Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к новым способам получения пиримидиновых производных, к их промежуточным соединениям и к получению промежуточных соединений. Настоящее изобретение также относится к новому способу получения конкретных твердых форм пиримидинового производного, 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина (соединение A, смотри ниже), его гидратов, его солей и гидратов и сольватов его солей, к его указанным конкретным твердым формам, к фармацевтическим композициям, содержащим указанные твердые формы, к способам получения фармацевтических композиций, содержащих указанные твердые формы, к способам применения указанных твердых форм и к фармацевтическим композициям для терапевтического лечения теплокровных животных, в частности людей.
Уровень техники настоящего изобретения
WO 2007/084786 (дата приоритета: 20 января 2006) описывает определенные пиримидиновые производные, обладающие PI3K ингибирующими свойствами, их применение в качестве фармацевтических средств и способы их получения. Одно пиримидиновое производное, описанное в WO 2007/084786, представляет собой селективный ингибитор форсфатидилиназитол 3-киназы, соединение 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амин, называемое в настоящем изобретение далее "соединение A" или "соединение формулы A".
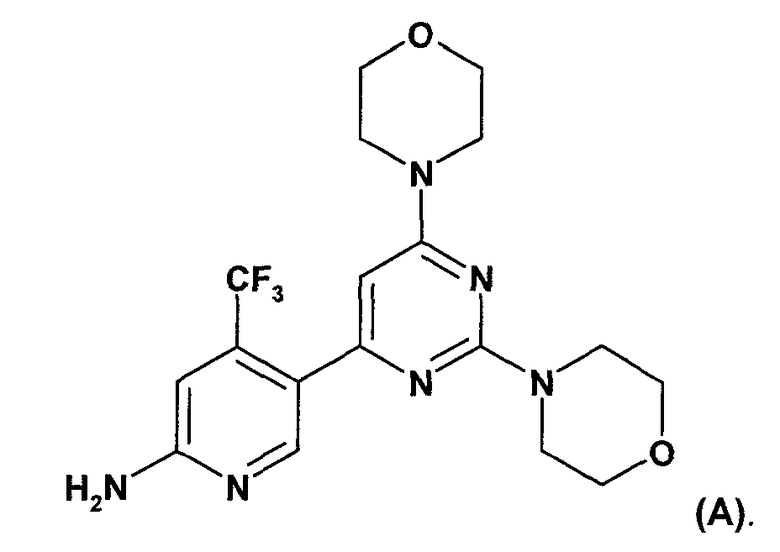
Соединение A описано в WO 2007/084786 в свободной форме и в виде соли хлористоводородной кислоты. Способ получения соединения A описан в примере 10 данного документа. Способы получения, описанные в настоящем документе, хотя и являются пригодными, рассматриваются как непригодные для промышленного получения.
Благодаря высокой активности пиримидиновых производных, в частности PI3K ингибиторов, существует необходимость в улучшенных способах получения данных соединений. В частности, существует необходимость в обеспечении способами, которые соответствуют одному или более из следующих критериев: маштабируемость, безопасность; простота; более высокие выходы и более экономичные по сравнению с известными способами.
Также остается необходимость в новых твердых формах для лечения рака.
Сущность настоящего изобретения
Таким образом, соответственно, настоящее изобретение относится к улучшенным способам получения пиримидиновых производных формулы 5, новых промежуточных соединений, пригодных в данных способах, и способам получения данных промежуточных соединений.
Таким образом, в одном аспекте, настоящее изобретение относится к способу получения соединения формулы

или его стереоизомера, таутомера или соли, где
W представляет собой CRw или N, где Rw выбран из группы, состоящей из (1) водорода, (2) циано, (3) галогена, (4) метила, (5) трифторметила, (6) сульфамидо;
R1 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) замещенного или незамещенного алкила, (6) замещенного или незамещенного алкенила, (7) замещенного или незамещенного алкинила, (8) замещенного и незамещенного арила, (9) замещенного или незамещенного гетероарила, (10) замещенного или незамещенного гетероциклила, (11) замещенного или незамещенного циклоалкила, (12) -COR1a, (13) -CO2R1a, (14) -CONR1aR1b, (15) -NR1aR1b, (17) -NR1aSO2R1b, (18) -OCOR1a, (19) -OR1a, (21) -SOR1a, где R1a и R1b независимо выбраны из группы, состоящей из (a) водорода, (b) замещенного или незамещенного алкила, (c) замещенного или незамещенного арила, (d) замещенного или незамещенного гетероарила, (e) замещенного или незамещенного гетероциклила и (f) замещенного или незамещенного циклоалкила;
R2 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) гидрокси, (6) амино, (7) замещенного или незамещенного алкила, (8) -COR2a и (9) -NR2aCOR2b, где R2a и R2b независимо выбраны из группы, состоящей из (a) водорода и (b) замещенного или незамещенного алкила;
R3 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) замещенного или незамещенного алкила, (6) замещенного или незамещенного алкенила, (7) замещенного или незамещенного алкинила, (8) замещенного и незамещенного арила, (9) замещенного или незамещенного гетероарила, (10) замещенного или незамещенного гетероциклила, 11) замещенного или незамещенного циклоалкила, (12) -COR3a, (13) -NR3aR3b, (14) -NR3aCOR3b, (15) -NR3aSO2R3b, (16) -OR3a, (17) -SR3a, (18) -SOR3a, (19) -SO2R3a, и где R3a и R3b независимо выбраны из группы, состоящей из (a) водорода, (b) замещенного или незамещенного алкила, (c) замещенного или незамещенного арила, (d) замещенного или незамещенного гетероарила, (e) замещенного или незамещенного гетероциклила и (f) замещенного или незамещенного циклоалкила; и
R4 выбран из группы, состоящей из (1) водорода и (2) галогена.
Данный способ («стадия c способа») включает стадию реакции соединения формулы 4
 ,
,
где Y2B- представляет собой ациклическую бороновую кислоту, ациклический бороновый эфир или циклический бороновый эфир, предпочтительно ациклический или циклический бороновый эфир, R1 и R2 представляют собой, как определено для формулы 5, с соединением формулы 4a
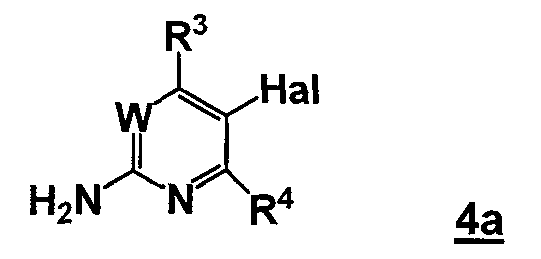 ,
,
где Hal представляет собой галоген, W, R3 и R4 представляют собой, как определено для соединения формулы 5.
Необязательно, за стадией с) способа может следовать одна или более реакций образования соли (т.е. стадия d) способа). Таким образом, стадию с) способа можно комбинировать со стадией d) способа, как описано ниже. Альтернативно или дополнительно, стадию c) способа можно комбинировать со стадией b) способа или стадиями a) и b) способа. Таким образом, настоящее изобретение относится к способам получения соединения формулы 5, включающим стадию c) способа или стадии b) и c) способа или стадии a) b) и c) способа, в каждом случае необязательно с последующей стадией d) способа. Под комбинированием способов подразумевают, что исходное вещество получают применением предшествующего способа, например, как показано на фигуре. 1. Данное исходное вещество можно применять непосредственно (т.е., без выделения и/или очистки) или после подходящих стадий обработки. Все данные альтернативные варианты включены в настоящее изобретение.
Было обнаружено, что способ, как описано в настоящем изобретении (также включающий конкретные стадии способа), соответствует одному или более из следующих критериев: безопасность; простота; большие выходы и большая экономичность по сравнению с известными способами получения соединений формулы 5. Кроме того, способ, описанный в настоящем изобретении, считают масштабируемым, что делает его подходящим для промышленного получения.
В другом аспекте, настоящее изобретение относится к способу получения соединения формулы 4
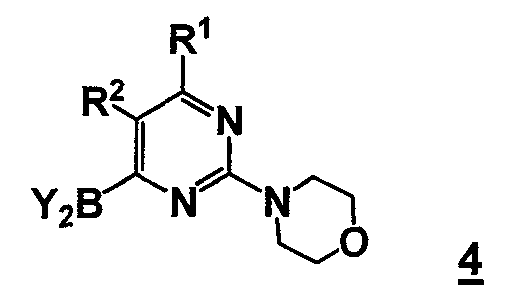 ,
,
где
Y2B- представляет собой бороновый эфир;
R1 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) замещенного или незамещенного алкила, (6) замещенного или незамещенного алкенила, (7) замещенного или незамещенного алкинила, (8) замещенного или незамещенного арила, (9) замещенного или незамещенного гетероарила, (10) замещенного или незамещенного гетероциклила, (11) замещенного или незамещенного циклоалкила, (12) -COR1a, (13) -CO2R1a, (14) - CONR1aR1b, (15) -NR1aR1b, (17) -NR1aSO2R1b, (18) -OCOR1a, (19) -OR1a, (21) -SOR1a, где R1a и R1b независимо выбраны из группы, состоящей из (a) водорода, (b) замещенного или незамещенного алкила, (c) замещенного или незамещенного арила, (d) замещенного или незамещенного гетероарила, (e) замещенного или незамещенного гетероциклила и (f) замещенного или незамещенного циклоалкила;
R2 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) гидрокси, (6) амино, (7) замещенного или незамещенного алкила, (8) -COR2a и (9) -NR2aCOR2b, где R2a и R2b независимо выбраны из группы, состоящей из (a) водорода и (b) замещенного или незамещенного алкила.
Данный способ ("стадия b способа") включает стадию реакции соединения формулы 3
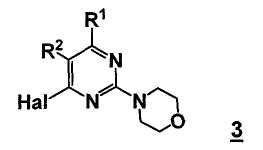 ,
,
где
R1 представляет собой, как определено для соединения формулы 5,
R2 представляет собой, как определено для соединения формулы 5, и
Hal представляет собой галоген,
с бороновым эфир или его производным формулы 6
 ,
,
где
Y2B представляет собой бороновый эфир,
X представляет собой водород, гидроксил, C1-C4 алкокси или Y2B, предпочтительно Y2B,
необязательно в присутствии катализатора, такого как Pd2(dba)3/PCy3, необязательно в присутствии разбавителя, необязательно в присутствии вспомогательного агента для реакции, получая соединение формулы 4.
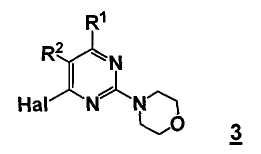
В еще другом аспекте, настоящее изобретение относится к способу получения соединения формулы 3
 ,
,
где
R1 представляет собой замещенный или незамещенный гетероцикл,
R2 представляет собой, как определено для соединения формулы 5,
Hal представляет собой галоген.
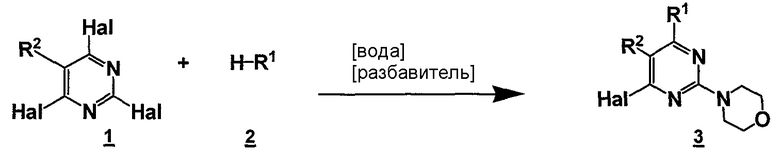
Данный способ ("стадия a способа") включает стадию реакции соединения формулы 1
 ,
,
где
R2 представляет собой, как определено для соединения формулы 5,
Hal представляет собой галоген,
с соединением формулы 2 или смесью различных соединений формулы 2
 ,
,
где R1 представляет собой замещенный или незамещенный гетероциклил или их смесь, в двухфазных условиях, необязательно в присутствии вспомогательного агента для реакции, необязательно в присутствии разбавителя, необязательно с последующими стадиями обработки и/или выделения.
В еще другом аспекте, настоящее изобретение относится к соединению формулы 4

или его стереоизомеру, таутомеру или соли, где
R1 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) замещенного или незамещенного алкила, (6) замещенного или незамещенного алкенила, (7) замещенного или незамещенного алкинила, (8) замещенного или незамещенного арила, (9) замещенного или незамещенного гетероарила, (10) замещенного или незамещенного гетероциклила, (11) замещенного или незамещенного циклоалкила, (12) -COR1a, (13) -CO2R1a, (14) -CONR1aR1b, (15) -NR1aR1b, (17) -NR1aSO2R1b, (18) -OCOR1a, (19) -OR1a, (21) -SOR1a, где R1a и R1b независимо выбраны из группы, состоящей из (a) водорода, (b) замещенного или незамещенного алкила, (c) замещенного или незамещенного арила, (d) замещенного или незамещенного гетероарила, (e) замещенного или незамещенного гетероциклила и (f) замещенного или незамещенного циклоалкила;
R2 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) гидрокси, (6) амино, (7) замещенного или незамещенного алкила, (8) -COR2a и (9) -NR2aCOR2b, где R2a и R2b независимо выбраны из группы, состоящей из (a) водорода и (b) замещенного или незамещенного алкила;
Y2B представляет собой бороновый эфир.
В другом аспекте, настоящее изобретение относится к реакции образования соли для получения соединения формулы 5a:

где
W, R1, R2, R3 и R4 представляют собой, как определено для соединения формулы 5, и
HX представляет собой кислое соединение для образования аддитивной соли кислоты.
В одном аспекте, настоящее изобретение относится к другому способу получения соединения формулы 5

или его стереоизомера, таутомера, или соли, где
W, R1, R2, R3 и R4 представляют собой, как определено выше для соединения формулы 5;
включающему стадию реакции соединения формулы 3
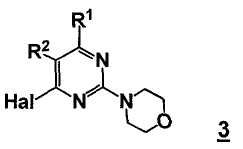 ,
,
где Hal представляет собой галоген, и R1 и R2 представляют собой, как определено для соединения формулы 5; с соединением формулы B3
 ,
,
где -BY2 представляет собой бороновую кислоту, ациклический бороновый эфир, циклический бороновый эфир или трифторборатную соль, и
W, R3 и R4 представляют собой, как определено для соединения формулы 5; и
где R5 выбран из группы, состоящей из (1) водорода, (2) замещенного или незамещенного алкила, (3) замещенного или незамещенного алкилокси, (4) замещенного или незамещенного арила, (5) замещенного или незамещенного арилокси, (6) замещенного или незамещенного арилалкилокси; в условиях реакции Сузуки, и с последующим удалением R5C(O)- группы, давая соединение формулы 5;
необязательно с последующей реакцией образования соли.
В другом аспекте, настоящее изобретение также относится к соединению формулы B3

или его стереоизомеру, таутомеру или соли, где W, R3, R4 и R5 представляют собой, как определено выше, и BY2 представляет собой бороновую кислоту, ациклический бороновый эфир, циклический бороновый эфир или трифторборатную соль.
В еще другом аспекте, настоящее изобретение также относится к способу получения соединения формулы 5,

или его стереоизомера, таутомера или соли, включающему одну или более из следующих стадий:
Стадия A: контакт соединения формулы B1

с реакционной смесью, содержащей растворитель и ангидрид кислоты (R5C=O)2O, так чтобы получить соединение формулы B2

Стадия B: i) контакт соединения формулы B2 с реакционной смесью, содержащей первый растворитель, первое основание и необязательно спиртовую добавку, ii) контакт смеси стадии (i) со вторым растворителем и вторым основанием, iii) контакт смеси стадии (ii) с производным борной кислоты, iv) необязательно контакт смеси стадии (iii) с третьим растворителем и третьим основанием, и затем контакт полученной в результате смеси с производным борной кислоты, и v) необязательно контакт смеси стадии (iii) или стадии (iv) с водой и кислотой, так что получают соединение формулы B3:

Стадия C: контакт соединения формулы B3 с реакционной смесью, содержащей растворитель, основание, катализатор и соединение формулы 3
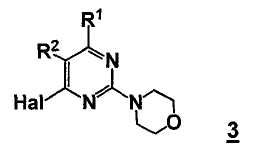
так что получают соединение формулы B5
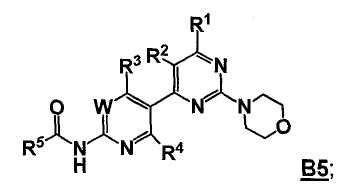
Стадия D: контакт соединения формулы B5 с реакционной смесью, содержащей растворитель и агент для удаления R5C(=O)- группы, так что получают соединение формулы 5; необязательно с последующей реакцией образования соли;
где W, R1, R2, R3, R4 и R5 представляют собой, как определено выше;
где Hal представляет собой галоген; и
где -BY2 представляет собой бороновую кислоту, ациклический бороновый эфир, циклический бороновый эфир или трифторборатную соль.
В еще других аспектах, настоящее изобретение относится к конкретным твердым, предпочтительно кристаллическим, формам соединения формулы A, его гидратов, его солей и гидратов и сольватов его солей, и способам получения данных конкретных твердых, предпочтительно кристаллических форм. Твердые формы соединения A настоящего изобретения определяют как полиморфная форма HA и безводная полиморфная форма A, и твердые формы моногидрохлоридной соли соединения A настоящего изобретения определяют как полиморфная форма Ha, полиморфная форма A, полиморфная форма B, полиморфная форма SA, полиморфная форма SB, полиморфная форма Sc, полиморфная форма SD и полиморфная форма SE.
В другом аспекте, настоящее изобретение относится к способу лечения состояний, расстройств или заболеваний, опосредованных активацией PI3K, таких как указанные выше, у нуждающегося в данном лечении субъекта, где способ включает введение указанному субъекту эффективного количества твердой, предпочтительно кристаллической, формы соединения формулы A или его моногидрохлоридной соли (например, полиморфная форма HA, безводный полиморф A, полиморфная форма Ha, полиморфная форма A, полиморфная форма B, полиморфная форма SA, полиморфная форма SB, полиморфная форма Sc, полиморфная форма SD и полиморфная форма SE).
Краткое описание чертежей
Приведенные выше аспекты и многие из сопутствующих преимуществ настоящего изобретения станут более ясны, когда они будут лучше поняты со ссылкой на следующее подробное описание, в сочетании с прилагаемыми чертежами, где:
ФИГУРА 1 показывает общий способ получения соединения формулы 5;
ФИГУРА 2 показывает способ согласно настоящему изобретению для конкретного соединения, 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина.
ФИГУРА 3 показывает известный способ для того же соединения, 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметил-пиридин-2-амина.
На рентгенограммах, обсуждаемых ниже, угол дифракции 2-тета откладывают на горизонтальной оси (x-ось) и относительную интенсивность линии (необработанная интенсивность пика) откладывают на вертикальной оси (y-ось).
ФИГУРА 4 показывает порошковую рентгеновскую дифрактограмму полиморфной формы HA гемигидрата 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина. Данные порошковой рентгеновской дифракции получали с помощью Bruker AXS Discover D8 прибора (Madison, WI, USA) с Cu K альфа источником излучения, шаг 0,02°, время цикла 2 минуты, 2 шаг, диапазон 2,00-40,00 (градусы тета) (все 2-тета величины приведены +/- 0,3).
ФИГУРА 5 показывает порошковую рентгеновскую дифрактограмму полиморфной формы A безводной 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина. Данные порошковой рентгеновской дифракции получали с помощью Bruker AXS Discover D8 прибора (Madison, WI, USA) с Cu K альфа источником излучения, шаг 0,02°, время цикла 2 минуты, 2 шаг, диапазон 2,00-40,00 (градусы тета) (все 2-тета величины приведены +/- 0,3).
ФИГУРА 6 показывает порошковую рентгеновскую дифрактограмму полиморфной формы Ha моногидрата моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина. Данные порошковой рентгеновской дифракции получали с помощью Bruker AXS Discover D8 прибора (Madison, WI, USA) с Cu K альфа источником излучения, шаг 0,02°, время цикла 2 минуты, 2 шаг, диапазон 2,00-40,00 (градусы тета) (все 2-тета величины приведены +/- 0,3).
ФИГУРА 7 показывает порошковую рентгеновскую дифрактограмму полиморфной формы A моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина (гидрохлоридная полиморфная форма A). Данные порошковой рентгеновской дифракции получали с помощью Bruker AXS Discover D8 прибора (Madison, WI, USA) с Cu K альфа источником излучения, шаг 0,02°, время цикла 2 минуты, 2 шаг, диапазон 2,00-40,00 (градусы тета) (все 2-тета величины приведены +/- 0,3).
ФИГУРА 8 показывает порошковую рентгеновскую дифрактограмму полиморфной формы B моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина. Данные порошковой рентгеновской дифракции получали с помощью Bruker AXS Discover D8 прибора (Madison, WI, USA) с Cu K альфа источником излучения, шаг 0,02°, время цикла 2 минуты, 2 шаг, диапазон 2,00-40,00 (градусы тета) (все 2-тета величины приведены +/- 0,3).
ФИГУРА 9 показывает порошковую рентгеновскую дифрактограмму полиморфной формы SA сольвата моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина. Данные порошковой рентгеновской дифракции получали с помощью Bruker AXS Discover D8 прибора (Madison, WI, USA) с Cu K альфа источником излучения, шаг 0,02°, время цикла 2 минуты, 2 шаг, диапазон 2,00-40,00 (градусы тета) (все 2-тета величины приведены +/- 0,3).
ФИГУРА 10 показывает порошковую рентгеновскую дифрактограмму полиморфной формы SB сольвата моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина. Данные порошковой рентгеновской дифракции получали с помощью Bruker AXS Discover D8 прибора (Madison, WI, USA) с Cu K альфа источником излучения, шаг 0,02°, время цикла 2 минуты, 2 шаг, диапазон 2,00-40,00 (градусы тета) (все 2-тета величины приведены +/- 0,3).
ФИГУРА 11 показывает порошковую рентгеновскую дифрактограмму полиморфной формы Sc сольвата моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина. Данные порошковой рентгеновской дифракции получали с помощью Bruker AXS Discover D8 прибора (Madison, WI, USA) с Cu K альфа источником излучения, шаг 0,02°, время цикла 2 минуты, 2 шаг, диапазон 2,00-40,00 (градусы тета) (все 2-тета величины приведены +/- 0,3).
ФИГУРА 12 показывает порошковую рентгеновскую дифрактограмму полиморфной формы SD сольвата моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина. Данные порошковой рентгеновской дифракции получали с помощью Bruker AXS Discover D8 прибора (Madison, WI, USA) с Cu K альфа источником излучения, шаг 0,02°, время цикла 2 минуты, 2 шаг, диапазон 2,00-40,00 (градусы тета) (все 2-тета величины приведены +/- 0,3).
ФИГУРА 13 показывает порошковую рентгеновскую дифрактограмму полиморфной формы SE сольвата моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина. Данные порошковой рентгеновской дифракции получали с помощью Bruker AXS Discover D8 прибора (Madison, WI, USA) с Cu K альфа источником излучения, шаг 0,02°, время цикла 2 минуты, 2 шаг, диапазон 2,00-40,00 (градусы тета) (все 2-тета величины приведены +/- 0,3).
ФИГУРА 14 показывает способ получения соединения 5.
Подробное описание
Известно, что соединения, описанные в настоящем изобретении, обладают PI3K ингибирующими свойствами. Соответственно, данные соединения являются ценными для лечения различных заболеваний, в частности для профилактики или лечения пролиферативных заболеваний. Таким образом, существует большая необходимость в обеспечении улучшенных способов получения данных соединений.
Настоящее изобретение будет более понятно, и цели, отличные от целей, приведенных выше, станут ясны при рассмотрении следующего его подробного описания, включая следующий словарь терминов, заключительные примеры и фигуры. Следующие общие определения следует применять в настоящем описании, если не указано особо:
"Галоген" обозначает фтор, бром, хлор или йод, в особенности бром или хлор. Замещенные галогеном группы и группировки, такие как алкил, замещенный галогеном (галогеналкил), могут быть моно-, поли- или пергалогенированными.
Гетероатомы представляют собой атомы, отличные от углерода и водорода, предпочтительно азот (N), кислород (O) или серу (S), в особенности азот.
Группы, группировки и молекулы, содержащие углерод, содержат 1-12, предпочтительно 1-6, более предпочтительно 1-4, самое предпочтительное 1 или 2, атомов углерода. Любая нециклическая группа или группировка, содержащая углерод, с более чем 1 атом углерода является нормальной или разветвленной.
Приставка "низший" или "C1-C7" обозначает радикал, содержащий вплоть до и включая максимум 7, особенно вплоть до и включая максимум 4 атома углерода, причем рассматриваемые радикалы являются или линейными или разветвленными с одним или несколькими разветвлениями.
"Алкил" относится к алкильным группам, которые не содержат гетероатомов. Таким образом, данная фраза включает нормальные алкильные группы, такие как метил, этил, пропил, бутил, пентил, гексил, гептил, октил, нонил, децил, ундецил, додецил и подобные. Данная фраза также включает разветвленные изомеры нормальных алкильных групп, включая, но не ограничиваясь, следующие, которые приводятся в качестве примеров: -CH(CH3)2, -CH(CH3)(CH2CH3), -CH(CH2CH3)2, -C(CH3)3, -C(CH2CH3)3, -CH2CH(CH3)2, -CH2CH(CH3)(CH2CH3), -CH2CH(CH2CH3)2, -CH2C(CH3)3, -CH2C(CH2CH3)3, -CH(CH3)-CH(CH3)(CH2CH3), -CH2CH2CH(CH3)2, -CH2CH2CH(CH3)(CH2CH3), -CH2CH2CH(CH2CH3)2, -CH2CH2C(CH3)2, -CH2CH2C(CH2CH3)3, -CH(CH3)CH2.CH(CH3)2, -CH(CH3)CH(CH3)CH(CH3)2, -CH(CH2CH3)CH(CH3)CH(CH3)(CH2CH3) и другие. Таким образом, фраза "алкильные группы" включает первичные алкильные группы, вторичные алкильные группы и третичные алкильные группы. Предпочтительные алкильные группы включают нормальные и разветвленные алкильные группы, содержащие 1-12 атомов углерода или 1-6 атомов углерода.
"Алкилен" относится к тем же самым остаткам, как указано выше для "алкила", но содержащим два положения присоединения. Примерные алкиленовые группы включают этилен (-CH2CH2-), пропилен (-CH2CH2CH2-), диметилпропилен (-CH2C(CH3)2CH2-) и циклогексилпропилен (-CH2CH2CH(C6H13)-).
"Алкенил" относится к нормальным, разветвленным или циклическим группам с от 2 до приблизительно 20 атомами углерода, таким как группы, описанные относительно алкильных групп, как определено выше, за исключением того, что они содержат одну или более углерод-углерод двойных связей. Примеры включают, но не ограничиваются, винил, -CH=C(H)(CH3), -CH=C(CH3)2, -C(CH3)=C(H)2) -C(CH3)=C(H)(CH3), -C(CH2CH3)=CH2, циклогексенил, циклопентенил, циклогексадиенил, бутадиенил, пентадиенил и гексадиенил среди других. Предпочтительные алкенильные группы включают нормальные и разветвленные алкенильные группы и циклические алкенильные группы, содержащие 2-12 атомов углерода или 2-6 атомов углерода.
"Алкинил" относится к нормальным, разветвленным или циклическим группам от 2 до приблизительно 20 атомов углерода, таким как группы, описанные относительно алкильных групп, как определено выше, за исключением того, что они содержат одну или более углерод-углерод тройных связей. Примеры включают, но не ограничиваются -C≡C(H), -C≡C(CH3), -C≡C(CH2CH3), -C(H2)C≡C(H), -C(H)2C≡C(CH3) и -C(H)2C≡C(CH2CH3) среди других. Предпочтительные алкинильные групп включают нормальные и разветвленные алкинильные группы, содержащие 2-12 атомов углерода или 2-6 атомов углерода.
Алкильные, алкиленовые, алкенильные и алкинильные группы могут быть замещенными. "Замещенный алкил" относится к алкильной группе, как определено выше, в которой одна или более связей с углеродом (углеродами) или водородом (водородами) заменены связью с атомом, не являющимся водородом или углеродом, таким как, но не ограничиваясь, атом галогена, такой как F, CI, Br и I; атом кислорода в группах, таких как гидроксильные группы, алкокси группы, арилокси группы и эфирные группы; атом серы в группах, таких как тиольные группы, алкил и арилсульфидные группы, сульфоновые группы, сульфонильные группы и сульфоксидные группы; атом азота в группах, таких как амины, амиды, алкиламины, диалкиламины, ариламины, алкилариламины, диариламины, N-оксиды, имиды и енамины; атом кремния в группах, таких как в триалкилсилильные группы, диалкиларилсилильные группы, алкилдиарилсилильные группы и триарилсилильные группы; и другие гетероатомы в различных других группах. Замещенные алкильные группы также включают группы, в которых одна или более связей с атомом углерода (атомами углерода) или водорода (атомами водорода) заменена связью большего порядка (например, двойной или тройной связью) с гетероатомом, таким как кислород в оксо, карбониле, карбоксиле и эфирных группах; азот в группах, таких как имины, оксимы, гидразоны и нитрилы. Замещенные алкильные группы дополнительно включают алкильные группы, в которых одна или более связей с атомом углерода (атомами углерода) или водорода (атомами водорода) заменена связью с арильной, гетероарильной, гетероциклильной или циклоалкильной группой. Предпочтительные замещенные алкильные группы включают, среди других, алкильные группы, в которых одна или более связей с атомом углерода или водорода заменена (заменены) одной или более связями со фтором, хлором или бром. Другой предпочтительной замещенной алкильной группой является трифторметильная группа и другие алкильные группы, которые содержат трифторметильную группу. Другие предпочтительные замещенные алкильные группы включают группы, в которых одна или более связей с атомом углерода или водорода заменена связью с атомом кислорода, так что замещенная алкильная группа содержит гидроксильную, алкокси или арилокси группу. Другие предпочтительные замещенные алкильные группы включают алкильные группы, которые содержат аминовую, или замещенную или незамещенную алкиламиновую, диалкиламиновую, ариламиновую, (алкил)(арил)аминовую, диариламиновую, гетероциклиламиновую, дигетероциклиламиновую, (алкил)(гетероциклил)аминовую или (арил)(гетероциклил)аминовую группу. Еще другие предпочтительные замещенные алкильные группы включают группы, в которых одна или более связей с атомами углерода или водорода заменена связью с арильной, гетероарильной, гетероциклильной или циклоалкильной группой. Примерами замещенного алкила являются: -(CH2)3NH2, -(CH2)3NH(CH3), -(CH2)3NH(CH3)2-CH2C(=CH2)CH2NH2, -CH2C(=O)CH2NH2, -CH2S(=O)2CH3, -CH2OCH2NH2, -CO2H, -CH2OH, -OH, -OCH3, -OC2H5, -OCF3, -OC(=O)CH3, -OC(=O)NH2), -OC(=O)N(CH3)2, -CN, -NO2, -C(=O)CH3, -CO2H, -CO2CH3, -CONH2, -NH2, -N(CH3)2, -NHSO2CH3, -NHCOCH3, -NHC(=O)OCH3, -NHSO2CH3, -SO2CH3, -SO2NH2, галоген.
"Замещенный алкенил" имеет то же значение относительно алкенильных групп, которое замещенные алкильные группы имели относительно незамещенных алкильных групп. Замещенная алкенильная группа включает алкенильные группы, в которых атом, не являющийся углеродом или водородом, соединен с углеродом, соединенным двойной связью с другим углеродом, и группы, в которых один из атомов, не являющихся углеродом или водородом, соединен с углеродом, не включенным в двойную связь с другим углеродом.
"Замещенный алкинил" имеет то же значение относительно алкинильных групп, которое замещенные алкильные группы имели относительно незамещенных алкильных групп. Замещенная алкинильная группа включает алкинильные группы, в которых атом, не являющийся углеродом или водородом, соединен с углеродом, соединенным тройной связью с другим углеродом и группы, в которых один из атомов, не являющихся углеродом или водородом, соединен с углеродом, не включенным в тройную связь с другим углеродом.
"Алкокси" относится к RO-, где R представляет собой алкил. Типичные примеры алкокси групп включают метокси, этокси, трет-бутокси, трифторметокси и подобные.
"Амино" относится в настоящем изобретении к группе -NH2. Термин "алкиламино" относится в настоящем изобретении к группе -NRR', где R представляет собой алкил, и R' представляет собой водород или алкил. Термин "ариламино" относится в настоящем изобретении к группе -NRR', где R представляет собой арил, и R' представляет собой водород, алкил или арил. Термин "аралкиламино" относится в настоящем изобретении к группе -NRR', где R представляет собой аралкил, и R' представляет собой водород, алкил, арил или аралкил.
"Алкоксиалкил" относится к группе -алкил-O-алкил2, где алкил представляет собой алкил или алкенил, и алкил2 представляет собой алкил или алкенил. Термин "арилоксиалкил" относится к группе -алкил-O-арил. Термин "аралкоксиалкил" относится к группе -алкиленил-O-аралкил.
"Алкоксиалкиламино" относится в настоящем изобретении к группе -NR-(алкоксиалкил), где R обычно представляет собой водород, аралкил или алкил.
"Аминокарбонил" относится в настоящем изобретении к группе -C(O)-NH2. "Замещенный аминокарбонил" относится в настоящем изобретении к группе -C(O)-NRR', где R представляет собой алкил, и R1 представляет собой водород или алкил. Термин "ариламинокарбонил" относится в настоящем изобретении к группе -C(O)-NRR', где R представляет собой арил, и R' представляет собой водород, алкил или арил. "Аралкиламинокарбонил" относится в настоящем изобретении к группе -C(O)-NRR1, где R представляет собой аралкил, и R1 представляет собой водород, алкил, арил или аралкил.
"Аминосульфонил" относится в настоящем изобретении к группе -S(O)2-NH2. "Замещенный аминосульфонил" относится в настоящем изобретении к группе -S(O)2-NRR', где R представляет собой алкил, и R' представляет собой водород или алкил. Термин "аралкиламиносульфониларил" относится в настоящем изобретении к группе -арил-S(O)2-NH-аралкил.
"Карбонил" относится к двухвалентной группе -C(O)-.
"Карбонилокси" относится в общем к группе -C(O)-O. Данные группы включают эфиры, -C(O)-O-R, где R представляет собой алкил, циклоалкил, арил или аралкил. Термин "карбонилоксициклоалкил" относится в общем в настоящем изобретении и к "карбонилоксикарбоциклоалкилу" и к "карбонилоксигетероциклоалкилу", т.е., где R представляет собой карбоциклоалкил или гетероциклоалкил, соответственно. Термин "арилкарбонилокси" относится в настоящем изобретении к группе -C(O)-O-арил, где арил представляет собой моно- или полициклический, карбоциклоарил или гетероциклоарил. Термин "аралкилкарбонилокси" относится в настоящем изобретении к группе -C(O)-O-аралкил.
"Сульфонил" относится в настоящем изобретении к группе -SO2-. "Алкилсульфонил" относится к замещенному сульфонилу структуры -SO2R-, в котором R представляет собой алкил. Алкилсульфонильные группы, применяемые в соединениях настоящего изобретения, обычно представляют собой алкилсульфонильные группы, содержащие от 1 до 6 атомов углерода в структуре его остова. Таким образом, типичные алкилсульфонильные группы, применяемые в соединениях настоящего изобретения, включают, например, метилсульфонил (т.е., где R представляет собой метил), этилсульфонил (т.е., где R представляет собой этил), пропилсульфонил (т.е., где R представляет собой пропил) и подобные. Термин "арилсульфонил" относится в настоящем изобретении к группе -SO2-арил. Термин "аралкилсульфонил" относится в настоящем изобретении к группе -SO2-аралкил. Термин "сульфамидо" относится в настоящем изобретении к -SO2NH2.
"Карбониламино" относится к двухвалентной группе -NH-C(O)-, в которой атом водорода амидного азота карбониламиногруппы можно замещать алкильной, арильной или аралкильной группой. Данные группы включают группировки, такие как карбаматные эфиры (-NH-C(O)-O-R) и амиды -NH-C(О)-R, где R представляет собой нормальный или разветвленный алкил, циклоалкил или арил или аралкил. Термин "алкилкарбониламино" относится к алкилкарбониламино, где R представляет собой алкил, содержащий от 1 до приблизительно 6 атомов углерода в структуре его остова. Термин "арилкарбониламино" относится к группе -NH-C(O)-R, где R представляет собой арил. Аналогично, термин "аралкилкарбониламино" относится к карбониламино, где R представляет собой аралкил.
"Гуанидино" или "гуанидил" относится к группировкам, полученным из гуанидина, H2N-C(=NH)-NH2. Данные группировки включают группировки, присоединенные через атом азота, несущий формальную двойную связь ("2"-положение гуанидина, например, диаминометиленамино, (H2N)2C=NH-)) и группировки, присоединенные через один из двух атомов азота, несущих формальную одинарную связь (1 и/или "3"-положения гуанидина, например, H2N-C=NH)-NH-)). Атомы водорода при любом из азотов могут быть заменены подходящим заместителем, таким как алкил, арил или аралкил.
"Амидино" относится к группировкам R-C(=N)-NR'- (причем радикал находится при "N1" азоте) и R(NR')C=N- (радикал находится при "N2" азоте), где R и R' могут представлять собой водород, алкил, арил или аралкил.
"Циклоалкил" относится к моно- или полициклическому, гетероциклическому или карбоциклическому алкильному заместителю.
Типичные циклоалкильные группы включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил и данные кольца, замещенные нормальными и разветвленными алкильными группами, как определено выше. Типичные циклоалкильные заместители содержат от 3 до 8 атомов в остове (т.е., кольцевых), где каждый атом остова представляет собой или углерод или гетероатом. Термин "гетероциклоалкил" относится в настоящем изобретении к циклоалкильным заместителям, которые содержат 1-5, и более обычно 1-4 гетероатома в кольцевой структуре. Подходящие гетероатомы, применяемые в соединениях настоящего изобретения, представляют собой азот, кислород и серу. Типичные гетероциклоалкильные группировки включают, например, морфолино, пиперазинил, пиперадинил и подобные. Карбоциклоалкильные группы представляют собой циклоалкильные группы, в которых все кольцевые атомы представляют собой углерод. При применении в связи с циклоалкильными заместителями, термин "полициклический" относится в настоящем изобретении к конденсированным и неконденсированным алкильным циклическим структурам.
"Замещенный гетероцикл", "гетероциклическая группа", "гетероцикл" или "гетероциклил", как применяют в настоящем изобретении, относится к любому 3- или 4-членному кольцу, содержащему гетероатом, выбранный из азота, кислорода и серы, или 5- или 6-членному кольцу, содержащему от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода или серы; где 5-членное кольцо содержит 0-2 двойные связи, и 6-членное кольцо содержит 0-3 двойные связи; где атом азота и серы может быть необязательно окислен; где гетероатом азота и серы может быть необязательно кватернизирован и включая любую бициклическую группу, в которой любое из приведенных выше гетероциклических колец конденсировано с бензольным кольцом или другим 5- или 6-членным гетероциклическим кольцом, независимо определенным выше. Примеры гетероциклильных групп включают, но не ограничиваются: ненасыщенные 3- - 8-членные кольца, содержащие 1-4 атома азота, такие как, но не ограничиваясь, пирролил, дигидропиридил, пиримидил, пиразинил, тетразолил, (например, 1H-тетразолил, 2H-тетразолил); конденсированные ненасыщенные гетероциклические группы, содержащие 1 до 4 атома азота, такие как, но не ограничиваясь, изоиндолил, индолинил, индолизинил, хинолил, индазолил; ненасыщенные 3- - 8-членные кольца, содержащие 1-2 атома кислорода и 1-3 атома азота, такие как, но не ограничиваясь, оксадиазолил (например, 1,2,4-оксадиазолил, 1,3,4-оксадиазолил, 1,2,5-оксадиазолил); насыщенные 3- - 8-членные кольца, содержащие 1-2 атома кислорода и 1-3 атома азота, такие как, но не ограничиваясь, морфолинил; ненасыщенные конденсированные гетероциклические группы, содержащие 1-2 атома кислорода и 1-3 атома азота, например, бензоксадиазолил, бензоксазинил (например, 2H-1,4-бензоксазинил); ненасыщенные 3- - 8-членные кольца, содержащие 1-3 атома серы и 1-3 атома азота, такие как, но не ограничиваясь, тиадиазолил (например, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,3,4-тиадиазолил, 1,2,-тиадиазолил); насыщенные 3- - 8-членные кольца, содержащие 1-2 атома серы и 1-3 атома азота, такие как, но не ограничиваясь, тиазолидинил; насыщенные и ненасыщенные 3- - 8-членные кольца, содержащие 1-2 атома серы, такие как, но не ограничиваясь, дигидродитиенил, дигидродитионил, тетрагидротиофен, тетрагидротиопиран; незамещенные конденсированные гетероциклические кольца, содержащие 1-2 атома серы и 1-3 атома азота, такие как, но не ограничиваясь, бензотиадиазолил, бензотиазинил (например, 2H-1,4-бензотиазинил), дигидробензотиазинил (например, 2H-3,4-дигидробензотиазинил), ненасыщенные 3- - 8-членные кольца, содержащие атомы кислорода, такие как, но не ограничиваясь, фурил; ненасыщенные конденсированные гетероциклические кольца, содержащие 1-2 атома кислорода, такие как бензодиоксоил (например, 1,3-бензодиоксоил); ненасыщенные 3- - 8-членные кольца, содержащие атом кислорода и 1-2 атома серы, такие как, но не ограничиваясь, дигидрооксатиенил; насыщенные 3- - 8-членные кольца, содержащие 1-2 атома кислорода и 1-2 атома серы, такие как 1,4-оксатиан; ненасыщенные конденсированные кольца, содержащие 1-2 атома серы, такие как бензодитиенил; и ненасыщенные конденсированные гетероциклические кольца, содержащие атом кислорода и 1-2 атома кислорода, такие как бензоксатиенил. Предпочтительные гетероциклы включают, например: диазапинил, пиррил, пирролинил, пирролидинил, пиразолил, пиразолинил, пиразолидинил, имидазоил, имидазолинил, имидазолидинил, пиридил, пиперидинил, пиразинил, пиперазинил, N-метилпиперазинил, азетидинил, N-метилазетидинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, изоксазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, изотиазолидинил, индолил, хинолинил, изохинолинил, бензимидазолил, бензотиазолил, бензоксазолил, фурил, тиенил, триазолил и бензотиенил. Гетероциклильные группы также включают группы, описанные выше, в которых один или более S атомов в кольце соединены двойной связью с одним или двумя атомами кислорода (сульфоксиды и сульфоны). Например, гетероциклильные группы включают тетрагидротиофен, тетрагидротиофеноксид и тетрагидротиофен 1,1-диоксид. Предпочтительные гетероциклильные группы содержат 5 или 6 кольцевых членов. Более предпочтительные гетероциклильные группы включают пиперазин, 1,2,3-триазол, 1,2,4-триазол, тетразол, тиоморфолин, морфолин, гомопиперазин, оксазолидин-2-он, пирролидин-2-он, хинуклидин и тетрагидрофуран.
Гетероциклические группы могут быть незамещенными, монозамещенными или дизамещенными различными заместителями, независимо выбранными из гидрокси, галогена, оксо (C=O), алкилимино (RN=, где R представляет собой алкильную или алкокси группу), амино, алкиламино, диалкиламино, ациламиноалкила, алкокси, тиоалкокси, полиалкокси, алкила, циклоалкила или галогеналкила. "Незамещенный гетероциклил" включает конденсированные гетероциклические кольца, такие как бензимидазолил, но не включает гетероциклильные группы, которые содержат другие группы, такие как алкильные или галогеновые группы, соединенные с одним из кольцевых членов, поскольку соединения, такие как 2-метилбензимидазолил, представляют собой замещенные гетероциклильные группы.
Гетероциклические группы могут быть присоединены в различных положениях, как ясно специалисту в области органической и медицинской химии в сочетании с описанием настоящего изобретения. Типичные гетероциклы включают, например, имидазолил, пиридил, пиперазинил, азетидинил, тиазолил, фуранил, триазолил, бензимидазолил, бензотиазолил, бензоксазолил, хинолинил, изохинолинил, хиназолинил, хиноксалинил, фталазинил, индолил, нафтпиридинил, индазолил, хинолизинил и гетероциклы, описанные в WO 2007/084786, параграф 154, где R представляет собой H или гетероциклический заместитель, как описывают в настоящем изобретении.
"Арил" относится к необязательно замещенным моноциклическим и полициклическим ароматическим группам, содержащим от 3 до 14 атомов углерода или гетероатомов в остове, и включает и карбоциклические арильные группы, и гетероциклические арильные группы. Термин относится, но не ограничивается, к группам, таким как фенил, бифенил, антраценил, нафтенил, в качестве примера. Карбоциклические арильные группы представляют собой арильные группы, в которых все кольцевые атомы в ароматическом кольце представляют собой углерод. Термин "гетероарил" относится в настоящем изобретении к арильным группам, содержащим от 1 до 4 гетероатомов в качестве кольцевых атомов в ароматическом кольце, причем оставшиеся кольцевые атомы представляют собой атомы углерода.
"Незамещенный арил" включает группы, содержащие конденсированные кольца, такие как нафталин. Он не включает арильные группы, которые содержат другие группы, такие как алкильные или галогеновые группы, соединенные с одним из кольцевых членов, поскольку арильные группы, такие как толил, считаются в настоящем изобретении замещенными арильными группами, как описано ниже. Предпочтительной незамещенной арильной группой является фенил. Однако, незамещенные арильные группы могут быть соединены с одним или более атомами углерода, атомами кислорода, атомами азота и/или атомами серы в исходном соединением.
"Замещенная арильная группа" имеет то же значение относительно незамещенных арильных групп, которое замещенные алкильные группы имели относительно незамещенных алкильных групп. Однако, замещенная арильная группа также включает арильные группы, в которых один из ароматических атомов углерода соединен с одним из атомов, не являющихся углеродом или водородом, описанных выше, и также включает арильные группы, в которых один или более ароматических атомов углерода соединены с замещенной и/или незамещенной алкильной, алкенильной или алкинильной группой, как определено в настоящем изобретении. Оно включает расположение связей, в котором два атома углерода арильной группы соединены с двумя атомами алкильной, алкенильной или алкинильной группы, определяя конденсированную кольцевую систему (например, дигидронафтил или тетрагидронафтил). Таким образом, фраза "замещенный арил" включает, но не ограничивается, среди других толил и гидроксифенил.
"Замещенный гетероарил", как применяют в настоящем изобретении, относится к гетероарильной группе, как определено в настоящем изобретении, в которой один, два или три атома водорода независимо замещены Cl, Br, F, I, -OH, -CN, C1-C6-алкилом, C1-C6-алкокси, C1-C6-алкокси, замещенным арилом, галогеналкилом, тиоалкокси, амино, алкиламино, диалкиламино, меркапто, нитро, карбоксальдегидом, карбокси, алкоксикарбонилом и карбоксамидом. Кроме того, любой один заместитель может представлять собой арильную, гетероарильную или гетероциклоалкильную группу.
При применении по отношению к арильным заместителям, термин "полициклический арил" относится в настоящем изобретении к конденсированным и неконденсированным циклическим структурам, в которых, по меньшей мере, одна циклическая структура является ароматической, таким как, например, бензодиоксол (который содержит гетероциклическую структуру, конденсированную с фенильной группой), нафтил и подобные. Примерные арильные или гетероарильные группы, применяемые в качестве заместителей в соединениях настоящего изобретения, включают фенил, пиридил, пиримидинил, тиазолил, индолил, имидазолил, оксадиазолил, тетразолил, пиразинил, триазолил, тиофенил, фуранил, хинолинил, пуринил, нафтил, бензотиазолил, бензопиридил и бензимидазолил, и подобные.
"Аралкил" или "арилалкил" относится к алкильной группе, замещенной арильной группой. Обычно, аралкильные группы, применяемые в соединениях настоящего изобретения, содержат от 1 до 6 атомов углерода, содержащихся в алкильной части аралкильной группы. Подходящие аралкильные группы, применяемые в соединениях настоящего изобретения, включают, например, бензил, пиколил и подобные.
Типичные гетероарильные группы включают, например, группы, показанные ниже. Данные гетероарильные группы могут быть дополнительно замещены и могут быть присоединены в различных положениях, как очевидно специалисту в области органической и медицинской химии в сочетании с описанием настоящего изобретения. Типичные гетероарилы включают, например, имидазолил, пиридил, тиазолил, триазолил, бензимидазолил, бензотиазолил и бензоксазолил и группы, описанные в WO 2007/084786, параграф 162, где R представляют собой H или гетероциклический заместитель, как описывают в настоящем изобретении.
"Биарил" относится к группе или заместителю, в котором соединены две арильные группы, которые не конденсированы друг с другом,. Примеры биарильных соединений включают, например, фенилбензол, дифенилдиазен, 4-метилтио-1-фенилбензол, феноксибензол, (2-фенилэтинил)бензол, дифенилкетон, (4-фенилбута-1,3-диинил)бензол, фенилбензиламин, (фенилметокси)бензол и подобные. Предпочтительные необязательно замещенные биарильные группы включают: 2-(фениламино)-N-[4-(2-фенилэтинил)фенил]ацетамид, 1,4-дифенилбензол, N-[4-(2-фенилэтинил)фенил]-2-[бензиламино]ацетамид, 2-амино-N-[4-(2-фенилэтинил)фенил]пропанамид, 2-амино-N-[4-(2-фенил-этинил)фенил]ацетамид, 2-(циклопропиламино)-N-[4-(2-фенилэтинил)фенил]ацетамид, 2-(этиламино)-N-[4-(2-фенилэтинил)фенил]ацетамид, 2-[(2-метилпропил)амино]-N-[4-(2-фенилэтинил)фенил]ацетамид, 5-фенил-2H-бензо-[d]1,3-диоксолен, 2-хлор-1-метокси-4-фенилбензол, 2-[(имидазолилметил)амино]-N-[4-(2-фенилэтинил)фенил]ацетамид, 4-фенил-1-феноксибензол, N-(2-аминоэтил)-[4-(2-фенилэтинил)фенил]карбоксамид, 2-{[(4-фторфенил)метил]амино}-N-[4-(2-фенилэтинил)фенил]ацетамид, 2-{[(4-метилфенил)метил]амино}-N-[4-(2-фенилэтинил)фенил]ацетамид, 4-фенил-1-(трифторметил)бензол, 1-бутил-4-фенилбензол, 2-(циклогексиламино)-N-[4-(2-фенилэтинил)фенил]ацетамид, 2-(этилметиламино)-N-[4-(2-фенилэтинил)фенил]ацетамид, 2-(бутиламино)-N-[4-(2-фенилэтинил)фенил]ацетамид, N-[4-(2-фенилэтинил)фенил]-2-(4-пиридиламино)ацетамид, N-[4-(2-фенилэтинил)фенил]-2-(хинуклидин-3-иламино)ацетамид, N-[4-(2-фенилэтинил)фенил]пирролидин-2-илкарбоксамид, 2-амино-3-метил-N-[4-(2-фенилэтинил)фенил]бутанамид, 4-(4-фенилбута-1,3-диинил)фениламин, 2-(диметиламино)-N-[4-(4-фенилбута-1,3-диинил)фенил]ацетамид, 2-(этиламино)-N-[4-(4-фенилбута-1,3-диинил)фенил]ацетамид, 4-этил-1-фенилбензол, 1-[4-(2-фенилэтинил)фенил]этан-1-он, N-(1-карбамоил-2-гидроксипропил)[4-(4-фенилбута-1,3-диинил)фенил]карбоксамид, N-[4-(2-фенил)этинил)фенил]пропанамид, 4-метоксифенилфенилкетон, фенил-N-бензамид, (трет-бутокси)-N-[(4-фенилфенил)метил]карбоксамид, 2-(3-фенилфенокси)этангидроксамовую кислоту, 3-фенилфенилпропаноат, 1-(4-этоксифенил)-4-метоксибензол и [4-(2-фенилэтинил)фенил]пиррол.
"Необязательно замещенный" или "замещенный" относится к замещению водорода одним или более моновалентным или двухвалентным радикалом. Подходящие замещающие группы включают, например, гидроксил, нитро, амино, имино, циано, галоген, тио, сульфонил, тиоамидо, амидино, имидино, оксо, оксамидино, метоксамидино, имидино, гуанидино, сульфамидо, карбоксил, формил, алкил, замещенный алкил, галогеналкил, алкиламино, галогеналкиламино, алкокси, галогеналкокси, алкоксиалкил, алкилкарбонил, аминокарбонил, арилкарбонил, аралкилкарбонил, гетероарилкарбонил, гетероаралкилкарбонил, алкилатио, аминоалкил, цианоалкил, арил, бензил, пиридил, пиразолил, пиррол, тиофен, имидазолил и подобные.
Замещающая группа может сама быть замещенной. Группа, замещающая замещающую группу, может представлять собой карбоксил, галоген, нитро, амино, циано, гидроксил, алкил, алкокси, аминокарбонил, -SR, тиоамидо, -SO3H, -SO2R или циклоалкил, где R обычно представляет собой водород, гидроксил или алкил.
Когда замещенный заместитель содержит группу с нормальной цепью, замещение может осуществляться или внутри цепи (например, 2-гидроксипропил, 2-аминобутил и подобные) или на концах цепи (например, 2-гидроксиэтил, 3-цианопропил и подобные). Замещенные заместители могут иметь нормальное, разветвленное или циклическое расположение ковалентно соединенных атомов углерода или гетероатомов.
Типичные замещенные аминокарбонильные группы включают, например, группы, показанные ниже. Они могут быть дополнительно замещены гетероциклильными группами и гетероарильными группами, как очевидно специалисту в области органической и медицинской химии в сочетании с описанием настоящего изобретения. Предпочтительные аминокарбонильные группы включают: N-(2-цианоэтил)карбоксамид, N-(3-метоксипропил)карбоксамид, N-циклопропилкарбоксамид, N-(2-гидроксиизопропил)карбоксамид, метил 2-карбониламино-3-гидроксипропаноат, N-(2-гидроксипропил)карбоксамид, N-(2-гидроксиизопропил)карбоксамид, N-[2-гидрокси-1-(гидроксиметил)этил]карбоксамид, N-(2-карбониламиноэтил)ацетамид, N-(2-(2-пиридил)этил)карбоксамид, N-(2-пиридилметил)карбоксамид, N-(оксолан-2-илметил)карбоксамид, N-(4-гидроксипирролидин-2-ил)карбоксамид, N-[2-(2-гидроксиэтокси)этил]карбоксамид, N-(4-гидроксициклогексил)карбоксамид, N-[2-(2-оксо-4-имидазолинил)этил]карбоксамид, N-карбониламинометилацетамид, N-(3-пирролидинилпропил)карбоксамид, N-[1-(карбониламинометил)пирролидин-3-ил]ацетамид, N-(2-морфолин-4-илэтил)карбоксамид, N-[3-(2-оксопирролидинил)пропил]карбоксамид, 4-метил-2-оксопиперазинкарбальдегид, N-(2-гидрокси-3-пирролидинилпропил)карбоксамид, N-(2-гидрокси-3-морфолин-4-илпропил)карбоксамид, N-{2-[(5-циано-2-пиридил)амино]этил}карбоксамид, 3-(диметиламино)пирролидинкарбальдегид, N-[(5-метилпиразин-2-ил)метил]карбоксамид, 2,2,2-трифтор-N-(1-формилпирролидин-3-ил)-ацетамид, и группы, показаные в WO2007/084786 параграф 170.
Типичные замещенные алкоксикарбонильные группы включают, например, группы, показанные в WO2007/084786, параграфы 171 и 172. Данные алкоксикарбонильные группы можно дополнительно заместить, как очевидно специалисту в области органической и медицинской химии в сочетании с описанием настоящего изобретения.
Термин "защищенный" относительно гидроксильных групп, аминогрупп и сульфгидрильных группы относится к формам данных групп, которые защищены от нежелательной реакции защитной группой, известной специалисту в данной области техники, такой как группы, описанные в Protective Groups in Organic Synthesis, Greene, T.W.; Wuts, P. G. M., John Wiley & Sons, New York, NY, (3rd Edition, 1999), которые можно вводить или удалять, применяя способы, описанные в настоящем изобретении. Примеры защищенных гидроксильных групп включают, но не ограничиваются, силильные эфиры, такие как эфиры, полученные реакцией гидроксильной группы с реагентом, таким как, но не ограничиваясь, трет-бутилдиметилхлорсилан, триметилхлорсилан, триизопропилхлорсилан, триэтилхлорсилан; замещенные метиловые и этиловые эфиры, такие как, но не ограничиваясь метоксиметиловый эфир, метилтиометиловый эфир, бензилоксиметиловый эфир, трет-бутоксиметиловый эфир, 2-метоксиэтоксиметиловый эфир, тетрагидропираниловые эфиры, 1-этоксиэтиловый эфир, аллиловый эфир, бензиловый эфир; эфиры, такие как, но не ограничиваясь, бензоилформиат, формиат, ацетат, трихлорацетат и трифторацетат. Примеры защищенных аминогрупп включают, но не ограничиваются, амиды, такие как формамид, ацетамид, трифторацетамид и бензамид; имиды, такие как фталимид и дитиосукцинимид; и другие. Примеры защищенных сульфгидрильных групп включают, но не ограничиваются, тиоэфиры, такие как S-бензилтиоэфир и S-4-пиколилтиоэфир; замещенные S-метиловые производные, такие как гемитио, дитио и аминотиоацетали; и другие.
"Карбокси-защитные группы" относится к карбонильной группе, которая этерифицирована одной из обычно применяемых карбокси-защитных эфирных групп, применяемых для блокирования или защиты карбоксильной функции, в то время как осуществляют реакции, в которых участвуют другие функциональные группы соединения. Кроме того, карбокси-защитная группа может быть присоединена к твердой подложке, посредством чего соединение остается соединенным с твердой подложкой в виде карбоксилата до отщепления гидролитическими способами, высвобождая соответствующую свободную кислоту. Типичные карбокси-защитные группы включают, например, алкиловые эфиры, вторичные амиды и подобные.
Как применяют в настоящем изобретении, термин "фармацевтически приемлемые соли" относится к нетоксичным кислым солям или солям щелочноземельных металлов пиримидиновых соединений настоящего изобретения. Данные соли можно получить in situ в процессе конечного выделения и очистки пиримидиновых соединений, или отдельно реакцией основных или кислых функциональных групп с подходящей органической или неорганической кислотой или основанием, соответственно. Типичные соли включают, но не ограничиваются, следующие: ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, циклопентанпропионат, додецилсульфат, этансульфонат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат и ундеканоат. Кроме того, основные содержащие азот группы могут быть кватернизованы такими агентами, как алкилгалогениды, такие как метил-, этил-, пропил- и бутил-хлорид, бромиды и йодиды; диалкилсульфаты, такие как диметил, диэтил, дибутил и диамилсульфаты, галиды с длинной цепью, такие как децил-, лаурил-, миристил- и стеарил-хлориды, бромиды и йодиды, аралкилгалогениды, такие как бензил и фенэтилбромиды, и другие. Посредством этого получают растворимые в воде или масле или диспергируемые продукты.
Аддитивные соли оснований можно получить in situ в процессе конечного выделения и очистки пиримидиновых соединений, или отдельно реакцией карбоксильных групп с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого металла, или с аммиаком, или органическим первичным, вторичным или третичным амином. Фармацевтически приемлемые соли содержат, но не ограничиваются, катионы на основе щелочных и щелочноземельных металлов, такие как соли натрия, лития, калия, кальция, магния, алюминия и подобные, а также нетоксичные аммониевые, четвертичные аммониевые и аминовые катионы, включая, но не ограничиваясь, аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, этиламин и подобные. Другие типичные органические амины, пригодные для образования аддитивных солей оснований, включают диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин, пиридин, пиколин, триэтаноламин и подобные, и основные аминокислоты, такие как аргинин, лизин и орнитин.
Как применяют в настоящем изобретении, термин "фармацевтически приемлемый эфир" относится к эфирам, которые гидролизуются in vivo и включают эфиры, которые легко расщепляются в человеческом теле, давая исходное соединение или его соль. Подходящие эфирные группы включают, например, группы, полученные из фармацевтически приемлемых алифатических карбоновых кислот, в частности алкановых, алкеновых, циклоалкановых и алкандионовых кислот, в которых каждая алкильная или алкенильная группа предпочтительно содержит не более 6 атомов углерода. Типичные примеры конкретных эфиров включают, но не ограничиваются, формиаты, ацетаты, пропионаты, бутираты, акрилаты и этилсукцинаты.
Предполагается, что любая формула, приведенная в настоящем изобретении, представляет соединения, имеющие структуры, показанные структурной формулой, а также определенные варианты или формы. В частности, соединения любой формулы, приведенной в настоящем изобретении, могут содержать асимметрические центры и, следовательно, существовать в виде различных энантиомерных форм. Если, по меньшей мере, один асимметрический атом углерода присутствует в соединении формулы A, то данное соединение может существовать в оптически активной форме или в виде смеси оптических изомеров, например, в виде рацемической смеси. Все оптические изомеры и их смеси, включая рацемические смеси, являются частью настоящего изобретения. Таким образом, предполагается, что любая указанная формула, приведенная в настоящем изобретении, представляет рацемат, одну или более энантиомерных форм, одну или более диастереомерных форм, одну или более атропоизомерных форм, и их смеси. Кроме того, определенные структуры могут существовать в виде геометрических изомеров (т.е., цис- и транс-изомеров), в виде таутомеров или в виде атропоизомеров.
Предполагается, что любая формула, приведенная в настоящем изобретении, представляет гидраты, сольваты и полиморфы данных соединений, и их смеси, за исключением специально указанных случаев в настоящем изобретении.
Предполагается, что любая формула, приведенная в настоящем изобретении, представляет немеченые формы, а также формы соединений с изотопной меткой. Соединения с изотопной меткой имеют структуры, показанные формулами, приведенными в настоящем изобретении, за исключением того, что один или более атомов замещены атомом, имеющим выбранную атомную массу или массовое число. Примеры изотопов, которые можно вводить в соединения настоящего изобретения, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 11C, 13C, 14C, 15N, 18F, 31P, 32P, 35S, 36Cl, 125I соответственно. Различные соединения настоящего изобретения с изотопной меткой, например, соединения, в которые вводят радиоактивные изотопы, такие как 3H, 3C и 13C. Данные соединения с изотопной меткой являются пригодными в исследованиях метаболизма (предпочтительно с 13C), исследованиях кинетики реакции (например, с 2H или 3H), способах обнаружения или получения изображений [таких как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), включая анализ на распределение лекарственного средства или субстрата в тканях], или в радиоактивном лечении пациентов. В частности, меченное 18F соединение может быть особенно предпочтительно для PET или SPECT исследований. Далее, замещение более тяжелыми изотопами, такими как дейтерий (т.е., 2H), может давать определенные терапевтические преимущества, являющиеся результатом большей метаболической стабильности, например, повышенный in vivo период полужизни или сниженные требуемые дозы. Соединения настоящего изобретения с изотопной меткой и их пролекарства можно обычно получить осуществлением способов, показанных на схемах или в примерах и примерах получения, описанных ниже, замещением немеченного изотопом реагента легко доступным изотопномеченым реагентом.
Что касается любой формулы, приведенной в настоящем изобретении, не предполагается, что выбор конкретной группы из списка возможных групп для конкретной переменной определяет группу для переменной, находящейся в другом месте. Другими словами, там где переменная появляется более одного раза, выбор групп из указанного списка является независимым от выбора групп для той же переменной в другом месте в формуле (где одно или более и вплоть до всех более общих выражений в вариантах осуществления, характеризующихся как предпочтительные выше или ниже, можно заменить более конкретными значениями, таким образом, получая более предпочтительный вариант осуществления настоящего изобретения, соответственно).
Где применяют множественную форму (например, соединения, соли), она включает единичную (например, одно соединение, одна соль). "Соединение" не исключает того, что (например, в фармацевтическом составе) присутствует более одного соединения формулы A (или его соли).
Где применяют единичную форму (например, растворитель, основание), оно включает множественную форму (например, растворители, основание). "Растворитель" и "основание" не исключают того, что (например, в реакционной смеси) присутствует более одного растворителя или основания.
Соли соединений формулы A представляют собой предпочтительно фармацевтически приемлемые соли; данные соли являются известными в данной области техники.
Способы получения соединений формулы 5
В одном аспекте, настоящее изобретение относится к способу получения соединения формулы 5
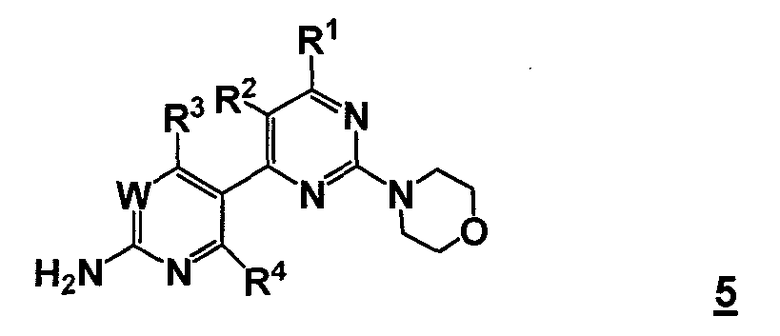
или его стереоизомера, таутомера или соли, где
W представляет собой CRW или N, где Rw выбран из группы, состоящей из (1) водорода, (2) циано, (3) галогена, (4) метила, (5) трифторметила, (6) сульфамида;
R1 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) замещенного или незамещенного алкила, (6) замещенного или незамещенного алкенила, (7) замещенного или незамещенного алкинила, (8) замещенного или незамещенного арила, (9) замещенного или незамещенного гетероарила, (10) замещенного или незамещенного гетероциклила, (11) замещенного или незамещенного циклоалкила, (12) -COR1a, (13) -CO2R1a, (14) -CONR1aR1b, (15) -NR1aR1b, (17) -NR1aSO2R1b, (18) -OCOR1a, (19) -OR1a, (21) -SOR1a, где R1a и R1b независимо выбраны из группы, состоящей из (a) водорода, (b) замещенного или незамещенного алкила, (c) замещенного или незамещенного арила, (d) замещенного или незамещенного гетероарила, (e) замещенного или незамещенного гетероциклила и (f) замещенного или незамещенного циклоалкила;
R2 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) гидрокси, (6) амино, (7) замещенного или незамещенного алкила, (8) -COR2a и (9) -NR2aCOR2b, где R2a и R2b независимо выбраны из группы, состоящей из (a) водорода и (b) замещенного или незамещенного алкила;
R3 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) замещенного или незамещенного алкила, (6) замещенного или незамещенного алкенила, (7) замещенного или незамещенного алкинила, (8) замещенного или незамещенного арила, (9) замещенного или незамещенного гетероарила, (10) замещенного или незамещенного гетероциклила, (11) замещенного или незамещенного циклоалкила, (12) -COR3a, (13) -NR3aR3b, (14) -NR3aCOR3b, (15) -NR3aSO2R3b, (16) -OR3a, (17) -SR3a, (18) -SOR3a, (19) -SO2R3a, и где R3a и R3b независимо выбраны из группы, состоящей из (a) водорода, (b) замещенного или незамещенного алкила, (c) замещенного или незамещенного арила, (d) замещенного или незамещенного гетероарила, (e) замещенного или незамещенного гетероциклила и (f) замещенного или незамещенного циклоалкила; и
R4 выбран из группы, состоящей из (1) водорода и (2) галогена.
Данный способ называют "стадия c) способа". Стадия c) способа может быть изображена следующей схемой:
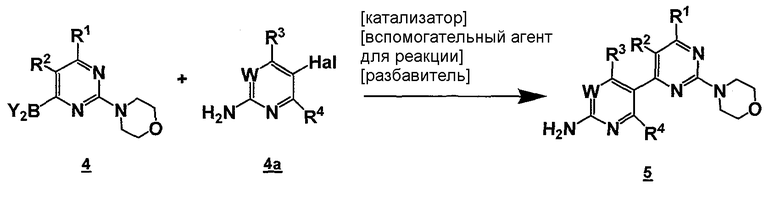
Стадия c) способа включает стадию реакции соединение формулы 4
 ,
,
где Y2B- представляет собой ациклическую бороновую кислоту, ациклический бороновый эфир, циклический бороновый эфир, предпочтительно ациклический или циклический бороновый эфир, R1 и R2 представляют собой, как определено для формулы 5, с соединением формулы 4a
 ,
,
где Hal представляет собой галоген, W, R3 и R4 представляют собой, как определено выше для соединения формулы 5 в условиях реакции Сузуки, давая соединение формулы 5.
Необязательно, за стадией c) способа может следовать одна или более реакций образования соли (т.е., стадия d) способа). Таким образом, данную стадию c) способа можно комбинировать со стадией d) способа, как описано ниже. Альтернативно или дополнительно, стадию c) способа можно комбинировать со стадией b) способа или стадиями a) и b) способа. Таким образом, настоящее изобретение относится к способам получения соединения 5, включающим стадию c) способа или стадии b) и c) способа или стадии a), b) и c) способа, в каждом случае необязательно с последующей стадией d) способа. Под комбинированием способов подразумевается, что исходные соединения получают предшествующим способом, например, как показано на фигуре 1. Данные исходные вещества можно применять непосредственно (т.е., без выделения и/или очистки) или после подходящих стадий обработки. Все данные альтернативные варианты включены в настоящее изобретение.
Предпочтительно, катализаторы/прекатализаторы для условий Сузуки выбирают из Pd(0) и Pd(II) соединений, необязательно в присутствии фосфинов. Особенно пригодными являются Pd(dbpf)Cl2 и Pd(dppf)Cl2, причем предпочтение отдается Pd(dbpf)Cl2. Подходящее количество катализатора находится в диапазоне 0,1-10 моль%, предпочтительно 3-6 моль%.
Предпочтительно, разбавители выбирают из группы полярных органических растворителей, предпочтительно эфира (такого как THF, диоксан, циклопентилметиловый эфир, 2-метил THF, DMF).
Предпочтительно, вспомогательные агенты для реакции выбирают из группы одного или более оснований, таких как карбонаты щелочных металлов, карбонаты щелочноземельных металлов, фосфатов щелочных металлов, алкоксидов, органических аминов, причем предпочтение отдается карбонату цезия.
Типичная продолжительность реакции находится в диапазоне от 1 минуты до 2 дней, предпочтительно от 10 минут до 10 часов, особенно предпочтительно 1-3 часа.
Типичные температуры реакции находятся в диапазоне от 20°C до температуры кипения, предпочтительно 30-90°C, особенно предпочтительно 40-60°C.
В одном варианте осуществления, настоящее изобретение относится к способу согласно стадии c) способа, где W представляет собой CH;
R1 представляет собой замещенный или незамещенный гетероциклил;
R2 представляет собой водород;
R3 представляет собой замещенный или незамещенный алкил;
R4 представляет собой водород.
В предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии c) способа, где
W представляет собой CH;
R1 представляет собой N-морфолинил;
R2 представляет собой водород;
R3 представляет собой трифторметил;
R4 представляет собой водород.
В следующем предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии c) способа, где Y2B- представляет собой циклический бороновый эфир, в частности 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил, и ациклическую бороновую кислоту и их эфиры.
В следующем предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии c) способа, где Hal представляет собой хлор или бром, в частности бром.
В следующем предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии c) способа, где условия Сузуки включают присутствие Pd-катализатора, в частности Pd(dbpf)Cl2.
В следующем предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии c) способа, где 4, 4a и катализатор суспендируют в разбавителе, как определено выше, и добавляют вспомогательный агент для реакции, как определено выше.
В следующем предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии c) способа, где обработка первоначально полученной реакционной смеси включает стадии i) отделения нерастворимых веществ (например, фильтрацией нерастворимых веществ, предпочтительно фильтрацией, применяя вспомогательное вещество для фильтрования, такое как слой целита), ii) отделения органической фазы, и необязательно замену растворителя другим растворителем (таким как изопропилацетат) iii) удаления остаточного палладия и iv) кристаллизации продукта (предпочтительно после водной кислотной экстракции и pH контролируемого осаждения).
Исходные вещества, вспомогательные вещества для реакции и катализаторы, применяемые на данной стадии способа, являются известными, или их можно получить аналогично известным способам. Предпочтительно, исходные вещества получают, как описано в настоящем изобретении.
В другом аспекте настоящее изобретение относится к способу получения соединения формулы 4
 ,
,
где
Y2B- представляет собой бороновый эфир;
R1 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) замещенного или незамещенного алкила, (6) замещенного или незамещенного алкенила, (7) замещенного или незамещенного алкинила, (8) замещенного или незамещенного арила, (9) замещенного или незамещенного гетероарила, (10) замещенного или незамещенного гетероциклила, (11) замещенного или незамещенного циклоалкила, (12) -COR1a, (13) -CO2R1a, (14) -CONR1aR1b, (15) -NR1aR1b, (17) -NR1aSO2R1b, (18) -OCOR1a, (19) -OR1a, (21) -SOR1a, где R1a и R1b независимо выбраны из группы, состоящей из (a) водорода, (b) замещенного или незамещенного алкила, (c) замещенного или незамещенного арила, (d) замещенного или незамещенного гетероарила, (e) замещенного или незамещенного гетероциклила и (f) замещенного или незамещенного циклоалкила;
R2 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) гидрокси, (6) амино, (7) замещенного или незамещенного алкила, (8) -COR2a и (9) -NR2aCOR2b, где R2a и R2b независимо выбраны из группы, состоящей из (a) водорода и (b) замещенного или незамещенного алкила.
Данный способ получения соединения формулы 4 называют "стадия b) способа". Стадия b) способа для получения соединений формулы 4 может быть изображена следующей схемой:

Стадия b) способа включает стадию реакции соединение формулы 3
 ,
,
где
R1 представляет собой, как определено для соединения формулы 5,
R2 представляет собой, как определено для соединения формулы 5, и
Hal представляет собой галоген,
с бороновым эфиром или его производным формулы 6
 ,
,
где
Y2B представляет собой бороновый эфир,
X представляет собой водород, гидроксил, C1-C4 алкокси или Y2B, предпочтительно Y2B,
необязательно в присутствии катализатора, такого как Pd2(dba)3/PCy3, необязательно в присутствии разбавителя, необязательно в присутствии вспомогательного вещества реакции, давая соединение формулы 4.
В предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии b) способа, где R1 представляет собой N-морфолинил.
В предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии b) способа, где Hal представляет собой хлор.
В предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии b) способа, где соединение 6 имеет формулу 6a:
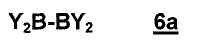 ,
,
где заместители представляют собой, как определено в настоящем изобретении.
Предпочтительно, катализаторы/прекатализаторы выбирают из Pd(0) и Pd(II) соединений, необязательно в присутствии фосфинов. Особенно пригодными являются Pd2(dba)3/PCy3. Подходящие количества катализатора находятся в диапазоне 0,1-20 моль%, предпочтительно 1-10 моль%.
Предпочтительно, разбавители выбирают из группы органических растворителей, предпочтительно THF, диоксана, ацетонитрила, пропионитрила и т.д.
Предпочтительно, дополнительные вспомогательные агенты для реакции выбирают из группы одного или более оснований, таких как карбонаты щелочных металлов, карбонаты щелочноземельных металлов, причем предпочтение отдается ацетату калия.
Типичные продолжительности реакции находятся в диапазоне 1 минута - 2 дней, предпочтительно 10 минут - 10 часов, особенно предпочтительно 2 - 4 часа.
Типичные температуры реакции находятся в диапазоне от 20°C до температуры кипения, предпочтительно 30-90°C, особенно предпочтительно 80-90°C.
Исходные вещества, вспомогательные агенты для реакции и катализаторы, применяемые на данной стадии способа, являются известными, или их можно получить аналогично известным способам. Предпочтительно, исходные вещества получают, как описывают в настоящем изобретении.
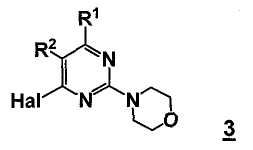
В еще другом аспекте настоящее изобретение относится к способу получения соединения формулы 3
 ,
,
где
R1 представляет собой замещенный или незамещенный гетероцикл,
R2 представляет собой, как определено для соединения формулы 5,
Hal представляет собой галоген.
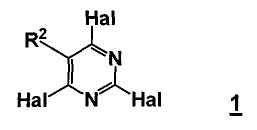
Данный способ получения соединения формулы 3 называют "стадия a) способа". Стадия a) способа для получений соединений формулы 3 может быть изображена следующей схемой:
Стадия а) способа включает стадию реакции соединение формулы 1
 ,
,
где
R2 представляет собой, как определено для соединения формулы 5,
Hal представляет собой галоген,
с соединением формулы 2 или смесью различных соединений формулы 2
 ,
,
где R1 представляет собой замещенный или незамещенный гетероциклил или их смесь, в двухфазных условиях, необязательно в присутствии вспомогательного агента для реакции, необязательно в присутствии разбавителя, необязательно с последующими стадиями обработки и/или выделения.
Настоящее изобретение также относится к способу согласно стадии a) способа, где R1 представляет собой замещенный или незамещенный гетероциклил.
Предпочтительно, стадию a) способа можно проводить в двухфазных условиях. Данный термин обозначает условия реакции, где присутствуют первая и вторая жидкая фаза. Указанная первая фаза содержит воду ("водная фаза") и указанная вторая фаза содержит органический растворитель/разбавитель ("органическая фаза"). Данные двухфазные системы являются известными в данной области техники и предпочтительно представляют собой систему вода/толуол. Ясно, что исходные вещества, промежуточные соединения, продукт и побочные продукты присутствуют в обеих фазах согласно их коэффициенту распределения. Обнаружено, что данные двухфазные условия дают большие выходы, большие селективности и облегчают выделение по сравнению с недвухфазными условиями.
В предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии a) способа, где R2 представляет собой водород.
В следующем предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии a) способа, где R1 представляет собой морфолинил. Таким образом, предпочтительное соединение формулы 2 представляет собой морфолин.
В следующем предпочтительном варианте осуществления настоящее изобретение относится к способу согласно стадии a) способа, где Hal представляет собой хлор.
Предпочтительно, 2 добавляют в избытке, предпочтительно, по меньшей мере, 4 эквивалента по сравнению с 1.
В альтернативном варианте осуществления, также можно применять в качестве исходного соединения 2 смесь двух компонентов 2-1 и 2-2, где 2-1 представляет собой морфолинил, и 2-2 представляет собой замещенный или незамещенный гетероциклил, предпочтительно замещенное или незамещенное 5- или 6-членное кольцо, содержащее от одного до трех гетероатомов, выбранных из группы, состоящей из азота, кислорода или серы; где 5-членное кольцо содержит 0-1 двойную связь, и 6-членное кольцо содержит 0-2 двойные связи. В данном случае, получают обычно смешанные замещенные производные формулы 3, которые можно разделить согласно известным способам, например, хроматографией или кристаллизацией. Из-за необходимости способов обработки, данный способ является менее предпочтительным.
Типичная продолжительность реакции находится в диапазоне 1 минута - 2 дня, предпочтительно 10 минут - 10 часов, особенно предпочтительно 1-3 часа.
Типичные температуры реакции находятся в диапазоне 0°C-100°C, предпочтительно 20°C - температура кипения, особенно предпочтительно 80-85°C.
Вещество, полученное на данной стадии, можно непосредственно применяют в следующей реакции, например, в стадии b) способа, как описывают в настоящем изобретении. Альтернативно, вещество можно очистить и выделить, например, превращением в растворимую в воде соль, такую как гидрохлорид, с последующим осаждением после добавления основания, такого как водный раствор NaOH.
Исходные вещества и вспомогательные вещества реакции, применяемые на данной стадии, являются известными и имеющимися в продаже, или их можно получить аналогично известным способам.
В еще другом аспекте, настоящее изобретение относится к соединению формулы 4

или его стереоизомеру, таутомеру или соли, где
R1 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) замещенного или незамещенного алкила, (6) замещенного или незамещенного алкенила, (7) замещенного или незамещенного алкинила, (8) замещенного или незамещенного арила, (9) замещенного или незамещенного гетероарила, (10) замещенного или незамещенного гетероциклила, (11) замещенного или незамещенного циклоалкила, (12) -COR1a, (13) -CO2R1a, (14) -CONR1aR1b, (5) -NR1aR1b (17) -NR1aSO2R1b, (18) -OCOR1a, (19) -OR1a, (21) -SOR1a, где R1a и R1b независимо выбраны из группы, состоящей из (a) водорода, (b) замещенного или незамещенного алкила, (c) замещенного или незамещенного арила, (d) замещенного или незамещенного гетероарила, (e) замещенного или незамещенного гетероциклила и (f) замещенного или незамещенного циклоалкила;
R2 выбран из группы, состоящей из (1) водорода, (2) циано, (3) нитро, (4) галогена, (5) гидрокси, (6) амино, (7) замещенного или незамещенного алкила, (8) -COR2a и (9) -NR2aCOR2b, где R2a и R2b независимо выбраны из группы, состоящей из (a) водорода и (b) замещенного или незамещенного алкила;
Y2B представляет собой бороновый эфир.
В предпочтительном варианте осуществления предпочтительные значения для соединения формулы 4 являются следующими:
R1 предпочтительно представляет собой замещенный или незамещенный гетероциклил.
R1 особенно предпочтительно представляет собой N-морфолинил.
R2 предпочтительно представляет собой водород;
Y2B предпочтительно представляет собой циклический бороновый эфир.
Y2B особенно предпочтительно представляет собой 4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил.
Данные соединения являются, например, пригодными для получения PI3K ингибиторов формулы 5. Таким образом, настоящее изобретение также относится к применению соединений формулы 4 для получения соединения формулы 5. Кроме того, настоящее изобретение относится к соединению формулы 4, как определено в настоящем изобретении, в качестве промежуточного соединения.
В другом аспекте настоящее изобретение относится к реакциям образования соли для получения соединения формулы 5a:
 ,
,
где
W, R1, R2, R3 и R4 представляют собой, как определено для соединения формулы 5, и
HX представляет собой кислое соединение для образования кислотно-аддитивной соли.
Данный способ ("стадия d способа") для получения соединений формулы 5a может быть изображен следующей схемой:
 .
.
Превращение свободных соединений в их соответствующие соли является хорошо известным в органической химии. Основные соединения, как в настоящем изобретении, можно превратить в соответствующие соли добавлением кислых соединений (HX), например, растворенных в органической или водной среде, в виде газа или вещества. Данную реакцию еще не применяли, применяя конкретные исходные вещества/условия реакции, как описывают в настоящем изобретении, где она, таким образом, образует новый и изобретательский способ.
Данную стадию предпочтительно применяют для получения фармацевтически приемлемых кислотно-аддитивных солей из соединения формулы 5. Предпочтительные фармацевтически приемлемые кислотно-аддитивные соли включают i) неорганические кислоты, в частности кислоты, выбранные из группы, состоящей из хлористоводородной кислоты, бромистоводородной кислоты, азотной кислоты, серной кислоты и фосфорной кислоты, ii) органические кислоты, в частности кислоты, выбранные из группы, состоящей из муравьиной кислоты, уксусной кислоты, трифторуксусной кислоты, фумаровой кислоты, винной кислоты, щавелевой кислоты, малеиновой кислоты, метансульфокислоты, янтарной кислоты, яблочной кислоты, метансульфокислоты, бензолсульфокислоты и п-толуолсульфокислоты, лимонной кислоты и iii) кислые аминокислоты, в частности аминокислоты, выбранные из группы, состоящей из аспарагиновой кислоты и глютаминовой кислоты. Особенно предпочтительной кислотой является хлористоводородная кислота.
Исходные вещества, вспомогательные агенты для реакции, применяемые на данной стадии реакции, являются известными, или их можно получить аналогично известным способам. Предпочтительно, исходные вещества получают, как описывают в настоящем изобретении.
Настоящее изобретение также относится еще к способу получения соединения формулы 5
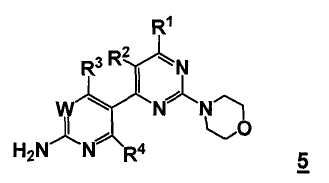
или его стереоизомера, таутомера или соли, где W, R1, R2, R3 и R4 представляют собой, как определено выше для соединения формулы 5; включающему стадию реакции соединение формулы 3
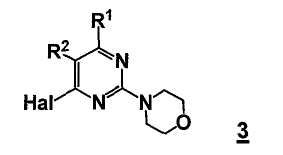 ,
,
где Hal представляет собой галоген, и R1 и R2 представляют собой, как определено для соединения формулы 5; с соединением формулы B3
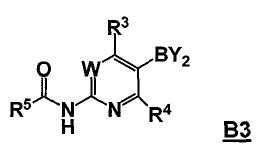 ,
,
где -BY2 представляет собой бороновую кислоту, ациклический бороновый эфир, циклический бороновый эфир или трифторборатную соль, и
W, R3 и R4 представляют собой, как определено для соединения формулы 5; и где R5 выбран из группы, состоящей из (1) водорода, (2) замещенного или незамещенного алкила, (3) замещенного или незамещенного алкилокси, (4) замещенного или незамещенного арила, (5) замещенного или незамещенного арилокси, (6) замещенного или незамещенного арилалкилокси; в условиях реакции Сузуки, и последующее удаление R5C(O)- группировки, давая соединение формулы 5; необязательно с последующей реакцией образования соли.
В конкретном варианте осуществления трифторборатная соль представляет собой калиевую соль.
Реакция Сузуки, которую применяют во многих из реакций, описанных выше, представляет собой, в принципе, известную реакцию в органической химии и обозначает катализируемую палладием конденсацию двух реагентов, где один из реагентов содержит галогенидную группу и другой реагент содержит реакционноспособную группу, представляющую собой бороновый эфир или бороновую кислоту. Подходящие условия для данной реакции ("условия Сузуки") являются известными специалисту в данной области техники и относятся в частности к выбору катализатора, разбавителя, дополнительных вспомогательных агентов для реакции, продолжительности реакции и температуры реакции. Данную реакцию еще не применяли, применяя конкретные исходные вещества, как описывают в настоящем изобретении, где она, таким образом, образует новый и изобретательский способ. В конкретном варианте осуществления способа, Pd-катализатор представляет собой Pd(PPh3)4.
В одном варианте осуществления способа, B3 получают реакцией соединение формулы B1

с ангидридом кислоты (R5C=O)2O, так что получают соединение формулы B2

реакцией соединения формулы B2 с реакционной смесью, содержащей первый растворитель, первое основание и необязательно спиртовую добавку; реакцией полученной в результате смеси со вторым растворителем и вторым основанием; реакцией таким образом образовавшейся смеси с производным борной кислоты; необязательно реакцией таким образом образовавшейся смеси с третьим растворителем и третьим основанием, с последующей реакцией с производным борной кислоты; и необязательно реакцией таким образом образовавшейся смеси с водой и кислотой, так что получают соединение формулы B3.
Неограничивающие примеры подходящего производного борной кислоты включают эфиры борной кислоты, такие как триизопропилборат.
В другом варианте осуществления способ получения B3 включает дополнительную стадию превращения остатка бороновой кислоты или боратного эфира B3 в трифторборатную соль.
В другом варианте осуществления B1 реагирует с производным карбоновой кислоты (R5C=O)-Z, где Z выбран из Hal и O(C=OR5).
В еще другом варианте осуществления данного способа, W представляет собой CRW; R1 представляет собой замещенный или незамещенный гетероциклил; R2 представляет собой водород; R3 представляет собой замещенный или незамещенный алкил; R4 представляет собой водород; и R5 представляет собой замещенный или незамещенный алкил или замещенный или незамещенный алкилокси. В еще другом варианте осуществления W представляет собой CH; R1 представляет собой N-морфолинил; R2 представляет собой водород; R3 представляет собой трифторметил; R4 представляет собой водород; и R5 представляет собой метил. В конкретном варианте осуществления -BY2 представляет собой бороновую кислоту.
В определенных вариантах осуществления Hal представляет собой хлор или бром. В конкретном варианте осуществления Hal представляет собой хлор.
В другом аспекте настоящее изобретение также относится к соединению формулы B3
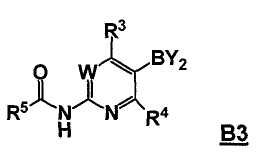
или его стереоизомеру, таутомеру или соли, где W, R3, R4 и R5 представляют собой, как определено выше, и BY2 представляет собой бороновую кислоту, ациклический бороновый эфир, циклический бороновый эфир или трифторборатную соль.
В одном варианте осуществления W представляет собой CRW; R1 представляет собой замещенный или незамещенный гетероциклил; R2 представляет собой водород; R3 представляет собой замещенный или незамещенный алкил; R4 представляет собой водород; и R5 представляет собой замещенный или незамещенный алкил или замещенный или незамещенный алкилокси. В другом варианте осуществления, W представляет собой CH; R1 представляет собой N-морфолинил; R2 представляет собой водород; R3 представляет собой трифторметил; R4 представляет собой водород; R5 представляет собой метил, и -BY2 представляет собой бороновую кислоту.
Настоящее изобретение также относится к следующему способу получения соединения формулы 5,
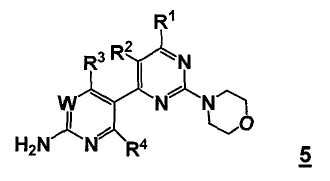
или его стереоизомера, таутомера или соли, включающему одну или более из следующих стадий:
стадия A: контакт соединения формулы B1

с реакционной смесью, содержащей растворитель и ангидрид кислоты (R5C=O)2O, так что получают соединение формулы B2
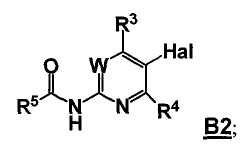
Стадия B: i) контакт соединения формулы B2 с реакционной смесью, содержащей первый растворитель, первое основание и необязательно спиртовую добавку, ii) контакт смеси стадии (i) со вторым растворителем и вторым основанием, iii) контакт смеси стадии (ii) с производным борной кислоты, iv) необязательно контакт смеси стадии (iii) с третьим растворителем и с третьим основанием, и затем контакт полученной в результате смеси с производным борной кислоты, и v) необязательно контакт смеси стадии (iii) или стадии (iv) с водой и кислотой, так что получают соединение формулы B3:
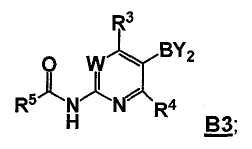
стадия C: контакт соединения формулы B3 с реакционной смесью, содержащей растворитель, основание, катализатор и соединение формулы 3

так что получают соединение B5:

стадия D: контакт соединения формулы B5 с реакционной смесью, содержащей растворитель и реагент для удаления R5C(=O)- группы, так что получают соединение формулы 5; необязательно с последующей реакцией образования соли; где W, R1, R2, R3 R4 представляют собой, как определено выше для соединения формулы 5; где Hal представляет собой галоген; и где -BY2 представляет собой бороновую кислоту, ациклический бороновый эфир, циклический бороновый эфир или трифторборатную соль.
В одном варианте осуществления стадии A, R5 представляет собой замещенный или незамещенный C1-6 алкил, замещенный или незамещенный C1-6 алкилокси, замещенный или незамещенный бензилокси или замещенный или незамещенный фенил. В другом варианте осуществления R5 представляет собой метил. В альтернативном варианте осуществления стадии A B1 реагирует с производным карбоновой кислоты (R5C=О)-Z, где Z выбран из Hal и O(O=CR5).
В другом варианте осуществления растворители стадий A и B независимо содержат один или более растворителей, выбранных из ароматических растворителей, алифатических растворителей, галогенированных растворителей, полярных апротонных растворителей и эфирных растворителей.
В еще другом варианте осуществления растворитель стадии A содержит один или более растворителей, выбранных из ароматических растворителей, алифатических растворителей, галогенированных растворителей, полярных апротонных растворителей, сложноэфирных растворителей и эфирных растворителей. В другом варианте осуществления растворитель стадии A содержит один или более растворителей, выбранных из этилацетата и гептана.
В еще другом варианте осуществления стадии A реакционная смесь содержит диметиламинопиридин (DMAP).
В одном варианте осуществления растворитель стадии B содержит один или более растворителей, выбранных из ароматических растворителей, алифатических растворителей, полярных апротонных растворителей и эфирных растворителей. В другом варианте осуществления, первый и второй растворители стадии B независимо содержат один или более растворителей, выбранных из THF и гексана. В еще другом варианте осуществления первый, второй и третий растворители стадии B, если они присутствуют, независимо содержат один или более растворителей, выбранных из THF и гексана.
В конкретном варианте осуществления первый растворитель стадии B представляет собой THF. В еще другом конкретном варианте осуществления, второй растворитель стадии B представляет собой гексан. В еще другом конкретном варианте осуществления третий растворитель стадии B представляет собой гексан.
В другом варианте осуществления первое основание стадии B выбрано из оснований, сопряженных с кислотой, углеводородов, аммиака, аминов, спиртов и диводорода. Неограничивающие примеры данных оснований включают н-бутиллитий, н-гексиллитий, гидрид натрия, трет-бутилмагнийхлорид, амид лития, изопропоксид лития и диизопропиламид лития. Другие данные основания являются известными специалисту в данной области техники. В конкретном варианте осуществления первое основание стадии B содержит одно или более оснований, выбранных из амида лития, диалкиламидов лития, алкоксидов лития и изомеров бутиллития. В другом конкретном варианте осуществления первое основание стадии B содержит амид лития. В еще другом конкретном варианте осуществления первое основание стадии B включает амид лития и изопропоксид лития.
В еще другом варианте осуществления второе и третье основания стадии B выбирают из оснований, сопряженных с кислотой, углеводородов. Неограничивающие примеры данных оснований включают н-бутиллитий, н-гексиллитий и трет-бутилмагнийхлорид. Другие данные основания являются известными специалисту в данной области техники. В определенных вариантах осуществления второе и третье основание стадии B, если они присутствуют, выбирают из изомеров бутиллития и реагентов Гриньяра. Неограничивающие примеры реагентов Гриньяра включают трет-бутилмагнийхлорид. В конкретном варианте осуществления второе и третье основание стадии B, если они присутствуют, представляют собой н-бутиллитий.
Ясно, что второе и третье основания стадии B представляют собой органометаллические реагенты, которые способствуют обмену атома галогена B2 на атом металла органометаллического реагента.
В конкретном варианте осуществления первое основание стадии B содержит амид лития и изопропоксид лития, второе основание стадии B представляет собой н-бутиллитий, и третье основание стадии B представляет собой н-бутиллитий. В предпочтительном варианте осуществления, добавление н-бутиллития осуществляют при температуре, которая является меньшей чем -75°C.
В еще другом варианте осуществления производное борной кислоты стадии B представляет собой триизопропилборат.
В еще другом варианте осуществления спиртовая добавка стадии B выбрана из метанола, этанола, 1-пропанола, 2-пропанола, н-бутанола, 2-бутанола и трет-бутанола. В конкретном варианте осуществления спиртовая добавка представляет собой 2-пропанол.
В предпочтительном варианте осуществления первый растворитель стадии B содержит тетрагидрофуран, первое основание стадии B содержит амид лития, второй и третий растворители стадии B представляют собой гексан, и второе и третье основание стадии B представляют собой н-бутиллитий. Предпочтительно, последовательное добавление н-бутиллития, триизопропилбората, н-бутиллития и триизопропилбората приводит в результате к сниженным количествам побочных продуктов.
В другом варианте осуществления стадии B способ получения B3 включает дополнительную стадию превращения бороновой кислоты или боратэфирной группы B3 в трифторборатную соль.
В одном варианте осуществления растворитель стадии C содержит один или более растворителей, выбранных из ароматических растворителей, алифатических растворителей, галогенированных растворителей, полярных апротонных растворителей, сложноэфирных растворителей, эфирных растворителей и воды. В другом варианте осуществления растворитель стадии C содержит один или более растворителей, выбранных из диметоксиэтана, тетрагидрофурана, 1,4-диоксана, 2-метилтетрагидрофурана и воды. В конкретном варианте осуществления растворитель стадии C включает диметоксиэтан и воду.
В другом варианте осуществления основание стадии C выбрано из ацетатов, фосфатов и карбонатов. В конкретном варианте осуществления основание стадии C представляет собой карбонат калия.
В еще другом варианте осуществления катализатор стадии C содержит палладий. В определенных вариантах осуществления катализатор стадии C выбран из тетракис(трифенилфосфин)палладия (0) и дихлорида бис(трифенилфосфин)палладия (II). В других вариантах осуществления палладиевый катализатор стадии C образуется смешением Pd(OAc)2 с фосфиновым лигандом. Подходящие фосфиновые лиганды являются известными специалисту в данной области техники; неорганичивающие примеры включают трифенилфосфин и трис(4-метокси-3,5-диметилфенил)фосфин. В конкретном варианте осуществления катализатор стадии C представляет собой тетракис(трифенилфосфин)палладий (0).
В еще другом варианте осуществления стадии C соединение формулы B3 добавляют непрерывно к реакционной смеси в процессе реакции.
В одном варианте осуществления растворитель стадии D содержит один или более растворителей, выбранных из ароматических растворителей, алифатических растворителей, галогенированных растворителей, полярных апротонных растворителей, сложноэфирных растворителей, эфирных растворителей и воды. В конкретном варианте осуществления растворитель стадии D представляет собой воду. В другом конкретном варианте осуществления растворитель стадии D представляет собой диоксан.
На стадии D удаление R5C(=O)- группы также влечет за собой замену данной группы атомом водорода. Удаление R5C(=O)- группы можно проводить способами, известными специалисту в данной области техники. Неограничивающие примеры данных способов включают реакции, опосредованные кислотой, основанием и металлом. Конкретным примером данных способов является гидролиз, опосредованный кислотой. В одном варианте осуществления стадии D, реагент для удаления R5C(=O)- группы выбран из кислот, оснований и металлических катализаторов. В конкретном варианте осуществления стадии D реагент для удаления R5C(=O)- группы представляет собой хлористоводородную кислоту. В другом конкретном варианте осуществления стадии D реагент для удаления R5C(=O)- группы представляет собой серную кислоту.
В определенных вариантах осуществления стадии A-D независимо включают дополнительные стадии или способы (например, для удаления побочных продуктов реакций или для обработки, выделения или очистки продуктов реакции), как подробно описано в примерах в настоящем изобретении.
В определенных вариантах осуществления за стадиями A-D может следовать стадия d) способа.
Специалистам известны несколько параметров приведенных выше способов, которые можно предпочтительно изменять для того, чтобы получить требуемый результат. Данные параметры включают, например, методики и способы очистки реакционных компонентов и растворителей; порядок добавления указанных реакционных компонентов и растворителей к реакционной смеси; продолжительность реакции указанных реакционных компонентов и растворителей; и температуру и скорость перемешивания, смешения или встряхивания реакционных компонентов и растворителей в процессе указанной реакции.
Обнаружено, что способ, осуществленный стадиями A-D (также включая конкретные стадии способа) удовлетворяет одному или более из следующих критериев: безопасность; простота; большие выходы и большая экономичность по сравнению с известными способами получения соединений формулы 5. Кроме того, способ, как описывают в настоящем изобретении, считают масштабируемым, что делает его пригодным для промышленного получения.
Твердые формы 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина
В еще других аспектах настоящее изобретение относится к конкретным твердым, предпочтительно кристаллическим, формам пиримидинового производного, 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина ("соединение A" или "соединение формулы A")

его гидратов, его солей и гидратов и сольватов его солей, и к способу образования данных конкретных, предпочтительно кристаллических, форм.
Обнаружено, что твердые формы соединения формулы A и его солей неожиданно обладают выгодными фармакокинетическими свойствами, которые делают их особенно пригодными для получения фармацевтических композиций, содержащих соединение формулы A и его соли. Различные кристаллические формы обладают различными физическими свойствами, такими как температура плавления, гигроскопичность, растворимость, свойства текучести или термодинамическая стабильность, и, следовательно, различные кристаллические формы позволяют выбрать самую пригодную форму для определенного применения или аспекта, например, применения в качестве промежуточного соединения в способе получения лекарственного средства или в различных формах введения, таких как таблетки, капсулы, мази или растворы.
Соединение A было впервые описано в WO2007/084786, содержание которого включено в настоящее изобретение с помощью ссылки. Соединение A представляет собой ингибитор PI3K (фосфатидилинохитол 3-киназы) и регулирует фосфорилирование AKT в биохимических, а также клеточных анализах.
Соответственно, соединение A и его фармацевтически приемлемые соли и фармацевтические композиции, содержащие соединение A или его фармацевтически приемлемую соль, можно применять для предотвращения, уменьшения интенсивности или лечения заболеваний, зависящих от PI3K. Как описывают в настоящем изобретении, свободное основание соединения A может представлять собой твердую форму, которая существует в виде одной или более полиморфных форм, включая безводные и гидратные формы. Моногидрохлоридная соль соединения A может представлять собой твердую форму, которая существует в виде одной или более полиморфных форм, включая безводные формы, гидраты и сольваты. Данные полиморфные формы (альтернативно известные в данной области техники как полиморфные формы или кристаллические формы) отличаются их порошковыми рентгеновскими дифрактограммами, спектроскопическими, физико-химическими и фармакокинетическими свойствами, а также их термодинамической стабильностью.
В настоящее время было неожиданно обнаружено, что в определенных условиях можно получить новые конкретные твердые формы соединения A, его гидратов, его солей и гидратов или сольватов его солей, которые описывают в настоящем изобретении далее, и которые обладают полезными применениями и свойствами. Желательно иметь доступ к различным полиморфным формам твердого соединения A, его гидратов, его солей и гидратов или сольватов его солей по многим соображениям. Например, различные полиморфные формы могут включать различные примеси при кристаллизации, т.е., примесь, включенная в гемигидратную форму соединения A, необязательно включена в безводную полиморфную форму A соединения. Примесь, включенная в полиморфную форму A или B моногидрохлоридной соли соединения A также необязательно включена в полиморфные формы SA, SB, Sc, SD или SE. Таким образом, многократное получение различных полиморфных форм соединения A можно применять для увеличения чистоты конечной полученной формы. Кроме того, различные полиморфные формы могут обладать различными физическими свойствами, такими как температура плавления, гигроскопичность, растворимость, свойства текучести или термодинамическая стабильность и, следовательно, различные полиморфные формы позволяют выбрать самую подходящую форму для указанного применения или аспекта, например, применения в качестве промежуточного соединения в способе получения лекарственного средства, в различных формах для введения, таких как таблетки, капсулы, мази, суспензии или растворы, или при получении лекарственной формы, обладающей оптимальными фармакокинетическими свойствами.
Таким образом, в одном аспекте, настоящее изобретение относится к твердой, предпочтительно кристаллической, форме соединения формулы A, или гидрату соединения формулы A, или соли соединения формулы A, или гидрату или сольвату соли соединения формулы A.
В одном варианте осуществления соединение формулы A представляет собой полиморфную форму HA соединения A.
Полиморфная форма HA представляет собой гемигидрат и может быть определена в соответствии с одним или более характеристическими сигналами, которые получены в результате аналитических измерений, включая, но необязательно ограничиваясь, порошковую рентгеновскую дифрактограмму фигуры 4. Полиморфную форму HA можно также определить в соответствии с одним или более следующими характеристическими сигналами:
В одном варианте осуществления полиморфная форма HA имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 19,2° +/- 0,3° и 18,7° +/- 0,3°. В другом варианте осуществления полиморфная форма HA имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 15,5° +/- 0,3° и 16,7° +/- 0,3°. В еще другом варианте осуществления форма HA имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 7,7° +/- 0,3, 22,0° +/- 0,3°, и 22,0° +/- 0,3°. В еще другом варианте осуществления полиморфная форма HA имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 19,2° +/- 0,3° и 18,7° +/- 0,3°, 15,5° +/- 0,3° и 16,7° +/- 0,3°, и 7,7° +/- 0,3, 22,0° +/- 0,3°, и 22,0° +/- 0,3°. В следующем варианте осуществления полиморфная форма HA имеет порошковую рентгеновскую дифрактограмму по существу в соответствии с фигурой 4 и таблицей 1.
Было обнаружено, что полиморфная форма A моногидрохлоридной соли соединения формулы A обладает меньшим влагопоглощением, чем или свободное основание или моногидрат моногидрохлорида. В экспериментах, полиморфная форма A моногидрохлоридной соли соединения формулы A обладало максимальным влагопоглощением, меньшим чем 0,1% при 25°C и вплоть до 92% относительной влажности. Гемигидратная полиморфная форма HA соединения формулы A обладала влагопоглощением 1,9% при 25% относительной влажности, безводная полиморфная форма A соединения формулы A обладала влагопоглощением 8,9% при 85% относительной влажности, и моногидрат моногидрохлорида соединения формулы A обладал влагопоглощением 4,4% при 75% относительной влажности и 4,9% при 95% относительной влажности. Полиморфная форма A моногидрохлоридной соли соединения формулы A была неожиданно только слегка гигроскопичной, таким образом, обеспечивая стабильными составами при минимальном риске собственного химического разрушения.
В одном варианте осуществления соединение формулы A представляет собой безводную полиморфную форму A соединения A. Безводную полиморфную форму A можно определить в соответствии с одним или более характеристическими сигналами, которые получены в результате аналитических измерений, включая, но необязательно ограничиваясь, порошковую рентгеновскую дифрактограмму фигуры 5. Безводную полиморфную форму можно также определить в соответствии с одним или более следующими характеристическими сигналами:
В одном варианте осуществления безводная полиморфная форма A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 14,8° +/- 0,3° и 10,2° +/- 0,3°. В другом варианте осуществления безводная полиморфная форма A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 17,4° +/- 0,3 и 21,8° +/- 0,3°. В еще другом варианте осуществления безводная полиморфная форма A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 14,8° +/- 0,3° и 10,2° +/- 0,3° и 17,4° +/- 0,3 и 21,8° +/- 0,3°. В еще другом варианте осуществления безводная полиморфная форма A имеет порошковую рентгеновскую дифрактограмму по существу в соответствии с фигурой 5 и таблицей 2.
В одном варианте осуществления, моногидрат моногидрохлорида соединения формулы A имеет полиморфную форму Ha. Полиморфную форму Ha можно определить в соответствии с одним или более характеристическими сигналами, которые получены в результате аналитических измерений, включая, но необязательно ограничиваясь, порошковую рентгеновскую дифрактограмму фигуры 6. Полиморфную форму Ha можно также определить в соответствии с одним или более следующими характеристическими сигналами:
В одном варианте осуществления полиморфная форма Ha имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 9,3° +/- 0,3° и 15,8° +/- 0,3°. В другом варианте осуществления полиморфная форма Ha имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 7,2° +/- 0,3 и 18,6° +/- 0,3°. В еще другом варианте осуществления полиморфная форма Ha имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 9,3° +/- 0,3° и 15,8° +/-0,3° и 7,2° +/- 0,3 и 18,6° +/- 0,3°. В еще другом варианте осуществления полиморфная форма Ha имеет порошковую рентгеновскую дифрактограмма по существу в соответствии с фигурой 6 и таблицей 3.
В одном варианте осуществления моногидрохлорид соединения формулы A имеет полиморфную форму A. Полиморфная форма A моногидрохлорида соединения формулы A является безводной, и ее можно определить в соответствии с одним или более характеристическими сигналами, которые получены в результате аналитических измерений, включая, но необязательно ограничиваясь, порошковую рентгеновскую дифрактограмму фигуры 7. Полиморфную форму A моногидрохлорида соединения формулы A можно также определить в соответствии с одним или более следующими характеристическими сигналами:
В одном варианте осуществления полиморфная форма A моногидрохлорида соединения формулы A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 9,9° +/- 0,3° и 20,0° +/- 0,3°. В другом варианте осуществления полиморфная форма A моногидрохлорида соединения формулы A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 18,0° +/- 0,3 и 20,7° +/- 0,3°. В еще другом варианте осуществления полиморфная форма A моногидрохлорида соединения формулы A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 8,8° +/- 0,3 и 25,0° +/- 0,3°. В еще другом варианте осуществления полиморфная форма A моногидрохлорида соединения формулы A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 9,9° +/- 0,3° и 20,0° +/- 0,3°, 18,0° +/- 0,3 и 20,7° +/- 0,3°, и 8,8° +/-0,3 и 25,0° +/- 0,3°. В следующем варианте осуществления полиморфная форма A моногидрохлорида соединения формулы A имеет порошковую рентгеновскую дифрактограмма по существу в соответствии с фигурой 7 и таблицей 4.
В одном варианте осуществления моногидрохлорид соединения формулы A имеет полиморфную форму B. Полиморфная форма B моногидрохлорида соединения формулы A является безводной, и ее можно определить в соответствии с одним или более характеристическими сигналами, которые получены в результате аналитических измерений, включая, но необязательно ограничиваясь, порошковую рентгеновскую дифрактограмму фигуры 8. Полиморфную форму B моногидрохлорида соединения формулы A можно также определить в соответствии с одним или более следующими характеристическими сигналами:
В одном варианте осуществления полиморфная форма B моногидрохлорида соединения формулы A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 18,7° +/- 0,3° и 21,8 +/- 0,3°. В другом варианте осуществления полиморфная форма B моногидрохлорида соединения формулы A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 18,3° +/- 0,3 и 20,1 +/- 0,3°. В еще другом варианте осуществления полиморфная форма B моногидрохлорида соединения формулы A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 16,7° +/- 0,3 и 26,8° +/- 0,3°. В еще другом варианте осуществления полиморфная форма B моногидрохлорида соединения формулы A имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 18,7° +/- 0,3° и 21,8° +/- 0,3°, 18,3° +/- 0,3 и 20,1° +/- 0,3° и 16,7° +/- 0,3 и 26,8° +/- 0,3°. В следующем варианте осуществления, полиморфная форма B моногидрохлорида соединения формулы A имеет порошковую рентгеновскую дифрактограмма по существу в соответствии с фигурой 8 и таблицей 5.
В одном варианте осуществления моногидрохлорид соединения формулы A имеет полиморфную форму SA. Полиморфная форма SA представляет собой сольват, и ее можно определить в соответствии с одним или более характеристическими сигналами, которые получены в результате аналитических измерений, включая, но необязательно ограничиваясь, порошковую рентгеновскую дифрактограмму фигуры 9. Полиморфную форму SA можно также определить в соответствии с одним или более следующими характеристическими сигналами:
В одном варианте осуществления полиморфная форма SA имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 16,6° +/- 0,3° и 28,4° +/- 0,3°. В другом варианте осуществления полиморфная форма SA имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 15,2° +/- 0,3 и 22,4° +/- 0,3°. В еще другом варианте осуществления полиморфная форма SA имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 14,8° +/- 0,3 и 23,8° +/- 0,3°. В еще другом варианте осуществления полиморфная форма SA имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 16,6° +/- 0,3° и 28,4° +/- 0,3°, 15,2° +/- 0,3 и 22,4° +/- 0,3°, и 14,8° +/- 0,3 и 23,8° +/- 0,3°. В следующем варианте осуществления полиморфная форма SA имеет порошковую рентгеновскую дифрактограмма по существу в соответствии с фигурой 9 и таблицей 6.
В одном варианте осуществления моногидрохлорид соединения формулы A имеет полиморфную форму SB. Полиморфная форма SB представляет собой сольват, и ее можно определить в соответствии с одним или более характеристическими сигналами, которые получены в результате аналитических измерений, включая, но необязательно ограничиваясь, порошковую рентгеновскую дифрактограмму фигуры 10. Полиморфную форму SB можно также определить в соответствии с одним или более следующими характеристическими сигналами:
В одном варианте осуществления полиморфная форма SB имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 19,8° +/- 0,3° и 17,5° +/- 0,3°. В другом варианте осуществления полиморфная форма SB имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 14,8° +/- 0,3 и 23,5° +/- 0,3°. В еще другом варианте осуществления полиморфная форма SB имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 19,8° +/- 0,3° и 17,5° +/- 0,3° и 14,8° +/- 0,3 и 23,5° +/- 0,3°. В еще другом варианте осуществления полиморфная форма SB имеет порошковую рентгеновскую дифрактограмма по существу в соответствии с фигурой 10 и таблицей 7.
В одном варианте осуществления моногидрохлорид соединения формулы A имеет полиморфную форму Sc. Полиморфная форма Sc представляет собой сольват, и ее можно определить в соответствии с одним или более характеристическими сигналами, которые получены в результате аналитических измерений, включая, но необязательно ограничиваясь, порошковую рентгеновскую дифрактограмму фигуры 11. Полиморфную форму SC можно также определить в соответствии с одним или более следующими характеристическими сигналами:
В одном варианте осуществления полиморфная форма Sc имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 9,9° +/- 0,3° и 20,0° +/- 0,3°. В другом варианте осуществления, полиморфная форма Sc имеет порошковую рентгеновскую дифрактограмма по существу в соответствии с фигурой 11 и таблицей 8.
В одном варианте осуществления, моногидрохлорид соединения формулы A имеет полиморфную форму SD. Полиморфная форма SD представляет собой сольват, и ее можно определить в соответствии с одним или более характеристическими сигналами, которые получены в результате аналитических измерений, включая, но необязательно ограничиваясь, порошковую рентгеновскую дифрактограмму фигуры 12. Полиморфную форму SD можно также определить в соответствии с одним или более следующими характеристическими сигналами:
В одном варианте осуществления полиморфная форма SD имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 9,9° +/- 0,3° и 23,5° +/- 0,3°. В другом варианте осуществления полиморфная форма SD имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 8,1° +/- 0,3 и 17,6° +/- 0,3°. В еще другом варианте осуществления полиморфная форма SD имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 9,9° +/- 0,3° и 23,5° +/-0,3°, и 8,1° +/- 0,3 и 17,6° +/- 0,3°. В другом варианте осуществления полиморфная форма SD имеет порошковую рентгеновскую дифрактограмма по существу в соответствии с фигурой 12 и таблицей 9.
В одном варианте осуществления моногидрохлорид соединения формулы A имеет полиморфную форму SE. Полиморфная форма SE представляет собой сольват, и ее можно определить в соответствии с одним или более характеристическими сигналами, которые получены в результате аналитических измерений, включая, но необязательно ограничиваясь, порошковую рентгеновскую дифрактограмму фигуры 13. Полиморфную форму SE можно также определить в соответствии с одним или более следующими характеристическими сигналами:
В одном варианте осуществления полиморфная форма SE имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 4,3° +/- 0,3° и 17,6° +/- 0,3°. В другом варианте осуществления полиморфная форма SE имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 7,3° +/- 0,3 и 9,9° +/- 0,3°. В еще другом варианте осуществления полиморфная форма SE имеет порошковую рентгеновскую дифрактограмму, имеющую характеристические пики, выраженные в градусах 2-тета при углах 4,3° +/- 0,3° и 17,6° +/- 0,3°, и 8,3° +/- 0,3 и 9,9° +/- 0,3°. В другом варианте осуществления полиморфная форма SD имеет порошковую рентгеновскую дифрактограмма по существу в соответствии с фигурой 13 и таблицей 10.
В одном варианте осуществления полиморфная форма A содержит менее чем 1% по весу в сумме примесей. В другом варианте осуществления полиморфная форма A содержит меньше чем 0,5% по весу в сумме примесей. В еще другом варианте осуществления полиморфная форма A содержит меньше чем 0,1% по весу в сумме примесей.
Подразумевается, что термин "по существу чистый" в контексте настоящего изобретения обозначает в частности, что, по меньшей мере, 90, предпочтительно, по меньшей мере, 95, и самое предпочтительное, по меньшей мере, 99 процентов по весу кристаллов соединения формулы A, его гидрата, его солей или гидратов или сольватов его солей присутствует в конкретной кристаллической форме согласно настоящему изобретению.
В контексте утверждения, что кристаллическая форма соединения формулы A, его гидратов, его солей или гидратов или сольватов его солей имеет рентгеновскую дифрактограмму, по существу как показано на одной из фигур, термин "по существу" обозначает, что должны присутствовать, по меньшей мере, основные линии диаграммы, показанной на указанной фигуре, т.е., линии, имеющие относительную интенсивность линий, большую чем 20%, особенно большую чем 30%, по сравнению с большинством интенсивных линий на диаграмме.
В одном предпочтительном варианте осуществления, кристаллическая форма соединения формулы A, его гидратов, его солей или его гидратов или сольваты его солей обладает рентгеновской диаграммой, по существу как показано на одной из фигур.
Особенно предпочтительной является твердая, предпочтительно кристаллическая, форма соединения формулы A, или гидратов, его солей или гидратов или сольватов его солей, которые можно получить, как описано в примерах. Термин "твердая форма" согласно настоящему изобретению включает кристаллические формы. Предпочтительные твердые формы являются кристаллическими формами.
Настоящее изобретение также относится к способу получения конкретных твердых, предпочтительно кристаллических, форм соединения формулы A, его гидратов, его солей и гидратов или сольватов его солей. Точные условия, при которых образуются конкретные твердые формы, можно в настоящее время определить эмпирически, и ряд способов является пригодным на практике, включая способ получения соединения формулы 5, как описывают в настоящем изобретении и при дополнительных условиях, описанных в примерах 4-12 в настоящем изобретении.
В другом аспекте настоящее изобретение также относится к способу лечения состояний, расстройств или заболеваний, опосредованных активацией PI3K, таких как указанные выше, у нуждающегося в лечении субъекта, где способ включает введение указанному субъекту эффективного количества твердой формы, предпочтительно кристаллической, формы соединения формулы A или его моногидрохлоридной соли. Твердая форма соединения формулы A или его моногидрохлоридной соли, которую можно применять для данного лечения, включает формы, описанные в настоящем изобретении, включая, но не ограничиваясь, полиморфную форму HA и безводную форму A соединения формулы A и полиморфную форму Ha, форму A, форму B, форму SA, форму SA, форму SB, форму Sc, форму SD и форму SE моногидрохлоридной соли соединения формулы A. Предпочтительной является полиморфная форма HA и безводная форма A соединения формулы A и полиморфная форма Ha, форма A и форма B SE моногидрохлоридной соли соединения формулы A. Самой предпочтительной является форма A моногидрохлоридной соли соединения формулы A.
Твердые, предпочтительно кристаллические, формы соединения формулы A, его гидратов, его солей и гидратов или сольватов его солей можно предпочтительно применять для лечения заболеваний клеточной пролиферации, таких как опухоль и/или рост раковых клеток, опосредованный P13. В частности, соединения формулы A, его гидраты, его соли и гидраты или сольваты его солей являются пригодными для лечения человеческих или животных (например, у мышей) раков, включая, например, рак легкого и бронх; простаты, груди, поджелудочной железы, толстой и прямой кишки; щитовидной железы, печени и внутрипеченочных желчных протоков; гепатоцеллюлярный рак; желудка; глиому/глиобластому; эндометрию; меланому, почки и почечной лоханки, мочевого пузыря, тела матки, шейки матки, яичников, множественную миелому, пищевода, острый миелобластный лейкоз, хронический миелолейкоз; лимфолейкоз; миелоидный лейкоз; мозга; полости рта и глотки, гортани; тонкой кишки; неходжкинскую лимфому, меланому и ворсинчатую аденому толстой кишки.
В одном варианте осуществления настоящее изобретение относится к применению полиморфной формы A моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина в лечении рака.
Настоящее изобретение особенно относится к полиморфной форме A моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина для лечения одного из указанных заболеваний, приведенных в настоящем изобретении или для получения фармацевтической композиции для лечения нуждающегося в данном лечении субъекта.
В одном варианте осуществления настоящее изобретение относится к применению кристаллического соединения формулы A или его моногидрохлоридной соли, в особенности формы A моногидрохлоридной соли, для получения лекарственного средства для лечения расстройств, опосредованных PI3K. В одном варианте осуществления применения заболевание представляет собой клеточное пролиферативное заболевание, такое как заболевания, приведенные выше.
В другом варианте осуществления настоящее изобретение относится к применению кристаллического соединения формулы A или его моногидрохлоридной соли, в особенности формы A моногидрохлоридной соли, и фармацевтически приемлемого носителя или разбавителя, для применения в лечении рака.
Настоящее изобретение также относится к способу лечения теплокровных животных, включая людей, страдающих от указанных заболеваний, где количество твердой, предпочтительно кристаллической, формы соединения формулы A, его гидратов, его солей или гидратов или сольватов его солей, которое является эффективным против указанного заболевания, особенно количество с антипролиферативной активностью, вводят нуждающимся в данном лечении теплокровным животным.
"Лечение" в контексте настоящего изобретения обозначает облегчение симптомов, связанных с расстройством или заболеванием, или остановку дальнейшего развития или осложнения данных симптомов, или предотвращение или профилактику заболевания или расстройства. Например, в контексте лечения пациентов, нуждающихся в ингибиторе PI3K, успешное лечение может включать ослабление пролиферации капилляров, питающих опухоль или пораженную ткань, облегчение симптомов, связанных с раковой опухолью или опухолью, пролиферацией капилляров или пораженной тканью, остановку капиллярной пролиферации, или остановку развития заболевания, такого как рак, или роста раковых клеток. Лечение может также включать введение фармацевтических составов настоящего изобретения в комбинации с другими терапиями. Например, соединения и фармацевтические составы настоящего изобретения можно вводить перед, в процессе или после хирургической процедуры и/или радиационной терапии. Соединения настоящего изобретения можно также вводить в сочетании с другими противораковыми агентами, включая агенты, применяемые в антисенс и генной терапии.
Как применяют в настоящем изобретении, "лечить" и "лечение" представляют собой взаимозаменяемые термины, как являются "ограничивающий" и "лечащий" и, как применяют в настоящем изобретении, они включают превентативное (например, профилактическое) и паллиативное лечение или действие, обеспечивающее превентативное или паллиативное лечение.
Термины "PI3K-опосредованное расстройство" и "заболевание, опосредованное PI3K," относится к расстройству, которое можно успешно лечить ингибированием PI3K.
Термин "клеточные пролиферативные заболевания" относится к заболеваниям, включающим, например, рак, опухоль, гиперплазию, рестеноз, гипертрофию сердца, иммунное расстройство и воспаление.
Термин "рак" относится к раковым заболеваниям, которые можно успешно лечить ингибированием PI3K, включая, например, рак легкого и бронх; простаты, груди, поджелудочной железы, толстой и прямой кишки; щитовидной железы, печени и внутрипеченочных желчных протоков; гепатоцеллюлярный рак; желудка; глиому/глиобластому; эндометрию; меланому, почки и почечной лоханки, мочевого пузыря, тела матки, шейки матки, яичников, множественную миелому, пищевода, острый миелобластный лейкоз, хронический миелолейкоз; лимфолейкоз; миелоидный лейкоз; мозга; полости рта и глотки, гортани; тонкой кишки; неходжкинскую лимфому, меланому и ворсинчатую аденому толстой кишки.
Твердые, предпочтительно кристаллические, формы соединения формулы A, его гидратов, его солей или гидратов или сольватов его солей, описанных в настоящем изобретении, можно применять для получения стабильных фармацевтических лекарственных форм. Следовательно, настоящее изобретение также относится к фармацевтическим композициям, которые содержат количество, особенно терапевтически эффективное количество, для лечения одного из заболеваний, приведенных выше, твердой, предпочтительно кристаллической, формы соединения формулы A, его гидрата, его солей, или гидрата или сольвата его солей, вместе с другими фармацевтически приемлемыми вспомогательными веществами, носителями, наполнителями, разбавителями и подобными.
Как применяют в настоящем изобретении, выражение "фармацевтическая композиция" включает препараты, пригодные для введения млекопитающим, например, людям. Когда соединения настоящего изобретения вводят в виде фармацевтических препаратов млекопитающим, например, людям, они могут быть предоставлены сами по себе или в виде фармацевтической композиции, содержащей, например, 0,1%-99,9% (более предпочтительно 0,5-90%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
Соединения можно применять отдельно или в виде композиций вместе с фармацевтически приемлемым носителем или вспомогательным веществом. Фармацевтические композиции настоящего изобретения содержат терапевтически эффективное количество соединения, ингибирующего фосфатидилинозитол 3-киназы, описанного в настоящем изобретении, смешанного с одним или более фармацевтически приемлемых носителей. Как применяют в настоящем изобретении, термин "фармацевтически приемлемый носитель" обозначает нетоксичный, инертный твердый, полутвердый или жидкий наполнитель, разбавитель, материал для инкапсулирования или вспомогательный агент для составов любого типа. Некоторыми примерами веществ, которые могут служить в качестве фармацевтически приемлемых носителей, являются сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин, тальк, вспомогательные вещества, таких как какао-масло и воск для суппозиторий, масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль, сложные эфиры, такие как этилолеат и этиллаурат; агар, буферные агенты, такие как гидроксид магния и гидроксид алюминия, альгиновая кислота; апирогенная вода; изотонический солевой раствор, раствор Рингера, этиловый спирт, и фосфатно-буферные растворы, а также другие нетоксичные совместимые смазочные материалы, такие как лаурилсульфат натрия и стеарат магния, а также красители, агенты, контролирующие высвобождение, агенты для покрытия, подсластители, ароматизаторы и душистые вещества, консерванты и антиоксиданты также могут присутствовать в композиции, в соответствии с решением составителя рецептуры. Другие подходящие фармацевтически приемлемые вспомогательные вещества описаны в "Remington's Pharmaceutical Sciences," Mack Pub. Co., New Jersey, 1991, введенной в настоящее изобретение с помощью ссылки.
Увлажняющие агенты, эмульгаторы и смазочные материалы, такие как лаурилсульфат натрия и стеарат магния, а также красители, агенты, контролирующие высвобождение, агенты для нанесения покрытия, подсластители, ароматизаторы и душистые вещества, консерванты и антиоксиданты также могут присутствовать в композициях.
Примеры фармацевтически приемлемых антиоксидантов включают: растворимые в воде антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфат натрия, сульфит натрия и подобные; жирорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (BHA), бутилированный гидрокситолуол (BHT), лецитин, пропилгаллат, α-токоферол и подобные; и агенты, образующие комплексы с металлами, такие как лимонная кислота, этилендиаминтетрауксусная кислота (EDTA), сорбитол, винная кислота, фосфорная кислота и подобные.
Соединения настоящего изобретения можно вводить людям и другим животным перорально, парентерально, сублингвально, распылением в виде аэрозоля или ингаляцией, ректально, интрацистернально, интравагинально, внутрибрюшинно, буккально или местно в виде дозированных единиц составов, содержащих общепринятые нетоксичные фармацевтически приемлемые носители, адъюванты и среды, при необходимости. Местное введение может также включать применение трансдермального введения, такого как трансдермальные пластыри или устройства для ионофореза. Термин парентеральный, как применяют в настоящем изобретении, включает подкожные инъекции, внутривенную, внутримышечную, интрастемальную инъекцию или вливание.
Способы формулирования являются хорошо известными в данной области техники и описаны, например, в Remington: The Science and Practice of Pharmacy, Mack Publishing Company, Easton, Pa., 19th Edition (1995). Фармацевтические композиции для применения в настоящее изобретение могут быть в виде стерильных, апирогенных жидких растворов или суспензий, капсул с покрытием, суппозиторий, лиофилизированных порошков, трансдермальных пластырей или других форм, известных в данной области техники.
Инъецируемые препараты, например, стерильные инъецируемые водные или масляные суспензии можно формулировать согласно известному уровню техники, применяя подходящие диспергирующие или увлажняющие агенты и суспендирующие агенты. Стерильный инъецируемый препарат может также представлять собой стерильный инъецируемый раствор, суспензию или эмульсию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-пропандиоле или 1,3-бутандиоле. К приемлемым средам и растворителям, которые можно применять, относятся вода, раствор Рингера, U.S.P. и изотонический раствор хлорида натрия. Кроме того, стерильные, нелетучие масла общепринято применяют в качестве растворителя или суспендирующей среды. С этой целью можно применять любое безвкусное нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, жирные кислоты, такие как олеиновая кислота, находят применение в инъецируемых препаратах. Инъецируемые составы можно стерилизовать, например, фильтрацией через фильтр, задерживающий бактерии, или введением стерилизующих агентов в виде стерильных твердых композиций, которые можно растворять или диспергировать в стерильной воде или другой стерильной инъецируемой среде перед применением.
Для того чтобы продлить эффект лекарственного средства, часто желательно замедлить поглощение лекарственного средства в результате подкожной или внутримышечной инъекции. Это можно осуществить применением жидкой суспензии кристаллического или аморфного вещества с низкой растворимостью в воде. Затем, скорость поглощения лекарственного средства зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, замедленного поглощения введенной парентерально лекарственной формы можно достигать растворением или суспендированием лекарственного средства в маслянистой среде.
Инъецируемые депо-формы можно получить получением матрикса для микроинкапсулирования лекарственного средства в биоразлагаемых полимерах, таких как полиактид-полигликолид. В зависимости от отношения лекарственного средства к полимеру и свойств конкретного применяемого полимера, можно контролировать скорость высвобождения лекарственного средства. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъецируемые депо-составы можно также получить включением лекарственного средства в липосомы или микроэмульсии, которые являются совместимыми с тканями тела.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые можно получить смешением соединений настоящего изобретения с подходящими нераздражающими вспомогательными веществами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиторий, который является твердым при температуре окружающей среды, но является жидким при температуре тела и, следовательно, плавится в прямой кишке или влагалищной полости и высвобождает активное соединение.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В данных твердых лекарственных формах, активное соединение смешивают, по меньшей мере, с одним инертным, фармацевтически приемлемым вспомогательным веществом или носителем, таким как цитрат натрия или фосфат дикальция и/или a) наполнителями или разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннитол и кремниевая кислота, b) связующими, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидинон, сахароза и камедь, c) увлажнителями, такими как глицерин, d) разрыхлителями, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, е) агентами, замедляющими растворение, такими как парафин, f) ускорителями абсорбции, такими как четвертичные аммониевые соединения, г) увлажняющими агентами, такими как, например, ацетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолин и бентонитовая глина, и i) смазывающими агентами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смесями. В случае капсул, таблеток и пилюль, лекарственная форма может также содержать буферные агенты.
Твердые композиции аналогичного типа можно также применять в качестве наполнителей для мягких и твердых желатиновых капсул, применяя такие вспомогательные веществ, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и подобные.
Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, можно получить с покрытиями и оболочками, такими как кишечнорастворимая оболочка и другие покрытия, известные в области фармацевтического формулирования. Они могут необязательно содержать затемняющие агенты и могут также быть такого состава, что они высвобождают активный ингредиент (ингредиенты) только или предпочтительно в определенной части желудочно-кишечного тракта, необязательно, замедленным способом. Примеры включающих композиций, которые можно применять, включают полимерные вещества и воски.
Активные соединения могут также быть в микроинкапсулированной форме с одним или более вспомогательными веществами, как приведено выше. Твердые лекарственные формы, такие как таблетки, драже, капсулы, пилюли и гранулы, можно получить с покрытиями и оболочками, такими как кишечнорастворимые оболочки, покрытия, контролирующие высвобождение, и другими покрытиями, хорошо известными в области фармацевтического формулирования. В данных твердых лекарственных формах активное соединение можно смешивать, по меньшей мере, с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Данные лекарственные формы могут также содержать, как в нормальной практике, дополнительные вещества, отличные от инертных разбавителей, например, смазывающие вещества для таблетирования и другие добавки для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль, лекарственные формы могут также содержать буферные агенты. Они могут необязательно содержать затемняющие агенты и иметь также такой состав, что они высвобождают активный ингредиент (ингредиенты) только или предпочтительно в определенной части желудочно-кишечного тракта, необязательно, замедленным способом. Примеры включающих композиций, которые можно применять, включают полимерные вещества и воски.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В добавление к активным соединениям, жидкие лекарственные формы могут содержать инертные разбавители, обычно применяемые в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, EtOAc, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, проростков, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и эфиры сорбитана и жирных кислот, и их смеси. Помимо инертных разбавителей, пероральные композиции могут также содержать адъюванты, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, подсластители, ароматизаторы и ароматизирующие добавки.
Лекарственные формы для местного или трансдермального введения соединения настоящего изобретения включают мази, пасты, крема, лосьоны, гели, порошки, растворы, спреи, ингаляторы или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми требуемыми консервантами или буферами, как может потребоваться. Также предполагается, что офтальмические составы, ушные капли и подобные включены в объем настоящего изобретения.
Мази, пасты, крема и гели могут содержать, в добавление к активному соединению настоящего изобретения, вспомогательные вещества, такие как животные и растительные жиры, масла, воски, парафин, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, кремнийорганические соединения, бентониты, кремниевую кислоту, тальк и оксид цинка, или их смеси.
Композиции настоящего изобретения можно также формулировать для доставки в виде жидкого аэрозоля или ингалируемого сухого порошка. Жидкие аэрозольные составы можно преимущественно распылять до размеров частиц, которые могут осуществлять доставку в концевые и респираторные бронхиолы.
Аэрозольные составы настоящего изобретения можно доставлять, применяя устройство для получения аэрозолей, такое как распыляющий наконечник, вибрирующая пористая пластинка или ультразвуковой распылитель, предпочтительно выбранный так, чтобы обеспечить образование аэрозольных частиц, имеющих средневесовой диаметр преимущественно между 1 и 5 мкм. Далее, состав предпочтительно имеет сбалансированную осмолярность, ионную силу и концентрацию хлорида, и наименьший объем, который можно распылять в виде аэрозоля, способный доставлять эффективную дозу соединений настоящего изобретения в область инфицирования. Дополнительно, аэрозольные составы предпочтительно не нарушают функционирование дыхательных путей и не вызывают нежелательных побочных эффектов.
Устройства для распыления в виде аэрозоля, подходящие для введения аэрозольных составов настоящего изобретения, включают, например, распыляющий наконечник, вибрирующую пористую пластинку, ультразвуковой распылитель и электрический порошковый ингалятор, которые способны распылять состав настоящего изобретения до размера аэрозольных частиц, преимущественно в диапазоне размеров 1-5 мкм. Преимущественно в данной заявке обозначает то, что, по меньшей мере, 70%, но предпочтительно более чем 90% всех полученных аэрозольных частиц, находятся в 1-5 мкм диапазоне. Струйный распылитель функционирует под давлением воздуха, раздробляя жидкий раствор на аэрозольные капли. Распылители с вибрирующей пористой пластинкой функционируют с применением звукового вакуума, генерируемого быстрым вибрированием пористой пластинки, проталкивая каплю растворителя через пористую пластинку. Ультразвуковой распылитель функционирует с помощью пьезоэлектрического кристалла, который разделяет жидкость на небольшие капли аэрозоля. Ряд подходящих устройств имеются в наличии, включая, например, AERONEB и AERODOSE распылители с вибрирующей пористой пластинкой (AeroGen, Inc., Sunnyvale, California), SIDESTREAM распылители (Medic-Aid Ltd., West Sussex, England), PARI LC и PARI LC STAR струйные распылители (Pari Respiratory Equipment, Inc., Richmond, Virginia), и AEROSONIC (DeVilbiss Medizinische Produkte (Deutschland) GmbH, Heiden, Germany) и ULTRAAIRE (Omron Healthcare, Inc., Vernon Hills, Illinois) ультразвуковые распылители.
Соединения настоящего изобретения можно также формулировать для применения в виде порошков и спреев для местного применения, которые могут содержать, в добавление к соединениям настоящего изобретения, вспомогательные вещества, такие как лактоза, тальк, кремниевая кислота, гидроксид алюминия, силикаты кальция и полиамидный порошок, или смеси данных веществ. Спреи могут дополнительно содержать общепринятые пропелленты, такие как хлорфторуглеводороды.
Трансдермальные пластыри имеют дополнительное преимущество, заключающееся в обеспечении контролируемой доставки соединения в тело. Данные лекарственные формы можно получить растворением или распределением соединения в подходящей среде. Агенты, улучшающие поглощение, можно также применять для улучшения проникновения соединения через кожу. Скорость можно контролировать или обеспечением мембраны, контролирующей скорость, или распределением соединения в полимерном матриксе или геле. Соединения настоящего изобретения можно также вводить в виде липосом. Как известно в данной области техники, липосомы обычно получают из фосфолипидов или других липидных веществ. Липосомы образуются моно- или многослойными гидратированными жидкими кристаллами, которые диспергированы в водной среде. Можно применять любой нетоксичный, физиологически приемлемый и метаболизируемый липид, способный образовывать липиды. Композиции настоящего изобретения в виде липосом могут содержать, в добавление к соединениям настоящего изобретения, стабилизаторы, консерванты, вспомогательные вещества и подобные. Предпочтительными липидами являются фосфолипиды и фосфатидилхолины (лецитины), и натуральные и синтетические. Способы получения липосом являются известными в данной области техники. Смотри, например, Prescott (ed.), "Methods in Cell Biology," Volume XIV, Academic Press, New York, 1976, p. 33 et seq
Эффективные количества соединений настоящего изобретения обычно включает любое количество, достаточные для обнаруживаемого ингибирования PI3K активности любым из испытаний, описанных в настоящем изобретении, любыми другими испытаниями на PI3K активность, известными специалисту в данной области техники, или обнаружением ингибирования или ослабления симптомов рака. Количество активного ингредиента, которое можно комбинировать с веществами, являющимися носителями, для получения единичной лекарственной формы, будет изменяться в зависимости от подвергающегося лечению пациента и конкретного пути введения. Однако, ясно, что конкретная доза для любого конкретного пациента будет зависеть от ряда факторов, включая активность конкретного применяемого соединения, возраста, веса тела, общего состояния здоровья, пола, рациона, времени введения, пути введения, скорости выведения, комбинации лекарственных средств и тяжести конкретного заболевания, подвергающегося терапии. Терапевтически эффективное количество для указанной ситуации можно легко определить стандартными экспериментами и это находится в пределах компетенции клинициста.
Согласно способам лечения настоящего изобретения, рост опухоли снижается или предотвращается у пациента, такого как человек или млекопитающее на более низкой ступени развития, введением пациенту терапевтически эффективного количества соединения настоящего изобретения, в таких количествах и в течение такого промежутка времени, которые необходимы для достижения требуемого результата. Под "терапевтически эффективным количеством" соединения настоящего изобретения подразумевается достаточное количество соединения для лечения роста опухоли при приемлемом отношении риск-польза, применимом для любого терапевтического лечения. Однако, ясно, что суммарное ежедневное количество соединений и композиций настоящего изобретения будет определяться лечащим врачом по результатам тщательной медицинской оценки. Конкретная терапевтически эффективная доза для любого конкретного пациента будет зависеть от ряда факторов, включая заболевание, подвергаемое лечению, и тяжесть заболевания; активность конкретного применяемого соединения; конкретную применяемую композицию; возраст, вес тела, общее состояние здоровья, пол и рацион пациента; время введения, путь введения и скорость выведения конкретного применяемого соединения; продолжительность лечения; лекарственные средства, применяемые в комбинации или совмещаемые с конкретным применяемым соединением; и подобные факторы, известные в области медицинской области техники.
Для целей настоящего изобретения терапевтически эффективная доза будет обычно составлять суммарную дневную дозу, введенную пациенту в виде одной или нескольких отдельных доз, и может составлять количество, например, от 0,001 до 1000 мг/кг веса тела в день и более предпочтительно от 1,0 до 30 мг/кг веса тела в день. Единичная доза композиции может содержать данные количества дольных единиц, образующие дневную дозу. В общем, режимы лечения согласно настоящему изобретению включают введение нуждающемуся в данном лечении пациенту от приблизительно 10 мг до приблизительно 2000 мг соединения (соединений) настоящего изобретения в день в виде одной или нескольких доз.
Фраза "парентеральное введение", как применяют в настоящем изобретении, обозначает способы введения, отличные от энтерального и местного введения, обычно инъекцией, и включает, без ограничения, внутривенную, внутримышечную, внутриартериальную, интратекальную, интракапсулярную, внутриглазничную, внутрисердечную, внутрикожную, внутрибрюшинную, транстрахеальную, подкожную, внутрикожную, внутрисуставную, субкапсулярную, субарахноидальную, интраспинальную и интрастернальную инъекцию и вливание.
Данные соединения можно вводить людям и другим животным для терапии любым подходящим путем введения, включая пероральный, назальный, также, например, спреем, ректальный, интравагинальный, парентеральный, интрацистернальный и местный, в виде порошков, мазей или капель, включая буккальный и сублингвальный.
Фармацевтические препараты настоящего изобретения, которые, по желанию, могут содержать дополнительные фармакологически активные вещества, получают, по сути, известным способом, например, посредством общепринятого смешения, гранулирования, нанесения покрытия, растворения или лиофилизации, и они содержат от приблизительно 1% до 100%, особенно от приблизительно 1% до приблизительно 20%, активного вещества или веществ.
Настоящее изобретение также относится к способу получения фармацевтической композиции, который включает смешение твердой, предпочтительно кристаллической, формы соединения формулы A, его гидратов или сольватов, его солей или гидратов или сольватов его солей настоящего изобретения, по меньшей мере, вместе с одним фармацевтически приемлемым носителем или разбавителем.
В другом аспекте настоящего изобретения, обеспечивают наборы, которые содержат одно или более соединений настоящего изобретения. Типичные наборы содержат PI3K ингибирующее соединение настоящего изобретения (например, соединение формулы 5) и листок-вкладыш или другие этикетки, включая руководства по лечению клеточного пролиферативного заболевания введением PI3K ингибирующего количества соединения.
Термин "набор", как применяют в настоящем изобретении, включает контейнер для хранения фармацевтических композиций, и он может также содержать разделенные контейнеры, такие как разделенный на части флакон или разделенную на части упаковку из фольги. Контейнер может иметь общепринятую форму или форму, которая является известной в данной области техники, которую получают из фармацевтически приемлемого материала, например, бумажная или картонная коробка, стеклянный или пластиковый пузырек или фляга, повторно герметизируемый пакет (например, для хранения "дополнения" таблеток для помещения в другой контейнер) или блистерная упаковка с отдельными дозами для выдавливания из упаковки согласно терапевтическому режиму. Применяемый контейнер может зависеть от точной применяемой лекарственной формы, например, общепринятую картонную коробку обычно не будут применять для хранения жидкой суспензии. Возможно применение более одного контейнера вместе в одной упаковке для продажи единичной лекарственной формы. Например, таблетки могут содержаться в пузырьке, который, в свою очередь, содержится в коробке.
Наборы настоящего изобретения могут также содержать, в добавление к PI3K ингибитору, один или более дополнительных фармацевтически активных соединений. Предпочтительно, дополнительное соединение является другим PI3K ингибитором или другим соединением, пригодным для лечения рака, ангиогенеза или роста опухоли. Дополнительные соединения можно вводить в той же лекарственной форме как PI3K ингибитор или в отличной лекарственной форме. Аналогично, дополнительные соединения можно вводить одновременно с PI3K ингибитором или в различные моменты времени.
Примеры
Следующие примеры иллюстрируют настоящее изобретение без ограничения его объема. Ясно, что настоящее изобретение не ограничивается вариантами осуществления, описанными в настоящем изобретении, но включает все их формы, которые попадают в объем настоящего описания.
Пример 1: 4,4'-(6-Хлорпиримидин-2,4-диил)ди[морфолин] (3)

Получали раствор 22 г (0,12 моль) 2,4,6-трихлорпиримидина 1 в 95,2 г (110 мл) толуола и загружали его в 25 мл капельную воронку. Загружали в 4-горлую 500 мл круглодонную колбу, продутую азотом, которая снабжена холодильником, колбонагревателем, термопарой, 125 мл капельной воронкой, механической мешалкой и впускным отверстием/выпускным отверстием для азота, 62,7 г (63 мл, 0,72 моль) морфолина 2, 95,2 г (110 мл) толуола и 44 г (44 мл) воды. Добавляли толуольный раствор 1 в течение 10 минут. Нагревали реакционную смесь до 83±3°C. Перемешивали при 83±3°C в течение 2 часов. Контролировали протекание реакции. Охлаждали до 30+3°C. Переносили 2-фазную смесь в 1 л делительную воронку. Разделяли фазы. Промывали органическую фазу (верхняя) дважды 200 мл (2×100 мл) теплой (30°C) воды. Разделяли фазы после каждой промывки. Переносили органическую (верхнюю) фазу назад в 500 мл реакционную колбу, которая снабжена холодильником, колбонагревателем, термопарой, 125 мл капельной воронкой, механической мешалкой и впускным отверстием/выпускным отверстием для азота. Перемешивали и добавляли 50,0 мл 10,0н водного раствора хлороводородной кислоты. Нагревали раствор до 53±3°C и перемешивали в течение 12-18 часов. Контролировали протекание реакции. Охлаждали до 22+3°C. Переносили 2-фазную смесь в 1 л делительную воронку. Разделяли фазы. Переносили водную (нижнюю) фазу в 500 мл круглодонную 4-горлую колбу, снабженную охлаждающей баней, термопарой, капельная воронка, pH зондом, механической мешалкой и впускным отверстием/выпускным отверстием для азота. Перемешивали и охлаждали до 0±3°C. Добавляли 85,0 г 25% водного раствора гидроксида натрия по каплям в течение 30 минут, поддерживая температуру смеси 10±10°C в процессе добавления. Нагревали до 20±3°C и перемешивали в течение 30 минут. Отделяли твердые вещества вакуумной фильтрацией. Промывали остаток на фильтре 3×100 мл воды. Сушили твердый остаток (55°C, 30 мбар) в течение 24 часов, получая 30,9 г (91,9% выход) 3 в виде белого кристаллического остатка.
Пример 2:
4,4'-[6-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2,4-диил]ди[морфолин] (4)

Загружали в 2 л круглодонную 4-горлую колбу, продутую азотом, которая снабжена холодильником, колбонагревателем, термопарой, резиновой септой, механической мешалкой и впускным отверстием/выпускным отверстием для азота, 100,0 г (0,351 моль) 4,4'-(6-хлорпиримидин-2,4-диил)ди[морфолина] 3 и 943 г (1200 мл) ацетонитрила. Перемешивали и нагревали до 60+3°C. Выдерживали данный раствор при 60+3°C для загрузки к порции. Загружали в 3 л колбу, продутую азотом, которая снабжена верхнеприводной мешалкой, холодильником, впускным отверстием/выпускным отверстием для азота и резиновой септой 115,9 г (0,457 моль) бис(пинаколато)дибора, 51,7 г (0,527 моль) ацетата калия, 12,9 г (0,014 моль) трис(дибензилиденацетон)дипалладия (0), 7,9 г (0,029 моль) трициклогексилфосфина и 393 г (500 мл) ацетонитрила. Перемешивали и нагревали суспензию до 84±3°C (температура кипения). Собирали 100 мл дистиллята. Переносили нагретый 3 ацетонитрильный раствор с помощью перистальтического насоса в 3 л колбу, содержащую реакционную смесь, в течение 30 минут и продолжали собирать дистиллят. Промывали 2 л колбу и линии загрузки 79 г (100 мл) ацетонитрила и переносили промывки к реакции. Продолжали перегонку при 84±3°C и собирали дополнительные 900 мл дистиллята (объем реакции ~1100 мл). Контролировали протекание реакции в течение 2 часов после начала добавления 3. Охлаждали реакционную смесь до 70±3°C и загружали 693 г (800 мл) толуола в течение 1-2 минут. Реакционную смесь охлаждали при добавлении толуола. Далее, охлаждали реакционную смесь до 50±3°C. Загружали в чистую 1 л колбу 347 г (400 мл) толуола и нагревали ее до 50°C. Это будут применять в качестве промывки остатка на фильтре. Фильтровали реакционную смесь через 15 г слой целита 545. Промывали остаток на фильтре теплым (50°C) толуолом (400 мл), и собирали данную промывку отдельно от основной порции. Данную промывку добавляли к остатку после перегонки, в конце способа. Переносили фильтрат назад в 3 л колбу. Концентрировали порцию (25°C-40°C температура внутри колбы, 50 мбар) до достижения объема порции 250 мл. Загружали толуольную промывку остатка на фильтре, хранимую в резерве (~400 мл), и продолжали концентрировать порцию (37°C-43°C температура внутри колбы, 50 мбар) до достижения объема порции 250 мл. Контролировали полное удаление ацетонитрила, применяя описанный Process Steering Control. Нагревали до 50°C и перемешивали в течение 15 минут. Добавляли 164 г (240 мл) гептана в течение 30 минут, поддерживая температуру равной 50°C в процессе добавления. Перемешивали полученную в результате суспензию в течение 1 часа. Охлаждали суспензию до 23±3°C в течение 1 часа и выдерживали при данной температуре, по меньшей мере, в течение 1 часа. Заполняли фильтровальную воронку, применяемую для выделения продукта, азотом (для избегания влаги) и быстро отфильтровывали твердые вещества. Промывали остаток на фильтре дважды смесью 22 г (25 мл) толуола и 51 г (75 мл) гептана. Сушили твердый остаток при 50°C, 35 мбар в течение 16 часов, получая 4,4 г (72,7% уточненный выход) 4 в виде песочного, бежевого остатка.
Пример 3: 5-бром-4-(трифторметил)пиридин-2-амин (4a)

Загружали в 3 л колбу, продутую азотом, которая снабжена верхнеприводной мешалкой, холодильником, впускным отверстием/выпускным отверстием для азота и резиновой септой, 112,14 г (0,63 моль) N-бромсукцинимида (NBS) и 645 г (725 мл) тетрагидрофурана. Перемешивали и охлаждали суспензию до -5±3°C. Загружали в продутую азотом 1 л круглодонную 4-горлую колбу, которая снабжена термопарой, механической мешалкой и впускным отверстием/выпускным отверстием для азота, 97,26 г (0,6 моль) 2-амино-4-(трифторметил)пиридина 4b и 511 г (575 мл) тетрагидрофурана. Перемешивали для растворения 4b. Переносили 4b раствор в капельную воронку над колбой и добавляли раствор к NBS суспензии в течение 2 часов, поддерживая температуру внутри колбы равной 0±3°C в процессе добавления. Промывали 1 л колбу и капельную воронку 44 г (50 мл) тетрагидрофурана и добавляли промывку к реакционной смеси. Нагревали раствор до 20+3°C в течение 30 минут. Контролировали на завершение реакции. Гасили добавлением раствора 24,6 г пентагидрата тиосульфата натрия, растворенного в 475 мл воды, в течение 10 минут, поддерживая температуру реакционной смеси равной 20±3°C в процессе добавления. Перемешивали в течение 1 часа после прекращения реакции. Концентрировали (температура внутри колбы = 25°C, 50 мбар) для удаления тетрагидрофурана. Добавляли 379 г (500 мл) трет-бутилметилового эфира. Перемешивали и нагревали полученный в результате раствор/суспензию до 30±3°C и перемешивали в течение 15 минут. Разделяли фазы. Промывали экстракт четыре раза раствором 32 г хлорида натрия, растворенного в 768 г (768 мл) воды (4×200 мл на промывку), разделяя фазы после каждой промывки. Наконец, промывали экстракт 150 г (150 мл) воды. Разделяли фазы. Загружали 152 г (200 мл) трет-бутилметилового эфира. Частично концентрировали (57±3°C) до объема 350 мл. Охлаждали до 50°C и добавляли 265 г (350 мл) трет-бутилметилового эфира. Продолжали концентрирование (57±3°C) до достижения объема порции 350 мл. Охлаждали до 50°C и добавляли 265 г (350 мл) трет-бутилметилового эфира. Снова, продолжали концентрирование (57±3°C) до достижения объема порции 350 мл. Охлаждали до 50°C и добавляли 103 г (150 мл) трет-бутил метилового эфира для повышения объема порции до 500 мл. Загружали 1026 г (1500 мл) гептана в течение 15 минут, поддерживая температуру равной 45±3°C в процессе добавления. Медленно увеличивали вакуум и концентрировали (температура внутри колбы = 40°C-50°C) до объема порции 1000 мл. Разряжали вакуум и вносили затравку. Продолжали дистилляцию, дополнительно увеличивали вакуум (медленно) и концентрировали (температура внутри колбы = 25°C-40°C) до объема порции 500 мл. Перемешивали полученную в результате суспензию при 0°C в течение 30 минут. Фильтровали твердые вещества. Промывали остаток на фильтре 68 г (100 мл) холодного (0°C) гептана (содержащего 30 ppm Octastat). Сушили твердый остаток (40°C, 50 мбар) в течение 16 часов, получая 109,8 г (78,0% выход) 4a в виде оранжевого твердого остатка.
Пример 4: 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амин (5)
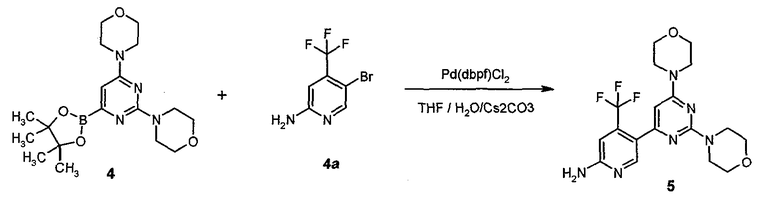
Загружали в 500 мл круглодонную 3-горлую колбу, которая снабжена термопарой, механической мешалкой, впускным отверстием/выпускным отверстием для азота и охлаждающей баней, 202,8 г (0,622 моль) карбоната цезия и 260 г (260 мл) воды. Перемешивали и охлаждали полученный в результате раствор до 22±3°C. Переносили раствор в капельную воронку. Загружали в продутую азотом 3 л колбу, которая снабжена верхнеприводной мешалкой, холодильником, pH зондом, впускным отверстием/выпускным отверстием для азота и 500 мл капельной воронкой, 50,0 г (0,207 моль) 5-бром-4-(трифторметил)пиридин-2-амина 4a, 190,9 г (0,456 моль) 4,4'-[6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2,4-диил]ди[морфолина] 4, 6,75 г (0,0103 моль) дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия и 556 г (625 мл) тетрагидрофурана. Перемешивали суспензию при 22±3°C. Добавляли водный раствор карбоната цезия через капельную воронку к суспензии в течение 1-2 минут. Перемешивали быстро (для обеспечения хорошего смешения), нагревали до 45±3°C в течение 15 минут и выдерживали при данной температуре, по меньшей мере, в течение 30 минут. Контролировали завершение реакции. Охлаждали до 22±3°C. Разделяли фазы. Частично концентрировали THF (25°C, 90 мбар) до объема 400 мл. Добавляли 654 г (750 мл) изопропилацетата, продолжая вакуумную дистилляцию, и концентрировали до объема 400 мл. Добавляли 610 г (700 мл) изопропилацетата, перемешивали и фильтровали мутный раствор через 25 г слой целита. Промывали колбу и остаток на фильтре 87 г (100 мл) изопропилацетата и добавляли промывку к порции. Добавляли 1 л 0,25н водного раствора N-ацетил-L-цистеина и перемешивали при 60±3°C в течение 1 часа. Охлаждали до 22±3°C и отделяли водную промывку. Добавляли 1 л 0,25н водного раствора N-ацетил-L-цистеина pH=7 и перемешивали при 60±3°C в течение 1 часа. Охлаждали до 22±3°C и отделяли водную промывку. Снова, добавляли 1 л 0,25н водного раствора N-ацетил-L-цистеина pH=7 и перемешивали при 60±3°C в течение 1 часа. Охлаждали до 22±3°C и отделяли водную промывку. Загружали 34,5 г Si-тиол функционализированного силикагеля и перемешивали суспензию при 60±3°C в течение 1 часа. Охлаждали до 22±3°C и фильтровали для удаления силикагеля. Добавляли 1 л 1н водного раствора хлористоводородной кислоты и перемешивали в течение 15 минут. Разделяли фазы и сохраняли водную фазу, которая на данный момент содержит продукт. Экстрагировали органическую фазу снова добавлением 500 мл 1н водного HCl раствора и перемешивали в течение 15 минут. Разделяли фазы и объединяли водные экстракты. Доводили pH до 2,3±0,2 добавлением ~280 мл 4н водного раствора гидроксида натрия. Загружали 17,2 г Si-тиол функционализированного силикагеля и перемешивали суспензию при 50±3°C в течение 1 часа. Охлаждали до 22±3°C и фильтровали для удаления силикагеля. Доводили pH до 5,0±0,2 медленным добавлением ~75 мл 4н водного раствора гидроксида натрия, поддерживая температуру реакции равной 15±3°C. Перемешивали суспензию, по меньшей мере, в течение 16 часов при 22±3°C для обеспечения полного затвердевания продукта. Фильтровали твердый остаток и промывали остаток на фильтре один раз 250 г (250 мл) воды. Сушили твердый остаток (50°C, 35 мбар) в течение 16 часов, получая 75 г (89% выход) 5 в виде коричневого твердого остатка. После данного способа соединение 5 представляет собой гемигидратную полиморфную форму HA соединения формулы A.
Альтернативный способ:
Загружали в 500 мл круглодонную 3-горлую колбу, которая снабжена термопарой, механической мешалкой, впускным отверстием/выпускным отверстием для азота и охлаждающей баней, 202,8 г (0,622 моль) карбоната цезия и 260 г (260 мл) воды. Перемешивали и охлаждали полученный в результате раствор до 22±3°C. Переносили раствор в капельную воронку. Загружали в продутую азотом 3 л колбу, которая снабжена верхнеприводной мешалкой, холодильником, pH зондом, впускным отверстием/выпускным отверстием для азота и 500 мл капельной воронкой, 50,0 г (0,207 моль) 5-бром-4-(трифторметил)пиридин-2-амина 4a, 190,9 г (0,456 моль) 4,4'-[6(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидин-2,4-диил]ди[морфолина] 4, 6,75 г (0,0103 моль) дихлорида 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладия и 556 г (625 мл) тетрагидрофурана. Перемешивали суспензию при 22±3°C. Добавляли водный раствор карбоната цезия через капельную воронку к суспензии в течение 1-2 минут. Перемешивали быстро (для обеспечения хорошего смешения), нагревали до 45±3°C в течение 15 минут и выдерживали при данной температуре, по меньшей мере, в течение 30 минут. Контролировали завершенность реакции. Охлаждали до 22+3°C. Разделяли фазы. Частично концентрировали THF (25 C, 90 мбар) до объема 400 мл. Добавляли 654 г (750 мл) изопропилацетата, продолжая вакуумную дистилляцию, и концентрировали до объема 400 мл. Добавляли 610 г (700 мл) изопропилацетата, перемешивали и фильтровали мутный раствор через 25 г слой целита. Промывали колбу и остаток на фильтре 87 г (100 мл) изопропилацетата и добавляли промывку к порции. Добавляли 1 л 0,125н водного раствора N-ацетил-L-цистеина и перемешивали при 60±3°C в течение 1 часа. Охлаждали до 22+3°C C и отделяли водную промывку. Добавляли 1 л 0,25н водного раствора N-ацетил-L-цистеина pH=7 и перемешивали при 60+3°C в течение 1 часа. Охлаждали до 22+3°C и отделяли водную промывку. Снова, добавляли 1 л 0,25н водного раствора N-ацетил-L-цистеина pH=7 и перемешивали при 60+3°C в течение 1 часа. Охлаждали до 22±3°C и отделяли водную промывку. Загружали 34,5 г Si-тиол функционализированного силикагеля и перемешивали суспензия при 60+3°C в течение 1 часа. Охлаждали до 22±3°C и фильтровали для удаления силикагеля. Добавляли 1 л 1н водного раствора хлористоводородной кислоты и перемешивали в течение 15 минут. Разделяли фазы и сохраняли водную фазу, которая содержит на данный момент продукт. Экстрагировали органическую фазу снова добавлением 500 мл 1н водного раствора хлористоводородной кислоты и перемешивали в течение 15 минут. Разделяли фазы и объединяли органические экстракты. Доводили pH до 2,3+0,2 добавлением ~280 мл 4н водного раствора гидроксида натрия. Загружали 17,2 г Si-тиол функционализированного силикагеля и перемешивали суспензия при 50±3°C в течение 1 часа. Охлаждали до 22±3°C и фильтровали для удаления силикагеля. Доводили pH до 5,0±0,2 медленным добавлением ~75 мл 4н водного раствора гидроксида натрия, поддерживая температуру реакции равной 15±3°C. Перемешивали суспензию, по меньшей мере, в течение 16 часов при 22±3°C для обеспечения полного затвердевания продукта. Фильтровали твердый остаток и промывали остаток на фильтре один раз 250 г (250 мл) воды. Сушили твердый остаток (50°C, 35 мбар) в течение 16 часов, получая 75 г (89% выход) 5 в виде коричневого твердого остатка. После данного способа соединение 5 представляет собой гемигидратную полиморфную форму HA соединения формулы A.
Пример 5: моногидрохлорид 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина (6)

Загружали в продутую азотом 3 л колбу, которая снабжена верхнеприводной мешалкой, холодильником, впускным отверстием/выпускным отверстием для азота и 500 мл капельной воронкой, 5,3 г (0,125 моль, 1 экв.) 5 и 247 г (312 мл) ацетона. Перемешивали суспензию при 25°C в течение 15 минут. Фильтровали через целит (2-5 г). Промывали колбу и остаток на фильтре 30 г (37 мл) ацетона и объединяли промывку с фильтратом. Промывали колбу метанолом и сушили ее при нагревании и вакууме. Охлаждали колбу и загружали в нее фильтрат. Нагревали раствор до 50°C. Добавляли раствор 25,7 мл (0,125 моль, 1,03 экв.) 5н хлористоводородной кислоты в изопропаноле и 198 г (250 мл) ацетона в течение 2 часов. Добавляли затравку после добавления первого 5% раствора кислоты (приблизительно 25 мл). Поддерживали температуру в течение 15 минут. Охлаждали до 10°C и фильтровали твердый остаток. Промывали остаток на фильтре 47 г (60 мл) ацетона и сушили твердый остаток при 50°C, 35 мбар в течение 16 часов, получая 49,4 г (88,1% выход) 6 в виде желтого кристаллического остатка. После данного способа соединение 6 представляет собой полиморфную форму A моногидрохлоридной соли соединения формулы A.
Пример 6: моногидрохлорид моногидрата 5-(2,6-ди-4-морфолинил-4-пиримидил)-4-(трифторметил)пиридин-2-амина
Загружали в продутую азотом 2 л колбу, которая снабжена верхнеприводной мешалкой, холодильником, впускным отверстием/выпускным отверстием для азота и 500 мл капельной воронкой, 82,08 г (0,20 моль, 1 экв.) 5 и 792 г (1,0 л) ацетона. Перемешивали суспензию при 25°C в течение 15 минут. Добавляли раствор 34,3 мл (0,206 моль, 1,03 экв.) 6н хлористоводородной кислоты в воде. Добавляли 3 мг затравки. Образовывался твердый остаток. Добавляли 20 мл воды. Нагревали реакционную смесь до температуры кипения (55-56°C) и выдерживали при температуре кипения в течение 15 минут. Охлаждали до 20°C в течение 1,5 часа. Охлаждали до 5°C в течение 1 часа и фильтровали твердый остаток. Промывали остаток на фильтре 80 мл 5°C ацетона и сушили твердый остаток при 40°C, 35 мбар в течение 16 часов, получая 70,4 г (75,7% выход) заявленного в заголовке соединения в виде желтого кристаллического остатка. После данного способа полученное в результате соединение представляет собой моногидрат моногидрохлоридной соли (полиморфная форма Ha) соединения формулы A.
Сравнительные данные
Получение 4,4'-(6-Хлорпиримидин-2,4-диил)ди[морфолина]
Получение моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина
Пример 7: Получение N-(5-бром-4-(трифторметил)пиридин-2-ил)ацетамида (B2)

Вариант A
В сухую колбу в атмосфере азота при комнатной температуре загружали 5-бром-4-(трифторметил)пиридин-2-амин B1 (200 г, 830 ммоль) и этилацетат (200 мл). Реакционную смесь охлаждали до 0°C. К раствору добавляли N,N-диметилпиридин-4-амин (1,01 г, 8,29 ммоль). Добавляли гептан (400 мл), и смесь охлаждали до 0°C. Добавляли уксусный ангидрид (109,6 мл, 1162 ммоль) в течение 60 минут. Реакционную смесь нагревали до 50°C в течение 60 минут. Реакционную смесь перемешивали при 50°C в течение 18 часов. Растворитель (330 г) удаляли дистилляцией (540-250 мбар, 50°C) до получения остатка ~300 мл. Реакционную смесь охлаждали до 20°C. Добавляли гептан (800 мл), и смесь перемешивали в течение 30 минут. Осадок собирали фильтрацией. Остаток на фильтре промывали гептаном (100 мл). Продукт сушили в центробежной сушилке в течение 16 часов при 40°C, <20 мбар, получая 212 г (90%) B2 в виде слегка коричневого твердого остатка.
Вариант B
В колбу загружали 5-бром-4-(трифторметил)пиридин-2-амин (197,04 г, 817,564 ммоль) и N,N-диметилпиридин-4-амин (0,999 г, 8,176 ммоль). Добавляли этилацетат (200 мл), и смесь перемешивали в течение 10 минут. Добавляли гептан (400 мл). Смесь нагревали до 80°C в течение 20 минут. Непрерывно добавляли уксусный ангидрид (107,994 мл, 1144,590 ммоль) в течение 4 часов. Реакционную смесь перемешивали при 80°C до того момента, как исходные соединения более не обнаруживались. Растворитель удаляли дистилляцией (80°C, 700-450 мбар) до получения остаточного объема 300 мл. Смесь охлаждали до 0°C. Добавляли гептан (800 мл), и смесь перемешивали при 0°C в течение ночи. Продукт собирали фильтрацией. Остаток промывали гептаном (100 мл) и сушили в центробежной сушилке в течение 16 часов при 40°C, <20 мбар, получая 215 г (92%) B2 в виде слегка коричневого твердого остатка.
Пример 8: получение 6-ацетамидо-4-(трифторметил)пиридин-3-илбороновой кислоты (B3)
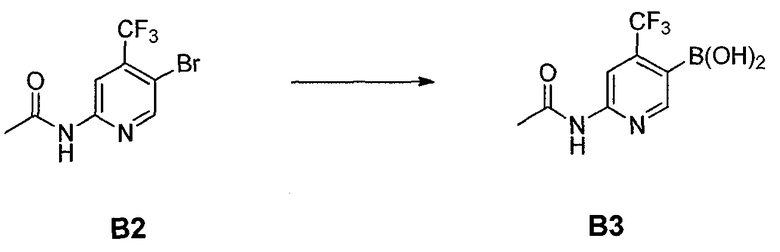
Вариант A способа
В колбу загружали 25,6 г амида лития и 60 мл THF при 20°C. Раствор 60,0 г N-(5-бром-4-(трифторметил)пиридин-2-ил)ацетамида B2 в 280 мл THF добавляли в течение 30 минут при 0°C. Суспензию перемешивали при 20°C в течение 4,5 часов. Остаточный амид лития удаляли фильтрацией. Остаток на фильтре промывали 240 мл THF. Растворитель удаляли дистилляцией (30°C, 250-85 мбар) до остаточного объема 290 мл. Добавляли 360 мл THF. Растворитель удаляли дистилляцией (30°C, 180-85 мбар) до остаточного объема 200 мл. Добавляли 160 мл THF. Реакционную смесь охлаждали до -78°C и 93,3 мл н-BuLi (2,5M в гексане) добавляли в течение 4 часов при -78≤T<-75°C. Раствор 87,7 г триизопропилбората в 30 мл THF добавляли в течение 30 минут при -78≤T≤-75°C. Реакционную смесь перемешивали при -78°C в течение 30 минут. Реакционную смесь нагревали до 20°C в течение 4 часов и перемешивали при 20°C в течение 30 минут. Добавляли 5,3 г воды в течение 30 минут при 20°C. Добавляли 45,4 мл HCl (5-6 N в PrOH) в течение 15 минут при 20°C. Реакционную смесь перемешивали в течение 15 минут при 20°C. Добавляли 480 мл толуола. Растворитель удаляли дистилляцией (30°C, 150-60 мбар) до получения остаточного объема 360 мл. Добавляли 480 мл толуола. Растворитель удаляли дистилляцией (30°C, 150-60 мбар) до получения остаточного объема 360 мл. Добавляли 480 мл толуола. Растворитель удаляли дистилляцией (30°C, 150-60 мбар) до получения остаточного объема 360 мл.
Добавляли 600 мл 2-бутанона в течение 10 минут при 30°C. Реакционную смесь перемешивали при 30°C в течение 60 минут. Реакционную смесь охлаждали в течение 20 минут до 20°C. Добавляли 12,0 г диатомовой земли (также известной как кизельгур) ("HYO"). Реакционную смесь перемешивали при 20°C в течение 20 минут. Реакционную смесь охлаждали до 0°C в течение 60 минут. Реакционную смесь перемешивали при 0°C в течение 60 минут. Неочищенный продукт собирали фильтрацией. Неочищенный продукт промывали 120 мл холодного (0°C) 2-бутанона. Неочищенный продукт сушили в вакууме (30°C, 30-35 мбар) до получения постоянного веса. Неочищенный продукт механически измельчали. В колбу загружали неочищенный продукт. Добавляли 600 мл воды с максимальной скоростью. Реакционную смесь перемешивали в течение 30 минут. pH был >8,5. Побочный осадок удаляли фильтрацией. Осадок промывали 120 мл воды. pH фильтрата доводили до pH 3 добавлением 202 г 1н HCl в течение 10 минут. Реакционную смесь охлаждали до 0°C в течение 20 минут. Реакционную смесь перемешивали при 0°C в течение 60 минут. Продукт собирали фильтрацией. Продукт промывали 60 мл холодной (5°C) воды. Продукт сушили в вакууме (30°C, 45≤p<30 мбар) до получения постоянного веса. Получали 31,5 г (59,9%) 6-ацетамидо-4-(трифторметил)пиридин-3-илбороновой кислоты B3.
Вариант B способа
К суспензии амида лития (5,6 г, 233,2 ммоль) в 140 мл THF добавляли при комнатной температуре 2-пропанол (1,3 г, 21,2 ммоль). Реакционную смесь охлаждали до IT=2°C. Раствор B2 (60,0 г, 212 ммоль) в 280 мл THF добавляли при поддержании температуры IT равной 2°C. Реакционную смесь перемешивали при IT=2°C в течение 90 минут. Добавляли THF (240 мл), и растворитель (~380 мл) удаляли дистилляцией. Добавляли THF (360 мл), и растворитель (~380 мл) удаляли дистилляцией. Добавляли THF (200 мл), и реакционную смесь охлаждали до IT=-85°C. Добавляли раствор (76 мл, 190 ммоль) н-BuLi, 2,5 M в гексане при -85°C, с последующим добавлением раствора триизопропилбората (31,9 г, 170 ммоль) в THF (17 мл). Добавляли раствор (17,3 мл, 43,3 ммоль) н-BuLi, 2,5 M в гексан при -85°C, с последующим добавлением раствора триизопропилбората (23,9 г, 127 ммоль) в THF (13 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи при комнатной температуре. Добавляли воду (15,3 г) и HCl в 2-пропаноле, 5-6M (36,7 г) при комнатной температуре. Добавляли изопропилацетат (480 мл), и растворитель удаляли дистилляцией до получения объема ~360 мл. Добавляли изопропилацетат (480 мл), и растворитель удаляли дистилляцией до получения объема ~360 мл. Добавляли изопропилацетат (480 мл), и смесь охлаждали до IT=0°C. Добавляли воду (480 г) и при добавлении 1н NaOH pH доводили при IT=0°C pH до 11. Осадок удаляли фильтрацией. Водный слой отделяли при IT=0°C. Добавляли воду, и водный слой отделяли при IT=0°C. объединенные водные слои охлаждали до IT=0°C, и pH доводили добавлением 1н HCl до pH 3. Осадок собирали и сушили при 30°C/40-45 мбар до получения постоянного веса. Получали 48,9 г (93%) 6-ацетамидо-4-(трифторметил)пиридин-3-илбороновой кислоты B3.
Пример 9: 5-(2,6-ди-4-морфолин-4-пиримидинил)-4-трифторметилпиридин-2-амин (5)
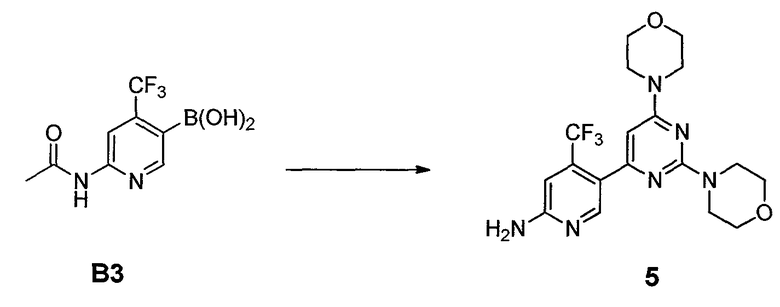
В колбу загружали при 20°C 111,7 г 4,4'-(6-хлорпиримидин-2,4-диил)диморфолина 3, 108,5 г K2CO3 и 4,54 г тетракис(трифенилфосфино)палладия (0). Добавляли 125 мл воды и 500 мл 1,2-диметоксиэтана. Белую суспензию нагревали в течение 30 минут до 90°C. Раствор 100 г 6-ацетамидо-4-(трифторметил)пиридин-3-илбороновой кислоты B3 в 450 мл 1,2-диметоксиэтана непрерывно добавляли в течение 4,5 часов. Воронку промывали 50 мл 1,2-диметоксиэтана. Реакционную смесь охлаждали до 70°C в течение 30 минут. Добавляли 1000 мл воды. 1000 мл растворителя удаляли дистилляцией (70°C, 400 мбар - 260 мбар). Добавляли 1000 мл воды. 1000 мл растворителя удаляли дистилляцией (70°C, 400 мбар - 260 мбар). Реакционную смесь охлаждали до 20°C в течение 30 минут. Осадок собирали фильтрацией и промывали 300 мл воды. В колбу загружали осадок, и добавляли 1900 г 1н HCl. Реакционную смесь нагревали до 75°C в течение 30 минут. Реакционную смесь перемешивали в течение 3 часов при 75°C. Реакционную смесь охлаждали до 20°C в течение 1 часа и перемешивали в течение ночи при 20°C. Осадок удаляли фильтрацией и промывали 200 мл воды. В колбу загружали фильтрат, и содержимое промывали 100 мл водой. pH доводили до pH 11,9. Добавляли 1800 мл изопропилацетата, и смесь перемешивали. Фазы разделяли, и органический слой собирали. Растворитель удаляли (70°C, 280 мбар) до снижения постоянного объема до 725 мл. Добавляли 1800 мл изопропилацетата, и смесь перемешивали. Фазы разделяли, и органический слой собирали. Растворитель удаляли (70°C, 280 мбар) до снижения постоянного объема до 725 мл. Реакционную смесь охлаждали до 20°C в течение 30 минут и хранили в течение выходных. Реакционную смесь фильтровали через слой HYO и промывали 225 мл изопропилацетата. В колбу загружали фильтрат, и добавляли 735,5 г раствора 1 (61,2 г N-ацетил-цистеина и 600 г воды, pH доведен до pH 7 4н NaOH). Реакционную смесь нагревали до 64°C в течение 15 минут. Реакционную смесь перемешивали при 64°C в течение 2 часов. Реакционную смесь охлаждали до 20°C в течение 45 минут. Фазы разделяли, и водный слой удаляли. Добавляли 926,5 мл 1н HCl, и смесь перемешивали в течение 15 минут. Фазы разделяли, и собирали водный слой. Добавляли 947 мл 1н HCl, и смесь перемешивали в течение 15 минут. Фазы разделяли, и собирали водный слой. Органический слой удаляли из колбы. В колбу загружали объединенные водные слои. pH доводили до pH 2,3. Добавляли 39 г Silabond. Реакционную смесь нагревали до 64°C в течение 20 минут. Реакционную смесь перемешивали в течение 2 часов при 64°C. Реакционную смесь охлаждали до 20°C в течение 30 минут. Осадок удаляли фильтрацией. Остаток промывали 500 г воды. В колбу загружали объединенные фильтраты. pH доводили до pH 5,0 4н NaOH. Реакционную смесь охлаждали до 0°C. Реакционную смесь перемешивали в течение 60 минут. Продукт собирали фильтрацией и промывали 500 г холодной (~10°C) воды. Продукт сушили до получения постоянного веса, получая 136,8 г (84,9%) 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина 5.
Пример 10: Полиморфная форма A 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина
40 мг 5 примера 4 приводили в состояние равновесия в 0,5 мл ацетонитрила в пробирке при 25°C±0,1 в течение 24 часового времени установления равновесия (с постоянным перемешиванием). Полученный в результате твердый остаток собирали фильтрацией, сушили на воздухе, удаляя остаток избытка растворителя, и затем анализировали порошковой рентгеновской дифракцией, получая безводную полиморфную форму A соединения формулы A.
Пример 11: Полиморфная форма B моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-трифторметилпиридин-2-амина
40 мг моногидрохлорида моногидрата 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина примера 6 приводили в состояние равновесия в 0,5 мл этанола в пробирке при 25°C±0,1 в течение 3 недельного времени установления равновесия (с постоянным перемешиванием). Полученный в результате твердый остаток собирали фильтрацией, сушили на воздухе, удаляя остаток избытка растворителя, и затем анализировали порошковой рентгеновской дифракцией, получая безводную полиморфную форму B моногидрохлоридной соли соединения формулы A.
Пример 12: Полиморфная форма SA моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина
40 мг моногидрохлорида моногидрата 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина примера 6 приводили в состояние равновесия в 0,5 мл ацетонитрила в пробирке при 25°C±0,1 в течение 3 дневного времени установления равновесия (с постоянным перемешиванием). Полученный в результате твердый остаток собирали фильтрацией, сушили на воздухе, удаляя остаток избытка растворителя, и затем анализировали порошковой рентгеновской дифракцией, получая полиморфную форму SA моногидрохлоридной соли соединения формулы A.
Пример 13: Полиморфная форма SB моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина
40 мг моногидрохлорида моногидрата 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина примера 6 приводили в состояние равновесия в 0,4 мл метанола в пробирке при 25°C±0,1 в течение 24 часового времени установления равновесия (с постоянным перемешиванием). Раствор упаривали досуха в потоке азота при комнатной температуре. Полученный в результате твердый остаток собирали, затем сушили и исследовали порошковой рентгеновской дифракцией, получая полиморфную форму SB моногидрохлоридной соли соединения формулы A.
Пример 14: Полиморфная форма Sc моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина
40 мг моногидрохлорида моногидрата 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина примера 6 приводили в состояние равновесия в 0,5 мл изопропилацетата в пробирке при 25°C±0,1 в течение 24 часового времени установления равновесия (с постоянным перемешиванием). Полученный в результате твердый остаток собирали фильтрацией, сушили на воздухе, удаляя остаток избытка растворителя, и затем анализировали порошковой рентгеновской дифракцией, получая полиморфную форму Sc моногидрохлоридной соли соединения формулы A.
Пример 15: Полиморфная форма SD моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина
40 мг 6 примера 5 перемешивали в растворе, содержащем этанол и воду в соотношении 1:1, или растворе, содержащем ацетонитрил и воду в соотношении 1:1, в пробирке при 25°C±0,1 в течение 24 часового времени установления равновесия (с постоянным перемешиванием). Полученный в результате твердый остаток собирали фильтрацией, получая полиморфную форму SD моногидрохлоридной соли соединения формулы A.
Пример 16: Полиморфная форма SE моногидрохлорида 5-(2,6-ди-4-морфолинил-4-пиримидинил)-4-(трифторметил)пиридин-2-амина
40 мг 6 примера 5 перемешивали в растворе, содержащем ацетон и воду в соотношении 1:1, в пробирке при 25°C+0,1 в течение 24 часового времени установления равновесия (с постоянным перемешиванием). Полученный в результате твердый остаток собирали фильтрацией, получая полиморфную форму SE моногидрохлоридной соли соединения формулы A.
Таблицы
Список наиболее значимых пиков фигуры 4 (Гемигидратная полиморфная форма HA)
Список наиболее значимых пиков фигуры 5 (Безводная полиморфная форма A)
Список наиболее значимых пиков фигуры 6 (Гидрохлоридная полиморфная форма Ha)
Список наиболее значимых пиков фигуры 7 (Гидрохлоридная полиморфная форма A)
Список наиболее значимых пиков фигуры 8 (Гидрохлоридная полиморфная форма B)
Список наиболее значимых пиков фигуры 9 (Полиморфная форма SA)
Список наиболее значимых пиков фигуры 10 (Полиморфная форма SB)
Список наиболее значимых пиков фигуры 11 (Полиморфная форма SC)
Список наиболее значимых пиков фигуры 12 (Полиморфная форма SD)
Список наиболее значимых пиков фигуры 13 (Полиморфная форма SE)


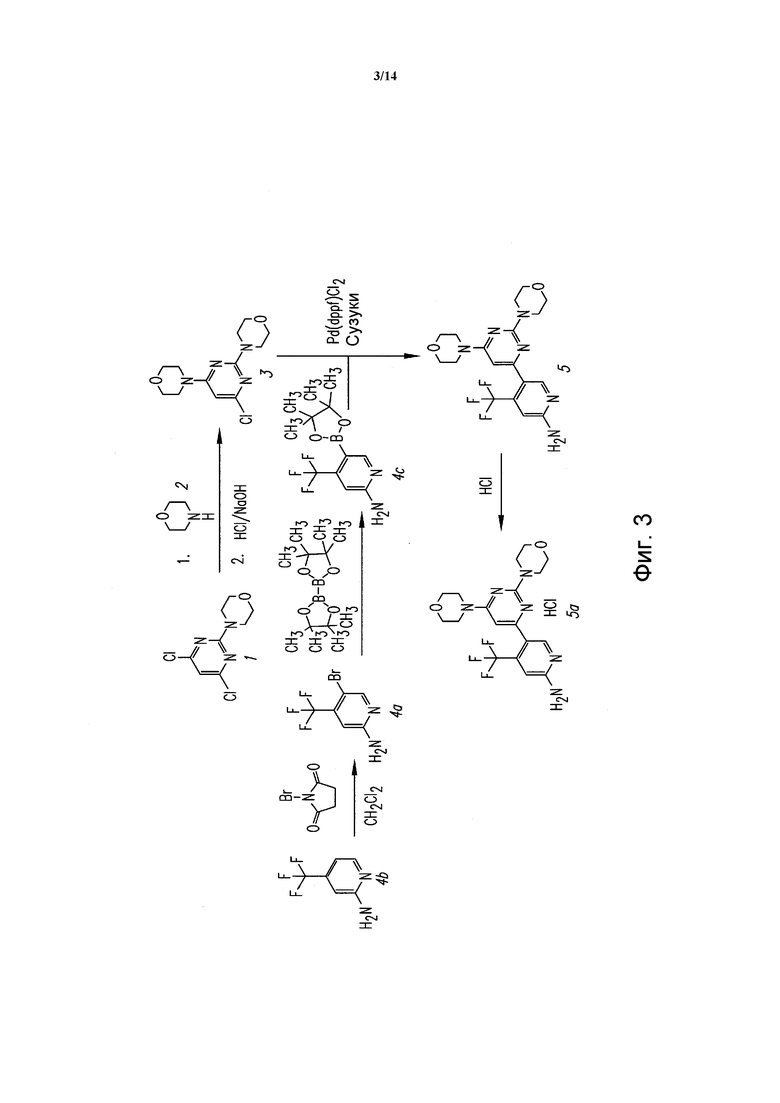


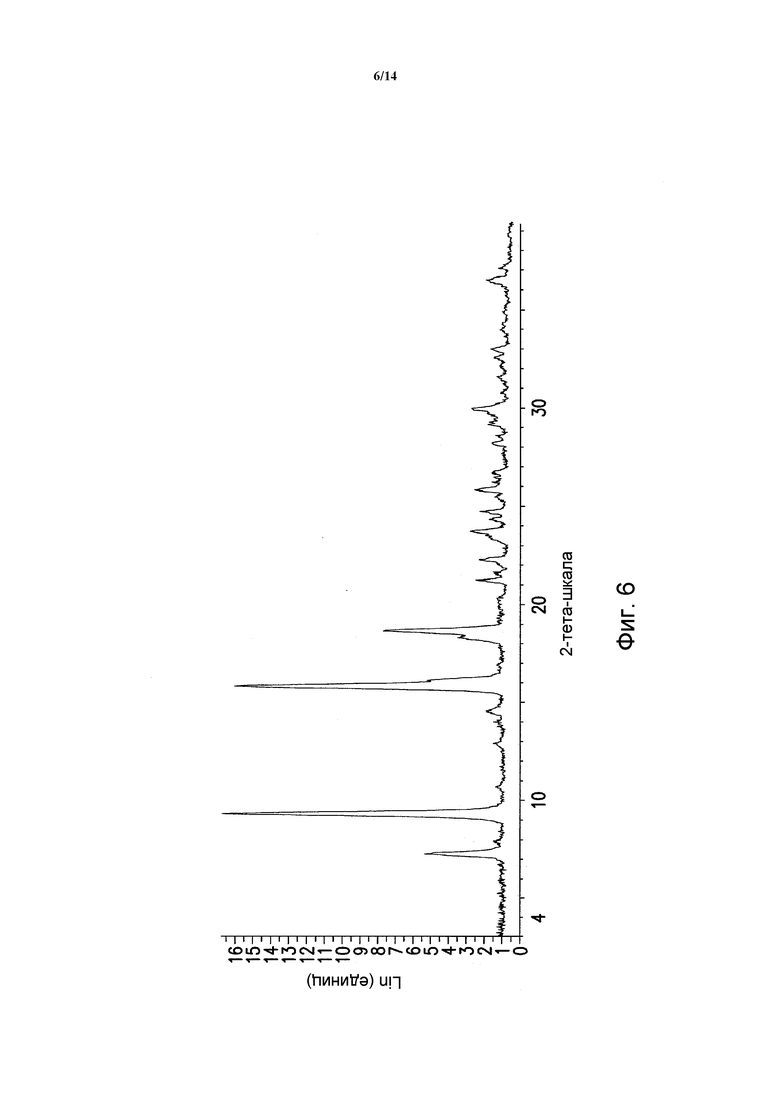

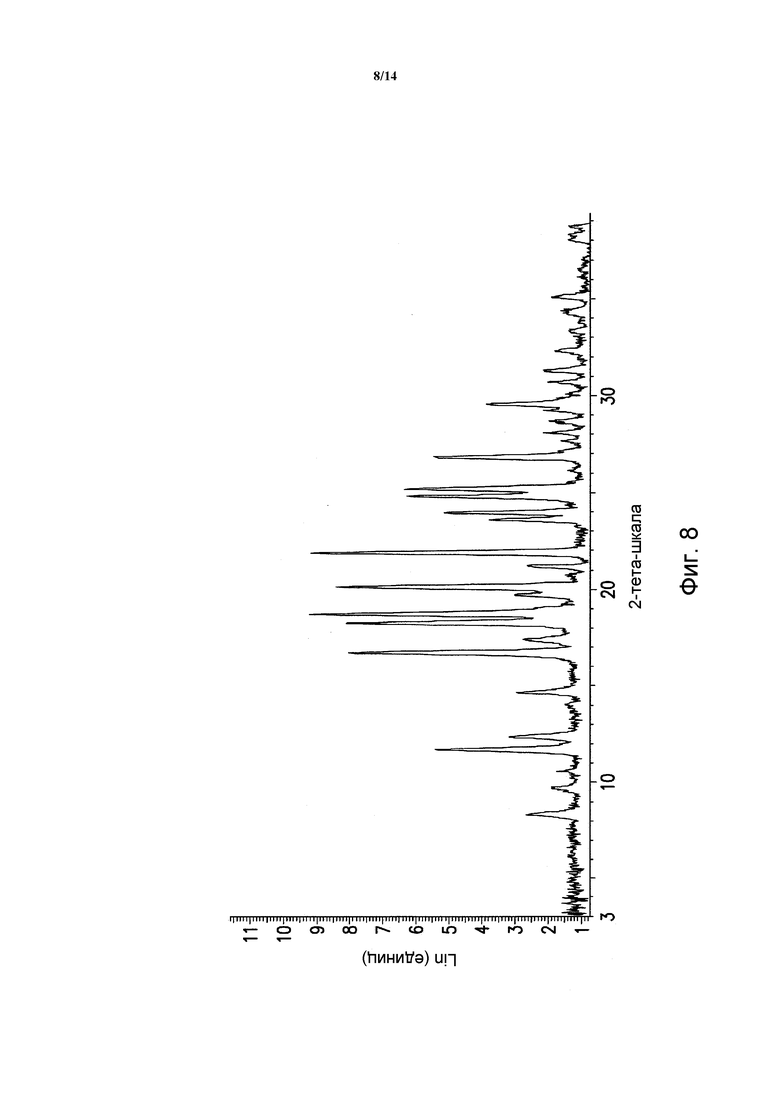






Изобретение относится к кристаллической форме моногидрохлоридной соли соединения формулы А 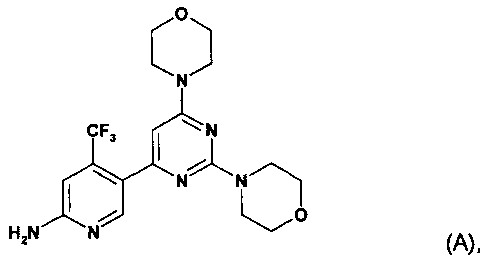
в полиморфной форме А, где указанная моногидрохлоридная соль имеет при дифракции рентгеновских лучей пик при угле дифракции 2θ 9,9±0,3° и 20,0±0,3°. Указанная кристаллическая форма имеет низкое влагопоглощение по сравнению со свободным основанием и моногидратом моногидрохлоридом, что позволяет обеспечивать стабильность состава при минимальном риске химического разрушения. 3 з.п. ф-лы, 10 табл., 14 ил., 16 пр.
1. Кристаллическая форма моногидрохлоридной соли соединения формулы А
в полиморфной форме А, где указанная моногидрохлоридная соль имеет при дифракции рентгеновских лучей пик при угле дифракции 2θ 9,9±0,3° и 20,0±0,3°.
2. Кристаллическая форма моногидрохлоридной соли соединения формулы А по п. 1 в полиморфной форме А, где указанная моногидрохлоридная соль имеет при дифракции рентгеновских лучей пик при угле дифракции 2θ 9,9±0,3°, 20,0±0,3°, 18,0±0,3°, 20,7±0,3°, 8,8±0,3° и 25±0,3°.
3. Кристаллическая форма моногидрохлоридной соли соединения формулы А по п. 1, где указанная моногидрохлоридная соль имеет при дифракции рентгеновских лучей пик при угле дифракции 2θ 8,8±0,3°, 9,9±0,3°, 12,8±0,3°, 14,0±0,3°, 14,9±0,3°, 15,4±0,3°, 18,0±0,3°, 18,8±0,3°, 20,0±0,3°, 20,7±0,3°, 22,5±0,3°, 23,9±0,3°, 24,3±0,3°, 25,0±0,3°, 27,5±0,3°, 29,1±0,3° и 31,3±0,3°.
4. Кристаллическая форма моногидрохлоридной соли соединения формулы А по п. 1, характеризующаяся порошковой рентгеновской дифрактограммой, представленной на фигуре 7.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| EA 200801680 A1, 30.12.2008. | |||

