Изобретение относится к области медицины, а именно к клинической фармакологии, и может быть использовано для количественного определения леводопы (3-гидрокси-L-тирозина) в плазме крови для решения задач лекарственного мониторинга при лечении пациентов, страдающих болезнью Паркинсона.
Среди всех дофаминергических агентов наиболее эффективным препаратом для лечения болезни Паркинсона по-прежнему остается такое соединение, как леводопа (3-гидрокси-L-тирозин). Считается, что леводопа особенно эффективна при наиболее ярких проявлениях этого заболевания, таких, как брадикинезия и ригидность. Фармакокинетика леводопы имеет ряд особенностей, которые негативно сказываются на реализации ее фармакологических эффектов. Леводопа имеет низкую гастроинтестинальную биодоступность, низкую жирорастворимость и транспортируется из кишечника в кровь, а также из крови в мозг посредством активного транспорта. Скорость опустошения желудка является принципиальным ограничивающим фактором в распределении леводопы. Леводопа сама влияет на процесс эвакуации содержимого желудка у здоровых волонтеров, что ведет к нарушению ее абсорбции в желудочно-кишечном тракте. Недавние исследования пациентов с болезнью Паркинсона показали, что одновременный прием леводопы с протеинами нередко вызывает извращение фармакологического эффекта препарата. У пожилых пациентов с болезнью Паркинсона наблюдается увеличение площади под кривой для леводопы после перорального приема, что обусловлено уменьшением перорального клиренса препарата (-24%). Более того, в данной подгруппе пациентов наблюдалась более высокая частота появления множественных всплесков концентрации леводопы в плазме крови. Эти, а также прочие причины, влияющие на уровень концентрации леводопы в крови могут вызывать у пациентов хореиформные гиперкинезы, причем зачастую бывает сложно отдифференцировать дискинезии, вызванные высокой концентрацией леводопы в крови от дискинезий вызванных резким спадом концентрации препарата. Таким образом, повышенная вариабельность фармакокинетики леводопы обуславливает необходимость индивидуализации лекарственной терапии с применением арсенала методов персонализованной медицины - а именно терапевтического лекарственного мониторинга и стратегии рационального дозирования.
Терапевтический лекарственный мониторинг требует применения адекватного метода количественного анализа. Существующие методы анализа, такие как газовая хроматография/масс-спектрометрия, как правило, довольно сложны в исполнении, по причине необходимости применения сложной многостадийной пробоподготовки с последующей дериватизацией леводопы. Это является причиной того, что рутинный анализ леводопы представлен в очень малом числе клинических лабораторий, а также того, что терапевтический лекарственный мониторинг этого препарата недостаточно распространен. Создание метода анализа леводопы на основе жидкостной тандемной хроматомасс-спектрометрии может позволить существенно упростить пробоподготовку, уменьшить продолжительность работы и стоимость анализа одного образца, а также улучшить селективность и специфичность метода.
Ближайшим техническим решением определения леводопы в плазме крови является способ, описанный в статье: I.C. Cesar, R.M. Byrro, E.S. Santana, I.M. Mundim, L. Souza Teixeira, S.A. Gomes, R.R. Bonfim, and G.A. Pianetti. Development and validation of a high-performance liquid chromatography-electrospray ionization-MS/MS method for the simultaneous quantitation of levodopa and carbidopa in human plasma. Journal of mass spectrometry: JMS. 46:943-948 (2011). Способ осуществляется путем высокоэффективной жидкостной тандемной хроматомасс-спектрометрии с электрораспылительной ионизацией для одновременного количественного определения леводопы и карбидопы в плазме крови. Для очистки плазмы использовали простую стадию осаждения белка с помощью хлорной кислоты, а метилдопу добавляли в качестве внутреннего стандарта. Анализ проводили с использованием колонки АСЕ С (18) (размер частиц размером 50×4,6 мм, размер частиц 5 мкм) и подвижной фазы, состоящей из 0,2% муравьиной кислоты и ацетонитрила (90:10). Трехквадрупольный масс-спектрометр работал при положительном электрораспылении в режиме мониторинга выбранных реакций для обнаружения ионных переходов m/z 198.1→m/z 107,0, m/z 227,2→m/z 181,0 и m/z 212,1→m/z 139,2 для леводопы, карбидопы и метилдопы соответственно. Метод был валидирован и оказался линейным и точным в диапазоне 50-5000 нг/мл для леводопы и 3-600 нг/мл для карбидопы. Однако описанный в указанной статье метод имеет ряд недостатков. В частности, описанный авторами метод пробоподготовки, основанный на преципитации белков плазмы перхлористой кислотой не позволяет направленно изолировать целевые соединения, в результате чего при хроматографировании биологических образцов происходит сильный матричный эффект, обусловленный присутствием коэкстрактивных веществ. Данный метод адаптирован авторами для применения на трехквадрупольных тандемных масс-детекторов типа API 5000 triple quadrupole mass spectrometer (MDS-SCIEX, Concord, Ontario, Canada), которые являются наиболее чувствительными приборами в своем классе. Созданный нами метод включает в себя стадию экстракции анализируемых соединений на оксиде алюминия с двухэтапной очисткой от коэкстрактивных веществ, что позволяет уменьшить влияние матричного эффекта. Кроме того, созданный нами метод подразумевает концентрирование образца, что делает его применимым для использования на приборах более бюджетного класса со средними показателями по чувствительности - например, на приборах с детекторами типа ионных ловушек. Авторами вышеуказанной публикации для хроматографирования используется короткая обращенно-фазная колонка с октадецилсилильной привитой фазой С18, в то время как мы используем фенилгиксильный сорбент, который позволяет добиваться лучшего и более селективного разделения аминов и других полярных соединений. Кроме того, определение леводопы в ближайшем аналоге возможно лишь в диапазоне от 0,05 до 5 мкг/мл, что подходит для применения, например, в исследованиях биоэквивалентности, но не подходит для терапевтического лекарственного мониторинга, поскольку известно, что верхняя граница диапазона терапевтических концентраций леводопы у больных паркинсонизмом может быть значительно выше 5 мкг/мл (J.I. Sage, М.Н. Mark, D.M. McHale, Р.K. Sonsalla, and D. Vitagliano. Benefits of monitoring plasma levodopa in Parkinson's disease patients with drug-induced chorea. Annals of neurology. 29:623-628 (1991)), что учитывалось нами при разработке метода (был заложен калибровочный диапазон градуировки 0,156-10 мкг/мл).
Нами разработан ВЭЖХ-масс-спектрометрический метод анализа содержания леводопы в плазме крови для фармакокинетического сопровождения лечения пациентов, страдающих болезнью Паркинсона, получающих леводопу как в виде монотерапии, так и в комбинации с другими противопаркинсоническими препаратами. Разработанный нами метод обладает высокой эффективностью в проведении анализа, не требует использования большого количества химических реактивов. Высокая точность и чувствительность представленного метода количественного определения леводопы в плазме крови обеспечивает идентификацию вещества с установленными характеристиками погрешности, что позволяет использовать данную методику, как в экспериментальных, так и в клинических фармакокинетических исследованиях.
Технический результат заключается в создании способа с высокой воспроизводимостью и точностью определения леводопы в плазме крови и наиболее пригодного для решения задач экспериментальной и клинической фармакокинетики.
Технический результат достигается тем, что определение леводопы в плазме крови проводят, анализируя кровь на ее наличие путем очистки плазмы крови, проведения хроматомасс-спектрометрии с использованием матрицы в виде плазмы крови с леводопой и внутреннего стандарта, вещества, близкого по строению молекулы к анализируемому веществу - метилдопы, при этом разделение продуктов экстракции проводят на обращенно-фазной хроматографической колонке, а в качестве элюента применяют муравьиную кислоту и ацетонитрил, с последующим расчетом концентрации леводопы, при этом очистку проводят методом твердофазной экстракции на активированном оксиде аллюминия, образовавшуюся смесь встряхивают в течение 5 минут на вортекс-миксере, а затем центрифугируют на скорости 3500 об/мин для осаждения взвеси оксида аллюминия. надосадочную жидкость осторожно декантируют и отбрасывают, а осадок двукратно промывают деионизированной водой, затем леводопу и метилдопу элюируют с оксида аллюминия 0,5 молярным водным раствором уксусной кислоты, а разделение продуктов экстракции проводят на обращенно-фазной хроматографической колонке 4,6×250 мм с фенилгексилом в качестве неподвижной фазы, содержащий линкерный гексильный фрагмент, соединяющий фенильную группу с поверхностью силикагеля, при этом в качестве элюента применяют 0,1% водный раствор муравьиной кислоты - раствор А и ацетонитрил с добавлением 0,1% муравьиной кислоты - раствор Б, взятых в процентном соотношении раствора А к раствору Б 85:15 соответственно, с температурой разделения 35°С, и скоростью подачи элюента 0,8 мл/мин, детектирование леводопы проводят по суммарному ионному току дочерних ионов в диапазоне от 70 до 200 а.е.м., образующихся в результате фрагментации родительского молекулярного иона леводопы с m/z 198.1, а его концентрацию рассчитывают по формуле: С=4,384714×AR, где С - концентрация леводопы (мкг/мл), AR - отношение площади хроматографического пика леводопы к площади пика внутреннего стандарта.
Способ осуществляется следующим образом.
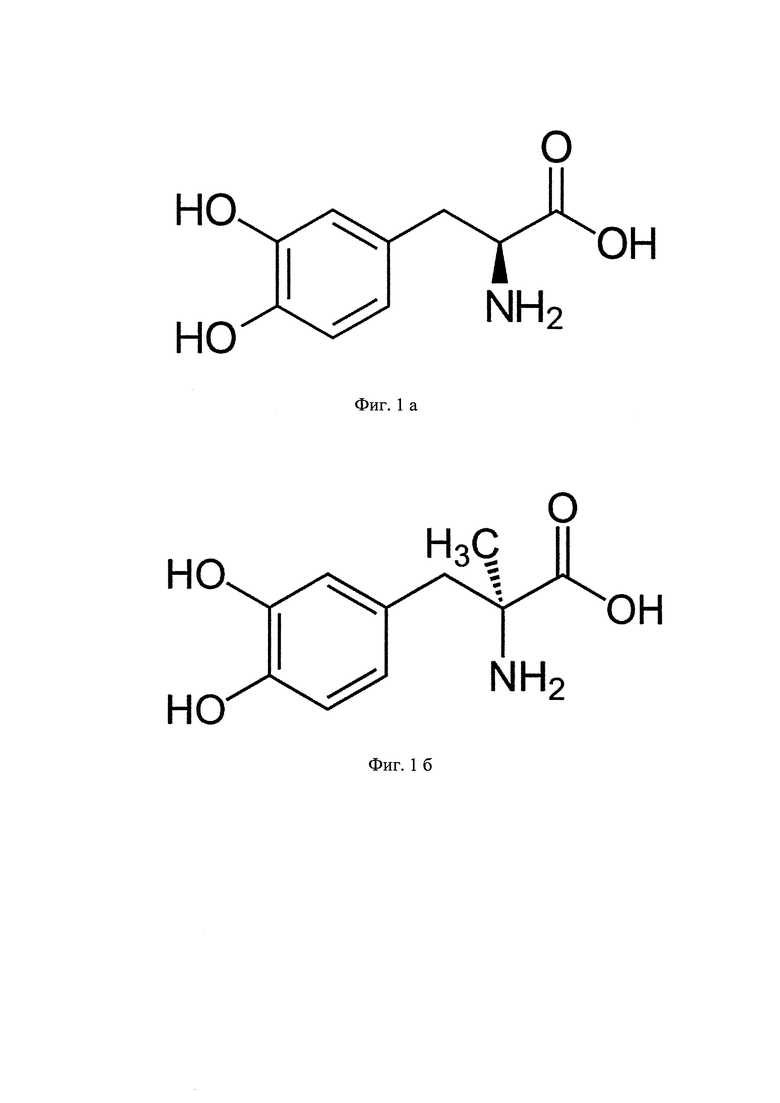
Все растворители имели квалификацию «Для хроматографии», реактивы - не ниже «ч.д.а.». Для приготовления растворов стандартных образцов использовали субстанции стандартов леводопы (производства Sigma-Aldrich, США) и метилдопы (см. фиг. 1а и б). На фиг 1а, б показаны структурные формулы леводопы (1а) и метилдопы (1б) стандарт леводопы (3-гидрокси-L-тирозина) - C9H11NO4. Молекулярная масса: 197.19 г/моль В качестве биологической матрицы использовали плазму крови. Д ля выделения леводопы из плазмы крови и ее очистки используют упрощенный метод твердофазной экстракции на активированном оксиде аллюминия. Для этого к образцу плазмы крови объемом 1 мл добавляют 50 мкл внутреннего стандарта (метилдопы, 100 мкг/мл), 50 мкл стабилизатора (метабисульфита) и 500 мкл щелочного трис-буфера (рН 8,6) с целью повышения коэффициента извлечения леводопы. Затем добавляют 30 мг активированного кислого оксида аллюминия. Образовавшуюся смесь встряхивают в течение 5 минут на вортекс-миксере Heidolph Ultra, а затем центрифугируют на скорости 3500 об/мин для осаждения взвеси оксида аллюминия. Надосадочную жидкость осторожно декантируют и отбрасывают. Затем производят двукратную промывку осадка деионизированной водой. После этого леводопу и внутренний стандарт элюируют с сорбента 150-ю микролитрами 0,5 М уксусной кислоты. Полученный раствор переносят в хроматографическую виалу, которую помещают в автосамплер хроматографа для дальнейшего хромато-масс-спектрометрического анализа. Раствор инжектируют в петлю хроматографа в объеме 10 мкл.
В данных условиях коэффициент экстракции для леводопы составляет 89,98±2,25%, для внутреннего стандарта - 91,86±1,73%.
Для высокоэффективной жидкостной хромато-масс-спектрометрии используют хроматограф - «Finnigan Surveyor LC Pump Plus», детектор - масс-спектрометрический детектор «LCQ Fleet MS» (квадрупольная ионная ловушка) и аналитическую колонку - обращенно-фазную фенилгексильную колонку Luna Phenyl-Hexyl фирмы Phenomenex, США (4,6×250 мм; 5 мкм).
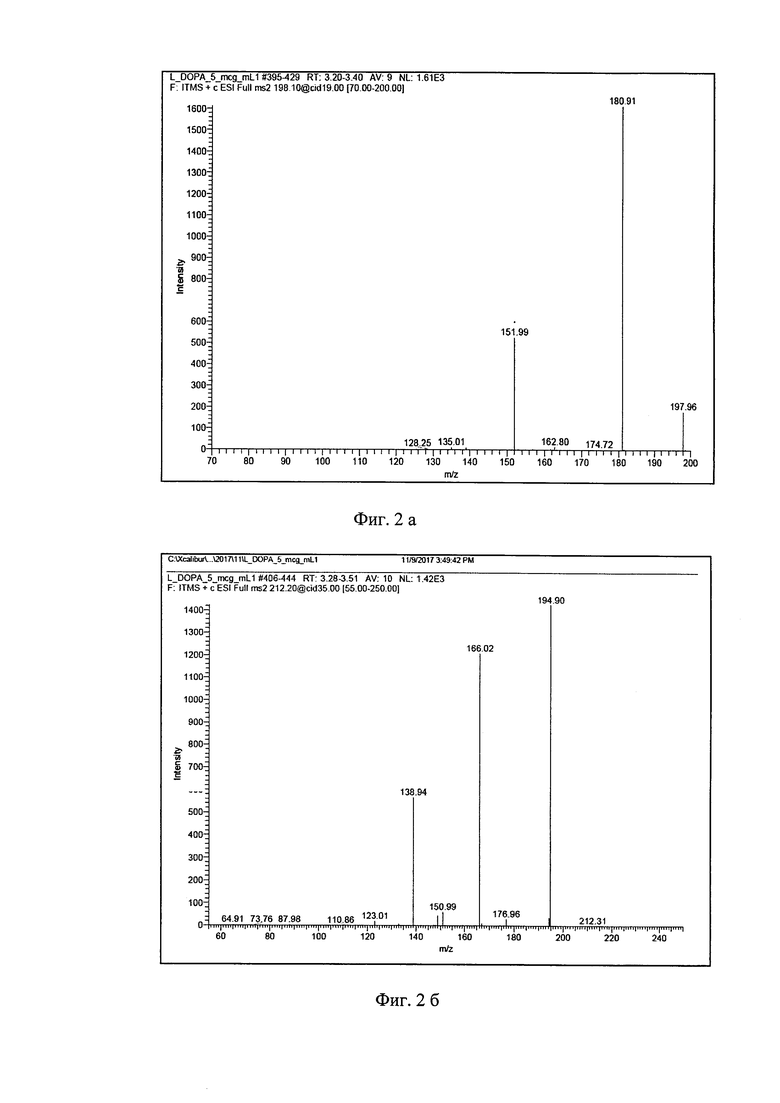
Масс-спектрометрическое детектирование леводопы проводят по суммарному ионному току дочерних ионов в диапазоне масс m/z 70-200 а.е.м., образующихся в результате фрагментации родительского молекулярного иона леводопы с m/z 198.1 при нормализованной энергии соударений 19 eV. Масс-спектр второго порядка для леводопы представлен на фиг. 2а и для метилдопы на фиг. 2б (по вертикали - интенсивность (Intensity), по горизонтали отношение молекулярной массы к заряду (m/z).
Внутренний стандарт детектируют по суммарному ионному току дочерних ионов в диапазоне масс 55-250 а.е.м., образующихся в результате распада молекулярного иона внутреннего стандарта с m/z 212.2. Масс-спектрометр работал в режиме регистрации ионов, положительно заряженных электроспреем (ESI), создаваемым напряжением в 5 кВ. Скорость потока газа-небулайзера (азота): 5 л/мин, давление на распылителе - 100 psi. Температура интерфейса капилляра составляла 350°С, температура нагревателя - 300°С. Амплитуда возбуждения на концевых электродах ловушки 0,1 В. В качестве демпфирующего газа в ионной ловушке используется гелий. Данные обрабатываются с помощью компьютерной хроматографической программы Xcalibur 2.1 w/Foundation 1.0.1. (Thermo Scientific, США).
Разделение анализируемых соединений осуществляют на обращенно-фазной хроматографической колонке Luna Phenyl-Hexyl производства фирмы Phenomenex, США (4,6×250 мм; 5 мкм). Подвижная фаза состоит из двух растворов: 0,1%-го водного раствора муравьиной кислоты - раствор А и ацетонитрила с добавлением 0,1% муравьиной кислоты - раствор Б. Растворы А и Б взяты в процентном соотношении 85А:15Б. Работа проводилась в изократическом режиме элюирования. Скорость потока подвижной фазы составляла 0,8 мл/мин. Объем пробы - 10 мкл. Температура разделения 35°С. Продолжительность хроматографирования - 10 минут. Время удерживания аналита - 3,33±0,05 минут. Время удерживания внутреннего стандарта (метилдопы) - 3,37±0,05 минут.
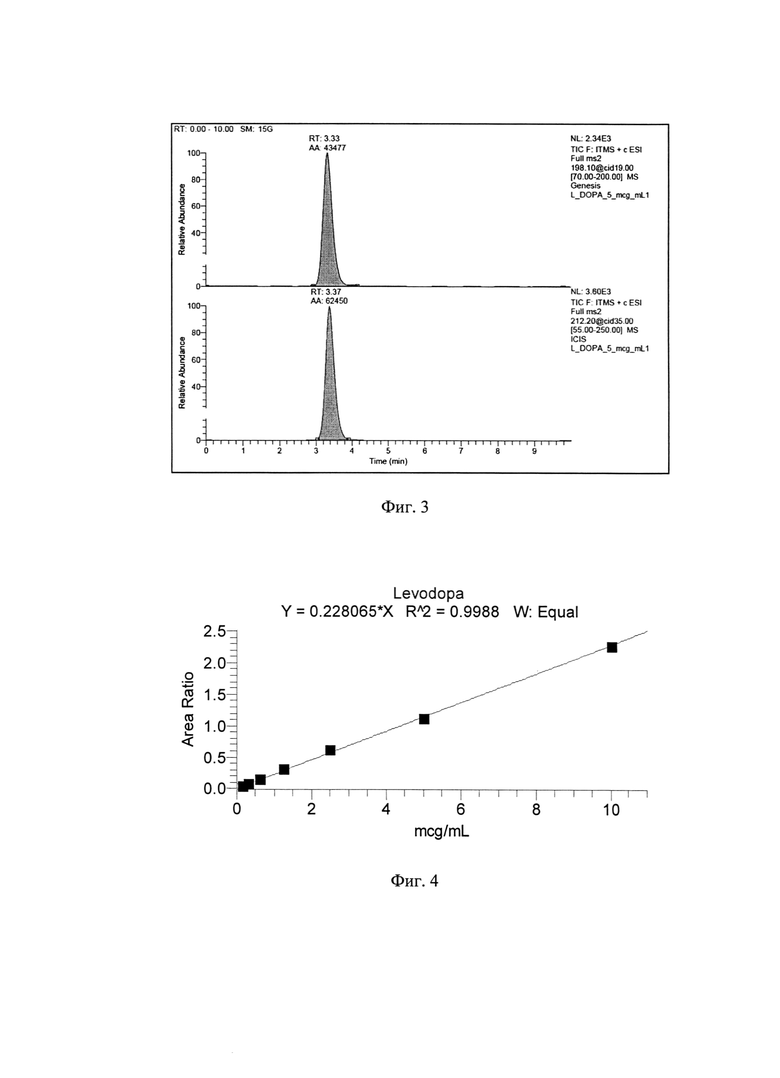
Демонстрационная хроматограмма образца плазмы крови с концентрацией леводопы 5 мкг/мл представлена на фиг. 3, на котором видна хроматограмма экстрагированного образца плазмы крови с концентрацией леводопы 5 мкг/мл, верхний пик - пик аналита, нижний пик - пик внутреннего стандарта (по вертикали - относительный отклик прибора в условных единицах интегрирования (Relative abundance), по горизонтали - время (Time) в минутах).
Для приготовления калибровки готовили маточные растворы стандартов леводопы и внутреннего стандарта в метаноле с концентрациями 1 мг/мл. Раствор леводопы применяли для приготовления растворов рабочих стандартных образцов на плазме крови с концентрациями 0,156 мкг/мл, 0,313 мкг/мл; 0,625 мкг/мл; 1,25 мкг/мл; 2,5 мкг/мл; 5 мкг/мл; 10 мкг/мл. Раствор внутреннего стандарта с концентрацией 1 мг/мл разбавляли в 10 раз для получения рабочего раствора внутреннего стандарта с концентрацией 100 мкг/мл. Калибровочная кривая леводопы в плазме крови показана на фиг. 4.
Количественное определение осуществляли по градуировочной зависимости для леводопы в плазме крови и рассчитывали по формуле: С=4,384714×AR, где С - концентрация леводопы (мкг/мл), AR (Area Ratio) - отношение площадей пиков аналита и внутреннего стандарта. Коэффициент корреляции составил R2=0,9988, что соответствует надлежащей аналитической аппроксимации. Предел количественного определения - 0,156 мкг/мл.
Прецизионность и правильность.
Прецизионность выражалась в виде коэффициента вариации (% C.V.) для каждой серии образцов согласно уравнению:
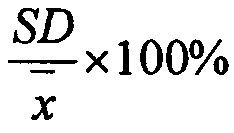 , где
, где
SD - стандартное отклонение серии определений;
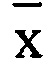 - среднее арифметическое значение полученных концентраций.
- среднее арифметическое значение полученных концентраций.
Правильность измерялась, как процент отклонения (% dev.) от теоретического значения по формуле: 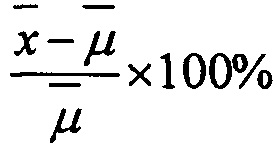 , где
, где
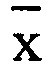 - среднее арифметическое значение полученных концентраций;
- среднее арифметическое значение полученных концентраций;
Для метрологической валидации полученной методики определяют точность в течение рабочего дня. Каждый из образцов, предназначенных для контроля качества, анализировали в течение 1 рабочего дня (6 определений). Результаты представлены в таблице 1.
Относительная ошибка определения леводопы не превышала 10%. Таким образом, представленный метод обладает высокой эффективностью в проведении анализа, не требует использования большого количества химических реактивов. Высокая точность и чувствительность данного метода количественного определения леводопы в плазме крови обеспечивает идентификацию вещества с установленными характеристиками погрешности, что позволяет использовать данную методику для решения задач по изучению как экспериментальной, так и клинической фармакокинетики препарата.
Разработанная технология может быть внедрена в медицинских учреждениях, осуществляющих фармакотерапевтическое лечение пациентов, страдающих болезнью Паркинсона для терапевтического лекарственного мониторинга леводопы.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного определения амантадина в плазме крови | 2017 |
|
RU2650968C1 |
| Способ определения топирамата в плазме крови | 2016 |
|
RU2631613C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КАРНОЗИНА В БИОЛОГИЧЕСКИХ МАТЕРИАЛАХ | 2015 |
|
RU2585115C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОКСИМА ПИНОСТРОБИНА В ПЛАЗМЕ КРОВИ | 2015 |
|
RU2568876C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ МЕТИЛДОПЫ В ПЛАЗМЕ КРОВИ ЧЕЛОВЕКА | 2016 |
|
RU2642593C1 |
| Способ количественного определения салицилатов в плазме крови | 2016 |
|
RU2622996C1 |
| Способ количественного определения ликарбазепина в плазме крови | 2017 |
|
RU2660364C1 |
| Способ количественного определения дисульфирама в биологических средах | 2019 |
|
RU2701524C1 |
| Способ количественного определения антиконвульсантов в плазме крови больных эпилепсией | 2021 |
|
RU2771430C1 |
| Способ определения дабигатрана в сыворотке крови человека | 2018 |
|
RU2683032C1 |
Изобретение относится к области медицины, а именно к клинической фармакологии, и может быть использовано для количественного определения леводопы в плазме крови для решения задач лекарственного мониторинга при лечении пациентов, страдающих болезнью Паркинсона. Способ определения леводопы в плазме крови включает анализ крови на ее наличие путем очистки плазмы крови, проведения хромато-масс-спектрометрии с использованием матрицы в виде плазмы крови с леводопой и внутреннего стандарта метилдопы, при этом разделение продуктов экстракции проводят на обращенно-фазной хроматографической колонке, а в качестве элюента применяют муравьиную кислоту и ацетонитрил, с последующим расчетом концентрации леводопы, при этом очистку проводят методом твердофазной экстракции на активированном оксиде аллюминия, разделение продуктов экстракции проводят на обращенно-фазной хроматографической колонке 4,6×250 мм с фенилгексилом в качестве неподвижной фазы, концентрацию леводопы рассчитывают по формуле: С=4,384714×AR, где С - концентрация леводопы (мкг/мл), AR - отношение площади хроматографического пика леводопы к площади пика внутреннего стандарта. 6 ил., 1 табл.
Способ количественного определения леводопы в плазме крови, включающий анализ крови на его наличие путем очистки плазмы крови, проведения хроматомасс-спектрометрии с использованием матрицы в виде плазмы крови с леводопой и внутреннего стандарта, вещества, близкого по строению молекулы к анализируемому веществу - метилдопы, при этом разделение продуктов экстракции проводят на обращенно-фазной хроматографической колонке, а в качестве элюента применяют муравьиную кислоту и ацетонитрил, с последующим расчетом концентрации леводопы, отличающийся тем, что очистку проводят методом твердофазной экстракции на активированном оксиде аллюминия, образовавшуюся смесь встряхивают в течение 5 минут на вортекс-миксере, а затем центрифугируют на скорости 3500 об/мин для осаждения взвеси оксида аллюминия, надосадочную жидкость осторожно декантируют и отбрасывают, а осадок двукратно промывают деионизированной водой, а леводопу и метилдопу элюируют с оксида аллюминия 0,1 молярным водным раствором уксусной кислоты, а разделение продуктов экстракции проводят на обращенно-фазной хроматографической колонке 4,6×250 мм с фенилгексилом в качестве неподвижной фазы, содержащим гексильный линкерный фрагмент, соединяющий фенильную группу с поверхностью силикагеля, при этом в качестве элюента применяют 0,1% водный раствор муравьиной кислоты - раствор А и ацетонитрил с добавлением 0,1% муравьиной кислоты - раствор Б, взятые в процентном соотношении раствора А к раствору Б 85:15 соответственно, с температурой разделения 35°С и скоростью подачи элюента 0,8 мл/мин, детектирование леводопы проводят по суммарному ионному току дочерних ионов в диапазоне от 70 до 200 а.е.м., образующихся в результате фрагментации родительского молекулярного иона леводопы с m/z 198.1, а его концентрацию рассчитывают по формуле: С=4,384714×AR, где С - концентрация леводопы (мкг/мл), AR - отношение площади хроматографического пика леводопы к площади пика внутреннего стандарта.
| MU Chun-Lei et al | |||
| Simultaneous and Sensitive Determination of Levodopa and Carbidopa in Pharmaceutical Formulation and Human Serum by High Performance Liquid Chromatography with On-Line Gold Nanoparticles-Catalyzed Luminol Chemiluminescence Detection / Chinese Journal of Analytical Chemistry, June 2017, 45(6): e1726-e1733 | |||
| V.Junnotula et al | |||
| Development and validation of a simple and sensitive method for quantification of levodopa and carbidopa in rat and monkey plasma using derivatization and UPLC-MS/MS / Journal of Chromatography B, 2013, vol | |||
| ЭЛЕКТРИЧЕСКИЙ ТРЕХФАЗНЫЙ КАБЕЛЬ | 1924 |
|
SU926A1 |
| Shu-Fang Li et al | |||
| Quantitative analysis of levodopa, carbidopa and methyldopa in human plasma samples using HPLC-DAD combined with second-order calibration based on alternating trilinear decomposition algorithm / Talanta, 2010, vol | |||
| Горный компас | 0 |
|
SU81A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| М.Р | |||
| Нодель и др | |||
| ЛЕВОДОПА: ПРОШЛОЕ, НАСТОЯЩЕЕ И БУДУЩЕЕ / КЛИНИЦИСТ, 2008, N 1, стр | |||
| Способ окисления боковых цепей ароматических углеводородов и их производных в кислоты и альдегиды | 1921 |
|
SU58A1 |

