ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Настоящая заявка испрашивает приоритет на основании предварительной заявки на патент США №61/611376, поданной 15 марта 2012 г., полное содержание которой включено в настоящую заявку посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
[0002] В настоящем изобретении предложены твердые формы соединения, подходящего для применения в качестве мутант-селективных ингибиторов киназы рецептора эпидермального фактора роста (EGFR). В настоящем изобретении также предложены фармацевтически приемлемые композиции, содержащие твердые формы соединений согласно настоящему изобретению, и способы применения указанных композиций для лечения различных расстройств.
[0003] Протеинтирозинкиназы представляют собой класс ферментов, которые катализируют перенос фосфатной группы от АТФ или ГТФ к остатку тирозина, находящемуся в белковом субстрате. Действие рецепторных тирозинкиназ заключается в том, что они передают сигналы с внешней стороны клетки внутрь путем активации эффекторов вторичного транспорта посредством фосфорилирования. Данные сигналы способствуют различным клеточным процессам, включая пролиферацию, утилизацию углеводов, синтез белка, ангиогенез, рост клеток и выживание клеток.
[0004] Существует серьезный прецедент в отношении участия EGFR в раке человека, поскольку более чем в 60% всех солидных опухолей происходит сверхэкспрессия по меньшей мере одного из этих белков или их лигандов. Сверхэкспрессию EGFR обычно обнаруживают в опухолях груди, легких, головы и шеи, а также мочевого пузыря.
[0005] Активирующие мутации в тирозинкиназном домене EGFR были идентифицированы у пациентов с немелкоклеточным раком легких (Lin, N.U.; Winer, Е.P., Breast Cancer Res 6: 204-210, 2004). Обратимые ингибиторы: Тарцева (эрлотиниб) и Иресса (гефатиниб) - в настоящее время являются терапией первой линии для пациентов, страдающих немелкоклеточным раком легких с активирующими мутациями. Наиболее распространенными активирующими мутациями являются L858R и delE746-A750.
[0006] Кроме того, у большинства пациентов с рецидивами приобретенная устойчивость к лекарственному средству, в том числе вызванная мутацией остатка «привратника» Т790М, была выявлена по меньшей мере у половины таких клинически устойчивых пациентов. Кроме того, Т790М может также предварительно существовать; мутация Т790М может иметь независимую онкогенную роль. Например, существуют пациенты с мутацией L858R/T790M, которые никогда не получали лечения гефитинибом. Кроме того, мутации EGFR Т790М у зародышей связаны с некоторыми наследственными формами рака легких.
[0007] Разрабатываемые в настоящее время лекарственные средства, включающие ковалентные ингибиторы второго поколения, такие как BIBW2992, HKI-272 и PF-0299804, эффективны против устойчивости, вызванной мутацией Т790М, но обладают дозолимитирующей токсичностью ввиду конкурентного ингибирования EGFR дикого типа. Соответственно, остается потребность в нахождении мутант-селективных ингибиторов киназы EGFR, подходящих для применения в качестве терапевтических агентов.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0007] Авторами настоящего изобретения было обнаружено, что новые твердые формы согласно настоящему изобретению и содержащие их композиции подходят для применения в качестве мутант-селективных ингибиторов одной или более киназ EGFR и имеют желаемые для этого характеристики. В общем, данные твердые формы и содержащие их фармацевтические композиции подходят для применения для лечения или уменьшения тяжести различных заболеваний или расстройств, подробно описанных в настоящем документе.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0008] На фигуре 1 изображена дифрактограмма рентгеновской порошковой дифракции (XRPD) для Формы А Соединения 1.
[0009] На фигуре 2 изображены кривые термогравиметрического анализа/дифференциального термического анализа (ТГА/ДТА) для Формы А Соединения 1.
[0010] На фигуре 3 изображена термограмма дифференциальной сканирующей калориметрии (ДСК) для Формы А Соединения 1.
[0011] На фигуре 4 изображен инфракрасный (ИК) спектр для Формы А Соединения 1.
[0012] На фигуре 5 изображен график динамической сорбции пара (ДСП) для Формы А Соединения 1.
[0013] На фигуре 6 изображена дифрактограмма XRPD для Формы В Соединения 1.
[0014] На фигуре 7 изображены кривые ТГА/ДТА для Формы В Соединения 1.
[0015] На фигуре 8 изображена термограмма ДСК для Формы В Соединения 1.
[0016] На фигуре 9 изображен ИК-спектр Формы В Соединения 1.
[0017] На фигуре 10 изображен график ДСП для Формы В Соединения 1.
[0018] На фигуре 11 изображено изменение дифрактограммы XRPD для Формы В Соединения 1 после нагревания до 120°С и 160°С, соответственно, по сравнению с Формами А и В при комнатной температуре.
[0019] На фигуре 12 изображена дифрактограмма XRPD для Формы С Соединения 1.
[0020] На фигуре 13 изображены кривые ТГА/ДТА для Формы С Соединения 1.
[0021] На фигуре 14 изображена термограмма ДСК для Формы С Соединения 1.
[0022] На фигуре 15 изображен ИК-спектр Формы С Соединения 1.
[0023] На фигуре 16 изображен график ДСП для Формы С Соединения 1.
[0024] На фигуре 17 изображено изменение дифрактограммы XRPD для Формы С Соединения 1 после выдерживания при 40°С/ОВ75% в течение 1 недели или 2 недель, соответственно, по сравнению с исходными формами Формы А и Формы С.
[0025] На фигуре 18 изображено изменение дифрактограммы XRPD для Формы С Соединения 1 после нагревания до 40°С и 120°С по сравнению с Формами А и С до сушки.
[0026] На фигуре 19 изображена дифрактограмма XRPD для Формы D Соединения 1, которая присутствует в смеси с Формой В.
[0027] На фигурах 20А и В изображены графики ТГА/ДТА для Формы D Соединения 1.
[0028] На фигуре 21 изображена термограмма ДСК для Формы D Соединения 1.
[0029] На фигуре 22 изображен ИК-спектр Формы D Соединения 1.
[0030] На фигуре 23 изображен график ДСП для Формы D Соединения 1.
[0031] На фигуре 24 изображено изменение дифрактограммы XRPD для Формы D Соединения 1 после выдерживания при 40°С/ОВ75% в течение 1 недели или 2 недель соответственно, по сравнению с исходными формами Формы А и Формы D.
[0032] На фигуре 25 изображена дифрактограмма XRPD для Формы Е Соединения 1.
[0033] На фигуре 26 изображены графики ТГА/ДТА для Формы Е Соединения 1.
[0034] На фигуре 27 изображена термограмма ДСК для Формы Е Соединения 1.
[0035] На фигуре 28 изображен ИК-спектр Формы Е Соединения 1.
[0036] На фигуре 29 изображен график ДСП для Формы Е Соединения 1.
[0037] На фигуре 30 изображено изменение дифрактограммы XRPD для Формы Е Соединения 1 после выдерживания при 40°С/ОВ75% в течение 1 недели или 2 недель соответственно, по сравнению с исходными формами Формы А и Формы Е.
[0038] На фигуре 31 изображено изменение дифрактограммы XRPD для Формы Е Соединения 1 после нагревания до 40°С и 120°С, по сравнению с Формами А и Е до сушки.
[0039] На фигуре 32 изображена дифрактограмма XRPD для Формы F Соединения 1.
[0040] На фигуре 33 изображены графики ТГА/ДТА для Формы F Соединения 1.
[0041] На фигуре 34 изображена термограмма ДСК для Формы F Соединения 1.
[0042] На фигуре 35 изображен ИК-спектр Формы F Соединения 1.
[0043] На фигуре 36 изображен график ДСП для Формы F Соединения 1.
[0044] На фигуре 37 изображено изменение дифрактограммы XRPD для Формы F Соединения 1 после выдерживания при 40°С/ОВ75% в течение 1 недели или 2 недель соответственно, по сравнению с исходными формами Формы А и Формы F.
[0045] На фигуре 38 изображено изменение дифрактограммы XRPD для Формы F Соединения 1 после нагревания до 40°С и 120°С, по сравнению с Формами А и F до сушки.
[0046] На фигуре 39 изображена дифрактограмма XRPD для Формы G Соединения 1.
[0047] На фигурах 40А, 40В и 40С изображены графики ТГА/ДТА для Формы G Соединения 1.
[0048] На фигуре 41 изображена термограмма ДСК для Формы G Соединения 1.
[0049] На фигуре 42 изображен ИК-спектр Формы G Соединения 1.
[0050] На фигуре 43 изображен график ДСП для Формы G Соединения 1.
[0051] На фигуре 44 изображено изменение дифрактограммы XRPD для Формы G Соединения 1 после выдерживания при 40°С/ОВ75% в течение 1 недели или 2 недель соответственно, по сравнению с исходными формами Формы А и Формы G.
[0052] На фигуре 45 изображено изменение дифрактограммы XRPD для Формы G Соединения 1 после нагревания до 40°С и 80°С, по сравнению с Формами А и G до сушки.
[0053] На фигуре 46 изображена дифрактограмма XRPD для Формы Н Соединения 1.
[0054] На фигуре 47 изображены графики ТГА/ДТА для Формы Н Соединения 1.
[0055] На фигуре 48 изображена дифрактограмма XRPD для Формы Н Соединения 1 после нагревания до 115°С.
[0056] На фигуре 49 изображена термограмма ДСК для Формы Н Соединения 1.
[0057] На фигуре 50 изображены графики ТГА/ДТА для Формы Н Соединения 1 после нагревания до 115°С.
[0058] На фигуре 51 изображены графики ТГА/ДТА для Формы Н Соединения 1, после того, как нагретый материал оставляли приблизительно на один час.
[0059] На фигуре 52 изображена дифрактограмма XRPD для Формы I Соединения 1.
[0060] На фигуре 53 изображен ИК-спектр для Формы I Соединения 1.
[0061] На фигуре 54 изображен спектр 1Н ЯМР для Формы I Соединения 1.
[0062] На фигуре 55 изображена термограмма ТГА/ДТА для Формы I Соединения 1 по истечении 3 суток сушки под вакуумом.
[0063] На фигуре 56 изображена термограмма ТГА/ДТА для Формы I Соединения 1 после сушки и выдерживания при температуре окружающей среды в течение приблизительно 1 часа.
[0064] На фигуре 57 изображена термограмма ДСК для Формы I Соединения 1.
[0065] На фигуре 58 изображены результаты анализа посредством ДСП для Формы I Соединения 1.
[0066] На фигуре 59 изображены результаты анализа посредством XRPD после ДСП для Формы I Соединения 1.
[0067] На фигуре 60 изображены результаты анализа посредством ВЭЖХ для Формы I Соединения 1.
[0068] На фигуре 61 изображены результаты XRPD-анализа твердых веществ, оставшихся после термодинамических исследований растворимости Формы I Соединения 1.
[0069] На фигуре 62 изображены результаты анализа посредством XRPD образцов из 1-недельного исследования стабильности Формы I Соединения 1, проведенного с применением открытых емкостей.
[0070] На фигуре 63 изображены результаты анализа посредством XRPD образцов из 1-недельного исследования стабильности Формы I Соединения 1, проведенного с применением закрытых емкостей.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Общее описание некоторых спектов изобретения:
[0071] В заявке на патент США №13/286061 (далее - заявка '061), поданной 31 октября 2011 г., полное содержание которой включено в настоящую заявку посредством ссылки, описаны некоторые 2,4-дизамещенные пиримидиновые соединения, которые ковалентно и необратимо ингибируют активности киназы EGFR. Такие соединения включают Соединение 1:
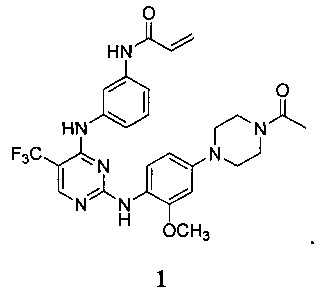
[0072] Соединение 1 (N-(3-(2-(4-(4-ацетилпиперазин-1-ил)-2-метоксифениламино)-5-(трифторметил)пиримидин-4-иламино)фенил)акриламид)) обозначено как соединение 1-4, и синтез Соединения 1 подробно описан в примере 3 заявки '061.
[0073] Соединение 1 проявило активность в различных исследованиях и терапевтических моделях, демонстрирующих селективное ковалентное необратимое ингибирование мутантной киназы EGFR (по данным ферментного анализа и анализа на клетках). Следует отметить, что Соединение 1, как было обнаружено, ингибирует пролиферацию клеток немелкоклеточного рака легких человека как in vitro, так и in vivo. Соответственно, Соединение 1 является подходящими для лечения одного или более расстройств, связанных с активностью мутантной киназы EGFR.
[0074] Было бы желательно предложить твердую форму Соединения 1, которая, по сравнению с Соединением 1, обладает такими характеристиками, как улучшенная растворимость в воде, стабильность и легкость введения в составы. Соответственно, в настоящем изобретении предложено несколько твердых форм Соединения 1.
[0075] Согласно одному из вариантов реализации в настоящем изобретении предложена аморфная форма, кристаллическая форма или их смесь. Примеры твердых форм более подробно описаны ниже.
[0076] Согласно другим вариантам реализации в настоящем изобретении предложено Соединение 1, по существу не содержащее примесей. В настоящем документе термин «по существу не содержит примесей» означает, что соединение не содержит значительного количества посторонних веществ. Такое постороннее вещество может включать исходные вещества, остаточные растворители или любые другие примеси, которые могут появиться при получении и/или выделении Соединения 1. В некоторых вариантах реализации настоящего изобретения присутствует по меньшей мере примерно 90% по массе Соединения 1. В некоторых вариантах реализации настоящего изобретения присутствует по меньшей мере примерно 95% по массе Соединения 1. В других вариантах реализации настоящего изобретения присутствует по меньшей мере примерно 99% по массе Соединения 1.
[0077] Согласно одному из вариантов реализации настоящего изобретения, Соединение 1 присутствует в количестве по меньшей мере примерно 95, 97, 97,5, 98,0, 98,5, 99, 99,5, 99,8 массовых процентов из расчета на общую массу композиции. Согласно другому варианту реализации настоящего изобретения Соединение 1 содержит общее количество органических примесей не более чем примерно 5,0% по площади пиков ВЭЖХ и в некоторых вариантах реализации настоящего изобретения содержит общее количество органических примесей не более чем примерно 3,0% по площади пиков ВЭЖХ, а в некоторых вариантах реализации настоящего изобретения содержит общее количество органических примесей не более чем примерно 1,5% по площади пиков ВЭЖХ по отношению к общей площади хроматограммы ВЭЖХ. В других вариантах реализации настоящего изобретения Соединение 1 содержит не более чем примерно 1,0% любой одной примеси по площади ВЭЖХ; не более чем примерно 0,6% любой одной примеси по площади ВЭЖХ и в некоторых вариантах реализации настоящего изобретения - не более чем примерно 0,5% любой одной примеси по площади ВЭЖХ по отношению к общей площади хроматограммы ВЭЖХ.
[0078] Также предполагается, что изображенная структура Соединения 1 включает все таутомерные формы Соединения 1. Кроме того, также предполагается, что структуры, изображенные в настоящем документе, включают соединения, которые отличаются только наличием одного или более изотопно-обогащенных атомов. Например, соединения, имеющие представленную структуру, за исключением замены водорода на дейтерий или тритий или замены углерода на 13С- или 14С-обогащенный углерод, находятся в рамках настоящего изобретения.
Твердые формы Соединения 1:
[0079] Было обнаружено, что Соединение 1 может существовать в различных твердых формах. Такие формы включают полиморфы и аморфные формы. Твердые формы могут представлять собой сольваты, гидраты и несольватированные формы Соединения 2. Все такие формы включены в настоящее изобретение. Согласно некоторым вариантам реализации, в настоящем изобретении предложено Соединение 1 в виде смеси одной или более твердых форм Соединения 1.
[0080] В настоящем документе термин «полиморф» относится к различным кристаллическим структурам (сольватированным или несольватированным формам), в которые соединение может кристаллизоваться.
[0081] В настоящем документе термин «сольват» относится к твердой форме либо со стехиометрическом, либо с нестехиометрическим количеством растворителя (сольват канального типа). В случае полиморфов растворитель включен в кристаллическую структуру. Аналогичным образом термин «гидрат» относится к твердой форме либо со стехиометрическом, либо с нестехиометрическим количеством воды. В случае полиморфов вода включена в кристаллическую структуру.
[0082] В настоящем документе термин «примерно» при использовании в отношении значения в градусах 2-тета относится к указанному значению ±0,3 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения термин «примерно» относится к ±0,2 градуса 2-тета или ±0,1 градуса 2-тета.
[0083] В некоторых вариантах реализации настоящего изобретения Соединение 1 представляет собой кристаллическое твердое вещество. В других вариантах реализации настоящего изобретения Соединение 1 представляет собой кристаллическое твердое вещество, по существу не содержащее аморфного Соединения 1. В настоящем документе термин «по существу не содержащее аморфного Соединения 1» означает, что соединение не содержит значительного количества аморфного Соединения 1. В некоторых вариантах реализации настоящего изобретения присутствует по меньшей мере примерно 90% по массе кристаллического Соединения 1 или по меньшей мере примерно 95% по массе кристаллического Соединения 1. В других вариантах реализации настоящего изобретения примерно 97%, 98% или 99% по массе кристаллического Соединения 1.
[0084] В некоторых вариантах реализации настоящего изобретения Соединение 1 представляет собой чистую или несольватированную кристаллическую форму и поэтому не содержит какой-либо воды или растворителя, входящих в кристаллическую структуру. Было обнаружено, что Соединение 1 может существовать по меньшей мере в виде двух чистых (то есть безводных) кристаллических форм, или полиморфов. В некоторых вариантах реализации настоящего изобретения предложена безводная полиморфная форма Соединения 1, обозначенная в настоящем документе как Форма А. В других вариантах реализации настоящего изобретения предложена безводная полиморфная форма Соединения 1, обозначенная в настоящем документе как Форма В.
[0085] В некоторых вариантах реализации настоящего изобретения предложена Форма А Соединения 1. Согласно одному из вариантов реализации настоящего изобретения Форма А Соединения 1 характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,73, примерно 18,30, примерно 18,96 и примерно 25,48 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма А Соединения 1 характеризуется двумя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,73, примерно 18,30, примерно 18,96 и примерно 25,48 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма А Соединения 1 характеризуется тремя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,73, примерно 18,30, примерно 18,96 и примерно 25,48 градуса 2-тета. В частных вариантах реализации настоящего изобретения Форма А Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,73, 14,24, 16,13, 18,30, 18,96, 20,59, 21,02, 21,23, 23,99 и 25,48 градуса 2-тета. В иллюстративном варианте реализации настоящего изобретения Форма А Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно:
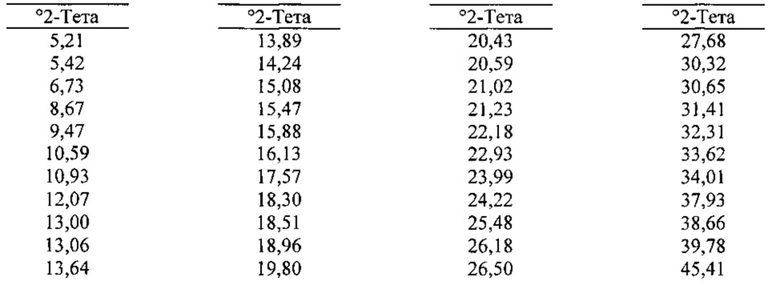
[0086] Согласно одному из аспектов Форма А Соединения 1 характеризуется дифрактограммой рентгеновской порошковой дифракции, по существу схожей с дифрактограммой, изображенной на фигуре 1. Согласно другому аспекту Форма А Соединения 1 характеризуется кривыми термогравиметрического анализа, по существу схожими с кривыми, изображенными на фигуре 2. Согласно еще одному аспекту Форма А Соединения 1 характеризуется термограммой дифференциальной сканирующей калориметрии, по существу схожей с термограммой, изображенной на фигуре 3. Согласно другому варианту реализации настоящего изобретения Форма А Соединения 1 характеризуется инфракрасным спектром, по существу схожим со спектром, изображенным на фигуре 4. Согласно другому варианту реализации настоящего изобретения Форма А Соединения 1 характеризуется графиком динамической сорбции пара, по существу схожим с графиком, изображенным на фигуре 5. Форма А Соединения 1 может характеризоваться тем, что ее характеристики по существу схожи с характеристиками, приведенными на двух или более из указанных фигур, одновременно.
[0087] В некоторых вариантах реализации настоящего изобретения предложена Форма В Соединения 1. Согласно другому варианту реализации настоящего изобретения Форма В Соединения 1 характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 10,67, примерно 12,21, примерно 18,11, примерно 19,24 и примерно 21,53 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма В Соединения 1 характеризуется двумя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 10,67, примерно 12,21, примерно 18,11, примерно 19,24 и примерно 21,53 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма В Соединения 1 характеризуется тремя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 10,67, примерно 12,21, примерно 18,11, примерно 19,24 и примерно 21,53 градуса 2-тета. В частных вариантах реализации настоящего изобретения Форма В Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 8,96, 10,67, 12,21, 14,56, 16,49, 17,74, 18,11, 19,24, 19,90, 21,53 и 23,93 градуса 2-тета. В иллюстративном варианте реализации настоящего изобретения Форма В Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно:
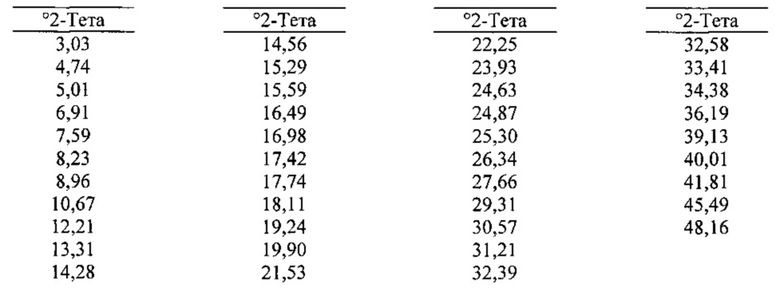
[0088] Согласно одному из аспектов Форма В Соединения 1 характеризуется дифрактограммой рентгеновской порошковой дифракции, по существу схожей с дифрактограммой, изображенной на фигуре 6. Согласно другому аспекту Форма В Соединения 1 характеризуется кривыми термогравиметрического анализа, по существу схожими с кривыми, изображенными на фигуре 7. Согласно еще одному аспекту Форма В Соединения 1 характеризуется термограммой дифференциальной сканирующей калориметрии, по существу схожей с термограммой, изображенной на фигуре 8. Согласно другому варианту реализации настоящего изобретения Форма В Соединения 1 характеризуется инфракрасным спектром, по существу схожим со спектром, изображенным на фигуре 9. Согласно другому варианту реализации настоящего изобретения Форма В Соединения 1 характеризуется графиком динамической сорбции пара, по существу схожим с графиком, изображенным на фигуре 10. Форма В Соединения 1 может характеризоваться тем, что ее характеристики по существу схожи с характеристиками, приведенными на двух или более из указанных фигур, одновременно.
[0089] В некоторых вариантах реализации настоящего изобретения Соединение 1 представляет собой кристаллическую форму сольвата в диметилформамиде (ДМФА). В некоторых вариантах реализации настоящего изобретения предложена полиморфная форма сольвата Соединения 1 в ДМФА, обозначенная в настоящем документе как Форма С.
[0090] В некоторых вариантах реализации настоящего изобретения предложена Форма С Соединения 1. Согласно одному из вариантов реализации настоящего изобретения Форма С Соединения 1 характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 16,32, примерно 18,82, примерно 20,26, примерно 22,58 и примерно 25,36 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма С Соединения 1 характеризуется двумя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 16,32, примерно 18,82, примерно 20,26, примерно 22,58 и примерно 25,36 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма С Соединения 1 характеризуется тремя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 16,32, примерно 18,82, примерно 20,26, примерно 22,58 и примерно 25,36 градуса 2-тета. В частных вариантах реализации настоящего изобретения Форма С Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 8,14, 14,45, 15,37, 16,33, 18,16, 18,82, 20,26, 22,58, 22,96, 24,33, 25,36 и 26,36 градуса 2-тета. В иллюстративном варианте реализации настоящего изобретения Форма С Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно:


[0091] Согласно одному из аспектов Форма С Соединения 1 характеризуется дифрактограммой рентгеновской порошковой дифракции, по существу схожей с дифрактограммой, изображенной на фигуре 12. Согласно другому аспекту Форма С Соединения 1 характеризуется кривыми термогравиметрического анализа, по существу схожими с кривыми, изображенными на фигуре 13. Согласно еще одному аспекту Форма С Соединения 1 характеризуется термограммой дифференциальной сканирующей калориметрии, по существу схожей с термограммой, изображенной на фигуре 14. Согласно другому варианту реализации настоящего изобретения Форма С Соединения 1 характеризуется инфракрасным спектром, по существу схожим со спектром, изображенным на фигуре 15. Согласно другому варианту реализации настоящего изобретения, Форма С Соединения 1 характеризуется графиком динамической сорбции пара, по существу схожим с графиком, изображенным на фигуре 16. Форма С Соединения 1 может характеризоваться тем, что ее характеристики по существу схожи с характеристиками, приведенными на двух или более из указанных фигур, одновременно.
[0092] В некоторых вариантах реализации настоящего изобретения Соединение 1 представляет собой кристаллическую форму сольвата в 1,4-диоксане. В некоторых вариантах реализации настоящего изобретения предложена полиморфная форма сольвата Соединения 1 в 1,4-диоксане, обозначенная в настоящем документе как Форма D.
[0093] В некоторых вариантах реализации настоящего изобретения предложена Форма D Соединения 1. Согласно одному из вариантов реализации настоящего изобретения Форма D Соединения 1 характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 18,40, примерно 19,31, примерно 20,14, примерно 20,53 и примерно 25,25 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма D Соединения 1 характеризуется двумя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 18,40, примерно 19,31, примерно 20,14, примерно 20,53 и примерно 25,25 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма D Соединения 1 характеризуется тремя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 18,40, примерно 19,31, примерно 20,14, примерно 20,53 и примерно 25,25 градуса 2-тета. В частных вариантах реализации настоящего изобретения Форма D Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 13,51, 16,97, 17,86, 18,40, 19,31, 20,14, 20,53, 21,04, 22,50, 24,98 и 25,25 градуса 2-тета. В иллюстративном варианте реализации настоящего изобретения Форма D Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно:
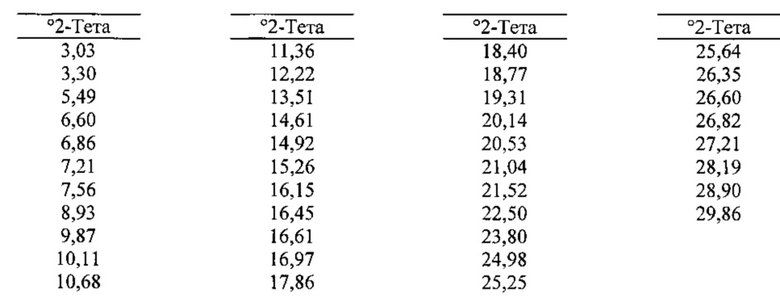
[0094] Согласно одному из аспектов Форма D Соединения 1 характеризуется дифрактограммой рентгеновской порошковой дифракции, по существу схожей с дифрактограммой, изображенной на фигуре 19. Согласно другому аспекту Форма D Соединения 1 характеризуется кривыми термогравиметрического анализа, по существу схожими с кривыми, изображенными на фигуре 20А или 20В. Согласно еще одному аспекту Форма D Соединения 1 характеризуется термограммой дифференциальной сканирующей калориметрии, по существу схожей с термограммой, изображенной на фигуре 21. Согласно другому варианту реализации настоящего изобретения Форма D Соединения 1 характеризуется инфракрасным спектром, по существу схожим со спектром, изображенным на фигуре 22. Согласно другому варианту реализации настоящего изобретения Форма D Соединения 1 характеризуется графиком динамической сорбции пара, по существу схожим с графиком, изображенным на фигуре 23. Форма D Соединения 1 может характеризоваться тем, что ее характеристики по существу схожи с характеристиками, приведенными на двух или более из указанных фигур, одновременно.
[0095] В некоторых вариантах реализации настоящего изобретения Соединение 1 представляет собой кристаллическую форму в метилэтилкетоне (МЭК). В некоторых вариантах реализации настоящего изобретения предложена полиморфная форма сольвата Соединения 1 в МЭК, обозначенная в настоящем документе как Форма Е.
[0096] В некоторых вариантах реализации настоящего изобретения предложена Форма Е Соединения 1. Согласно одному из вариантов реализации настоящего изобретения Форма Е Соединения 1 характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 5,78, примерно 12,57, примерно 15,34, примерно 19,10 и примерно 24,80 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма Е Соединения 1 характеризуется двумя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 5,78, примерно 12,57, примерно 15,34, примерно 19,10 и примерно 24,80 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма Е Соединения 1 характеризуется тремя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 5,78, примерно 12,57, примерно 15,34, примерно 19,10 и примерно 24,80 градуса 2-тета. В частных вариантах реализации настоящего изобретения Форма Е Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 5,78, 12,38, 12,57, 14,14, 15,34, 18,22, 19,10, 20,05, 24,36 и 24,80 градуса 2-тета. В иллюстративном варианте реализации настоящего изобретения Форма Е Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно:
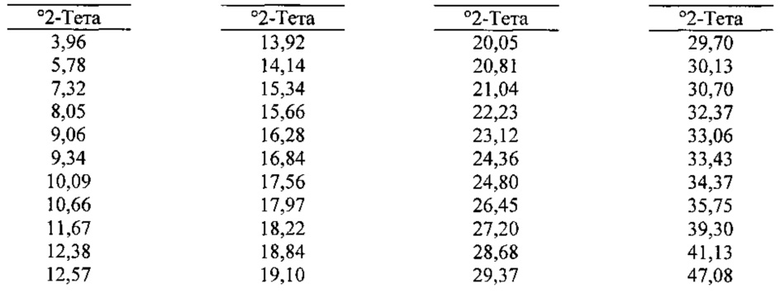
[0097] Согласно одному из аспектов Форма Е Соединения 1 характеризуется дифрактограммой рентгеновской порошковой дифракции, по существу схожей с дифрактограммой, изображенной на фигуре 25. Согласно другому аспекту Форма Е Соединения 1 характеризуется кривыми термогравиметрического анализа, по существу схожими с кривыми, изображенными на фигуре 26. Согласно еще одному аспекту Форма Е Соединения 1 характеризуется термограммой дифференциальной сканирующей калориметрии, по существу схожей с термограммой, изображенной на фигуре 27. Согласно другому варианту реализации настоящего изобретения Форма Е Соединения 1 характеризуется инфракрасным спектром, по существу схожим со спектром, изображенным на фигуре 28. Согласно другому варианту реализации настоящего изобретения Форма Е Соединения 1 характеризуется графиком динамической сорбции пара, по существу схожим с графиком, изображенным на фигуре 29. Форма Е Соединения 1 может характеризоваться тем, что ее характеристики по существу схожи с характеристиками, приведенными на двух или более из указанных фигур, одновременно.
[0098] В некоторых вариантах реализации настоящего изобретения Соединение 1 представляет собой кристаллическую полиморфную форму сольвата в N-метил-2-пирролидоне (NMP). Было обнаружено, что Соединение 1 может существовать по меньшей мере в виде двух различных NMP-кристаллических форм, или полиморфов. В некоторых вариантах реализации настоящего изобретения предложена полиморфная форма сольвата Соединения 1 в NMP, обозначенная в настоящем документе как Форма F. В других вариантах реализации настоящего изобретения предложена полиморфная форма сольвата Соединения 1 в NMP, обозначенная в настоящем документе как Форма G.
[0099] В некоторых вариантах реализации настоящего изобретения предложена Форма F Соединения 1. Согласно одному из вариантов реализации настоящего изобретения Форма F Соединения 1 характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 15,51, примерно 16,86, примерно 18,80, примерно 20,97 и примерно 23,32 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма F Соединения 1 характеризуется двумя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 15,51, примерно 16,86, примерно 18,80, примерно 20,97 и примерно 23,32 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма F Соединения 1 характеризуется тремя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 15,51, примерно 16,86, примерно 18,80, примерно 20,97 и примерно 23,32 градуса 2-тета. В частных вариантах реализации настоящего изобретения Форма F Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 5,64, 10,32, 12,97, 13,54, 15,51, 16,39, 16,86, 18,80, 19,16, 20,97, 23,32 и 24,55 градуса 2-тета. В иллюстративном варианте реализации настоящего изобретения Форма F Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно:
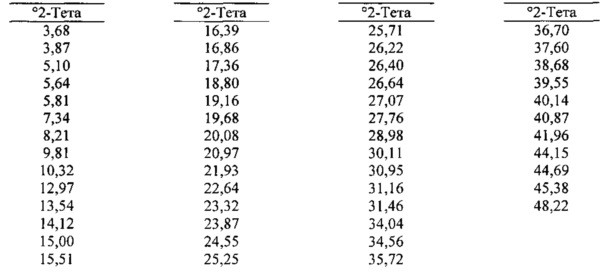
[00100] Согласно одному из аспектов Форма F Соединения 1 характеризуется дифрактограммой рентгеновской порошковой дифракции, по существу схожей с дифрактограммой, изображенной на фигуре 32. Согласно другому аспекту Форма F Соединения 1 характеризуется кривыми термогравиметрического анализа, по существу схожими с кривыми, изображенными на фигуре 33. Согласно еще одному аспекту Форма F Соединения 1 характеризуется термограммой дифференциальной сканирующей калориметрии, по существу схожей с термограммой, изображенной на фигуре 34. Согласно другому варианту реализации настоящего изобретения Форма F Соединения 1 характеризуется инфракрасным спектром, по существу схожим со спектром, изображенным на фигуре 35. Согласно другому варианту реализации настоящего изобретения Форма F Соединения 1 характеризуется графиком динамической сорбции пара, по существу схожим с графиком, изображенным на фигуре 36. Форма F Соединения 1 может характеризоваться тем, что ее характеристики по существу схожи с характеристиками, приведенными на двух или более из указанных фигур, одновременно.
[00101] В некоторых вариантах реализации настоящего изобретения предложена Форма G Соединения 1. Согласно другому варианту реализации настоящего изобретения Форма G Соединения 1 характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,79, примерно 17,86, примерно 19,43, примерно 19,98 и примерно 22,35 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма G Соединения 1 характеризуется двумя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,79, примерно 17,86, примерно 19,43, примерно 19,98 и примерно 22,35 градуса 2-тета. В дополнительных вариантах реализации изобретения Форма G Соединения 1 характеризуется тремя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,79, примерно 17,86, примерно 19,43, примерно 19,98 и примерно 22,35 градуса 2-тета. В частных вариантах реализации настоящего изобретения Форма G Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,79, 6,89, 16,50, 17,86, 19,43, 19,98, 22,35, 23,77 и 24,06 градуса 2-тета. В иллюстративном варианте реализации настоящего изобретения Форма G Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно:
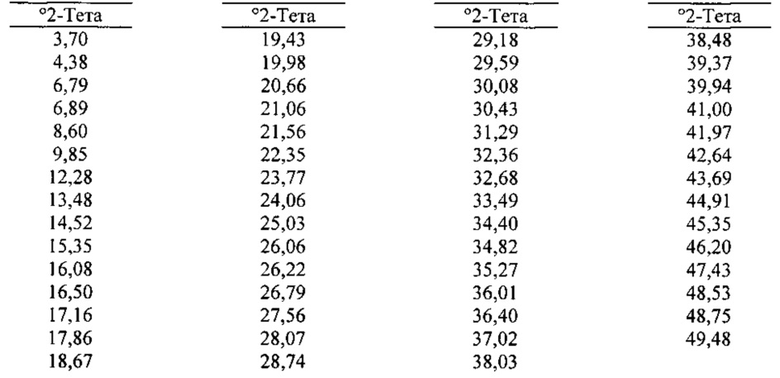
[00102] Согласно одному из аспектов Форма G Соединения 1 характеризуется дифрактограммой рентгеновской порошковой дифракции, по существу схожей с дифрактограммой, изображенной на фигуре 39. Согласно другому аспекту Форма G Соединения 1 характеризуется кривыми термогравиметрического анализа, по существу схожими с кривыми, изображенными на любой из фигуре 40А, 40В или 40С. Согласно еще одному аспекту Форма G Соединения 1 характеризуется термограммой дифференциальной сканирующей калориметрии, по существу схожей с термограммой, изображенной на фигуре 41. Согласно другому варианту реализации настоящего изобретения Форма G Соединения 1 характеризуется инфракрасным спектром, по существу схожим со спектром, изображенным на фигуре 42. Согласно другому варианту реализации настоящего изобретения Форма G Соединения 1 характеризуется графиком динамической сорбции пара, по существу схожим с графиком, изображенным на фигуре 43. Форма G Соединения 1 может характеризоваться тем, что ее характеристики по существу схожи с характеристиками, приведенными на двух или более из указанных фигур, одновременно.
[00103] В некоторых вариантах реализации настоящего изобретения Соединение 1 представляет собой гидратированную кристаллическую форму. Было обнаружено, что Соединение 1 может существовать по меньшей мере в виде двух различных кристаллических форм, или полиморфов. В некоторых вариантах реализации настоящего изобретения предложена гидратированная полиморфная форма Соединения 1, обозначенная в настоящем документе как Форма Н. В некоторых вариантах реализации настоящего изобретения предложена гидратированная полиморфная форма Соединения 1, обозначенная в настоящем документе как Форма I.
[00104] В некоторых вариантах реализации настоящего изобретения предложена Форма Н Соединения 1. Согласно одному из вариантов реализации настоящего изобретения, Форма Н Соединения 1 характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 10,82, примерно 11,08, примерно 18,45, примерно 22,85 и примерно 25,06 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма Н Соединения 1 характеризуется двумя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 10,82, примерно 11,08, примерно 18,45, примерно 22,85 и примерно 25,06 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма Н Соединения 1 характеризуется тремя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 10,82, примерно 11,08, примерно 18,45, примерно 22,85 и примерно 25,06 градуса 2-тета. В частных вариантах реализации настоящего изобретения Форма Н Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 10,14, 10,82, 11,08, 18,45, 22,85, 24,33, 25,06 и 26,54 градуса 2-тета. В иллюстративном варианте реализации настоящего изобретения Форма Н Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно:
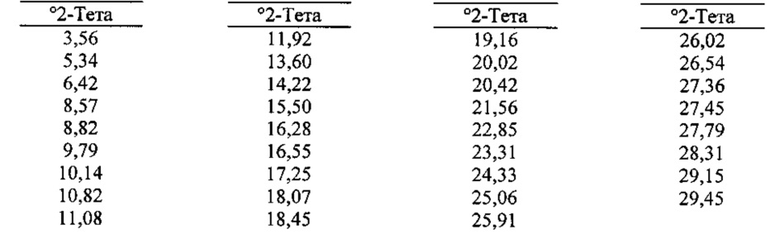
[00105] Согласно одному из аспектов Форма Н Соединения 1 характеризуется дифрактограммой рентгеновской порошковой дифракции, по существу схожей с дифрактограммой, изображенной на фигуре 46. Согласно другому аспекту Форма Н Соединения 1 характеризуется кривыми термогравиметрического анализа, по существу схожими с кривыми, изображенными на фигуре 47. Согласно еще одному аспекту Форма Н Соединения 1 характеризуется термограммой дифференциальной сканирующей калориметрии, по существу схожей с термограммой, изображенной на фигуре 49. В некоторых вариантах реализации настоящего изобретения Форма Н Соединения 1 характеризуется кривыми термогравиметрического анализа, по существу схожими с кривыми, изображенными на фигуре 50 или фигуре 51. Форма Н Соединения 1 может характеризоваться тем, что ее характеристики по существу схожи с характеристиками, приведенными на двух или более из указанных фигур, одновременно.
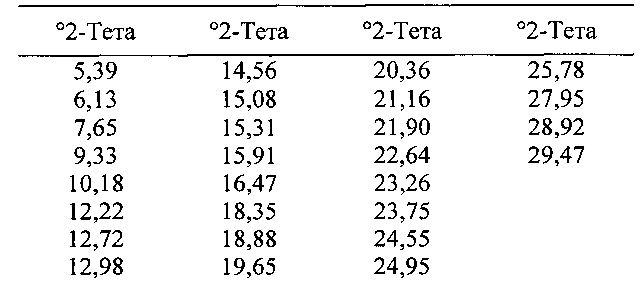
[00106] В некоторых вариантах реализации настоящего изобретения предложена Форма I Соединения 1. Согласно одному из вариантов реализации настоящего изобретения Форма I Соединения 1 характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,13, примерно 12,22, примерно 15,91, примерно 18,35, примерно 18,88 и примерно 21,90 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма I Соединения 1 характеризуется двумя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,13, примерно 12,22, примерно 15,91, примерно 18,35, примерно 18,88 и примерно 21,90 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма I Соединения 1 характеризуется тремя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,13, примерно 12,22, примерно 15,91, примерно 18,35, примерно 18,88 и примерно 21,90 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма I Соединения 1 характеризуется четырьмя или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,13, примерно 12,22, примерно 15,91, примерно 18,35, примерно 18,88 и примерно 21,90 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма I Соединения 1 характеризуется по существу всеми пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при примерно 6,13, примерно 12,22, примерно 15,91, примерно 18,35, примерно 18,88 и примерно 21,90 градуса 2-тета. В некоторых вариантах реализации настоящего изобретения Форма I Соединения 1 характеризуется по существу всеми пиками, выбранными из пиков при примерно:
5684173v1
[00107] Согласно одному из аспектов Форма I Соединения 1 характеризуется дифрактограммой рентгеновской порошковой дифракции, по существу схожей с дифрактограммой, изображенной на фигуре 52. В некоторых вариантах реализации настоящего изобретения Форма I Соединения 1 характеризуется инфракрасным спектром, по существу схожим со спектром, изображенным на фигуре 53. В некоторых вариантах реализации настоящего изобретения Форма I Соединения 1 характеризуется спектром 1Н-ЯМР, по существу схожим со спектром, изображенным на фигуре 54. Согласно другому аспекту Форма I Соединения 1 характеризуется кривыми термогравиметрического анализа, по существу схожими с кривыми, изображенными на фигуре 55 или фигуре 56. Согласно еще одному аспекту Форма I Соединения 1 характеризуется термограммой дифференциальной сканирующей калориметрии, по существу схожей с термограммой, изображенной на фигуре 57. В некоторых вариантах реализации настоящего изобретения Форма I Соединения 1 характеризуется графиком динамической сорбции пара, по существу схожим с графиком, изображенным на фигуре 58. Форма I Соединения 1 может характеризоваться тем, что ее характеристики по существу схожи с характеристиками, приведенными на двух или более из указанных фигур, одновременно.
[00108] Очевидно, что любая из вышеуказанных полиморфных форм может быть охарактеризована, например, путем ссылки на любые из пиков на их соответствующих дифрактограммах рентгеновской порошковой дифракции. Соответственно, в некоторых вариантах реализации настоящего изобретения полиморф, описанный в настоящем документе, характеризуется одним, двумя, тремя, четырьмя, пятью, шестью, семью, восьмью, девятью, десятью, одиннадцатью, двенадцатью, тринадцатью, четырнадцатью, пятнадцатью, шестнадцатью, семнадцатью, восемнадцатью, девятнадцатью, двадцатью или более пиками XRPD (°2θ). Согласно другому варианту реализации в настоящем изобретении предложено соединение 1 в виде аморфного твердого вещества. Аморфные твердые вещества хорошо известны среднему специалисту в данной области техники и их, как правило, получают такими методами, как, среди прочих, лиофилизация, плавление и осаждение из сверхкритической жидкости.
Общие способы получения Соединения 1:
[00109] Соединение 1 получают согласно способам, подробно описанным в заявке '061, полное содержание которой включено в настоящее описание посредством ссылки. Различные твердые формы Соединения 1 могут быть получены путем растворения Соединения 1 в различных подходящих растворителях и дальнейшего обеспечения возврата Соединения 1 в твердую фазу. Конкретные комбинации растворителей и условий, при которых Соединение 1 возвращается в твердую фазу, обсуждается более подробно в разделе «Примеры».
[00110] Подходящий растворитель может солюбилизировать Соединение 1 либо частично, либо полностью. Примерами подходящих растворителей, которые могут быть применены в настоящем изобретении, являются протонные растворители, полярные апротонные растворители или их смеси. В некоторых вариантах реализации настоящего изобретения подходящие растворители включают простые эфиры, сложные эфиры, спирты, кетоны или их смеси. В некоторых вариантах реализации настоящего изобретения подходящий растворитель представляет собой метанол, этанол, изопропанол или ацетон, при этом указанный растворитель является безводным или находится в комбинации с водой, метил-трет-бутиловым эфиром (МТБЭ) или гептаном. В других вариантах реализации настоящего изобретения подходящие растворители включают тетрагидрофуран, 1,4-диоксан, диметилформамид, диметилсульфоксид, глим, диглим, метилэтилкетон, N-метил-2-пирролидон, метил-т-бутиловый эфир, т-бутанол, н-бутанол и ацетонитрил. В другом варианте реализации настоящего изобретения подходящий растворитель представляет собой безводный этанол. В некоторых вариантах реализации настоящего изобретения подходящий растворитель представляет собой МТБЭ.
[00111] Согласно другому варианту реализации в настоящем изобретении предложен способ получения твердой формы Соединения 1, включающий стадии растворения Соединения 1 в подходящем растворителе и, необязательно, нагревания с получением раствора указанного соединения; и выделение Соединения 1.
[00112] Как в общем описано выше, Соединение 1 растворяют в подходящем растворителе, необязательно, при нагревании. В некоторых вариантах реализации настоящего изобретения Соединение 1 растворяют при температуре от примерно 50 до примерно 60°С. В других вариантах реализации настоящего изобретения Соединение 1 растворяют при температуре от примерно 50 до примерно 55°С. В других вариантах реализации настоящего изобретения Соединение 1 растворяют при температуре кипения растворителя. В других вариантах реализации настоящего изобретения Соединение 1 растворяют без нагревания (например, при температуре окружающей среды, составляющей приблизительно 20-25°С).
[00113] В некоторых вариантах реализации настоящего изобретения Соединение 1 осаждают из смеси. В другом варианте реализации настоящего изобретения Соединение 1 кристаллизуют из смеси. В других вариантах реализации настоящего изобретения Соединение 1 кристаллизуют из раствора после внесения в раствор затравки (то есть добавления в раствор кристаллов Соединения 1).
[00114] Кристаллическое Соединение 1 может выпадать в осадок из реакционной смеси или может быть получено путем удаления части или всего растворителя посредством таких методов, таких как выпаривание, дистилляция, фильтрование (например, нанофильтрация, ультрафильтрация), обратный осмос, абсорбция и реакция, путем добавления антирастворителя (например, воды, МТБЭ и/или гептана), путем охлаждения (например, резкого охлаждения) или с помощью различных комбинаций этих методов.
[00115] Как в общем описано выше, Соединение 1 необязательно может быть выделено. Очевидно, что Соединение 1 может быть выделено любыми подходящими физическими средствами, известными специалисту в данной области техники. В некоторых вариантах реализации настоящего изобретения осажденное твердое Соединение 1 отделяют от надосадочной жидкости путем фильтрования. В других вариантах реализации настоящего изобретения осажденное твердое Соединение 1 отделяют от надосадочной жидкости путем декантации надосадочной жидкости.
[00116] В некоторых вариантах реализации настоящего изобретения осажденное твердое Соединение 1 отделяют от надосадочной жидкости путем фильтрования.
[00117] В некоторых вариантах реализации настоящего изобретения выделенное Соединение 1 сушат на воздухе. В других вариантах реализации настоящего изобретения выделенное Соединение 1 сушат при пониженном давлении, необязательно при повышенной температуре.
Применение, приготовление форм и введение
Фармацевтически приемлемые композиции
[00118] Согласно другому варианту реализации, в настоящем изобретении предложена композиция, содержащая Соединение 1 и фармацевтически приемлемый носитель, адъювант или среду. Количество Соединения 1 в композициях согласно настоящему изобретению является таким, которое эффективно для измеримого ингибирования протеинкиназы, в частности киназы EGFR, или ее мутанта, в биологическом образце или у пациента. В некоторых вариантах реализации настоящего изобретения композиция согласно настоящему изобретению приготовлена для введения пациенту, нуждающемуся в такой композиции. В некоторых вариантах реализации настоящего изобретения композиция согласно настоящему изобретению приготовлена для перорального введения пациенту.
[00119] Термин «пациент» в настоящем документе означает животное, предпочтительно млекопитающее и наиболее предпочтительно человека.
[00120] Термин «фармацевтически приемлемый носитель, вспомогательное вещество или среда» относится к нетоксичным носителям, адъювантам или средам, которые не ухудшают фармакологическую активность соединения, с которым они находятся в композиции. Фармацевтически приемлемые носители, адъюванты или среды, которые могут быть использованы в композициях согласно настоящему изобретению, включают, но не ограничиваются ими, ионообменники, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси неполных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат протамина, гидрофосфат натрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, витамин Е-полиэтиленгликоля сукцинат (d-альфа-токоферил-полиэтиленгликоля 1000 сукцинат), натрий-карбоксиметилцеллюлозу, полиакрилаты, воски, блок-полимеры полиэтилен-полиоксипропилен и ланолин.
[00121] Композиции согласно настоящему изобретению могут быть введены перорально, парентерально, посредством ингаляционного спрея, местно, ректально, назально, трансбуккально, вагинально или через имплантированный резервуар. Термин «парентеральный» в настоящем документе включает методы подкожных, внутривенных, внутримышечных, внутрисуставных, внутрисиновиальных, интрастернальных, интратекальных, внутрипеченочных, внутриочаговых (внутрь пораженной ткани) и внутричерепных инъекций или инфузий. Предпочтительно, композиции вводят перорально, внутрибрюшинно или внутривенно. Стерильные инъецируемые формы композиций согласно настоящему изобретению могут представлять собой водные или масляные суспензии. Эти суспензии могут быть получены в соответствии со способами, известными в данной области техники, с применением подходящих разрыхляющих или смачивающих агентов и суспендирующих агентов. Стерильный инъецируемый препарат может также представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном приемлемом для парентерального введения разбавителе или растворителе, например, может представлять собой раствор в 1,3-бутандиоле. К приемлемым носителям и растворителям, которые могут быть использованы, относятся, среди прочих, вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла.
[00122] Для этой цели может быть использовано любое мягкое нелетучее масло, включая синтетические моно или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, подходят для получения инъецируемых препаратов, также подходят природные фармацевтически приемлемые масла, такие как оливковое масло или касторовое масло, в частности в их полиоксиэтилированных вариантах. Такие масляные растворы или суспензии могут также содержать длинноцепочечный спиртовой разбавитель или диспергатор, такие как карбоксиметилцеллюлоза или подобные разрыхляющие агенты, которые обычно используются для приготовления фармацевтически приемлемых лекарственных форм, включая эмульсии и суспензии. Для получения лекарственных форм также могут быть использованы и другие обычно используемые поверхностно-активные вещества, такие как Твины (Tweens), Спаны (Spans) и другие эмульгаторы или усилители биодоступности, которые обычно используются при приготовлении фармацевтически приемлемых твердых, жидких или других лекарственных форм.
[00123] Фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены перорально в любой перорально приемлемой лекарственной форме, включая, но не ограничиваясь ими, капсулы, таблетки, водные и неводные суспензии или растворы. В случае таблеток для перорального применения, обычно используемые носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие агенты, такие как стеарат магния. Для перорального введения в форме капсулы подходящие растворители включают лактозу и высушенный кукурузный крахмал. Когда требуются водные суспензии для перорального применения, активный ингредиент обычно комбинируют с эмульгирующими и суспендирующими агентами. При желании, также могут быть добавлены определенные подсластители, ароматизаторы или красители.
[00124] В качестве альтернативы, фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены в форме суппозиториев для ректального введения. Они могут быть получены путем смешивания агента с подходящим, не вызывающим раздражения вспомогательным веществом, которое является твердым при комнатной температуре, но жидким при ректальной температуре и, следовательно, будет плавиться в прямой кишке с высвобождением лекарственного средства. Такие материалы включают масло какао, пчелиный воск и полиэтиленгликоли.
[00125] Фармацевтически приемлемые композиции согласно настоящему изобретению можно также вводить местно, в частности, когда цель лечения включает области или органы, легко доступные для местного применения, включая заболевания глаз, кожи или нижней части кишечника. Подходящие составы для местного применения могут быть легко получены для каждой из этих областей или органов.
[00126] Местное применение для нижней части кишечника может быть осуществлено в виде ректальных суппозиториев (см. выше) или в виде подходящего состава для клизмы. Также могут быть использованы трансдермальные пластыри местного действия.
[00127] Для местного применения предложенные фармацевтически приемлемые композиции могут быть приготовлены в виде подходящей мази, содержащей активный компонент, суспендированный или растворенный в одном или более носителей. Носители для местного введения Соединения 1 включают, но не ограничиваются ими, минеральное масло, жидкий петролатум, белый петролатум, пропиленгликоль, полиоксиэтилен, полиоксипропиленовые соединения, эмульгирующий воск и воду. В качестве альтернативы, предложенные фармацевтически приемлемые композиции могут быть приготовлены в виде подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или более фармацевтически приемлемых носителей. Подходящие носители включают, но не ограничиваются ими, минеральное масло, моностеарат сорбитана, полисорбат 60, воски из цетиловых сложных эфиров, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
[00128] Для офтальмологического применения предложенные фармацевтически приемлемые композиции могут быть приготовлены в виде микронизированных суспензий в изотоническом стерильном физиологическом растворе с доведенным pH или предпочтительно в виде растворов в изотоническом стерильном физиологическом растворе с доведенным pH с консервантом, таким как хлорид бензилалкония, или без него. В качестве альтернативы, для офтальмологического применения фармацевтически приемлемые композиции могут быть приготовлены в виде мази, такой как петролатум.
[00129] Фармацевтически приемлемые композиции согласно настоящему изобретению также можно вводить в виде назального аэрозоля или путем ингаляции. Такие композиции получают в соответствии с методиками, хорошо известными в области фармацевтических составов, и могут быть приготовлены в виде растворов в физиологическом растворе с использованием бензилового спирта или других подходящих консервантов, усилителей всасывания для повышения биодоступности, фторуглеродов и/или других традиционных солюбилизирующих или разрыхляющих агентов.
[00130] В некоторых вариантах реализации настоящего изобретения фармацевтически приемлемые композиции согласно настоящему изобретению приготовлены для перорального введения.
[00131] Количество Соединения 1, которое может быть объединено с материалами-носителями для получения композиции в лекарственной форме для однократного введения, будет варьироваться в зависимости от подвергаемого лечению пациента и конкретного способа введения. В некоторых вариантах реализации настоящего изобретения предложенные композиции приготовлены таким образом, чтобы пациенту, получающему данные композиции, можно было бы ввести дозу Соединения 1 от 0,01 до 100 мг/кг массы тела/день.
[00132] Кроме того, следует понимать, что конкретная доза и схема лечения для любого конкретного пациента будет зависеть от множества факторов, включая активность конкретного применяемого соединения, возраст, массу тела, общее состояние здоровья, пол, рацион, время введения, скорость выведения, комбинацию лекарственных средств, а также мнение лечащего врача и тяжесть конкретного заболевания, подлежащего лечению.
Применение соединений и фармацевтически приемлемых композиций
[00133] Соединение 1 и композиции, описанные в настоящем документе, как правило, подходят для ингибирования протеинкиназной активности одного или более ферментов. Примеры киназ, которые ингибирует Соединение 1 и композиции, описанные в настоящем документе, и в отношении которых описанные в настоящем документе способы могут найти применение, включают киназу EGFR или ее мутант. Было обнаружено, что Соединение 1 представляет собой селективный ингибитор по меньшей мере одного мутанта EGFR по сравнению с EGFR дикого типа («ДТ»). В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой Т790М. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой активирующую мутацию. В некоторых вариантах реализации настоящего изобретения Соединение 1 селективно ингибирует по меньшей мере одну устойчивую мутацию и по меньшей мере одну активирующую мутацию по сравнению с EGFR ДТ. В некоторых вариантах реализации настоящего изобретения Соединение 1 селективно ингибирует по меньшей мере одну делеционную мутацию и/или по меньшей мере одну точечную мутацию и не оказывает ингибирующего действия на EGFR ДТ.
[00134] Мутация EGFR может быть выбрана из Т790М (устойчивая или онкогенная), L858R (активирующая), delE746-A750 (активирующая), G719S (активирующая) или их комбинаций.
[00135] В настоящем документе термин «селективно ингибирует», используемый по сравнению с ингибированием EGFR ДТ, означает, что Соединение 1 ингибирует по меньшей мере одну мутацию EGFR (то есть по меньшей мере одну делеционную мутацию, по меньшей мере одну активирующую мутацию, по меньшей мере одну устойчивую мутацию или комбинации по меньшей мере одной делеционной мутации и по меньшей мере одной точечной мутации) по меньшей мере в одном анализе, описанном в настоящем документе (например, биохимическом анализе или анализе на клетках). В некоторых вариантах реализации настоящего изобретения термин «селективно ингибирует», используемый по сравнению с ингибированием EGFR ДТ, означает, что Соединение 1 является по меньшей мере в 50 раз, по меньшей мере в 45 раз, по меньшей мере в 40 раз, по меньшей мере в 35 раз, по меньшей мере в 30 раз, по меньшей мере в 25 раз или по меньшей мере в 20 раз более сильным ингибитором по меньшей мере одной мутации EGFR, определенной и описанной в настоящем документе, по сравнению с ингибитором EGFR ДТ.
[00136] В настоящем документе термин «не действует на EGFR ДТ» означает, что селективный ингибитор по меньшей мере одной мутации EGFR, определенный и описанный выше и здесь, ингибирует EGFR на уровне, соответствующем верхнему пределу определения по меньшей мере одного анализа, такого как описано в заявке '061 (например, биохимического анализа или анализа на клетках, подробно описанных в Примерах 56-58). Анализы in vitro включают анализы, в которых определяют ингибирование активности фосфорилирования и/или последующие функциональные проявления, или АТФазную активность активированного EGFR (ДТ или мутанта). Альтернативные анализы in vitro позволяют количественно оценить способность ингибитора связываться с EGFR (ДТ или мутанта). Связывание ингибитора может быть измерено путем радиоактивного мечения ингибитора перед связыванием, выделения комплекса ингибитор/EGFR (ДТ или мутанта) и определения количества связанной радиоактивной метки. В качестве альтернативы, связывание ингибитора можно определить путем проведения конкурентного эксперимента, в котором новые ингибиторы инкубируют с EGFR (ДТ или мутантом), связанным с известными радиоактивно меченными лигандами. В некоторых вариантах реализации настоящего изобретения термин «не действует на EGFR ДТ» означает, что Соединение 1 ингибирует EGFR ДТ при IC50, составляющей по меньшей мере 10 мкМ, по меньшей мере 9 мкМ, по меньшей мере 8 мкМ, по меньшей мере 7 мкМ, по меньшей мере 6 мкМ, по меньшей мере 5 мкМ, по меньшей мере 3 мкМ, по меньшей мере 2 мкМ или по меньшей мере 1 мкМ.
[00137] В некоторых вариантах реализации настоящего изобретения Соединение 1 селективно ингибирует (а) по меньшей мере одну активирующую мутацию; и (b) Т790М; и (с) не действует на ДТ. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой delE746-А750. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой G719S.
[00138] В некоторых вариантах реализации настоящего изобретения по меньшей мере одна мутация EGFR представляет собой L858R и/или Т790М.
[00139] Не желая связываться какой-либо конкретной теорией, предполагается, что введение Соединения 1 у пациента, содержащего по меньшей мере одну активирующую мутацию, можно предупредить образование устойчивой мутации Т790М. Таким образом, в некоторых вариантах реализации настоящего изобретения предложен способ ингибирования активирующей мутации у пациента, включающий введение указанному пациенту Соединения 1 или содержащей его композиции, описанных в настоящем документе.
[00140] Специалисту в данной области техники очевидно, что некоторые пациенты имеют онкогенную форму мутации Т790М, то есть мутация Т790М присутствует до введения любого ингибитора киназы EGFR пациенту и поэтому является онкогенной. Соответственно, в некоторых вариантах реализации настоящего изобретения предложен способ ингибирования онкогенной Т790М у пациента, включающий введение указанному пациенту предложенного соединения или содержащей его композиции, описанных в настоящем документе.
[00141] В некоторых вариантах реализации настоящего изобретения количество Соединения 1 в композиции является эффективным, чтобы измеримо ингибировать по меньшей мере один мутант EGFR селективно по сравнению с EGFR ДТ и другими протеинкиназами (например, ErbB2, ErbB4, ТЕС-киназой и/или JAK3) в биологическом образце или у пациента.
[00142] В настоящем документе термины «лечение», «лечить» и «проведение лечения» относятся к обращению, облегчению, задержке начала или подавлению развития заболевания или расстройства, или одного или более симптомов, которые описаны в настоящем документе. В некоторых вариантах реализации настоящего изобретения введение для лечения можно осуществлять после того, как развились один или более симптомов. В других вариантах введение для лечения можно осуществлять в отсутствие симптомов. Например, введение для лечения можно осуществлять чувствительному индивидууму до появления симптомов (например, с учетом истории симптомов и/или с учетом генетических или других факторов предрасположенности). Лечение также может быть продолжено после устранения симптомов, например, чтобы предотвратить или задержать их повторное появление.
[00143] Соединение 1 является ингибитором по меньшей мере одного мутанта EGFR и поэтому является подходящим для лечения одного или более расстройств, связанных с активностью одного или более мутантов EGFR (например, делеционной мутации, активирующей мутации, устойчивой мутации или их комбинации). Таким образом, в некоторых вариантах реализации настоящего изобретения предложен способ лечения расстройства, опосредованного мутантным EGFR, включающий стадию введения нуждающемуся в этом пациенту Соединения 1 или содержащей его фармацевтически приемлемой композиции.
[00144] В настоящем документе термин «опосредованные мутантным EGFR» расстройства или состояния означает любое заболевание или другое болезненное состояние, для которого известно, что в нем участвует по меньшей мере один мутант EGFR. В некоторых вариантах реализации настоящего изобретения по меньшей мере один мутант EGFR представляет собой Т790М. В некоторых вариантах реализации настоящего изобретения указанный по меньшей мере один мутант EGFR представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения указанный по меньшей мере один мутант EGFR представляет собой активирующую мутацию. В некоторых вариантах реализации настоящего изобретения указанный по меньшей мере один мутант EGFR представляет собой L858R и/или Т790М. В некоторых вариантах реализации настоящего изобретения Соединение 1 селективно ингибирует (а) по меньшей мере одну активирующую мутацию, (b) Т790М и (с) не действует на ДТ. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой delE746-A750. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой G719S.
[00145] Соответственно, другой вариант реализации настоящего изобретения относится к лечению или уменьшению тяжести одного или более заболеваний, для которых известно, что в них участвует по меньшей мере один мутант EGFR. В частности, настоящее изобретение относится к способу лечения или уменьшения тяжести заболевания или состояния, выбранного из пролиферативного расстройства, при этом указанный способ включает введение нуждающемуся в этом пациенту соединения или композиции согласно настоящему изобретению.
[00146] В некоторых вариантах реализации настоящего изобретения предложен способ лечения или уменьшения тяжести одного или более расстройств, выбранных из рака. В некоторых вариантах реализации настоящего изобретения рак связан с солидной опухолью. В некоторых вариантах реализации настоящего изобретения рак представляет собой рак молочной железы, глиобластому, рак легкого, рак головы и шеи, колоректальный рак, рак мочевого пузыря или немелкоклеточный рак легкого. В некоторых вариантах реализации настоящего изобретения предложен способ лечения или уменьшения тяжести одного или более расстройств, выбранных из плоскоклеточной карциномы, карциномы слюнных желез, карциномы яичника или рака поджелудочной железы.
[00147] В некоторых вариантах реализации настоящего изобретения предложен способ лечения или уменьшения тяжести нейрофиброматоза типа I (NF1), нейрофиброматоза типа II (NF2), неоплазм шванновских клеток (например, MPNST's), или шванном.
[00148] Соединение 1 и композиции на его основе, в соответствии со способом согласно настоящему изобретению, могут быть введены с применением любого количества и любого пути введения, эффективного для лечения или уменьшения тяжести рака. Точное требуемое количество будет варьироваться от субъекта к субъекту в зависимости от вида, возраста и общего состояния субъекта, тяжести инфекции, конкретного агента, способа его введения и тому подобного. Соединение 2 предпочтительно готовят в виде стандартной лекарственной формы для облегчения введения и равномерности дозировки. Выражение «лекарственная форма», используемое в настоящем документе, относится к физически дискретной единице агента, подходящей для пациента, подвергаемого лечению. Следует иметь в виду, однако, что общая суточная доза из соединений и композиций согласно настоящему изобретению будет определяться лечащим врачом в рамках здравого медицинского суждения. Уровень конкретной эффективной дозы для любого конкретного пациента или организма будет зависеть от различных факторов, включая расстройство, подлежащее лечению и тяжесть данного расстройства; активность конкретного применяемого соединения; конкретную применяемую композицию; возраст, массу тела, общее состояние здоровья, пол и рацион пациента; время введения, способ введения и скорость выведения конкретного применяемого соединения; продолжительность лечения; лекарства, применяемые в комбинации или случайно с конкретным применяемым соединением и подобные факторы, хорошо известные в области медицины.
[00149] Фармацевтически приемлемые композиции согласно настоящему изобретению могут быть введены людям и другим животным перорально, ректально, парентерально, интрастернально, интравагинально, внутрибрюшинно, местно (в виде порошков, мазей или капель), буккально, в виде перорального или назального спрея или тому подобного в завивимости от тяжести инфекции, подлежащей лечению. В некоторых вариантах реализации настоящего изобретения Соединение 1 может быть введено перорально или парентерально при уровнях доз от примерно 0,01 мг/кг до примерно 50 мг/кг и предпочтительно от примерно 0,5 мг/кг до примерно 25 мг/кг массы тела субъекта в день один или более раз в день с получением желательного терапевтического эффекта.
[00150] Жидкие лекарственные формы для перорального введения включают, но не ограничиваются ими, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к Соединению 1 жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, полиэтиленгликоль (например, ПЭГ 200, ПЭГ 400, ПЭГ 1000, ПЭГ 2000), пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, масло из ростков, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, витамин Е- полиэтиленгликоля сукцинат (d-альфа-токоферолполиэтиленгликоля 1000 сукцинат), полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана и их смеси. Кроме инертных разбавителей пероральные композиции также могут содержать адъюванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители, ароматизаторы и отдушки. Жидкие формы, представленные выше, также могут быть помещены в мягкую или твердую капсулы с получением твердой лекарственной формы. Подходящие капсулы могут быть получены из, например, желатина, крахмала и производных целлюлозы (например, гидроксицеллюлозы, гидропропилметилцеллюлозы).
[00151] Инъецируемые препараты, например стерильные инъецируемые водные или масляные суспензии могут быть приготовлены согласно известным способам с применением подходящих разрыхляющих или смачивающих агентов и суспендирующих агентов. Стерильный инъецируемый препарат может также представлять собой стерильный инъецируемый раствор, суспензию или эмульсию в нетоксичном приемлемом для парентерального введения разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. К приемлемым носителям и растворителям, которые могут быть использованы, относятся, среди прочих, вода, раствор Рингера согласно Фармакопее США и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. С этой целью может быть использовано любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для получения инъецируемых препаратов применяют жирные кислоты, такие как олеиновая кислота.
[00152] Инъецируемые составы могут быть стерилизованы, например, путем фильтрования через фильтр, удерживающий бактерии, или путем включения стерилизующих агентов в форме стерильных твердых композиций, которые перед применением могут быть растворены или диспергированы в стерильной воде или другой стерильной инъецируемой среде.
[00153] Для того чтобы продлить действие Соединения 1 согласно настоящему изобретению, часто желательно замедлить всасывание соединения, введенного путем подкожной или внутримышечной инъекции. Это может быть достигнуто путем применения жидкой суспензии кристаллического или аморфного материала с плохой растворимостью в воде. Скорость всасывания соединения, таким образом, зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Кроме того, замедленное всасывание парентерально вводимой формы соединение достигается путем растворения или суспендирования соединения в масляном носителе. Инъецируемые депо-формы изготавливают путем получения микроинкапсулированных матриц соединения в биоразлагаемых полимерах, таких как полилактид-полигликолид. В зависимости от отношения соединения к полимеру и природы конкретного используемого полимера, регулируют скорость высвобождения соединения. Примеры других биоразлагаемых полимеров включают поли(ортоэфиры) и поли(ангидриды). Инъецируемые депо-составы также получают путем включения соединения в липосомы или микроэмульсии, которые совместимы с тканями организма.
[00154] Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые могут быть получены путем смешивания Соединения 1 согласно изобретению с подходящими нераздражающими вспомогательными веществами или носителями, такими как какао-масло, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при температуре окружающей среды, но жидкими при температуре тела и поэтому плавятся в прямой кишке или вагинальной полости и высвобождают активное соединение.
[00155] Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах Соединение 1 смешано по меньшей мере с одним инертным, фармацевтически приемлемым вспомогательным веществом или носителем, таким как цитрат натрия или дикальцийфосфат, и/или a) наполнителями или увеличителями объема, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связующими веществами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон (ПВП), сахароза и гуаровая камедь, c) увлажнителями, такими как глицерин, d) дезинтегрирующими агентами, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, e) замедляющими растворение агентами, такими как парафин, f) ускорителями всасывания, такими как четвертичные аммониевые соединения, g) смачивающими агентами, такими как, например, цетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолин и бентонитовая глина, и i) смазывающими веществами, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае капсул, таблеток и пилюль лекарственная форма также может содержать буферные агенты.
[00156] Твердые композиции подобного типа могут также быть использованы в качестве наполнителей для мягких и твердых заполненных капсул, используя такие вспомогательные вещества, как лактоза или молочный сахар, а также полиэтиленгликоли с высокой молекулярной массой и тому подобное. Твердые лекарственные формы: таблетки, драже, капсулы, пилюли и гранулы - могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в области получения фармацевтических препаратов. Они могут необязательно содержать замутнители и также могут представлять собой композицию, из которой активный(ые) ингредиент(ы) высвобождает(ют)ся исключительно или предпочтительно в определенной части кишечного тракта, необязательно, замедленным образом. Примеры вспомогательных веществ для заливки, которые могут быть использованы, включают полимерные вещества и воски. Твердые композиции подобного типа могут также быть использованы в качестве наполнителей в мягких и твердых заполненных капсулах, в которых использованы такие вспомогательные вещества, как лактоза или молочный сахар, а также полиэтиленгликоли с высокой молекулярной массой и тому подобное.
[00157] Соединение 1 также может быть представлено в форме микрокапсул с одним или более вспомогательных веществ, как отмечалось выше. Твердые лекарственные формы: таблетки, драже, капсулы, пилюли и гранулы - могут быть получены с покрытиями и оболочками, такими как энтеросолюбильные покрытия, покрытия, регулирующие высвобождение, и другие покрытия, хорошо известные в области получения фармацевтических препаратов. В таких твердых лекарственных формах активное соединение может быть смешано по меньшей мере с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы могут также включать, как в обычной практике, дополнительные вещества, отличные от инертных разбавителей, например смазывающие вещества и другие средства для таблетирования, такие как стеарат магния и микрокристаллическая целлюлоза. В случае капсул, таблеток и пилюль, лекарственные формы могут также содержать буферные агенты. Они могут необязательно содержать замутнители и также могут представлять собой композицию, из которой активный(ые) ингредиент(ы) высвобождает(ют)ся исключительно или предпочтительно в определенной части кишечного тракта, необязательно, замедленным образом. Примеры вспомогательных веществ для заливки, которые могут быть использованы, включают полимерные вещества и воски.
[00158] Лекарственные формы для местного или трансдермального введения Соединения 1 включают мази, пасты, кремы, лосьоны, гели, порошки, растворы, спреи, формы для ингаляций или пластыри. Активный компонент смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами или буферами, которые могут потребоваться. Состав для глаз, ушные капли и глазные капли также, как предполагается, входят в объем настоящего изобретения. Кроме того, в настоящем изобретении предполагается применение трансдермальных пластырей, которые имеют дополнительное преимущество, заключающееся в обеспечении регулируемой доставки соединения в организм. Такие лекарственные формы могут быть получены путем растворения или диспергирования соединения в соответствующей среде. Также могут быть использованы усилители всасывания для увеличения потока соединения через кожу. Скорость можно регулировать либо путем обеспечения регулирующей скорость мембраны, либо путем диспергирования соединения в полимерной матрице или геле.
[00159] Согласно другому варианту реализации, настоящее изобретение относится к способу ингибирования активности по меньшей мере одного мутанта EGFR (например, делеционной мутации, активирующей мутации, устойчивых мутаций или их комбинации) в биологическом образце, включающему стадию приведения указанного биологического образца в контакт с Соединением 1 или композицией, содержащей указанное соединение. В некоторых вариантах реализации настоящее изобретение относится к способу необратимого ингибирования активности по меньшей мере одного мутанта EGFR (например, делеционной мутации, активирующей мутации, устойчивой мутации или их комбинации) в биологическом образце, включающиему стадию приведения указанного биологического образца в контакт с Соединением 1 или композицией, содержащей заявленное соединение.
[00160] В некоторых вариантах реализации настоящего изобретения Соединение 1 селективно ингибирует в биологическом образце (a) по меньшей мере одну активирующую мутацию и (b) Т790М и (с) не действует на ДТ. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой delE746-A750. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения активирующая мутация представляет собой G719S.
[00161] Используемый в настоящем документе термин «биологический образец» включает, без ограничения, клеточные культуры или экстракты из них; материал биопсии, полученный от млекопитающего, или экстракты из него; и кровь, слюну, мочу, кал, сперму, слезы или другие жидкости организма или экстракты из них.
[00162] Ингибирование активности по меньшей мере одного мутанта EGFR (например, делеционной мутации, активирующей мутации, устойчивой мутации или их комбинации) в биологическом образце является ценным для различных целей, которые известны специалистам в данной области техники. Примеры таких целей включают, но не ограничиваются ими, переливание крови, трансплантация органов, хранение биологических образцов и биологические исследования.
[00163] Другой вариант реализации настоящего изобретения относится к способу ингибирования активности по меньшей мере одного мутанта EGFR (например, делеционной мутации, активирующей мутации, устойчивой мутации или их комбинации) у пациента, включающему стадию введения указанному пациенту Соединения 1 или композиции, содержащей данное соединение. В некоторых вариантах реализации настоящего изобретения предложен способ ингибирования (a) по меньшей мере одной активирующей мутации и (b) Т790М у пациента и (c) неоказания действия на ДТ, причем указанный способ включает введение указанному пациенту Соединения 1 или содержащей его композиции. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения по меньшей мере одна активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения предложен способ ингибирования по меньшей мере одного мутанта EGFR у пациента, при этом активирующая мутация представляет собой delE746-A750. В некоторых вариантах реализации настоящего изобретения предложен способ ингибирования по меньшей мере одного мутанта EGFR у пациента, при этом активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения предложен способ ингибирования по меньшей мере одного мутанта EGFR у пациента, при этом активирующая мутация представляет собой G719S.
[00164] Согласно другому варианту реализации настоящее изобретение относится к способу ингибирования активности по меньшей мере одного мутанта EGFR (например, делеционной мутации, активирующей мутации, устойчивой мутации или их комбинации) у пациента, включающему стадию введения указанному пациенту Соединения 1 или композиции, содержащей указанное соединение. Согласно некоторым вариантам реализации настоящее изобретение относится к способу ингибирования активности по меньшей мере одного мутанта EGFR (например, делеционной мутации, активирующей мутации, устойчивой мутации или их комбинации) у пациента, включающему введение указанному пациенту Соединения 1 или композиции, содержащей указанное соединение. В некоторых вариантах реализации настоящего изобретения предложен способ необратимого ингибирования (а) по меньшей мере одной активирующей мутации и (b) Т790М у пациента и (с) неоказания действия на ДТ, причем указанный способ включает введение указанному пациенту Соединения 1 или содержащей его композиции. В некоторых вариантах реализации настоящего изобретения необратимо ингибированная по меньшей мере одна активирующая мутация представляет собой делеционную мутацию. В некоторых вариантах реализации настоящего изобретения необратимо ингибированная по меньшей мере одна активирующая мутация представляет собой точечную мутацию. В некоторых вариантах реализации настоящего изобретения предложен способ необратимого ингибирования по меньшей мере одного мутанта EGFR у пациента, в котором активирующая мутация представляет собой delE746-A750. В некоторых вариантах реализации настоящего изобретения предложен способ необратимого ингибирования по меньшей мере одного мутанта EGFR у пациента, в котором активирующая мутация представляет собой L858R. В некоторых вариантах реализации настоящего изобретения предложен способ необратимого ингибирования по меньшей мере одного мутанта EGFR у пациента, в котором активирующая мутация представляет собой G719S.
[00165] Согласно другим вариантам реализации в настоящем изобретении предложен способ лечения расстройства, опосредованного одним или более из по меньшей мере одного мутанта EGFR (например, делеционной мутации, активирующей мутации, устойчивой мутации или их комбинации) у нуждающегося в этом пациента, включающий стадию введения указанному пациенту Соединения 1 или содержащей его фармацевтически приемлемой композиции. Такие расстройства подробно описаны в настоящем документе.
[00166] В зависимости от конкретного состояния или заболевания, подлежащих лечению, в композициях согласно настоящему изобретению или в качестве части лечебной схемы, включающей Соединение 1, могут присутствовать дополнительные терапевтические агенты, которые обычно вводят для лечения такого состояния. В настоящем документе дополнительные терапевтические агенты, которые обычно вводят для лечения конкретного заболевания или состояния, известны как «подходящие для заболевания или состояния, подлежащего лечению».
[00167] Например, Соединение 1 или содержащую его фармацевтически приемлемую композицию вводят в комбинации с химиотерапевтическими агентами для лечения пролиферативных заболеваний и рака. Примеры известных химиотерапевтических агентов включают, среди прочих, но не ограничиваются ими, адриамицин, дексаметазон, винкристин, циклофосфамид, фторурацил, топотекан, таксол, интерфероны, производные платины, таксан (например, паклитаксел), алкалоиды барвинка (например, винбластин), антрациклины (например, доксорубицин), эпиподофиллотоксины (например, этопозид), цисплатин, ингибитор mTOR (например, рапамицин), метотрексат, актиномицин D, доластатин 10, колхицин, эметин, триметрексат, метоприн, циклоспорин, даунорубицин, тенипозид, амфотерицин, алкилирующие агенты (например, хлорамбуцил), 5-фторурацил, камптотецин, цисплатин, метронидазол и Гливек (Gleevec™). В других вариантах реализации настоящего изобретения Соединение 1 вводят в комбинации с биологической агентом, таким как Авастин или ВЕКТИБИКС (VECTIBIX).
[00168] В некоторых вариантах реализации настоящего изобретения Соединение 1 или содержащую его фармацевтически приемлемую композицию вводят в комбинации с антипролиферативным или химиотерапевтическим агентом, выбранным из любого одного или более из следующих агентов: абареликс, алдеслейкин, алемтузумаб, алитретиноин, аллопуринол, алтретамин, амифостин, анастрозол, триоксид мышьяка, аспарагиназа, азацитидин, живые бациллы Кальметта-Герена (БЦЖ), бевакузимаб, фторурацил, бексаротен, блеомицин, бортезомиб, бусульфан, калустерон, капецитабин, камптотецин, карбоплатин, кармустин, целекоксиб, цетуксимаб, хлорамбуцил, кладрибин, клофарабин, циклофосфамид, цитарабин, дактиномицин, дарбэпоэтин альфа, даунорубицин, денилейкин, дексразоксан, доцетаксел, доксорубицин (нейтральный), доксорубицина гидрохлорид, дромостанолона пропионат, эпирубицин, эпоэтин альфа, эрлотиниб, эстрамустин, этопозида фосфат, этопозид, эксеместан, филграстим, флоксуридин флударабин, фульвестрант, гефитиниб, гемцитабин, гемтузумаб, гозерелина ацетат, гистрелина ацетат, гидроксимочевину, ибритумомаб, идарубицин, ифосфамид, иматиниба мезилат, интерферон альфа-2а, интерферон альфа-2b, иринотекан, леналидомид, летрозол, лейковорин, лейпролида ацетат, левамизол, ломустин, мегестрола ацетат, мелфалан, меркаптопурин, 6-МР, месну, метотрексат, метокссален, митомицин С, митотан, митоксантрон, нандролон, неларабин, нофетумомаб, опрелвекин, оксалиплатин, паклитаксел, палифермин, памидронат, пэгадемазу, пэгаспаргазы, пэгфилграстим, пеметрексед динатрия, пентостатин, пипоброман, пликамицин, натрия порфимер, прокарбазин, хинакрин, расбуриказу, ритуксимаб, сарграмостим, сорафениб, стрептозоцин, сунитиниба малеат, тальк, тамоксифен, темозоломид, тенипозид, VM-26, тестолактон, тиогуанин, 6-TG, тиотепу, топотекан, торемифен, тозитумомаб, трастузумаб, третиноин, ATRA, урамустин, валрубицин, винбластин, винкристин, винорелбин, золедронат или золедроновую кислоту.
[00169] Другие примеры агентов, с которыми также могут быть объединены ингибиторы согласно настоящему изобретению, включают, без ограничения: агенты для лечения болезни Альцгеймера, такие как донепезила гидрохлорид (Арисепт (Aricept®)) и ривастигмина (Экселон (Exelon®)); агенты для лечения болезни Паркинсона, такие как L-ДОПА/карбидопа, энтакапон, ропинрол, прамипексол, бромокриптин, перголид, тригексефендил и амантадин; агенты для лечения рассеянного склероза (PC), такие как бета-интерферон (например, Авонекс (Avonex®) и Ребиф (Rebif®)), глатиромера ацетат (Копаксон (Copaxone®)) и митоксантрон; агенты для лечения астмы, такие как альбутерол и монтелукаст (Сингуляр (Singulair®)); агенты для лечения шизофрении, такие как зипрекса, риспердал, сероквель и галоперидол; противовоспалительные агенты, такие как кортикостероиды, блокаторы фактора некроза опухоли (ФНО), ИЛ-1 RA, азатиоприн, циклофосфамид и сульфасалазин; иммуномодулирующие и иммуносупрессивные агенты, такие как циклоспорин, такролимус, рапамицин, микофенолата мофетил, интерфероны, кортикостероиды, циклофосфамид, азатиоприн и сульфасалазин; нейротрофические факторы, такие как ингибиторы ацетилхолинэстеразы, ингибиторы моноаминоксидазы (МАО), интерфероны, антиконвульсанты, блокаторы ионных каналов, рилузол и агенты против болезни Паркинсона; агенты для лечения сердечно-сосудистых заболеваний, такие как бета-блокаторы, ингибиторы ангиотензинпревращающего фермента (АПФ), диуретики, нитраты, блокаторы кальциевых каналов и статины; агенты для лечения заболеваний печени, такие как кортикостероиды, холестирамин, интерфероны и противовирусные агенты; агенты для лечения расстройств, связанных с кровью, такие как кортикостероиды, антилейкозные агенты и факторы роста; и агенты для лечения расстройств, связанных с иммунодефицитом, такие как гамма-глобулин.
[00170] В некоторых вариантах реализации настоящего изобретения Соединение 1 или содержащую его фармацевтически приемлемую композицию вводят в комбинации с моноклональным антителом или терапевтическим агентом, содержащим миРНК.
[00171] Дополнительные агенты могут быть введены отдельно от композиции, содержащей Соединение 1, как часть многократного режима дозирования. Кроме того, эти агенты могут быть частью лекарственной формы для однократного введения: их смешивают с Соединением 1 в единую композицию. Если осуществлять введение в виде части многократного режима дозирования, указанные два активных агента могут быть введены одновременно, последовательно или один после другого с перерывом в течение некоторого периода времени (например, один час, два часа, шесть часов, двенадцать часов, один день, одна неделя, две недели, один месяц).
[00172] В настоящем документе, термины «комбинация», «в комбинации» и связанные с ними термины относятся к одновременному или последовательному введению терапевтических агентов в соответствии с настоящим изобретением. Например, Соединение 1 может быть введено с другим терапевтическим агентом одновременно или последовательно в отдельных единичных лекарственных формах или совместно в единичной лекарственной форме для однократного введения. Соответственно, в настоящем изобретении предложена единичная лекарственная форма для однократного введения, содержащая Соединение 1, дополнительный терапевтический агент и фармацевтически приемлемый носитель, адъювант или среду.
[00173] Количество Соединения 1 и дополнительного терапевтического агента (в тех композициях, которые содержат дополнительный терапевтический агент, описанный выше), которые могут быть объединены с материалами-носителями для получения лекарственной формы для однократного введения, будет варьироваться в зависимости от субъекта, подвергаемого лечению, и конкретного способа введения. Предпочтительно, композиции согласно настоящему изобретению должны быть приготовлены таким образом, чтобы можно было бы ввести дозу Соединения 1 0,01-100 мг/кг массы тела/день.
[00174] В композициях, содержащих дополнительный терапевтический агент, данный дополнительный терапевтический агент и Соединение 1 могут действовать синергически. Таким образом, количество дополнительного терапевтического агента в таких композициях может быть меньше, чем требуется для монотерапии, в которой применяют только данный терапевтический агент. В таких композициях, может быть введена дозировка дополнительного терапевтического агента 0,01-1000 мкг/кг массы тела/день.
[00175] Количество дополнительного терапевтического агента, присутствующего в композициях согласно настоящему изобретению, не будет превышать количество, которое, как правило, вводили бы в виде композиции, содержащей данный терапевтический агент в качестве единственного активного агента. Предпочтительно количество дополнительного терапевтического агента в описанных в настоящем документе композициях находится в диапазоне от примерно 50% до 100% от количества, как правило, присутствующего в композиции, содержащей данный агент в качестве единственного терапевтически активного агента.
[00176] Соединение 1 или содержащие его фармацевтические композиции могут быть также включены в композиции для нанесения покрытия на имплантируемое медицинское устройство, такое как протезы, искусственные клапаны, сосудистые трансплантаты, стенты и катетеры. Сосудистые стенты, например, применяют для преодоления рестеноза (повторного сужения стенки сосуда после травмы). Однако, пациенты, использующие стенты или другие имплантируемые устройства, имеют риск образования сгустков или активации тромбоцитов. Эти нежелательные эффекты могут быть предотвращены или уменьшены путем предварительного покрытия устройства фармацевтически приемлемой композицией, содержащей ингибитор киназы. Имплантируемые устройства, покрытые Соединением 1, представляют собой еще один вариант реализации настоящего изобретения.
[00177] Все признаки каждого из аспектов изобретения относятся ко всем другим аспектам с соответствующими изменениями.
[00178] Для того чтобы более полно понять описанное в настоящем документе изобретение, представлены следующие примеры. Следует понимать, что эти примеры приведены только для иллюстративных целей и их не следует рассматривать как ограничивающие данное изобретение каким-либо образом.
ПРИМЕРЫ
[00179] Как показано в приведенных ниже примерах, в некоторых иллюстративных вариантах реализации настоящего изобретения соединения получают согласно следующим общим методикам. Очевидно, что хотя приведенные общие методы отображают синтез отдельных соединений согласно настоящему изобретению, приведенные методы и другие методы, известные среднему специалисту в данной области, могут быть применены ко всем соединениям и подклассам и разновидностям каждого из соединений, описанных в настоящем документе.
Получение Соединения 1
[00180] Синтез Соединения 1 подробно описан в примере 3 заявки '061.
[00181] В 3-горлую круглодонную колбу вместимостью 25 мл, предварительно снабженную магнитной мешалкой, рубашкой (Thermo pocket) и защитной трубкой с CaCl2, помещали N-Boc-1,3-диаминобензол (0,96 г) и н-бутанол (9,00 мл). Реакционную смесь охлаждали до 0°С. К указанной реакционной смеси по каплям добавляли 2,4-дихлор-5-трифторметилпиримидин (1,0 г) при 0°С. К полученной реакционной смеси по каплям добавляли диизопропилэтиламин (DIPEA) (0,96 мл) при 0°С, и реакционную смесь перемешивали в течение 1 ч при температуре от 0°С до 5°С. После этого реакционную смесь оставляли для нагревания до комнатной температуры. Реакционную смесь перемешивали еще в течение 4 ч при комнатной температуре. Завершение реакции контролировали посредством тонкослойной хроматографии (ТСХ) с использованием смеси гексан : этилацетат (7:3). Осажденное твердое вещество отофильтровывали и промывали 1-бутанолом (2 мл). Полученное твердое вещество сушили при пониженном давлении при 40°С в течение 1 ч. 1Н-ЯМР (ДМСО-d6, 400 МГц) δ 1,48 (S, 9 Н), 7,02 (m, 1 Н), 7,26 (m, 2 Н), 7,58 (S, 1 Н), 8,57 (S, 1 Н), 9,48 (S, 1 Н), 9,55 (S, 1 Н).
Стадия 2:
[00182] К указанному выше неочищенному твердому веществу (3,1 г) в дихлорметане (ДХМ) (25 мл) медленно добавляли трифторуксусную кислоту (ТФУ) (12,4 мл) при 0°С. Реакционной смеси позволяли нагреться до комнатной температуры. Реакционную смесь перемешивали еще в течение 10 мин при комнатной температуре. Неочищенное вещество концентрировали при пониженном давлении.
Стадия 3:
[00183] Концентрированное неочищенное вещество растворяли в DIPEA (2,0 мл) и дихлорметане (25 мл) и далее охлаждали до -30°С. К реакционной смеси медленно добавляли акрилоилхлорид (0,76 г) при -30°С. Реакционную массу нагревали до комнатной температуры в течение 1,0 ч. Ход реакции контролировали посредством ТСХ с применением смеси гексан : этил ацетат (7:3) в качестве подвижной фазы. Реакция была завершена через 1 ч. На стадии 3 получали промежуточное соединение 1.
Стадия 4:
[00184] Для получения соли Соединения 1 смесь промежуточного соединения 1 (16 мг) и 2-метокси-4-(4-ацетилпиперазинил)анилина в диоксане (1,0 мл) с каталитическим количеством трифторуксусной кислоты перемешивали в течение ночи при 50°С. Полученное неочищенное вещество концентрировали при пониженном давлении и очищали посредством ВЭЖХ (ТФУ-модификатор) с получением соединения 1 в виде соли ТФУ. 1Н-ЯМР (ДМСО-d6, 400 МГц) δ 10,2 (S, 1 Н), 8,2 (ушир., 1 Н), 8,30 (S, 1 Н), 7,73 (ушир., 1 Н), 7,52 (d, J=7,8 Гц, 1 Н), 7,45 (d, J=7,8 Гц, 1 Н), 7,26 (J=8,2 Гц, 1 Н), 7,14 (be, 1 Н), 6,60 (S, 1 Н), 6,42 (dd, J=11,4, 16,9 Гц, 1 Н), 6,24 (d, J=16,9 Гц, 1 Н), 5,75 (d, J=11,4 Гц, 1 Н), 3,76 (S, 3 Н), 3,04 (ушир., 4 Н), 2,04 (S, 3 Н); расчетная масса для C27H28F3N7O3: 555,2, обнаружено: 556,2 (М+Н+).
Стадия 5:
[00185] Для получения формы свободного основания Соединения 1 из соли ТФУ данную соль добавляли к ДХМ и охлаждали до 0°С. Добавляли раствор Na2CO3 (9,6% масс./масс.) при 0°С. Смесь нагревали до 20°С и перемешивали в течение 35 мин pH водного слоя был >8. Слои разделяли. Водный слой подвергали экстракции с применением ДХМ. Органические слои объединяли и промывали солевым раствором. Органический слой собирали и упаривали с получением твердого Соединения 1.
Общие методики
[00186] Анализ посредством рентгеновской порошковой дифракции (XRPD) проводили на приборе Siemens D5000, сканируя образцы при значениях °2-тета от 3 до 30 или 50 °2-тета. Для образцов <100 мг, приблизительно 5-10 мг образца аккуратно прессовали на предметном стекле, которое вставляется в держатель образца. Для образцов >100 мг приблизительно 100 мг образца аккуратно прессовали на пластиковом держателе образца таким образом, что поверхность образца была гладкой и была немного выше уровня держателя образца. Измерения проводили следующим образом:
[00187] В поляризационной микроскопии (микроскопии в поляризованном свете МПС), наличие кристалличности (двулучепреломления) определяли с помощью поляризационного микроскопа Olympus ВХ50, оснащенного камерой Motic и программным обеспечением для съемки (Motic Images Plus 2.0). Все изображения были получены с помощью объектива 20х цели, если не указано иное.
[00188] Для термогравиметрического анализа (ТГА) приблизительно 5-10 мг вещества точно взвешивали в открытом алюминиевом тигле и загружали в одновременно термогравиметрический/дифференциальный термический анализатор (ТГ/ДТА) и выдерживали при комнатной температуре. Затем образец нагревали со скоростью 10°С/мин от 25°С до 300°С, и в течение этого времени снимали показания изменения массы образца, а также любых дифференциальных термических изменений (ДТА). В качестве продувочного газа использовали азот с расходом 100 см3/мин.
[00189] Для дифференциальной сканирующей калориметрии (ДСК) приблизительно 5-10 мг вещества взвешивали в алюминиевом тигле для ДСК и негерметично закрывали перфорированной алюминиевой крышкой. Тигель с образцом затем загружали в прибор Seiko DSC6200 (оснащенный охладителем), охлаждали и выдерживали при 25°С. После того, как был получен ответ на тепловой поток, исследуемый и эталонный образцы нагревали приблизительно до 260°С-280°С при скорости сканирования 10°С/мин и контролировали полученные значения теплового потока.
[00190] Эксперименты 1Н-ЯМР проводили на приборе Bruker AV400 (частота 1Н: 400 МГц). Эксперименты с 1Н для каждого образца проводили в дейтерированном ДМСО, и каждый образец готовили в концентрации приблизительно 10 мг/мл.
[00191] Для динамической сорбции пара (ДСП), приблизительно 10 мг образца помещали в тигель из проволочной сетки для баланса сорбции пара и помещали в баланс динамической сорбции пара ДСП-1 компании Surface Measurment Systems. Образец подвергали воздействию профиля повышения относительной влажности (ОВ) от 20 до 90% (RH) с шагом 10%, сохраняя образец при каждой стадии до достижения стабильной массы (99,5% от массы по окончании предыдущей стадии). После завершения цикла сорбции, образец сушили по той же методике, но снижали ОВ до 0% и, наконец, возвращали обратно к исходной точке ОВ 20%. Изменение массы во время циклов сорбции/десорбции наносили на график, что позволяло определить гигроскопические свойства образца.
[00192] Анализ методом инфракрасной спектроскопии (ИК) проводили на спектрометре Bruker ALPHA Р. Достаточное количество вещества помещали в центр пластины спектрометра, и снимали спектры при следующих параметрах: разрешение - 4 см-1, время сканирования фона - 16 сканирований, время сканирования образца - 16 сканирований, сбор данных от 4000 до 400 см-1, полученный спектр - пропускание.
[00193] Для кулонометрического титрования методом Карла Фишера (КФ) 10-15 мг твердого продукта точно взвешивали во флаконе. Твердое вещество затем вручную вводили в ячейку для титрования компактного титратора Mettler Toledo С30. Флакон снова взвешивали после добавления твердого вещества и вводили в прибор массу добавленного твердого вещества. Титрование начинали после того, как образец в ячейке был полностью растворен. Содержание воды прибор рассчитывал автоматически в процентах, и полученные данные распечатывали.
[00194] Анализ методом обращенно-фазовой градиентной высокоэффективной жидкостной хроматографии (ВЭЖХ) проводили на приборе Agilent 1100, снабженном колонкой С18, 3,0 × 100 мм × 3,5 мкм. Определение проводили на длине волны 240 нм.
[00195] Для исследования растворения использовали баню для растворения Sotax АТ7 (устройство согласно 2й Фармакопее США (USP 2), 2й Европейской Фармакопее (ЕР 2)) с лопастями для перемешивания среды. Все исследования проводили при 37°С и скорости мешалки 100 об/мин.
[00196] Образцы каждой формы подвергали воздействию условий 40°С/ОВ75% в течение периодов, составляющих 1 и 2 недели, для определения стабильности. Полученные твердые вещества анализировали посредством XRPD и ВЭЖХ для установления того, произошли ли какие-либо изменения.
[00197] Суспензии каждой полиморфной формы готовили в деионизованной воде и перемешивали встряхиванием в течение приблизительно 24 часов. Далее полученное твердое вещество анализировали посредством ВЭЖХ для определения концентрации растворенного вещества.
Пример 1
Получение Формы А
[00198] Приблизительно 120 мг Соединения 1 взвешивали во флаконе и суспендировали приблизительно в 2 мл ацетонитрила. Полученную суспензию подвергали циклическому изменению температуры от приблизительно 0°С до температуры окружающей среды (приблизительно 22°С) при перемешивании в циклах по 2 часа в течение периода, составляющего 2-3 суток. В течение ночи образец выдерживали при температуре приблизительно 2-5°С. Твердое вещество выделяли и оставляли сушиться под вакуумом в течение 7 суток.
[00199] Анализ посредством XRPD (фигура 1) показал, что вещество является кристаллическим. Анализ посредством МПС (не показано) показал наличие очень мелких игольчатых кристаллов с двойным лучепреломлением. ТГА/ДТА (фигура 2) показал потерю массы 0,4% от конечной температуры до приблизительно 150°С, вероятно имеющую место из-за несвязанного растворителя. Значительных потерь массы до разложения не наблюдалось. Анализ посредством ДСК (фигура 3) показал одну эндотерму с началом приблизительно при 203,2°С (пик при 207,5°С), обусловленную плавлением. Результаты ИК-анализа (фигура 4) соответствуют результатам для исходного свободного основания. В результате анализа посредством 1Н ЯМР (не показано), проведенного в дейтерированном ДМСО, был получен спектр, который соответствовал спектру исходного свободного основания. Как представляется, ацетонитрил не присутствует. Анализ посредством ДСП (фигура 5) показал, что поглощение воды составило 0,87% при ОВ от 20 до 70%, что означает, что вещество не является гигроскопичным. Анализ посредством XRPD после ДСП показал, что вещество оставалось в Форме А (данные не показаны). Изменений полиморфной формы не наблюдалось. Наблюдалась некоторая потеря кристалличности. Анализ методом КФ не показал присутствие воды. Анализ чистоты посредством ВЭЖХ показал, что чистота вещества составила приблизительно 97,6%. Растворимость Формы А в воде нельзя было установить с помощью анализа посредством ВЭЖХ. Поэтому ее растворимость в воде считается низкой.
[00200] Анализ посредством XRPD после выдерживания в течение 1 недели (открытая емкость) при 40°С/ОВ75% показал, что вещество все еще соответствует Форме А при некоторой потере кристалличности. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 97,5%. Анализ посредством XRPD после выдерживания в течение 2 недель (открытая емкость) при 40°С/ОВ75% показал, что вещество все еще соответствует Форме А, однако, оно стало низкокристалличным. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 96,3%.
Пример 2
Получение Формы В
[00201] Приблизительно 120 мг Соединения 1 взвешивали во флаконе и суспендировали приблизительно в 2 мл тетрагидрофурана. Полученную суспензию подвергали циклическому изменению температуры от приблизительно 0°С до температуры окружающей среды (приблизительно 22°С) при перемешивании в циклах по 2 часа в течение периода, составляющего 2-3 суток. В течение ночи образец выдерживали при температуре приблизительно 2-5°С. Твердое вещество выделяли и оставляли сушиться под вакуумом при температуре окружающей среды в течение 7 суток и при 40°С еще в течение 2 суток.
[00202] Анализ посредством XRPD (фигура 6) показал, что полученное вещество является кристаллическим. Анализ посредством МПС (не показано) показал наличие палочковидных кристаллов с двойным лучепреломлением. ТГА/ДТА (фигура 7) показал, отсутствие значительных потерь массы до разложения после сушки в течение 7 суток при температуре окружающей среды под вакуумом и еще 2 суток при 40°С. Анализ посредством ДСК (фигура 8) показал эндотерму с началом при 153,6°С (пик при 157,6°), после которой сразу же следовала экзотерма с пиком 161,3°С что означает полиморфное превращение. Также присутствовала другая небольшая эндотерма с пиком 186,0°С, после чего следовала последняя эндотерма с началом при 203,9°С (пик при 207,9°С), которая, как представляется, соответствует расплавленной Форме А. Результаты ИК-анализа (фигура 9) показали значительное количество различий и сдвигов по сравнению с Формой А. В результате анализа посредством 1Н ЯМР (не показано), проведенного в дейтерированном ДМСО, был получен спектр, который соответствовал спектру исходного свободного основания. Как представляется, присутствуют следы ТГФ. Анализ посредством ДСП (фигура 10) показал, что поглощение воды составило 0,74% при ОВ от 20 до 70%, что означает, что вещество не является гигроскопичным. Анализ посредством XRPD после ДСП (не показано) показал, что вещество оставалось в Форме В. Изменений в полиморфной форме не наблюдалось. Анализ методом КФ (не показано) показал присутствие приблизительно 0,97% воды. Анализ чистоты посредством ВЭЖХ показал, что чистота составила приблизительно 97,0%. Растворимость Формы В в воде нельзя было установить с помощью анализа посредством ВЭЖХ. Поэтому растворимость в воде у данного вещества считается низкой.
[00203] Анализ посредством XRPD после выдерживания в течение 1 недели (открытая емкость) при 40°С/ОВ75% показал, что вещество является преимущественно аморфным. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 96,8%. Анализ посредством XRPD после выдерживания в течение 2 недель (открытая емкость) при 40°С/ОВ75% показал, что вещество является преимущественно аморфным. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 95,9%.
[00204] Чтобы определить взаимосвязь между Формами А и В, образец Формы В нагревали до 120°С, и затем проводили анализ посредством XRPD. XRPD показала присутствие смеси Формы В и Формы А, что означает, что при нагревании Формы В она начинает превращаться в Форму А. Форму В также нагревали до 160°С (температура выше температуры фазового перехода, наблюдаемой согласно ДСК), и далее проводили анализ посредством XRPD, который показал, что при данной температуре вещество преимущественно превращалось в Форму А. Сравнительные результаты XRPD показаны на фигуре 11.
Пример 3
Получение Формы С
[00205] Приблизительно 120 мг Соединения 1 взвешивали во флаконе и суспендировали приблизительно в 100 мкл ДМФА. Полученную суспензию подвергали циклическому изменению температуры от приблизительно 0°С до температуры окружающей среды (приблизительно 22°С) при перемешивании в циклах по 2 часа. Приблизительно через 2 часа добавляли еще 300 мкл ДМФА. Циклическое изменение температуры продолжали в течение периода, составляющего 2-3 суток. В течение ночи образец выдерживали при температуре приблизительно 2-5°С. Твердое вещество выделяли и оставляли сушиться под вакуумом при температуре окружающей среды в течение 7 суток и при 40°С еще в течение 2 суток.
[00206] Анализ посредством XRPD (фигура 12) показал, что вещество является кристаллическим. Анализ посредством МПС (не показано) показал наличие палочковидных кристаллов с двойным лучепреломлением. ТГА/ДТА (фигура 13) показал потерю массы приблизительно 8,2% (11,6 масс % ДМФА требовалось для моносольвата) после сушки в течение 7 суток при температуре окружающей среды под вакуумом и еще 2 суток при 40°С. Анализ посредством ДСК (фигура 14) показал широкую эндотерму при температуре от 85 до 125°С, соответствующую потере массы в ТГА. Начало конечной эндотермы наблюдали приблизительно при 204,4°С (пик при 208,3°С), что соответствовало расплавленной Форме А. Результаты ИК-анализа (фигура 15) показали некоторые различия и сдвиги по сравнению с Формой А. В результате анализа посредством 1Н ЯМР (не показано), проведенного в дейтерированном ДМСО, был получен спектр, который соответствует спектру исходного свободного основания с присутствием некоторого количества ДМФА (приблизительно 3:1 АФИ : ДМФА). Результаты анализа посредством ДСП (фигура 16) соответствовали результатам ТГА, которые показали, что вещество содержит растворитель, который теряется при повышении относительной влажности. Анализ посредством XRPD после ДСП (не показано) показал, что вещество в процессе анализа посредством ДСП превратилось в Форму А. Анализ методом КФ (не показано) показал присутствие приблизительно 0,01% воды. Анализ чистоты посредством ВЭЖХ показал, что чистота вещества составила приблизительно 97,3%. Растворимость Формы С в воде нельзя было установить с помощью анализа посредством ВЭЖХ. Поэтому ее растворимость в воде считается низкой.
[00207] Анализ посредством XRPD после выдерживания в течение 1 недели (открытая емкость) при 40°С/ОВ75% показал, что вещество превратилось в Форму А с некоторой потерей кристалличности. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 97,1%. Анализ посредством XRPD после выдерживания в течение 2 недель (открытая емкость) при 40°С/ОВ75% показал, что вещество имеет низкую кристалличность с присутствием пиков, соответствующих Форме А. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 96,8%). Сравнительные результаты XRPD показаны на фигуре 17.
[00208] Для определения того, сохраняется ли Форма С после потери присутствующего в ней растворителя, образец Формы С нагревали до 120°С (то есть до температуры, чуть большей, чем температура при которой удаляют растворитель) и затем проводили анализ посредством XRPD. XRPD показала, что вещество превратилось в Форму А. Аналогичным образом, после сушки при температуре окружающей среды в вакууме в течение 7 суток и затем еще в течение 2 суток при 40°С, для Формы С наблюдалась некоторая потеря кристалличности, и данная форма превращалась в Форму А. Сравнительные результаты XRPD показаны на фигуре 18.
Пример 4
Получение Формы D
[00209] Приблизительно 120 мг Соединения 1 взвешивали во флаконе и суспендировали приблизительно в 2 мл 1,4-диоксана. Полученную суспензию подвергали циклическому изменению температуры от приблизительно 0°С до температуры окружающей среды (приблизительно 22°С) при перемешивании в циклах по 2 часа в течение периода, составляющего 2-3 суток. В течение ночи образец выдерживали при температуре приблизительно 2-5°С. Твердое вещество выделяли и оставляли сушиться под вакуумом при температуре окружающей среды в течение 7 суток и при 40°С еще в течение 2 суток.
[00210] Анализ посредством XRPD показал, что при указанном выше получении Формы D была получена смесь Формы В и Форма D (фигура 19). Анализ посредством МПС показал наличие палочковидных кристаллов с двойным лучепреломлением (не показано). ТГА/ДТА (фигура 20А) показал потерю массы приблизительно 5,5% при температуре от 60 до 100°С после сушки в течение 7 суток при температуре окружающей среды под вакуумом. После сушки в течение 7 суток при температуре окружающей среды под вакуумом и еще 2 суток при 40°С ТГА (фигура 20В) показал потерю массы приблизительно 1,4% при температуре от 100 до 150°С. Анализ посредством ДСК после сушки при 40°С (фигура 21) показал очень маленькую экзотерму приблизительно при 146°С. Далее наблюдали конечную эндотерму с началом приблизительно при 202,6°С (пик при 207,4°С), соответствующую расплавленной Форме А. Результаты ИК-анализа (фигура 22) соответствуют спектру Формы В с присутствием небольших пиков 1,4-диоксана. В результате анализа посредством 1Н-ЯМР (не показано), проведенного в дейтерированном ДМСО после 7 суток сушки при температуре окружающей среды под вакуумом, был получен спектр, который соответствует спектру исходного свободного основания с присутствием небольшого количества 1,4-диоксана (приблизительно 2:1 АФИ : 1,4-диоксан). Результаты анализа посредством ДСП (фигура 23) соответствовали результатам ТГА, которые показали, что вещество содержит растворитель, который теряется при повышении относительной влажности. Анализ посредством XRPD после ДСП (не показано) показал, что в процессе анализа посредством ДСП вещество полностью превратилось в Форму В. Анализ методом КФ (не показано) показал присутствие приблизительно 1,1% воды. Анализ чистоты посредством ВЭЖХ показал, что чистота составила приблизительно 96,6%. Растворимость Формы D в воде нельзя было установить с помощью анализа посредством ВЭЖХ. Поэтому ее растворимость в воде считается крайне низкой.
[00211] Анализ посредством XRPD после выдерживания в течение 1 недели (открытая емкость) при 40°С/ОВ75% показал, что вещество полностью превратилось в Форму В с некоторой потерей кристалличности. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 96,5%. Анализ посредством XRPD после выдерживания в течение 2 недель (открытая емкость) при 40°С/ОВ75% показал, что вещество имеет низкую кристалличность с присутствием пиков, соответствующих Форме В. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 95,5%. Сравнительные результаты XRPD показаны на фигуре 24.
Пример 5
Получение Формы Е
[00212] Приблизительно 120 мг Соединения 1 взвешивали во флаконе и затем в попытках растворить данное вещество добавляли приблизительно 12 мл МЭК. Получилась суспензия с очень мелкими частицами, и данную суспензию затем фильтровали с получением насыщенного раствора. Раствор помещали в условия с температурой приблизительно -18°С для резкого охлаждения в течение 2-3 суток. Твердое вещество выделяли и оставляли сушиться под вакуумом при температуре окружающей среды в течение 7 суток и при 40°С еще в течение 2 суток.
[00213] Анализ посредством XRPD (фигура 25) показал, что вещество является кристаллическим. Анализ посредством МПС (не показано) показал наличие мелких палочковидных кристаллов с двойным лучепреломлением. ТГА/ДТА (фигура 26) показал потерю массы приблизительно 5,2%) при температуре приблизительно от 70 до 110°С после сушки в течение 7 суток при температуре окружающей среды под вакуумом. Анализ посредством ДСК (фигура 27) показал широкую эндотерму при температуре от 70 до 110°С, соответствующую потере массы в ТГА. Начало конечной эндотермы наблюдали приблизительно при 198,4°С (пик при 203,3°С). Результаты ИК-анализа (фигура 28) показали некоторые небольшие сдвиги по сравнению с Формой А. В результате анализа посредством 1Н-ЯМР (не показано), проведенного в дейтерированном ДМСО после 7 суток сушки при температуре окружающей среды под вакуумом, был получен спектр, который соответствует исходному свободному основанию с присутствием нестехиометрического количества МЭК. Анализ посредством ДСП (фигура 29) показал, что поглощение воды составило 0,25% при ОВ от 20 до 70%. Анализ посредством XRPD после ДСП (не показано) показал, что в процессе анализ посредством ДСП вещество превратилось в Форму А. Анализ методом КФ (не показано) показал присутствие приблизительно 1,2% воды. Анализ чистоты посредством ВЭЖХ показал, что чистота составила приблизительно 97,8%. Растворимость Формы Е в воде нельзя было установить с помощью анализа посредством ВЭЖХ. Поэтому ее растворимость в воде считается крайне низкой.
[00214] Анализ посредством XRPD после выдерживания в течение 1 недели (открытая емкость) при 40°С/ОВ75% показал, что вещество является преимущественно аморфным с присутствием видимых пиков, соответствующих Форме А. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 97,7%. Анализ посредством XRPD после выдерживания в течение 2 недель (открытая емкость) при 40°С/ОВ75% показал, что вещество является преимущественно аморфным с присутствием видимых пиков, соответствующих Форме А. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 97,3%. Сравнительные результаты XRPD показаны на фигуре 30.
[00215] Для того чтобы определить, сохраняется ли Форма Е после потери присутствующего растворителя, образец Формы Е нагревали до 120°С (то есть до температуры, чуть большей, чем температура при которой удаляют растворитель) и затем проводили анализ посредством XRPD. XRPD показала, что вещество превратилось в Форму А. Аналогичным образом, после сушки при температуре окружающей среды под вакуумом в течение 7 суток и затем еще в течение 2 суток при 40°С, для Формы Е наблюдалась некоторая потеря кристалличности, и данная форма превращалась в Форма А. Сравнительные результаты XRPD показаны на фигуре 31.
Пример 6
Получение Формы F
[00216] Приблизительно 120 мг Соединения 1 взвешивали во флаконе и затем для растворения данного вещества добавляли приблизительно 200 мкл NMP. Далее добавляли антирастворитель путем добавления МТБЭ, общее количество которого составило приблизительно 4,5 мл, в аликвотах по 500 мкл с получением мутного раствора. Далее образец выдерживали в течение приблизительно 2 часов, после чего выделяли полученное твердое вещество. Далее твердое вещество оставляли сушиться под вакуумом при температуре окружающей среду в течение 7 суток и при 40°С еще в течение 2 суток.
[00217] Анализ посредством XRPD (фигура 32) показал, что вещество является кристаллическим. Анализ посредством МПС (не показано) показал наличие очень мелких пластинчатых кристаллов с двойным лучепреломлением, которые, как оказалось, растут в кластерах. ТГА/ДТА (фигура 33) показал потерю массы приблизительно 10,2% при температуре от 70 до 120°С (15,1 масс % NMP требовалось для моносольвата) после сушки в течение 7 суток при температуре окружающей среды под вакуумом. Анализ посредством ДСК (фигура 34) показал эндотерму с температурой начала 97°С (пик при 100,9°С), соответствующую потере массы в ТГА. Наблюдали конечную эндотерму с началом приблизительно при 204,2°С (пик при 208,3°С), соответствующую расплавленной Форме А. Результаты ИК-анализа (фигура 35) показали некоторые небольшие сдвиги по сравнению с Формой А. В результате анализа посредством 1Н-ЯМР (не показано), проведенного в дейтерированном ДМСО через 7 суток сушки при температуре окружающей среды под вакуумом, был получен спектр, который соответствует спектру исходного свободного основания с присутствием нестехиометрического количества NMP. Результаты анализа посредством ДСП (фигура 36) соответствовал результатам ТГА, которые показали, что вещество содержит растворитель, который теряется при повышении относительной влажности. Анализ посредством XRPD после ДСП (не показано) показал, что вещество преимущественно превратилось в Форму А (остались следы Формы F). Анализ методом КФ (не показано) показал присутствие приблизительно 0,87% воды. Анализ чистоты посредством ВЭЖХ показал, что чистота составила приблизительно 97,0%. Форма F в воде нельзя было установить с помощью анализа посредством ВЭЖХ. Поэтому ее растворимость в воде считается низкой.
[00218] Анализ посредством XRPD после выдерживания в течение 1 недели (открытая емкость) при 40°С/ОВ75% показал, что вещество превратилось в Форму А с некоторой потерей кристалличности. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 96,8%. Анализ посредством XRPD после выдерживания в течение 2 недель (открытая емкость) при 40°С/ОВ75% показал, что вещество является частично кристаллическим с присутствием пиков, соответствующих Форме А. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 96,3%. Сравнительные результаты XRPD показаны на фигуре 37.
[00219] Для определения того, сохраняется ли Форма F после потери присутствующего в ней растворителя, образец Формы F нагревали до 120°С (то есть до температуры, чуть большей, чем температура при которой удаляют растворитель) и затем проводили анализ посредством XRPD. XRPD показала, что вещество преимущественно превратилось в Форму А. Аналогичным образом, после сушки при температуре окружающей среды под вакуумом в течение 7 суток и затем еще в течение 2 суток при 40°С, для Формы F наблюдалась некоторая потеря кристалличности, и данная форма преимущественно превращалась в Форму А. Сравнительные результаты XRPD показаны на фигуре 38.
Пример 7
Получение Формы G
[00220] Приблизительно 120 мг Соединения 1 взвешивали во флаконе и суспендировали приблизительно в 100 мкл NMP. Полученную суспензию подвергали циклическому изменению температуры от приблизительно 0°С до температуры окружающей среды (приблизительно 22°С) при перемешивании в циклах по 2 часа в течение периода, составляющего 2-3 суток. В течение ночи образец выдерживали при температуре приблизительно 2-5°С. Твердое вещество выделяли и оставляли сушиться под вакуумом при температуре окружающей среды в течение 7 суток и при 40°С еще в течение 2 суток. Часть полученного твердого вещества также сушили при 80°С в течение приблизительно 4 суток.
[00221] Анализ посредством XRPD (фигура 39) показал, что вещество является кристаллическим. Анализ посредством МПС (не показано) показал наличие пластинчатых кристаллов с двойным лучепреломлением. ТГА/ДТА (фигура 40А) потери массы приблизительно 23,6% и 10,2% после сушки в течение 4 суток при температуре окружающей среды под вакуумом. После сушки в течение 7 суток при температуре окружающей среды под вакуумом и еще 2 суток при 40°С ТГА (фигура 40В) показал потерю массы приблизительно 6,3% при температуре приблизительно от 70 до 110°С. После сушки в течение 4 суток при 80°С, ТГА (фигура 40С) показал потерю массы приблизительно 2,8% при температуре приблизительно от 70 до 110°С. Анализ посредством ДСК после сушки при 80°С (фигура 41) показал эндотерму с температурой начала приблизительно 205,3°С (пик при 210,0°С). Результаты ИК-анализа после 4 суток сушки при температуре окружающей среды (фигура 42) показал некоторые сдвиги по сравнению с Формой А, а также присутствие NMP. Спектр, полученный в результате анализа посредством 1Н-ЯМР (не показано), проведенного в дейтерированном ДМСО через 4 суток сушки при температуре окружающей среды под вакуумом, соответствовал спектру исходного свободного основания с присутствием значительного количества NMP. Результаты анализа посредством ДСП (фигура 43) соответствовали результатам ТГА, которые показали, что значительные количества растворителя теряются при повышении относительной влажности. Анализ посредством XRPD после ДСП (не показано) показал, что вещество превратилось в Форму А. Анализ методом КФ (не показано) показал присутствие приблизительно 0,45% воды. Анализ чистоты посредством ВЭЖХ показал, что чистота составила приблизительно 97,0%. При определении растворимости Формы F в воде на хроматограмме ВЭЖХ Форма F находилась ниже предела количественного определения. Поэтому ее растворимость в воде считается низкой.
[00222] Анализ посредством XRPD после выдерживания в течение 1 недели (открытая емкость) при 40°С/ОВ75% показал, что вещество преимущественно превратилось в Форму А с некоторой потерей кристалличности. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 96,8%. Анализ посредством XRPD после выдерживания в течение 2 недель (открытая емкость) при 40°С/ОВ75% показал, что вещество преимущественно превратилось в Форму А с некоторой потерей кристалличности. Анализ посредством ВЭЖХ показал, что чистота вещества составила приблизительно 96,2%. Сравнительные результаты XRPD показаны на фигуре 44.
[00223] После сушки при температуре окружающей среды под вакуумом в течение 7 суток и затем еще в течение 2 суток при 40°С, Форма G превращалась в Форму А. После сушки при 80°С в течение 4 суток, Форма G превращалась в Форму А. Сравнительные результаты XRPD показаны на фигуре 45.
Пример 8
Исследования растворимости полиморфов
[00224] Результаты сравнения суспензий, полученные при температуре окружающей среды (приблизительно 22°С) и 60°С приведены в таблице ниже.
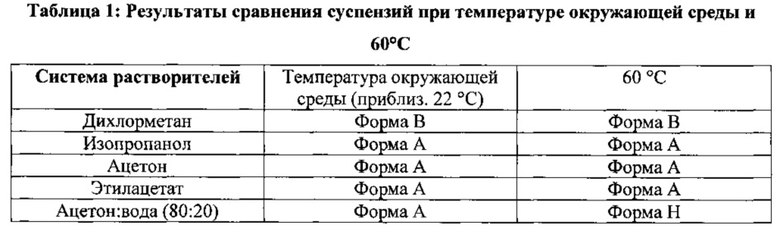
При суспендировании как при температуре окружающей среды, так и при 60°С, большинство экспериментов показали превращение в Форму А, что означает, что Форма А, вероятно, является более стабильной формой, чем Форма В. Форма В была получена соответственно в исследованиях с дихлорметаном и тетрагидрофураном. Новая полиморфная форма, обозначенная как Форма Н, была получена при сравнении суспензий из суспензии в смеси ацетон : вода (80:20) при 60°С.
Пример 9
Получение Формы Н
[00225] Приблизительно 10 мг Формы А и приблизительно 10 мг Формы В взвешивали во флаконе. Во флакон добавляли приблизительно 200 мкл смеси ацетон : вода (80:20%) с получением суспензии. Образец перемешивали приблизительно при 50°С в течение 3 суток. Полученное твердое вещество выделяли и оставляли сушиться при температуре окружающей среды, а затем подвергали анализу. Также проводили дополнительную сушку при 40°С в течение 2 суток.
[00226] Анализ посредством XRPD (фигура 46) показал, что вещество является кристаллическим, при этом дифрактограмма данного вещества отличалась от дифрактограмм всех идентифицированных полиморфных форм. Анализ посредством МПС (не показано) показал наличие кубовидных кристаллов с двойным лучепреломлением. Визуально вещество оказалось желтого цвета. ТГА/ДТА (фигура 47) через 2 суток сушки при 40°С показал потерю массы 4,4% от конечной температуры до приблизительно 120°С. Конечная эндотерма на графике ДТА, как представляется соответствует расплавленной Форме А (3,14 масс % требовалось для 1 моль-эквивалента воды).
[00227] Чтобы определить, изменяется ли Форма Н до Формы А после нагревания до 115°С (температура после удаления растворителя), вещество Формы Н нагревали до 115°С и затем проводили анализ посредством XRPD и анализ посредством ДСК. Дифрактограмма XRPD (фигура 48) после нагревания осталась соответствующей Форме Н с некоторой потерей кристалличности, вероятно из-за жестких условий нагревания. В результате анализа посредством ДСК (фигура 49) были получены перекрывающиеся изотермы при температуре приблизительно от 115 до 135°C с последующей экзотермой с пиком 143,9°С, вероятно означающей полиморфный переход. Присутствовала конечная эндотерма с началом при 202,7°С (пик при 206,6°С), соответствующая расплавленной Форме А.
[00228] Чтобы определить, поглощает ли Форма Н воду после десольватирования/дегидратации вещества при нагревании до 115°С, проводили дополнительное испытание, в котором Форму Н нагревали до 115°С в тигле для ТГА. Образец извлекали из тигля для ТГА и оставляли его приблизительно на 1 час. Через 1 час проводили дополнительный ТГА с нагреванием до 115°С. ТГА для образца, нагретого до 115°С, показал потерю массы присутствовавшего растворителя/воды приблизительно 4,2% (фигура 50). ТГА после выдерживания десольватированного/дегидратированного вещества в течение приблизительно 1 часа показал потерю массы приблизительно 3,7% при нагревании до 115°С (фигура 51). Таким образом, вещество поглощало воду при выдерживании в условиях окружающей среды. Это означает, что вещество либо является гигроскопичным, либо быстро возвращается в гидратированное состояние после дегидратации/десольватации.
Пример 10
Получение Формы I
[00229] Приблизительно к 1 г свободного основания Соединения 1 добавляли приблизительно 5 мл смеси ацетонитрил: вода (10%) с получением суспензии. В отдельном флаконе к 1 эквиваленту бромоводородной кислоты (48%) добавляли приблизительно 3 мл смеси ацетонитрил : вода (10%). Далее полученный раствор кислоты по каплям в течение 1 часа добавляли к суспензии свободного основания при перемешивании и при поддержании температуры от 0 до 5°С. После полного добавления кислоты добавляли еще 3 мл смеси ацетонитрил: вода (10%). Реакционную смесь перемешивали в течение приблизительно 1 дня, после чего полученное вещество выделяли и сушили под вакуумом при температуре окружающей среды (приблизительно 22°С). Выход составил приблизительно 79%.
[00230] Гидробромидную соль Соединения 1 измельчали на шаровой мельнице Retsch в течение приблизительно 25 минут с 5-минутным перерывом в середине для предотвращения перегрева образца.
[00231] Приблизительно 500 мг аморфной гидробромидной соли Соединения 1 суспендировали приблизительно в 18 мл смеси ацетон : вода (90:10). Полученную суспензию подвергали циклическому изменению температуры от 4 до 25°С в циклах по четыре часа в течение приблизительно 2 суток, после чего полученное вещество выделяли и сушили под вакуумом при температуре окружающей среды (приблизительно 22°С). Для Формы I после сушки проводили вторичный скрининговый анализ.
[00232] Проводили масштабирование получения Формы I для дальнейшего анализа. В процессе масштабирования наблюдали изменение цвета от желтого до светло-бежевого/кремового. Анализ посредством XRPD (фигура 52) показал, что вещество является кристаллическим, и его дифрактограмма преимущественно соответствует дифрактограмме Формы I, полученной в малом масштабе. Результаты ИК и 1Н ЯМР изображены на фигуре 53 и фигуре 54, соответственно. Анализ влажного образца посредством МПС показал наличие волокнистых игольчатых кристаллов с двойным лучепреломлением. Оказалось, что в результате сушки вещество потеряло его игольчатую морфологию, демонстрируя наличие мелких частиц с отсутствием четко определенной морфологии. Результаты анализа посредством высокотемпературной микроскопии показали, что температура плавления составляет приблизительно 135°С, и приблизительно при 180°С возникала некоторая рекристаллизация с последующим полным расплавлением при достижении температуры приблизительно 210°С. После сушки под вакуумом в течение приблизительно 72 часов ТГА/ДТА показал потерю массы 2,8% приблизительно от 80 до 120°С, соответствующую эндотерме на графике ДТА (фигура 55). Экзотерму наблюдали на графике ДТА с началом приблизительно при 149°С (пик приблизительно при 166°С) с последующей дальнейшей эндотермой с началом приблизительно при 197°С (пик приблизительно при 201°С). После выдерживания в условиях окружающей среды повторно проведенный ТГА/ДТА показал потерю массы 2,6% от конечной температуры до приблизительно 80°C с последующей дальнейшей потерей массы 2,8% при температуре приблизительно от 80°С до 120°С, соответствующей двум эндотермам на графике ДТА (фигура 56). Экзотерму наблюдали на графике ДТА с началом приблизительно при 155°С (пик приблизительно при 167°С) с последующей дальнейшей эндотермой с началом приблизительно при 196°С (пик приблизительно при 202°С). Анализ посредством ДСК показал наличие перекрывающихся эндотерм, начинающихся при начальной температуре, с последующей экзотермой с началом приблизительно при 138°С (пик приблизительно при 149°С) и последующей эндотермой с началом приблизительно при 92°С (пик приблизительно при 200°С) (фигура 57). В процессе анализа посредством ДСП (фигура 58) были сделаны следующие наблюдения:
- Цикл 1 - Сорбция при ОВ 20-90%
Образец постепенно поглощал приблизительно 0,66% от своей массы.
- Цикл 2 - Десорбция при ОВ 90-0%
При изменении ОВ от 90 до 10% масса образца постепенно уменьшалась приблизительно на 1,2%.
Резкая потеря массы, составляющая приблизительно 2,7%, наблюдалась при изменении ОВ от 10 до 0%.
- Цикл 3 - Сорбция при ОВ 0-20%
Поглощение влаги в количестве приблизительно 2,8% при изменении ОВ от 0 до 20%.
[00233] Исходное вещество, содержащее приблизительно 5,6% воды, как оказалось, является относительно негигроскопичным. При меньших значениях ОВ потеря воды составила приблизительно 1 эквивалент. Анализ посредством XRPD после ДСП показал, что вещество оставалось в Форме I (фигура 59). Изменений в полиморфной форме не наблюдалось. Анализ методом КФ показал присутствие приблизительно 5,4% воды. Анализ чистоты посредством ВЭЖХ показал, что чистота вещества составила приблизительно 99,66% (фигура 60). Ионная хроматография показала присутствие 1,64% бромида (приблизительно 12,57% требовалось для 1 эквивалента). Анализ посредством XRPD, проведенный для твердых веществ, оставшихся через 24 часа при термодинамическом исследовании растворимости, показал, что при pH 6,6, 4,5 и 3,0 вещество оставалось в Форме I (фигура 61). При pH 1, как оказалось, вещество представляет собой смесь Формы I и, возможно, гидрохлоридной (HCl) соли, полученной в процессе скрининга.
[00234] В процессе определения характеристик Формы I, было установлено, что данная форма представляет собой гидратированную версию свободного основания, а не форму бромидной соли. Результаты ТГА/ДТА и ДСП, как представляется, позволяют предположить, что данное вещество может представлять собой либо гигроскопичный моногидрат, либо форму дигидрата.
[00235] Исследования стабильности в течение 7 дней при 25°С, 80°С, 40°С/ОВ75% (закрытые и открытые условия). Для определения стабильности приблизительно по 15 мг Формы I помещали в отдельные флаконы и подвергали действию условий 25°С, 80°С и 40°С/ОВ75% (открытый и закрытый флаконы) в течение 1 недели. Полученные твердые вещества подвергали анализу посредством XRPD и ВЭЖХ для установления того, произошли ли какие-либо изменения. Результаты представлены в таблицах 2 и 3 и фигурах 62 и 63.


[00236] Термодинамические исследования растворимости. Суспензии Формы I готовили в средах с различным pH (pH 1; pH 3; pH 4,5 и pH 6,6) и перемешивали встряхиванием в течение приблизительно 24 часов. Через 24 часа суспензии фильтровали, и полученный раствор анализировали посредством ВЭЖХ для определения растворимости при различных уровнях pH. Для приготовления буферных растворов использовали KCl/HCl для получения pH 1 и комбинации цитрат/фосфат для pH 3, 4,5 и 6,6 (10 мМ). pH растворов также измеряли перед анализом посредством ВЭЖХ. Анализ посредством XRPD проводили для твердых веществ, оставшихся после перемешивания встряхиванием в течение 24 часов. Результаты представлены в таблице 4:

| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛИ ИНГИБИТОРА КИНАЗЫ РЕЦЕПТОРА ЭПИДЕРМАЛЬНОГО ФАКТОРА РОСТА | 2013 |
|
RU2711077C2 |
| ЦИТРАТНАЯ СОЛЬ 9Е-15-(2-ПИРРОЛИДИН-1-ИЛ-ЭТОКСИ)-7,12,25-ТРИОКСА-19,21,24-ТРИАЗАТЕТРАЦИКЛО[18.3.1.1(2,5).1(14,18)]ГЕКСАКОЗА-1(24),2,4,9,14,16,18(26),20,22-НОНАЕНА | 2010 |
|
RU2543721C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ { [1-ЦИАНО-5-(4-ХЛОРОФЕНОКСИ)-4-ГИДРОКСИИЗОХИНОЛИН-3-КАРБОНИЛ]-АМИНО} -УКСУСНОЙ КИСЛОТЫ | 2014 |
|
RU2666144C2 |
| ПОЛИМОРФЫ ДОЦЕТАКСЕЛА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2437875C2 |
| ТВЕРДЫЕ ФОРМЫ СОЕДИНЕНИЯ, МОДУЛИРУЮЩЕГО КИНАЗЫ | 2016 |
|
RU2730506C2 |
| НОВЫЕ ПОЛИМОРФЫ И СОЛИ | 2011 |
|
RU2573384C2 |
| СОЛЬ ИНГИБИТОРА LSD1 И ЕЁ ПОЛИМОРФНАЯ ФОРМА | 2019 |
|
RU2794977C2 |
| СПОСОБ ПОЛУЧЕНИЯ НОВЫХ КРИСТАЛЛИЧЕСКИХ ФОРМ-4(ЦИКЛОПРОПИЛМЕТОКСИ)-N-(3, 5-ДИХЛОР-1-ОКСИДОПИРИДИН-4-ИЛ)-5-МЕТОКСИПИРИДИН-2-КАРБОКСАМИДА И ЕГО КРИСТАЛЛИЧЕСКИЕ ФОРМЫ | 2013 |
|
RU2621894C2 |
| КРИСТАЛЛИЧЕСКАЯ ФОРМА 13-[(N-ТРЕТ-БУТОКСИКАРБОНИЛ)-2'-О-ГЕКСАНОИЛ-3-ФЕНИЛИЗОСЕРИНИЛ]-10-ДИАЦЕТИЛБАККАТИНА III | 2011 |
|
RU2546661C2 |
| Твёрдые формы цефтолозана | 2014 |
|
RU2703457C2 |
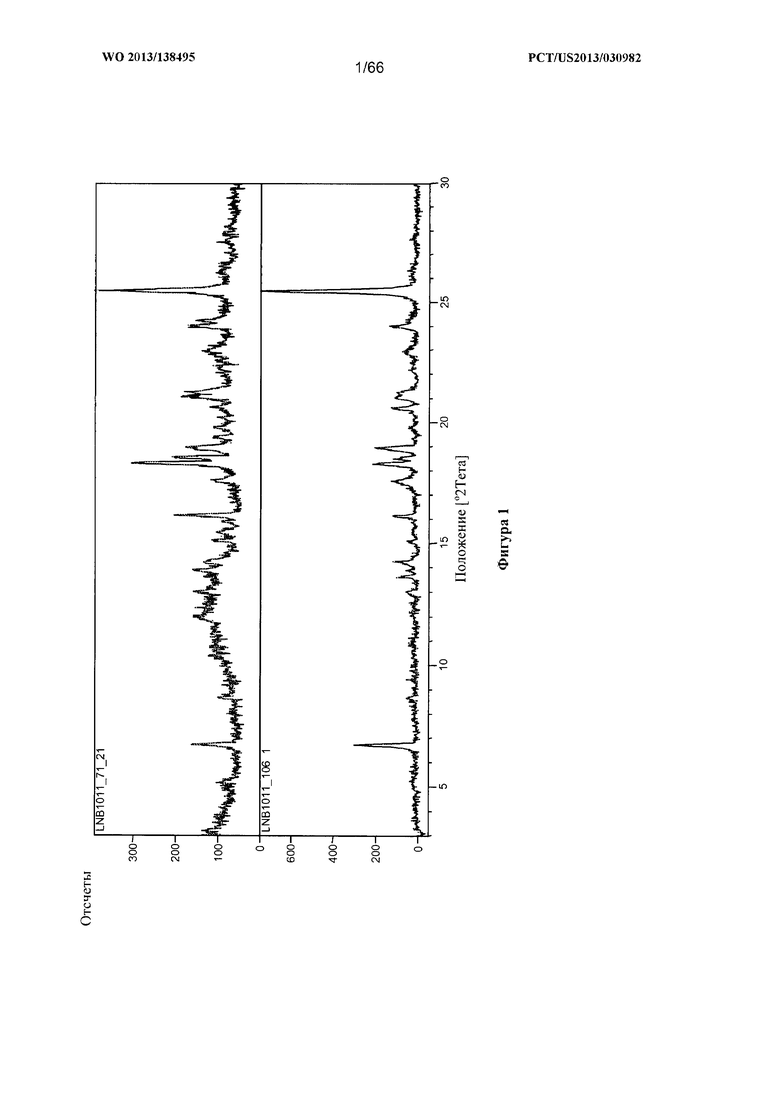
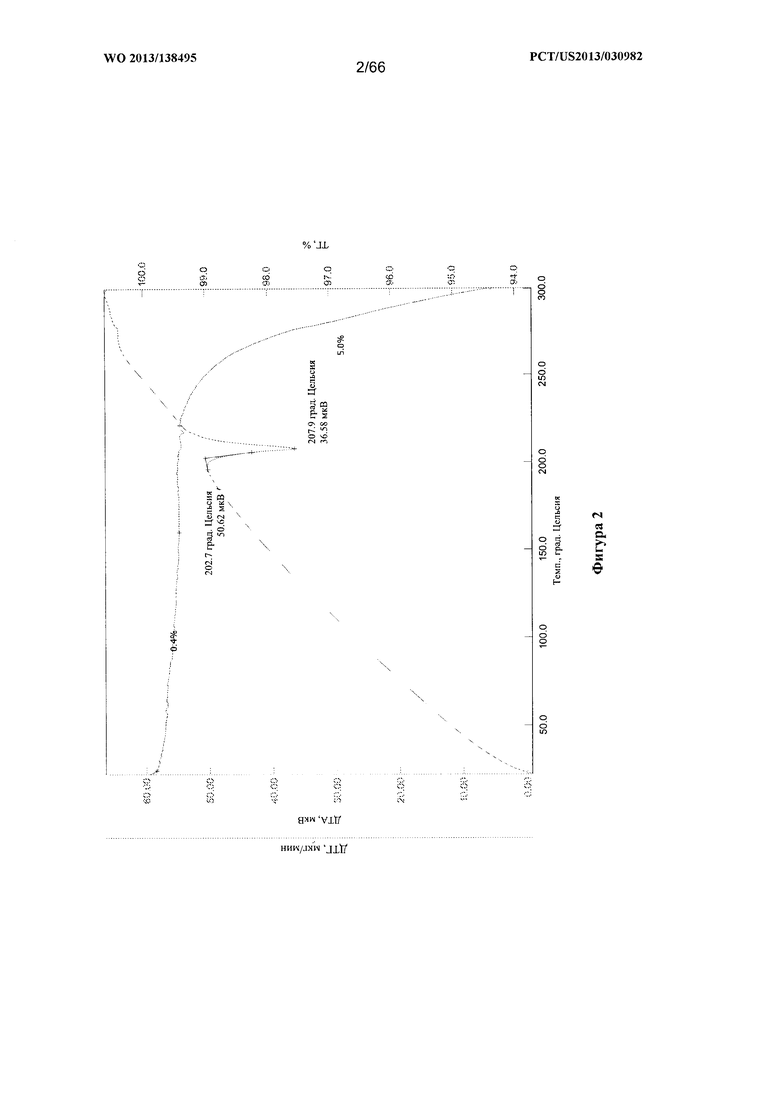
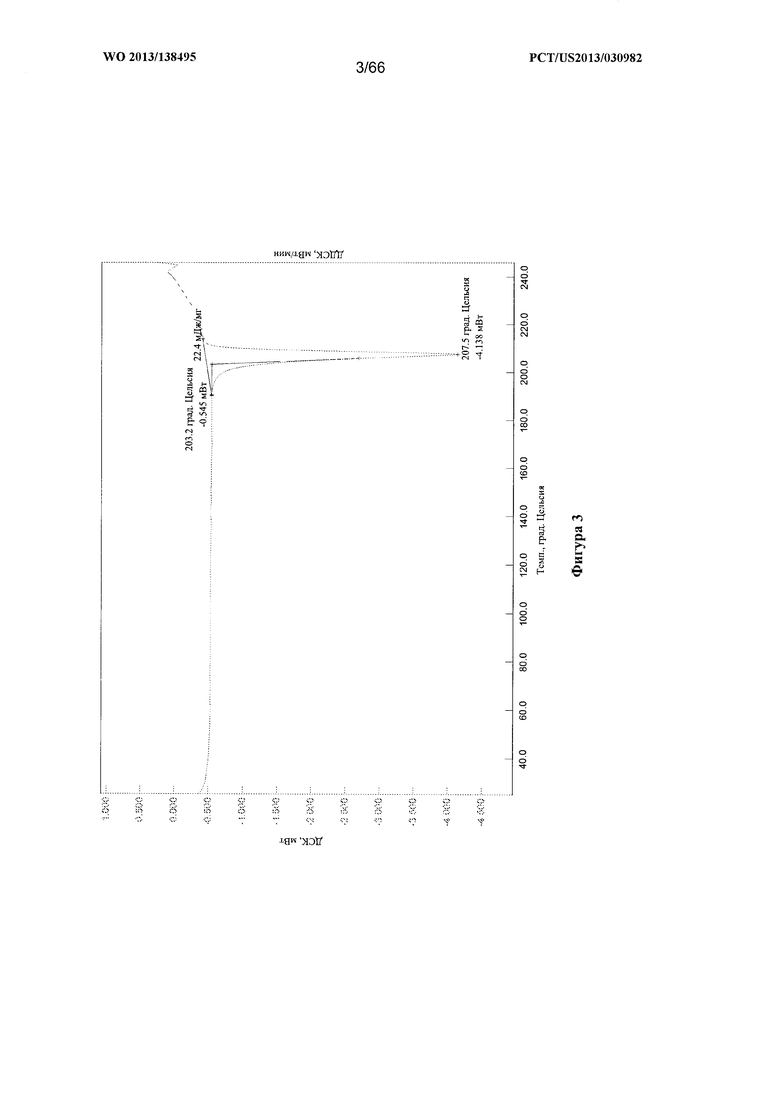
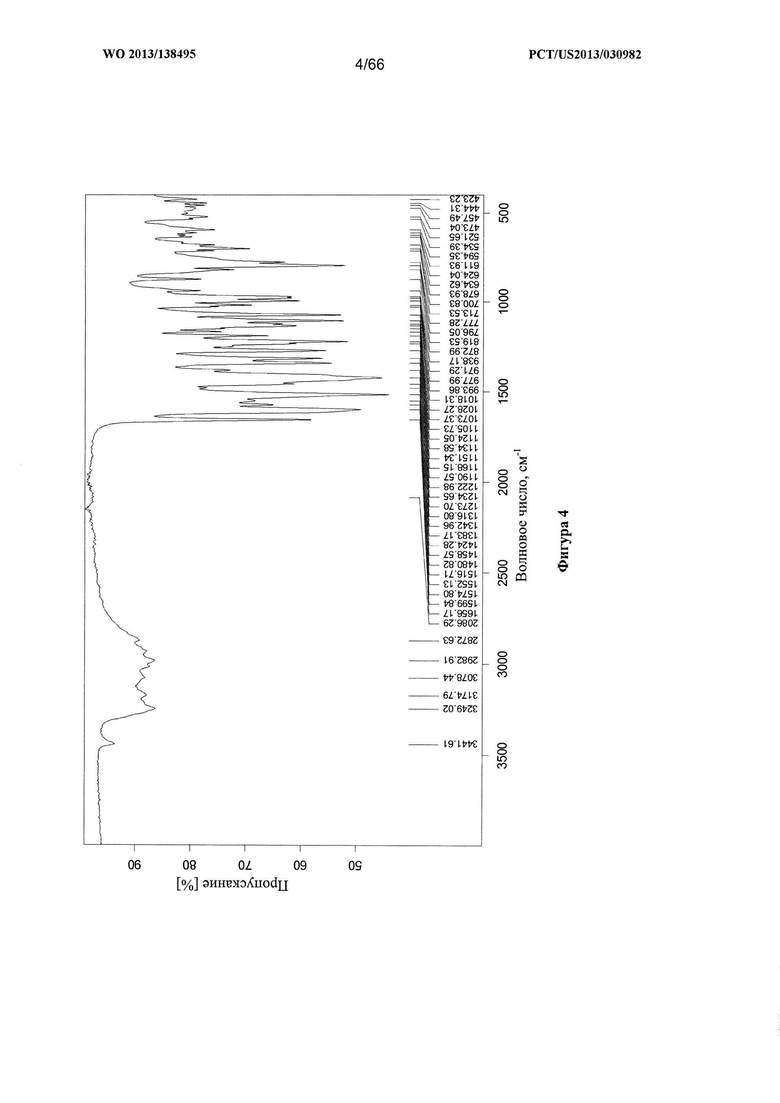
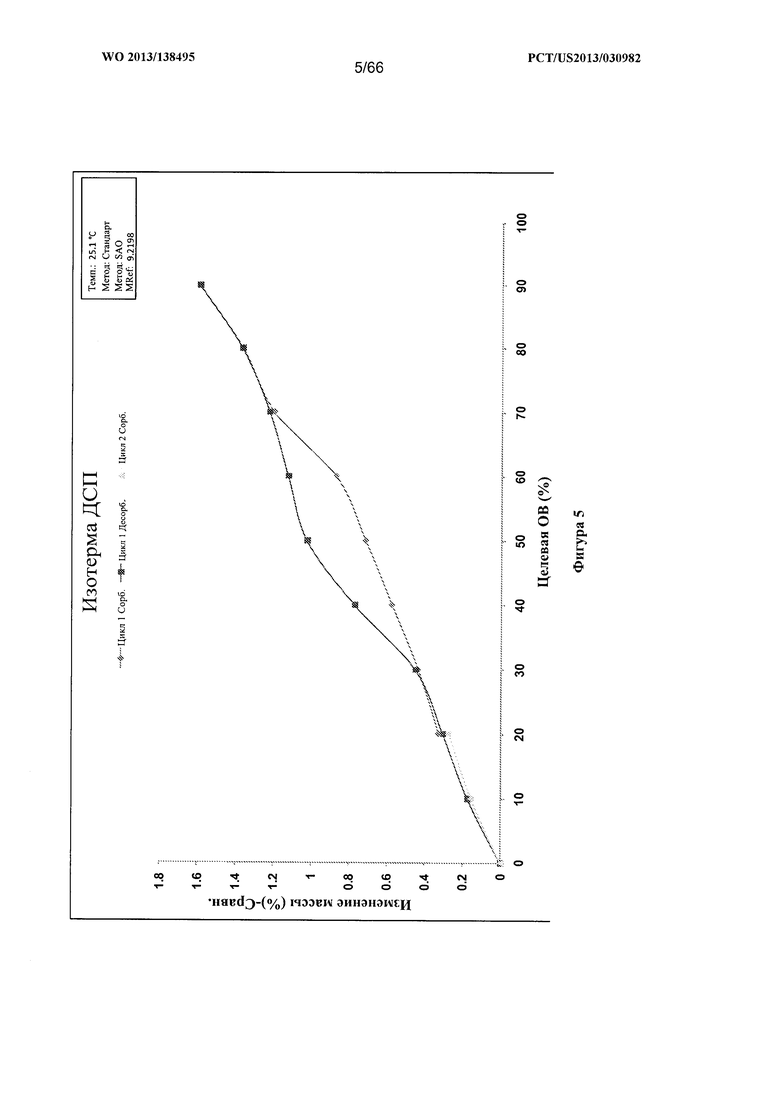
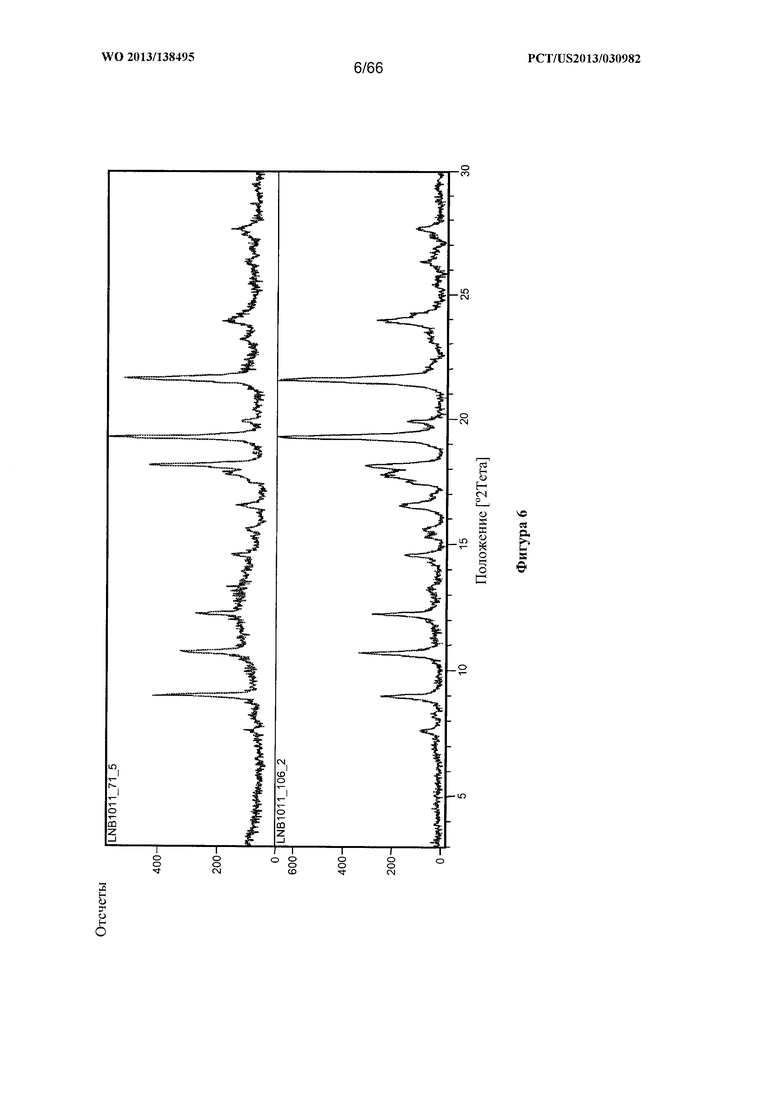
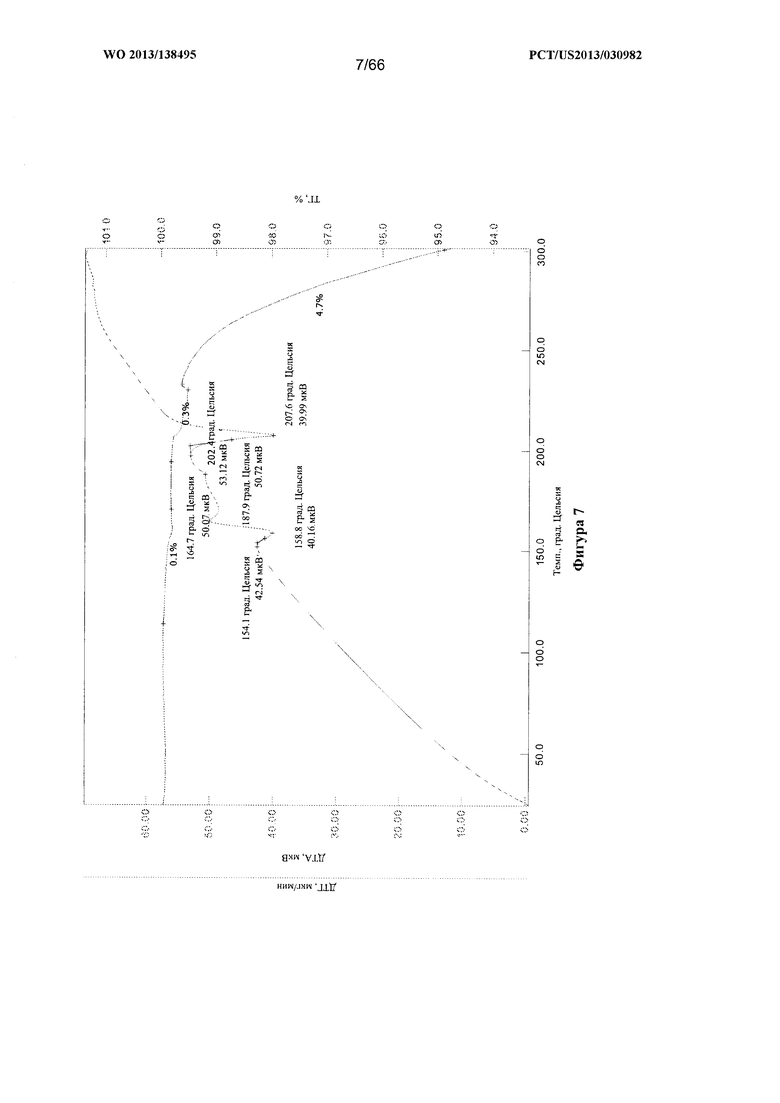

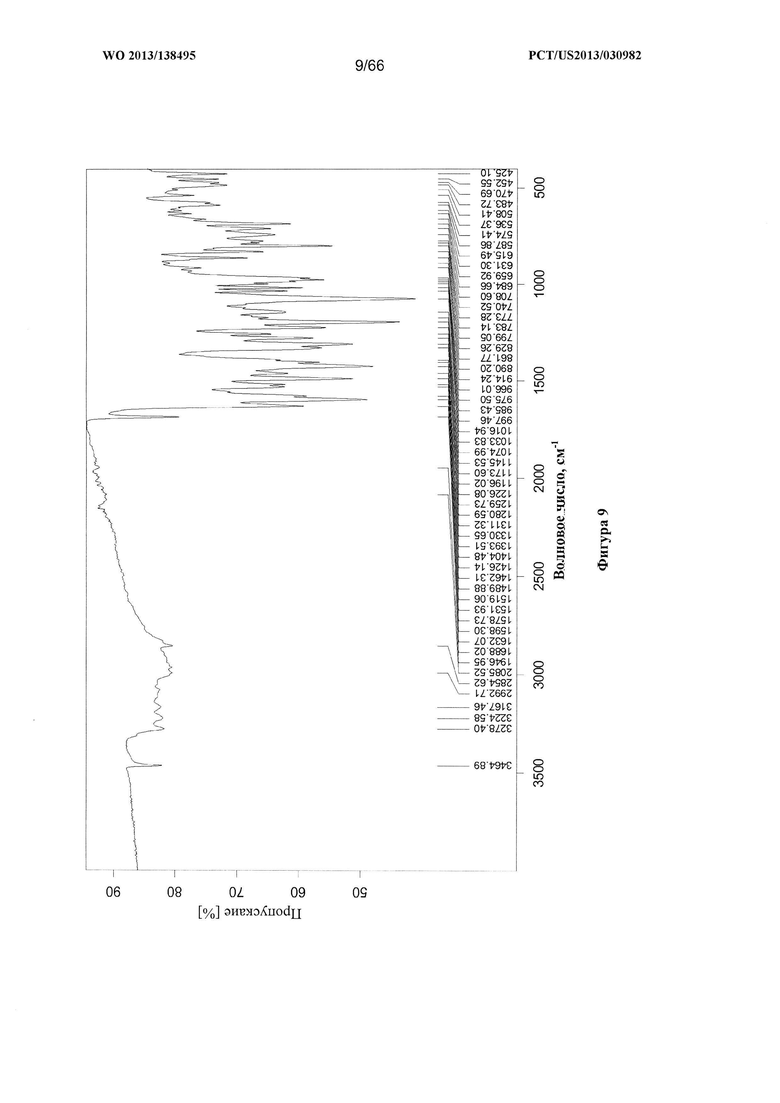
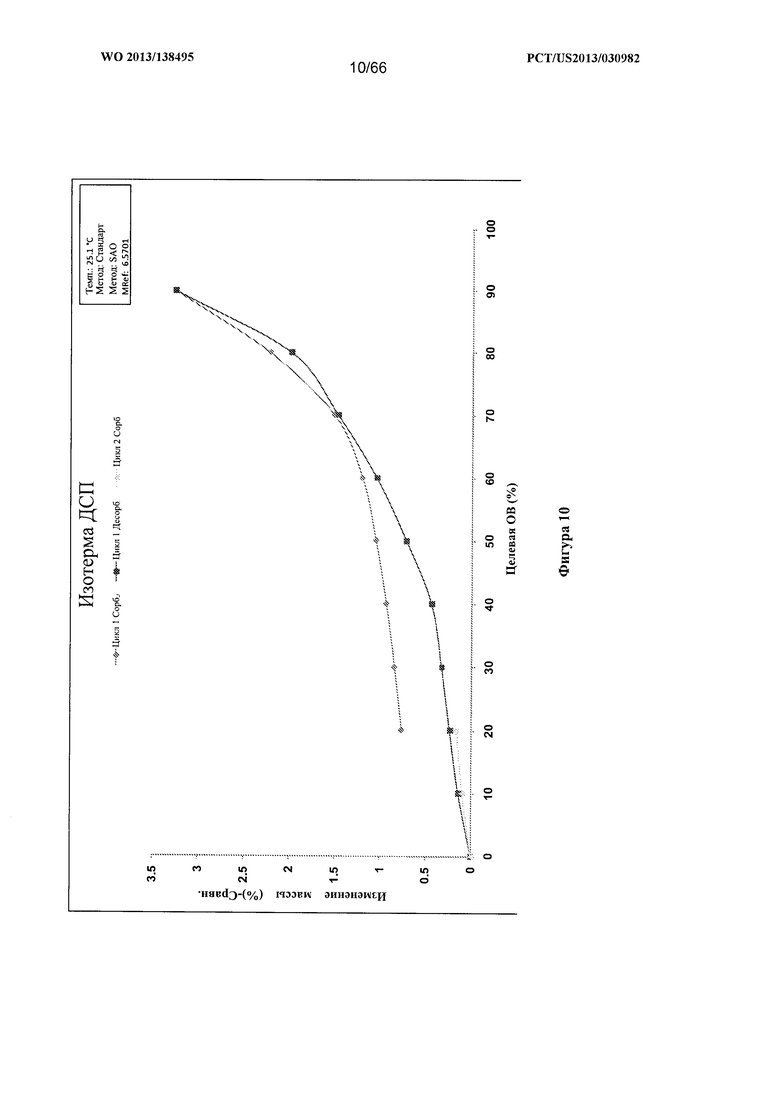
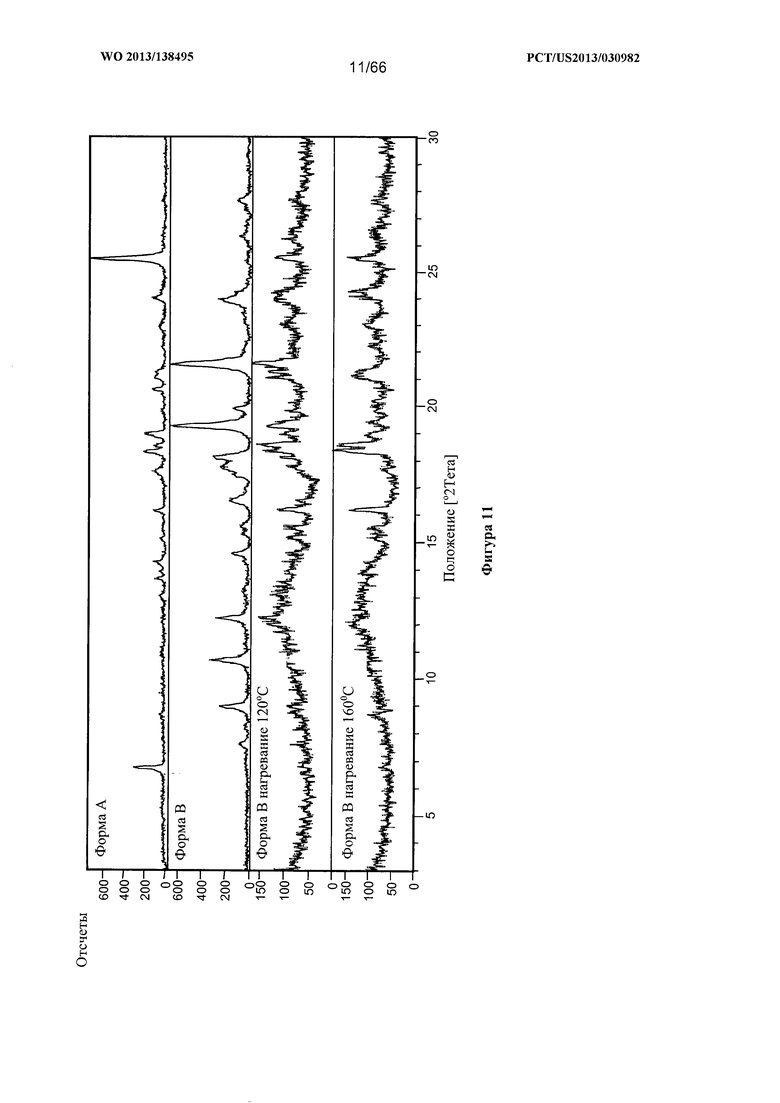
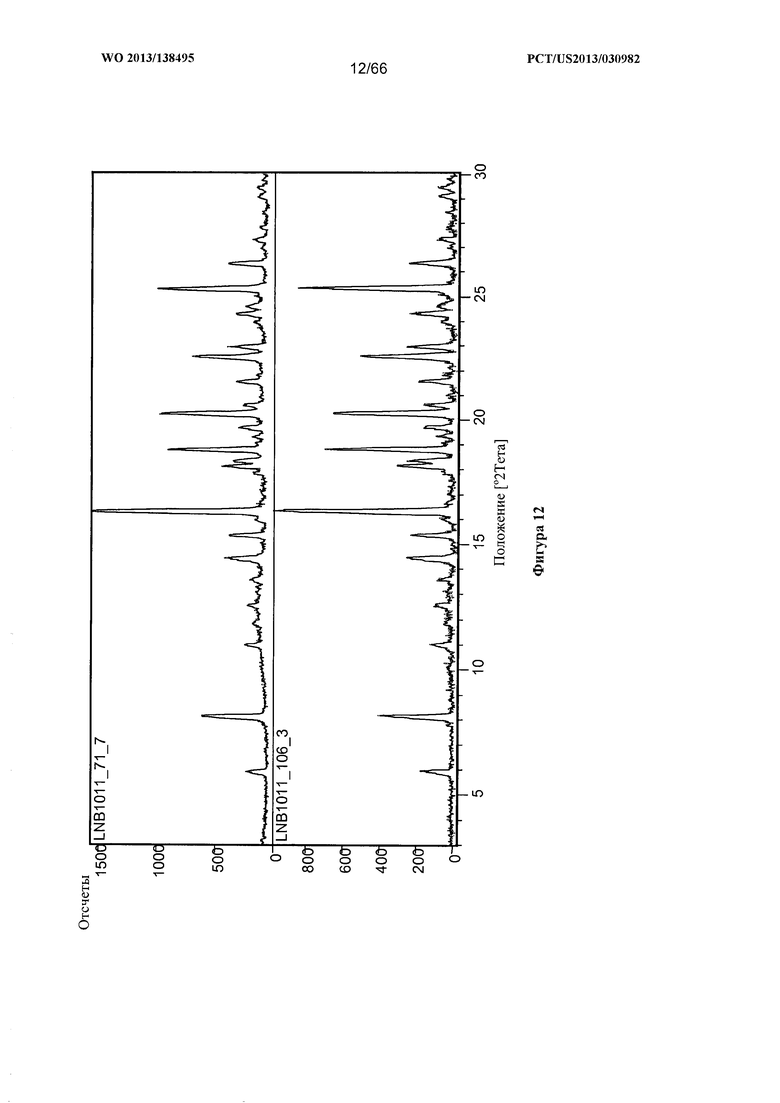
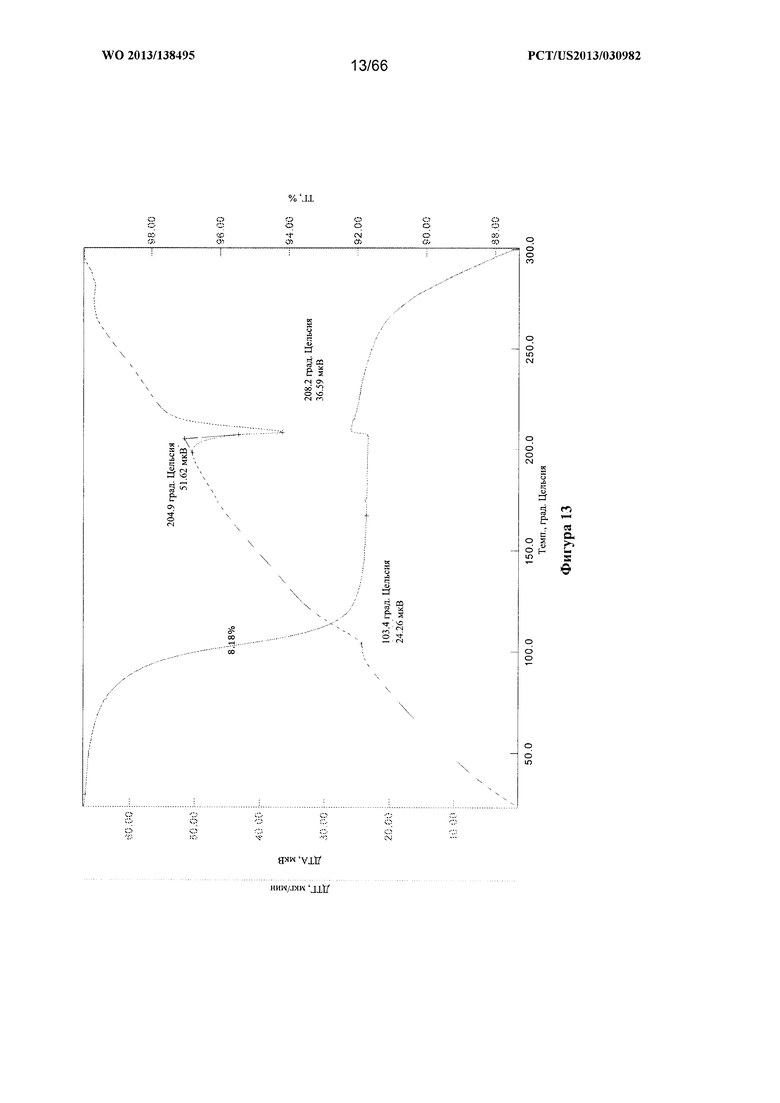
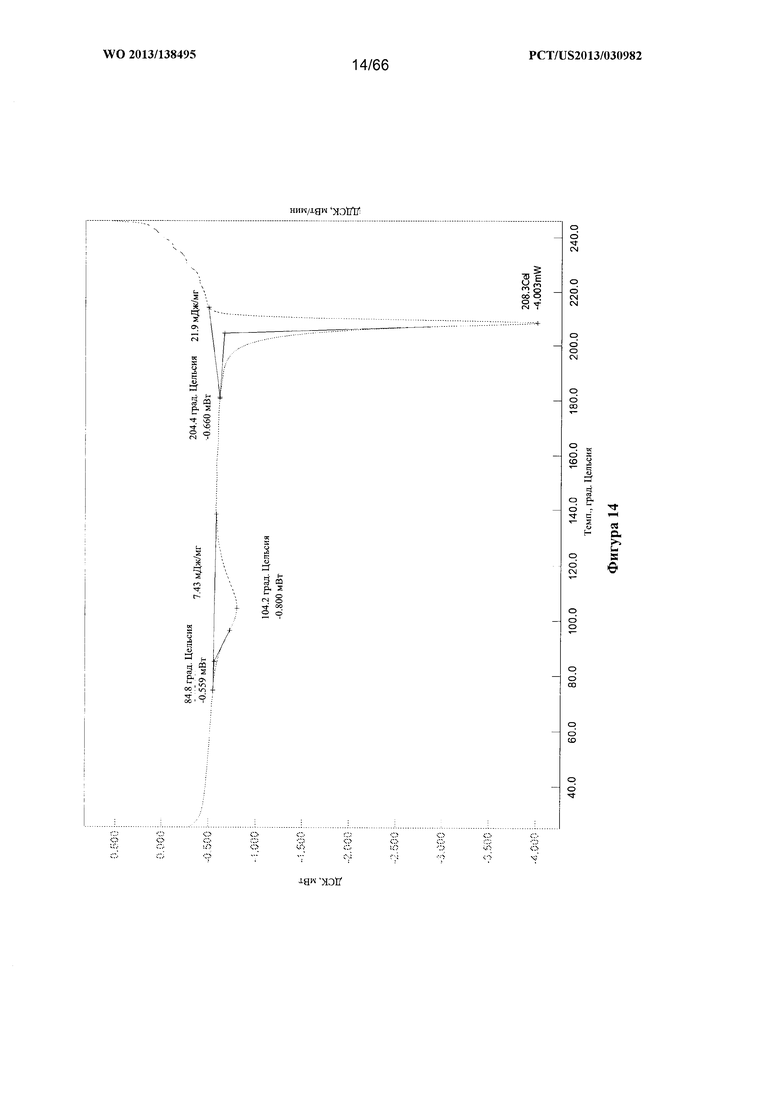
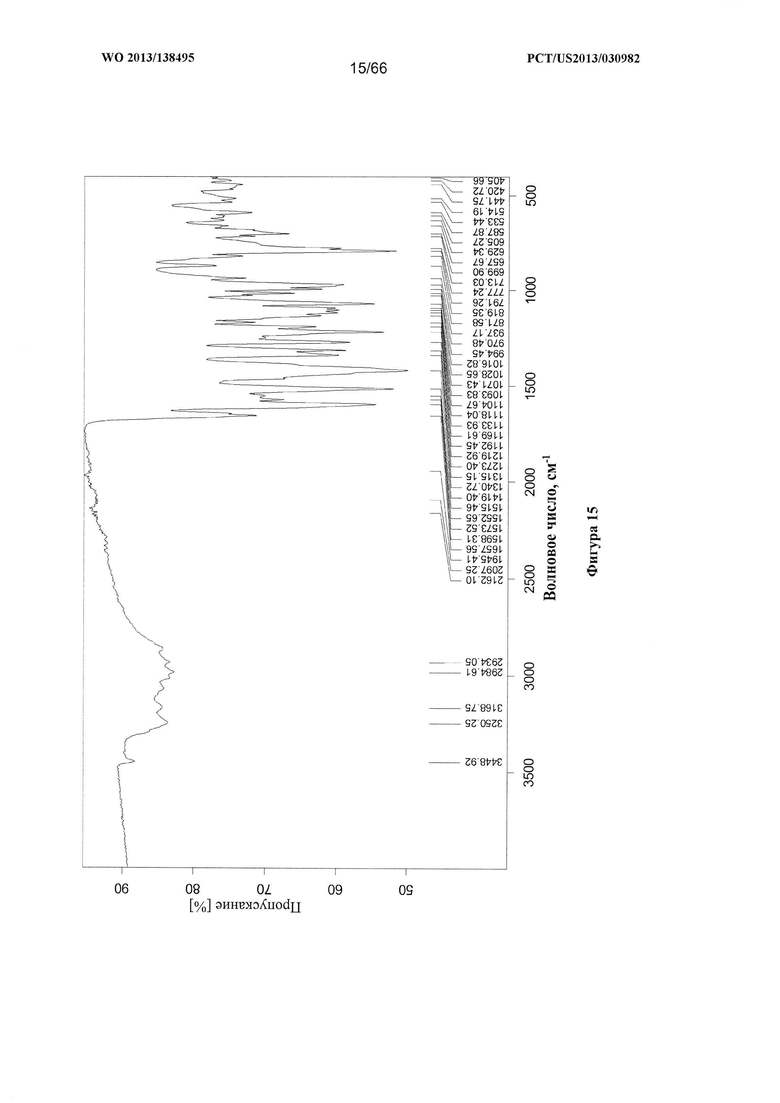
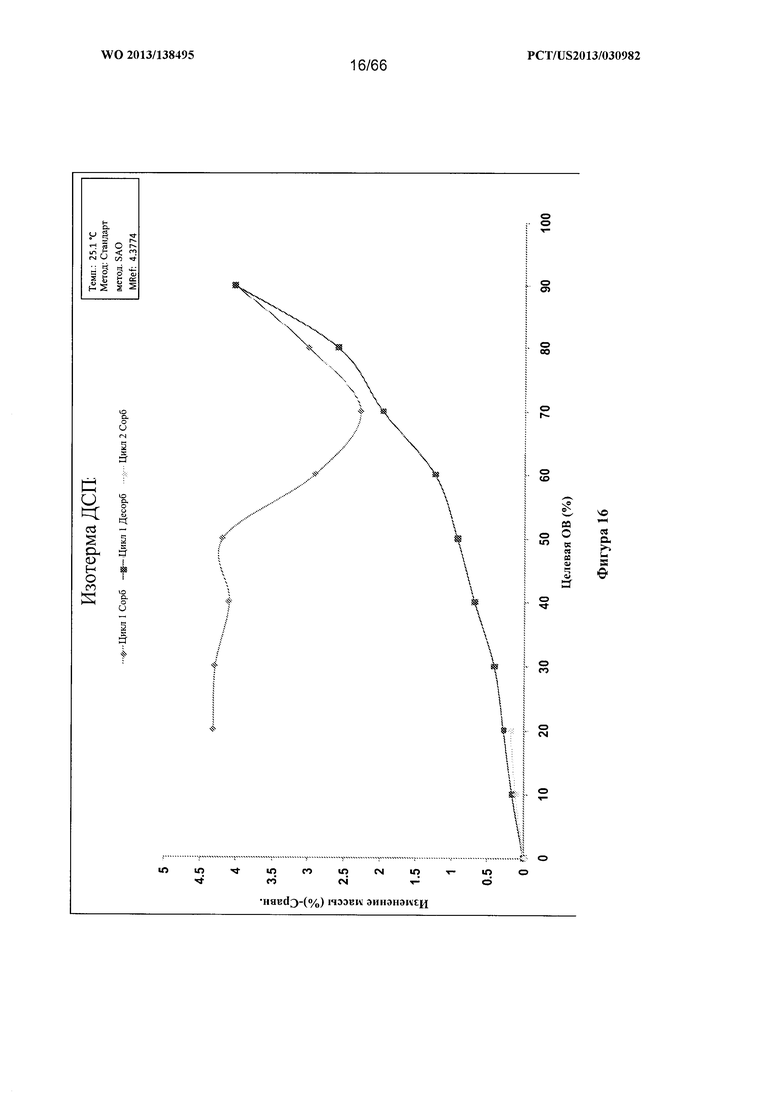
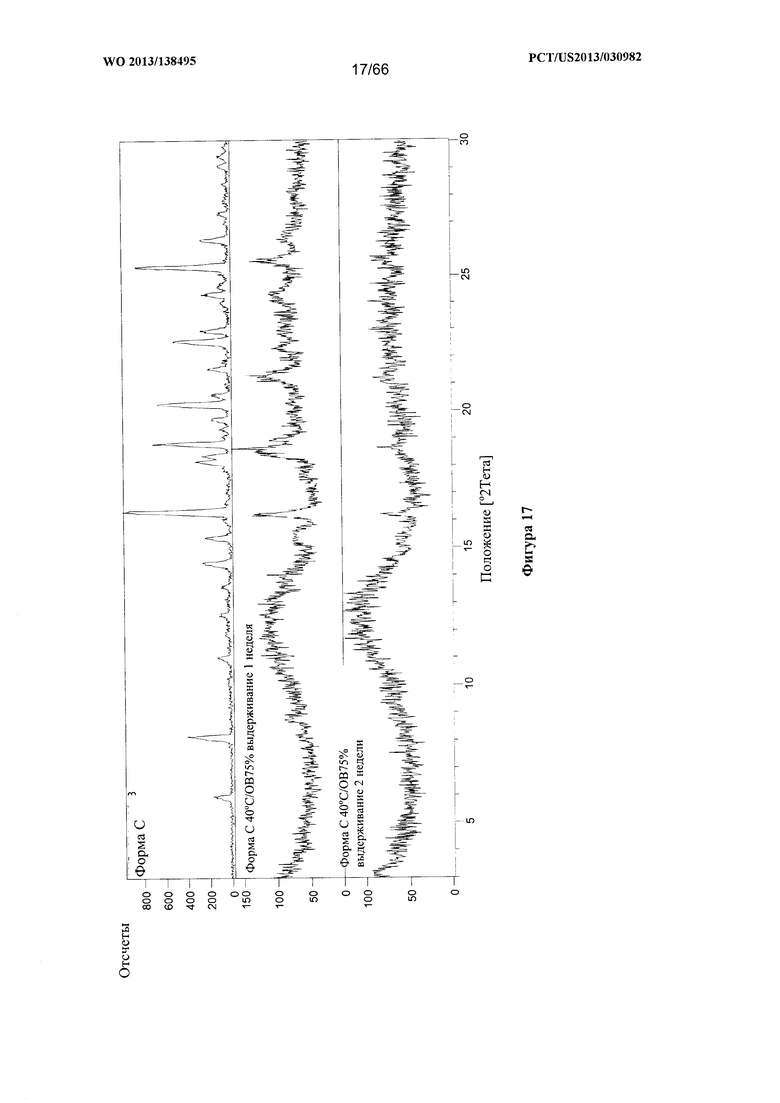
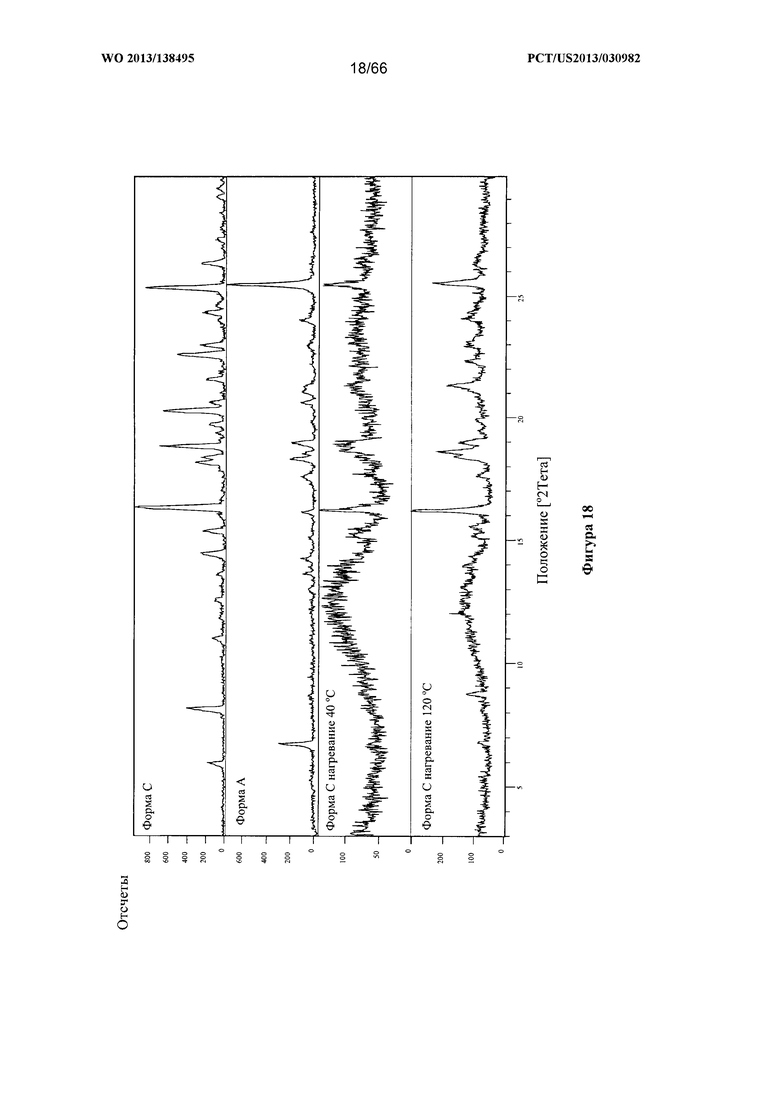
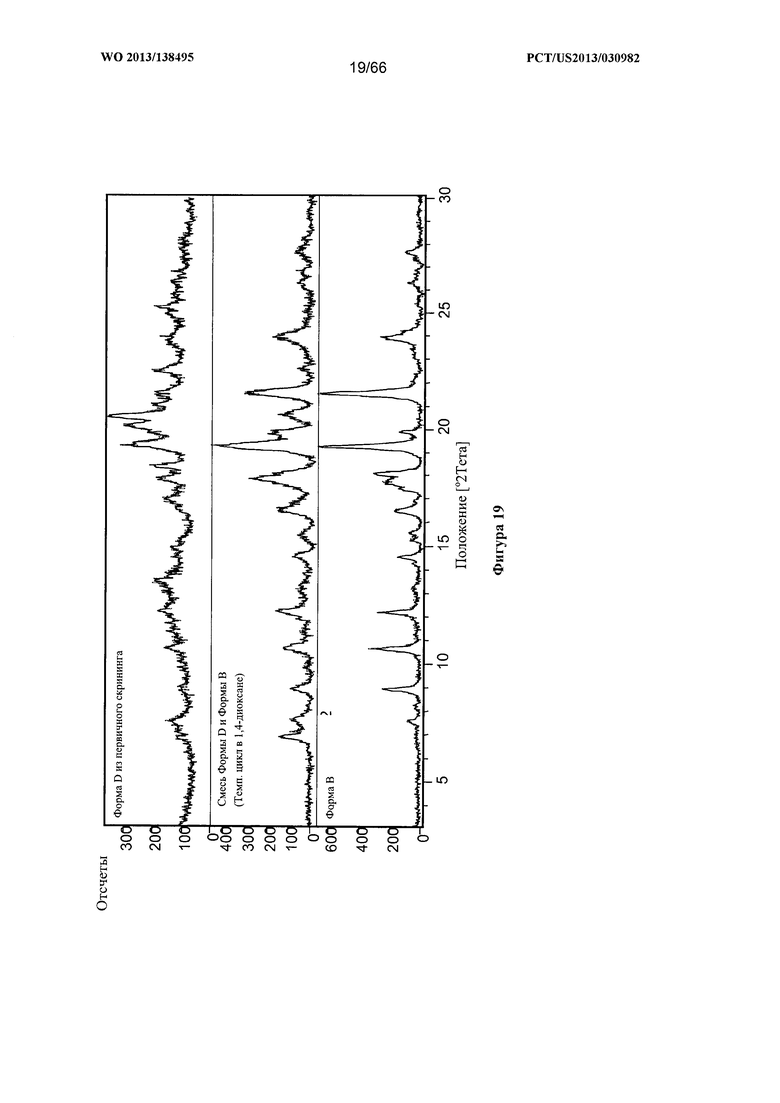
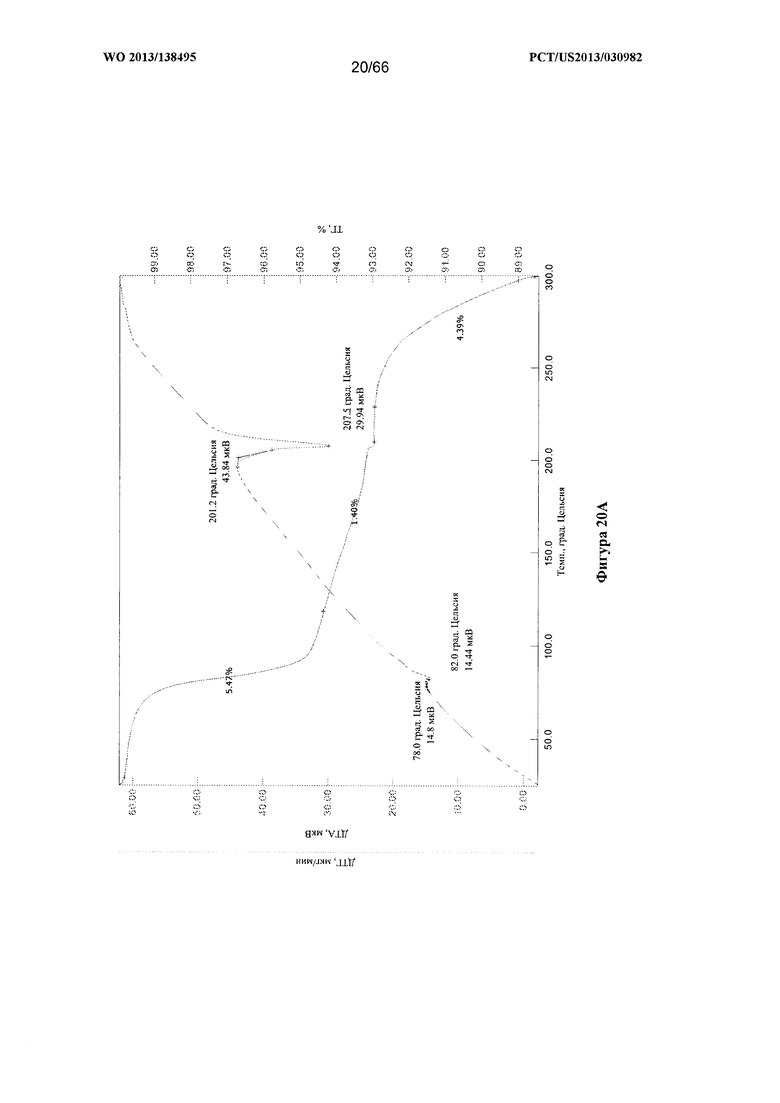
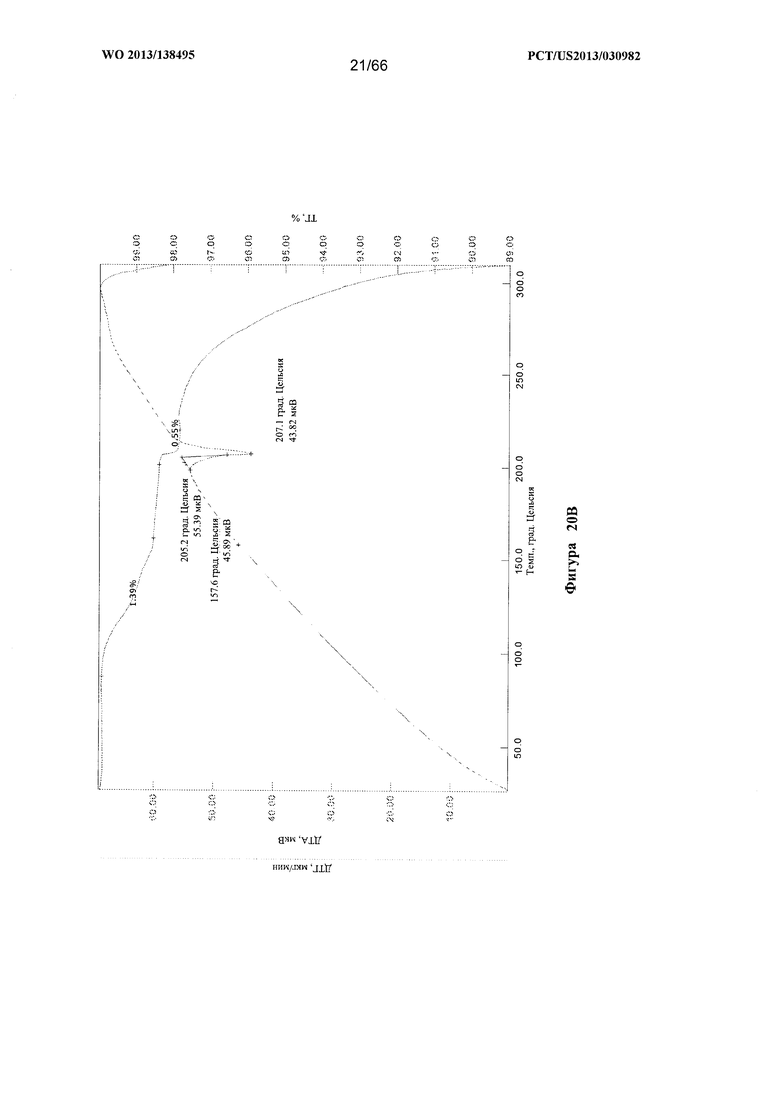

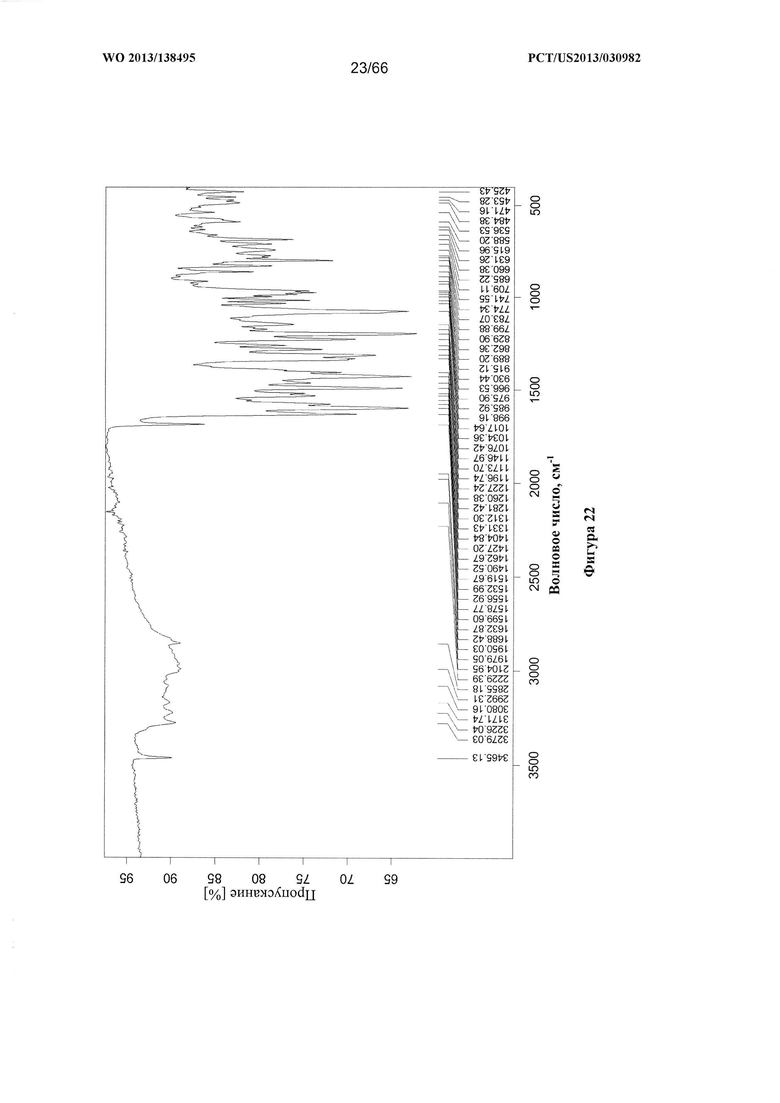
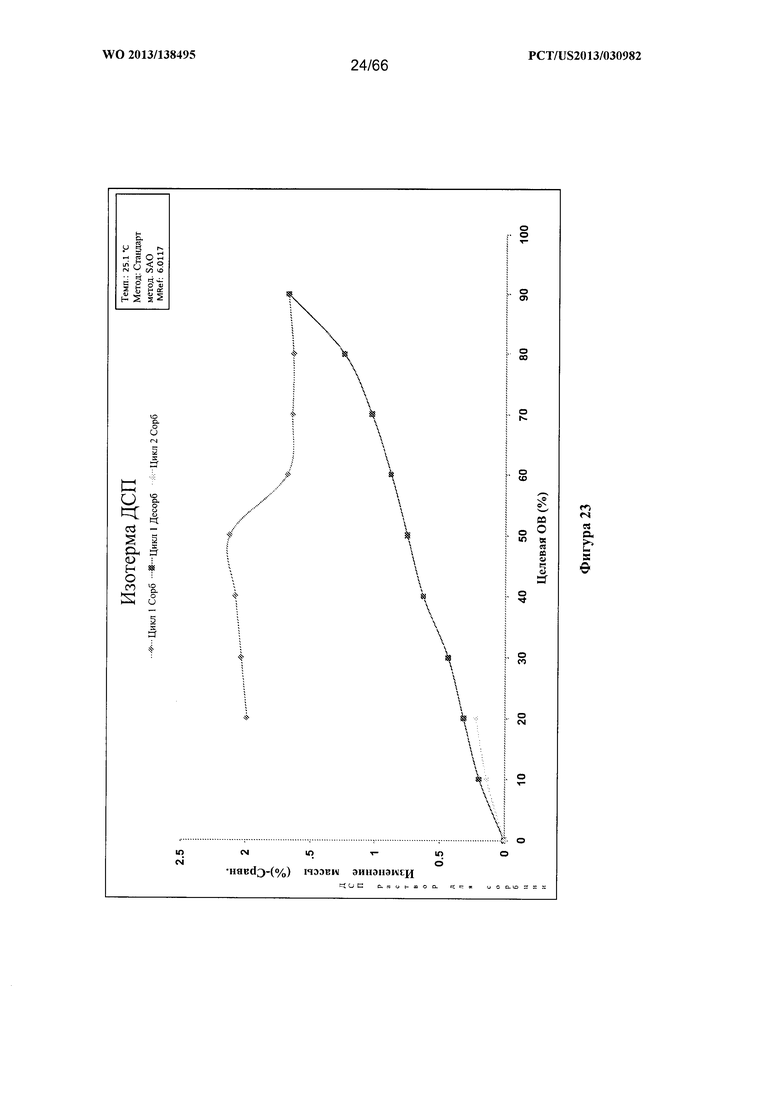
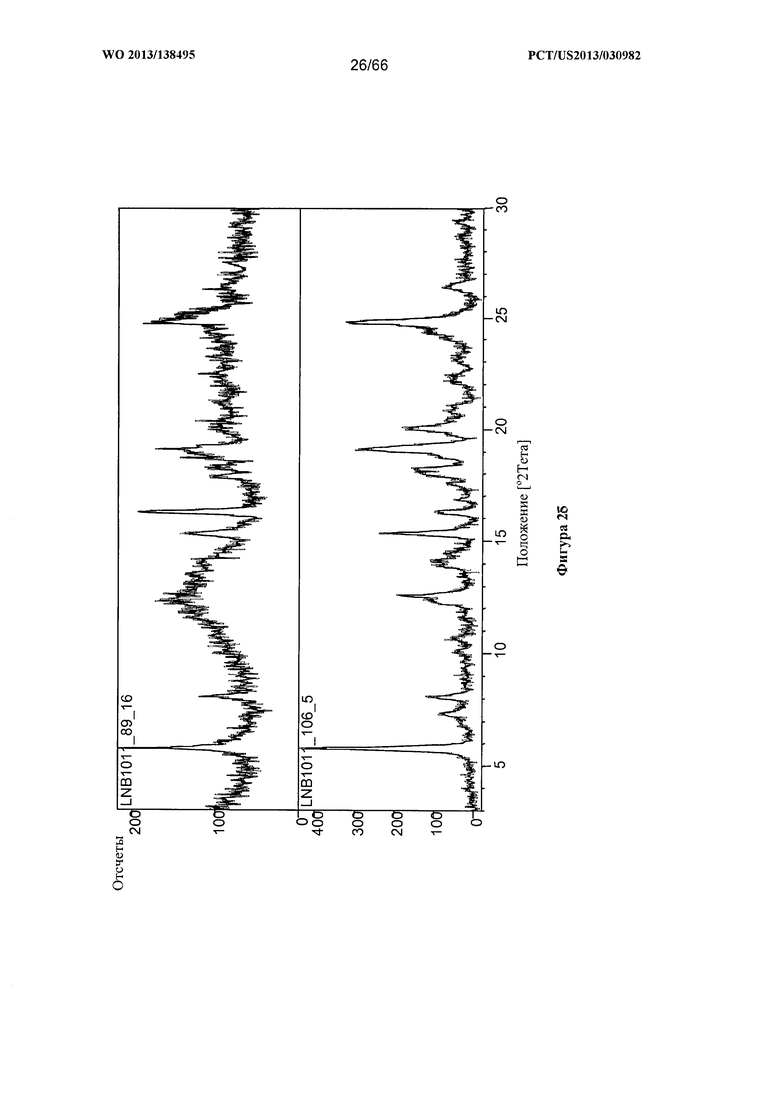
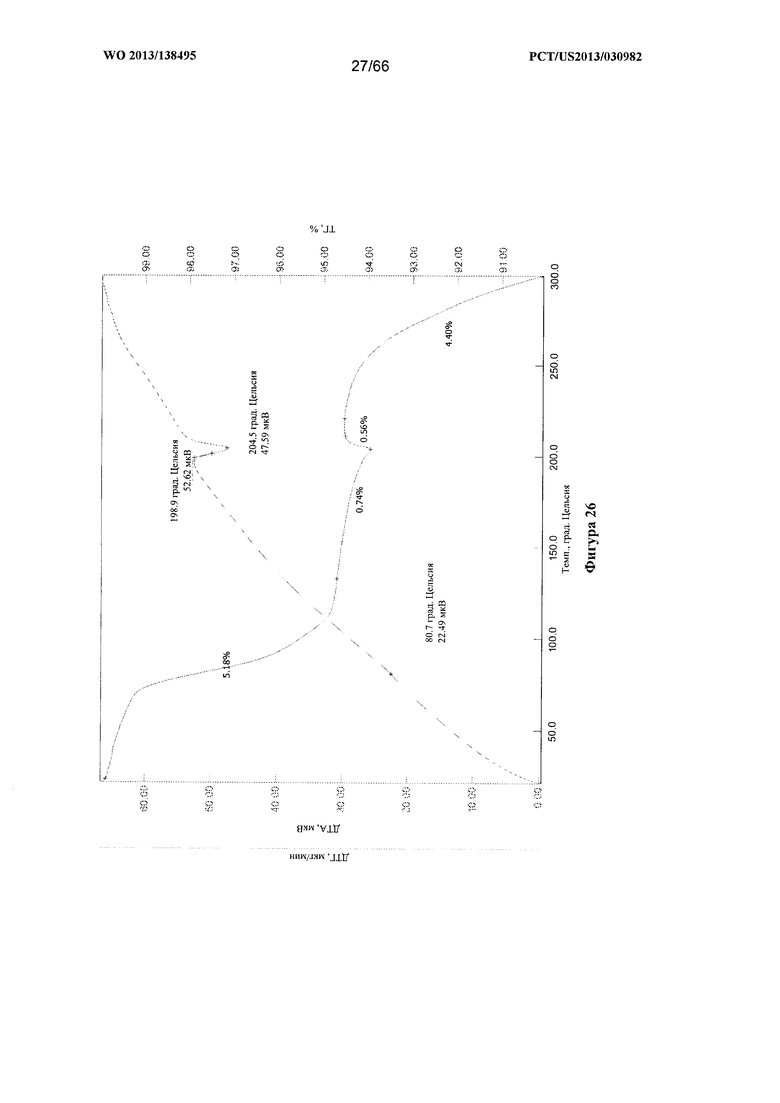
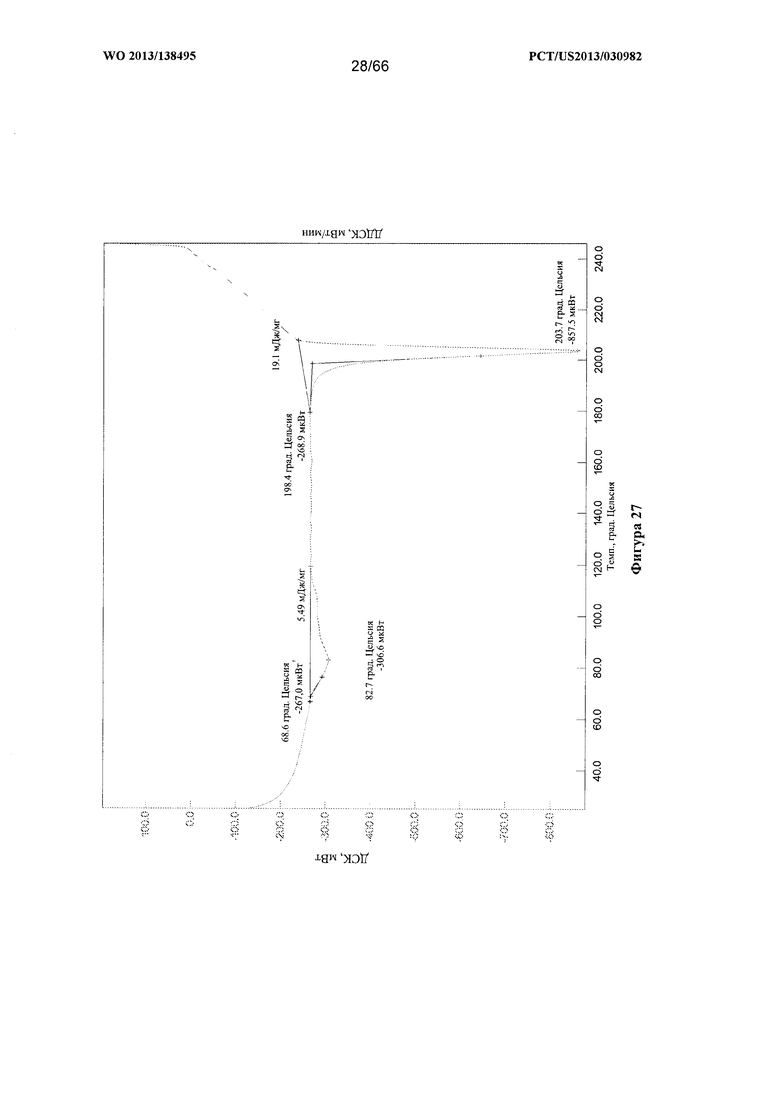

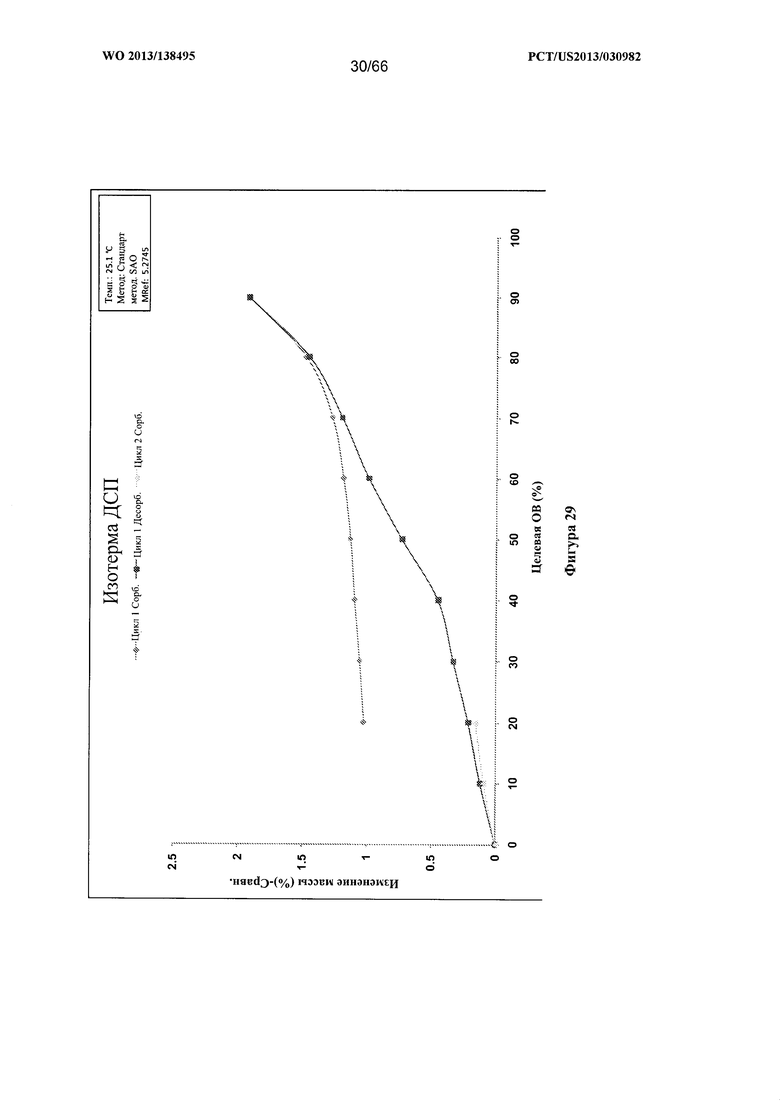
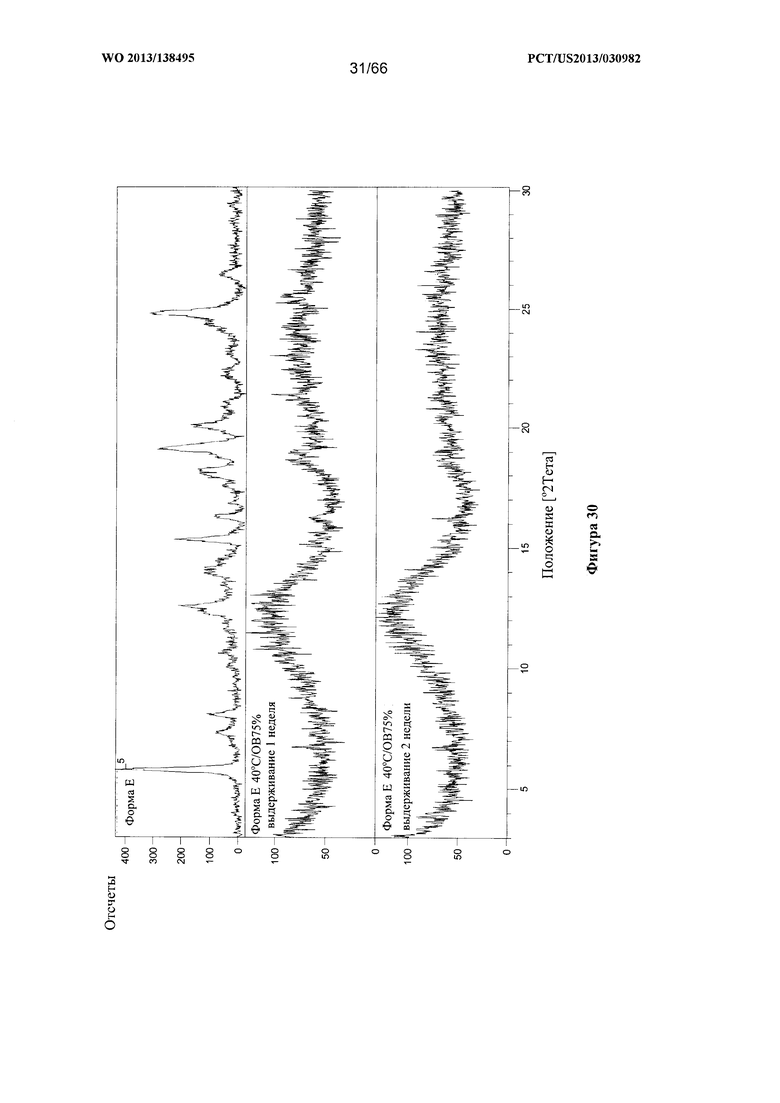
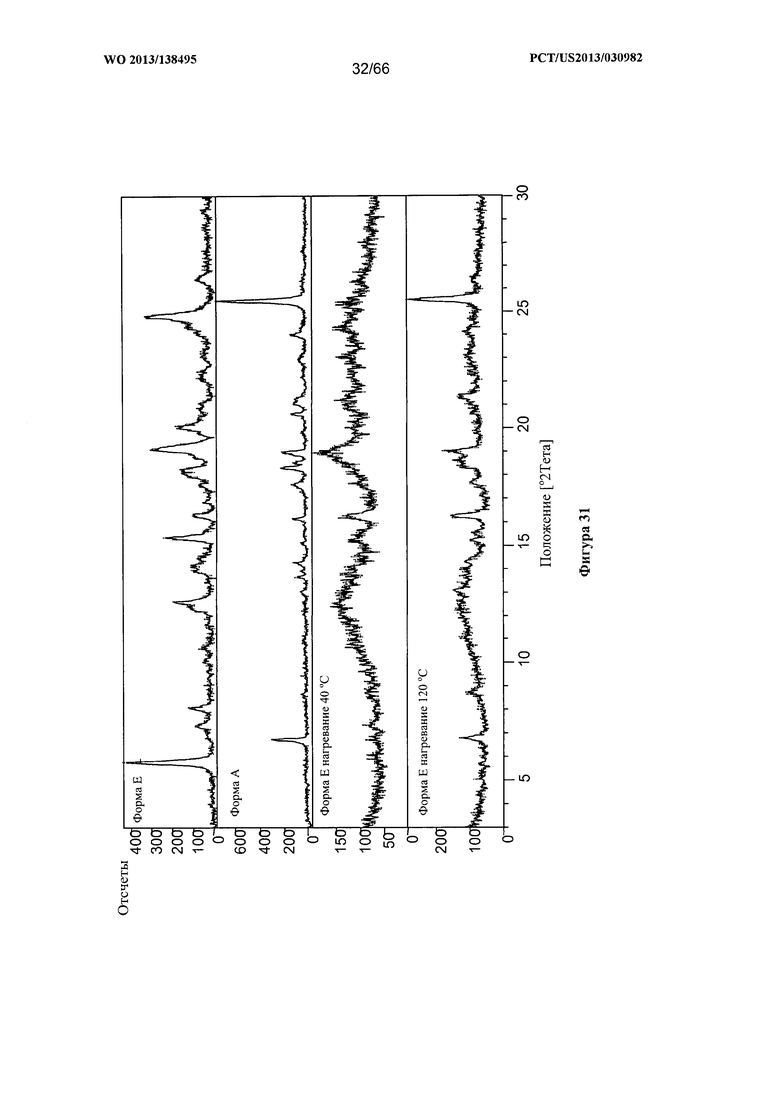
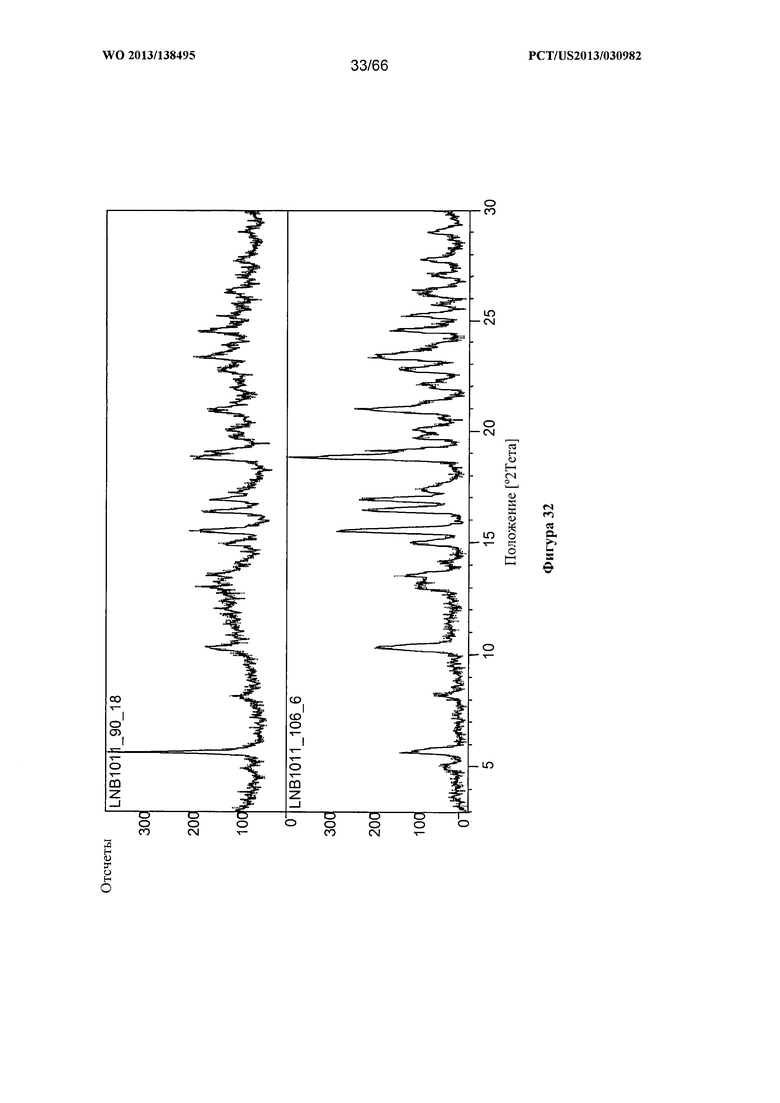
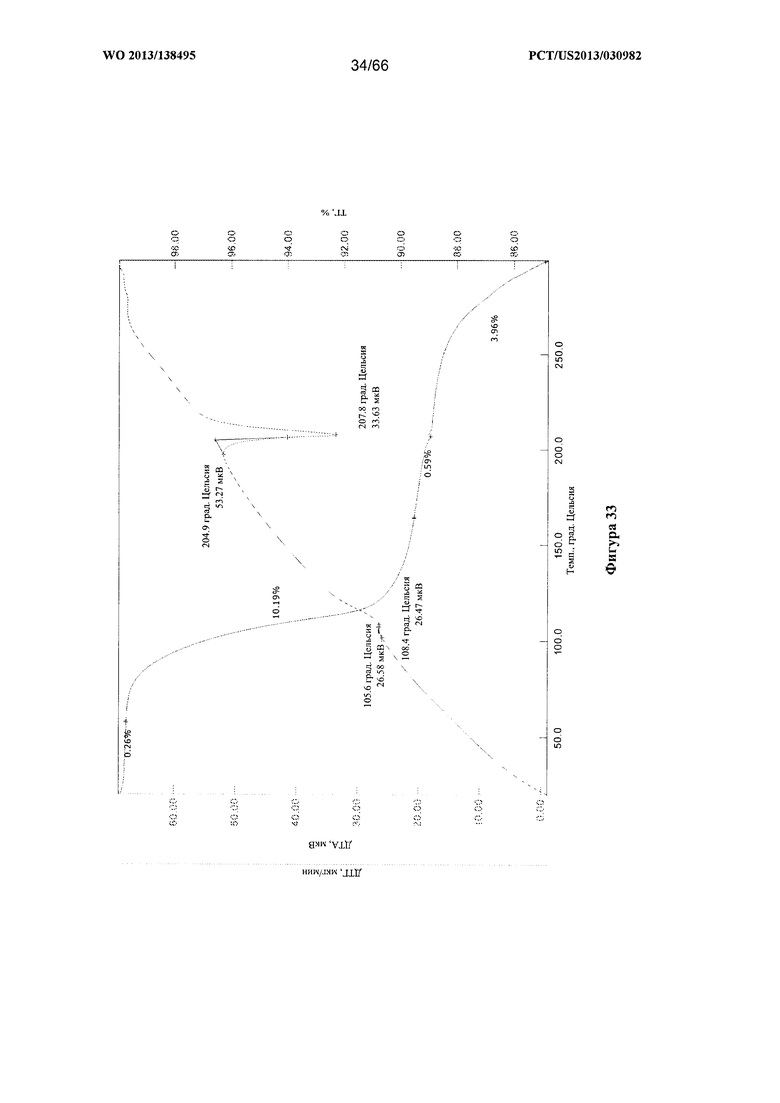
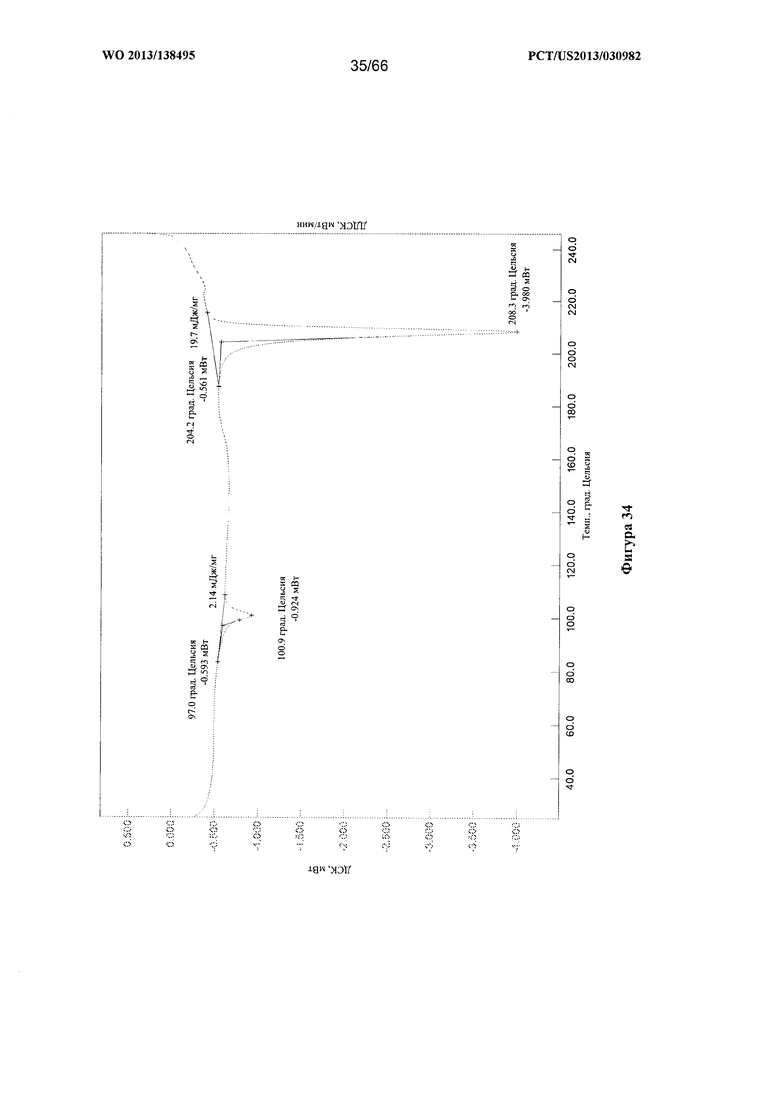
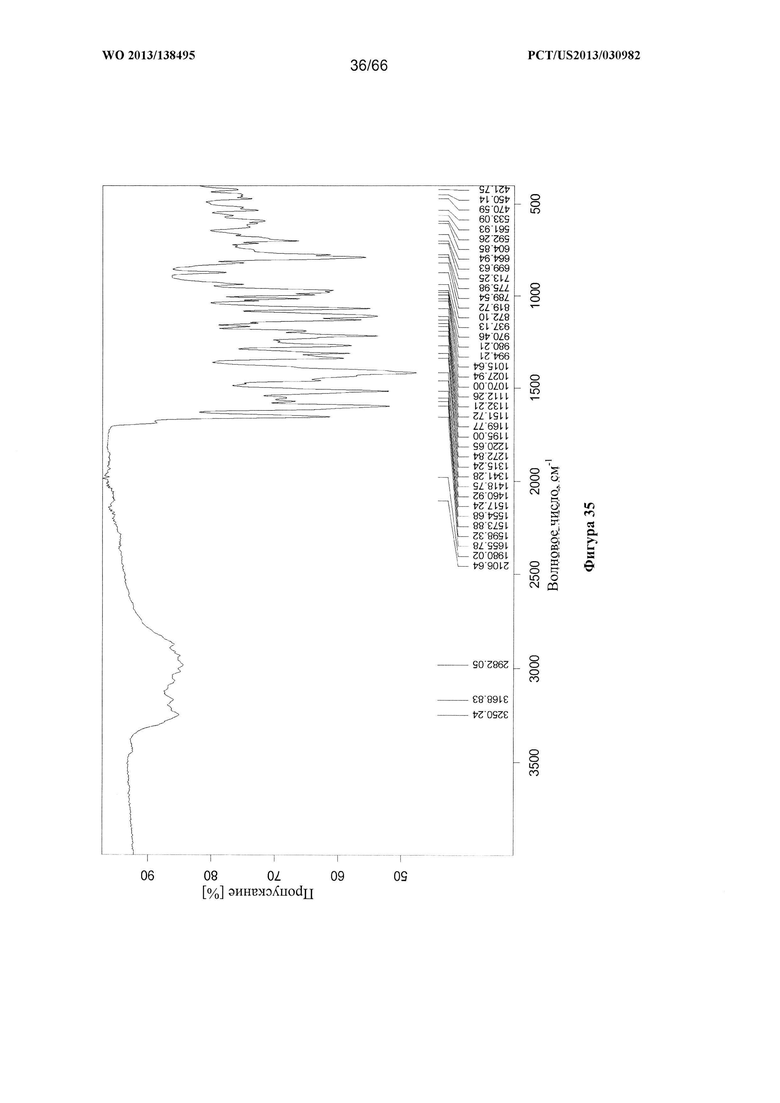
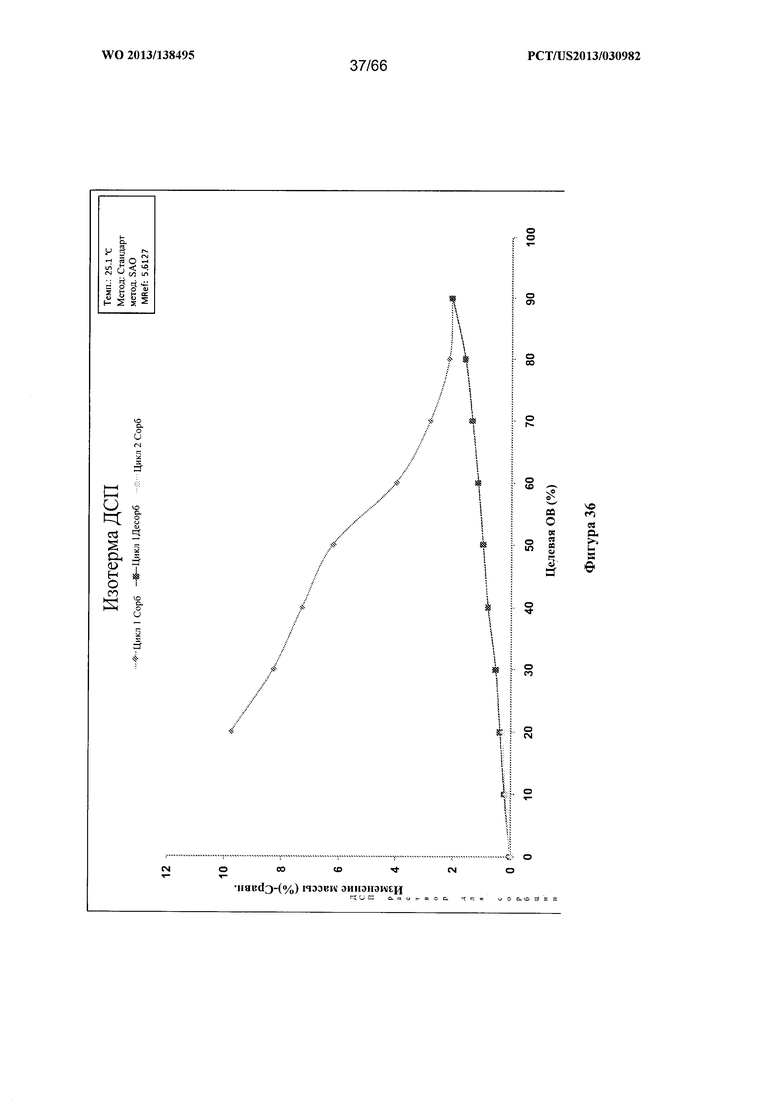
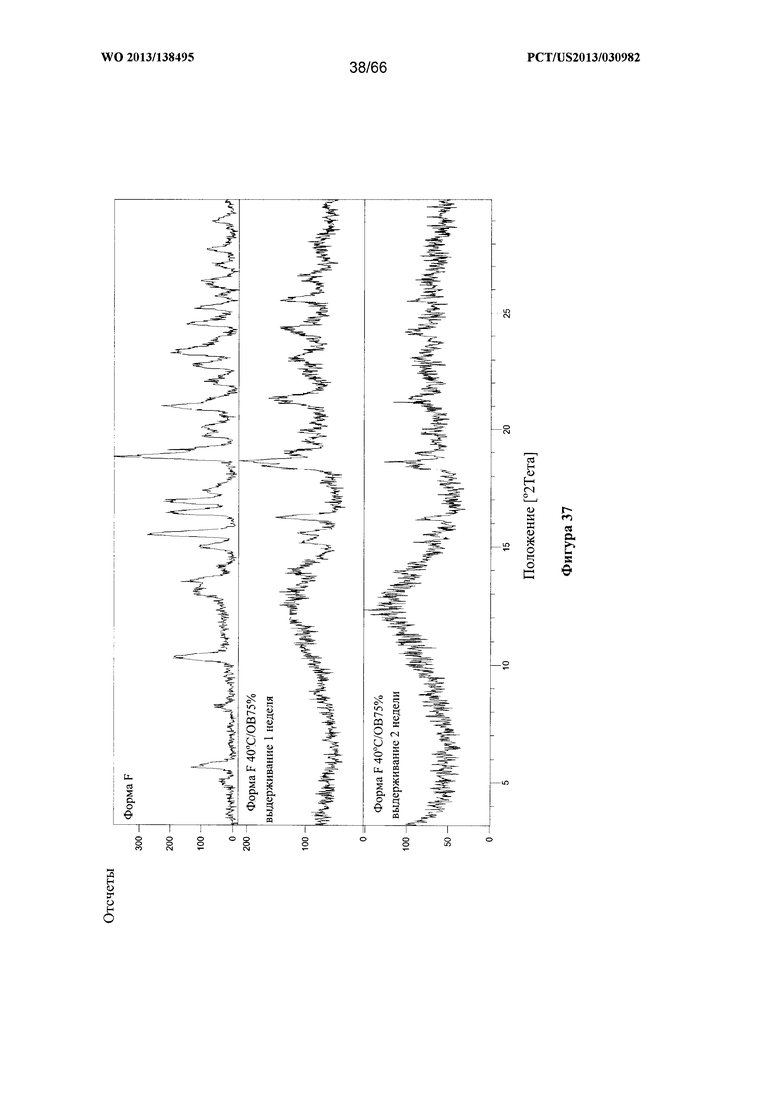
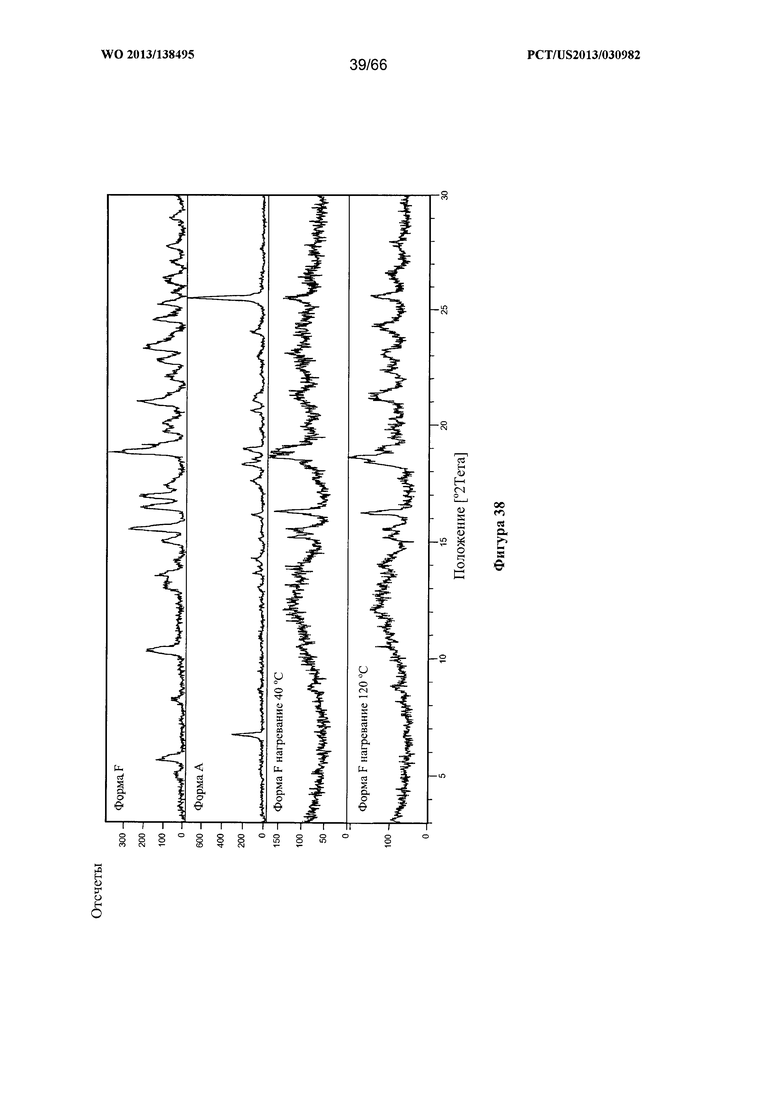
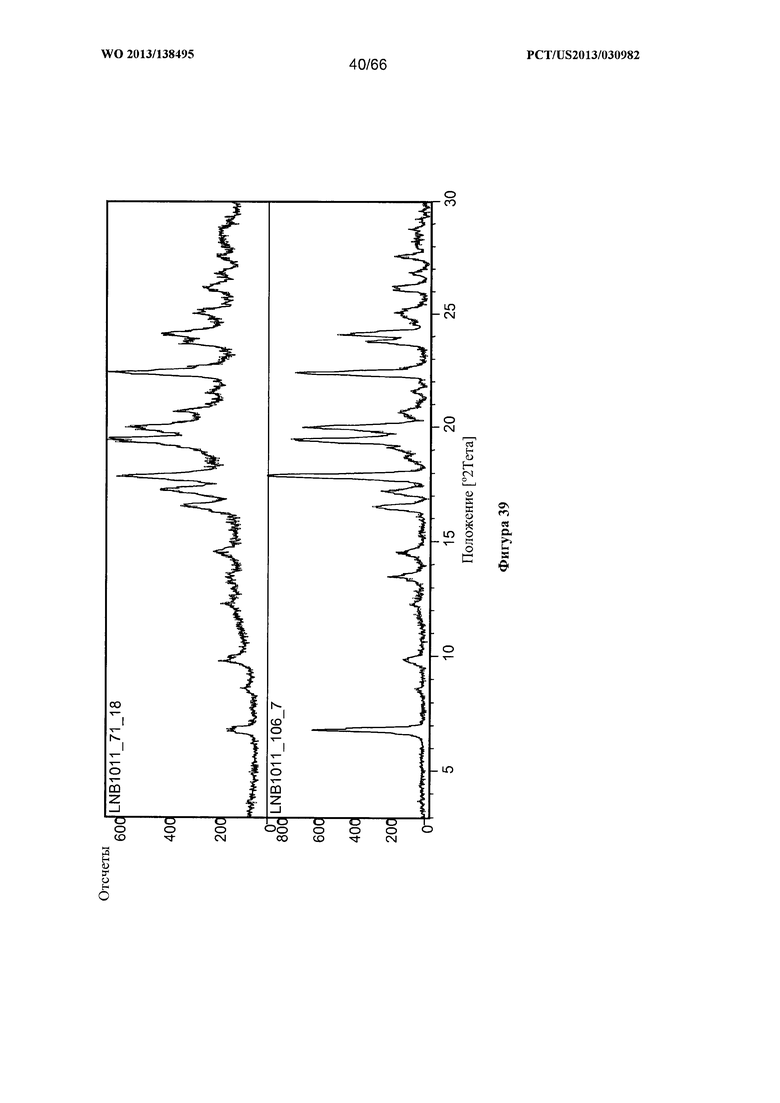
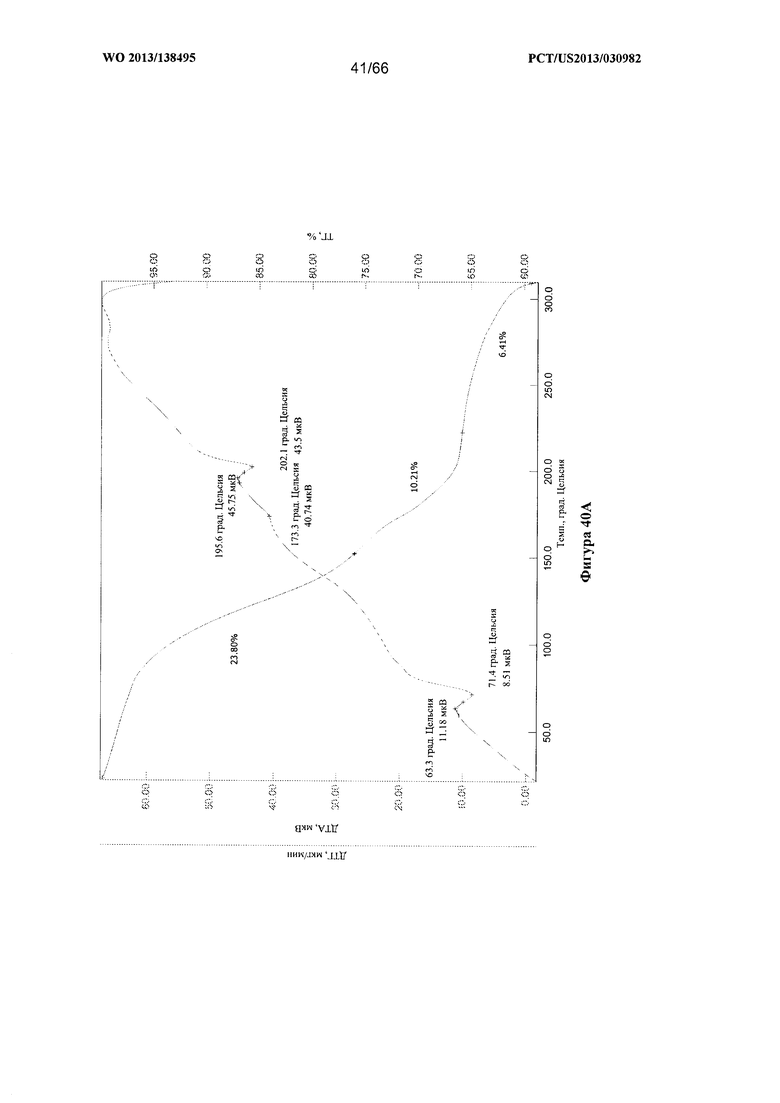
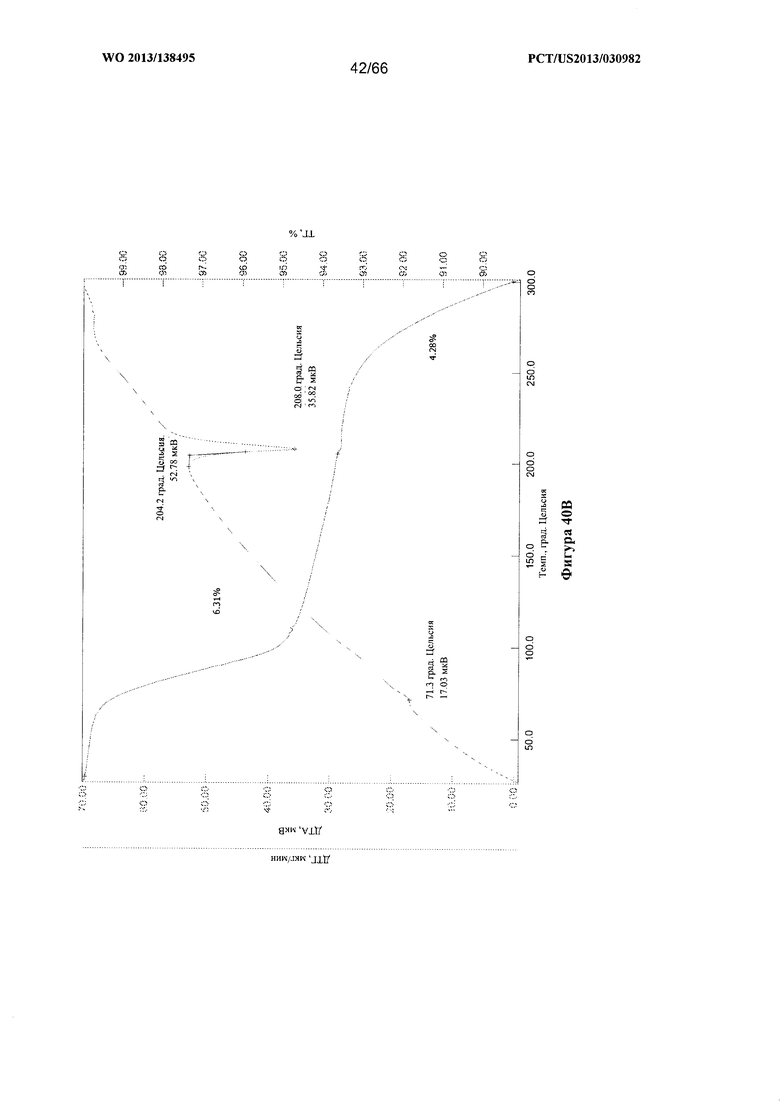
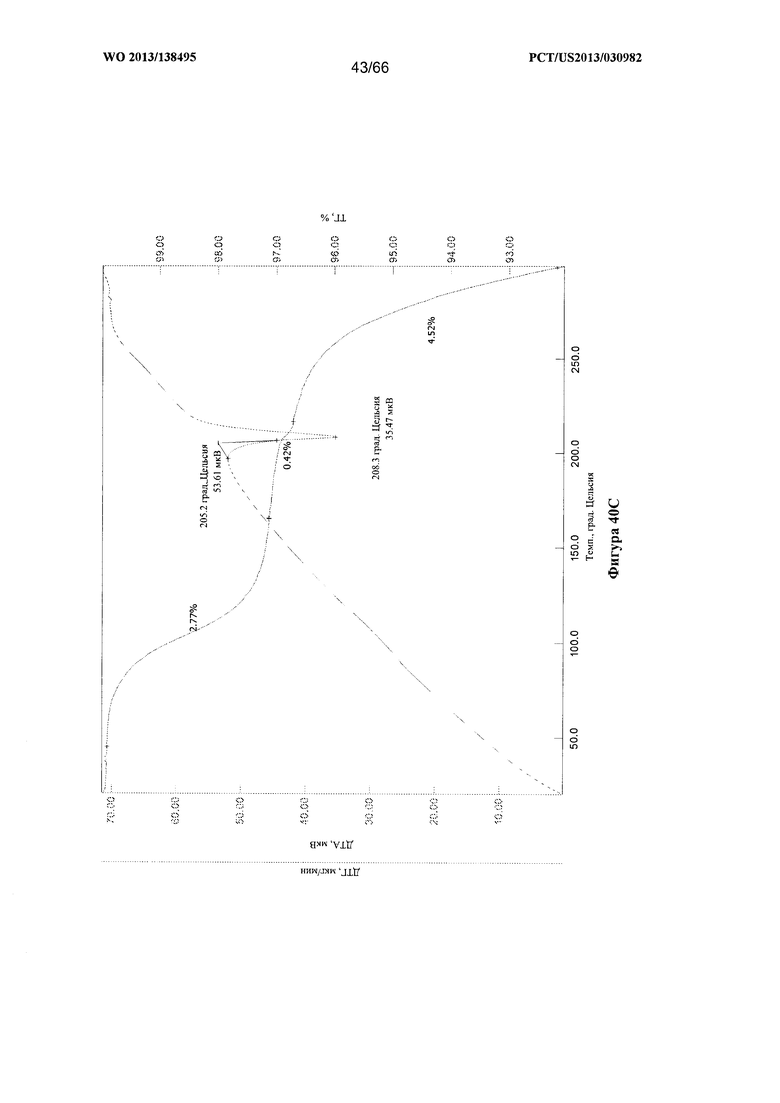
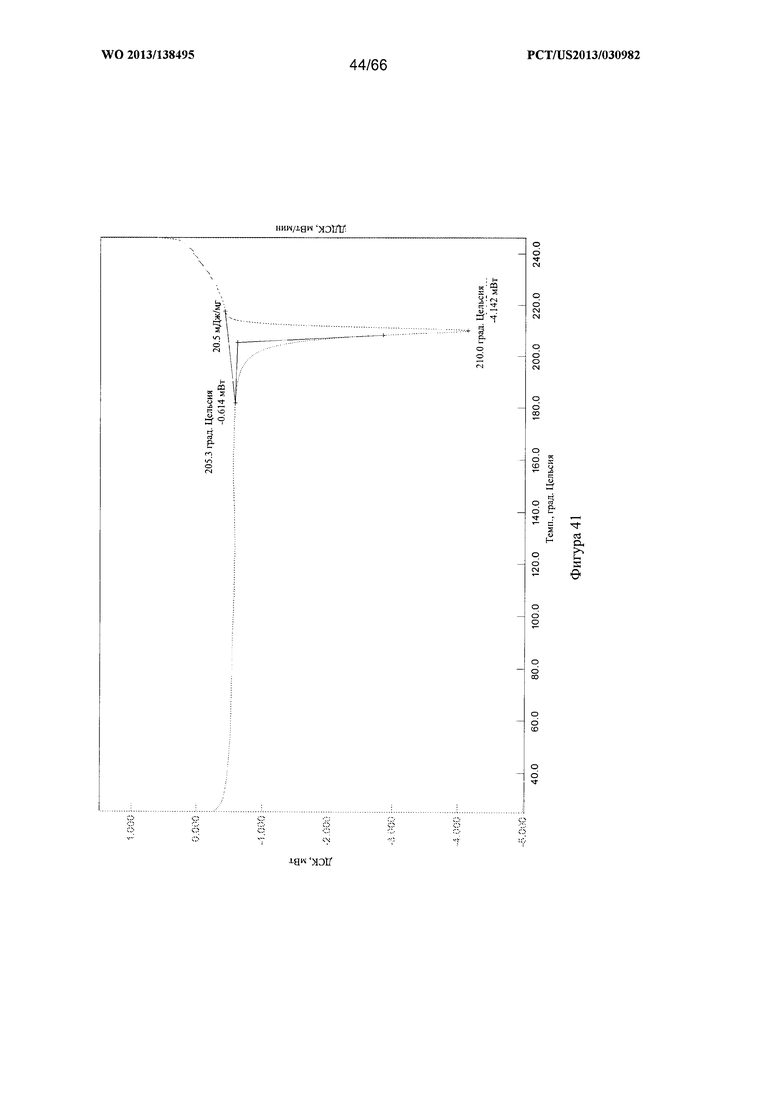
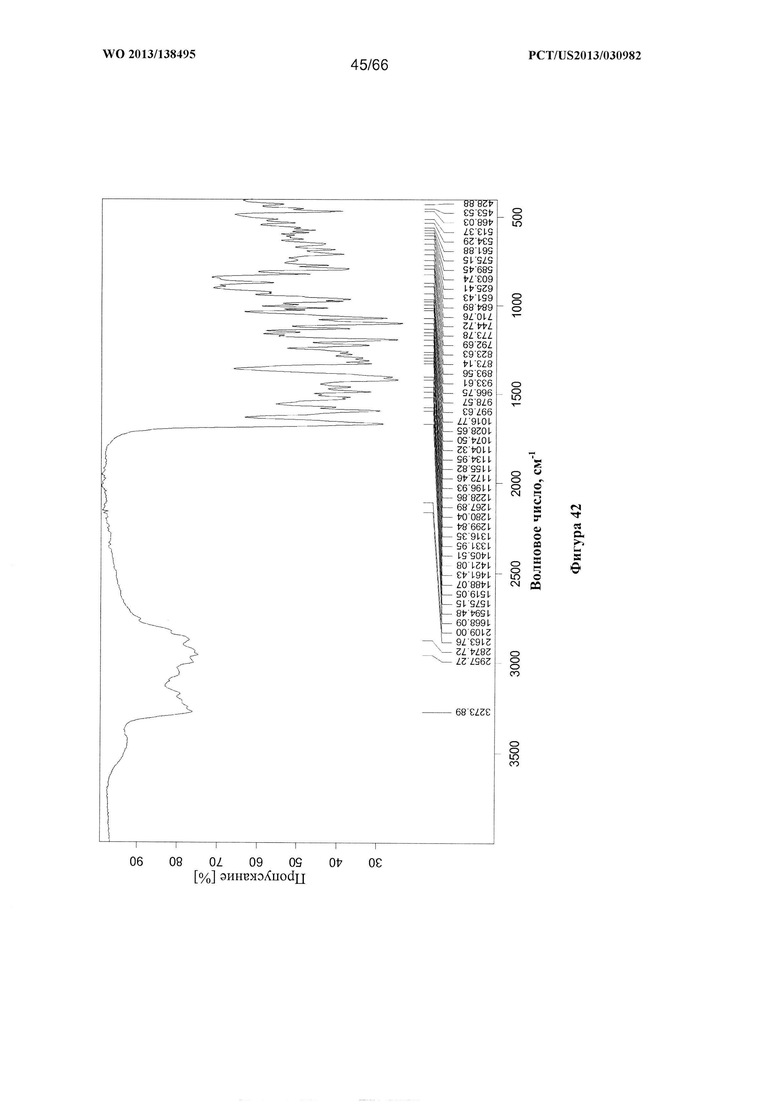
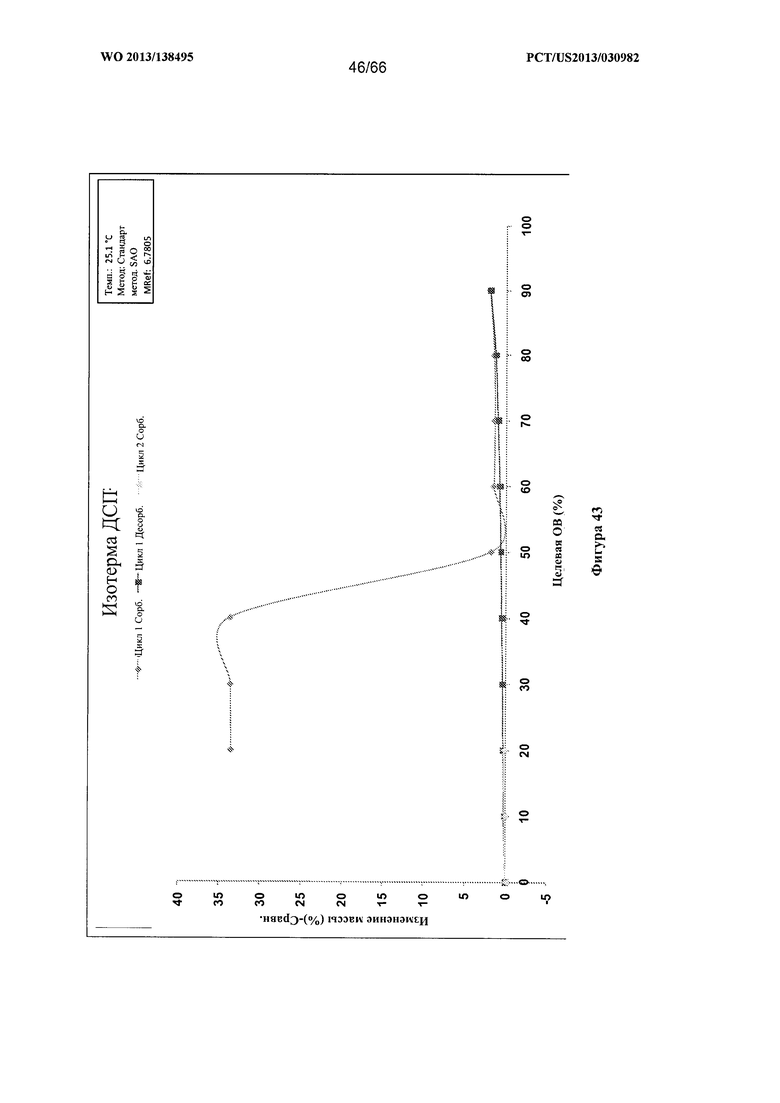
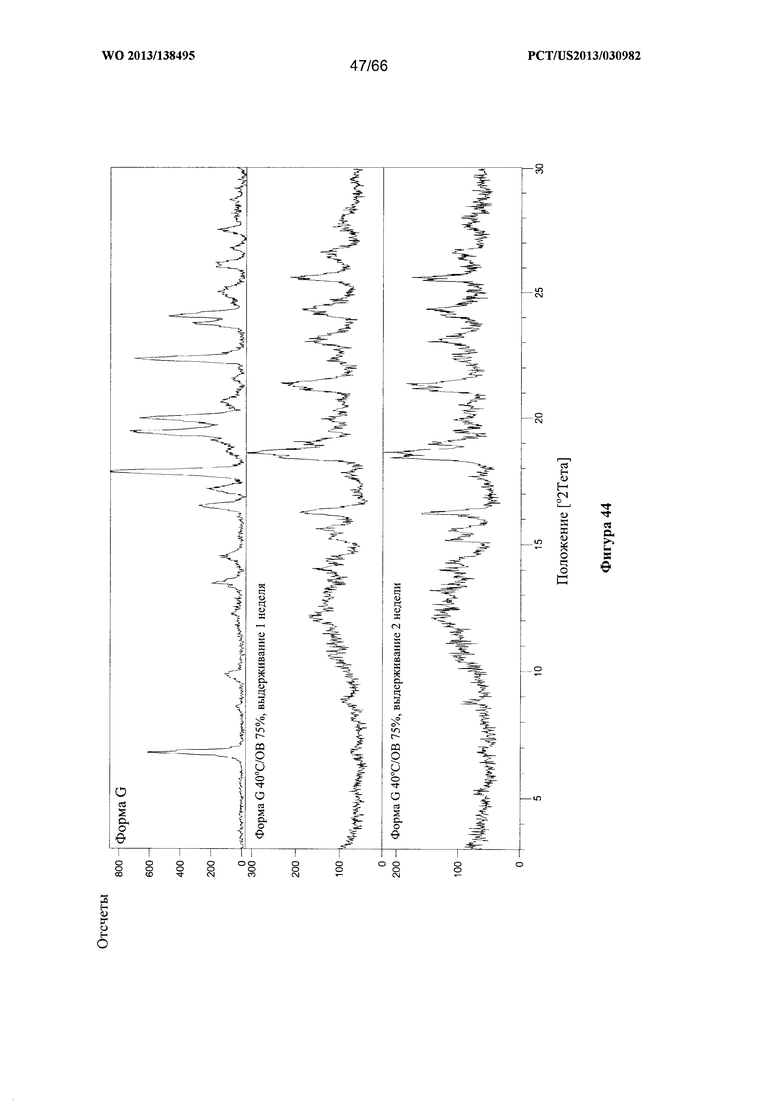
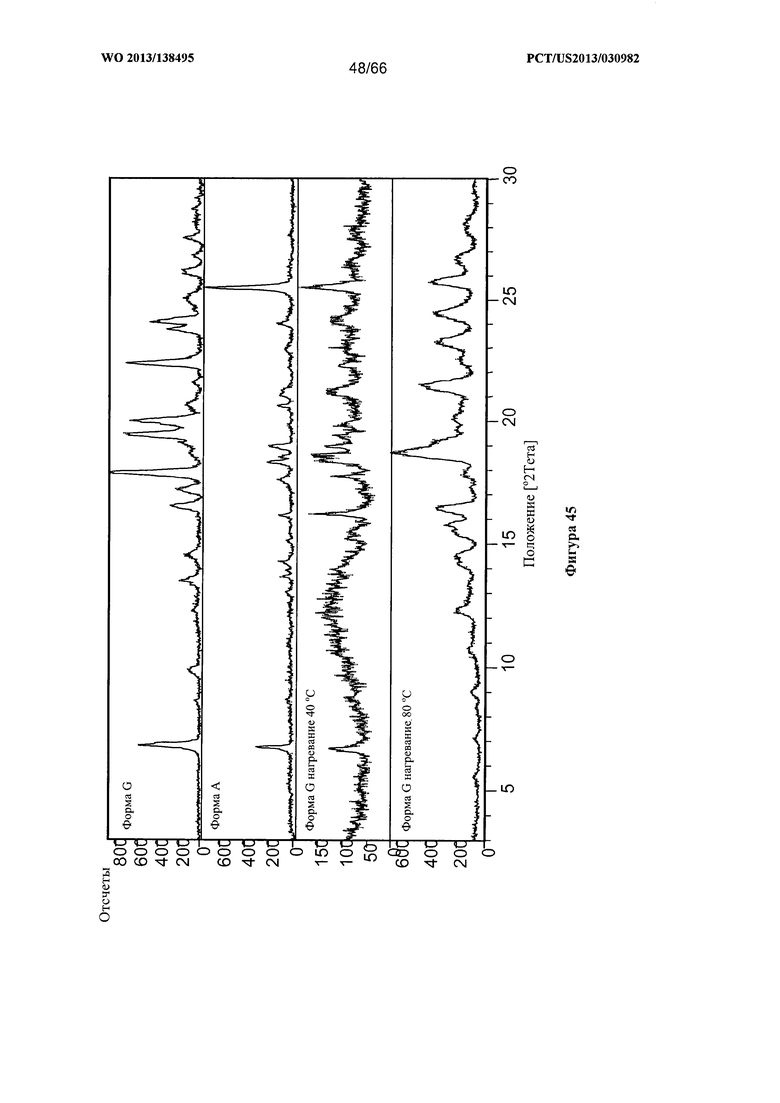
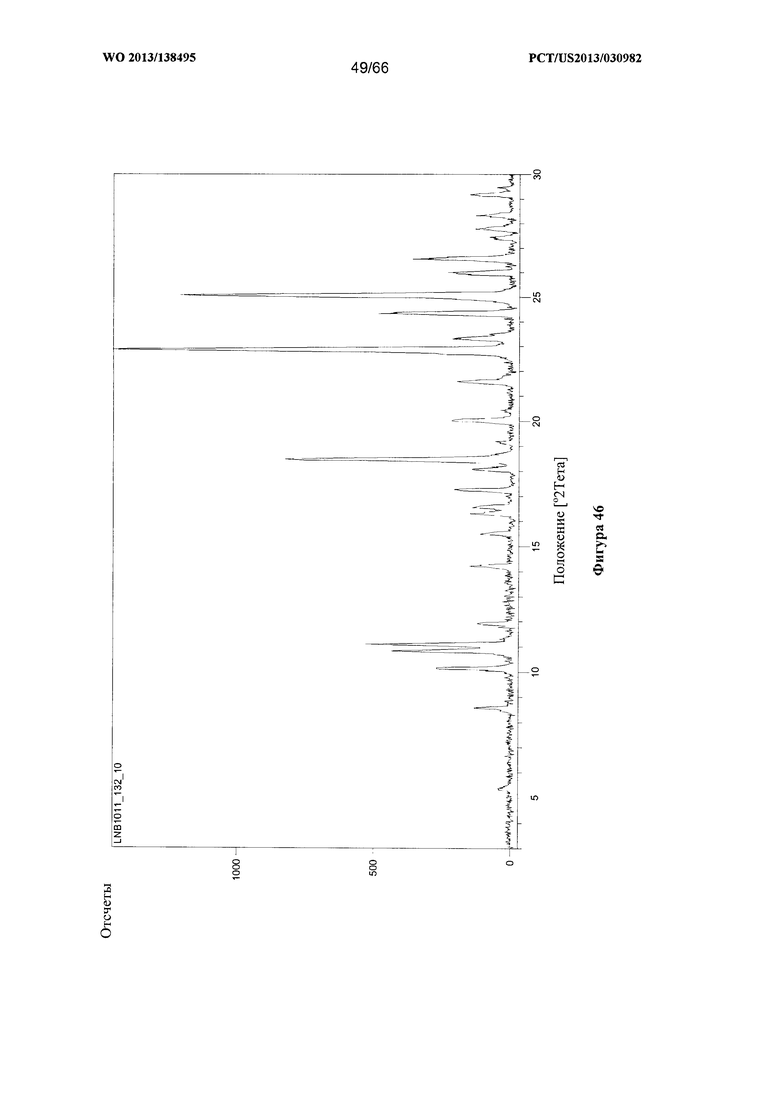
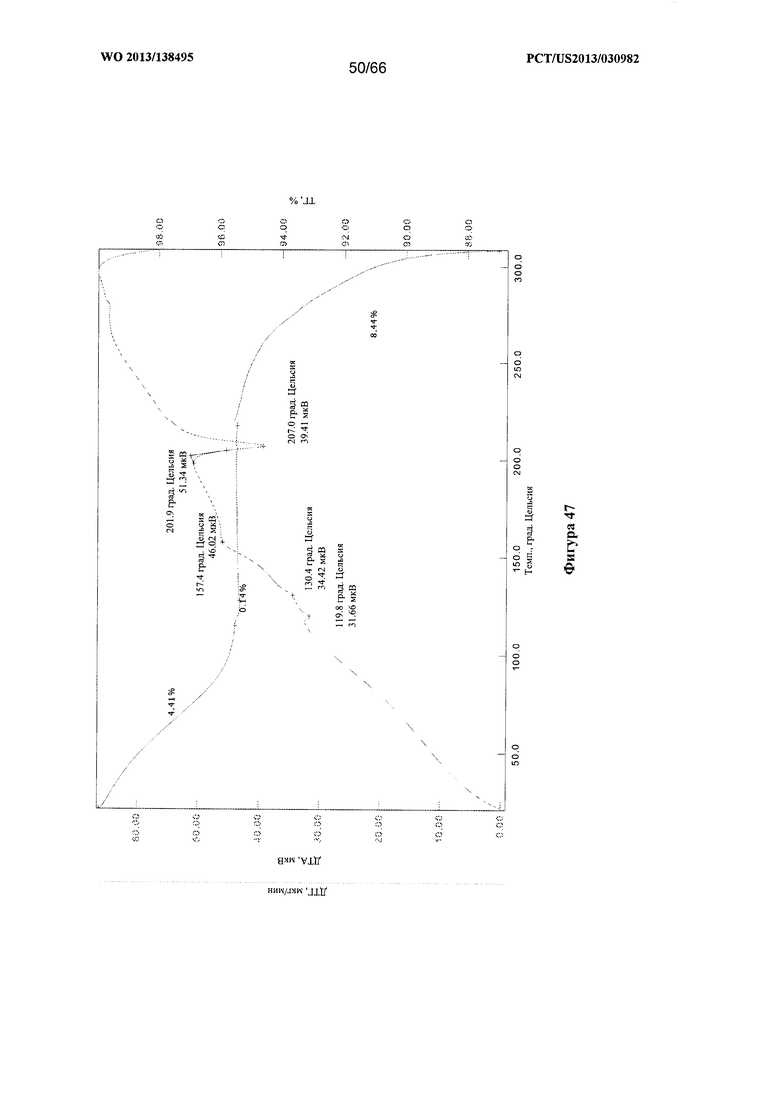
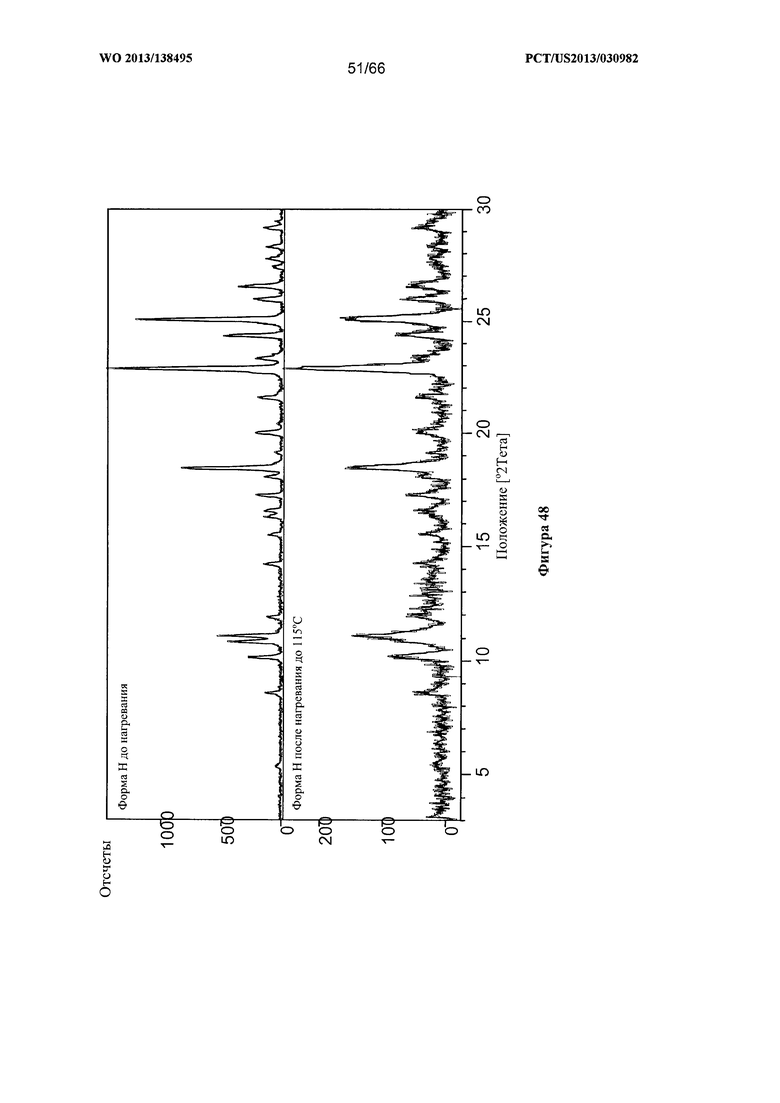
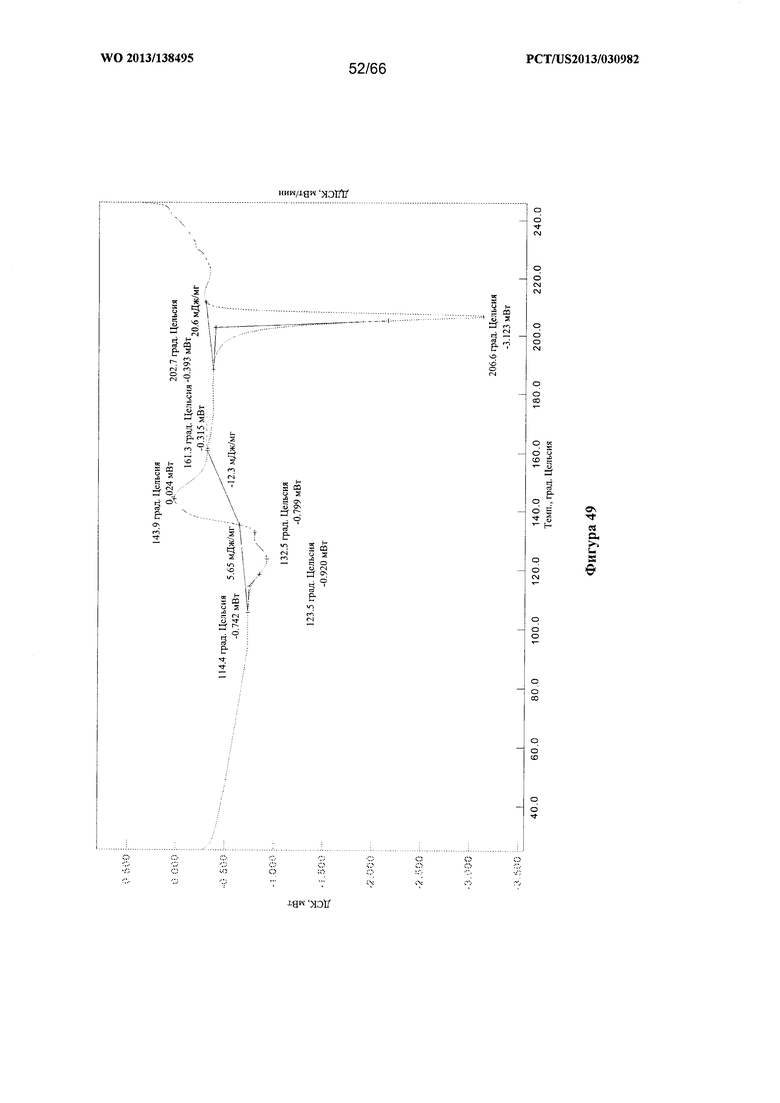
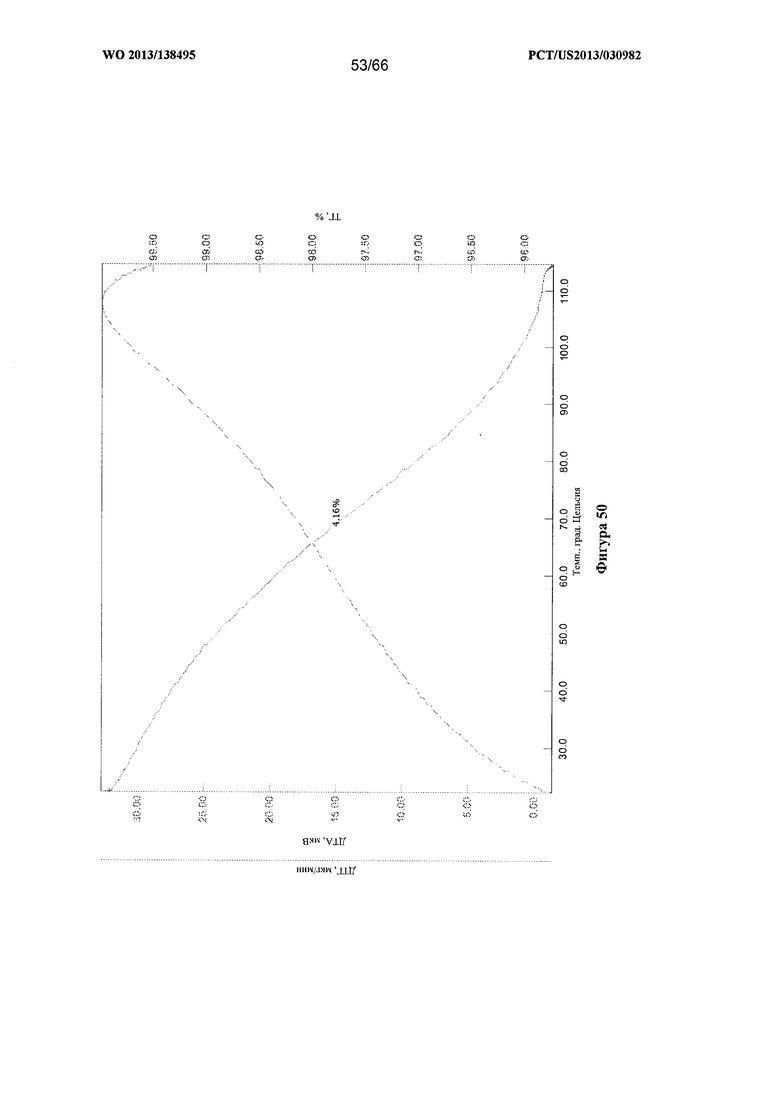
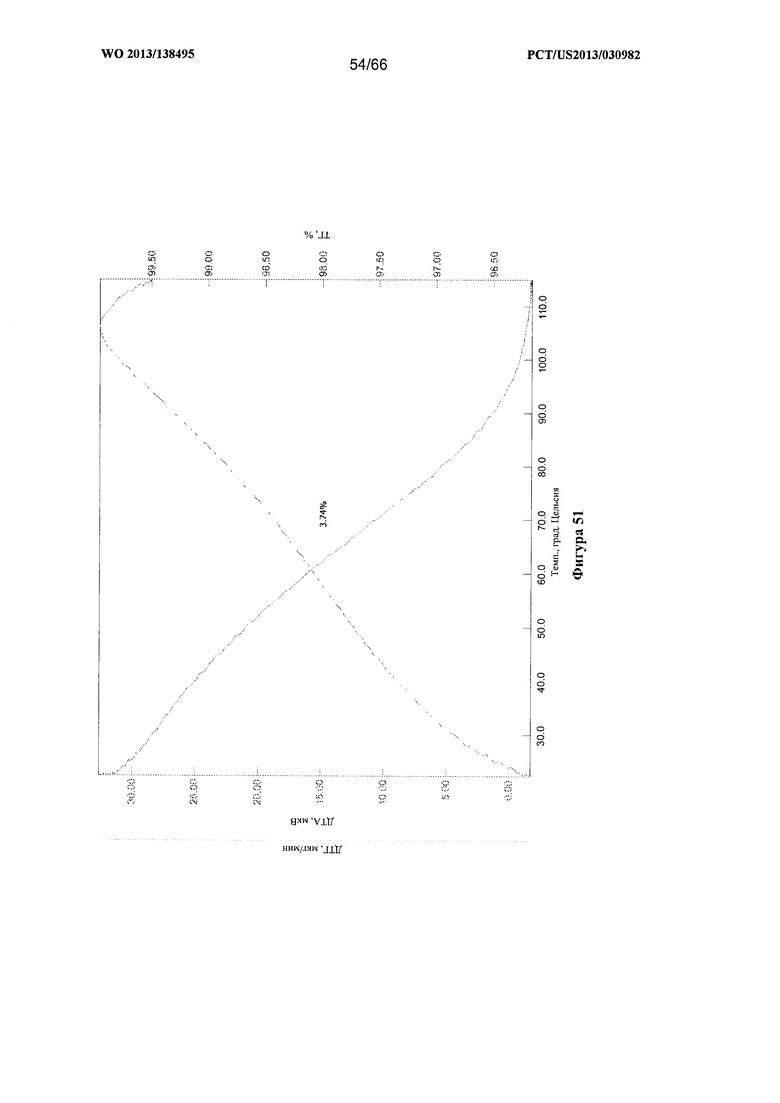
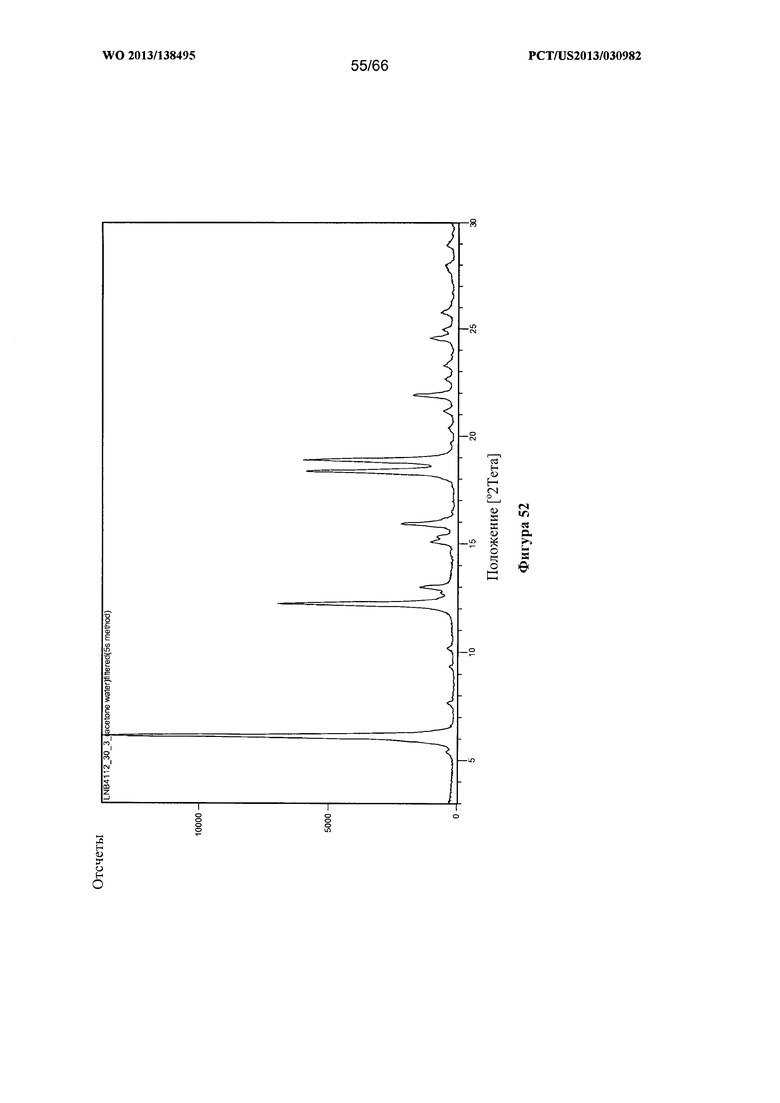
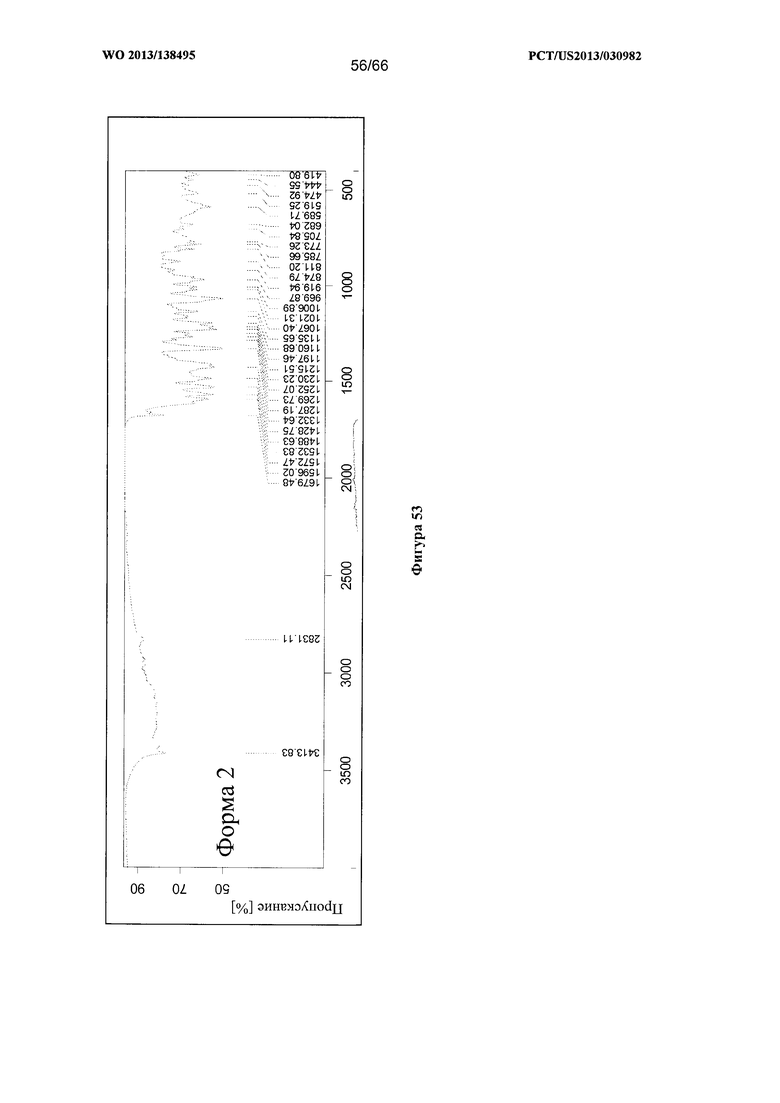
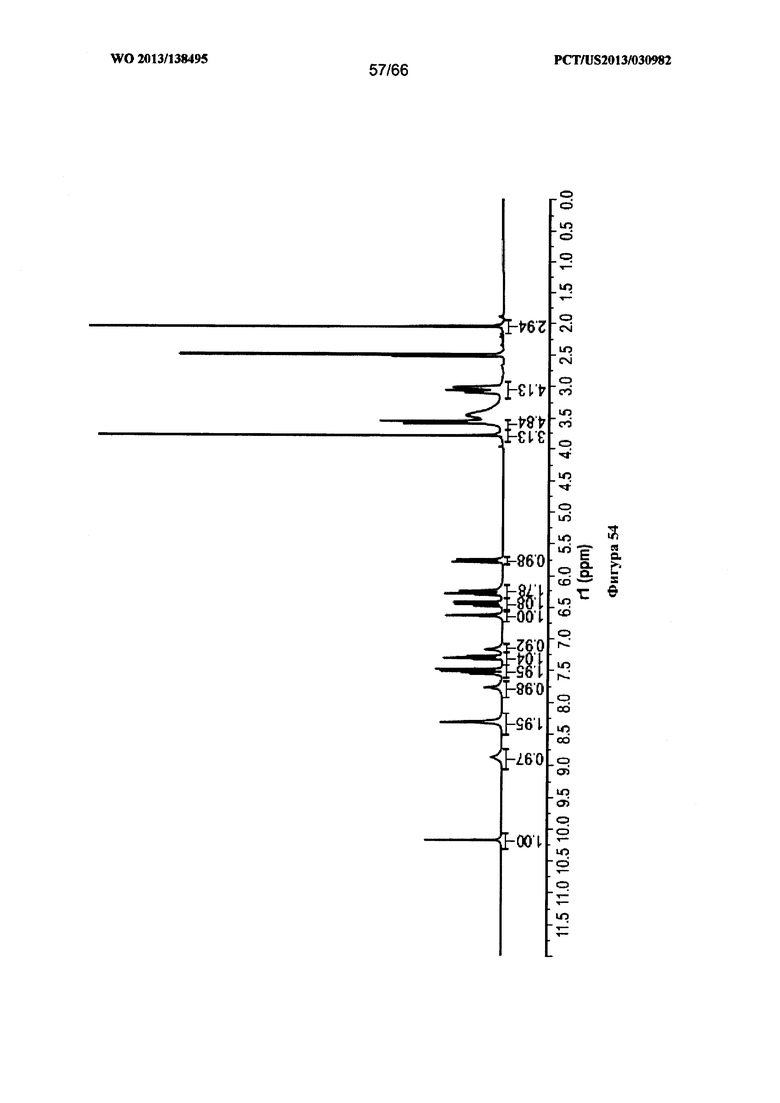
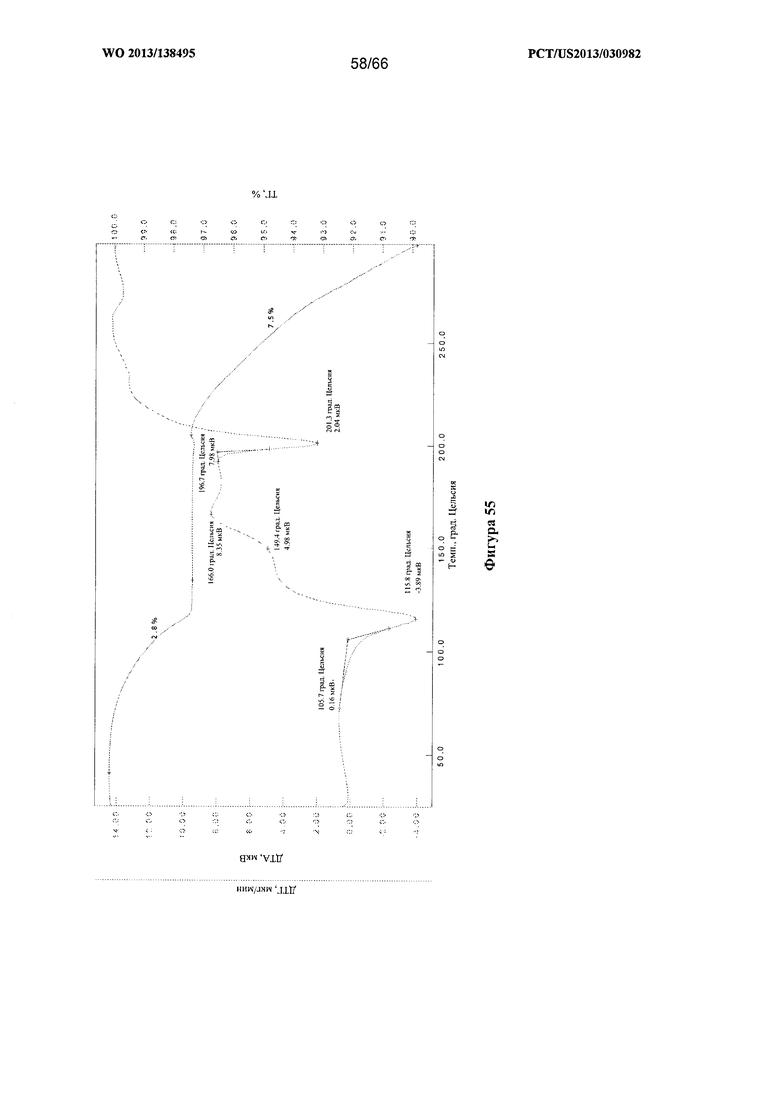
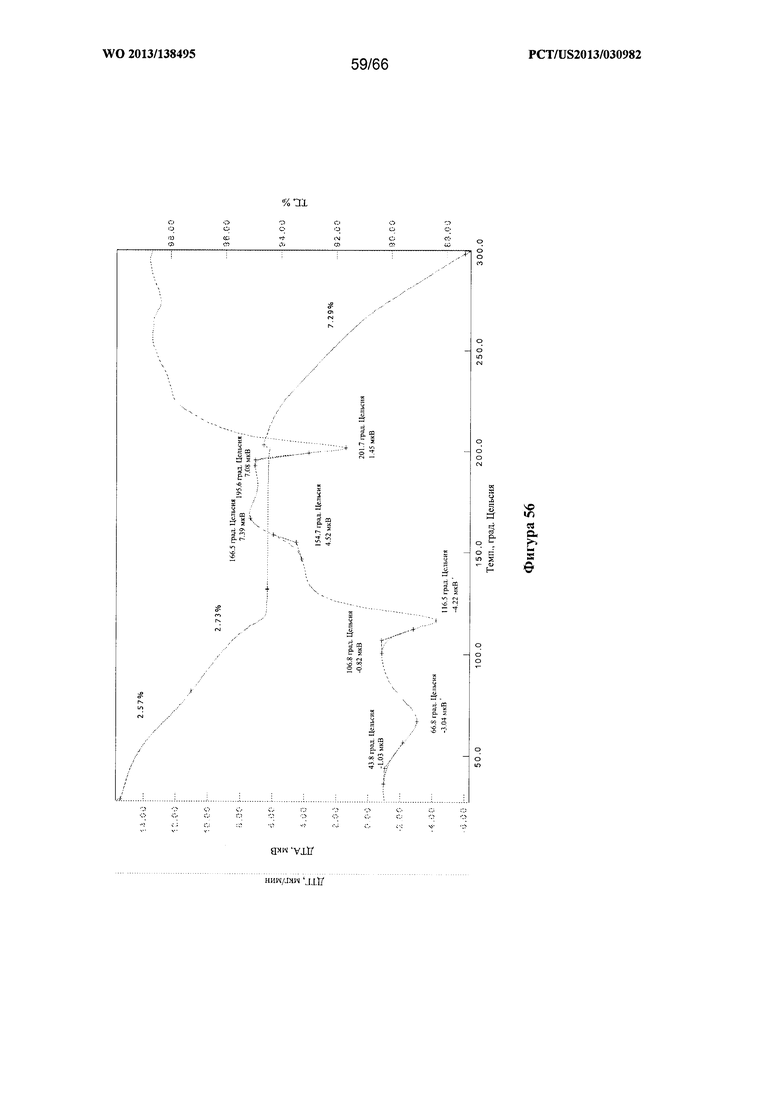

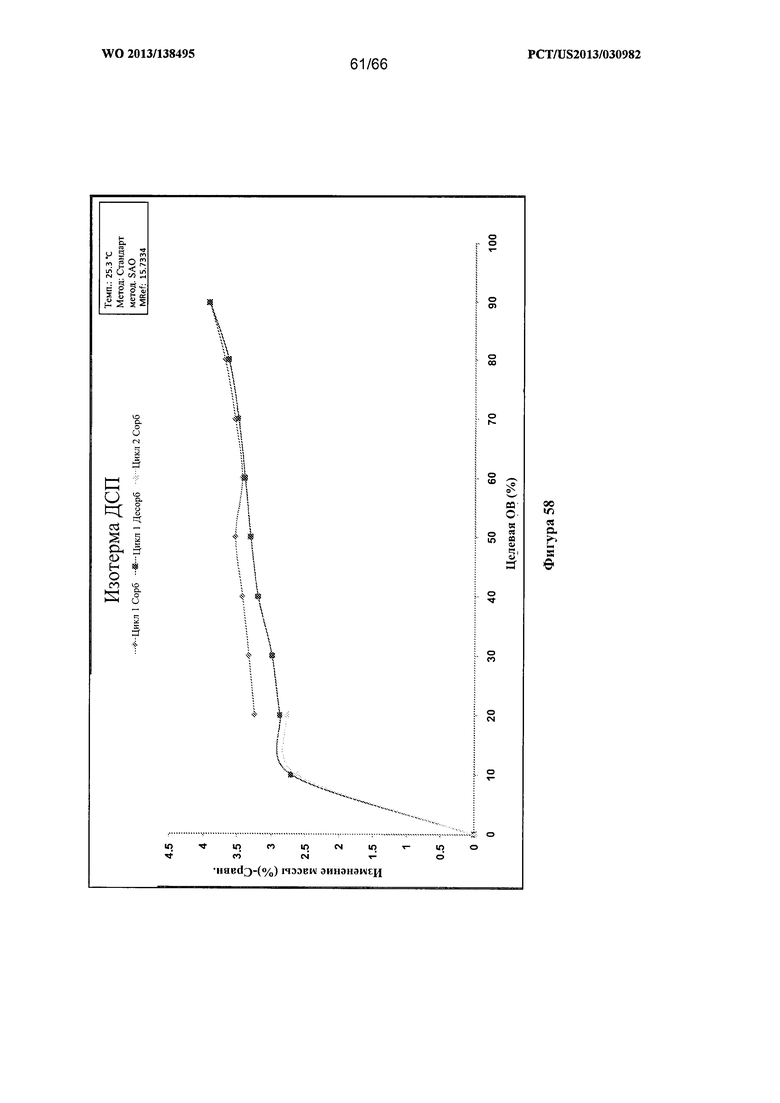
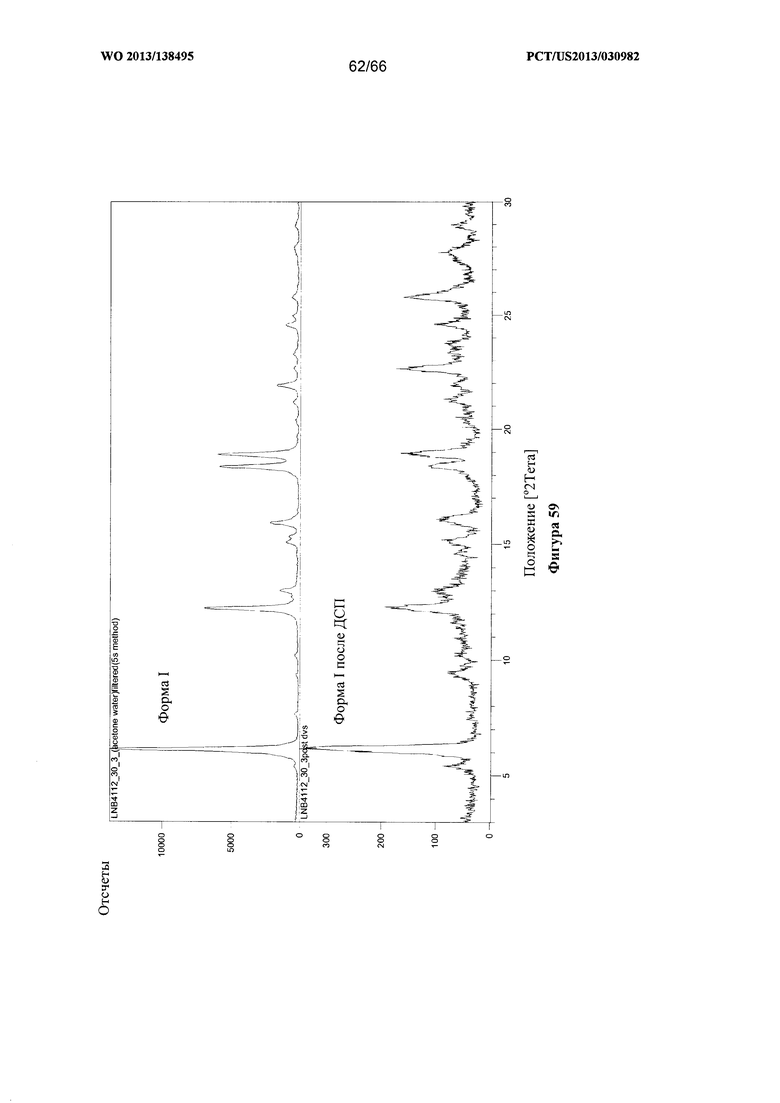
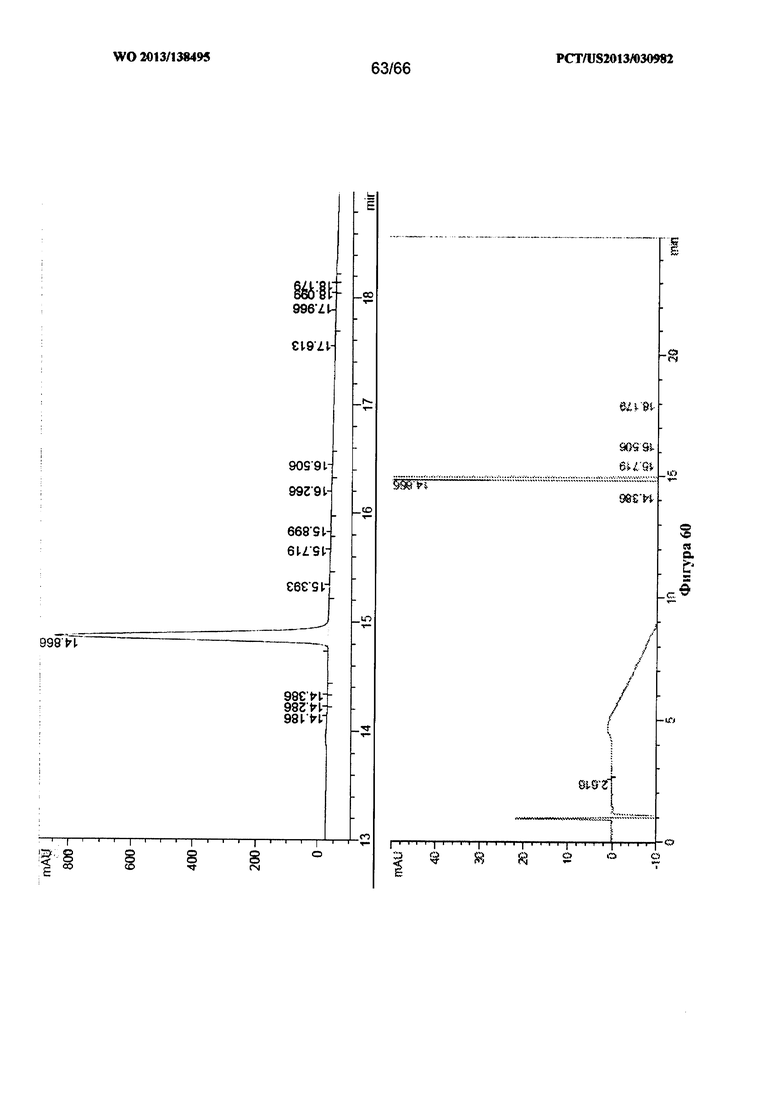
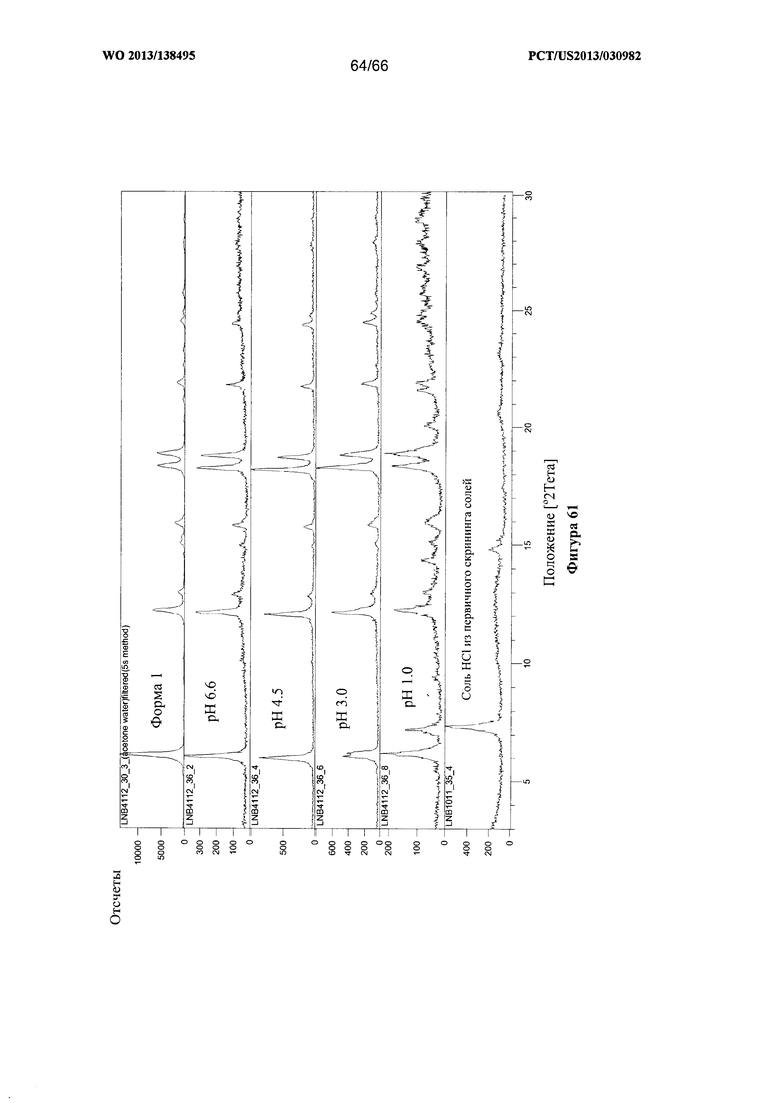
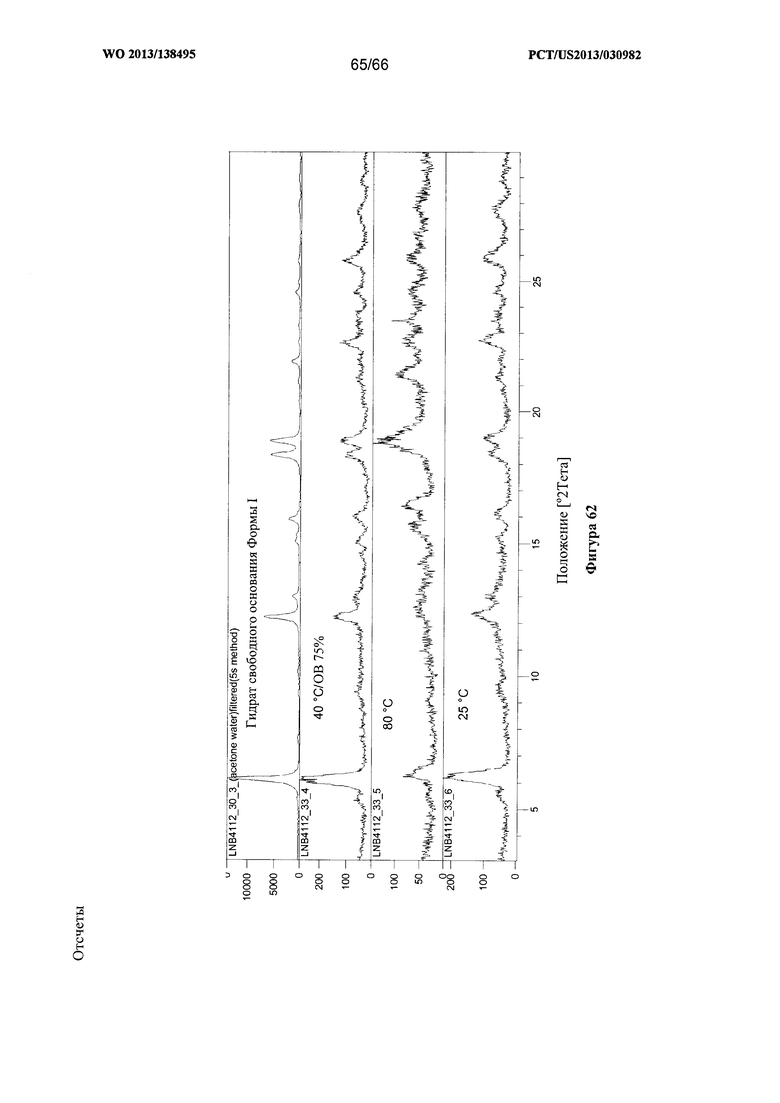
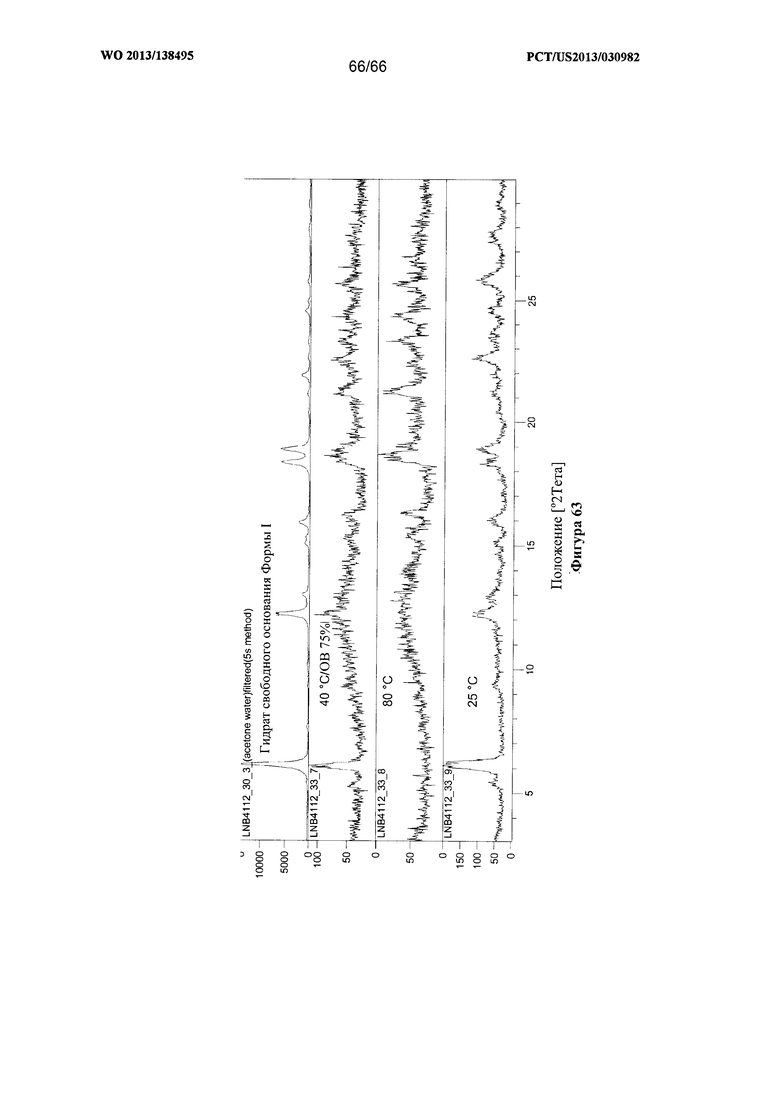
Изобретение относится к кристаллической форме соединения 1, которое представляет собой свободное основание и может быть сольватированным. Соединение обладает свойствами ингибитора киназ рецептора эпидермального фактора роста (EGFR) и может найти применение для лечения заболеваний, связанных с клеточной пролиферацией опосредованной указанной активностью соединения. Кристаллическая форма соединения 1 в виде свободного основания
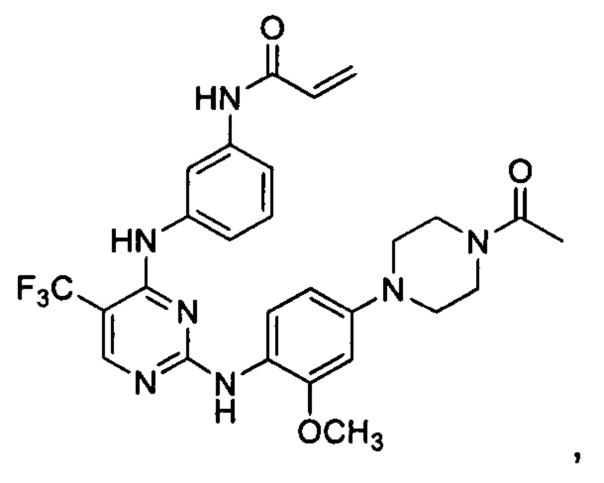 (I)
(I)
характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 6,73±0,3, 18,30±0,3, 18,96±0,3 и 25,48±0,3 градуса 2-тета (форма А) или пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 10,67±0,3, 12,21±0,3, 18,11±0,3, 19,24±0,3 и 21,53±0,3 градуса 2-тета (форма В).
Кристаллическая форма свободного основания соединения 1 также может быть кристаллической сольватированной формой с диметилформамидом и характеризоваться одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 16,32±0,3, 18,82±0,3, 20,26±0,3, 22,58±0,3 и 25,36±0,3 градуса 2-тета (Формой С), или представлять собой сольват с 1,4-диоксаном, который характеризуется пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 18,40±0,3, 19,31±0,3, 20,14±0,3, 20,53±0,3 и 25,25±0,3 градуса 2-тета (Форму D), или сольват с метилэтилкетоном, характеризующийся пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 5,78±0,3, 12,57±0,3, 15,34±0,3, 19,10±0,3 и 24,80±0,3 градуса 2-тета (Форму Е), или сольват с N-метил-2-пирролидоном, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 15,51±0,3, 16,86±0,3, 18,80±0,3, 20,97±0,3 и 23,32±0,3 градуса 2-тета (Форму F) или пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 6,79±0,3, 17,86±0,3, 19,43±0,3, 19,98±0,3 и 22,35±0,3 градуса 2-тета (Форму G). Соединение 1 может представлять собой гидрат, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 10,82±0,3, 11,08±0,3, 18,45±0,3, 22,85±0,3 и 25,06±0,3 градуса 2-тета (Форму Н) или пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 6,13±0,3, 12,22±0,3, 15,91±0,3, 18,35±0,3, 18,88±0,3 и 21,90±0,3 градуса 2-тета (Форму I). 3 н. и 29 з.п. ф-лы, 63 ил., 4 табл., 10 пр.
1. Кристаллическая форма Соединения 1:
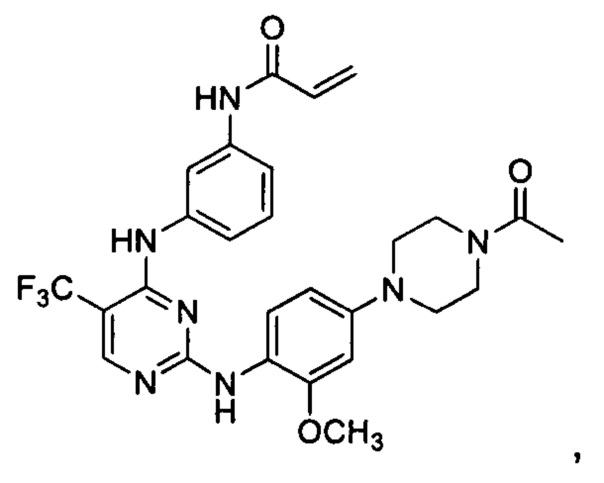
где Соединение 1 представляет собой свободное основание.
2. Кристаллическая форма по п. 1, где Соединение 1 не является сольватированным.
3. Кристаллическая форма по п. 2, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 6,73±0,3, 18,30±0,3, 18,96±0,3 и 25,48±0,3 градуса 2-тета.
4. Кристаллическая форма по п. 3, где указанная кристаллическая форма представляет собой Форму А, и при этом Форма А характеризуется следующими пиками на дифрактограмме рентгеновской порошковой дифракции:
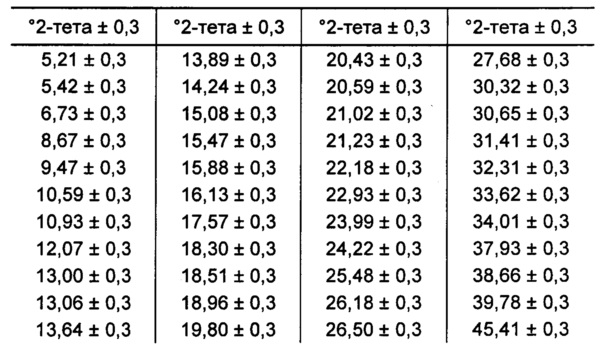
5. Кристаллическая форма по п. 2, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 10,67±0,3, 12,21±0,3, 18,11±0,3, 19,24±0,3 и 21,53±0,3 градуса 2-тета.
6. Кристаллическая форма по п. 5, где указанная кристаллическая форма представляет собой Форму В, и при этом Форма В характеризуется следующими пиками на дифрактограмме рентгеновской порошковой дифракции:
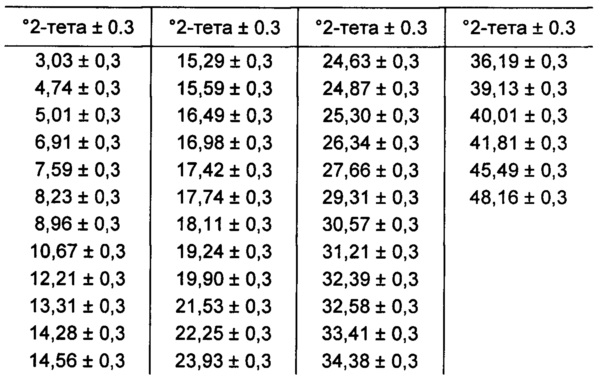
7. Кристаллическая форма по п. 1, где Соединение 1 представляет собой сольват в диметилформамиде.
8. Кристаллическая форма по п. 7, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 16,32±0,3, 18,82±0,3, 20,26±0,3, 22,58±0,3 и 25,36±0,3 градуса 2-тета.
9. Кристаллическая форма по п. 8, где указанная кристаллическая форма представляет собой Форму С, и при этом Форма С характеризуется следующими пиками на дифрактограмме рентгеновской порошковой дифракции:

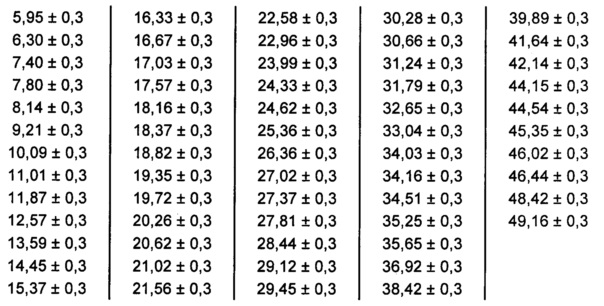
10. Кристаллическая форма по п. 1, где Соединение 1 представляет собой сольват в 1,4-диоксане.
11. Кристаллическая форма по п. 10, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 18,40±0,3, 19,31±0,3, 20,14±0,3, 20,53±0,3 и 25,25±0,3 градуса 2-тета.
12. Кристаллическая форма по п. 11, где указанная кристаллическая форма представляет собой Форму D, и при этом Форма D характеризуется следующими пиками на дифрактограмме рентгеновской порошковой дифракции:
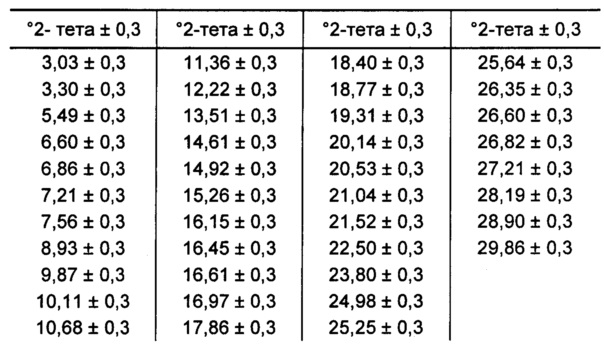
13. Кристаллическая форма по п. 1, где Соединение 1 представляет собой сольват в метилэтилкетоне.
14. Кристаллическая форма по п. 13, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 5,78±0,3, 12,57±0,3, 15,34±0,3, 19,10±0,3 и 24,80±0,3 градуса 2-тета.
15. Кристаллическая форма по п. 14, где указанная кристаллическая форма представляет собой Форму Е, и при этом Форма Е характеризуется следующими пиками на дифрактограмме рентгеновской порошковой дифракции:
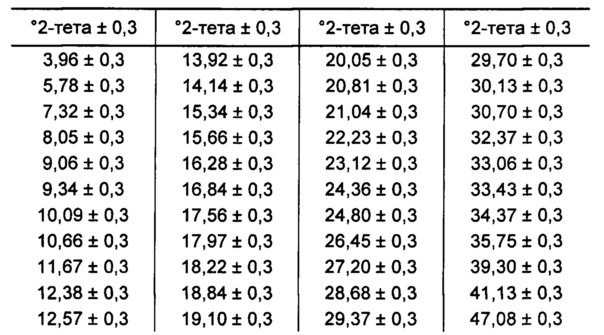
16. Кристаллическая форма по п. 1, где Соединение 1 представляет собой сольват в N-метил-2-пирролидоне.
17. Кристаллическая форма по п. 16, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 15,51±0,3, 16,86±0,3, 18,80±0,3, 20,97±0,3 и 23,32±0,3 градуса 2-тета.
18. Кристаллическая форма по п. 17, где указанная кристаллическая форма представляет собой Форму F, и при этом Форма F характеризуется следующими пиками на дифрактограмме рентгеновской порошковой дифракции:
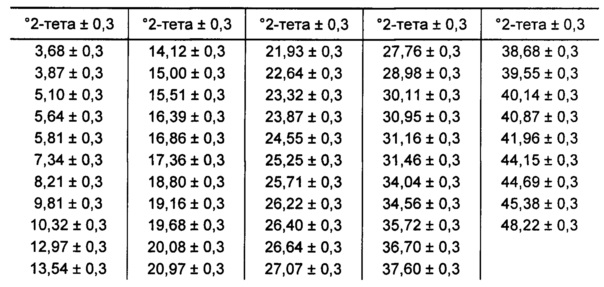
19. Кристаллическая форма по п. 16, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 6,79±0,3, 17,86±0,3, 19,43±0,3, 19,98±0,3 и 22,35±0,3 градуса 2-тета.
20. Кристаллическая форма по п. 19, где указанная кристаллическая форма представляет собой Форму G, и при этом Форма G характеризуется следующими пиками на дифрактограмме рентгеновской порошковой дифракции:
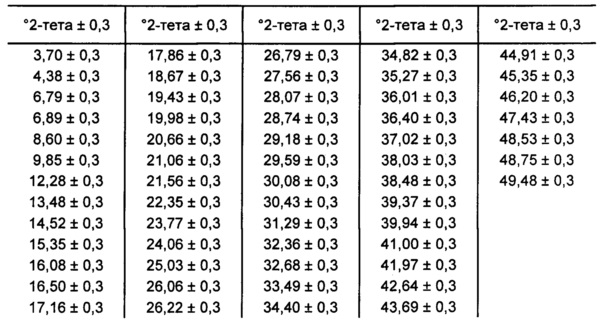
21. Кристаллическая форма по п. 1, где Соединение 1 представляет собой гидрат.
22. Кристаллическая форма по п. 21, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 10,82±0,3, 11,08±0,3, 18,45±0,3, 22,85±0,3 и 25,06±0,3 градуса 2-тета.
23. Кристаллическая форма по п. 22, где указанная кристаллическая форма представляет собой Форму Н, и при этом Форма Н характеризуется следующими пиками на дифрактограмме рентгеновской порошковой дифракции:
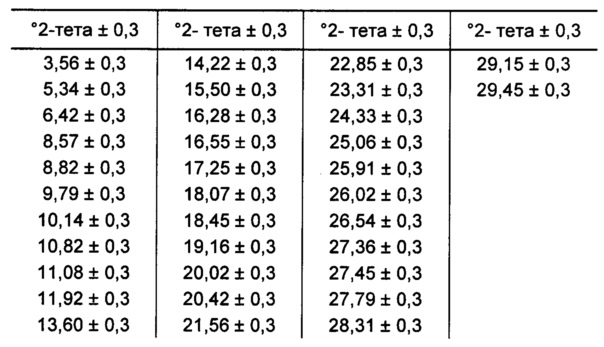
24. Кристаллическая форма по п. 21, где указанная кристаллическая форма характеризуется одним или более пиками на дифрактограмме рентгеновской порошковой дифракции, выбранными из пиков при 6,13±0,3, 12,22±0,3, 15,91±0,3, 18,35±0,3, 18,88±0,3 и 21,90±0,3 градуса 2-тета.
25. Кристаллическая форма по п. 24, где указанная кристаллическая форма представляет собой Форму I, и при этом Форма I характеризуется следующими пиками на дифрактограмме рентгеновской порошковой дифракции:
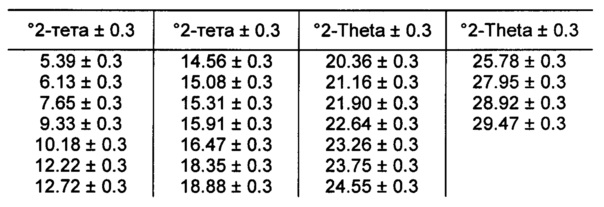
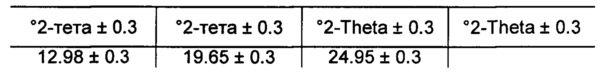
26. Композиция, обладающая свойствами селективного ингибитора по меньшей мере одного мутанта рецептора эпидермельного фактора роста (EGFR) по сравнению с EGFR дикого типа (EGFR ДТ), причем композиция содержит кристаллическую форму по п. 1 и фармацевтически приемлемый носитель или вспомогательное вещество.
27. Применение кристаллической формы по п. 1 для селективного ингибирования по меньшей мере одного мутанта рецептора эпидермального фактора роста (EGFR) по сравнению с EGFR дикого типа (EGFR ДТ).
28. Применение по п. 27, где указанная кристаллическая форма по существу не действует на EGFR ДТ.
29. Применение по п. 27, где указанный по меньшей мере один мутант EGFR представляет собой активирующий мутант, мутант с делецией, мутант с точечной мутацией или мутант, выбранный из Т790М, delE746-A750, L858R или G719S.
30. Применение композиции по п. 26 для лечения опосредованного мутантным EGFR расстройства или состояния.
31. Применение по п. 30, где указанное расстройство или состояние представляет собой рак.
32. Применение по п. 31, где указанный рак представляет собой немелкоклеточный рак легких.
| WO 2010129053 A2, 11.11.2010 | |||
| OGISO H; ET AL Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains CELL, 20020920 CELL PRESS, USVol:110, Nr:6, Page(s): 775 - 787 | |||
| WO 2009158571 A1, 30.12.2009 & RU2536584C2, 27.12.2014 | |||
| WO 2012021444 A1, 16.02.2012 & RU2013109393 (A), 20.09.2014 | |||
| US 6908906 В2, 21.06.2005 | |||
| RU 2013118648A1, приоритет 31.11.2011, опубл.10.12.2014 & US 8975249 B2, 10.03.2015. |

