке соединение получали с использованием изомера A, полученного на стадии 2 примера 23, и с использованием процедуры, описанной в примере 23, и с использованием изомера, полученного на стадии 3.
Анализ:
1H ЯМР (ДМСО-d6 после обмена D2O) = 4,82 (д, 1H), 4,04 (с, 3H), 2,83 (шир.д, 1H), 2,50 (д, 1H), 2,22-2,18 (м, 1H), 2,08-2,03 (м, 1H), 2,00-1,96 (м, 2H).
Пример 25
Натриевая соль транс-6-сульфоокси-2-(3-этоксикарбонил-[1,2,4]оксадиазол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
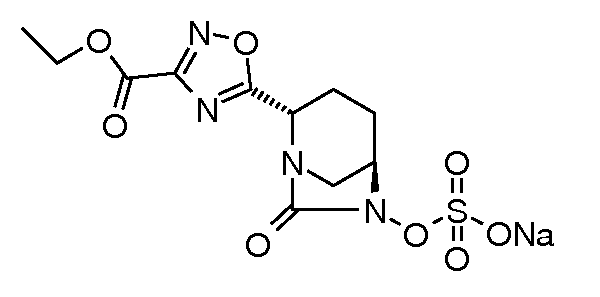
Стадия 1: Получение транс-6-бензилокси-2-(этоксикарбонилaформамидино-N-оксикарбонил)-7-оксо-1,6-диазабицикло[3.2.1]октана
К раствору транс-6-бензилокси-7-оксо-1,6-диазабицикло[3.2.1]октан-2-карбоновой кислоты (2,5 г, 9,05 ммоль) в N,N-диметилформамиде (25 мл) последовательно добавляли EDC гидрохлорид (2,56 г, 13,58 ммоль), HOBt (1,22 г, 9,05 ммоль), N-гидроксиэтоксикарбонилформамидин (669 мг, 9,05 ммоль) и N-метилморфолин (3 мл, 27,15 ммоль) при температуре от 10°С до 15°С при перемешивании. Реакционной смеси давали возможность нагреться до температуры от 25°С до 35°С и перемешивали в течение 24 часов. Ее выливали в воду (250 мл) для получения суспензии. Суспензию фильтровали и фильтрат экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты сушили над сульфатом натрия и выпаривали в вакууме с получением остатка. Остаток очищали колоночной хроматографией, используя этилацетат, гексан (5:5), с получением продукта стадии 1 в виде твердого вещества белого цвета в количестве 1,5 г (выход 42%).
Анализ:
MS: 391 (M+H); MW: 390; M.F: C18H22N4O6.
Стадия 2: Получение транс-6-бензилокси-2-(3-этоксикарбонил[1,2,4]оксадиазол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
Раствор продукта стадии 1 (1,5 г) в пиридине (15 мл) нагревали до 110°С-115°С в атмосфере аргона в течение 2,5 часов. Растворитель выпаривали в вакууме с получением остатка. Остаток очищали колоночной хроматографией, используя этилацетат, гексан (5:5), с получением продукта стадии 2 в виде твердого вещества белого цвета в количестве 1 г (выход 70%).
Анализ:
MS: 373,2 (M+H); M.W: 372; M.F.: C18H20N4O5.
Стадия 3: Получение тетрабутиламмониевой соли транс-6-сульфоокси-2-(3-этоксикарбонил[1,2,4]оксадиазол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
К раствору продукта стадии 2 (100 мг, 0,268 ммоль) в смеси дихлорметана и N,N'-диметилформамида (по 250 мкл каждого) добавляли 10% палладий на углероде (25 мг), и полученную смесь перемешивали в атмосфере водорода в течение 1 часа при 25°С-35°С. Катализатор фильтровали через микронный фильтр, и фильтрат концентрировали в вакууме при температуре ниже 40°С для получения остатка. Остаток растворяли в N,N-диметилформамиде (500 мкл) и одной партией добавляли комплекс N,N-диметилформамида и триоксида серы (50 мг, 0,321 ммоль) при температуре 0°С. Смесь перемешивали в течение 1 часа. Затем к ней добавляли водный раствор ацетата тетрабутиламмония (97 мг, 0,321 ммоль, растворенного в 350 мкл воды). Реакционной смеси давали возможность нагреться до 25°С-35°С и перемешивали еще в течение 1 часа. Летучие вещества удаляли в вакууме с получением остатка, и остаток растирали в порошок с ксилолом (10 мл) для удаления следов N,N-диметилформамида. Остаток разделяли между водой (10 мл) и дихлорметаном (10 мл). Водный слой повторно экстрагировали дихлорметаном (10 мл). Объединенные органические экстракты промывали водой (10 мл) и солевым раствором (10 мл). Органический слой сушили над сульфатом натрия и концентрировали в вакууме с получением желтого масла в качестве продукта стадии 3 в количестве 100 мг (выход 62%).
Анализ:
MS: 361,2 (M-H) свободной сульфоновой кислоты; M.W: 603: M.F: C11H13N4O8S:C16H36N.
Стадия 4: Натриевая соль транс-6-сульфоокси-2-(3-этоксикарбонил-[1,2,4]оксадиазол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
Продукт стадии 3 (100 мг) растворяли в 50% водном тетрагидрофуране, и раствор загружали на колонку со смолой Амберлит 200 Na+ (длина 20 см, диаметр 3 см). Колонку элюировали 50% водным тетрагидрофураном. Желательные фракции выпаривали в вакууме при температуре ниже 40°С для получения указанного в заголовке инновационного соединения в виде твердого вещества белого цвета в количестве 30 мг (выход 47%).
Анализ:
Т.пл.: 184-189°С (разложение);
MS: 361,2 (M-H) в виде свободной сульфоновой кислоты; M.F: C11H13N4O8S Na.
1H ЯМР (ДМСО-d6 после обмена D2O) = 4,81 (д, 1H), 4,42 (кв, 2H), 4,03 (шир.c, 1H), 3,02 (шир.д, 1H), 2,79 (д, 1H), 2,22-2,16 (м, 1H), 2,09-1,96 (м, 2H), 1,85-1,82 (м, 1H), 1,32 (т, 3H).
Пример 26
Транс-6-(сульфоокси)-2-(5-(пиперидин-4-ил)[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан
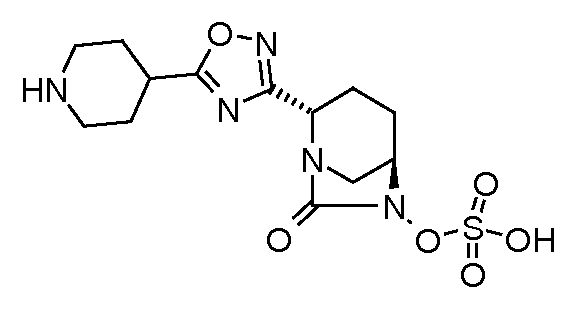
Соблюдая процедуру, описанную в примере 21, и используя хлорид пиперидин-4-карбоновой кислоты вместо уксусного ангидрида на стадии 4, получали указанное в заголовке соединение в количестве 52 мг.
Анализ:
MS: 373,1 (M-H) сульфата; M.F: C13H19N5O6S;
1H ЯМР (ДМСО-d6): 8,52 (шир.c, 1H), 8,29 (шир.c, 1H), 4,50 (дд, 1H), 4,04 (дд, 1H), 3,47-3,30 (м, 1H), 3,05-3,034 (м, 3H), 2,94-2,86 (м, 3H), 2,22-2,19 (м, 2H), 2,03-1,81 (м, 6H).
Пример 27
Натриевая соль транс-6-(сульфоокси)-2-(5-карбоксамидо[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
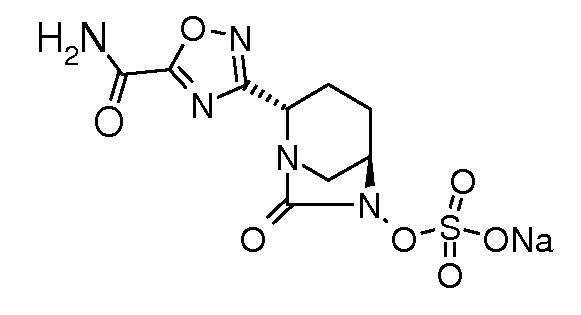
Соблюдая процедуру, описанную в примере 21, и используя карбамоилхлорид вместо уксусного ангидрида на стадии 4, получали указанное в заголовке соединение в количестве 102 мг.
Анализ:
MS: 332 (M-H); MF C9H11N5O7SNa; MW 333,28
1H ЯМР (ДМСО-d6): 8,766 (с, 1H), 8,359 (с, 1H), 4,59 (д, 1H), 4,03 (с, 1H), 2,91 (м, 2H), 2,0-2,48 (м, 3H), 1,85 (м, 1H).
Пример 28
Натриевая соль транс-6-(сульфоокси)-2-(5-(R)-пиперидин-3-ил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана
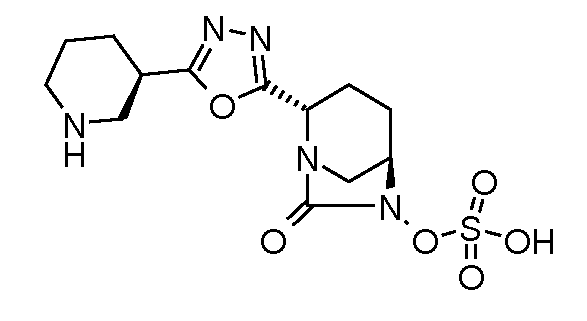
Соблюдая процедуру, описанную в примере 13, и используя гидразид (R)-N-трет-бутоксикарбонилпиперидин-3-карбоновой кислоты вместо гидразида трет-бутоксикарбониламиноуксусной кислоты, получали указанное в заголовке соединение в количестве 35 мг.
Анализ:
MS: 372,1 (M-H); MF C13H19N5О6S; MW 373,38
1H ЯМР (ДМСО-d6): 8,70 (с, 1H), 8,54 (с, 1H), 4,59 (дд, 1H), 4,06 (шир.c, 1H), 3,58 (шир.дд, 1H), 3,15-3,24 (м, 4H), 2,91-3,05 (м, 1H), 2,0-2,14 (м, 4H), 1,70-1,90 (м, 5H).
Соединения по изобретению, описанные в примерах 1-28, получали, используя (S)-пирроглутаминовую кислоту в качестве исходного соединения. Поэтому абсолютная стереохимия представляет собой кольцо (2S,5R)-7-оксо-1,6-диазабицикло[3.2.1]октана. Таким образом, соединение примера 4, натриевая соль транс-6-(сульфоокси)-2-(5-карбоксамидо[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана, имеет абсолютную стереохимию как натриевая соль (2S,5R)-6-(сульфоокси)-2-(5-карбоксамидо[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана. Альтернативно, если используемое исходное соединение представляет собой (R)-пирроглутаминовую кислоту, то полученное соединение будет иметь (2R, 5S) стереохимию в кольце 7-оксо-1,6-диазабицикло[3.2.1]октана. Ссылка на соединение в соответствии с изобретением также включает соответствующие соединения, имеющие стереохимию (2S, 5R) и (2R, 5S).
Биологическая активность
Исследовали биологическую активность репрезентативных соединений в соответствии с изобретением против различных бактериальных штаммов. В типичном исследовании выращенные в течение ночи бактериальные культуры соответствующим образом разбавляли и инокулировали на агаровую среду, содержащую дублирующие разведения тестируемых соединений. Наблюдения роста или отсутствия роста проводили после 16-20 часов инкубации при температуре окружающего воздуха 35±2°С. В целом процедуру выполняли в соответствии с рекомендациями Института клинических и лабораторных стандартов (CLSI) (Clinical and Laboratory Standards Institute (CLSI), Стандарты Института клинических и лабораторных стандартов (CLSI) по выполнению тестов восприимчивости к антимикробным средствам, 20-ое Информационное дополнение, M 100 - S20, том 30, № 1, 2010). Результаты этих исследований суммированы в таблицах 3 и 4. В целом, было обнаружено, что индивидуальная активность соединений по изобретению составила >32 мкг/мл.
Антибактериальную активность репрезентативных соединений в соответствии с изобретением исследовали также в комбинации по меньшей мере с одним антибактериальным средством с использованием описанного выше протокола, и результаты представлены в таблицах 3 и 4. Как видно, применение соединений в соответствии с изобретением значимо снижало величины MIC (минимальной ингибирующей концентрации) антибактериального средства (например, в данном случае цефтазидима). Результаты также свидетельствуют о том, что соединения в соответствии с изобретением увеличивают антибактериальную эффективность антибактериального средства, когда указанное антибактериальное средство совместно вводится с фармацевтические эффективным количеством соединения формулы (I), или его стереоизомера, или фармацевтически приемлемой соли.
NCTC 13351
M50
7MP
W 13353
W 13351
W 13352
M50
7MP
B89
H520
H521
H522
13301
13304
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-7-ОНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2614418C2 |
| ПРОИЗВОДНЫЕ 1,6- ДИАЗАБИЦИКЛО [3,2,1] ОКТАН-7-ОНА И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ БАКТЕРИАЛЬНЫХ ИНФЕКЦИОННЫХ БОЛЕЗНЕЙ | 2012 |
|
RU2578370C2 |
| АЗОТСОДЕРЖАЩИЕ БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2835672C1 |
| ПРОИЗВОДНЫЕ 1,6-ДИАЗАБИЦИКЛО[3,2,1]ОКТАН-7-ОНА И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ БАКТЕРИАЛЬНЫХ ИНФЕКЦИОННЫХ БОЛЕЗНЕЙ | 2012 |
|
RU2636147C1 |
| АЗОТСОДЕРЖАЩИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2012 |
|
RU2569307C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 2015 |
|
RU2705375C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 2014 |
|
RU2702361C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ СУЛБАКТАМ И ИНГИБИТОР БЕТА-ЛАКТАМАЗЫ | 2011 |
|
RU2560846C1 |
| (2S,5R)-5-[(бензилокси)амино]пиперидин-2-карбоксамид | 2012 |
|
RU2610091C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ВКЛЮЧАЯ ТРАНС-7-ОКСО-6-(СУЛЬФОКСИ)-1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-2-КАРБОКСАМИД И ЕГО СОЛИ | 2012 |
|
RU2769076C2 |
Изобретение относится к новому гетероциклическому соединению общей формулы (I) или к его стереоизомеру, или к его фармацевтически приемлемой соли, где Q обозначает оксадиазол, тиадиазол или тетразол; R1 обозначает: (b) (CO)n-R3 или (с) COOR4, n=0 или 1; R2 обозначает SO3M, М обозначает водород или катион; R3 обозначает: (а) C1-С6-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, NR6R7, гетероциклила или арила, (b) NR6R7, (с) CONR6R7, (d) арил, (e) гетероциклил, или (f) гетероарил, необязательно замещенный CONR6R7, R4 обозначает (а) водород, или (b) C1-С6-алкил; R6 и R7, каждый независимо, обозначает (а) водород, или (b) R6 и R7 соединены вместе для образования от четырех- до семичленного кольца, где циклоакил представляет собой 3-7-членный циклический углеводородный радикал; гетероциклил представляет собой 4-7-членную циклоалкильную группу, содержащую один или более гетероатомов, выбранных из азота, кислорода или серы; арил представляет собой моноциклический или полициклический углеводород, содержащий 6-14 атомов в кольце; гетероарил представляет собой моноциклический или полициклический гетероарил, где один или более атомов углерода заменены гетероатомами, выбранными из азота, кислорода или серы. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I) и способу предотвращения или лечения бактериальной инфекции, основанному на использовании соединения формулы (I). Технический результат: получены новые гетероциклические соединения, полезные при лечении бактериальных инфекций. 13 н. и 14 з.п. ф-лы, 4 табл., 28 пр.
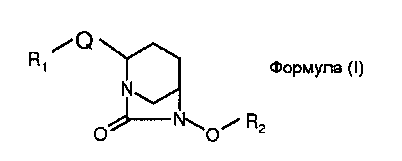
1. Соединение формулы (I)
или его стереоизомер, или фармацевтически приемлемая соль;
где:
Q обозначает оксадиазол, тиадиазол или тетразол;
R1 обозначает:
(b) (CO)n-R3 или
(с) COOR4,
n=0 или 1;
R2 обозначает SO3M,
М обозначает водород или катион;
R3 обозначает:
(а) C1-С6-алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, NR6R7, гетероциклила или арила,
(b) NR6R7,
(с) CONR6R7,
(d) арил,
(e) гетероциклил, или
(f) гетероарил, необязательно замещенный CONR6R7;
R4 обозначает:
(а) водород, или
(b) C1-С6-алкил;
R6 и R7, каждый независимо, обозначает:
(а) водород,
или
(b) R6 и R7 соединены вместе для образования от четырех- до семичленного кольца,
где циклоакил представляет собой 3-7-членный циклический углеводородный радикал;
гетероциклил представляет собой 4-7-членную циклоалкильную группу, содержащую один или более гетероатомов, выбранных из азота, кислорода или серы;
арил представляет собой моноциклический или полициклический углеводород, содержащий 6-14 атомов в кольце;
гетероарил представляет собой моноциклический или полициклический гетероарил, где один или более атомов углерода заменены гетероатомами, выбранными из азота, кислорода или серы.
2. Соединение по п.1, выбранное из
сложного моно-[2-(5-аминометил[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-илового]эфира транс-серной кислоты;
сложного моно-[2-(5-((S)-1-аминоэтил)-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-илового]эфира транс-серной кислоты;
сложного моно-[2-(5-((R)-1-аминоэтил)-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-илового]эфира транс-серной кислоты;
сложного моно-[2-(5-(пиперидин-4-ил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-илового]эфира транс-серной кислоты;
сложного моно-[2-(5-((S)-пирролидин-2-ил)-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-илового]эфира транс-серной кислоты;
сложного моно-[2-(5-(пиперазин-1-илметил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-илового]эфира транс-серной кислоты;
сложного моно-[2-(5-((RS)-1-амино-1-фенил-метил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-илового]эфира транс-серной кислоты;
моно-[2-(5-(пиперидин-4-ил)-[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана транс-серной кислоты;
моно-[2-(5-((R)-пиперидин-3-ил)-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана транс-серной кислоты
или их стереоизомера, или фармацевтически приемлемой соли.
3. Соединение по п.1, выбранное из
натриевой соли транс-6-(сульфоокси)-2-(5-метил-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-этил-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-трифторметил-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-карбоксамидо-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-(изооксазол-3-ил)-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-(фуран-2-ил)-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-фенил-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-(пиридин-2-ил)-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-(6-карбоксамидо-пиридин-2-ил)-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-(5,6-дигидро-8Н-имидазо[2,1-с][1,4]оксазин-2-ил)[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-(морфолино-4-метил)-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли сложного моно-[2-(5-(морфолин-4-илкарбонил)-[1,3,4]оксадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октан-6-илового]эфира транс-серной кислоты;
натриевой соли транс-6-(сульфоокси)-2-(5-метил-[1,3,4]-тиадиазол-2-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-метил-[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-этоксикарбонил-[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(2-метил-2Н-[1,2,3,4]-тетразол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(1-метил-1Н-[1,2,3,4]-тетразол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(3-этоксикарбонил-[1,2,4]оксадиазол-5-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
натриевой соли транс-6-(сульфоокси)-2-(5-карбоксамидо-[1,2,4]оксадиазол-3-ил)-7-оксо-1,6-диазабицикло[3.2.1]октана;
или их стереоизомера.
4. Фармацевтическая композиция для предотвращения или лечения бактериальной инфекции, включающая соединение по любому из пп.1-3 в эффективном количестве.
5. Способ предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества соединения по любому из пп.1-3.
6. Способ предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества соединения по любому из пп.1-3.
7. Фармацевтическая композиция по п.4, дополнительно включающая по меньшей мере один ингибитор бета-лактамазы, выбранный из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
8. Фармацевтическая композиция по пп.4 или 7, дополнительно включающая по меньшей мере одно антибактериальное средство или его фармацевтически приемлемое производное.
9. Способ предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции по пп.4, 7 или 8.
10. Способ предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем способ включает введение указанному субъекту фармацевтически эффективного количества фармацевтической композиции по пп.4, 7 или 8.
11. Способ предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (а) соединения формулы (I) по п.1, или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
12. Способ предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (а) соединения формулы (I) по п.1, или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного.
13. Способ предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (а) соединения формулы (I) по п.1, или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного антибактериального средства или его фармацевтически приемлемого производного.
14. Способ предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (а) соединения формулы (I) по п.1, или его стереоизомера, или фармацевтически приемлемой соли, и (b) по меньшей мере одного антибактериального средства или его фармацевтически приемлемого производного.
15. Способ предотвращения или лечения бактериальной инфекции у субъекта, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (а) соединения формулы (I) по п.1, или его стереоизомера, или фармацевтически приемлемой соли; (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (с) по меньшей мере одного антибактериального средства или его фармацевтически приемлемого производного.
16. Способ предотвращения или лечения бактериальной инфекции у субъекта, причем указанная инфекция вызвана бактериями, продуцирующими один или несколько бета-лактамазных ферментов, причем указанный способ включает введение указанному субъекту фармацевтически эффективного количества: (а) соединения формулы (I) по п.1, или его стереоизомера, или фармацевтически приемлемой соли, (b) по меньшей мере одного ингибитора бета-лактамазы, выбранного из сульбактама, тазобактама, клавулановой кислоты или ее фармацевтически приемлемого производного, и (с) по меньшей мере одного антибактериального средства или его фармацевтически приемлемого производного.
17. Способ увеличения антибактериальной эффективности антибактериального средства у субъекта, причем указанный способ включает совместное введение указанного антибактериального средства или его фармацевтически приемлемого производного с фармацевтически эффективным количеством соединения формулы (I) по п.1, или его стереоизомера, или фармацевтически приемлемой соли.
18. Фармацевтическая композиция по п.8, где антибактериальное средство выбрано из группы, состоящей из аминогликозидных, ансамициновых, карбацефемовых, цефалоспориновых, цефамициновых, линкозамидных, липопептидных, макролидных, монобактамовых, нитрофурановых, пенициллиновых, полипептидных, хинолоновых, сульфонамидных, тетрациклиновых или оксазолидиноновых антибактериальных средств.
19. Фармацевтическая композиция по п.8, где антибактериальное средство представляет собой бета-лактамовое антибактериальное средство.
20. Фармацевтическая композиция по п.8, где указанное антибактериальное средство выбрано из группы, состоящей из пенициллинов, пенемов, карбапенемов, цефалоспоринов и монобактамов.
21. Фармацевтическая композиция по п.8, где антибактериальное средство представляет собой цефалоспориновый антибиотик, выбранный из группы, состоящей из цефалотина, цефалоридина, цефаклора, цефадроксила, цефамандола, цефазолина, цефалексина, цефрадина, цефтизоксима, цефокситина, цефацетрила, цефотиама, цефотаксима, цефсулодина, цефоперазона, цефтизоксима, цефменоксима, цефметазола, цефалоглицина, цефоницида, цефодизима, цефпирома, цефтазидима, цейфриаксона, цефпирамида, цефбуперазона, цефозопрана, цефепима, цефоселиса, цефлупренама, цефузонама, цефпимизола, цефклидина, цефиксима, цефтибутена, цефдинира, цефподоксима аксетила, цефподоксима проксетила, цефтерама пивоксила, цефетамета пивоксила, цефкапена пивоксила или цефдиторена пивоксила, цефуроксима, цефуроксима аксетила, лоракарбацефа, цефтаролина и латамоксефа.
22. Фармацевтическая композиция по п.8, где антибактериальное средство выбрано из группы, состоящей из цефтазидима, цефепима, цефпирома, пиперациллина дорипенема, меропенема, имипенема, цефтаролина и цефтолозана.
23. Способ по любому из пп.9, 10, 13, 14, 15, 16 или 17, где антибактериальное средство выбрано из группы, состоящей из аминогликозидных, ансамициновых, карбацефемовых, цефалоспориновых, цефамициновых, линкозамидных, липопептидных, макролидных, монобактамовых, нитрофурановых, пенициллиновых, полипептидных, хинолоновых, сульфонамидных, тетрациклиновых или оксазолидиноновых антибактериальных средств.
24. Способ по любому из пп.9, 10, 13, 14, 15, 16 или 17, где антибактериальное средство представляет собой бета-лактамовое антибактериальное средство.
25. Способ по любому из пп.9, 10, 13, 14, 15, 16 или 17, где указанное антибактериальное средство выбрано из группы, состоящей из пенициллинов, пенемов, карбапенемов, цефалоспоринов и монобактамов.
26. Способ по любому из пп.9, 10, 13, 14, 15, 16 или 17, где антибактериальное средство представляет собой цефалоспориновый антибиотик, выбранный из группы, состоящей из цефалотина, цефалоридина, цефаклора, цефадроксила, цефамандола, цефазолина, цефалексина, цефрадина, цефтизоксима, цефокситина, цефацетрила, цефотиама, цефотаксима, цефсулодина, цефоперазона, цефтизоксима, цефменоксима, цефметазола, цефалоглицина, цефоницида, цефодизима, цефпирома, цефтазидима, цейфриаксона, цефпирамида, цефбуперазона, цефозопрана, цефепима, цефоселиса, цефлупренама, цефузонама, цефпимизола, цефклидина, цефиксима, цефтибутена, цефдинира, цефподоксима аксетила, цефподоксима проксетила, цефтерама пивоксила, цефетамета пивоксила, цефкапена пивоксила или цефдиторена пивоксила, цефуроксима, цефуроксима аксетила, лоракарбацефа, цефтаролина и латамоксефа.
27. Способ по любому из пп.9, 10, 13, 14, 15, 16 или 17, где антибактериальное средство выбрано из группы, состоящей из цефтазидима, цефепима, цефпирома, пиперациллина, дорипенема, меропенема, имипенема, цефтаролина и цефтолозана.
| Колосоуборка | 1923 |
|
SU2009A1 |
| US 20030199541 A1, 23.10.2003 | |||
| КОПИРОВАЛЬНЫЙ СТАНОК ДЛЯ ДЕРЕВА | 1925 |
|
SU7220A1 |
| ДИАЗАБИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2124014C1 |

