Область техники
Настоящее изобретение относится к кристаллической полиморфной модификации 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила и к способу ее получения.
Предшествующий уровень техники
Соединение (1), 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрил, известно в качестве лекарственного средства, обладающего ксантиноксидазы ингибирующим действием и способного снизить уровень мочевой кислоты в сыворотке крови (патентный документ 1).
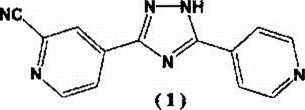
Известно несколько способов получения вышеуказанного соединения (1). В одном способе получения N-оксид метилизоникотината подвергают реакции Рейсерта-Хензе, чтобы таким образом сформировать метил-2-цианоизоникотинат, который превращают в гидразид, и гидразид конденсируют с 4-цианопиридином (патентный документ 1, пример 12). В другом способе получения N-оксид изоникотиновой кислоты превращают в гидразид, в который цианогруппу вводят с помощью реакции Рейссерта-Хензе, и полученный продукт конденсируют с 4-цианопиридином (патентный документ 1, пример 39). В альтернативном способе получения N-оксид 4-цианопиридина (исходный реагент) конденсируют с гидразидом изоникотиновой кислоты, чтобы таким образом сформировать триазольное кольцо, которое затем защищают (патентный документ 2) или не защищают (патентный документ 3), и вводят цианогруппу в полученный продукт с помощью реакции Рейссерта-Хензе, чтобы таким образом получить соединение (1).
Между тем кристаллический полиморфизм означает такое условие, при котором соединение, представляющее собой уникальную молекулу, обладающую уникальным химическим составом, существует в двух или более кристаллических формах, содержащих различные молекулярные группировки. Когда фармацевтическое соединение является таким соединением, фармакологическая активность, растворимость, биодоступность, стабильность и другие свойства такого соединения, как известно, различаются в зависимости от физико-химических свойств, присущих данной полиморфной модификации. Таким образом, когда полезное фармацевтическое соединение включает в себя кристаллические полиморфные модификации, предпочтительно получают соединение в кристаллической форме, обеспечивающей высокую полезность.
Перечень ссылок
Патентный документ
Патентный документ 1: WO 2003/064410.
Патентный документ 2: WO 2005/009991.
Патентный документ 3: JP-A-2005-41802.
Сущность изобретения
Проблемы, решаемые изобретением
Хотя вышеупомянутые патентные документы раскрывают способы получения 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, они не раскрывают кристаллический полиморфизм данного соединения. Раскрытые способы получения обеспечивают повышение выхода и химической чистоты. То есть эти патентные документы не описывают кристаллографические аспекты данного соединения.
Таким образом, задачей настоящего изобретения является обеспечение новой фармацевтически полезной кристаллической формы 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, кристаллический полиморфизм которого до сих пор не был освещен. Другой целью изобретения является обеспечение способа ее получения.
Средство решения проблем
Авторы настоящего изобретения провели обширные исследования для решения вышеупомянутых проблем и обнаружили, что обработка свободного 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила кислотой с образованием соответствующей соли, обработка данной соли основанием и нейтрализация данного обработанного основанием продукта кислотой может приводить к образованию его кристаллов типа I. Авторы изобретения также обнаружили, что перекристаллизация свободного 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила из органического растворителя может приводить к образованию его кристаллов типа II. Авторы изобретения также обнаружили, что хранение свободного 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила в увлажненных условиях может приводить к образованию его гидрата.
В связи с этим настоящее изобретение обеспечивает следующие пункты с [1] по [9].
[1] Кристаллы типа I 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила демонстрируют характеристические пики в рентгеновской порошковой дифрактометрии при дифракционных углах (2θ) около 10,1°, 16,0°, 20,4°, 25,7° и 26,7°.
[2] Кристаллы типа II 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]-пиридин-2-карбонитрила демонстрируют характеристические пики в рентгеновской порошковой дифрактометрии при дифракционных углах (2θ) около 9,9°, 16,3°, 18,2° и 22,4°.
[3] Гидрат 4-(5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила демонстрирует характеристические пики в рентгеновской порошковой дифрактометрии при дифракционных углах (2θ) около 8,1°, 14,9°, 16,4°, 25,3°, 26,9° и 27,6°.
[4] Способ получения кристаллов типа I 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, способ, содержащий обработку кислой соли 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрила основанием и последующую нейтрализацию обработанного продукта кислотой.
[5] Способ получения кристаллов типа II 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, способ, содержащий перекристаллизацию 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрила из органического растворителя.
[6] Способ получения гидрата 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, способ, содержащий хранение 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила в увлажненных условиях.
[7] Фармацевтическая композиция, содержащая кристаллы типа I, описанные выше в п. [1], и фармацевтически приемлемый носитель.
[8] Фармацевтическая композиция, содержащая кристаллы типа II, описанные выше в п. [2], и фармацевтически приемлемый носитель.
[9] Фармацевтическая композиция, содержащая гидрат, описанный выше в п. [3], и фармацевтически приемлемый носитель.
Действия изобретения
Настоящее изобретение открывает доступ к кристаллам типа I, кристаллам типа II и гидрату 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, которые являются полезными лекарственными препаратами.
Настоящее изобретение открывает доступ к способам раздельного получения кристаллов типа I, кристаллов типа II и гидрата 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила.
В частности, кристаллы типа I данного соединения являются более полезными, чем другие кристаллические формы, с точки зрения промышленного превосходства, растворимости и стабильности кристаллической формы.
Краткое описание чертежей
Фиг. 1 - порошковая рентгеновская дифрактограмма кристаллов типа I.
Фиг. 2 - порошковая рентгеновская дифрактограмма кристаллов типа II.
Фиг. 3 - порошковая рентгеновская дифрактограмма гидрата.
Фиг. 4 - термограмма дифференциальной сканирующей калориметрии (ДСК) кристаллов типа I.
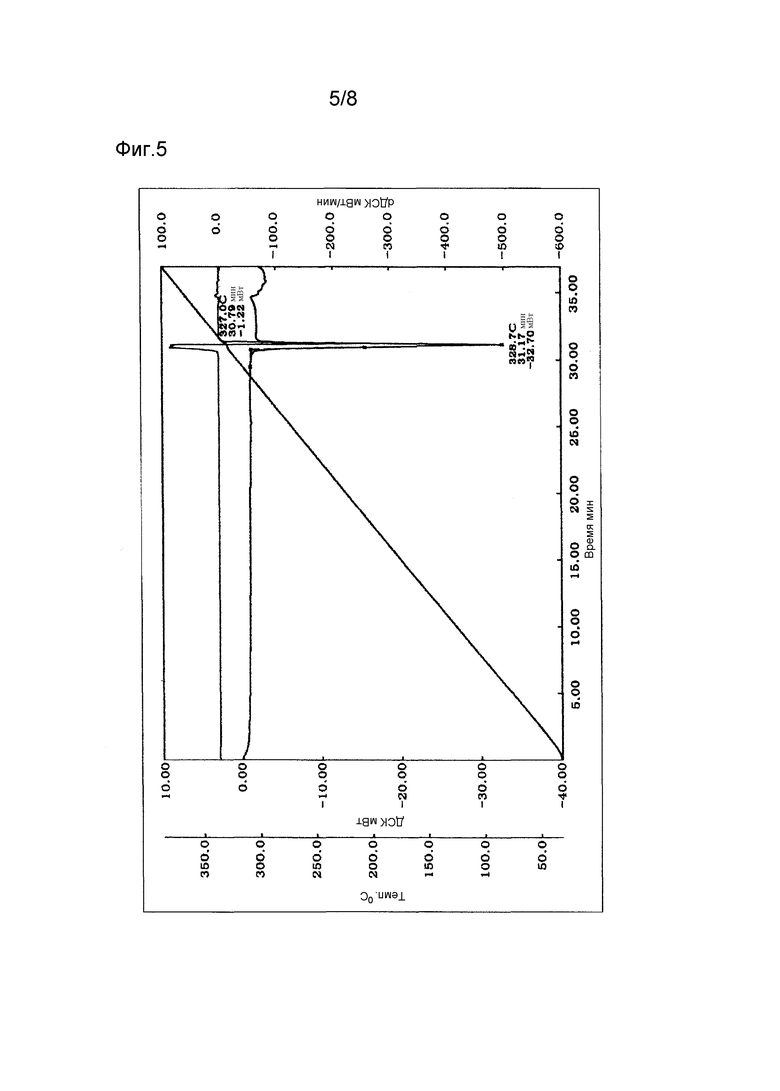
Фиг. 5 - термограмма дифференциальной сканирующей калориметрии (ДСК) кристаллов типа II.
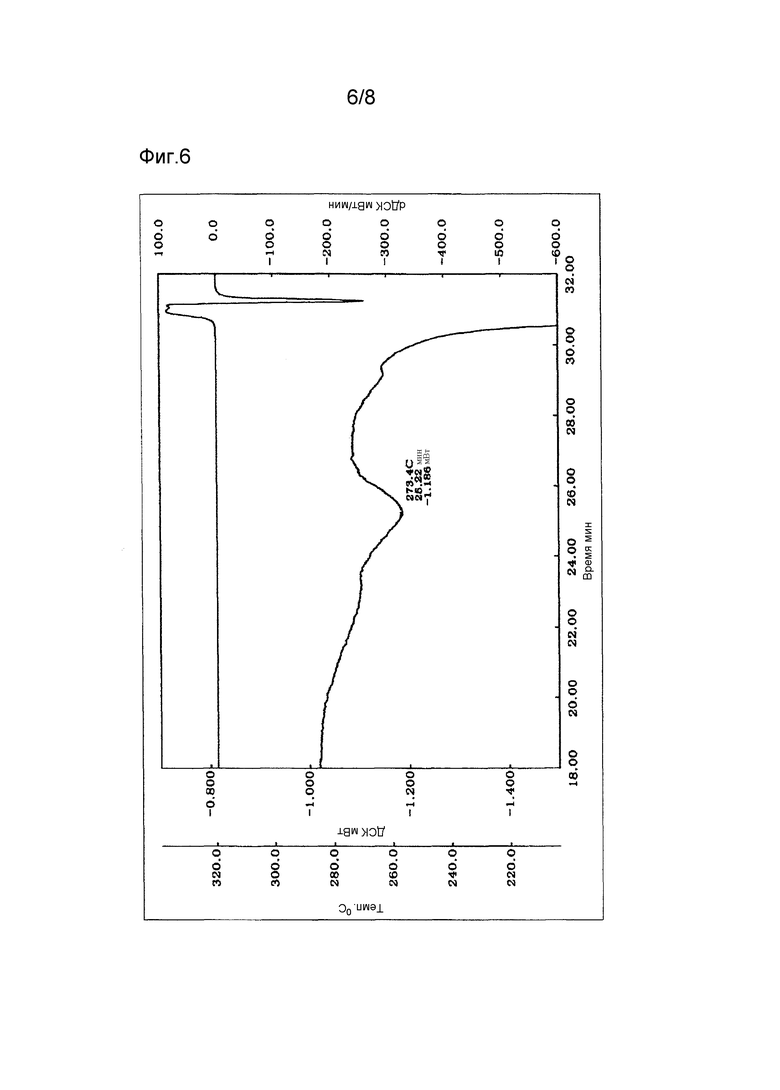
Фиг. 6 - термограмма (увеличенная) дифференциальной сканирующей калориметрии (ДСК) кристаллов типа II.
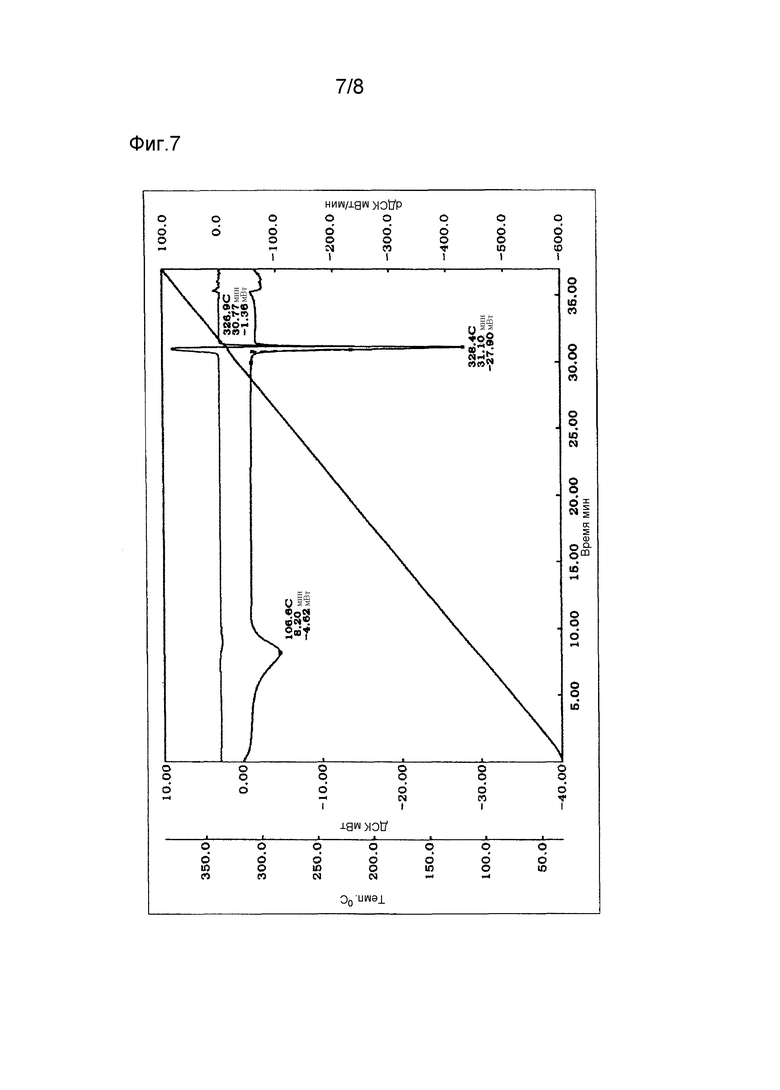
Фиг. 7 - термограмма дифференциальной сканирующей калориметрии (ДСК) гидрата.
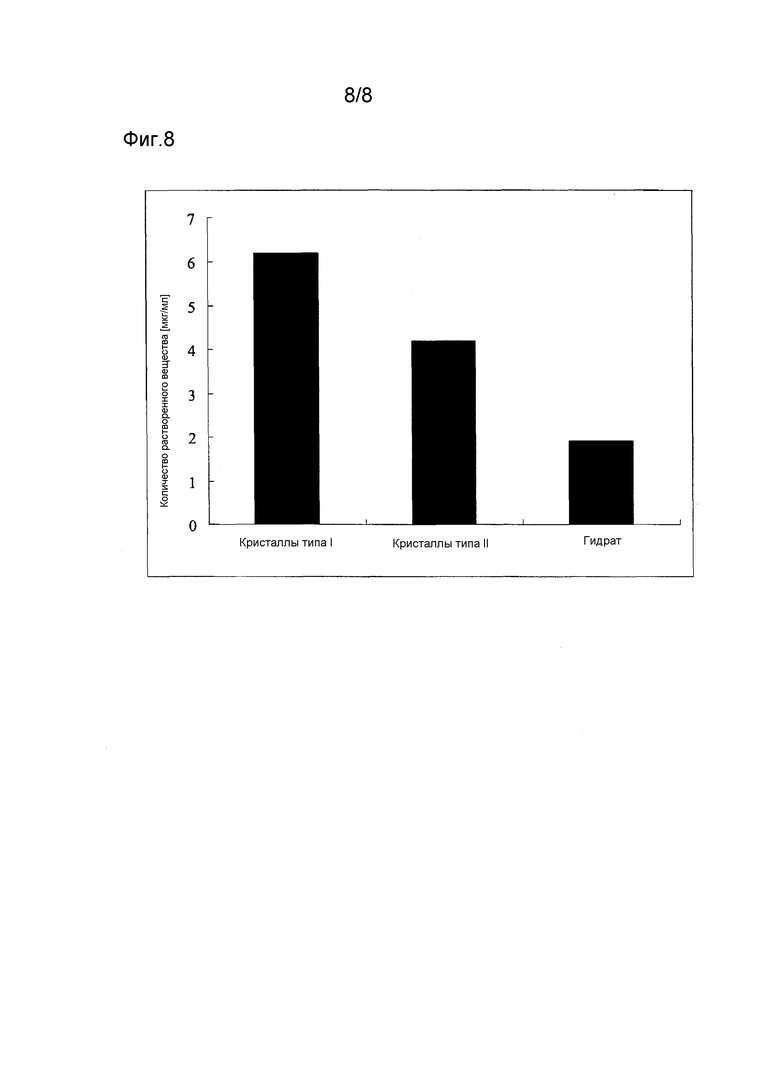
Фиг. 8 - результаты испытания растворимости различных кристаллических форм.
Способы осуществления изобретения
Кристаллы типа I 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила (далее как соединение (1)) получают с помощью обработки кислой соли соединения (1) основанием и нейтрализации обработанного продукта кислотой.
Примеры такой кислой соли соединения (1) включают в себя соли неорганических кислот, такие как гидрохлорид, сульфат и фосфат; и соли органических кислот, такие как оксалат, малонат, сукцинат, ацетат и п-толуолсульфонат. Из них предпочтительным является п-толуолсульфонат. Эти кислые соли могут быть получены с помощью любого способа, раскрытого в патентных документах с 1 по 3.
В предпочтительном варианте обработки основанием кислой соли соединения (1) основание растворяют в растворителе, а кислую соль соединения (1) добавляют к раствору. Примеры растворителя, который может растворить кислые соли соединения (1), включают в себя протонные растворители, такие как вода, метанол, этанол, изопропанол, 1-бутиловый спирт, 2-метил-1-пропанол, 2-бутанол, 2-метил-2-пропанол и этиленгликоль. При использовании эти растворители могут быть смешаны в любом соотношении, обеспечивая таким образом смешанный растворитель. Среди этих растворителей предпочтительным является водно-спиртовой смешанный растворитель, более предпочтительным смешанным растворителем является вода-этанол (от 3:1 до 10:1).
Не накладывается особых ограничений на количество, температуру и т.д. вышеуказанного растворителя, при условии, что количество, температура и т.д. позволяют кислой соли соединения (1) растворяться в нем.
Любое основание может быть использовано для обработки основанием кислой соли соединения (1), при условии, что основание может доводить раствор кислой соли соединения (1) до слабо основного состояния. Примеры оснований включают в себя неорганические основания, такие как гидроксид натрия, гидроксид калия, карбонат натрия, карбонат калия, тринатрийфосфат и трикалийфосфат; и третичные амины, такие как триэтиламин и диизопропилэтиламин. Из них предпочтительными являются карбонат калия и трикалийфосфат.
Данные основания предпочтительно используют в количестве от 2 до 5 моль, более предпочтительно в количестве от 2 до 4 моль по отношению к 1 моль кислой соли соединения (1).
Для нейтрализации обработанного основанием раствора могут быть использованы кислоты, такие как лимонная кислота, соляная кислота, серная кислота или фосфорная кислота. Из них предпочтительной является соляная кислота.
Не накладывается особых ограничений на температуру реакции нейтрализации кислотой. Тем не менее предпочтительно температура находится в диапазоне -10°С - 30°С, более предпочтительно в диапазоне 20-30°С.
При нейтрализации кислотой осаждаются кристаллы типа I соединения (1). Кристаллы типа I соединения (1) могут быть выделены при помощи сушки при пониженном давлении и нагревании.
Кристаллы типа II соединения (1) могут быть получены с помощью перекристаллизации соединения (1) из органического растворителя. Примеры растворителя для перекристаллизации включают в себя метанол, этанол, 1-пропанол, изопропанол, 1-бутиловый спирт, 2-метил-1-пропанол, 2-бутанол, 2-метил-2-пропанол, тетрагидрофуран, ацетон, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, этилацетат, эфир, диизопропиловый эфир, хлороформ, гексан, циклогексан, гептан, октан, бензол, толуол и ксилол. Эти растворители могут быть использованы по отдельности или в комбинации из двух или более видов. Вышеупомянутый растворитель для перекристаллизации предпочтительно представляет собой растворитель амидного типа, более предпочтительно N,N-диметилформамид. Перекристаллизация может быть осуществлена при помощи растворения соединения (1) при 60-160°С, предпочтительно при 120-150°С, и последующего охлаждения раствора до 15-40°С, предпочтительно до 20-30°С.
Гидрат соединения (1) может быть получен путем хранения соединения (1) в условиях высокой влажности (например, в диапазоне 20-30°С, относительная влажность (ОВ) в диапазоне 85% - 97%). Срок хранения составляет не менее 10 дней.
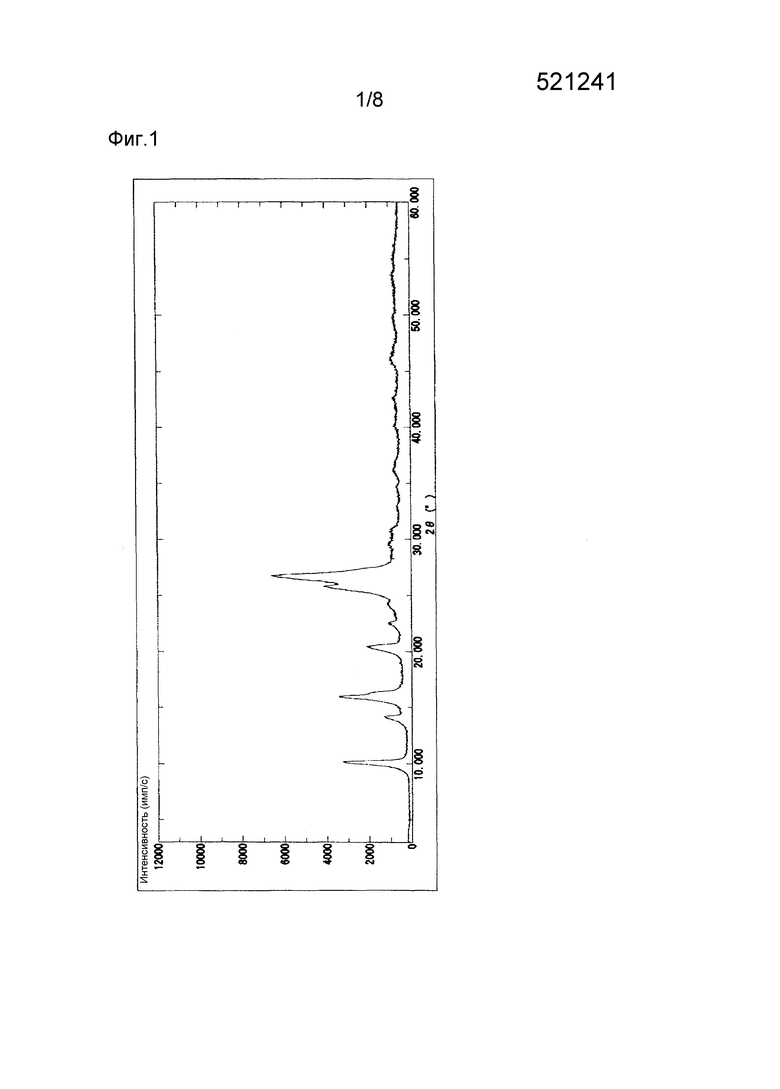
Полученные, как описано выше, кристаллы типа I соединения (1) демонстрируют характеристические пики в рентгеновской порошковой дифрактограмме при дифракционных углах (2θ) около 10,1°, 16,0°, 20,4°, 25,7° и 26,7; порошковая рентгеновская дифрактограмма показана на Фиг. 1.
Термограмма ДСК на Фиг. 4 имеет эндотермический пик примерно при 327°С.
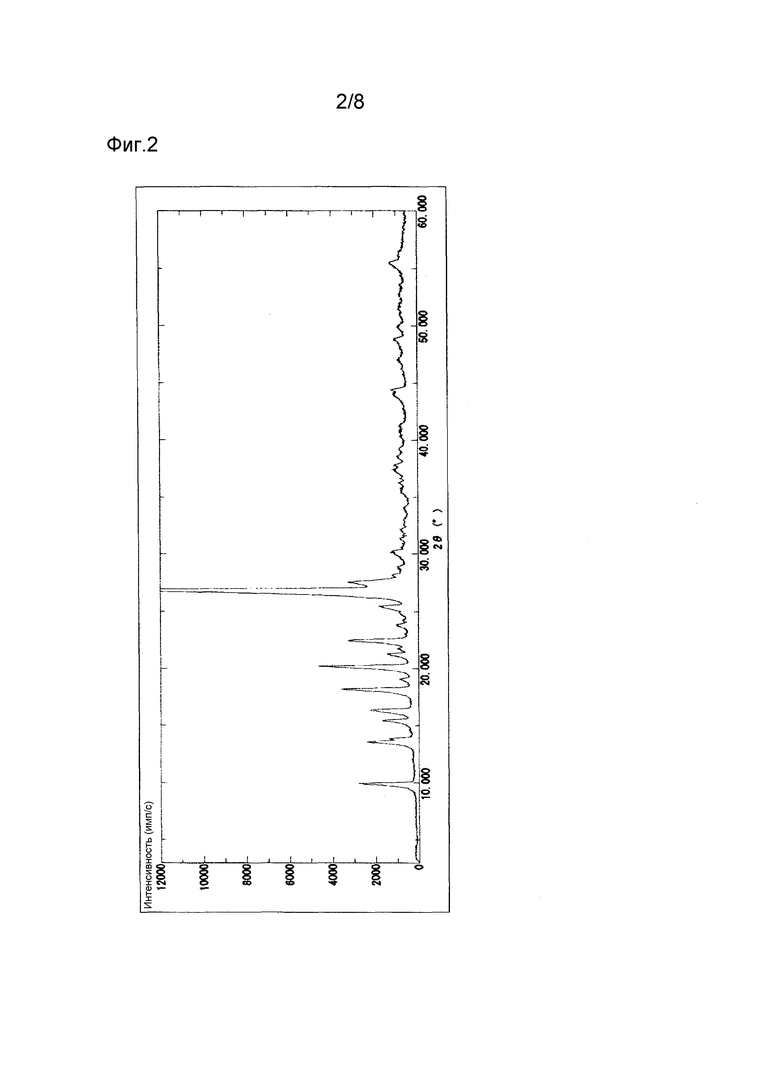
Полученные, как описано выше, кристаллы типа II соединения (1) демонстрируют характеристические пики в рентгеновской порошковой дифрактограмме при дифракционных углах (2θ) около 9,9°, 16,3°, 18,2° и 22,4°. Порошковая рентгеновская дифрактограмма показана на Фиг. 2. Термограмма ДСК на Фиг. 5 имеет эндотермический пик примерно при 327°С, а на Фиг. 6 имеет эндотермический пик примерно при 273°С.
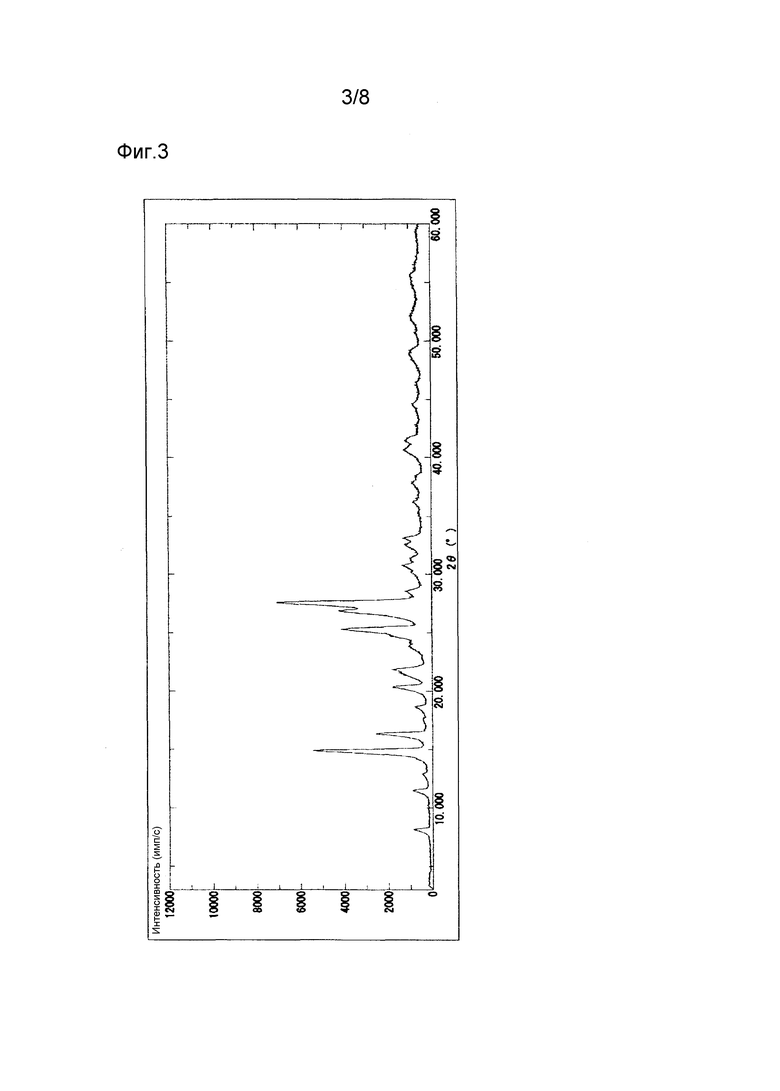
Гидрат соединения (1) демонстрирует характеристические пики в рентгеновской порошковой дифрактограмме при дифракционных углах (2θ) около 8,1°, 14,9°, 16,4°, 25,3°, 26,9° и 27,6°. Порошковая рентгеновская дифрактограмма показана на Фиг. 3.
Термограмма ДСК на Фиг. 7 имеет эндотермические пики примерно при 107°С и 327°С.
Гидрат соединения (1) предпочтительно представляет собой его моногидрат.
В настоящем изобретении спектр порошковой рентгеновской дифракции относится к спектру, измеренному с помощью прибора Mini Flex (производства Rigaku Corporation) при следующих условиях.
Источник рентгеновского излучения: Cu.
Гониометр: вертикальный.
Щель расходимости: переменная.
Щель рассеивания: 4,2 градуса.
Приемная щель: 0,3 мм.
Режим сканирования: непрерывный.
Скорость сканирования: 2°/мин.
Шаг сканирования: 0,02°.
Ось сканирования: θ/2°.
Диапазон сканирования: от 3 до 60°.
Эндотермические пики ДСК относятся к пикам, измеренным с помощью прибора DSC 220U (производства Seiko Instruments Inc.) при следующих условиях.
Скорость повышения температуры: 10°C/мин.
Атмосфера: азот.
Диапазон измерения температуры: от 30 до 400°С.
Когда кристаллы соединения (1) анализируют с помощью указанных выше приборов, кристаллические формы соединения (1), которые имеют сходные друг к другу данные и спектральные характеристики, относят к одной и той же кристаллической форме настоящего изобретения. Кроме того, если кристаллы типа I соединения (1), кристаллы типа II соединения (1) или гидрат соединения (1) по настоящему изобретению содержат другую кристаллическую форму в таком малом количестве, что его невозможно обнаружить с помощью установленного способа измерения, их также относят к одной и той же кристаллической форме настоящего изобретения.
Кроме того, данные о физических свойствах из спектров порошковой рентгеновской дифракции, ДСК и т.д. могут несколько отличаться из-за различий в условиях измерения, таких как направление роста кристаллов и размера частиц. Кристаллическая форма соединения (1) настоящего изобретения главным образом должна определяться по данным о физических свойствах, раскрытых в данном описании. Тем не менее, как описано выше, это положение не должно быть строгим, и может быть допустимо небольшое изменение в данных о физических свойствах. Например, изменение угла ±0,5° в рентгеновской дифракции, попадающее в допустимый диапазон, должно быть включено в объем прав настоящего изобретения.
Среди кристаллических форм соединения (1) по настоящему изобретению кристаллы типа I являются особенно предпочтительными с точки зрения высокой растворимости в воде и превосходной термической стабильности.
Кристаллические формы соединения (1) по настоящему изобретению обладают превосходной растворимостью в воде и термической стабильностью. Таким образом, любая из кристаллических форм может производиться в виде различных фармацевтических композиций в смеси с фармацевтически приемлемым носителем. Такая фармацевтическая композиция предпочтительно представляет собой твердый препарат, особенно предпочтительно твердый препарат для перорального применения.
При получении твердого препарата для перорального применения кристаллы соединения (1) смешивают с необходимыми добавками, такими как наполнитель, связывающее вещество, разрыхлитель, смазывающее вещество, краситель, смачивающий агент, сахарный покрывающий агент, антисептический агент, консервант, антиоксидант или вкусовой агент/корригент. Полученную таким образом смесь формируют в препараты в виде таблеток, таблеток с покрытием, гранул, порошков, капсул и т.п.
Фармацевтическая композиция по настоящему изобретению является полезной в качестве агента, сокращающего уровень мочевой кислоты или агента профилактики/терапии подагры.
Примеры
Настоящее изобретение далее будет описано более подробно с помощью примеров и примера испытания, которые не следует рассматривать как ограничивающие данное изобретение.
В примерах используются следующие сокращения: 1H ЯМР: спектр протонного ядерного магнитного резонанса, ДМСО-d6: дейтерированный диметилсульфоксид, Гц: герц, J: константа взаимодействия, с: синглет, дд: дублет дублетов и м: мультиплет. Термин "ЯМР" относится к 270 МГц спектру ядерного магнитного резонанса, измеренному с использованием ТМС (тетраметилсилан) в качестве внутреннего стандарта.
Пример 1: Синтез 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил] пиридин-2-карбонитрил п-толуолсульфоната
Моногидрат п-толуолсульфоновой кислоты (6,62 г) добавляют к смеси вода-2-бутанол (10:1) (55 мл). Далее к смеси добавляют 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрил (7,85 г) при 80°С, и полученную в результате смесь перемешивают при 80°С в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры, и осажденные кристаллы выделяют фильтрованием. Кристаллы промывают смесью вода-2-бутанол (10:1) (40 мл) и сушат при 80°С в течение 10 часов при пониженном давлении, получая таким образом 12,6 г 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрил п-толуолсульфоната.
1H-ЯМР (ДМСО-d6) δ (м.д.): 2,29 (с, 3H), 7,11 (м, 2H), 7,48 (дд, 2Н, J=6,48, 1,62 Гц), 8,32-8,35 (м, 3H), 8,57 (дд, 1Н, J=1,62, 0,81 Гц), 8,94-8,98 (м, 3H).
Пример 2: Приготовление кристаллов типа I
Карбонат калия (8,22 г) и 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрил п-толуолсульфонат (10,0 г) растворяют в смеси вода-этанол (9:1) (80 мл). К раствору добавляют 6M соляную кислоту (15 мл), и полученную смесь перемешивают при 20°С в течение 5 часов. Осажденные кристаллы выделяют фильтрованием и промывают водой (100 мл). Кристаллы сушат при 80°С в течение 23 часов при пониженном давлении, получая таким образом 5,78 г 4-[5-(пиридин-4-ил)-1H-1,2,4-триазол-3-ил]пиридин-2-карбонитрила. Полученные таким образом кристаллы демонстрируют порошковую рентгеновскую дифрактограмму, показанную на Фиг. 1, и профиль ДСК, показанный на фиг. 4, свидетельствующие о том, что полученные кристаллы являются кристаллами типа I.
1H-ЯМР (ДМСО-d6) δ (м.д.): 8,02 (дд, 2Н, J=4,59, 1,62 Гц), 8,32 (дд, 1Н, J=5,13, 1,62 Гц), 8,55 (дд, 1Н, J=1,62, 1,08 Гц), 8,80 (дд, 2Н, J=4,59, 1,62 Гц), 8,93 (дд, 1H, 5,13, 1,08 Гц).
Температура плавления: 327°С.
Пример 3: Приготовление кристаллов типа II
N,N-диметилформамид (300 мл) добавляют к 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрилу (40,0 г), и данную смесь перемешивают при 150°С в течение 25 минут. Полученный таким образом раствор охлаждают до комнатной температуры, и осажденные кристаллы выделяют фильтрованием. Кристаллы дважды промывают водой (200 мл) и сушат в течение ночи при 80°С при пониженном давлении, получая таким образом 30,4 г 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила. Полученные таким образом кристаллы демонстрируют порошковую рентгеновскую дифрактограмму, показанную на Фиг. 2, и профиль ДСК, показанный на Фиг. 5, свидетельствующие о том, что полученные кристаллы являются кристаллами типа II.
1H-ЯМР (ДМСО-d6) δ (м.д.): 8,02 (дд, 2Н, J=4,59, 1,62 Гц), 8,32 (дд, 1H, J=5,13, 1,62 Гц), 8,55 (дд, 1H, J=1,62, 1,08 Гц), 8,80 (дд, 2Н, J=4,59, 1,62 Гц), 8,93 (дд, 1H, 5,13, 1,08 Гц).
Температура плавления: 327°С.
Пример 4: Приготовление гидрата
4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрил (около 2 г) хранили при 25°С и относительной влажности воздуха 97% в течение 14 дней. Полученные таким образом кристаллы демонстрируют порошковую рентгеновскую дифрактограмму, показанную на Фиг. 3, и профиль ДСК, показанный на Фиг. 7, свидетельствующие о том, что полученные кристаллы находятся в форме гидрата.
1H-ЯМР (ДМСО-d6) δ (м.д.): 8,02 (дд, 2Н, J=4,59, 1,62 Гц), 8,32 (дд, 1H, J=5,13, 1,62 Гц), 8,55 (дд, 1H, J=1,62, 1,08 Гц), 8,80 (дд, 2Н, J=4,59, 1,62 Гц), 8,93 (дд, 1H, 5,13, 1,08 Гц).
Температура плавления: 327°С.
Пример испытания: Испытание растворимости различных форм кристаллов
Растворимость в воде кристаллов типа I, кристаллов типа II и гидрата соединения (1) определяют путем расчета концентрации насыщенного раствора каждого образца, определяемой путем измерения поглощения. На Фиг. 8 показаны результаты данного испытания. Растворимость в воде для кристаллов типа I составляет 6,2 мкг/мл, для кристаллов типа II - 4,2 мкг/мл, и 1,9 мкг/мл для гидрата.
Как видно из Фиг. 8, кристаллы типа I и кристаллы типа II имеют отличную растворимость в воде. В частности, растворимость в воде кристаллов типа I удивительно превосходна.

Изобретение относится к кристаллам типа I 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, демонстрирующим характеристические пики в рентгеновской порошковой дифрактометрии при дифракционных углах 2θ около 10,1°, 16,0°, 20,4°, 25,7° и 26,7°. Изобретение также относится к способу получения кристаллов типа I. Технический результат: получены новые кристаллы типа I 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, имеющие более высокую растворимость и более высокую термическую стабильность, по сравнению с его гидратом и кристаллами типа II. 2 н.п. ф-лы, 8 ил., 4 пр.
1. Кристаллы типа I 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила, демонстрирующие характеристические пики в рентгеновской порошковой дифрактометрии при дифракционных углах 2θ около 10,1°, 16,0°, 20,4°, 25,7° и 26,7°.
2. Способ получения кристаллов типа I 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила по п. 1, включающий обработку кислой соли 4-[5-(пиридин-4-ил)-1Н-1,2,4-триазол-3-ил]пиридин-2-карбонитрила основанием и последующую нейтрализацию обработанного продукта кислотой.
| НОВЫЕ 1,2,4-ТРИАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2002 |
|
RU2293733C2 |
| Влагоотделитель | 1989 |
|
SU1650204A1 |

