Изобретение относится к области медицины, а именно к клинической фармакологии, и может быть использовано для количественного определения ликарбазепина в плазме крови для решения задач лекарственного мониторинга антиконвульсанта второго поколения при лечении парциальной эпилепсии.
Ликарбазепин (10,11-дигидро-10-гидроксикарбамазепин) является основным фармакологически активным метаболитом противосудорожного пролекарства окскарбазепина, а S-энантиомер ликарбазепина является действующим веществом антиконвульсанта эсликарбазепина ацетата. Окскарбазепин является широко используемым антиконвульантом. В настоящее время одобрено его применение как в качестве монотерапии, так и в виде адъюнктивного лечения парциальной эпилепсии. После перорального введения, окскарбазепин быстро и интенсивно метаболизируется цитозольной альдокетодеструктазой в печени до моногидроксикарбазепина (МГД) (Faught Е., Limdi N., Shorvon S., Perucca E., Engrl J. Jr. Oxcarbazepine. The treatment of epilepsy. Chichester: Wiley-Blackwell; 2009; 575-584), который представляет из себя энантиомерную смесь эсликарбазепина (известного как (S)-ликарбазепин или (S)-MHD) и (R)-ликарбазепин (известного как (R)-MHD).
Данные препараты пользуются все большей популярностью у пациентов, поскольку они близки по эффективности к карбамазепину, но в отличие от последнего, они не метаболизируются до токсичного карбамазепин-10,11-эпоксида и не являются индукторами микросомального метаболизма, что обуславливает закономерное уменьшение числа нежелательных лекарственных реакций и лучшую их переносимость (Bialer M1, Soares-da-Silva P. Pharmacokinetics and drug interactions of eslicarbazepine acetate. Epilepsia. 2012 Jun; 53(6):935-46). В этой связи высокую актуальность приобретает разработка нового метода количественного определения 10,11-дигидро-10-гидроксикарбамазепина, поскольку данный метод является универсальным способом фармакокинетического сопровождения лечения как пациентов, принимающих ликарбазепин, так и эсликарбазепина ацетат и окскарбазепин.
Известен способ одновременного определения окскарбазепина и его основных метаболитов методом высокоэффективной жидкостной хроматографии со спектрофотометрическим детектированием на длине волны 237 нм (V. Kimiskidis, М. Spanakis, I. Niopas. Development and validation of a high performance liquid chromatographic method for the determination of oxcarbazepine and its main metabolites in human plasma and cerebrospinal fluid and its application to pharmacokinetic study Journal of Pharmaceutical and Biomedical Analysis 43 (2007) 763-768). Недостатками данного метода является то, что спектрофотометрическое детектирование имеет низкую избирательность в отношении исследуемых соединений, ввиду чего нередко происходит интерференция хроматографических пиков коэкстрактивных веществ биологической матрицы с аналитами, в том числе и с образованием так называемых «критических пар», что делает данный метод недостаточно селективным и ограничивает сферу его применения. Данные недостатки устраняются применением масс-спектрометрического детектирования.
Известен способ количественного определения ликарбамазепина путем газовой хроматографии. При этом проводят жидкостно-жидкостную экстракцию из плазмы крови, дериватизацию продуктов экстракции с последующей газовой хромато-масс-спектрометрией продуктов дериватизации и расчетом концентрации дериватизированного ликарбазепина (G.E. von Unruht and W.D. Paar Gas Chromatsgraphic / Mass Spectrometric Assays for Oxcarbazepine and Its Main Metabolites, 10-Hydroxy-Carbazepine and Carbazepine-10,11-trans-diol. BIOMEDICAL AND ENVIRONMENTAL MASS SPECTROMETRY, 1986, Vol. 13, 651-656). Недостатками способа является применение сложной дериватизации с использованием силилирующего агента MSTFA. Наличие данной стадии дериватизации негативным образом сказывается на робастности метода, поскольку дериватизированное производное ликарбазепина стабильно при комнатной температуре только в течение несколько часов. Кроме того, сами авторы метода указывают, что если образец, дериватизированный 30 мкл MSTFA, вводится неоднократно, то раствор с образцом может частично затвердевать в хроматографической виале, особенно если влажность в помещении высока, что также негативно сказывается на рабочих характеристиках указанной методики. Данный источник информации рассмотрен в качестве ближайшего аналога.
Технический результат заключается в создании способа количественного определения ликарбазепина с высокой воспроизводимостью и точностью, и наиболее адаптированного для решения задач экспериментальной и клинической фармакокинетики.
Технический результат достигается тем, что создан способ количественного определения ликарбазепина в плазме крови, включающий анализ крови на его наличие путем жидкостно-жидкостной экстракции из плазмы крови с последующей газовой хромато-масс-спектрометрией продуктов экстракции и расчетом концентрации ликарбазепина, при этом экстракцию анализируемого соединения и внутреннего стандарта - нордазепама, проводят с концентрированным раствором бикарбоната натрия и органическим экстрагентом этилацетатом, смесь встряхивают в течение 5 мин на многопозиционном вортекс-миксере, а затем центрифугируют на скорости 3500 об/мин для разделения органического и гидрофильного слоя, затем верхний органический слой декантируют и упаривают в центрифужном вакуумном концентраторе при температуре 60°C, полученный сухой остаток растворяют в 500 мкл метанола, а газовую хромато-масс-спектрометрию данного раствора продуктов экстракции, включающую анализируемое соединение и внутренний стандарт, осуществляют на неполярной капиллярной колонке HP-5MS, при температуре инжектора 350°C и начальной температуре печи хроматографа - 170°C, затем колонку с анализируемыми продуктами термостатируют в течение 1 мин, после чего осуществляют ее нагрев с температурным градиентом 20°C/мин до 210°C, вновь проводят термостатирование на 210°C в течение 4 мин и осуществляют регистрацию масс-спектров анализируемых продуктов в следующем режиме: энергии ионизации 70 эВ, температуры источника ионов - 230°C, ионизации типа «электронный удар» с регистрацией положительных ионов и сканированием в режиме мониторинга выбранных ионов с m/z 193.0, 210.0 и 254.0 для ликарбазепина и 269.1 и 242.2 для внутреннего стандарта - нордазепама со скоростью 2 скан/с, с последующим расчетом концентрации ликарбазепина по формуле Y=6,710⋅Х+0,7567, где Y - концентрация ликарбазепина (мкг/мл), X - отношение площади хроматографического пика ликарбрзепина к площади пика внутреннего стандарта - нордазепама.
Способ осуществляется следующим образом.
Для извлечения ликарбазепина (10,11-дигидро-10-гидроксикарбамазепина) из плазмы применяют метод жидкостно-жидкостной экстракции с последующим концентрированием. К образцу плазмы крови объемом 500 мкл добавляют 50 мкл внутреннего стандарта (нордазепам, 10 мкг/мл) и 500 мкл концентрированного раствора бикарбоната натрия с целью повышения коэффициента извлечения анализируемых веществ, а затем приливают 5 мл органического экстрагента (этилацетата). Образовавшуюся смесь встряхивают в течение 5 мин на многопозиционном вортекс-миксере типа «Heidolph Ultra», а затем центрифугируют на скорости 3500 об/мин для разделения органического и гидрофильного слоя. После разделения, верхний органический слой осторожно декантируют и упаривают в центрифужном вакуумном концентраторе типа «Eppendorf Concentrator 5301» под вакуумом при температуре 60°C. Полученный сухой остаток растворяют в 500 мкл метанола. Образец переносят в хроматографическую виалу, которую затем помещают в автосемплер газового хроматографа для дальнейшего хромато-масс-спектрометрического анализа. Раствор инжектируют в хроматограф в объеме 1 мкл. Использовался хроматограф «Agilent 6850 Series II Network GC System» оснащенный моноквадрупольным масс-спектрометрическим детектором «Agilent 5975В inert XL MSD» (США). В работе возможно использование любого коммерчески доступного хроматомасс-спектрометра аналогичного класса.
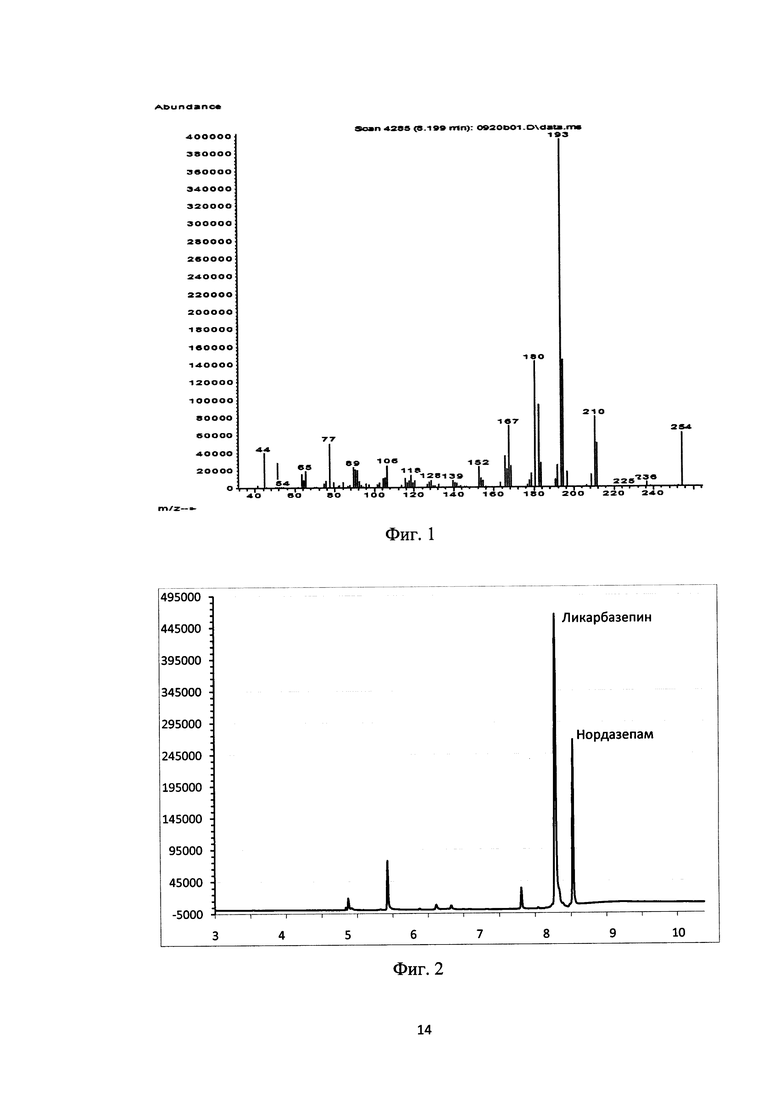
Для хроматографического разделения используют неполярную капиллярную колонку HP-5MS или любую другую капиллярную колонку с аналогичными характеристиками (привитая фаза - (5%-фенил) метилполисилокеан, длина 30 м, внутренний диаметр 0,25 мм, толщина фазы 0,25 мкм, газ-носитель - гелий со степенью чистоты не ниже 6.0, давление газа-носителя 5 psi). Для разделения смеси веществ применяют следующий режим работы хроматографа: температура инжектора составляла 350°C, начальная температура печи хроматографа - 170°C. Термостатирование проводят в течение 1 мин, после чего осуществляют нагрев с температурным градиентом 20°C/мин до 210°C. Затем проводят повторное термостатирование на 210°C в течение 4 мин, и повторный нагрев до 280°C с температурным градиентом 25°C. На темпертатурной точке 280°C печь термостатируется в течение 2-х мин. Общее время анализа составляет в данных условиях 11,8 мин. Выбирают следующий режим регистрации масс-спектров: энергия ионизации 70 эВ, температура источника ионов - 230°C, ионизация типа «электронный удар», регистрация положительных ионов, сканирование в диапазоне 50-400 Да со скоростью 2 скан/с. Объем вводимой пробы - 1 мкл, пробы вводили в инжектор в режиме "Без деления потока" (Splitless). Для разработки метода мониторинга выбранных ионов (SIM-метода) снимают масс-спектры целевых соединений в сканирующем режиме в диапазоне 50-400 Да. На фиг. 1 представлен масс-спектр ликарбазипина (10,11-дигидро-10-гидроксикарбамазепина).
Для регистрации положительных ионов и сканирования в режиме мониторинга выбирают следующие характеристические ионы 10,11-дигидро-10-гидроксикарбамазепина: ион с m/z 193.0 (основной ион), ион с m/z 210.0 и ион с m/z 254.0 (вспомогательные ионы), для внутреннего стандарта-нордазепама. ион с m/z 269.1 (основной ион) и ион с m/z 242.2 (вспомогательный ион). В указанных условиях время удерживания анализируемого соединения ликарбазепина и внутреннего стандарта - нордазепама составляло 8,14 и 8, 73 соответственно.
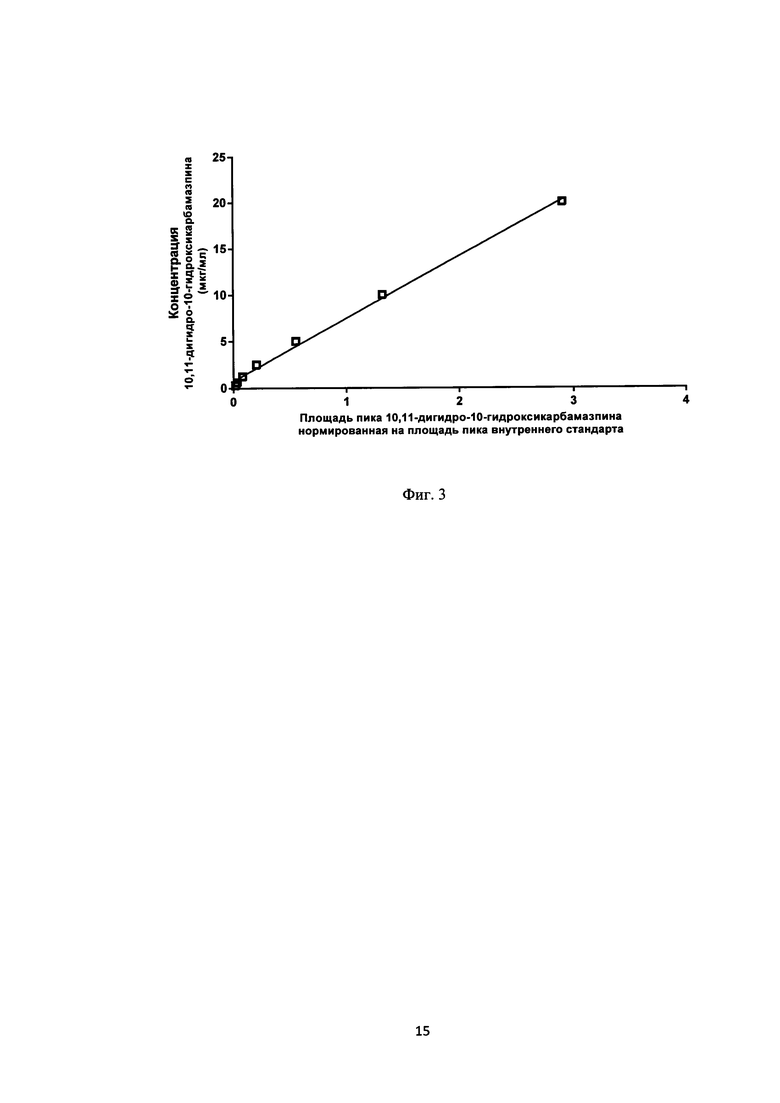
На каждой хроматограмме измеряют площадь пика основного характеристического иона анализируемого соединения и "внутреннего стандарта". На фиг. 2 показана хроматограмма ликарбазепина с внутренним стандартом (нордазепамом). Концентрация целевого соединения - 15 мкг/мл.
Записывают хроматограммы в виде файлов данных. Для основного характеристического и дополнительных подтверждающих ионов получают хроматограмму по реконструированному полному ионному току. Качественную идентификацию осуществляют по времени удерживания и относительной интенсивности одного основного и одного/двух дополнительных подтверждающих ионов (m/z). Количественное определение идентифицированного соединения выполняют методом "внутренней градуировки с добавкой известного количества постороннего вещества", называемого "внутренним стандартом", относительно которого предварительно определяют градуировочный поправочный коэффициент (F), показывающий во сколько раз отклик масс-селективного детектора (площадь хроматографического пика, соответствующая основному для данного конкретного соединения иону m/z) на единицу массы вещества отличается от отклика масс-селективного детектора на единицу массы "внутреннего стандарта". В качестве внутреннего стандарта используют нордазепам, как термостабильное низкоплавкое вещество, близкое по своим экстракционным характеристикам к анализируемому соединению - ликарбазепину.
Для приготовления калибровки готовили маточные растворы стандартов ликарбазепина (10,11-дигидро-10-гидроксикарбамазепина) и внутреннего стандарта нордазепама в метаноле с концентрацией 1 мг/мл. Из маточных растворов в метаноле путем каскадного разбавления изготавливают стартовые растворы для получения калибровочных образцов в концентрационном диапазоне - 3,13-200 мкг/мл. Калибровочные стандарты приготавливают в донорской интактной плазме крови для получения конечных концентраций анализируемых соединений 0,313; 0,625; 1,25; 2,5; 5; 10 и 20 мкг/мл. Холостые образцы приготавливают в плазме крови без аналита и внутреннего стандарта. Нулевые образцы приготавливают в плазме крови без аналита, но с внутренним стандартом. Линейные калибровочные кривые анализируемого соединения представлены на фиг. 3.
Градуировочная зависимость для ликарбазепина в плазме крови описывалась формулой: Y=6,710⋅Х+0,7567, где Y - концентрация ликарбазепина (мкг/мл), X - отношение площади хроматографического пика ликарбрзепина к площади пика внутреннего стандарта - нордазепама.
При вышеописанных параметрах метода калибровочные характеристики должны приближаться к следующим значениям, указанным в таблице 1.


Нижний концентрационный предел количественного определения данного метода составил 313 нг/мл.
Для метрологической валидации полученной методики определяли правильность и прецизионность методики в течение рабочего дня.
Прецизионность (precision), выражается в виде коэффициента вариации (% C.V.) для каждой серии образцов согласно уравнению:
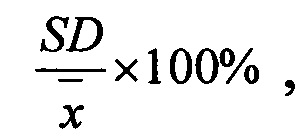
где
SD - стандартное отклонение серии определений;
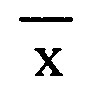 - среднее арифметическое значение полученных концентраций.
- среднее арифметическое значение полученных концентраций.
Правильность (accuracy) аналитической методики характеризуется близостью полученных с помощью нее значений к номинальным концентрациям анализируемого соединения.
Правильность измеряется, как процент отклонения (% dev.) от теоретического значения по формуле 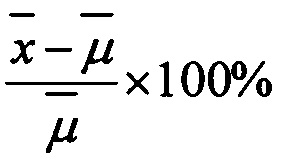 ,
,
где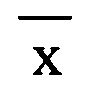 - среднее арифметическое значение полученных концентраций,
- среднее арифметическое значение полученных концентраций,
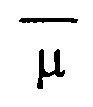 - отклонение измеряемой величины от среднего значения
- отклонение измеряемой величины от среднего значения
Каждый из образцов, предназначенных для контроля качества, анализировали в течение 1 рабочего дня (6 определений).
Ниже представлены эмпирические параметры разработанного метода для ликарбазепина:
Результаты представлены в таблице 2.
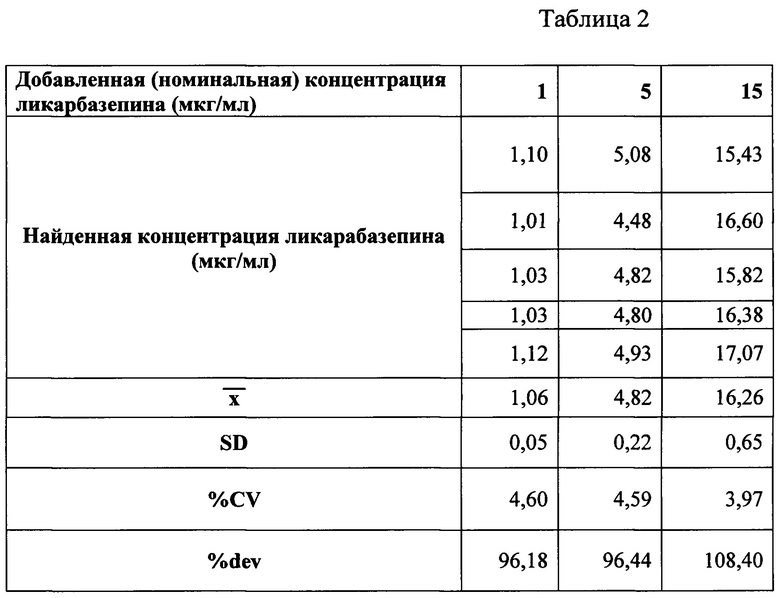
Таким образом, процент ошибки метода не превышал 10%. Разработанный газовый хроматомасс-спектрометрический метод количественного определения ликарбазепина (10,11-дигидро-10-гидроксикарбамазепина) имеет чувствительность до 313 нг/мл с верхним пределом количественного определения до 20 мкг/мл. Следовательно, заявленный способ позволяет с надлежащей воспроизводимостью и точностью производить количественное определение ликарбазепина.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного определения антиконвульсантов в плазме крови больных эпилепсией | 2021 |
|
RU2771430C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОКСИМА ПИНОСТРОБИНА В ПЛАЗМЕ КРОВИ | 2015 |
|
RU2568876C1 |
| Способ количественного определения амантадина в плазме крови | 2017 |
|
RU2650968C1 |
| Способ определения топирамата в плазме крови | 2016 |
|
RU2631613C1 |
| Способ количественного определения салицилатов в плазме крови | 2016 |
|
RU2622996C1 |
| Способ количественного определения леводопы в плазме крови | 2017 |
|
RU2665164C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КАРНОЗИНА В БИОЛОГИЧЕСКИХ МАТЕРИАЛАХ | 2015 |
|
RU2585115C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЭНДОГЕННЫХ СТЕРОИДОВ В ПЛАЗМЕ КРОВИ ЧЕЛОВЕКА | 2010 |
|
RU2451292C2 |
| Способ контроля содержания противотуберкулёзных препаратов основного ряда и их токсичных метаболитов в плазме крови | 2018 |
|
RU2702998C1 |
| Способ количественного определения фурана и метилфурана в крови методом газохроматографического анализа с масс-селективным детектированием | 2023 |
|
RU2813866C1 |
Изобретение относится к области медицины, а именно к клинической фармакологии, и может быть использовано для количественного определения ликарбазепина в плазме крови для решения задач лекарственного мониторинга антиконвульсанта второго поколения при лечении парциальной эпилепсии. Способ количественного определения ликарбазепина в плазме крови путем проведения жидкостно-жидкостной экстракции ликарбазепина из плазмы крови с последующей газовой хромато-масс-спектрометрией продуктов экстракции и расчетом концентрации ликарбазепина включает экстракцию анализируемого соединения и внутреннего стандарта – нордазепама, газовую хромато-масс-спектрометрию раствора продуктов экстракции, включающего анализируемое соединение и внутренний стандарт, с последующим расчетом концентрации ликарбазепина по формуле: Y=6,710⋅Х+0,7567, где Y - концентрация ликарбазепина (мкг/мл), X - отношение площади хроматографического пика ликарбазепина к площади пика внутреннего стандарта - нордазепама. 2 табл., 3 ил.
Способ количественного определения ликарбазепина в плазме крови, включающий анализ крови на его наличие путем жидкостно-жидкостной экстракции из плазмы крови с последующей газовой хромато-масс-спектрометрией продуктов экстракции и расчетом концентрации ликарбазепина, отличающийся тем, что экстракцию анализируемого соединения и внутреннего стандарта - нордазепама проводят с концентрированным раствором бикарбоната натрия и органическим экстрагентом этилацетатом, смесь встряхивают в течение 5 мин на многопозиционном вортекс-миксере, а затем центрифугируют на скорости 3500 об/мин для разделения органического и гидрофильного слоя, затем верхний органический слой декантируют и упаривают в центрифужном вакуумном концентраторе при температуре 60°С, полученный сухой остаток растворяют в 500 мкл метанола, а газовую хромато-масс-спектрометрию данного раствора продуктов экстракции, включающую анализируемое соединение и внутренний стандарт, осуществляют на неполярной капиллярной колонке HP-5MS при температуре инжектора 350°С и начальной температуре печи хроматографа - 170°С, затем колонку с анализируемыми продуктами термостатируют в течение 1 мин, после чего осуществляют ее нагрев с температурным градиентом 20°С/мин до 210°С, вновь проводят термостатирование на 210°С в течение 4 мин и осуществляют регистрацию масс-спектров анализируемых продуктов в следующем режиме: энергии ионизации 70 эВ, температуры источника ионов - 230°С, ионизации типа «электронный удар» с регистрацией положительных ионов и сканированием в режиме мониторинга выбранных ионов с m/z 193, 210 и 254 для ликарбазепина и 269.1 и 242.2 для внутреннего стандарта - нордазепама со скоростью 2 скан/с, с последующим расчетом концентрации ликарбазепина по формуле Y=6,710⋅Х+0,7567, где Y - концентрация ликарбазепина (мкг/мл), X - отношение площади хроматографического пика ликарбрзепина к площади пика внутреннего стандарта - нордазепама.
| KASHIF UL HAQ et al | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| G.Paglia et al | |||
| Development and validation of a LC/MS/MS method for simultaneous quantification of oxcarbazepine and its main metabolites in human serum / J Chromatogr B Analyt Technol Biomed Life Sci., 2007, 860(2), pages 153-159 (abstract) | |||
| Способ определения карбамазепина | 1982 |
|
SU1120240A1 |

