ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к безводным полиморфным формам, гидратированным полиморфным формам и аморфным формам 3-(2,6-дихлор-3,5-диметоксифенил)-1-{6-[4-(4-этилпиперазин-1-ил)-фениламино]-пиримидин-4-ил}-1-метилмочевины, к их солям, способам их получения, содержащим их композициям и способам лечения с их использованием.
УРОВЕНЬ ТЕХНИКИ
3-(2,6-Дихлор-3,5-диметоксифенил)-1-{6-[4-(4-этилпиперазин-1-ил)-фениламино]-пиримидин-4-ил}-1-метилмочевина (описана в USSN 11/570983, поданном 23 июня 2005 г., и который во всей своей полноте включен в настоящее изобретение в качестве ссылки) обладает структурой формулы I:
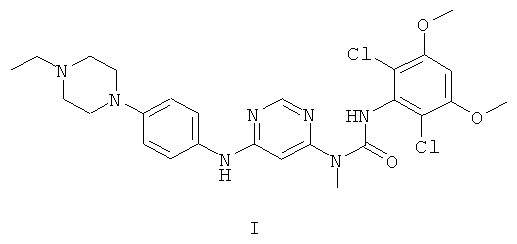
Соединение формулы I является ингибитором протеинкиназы и применимо для лечения пролиферативных заболеваний, опосредуемых протеинкиназами. В частности, соединение формулы I ингибирует киназы FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2 и Ret. Поэтому оно применимо для лечения раковых заболеваний, включая острый миелоцитарный лейкоз, меланоцитарную неоплазию, рак молочной железы, рак толстой кишки, рак легких (в особенности мелкоклеточный рак легких), рак предстательной железы или саркому Капоши.
Хорошо известно, что кристаллическая форма активного фармацевтического ингредиента (АФИ) конкретного лекарственного средства часто является важным фактором, определяющим легкость приготовления, гигроскопичность, стабильность, растворимость, стабильность при хранении, легкость приготовления препарата, скорость растворения в желудочно-кишечном тракте и биологическую доступность in vivo лекарственного средства. Кристаллические формы образуются, когда одна и та же композиция кристаллизуется в различных решетках, что приводит к разным термодинамическим характеристикам и стабильностям, характерным для конкретной кристаллической формы. Кристаллические формы также могут представлять собой различные гидраты или сольваты одного и того же соединения. При решении, какая форма является предпочтительной, сопоставляют многочисленные характеристики форм и предпочтительную форму выбирают с учетом многих физических характеристик. Вполне возможно, что одна форма может быть предпочтительной при некоторых обстоятельствах, когда критически важными считают некоторые характеристики, такие как легкость приготовления, стабильность и т.п. В других ситуациях может быть предпочтительной другая форма вследствие большей скорости растворения и/или лучшей биологической доступности. Пока невозможно предсказать, какие полиморфные формы образует конкретное соединение или соль соединения и какие из таких полиморфных форм будут подходящими для коммерческого применения в терапевтической композиции или какие полиморфные формы будут обладать необходимыми характеристиками.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к кристаллическим и аморфным формам соединения формулы I, композициям, содержащим кристаллические и аморфные формы, и к способам получения кристаллических и аморфных форм и композиций. Настоящее изобретение также относится к способам применения кристаллических или аморфных форм соединений формулы I и содержащих их композиций для лечения различных заболеваний, включая, но не ограничиваясь только ими, те, которые можно предупредить, подавить или облегчить их протекание путем ингибирования активности киназы, выбранной из группы, включающей киназы FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2 и Ret.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
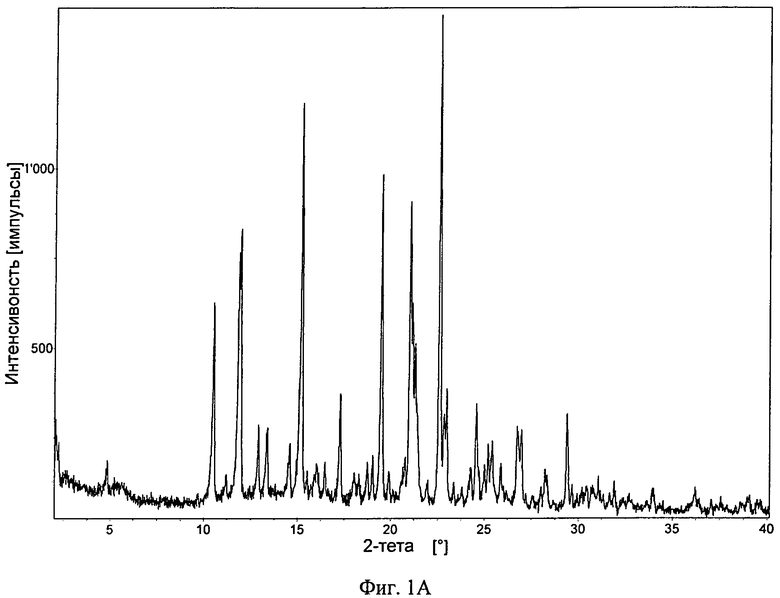
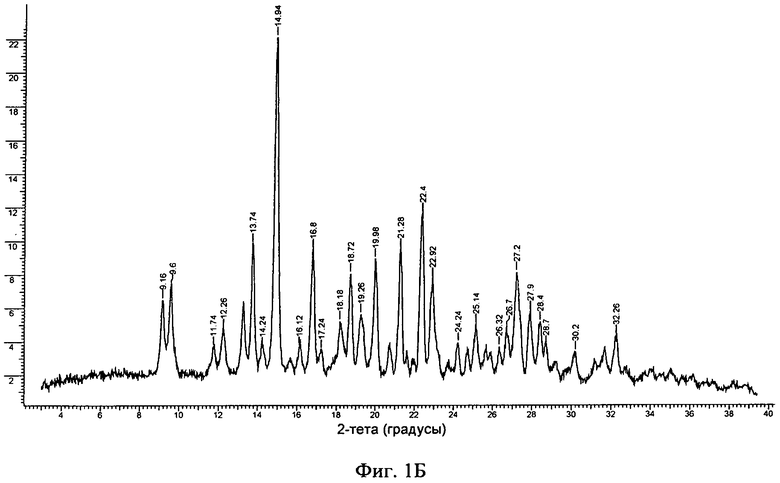
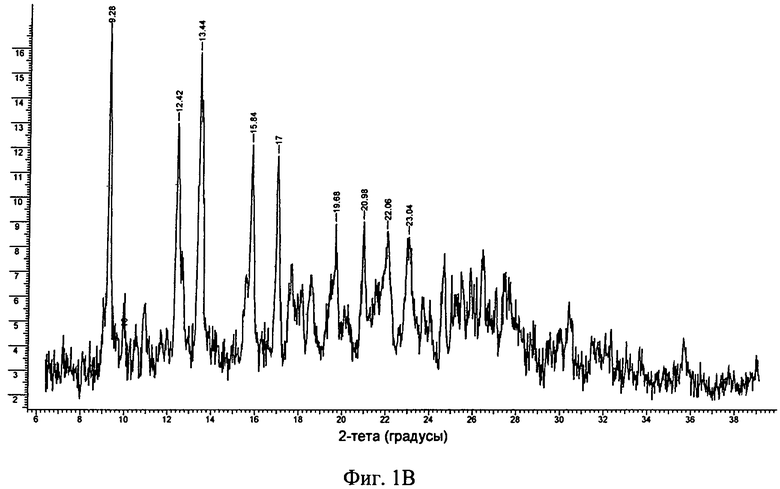
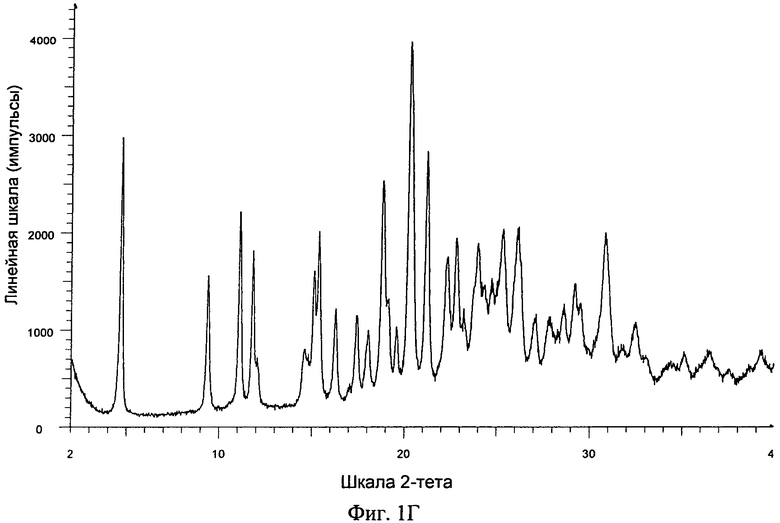
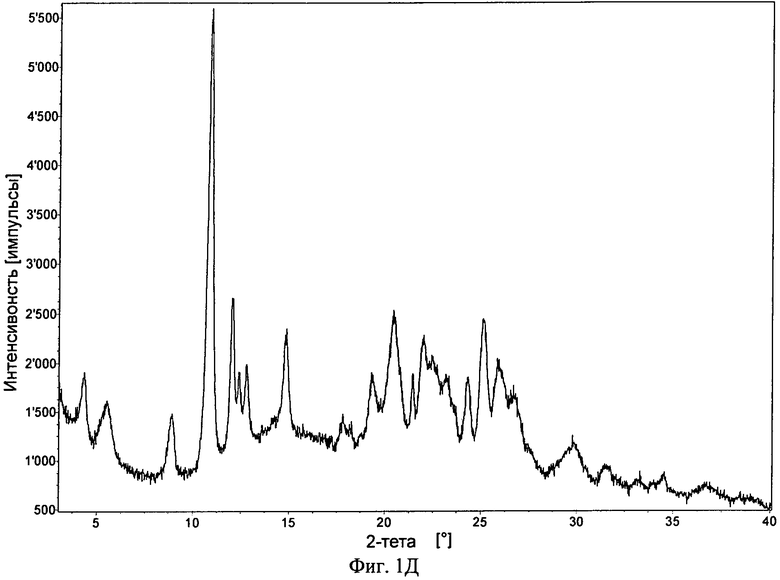
Фиг.1А-Д. ПРРГ свободного основания (фиг.1А) соединения формулы I и его фосфатов: форма А (фиг.1Б); и форма В (фиг.1В), моногидрат свободного основания (фиг.1Г) и дигидрат гидрохлорида (фиг.1Д).
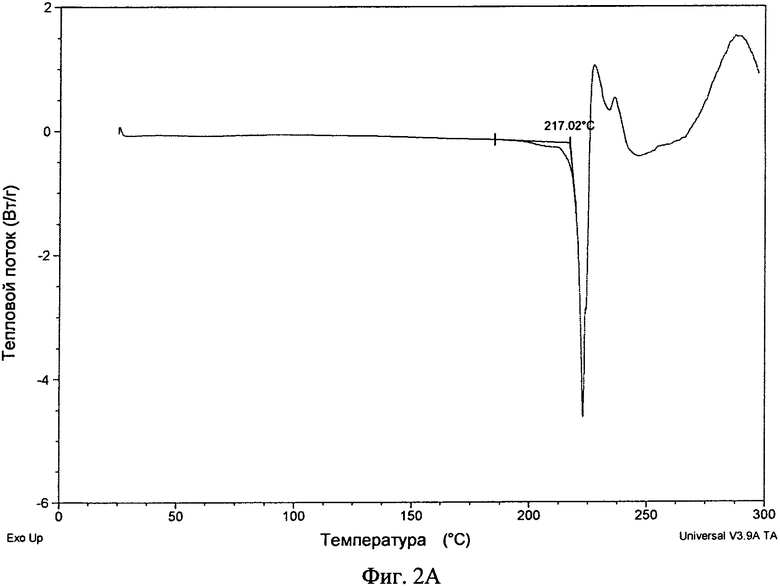
Фиг.2А и 2Б. Полученные с помощью ДСК термограммы соединения формулы I в виде безводного свободного основания (фиг.2А и в виде фосфата (форма А, фиг.2Б).
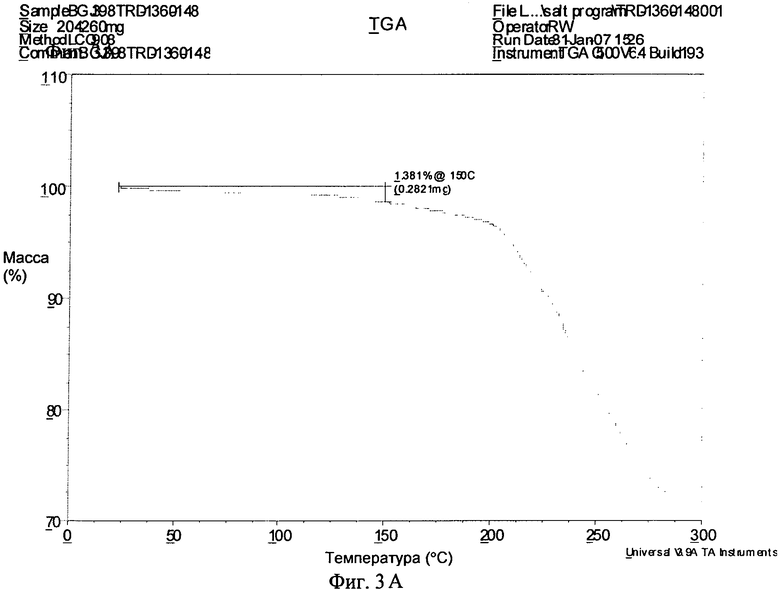
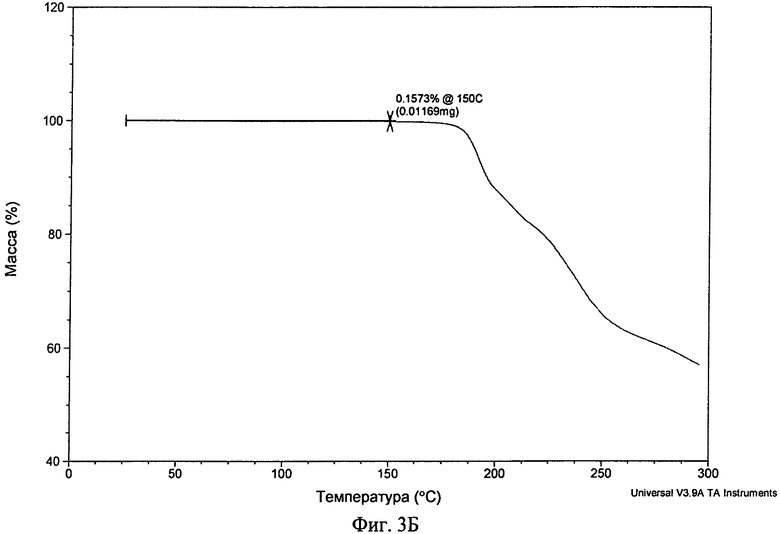
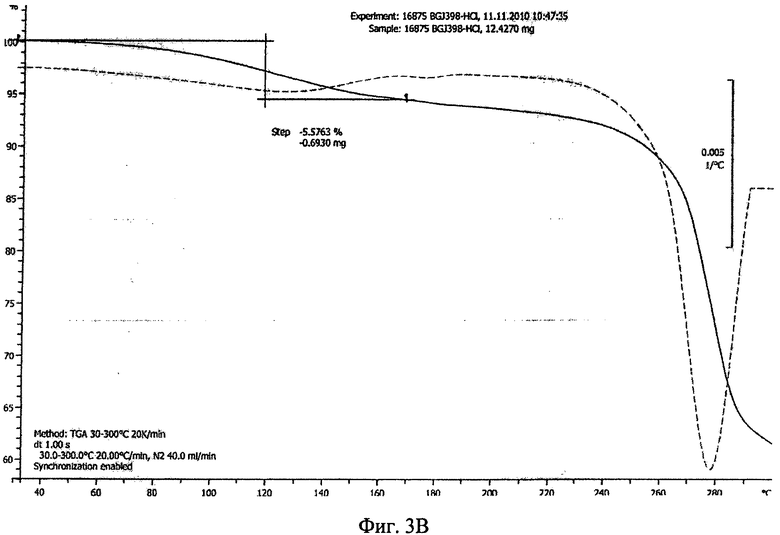
Фиг.3А-3В. Полученные с помощью ТГА термограммы соединения формулы I в виде безводного свободного основания (фиг.3А), в виде фосфата (форма А, фиг.3Б) и в виде гидрохлорида (дигидрат, фиг.3В).
Фиг.4А-4В. Изотермы сорбции-десорбции соединения формулы I в виде безводного свободного основания (фиг.4А), в виде фосфата (форма А, фиг.4Б) и в виде моногидрата свободного основания (фиг.4В) получали с помощью ДСП при 25°C.
Фиг.5А и 5Б. Полученные с помощью СЭМ микрофотографии соединения формулы I в виде кристаллического свободного основания (фиг.5А) и в виде фосфата (форма А, фиг.5Б).
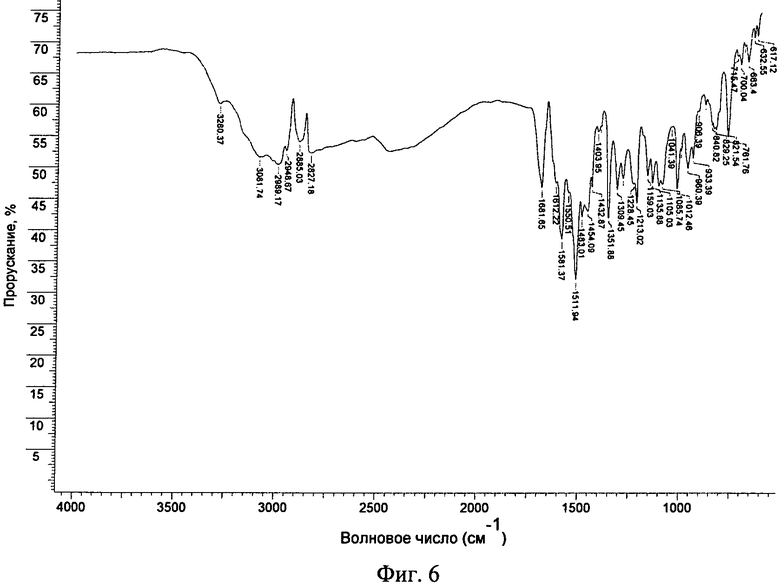
Фиг.6. Ф-ИК-спектр фосфата (форма А) соединения формулы I.
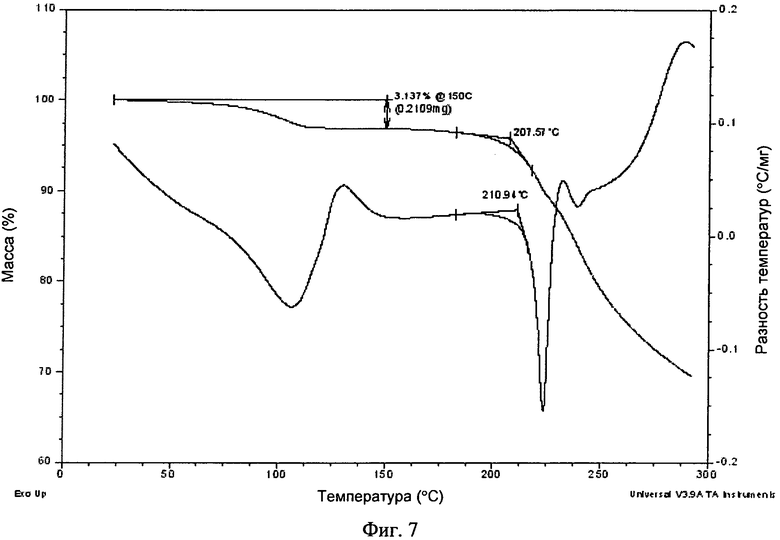
Фиг.7. Полученные с помощью ДСК и ТГА термограммы моногидрата свободного основания.
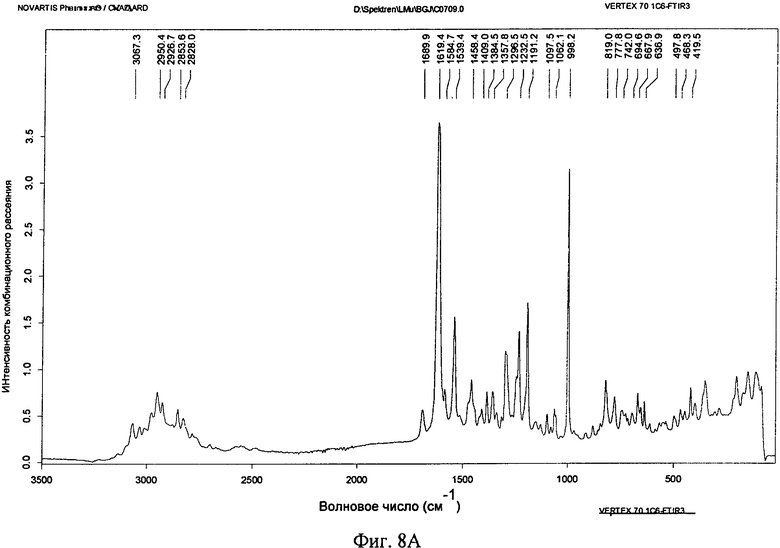
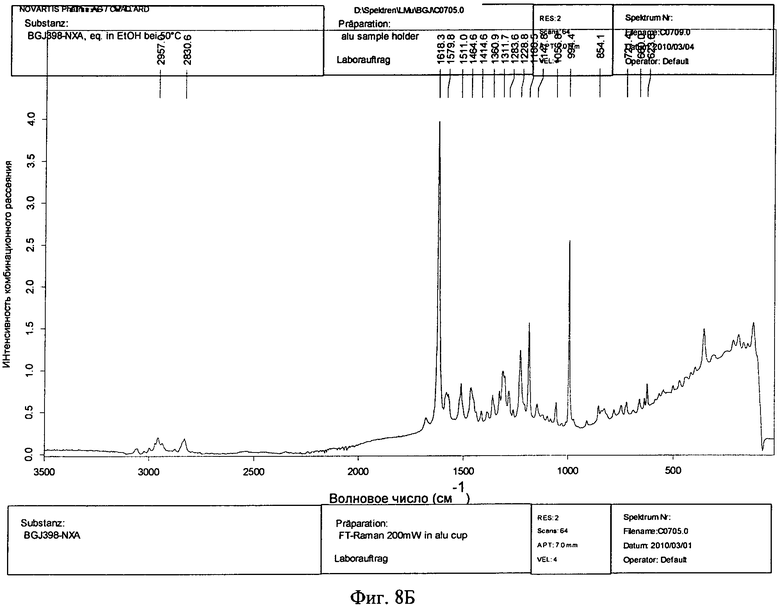
Фиг.8А и 8Б. Спектры комбинационного рассеяния соединения формулы I в виде безводного свободного основания (фиг.8А) и в виде моногидрата свободного основания (фиг.8Б).
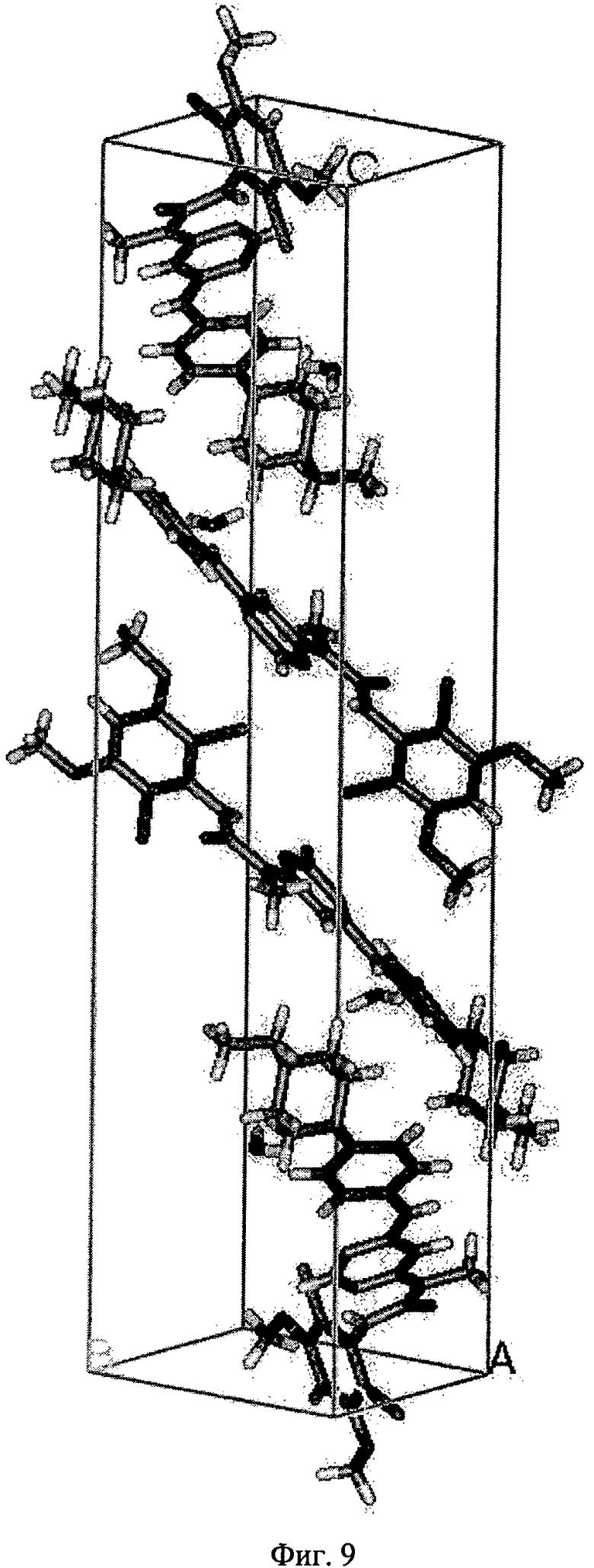
Фиг.9. Рентгеновская структура монокристалла элементарной формы моногидрата свободного основания соединения формулы I.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Одним объектом настоящего изобретения являются безводные и гидратированные кристаллические формы соединения формулы I (свободного основания):
и их фосфаты.
В одном варианте осуществления настоящее изобретение относится к безводной кристаллической форме свободного основания, обладающей порошковой рентгенограммой, содержащей характеристический пик, выраженный в 2θ, расположенный примерно при 12°. В другом варианте осуществления порошковая рентгенограмма дополнительно содержит один или большее количество характеристических пиков, выраженных в 2θ, расположенных при 10,5°, примерно при 15,2°, примерно при 19,5° и примерно при 21,0°. Таким образом, порошковая рентгенограмма безводной формы свободного основания может содержать 1, 2, 3, 4 или 5 характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 10,5°, примерно при 12,0°, примерно при 15,2°, примерно при 19,5° и примерно при 21,0°. Порошковая рентгенограмма может дополнительно содержать 1, 2 или 3 дополнительных характеристических пика, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 11,9°, примерно при 21,3° и примерно при 22,6°. В другом варианте осуществления безводная кристаллическая форма свободного основания обладает порошковой рентгенограммой, в основном такой, как представленная на фиг.1А. При использовании в настоящем изобретении термины "примерно" и "в основном" указывают применительно к значениям 2θ, что такие значения для отдельных пиков могут меняться на ±0,4°. В некоторых вариантах осуществления значения 2θ для отдельных пиков могут меняться на ±0,2°.
Безводную кристаллическую форму свободного основания можно охарактеризовать термически. В одном варианте осуществления безводная кристаллическая форма свободного основания обладает полученной с помощью дифференциальной сканирующей калориметрии (ДСК) термограммой, на которой начало эндотермы расположено примерно при 217°C. В другом варианте осуществления безводная кристаллическая форма свободного основания обладает полученной с помощью дифференциальной сканирующей калориметрии термограммой, в основном такой, как представленная на фиг.2А. При использовании в настоящем изобретении термины "примерно" и "в основном" указывают применительно к особенностям, таким как эндотермы, экзотермы, сдвиги базовых линий и т.п., что их значения могут меняться на ±2°C. Для ДСК наблюдающиеся изменения температур зависят от скорости изменения температуры, а также методики приготовления образца и конкретного использующегося прибора. Таким образом, значения, приведенные в настоящем изобретении, относящиеся к полученным с помощью ДСК термограммам, могут меняться на ±4°C.
Безводную кристаллическую форму свободного основания также можно охарактеризовать с помощью спектроскопии комбинационного рассеяния. В одном варианте осуществления безводная кристаллическая форма свободного основания обладает характеристическими пиками комбинационного рассеяния, расположенными примерно при 2950, примерно при 1619, примерно при 1539, примерно при 1297, примерно при 1233, примерно при 1191, и примерно при 998 см-1. В некоторых вариантах осуществления безводная кристаллическая форма свободного основания обладает одним или большим количеством дополнительных характеристических пиков комбинационного рассеяния, расположенных примерно при 3067, примерно при 2927, примерно при 2828, примерно при 1690, примерно при 1585, примерно при 1458, примерно при 1385, примерно при 1358, примерно при 1098, примерно при 1062, примерно при 819, примерно при 778, примерно при 695, примерно при 668, примерно при 637, примерно при 498, примерно при 468 и примерно при 420 см-1. В другом варианте осуществления безводная кристаллическая форма свободного основания обладает спектром комбинационного рассеяния, в основном таким, как представленный на фиг.8А. При использовании в настоящем изобретении термины "примерно" и "в основном" указывают применительно к значениям волновых чисел, что такие значения для отдельных пиков могут меняться на ±2 см-1.
Одним объектом настоящего изобретения является кристаллический моногидрат свободного основания, обладающий порошковой рентгенограммой, содержащей характеристический пик, выраженный в 2θ, расположенный примерно при 20,3°. В другом варианте осуществления порошковая рентгенограмма дополнительно содержит один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 21,2° и 19°. В другом варианте осуществления порошковая рентгенограмма дополнительно содержит один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 4,7°, примерно при 9,4° и примерно при 11,0°. Таким образом, порошковая рентгенограмма формы моногидрата свободного основания может содержать 1, 2, 3, 4, 5 или 6 характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 4,7°, примерно при 9,4°, примерно при 11,0°, примерно при 18,8°, примерно при 20,3° и примерно при 21,2°. Порошковая рентгенограмма формы моногидрата свободного основания может дополнительно содержать 1, 2, 3, 4, 5, 6 или 7 характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 11,8°, примерно при 15,3°, примерно при 16,2°, примерно при 19,1°, примерно при 22,3°, примерно при 22,8° и примерно при 25,3°. В другом варианте осуществления кристаллическая форма моногидрата свободного основания обладает порошковой рентгенограммой, в основном такой, как представленная на фиг.1Г.
Кристаллическую форму моногидрата свободного основания можно охарактеризовать термически. В одном варианте осуществления кристаллическая форма моногидрата свободного основания обладает полученной с помощью ДСК термограммой, на которой начало эндотермы расположено примерно при 211°C. В другом варианте осуществления кристаллическая форма моногидрата свободного основания обладает полученной с помощью ДСК термограммой и/или полученной с помощью ТГА термограммой, в основном такой, как представленная на фиг.7. Полученная с помощью ТГА термограмма, представленная на фиг.7, показывает, что по данным потери, составляющей примерно 0,2 мг для образца массой 6,7 мг, или потери массы, составляющей 3,1%, потеря воды (молекулярная масса 18) моногидратом (молекулярная масса 578) составляет 18/578×100=3,1%.
Моногидрат кристаллической формы свободного основания также можно охарактеризовать с помощью спектроскопии комбинационного рассеяния. В одном варианте осуществления безводная кристаллическая форма свободного основания обладает характеристическими пиками комбинационного рассеяния, расположенными примерно при 2957, примерно при 2831, примерно при 1618, примерно при 1511, примерно при 1465, примерно при 1361, примерно при 1229, примерно при 1186 и примерно при 994 см-1. В некоторых вариантах осуществления безводная кристаллическая форма свободного основания обладает дополнительными характеристическими пиками комбинационного рассеяния, расположенными примерно при примерно 1580, примерно при 1415, примерно при 1312, примерно при 1284, примерно при 1184, примерно при 1057, примерно при 854, примерно при 721, примерно при 661 и примерно при 624 см-1. В другом варианте осуществления моногидрат кристаллической формы свободного основания обладает спектром комбинационного рассеяния, в основном таким, как представленный на фиг.8Б.
Другим объектом настоящего изобретения является безводная кристаллическая соль монофосфорной кислоты (или фосфат) соединения формулы I:
В другом варианте осуществления безводная кристаллическая соль монофосфорной кислоты находится в форме A, которая обладает порошковой рентгенограммой, содержащей характеристический пик, выраженный в 2θ, расположенный примерно при 15°. В другом варианте осуществления порошковая рентгенограмма дополнительно содержит один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 13,7°, примерно при 16,8°, примерно при 21,3° и примерно при 22,4°. В другом варианте осуществления порошковая рентгенограмма дополнительно содержит один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 9,2°, примерно при 9,6°, примерно при 18,7°, примерно при 20,0°, примерно при 22,9° и примерно при 27,2°. В другом варианте осуществления безводная кристаллическая форма (форма А) соли монофосфорной кислоты (или фосфата) соединения формулы I обладает порошковой рентгенограммой, содержащей не менее трех характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 13,7°, примерно при 15°, примерно при 16,8°, примерно при 21,3° и примерно при 22,4°. Таким образом, порошковая рентгенограмма формы А фосфата может содержать 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или 11 характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 9,2°, примерно при 9,6°, примерно при 13,7°, примерно при 15°, примерно при 16,8°, примерно при 18,7°, примерно при 20,0°, примерно при 21,3° и примерно при 22,4°, примерно при 22,9° и примерно при 27,2°. В другом варианте осуществления безводная кристаллическая форма (форма А) фосфата обладает порошковой рентгенограммой, в основном такой, как представленная на фиг.1Б.
Безводная кристаллическая форма фосфата (форма А) можно охарактеризовать термически. В одном варианте осуществления безводная кристаллическая форма фосфата (форма А) обладает полученной с помощью дифференциальной сканирующей калориметрии термограммой, на которой начало эндотермы расположено примерно при 184°C. В другом варианте осуществления безводная кристаллическая форма фосфата (форма А) обладает полученной с помощью дифференциальной сканирующей калориметрии термограммой, в основном такой, как представленная на фиг.2Б.
Другим объектом настоящего изобретения является безводная кристаллическая соль монофосфорной кислоты в форме B, которая обладает порошковой рентгенограммой, содержащей один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 9,3°, примерно при 12,5°, примерно при 13,4°, примерно при 15,8° и примерно при 17°. В одном варианте осуществления форма B фосфата обладает порошковой рентгенограммой, в основном такой, как представленная на фиг.1В.
Одним объектом настоящего изобретения является аморфная форма соли монофосфорной кислоты соединения формулы I:
ПРРГ аморфной формы соли монофосфорной кислоты соединения формулы I не содержит характеристических пиков. С помощью ДСК для аморфной формы не обнаружено стеклования, однако при исследовании с помощью ТГА образец начинает разлагаться при температуре, равной примерно 115°C, которая намного ниже, чем для формы A кристаллического фосфата.
Еще одним объектом настоящего изобретения является соль хлористоводородной кислоты (или гидрохлорид) соединения формулы I:
В одном варианте осуществления гидрохлоридом является кристаллический дигидрат. Дигидрат может обладать порошковой рентгенограммой, содержащей один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 10,9°, примерно при 12,1°, примерно при 14,8°, примерно при 20,5°, примерно при 22° и примерно при 25,1°. В некоторых вариантах осуществления гидрохлорид обладает порошковой рентгенограммой, в основном такой, как представленная на фиг.1Д.
В дополнение к описанным выше методикам характеризации кристаллических форм, предлагаемых в настоящем изобретении, для характеризации других кристаллических или аморфных форм, предлагаемых в настоящем изобретении, также можно использовать ПРРГ, рентгенографию монокристаллов, ДСК, динамическую сорбцию паров (ДСП), морфологию кристаллов, твердофазный ядерный магнитный резонанс, комбинационное рассеяние, инфракрасную (ИК) спектроскопию.
Другим объектом настоящего изобретения является способ получения безводной кристаллической формы фосфата в форме A, включающий взаимодействие суспензии соединения формулы I (свободного основания):
с фосфорной кислотой с получением безводной кристаллической формы фосфата в форме А. В одном варианте осуществления суспензия соединения формулы I содержит изопропанол. В другом варианте осуществления способ дополнительно включает кристаллизацию безводной кристаллической формы фосфата в форме A из суспензии. В другом варианте осуществления способ дополнительно включает выделение закристаллизовавшейся безводной кристаллической формы А.
В некоторых вариантах осуществления способа фосфорная кислота, с которой проводится взаимодействие, содержится в количестве, составляющем от примерно 1 до примерно 10 экв., от примерно 2 до примерно 9 экв., от примерно 3 до примерно 8 экв. или от примерно 4 до примерно 7 экв. в пересчете на молярное содержание свободного основания, с которым проводится взаимодействие. В других вариантах осуществления фосфорная кислота, с которой проводится взаимодействие, содержится в количестве, составляющем от примерно 2 до примерно 4 экв. или примерно 3 экв. в пересчете на молярное содержание свободного основания, с которым проводится взаимодействие. В некоторых вариантах осуществления суспензию свободного основания и фосфорной кислоты вводят в реакцию или во взаимодействие в количестве, составляющем от примерно 2 до примерно 40 ч., от примерно 4 до примерно 20 ч. или от примерно 8 до примерно 10 ч. В некоторых вариантах осуществления свободное основание и фосфорную кислоту вводят в реакцию при температуре, находящейся в диапазоне от примерно 25 до примерно 100°C, от примерно 40 до примерно 85°C или от примерно 55 до примерно 70°C. В некоторых вариантах осуществления свободное основание и фосфорную кислоту вводят в реакцию при температуре кипения растворителя, использующегося в реакции. В некоторых вариантах осуществления реакцию проводят с использованием реагентов и одного или большего количества растворителей, которые в основном не содержат воды (т.е. совсем не содержат воды).
Безводную кристаллическую форму фосфата в форме A, полученную способами, предлагаемыми в настоящем изобретении, можно дополнительно направить на такие стадии, как, например, сушка, очистка и т.п. Выделенные кристаллы можно подвергнуть сушке при подходящей температуре. В одном варианте осуществления кристаллы сушат при температуре, находящейся в диапазоне от примерно 20 до примерно 80°C. В некоторых вариантах осуществления кристаллы сушат при температуре, находящейся в диапазоне от примерно 30 до примерно 70°C. В некоторых вариантах осуществления кристаллы сушат при температуре, находящейся в диапазоне от примерно 40 до примерно 60°C. В одном варианте осуществления кристаллы сушат при пониженном давлении, находящемся в диапазоне, например, от примерно 10 до примерно 40 мбар. Стадию сушки можно проводить в течение подходящего периода времени. Так, в одном варианте осуществления кристаллы сушат в течение от примерно 1 до примерно 72 ч, от примерно 2 до примерно 36 ч или от примерно 4 до примерно 18 ч. В некоторых вариантах осуществления кристаллы сушат в течение примерно 48 ч.
Фосфат соединения формулы I можно использовать для получения моногидрата свободного основания. Например, фосфат можно растворить в водном растворе неорганического основания, такого как водный раствор гидроксида аммония. С течением времени свободное основание осаждается в виде кристаллического моногидрата. Необязательно можно использовать дополнительные стадии, такие как фильтрование, промывка и сушка и выделит кристаллический моногидрат свободного основания, так же, как выделяли и фосфат, как это описано выше.
Кристаллические формы, предлагаемые в настоящем изобретении, можно выделить в основном в чистом форме. "В основном чистые" означает, что более 50 мас.% 3-(2,6-дихлор-3,5-диметоксифенил)-1-{6-[4-(4-этилпиперазин-1-ил)-фениламино]-пиримидин-4-ил}-1-метилмочевин содержится в одной из кристаллических форм, раскрытых в настоящем изобретении. В некоторых вариантах осуществления выделенных или в основном чистых кристаллических форм, предлагаемых в настоящем изобретении, 3-(2,6-дихлор-3,5-диметоксифенил)-1-{6-[4-(4-этилпиперазин-1-ил)-фениламино]-пиримидин-4-ил}-1-метилмочевина содержится в количестве, составляющем не менее примерно 60%, не менее примерно 70%, не менее примерно 80%, не менее примерно 90%, не менее примерно 95%, не менее примерно 97%, не менее примерно 98% или не менее примерно 99 мас.% в пересчете на указанную форму. Например, в некоторых вариантах осуществления настоящее изобретение относится к фосфатам свободного основания, в которых не менее примерно 50%, не менее примерно 70%, не менее примерно 80%, не менее примерно 90%, не менее примерно 95%, не менее примерно 97%, не менее примерно 98% или не менее примерно 99 мас.% всего фосфата свободного основания находится в виде формы A, формы B или аморфной формы. В других вариантах осуществления настоящее изобретение относится к свободному основанию, такому что не менее примерно 50%, не менее примерно 70%, не менее примерно 80%, не менее примерно 90%, не менее примерно 95%, не менее примерно 97%, не менее примерно 98% или не менее примерно 99 мас.% всего свободного основания находится в виде безводного кристаллического свободного основания, кристаллического моногидрата свободного основания или аморфной формы свободного основания. В других вариантах осуществления настоящее изобретение относится к гидрохлоридам свободного основания, в которых не менее примерно 50%, не менее примерно 70%, не менее примерно 80%, не менее примерно 90%, не менее примерно 95%, не менее примерно 97%, не менее примерно 98% или не менее примерно 99 мас.% всего соединения представляет собой аморфную или кристаллическую форму дигидрата гидрохлорида.
Настоящее изобретение также относится к фармацевтическим композициям и лекарственным средствам, которые можно приготовить путем смешивания одной или большего количества безводных или гидратированных кристаллических или аморфных форм (например, свободного основания, его фосфатов или гидрохлоридов), предлагаемых в настоящем изобретении, с фармацевтически приемлемыми носителями, инертными наполнителями, связующими, разбавителями и т.п. для лечения некоторых заболеваний, описанных в настоящем изобретении.
Таким образом, другим объектом настоящего изобретения являются композиции, содержащие безводную или гидратированную кристаллическую форму или аморфную форму, предлагаемую в настоящем изобретении, и фармацевтически приемлемый носитель, инертный наполнитель и/или разбавитель. В одном варианте осуществления безводная кристаллическая форма представляет собой соединение формулы I в виде свободного основания. В одном варианте осуществления гидратированная кристаллическая форма представляет собой моногидрат соединения формулы I в виде свободного основания. В другом варианте осуществления безводная кристаллическая форма представляет собой соль монофосфорной кислоты соединения формулы I. В другом варианте осуществления безводная кристаллическая форма соли монофосфорной кислоты находится в форме А. В другом варианте осуществления безводная кристаллическая форма соли монофосфорной кислоты находится в форме В. В одном варианте осуществления соль монофосфорной кислоты соединения формулы I является аморфной. В другом варианте осуществления форма представляет собой гидрохлорид соединения формулы I. В иллюстративном варианте осуществления гидрохлорид представляет собой кристаллический дигидрат или аморфную форму.
Такие композиции могут находиться в форме, например, гранул, порошков, таблеток, капсул, сиропов, суппозиториев, препаратов для инъекции, эмульсий, эликсиров, суспензий или растворов. В одном варианте осуществления композиция в основном не содержит воды. Композиции, предлагаемые в настоящем изобретении, можно приготовить для различных путей введения, например перорального и парентерального введения. Парентеральное или системное введение включает, но не ограничивается только ими, подкожные, внутривенные, внутрибрюшинные и внутримышечные инъекции. Указанные ниже дозированные формы приведены в качестве примеров и их не следует рассматривать в качестве ограничивающих настоящее изобретение.
Дозированные формы для инъекции обычно включают суспензии в масле или водные суспензии, которые можно приготовить с использованием подходящего диспергирующего или смачивающего агента и суспендирующего агента. Формы для инъекции могут представлять собой растворенную фазу или суспензию, которую готовят с использованием растворителя или разбавителя. Приемлемые растворители или разбавитель включают стерилизованную воду, раствор Рингера или изотонический водный раствор соли. В некоторых вариантах осуществления такие водные формы для инъекции готовят (или восстанавливают) непосредственно перед введением субъекту. Альтернативно, в качестве растворителей или суспендирующих агентов можно использовать стерильные масла. Обычно масло или жирная кислота является нелетучей и они включают природные или синтетические масла, жирные кислоты, моно-, ди- или триглицериды.
Предназначенные для инъекции фармацевтические композиции и/или лекарственное средство могут представлять собой порошок, применимый для восстановления с помощью подходящего, описанного выше. Их примеры включают, но не ограничиваются только ими, полученные сушкой вымораживанием, с помощью барабанной сушилки или распылительной сушкой порошки, аморфные порошки, гранулы, осадки или измельченные вещества. Предназначенные для инъекции фармацевтические композиции необязательно могут содержать стабилизаторы, модификаторы рН, поверхностно-активные вещества, модификаторы биологической доступности и их комбинации.
Для перорального, трансбуккального и сублингвального введения в виде твердых дозированных форм приемлемы порошки, суспензии, гранулы, таблетки, пилюли, капсулы, желатиновые капсулы и каплеты. Их можно изготовить, например, путем смешивания одной или большего количества безводных кристаллических форм, предлагаемых в настоящем изобретении, по меньшей мере с одной добавкой, такой как крахмал или другая добавка. Подходящими добавками являются сахароза, лактоза, целлюлоза сахар, маннит, мальтит, декстран, крахмал, агар-агар, альгинаты, хитины, хитозаны, пектины, трагакантовая камедь, гуммиарабик, желатины, коллагены, казеин, альбумин, синтетические или полусинтетические полимеры или глицериды. Пероральные дозированные формы необязательно могут содержать другие ингредиенты, содействующие введению, такие как неактивный разбавитель или смазывающие вещества, такие как стеарат магния, или консерванты, такие как парабен или сорбиновая кислота, или антиоксиданты, такие как аскорбиновая кислота, токоферол или цистеин, разрыхлитель, связующие, загустители, буферы, подсластители, ароматизаторы или отдушки. Таблетки и пилюли можно дополнительно обработать подходящими образующими покрытия материалами, известными в данной области техники.
Жидкие дозированные формы для перорального введения могут находиться в форме фармацевтически приемлемых эмульсий, сиропов, эликсиров, суспензий и растворов, которые могут содержать неактивный разбавитель, такой как вода. Фармацевтические композиции и лекарственные средства можно приготовить в виде жидких суспензий или растворов с использованием стерильной жидкости, такой как, но не ограничиваясь только ими, масло, вода, спирт и их комбинации. Для перорального или парентерального введения можно добавить фармацевтически приемлемые поверхностно-активные вещества, суспендирующие агенты, эмульгирующие агенты. В некоторых вариантах осуществления жидкие дозированные формы, в частности кристаллические формы, монофосфата свободного основания в основном не содержат воды.
Как отмечено выше, суспензии могут содержать масла. Такие масла включают, но не ограничиваются только ими, арахисовое масло, кунжутное масло, хлопковое масло, кукурузное масло и оливковое масло. Препарат суспензии также может содержать эфиры жирных кислот, такие как этилолеат, изопропилмиристат, жирные кислоты глицериды и ацетилированные глицериды жирных кислот. Суспензии фармацевтических композиций могут содержать спирты, такие как, но не ограничиваясь только ими, этанол, изопропиловый спирт, гексадециловый спирт, глицерин и пропиленгликоль. В суспензиях фармацевтических композиций также можно использовать простые эфиры, такие как, но не ограничиваясь только ими, поли(этиленгликоль), углеводороды нефти, такие как минеральное масло и жидкий парафин; и воду.
Кроме этих типичных дозированных форм, описанных выше, фармацевтически приемлемые инертные наполнители и носители обычно известны специалистам в данной области техники и входят в объем настоящего изобретения. Такие инертные наполнители и носители описаны, например, в публикации "Remingtons Pharmaceutical Sciences" Mack Pub. Co., New Jersey (1991), которая включена в настоящее изобретение в качестве ссылки.
Фармацевтические композиции, предлагаемые в настоящем изобретении, могут быть приготовлены в виде форм кратковременного действия, быстрого высвобождения, длительного действия и пролонгированного высвобождения, как это описано ниже. Таким образом, фармацевтические высвобождения композиции также могут быть приготовлены в виде форм регулируемого или медленного высвобождения.
Композиции, предлагаемые в настоящем изобретении, можно вводить в форме продолжительного высвобождения для обеспечения пролонгированного хранения и/или доставки. Поэтому фармацевтические композиции и лекарственные средства можно спрессовать в пеллеты или цилиндры и имплантировать внутримышечно или подкожно в качестве форм-депо для инъекций или в виде имплантатов, таких как стенты. В таких имплантатах можно использовать известные инертные материалы, такие как силиконы и биологически разлагающиеся полимеры.
Конкретные дозы можно подобрать в зависимости от условий протекания заболевания, возраста, массы тела, общего состояния здоровья, пола и диеты субъекта, интервалов между введением доз, путей введения, скорости выведения и комбинаций лекарственных средств. Величины любой из описанных выше дозированных форм, содержащих терапевтически эффективные количества, можно определить с помощью стандартных экспериментов и поэтому они входят в объем настоящего изобретения.
В некоторых других вариантах осуществления настоящее изобретение относится к композициям, содержащим свободное основание соединения формулы I, в которых не менее примерно 50%, не менее примерно 70%, не менее примерно 80%, не менее примерно 90%, не менее примерно 95%, не менее примерно 97%, не менее примерно 98% или не менее примерно 99 мас.% всего свободного основания находится в композиции в виде безводной формы или в виде моногидрата. В других вариантах осуществления композиции, предлагаемые в настоящем изобретении, в основном состоят из свободного основания, причем не менее примерно 95%, не менее примерно 97%, не менее примерно 98% или не менее примерно 99% свободного основания находится в композиции в виде безводной формы или в виде моногидрата.
В некоторых других вариантах осуществления настоящее изобретение относится к композициям, содержащим фосфат свободного основания, в которых не менее примерно 50%, не менее примерно 70%, не менее примерно 80%, не менее примерно 90%, не менее примерно 95%, не менее примерно 97%, не менее примерно 98%» или не менее примерно 99 мас.% всего фосфата свободного основания в композиции находится в виде формы A, в виде формы B или в виде аморфной формы. В других вариантах осуществления композиции, предлагаемые в настоящем изобретении, в основном состоят из фосфата свободного основания, причем не менее примерно 95%, не менее примерно 97%, не менее примерно 98% или не менее примерно 99% фосфата свободного основания находится в композиции в виде формы A, в виде формы В или в виде аморфной формы.
В некоторых вариантах осуществления настоящее изобретение относится к композициям, содержащим гидрохлорид свободного основания, в которых не менее примерно 50%, не менее примерно 70%, не менее примерно 80%, не менее примерно 90%, не менее примерно 95%, не менее примерно 97%, не менее примерно 98% или не менее примерно 99 мас.% всего гидрохлорида свободного основания в композиции находится в виде дигидрата или в виде аморфной формы. В других вариантах осуществления композиции, предлагаемые в настоящем изобретении, в основном состоят из гидрохлорида свободного основания, причем не менее примерно 95%, не менее примерно 97%, не менее примерно 98% или не менее примерно 99% фосфата свободного основания находится в композиции в виде дигидрата или в виде аморфной формы.
Другим объектом настоящего изобретения является способ лечения, включающий введение терапевтически эффективного количества безводной или гидратированной кристаллической формы или аморфной формы или композиции, содержащей безводную или гидратированную кристаллическую форму или аморфную форму, предлагаемую в настоящем изобретении, субъекту, страдающему от заболевания, патологии и/или симптомов, когда заболевание можно предупредить, подавить или облегчить его протекание путем ингибирования активности киназы, где киназа выбрана из группы, включающей FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2 и Ret. В другом варианте осуществления настоящее изобретение относится к применению безводных или гидратированных кристаллических форм, аморфных форм или композиций, содержащих безводные или гидратированные кристаллические формы или аморфные формы, предлагаемые в настоящем изобретении, для приготовления лекарственного средства, предназначенного для лечения у субъекта заболевания, при котором активность киназы FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2 и Ret способствует патологии и/или симптоматике заболевания. В некоторых вариантах осуществления настоящего изобретения киназа выбрана из группы, включающей FGFR1, FGFR2, FGFR3, FGFR4.
Лечение в контексте настоящего изобретения означает полное или частичное ослабление симптомов, связанных с нарушением или заболеванием, или замедление или остановку дальнейшего прогрессирования этих симптомов, или содействие предупреждению или предотвращению заболевания или нарушения у субъекта, для которого существует опасность развития заболевания или нарушения. Такие заболевания или нарушения включают, но не ограничиваются только ими, карциному почек, печени, надпочечников, мочевого пузыря, молочной железы, желудка, яичников, толстой кишки, прямой кишки, предстательной железы, поджелудочной железы, легких, влагалища или щитовидной железы; саркому; глиобластому; лейкоз; опухоли головы или шеи; псориаз; гиперплазию предстательной железы; или неоплазию.
Например, в контексте лечения заболеваний, включая опосредуемое киназой нарушение, описанное выше, успешное лечение может включать ослабление симптомов или замедление или остановку прогрессирования заболевания, характеризующееся уменьшением содержания или активности одной или большего количества соответствующих киназ. При использовании в настоящем изобретении "терапевтически эффективное количество" означает количество безводной или гидратированной кристаллической формы или аморфной формы соединения формулы I (включая свободное основание), или безводной кристаллической формы или аморфной формы фосфата соединения формулы I, гидрохлорида соединения формулы I (включая дигидрат или аморфную форму) или содержащих их композиций, которое полностью или частично ослабляет симптомы, связанные с подвергающимся лечению нарушением или заболеванием, или замедляет или останавливает дальнейшее прогрессирование или ухудшение его симптомов, или предупреждает или обеспечивает профилактику заболевания или нарушения у субъекта, для которого существует опасность развития заболевания или нарушения. Субъектом является любое животное, для которого благоприятно введение любой из форм соединения формулы I или содержащей его композиции, раскрытой в настоящем изобретении. В некоторых вариантах осуществления субъектом является млекопитающее, например человек, примат, собака, кошка, лошадь, корова, свинья, грызун, такой как, например, крыса или мышь. Обычно млекопитающим является человек.
Терапевтически эффективное количество безводной или гидратированной кристаллической формы или аморфной формы, или композиции, содержащей любую такую форму, предлагаемую в настоящем изобретении, может меняться в зависимости от пути введения и дозированных форм. Эффективные количества безводных или гидратированных кристаллических форм или аморфных форм соединений формулы I обычно находятся в диапазоне от примерно 0,01 до 100 мг/кг/сутки и чаще в диапазоне от примерно 0,05 до 25 мг/кг/сутки. Обычно безводная или гидратированная кристаллическая форма, аморфная форма и композиция, содержащая такие формы, предлагаемая в настоящем изобретении, выбрана так, чтобы получить фармацевтические композиции, которые характеризуются большим терапевтическим индексом. Терапевтический индекс представляет собой отношение доз, приводящих к токсическому и терапевтическому эффектам, и его можно представить в виде отношения значений LD50 и ED50. LD50 означает дозу, летальную для 50% популяции, и ED50 означает дозу, терапевтически эффективную для 50% популяции. LD50 и ED50 определяют по стандартным фармацевтическим методикам с использованием культур клеток животных или экспериментальных животных.
Все публикации, заявки на патенты, выданные патенты и другие документы, указанные в настоящем описании, включены в настоящее изобретение в качестве ссылки, как если бы каждая отдельная публикация, заявка на патент, выданный патент или другой документ специально и отдельно был указан, как во всей своей полноте включенный в настоящее изобретение в качестве ссылки. Определения, содержащиеся в тексте, включенном в качестве ссылки, исключены в той степени, в которой они противоречат определениям, содержащимся в настоящем раскрытии.
Настоящее изобретение, описанное в общем виде, легче понять с использованием приведенных ниже примеров, которые представлены для иллюстрации, а не для ограничения настоящего изобретения.
ПРИМЕРЫ
Приведенные ниже аббревиатуры используются в настоящем раскрытии для описания химических и биологических терминов.
Использующиеся общие методики, приборы и стандарты
Значение pH
Значение pH раствора определяли, как указано в примере: примерно 10 мг свободного основания или его соли переносили в сосуд объемом 20 мл и к нему добавляли 10 мл соответствующего буфера или воды. Во время измерения рН раствор непрерывно перемешивали. Измерение pH можно провести по многим методикам, таким как в которых для определения pH используются микроэлектроды.
Определение растворимости
Избыток свободного основания (свободного основания формулы I), его фосфата (соли монофосфорной кислоты свободного основания) или соли другой кислоты приводили в равновесие в растворителях в течение 1 дня при 25±0,5°C. Полученную взвесь фильтровали и фильтрат использовали для определения растворимости с помощью ВЭЖХ.
Характеристическое растворение
Характеристическую скорость растворения определяли с использованием пуансона и матрицы в сборе VanKel площадью 0,5 см2 при давлении на пеллету, равном 2000 фунтов. Растворение исследовали с помощью волоконно-оптической системы изучения растворения (С technologies Inc.) и спектрофотометра Сагу в УФ/видимой области спектра при скорости перемешивания, равной 200 об/мин. Температуру растворяющей среды поддерживали равной 37±0,5°C и измерение концентрации проводили при длине волны, равной 260 нм.
Гигроскопичность
Гигроскопичность исследовали по изотермам сорбции/десорбции с использованием системы исследования поверхности DVS-1 при 25±0,5°C. Исследовали образцы различной влажности, включая 75%, 85% и 95%.
Полиморфизм
Взвеси образцов перемешивали с высокой скоростью в течение 24 ч при 25±0,5°C, фильтровали и собранное твердое вещество анализировали с помощью ПРРГ.
Методика ВЭЖХ
Прибор: Waters; Колонка: Symmetry С18, диаметр частиц 3,5 мкм, 4,6x75 мм; температура колонки: 35°C; скорость потока: 1 мл/мин; подвижная фаза: A=0,1% ТФК (трифторуксусная кислота) в воде и B=ацетонитрил; детектирование: УФ при 254 нм; инжектируемое количество: примерно 2 мкг; разбавитель: 60:40 (об./об.) ацетонитрил/0,2% водный раствор фосфорной кислоты; и градиентный режим: от 10 до 70% В за 10 мин.
Методика ТГ
Прибор: ТА instrument Q500; диапазон температур: от комнатной температуры до 300°C; скорость сканирования: 10°C/мин; скорость потока азота: 60 мл/мин. Mettler ТГА 850; диапазон температур: от комнатной температуры до 300°C; скорость сканирования: 20°C/мин; скорость потока азота: 40 мл/мин.
Методика СЭМ
Прибор: Jeol JSM 6300.
Методика ДСК
Прибор: ТА instrument Q1000; диапазон температур: от комнатной температуры до 210°C; скорость сканирования: 10°C/мин; скорость потока азота: 60 мл/мин.
Рентгенография кристаллов
Использовали дифрактометр Bruker AXS с тремя лимбами с графитовым монохроматором для излучения Cu(Kα) с использованием генератора с запаянной трубкой с тонкой фокусировкой и детектора Smart 6000 CCD и с использованием программного обеспечения SMART. Обработку данных и глобальное уточнение ячейки проводили с использованием программного обеспечения Saint. Использовали полуэмпирическую коррекцию поглощения на основе интенсивностей симметрично связанных отражений при разных угловых установках. Структуры разрешали по методикам повторения циклов в двойном пространстве с последующим синтезом DF и уточняли с помощью полного матричного метода наименьших квадратов с критерием F2 с использованием комплекта программ SHELXTL.
Методика ПРРГ
Прибор: Bruker D8 Advance; излучение: Cu Kα, (30 кВ, 40 мА); переменная щель V12 мм; диапазон сканирования 2-40° (значение 2-тета); скорость сканирования 0,3 с/шаг. Альтернативно, использовали XDS2000, выпускающийся фирмой Scintag, Inc.; излучение: Cu Kα (45 кВ, 40 мА); щель с переменным отклонением: 3 мм и 2 мм; измерительная щель: 0,3 мм и 0,2 мм; прерыватель 0,02°; длительность сканирования: 6 мин (3 мин на рамку); диапазон сканирования: 2°-35° или 40° (значение 2-тета); скорость сканирования 0,5°/мин (значение 2-тета).
Методика ИК
Прибор: Thermo Magna 560; режим: пропускание; диапазон сканирования: 4000 см-1 - 600 см-1.
Методика комбинационного рассеяния
Прибор: Bruker Vertex 70 FTIR; APT: 3,5 мм; 64 сканирования; RES 2.
Скрининг солей
Пример 1: Скрининг солей с кислотами свободного основания формулы I.
Скрининг солей проводили с использованием свободного основания формулы I (полученного, как это описано ниже в примере 2) и различных кислот: уксусной кислоты, бензойной кислоты, лимонной кислоты, фумаровой кислоты, гиппуровой кислоты, бромистоводородной кислоты, хлористоводородной кислоты, молочной кислоты (D,L), малеиновой кислоты, яблочной кислоты (L), малоновой кислоты, метансульфоновой кислоты, фосфорной кислоты, нафталин-1,5-дисульфоновой кислоты, фосфорной кислоты, янтарной кислоты, серной кислоты, винной кислоты (D) и винной кислоты (L). Свободное основание (30 мг) растворяли в 12 мл ТГФ (тетрагидрофуран). Аликвоты по 0,1 мл этого раствора помещали в лунки 96-луночного планшета (Zinsser Crissy block) блока кристаллизации. В каждую лунку помещали примерно по 0,25 мг свободного основания; в каждую лунку также добавляли 0,044 мл 0,01 М раствора соответствующих кислот в ацетоне. Блок накрывали крышкой с отверстиями и затем смеси растворителей давали испаряться в течение примерно 10 ч при комнатной температуре.
Исследования с использованием уксусной кислоты, бензойной кислоты, лимонной кислоты, гиппуровой кислоты, молочной кислоты, малеиновой кислоты и яблочной кислоты приводили к образованию твердых веществ, обладающих такими же ПРРГ, как свободное основание или аморфные твердые вещества. При использовании фумаровой кислоты, бромистоводородной кислоты, нафталин-1,5-дисульфоновой кислоты, серной кислоты или винной кислоты получали твердые вещества с низкой степенью кристалличности. Для фосфорной кислоты кристаллическую форму соли получали из 95% этанола. Вследствие кристалличности или легкости синтеза для твердых веществ, образовавшихся из фосфорной кислоты, хлористоводородной кислоты, малоновой кислоты, метансульфоновой кислоты и янтарной кислоты, дополнительно исследовали степень кристалличности и гигроскопичность.
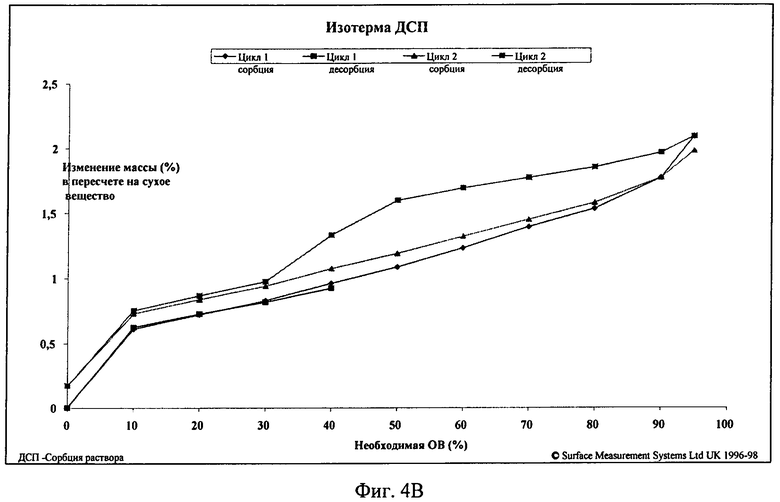
Обнаружено, что фосфат является высококристаллическим. Гигроскопичность, исследованная с помощью ДСП по изотермам сорбции/десорбции, показала, что даже при 95% ОВ вследствие абсорбции воды увеличение массы составило менее 2%. При 75% и 85% ОВ увеличение массы составило менее 1%.
Для гидрохлорида с помощью ПРРГ получены несколько рентгенограмм, что указывает на наличие более одной полиморфной формы. Большинство из этих солей обладало низкой степенью кристалличности. Установлено, что дигидрат обладает наибольшей степенью кристалличности. Гидрохлорид являлся более гигроскопичным, чем фосфат. Для одной формы гидрохлорида по данным ДСП увеличение массы составило от примерно 4,1% (75% ОВ) до примерно 16,9% (95% ОВ).
Для малонатов по данным рентгенограмм ПРРГ идентифицированы 4 формы. Одна дополнительно один раз исследованная представляет собой хрупкий кристалл с рентгенограммой, соответствующей влажному веществу и являлась гигроскопичной (увеличение массы составило примерно 10% при всех использованных влажностях).
Для соли метансульфоновой кислоты (или мезилата) с помощью ПРРГ получены несколько рентгенограмм, и которой только одна хорошо воспроизводилась, но вещество являлось гигроскопичным (по данным ДСП увеличение массы составило примерно от 2,6 до 10%). Аналогичным образом, сукцинат также являлся гигроскопичным (по данным ДСП увеличение массы составило примерно от 4,1 до 10%).
Результаты исследования начальной растворимости в буфере с рН 3 и в воде приведены ниже в таблице 1. В буфере с рН 3 и в воде фосфат растворялся лучше, чем метансульфонат, малонат и сукцинат. Фосфат также растворялся в воде лучше, чем свободное основание.
Химический синтез
Пример 2: Получение свободного основания соединения формулы I
A. N-[4-(4-Этилпиперазин-1-ил)-фенил]-N'-метилпиримидин-4,6-диамин
Смесь 4-(4-этилпиперазин-1-ил)-анилина (1 г, 4,88 ммоля), (6-хлорпиримидин-4-ил)-метиламина (1,81 г, 12,68 ммоля, 1,3 экв.) и 4 н. HCl в диоксане (15 мл) нагревали в герметизированной пробирке при 150°C в течение 5 ч. Реакционную смесь концентрировали, разбавляли с помощью ДХМ и насыщенным водным раствором бикарбоната натрия. Водный слой отделяли и экстрагировали с помощью ДХМ. Органическую фазу промывали рассолом, сушили (сульфат натрия), фильтровали и концентрировали. Очистка остатка с помощью колоночной хроматографии на силикагеле (ДХМ/MeOH, 93:7) с последующим растиранием в диэтиловом эфире давала искомое соединение в виде белого твердого вещества: ИЭР-МС: 313,2 [МН]+; tR=1,10 мин (градиентный режим J); ТСХ (тонкослойная хроматография): Rf=0,21 (ДХМ/MeOH, 93:7).
B. 4-(4-Этилпиперазин-1-ил)-анилин
Суспензию 1-этил-4-(4-нитрофенил)-пиперазина (6,2 г, 26,35 ммоля) и никеля Ренея (2 г) в MeOH (120 мл) перемешивали в атмосфере водорода в течение 7 ч при КТ. Реакционную смесь фильтровали через слой целита и концентрировали и получали 5,3 г искомого соединения в виде фиолетового твердого вещества: ИЭР-МС: 206,1 [МН]+; ТСХ: Rf=0,15 (ДХМ/MeOH+1% NH3 aq, 9:1).
C. 1 -Этил-4-(4-нитрофенил)-пиперазин
Смесь 1-бром-4-нитробензола (6 г, 29,7 ммоля) и 1-этилпиперазина (7,6 мл, 59,4 ммоля, 2 экв.) нагревали при 80°C в течение 15 ч. После охлаждения до КТ реакционную смесь разбавляли водой и смесью ДХМ/МеОН, 9:1. Водный слой отделяли и экстрагировали смесью ДХМ/MeOH, 9:1. Органическую фазу промывали рассолом, сушили (сульфат натрия), фильтровали и концентрировали. Очистка остатка с помощью колоночной хроматографии на силикагеле (ДХМ/MeOH+1% NH3 aq, 9:1) давала 6,2 г искомого соединения в виде желтого твердого вещества: ИЭР-МС: 236,0 [МН]+; tR=2,35 мин (чистота: 100%, градиентный режим J); ТСХ: Rf-0,50 (ДХМ/МеОН+1% NH3aq, 9:1).
D. (6-хлорпиримидин-4-ил)-метиламин
Это вещество получали по модифицированной методике, опубликованной в литературе (J. Appl. Chem. 1955, 5, 358): К суспензии имеющегося в продаже 4,6-дихлорпиримидина (20 г, 131,6 ммоля, 1,0 экв.) в изопропаноле (60 мл) добавляли 33% метиламин в этаноле (40,1 мл, 328,9 ммоля, 2,5 экв.) с такой скоростью, чтобы внутренняя температура не превышала 50°C. После завершения добавления реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Затем добавляли воду (50 мл) и образовавшуюся суспензию охлаждали в бане со льдом до 5°C. Осадившийся продукт отфильтровывали, промывали холодной смесью изопропанол/вода 2:1 (45 мл) и водой. Собранное вещество сушили в вакууме в течение ночи при 45°C и получали искомое соединение в виде бесцветного порошкообразного вещества: tR=3,57 мин (чистота:>99%, градиентный режим А), ИЭР-МС: 144,3/146,2 [МН]+.
Е. (3-(2,6-Дихлор-3,5-диметоксифенил)-1 -{6-[4-(4-этилпиперазин-1 -ил)-фениламино]-пиримидин-4-ил}-1-метилмочевина)
Искомое соединение получали путем добавления 2,6-дихлор-3,5-диметоксифенилизоцианата (1,25 экв.) к раствору N-[4-(4-этилпиперазин-1-ил)-фенил]-N'-метилпиримидин-4,6-диамина (2,39 г, 7,7 ммоля, 1 экв.) в толуоле и перемешивания реакционной смеси в течение 1,5 ч при кипячении с обратным холодильником. Очистка неочищенного продукта с помощью колоночной хроматографии на силикагеле (ДХМ/MeOH+1% NH3 aq, 95:5) давала искомое соединение в виде белого твердого вещества: ИЭР-МС: 560,0/561,9 [МН]+; tR=3,54 мин (чистота: 100%, градиентный режим J); ТСХ: Rf=0,28 (ДХМ/MeOH+1% NH3aq, 95:5). Анализ: C26H31N7O3Cl2, рассчитано C 55,72%, H 5,57%, N 17,49%, О 8,56%, Cl 12,65%; найдено C 55,96%, H 5,84%, N 17,17%, O 8,46%, Cl 12,57%. Искомое соединение охарактеризовывали с помощью ПРРГ, термических и других методик, описанных ниже.
Пример 3: Получение формы А соли монофосфорной кислоты соединения формулы I.
В круглодонную колбу добавляли 3-(2,6-дихлор-3,5-диметоксифенил)-1-{6-[4-(4-этилпиперазин-1-ил)фениламино]-пиримидин-4-ил}-1-метилмочевину (134 г, 240 ммоля) и ИПА (2000 мл). Суспензию перемешивали и нагревали до 50°C и порциями добавляли раствор фосфорной кислоты (73,5 г, 750 ммоля) в воде (2000 мл). Смесь перемешивали при 60°С в течение 30 мин и фильтровали через слой полипропилена. Слой промывали теплой смесью ИПА/вода (1:1, 200 мл) и фильтраты объединяли. К этому прозрачному раствору добавляли ИПА (6000 мл) и смесь перемешивали при кипячении с обратным холодильником в течение 20 мин, медленно охлаждали до комнатной температуры (25°C) и перемешивали в течение 24 ч. Полученную белую соль собирали фильтрованием, промывали с помощью ИПА (2×500 мл) и сушили в сушильном шкафу при 60°C при пониженном давлении в течение 2 дней и получали фосфат (форма А) 110 г. Выход 70%. Чистота >98% по данным ВЭЖХ. Анализ: C26H34N7O7Cl2P, рассчитано C 47,42%, H 5,20%, N 14,89%, O 17,01%, Cl 10,77%, P 4,70%; найдено C 47,40%, H 5,11%, N 14,71%, O 17,18%, Cl 10,73%, P 4,87%. Искомое соединение охарактеризовывали с помощью ПРРГ, термических и других методик, описанных ниже.
Пример 4: Получение моногидрата свободного основания соединения формулы I.
В 4-горлую круглодонную колбу объемом 22 л при 18°C в атмосфере азота помещали 15М гидроксид аммония (10 л, 15 моля). К раствору порциями при 18°C в течение 1 ч добавляли фосфат 3-(2,6-дихлор-3,5-диметоксифенил)-1-{6-[4-(4-этилпиперазин-1-ил)фениламино]-пиримидин-4-ил}-1-метилмочевины (660 г, 1,002 моля; полученный по методике примера 3) в виде твердого вещества. Полученную белую суспензию перемешивали при 18°C в течение 16 ч. Отбирали образец и с помощью 1H ЯМР проверяли полноту превращения фосфата в свободное основание. ЯМР указывал на полное превращение в свободное основание. Смесь фильтровали через воронку объемом 3 л, содержащую крупнопористый (40-60 ASTM) стеклянный фильтр. Фильтрование было медленным и потребовало ~ 3 ч вследствие небольшого размера частиц твердого свободного основания. Твердое вещество промывали деионизированной водой (1 л). Эта промывка также была очень медленной (потребовалось 1,5 ч). Твердое вещество сушили при 60°C в вакууме (5 торр) в течение 16 ч и получали 642 г свободного основания (114% от теоретического значения, теоретическое значение=561,1 г). Высушенное твердое вещество повторно диспергировали в деионизированной воде (10,5 л) при 18°C в течение 3 ч. Твердое вещество фильтровали через воронку объемом 3 л, содержащую крупнопористый (40-60 ASTM) стеклянный фильтр. Осадок на фильтре промывали деионизированной водой (2 л). Твердое вещество сушили при 60°C в вакууме (5 торр) в течение 16 ч и получали свободное основание в виде моногидрата (559 г, 96,7% от теоретического значения для моногидрата).
Характеризация кристаллических форм
Пример 5: Физико-химические характеристики свободного основания, фосфата (форма А) и гидрохлорида.
A. Свободное основание
Безводное свободное основание обладало хорошей степенью кристалличности (по данным ПРРГ, см. фиг.1А). Оно было немного гигроскопичным и при нагревании со скоростью, равной 10°C/мин, плавилось при 217°C (начало) с последующим разложением (полученная с помощью ДСК термограмма приведена на фиг.2А). Термограмма ТГА безводного основания приведена на фиг.3А и наблюдающаяся потеря массы согласуется с безводной формой. Оно нерастворимо в водных средах при высоких значениях pH (примерно 0,00004 мг/мл в буфере с pH 6,8 и примерно 0,00009 мг/мл при pH 6,67 в воде) и обладает растворимостью от плохой до небольшой в обычных органических растворителях. B 0,1 н. HCl безводное свободное основание переходит в другую форму (вероятно, в форму гидрохлорида). Моногидрат свободного основания также обладал хорошей степенью кристалличности (по данным ПРРГ, см. фиг.1Г). Полученная с помощью СЭМ микрофотография безводного свободного основания приведена на фиг.5А.
B. Фосфат (форма А)
Фосфат обладал хорошей степенью кристалличности (по данным ПРРГ, см. фиг.1B (фосфат (форма А)). Он был немного гигроскопичным и при нагревании со скоростью, равной 10°C/мин, плавился при 184,0°C (начало) с последующим разложением (см. фиг.2Б (фосфат (форма А)). Термограмма ТГА фосфата приведена на фиг.3Б и наблюдающаяся потеря массы согласуется с безводной формой. Он нерастворим в буфере с pH 6,8 (примерно 0,00009 мг/мл), но, в отличие от свободного основания, растворим в воде (1,3 мг/мл). Он обладает плохой растворимостью в обычных органических растворителях. Данные дополнительных исследований растворимости приведены ниже. Ф-ИК-спектр фосфата (форма А) приведен на фиг.6. В фосфате обнаружено до 0,6% остаточного растворителя (изопропанола). Полученная с помощью СЭМ микрофотография формы А фосфата приведена на фиг.5Б.
С. Гидрохлорид свободного основания
Гидрохлорид в форме дигидрата обладает средней степенью кристалличности (по данным ПРРГ, см. фиг.1Д). Термограмма ТГА приведена на фиг.3В и наблюдающаяся потеря массы согласуется с формой дигидрата. Гидрохлорид является гигроскопичным и находится в форме дигидрата. Данные исследования растворимости приведены в примере 8, таблица 3Г.
Пример 6: Аморфная форма фосфата свободного основания.
Примерно 40 мг фосфата (форма А) при 25°C смешивали с 4 мл смеси тетрагидрофуран/вода (1:1). После перемешивания в течение ночи раствор фильтровали. Без перемешивания прозрачный фильтрат помещали в ток азота и сушили. Затем твердое вещество, полученное после выпаривания растворителя, исследовали с помощью ДСК и ТГА. На термограмме ДСК при нагревании от -40 до 140°C стеклование не обнаружено. На термограмме ТГА обнаружено, что образец начинает разлагаться при ~115°C, т.е. при намного более низкой температуре, чем форма A кристаллического фосфата. См. также ниже пример 15.
Пример 7: Определение характеристических скоростей растворения.
Характеристическую скорость растворения определяли для фосфата в воде при рН=6,8 и в 0,1 н. HCl и для моногидрата свободного основания при рН=6,8, 4,5 и в 0,1 н. HCl. Исследования проводили на приборе VanKel с использованием спектрофотометра Сагу в УФ/видимой области спектра (таблица 2). Фосфат обладает более высокими характеристическими скоростями растворения в воде и 0,1 н. HCl, чем свободное основание.
HO - не определяли
Пример 8: Растворимость в водных средах и растворимость безводного моногидрата и моногидрата свободного основания определяли в различных водных средах (например, в буфере с рН 6,8, в буфере с рН 4,5, в буфере с рН 1), включая искусственные и реальные жидкости человека. Состав искусственной жидкости приведен ниже в таблице 3А-3С.
Жидкости человека брали у 12 человек. Растворимость определяли, как это описано выше, через 24 ч. Результаты приведены ниже в таблицах 4А и 4Б.
*<НПО: нижний предел количественного определения.
*<НПО: нижний предел количественного определения.
Исследование растворимости фосфата проводили в водных средах и в жидкостях человека при 24 ч. Приведены данные для фосфата без предварительного приведения в равновесие (таблица 4С) и с приведением в равновесие в течение 24 ч в HCl при pH 1 (таблица 4D). При последних условиях обнаружено, что за это время фосфат превращается в соль с HCl.
*<НПО: нижний предел количественного определения.
*<НПО: нижний предел количественного определения.
Пример 9: Спектроскопия Ф-комбинационного рассеяния свободного основания соединения формулы I
Спектроскопию Ф-комбинационного рассеяния моногидрата и безводных кристаллических форм свободного основания соединения формулы I исследовали, как это описано выше. Спектр комбинационного рассеяния безводной формы свободного основания соединения формулы I приведен на фиг.8А. Пики на фиг.8А включают 3067, 2950, 2927, 2854, 2828, 1690, 1619, 1585, 1539, 1458, 1409, 1385, 1358, 1297, 1233, 1191, 1098, 1062, 998, 819, 778, 742, 695, 668, 637, 498, 468, 420 см-1. Спектр комбинационного рассеяния моногидрата свободного основания приведен на фиг.8Б. Пики на фиг.8Б включают 2957, 2831, 1618, 1580, 1511, 1465, 1415, 1361, 1312, 1284, 1229, 1186, 1148, 1057, 994, 854, 721, 661, 624 см-1.
Пример 10: Рентгеноструктурный анализ монокристалла кристаллической формы моногидрата свободного основания соединения формулы I
Рентгеноструктурный анализ монокристалла кристаллической формы моногидрата свободного основания соединения формулы I проводили, как это описано выше. Результаты анализа приведены ниже в таблице 5 и на фиг.9.
Демонстрация стабильности кристаллических форм Пример 11: Стабильность раствора.
Стабильность раствора свободного основания исследовали в буферах в диапазоне от pH 1 до pH 9 в течение 1 недели при 50°C. При pH 5 и выше свободное основание являлось стабильным и после учета поправки на небольшое начальное количество примесей степень разложения составляла менее 2%. При pH 3 степень разложения составляла примерно 4% и при pH 1 степень разложения составляла 15%. Стабильность также исследовали в воде и различных органических растворителях (в течение 1 недели при 50°C). Свободное основание являлось стабильным в изопропаноле и степень разложения в ацетонитриле и метаноле составляла примерно от 6 до 7%.
Фосфат, форма A, являлся стабильным в изопропаноле и ацетонитриле и степень разложения в метаноле составляла примерно 8%. В воде свободное основание являлось стабильным, но степень разложения фосфата составляла примерно 45%. Это не влияет на пероральное введение фосфата; в действительности, как показано ниже, фосфат обладает примерно в 2 раза более значительной биологической доступностью, чем свободное основание. Если не ограничиваться конкретным механизмом, то можно полагать, что различия стабильностей свободного основания и фосфата могут быть обусловлены большей растворимостью фосфата в воде по сравнению со свободным основанием, а также склонностью фосфата уменьшать значение pH воды, что может увеличивать скорость разложения. И для свободного основания, и для фосфата наибольшая степень разложения наблюдается в смеси 1:1 ацетонитрил/вода (примерно 8% и 70% соответственно).
По данным анализа с помощью ВЭЖХ-МС представляется, что мочевинная группа свободного основания гидролизуется, как показано ниже, с образованием двух основных продуктов гидролиза. Два основных пика разложения, обнаруженные с помощью ВЭЖХ-МС, соответствуют ожидаемым молекулярным ионам (МН+=313 для пика ВЭЖХ при 2,1 мин и МН+=222 для пика ВЭЖХ при 8,6 мин). Для дополнительного подтверждения в прибор для ВЭЖХ вводили аутентичный образец 2,6-дихлор-3,5-диметоксианилина и время удерживания и УФ-спектр обнаруживали удовлетворительное согласие с данными для последнего элюирующегося пика разложения в ВЭЖХ.
Схема 1: Предположительный путь разложения в воде
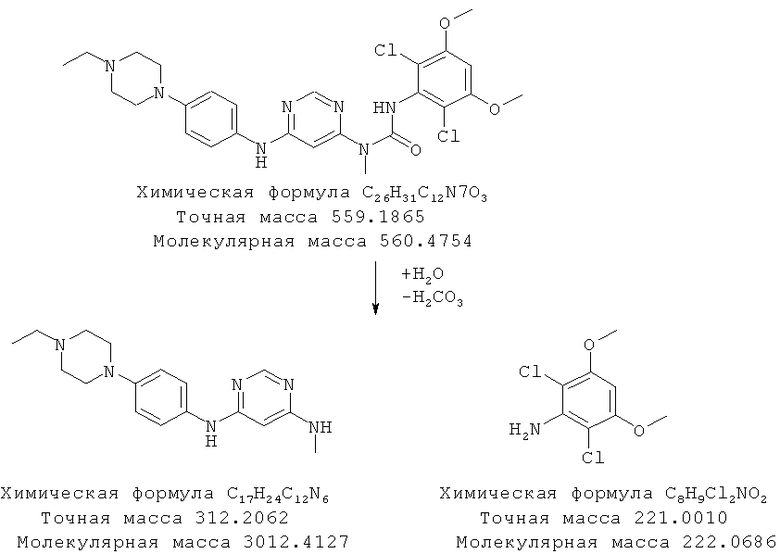
Пример 12: Стабильность в твердом состоянии
В твердом состоянии образцы выдерживали в течение 1 недели при 80°C в герметичных контейнерах и при относительной влажности, составляющей 75%. Кристаллические формы безводного свободного основания и фосфата (форма А) являлись стабильными в твердом состоянии в чистом виде и в присутствии инертных наполнителей, количество которых составляло 1% смеси. Количество продуктов разложения безводного свободного основания, определенное с помощью ВЭЖХ, составляло 1% или менее (по данным о площадях пиков), тогда как степень разложения фосфата составляла 1,4% или менее. На ПРРГ каждой полиморфной формы не обнаружено изменение формы.
Пример 13: Влияние приведения в равновесие в растворителях
А. Комнатная температура
Примерно 20 мг фосфата (форма А) приводили в равновесие с 2 мл различных растворителей в течение не менее 24 ч при комнатной температуре. Затем раствор фильтровали и остаток сушили в течение ночи при 50°C в вакууме, создаваемом лабораторной установкой. Полученное твердое вещество исследовали с помощью ПРРГ. Для растворителей, перечисленных в таблице 6, новые ПРРГ не обнаруживались. Эти результаты свидетельствуют о полиморфной стабильности фосфата (форма А).
Свободное основание приводили в равновесие в воде, этаноле, изопропаноле, этилацетате и ацетоне и, как указано выше, анализировали с помощью ПРРГ. Новые ПРРГ не обнаруживались.
В. 50°C
Примерно 20 мг фосфата (форма А) также приводили в равновесие примерно при 50°C с 2 мл растворителя в течение не менее 24 ч. Затем раствор фильтровали и остаток сушили в течение ночи при 50°C в вакууме, создаваемом лабораторной установкой. Полученное твердое вещество исследовали с помощью ПРРГ. Для растворителей, перечисленных в таблице 7, новые ПРРГ не обнаруживались. Эти результаты свидетельствуют о полиморфной стабильности фосфата (форма А).
Пример 14: Влияние кристаллизации из горящих насыщенных растворов
Примерно 20 мг фосфата (формула A) растворяли в 2 мл растворителя при 60°C. Раствор фильтровали. Полученный прозрачный фильтрат охлаждали в бане со льдом и перемешивали. Осадок собирали на фильтре, сушили и исследовали с помощью ПРРГ. При кристаллизации из 1:1 растворов воды в метаноле, этаноле, 2-пропаноле, ацетоне и тетрагидрофуране изменение полиморфной формы не обнаруживали. Только при использовании ДМСО ПРРГ указывала на изменение формы. Эти результаты свидетельствуют о полиморфной стабильности фосфата (форма А).
Пример 15: Влияние кристаллизации путем выпаривания
А. 25°C
Примерно 20 мг фосфата (форма А) смешивали с 2 мл растворителя при 25°C. После перемешивания в течение ночи раствор фильтровали. Фильтрат помещали в ток азота для выпаривания растворителя. Оставшееся твердое вещество собирали и исследовали. Указанное выше выпаривание проводили без перемешивания. После выпаривания из смеси ТГФ/вода (1:1) без перемешивания один раз получали твердое вещество, обладающее рентгенограммой ПРРГ, сходной с рентгенограммой формы В (см. фиг.1В). Затем при этих условиях получали аморфное вещество, а не форму В. Аналогичным образом, аморфное вещество получали при кристаллизации выпариванием из 1:1 растворов воды в этаноле, 2-пропаноле, ацетоне и тетрагидрофуране. Если раствор перемешивали во время выпаривания, постоянно получали форму A, что свидетельствует о полиморфной стабильности кристаллической формы фосфата (форма А).
В. 50°C
Такие же эксперименты (описанные выше для выпаривания при 25°С) проводили при 50°C. При этих условиях аморфное вещество получали только из некоторых неперемешиваемых растворов. Как показано в таблице 8, другие неперемешиваемые растворы давали форму А. Форму А также получали, когда раствор перемешивали, что свидетельствует о полиморфной стабильности кристаллической формы фосфата (форма А).
”-”: Изменение не обнаружено.
Пример 16: Влияние осаждения вследствие добавления растворителя
Исследовали 2 разные комбинации растворителей. Фосфат (форма А) растворяли в ДМСО, в котором его растворимость была высокой. К раствору добавляли растворитель (антирастворитель), в котором форма A нерастворима. Осадок собирали на фильтре, сушили и исследовали с помощью ПРРГ. Изменение формы не наблюдали для всех следующих растворителей: метанол, этанол, изопропанол, ацетон, ацетонитрил, ТГФ, этилацетат, пропилацетат, толуол, метил-трет-бутиловый эфир, 1,4-диоксан, метиленхлорид. Эти результаты свидетельствуют о полиморфной стабильности кристаллической формы фосфата (форма А).
Пример 17: Стабильность при сжатии
Фосфат (форма A, 300 мг) сжимали в течение 5 мин при 4000 фунтах в гидравлическом прессе (диаметр таблеток 8 мм). С помощью ПРРГ не наблюдали измерения кристаллической модификации. Эти результаты свидетельствуют о стабильности кристаллической формы фосфата (форма А).
Пример 18: Стабильность при гранулировании
Растворитель (воду или этанол) по каплям добавляли к фосфату (форма А), помещенному в ступку, пока твердое вещество в ступке не становилось достаточно влажным, и размалывали пестиком до пастообразной консистенции. Пасту сушили примерно при 40°C и при пониженном давлении в течение >8 ч. Твердые вещества исследовали с помощью ПРРГ и не обнаружено изменений рентгенограмм ПРРГ. Эти результаты свидетельствуют о стабильности кристаллической формы фосфата (форма А).
Пример 19: Стабильность при нагревании и охлаждении
Ее исследовали после второго эксперимента с помощью ДСК после резкого охлаждения расплава. Начало плавления обнаруживалось при 183,2°C. Образец начинал разлагаться при 185,9°C, вскоре после начала плавления. Дополнительные исследования не проводили.
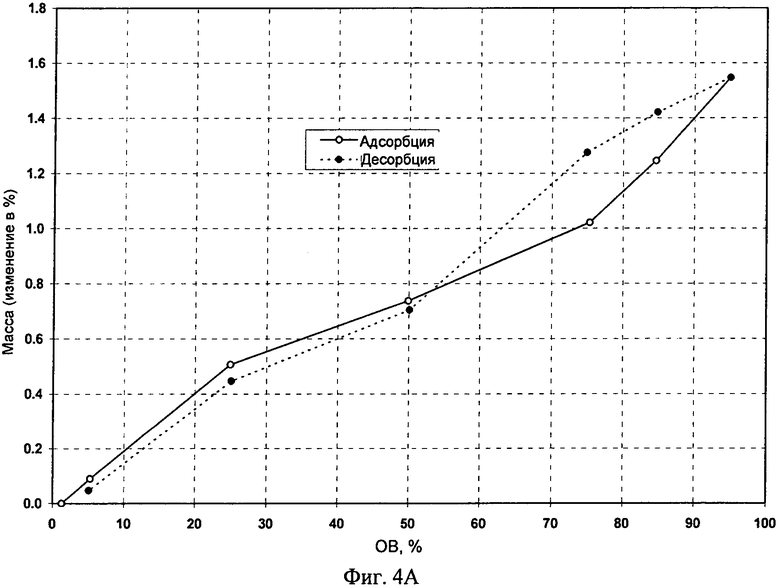
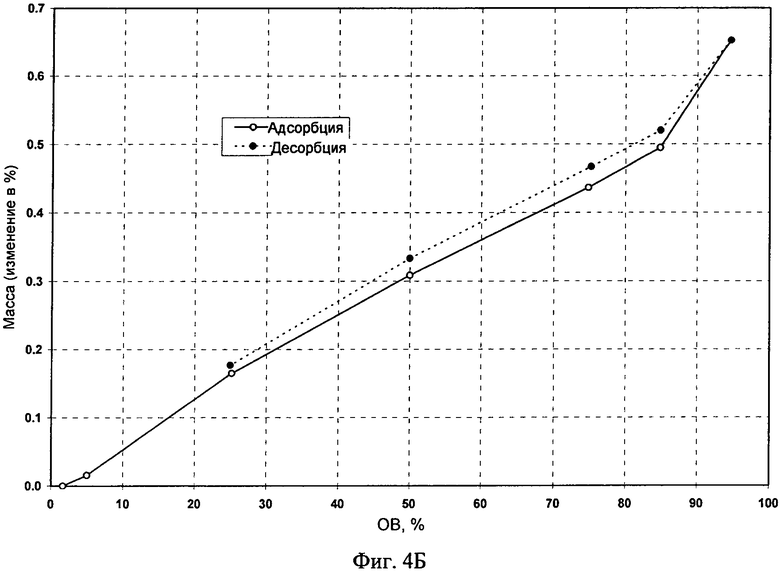
Пример 20: Эксперименты по сорбции и десорбции воды Свободное основание (безводное и моногидрат) и его фосфат (форма А) вводили в цикл сорбции и десорбции с использованием прибора VTI. После проведения эксперимента твердые вещества собирали и анализировали. На фиг.3А (свободное основание) и 3Б (фосфат (форма А)) приведены изотермы сорбции-десорбции воды, полученные с помощью прибора ТГА. Диаграммы ДСП приведены на фиг.4А (свободное основание, безводное), 4Б (фосфат, форма A) и 4В (свободное основание, моногидрат). Максимальное поглощение воды для фосфата (форма А) составляло менее 2% (т.е. 0,7%) при 25°C и относительной влажности (ОВ) до 95% (см. таблицу 9). Максимальное поглощение воды для моногидрата составляло менее 1,4% при 25°C и ОВ до 95%. Моногидрат, содержащий примерно 3% воды, при проведении эксперимента было невозможно полностью высушить, поскольку вода, которую можно было бы удалить, повторно абсорбировалась. Установлено, что плато существования моногидрата расположено в диапазоне от 10% до 95% ОВ с небольшим гистерезисом при втором цикле десорбции.
Превосходные терапевтические характеристики фосфата
Пример 21: Фармакокинетические характеристики фосфата
Более высокая растворимость в воде фосфата, чем свободного основания (и других солей с кислотами, описанных выше) позволила приготовить препарат раствора (или композиции), который невозможно было приготовить со свободным основанием вследствие его ограниченной растворимости. Для этого эксперимента фосфат получали следующим образом. В стеклянном сосуде 10,7 мг свободного основания растворяли в 4 мл ТГФ. Добавляли фосфорную кислоту (1,76 мл) в ацетоне (0,01 М). Смесь нагревали при 50°С в течение примерно 2 мин. Сосуд оставляли открытым при комнатной температуре для частичного испарения. В течение ночи образовывалась суспензия, которую фильтровали и получали примерно 5,5 мг почти белого порошка. Биологические доступности различных препаратов активных средств (свободного основания и его фосфата) для крыс приведены ниже (таблица 10).
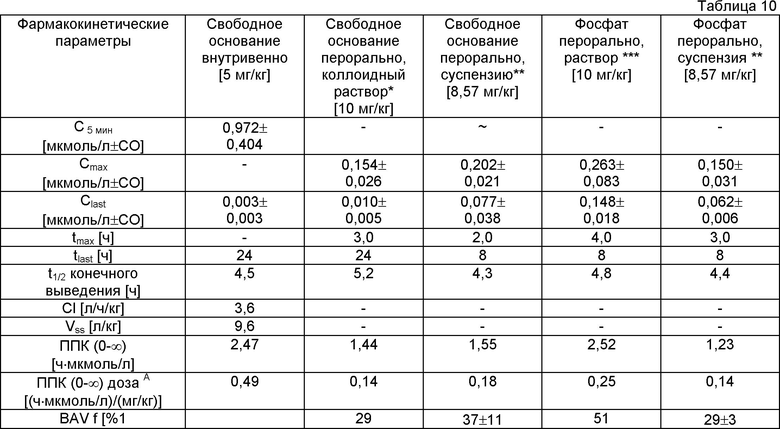
* Раствор свободного основания в смеси воды, этанола и кремофора EL.
** Суспензии готовили в воде, содержащей 0,5% гидроксипропилметилцеллюлозы.
*** Раствор фосфата готовили в смеси РЕG300/ацетатный буфер с рН 4,7 (1:1 по
объему).
**** Стандартное отклонение.
***** ППК - площадь под кривой.



Изобретение относится к новым гидратированным и безводным кристаллическим формам, а также к аморфной форме 3-(2,6-дихлор-3,5-диметоксифенил)-1-{6-[4-(4-этилпиперазин-1-ил)-фениламино]-пиримидин-4-ил}-1-метилмочевины формулы (I) и ее монофосфорной и гидрохлоридной соли. Соединения обладают свойствами ингибиторов активности киназы, выбранной из группы, включающей FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2 и Ret, и могут быть использованы при лечении заболеваний, опосредованных активностью указанных киназ. Заболевания выбраны из группы, включающей карциному почек, печени, надпочечников, мочевого пузыря, молочной железы, желудка, яичников, толстой кишки, прямой кишки, предстательной железы, поджелудочной железы, легких, влагалища или щитовидной железы; саркому; глиобластому; лейкоз; опухоли головы или шеи; псориаз; гиперплазию предстательной железы или неоплазию. В частности изобретение относится к безводной кристаллической форме соединения формулы (I) и ее моногидрату; к соли фосфорной кислоты соединения формулы (I), ее безводным кристаллическим формам А и В и ее аморфной форме; кристаллическому дигидрату гидрохлорида. Кристаллические формы охарактеризованы соответствующими данными порошковой рентгенограммы, показателями термограммы дифференциальной сканирующей калориметрии, характеристическими пиками комбинационного рассеяния. Аморфная форма соли монофосфорной кислоты не содержит характеристических пиков на порошковой рентгенограмме, не проявляет стеклования при определении ДСК и имеет температуру разложения при 115°C, определенную ТГА. Изобретение также относится к способу получения безводной кристаллической формы А соли монофосфорной кислоты. 9 н. и 25 з.п. ф-лы, 9 ил., 10 табл., 21 пр.
1. Безводная кристаллическая форма соединения формулы I
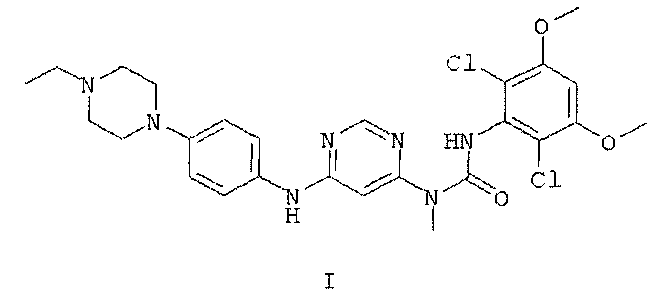
где безводная кристаллическая форма обладает порошковой рентгенограммой, содержащей характеристический пик, выраженный в 2θ, расположенный примерно при 12°.
2. Безводная кристаллическая форма по п. 1, для которой порошковая рентгенограмма дополнительно содержит один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 10,5°, примерно при 15,2°, примерно при 19,5° и примерно при 21,0°.
3. Безводная кристаллическая форма по п. 1, обладающая порошковой рентгенограммой, представленной на фиг. 1А.
4. Безводная кристаллическая форма по п. 1, обладающая полученной с помощью дифференциальной сканирующей калориметрии термограммой, на которой начало эндотермы расположено примерно при 217°C.
5. Безводная кристаллическая форма по п. 1, обладающая полученной с помощью дифференциальной сканирующей калориметрии термограммой, представленной на фиг. 2А.
6. Кристаллическая форма моногидрата соединения формулы I
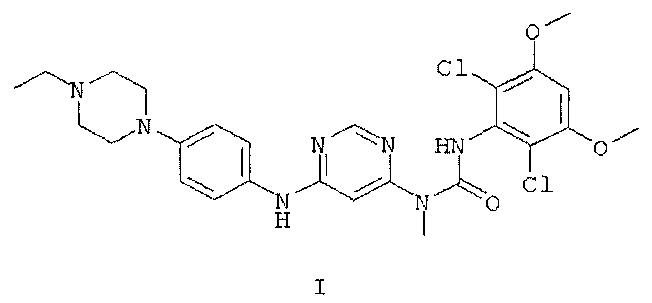
где кристаллическая форма моногидрата обладает порошковой рентгенограммой, содержащей характеристический пик, выраженный в 2θ, расположенный примерно при 20,3°.
7. Кристаллическая форма моногидрата по п. 6, для которой порошковая рентгенограмма дополнительно содержит один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 21,2° и примерно при 18,8°.
8. Кристаллическая форма моногидрата по п. 7, для которой порошковая рентгенограмма дополнительно содержит один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 4,7°, примерно при 9,4° и примерно при 11,0°.
9. Кристаллическая форма моногидрата по любому из пп. 6-8, обладающая порошковой рентгенограммой, представленной на фиг. 1Г.
10. Кристаллическая форма моногидрата по любому из пп. 6-8, обладающая полученной с помощью дифференциальной сканирующей калориметрии термограммой, на которой начало эндотермы расположено примерно при 211°C.
11. Кристаллическая форма моногидрата по любому из пп. 6-8, обладающая полученной с помощью дифференциальной сканирующей калориметрии термограммой, представленной на фиг. 7.
12. Кристаллическая форма моногидрата по любому из пп. 6-8, обладающая характеристическими пиками комбинационного рассеяния, расположенными примерно при 2957, примерно при 2831, примерно при 1618, примерно при 1511, примерно при 1465, примерно при 1361, примерно при 1229, примерно при 1186 и примерно при 994 см-1.
13. Соль монофосфорной кислоты соединения формулы I

14. Соль монофосфорной кислоты по п. 13, которая представляет собой безводную кристаллическую соль монофосфорной кислоты.
15. Безводная кристаллическая соль монофосфорной кислоты по п. 14 в форме А, где форма А обладает порошковой рентгенограммой, содержащей характеристический пик, выраженный в 2θ, расположенный примерно при 15°.
16. Безводная кристаллическая соль монофосфорной кислоты в форме А по п. 15, для которой порошковая рентгенограмма дополнительно содержит один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 13,7°, примерно при 16,8°, примерно при 21,3° и примерно при 22,4°.
17. Безводная кристаллическая соль монофосфорной кислоты в форме А по п. 16, для которой порошковая рентгенограмма дополнительно содержит один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 9,2°, примерно при 9,6°, примерно при 18,7°, примерно при 20,0°, примерно при 22,9° и примерно при 27,2°.
18. Безводная кристаллическая форма по любому из пп. 15-17, обладающая порошковой рентгенограммой, представленной на фиг. 1Б.
19. Безводная кристаллическая форма по любому из пп. 15-17, обладающая полученной с помощью дифференциальной сканирующей калориметрии термограммой, на которой начало эндотермы расположено примерно при 184°C.
20. Безводная кристаллическая форма по любому из пп. 15-17, обладающая полученной с помощью дифференциальной сканирующей калориметрии термограммой, представленной на фиг. 2Б.
21. Безводная кристаллическая форма по любому из пп. 15-17, обладающая Ф-ИК спектром, как он представлен на фиг. 6.
22. Безводная кристаллическая соль монофосфорной кислоты по п. 14 в форме В, где форма В обладает порошковой рентгенограммой, содержащей один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 9,3°, примерно при 12,5°, примерно при 13,4°, примерно при 15,8 и примерно при 17°.
23. Безводная кристаллическая форма по п. 22, обладающая порошковой рентгенограммой, представленной на фиг. 1B.
24. Аморфная форма соли монофосфорной кислоты соединения формулы I
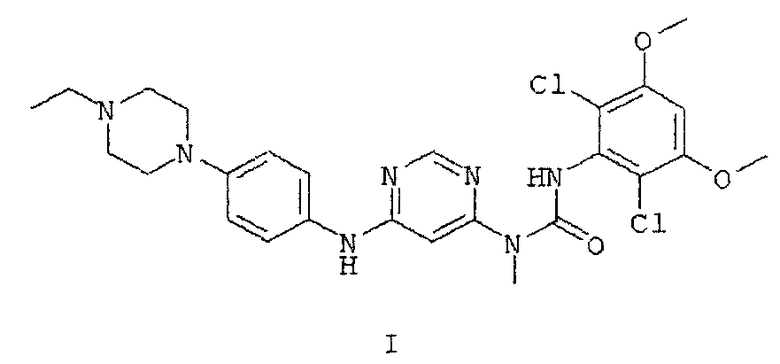
которая не содержит характеристических пиков на порошковой рентгенограмме, не проявляет стеклования при определении ДСК и имеет температуру разложения при 115°C, определенную ТГА.
25. Гидрохлорид соединения формулы I
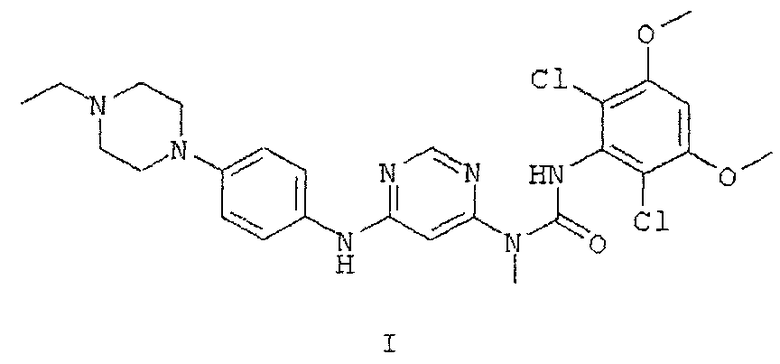
который представляет собой кристаллический дигидрат.
26. Кристаллический гидрохлорид по п. 25, обладающий порошковой рентгенограммой, содержащей один или большее количество характеристических пиков, выраженных в 2θ, выбранных из числа пиков, расположенных примерно при 10,9°, примерно при 12,1°, примерно при 14,8°, примерно при 20,5°, примерно при 22° и примерно при 25,1°.
27. Кристаллический гидрохлорид по п. 25, обладающий порошковой рентгенограммой, в основном такой, как представленная на фиг. 1Д.
28. Способ получения безводной кристаллической формы А по любому из пп. 14-17, включающий взаимодействие суспензии соединения формулы I
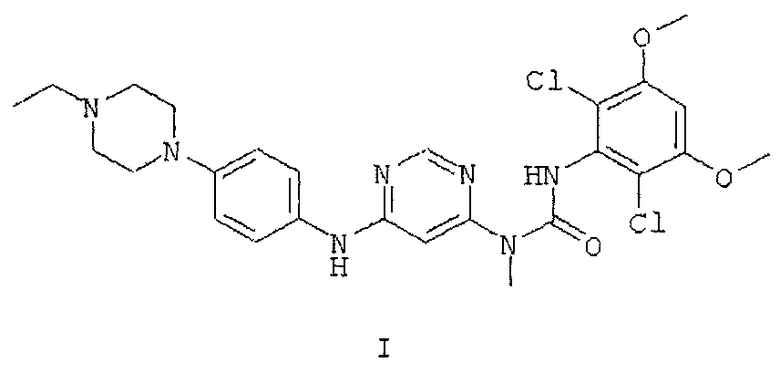
в растворителе с фосфорной кислотой с получением безводной кристаллической формы А, дополнительно включающий кристаллизацию безводной кристаллической формы А из суспензии.
29. Способ по п. 28, в котором суспензия соединения формулы I содержит изопропанол.
30. Способ по п. 28, дополнительно включающий выделение закристаллизовавшейся безводной кристаллической формы А.
31. Фармацевтическая композиция, ингибирующая активность киназы, где киназа выбрана из группы, включающей FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2 и Ret, и содержащая терапевтически активное количество кристаллической или аморфной формы по любому из пп. 1-27 и фармацевтически приемлемый носитель, инертный наполнитель или разбавитель.
32. Способ лечения, включающий введение терапевтически эффективного количества кристаллической или аморфной формы по любому из пп. 1-27 или композиции по п. 31 субъекту, страдающему от заболевания, патологии и/или симптомов, когда заболевание можно предупредить, подавить или облегчить его протекание путем ингибирования активности киназы, где киназа выбрана из группы, включающей FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2 и Ret.
33. Способ по п. 32, в котором заболевание выбрано из группы, включающей карциному почек, печени, надпочечников, мочевого пузыря, молочной железы, желудка, яичников, толстой кишки, прямой кишки, предстательной железы, поджелудочной железы, легких, влагалища или щитовидной железы; саркому; глиобластому; лейкоз; опухоли головы или шеи; псориаз; гиперплазию предстательной железы или неоплазию.
34. Применение кристаллической или аморфной формы по любому из пп. 1-27 или композиции по п. 31 для приготовления лекарственного средства, предназначенного для лечения у субъекта заболевания, при котором активность киназы FGFR1, FGFR2, FGFR3, FGFR4, KDR, HER1, HER2, Bcr-Abl, Tie2 и/или Ret способствует патологии и/или симптоматике заболевания.

