Изобретение относится к области медицины, а именно к экспериментальной фармакологии, и может быть использовано для количественного определения карнозина в тканях и физиологических жидкостях для решения задач экспериментальной и клинической фармакокинетики.
Природный карнозин экстрагируется из мышц крупного рогатого скота. Один из наиболее используемых способов химического синтеза карнозина - это конденсация при низкой температуре фталоил-β-аланилхлорида с L-гистидином в присутствии триэтиламина, что приводит к синтезу фталоил-β-аланилгистидина. При удалении фталоильной группировки гидразином образуется карнозин. В литературе описаны различные способы разделения и определения карнозина, в частности с использованием титрометрического метода, тонкослойной хроматографии и жидкостной хроматографии высокого давления, позволяющей определять следовые количества карнозина и его производных.
На сегодняшний день известен способ количественного определения карнозина путем прямого кислотного титрования 0,1 M раствором хлороводородной кислоты с последующим потенциометрическим определением точки эквивалентности (Фадеева Д.А., Халикова М.А., Новиков О.О. Применение прямой ацидометрии для количественного определения карнозина., Научные ведомости Белгородского государственного университета, Серия: Медицина. Фармация, 2010, вып. №12-2 (93), том №22, с. 98-100). Однако данный метод не адаптирован для работы с биологическим материалом.
Наиболее близким техническим решением является способ количественного определения карнозина путем масс-спектрометрии MALDI/TOF/MS, при этом получают водный раствор карнозина, который в количестве 0,5 µl наносят на мишень и после высыхания сверху капают каплю матрицы - α-цианокоричной кислоты. При регистрации масс-спектра наблюдается наиболее интенсивный пик молекулярного иона с зарядом m/z=227,489, что соответствует протонированной форме карнозина. Чувствительность методики составляет (0,221 пикомоль (2,21*1012 моль) (Писарев Д.И., Новиков О.О., Васильев Г.В., Селютин О.А. Опыт использования метода MALDI/TOF/MS в фармацевтическом анализе., Научные ведомости Белгородского государственного университета. Серия: Медицина. Фармация, 2012, вып. №10-2 (129), том 18, с. 1-11). Однако данный метод не учитывает влияние на эффективность ионизации компонентов биологического субстрата (плазма крови, ткань мозга и др.). Таким образом, данный метод также не адаптирован для работы с биоматериалами.
Технический результат заявленного нами способа заключается в создании высокоселективного и чувствительного хроматомасс-спектрометрического метода количественного определения карнозина в биологических субстратах, высокоселективность и чувствительность которого, в частности, обусловлена учетом влияния компонентов биологических материалов на эффективность ионизации. Надежность и точность количественного определения обеспечивается применением внутреннего стандарта L-аланил-карнозина. Все перечисленное делает данный метод удобным для изучения фармакокинетики карнозина в эксперименте и в клинике.
Технический результат достигается тем, что определение карнозина в биологических материалах осуществляют путем анализа вещества высокоселективным методом масс-спектрометрии с применением электроспрейной ионизации, при этом предварительно проводят депротеинизацию плазмы крови 10% водным раствором трихлоруксусной кислоты, затем к депротеинизированному образцу добавляют аликвоту раствора внутреннего стандарта L-аланил-карнозина, разделение продуктов экстракции проводят на обращенно-фазной хроматографической колонке 4,6×150 мм с температурой разделения 35°C и скоростью подачи элюента 0,7 мл/мин, при этом в качестве элюента применяют 10 мМ ацетат аммония, подкисленный ледяной уксусной кислотой до pH 3.7, и смесь ацетонитрила с 10 мМ ацетатом аммония в соотношении 90:10, взятыми в процентном соотношении 10:90 соответственно, детектирование карнозина проводят по четырем дочерним ионам с m/z 110.0, 156.1, 180.0, 210.1, образующимся в результате распада молекулярного иона карнозина с m/z 227.1, а его концентрацию рассчитывают по отношению площади хроматографического пика карнозина к площади пика внутреннего стандарта - L-аланил-карнозина.
Способ осуществляется следующим образом.
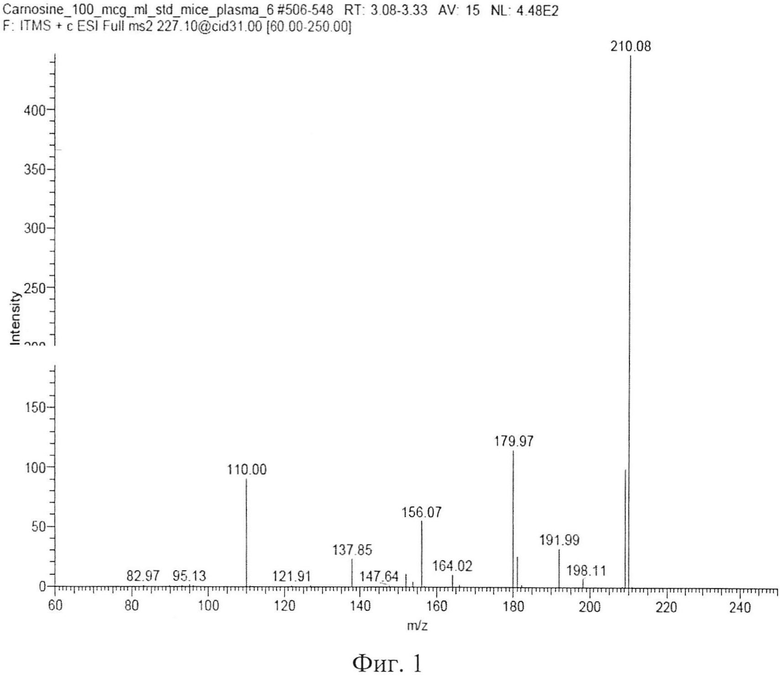
В качестве биологической матрицы используют плазму крови и гомогенат ткани головного мозга мышей. Извлечение карнозина осуществляли методом депротеинизации. К образцу плазмы крови или образцу гомогената ткани мозга (1:2, m:v, мозг : бидистиллят) объемом 100 мкл добавляют 10 мкл раствора внутреннего стандарта L-аланил-карнозина с концентрацией 200 мкг/мл. К полученной смеси приливают 400 мкл 10% водного раствора трихлоруксусной кислоты с целью преципитации протеинов плазмы. Образовавшуюся взвесь денатурированных белков осаждают на ультрацентрифуге со скоростью 13000 об/мин. Надосадочную жидкость осторожно декантируют и переносят в хроматографическую виалу, которую помещают в автосамплер хроматографа для дальнейшего хромато-масс-спектрометрического анализа. При этом для анализа используют Хроматограф - «Finnigan Surveyor LC Pump Plus». Детектор - масс-спектрометрический детектор «LCQ Fleet MS» (квадрупольная ионная ловушка). Аналитическую колонку - Ultrasphere 5 ODS фирмы Hichrom Ltd., Великобритания (150×4,6 мм; 5 мкм). Супернатант инжектируют в петлю хроматографа в объеме 10 мкл. Подвижная фаза состоит из двух растворов: 10 мМ ацетат аммония, подкисленный ледяной уксусной кислотой до pH 3.7 (раствор А) и ацетонитрил с 10 мМ ацетатом аммония в соотношении (90:10) (раствор Б), взятых в процентном соотношении растворов А:Б 90:10 соответственно. Работу проводят в изократическом режиме элюирования. При этом скорость потока подвижной фазы составляет 0,7 мл/мин. Объем пробы - 25 мкл. Температура разделения 35°C. Продолжительность хроматографирования - 10 минут. Время удерживания аналита - 3,15±0,05 минут. Время удерживания внутреннего стандарта L-аланил-карнозин - 3,28±0,05 минут. Детектирование: масс-спектрометрическое, по дочерним ионам с m/z 110.0, 156.1, 180.0, 210.1, образующимся в результате распада молекулярного иона карнозина с m/z 227.1 при нормализованной энергии соударений 35 eV (масс-спектр второго порядка для карнозина представлен на фигуре 1). Внутренний стандарт L-аланил-карнозин детектируют по суммарному ионному току дочерних ионов в диапазоне m/z 75-300, образующихся в результате распада молекулярного иона L-аланил-карнозина с m/z 298.3. Масс-спектрометр работает в режиме регистрации ионов, положительно заряженных электроспреем (ESI), создаваемым напряжением в 5 кВ. Скорость потока газа-небулайзера (азота): 5 л/мин, давление на распылителе - 100 psi. Температура интерфейса капилляра составляет 350°C, температура нагревателя - 300°C. Амплитуда возбуждения на концевых электродах ловушки 0,1 В. В качестве демпфирующего газа в ионной ловушке использовался гелий. Данные обрабатывались с помощью Xcalibur 2.1 w/Foimdation 1.0.1.
Были проведены определения карнозина в плазме крови и гомогенате ткани мозга.
Приготовление реактивов.
Все растворители имели квалификацию «Для хроматографии», реактивы - не ниже «ч.д.а.». Для приготовления растворов стандартных образцов использовали субстанции стандартов карнозина и аланил-карнозина производства Hamati Chemicals Ltd. Для количественного определения готовили маточные растворы стандартов карнозина и аланил-карнозина в метаноле с концентрациями 1 мг/мл. Раствор карнозина применяли для приготовления растворов рабочих стандартных образцов на плазме крови с концентрациями 3,13 мкг/мл; 6,25 мкг/мл; 12,5 мкг/мл; 25 мкг/мл; 50 мкг/мл; 100 мкг/мл; 200 мкг/мл; 400 мкг/мл; 800 мкг/мл; 1600 мкг/мл.
На основании пилотного исследования было установлено, что в мозге максимальная тканевая концентрация карнозина была значительно меньше, чем в крови. Соответственно градуировку для количественного определения карнозина получали в концентрационном диапазоне: 0,39-50 мкг/мл: 0,39 мкг/мл; 0,78 мкг/мл; 1,56 мкг/мл; 3,13 мкг/мл; 6,25 мкг/мл, 12,5 мкг/мл, 25 мкг/мл, 50 мкг/мл. Раствор внутреннего стандарта с концентрацией 1 мг/мл разбавляли в 5 раз для получения рабочего раствора аланил-карнозина с концентрацией 200 мкг/мл.
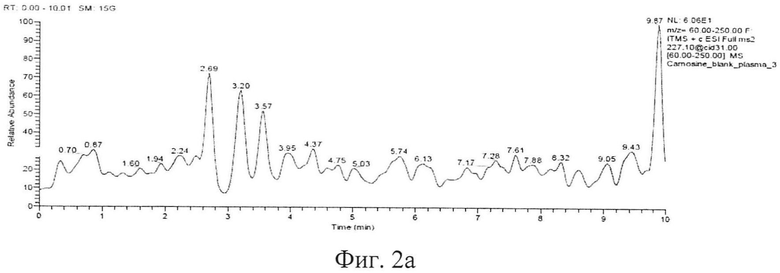
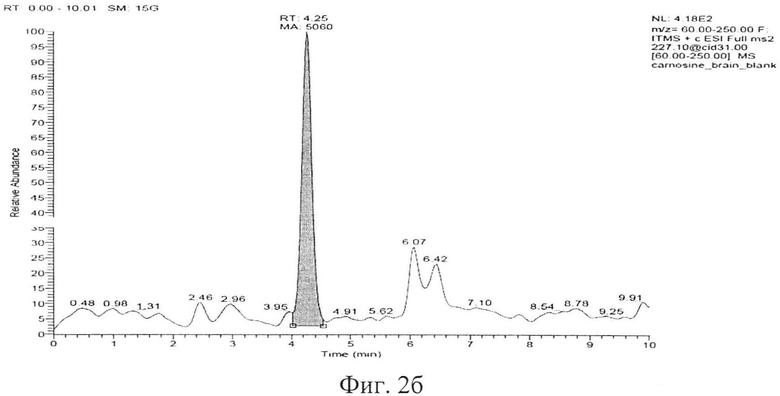
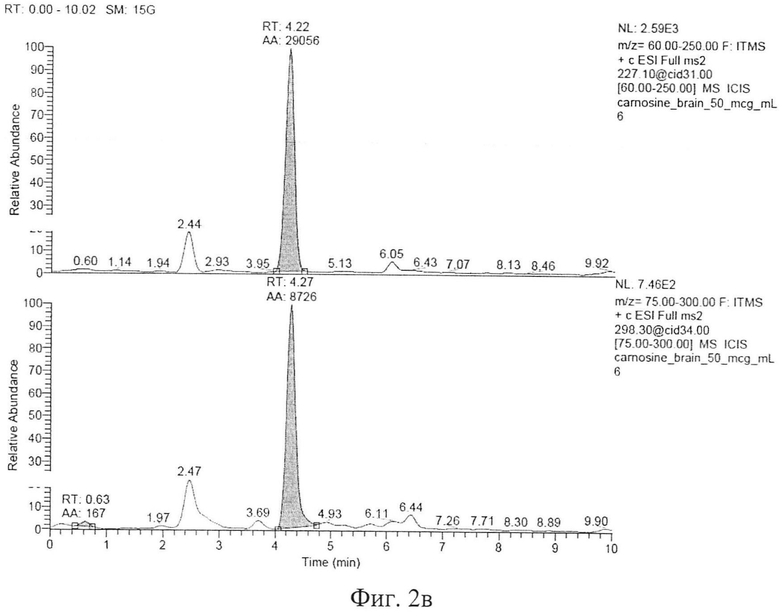
Типичные хроматограммы образцов интактной плазмы крови, гомогената интактного мозга и демонстрационная хроматограмма образца гомогената ткани мозга с концентрацией карнозина 50 мкг/мл представлены на фигуре 2а, б, в. На фиг. 2а представлена хроматограмма депротеинизированного образца интактной плазмы крови. На фиг. 2б, представлена хроматограмма депротеинизированного образца ткани мозга (пик с TR=4,25 мин - пик базального карнозина). На фигуре 2в представлена хроматограмма депротеинизированного образца гомогената ткани мозга с концентрацией карнозина 50 мкг/мл, верхний пик - пик аналита, нижний пик - пик внутреннего стандарта.
Количественное определение карнозина.
По причине того, что в интактной крови мышей карнозин отсутствует, а в мозге его базальная концентрация довольно высока, а также по причине существенных отличий в матричном эффекте со стороны разных биосубстратов, было принято решение сделать две самостоятельные градуировки для карнозина в мозге и карнозина в крови. Градуировочная зависимость для карнозина в плазме крови описывалась формулой:
С=158,230602×AR, где С - концентрация карнозина (мкг/мл), AR (Area Ratio) - отношение площадей пиков аналита и внутреннего стандарта. Коэффициент корреляции составил R2=0.9986, что соответствует надлежащей аналитической аппроксимации.
Поскольку в работе использовался гомогенат ткани мозга, полученный при гомогенизации мозга мышей в дистиллированной воде, взятой в соотношении 1:2 (m:v), концентрация, полученная при определении содержания вещества в гомогенате, должна быть перемножена на 3 (С(мкг/г)=3*С(мкг/мл). Кроме того, в связи с наличием довольно высокого содержания карнозина в ткани интактного мозга, для определения содержания экзогенного карнозина был использован следующий метод перерасчета, с учетом поправки на базальный карнозин:
Сэкз=(Sобщ-Sбаз)/SIS*0,0746885, где Сэкз - концентрация экзогенного карнозина, Sобщ - площадь хроматографического пика карнозина в образце гомогената мозга животного, получавшего экзогенный карнозин, Sбаз - рассчитанная средняя площадь пика базального карнозина, SIS - площадь пика внутреннего стандарта. За площадь пика базального карнозина принимали среднее значение, рассчитанное из площадей пиков карнозина в гомогенате мозга 8-ми интактных мышей.
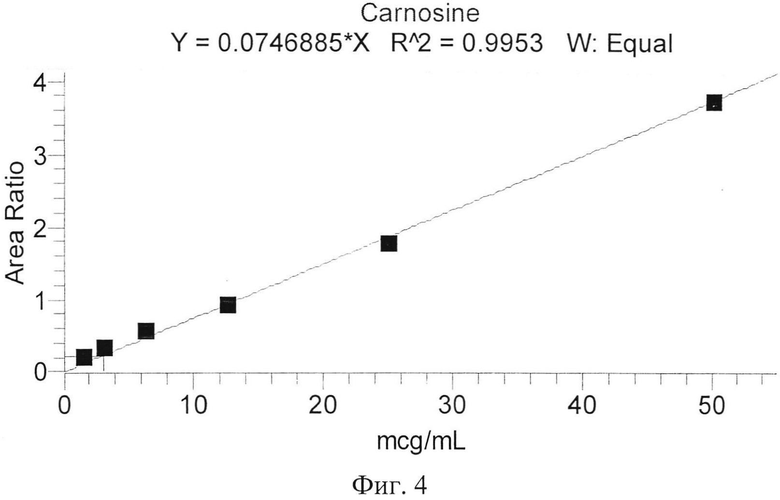
Калибровочные кривые представлены на фиг. 3, 4. На фиг. 3 представлена калибровочная зависимость карнозина в плазме крови. На фиг. 4 представлена калибровочная зависимость карнозина в гомогенате ткани мозга.
Точность и воспроизводимость определения карнозина в биологических материалах.
Точность выражалась в виде коэффициента вариации (% C.V.) для каждой серии образцов согласно уравнению: 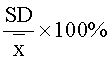
SD - стандартное отклонение серии определений карнозина;

Воспроизводимость измерялась как процент отклонения (% dev.) от теоретического значения по формуле: 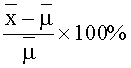
Для метрологической валидации полученной методики определяли точность в течение рабочего дня. Каждый из образцов, предназначенных для контроля качества, анализировали в течение 1 рабочего дня (6 определений). Также провели контроль во время исследования и после его окончания.
Результаты представлены в таблице 1 и 2.

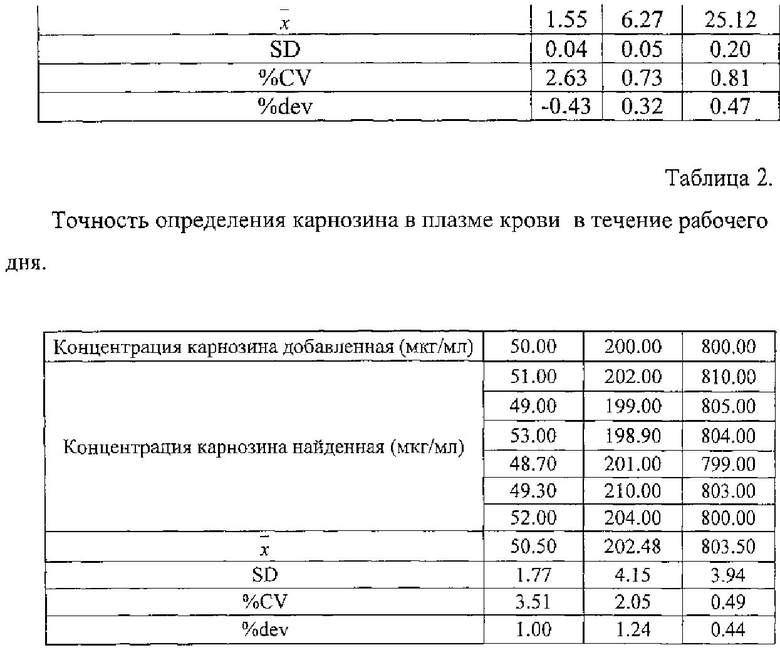
Относительная ошибка определения карнозина не превышала 10%.
Таким образом, заявленный способ обладает высокой селективностью и чувствительностью, не требует проведения многочисленных предварительных химических реакций и не предусматривает использование большого количества химических реактивов.
Высокая точность и воспроизводимость биоаналитического метода количественного определения карнозина в биологических жидкостях и тканях обеспечивает идентификацию вещества с установленными характеристиками погрешности, что позволяет использовать данную методику как в экспериментальной, так и клинической фармакокинетике.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения топирамата в плазме крови | 2016 |
|
RU2631613C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОКСИМА ПИНОСТРОБИНА В ПЛАЗМЕ КРОВИ | 2015 |
|
RU2568876C1 |
| Способ количественного определения амантадина в плазме крови | 2017 |
|
RU2650968C1 |
| Способ количественного определения леводопы в плазме крови | 2017 |
|
RU2665164C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ 2-ФЕНОКСИЭТАНОЛА В БИОЛОГИЧЕСКИХ СРЕДАХ | 2021 |
|
RU2776730C1 |
| Способ количественного определения салицилатов в плазме крови | 2016 |
|
RU2622996C1 |
| Способ количественного определения ликарбазепина в плазме крови | 2017 |
|
RU2660364C1 |
| Способ количественного определения дисульфирама в биологических средах | 2019 |
|
RU2701524C1 |
| Средство, обладающее антиагрегантной, цитопротекторной и антиоксидантной активностью | 2018 |
|
RU2694061C1 |
| Способ определения концентрации противовирусных препаратов в биоматериалах лабораторных животных | 2020 |
|
RU2748249C1 |

Изобретение относится к области медицины, а именно к экспериментальной фармакологии, и может быть использовано для количественного определения карнозина в тканях и физиологических жидкостях. Определение карнозина в биологических материалах осуществляют высокоселективным методом масс-спектрометрии с применением электроспрейной ионизации. При этом предварительно осуществляют депротеинизацию плазмы крови с помощью 10% водного раствора трихлоруксусной кислоты. Затем к депротеинизированному образцу добавляют аликвоту раствора внутреннего стандарта L-аланил-карнозина. А разделение продуктов экстракции проводят на обращенно-фазной хроматографической колонке 4,6×150 мм с температурой разделения 35°C и скоростью подачи элюента 0,7 мл/мин. Причем в качестве элюента применяют 10 мМ ацетат аммония, подкисленный ледяной уксусной кислотой до pH 3.7, и смесь ацетонитрила с 10 мМ ацетатом аммония в соотношении 90:10, взятые в процентном соотношении 10:90 соответственно. Детектирование карнозина проводят по четырем дочерним ионам с m/z 110.0, 156.1, 180.0, 210.1, образующимся в результате распада молекулярного иона карнозина с m/z 227.1. А концентрацию карнозина рассчитывают по отношению площади хроматографического пика карнозина к площади пика внутреннего стандарта - L-аланил-карнозина. Изобретение обеспечивает высокоселективный и чувствительный хроматомасс-спектрометрический метод количественного определения карнозина в биологических субстратах. 6 ил., 2 табл., 1 пр.
Способ определения карнозина в биологических материалах, включающий анализ вещества высокоселективным методом масс-спектрометрии с применением электроспрейной ионизации, отличающийся тем, что предварительно осуществляют депротеинизацию плазмы крови с помощью 10% водного раствора трихлоруксусной кислоты, затем к депротеинизированному образцу добавляют аликвоту раствора внутреннего стандарта L-аланил-карнозина, разделение продуктов экстракции проводят на обращенно-фазной хроматографической колонке 4,6×150 мм с температурой разделения 35°C и скоростью подачи элюента 0,7 мл/мин, при этом в качестве элюента применяют 10 мМ ацетат аммония, подкисленный ледяной уксусной кислотой до pH 3.7, и смесь ацетонитрила с 10 мМ ацетатом аммония в соотношении 90:10, взятые в процентном соотношении 10:90 соответственно, детектирование карнозина проводят по четырем дочерним ионам с m/z 110.0, 156.1, 180.0, 210.1, образующимся в результате распада молекулярного иона карнозина с m/z 227.1, а его концентрацию рассчитывают по отношению площади хроматографического пика карнозина к площади пика внутреннего стандарта - L-аланил-карнозина.
| Фадеева Д.А., Халикова М.А., Новиков О.О Применение прямой ацидометрии для количественного определения карнозина., Научные ведомости Белгородского государственного университета, Серия: Медицина | |||
| Фармация, 2010, вып | |||
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Писарев Д.И., Новиков О.О., Васильев Г.В., Селютин О.А | |||
| Опыт использования метода MALDI/TOF/MS в | |||

